JP2017155195A - パラミロン水性分散液及び細胞増殖促進剤 - Google Patents
パラミロン水性分散液及び細胞増殖促進剤 Download PDFInfo
- Publication number
- JP2017155195A JP2017155195A JP2016042619A JP2016042619A JP2017155195A JP 2017155195 A JP2017155195 A JP 2017155195A JP 2016042619 A JP2016042619 A JP 2016042619A JP 2016042619 A JP2016042619 A JP 2016042619A JP 2017155195 A JP2017155195 A JP 2017155195A
- Authority
- JP
- Japan
- Prior art keywords
- paramylon
- cellulose
- dispersion
- nanofibers
- aqueous
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images


Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Medicinal Preparation (AREA)
- Cosmetics (AREA)
Abstract
Description
ユーグレナから抽出したパラミロンは、水や熱水には不溶性である。従って、生体に摂取・投与されたときにより高い効果を奏するよう、アルカリ処理,化学修飾,架橋結合等の処理を施した処理パラミロンが、種々提案されている(例えば特許文献1,2)。
また、特許文献2は、パラミロンをアルカリ処理後酸で中和処理をすることにより、パラミロンよりも結晶化度の低いアモルファスパラミロンが得られ、このアモルファスパラミロンが、パラミロンよりも高いアレルギー抑制効果を備えることが記載されている。
処理パラミロンの種類が増えれば、改善できる体質や治療・予防できる疾患の種類も増えることが予想される。生体組織において、疾患の治療・予防や体質改善等の新たな効果を発現できる新規な処理パラミロンの開発が望まれていた。
本発明の他の目的は、パラミロンを水性溶媒に分散する新規な方法を提供するパラミロン水性分散液及び細胞増殖促進剤を提供することにある。
前記目的を達成するために、本発明のパラミロン水性分散液又は細胞増殖促進剤は、少なくとも下記の(A)〜(C)成分を含有することを特徴とするパラミロン水性分散液により解決される。
(A)パラミロン。
(B)アニオン変性セルロースナノファイバー、セルロースナノファイバー、キチンナノファイバー、およびキトサンナノファイバーから選ばれる一種以上の繊維。
(C)水。
これらの成分を含有するパラミロン水性分散液は、分散性がよく、1週間放置しても分散度が低下しない良好な分散安定性を有する。また、本発明のパラミロン水性分散液は、ヒトの皮膚に適用した場合でもべたつきがない。従って、皮膚に良好に適用でき、皮膚創傷治癒促進剤,皮膚外用剤,化粧品,皮膚の老化防止剤又は皮膚の老化改善剤として皮膚に適用した場合も、べたつきなく良好な使用感が得られる。
皮膚の線維芽細胞の増殖促進効果と、皮膚に適用したときのべたつきのない優れた使用感とを併せて発揮するため、皮膚創傷治癒促進剤,皮膚外用剤,化粧品,皮膚の老化防止剤及び皮膚の老化改善剤として利用できる。
前記(B)成分の濃度が、6.0×10−5重量%〜5.0重量%であるとよい。
前記(B)成分がアニオン変性セルロースナノファイバーであるとよい。
前記細胞増殖促進剤は、線維芽細胞の増殖促進剤として用いられてもよい。皮膚創傷治癒促進剤,皮膚外用剤,化粧品,皮膚の老化防止剤又は皮膚の老化改善剤として用いられてもよい。
これらの成分を含有するパラミロン水性分散液は、分散性がよく、1週間放置しても分散度が低下しない良好な分散安定性を有する。また、本発明のパラミロン水性分散液は、ヒトの皮膚に適用した場合でもべたつきがない。従って、皮膚に良好に適用でき、皮膚創傷治癒促進剤,皮膚外用剤,化粧品,皮膚の老化防止剤又は皮膚の老化改善剤として皮膚に適用した場合も、べたつきなく良好な使用感が得られる。
皮膚の線維芽細胞の増殖促進効果と、皮膚に適用したときのべたつきのない優れた使用感とを併せて発揮するため、皮膚創傷治癒促進剤,皮膚外用剤,化粧品,皮膚の老化防止剤及び皮膚の老化改善剤として利用できる。
本実施形態のパラミロン水性分散液は、少なくとも下記の(A)〜(C)成分を含有する。
(A)パラミロン。
(B)アニオン変性セルロースナノファイバー、セルロースナノファイバー、キチンナノファイバー、およびキトサンナノファイバーから選ばれる一種以上の繊維。
(C)水。
以下、(A)〜(C)の各成分について説明する。
パラミロン(Paramylon)は、約700個のグルコースがβ−1,3−結合により重合した高分子体(β−1,3−グルカン)であり、ユーグレナ属が含有する貯蔵多糖である。パラミロン粒子は、扁平な回転楕円体粒子であり、β−1,3−グルカン鎖がらせん状に絡まりあって形成されている。
パラミロンは、グルコースのみからなり、E.gracilis Zの野生株と葉緑体欠損株SM−ZKから得られたパラミロンの平均重合度は、グルコース単位で約700である。
パラミロンは、水,熱水には不溶性であるが、希アルカリ,濃い酸,ジメチルスルホキシド,ホルムアルデヒド,ギ酸に溶ける。
パラミロンの平均密度は、E.gracilis Zでは、1.53、E.gracilis var.bacillaris SM−L1では、1.63である。
なお、パラミロン((株)ユーグレナ製)の粒度分布は、レーザ回折/散乱式粒度分布測定装置で測定したときのメジアン径が、1.5〜2.5μmである。
例えば、パラミロン粒子は、(1)任意の適切な培地中でのユーグレナ細胞の培養;(2)当該培地からのユーグレナ細胞の分離;(3)分離されたユーグレナ細胞からのパラミロンの単離;(4)単離されたパラミロンの精製;および必要に応じて(5)冷却およびその後の凍結乾燥により得ることができる。
パラミロンの単離は、例えば、大部分が生物分解される種類の非イオン性または陰イオン性の界面活性剤を用いて行われ得る。パラミロンの精製は、実質的には単離と同時に行われ得る。
成分(B)は、アニオン変性セルロースナノファイバー、セルロースナノファイバー、キチンナノファイバー、およびキトサンナノファイバーから選ばれる一種以上の繊維である。
これらの繊維は、単独で(B)成分として用いてもよいし、二種以上を組み合わせて用いてもよい。以下、これらの繊維について説明する。
(B)成分として、アニオン変性セルロースナノファイバーを用いることができる。以下、アニオン変性セルロースナノファイバーについて説明する。
セルロース系原料は、木材由来のクラフトパルプまたはサルファイトパルプ、それらを高圧ホモジナイザーやミル等で粉砕した粉末セルロース、あるいはそれらを酸加水分解などの化学処理により精製した微結晶セルロース粉末等を含む。この他に、ケナフ、麻、イネ、バカス、竹等の植物由来のセルロース系原料も使用できる。
また、上記したセルロース系原料を高速回転式、コロイドミル式、高圧式、ロールミル式、超音波式などの分散装置、湿式の高圧または超高圧ホモジナイザーなどで微細化したものをセルロース系原料として使用することもできる。
上記のセルロース原料に、下記に例示する公知の方法を用いてアニオン性の基を導入(アニオン変性)することで、アニオン変性セルロースを得ることができる。
上記のセルロース系原料を発底原料にし、3〜20質量倍の低級アルコール、具体的にはメタノール、エタノール、N−プロピルアルコール、イソプロピルアルコール、N−ブタノール、イソブタノール、第3級ブタノール等の単独、又は2種以上の混合物と水の混合媒体を溶媒として使用する。混合媒体における低級アルコールの混合割合は、60〜95質量%である。発底原料のグルコース残基当たり0.5〜20倍モルの水酸化アルカリ金属、具体的には水酸化ナトリウム又は水酸化カリウムをマーセル化剤として使用し、発底原料、溶媒、及びマーセル化剤を混合し、反応温度を0〜70℃、かつ反応時間を15分〜8時間としてマーセル化処理を行う。その後、モノクロロ酢酸又はモノクロロ酢酸ナトリウム(カルボキシメチル化剤)をグルコース残基当たり0.05〜10.0倍モル添加し、反応温度30〜90℃、かつ反応時間30分〜10時間としてエーテル化反応を行うことにより、カルボキシメチル基を導入したセルロースを得ることができる。
試料約2.0gを精秤して、300ml共栓三角フラスコに入れる。硝酸メタノール(無水メタノール1Lに特級濃硝酸100mlを加えた液)100mlを加え、3時間振盪して、カルボキシメチルセルロースナトリウム(Na−CMC)をカルボキシメチルセルロース(H−CMC)にする。その絶乾H−CMC1.5〜2.0gを精秤し、300ml共栓三角フラスコに入れる。80%メタノール15mlでH−CMCを湿潤し、0.1NのNaOH100mlを加えて室温で3時間振盪する。指示薬としてフェノールフタレインを用いて、0.1NのH2SO4で過剰のNaOHを逆滴定する。次式:
[{100×F’−(0.1NのH2SO4(ml))×F}/(H−CMCの絶乾質量(g))]×0.1=A
カルボキシルメチル置換度=0.162A/(1−0.058A)
A:1gのH−CMCを中和するのに必要な1NのNaOHの量(ml)
F’:0.1NのH2SO4のファクター
F:0.1NのNaOHのファクター
を用いてカルボキシルメチル置換度を算出する。
上記のセルロース原料を、N−オキシル化合物、及び、臭化物、ヨウ化物若しくはこれらの混合物からなる群から選択される化合物の存在下で酸化剤を用いて水中で酸化することにより、カルボキシル基を導入したセルロース(以下、「酸化セルロース」とも呼ぶ。)を得ることができる。
酸化セルロースのカルボキシル基量が、セルロースの絶乾質量に対して、0.2〜2.0mmol/g以上、好ましくは1.0mmol/g〜2.0mmol/gとなるように条件を設定するとよい。
カルボキシル基量〔mmol/g酸化セルロース又はセルロースナノファイバー〕=a〔ml〕×0.05/酸化セルロース又はセルロースナノファイバー質量〔g〕。
前記で得たアニオン変性セルロースを含む分散液を調製し、分散液中でアニオン変性セルロースを解繊してナノファイバー化する。「ナノファイバー化する」とは、セルロースを、平均繊維幅1〜1000nm、又は2〜300nm、好ましくは2〜150nm、平均繊維長50〜5000nm、好ましくは0.1〜5μmのセルロースファイバーへと加工することを意味する。分散液とは前記アニオン変性セルロースが水等の分散媒に分散している液である。
アルカリ加水分解により、アニオン変性セルロースをナノファイバー化する際に要するエネルギーを低減させることができる。
上記アニオン変性セルロースの中でも、TEMPO酸化セルロースを、好適に用いることができる。
TEMPO酸化セルロースとは、パルプを、水溶性の安定ニトロキシルラジカルであるTEMPO(2,2,6,6−テトラメチルピペリジニル−1−オキシルの略)触媒酸化したセルロースシングルナノファイバーである。
TEMPO酸化セルロースは、例えば、(1)酸化反応工程、(2)精製工程、(3)分散工程(微細化処理工程)等を行うことにより得ることができる。以下、各工程を順に説明し、最後に、(4)他の添加剤の添加について説明する。
天然セルロースと、TEMPOとを水(分散媒体)に分散させた後、共酸化剤を添加して、反応を開始する。反応中は0.5Mの水酸化ナトリウム水溶液を滴下してpHを10〜11に保ち、pHに変化が見られなくなった時点で反応終了と見なす。ここで、共酸化剤とは、直接的にセルロース水酸基を酸化する物質ではなく、酸化触媒として用いられるN−オキシル化合物を酸化する物質のことである。
そして、上記反応終了後、塩酸を添加して中性(pH6.0〜8.0)に調整する。また、長期保存安定性を向上させる目的で、上記反応終了後に、水素化ホウ素ナトリウム等により還元処理を行っても良い。
つぎに、未反応の共酸化剤(次亜塩素酸等)や、各種副生成物等を除く目的で、適宜、精製を行う。反応物繊維は通常、この段階ではナノファイバー単位までばらばらに分散しているわけではないため、通常の精製法、すなわち水洗とろ過を繰り返すことで高純度(99重量%以上)の反応物繊維と水の分散体とする。
上記精製工程にて得られる水を含浸した反応物繊維(水分散体)を、水等の分散媒体中に分散させ分散処理を行う。処理に伴って粘度が上昇し、微細化処理されたセルロース繊維の分散体を得ることができる。その後、上記セルロース繊維の分散体を乾燥することによって、TEMPO酸化セルロースを得ることができる。なお、上記セルロース繊維の分散体を乾燥することなく、分散体の状態で用いても差し支えない。
また、本発明の細胞増殖促進剤は、上記特定のセルロース繊維及び分散媒体のほかに、他の成分材料として、機能性添加剤を用いることも可能である。上記機能性添加剤としては、例えば、増粘促進剤、無機塩類、有機塩類、界面活性剤、オイル類、保湿剤、防腐剤、有機微粒子、無機微粒子、消臭剤、香料、有機溶媒等があげられる。これらは単独でもしくは二種以上併せて用いられる。
セルロースナノファイバー(CNF)は、パルプ等のセルロースファイバー含有材料をリファイナー、グラインダー(石臼式磨砕機)、一軸又は二軸混練機(押出機)、高圧ホモジナイザー、ビーズミル等によって磨砕、叩解等の機械処理をすることによって解繊、微細化して製造される。必要に応じて、これらの解繊方法を組み合わせて処理したものであってもよい。一般的には、平均幅が数〜100nm程度、平均長さが0.5〜数μm程度のサイズの繊維状物質である。
本実施形態のキチンナノファイバーは、キチン含有生物由来の材料を少なくとも1回の脱蛋白工程及び少なくとも1回の脱灰工程に付し、次いで、解繊工程に付すことにより得られ、幅(または径)が約2nm〜約30nmの伸びきり鎖の結晶の繊維であって、生体内のキチンナノファイバーがありのままの状態で単離・抽出されたものである。水中で完全にナノ分散して透明高粘度になる性質を有する。キチンナノファイバーには、カニ等由来のα型結晶構造を有するα−キチンナノファイバー、イカ等由来のβ型結晶構造を有するβ−キチンナノファイバーの双方を含む。
キチンナノファイバーの幅(または径)は、通常は、約2nm〜約30nm、好ましくは約2nm〜約20nm、例えば、5nm〜20nmである。ここで、例えば、「キチンナノファイバーの幅(または径)は約2nm〜約20nm」とは、電子顕微鏡観察にて観察した場合に,幅(または径)が約2nm〜約20nm以下であるファイバーが全体の約50%以上、好ましくは約60%以上、さらに好ましくは約70%以上を占める状態をいう。
キチンナノファイバーは、生体内のキチンナノファイバーをありのままの状態で単離・抽出することが可能である。そのため、キチンナノファイバーは、細くて均質であり、長く、繊維の分子が伸びきり鎖結晶で強度が高いものとして得ることができる。伸びきり鎖結晶とは、剛直性の高分子が伸びきった状態で規則正しく配列し、束になった繊維状の結晶のことであり、欠陥が少ないため強靭な物性を発揮することが可能である。特に、エビやカニなどの甲殻類のキチンは結晶性の高いアルファキチンであるため、エビやカニなどの甲殻類の殻を原料にして得られるキチンナノファイバーは、上記の優れた特性が顕著である。
本実施形態のキトサンナノファイバーは、キチン含有生物由来の材料を、少なくとも1回の脱蛋白工程および少なくとも1回の脱灰工程および少なくとも1回の脱アセチル化工程に付し、次いで、解繊工程に付すことにより得られる。脱蛋白工程、脱灰工程、解繊工程については、キチンナノファイバーの製造工程に関して上で説明したのと同様である。なお、本実施形態において、脱蛋白工程と脱アセチル化工程を同時に行うことも可能である。さらに、既に脱蛋白工程および脱灰工程を行った市販のキチン粉末を脱アセチル化工程に付すことによって、キトサンナノファイバーを製造することも可能である。脱アセチル化方法はいくつかの方法が公知であるが、アルカリ処理法が好適である。アルカリ処理による脱アセチル化において、水酸化カリウム、水酸化ナトリウム、水酸化リチウムなどのアルカリの水溶液が好ましく用いられ、その濃度は、通常は約20〜約50%(w/v)、好ましくは約30〜約40%(w/v)、例えば約40%(w/v)である。アルカリ処理による脱アセチル化の温度は、キチン含有生物由来の材料の量、キチン含有生物の種類、部位などに応じて適宜選択されうるが、通常は約80℃以上、好ましくは約90℃以上、さらに好ましくはアルカリ水溶液を還流しながら行う。処理時間も、キチン含有生物由来の材料の量、キチン含有生物の種類、部位などに応じて適宜選択されうるが、通常は30分〜約3日間、好ましくは30分〜一晩行ってもよい。なお、キトサンナノファイバーは乾燥すると水素結合して強固に凝集するため、キトサンナノファイバーの製造工程を、材料を常に乾燥させずに行うことが非常に好ましい。
キトサンナノファイバーの幅(または径)は、上述したキチンナノファイバーの幅(または径)と同様である。
本実施形態のパラミロン水性分散液は、(A)パラミロンと、(B)アニオン変性セルロースナノファイバー、セルロースナノファイバー、キチンナノファイバー、およびキトサンナノファイバーから選ばれる一種以上の繊維と、(C)水を含有する分散液である。
パラミロン水性分散液におけるパラミロンの含有量は、パラミロン水性分散液全量の60重量%以下である。パラミロンの含有量が、60重量%を超えると、上記特定のセルロース繊維の存在下であっても、水性溶媒に分散しなくなるからである。
パラミロン水性分散液における(B)成分の濃度は、6.0×10−5重量%〜5.0重量%の範囲内であるとよい。(B)成分の濃度の含有量が、上記範囲内であると、パラミロンを水性液中において分散させることができると同時に、パラミロンと繊維を含む水性分散液が、細胞増殖促進効果を得ることが可能となる。
本実施形態のパラミロン水性分散液は、さらに、細胞増殖を促進する機能を有することから、細胞増殖促進剤としても用いることができる。
〔試験1:パラミロン水性分散液の物性試験:パラミロンとTOCの分散液〕
本発明のパラミロン水性分散液として、実施例1〜9を調製し、各実施例のパラミロン水性分散液の対比例として、比較例1〜3を調整した。
(実施例1〜3:パラミロンとTOCの分散液)
(A)成分のパラミロン((株)ユーグレナ製)10重量%と、(B)成分の、下記合成例に基づいて合成したTEMPO酸化セルロース(TOC)水性分散液に含まれるTEMPO酸化セルロース(TOC)0.2重量%,0.5重量%又は1.0重量%と、(C)成分の水を89.8重量%,89.5重量%又は89.0重量%をそれぞれ含む混合液を調整した。ホモディスパーを用いて回転数3000rpm、25℃で5分間攪拌し、実施例1〜3のパラミロン水性分散液を調製した。
合成例:
針葉樹パルプ2gに、水150mlと、臭化ナトリウム0.25gと、TEMPOを0.025gとを加え、充分撹拌して分散させた後、13重量%次亜塩素酸ナトリウム水溶液(共酸化剤)を、上記パルプ1.0gに対して次亜塩素酸ナトリウム量が6.5mmol/gとなるように加え、反応を開始した。反応の進行に伴いpHが低下するため、pHを10〜11に保持するように0.5N水酸化ナトリウム水溶液を滴下しながら、pHの変化が見られなくなるまで反応させた(反応時間:120分)。反応終了後、0.1N塩酸を添加してpHを7.0に調整し、ろ過と水洗を繰り返して精製し、繊維表面が酸化されたセルロース繊維を得た。このセルロース繊維を、固形分濃度が2重量%となるように純水で希釈し、超高圧ホモジナイザーで処理し、セルロース繊維の水性分散液を得た。数平均繊維径が6nmであった。
針葉樹漂白クラフトパルプ(NBKP)50gを水4950gに分散させ、パルプ濃度1重量%の分散液を調整した。この分散液をセレンディピターMKCA6−3(増幸産業(株)製)で30回処理し、セルロースナノファイバー(CNF)を得た。得られたCNFの繊維径の平均値は70nmであった。
このCNFを(B)成分として用い、(A)成分のパラミロン((株)ユーグレナ製)10重量%と、(B)成分のCNFを0.5重量%又は1.0重量%と、(C)成分の水を89.5重量%又は89.0重量%をそれぞれ含む混合液を調整した。ホモディスパーを用いて回転数3000rpm、25℃で5分間攪拌し、実施例4,5のパラミロン水性分散液を調整した。
市販キチンの水分散体を調整し、100〜245MPaの超高圧に加圧し、微細なオリフィスノズル(φ0.1〜0.8mm)から高圧で噴射して製造された(株)スギノマシン製BiNFi−s(ビンフィス)(登録商標)SFo−20002を、(B)成分のキチンナノファイバー(キチンNF)として用い、(A)成分のパラミロン((株)ユーグレナ製)10重量%と、(B)成分のキチンNFを0.5重量%又は1.0重量%と、(C)成分の水を89.5重量%又は89.0重量%をそれぞれ含む混合液を調整した。ホモディスパーを用いて回転数3000rpm、25℃で5分間攪拌し、実施例6,7のパラミロン水性分散液を調整した。
市販キトサンの水分散体を調整し、100〜245MPaの超高圧に加圧し、微細なオリフィスノズル(φ0.1〜0.8mm)から高圧で噴射して製造された(株)スギノマシン製BiNFi−s(ビンフィス)(登録商標)EFo−08002を、(B)成分のキトサンナノファイバー(キトサンNF)として用い、(A)成分のパラミロン((株)ユーグレナ製)10重量%と、(B)成分のキトサンNFを0.5重量%又は1.0重量%と、(C)成分の水を89.5重量%又は89.0重量%をそれぞれ含む混合液を調整した。ホモディスパーを用いて回転数3000rpm、25℃で5分間攪拌し、実施例8のパラミロン水性分散液を調整した。
撹拌機に、パルプ(LBKP、日本製紙(株)製)を乾燥質量で200g、水酸化ナトリウムを乾燥質量で18g加え、パルプ固形分濃度が15重量%になるように水を加えた。その後、30℃で30分攪拌した後に70℃まで昇温し、モノクロロ酢酸ナトリウムを117g(有効成分換算)添加した。1時間反応した後に、反応物を取り出して中和、洗浄して、グルコース単位当たりの置換度0.05のアニオン変性されたセルロースを得た。その後、アニオン変性されたセルロースに水を添加して固形分濃度5重量%とし、高圧ホモジナイザーにより20℃、140MPaの圧力で5回処理し、数平均繊維径30nmのセルロース繊維の分散液を、低置換度カルボキシメチルセルロースナノファイバー(CMCNF)の分散液として得た。
この分散液中の低置換度CMCNFを(B)成分として用い、(A)成分のパラミロン((株)ユーグレナ製)10重量%と、調整した分散液中の(B)成分低置換度CMCNFを0.5重量%又は1.0重量%と、(C)成分の水を89.5重量%又は89.0重量%をそれぞれ含む混合液を調整した。ホモディスパーを用いて回転数3000rpm、25℃で5分間攪拌し、実施例9のパラミロン水性分散液を調整した。
比較例1〜3には、(B)成分の代わりに、それぞれ、キサンタンガム(KELTROL CG−T),カルボキシメチルセルロース(CMC),ポリビニルピロリドン(PVP)を用い、これらの成分を1.0重量%と、(A)成分のパラミロン((株)ユーグレナ製)10重量%と、(C)成分の水を89.0重量%をそれぞれ含む混合液を調整した。ホモディスパーを用いて回転数3000rpm、25℃で5分間攪拌し、比較例1〜3のパラミロン水性分散液を調整した。
比較例1〜3において(B)成分の代わりにキサンタンガムは、化粧品によく利用される分散剤である。また、CMC(カルボキシメチルセルロース)は、各(B)成分の類似化合物である。PVP(ポリビニルピロリドン)はよく利用される合成系の増粘剤である。
調製した実施例1〜9及び比較例1〜3について、分散性,分散安定性,べたつき感の試験を行った。
分散性の試験では、実施例1〜9及び比較例1〜3のパラミロン水性分散液を調整後、25℃で5分間放置し、分散状態を評価した。分散状態は下記式で分散度を計算し、分散度が100である場合「○」、80以上である場合「△」、80未満である場合「×」と評価した。
分散度[%]=パラミロンが分散している部分の高さ/液全体の高さ×100 … 式
分散度[%]=パラミロンが分散している部分の高さ/液全体の高さ×100 … 式
実施例1〜9及び比較例1〜3についての分散性,分散安定性及びべたつき感の試験結果を、表1に示す。
〔試験2:パラミロン水性分散液の細胞増殖促進効果確認試験〕
本発明のパラミロン水性分散液の一実施例として、(A)成分に(株)ユーグレナ製のパラミロン、(B)成分にアニオン変性セルロースナノファイバーの一例のTEMPO酸化セルロース(TOC)水性分散液10重量%希釈溶液を用いたパラミロン水性分散液を使用し、パラミロン水性分散液の線維芽細胞増殖促進作用を試験した。
TEMPO酸化セルロース(TOC)水性分散液10重量%希釈溶液は、本発明の実施例に係るTEMPO酸化セルロース0.2重量%と、水からなる分散体である。
まず、対数増殖期にあるヒト正常皮膚線維芽細胞(DSファーマ製,Cell System−Fb Cells)を、96ウェルプレートにウェルあたり2,000セルとなるよう0.5%ウシ胎児血清(FBS),ダルベッコ改変イーグル培地(DMEM)に懸濁して播種し、1日培養した。
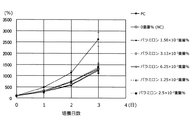
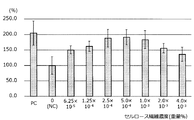
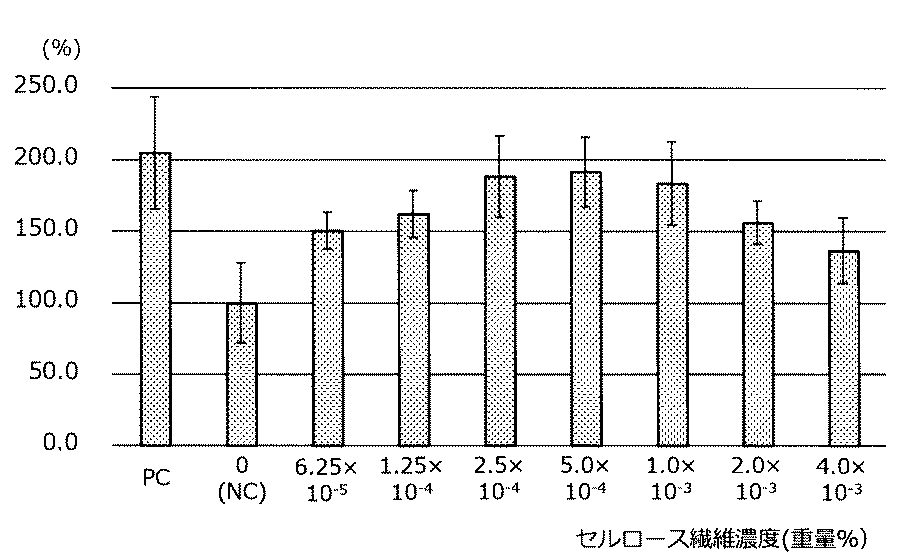
図1は、培養0日目(パラミロン水性分散液添加前)の細胞数を100として、培養1,2,3日後の各サンプルの細胞数の増加率を示すグラフである。図2は、培養3日後の各サンプルの細胞数を示すグラフであって、ネガティブコントロール(NC)の3日目の細胞数を、100%とした割合を示している。
図1,2の結果より、TEMPO酸化セルロース(TOC)水性分散液10重量%希釈溶液の濃度が0.03125重量%(セルロース繊維濃度6.25×10−5重量%,パラミロン1.56×10−3重量%)以上のときに、細胞の増殖が促進されていた。
また、パラミロン水性分散液濃度0重量%〜16重量%のサンプルは、濃度が上昇するに従って白濁していたが、すべてのサンプルにおいて、沈殿はなく、パラミロンが液中に分散していた。
試験2のパラミロン水性分散液の細胞増殖促進効果を確認するための対照試験として、TEMPO酸化セルロース(TOC)を含まない対比例のパラミロン液を用いて、試験2と同様の手順で実験をおこなった。
本対照試験1では、試験2のパラミロン水性分散液の代わりに、全量に対して5重量%のパラミロンを含むよう、水にパラミロンを添加した対比例のパラミロン混合水を用いたことを除いては、試験2と同様の手順とした。
図3は、培養0日目(パラミロン混合液添加前)の細胞数を100として、培養1,2,3日後の各サンプルの細胞数の増加率を示すグラフである。図4は、培養3日後の各サンプルの細胞数を示すグラフであって、ネガティブコントロール(NC)の3日目の細胞数を、100%としている。
図3,図4の結果より、試験2で見られたような顕著な細胞増殖の促進効果は見られなかった。
また、濃度0.03125重量%〜16重量%の各パラミロン混合液は、パラミロンが水中に分散しておらず、撹拌を止めるとパラミロンが沈殿した。
Claims (7)
- 少なくとも下記の(A)〜(C)成分を含有することを特徴とするパラミロン水性分散液。
(A)パラミロン。
(B)アニオン変性セルロースナノファイバー、セルロースナノファイバー、キチンナノファイバー、およびキトサンナノファイバーから選ばれる一種以上の繊維。
(C)水。 - 前記(A)成分と前記(B)成分との比率が、(A)/(B)=1〜200である、請求項1に記載のパラミロン水性分散液。
- 前記パラミロン水性分散液中の前記(B)成分の濃度が、6.0×10−5重量%〜5.0重量%である、請求項1又は2記載のパラミロン水性分散液。
- 前記(B)成分がアニオン変性セルロースナノファイバーである、請求項1〜3のいずれか1項に記載のパラミロン水性分散液。
- 少なくとも下記の(A)〜(C)成分を含有するパラミロン水性分散液からなることを特徴とする細胞増殖促進剤。
(A)パラミロン。
(B)アニオン変性セルロースナノファイバー、セルロースナノファイバー、キチンナノファイバー、およびキトサンナノファイバーから選ばれる一種以上の繊維。
(C)水。 - 線維芽細胞の増殖促進剤として用いられる、請求項5記載の細胞増殖促進剤。
- 皮膚創傷治癒促進剤,皮膚外用剤,化粧品,皮膚の老化防止剤又は皮膚の老化改善剤として用いられる、請求項5又は6記載の細胞増殖促進剤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016042619A JP6712875B2 (ja) | 2016-03-04 | 2016-03-04 | 繊維芽細胞増殖促進剤 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016042619A JP6712875B2 (ja) | 2016-03-04 | 2016-03-04 | 繊維芽細胞増殖促進剤 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017155195A true JP2017155195A (ja) | 2017-09-07 |
| JP6712875B2 JP6712875B2 (ja) | 2020-06-24 |
Family
ID=59809357
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016042619A Expired - Fee Related JP6712875B2 (ja) | 2016-03-04 | 2016-03-04 | 繊維芽細胞増殖促進剤 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6712875B2 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019235318A1 (ja) * | 2018-06-05 | 2019-12-12 | 国立大学法人福井大学 | 免疫測定用組成物、免疫測定用診断薬、及び、免疫測定用デバイス |
| JP2020066658A (ja) * | 2018-10-22 | 2020-04-30 | 株式会社Kri | 硫酸エステル化セルロースナノファイバー及びその乾燥物 |
| JP2021046392A (ja) * | 2019-09-13 | 2021-03-25 | ユー ピー エム キュンメネ コーポレーション | 医薬組成物を調製する方法及び医薬組成物 |
| WO2023149408A1 (ja) * | 2022-02-02 | 2023-08-10 | 株式会社ユーグレナ | 化粧料組成物、表皮角化細胞増殖用化粧料組成物、表皮角化細胞増殖剤、抗酸化剤、毛乳頭細胞増殖用化粧料組成物、毛乳頭細胞増殖剤、育毛剤、ロリクリン遺伝子発現促進剤、ヘムオキシゲナーゼ-1遺伝子発現促進剤及びコーニファイドエンベロープ形成促進剤 |
| WO2025070618A1 (ja) * | 2023-09-28 | 2025-04-03 | 株式会社日本触媒 | ナノファイバー、皮膚外用剤用添加剤、皮膚外用剤及びナノファイバーの製造方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0459715A (ja) * | 1990-06-29 | 1992-02-26 | Harima Chem Inc | 皮膚化粧料 |
| JP2004307438A (ja) * | 2003-04-10 | 2004-11-04 | Unitika Ltd | 皮膚外用剤 |
| JP2012092223A (ja) * | 2010-10-27 | 2012-05-17 | National Institute Of Advanced Industrial Science & Technology | 多孔質膜 |
| JP2012193139A (ja) * | 2011-03-16 | 2012-10-11 | Daicel Corp | 化粧料 |
| WO2014088072A1 (ja) * | 2012-12-07 | 2014-06-12 | 日本製紙株式会社 | カルボキシメチル化セルロースの繊維 |
| JP2014139303A (ja) * | 2012-12-18 | 2014-07-31 | Mitsubishi Chemicals Corp | ゴム改質剤、ゴムラテックス分散液及びゴム組成物 |
| JP2014231479A (ja) * | 2013-05-02 | 2014-12-11 | 株式会社ユーグレナ | 創傷治療剤 |
-
2016
- 2016-03-04 JP JP2016042619A patent/JP6712875B2/ja not_active Expired - Fee Related
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0459715A (ja) * | 1990-06-29 | 1992-02-26 | Harima Chem Inc | 皮膚化粧料 |
| JP2004307438A (ja) * | 2003-04-10 | 2004-11-04 | Unitika Ltd | 皮膚外用剤 |
| JP2012092223A (ja) * | 2010-10-27 | 2012-05-17 | National Institute Of Advanced Industrial Science & Technology | 多孔質膜 |
| JP2012193139A (ja) * | 2011-03-16 | 2012-10-11 | Daicel Corp | 化粧料 |
| WO2014088072A1 (ja) * | 2012-12-07 | 2014-06-12 | 日本製紙株式会社 | カルボキシメチル化セルロースの繊維 |
| JP2014139303A (ja) * | 2012-12-18 | 2014-07-31 | Mitsubishi Chemicals Corp | ゴム改質剤、ゴムラテックス分散液及びゴム組成物 |
| JP2014231479A (ja) * | 2013-05-02 | 2014-12-11 | 株式会社ユーグレナ | 創傷治療剤 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019235318A1 (ja) * | 2018-06-05 | 2019-12-12 | 国立大学法人福井大学 | 免疫測定用組成物、免疫測定用診断薬、及び、免疫測定用デバイス |
| JP2019211347A (ja) * | 2018-06-05 | 2019-12-12 | 国立大学法人福井大学 | 免疫測定用組成物、免疫測定用診断薬、及び、免疫測定用デバイス |
| JP2020066658A (ja) * | 2018-10-22 | 2020-04-30 | 株式会社Kri | 硫酸エステル化セルロースナノファイバー及びその乾燥物 |
| JP2021046392A (ja) * | 2019-09-13 | 2021-03-25 | ユー ピー エム キュンメネ コーポレーション | 医薬組成物を調製する方法及び医薬組成物 |
| JP7640065B2 (ja) | 2019-09-13 | 2025-03-05 | ユー ピー エム キュンメネ コーポレーション | 医薬組成物を調製する方法及び医薬組成物 |
| WO2023149408A1 (ja) * | 2022-02-02 | 2023-08-10 | 株式会社ユーグレナ | 化粧料組成物、表皮角化細胞増殖用化粧料組成物、表皮角化細胞増殖剤、抗酸化剤、毛乳頭細胞増殖用化粧料組成物、毛乳頭細胞増殖剤、育毛剤、ロリクリン遺伝子発現促進剤、ヘムオキシゲナーゼ-1遺伝子発現促進剤及びコーニファイドエンベロープ形成促進剤 |
| WO2025070618A1 (ja) * | 2023-09-28 | 2025-04-03 | 株式会社日本触媒 | ナノファイバー、皮膚外用剤用添加剤、皮膚外用剤及びナノファイバーの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6712875B2 (ja) | 2020-06-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5502712B2 (ja) | 粘性水系組成物 | |
| JP6308531B2 (ja) | 皮膚外用剤 | |
| JP6712875B2 (ja) | 繊維芽細胞増殖促進剤 | |
| JP2010037348A (ja) | ゲル状組成物 | |
| JP5269512B2 (ja) | 化粧料組成物 | |
| JP5795094B2 (ja) | 増粘剤組成物 | |
| JP5701570B2 (ja) | 粘性水系組成物およびその製法 | |
| WO2019221272A1 (ja) | カルボキシメチル化パルプの粉砕物及び該粉砕物を含む添加剤 | |
| JP6229090B1 (ja) | H型カルボキシル化セルロースナノファイバー | |
| JP5628018B2 (ja) | 水性ゲル組成物 | |
| JP6276812B2 (ja) | 日焼け止め化粧料 | |
| Souza et al. | Study of morphological properties and rheological parameters of cellulose nanofibrils of cocoa shell (Theobroma cacao L.) | |
| JP7605945B2 (ja) | フィブリル化された化学変性セルロース繊維 | |
| JP6645917B2 (ja) | ゲル状シート組成物およびその製造方法 | |
| WO2018143149A1 (ja) | 乾燥セルロースナノファイバーの製造方法 | |
| EP3722324B1 (en) | Carboxymethylated cellulose nanofibers | |
| CN104892772B (zh) | 以禾本科植物为原料生产纳米纤维的方法 | |
| Popa | Polysaccharides in medicinal and pharmaceutical applications | |
| EP3722328A1 (en) | Carboxymethylated cellulose nanofibers | |
| US11578142B2 (en) | Acid type carboxylated cellulose nanofiber | |
| Wu et al. | One-pot fabrication of sacchachitin for production of TEMPO-oxidized sacchachitin nanofibers (TOSCNFs) utilized as scaffolds to enhance bone regeneration | |
| WO2024154815A1 (ja) | 水性組成物、抗炎症剤、保湿剤、抗炎症用又は保湿用の食品組成物及び水性組成物の製造方法 | |
| JP2017155024A (ja) | 細胞増殖促進剤 | |
| JP6139754B1 (ja) | 細胞増殖促進剤 | |
| EP3936167B1 (en) | Subcutaneous or submucosal expansion agent |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7426 Effective date: 20160421 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190226 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20191218 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200114 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200311 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200519 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200602 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6712875 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |