JP2017002231A - 微細繊維状セルロース濃縮物の製造方法、および微細繊維状セルロースを含む組成物 - Google Patents
微細繊維状セルロース濃縮物の製造方法、および微細繊維状セルロースを含む組成物 Download PDFInfo
- Publication number
- JP2017002231A JP2017002231A JP2015119587A JP2015119587A JP2017002231A JP 2017002231 A JP2017002231 A JP 2017002231A JP 2015119587 A JP2015119587 A JP 2015119587A JP 2015119587 A JP2015119587 A JP 2015119587A JP 2017002231 A JP2017002231 A JP 2017002231A
- Authority
- JP
- Japan
- Prior art keywords
- fine fibrous
- fibrous cellulose
- pulp
- mass
- cellulose
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images

Landscapes
- Polysaccharides And Polysaccharide Derivatives (AREA)
Abstract
【解決手段】(1)イオン性置換基と、炭素数がn(但しnは1以上の整数)である対イオンAと、を有するセルロース繊維を、25℃における比誘電率が70以下の溶媒中で微細化し、微細繊維状セルロース分散液を得る工程;および
(2)微細繊維状セルロース分散液に含まれる対イオンAの一部または全部を、炭素数がn−1以下である対イオンBに置換して、微細繊維状セルロースの濃縮物を得る工程
を含む、微細繊維状セルロース濃縮物の製造方法を提供する。
【選択図】なし
Description
[1](1)イオン性置換基と、炭素数がn(但しnは1以上の整数)である対イオンAと、を有するセルロース繊維を、25℃における比誘電率が70以下の溶媒中で微細化し、微細繊維状セルロース分散液を得る工程;および
(2)微細繊維状セルロース分散液に含まれる対イオンAの一部または全部を、炭素数がn−1以下である対イオンBに置換して、微細繊維状セルロースの濃縮物を得る工程
を含む、微細繊維状セルロース濃縮物の製造方法。
[2] イオン性置換基が、アニオン性置換基を含む、1に記載の製造方法。
[3] アニオン性置換基が、リン酸由来の基、カルボン酸由来の基および硫酸由来の基からなる群より選択される1種または2種以上を含む、2に記載の製造方法。
[4] 対イオンAが、テトラアルキルアンモニウムイオンを含む、2または3に記載の製造方法。
[5] イオン性置換基が、カチオン性置換基含む、1に記載の製造方法。
[6] カチオン性置換基が、4級アンモニウム塩由来の基およびホスホニウム塩由来の基のうちの1種または2種を含む、5に記載の製造方法。
[7] 対イオンBが、1価のイオンである、1〜6のいずれか1項に記載の製造方法。
[8] イオン性置換基を有する微細繊維状セルロースと、
炭素数n(ただしnは1以上の整数)である対イオンAと、
炭素数n−1以下である対イオンBと、
有機溶媒と、
を含む組成物。
[9] 組成物全体に対する微細繊維状セルロースの含有量が、5質量%以上である8に記載の組成物。
[10] 微細繊維状セルロースが、イオン性置換基としてリン酸由来の基、カルボン酸由来の基および硫酸由来の基からなる群より選択される1種または2種以上を有する、8または9に記載の組成物。
[11] 微細繊維状セルロースが、イオン性置換基として4級アンモニウム塩由来の基およびホスホニウム塩由来の基のうちの1種または2種を有する、8または9に記載の組成物。
[12] 対イオンBが、1価のイオンである、8〜11のいずれか1項に記載の組成物。
[13] 組成物が、有機溶媒を含有する溶媒であって25℃における比誘電率が70以下である溶媒中に微細繊維状セルロースを含む形態である、8〜12のいずれか1項に記載の組成物。
本発明により、溶剤の使用量が削減できる。
(1)イオン性置換基と、炭素数がn(但しnは1以上の整数)である対イオンAと、を有するセルロース繊維を、25℃における比誘電率が70以下の溶媒中で微細化し、微細繊維状セルロース分散液を得る工程;および
(2)微細繊維状セルロース分散液に含まれる対イオンAの一部または全部を、炭素数がn−1以下である対イオンBに置換して、微細繊維状セルロースの濃縮物を得る工程。
工程(1)では、イオン性置換基が導入されたセルロース繊維が原料として用いられる。
イオン性置換基が導入されたセルロース繊維得るためのセルロース繊維原料としては特に限定されないが、入手しやすく安価である点から、パルプを用いることが好ましい。パルプとしては、木材パルプ、非木材パルプ、脱墨パルプから選ばれる。木材パルプとしては例えば、広葉樹クラフトパルプ(LBKP)、針葉樹クラフトパルプ(NBKP)、サルファイトパルプ(SP)、ソーダパルプ(AP)、未晒しクラフトパルプ(UKP)、酸素漂白クラフトパルプ(OKP)等の化学パルプ等が挙げられる。また、セミケミカルパルプ(SCP)、ケミグラウンドウッドパルプ(CGP)等の半化学パルプ、砕木パルプ(GP)、サーモメカニカルパルプ(TMP、BCTMP)等の機械パルプ、等が挙げられるが、特に限定されない。非木材パルプとしてはコットンリンターやコットンリント等の綿系パルプ、麻、麦わら、バガス等の非木材系パルプ、ホヤや海草等から単離されるセルロース、キチン、キトサン等が挙げられるが、特に限定されない。脱墨パルプとしては古紙を原料とする脱墨パルプが挙げられるが、特に限定されない。本実施態様のパルプは上記1種を単独で用いてもよいし、2種以上混合して用いてもよい。上記パルプの中で、入手のしやすさという点で、セルロースを含む木材パルプ、脱墨パルプが好ましい。木材パルプの中でも化学パルプはセルロース比率が大きいため、繊維微細化(解繊)時の微細セルロース繊維の収率が高く、また、パルプ中のセルロースの分解が小さく、軸比の大きい長繊維の微細セルロース繊維が得られる点で特に好ましいが、特に限定されない。中でもクラフトパルプ、サルファイトパルプが最も好ましく選択されるが、特に限定されない。この軸比の大きい長繊維の微細セルロース繊維を含有するシートは高強度が得られる。
セルロース繊維には、イオン性置換基が導入される。繊維への置換基を導入する方法は、特に限定されないが、例えば、酸化処理、セルロース中の官能基と共有結合を形成し得る化合物による処理などが挙げられる。酸化処理とは、セルロース中のヒドロキシ基をアルデヒド基やカルボキシ基に変換する処理であり、例えばTEMPO酸化処理や各種酸化剤(亜塩素酸ナトリウム、オゾンなど)を用いた処理が挙げられる。
アニオン性置換基を導入する場合、繊維原料と反応する化合物としては、例えば、リン酸由来の基を有する化合物、カルボン酸由来の基を有する化合物、硫酸由来の基を有する化合物、スルホン酸由来の基を有する化合物等が挙げられる。取扱いの容易さ、繊維との反応性から、リン酸由来の基、カルボン酸由来の基および硫酸由来の基からなる群より選択される少なくとも1種を有する化合物が好ましい。これらの化合物が繊維とエステルまたは/およびエーテルを形成することがより好ましいが、特に限定されない。
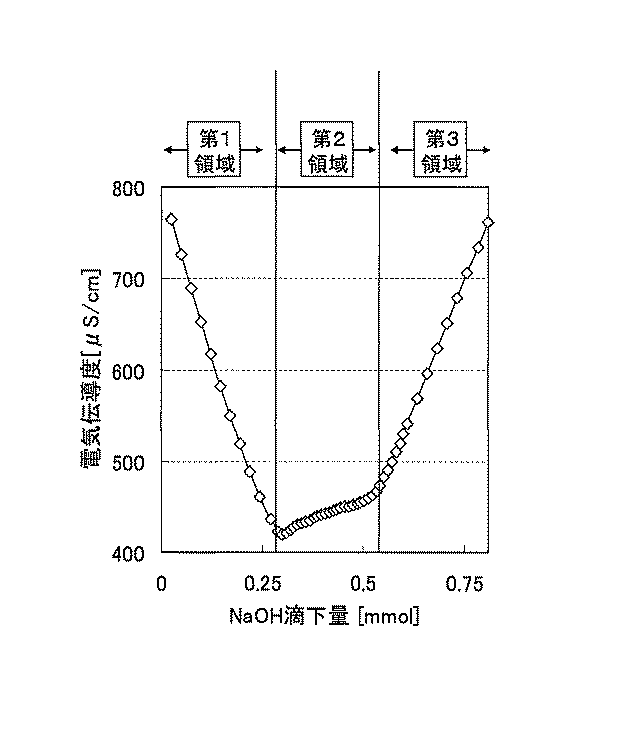
なお、繊維表面の置換基の導入量(滴定法)の測定は、特に記載した場合を除き、次の方法で行うことができる:
絶乾質量で0.04g程度の固形分を含む微細繊維含有スラリーを分取し、イオン交換水を用いて50g程度に希釈する。この溶液を撹拌しながら、0.01Nの水酸化ナトリウム水溶液を滴下した場合の電気伝導度の値の変化を測定し、その値が極小となる時の0.01N水酸化ナトリウム水溶液の滴下量を、滴定終点における滴下量とする。セルロース表面の置換基量XはX(mmol/g)=0.01(mol/l)×V(ml)/W(g)で表される。ここで、V:0.01N水酸化ナトリウム水溶液の滴下量(ml)、W:微細セルロース繊維含有スラリーが含む固形分(g)である。
これらのうち、リン酸基導入の効率が高く、工業的に適用しやすい観点から、リン酸、リン酸のナトリウム塩、リン酸のカリウム塩、リン酸のアンモニウム塩が好ましく、リン酸二水素ナトリウム、リン酸水素二ナトリウムがより好ましいが、特に限定されない。
カチオン化剤の具体例としては、グリシジルトリメチルアンモニウムクロリド、3−クロロ−2−ヒドロキシプロピルトリメチルアンモニウムクロリドなどのグリシジルトリアルキルアンモニウムハライド或いはそのハロヒドリン型の化合物が挙げられる。
アルカリ金属の水酸化物としては、水酸化リチウム、水酸化ナトリウム、水酸化カリウムが挙げられる。アルカリ土類金属の水酸化物としては、水酸化カルシウムが挙げられる。
アルカリ金属の炭酸塩としては炭酸リチウム、炭酸水素リチウム、炭酸カリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸水素ナトリウムが挙げられる。アルカリ土類金属の炭酸塩としては、炭酸カルシウムなどが挙げられる。
アルカリ金属のリン酸塩としては、リン酸リチウム、リン酸カリウム、リン酸3ナトリウム、リン酸水素2ナトリウムなどが挙げられる。アルカリ土類金属のリン酸塩としては、リン酸カルシウム、リン酸水素カルシウムなどが挙げられる。
上記アルカリ化合物は1種単独でもよいし、2種以上を組み合わせてもよい。
必要に応じ、イオン性置換基を導入した後であって後述する対イオンAを付加する工程の前において、アニオン性セルロースに対しては酸処理を、カチオン性セルロースに対しては塩基処理を行うことができる。酸処理に用いられる酸は、セルロースに導入されたアニオン性置換基以上の電離度を有する酸を用いることが好ましいが、特に限定されない。。酸処理は、例えば、塩酸、硝酸および硫酸からなる群より選択される1種または2種以上を用いて行うことができる。また塩基処理に用いられる塩基は、セルロースに導入されたカチオン性置換基以上の電離度を有する塩基を用いることが好ましいが、特に限定されない。塩基処理は、例えば、水酸化ナトリウム、水酸化カリウム、水酸化バリウムおよび水酸化カルシウムからなる群より選択される1種または2種以上を用いて行うことができる。このような処理より、導入されたイオン性置換基が十分にH型またはOH型となり、後述する対イオンAを、イオン性置換基に対してより容易に付加させることが可能となる。
工程(1)では、イオン性置換基が導入されたセルロース繊維は、炭素数がnであるイオン性置換基の対イオンAが付加される。
本発明においては、対イオンAとして、炭素数がnであるものが用いられる。nは1以上の整数であり、次の微細化処理が十分に行える限り特に限定されないが、4以上であることが好ましく、8以上であることがより好ましく、16以上であることがさらに好ましい。nの数が大きく、ある程度嵩高いものが微細化処理上、効果的だからである。
Mは、窒素原子またはリン原子であり;
R1〜R4は、ヘテロ原子を含んでいてもよい炭素水素基であり、同一でもよく、それぞれ異なっていてもよい。R1〜R4の炭素数の合計は4以上であり、次の微細化処理が十分に行える限り特に限定されない。8以上であることが好ましく、12以上であることがより好ましく、16以上であることがさらに好ましい。nの数が大きく、ある程度嵩高いものが微細繊維分散上、効果的だからである。
イオン性置換基への対イオンAの付加のための条件は、対イオンAが十分に付加される限り特に限定されない。例えば、上述の工程により得られたイオン性置換基が導入されたセルロース繊維を適切な溶媒に分散し、必要に応じ攪拌しながら対イオンAを含む溶液を添加することにより、行うことができる。
工程(1)では、上述の工程で得られたイオン性置換基導入セルロース繊維の対イオンA型のものが、微細化(解繊)処理に供される。
微細化処理に際し、セルロース繊維は溶媒に分散される。使用される溶媒については、後述する。分散濃度は、0.1〜20質量%であることが好ましく、0.5〜10質量%であることがより好ましい。含有量が前記下限値以上であれば、解繊処理の効率が向上し、前記上限値以下であれば、解繊処理装置内での閉塞を防止できるからである。
(1)観察画像内の任意箇所に一本の直線Xを引き、該直線Xに対し、20本以上の繊維が交差する。
(2)同じ画像内で該直線と垂直に交差する直線Yを引き、該直線Yに対し、20本以上の繊維が交差する。
上記条件を満足する観察画像に対し、直線X、直線Yと交錯する繊維の幅を目視で読み取る。こうして少なくとも重なっていない表面部分の画像を3組以上観察し、各々の画像に対して、直線X、直線Yと交錯する繊維の幅を読み取る。このように少なくとも20本×2×3=120本の繊維幅を読み取る。平均繊維幅とはこのように読み取った繊維幅の平均値である。
微細化処理の際のセルロース繊維を分散させる溶媒は、25℃における比誘電率が70以下であるものを用いる。これにより、次の工程で対イオンAを対イオンBに置換したときに、微細繊維状セルロースを濃縮することができるからである。溶媒の比誘電率は、好ましくは60以下であり、より好ましくは50以下である。溶媒の比誘電率の下限値は特に限定されないが、例えば20以上のものを用いることができる。なお、比誘電率の値を示すときは、特に記載した場合を除き、25℃における値である。比誘電率は、誘電率の電気定数(真空の誘電率)に対する比である。純粋な溶媒の誘電率および比誘電率はよく知られている。混合溶媒の誘電率は、液体用誘電率計等の公知の方法で測定することができる。混合溶媒のおよその誘電率および比誘電率としては混合比に基づく比例計算で求めた値を参考としてもよい。
工程(2)では、得られた微細繊維状セルロース分散液に含まれる対イオンAの一部または全部が対イオンBに置換される。これにより、対イオンAの作用により分散していた微細繊維状セルロースが濃縮され、目的の微細繊維状セルロースの濃縮物が得られる。
対イオンBは、微細繊維状セルロースに導入されているイオン性置換基とイオン対を形成可能であり、炭素数がn−1以下(ここで、nは対イオンAに関して述べたとおり、1以上の整数である。)のものである。対イオンA(炭素数n)がいずれの場合であっても、対イオンBの炭素数はn−1以下である限り特に限定されない。微細繊維状セルロースに導入された置換基がアニオン性置換基である場合、対イオンBとしては、例えば、水素イオン、ナトリウムイオン、カリウムイオン、リチウムイオン、セシウムイオン、アンモニウムイオンなどが挙げられるが、特に限定されない。微細繊維状セルロースに導入された置換基がカチオン性置換基である場合、対イオンBとしては、例えば、水酸化物イオン、フッ化物イオン、塩化物イオン、酢酸イオン、臭化物イオン、ヨウ化物イオンなどが挙げられるが、特に限定されない。本実施形態においては、対イオンBの炭素数は、例えば3以下であることがより好ましく、2以下であることがとくに好ましい。
対イオンAの対イオンBへの置換は、置換が十分に行われる限り特に限定されず、例えば、上述の工程により得られた対イオンA型のイオン性置換基が導入されたセルロース繊維の分散液に対し、必要に応じ攪拌しながら対イオンBを含む溶液を添加することにより、行うことができる。なお、対イオンAを対イオンBにより置換できる理由は定かではないが、炭素数の少ない対イオンBのほうが対イオンAよりも立体障害が小さいこと、対イオンBとして対イオンAよりも強酸または強塩基を使用すること等が寄与していると推測される。
また、対イオンBを含む溶液とともに、有機溶媒を添加してもよい。有機溶媒としては、例えば微細化処理工程において用いたものを使用できる。本工程において有機溶媒を添加する場合、添加される有機溶媒の量は、微細繊維状セルロース分散液中のセルロース全体に対して1000質量%以下であることが好ましく、100質量%以下であることがより好ましく、10質量%以下であることがさらに好ましい。なお、本工程においては、有機溶媒は添加しなくともよい。
対イオンBを添加することにより、微細繊維状セルロースを凝集させることができる。この際、凝集を促進させる観点から、微細繊維状セルロースの分散液を撹拌することができる。撹拌時間は、特に限定されないが、例えば1分以上2時間以下とすることができる。微細繊維状セルロースが凝集した液から、適切な方法で必要な程度、溶媒を除去することにより、微細繊維状セルロースの濃縮物を得ることができる。従来法では、再分散性が良好な濃縮物を得るためには、微細繊維状セルロースを凝集させるために大量のアルコール等の有機溶媒を要したが、本実施態様によれば、その必要はない。
このような、本実施態様により得られる、イオン性置換基が導入された微細繊維状セルロースの濃縮物は、新規なものである。この濃縮物は、イオン性置換基の対イオンBを含み、場合によりイオン性置換基の対イオンAを含んでいてもよい。また本実施態様により得られる、イオン性置換基が導入された微細繊維状セルロースの濃縮物は、水系溶媒に分散可能である。なお、濃縮物中に対イオンAおよび対イオンBが含まれるか否かは、例えば、イオンクロマトグラフィ、微量窒素分析装置、蛍光X線分析、またはICP発光分析により確認することができる。
得られた微細繊維状セルロースを含有する組成物は、水系溶媒に再分散することができる。水系溶媒は、できる限り有機溶媒を含まないことが好ましい。水系溶媒中の有機溶媒の量は、例えば10%以下であり、5%以下であることが好ましく、1%以下であることがより好ましい。水系溶媒として、有機溶媒を含まない水を用い得る。再分散液における繊維の含有量は、特に限定されないが、0.02〜10質量%とすることができ、0.1〜5質量%とすることもできる。このような濃度の微細繊維状セルロースは、シートを得るために好ましく、塗工の際の取り扱いおよび分散安定性に優れる。なお、水系溶媒としては、pHの調整が行われていないものを用いることができる。このような水系溶媒のpHは、特に限定されないが、例えばpH5〜7である。
工程(1)で得られる微細セルロース繊維の分散液、工程(2)で得られる濃縮物、または濃縮物からの再分散液には、界面活性剤が含まれてもよい。微細セルロース繊維の含有液に界面活性剤が含まれると、表面張力が低下して、工程基材に対する濡れ性を高めることができ、微細セルロース繊維含有シートをより容易に形成できる。具体的に、微細セルロース繊維含有液の表面張力は25〜45mN/mであることが好ましく、27〜40mN/mであることがより好ましく、30〜38mN/mであることが最も好ましい。微細セルロース繊維含有液の表面張力が前記下限値以上であれば、水を保持しやすい界面活性剤による微細セルロース繊維含有液の乾燥性の低下を防ぐことができ、前記上限値以下であれば、工程基材に対する微細セルロース繊維含有液の濡れ性を充分に向上させることができる。
・リン酸基の導入
針葉樹クラフトパルプとして、王子製紙製のパルプ(固形分93% 米坪208g/m2シート状 離解してJIS P8121に準じて測定されるカナダ標準濾水度(CSF)700ml)を使用した。上記針葉樹クラフトパルプの絶乾質量として100質量部に、リン酸二水素アンモニウムと尿素の混合水溶液を含浸し、リン酸二水素アンモニウム49質量部、尿素130質量部となるように圧搾し、薬液含浸パルプを得た。得られた薬液含浸パルプを105℃の乾燥機で乾燥し、水分を蒸発させてプレ乾燥させた。その後、140℃に設定した送風乾燥機で、10分間加熱し、パルプ中のセルロースにリン酸基を導入し、リン酸化パルプを得た。
得られたリン酸化パルプの絶乾質量として100質量部に対して10000質量部のイオン交換水を注ぎ、攪拌して均一に分散させた後、濾過脱水して、脱水シートを得る工程を2回繰り返し、リン酸化パルプの脱水シートAを得た。
得られたリン酸化パルプの脱水シートAを原料にし、先と同様にして、リン酸基を導入する工程、濾過脱水する工程をさらに2回繰り返して(リン酸化および濾過脱水の合計回数は3回)、リン酸化パルプの脱水シートBを得た。
該リン酸化パルプの脱水シートBは、次に示す滴定法で求められるリン酸基の導入量が1.435mmol/gであった。
置換基導入量は、繊維原料へのリン酸基の導入量であり、この値が大きいほど、多くのリン酸基が導入されている。置換基導入量は、対象となる微細繊維状セルロースをイオン交換水で含有量が0.2質量%となるように希釈した後、イオン交換樹脂による処理、アルカリを用いた滴定によって測定した。イオン交換樹脂による処理では、0.2質量%セルロース繊維含有スラリーに体積で1/10の強酸性イオン交換樹脂(アンバージェット1024;オルガノ株式会社、コンディショング済)を加え、1時間振とう処理を行った。その後、目開き90μmのメッシュ上に注ぎ、樹脂とスラリーを分離した。アルカリを用いた滴定では、イオン交換後のセルロース繊維含有スラリーに、0.1Nの水酸化ナトリウム水溶液を加えながら、スラリーが示す電気伝導度の値の変化を計測した。すなわち、図1に示した曲線の第1領域で必要としたアルカリ量(mmol)を、滴定対象スラリー中の固形分(g)で除して、置換基導入量(mmol/g)とした。
リン酸化パルプの脱水シートBの絶乾質量として100質量部に5000質量部のイオン交換水を加え、希釈した。次いで、攪拌しながら、1N塩酸を少しずつ添加し、pHが2〜3のパルプスラリーを得た。その後、このパルプスラリーを脱水し、脱水シートを得た後、再びイオン交換水を注ぎ、攪拌して均一に分散させた。次いで、濾過脱水して脱水シートを得る操作を繰り返すことにより、余剰の塩酸を十分に洗い流し、リン酸化パルプ(H型)を得た。
製造例1で得られたリン酸化パルプ(H型)の絶乾質量として100質量部に5000質量部のイオン交換水を加え、希釈した。次いで、攪拌しながら、10%テトラブチルアンモニウムヒドロキシド水溶液を少しずつ添加し、pHが10〜12のパルプスラリーを得た。その後、このパルプスラリーを脱水し、脱水シートを得た後、再びイオン交換水を注ぎ、攪拌して均一に分散させた。次いで、濾過脱水して脱水シートを得る操作を繰り返すことにより、余剰のテトラブチルアンモニウムヒドロキシド水溶液を十分に洗い流し、リン酸化パルプ(TBA型)(対イオンの炭素数16)を得た。
10%テトラブチルアンモニウムヒドロキシド水溶液の代わりに、1N NaOH水溶液を用いた以外は製造例2と同様にして、リン酸化パルプ(Na型)(対イオンの炭素数0)を得た。
10%テトラブチルアンモニウムヒドロキシド水溶液の代わりに、10%テトラプロピルアンモニウムヒドロキシド水溶液を用いた以外は製造例2と同様にして、リン酸化パルプ(TPA型)(対イオンの炭素数12)を得た。
10%テトラブチルアンモニウムヒドロキシド水溶液の代わりに、10%テトラエチルアンモニウムヒドロキシド水溶液を用いた以外は製造例2と同様にして、リン酸化パルプ(TEA型)(対イオンの炭素数8)を得た。
・TEMPO酸化反応
乾燥質量100質量部相当の未乾燥の針葉樹晒クラフトパルプとTEMPO1.25質量部と、臭化ナトリウム12.5質量部とを水10000質量部に分散させた。次いで、13質量%次亜塩素酸ナトリウム水溶液を、1.0gのパルプに対して次亜塩素酸ナトリウムの量が8.0mmolになるように加えて反応を開始した。反応中は0.5Mの水酸化ナトリウム水溶液を滴下してpHを10〜11に保ち、pHに変化が見られなくなった時点で反応終了と見なした。
その後、このパルプスラリーを脱水し、脱水シートを得た後、5000質量部のイオン交換水を注ぎ、攪拌して均一に分散させた後、濾過脱水して、脱水シートを得る工程を2回繰り返した。
滴定法により測定される置換基(カルボキシ基)の導入量は1.5mmol/gであった。
さらに、得られた脱水シートに、5000質量部のイオン交換水を加えて希釈した。次いで、攪拌しながら、1N塩酸を少しずつ添加し、pHが2〜3のパルプスラリーを得た。その後、このパルプスラリーを脱水し、脱水シートを得た後、再びイオン交換水を注ぎ、攪拌して均一に分散させた。次いで、濾過脱水して脱水シートを得る操作を繰り返すことにより、余剰の塩酸を十分に洗い流し、TEMPO酸化パルプ(H型)を得た。
得られたTEMPO酸化パルプ(H型)の絶乾質量として100質量部に5000質量部のイオン交換水を加え、希釈した。次いで、攪拌しながら、10% テトラブチルアンモニウムヒドロキシド水溶液を少しずつ添加し、pHが10〜12のパルプスラリーを得た。その後、このパルプスラリーを脱水し、脱水シートを得た後、再びイオン交換水を注ぎ、攪拌して均一に分散させた。次いで、濾過脱水して脱水シートを得る操作を繰り返すことにより、余剰のテトラブチルアンモニウムヒドロキシド水溶液を十分に洗い流し、TEMPO酸化パルプ(TBA型)(対イオンの炭素数16)を得た。
・パルプのフラッフィング
針葉樹晒クラフトパルプ(NBKP)を抄き上げたシート(パルプ含有量90質量%)をハンドミキサー(大阪ケミカル製、ラボミルサーPLUS)を用い、回転数20,000rpmで15秒処理して綿状のフラッフィングパルプ(パルプ含有量90質量%)にした。
次いで、カチオン化剤(カチオマスターG、四日市合成株式会社製、グリシジルトリメチルアンモニウムクロリド、純分73.1質量%、含水率20.2質量%)100質量部と1.5Nの水酸化ナトリウム水溶液70質量部とを混合したカチオン化剤混合液を、スプレーを用いて、前記フラッフィングパルプ100質量部に添加し、ポリ塩化ビニリデン製の袋の中に入れ、その袋を手で揉むことにより、混合液をパルプに均一に浸透させて、反応用試料を調製した。
その後、袋内の空気を除去し、80℃で1時間反応させ、カチオン化パルプAを得た。
得られたカチオン化パルプAに、前記カチオン化剤混合液を、前記同様に添加し、前記同様に反応させカチオン化パルプBを得た。
得られたカチオン化パルプBに5000質量部のイオン交換水を加え、攪拌しながら洗浄した後、脱水した。その洗浄・脱水の処理を4回繰り返し、洗浄済カチオン化パルプを得た。
微量窒素分析法により、洗浄済みカチオン化パルプに含まれるカチオン基量を測定したところ、1.1mmol/gであった。
得られた洗浄済みカチオン化パルプに5000質量部のイオン交換水を加え、さらに1NNaOH水溶液を少しずつ加え、pH11〜12のパルプスラリーを得た。その後、このパルプスラリーを脱水し、脱水シートを得た後、再びイオン交換水を注ぎ、攪拌して均一に分散させた。次いで、濾過脱水して脱水シートを得る操作を繰り返すことにより、余剰の水酸化ナトリウムを十分に洗い流し、カチオン化パルプ(OH型)を得た。
得られたカチオン化パルプ(OH型)の絶乾質量として100質量部に5000質量部のイオン交換水を加え、希釈した。次いで、攪拌しながら、ジブチルリン酸溶液を少しずつ添加し、pHが2〜3のパルプスラリーを得た。その後、このパルプスラリーを脱水し、脱水シートを得た後、再びイオン交換水を注ぎ、攪拌して均一に分散させた。次いで、濾過脱水して脱水シートを得る操作を繰り返すことにより、余剰のジブチルリン酸溶液を十分に洗い流し、カチオン化パルプ(DBP型)(対イオンの炭素数8)を得た。
・微細化前希釈
得られたリン酸化パルプ(TBA型)に、イソプロパノール(IPA)/水質量比率が70/30、リン酸化パルプ(TBA)の含有量が0.7質量%となるようイソプロパノール(対パルプ9900%)、およびイオン交換水(対パルプ4300%)を添加し、微細化前スラリーを得た。
得られた微細化前スラリーAを、高圧ホモジナイザー(NiroSoavi社「Panda Plus 2000」)で、操作圧力60MPaで5パス処理を行い、微細繊維状セルローススラリーを得た。
得られた微細繊維状セルローススラリーA100質量部(0.7質量%)に、撹拌しながら48%水酸化ナトリウム水溶液(炭素数0)0.17質量部、イオン交換水0.6質量部(対パルプ0.9%)、イソプロパノール1.8質量部(対パルプ2.6%)の混合液を加えた。
さらに30分撹拌を続けたところ、微細繊維状セルロースの濃縮物の凝集物が認められた。凝集物が生じた微細繊維状セルローススラリーを、良く撹拌しながら116.6g分取し、フィルターホルダーに装着したメンブレンフィルター(ADVANTEC、φ90mm、PTFE、孔径0.5μm)をろ紙を敷き、アスピレーターで減圧ろ別し、微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。なお、得られた濃縮物中のNa量はイオンクロマトで、TBA量は微量窒素分析装置で確認した(実施例2において同じ。)。
得られた微細繊維状セルロースの濃縮物を、含有量が0.7質量%となるように、イオン交換水で希釈し、ホモディスパー(特殊機化工業製 4000rpm 3分)処理で再分散させた。
表1、表2に、微細化時の溶媒の比誘電率、パルプ重量に対して使用した溶媒量、微細化後のスラリーの全光線透過率、およびヘーズ、凝集物のろ別に掛かった時間、再分散後のスラリーの全光線透過率、およびヘーズを記載した。
JIS規格K7361に準拠し、ヘーズメータ(村上色彩技術研究所社製「HM−150」)を用いて全光線透過率を測定した。
JIS規格K7136に準拠し、ヘーズメータ(村上色彩技術研究所社製「HM−150」)を用いてヘーズを測定した。
上述のようにホモディスパー(特殊機化工業製 8000rpm 3分)処理で再分散処理した直後液を、目視により評価した。
良好:沈殿が認められない。
不良:沈殿が認められる。
微細化前希釈に際し、 分散溶媒をイソプロパノール/水比率が50/50となるようにした以外は実施例1と同様にして、微細繊維状セルロースの濃縮物(微細繊維状セルロースの含有量5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。さらに、再分散性の評価を行った。結果を表1、表2に記載した。
48%水酸化ナトリウム水溶液(炭素数0)0.17質量部の代わりに、水酸化リチウム0.05質量部、イオン交換水0.6質量部(対パルプ0.9%)、イソプロパノール1.8質量部(対パルプ2.6%)の混合液を加えた以外は実施例1と同様にして、微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
48%水酸化ナトリウム水溶液(炭素数0)0.17質量部の代わりに、水酸化カリウム0.11質量部、イオン交換水0.6質量部(対パルプ0.9%)、イソプロパノール1.8質量部(対パルプ2.6%)の混合液を加えた以外は実施例1と同様にして、微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
48%水酸化ナトリウム水溶液(炭素数0)0.17質量部の代わりに、1N塩酸2質量部を加え、対イオンの炭素数低減時にイオン交換水とイソプロパノールを加えなかった以外は実施例1と同様にして、微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
リン酸化パルプ(TBA型)の代わりにリン酸化パルプ(TEA型)を用い、高圧ホモジナイザーの操作圧力を120MPaとした以外は、実施例1と同様にして微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
リン酸化パルプ(TBA型)の代わりにリン酸化パルプ(TPA型)を用い、高圧ホモジナイザーの操作圧力を120MPaとした以外は、実施例1と同様にして微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
リン酸化パルプ(TBA型)の代わりにTEMPO酸化パルプ(TBA型)を用い、高圧ホモジナイザーの操作圧力を120MPaとした以外は、実施例1と同様にして微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
リン酸化パルプ(TBA型)の代わりにカチオン化パルプ(DBP型)を用い、高圧ホモジナイザーの操作圧力を120MPaとし、さらに、対イオンの炭素数低減時に酢酸ナトリウム0.1質量部、イオン交換水0.5質量部を添加した以外は、実施例1と同様にして微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
対イオンの炭素数低減時に塩酸0.04質量部を添加した以外は、実施例9と同様にして微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
対イオンの炭素数低減時に水酸化ナトリウム0.05質量部、イオン交換水0.5質量部を添加した以外は、実施例9と同様にして微細繊維状セルロースの濃縮物(濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上)を得た。また、濃縮物中に含まれる溶媒の25℃における比誘電率が70以下であることを確認した。結果を表1に記載した。
得られた濃縮物のイオン交換水に対する再分散性(目視)も良好であった(表2)。
対イオンの炭素数の低減工程を省略した以外は実施例1と同様にしたところ、凝集物は生成せず、極めて濾水性が悪く、濾過時間は1時間以上であった。このため、比較例1では、濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上である濃縮物は得られなかった。
リン酸化パルプ(TBA型)の代わりにリン酸化パルプ(Na型)を用いた以外は実施例1と同様にしたところ、微細化時に、高圧ホモジナイザーの閉塞が頻発したため、微細化処理が出来なかった。
微細化前の希釈段階で、有機溶媒(イソプロパノール)を用いず、溶媒を水のみとした以外は、実施例1と同様にしたところ、凝集物が生成せず、極めて濾水性が悪く、濾過時間は1時間以上であった。このため、比較例3では、濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上である濃縮物は得られなかった。
リン酸化パルプ(TBA型)の代わりにリン酸化パルプ(Na型)を用い、微細化前の希釈段階で、有機溶媒(イソプロパノール)を用いず、溶媒を水のみとし、炭素数の低減工程の代わりに、微細繊維状セルローススラリーに、有機溶媒(イソプロパノール)を298質量部(対パルプ28400質量%)加えた以外は実施例1と同様にし、微細繊維状セルロースの濃縮物を得て、再分散性の評価を行った。結果を表1、表2に記載した。
微細繊維状セルローススラリーに有機溶媒(イソプロパノール)を71.3質量部(対パルプ9900質量%)加えた以外は比較例4と同様にしたところ、僅かに凝集が生じたが、極めて濾水性が悪く、濾過時間は1時間以上であった。このため、比較例5では、濃縮物全体に対する微細繊維状セルロースの含有量が5質量%以上である濃縮物は得られなかった。
微細繊維状セルローススラリーに、有機溶媒(イソプロパノール)を加える代わりに、塩化アルミニウムを固体で0.08質量部加えた以外は比較例4と同様にし、微細繊維状セルロースの濃縮物を得て、再分散性の評価を行ったが、凝集物は再分散しなかった。結果を表1、表2に記載した。
Claims (13)
- (1)イオン性置換基と、炭素数がn(但しnは1以上の整数)である対イオンAと、を有するセルロース繊維を、25℃における比誘電率が70以下の溶媒中で微細化し、微細繊維状セルロース分散液を得る工程;および
(2)微細繊維状セルロース分散液に含まれる対イオンAの一部または全部を、炭素数がn−1以下である対イオンBに置換して、微細繊維状セルロースの濃縮物を得る工程
を含む、微細繊維状セルロース濃縮物の製造方法。 - イオン性置換基が、アニオン性置換基を含む、請求項1に記載の製造方法。
- アニオン性置換基が、リン酸由来の基、カルボン酸由来の基および硫酸由来の基からなる群より選択される1種または2種以上を含む、請求項2に記載の製造方法。
- 対イオンAが、テトラアルキルアンモニウムイオンを含む、請求項2または3に記載の製造方法。
- イオン性置換基が、カチオン性置換基含む、請求項1に記載の製造方法。
- カチオン性置換基が、4級アンモニウム塩由来の基およびホスホニウム塩由来の基のうちの1種または2種を含む、請求項5に記載の製造方法。
- 対イオンBが、1価のイオンである、請求項1〜6のいずれか1項に記載の製造方法。
- イオン性置換基を有する微細繊維状セルロースと、
炭素数n(ただしnは1以上の整数)である対イオンAと、
炭素数n−1以下である対イオンBと、
有機溶媒と、
を含む組成物。 - 組成物全体に対する微細繊維状セルロースの含有量が、5質量%以上である請求項8に記載の組成物。
- 微細繊維状セルロースが、イオン性置換基としてリン酸由来の基、カルボン酸由来の基および硫酸由来の基からなる群より選択される1種または2種以上を有する、請求項8または9に記載の組成物。
- 微細繊維状セルロースが、イオン性置換基として4級アンモニウム塩由来の基およびホスホニウム塩由来の基のうちの1種または2種を有する、請求項8または9に記載の組成物。
- 対イオンBが、1価のイオンである、請求項8〜11のいずれか1項に記載の組成物。
- 組成物が、有機溶媒を含有する溶媒であって25℃における比誘電率が70以下である溶媒中に微細繊維状セルロースを含む形態である、請求項8〜12のいずれか1項に記載の組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015119587A JP6561607B2 (ja) | 2015-06-12 | 2015-06-12 | 微細繊維状セルロース濃縮物の製造方法、および微細繊維状セルロースを含む組成物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015119587A JP6561607B2 (ja) | 2015-06-12 | 2015-06-12 | 微細繊維状セルロース濃縮物の製造方法、および微細繊維状セルロースを含む組成物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017002231A true JP2017002231A (ja) | 2017-01-05 |
| JP6561607B2 JP6561607B2 (ja) | 2019-08-21 |
Family
ID=57751409
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015119587A Active JP6561607B2 (ja) | 2015-06-12 | 2015-06-12 | 微細繊維状セルロース濃縮物の製造方法、および微細繊維状セルロースを含む組成物 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6561607B2 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018143149A1 (ja) * | 2017-02-03 | 2018-08-09 | 株式会社片山化学工業研究所 | 乾燥セルロースナノファイバーの製造方法 |
| WO2018159743A1 (ja) * | 2017-03-01 | 2018-09-07 | 王子ホールディングス株式会社 | 繊維状セルロース、繊維状セルロース含有組成物、繊維状セルロース分散液及び繊維状セルロースの製造方法 |
| WO2018159473A1 (ja) * | 2017-02-28 | 2018-09-07 | 大王製紙株式会社 | セルロース微細繊維及びその製造方法 |
| JP6404415B1 (ja) * | 2017-07-24 | 2018-10-10 | 大王製紙株式会社 | セルロース微細繊維含有物及びその製造方法、並びにセルロース微細繊維分散液 |
| JP2019023296A (ja) * | 2017-07-24 | 2019-02-14 | 大王製紙株式会社 | セルロース微細繊維含有物及びその製造方法、並びにセルロース微細繊維分散液 |
| JP2020033476A (ja) * | 2018-08-30 | 2020-03-05 | 王子ホールディングス株式会社 | 固形状体及び繊維状セルロース含有組成物 |
| JP2020041255A (ja) * | 2017-01-16 | 2020-03-19 | 株式会社Kri | 硫酸エステル化修飾セルロースナノファイバーの製造方法 |
| EP3553123A4 (en) * | 2016-12-12 | 2020-07-15 | Oji Holdings Corporation | COMPOSITION WITH FIBROUS CELLULOSE |
| CN112739722A (zh) * | 2018-09-21 | 2021-04-30 | 东洋制罐集团控股株式会社 | 纳米纤维素及其生产方法 |
| US11441243B2 (en) | 2017-05-15 | 2022-09-13 | Daio Paper Corporation | Fine cellulose fiber and method for producing same |
| US11578456B2 (en) | 2017-03-01 | 2023-02-14 | Oji Holdings Corporation | Cellulose fibers, cellulose fiber-containing composition, cellulose fiber dispersion, and method for producing cellulose fibers |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003528935A (ja) * | 1999-06-14 | 2003-09-30 | サントル・ナショナル・ドゥ・ラ・ルシェルシュ・シャンティフィク | 有機溶媒中の特にセルロースの微細繊維および/または微結晶分散物 |
| JP2011127067A (ja) * | 2009-12-21 | 2011-06-30 | Teijin Ltd | 微細修飾セルロースの製造方法 |
| JP2011140738A (ja) * | 2009-12-11 | 2011-07-21 | Kao Corp | 微細セルロース繊維複合体、微細セルロース繊維分散液及び複合材料 |
| WO2011111612A1 (ja) * | 2010-03-09 | 2011-09-15 | 凸版印刷株式会社 | 微細セルロース繊維分散液およびその製造方法、セルロースフィルムならびに積層体 |
| JP2012021081A (ja) * | 2010-07-14 | 2012-02-02 | Univ Of Tokyo | セルロースナノファイバー分散液の製造方法、セルロースナノファイバー分散液、セルロースナノファイバー成形体、及びセルロースナノファイバー複合体 |
| JP2013241702A (ja) * | 2012-05-21 | 2013-12-05 | Bridgestone Corp | 多糖類繊維の製造方法、多糖類繊維、コード、繊維−ゴム複合体、及びタイヤ |
| JP2015101694A (ja) * | 2013-11-27 | 2015-06-04 | 凸版印刷株式会社 | セルロースナノファイバー分散体及びセルロース修飾体、ならびに製造方法 |
-
2015
- 2015-06-12 JP JP2015119587A patent/JP6561607B2/ja active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003528935A (ja) * | 1999-06-14 | 2003-09-30 | サントル・ナショナル・ドゥ・ラ・ルシェルシュ・シャンティフィク | 有機溶媒中の特にセルロースの微細繊維および/または微結晶分散物 |
| JP2011140738A (ja) * | 2009-12-11 | 2011-07-21 | Kao Corp | 微細セルロース繊維複合体、微細セルロース繊維分散液及び複合材料 |
| JP2011127067A (ja) * | 2009-12-21 | 2011-06-30 | Teijin Ltd | 微細修飾セルロースの製造方法 |
| WO2011111612A1 (ja) * | 2010-03-09 | 2011-09-15 | 凸版印刷株式会社 | 微細セルロース繊維分散液およびその製造方法、セルロースフィルムならびに積層体 |
| JP2012021081A (ja) * | 2010-07-14 | 2012-02-02 | Univ Of Tokyo | セルロースナノファイバー分散液の製造方法、セルロースナノファイバー分散液、セルロースナノファイバー成形体、及びセルロースナノファイバー複合体 |
| JP2013241702A (ja) * | 2012-05-21 | 2013-12-05 | Bridgestone Corp | 多糖類繊維の製造方法、多糖類繊維、コード、繊維−ゴム複合体、及びタイヤ |
| JP2015101694A (ja) * | 2013-11-27 | 2015-06-04 | 凸版印刷株式会社 | セルロースナノファイバー分散体及びセルロース修飾体、ならびに製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| FUJISAWA, S. ET AL., CELLULOSE, vol. 19, no. 2, JPN6018027293, 2012, pages 459 - 466, ISSN: 0004014922 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3553123A4 (en) * | 2016-12-12 | 2020-07-15 | Oji Holdings Corporation | COMPOSITION WITH FIBROUS CELLULOSE |
| JP2020041255A (ja) * | 2017-01-16 | 2020-03-19 | 株式会社Kri | 硫酸エステル化修飾セルロースナノファイバーの製造方法 |
| WO2018143149A1 (ja) * | 2017-02-03 | 2018-08-09 | 株式会社片山化学工業研究所 | 乾燥セルロースナノファイバーの製造方法 |
| JP7270194B2 (ja) | 2017-02-03 | 2023-05-10 | 株式会社片山化学工業研究所 | 乾燥セルロースナノファイバーの製造方法 |
| JP2021143343A (ja) * | 2017-02-03 | 2021-09-24 | 株式会社片山化学工業研究所 | 乾燥セルロースナノファイバーの製造方法 |
| JPWO2018143149A1 (ja) * | 2017-02-03 | 2019-11-21 | 株式会社片山化学工業研究所 | 乾燥セルロースナノファイバーの製造方法 |
| WO2018159473A1 (ja) * | 2017-02-28 | 2018-09-07 | 大王製紙株式会社 | セルロース微細繊維及びその製造方法 |
| JP2018141249A (ja) * | 2017-02-28 | 2018-09-13 | 大王製紙株式会社 | セルロース微細繊維及びその製造方法 |
| US10975518B2 (en) | 2017-02-28 | 2021-04-13 | Daio Paper Corporation | Cellulose microfibers and method for manufacturing the same |
| WO2018159743A1 (ja) * | 2017-03-01 | 2018-09-07 | 王子ホールディングス株式会社 | 繊維状セルロース、繊維状セルロース含有組成物、繊維状セルロース分散液及び繊維状セルロースの製造方法 |
| US11578456B2 (en) | 2017-03-01 | 2023-02-14 | Oji Holdings Corporation | Cellulose fibers, cellulose fiber-containing composition, cellulose fiber dispersion, and method for producing cellulose fibers |
| US11441243B2 (en) | 2017-05-15 | 2022-09-13 | Daio Paper Corporation | Fine cellulose fiber and method for producing same |
| CN110959016A (zh) * | 2017-07-24 | 2020-04-03 | 大王制纸株式会社 | 纤维素微细纤维含有物及其制造方法以及纤维素微细纤维分散液 |
| JP2019023365A (ja) * | 2017-07-24 | 2019-02-14 | 大王製紙株式会社 | セルロース微細繊維含有物及びその製造方法、並びにセルロース微細繊維分散液 |
| KR102616191B1 (ko) | 2017-07-24 | 2023-12-20 | 다이오 페이퍼 코퍼레이션 | 셀룰로오스 미세섬유 함유물 및 그 제조 방법, 그리고 셀룰로오스 미세섬유 분산액 |
| JP6404415B1 (ja) * | 2017-07-24 | 2018-10-10 | 大王製紙株式会社 | セルロース微細繊維含有物及びその製造方法、並びにセルロース微細繊維分散液 |
| JP2019023296A (ja) * | 2017-07-24 | 2019-02-14 | 大王製紙株式会社 | セルロース微細繊維含有物及びその製造方法、並びにセルロース微細繊維分散液 |
| US11220787B2 (en) | 2017-07-24 | 2022-01-11 | Daio Paper Corporation | Fine cellulose fiber-containing substance, method for manufacturing the same, and fine cellulose fiber dispersion |
| CN110959016B (zh) * | 2017-07-24 | 2022-04-01 | 大王制纸株式会社 | 纤维素微细纤维含有物及其制造方法以及纤维素微细纤维分散液 |
| KR20200034709A (ko) * | 2017-07-24 | 2020-03-31 | 다이오 페이퍼 코퍼레이션 | 셀룰로오스 미세섬유 함유물 및 그 제조 방법, 그리고 셀룰로오스 미세섬유 분산액 |
| WO2019021619A1 (ja) * | 2017-07-24 | 2019-01-31 | 大王製紙株式会社 | セルロース微細繊維含有物及びその製造方法、並びにセルロース微細繊維分散液 |
| JP7255106B2 (ja) | 2018-08-30 | 2023-04-11 | 王子ホールディングス株式会社 | 固形状体及び繊維状セルロース含有組成物 |
| JP2020033476A (ja) * | 2018-08-30 | 2020-03-05 | 王子ホールディングス株式会社 | 固形状体及び繊維状セルロース含有組成物 |
| CN112739722A (zh) * | 2018-09-21 | 2021-04-30 | 东洋制罐集团控股株式会社 | 纳米纤维素及其生产方法 |
| CN112739722B (zh) * | 2018-09-21 | 2023-08-08 | 东洋制罐集团控股株式会社 | 纳米纤维素及其生产方法 |
| US11905340B2 (en) | 2018-09-21 | 2024-02-20 | Toyo Seikan Group Holdings, Ltd. | Nanocellulose and method for producing the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6561607B2 (ja) | 2019-08-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6561607B2 (ja) | 微細繊維状セルロース濃縮物の製造方法、および微細繊維状セルロースを含む組成物 | |
| JP7088273B2 (ja) | 微細繊維状セルロース再分散スラリーの製造方法および微細繊維状セルロース再分散スラリー | |
| JP6613771B2 (ja) | 微細繊維状セルロース含有物 | |
| WO2017047768A1 (ja) | 微細繊維状セルロース含有物 | |
| JP5910786B1 (ja) | 微細繊維状セルロース含有物 | |
| JP6269617B2 (ja) | 微細繊維状セルロース凝集物 | |
| JP6601088B2 (ja) | 微細繊維状セルロース含有物 | |
| JP6907489B2 (ja) | 微細繊維状セルロース含有物 | |
| JP6418212B2 (ja) | 微細繊維状セルロース含有物 | |
| JP6361836B1 (ja) | 繊維状セルロース含有物及び繊維状セルロース含有物の製造方法 | |
| KR20160008607A (ko) | 인산에스테르화 미세 셀룰로오스 섬유 및 그 제조 방법 | |
| WO2017141800A1 (ja) | 繊維状セルロース含有物及び繊維状セルロース含有物の製造方法 | |
| JP7355028B2 (ja) | 微細繊維状セルロース含有組成物およびその製造方法 | |
| WO2020059859A1 (ja) | セルロースファイバーボールおよびこれを含有する紙 | |
| JP6418213B2 (ja) | 微細繊維状セルロース含有物 | |
| JP2020033398A (ja) | 微細繊維状セルロース含有組成物およびその製造方法 | |
| JP6579286B1 (ja) | 樹脂組成物および成形体 | |
| JP6504094B2 (ja) | 微細繊維状セルロース含有物 | |
| JP7127005B2 (ja) | 微細繊維状セルロース含有物 | |
| WO2018110525A1 (ja) | 繊維状セルロース含有組成物 | |
| WO2021107147A1 (ja) | 繊維状セルロース、繊維状セルロース分散液及びシート | |
| WO2021107146A1 (ja) | 繊維状セルロース、繊維状セルロース分散液及びシート | |
| JP2021155738A (ja) | 粉塵飛散防止剤及び粉塵飛散防止方法 | |
| WO2020050348A1 (ja) | 繊維状セルロース含有物、フラッフ化セルロース及び組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170906 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180717 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180918 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181115 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190416 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190611 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190625 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190708 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6561607 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |