JP2017001990A - 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 - Google Patents
1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 Download PDFInfo
- Publication number
- JP2017001990A JP2017001990A JP2015118436A JP2015118436A JP2017001990A JP 2017001990 A JP2017001990 A JP 2017001990A JP 2015118436 A JP2015118436 A JP 2015118436A JP 2015118436 A JP2015118436 A JP 2015118436A JP 2017001990 A JP2017001990 A JP 2017001990A
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- base
- activated carbon
- production
- trichloro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- OQISUJXQFPPARX-UHFFFAOYSA-N C=C(C(F)(F)F)Cl Chemical compound C=C(C(F)(F)F)Cl OQISUJXQFPPARX-UHFFFAOYSA-N 0.000 description 1
- OMMADTGFNSRNEJ-UHFFFAOYSA-N FC(C(CCl)(Cl)Cl)(F)F Chemical compound FC(C(CCl)(Cl)Cl)(F)F OMMADTGFNSRNEJ-UHFFFAOYSA-N 0.000 description 1
Images

Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【課題】工業的に実施可能な方法で、1,2−ジクロロ−3,3,3−トリフルオロプロペンを効率的に製造する方法の提供。【解決手段】1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する、または、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、ことを特徴とする1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法。【選択図】図1
Description
本発明は、1,2−ジクロロ−3,3,3−トリフルオロプロペンを製造する方法に関する。
1,2−ジクロロ−3,3,3−トリフルオロプロペン(CClH=CCl−CF3、HCFO−1223xd。以下、1223xdとも記す。)は、3,3−ジクロロ−1,1,1,2,2−ペンタフルオロプロパン(CF3−CF2−CHCl2、HCFC−225ca)や1,3−ジクロロ−1,1,2,2,3−ペンタフルオロプロパン(CClF2−CF2−CClFH、HCFC−225cb)に代わる地球温暖化係数(GWP)の小さい新しい洗浄剤、冷媒、発泡剤、溶剤、およびエアゾール用途に用いられる化合物である。
本明細書において、ハロゲン化炭化水素については、化合物名の後の括弧内にその化合物の略称を記すが、本明細書では必要に応じて化合物名に代えてその略称を用いる。
1223xdは二重結合上の置換基の位置により、幾何異性体であるZ体とE体が存在する。本明細書中では特に断らずに化合物名や化合物の略称を用いた場合には、Z体およびE体から選ばれる少なくとも1種を示し、化合物名や化合物の略称の後ろに(E)または(Z)を付した場合には、其々の化合物のE体またはZ体であることを示す。例えば、1223xd(Z)はZ体を示し、1223xd(E)はE体を示す。
1223xdは二重結合上の置換基の位置により、幾何異性体であるZ体とE体が存在する。本明細書中では特に断らずに化合物名や化合物の略称を用いた場合には、Z体およびE体から選ばれる少なくとも1種を示し、化合物名や化合物の略称の後ろに(E)または(Z)を付した場合には、其々の化合物のE体またはZ体であることを示す。例えば、1223xd(Z)はZ体を示し、1223xd(E)はE体を示す。
1223xdを製造する方法として、例えば、1,1,2−トリクロロ−3,3,3−トリフルオロプロパン(CCl2H−CClH−CF3、HCFC−233da)を気相中で活性炭と接触させて脱塩化水素反応させる方法や、液相中で塩基の存在下で接触させて脱HCl反応させる方法、等が開示されている。
これらの方法は、いずれもHCFC−233daを原料とするため、目的物である1223xd(Z)(沸点53℃)、1223xd(E)(沸点60℃)の他、1,1−ジクロロ−3,3,3−トリフルオロプロペン(CCl2=CH−CF3、HCFO−1223za、沸点55℃)が副生する。沸点差が小さい副生物を除いて高純度な1223xdを得るためには蒸留操作等による高度な精製工程が必要となり、その工程において多量の収量のロスがありうるため、生産効率が悪い。
1,2,2−トリクロロ−3,3,3−トリフルオロプロパン(CClH−CCl2−CF3、HFO−233ab。以下、233abとも記す。)を原料とし、これを脱塩化水素反応させる1223xdの製造方法も検討されている。
特許文献1には、特定の金属触媒を用いて、233abを脱塩化水素反応させて1223xdを製造する方法が開示されている。しかし、この方法は、特定の金属触媒が高価であり、かつ、連続反応時には金属触媒の交換が必要となる方法であることから、工業的規模で生産しようとする場合の経済性と生産性が劣る。
非特許文献1には、液体状態の233abに粉末状の水酸化カリウムを分散させて脱塩化水素反応を行い、1223xdを得る方法が開示されている。しかし、この方法は、収率が低く(48%)、不均一反応であるために反応のコントロールが難しく、工業的な製造方法として実施しにくい。
R.N.Haszeldine et al.,J.Chem.Soc.,1951,p.2495−2504
本発明は、1223xdを高純度で製造するための原料として1,2,2−トリクロロ−3,3,3−トリフルオロプロパン(233ab)を選択し、特定の反応条件を採用して脱HCl反応を起こすことにより、工業的に実施可能で、効率的に目的化合物を得ることができ、かつ、経済的に有利な製造方法を提供する。
本発明は、以下の[1]〜[15]の構成を有する1,2−ジクロロ−3,3,3−トリフルオロプロペン(1223xd)の製造方法を提供する。
[1]1,2,2−トリクロロ−3,3,3−トリフルオロプロパン(233ab)を、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する、または、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、ことを特徴とする1,2−ジクロロ−3,3,3−トリフルオロプロペン(1223xd)の製造方法。
[2]前記塩基が、金属水酸化物、金属酸化物および金属炭酸塩からなる群より選ばれる少なくとも1種の塩基である、[1]に記載の製造方法。
[3]前記塩基を、233abの1モルに対して0.5〜2.0モル用いる、[1]または[2]に記載の製造方法。
[4]前記233abを、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する[1]〜[3]のいずれかに記載の製造方法。
[5]前記脱塩化水素反応の反応温度が100〜350℃である[4]に記載の製造方法。
[6]ガス線速を毎秒0.5〜3.0cmとしながら233abを反応系中に導入する[4]または[5]に記載の製造方法。
[7]前記233abを、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、[1]〜[3]のいずれかに記載の製造方法。
[8]前記脱塩化水素反応の反応温度が0〜100℃である[7]に記載の製造方法。
[9]前記脱塩化水素反応の反応温度が、5〜100℃である、[7]または[8]に記載の製造方法。
[10]前記溶液状態の塩基が、水溶液の状態の塩基である[7]〜[9]のいずれかに記載の製造方法。
[11]前記溶液状態の塩基が、溶液全量に対して塩基を0.5質量%〜40質量%含む溶液状態の塩基である[7]〜[10]のいずれかに記載の製造方法。
[12]前記脱塩化水素反応を、相間移動触媒の存在下に行う、[7]〜[11]のいずれかに記載の製造方法。
[13]前記相間移動触媒が、第4級アンモニウム塩である、[12]に記載の製造方法。
[14]前記第4級アンモニウム塩が、テトラ−n−ブチルアンモニウムクロリド、テトラ−n−ブチルアンモニウムブロミドおよびメチルトリ−n−オクチルアンモニウムクロリドからなる群より選ばれる少なくとも1種である、[13]に記載の製造方法。
[15]1223xdを、2−クロロ−3,3,3−トリフルオロプロペン(HCFO−1233xf)を塩素化して233abを得て、つぎに該233abを脱塩化水素反応して得る、[1]〜[14]のいずれかに記載の製造方法。
[1]1,2,2−トリクロロ−3,3,3−トリフルオロプロパン(233ab)を、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する、または、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、ことを特徴とする1,2−ジクロロ−3,3,3−トリフルオロプロペン(1223xd)の製造方法。
[2]前記塩基が、金属水酸化物、金属酸化物および金属炭酸塩からなる群より選ばれる少なくとも1種の塩基である、[1]に記載の製造方法。
[3]前記塩基を、233abの1モルに対して0.5〜2.0モル用いる、[1]または[2]に記載の製造方法。
[4]前記233abを、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する[1]〜[3]のいずれかに記載の製造方法。
[5]前記脱塩化水素反応の反応温度が100〜350℃である[4]に記載の製造方法。
[6]ガス線速を毎秒0.5〜3.0cmとしながら233abを反応系中に導入する[4]または[5]に記載の製造方法。
[7]前記233abを、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、[1]〜[3]のいずれかに記載の製造方法。
[8]前記脱塩化水素反応の反応温度が0〜100℃である[7]に記載の製造方法。
[9]前記脱塩化水素反応の反応温度が、5〜100℃である、[7]または[8]に記載の製造方法。
[10]前記溶液状態の塩基が、水溶液の状態の塩基である[7]〜[9]のいずれかに記載の製造方法。
[11]前記溶液状態の塩基が、溶液全量に対して塩基を0.5質量%〜40質量%含む溶液状態の塩基である[7]〜[10]のいずれかに記載の製造方法。
[12]前記脱塩化水素反応を、相間移動触媒の存在下に行う、[7]〜[11]のいずれかに記載の製造方法。
[13]前記相間移動触媒が、第4級アンモニウム塩である、[12]に記載の製造方法。
[14]前記第4級アンモニウム塩が、テトラ−n−ブチルアンモニウムクロリド、テトラ−n−ブチルアンモニウムブロミドおよびメチルトリ−n−オクチルアンモニウムクロリドからなる群より選ばれる少なくとも1種である、[13]に記載の製造方法。
[15]1223xdを、2−クロロ−3,3,3−トリフルオロプロペン(HCFO−1233xf)を塩素化して233abを得て、つぎに該233abを脱塩化水素反応して得る、[1]〜[14]のいずれかに記載の製造方法。
本発明によれば、金属触媒に比べ安価かつ連続反応においてもより安定と考えられる活性炭を用いることにより、より長時間の脱塩化水素反応を維持でき、経済的であり工業的規模での生産において有利である。また、塩基または溶媒に溶解した塩基を用いることにより、安価な塩基を存在させる(不足した場合は供給する)ことで、脱塩化水素反応を維持でき、経済的である。このように、工業的に実施可能な簡便な方法で、233abから高純度の1223xdを効率的に製造することができ、経済的に有利である。
本発明の1223xdの製造方法は、活性炭もしくは塩基の存在下に気相中で233abの脱塩化水素反応を行う、または、活性炭もしくは溶液状態の塩基の存在下に液相中で233abの脱塩化水素反応を行う、ことを特徴とする。本発明の製造方法は、活性炭もしくは塩基が気相中で233abに接触する条件下で脱塩化水素反応を行う、または、活性炭もしくは溶液状態の塩基が液相中で233abに接触する条件下で脱塩化水素反応を行う、ことが好ましい。
本発明において提供される1223xdの製造方法は、気相中での脱塩化水素反応(以下、A法という)、および、液相中での脱塩化水素反応(以下、B法という)である。
A法としては、233abを気相中で活性炭または塩基と接触させる方法によることが好ましい。
B法としては、233abを液相中で活性炭または、溶媒に溶解した塩基、すなわち溶液状態の塩基と接触させることが好ましい。
A法としては、233abを気相中で活性炭または塩基と接触させる方法によることが好ましい。
B法としては、233abを液相中で活性炭または、溶媒に溶解した塩基、すなわち溶液状態の塩基と接触させることが好ましい。
本発明において、気相中での脱塩化水素反応とは、233abが気相状態で脱塩化水素反応することをいい、液相中での脱塩化水素反応とは233abが液相状態で脱塩化水素反応することをいう。
本発明の脱塩化水素化反応においては、反応の系中を均一系の状態にして反応させることが好ましい。均一系とは、反応系中に反応基質(原料、生成物、反応に用いる反応資材等)が、ほぼ均一に分布している系をいう。
本発明の製造方法に係る233abの脱塩化水素反応は、下式(1)で示される。

本発明の製造方法で得られる1223xdは、Z体およびE体の混合物であってもよく、Z体のみであってもよく、E体のみでもよい。1223xdの有用性の観点からは、1223xdはZ体が好ましい。本発明の製造方法によれば、1223xd(Z)を必須とする1223xdを効率的に製造できる。さらに、本発明の製造方法によれば、1223xd(E)に対する1223xd(Z)の割合が多い1223xdを得ることができる。
本発明の製造方法に用いる233abは、含フッ素化合物の製造原料または中間体として知られる公知の化合物であり、公知の製造方法により製造できる。例えば、下式(2)に示される反応により、2−クロロ−3,3,3−トリフルオロプロペン(HCFO−1233xf)と塩素を反応させることにより、233abを製造できる。
式(2)の反応の出発物質であるHCFO−1233xfは、2,3,3,3−テトラフルオロ−1−プロペン(HFO−1234yf)の製造原料または中間体として知られる公知の化合物であり、公知の製造方法により入手できる。
HCFO−1233xfの製造方法としては、1,1,2,3−テトラクロロプロペン、1,1,1,2,3−ペンタクロロプロパン、および/または、2,3,3,3−テトラクロロプロペンをフッ素化触媒および安定剤の存在下に気相でフッ化水素と反応させる方法が挙げられる。
HCFO−1233xfの製造方法としては、1,1,2,3−テトラクロロプロペン、1,1,1,2,3−ペンタクロロプロパン、および/または、2,3,3,3−テトラクロロプロペンをフッ素化触媒および安定剤の存在下に気相でフッ化水素と反応させる方法が挙げられる。
式(2)の反応において、HCFO−1233xfと塩素を反応させる際の塩素量としては、副生成物を抑制の観点から、HCFO−1233xf:塩素(Cl2、モル比)を0.2:1〜5:1とするのが好ましく、1:1〜5:1とするのがより好ましい。
式(2)の反応温度は、反応速度を上げる観点から、0〜100℃が好ましく、10〜50℃がより好ましい。式(2)の反応は、反応速度を上げる観点から、光照射下で行うことが好ましい。光照射に用いる光は、過塩素化体である1,1,2,2−テトラクロロ−3,3,3−トリフルオロプロパン(HCFC−223aa)および1,1,1,2,2−クロロ−3,3,3−トリフルオロプロパン(CFC−213ab)の副生を抑制する観点から、波長が300〜780nmの光が好ましく、波長が380〜780nmの光がより好ましい。
式(2)の反応圧力は0〜1MPa(ゲージ圧)が好ましく、製造効率の観点から0.1〜1MPa(ゲージ圧)がより好ましい。本明細書においては、特に記載しない限り、圧力はゲージ圧で示す。HCFO−1233xfと塩素の反応器内の滞留時間は、HCFO−1233xfの転化率の観点から30〜300分間が好ましく、45〜90分間がより好ましい。
上記反応により得られた反応粗生成物は、水またはアルカリで洗浄することで、233abを含む生成物が得られる。該生成物中には、233ab以外に、副生物として1,1,2,2−テトラクロロ−3,3,3−トリフルオロプロパン(CCl2H−CCl2−CF3、HCFC−223aa)、CFC−213ab等が含まれる場合がある。副生成物が含まれる場合は、必要に応じて生成物を精製するのが好ましい。精製方法としては、通常の分離方法を適用でき、該分離方法としては、例えば蒸留等の方法が挙げられる。
本発明の製造方法における233abとしては、前記方法により得た233abを用いるのが好ましい。233abとしては、233abが100%であるものが概念上は好ましいが、経済性の観点からは不純物を含む233abであってもよい。ただし、不純物は、233abの脱塩化水素反応を阻害しないような化合物が好ましい。不純物としては233ab以外のHCFO−1233xfの塩素化物が挙げられ、HCFC−223aa、CFC−213ab等が例示できる。
不純物を含む233abの純度は、不純物と233abの総量を100質量%とした場合、85質量%以上100質量%未満が好ましく、95質量%以上100質量%未満がより好ましい。
不純物を含む233abとしては、233abを主成分とし、HCFC−223aaおよびCFC−213abから選ばれる少なくとも1種の化合物を含むものが好ましい。HCFC−223aaおよびCFC−213abの総量の割合は、不純物を含む233abを100モル%とした場合に、0モル%超15モル%以下が好ましく、効率よく1223xdを製造するため0モル%超5モル%以下がより好ましい。
本発明の製造方法を、気相中で行う場合(A法)と、液相中で行う場合(B法)について、それぞれ以下に説明する。
転化率は、反応に使用した原料(特に記載しない限り、233ab)量に対する、反応で消費された原料(233ab)量の割合(単位:%)を示し、選択率は、生成物の全量に対する、目的物(1223xd)の生成量の割合(単位:%)を示す。
転化率は、反応に使用した原料(特に記載しない限り、233ab)量に対する、反応で消費された原料(233ab)量の割合(単位:%)を示し、選択率は、生成物の全量に対する、目的物(1223xd)の生成量の割合(単位:%)を示す。
[A法]233abを気相中で脱塩化水素反応する方法
A法は、以下のいずれかの方法であり、A−1法が好ましい。
A−1法:活性炭の存在下に気相中で脱塩化水素反応する方法。
A−2法:塩基の存在下に気相中で脱塩化水素反応する方法。
A法は、以下のいずれかの方法であり、A−1法が好ましい。
A−1法:活性炭の存在下に気相中で脱塩化水素反応する方法。
A−2法:塩基の存在下に気相中で脱塩化水素反応する方法。
A−1法は、233abを活性炭の存在下に気相中で脱塩化水素反応する方法である。A−1法で用いる活性炭の入手方法は特に限定されない。活性炭の比表面積は、反応変換率の向上、および副生物の抑制の両立が容易である点から、10〜3000m2/gが好ましく、20〜2500m2/gがより好ましく、50〜2000m2/gがさらに好ましい。活性炭の比表面積の下限を前記値にすると、233abの反応率が向上する。活性炭の比表面積の上限を前記値にすると、活性点が減少し、副生物の生成を抑制しやすい。活性炭の比表面積は、BET法に準拠した方法で測定される。
活性炭の種類としては、木炭、石炭、ヤシ殻等から調製された活性炭等が挙げられる。活性炭の形状としては、長さ2〜5mm程度の成形炭、4〜50メッシュ程度の破砕炭、粒状炭、粉末炭等が挙げられる。233abを気相で反応させる場合は、4〜20メッシュの破砕炭または成形炭が好ましい。
活性炭の灰分は、15%以下が好ましく、10%以下がより好ましく、8%以下がさらに好ましい。活性炭の灰分が上限を超えると、副反応が起こりやすくなる。活性炭の灰分は、ASTM D2866に準じて測定される。活性炭の灰分は、酸による洗浄等の公知の方法で除去できる。活性炭の灰分量は、石炭等を原料とした活性炭においては15%を超える場合がありうるが、塩酸等の酸で洗浄することにより、灰分を15%以下にできる。
活性炭は、反応に用いる前に充分に乾燥させることが好ましい。該活性炭中の水分量は、活性炭と水分の総量を100質量%とした場合に、10質量%以下が好ましく、5質量%以下がより好ましく、1質量%以下が特に好ましい。
A−1法を実施する際には、まず、活性炭を充填した触媒層を形成し、該触媒層に233abをガス状で導入する方法により実施するのが好ましい。
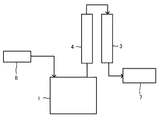
図1は、A−1法を実施するフローの概念図であり、233abを活性炭の存在下に気相中で脱塩化水素反応する例を示すフロー図である。加熱手段(図示されない)によって加熱された反応器1に、ガス状の233ab(図1中、符号5で示す。)、および必要に応じて希釈ガス6を導入する。目的物である1223xdを含むガス状の反応組成物を反応器1の下部から連続的に取り出す。反応器1の下部から取り出された反応組成物の一部は、組成管理をするために一部を採取し、ガスクロマトグラフィ2(GC)による組成分析を行ってもよい。また、反応組成物は、必要に応じて塩化水素を取り除くために脱酸塔3に通した後、生成物7を得る。
図1においては、得られたガス状の反応組成物を冷却等により液化して(図示されない)粗液として回収してもよい。液化する場合は、脱酸塔3の前に行うのが好ましく、液化した回収した粗液は、つぎに脱酸塔3に導入して塩化水素を取り除くのが好ましい。
図1の反応器1としては、触媒層を形成できる公知の反応器を採用するのが好ましい。該反応器1としては、例えば、固定床型反応器、流動床型反応器が挙げられる。反応器1の材質としては、鉄、ニッケル、これらを主成分とする合金、ガラス等が挙げられる。
反応器1に触媒層を形成する場合、活性炭を反応器1に充填することによって形成されうる。触媒層は、反応器1内に1層あっても、2層以上あってもよい。触媒層における活性炭の充填密度は、0.2〜1.0g/cm3が好ましく、0.25〜0.7g/cm3がより好ましい。活性炭の充填密度が下限以上であれば、単位容積あたりの活性炭の充填量が多く、接触させるガス量を多くすることができるため、生産性が向上する。活性炭の充填密度が上限以下であれば、触媒層の温度上昇を抑制しやすく、接触温度の管理が容易になる。
活性炭の触媒寿命を延ばし、転化率を向上し、選択率を向上するためには、ガス状の233abとともに、希釈ガスを導入することが好ましい。希釈ガスとしては、不活性ガス(窒素ガス、希ガス、脱塩化水素化に不活性なフロン類のガス等)や、塩化水素等が挙げられる。希釈ガスの割合は、不活性ガスの回収率の観点から、233abの1モルに対して、10モル以下が好ましく、4モル以下がより好ましい。
脱塩化水素反応は加熱条件下に行うのが好ましく、反応温度は、脱塩化水素反応の反応率の観点から、50〜500℃が好ましく、100〜350℃がより好ましく、160〜330℃がさらに好ましく、200〜300℃が最も好ましい。加熱手段としては、電気炉、オイルバス等が挙げられる。
脱塩化水素反応の圧力は、常圧、加圧、および減圧のいずれであってもよく、常圧または加圧が好ましい。脱塩化水素反応の圧力は、0〜10MPaが好ましく、0.05〜5MPaがより好ましく、0.05〜1MPaがさらに好ましい。ただし、気相中での反応を行うために、該圧力は、脱塩化水素反応の反応温度において、233abの蒸気圧以下であることが好ましい。
脱塩化水素反応の時間は、1〜1000秒が好ましく、5〜300秒がさらに好ましく、10〜100秒が特に好ましい。反応の時間を前記範囲とすることにより、原料の転化率と選択率を制御することができる。
触媒層中の233abの線速度は、0.1〜100cm/秒が好ましい。上記線速度の下限以上であれば、生産性が向上する。上記線速度の上限以下であれば、233abの反応率が向上する。ガス状の233abを希釈ガスと混合して反応に用いる場合、線速度は、成分の比率によって上記範囲で任意に調整することができる。
線速度uは、反応器に導入される233ab量と触媒層の体積とから、下式(3)によって計算される。
u=(W/100)×V/S 式(3)
W:触媒層に導入される全ガス状物質中の233abの濃度(モル%)。
V:触媒層に導入される全ガスの流量(cm3/秒)。
S:触媒層のガスの流通方向に対する断面積(cm2)。
線速度uは、反応器に導入される233ab量と触媒層の体積とから、下式(3)によって計算される。
u=(W/100)×V/S 式(3)
W:触媒層に導入される全ガス状物質中の233abの濃度(モル%)。
V:触媒層に導入される全ガスの流量(cm3/秒)。
S:触媒層のガスの流通方向に対する断面積(cm2)。
反応組成物には、目的物の他に、未反応の原料である233abや副生物が含まれ、該副生物には、塩化水素が含まれる。塩化水素は、脱酸塔3を通す等の脱酸工程、または蒸留工程等の後処理工程により容易に除去できる。脱酸工程としては金属水酸化物またはその水溶液と接触させて中和する方法も挙げられる。金属水酸化物としては、水酸化ナトリウム、水酸化カリウム等が挙げられる。
A−2法は、233abを塩基の存在下に気相中で脱塩化水素反応する方法である。
A−2法で用いる塩基は、前記脱塩化水素反応が実行可能な塩基を特に限定せずに用い得る。塩基としては、金属水酸化物、金属酸化物および金属炭酸塩からなる群より選ばれる少なくとも1種が好ましい。
A−2法で用いる塩基は、前記脱塩化水素反応が実行可能な塩基を特に限定せずに用い得る。塩基としては、金属水酸化物、金属酸化物および金属炭酸塩からなる群より選ばれる少なくとも1種が好ましい。
上記金属水酸化物としては、アルカリ土類金属水酸化物、アルカリ金属水酸化物などが挙げられる。アルカリ土類金属水酸化物としては、水酸化マグネシウム、水酸化カルシウム、水酸化ストロンチウム、水酸化バリウムが好ましく、アルカリ金属水酸化物としては、水酸化リチウム、水酸化ナトリウム、水酸化カリウムが好ましい。
上記金属酸化物としては、アルカリ金属、アルカリ土類金属等の金属の酸化物が挙げられる。アルカリ金属酸化物としては、酸化ナトリウムが好ましく、アルカリ土類金属酸化物としては、酸化カルシウムが好ましい。
また金属酸化物は、1種の金属の酸化物であってもよく、2種以上の金属の複合酸化物であってもよい。
また金属酸化物は、1種の金属の酸化物であってもよく、2種以上の金属の複合酸化物であってもよい。
金属炭酸塩としては、アルカリ土類金属炭酸塩、アルカリ金属炭酸塩などが挙げられる。アルカリ土類金属炭酸塩としては、ベリリウム、マグネシウム、カルシウム、ストロンチウム、バリウム、ラジウムの炭酸塩が挙げられる。アルカリ金属炭酸塩としては、リチウム、ナトリウム、カリウム、ルビジウム、セシウム、フランシウムの炭酸塩が挙げられる。
上記塩基としては、金属水酸化物からなる群から選ばれる少なくとも1種が好ましく、水酸化カリウムおよび/または水酸化ナトリウムが特に好ましい。A−2法に用いる塩基は、1種であっても、2種以上の併用であってもよい。
A−2法を実施する際には、例えば、固体の塩基を充填した充填層を形成し、充填層に233abを含むガスを導入する方法が挙げられる。固体の塩基は、粉末状、ペレット状、ビーズ状等の種々の形態のものが使用できる。
A−1法における脱塩化水素反応のフローは、図1に示すフローと同様であり、A−1法において説明した活性炭および触媒層を塩基に変えて実施できる。
反応器の種類および材質はA−1法と同様のものを使用できる。
A−2における塩基は、充填層に充填して用いることが好ましい。充填層中の塩基の充填密度は、効率よく233abと接触させる観点から、0.2〜1.0g/cm3が好ましく、0.25〜0.7g/cm3がより好ましい。
反応器の種類および材質はA−1法と同様のものを使用できる。
A−2における塩基は、充填層に充填して用いることが好ましい。充填層中の塩基の充填密度は、効率よく233abと接触させる観点から、0.2〜1.0g/cm3が好ましく、0.25〜0.7g/cm3がより好ましい。
脱塩化水素反応の反応温度は、脱塩化水素反応の反応率の観点から、50〜500℃が好ましく、100〜350℃がより好ましい。
反応器内の圧力は、常圧、加圧、および減圧のいずれであってもよく、常圧または加圧が好ましい。ただし、気相中での反応を行うために、該圧力は、脱塩化水素反応の反応温度において、233abの蒸気圧以下であることが好ましい。
反応時間は、転化率と選択率の制御のしやすさの観点から、1〜1000秒が好ましく、5〜300秒がさらに好ましい。
A−2法によれば、反応により生成する塩化水素は、反応に用いた塩基によって中和されことから、脱酸設備を必須としない方法であり、好ましい。
反応器内の圧力は、常圧、加圧、および減圧のいずれであってもよく、常圧または加圧が好ましい。ただし、気相中での反応を行うために、該圧力は、脱塩化水素反応の反応温度において、233abの蒸気圧以下であることが好ましい。
反応時間は、転化率と選択率の制御のしやすさの観点から、1〜1000秒が好ましく、5〜300秒がさらに好ましい。
A−2法によれば、反応により生成する塩化水素は、反応に用いた塩基によって中和されことから、脱酸設備を必須としない方法であり、好ましい。
[B法]233abを液相中で脱塩酸反応させる方法
B法は以下のいずれかの方法であり、B−2法が好ましい。
B−1法:活性炭の存在下に液相中で脱塩素化反応する方法
B−2法:溶液状態の塩基の存在下に液相中で脱塩素化反応する方法。
B法は以下のいずれかの方法であり、B−2法が好ましい。
B−1法:活性炭の存在下に液相中で脱塩素化反応する方法
B−2法:溶液状態の塩基の存在下に液相中で脱塩素化反応する方法。
B−1法は、233abを活性炭の存在下に液相中で脱塩化水素反応する方法である。B−1法で用いる活性炭は、特に限定されず、A−1法において説明した活性炭と同様のものを用いることができる。活性炭の比表面積、種類、形状、灰分、水分、および形状についても、A−1法に挙げたものと同様のものを用いることができる。B−1法における活性炭は、粉末炭または粒状炭が特に好ましい。
B−1法としては、液相中で233abと活性炭とを接触させる方法が挙げられる。B−1法は、バッチ式で実施しても、連続式で実施してもよく、生産効率の点から、連続式で実施するのが好ましい。
図2は、B−1法を実施するフローの概念図である。活性炭、233ab、および必要に応じ導入する他の反応資材を入れ反応器1に、233ab(図1中、符号8で示す。)を連続的に供給する。生成した1223xdを含む反応組成物は、反応器1から回収するが、必要に応じて冷却器4を経由して冷却する。さらに必要に応じて脱酸塔3に通して塩化水素を取り除いたものを生成物7として回収するのが好ましい。
図2の反応器1としては、液相反応での脱塩化水素反応に用いうる公知の反応器を使うのが好ましい。反応器1の材質としては、A−1法と同様のものが挙げられ、必要に応じて、樹脂ライニング、ガラスライニング等のライニング処理を反応器1に行ってもよい。
活性炭は、充填して触媒層として、または、反応器に添加して、用いるのが好ましい。
活性炭を充填して触媒層として用いる場合、触媒層は、活性炭を反応器1に充填することによって形成できる。触媒層は、反応器1内に1か所の設置であってもよく、2か所を設置してもよい。触媒層における活性炭の充填密度は、A−1法と同じである。
活性炭を充填して触媒層として用いる場合、触媒層は、活性炭を反応器1に充填することによって形成できる。触媒層は、反応器1内に1か所の設置であってもよく、2か所を設置してもよい。触媒層における活性炭の充填密度は、A−1法と同じである。
活性炭を反応器に添加して用いる場合の、活性炭の量は、脱塩化水素反応の反応速度の観点から、液相量の質量に対する活性炭の質量を0.5〜40質量%にするのが好ましく、0.5〜30質量%にするのがより好ましい。活性炭の量が少ないと充分な反応速度が得られない場合があり、活性炭量が多いと、経済的に不利であり工業プロセスに向かない場合がある。
B−1法の反応温度は、0〜100℃が好ましく、20〜80℃がより好ましい。反応温度を該範囲にすることにより、反応率が向上し、副生物を抑制しやすい利点がある。ただし、B−1法は液相中での反応を行うことから、反応温度の上限は、常圧における233abの沸点以下に設定することが好ましい。
B−1法の反応圧力は、0〜10MPaが好ましく、0.05〜5MPaがより好ましく、0.15〜1MPaがさらに好ましい。ただし、反応圧力は、反応温度において、233abの蒸気圧以上であることが好ましい。
B−1法の反応時間は、原料の転化率と選択率を制御のしやすさの観点から、バッチ式であれば1〜50時間が好ましく、連続式であれば1〜3000秒が好ましい。
B−1法では、233abを液体状態で反応に用いることにより液相中での反応が実施できるが、他の液状媒体を用いてもよいが、媒体は必須ではなく、むしろ用いないことが好ましい。媒体を用いる場合は、水、有機溶媒(アルコール等)等が挙げられる。媒体を用いる場合の量は、233abの100質量部に対して、10〜100質量部が好ましい。
B−1法で生成した1223xdは、気体として、または液相中に溶解した液体として反応系中に存在しうる。B−1法においては、気体の1223xdの気体を抜き出し、冷却器3により冷却して液体にしたものを回収することが好ましい。冷却器3を取り付けた場合は、未反応の233abを反応器に戻し、沸点の低い副生物(例えば1−クロロ−3,3,3−トリフルオロプロピン等の副生物)および塩化水素を選択的に反応系内から取り出すことができ、転化率や選択率が向上する利点がある。
反応組成物には、目的とする1223xdの他に、未反応の233ab、塩化水素、副生物が含まれうる。これらは、脱酸塔を通して除去する、または蒸留して分離する等の後処理工程により容易に除去することができる。塩化水素は、金属水酸化物またはその水溶液と接触させて中和することによっても除去できる。金属水酸化物としては、水酸化ナトリウム、水酸化カリウム等が挙げられる。
B−2法は、233abを溶液状態の塩基の存在下に液相中で脱塩化水素反応する方法である。
B−2法で用いる塩基は、A−2法において記載した塩基と同様のものを用いることができる。B−2法における脱塩化水素反応のフローは、図2に示すフローと同様であり、B−1法において説明した活性炭を、溶液状態の塩基に変えて実施できる。反応器の種類および材質はA−2法と同様のものを使用できる。
B−2法においては、均一な反応系での反応が行われるように、反応器1に撹拌手段を設け、撹拌しながら反応を行うことが好ましい。
B−2法で用いる塩基は、A−2法において記載した塩基と同様のものを用いることができる。B−2法における脱塩化水素反応のフローは、図2に示すフローと同様であり、B−1法において説明した活性炭を、溶液状態の塩基に変えて実施できる。反応器の種類および材質はA−2法と同様のものを使用できる。
B−2法においては、均一な反応系での反応が行われるように、反応器1に撹拌手段を設け、撹拌しながら反応を行うことが好ましい。
B−2法では、溶液状態の塩基を用いる。溶液状態の塩基は、塩基を溶媒に溶解させた溶液が好ましい。
溶媒としては、上記塩基の溶解性が高く、脱塩化水素反応に対して不活性な溶媒が好ましい。溶媒としては、溶解性の観点から、水が好ましい。また塩基としてはA−2法に記載した前記と同様の塩基を用いうるが、アルカリ金属水酸化物が好ましい。すなわち、溶媒状態の塩基としては、アルカリ金属水酸化物の水溶液が好ましい。
上記溶液状態の塩基は、塩基と溶媒の総量を100質量%とした場合の塩基量が、0.5〜40質量%が好ましく、0.5〜20質量%がより好ましい。塩基量が少ないと十分な反応速度が得られないことがあり、塩基量が多いと塩基が十分に溶解できないおそれや、金属塩が析出する場合があり、工業的なプロセスにおいて不利になる場合がある。一方、前記塩基量が上記範囲であると、2層分離を簡便に行うことができる利点がある。
B−2法の反応に用いる塩基量は、転化率および選択率の観点から、233abの1モルに対して、0.5〜2.0モルが好ましく、0.8〜1.2モルがより好ましい。
B−2法の反応温度は、反応速度、収率の観点から、5〜100℃が好ましく、20〜80℃がより好ましい。
B−2法においては、本発明の反応に影響を与えない範囲で、相関移動触媒やテトラグライム等の水溶性有機溶媒等を反応系中に存在させてもよく、相関移動触媒を存在させるのが好ましい。
相間移動触媒としては、第4級アンモニウム塩、第4級ホスホニウム塩、第4級アルソニウム塩、スルホニウム塩、クラウンエーテルなどが挙げられ、第4級アンモニウム塩が好ましく、第4級アンモニウム塩が特に好ましい。
第4級アンモニウム塩としては、下式(i)で表される化合物(以下、「化合物(i)」と称することがある)が挙げられる。
R11〜R14が炭化水素基である場合、アルキル基、シクロアルキル基、アルケニル基、シクロアルケニル基、アリール基などが挙げられ、アルキル基、アリール基が好ましい。R11〜R14の炭素原子数は、4〜100が好ましい。R11〜R14は、それぞれ同じ基であってもよいし、異なる基であってもよい。
R11〜R14が、反応に不活性な官能基が結合した1価の炭化水素基である場合の官能基は、反応条件に応じて適宜選択されるが、ハロゲン原子、アルコキシカルボニル基、アシルオキシ基、ニトリル基、アシル基、カルボキシル基、アルコキシル基などが挙げられる。
R11〜R14が、反応に不活性な官能基が結合した1価の炭化水素基である場合の官能基は、反応条件に応じて適宜選択されるが、ハロゲン原子、アルコキシカルボニル基、アシルオキシ基、ニトリル基、アシル基、カルボキシル基、アルコキシル基などが挙げられる。
R11R12R13R14N+としては、テトラメチルアンモニウム、テトラエチルアンモニウム、テトラ−n−プロピルアンモニウム、テトラ−n−ブチルアンモニウム、メチル(トリ−n−オクチル)アンモニウム、セチルトリメチルアンモニウム、ベンジルトリメチルアンモニウム、ベンジルトリエチルアンモニウム、セチルベンジルジメチルアンモニウム、セチルピリジニウム、n−ドデシルピリジニウム、フェニルトリメチルアンモニウム、フェニルトリエチルアンモニウム、N−ベンジルピコリニウム、ペンタメトニウム、ヘキサメトニウムなどが挙げられる。
Y−としては、塩素イオン、フッ素イオン、臭素イオン、ヨウ素イオン、硫酸イオン、硝酸イオン、リン酸イオン、過塩素酸イオン、硫酸水素イオン、水酸化物イオン、酢酸イオン、安息香酸イオン、ベンゼンスルホン酸イオン、p−トルエンスルホン酸イオンなどが挙げられ、塩素イオン、臭素イオン、ヨウ素イオン、硫酸水素イオン、水酸化物イオンが好ましい。
化合物(i)としては、汎用性および反応性の観点から、下記R11R12R13R14N+と、下記Y−との組合せが好ましい。
R11R12R13R14N+:テトラメチルアンモニウム、テトラエチルアンモニウム、テトラ−n−プロピルアンモニウム、テトラ−n−ブチルアンモニウム、トリ−n−オクチルメチルアンモニウム。
Y−:フッ素イオン、塩素イオン、臭素イオン、ヨウ素イオン、水酸化物イオン。
R11R12R13R14N+:テトラメチルアンモニウム、テトラエチルアンモニウム、テトラ−n−プロピルアンモニウム、テトラ−n−ブチルアンモニウム、トリ−n−オクチルメチルアンモニウム。
Y−:フッ素イオン、塩素イオン、臭素イオン、ヨウ素イオン、水酸化物イオン。
4級アンモニウム塩としては、テトラ−n−ブチルアンモニウムクロリド(TBAC)、テトラ−n−ブチルアンモニウムブロミド(TBAB)、メチルトリ−n−オクチルアンモニウムクロリド(TOMAC)が好ましい。
第4級ホスホニウム塩としては、下式(ii)で表される化合物が挙げられる。
R21〜R24における炭化水素基としては、アルキル基、シクロアルキル基、アルケニル基、シクロアルケニル基、アリール基などが挙げられ、アルキル基、アリール基が好ましい。
式(ii)における第4級ホスホニウム(R21R22R23R24P+)としては、テトラエチルホスホニウム、テトラ−n−ブチルホスホニウム、トリ−n−オクチルエチルホスホニウム、セチルトリエチルホスホニウム、セチルトリ−n−ブチルホスホニウム、n−ブチルトリフェニルホスホニウム、n−アミルトリフェニルホスホニウム、メチルトリフェニルホスホニウム、ベンジルトリフェニルホスホニウム、テトラフェニルホスホニウムなどが挙げられる。
Y−としては、塩素イオン、フッ素イオン、臭素イオン、ヨウ素イオン、硫酸イオン、硝酸イオン、リン酸イオン、過塩素酸イオン、硫酸水素イオン、水酸化物イオン、酢酸イオン、安息香酸イオン、ベンゼンスルホン酸イオン、p−トルエンスルホン酸イオンなどが挙げられ、フッ素イオン、塩素イオン、臭素イオンが好ましい。
第4級アルソニウム塩としては、下式(iii)で表される化合物が挙げられる。
R31〜R34における炭化水素基としては、例えば、アルキル基、シクロアルキル基、アルケニル基、シクロアルケニル基、アリール基などが挙げられ、アルキル基、アリール基が好ましい。
Y−としては、ハロゲンイオンが好ましく、フッ素イオン、塩素イオン、臭素イオンがより好ましい。
式(iii)で表わされる第4級アルソニウム塩としては、トリフェニルメチルアルソニウムフロライド、テトラフェニルアルソニウムフロライド、トリフェニルメチルアルソニウムクロライド、テトラフェニルアルソニウムクロライド、テトラフェニルアルソニウムブロマイドなどが挙げられる。
第4級アルソニウム塩としては、トリフェニルメチルアルソニウムクロライドが特に好ましい。
第4級アルソニウム塩としては、トリフェニルメチルアルソニウムクロライドが特に好ましい。
スルホニウム塩としては、下式(iv)で表される化合物が挙げられる。
R41〜R43における炭化水素基としては、例えば、アルキル基、シクロアルキル基、アルケニル基、シクロアルケニル基、アリール基などが挙げられ、アルキル基、アリール基が好ましい。
Y−としては、ハロゲンイオンが好ましく、フッ素イオン、塩素イオン、臭素イオンがより好ましい。
式(iv)で表されるスルホニウム塩としては、ジ−n−ブチルメチルスルホニウムアイオダイド、トリ−n−ブチルスルホニウムテトラフルオロボレート、ジヘキシルメチルスルホニウムアイオダイド、ジシクロヘキシルメチルスルホニウムアイオダイド、ドデシルメチルエチルスルホニウムクロライド、トリス(ジエチルアミノ)スルホニウムジフルオロトリメチルシリケートなどが挙げられる。
スルホニウム塩としては、ドデシルメチルエチルスルホニウムクロライドが特に好ましい。
スルホニウム塩としては、ドデシルメチルエチルスルホニウムクロライドが特に好ましい。
クラウンエーテルとしては、18−クラウン−6、ジベンゾ−18−クラウン−6、ジシクロヘキシル−18−クラウン−6などが挙げられる。
相間移動触媒の使用量は、233abの100質量部に対して、0.001〜5質量部が好ましく、0.01〜1質量部がより好ましい。相間移動触媒の量が少なすぎると、充分な反応速度が得られないことがあり、多く用いても、使用量に応じた反応促進効果は得られず、コスト面で不利である。
相間移動触媒を使用する場合の、反応工程、反応装置、および反応器の材質は、相間移動触媒を使用しない場合と同様であってよい。また、塩基の濃度、使用量、または反応温度などの反応条件も、相間移動触媒を使用しない場合と同様の条件であってよい。
B−2法の反応は、反応に関与する化合物(例えば、233ab、塩基、溶媒さらに必要に応じて相間移動触媒)を、反応器に導入し、これらが均一になるように撹拌し、所望の温度条件、圧力条件にすることで進行させうる。
B−2法においては、上記溶液状態の塩基を調製する際に溶媒として親水性が高い溶媒を用いた場合、(例えば、溶液状態の塩基として、アルカリ金属水酸化物の水溶液を用いた場合)、B−2の反応系が、水相と有機相に分離する。そのような場合は、相間移動触媒の代わりに水溶性有機溶媒(たとえば、テトラグライム等)を反応系中に存在させて、有機相と、塩基を含む水相を相溶化することにより反応を実施できる。水溶性有機溶媒を用いる場合は、反応系を均一にするために、撹拌を充分に行うのが好ましい。また、該場合に反応終了後の反応液を放置すると、有機相と水相に分離する。有機相中には、未反応の233ab、生成物の1223xd以外に、副生物が含まれ得る。
副生物の例としては、1223xdの脱塩化水素体である1−クロロ−3,3,3−トリフルオロプロピン等が挙げられる。また、原料の233abとして、HCFC−223aaやCFC−213abが含まれる場合、反応組成物中には、HCFC−223aaが脱塩化水素した1,1,2−トリクロロ−3,3,3−トリフルオロプロぺン(CFO−1213xa)が含まれることがある。
B−2法においては、1223xdを含む有機相を回収し、つぎに副生成物を分離することによって、高純度の1223xdを得るのが好ましい。分離において、有機相中に含まれる成分が分離可能な沸点差である場合、一般的な蒸留等による分離できる。
A法およびB法において得た1223xdから、さらに、高純度の1223xdを得たい場合や、E体、Z体を分離したい場合には、さらに蒸留等の通常の方法で分離精製を行うことで、1223xdを高純度で得ることができる。また分離条件を調整することで、1223xdのE体とZ体の分離ができる。
本発明の製造方法によれば、工業的に実施可能な簡便な方法で、233abから、高反応率および高選択率で1223xdを製造することができる。さらに、液相で脱塩化水素を行うと、気相で行った場合と比較して同量の1223xdを製造する場合に反応器サイズを縮小することができる。つまり、本発明によれば原料および製造設備に要するコストを大幅に低減することができる。
1223xdは、地球温暖化係数(GWP)の小さい新しい洗浄剤、冷媒、発泡剤、溶剤、噴射剤、相溶剤およびエアゾール用途に用いられる冷媒として有用である。
以下に、本発明を実施例によって具体的に説明するが、本発明はこれらの実施例によって限定されるものではない。
[分析条件]
以下の各種化合物の製造において、得られた反応組成物の組成分析はガスクロマトグラム(GC)を用いた。カラムはDB−1301(長さ60m×内径250μm×厚み1μm、アジレント・テクノロジー株式会社製)を用いた。
以下の各種化合物の製造において、得られた反応組成物の組成分析はガスクロマトグラム(GC)を用いた。カラムはDB−1301(長さ60m×内径250μm×厚み1μm、アジレント・テクノロジー株式会社製)を用いた。
(233abの製造例1)
公知の方法(特表2013−519631号公報に記載の方法)で得たHCFO−1233xfを、式(2)にしたがって塩素化して233abを製造した。
公知の方法(特表2013−519631号公報に記載の方法)で得たHCFO−1233xfを、式(2)にしたがって塩素化して233abを製造した。
すなわち、光源からの光を透過する石英管およびジャケットを取り付けたステンンレス製オートクレーブ(内容積2.3リットル)を0℃に冷却した。オートクレーブに四塩化炭素(CCl4)の1580gを入れ、続いてHCFO−1233xfの846gを加えた。反応器内の圧力は0.08MPaであった。
光照射下(蛍光灯:東芝社製ネオコンパクトEFP12EL、出力12W)で、塩素(Cl2)をオートクレーブに600ml/分で466gになるまで導入した。反応の進行に伴い、反応熱が生じると共に、HCFO−1233xf(0MPaにおける沸点:15℃)よりも沸点の高い233ab(233abの0MPaにおける沸点:104℃)が生成することで圧力が低下した。
反応中のオートクレーブ内の温度は15.4〜18.6℃であった。反応時間は4時間であり、反応終了時の圧力は0MPa(大気圧)であった。
光照射下(蛍光灯:東芝社製ネオコンパクトEFP12EL、出力12W)で、塩素(Cl2)をオートクレーブに600ml/分で466gになるまで導入した。反応の進行に伴い、反応熱が生じると共に、HCFO−1233xf(0MPaにおける沸点:15℃)よりも沸点の高い233ab(233abの0MPaにおける沸点:104℃)が生成することで圧力が低下した。
反応中のオートクレーブ内の温度は15.4〜18.6℃であった。反応時間は4時間であり、反応終了時の圧力は0MPa(大気圧)であった。
反応終了後、粗液を炭酸水素カリウムの20質量%水溶液と混合し、中和、分液操作を実施した。静置後分離した下層から回収した粗生成物(1)は2921gであった。回収した粗生成物のGC分析を行った。
(233abの製造例2)
四塩化炭素を1432g、HCFO−1233xfを458g使用し、塩素を600mL/分で351gになるまで導入したこと以外は、製造例1と同様の操作を行い、粗生成物の731gを得て、GC分析を行った。
四塩化炭素を1432g、HCFO−1233xfを458g使用し、塩素を600mL/分で351gになるまで導入したこと以外は、製造例1と同様の操作を行い、粗生成物の731gを得て、GC分析を行った。
(233abの製造例3)
タイマーによって自動で開閉できる電磁弁を接続した上記オートクレーブに、233ab(純度99.8%)の1500gを溶媒として導入した。
光照射下で、塩素とHCFO−1233xfを、それぞれ2.5L/分でオートクレーブに供給し、850g/時で連続的に反応組成物を抜き出しながら、90分間反応を行った。抜き出した組成物を、20%炭酸水素カリウム水溶液に加え中和した後に、分液操作を行い、粗組成物(3541g)を回収し、GC分析を行った。
タイマーによって自動で開閉できる電磁弁を接続した上記オートクレーブに、233ab(純度99.8%)の1500gを溶媒として導入した。
光照射下で、塩素とHCFO−1233xfを、それぞれ2.5L/分でオートクレーブに供給し、850g/時で連続的に反応組成物を抜き出しながら、90分間反応を行った。抜き出した組成物を、20%炭酸水素カリウム水溶液に加え中和した後に、分液操作を行い、粗組成物(3541g)を回収し、GC分析を行った。
製造例1〜3の反応条件、得られた粗組成物のGC分析結果を表1に示す。表1中、製造例1〜2の転化率、選択率は、反応器に供給したHCFO−1233xfに対する値であり、モル換算値(単位:モル%)である。製造例3においては、粗生成物中の233abのmolから、初期に導入した233abのmolを除いた量を、233abの生成量として選択率を求めた。
製造1〜3において、得られた粗生成物は、通常の蒸留操作を実施することで、純度99.8%の233abを得た。
[1223xdの製造例(実施例1〜6)]
実施例1〜6においては、気相中での脱塩化水素反応により、実施例7〜9においては、液相中での脱塩化水素反応により、1223xdを製造した。
実施例1〜6においては、気相中での脱塩化水素反応により、実施例7〜9においては、液相中での脱塩化水素反応により、1223xdを製造した。
(実施例1:気相中での製造例)
反応器として垂直固定床反応器(材質:SUS316、内径22.6mm×高さ400mm)を用いた。反応器の中心に差込管(材質:SUS316、直径:4mm)を導入し、その中にK型熱電対を挿入し、内温を測定した。反応器の中央部に活性炭(日本エンバイロケミカルズ社製、白鷺活性炭C2x、比表面積:1260m2/g、灰分:1.2質量%)の84mL(44g)を充填し、触媒層とした。触媒層は電気式ヒーターによって250℃に加熱した。窒素ガスフィードラインおよび原料フィードラインを接続した原料余熱混合ラインを、反応器の上部に接続した。原料フィードラインは100℃に加熱した。
反応器として垂直固定床反応器(材質:SUS316、内径22.6mm×高さ400mm)を用いた。反応器の中心に差込管(材質:SUS316、直径:4mm)を導入し、その中にK型熱電対を挿入し、内温を測定した。反応器の中央部に活性炭(日本エンバイロケミカルズ社製、白鷺活性炭C2x、比表面積:1260m2/g、灰分:1.2質量%)の84mL(44g)を充填し、触媒層とした。触媒層は電気式ヒーターによって250℃に加熱した。窒素ガスフィードラインおよび原料フィードラインを接続した原料余熱混合ラインを、反応器の上部に接続した。原料フィードラインは100℃に加熱した。
原料予熱混合ラインを100℃に加熱した。マスフローコントローラを用いて窒素ガス流量を毎分50Nmlに調整し、窒素ガスフィードラインから原料予熱混合ラインに供給した。233ab(純度99.8%)を含む原料は、原料フィードラインを通して気化させた後、そのガスを毎分50Nmlで原料予熱混合ラインに供給した。233abの原料余熱混合ラインへの供給速度は、233abの原料フィードラインへの供給量をプランジャーポンプで調整することで制御した。窒素ガスおよび気化させた233abを反応器に導入し、滞留時間は26.5秒、線速毎秒0.8cmの条件下で、連続6時間反応させた。
反応生成物は、反応器の下部から連続的に取り出した。反応生成物の一部を採取し、GCによる組成分析を行った。結果を表2に示す。
表2中の「HCFO−1223za/HCFO−1223xd」は、目的物である1223xd(E体およびZ体のモル総量)に対する、副生物であるHCFO−1223zaの生成量(モル量)の割合(モル%)を示す。
表2中の「HCFO−1223za/HCFO−1223xd」は、目的物である1223xd(E体およびZ体のモル総量)に対する、副生物であるHCFO−1223zaの生成量(モル量)の割合(モル%)を示す。
(実施例2:気相中での製造例)
触媒層を200℃に加熱すること、窒素ガス流量を毎分55Nmlに調整すること、および233abを含む原料ガスの供給量を毎分55Nmlとすること、以外は実施例1と同様の方法で反応を行った。反応生成物の分析結果を表2に示す。
触媒層を200℃に加熱すること、窒素ガス流量を毎分55Nmlに調整すること、および233abを含む原料ガスの供給量を毎分55Nmlとすること、以外は実施例1と同様の方法で反応を行った。反応生成物の分析結果を表2に示す。
(実施例3:気相中での製造例)
触媒層を300℃に加熱すること、窒素ガス流量を毎分46Nmlに調整すること、および233abを含む原料ガスの供給量を毎分46Nmlとすること、以外は実施例1と同様の方法で反応させた。反応生成物の分析結果を表2に示す。
触媒層を300℃に加熱すること、窒素ガス流量を毎分46Nmlに調整すること、および233abを含む原料ガスの供給量を毎分46Nmlとすること、以外は実施例1と同様の方法で反応させた。反応生成物の分析結果を表2に示す。
(実施例4:気相中での製造例)
触媒層を150℃に加熱すること、窒素ガス流量を毎分62Nmlに調整すること、および233abを含む原料ガスの供給量を毎分62Nmlとすること、以外は実施例1と同様の方法で反応させた。反応生成物の分析結果を表2に示す。
触媒層を150℃に加熱すること、窒素ガス流量を毎分62Nmlに調整すること、および233abを含む原料ガスの供給量を毎分62Nmlとすること、以外は実施例1と同様の方法で反応させた。反応生成物の分析結果を表2に示す。
(実施例5:気相中での製造例)
窒素ガスの流量を毎分101Nmlとすること、233abを含む原料ガスの供給量を毎分101Nmlとすること、以外は実施例1と同様の方法で反応させた。反応の滞留時間は13.1秒、線速毎秒1.6cmであった。反応生成物の分析結果を表2に示す。
窒素ガスの流量を毎分101Nmlとすること、233abを含む原料ガスの供給量を毎分101Nmlとすること、以外は実施例1と同様の方法で反応させた。反応の滞留時間は13.1秒、線速毎秒1.6cmであった。反応生成物の分析結果を表2に示す。
(実施例6:気相中での製造例)
希釈剤として窒素ガスを用いないこと、気化させた233abを毎分101Nml反応器に供給すること、以外は実施例1と同様の方法で反応させた。反応の滞留時間は26.2秒、線速毎秒0.8cmであった。反応生成物のGC分析結果を表2に示す。
希釈剤として窒素ガスを用いないこと、気化させた233abを毎分101Nml反応器に供給すること、以外は実施例1と同様の方法で反応させた。反応の滞留時間は26.2秒、線速毎秒0.8cmであった。反応生成物のGC分析結果を表2に示す。
(比較例1:HCFC−233daを気相中で活性炭触媒を用いて反応させた例)
実施例1の233abをHCFC−233daに変更すること以外は、同様の方法で反応させた。得られた反応生成物のGC分析結果を表3に示す。
実施例1の233abをHCFC−233daに変更すること以外は、同様の方法で反応させた。得られた反応生成物のGC分析結果を表3に示す。
(比較例2:HCFC−233daを気相中で活性炭触媒を用いて反応させた例)
実施例2のHCFC−233をHCFC−233daに変更すること以外は、同様の方法で反応させた。得られた反応生成物のGC分析結果を表3に示す。
実施例2のHCFC−233をHCFC−233daに変更すること以外は、同様の方法で反応させた。得られた反応生成物のGC分析結果を表3に示す。
気相で反応を行う本発明の方法(表2)の結果によれば、高選択率で目的とする1223xdを製造できる。さらに反応温度を調整することで、高収率で1223xdを製造できる。
(実施例7:液相中での製造例)
撹拌機、ジムロート冷却器を設置した0.3リットルの三口フラスコに、233abを50.3g、およびテトラ−n−ブチルアンモニウムクロリド(TBAC)の503mgを入れ、20質量%の水酸化カリウム水溶液76.4gを加え、オイルバスにて40℃に昇温した後、回転数400rpmで撹拌を続けた。反応中の反応器内温を39〜42℃の範囲に保った。撹拌を停止し、静置し、水相と有機相が自然に分離するまで放置した後、有機相を回収しGC分析を行い、233abの転化率が90%以上になっていることを確認し、反応を終了した。反応終了までに要した時間は、20時間であった。得られた反応生成物のGC分析結果を表4に示す。表4中、CTFPは、1−クロロ‐3,3,3−トリフルオロプロピンを示す。
撹拌機、ジムロート冷却器を設置した0.3リットルの三口フラスコに、233abを50.3g、およびテトラ−n−ブチルアンモニウムクロリド(TBAC)の503mgを入れ、20質量%の水酸化カリウム水溶液76.4gを加え、オイルバスにて40℃に昇温した後、回転数400rpmで撹拌を続けた。反応中の反応器内温を39〜42℃の範囲に保った。撹拌を停止し、静置し、水相と有機相が自然に分離するまで放置した後、有機相を回収しGC分析を行い、233abの転化率が90%以上になっていることを確認し、反応を終了した。反応終了までに要した時間は、20時間であった。得られた反応生成物のGC分析結果を表4に示す。表4中、CTFPは、1−クロロ‐3,3,3−トリフルオロプロピンを示す。
(実施例8:液相中での製造例)
233abを50.1gをすること、およびTBACを500mgとすること、20質量%の水酸化カリウム水溶液を76.3g使用すること、反応温度を60℃すること、以外は実施例7と同様の方法で反応させた。反応終了までに要した時間は8時間であった。反応生成物のGC分析結果を表4に示す。
233abを50.1gをすること、およびTBACを500mgとすること、20質量%の水酸化カリウム水溶液を76.3g使用すること、反応温度を60℃すること、以外は実施例7と同様の方法で反応させた。反応終了までに要した時間は8時間であった。反応生成物のGC分析結果を表4に示す。
(実施例9:液相中での製造例)
撹拌機を備えた0.3リットルの三口フラスコを反応器とした。充填物(ヘリパックNo.3)を充填したガラス管(内径29mm、長さ100mm)を、反応器と冷却器の間に設置した。三口フラスコに、233abを49.9g、およびTBACを501mg、20質量%の水酸化カリウム水溶液を77.0g加え、オイルバスにて80℃に昇温し、回転数400rpmで撹拌を行った。反応生成物は気体として取り出し、ドライアイストラップを用いて、液化したものを回収した。反応が進行するにつれ、反応器内の有機相が減少する。反応器内の有機相が消失することを確認し、反応を終了した。反応終了までに要した時間は3時間であった。得られた液化した反応生成物のGC分析結果を表4に示す。
撹拌機を備えた0.3リットルの三口フラスコを反応器とした。充填物(ヘリパックNo.3)を充填したガラス管(内径29mm、長さ100mm)を、反応器と冷却器の間に設置した。三口フラスコに、233abを49.9g、およびTBACを501mg、20質量%の水酸化カリウム水溶液を77.0g加え、オイルバスにて80℃に昇温し、回転数400rpmで撹拌を行った。反応生成物は気体として取り出し、ドライアイストラップを用いて、液化したものを回収した。反応が進行するにつれ、反応器内の有機相が減少する。反応器内の有機相が消失することを確認し、反応を終了した。反応終了までに要した時間は3時間であった。得られた液化した反応生成物のGC分析結果を表4に示す。
液相で反応を行う本発明の方法(表4)の結果によれば、副生成物であるHCFO−1223za等の生成を顕著に抑制し、高選択率、高収率で目的とする1223xdを製造できる。
(実施例10:気相での製造例)
実施例1の活性炭の触媒層を、水酸化カリウムを充填した充填層に変えて、同様に反応させると、1223xdを含む反応生成物を得る。
(実施例11:液相での製造例)
実施例7の20質量%の水酸化カリウム水溶液を、活性炭に変えて同様に反応させると、1223xdを含む反応生成物を得る。
実施例1の活性炭の触媒層を、水酸化カリウムを充填した充填層に変えて、同様に反応させると、1223xdを含む反応生成物を得る。
(実施例11:液相での製造例)
実施例7の20質量%の水酸化カリウム水溶液を、活性炭に変えて同様に反応させると、1223xdを含む反応生成物を得る。
本発明の製造方法においては、脱塩化水素反応の基質を233abとして選択する。233abを用いて反応を行うことにより、他の原料(HCFC−233da等)を用いた場合に比べて副生物であるHCFO−1223za(沸点55℃)等の量を低減できる。副生成物は、反応生成物の収率を低下させるだけでなく、分離の工程においても目的物の収率を下げる原因になる、また、副生成物の物性(例えば沸点差)によっては、目的物とは容易に分離精製しにくくさせる場合がある。これらの副生成物の生成を防ぐ本発明方法は、収率を高くし、かつ、生産効率も高くさせることから、工業的に有利な製造方法である。
1…反応器、2…ガスクロマトグラフィ、3…脱酸塔、4…冷却器、5…ガス状の233ab、6…希釈ガス、7…生成物、8…233ab
Claims (15)
- 1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する、または、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、ことを特徴とする1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法。
- 前記塩基が、金属水酸化物、金属酸化物および金属炭酸塩からなる群より選ばれる少なくとも1種の塩基である、請求項1に記載の製造方法。
- 前記塩基を、1,2,2−トリクロロ−3,3,3−トリフルオロプロパンの1モルに対して0.5〜2.0モル用いる、請求項1または2に記載の製造方法。
- 前記1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを、活性炭もしくは塩基の存在下に気相中で脱塩化水素反応する請求項1〜3のいずれか一項に記載の製造方法。
- 前記脱塩化水素反応の反応温度が100〜350℃である請求項4に記載の製造方法。
- ガス線速を毎秒0.5〜3.0cmとしながら1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを反応系中に導入する請求項4または5に記載の製造方法。
- 前記1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを、活性炭もしくは溶液状態の塩基の存在下に液相中で脱塩化水素反応する、請求項1〜3のいずれか一項に記載の製造方法。
- 前記脱塩化水素反応の反応温度が0〜100℃である請求項7に記載の製造方法。
- 前記脱塩化水素反応の反応温度が、5〜100℃である、請求項7または8に記載の製造方法。
- 前記溶液状態の塩基が、水溶液の状態の塩基である請求項7〜9のいずれか一項に記載の製造方法。
- 前記溶液状態の塩基が、溶液全量に対して塩基を0.5質量%〜40質量%含む溶液状態の塩基である請求項7〜10のいずれか一項に記載の製造方法。
- 前記脱塩化水素反応を、相間移動触媒の存在下に行う、請求項7〜11のいずれか一項に記載の製造方法。
- 前記相間移動触媒が、第4級アンモニウム塩である、請求項12に記載の製造方法。
- 前記第4級アンモニウム塩が、テトラ−n−ブチルアンモニウムクロリド、テトラ−n−ブチルアンモニウムブロミドおよびメチルトリ−n−オクチルアンモニウムクロリドからなる群より選ばれる少なくとも1種である、請求項13に記載の製造方法。
- 1,2−ジクロロ−3,3,3−トリフルオロプロペンを、2−クロロ−3,3,3−トリフルオロプロペンを塩素化して1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを得て、つぎに該1,2,2−トリクロロ−3,3,3−トリフルオロプロパンを脱塩化水素反応して得る、請求項1〜14のいずれか一項に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015118436A JP2017001990A (ja) | 2015-06-11 | 2015-06-11 | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015118436A JP2017001990A (ja) | 2015-06-11 | 2015-06-11 | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017001990A true JP2017001990A (ja) | 2017-01-05 |
Family
ID=57751269
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015118436A Pending JP2017001990A (ja) | 2015-06-11 | 2015-06-11 | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017001990A (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019189024A1 (ja) * | 2018-03-30 | 2019-10-03 | Agc株式会社 | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 |
| WO2019203319A1 (ja) * | 2018-04-19 | 2019-10-24 | Agc株式会社 | フルオロオレフィンの製造方法 |
| WO2020075727A1 (ja) * | 2018-10-12 | 2020-04-16 | セントラル硝子株式会社 | 液体組成物の保存方法および製品 |
| CN112125776A (zh) * | 2020-10-20 | 2020-12-25 | 淄博雷玛国际贸易有限公司 | 一种1-氯-2,3,3-三氟丙烯的制备方法 |
| JP2021059498A (ja) * | 2019-10-03 | 2021-04-15 | セントラル硝子株式会社 | 不飽和クロロフルオロカーボンの製造方法 |
-
2015
- 2015-06-11 JP JP2015118436A patent/JP2017001990A/ja active Pending
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019189024A1 (ja) * | 2018-03-30 | 2019-10-03 | Agc株式会社 | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 |
| JPWO2019189024A1 (ja) * | 2018-03-30 | 2021-04-08 | Agc株式会社 | 1−クロロ−2,3,3−トリフルオロプロペンの製造方法 |
| WO2019203319A1 (ja) * | 2018-04-19 | 2019-10-24 | Agc株式会社 | フルオロオレフィンの製造方法 |
| WO2020075727A1 (ja) * | 2018-10-12 | 2020-04-16 | セントラル硝子株式会社 | 液体組成物の保存方法および製品 |
| JPWO2020075727A1 (ja) * | 2018-10-12 | 2021-09-02 | セントラル硝子株式会社 | 液体組成物の保存方法および製品 |
| JP7356041B2 (ja) | 2018-10-12 | 2023-10-04 | セントラル硝子株式会社 | 液体組成物の保存方法および製品 |
| JP2021059498A (ja) * | 2019-10-03 | 2021-04-15 | セントラル硝子株式会社 | 不飽和クロロフルオロカーボンの製造方法 |
| CN112125776A (zh) * | 2020-10-20 | 2020-12-25 | 淄博雷玛国际贸易有限公司 | 一种1-氯-2,3,3-三氟丙烯的制备方法 |
| CN112125776B (zh) * | 2020-10-20 | 2021-06-29 | 淄博雷玛国际贸易有限公司 | 一种1-氯-2,3,3-三氟丙烯的制备方法 |
| WO2022083017A1 (zh) * | 2020-10-20 | 2022-04-28 | 淄博雷玛国际贸易有限公司 | 一种 1- 氯 -2 , 3 , 3- 三氟丙烯的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6860057B2 (ja) | 1−クロロ−2,3,3−トリフルオロプロペンの製造方法 | |
| JP6426795B2 (ja) | 塩素化プロペンの製造方法 | |
| JP6157568B2 (ja) | テトラフルオロプロペンの製造方法 | |
| JP7081596B2 (ja) | 2-クロロ-1,1,1,2-テトラフルオロプロパンおよび/または3-クロロ-1,1,1,2-テトラフルオロプロパンの製造方法、ならびに2,3,3,3-テトラフルオロプロペンの製造方法 | |
| JP6449791B2 (ja) | クロロアルカンの製造方法 | |
| JP6812988B2 (ja) | 1−クロロ−2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP2017001990A (ja) | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 | |
| JP6245259B2 (ja) | (e)−1−クロロ−3,3,3−トリフルオロプロペンの製造方法 | |
| WO2019189024A1 (ja) | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| JP2015120670A (ja) | 1−クロロ−1,2−ジフルオロエチレンの製造方法 | |
| JP7151257B2 (ja) | 2-クロロ-3,3-ジフルオロプロペンの製造方法、2-クロロ-1,1,2-トリフルオロプロパンの製造方法、2,3,3-トリフルオロプロペンの製造方法、1,2-ジクロロ-2,3,3-トリフルオロプロパンの製造方法、1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| JP7331700B2 (ja) | 1-クロロ-2,3,3,4,4,5,5-ヘプタフルオロペンテンの製造方法 | |
| JP5858830B2 (ja) | ポリクロロプロパンの製造方法 | |
| WO2015166847A1 (ja) | トランス-1-クロロ-3,3,3-トリフルオロプロペンの製造方法 | |
| JP2019151629A (ja) | 化合物の製造方法 | |
| WO2016111227A1 (ja) | (e)-1-クロロ-3,3,3-トリフルオロプロペンの製造方法 | |
| JP7070220B2 (ja) | 2-クロロ-3,3-ジフルオロプロペンの製造方法、2-クロロ-1,1,2-トリフルオロプロパンの製造方法、2,3,3-トリフルオロプロペンの製造方法、1,2-ジクロロ-2,3,3-トリフルオロプロパンの製造方法、1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| WO2019168115A1 (ja) | 1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンの製造方法及び1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 | |
| CN107235822B (zh) | 制备四氟丙烯的方法 | |
| JPWO2019124220A1 (ja) | 5−クロロ−1,1,2,2,3,3,4,4−オクタフルオロペンタンの製造方法及び1−クロロ−2,3,3,4,4,5,5−ヘプタフルオロペンテンの製造方法 | |
| JP2013018722A (ja) | 1−クロロ−3,3,3−トリフルオロプロピンの製造方法 | |
| JP2015224236A (ja) | (e)−1−クロロ−3,3,3−トリフルオロプロペンの製造方法 |