JP2013529196A - キナーゼlrrk2の阻害剤としてのピラゾロピリジン - Google Patents
キナーゼlrrk2の阻害剤としてのピラゾロピリジン Download PDFInfo
- Publication number
- JP2013529196A JP2013529196A JP2013509631A JP2013509631A JP2013529196A JP 2013529196 A JP2013529196 A JP 2013529196A JP 2013509631 A JP2013509631 A JP 2013509631A JP 2013509631 A JP2013509631 A JP 2013509631A JP 2013529196 A JP2013529196 A JP 2013529196A
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- heterocycloalkyl
- aryl
- heteroaryl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 CC(C)[C@@](CN)[C@@](C)C(C)* Chemical compound CC(C)[C@@](CN)[C@@](C)C(C)* 0.000 description 1
- RUOPESHGFXYMCE-PKPIPKONSA-N CC(N1N=C(C)C[C@@H]1C)I Chemical compound CC(N1N=C(C)C[C@@H]1C)I RUOPESHGFXYMCE-PKPIPKONSA-N 0.000 description 1
- YEBBCAXAPQQICH-UHFFFAOYSA-N CC(c(c(Cl)cc(Cl)n1)c1Cl)=O Chemical compound CC(c(c(Cl)cc(Cl)n1)c1Cl)=O YEBBCAXAPQQICH-UHFFFAOYSA-N 0.000 description 1
- OVRKATYHWPCGPZ-UHFFFAOYSA-N CC1CCOCC1 Chemical compound CC1CCOCC1 OVRKATYHWPCGPZ-UHFFFAOYSA-N 0.000 description 1
- SXVSQYAEJMBGBN-MUMRKEEXSA-N CCCC([C@@H](Cc1c2c(-c3c[n](C(C)C)nc3)n[nH]1)N=C2OC(CCOC)C(C)C)(F)F Chemical compound CCCC([C@@H](Cc1c2c(-c3c[n](C(C)C)nc3)n[nH]1)N=C2OC(CCOC)C(C)C)(F)F SXVSQYAEJMBGBN-MUMRKEEXSA-N 0.000 description 1
- UIIOSUKUDFDWQA-UHFFFAOYSA-N Cc1n[nH]c2cc(Cl)nc(NC3CCCCC3)c12 Chemical compound Cc1n[nH]c2cc(Cl)nc(NC3CCCCC3)c12 UIIOSUKUDFDWQA-UHFFFAOYSA-N 0.000 description 1
- LMYJGUNNJIDROI-UHFFFAOYSA-N OC1CCOCC1 Chemical compound OC1CCOCC1 LMYJGUNNJIDROI-UHFFFAOYSA-N 0.000 description 1
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Abstract
式Ia若しくは式Ibの化合物、又は薬学的に許容されるその塩若しくはエステル[式中、R1は、アリール、ヘテロアリール、−NHR3、縮合アリール−C4−7−ヘテロシクロアルキル、−CONR4R5、−NHCOR6、−C3−7−シクロアルキル、−O−C3−7−シクロアルキル、−NR3R6及び置換されていてもよい−C1−6アルキルから選択され、ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、それぞれ置換されていてもよく、Qは、CN、ハロゲンであるか、又は、C1−6−アルキル、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール及びヘテロアリールから選択され、そのそれぞれは、1又は2以上の置換基Aで置換されていてもよく、R2は、水素、アリール、C1−6−アルキル、C2−6−アルケニル、C3−7−シクロアルキル、ヘテロアリール、C4−7ヘテロシクロアルキル及びハロゲンから選択され、ここで、前記C1−6−アルキル、C2−6−アルケニル、アリール、ヘテロアリール及びC4−7−ヘテロシクロアルキルは、それぞれ置換されていてもよく、R3は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、C3−7−シクロアルキル、縮合アリール−C−ヘテロシクロアルキル及びC1−6−アルキルから選択され、そのそれぞれは、置換されていてもよく、R4及びR5は、それぞれ独立に、水素、又は置換されていてもよいC3−7−シクロアルキル、アリール、ヘテロアリール、C1−6−アルキル若しくはC3−6−ヘテロシクロアルキルであるか、或いはR4及びR5は、それらが結合したNと一緒になって、C3−6−ヘテロシクロアルキル環を形成し、各R6は、C1−6−アルキル、C3−7シクロアルキル、C−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれは、置換されていてもよく、各R7は、水素、置換されていてもよいC1−6−アルキル及びC3−7−シクロアルキルから選択され、R8及びR9のそれぞれは、独立に、水素又は置換されていてもよいC1−6−アルキルであるか、或いはR8及びR9は、それらが結合したNと一緒になって、C4−6−ヘテロシクロアルキルを形成し、各R10は、C3−7−シクロアルキル及び置換されていてもよいC1−6−アルキルから選択され、各R11は、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれは、置換されていてもよく、Aは、ハロゲン、−NR4SO2R5、−CN、−OR6、−NR4R5、−NR7R11、ヒドロキシル、−CF3、−CONR4R5、−NR4COR5、−NR7(CO)NR4R5、−NO2、−CO2H、−CO2R6、−SO2R6、−SO2NR4R5、−NR4COR5、−NR4COOR5、C1−6−アルキル及び−COR6から選択される]。さらなる態様は、医薬組成物、治療的使用、並びに式Ia及びIbの化合物を調製するための方法に関する。
Description
本発明は、1又は2以上のキナーゼ、より詳細にはLRRK2を阻害することができるピラゾロピリジン化合物に関する。該化合物は、がん、及びパーキンソン病等の神経変性疾患を含む様々な障害の治療における用途が見出されている。
LRRK2をコードしている遺伝子内の異なる常染色体優性点突然変異は、ヒトに、特発性PDと識別不能な臨床的所見を伴う遅発性PD(OMIM受託番号609007)を発症する素因を与えるという最近の発見により、非常に関心が高まってきている(Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W. and Singleton, A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 44, 595-600、Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P. and Gallo, K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286-293、Taylor, J. P., Mata, I.F. and Farrer, M. J. (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 12, 76-82)。今までに行われた遺伝子分析は、LRRK2における突然変異が比較的頻度の高いものであり、家族性PDの5〜10%を占めるだけでなく、散発性PD症例のかなりの割合においても見られることを示している(Farrer, M., Stone, J., Mata, I. F., Lincoln, S., Kachergus, J., Hulihan, M., Strain, K. J. and Maraganore, D. M. (2005) LRRK2 mutations in Parkinson disease. Neurology. 65, 738-740、Zabetian, C. P., Samii, A., Mosley, A. D., Roberts, J. W., Leis, B. C., Yearout, D., Raskind, W. H. and Griffith, A. (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 65, 741-744)。LRRK2が細胞中でいかにして調節されるのか、その生理学的基質は何であるか、及び突然変異がいかにしてPDのリスクを引き起こし又は増加させるかについては、ほとんど分かっていない。
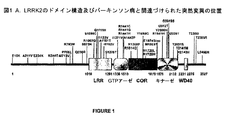
LRRK2のドメイン構造は図1に示されており、図1は、PD患者においてこれまでに報告されてきた突然変異も描写している。LRRK2酵素の定義的特徴は、ロイシンリッチリピート(LRR,Leucine Rich Repeat)モチーフ(残基1010〜1291)、Ras様低分子量GTPアーゼ(残基1336〜1510)、複雑なRasのC末端(COR,C-terminal Of Ras)ドメインと呼ばれている高アミノ酸保存の領域(残基1511〜1878)、タンパク質キナーゼ触媒ドメイン(残基1879〜2132)及びC末端WD40モチーフ(2231〜2276)である(Bosgraaf, L. and Van Haastert, P. J. (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 1643, 5-10、Marin, I. (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. 23, 2423-2433)。LRRK2のタンパク質キナーゼドメインは、チロシン様セリン/トレオニンタンパク質キナーゼに属し、先天性免疫シグナル伝達経路において中心的な役割を果たすキナーゼ受容体共役タンパク質(RIP,Receptor Interacting Protein)に極めて類似している(Manning, G., Whyte, D. B., Martinez, R., Hunter, T. and Sudarsanam, S. (2002) The protein kinase complement of the human genome. Science. 298, 1912-1934)。今までに、ほぼ40種の単一アミノ酸置換変異が常染色体優性PDと関連づけられており、これらの突然変異の位置を図1に例証する(Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P. and Gallo, K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286-293、Taylor, J. P., Mata, I.F. and Farrer, M. J. (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 12, 76-82)。ヨーロッパにおいて家族性PD症例の約6%及び散発性PD症例の3%を占めるLRRK2の最も蔓延している突然変異型は、Gly2019からSer残基へのアミノ酸置換を含む。Gly2019は、キナーゼドメインのサブドメインVIIにおける保存DYG−Mg2+結合モチーフ内に位置している(Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P. and Gallo, K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286-293)。最近の報告は、この突然変異が、LRRK2の自己リン酸化、及びミエリン塩基性タンパク質をリン酸化するその能力を2〜3倍増強することを示唆しており(West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., Dawson, V. L. and Dawson, T. M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 102, 16842-16847、Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., van der Brug, M. P., Beilina, A., Blackinton, J., Thomas, K. J., Ahmad, R., Miller, D. W., Kesavapany, S., Singleton, A., Lees, A., Harvey, R. J., Harvey, K. and Cookson, M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 23, 329-341)、本出願人によって知見が確認された(Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317)。これらの観察は、LRRK2の過活性化がヒトにPDを発症する素因を与えることを示唆し、LRRK2を阻害した薬物を利用して、いくつかの形態のPDの症状の進行を停止させ得ること、又はことによると逆行すらさせ得ることを暗示している。
LRRK2の研究は、活性組換え酵素を発現させることの困難さによって、及びロバストな定量的アッセイの欠如によって妨げられてきた。本出願人が行った研究では、GTPアーゼ−CORと残基1326〜2527を包含するキナーゼドメインとを含有するLRRK2の活性組換え断片を、293の細胞において発現させた(Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317)。生理学的基質を同定するための最初の試みにおいて、このLRRK2断片のより活性なG2019S変異体をキナーゼ基質の追跡及び解明(KESTREL,KinasE Substrate TRacking and ELucidation)スクリーニングに利用した(Cohen, P. and Knebel, A. (2006) KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem J. 393, 1-6において総説されている)。これは、LRRK2によってインビトロで効率的にリン酸化される、モエシンと呼ばれるタンパク質の同定に至った(Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317)。モエシンは、アクチン細胞骨格を原形質膜に固定するように機能し、膜構造及び組織を調節する上で重要な役割を果たす、タンパク質のエズリン/ラディキシン/モエシン(ERM,Ezrin/Radixin/Moesin)ファミリーのメンバーである(Bretscher, A., Edwards, K. and Fehon, R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 3, 586-599、Polesello, C. and Payre, F. (2004) Small is beautiful: what flies tell us about ERM protein function in development. Trends Cell Biol. 14, 294-302)。LRRK2は、先に特徴づけられた生理学的に妥当なリン酸化部位(Bretscher, A., Edwards, K. and Fehon, R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 3, 586-599、Polesello, C. and Payre, F. (2004) Small is beautiful: what flies tell us about ERM protein function in development. Trends Cell Biol. 14, 294-302)であるThr558においてモエシンをリン酸化することが分かった(Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317)。LRRK2は、同等のThr残基においてエズリン及びラディキシンもリン酸化した。Thr558と同等の残基におけるERMタンパク質のリン酸化は、これらのタンパク質の構造を開き、N末端FERMドメインを介して、それらのC末端残基におけるアクチンマイクロフィラメント並びにホスホイノシチド及び原形質膜タンパク質と相互作用することを可能にする。これらの知見を利用して、モエシンの、又はLRRK2によっても効率的にリン酸化されるモエシンのThr558残基を包含する短ペプチドのリン酸化に基づく、LRRK2のためのロバストで定量的なアッセイを開発した(Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317)。これらのアッセイを、ニクチド(Nictide)ペプチドの使用に基づく改善されたアッセイを開発するためにさらに適応させた(Nichols, R. J., Dzamko, N., Hutti, J. E., Cantley, L. C., Deak, M., Moran, J., Bamborough, P., Reith, A. D. and Alessi, D. R. (2009) Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 424, 47-60)。
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W. and Singleton, A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 44, 595-600
Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P. and Gallo, K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286-293
Taylor, J. P., Mata, I. F. and Farrer, M. J. (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 12, 76-82
Farrer, M., Stone, J., Mata, I. F., Lincoln, S., Kachergus, J., Hulihan, M., Strain, K. J. and Maraganore, D. M. (2005) LRRK2 mutations in Parkinson disease. Neurology. 65, 738-740
Zabetian, C. P., Samii, A., Mosley, A. D., Roberts, J. W., Leis, B. C., Yearout, D., Raskind, W. H. and Griffith, A. (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 65, 741-744
Bosgraaf, L. and Van Haastert, P. J. (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 1643, 5-10
Marin, I. (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. 23, 2423-2433
Manning, G., Whyte, D. B., Martinez, R., Hunter, T. and Sudarsanam, S. (2002) The protein kinase complement of the human genome. Science. 298, 1912-1934
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., Dawson, V. L. and Dawson, T. M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 102, 16842-16847
Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., van der Brug, M. P., Beilina, A., Blackinton, J., Thomas, K. J., Ahmad, R., Miller, D. W., Kesavapany, S., Singleton, A., Lees, A., Harvey, R. J., Harvey, K. and Cookson, M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 23, 329-341
Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317
Cohen, P. and Knebel, A. (2006) KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem J. 393, 1-6
Bretscher, A., Edwards, K. and Fehon, R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 3, 586-599
Polesello, C. and Payre, F. (2004) Small is beautiful: what flies tell us about ERM protein function in development. Trends Cell Biol. 14, 294-302
Nichols, R. J., Dzamko, N., Hutti, J. E., Cantley, L. C., Deak, M., Moran, J., Bamborough, P., Reith, A. D. and Alessi, D. R. (2009) Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 424, 47-60
本発明は、1又は2以上のキナーゼ、より詳細にはLRRK、一層好ましくはLRRK2を阻害することができる化合物を提供しようとするものである。
本発明の第一の態様は、式Ia若しくは式Ibの化合物、又は薬学的に許容されるその塩若しくはエステル

[式中、
R1は、
アリール、
ヘテロアリール、
C4−7−ヘテロシクロアルキル、
−NHR3、
縮合アリール−C4−7−ヘテロシクロアルキル、
−CONR4R5
−NHCOR6、
−C3−7−シクロアルキル、
−NR3R6、
OR3、
OH、
NR4R5、並びに
R11及び基Aから選択される置換基で置換されていてもよい−C1−6アルキルから選択され、
ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基でそれぞれ置換されていてもよく、
R2は、水素、アリール、C1−6−アルキル、C2−6−アルケニル、C3−7−シクロアルキル、ヘテロアリール、C4−7ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及びハロゲンから選択され、ここで、前記C1−6−アルキル、C2−6−アルケニル、アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、R11及びAから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、
Qは、ハロゲン、CNであるか、又は、C1−6−アルキル、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール及びヘテロアリールから選択され、そのそれぞれが、1又は2以上の置換基Aで置換されていてもよく、
各R3は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、C3−7−シクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及びC1−6−アルキルから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基で置換されていてもよく、
R4及びR5は、水素、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、アリール、ヘテロアリール、C1−6−アルキル、並びに酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよく、かつ1又は2以上のR10基によって置換されていてもよいC3−6−ヘテロシクロアルキル環からそれぞれ独立に選択され、ここで、各C1−6−アルキル、ヘテロアリール及びアリールは、C1−6−アルキル、ハロゲン、シアノ、ヒドロキシル、アリール、ハロ置換アリール、ヘテロアリール、−NR8R9、−NR6R7、NR7(CO)R6、−NR7COOR6、−NR7(SO2)R6、−COOR6、−CONR8R9、OR6、−SO2R6、並びに酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよく、かつ1又は2以上のR10基によって置換されていてもよいC3−6−ヘテロシクロアルキル環から選択される1又は2以上の置換基によって置換されていてもよく、或いは
R4及びR5は、それらが結合したNと一緒になって、酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよいC3−6−ヘテロシクロアルキル環を形成し、ここで、前記C3−6−ヘテロシクロアルキル環は、飽和又は不飽和であり、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよく、
各R6は、C1−6−アルキル、C3−7シクロアルキル、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれは、R10、R11及びAから選択される1又は2以上の置換基によって置換されていてもよく、
各R7は、水素、C1−6−アルキル及びC3−7−シクロアルキルから選択され、ここで、前記C1−6−アルキルは、1又は2以上のハロゲンによって置換されていてもよく、
R8及びR9のそれぞれは、水素及びC1−6−アルキルから独立に選択され、ここで、前記C1−6−アルキル基は、1又は2以上のハロゲンによって置換されていてもよく、或いは
R8及びR9は、それらが結合したNと一緒になって、酸素及び硫黄から選択される1又は2以上のヘテロ原子をさらに含有していてもよいC4−6−ヘテロシクロアルキル環を形成し、ここで、前記C4−6−ヘテロシクロアルキル環は、1又は2以上のR10基によって置換されていてもよく、
各R10は、C3−7−シクロアルキル、アリール、ヘテロアリール、O−ヘテロアリール、アラルキル及びC1−6−アルキルから選択され、そのそれぞれが、1又は2以上のA基によって置換されていてもよく、ここで、R10がC1−6−アルキルであり、2又は3以上のR10基が同じ炭素原子に結合している場合、該R10基は連結してスピロアルキル基を形成してよく、
各R11は、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、C1−6−アルキル−ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれが、Aから選択される1又は2以上の置換基で置換されていてもよく、
Aは、ハロゲン、−NR4SO2R5、−CN、−OR6、−NR4R5、−NR7R11、ヒドロキシル、−CF3、−CONR4R5、−NR4COR5、−NR7(CO)NR4R5、−NO2、−CO2H、−CO2R6、−SO2R6、−SO2NR4R5、−NR4COR5、−NR4COOR5、C1−6−アルキル、アリール及び−COR6から選択される]
に関する。
R1は、
アリール、
ヘテロアリール、
C4−7−ヘテロシクロアルキル、
−NHR3、
縮合アリール−C4−7−ヘテロシクロアルキル、
−CONR4R5
−NHCOR6、
−C3−7−シクロアルキル、
−NR3R6、
OR3、
OH、
NR4R5、並びに
R11及び基Aから選択される置換基で置換されていてもよい−C1−6アルキルから選択され、
ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基でそれぞれ置換されていてもよく、
R2は、水素、アリール、C1−6−アルキル、C2−6−アルケニル、C3−7−シクロアルキル、ヘテロアリール、C4−7ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及びハロゲンから選択され、ここで、前記C1−6−アルキル、C2−6−アルケニル、アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、R11及びAから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、
Qは、ハロゲン、CNであるか、又は、C1−6−アルキル、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール及びヘテロアリールから選択され、そのそれぞれが、1又は2以上の置換基Aで置換されていてもよく、
各R3は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、C3−7−シクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及びC1−6−アルキルから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基で置換されていてもよく、
R4及びR5は、水素、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、アリール、ヘテロアリール、C1−6−アルキル、並びに酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよく、かつ1又は2以上のR10基によって置換されていてもよいC3−6−ヘテロシクロアルキル環からそれぞれ独立に選択され、ここで、各C1−6−アルキル、ヘテロアリール及びアリールは、C1−6−アルキル、ハロゲン、シアノ、ヒドロキシル、アリール、ハロ置換アリール、ヘテロアリール、−NR8R9、−NR6R7、NR7(CO)R6、−NR7COOR6、−NR7(SO2)R6、−COOR6、−CONR8R9、OR6、−SO2R6、並びに酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよく、かつ1又は2以上のR10基によって置換されていてもよいC3−6−ヘテロシクロアルキル環から選択される1又は2以上の置換基によって置換されていてもよく、或いは
R4及びR5は、それらが結合したNと一緒になって、酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよいC3−6−ヘテロシクロアルキル環を形成し、ここで、前記C3−6−ヘテロシクロアルキル環は、飽和又は不飽和であり、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよく、
各R6は、C1−6−アルキル、C3−7シクロアルキル、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれは、R10、R11及びAから選択される1又は2以上の置換基によって置換されていてもよく、
各R7は、水素、C1−6−アルキル及びC3−7−シクロアルキルから選択され、ここで、前記C1−6−アルキルは、1又は2以上のハロゲンによって置換されていてもよく、
R8及びR9のそれぞれは、水素及びC1−6−アルキルから独立に選択され、ここで、前記C1−6−アルキル基は、1又は2以上のハロゲンによって置換されていてもよく、或いは
R8及びR9は、それらが結合したNと一緒になって、酸素及び硫黄から選択される1又は2以上のヘテロ原子をさらに含有していてもよいC4−6−ヘテロシクロアルキル環を形成し、ここで、前記C4−6−ヘテロシクロアルキル環は、1又は2以上のR10基によって置換されていてもよく、
各R10は、C3−7−シクロアルキル、アリール、ヘテロアリール、O−ヘテロアリール、アラルキル及びC1−6−アルキルから選択され、そのそれぞれが、1又は2以上のA基によって置換されていてもよく、ここで、R10がC1−6−アルキルであり、2又は3以上のR10基が同じ炭素原子に結合している場合、該R10基は連結してスピロアルキル基を形成してよく、
各R11は、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、C1−6−アルキル−ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれが、Aから選択される1又は2以上の置換基で置換されていてもよく、
Aは、ハロゲン、−NR4SO2R5、−CN、−OR6、−NR4R5、−NR7R11、ヒドロキシル、−CF3、−CONR4R5、−NR4COR5、−NR7(CO)NR4R5、−NO2、−CO2H、−CO2R6、−SO2R6、−SO2NR4R5、−NR4COR5、−NR4COOR5、C1−6−アルキル、アリール及び−COR6から選択される]
に関する。
本発明の第二の態様は、少なくとも1の上述した通りの化合物と、薬学的に許容される担体、賦形剤(diluent)又は添加剤とを含む医薬組成物に関する。
本発明の第三の態様は、医療において使用するための、上述した通りの化合物に関する。
本発明の第四の態様は、がん、及びパーキンソン病等の神経変性疾患から選択される障害を治療するのに使用するための、上述した通りの化合物に関する。
本発明の第五の態様は、がん、及びパーキンソン病等の神経変性疾患から選択される障害を治療又は予防するための薬剤の調製における、上述した通りの化合物の使用に関する。
本発明の第六の態様は、何らかの異常なキナーゼ活性によって引き起こされ、それに関連し、又はそれを伴い、該キナーゼが好ましくはLRRK、より好ましくはLRRK2である障害の予防又は治療のための薬剤の調製における、上述した通りの化合物の使用に関する。
本発明の第七の態様は、哺乳動物に、治療有効量の上述した通りの化合物を投与するステップを含む、キナーゼ(好ましくはLRRK、より好ましくはLRRK2)の阻害によって軽減される病状を有する哺乳動物を治療する方法に関する。
本発明の第八の態様は、キナーゼ、好ましくはLRRK、より好ましくはLRRK2の阻害ができるさらなる候補化合物を同定するためのアッセイにおける、上述した通りの化合物の使用に関する。
本発明の第九の態様は、式Ia及び式Ibの化合物を調製するための方法に関する。
本発明は、1又は2以上のキナーゼ、より詳細にはLRRK、一層詳細にはLRRK2を阻害することができるピラゾロピリジン化合物に関する。特に、本発明は、置換ピラゾロ[4,3−c]ピリジン誘導体に関する。
「アルキル」は、本明細書において、直鎖状又は分岐状アルキルラジカル、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、tert−ブチル、ペンチル、ヘキシルとして定義される。
「シクロアルキル」は、本明細書において、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル若しくはシクロヘプチル等の単環式アルキル環、又はノルボルナン等の縮合二環式環系として定義される。
「ハロゲン」は、本明細書において、クロロ、フルオロ、ブロモ又はヨードとして定義される。
本明細書において使用される場合、用語「アリール」は、C6−12芳香族基を指し、これは、ベンゾ縮合した、例えばフェニル又はナフチルであってよい。
「ヘテロアリール」は、本明細書において、酸素、窒素又は硫黄等の(同じであっても異なっていてもよい)1又は2以上のヘテロ原子を含む単環式又は二環式C2−12芳香族環として定義される。適切なヘテロアリール基の例は、チエニル、フラニル、ピロリル、ピリジニル、オキサゾリル、チアゾリル、イミダゾリル、ピラゾリル、イソオキサゾリル、イソチアゾリル、オキサジアゾリル、トリアゾリル、チアジアゾリル等、及びベンゾフラニル、ベンゾチエニル、ベンズイミダゾリル、インドリル、イソインドリル、インダゾリル等のそのベンゾ誘導体;又は、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、トリアジニル等、及びキノリニル、イソキノリニル、シンノリニル、フタラジニル、キナゾリニル、キノキサリニル、ナフチリジニル等のそのベンゾ誘導体を含む。
「ヘテロシクロアルキル」は、環中で1若しくは2以上の−(CO)−基によって中断されていてもよく、及び/又は環中に1若しくは2以上の二重結合を含有していてもよい、窒素、酸素及び硫黄から選択される1又は2以上のヘテロ原子を含有する環式脂肪族基を指す。好ましくは、ヘテロシクロアルキル基は、C3−7−ヘテロシクロアルキル、より好ましくはC3−6−ヘテロシクロアルキルである。代替として、ヘテロシクロアルキル基は、C4−7−ヘテロシクロアルキル、より好ましくはC4−6−ヘテロシクロアルキルである。好ましいヘテロシクロアルキル基は、ピペラジニル、ピペリジニル、モルホリニル、チオモルホリニル、ピロリジニル、テトラヒドロフラニル及びテトラヒドロピラニルを含むがこれらに限定されない。
1つの好ましい実施形態において、本発明は、式Iaの化合物に関する。
別の好ましい実施形態において、本発明は、式Ibの化合物に関する。
以下で説明する好ましい定義は、式Ia及び式Ibに当てはまる。
本発明の1つの好ましい実施形態において、R2は、
水素、
ハロゲン、より好ましくは臭素、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいアリール、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいC1−6−アルキル、
1又は2以上のA置換基によって置換されていてもよいC2−6−アルケニル、
C3−7−シクロアルキル、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール、
C4−7−ヘテロシクロアルキル、並びに
縮合アリール−C4−7−ヘテロシクロアルキル
から選択される。
水素、
ハロゲン、より好ましくは臭素、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいアリール、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいC1−6−アルキル、
1又は2以上のA置換基によって置換されていてもよいC2−6−アルケニル、
C3−7−シクロアルキル、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール、
C4−7−ヘテロシクロアルキル、並びに
縮合アリール−C4−7−ヘテロシクロアルキル
から選択される。
本発明の1つの好ましい実施形態において、R2は、
−NR4COR5、−CONR4R5、OR6、ハロゲン、置換されていてもよいC1−6−アルキル、CN、C4−7−ヘテロシクロアルキル及びヘテロアリールから選択される1又は2以上の置換基によって置換されていてもよいアリール、
−NR4COR5、−CONR4R5、−NR4R5、OR6、置換されていてもよいアリール、置換されていてもよいヘテロアリール及びC4−7−ヘテロシクロアルキルから選択される1又は2以上の置換基によって置換されていてもよいC1−6−アルキル、
1又は2以上の−CONR4R5置換基によって置換されていてもよいC2−6−アルケニル、
C3−7−シクロアルキル、
C4−7−ヘテロシクロアルキル、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル及びOR6から選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール、
C4−7−ヘテロシクロアルキル、並びに
縮合アリール−C4−7−ヘテロシクロアルキル
から選択される。
−NR4COR5、−CONR4R5、OR6、ハロゲン、置換されていてもよいC1−6−アルキル、CN、C4−7−ヘテロシクロアルキル及びヘテロアリールから選択される1又は2以上の置換基によって置換されていてもよいアリール、
−NR4COR5、−CONR4R5、−NR4R5、OR6、置換されていてもよいアリール、置換されていてもよいヘテロアリール及びC4−7−ヘテロシクロアルキルから選択される1又は2以上の置換基によって置換されていてもよいC1−6−アルキル、
1又は2以上の−CONR4R5置換基によって置換されていてもよいC2−6−アルケニル、
C3−7−シクロアルキル、
C4−7−ヘテロシクロアルキル、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル及びOR6から選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール、
C4−7−ヘテロシクロアルキル、並びに
縮合アリール−C4−7−ヘテロシクロアルキル
から選択される。
本発明の1つの好ましい実施形態において、R2は、
−NHCO−C1−6−アルキル、−CONHC1−6−アルキル、CO−(N−モルホリニル)、Cl、F、−OC1−6−アルキル、−CONMe2、OCF3、CN、CF3、C1−6−アルキル−(A)、N−モルホリニル及びピラゾリルから選択される1又は2以上の置換基によって置換されていてもよいフェニル基、
ピリジニル、キノリニル、ピラゾイル(pyrazoyl)、フラニル及びピリミジニル[そのそれぞれは、C1−6−アルキル、アラルキル、OC1−6−アルキル、N−モルホリニルから選択される1又は2以上の置換基によって置換されていてもよい]から選択されるヘテロアリール基、
−CONR4R5、フェニル、ピリジニル及びオキサジアゾリル及びピペリジニルから選択される1又は2以上の置換基[ここで、前記フェニル、ピリジニル及びオキサジアゾリル及びピペリジニル基はそれぞれ、1又は2以上の−NR4COR5、−CONR4R5、COR6、SO2R6又はアリール基によってさらに置換されていてもよい]によって置換されていてもよいC1−6−アルキル基
から選択される。
−NHCO−C1−6−アルキル、−CONHC1−6−アルキル、CO−(N−モルホリニル)、Cl、F、−OC1−6−アルキル、−CONMe2、OCF3、CN、CF3、C1−6−アルキル−(A)、N−モルホリニル及びピラゾリルから選択される1又は2以上の置換基によって置換されていてもよいフェニル基、
ピリジニル、キノリニル、ピラゾイル(pyrazoyl)、フラニル及びピリミジニル[そのそれぞれは、C1−6−アルキル、アラルキル、OC1−6−アルキル、N−モルホリニルから選択される1又は2以上の置換基によって置換されていてもよい]から選択されるヘテロアリール基、
−CONR4R5、フェニル、ピリジニル及びオキサジアゾリル及びピペリジニルから選択される1又は2以上の置換基[ここで、前記フェニル、ピリジニル及びオキサジアゾリル及びピペリジニル基はそれぞれ、1又は2以上の−NR4COR5、−CONR4R5、COR6、SO2R6又はアリール基によってさらに置換されていてもよい]によって置換されていてもよいC1−6−アルキル基
から選択される。
本発明のより好ましい実施形態において、各−CONR4R5基は、
−CO(N−モルホリニル)、−CO(N−ピペリジニル)、−CO(N−ピロリジニル)、−CO−(N−ピペラジニル)[そのそれぞれは、アリール、ヘテロアリール、−OR6、CF3、アラルキル、−NR4COR5−CONR4R5、−NR4R5、ハロゲン、C1−6−アルキルから選択される1又は2以上の置換基によってさらに置換されていてもよい]、及び
−CON(C1−6−アルキル)2、CONH(C1−6−アルキル)、CON(C1−6−アルキル)(アラルキル)、CONH(C3−7−シクロアルキル)、−CONH(アリール)、−CONH(ヘテロアリール)[ここで、前記C1−6−アルキル、アラルキル、アリール及びヘテロアリール基はそれぞれ、1又は2以上のR11又はA基によってさらに置換されていてもよい]
から独立に選択される。
−CO(N−モルホリニル)、−CO(N−ピペリジニル)、−CO(N−ピロリジニル)、−CO−(N−ピペラジニル)[そのそれぞれは、アリール、ヘテロアリール、−OR6、CF3、アラルキル、−NR4COR5−CONR4R5、−NR4R5、ハロゲン、C1−6−アルキルから選択される1又は2以上の置換基によってさらに置換されていてもよい]、及び
−CON(C1−6−アルキル)2、CONH(C1−6−アルキル)、CON(C1−6−アルキル)(アラルキル)、CONH(C3−7−シクロアルキル)、−CONH(アリール)、−CONH(ヘテロアリール)[ここで、前記C1−6−アルキル、アラルキル、アリール及びヘテロアリール基はそれぞれ、1又は2以上のR11又はA基によってさらに置換されていてもよい]
から独立に選択される。
本発明の1つの好ましい実施形態において、R2は、−NR4COR5、−CONR4R5、−NR4R5、OR6、C4−7−ヘテロシクロアルキル、ヘテロアリール及びアリールから選択される1又は2以上の置換基によって選択されていてもよいC1−6−アルキル基であり、ここで、前記アリール基は、−NR4COR5及び−CONR4R5から選択される1又は2以上の置換基によって置換されていてもよい。
本発明の1つの好ましい実施形態において、R2は、−CH2CH2CO−NR4R5、C1−6−アルキル、C3−7シクロアルキル、並びにフラニル及びピラゾリルから選択されるヘテロアリールから選択され、ここで、前記フラニル及びピラゾリル基は、C1−6−アルキル、C3−7−シクロアルキル及びC1−6−アルキル−C3−7−シクロアルキルから選択される1又は2以上の置換基によって置換されていてもよい。
本発明の1つの好ましい実施形態において、R2は、Me
[式中、R4及びR5は、それらが結合したNと一緒になって、酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよいC3−6−ヘテロシクロアルキル環を形成し、ここで、前記C3−6−ヘテロシクロアルキル環は、飽和又は不飽和であり、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよい]から選択される。一層好ましくは、R4及びR5は、それらが結合したNと一緒になって、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよい6員ヘテロシクロアルキル環を形成する。さらに好ましくは、R4及びR5は、それらが結合したNと一緒になって、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよい飽和6員環(より好ましくは、ピペリジニル環)を形成する。
本発明の1つの非常に好ましい実施形態において、R2は、Me
から選択される。
本発明の1つの好ましい実施形態において、R2は、アリール、C1−6−アルキル及びヘテロアリールから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基で置換されていてもよい。
本発明の別の好ましい実施形態において、R2は、アリール、C1−6−アルキル及びヘテロアリールから選択され、そのそれぞれは、CONR4R5、CF3、C1−6−アルキル、OR6及びC4−7−ヘテロシクロアルキルから選択される1又は2以上の置換基で置換されていてもよい。
本発明の別の好ましい実施形態において、R2は、C1−6−アルキル、フェニル、ピリジニル、ピリミジニル及びピラゾリルから選択され、そのそれぞれは、CONR4R5、CF3、C1−6−アルキル、OR6及びC4−7−ヘテロシクロアルキルから選択される1又は2以上の置換基によって置換されていてもよい。
本発明の別の好ましい実施形態において、R2は、C1−6−アルキル、フェニル、ピリジニル、ピリミジニル及びピラゾリルから選択され、そのそれぞれは、CONMe2、CF3、イソ−ブチル、イソ−プロピル、OEt及びモルホリニルから選択される1又は2以上の置換基によって置換されていてもよい。
本発明の非常に好ましい実施形態において、R2は、下記:Me、
から選択される。
本発明の1つの好ましい実施形態において、R2は、非置換C1−6−アルキル基、より好ましくはメチルである。
本発明の1つの好ましい実施形態において、R1は、
−NHR3、
アリール、
ヘテロアリール、
C4−7−ヘテロシクロアルキル、
縮合アリール−C4−7−ヘテロシクロアルキル、
−C3−7−シクロアルキル、
−NR3R6、
OR3、
NR4R5、並びに
R11及び基Aから選択される置換基で置換されていてもよい−C1−6アルキルから選択され、
ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基によってそれぞれ置換されていてもよい。
−NHR3、
アリール、
ヘテロアリール、
C4−7−ヘテロシクロアルキル、
縮合アリール−C4−7−ヘテロシクロアルキル、
−C3−7−シクロアルキル、
−NR3R6、
OR3、
NR4R5、並びに
R11及び基Aから選択される置換基で置換されていてもよい−C1−6アルキルから選択され、
ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基によってそれぞれ置換されていてもよい。
本発明の1つの好ましい実施形態において、R1は−NHR3であり、R3は、
1又は2以上の−OR6、NR4COR5、ヘテロアリール、アリール、C4−7−ヘテロシクロアルキル及びC3−7−シクロアルキル基[ここで、前記アリール及びヘテロアリール基は、それぞれ独立に、CF3、ハロゲン、C1−6−アルキル、−OR6及び−NR4R5から選択される1又は2以上の基によってさらに置換されていてもよい]によって置換されていてもよいC1−6−アルキル、
−OR6、NR4COR5、−CONR4R5、アリール、−NR4R5、C1−6−アルキル−ヘテロアリール、ヘテロアリール、ハロゲン、−SO2R6、CN、CF3、C1−6−アルキル、−SO2NR4R5、−NR4SO2R5から選択される1又は2以上の置換基[ここで、前記C1−6−アルキル、ヘテロアリール及びアリール基は、それぞれ独立に、CN、CF3、ハロゲン、C1−6−アルキル、−OR6及び−NR4R5から選択される1又は2以上の基によってさらに置換されていてもよい]によって置換されていてもよいフェニル基、
アリール、C1−6−アルキル及び−NR4R5から選択される1又は2以上の置換基[ここで、前記アリール基は、1又は2以上のA基によってさらに置換されていてもよい]によって置換されていてもよいヘテロアリール基、
1又は2以上の−COR6基によって置換されていてもよいC4−7−ヘテロシクロアルキル、
1又は2以上のハロゲン又はC1−6−アルキル基によって置換されていてもよいC3−7−シクロアルキル基
から選択される。
1又は2以上の−OR6、NR4COR5、ヘテロアリール、アリール、C4−7−ヘテロシクロアルキル及びC3−7−シクロアルキル基[ここで、前記アリール及びヘテロアリール基は、それぞれ独立に、CF3、ハロゲン、C1−6−アルキル、−OR6及び−NR4R5から選択される1又は2以上の基によってさらに置換されていてもよい]によって置換されていてもよいC1−6−アルキル、
−OR6、NR4COR5、−CONR4R5、アリール、−NR4R5、C1−6−アルキル−ヘテロアリール、ヘテロアリール、ハロゲン、−SO2R6、CN、CF3、C1−6−アルキル、−SO2NR4R5、−NR4SO2R5から選択される1又は2以上の置換基[ここで、前記C1−6−アルキル、ヘテロアリール及びアリール基は、それぞれ独立に、CN、CF3、ハロゲン、C1−6−アルキル、−OR6及び−NR4R5から選択される1又は2以上の基によってさらに置換されていてもよい]によって置換されていてもよいフェニル基、
アリール、C1−6−アルキル及び−NR4R5から選択される1又は2以上の置換基[ここで、前記アリール基は、1又は2以上のA基によってさらに置換されていてもよい]によって置換されていてもよいヘテロアリール基、
1又は2以上の−COR6基によって置換されていてもよいC4−7−ヘテロシクロアルキル、
1又は2以上のハロゲン又はC1−6−アルキル基によって置換されていてもよいC3−7−シクロアルキル基
から選択される。
本発明の1つの好ましい実施形態において、R1は−NHR3であり、R3は、C1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びアリールから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい。
本発明の1つの好ましい実施形態において、R1は−OR3であり、ここで、R3は、C1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びアリールから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい。
本発明の1つの好ましい実施形態において、R1は−OR3であり、ここで、R3は、C1−6−アルキル、C3−7−シクロアルキル又はC4−7−ヘテロシクロアルキルであり、そのそれぞれは、1又は2以上のA置換基によって置換されていてもよい。本発明の1つの特に好ましい実施形態において、R1は、−O−C3−7−シクロアルキル、より好ましくは−O−シクロヘキシルである。
本発明の1つの好ましい実施形態において、R1は、ヘテロアリール、−NHR3及びOR3から選択され、ここで、前記ヘテロアリール基は、基Aから選択される1又は2以上の置換基で置換されていてもよい。
本発明の1つの好ましい実施形態において、R1はアリール又はヘテロアリールであり、そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよく、より好ましくは、R1はフリルである。
本発明の1つの好ましい実施形態において、R1は−NH−C3−7−シクロアルキル又はNH−C4−7−ヘテロシクロアルキルであり、そのそれぞれは、1又は2以上のA置換基によって置換されていてもよい。
好ましくは、すべての実施形態に関して、Aはハロゲン又はC1−6−アルキルである。
本発明の1つの好ましい実施形態において、R3はシクロヘキシル又はテトラヒドロピラニルであり、そのそれぞれは、1又は2以上のA置換基によって置換されていてもよい。
本発明の1つの好ましい実施形態において、R1は、下記:
から選択される。
本発明の1つの好ましい実施形態において、R1は−OR3又はNHR3であり、R3は、シクロヘキシル、Me又はテトラヒドロピラン−4−イルである。
本発明の1つの好ましい実施形態において、R1は−NH−シクロヘキシルである。
本発明の1つの好ましい実施形態において、R1は−NHR3であり、R2は非置換C1−6−アルキル基、より好ましくはメチルである。
本発明の1つの好ましい実施形態において、R1は−NHR3であり、R2は、1又は2以上の−CONR4R5基によって置換されているC1−6−アルキル基である。
本発明の1つの好ましい実施形態において、R1は−NHR3であり、R2はアリール又はヘテロアリール基であり、そのそれぞれは、C4−7−ヘテロシクロアルキル、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル及びOR6から選択される1又は2以上の置換基によって置換されていてもよい。
本発明の1つの好ましい実施形態において、R1は−OR3であり、R2はC1−6−アルキル基、より好ましくはメチルである。
本発明の1つの好ましい実施形態において、R1は、
から選択され、R2は、
[式中、R4及びR5は、それらが結合したNと一緒になって、酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよいC3−6−ヘテロシクロアルキル環を形成し、ここで、前記C3−6−ヘテロシクロアルキル環は、飽和又は不飽和であり、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよい]から選択される。一層好ましくは、R4及びR5は、それらが結合したNと一緒になって、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよい6員ヘテロシクロアルキル環を形成する。さらに好ましくは、R4及びR5は、それらが結合したNと一緒になって、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよい飽和6員環(より好ましくは、ピペリジニル環)を形成する。
より好ましくは、R1は上記で定義した通りであり、R2は、Me
から選択される。
本発明の1つの好ましい実施形態において、
R1は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及び−NHR3から選択され、ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基でそれぞれ置換されていてもよく、
R2は、水素、アリール、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7ヘテロシクロアルキル及びハロゲンから選択され、ここで、前記C1−6−アルキル、アリール、ヘテロアリール及びC4−7ヘテロシクロアルキルは、R11及びAから選択される1又は2以上の置換基でそれぞれ置換されていてもよい。
R1は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及び−NHR3から選択され、ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基でそれぞれ置換されていてもよく、
R2は、水素、アリール、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7ヘテロシクロアルキル及びハロゲンから選択され、ここで、前記C1−6−アルキル、アリール、ヘテロアリール及びC4−7ヘテロシクロアルキルは、R11及びAから選択される1又は2以上の置換基でそれぞれ置換されていてもよい。
本発明の別の好ましい実施形態において、R2は、R11及びAから選択される1又は2以上の置換基で置換されていてもよいC1−6−アルキル基である。
本発明の1つの好ましい実施形態において、R1は、
NH−R3[ここで、R3は、C1−6−アルキル、モルホリニル、C3−7−シクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル、ピペリジニル、テトラヒドロピラニル、ピペラジニル、フェニル、ピリジニル、インダゾリル及びピラゾリルから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい]、並びに
フリル、ピラゾリル及びフェニル[そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい]
から選択される。
NH−R3[ここで、R3は、C1−6−アルキル、モルホリニル、C3−7−シクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル、ピペリジニル、テトラヒドロピラニル、ピペラジニル、フェニル、ピリジニル、インダゾリル及びピラゾリルから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい]、並びに
フリル、ピラゾリル及びフェニル[そのそれぞれは、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい]
から選択される。
本発明の1つの好ましい実施形態において、R1は、
NH−C1−6−アルキル[ここで、前記C1−6−アルキルは、OR6、OH、C4−7ヘテロシクロアルキル、NR4R5、ヘテロアリール、C3−7−シクロアルキル、フェニルから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記フェニル基は、1又は2以上のハロ基によって置換されていてもよく、前記C4−7ヘテロシクロアルキル基は、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、
NH−ピペラジニル[ここで、前記ピペラジニルは、C1−6−アルキル、アリール、C1−6−アルキル−アリール及びヘテロアリールから選択される1又は2以上の置換基によって置換されていてもよく、そのそれぞれは、1又は2以上のハロ基によってさらに置換されていてもよい]、
NH−モルホリニル、
NH−C3−7−シクロアルキル[ここで、前記C3−7−シクロアルキルは、OH及びハロから選択される1又は2以上の置換基によって置換されていてもよい]、
NH−縮合アリール−C4−7−ヘテロシクロアルキル[ここで、前記縮合アリール−C4−7−ヘテロシクロアルキルは、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、
NH−ピペリジニル[ここで、前記ピペリジニルは、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、
NH−テトラヒドロピラニル、
フリル基、
1又は2以上のC1−6−アルキル基によって置換されていてもよいピラゾリル基、
NH−フェニル[ここで、前記フェニルは、ハロ、CF3、OH、OR6、NR4SO2R5、NR4R5、C4−7ヘテロシクロアルキル、CONR4R5及び−NR4COR5から選択される1又は2以上の置換基によって置換されていてもよい]、
NH−ピリジニル[ここで、前記ピリジニルは、C4−7ヘテロシクロアルキル及びアリールから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記アリール基は、1又は2以上のハロ基でさらに置換されていてもよい]、
ハロ、OR6、−NR4SO2R5、CN、C4−7ヘテロシクロアルキル及びC1−6−アルキル−NR4SO2R5から選択される1又は2以上の置換基によって置換されていてもよいフェニル、
NH−インダゾリル[ここで、前記インダゾリルは、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、並びに
NH−ピラゾリル
から選択される。
NH−C1−6−アルキル[ここで、前記C1−6−アルキルは、OR6、OH、C4−7ヘテロシクロアルキル、NR4R5、ヘテロアリール、C3−7−シクロアルキル、フェニルから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記フェニル基は、1又は2以上のハロ基によって置換されていてもよく、前記C4−7ヘテロシクロアルキル基は、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、
NH−ピペラジニル[ここで、前記ピペラジニルは、C1−6−アルキル、アリール、C1−6−アルキル−アリール及びヘテロアリールから選択される1又は2以上の置換基によって置換されていてもよく、そのそれぞれは、1又は2以上のハロ基によってさらに置換されていてもよい]、
NH−モルホリニル、
NH−C3−7−シクロアルキル[ここで、前記C3−7−シクロアルキルは、OH及びハロから選択される1又は2以上の置換基によって置換されていてもよい]、
NH−縮合アリール−C4−7−ヘテロシクロアルキル[ここで、前記縮合アリール−C4−7−ヘテロシクロアルキルは、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、
NH−ピペリジニル[ここで、前記ピペリジニルは、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、
NH−テトラヒドロピラニル、
フリル基、
1又は2以上のC1−6−アルキル基によって置換されていてもよいピラゾリル基、
NH−フェニル[ここで、前記フェニルは、ハロ、CF3、OH、OR6、NR4SO2R5、NR4R5、C4−7ヘテロシクロアルキル、CONR4R5及び−NR4COR5から選択される1又は2以上の置換基によって置換されていてもよい]、
NH−ピリジニル[ここで、前記ピリジニルは、C4−7ヘテロシクロアルキル及びアリールから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記アリール基は、1又は2以上のハロ基でさらに置換されていてもよい]、
ハロ、OR6、−NR4SO2R5、CN、C4−7ヘテロシクロアルキル及びC1−6−アルキル−NR4SO2R5から選択される1又は2以上の置換基によって置換されていてもよいフェニル、
NH−インダゾリル[ここで、前記インダゾリルは、1又は2以上のC1−6−アルキル基によって置換されていてもよい]、並びに
NH−ピラゾリル
から選択される。
本発明の1つの好ましい実施形態において、R1は、
NH−C1−6−アルキル[ここで、前記C1−6−アルキルは、OMe、OH、テトラヒドロピラニル、ピロリジニル、NEt2、イミダゾリル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、フェニルから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記フェニル基は、1又は2以上のクロロ基によって置換されていてもよく、前記ピロリジニル基は、1又は2以上のメチル基によって置換されていてもよい]、
NH−ピペラジニル[ここで、前記ピペラジニルは、メチル、フェニル、CH2−フェニル及びピリジニルから選択される1又は2以上の置換基よって置換されていてもよく、ここで、該フェニル基は、1又は2以上のF又はCl基によってさらに置換されていてもよい]、
NH−モルホリニル、
NH−シクロプロピル、NH−シクロブチル、NH−シクロペンチル及びNH−シクロヘキシル[ここで、前記シクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシル基は、OH及びFから選択される1又は2以上の置換基によって置換されていてもよい]、
NH−(1,2,3,4−テトラヒドロイソキノリニル)[ここで、前記1,2,3,4−テトラヒドロイソキノリニル基は、1又は2以上のメチル基によって置換されていてもよい]、
NH−ピペリジニル[ここで、前記ピペリジニルは、1又は2以上のメチル基によって置換されていてもよい]、
NH−テトラヒドロピラニル、
フリル基、
1又は2以上のメチル基によって置換されていてもよいピラゾリル基、
NH−フェニル[ここで、前記フェニルは、F、Cl、Br、CF3、OH、OEt、NHSO2Me、NMe2、モルホリニル、CONMe2、CONH2及び−NHCOMeから選択される1又は2以上の置換基によって置換されていてもよい]、
NH−ピリジニル[ここで、前記ピリジニルは、モルホリニル及びフェニルから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記フェニル基は、1又は2以上のCN基でさらに置換されていてもよい]、
F、Cl、OMe、−NHSO2Me、CN、モルホリニル及びCH2−NHSO2Meから選択される1又は2以上の置換基によって置換されていてもよいフェニル、
NH−インダゾリル[ここで、前記インダゾリルは、1又は2以上のメチル基によって置換されていてもよい]、並びに
NH−ピラゾリル
から選択される。
NH−C1−6−アルキル[ここで、前記C1−6−アルキルは、OMe、OH、テトラヒドロピラニル、ピロリジニル、NEt2、イミダゾリル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、フェニルから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記フェニル基は、1又は2以上のクロロ基によって置換されていてもよく、前記ピロリジニル基は、1又は2以上のメチル基によって置換されていてもよい]、
NH−ピペラジニル[ここで、前記ピペラジニルは、メチル、フェニル、CH2−フェニル及びピリジニルから選択される1又は2以上の置換基よって置換されていてもよく、ここで、該フェニル基は、1又は2以上のF又はCl基によってさらに置換されていてもよい]、
NH−モルホリニル、
NH−シクロプロピル、NH−シクロブチル、NH−シクロペンチル及びNH−シクロヘキシル[ここで、前記シクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシル基は、OH及びFから選択される1又は2以上の置換基によって置換されていてもよい]、
NH−(1,2,3,4−テトラヒドロイソキノリニル)[ここで、前記1,2,3,4−テトラヒドロイソキノリニル基は、1又は2以上のメチル基によって置換されていてもよい]、
NH−ピペリジニル[ここで、前記ピペリジニルは、1又は2以上のメチル基によって置換されていてもよい]、
NH−テトラヒドロピラニル、
フリル基、
1又は2以上のメチル基によって置換されていてもよいピラゾリル基、
NH−フェニル[ここで、前記フェニルは、F、Cl、Br、CF3、OH、OEt、NHSO2Me、NMe2、モルホリニル、CONMe2、CONH2及び−NHCOMeから選択される1又は2以上の置換基によって置換されていてもよい]、
NH−ピリジニル[ここで、前記ピリジニルは、モルホリニル及びフェニルから選択される1又は2以上の置換基によって置換されていてもよく、ここで、前記フェニル基は、1又は2以上のCN基でさらに置換されていてもよい]、
F、Cl、OMe、−NHSO2Me、CN、モルホリニル及びCH2−NHSO2Meから選択される1又は2以上の置換基によって置換されていてもよいフェニル、
NH−インダゾリル[ここで、前記インダゾリルは、1又は2以上のメチル基によって置換されていてもよい]、並びに
NH−ピラゾリル
から選択される。
本発明の1つの好ましい実施形態において、Qは、ハロゲン、CN、C1−6−アルキル、C3−7−シクロアルキル、並びにC4−7−ヘテロシクロアルキル及びヘテロアリールから選択され、ここで、前記C1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びヘテロアリールは、それぞれ独立に、基Aからの1又は2以上の置換基で置換されていてもよい。好ましくは、Aはハロ又はC1−6−アルキルである。
本発明のより好ましい実施形態において、Qは、CN、シクロプロピル、CF3、クロロ、メチル、N−モルホリニル及び1−メチルピラゾール−4−イルから選択される。
1つの好ましい実施形態において、Qは、ハロゲンであるか、又はC1−6−アルキル、ヘテロシクロアルキル及びヘテロアリールから選択され、そのそれぞれは、1又は2以上の置換基Aで置換されていてもよい。
より好ましくは、Qは、クロロ、メチル、N−モルホリニル及び1−メチルピラゾール−4−イルから選択される。
本発明の1つの非常に好ましい実施形態において、式Ia又は式Ibの化合物は、下記:
又は薬学的に許容されるその塩から選択される。
生物学的活性
本発明の化合物は、1又は2以上のキナーゼ、好ましくはLRRK、さらに一層好ましくはLRRK2を阻害することができる。
本発明の化合物は、1又は2以上のキナーゼ、好ましくはLRRK、さらに一層好ましくはLRRK2を阻害することができる。
1つの好ましい実施形態において、本発明の化合物は、添付の実施例の項において記述されているアッセイによって測定される通り、LRRK2を阻害することができる。好ましくは、本発明の化合物は、10μM未満、より好ましくは5μM未満、一層好ましくは1μM未満又は0.5μM未満、さらに一層好ましくは0.1μM未満のIC50値を呈する。
好ましくは、本発明の化合物は、10μM未満、より好ましくは5μM未満、一層好ましくは1μM未満又は0.5μM未満、さらに一層好ましくは0.1μM未満のKI値を呈する。
特に好ましい化合物は、下記:[1]、[2]、[6]〜[10]及び[11〜25]を含む。
非常に好ましい化合物は、下記:[11]、[12]、[15]、[17]、[18]、[19]、[22]、[24]及び[25]を含む。
治療用途
本発明のさらなる態様は、医療において使用するための上述した通りの化合物に関する。
本発明のさらなる態様は、医療において使用するための上述した通りの化合物に関する。
本発明の別の態様は、がん又は神経変性障害を治療するのに使用するための、上述した通りの化合物に関する。
別の態様は、神経変性障害を治療又は予防するための薬剤の調製における、上述した通りの化合物の使用に関する。好ましくは、神経変性障害はパーキンソン病である。
別の態様は、増殖性障害、例えばがんを治療又は予防するための薬剤の調製における、上述した通りの化合物の使用に関する。
好ましくは、化合物は、1又は2以上のキナーゼ、好ましくはLRRK、一層好ましくはLRRK2を阻害するのに十分な量で投与される。
また別の態様は、生物学的標的に対する何らかの異常な活性によって引き起こされ、それに関連し、又はそれを伴い、該標的が、キナーゼ、より好ましくはLRRK、一層好ましくはLRRK2である障害の予防又は治療のための薬剤の調製における、本発明の化合物の使用に関する。
好ましくは、障害はパーキンソン病である。
本発明の別の態様は、タンパク質キナーゼに関係する疾患又は障害を治療する方法に関する。本発明のこの態様による方法は、それを必要とする対象に、治療有効量の以上に記載した通りの本発明の化合物を、それ自体で、又は、より好ましくは、例えば、以後詳述するように薬学的に許容される担体と混合された医薬組成物の一部としてのいずれかで投与することによって実現される。
本発明のまた別の態様は、哺乳動物に、治療有効量の本発明による化合物を投与するステップを含む、タンパク質キナーゼの阻害によって軽減される病状を有する哺乳動物を治療する方法に関する。
好ましくは、病状は、タンパク質キナーゼLRRK、より好ましくはLRRK2の阻害によって軽減される。
好ましくは、哺乳動物はヒトである。
用語「方法」は、化学、薬理学、生物学、生化学及び医学技術分野の実務家に公知であるか、又は該実務家によって公知の様式、手段、技術及び手順から容易に開発されるかのいずれかである様式、手段、技術及び手順を含むがこれらに限定されない、所与の作業を遂行するための様式、手段、技術及び手順を指す。
用語「投与すること」は、本明細書において使用される場合、化合物が、直接的に、すなわち、タンパク質キナーゼ自体と相互作用することによって、又は間接的に、すなわち、タンパク質キナーゼの触媒活性が依存している別の分子と相互作用することによって、タンパク質キナーゼの酵素活性に影響を及ぼし得るような様式で、本発明の化合物とタンパク質キナーゼとを結びつけるための方法を指す。本明細書において使用される場合、投与は、インビトロ、すなわち試験管内、又はインビボ、すなわち生体の細胞若しくは組織内のいずれかで遂行され得る。
本明細書において、用語「治療すること」は、疾患若しくは障害の進行を、抑止し、実質的に阻害し、遅らせ、若しくは逆行させること、疾患若しくは障害の臨床症状を実質的に寛解させること、又は疾患若しくは障害の臨床症状の出現を実質的に予防することを含む。
本明細書において、用語「予防すること」は、生物が障害又は疾患を獲得することを最初の段階で阻止するための方法を指す。
用語「治療有効量」は、治療されている疾患又は障害の症状の1又は2以上をある程度緩和するであろう、投与される化合物の量を指す。
本発明において使用されるいかなる化合物についても、本明細書において治療有効用量とも称される治療有効量は、細胞培養アッセイから最初に推定され得る。例えば、動物モデルでは、細胞培養において決定された通りのIC50又はIC100を含む循環濃度範囲を達成するように用量を処方することができる。そのような情報を使用して、ヒトにおける有用な用量をより正確に決定することができる。初期投薬量は、インビボデータから推定することもできる。これらの初期ガイドラインを使用して、当業者はヒトにおける有効投薬量を決定することができる。
その上、本明細書において記載されている化合物の毒性及び治療有効性は、細胞培養又は実験動物における標準的な薬学的手順によって、例えばLD50及びED50を決定することによって決定され得る。毒性効果と治療効果との用量比は、治療指数であり、LD50とED50との比として表現できる。高い治療指数を呈する化合物が好ましい。これらの細胞培養アッセイ及び動物試験から取得されるデータは、ヒトにおける使用のための毒性でない投薬量範囲を処方するのに使用され得る。そのような化合物の投薬量は、好ましくは、毒性がほとんど又はまったくないED50を含む循環濃度の範囲内にある。投薬量は、用いられる剤形及び利用される投与経路に応じて、この範囲内で変動し得る。正確な処方、投与経路及び投薬量は、患者の状態を考慮して個々の医師によって選択され得る(例えば、Fingl et al, 1975, In: The Pharmacological Basis of Therapeutics, chapter 1, page 1を参照)。
投薬量及び間隔は、治療効果を維持するのに十分な活性化合物の血漿中レベルを提供するために、個々に調整され得る。経口投与のための通常の患者投薬量は、約50〜2000mg/kg/日、一般に約100〜1000mg/kg/日、好ましくは約150〜700mg/kg/日、最も好ましくは約250〜500mg/kg/日の範囲である。好ましくは、治療有効血清中レベルは、複数回用量を毎日投与することによって達成される。局所投与又は選択的取り込みの場合、薬物の有効局所濃度は血漿中濃度と関係ないことがある。当業者であれば、必要以上の実験をすることなく治療有効局所投薬量を最適化することができよう。
本明細書において使用される場合、「キナーゼに関係する疾患又は障害」は、本明細書において定義される通りの不適切なキナーゼ活性又はキナーゼの過活性を特徴とする疾患又は障害を指す。不適切な活性は、(i)あるキナーゼを通常は発現しない細胞における前記キナーゼ発現、(ii)望ましくない細胞増殖、分化及び/若しくは成長につながるキナーゼ発現の増加、又は(iii)細胞増殖、分化及び/若しくは成長における望ましくない低減につながるキナーゼ発現の減少のいずれかを指す。キナーゼの過活性は、特定のキナーゼをコードする遺伝子の増幅、又は細胞増殖、分化及び/若しくは成長障害と相関し得るレベルのキナーゼ活性の産生(すなわち、キナーゼのレベルが増加するにつれて、細胞障害の1又は2以上の症状の重症度が増加する)のいずれかを指す。過活性は、リガンド結合を司るキナーゼ断片の欠失等の突然変異の結果としての、リガンド非依存性又は構成的活性化の結果でもあり得る。
予防するのに本明細書において記載されている化合物が有用となり得る好ましい疾患又は障害は、がん、及びパーキンソン病等の神経変性障害を含む。
故に、本発明は、LRRK2を阻害することが望ましい疾患の治療用の薬剤の製造のための、本明細書において定義される通りの化合物の使用をさらに提供する。そのような疾患は、パーキンソン病を含む。
医薬組成物(COMPOSTIONS)
本発明による使用のために、本明細書において記載されている化合物、又はその生理学的に許容される塩、エステル若しくは他の生理学的機能性誘導体は、化合物、又はその生理学的に許容される塩、エステル若しくは他の生理学的機能性誘導体を、1又は2以上の薬学的に許容される担体、したがって並びに場合により他の治療及び/又は予防成分と一緒に含む医薬製剤として提示され得る。担体(複数可)は、製剤の他の成分と適合し、そのレシピエントに有害でないという意味で許容されるものでなくてはならない。医薬組成物は、ヒト及び獣医学におけるヒト又は動物用法のためのものであってよい。
本発明による使用のために、本明細書において記載されている化合物、又はその生理学的に許容される塩、エステル若しくは他の生理学的機能性誘導体は、化合物、又はその生理学的に許容される塩、エステル若しくは他の生理学的機能性誘導体を、1又は2以上の薬学的に許容される担体、したがって並びに場合により他の治療及び/又は予防成分と一緒に含む医薬製剤として提示され得る。担体(複数可)は、製剤の他の成分と適合し、そのレシピエントに有害でないという意味で許容されるものでなくてはならない。医薬組成物は、ヒト及び獣医学におけるヒト又は動物用法のためのものであってよい。
本明細書において記載されている医薬組成物の種々の異なる形態に適するそのような添加剤の例は、"Handbook of Pharmaceutical Excipients, 2nd Edition, (1994), Edited by A Wade and PJ Wellerにおいて見ることができる。
治療的使用に許容される担体又は賦形剤は薬学技術分野において周知であり、例えば、Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985)に記載されている。
適切な担体の例は、ラクトース、デンプン、グルコース、メチルセルロース、ステアリン酸マグネシウム、マンニトール、ソルビトール等を含む。適切な賦形剤の例は、エタノール、グリセロール及び水を含む。
医薬担体、添加剤又は賦形剤の選択は、意図されている投与経路及び標準的な医薬実務に関連して選択され得る。医薬組成物は、担体、添加剤又は賦形剤として、又はそれに加えて、任意の適切な結合剤(複数可)、滑沢剤(複数可)、懸濁化剤(複数可)、コーティング剤(複数可)、可溶化剤(複数可)、緩衝剤(複数可)、香味剤(複数可)、表面活性剤(複数可)、増粘剤(複数可)、保存剤(複数可)(酸化防止剤を含む)等、及び製剤を意図されているレシピエントの血液と等張にすることを目的として含まれる物質を含み得る。
適切な結合剤の例は、デンプン、ゼラチン、グルコース、無水ラクトース、易流動性ラクトース、ベータ−ラクトース等の天然糖、トウモロコシ甘味料、アカシア、トラガカント又はアルギン酸ナトリウム等の天然及び合成ゴム、カルボキシメチルセルロース並びにポリエチレングリコールを含む。
適切な滑沢剤の例は、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウム等を含む。
保存剤、安定剤、色素、さらには香味剤が医薬組成物中に提供され得る。保存剤の例は、安息香酸ナトリウム、ソルビン酸、及びp−ヒドロキシ安息香酸のエステルを含む。酸化防止剤及び懸濁化剤を使用してもよい。
医薬製剤は、経口、局所(真皮、口腔及び舌下を含む)、直腸又は非経口(皮下、皮内、筋肉内及び静脈内を含む)、例えば吸入による経鼻及び経肺投与に適するものを含む。製剤は、適切な場合には、好都合なことに個別の投薬単位で提示されてよく、薬学技術分野において周知である方法のいずれかによって調製できる。いずれの方法も、活性化合物を液体担体若しくは微粉化した固体担体又は両方と会合させるステップと、次いで、必要ならば、生成物を望ましい製剤に成形するステップとを含む。
担体が固体である経口投与に適する医薬製剤は、最も好ましくは、それぞれ所定量の活性化合物を含有するボーラス剤、カプセル剤又は錠剤等の単位用量製剤として提示される。錠剤は、場合により1又は2以上の副成分を用い、圧縮又は成型によって作製できる。圧縮錠剤は、結合剤、滑沢剤(lubricant)、不活性賦形剤、滑沢剤(lubricating agent)、表面活性剤又は分散剤と混合されていてもよい粉末又は顆粒等の易流動性形態の活性化合物を適切なマシン内で圧縮することによって調製できる。成型錠剤は、活性化合物を不活性液体賦形剤とともに成型することによって作製できる。錠剤は、コーティングされていてもよく、コーティングされていないならば、切り込み線が入れられていてもよい。カプセル剤は、活性化合物を、単独で又は1若しくは2以上の副成分と混和してのいずれかでカプセル殻に充填し、次いでそれらを通常の様式で密封することによって調製できる。カシェ剤はカプセル剤に類似しており、ここでは、任意の副成分(複数可)と一緒になった活性化合物がライスペーパー外皮内に密封される。活性化合物を、例えば投与前に水に懸濁させるか又は食品に振りかけることができる分散性顆粒剤として製剤化してもよい。顆粒剤は、例えば小袋内に包装されていてよい。担体が液体である経口投与に適する製剤は、水性若しくは非水性の液体中の液剤若しくは懸濁剤として、又は水中油型液体乳剤として提示され得る。
経口投与用の製剤は、制御放出剤形、例えば、活性化合物が適切な放出制御マトリックス中で製剤化されている、又は適切な放出制御フィルムでコーティングされている錠剤を含む。そのような製剤は、予防的使用に特に好都合となり得る。
担体が固体である直腸投与に適する医薬製剤は、最も好ましくは単位用量坐剤として提示される。適切な担体は、ココアバター及び当技術分野において一般に使用される他の材料を含む。坐剤は、好都合なことに、活性化合物と軟化又は融解された担体(複数可)との混和、続いて低温化及び鋳型内での成形によって形成され得る。非経口投与に適する医薬製剤は、水性又は油性媒体中の活性化合物の滅菌液剤又は懸濁剤を含む。
注射用調製物は、ボーラス注射又は持続注入に適応し得る。そのような調製物は、好都合なことに、製剤の導入後から使用が必要になるまで密封されている単位用量又は複数回用量容器内で提示される。代替として、活性化合物は、滅菌パイロジェンフリー水等の適切な媒体を用いて使用前に構成される粉末形態であってよい。
活性化合物は、筋肉内注射によって、又は移植によって、例えば皮下に若しくは筋肉内に投与され得る、長時間作用型デポー調製物として製剤化されてもよい。デポー調製物は、例えば、適切なポリマー材料若しくは疎水性材料、又はイオン交換樹脂を含み得る。そのような長時間作用型製剤は、予防的使用に特に好都合である。
口腔を介する経肺投与に適する製剤は、活性化合物を含有し、望ましくは0.5〜7ミクロンの範囲内の直径を有する粒子が、レシピエントの気管支樹中を送達されるようなものが提示される。
1つの可能性として、そのような製剤は、好都合なことに、吸入デバイスにおける使用のための、例えば適宜ゼラチン製の貫通可能なカプセル中で、又は、活性化合物、適切な液体若しくはガス状噴射剤、並びに場合により界面活性剤及び/若しくは固体賦形剤等の他の成分を含む自己噴射式製剤としてのいずれかで提示され得る、細かく破砕された粉末の形態である。適切な液体噴射剤はプロパン及びクロロフルオロカーボンを含み、適切なガス状噴射剤は二酸化炭素を含む。活性化合物が液剤又は懸濁剤の液滴の形態で分注される場合、自己噴射式製剤を用いてもよい。
そのような自己噴射式製剤は、当技術分野において公知の製剤に類似しており、確立した手順によって調製できる。適宜、製剤は、望ましい噴射特性を有する手動操作式又は自動機能式弁のいずれかが備わっている容器内で提示され、有利なことに、弁は、その各操作時に、固定体積、例えば25〜100マイクロリットルを送達する定量型のものである。
さらなる可能性として、活性化合物は、アトマイザー又はネブライザーにおいて使用するための液剤又は懸濁剤の形態であってよく、それによって、加速気流又は超音波撹拌を用いて吸入用の微細液滴ミストを産生する。
経鼻投与に適する製剤は、経肺投与について上述したものと概して同様の調製物を含む。分注される際、そのような製剤は、望ましくは鼻腔における滞留を可能にするために10〜200ミクロンの範囲内の粒径を有するべきであり、これは、必要に応じて、適切な粒子サイズの粉末の使用又は適切な弁の選択によって達成され得る。他の適切な製剤は、鼻に接近して保持した容器から鼻孔を経由する急速吸入による投与のための、20〜500ミクロンの範囲の粒径を有する粗粉末剤、及び水性又は油性の液剤又は懸濁剤中0.2〜5%w/vの活性化合物を含む点鼻剤を含む。
薬学的に許容される担体は、当業者に周知であり、0.1M、好ましくは0.05Mのリン酸緩衝液又は0.8%の生理食塩水を含むがこれらに限定されない。加えて、そのような薬学的に許容される担体は、水性又は非水性の溶液、懸濁液及び乳液であってよい。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコール、オリーブ油等の植物油、及びオレイン酸エチル等の注射用有機エステルである。水性担体は、水、アルコール性/水性の溶液、乳液又は生理食塩水及び緩衝媒体を含む懸濁液を含む。非経口媒体は、塩化ナトリウム溶液、リンゲルデキストロース、デキストロース及び塩化ナトリウム、乳酸加リンゲル液又は固定油を含む。保存剤及び他の添加物、例えば抗菌剤、酸化防止剤、キレート剤、不活性ガス等が存在してもよい。
局所製剤に適する製剤は、例えばゲル剤、クリーム剤又は軟膏剤として提供され得る。そのような調製物は、例えば、創傷若しくは潰瘍の表面に直接塗布されるか、又は治療される領域にそれを覆って適用され得る絆創膏、ガーゼ、メッシュ等の適切な支持体上に担持されるかのいずれかで、創傷又は潰瘍に適用され得る。
治療される部位、例えば創傷又は潰瘍に直接スプレーするか又は撒き散らすことができる液体又は粉末製剤も提供され得る。代替として、絆創膏、ガーゼ、メッシュ等の担体に製剤をスプレーするか又は撒き散らし、次いで、治療される部位に適用してもよい。
本発明のさらなる態様によれば、活性化合物(複数可)を、例えば混和によって担体と会合させるステップを含む、上述した通りの医薬又は獣医学組成物の調製のための方法が提供される。
概して、製剤は、活性剤を液体担体若しくは微粉化した固体担体又は両方と均一で密接に会合させ、次いで、必要ならば、生成物を成形することによって調製される。本発明は、一般式(I)の化合物を薬学的に又は獣医学的に許容される担体又は媒体と接合又は会合させるステップを含む、医薬組成物を調製する方法にまで及ぶ。
塩/エステル
本発明の化合物は、塩又はエステル、特に薬学的に及び獣医学的に許容される塩又はエステルとして存在し得る。
本発明の化合物は、塩又はエステル、特に薬学的に及び獣医学的に許容される塩又はエステルとして存在し得る。
本発明の化合物の薬学的に許容される塩は、その適切な酸付加塩又は塩基塩を含む。適切な薬学的塩についての総説は、Berge et al, J Pharm Sci, 66, 1-19 (1977)において見ることができる。塩は、例えば、無機強酸[鉱酸、例えば、塩化水素、臭化水素及びヨウ化水素等のハロゲン化水素酸、硫酸、リン酸硫酸、重硫酸、ヘミ硫酸、チオシアン酸、過硫酸、並びにスルホン酸等]と;有機強カルボン酸[酢酸等、置換されていない又は(例えばハロゲンによって)置換されている1〜4個の炭素原子のアルカンカルボン酸等]と;飽和又は不飽和ジカルボン酸、例えば、シュウ酸、マロン酸、コハク酸、マレイン酸、フマル酸、フタル酸又はテトラフタル酸と;ヒドロキシカルボン酸、例えば、アスコルビン酸、グリコール酸、乳酸、リンゴ酸、酒石酸又はクエン酸と;アミノ酸、例えばアスパラギン酸又はグルタミン酸と;安息香酸と;或いは、有機スルホン酸[メタン−又はp−トルエンスルホン酸等、置換されていない又は(例えばハロゲンによって)置換されている(C1−C4)−アルキル−又はアリール−スルホン酸等]と形成される。薬学的に又は獣医学的に許容されない塩であっても、中間体として価値のあるものとなり得る。
好ましい塩は、例えば、酢酸塩、トリフルオロ酢酸塩、乳酸塩、グルコン酸塩、クエン酸塩、酒石酸塩、マレイン酸塩、リンゴ酸塩、パントテン酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、酪酸塩、ジグルコン酸塩、シクロペンタン酸塩、グルコヘプタン酸塩、グリセロリン酸塩、シュウ酸塩、ヘプタン酸塩、ヘキサン酸塩、フマル酸塩、ニコチン酸塩、パルモ酸塩(palmoate)、ペクチン酸塩(pectinate)、3−フェニルプロピオン酸塩、ピクリン酸塩、ピバル酸塩、プロピオン酸塩(proprionate)、酒石酸塩、ラクトビオン酸塩、ピボル酸塩(pivolate)、樟脳酸塩、ウンデカン酸塩及びコハク酸塩;メタンスルホン酸塩、エタンスルホン酸塩、2−ヒドロキシエタンスルホン酸塩、カンファースルホン酸塩、2−ナフタレンスルホン酸塩、ベンゼンスルホン酸塩、p−クロロベンゼンスルホン酸塩及びp−トルエンスルホン酸塩等の有機スルホン酸;並びに、塩酸、臭素酸、ヨウ素酸、硫酸塩、重硫酸塩、ヘミ硫酸塩(hemisulphate)、チオシアン酸塩、過硫酸塩、リン酸及びスルホン酸等の無機酸を含む。
エステルは、エステル化される官能基に応じて、有機酸又はアルコール/水酸化物のいずれかを使用して形成される。有機酸は、カルボン酸[酢酸等、置換されていない又は(例えばハロゲンによって)置換されている1〜12個の炭素原子のアルカンカルボン酸等]を;飽和又は不飽和ジカルボン酸、例えば、シュウ酸、マロン酸、コハク酸、マレイン酸、フマル酸、フタル酸又はテトラフタル酸と;ヒドロキシカルボン酸、例えば、アスコルビン酸、グリコール酸、乳酸、リンゴ酸、酒石酸又はクエン酸と;アミノ酸、例えばアスパラギン酸又はグルタミン酸と;安息香酸と;或いは、有機スルホン酸[メタン−又はp−トルエンスルホン酸等、置換されていない又は(例えばハロゲンによって)置換されている(C1−C4)−アルキル−又はアリール−スルホン酸等]とともに含む。適切な水酸化物は、水酸化ナトリウム、水酸化カリウム、水酸化カルシウム、水酸化アルミニウム等の無機水酸化物を含む。アルコールは、置換されていなくても例えばハロゲンによって)置換されていてもよい1〜12個の炭素原子のアルカンアルコールを含む。
鏡像異性体/互変異性体
先に論じた本発明のすべての態様において、本発明は、適切な場合、本発明の化合物のすべての鏡像異性体、ジアステレオ異性体及び互変異性体を含む。当業者であれば、光学的性質(1又は2以上のキラル炭素原子)又は互変異性特性を保有する化合物を認識するであろう。対応する鏡像異性体及び/又は互変異性体は、当技術分野において公知の方法によって単離/調製できる。
先に論じた本発明のすべての態様において、本発明は、適切な場合、本発明の化合物のすべての鏡像異性体、ジアステレオ異性体及び互変異性体を含む。当業者であれば、光学的性質(1又は2以上のキラル炭素原子)又は互変異性特性を保有する化合物を認識するであろう。対応する鏡像異性体及び/又は互変異性体は、当技術分野において公知の方法によって単離/調製できる。
鏡像異性体は、そのキラル中心の絶対配置によって特徴づけられ、Cahn, Ingold and PrelogのR及びSの優先順位ルールにより表わされる。そのような慣習は、当技術分野においては周知である(例えば、'Advanced Organic Chemistry', 3rd edition, ed. March, J., John Wiley and Sons, New York, 1985を参照)。
キラル中心を含有する本発明の化合物は、ラセミ混合物として、又は鏡像異性体が富化された混合物として使用され得、又は該ラセミ混合物が、周知の技術を使用して分離され得、個々の鏡像異性体を単独で使用することができる。
立体異性体及び幾何異性体
本発明の化合物のいくつかは、立体異性体及び/又は幾何異性体として存在し得、例えば、該化合物は、1又は2以上の不斉及び/又は幾何中心を保有し得るため、2以上の立体異性及び/又は幾何形態で存在し得る。本発明は、それらの阻害剤の個々の立体異性体及び幾何異性体すべて、並びにそれらの混合物の使用を企図している。特許請求の範囲において使用される用語は、これらの形態が(必ずしも同程度までとは限らないが)適切な機能的活性を保持する限り、前記形態を包含する。
本発明の化合物のいくつかは、立体異性体及び/又は幾何異性体として存在し得、例えば、該化合物は、1又は2以上の不斉及び/又は幾何中心を保有し得るため、2以上の立体異性及び/又は幾何形態で存在し得る。本発明は、それらの阻害剤の個々の立体異性体及び幾何異性体すべて、並びにそれらの混合物の使用を企図している。特許請求の範囲において使用される用語は、これらの形態が(必ずしも同程度までとは限らないが)適切な機能的活性を保持する限り、前記形態を包含する。
本発明は、作用物質又は薬学的に許容されるその塩のすべての適切な同位体標識変種も含む。本発明の作用物質又は薬学的に許容されるその塩の同位体標識変種は、少なくとも1個の原子が、同じ原子番号を有するが自然界において通常見られる原子質量とは異なる原子質量を有する原子によって置き換えられているものとして定義される。作用物質及び薬学的に許容されるその塩に組み込まれ得る同位体の例は、2H、3H、13C、14C、15N、17O、18O、31P、32P、35S、18F及び36Cl等、それぞれ水素、炭素、窒素、酸素、リン、硫黄、フッ素及び塩素の同位体を含む。作用物質及び薬学的に許容されるその塩のある特定の同位体標識変種、例えば、3H又は14C等の放射性同位体が組み込まれている変種は、薬物及び/又は基質組織分布研究において有用である。トリチウム化、すなわち3H、及び炭素−14、すなわち14C同位体は、それらの調製の容易さ及び検出性によって特に好ましい。さらに、重水素、すなわち2H等の同位体による置換は、より優れた代謝安定性から生じるある特定の治療上の利点、例えば、インビボ半減期の増大又は必要投薬量の低減を生じさせることができ、それ故、いくつかの状況においては好ましいことがある。例えば、本発明は、任意の水素原子が重水素原子によって置き換えられた一般式(I)の化合物を含む。本発明の作用物質及び本発明の薬学的に許容されるその塩の同位体変化物は、概して、適切な試薬の適切な同位体標識変種を使用し、従来の手順によって調製され得る。
プロドラッグ
本発明は、プロドラッグ形態の本発明の化合物、すなわち、一般式(I)による活性親薬物をインビボで放出する共有結合化合物をさらに含む。そのようなプロドラッグは、概して、ヒト又は哺乳類対象への投与時に修飾を逆転(reverse)できるように1又は2以上の適切な基が修飾された本発明の化合物である。逆転は通常、そのような対象において自然に存在する酵素によって実施されるが、逆転をインビボで実施するために、そのようなプロドラッグと一緒に第二の作用物質を投与することが可能である。そのような修飾の例は、エステル(例えば、上述したものいずれか)を含み、ここで、逆転はエステラーゼ等によって行われ得る。他のそのようなシステムは、当業者には周知であろう。
本発明は、プロドラッグ形態の本発明の化合物、すなわち、一般式(I)による活性親薬物をインビボで放出する共有結合化合物をさらに含む。そのようなプロドラッグは、概して、ヒト又は哺乳類対象への投与時に修飾を逆転(reverse)できるように1又は2以上の適切な基が修飾された本発明の化合物である。逆転は通常、そのような対象において自然に存在する酵素によって実施されるが、逆転をインビボで実施するために、そのようなプロドラッグと一緒に第二の作用物質を投与することが可能である。そのような修飾の例は、エステル(例えば、上述したものいずれか)を含み、ここで、逆転はエステラーゼ等によって行われ得る。他のそのようなシステムは、当業者には周知であろう。
溶媒和物
本発明は、本発明の化合物の溶媒和物形態も含む。特許請求の範囲において使用される用語は、これらの形態を包含する。
本発明は、本発明の化合物の溶媒和物形態も含む。特許請求の範囲において使用される用語は、これらの形態を包含する。
多形
本発明は、その種々の結晶形態、多形形態及び(無)水和形態である本発明の化合物にさらに関する。そのような化合物の合成的調製において使用される精製及び又は溶媒からの単離の方法をわずかに変更することにより、化学化合物がそのような形態のいずれかで単離され得ることは、製薬業界内で十分に確立されている。
本発明は、その種々の結晶形態、多形形態及び(無)水和形態である本発明の化合物にさらに関する。そのような化合物の合成的調製において使用される精製及び又は溶媒からの単離の方法をわずかに変更することにより、化学化合物がそのような形態のいずれかで単離され得ることは、製薬業界内で十分に確立されている。
投与
本発明の医薬組成物は、直腸、経鼻、気管支内、局所(口腔及び舌下を含む)、膣内又は非経口(皮下、筋肉内、静脈内、動脈内及び皮内を含む)、腹腔内又は髄腔内投与に適応し得る。好ましくは、製剤は、経口投与される製剤である。製剤は、好都合なことに、単位剤形で、すなわち、単位用量、又は単位用量の複数若しくはサブユニットを含有する個別の部分の形態で提示され得る。例として、製剤は、錠剤及び持続放出カプセル剤の形態であってよく、薬学技術分野において周知である任意の方法によって調製され得る。
本発明の医薬組成物は、直腸、経鼻、気管支内、局所(口腔及び舌下を含む)、膣内又は非経口(皮下、筋肉内、静脈内、動脈内及び皮内を含む)、腹腔内又は髄腔内投与に適応し得る。好ましくは、製剤は、経口投与される製剤である。製剤は、好都合なことに、単位剤形で、すなわち、単位用量、又は単位用量の複数若しくはサブユニットを含有する個別の部分の形態で提示され得る。例として、製剤は、錠剤及び持続放出カプセル剤の形態であってよく、薬学技術分野において周知である任意の方法によって調製され得る。
本発明における経口投与用の製剤は、それぞれ所定量の活性剤を含有するカプセル剤、ジェル剤(gellules)、点滴剤、カシェ剤(cachets)、丸剤又は錠剤等の個別の単位として;散剤又は顆粒剤として;水性液体又は非水性液体中の活性剤の液剤、乳剤又は懸濁剤として;或いは、水中油型液体乳剤又は油中水型液体乳剤として;或いはボーラス剤等として提示され得る。好ましくは、これらの組成物は、1用量当たり1〜250mg、より好ましくは10〜100mgの活性成分を含有する。
経口投与用の組成物(例えば錠剤及びカプセル剤)では、用語「許容される担体」は、一般的な添加剤、例えば結合剤、例えば、シロップ、アカシア、ゼラチン、ソルビトール、トラガカント、ポリビニルピロリドン(ポビドン)、メチルセルロース、エチルセルロース、ナトリウムカルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、スクロース及びデンプン等の媒体;充填剤及び担体、例えば、コーンスターチ、ゼラチン、ラクトース、スクロース、微結晶性セルロース、カオリン、マンニトール、第二リン酸カルシウム(dicalcium phosphate)、塩化ナトリウム及びアルギン酸;並びに、ステアリン酸マグネシウム、ステアリン酸ナトリウム及び他のステアリン酸金属塩、ステアリン酸グリセロールステアリン酸、シリコーン流体、タルクワックス、油及びコロイド状シリカ等の滑沢剤を含む。ペパーミント、冬緑油、サクランボ香味剤等の香味剤を使用してもよい。容易に識別可能な剤形を作製するために、着色剤を添加することが望ましい場合がある。錠剤は、当技術分野において周知の方法によってコーティングしてもよい。
錠剤は、場合により1又は2以上の副成分を用い、圧縮又は成型によって作製できる。圧縮錠剤は、結合剤、滑沢剤、不活性賦形剤、保存剤、表面活性剤又は分散剤と混合されていてもよい粉末又は顆粒等の易流動性形態の活性化合物を適切なマシン内で圧縮することによって調製できる。成型錠剤は、不活性液体賦形剤で湿らせた粉末状化合物の混合物を適切なマシン内で成型することによって作製できる。錠剤は、コーティングされていても切り込み線が入れられていてもよく、活性剤の緩徐又は制御放出を提供するように製剤化され得る。
経口投与に適する他の製剤は、香味付けされた基剤、通常はスクロース及びアカシア又はトラガカント中に活性剤を含むロゼンジ剤(lozenges);ゼラチン及びグリセリン、又はスクロース及びアカシア等の不活性基剤中に活性剤を含むパステル剤(pastilles);並びに、適切な液体担体中に活性剤を含む洗口剤を含む。
他の投与形態は、静脈内、動脈内、髄腔内、皮下、皮内、腹腔内又は筋肉内に注射され得、滅菌溶液又は滅菌可能な溶液から調製できる、液剤又は乳剤を含む。注射用形態は、典型的には、1用量当たり10〜1000mg、好ましくは10〜250mgの活性成分を含有する。
本発明の医薬組成物は、坐剤、ペッサリー、懸濁剤、乳剤、ローション剤、軟膏剤、クリーム剤、ゲル剤、スプレー剤、液剤又は撒布剤の形態であってもよい。
経皮投与の代替手段は、皮膚パッチ剤の使用によるものである。例えば、活性成分を、ポリエチレングリコール又は流動パラフィンの水性乳剤からなるクリーム剤に組み込んでよい。活性成分を、1〜10重量%の濃度で、必要となり得るような安定剤及び保存剤と一緒に白色ワックス又は白色軟パラフィン基剤からなる軟膏剤に組み込んでもよい。
投薬量
当業者であれば、対象に投与する本組成物の1つの適切な用量を、必要以上の実験をすることなく簡単に決定することができるであろう。典型的には、医師は、個々の患者に最も適するであろう実際の投薬量を決定し、該投薬量は、用いられる特定化合物の活性、該化合物の代謝安定性及び作用長さ、年齢、体重、全般的健康、性別、食習慣、投与モード及び投与時期、排泄率、薬物組合せ、特定の状態の重症度、並びに療法を受けている個体を含む様々な要因によって決まることになる。本明細書において開示されている投薬量は、平均的事例の例示である。当然ながら、より高い又は低い投薬量範囲がふさわしい個々の場合があり得、それらは本発明の範囲内である。
当業者であれば、対象に投与する本組成物の1つの適切な用量を、必要以上の実験をすることなく簡単に決定することができるであろう。典型的には、医師は、個々の患者に最も適するであろう実際の投薬量を決定し、該投薬量は、用いられる特定化合物の活性、該化合物の代謝安定性及び作用長さ、年齢、体重、全般的健康、性別、食習慣、投与モード及び投与時期、排泄率、薬物組合せ、特定の状態の重症度、並びに療法を受けている個体を含む様々な要因によって決まることになる。本明細書において開示されている投薬量は、平均的事例の例示である。当然ながら、より高い又は低い投薬量範囲がふさわしい個々の場合があり得、それらは本発明の範囲内である。
本発明に従って、有効量の一般式(I)の化合物を投与して、特定の状態又は疾患に関与するキナーゼを阻害することができる。当然ながら、この投薬量は、化合物の投与の種類によってさらに修正されることになる。例えば、急性療法のための「有効量」を達成するためには、一般式(I)の化合物の非経口投与が好ましい。水若しくは生理食塩水中5%デキストロース中の化合物、又は適切な添加剤を加えた同様の製剤の静脈内注入が最も有効であるが、筋肉内ボーラス注射も有用である。典型的には、非経口用量は、血漿中の薬物濃度を、キナーゼを阻害するのに有効な濃度に維持するための様式で、約0.01〜約100mg/kg、好ましくは0.1〜20mg/kgとなる。化合物は、約0.4〜約400mg/kg/日の総日用量を達成するレベルで、1日1〜4回投与され得る。治療上有効である発明化合物の正確な量及びそのような化合物を投与するのに最適な経路は、作用物質の血中レベルを、治療効果を有するために必要とされる濃度と比較することにより、当業者によって容易に決定される。
本発明の化合物を、本明細書において開示されている治療指標の1又は2以上を達成するために薬物濃度が十分であるような様式で、患者に経口投与してもよい。典型的には、化合物を含有する医薬組成物は、約0.1〜約50mg/kgの経口用量で、患者の状態に合致する様式にて投与される。好ましくは、経口用量は約0.5〜約20mg/kgであろう。
本発明の化合物を本発明に従って投与する場合、許容されない毒物学的効果は予測されない。本発明の化合物は、良好なバイオアベイラビリティを有し得、所与の薬理学的作用を有するために必要とされる化合物濃度を決定するための数種の生物学的アッセイの1つにおいて試験され得る。
組合せ
特に好ましい実施形態において、1又は2以上の本発明の化合物は、1又は2以上の他の活性剤、例えば、市場に流通している既存の薬物と組み合わせて投与される。そのような事例では、本発明の化合物は、1又は2以上の他の活性剤と連続的に、同時に又は順次に投与され得る。
特に好ましい実施形態において、1又は2以上の本発明の化合物は、1又は2以上の他の活性剤、例えば、市場に流通している既存の薬物と組み合わせて投与される。そのような事例では、本発明の化合物は、1又は2以上の他の活性剤と連続的に、同時に又は順次に投与され得る。
概して、薬物は組み合わせて使用するとより有効である。特に、主要な毒性、作用機序及び耐性機序(複数可)の重複を回避するために、併用療法が望ましい。さらに、ほとんどの薬物を、その最大忍容用量で、そのような用量の時間間隔を最小にして投与することが望ましい。化学療法薬物を組み合わせることの主要な利点は、生化学的相互作用により相加作用又は考えられる相乗作用を促進し得ること、また耐性の発生を減少させ得ることである。
有益な組合せは、特定の障害の治療において価値があることが公知の又は疑われる作用物質を用い、試験化合物の阻害活性を研究することによって示唆され得る。この手順を使用して、作用物質の投与順序、すなわち、送達の前か、同時か、後かを決定することもできる。そのようなスケジューリングは、本明細書において同定されるすべての活性剤の特色となり得る。
アッセイ
本発明のさらなる態様は、1又は2以上のキナーゼ、より好ましくはLRRK、一層好ましくはLRRK2を阻害することができるさらなる候補化合物を同定するためのアッセイにおける、上述した通りの化合物の使用に関する。
本発明のさらなる態様は、1又は2以上のキナーゼ、より好ましくはLRRK、一層好ましくはLRRK2を阻害することができるさらなる候補化合物を同定するためのアッセイにおける、上述した通りの化合物の使用に関する。
好ましくは、アッセイは競合的結合アッセイである。
より好ましくは、競合的結合アッセイは、本発明の化合物を、キナーゼ、好ましくはLRRK、より好ましくはLRRK2、及び候補化合物と接触させること、並びに、本発明による化合物とキナーゼとの相互作用におけるいかなる変化も検出することを含む。
好ましくは、候補化合物は、本発明の化合物の従来のSAR修飾によって生成される。
本明細書において使用される場合、用語「従来のSAR修飾」は、化学誘導体化を利用して所与の化合物を変化させるための、当技術分野において公知である標準的な方法を指す。
故に、一態様において、同定された化合物は、他の化合物の開発のためのモデル(例えばテンプレート)として作用し得る。そのような試験において用いられる化合物は、溶液中で遊離している、固体支持体に付着している、細胞表面上にある、又は細胞内に位置しているものであってよい。活性の消失、又は化合物と試験される作用物質との間における結合複合体の形成を測定することができる。
本発明のアッセイはスクリーニングであってよく、これによって多くの(a number of)作用物質を試験する。一態様において、本発明のアッセイ方法はハイスループットスクリーニングである。
本発明は、化合物と結合することができる中和抗体が、化合物との結合について試験化合物と特異的に競合する、競合的薬物スクリーニングアッセイの使用も企図している。
スクリーニングのための別の技術は、物質に対する適切な結合親和性を有する作用物質のハイスループットスクリーニング(HTS)を提供し、国際公開第84/03564号パンフレットにおいて詳細に記述されている方法に基づく。
本発明のアッセイ方法は、試験化合物の小規模及び大規模スクリーニングの両方、並びに定量的アッセイに適することが予測される。
好ましくは、競合的結合アッセイは、本発明の化合物をキナーゼと、前記キナーゼの公知の基質の存在下で接触させること、及び、前記キナーゼと前記公知の基質との相互作用におけるいかなる変化も検出することを含む。
本発明のさらなる態様は、
(i)リガンドをキナーゼと、前記キナーゼの公知の基質の存在下で接触させるステップと、
(ii)前記キナーゼと前記公知の基質との相互作用におけるいかなる変化も検出するステップを含み、
ここで、前記リガンドは本発明の化合物である、
キナーゼとのリガンドの結合を検出する方法を提供する。
(i)リガンドをキナーゼと、前記キナーゼの公知の基質の存在下で接触させるステップと、
(ii)前記キナーゼと前記公知の基質との相互作用におけるいかなる変化も検出するステップを含み、
ここで、前記リガンドは本発明の化合物である、
キナーゼとのリガンドの結合を検出する方法を提供する。
本発明の一態様は、
(a)以上に記載したアッセイ方法を実施するステップと、
(b)リガンド結合ドメインに結合することができる1又は2以上のリガンドを同定するステップと、
(c)ある分量の前記1又は2以上のリガンドを調製するステップと
を含む方法に関する。
(a)以上に記載したアッセイ方法を実施するステップと、
(b)リガンド結合ドメインに結合することができる1又は2以上のリガンドを同定するステップと、
(c)ある分量の前記1又は2以上のリガンドを調製するステップと
を含む方法に関する。
本発明の別の態様は、
(a)以上に記載したアッセイ方法を実施するステップと、
(b)リガンド結合ドメインに結合することができる1又は2以上のリガンドを同定するステップと、
(c)前記1又は2以上のリガンドを含む医薬組成物を調製するステップと
を含む方法を提供する。
(a)以上に記載したアッセイ方法を実施するステップと、
(b)リガンド結合ドメインに結合することができる1又は2以上のリガンドを同定するステップと、
(c)前記1又は2以上のリガンドを含む医薬組成物を調製するステップと
を含む方法を提供する。
本発明の別の態様は、
(a)以上に記載したアッセイ方法を実施するステップと、
(b)リガンド結合ドメインに結合することができる1又は2以上のリガンドを同定するステップと、
(c)リガンド結合ドメインに結合することができる前記1又は2以上のリガンドを修飾するステップと、
(d)以上に記載したアッセイ方法を実施するステップと、
(e)前記1又は2以上のリガンドを含む医薬組成物を場合により調製するステップと
を含む方法を提供する。
(a)以上に記載したアッセイ方法を実施するステップと、
(b)リガンド結合ドメインに結合することができる1又は2以上のリガンドを同定するステップと、
(c)リガンド結合ドメインに結合することができる前記1又は2以上のリガンドを修飾するステップと、
(d)以上に記載したアッセイ方法を実施するステップと、
(e)前記1又は2以上のリガンドを含む医薬組成物を場合により調製するステップと
を含む方法を提供する。
本発明は、以上に記載した方法によって同定されるリガンドにも関する。
本発明のまた別の態様は、以上に記載した方法によって同定されるリガンドを含む医薬組成物に関する。
本発明の別の態様は、上述した1又は2以上の障害の治療において使用するための医薬組成物の調製における、以上に記載した方法によって同定されるリガンドの使用に関する。
上記の方法を使用して、1又は2以上のキナーゼの阻害剤として有用なリガンドをスクリーニングすることができる。
本発明の化合物は、研究室のツールとして及び治療剤としての両方で有用である。研究室において、本発明のある特定の化合物は、公知の又は新たに発見されたキナーゼが、病状の定着又は進行中に、決定的な又は少なくとも有意な生化学的機能に寄与するのかを確認する、一般に「ターゲットバリデーション」と称されるプロセスにおいて有用である。
合成
本発明の別の態様は、式Ia及び式Ibの化合物を調製するための方法に関する。
本発明の別の態様は、式Ia及び式Ibの化合物を調製するための方法に関する。
より具体的には、本発明は、式IIa’の化合物を式Ia’の化合物に変換するステップ:
を含む、式Ia’の化合物[式中、Q’はハロゲン又はC1−6−アルキルであり、R1及びR2は上記で定義した通りである]を調製するための方法を提供する。
本発明の1つの好ましい実施形態において、該方法は、式IIIa’の化合物をヒドラジン一水和物で処理することによって式IIa’の化合物を調製するステップ:
をさらに含む。
本発明の1つの好ましい実施形態において、該方法は、式IVa’の化合物を酸化剤で処理することによって式IIIa’の化合物を調製するステップ:
をさらに含む。
本発明の1つの好ましい実施形態において、該方法は、式Va’の化合物をR2−Mg−Clで処理することによって式IVa’の化合物を調製するステップ:
をさらに含む。
本発明の1つの好ましい実施形態において、R1は−NHR3であり、該方法は、式IIa’の化合物を式NH2R3のアミンと反応させるステップを含む。
本発明の別の好ましい実施形態において、R1は、NH含有C4−7−ヘテロシクロアルキル又はNH含有縮合アリール−C4−7−ヘテロシクロアルキルであり、該方法は、式IIa’の化合物を、前記C4−7−ヘテロシクロアルキル又は縮合アリール−C4−7−ヘテロシクロアルキルのNH基と反応させるステップを含む。
本発明の別の好ましい実施形態において、R1は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル、−C3−7シクロアルキル及び−C1−6アルキルから選択され、前記方法は、式IIa’の化合物を、X−R1[ここで、Xは4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル基である]と、カップリング剤の存在下で反応させるステップを含む。
好ましくは、カップリング剤はパラジウムジフェニルホスフィノフェロセンジクロリドである。
本発明の別の態様は、式VIa”の化合物を式Ia”の化合物に変換するステップ:
を含む、式Ia”の化合物[式中、Q”は、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール又はヘテロアリールであり、そのそれぞれは、1又は2以上の置換基Aで置換されていてもよく、R1及びR2は上記で定義した通りである]を調製するための方法に関する。
1つの好ましい実施形態において、該方法は、式VIa”の化合物をC4−7−ヘテロシクロアルキルのNH−基と、カップリング剤の存在下で反応させるステップを含む。
別の好ましい実施形態において、該方法は、式VIa”の化合物を化合物Q”−Y[ここで、Q”は、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール又はヘテロアリールであり、Yはボロン酸又はボロン酸エステル部分である]と、カップリング剤の存在下で反応させるステップを含む。
本発明の別の態様は、式IIbの化合物を式Ibの化合物に変換するステップ:
を含む、上記で定義した通りの式Ibの化合物を調製するための方法に関する。
好ましくは、前記式IIbの化合物は、式IIIbの化合物から、
前記式IIIbの化合物を、ヒドラジン、好ましくはヒドラジン一水和物で処理することによって調製される。
本発明の1つの好ましい実施形態において、該方法は、式IVbの化合物を酸化剤で処理することによって前記式IIIbの化合物を調製するステップ:
をさらに含む。
本発明の1つの好ましい実施形態において、該方法は、式Vbの化合物をR2−Mg−Clで処理することによって前記式IVbの化合物を調製するステップ:
をさらに含む。
1つの好ましい実施形態において、R1はアリール又はヘテロアリールであり、該方法は、前記式IIbの化合物を化合物R1−Y[ここで、Yはボロン酸又はボロン酸エステル部分である]と、カップリング剤の存在下で反応させるステップを含む。
別の好ましい実施形態において、R1は−NHR3であり、該方法は、前記式IIbの化合物を式NH2R3のアミンと反応させるステップを含む。
本発明の別の態様は、式IIIbの化合物を式Ibの化合物に変換するステップ:
を含む、上記で定義した通りの式Ibの化合物[式中、R1はOR3である]を調製するための方法に関する。
好ましくは、この実施形態について、該方法は、前記式IIIbの化合物を、ヒドラジン、好ましくはヒドラジン一水和物で処理するステップと、反応混合物をマイクロ波放射に供するステップとを含む。
式Ia及び式Ibの化合物を調製するための方法に関するさらなる態様を、添付の実施例の項において記述する。
下記の非限定的な実施例により、図面を参照して、本発明をさらに記述する。
[実施例]
材料及び方法
キナーゼの供給源及び精製
すべてのLRRK2タンパク質キナーゼは、別段の指示がない限り、ヒト由来のものとし、Invitrogen Corporation(Carlsbad、CA 92008 USA)から調達した。使用した活性突然変異体は、昆虫細胞において発現させた、G2019S突然変異を含有しGST標識した組換えヒト触媒ドメイン(アミノ酸970〜2527)(Invitrogen社製、カタログ番号PV4881)であった。使用した野生型は、昆虫細胞において発現させた、GST標識した組換えヒト触媒ドメイン(アミノ酸970〜2527)(Invitrogen社製、カタログ番号PV4873)であった。使用したキナーゼ死滅突然変異体は、昆虫細胞において発現させた、D1994A突然変異を含有しGST標識した組換えヒト触媒ドメイン(アミノ酸970〜2527)(Invitrogen社製、カタログ番号PM4041AE)であった。キナーゼのいずれを活性化させるためにも特別な措置を取らなかった。
材料及び方法
キナーゼの供給源及び精製
すべてのLRRK2タンパク質キナーゼは、別段の指示がない限り、ヒト由来のものとし、Invitrogen Corporation(Carlsbad、CA 92008 USA)から調達した。使用した活性突然変異体は、昆虫細胞において発現させた、G2019S突然変異を含有しGST標識した組換えヒト触媒ドメイン(アミノ酸970〜2527)(Invitrogen社製、カタログ番号PV4881)であった。使用した野生型は、昆虫細胞において発現させた、GST標識した組換えヒト触媒ドメイン(アミノ酸970〜2527)(Invitrogen社製、カタログ番号PV4873)であった。使用したキナーゼ死滅突然変異体は、昆虫細胞において発現させた、D1994A突然変異を含有しGST標識した組換えヒト触媒ドメイン(アミノ酸970〜2527)(Invitrogen社製、カタログ番号PM4041AE)であった。キナーゼのいずれを活性化させるためにも特別な措置を取らなかった。
タンパク質キナーゼアッセイ
すべてのアッセイを室温(約21℃)で行い、使用される条件下における時間及び酵素濃度に対して線形とした。アッセイは、96ウェルフォーマットで180分間実施した。LRRK2は約5nMの濃度で存在した。酵素を、50mMのトリス−HCl pH7.5、0.1mMのEGTA、1mMのDTT及び10mMのMgCl2中で希釈しアッセイした。アッセイにおける塩化マグネシウムの濃度は10mMであった。[γ−33P]ATP(0.4μCi/ウェル)を、Kmとなるように、G2019S突然変異体については134uMで、野生型キナーゼについては57μMで使用した。アッセイにおけるペプチド基質は、100μMのRLGWWRFYTLRRARQGNTKQRであった。
すべてのアッセイを室温(約21℃)で行い、使用される条件下における時間及び酵素濃度に対して線形とした。アッセイは、96ウェルフォーマットで180分間実施した。LRRK2は約5nMの濃度で存在した。酵素を、50mMのトリス−HCl pH7.5、0.1mMのEGTA、1mMのDTT及び10mMのMgCl2中で希釈しアッセイした。アッセイにおける塩化マグネシウムの濃度は10mMであった。[γ−33P]ATP(0.4μCi/ウェル)を、Kmとなるように、G2019S突然変異体については134uMで、野生型キナーゼについては57μMで使用した。アッセイにおけるペプチド基質は、100μMのRLGWWRFYTLRRARQGNTKQRであった。
アッセイは、Mg/ATPを用いて開始し、25μl/ウェルの50%オルトリン酸の添加によって停止した。Tomtecハーベスター(Tomtec Hamden社製、Ct 06514. USA)を使用して、反応物をWhatman P81 Unifilter Plates(Fisher Scientific社製、Loughborough、LE115RG、UK.、カタログ番号FDU-105-020U)上に採取した。Perkin Elmer Top Count NX7(Perkin Elmer社製、Shelton CT 06484-4794 USA)を使用してプレートを計数した。
各化合物について10の異なる濃度で二通りのアッセイを行った後、阻害剤のIC50値を決定した。
化合物合成のための一般的手順
クロマトグラフィー
分取高圧液体クロマトグラフィーは、Agilent社製の装置を使用して行った。該装置は、クロマトグラフィーが、直列につながれた多波長UV検出器(G1365B、Agilent社製)及びMM−ES+APCI質量分析計(G-1956A、Agilent社製)によってモニターされ、適正基準が満たされれば、自動画分収集器(G1364B、Agilent社製)によって試料が収集されるように構成されている。収集は、UV若しくは質量分析の任意の組合せによってトリガーされ(triggered)得、又は時間に基づいてよい。分離プロセスのための典型的な条件は、下記の通りである。勾配は、10分間にわたって実行する(開始時の勾配:メタノール10%及び水90%、終了時の勾配:メタノール100%及び水0%;緩衝液として、0.1%トリフルオロ酢酸を水に添加する(低pH緩衝液)、又は重炭酸アンモニウム(10mmol/l)及び35%水酸化アンモニウム(1.6ml/l)を水に添加する(高pH緩衝)のいずれか。当業者には、例えば、開始時又は終了時の溶媒組成を変更すること、溶媒又は緩衝液を修正すること、実行時間を変更すること、流速及び/又はクロマトグラフィーカラムを変更することにより、各特定化合物について条件を修正することが必要な又は望ましい場合があることが理解されよう。
クロマトグラフィー
分取高圧液体クロマトグラフィーは、Agilent社製の装置を使用して行った。該装置は、クロマトグラフィーが、直列につながれた多波長UV検出器(G1365B、Agilent社製)及びMM−ES+APCI質量分析計(G-1956A、Agilent社製)によってモニターされ、適正基準が満たされれば、自動画分収集器(G1364B、Agilent社製)によって試料が収集されるように構成されている。収集は、UV若しくは質量分析の任意の組合せによってトリガーされ(triggered)得、又は時間に基づいてよい。分離プロセスのための典型的な条件は、下記の通りである。勾配は、10分間にわたって実行する(開始時の勾配:メタノール10%及び水90%、終了時の勾配:メタノール100%及び水0%;緩衝液として、0.1%トリフルオロ酢酸を水に添加する(低pH緩衝液)、又は重炭酸アンモニウム(10mmol/l)及び35%水酸化アンモニウム(1.6ml/l)を水に添加する(高pH緩衝)のいずれか。当業者には、例えば、開始時又は終了時の溶媒組成を変更すること、溶媒又は緩衝液を修正すること、実行時間を変更すること、流速及び/又はクロマトグラフィーカラムを変更することにより、各特定化合物について条件を修正することが必要な又は望ましい場合があることが理解されよう。
フラッシュクロマトグラフィーは、シリカゲルクロマトグラフィーを指し、SP4又はIsolara 4 MPLCシステム(Biotage社製)、プレパックシリカゲルカートリッジ(Biotage社供給)を使用して、又は従来のガラスカラムクロマトグラフィーを使用して行われる。
分析方法
1H核磁気共鳴(NMR,Nuclear magnetic resonance)分光法は、別段の規定がない限り、ECX400質量分析計(JEOL社製)を使用し、規定溶媒中、室温前後で行った。いずれの場合も、NMRデータは推定構造に合致していた。特徴的な化学シフト(δ)は、主要なピークの呼称を表す従来の略号:例えば、s、一重線;d、二重線;t、三重線;q、四重線;dd、二重線の二重線;br、ブロード(broad)を使用して、パーツパーミリオンで示される。質量スペクトルは、MM−ES+APCI質量分析計(G-1956A、Agilent社製)を使用して記録した。薄層クロマトグラフィー(TLC,thin layer chromatography)が使用された場合、それは、シリカゲルMK6F 60Åプレートを使用するシリカゲルTLCを指し、Rfは、TLCプレート上で化合物が移動した距離を、溶媒が移動した距離で割ったものである。
1H核磁気共鳴(NMR,Nuclear magnetic resonance)分光法は、別段の規定がない限り、ECX400質量分析計(JEOL社製)を使用し、規定溶媒中、室温前後で行った。いずれの場合も、NMRデータは推定構造に合致していた。特徴的な化学シフト(δ)は、主要なピークの呼称を表す従来の略号:例えば、s、一重線;d、二重線;t、三重線;q、四重線;dd、二重線の二重線;br、ブロード(broad)を使用して、パーツパーミリオンで示される。質量スペクトルは、MM−ES+APCI質量分析計(G-1956A、Agilent社製)を使用して記録した。薄層クロマトグラフィー(TLC,thin layer chromatography)が使用された場合、それは、シリカゲルMK6F 60Åプレートを使用するシリカゲルTLCを指し、Rfは、TLCプレート上で化合物が移動した距離を、溶媒が移動した距離で割ったものである。
化合物調製
出発材料の調製について記載されていない場合、これらは市販されているか、文献において公知であるか、又は標準的手順を使用して当業者によって容易に入手可能である。化合物が前の実施例又は中間体と同様にして調製されたと述べられている場合、当業者には、反応時間、試薬の当量数及び温度は特定の反応ごとに修正され得ること、並びに異なるワークアップ又は精製技術を用いることが必要な又は望ましい場合があることが理解されよう。反応がマイクロ波照射を使用して行われる場合、使用されるマイクロ波は、Biotage社によって供給されるInitiator 60である。実際に供給される出力は、一定温度を維持するために、反応経過中に変動する。
出発材料の調製について記載されていない場合、これらは市販されているか、文献において公知であるか、又は標準的手順を使用して当業者によって容易に入手可能である。化合物が前の実施例又は中間体と同様にして調製されたと述べられている場合、当業者には、反応時間、試薬の当量数及び温度は特定の反応ごとに修正され得ること、並びに異なるワークアップ又は精製技術を用いることが必要な又は望ましい場合があることが理解されよう。反応がマイクロ波照射を使用して行われる場合、使用されるマイクロ波は、Biotage社によって供給されるInitiator 60である。実際に供給される出力は、一定温度を維持するために、反応経過中に変動する。
略号
DCM=ジクロロメタン
DMF=N,N−ジメチルホルムアミド
THF=テトラヒドロフラン
MeOH=メタノール
TFA=トリフルオロ酢酸
キサントホス=4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン
HATU=N,N,N’,N’−テトラメチル−O−(7−アザベンゾトリアゾール−1−イル)ウロニウム−ヘキサフルオロホスフェート
EDCI=1,3−プロパンジアミン,N3−(エチルカルボンイミドイル)−N1,N1−ジメチル−,塩酸塩
DCC=1,3−ジシクロヘキシルカルボジイミド
Pd2(dba)3=トリス(ジベンジリデンアセトン)ジパラジウム(0)
TEA=トリエチルアミン
rm=反応混合物
rt=室温
AcOH=酢酸
IPA=イソプロパノール
DIPEA=N,N−ジイソプロピルエチルアミン
TBSMSCl=第三級ブチルジメチルシリルクロリド
MeCN=アセトニトリル
NH3=アンモニア
EtOH=エタノール
EtOAc=酢酸エチル
LCMS=質量分析指向性高圧液体クロマトグラフィー
UV=紫外線
SCX=強カチオン交換
TPAP=過ルテニウム酸テトラプロピルアンモニウム
DMSO=ジメチルスルホキシド
BINAP=2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル
TPAP=過ルテニウム酸テトラプロピルアンモニウム
DIAD=アゾジカルボン酸ジイソプロピル
NMO=N−メチルモルホリンN−オキシド
DCM=ジクロロメタン
DMF=N,N−ジメチルホルムアミド
THF=テトラヒドロフラン
MeOH=メタノール
TFA=トリフルオロ酢酸
キサントホス=4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン
HATU=N,N,N’,N’−テトラメチル−O−(7−アザベンゾトリアゾール−1−イル)ウロニウム−ヘキサフルオロホスフェート
EDCI=1,3−プロパンジアミン,N3−(エチルカルボンイミドイル)−N1,N1−ジメチル−,塩酸塩
DCC=1,3−ジシクロヘキシルカルボジイミド
Pd2(dba)3=トリス(ジベンジリデンアセトン)ジパラジウム(0)
TEA=トリエチルアミン
rm=反応混合物
rt=室温
AcOH=酢酸
IPA=イソプロパノール
DIPEA=N,N−ジイソプロピルエチルアミン
TBSMSCl=第三級ブチルジメチルシリルクロリド
MeCN=アセトニトリル
NH3=アンモニア
EtOH=エタノール
EtOAc=酢酸エチル
LCMS=質量分析指向性高圧液体クロマトグラフィー
UV=紫外線
SCX=強カチオン交換
TPAP=過ルテニウム酸テトラプロピルアンモニウム
DMSO=ジメチルスルホキシド
BINAP=2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル
TPAP=過ルテニウム酸テトラプロピルアンモニウム
DIAD=アゾジカルボン酸ジイソプロピル
NMO=N−メチルモルホリンN−オキシド
中間体1
THF(100ml)中の2,4,6−トリクロロピリジン(9.85g、53.8mmol)の溶液を、THF(40ml)中のn−ブチルリチウム、ヘキサン中1.6M(33.6ml、53.8mmol)の溶液に、温度を−73℃より低温に維持しながら滴下添加した。添加が完了した後、混合物を−78℃で1時間撹拌した。THF(10ml)中の1−ホルミルピペリジン(6.0ml、53.8mmol)の溶液を滴下添加し、混合物を−78℃で1時間撹拌した。反応混合物を−78℃で飽和NH4Cl(水溶液)でクエンチし、室温に加温させた。混合物をエーテルで抽出し、1M HCl(水溶液)、水、飽和NH4HCO3(水溶液)、水及びブラインで連続的に洗浄した。有機相を乾燥させ、濃縮した。10:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色固体(5.18g、45%)を提供した。1H NMR(400MHz,DMSO−d6)δ ppm 8.09(s,1H)、10.27(s,1H)。
中間体2
THF(10ml)中のTHF中3M塩化メチルマグネシウム(9.0ml、26.9mmol)の溶液を、−78℃でTHF(110ml)中の中間体1(5.18g、24.4mmol)の撹拌溶液に滴下添加した。添加後、反応混合物を−78℃で30分間撹拌し、次いで室温に加温させた。混合物を飽和NH4Cl(水溶液)でクエンチし、酢酸エチルで抽出した。有機相をブラインで洗浄し、乾燥させ、濃縮して、黄色油(5.57g、100%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.46(d,J=6.9Hz,3H)、5.36(m,1H)、5.57(d,J=4.1Hz,1H)、7.84(s,1H)。
中間体3
NMO(4.3g、36.6mmol)及び4Åモレキュラーシーブ(6.4g)を、DCM(100ml)中の中間体2(5.57g、24.4mmol)の撹拌溶液に添加した。15分後、TPAP(288mg、0.82mmol)を添加し、反応混合物を室温で1時間撹拌した。反応混合物をセライト(Celite)に通して濾過し、濾液を濃縮し、7:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色油(4.6g、83%)を提供した。1H NMR(400MHz,DMSO−d6)δ ppm 2.59(s,3H)、8.08(s,1H)。
中間体4
エタノール(35ml)中の中間体3(3.55g、15.8mmol)及び35%ヒドラジン水溶液(35ml)の混合物を、室温で3.5時間撹拌した。反応混合物を氷に添加し、酢酸エチルで抽出した。有機相をブラインで洗浄し、乾燥させ、濃縮した。3:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、オフホワイト色の固体(1.71g、54%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 2.64(s,3H)、7.63(s,1H)。
中間体5
THF(80ml)中の2,4−ジクロロ−6−メチルニコチン酸塩(15g、64.1mmol)の溶液を、−78℃でTHF中2M水素化アルミニウムリチウム(160ml、320mmol)、に滴下添加した。エステル添加前に沈殿物が観察され、添加中にさらなる沈殿物が形成されたため、混合物を可動化するためにさらなるTHF(100ml)を添加した。反応混合物を−78℃で5時間、次いで−30℃で1時間撹拌した。次いで、水(10.3ml)を−30℃で極めてゆっくり添加し、続いて15%水酸化ナトリウム水溶液(10.3ml)を極めてゆっくり添加し、最後にさらなる水(32ml)を添加した。混合物を室温に加温させ、終夜撹拌した。混合物をセライトに通して濾過し、濾液を濃縮した。4:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、オフホワイトの固体(9.35g、76%)を生じさせた。1H NMR(400MHz,DMSO−d6)δ ppm 2.43(s,3H)、4.63(d,J=5.5Hz,2H)、5.30〜5.34(m,1H)、7.49(s,1H)。m/z(ES+APCI)+:192/194/196[M+H]+。
中間体6
DMSO(20ml、282mmol)を、−78℃のDCM(140ml)中の塩化オキサリル(12.1ml、140mmol)の撹拌溶液に添加した。添加後、次いでDCM(35ml)中の中間体5(9.32g、49mmol)を添加し、続いて温度を−70℃より低温に維持しながらEt3N(79ml、568mmol)を添加した。反応混合物を室温に加温させ、1時間撹拌した。反応混合物をNaHCO3(水溶液)溶液で洗浄し、有機相を乾燥させ、濃縮した。10:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、オフホワイトの固体(7.88g、85%)を得た。1H NMR(400MHz,クロロホルム−d)δ ppm 2.60(s,3H)、7.27(s,1H)、10.46(s,1H)。
中間体7
THF中3M塩化メチルマグネシウム(5.8ml、17.4mmol)を、−78℃でTHF(60ml)中の中間体6(3.0g、15.8mmol)の溶液に添加した。混合物を−78℃で45分間撹拌し、次いで室温に加温させた。混合物を飽和NH4Cl(水溶液)でクエンチし、水で希釈し、酢酸エチルで抽出した。有機相をブラインで洗浄し、乾燥させ、濃縮して、黄色油(3.23g、99%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.45(d,J=6.4Hz,3H)、2.40(s,3H)、5.31〜5.40(m,1H)、5.44(d,J=4.1Hz,1H)、7.43(s,1H)。m/z(ES+APCI)+:206[M+H]+。
中間体8
新たに活性化させた4Åモレキュラーシーブ(5.75g)及びNMO(4.19g、35.8mmol)を、DCM(100ml)中の中間体7(2.95g、14.3mmol)の溶液に添加した。混合物を室温で15分間撹拌し、続いてTPAP(256mg、0.729mmol)を添加した。反応混合物を室温で45分間撹拌し、次いでセライトに通して濾過し、濾液を濃縮乾固させた。6:1 ガソリン−酢酸エチル中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色油(2.31g、79%)を得た。1H NMR(400MHz,クロロホルム−d)δ ppm 2.55(s,3H)、2.60(s,3H)、7.19(s,1H)。
中間体9
65%ヒドラジン水溶液(2ml)中の中間体7(232mg、1.14mmol)の混合物を、室温で終夜撹拌した。混合物をDCM及び水で希釈し、MeOHを添加して、存在している固体を溶解した。有機相を分離し、水性物をDCMで再抽出した。合わせた有機抽出物を乾燥させ、濃縮して、白色固体を得た。固体を酢酸エチルから再結晶させて、オフホワイトの結晶性固体(50mg、24%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 2.49(s,3H)、2.61(s,3H)、7.28(d,J=0.9Hz,1H)。m/z(ES+APCI)+:182/184[M+H]+。
中間体10
THF(10ml)中のTHF中3M塩化メチルマグネシウム(11.5ml、34.4mmol)を、−78℃でTHF(110ml)中の3,5−ジクロロピリジン−4−カルボキサルデヒド(5.51g、31.3mmol)の撹拌懸濁液に滴下添加した。混合物を−78℃で1時間撹拌し、次いで室温に加温させ、その温度でさらに1時間撹拌した。氷冷を適用しながら、混合物を飽和NH4Cl(水溶液)でクエンチした。混合物を酢酸エチルで抽出し、有機相をブラインで洗浄し、乾燥させ、濃縮した。5:1〜3:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、オフホワイトの固体(3.65g、61%)を生じさせた。1H NMR(400MHz,DMSO−d6)δ ppm 1.41(d,J=6.4Hz,3H)、5.31〜5.37(m,1H)、5.59(d,J=4.1Hz,1H)、8.51(s,2H)。m/z(ES+APCI)+:192/194[M+H]+。
中間体11
新たに活性化させた4Åモレキュラーシーブ(7.08g)及びNMO(5.53g、47.3mmol)を、DCM(125ml)中の中間体10(3.63g、18.9mmol)の撹拌溶液に添加した。15分後、TPAP(332mg、0.945mmol)を添加し、混合物を室温で45分間撹拌した。反応混合物をセライトに通して濾過し、濾液を濃縮した。5:1 ガソリン−酢酸エチル中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色油(2.57g、72%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 2.59(s,3H)、8.76(s,2H)。
中間体12
中間体11(500mg、2.63mmol)及び65%ヒドラジン水溶液(1.91ml、39.5mmol)及びn−ブタノール(10ml)の混合物を、I-60マイクロ波反応器内、200℃で30分間照射した。反応を同じ規模で3回繰り返した。反応混合物を合わせ、水及び酢酸エチルで希釈し、有機相をブラインで洗浄し、乾燥させ、濃縮した。40:1〜10:1 DCM−MeOHで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、オフホワイトの固体(1.05g、59%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 2.68(s,3H)、8.17(s,1H)、8.92(s,1H)。
中間体13
4−メトキシベンジルクロリド(242μl、1.79mmol)を、DMF(20ml)中の中間体12(300mg、1.79mmol)及び水酸化カリウム(150mg、2.67mmol)の撹拌混合物に添加し、得られた混合物を室温で終夜撹拌した。反応混合物を水でクエンチし、酢酸エチルで抽出した。有機相を水(×3)及びブライン(×1)で洗浄し、乾燥させ、濃縮した。2:1〜1:2 ガソリン−酢酸エチル中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、生成物(361mg、70%)を生じさせた。1H NMR(400MHz,DMSO−d6)δ ppm 2.66(s,3H)、3.70(s,3H)、5.63(s,2H)、6.88(m,2H)、7.26(m,2H)、8.20(s,1H)、9.16(s,1H)。m/z(ES+APCI)+:288/290[M+H]+。
中間体14
THF(40ml)中のDIAD(11.3ml、57.7mmol)の溶液を、THF(160ml)中の、トリフェニルホスフィン(15.1g、57.7mmol)、シクロヘキサノール(5.8g、57.7mmol)及び3−クロロ−5−ヒドロキシピリジン(5.0g、38.5mmol)の氷冷溶液に徐々に添加した。反応混合物を室温で48時間撹拌した。混合物を濃縮乾固させ、10:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色油(4.6g、56%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.20〜1.31(m,1H)、1.32〜1.57(m,5H)、1.63〜1.76(m,2H)、1.85〜1.97(m,2H)、4.46〜4.54(m,1H)、7.62(t,J=2.3Hz,1H)、8.17(d,J=1.8Hz,1H)、8.24(d,J=2.3Hz,1H)。m/z(ES+APCI)+:212/214[M+H]+。
中間体15
ヘキサン中1.6Mn−ブチルリチウム(11.1ml、17.7mmol)を、−78℃でTHF(55ml)に添加した。温度を−74℃より低温に維持しながら、THF(10ml)中の中間体14(2.5g、11.8mmol)の溶液を滴下添加し、次いで混合物を−78℃で1.5時間撹拌した。この期間の後、次いでTHF(10ml)中のギ酸エチル(2.85ml、35.4mmol)の溶液を−78℃で滴下添加した。次いで、反応混合物をこの温度で3.5時間撹拌した後、飽和NH4Cl(水溶液)でクエンチした。混合物を室温に加温させ、酢酸エチル及び水で希釈した。有機相をブラインで洗浄し、乾燥させ、濃縮した。5:1 ガソリン−酢酸エチル中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、橙色油(950mg、34%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.29〜1.61(m,6H)、1.63〜1.77(m,2H)、1.81〜1.99(m,2H)、4.76(dt,J=8.0,4.2Hz,1H)、8.35(s,1H)、8.68(s,1H)、10.36(s,1H)。
中間体16
THF(5ml)中のTHF中3M塩化メチルマグネシウム(1.45ml、4.35mmol)を、−78℃でTHF(70ml)中の中間体15(950mg、3.96mmol)の撹拌溶液に滴下添加した。反応混合物を−78℃で45分間撹拌し、次いで室温に加温させ、ここでさらに1時間撹拌した。次いで、混合物を−20℃に冷却し、飽和NH4Cl(水溶液)でクエンチした。混合物を水及び酢酸エチルで希釈した。有機相をブラインで洗浄し、乾燥させ、濃縮し、3:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色油(667mg、66%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.28〜1.59(m,9H)、1.62〜1.76(m,2H)、1.83〜1.96(m,2H)、4.58(dt,J=8.0,4.2Hz,1H)、5.05(d,J=5.5Hz,1H)、5.27〜5.35(m,1H)、8.14(s,1H)、8.32(s,1H)。
中間体17
DCM(30ml)中の、中間体16(615mg、2.40mmol)、NMO(422mg、3.60mmol)及び4Åモレキュラーシーブ(800mg)を、室温で15分間撹拌した。次いでTPAP(60mg、0.171mmol)を添加し、反応混合物を室温で3時間撹拌した。混合物をセライトに通して濾過し、濾液を濃縮した。3:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、黄色油(488mg、80%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.24〜1.53(m,6H)、1.56〜1.71(m,2H)、1.84〜1.94(m,2H)、2.48(s,3H)、4.66(m 1H)、8.31(s,1H)、8.53(s,1H)。
n−ブタノール(1ml)中の中間体4(33mg、0.163mmol)及びシクロヘキシルアミン(19μl、0.163mmol)の混合物を、LC/MSによって反応をモニターしながら、室温で終夜撹拌した。混合物を100℃に終夜加熱し、さらなるシクロヘキシルアミン(57μl、0.499mmol)を添加し、撹拌を100℃で終夜続けた。次いで、混合物を酢酸エチルで希釈し、水及びブラインで洗浄した。有機相を乾燥させ、濃縮した。2:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、オフホワイト色の固体(10mg、23%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.13〜1.25(m,1H)、1.28〜1.47(m,4H)、1.58〜1.66(m,1H)、1.69〜1.79(m,2H)、1.90〜1.97(m,2H)、2.56(s,3H)、3.91〜4.01(m,1H)、5.85(d,J=7.8Hz,1H)、6.56(s,1H)。m/z(ES+APCI)+:265/267[M+H]+
水素化ナトリウム(油中60%分散、426mg、10.64mmol)を、マイクロ波反応器バイアル内、ジオキサン(15ml)中のシクロヘキサノール(1.24g、12.38mmol)の撹拌溶液に小分けにして添加した。混合物を室温で45分間撹拌した後、中間体4(500mg、2.48mmol)を添加した。混合物を室温で終夜撹拌し、続いて、Biotage社製I-60マイクロ波反応器内、190℃で1時間照射した。反応混合物を氷に添加し、酢酸エチルで抽出した。有機相をブラインで洗浄し、乾燥させ、濃縮した。3:1 ガソリン−酢酸エチルで溶離するシリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して油性固体を生じさせ、これを石油エーテルで粉砕して、白色固体(312mg、47%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.35〜1.56(m,4H)、1.56〜1.78(m,4H)、1.86〜1.96(m,2H)、2.52(s,3H)、5.21(dt,J=7.7,3.7Hz,1H)、7.05(s,1H)。m/z(ES+APCI)+:266/268[M+H]+。
実施例2(30mg、0.113mmol)及びモルホリン(1ml)を、Biotage社製I-60マイクロ波反応器内、200℃で5時間照射した。混合物を濃縮乾固させ、分取HPLC(高pH緩衝液)によって精製して、オフホワイトの固体(5mg、14%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.37〜1.53(m,4H)、1.57〜1.77(m,4H)、1.85〜1.93(m,2H)、2.43(s,3H)、3.25〜3.33(m,4H)、3.68〜3.74(m,4H)、5.17(dt,J=7.4,3.8Hz,1H)、5.99(s,1H)。m/z(ES+APCI)+:317[M+H]+。
マイクロ波反応器管に、ジオキサン(2ml)中の、実施例2(40mg、0.150mmol)、1−メチルピラゾール−4−ボロン酸ピナコールエステル(47mg、0.226mmol)、Pd(dppf)Cl2(6.1mg、0.0075mmol)及び2M Na2CO3(水溶液)(263μl、0.526mmol)を投入した。管の内容物を脱気し、窒素雰囲気下に置き、Biotage社製I-60マイクロ波反応器内、160℃で30分間照射した。反応混合物を酢酸エチル及び水で希釈した。有機相をブラインで洗浄し、乾燥させ、濃縮した。分取HPLC(高pH緩衝液)による精製により、淡褐色固体(7mg、15%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.38〜1.58(m,4H)、1.60〜1.81(m,4H)、1.91〜2.00(m,2H)、2.52(s,3H)、3.87(s,3H)、5.39(dt,J=7.6,4.0Hz,1H)、7.13(s,1H)、7.95(s,1H)、8.18(s,1H)。m/z(ES+APCI)+:312[M+H]+
n−ブタノール(1ml)中の中間体9(50mg、0.275mmol)及びシクロヘキシルアミン(63μl、0.549mmol)を、密封したマイクロ波反応器管に入れ、I-60マイクロ波反応器内、190℃で2時間照射した。混合物を濃縮乾固させ、残渣を分取HPLC(高pH緩衝液)によって精製して、オフホワイトの固体を得た。分取HPLC(低pH緩衝液)によるさらなる精製により、白色固体(16mg、24%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.14〜1.25(m,1H)、1.28〜1.42(m,4H)、1.57〜1.64(m,1H)、1.67〜1.77(m,2H)、1.91〜2.01(m,2H)、2.27(s,3H)、2.54(s,3H)、4.00〜4.10(m,1H)、5.34(d,J=7.8Hz,1H)、6.37(s,1H)、12.33(s,1H)。m/z(ES+APCI)+:245[M+H]+。
水素化ナトリウム、油中60%分散(38mg、0.961mmol)を、ジオキサン(3ml)中のシクロヘキサノール(116μl、1.10mmol)の撹拌溶液に添加し、得られた混合物を室温で1時間撹拌した。中間体9(50mg、0.275mmol)を添加し、反応混合物を、I-60マイクロ波反応器内、180℃で1.5時間照射した。混合物を水でクエンチし、酢酸エチルで抽出した。有機相をブラインで洗浄し、乾燥させ、濃縮した。分取HPLC(高pH緩衝液)による精製により、淡桃色固体(18mg、27%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.36〜1.55(m,4H)、1.55〜1.78(m,4H)、1.90(m,2H)、2.37(d,J=0.9Hz,3H)、2.51(s,3H)、5.29(m,1H)、6.75(d,J=0.9Hz,1H)。m/z(ES+APCI)+:246[M+H]+。
ジオキサン(2ml)中の、中間体9(50mg、0.275mmol)、フラン−2−ボロン酸(46mg、0.412mmol)、Pd(dppf)Cl2(11mg、0.014mmol)及び2M NaHCO3(水溶液)(481μl、0.962mmol)の混合物を、密封したマイクロ波反応器管に入れ、脱気し、窒素雰囲気下に置いた。混合物を、I-60マイクロ波反応器内、160℃で30分間照射した。混合物を酢酸エチル及び水で希釈し、有機相をブラインで洗浄し、乾燥させ、濃縮した。残渣をDMSO(1ml)に溶解し、分取HPLC(高pH)によって精製して、淡褐色固体(30mg、51%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 2.54(s,3H)、2.62(s,3H)、6.69〜6.72(m,1H)、7.11(dd,J=3.2,0.9Hz,1H)、7.18〜7.21(m,1H)、7.94(dd,J=1.8,0.9Hz,1H)。m/z(ES+APCI)+:214[M+H]+。
中間体12(40mg、0.238mmol)、フラン−2−ボロン酸(40mg、0.357mmol)、Pd(dppf)Cl2(10mg、0.012mmol)及び2M NaHCO3(水溶液)(417μl、0.833mmol)及びジオキサン(2ml)を、密封したマイクロ波反応器バイアルに入れた。内容物を脱気し、窒素雰囲気下に置き、I-60マイクロ波反応器内、160℃で20分間照射した。混合物を酢酸エチル及び水で希釈した。有機相をブラインで洗浄し、乾燥させ、濃縮した。分取HPLC(高pH緩衝液)による精製により、淡褐色固体(14mg、30%)を生じさせた。1H NMR(400MHz,DMSO−d6)δ ppm 2.55(s,3H)、6.70〜6.72(m,1H)、6.99〜7.02(m,1H)、7.95(dd,J=1.8,0.9Hz,1H)、8.38(s,1H)、8.94(s,1H)。m/z(ES+APCI)+:200[M+H]+。
ステップ1
フラスコに、ジオキサン(6ml)中の、中間体13(150mg、0.521mmol)、キサントホス(24mg、0.041mmol)、Pd2(dba)3(28.5mg、0.031mmol)、シクロヘキシルアミン(71μl、0.622mmol)及びナトリウムt−ブトキシド(150mg、1.56mmol)を投入した。得られた混合物を脱気し、窒素雰囲気下に置き、次いで100℃で終夜加熱した。室温に冷却した上で、混合物を水及び酢酸エチルで希釈し、有機相を乾燥させ、濃縮した。3:1 酢酸エチル−ガソリン中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して黄色固体(78mg)を提供し、これは不純なものであったが、さらに精製することなくステップ2において使用した。
フラスコに、ジオキサン(6ml)中の、中間体13(150mg、0.521mmol)、キサントホス(24mg、0.041mmol)、Pd2(dba)3(28.5mg、0.031mmol)、シクロヘキシルアミン(71μl、0.622mmol)及びナトリウムt−ブトキシド(150mg、1.56mmol)を投入した。得られた混合物を脱気し、窒素雰囲気下に置き、次いで100℃で終夜加熱した。室温に冷却した上で、混合物を水及び酢酸エチルで希釈し、有機相を乾燥させ、濃縮した。3:1 酢酸エチル−ガソリン中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して黄色固体(78mg)を提供し、これは不純なものであったが、さらに精製することなくステップ2において使用した。
ステップ2
トルエン(7ml)中のステップ1からの生成物(78mg、0.223mmol)及び塩化アルミニウム(III)(119mg、0.891mmol)を、50℃で4時間加熱した。反応混合物を室温に冷却させ、濃縮乾固させた。20:1 DCM−MeOH中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、橙色油を生じさせた。分取HPLC(高pH緩衝液)によるさらなる精製により、オフホワイトの固体(6mg、12%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.16〜1.45(m,5H)、1.58〜1.66(m,1H)、1.67〜1.77(m,2H)、1.98〜2.08(m,2H)、2.60〜2.68(m,3H)、3.35〜3.46(m,1H)、4.85(d,J=7.8Hz,1H)、7.41(s,1H)、8.13(s,1H)。m/z(ES+APCI)+:231[M+H]+。
トルエン(7ml)中のステップ1からの生成物(78mg、0.223mmol)及び塩化アルミニウム(III)(119mg、0.891mmol)を、50℃で4時間加熱した。反応混合物を室温に冷却させ、濃縮乾固させた。20:1 DCM−MeOH中、シリカゲル上でのフラッシュカラムクロマトグラフィーによって残渣を精製して、橙色油を生じさせた。分取HPLC(高pH緩衝液)によるさらなる精製により、オフホワイトの固体(6mg、12%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.16〜1.45(m,5H)、1.58〜1.66(m,1H)、1.67〜1.77(m,2H)、1.98〜2.08(m,2H)、2.60〜2.68(m,3H)、3.35〜3.46(m,1H)、4.85(d,J=7.8Hz,1H)、7.41(s,1H)、8.13(s,1H)。m/z(ES+APCI)+:231[M+H]+。
n−ブタノール(2.5ml)中の中間体17(100mg、0.394mmol)及び65%ヒドラジン水溶液(282μl、0.582mmol)の混合物を、I-60マイクロ波反応器内、200℃で1時間照射した。反応混合物を濃縮乾固させた。残渣をDMSO(1ml)に溶解し、分取HPLC(高pH)によって精製して、白色固体(6mg、7%)を得た。1H NMR(400MHz,DMSO−d6)δ ppm 1.33〜1.57(m,4H)、1.57〜1.79(m,4H)、1.87〜2.00(m,2H)、2.59(s,3H)、4.63〜4.72(m,1H)、7.81(s,1H)、8.47(s,1H)。m/z(ES+APCI)+:232[M+H]+。
中間体18
1,4−ジオキサン(8mL)中の3−ヨード−6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(302mg、0.502mmol)の溶液に、二酸化セレン(201mg、1.81mmol)を添加した。反応物を100℃で5時間撹拌した。追加の二酸化セレン(200mg、1.8mmol)を添加した。反応物を100℃で18時間さらに撹拌した。次いで、反応物を濾過し、濃縮した。粗生成物をフラッシュクロマトグラフィーによって精製して、所望のアルデヒド中間体(0.2g、60%)を得た。
中間体19
DMF(20mL)中の3−ヨード−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−6−カルバルデヒド(0.31g、0.50mmol)の溶液に、ヒドロキシルアミン塩酸塩(38mg、0.55mmol)、トリエチルアミン(0.078mL、0.55mmol)及びプロパンホスホン酸環状三量体(0.400g、1.1mmol)を添加した。反応物を100℃で2時間撹拌した。反応物を飽和NaHCO3で希釈し、EtOAc(3×)で抽出した。合わせた抽出物をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。粗生成物をフラッシュクロマトグラフィーによって精製して、所望の生成物(0.31g、定量的)を得た。
中間体20
乾燥CH2Cl2(450mL)中の2,4−ジクロロ−6−メチルニコチン酸メチル(45g、0.19mol)の撹拌溶液に、−78℃でDIBAL−H(トルエン中1M溶液)(576mL、0.57mol)を30分間かけて添加した。撹拌を−78℃でもう2時間続けた。同じ温度で飽和NH4Cl溶液(100mL)で反応混合物をクエンチし、室温に加温させた。反応混合物を0.2N HCl溶液(1000mL)で希釈し、CH2Cl2(3×500mL)で抽出し、合わせた有機物を、H2O(2×200mL)、ブライン溶液(2×250mL)で洗浄し、乾燥させ(Na2SO4)、濃縮した。粗化合物をフラッシュカラムクロマトグラフィー(シリカゲル、100〜200メッシュ)によって精製し、所望のアルデヒド(XX)を5%EtOAc−石油エーテルで溶離して、16gをオフホワイトの固体として生じさせた。Rf:0.6(20%EtOAc/石油エーテル)。(2,4−ジクロロ−6−メチルピリジン−3−イル)メタノールを15%EtOAc−石油エーテルで溶離して、15gをオフホワイトの固体として生じさせた。Rf:0.25(20%EtOAc/石油エーテル)、全体にわたる収率(31g、84%)。
CH2Cl2(200mL)中の(2,4−ジクロロ−6−メチルピリジン−3−イル)メタノール(15g、78.12mmol)の溶液に、PCC(42g、195.3mmol)を0℃で添加し、室温で16時間撹拌した。反応混合物を濃縮し、残渣をフラッシュカラムクロマトグラフィー(シリカゲル、100〜200メッシュ、5%EtOAc−石油エーテルで溶離)によって精製して、2,4−ジクロロ−6−メチルニコチンアルデヒド(XX、11.6g、78%)をオフホワイトの固体として生じさせた。
1,2−ジメトキシエタン(400mL)中の化合物2,4−ジクロロ−6−メチルニコチンアルデヒド(18g、94.7mmol)の溶液に、98%N2H4.H2O(14.2g、284.2mmol)を室温で添加し、80℃で16時間加熱した。反応混合物を濃縮し、残渣を水(250mL)に懸濁し、30分間撹拌した。沈殿した固体を濾過によって収集し、石油エーテル(2×250mL)で洗浄した。微量の7−アザ位置異性体を含有する取得された固体を、さらなる精製にかけた。混合物をCHCl3(150mL)に懸濁し、30分間撹拌し、濾過し(このプロセスを2回繰り返した)、乾燥させて、4−クロロ−6−メチル−1H−ピラゾロ[4,3−c]ピリジン(8g、50%)を白色固体として取得した。
1,4−ジオキサン(120mL)中の4−クロロ−6−メチル−1H−ピラゾロ[4,3−c]ピリジン(8g、47.9mmol)及びKOH(9.92g、177.2mmol)の撹拌懸濁液に、I2(24.2g、95.8mmol)を室温で添加し、70℃で4時間加熱した。反応混合物を氷浴中で冷却し、飽和メタ重亜硫酸ナトリウム溶液に添加し、30分間撹拌し、沈殿した固体を濾過によって収集し、水(600mL)、石油エーテル(2×200mL)で洗浄し、乾燥させて、4−クロロ−3−ヨード−6−メチル−1H−ピラゾロ[4,3−c]ピリジン(11g、78%)を白色固体として取得した。
中間体21
DMF(30mL)中の4−クロロ−3−ヨード−6−メチル−1H−ピラゾロ[4,3−c]ピリジン(3.00g、10.2mmol)の溶液に、KOH(1.14g、20.4mmol)及び1−(クロロメチル)−4−メトキシベンゼン(3.20g、20.4mol)を添加した。混合物を室温で終夜撹拌した。次いで、反応混合物を減圧下で濃縮し、石油エーテル/酢酸エチル(10:1〜8:1)で溶離するフラッシュカラムクロマトグラフィーによって残渣を精製して、4−クロロ−3−ヨード−1−(4−メトキシベンジル)−6−メチル−1H−ピラゾロ[4,3−c]ピリジンを白色固体(3.30g、62%)として生じさせた。
磁気撹拌器を備えたマイクロ波バイアルに、4−クロロ−3−ヨード−1−(4−メトキシベンジル)−6−メチル−1H−ピラゾロ[4,3−c]ピリジン(2.60g、6.30mmol)、テトラヒドロ−2H−ピラン−4−アミン(1.91g、18.9mmol)、n−BuOH(10mL)及びジイソプロピルエチルアミン(2.44g、18.9mmol)を投入した。反応混合物を、マイクロ波照射下、170℃で2時間加熱した。次いでこれを室温に冷却し、減圧下で濃縮した。得られた残渣を、DCM/石油エーテル/TEA(2:1:0.01)で溶離するフラッシュカラムクロマトグラフィーによって精製して、3−ヨード−1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミンを白色固体(2.35g、66%)として生じさせた。
磁気撹拌器を備えた密封管に、3−ヨード−1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(600mg、1.26mmol)、1,1,1,2,2,2−ヘキサメチルジスタンナン(618mg、1.88mmol)、トルエン(12mL)及びtrans−Pd(PPh3)2Cl2(24mg、0.0314mol)を投入した。3サイクルの真空/アルゴンフラッシュ後、反応混合物を110℃で15時間加熱した。次いでこれを室温に冷却し、濾過した。濾液を減圧下で濃縮して粗生成物(21,860mg)を生じさせ、これをさらに精製することなくその後の転換において使用した。
中間体22
磁気撹拌器を備えたマイクロ波バイアルに、粗製の1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(トリメチルスタンニル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(550mg、1.07mmol)、2−ブロモイソニコチン酸メチル(276mg、1.28mmol)、LiCl(184mg、4.28mmol)、CuI(20mg、0.107mmol)、Pd(PPh3)4(124mg、0.107mmol)及びTHF(15mL)を投入した。3サイクルの真空/アルゴンフラッシュ後、反応混合物を、マイクロ波照射下、100℃で1時間加熱した。次いでこれを室温に冷却し、濾過した。濾液を減圧下で濃縮し、石油エーテル/酢酸エチル(1:2)で展開する分取TLCによって残渣を精製して、2−(1−(4−メトキシベンジル)−6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)イソニコチン酸メチルを黄色固体(160mg、41%、2ステップ)として生じさせた。
THF(10mL)、メタノール(10mL)及びH2O(5mL)の溶液に、2−(1−(4−メトキシベンジル)−6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)イソニコチン酸メチル(160mg、0.329mmol)及びLiOH(131mg、3.29mmol)を添加した。混合物を室温で4時間撹拌し、次いで減圧下で濃縮した。H2O(5mL)及び酢酸エチル(8mL)を残渣に添加し、得られた沈殿物を収集して、2−(1−(4−メトキシベンジル)−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)イソニコチン酸(140mg、90%)を白色固体として生じさせた。
2−(1−(4−メトキシベンジル)−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)イソニコチン酸(120mg、0.254mmol)を投入した25mLの丸底フラスコに、ジメチルアミン塩酸塩(107mg、1.27mmol)、HATU(193mg、0.508mmol)、ジイソプロピルエチルアミン(0.5mL)及びDMF(3mL)を添加した。反応混合物を室温で12時間撹拌した。次いでこれを、1:3 水/CH3CN中0.3%NH4HCO3で溶離する逆相Combi-flashによって精製して、2−(1−(4−メトキシベンジル)−6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)−N,N−ジメチルイソニコチンアミドを黄色固体(90mg、71%)として生じさせた。
中間体23
磁気撹拌器を備えたマイクロ波バイアルに、1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(トリメチルスタンニル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(600mg、1.16mmol)、4,6−ジクロロピリミジン(208mg、1.40mmol)、LiCl(195mg、4.64mmol)、CuI(22mg、0.116mmol)、Pd(PPh3)4(134mg、0.116mmol)及びTHF(10mL)を投入した。3サイクルの真空/アルゴンフラッシュ後、反応混合物を、マイクロ波照射下、100℃で30分間加熱した。次いでこれを室温に冷却し、減圧下で濃縮した。得られた残渣を、石油エーテル/EA(2:1)で溶離するフラッシュカラムクロマトグラフィーによって精製して、N−イソプロピル−3−(ピリジン−2−イル)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−4−アミンを黄色固体(130mg、24%)として生じさせた。
磁気撹拌器を備えたマイクロ波バイアルに、N−イソプロピル−3−(ピリジン−2−イル)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(120mg、0.26mmol)及びTFA(15mL)を投入した。反応混合物を、マイクロ波照射下、150℃で2時間加熱した。次いでこれを室温に冷却し、減圧下で濃縮した。得られた残渣のpHを、飽和NaHCO3溶液をゆっくり導入することによって7に調整した。次いでこれを酢酸エチル(2×50mL)で抽出した。合わせた有機相を水(50mL)及びブラインで洗浄し、無水Na2SO4で乾燥させ、濾過し、濃縮し、6−(6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリミジン−4−オール(80mg、94%)を生じさせた。
POCl3(20mL)中の6−(6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリミジン−4−オール(70mg、0.21mmol)の混合物を、5時間撹拌還流した。次いでこれを室温に冷却し、減圧下で濃縮した。得られた残渣を、飽和NaHCO3溶液をゆっくり導入することによってpH7に調整し、酢酸エチル(2×50mL)で抽出した。合わせた有機相を水(50mL)及びブラインで洗浄し、無水Na2SO4で乾燥させ、濾過し、濃縮し、所望生成物を白色固体(50mg、69%)として生じさせた。
中間体24
−78℃で乾燥THF(5mL)中のジイソプロピルアミン(1.6g、15.8mmol)の溶液に、n−BuLi(ヘキサン中2.5M、5.1mL)を添加し、得られた混合物を−10℃で1時間撹拌した。上記の混合物に、THF(2mL)中の2,4−ジクロロ−6−(トリフルオロメチル)ピリジン(1.9g、8.84mmol)の溶液を添加し、混合物を−78℃で40分間撹拌した。THF(2mL)中の新たに蒸留したギ酸エチル(0.37g、5mmol)の溶液を、−78℃の上記の反応混合物に30分間かけて添加し、1時間撹拌した。反応混合物を飽和NH4Cl水溶液(10mL)でクエンチし、室温に加温させ、EtOAc(2×8mL)で抽出し、合わせた有機物を、水(2×5mL)、ブライン溶液(2×5mL)で洗浄し、Na2SO4で乾燥させ、濃縮して、粗製の2,4−ジクロロ−6−(トリフルオロメチル)ニコチンアルデヒド(1.7g、81%)を淡黄色固体として生じさせた。1,2−ジメトキシエタン(20mL)に溶解した粗生成物に、50%N2H4.H2O(1.42g、13.95mmol)を室温で添加し、次いで80℃に2時間加熱した。反応混合物を濃縮して、粗製の4−クロロ−6−(トリフルオロメチル)−1H−ピラゾロ[4,3−c]ピリジン(200mg、13%)を黄色固体として生じさせた。1,4−ジオキサン(40mL)中の粗生成物及びKOH(270mg、4.84mmol)の撹拌懸濁液に、I2(663mg、2.61mmol)を室温で添加した。次いで、混合物を70℃で3時間加熱した。次いで、反応混合物を氷浴中で冷却し、飽和メタ重亜硫酸ナトリウム水溶液を添加した。混合物を30分間撹拌し、沈殿物を濾過によって収集した。濾液を水で洗浄し、乾燥させ(Na2SO4)、フラッシュカラムクロマトグラフィーによって精製して、4−クロロ−3−ヨード−6−(トリフルオロメチル)−1H−ピラゾロ[4,3−c]ピリジン(70mg、3%、3ステップ)を黄色固体として得た。
塩化メチレン(1.0mL)中の4−クロロ−3−ヨード−6−(トリフルオロメチル)−1H−ピラゾロ[4,3−c]ピリジン(0.06g、0.17mmol)の溶液に、トリエチルアミン(0.036mL、0.26mmol)及びクロロトリフェニルメタン(0.052g、0.18mmol)を添加し、反応混合物を室温で12時間撹拌した。次いで、反応物を濃縮し、フラッシュクロマトグラフィーによって精製して、4−クロロ−3−ヨード−6−(トリフルオロメチル)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(0.061g、60%)を得た。
マイクロ波管中、1,4−ジオキサン(0.5mL)に懸濁した水素化ナトリウム(4.96mg、0.12mmol、60%)に、テトラヒドロ−4−ピラノール(12.9mg、0.12mmol)を室温で添加し、反応混合物を30分間撹拌した。次いで、1,4−ジオキサン(1.0mL)中の4−クロロ−3−ヨード−6−(トリフルオロメチル)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(0.061g、0.1mmol)をキャニュラー(cannula)によって添加し、得られた混合物を130℃に12時間加熱した。次いで、反応物を塩化メチレンで希釈し、濾過し、濃縮し、フラッシュクロマトグラフィーによって精製して、所望の生成物(0.012g、18%)を得た。
中間体25
トリエチルアミン(499uL、0.00358mol)を、室温で塩化メチレン(19.7mL、0.308mol)中の4−クロロ−3−ヨード−6−メチル−1H−ピラゾロ[4,3−c]ピリジン(0.700g、0.00238mol)及び塩化トリフェニルメチル(0.698g、0.00250mol)の懸濁液に添加した。反応混合物は約5分後に均質になった。反応物を室温で3時間撹拌し、水で希釈し、塩化メチレンで抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮して、4−クロロ−3−ヨード−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジンをオフホワイトの固体(1.24g、97%)として提供した。1H−NMR(400MHz,CDCl3)δ 7.30(m,9H)、7.18〜7.08(m,6H)、5.93(s,1H)、2.26(s,3H)。
中間体26
ナトリウムメトキシドのメタノール中25%溶液(25:75、ナトリウムメトキシド:メタノール、0.282mL、1.23mmol)を、テトラヒドロフラン(5.4mL、67mmol)中の4−クロロ−3−ヨード−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(600mg、1mmol)の懸濁液に滴下添加した。得られた溶液を50℃で2時間加熱した。揮発性材料を蒸発によって除去し、得られた固体を水と塩化メチレンとに分配した。有機層を乾燥させ(Na2SO4)、濾過し、濃縮し、3−ヨード−4−メトキシ−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(564mg、90%)を提供した。1H−NMR(400MHz,CDCl3)δ 7.27(dd,J = 6.7,2.3Hz,9H)、7.15(dd,J = 6.8,2.9Hz,6H)、5.55(s,1H)、4.05(s,3H)、2.15(s,3H)。
中間体27
テトラヒドロ−2H−ピラン−4−オール(108uL、1.13mmol)を、1,4−ジオキサン(1mL、20mmol)中の水素化ナトリウム(61.0mg、1.52mmol)の懸濁液に滴下添加した。発泡が止まった後、懸濁液を10分間撹拌し、次いで、1,4−ジオキサン(5mL、60mmol)中の4−クロロ−3−ヨード−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(0.505g、0.942mmol)の溶液を添加し、溶液をマイクロ波中180℃に1時間加熱した。反応混合物をEtOAcで希釈し、セライトに通して濾過した。溶媒を除去し、5mLのEtOAcに再溶解し、15分間静置させた。10mLのヘプタンを添加し、溶液を−10℃で5時間貯蔵した。得られた固体を濾過によって収集した(約250mgs)。濾液の濃縮からの残渣をシリカにロード(loaded)し、フラッシュカラムクロマトグラフィー(24gカラム、0%〜50%EtOAc/ヘプタン)を使用し生成物を精製して、追加で180mgを提供した。1H−NMR(400MHz,CDCl3)δ 7.39〜7.21(m,10H)、7.16(dd,J = 6.8,2.9Hz,5H)、5.63〜5.54(m,1H)、5.53(s,1H)、4.13(ddd,J=11.5,8.3,3.3Hz,2H)、3.83〜3.63(m,2H)、2.12(s,3H)、2.09〜2.00(m,2H)、1.97〜1.82(m,2H)。
中間体28
3−ヨード−4−メトキシ−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(175mg、0.329mmol)、2−モルホリノピリジン−4−イルボロン酸(95.9mg、0.461mmol)、ビス(ジ−tert−ブチル(4−ジメチルアミノフェニル)ホスフィン)ジクロロパラジウム(II)(23.3mg、0.0329mmol)、酢酸カリウム(45.2mg、0.461mmol)及び炭酸ナトリウム(48.9mg、0.461mmol)を、マイクロ波バイアル内に充填し、これを窒素でパージした。アセトニトリル(2.41mL、46.1mmol)及び水(0.593mL、32.9mmol)を添加し、溶液を窒素で10分間パージした。混合物を150℃に30分間加熱した。反応が完了したら、混合物を15mLのEtOAcで希釈し、セライトに通して濾過し、濃縮して、4−(4−(4−メトキシ−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリジン−2−イル)モルホリンを得た。
中間体29
3−ヨード−6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(500mg、0.8mmol)、4,4,5,5−テトラメチル−2−(1H−ピラゾール−4−イル)−1,3,2−ジオキサボロラン(226mg、1.16mmol)、ビス(ジ−tert−ブチル(4−ジメチルアミノフェニル)ホスフィン)ジクロロパラジウム(II)(58.9mg、0.0831mmol)、酢酸カリウム(114mg、1.16mmol)及び炭酸ナトリウム(123mg、1.16mmol)を、マイクロ波バイアル内に充填し、これを窒素でパージした。アセトニトリル(6.08mL、116mmol)及び水(1.50mL、83.1mmol)を添加し、溶液を窒素で10分間パージした。反応混合物をマイクロ波中150℃に30分間加熱した。反応混合物を濾過し、EtOAcで洗浄し、濃縮し、CH2Cl2と水とに分配した。有機層を収集し、Na2SO4で乾燥させ、次いで濾過し、濃縮した。得られた6−メチル−3−(1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(462mg、92%)を、その後の反応において直接使用した。
中間体30
6−メチル−3−(1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(464mg、0.857mmol)、テトラヒドロ−2H−ピラン−4−イルメタンスルホン酸塩(290mg、1.6mmol)、炭酸セシウム(399mg、1.22mmol)及びヨウ化テトラ−n−ブチルアンモニウム(60.4mg、0.163mmol)を、撹拌子を備えたバイアルに充填した。N,N−ジメチルホルムアミド(1.90mL、24.5mmol)を添加し、混合物を90℃で加熱した。2時間後、反応物を室温に冷却し、EtOAcで希釈し、セライトに通して濾過した。得られた6−メチル−3−(1−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジンを、次の反応において直接使用した。
マイクロ波管に、3−ヨード−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−6−カルボニトリル(0.112g、0.18mmol)、1−イソ−ブチル−1H−ピラゾール−4−ボロン酸ピナコールエステル(0.069g、0.27mmol)、ビス(ジ−tert−ブチル−(4−ジメチルアミノフェニル)ホスフィン)ジクロロパラジウム(13mg 0.018mmol)、2M炭酸ナトリウム(0.18mL)及びアセトニトリル(2.3mL)を添加した。反応物を密封し、マイクロ波中140℃で20分間加熱した。反応物を水で希釈し、EtOAc(3×)で抽出した。合わせた抽出物をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。
次いで、粗生成物をDCM(3.4mL)及びTFA(0.028mL)に溶解した。その後、トリエチルシラン(0.058mL)を添加した。次いで、反応物を室温で30分間撹拌した。反応物を濾過し、濃縮した。粗生成物を逆相HPLCによって精製して、3−(1−イソブチル−1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1H−ピラゾロ[4,3−c]ピリジン−6−カルボニトリル(26.5mg、39.5%)を得た。1H−NMR(400MHz,DMSO−d6)δ ppm 8.56(s,1H)、8.19(s,1H)、7.89(s,1H)、7.32(s,1H)、7.24(d,J=4.3,1H)、3.81(m,3H)、3.60(m,4H)、3.52(m,2H)、3.17(m,2H)、2.91(d,J=4.3,3H)。m/z(ES+APCI)+:367.2[M+H]+。
マイクロ波管に、6−クロロ−3−ヨード−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(0.102g、0.16mmol)、4−(4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピリジン−2−イル)モルホリン(0.057g、0.19mmol)、ビス(ジ−tert−ブチル−(4−ジメチルアミノフェニル)ホスフィン)ジクロロパラジウム(11mg 0.016mmol)、2M炭酸ナトリウム(0.16mL)及びアセトニトリル(2mL)を添加した。反応物を密封し、マイクロ波中140℃で30分間加熱した。反応物を水で希釈し、EtOAc(3×)で抽出した。合わせた抽出物をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。
次いで、粗生成物をDCM(3mL)及びTFA(0.5mL)に溶解した。その後、トリエチルシラン(0.1mL)を添加した。次いで、反応物を室温で3時間撹拌した。反応物を濾過し、濃縮した。粗生成物を逆相HPLCによって精製して、4−(4−(6−クロロ−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリジン−2−イル)モルホリン(26.5mg、39.5%)を得た。1H−NMR(400MHz,DMSO−d6)δppm 8.23(d,J=5.2,1H)、7.26(s,2H)、7.21(d,J=5.2,1H)、5.42(m,1H)、3.73(m,4H)、3.50(m,4H)、2.05(m,2H)、1.67(m,2H)。m/z(ES+APCI)+:416.1[M+H]+。
マイクロ波管に、3−ヨード−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−6−カルボニトリル(0.099g、0.16mmol)、4−(4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピリジン−2−イル)モルホリン(0.070g、0.24mmol)、ビス(ジ−tert−ブチル−(4−ジメチルアミノフェニル)ホスフィン)ジクロロパラジウム(11mg 0.016mmol)、2M炭酸ナトリウム(0.16mL)及びアセトニトリル(2mL)を添加した。反応物を密封し、マイクロ波中140℃で30分間加熱した。反応物を水で希釈し、EtOAc(3×)で抽出した。合わせた抽出物をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。次いで、粗生成物をDCM(3mL)及びTFA(0.25mL)に溶解した。その後、トリエチルシラン(0.05mL)を添加した。次いで、反応物を室温で1時間撹拌した。反応物を濾過し、濃縮した。粗生成物を逆相HPLCによって精製して、所望生成物(26.5mg、39.5%)を得た。1H−NMR(400MHz,DMSO−d6)δppm 8.24(d,J=5.1,1H)、8.00(s,1H)、7.26(s,1H)、7.20(d,J=5.2,1H)、5.47(s,1H)、3.72(m,6H)、3.52(m,6H)、2.05(m,2H)、1.69(m,2H)。m/z(ES+APCI)+:427.1[M+H]+。
マイクロ波反応バイアルに、6−クロロ−4−(シクロヘキシルオキシ)−3−メチル−1H−ピラゾロ[4,3−c]ピリジン(49mg、0.18mmol)、シクロプロピルボロン(cycloproylboronic)酸(80mg、0.9mmol)、Pd(dppf)2Cl2(15mg、0.018mmol)、2M Na2CO3(0.28mL、0.6mmol)及び1,4−ジオキサン(2mL)を添加した。次いで、反応物を密封し、マイクロ波中160℃で25分間撹拌しながら加熱した。次いで、反応物を濾過し、濃縮した。粗生成物を逆相HPLCによって精製して、4−(シクロヘキシルオキシ)−6−シクロプロピル−3−メチル−1H−ピラゾロ[4,3−c]ピリジン(8.7mg 17%)を得た。1H−NMR(400MHz,DMSO−d6)δppm 12.57(s,1H)、6.83(s,1H)、5.15(m,1H)、2.02(m,1H)、1.87(m,2H)、1.71(m,2H)、1.62(m,2H)、1.46(m,4H)、0.87(m,4H)。m/z(ES+APCI)+:272.1[M+H]+。
マイクロ波管に、3−(1−イソプロピル−1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−6−(トリフルオロメチル)−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン(12mg、0.018mmol)、1−イソプロピル−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピラゾール(5.2mg、0.02mmol)、1,1’−ビス(ジフェニルホスフィノ)フェロセンパラジウム(II)クロリド(1.5mg 0.002mmol)、1M酢酸カリウム(0.4mL)及びアセトニトリル(1.6mL)を添加した。反応物を窒素ガスで脱気し、密封し、マイクロ波中150℃で40分間加熱した。反応物を水で希釈し、EtOAc(3×)で抽出した。合わせた抽出物をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。粗生成物を、室温の塩化メチレン(0.8mL)、トリエチルシラン(0.012mL、0.07mmol)及びトリフルオロ酢酸(0.7mL、9mmol)に溶解した。反応物を15分間撹拌し、トルエン(2mL)とともに濃縮し、粗生成物を逆相HPLCによって精製して、3−(1−イソプロピル−1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−6−(トリフルオロメチル)−1H−ピラゾロ[4,3−c]ピリジン(3.1mg、43%)を得た。m/z(ES+APCI)+:396.1[M+H]+。
磁気撹拌器を備えたマイクロ波バイアルに、2−(1−(4−メトキシベンジル)−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)−N,N−ジメチルイソニコチンアミド(90mg、0.18mmol)及びTFA(3mL)を投入した。反応混合物を、マイクロ波照射下、120℃で2時間加熱した。次いでこれを室温に冷却し、減圧下で濃縮した。得られた残渣を逆相分取HPLCによって精製して、N,N−ジメチル−2−(6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルアミノ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)イソニコチンアミドを黄色固体(25mg、37%)として生じさせた。1H−NMR(500MHz,MeOD)δ 8.74(d,J=5.0,1H)、8.36(s,1H)、7.43(d,J=4.5,1H)、6.49(s,1H)、4.33〜4.35(m,1H)、4.03〜4.07(m,2H)、3.65〜3.69(m,2H)、3.17(s,3H)、3.06(s,3H)、2.41(s,3H)、2.18〜2.20(m,2H)、1.70〜1.73(m,2H)。m/z(ES+APCI)+:381[M+H]+。
3−(6−クロロピリミジン−4−イル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(50mg、344mmol)並びにEtOH(10mL)中の1M NaOEtの溶液及びTHF(5mL)の混合物を、2時間撹拌還流した。次いでこれを室温に冷却し、減圧下で濃縮した。次いで、混合物を酢酸エチル(2×50mL)で抽出した。合わせた有機相を水(50mL)及びブラインで洗浄し、無水Na2SO4で乾燥させ、濃縮した。得られた残渣を逆相分取HPLCによって精製して、3−(6−エトキシピリミジン−4−イル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミンを黄色固体(7mg、12%)として生じさせた。1H−NMR(500MHz,CDCl3)δ 10.17(s,1H)、8.74(s,1H)、7.61(s,1H)、6.40(s,1H)、4.47〜4.51(m,2H)、4.41〜4.42(m,1H)、4.03〜4.07(m,2H)、3.64〜3.69(m,2H)、2.43(s,3H)、2.19〜2.21(m,2H)、1.67〜1.74(m,2H)、1.45(t,3H)。m/z(ES+APCI)+:355[M+H]+。
磁気撹拌器を備えたマイクロ波バイアルに、1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(トリメチルスタンニル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(190mg、0.369mmol)、2−ブロモ−4−(トリフルオロメチル)ピリジン(167mg、0.738mmol)、LiCl(64mg、1.48mmol)、CuI(7mg、0.037mmol)、Pd(PPh3)4(43mg、0.037mmol)及びTHF(10mL)を投入した。3サイクルの真空/アルゴンフラッシュ後、反応混合物を、マイクロ波照射下、100℃で1時間加熱した。次いでこれを室温に冷却し、濾過した。濾液を減圧下で濃縮し、得られた残渣を、1:3 水/CH3CN中0.3%NH4HCO3で溶離する逆相Combi-flashによって精製して、1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(4−(トリフルオロメチル)ピリジン−2−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミンを黄色固体(55mg)として生じさせた。m/z(ES+APCI)+:498[M+H]+。
磁気撹拌器を備えたマイクロ波バイアルに、1−(4−メトキシベンジル)−6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(4−(トリフルオロメチル)ピリジン−2−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミン(55mg、0.111mmol)及びTFA(3mL)を投入した。反応混合物を、マイクロ波照射下、120℃で2時間加熱した。次いでこれを室温に冷却し、減圧下で濃縮した。得られた残渣を、逆相分取HPLCによって精製して、6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(4−(トリフルオロメチル)ピリジン−2−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミンを黄色固体(21mg、50%)として生じさせた。1H−NMR(500MHz,CDCl3)δ 9.96(d,J=3.5,1H)、8.73(s,1H)、8.61(s,1H)、.51(d,J=4.5,1H)、6.43(s,1H)、4.41〜4.42(m,1H)、4.03〜4.07(m,2H)、3.67〜3.69(m,2H)、2.49(s,3H)、2.18〜2.30(m,2H)、1.65〜1.73(m,2H)。m/z(ES+APCI)+:378[M+H]+。
2−ブロモ−4−(トリフルオロメチル)ピリジンを4−ブロモ−6−(トリフルオロメチル)ピリミジンで代用することにより、化合物実施例19について記述した通りの手順に準じて、6−メチル−N−(テトラヒドロ−2H−ピラン−4−イル)−3−(6−(トリフルオロメチル)ピリミジン−4−イル)−1H−ピラゾロ[4,3−c]ピリジン−4−アミンを黄色固体として取得した。1H−NMR(500MHz,CDCl3)δ 10.20(s,1H)、9.54(s,1H)、7.31(s,1H)、8.64(s,1H)、6.46(s,1H)、4.40〜4.41(m,1H)、4.04〜4.08(m,2H)、3.67(t,2H)、2.44(s,1H)、2.20〜2.23(m,2H)、1.67〜1.76(m,2H)。m/z(ES+APCI)+:379[M+H]+。
トリフルオロ酢酸(2.0mL、26mmol)を、23℃で塩化メチレン(4.0mL、62mmol)中の4−(4−(4−メトキシ−6−メチル−1−トリチル−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリジン−2−イル)モルホリン(0.187g、0.329mmol)及びトリエチルシラン(158uL、0.987mmol)の溶液に滴下添加した。反応物が赤色になり、15分間撹拌した。揮発性材料を蒸発させ、残渣を逆相HPLCによって精製して、4−(4−(4−メトキシ−6−メチル−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリジン−2−イル)モルホリンを得た。1H−NMR(400MHz,DMSO)δ 13.26(s,1H)、7.57(s,1H)、7.39(d,J=7.6Hz,1H)、7.30(t,J=7.9Hz,1H)、6.99(d,J=7.9Hz,1H)、6.94(s,1H)、3.96(s,3H)、3.86〜3.67(m,4H)、3.22〜3.08(m,4H)、2.45(s,3H)。注記:プロトンNMRは、ギ酸付加物について補正している。m/z(ES+APCI)+:367[M+H]+。
3−(4−メトキシ−6−メチル−1H−ピラゾロ[4,3−c]ピリジン−3−イル)−N−メチルベンズアミドは、実施例21において記述した通りの手順に従って調製した。1H−NMR(400MHz,DMSO)δ 13.41(s,1H)、8.45(s,1H)、8.08(d,J=7.8Hz,1H)、7.84(d,J=7.8Hz,1H)、7.54(t,J=7.8Hz,1H)、6.97(s,1H)、3.96(s,3H)、2.81(d,J=4.5Hz,3H)、2.46(s,3H)。m/z(ES+APCI)+:297[M+H]+。
4−(4−(6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ピリジン−2−イル)モルホリンは、実施例21において記述した通りの手順に従って調製した。1H−NMR(400MHz,DMSO)δ 13.48(s,1H)、8.21(d,J=5.1Hz,1H)、7.29(s,1H)、7.26(d,J=5.2Hz,1H)、6.96(s,1H)、5.56〜5.42(m,1H)、3.82〜3.68(m,6H)、3.56〜3.47(m,6H)、2.44(s,3H)、2.10〜1.98(m,2H)、1.66(dtd,J=12.5,8.3,3.9Hz,2H)。m/z(ES+APCI)+:397[M+H]+。
N−メチル−3−(6−メチル−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1H−ピラゾロ[4,3−c]ピリジン−3−イル)ベンズアミドは、実施例21において記述した通りの手順に従って調製した。1H−NMR(400MHz,DMSO)δ 13.39(s,1H)、8.46(d,J=4.4Hz,1H)、8.43(d,J=1.5Hz,1H)、8.08(d,J=7.7Hz,1H)、7.90〜7.81(m,1H)、7.54(t,J=7.7Hz,1H)、6.95(s,1H)、5.47(tt,J=7.7,3.8Hz,1H)、3.75〜3.62(m,2H)、3.48(ddd,J=11.4,8.0,3.2Hz,2H)、2.80(d,J=4.5Hz,3H)、2.44(s,3H)、2.09〜1.94(m,2H)、1.69(dtd,J=12.0,8.0,3.8Hz,2H)。m/z(ES+APCI)+:367[M+H]+。
6−メチル−3−(1−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾール−4−イル)−4−(テトラヒドロ−2H−ピラン−4−イルオキシ)−1H−ピラゾロ[4,3−c]ピリジンは、実施例21において記述した通りの手順に従って調製した。1H−NMR(400MHz,DMSO)δ 13.04(s,1H)、8.26(s,1H)、8.02(s,1H)、6.87(s,1H)、5.59〜5.34(m,1H)、4.60〜4.41(m,1H)、3.99(d,J=11.4Hz,2H)、3.89(dt,J=11.5,4.1Hz,2H)、3.68〜3.44(m,4H)、2.16(d,J=9.5Hz,2H)、1.99(ddd,J=19.5,16.5,9.8Hz,4H)、1.76(qd,J=9.9,4.2Hz,2H)。m/z(ES+APCI)+:384[M+H]+。
4−メトキシ−6−メチル−3−(1−(テトラヒドロ−2H−ピラン−4−イル)−1H−ピラゾール−4−イル)−1H−ピラゾロ[4,3−c]ピリジンは、実施例21において記述した通りの手順に従って調製した。1H−NMR(400MHz,DMSO)δ 13.05(s,1H)、8.28(s,1H)、8.01(s,1H)、6.88(s,1H)、4.58〜4.39(m,1H)、4.04(s,3H)、4.02〜3.93(m,2H)、3.56〜3.44(m,2H)、2.43(s,3H)、2.03(dd,J=8.9,3.7Hz,4H)。m/z(ES+APCI)+:314[M+H]+。
本発明の記載されている態様の種々の修正形態及び変形形態は、本発明の範囲及び趣旨から逸脱することなく、当業者に明らかであろう。本発明を特定の好ましい実施形態との関連で記載してきたが、特許請求されている通りの本発明は、そのような特定の実施形態に過度に限定されるべきでないことを理解すべきである。実際に、関連分野の当業者には明白な、本発明を行う記載されているモードの種々の修正形態は、下記の特許請求の範囲内であることが意図されている。
(参考文献)
1 Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W. and Singleton, A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 44, 595-600
2 Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P. and Gallo, K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286-293
3 Taylor, J. P., Mata, I. F. and Farrer, M. J. (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 12, 76-82
4 Farrer, M., Stone, J., Mata, I. F., Lincoln, S., Kachergus, J., Hulihan, M., Strain, K. J. and Maraganore, D. M. (2005) LRRK2 mutations in Parkinson disease. Neurology. 65, 738-740
5 Zabetian, C. P., Samii, A., Mosley, A. D., Roberts, J. W., Leis, B. C., Yearout, D., Raskind, W. H. and Griffith, A. (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 65, 741-744
6 Bosgraaf, L. and Van Haastert, P. J. (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 1643, 5-10
7 Marin, I. (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. 23, 2423-2433
8 Manning, G., Whyte, D. B., Martinez, R., Hunter, T. and Sudarsanam, S. (2002) The protein kinase complement of the human genome. Science. 298, 1912-1934
9 West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., Dawson, V. L. and Dawson, T. M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 102, 16842-16847
10 Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., van der Brug, M. P., Beilina, A., Blackinton, J., Thomas, K. J., Ahmad, R., Miller, D. W., Kesavapany, S., Singleton, A., Lees, A., Harvey, R. J., Harvey, K. and Cookson, M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 23, 329-341
11 Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317
12 Goldberg, J. M., Bosgraaf, L., Van Haastert, P. J. and Smith, J. L. (2002) Identification of four candidate cGMP targets in Dictyostelium. Proc Natl Acad Sci U S A. 99, 6749-6754
13 Bosgraaf, L., Russcher, H., Smith, J. L., Wessels, D., Soll, D. R. and Van Haastert, P. J. (2002) A novel cGMP signalling pathway mediating myosin phosphorylation and chemotaxis in Dictyostelium. Embo J. 21, 4560-4570
14 Cohen, P. and Knebel, A. (2006) KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem J. 393, 1-6
15 Bretscher, A., Edwards, K. and Fehon, R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 3, 586-599
16 Polesello, C. and Payre, F. (2004) Small is beautiful: what flies tell us about ERM protein function in development. Trends Cell Biol. 14, 294-302
17 Nichols, R. J., Dzamko, N., Hutti, J. E., Cantley, L. C., Deak, M., Moran, J., Bamborough, P., Reith, A. D. and Alessi, D. R. (2009) Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 424, 47-60
1 Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W. and Singleton, A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 44, 595-600
2 Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P. and Gallo, K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286-293
3 Taylor, J. P., Mata, I. F. and Farrer, M. J. (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 12, 76-82
4 Farrer, M., Stone, J., Mata, I. F., Lincoln, S., Kachergus, J., Hulihan, M., Strain, K. J. and Maraganore, D. M. (2005) LRRK2 mutations in Parkinson disease. Neurology. 65, 738-740
5 Zabetian, C. P., Samii, A., Mosley, A. D., Roberts, J. W., Leis, B. C., Yearout, D., Raskind, W. H. and Griffith, A. (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 65, 741-744
6 Bosgraaf, L. and Van Haastert, P. J. (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 1643, 5-10
7 Marin, I. (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. 23, 2423-2433
8 Manning, G., Whyte, D. B., Martinez, R., Hunter, T. and Sudarsanam, S. (2002) The protein kinase complement of the human genome. Science. 298, 1912-1934
9 West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., Dawson, V. L. and Dawson, T. M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 102, 16842-16847
10 Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., van der Brug, M. P., Beilina, A., Blackinton, J., Thomas, K. J., Ahmad, R., Miller, D. W., Kesavapany, S., Singleton, A., Lees, A., Harvey, R. J., Harvey, K. and Cookson, M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 23, 329-341
11 Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A. and Alessi, D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 405, 307-317
12 Goldberg, J. M., Bosgraaf, L., Van Haastert, P. J. and Smith, J. L. (2002) Identification of four candidate cGMP targets in Dictyostelium. Proc Natl Acad Sci U S A. 99, 6749-6754
13 Bosgraaf, L., Russcher, H., Smith, J. L., Wessels, D., Soll, D. R. and Van Haastert, P. J. (2002) A novel cGMP signalling pathway mediating myosin phosphorylation and chemotaxis in Dictyostelium. Embo J. 21, 4560-4570
14 Cohen, P. and Knebel, A. (2006) KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem J. 393, 1-6
15 Bretscher, A., Edwards, K. and Fehon, R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 3, 586-599
16 Polesello, C. and Payre, F. (2004) Small is beautiful: what flies tell us about ERM protein function in development. Trends Cell Biol. 14, 294-302
17 Nichols, R. J., Dzamko, N., Hutti, J. E., Cantley, L. C., Deak, M., Moran, J., Bamborough, P., Reith, A. D. and Alessi, D. R. (2009) Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 424, 47-60
Claims (46)
- 式Ia若しくはIbの化合物、又は薬学的に許容されるその塩若しくはエステル
[式中、
R1は、
アリール、
ヘテロアリール、
−NHR3、
縮合アリール−C4−7−ヘテロシクロアルキル、
−CONR4R5、
−NHCOR6、
−C3−7−シクロアルキル、
−NR3R6、
OR3、
OH、
NR4R5、並びに
R11及び基Aから選択される置換基で置換されていてもよい−C1−6アルキルから選択され、
ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基でそれぞれ置換されていてもよく、
R2は、水素、アリール、C1−6−アルキル、C2−6−アルケニル、C3−7−シクロアルキル、ヘテロアリール、C4−7ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及びハロゲンから選択され、ここで、前記C1−6−アルキル、C2−6−アルケニル、アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、R11及びAから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、
Qは、ハロゲン、CNであるか、又は、C1−6−アルキル、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール及びヘテロアリールから選択され、そのそれぞれが、1又は2以上の置換基Aで置換されていてもよく、
各R3は、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、C3−7−シクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル及びC1−6−アルキルから選択され、そのそれぞれは、R11及びAから選択される1又は2以上の置換基で置換されていてもよく、
R4及びR5は、水素、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、アリール、ヘテロアリール、C1−6−アルキル、並びに酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよく、かつ1又は2以上のR10基によって置換されていてもよいC3−6−ヘテロシクロアルキル環からそれぞれ独立に選択され、ここで、各C1−6−アルキル、ヘテロアリール及びアリールは、C1−6−アルキル、ハロゲン、シアノ、ヒドロキシル、アリール、ハロ置換アリール、ヘテロアリール、−NR8R9、−NR6R7、NR7(CO)R6、−NR7COOR6、−NR7(SO2)R6、−COOR6、−CONR8R9、OR6、−SO2R6、並びに酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよく、かつ1又は2以上のR10基によって置換されていてもよいC3−6−ヘテロシクロアルキル環から選択される1又は2以上の置換基によって置換されていてもよく、或いは
R4及びR5は、それらが結合したNと一緒になって、酸素、硫黄、窒素及びCOから選択される1又は2以上の基をさらに含有していてもよいC3−6−ヘテロシクロアルキル環を形成し、ここで、前記C3−6−ヘテロシクロアルキル環は、飽和又は不飽和であり、A、NR8R9及びR10から選択される1又は2以上の基で置換されていてもよく、
各R6は、C1−6−アルキル、C3−7シクロアルキル、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれが、R10、R11及びAから選択される1又は2以上の置換基によって置換されていてもよく、
各R7は、水素、C1−6−アルキル及びC3−7−シクロアルキルから選択され、ここで、前記C1−6−アルキルは、1又は2以上のハロゲンによって置換されていてもよく、
R8及びR9のそれぞれは、水素及びC1−6−アルキルから独立に選択され、ここで、前記C1−6−アルキル基は、1又は2以上のハロゲンによって置換されていてもよく、或いは
R8及びR9は、それらが結合したNと一緒になって、酸素及び硫黄から選択される1又は2以上のヘテロ原子をさらに含有していてもよいC4−6−ヘテロシクロアルキル環を形成し、ここで、前記C4−6−ヘテロシクロアルキル環は、1又は2以上のR10基によって置換されていてもよく、
各R10は、C3−7−シクロアルキル、アリール、ヘテロアリール、O−ヘテロアリール、アラルキル及びC1−6−アルキルから選択され、そのそれぞれが、1又は2以上のA基によって置換されていてもよく、ここで、R10がC1−6−アルキルであり、2又は3以上のR10基が同じ炭素原子に結合している場合、前記R10基は連結してスピロアルキル基を形成してよく、
各R11は、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル、C1−6−アルキル−ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及びヘテロアリールから独立に選択され、そのそれぞれが、Aから選択される1又は2以上の置換基で置換されていてもよく、
Aは、ハロゲン、−NR4SO2R5、−CN、−OR6、−NR4R5、−NR7R11、ヒドロキシル、−CF3、−CONR4R5、−NR4COR5、−NR7(CO)NR4R5、−NO2、−CO2H、−CO2R6、−SO2R6、−SO2NR4R5、−NR4COR5、−NR4COOR5、C1−6−アルキル、アリール及び−COR6から選択される]。 - R2が、
水素、
ハロゲン、より好ましくは臭素、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいアリール、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいC1−6−アルキル、
1又は2以上のA置換基によって置換されていてもよいC2−6−アルケニル、
C3−7−シクロアルキル、
R11及びAから選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール、
C4−7−ヘテロシクロアルキル、並びに
縮合アリール−C4−7−ヘテロシクロアルキル
から選択される、請求項1に記載の化合物。 - R2が、
−NR4R5、−NR4COR5、−CONR4R5、OR6、ハロゲン、置換されていてもよいC1−6−アルキル、CN、C4−7−ヘテロシクロアルキル及びヘテロアリールから選択される1又は2以上の置換基によって置換されていてもよいアリール、
−NR4COR5、−CONR4R5、−NR4R5、OR6、置換されていてもよいアリール、置換されていてもよいヘテロアリール及びC4−7−ヘテロシクロアルキルから選択される1又は2以上の置換基によって置換されていてもよいC1−6−アルキル、
1又は2以上の−CONR4R5置換基によって置換されていてもよいC2−6−アルケニル、
C3−7−シクロアルキル、より好ましくはシクロプロピル、
−NR4R5、C4−7−ヘテロシクロアルキル、C1−6−アルキル、C3−7−シクロアルキル、C1−6−アルキル−C3−7−シクロアルキル及びOR6から選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール、
C4−7−ヘテロシクロアルキル、並びに
縮合アリール−C4−7−ヘテロシクロアルキル
から選択される、請求項1又は2に記載の化合物。 - R2が、
−NHCO−C1−6−アルキル、−CONHC1−6−アルキル、CO−(N−モルホリニル)、Cl、F、−OC1−6−アルキル、−CONMe2、OCF3、CN、CF3、C1−6−アルキル−(A)、N−モルホリニル及びピラゾリルから選択される1又は2以上の置換基によって置換されていてもよいフェニル基、
ピリジニル、キノリニル、ピラゾイル、フラニル及びピリミジニルから選択され、そのそれぞれが、C1−6−アルキル、アラルキル、OC1−6−アルキル、N−モルホリニルから選択される1又は2以上の置換基によって置換されていてもよいヘテロアリール基、
−CONR4R5、フェニル、ピリジニル及びオキサジアゾリル及びピペリジニルから選択される1又は2以上の置換基によって置換されていてもよく、前記フェニル、ピリジニル及びオキサジアゾリル及びピペリジニル基がそれぞれ、1又は2以上の−NR4COR5、−CONR4R5、COR6、SO2R6又はアリール基によってさらに置換されていてもよいC1−6−アルキル基
から選択される、請求項1〜3のいずれかに記載の化合物。 - R2が、アリール、C1−6−アルキル及びヘテロアリールから選択され、そのそれぞれが、R11及びAから選択される1又は2以上の置換基で置換されていてもよい、請求項1〜4のいずれかに記載の化合物。
- R2が、アリール、C1−6−アルキル及びヘテロアリールから選択され、そのそれぞれは、CONR4R5、CF3、C1−6−アルキル、OR6及びC4−7−ヘテロシクロアルキルから選択される1又は2以上の置換基で置換されていてもよい、請求項1〜5のいずれかに記載の化合物。
- R2が、C1−6−アルキル、フェニル、ピリジニル、ピリミジニル及びピラゾリルから選択され、そのそれぞれが、CONMe2、CF3、イソ−ブチル、イソ−プロピル、OEt及びモルホリニルから選択される1又は2以上の置換基によって置換されていてもよい、請求項1〜6のいずれかに記載の化合物。
- R2が、下記:Me、
から選択される、請求項1〜7のいずれかに記載の化合物。 - R2が、非置換C1−6−アルキル基、より好ましくはメチルである、請求項1〜7のいずれかに記載の化合物。
- R1が、
−NHR3、
アリール、
ヘテロアリール、
C4−7−ヘテロシクロアルキル、
縮合アリール−C4−7−ヘテロシクロアルキル、
−C3−7−シクロアルキル、
−NR3R6、
OR3、
NR4R5、並びに
R11及び基Aから選択される置換基で置換されていてもよい−C1−6アルキルから選択され、
ここで、前記アリール、ヘテロアリール、縮合アリール−C4−7−ヘテロシクロアルキル及びC4−7−ヘテロシクロアルキルは、C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル、アリール及び基Aから選択される1又は2以上の置換基でそれぞれ置換されていてもよく、前記C1−6−アルキル、C3−7−シクロアルキル、ヘテロアリール、C4−7−ヘテロシクロアルキル及びアリール置換基は、同様にR11及び基Aから選択される1又は2以上の基によってそれぞれ置換されていてもよい、
請求項1〜9のいずれかに記載の化合物。 - R1が−NHR3であり、ここで、R3は、C1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びアリールから選択され、そのそれぞれがR11及びAから選択される1又は2以上の置換基によって置換されていてもよい、請求項1〜10のいずれかに記載の化合物。
- R1が−NHR3であり、R3が、
1又は2以上の−OR6、NR4COR5、ヘテロアリール、アリール、C4−7−ヘテロシクロアルキル及びC3−7−シクロアルキル基によって置換されていてもよく、前記アリール及びヘテロアリール基が、それぞれ独立に、CF3、ハロゲン、C1−6−アルキル、−OR6及び−NR4R5から選択される1又は2以上の基によってさらに置換されていてもよいC1−6−アルキル、
−OR6、NR4COR5、−CONR4R5、アリール、−NR4R5、C1−6−アルキル−ヘテロアリール、ヘテロアリール、ハロゲン、−SO2R6、CN、CF3、C1−6−アルキル、−SO2NR4R5、−NR4SO2R5から選択される1又は2以上の置換基によって置換されていてもよく、前記C1−6−アルキル、ヘテロアリール及びアリール基が、それぞれ独立に、CN、CF3、ハロゲン、C1−6−アルキル、−OR6及び−NR4R5から選択される1又は2以上の基によってさらに置換されていてもよいフェニル基、
アリール、C1−6−アルキル及び−NR4R5から選択される1又は2以上の置換基によって置換されていてもよく、前記アリール基が、1又は2以上のA基によってさらに置換されていてもよいヘテロアリール基、
1又は2以上の−COR6基によって置換されていてもよいC4−7−ヘテロシクロアルキル、
1又は2以上のハロゲン又はC1−6−アルキル基によって置換されていてもよいC3−7−シクロアルキル基
から選択される、請求項1〜11のいずれかに記載の化合物。 - R1が−OR3であり、ここで、R3はC1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びアリールから選択され、そのそれぞれが、R11及びAから選択される1又は2以上の置換基によって置換されていてもよい、請求項1〜10のいずれかに記載の化合物。
- R1が−OR3であり、ここで、R3はC1−6−アルキル、C3−7−シクロアルキル又はC4−7−ヘテロシクロアルキルであり、そのそれぞれが、1又は2以上のA置換基によって置換されていてもよい、請求項13に記載の化合物。
- R1が、ヘテロアリール、−NHR3及びOR3から選択され、ここで、前記ヘテロアリール基が、基Aから選択される1又は2以上の置換基で置換されていてもよい、請求項1〜10のいずれかに記載の化合物。
- R1がアリール又はヘテロアリールであり、そのそれぞれが、R11及びAから選択される1又は2以上の置換基によって置換されていてもよく、より好ましくは、R1がフリルである、請求項1〜10のいずれかに記載の化合物。
- R1が−NH−C3−7−シクロアルキル又はNH−C4−7−ヘテロシクロアルキルであり、そのそれぞれが、1又は2以上のA置換基によって置換されていてもよい、請求項1〜10のいずれかに記載の化合物。
- R3がシクロヘキシル又はテトラヒドロピラニルであり、そのそれぞれが、1又は2以上のA置換基によって置換されていてもよい、請求項1〜17のいずれかに記載の化合物。
- R1が、下記:
から選択される、請求項1〜18のいずれかに記載の化合物。 - R1が−OR3又はNHR3であり、R3がシクロヘキシル、Me又はテトラヒドロピラン−4−イルである、請求項1〜15のいずれかに記載の化合物。
- Qが、ハロゲン、CN、C1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びヘテロアリールから選択され、ここで、前記C1−6−アルキル、C3−7−シクロアルキル、C4−7−ヘテロシクロアルキル及びヘテロアリールが、それぞれ独立に、基Aからの1又は2以上の置換基で置換されていてもよい、請求項1〜20のいずれかに記載の化合物。
- Qが、CN、シクロプロピル、CF3、クロロ、メチル、N−モルホリニル及び1−メチルピラゾール−4−イルから選択される、請求項1〜21のいずれかに記載の化合物。
- 下記:
又は薬学的に許容されるその塩から選択される、請求項1〜22のいずれかに記載の化合物。 - 請求項1〜23のいずれかに記載の化合物と、薬学的に許容される担体、賦形剤又は添加剤とを含む医薬組成物。
- 医療において使用するための、請求項1〜23のいずれかに記載の化合物。
- がん及び神経変性疾患から選択される障害を治療するのに使用するための、請求項1〜23のいずれかに記載の化合物。
- がん及び神経変性疾患から選択される障害を治療又は予防するための薬剤の調製における、請求項1〜23のいずれかに記載の化合物の使用。
- 異常なキナーゼ活性、好ましくは異常なLRRK2活性によって引き起こされる、それに関連する、又はそれを伴う障害の予防又は治療のための薬剤の調製における、請求項1〜23のいずれかに記載の化合物の使用。
- 哺乳動物に、治療有効量の請求項1〜23のいずれかに記載の化合物を投与するステップを含む、LRRK2の阻害によって軽減される病状を有する哺乳動物を治療する方法。
- LRRK、より好ましくはLRRK2を阻害することができるさらなる候補化合物を同定するためのアッセイにおける、請求項1〜23のいずれかに記載の化合物の使用。
- 式IIa’の化合物を式Ia’の化合物に変換するステップ:
を含む、式Ia’の化合物[式中、Q’はハロゲン又はC1−6−アルキルであり、R1及びR2は請求項1で定義した通りである]を調製する方法。 - 式IIIa’の化合物をヒドラジン一水和物で処理することによって式IIa’の化合物を調製するステップ:
をさらに含む、請求項31に記載の方法。 - 式IVa’の化合物を酸化剤で処理することによって式IIIa’の化合物を調製するステップ:
をさらに含む、請求項32に記載の方法。 - 式Va’の化合物をR2−Mg−Clで処理することによって式IVa’の化合物を調製するステップ:
をさらに含む、請求項33に記載の方法。 - R1が−NHR3であり、式IIa’の化合物を式NH2R3のアミンと反応させるステップを含む、請求項31〜34のいずれかに記載の方法。
- R1が、NH含有C4−7−ヘテロシクロアルキル又はNH含有縮合アリール−C4−7−ヘテロシクロアルキルであり、式IIa’の化合物を、前記C4−7−ヘテロシクロアルキル又は縮合アリール−C4−7−ヘテロシクロアルキルのNH基と反応させるステップを含む、請求項31〜34のいずれかに記載の方法。
- R1が、アリール、ヘテロアリール、C4−7−ヘテロシクロアルキル、縮合アリール−C4−7−ヘテロシクロアルキル、−C3−7シクロアルキル及び−C1−6アルキルから選択され、式IIa’の化合物を、X−R1[ここで、Xは4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル基である]と、カップリング剤の存在下で反応させるステップを含む、請求項31〜34のいずれかに記載の方法。
- 式VIa”の化合物を式Iの化合物に変換するステップ:
を含む、式Ia”の化合物[式中、Q”は、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール又はヘテロアリールであり、そのそれぞれは、1又は2以上の置換基Aで置換されていてもよく、R1及びR2は請求項1で定義した通りである]を調製する方法。 - 式VIa”の化合物をC4−7−ヘテロシクロアルキルのNH−基と、カップリング剤の存在下で反応させるステップを含む、請求項38に記載の方法。
- 式VIa”の化合物を化合物Q”−Y[ここで、Q”は、C3−7−シクロアルキル、ヘテロシクロアルキル、アリール又はヘテロアリールであり、Yはボロン酸又はボロン酸エステル部分である]と、カップリング剤の存在下で反応させるステップを含む、請求項38に記載の方法。
- 式IIbの化合物を式Ibの化合物に変換するステップ:
を含む、請求項1で定義した通りの式Ibの化合物を調製する方法。 - R1がアリール又はヘテロアリールであり、式IIbの化合物を化合物R1−Y[ここで、Yはボロン酸又はボロン酸エステル部分である]と、カップリング剤の存在下で反応させるステップを含む、請求項41に記載の方法。
- R1が−NHR3であり、式IIbの化合物を式NH2R3のアミンと反応させるステップを含む、請求項41に記載の方法。
- 式IIIbの化合物を式Ibの化合物に変換するステップ:
を含む、請求項1で定義した通りの式Ibの化合物[式中、R1はOR3である]を調製する方法。 - 請求項1〜23のいずれかに記載の化合物及びさらなる治療剤を含む組合せ。
- 第二の治療剤をさらに含む、請求項24に記載の医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1008134.7 | 2010-05-14 | ||
| GBGB1008134.7A GB201008134D0 (en) | 2010-05-14 | 2010-05-14 | Compounds |
| PCT/GB2011/050937 WO2011141756A1 (en) | 2010-05-14 | 2011-05-16 | Pyrazolopyridines as inhibitors of the kinase lrrk2 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013529196A true JP2013529196A (ja) | 2013-07-18 |
| JP2013529196A5 JP2013529196A5 (ja) | 2014-07-03 |
Family
ID=42334821
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013509631A Withdrawn JP2013529196A (ja) | 2010-05-14 | 2011-05-16 | キナーゼlrrk2の阻害剤としてのピラゾロピリジン |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20130267513A1 (ja) |
| EP (1) | EP2569293A1 (ja) |
| JP (1) | JP2013529196A (ja) |
| CN (1) | CN102971306A (ja) |
| AU (1) | AU2011251733A1 (ja) |
| CA (1) | CA2798222A1 (ja) |
| GB (1) | GB201008134D0 (ja) |
| WO (1) | WO2011141756A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016516791A (ja) * | 2013-04-18 | 2016-06-09 | ハー・ルンドベック・アクチエゼルスカベット | Lrrk2阻害剤としてのアリールピロロピリジン誘導化合物 |
| JP2016540794A (ja) * | 2013-12-17 | 2016-12-28 | ファイザー・インク | LRRK2阻害薬としての新規の3,4−二置換−1H−ピロロ[2,3−b]ピリジンおよび4,5−二置換−7H−ピロロ[2,3−c]ピリダジン |
| JP2018524390A (ja) * | 2015-07-23 | 2018-08-30 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | 化合物 |
| KR20190067795A (ko) * | 2016-09-09 | 2019-06-17 | 인사이트 코포레이션 | Hpk1 조절제로서의 피라졸로피리딘 유도체 및 암을 치료하기 위한 이의 용도 |
| JP2020519667A (ja) * | 2017-05-15 | 2020-07-02 | ザ・リージェンツ・オブ・ザ・ユニバーシティ・オブ・ミシガンThe Regents Of The University Of Michigan | LSD−1インヒビターとしてのピロロ〔2,3−c〕ピリジン及び関連類似体 |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012088266A2 (en) | 2010-12-22 | 2012-06-28 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of fgfr3 |
| US8791112B2 (en) | 2011-03-30 | 2014-07-29 | Arrien Pharmaceuticals Llc | Substituted 5-(pyrazin-2-yl)-1H-pyrazolo [3, 4-B] pyridine and pyrazolo [3, 4-B] pyridine derivatives as protein kinase inhibitors |
| GB201204985D0 (en) | 2012-03-21 | 2012-05-02 | Genentech Inc | Compounds |
| US9187484B2 (en) | 2012-05-02 | 2015-11-17 | Southern Research Institute | Triazolopyridazine compounds, use as inhibitors of the kinase LRRK2, and methods for preparation thereof |
| CN102675310B (zh) * | 2012-05-09 | 2014-12-24 | 林辉 | 制备吡唑并芳杂环化合物的方法 |
| PT3176170T (pt) | 2012-06-13 | 2019-02-05 | Incyte Holdings Corp | Compostos tricíclicos substituídos como inibidores de fgfr |
| MD20140130A2 (ro) * | 2012-06-29 | 2015-04-30 | Pfizer Inc. | 4-(amino-substituite)-7H-pirolo[2,3-d]pirimidine noi ca inhibitori de LRRK2 |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| CN104837839A (zh) | 2012-11-08 | 2015-08-12 | 辉瑞公司 | 作为多巴胺d1配体的杂芳族化合物 |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| WO2014134774A1 (en) * | 2013-03-04 | 2014-09-12 | Merck Sharp & Dohme Corp. | Compounds inhibiting leucine-rich repeat kinase enzyme activity |
| WO2014134772A1 (en) | 2013-03-04 | 2014-09-12 | Merck Sharp & Dohme Corp. | Compounds inhibiting leucine-rich repeat kinase enzyme activity |
| WO2014137728A1 (en) | 2013-03-04 | 2014-09-12 | Merck Sharp & Dohme Corp. | Compounds inhibiting leucine-rich repeat kinase enzyme activity |
| WO2014134776A1 (en) | 2013-03-04 | 2014-09-12 | Merck Sharp & Dohme Corp. | Compounds inhibiting leucine-rich repeat kinase enzyme activity |
| EP2970333B1 (en) * | 2013-03-15 | 2017-05-03 | Ipsen Pharma S.A.S. | Macrocyclic lrrk2 kinase inhibitors |
| US20140288043A1 (en) * | 2013-03-19 | 2014-09-25 | Genentech, Inc. | Pyrazolopyridine compounds |
| PE20152033A1 (es) | 2013-04-19 | 2016-01-21 | Incyte Holdings Corp | Heterociclos bicicliclos como inhibidores de fgfr |
| WO2015026683A1 (en) | 2013-08-22 | 2015-02-26 | Merck Sharp & Dohme Corp. | Compounds inhibiting leucine-rich repeat kinase enzyme activity |
| CN103819396B (zh) * | 2014-02-26 | 2016-06-15 | 四川大学 | 一种手性的1-(3,5-二氯吡啶-4-基)-乙醇的合成方法 |
| US9868744B2 (en) | 2014-04-25 | 2018-01-16 | Pfizer Inc. | Heteroaromatic compounds and their use as dopamine D1 ligands |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| EP3617205B1 (en) | 2015-02-20 | 2021-08-04 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| MA41551A (fr) | 2015-02-20 | 2017-12-26 | Incyte Corp | Hétérocycles bicycliques utilisés en tant qu'inhibiteurs de fgfr4 |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| AU2016322813B2 (en) | 2015-09-14 | 2021-04-01 | Pfizer Inc. | Novel imidazo (4,5-c) quinoline and imidazo (4,5-c)(1,5) naphthyridine derivatives as LRRK2 inhibitors |
| WO2017087905A1 (en) | 2015-11-20 | 2017-05-26 | Denali Therapeutics Inc. | Compound, compositions, and methods |
| WO2017106771A1 (en) | 2015-12-16 | 2017-06-22 | Southern Research Institute | Pyrrolopyrimidine compounds, use as inhibitors of the kinase lrrk2, and methods for preparation thereof |
| US11028080B2 (en) | 2016-03-11 | 2021-06-08 | Denali Therapeutics Inc. | Substituted pyrimidines as LRKK2 inhibitors |
| KR20230107407A (ko) | 2016-06-16 | 2023-07-14 | 데날리 테라퓨틱스 인크. | 신경퇴행성 장애의 치료에서 사용하기 위한 lrrk2 저해제로서피리미딘-2-일아미노-1h-피라졸 |
| TW201811799A (zh) | 2016-09-09 | 2018-04-01 | 美商英塞特公司 | 吡唑并嘧啶化合物及其用途 |
| WO2018049191A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridone derivatives as hpk1 modulators and uses thereof for the treatment of cancer |
| WO2018049214A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridine derivatives as hpk1 modulators and uses thereof for the treatment of cancer |
| US10040211B2 (en) | 2016-12-09 | 2018-08-07 | Bettcher Industries, Inc. | Power operated rotary knife |
| US20180228786A1 (en) | 2017-02-15 | 2018-08-16 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| KR101923852B1 (ko) * | 2017-02-24 | 2018-11-29 | 재단법인 대구경북첨단의료산업진흥재단 | 혈액 뇌관문을 통과할 수 있는 화합물을 유효성분으로 함유하는 뇌암의 예방 또는 치료용 약학적 조성물 |
| IL269215B (en) | 2017-03-10 | 2022-09-01 | Pfizer | Disubstituted imidazole[4,5-c]quinoline derivatives |
| MX2019010756A (es) | 2017-03-10 | 2020-01-20 | Pfizer | Derivados novedosos de imidazo[4,5-c]quinolina como inhibidores de cinasa 2 rica en repetición de leucina (lrrk2). |
| AR111960A1 (es) | 2017-05-26 | 2019-09-04 | Incyte Corp | Formas cristalinas de un inhibidor de fgfr y procesos para su preparación |
| WO2019051199A1 (en) | 2017-09-08 | 2019-03-14 | Incyte Corporation | 6-CYANO-INDAZOLE COMPOUNDS AS HEMATOPOIETIC PROGENITOR KINASE 1 (HPK1) MODULATORS |
| LT3755703T (lt) | 2018-02-20 | 2022-10-10 | Incyte Corporation | N-(fenil)-2-(fenil)pirimidin-4-karboksamido dariniai ir susiję junginiai, kaip hpk1 inhibitoriai, skirti vėžio gydymui |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| WO2019164847A1 (en) | 2018-02-20 | 2019-08-29 | Incyte Corporation | Indazole compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| CA3099116A1 (en) | 2018-05-04 | 2019-11-07 | Incyte Corporation | Salts of an fgfr inhibitor |
| CN112867716A (zh) | 2018-05-04 | 2021-05-28 | 因赛特公司 | Fgfr抑制剂的固体形式和其制备方法 |
| ES2738911A1 (es) | 2018-07-25 | 2020-01-27 | Consejo Superior Investigacion | Procedimiento para depositar elementos sobre un sustrato de interes y dispositivo |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| ES2744304B2 (es) | 2018-08-24 | 2020-06-22 | Consejo Superior Investigacion | Compuestos inhibidores de LRRK2 y su uso para el tratamiento de enfermedades neurodegenerativas |
| JP7399968B2 (ja) | 2018-09-25 | 2023-12-18 | インサイト・コーポレイション | Alk2及び/またはfgfr調節剤としてのピラゾロ[4,3-d]ピリミジン化合物 |
| WO2020185532A1 (en) | 2019-03-08 | 2020-09-17 | Incyte Corporation | Methods of treating cancer with an fgfr inhibitor |
| WO2021007269A1 (en) | 2019-07-09 | 2021-01-14 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| JP2022543155A (ja) | 2019-08-06 | 2022-10-07 | インサイト・コーポレイション | Hpk1阻害剤の固体形態 |
| JOP20220083A1 (ar) | 2019-10-14 | 2023-01-30 | Incyte Corp | حلقات غير متجانسة ثنائية الحلقة كمثبطات لـ fgfr |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| CA3163875A1 (en) | 2019-12-04 | 2021-06-10 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| CA3162010A1 (en) | 2019-12-04 | 2021-06-10 | Incyte Corporation | Derivatives of an fgfr inhibitor |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| WO2023076404A1 (en) | 2021-10-27 | 2023-05-04 | Aria Pharmaceuticals, Inc. | Methods for treating systemic lupus erythematosus |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ207394A (en) | 1983-03-08 | 1987-03-06 | Commw Serum Lab Commission | Detecting or determining sequence of amino acids |
| GB9216783D0 (en) * | 1992-08-07 | 1992-09-23 | Ici Plc | Heterocyclic derivatives |
| US5300478A (en) * | 1993-01-28 | 1994-04-05 | Zeneca Limited | Substituted fused pyrazolo compounds |
| WO2002094790A1 (fr) * | 2001-05-23 | 2002-11-28 | Mitsubishi Pharma Corporation | Compose heterocyclique condense et son utilisation medicale |
| PE20061119A1 (es) * | 2005-01-19 | 2006-11-27 | Aventis Pharma Sa | PIRAZOLO PIRIDINAS SUSTITUIDAS COMO INHIBIDORES DE CINASAS FAK, KDR Y Tie |
| BRPI0708507A2 (pt) * | 2006-03-03 | 2011-05-31 | Torrent Pharmaceuticals Ltd | Novos antagonistas de receptores de dupla ação (dara) para os receptores at1 eta |
| BRPI0709699A2 (pt) * | 2006-03-29 | 2011-07-26 | Foldrx Pharmaceuticals Inc | inibiÇço da toxidez da alfa-sinucleina |
| EP1932845A1 (en) * | 2006-12-15 | 2008-06-18 | Bayer Schering Pharma Aktiengesellschaft | 3-H-pyrazolopyridines and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same |
| WO2009030270A1 (en) * | 2007-09-03 | 2009-03-12 | Novartis Ag | Dihydroindole derivatives useful in parkinson's disease |
| CN102428084B (zh) * | 2009-03-19 | 2016-05-18 | 医疗技术研究局 | 化合物 |
| JP5819831B2 (ja) * | 2009-08-17 | 2015-11-24 | インテリカイン, エルエルシー | 複素環式化合物およびそれらの使用 |
-
2010
- 2010-05-14 GB GBGB1008134.7A patent/GB201008134D0/en not_active Ceased
-
2011
- 2011-05-16 WO PCT/GB2011/050937 patent/WO2011141756A1/en active Application Filing
- 2011-05-16 CN CN201180033483XA patent/CN102971306A/zh active Pending
- 2011-05-16 JP JP2013509631A patent/JP2013529196A/ja not_active Withdrawn
- 2011-05-16 EP EP11720847A patent/EP2569293A1/en not_active Withdrawn
- 2011-05-16 AU AU2011251733A patent/AU2011251733A1/en not_active Abandoned
- 2011-05-16 US US13/697,878 patent/US20130267513A1/en not_active Abandoned
- 2011-05-16 CA CA2798222A patent/CA2798222A1/en not_active Abandoned
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016516791A (ja) * | 2013-04-18 | 2016-06-09 | ハー・ルンドベック・アクチエゼルスカベット | Lrrk2阻害剤としてのアリールピロロピリジン誘導化合物 |
| JP2016540794A (ja) * | 2013-12-17 | 2016-12-28 | ファイザー・インク | LRRK2阻害薬としての新規の3,4−二置換−1H−ピロロ[2,3−b]ピリジンおよび4,5−二置換−7H−ピロロ[2,3−c]ピリダジン |
| JP2018524390A (ja) * | 2015-07-23 | 2018-08-30 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | 化合物 |
| KR20190067795A (ko) * | 2016-09-09 | 2019-06-17 | 인사이트 코포레이션 | Hpk1 조절제로서의 피라졸로피리딘 유도체 및 암을 치료하기 위한 이의 용도 |
| JP2019530670A (ja) * | 2016-09-09 | 2019-10-24 | インサイト・コーポレイションIncyte Corporation | Hpk1調節薬としてのピラゾロピリジン誘導体及びがんの治療のためのその用法 |
| JP7076432B2 (ja) | 2016-09-09 | 2022-05-27 | インサイト・コーポレイション | Hpk1調節薬としてのピラゾロピリジン誘導体及びがんの治療のためのその用法 |
| KR102507967B1 (ko) | 2016-09-09 | 2023-03-09 | 인사이트 코포레이션 | Hpk1 조절제로서의 피라졸로피리딘 유도체 및 암을 치료하기 위한 이의 용도 |
| JP2020519667A (ja) * | 2017-05-15 | 2020-07-02 | ザ・リージェンツ・オブ・ザ・ユニバーシティ・オブ・ミシガンThe Regents Of The University Of Michigan | LSD−1インヒビターとしてのピロロ〔2,3−c〕ピリジン及び関連類似体 |
| JP7352284B2 (ja) | 2017-05-15 | 2023-09-28 | ザ・リージェンツ・オブ・ザ・ユニバーシティ・オブ・ミシガン | LSD-1インヒビターとしてのピロロ〔2,3-c〕ピリジン及び関連類似体 |
Also Published As
| Publication number | Publication date |
|---|---|
| GB201008134D0 (en) | 2010-06-30 |
| AU2011251733A1 (en) | 2012-12-20 |
| CN102971306A (zh) | 2013-03-13 |
| CA2798222A1 (en) | 2011-11-17 |
| US20130267513A1 (en) | 2013-10-10 |
| EP2569293A1 (en) | 2013-03-20 |
| WO2011141756A1 (en) | 2011-11-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2013529196A (ja) | キナーゼlrrk2の阻害剤としてのピラゾロピリジン | |
| JP5732036B2 (ja) | 化合物 | |
| US8569281B2 (en) | Compounds and their administration for treating a neurodegenerative disease as well as a method for identifying a compound capable of inhibiting a kinase, such as LRRK | |
| RU2745035C1 (ru) | Ингибитор fgfr и его применение | |
| US7723347B2 (en) | Substituted phenylamino-pyrimidines | |
| JP6175484B2 (ja) | 新規Syk阻害剤としての置換ピリドピラジン | |
| JP6096879B2 (ja) | 二環式ピラジノン誘導体 | |
| WO2013139882A1 (en) | Pyrazolopyridines for treatment of parkinsons disease | |
| MX2010014005A (es) | Compuestos de triazolopiridina inhibidores de jak y los metodos. | |
| JP2016520118A (ja) | Bet阻害剤としてのピラゾロ−ピロリジン−4−オン誘導体および疾患の処置におけるその使用 | |
| JP6283688B2 (ja) | カゼインキナーゼ1d/e阻害剤としての新規なピラゾール置換のイミダゾピラジン | |
| JPWO2015022926A1 (ja) | 新規な縮合ピリミジン化合物又はその塩 | |
| JP7247092B2 (ja) | キナーゼ阻害剤としての置換された縮合ヘテロアリール化合物及びその用途 | |
| JP2016520119A (ja) | ピラゾロ−ピロリジン−4−オン誘導体および疾患の処置におけるその使用 | |
| JP6946290B2 (ja) | Mnk阻害剤としてのピロロピリミジン化合物 | |
| JP2021518379A (ja) | Jak阻害剤 | |
| JP6885962B2 (ja) | 1,4−ジカルボニル−ピペリジル誘導体 | |
| US9550796B2 (en) | Pyrrolopyrrolone derivatives and their use as BET inhibitors | |
| CN104822658B (zh) | 作为多种激酶抑制剂的稠合三环酰胺类化合物 | |
| JP6847942B2 (ja) | 二環式複素環式誘導体 | |
| US20140288043A1 (en) | Pyrazolopyridine compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140514 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140514 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20140709 |