JP2011132500A - 樹脂粒子 - Google Patents
樹脂粒子 Download PDFInfo
- Publication number
- JP2011132500A JP2011132500A JP2010247246A JP2010247246A JP2011132500A JP 2011132500 A JP2011132500 A JP 2011132500A JP 2010247246 A JP2010247246 A JP 2010247246A JP 2010247246 A JP2010247246 A JP 2010247246A JP 2011132500 A JP2011132500 A JP 2011132500A
- Authority
- JP
- Japan
- Prior art keywords
- resin
- resin particles
- acid
- aqueous dispersion
- particles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Processes Of Treating Macromolecular Substances (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polyesters Or Polycarbonates (AREA)
- Polyurethanes Or Polyureas (AREA)
Abstract
【解決手段】3価又は4価の芳香族カルボン酸(c)と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)及びポリアミン(b)を構成単位として有する樹脂(A)を含有することを特徴とする樹脂粒子(R)並びに上記樹脂粒子(R)の表面に樹脂(C)を含有する樹脂粒子(S)が付着されてなる構造の樹脂粒子(T)。
【選択図】なし
Description
しかしながら、熱定着方式・熱加工方式に用いられる樹脂粒子では、低温溶融性と耐ブロッキング性の両立が求められており、特許文献1に記載の樹脂粒子を、スラッシュ成型用樹脂、粉体塗料、電子写真トナー用母体粒子、静電記録トナー用母体粒子、静電印刷トナー用母体粒子又はホットメルト接着剤として使用した場合には、低温溶融性と耐ブロッキング性が両立できるという効果が十分ではなかった。
(I):3価又は4価の芳香族カルボン酸(c)と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)及びポリアミン(b)を構成単位として有する樹脂(A)を含有することを特徴とする樹脂粒子(R)。
(II):(I)の樹脂粒子(R)の表面に樹脂(C)を含有する樹脂粒子(S)が付着されてなる構造の樹脂粒子(T)。
(III):樹脂(C)を含有する樹脂粒子(S)の水性分散液(W)と、樹脂(A)及び/又はその前駆体、並びに必要により有機溶剤を含有する油性液(OL)とを混合し、(W)中に(OL)を分散させ、(A)の前駆体を用いる場合は(W)中で前駆体を反応させて、(A)を含有する樹脂粒子(R)を形成させることにより、(R)の表面に(S)が付着された構造の樹脂粒子(T)の水性分散体(X1)を得る工程を含み、樹脂(A)が、3価又は4価の芳香族カルボン酸と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)及びポリアミン(b)を構成単位として有する樹脂であることを特徴とする水性分散体(X1)の製造方法。
(IV):(III)で得られた水性分散体(X1)から水性溶剤を除去して樹脂粒子(T)を得る工程を含む樹脂粒子の製造方法。
(V):(III)で得られた水性分散体(X1)中において、付着している樹脂粒子(S)を樹脂粒子(R)から脱離させた後、水性分散体から(S)を分離除去して(R)の水性分散体(X2)を得る工程、又は(S)を溶解させ、必要により(S)の溶解物を分離除去して(R)の水性分散体(X2)を得る工程を含む水性分散体(X2)の製造方法。
(VI):(V)の製造方法により得られた水性分散体(X2)から水性溶剤を除去して樹脂粒子(R)を得る工程を含む樹脂粒子の製造方法。
(VII):樹脂(C)を含有する樹脂粒子(S)が、樹脂粒子(R)の表面に付着さ
れてなる構造の樹脂粒子(T)であり、
[1][(S)の体積平均粒径/(T)の体積平均粒径]が0.001〜0.3であり、
[2](S)の体積平均粒径が0.0005〜30μm、かつ(T)の体積平均粒径が0.1〜300μmであり、
[3](R)の表面の5%以上が(S)で覆われており、
[4](T)の[体積平均粒径/個数平均粒径]が1.0〜1.5であり、
[5](R)が樹脂(A)及び必要により架橋構造を有さない樹脂(B)を含有し、樹脂(A)が3価又は4価の芳香族カルボン酸と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)及びポリアミン(b)を構成単位として有する樹脂である樹脂粒子。
1.低温溶融性及び耐ブロッキング性のいずれにも優れた樹脂粒子を得ることができる。従って、例えば本発明の製造方法によって得られる本発明の樹脂粒子をトナーの母体粒子として用いた場合、低温定着性及び耐熱保存安定性に優れる。
2.粒径が均一な樹脂粒子分散体及び樹脂粒子を安定的に製造できる。
3.水性分散液中で樹脂粒子が得られるため、安全かつ低コストで樹脂粒子を製造できる。
本発明の樹脂粒子(R)が含有する樹脂(A)は、3価又は4価の芳香族カルボン酸(c)と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)を必須構成単位として含有する。
3価又は4価の芳香族カルボン酸(c)としては、1,2,3−ベンゼントリカルボン酸、1,2,4−ベンゼントリカルボン酸、1,3,5−ベンゼントリカルボン酸、1,2,3,4−ベンゼンテトラカルボン酸、1,2,3,5−ベンゼンテトラカルボン酸及び1,2,4,5−ベンゼンテトラカルボン酸等が挙げられる。
これらのうち、低温溶融性及び耐ブロッキング性の観点から、好ましいのは1,2,4−ベンゼントリカルボン酸及び1,2,4,5−ベンゼンテトラカルボン酸であり、更に好ましいのは1,2,4−ベンゼントリカルボン酸である。
(d1)のうち、3価又は4価の芳香族カルボン酸(c)との反応性の観点から、好ましいのは分子末端に1級水酸基を有する分岐のない脂肪族ジオール(エチレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール、1,9−ノナンジオール及び1,10−デカンジオール等)であり、更に好ましいのは、エチレングリコール、1,3−プロピレングリコール及び1,4−ブタンジオールであり、特に好ましいのは1,3−プロピレングリコール及びエチレングリコールである。
HO−R1−N−R2−OH
| (1)
R3
式中、R1及びR2は、それぞれ独立に炭素数が2〜10の脂肪族アルキレン基、R3は、水素原子又は炭素数1〜3のアルキル基である。
炭素数2〜10のアルキレン基としては、エチレン基、トリメチレン基、メチルエチレン基、テトラメチレン基、ペンタメチレン基、2,2−ジメチル−トリメチレン基、ヘキサメチレン基、ヘプタメチレン基、オクタメチレン基、ノナメチレン基及びデカメチレン基等が挙げられる。R1及びR2のうち、1,2,4−ベンゼントリカルボン酸(c)との反応性の観点から好ましいのは、エチレン基、トリメチレン基、メチルエチレン基及びテトラメチレン基であり、更に好ましいのはエチレン基及び/又はトリメチレン基である。
炭素数1〜3のアルキル基としては、メチル基、エチル基及びn−又はイソプロピル基である。
R3のうち、1,2,4−ベンゼントリカルボン酸(c)との反応性の観点から好ましいのは、水素原子及びメチル基である。
ジアルカノールアミン化合物(d2)の具体例としては、ジエタノールアミン、N−メチルジエタノールアミン、N−エチルジエタノールアミン、N−n−プロピルジエタノールアミン、1−ヒドロキシエチル−1−ヒドロキシ−n−プロピルアミン、ジ(1−ヒドロキシ−n−プロピル)アミン、ジ(1−ヒドロキシ−n−ヘキシル)−N−メチルアミン、ジ(1−ヒドロキシ−n−プロピル)アミン、ジ(1−ヒドロキシ−n−ヘキシル)アミン、ジ(1−ヒドロキシ−n−オクチル)アミン及びジ(1−ヒドロキシ−n−デシル)アミン等が挙げられ、これらの2種以上を併用してもよい。
(d2)のうち、1,2,4−ベンゼントリカルボン酸(c)との反応性の観点から好ましいのは、ジエタノールアミン及びN−メチルジエタノールアミンである。
エステル(e)を構成する4価の芳香族カルボン酸(c)と、ジアルカノールアミン化合物(d2)とのモル比は、耐ホットオフセット性の観点から、好ましくは(c)1モルに対して(d2)が1〜4モルであり、更に好ましくは1.2〜4モル、特に好ましくは2〜4モルである。
装置(一例) :「HLC−8120」[東ソー(株)製]
カラム(一例) :「TSK GEL GMH6」2本[東ソー(株)製]
測定温度 :40℃
試料溶液 :0.25重量%のTHF(テトラヒドロフラン)溶液
溶液注入量 :100μl
検出装置 :屈折率検出器
基準物質 :標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500、1,050、2,800、5,970、9,100、18,100、37,900、96,400、190,000、355,000、1,090,000、2,890,000)[東ソー(株)製]
なお、分子量の測定には、試料をTHFに溶解し、不溶解分をグラスフィルターでろ別したものを試料溶液とし用いる。
(e)の水酸基価は、ポリイソシアネート(i)との反応率の観点から、好ましくは10〜1,000(mgKOH/g、以下同じ)であり、更に好ましくは50〜800、特に好ましくは100〜700である。
なお、試料に架橋に伴う溶剤不溶解分がある場合は、以下の方法で溶融混練後のものを試料として用いる。
混練装置:ラボプラストミル「MODEL4M150」[東洋精機(株)製]
混練条件:130℃、70rpmで30分間
芳香族ポリエーテルポリオールとしては、ビスフェノール骨格を有するポリオール(例えばビスフェノールAのエチレオキサイド(以下、EOと略記する)及び/又はプロピレンオキサイド(以下、POと略記する)2〜20モル付加物)、並びにレゾルシンのEO及び/又はPO付加物等が挙げられる。
ポリエーテルポリオールは、脂肪族又は芳香族活性水素原子含有化合物に、付加触媒(アルカリ金属水酸化物及びルイス酸等の公知の触媒)の存在下にアルキレンオキサイド(以下、AOと略記する)を開環付加反応させることで得られる。
縮合型ポリエステルとしては、低分子量(Mn300以下)の多価アルコールと多価カルボン酸又はそのエステル形成性誘導体とのポリエステルが挙げられる。
低分子量の多価アルコールとしては、水酸基当量が30以上150未満の2〜8価又はそれ以上の脂肪族多価アルコール及び水酸基当量が30以上150未満の2〜8価又はそれ以上のフェノールのAO低モル付加物等が挙げられる。
縮合型ポリエステルを構成する低分子量の多価アルコールのうち、好ましいのは、エチレングリコール、プロピレングリコール、1,4−ブタンジオール、ネオペンチルグリコール、1,6−ヘキサングリコール、ビスフェノールAのEO及び/又はPO低モル付加物並びにこれらの併用である。
縮合型ポリエステルを構成する多価カルボン酸又はそのエステル形成性誘導体としては、脂肪族ジカルボン酸(コハク酸、アジピン酸、アゼライン酸、セバチン酸、フマル酸及びマレイン酸等)、脂環式ジカルボン酸(ダイマー酸等)、芳香族ジカルボン酸(テレフタル酸、イソフタル酸及びフタル酸等)、3価又はそれ以上のポリカルボン酸(1,2,4−ベンゼントリカルボン酸及び1,2,4,5−ベンゼンテトラカルボン酸等)、これらの無水物(無水コハク酸、無水マレイン酸、無水フタル酸及び無水1,2,4−ベンゼントリカルボン酸等)、これらの酸ハロゲン化物(アジピン酸ジクロライド等)、これらの低分子量アルキルエステル(コハク酸ジメチル及びフタル酸ジメチル等)及びこれらの併用が挙げられる。
ポリラクトンポリオールとしては、具体的にはポリカプロラクトンジオール、ポリバレロラクトンジオール及びポリカプロラクトントリオール等が挙げられる。
ポリカーボネートポリオールとしては、ポリヘキサメチレンカーボネートジオール等が挙げられる。
アルキルポリオールとしては、上記の低分子量の脂肪族多価アルコール等が挙げられる。
[1]脂肪族ジアミン{アルキレン(炭素数2〜6)ジアミン(エチレンジアミン、プロピレンジアミン、トリメチレンジアミン、テトラメチレンジアミン及びヘキサメチレンジアミン等)、ポリアルキレン(炭素数2〜6)ジアミン[ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン、トリエチレンテトラミン、テトラエチレンペンタミン及びペンタエチレンヘキサミン等]};
[2]脂肪族ジアミンのアルキル(炭素数1〜4)又はヒドロキシアルキル(炭素数2〜4)置換体[ジアルキル(炭素数1〜3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン及びメチルイミノビスプロピルアミン等];
[3]脂環又は複素環含有脂肪族ジアミン{脂環式ジアミン(炭素数4〜15)[1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン及び4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)等]、複素環式ジアミン(炭素数4〜15)[ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン及び3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカン等]};
[4]芳香環含有脂肪族アミン類(炭素数8〜15)(キシリレンジアミン及びテトラクロロp−キシリレンジアミン等);等が挙げられる。
[1]非置換芳香族ジアミン[1,2−、1,3−又は1,4−フェニレンジアミン、2,4´−及び4,4´−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4´,4”−トリアミン及びナフチレンジアミン等;
[2]核置換アルキル基(メチル基、エチル基、プロピル基及びブチル基等の炭素数1〜4のアルキル基)を有する芳香族ジアミン[例えば2,4−又は2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、4,4´−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン及び4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン等]及びこれらの異性体の種々の割合の混合物;
[3]核置換電子吸引基(フッ素、塩素、臭素及びヨウ素等のハロゲン原子;メトキシ基及びエトキシ基等のアルコキシ基;ニトロ基等)を有する芳香族ジアミン(メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロロ−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロロ−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン及び3−ジメトキシ−4−アミノアニリン等];
[4]2級アミノ基を有する芳香族ジアミン[上記[1]〜[3]の芳香族ジアミンの−NH2の一部又は全部が−NH−R´(R´はアルキル基;例えばメチル基及びエチル基等の低級アルキル基)で置換されたもの][4,4´−ジ(メチルアミノ)ジフェニルメタン及び1−メチル−2−メチルアミノ−4−アミノベンゼン等]が挙げられる。
上記モル比率は、樹脂(A)を製造する際に使用した、ポリイソシアネート(i)と、ポリアミン(b)の重量から、(A)中に含有されるウレタン基(―NHCOO―)のモル数とウレア基(―NHCONH―)のモル数の比を、計算により求めたものである。
なお、エステル(e)をジカルボン酸(好ましくは直鎖脂肪族ジカルボン酸)と反応させて得られる重縮合ポリエステルポリオールをポリイソシアネート(i)と反応させてプレポリマー(A0)を得てもよい。
[NCO]/[OH]が5以下であると低温溶融性が向上する。[NCO]/[OH]が1以上であれば、高温高湿下での耐ブロッキング性が向上する。
樹脂(A)のMwは、好ましくは1万以上であり、更に好ましくは2万〜1,000万、特に好ましくは3万〜100万である。
樹脂(B)としては、ポリエステル樹脂、ポリウレタン樹脂、ビニル樹脂、エポキシ樹脂、ポリアミド樹脂、ポリイミド樹脂、ケイ素樹脂、フェノール樹脂、メラミン樹脂、ユリア樹脂、アニリン樹脂、アイオノマー樹脂及びポリカーボネート樹脂等の、樹脂(A)以外の樹脂が挙げられる。
これらのうち好ましいのは、ポリエステル樹脂、ポリウレタン樹脂、ビニル樹脂及びエポキシ樹脂であり、更に好ましいのはポリエステル樹脂及びポリウレタン樹脂であり、特に好ましいのはポリエステル樹脂である。
ジオールとしては、炭素数2〜36のアルキレングリコール(エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール及びネオペンチルグリコール等);炭素数4〜36のアルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール及びポリテトラメチレンエーテルグリコール等);炭素数6〜36の脂環式ジオール(1,4−シクロヘキサンジメタノール及び水素添加ビスフェノールA等);上記脂環式ジオールの(ポリ)オキシアルキレン(アルキレン基の炭素数2〜4、以下のポリオキシアルキレン基も同じ)エーテル[オキシアルキレン(以下、OAと略記する)単位の数1〜30];ビスフェノールAのEO付加物、2価フェノール[単環2価フェノール(例えばハイドロキノン)及びビスフェノール類(ビスフェノールA、ビスフェノールF及びビスフェノールS等)]のポリオキシアルキレンエーテル等が挙げられる。
ジカルボン酸としては、炭素数4〜36のアルカンジカルボン酸(コハク酸、アジピン酸、セバシン酸、アゼライン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸及びデシルコハク酸等);炭素数6〜40の脂環式ジカルボン酸[ダイマー酸(2量化リノール酸)等]、炭素数4〜36のアルケンジカルボン酸[アルケニルコハク酸(ドデセニルコハク酸、ペンタデセニルコハク酸及びオクタデセニルコハク酸等)、マレイン酸、フマール酸及びシトラコン酸等];炭素数8〜36の芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、t−ブチルイソフタル酸、2,6−ナフタレンジカルボン酸及び4,4’−ビフェニルジカルボン酸等);及びこれらの無水物又は低級アルキル(炭素数1〜4)若しくはヒドロキシアルキル(炭素数1〜4)エステル[(無水)フタル酸、テレフタル酸ジメチル及びテレフタル酸1,2−プロピレングリコールジエステル等]等が挙げられる。これらは、単独で又は2種以上を併用して用いることができる。
重縮合反応は、脂肪族ジオール成分の一部又はポリカルボン酸の低級アルキルエステルに由来する炭素数1〜4のアルコールを系外に除去させながら行ってもよい。
更に反応末期の反応速度を向上させるために、反応系内を減圧にすることも有効である。
上記重縮合反応には、エステル化触媒を使用してもよい。エステル化触媒としては前記のものが挙げられる。
エステル化触媒の添加量としては、全原料に対し、好ましくは10ppm〜1.9重量%であり、更に好ましくは100ppm〜1.7重量%である。添加量を10ppm以上にすることで反応速度が大きくなる点で好ましい。
ポリチオールとしては、エチレンジチオール、1,4−ブタンジチオール及び1,6−ヘキサンジチオール等が挙げられる。重付加反応には、公知の重合触媒等が使用できる。
ビニルモノマーとしては、下記(1)〜(10)等が挙げられる。
(1−1)脂肪族ビニル炭化水素:炭素数2〜12のアルケン(例えばエチレン、プロピレン、ブテン、イソブチレン、ペンテン、ヘプテン、ジイソブチレン、オクテン、ドデセン、オクタデセン及び炭素数3〜24のα−オレフィン等);炭素数4〜12のアルカジエン(例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン及び1,7−オクタジエン等)。
(1−2)脂環式ビニル炭化水素:炭素数6〜15のモノ−又はジ−シクロアルケン(例えばシクロヘキセン、ビニルシクロヘキセン及びエチリデンビシクロヘプテン等)、炭素数5〜12のモノ−又はジ−シクロアルカジエン[例えば、(ジ)シクロペンタジエン等];及びテルペン(例えばピネン、リモネン及びインデン等)等。
(1−3)芳香族ビニル炭化水素:スチレン;スチレンの炭化水素(炭素数1〜24のアルキル、シクロアルキル、アラルキル及び/又はアルケニル)置換体(例えばα−メチルスチレン、ビニルトルエン、2,4−ジメチルスチレン、エチルスチレン、イソプロピルスチレン、ブチルスチレン、フェニルスチレン、シクロヘキシルスチレン、ベンジルスチレン、クロチルベンゼン、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン及びトリビニルベンゼン等);及びビニルナフタレン等。
炭素数3〜30の不飽和モノカルボン酸[例えば(メタ)アクリル酸(アクリル酸及び/又はメタクリル酸を表す。以下同様。)、クロトン酸イソクロトン酸及び桂皮酸等];炭素数3〜30の不飽和ジカルボン酸(無水物)[例えば(無水)マレイン酸、フマル酸、イタコン酸、(無水)シトラコン酸及びメサコン酸等];及び炭素数3〜30の不飽和ジカルボン酸のモノアルキル(炭素数1〜24)エステル(例えばマレイン酸モノメチルエステル、マレイン酸モノオクタデシルエステル、フマル酸モノエチルエステル、イタコン酸モノブチルエステル、イタコン酸グリコールモノエーテル及びシトラコン酸モノエイコシルエステル等)等。
カルボキシル基含有ビニルモノマーの塩としては、例えばアルカリ金属塩(ナトリウム塩及びカリウム塩等)、アルカリ土類金属塩(カルシウム塩及びマグネシウム塩等)、アンモニウム塩、アミン塩及び4級アンモニウム塩等が挙げられる。アミン塩としては、アミン化合物であれば特に限定されないが、例えば1級アミン塩(エチルアミン塩、ブチルアミン塩及びオクチルアミン塩等)、2級アミン塩(ジエチルアミン塩及びジブチルアミン塩等)、3級アミン塩(トリエチルアミン塩及びトリブチルアミン塩等)が挙げられる。4級アンモニウム塩としては、テトラエチルアンモニウム塩、トリエチルラウリルアンモニウム塩、テトラブチルアンモニウム塩及びトリブチルラウリルアンモニウム塩等)が挙げられる。
カルボキシル基含有ビニルモノマーの塩の具体例としては、アクリル酸ナトリウム、メタクリル酸ナトリウム、マレイン酸モノナトリウム、マレイン酸ジナトリウム、アクリル酸カリウム、メタクリル酸カリウム、マレイン酸モノカリウム、アクリル酸リチウム、アクリル酸セシウム、アクリル酸アンモニウム、アクリル酸カルシウム及びアクリル酸アルミニウム等が挙げられる。
炭素数2〜14のアルケンスルホン酸(例えばビニルスルホン酸、(メタ)アリルスルホン酸及びメチルビニルスルホン酸等);スチレンスルホン酸及びこのアルキル(炭素数2〜24)誘導体(例えばα−メチルスチレンスルホン酸等);炭素数5〜18のスルホ(ヒドロキシ)アルキル−(メタ)アクリレート[例えばスルホプロピル(メタ)アクリレート、2−ヒドロキシ−3−(メタ)アクリロキシプロピルスルホン酸、2−(メタ)アクリロイルオキシエタンスルホン酸及び3−(メタ)アクリロイルオキシ−2−ヒドロキシプロパンスルホン酸等];炭素数5〜18のスルホ(ヒドロキシ)アルキル(メタ)アクリルアミド[例えば2−(メタ)アクリロイルアミノ−2,2−ジメチルエタンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸及び3−(メタ)アクリルアミド−2−ヒドロキシプロパンスルホン酸等];アルキル(炭素数3〜18)アリルスルホコハク酸(例えばプロピルアリルスルホコハク酸、ブチルアリルスルホコハク酸及び2−エチルヘキシル−アリルスルホコハク酸等);ポリ[n(重合度を表す。以下同様。)=2〜30]キシアルキレン(オキシエチレン、オキシプロピレン及びオキシブチレン等:単独、ランダム又はブロックでもよい)モノ(メタ)アクリレートの硫酸エステル[例えばポリ(n=5〜15)オキシエチレンモノメタクリレート硫酸エステル及びポリ(n=5〜15)オキシプロピレンモノメタクリレート硫酸エステル等];下記一般式(2−1)〜(2−3)で表される化合物;及びこれらの塩等が挙げられる。
なお、塩としては、(6)カルボキシル基含有ビニルモノマーの塩と同様の塩等が用いられる。
|
CH2=CHCH2OCH2CHCH2O−Ar−R (2−1)
CH=CH−CH3
|
R’−Ar−O−(AO)nSO3H (2−2)
CH2COOR”
|
HO3SCHCOOCH2CH(OH)CH2OCH2CH=CH2 (2−3)
式中、R及びR’は、それぞれ独立に炭素数1〜15のアルキル基;AOは炭素数2〜
4のオキシアルキレン基であり、AOは単独でも2種以上を併用したものでもよく、2種以上を併用した場合は、結合形式はランダムでもブロックでもよい;m及びnは、それぞれ独立に1〜50の数;Arはベンゼン環;R”はフッ素原子で置換されていてもよい炭素数1〜15のアルキル基を表す。
(メタ)アクリロイルオキシアルキルリン酸モノエステル(アルキル基の炭素数1〜24)[例えば2−ヒドロキシエチル(メタ)アクリロイルホスフェート及びフェニル−2−アクリロイロキシエチルホスフェート等]、(メタ)アクリロイルオキシアルキルホスホン酸(アルキル基の炭素数1〜24)(例えば2−アクリロイルオキシエチルホスホン酸等)。
なお、塩としては、(2)カルボキシル基含有ビニルモノマーの塩と同様の塩等が挙げられる。
ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,
4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル及びショ糖アリルエーテル等。
(6−1)アミノ基含有ビニルモノマー:
アミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、t−ブチルアミノエチルメタクリレート、N−アミノエチル(メタ)アクリルアミド、(メタ)アリルアミン、モルホリノエチル(メタ)アクリレート、4−ビニルピリジン、2−ビニルピリジン、クロチルアミン、N,N−ジメチルアミノスチレン、メチル−α−アセトアミノアクリレート、ビニルイミダゾール、N−ビニルピロール、N−ビニルチオピロリドン、N−アリールフェニレンジアミン、アミノカルバゾール、アミノチアゾール、アミノインドール、アミノピロール、アミノイミダゾール、アミノメルカプトチアゾール及びこれらの塩等。
(6−2)アミド基(カルバモイル基)含有ビニルモノマー:
(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド、N−ブチルアクリルアミド、ジアセトンアクリルアミド、N−メチロール(メタ)アクリルアミド、N,N’−メチレン−ビス(メタ)アクリルアミド、桂皮酸アミド、N,N−ジメチルアクリルアミド、N,N−ジベンジルアクリルアミド、メタクリルホルムアミド、N−メチルN−ビニルアセトアミド及びN−ビニルピロリドン等。
(6−3)炭素数3〜10のニトリル基(シアノ基)含有ビニルモノマー:
(メタ)アクリロニトリル、シアノスチレン及びシアノアクリレート等。
(6−4)4級アンモニウムカチオンを有する基(第4級アンモニオ基)を含有するビニルモノマー:
トリメチルアンモニオエチル(メタ)アクリレートクロライド、メチルジエチルアンモニオエチル(メタ)アクリレートブロマイド、トリメチルアンモニオエチル(メタ)アクリルアミドメトサルフェート、ベンジルジエチルアンモニオエチル(メタ)アクリルアミドカーボネート、ジメチルジアリルアンモニウムクロライド及びトリメチルアリルアンモニウムクロライド等。
(6−5)炭素数8〜12のニトロ基含有ビニルモノマー:
ニトロスチレン等。
グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート及びp−ビニルフェニルフェニルオキサイド等。
塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロロスチレン、ブロモスチレン、ジクロロスチレン、クロロメチルスチレン、テトラフルオロスチレン及びクロロプレン等。
(9−1)炭素数4〜16のビニルエステル:
酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル−4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチル−α−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート及びエイコシル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の直鎖、分枝又は脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の直鎖、分枝又は脂環式の基である)、ポリ(メタ)アリロキシアルカン[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン及びテトラメタアリロキシエタン等]、ポリアルキレングリコール鎖を有するビニルモノマー[ポリエチレングリコール(Mn=300)モノ(メタ)アクリレート、ポリプロピレングリコール(Mn=500)モノアクリレート、メチルアルコールのEO10モル付加物(メタ)アクリレート及びラウリルアルコールのEO30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート[多価アルコールのポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート及びポリエチレングリコールジ(メタ)アクリレート等]等。
(9−2)炭素数3〜16のビニル(チオ)エーテル:
ビニルメチルエーテル、ビニルエチルエーテル、ビニルプロピルエーテル、ビニルブチルエーテル、ビニル2−エチルヘキシルエーテル、ビニルフェニルエーテル、ビニル2−メトキシエチルエーテル、メトキシブタジエン、ビニル2−ブトキシエチルエーテル、3,4−ジヒドロ−1,2−ピラン、2−ブトキシ−2’−ビニロキシジエチルエーテル、ビニル−2−エチルメルカプトエチルエーテル、アセトキシスチレン及びフェノキシスチレン等。
(9−3)炭素数4〜12のビニルケトン:
ビニルメチルケトン、ビニルエチルケトン及びビニルフェニルケトン等。
(9−4)炭素数2〜16のビニルスルホン:
ジビニルサルファイド、p−ビニルジフェニルサルファイド、ビニルエチルサルファイド、ビニルエチルスルホン、ジビニルスルホン及びジビニルスルホキサイド等。
イソシアナトエチル(メタ)アクリレート及びm−イソプロペニル−α,α−ジメチルベンジルイソシアネート等。
芳香族ポリエポキシドとしては、多価フェノールのグリシジルエーテル、多価フェノールグリシジルエステル、グリシジル芳香族ポリアミン及びアミノフェノールのグリシジル化物等が挙げられる。
多価フェノールのグリシジルエーテルとしては、ビスフェノールFジグリシジルエーテル、ビスフェノールAジグリシジルエーテル、ビスフェノールBジグリシジルエーテル、ビスフェノールADジグリシジルエーテル、ビスフェノールSジグリシジルエーテル、ハロゲン化ビスフェノールAジグリシジルエーテル、テトラクロロビスフェノールAジグリシジルエーテル、カテキンジグリシジルエーテル、レゾルシノールジグリシジルエーテル、ハイドロキノンジグリシジルエーテル、ピロガロールトリグリシジルエーテル、1,5−ジヒドロキシナフタリンジグリシジルエーテル、ジヒドロキシビフェニルジグリシジルエーテル、オクタクロロ−4,4’−ジヒドロキシビフェニルジグリシジルエーテル、テトラメチルビフェニルジグリシジルエーテル、ジヒドロキシナフチルクレゾールトリグリシジルエーテル、トリス(ヒドロキシフェニル)メタントリグリシジルエーテル、ジナフチルトリオールトリグリシジルエーテル、テトラキス(4−ヒドロキシフェニル)エタンテトラグリシジルエーテル、p−グリシジルフェニルジメチルトリールビスフェノールAグリシジルエーテル、トリスメチル−t−ブチル−ブチルヒドロキシメタントリグリシジルエーテル、9,9’−ビス(4−ヒドキシフェニル)フロオレンジグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)テトラクレゾールグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)フェニルグリシジルエーテル、ビス(ジヒドロキシナフタレン)テトラグリシジルエーテル、フェノール又はクレゾールノボラック樹脂のグリシジルエーテル、リモネンフェノールノボラック樹脂のグリシジルエーテル、ビスフェノールA2モルとエピクロロヒドリン3モルの反応から得られるジグリシジルエーテル、フェノールとグリオキザール、グルタールアルデヒド又はホルムアルデヒドの縮合反応によって得られるポリフェノールのポリグリシジルエーテル及びレゾルシンとアセトンの縮合反応によって得られるポリフェノールのポリグリシジルエーテル等が挙げられる。
グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン及びN,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。
アミノフェノールのグリシジル化物としては、P−アミノフェノールのトリグリシジルエーテル等が挙げられる。
芳香族ポリエポキシ化合物には、トリレンジイソシアネート又はジフェニルメタンジイソシアネートとグリシドールとの付加反応によって得られるジグリシジルウレタン化合物、トリレンジイソシアネート又はジフェニルメタンジイソシアネートとグリシドールとポリオールとを反応させて得られるグリシジル基含有ポリウレタン(プレ)ポリマー及びビスフェノールAのAO付加物のジグリシジルエーテルも含まれる。
脂環族ポリエポキシドとしては、ビニルシクロヘキセンジオキサイド、リモネンジオキサイド、ジシクロペンタジエンジオキサイド、ビス(2,3−エポキシシクロペンチル)エーテル、エチレングリコールビスエポキシジシクロペンチルエーテル、3,4−エポキシ−6−メチルシクロヘキシルメチル−3’,4’−エポキシ−6’−メチルシクロヘキサンカルボキシレート、ビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)アジペート、ビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)ブチルアミン、ダイマー酸ジグリシジルエステル及び芳香族ポリエポキシドの核水添化物等が挙げられる。
脂肪族ポリエポキシドとしては、多価脂肪族アルコールのポリグリシジルエーテル、多価脂肪酸のポリグリシジルエステル及びグリシジル脂肪族アミン等が挙げられる。
多価脂肪酸のポリグリシジルエステルとしては、ジグリシジルオキサレート、ジグリシジルマレート、ジグリシジルスクシネート、ジグリシジルグルタレート、ジグリシジルアジペート及びジグリシジルピメレート等が挙げられる。
グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミン等が挙げられる。
脂肪族ポリエポキシドには、ジグリシジルエーテル及びグリシジル(メタ)アクリレートの(共)重合体も含まれる。
ポリエポキシド(11)のうち好ましいのは、脂肪族ポリエポキシド及び芳香族ポリエポキシドである。ポリエポキシド(11)は、2種以上を併用してもよい。
<軟化点>
降下式フローテスター{例えば「CFT−500D」[(株)島津製作所製]}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、プランジャーの降下量の最大値の1/2に対応する温度をグラフから読み取り、この値(測定試料の半分が流出したときの温度)を軟化点とする。
示差走査熱量計(DSC){例えば「DSC210」[セイコー電子工業(株)製]}を用いて、測定する。
融解熱の最大ピーク温度の測定に供する試料は、前処理として、130℃で溶融した後、130℃から70℃まで1.0℃/分の速度で降温し、次に70℃から10℃まで0.5℃/分の速度で降温する。ここで、一度DSCにより、昇温速度20℃/分で昇温して吸発熱変化を測定して、「吸発熱量」と「温度」とのグラフを描き、このとき観測される20℃〜100℃にある吸熱ピーク温度を「Ta*」とする。複数ある場合は最も吸熱量が大きいピークの温度をTa*とする。最後に試料を(Ta*−10)℃で6時間保管した後、(Ta*−15)℃で6時間保管する。
次いで上記試料を、DSCにより、降温速度10℃/分で0℃まで冷却した後、昇温速度20℃/分で昇温して吸発熱変化を測定して、同様のグラフを描き、吸発熱量の最大ピークに対応する温度を、融解熱の最大ピーク温度とする。
測定試料は、測定装置の冶具にセットした後、(Ta+30)℃まで昇温して冶具に密着させてから、(Ta+30)℃から(Ta−30)℃まで0.5℃/分の速度で降温し、(Ta−30)℃で1時間静置し、次いで(Ta−10)℃まで0.5℃/分の速度で降温し、更に(Ta−10)℃で1時間静置し、十分に結晶化を進行させたのち、これを用いて測定を行う。測定温度範囲は30〜200℃であり、この温度間の溶融粘弾性を測定することによって、温度−G’、温度−G”の曲線として得ることができる。
なお、結晶性ポリエステル樹脂(B1)を測定する場合、上記の方法で測定した結晶性樹脂粒子の融解熱の最大ピーク温度(Ta’)を、結晶性ポリエステル樹脂(B1)の融解熱の最大ピーク温度(Ta)に読み替えて行う。
(Ta+20)℃におけるG”が5×105(Pa)以下であると、低温定着時でコールドオフセットが起きにくくなり、低温定着性がより良好となる。また1×102(Pa)以上であると、定着温度領域が広くなる。
上記条件を満たす結晶性ポリエステル樹脂(B1)は、樹脂中の結晶性成分の比率を調整すること等により得ることができる。例えば、結晶化率や結晶性成分の比率を増加させると、G”(Ta+20)の値は小さくなる。結晶性成分としては、直鎖構造を有するポリオール等が挙げられる。
結晶化率が20重量%以上であると、低温定着性が更に良好となる。
なお、ポリエステル樹脂としては、重縮合ポリエステル樹脂以外に、ラクトン開環重合物及びポリヒドロキシカルボン酸も同様に好ましい。
なお、結晶性部位には、その他必要に応じて前記のポリオール成分を用いてもよい。
開環重合の開始剤としてグリコールを用いると、末端にヒドロキシル基を有するラクトン開環重合物が得られる。例えば、上記炭素数3〜12のモノラクトンと前記ジオール成分(エチレングリコール及びジエチレングリコール等)を触媒の存在下で反応させることにより得ることができる。触媒としては、有機スズ化合物、有機チタン化合物及び有機ハロゲン化スズ化合物等が一般的であり、0.1〜5,000ppm程度の割合で添加して、重合温度100〜230℃で、好ましくは不活性雰囲気下に重合させることにより、ラクトン開環重合物を得ることができる。ラクトン開環重合物は、その末端を例えばカルボキシル基になるように変性したものであってもよい。ラクトン開環重合物は、結晶性の高い熱可塑性脂肪族ポリエステル樹脂である。ラクトン開環重合物は、市販品を用いてもよく、例えば「PLACCEL」シリーズのH1P、H4、H5、H7等[ダイセル(株)製](いずれも、融点=約60℃、Tg=約−60℃の高結晶性ポリカプロラクトン)等が挙げられる。
ただし、低温定着性及び光沢性の観点から、樹脂(B)中の結晶性ポリエステル樹脂(B1)の含有率は、好ましくは65重量%以上であり、更に好ましくは68重量%以上、特に好ましくは70重量%以上である。
更に本第1発明の樹脂粒子(R)は、例えば、第3発明の製造方法により得られる水性分散体(X1)を用いる第5発明の水性分散体の製造方法により得られる水性分散体(X2)を用いて、第6発明の樹脂粒子の製造方法により得ることができる。
これらの樹脂の具体例としては、前記樹脂粒子(R)中に含有される樹脂として例示したものと同様のものが挙げられる。
[1]ビニル樹脂の場合、モノマーを出発原料として、懸濁重合法、乳化重合法、シード重合法又は分散重合法等の重合反応により、直接、樹脂粒子(S)の水性分散液を製造する方法。
[2]ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂等の重付加又は縮合樹脂の場合、前駆体(モノマー及びオリゴマー等)又はその有機溶剤(u)溶液を適当な分散剤存在下で水性溶剤[水と必要により有機溶剤(u)を含有する溶剤]中に分散させ、その後に加熱したり、硬化剤を加えたりして、前駆体を硬化させて樹脂粒子(S)の水性分散液を製造する方法。
[3]ポリエステル樹脂、ポリウレタン樹脂及びエポキシ樹脂等の重付加又は縮合樹脂の場合、前駆体(モノマー及びオリゴマー等)又はその有機溶剤(u)溶液(液体であることが好ましい。加熱により液状化してもよい)中に適当な乳化剤を溶解させた後、水を加えて転相乳化した後、加熱したり、硬化剤を加えたりして前駆体を硬化させて、樹脂粒子(S)の水性分散液を製造する方法。
[4]あらかじめ重合反応(付加重合、開環重合、重付加、付加縮合又は縮合重合等いずれの重合反応様式であってもよい。以下同様。)により作製した樹脂を機械回転式又はジェット式等の微粉砕機を用いて粉砕し、次いで分級することにより樹脂粒子を得た後、適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(S)の樹脂分散液を製造する方法。
[5]あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液を、霧状に噴霧することにより樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(S)の樹脂分散液を製造する方法。
[6]あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液に貧
溶剤を添加するか、又はあらかじめ有機溶剤(u)に加熱溶解した樹脂溶液を冷却することにより樹脂粒子を析出させ、次いで有機溶剤(u)を除去して樹脂粒子を得た後、前記樹脂粒子を適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(S)の樹脂分散液を製造する方法。
[7]あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液を、適当な分散剤存在下で水性溶剤中に分散させ、これを加熱又は減圧等によって有機溶剤(u)を除去して、樹脂粒子(S)の樹脂分散液を製造する方法。
[8]あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液中に適当な乳化剤を溶解させた後、水性溶剤を加えて転相乳化して樹脂粒子(S)の樹脂分散液を製造する方法。
加物等の無機酸塩又は有機酸塩が挙げられる。
カチオン界面活性剤(s−2)としては、第4級アンモニウム塩型界面活性剤及びアミン塩型界面活性剤等が挙げられる。
両性界面活性剤(s−3)としては、カルボン酸塩型両性界面活性剤、硫酸エステル塩型両性界面活性剤、スルホン酸塩型両性界面活性剤及びリン酸エステル塩型両性界面活性剤等が挙げられる。
非イオン界面活性剤(s−4)としては、AO付加型非イオン界面活性剤及び多価アルコ−ル型非イオン界面活性剤等が挙げられる。
これらの界面活性剤(s)の具体例としては、特開2002−284881号公報に記載のもの等が挙げられる。
有機溶剤(u)の具体例としては、芳香族炭化水素溶剤(トルエン、キシレン、エチルベンゼン及びテトラリン等);脂肪族又は脂環式炭化水素溶剤(n−ヘキサン、n−ヘプタン、ミネラルスピリット及びシクロヘキサン等);ハロゲン溶剤(塩化メチル、臭化メチル、ヨウ化メチル、メチレンジクロライド、四塩化炭素、トリクロロエチレン及びパークロロエチレン等);エステル又はエステルエーテル溶剤(酢酸エチル、酢酸ブチル、メトキシブチルアセテート、メチルセロソルブアセテート及びエチルセロソルブアセテート等);エーテル溶剤(ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチルセロソルブ、ブチルセロソルブ及びプロピレングリコールモノメチルエーテル等);ケトン溶剤(アセトン、メチルエチルケトン、メチルイソブチルケトン、ジ−n−ブチルケトン及びシクロヘキサノン等);アルコール溶剤(メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、t−ブタノール、2−エチルヘキシルアルコール及びベンジルアルコール等);アミド溶剤(ジメチルホルムアミド及びジメチルアセトアミド等);スルホキシド溶剤(ジメチルスルホキシド等);複素環式化合物溶剤(N−メチルピロリドン等)並びにこれらの2種以上の混合溶剤が挙げられる。なお、前記水性溶剤中に用いる有機溶剤(u)としては、これらのうち、25℃で水と任意の割合で混和する溶剤(アセトン及びメタノール等)が好ましい。
可塑剤(v)としては特に限定されず、(v1)フタル酸エステル(フタル酸ジブチル、フタル酸ジオクチル、フタル酸ブチルベンジル及びフタル酸ジイソデシル等);(v2)脂肪族2塩基酸エステル(アジピン酸ジ−2−エチルヘキシル及びセバシン酸−2−エチルヘキシル等);(v3)トリメリット酸エステル(トリメリット酸トリ−2−エチルヘキシル及びトリメリット酸トリオクチル等);(v4)リン酸エステル(リン酸トリエチル、リン酸トリ−2−エチルヘキシル及びリン酸トリクレジール等);(v5)脂肪酸エステル(オレイン酸ブチル等);(v6)及びこれらの2種以上の混合物が挙げられる。
樹脂粒子(S)の体積平均粒径は、好ましくは0.0005〜30μmである。上限は、更に好ましくは20μm、特に好ましくは10μm、最も好ましくは2μmであり、下限は、更に好ましくは0.01μm、特に好ましくは0.02μm、最も好ましくは0.04μmである。ただし、例えば、体積平均粒径1μmの樹脂粒子(T)を得たい場合、好ましくは0.0005〜0.3μm、更に好ましくは0.001〜0.2μmであり、10μmの樹脂粒子(T)を得たい場合、好ましくは0.005〜3μm、更に好ましくは0.04〜2μm、特に好ましくは0.05〜1μmであり、100μmの樹脂粒子(T)を得たい場合、好ましくは0.05〜30μm、更に好ましくは0.1〜20μmである。なお、体積平均粒径は、レーザー式粒度分布測定装置{例えば「LA−920」[(株)堀場製作所製]}やコールターカウンター[例えば「マルチサイザーIII」(コールター社製)]で測定できる。
なお、上記粒径比が得やすいことから、後述する樹脂粒子(R)の体積平均粒径は、0.1〜300μmが好ましい。この上限は、更に好ましくは250μm、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μm、特に好ましくは1μm、最も好ましくは4μmである。
樹脂粒子(T)又は樹脂粒子(R)を、電子部品(液晶ディスプレイ等)製造用スペーサー又は電子測定機の標準粒子として用いる場合、樹脂(A)のMnは、好ましくは2万〜1,000万であり、更に好ましくは4万〜200万である。(A)の融点は、好ましくは40〜300℃であり、更に好ましくは70〜250℃である。(A)のTgは、好ましくは−0〜250℃であり、更に好ましくは50〜200℃である。(A)のSP値は、好ましくは8〜18(cal/cm3)1/2であり、更に好ましくは9〜14(cal/cm3)1/2である。
樹脂粒子(T)又は樹脂粒子(R)をトナーの母体粒子(電子写真、静電記録又は静電印刷等に使用されるトナーの母体粒子)として用いる場合、樹脂(A)のMnは、好ましくは1,000〜500万であり、更に好ましくは2,000〜50万である。(A)の融点は、好ましくは20〜300℃であり、更に好ましくは80〜250℃である。(A)のTgは、好ましくは20〜200℃であり、更に好ましくは40〜100℃である。(A)のSP値は、好ましくは8〜16(cal/cm3)1/2であり、更に好ましくは9〜14(cal/cm3)1/2である。
分散装置としては、一般に乳化機や、分散機として市販されているものであれば特に限定されず、例えばバッチ式乳化機{ホモジナイザー(IKA社製)、ポリトロン(キネマティカ社製)及びTKオートホモミキサー[特殊機化工業(株)製]等}、連続式乳化機{エバラマイルダー[(株)荏原製作所製]、TKフィルミックス、TKパイプラインホモミキサー[特殊機化工業(株)製]、コロイドミル[神鋼パンテック(株)製]、スラッシャー、トリゴナル湿式微粉砕機[サンテック(株)製]、キャピトロン(ユーロテック社製)及びファインフローミル[太平洋機工(株)製]等}、高圧乳化機{マイクロフルイダイザー[みずほ工業(株)製]、ナノマイザー[エス・ジーエンジニアリング(株)製]及びAPVガウリン(ガウリン社製)等}、膜乳化機{膜乳化機[冷化工業(株)製]等}、振動式乳化機{バイブロミキサー[冷化工業(株)製]等}、超音波乳化機{超音波ホモジナイザー(ブランソン社製)等}等が挙げられる。これらのうち、粒径の均一化の観点から好ましいのは、バッチ式乳化機、連続式乳化機及び高圧乳化機であり、更に好ましいのはAPVガウリン、ホモジナイザー、TKオートホモミキサー、エバラマイルダー、TKフィルミックス及びTKパイプラインホモミキサーである。
油性液(OL)の粘度は、粒径均一性の観点から、好ましくは10〜5万mPa・s(分散時の温度でB型粘度計で測定したもの)であり、更に好ましくは100〜1万mPa・sである。
分散時の温度としては、好ましくは0〜150℃(加圧下)であり、更に好ましくは5〜98℃である。上記の粘度が高い場合、温度を上げて上記の好ましい範囲まで粘度を低下させて、乳化分散を行うことが好ましい。
油性液(OL)に必要により用いる有機溶剤は、樹脂(A)及びその前駆体を25℃〜分散時の温度で溶解し得る溶剤であれば特に限定されず、具体的には、有機溶剤(u)と同様のものが挙げられる。好ましいものは樹脂(A)の種類によって異なるが、樹脂(A)とのSP値差が3以下であるのが好適である。また、樹脂粒子(T)の粒径均一性の観点から、樹脂(A)を溶解させるが、樹脂(C)を含有する樹脂粒子(S)を溶解・膨潤させにくい溶剤が好ましい。
また、本発明においては、添加剤は必ずしも水性分散液(W)中で粒子を形成させる時に混合しておく必要はなく、粒子を形成せしめた後添加してもよい。例えば、着色剤を含まない樹脂粒子を形成させた後、公知の染着の方法で着色剤を添加したり、有機溶剤(u)及び/又は可塑剤(v)と共に前記添加剤を樹脂粒子に含浸させることもできる。
[1]水性分散体(X1)を減圧下又は大気圧下で乾燥する方法。
[2]遠心分離器、スパクラフィルター又はフィルタープレス等により、水性分散体(X1)を固液分離し、得られた粉末を乾燥する方法。
[3]水性分散体(X1)を凍結させて乾燥させる方法(いわゆる凍結乾燥)。
上記[1]又は[2]の方法において、得られた粉末を乾燥する際、流動層式乾燥機、減圧乾燥機及び循風乾燥機等の公知の設備を用いて行うことができる。また、必要に応じ、風力分級器等を用いて分級し、所定の粒度分布とすることもできる。
表面被覆率(%)=(SS)×100/[(SS)+(SR)]
(SS):樹脂粒子(S)に覆われている部分の面積
(SR):樹脂粒子(R)が露出している部分の面積
樹脂粒子(T)の体積平均粒径は、用途により異なるが、好ましくは0.1〜300μmである。上限は、更に好ましくは250μmであり、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μmであり、特に好ましくは1μm、最も好ましくは4μmである。
なお、体積平均粒径及び個数平均粒径は、コールターカウンターで同時に測定することができる。
粉体流動性を向上させたい場合、樹脂粒子(T)のBET値比表面積が0.5〜5.0m2/gであることが好ましい。BET比表面積は、比表面積計、例えば「QUANTASORB」[ユアサアイオニクス(株)製]を用いて測定(測定ガス:He/Kr=99.9/0.1vol%、検量ガス:窒素)することができる。
同様に粉体流動性の観点から、樹脂粒子(T)の表面平均中心線粗さRaが0.01〜0.8μmであることが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム[(株)東陽テクニカ製]で測定することができる。
更に、水性分散体(X2)から水性溶剤を除去することにより樹脂粒子(R)が得られる。水性溶剤の除去方法としては、水性分散体(X1)から樹脂粒子(T)を得る場合と同様の方法が挙げられる。
水性分散体(X1)中において、付着している樹脂粒子(S)を樹脂粒子(R)から脱離させる方法としては、以下の方法等が挙げられる。
[1]水性分散体(X1)を超音波処理する方法。
[2]水性分散体(X1)を大量の水又は水溶性有機溶剤{メタノール、エタノール及びアセトン等}で希釈し、撹拌により剪断を与える方法。
[3]水性分散体(X1)に酸、アルカリ又は無機塩等を添加し、撹拌により剪断を与える方法。
[4]水性分散体(X1)を加熱し、撹拌により剪断を与える方法。
[5]水性分散体(X1)に有機溶剤を含む場合に、有機溶剤を留去する方法。
[1]樹脂(C)がカルボキシル基、ホスホノ基、スルホ基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりのMwが1,000以下であることが好ましい)である場合、水性分散体(X1)中に塩基(水酸化ナトリウム、水酸化カリウム、アンモニア及びDBU等)又はそれらの水溶液を加える方法。
[2]樹脂(C)が1級アミノ基、2級アミノ基、3級アミノ基及び4級アンモニオ基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりのMwが1,000以下であることが好ましい)である場合、水性分散体(X1)中に酸(塩酸、硫酸、リン酸及び酢酸等)又はそれらの水溶液を加える方法。
[3]樹脂(C)が、特定の有機溶剤(u)に溶解する場合[一般に樹脂(C)と有機溶剤(u)のSP値の差が2.5以下であることが好ましい]に、水性分散体(X1)中に特定の有機溶剤(u)を加える方法。
[1]一定の目開きを有するろ紙、ろ布、メッシュ等を用いてろ過し、樹脂粒子(R)のみをろ別する方法。
[2]遠心分離により樹脂粒子(R)のみを沈降させ、上澄み中に含まれる樹脂粒子(S)又はその溶解物を除去する方法。
樹脂粒子(S)による樹脂粒子(R)表面の被覆率や、樹脂粒子(S)が樹脂粒子(R)側に埋め込まれている深さは、以下のような方法で制御することができる。
[1]水性分散体(X1)を製造する際に、樹脂粒子(S)と樹脂粒子(R)とが正負逆の電荷を持つようにすると被覆率及び深さが大きくなる。この場合、樹脂粒子(S)、樹脂粒子(R)の各々の電荷を大きくするほど、被覆率及び深さが大きくなる。
[2]水性分散体(X1)を製造する際に、樹脂粒子(S)と樹脂粒子(R)とが同極性(いずれも正又はいずれも負)の電荷を持つようにすると、被覆率は下がり、深さが小さくなる傾向にある。この場合、一般に界面活性剤(s)及び/又は水溶性ポリマー(h)[特に樹脂粒子(S)及び樹脂粒子(R)と逆電荷を有するもの]を使用すると被覆率が上がる。また、水溶性ポリマー(h)を使用する場合には、水溶性ポリマー(h)のMwが大きいほど深さが小さくなる。
[3]水性分散体(X1)を製造する際に、樹脂(C)がカルボキシル基、ホスホノ基及びスルホ基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりのMwが1,000以下であることが好ましい)である場合に、水性溶剤のpHが低いほど被覆率及び深さが大きくなる。逆に、pHを高くするほど被覆率及び深さが小さくなる。
[4]水性分散体(X1)を製造する際に、樹脂(C)が1級アミノ基、2級アミノ基、3級アミノ基及び4級アンモニオ基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であることが好ましい)である場合に、水性溶剤のpHが高いほど被覆率及び深さが大きくなる。逆に、pHを低くするほど被覆率及び深さが小さくなる。
[5]樹脂(C)と樹脂(A)とのSP値差を小さくするほど被覆率及び深さが大きくなる。
することが好ましい。
樹脂粒子(R)の形状は、粉体流動性及び溶融レベリング性の観点から球状であることが好ましく、Wadellの実用球形度が好ましくは0.85〜1.00であり、更に好ましくは0.90〜1.00である。
撹拌機、加熱冷却装置、温度計、冷却管及び窒素導入管を備えた反応容器(特に記載のない限り、以下の製造例で用いる反応容器も同様。)に、1,2,4−ベンゼントリカルボン酸109部、エチレングリコール291部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びエチレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ350になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−1]を得た。[エステルe−1]のMnは340、Mwは370、水酸基価は470であった。
反応容器に、1,2,4−ベンゼントリカルボン酸94部、1,3−プロピレングリコール306部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,3−プロピレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ380になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−2]を得た。[エステルe−2]のMnは390、Mwは410、水酸基価は420であった。
反応容器に、1,2,4−ベンゼントリカルボン酸82部、1,4−ブタンジオール318部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,4−ブタンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ420になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−3]を得た。[エステルe−3]のMnは430、Mwは460、水酸基価は380であった。
反応容器に、1,2,4−ベンゼントリカルボン酸66部、1,6−ヘキサンジオール334部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,6−ヘキサンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ500になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−4]を得た。[エステルe−4]のMnは500、Mwは530、水酸基価は340であった。
反応容器に、1,2,4−ベンゼントリカルボン酸107部、1,3−ベンゼンジカルボン酸8.5部、エチレングリコール285部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びエチレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ340になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−5]を得た。[エステルe−5]のMnは350、Mwは370、水酸基価は480であった。
反応容器に、1,2,4−ベンゼントリカルボン酸107部、1,4−ベンゼンジカルボン酸8.5部、エチレングリコール285部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びエチレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ340になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−6]を得た。[エステルe−6]のMnは340、Mwは360、水酸基価は490であった。
反応容器に、1,2,4−ベンゼントリカルボン酸109部、ジエタノールアミン493部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びジエタノールアミンを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ500になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−7]を得た。[エステルe−7]のMnは480、Mwは520、水酸基価は107であった。
反応容器に、1,2,4,5−ベンゼンテトラカルボン酸132部、エチレングリコール388部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びエチレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ466になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−8]を得た。[エステルe−8]のMnは、460、Mwは470、水酸基価は118であった。
反応容器に、1,2,4、5−ベンゼンテトラカルボン酸114部、1,3−プロピレングリコール408部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,3−プロピレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ500になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−9]を得た。[エステルe−9]のMnは500、Mwは510、水酸基価は106であった。
反応容器に、1,2,4,5−ベンゼンテトラカルボン酸99部、1,4−ブタンジオール424部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,4−ブタンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ540になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[エステルe−10]を得た。[エステルe−10]のMnは530、Mwは540、水酸基価は102であった。
反応容器に、セバシン酸145部、製造例4で得られたエステル(e−4)256部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,6−ヘキサンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ10,000になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、結晶性重縮合ポリエステル樹脂[結晶性樹脂a−1]を得た。[結晶性樹脂a−1]の融点は62℃、Mnは5,300、Mwは11,000、水酸基価は22であった。
反応容器に、セバシン酸170部、製造例3で得られたエステル(e−3)230部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,4−ブタンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ10,000になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、結晶性重縮合ポリエステル樹脂[結晶性樹脂a−2]を得た。[結晶性樹脂a−2]の融点は58℃、Mnは5,000、Mwは11,500、水酸基価は23であった。
反応容器に、セバシン酸145部、1,6−ヘキサンジオール255部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,6−ヘキサンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ10,000になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、結晶性重縮合ポリエステル樹脂[結晶性樹脂B−1]を得た。[結晶性樹脂B−1]の融点は67℃、Mnは5,200、Mwは10,000、水酸基価は22であった。
反応容器に、セバシン酸171部、1,4−ブタンジオール229部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,4−ブタンジオールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ10,000になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、結晶性重縮合ポリエステル樹脂[結晶性樹脂B−2]を得た。[結晶性樹脂B−2]の融点は57℃、Mnは5,000、Mwは10,000、水酸基価は24であった。
反応容器に、1,4−ベンゼンジカルボン酸159部、1,2−プロピレングリコール108部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及び1,2−プロピレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ10,000になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、非晶性重縮合ポリエステル樹脂[非結晶性樹脂p−1]を得た。[非結晶性樹脂p−1]のMnは4,800、Mwは12,000、水酸基価は26であった。
反応容器に、1,4−ベンゼンジカルボン酸120部、ビスフェノールAのPO2モル付加物280部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、酸価が1となった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、非晶性重縮合ポリエステル樹脂[非結晶性樹脂p−2]を得た。[非結晶性樹脂p−2]のMnは5,200、Mwは12,000、水酸基価は25であった。
オートクレーブに、製造例11で得られた[結晶性樹脂a−1]414部、IPDI36部及び酢酸エチル450部を投入し、密閉状態で100℃、5時間反応を行い、分子末端にイソシアネート基を有する[プレポリマーA0−1溶液]を得た。[プレポリマーA0−1溶液]のNCO含量は0.8%であった。
製造例17において、IPDI36部の替わりにヘキサメチレンジイソシアネート(以下、HDIと記載。)27部を使用した以外は製造例17と同様にして、[プレポリマーA0−2溶液]を得た。[プレポリマーA0−2溶液]のNCO含量は0.8%であった。
製造例17において、[結晶性樹脂a−1]414部の替わりに製造例12で得られた[結晶性樹脂a−2]395部を使用した以外は製造例17と同様にして、[プレポリマーA0−3溶液]を得た。[プレポリマーA0−3溶液]のNCO含量は0.8%であった。
製造例17において、IPDI36部の替わりにHDI27部を使用した以外は製造例17と同様にして、[プレポリマーA0−4溶液]を得た。[プレポリマーA0−4溶液]のNCO含量は0.8%であった。
オートクレーブに、製造例13で得られた[結晶性樹脂B−1]406部、IPDI35部及び酢酸エチル440部を投入し、密閉状態で100℃、5時間反応を行った後、25℃まで冷却し、更に製造例1で得られた[エステルe−1]8.1部及び酢酸エチル10部を投入し、再度密閉状態で100℃、5時間反応を行うことで分子末端にイソシアネート基を有する[プレポリマーA0−5溶液]を得た。[プレポリマーA0−5溶液]のNCO含量は0.4%であった。
製造例21において、IPDI35部の替わりにHDI27部を使用した以外は製造例21と同様にして、[プレポリマーA0−6溶液]を得た。[プレポリマーA0−6溶液]のNCO含量は0.4%であった。
製造例21において、IPDI35部の替わりに2,4−又は2,6−トリレンジイソシアネート(TDI)28部を使用した以外は製造例21と同様にして、[プレポリマーA0−7溶液]を得た。[プレポリマーA0−7溶液]のNCO含量は0.4%であった。
製造例21において、[結晶性樹脂B−1]406部の替わりに製造例14で得られた[結晶性樹脂B−2]367部を使用した以外は製造例21と同様にして、[プレポリマーA0−8溶液]を得た。[プレポリマーA0−8溶液]のNCO含量は0.5%であった。
製造例21において、[エステルe−1]の替わりに製造例2で得られた[エステルe−2]を使用した以外は製造例21と同様にして、[プレポリマーA0−9溶液]を得た。[プレポリマーA0−9溶液]のNCO含量は0.5%であった。
製造例21において、[エステルe−1]の替わりに製造例3で得られた[エステルe−3]を使用した以外は製造例21と同様にして、[プレポリマーA0−10溶液]を得た。[プレポリマーA0−10溶液]のNCO含量は0.5%であった。
製造例21において、[エステルe−1]の替わりに製造例4で得られた[エステルe−4]を使用した以外は製造例21と同様にして、[プレポリマーA0−11溶液]を得た。[プレポリマーA0−11溶液]のNCO含量は0.5%であった。
製造例21において、[エステルe−1]の替わりに製造例5で得られた[エステルe−5]を使用した以外は製造例21と同様にして、[プレポリマーA0−12溶液]を得た。[プレポリマーA0−12溶液]のNCO含量は0.4%であった。
製造例21において、[エステルe−1]の替わりに製造例6で得られた[エステルe−6]を使用した以外は製造例21と同様にして、[プレポリマーA0−13溶液]を得た。[プレポリマーA0−13溶液]のNCO含量は0.4%であった。
製造例21において、[エステルe−1]の替わりに製造例7で得られた[エステルe−7]を使用した以外は製造例21と同様にして、[プレポリマーA0−14溶液]を得た。[プレポリマーA0−14溶液]のNCO含量は0.4%であった。
製造例21において、[結晶性樹脂B−1]406部の替わりに製造例15で得られた[非結晶性樹脂p−1]339部を使用した以外は製造例21と同様にして、[プレポリマーA0−15溶液]を得た。[プレポリマーA0−15溶液]のNCO含量は0.5%であった。
製造例21において、[結晶性樹脂B−1]406部の替わりに製造例16で得られた[非晶性樹脂p−2]354部を使用した以外は製造例21と同様にして、[プレポリマーA0−16溶液]を得た。[プレポリマーA0−16溶液]のNCO含量は0.5%であった。
製造例21において、[エステルe−1]の替わりに製造例8で得られた[エステルe−8]を使用した以外は製造例21と同様にして、[プレポリマーA0−17溶液]を得た。[プレポリマーA0−17溶液]のNCO含量は0.5%であった。
製造例21において、[エステルe−1]の替わりに製造例9で得られた[エステルe−9]を使用した以外は製造例21と同様にして、[プレポリマーA0−18溶液]を得た。[プレポリマーA0−18溶液]のNCO含量は0.4%であった。
製造例21において、[エステルe−1]の替わりに製造例10で得られた[エステルe−10]を使用した以外は製造例21と同様にして、[プレポリマーA0−19溶液]を得た。[プレポリマーA0−19溶液]のNCO含量は0.4%であった。
反応容器に、1,2,4−ベンゼントリカルボン酸2.1部、1,4−ベンゼンジカルボン酸168部、プロピレングリコール230部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びエチレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、Mwがおよそ10,000になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[ポリエステル樹脂q−1]を得た。[ポリエステル樹脂q−1]のMnは5,200、Mwは10,000、水酸基価は23であった。
反応容器に、1,2,4−ベンゼントリカルボン酸1.5部、1,4−ベンゼンジカルボン酸119部、ビスフェノールAのPO2モル付加物279部及び縮合触媒としてチタニウムジヒドロキシビス(トリエタノールアミネート)0.5部を投入し、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで225℃まで徐々に昇温しながら、窒素気流下に、生成する水及びエチレングリコールを留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、酸価が1となった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、重縮合ポリエステル樹脂[ポリエステル樹脂q−2]を得た。[ポリエステル樹脂q−2]のMnは5,300、Mwは12,000、水酸基価は24であった。
温度計及び撹拌機を備えた反応容器に、製造例13で得られた[結晶性樹脂B−1]10部及び酢酸エチル10部を投入し、70℃まで昇温し撹拌して均一に分散させ、更に室温まで冷却して[樹脂溶液D−1]を得た。
比較製造例3において、[結晶性樹脂B−1]の替わりに製造例14で得られた[結晶性樹脂B−2]を使用した以外は比較製造例3と同様にして、[樹脂溶液D−2]を得た。
比較製造例3において、[結晶性樹脂B−1]の替わりに比較製造例1で得られた[ポリエステル樹脂q−1]を使用した以外は比較製造例3と同様にして、[樹脂溶液D−3]を得た。
比較製造例3において、[結晶性樹脂B−1]の替わりに比較製造例2で得られた[ポリエステル樹脂q−2]を使用した以外は比較製造例3と同様にして、[樹脂溶液D4]を得た。
温度計及び撹拌機を備えた反応容器に、水682部、メタクリル酸EO付加物硫酸エステルのナトリウム塩「エレミノールRS−30」[三洋化成工業(株)製]11部、スチレン138部、メタクリル酸138部及び過硫酸アンモニウム1部を投入し、25℃、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。この乳濁液を加熱して75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(C)(スチレン−メタクリル酸−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の水性分散体[樹脂粒子(S1)分散体]を得た。[樹脂粒子(S1)分散体]の体積平均粒径は、0.15μmであった。また、[樹脂粒子(S1)分散体]の一部を乾燥して単離した樹脂粒子(S1)のTgは148℃であった。
温度計及び撹拌機を備えた反応容器に、水634部、[樹脂粒子(S1)分散体]286部、カルボキシメチルセルロース「CMCダイセル1170」[ダイセル化学工業(株)製]2部及びドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液「エレミノールMON−7」[三洋化成工業(株)製]154部を投入し、25℃で撹拌し乳白色の液体[水性分散液W−1]を得た。
ビーカーに銅フタロシアニン20部と着色剤分散剤「ソルスパーズ28000」[アビシア(株)製]5部及び酢酸エチル75部を投入し、撹拌して均一に分散させた後、ビーズミルによって銅フタロシアニンを微分散して、[着色剤分散液1]を得た。[着色剤分散液1]の粒子径測定装置「LA−920」[(株)堀場製作所製]で測定した体積平均粒径は0.3μmであった。
オートクレーブに、キシレン454部及び低分子量ポリエチレン「サンワックス LEL−400」[軟化点128℃、三洋化成工業(株)製]150部を投入し、窒素置換後170℃に昇温して均一に溶解した後、スチレン595部、メタクリル酸メチル255部、ジ−t−ブチルパーオキシヘキサヒドロテレフタレート34部及びキシレン119部の混合溶液を、170℃で3時間かけて滴下して重合し、更に同温度で30分間保持した。次いで脱溶剤を行い、[変性ワックス1]を得た。[変性ワックス1]のMnは1,900、Mwは5,200、Tgは57℃であった。
温度計及び撹拌機を備えた反応容器に、パラフィンワックス(融点73℃)10部、[変性ワックス1]1部及び酢酸エチル33部を投入し、78℃に昇温して均一に溶解した後、1時間かけて30℃まで冷却し、ワックスを微粒子状に晶析させ、更にウルトラビスコミル(アイメックス製)で湿式粉砕し、[ワックス分散液1]を得た。[ワックス分散液1]の粒子径測定装置「LA−920」で測定した体積平均粒径は0.3μmであった。
ビーカーに[プレポリマーA0−1溶液]366部、[ワックス分散液1]31部、[着色剤分散液1]50部及びイソホロンジアミン3.0部を投入して溶解・混合均一化し、[樹脂溶液1]を得た。得られた[樹脂溶液1]中に[水性分散液W−1]550部を投入し、TKホモミキサー[特殊機化工業(株)製]を使用し、25℃、回転数12,000rpmで1分間分散操作を行い、更にフィルムエバポレータで減圧度−0.05MPa(ゲージ圧)、温度40℃、回転数100rpmの条件で240分間脱溶剤し、水性分散体(X−1)を得た。水性分散体(X−1)100部を遠心分離し、更に水60部を加えて遠心分離して固液分離する工程を2回繰り返した後、35℃で1時間乾燥して樹脂粒子(T1)を得た。
水性分散体(X−1)100部に、5%水酸化ナトリウム水溶液100部を加え、TKホモミキサーを使用し、40℃に温調し回転数12,000rpmで10分間混合して、(T1)の表面に付着した[水性分散液W−1]由来の微粒子を溶解させた。次いで遠心分離で上澄みを除去し、更に水100部を加えて遠心分離する工程を2回繰り返した後、乾燥して樹脂粒子(R1)を得た。なお、[プレポリマーA0−1溶液]とイソホロンジアミンの反応は、それらを溶解・混合均一化し[樹脂溶液1]を得る工程から、フィルムエバポレータでの脱溶剤工程にかけて徐々に進行し、ウレタン基及びウレア基を有する樹脂が得られる。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−2溶液]を使用する以外は実施例1と同様にして、水性分散体(X−2)、樹脂粒子(T2)及び樹脂粒子(R2)を得た。
実施例2において、イソホロンジアミンの替わりにヘキサメチレンジアミンを使用する以外は実施例1と同様にして、水性分散体(X−3)、樹脂粒子(T3)及び樹脂粒子(R3)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−3溶液]を使用する以外は実施例1と同様にして、水性分散体(X−4)、樹脂粒子(T4)及び樹脂粒子(R4)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−4溶液]を使用する以外は実施例1と同様にして、水性分散体(X−5)、樹脂粒子(T5)及び樹脂粒子(R5)を得た。
実施例5において、イソホロンジアミンの替わりにヘキサメチレンジアミンを使用する以外は実施例1と同様にして、水性分散体(X−6)、樹脂粒子(T6)及び樹脂粒子(R6)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−5溶液]を使用する以外は実施例1と同様にして、水性分散体(X−7)、樹脂粒子(T7)及び樹脂粒子(R7)を得た。
実施例7において、イソホロンジアミンの替わりにヘキサメチレンジアミンを使用する以外は実施例7と同様にして、水性分散体(X−8)、樹脂粒子(T8)及び樹脂粒子(R8)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−6溶液]を使用する以外は実施例1と同様にして、水性分散体(X−9)、樹脂粒子(T9)及び樹脂粒子(R9)を得た。
実施例9において、イソホロンジアミンの替わりにヘキサメチレンジアミンを使用する以外は実施例9と同様にして、水性分散体(X−10)、樹脂粒子(T10)及び樹脂粒子(R10)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−7溶液]を使用する以外は実施例1と同様にして、水性分散体(X−11)、樹脂粒子(T11)及び樹脂粒子(R11)を得た。
実施例11において、イソホロンジアミンの替わりにヘキサメチレンジアミンを使用する以外は実施例11と同様にして、水性分散体(X−12)、樹脂粒子(T12)及び樹脂粒子(R12)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[プレポリマーA0−8溶液]〜[プレポリマーA0−19溶液]を使用する以外は実施例1と同様にして、水性分散体(X−13)〜(X−24)、樹脂粒子(T13)〜(T24)及び樹脂粒子(R13)〜(R24)を得た。
実施例1において、[プレポリマーA0−1溶液]の替わりに[樹脂溶液D1]〜[樹脂溶液D4]を使用する以外は実施例1と同様にして、水性分散体(X’−1)〜(X’−4)、樹脂粒子(T’1)〜(T’4)及び樹脂粒子(R’1)〜(R’4)を得た。
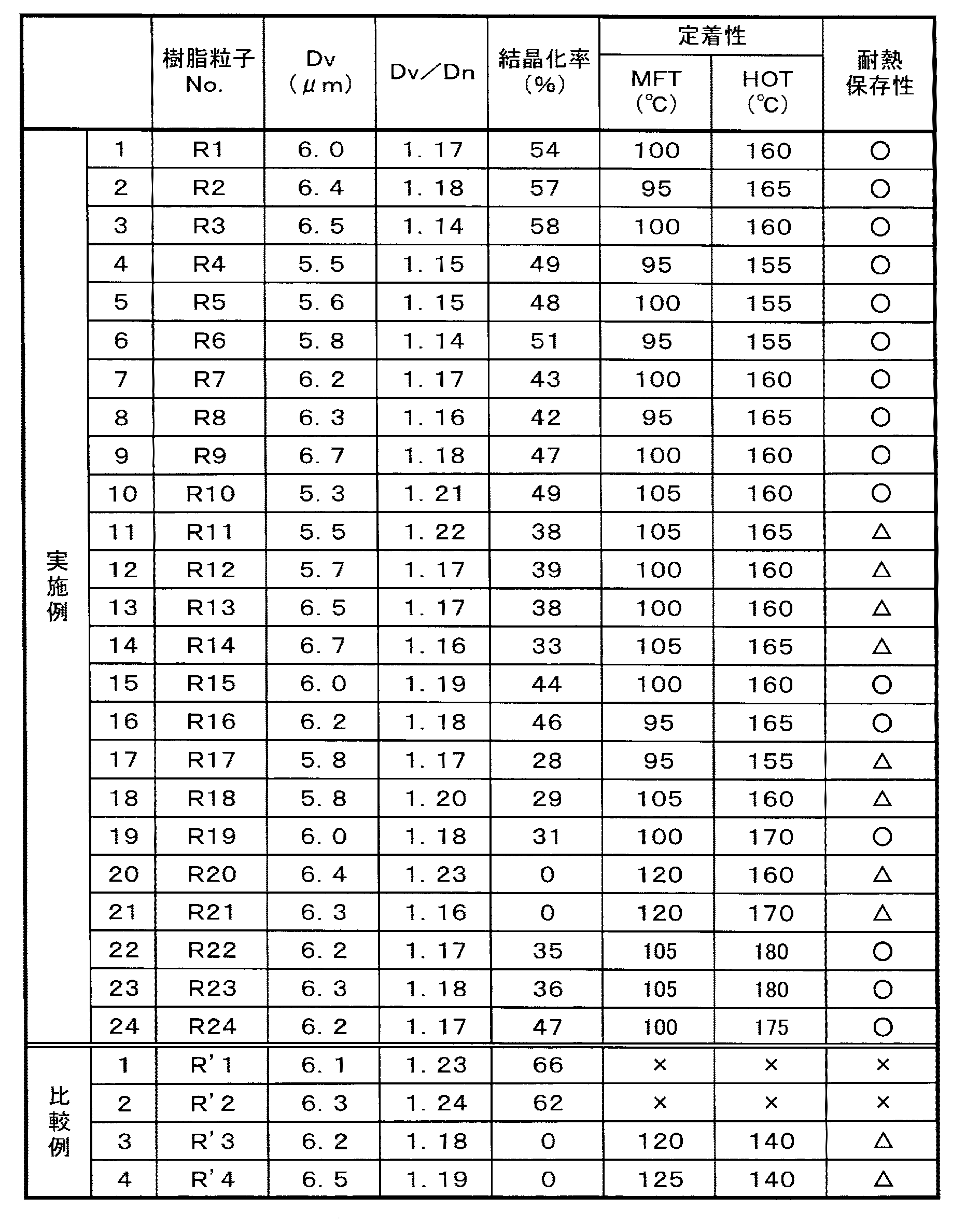
本発明の樹脂粒子(T1)〜(T24)、(R1)〜(R24)及び比較の樹脂粒子(T’1)〜(T’4)、(R’1)〜(R’4)それぞれについて、粒径、粒度分布、結晶化率、定着性及び耐熱保存安定性を以下に記載の方法で測定又は評価した。その結果を表1、2に示す。
樹脂粒子(T)及び樹脂粒子(R)の体積平均粒径(Dv)並びに粒度分布(体積平均粒径/個数平均粒径;Dv/Dn)は、コールカウンター「マルチサイザーIII」(コールター社製)で測定した(0.5%のイオン交換水の分散液、25℃)。
なお、Dv/Dnが1.25以下であれば、粒度分布が良好であると判断される。
樹脂粒子(T)及び樹脂粒子(R)の結晶化率は、X線回折装置「XD−D1」[(株)島津製作所製]で回折X線のピーク角度位置θに応じたピーク強度を測定し、結晶部からの回折線と非晶部からの散乱線のピーク強度比から求めた。
各樹脂粒子に、「アエロジルR972」[日本アエロジル(株)社製]を1.0%添加し、よく混ぜて均一にした後、この粉体を紙面上に0.6mg/cm2となるよう均一に載せた。このとき粉体を紙面に載せる方法として、熱定着機を外したプリンターを用いた(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)5kg/cm2の条件で通した時の最低定着温度(MFT)及びホットオフセット発生温度(HOT)を測定した。
MFT欄及びHOT欄が“×”は定着領域なしである。
50℃に温調された乾燥機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○:ブロッキングが発生しない。
△:ブロッキングが発生するが、力を加えると容易に分散する。
×:ブロッキングが発生し、力を加えても分散しない。
Claims (11)
- 3価又は4価の芳香族カルボン酸(c)と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)、及びポリアミン(b)を構成単位として有する樹脂(A)を含有する樹脂粒子(R)。
- 樹脂(A)が、更にポリオール(p)を構成単位として有する請求項1記載の樹脂粒子(R)。
- 樹脂(A)が、イソシアネート基含有ポリエステルプレポリマー(A0)とポリアミン(b)との伸長反応及び/又は架橋反応により形成された樹脂である請求項1又は2記載の樹脂粒子。
- 更に、架橋構造を有さない樹脂(B)を含有する請求項1〜3のいずれかに記載の樹脂粒子(R)。
- 樹脂(A)及び/又は樹脂(B)が結晶性ポリエステル樹脂であり、結晶化率が20〜60重量%である請求項1〜4のいずれかに記載の樹脂粒子(R)。
- 請求項1〜5のいずれかに記載の樹脂粒子(R)の表面に樹脂(C)を含有する樹脂粒子(S)が付着されてなる構造の樹脂粒子(T)。
- 樹脂(C)を含有する樹脂粒子(S)の水性分散液(W)と、樹脂(A)及び/又はその前駆体、並びに必要により有機溶剤を含有する油性液(OL)とを混合し、(W)中に(OL)を分散させ、(A)の前駆体を用いる場合は(W)中で前駆体を反応させて、(A)を含有する樹脂粒子(R)を形成させることにより、(R)の表面に(S)が付着された構造の樹脂粒子(T)の水性分散体(X1)を得る工程を含み、樹脂(A)が、3価又は4価の芳香族カルボン酸(c)と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)及びポリアミン(b)を構成単位として有する樹脂であることを特徴とする水性分散体(X1)の製造方法。
- 請求項7記載の製造方法により得られた水性分散体(X1)から、水性溶剤を除去して樹脂粒子(T)を得る工程を含む樹脂粒子の製造方法。
- 請求項7記載の製造方法で得られた水性分散体(X1)中において、付着している樹脂粒子(S)を樹脂粒子(R)から脱離させた後、水性分散体から(S)を分離除去して(R)の水性分散体(X2)を得る工程、又は(S)を溶解させ、必要により(S)の溶解物を分離除去して(R)の水性分散体(X2)を得る工程を含む水性分散体(X2)の製造方法。
- 請求項9記載の製造方法により得られた水性分散体(X2)から水性溶剤を除去して樹脂粒子(R)を得る工程を含む樹脂粒子の製造方法。
- 樹脂(C)を含有する樹脂粒子(S)が、樹脂粒子(R)の表面に付着されてなる構造の樹脂粒子(T)であり、
[1][(S)の体積平均粒径/(T)の体積平均粒径]が0.001〜0.3であり、
[2](S)の体積平均粒径が0.0005〜30μm、かつ(T)の体積平均粒径が0.1〜300μmであり、
[3](R)の表面の5%以上が(S)で覆われており、
[4](T)の[体積平均粒径/個数平均粒径]が1.0〜1.5であり、
[5](R)が樹脂(A)及び必要により架橋構造を有さない樹脂(B)を含有し、樹脂(A)が3価又は4価の芳香族カルボン酸と炭素数が2〜10の脂肪族ジオール(d1)又はジアルカノールアミン化合物(d2)とのエステル(e)、ポリイソシアネート(i)及びポリアミン(b)を構成単位として有する樹脂である樹脂粒子。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010247246A JP5705505B2 (ja) | 2009-11-24 | 2010-11-04 | 樹脂粒子 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009266026 | 2009-11-24 | ||
| JP2009266026 | 2009-11-24 | ||
| JP2010247246A JP5705505B2 (ja) | 2009-11-24 | 2010-11-04 | 樹脂粒子 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2011132500A true JP2011132500A (ja) | 2011-07-07 |
| JP5705505B2 JP5705505B2 (ja) | 2015-04-22 |
Family
ID=44345577
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010247246A Active JP5705505B2 (ja) | 2009-11-24 | 2010-11-04 | 樹脂粒子 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5705505B2 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5753836B2 (ja) * | 2010-02-23 | 2015-07-22 | 三洋化成工業株式会社 | ポリウレタンフォーム製造用強度向上剤 |
| JP2015171817A (ja) * | 2014-02-20 | 2015-10-01 | 三洋化成工業株式会社 | スラッシュ成形用粉末状樹脂組成物 |
| WO2018085483A1 (en) | 2016-11-04 | 2018-05-11 | Cabot Corporation | Nanocomposites containing crystalline polyester and organosilica |
| JP2019066538A (ja) * | 2017-09-28 | 2019-04-25 | 花王株式会社 | 電子写真用トナー用結着樹脂組成物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07324117A (ja) * | 1994-05-31 | 1995-12-12 | Dainippon Ink & Chem Inc | ポリウレタン微粒子 |
| JP2002284881A (ja) * | 2000-02-16 | 2002-10-03 | Sanyo Chem Ind Ltd | 粒径が均一である樹脂分散体、樹脂粒子およびそれらの製造方法 |
| JP2003327707A (ja) * | 2000-02-16 | 2003-11-19 | Sanyo Chem Ind Ltd | 粒径が均一である樹脂分散体およびその製造方法 |
| JP2008101204A (ja) * | 2006-09-20 | 2008-05-01 | Sanyo Chem Ind Ltd | 樹脂分散体の製造方法及び樹脂粒子 |
| JP2008156627A (ja) * | 2006-11-30 | 2008-07-10 | Sanyo Chem Ind Ltd | 樹脂粒子の製造方法及び樹脂粒子 |
-
2010
- 2010-11-04 JP JP2010247246A patent/JP5705505B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07324117A (ja) * | 1994-05-31 | 1995-12-12 | Dainippon Ink & Chem Inc | ポリウレタン微粒子 |
| JP2002284881A (ja) * | 2000-02-16 | 2002-10-03 | Sanyo Chem Ind Ltd | 粒径が均一である樹脂分散体、樹脂粒子およびそれらの製造方法 |
| JP2003327707A (ja) * | 2000-02-16 | 2003-11-19 | Sanyo Chem Ind Ltd | 粒径が均一である樹脂分散体およびその製造方法 |
| JP2008101204A (ja) * | 2006-09-20 | 2008-05-01 | Sanyo Chem Ind Ltd | 樹脂分散体の製造方法及び樹脂粒子 |
| JP2008156627A (ja) * | 2006-11-30 | 2008-07-10 | Sanyo Chem Ind Ltd | 樹脂粒子の製造方法及び樹脂粒子 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5753836B2 (ja) * | 2010-02-23 | 2015-07-22 | 三洋化成工業株式会社 | ポリウレタンフォーム製造用強度向上剤 |
| JP2015171817A (ja) * | 2014-02-20 | 2015-10-01 | 三洋化成工業株式会社 | スラッシュ成形用粉末状樹脂組成物 |
| WO2018085483A1 (en) | 2016-11-04 | 2018-05-11 | Cabot Corporation | Nanocomposites containing crystalline polyester and organosilica |
| JP2019066538A (ja) * | 2017-09-28 | 2019-04-25 | 花王株式会社 | 電子写真用トナー用結着樹脂組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5705505B2 (ja) | 2015-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5237902B2 (ja) | 結晶性樹脂粒子 | |
| JP5291649B2 (ja) | 樹脂粒子 | |
| KR100837646B1 (ko) | 수지 입자 | |
| JP5663007B2 (ja) | 非水系樹脂粒子分散液 | |
| JP5705493B2 (ja) | 樹脂粒子の製造方法 | |
| JP2011237790A (ja) | 樹脂粒子及びその製造方法 | |
| JP2011094136A (ja) | 樹脂粒子の製造方法 | |
| JP5214558B2 (ja) | 樹脂粒子およびその製造方法 | |
| JP2014080586A (ja) | 樹脂粒子の製造方法 | |
| WO2010021081A1 (ja) | 樹脂粒子およびその製造方法 | |
| JP5705505B2 (ja) | 樹脂粒子 | |
| JP2010254896A (ja) | 樹脂粒子およびその水性分散体、並びにその製造方法 | |
| JP2009057487A (ja) | 樹脂粒子および樹脂粒子の製造方法 | |
| JP5101574B2 (ja) | 樹脂粒子 | |
| JP6948282B2 (ja) | 複合粒子及び複合粒子を含んでなる分散液の製造方法 | |
| JP4629696B2 (ja) | 樹脂粒子および樹脂粒子の製造方法 | |
| JP2015078352A (ja) | 樹脂粒子 | |
| JP2006307195A (ja) | 樹脂粒子 | |
| JP2009040893A (ja) | 樹脂粒子および樹脂粒子の製造方法 | |
| JP2009030046A (ja) | 樹脂粒子 | |
| JP5020529B2 (ja) | 着色樹脂粒子 | |
| JP2016138257A (ja) | 樹脂粒子の製造方法 | |
| JP5627885B2 (ja) | 樹脂粒子およびその製造方法 | |
| JP2016135483A (ja) | 粒子の製造方法 | |
| JP2016130306A (ja) | 樹脂粒子の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130708 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20131210 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140617 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140818 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150224 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150225 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5705505 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |