JP2010504088A - グルタミニルシクラーゼに関連した新規遺伝子 - Google Patents
グルタミニルシクラーゼに関連した新規遺伝子 Download PDFInfo
- Publication number
- JP2010504088A JP2010504088A JP2009528732A JP2009528732A JP2010504088A JP 2010504088 A JP2010504088 A JP 2010504088A JP 2009528732 A JP2009528732 A JP 2009528732A JP 2009528732 A JP2009528732 A JP 2009528732A JP 2010504088 A JP2010504088 A JP 2010504088A
- Authority
- JP
- Japan
- Prior art keywords
- qpctl
- seq
- nos
- polypeptide
- inhibitor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC1C*CC1 Chemical compound CC1C*CC1 0.000 description 4
- LGDHZCLREKIGKJ-UHFFFAOYSA-N COc(c(OC)c1)ccc1N Chemical compound COc(c(OC)c1)ccc1N LGDHZCLREKIGKJ-UHFFFAOYSA-N 0.000 description 1
Images








































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1025—Acyltransferases (2.3)
- C12N9/104—Aminoacyltransferases (2.3.2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/45—Transferases (2)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
- C07D233/61—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms not forming part of a nitro radical, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/573—Immunoassay; Biospecific binding assay; Materials therefor for enzymes or isoenzymes
Abstract
【選択図】 なし
Description
本発明は、グルタミニルシクラーゼ(QC、EC 2.3.2.5)のアイソザイムである新規グルタミニル-ペプチドシクロトランスフェラーゼ様タンパク質(QPCTL)に、及びこれらのアイソザイムをコードする単離された核酸に関し、これらの全ては、新たな治療薬の発見のために、シクラーゼ活性を測定するために、及びこれらのグルタミニルシクラーゼアイソザイムに対する化合物の阻害活性を決定するために有用である。
グルタミニルシクラーゼ(QC、EC 2.3.2.5)は、N末端のグルタミン残基のピログルタミン酸(pGlu*)への分子内の環化を触媒してアンモニアを遊離させる。QCは、Messerによって1963年に熱帯植物パパイア(Carica papaya)のラテックスから最初に単離された(Messer, M.の文献 1963 Nature 4874、1299)。24年後、対応する酵素活性が、動物下垂体において発見された(Busby, W. H. J.らの文献、1987 J Biol Chem 262、8532-8536;Fischer, W. H.、及びSpiess, J.の文献、1987 Proc Natl Acad Sci U S A 84、3628-3632)。哺乳動物QCについては、QCによるGlnのpGluへの変換を、TRH、及びGnRHの前駆体について示すことができる(Busby, W. H. J.らの文献、1987 J Biol Chem 262、8532-8536;Fischer, W. H.、及びSpiess, J.の文献、1987 Proc Natl Acad sci USA 84、3628-3632)。加えて、QCの初期の局在実験により、ウシ下垂体における触媒作用のその推定産物との共局在が明らかになり、ペプチドホルモン合成において示唆される機能を更に改善する(Bockers, T. M.らの文献、1995 J Neuroendocrinol 7、445-453)。対照的に、植物QCの生理機能は、あまり明らかではない。C.パパイア由来の酵素の場合、病原微生物に対する植物防御における役割が示唆された(El Moussaoui, A.らの文献、2001 Cell Mol Life Sci 58、556-570)。その他の植物由来の推定上のQCが、最近配列比較によって同定された(Dahl, S. W.らの文献、2000 Protein Expr Purif 20、27-36)。しかし、これらの酵素の生理機能は、いまだあいまいである。
(酵素阻害剤)
可逆的酵素阻害剤:競合阻害剤、非競合的可逆的阻害剤、緩慢結合又は密接結合阻害剤、遷移状態類似体、及び多基質類似体を含む。
i)酵素との非共有結合性の相互作用、
ii)酵素活性部位に関して基質との競合、
を示す。

i)活性部位以外の部位(アロステリックな結合部位)に結合し、
ii)触媒活性を低減し、又は停止する酵素の高次構造上の変化を引き起こす。
i)阻害剤と酵素との間の平衡にゆっくりと到達する競合阻害剤であり、
ii)(konが、緩徐である)、おそらく、酵素、又は阻害剤において生じなければならない高次構造上の変化のために、
a)遷移状態類似体であることが多く、
b)酵素濃度と同様の濃度にて効率的であり(nM以下のKD値)、
c)koff値がとても低いため、この種の阻害剤は、「ほぼ」不可逆的である。
2つ以上の基質を含む反応については、2つ以上の基質に似た構造的特徴を含む、競合阻害剤、又は遷移状態類似体をデザインすることができる。
・(不可逆的阻害剤に向けられた活性部位)(競合的不可逆的阻害剤)は、酵素によって認識されて(可逆的、特異的な結合)、続いて共有結合形成をし、かつ
i)構造的に基質、遷移状態、又は産物に類似しており、薬物と標的酵素との間の特異的な相互作用が可能であり、
ii)反応性官能基(例えば、求核試薬、-COCH2Br)を含み、共有結合形成が可能である。
QC阻害との相関を考慮して、好ましい実施態様において、本方法、及び医学的使用では、10μM以下、より好ましくは1μM以下、更により好ましくは0.1μM以下、若しくは0.01μM以下、又は最も好ましくは0.01μM以下のQC阻害のためのKiをもつ薬剤を利用する。実際に、マイクロモル以下、好ましくはナノモル、及び更により好ましくはピコモルの範囲のKi値をもつ阻害剤が想定される。従って、活性薬剤は、便宜上、「QC阻害剤」として本明細書に記述してあるが、このような命名法は、本発明の対象を特定の作用機序に限定することを意図していないことが理解されるであろう。
一般に、本方法、又は医学的使用のQC阻害剤は、例えば1000g/モル以下、500g/モル以下、好ましくは400g/モル以下、及び更により好ましくは350g/モル以下、又は更に300g/モル以下の分子量をもつ小分子であるだろう。
別名は、ランドリーギランバレー症候群であり、急性特発性の多発性神経炎、感染性多発性神経炎、又は急性炎症性多発神経障害である。
GBSに似ているが、慢性経過によって特徴づけられる疾患は、慢性炎症性脱髄性多発神経根筋障害(CIDP)と呼ばれる。CIDPについて一般に適用できる定義は、GBSとは対照的に、進行期が4週より長く、たいてい6月より長く持続すること、及び欠陥が患者に残ったままであることが多いことという観察を除いて、未だない。GBS、及びCIDPを伴った重篤な不全麻痺を生じさせるメカニズムは、おそらくTリンパ球によって媒介される免疫反応、及び炎症、これに続く末梢ニューロンの脱髄を含む。この仮説は、GBS患者の血清、及び脳脊髄液で観察される補体化合物、及びサイトカインの量の増加によって確認される。脱髄の過程は、特に神経根の領域において、現在、神経伝導遮断の発症の決定的なメカニズムと考えられている。1つの理論は、疾患の発症における比較的初期の重要な段階としての、血液/脳脊髄液(CSF)バリアの障害に基づく。別の理論では、疾患の結果として血液/CSFバリアにおいて発生するその漏出を求め、かつCSFにおけるタンパク質含有量の増加を生じさせる。任意の割合にて、非特異的血清成分は、免疫系に直接関連することなく、血液からCSFに浸透し、ニューロン、又はグリアの機能障害を生じさせ、及び/又はニューロン活性を修飾し得る。代わりのメカニズムは、CSFの流速の低減であり、これによりCSFのタンパク質含有量の増加を説明し得る。この解釈には、血液/CSFバリアの障害、又は選択性の修飾を必要としない。言及した全ての効果は、GBS、及びCIDPの経過のために重要であり得るが、症候へのこれらの実際の貢献は、いまだ明らかにされていない。CSFにおけるタンパク質濃度の増加と特異的な電気生理学的知見、又は病像との間の関連を確立することはできていない。電位依存的ナトリウムチャンネルと相互作用する、GBS患者、及び多発性硬化症患者のCSFの因子が最近記述された(Wuzらの文献、1995、Muscle and Nerve 18,772-781)。Brinkmeierの文献(Brinkmeierらの文献、1996、Muscle and Nerve 19,54-62)は、因子が3kDa未満の、及びよりストリンジェントな試験条件下では1kDa分子量を有することを報告する。この観察、及びプロテアーゼとのCSFのインキュベーション後でさえも因子の活性が実質的に低減しなかったという事実に基づいて、著者らは、因子が抗体でもサイトカインでもないと結論した。
多発性硬化症は、中枢神経系(脳、及び脊髄)に影響を及ぼす自己免疫性疾患である。多発性硬化症は、通常男性以上に女性に発症する。本障害は、最も一般的には、20〜40歳の間に始まるが、任意の年齢にて襲い得る。正確な原因は、知られていないが、MSは、神経細胞を囲む髄鞘に対する損傷によって生じると考えられる。それは、進行性疾患であり、損傷が時間とともに更に悪化することを意味する。炎症は、ミエリンを破壊して、複数の瘢痕組織の領域が残る(硬化症)。体の自身の免疫細胞が神経系を襲うときに、炎症が生じる。炎症が、神経活動電位を遅らせて、又は遮断するようになり、MSの症候を引き起こす。炎症の繰り返される発症、又は突然の再発が、脳、及び脊髄の任意の領域に沿って生じ得る。それぞれの発作の位置、及び程度が変化するので、症候も変化する。通常、数日、数週、又は数月持続する症状発現は、症候の回数の低減、又は症候なし(緩解)と交互に現れる。再発(再燃)は、一般的であるが、緩解の期間を伴わないノンストップの進行も生じ得る。
本発明は、グルタミニルシクラーゼに関連したタンパク質のファミリーの新規メンバーを構成するグルタミニルシクラーゼ活性をもつタンパク質を提供し、全長タンパク質、選択的スプライス形態、サブユニット、及び突然変異体、並びに前述のものをコードするヌクレオチド配列を含む。また、本発明は、上記タンパク質の基質、相互作用タンパク質、アゴニスト、アンタゴニスト、又は阻害剤をスクリーニングする方法を、並びに更に、タンパク質、及び/又は突然変異体を含む医薬組成物、その誘導体、及び/又は類似体、及び/又はそれに対するリガンドを提供する。
本発明の態様に従って、異なる供与源からのQPCTLの推定されるアミノ酸配列(配列番号:11〜18、21、及び22)を有する成熟ポリペプチドをコードする配列番号:2〜9、19、及び20の単離された核酸配列(ポリヌクレオチド)が提供される。
溶媒の混合物の例には、10%(重量による)のエタノール水溶液、及び2%(重量による)DMSO水溶液を含む。溶液には、塩、緩衝剤、カオトロピック試薬、洗浄剤、防腐剤、その他を更に含んでいてもよい。或いは、タンパク質は、凍結乾燥粉末、又は結晶質固体などの固体の形態であってもよく、これには、また、残留溶媒、塩、又はその他を含んでいてもよい。
本発明に従った使用、及び方法のために適した潜在的QPCTL-阻害剤は、QC-阻害剤の構造、合成、及び使用方法に関してその全体が本明細書に組み込まれるWO2005/075436に開示されている。
アミロイドペプチド、例えばAβ 1-42(配列番号23)、及びAβ 1-40(配列番号24)が、例えばアミノペプチダーゼ、又はジペプチジルアミノペプチダーゼなどのタンパク質分解酵素によってN-末端で切断されて、Aβ-ペプチド3-42(配列番号25)、3-40(配列番号26)、11-42(配列番号27)、及び11-40(配列番号28)を生じることは、当該技術分野において公知である。これらの切断されたAβペプチドは、N末端にてグルタミン酸残基で始まり、従って、QCのための基質(また、WO2004/09862を参照されたい)、並びにおそらくまた配列番号11〜18、21及び22のQPCTL、好ましくは配列番号11、12、21及び22のヒトisoQC、最も好ましくは配列番号11及び12のヒトisoQCのための基質である。生じる配列番号29〜32のpGlu-Aβペプチドは、ピログルタミン酸化されていないペプチドよりもずっと疎水性であり、オリゴマー及び原繊維などのAβペプチド凝集体を形成する傾向がずっとあり、かつ高度に神経毒であることが示された。最後に、配列番号29-32のAβ-ペプチドは、アルツハイマー病、及びダウン症候群の発症において重要な役割を果たす。
前記疾患、及び/又は状態に苦しめられていることが疑われる対象から試料を収集する工程、
前記試料をQC-阻害剤と接触する工程、
前記対象が前記疾患、及び/又は状態によって苦しめられているかどうかを決定する工程。
(株化細胞、及び培地)
アフリカミドリザル腎臓株化細胞COS-7、ヒト神経芽細胞腫株化細胞SH-SY5Y、ヒト星細胞腫株化細胞LN405、ヒトケラチノサイト腫(keratinocytoma)株化細胞HaCaT、及びヒト肝臓癌株化細胞Hep-G2は、適切な細胞培地(Cos-7、SH-SY5Y、LN405、HaCaTのためには、DMEM、10% FBS)(Hep-G2のためには、RPMI1640、10%のFBS)中で、5% CO2(HaCaT、Hep-G2、COS-7)、又は10% CO2(SH-SY5Y、LN405)の加湿された雰囲気において、37℃にて培養した。
総RNAを、RNeasy Mini Kit(Qiagen)を使用してSH-SY5Y、LN405、HaCaT、及びHep-G2細胞から単離して、SuperScript II(Invitrogen)によって逆転写した。その後、ヒトisoQCを、プライマーisoQCh-1(センス、配列番号:53)、及びisoQCh-2、(アンチセンス、配列番号:54)を使用して、Herculase Enhanced DNA-Polymerase(Stratagene)での25μl反応において、産生されたcDNA産物の1:12,5希釈に対して増幅した。Hep-G2のPCR産物を、Strataprep PCR Purification Kit(Stratagene)を利用して精製し、シーケンシングによって確認した。
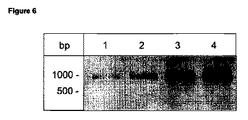
(RT-PCRを使用するヒトisoQC発現の解析)
ヒトisoQCの転写物は、株化細胞SH-SY5Y(図6、レーン1)、LN405(図6、レーン2)、HaCaT(図6、レーン3)、及びHep-G2(図6、レーン4)に存在することが見いだされた。Hep-G2のPCR産物をシーケンシングによって確認した。
ヒトisoQCの全長cDNAは、RT-PCRを使用してHep-G2細胞から単離した。簡潔には、Hep-G2細胞の総RNAをSuperScript II(Invitrogen)によって逆転写した。その後、ヒトisoQCをプライマーisoQChu-1(センス、配列番号:55)、及びisoQChu-2(アンチセンス、配列番号:56)を使用して、Herculase Enhanced DNA-Polymerase(Stratagene)での25μl反応において、産生されたcDNA産物の1:12,5希釈に対して増幅した。生じるPCR-産物をベクターpPCRScript CAM SK(+)(Stratagene)にサブクローニングして、シーケンシングによって確認した。
(ヒトisoQC-EGFP融合タンパク質をコードするプラスミドベクターの分子クローニング)
全てのクローニング手順は、標準的な分子生物学技術を適用して行った。ヒト細胞におけるヒトisoQC-EGFP融合タンパク質の発現については、ベクターpEGFP-N3(Invitrogen)を使用した。メチオニンIにて、又はメチオニンIIにて、いずれかで始まる天然のヒトisoQCのcDNAを、高感度緑色蛍光タンパク質(EGFP)をコードするプラスミドとインフレームでN-末端に融合した。プライマーisoQC EGFP-1Met I(配列番号:57)、及びisoQC EGFP-3、(配列列号:59)をメチオニンIで始まるヒトisoQCの増幅のために使用し、プライマーisoQC EGFP-2 Met II(配列番号:58)、及びisoQC EGFP-3(配列番号:59)をメチオニンIIで始まるヒトisoQCの増幅のために使用した。断片を、EcoRI、及びSalIの制限部位を使用して、ベクターpEGFP-N3(Invitrogen)に挿入し、正確な挿入をシーケンシングによって確認した。その後、ベクターを、EndoFree Maxi Kit(Qiagen)を使用して、細胞培養目的のために単離した。
加えて、ベクターpEGFP-N3(Invitrogen)のEGFP配列を、増幅のために、EGFP-1(センス)(配列番号:85)、及びEGFP-2(アンチセンス)(配列番号:86)を使用して、ベクターpcDNA 3.1(Invitrogen)に導入した。断片をpcDNA 3.1のXhoI部位に導入した。メチオニンI、及びIIで始まり、それぞれセリン53にて終わるhisoQCのN末端の配列を、メチオニンIで開始するhisoQCのN末端の断片については、isoQC EGFP-1 MetI(センス、配列番号:57)、及びhisoQC ss EGFP pcDNA as(アンチセンス)(配列番号:87)を使用して、並びにメチオニンIIで開始するhisoQCのN末端の断片については、isoQC EGFP-2 Met II(センス、配列番号:58)、及びhisoQC ss EGFP pcDNA as(アンチセンス)(配列番号:87)を使用して、ベクターpcDNA 3.1のEGFPとC末端に融合した。断片をベクターpcDNA 3.1のEcoRI、及びNotI制限部位に挿入した。その後、ベクターを、EndoFree Maxi Kit(Qiagen)を使用して、細胞培養目的のために単離した。
hQCについてプライマーhQC-1(センス)(配列番号:82)、及びhQC-2(アンチセンス)(配列番号:83)を、メチオニンIで開始するhisoQCについてisoQC EGFP-1 Met I(センス)(配列番号:57)、及びhisoQC pcDNA(アンチセンス)(配列番号:84)を、並びにメチオニンIIで開始するhisoQCについてisoQC EGFP-2 Met II(センス)(配列番号:58)、及びhisoQC pcDNA as(アンチセンス)(配列番号:84)を利用する増幅後に、天然のhQCは、ベクターpcDNA 3.1(+)(Invitrogen)のHindIII、及びNotI制限部位に挿入し、天然のhisoQCは、EcoRI、及びNotI制限部位に挿入した。
ヒトQCは、プライマーhQC-1(センス)(配列番号:82)、及びhQC C-フラグpcDNA as(アンチセンス)(配列番号:88)を適用して増幅後に、ベクターpcDNA 3.1のHindIII、及びNotI制限部位に、C末端のFLAG-タグと共にクローン化した。ヒトisoQCは、メチオニン1で開始するhisoQCについてプライマーisoQC EGFP-1 Met I(センス)(配列番号:57)、及びhisoQC C-フラグpcDNA as(アンチセンス)(配列番号:89)、並びにメチオニン2で開始するhisoQCについてプライマーisoQC EGFP-2 Met II(センス)(配列番号:58)、及びhisoQC C-フラグpcDNA as(アンチセンス)(配列番号:89)を使用する増幅後に、pcDNA 3.1にC末端のFLAG-タグと共に挿入した。
(COS-7、及びLN405のトランスフェクション、及び組織化学染色)
メチオニンI、又はメチオニンIIのいずれかで始まるヒトisoQC-EGFP融合タンパク質の発現のために、COS-7、及びLN405を、カバーガラスを含む6ウェルディッシュにおいて培養した。細胞は、80%の集密度まで培養して、製造業者のマニュアルに従ってLipofectamin2000(Invitrogen)を使用してトランスフェクトし、5時間の間トランスフェクション溶液中でインキュベートした。その後、溶液を適切な培養培地に置き換えて、細胞を一晩培養した。
(1. LN405のトランスフェクション、及び組織化学染色)
株化細胞LN405におけるメチオニンI、及びメチオニンIIで開始するヒトisoQC-EGFP融合タンパク質の発現(緑蛍光)は、生じるタンパク質の区画化を引き起こす。マンノシダーゼII抗体(赤蛍光)を使用するLN405のゴルジ帯域の対比染色、及びその後のマンノシダーゼIIとヒトisoQC-EGFPの重ね合せは、ゴルジコンパートメント内のヒトisoQC-EGFP融合タンパク質の局在化を示唆する(マージされたイメージの黄色の呈色)(図7,9)。これにより、メチオニンIIにて開始するヒトisoQCは、ヒトisoQC融合タンパク質のゴルジ局在化を生じるために十分であることが明らかである。
株化細胞LN405におけるメチオニンI、及びメチオニンIIで開始するヒトisoQC-EGFP融合タンパク質の発現に類似して、メチオニンI、及びメチオニンIIで開始するヒトisoQC-EGFP融合タンパク質の発現は、COS-7において、生じるタンパク質の区画化を引き起こす(緑蛍光)。マンノシダーゼII抗体(赤蛍光)を使用するCOS-7細胞のゴルジ帯域の対比染色、及びその後のマンノシダーゼIIとヒトisoQC-EGFPの重ね合せは、COS-7のゴルジコンパートメント内のヒトisoQC-EGFP融合タンパク質の局在化を示唆する(マージされたイメージの黄色の呈色)(図11,13)。また、COS-7細胞において、メチオニンIIにて開始するヒトisoQC-EGFP融合タンパク質の発現は、ゴルジ局在化を生じさせるために十分である。
(宿主株、及び培地)
大腸菌(Escherichia coli)株DH5をプラスミドの増殖のために使用し、大腸菌(E. coli)株BL21をヒトisoQCの発現のために使用した。大腸菌(E. coli)株は、製造業者の説明書(Qiagen(DH5α)Stratagene(BL21))に従って培養し、形質転換して、解析した。大腸菌(E. coli)のために必要とされる培地、すなわちルリア・バルダニ(Luria-Bertani)(LB)培地は、製造業者の推奨に従って製造した。
全てのクローニング手順には、標準的な分子生物学技術を適用した。大腸菌(E. coli)BL21における発現のためには、ベクターpET41a(Novagen)を使用した。コドン30(メチオニンIIから計数)で開始する成熟ヒトisoQCのcDNAを、GST-タグをコードするプラスミドとインフレームで融合した。プライマーhisoQC pET41a-1(配列番号:60)、及びhisoQC pET41a-2(配列番号:61)(表4)を利用する増幅の後、エンテロキナーゼ、及びC末端(His)6-タグのためのN末端のプロテアーゼ切断部位を導入した。サブクローニング後、断片を、Spe I、及びEcoR I制限部位を使用して発現ベクターに挿入した。
ヒトisoQCをコードする構築物をBL21細胞(Stratagene)に形質転換して、選択的LB寒天板上で37℃にて培養した。タンパク質発現は、1%グルコースを含むLB培地において37℃にて実施した。およそ0.8のOD600に到達後、isoQC発現を20μM IPTGで37℃にて4時間誘導した。細胞を遠心分離(4000×g、20分)によって培地から分離して、PBS(140mM NaCl、2,7mM KCl、10mM Na2HPO4、1,8mM KH2PO4、pH 7,3)に再懸濁して、凍結解凍の1サイクル、続いてフレンチプレスの1サイクルによって溶解した。細胞可溶化物を、リン酸含有緩衝液(50mM Na2HPO4、500mM NaCl pH 7,3)を使用して1.5lの最終容積に希釈して、13.400×gで4℃にて1時間遠心分離した。遠心分離後、生じる上清タンパク質濃度を、ブラッドフォード法を使用して決定した。必要に応じて、溶液を再び希釈して、0.6mg/mlの最終的な総タンパク質濃度を得た。GST-isoQC融合タンパク質は、4工程プロトコル(表5)を利用して精製した。精製は、図20におけるSDS-PAGE解析によって例証される。
(蛍光定量的アッセイ)
全ての測定は、30℃にてマイクロプレート(BMG Labtechnologies)のためのNovoStarリーダーで行った。QC活性は、最終的容積で、H-Gln-βNAを使用して蛍光定量的に評価した。試料は、0.2mM蛍光発生基質、0.05M Tris/HCl、pH 8.0中の0.25Uピログルタミルアミノペプチダーゼ(Qiagen、Hilden、Germany)、及び250μlのQCの適切に希釈された一定分量からなった。励起波長/放出波長は、320/410nmであった。アッセイ反応は、グルタミニルシクラーゼの添加によって開始した。QC活性は、アッセイ条件下でのβ-ナフチルアミンの標準曲線から決定した。1単位は、記述された条件下で1分あたりにH-Gln-βNAから1μモルpGlu-βNAの形成を触媒するQCの量として定義される。
このアッセイを使用して、大部分のQC基質についての動力学的パラメーターを決定した。QC活性は、補助的酵素としてグルタミン酸脱水素酵素を利用する連続法(Schilling, S.らの文献、2003 Biol Chem 384、1583-1592)を使用して、分光光度的に解析した。試料は、250μlの最終的な容積で、それぞれのQC基質、0.3mM NADH、14mM α-ケトグルタル酸、及び30U/mlグルタミン酸脱水素酵素からなった。反応をQCの添加によって開始して、340nmにて吸光度の低減のモニタリングによって8〜15分間追跡した。初速度を評価して、酵素活性をアッセイ条件下でのアンモニアの標準曲線から決定した。全ての試料は、マイクロプレートのためのSunriseリーダーを使用して30℃にて測定した。動力学的データは、GraFitソフトウェアを使用して評価した。
阻害剤試験については、試料組成は、推定上の阻害化合物を添加したこと以外、上記と同じであった。QC-阻害の迅速試験については、試料には、4mMのそれぞれの阻害剤、及び1KMの基質濃度を含んだ。阻害の詳細な研究、及びKi値の決定については、補助的酵素に対する阻害剤の影響を最初に調査した。全ての場合において、検出した酵素のいずれに対しても影響はなく、従って、QC阻害の信頼できる決定が可能であった。阻害定数は、GraFitソフトウェアを使用して、競合阻害についての一般的な方程式に対してプログレス曲線のセットをフィットすることによって評価した。
様々な異なる基質を、ヒトisoQCによる変換について評価した(表3)。全ての解析した基質は、isoQCによって変換され、ヒトQCと同様の比較的緩い全体的特異性を示した(Schilling, S.らの文献、2003 Biol Chem 384、1583-1592)。ヒトQCについて以前に観察されたように(Schilling, S.らの文献、2003 Biol Chem 384、1583-1592)、最高の特異性定数(kcat/KM)は、N末端のグルタミニル残基、例えばGln-AMCに隣接する大きな疎水性アミノ酸を有する基質について観察された。対照的に、Gln-Gluについて観察されたように、まさしくその位置における負に荷電した残基は、急激なドロップイン(drop in)特異性を生じ、isoQCの負に荷電した活性部位を示している。ヒトQCと比較して、両方の組換えiosQCは、より低い酵素活性を発揮した(図21)。相違は、1桁までであった。isoQCの特異性によれば、酵素がインビボで異なる基質の変換のための役割を担う、すなわちisoQCが多くの異なる生理学的基質の産生に関与すると仮定することは理にかなっている。
(宿主株、及び培地)
大腸菌(Escherichia coli)株DH5をプラスミドの増殖のために使用し、P.パストリス(P. pastoris)株X-33を酵母におけるヒトisoQCの発現のために使用した。大腸菌(E. coli)、及びP.パストリス(P. pastoris)株は、製造業者の説明書(Qiagen(DH5α)invitrogen(X-33))に従って培養し、形質転換して、解析した。大腸菌のために必要とされる培地、すなわちLuria-Bertani(LB)培地は、製造業者の推奨に従って製造した。ピチア・パストリス(Pichia pastoris)のために必要とされる培地、すなわちBMMY、BMGY、YPD、YPDS、及び抗生物質、すなわちZeocinの濃度は、ピチアマニュアルに記載されているように製造した(invitrogen、カタログ番号K1740-01)。また、マニュアルには、酵母の取扱いのための全ての関連した記述を含む。
全てのクローニング手順には、標準的な分子生物学技術を適用した。ピチア・パストリス(Pichia pastoris)X-33における発現のために、pPiCZαA(invitrogen)使用した。コドン30(メチオニンIIから計数)で開始する成熟ヒトisoQCのcDNAを、分泌経路にタンパク質を向けるα-ファクターをコードするプラスミドにインフレームで融合した。センスプライマーとしてプライマーhisoQC HIS C-Term pPICZAA-1(配列番号:62)、又はhisoQC N-Term pPICZAA-1(配列番号:63)、及びアンチセンスプライマとしてhisoQC His、N-Term pPICZAA-2(配列番号:64)、及びhisoQC His C-Term pPICZAA-2(配列番号:66)を利用する増幅の後、断片を、NotI、及びEcoR Iの制限部位を使用して、発現ベクターに挿入した(表4)。構築物に応じて、突然変異をコドン55(Ile)、及び351(Cys)に導入した。突然変異誘発は、標準的PCR技術に従って、続いてDpnI(quik-change II部位特異的変異誘発キット、Stratagene、Catalog No.200524)を使用する親DNAの消化によって行った。産生された構築物は、図17に模式的に図示してある。
1〜2μgのプラスミドDNAを、製造業者(BioRad)の説明書に従って、電気穿孔法によってコンピテントP.パストリス(P. pastoris)細胞の形質転換のために適用した。選択は、100μg/ml ゼオシンを含むプレートで行った。isQC発現による組換え酵母クローンを試験するために、組換え体を、2mlのBMGYを含む10mlの円錐チューブにおいて24時間培養した。その後、酵母を遠心分離して、0.5%のメタノールを含む2mlのBMMY中に再懸濁した。約72時間の間、24時間毎にメタノールを添加することにより、この濃度を維持した。その後、上清中のQC活性を決定した。最高の活性を示したクローンを、更なる実験、及び発酵のために選択した。発現された構築物に応じて、培地におけるisoQC-活性は、異なった(図18)。
ピチア・パストリス(Pichia pastoris)におけるisoQCの大規模発現のためには、ミニスケール発現に記載したように、条件を保持したが、総容積は、8Lであった。発現は、振盪フラスコにおいて行った。発現の後、細胞を遠心分離(1500×g、20分)によって培地から分離して、ペレットを廃棄した。上清のpHを中性に調整して、再び遠心し、最初の精製工程のために適用した。isoQCタンパク質は、3工程プロトコル(表7)を利用して精製した。精製は、図19におけるSDS-PAGE解析によって図示してある。
ヒトisoQCは、メチロトローフの酵母P.パストリス(P. pastoris)において首尾よく発現された。酵母における最高の発現条件を選択するために、いくつかの異なる構築物を産生した(図17)。図18に図示したように、発現され、及び発現する細胞の培地に存在するQC活性は、発現された構築物に応じて変化する。グリコシル化部位の導入により、適切な分泌を生じ、これは、構築物YSShisoQCN55IC351A C-His、及びYSShisoQCN55I C-Hisから観察することができる。培地において最高の活性ため、構築物YSShisoQCN55IC351A C-Hisを大規模に発現させて、精製した。精製は、表7に記載したように実施し、精製の収率は、59%であった。見かけ上均一なタンパク質は、より低い分子量に異動する変化によって証明されるように(図19)、グリコシル化された。グリコシル化は、酵素の触媒活性に影響を及ぼさなかった。
H-Gln-βNAを使用する蛍光定量的アッセイ(実施例5に記述した)を、触媒特異性のpH-依存性を調査するために適用した。反応は、7μMの基質濃度にて、すなわち[s]<<KMにて実施した。従って、観察された特異性定数は、基質変換のプログレス曲線の初速度から直接推定することができる。これらの研究では、反応緩衝液は、HCl、又はNaOHを使用して所望のpHに調整した0.075M 酢酸、0.075 M Mes、及び0.15M Trisからなった。緩衝液は、非常に広いpH範囲にわたって特定のイオン強度を保証する。獲得した酵素動力学的データの評価は、以下の方程式を使用して行った:
kcat/KM(pH)= kcat/KM(限定)*1/(1 +[H+]/KHS + KE1/[H+]+ KE1/[ H+]*KE2/[ H+])
式中、kcat/KM(pH)は、pH依存的(観察される)動力学的パラメーターを意味する。kcat/KM(限定)は、pH非依存的(「限定」)値を意味する。KHS、KE1、及びKE2は、それぞれ酸性pH範囲において解離する基、及び酵素の2つの解離する基の解離定数を意味する。全ての動力学的データの評価は、GraFitソフトウェア(バージョン5.0.4.、ウィンドウズ用、Erithacus software Ltd., Horley, UK)を使用して行った。
hisoQCは、pH 7〜8にて特異性の最適pHを示す。従って、触媒作用の最適pHは、ヒトQCに非常に類似する。3つの解離する基に基づくモデルに従ったデータのフィッティングにより、hisoQC、及びhQCのpH-依存性の満足な解釈を生じた(図22)。従って、両酵素反応の触媒作用は、同様の解離する基による影響を受け、一般に同様の触媒メカニズムを示唆する。
ヒトQCについて、該酵素は、N末端のグルタミン酸のピログルタミン酸への環化を触媒することが記述されている。従って、QCは、pGluで修飾されたアミロイドペプチドの産生に関与する。
マウスQC、及びマウスisoQCの組織分布を、定量的リアルタイムPCR技術を使用して調査した。いくつかの異なる器官、及び組織からのcDNAの解析の前に、マウスisoQCオープンリーディングフレームを、マウスisoQCの染色体のコード領域から推定した特異的プライマー(isoQCm MetI s(配列番号:68)、isoQCm MetIとして(配列番号:69)(表4)を適用して単離した。オープンリーディングフレームをベクターpPCR-Script CAM SK(+)(PCR-Script CAM Cloning Kit、Stratagene)にクローン化して、リアルタイムPCR決定における正の対照として、及びアッセイ条件下での標準曲線の作成のために使用した。misoQC発現の組織特異性の特性付けは、3〜6月齢のマウスからのcDNAを適用して達成した。総RNAは、RNA-単離キットII(Macherey、及びNagel)を使用して、30mgの組織から単離した。RNA濃度、及び純度は、ゲル電気泳動(アガロースゲル)、及び分光光度法によって評価した。cDNAの合成のためには、1μgのRNAを使用した。反応は、供給元の推奨に従って逆転写酵素Superscript II RT(Invitrogen)を適用して行い、cDNAは、-80℃に貯蔵した。
(Xg/μl DNA)/(bpのプラスミド長*660)*6.022*1023 = Y 分子/μl。DNA標準には、107-101分子/μl、及び限界濃度(100)の範囲の4つの濃度を含んだ。反応プロトコルを表8に示してある。結果は、図24に示してある。
図24に示したように、マウスQC、及びマウスisoQCは、試験した全ての器官において発現される。マウスQCとは対照的に、異なる器官の間でのマウスisoQCの発現の変動は、より小さく、転写の制御の厳密性が低いことを示している。mQCの発現についてのデータは、ノーザンブロットを使用して解析したウシQCの以前の解析に対応する(Pohl, T.らの文献、1991 Proc Natl Acad Sci U S A 88、10059-10063)。QCの最高の発現は、視床、海馬、及び皮質において観察された。従って、QC発現は、主にニューロン組織において検出される。QC発現は、脾臓、及び腎臓としての末梢器官ではほとんど検出されない。また、misoQCもニューロン組織に、しかしmQCと比較して低レベルで発現される。対照的に、末梢器官における発現レベルは、isoQCとQCとの間で非常に類似する。結果として、転写物濃度の結果に基づいて、組み合わさった活性(isoQC、及びQC)は、脳において最高であるはずである。従って、最高のQCタンパク質レベルは、アルツハイマー病、家族性英国痴呆、及び家族性デンマーク痴呆のようなアミロイド症によって苦しめられる器官に存在する。
(結果)
1,10-フェナントロリン、及びジピコリン酸などの複素環式キレート剤を使用した、異なる供与源からのQCの時間依存的阻害は、以前に調査されている。類似して、h-isoQCも、複素環式キレート剤1,10-フェナントロリン(図25)、及びジピコリン酸(図示せず)によって時間依存的に不活性化され、明らかに金属依存的活性を示す。更にまた、EDTAもまたhisoQCを阻害した(図25)。ヒトQCも、ブタQCも、マウスQCも、EDTAによって識別可能な阻害を示さないので、これは、QCとは全く対照的である。しかし、更に強力なEDTAによるhisoQCの阻害は、金属依存的触媒作用を示唆する。
(細胞分画)
トランスフェクション後の日に、発現するHEK293細胞をD-PBSで洗浄して、4℃にて5分間の500×gでの遠心分離によって収集した。その後に、D-PBSを廃棄して、細胞を1mlの破壊緩衝液(50mM Tris、50mM KCl、5mM EDTA、2mM MgCl2、HClでpH 7.6に調整)に再懸濁して、Potter細胞ホモジナイゼイター(homogenisator)において30回の粉砕によって分解した。懸濁液を4℃にて10分間700×gで遠心分離した。得られたペレットを300μlの破壊緩衝液に再懸濁して、細片画分(D)と命名した。生じる上清を4℃にて30分間20.000×gにて更に遠心分離した。ペレットは、重い膜画分(HM)を示し、200μl破壊緩衝液に再懸濁した。生じる上清を、超遠心機(Beckmann)を使用して、4℃にて1時間100.000×gにて遠心分離した。得られたペレットを200μl破壊緩衝液に再懸濁して、軽い膜画分(LM)と名付けた。上清は、可溶性画分(S)と命名した。細片、重い膜、及び軽い膜画分を10秒間超音波処理して、全ての画分のタンパク質含有量を、ブラッドフォード法を使用して決定した。その後、画分をQC活性について解析して、ウエスタンブロットを使用してマーカータンパク質を染色した。
更なる確認のために、hisoQC、及びhQC発現に由来するQC活性分布の生化学的解析を行った。メチオニンI、及びIIで開始する天然のhisoQC、並びにhQCを、それぞれHEK293細胞において発現させた。細胞分画の後、それぞれの画分のQC活性を、基質としてH-Gln-βNAを適用する蛍光アッセイを使用して決定した。空のベクター(pcDNA)をトランスフェクトした細胞において、特異的なQC活性をほとんど測定することができない。天然のhisoQC(MetI)、及びhisoQC(MetII)を発現するときに、QC活性は、重い膜画分(MetI:40±2 μモル/分/g;MetII:36±1.5μモル/分/g)、及び培地(MetI:30±2μモル/分/g;MetII:54±3μモル/分/g)において最高の比活性で容易に検出可能であった。対照的に、hQCは、培地(1339±76μモル/分/g)内で、続いて重い膜画分(251±21μモル/分/g)で最高の特異的なQC活性を示す(図26A)。
明確さのために、予測されるN末端の膜貫通ヘリックスが、ゴルジ複合体内におけるhisoQCの保持の役割を担うかどうか、膜貫通ヘリックスを含むMetI、及びMetIIにて開始するシグナルペプチドを、EGFPとインフレームにクローン化した。生じるベクターhisoQC(MetI)SS EGFP、及びhisoQC(MetII)SS EGFPをLN405細胞に発現させて、共焦点レーザースキャニング顕微鏡を使用して全長hisoQC EGFP融合タンパク質と同様に調べた。hisoQC(MetI)SS EGFPの発現は、全長hisoQC(MetI)EGFP融合タンパク質について観察される同じゴルジ複合体局在化を引き起こした。また、ミトコンドリアに対するhisoQC(MetI)SS EGFPの輸送は、観察されなかった(図27A)。加えて、N末端の切断されたペプチドhisoQC(MetII)SS EGFPの発現は、ゴルジ複合体内のタンパク質の濃縮も引き起こした。hisoQC(MetI)SS EGFPと類似して、ミトコンドリアのEGFP蛍光を記録することができなかった(図27B)。結果的に、hisoQCのN末端配列は、ER膜へのタンパク質の同時翻訳転位置を、及びゴルジ複合体内の保持を引き起こす。更にまた、hisoQC(MetII)SS EGFPの発現により、ゴルジ保持シグナルは、全体的にメチオニン19とセリン53(MetIにて開始したアミノ酸の計数)との間に存在するようにマップされた。
(qPCR解析分析)
ヒト癌腫株化細胞におけるヒトQPCTL発現は、本質的に実施例9に記載したように、定量的リアルタイムPCR(qPCR)技術を使用して行った。QPCTL mRNAを決定するために、ゲノムDNAの同時増幅を排除するためにエキソン/エキソン領域をカバーする、QuantiTect(登録商標)プライマーアッセイのプライマーを適用した。QPCRは、製造業者推奨に従って行った。反応混合物は、表9に示してあり、PCRプログラムは、表8に図示してある。
qPCRの結果は、ローター-遺伝子オペレーティングソフトウェア(Corbett reserch)を使用して評価した。
(異なる癌腫株化細胞におけるQPCTLの発現)
試験した癌株化細胞のなかで、ヒトメラノーマ細胞は、最高のQPCTL転写物の発現(約7000コピー/50ng総RNA)を示すが、ヒト軟部組織肉腫株化細胞は、最低のQPCTLの発現(365コピー/50ng総RNA)を示す。中央値で、膵臓癌腫は、2100コピーを示し、甲状腺癌腫は、3500コピー、及び胃癌腫は、4100コピーを有する(図29)。
最近、メラノーマ細胞が相当する高いQPCT発現を有することが示された(Gillis, J.S.の文献、J. Transl. Med. 4(2006) 4:27)。従って、異なるメラノーマ株化細胞におけるQPCTL発現を解析した。図30に図示したように、QPCTL発現は、試験した全てのメラノーマ株化細胞において検出された。株化細胞間の変動は、株Mel_ZL_11における2025コピー/50ng総RNA〜株Mel_ZL12における18043コピー/50ng総RNAまでで変動した。
QPCT、及びQPCTLの発現を軟部組織肉腫、胃癌腫、及び甲状腺癌腫の腫瘍組織において評価した。QPCTの最高の発現は、甲状腺癌腫、続いて胃癌腫、及び軟部組織癌腫において見いだされた(表11)。しかし、同じ順序をQPCTL発現について観察したが、しかし、スチューデントt検定によって明らかなように、QPCTL転写物のコピー数は、QPCT転写物について観察されるよりも常に低かった(P軟部組織癌腫= 0.001;P胃癌腫= 4.8E-7;P甲状腺癌腫= 0.04)(表11;図31)。
胃癌腫について、腫瘍分化の異なる段階を表す試料におけるQPCTL発現を調査した。対照は、腫瘍周囲正常組織として役立てた。正常と腫瘍組織との比較により、腫瘍組織における有意に高いQPCTL発現(p = 0.04)が明らかになった。未分化な胃癌腫は、正常組織よりも低いQPCTL発現を示す。低分化、及び十分から適度に分化した胃癌腫は、正常組織と比較して、中央値の相違は示さない(図32)。
異なる段階の甲状腺癌腫を、QPCT、及びQPCTL発現に関して調査した。段階は、世界健康組織(WHO)の命名法に従って濾胞性甲状腺癌腫(FTC)、乳頭甲状腺癌腫(PTC)、及び未分化甲状腺癌腫(UTC)として分類した。甲状腺腫を有する患者からの試料を対照として役立てた。
(株化細胞、及び培地)
刺激実験は、ヒト胚性腎臓株化細胞HEK293、ヒト急性単球性白血病株化細胞THP-1、及び濾胞性甲状腺癌腫株化細胞FTC-133を使用して行った。細胞は、37℃、及び5%のCO2に加湿された雰囲気において、適切な培地(HEK293についてDMEM、10% FBS、THP-1についてRPMI1640、10% FBS、及びFTC-133についてDMEM/F12、10% FBS)中で培養した。
HEK293、及びFTC-133細胞は、接着培養として培養し、THP-1細胞は、懸濁液中で培養した。刺激アッセイのためには、2×105細胞のFTC-133、及びHEK293細胞を24ウェルプレートに移した。HEK293の場合には、適当な接着を保証するために、プレートをコラーゲンIで被覆した。加えて、2×106細胞のTHP-1を24ウェル懸濁液プレートで培養した。全ての刺激実験は、無血清条件下で適用した。FTC-133は、一晩培養した。その後、細胞を更に24時間無血清の培地に適応させ、条件培地を新鮮な無血清培地によって置換することによって刺激を開始した。HEK293細胞を一晩培養し、その後、24時間を超えるの間の無血清条件下でのHEK293の培養の場合における形態学的変化のため、それぞれの薬剤を使用する刺激を無血清条件へ適応させることなく開始した。THP-1細胞は、それぞれの薬剤と共に無血清培地中のプレートにまいた。適用した刺激、及び終濃度は、表12に収載してある。
THP-1、HEK293、及びFTC-133細胞をそれぞれ2つの25cm2組織培養瓶にプレートにまいた。これにより、それぞれの株化細胞の1つのフラスコを負の対照として役立てて、通常の増殖条件下で24時間培養した。他方のフラスコは、嫌気的試薬(Anaerocult(登録商標)P、Merck)、及びインジケータと共に嫌気的バッグに置いた。バッグは、気密状態を保証するために封着した。また、細胞を24時間培養して、その後、総RNAをNucleo-Spin(登録商標)RNA II Kit(Macherey-Nagel)を使用して単離して、qPCRアッセイまで貯蔵した。
(HEK293、FTC-133、及びTHP-1におけるQPCT、及びQPCTLの基礎発現)
使用した株化細胞HEK293、FTC-133、及びTHP-1における基礎発現を、以下の刺激実験について評価した。QPCT、及びQPCTL転写物のコピー数を表13に要約してある。
QPCT、及びQPCTLのプロモーターの制御性結合部位、並びにこれらの制御を引き起こすシグナル伝達経路は、今までに記述されていない。従って、種々の株化細胞を使用する刺激実験、及び刺激を行った。HEK293細胞におけるQPCT mRNAレベルは、TNF-α、HGF、及び酪酸を使用する刺激によって増加した。加えて、QPCT/QPCTL基質としてのCCL2の制御を調査した。TNF-α、及び酪酸は、HEK293におけるCCL2転写物の量を増加させた。HGFは、CCL2発現に影響を有さなかった。対照的に、QPCTLは、TNF-α、HGF、及び酪酸によって調節されなかった(図35)。
QPCT発現は、多数の刺激によって誘導されたので、QPCT誘導が、QPCT基質CCL2、CCL7、CCL8、及びCCL13の誘導と組み合わせて生じるかどうかの問題が生じた。従って、LPS(1μg/ml)、LPS(10μg/ml)、TGF-β(100ng/ml)、及びTNF-α(100ng/ml)をそれぞれ使用する刺激を、THP-1単球を使用して行った。THP-1は、刺激された細胞と負の対照との比較のために重要である全てのケモカインを基礎レベルにて発現する。
QPCTL発現は、化学剤、生物活性、ペプチド、又はLPSによって調節することができなかった。従って、本発明者らはQPCTL発現が低酸素によって調節されるかどうかを試験した。図39に要約したとおり。低酸素は、選択的にQPCTLの発現を誘導したが、QPCTの発現を誘導しなかった。比較として、低酸素誘導因子1a(HIF1a)は、15%(図39A)、及び45%(図39C)まで抑制された。データは、低酸素に対するQPCTLの関連を示唆する。
合成スキーム1:実施例1〜53、96〜102、136〜137の合成
ESI-Massスペクトルは、SCIEX API 365分光計(Perkin Elmer)で得た。1H-NMR(500MHz)データは、溶媒としてDMSO-D6を使用してBRUKER AC 500で記録した。化学シフトは、テトラメチルシランからのppm低磁場として表してある。分割パターンは、以下の通りに示した:s(一重項)、d(二重項)、dd(二重項の二重項)、t(三重項)、m(多重項)、及びbr(広域シグナル)。
実施例1〜12、及び14〜53
1H-イミダゾール-1-プロパンアミンを、対応するイソチオシアナートのエタノール溶液と還流下で8時間反応させた。その後、溶媒を除去して、残りの油を塩化メチレンに溶解した。有機層をNaHCO3の飽和溶液で2回、続いてNaHSO4、及び鹹水で洗浄し、乾燥させ、次いで蒸発させた。残りの固体を酢酸エチルから再結晶して、80〜98% の収率で実施例チオ尿素を得た。
1-(3-(1H-イミダゾール-1-イル)プロピル)-3-(3,4-ジメトキシフェニル)チオ尿素
4.0mmolの3,4-ジメトキシフェネチルイソチオシアナート、及び4.0mmolの3-(1H-イミダゾール-1-イル)アルキル-1-アミンを10mLの無水エタノールに溶解した。還流下で2時間撹拌した後、溶媒を蒸発させて、生じた固体をエタノールから再結晶した。
収率:0.66 g(51.3%);mp:160.0-161.0℃
1H NMR δ 1.8 - 2.0 (m, 2H), 3.4 - 3.5 (m, 2H), 3.75 (s, 6H), 3.9 - 4.0 (m, 2H), 6.7 - 6.8 (m, 1H), 6.9 (br m, 2H), 6.95 (s, 1H), 7.15 (s, 1H), 7.55 (br s, 1H), 7.6 (s, 1H), 9.3 (s, 1H); MS m/z 321.2 (M+H), 253.3 (M-C3H3N2・)
1H-イミダゾール-1-プロパンアミンを対応するイソシアネートのエタノール溶液と還流下で8時間反応させた。その後、溶媒を除去して、残りの油を塩化メチレンに溶解した。有機層をNaHCO3の飽和溶液で2回、続いてNaHSO4、及び鹹水で洗浄し、乾燥させて、次いで蒸発させた。残りの固体を酢酸エチルから再結晶し、85〜90% の収率で実施例尿素を得た。
1H-イミダゾール-1-アルキルアミンは、文献に従って、□-臭素アルキルフタルイミド、及びイミダゾリウム塩、及びその後のヒドラジン分解により製造した。生じた精製物を実施例1〜53に従ってチオ尿素に変換し、88%(実施例136)の、及び95%(実施例137)の収率を示す。
全ての実施例は、対応するチオ尿素から、水溶性カルボジイミド(WSCD)、及び1H-イミダゾール-1-プロパンアミンと、乾燥ジメチル型アミド中で室温にて2時間反応させることによって作製され、40〜87%収率で三置換のグアニジンを与える。
イミダゾールを、対応する臭素メチルフェニルシアニドのDMF溶液と、1当量のNaHを利用して室温下で3時間反応させ、1H-イミダゾール-1-メチルフェニルシアニドを与えた。溶媒を除去して、生じた油をジオキサンに再融解した。シアニドを、1当量のLiAlH4を使用して対応するアミンに変換した。KHSO4の飽和溶液を添加した後、ジオキサンを蒸発させて、水層をCHCl3によって抽出した。有機層を真空中で濃縮して、アミンを実施例1〜53に従って、対応するチオ尿素に変換し、78%(実施例103)、及び65%(実施例104)、及び81%(実施例105)を与えた。
対応するメタンスルホナート-2-メチルプロピル-フタルイミドから開始して、アミンを実施例136〜137においてアミンについて記述したように合成した。生じた生成物を実施例1〜53に従ってチオ尿素に変換し、25〜30%の全収率で実施例106〜109を与えた。
1H-イミダゾール-1-プロパンアミンを、対応する2-クロロベンゾ[d]チアゾールのトルオール溶液と130℃の温度にて24時間反応させた。溶媒の除去、及びメタノールから再結晶後、実施例110〜112を55〜65% の量で得た。
1H-イミダゾール-1-プロパンアミンを0℃の温度にて1当量のCAIBE、及びN-メチルモルホリンを添加することによって、対応する2-フェニル酢酸の乾燥ジオキサン溶液と反応させた。2時間後、混合物を室温に温めて、混合物を12時間撹拌した。溶媒を除去後、生じた油を塩化メチレンに再融解し、有機層をNaHCO3の水溶液、及び水によって洗浄し、乾燥させて、溶媒を蒸発させた。残りの油をジオキサンに溶解してLaweson試薬を添加した。12時間撹拌後、NaHCO3の飽和溶液を添加した。ジオキサンを蒸発させて、水層を酢酸エチルによって抽出した。有機層を分離し、乾燥させて、溶媒を蒸発させた。残りの固体をアセチルアセテート/エーテルから結晶化し、62〜85%の全収率で113〜118、120〜124、及び126〜132を与えた。
N-(3-(1H-イミダゾール-1-イル)プロピル)-2-(3,4-ジメトキシフェニル)エタンチオアミド
4.0mmolのトリエチルアミン、及び4.0mmolの3-(1H-イミダゾール-1-イル)アルキル-1-アミンの混合物、20mLのジオキサンを、氷冷した4.0mmolの2-(3,4-ジメトキシフェニル)アセチルクロライドの30mLのジオキサン中の撹拌溶液に滴加した。混合物を室温に温て、次いで、1時間撹拌した。減圧によって溶媒を除去後、残渣を50mLのジクロロメタンに再融解した。有機層を30mLのNaHCO3の飽和水溶液、及び水によって洗浄した。濾過して、有機溶液を乾燥させて、溶媒を減圧下で除去した。50mLの乾燥ジオキサンに再融解後、2.2mmol のLawesson試薬を添加して、混合物を90℃まで加熱して、8時間撹拌した。溶媒を減圧によって除去して、残渣を50mLのジクロロメタンに再融解した。有機層をNaHCO3の飽和水溶液によって3回洗浄し、水によって3回行い、乾燥させて、濾過し、次いで、有機溶媒を除去した。化合物を、2mmの層の厚さのシリカプレート、及び溶出系としてCHCl3/MeOH勾配を利用する遠心-クロマトグラフィー装置(Harrison Research Ltd.)を使用するクロマトグラフィーによって精製した。
収率:0.14 g(10.6%);融点:148.0-150.0℃
1H NMR δ 2.0 - 2.15 (br m, 2H), 3.4 - 3.5 (m, 2H), 3.7 (s, 6H), 6.75 - 6.8 (m, 2H), 4.1 - 4.2 (m, 2H), 6.8 - 6.9 (m, 2H), 6.95 - 7.0 (m, 1H), 7.4 (s, 1H), 7.75 - 7.85 (br m, 1H), 8.6 (s, 1H), 10.2 (s, 1H); MS m/z 320.2 (M+H), 252.2 (M-C3H3N2・)。
N-(3-(1H-イミダゾール-1-イル)プロピル)-1-(3,4-ジメトキシフェニル)シクロプロパンカルボチオアミド
11.06mmolの3,4-ジメトキシフェニルアセトニトリル、34.8mmolの2-ブロモ-1-クロロエタノール、及び1.16mmolの塩酸トリエチルベンジルアンモニウムを、10mLのKOH(60%)の水溶液に溶解した。混合物を超音波浴に移して、室温で3時間勢いよく撹拌した。生じた懸濁液を40mLの水で希釈して、20mLのジクロロメタンによって3回抽出した。合わせた有機層を、ここで塩酸(1N)の水溶液によって洗浄し、Na2SO4上で乾燥させて、溶媒を減圧下で除去した。残りの油をシリカゲル、及び溶出系として酢酸エチル/ヘプタンを使用するフラッシュクロマトグラフィーによって精製し、0.81g(34.4%)の1-(3,4-ジメトキシフェニル)シクロプロパンカルボニトリルを生じて、3.9mmolの1-(3,4-ジメトキシフェニル)シクロプロパンカルボニトリル、及び11.2mmolのKOHを80mLのエチレングリコールに懸濁した。混合物を還流下で12時間撹拌した。次いで、80mLの水を添加して、水層をエーテルで2回抽出した。HCl(1N)を使用してpH = 4〜5の値にpH調整後、水層をエーテルによって3回抽出し、次いで合わせた有機層をNa2SO4上で乾燥させて、溶媒を除去し、0.81g(93.5%)の1-(3,4-ジメトキシフェニル)シクロプロパンカルボン酸を生じた。
収率:0.33 g(46.2%);融点:127.0 -127.5℃
1H NMR δ 1.1 - 1.2 (t, 2H), 1.55 - 1.6 (t, 2H), 2.0 - 2.1 (m, 2H), 3.5 - 3.6 (m, 2H), 3.7 - 3.8 (s, 6H), 4.1 - 4.2 (t, 2H), 6.8 - 6.9 (m, 3H), 7.65 (s, 1H), 7.75 (s, 1H), 8.8 (m, 1H), 9.05 (s, 1H; MS m/z 346.0 (M+H), 278.2 (M-C3H3N2・), 177.1 (M-C6H8N3S・)
1当量のトリエチルアミン、及び3,4-ジメトキシアニリンのジオキサン中の混合物を、対応するω-ブロモアルキル酸性塩化物の撹拌溶液に0℃の温度にて添加した。溶液を室温に温めて、2時間撹拌した。溶媒を蒸発させて、残りの油をジクロロメタンに再融解した。有機層を水によって洗浄して、乾燥させて、濾過して、溶媒を減圧下で除去した。
融点:122〜122.5℃
1H NMR δ 1.85 - 1.95 (m, 2H), 2.8 (s, 3H), 3.2 - 3.5 (br d, 2H), 3.8 - 3.9 (m, 2H), 6.85 (d, 1H), 7.15 (d, 1H), 7.3 - 7.5 (br d, 2H), 7.65 (s, 1H); MS m/z 199.1 (M+H), 221.3 (M+Na), 131.0 (M-C3H3N2・)。
融点:147.0-147.5℃
1H NMR δ 1.3 - 1.4 (s, 9H), 1.85 -1.95 (m, 2H), 3.5 (t, 2H), 3.8 (t, 2H), 6.85 (d, 1H), 7.15 (d, 1H), 7.3 - 7.5 (br d, 2H), 7.65 (s, 1H); MS m/z 241.1 (M+H), 173.1 (M-C3H3N2・)。
融点:127.0 -128.0℃
1H NMR δ 1.85 - 1.95 (m, 2H), 3.2 - 3.5 (br d, 2H), 3.8 - 3.9 (m, 2H), 4.6 (s, 2H), 6.8 (d, 1H), 7.15 (d, 1H), 7.19 - 7.35 (m, 5H), 7.5 - 7.6 (br d, 2H), 7.85 (s, 1H); MS m/z 275.3 (M+H), 207.1 (M-C3H3N2・)。
融点:166.5 -167.0℃
1H NMR δ 1.95 - 2.05 (m, 2H), 3.3 - 3.5 (br d, 2H), 3.9 - 4.0 (m, 2H), 6.85 (d, 1H), 7.05 (m, 1H) 7.15 (d, 1H), 7.25 (m, 2H), 7.35 (m, 2H), 7.6 (s, 1H), 7.8 (br s, 1H), 9.5 (br s, 1H); MS m/z 261.1 (M+H), 193.2 (M-C3H3N2・)。
融点:147.0 -148.0℃
1H NMR δ 1.95 - 2.05 (m, 2H), 3.3 - 3.5 (br d, 2H), 3.9 - 4.05 (m, 2H), 6.85 (d, 1H), 7.05 - 7.15 (m, 3H), 7.3 - 7.4 (m, 2H), 7.6 (s, 1H), 7.7 - 7.8 (br s, 1H), 9.4 (br s, 1H); MS m/z 279.3 (M+H), 211.2 (M-C3H3N2・)。
融点:100.0 -100.5℃
1H NMR δ 1.15 - 1.2 (t, 3H), 1.9 - 2.0 (m, 2H), 2.5 - 2.6 (m, 2H), 3.3 - 3.5 (br d, 2H), 3.9 - 4.05 (m, 2H), 6.85 (d, 1H), 7.1 - 7.2 (m, 3H), 7.25 - 7.3 (m, 2H), 7.6 (s, 1H), 7.7 - 7.8 (br s, 1H), 9.4 (br s, 1H); MS m/z 289.3 (M+H), 221.1 (M-C3H3N2・)。
融点:154.5 -155.0℃
1H NMR δ 1.9 - 2.1 (br m, 2H), 3.4 - 3.6 (br d, 2H), 3.95 - 4.1 (br m, 2H), 6.85 (d, 1H), 7.2 (d, 1H), 7.6 - 7.8 (m, 5H), 8.2 (br s, 1H), 9.9 (br s, 1H); MS m/z 329.3 (M+H), 261.2 (M-C3H3N2・)。
融点:170.0 -171.0℃
1H NMR δ 1.9 - 2.1 (br m, 2H), 2.4 - 2.5 (s, 3H), 3.2 3.5 (br m, 2H), 3.9 - 4.1 (m, 2H), 6.85 (d, 1H), 7.15 (d, 1H), 7.5 - 7.65 (br m, 3H), 7.8 - 7.9 (m, 2H), 8.1 (m, 2H), 9.8 (br s, 1H); MS m/z 303.2 (M+H), 235.1 (M-C3H3N2・)。
融点:125.0 -125.5℃
1H NMR δ 1.8 - 2.0 (br m, 2H), 3.2 - 3.5 (br m, 2H), 3.7 (s, 3H), 3.9 - 4.0 (m, 2H), 6.7 - 6.9 (m, 3H), 7.1 - 7.2 (m, 3H), 7.5 (s, 1H), 7.6 (s, 1H), 9.2 (s, 1H); MS m/z 291.1 (M+H), 223.2 (M-C3H3N2・)。
融点:120.0 -120.5℃
1H NMR δ 1.8 - 2.0 (br m, 2H), 3.4 - 3.5 (br m, 2H), 3.75 (s, 6H), 3.9 - 4.0 (m, 2H), 6.5 (d, 1H), 6.6 (s, 1H), 6.9 (s, 1H), 7.15 (s, 1H), 7.3 (d, 1H), 7.5 (br s, 1H), 7.6 (s, 1H), 9.75 (s, 1H); MS m/z 321.2 (M+H), 253.3 (M-C3H3N2・)。
融点:142.0 -143.0℃
1H NMR δ 1.8 - 2.0 (br m, 2H), 3.4 - 3.5 (br m, 2H), 3.6 (s, 6H), 3.95 - 4.0 (m, 2H), 6.25 (m, 1H), 6.6 (m, 2H), 6.9 (s, 1H), 7.2 (s, 1H), 7.6 (s, 1H), 7.8 (s, 1H), 9.5 (s, 1H); MS m/z 321.2 (M+H), 253.3 (M-C3H3N2・)。
融点:103.0 -103.5℃
1H NMR δ 1.9 - 2.0 (br m, 2H), 3.3 - 3.5 (br d, 2H), 3.9 - 4.0 (m, 2H), 4.2 - 4.3 (m, 4H), 6.7 (m, 1H), 6.8 - 6.8 (m, 1H), 6.9 (m, 2H), 7.2 (s, 1H), 7.6 (m, 2H), 9.3 (s, 1H); MS m/z 319.3 (M+H), 251.3 (M-C3H3N2・)。
融点:115.0 -115.6℃
1H NMR δ 1.9 - 2.1 (br m, 2H), 3.4 - 3.5 (br d, 2H), 4.05 - 4.15 (m, 2H), 6.0 (s, 2H), 6.7 (m, 1H), 6.8 - 6.85 (m, 1H), 6.95 (d, 1H), 7.25 (s, 1H), 7.45 (s, 1H), 7.7 (br s, 1H), 8.5 (br s, 1H), 9.4 (br s, 1H); MS m/z 305.2 (M+H), 237.2 (M-C3H3N2・)。
融点:124.5 -125.5℃
1H NMR δ 1.8 - 2.0 (m, 2H), 3.4 - 3.5 (br m, 2H), 3.6 (s, 3H), 3.7 (s, 6H), 3.9 - 4.0 (m, 2H), 6.65 (m, 2H), 6.85 (s, 1H), 7.2 (s, 1H), 7.6 (s, 1H), 7.7 (br s, 1H), 9.4 (s, 1H); MS m/z 351.3 (M+H), 283.2 (M-C3H3N2・)。
融点:89.5 -90.0℃
1H NMR δ 1.9 - 2.1 (br m, 2H), 3.4 - 3.5 (br m, 2H), 3.7 (s, 3H), 3.9 - 4.0 (m, 2H), 6.6 - 6.7 (m, 1H), 6.8 - 6.9 (m, 2H), 7.1 (m, 2H), 7.15 - 7.25 (br m, 1H), 7.6 (s, 1H), 7.8 (br s, 1H), 9.5 (s, 1H); MS m/z 291.1 (M+H), 223.2 (M-C3H3N2・)。
1H NMR δ 1.5 (br m, 3H), 1.9 - 2.0 (br m, 2H), 3.4 - 3.5 (br m, 2H), 3.9 - 4.0 (br m, 4H), 6.8 - 6.9 (m, 2H), 6.95 (s, 1H), 7.15 - 7.2 (m, 2H), 7.25 (s, 1H), 7.55 - 7.6 (br s, 1H), 7.8 (s, 1H), 9.3 (s, 1H); MS m/z 305.2 (M+H), 237.2 (M-C3H3N2・)。
融点:140.0 -140.5℃
1H NMR δ 1.8 - 2.05 (br m, 2H), 2.5 (s, 3H), 3.3 - 3.5 (br m, 2H), 3.9 - 4.1 (m, 2H), 6.9 (m, 1H), 7.1 - 7.3 (br m, 5H), 7.6 (s, 1H), 7.75 (br s, 1H), 9.4 (s, 1H); MS m/z 307.2 (M+H), 239.2 (M-C3H3N2・)。
融点:165.0 . 166.0 ℃
1H NMR δ 1.9 - 2.05 (m, 2H), 3.3 - 3.5 (br d, 2H), 3.95 - 4.05 (m, 2H), 6.85 (d, 1H), 7.15 (d, 1H), 7.6 (d, 1H), 7.7 (m, 2H), 8.1 (m, 2H), 8.3 (br s, 1H), 10.1 (br s, 1H); MS m/z 306.2 (M+H), 237.9 (M-C3H3N2・)。
融点:146.5 -147.0℃
1H NMR δ 1.9 - 2.0 (m, 2H), 2.9 (s, 6H), 3.4 (m, 2H), 3.9 - 4.0 (m, 2H), 6.7 (m, 2H), 6.9 (s, 1H), 7.05 - 7.1 (m, 2H), 7.15 (s, 1H), 7.4 (br s, 1H), 7.6 (s, 1H), 9.2 (s, 1H); MS m/z 304.2 (M+H), 236.0 (M-C3H3N2・)
融点:114.5 -115.0℃
1H NMR δ 1.7 - 1.9 (m, 2H), 2.9 - 3.1 (m, 2H), 3.7 (2s, 6H), 3.9 - 4.0 (m, 2H), 6.1 (t, 1H), 6.7 (s, 2H), 6.8 (s, 1H), 7.15 (d, 2H), 7.6 (s, 1H), 8.2 (s, 1H); MS m/z 321.2 (M+H), 253.3 (M-C3H3N2・)。
融点:150.5 -151.5℃
1H NMR δ 0.9 (d, 3H), 2.3 - 2.4 (m, 2H), 2.5 (s, 1H), 3.7 (d, 6H), 4.0 - 4.1 (br m, 1H), 4.15 - 4.25 (br m, 1H), 6.75 - 6.8 (m, 1H), 6.85 (m, 1H), 6.9 - 7.0 (m, 1H), 7.65 (s, 1 H), 7.75 (s, 2H), 9.1 (s,1H), 9.5 (s, 1H); MS m/z 335.6 (M+H), 267.1 (M-C3H3N2・)。
融点:155.0 〜157.5℃
1H NMR δ 0.9 (d, 3H), 2.3 - 2.4 (m, 2H), 2.5 (s, 1H), 3.7 (d, 6H), 4.0 - 4.1 (br m, 1H), 4.15 - 4.25 (br m, 1H), 6.75 - 6.8 (m, 1H), 6.85 (m, 1H), 6.9 - 7.0 (m, 1H), 7.65 (s, 1H), 7.75 (s, 2H), 9.1 (s,1H), 9.5 (s, 1H); MS m/z 335.4 (M+H), 267.2 (M-C3H3N2・)。
融点:166.5 -168.5℃
1H NMR δ 0.7 - 0.8 (br m, 2H), 1.85 - 1.9 (m, 1H), 2.15 - 2.2 (m, 1H), 2.2 - 2.3 (m, 1H), 3.4 - 3.5 (m, 1H), 3.7 (d, 6H), 4.2 (s, 1H), 4.95 (s, 1H), 6.75 - 6.8 (br m, 1H), 6.85 - 6.9 (br m, 1H), 7.0 (s, 1H), 7.5 (m, 1H), 7.6 (m, 1H), 7.7 (s, 0.5H), 7.8 (s, 0.5H), 8.85 (s, 0.5 H), 9.1 (s, 0.5H), 9.35 (s, 0.5H), 9.45 (s, 0.5H); MS m/z 347.2 (M+H), 279.2 (M-C3H3N2・), 137.5 (M-C9H13N4S・)。
1H NMR δ 1.95 - 2.15 (m, 2H), 3.25 - 3. 35 (m, 2H), 4.0 - 4.1 (t, 2H), 6.9 (s, 1H), 6.95 - 7.05 (t, 1H), 7.15 7.2 (m, 2H), 7.35 7.4 (d, 1H), 7.60 7.70 (m, 2H), 8.0 8.1 (br s, 1H); MS m/z 259.4 (M+H), 191.3 (M-C3H3N2・)。
1H NMR δ 1.95 - 2.15 (m, 2H), 3.25 - 3. 35 (m, 2H), 4.0 - 4.1 (t, 2H), 6.9 (s, 1H), 7.1 7.2 (d, 2H), 7.3 7.4 (d, 1H), 7.65 (s, 1H), 7.8 (s, 1H), 8.2 (s, 1H); MS m/z 293.3 (M+H), 225.3 (M-C3H3N2・)。
1H NMR δ 1.9 - 2.05 (m, 2H), 3.2 - 3. 3 (m, 2H), 3.7 (s, 3H), 4.0 - 4.1 (t, 2H), 6.7 6.8 (d, 1H), 6.9 (s, 1H), 7.15 7.2 (s, 1H), 7.2 7.3 (m, 2H), 7.65 (s, 1H), 7.8 (s, 1H); MS m/z 289.1 (M+H), 221.4 (M-C3H3N2・)。
融点:82.0 -82.5℃
1H NMR δ 1.4 - 1.55 (d, 3H), 1.9 - 2.0 (m, 2H), 3.4 - 3.5 (m, 2H), 3.85 - 3.95 (m, 2H), 4.0 - 4.1 (q, 1H), 6.8 - 6.9 (s, 1H), 7.1 (s, 1H), 7.15 - 7.2 (m, 1H), 7.2 - 7.3 (m, 2H), 7.35 - 7.4 (m, 2H), 7.55 (s, 1H), 10.1 (s, 1H); MS m/z 274.4 (M+H), 206.3 (M-C3H3N2・)。
融点:82.5 -83.5℃
1H NMR δ 1.4 - 1.55 (d, 3H), 1.9 - 2.0 (m, 2H), 3.4 - 3.5 (m, 2H), 3.85 - 3.95 (m, 2H), 4.0 - 4.1 (q, 1H), 6.8 - 6.9 (s, 1H), 7.1 (s, 1H), 7.15 - 7.2 (m, 1H), 7.2 - 7.3 (m, 2H), 7.35 - 7.4 (m, 2H), 7.55 (s, 1H), 10.1 (s, 1H); MS m/z 274.4 (M+H), 206.3 (M-C3H3N2・)。
融点:137.5 -139.0℃
1H NMR δ 1.55 - 1.75 (br m, 2H), 1.85 - 1.95 (br m, 2H), 2.4 - 2.5 (br m, 2H), 2.7 - 2.85 (br m, 2H), 3.3 - 3.5 (br m, 2H), 3.8 (m, 2H), 6.9 (s, 1H), 7.0 (s, 1H), 7.3 (m, 2H), 7.45 (s, 1H), 7.5 (m, 2H), 9.6 (t, 1H); MS m/z 334.3 (M+H), 266.1 (M-C3H3N2・)。
融点:140.0 -141.0℃
1H NMR δ 1.5 - 1.65 (br m, 4H), 1.8 - 1.9 ( m, 2H), 2.0 - 2.1 (m, 2H), 2.6 (m, 2H), 3.4 - 3.5 (m, 2H), 3.7 - 3.8 (m, 2H), 6.85 (s, 1H), 7.0 (s, 1H), 7.35 (m, 2H), 7.4 (m, 2H), 7.5 (s, 1H), 9.4 (t, 1H); MS m/z 348.2 (M+H), 280.2 (M-C3H3N2・)。
融点:162.5 -164.0℃
1H NMR δ 1.2 - 1.3 (m, 1H), 1.35 - 1.5 (br m, 5H), 1.85 - 2.0 (br m, 4H), 2.4 - 2.6 (br m, 2H), 3.4 - 3.5 (m, 2H), 3.7 (s, 3H), 3.8 (m, 2H), 6.8 (m, 3H), 7.0 (s, 1H), 7.3 (m, 2H), 7.5 (s, 1H), 9.2 (t, 1H); MS m/z 358.3 (M+H), 290.3 (M-C3H3N2・)。
融点:129.0 -129.5℃
1H NMR δ 1.0 - 1.1 (m, 2H), 1.5 - 1.6 (m, 2H), 1.9 - 2.0 (br m, 2H), 3.4 - 3.5 (m, 2H), 3.7 (s, 3H), 3.9 (m, 2H), 6.9 (m, 3H), 7.1 (s, 1H), 7.2 - 7.3 (m, 2H), 7.6 (s, 1H), 8.9 (br s, 1H); MS m/z 316.0 (M+H), 248.4 (M-C3H3N2・)。
融点:128.0 〜128.5℃
1H NMR δ 1.65 - 1.70 (m, 2H), 1.75 - 1.80 (m, 2H), 2.7 - 2.75 (m, 2H), 3.7 (s, 3H), 3.75 (s, 3H), 4.0 - 4.05 (t, 2H), 6.9 - 7.0 (m, 2H), 7.2 (s, 1H), 7.3 (d, 1H), 7.5 (s, 1H), 7.75 (s, 1H), 11.0 (s, 1H); MS m/z 320.2 (M+H), 252.2 (M-C3H3N2・)。
融点:157.5 -159.0℃
1H NMR δ 3.7 (2 s, 6H), 3.8 (m, 2H), 4.2 (m, 2H), 6.7 (m, 1H), 6.85 (m, 1H), 6.9 (m, 2H), 7.15 (s, 1H), 7.5 (br s, 1H), 7.6 (s, 1H), 9.5 (s, 1H); MS m/z 307.2 (M+H), 239.1 (M-C3H3N2・)。
Claims (44)
- (a)配列番号:11〜18の1つのアミノ酸配列を含むポリペプチド、若しくは(b)それに対して少なくとも約75%類似であり、かつ同じ生物学的機能を示すアミノ酸配列を有するポリペプチドをコードするか;又は配列番号:2〜9の1つの選択的スプライスバリアントであるか;又は前記(a)若しくは(b)をコードする核酸からの少なくとも14個の隣接するヌクレオチドを含むプローブであるか;又は前述のいずれか一つに対して相補的である、単離された核酸
- DNA又はRNAである、請求項1記載の単離された核酸。
- 配列番号:2〜9のいずれか1つの全長を含むDNA転写物であるか、又は配列番号:2〜9の1つの全コード領域に対して相補的である、請求項1記載の単離された核酸。
- 請求項3記載のDNAに対して向けられたアンチセンスオリゴヌクレオチド。
- 配列番号:2〜9のいずれか1つの全長を含む、RNA転写物である、請求項1記載の単離された核酸。
- 配列番号:2〜9の1つの選択的スプライスバリアントである、請求項1記載の単離された核酸。
- 配列番号:19、又は20に対応する、請求項6記載のスプライスバリアント。
- 請求項6、又は7記載の核酸によってコードされるポリペプチド。
- 配列番号:11〜18の1つに対して少なくとも約85%類似するアミノ酸配列を有するポリペプチドをコードする、請求項1記載の単離された核酸。
- 配列番号:11〜18の1つに対して少なくとも約95%類似するアミノ酸配列を有するポリペプチドをコードする、請求項1記載の単離された核酸。
- 配列番号:11〜18の1つと少なくとも約50%の同一性を有するポリペプチドをコードする、請求項1記載の単離された核酸。
- 配列番号:2〜9の1つからの少なくとも14個の隣接するヌクレオチドを含む、請求項1記載の核酸プローブ。
- 配列番号:53〜61のいずれか1つに対応する、請求項12記載の核酸プローブ。
- 請求項1記載の核酸プラスその発現を駆動するように前記核酸と作動可能に連結された発現制御エレメントを含む、単離された組換えポリヌクレオチド分子。
- プロモーターに作動可能に連結された、配列番号:11〜18のいずれか1つに記載された全アミノ酸配列を有するポリペプチドをコードする、請求項1の核酸を含む発現ベクターであって、適合性宿主細胞に存在する、前記発現ベクター。
- 配列番号:11〜18の1つのアミノ酸配列の少なくとも成熟タンパク質部分をコードする請求項1記載の核酸の挿入によって遺伝子操作された哺乳動物、昆虫、又は細菌の宿主細胞。
- 配列番号:11〜18の1つの成熟タンパク質部分を含むポリペプチドを産生するための方法であって、前記ポリペプチドの産生に十分な条件下で請求項16記載の宿主細胞を培養すること含む、前記産生方法。
- 前記ポリペプチドが前記細胞の表面に発現されて、前記培養から該ポリペプチド、又はその断片を回収することを更に含む、請求項17記載の産生方法。
- 任意にグリコシル化されていてもよく、かつ(a)配列番号:11〜18のいずれか1つに記載された成熟タンパク質のアミノ酸配列を有するか;(b)(a)の成熟タンパク質の1つに対して少なくとも約75%の類似性を有する成熟タンパク質のアミノ酸配列を有し、かつ同じ生物学的機能を示すか;(c)配列番号:11〜18のいずれか1つの成熟タンパク質と少なくとも約50%の同一性を有する成熟タンパク質のアミノ酸配列を有するか;又は(d)(a)の免疫学的に反応性の断片である、ポリペプチド。
- (a)の成熟タンパク質に対して少なくとも約85%の類似性を有する成熟タンパク質である、請求項19記載のポリペプチド。
- (a)の成熟タンパク質に対して少なくとも約95%の類似性を有する成熟タンパク質である、請求項19記載のポリペプチド。
- 配列番号:11〜18の1つの成熟タンパク質のアミノ酸配列を有するか、又はそれぞれの成熟タンパク質と同じ生物学的機能を示すその断片である、請求項19記載のポリペプチド。
- 請求項の19〜22いずれか1項記載のポリペプチド又は断片を認識する抗体。
- 配列番号:11〜18の1つのアミノ酸配列を有するポリペプチドを認識する、請求項23記載の抗体。
- 請求項19記載の少なくとも1つの成熟タンパク質の酵素活性を阻害することができる化合物をスクリーニングする方法であって、前記成熟タンパク質、及び前記成熟タンパク質のために適した基質を、1つ以上の試験化合物又はその塩の存在下においてインキュベートすること、前記成熟タンパク質の酵素活性を測定すること、前記活性を試験化合物の非存在下で決定された相当する活性と比較すること、及び酵素活性を低減させる試験化合物又は化合物群を選択することを含む、前記方法。
- 請求項19記載の少なくとも1つの成熟タンパク質の酵素活性を阻害しない選択的QC-阻害剤をスクリーニングする方法であって、前記成熟タンパク質、及び適切な基質をQCの1つ以上の阻害剤又はその塩の存在下においてインキュベートすること、前記成熟タンパク質の酵素活性を測定すること、前記活性をQC阻害剤の非存在下で決定された相当する活性と比較すること、及び前記成熟タンパク質の酵素活性を低減させない化合物を選択することを含む、前記方法。
- QCの酵素活性を阻害しない選択的QPCTL-阻害剤をスクリーニングする方法であって、前記QCをQPCTLの1つ以上の阻害剤又はその塩の存在下においてインキュベートすること、QCの酵素活性を測定すること、前記活性をQPCTL阻害剤の非存在下で決定された相当する活性を比較すること、及び前記QPCTLタンパク質の酵素活性を低減させない化合物を選択することを含む、前記方法。
- 請求項19〜22のいずれか1項記載の前記成熟タンパク質の1つの生物学的機能を阻害する、QPCTLアンタゴニスト。
- 小分子阻害剤である、請求項28記載のQPCTLアンタゴニスト。
- 請求項25又は27記載のスクリーニング法によって同定された、請求項29記載の阻害剤。
- 請求項26記載のスクリーニング法によって同定された、選択的QC阻害剤。
- 少なくとも1つQPCTL阻害剤、又はその薬学的許容し得る塩を、任意に従来の担体及び/又は賦形剤と組み合わせて含む、非経口、経腸、又は経口投与のための医薬組成物。
- アルツハイマー病、家族性英国痴呆症、家族性デンマーク痴呆症、ダウン症候群、ハンチントン病、ケネディ病、潰瘍疾患、ヘリコバクターピロリ(Helicobacter pylori)感染を伴う又は伴わない十二指腸癌、結直腸癌、ゾリンジャー・エリソン症候群、ヘリコバクターピロリ感染を伴う又は伴わない胃癌、病原性精神病状態、統合失調症、不妊症、新形成、炎症性宿主反応、癌、悪性転移、メラノーマ、乾癬、リウマチ様関節炎、アテローム性動脈硬化症、体液性及び細胞媒介免疫応答の障害、内皮における白血球接着及び遊走プロセス、摂食障害、睡眠-覚醒状態障害、エネルギー代謝の恒常性制御の障害、自律神経機能障害、ホルモンバランス障害、又は体液の制御の障害、多発性硬化症、ギランバレー症候群、並びに慢性炎症性脱髄性多発神経根筋障害から選択される疾患の予防又は治療のための医薬の製造のための、請求項32記載の医薬組成物の使用。
- アルツハイマー病、家族性英国痴呆症、家族性デンマーク痴呆症、ダウン症候群、ハンチントン病、ケネディ病、潰瘍疾患、ヘリコバクターピロリ感染を伴う又は伴わない十二指腸癌、結直腸癌、ゾリンジャー・エリソン症候群、ヘリコバクターピロリ感染を伴う又は伴わない胃癌、病原性精神病状態、統合失調症、不妊症、新形成、炎症性宿主反応、癌、悪性転移、メラノーマ、乾癬、リウマチ様関節炎、アテローム性動脈硬化症、体液性及び細胞媒介免疫応答の障害、内皮における白血球接着及び遊走プロセス、摂食障害、睡眠-覚醒状態障害、エネルギー代謝の恒常性制御の障害、自律神経機能障害、ホルモンバランス障害、又は体液の制御の障害、多発性硬化症、ギランバレー症候群、及び慢性炎症性脱髄性多発神経根筋障害から選択される疾患から選択される疾患の予防又は治療のための医薬の製造のための、QPCTL阻害剤、又はその医薬として許容し得る塩の使用。
- QPCTL阻害剤が競合阻害剤である、請求項32〜34のいずれか1項記載の医薬組成物又は使用。
- QPCTL阻害剤が、QPCTLの活性部位に結合した金属イオンに結合する、請求項32〜35のいずれか1項記載の医薬組成物又は使用。
- QPCTLが、配列番号:11〜18、21、及び22の1つから選択される、請求項32〜36のいずれか1項記載の医薬組成物又は使用。
- QPCTLが、配列番号:11及び12の1つから選択される、請求項37記載の医薬組成物又は使用。
- 請求項28〜30のいずれか1項記載の阻害剤が使用される、請求項32〜38のいずれか1項記載の医薬組成物又は使用。
- QPCTL阻害剤を含む、診断アッセイ。
- 請求項33又は34において定義した疾患及び/又は状態のいずれか1つを診断する方法であって、
-前記疾患及び/又は状態で苦しめられていることが疑われる対象から試料を収集する工程、
-前記試料をグルタミニルペプチドシクロトランスフェラーゼの阻害剤と接触させる工程、及び
-前記対象が前記疾患及び/又は状態によって苦しめられているか否かを決定する工程、
を含む、前記方法。 - 前記対象がヒトである、請求項41記載の方法。
- 前記試料が、血液試料、血清試料、脳脊髄溶液の試料、又は尿試料である、請求項41又は42記載の方法。
- 検出手段として請求項40記載の診断アッセイ、及び決定手段を含む、請求項41〜43の方法を実施するための診断キット。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US84624406P | 2006-09-21 | 2006-09-21 | |
| US94778007P | 2007-07-03 | 2007-07-03 | |
| PCT/EP2007/060013 WO2008034891A2 (en) | 2006-09-21 | 2007-09-21 | Novel genes related to glutaminyl cyclase |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010504088A true JP2010504088A (ja) | 2010-02-12 |
| JP2010504088A5 JP2010504088A5 (ja) | 2010-11-11 |
Family
ID=38779576
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009528732A Pending JP2010504088A (ja) | 2006-09-21 | 2007-09-21 | グルタミニルシクラーゼに関連した新規遺伝子 |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US20080249083A1 (ja) |
| EP (2) | EP2581449B1 (ja) |
| JP (1) | JP2010504088A (ja) |
| KR (1) | KR101432848B1 (ja) |
| AU (1) | AU2007298929B2 (ja) |
| BR (1) | BRPI0718507A2 (ja) |
| CA (1) | CA2663635A1 (ja) |
| EA (1) | EA016584B1 (ja) |
| HK (1) | HK1136603A1 (ja) |
| IL (2) | IL197090A (ja) |
| MX (1) | MX2009003090A (ja) |
| NZ (2) | NZ590631A (ja) |
| WO (1) | WO2008034891A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013519891A (ja) * | 2010-02-18 | 2013-05-30 | プロビオドルグ エージー | ピログルタミン酸修飾mcp−1を決定することにより炎症性疾患を診断する方法及びグルタミニルシクラーゼの阻害剤のスクリーニング方法 |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2206496B1 (en) * | 2003-05-05 | 2014-09-17 | Probiodrug AG | Screening of inhibitors of formation of pyroglutamic acid in amyloid beta peptide |
| US9277737B2 (en) | 2006-09-21 | 2016-03-08 | Probiodrug Ag | Mouse models carrying a knock-out mutation of the QPCTL-gene |
| US8889709B2 (en) | 2006-09-21 | 2014-11-18 | Probiodrug Ag | Use of isoQC inhibitors in the treatment and prevention of inflammatory diseases or conditions |
| NZ590631A (en) * | 2006-09-21 | 2011-12-22 | Probiodrug Ag | Novel genes related to glutaminyl cyclase |
| WO2008055945A1 (en) * | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| US8420684B2 (en) * | 2006-11-09 | 2013-04-16 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| WO2008055950A1 (en) * | 2006-11-09 | 2008-05-15 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| SI2091948T1 (sl) * | 2006-11-30 | 2012-07-31 | Probiodrug Ag | Novi inhibitorji glutaminil ciklaze |
| ZA200904353B (en) * | 2007-01-19 | 2010-09-29 | Probiodrug Ag | In vivo screening models for treatment of alzheimer's disease and other Qpct-related disorders |
| JP5612860B2 (ja) * | 2007-03-09 | 2014-10-22 | プロビオドルグ エージー | グルタミニルシクラーゼ阻害剤としてのイミダゾ[1,5−a]ピリジン誘導体 |
| DK2142514T3 (da) * | 2007-04-18 | 2015-03-23 | Probiodrug Ag | Thioureaderivater som glutaminylcyclase-inhibitorer |
| EP2142515B1 (en) * | 2007-04-18 | 2014-03-26 | Probiodrug AG | Nitrovinyl-diamine derivatives as glutaminyl cyclase inhibitors |
| DK2160380T3 (da) * | 2007-04-18 | 2014-06-30 | Probiodrug Ag | Cyano-guanidin derivater som glutaminyl cyclase inhibitorer |
| WO2008128986A1 (en) * | 2007-04-18 | 2008-10-30 | Probiodrug Ag | Urea derivatives as glutaminyl cyclase inhibitors |
| WO2008128982A1 (en) * | 2007-04-18 | 2008-10-30 | Probiodrug Ag | Thioxoquinazolinone derivatives as glutaminyl cyclase inhibitors |
| DK2142513T3 (da) * | 2007-04-18 | 2014-06-10 | Probiodrug Ag | Nitrovinyl-diamin-derivater som glutaminyl-cyclase-inhibitorer |
| EP2142536B1 (en) * | 2007-04-20 | 2015-10-21 | Probiodrug AG | Aminopyrimidine derivatives as glutaminyl cyclase inhibitors |
| WO2009034158A2 (en) * | 2007-09-12 | 2009-03-19 | Probiodrug Ag | Transgenic mice |
| NZ586665A (en) * | 2008-01-14 | 2011-12-22 | Probiodrug Ag | MOUSE MODELS CARRYING A KNOCK-OUT MUTATION OF THE GLUTAMINYL CYCLASE GENE (Qpct) |
| EA025107B1 (ru) * | 2008-07-31 | 2016-11-30 | Пробиодруг Аг | Способ диагностирования болезни альцгеймера, нейродегенерации при синдроме дауна или легкого когнитивного нарушения у субъекта (варианты) и набор для осуществления способа |
| EP2344158B1 (en) * | 2008-09-04 | 2017-12-27 | Probiodrug AG | Use of isoqc inhibitors |
| EA021150B1 (ru) * | 2008-09-04 | 2015-04-30 | Пробиодруг Аг | Производные имидазолидина в качестве ингибиторов глутаминилциклазы |
| AU2010251176A1 (en) * | 2009-05-19 | 2011-12-08 | Probiodrug Ag | Mouse models carrying a knock-out mutation of the QPCTL-gene |
| WO2012022804A1 (en) | 2010-08-19 | 2012-02-23 | Probiodrug Ag | Crystal structure of glutaminyl cyclase |
| JP2013541938A (ja) | 2010-09-02 | 2013-11-21 | プロビオドルグ エージー | アイソqc関連障害の治療のためのインビボスクリーニングモデル |
| WO2012059413A1 (en) | 2010-11-02 | 2012-05-10 | Probiodrug Ag | Crystal structure of isoglutaminyl cyclase |
| CA2879454A1 (en) | 2012-07-19 | 2014-01-23 | Drexel University | Novel sigma receptor ligands and methods of modulating cellular protein homeostasis using same |
| WO2017079547A2 (en) * | 2015-11-04 | 2017-05-11 | The Scripps Research Institute | Compositions and methods for treating cystic fibrosis |
| EP3747437A1 (en) * | 2017-07-24 | 2020-12-09 | Stichting Het Nederlands Kanker Instituut- Antoni van Leeuwenhoek Ziekenhuis | Treating pathological conditions by direct and indirect targeting of sirpa - cd47 interaction |
| WO2019089902A1 (en) | 2017-11-01 | 2019-05-09 | Drexel University | Compounds, compositions, and methods for treating diseases |
| GB201918108D0 (en) * | 2019-12-10 | 2020-01-22 | Vib Vzw | Improved aminopeptiadases for single molecule peptide sequencing |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004098625A2 (en) * | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| WO2005108415A2 (en) * | 2004-04-30 | 2005-11-17 | Biogen Idec Ma Inc. | Membrane associated molecules |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ207394A (en) | 1983-03-08 | 1987-03-06 | Commw Serum Lab Commission | Detecting or determining sequence of amino acids |
| IT1183575B (it) | 1985-05-08 | 1987-10-22 | Eurand Spa | Formulazione deodorando ad effetto modulante sulla trfaspirazione |
| DD293584A5 (de) | 1990-03-23 | 1991-09-05 | Adw,Zi Fuer Organische Chemie,De | Verfahren zur herstellung von neuen 3,5,5-trisubstituierten 1-(benzimidazol-2-yl)-imidazolidin-2,4-dionen bzw. -4-on-2-thionen |
| US20040006011A1 (en) * | 1996-07-12 | 2004-01-08 | Gour Barbara J. | Peptidomimetic modulators of cell adhesion |
| US6247293B1 (en) * | 1998-11-03 | 2001-06-19 | Klockner Bartelt, Inc. | Modular packaging machine with web tension control |
| FR2796945B1 (fr) | 1999-07-30 | 2002-06-28 | Sod Conseils Rech Applic | Nouveaux derives d'hydantoines, de thiohydantoines, de pyrimidinediones et de thioxopyrimidinones, leurs procedes de preparation et leur application a titre de medicaments |
| WO2001053331A2 (en) | 2000-01-24 | 2001-07-26 | Adherex Technologies, Inc. | Peptidomimetic modulators of cell adhesion |
| FR2808711B1 (fr) * | 2000-05-10 | 2002-08-09 | Poudres & Explosifs Ste Nale | Procede de fabrication d'elements composites etain-tungstene de faible epaisseur |
| WO2002093436A1 (en) * | 2001-05-11 | 2002-11-21 | Swisscom Mobile Ag | Method for transmitting an anonymous request from a consumer to a content or service provider through a telecommunication network |
| US6948038B2 (en) * | 2001-07-24 | 2005-09-20 | Microsoft Corporation | System and method for backing up and restoring data |
| KR100474689B1 (ko) * | 2001-08-30 | 2005-03-08 | 삼성전자주식회사 | 이동통신 시스템에서 소프트 핸드오프 도중의 전력제어 방법 |
| WO2003045321A2 (en) * | 2001-11-26 | 2003-06-05 | Exelixis, Inc. | MBMs AS MODIFIERS OF BRANCHING MORPHOGENESIS AND METHODS OF USE |
| US7677859B2 (en) | 2002-07-22 | 2010-03-16 | Brooks Automation, Inc. | Substrate loading and uploading station with buffer |
| GB0220501D0 (en) * | 2002-09-04 | 2002-10-09 | Hall Geoffrey W | Push 22 |
| US20040098591A1 (en) * | 2002-11-15 | 2004-05-20 | Fahrny James W. | Secure hardware device authentication method |
| US6851324B2 (en) * | 2002-12-16 | 2005-02-08 | Delphi Technologies, Inc. | Non-contacting compliant torque sensor |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| US7732162B2 (en) * | 2003-05-05 | 2010-06-08 | Probiodrug Ag | Inhibitors of glutaminyl cyclase for treating neurodegenerative diseases |
| US8338120B2 (en) * | 2003-05-05 | 2012-12-25 | Probiodrug Ag | Method of treating inflammation with glutaminyl cyclase inhibitors |
| EP2206496B1 (en) * | 2003-05-05 | 2014-09-17 | Probiodrug AG | Screening of inhibitors of formation of pyroglutamic acid in amyloid beta peptide |
| US7192995B2 (en) * | 2003-10-03 | 2007-03-20 | Virginia Commonwealth University | Homogenous compositions of polymers and crystalline solids or cross-linking agents and methods of making the same |
| BRPI0415409A (pt) * | 2003-10-15 | 2006-12-05 | Probiodrug Ag | uso de efetuadores de ciclases de glutaminila e glutamato |
| US20050171112A1 (en) * | 2003-11-03 | 2005-08-04 | Probiodrug Ag | Combinations useful for the treatment of neuronal disorders |
| US7667044B2 (en) * | 2003-11-03 | 2010-02-23 | Probiodrug Ag | Compounds for the treatment of neurological disorders |
| EP1680120A2 (en) * | 2003-11-03 | 2006-07-19 | Probiodrug AG | Combinations useful for the treatment of neuronal disorders |
| JP2007511543A (ja) * | 2003-11-19 | 2007-05-10 | アクルックス・ディ・ディ・エス・プロプライエタリー・リミテッド | 皮膚病変治療のための組成物および方法 |
| CN1918131B (zh) * | 2004-02-05 | 2011-05-04 | 前体生物药物股份公司 | 谷氨酰胺酰基环化酶抑制剂 |
| CN101014836B (zh) * | 2004-07-01 | 2010-04-28 | 微动公司 | 用于消除流动中的密度效应的分离式平衡配重 |
| US7663268B2 (en) * | 2006-08-30 | 2010-02-16 | The Regents of the University of Cailfornia | Converters for high power applications |
| NZ590631A (en) | 2006-09-21 | 2011-12-22 | Probiodrug Ag | Novel genes related to glutaminyl cyclase |
| EP2091945B1 (en) | 2006-11-09 | 2014-01-15 | Probiodrug AG | Novel inhibitors of glutaminyl cyclase |
| EP2142536B1 (en) * | 2007-04-20 | 2015-10-21 | Probiodrug AG | Aminopyrimidine derivatives as glutaminyl cyclase inhibitors |
-
2007
- 2007-09-21 NZ NZ590631A patent/NZ590631A/en not_active IP Right Cessation
- 2007-09-21 JP JP2009528732A patent/JP2010504088A/ja active Pending
- 2007-09-21 MX MX2009003090A patent/MX2009003090A/es active IP Right Grant
- 2007-09-21 KR KR1020097008086A patent/KR101432848B1/ko active IP Right Grant
- 2007-09-21 NZ NZ575727A patent/NZ575727A/en not_active IP Right Cessation
- 2007-09-21 US US11/859,217 patent/US20080249083A1/en not_active Abandoned
- 2007-09-21 EP EP13150579.4A patent/EP2581449B1/en active Active
- 2007-09-21 EA EA200900404A patent/EA016584B1/ru not_active IP Right Cessation
- 2007-09-21 AU AU2007298929A patent/AU2007298929B2/en not_active Ceased
- 2007-09-21 WO PCT/EP2007/060013 patent/WO2008034891A2/en active Application Filing
- 2007-09-21 CA CA002663635A patent/CA2663635A1/en not_active Abandoned
- 2007-09-21 BR BRPI0718507-3A patent/BRPI0718507A2/pt not_active IP Right Cessation
- 2007-09-21 EP EP07820442.7A patent/EP2082041B1/en active Active
-
2009
- 2009-02-17 IL IL197090A patent/IL197090A/en active IP Right Grant
- 2009-07-02 US US12/497,082 patent/US8129160B2/en active Active
-
2010
- 2010-04-28 HK HK10104168.6A patent/HK1136603A1/zh unknown
-
2011
- 2011-08-25 IL IL214820A patent/IL214820A/en active IP Right Grant
- 2011-12-13 US US13/325,015 patent/US8647834B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004098625A2 (en) * | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| WO2005108415A2 (en) * | 2004-04-30 | 2005-11-17 | Biogen Idec Ma Inc. | Membrane associated molecules |
Non-Patent Citations (1)
| Title |
|---|
| JPN6012055658; Kawabata,A.et al.: '"Accession:AK000091[gi:7019952],Definition:Homo sapiens cDNA FLJ20084 fis, clone COL03526."' NCBI Entrez Nucleotide[online];12-SEP-2006 uploaded,NCBI,[retrieved on 19 October 2012] * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013519891A (ja) * | 2010-02-18 | 2013-05-30 | プロビオドルグ エージー | ピログルタミン酸修飾mcp−1を決定することにより炎症性疾患を診断する方法及びグルタミニルシクラーゼの阻害剤のスクリーニング方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| HK1136603A1 (zh) | 2010-07-02 |
| EP2581449A2 (en) | 2013-04-17 |
| EA200900404A1 (ru) | 2009-10-30 |
| EP2581449A3 (en) | 2013-07-31 |
| EA016584B1 (ru) | 2012-06-29 |
| CA2663635A1 (en) | 2008-03-27 |
| US20100009337A1 (en) | 2010-01-14 |
| NZ590631A (en) | 2011-12-22 |
| EP2581449B1 (en) | 2016-09-14 |
| WO2008034891A2 (en) | 2008-03-27 |
| US20080249083A1 (en) | 2008-10-09 |
| AU2007298929B2 (en) | 2012-09-27 |
| EP2082041A2 (en) | 2009-07-29 |
| US20120183974A1 (en) | 2012-07-19 |
| IL214820A (en) | 2016-06-30 |
| KR20090059163A (ko) | 2009-06-10 |
| IL197090A0 (en) | 2011-08-01 |
| IL197090A (en) | 2016-03-31 |
| US8129160B2 (en) | 2012-03-06 |
| US8647834B2 (en) | 2014-02-11 |
| NZ575727A (en) | 2011-12-22 |
| KR101432848B1 (ko) | 2014-08-27 |
| MX2009003090A (es) | 2009-04-02 |
| IL214820A0 (en) | 2011-09-27 |
| AU2007298929A1 (en) | 2008-03-27 |
| WO2008034891A3 (en) | 2008-05-08 |
| BRPI0718507A2 (pt) | 2013-11-12 |
| EP2082041B1 (en) | 2018-03-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2010504088A (ja) | グルタミニルシクラーゼに関連した新規遺伝子 | |
| RU2305133C2 (ru) | Ген новой сериновой протеазы, родственной dppiv | |
| JP4713341B2 (ja) | Ca−ix特異的阻害剤 | |
| MX2012005342A (es) | Uso de derivados benzo-heterociclico para prevenir y tratar el cancer o para inhibir metastasis de cancer. | |
| US10889585B2 (en) | Inhibitors of kidney-type glutaminase, GLS-1 | |
| JP2003135064A (ja) | アッセイ方法 | |
| JP5788320B2 (ja) | イソqc阻害剤の使用 | |
| JP2005526051A (ja) | Ptp−1bおよびtc−ptpの活性を調節する化合物 | |
| CN101573450B (zh) | 与谷氨酰环化酶相关的新基因 | |
| AU2012241108B2 (en) | Novel genes related to glutaminyl cyclase | |
| US10077479B2 (en) | Fusion gene as therapeutic target in proliferative diseases | |
| JP2006180813A (ja) | アミノペプチダーゼoおよびその利用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100917 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100917 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20121113 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130205 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130213 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130508 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130813 |