JP2007182567A - 特定の担体を有する触媒を利用する選択的水素化法 - Google Patents
特定の担体を有する触媒を利用する選択的水素化法 Download PDFInfo
- Publication number
- JP2007182567A JP2007182567A JP2006345142A JP2006345142A JP2007182567A JP 2007182567 A JP2007182567 A JP 2007182567A JP 2006345142 A JP2006345142 A JP 2006345142A JP 2006345142 A JP2006345142 A JP 2006345142A JP 2007182567 A JP2007182567 A JP 2007182567A
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- support
- metal
- pore volume
- weight
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 200
- 238000000034 method Methods 0.000 title claims abstract description 65
- 238000005984 hydrogenation reaction Methods 0.000 title claims abstract description 40
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims abstract description 88
- 229910052751 metal Inorganic materials 0.000 claims abstract description 60
- 239000002184 metal Substances 0.000 claims abstract description 60
- 150000001875 compounds Chemical class 0.000 claims abstract description 50
- 229910052759 nickel Inorganic materials 0.000 claims abstract description 44
- 238000006243 chemical reaction Methods 0.000 claims abstract description 37
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 34
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 34
- 239000011593 sulfur Substances 0.000 claims abstract description 34
- 229910000510 noble metal Inorganic materials 0.000 claims abstract description 12
- 229910017052 cobalt Inorganic materials 0.000 claims abstract description 10
- 239000010941 cobalt Substances 0.000 claims abstract description 10
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims abstract description 10
- 239000007788 liquid Substances 0.000 claims abstract description 7
- 150000004645 aluminates Chemical class 0.000 claims abstract description 5
- 239000011148 porous material Substances 0.000 claims description 47
- 230000008569 process Effects 0.000 claims description 15
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims description 13
- 229910052750 molybdenum Inorganic materials 0.000 claims description 12
- 239000011733 molybdenum Substances 0.000 claims description 12
- 238000005486 sulfidation Methods 0.000 claims description 11
- 229910000480 nickel oxide Inorganic materials 0.000 claims description 9
- GNRSAWUEBMWBQH-UHFFFAOYSA-N oxonickel Chemical compound [Ni]=O GNRSAWUEBMWBQH-UHFFFAOYSA-N 0.000 claims description 9
- 150000002739 metals Chemical class 0.000 claims description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 7
- -1 nickel aluminate Chemical class 0.000 claims description 7
- 229910044991 metal oxide Inorganic materials 0.000 claims description 6
- 150000004706 metal oxides Chemical class 0.000 claims description 6
- 239000000470 constituent Substances 0.000 claims description 3
- 229910021472 group 8 element Inorganic materials 0.000 claims description 3
- 229910052742 iron Inorganic materials 0.000 claims description 3
- 229920006395 saturated elastomer Polymers 0.000 claims description 3
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 3
- 229910052721 tungsten Inorganic materials 0.000 claims description 3
- 239000010937 tungsten Substances 0.000 claims description 3
- 229910000476 molybdenum oxide Inorganic materials 0.000 claims description 2
- PQQKPALAQIIWST-UHFFFAOYSA-N oxomolybdenum Chemical compound [Mo]=O PQQKPALAQIIWST-UHFFFAOYSA-N 0.000 claims description 2
- 230000002194 synthesizing effect Effects 0.000 claims 3
- 239000000969 carrier Substances 0.000 claims 1
- 239000000463 material Substances 0.000 abstract 1
- 150000001993 dienes Chemical class 0.000 description 31
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 27
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 26
- 150000003573 thiols Chemical class 0.000 description 25
- 229910052739 hydrogen Inorganic materials 0.000 description 23
- 239000001257 hydrogen Substances 0.000 description 21
- 230000000694 effects Effects 0.000 description 20
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 18
- KJRCEJOSASVSRA-UHFFFAOYSA-N propane-2-thiol Chemical compound CC(C)S KJRCEJOSASVSRA-UHFFFAOYSA-N 0.000 description 18
- 150000001336 alkenes Chemical class 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- LIKMAJRDDDTEIG-UHFFFAOYSA-N 1-hexene Chemical compound CCCCC=C LIKMAJRDDDTEIG-UHFFFAOYSA-N 0.000 description 13
- 238000012360 testing method Methods 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- 239000000203 mixture Substances 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 9
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 229910001151 AlNi Inorganic materials 0.000 description 8
- 230000009467 reduction Effects 0.000 description 8
- 238000005987 sulfurization reaction Methods 0.000 description 8
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 7
- 238000005470 impregnation Methods 0.000 description 7
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 6
- 238000004523 catalytic cracking Methods 0.000 description 6
- 230000008859 change Effects 0.000 description 6
- WQOXQRCZOLPYPM-UHFFFAOYSA-N dimethyl disulfide Chemical compound CSSC WQOXQRCZOLPYPM-UHFFFAOYSA-N 0.000 description 6
- 238000004231 fluid catalytic cracking Methods 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 5
- 229910052753 mercury Inorganic materials 0.000 description 5
- KBJMLQFLOWQJNF-UHFFFAOYSA-N nickel(ii) nitrate Chemical compound [Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O KBJMLQFLOWQJNF-UHFFFAOYSA-N 0.000 description 5
- 238000002459 porosimetry Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 150000003568 thioethers Chemical class 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- QENGPZGAWFQWCZ-UHFFFAOYSA-N Methylthiophene Natural products CC=1C=CSC=1 QENGPZGAWFQWCZ-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- UFMZWBIQTDUYBN-UHFFFAOYSA-N cobalt dinitrate Chemical compound [Co+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O UFMZWBIQTDUYBN-UHFFFAOYSA-N 0.000 description 3
- 229910001981 cobalt nitrate Inorganic materials 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 229930192474 thiophene Natural products 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 2
- 229910003294 NiMo Inorganic materials 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- 238000001994 activation Methods 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 238000001354 calcination Methods 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- LJSQFQKUNVCTIA-UHFFFAOYSA-N diethyl sulfide Chemical compound CCSCC LJSQFQKUNVCTIA-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethanethiol Chemical compound CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 229910001960 metal nitrate Inorganic materials 0.000 description 2
- 229910052976 metal sulfide Inorganic materials 0.000 description 2
- 150000005673 monoalkenes Chemical class 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- QMMOXUPEWRXHJS-UHFFFAOYSA-N pent-2-ene Chemical compound CCC=CC QMMOXUPEWRXHJS-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- PMJHHCWVYXUKFD-SNAWJCMRSA-N (E)-1,3-pentadiene Chemical compound C\C=C\C=C PMJHHCWVYXUKFD-SNAWJCMRSA-N 0.000 description 1
- MJRCCWJSYFOGBX-UHFFFAOYSA-N 1-propylsulfanylpentane Chemical compound CCCCCSCCC MJRCCWJSYFOGBX-UHFFFAOYSA-N 0.000 description 1
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical compound CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 1
- XQQBUAPQHNYYRS-UHFFFAOYSA-N 2-methylthiophene Chemical compound CC1=CC=CS1 XQQBUAPQHNYYRS-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 101710178035 Chorismate synthase 2 Proteins 0.000 description 1
- 101710152694 Cysteine synthase 2 Proteins 0.000 description 1
- 238000005698 Diels-Alder reaction Methods 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 229910000943 NiAl Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 238000004939 coking Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000006477 desulfuration reaction Methods 0.000 description 1
- 230000023556 desulfurization Effects 0.000 description 1
- XYWDPYKBIRQXQS-UHFFFAOYSA-N di-isopropyl sulphide Natural products CC(C)SC(C)C XYWDPYKBIRQXQS-UHFFFAOYSA-N 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000011133 lead Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- WXEHBUMAEPOYKP-UHFFFAOYSA-N methylsulfanylethane Chemical compound CCSC WXEHBUMAEPOYKP-UHFFFAOYSA-N 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- PMJHHCWVYXUKFD-UHFFFAOYSA-N piperylene Natural products CC=CC=C PMJHHCWVYXUKFD-UHFFFAOYSA-N 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 229920001021 polysulfide Polymers 0.000 description 1
- 239000005077 polysulfide Substances 0.000 description 1
- 150000008117 polysulfides Polymers 0.000 description 1
- 239000010970 precious metal Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000000197 pyrolysis Methods 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000000066 reactive distillation Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical compound [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000001577 simple distillation Methods 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000011029 spinel Substances 0.000 description 1
- 229910052596 spinel Inorganic materials 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 150000003463 sulfur Chemical class 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- 239000011135 tin Substances 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 229910001928 zirconium oxide Inorganic materials 0.000 description 1
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/75—Cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/755—Nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/883—Molybdenum and nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/20—Sulfiding
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/32—Selective hydrogenation of the diolefin or acetylene compounds
- C10G45/34—Selective hydrogenation of the diolefin or acetylene compounds characterised by the catalyst used
- C10G45/36—Selective hydrogenation of the diolefin or acetylene compounds characterised by the catalyst used containing nickel or cobalt metal, or compounds thereof
- C10G45/38—Selective hydrogenation of the diolefin or acetylene compounds characterised by the catalyst used containing nickel or cobalt metal, or compounds thereof in combination with chromium, molybdenum or tungsten metals, or compounds thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/615—100-500 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/63—Pore volume
- B01J35/635—0.5-1.0 ml/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/66—Pore distribution
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1037—Hydrocarbon fractions
- C10G2300/104—Light gasoline having a boiling range of about 20 - 100 °C
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1037—Hydrocarbon fractions
- C10G2300/1044—Heavy gasoline or naphtha having a boiling range of about 100 - 180 °C
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/201—Impurities
- C10G2300/202—Heteroatoms content, i.e. S, N, O, P
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/40—Characteristics of the process deviating from typical ways of processing
- C10G2300/4018—Spatial velocity, e.g. LHSV, WHSV
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
- Catalysts (AREA)
Abstract
【解決手段】硫化された形態で用いられ、MAl2O4タイプの金属アルミナートを含む特定の担体上に担持される第VIB族からの少なくとも1種の金属と、第VIII族からの少なくとも1種の非貴金属と含む担持触媒を用い、金属Mは、ニッケルおよびコバルトによって構成される群から選択される。80〜220℃の温度、1〜10h−1の液空間速度および0.5〜5MPaの圧力で供給材料を触媒と接触させることからなる方法。
【選択図】なし
Description
・第VIB族元素の酸化物の重量による量は、全触媒重量に対する重量で厳格に12%超であり;
・第VIII族元素の酸化物の重量による量は、全触媒重量に対して15重量%未満であり;
・前記触媒の構成金属の硫化度は少なくとも60%であり;
・直径が0.05ミクロン超である細孔の容積は総細孔容積の10〜40%である、方法が記載される。
・ジオレフィンをオレフィンに選択的に水素化すること
・オレフィンとの反応によりより軽質飽和硫黄含有化合物、主としてチオールを重質のスルフィドまたはチオールに変換すること
からなる。

・温度立ち上げ:1000℃まで5℃/分;そして、
・1時間にわたる1000℃での一定の温度段階。
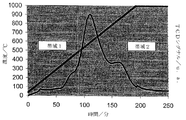
・帯域1は、低温で還元され得る、すなわち、低温でアルミナと弱く相互作用しているニッケルの還元部分に相当する;この部分は、上記の条件下に500℃未満の温度で還元されることが可能である;このため、それは、担体に組み込まれないフリーなニッケル酸化物である;
・帯域2は、高温で還元され得る、すなわち、アルミナと強く相互作用しているニッケルの部分、すなわち、アルミナと強く相互作用し、かつ、上記条件下に500〜750℃の温度で還元され得るニッケル酸化物粒子またはスピネルタイプのニッケルアルミナート構造(NiAl2O4)中に拘束されるニッケル酸化物(上記条件下に750℃超の温度で還元されることが可能である)のいずれかに相当する。
帯域1(弱い相互作用)の領域:24%;
帯域2(強い相互作用)の領域:76%。
(工程1:担体の合成)
本発明の担体は、アルミナ、例えば、高い比表面積を有するアルミナを含む。担体はまた、アルミナと、当業者に知られている任意の他の酸化物、例えばシリカ、酸化チタン、酸化マグネシウム、酸化亜鉛、酸化ジルコニウム等との混合物によって構成されてもよい。用いられるアルミナは、ニッケル、コバルト等の金属Mを組み込み得るアルミナの群から選択される。特に、それは、ガンマアルミナ、デルタアルミナまたはアルミナであるが、好ましくはガンマアルミナである。このアルミナは、適切な量の硝酸金属、例えば、硝酸ニッケル、硝酸コバルト等を含有する水溶液を乾式含浸させられる。本発明によると、硝酸金属の量は、固体上で全触媒重量に対して0.5〜10重量%、好ましくは0.7〜8重量%、より好ましくは1〜5重量%の金属含有量(酸化物MO等価量;Mはニッケルおよびコバルトによって構成される群から選択される)である。含浸後、固体は熟成のため室温で12時間にわたり放置され、120℃で12時間にわたり乾燥させられる。最後に、固体は、750℃で2時間にわたりマッフル炉中でか焼される:この固体は、以降では、AlNiまたはAlCoと表される。好ましくは、担体の合成のためのこの工程において用いられる金属Mの量の最大40%、好ましくは最大30%、より好ましくは最大25%は、前記担体と弱く相互作用する。
本発明の触媒は、当業者に知られているあらゆる技術を用いて調製され得るが、特に、第VIII族および第VIB族からの元素を選択された担体上に含浸させることが用いられる。前記含浸は、可及的に正確に担体の多孔度を満たすために、例えば、乾式含浸のような、所望量の元素が選択された溶媒(例えば脱塩水)に可溶な塩の形態で正確に導入される当業者に知られた技術を用いて行われてよい。その後溶液で満たされた担体は、好ましくは乾燥させられる。好ましい担体はアルミナであり、これは、当業者に知られたあらゆるタイプの前駆体および形状化ツールから調製され得る。
か焼後、担体上に担持された金属は酸化物の形態にある。ニッケルおよびモリブデンの場合、金属は、主として、MoO3およびNiOの形態にある。処理されるべき供給材料と接触させる前に、触媒は、硫化工程を経る。硫化は、好ましくは、硫化還元媒体(sulphoreducing medium)中、すなわち、H2Sおよび水素の存在下に行われ、金属酸化物がスルフィド、例えば、MoS2およびNi3S2に変換される。硫化は、H2Sおよび水素を含有する流れまたは触媒の存在下にH2Sに分解し得る硫黄含有化合物および水素を触媒上に注入することによって行われる。ジメチルジスルフィド等のポリスルフィドは、触媒を硫化するために日常的に用いられるH2S前駆体である。温度は、H2Sが金属酸化物と反応して金属スルフィドが形成されるように調整される。前記硫化は、水素化脱硫反応器に対して現場または現場外(反応器の内側または外側)で行われてよく、その際の温度は200〜600℃、より好ましくは300〜500℃である。活性であるようにするために、金属は十分に硫化される必要がある。触媒上に存在する硫黄(S)と前記元素とのモル比が考慮中の元素の全硫化に相当する理論モル比の少なくとも60%:
(S/元素)触媒≧0.6×(S/元素)理論
である場合に、元素は「十分に」硫化されたと考えられる
ここで:
(S/元素)触媒は、触媒上に存在する硫黄(S)と元素との間のモル比であるが、担体の調製の間に用いられた金属(NiまたはCo)は除かれている;
(S/元素)理論は、元素のスルフィドへの全硫化に相当する硫黄と元素との間のモル比である。
(S/Fe)理論=1
(S/Co)理論=8/9
(S/Ni)理論=2/3
(S/Mo)理論=2/1
(S/W)理論=2/1。
(S/Mo+Ni)触媒=0.6×{(0.7×2)+(0.3×(2/3))
によって与えられる。
(担体の合成)
ニッケルの場合、Axensによって供給された高比表面積アルミナ(Al−1)が用いられた。このアルミナは、適切な量の硝酸ニッケルを含有する水溶液を乾式含浸させられた。硝酸ニッケルの量は、固体上の4重量%のニッケル含有量(酸化物等価量,NiO)に対応していた。含浸後、固体は、室温で12時間にわたり熟成のため放置され、次いで、120℃で12時間にわたり乾燥させられた。最後に、固体は、750℃で2時間にわたりマッフル炉内においてか焼された。この固体は、以降の記載では、AlNiと記される。
触媒A、B、C、D、EおよびFは、乾式含浸法によりAlNi担体を用いて調製された。合成プロトコールは、七モリブデン酸六アンモニウムおよび硝酸ニッケルの溶液の乾式含浸を行うことからなっていた。金属前駆体を含有する水溶液の容積は、含浸させられるべき担体の質量に対応する水の吸い上げ容積に等しい。溶液中の前駆体の濃度は、金属酸化物の重量で所望量を担体上に担持するように調整された。次いで、固体は、室温で12時間にわたり熟成のため放置され、120℃で12時間にわたり乾燥させられた。最後に、固体は、500℃で2時間にわたり、空気流(1リットル/g・h)の中でか焼された。調製された触媒の特徴は、下記表2に示される。調製された触媒は、それらの活性相含有量によって区別された。
触媒A、B、C、D(本発明に合致しない)および触媒E、F(本発明に合致する)の活性は、攪拌型の500mLのオートクレーブ反応器において行われるモデル分子の混合物の選択的水素化についての試験を用いて評価された。典型的には、2〜6gの触媒が、大気圧で硫化装置において、15容積%のH2Sで構成されたH2S/H2混合物中で、1時間当たりかつ触媒1g当たり混合物1lとして、400℃で2時間にわたり(5℃/分の立ち上げ)硫化され、次いで、200℃での高純度水素中2時間の一定温度段階が行われた。このプロトコールにより、本発明の触媒の全てについて80%超の硫化度が引き出された。硫化された触媒は反応器に移され、空気から密閉され、次いで、1.5MPaの全圧、160℃の温度で250mLのモデル供給材料と接触させられた。圧力は、水素を加えることにより試験の間一定に維持された。活性試験のために用いられた供給材料は、以下の組成を有していた:n−ヘプタン中、3−メチルチオフェンの形態にある硫黄 1000重量ppm、プロパン−2−チオールの形態にある硫黄 100重量ppm、1−ヘキセン形態にあるオレフィン 10重量%。
A(X)=k(X)/m
ここで、A(X)=反応Xについての触媒の活性(分−1/触媒のg);
k=考慮中の反応についての速度定数(分−1)。
k(X)=(1/45)*ln(100/(100−conv(X)))
を用いて計算される。
conv(X)(化合物Xの転化);X=イソプレンまたはプロパン−2−チオールまたは1−ヘキサンである)
m=試験において用いられた触媒の質量(酸化物の形態);
X:考慮中の反応
X=イソプレン:イソプレンの水素化
X=1−ヘキセン:1−ヘキセンの水素化
X=プロパン−2−チオール:プロパン−2−チオールの転化。
上記触媒Eは、同一のモデル分子試験で評価されたが、従前の硫化工程が行われなかった。したがって、固体の硫化度はゼロであった。硫化ベンチ上での触媒硫化プロトコールの間のH2S/H2混合物による一定温度段階の温度の低減(400から典型的には100〜150℃)も、触媒Eに中間的な硫化度を引き出し得た。表4は、前記触媒について得られた、硫化度に応じた触媒の結果を記録している。触媒の従前の硫化が、イソプレンの水素化およびプロパン−2−チオールの転化における触媒の活性並びにその選択性に関して有用性の大きい効果を有することが理解され得る。
本実施例では、実施例1において記載された操作プロトコールを用いて触媒GおよびHが調製された。これらの触媒が触媒Eと実質的に異なっているのは、それらのニッケル含有量、それ故にNi/Moモル比(表5)においてのみである。このため、それらは、本発明に合致していない。
触媒調製法の工程2を用い、Axensによって供給された、性質が下記表7に示された異なるアルミナ担体Al−3、Al−4およびAl−5を用いて触媒I、JおよびKは調製された。
物は、基準硫黄含有化合物の保持時間との比較によって識別された。
Claims (29)
- 多価不飽和化合物を一価不飽和化合物に選択的に水素化すると共に、ガソリンに含まれる不飽和化合物との反応により飽和軽質硫黄含有化合物をより重質の化合物に変換し得る方法であって、
該方法は、第VIB族からの少なくとも1種の金属と、第VIII族からの少なくとも1種の非貴金属とを担体上に担持されて含有する触媒を用い、該担体は、MAl2O4タイプの金属アルミナートを含み、金属Mは、ニッケルおよびコバルトによって構成される群から選択され、担体と強く相互作用する金属Mの量は、酸化物等価体の重量で0.5〜10%になるようにされ、
・第VIB族元素の酸化物の重量による量は、全触媒重量に対して厳格に12重量%超であり;
・第VIII族元素の酸化物重量による量は、全触媒重量に対して15重量%未満であり;
・前記触媒の構成金属の硫化度は、少なくとも60%であり;
・直径が0.05ミクロン超である細孔の容積は、総細孔容積の10〜40%である、方法。 - 触媒は、モリブデンおよびタングステンから選択される第VIB族からの金属を含む、請求項1に記載の方法。
- 第VIB族金属はモリブデンである、請求項2に記載の方法。
- 触媒は、ニッケル、コバルトおよび鉄から選択される第VIII族からの非貴金属を含む、請求項1に記載の方法。
- 第VIII族非貴金属はニッケルである、請求項4に記載の方法。
- 触媒は、第VIII族元素の酸化物を、触媒重量に対して1〜10重量%の量で含む、請求項1〜5のいずれか1つに記載の方法。
- 触媒の構成金属の硫化度は80%超である、請求項1に記載の方法。
- 触媒のNi/Moモル比は0.2〜0.5である、請求項1〜7のいずれか1つに記載の方法。
- Ni/Moモル比は0.25〜0.45である、請求項8に記載の方法。
- 触媒の総細孔容積は0.3〜0.7cm3/gである、請求項1〜9のいずれか1つに記載の方法。
- 触媒の総細孔容積は0.35〜0.65cm3/gである、請求項10に記載の方法。
- 直径が0.1ミクロン超である触媒の細孔の容積は総細孔容積の5〜35%である請求項1〜11のいずれか1つに記載の方法。
- 直径が0.1ミクロン超である触媒の細孔の容積は、総細孔容積の10〜30%である、請求項12に記載の方法。
- 直径が0.05ミクロン超である触媒の細孔の容積は、総細孔容積の15〜35%を示す、請求項1に記載の方法。
- 触媒の比表面積は、250m2/g未満、好ましくは30〜150m2/gである、請求項1〜14のいずれか1つに記載の方法。
- 担体は、MAl2O4タイプの金属アルミナートを含み、Mはニッケルである、請求項1〜15のいずれか1つに記載の方法。
- 担体と強く相互作用する金属Mの量は、酸化物等価体重量で0.7〜8%である、請求項16に記載の方法。
- 担体と強く相互作用する金属Mの量は、酸化物等価体重量で1〜5%である、請求項17に記載の方法。
- 担体を合成する工程において用いられた金属の量の最大40%が前記担体と弱く相互作用している、請求項17または18に記載の方法。
- 担体を合成する工程において用いられた金属の量の最大30%が前記担体と弱く相互作用している、請求項19に記載の方法。
- 担体を合成する工程において用いられた金属の量の最大25%が前記担体と弱く相互作用している、請求項19または20に記載の方法。
- 触媒担体の細孔容積は0.3〜0.7cm3/gである、請求項16〜21のいずれか1つに記載の方法。
- 触媒担体の細孔容積は0.35〜0.65cm3/gである、請求項22に記載の方法。
- 直径が0.1ミクロン超である担体の細孔の容積は、総細孔容積の5〜35%を示す、請求項16〜23のいずれか1つに記載の方法。
- 直径が0.1ミクロン超である担体の細孔の容積は、総細孔容積の5〜30%を示す、請求項24に記載の方法。
- 直径が0.05ミクロン超である担体の細孔の容積は、総細孔容積の5〜50%を示す、請求項16〜25のいずれか1つに記載の方法。
- 直径が0.05ミクロン超である担体の細孔の容積は、総細孔容積の10〜40%を示す、請求項26に記載の方法。
- 触媒は、全触媒重量に対して1〜10重量%の含有量のニッケル酸化物と、全触媒重量に対して12重量%超の含有量のモリブデン酸化物とを含有し、ニッケル/モリブデンのモル比は0.25〜0.45であり、これら金属は、ニッケルアルミナートを含む担体上に担持され、担体と強く相互作用している金属Mの量は、全触媒重量に対する金属酸化物の重量で1〜5%であり、触媒を構成する金属の硫化度は80%超であり、直径が0.05ミクロン超である前記触媒の細孔の容積は15〜35%である、請求項1〜27のいずれか1つに記載の方法。
- 供給材料は、80〜220℃の温度、1〜10h−1の液空間速度および0.5〜5MPaの圧力で触媒と接触させられる、請求項1〜28のいずれか1つに記載の選択的水素化方法。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0513173A FR2895415B1 (fr) | 2005-12-22 | 2005-12-22 | Procede d'hydrogenation selective mettant en oeuvre un catalyseur presentant un support specifique |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007182567A true JP2007182567A (ja) | 2007-07-19 |
| JP2007182567A5 JP2007182567A5 (ja) | 2010-02-12 |
Family
ID=37052763
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006345142A Ceased JP2007182567A (ja) | 2005-12-22 | 2006-12-22 | 特定の担体を有する触媒を利用する選択的水素化法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7736492B2 (ja) |
| EP (1) | EP1800749B1 (ja) |
| JP (1) | JP2007182567A (ja) |
| KR (1) | KR20070066980A (ja) |
| CN (1) | CN101205483B (ja) |
| BR (1) | BRPI0605369A (ja) |
| FR (1) | FR2895415B1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20100028502A (ko) * | 2008-09-04 | 2010-03-12 | 아이에프피 | 특성 조성을 갖는 황화 촉매를 사용하는 선택적 수소화 방법 |
| JP2011512247A (ja) * | 2008-02-07 | 2011-04-21 | イエフペ エネルジ ヌヴェル | 選択的水素化触媒およびその調製方法 |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2895414B1 (fr) * | 2005-12-22 | 2011-07-29 | Inst Francais Du Petrole | Procede d'hydrogenation selective mettant en oeuvre un catalyseur presentant une porosite controlee |
| US8017545B2 (en) * | 2008-12-04 | 2011-09-13 | Uop Llc | Dynamic composition for the removal of sulfur from a gaseous stream |
| US7811474B2 (en) | 2008-12-04 | 2010-10-12 | Uop Llc | Simultaneous warm gas desulfurization and CO-shift for improved syngas cleanup |
| CN103146429B (zh) | 2011-12-06 | 2015-08-19 | 中国石油天然气股份有限公司 | 一种液化气加氢处理的方法 |
| FR2993570B1 (fr) * | 2012-07-17 | 2015-12-04 | IFP Energies Nouvelles | Procede de production d'une essence legere basse teneur en soufre |
| FR2993569B1 (fr) * | 2012-07-17 | 2015-12-04 | IFP Energies Nouvelles | Procede de desulfuration d'une essence |
| US9806164B1 (en) * | 2013-03-26 | 2017-10-31 | The Penn State Research Foundation | Controlled synthesis and transfer of large area heterostructures made of bilayer and multilayer transition metal dichalocogenides |
| EP2816094B1 (fr) * | 2013-06-19 | 2020-04-29 | IFP Energies nouvelles | Procédé de production d'une essence à basse teneur en soufre et en mercaptans |
| FR3080117B1 (fr) * | 2018-04-11 | 2020-04-03 | IFP Energies Nouvelles | Procede de captation de l'arsenic mettant en œuvre une masse de captation a base de particules d'oxyde de nickel |
| EP3822332A1 (en) | 2019-11-13 | 2021-05-19 | Indian Oil Corporation Limited | Catalyst for selective hydrogenation of diolefins and method for preparing catalyst |
| CN113559870A (zh) * | 2020-04-28 | 2021-10-29 | 中国石油化工股份有限公司 | 重油加氢催化剂及其制备方法和应用 |
| FR3109897B1 (fr) | 2020-05-07 | 2023-11-24 | Ifp Energies Now | Catalyseur d’hydrogénation sélective comprenant un support spécifique en partie sous forme aluminate |
| FR3109899B1 (fr) | 2020-05-07 | 2023-11-24 | Ifp Energies Now | Catalyseur d’hydrogénation comprenant un support et un ratio NiMo spécifique |
| FR3109898B1 (fr) * | 2020-05-07 | 2023-04-14 | Ifp Energies Now | Catalyseur d’hydrogénation sélective comprenant une répartition particulière du nickel et du molybdène |
| FR3116831B1 (fr) | 2020-11-27 | 2023-11-03 | Ifp Energies Now | Procede d’hydrogenation selective d’une essence en presence d’un catalyseur sur support meso-macroporeux |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5154087A (ja) * | 1974-09-24 | 1976-05-12 | American Cyanamid Co | |
| JPS58205547A (ja) * | 1982-05-07 | 1983-11-30 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | 高表面積スピネル型アルミン酸ニツケル触媒及びこれを用いたスチ−ムリホ−ミング法 |
| JP2000239668A (ja) * | 1999-02-24 | 2000-09-05 | Inst Fr Petrole | 低硫黄含有量のガソリン製造方法 |
| JP2001055584A (ja) * | 1999-08-19 | 2001-02-27 | Inst Fr Petrole | 硫黄含有量の低いガソリンの製造方法 |
| JP2003327970A (ja) * | 2002-03-29 | 2003-11-19 | Inst Fr Petrole | 硫黄およびメルカプタンの含量の低い炭化水素を製造する方法 |
| JP2004010892A (ja) * | 2002-06-03 | 2004-01-15 | Inst Fr Petrole | 第8族および第6b族の元素を含む担持触媒の存在下に、硫黄含有化合物およびオレフィンを含む留分を水素化脱硫するための方法 |
| JP2004230383A (ja) * | 2003-01-29 | 2004-08-19 | Inst Fr Petrole | 硫黄化合物及びオレフィンを含む留分の水素処理に使用可能な部分的にコーキングされた触媒 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| OA02047A (fr) | 1965-02-13 | 1970-05-05 | Inst Francais Du Petrole | Procédé d'hydrogenation selective des diolefines. |
| GB1379202A (en) * | 1970-11-30 | 1975-01-02 | Shell Int Research | Two-stage hydrocracking process employing a supported nickel- molybdenum catalyst |
| GB1415417A (en) * | 1973-10-12 | 1975-11-26 | American Cyanamid Co | Hydrotreating of petroleum distillates using shaped catalyst prtticles |
| FR2840316B1 (fr) * | 2002-06-03 | 2005-08-26 | Inst Francais Du Petrole | Procede d'hydrodesulfuration de coupes contenant des composes soufres et des olefines en presence d'un catalyseur comprenant un element du groupe viii et du tungstene |
| FR2857974B1 (fr) * | 2003-07-25 | 2008-01-18 | Inst Francais Du Petrole | Procede de desulfuration d'une charge d'hydrocarbures par adsorption/desorption |
| FR2895416B1 (fr) | 2005-12-22 | 2011-08-26 | Inst Francais Du Petrole | Procede d'hydrogenation selective mettant en oeuvre un catalyseur sulfure |
| FR2895414B1 (fr) | 2005-12-22 | 2011-07-29 | Inst Francais Du Petrole | Procede d'hydrogenation selective mettant en oeuvre un catalyseur presentant une porosite controlee |
-
2005
- 2005-12-22 FR FR0513173A patent/FR2895415B1/fr not_active Expired - Fee Related
-
2006
- 2006-12-14 EP EP06291972.5A patent/EP1800749B1/fr not_active Not-in-force
- 2006-12-21 US US11/642,974 patent/US7736492B2/en not_active Expired - Fee Related
- 2006-12-22 JP JP2006345142A patent/JP2007182567A/ja not_active Ceased
- 2006-12-22 KR KR1020060132517A patent/KR20070066980A/ko not_active Application Discontinuation
- 2006-12-22 BR BRPI0605369-6A patent/BRPI0605369A/pt not_active IP Right Cessation
- 2006-12-22 CN CN2006100643976A patent/CN101205483B/zh not_active Expired - Fee Related
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5154087A (ja) * | 1974-09-24 | 1976-05-12 | American Cyanamid Co | |
| JPS58205547A (ja) * | 1982-05-07 | 1983-11-30 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | 高表面積スピネル型アルミン酸ニツケル触媒及びこれを用いたスチ−ムリホ−ミング法 |
| JP2000239668A (ja) * | 1999-02-24 | 2000-09-05 | Inst Fr Petrole | 低硫黄含有量のガソリン製造方法 |
| JP2001055584A (ja) * | 1999-08-19 | 2001-02-27 | Inst Fr Petrole | 硫黄含有量の低いガソリンの製造方法 |
| JP2003327970A (ja) * | 2002-03-29 | 2003-11-19 | Inst Fr Petrole | 硫黄およびメルカプタンの含量の低い炭化水素を製造する方法 |
| JP2004010892A (ja) * | 2002-06-03 | 2004-01-15 | Inst Fr Petrole | 第8族および第6b族の元素を含む担持触媒の存在下に、硫黄含有化合物およびオレフィンを含む留分を水素化脱硫するための方法 |
| JP2004230383A (ja) * | 2003-01-29 | 2004-08-19 | Inst Fr Petrole | 硫黄化合物及びオレフィンを含む留分の水素処理に使用可能な部分的にコーキングされた触媒 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011512247A (ja) * | 2008-02-07 | 2011-04-21 | イエフペ エネルジ ヌヴェル | 選択的水素化触媒およびその調製方法 |
| KR20100028502A (ko) * | 2008-09-04 | 2010-03-12 | 아이에프피 | 특성 조성을 갖는 황화 촉매를 사용하는 선택적 수소화 방법 |
| JP2010090366A (ja) * | 2008-09-04 | 2010-04-22 | Ifp | 特定の組成を有する硫化触媒を用いる選択的水素化方法 |
| KR101668481B1 (ko) | 2008-09-04 | 2016-10-21 | 아이에프피 에너지 누벨르 | 특성 조성을 갖는 황화 촉매를 사용하는 선택적 수소화 방법 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1800749B1 (fr) | 2013-05-15 |
| FR2895415A1 (fr) | 2007-06-29 |
| EP1800749A2 (fr) | 2007-06-27 |
| KR20070066980A (ko) | 2007-06-27 |
| CN101205483B (zh) | 2012-05-23 |
| US7736492B2 (en) | 2010-06-15 |
| CN101205483A (zh) | 2008-06-25 |
| EP1800749A3 (fr) | 2011-04-20 |
| US20070187297A1 (en) | 2007-08-16 |
| BRPI0605369A (pt) | 2007-10-16 |
| FR2895415B1 (fr) | 2011-07-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2007182567A (ja) | 特定の担体を有する触媒を利用する選択的水素化法 | |
| JP5392979B2 (ja) | 硫化触媒を利用する選択的水素化法 | |
| JP5563259B2 (ja) | 特定の組成を有する硫化触媒を用いる選択的水素化方法 | |
| JP4871717B2 (ja) | 制御された多孔度を有する触媒を利用する選択的水素化法 | |
| JP5329926B2 (ja) | ヒ素を含むオレフィンガソリンの脱硫の二工程方法 | |
| JP4547922B2 (ja) | 硫黄化合物及びオレフィンを含む留分の水素処理に使用可能な部分的にコーキングされた触媒 | |
| JP2008030036A (ja) | 少なくとも1種の担体と、少なくとも1種の第viii族元素と、少なくとも1種の第vib族元素とを含む触媒の存在下に硫黄およびオレフィンを含有するガソリン留分を水素化脱硫する方法 | |
| JP6244094B2 (ja) | ガソリンの選択的水素化方法 | |
| Klimova et al. | NiMo/HNaY (x)-Al2O3 Catalysts for hydrodesulphurization of Hindered Dibenzothiophenes: Effect of the Preparation Method | |
| JP2023524797A (ja) | アルミン酸塩の形態にある特定の担体を含んでいる選択的水素化触媒 | |
| CN115443187A (zh) | 包含载体和特定镍钼比的加氢催化剂 | |
| JP2023550821A (ja) | メソ多孔性・マクロ多孔性担体上の触媒の存在下におけるガソリンの選択的水素化のための方法 | |
| JP2023550822A (ja) | メソ多孔性・マクロ多孔性担体上の捕捉塊の存在中での有機金属不純物の捕捉方法 | |
| CN115461144A (zh) | 具有镍和钼的特定分布的选择性加氢催化剂 | |
| JP2023550823A (ja) | メソ-マクロ多孔性担体上の触媒の存在中での仕上げ水素化脱硫の実施方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091218 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20091218 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120928 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20121002 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20121213 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20121218 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130201 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131119 |
|
| A045 | Written measure of dismissal of application [lapsed due to lack of payment] |
Free format text: JAPANESE INTERMEDIATE CODE: A045 Effective date: 20140325 |