JP2004217543A - アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法 - Google Patents
アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法 Download PDFInfo
- Publication number
- JP2004217543A JP2004217543A JP2003004820A JP2003004820A JP2004217543A JP 2004217543 A JP2004217543 A JP 2004217543A JP 2003004820 A JP2003004820 A JP 2003004820A JP 2003004820 A JP2003004820 A JP 2003004820A JP 2004217543 A JP2004217543 A JP 2004217543A
- Authority
- JP
- Japan
- Prior art keywords
- palladium
- antimony
- reaction
- bismuth
- acetate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

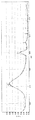
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【課題】アルコキシトルエンをカルボン酸及び分子状酸素により液相で酸化するにあたり、特定の金属元素からなる高活性な触媒を用いて高収率でアルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートを製造する方法を提供する。
【解決手段】アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いて、アルコキシトルエンとカルボン酸及び分子状酸素を液相状態で接触させて反応させる。
【選択図】 選択図なし
【解決手段】アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いて、アルコキシトルエンとカルボン酸及び分子状酸素を液相状態で接触させて反応させる。
【選択図】 選択図なし
Description
【0001】
【発明の属する技術分野】
本発明は、アルコキシトルエン、カルボン酸及び分子状酸素を液相で反応させるアルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法に関する。
【0002】
【従来の技術】
アルコキシトルエン類をカルボン酸及び酸素により酸化する方法として、スズ、ゲルマニウム、テルル及び銅から選ばれた元素及びパラジウムを活性炭に担持した触媒により、水/有機溶媒混合物中で反応を行う方法が開示されている。しかし、この方法ではアルコキシ安息香酸等の副生成物の選択率が高いため、工業的に満足できるものではない。(例えば、特許文献1参照。)
また、パラジウムを活性炭に担持した触媒に、スズ化合物及び酢酸カリウムを添加して、酢酸中で反応を行う方法が報告されている。しかし、この方法では、スズ化合物及び酢酸カリウムが反応系に可溶であるため、スズ化合物及び酢酸カリウムの分離・回収工程が必要となり、工業的に実用的ではない。(例えば、非特許文献1参照。)
【特許文献1】
特開平6−100480号公報(第13頁)
【非特許文献1】
「ジャーナル・オブ・キャタリシス」(J.Cat.)(140),(1993)(第311頁)
【0003】
【発明が解決しようとする課題】
本発明は上記の課題に鑑みてなされたものであり、高活性な触媒を用いて高選択的にアルコキシトルエンを酸化し、アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートを高収率で得る方法を提供するものである。
【0004】
【課題を解決するための手段】
本発明者らは、上記問題点を解決すべく鋭意研究を行った結果、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが担持されている特定の触媒を液相で用いることにより、アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートが高収率で得られることを見出し、本発明を完成するに至った。
【0005】
すなわち、本発明はアルコキシトルエンをカルボン酸及び分子状酸素により液相で酸化するにあたり、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いることを特徴とする。
【0006】
以下本発明について詳細に説明する。
【0007】
本発明の触媒を調製するにあたり、使用するパラジウム、アンチモン、ビスマス及びテルルの原料は、特に限定するものではない。
【0008】
パラジウム原料としては、パラジウム金属、ヘキサクロロパラジウム酸アンモニウム、へキサクロロパラジウム酸カリウム、ヘキサクロロパラジウム酸ナトリウム、テトラクロロパラジウム酸アンモニウム、テトラクロロパラジウム酸カリウム、テトラクロロパラジウム酸ナトリウム、テトラブロモパラジウム酸カリウム、酸化パラジウム、塩化パラジウム、臭化パラジウム、ヨウ化パラジウム、硝酸パラジウム、硫酸パラジウム、酢酸パラジウム、ジニトロサルファイトパラジウム酸カリウム、クロロカルボニルパラジウム、ジニトロジアンミンパラジウム、テトラアンミンパラジウム塩化物、テトラアンミンパラジウム硝酸塩、cis−ジクロロジアミンパラジウム、trans−ジクロロジアミンパラジウム、ジクロロ(エチレンジアミン)パラジウム、テトラシアノパラジウム酸カリウム等を例示できる。
【0009】
さらに、アンチモンの原料としては、アンチモン金属、フッ化アンチモン、塩化アンチモン、臭化アンチモン、ヨウ化アンチモン、酢酸アンチモン、アンチモニメトキシド、アンチモニエトキシド、アンチモニイソプロポキシド、アンチモニブトキシド、アンチモニエチレングリコシド、アンチモニポタシウムタータレイト、酸化アンチモン、硫化アンチモン、酒石酸やシュウ酸等の有機酸とのアンチモン錯化合物等を例示することができる。
【0010】
さらに、ビスマスの原料としては、ビスマス金属、塩化ビスマス、硝酸ビスマス、オキシ塩化ビスマス、酢酸ビスマス、オキシ酢酸ビスマス、酸化ビスマス等を例示することができる。
【0011】
さらに、テルルの原料としては、金属テルル、塩化テルル、酸化テルル、テルル酸、テルル酸カリウム、テルル酸ナトリウム等を例示することができる。
【0012】
アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムは、それ自体反応に不活性な担体に担持されて使用される。好ましい担体としてはシリカや活性炭を例示できる。
【0013】
パラジウムの担体への担持量は、担体の重量に対して、0.01〜10重量%、好ましくは0.1〜5重量%である。0.01重量%より少ないと実質的な反応速度を得られず、また、10重量%より多いと経済的に不利となる。
【0014】
又、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの担体への担持は、高選択的にアルコキシトルエンを酸化するために、原子比で(パラジウム)/(アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素)=1〜100の範囲で行うことが好ましい。
【0015】
本発明において、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムは0価の金属状態で担体に担持され、且つアンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有して担体に担持される。
【0016】
本発明の触媒調製方法について述べる。
【0017】
本発明の触媒の形状には特に制限はなく、反応形式に応じて粉末状のもの又は成形品を用いることができる.懸濁床における反応では粉末又は顆粒を、固定床における反応ではタブレットの打錠成形品、球状又は棒柱状の押し出し成形品等が好ましく用いられる。
【0018】
アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの原料を担体に担持する方法は特に限定されるものではなく、公知の方法で担持することができる。例えば沈殿法、イオン交換法、含浸法、沈着法、混練法で担持することができる。含浸法で担持する場合には、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの原料を同時に含浸担持する、もしくは、一方の原料を含浸担持した後、残りの原料を含浸担持してもよい。しかし、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムをより均一に担持するためには、両者を同時に含浸担持するのが好ましい。
【0019】
例えば含浸法によりパラジウムとアンチモンの合金を調製する際には、乾燥工程において真空下で乾燥させるのが好ましい。乾燥温度は、好ましくは0〜700℃、さらに好ましくは10〜500℃である。
【0020】
含浸法についてさらに例示するならば、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの原料を適当な溶媒に溶解し、これを担体と混合し、必要ならば所定の時間静置した後、公知の方法で溶媒を除去し、乾燥し、触媒前駆体を得る。得られた触媒前駆体を水素又は水素を含む不活性ガス中で還元処理することにより本発明の触媒を得ることができる。還元処理の温度は、通常100〜700℃、好ましくは200〜500℃の範囲である。
【0021】
なお、還元処理を行う場合の還元剤としては、水素以外にも、一酸化炭素、エチレン等のガス類、アルコール、ヒドラジン水和物等も使用することができ、用いる還元剤の種類に応じて気相又は液相のいずれでも還元処理を行うことができる。
【0022】
なお、水素等による還元処理前に酸素雰囲気下で焼成しても構わない。還元処理前に焼成を行う場合には、酸素又は窒素、ヘリウム、アルゴン等で希釈した酸素、さらには空気の雰囲気中で、200〜700℃で焼成するのがよい。
【0023】
本発明で用いられる原料のアルコキシトルエンは特に限定されるものではなく、p−メトキシトルエン、m−メトキシトルエン、o−メトキシトルエン、p−エトキシトルエン、m−エトキシトルエン、o−エトキシトルエン、p−n−プロポキシトルエン、m−n−プロポキシトルエン、o−n−プロポキシトルエン、p−イソプロポキシトルエン、m−イソプロポキシトルエン、o−イソプロポキシトルエン等を例示することができる。また、本発明で用いられる原料のカルボン酸は特に限定されるものではなく、酢酸、プロピオン酸、酪酸等を例示することができる。これらアルコキシトルエンとカルボン酸の混合比としては、任意の混合比で反応を行うことができる。
【0024】
本反応における溶媒は、反応条件によりカルボン酸もしくはカルボン酸/水の混合物を用いることができる。
【0025】
本発明で用いられる反応器は、反応が液相で行われるために触媒の表面が原料液で覆われる構造であれば特に限定するものではなく、例示として、懸濁床による回分、半回分、連続式反応器、又は固定床流通式反応器が挙げられる。
【0026】
本発明の反応温度は、通常80〜230℃、好ましくは100〜200℃である。反応温度が230℃より高いと副反応の進行が増し、80℃より低いと反応速度の点で不利になる。
【0027】
本発明の反応圧力は、反応温度下で原料のアルコキシトルエン及びカルボン酸が触媒表面で液相に保たる圧力であればよく、好ましくは常圧〜10MPaである。
【0028】
本発明の方法においては、分子状酸素を酸化剤として用いる。分子状酸素は、窒素等の不活性ガスで希釈されていてもよく、空気であってもよい。酸素の供給量は、反応温度、触媒量等によって最適量が変わるが、反応器内の気相部のガス組成が爆発範囲以下であればよい。
【0029】
反応時間は、反応温度、圧力、触媒量等の設定、及び反応器の形式等に応じて適宜最適な時間を設定すればよく、特に限定するものではないが、懸濁床での回分式、半回分式反応においては0.5時間以上、好ましくは1〜10時間である。また、懸濁床による連続式反応又は固定床流通式反応においては、滞留時間は0.03〜10時間が好ましい。
【0030】
【実施例】
以下、本反応を実施例によりさらに詳しく説明するが、本発明はこれら実施例に限定されるものではない。
【0031】
(X線回折測定方法)
X線回折測定は、X線回折測定装置(マックサイエンス社製 M18XHF)を用いてX線源CuKα1、管電流200mA、管電圧40kVで行った。
【0032】
実施例1
蒸留水16gに酒石酸3.6gを溶解し、ここに酸化アンチモン0.49gを加え溶解させた。続いて8.3重量%硝酸パラジウム水溶液7.27gを加えた。この水溶液に担体(富士シリシア社製、商品名キャリアクトQ−30)を20g加えて含浸した。その後、50℃で減圧下乾燥して、100℃で3時間真空乾燥、400℃で5時間空気焼成を経て、400℃で5時間水素還元した。パラジウムのシリカに対する担持量は3重量%、アンチモンのパラジウムに対する添加量は、アンチモン/パラジウム原子比で0.6であった。
【0033】
また、X線回折の結果から、パラジウムとアンチモンが共に酸化状態ではなく、0価の金属状態で合金を形成していることを確認した(図1)。
【0034】
温度計、撹拌装置、ガス導入管及び還流冷却装置を取り付けた三つ口フラスコに、上記触媒0.7g、p−メトキシトルエン 2g及び酢酸8mlを加え、内温100℃、常圧で、酸素を60ml/分(0℃、1気圧換算)で連続的に供給し、5時間撹拌することで反応を行った。反応終了後、反応液をガスクロマトグラフィーで分析した結果、p−メトキシトルエンの転化率86.9%、p−メトキシベンジルアセテートの選択率40.5%、p−メトキシベンズアルデヒドの選択率42.2%、アニス酸の選択率17.3%であった。
【0035】
実施例2
蒸留水10gに酒石酸2.3gを溶解させた後、三酸化アンチモン0.33gを添加し溶解させた。続いて8.3重量%のジニトロジアンミンパラジウム硝酸溶液を7.27g添加し、さらに蒸留水を加え全量を21mlとした。この水溶液に担体(富士シリシア社製、商品名キャリアクトQ−30)を20g加えて含浸した。その後、50℃で減圧下乾燥して、100℃で3時間真空乾燥、400℃で5時間空気焼成を経て、400℃で水素還元した。得られた触媒は、元素分析の結果、パラジウム担持量は3重量%で、アンチモンのパラジウムに対する添加量は、アンチモン/パラジウム原子比で0.4であった。また、X線回折の結果から、パラジウムとアンチモンが合金を形成していることを確認した。
【0036】
温度計、撹拌装置、ガス導入管及び還流冷却装置を取り付けた三つ口フラスコに、上記触媒0.7g、p−メトキシトルエン 2g及び酢酸8mlを加え、内温100℃、常圧で、酸素を30ml/分(0℃、1気圧換算)で連続的に供給し、5時間撹拌することで反応を行った。反応終了後、反応液をガスクロマトグラフィーで分析した結果、p−メトキシトルエンの転化率81.5%、p−メトキシベンジルアセテートの選択率76.3%、p−メトキシベンズアルデヒドの選択率15.3%、アニス酸の選択率8.4%であった。
【0037】
比較例1
蒸留水16gに酒石酸3.6gを溶解し、ここに酸化アンチモン0.49gを加え溶解させた。続いて8.3重量%硝酸パラジウム水溶液7.27gを加えた。この水溶液に担体(富士シリシア社製、商品名キャリアクトQ−30)を20g加えて含浸した。その後、50℃で減圧下乾燥して、300℃で5時間空気焼成を経て、400℃で5時間水素還元した。パラジウムのシリカに対する担持量は3重量%、アンチモンのパラジウムに対する添加量は、アンチモン/パラジウム原子比で0.6であった。また、X線回折の結果から、パラジウムとアンチモンは合金を形成していないことを確認した(図2)。
【0038】
温度計、撹拌装置、ガス導入管及び還流冷却装置を取り付けた三つ口フラスコに、上記触媒0.7g、p−メトキシトルエン 2g及び酢酸8mlを加え、内温100℃、常圧で、酸素を60ml/分(0℃、1気圧換算)で連続的に供給し、5時間撹拌することで反応を行った。反応終了後、反応液をガスクロマトグラフィーで分析した結果、p−メトキシトルエンの転化率77.6%、p−メトキシベンジルアセテートの選択率21.5%、p−メトキシベンズアルデヒドの選択率24.0%、アニス酸の選択率54.5%であった。
【0039】
【発明の効果】
アルコキシトルエンをカルボン酸及び分子状酸素により液相で酸化するにあたり、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いることにより、アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートを高収率で得ることができる。
【図面の簡単な説明】
【図1】実施例1により得られた触媒のX線回折結果
【図2】比較例1により得られた触媒のX線回折結果
【発明の属する技術分野】
本発明は、アルコキシトルエン、カルボン酸及び分子状酸素を液相で反応させるアルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法に関する。
【0002】
【従来の技術】
アルコキシトルエン類をカルボン酸及び酸素により酸化する方法として、スズ、ゲルマニウム、テルル及び銅から選ばれた元素及びパラジウムを活性炭に担持した触媒により、水/有機溶媒混合物中で反応を行う方法が開示されている。しかし、この方法ではアルコキシ安息香酸等の副生成物の選択率が高いため、工業的に満足できるものではない。(例えば、特許文献1参照。)
また、パラジウムを活性炭に担持した触媒に、スズ化合物及び酢酸カリウムを添加して、酢酸中で反応を行う方法が報告されている。しかし、この方法では、スズ化合物及び酢酸カリウムが反応系に可溶であるため、スズ化合物及び酢酸カリウムの分離・回収工程が必要となり、工業的に実用的ではない。(例えば、非特許文献1参照。)
【特許文献1】
特開平6−100480号公報(第13頁)
【非特許文献1】
「ジャーナル・オブ・キャタリシス」(J.Cat.)(140),(1993)(第311頁)
【0003】
【発明が解決しようとする課題】
本発明は上記の課題に鑑みてなされたものであり、高活性な触媒を用いて高選択的にアルコキシトルエンを酸化し、アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートを高収率で得る方法を提供するものである。
【0004】
【課題を解決するための手段】
本発明者らは、上記問題点を解決すべく鋭意研究を行った結果、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが担持されている特定の触媒を液相で用いることにより、アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートが高収率で得られることを見出し、本発明を完成するに至った。
【0005】
すなわち、本発明はアルコキシトルエンをカルボン酸及び分子状酸素により液相で酸化するにあたり、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いることを特徴とする。
【0006】
以下本発明について詳細に説明する。
【0007】
本発明の触媒を調製するにあたり、使用するパラジウム、アンチモン、ビスマス及びテルルの原料は、特に限定するものではない。
【0008】
パラジウム原料としては、パラジウム金属、ヘキサクロロパラジウム酸アンモニウム、へキサクロロパラジウム酸カリウム、ヘキサクロロパラジウム酸ナトリウム、テトラクロロパラジウム酸アンモニウム、テトラクロロパラジウム酸カリウム、テトラクロロパラジウム酸ナトリウム、テトラブロモパラジウム酸カリウム、酸化パラジウム、塩化パラジウム、臭化パラジウム、ヨウ化パラジウム、硝酸パラジウム、硫酸パラジウム、酢酸パラジウム、ジニトロサルファイトパラジウム酸カリウム、クロロカルボニルパラジウム、ジニトロジアンミンパラジウム、テトラアンミンパラジウム塩化物、テトラアンミンパラジウム硝酸塩、cis−ジクロロジアミンパラジウム、trans−ジクロロジアミンパラジウム、ジクロロ(エチレンジアミン)パラジウム、テトラシアノパラジウム酸カリウム等を例示できる。
【0009】
さらに、アンチモンの原料としては、アンチモン金属、フッ化アンチモン、塩化アンチモン、臭化アンチモン、ヨウ化アンチモン、酢酸アンチモン、アンチモニメトキシド、アンチモニエトキシド、アンチモニイソプロポキシド、アンチモニブトキシド、アンチモニエチレングリコシド、アンチモニポタシウムタータレイト、酸化アンチモン、硫化アンチモン、酒石酸やシュウ酸等の有機酸とのアンチモン錯化合物等を例示することができる。
【0010】
さらに、ビスマスの原料としては、ビスマス金属、塩化ビスマス、硝酸ビスマス、オキシ塩化ビスマス、酢酸ビスマス、オキシ酢酸ビスマス、酸化ビスマス等を例示することができる。
【0011】
さらに、テルルの原料としては、金属テルル、塩化テルル、酸化テルル、テルル酸、テルル酸カリウム、テルル酸ナトリウム等を例示することができる。
【0012】
アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムは、それ自体反応に不活性な担体に担持されて使用される。好ましい担体としてはシリカや活性炭を例示できる。
【0013】
パラジウムの担体への担持量は、担体の重量に対して、0.01〜10重量%、好ましくは0.1〜5重量%である。0.01重量%より少ないと実質的な反応速度を得られず、また、10重量%より多いと経済的に不利となる。
【0014】
又、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの担体への担持は、高選択的にアルコキシトルエンを酸化するために、原子比で(パラジウム)/(アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素)=1〜100の範囲で行うことが好ましい。
【0015】
本発明において、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムは0価の金属状態で担体に担持され、且つアンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有して担体に担持される。
【0016】
本発明の触媒調製方法について述べる。
【0017】
本発明の触媒の形状には特に制限はなく、反応形式に応じて粉末状のもの又は成形品を用いることができる.懸濁床における反応では粉末又は顆粒を、固定床における反応ではタブレットの打錠成形品、球状又は棒柱状の押し出し成形品等が好ましく用いられる。
【0018】
アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの原料を担体に担持する方法は特に限定されるものではなく、公知の方法で担持することができる。例えば沈殿法、イオン交換法、含浸法、沈着法、混練法で担持することができる。含浸法で担持する場合には、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの原料を同時に含浸担持する、もしくは、一方の原料を含浸担持した後、残りの原料を含浸担持してもよい。しかし、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムをより均一に担持するためには、両者を同時に含浸担持するのが好ましい。
【0019】
例えば含浸法によりパラジウムとアンチモンの合金を調製する際には、乾燥工程において真空下で乾燥させるのが好ましい。乾燥温度は、好ましくは0〜700℃、さらに好ましくは10〜500℃である。
【0020】
含浸法についてさらに例示するならば、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムの原料を適当な溶媒に溶解し、これを担体と混合し、必要ならば所定の時間静置した後、公知の方法で溶媒を除去し、乾燥し、触媒前駆体を得る。得られた触媒前駆体を水素又は水素を含む不活性ガス中で還元処理することにより本発明の触媒を得ることができる。還元処理の温度は、通常100〜700℃、好ましくは200〜500℃の範囲である。
【0021】
なお、還元処理を行う場合の還元剤としては、水素以外にも、一酸化炭素、エチレン等のガス類、アルコール、ヒドラジン水和物等も使用することができ、用いる還元剤の種類に応じて気相又は液相のいずれでも還元処理を行うことができる。
【0022】
なお、水素等による還元処理前に酸素雰囲気下で焼成しても構わない。還元処理前に焼成を行う場合には、酸素又は窒素、ヘリウム、アルゴン等で希釈した酸素、さらには空気の雰囲気中で、200〜700℃で焼成するのがよい。
【0023】
本発明で用いられる原料のアルコキシトルエンは特に限定されるものではなく、p−メトキシトルエン、m−メトキシトルエン、o−メトキシトルエン、p−エトキシトルエン、m−エトキシトルエン、o−エトキシトルエン、p−n−プロポキシトルエン、m−n−プロポキシトルエン、o−n−プロポキシトルエン、p−イソプロポキシトルエン、m−イソプロポキシトルエン、o−イソプロポキシトルエン等を例示することができる。また、本発明で用いられる原料のカルボン酸は特に限定されるものではなく、酢酸、プロピオン酸、酪酸等を例示することができる。これらアルコキシトルエンとカルボン酸の混合比としては、任意の混合比で反応を行うことができる。
【0024】
本反応における溶媒は、反応条件によりカルボン酸もしくはカルボン酸/水の混合物を用いることができる。
【0025】
本発明で用いられる反応器は、反応が液相で行われるために触媒の表面が原料液で覆われる構造であれば特に限定するものではなく、例示として、懸濁床による回分、半回分、連続式反応器、又は固定床流通式反応器が挙げられる。
【0026】
本発明の反応温度は、通常80〜230℃、好ましくは100〜200℃である。反応温度が230℃より高いと副反応の進行が増し、80℃より低いと反応速度の点で不利になる。
【0027】
本発明の反応圧力は、反応温度下で原料のアルコキシトルエン及びカルボン酸が触媒表面で液相に保たる圧力であればよく、好ましくは常圧〜10MPaである。
【0028】
本発明の方法においては、分子状酸素を酸化剤として用いる。分子状酸素は、窒素等の不活性ガスで希釈されていてもよく、空気であってもよい。酸素の供給量は、反応温度、触媒量等によって最適量が変わるが、反応器内の気相部のガス組成が爆発範囲以下であればよい。
【0029】
反応時間は、反応温度、圧力、触媒量等の設定、及び反応器の形式等に応じて適宜最適な時間を設定すればよく、特に限定するものではないが、懸濁床での回分式、半回分式反応においては0.5時間以上、好ましくは1〜10時間である。また、懸濁床による連続式反応又は固定床流通式反応においては、滞留時間は0.03〜10時間が好ましい。
【0030】
【実施例】
以下、本反応を実施例によりさらに詳しく説明するが、本発明はこれら実施例に限定されるものではない。
【0031】
(X線回折測定方法)
X線回折測定は、X線回折測定装置(マックサイエンス社製 M18XHF)を用いてX線源CuKα1、管電流200mA、管電圧40kVで行った。
【0032】
実施例1
蒸留水16gに酒石酸3.6gを溶解し、ここに酸化アンチモン0.49gを加え溶解させた。続いて8.3重量%硝酸パラジウム水溶液7.27gを加えた。この水溶液に担体(富士シリシア社製、商品名キャリアクトQ−30)を20g加えて含浸した。その後、50℃で減圧下乾燥して、100℃で3時間真空乾燥、400℃で5時間空気焼成を経て、400℃で5時間水素還元した。パラジウムのシリカに対する担持量は3重量%、アンチモンのパラジウムに対する添加量は、アンチモン/パラジウム原子比で0.6であった。
【0033】
また、X線回折の結果から、パラジウムとアンチモンが共に酸化状態ではなく、0価の金属状態で合金を形成していることを確認した(図1)。
【0034】
温度計、撹拌装置、ガス導入管及び還流冷却装置を取り付けた三つ口フラスコに、上記触媒0.7g、p−メトキシトルエン 2g及び酢酸8mlを加え、内温100℃、常圧で、酸素を60ml/分(0℃、1気圧換算)で連続的に供給し、5時間撹拌することで反応を行った。反応終了後、反応液をガスクロマトグラフィーで分析した結果、p−メトキシトルエンの転化率86.9%、p−メトキシベンジルアセテートの選択率40.5%、p−メトキシベンズアルデヒドの選択率42.2%、アニス酸の選択率17.3%であった。
【0035】
実施例2
蒸留水10gに酒石酸2.3gを溶解させた後、三酸化アンチモン0.33gを添加し溶解させた。続いて8.3重量%のジニトロジアンミンパラジウム硝酸溶液を7.27g添加し、さらに蒸留水を加え全量を21mlとした。この水溶液に担体(富士シリシア社製、商品名キャリアクトQ−30)を20g加えて含浸した。その後、50℃で減圧下乾燥して、100℃で3時間真空乾燥、400℃で5時間空気焼成を経て、400℃で水素還元した。得られた触媒は、元素分析の結果、パラジウム担持量は3重量%で、アンチモンのパラジウムに対する添加量は、アンチモン/パラジウム原子比で0.4であった。また、X線回折の結果から、パラジウムとアンチモンが合金を形成していることを確認した。
【0036】
温度計、撹拌装置、ガス導入管及び還流冷却装置を取り付けた三つ口フラスコに、上記触媒0.7g、p−メトキシトルエン 2g及び酢酸8mlを加え、内温100℃、常圧で、酸素を30ml/分(0℃、1気圧換算)で連続的に供給し、5時間撹拌することで反応を行った。反応終了後、反応液をガスクロマトグラフィーで分析した結果、p−メトキシトルエンの転化率81.5%、p−メトキシベンジルアセテートの選択率76.3%、p−メトキシベンズアルデヒドの選択率15.3%、アニス酸の選択率8.4%であった。
【0037】
比較例1
蒸留水16gに酒石酸3.6gを溶解し、ここに酸化アンチモン0.49gを加え溶解させた。続いて8.3重量%硝酸パラジウム水溶液7.27gを加えた。この水溶液に担体(富士シリシア社製、商品名キャリアクトQ−30)を20g加えて含浸した。その後、50℃で減圧下乾燥して、300℃で5時間空気焼成を経て、400℃で5時間水素還元した。パラジウムのシリカに対する担持量は3重量%、アンチモンのパラジウムに対する添加量は、アンチモン/パラジウム原子比で0.6であった。また、X線回折の結果から、パラジウムとアンチモンは合金を形成していないことを確認した(図2)。
【0038】
温度計、撹拌装置、ガス導入管及び還流冷却装置を取り付けた三つ口フラスコに、上記触媒0.7g、p−メトキシトルエン 2g及び酢酸8mlを加え、内温100℃、常圧で、酸素を60ml/分(0℃、1気圧換算)で連続的に供給し、5時間撹拌することで反応を行った。反応終了後、反応液をガスクロマトグラフィーで分析した結果、p−メトキシトルエンの転化率77.6%、p−メトキシベンジルアセテートの選択率21.5%、p−メトキシベンズアルデヒドの選択率24.0%、アニス酸の選択率54.5%であった。
【0039】
【発明の効果】
アルコキシトルエンをカルボン酸及び分子状酸素により液相で酸化するにあたり、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いることにより、アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートを高収率で得ることができる。
【図面の簡単な説明】
【図1】実施例1により得られた触媒のX線回折結果
【図2】比較例1により得られた触媒のX線回折結果
Claims (1)
- アルコキシトルエンをカルボン酸及び分子状酸素により液相で酸化するにあたり、アンチモン、ビスマス、テルルから選ばれる少なくとも1種類以上の元素とパラジウムが合金構造を有しており、且つ担体に担持されている触媒を用いることを特徴とするアルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003004820A JP2004217543A (ja) | 2003-01-10 | 2003-01-10 | アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003004820A JP2004217543A (ja) | 2003-01-10 | 2003-01-10 | アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004217543A true JP2004217543A (ja) | 2004-08-05 |
Family
ID=32895680
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003004820A Pending JP2004217543A (ja) | 2003-01-10 | 2003-01-10 | アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004217543A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007136434A (ja) * | 2005-10-20 | 2007-06-07 | Mitsubishi Rayon Co Ltd | パラジウム含有触媒、その製造方法、およびα,β−不飽和カルボン酸の製造方法 |
-
2003
- 2003-01-10 JP JP2003004820A patent/JP2004217543A/ja active Pending
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007136434A (ja) * | 2005-10-20 | 2007-06-07 | Mitsubishi Rayon Co Ltd | パラジウム含有触媒、その製造方法、およびα,β−不飽和カルボン酸の製造方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4750283B2 (ja) | エチレンと酢酸の気相酸化による酢酸ビニルの製造用触媒、その製法およびその使用方法 | |
| US8044250B2 (en) | Manufacture of 1,1,1,2,3,3-hexafluoropropane and 1,1,1,2-tetrafluoropropane via catalytic hydrogenation | |
| CN86102452A (zh) | 通过羧酸加氢制备醇 | |
| JPH0644998B2 (ja) | 銀触媒及びそれの製造方法 | |
| JP2004217543A (ja) | アルコキシベンズアルデヒド及び/又はアルコキシベンジルアセテートの製造方法 | |
| JP5017092B2 (ja) | パラジウム含有触媒、その製造方法、およびα,β−不飽和カルボン酸の製造方法 | |
| JPH03178335A (ja) | エチレンをエチレンオキシドに酸化するための銀触媒、その製造方法およびエチレンを酸化する方法 | |
| JP2887278B2 (ja) | 1―クロロ―1,2,2―トリフルオロエチレンおよび1,2,2―トリフルオロエチレンの製造方法 | |
| JP4599780B2 (ja) | 含フッ素アルコール化合物の製造用触媒及び含フッ素アルコールの製造方法 | |
| JP2004137234A (ja) | キシレンのカルボン酸エステルの製造方法 | |
| JP5781431B2 (ja) | 1,3−プロパンジオールの製造方法、及びグリセリンの水素化反応用触媒 | |
| JP4284740B2 (ja) | パラジウム−アンチモン合金触媒の調製方法 | |
| JPS608864B2 (ja) | ジアシルオキシアルケン合成用触媒の製造法 | |
| JP4062763B2 (ja) | キシリレンジアセテートの製造用触媒 | |
| JP3422062B2 (ja) | ジアセトキシブテンの製造方法 | |
| CN121085811A (zh) | 一种制备(甲基)丙烯腈的方法 | |
| JP6988642B2 (ja) | アリル化合物の異性化方法 | |
| JP4232266B2 (ja) | フェニルエステル製造方法 | |
| JP4158217B2 (ja) | 酢酸ベンジルの製造方法 | |
| JP4273784B2 (ja) | フェニルエステルの製造方法 | |
| JP2001097922A (ja) | フェニルエステルの製造方法及び触媒の調製方法 | |
| JPH08231466A (ja) | キシリレンジアセテートの製造方法 | |
| RU2156756C2 (ru) | Способ получения гексафторацетона | |
| JP4009766B2 (ja) | カルボン酸ベンジルの製造方法 | |
| JP2000281621A (ja) | フェニルエステルの製造方法 |