JP2004131395A - アミノウラシルの製造方法 - Google Patents
アミノウラシルの製造方法 Download PDFInfo
- Publication number
- JP2004131395A JP2004131395A JP2002294767A JP2002294767A JP2004131395A JP 2004131395 A JP2004131395 A JP 2004131395A JP 2002294767 A JP2002294767 A JP 2002294767A JP 2002294767 A JP2002294767 A JP 2002294767A JP 2004131395 A JP2004131395 A JP 2004131395A
- Authority
- JP
- Japan
- Prior art keywords
- amino
- dialkyluracil
- general formula
- producing
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 C*NC(N*)=O Chemical compound C*NC(N*)=O 0.000 description 1
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【課題】従来技術における、水溶媒の使用により、収率が低下するという実用面での欠点を解消し、効率的な4−アミノ−1、3−ジアルキルウラシルの製造方法を提供する。
【解決手段】縮合後の環化反応に際して特定のアルコール水溶液を使用することにより、溶解性を改善し、収率を向上させて効率的に4−アミノ−1、3−ジアルキルウラシルを製造する。
【効果】反応器の内容物の溶媒への溶解性低下が回避され、攪拌性および4−アミノ−1、3−ジアルキルウラシルの収率の向上が達成でき、効率的な工業的製造法が提供される。
【選択図】なし
【解決手段】縮合後の環化反応に際して特定のアルコール水溶液を使用することにより、溶解性を改善し、収率を向上させて効率的に4−アミノ−1、3−ジアルキルウラシルを製造する。
【効果】反応器の内容物の溶媒への溶解性低下が回避され、攪拌性および4−アミノ−1、3−ジアルキルウラシルの収率の向上が達成でき、効率的な工業的製造法が提供される。
【選択図】なし
Description
【0001】
【発明の属する技術分野】
本発明は一般式〔3〕[化4]
【0002】
【化4】
 【0003】
【0003】
(式中、R1 、R2は同一、もしくは異なっていてもよく、炭素数1〜5の直鎖あるいは分岐鎖のアルキル基を示す。)で表わされる4−アミノ−1、3−ジアルキルウラシルの製造方法に関する。近年、4−アミノ−1、3−ジアルキルウラシルは、抗パーキンソン治療薬、うっ血性心不全治療薬、喘息治療薬などとして開発されているアデノシンアンタゴニスト、アデノシンアゴニスト向け医薬品の製造中間体として、有用な化合物として注目されている。
【0004】
【従来の技術】
従来、4−アミノ−1、3−ジアルキルウラシルの製造法は、例えば、N、N’−ジアルキルウレアにシアノ酢酸と無水酢酸を加え、水溶媒中で反応させて縮合した後、これに水酸化ナトリウムを加えてアルカリ性として環化反応を行い、その溶液を冷却、晶析、濾過して得る方法(非特許文献1参照。)が知られている。
【0005】
【非特許文献1】
J.Am.Chem.Soc.,76,2798(1954)
【0006】
【発明が解決しようとする課題】
しかしながら、上記の製造法では工業的に製造する場合、次のような問題点があった。
【0007】
すなわち、溶媒として水を用いているため、出発原料化合物の1つであるN、N’−ジアルキルウレアのアルキル基の炭素数増加に伴い、脂溶性が増し、水溶性が低下してしまうため、反応系の粘性が増大し、反応収率が低下していた。
【0008】
このため、4−アミノ−1、3−ジアルキルウラシルの工業的製造法の開発が望まれていた。
【0009】
よって、本発明の課題は、より攪拌性が良好で、反応収率の高い4−アミノ−1、3−ジアルキルウラシルの工業的製造法を提供することにある。
【0010】
【課題を解決するための手段】
そこで本発明者等は、上記の課題を解決するために鋭意検討した結果、縮合後の反応液を濃縮し、反応溶媒としてアルコール水溶液を添加することにより、効率的に4−アミノ−1、3−ジアルキルウラシルを製造することを見出し、本発明を完成するに至った。
【0011】
すなわち、本発明は以下のとおりである。
[1] 一般式〔1〕[化5]
【0012】
【化5】
【0013】
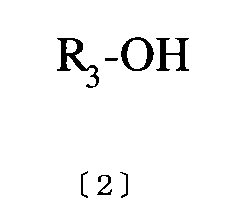
(式中、R1 、R2は同一、もしくは異なっていてもよく、炭素数1〜5の直鎖あるいは分岐鎖のアルキル基を示す。)で表わされるN、N’−ジアルキルウレアとシアノ酢酸とを、無水酢酸を用いて縮合し、塩基を加えて環化するに際して、縮合後の反応液を濃縮し、反応溶媒として一般式〔2〕[化6]
【0014】
【化6】
【0015】
(式中、R3は炭素数1〜5の直鎖あるいは分岐鎖のアルキル基を示す。)で表されるアルコール水溶液を添加して環化することを特徴とする一般式〔3〕[化7]
【0016】
【化7】
【0017】
(式中、R1 、R2は前記と同様。)で表される4−アミノ−1、3−ジアルキルウラシルの製造方法、
[2] 一般式〔2〕で表されるアルコール水溶液が、メタノール水溶液である[1]記載の4−アミノ−1、3−ジアルキルウラシルの製造方法、
[3] 一般式〔2〕で表されるアルコール水溶液が、1〜50vol%である[1]又は[2]記載の4−アミノ−1、3−ジアルキルウラシルの製造方法、
[4] 一般式〔2〕で表されるアルコール水溶液が、1〜30vol%である[1]〜[3]記載のいずれか一項に4−アミノ−1、3−ジアルキルウラシルの製造方法、
[5] 一般式〔2〕で表されるアルコール水溶液が、10〜25vol%である[1]〜[4]のいずれか一項に記載の4−アミノ−1、3−ジアルキルウラシルの製造方法。
【0018】
【発明の実施の形態】
以下、本発明を詳細に説明する。
本発明において、炭素数1〜5の直鎖あるいは分岐鎖のアルキル基としては、例えば、メチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、i−ブチル基、t−ブチル基、n−ペンチル基、i−ペンチル基、ネオペンチル基を挙げることができるが、これらに限定されるものではない。
【0019】
一般式〔1〕で表されるN、N’−ジアルキルウレアとしては、例えば、N、N’−ジメチルウレア、N、N’−ジエチルウレア、N、N’−ジ−n−プロピルウレア、N、N’−ジ−i−プロピルウレア、N、N’−ジ−n− ブチルウレア、N、N’−ジ−i−ブチルウレア、N、N’−ジ−t−ブチルウレア、N、N’−ジ−n−ペンチルウレア、N、N’−ジ−i− ペンチウレア、N、N’−ジ−ネオ−ペンチウレア、N−メチル−N’−エチルウレア、N−メチル−N’− n−プロピルウレア、N−エチル−N’− n−プロピルウレアなどを挙げることができるが、これらに限定されるものではない。
【0020】
また、一般式〔2〕で表されるアルコール水溶液としては、水溶媒にアルコールを添加したメタノール水溶液、エタノール水溶液、n−プロピルアルコール水溶液、i−プロピルアルコール水溶液、n−ブチルアルコール水溶液、i−ブチルアルコール水溶液、t−ブチルアルコール水溶液、n−ペンチルアルコール水溶液、i−ペンチルアルコール水溶液、ネオペンチルアルコール水溶液が挙げられるが、好ましくはメタノール水溶液である。
【0021】
本発明の一般式〔1〕で表されるN、N’−ジアルキルウレアの縮合および環化反応は、例えば、次に述べる条件において行うことができる。
【0022】
N、N’−ジアルキルウレアに対するシアノ酢酸および無水酢酸の使用量は特に限定されないが、それぞれ等モル、等モルから2倍モルが好ましい。反応温度は、室温から80℃まで可能であるが、好ましくは50℃から70℃の間である。反応時間は、1時間から5時間の間であれば良く、好ましくは2時間から3時間である。
【0023】
上記操作で得られた反応液を、4−アミノ−1、3−ジアルキルウラシルの理論収量の1倍重量から10倍重量、好ましくは1.5倍重量から5倍重量の間になるよう濃縮する。濃縮は、反応で生じた揮発成分、酸性成分を除去する目的で行い、必要に応じて水を添加して何回か濃縮することもできる。
【0024】
濃縮した反応液に、反応溶媒としてアルコール水溶液を添加し、塩基を滴下することにより環化反応を行い、4−アミノ−1、3−ジアルキルウラシルを合成する。
【0025】
反応溶媒の濃度は、1〜50vol%アルコール水溶液であればよく、好ましくは1〜30vol%アルコール水溶液であり、さらに好ましくは10〜25vol%メタノール水溶液である。
【0026】
この時滴下する塩基としては、特に限定されないが、水酸化ナトリウム、水酸化カリウム、炭酸水素ナトリウムなど、または、これらの水溶液が挙げられるが、好ましくは水酸化ナトリウム水溶液である。滴下する塩基の濃度は、10〜20%であればよく、好ましくは15〜20%の間である。滴下する塩基の使用量は、N、N’−ジアルキルウレアに対して、1.0倍モルから3.0倍モルであり、好ましくは1.5倍モルから2.0培モルの間である。塩基を滴下する温度は、−10℃から20℃まで可能であるが、好ましくは0℃から10℃の間である。
【0027】
さらに晶析して析出する4−アミノ−1、3−ジアルキルウラシルは、ろ過することにより容易に単離できる。
【0028】
以上、本発明により、4−アミノ−1、3−ジアルキルウラシルをより効率的に製造できるようになった。
【0029】
【実施例】
以下において、実施例と比較例をあげて本発明を説明するが、本発明はこれらにより限定されるものではない。
【0030】
実施例1
4−アミノ−1、3−ジ−n−プロピルウラシル一般式〔4〕[化8]の製造
【0031】
【化8】
【0032】
反応器に、市販(Aldrich社製など)のN、N’−ジ −n−プロピルウレア7.4g(49.7ミリモル、純度97.0%)、シアノ酢酸4.4g(49.7ミリモル、純度96.0%)、無水酢酸7.6g(75.0ミリモル、純度100%)を装入し、60℃にて1.5時間攪拌した。減圧濃縮後、水10gを加え、さらに、60℃で減圧濃縮し、13vol%メタノール水溶液56.9gを添加した。17%水酸化ナトリウム水溶液3.9gを用いて、氷冷下でpH8.5に調整し、60℃にて1.5時間攪拌した。その後、反応器を5℃まで冷却した。生じた析出物を濾取し、少量の冷10%メタノール水溶液を用いて洗浄した。洗浄結晶を室温で30分風乾した後、40℃で2時間減圧乾燥して、4−アミノ−1、3−ジ−n−プロピルウラシル9.6gを得た。N、N’−ジ−n−プロピルウレアを基準とする収率は97.0%(純度99.5%)であった。
【0033】
実施例2
4−アミノ−1、3−ジ−n−プロピルウラシルの製造
減圧濃縮後、水酸化ナトリウム水溶液を加えて反応する際、反応溶媒の濃度を10vol%メタノール水溶液にした以外、実施例1と同様の操作を行った。結果を表1に示す。
【0034】
実施例3
4−アミノ−1、3−ジ−n−プロピルウラシルの製造
減圧濃縮後、水酸化ナトリウム水溶液を加えて反応する際、反応溶媒の濃度を25vol%メタノール水溶液にした以外、実施例1と同様の操作を行った。結果を表1に示す。
【0035】
比較例1
4−アミノ−1、3−ジ−n−プロピルウラシルの製造
減圧濃縮後、水酸化ナトリウム水溶液を加えて反応する際、反応溶媒としてメタノール水溶液を用いず、水のみを使用した以外、実施例1と同様の操作を行った。結果を表1に示す。
【0036】
【表1】
【0037】
尚、実施例中の反応器内は、比較例に比べ粘性が低く、攪拌性が低下しないため、溶解性が良好であった。
【0038】
【発明の効果】
このように、本発明の方法では反応器内の攪拌性を低下させず、溶解性が良好であるため、4−アミノ−1、3−ジアルキルウラシルの収率が向上し、効率的な工業的製造法を提供できる。
【発明の属する技術分野】
本発明は一般式〔3〕[化4]
【0002】
【化4】
(式中、R1 、R2は同一、もしくは異なっていてもよく、炭素数1〜5の直鎖あるいは分岐鎖のアルキル基を示す。)で表わされる4−アミノ−1、3−ジアルキルウラシルの製造方法に関する。近年、4−アミノ−1、3−ジアルキルウラシルは、抗パーキンソン治療薬、うっ血性心不全治療薬、喘息治療薬などとして開発されているアデノシンアンタゴニスト、アデノシンアゴニスト向け医薬品の製造中間体として、有用な化合物として注目されている。
【0004】
【従来の技術】
従来、4−アミノ−1、3−ジアルキルウラシルの製造法は、例えば、N、N’−ジアルキルウレアにシアノ酢酸と無水酢酸を加え、水溶媒中で反応させて縮合した後、これに水酸化ナトリウムを加えてアルカリ性として環化反応を行い、その溶液を冷却、晶析、濾過して得る方法(非特許文献1参照。)が知られている。
【0005】
【非特許文献1】
J.Am.Chem.Soc.,76,2798(1954)
【0006】
【発明が解決しようとする課題】
しかしながら、上記の製造法では工業的に製造する場合、次のような問題点があった。
【0007】
すなわち、溶媒として水を用いているため、出発原料化合物の1つであるN、N’−ジアルキルウレアのアルキル基の炭素数増加に伴い、脂溶性が増し、水溶性が低下してしまうため、反応系の粘性が増大し、反応収率が低下していた。
【0008】
このため、4−アミノ−1、3−ジアルキルウラシルの工業的製造法の開発が望まれていた。
【0009】
よって、本発明の課題は、より攪拌性が良好で、反応収率の高い4−アミノ−1、3−ジアルキルウラシルの工業的製造法を提供することにある。
【0010】
【課題を解決するための手段】
そこで本発明者等は、上記の課題を解決するために鋭意検討した結果、縮合後の反応液を濃縮し、反応溶媒としてアルコール水溶液を添加することにより、効率的に4−アミノ−1、3−ジアルキルウラシルを製造することを見出し、本発明を完成するに至った。
【0011】
すなわち、本発明は以下のとおりである。
[1] 一般式〔1〕[化5]
【0012】
【化5】
(式中、R1 、R2は同一、もしくは異なっていてもよく、炭素数1〜5の直鎖あるいは分岐鎖のアルキル基を示す。)で表わされるN、N’−ジアルキルウレアとシアノ酢酸とを、無水酢酸を用いて縮合し、塩基を加えて環化するに際して、縮合後の反応液を濃縮し、反応溶媒として一般式〔2〕[化6]
【0014】
【化6】
(式中、R3は炭素数1〜5の直鎖あるいは分岐鎖のアルキル基を示す。)で表されるアルコール水溶液を添加して環化することを特徴とする一般式〔3〕[化7]
【0016】
【化7】
(式中、R1 、R2は前記と同様。)で表される4−アミノ−1、3−ジアルキルウラシルの製造方法、
[2] 一般式〔2〕で表されるアルコール水溶液が、メタノール水溶液である[1]記載の4−アミノ−1、3−ジアルキルウラシルの製造方法、
[3] 一般式〔2〕で表されるアルコール水溶液が、1〜50vol%である[1]又は[2]記載の4−アミノ−1、3−ジアルキルウラシルの製造方法、
[4] 一般式〔2〕で表されるアルコール水溶液が、1〜30vol%である[1]〜[3]記載のいずれか一項に4−アミノ−1、3−ジアルキルウラシルの製造方法、
[5] 一般式〔2〕で表されるアルコール水溶液が、10〜25vol%である[1]〜[4]のいずれか一項に記載の4−アミノ−1、3−ジアルキルウラシルの製造方法。
【0018】
【発明の実施の形態】
以下、本発明を詳細に説明する。
本発明において、炭素数1〜5の直鎖あるいは分岐鎖のアルキル基としては、例えば、メチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、i−ブチル基、t−ブチル基、n−ペンチル基、i−ペンチル基、ネオペンチル基を挙げることができるが、これらに限定されるものではない。
【0019】
一般式〔1〕で表されるN、N’−ジアルキルウレアとしては、例えば、N、N’−ジメチルウレア、N、N’−ジエチルウレア、N、N’−ジ−n−プロピルウレア、N、N’−ジ−i−プロピルウレア、N、N’−ジ−n− ブチルウレア、N、N’−ジ−i−ブチルウレア、N、N’−ジ−t−ブチルウレア、N、N’−ジ−n−ペンチルウレア、N、N’−ジ−i− ペンチウレア、N、N’−ジ−ネオ−ペンチウレア、N−メチル−N’−エチルウレア、N−メチル−N’− n−プロピルウレア、N−エチル−N’− n−プロピルウレアなどを挙げることができるが、これらに限定されるものではない。
【0020】
また、一般式〔2〕で表されるアルコール水溶液としては、水溶媒にアルコールを添加したメタノール水溶液、エタノール水溶液、n−プロピルアルコール水溶液、i−プロピルアルコール水溶液、n−ブチルアルコール水溶液、i−ブチルアルコール水溶液、t−ブチルアルコール水溶液、n−ペンチルアルコール水溶液、i−ペンチルアルコール水溶液、ネオペンチルアルコール水溶液が挙げられるが、好ましくはメタノール水溶液である。
【0021】
本発明の一般式〔1〕で表されるN、N’−ジアルキルウレアの縮合および環化反応は、例えば、次に述べる条件において行うことができる。
【0022】
N、N’−ジアルキルウレアに対するシアノ酢酸および無水酢酸の使用量は特に限定されないが、それぞれ等モル、等モルから2倍モルが好ましい。反応温度は、室温から80℃まで可能であるが、好ましくは50℃から70℃の間である。反応時間は、1時間から5時間の間であれば良く、好ましくは2時間から3時間である。
【0023】
上記操作で得られた反応液を、4−アミノ−1、3−ジアルキルウラシルの理論収量の1倍重量から10倍重量、好ましくは1.5倍重量から5倍重量の間になるよう濃縮する。濃縮は、反応で生じた揮発成分、酸性成分を除去する目的で行い、必要に応じて水を添加して何回か濃縮することもできる。
【0024】
濃縮した反応液に、反応溶媒としてアルコール水溶液を添加し、塩基を滴下することにより環化反応を行い、4−アミノ−1、3−ジアルキルウラシルを合成する。
【0025】
反応溶媒の濃度は、1〜50vol%アルコール水溶液であればよく、好ましくは1〜30vol%アルコール水溶液であり、さらに好ましくは10〜25vol%メタノール水溶液である。
【0026】
この時滴下する塩基としては、特に限定されないが、水酸化ナトリウム、水酸化カリウム、炭酸水素ナトリウムなど、または、これらの水溶液が挙げられるが、好ましくは水酸化ナトリウム水溶液である。滴下する塩基の濃度は、10〜20%であればよく、好ましくは15〜20%の間である。滴下する塩基の使用量は、N、N’−ジアルキルウレアに対して、1.0倍モルから3.0倍モルであり、好ましくは1.5倍モルから2.0培モルの間である。塩基を滴下する温度は、−10℃から20℃まで可能であるが、好ましくは0℃から10℃の間である。
【0027】
さらに晶析して析出する4−アミノ−1、3−ジアルキルウラシルは、ろ過することにより容易に単離できる。
【0028】
以上、本発明により、4−アミノ−1、3−ジアルキルウラシルをより効率的に製造できるようになった。
【0029】
【実施例】
以下において、実施例と比較例をあげて本発明を説明するが、本発明はこれらにより限定されるものではない。
【0030】
実施例1
4−アミノ−1、3−ジ−n−プロピルウラシル一般式〔4〕[化8]の製造
【0031】
【化8】
反応器に、市販(Aldrich社製など)のN、N’−ジ −n−プロピルウレア7.4g(49.7ミリモル、純度97.0%)、シアノ酢酸4.4g(49.7ミリモル、純度96.0%)、無水酢酸7.6g(75.0ミリモル、純度100%)を装入し、60℃にて1.5時間攪拌した。減圧濃縮後、水10gを加え、さらに、60℃で減圧濃縮し、13vol%メタノール水溶液56.9gを添加した。17%水酸化ナトリウム水溶液3.9gを用いて、氷冷下でpH8.5に調整し、60℃にて1.5時間攪拌した。その後、反応器を5℃まで冷却した。生じた析出物を濾取し、少量の冷10%メタノール水溶液を用いて洗浄した。洗浄結晶を室温で30分風乾した後、40℃で2時間減圧乾燥して、4−アミノ−1、3−ジ−n−プロピルウラシル9.6gを得た。N、N’−ジ−n−プロピルウレアを基準とする収率は97.0%(純度99.5%)であった。
【0033】
実施例2
4−アミノ−1、3−ジ−n−プロピルウラシルの製造
減圧濃縮後、水酸化ナトリウム水溶液を加えて反応する際、反応溶媒の濃度を10vol%メタノール水溶液にした以外、実施例1と同様の操作を行った。結果を表1に示す。
【0034】
実施例3
4−アミノ−1、3−ジ−n−プロピルウラシルの製造
減圧濃縮後、水酸化ナトリウム水溶液を加えて反応する際、反応溶媒の濃度を25vol%メタノール水溶液にした以外、実施例1と同様の操作を行った。結果を表1に示す。
【0035】
比較例1
4−アミノ−1、3−ジ−n−プロピルウラシルの製造
減圧濃縮後、水酸化ナトリウム水溶液を加えて反応する際、反応溶媒としてメタノール水溶液を用いず、水のみを使用した以外、実施例1と同様の操作を行った。結果を表1に示す。
【0036】
【表1】
尚、実施例中の反応器内は、比較例に比べ粘性が低く、攪拌性が低下しないため、溶解性が良好であった。
【0038】
【発明の効果】
このように、本発明の方法では反応器内の攪拌性を低下させず、溶解性が良好であるため、4−アミノ−1、3−ジアルキルウラシルの収率が向上し、効率的な工業的製造法を提供できる。
Claims (5)
- 一般式〔1〕[化1]
- 一般式〔2〕で表されるアルコール水溶液が、メタノール水溶液である請求項1記載の4−アミノ−1、3−ジアルキルウラシルの製造方法。
- 一般式〔2〕で表されるアルコール水溶液が、1〜50vol%である請求項1又は2記載の4−アミノ−1、3−ジアルキルウラシルの製造方法。
- 一般式〔2〕で表されるアルコール水溶液が、1〜30vol%である請求項1〜3のいずれか一項に記載の4−アミノ−1、3−ジアルキルウラシルの製造方法。
- 一般式〔2〕で表されるアルコール水溶液が、10〜25vol%である請求項1〜4のいずれか一項に記載の4−アミノ−1、3−ジアルキルウラシルの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002294767A JP2004131395A (ja) | 2002-10-08 | 2002-10-08 | アミノウラシルの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002294767A JP2004131395A (ja) | 2002-10-08 | 2002-10-08 | アミノウラシルの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004131395A true JP2004131395A (ja) | 2004-04-30 |
Family
ID=32285210
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002294767A Pending JP2004131395A (ja) | 2002-10-08 | 2002-10-08 | アミノウラシルの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004131395A (ja) |
-
2002
- 2002-10-08 JP JP2002294767A patent/JP2004131395A/ja active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20060052532A (ko) | 히드록시나프토산 히드라지드 화합물 및 그의 제조 방법 | |
| JP2009503034A (ja) | 7H−ピロロ[2,3−d]ピリミジン誘導体の製造方法 | |
| RU2272036C2 (ru) | Способ получения мезилатов производных пиперазина | |
| EP1873151B1 (en) | Improved process for producing moxonidine | |
| WO2003076374A1 (fr) | Procede de production de derive d'acide trans-4-amino-1-cyclohexanecarboxylique | |
| JP4770826B2 (ja) | 2−オキシインドール誘導体の製造法 | |
| JP2000026378A (ja) | 塩酸セルトラリンの製法 | |
| KR20080031910A (ko) | 1-[시아노(4-하이드록시페닐)메틸]사이클로헥사놀화합물의 제조 방법 | |
| JP2004131395A (ja) | アミノウラシルの製造方法 | |
| JP4406482B2 (ja) | 光学活性2−アミノシクロヘキサノール誘導体の製造法 | |
| WO2014103812A1 (ja) | ピラゾール化合物の結晶の製造方法 | |
| JP4032861B2 (ja) | β−オキソニトリル誘導体又はそのアルカリ金属塩の製法 | |
| JP2005097149A (ja) | [3−[(2r)−[[(2r)−(3−クロロフェニル)−2−ヒドロキシエチル]アミノ]プロピル]−1h−インドール−7−イルオキシ]酢酸の溶媒和物 | |
| EA011763B1 (ru) | Способы получения венлафаксина и формы i венлафаксина гидрохлорида | |
| JP5234856B2 (ja) | Npyy5受容体拮抗作用を有する化合物の結晶 | |
| JP4078328B2 (ja) | 2−オキシインドール誘導体の製造法 | |
| JP2004131414A (ja) | ニトロソウラシルの製造方法 | |
| JP6336166B2 (ja) | イミダフェナシンの新規中間体、その製造方法及びそれを用いたイミダフェナシンの製造方法 | |
| US6008413A (en) | Process for recrystallizing 1,3-bis(aminophenoxy benzene) | |
| KR20220039116A (ko) | 고순도 펠루비프로펜의 제조방법 | |
| JP2006124325A (ja) | dl−1,2−ジフェニルエチレンジアミンの製造方法 | |
| JP4329325B2 (ja) | ジアリルシアヌレートのモノアルカリ金属塩の製造方法 | |
| JP2010018595A (ja) | N,n’−ジアルキルヒドラジン化合物の製造方法およびピラゾリジンジオン化合物 | |
| KR101152607B1 (ko) | 6-[4-(2-피페리딘-1-일-에톡시)-페닐]-3-피리딘-4-일-피라졸로[1,5-a]피리미딘의 제조방법 | |
| WO2002085880A1 (fr) | Procede de fabrication d'un compose de nitrile |