ES2951212T3 - Fenilacetamidas como inhibidores de ROCK - Google Patents
Fenilacetamidas como inhibidores de ROCK Download PDFInfo
- Publication number
- ES2951212T3 ES2951212T3 ES18746509T ES18746509T ES2951212T3 ES 2951212 T3 ES2951212 T3 ES 2951212T3 ES 18746509 T ES18746509 T ES 18746509T ES 18746509 T ES18746509 T ES 18746509T ES 2951212 T3 ES2951212 T3 ES 2951212T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- nicotinamide
- amino
- oxoethyl
- phenoxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000011435 rock Substances 0.000 title abstract description 44
- 239000003112 inhibitor Substances 0.000 title abstract description 14
- LSBDFXRDZJMBSC-UHFFFAOYSA-N 2-phenylacetamide Chemical class NC(=O)CC1=CC=CC=C1 LSBDFXRDZJMBSC-UHFFFAOYSA-N 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 128
- 150000003839 salts Chemical class 0.000 claims abstract description 48
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 18
- 230000002526 effect on cardiovascular system Effects 0.000 claims abstract description 7
- 210000002460 smooth muscle Anatomy 0.000 claims abstract description 6
- 230000003176 fibrotic effect Effects 0.000 claims abstract description 5
- 208000027866 inflammatory disease Diseases 0.000 claims abstract description 3
- 230000000771 oncological effect Effects 0.000 claims abstract description 3
- 230000002981 neuropathic effect Effects 0.000 claims abstract 2
- 125000000217 alkyl group Chemical group 0.000 claims description 255
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 110
- 125000000623 heterocyclic group Chemical group 0.000 claims description 83
- 229960003966 nicotinamide Drugs 0.000 claims description 55
- 235000005152 nicotinamide Nutrition 0.000 claims description 55
- 239000011570 nicotinamide Substances 0.000 claims description 55
- 229910052736 halogen Inorganic materials 0.000 claims description 52
- 150000002367 halogens Chemical class 0.000 claims description 50
- 125000003545 alkoxy group Chemical group 0.000 claims description 44
- 229910052757 nitrogen Inorganic materials 0.000 claims description 44
- 229910052799 carbon Inorganic materials 0.000 claims description 42
- 229910052739 hydrogen Inorganic materials 0.000 claims description 41
- 125000004432 carbon atom Chemical group C* 0.000 claims description 39
- 229910052801 chlorine Inorganic materials 0.000 claims description 30
- 229910052731 fluorine Inorganic materials 0.000 claims description 30
- 229910052760 oxygen Inorganic materials 0.000 claims description 29
- 125000005842 heteroatom Chemical group 0.000 claims description 28
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 27
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 claims description 27
- 125000002947 alkylene group Chemical group 0.000 claims description 24
- 238000011282 treatment Methods 0.000 claims description 24
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 22
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 21
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 20
- 229910052794 bromium Inorganic materials 0.000 claims description 18
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 claims description 18
- 229910052701 rubidium Inorganic materials 0.000 claims description 18
- 230000000694 effects Effects 0.000 claims description 17
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 17
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 16
- 125000003342 alkenyl group Chemical group 0.000 claims description 14
- 125000000304 alkynyl group Chemical group 0.000 claims description 14
- 201000010099 disease Diseases 0.000 claims description 13
- 239000003937 drug carrier Substances 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 11
- 206010020772 Hypertension Diseases 0.000 claims description 10
- 108010041788 rho-Associated Kinases Proteins 0.000 claims description 10
- 102000000568 rho-Associated Kinases Human genes 0.000 claims description 10
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 9
- 208000035475 disorder Diseases 0.000 claims description 9
- 208000006011 Stroke Diseases 0.000 claims description 8
- 238000011321 prophylaxis Methods 0.000 claims description 7
- 206010019280 Heart failures Diseases 0.000 claims description 5
- 230000001594 aberrant effect Effects 0.000 claims description 5
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 4
- 206010002383 Angina Pectoris Diseases 0.000 claims description 4
- 201000001320 Atherosclerosis Diseases 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 4
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 4
- 238000002560 therapeutic procedure Methods 0.000 claims description 4
- WJMACOOANXCIEN-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1C(C)C)C=CC=C2 Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1C(C)C)C=CC=C2 WJMACOOANXCIEN-UHFFFAOYSA-N 0.000 claims description 3
- 208000029078 coronary artery disease Diseases 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- FGRAKCPDDQNYMR-UHFFFAOYSA-N CC(C)(O)COC1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 Chemical compound CC(C)(O)COC1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 FGRAKCPDDQNYMR-UHFFFAOYSA-N 0.000 claims description 2
- OLWOEZWROPDSMA-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN(C(F)F)C(=O)C=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN(C(F)F)C(=O)C=C1 OLWOEZWROPDSMA-UHFFFAOYSA-N 0.000 claims description 2
- OEHXXFUOLJBQMM-UHFFFAOYSA-N COC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OC Chemical compound COC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OC OEHXXFUOLJBQMM-UHFFFAOYSA-N 0.000 claims description 2
- FLCGNSSUFYOAAW-UHFFFAOYSA-N COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)C(C)C1=CC=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 Chemical compound COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)C(C)C1=CC=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 FLCGNSSUFYOAAW-UHFFFAOYSA-N 0.000 claims description 2
- UMYRLNVFCTYLFL-UHFFFAOYSA-N COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)CC1=CC(F)=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 Chemical compound COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)CC1=CC(F)=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 UMYRLNVFCTYLFL-UHFFFAOYSA-N 0.000 claims description 2
- WDRJMNLXRNPDPR-UHFFFAOYSA-N COC1=CC=C2SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=NC2=C1 Chemical compound COC1=CC=C2SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=NC2=C1 WDRJMNLXRNPDPR-UHFFFAOYSA-N 0.000 claims description 2
- BETLIUXUURPAIC-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1=NC(=CC=C1)C)C Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1=NC(=CC=C1)C)C BETLIUXUURPAIC-UHFFFAOYSA-N 0.000 claims description 2
- AGVPQFQVDMZWBJ-UHFFFAOYSA-N FC(CN1C(=NC2=C1C=CC=C2)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F Chemical compound FC(CN1C(=NC2=C1C=CC=C2)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F AGVPQFQVDMZWBJ-UHFFFAOYSA-N 0.000 claims description 2
- UOYGVFICGHVSJN-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1=NC(=CC=C1)C)C Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1=NC(=CC=C1)C)C UOYGVFICGHVSJN-UHFFFAOYSA-N 0.000 claims description 2
- NPMAZYOZUNOVKR-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1=NC(=CC=C1)F)C Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1=NC(=CC=C1)F)C NPMAZYOZUNOVKR-UHFFFAOYSA-N 0.000 claims description 2
- XQTRUWLRTRKZIF-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OCC(C)(C)O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OCC(C)(C)O XQTRUWLRTRKZIF-UHFFFAOYSA-N 0.000 claims description 2
- BENHAPZDHMSBKV-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=C(OC(F)(F)F)C=CC3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=C(OC(F)(F)F)C=CC3=N2)C=C1 BENHAPZDHMSBKV-UHFFFAOYSA-N 0.000 claims description 2
- NUJYRLMIGKQKJL-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC(Br)=CC3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC(Br)=CC3=N2)C=C1 NUJYRLMIGKQKJL-UHFFFAOYSA-N 0.000 claims description 2
- WUJQSIODAAFXTL-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC(OCC4=CC=CC=C4)=CC3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC(OCC4=CC=CC=C4)=CC3=N2)C=C1 WUJQSIODAAFXTL-UHFFFAOYSA-N 0.000 claims description 2
- CIAXFFNVSXFZRA-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC=C(OC(F)F)C3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC=C(OC(F)F)C3=N2)C=C1 CIAXFFNVSXFZRA-UHFFFAOYSA-N 0.000 claims description 2
- NMDVLMINCZSFTA-UHFFFAOYSA-N NC(=O)c1cccnc1Oc1ccc(CC(=O)Nc2nc3ccccc3s2)cc1F Chemical compound NC(=O)c1cccnc1Oc1ccc(CC(=O)Nc2nc3ccccc3s2)cc1F NMDVLMINCZSFTA-UHFFFAOYSA-N 0.000 claims description 2
- GIQASVOCJIECSZ-UHFFFAOYSA-N OC(COC1=CC2=C(N=C(S2)NC(CC2=CC=C(OC3=C(C(=O)N)C=CC=N3)C=C2)=O)C=C1)(C)C Chemical compound OC(COC1=CC2=C(N=C(S2)NC(CC2=CC=C(OC3=C(C(=O)N)C=CC=N3)C=C2)=O)C=C1)(C)C GIQASVOCJIECSZ-UHFFFAOYSA-N 0.000 claims description 2
- KDEUCYKTPGYEBQ-UHFFFAOYSA-N OC(COC=1C=CC2=C(N=C(S2)NC(CC2=CC=C(OC3=C(C(=O)N)C=CC=N3)C=C2)=O)C=1)(C)C Chemical compound OC(COC=1C=CC2=C(N=C(S2)NC(CC2=CC=C(OC3=C(C(=O)N)C=CC=N3)C=C2)=O)C=1)(C)C KDEUCYKTPGYEBQ-UHFFFAOYSA-N 0.000 claims description 2
- 229910004013 NO 2 Inorganic materials 0.000 claims 10
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 claims 7
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims 4
- DCUUMKYVBWZTOB-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=CC=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=CC=C1 DCUUMKYVBWZTOB-UHFFFAOYSA-N 0.000 claims 2
- NLKDACZSQAKNSW-UHFFFAOYSA-N 2-[2-chloro-4-[2-[[1-(cyclopropylmethyl)-6-(trifluoromethyl)benzimidazol-2-yl]amino]-2-oxoethyl]phenoxy]pyridine-3-carboxamide Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CC1CC1)C=C(C=C2)C(F)(F)F NLKDACZSQAKNSW-UHFFFAOYSA-N 0.000 claims 1
- ILQOVKOFRQPVAS-UHFFFAOYSA-N 2-[2-chloro-4-[2-[[4-(2-fluorophenyl)-5-methyl-1,3-thiazol-2-yl]amino]-2-oxoethyl]phenoxy]pyridine-3-carboxamide Chemical compound CC1=C(N=C(NC(=O)CC2=CC(Cl)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=CC=C1F ILQOVKOFRQPVAS-UHFFFAOYSA-N 0.000 claims 1
- HTRHXYBUBWWNME-UHFFFAOYSA-N 2-[2-fluoro-4-[2-oxo-2-[(5-pyrazin-2-yl-1,3,4-thiadiazol-2-yl)amino]ethyl]phenoxy]pyridine-3-carboxamide Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C2=CN=CC=N2)C=C1 HTRHXYBUBWWNME-UHFFFAOYSA-N 0.000 claims 1
- FJANTVRQQYECNB-UHFFFAOYSA-N 2-[2-fluoro-4-[2-oxo-2-[[6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl]amino]ethyl]phenoxy]pyridine-3-carboxamide Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC=1N=C2N(C=C(C=C2)C(F)(F)F)C=1)=O FJANTVRQQYECNB-UHFFFAOYSA-N 0.000 claims 1
- JFRARMADNIYDLR-UHFFFAOYSA-N 2-[4-[2-[(4-cyclopropyl-5-methyl-1,3-thiazol-2-yl)amino]-2-oxoethyl]-2-fluorophenoxy]pyridine-3-carboxamide Chemical compound C1(CC1)C=1N=C(SC=1C)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O JFRARMADNIYDLR-UHFFFAOYSA-N 0.000 claims 1
- NMZNIDKIUYCRPN-UHFFFAOYSA-N 2-[4-[2-[[1-(2,2-difluoroethyl)-6-fluorobenzimidazol-2-yl]amino]-2-oxoethyl]-2-fluorophenoxy]pyridine-3-carboxamide Chemical compound FC(CN1C(=NC2=C1C=C(C=C2)F)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F NMZNIDKIUYCRPN-UHFFFAOYSA-N 0.000 claims 1
- ZUDVUEMPJYSALR-UHFFFAOYSA-N 2-[4-[2-[[4-(4-fluorophenyl)-5-methyl-1,3-thiazol-2-yl]amino]-2-oxoethyl]phenoxy]pyridine-3-carboxamide Chemical compound CC1=C(N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=C(F)C=C1 ZUDVUEMPJYSALR-UHFFFAOYSA-N 0.000 claims 1
- GKUGBWPBELMECY-UHFFFAOYSA-N 2-[4-[2-[[5-(2-hydroxy-2-methylpropoxy)-1,3-benzothiazol-2-yl]amino]-2-oxoethyl]-2-methoxyphenoxy]pyridine-3-carboxamide Chemical compound OC(COC=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)OC)=O)C=1)(C)C GKUGBWPBELMECY-UHFFFAOYSA-N 0.000 claims 1
- 208000023275 Autoimmune disease Diseases 0.000 claims 1
- IOEJLPFEYZVPFK-UHFFFAOYSA-N C(#N)C1=CC=CC(=N1)C=1N=C(SC=1C)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O Chemical compound C(#N)C1=CC=CC(=N1)C=1N=C(SC=1C)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O IOEJLPFEYZVPFK-UHFFFAOYSA-N 0.000 claims 1
- LKICIJOUXZQPCX-UHFFFAOYSA-N C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC1=NC2=C(N1CCOC)C=C(C=C2)C(=O)N)F Chemical compound C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC1=NC2=C(N1CCOC)C=C(C=C2)C(=O)N)F LKICIJOUXZQPCX-UHFFFAOYSA-N 0.000 claims 1
- VZMWCVNZSDORBK-UHFFFAOYSA-N C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC1=NC2=C(N1CCOC)C=CC(=C2)C(=O)N)F Chemical compound C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC1=NC2=C(N1CCOC)C=CC(=C2)C(=O)N)F VZMWCVNZSDORBK-UHFFFAOYSA-N 0.000 claims 1
- CSUYFZHDYQLQKV-UHFFFAOYSA-N C1(CC1)C1=NN(C=C1C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)C Chemical compound C1(CC1)C1=NN(C=C1C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)C CSUYFZHDYQLQKV-UHFFFAOYSA-N 0.000 claims 1
- ALAJXJUZIIHZFR-UHFFFAOYSA-N C1=CC(=C(N=C1)OC2=C(C=C(C=C2)CC(=O)NC3=NC4=C(S3)C=CC(=C4)C5=C(NN=C5)C(F)(F)F)F)C(=O)N Chemical compound C1=CC(=C(N=C1)OC2=C(C=C(C=C2)CC(=O)NC3=NC4=C(S3)C=CC(=C4)C5=C(NN=C5)C(F)(F)F)F)C(=O)N ALAJXJUZIIHZFR-UHFFFAOYSA-N 0.000 claims 1
- ITYQIZLXHYRCPU-UHFFFAOYSA-N CC(C)(O)COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)CC1=CC=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 Chemical compound CC(C)(O)COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)CC1=CC=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 ITYQIZLXHYRCPU-UHFFFAOYSA-N 0.000 claims 1
- CMALLRUMKVEVTR-UHFFFAOYSA-N CC(C)S(=O)(=O)C1=CC=C(C=C1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound CC(C)S(=O)(=O)C1=CC=C(C=C1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 CMALLRUMKVEVTR-UHFFFAOYSA-N 0.000 claims 1
- YMPVCSIHWDNVMG-UHFFFAOYSA-N CC1=C(C(C)=NN1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound CC1=C(C(C)=NN1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 YMPVCSIHWDNVMG-UHFFFAOYSA-N 0.000 claims 1
- JSRIJVNIHCYCEV-UHFFFAOYSA-N CC1=C(C)N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1 Chemical compound CC1=C(C)N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1 JSRIJVNIHCYCEV-UHFFFAOYSA-N 0.000 claims 1
- VNWRNJMBGSEWNR-UHFFFAOYSA-N CC1=C(N2C=CSC2=N1)C1=CSC(NC(=O)CC2=CC(Cl)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound CC1=C(N2C=CSC2=N1)C1=CSC(NC(=O)CC2=CC(Cl)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 VNWRNJMBGSEWNR-UHFFFAOYSA-N 0.000 claims 1
- ISHALBHSXIFKBM-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=C(C=C1)S(C)(=O)=O Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=C(C=C1)S(C)(=O)=O ISHALBHSXIFKBM-UHFFFAOYSA-N 0.000 claims 1
- JJWZSQHXQMAKHO-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=NC=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC=NC=C1 JJWZSQHXQMAKHO-UHFFFAOYSA-N 0.000 claims 1
- KXDLZZWKXNLQPO-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN=C(F)C=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN=C(F)C=C1 KXDLZZWKXNLQPO-UHFFFAOYSA-N 0.000 claims 1
- QHTULYOLPNTOEI-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN=C(N=C1)C(C)(C)O Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN=C(N=C1)C(C)(C)O QHTULYOLPNTOEI-UHFFFAOYSA-N 0.000 claims 1
- BDHZJIHRUVMUEL-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN=CN=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CN=CN=C1 BDHZJIHRUVMUEL-UHFFFAOYSA-N 0.000 claims 1
- VIBXZYWLNXEONR-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=NC(C)=NC=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=NC(C)=NC=C1 VIBXZYWLNXEONR-UHFFFAOYSA-N 0.000 claims 1
- OZJBEHWWTXTDJE-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=NC=C(F)C=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=NC=C(F)C=C1 OZJBEHWWTXTDJE-UHFFFAOYSA-N 0.000 claims 1
- WALWBIMHLIPEED-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=NC=CC=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=NC=CC=C1 WALWBIMHLIPEED-UHFFFAOYSA-N 0.000 claims 1
- QWYDREQCEWLFRJ-UHFFFAOYSA-N CC1=C(N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC(Cl)=C(C)C=C1 Chemical compound CC1=C(N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1)C1=CC(Cl)=C(C)C=C1 QWYDREQCEWLFRJ-UHFFFAOYSA-N 0.000 claims 1
- VSAPNUAXEQEEMW-UHFFFAOYSA-N CC1=C(SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1)C1=NC=CC=C1 Chemical compound CC1=C(SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1)C1=NC=CC=C1 VSAPNUAXEQEEMW-UHFFFAOYSA-N 0.000 claims 1
- OEEJNHDLISBUNU-UHFFFAOYSA-N CC1=CC2=C(NC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 Chemical compound CC1=CC2=C(NC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 OEEJNHDLISBUNU-UHFFFAOYSA-N 0.000 claims 1
- PXJZABZUZNEQFX-UHFFFAOYSA-N CC1=CC=CC(NC2=NC=NC3=CC=C(NC(=O)CC4=CC=C(OC5=NC=CC=C5C(N)=O)C=C4)C=C23)=C1 Chemical compound CC1=CC=CC(NC2=NC=NC3=CC=C(NC(=O)CC4=CC=C(OC5=NC=CC=C5C(N)=O)C=C4)C=C23)=C1 PXJZABZUZNEQFX-UHFFFAOYSA-N 0.000 claims 1
- ASHMPYNUZOTQJH-UHFFFAOYSA-N CC1=CC=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN12 Chemical compound CC1=CC=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN12 ASHMPYNUZOTQJH-UHFFFAOYSA-N 0.000 claims 1
- TZLZBPJMYZNNBU-UHFFFAOYSA-N CC1=CN=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1 Chemical compound CC1=CN=C(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)S1 TZLZBPJMYZNNBU-UHFFFAOYSA-N 0.000 claims 1
- DDUQLKLNRHIKJX-UHFFFAOYSA-N CC1=NN=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1 Chemical compound CC1=NN=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)S1 DDUQLKLNRHIKJX-UHFFFAOYSA-N 0.000 claims 1
- CYECZCLVAZKDKL-UHFFFAOYSA-N CC=1C(=CC=2N(C=1)N=C(N=2)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)C Chemical compound CC=1C(=CC=2N(C=1)N=C(N=2)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)C CYECZCLVAZKDKL-UHFFFAOYSA-N 0.000 claims 1
- OWKXNXOOUZKTTH-UHFFFAOYSA-N CCCS(=O)(=O)C1=CC2=C(NC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 Chemical compound CCCS(=O)(=O)C1=CC2=C(NC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 OWKXNXOOUZKTTH-UHFFFAOYSA-N 0.000 claims 1
- WFBKCGWFRTZMDP-UHFFFAOYSA-N CN1C=C(C(C)=N1)C1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 Chemical compound CN1C=C(C(C)=N1)C1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 WFBKCGWFRTZMDP-UHFFFAOYSA-N 0.000 claims 1
- ARNLPRYEFTZDOQ-UHFFFAOYSA-N CN1C=C(C=CC1=O)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound CN1C=C(C=CC1=O)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 ARNLPRYEFTZDOQ-UHFFFAOYSA-N 0.000 claims 1
- UMBFOZNPCRXFSJ-UHFFFAOYSA-N CN1C=C(C=N1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound CN1C=C(C=N1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 UMBFOZNPCRXFSJ-UHFFFAOYSA-N 0.000 claims 1
- AMQZAYMOTMWMDO-UHFFFAOYSA-N CN1C=C(C=N1)C1=CC=C2N(CC(F)(F)F)C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NC2=C1 Chemical compound CN1C=C(C=N1)C1=CC=C2N(CC(F)(F)F)C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NC2=C1 AMQZAYMOTMWMDO-UHFFFAOYSA-N 0.000 claims 1
- PHMOOYMOIMBJMK-UHFFFAOYSA-N CN1C=C(C=N1)C1=CN2C=C(NC(=O)CC3=CC(Cl)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C2C=C1 Chemical compound CN1C=C(C=N1)C1=CN2C=C(NC(=O)CC3=CC(Cl)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C2C=C1 PHMOOYMOIMBJMK-UHFFFAOYSA-N 0.000 claims 1
- QLOPZDPGEUSQNB-UHFFFAOYSA-N CN1C=C(C=N1)C1=CN2C=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C2C=C1 Chemical compound CN1C=C(C=N1)C1=CN2C=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C2C=C1 QLOPZDPGEUSQNB-UHFFFAOYSA-N 0.000 claims 1
- XIIOVNWLZHJORE-UHFFFAOYSA-N CN1C=CC(=CC1=O)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound CN1C=CC(=CC1=O)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 XIIOVNWLZHJORE-UHFFFAOYSA-N 0.000 claims 1
- HVKZQJAWLUPKAA-UHFFFAOYSA-N CN1C=CC(=N1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound CN1C=CC(=N1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 HVKZQJAWLUPKAA-UHFFFAOYSA-N 0.000 claims 1
- QVUBXVQQEVSOAL-UHFFFAOYSA-N CN1CCC2=C(C1)N=C(NC(=O)CC1=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 Chemical compound CN1CCC2=C(C1)N=C(NC(=O)CC1=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 QVUBXVQQEVSOAL-UHFFFAOYSA-N 0.000 claims 1
- YEWRBZAPRVPUNA-UHFFFAOYSA-N CN1CCN(CC1)C1=NC=C(C=N1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound CN1CCN(CC1)C1=NC=C(C=N1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 YEWRBZAPRVPUNA-UHFFFAOYSA-N 0.000 claims 1
- MYKHUDZQUJTSEB-UHFFFAOYSA-N CN1N=C(C(=C1)C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)C Chemical compound CN1N=C(C(=C1)C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)C MYKHUDZQUJTSEB-UHFFFAOYSA-N 0.000 claims 1
- HPYFKAUMNHKUEN-UHFFFAOYSA-N CN1N=C(C)C(C)=C1C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound CN1N=C(C)C(C)=C1C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 HPYFKAUMNHKUEN-UHFFFAOYSA-N 0.000 claims 1
- ADKGNHCRDPHGLF-UHFFFAOYSA-N CN1N=C(C=C1C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1)C(F)(F)F Chemical compound CN1N=C(C=C1C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1)C(F)(F)F ADKGNHCRDPHGLF-UHFFFAOYSA-N 0.000 claims 1
- SRDDKWQYRCOIFA-UHFFFAOYSA-N CN1N=C(C=C1C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)C Chemical compound CN1N=C(C=C1C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)C SRDDKWQYRCOIFA-UHFFFAOYSA-N 0.000 claims 1
- MYSGGYWFSXTVGD-UHFFFAOYSA-N CN1N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)C2=C1C=CC=C2 Chemical compound CN1N=C(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)C2=C1C=CC=C2 MYSGGYWFSXTVGD-UHFFFAOYSA-N 0.000 claims 1
- VYXPVWKCCYNUPS-UHFFFAOYSA-N CN1N=CC=C1C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound CN1N=CC=C1C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 VYXPVWKCCYNUPS-UHFFFAOYSA-N 0.000 claims 1
- ZUUVNLGFYOOGOX-UHFFFAOYSA-N CN1N=CC=C1C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound CN1N=CC=C1C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 ZUUVNLGFYOOGOX-UHFFFAOYSA-N 0.000 claims 1
- WLYQIEVTXGXUDE-UHFFFAOYSA-N CN1N=CC=C1C1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 Chemical compound CN1N=CC=C1C1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 WLYQIEVTXGXUDE-UHFFFAOYSA-N 0.000 claims 1
- INBOAJMPEKOPFU-UHFFFAOYSA-N COC(=O)C1=CC2=C(C=C1)N(C)C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)=N2 Chemical compound COC(=O)C1=CC2=C(C=C1)N(C)C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)=N2 INBOAJMPEKOPFU-UHFFFAOYSA-N 0.000 claims 1
- ODCAJVLFSILQGK-UHFFFAOYSA-N COC(=O)C1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 Chemical compound COC(=O)C1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 ODCAJVLFSILQGK-UHFFFAOYSA-N 0.000 claims 1
- OAJBDNXKZPRSGU-UHFFFAOYSA-N COC(=O)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 Chemical compound COC(=O)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 OAJBDNXKZPRSGU-UHFFFAOYSA-N 0.000 claims 1
- RBFVZMBOOKHVHG-UHFFFAOYSA-N COC1=C(OC)C=C(C=C1)C1=CSC(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound COC1=C(OC)C=C(C=C1)C1=CSC(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 RBFVZMBOOKHVHG-UHFFFAOYSA-N 0.000 claims 1
- FDJRSCACUKTRBI-UHFFFAOYSA-N COC1=C(OC)C=C(CC2=NN=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)S2)C=C1 Chemical compound COC1=C(OC)C=C(CC2=NN=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)S2)C=C1 FDJRSCACUKTRBI-UHFFFAOYSA-N 0.000 claims 1
- UQKMLEXGSZTUHS-UHFFFAOYSA-N COC1=CC2=C(C=C1)C1=C(CC2)SC(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound COC1=CC2=C(C=C1)C1=C(CC2)SC(NC(=O)CC2=CC=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 UQKMLEXGSZTUHS-UHFFFAOYSA-N 0.000 claims 1
- LTLDHOVEMPXTNE-UHFFFAOYSA-N COC1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 Chemical compound COC1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 LTLDHOVEMPXTNE-UHFFFAOYSA-N 0.000 claims 1
- IQJIMKMYMFQZSA-UHFFFAOYSA-N COC1=CC2=NC(NC(=O)CC3=CC(Br)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 Chemical compound COC1=CC2=NC(NC(=O)CC3=CC(Br)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 IQJIMKMYMFQZSA-UHFFFAOYSA-N 0.000 claims 1
- KPJDONJTWCSXAX-UHFFFAOYSA-N COC1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 Chemical compound COC1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN2C=C1 KPJDONJTWCSXAX-UHFFFAOYSA-N 0.000 claims 1
- STUBEZYSLHJOQZ-UHFFFAOYSA-N COC1=CC=2N(C=C1)N=C(N=2)NC(CC1=CC=C(OC2=C(C(=O)N)C=CC=N2)C=C1)=O Chemical compound COC1=CC=2N(C=C1)N=C(N=2)NC(CC1=CC=C(OC2=C(C(=O)N)C=CC=N2)C=C1)=O STUBEZYSLHJOQZ-UHFFFAOYSA-N 0.000 claims 1
- LMYJRRTVCYXYMX-UHFFFAOYSA-N COC1=CC=CN2N=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C12 Chemical compound COC1=CC=CN2N=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C12 LMYJRRTVCYXYMX-UHFFFAOYSA-N 0.000 claims 1
- QSEWWUQBGGRGIH-UHFFFAOYSA-N COC1=NC(=CC=C1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound COC1=NC(=CC=C1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 QSEWWUQBGGRGIH-UHFFFAOYSA-N 0.000 claims 1
- CKMPSSWWOWGDDN-UHFFFAOYSA-N COC1=NC=C(C=N1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 Chemical compound COC1=NC=C(C=N1)C1=C(C)SC(NC(=O)CC2=CC(F)=C(OC3=NC=CC=C3C(N)=O)C=C2)=N1 CKMPSSWWOWGDDN-UHFFFAOYSA-N 0.000 claims 1
- DXEGJCVJSJSGAX-UHFFFAOYSA-N CP(=O)(C)C1=CC2=C(N(C(=N2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)CC(F)(F)F)C=C1 Chemical compound CP(=O)(C)C1=CC2=C(N(C(=N2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)CC(F)(F)F)C=C1 DXEGJCVJSJSGAX-UHFFFAOYSA-N 0.000 claims 1
- ZPHWWBHMXBITDJ-UHFFFAOYSA-N CP(=O)(C)C=1C=CC=2N(C=1)C=C(N=2)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O Chemical compound CP(=O)(C)C=1C=CC=2N(C=1)C=C(N=2)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O ZPHWWBHMXBITDJ-UHFFFAOYSA-N 0.000 claims 1
- QVURHIUULNOFTP-UHFFFAOYSA-N CP(C)(=O)C1=CC2=C(SC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 Chemical compound CP(C)(=O)C1=CC2=C(SC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=N2)C=C1 QVURHIUULNOFTP-UHFFFAOYSA-N 0.000 claims 1
- ROMUVCSCANQGIQ-UHFFFAOYSA-N CP(C)(=O)C1=CC=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN12 Chemical compound CP(C)(=O)C1=CC=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)=NN12 ROMUVCSCANQGIQ-UHFFFAOYSA-N 0.000 claims 1
- YXSZZMDREJYCAP-UHFFFAOYSA-N CS(=O)(=O)C1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 Chemical compound CS(=O)(=O)C1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 YXSZZMDREJYCAP-UHFFFAOYSA-N 0.000 claims 1
- HTWAHMGKLJCWJA-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CC1CC1)C=CC(=C2)C(F)(F)F Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CC1CC1)C=CC(=C2)C(F)(F)F HTWAHMGKLJCWJA-UHFFFAOYSA-N 0.000 claims 1
- NVHZCPYTLPVUIG-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CC1CC1)C=CC=C2 Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CC1CC1)C=CC=C2 NVHZCPYTLPVUIG-UHFFFAOYSA-N 0.000 claims 1
- CYYWNSAURHEPIY-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NN(C2=NC=CC=C21)C Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NN(C2=NC=CC=C21)C CYYWNSAURHEPIY-UHFFFAOYSA-N 0.000 claims 1
- MLPXZRRIWJDBMB-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1CCC1)C1=NC=CC=C1 Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC(=C(N=1)C1CCC1)C1=NC=CC=C1 MLPXZRRIWJDBMB-UHFFFAOYSA-N 0.000 claims 1
- DSAUQYLXJQKSOA-UHFFFAOYSA-N FC(CN1C(=NC2=C1C=C(C=C2)OC)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F Chemical compound FC(CN1C(=NC2=C1C=C(C=C2)OC)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F DSAUQYLXJQKSOA-UHFFFAOYSA-N 0.000 claims 1
- WOKWTRLJJGXTKX-UHFFFAOYSA-N FC(CN1C(=NC2=C1C=C(C=C2)P(=O)(C)C)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F Chemical compound FC(CN1C(=NC2=C1C=C(C=C2)P(=O)(C)C)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F WOKWTRLJJGXTKX-UHFFFAOYSA-N 0.000 claims 1
- HTOGGPPMVUMBCH-UHFFFAOYSA-N FC(CN1C(=NC2=C1C=CC=C2F)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F Chemical compound FC(CN1C(=NC2=C1C=CC=C2F)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O)F HTOGGPPMVUMBCH-UHFFFAOYSA-N 0.000 claims 1
- PLGADNZPBCGJPK-UHFFFAOYSA-N FC(N1N=CC(=C1)C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)F Chemical compound FC(N1N=CC(=C1)C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1)F PLGADNZPBCGJPK-UHFFFAOYSA-N 0.000 claims 1
- RTCILOYBMNRTPU-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CCOC)C=C(C=C2)OC Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CCOC)C=C(C=C2)OC RTCILOYBMNRTPU-UHFFFAOYSA-N 0.000 claims 1
- IFRMGYMBGOCZMW-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CCOC)C=CC=C2 Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NC2=C(N1CCOC)C=CC=C2 IFRMGYMBGOCZMW-UHFFFAOYSA-N 0.000 claims 1
- CRYOQJHRMIZCTK-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NN2C(C(=CC=C2)F)=N1 Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC1=NN2C(C(=CC=C2)F)=N1 CRYOQJHRMIZCTK-UHFFFAOYSA-N 0.000 claims 1
- HZNHOFOQHWJVLZ-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NC2=C(N1CC1CCOCC1)C=CC=C2)=O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NC2=C(N1CC1CCOCC1)C=CC=C2)=O HZNHOFOQHWJVLZ-UHFFFAOYSA-N 0.000 claims 1
- MRNCVIYXWOTILZ-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NC2=C(N1CCC(F)(F)F)C=CC=C2)=O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NC2=C(N1CCC(F)(F)F)C=CC=C2)=O MRNCVIYXWOTILZ-UHFFFAOYSA-N 0.000 claims 1
- IUKBLHZTGYRRFN-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NN2C(C=C(C=C2)C(F)(F)F)=N1)=O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NN2C(C=C(C=C2)C(F)(F)F)=N1)=O IUKBLHZTGYRRFN-UHFFFAOYSA-N 0.000 claims 1
- GTZZQIZTMGSLEA-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NN2C(C=CC(=C2)C(F)(F)F)=N1)=O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NN2C(C=CC(=C2)C(F)(F)F)=N1)=O GTZZQIZTMGSLEA-UHFFFAOYSA-N 0.000 claims 1
- WAYMOBQPDIYBJE-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NN2C(C=CC=C2C(F)(F)F)=N1)=O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(NC1=NN2C(C=CC=C2C(F)(F)F)=N1)=O WAYMOBQPDIYBJE-UHFFFAOYSA-N 0.000 claims 1
- GREYDNPXYVFIKF-UHFFFAOYSA-N N1N=CC(=C1)C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1 Chemical compound N1N=CC(=C1)C=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(OC3=C(C(=O)N)C=CC=N3)C=C2)F)=O)C=1 GREYDNPXYVFIKF-UHFFFAOYSA-N 0.000 claims 1
- RQUGKTILHPGERS-UHFFFAOYSA-N NC(=O)C1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 Chemical compound NC(=O)C1=CC2=C(C=C1)N=C(NC(=O)CC1=CC=C(OC3=NC=CC=C3C(N)=O)C=C1)S2 RQUGKTILHPGERS-UHFFFAOYSA-N 0.000 claims 1
- IQFQLZRYZJORCR-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(Cl)C=C(CC(=O)NC2=CN3C=C(Br)C=CC3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(Cl)C=C(CC(=O)NC2=CN3C=C(Br)C=CC3=N2)C=C1 IQFQLZRYZJORCR-UHFFFAOYSA-N 0.000 claims 1
- GUZAZDWYQNKVFG-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(Cl)C=C(CC(=O)NC2=NC3=CC=CC=C3N2CC2=CC=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(Cl)C=C(CC(=O)NC2=NC3=CC=CC=C3N2CC2=CC=CC=C2)C=C1 GUZAZDWYQNKVFG-UHFFFAOYSA-N 0.000 claims 1
- NTBHSCPBMKSGIW-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NC(=CS2)C2=CC(=O)NC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NC(=CS2)C2=CC(=O)NC=C2)C=C1 NTBHSCPBMKSGIW-UHFFFAOYSA-N 0.000 claims 1
- YTEOCLACNZQFIT-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN(CC(F)(F)F)C=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN(CC(F)(F)F)C=C2)C=C1 YTEOCLACNZQFIT-UHFFFAOYSA-N 0.000 claims 1
- YACLJXDEHQXBAE-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC(F)=CC3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC(F)=CC3=N2)C=C1 YACLJXDEHQXBAE-UHFFFAOYSA-N 0.000 claims 1
- KRIYEGIRGBWTEE-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC=C(C3=N2)C(F)(F)F)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=CC=C(C3=N2)C(F)(F)F)C=C1 KRIYEGIRGBWTEE-UHFFFAOYSA-N 0.000 claims 1
- NBDYFVZIFGUKGT-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C(F)(F)F)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C(F)(F)F)C=C1 NBDYFVZIFGUKGT-UHFFFAOYSA-N 0.000 claims 1
- IXINLPWPDGEBBT-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C2=CC=CN=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C2=CC=CN=C2)C=C1 IXINLPWPDGEBBT-UHFFFAOYSA-N 0.000 claims 1
- WXEVLPIVNQFQBE-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C2=CC=NC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN=C(S2)C2=CC=NC=C2)C=C1 WXEVLPIVNQFQBE-UHFFFAOYSA-N 0.000 claims 1
- BLFWDJUVDSXOBZ-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=C3C=CC(=O)OC3=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=C3C=CC(=O)OC3=CC=C2)C=C1 BLFWDJUVDSXOBZ-UHFFFAOYSA-N 0.000 claims 1
- LGKIMVHZNBLUBX-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC(=CC=C2)C#N)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC(=CC=C2)C#N)C=C1 LGKIMVHZNBLUBX-UHFFFAOYSA-N 0.000 claims 1
- CREYLROHUARRGR-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC(F)=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC(F)=CC=C2)C=C1 CREYLROHUARRGR-UHFFFAOYSA-N 0.000 claims 1
- NJRROBWVCUPSBQ-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC(OC(F)(F)F)=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC(OC(F)(F)F)=CC=C2)C=C1 NJRROBWVCUPSBQ-UHFFFAOYSA-N 0.000 claims 1
- PTEPLOWICYAFIF-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC3=C(OCCO3)C=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC3=C(OCCO3)C=C2)C=C1 PTEPLOWICYAFIF-UHFFFAOYSA-N 0.000 claims 1
- ARKOCTSTLYSYIT-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC(=CS2)C2=CC=CC=C2)C=C1 ARKOCTSTLYSYIT-UHFFFAOYSA-N 0.000 claims 1
- XIMDQJXVYCARGX-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(N2)C(Cl)=CC=C3)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(N2)C(Cl)=CC=C3)C=C1 XIMDQJXVYCARGX-UHFFFAOYSA-N 0.000 claims 1
- JHWAOVKVLMBISH-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(N2)C=CC(=C3)C#N)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(N2)C=CC(=C3)C#N)C=C1 JHWAOVKVLMBISH-UHFFFAOYSA-N 0.000 claims 1
- JQEVMVJGCADAIH-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(N2)C=CC(=C3)C(=O)C2=CC=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(N2)C=CC(=C3)C(=O)C2=CC=CC=C2)C=C1 JQEVMVJGCADAIH-UHFFFAOYSA-N 0.000 claims 1
- DBTBGCWVUFUCQI-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(O2)C=CC=C3)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(O2)C=CC=C3)C=C1 DBTBGCWVUFUCQI-UHFFFAOYSA-N 0.000 claims 1
- SOAXROLJSRMTPN-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(S2)C=CC(=C3)C2=CC(=O)NC=C2)C=C1F Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(S2)C=CC(=C3)C2=CC(=O)NC=C2)C=C1F SOAXROLJSRMTPN-UHFFFAOYSA-N 0.000 claims 1
- JYGSNRUNPZWKHB-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(S2)C=CC=C3)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NC3=C(S2)C=CC=C3)C=C1 JYGSNRUNPZWKHB-UHFFFAOYSA-N 0.000 claims 1
- DVOUQVNKXXGNKJ-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NN(C=C2)C2=CC=C(C=N2)C(F)(F)F)C=C1F Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NN(C=C2)C2=CC=C(C=N2)C(F)(F)F)C=C1F DVOUQVNKXXGNKJ-UHFFFAOYSA-N 0.000 claims 1
- SNMNLHMDAKWHBY-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NN=C(S2)C2=CC=CC=C2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(=O)NC2=NN=C(S2)C2=CC=CC=C2)C=C1 SNMNLHMDAKWHBY-UHFFFAOYSA-N 0.000 claims 1
- 208000031481 Pathologic Constriction Diseases 0.000 claims 1
- 208000018262 Peripheral vascular disease Diseases 0.000 claims 1
- 206010047163 Vasospasm Diseases 0.000 claims 1
- HVKZQJAWLUPKAA-FIBGUPNXSA-N [2H]C([2H])([2H])N1C=CC(=N1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 Chemical compound [2H]C([2H])([2H])N1C=CC(=N1)C1=CC2=C(SC(NC(=O)CC3=CC=C(OC4=NC=CC=C4C(N)=O)C(F)=C3)=N2)C=C1 HVKZQJAWLUPKAA-FIBGUPNXSA-N 0.000 claims 1
- 208000026106 cerebrovascular disease Diseases 0.000 claims 1
- FGCMEBLQCLCIQQ-UHFFFAOYSA-N methyl N-[4-[2-[[2-[4-(3-carbamoylpyridin-2-yl)oxy-3-fluorophenyl]acetyl]amino]-5-methyl-1,3-thiazol-4-yl]pyridin-2-yl]carbamate Chemical compound C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC=1SC(=C(N=1)C1=CC(=NC=C1)NC(OC)=O)C)F FGCMEBLQCLCIQQ-UHFFFAOYSA-N 0.000 claims 1
- 208000010125 myocardial infarction Diseases 0.000 claims 1
- 208000037804 stenosis Diseases 0.000 claims 1
- 230000036262 stenosis Effects 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 81
- 230000001363 autoimmune Effects 0.000 abstract 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 97
- -1 heterocyclic carboxamide compounds Chemical class 0.000 description 85
- 239000011541 reaction mixture Substances 0.000 description 80
- 238000005160 1H NMR spectroscopy Methods 0.000 description 78
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 75
- 239000000543 intermediate Substances 0.000 description 61
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 55
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 52
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 52
- 230000014759 maintenance of location Effects 0.000 description 46
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 44
- 239000007787 solid Substances 0.000 description 44
- 238000004128 high performance liquid chromatography Methods 0.000 description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 42
- 230000002829 reductive effect Effects 0.000 description 41
- 235000002639 sodium chloride Nutrition 0.000 description 38
- 235000019439 ethyl acetate Nutrition 0.000 description 33
- 239000000203 mixture Substances 0.000 description 30
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 29
- 239000002904 solvent Substances 0.000 description 29
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 28
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 28
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 239000012453 solvate Substances 0.000 description 22
- 239000012267 brine Substances 0.000 description 21
- 239000000460 chlorine Substances 0.000 description 21
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 20
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 19
- 239000003814 drug Substances 0.000 description 19
- 239000012044 organic layer Substances 0.000 description 19
- 239000000047 product Substances 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 17
- 230000000670 limiting effect Effects 0.000 description 16
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 14
- 125000001424 substituent group Chemical group 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 13
- 238000004007 reversed phase HPLC Methods 0.000 description 13
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 13
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 12
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 12
- 239000010410 layer Substances 0.000 description 12
- 239000012071 phase Substances 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 125000004429 atom Chemical group 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- 238000010898 silica gel chromatography Methods 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 238000004519 manufacturing process Methods 0.000 description 10
- 239000000243 solution Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 125000002950 monocyclic group Chemical group 0.000 description 9
- 230000007170 pathology Effects 0.000 description 9
- 239000000651 prodrug Substances 0.000 description 9
- 229940002612 prodrug Drugs 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 8
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 230000008878 coupling Effects 0.000 description 8
- 238000010168 coupling process Methods 0.000 description 8
- 238000005859 coupling reaction Methods 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 8
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 7
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 description 7
- 108010081348 HRT1 protein Hairy Proteins 0.000 description 7
- 102100021881 Hairy/enhancer-of-split related with YRPW motif protein 1 Human genes 0.000 description 7
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 101100356682 Caenorhabditis elegans rho-1 gene Proteins 0.000 description 6
- 101150111584 RHOA gene Proteins 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 235000011114 ammonium hydroxide Nutrition 0.000 description 6
- 125000002619 bicyclic group Chemical group 0.000 description 6
- 150000001721 carbon Chemical group 0.000 description 6
- 125000000753 cycloalkyl group Chemical group 0.000 description 6
- 125000004438 haloalkoxy group Chemical group 0.000 description 6
- 125000001188 haloalkyl group Chemical group 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 235000015320 potassium carbonate Nutrition 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- URJOUYRTHKWERB-UHFFFAOYSA-N 3-(difluoromethoxy)pyridin-2-amine Chemical compound NC1=NC=CC=C1OC(F)F URJOUYRTHKWERB-UHFFFAOYSA-N 0.000 description 5
- OMIHQJBWAPWLBO-UHFFFAOYSA-N 5-methoxy-1,3-benzothiazol-2-amine Chemical compound COC1=CC=C2SC(N)=NC2=C1 OMIHQJBWAPWLBO-UHFFFAOYSA-N 0.000 description 5
- KJZJEUCIXBKYCH-UHFFFAOYSA-N ClC=1C=C(C=CC=1OC1=NC=CC=C1C#N)CC(=O)O Chemical compound ClC=1C=C(C=CC=1OC1=NC=CC=C1C#N)CC(=O)O KJZJEUCIXBKYCH-UHFFFAOYSA-N 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 125000005605 benzo group Chemical group 0.000 description 5
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 5
- 229910052796 boron Inorganic materials 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 125000001072 heteroaryl group Chemical group 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 125000001041 indolyl group Chemical group 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 208000017169 kidney disease Diseases 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000019491 signal transduction Effects 0.000 description 5
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 5
- 125000001544 thienyl group Chemical group 0.000 description 5
- WIESEJCMPWONJZ-UHFFFAOYSA-N 1-(cyclobutylmethyl)benzimidazol-2-amine Chemical compound NC1=NC2=CC=CC=C2N1CC1CCC1 WIESEJCMPWONJZ-UHFFFAOYSA-N 0.000 description 4
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- XQXPVVBIMDBYFF-UHFFFAOYSA-N 4-hydroxyphenylacetic acid Chemical compound OC(=O)CC1=CC=C(O)C=C1 XQXPVVBIMDBYFF-UHFFFAOYSA-N 0.000 description 4
- RCVDCZOEHBRZGK-UHFFFAOYSA-N 7-methoxy-4,5-dihydrobenzo[e][1,3]benzothiazol-2-amine Chemical compound C1CC2=CC(OC)=CC=C2C2=C1SC(N)=N2 RCVDCZOEHBRZGK-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- WWNWNNKPTBFUAN-UHFFFAOYSA-N C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)Cl)F Chemical compound C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)Cl)F WWNWNNKPTBFUAN-UHFFFAOYSA-N 0.000 description 4
- MPDJSTDQHAHMGN-UHFFFAOYSA-N CC(C)(O)COC1=CC=C2SC(N)=NC2=C1 Chemical compound CC(C)(O)COC1=CC=C2SC(N)=NC2=C1 MPDJSTDQHAHMGN-UHFFFAOYSA-N 0.000 description 4
- ANRFNVFWNXDAPU-UHFFFAOYSA-N CC1=C(N=C(S1)N)C1=NC(=CC=C1)C Chemical compound CC1=C(N=C(S1)N)C1=NC(=CC=C1)C ANRFNVFWNXDAPU-UHFFFAOYSA-N 0.000 description 4
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 208000010228 Erectile Dysfunction Diseases 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 238000006069 Suzuki reaction reaction Methods 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 125000002527 bicyclic carbocyclic group Chemical group 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 229910052805 deuterium Inorganic materials 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- NGOGFTYYXHNFQH-UHFFFAOYSA-N fasudil Chemical compound C=1C=CC2=CN=CC=C2C=1S(=O)(=O)N1CCCNCC1 NGOGFTYYXHNFQH-UHFFFAOYSA-N 0.000 description 4
- 229960002435 fasudil Drugs 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 125000002541 furyl group Chemical group 0.000 description 4
- 125000002883 imidazolyl group Chemical group 0.000 description 4
- 201000001881 impotence Diseases 0.000 description 4
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 208000001286 intracranial vasospasm Diseases 0.000 description 4
- 125000000842 isoxazolyl group Chemical group 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 125000001624 naphthyl group Chemical group 0.000 description 4
- 125000002971 oxazolyl group Chemical group 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 229920003023 plastic Polymers 0.000 description 4
- 125000003373 pyrazinyl group Chemical group 0.000 description 4
- 125000003226 pyrazolyl group Chemical group 0.000 description 4
- 125000004076 pyridyl group Chemical group 0.000 description 4
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 125000003831 tetrazolyl group Chemical group 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 125000004306 triazinyl group Chemical group 0.000 description 4
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 4
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- 125000005955 1H-indazolyl group Chemical group 0.000 description 3
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- YRFBZAHYMOSSGX-UHFFFAOYSA-N 2-(3-fluoro-4-hydroxyphenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(O)C(F)=C1 YRFBZAHYMOSSGX-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- RLIPCWGTDDONRJ-UHFFFAOYSA-N 2-[4-(3-carbamoylpyridin-2-yl)oxyphenyl]acetic acid Chemical compound NC(=O)C1=CC=CN=C1OC1=CC=C(CC(O)=O)C=C1 RLIPCWGTDDONRJ-UHFFFAOYSA-N 0.000 description 3
- JAUPUQRPBNDMDT-UHFFFAOYSA-N 2-chloropyridine-3-carbonitrile Chemical compound ClC1=NC=CC=C1C#N JAUPUQRPBNDMDT-UHFFFAOYSA-N 0.000 description 3
- MGIXALWFXBQXRE-UHFFFAOYSA-N 7-methoxy-[1,2,4]triazolo[1,5-a]pyridin-2-amine Chemical compound C1=C(OC)C=CN2N=C(N)N=C21 MGIXALWFXBQXRE-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 206010003225 Arteriospasm coronary Diseases 0.000 description 3
- UATUQAOBFPRKCK-UHFFFAOYSA-N BrC=1N=C(SC=1C)NC(CC1=CC(=C(C=C1)OC1=NC=CC=C1C#N)F)=O Chemical compound BrC=1N=C(SC=1C)NC(CC1=CC(=C(C=C1)OC1=NC=CC=C1C#N)F)=O UATUQAOBFPRKCK-UHFFFAOYSA-N 0.000 description 3
- FAPFBYZVHNMBFH-UHFFFAOYSA-N CC(C)(O)COC1=CC=C2C3=C(CCC2=C1)SC(N)=N3 Chemical compound CC(C)(O)COC1=CC=C2C3=C(CCC2=C1)SC(N)=N3 FAPFBYZVHNMBFH-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 208000003890 Coronary Vasospasm Diseases 0.000 description 3
- 208000010412 Glaucoma Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000282414 Homo sapiens Species 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 206010063837 Reperfusion injury Diseases 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- CWGJBBRZFIPXNZ-UHFFFAOYSA-N [C-]#N.Br Chemical compound [C-]#N.Br CWGJBBRZFIPXNZ-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 208000006673 asthma Diseases 0.000 description 3
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 230000036772 blood pressure Effects 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 3
- 201000011634 coronary artery vasospasm Diseases 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical compound C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 125000002632 imidazolidinyl group Chemical group 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 208000028867 ischemia Diseases 0.000 description 3
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 125000001715 oxadiazolyl group Chemical group 0.000 description 3
- 125000000160 oxazolidinyl group Chemical group 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000000123 paper Substances 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 125000004193 piperazinyl group Chemical group 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 3
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000010265 sodium sulphite Nutrition 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 2
- AAZCFOYBTCRVEJ-YUMQZZPRSA-N (2S,5S)-2,5-diethylphospholane Chemical compound CC[C@H]1CC[C@H](CC)P1 AAZCFOYBTCRVEJ-YUMQZZPRSA-N 0.000 description 2
- IYTUKSIOQKTZEG-UHFFFAOYSA-N (3-chloro-4-hydroxyphenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(O)C(Cl)=C1 IYTUKSIOQKTZEG-UHFFFAOYSA-N 0.000 description 2
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 2
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- ITZMXPVBBFYECZ-UHFFFAOYSA-N 1-(2,2-difluoroethyl)-5-methoxybenzimidazol-2-amine Chemical compound COC1=CC=C2N(CC(F)F)C(N)=NC2=C1 ITZMXPVBBFYECZ-UHFFFAOYSA-N 0.000 description 2
- PTGUURFTJDPTAC-UHFFFAOYSA-N 1-(2,3-dihydro-1h-indol-6-yloxy)-2-methylpropan-2-ol Chemical compound CC(C)(O)COC1=CC=C2CCNC2=C1 PTGUURFTJDPTAC-UHFFFAOYSA-N 0.000 description 2
- JFYRTFZFHWBODA-UHFFFAOYSA-N 1-(6-fluoropyridin-2-yl)propan-1-one Chemical compound C1=C(F)N=C(C(=O)CC)C=C1 JFYRTFZFHWBODA-UHFFFAOYSA-N 0.000 description 2
- NPTHNNQFYCAGKP-UHFFFAOYSA-N 1-(6-methylpyridin-2-yl)propan-1-one Chemical compound CCC(=O)C1=CC=CC(C)=N1 NPTHNNQFYCAGKP-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- DFPYXQYWILNVAU-UHFFFAOYSA-N 1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1=CC=C2N(O)N=NC2=C1 DFPYXQYWILNVAU-UHFFFAOYSA-N 0.000 description 2
- UDJSHISPNDRJNE-UHFFFAOYSA-N 1-propan-2-ylbenzimidazol-2-amine Chemical compound C1=CC=C2N(C(C)C)C(N)=NC2=C1 UDJSHISPNDRJNE-UHFFFAOYSA-N 0.000 description 2
- GELKGHVAFRCJNA-UHFFFAOYSA-N 2,2-Dimethyloxirane Chemical compound CC1(C)CO1 GELKGHVAFRCJNA-UHFFFAOYSA-N 0.000 description 2
- MDNDJMCSXOXBFZ-UHFFFAOYSA-N 2-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-5,5-dimethyl-1,3,2-dioxaborinane Chemical compound O1CC(C)(C)COB1B1OCC(C)(C)CO1 MDNDJMCSXOXBFZ-UHFFFAOYSA-N 0.000 description 2
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 2
- XLPKMCJZEJFNBE-UHFFFAOYSA-N 2-[4-(3-cyanopyridin-2-yl)oxyphenyl]acetic acid Chemical compound C1=CC(CC(=O)O)=CC=C1OC1=NC=CC=C1C#N XLPKMCJZEJFNBE-UHFFFAOYSA-N 0.000 description 2
- PIERIBXILPXPFL-UHFFFAOYSA-N 2-amino-1,3-benzothiazol-5-ol Chemical compound OC1=CC=C2SC(N)=NC2=C1 PIERIBXILPXPFL-UHFFFAOYSA-N 0.000 description 2
- BFYSLYUNODVYKG-UHFFFAOYSA-N 2-amino-4,5-dihydrobenzo[e][1,3]benzothiazol-7-ol Chemical compound OC1=CC=C2C(N=C(S3)N)=C3CCC2=C1 BFYSLYUNODVYKG-UHFFFAOYSA-N 0.000 description 2
- RAFRWGUJJXZTNF-UHFFFAOYSA-N 2-bromo-1-(6-fluoropyridin-2-yl)propan-1-one Chemical compound BrC(C(=O)C1=NC(=CC=C1)F)C RAFRWGUJJXZTNF-UHFFFAOYSA-N 0.000 description 2
- OFINLCWPTZLDOZ-UHFFFAOYSA-N 2-bromo-1-cyclobutyl-2-pyridin-2-ylethanone Chemical compound BrC(C(=O)C1CCC1)C1=NC=CC=C1 OFINLCWPTZLDOZ-UHFFFAOYSA-N 0.000 description 2
- JUGGVCLJDPHFQC-UHFFFAOYSA-N 2-fluoro-1-nitro-4-phenylmethoxybenzene Chemical compound C1=C(F)C([N+](=O)[O-])=CC=C1OCC1=CC=CC=C1 JUGGVCLJDPHFQC-UHFFFAOYSA-N 0.000 description 2
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 2
- KGBRVJLHFOVILU-UHFFFAOYSA-N 4-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-1H-pyridin-2-one Chemical compound CC1(C)COB(OC1)C1=CC(=O)NC=C1 KGBRVJLHFOVILU-UHFFFAOYSA-N 0.000 description 2
- IDDDVXIUIXWAGJ-DDSAHXNVSA-N 4-[(1r)-1-aminoethyl]-n-pyridin-4-ylcyclohexane-1-carboxamide;dihydrochloride Chemical compound Cl.Cl.C1CC([C@H](N)C)CCC1C(=O)NC1=CC=NC=C1 IDDDVXIUIXWAGJ-DDSAHXNVSA-N 0.000 description 2
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 2
- COVREJPVUUCVMP-UHFFFAOYSA-N 4-bromo-2-n-(cyclopropylmethyl)benzene-1,2-diamine Chemical compound NC1=CC=C(Br)C=C1NCC1CC1 COVREJPVUUCVMP-UHFFFAOYSA-N 0.000 description 2
- MOGMAODWVQPRIC-UHFFFAOYSA-N 5-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-1-methylpyridin-2-one Chemical compound CN1C=C(C=CC1=O)B1OCC(C)(C)CO1 MOGMAODWVQPRIC-UHFFFAOYSA-N 0.000 description 2
- HHVFVHIUOMMWRN-UHFFFAOYSA-N 5-bromo-1-methylpyridin-2-one Chemical compound CN1C=C(Br)C=CC1=O HHVFVHIUOMMWRN-UHFFFAOYSA-N 0.000 description 2
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 2
- NTKQPXFXPMRBKY-UHFFFAOYSA-N 6-bromo-1-(cyclopropylmethyl)benzimidazol-2-amine Chemical compound NC1=NC2=CC=C(Br)C=C2N1CC1CC1 NTKQPXFXPMRBKY-UHFFFAOYSA-N 0.000 description 2
- OSMYOADKQWQBQT-UHFFFAOYSA-N 6-fluoro-n-methoxy-n-methylpyridine-2-carboxamide Chemical compound CON(C)C(=O)C1=CC=CC(F)=N1 OSMYOADKQWQBQT-UHFFFAOYSA-N 0.000 description 2
- DAYYXZOMYRSLBX-UHFFFAOYSA-N 7-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-amine Chemical compound C1=C(Br)C=CN2N=C(N)N=C21 DAYYXZOMYRSLBX-UHFFFAOYSA-N 0.000 description 2
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- CQVVVHVFXJTVGX-UHFFFAOYSA-N BrC=1C=C(C=CC=1OC1=NC=CC=C1C#N)CC(=O)O Chemical compound BrC=1C=C(C=CC=1OC1=NC=CC=C1C#N)CC(=O)O CQVVVHVFXJTVGX-UHFFFAOYSA-N 0.000 description 2
- FMTVIXJPYCQPLU-UHFFFAOYSA-N BrC=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(C=C2)OC2=NC=CC=C2C#N)F)=O)C=1 Chemical compound BrC=1C=CC2=C(N=C(S2)NC(CC2=CC(=C(C=C2)OC2=NC=CC=C2C#N)F)=O)C=1 FMTVIXJPYCQPLU-UHFFFAOYSA-N 0.000 description 2
- TXRLQAJNGLYNNS-UHFFFAOYSA-N BrC=1N=C(SC=1)NC(CC1=CC(=C(C=C1)OC1=NC=CC=C1C#N)F)=O Chemical compound BrC=1N=C(SC=1)NC(CC1=CC(=C(C=C1)OC1=NC=CC=C1C#N)F)=O TXRLQAJNGLYNNS-UHFFFAOYSA-N 0.000 description 2
- PFOMVKBVWFRHHR-UHFFFAOYSA-N C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OC)OC Chemical compound C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OC)OC PFOMVKBVWFRHHR-UHFFFAOYSA-N 0.000 description 2
- LPNNFVXAFMPCAH-UHFFFAOYSA-N C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)OCC)OC Chemical compound C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)OCC)OC LPNNFVXAFMPCAH-UHFFFAOYSA-N 0.000 description 2
- AVPSHYIHCDJXQV-UHFFFAOYSA-N C(C1=CC=CC=C1)OC=1C=CC2=C(N(C(=N2)N)CC2CC2)C=1 Chemical compound C(C1=CC=CC=C1)OC=1C=CC2=C(N(C(=N2)N)CC2CC2)C=1 AVPSHYIHCDJXQV-UHFFFAOYSA-N 0.000 description 2
- DNRLJQYIBLFUPP-UHFFFAOYSA-N C(N)(=O)C=1C(=NC=CC=1)OC1=CC=C(C=C1)C(C(=O)O)C Chemical compound C(N)(=O)C=1C(=NC=CC=1)OC1=CC=C(C=C1)C(C(=O)O)C DNRLJQYIBLFUPP-UHFFFAOYSA-N 0.000 description 2
- DVHOHOXWQXNTPT-UHFFFAOYSA-N C1(CCC1)C=1N=C(SC=1C1=NC=CC=C1)N Chemical compound C1(CCC1)C=1N=C(SC=1C1=NC=CC=C1)N DVHOHOXWQXNTPT-UHFFFAOYSA-N 0.000 description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 2
- WKLCYFKVTMZDIF-UHFFFAOYSA-N CC(C)(O)COC1=CC2=NC(N)=NN2C=C1 Chemical compound CC(C)(O)COC1=CC2=NC(N)=NN2C=C1 WKLCYFKVTMZDIF-UHFFFAOYSA-N 0.000 description 2
- 241001070941 Castanea Species 0.000 description 2
- 235000014036 Castanea Nutrition 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 201000000057 Coronary Stenosis Diseases 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- GSCCALZHGUWNJW-UHFFFAOYSA-N N-Cyclohexyl-N-methylcyclohexanamine Chemical compound C1CCCCC1N(C)C1CCCCC1 GSCCALZHGUWNJW-UHFFFAOYSA-N 0.000 description 2
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- LIHFPFZRWOZNMN-UHFFFAOYSA-N NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=C(Br)C=CC3=N2)C=C1 Chemical compound NC(=O)C1=CC=CN=C1OC1=C(F)C=C(CC(=O)NC2=NN3C=C(Br)C=CC3=N2)C=C1 LIHFPFZRWOZNMN-UHFFFAOYSA-N 0.000 description 2
- OQUWVUNAEIZLHL-UHFFFAOYSA-N NC1=NC2=C(N1CC1CC1)C=C(C=C2)OCC(C)(O)C Chemical compound NC1=NC2=C(N1CC1CC1)C=C(C=C2)OCC(C)(O)C OQUWVUNAEIZLHL-UHFFFAOYSA-N 0.000 description 2
- CJKHBYCVAKDING-UHFFFAOYSA-N NC1=NN2C=CC=C(OC(F)F)C2=N1 Chemical compound NC1=NN2C=CC=C(OC(F)F)C2=N1 CJKHBYCVAKDING-UHFFFAOYSA-N 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 208000025966 Neurological disease Diseases 0.000 description 2
- 229920000954 Polyglycolide Polymers 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- HRAHSHBSDCCHEO-UHFFFAOYSA-N [2-(methoxycarbonylamino)pyridin-4-yl]boronic acid Chemical compound COC(=O)NC1=NC=CC(=C1)B(O)O HRAHSHBSDCCHEO-UHFFFAOYSA-N 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 125000004450 alkenylene group Chemical group 0.000 description 2
- 125000004419 alkynylene group Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 2
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- FLHFTXCMKFVKRP-UHFFFAOYSA-N bromomethylcyclobutane Chemical compound BrCC1CCC1 FLHFTXCMKFVKRP-UHFFFAOYSA-N 0.000 description 2
- YSHOWEKUVWPFNR-UHFFFAOYSA-N burgess reagent Chemical compound CC[N+](CC)(CC)S(=O)(=O)N=C([O-])OC YSHOWEKUVWPFNR-UHFFFAOYSA-N 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- IGSKHXTUVXSOMB-UHFFFAOYSA-N cyclopropylmethanamine Chemical compound NCC1CC1 IGSKHXTUVXSOMB-UHFFFAOYSA-N 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000003292 glue Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- 230000004047 hyperresponsiveness Effects 0.000 description 2
- 230000001631 hypertensive effect Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- XGDZEDRBLVIUMX-UHFFFAOYSA-N methyl 2-(4-hydroxyphenyl)acetate Chemical compound COC(=O)CC1=CC=C(O)C=C1 XGDZEDRBLVIUMX-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 208000004296 neuralgia Diseases 0.000 description 2
- 208000021722 neuropathic pain Diseases 0.000 description 2
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 239000003182 parenteral nutrition solution Substances 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 2
- 229920000747 poly(lactic acid) Polymers 0.000 description 2
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000004633 polyglycolic acid Substances 0.000 description 2
- 239000004626 polylactic acid Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000009862 primary prevention Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 230000009863 secondary prevention Effects 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 150000003413 spiro compounds Chemical class 0.000 description 2
- 238000011699 spontaneously hypertensive rat Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 235000011150 stannous chloride Nutrition 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- 125000004525 thiadiazinyl group Chemical group S1NN=C(C=C1)* 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 238000003828 vacuum filtration Methods 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- XFQNWPYGEGCIMF-HCUGAJCMSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].[Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 XFQNWPYGEGCIMF-HCUGAJCMSA-N 0.000 description 1
- RRKODOZNUZCUBN-CCAGOZQPSA-N (1z,3z)-cycloocta-1,3-diene Chemical compound C1CC\C=C/C=C\C1 RRKODOZNUZCUBN-CCAGOZQPSA-N 0.000 description 1
- XDIYNQZUNSSENW-UUBOPVPUSA-N (2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O XDIYNQZUNSSENW-UUBOPVPUSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- YUXKOWPNKJSTPQ-AXWWPMSFSA-N (2s,3r)-2-amino-3-hydroxybutanoic acid;(2s)-2-amino-3-hydroxypropanoic acid Chemical compound OC[C@H](N)C(O)=O.C[C@@H](O)[C@H](N)C(O)=O YUXKOWPNKJSTPQ-AXWWPMSFSA-N 0.000 description 1
- CUKWUWBLQQDQAC-VEQWQPCFSA-N (3s)-3-amino-4-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2s,3s)-1-[[(2s)-1-[(2s)-2-[[(1s)-1-carboxyethyl]carbamoyl]pyrrolidin-1-yl]-3-(1h-imidazol-5-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-methyl-1-ox Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C1=CC=C(O)C=C1 CUKWUWBLQQDQAC-VEQWQPCFSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 description 1
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- FIARMZDBEGVMLV-UHFFFAOYSA-N 1,1,2,2,2-pentafluoroethanolate Chemical group [O-]C(F)(F)C(F)(F)F FIARMZDBEGVMLV-UHFFFAOYSA-N 0.000 description 1
- VMFCTZUYOILUMY-UHFFFAOYSA-N 1,1-difluoro-2-iodoethane Chemical compound FC(F)CI VMFCTZUYOILUMY-UHFFFAOYSA-N 0.000 description 1
- 125000004605 1,2,3,4-tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000004607 1,2,3,4-tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-Bis(diphenylphosphino)propane Substances C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 1
- FBNAMBTYMSWTIB-UHFFFAOYSA-N 1,3-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound CC1=NN(C)C=C1B1OC(C)(C)C(C)(C)O1 FBNAMBTYMSWTIB-UHFFFAOYSA-N 0.000 description 1
- GORGFCBYOBPURU-UHFFFAOYSA-N 1-(2,2-difluoroethyl)-6-methoxybenzimidazol-2-amine Chemical compound COc1ccc2nc(N)n(CC(F)F)c2c1 GORGFCBYOBPURU-UHFFFAOYSA-N 0.000 description 1
- MLLXQRVIHBKKEZ-UHFFFAOYSA-N 1-(2,2-difluoroethyl)benzimidazol-2-amine Chemical compound Nc1nc2ccccc2n1CC(F)F MLLXQRVIHBKKEZ-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- RFJCEINMRPKWOZ-UHFFFAOYSA-N 1-cyclobutyl-2-pyridin-2-ylethanone Chemical compound C1CCC1C(=O)CC1=CC=CC=N1 RFJCEINMRPKWOZ-UHFFFAOYSA-N 0.000 description 1
- 125000006432 1-methyl cyclopropyl group Chemical group [H]C([H])([H])C1(*)C([H])([H])C1([H])[H] 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- BFVCOQXPSXVGPS-UHFFFAOYSA-N 2-(3-bromo-4-hydroxyphenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(O)C(Br)=C1 BFVCOQXPSXVGPS-UHFFFAOYSA-N 0.000 description 1
- HSYYELKVUOSOFF-UHFFFAOYSA-N 2-N-(cyclopropylmethyl)-4-phenylmethoxybenzene-1,2-diamine Chemical compound C(C1=CC=CC=C1)OC1=CC=C(C(=C1)NCC1CC1)N HSYYELKVUOSOFF-UHFFFAOYSA-N 0.000 description 1
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- MLONYBFKXHEPCD-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(N)(CO)CO.OCC(N)(CO)CO MLONYBFKXHEPCD-UHFFFAOYSA-N 0.000 description 1
- JWYUFVNJZUSCSM-UHFFFAOYSA-N 2-aminobenzimidazole Chemical compound C1=CC=C2NC(N)=NC2=C1 JWYUFVNJZUSCSM-UHFFFAOYSA-N 0.000 description 1
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- VSCBATMPTLKTOV-UHFFFAOYSA-N 2-tert-butylimino-n,n-diethyl-1,3-dimethyl-1,3,2$l^{5}-diazaphosphinan-2-amine Chemical compound CCN(CC)P1(=NC(C)(C)C)N(C)CCCN1C VSCBATMPTLKTOV-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- YWOWJQMFMXHLQD-UHFFFAOYSA-N 3-(trifluoromethyl)pyridin-2-amine Chemical group NC1=NC=CC=C1C(F)(F)F YWOWJQMFMXHLQD-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- CSSGKHVRDGATJL-UHFFFAOYSA-N 3-fluoro-4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C(F)=C1 CSSGKHVRDGATJL-UHFFFAOYSA-N 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- IYOZTVGMEWJPKR-VOMCLLRMSA-N 4-[(1R)-1-aminoethyl]-N-pyridin-4-yl-1-cyclohexanecarboxamide Chemical compound C1CC([C@H](N)C)CCC1C(=O)NC1=CC=NC=C1 IYOZTVGMEWJPKR-VOMCLLRMSA-N 0.000 description 1
- QJPQUNWGOHADOB-UHFFFAOYSA-N 4-bromo-1,3-thiazol-2-amine Chemical compound NC1=NC(Br)=CS1 QJPQUNWGOHADOB-UHFFFAOYSA-N 0.000 description 1
- SSLMGOKTIUIZLY-UHFFFAOYSA-N 4-bromo-1h-pyridin-2-one Chemical compound OC1=CC(Br)=CC=N1 SSLMGOKTIUIZLY-UHFFFAOYSA-N 0.000 description 1
- VQCWSOYHHXXWSP-UHFFFAOYSA-N 4-bromo-2-fluoro-1-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(Br)C=C1F VQCWSOYHHXXWSP-UHFFFAOYSA-N 0.000 description 1
- KPTFKWPRFDQHJL-UHFFFAOYSA-N 4-bromo-5-methyl-1,3-thiazol-2-amine Chemical compound CC=1SC(N)=NC=1Br KPTFKWPRFDQHJL-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- WCCFLOQQACDOAX-UHFFFAOYSA-N 4-fluoropyridin-2-amine Chemical compound NC1=CC(F)=CC=N1 WCCFLOQQACDOAX-UHFFFAOYSA-N 0.000 description 1
- 125000006042 4-hexenyl group Chemical group 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003119 4-methyl-3-pentenyl group Chemical group [H]\C(=C(/C([H])([H])[H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- RAFCWIXBEWVPGL-UHFFFAOYSA-N 4-phenylmethoxypyridin-2-amine Chemical group C1=NC(N)=CC(OCC=2C=CC=CC=2)=C1 RAFCWIXBEWVPGL-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- 125000004608 5,6,7,8-tetrahydroquinolinyl group Chemical group N1=C(C=CC=2CCCCC12)* 0.000 description 1
- HCYQWUCBICRJGO-UHFFFAOYSA-N 5-(trifluoromethoxy)pyridin-2-amine Chemical group NC1=CC=C(OC(F)(F)F)C=N1 HCYQWUCBICRJGO-UHFFFAOYSA-N 0.000 description 1
- ZPUJTWBWSOOMRP-UHFFFAOYSA-N 5-bromo-1,3-benzothiazol-2-amine Chemical compound BrC1=CC=C2SC(N)=NC2=C1 ZPUJTWBWSOOMRP-UHFFFAOYSA-N 0.000 description 1
- KMAQQDDPRGOPRF-UHFFFAOYSA-N 5-bromo-1-(difluoromethyl)pyridin-2-one Chemical compound FC(F)N1C=C(Br)C=CC1=O KMAQQDDPRGOPRF-UHFFFAOYSA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- XHBQNLHFUPSZNL-UHFFFAOYSA-N 6-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-amine Chemical group C1=CC(Br)=CN2N=C(N)N=C21 XHBQNLHFUPSZNL-UHFFFAOYSA-N 0.000 description 1
- LAUJNLOJYLUKCP-UHFFFAOYSA-N 6-bromoimidazo[1,2-a]pyridin-2-amine Chemical compound C1=C(Br)C=CC2=NC(N)=CN21 LAUJNLOJYLUKCP-UHFFFAOYSA-N 0.000 description 1
- DIEMCUFYSOEIDU-UHFFFAOYSA-N 6-fluoropyridine-2-carboxylic acid Chemical compound OC(=O)C1=CC=CC(F)=N1 DIEMCUFYSOEIDU-UHFFFAOYSA-N 0.000 description 1
- FXFSGALQLLEBJD-UHFFFAOYSA-N 6-methoxy-1h-benzimidazol-2-amine Chemical compound COC1=CC=C2NC(N)=NC2=C1 FXFSGALQLLEBJD-UHFFFAOYSA-N 0.000 description 1
- GJNUCHYUWAFTGQ-UHFFFAOYSA-N 7-bromoimidazo[1,2-a]pyridin-2-amine Chemical compound C1=CC(Br)=CC2=NC(N)=CN21 GJNUCHYUWAFTGQ-UHFFFAOYSA-N 0.000 description 1
- FXXPKXPMSFTVBM-UHFFFAOYSA-N 7-methoxy-4,5-dihydrobenzo[e][1,3]benzothiazol-2-amine;hydrobromide Chemical compound Br.C1CC2=CC(OC)=CC=C2C2=C1SC(N)=N2 FXXPKXPMSFTVBM-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 102100023818 ADP-ribosylation factor 3 Human genes 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 102400000345 Angiotensin-2 Human genes 0.000 description 1
- 101800000733 Angiotensin-2 Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- NCJXUCPXDPIAOK-UHFFFAOYSA-N BrC=1C=C(C=CC=1OC1=NC=CC=C1C#N)CC(=O)Cl Chemical compound BrC=1C=C(C=CC=1OC1=NC=CC=C1C#N)CC(=O)Cl NCJXUCPXDPIAOK-UHFFFAOYSA-N 0.000 description 1
- CJKLQNXNIMBXPM-UHFFFAOYSA-N BrC=1N=C(SC=1)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O Chemical compound BrC=1N=C(SC=1)NC(CC1=CC(=C(OC2=C(C(=O)N)C=CC=N2)C=C1)F)=O CJKLQNXNIMBXPM-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- ILYGHAYAXMQOQQ-UHFFFAOYSA-N C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)O)F Chemical compound C(#N)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)O)F ILYGHAYAXMQOQQ-UHFFFAOYSA-N 0.000 description 1
- ZRBDLHGSRNLMRZ-UHFFFAOYSA-N C(#N)C=1C(=NC=CC=1)OC1=CC=C(C=C1)C(C(=O)OC)C Chemical compound C(#N)C=1C(=NC=CC=1)OC1=CC=C(C=C1)C(C(=O)OC)C ZRBDLHGSRNLMRZ-UHFFFAOYSA-N 0.000 description 1
- UTZHDWKVQXMULC-UHFFFAOYSA-N C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)O)F Chemical compound C(N)(=O)C=1C(=NC=CC=1)OC1=C(C=C(C=C1)CC(=O)O)F UTZHDWKVQXMULC-UHFFFAOYSA-N 0.000 description 1
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 1
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- 125000000041 C6-C10 aryl group Chemical group 0.000 description 1
- ANLLVHOXEAASNF-UHFFFAOYSA-N CN1N=CC=C1C1=CN2N=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C2C=C1 Chemical compound CN1N=CC=C1C1=CN2N=C(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C(N)=O)C=C3)N=C2C=C1 ANLLVHOXEAASNF-UHFFFAOYSA-N 0.000 description 1
- GPLYPWUWGNGFAD-UHFFFAOYSA-N COC1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C#N)C=C3)=NN2C=C1 Chemical compound COC1=CC2=NC(NC(=O)CC3=CC(F)=C(OC4=NC=CC=C4C#N)C=C3)=NN2C=C1 GPLYPWUWGNGFAD-UHFFFAOYSA-N 0.000 description 1
- PJQLCHIHJAMTEK-UHFFFAOYSA-N COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)CC1=CC(Cl)=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 Chemical compound COC1=CC=C2C3=C(CCC2=C1)SC(NC(=O)CC1=CC(Cl)=C(OC2=NC=CC=C2C(N)=O)C=C1)=N3 PJQLCHIHJAMTEK-UHFFFAOYSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RFBMNKDAVVJSTG-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OC Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OC RFBMNKDAVVJSTG-UHFFFAOYSA-N 0.000 description 1
- VDEWYYVWMVGTRD-UHFFFAOYSA-N ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OCC(C)(C)O Chemical compound ClC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)NC=1SC2=C(N=1)C=C(C=C2)OCC(C)(C)O VDEWYYVWMVGTRD-UHFFFAOYSA-N 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 102400000686 Endothelin-1 Human genes 0.000 description 1
- 101800004490 Endothelin-1 Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- BYJYGMOYGSYEAN-UHFFFAOYSA-N FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)N1CCC2=CC=C(C=C12)OCC(C)(C)O Chemical compound FC1=C(OC2=C(C(=O)N)C=CC=N2)C=CC(=C1)CC(=O)N1CCC2=CC=C(C=C12)OCC(C)(C)O BYJYGMOYGSYEAN-UHFFFAOYSA-N 0.000 description 1
- LYUHZRWVTLIVHQ-UHFFFAOYSA-N FC1=CC=CC(=N1)C=1N=C(SC=1C)N Chemical group FC1=CC=CC(=N1)C=1N=C(SC=1C)N LYUHZRWVTLIVHQ-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 101000684275 Homo sapiens ADP-ribosylation factor 3 Proteins 0.000 description 1
- 101001000061 Homo sapiens Protein phosphatase 1 regulatory subunit 12A Proteins 0.000 description 1
- 101001130437 Homo sapiens Ras-related protein Rap-2b Proteins 0.000 description 1
- 101000669917 Homo sapiens Rho-associated protein kinase 1 Proteins 0.000 description 1
- 101000669921 Homo sapiens Rho-associated protein kinase 2 Proteins 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 102000003777 Interleukin-1 beta Human genes 0.000 description 1
- 108090000193 Interleukin-1 beta Proteins 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229920001410 Microfiber Polymers 0.000 description 1
- 108010037801 Myosin-Light-Chain Phosphatase Proteins 0.000 description 1
- 102000011131 Myosin-Light-Chain Phosphatase Human genes 0.000 description 1
- OFMWZNPAAYCXKA-UHFFFAOYSA-N NC(=O)c1cccnc1Oc1ccc(CC(=O)Nc2nc3ccc(Br)cc3n2CC2CC2)cc1F Chemical compound NC(=O)c1cccnc1Oc1ccc(CC(=O)Nc2nc3ccc(Br)cc3n2CC2CC2)cc1F OFMWZNPAAYCXKA-UHFFFAOYSA-N 0.000 description 1
- HGKMGSSERGTPBS-UHFFFAOYSA-N NC1=NN2C(C=C(C=C2)O)=N1 Chemical compound NC1=NN2C(C=C(C=C2)O)=N1 HGKMGSSERGTPBS-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 1
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102100036547 Protein phosphatase 1 regulatory subunit 12A Human genes 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 206010062237 Renal impairment Diseases 0.000 description 1
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 description 1
- 102100039313 Rho-associated protein kinase 1 Human genes 0.000 description 1
- 102100039314 Rho-associated protein kinase 2 Human genes 0.000 description 1
- 102100027609 Rho-related GTP-binding protein RhoD Human genes 0.000 description 1
- 229910006124 SOCl2 Inorganic materials 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102000050488 Urotensin II Human genes 0.000 description 1
- 108010018369 Urotensin II Proteins 0.000 description 1
- 206010047295 Ventricular hypertrophy Diseases 0.000 description 1
- 239000012391 XPhos Pd G2 Substances 0.000 description 1
- IYOZTVGMEWJPKR-IJLUTSLNSA-N Y-27632 Chemical compound C1C[C@@H]([C@H](N)C)CC[C@@H]1C(=O)NC1=CC=NC=C1 IYOZTVGMEWJPKR-IJLUTSLNSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 1
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 159000000021 acetate salts Chemical class 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 208000037883 airway inflammation Diseases 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 125000006177 alkyl benzyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- XKMRRTOUMJRJIA-UHFFFAOYSA-N ammonia nh3 Chemical compound N.N XKMRRTOUMJRJIA-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229950006323 angiotensin ii Drugs 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- FCDPQMAOJARMTG-UHFFFAOYSA-L benzylidene-[1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichlororuthenium;tricyclohexylphosphane Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1.CC1=CC(C)=CC(C)=C1N(CCN1C=2C(=CC(C)=CC=2C)C)C1=[Ru](Cl)(Cl)=CC1=CC=CC=C1 FCDPQMAOJARMTG-UHFFFAOYSA-L 0.000 description 1
- FCDPQMAOJARMTG-UHFFFAOYSA-M benzylidene-[1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichlororuthenium;tricyclohexylphosphanium Chemical compound C1CCCCC1[PH+](C1CCCCC1)C1CCCCC1.CC1=CC(C)=CC(C)=C1N(CCN1C=2C(=CC(C)=CC=2C)C)C1=[Ru](Cl)(Cl)=CC1=CC=CC=C1 FCDPQMAOJARMTG-UHFFFAOYSA-M 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000011132 calcium sulphate Nutrition 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001735 carboxylic acids Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- ARQRPTNYUOLOGH-UHFFFAOYSA-N chcl3 chloroform Chemical compound ClC(Cl)Cl.ClC(Cl)Cl ARQRPTNYUOLOGH-UHFFFAOYSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910001850 copernicium Inorganic materials 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- XSYZCZPCBXYQTE-UHFFFAOYSA-N cyclodecylcyclodecane Chemical compound C1CCCCCCCCC1C1CCCCCCCCC1 XSYZCZPCBXYQTE-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 230000021953 cytokinesis Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 238000005695 dehalogenation reaction Methods 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- ASWXNYNXAOQCCD-UHFFFAOYSA-N dichloro(triphenyl)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(Cl)(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 ASWXNYNXAOQCCD-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- WQAWEUZTDVWTDB-UHFFFAOYSA-N dimethyl(oxo)phosphanium Chemical compound C[P+](C)=O WQAWEUZTDVWTDB-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 230000001856 erectile effect Effects 0.000 description 1
- OLAMWIPURJGSKE-UHFFFAOYSA-N et2o diethylether Chemical compound CCOCC.CCOCC OLAMWIPURJGSKE-UHFFFAOYSA-N 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- AWPMWZHWVKXADV-UHFFFAOYSA-N ethyl 2-(4-hydroxy-3-methoxyphenyl)acetate Chemical compound CCOC(=O)CC1=CC=C(O)C(OC)=C1 AWPMWZHWVKXADV-UHFFFAOYSA-N 0.000 description 1
- BDTDECDAHYOJRO-UHFFFAOYSA-N ethyl n-(sulfanylidenemethylidene)carbamate Chemical compound CCOC(=O)N=C=S BDTDECDAHYOJRO-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000004995 haloalkylthio group Chemical group 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 150000004680 hydrogen peroxides Chemical class 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 230000035874 hyperreactivity Effects 0.000 description 1
- 238000013299 hypertensive rat model Methods 0.000 description 1
- SKOWZLGOFVSKLB-UHFFFAOYSA-N hypodiboric acid Chemical compound OB(O)B(O)O SKOWZLGOFVSKLB-UHFFFAOYSA-N 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000005438 isoindazolyl group Chemical group 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- IWDLQINOGAKSJF-UHFFFAOYSA-N methyl 2-[4-(3-cyanopyridin-2-yl)oxyphenyl]acetate Chemical compound C1=CC(CC(=O)OC)=CC=C1OC1=NC=CC=C1C#N IWDLQINOGAKSJF-UHFFFAOYSA-N 0.000 description 1
- KZEFCECVTCHENL-UHFFFAOYSA-N methyl n-(4-chloropyridin-2-yl)carbamate Chemical compound COC(=O)NC1=CC(Cl)=CC=N1 KZEFCECVTCHENL-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 239000003658 microfiber Substances 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003506 n-propoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000008692 neointimal formation Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000007971 neurological deficit Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- 238000004958 nuclear spectroscopy Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 208000030613 peripheral artery disease Diseases 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000005954 phenoxathiinyl group Chemical group 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 125000001557 phthalyl group Chemical group C(=O)(O)C1=C(C(=O)*)C=CC=C1 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- LYKMMUBOEFYJQG-UHFFFAOYSA-N piperoxan Chemical compound C1OC2=CC=CC=C2OC1CN1CCCCC1 LYKMMUBOEFYJQG-UHFFFAOYSA-N 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000012041 precatalyst Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 238000009117 preventive therapy Methods 0.000 description 1
- 150000003140 primary amides Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 210000001243 pseudopodia Anatomy 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 230000016487 regulation of smooth muscle contraction Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 239000008299 semisolid dosage form Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 208000020431 spinal cord injury Diseases 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- PHCBRBWANGJMHS-UHFFFAOYSA-J tetrasodium;disulfate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O PHCBRBWANGJMHS-UHFFFAOYSA-J 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 150000003557 thiazoles Chemical class 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 125000005403 thiohaloalkoxy group Chemical group 0.000 description 1
- 150000003577 thiophenes Chemical class 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- HFNHAPQMXICKCF-USJMABIRSA-N urotensin-ii Chemical compound N([C@@H](CC(O)=O)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@@H](C(C)C)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@@H](N)CCC(O)=O)[C@@H](C)O HFNHAPQMXICKCF-USJMABIRSA-N 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 230000002227 vasoactive effect Effects 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Rheumatology (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Cardiology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
La presente invención proporciona compuestos de Fórmula (I): Fórmula (I) o estereoisómeros, tautómeros o sales farmacéuticamente aceptables de los mismos, en los que todas las variables son como se definen en el presente documento. Estos compuestos son inhibidores selectivos de ROCK. Esta invención también se refiere a composiciones farmacéuticas que comprenden estos compuestos y a métodos para tratar trastornos cardiovasculares, del músculo liso, oncológicos, neuropatológicos, autoinmunes, fibróticos y/o inflamatorios utilizando los mismos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Fenilacetamidas como inhibidores de ROCK
Campo de la invención
La presente invención se refiere en general a nuevas fenilacetamidas, que son inhibidores de Rho cinasas, composiciones que los contienen y su uso, por ejemplo, para el tratamiento o la profilaxis de trastornos asociados a la actividad aberrante de la Rho cinasa.
Antecedentes de la invención
La Rho-cinasa (ROCK) es un miembro de la familia de proteínas cinasas de serina-treonina. ROCK existe en dos isoformas, ROCK1 y ROCK2 (Ishizaki, T. et al., EMBO J., 15:1885-1893 (1996)). ROCK se ha identificado como una molécula efectora de RhoA, una pequeña proteína de unión a GTP (proteína G) que desempeña una función clave en múltiples rutas de señalización celular. ROCK y RhoA se expresan de forma ubicua a través de los tejidos. La ruta de señalización RhoA/ROCK está implicada en varias funciones celulares, tales como la organización ACTIN®, adhesión celular, migración celular y citocinesis (Riento, K. et al., Nat. Rev. Mol. Cell Biol., 4:446-456 (2003)). También está directamente implicada en la regulación de la contracción del músculo liso (Somlyo, A.P., Nature, 389:908-911 (1997)). Tras la activación de su receptor, RhoA se activa y, a su vez, activa a ROCK. La ROCK activada fosforila la subunidad de unión a miosina de la fosfatasa de cadena ligera de miosina, que inhibe la actividad de la fosfatasa y da lugar a la contracción. La contracción del músculo liso en la vasculatura aumenta la presión sanguínea, dando lugar a hipertensión.
En la bibliografía hay pruebas considerables de que la ruta de señalización Rho A/ROCK desempeña una función importante en la transducción de señales iniciada por varios factores vasoactivos, por ejemplo, la angiotensina II (Yamakawa, T. et al., Hypertension, 35:313-318 (2000)), urotensina II (Sauzeau, V. et al., Circ. Res., 88:1102-1104 (2001)), endotelina-1 (Tangkijvanich, P. et al., Hepatology, 33:74-80 (2001)), serotonina (Shimokawa, H., Jpn. Circ. J., 64:1-12 (2000)), noradrenalina (Martinez, M.C. et al., Am. J. Physiol., 279:H1228-H1238 (2000)) y el factor de crecimiento derivado de plaquetas (PGDF) (Kishi, H. et al., J. Biochem., 128:719-722 (2000)). Muchos de estos factores están implicados en la patogénesis de la enfermedad cardiovascular.
Estudios adicionales en la bibliografía, algunos usando los inhibidores de ROCK conocidos fasudil (Asano, T. et al., J. Pharmacol. Exp. Ther., 241:1033-1040 (1987)) o Y-27632 (Uehata, M. et al., Nature, 389:990-994 (1997)), ilustran además la conexión entre ROCK y la enfermedad cardiovascular. Por ejemplo, se ha mostrado que la expresión y la actividad de ROCK son elevadas en ratas espontáneamente hipertensas, lo que sugiere una conexión con el desarrollo de hipertensión en estos animales (Mukai, Y. et al., FASEB J., 15:1062-1064 (2001)). El inhibidor de ROCK Y-27632 (Uehata, M. et al., Nature, ibid.) se mostró que reducía significativamente la presión sanguínea en tres modelos de hipertensión de ratas, incluyendo los modelos de rata espontáneamente hipertensa, rata hipertensa por enfermedad renal y rata hipertensa por sal de acetato de desoxicortisona, aunque solo tenía un efecto leve sobre la presión sanguínea en ratas de control. Esto refuerza la conexión entre ROCK y la hipertensión.
Otros estudios sugieren una conexión entre ROCK y la ateroesclerosis. Por ejemplo, la transferencia génica de una forma dominante negativa de ROCK suprimió la formación de la neαntima después del daño por globo en arterias femorales de porcino (Eto, Y. et al., Am. J. Physiol. Heart Circ. Physiol., 278:H1744-H1750 (2000)). En un modelo similar, el inhibidor de ROCK Y-27632 también inhibió la formación de la neoíntima en ratas (Sawada, N. et al., Circulation, 101:2030-2033 (2000)). En un modelo porcino de estenosis coronaria inducida por IL-1 beta, se demostró que el tratamiento a largo plazo con el inhibidor de ROCK, fasudil, reducía progresivamente la estenosis coronaria, así como promovía una regresión del remodelado coronario constrictivo (Shimokawa, H. et al., Cardiovascular Res., 51:169-177 (2001)).
Las investigaciones adicionales sugieren que un inhibidor de ROCK sería útil en el tratamiento de otras enfermedades cardiovasculares. Por ejemplo, en un modelo de ictus en rata, se demostró que el fasudil reducía tanto la magnitud del infarto como el déficit neurológico (Toshima, Y., Stroke, 31:2245-2250 (2000)). Se demostró que el inhibidor de ROCK, Y-27632, mejoraba la hipertrofia ventricular, la fibrosis y la función en un modelo de insuficiencia cardíaca congestiva de ratas sensibles a la sal Dahl (Kobayashi, N. et al., Cardiovascular Res., 55:757-767 (2002)).
Otros estudios en animales o clínicos han implicado a ROCK en enfermedades adicionales, incluyendo el vasoespasmo coronario (Shimokawa, H. et al., Cardiovasc. Res., 43:1029-1039 (1999)), el vasoespasmo cerebral (Sato, M. et al., Circ. Res., 87:195-200 (2000)), lesión por isquemia/reperfusión (Yada, T. et al., J. Am. Coll. Cardiol., 45:599-607 (2005)), la hipertensión pulmonar (Fukumoto, Y. et al., Heart, 91:391-392 (2005)), la angina (Shimokawa, H. et al., J. Cardiovasc. Pharmacol., 39:319-327 (2002)), la nefropatía (Satoh, S. et al., Eur. J. Pharmacol., 455:169-174 (2002)) y la disfunción eréctil (Gonzalez-Cadavid, N.F. et al., Endocrine, 23:167-176 (2004)).
En otro estudio, se ha demostrado que la inhibición de la ruta de señalización RhoA/ROCK permite la formación de múltiples lamelipodios competidores que interrumpen la migración productiva de los monocitos (Worthylake, R.A. et
al., J. Biol. Chem., 278:13578-13584 (2003)). También se ha informado que los inhibidores de molécula pequeña de Rho cinasa son capaces de inhibir la quimiotaxis mediada por MCP-1 in vitro (Iijima, H., Bioorg. Med. Chem., 15:1022-1033 (2007)). Debido a la dependencia de la migración de las células inmunitarias de la ruta de señalización RhoA/ROCK se podría prever que la inhibición de la Rho cinasa también sería beneficiosa para enfermedades tales como la artritis reumatoide, la psoriasis y la enfermedad intestinal inflamatoria.
Los estudios anteriores proporcionan pruebas de una conexión entre ROCK y enfermedades cardiovasculares incluyendo hipertensión, ateroesclerosis, reestenosis, ictus, insuficiencia cardíaca, vasoespasmo coronario, vasoespasmo cerebral, lesión por isquemia/reperfusión, hipertensión pulmonar y angina de pecho, así como enfermedad renal y disfunción eréctil. Dado el efecto demostrado de ROCK sobre el músculo liso, los inhibidores de ROCK también pueden ser útiles en otras enfermedades que implican hiperreactividad del músculo liso, incluyendo asma y glaucoma (Shimokawa, H. et al., Arterioscler. Thromb. Vase. Biol., 25:1767-1775 (2005)). Además, la Rhocinasa se ha indicado como una diana farmacológica para el tratamiento de diversas otras enfermedades, incluyendo la inflamación y la hiperreactividad de las vías respiratorias (Henry, P.J. et al., Pulm. Pharmacol Ther., 18:67-74 (2005)), el cáncer (Rattan, R. et al., J. Neurosci. Res., 83:243-255 (2006); Lepley, D. et al., Cancer Res., 65:3788-3795 (2005)), las enfermedades fibróticas (Jiang, C. et al., Int. J. Mol. Sci., 13:8293-8307 (2012); Zhou, L. et al., Am. J. Nephrol., 34:468-475 (2011)), así como trastornos neurológicos, tales como lesión de la médula espinal, enfermedad de Alzheimer, esclerosis múltiple, ictus y dolor neuropático (Mueller, B.K. et al., Nat. Rev. Drug Disc., 4:387-398 (2005); Sun, X. et al., J. Neuroimmunol., 180:126-134 (2006)).
Sigue habiendo una necesidad médica no satisfecha de nuevos fármacos para tratar la enfermedad cardiovascular. En la actualización de 2012 de las Estadísticas sobre Cardiopatías e Ictus de la Asociación Americana del Corazón (Circulation, 125:e2-e220 (2012)), se informó que las enfermedades cardiovasculares representaban el 32,8% de todas las muertes en los Estados Unidos, representando la cardiopatía coronaria ~1 de cada 6 fallecimientos totales en los Estados Unidos. Contribuyendo a estos números, se descubrió que el ~33,5 % de la población adulta de los EE. UU era hipertensa y se estimó que en 2010 ~6,6 millones de adultos en EE. UU tendrían insuficiencia cardíaca. Por lo tanto, a pesar de la cantidad de medicamentos disponibles para tratar las enfermedades cardiovasculares (CVD, por sus siglas en inglés), incluyendo diuréticos, betabloqueantes, inhibidores de la enzima convertidora de angiotensina, bloqueantes de angiotensina y bloqueantes del canal de calcio, las CVD se mantienen poco controladas o resistentes a la medicación actual en muchos pacientes.
Hay muchos informes de inhibidores de ROCK en investigación (véanse, por ejemplo, los documentos US 2012/0122842 A1, US 2010/0041645 A1, US 2008/0161297 A1, y Hu, E. et al., Exp. Opin. Ther. Targets, 9:715-736 (2005)). Los informes también incluyen los documentos WO2014/113620, WO 2014/134388, WO 2014/134391, WO2015/002915, WO2015/002926, WO2016/010950, WO2016/028971, WO2016/112236 y WO2016/144936, todos los cuales están asignados al presente solicitante. Los documentos WO 2004/041813 y WO 2010/032875 divulgan derivados de tiazol y tiofeno disustituidos útiles como inhibidores de ROCK y compuestos de carboxamida heterocíclica. Sin embargo, el fasudil es el único inhibidor de ROCK comercializado en este momento. En Japón se aprobó una formulación i.v. para el tratamiento de vasoespasmo cerebral. Por lo tanto, sigue existiendo la necesidad de nuevos productos terapéuticos, incluyendo inhibidores de ROCK, para el tratamiento de enfermedades cardiovasculares, cáncer, enfermedades neurológicas, enfermedades renales, enfermedades fibróticas, asma bronquial, disfunción eréctil y glaucoma.
Sumario de la invención
La presente invención proporciona nuevas fenilacetamidas, estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, que son útiles como inhibidores selectivos de Rho cinasas.
La presente invención también proporciona procesos e intermedios para fabricar los compuestos de la presente invención.
La presente invención también proporciona composiciones farmacéuticas que comprenden un transportador farmacéuticamente aceptable y al menos uno de los compuestos de la presente invención o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos.
Los compuestos de la invención pueden usarse en el tratamiento y/o la profilaxis de afecciones asociadas a la actividad aberrante de ROCK.
Los compuestos de la presente invención pueden usarse en terapia.
Los compuestos de la presente invención se pueden usar para la fabricación de un medicamento para el tratamiento y/o la profilaxis de una afección asociada a la actividad aberrante de ROCK.
En otro aspecto, la presente invención se refiere a un compuesto de la presente invención como se ha descrito anteriormente para su uso en el tratamiento de una enfermedad cardiovascular o relacionada en un paciente que necesita dicho tratamiento. Los ejemplos de dichas enfermedades que pueden tratarse incluyen, por ejemplo,
hipertensión, ateroesclerosis, reestenosis, ictus, insuficiencia cardíaca, insuficiencia renal, arteriopatía coronaria, arteriopatía periférica, vasoespasmo coronario, vasoespasmo cerebral, lesión por isquemia/reperfusión, hipertensión pulmonar, angina de pecho, disfunción eréctil y enfermedad renal.
En otro aspecto, la presente invención se refiere a un compuesto de la presente invención, como se ha descrito anteriormente, para su uso en el tratamiento de enfermedades que implican hiperreactividad del músculo liso incluyendo asma, disfunción eréctil y glaucoma, en un paciente que necesita dicho tratamiento.
En otro aspecto, la presente invención se refiere a un compuesto de la presente invención, como se ha descrito anteriormente, para su uso en el tratamiento de enfermedades mediadas, al menos parcialmente, por Rho cinasa, incluyendo enfermedades fibróticas, oncológicas, lesión de la médula espinal, enfermedad de Alzheimer, esclerosis múltiple, ictus, dolor neuropático, artritis reumatoide, psoriasis y enfermedad inflamatoria intestinal, en un paciente que necesita dicho tratamiento.
En aún otros aspectos, la presente invención se refiere a composiciones farmacéuticas que comprenden los compuestos mencionados anteriormente, a procesos para preparar los compuestos e intermedios mencionados anteriormente usados en estos procesos.
Los compuestos de la invención pueden usarse en solitario, en combinación con otros compuestos de la presente invención, o en combinación con uno o más, preferentemente de uno a dos agentes distintos.
Estas y otras características de la invención se expondrán ampliamente conforme continúe la divulgación.
Descripción detallada de la invención
I. COMPUESTOS DE LA INVENCIÓN
En un aspecto, la presente invención proporciona compuestos de Fórmula (II):

o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos,
en donde
R2 se selecciona independientemente entre halógeno y alquilo C-i-a;
R5 es un heterociclo de 4-15 miembros que comprende átomos de carbono y 1-4 heteroátomos seleccionados entre N, NR8, O y S(O)p, en donde dicho heterociclo está sustituido con 1-4 R7;
R7, en cada caso, se selecciona independientemente de H, =O, NO2, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2V C O 2H, -(CH2)n-CO2(alquilo C1.4), -(CH2)n-NR8R8, -NHCO(alquilo C1.4), -NHCOCF3, -NHCO2(alquilo C1 4), -NHCO2(CH2)2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2)2OH, -NHCO2(CH2)2NH2, -NH-CO2(CH2)2N(alquilo C1-4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1-4), -NHC(O)NR8R8, -NHSO2(alquilo C1-4), -SO2NH2, -SO2NH(alquilo C1-4), -SO2N(alquilo C1-4)2, -SO2NH(CH2)2OH, -SO2NH(CH2)2O(alquilo C1-4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, -NHCO-carbociclo, -NHCO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dicho alquilo, alquenilo, alquinilo, alcoxi, carbociclo y heterociclo están sustituidos con 0-4 R9; R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, C(O)alquilo C1-4, -(CH2)nC(O)NRaRa, C(O)O-alquilo, SO2alquilo, SO2NRaRa, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dicho alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO2H, CO2(alquilo C1.4), CONH2, -(CH2)nNRaRa, -(CH2)nCONRaRa, -O(CH2)nheterociclo, -O(CH2)(2-4)NRa-Ra, -(CH2)n-heterociclo de 4-10 miembros, en donde dicho alquilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 Rb;
Ra, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)nOH, CO(alquilo C1-4), COCF3,
CO2(alquilo C1-4), -CONH2, -CONH-alquilen C i-4-CO2(alquilo C1-4), alquilen C i-4-CO2(alquilo C1-4), Rc, CONHRc; como alternativa, Ra y Ra se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros, en donde dicho alquilo, alquileno y heterociclo están sustituidos por 0-4 Rb;
Rb, en cada caso, se selecciona independientemente de =O, halógeno, alquilo C1-4, alcoxi C1-4, OCF3, NH2, NO2, N(alquilo C1-4)2, CO(alquilo C1-4), CO(haloalquilo C1-4), CO2(alquilo C1-4), CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, -CONH-alquilen C1-4-O(alquilo C1.4), -CONH-alquilen C1-4-N(alquilo C1-4)2, -CONH-alquilen C1-4-N(alquilo C1. 4)2, -alquilen C1-4-O-P(O)(OH)2, -NHCO2(alquilo C1.4), -Rc, CORc, CO2Rc y CONHRc;
Rc, en cada caso, se selecciona independientemente de -(CH2)n-cicloalquilo C3-6, -(CH2)n-fenilo y -(CH2)nheterociclo de 5 a 6 miembros que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p; en donde cada resto del anillo está sustituido por 0-2 Rd;
Rd, en cada caso, se selecciona independientemente de =O, halógeno, -OH, alquilo C1-4, NH2, NH(alquilo C1.4), N(alquilo C1-4)2, alcoxi C1.4 y -NHCO(alquilo C1.4) y heterociclo que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p;
n, en cada caso, se selecciona independientemente de 0, 1, 2, 3 y 4;
p, en cada caso, se selecciona independientemente de 0, 1 y 2.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (II) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona independientemente entre F, Cl y Br;
R5 se selecciona entre

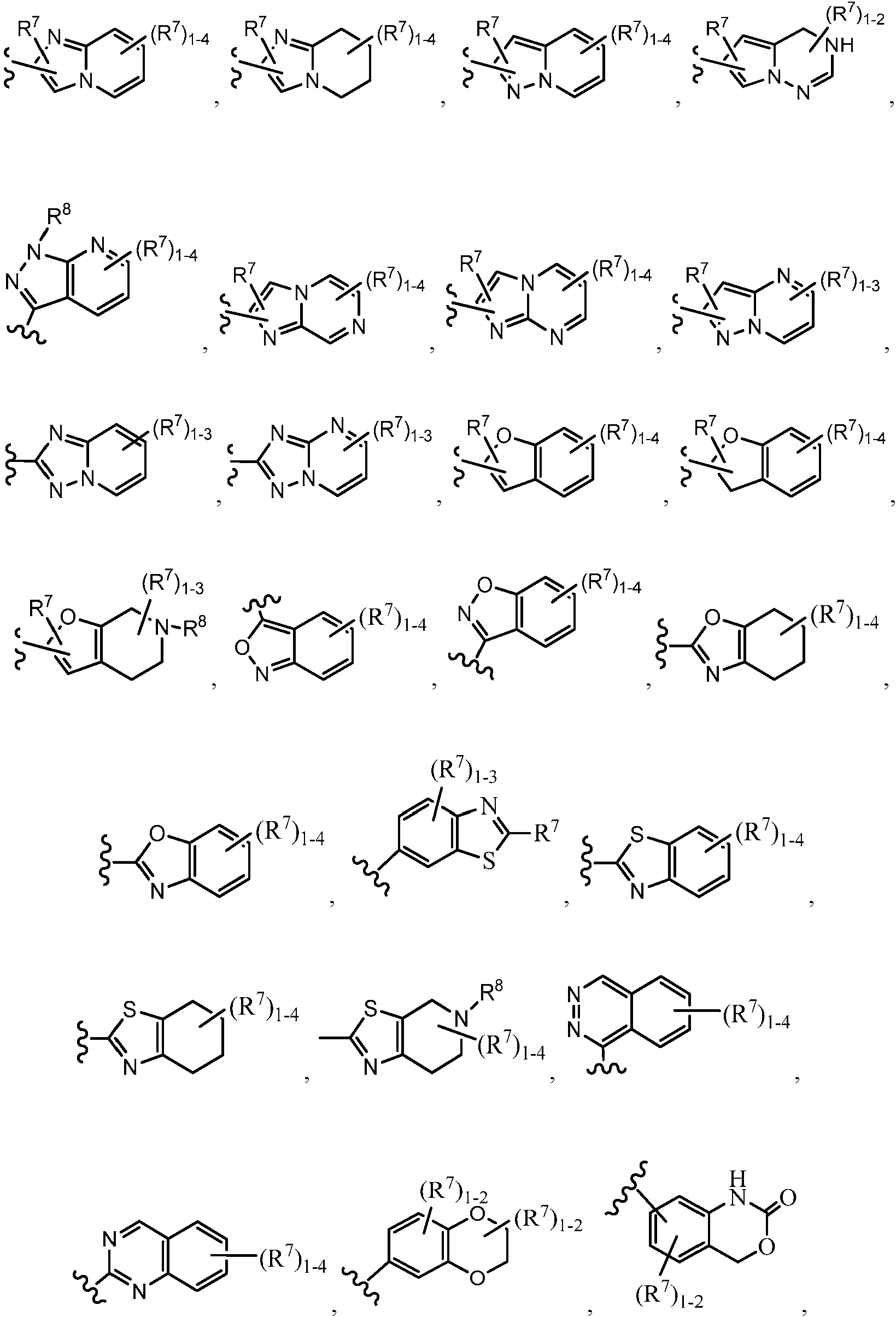

R7, en cada caso, se selecciona independientemente de H, =O, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2)n-CO2H, -(CH2)n-CO2(alquilo C1-4), -(CH2)n-NR8R8, -NHCO(alquilo C1-4), -NHCOCF3, -NHCO2(alquilo C1-4), -NHCO2(CH2)2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2)2OH, -NHCO2(CH2)2NH2, -NHCO2(CH2)2N(alquilo ^ .4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1.4), -NHC(O)NR8R8, -NHSO2(alquilo C1.4), -SO2NH2, -SO2NH(alquilo C1-4), -SO2N(alquilo C1-4)2, -SO2NH(CH2)2OH, -SO2NH(CH2)2O(alquilo C1-4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, -NHCO-carbociclo, -NHCO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dicho alquilo, alquenilo, alquinilo, alcoxi, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dicho alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO2H, CO2(alquilo C ^ ), CONH2, -(CH2)nNH2, -(CH2)nCONH2, -O(CH2)nheterociclo, -O(CH2)(2-4)NH2, -(CH2)n-heterociclo de 4-10 miembros; y
otras variables son como se han definido en la Fórmula (II) anterior.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (II) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona independientemente entre F y Cl;
R5 se selecciona entre

R7, en cada caso, se selecciona independientemente de H, halógeno, alquilo C1-4, alcoxi C1-4, un carbociclo y un heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dicho alquilo, alquenilo, alquinilo, alcoxi, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4 y -(CH2)n-cicloalquilo C3-6;
R9, en cada caso, se selecciona independientemente de halógeno, OH, CHF2, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO2H, CO2(alquilo C1-4) y CONH2; y
otras variables son como se han definido en la Fórmula (II) anterior.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (II) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R7, en cada caso, se selecciona independientemente de H, F, Cl, Br, Me, Et, OMe, OEt, OCH2C(CH2)2OH,

R9, en cada caso, se selecciona independientemente de F, Cl, Br, OH, CHF2, CF3, alquilo C1-4 y alcoxi C1-4; y otras variables son como se definen en la Fórmula (II) anterior.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (III):

o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona entre F, Cl, Br y Oalquilo C1-4;
R4 se selecciona entre H y alquilo C1-4;
R5 se selecciona entre

y

R7, en cada caso, se selecciona independientemente de H, =O, NO2, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2)n-CO(alquilo C1-4), -(CH2)n-CO2H, -(CH2)n-CO2(alquilo C1-4), -(CH2)n-NR8R8, -NHCO(alquilo C1-4), -NHCOCF3, -NHCO2(alquilo C1.4), -NHCO2(CH2)2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2)2OH, -NHCO2(CH2)2NH2, -NHCO2(CH2)2N(alquilo C1-4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1-4), -NHC(O)NR8R8, -NHSO2(alquilo C1.4), -P(=O)(alquilo C1-3)2, -SO2alquilo C1.4, -SO2NH2, -SO2NH(alquilo C1.4), -SO2N(alquilo C1-4)2, -SO2NH(CH2)2OH, -SO2NH(CH2)2O(alquilo C1.4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, -NHCO-carbociclo, -NHCO-heterociclo, -CO-carbociclo, -CO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dicho alquilo, alquenilo, alquinilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, -(CH2)n-C(O)alquilo C1.4, -(CH2)n-C(O)carbociclo, -(CH2)n-C(O)heterociclo, -(CH2)n-C(O)NRaRa, -(CH2)n-C(O)O-alquilo, -(CH2)n-C(O)O-carbociclo, -(CH2)n-C(O)O-heterociclo, -(CH2)n-SO2alquilo, -(CH2)n-SO2carbociclo, -(CH2)n-SO2heterociclo, -(CH2)n-SO2NRaRa, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dicho alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
como alternativa, R8 y R8 se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros sustituido con 0-4 R9;
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CN, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO(alquilo C1-4), CO2H, CO2(alquilo C1-4), -(CH2)nNRaRa, -(CH2)nCONRaRa, -O(CH2)ncarbociclo, -O(CH2)nheterociclo, -O(CH2)nNRaRa, NRaCO2(alquilo C1.4), S(O)palquilo C1.4, -(CR10R10)n-carbociclo, -(CR10R10)nheterociclo de 4-10 miembros, en donde dicho alquilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 Rb; R10 se selecciona entre H y alquilo C1-4;
Ra, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)nOH, CO(alquilo C1.4), COCF3, CO2(alquilo C1.4), -CONH2, -CONH-alquilen C1-4-CO2(alquilo C1.4), alquilen C1-4-CO2(alquilo C1.4), Rc, CO2Rc y CONHRc; como alternativa, Ra y Ra se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros, en donde dicho alquilo, alquileno y heterociclo están sustituidos por 0-4 Rb; Rb, en cada caso, se selecciona independientemente de =O, OH, halógeno, alquilo C1-4, alcoxi C1-4, OCF3, NH2, NO2, N(alquilo C1-4)2, CO(alquilo C1-4), CO(haloalquilo C1.4), CO2(alquilo C1.4), CONH2, -CONH(alquilo C1.4), -CON(alquilo ^ -4)2, -CONH-alquilen C1-4-O(alquilo C1.4), -CONH-alquilen C1-4-N(alquilo C1-4)2, -CONH-alquilen C1.
4-N(alquilo ^ .4)2, -alquilen C1-4-O-P(O)(OH)2, -NHCO2(alquilo C1.4), -Rc, CORc, CO2Rc y CONHRc;
Rc, en cada caso, se selecciona independientemente de -(CH2)n-cicloalquilo C3-6, -(CH2)n-fenilo y -(CH2)nheterociclo de 5 a 6 miembros que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p; en donde cada resto del anillo está sustituido por 0-2 Rd;
Rd, en cada caso, se selecciona independientemente de =O, halógeno, -OH, alquilo C1-4, NH2, NH(alquilo C1.4), N(alquilo C1-4)2, alcoxi C1-4 y -NHCO(alquilo C1.4) y heterociclo que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p;
n, en cada caso, se selecciona independientemente de 0, 1,2, 3 y 4; y
p, en cada caso, se selecciona independientemente de 0, 1 y 2.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (III) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona entre F, Cl y OCH3;
R5 se selecciona entre
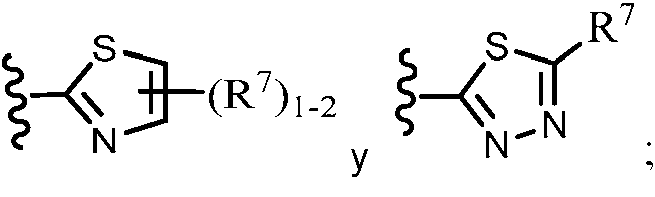
R7 se selecciona entre H, alquilo C1-3 sustituido con halógeno,

y 
R8 se selecciona entre H y alquilo C1-3 opcionalmente sustituido con halógeno;
R9 se selecciona entre F, Cl, Cn , alquilo C1-4 sustituido con 0-2 Rb, alcoxi C1-3, cicloalquilo C3-6, NHCO2(alquilo C1-4), SO2alquilo C1-3 y

y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (III) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona independientemente entre F, Cl y OCH3;
R5 se selecciona entre

R7 se selecciona entre H, F, Cl, Br, alquilo C1-3 sustituido con 0-4 R9, alcoxi C1-3 sustituido con 0-4 R9, CN, CO2alquilo C1-4, P(=O)(alquilo C1-3)2, SO2alquilo C1-4,

R8 se selecciona entre H y alquilo C1-3 sustituido con 0-4 R9;
R9 se selecciona entre halógeno, OH, CN, alquilo C1-3 sustituido con 0-4 Rb, alcoxi C1-3 sustituido con 0-4 Rb CO2(alquilo C1.4), CONH2, NHCO2(alquilo C1.3), cicloalquilo C3-6, fenilo y heterociclo; y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (III) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona independientemente entre F, Cl y OCH3;
R5 se selecciona entre,

y

R7 se selecciona entre H, CN, alquilo C1-3 sustituido con 0-4 R9, alcoxi C1-3 sustituido con 0-4 R9, CO2alquilo C1-4, CONH2, P(=O)(alquilo C1-3)2, SO2alquilo C1.3,

R8 se selecciona entre H y alquilo C1-3 sustituido con 0-4 R9;
R9 se selecciona entre halógeno, OH, CN, alquilo C1-3 sustituido con 0-4 Rb, alcoxi C1-3 sustituido con 0-4 Rb, CO2(alquilo C1.4), CONH2, NHCO2(alquilo C1.3), cicloalquilo C3-6, fenilo y heterociclo; y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
En otro aspecto, la presente invención proporciona compuestos de Fórmula (III) o estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos, en donde
R2 se selecciona entre F, Cl y OCH3;
R5 es

R7 se selecciona entre H, F, Cl, Br, CN, alquilo C1-3 sustituido con 0-4 R9, alcoxi C1-3 sustituido con 0-4 R9, CO2alquilo C1-4, CONH2, P(=O)(alquilo C1-3)2, S o 2alquilo C1-3y

R8 se selecciona entre H y alquilo C1-3 sustituido con 0-4 R9;
R9 se selecciona entre halógeno, OH, CN, alquilo C1-3 sustituido con 0-4 Rb, alcoxi C1-3 sustituido con 0-4 Rb, CO2(alquilo C1.4), CONH2, NHCO2(alquilo C1.3), cicloalquilo C3-6, fenilo y heterociclo; y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
La invención puede realizarse de otras formas específicas. Esta invención también incluye todas las combinaciones de aspectos alternativos de la invención indicados en el presente documento. Se entiende que cualquiera y todas las realizaciones de la presente invención se pueden tomar en combinación con cualquier otra realización para describir realizaciones adicionales de la presente invención. Además, se pretende que cualquier elemento (incluyendo definiciones individuales variables) de una realización se combine con cualquiera y todos los demás elementos de cualquiera de las realizaciones para describir realizaciones adicionales.
Por ejemplo, en una realización no limitante de Fórmula (II), R5 es

R7 se selecciona entre H y alcoxi C1-4 opcionalmente sustituido con OH.
En otra realización no limitante de Fórmula (II), R5 es

R7 se selecciona entre H y alcoxi C1-4 opcionalmente sustituido con OH

y

En otra realización no limitante de Fórmula (II), R5 es

En otra realización no limitante de Fórmula (II),

R7 se selecciona entre H y alcoxi C1-4 opcionalmente sustituido con OH.
En otra realización no limitante de Fórmula (II), R5 es
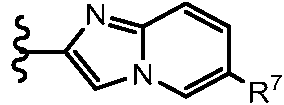
R7 se selecciona entre H y halógeno.
En otra realización no limitante de Fórmula (II), R5 es
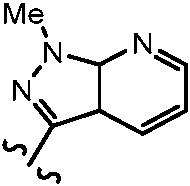
En otra realización no limitante de Fórmula (II), R5 es

R7 se selecciona entre H, alquilo C1-4,

y R9 es halógeno.
En otra realización no limitante de Fórmula (III), R5 es

un R7 es

el otro es cicloalquilo C3-6.
En otra realización no limitante de Fórmula (III), R5 es

un R7 es CH3, el otro es

R8 es CH3.
En otra realización no limitante de Fórmula (III), R5 es

R7 se selecciona entre F, CF3 y

R8 se selecciona entre CH3, CHF2 y CH2-ciclopropilo.
En otra realización no limitante de Fórmula (III), R5 es
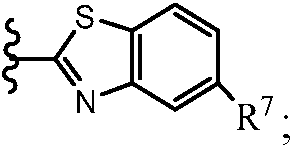
R7 es

En otra realización no limitante de Fórmula (III), R5 es

R7 es

R8 se selecciona entre H, CH3, CD3 y CHF2; R9 se selecciona entre CH3, CF3 y ciclopropilo.
En otra realización no limitante de Fórmula (III), R5 es

R7 es alcoxi C1-4 opcionalmente sustituido con OH.
En otra realización no limitante de Fórmula (III), R2 se selecciona entre F y Cl R5 es

R7 es alcoxi C1-4 opcionalmente sustituido con OH.
En otra realización no limitante de Fórmula (III), R2 se selecciona entre F, Cl y Br; R5 se selecciona entre

y
R7 es alcoxi C1-4 opcionalmente sustituido con fenilo.
En otra realización no limitante de Fórmula (III), R2 es Cl; R5 es

R7 es alcoxi C1-4.
En otro aspecto, la presente invención proporciona un compuesto seleccionado de cualquier subconjunto de la lista de compuestos ilustrados en la presente solicitud.
En otra realización, los compuestos de la presente invención tienen valores de CI50 de ROCK <10 μM.
En otra realización, los compuestos de la presente invención tienen valores de CI50 de ROCK <1 μM.
En otra realización, los compuestos de la presente invención tienen valores de CI50 de ROCK <0,1 μM.
En otra realización, los compuestos de la presente invención tienen valores de CI50 de ROCK <0,05 μM.
En otra realización, los compuestos de la presente invención tienen valores de CI50 de ROCK <0,01 μM.
II. OTRAS REALIZACIONES DE LA INVENCION
En otra realización, la presente invención proporciona una composición que comprende al menos uno de los compuestos de la presente invención o un estereoisómero, un tautómero, una sal farmacéuticamente aceptable o un solvato de los mismos.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende un transportador farmacéuticamente aceptable y al menos uno de los compuestos de la presente invención o un estereoisómero, un tautómero, una sal farmacéuticamente aceptable o un solvato, de los mismos.
En otra realización, la presente invención proporciona una composición farmacéutica, que comprende: un
transportador farmacéuticamente aceptable y una cantidad terapéuticamente eficaz de al menos uno de los compuestos de la presente invención o un estereoisómero, un tautómero, una sal farmacéuticamente aceptable o un solvato de los mismos.
En otra realización, la presente invención proporciona un proceso para fabricar un compuesto de la presente invención.
En otra realización, la presente invención proporciona un intermedio para elaborar un compuesto de la presente invención.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende además uno o más agentes terapéuticos adicionales.
En otra realización, la presente invención proporciona al menos uno de los compuestos de la presente invención o un estereoisómero, un tautómero, una sal farmacéuticamente aceptable o un solvato del mismo, para su uso en el tratamiento y/o profilaxis de una afección asociada a la actividad aberrante de ROCK en un paciente que necesita dicho tratamiento y/o profilaxis. Como se usa en el presente documento, el término "paciente" abarca todas las especies de mamíferos.
Como se usa en el presente documento, "tratar" o "tratamiento" incluyen el tratamiento de una patología en un mamífero, particularmente en un ser humano, e incluyen: (a) inhibir la patología, es decir, detener su desarrollo; y/o (b) aliviar la patología, es decir, provocar la regresión de la patología.
Como se usa en el presente documento, "profilaxis" o "prevención" incluyen el tratamiento preventivo de una patología subclínica en un mamífero, particularmente en un ser humano, con el objetivo de reducir la probabilidad de la aparición de una patología clínica. Los pacientes se seleccionan para la terapia preventiva basándose en factores que se sabe que aumentan el riesgo de padecer una patología clínica en comparación con la población general. Las terapias "profilácticas" pueden dividirse en (a) prevención primaria y (b) prevención secundaria. La prevención primaria se define como el tratamiento en un paciente que todavía no ha presentado una patología clínica, mientras que la prevención secundaria se define como la prevención de una segunda aparición de la misma patología clínica, o una similar. En otra realización, la presente invención proporciona una preparación combinada de un compuesto de la presente invención y uno o más agentes terapéuticos adicionales para su uso simultáneo, por separado o secuencial en terapia.
La presente invención puede realizarse de otras formas específicas. Esta invención abarca todas las combinaciones de los aspectos preferidos de la invención indicados en el presente documento. Se entiende que cualquiera y todas las realizaciones de la presente invención pueden tomarse junto con cualquier otra realización o realizaciones para describir realizaciones adicionales. También ha de entenderse que cada elemento individual de las realizaciones es su propia realización independiente. Además, se pretende que cualquier elemento de una realización se combine con cualquiera y todos los demás elementos de cualquier realización para describir una realización adicional.
III. QUÍMICA
A lo largo de la presente memoria descriptiva y las reivindicaciones adjuntas, una fórmula o nombre químico dado puede abarcar todos los estereoisómeros e isómeros ópticos y los racematos de los mismos cuando existan dichos isómeros. A menos que se indique de otro modo, todas las formas quirales (enantioméricas y diastereoméricas) y racémicas están dentro del alcance de la presente invención. Muchos isómeros geométricos de dobles enlaces C=C, dobles enlaces C=N, sistemas anulares y similares también pueden estar presentes en los compuestos y todos estos isómeros estables se contemplan en la presente invención. Se describen los isómeros geométricos cis y trans (o E y Z) de los compuestos de la presente invención y se pueden aislar en forma de una mezcla de isómeros o como formas isoméricas separadas. Los presentes compuestos pueden aislarse en formas ópticamente activas o racémicas. Las formas ópticamente activas pueden prepararse por resolución de formas racémicas o por síntesis de materiales de partida ópticamente activos. Se considera que todos los procesos usados para preparar los compuestos de la presente invención y los intermedios elaborados con los mismos forman parte de la presente invención. Cuando se preparan productos enantioméricos o diastereoméricos, pueden separarse por métodos convencionales, por ejemplo, por cromatografía o cristalización fraccionada. Dependiendo de las condiciones del proceso, los productos finales de la presente invención se obtienen en forma libre (neutra) o de sal. Tanto la forma libre como las sales de estos productos finales están dentro del alcance de la invención. Si así se desea, puede convertirse una forma de un compuesto en otra forma. Puede convertirse una base o un ácido libres en una sal; puede convertirse una sal en el compuesto libre u otra sal; puede separarse una mezcla de compuestos isoméricos de la presente invención en los isómeros individuales. Los compuestos de la presente invención, la forma libre y las sales de los mismos, pueden existir en múltiples formas tautoméricas, en las que los átomos de hidrógeno se transponen a otras partes de las moléculas y, por consiguiente, se reordenan los enlaces químicos entre los átomos de las moléculas. Debe entenderse que todas las formas tautoméricas, en la medida en que puedan existir, se incluyen dentro de la invención.
El término "estereoisómero" se refiere a isómeros de constitución idéntica que difieren en la disposición espacial de sus átomos. Los enantiómeros y diastereómeros son ejemplos de estereoisómeros. El término "enantiómero" se refiere
a uno de un par de especies moleculares que son imágenes especulares entre sí y no son superponibles. El término "diastereómero" se refiere a estereoisómeros que no son imágenes especulares. El término "racemato" o la expresión "mezcla racémica" se refieren a una composición compuesta por cantidades equimolares de dos especies enantioméricas, en donde la composición está desprovista de actividad óptica.
Los símbolos "R" y "S" representan la configuración de los sustituyentes en torno a uno o más átomos de carbono quirales. Los descriptores isoméricos "R" y "S" se usan como se describe en el presente documento para indicar una o más configuraciones de átomos con respecto a una molécula central y se pretende que se usen como se define en la bibliografía (IUPAC Recommendations 1996, Pure and Applied Chemistry, 68:2193-2222 (1996)).
El término "quiral" se refiere a la característica estructural de una molécula que hace imposible que se superponga sobre su imagen especular. El término "homoquiral" se refiere a un estado de pureza enantiomérica. La expresión "actividad óptica" se refiere al grado en que una molécula homoquiral o una mezcla no racémica de moléculas quirales rota un plano de luz polarizada.
Como se usa en el presente documento, el término "alquilo" o "alquileno" pretende incluir grupos hidrocarburo alifáticos saturados de cadena tanto ramificada como lineal que tienen el número especificado de átomos de carbono. Por ejemplo, "alquilo C1 a C10" o "alquilo C1-10" (o alquileno), pretende incluir los grupos alquilo C1, C2, C3, C4, C5, Ce, C7,
Ce, Cg y C10. Además, por ejemplo, "alquilo C1 a Ce" o "alquilo C1-C6" representa alquilo que tiene de 1 a 6 átomos de carbono. El grupo alquilo puede estar sin sustituir o sustituido por al menos un hidrógeno que está reemplazado por otro grupo químico. Los ejemplos de grupos alquilo incluyen, pero sin limitación, metilo (Me), etilo (Et), propilo (por ejemplo, n-propilo e isopropilo), butilo (por ejemplo, n-butilo, isobutilo, f-butilo) y pentilo (por ejemplo, n-pentilo, isopentilo, neopentilo). Cuando se usa "alquilo Co" o "alquileno Co", se pretende indicar un enlace directo.
"Alquenilo" o "alquenileno" pretende incluir cadenas de hidrocarburo tanto de configuración lineal como ramificada que tienen el número especificado de átomos de carbono y uno o más, preferentemente de uno a dos, dobles enlaces carbono-carbono que pueden aparecer en cualquier punto estable a lo largo de la cadena. Por ejemplo, "alquenilo C2 a C6" o "alquenilo C2-6" (o alquenileno), pretende incluir los grupos alquenilo C2, C3, C4, C5 y C6. alquenilo incluyen, pero sin limitación, etenilo, 1-propenilo, 2-propenilo, 2-butenilo, 3-butenilo, 2-pentenilo, 3-pentenilo,
4-pentenilo, 2-hexenilo, 3-hexenilo, 4-hexenilo, 5-hexenilo, 2-metil-2-propenilo y 4-metil-3-pentenilo.
"Alquinilo" o "alquinileno" pretende incluir cadenas de hidrocarburo de configuración tanto lineal como ramificada que tienen uno o más, preferentemente de uno a tres, triples enlaces carbono-carbono que pueden aparecer en cualquier punto estable a lo largo de la cadena. Por ejemplo, "alquinilo C2 a C6" o "alquinilo C2-6" (o alquinileno), pretende incluir los grupos alquinilo C2, C3, C4, C5 y C6; tales como etinilo, propinilo, butinilo, pentinilo y hexinilo.
El término "alcoxi" o "alquiloxi" se refiere a un grupo -O-alquilo. "Alcoxi C1 a C6" o "alcoxi C1-6" (o alquiloxi), pretende incluir los grupos alcoxi C1, C2, C3, C4, C5 y C6. Los ejemplos de grupos alcoxi incluyen, pero sin limitación, metoxi, etoxi, propoxi (por ejemplo, n-propoxi e isopropoxi) y f-butoxi. De manera similar, "alquiltio" o "tioalcoxi" representa un grupo alquilo como se ha definido anteriormente con el número de átomos de carbono indicado unidos a través de un puente de azufre; por ejemplo, metil-S- y etil-S-.
"Halo" o "halógeno" incluye flúor (F), cloro (Cl), bromo (Br) y yodo (I). "Haloalquilo" pretende incluir grupos hidrocarburo alifáticos saturados tanto de cadena ramificada como lineal que tienen el número especificado de átomos de carbono, sustituidos con 1 o más halógenos. Los ejemplos de haloalquilo incluyen, pero sin limitación, fluorometilo, difluorometilo, trifluorometilo, triclorometilo, pentafluoroetilo, pentacloroetilo, 2,2,2-trifluoroetilo, heptafluoropropilo y heptacloropropilo. Los ejemplos de haloalquilo también incluyen "fluoroalquilo" que pretende incluir grupos hidrocarburo alifáticos saturados tanto de cadena ramificada como lineal que tienen el número de átomos de carbono especificado, sustituidos por 1 o más átomos de flúor.
"Haloalcoxi" o "haloalquiloxi" representa un grupo haloalquilo como se ha definido anteriormente con el número indicado de átomos de carbono, unido a través de un puente de oxígeno. Por ejemplo, "haloalcoxi C1 a C6" o "haloalcoxi
C1-6", pretende incluir los grupos haloalcoxi C1, C2, C3, C4, C5 y C6. Los ejemplos de haloalcoxi incluyen, pero sin limitación, trifluorometoxi, 2,2,2-trifluoroetoxi y pentafluoroetoxi. De manera similar, "haloalquiltio" o "tiohaloalcoxi" representa un grupo haloalquilo como se ha definido anteriormente con el número indicado de átomos de carbono unido a través de un puente de azufre; por ejemplo, trifluorometil-S- y pentafluoroetil-S-.
El término "cicloalquilo" se refiere a grupos alquilo ciclados, incluyendo sistemas anulares monocíclicos, bicíclicos o policíclicos. "Cicloalquilo C3 a C7" o "cicloalquilo C3-7" pretenden incluir grupos cicloalquilo C3, C4, C5, C6 y C7. Los ejemplos de grupos cicloalquilo incluyen, pero sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y norbornilo. Se incluyen en la definición de "cicloalquilo" los grupos cicloalquilo ramificados tales como 1-metilciclopropilo y 2-metilciclopropilo.
Como se usa en el presente documento, "carbociclo" o "residuo carbocíclico" pretende indicar cualquier anillo hidrocarburo estable, monocíclico o bicíclico de 3, 4, 5, 6, 7 u 8 miembros o bicíclico o tricíclico de 7, 8, 9, 10, 11, 12 o 13 miembros, cualquiera de los cuales puede estar saturado, parcialmente insaturado, insaturado o ser aromático.
Los ejemplos de tales carbociclos incluyen, pero sin limitación, ciclopropilo, ciclobutilo, ciclobutenilo, ciclopentilo, ciclopentenilo, ciclohexilo, cicloheptenilo, cicloheptilo, cicloheptenilo, adamantilo, ciclooctilo, ciclooctenilo, ciclooctadienilo, [3.3.0]biciclooctano, [4.3.0]biciclononano, [4.4.0]biciclodecano (decalina), [2.2.2]biciclooctano, fluorenilo, fenilo, naftilo, indanilo, adamantilo, antracenilo y tetrahidronaftilo (tetralina). Como se ha mostrado anteriormente, los anillos puenteados también están incluidos en la definición de carbociclo (por ejemplo, [2.2.2]biciclooctano). Los carbociclos preferidos, a menos que se especifique de otro modo, son ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, fenilo e indanilo. Cuando se usa el término "carbociclo", se pretende incluir "arilo". Un anillo puenteado se produce cuando uno o más átomos de carbono conectan dos átomos de carbono no adyacentes. Los puentes preferidos son uno o dos átomos de carbono. Cabe apreciar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes citados para el anillo también pueden estar presentes en el puente.
Como se usa en el presente documento, la expresión "carbociclo bicíclico" o "grupo carbocíclico bicíclico" pretende indicar un sistema anular carbocíclico estable de 9 o 10 miembros que contiene dos anillos condensados y consiste en átomos de carbono. De los dos anillos condensados, un anillo es un anillo benzo condensado a un segundo anillo; y el segundo anillo es un anillo de carbono de 5 o 6 miembros que está saturado, parcialmente insaturado o insaturado. El grupo carbocíclico bicíclico puede estar unido a su grupo colgante en cualquier átomo de carbono que dé como resultado una estructura estable. El grupo carbocíclico bicíclico descrito en el presente documento puede estar sustituido en cualquier carbono si el compuesto resultante es estable. Los ejemplos de un grupo carbocíclico bicíclico son, pero sin limitación, naftilo, 1,2-dihidronaftilo, 1,2,3,4-tetrahidronaftilo e indanilo.
Los grupos "arilo" se refieren a hidrocarburos aromáticos monocíclicos o policíclicos, incluyendo, por ejemplo, fenilo, naftilo y fenantranilo. Los restos de arilo se conocen bien y se describen, por ejemplo, en Lewis, R. J., ed., Hawley's Condensed Chemical Dictionary, 13a Edición, John Wiley & Sons, Inc., Nueva York (1997). "Arilo C6 o C10" o "arilo C6-10" se refiere a fenilo y naftilo. A menos que se especifique de otro modo, "arilo", "arilo C6 o C10" o "arilo C6-10" o "residuo aromático" pueden estar sin sustituir o sustituidos con de 1 a 5 grupos, preferentemente de 1 a 3 grupos, OH, OCH3, Cl, F, Br, I, CN, NO2, NH2, N(CH3)H, N(CH3)2, CF3, OCF3, C(=O)CH3, SCH3, S(=O)CH3, S(=O)2CH3, CH3, CH2CH3, CO2H y CO2CH3.
El término "bencilo", como se usa en el presente documento, se refiere a un grupo metilo en el que uno de los átomos de hidrógeno se reemplaza por un grupo fenilo, en donde dicho grupo fenilo puede estar opcionalmente sustituido por de 1 a 5 grupos, preferentemente de 1 a 3 grupos, OH, OCH3, Cl, F, Br, I, CN, NO2, NH2, N(CH3)H, N(CH3)2, CF3, OCF3, C(=O)CH3, SCH3, S(=O)CH3, S(=O)2CH3, CH3, CH2CH3, CO2H y CO2CH3.
Como se usa en el presente documento, la expresión "heterociclo" o "grupo heterocíclico" pretende indicar un anillo heterocíclico estable, monocíclico o bicíclico de 3, 4, 5, 6 o 7 miembros o policíclico de 7, 8, 9, 10, 11, 12, 13 o 14 miembros, que está saturado, parcialmente insaturado o totalmente insaturado y que contiene átomos de carbono y 1, 2, 3 o 4 heteroátomos seleccionados independientemente del grupo que consiste en N, O y S; y que incluye cualquier grupo policíclico en el que cualquiera de los anillos heterocíclicos definidos anteriormente está condensado con un anillo de benceno. Opcionalmente, los heteroátomos de nitrógeno y azufre pueden estar oxidados (es decir, N ^O y S(O)p, en donde p es 0, 1 o 2). El átomo de nitrógeno puede estar sustituido o sin sustituir (es decir, N o NR, en donde R es H u otro sustituyente, si se define). El anillo heterocíclico puede estar unido a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. Los anillos heterocíclicos descritos en el presente documento pueden estar sustituidos en un átomo de carbono o en uno de nitrógeno en caso de que el compuesto resultante sea estable. Un nitrógeno del heterociclo puede estar opcionalmente cuaternizado. Se prefiere que cuando el número total de átomos S y O en el heterociclo exceda de 1, entonces estos heteroátomos no sean adyacentes entre sí. Se prefiere que el número total de átomos de S y O en el heterociclo no sea mayor de 1. Cuando se usa el término "heterociclo", se pretende incluir heteroarilo.
Los ejemplos de heterociclos incluyen, pero sin limitación, acridinilo, azetidinilo, azocinilo, benzoimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, benzoxazolilo, benzoxazolinilo, benzotiazolilo, benzotriazolilo, benzotetrazolilo, benzoisoxazolilo, benzoisotiazolilo, benzoimidazolinilo, carbazolilo, 4aH-carbazolilo, carbolinilo, cromanilo, cromenilo, cinolinilo, decahidroquinolinilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro[2,3-6]tetrahidrofurano, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1H-indazolilo, imidazolopiridinilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isatinαlo, isobenzofuranilo, isocromanilo, isoindazolilo, isoindolinilo, isoindolilo, isoquinolinilo, isotiazolilo, isotiazolopiridinilo, isoxazolilo, isoxazolopiridinilo, metilenodioxifenilo, morfolinilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo, 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, oxazolopiridinilo, oxazolidinilperimidinilo, oxindolilo, pirimidinilo, fenantridinilo, fenantrolinilo, fenazinilo, fenotiazinilo, fenoxatiinilo, fenoxazinilo, ftalazinilo, piperazinilo, piperidinilo, piperidonilo, 4-piperidonilo, piperonilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolopiridinilo, pirazolilo, piridazinilo, piridooxazolilo, piridoimidazolilo, piridotiazolilo, piridinilo, pirimidinilo, pirrolidinilo, pirrolinilo, 2-pirrolidonilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrazolilo, tetrahidrofuranilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantrenilo, tiazolilo, tienilo, tiazolopiridinilo, tienotiazolilo, tienooxazolilo, tienoimidazolilo, tiofenilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1,3,4-triazolilo y xantenilo. También se incluyen anillos condensados y compuestos espiro que contienen, por ejemplo, los heterociclos anteriores.
Los ejemplos de heterociclos de 5 a 10 miembros incluyen, pero sin limitación, piridinilo, furanilo, tienilo, pirrolilo, pirazolilo, pirazinilo, piperazinilo, piperidinilo, imidazolilo, imidazolidinilo, indolilo, tetrazolilo, isoxazolilo, morfolinilo, oxazolilo, oxadiazolilo, oxazolidinilo, tetrahidrofuranilo, tiadiazinilo, tiadiazolilo, tiazolilo, triazinilo, triazolilo, benzoimidazolilo, 1H-indazolilo, benzofuranilo, benzotiofuranilo, benzotetrazolilo, benzotriazolilo, benzoisoxazolilo, benzoxazolilo, oxindolilo, benzoxazolinilo, benzotiazolilo, benzoisotiazolilo, isatinαlo, isoquinolinilo, octahidroisoquinolinilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, isoxazolopiridinilo, quinazolinilo, quinolinilo, isotiazolopiridinilo, tiazolopiridinilo, oxazolopiridinilo, imidazolopiridinilo y pirazolopiridinilo.
Los ejemplos de heterociclos de 5 a 6 miembros incluyen, pero sin limitación, piridinilo, furanilo, tienilo, pirrolilo, pirazolilo, pirazinilo, piperazinilo, piperidinilo, imidazolilo, imidazolidinilo, indolilo, tetrazolilo, isoxazolilo, morfolinilo, oxazolilo, oxadiazolilo, oxazolidinilo, tetrahidrofuranilo, tiadiazinilo, tiadiazolilo, tiazolilo, triazinilo y triazolilo. También se incluyen anillos condensados y compuestos espiro que contienen, por ejemplo, los heterociclos anteriores.
Como se usa en el presente documento, la expresión "heterociclo bicíclico" o "grupo heterocíclico bicíclico" pretende indicar un sistema anular heterocíclico estable de 9 o 10 miembros que contiene dos anillos condensados y que consiste en átomos de carbono y 1, 2, 3 o 4 heteroátomos seleccionados independientemente del grupo que consiste en N, O y S. De los dos anillos condensados, un anillo es un anillo aromático monocíclico de 5 o 6 miembros que comprende un anillo heteroarilo de 5 miembros, un anillo heteroarilo de 6 miembros o un anillo benzo, cada uno condensado a un segundo anillo. El segundo anillo es un anillo monocíclico de 5 o 6 miembros el cual está saturado, parcialmente insaturado o insaturado y comprende un heterociclo de 5 miembros, un heterociclo de 6 miembros o un carbociclo (con la condición de que el primer anillo no sea benzo cuando el segundo anillo es un carbociclo).
El grupo heterocíclico bicíclico puede estar unido a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. El grupo heterocíclico bicíclico descrito en el presente documento puede estar sustituido en un átomo de carbono o en uno de nitrógeno si el compuesto resultante es estable. Se prefiere que cuando el número total de átomos S y O en el heterociclo exceda de 1, entonces estos heteroátomos no sean adyacentes entre sí. Se prefiere que el número total de átomos de S y O en el heterociclo no sea superior a 1.
Son ejemplos de un grupo heterocíclico bicíclico, pero sin limitación, quinolinilo, isoquinolinilo, ftalazinilo, quinazolinilo, indolilo, isoindolilo, indolinilo, 1H-indazolilo, benzoimidazolilo, 1,2,3,4-tetrahidroquinolinilo, 1,2,3,4-tetrahidroisoquinolinilo, 5,6,7,8-tetrahidro-quinolinilo, 2,3-dihidro-benzofuranilo, cromanilo, 1,2,3,4-tetrahidroquinoxalinilo y 1,2,3,4-tetrahidro-quinazolinilo.
Como se usa en el presente documento, se pretende que la expresión "grupo heterocíclico aromático" o "heteroarilo" signifique hidrocarburos aromáticos monocíclicos y policíclicos estables que incluyen al menos un miembro de anillo de heteroátomos tal como azufre, oxígeno o nitrógeno. Los grupos heteroarilo incluyen, sin limitación, piridilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, furilo, quinolilo, isoquinolilo, tienilo, imidazolilo, tiazolilo, indolilo, pirrαlo, oxazolilo, benzofurilo, benzotienilo, benzotiazolilo, isoxazolilo, pirazolilo, triazolilo, tetrazolilo, indazolilo, 1,2,4-tiadiazolilo, isotiazolilo, purinilo, carbazolilo, benzoimidazolilo, indolinilo, benzodioxolanilo y benzodioxano. Los grupos heteroarilo están sustituidos o sin sustituir. El átomo de nitrógeno está sustituido o sin sustituir (es decir, N o NR, en donde R es H u otro sustituyente, si se define). Opcionalmente, los heteroátomos de nitrógeno y azufre pueden estar oxidados (es decir, N ^ O y S(O)p, en donde p es 0, 1 o 2).
Los anillos con puentes también están incluidos en la definición de heterociclo. Un anillo puenteado aparece cuando uno o más átomos (es decir, C, O, N o S) unen dos átomos de carbono o nitrógeno no adyacentes. Los ejemplos de anillos puenteados incluyen, pero sin limitación, un átomo de carbono, dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo de carbono-nitrógeno. Cabe apreciar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes citados para el anillo también pueden estar presentes en el puente.
El término "contraión" se usa para representar una especie cargada negativamente tal como cloruro, bromuro, hidróxido, acetato y sulfato.
Cuando se usa un anillo discontinuo dentro de una estructura anular, esto indica que la estructura anular puede estar saturada, parcialmente saturada o insaturada.
Como se cita en el presente documento, el término "sustituido" significa que al menos un átomo de hidrógeno se reemplaza con un grupo distinto de hidrógeno, con la condición de que las valencias normales se mantengan y que la sustitución dé como resultado un compuesto estable. Cuando un sustituyente es ceto (es decir, =O), entonces se reemplazan 2 hidrógenos en el átomo. Los sustituyentes ceto no están presentes en restos aromáticos. Cuando se dice que un sistema anular (por ejemplo, carbocíclico o heterocíclico) está sustituido con un grupo carbonilo o un doble enlace, se pretende que el grupo carbonilo o doble enlace forme parte (es decir, esté dentro) del anillo. Los dobles enlaces de anillo, como se usan en el presente documento, son dobles enlaces que se forman entre dos átomos adyacentes del anillo (por ejemplo, C=C, C=N o N=N).
En los casos en donde hay átomos de nitrógeno (por ejemplo, aminas) en los compuestos de la presente invención, estos pueden convertirse en N-óxidos mediante tratamiento con un agente oxidante (por ejemplo, mCPBA y/o peróxidos de hidrógeno). Por lo tanto, se considera que los átomos de nitrógeno mostrados y reivindicados incluyen tanto el nitrógeno mostrado como su derivado de N-óxido (N^O ).
Cuando aparece cualquier variable más de una vez en cualquier constituyente o fórmula de un compuesto, su definición cada vez que aparece es independiente de su definición en cualquier otra aparición. Por lo tanto, por ejemplo, si se muestra que un grupo está sustituido por 0-3 grupos R, a continuación, dicho grupo puede sustituirse opcionalmente por hasta tres grupos R y en cada caso, R se selecciona independientemente de la definición de R. Además, solo se permiten las combinaciones de sustituyentes y/o variables en caso de que dichas combinaciones den como resultado compuestos estables.
Cuando se muestra un enlace a un sustituyente que cruza un enlace que conecta dos átomos en un anillo, entonces tal sustituyente puede unirse a cualquier átomo del anillo. Cuando se enumera un sustituyente sin indicar el átomo en donde se une dicho sustituyente al resto del compuesto de una fórmula dada, entonces tal sustituyente puede unirse a través de cualquier átomo en dicho sustituyente. Solo se permiten las combinaciones de sustituyentes y/o variables en caso de que dichas combinaciones den como resultado compuestos estables.
En el presente documento la expresión "farmacéuticamente aceptable" se emplea para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que son, dentro del alcance del buen criterio médico, adecuados para su uso en contacto con los tejidos de seres humanos y animales sin producir demasiada toxicidad, irritación, respuesta alérgica y/u otro problema o complicación, en proporción con una relación beneficio/riesgo razonable.
Como se usa en el presente documento, "sales farmacéuticamente aceptables" se refieren a derivados de los compuestos desvelados en donde el compuesto precursor se modifica elaborando sales ácidas o básicas del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero sin limitación, sales de ácidos minerales u orgánicos de grupos básicos tales como aminas; y sales alcalinas u orgánicas de grupos ácidos tales como ácidos carboxílicos. Las sales farmacéuticamente aceptables incluyen las sales no tóxicas convencionales o las sales de amonio cuaternario del compuesto precursor formadas, por ejemplo, a partir de ácidos orgánicos o inorgánicos no tóxicos. Por ejemplo, dichas sales no tóxicas convencionales incluyen las derivadas de ácidos inorgánicos tales como, clorhídrico, bromhídrico, sulfúrico, sulfámico, fosfórico y nítrico; y las sales preparadas a partir de ácidos orgánicos, tales como acético, propiónico, succínico, glicólico, esteárico, láctico, málico, tartárico, cítrico, ascórbico, pamoico, maleico, hidroximaleico, fenilacético, glutámico, benzoico, salicílico, sulfanílico, 2-acetoxibenzoico, fumárico, toluenosulfónico, metanosulfónico, etanodisulfónico, oxálico e isetiónico.
Las sales farmacéuticamente aceptables de la presente invención pueden sintetizarse a partir del compuesto precursor que contiene un resto básico o ácido por métodos químicos convencionales. Generalmente, dichas sales se pueden preparar haciendo reaccionar las formas de ácido o base libres de estos compuestos con una cantidad estequiométrica de la base o ácido apropiado en agua o en un disolvente orgánico, o en una mezcla de los dos; generalmente, medios no acuosos como éter, acetato de etilo, etanol, isopropanol o acetonitrilo. Se encuentran listas de sales adecuadas en Remington's Pharmaceutical Sciences, 18a edición, Mack Publishing Company, Easton, PA (1990).
Además, los compuestos de fórmula I pueden tener formas de profármaco. Cualquier compuesto que se convierta in vivo para proporcionar el principio bioactivo (es decir, un compuesto de fórmula I) es un profármaco. Se conocen bien en la técnica diversas formas de profármacos. Para ejemplos de dichos derivados de profármacos, véanse:
a) Bundgaard, H., ed., Design of Prodrugs, Elsevier (1985) y Widder, K. et al., eds., Methods in Enzymology, 112:309-396, Academic Press (1985);
b) Bundgaard, H., Capítulo 5, "Design and Application of Prodrugs", Krosgaard-Larsen, P. et al., eds., A Textbook of Drug Design and Development, págs. 113-191, Harwood Academic Publishers (1991);
c) Bundgaard, H., Adv. Drug Deliv. Rev., 8:1-38 (1992);
d) Bundgaard, H. et al., J. Pharm. Sci., 77:285 (1988); y
e) Kakeya, N. et al., Chem. Pharm. Bull., 32:692 (1984).
Los compuestos que contienen un grupo carboxi pueden formar ésteres fisiológicamente hidrolizables que sirven como profármacos al hidrolizarse en el cuerpo para proporcionar los compuestos de fórmula I perse. Tales profármacos se administran preferentemente por vía oral, ya que la hidrólisis en muchos casos se produce principalmente bajo la influencia de las enzimas digestivas. Puede usarse la administración parenteral cuando el éster es activo por sí mismo o en aquellos casos en los que la hidrólisis se produce en la sangre. Los ejemplos de ésteres fisiológicamente hidrolizables de compuestos de fórmula I incluyen alquilo C1-6, alquilbencilo C1-6, 4-metoxibencilo, indanilo, ftalilo, metoximetilo, alcanoiloxi C1-6-alquilo C1-6 (por ejemplo, acetoximetilo, pivaloiloximetilo o propioniloximetilo), alcoxicarboniloxi C1-6-alquilo C1-6 (por ejemplo, metoxicarbonil-oximetilo o etoxicarboniloximetilo, gliciloximetilo, fenilgliciloximetilo, (5-metil-2-oxo-1,3-dioxolen-4-il)-metilo) y otros ésteres fisiológicamente hidrolizables bien conocidos usados, por ejemplo, en las técnicas de las penicilinas y cefalosporinas. Tales ésteres pueden prepararse mediante técnicas convencionales conocidas en la técnica.
La preparación de profármacos se conoce bien en la técnica y se describe en, por ejemplo, King, F. D., ed., Medicinal Chemistry: Principles and Practice, The Royal Society of Chemistry, Cambridge, Reino Unido (1994); Testa, B. et al., Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry and Enzymology, VCHA y Wiley-VCH, Zúrich, Suiza (2003); Wermuth, C. G., ed., The Practice of Medicinal Chemistry, Academic Press, San Diego, Ca (1999).
Se pretende que la presente invención incluya todos los isótopos de los átomos que aparecen en los presentes compuestos. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico pero diferentes números másicos. A modo de ejemplo general y sin limitación, los isótopos de hidrógeno incluyen deuterio y tritio. El deuterio tiene un protón y un neutrón en su núcleo y tiene dos veces la masa del hidrógeno habitual. El deuterio puede representarse por símbolos tales como "2H" o "D". El término "deuterado" en el presente documento, en sí mismo o usado para modificar un compuesto o un grupo, se refiere al reemplazo de uno o más átomos de hidrógeno, que están unidos a uno o más átomos de carbono, por un átomo de deuterio. Los isótopos de carbono incluyen 13C y 14C.
Los compuestos marcados con isótopos de la invención pueden prepararse generalmente por técnicas convencionales conocidas por los expertos en la técnica o por procesos análogos a los descritos en el presente documento, usando un reactivo marcado con isótopos apropiado en lugar del reactivo no marcado empleado de otro modo. Tales compuestos tienen una diversidad de usos potenciales, por ejemplo, como patrones y reactivos para determinar la capacidad de un compuesto farmacéutico potencial para unirse a proteínas o receptores diana o para obtener imágenes de compuestos de esta invención unidos a receptores biológicos in vivo o in vitro.
"Compuesto estable" y "estructura estable" pretenden incluir un compuesto que es suficientemente robusto como para sobrevivir al aislamiento hasta un grado útil de pureza a partir de una mezcla de reacción y a su formulación en un agente terapéutico eficaz. Se prefiere que los compuestos de la presente invención no contengan un grupo N-halo, S(O)2H o S(O)H.
El término "solvato" significa una asociación física de un compuesto de la presente invención con una o más moléculas de disolvente, ya sean orgánicas o inorgánicas. Esta asociación física incluye enlaces de hidrógeno. En determinados casos, el solvato podrá aislarse, por ejemplo, cuando se incorporan una o más moléculas de disolvente a la red cristalina del sólido cristalino. Las moléculas de disolvente en el solvato pueden estar presentes en una disposición regular y/o una disposición no ordenada. El solvato puede comprender una cantidad tanto estequiométrica como no estequiométrica de las moléculas de disolvente. "Solvato" abarca solvatos tanto en fase de solución como aislables. Los solvatos de ejemplo incluyen, pero sin limitación, hidratos, etanolatos, metanolatos e isopropanolatos. Los métodos de solvatación se conocen generalmente en la técnica.
Las abreviaturas, como se usan en el presente documento, se definen de la siguiente manera: "1 x" para una vez, "2 x" para dos veces, "3 x" para tres veces, "°C" para grados Celsius, "equiv." para equivalente o equivalentes, "g" para gramo o gramos, "mg" para miligramo o miligramos, "l" para litro o litros, "ml" para mililitro o mililitros, " jl" para microlitro o microlitros, "N" para normal, "M" para molar, "mmol" para milimol o milimoles, "min" para minuto o minutos, "h" para hora u horas, "ta" para temperatura ambiente, "TR" para tiempo de retención, "atm" para atmósfera, "MPa (psi)" para megapascales (libras por pulgada cuadrada), "conc." para concentrado, "sat." o "saturado" para saturado, "PM" para peso molecular, "p.f." para punto de fusión, "e.e." para exceso enantiomérico, "MS" o "Espec. Masas" para espectrometría de masas, "ESI" para espectroscopía de masas con ionización por electronebulización, "HR" para alto rendimiento, "HRMS" para espectrometría de masas de alto rendimiento, "LCMS" para cromatografía líquidaespectrometría de masas, "HPLC" para cromatografía líquida de alto rendimiento, "RP HPLC" para HPLC de fase inversa, "TLC" o "tlc" para cromatografía de capa fina, "r Mn " para espectroscopía de resonancia magnética nuclear, "nOe" para espectroscopía nuclear de efecto Overhauser, "1H" para protón, "8" para delta, "s" para singlete, "d" para doblete, "t" para triplete, "c" para cuadruplete, "m" para multiplete, "a" para ancho, "Hz" para hertzio y "a", "p", "R", "S", "E" y "Z" son designaciones estereoquímicas conocidas por un experto en la técnica.
Me Metilo
Et Etilo
Pr Propilo
i-Pr Isopropilo
Bu Butilo
i-Bu Isobutilo
t-Bu tere-butilo
Ph Fenilo
Bn Bencilo
Boc tere-butiloxicarbonilo
AcOH u HOAc ácido acético
AlCl3 cloruro de aluminio
AIBN Azobisisobutironitrilo
BBr3 tribromuro de boro
BCl3 tricloruro de boro
BEMP 2-ferc-butilimino-2-dietilamino-1,3-dimetilperhidro-1,3,2-diazafosforina
reactivo BOP hexafluorofosfato de benzotriazol-1-iloxitris(dimetilamino)fosfonio
B0C2O Dicarbonato de di-ferc-butilo
reactivo de Burgess 1-metoxi-N-trietilammoniosulfonil-metanimidato
CBz Carbobenciloxi
CH2CI2 Diclorometano
CH3CN o CAN Acetonitrilo
CDCI3 deutero-cloroformo
CHCl3 Cloroformo
mCPBA o m-CPBA ácido mefa-cloroperbenzoico
CS2CO3 carbonato de cesio
Cu(OAc)2 acetato de cobre (II)
Cy2NMe N-ciclohexil-N-metilciclohexanamina
DBU 1,8-diazabiciclo[5.4.0]undec-7-eno
DCE 1.2- dicloroetano
DCM diclorometano
DEA dietilamina
Dess-Martin 1,1,1-tris(acetiloxi)-1,1-dihidro-1,2-beniziodoxol-3-(1H)-ona
DIC o DIPCDI diisopropilcarbodiimida
DIEA, DIPEA o diisopropiletilamina
base de Hunig, 4-dimetilaminopiridina
DMAP DME 1.2- dimetoxietano
DMF dimetilformamida
DMSO dimetilsulfóxido
ADNc ADN complementario
Dppp (RM+)-1,2-bis(difen¡lfosf¡no)propano
DuPhos (+)-1,2-bis((2S,5S)-2,5-dietilfosfolano)benceno
EDC N-(3-dimetilaminopropil)-N-etilcarbodiimida
EDCI clorhidrato de N-(3-dimetilaminopropil)-W'-etilcarbodiimida
EDTA ácido etilendiaminotetraacético
(S,S)- trifluorometanosulfonato de (+)-1,2-bis((2S,5S)-2,5-dietilfosfolano)benceno(1,5-EtDuPhosRh(I) ciclooctadieno)rodio (I)
Et3N o TEA trietilamina
EtOAc acetato de etilo
Et2O éter dietílico
EtOH Etanol
precatalizador G3 catalizador Buchwald G3 (3a gen.)
GMF filtro de microfibra de vidrio
Grubbs (II) (1,3-bis(2,4,6-trimetilfenil)-2-imidazolidiniliden)dicloro(fenilmetilen)(triciclohexilfosfina)rutenio
HCl ácido clorhídrico
HATU hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio
HEPES ácido 4-(2-hidroxietil)piperazin-1-etanosulfónico
Hex Hexano
HOBt o HOBT 1-hidroxibenzotriazol
H2SO4 ácido sulfúrico
K2CO3 carbonato potásico
KOAc acetato potásico
K3PO4 fosfato potásico
LAH hidruro de litio y aluminio
LG grupo saliente
LiOH hidróxido de litio
MeOH Metanol
MgSO4 sulfato de magnesio
MsOH o MSA ácido metilsulfónico
NaCl cloruro sódico
NaH hidruro sódico
NaHCOa bicarbonato sódico
Na3CO3 carbonato sódico
NaOH hidróxido de sodio
Na2SO3 sulfito de sodio
Na2SO4 sulfato sódico
NBS N-bromosuccinimida
NCS N-clorosuccinimida
NH3 Amoniaco
NH4Cl cloruro de amonio
NH4OH hidróxido de amonio
OTf triflato o trifluorometanosulfonato
Pd2(dba)3 tris(dibencilidenoacetona)dipaladio (0 )
Pd(OAc)2 acetato de paladio (II)
Pd/C paladio sobre carbono
Pd(dppf)Cl2 [1,1'-bis(difenilfosfino)-ferroceno]dicloropaladio (II)
Ph3PCl2 dicloruro de trifenilfosfina
PG grupo protector
POCl3 oxicloruro de fósforo
i-PrOH o IPA isopropanol
PS poliestireno
SEM-Cl cloruro de 2 -(trimetilsilil)etoximetilo
SiO2 óxido de sílice
SnCl2 cloruro de estaño (II)
TBAI yoduro de tetra-n-butilamonio
TEA trietilamina
TFA ácido trifluoroacético
THF tetrahidrofurano
TMSCHN2 trimetilsilildiazometano
T3P anhídrido de ácido propanofosfónico
Tris tris(hidroximetil)aminometano
Los compuestos de la presente invención pueden prepararse de diversas formas conocidas por un experto en la técnica de la síntesis orgánica.
IV. BIOLOGÍA
Ensayos in vitro
La eficacia de los compuestos de la presente invención como inhibidores de ROCK puede determinarse en un ensayo de 30 |jl que contiene HEPES 20 mM, pH 7,5, MgCh 20 mM, Brij-35 al 0,015 %, DTT 4 mM, ATP 5 |jM y sustrato peptídico 1,5 jM (FITC-AHA-AKRRRLSSLRA-OH) (SEQ ID No. 1). Los compuestos se disolvieron en d Ms O de forma que la concentración final de DMSO era <2 % y la reacción comenzó con variantes de Rho cinasa. Después de la incubación, la reacción finalizó mediante la adición de EDTA y los péptidos fosforilados y no fosforilados se separaron usando un Lector LABCHIP® 3000 (Caliper Life Sciences). Los controles consistieron en ensayos que no contenían compuesto y los fondos consistieron en ensayos que contenían enzima y sustrato pero tenían EDTA desde el inicio de la reacción para inhibir la actividad cinasa. Los compuestos se analizaron en un formato de respuesta a la dosis y la inhibición de la actividad cinasa se calculó a cada concentración del compuesto. Los datos de inhibición se ajustaron usando un programa de ajuste de curvas para determinar la CI50; es decir, la concentración de compuesto necesaria para inhibir el 50 % de la actividad de cinasa.
Los ejemplos representativos se probaron en el ensayo de ROCK descrito anteriormente y se encontró que tenían actividad inhibidora de ROCK. Se observó un intervalo de actividad inhibitoria de ROCK (valores CI50) de < 50 jM (50000 nM). La tabla A a continuación enumera los valores de CI50 de ROCK medidos para los siguientes ejemplos.
Tabla A

continuación

continuación

continuación

V. COMPOSICIONES FARMACEUTICAS, FORMULACIONES Y COMBINACIONES
Los compuestos de la presente invención se pueden administrar en formas de dosificación orales como comprimidos,
cápsulas (casa una de las cuales incluye formulaciones de liberación sostenida o de liberación programada), píldoras, polvos, gránulos, elixires, tinturas, suspensiones, jarabes y emulsiones. También pueden administrarse en forma intravenosa (bolo o infusión), intraperitoneal, subcutánea o intramuscular, usando todas las formas de dosificación bien conocidas por los expertos en la técnica farmacéutica. Se pueden administrar en solitario, pero generalmente se administrarán con un transportador farmacéutico seleccionado sobre la base de la vía de administración elegida y la práctica farmacéutica convencional.
La expresión "composición farmacéutica" significa una composición que comprende un compuesto de la invención junto con al menos un transportador farmacéuticamente aceptable adicional. Un "portador farmacéuticamente aceptable" se refiere a medios generalmente aceptados en la técnica para el suministro de agentes biológicamente activos a animales, en particular, mamíferos, incluyendo, es decir, adyuvante, excipiente o vehículo, tales como diluyentes, agentes conservantes, cargas, agentes reguladores de flujo, agentes disgregantes, agentes humectantes, agentes emulsionantes, agentes de suspensión, agentes edulcorantes, agentes saporíferos, agentes perfumantes, agentes antibacterianos, agentes antifúngicos, agentes lubricantes y agentes dispensadores, dependiendo de la naturaleza del modo de administración y las formas de dosificación. Los transportadores farmacéuticamente aceptables se formulan de acuerdo con una serie de factores incluidos dentro del alcance de los expertos en la técnica. Estos incluyen, sin limitación: el tipo y la naturaleza del agente activo que se formule; el paciente al que ha de administrarse la composición que contiene el agente; la vía de administración prevista de la composición; y la indicación terapéutica a la que se dirigen. Los transportadores farmacéuticamente aceptables incluyen medios líquidos tanto acuosos como no acuosos, así como una diversidad de formas de dosificación sólidas o semisólidas. Dichos transportadores pueden incluir varios ingredientes y aditivos diferentes además del agente activo, incluyéndose dichos ingredientes adicionales en la formulación por diversos motivos, por ejemplo, estabilización del agente activo, aglutinantes, etc., bien conocidos por los expertos en la técnica. Las descripciones de transportadores farmacéuticamente aceptables y los factores implicados en su selección, se encuentran en una diversidad de fuentes fácilmente disponibles tales como, por ejemplo, Remington's Pharmaceutical Sciences, 18a edición (1990).
El régimen de dosificación de los compuestos de la presente invención, por supuesto, variará dependiendo de factores conocidos, tales como las características farmacodinámicas del agente particular y su modo y vía de administración; la especie, la edad, el sexo, el estado de salud, la condición médica y el peso del receptor; la naturaleza y extensión de los síntomas; el tipo de tratamiento concurrente; la frecuencia del tratamiento; la vía de administración, la función renal y hepática del paciente y el efecto deseado. Un médico o un veterinario puede determinar y prescribir la cantidad eficaz del fármaco que es necesaria para prevenir, contrarrestar o detener la evolución del trastorno.
A modo de orientación general, la dosificación oral diaria de cada principio activo, cuando se usa para los efectos indicados, variará entre aproximadamente 0,001 y aproximadamente 1000 mg/kg de peso corporal, preferentemente entre aproximadamente 0,01 y aproximadamente 100 mg/kg de peso corporal al día, y mucho más preferentemente entre aproximadamente 0,1 y aproximadamente 20 mg/kg/día. Por vía intravenosa, las dosis más preferidas variarán de aproximadamente 0,001 a aproximadamente 10 mg/kg/minuto durante una infusión a velocidad constante. Los compuestos de la presente invención pueden administrarse en una única dosis diaria o la dosificación diaria total puede administrarse en dosis divididas de dos, tres o cuatro veces al día.
Los compuestos de la presente invención también pueden administrarse mediante administración parenteral (por ejemplo, intravenosa, intraarterial, intramuscular o subcutánea. Cuando se administra por vía intravenosa o intraarterial, la dosis se puede proporcionar de manera continua o intermitente. Además, la formulación puede desarrollarse para el suministro intramuscular y subcutáneo que garantice una liberación gradual del principio farmacéuticamente activo.
Los compuestos de la presente invención pueden administrarse de forma intranasal a través del uso tópico de vehículos intranasales adecuados o a través de vías transdérmicas, usando parches cutáneos transdérmicos. Cuando se administra en forma de un sistema de suministro transdérmico, la administración de la dosis será, por supuesto, continua en lugar de intermitente a lo largo del régimen de dosificación.
Los compuestos se administran típicamente mezclados con diluyentes, excipientes o transportadores farmacéuticamente adecuados (denominados colectivamente en el presente documento como transportadores farmacéuticos) seleccionados adecuadamente con respecto a la forma de administración prevista, por ejemplo, comprimidos orales, cápsulas, elixires y jarabes y conformes con las prácticas farmacéuticas convencionales.
Por ejemplo, para la administración oral en forma de un comprimido o una cápsula, el componente de fármaco activo puede combinarse con un vehículo inerte oral, no tóxico, farmacéuticamente aceptable, vehículo inerte, tal como lactosa, almidón, sacarosa, glucosa, metilcelulosa, estearato de magnesio, fosfato de dicalcio, sulfato de calcio, manitol, sorbitol y similares; para la administración oral en forma líquida, los componentes del fármaco oral se pueden combinar con cualquier transportador inerte oral, no tóxico y farmacéuticamente aceptable, tal como etanol, glicerol, agua y similares. Además, cuando se desee o sea necesario, se pueden incorporar también aglutinantes, lubricantes, agentes disgregantes y agentes colorantes adecuados en la mezcla. Los aglutinantes adecuados incluyen almidón, gelatina, azúcares naturales tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares. Los
lubricantes usados en estas formas de dosificación incluyen oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y similares. Los disgregantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares.
Los compuestos de la presente invención también pueden administrarse en forma de sistemas de suministro en liposomas, tales como vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas pueden formarse a partir de una diversidad de fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas.
Los compuestos de la presente invención también pueden acoplarse a polímeros solubles como transportadores farmacéuticos que pueden marcarse como diana. Dichos polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietilaspartamidafenol u óxido de polietileno-polilisina sustituido por restos palmitoílo. Además, los compuestos de la presente invención pueden acoplarse a una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, ácido poliglicólico, copolímeros de ácido poliláctico y poliglicólico, poliépsilon caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polihidropiranos, policianoacilatos y copolímeros de bloque de hidrogeles reticulados o anfipáticos.
Las formas de dosificación (composiciones farmacéuticas) adecuadas para la administración pueden contener de aproximadamente 1 miligramo a aproximadamente 1000 miligramos de principio activo por unidad de dosificación. En estas composiciones farmacéuticas el principio activo estará habitualmente presente en una cantidad de aproximadamente el 0,1-95 % en peso basado en el peso total de la composición.
Las cápsulas de gelatina pueden contener el principio activo y transportadores en polvo, tales como lactosa, almidón, derivados de celulosa, estearato de magnesio, ácido esteárico y similares. Para elaborar comprimidos compactados pueden usarse diluyentes similares. Tanto los comprimidos como las cápsulas pueden fabricarse como productos de liberación sostenida para proporcionar la liberación continua de la medicación durante un período de horas. Los comprimidos compactados pueden recubrirse con azúcar o con una película para ocultar cualquier sabor desagradable y proteger al comprimido de la atmósfera, o pueden tener un recubrimiento entérico para la disgregación selectiva en el tubo digestivo.
Las formas farmacéuticas líquidas para la administración oral pueden contener colorantes y saporíferos para aumentar la aceptación del paciente.
En general, agua, un aceite adecuado, solución salina, solución acuosa de dextrosa (glucosa) y soluciones de azúcares relacionados y glicoles tales como propilenglicol o polietilenglicoles son portadores adecuados para las soluciones parenterales. Las soluciones para administración parenteral contienen preferentemente una sal soluble en agua del principio activo, agentes estabilizantes adecuados y, si es necesario, sustancias tamponantes. Los agentes antioxidantes tales como, bisulfito de sodio, sulfito de sodio o ácido ascórbico, en solitario o en combinación, son agentes estabilizantes adecuados. También se usan ácido cítrico y sus sales y EDTA sódico. Además, las soluciones parenterales pueden contener conservantes, tales como cloruro de benzalconio, metil o propilparabeno y clorobutanol.
Los compuestos de la presente invención pueden administrarse en solitario o en combinación con uno o más agentes terapéuticos adicionales. Por "administrado en combinación" o "terapia de combinación" se entiende que el compuesto de la presente invención y uno o más agentes terapéuticos adicionales se administran a la vez al mamífero a tratar. Cuando se administran en combinación, cada componente se puede administrar al mismo tiempo o de manera secuencial en cualquier orden en distintos momentos en el tiempo. Por lo tanto, cada componente se puede administrar por separado pero lo suficientemente cerca en el tiempo como para proporcionar el efecto terapéutico deseado.
Los compuestos de la presente invención también son útiles como patrones o compuestos de referencia, por ejemplo, como un patrón de calidad o control, en ensayos o pruebas que implican la inhibición de ROCK. Dichos compuestos pueden proporcionarse en un kit comercial, por ejemplo, para su uso en investigación farmacéutica que implica ROCK. Por ejemplo, un compuesto de la presente invención podría usarse como referencia en un ensayo para comparar su actividad conocida con un compuesto con una actividad desconocida. Esto aseguraría al experimentador que el ensayo se estaría realizando de manera apropiada y proporcionaría una base para la comparación, especialmente si el compuesto de ensayo era un derivado del compuesto de referencia. Cuando se desarrollan nuevos ensayos o protocolos, los compuestos de acuerdo con la presente invención podrían usarse para probar su eficacia.
La presente invención también abarca un artículo de fabricación. Como se usa en el presente documento, se pretende que el artículo de fabricación incluya, pero sin limitación, kits y paquetes. El artículo de fabricación de la presente invención, comprende: (a) un primer recipiente; (b) una composición farmacéutica ubicada dentro del primer recipiente, en donde la composición, comprende: un primer agente terapéutico, que comprende: un compuesto de la presente invención o una forma de sal farmacéuticamente aceptable del mismo; y, (c) un prospecto que indica que la composición farmacéutica puede usarse para el tratamiento de un trastorno cardiovascular y/o inflamatorio (como se ha definido previamente). En otra realización, el prospecto indica que la composición farmacéutica puede usarse en combinación (como se ha definido previamente) con un segundo agente terapéutico para tratar un trastorno
cardiovascular y/o inflamatorio. El artículo de fabricación puede comprender, además: (d) un segundo recipiente, en donde los componentes (a) y (b) se localizan dentro del segundo recipiente y el componente (c) se localiza dentro o fuera del segundo recipiente. Situado dentro del primer y segundo recipientes significa que el recipiente respectivo contiene el artículo dentro de sus límites.
El primer recipiente es un receptáculo usado para contener una composición farmacéutica. Este recipiente puede ser para fabricación, almacenamiento, envío y/o venta individual/a granel. Se pretende que el primer recipiente guarde una botella, tarro, vial, matraz, jeringa, tubo (por ejemplo para una preparación en crema) o cualquier otro recipiente usado para fabricar, contener, almacenar o distribuir un producto farmacéutico.
El segundo recipiente es uno que se utiliza para contener el primer recipiente y, opcionalmente, el prospecto. Los ejemplos del segundo recipiente incluyen, pero sin limitación, cajas (por ejemplo, de cartón o plástico), cajones, cartones, bolsas (por ejemplo, bolsas de papel o plástico), bolsitas y sacos. El prospecto puede estar físicamente unido al exterior del primer recipiente mediante cinta adhesiva, pegamento, grapas u otro método de unión o puede estar dentro del segundo recipiente sin ningún medio físico de unión al primer recipiente. Como alternativa, el prospecto se localiza en el exterior del segundo recipiente. Cuando se localiza en el exterior del segundo recipiente, se prefiere que el prospecto esté unido físicamente mediante cinta adhesiva, pegamento, grapas u otro método de unión. Como alternativa, puede estar adyacente o en contacto con el exterior del segundo recipiente sin estar unido físicamente.
El prospecto es un rótulo, una etiqueta, un marcador, etc. que enumera información relacionada con la composición farmacéutica localizada en el primer recipiente. La información citada normalmente será determinada por el organismo regulador gubernamental de la zona geográfica en que se va a comercializar el artículo de fabricación (por ejemplo, la Administración de Alimentos y Medicamentos de los Estados Unidos). Preferentemente, el prospecto describe específicamente las indicaciones para las que se ha aprobado la composición farmacéutica. El prospecto puede fabricarse con cualquier material sobre el que una persona pueda leer información contenida en el mismo o sobre el mismo. Preferentemente, el prospecto es un material imprimible (por ejemplo, papel, plástico, cartulina, lámina, papel o plástico adhesivo, etc.) en el que se ha plasmado (por ejemplo, impreso o aplicado) la información deseada.
Otras características de la invención serán obvias en el transcurso de las siguientes descripciones de realizaciones de ejemplo que se ofrecen para ilustrar la invención y que no pretenden limitarla. Los siguientes ejemplos se han preparado, aislado y caracterizado usando los métodos desvelados en el presente documento.
VI. SÍNTESIS GENERAL QUE INCLUYE ESQUEMAS
Los ejemplos de compuestos de la presente invención preparados por los métodos descritos en los esquemas generales se dan en las secciones de intermedios y ejemplos expuestas en lo sucesivo en el presente documento. La preparación de ejemplos homoquirales puede realizarse por técnicas conocidas por un experto en la técnica. Por ejemplo, pueden prepararse compuestos homoquirales mediante separación de productos racémicos por HPLC preparativa de fase quiral. Como alternativa, los compuestos de ejemplo pueden prepararse mediante métodos conocidos para dar productos enantioméricamente enriquecidos. Estos incluyen, pero sin limitación, la incorporación de funcionalidades auxiliares quirales a intermedios racémicos que sirven para controlar la diastereoselectividad de las transformaciones, proporcionando productos enantioenriquecidos tras la escisión del auxiliar quiral.
También se reconocerá que otra consideración principal al planear cualquier ruta sintética en este campo es la elección juiciosa del grupo protector usado para la protección de grupos funcionales reactivos presentes en los compuestos descritos en la presente invención. Un informe autorizado que describe las muchas alternativas al médico capacitado es Greene et al., (Protective Groups in Organic Synthesis, 4a Edición, Wiley-Interscience (2006)).
Los compuestos de esta invención se pueden preparar mediante desplazamiento del haluro de arilo o heteroarilo (A) mediante un ácido 2-(4-hidroxifenil)acético (B) seguido de acoplamiento estándar de ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acético (C) con aminas bicíclicas o monocíclicas-heterocíclicas (D o F)usando dichos agentes de acoplamiento como BOP. Las amidas (E o G) con sustituyentes halógeno en R7 también se pueden elaborar mediante metodologías de acoplamiento cruzado de Suzuki conocidas en la materia.


La purificación de intermedios y productos finales se realizó por cromatografía de fase normal o inversa. La cromatografía de fase normal se realizó usando cartuchos de SO 2 precargados eluyendo con gradientes de hexanos y EtOAc, DCM y MeOH a menos que se indique de otro modo. La HPLC preparativa de fase inversa se realizó usando columnas C18 eluyendo con gradientes de Disolvente A (agua al 90 %, MeOH al 10 %, TFA al 0,1 %) y Disolvente B (agua al 10 %, MeOH al 90 %, TFA al 0,1 %, UV 220 nm) o con gradientes de Disolvente A (agua al 90 %, CH3CN al 10 %, TFA al 0,1 %) y Disolvente B (agua al 10 %, CH3CN al 90 %, TFA al 0,1 %, UV 220 nm) o con gradientes de Disolvente A (agua al 98 %, CH3CN al 2 %, TFA al 0,05 %) y Disolvente B (CH3CN al 98 %, agua al 2 %, TFA al 0,05 %, UV 220 nm) (o) Sunfire Prep C18 OBD 5u 30x100 mm, 25 min de gradiente de B al 0-100 %. A = 90:10:0,1 de H2O/CH3CN/TFA. B = CH3CN/H20/TFA 90:10:0.1
A menos que se indique de otro modo, el análisis de los productos finales se realizó por HPLC analítica de fase inversa.
Método A: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 μm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: B al 0-100 % durante 3 minutos, a continuación un mantenimiento de 0,75 minutos al 100 % de B; Flujo: 1,11 ml/min.
Método B: Waters Acquity UPLC BEH C18, 2,1 x 50 mm, partículas de 1,7 μm; Fase móvil A: 5:95 de acetonitrilo:agua con TFA al 0,1 %; Fase móvil B: 95:5 de acetonitrilo:agua con TFA al 0,1 %; Temperatura: 50 °C; Gradiente: B al 0 100 % durante 3 minutos, a continuación un mantenimiento de 0,75 minutos al 100 % de B; Flujo: 1,11 ml/min.
Método C SunFire (4,6 x 150 mm) (gradiente de 15 min 95:5 H2O/acetonitrilo a 95:5 de acetonitrilo/H2O-TFA al 0,05 %).
Intermedio 1. Ácido 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acético

A 2-cloronicotinonitrilo (0,489 g, 3,53 mmol) en DMSO (5 ml) se le añadió ácido 2-(3-fluoro-4-hidroxifenil)acético (0,6 g, 4 mmol) y K2CO3 (1,072 g, 7,760 mmol) y la mezcla de reacción se calentó a 60 °C durante 14 h. La mezcla de reacción se dejó enfriar a temperatura ambiente, se inactivó mediante la adición de HCl 1 N y el sólido resultante se filtró y se secó al vacío para proporcionar (0,9 g, 90 % de rendimiento) del intermedio 1 en forma de un sólido de color castaño. RMN 1H (400 MHz, CD3OD) 88,32 (dd, J = 5,0, 1,9 Hz, 1H), 8,25 (dd, J = 7,7, 2,0 Hz, 1H), 7,35-7,23 (m, 3H), 7,23 7,10 (m, 1H), 3,70 (s, 2H). LCMS m/z = 273.0 (M+H).+
Intermedio 2. Cloruro de 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetilo

A una suspensión del intermedio 1 (1.6 g, 5.9 mmol) en DCE (11.7 ml) se le añadió SOCI2 (4,29 ml, 58,8 mmol) y la mezcla de reacción se calentó a 50 °C. La mezcla de reacción se hizo transparente y se agitó durante 1 h. La evaporación de los disolventes al vacío proporcionó el cloruro de 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetilo (1,71 g, 100 %) en forma de un sólido de color blanco. LCMS m/z = 286,9 (M+H)+ para el éster metílico.
Intermedio 3. Ácido 2-(4-((3-cianopiridin-2-il)oxi)fenil)acético, HCl.

El ácido 2-(4-((3-cianopiridin-2-il)oxi)fenil)acético, HCl (14,7 g, 99% de rendimiento), un sólido de color pardo; se preparó según se describe para el intermedio 1, sustituyendo el ácido 2-(4-hidroxifenil)acético por ácido 2-(3-fluoro-4-hidroxifenil)acético RMN 1H (400 MHz, DMSO-da) 812,39 (s a, 1H), 8,88-8,18 (m, 2H), 7,42-7,29 (m, 3H), 7,23-7,15 (m, 2H), 2,63 (s, 2H). LCMS m/z = 255.1 (M+H).+
Intermedio 4. Ácido 2-(3-cloro-4-((3-cianopiridin-2-il)oxi)fenil)acético

Se preparó ácido 2-(3-cloro-4-((3-cianopiridin-2-il)oxi)fenil)acético (1,5 g, 97 % de rendimiento) se preparó según se describe para el intermedio 1, sustituyendo ácido 2-(3-cloro-4-hidroxifenil)acético por ácido 2-(3-fluoro-4-hidroxifenil)acético RMN 1H (400 MHz, CD3OD) 88,35-8,20 (m, 2H), 7,52 (d, J = 2,0 Hz, 1H), 7,40-7,33 (m, 1H), 7,32 7,21 (m, 2H), 3,70 (s, 2H). LCMS (ESI) m/z: 289-290,9 (M+H).+
Intermedio 5. Ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acético, HCl

El intermedio 1 (3 g, 10 mmol) se disolvió en DMSO (55,1 ml), después se añadió K2CO3 (6,09 g, 44,1 mmol). La mezcla de reacción se enfrió ligeramente en un baño de agua (no DMSO congelado). A la mezcla de reacción se le añadió peróxido de hidrógeno (ac. al 30% en peso) (11,26 ml, 110,0 mmol) gota a gota durante 5 min (ligera exotermia) y la mezcla de reacción se agitó a temperatura ambiente. Después de 72 h, se añadió cuidadosamente HCl conc. y el sólido de color blanco resultante se filtró, se lavó con exceso de agua y se secó al vacío para proporcionar (2,86 g, 79,0 % de rendimiento) el intermedio 5. RMN 1H (400 MHz, DMSO-d6) 8 12,46 (s a, 1H), 8,47 8,09 (m, 2H), 7,81 (d a, J = 8,1 Hz, 2H), 7,40-7,23 (m, 3H), 7,16 (dd, J = 8,1, 1,3 Hz, 1H), 3,66 (s, 2H). LCMS m/z = 291.1 (M+H).+
Intermedio 6. Ácido 2-(4-((3-carbamoMpiridin-2-N)oxi)fenM)acético, HCl

Ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acético, HCl (1,3 g, 87 % de rendimiento), un sólido de color amarillo claro, se preparó de una manera similar a la del intermedio 5. RMN 1H (500 MHz, DMSO-d6) 88,26-8,11 (m, 2H), 7,78 (s a, 1H), 7,75 (s a, 1H), 7,36-7,27 (m, 2H), 7,22 (dd, J = 7,4, 4,7 Hz, 1H), 7,16-7,05 (m, 2H), 3,48-3,32 (m, 1H), 2,59-2,52 (m, 2H). LCMS m/z = 273.1 (M+H).+
Intermedio 7 Ácido 2-(3-cloro-4-((3-cianopiridin-2-il)oxi)fenil)acético, HCl

Ácido 2-(3-cloro-4-((3-cianopiridin-2-il)oxi)fenil)acético, HCl (0,45 g, 100 % de rendimiento), un sólido de color blanco, se preparó según se describe para el intermedio 5 , sustituyendo el ácido 2-(3-cloro-4-((3-cianopiridin-2-il)oxi)fenil)acético por ácido 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acético. RMN 1H (400 MHz, DMsO-cfe) 8 12,48
(s a, 1H), 8,40-8,03 (m, 2H), 7,97-7,69 (m, 2H), 7,49 (d, J = 2,0 Hz, 1H), 7,44-6,89 (m, 3H), 3,32 (s, 2H). LCMS m/z: 306,9 (M+H).+
Intermedio 8.2-(4-(2-((5-bromobenzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida, HCl
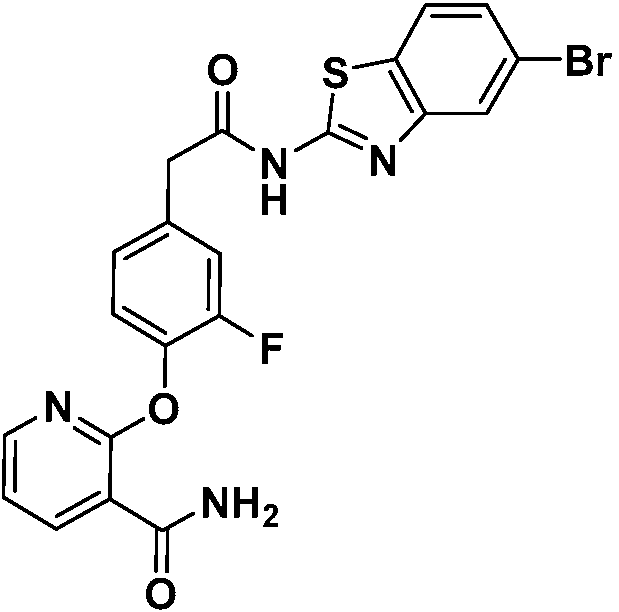
Etapa 1. A una suspensión del intermedio 1 (0,35 g, 1,3 mmol) en DCE (4 ml) se le añadió SOCh (0,925 ml, 12,7 mmol) y la mezcla de reacción se calentó a 50 °C. La reacción se hizo transparente y se agitó durante 1 h. Los disolventes se eliminaron a presión reducida y al sólido resultante se le añadió 5-bromobenzo[d]tiazol-2-amina (0,32 g, 1,4 mmol), THF (3 ml) y DIEa (0,66 ml, 3,8 mmol). Después de 24 h, los disolventes se evaporaron a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice eluyendo con DCM/acetato de etilo al 0-100% para proporcionar (0,385 g, 62,9 % de rendimiento) N-(5-bromobenzo[d]tiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCh) 6 9,06 (s a, 1H), 8,34 (dd, J = 4,8, 2,0 Hz, 1H), 8,07 (dd, J = 7,7, 2,0 Hz, 1H), 7,93 (d, J = 1,8 Hz, 1H), 7,72 (d, J = 8,6 Hz, 1H), 7,55-7,46 (m, 1H), 7,51-7,45 (m, 1H), 7,36 (t, J = 8,0 Hz, 1H), 7,28-7,14 (m, 2H), 3,92 (s, 2H). LCMS m/z = 484.9 (M+H).+
Etapa 2. Se disolvió N-(5-bromobenzo[d]tiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida (0,38 g, 0,78 mmol) en DMSO (3,93 ml), después se añadió K2CO3 (0,435 g, 3,14 mmol). La mezcla de reacción se enfrió ligeramente en un baño de agua y se añadió peróxido de hidrógeno al 30 % (0,80 ml, 7,9 mmol) gota a gota durante 5 min (exotermia ligera) y la mezcla de reacción se agitó a temperatura ambiente durante 14 h. La mezcla de reacción se inactivó mediante la adición cuidadosa de HCl y el sólido resultante se filtró y se secó al vacío para proporcionar (0,45 g, 106 % de rendimiento) del intermedio 8 en forma de un sólido de color castaño. RMN 1H (400 MHz, DMSO-cfe) 6 12,82 (s a, 1H), 8,29-8,14 (m, 2H), 7,98 (dd, J = 5,2, 3,2 Hz, 2H), 7,82 (s a, 1H), 7,80 (s a, 1H), 7,53-7,46 (m, 1H), 7,42-7,33 (m, 2H), 7,25 (dd, J = 7,4, 5,0 Hz, 2H), 3,93 (s, 2H). LCMS m/z = 500,9-501,9 (M+H).+
Ejemplo 1. 2-(2-fluoro-4-(2-((7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

En un vial, a una mezcla de intermedio 5 (0,02 g, 0,07 mmol), 7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2- amina (0,02 g, 0,07 mmol) se le añadió BOP (0,030 g, 0,069 mmol), DMF (0,3 ml) y DIEA (0,060 ml, 0,35 mmol). Después de 72 h, la mezcla de reacción se filtró y se sometió a purificación por HPLC de fase inversa para proporcionar el Ejemplo 1 (4,4 mg, 9,9 %). RMN 1H (500 MHz, DMSO-d6) 68,34-8,04 (m, 2H), 7,85-7,81 (m, 1H), 7,80-7,73 (m, 1H), 7,58 (d, J = 8,2 Hz, 1H), 7,42-7,29 (m, 2H), 7,27-7,12 (m, 2H), 6,91-6,79 (m, 2H), 3,83 (s, 2H), 3,33 (s a, 1H), 3,02-2,93 (m, 2H), 2,92-2,84 (m, 2H), 3,77 (s, 3H). LCMS m/z = 505,0 (M+H)+; pureza por Hp Lc > 96 % con un tiempo de retención de 1,81 min. [Método A]
Ejemplo 2. 2-(4-(2-((5-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

A una mezcla de intermedio 6 (20 mg, 0,065 mmol), 5-metoxibenzo[d]tiazol-2-amina (11,68 mg, 0,06500 mmol), se le añadió DMF (0,3 ml), una solución en DMF (0,5 ml) que contenía BOP (28,7 mg, 0,0650 mmol) y DIEA (0,057 ml, 0,32 mmol) se añadieron. Después de 24 h, la mezcla de reacción se filtró y se sometió a purificación mediante HPLC de fase inversa para proporcionar el ejemplo 2 (1,6 mg, 4,2 % de rendimiento), RMN 1H (500 MHz, DMSO-cfe) 8 RMN 1H (500 MHz, DMSO-d6) 8 12,58 (s a, 1H), 8,29-8,00 (m, 2H), 7,82 (d a, J = 8,5 Hz, 1H), 7,79 (s a, 1H), 7,75 (s a, 1H), 7,40 (d a, J = 8,2 Hz, 2H), 7,28 (s, 1H), 7,21 (dd a, J = 7,3, 4,9 Hz, 1H), 7,16 (d a, J = 8,2 Hz, 2H), 6,94 (dd, J = 8,5, 1,8 Hz, 1H), 3,86 (s, 2H) 3,82 (s, 3H). LCMS m/z = 434,9 (M+H)+; pureza por HPLC > 94 % con un tiempo de retención de 1,52 min. [Método B]
Ejemplo 3. 2-(2-fluoro-4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

E ta p a 1. Se suspendió 2-aminobenzo[d]tiazol-5-ol (0,066 g, 0,40 mmol) en MeCN (4,5 ml), después se añadieron 2,2-dimetiloxirano (0,35 ml, 4,0 mmol), K2CO3 (0,220 g, 1,59 mmol) y agua (0,300 ml). La mezcla de reacción se agitó con irradiación con microondas a 120 °C durante 30 min. La mezcla de reacción se diluyó con MeOH, se concentró a presión reducida y se purificó por cromatografía sobre gel de sílice usando DCM/MeOH 0-al 15 % como eluyentes para proporcionar (73 mg, 77 % de rendimiento) 1-((2-aminobenzo[d]tiazol-5-il)oxi)-2-metilpropan-2-ol en forma de un aceite transparente. RMN 1H (400 MHz, CDCh) 87,47 (d, J = 8,8 Hz, 1H), 7,14 (d, J = 2,4 Hz, 1H), 6,82 (dd, J = 8 ,6 , 2,4 Hz, 1H), 5,34 (d, J = 7,7 Hz, 2H), 3,85 (s, 2H), 3,52 (s, 1H), 1,41-1,37 (m, 6 H). LCMS m/z = 238.9 (M+H).+
E ta p a 2. En un vial, al intermedio 5 (12,18 mg, 0,04200 mmol) y 1-((2-aminobenzo[d]tiazol-5-il)oxi)-2-metilpropan-2-ol (10 mg, 0,042 mmol) se le añadió Dm F (0,3 ml), BOP (18,56 mg, 0,04200 mmol) y DIEA (0,037 ml, 0,21 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 72 h. La mezcla de reacción se filtró y se sometió a purificación mediante HPLC de fase inversa para proporcionar el ejemplo 3 (1,5 mg, 5,4 % de rendimiento). RMN 1H (500 MHz, DMSO-cfe) 88,30-8,11 (m, 2H), 7,84 (s a, 1H), 7,82 (s a, 1H), 7,79 (d a, J = 8,7 Hz, 1H), 7,47-7,30 (m, 2H), 7,30-7,17 (m, 3H), 6,93 (d a, J = 8,8 Hz, 1H), 3,86 (s, 2H), 3,78 (s, 2H), 2,55 (s, 1H), 1,83 (s a, 1H), 1,23 (s, 6 H). LCMS m/z = 511,0 (M+H)+; pureza por HPLC > 94 % con un tiempo de retención de 1,45 min. [Método B]
Ejemplo 4. 2-(4-(2-((5-(1,3-dimetiMH-pirazol-4-N)benzo[d]tiazol-2-N)ammo)-2-oxoetil)-2-fluorofenoxi)nicotinamida

A un vial se le añadió el intermedio 8 (25 mg, 0,050 mmol), 1,3-dimetil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (16,61 mg, 0,07500 mmol), fosfato potásico tribásico 3 M (83 μl, 0,25 mmol) y DMF (1,5 ml). La mezcla de reacción se desgasificó con N2 y se le añadió cloro(2-diciclohexilfosfino-2',4',6'-trH-propil-1,1'-bifenil)(2'-amino-1,1'-bifenil-2-il)paladio (II) (3,92 mg, 4,99 μmol) y la mezcla de reacción se calentó a 70 °C. Después de 1 h, la mezcla de reacción se dejó enfriar a temperatura ambiente, se filtró y se sometió a purificación mediante HPLC de fase inversa para proporcionar el ejemplo 4 (4,4 mg, 14 % de rendimiento). RMN 1H (500 MHz, DMSO-cfe) 58,21 (d a, J = 7,5 Hz, 1H), 8,20-8,16 (m, 1H), 7,95 (d, J = 8,2 Hz, 1H), 7,92 (s, 1H), 7,75 (s, 1H), 7,72 (s a, 1H), 7,66 (s a, 1H), 7,37 (t a, J = 8,2 Hz, 3H), 7,25 (dd a, J = 7,3, 4,4 Hz, 2H), 3,92 (s, 2H), 3,81 (s, 3H), 2,34 (s, 3H), 1,02 (m, 1H). LCMS m/z = 517,1 (M+H).+ Pureza por HPLC > 98 % con un tiempo de retención de 1,41 min. [Método B]
Los ejemplos 5 y 6 se prepararon como se ha descrito siguiendo el procedimiento general para el ejemplo 4

(continuación)

Ejemplo 7. 2-(2-doro-4-(2-((1-(ddoproμMmetil)-1H-benzo[d]imidazol-2-il)ammo)-2-oxoetil)fenoxi)mcotmamida

Etapa 1. A W1-(dcloprop¡lmetil)bencen-1,2-d¡am¡na (0,18 g, 1,1 mmol) disponible en el mercado en MeOH (4 ml)/H2O (1 ml) se le añadió bromuro ciánico (0,17 g, 1,7 mmol). Después de agitar durante 24 h, la mezcla de reacción se concentró a presión reducida y se trató con NH4OH conc. El sólido precipitado se recogió y se secó al vacío para proporcionar 1-(c¡cloprop¡lmet¡l)-1H-benzo[d]¡m¡dazol-2-am¡na (0,138 g, 66,4 % de rendimiento) en forma de un sólido de color rojo/pardo. RMN 1H (400 MHz, DMSO-da) 57,17 (dt, J=7,6, 0,7 Hz, 1H), 7,12 (d, J = 7,4 Hz, 1H), 6,97-6,84 (m, 2H), 6,34 (s, 2H), 3,91 (d, J = 7,0 Hz, 2H), 2,73-2,54 (m, 1H), 1,27-1,15 (m, 1H), 0,50-0,37 (m, 4H). LCMS m/z = 188.1 (M+H).+
El ejemplo 7 se preparó mediante un acoplamiento de BOP del producto de la etapa 1 y el intermedio 7 (9,6 mg, 32,8 % de rendimiento). RMN 1H (500 MHz, DMSO-cfe) 88,48-8,04 (m, 2H), 7,96-7,74 (m, 2H), 7,66-7,48 (m, 3H), 7,34 (d a, J = 16,9 Hz, 2H), 7,30-6,85 (m, 3H), 4,00 (s a, 2H), 3,69 (s a, 2H), 1,23 (s a, 2H), 0,43 (s a, 4H). LCMS m/z = 476,1 (M+H).+ Pureza por HPLC > 92 % con un tiempo de retención de 1,38 min. [Método B]
Ejemplo 8.2-(2-fluoro-4-(2-((7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

Etapa 1. Al intermedio 2 (0,25 g, 0,86 mmol) y 7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-amina (0,14 g, 0,86 mmol) se le añadió THF (3 ml) y piridina (0,21 ml, 2,6 mmol). Después de 24 h, los disolventes se evaporaron y el residuo se purificó por cromatografía sobre gel de sílice, eluyendo con DCM/EtOAc al 0-100 %, después DCM/10 %MeOH para proporcionar 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)-N-(7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)acetamida (35 mg, 9,7 % de rendimiento) en forma de un sólido de color castaño. RMN 1H (400 MHz, CD3OD) 88,38-8,28 (m, 2H), 8,27 8,17 (m, 2H), 7,34-7,18 (m, 5H), 4,00-3,89 (m, 2H), 3,34 (s, 3H). LCMS m/z = 419.0 (M+H).+
Ejemplo 8. El producto de la etapa 1 se hidrolizó según se describe para el intermedio 5 seguido de purificación mediante HPLC de fase inversa (14 mg, 28,9% de rendimiento) en forma de un sólido de color blanco. RMN 1H (400 MHz, DMSO-d6) 810,96 (s a, 1H), 8,67 (d, J = 7,5 Hz, 1H), 8,30-8,15 (m, 2H), 7,86-7,81 (m, 1H), 7,80-7,71 (m, 1H), 7,45-7,30 (m, 2H), 7,27-7,16 (m, 2H), 7,09 (d, J = 2,4 Hz, 1H), 6,78 (dd, J = 7,5, 2,6 Hz, 1H), 3,90 (s, 3H), 3,88 3,67 (m, 2H). LCMS m/z = 436,9 (M+H).+ Pureza por HPLC > 95 % con un tiempo de retención de 6,07 min. [Método C]
Ejemplo 9. 2-(4-(2-((6-bromo¡m¡dazo[1,2-a]μmdm-2-¡l)ammo)-2-oxoet¡l)-2-clorofenox¡)mcotmam¡da
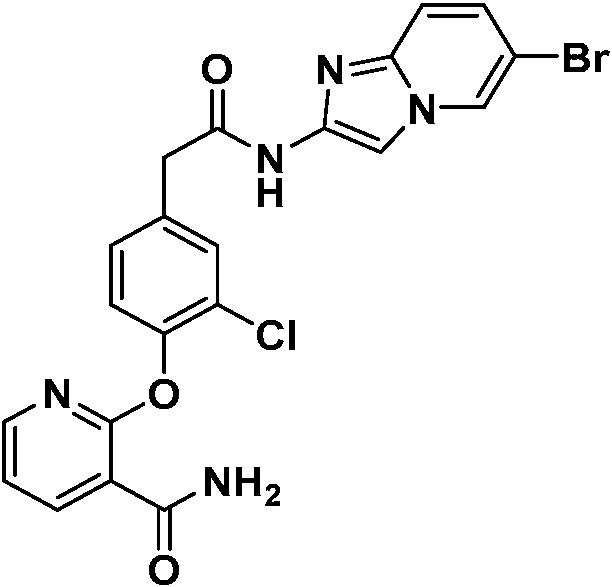
El ejemplo 9 se preparó mediante un acoplamiento de BOP, según se describe en el ejemplo 1, del intermedio 7 y 6-bromoimidazo[1,2-a]piridin-2-amina, y el residuo se purificó por HPLC de fase inversa para proporcionar el producto deseado (2,6 mg, 1,1 % de rendimiento). RMN 1H (500 MHz, DMSO-d6) 88,89 (s a, 1H), 8,21 (dd, J = 7,4, 1,7 Hz, 1H), 8,19-8,13 (m, 1H), 8,10 (s, 1H), 7,84 (s a, 1H), 7,79 (s a, 1H), 7,56 (s, 1H), 7,43 (d, J = 9,4 Hz, 1H), 7,41-7,28 (m, 3H), 7,24 (dd, J = 7,4, 4,9 Hz, 1H), 3,76 (s, 2H). LCMS m/z = 500-501,9 (M+H).+ Pureza por HPLC > 95 % con un tiempo de retención de 1,20 min. [Método B]
Ejemplo 10. 2-(2-cloro-4-(2-((1-met¡MH-p¡razolo[3,4-b]μmdm-3-il)ammo)-2-oxoetil)fenox¡)mcotmam¡da

Etapa 1. El intermedio 4 (28 mg, 0,097 mmol) se acopló a 1-met¡l-1H-p¡razolo[3,4-b]p¡r¡d¡n-3-am¡na (14 mg, 0,097 mmol) usando BOP según se describe en, por ejemplo 1, para proporcionar 2-(3-cloro-4-((3-c¡anop¡r¡d¡n-2-¡l)ox¡)feml)-N-(1-met¡l-1H-p¡razolo[3,4-b]p¡r¡d¡n-3-¡l)acetam¡da, que se usó sin más purificación. LCMS m/z = 419.0 (M+H).+
Etapa 2. Al intermedio de la etapa 1 se le añadió exceso de K2CO3 (0,054 g, 0,39 mmol), DMSO (0,3 ml) y H2O2 al 30 % (0,040 ml, 0,39 mmol). Después de 72 h, la mezcla de reacción se inactivó mediante la adición de NaSO3 ac., se filtró y se purificó por HPLC de fase inversa para proporcionar el ejemplo 10 (8 mg, 10 % de rendimiento). RMN 1H (500 MHz, DMSO-da) 8 11,04 (s, 1H), 8,53 (d a, J = 4,0 Hz, 1H), 8,39 (d a, J = 7,9 Hz, 1H), 8,22 (dd, J = 7,5, 1,7 Hz, 1H), 8,17 (dd, J = 4,7, 1,7 Hz, 1H), 7,81 (s a, 1H), 7,77 (s a, 1H), 7,59 (s, 1H), 7,48-7,31 (m, 2H), 7,24 (dd, J = 7,3, 4,9 Hz, 1H), 7,15 (dd, J = 8,2, 4,3 Hz, 1H), 3,98 (s, 3H), 3,83 (s, 2H). LCMS m/z = 437,1 (M+H).+ Pureza por HPLC > 99 % con un tiempo de retención de 1,23 min. [Método B]
Los ejemplos 11, 12, 14 y 16 se prepararon como se ha descrito siguiendo el procedimiento general para el ejemplo 1

continuación

Ejemplo 13. 2-(2-fluoro-4-(2-((4-(6-fluoropiridin-2-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

Etapa 1. A ácido 6-fluoropicolínico (980 mg, 6,95 mmol) en DCM (50 ml) se le añadió DMF (50 μl, 0,65 mmol), seguido de la adición gota a gota de cloruro de oxalilo (650 μl, 7,43 mmol). Después de 1,2 h, se añadió N,O-dimetilhidroxilamina HCl (1,05 g, 10,8 mmol) y la mezcla de reacción se enfrió a 0 °C. Se añadió piridina (2,6 ml, 32 mmol) y la mezcla de reacción se dejó calentar a temperatura ambiente y se agitó 72 h. La mezcla de reacción se inactivó mediante la adición de agua y las capas se separaron. La fase acuosa se extrajo con diclorometano y la capa orgánica combinada se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a presión reducida para proporcionar 6-fluoro-N-metoxi-N-metilpicolinamida (1,09 g, 85,0% de rendimiento) en forma de un aceite transparente de color amarillo claro. RMN 1H (500 MHz, CDaCl) 87,88 (c, J = 7,8 Hz, 1H), 7,56 (s a, 1H), 7,02 (dd, J = 8,2, 2,4 Hz, 1H), 3,79 (s a, 3H), 3,37 (s a, 3H). LCMS m/z = 185.0 (M+H).+
Etapa 2. A una solución a 0 °C de 6-fluoro-N-metoxi-N-metilpicolinamida (1,23 g, 6,68 mmol) en THF (40 ml) se le añadió gota a gota a gota CH3CH2MgBr (9 ml, 30 mmol). Después de 1,5 h a 0 °C la mezcla de reacción se inactivó cuidadosamente mediante la adición de NH4Cl acuoso. Las capas se repartieron y la capa acuosa se extrajo con Et2O. La capa orgánica combinada se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía sobre gel de sílice usando 30 % de EtOAc en hexanos como eluyentes. Se obtuvo 1-(6-fluoropiridin-2-il)propan-1-ona (684 mg, 66,9% de rendimiento) en forma de un aceite transparente incoloro. RMN 1H (500 MHz, CDaCl) 87,86-8,05 (m, 2H), 7,12 (ddd, J = 5,6, 3,1, 2,9 Hz, 1H), 3,16 (c, J = 7,3 Hz, 2H), 1,19 (t, J = 7,3 Hz, 3H). LCMS m/z = 154.0 (M+H).+
Etapa 3. A 1-(6-fluoropiridin-2-il)propan-1-ona (680 mg, 4,44 mmol) en AcOH (30 ml, 524 mmol), calentada a 90 °C, se le añadió Br2 (230 μl, 4,46 mmol). Después de 15 min, la mezcla de reacción se concentró al vacío. Al residuo se le añadió NaHCO3 ac. La capa acuosa se extrajo con Et2O. La capa orgánica combinada se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad para proporcionar 2-bromo-1-(6-fluoropiridin-2-il)propan-1-ona (1,01 g, 98,0 % de rendimiento) en forma de un aceite de color amarillo claro. RMN 1H (500 Mh z , CD3C087,88-8,05 (m, 2H), 7,17 (dd, J = 7,8, 2,6 Hz, 1H), 5,85 (c, J = 6,7 Hz, 1H), 1,88 (d, J = 6,7 Hz, 3H).
Etapa 4. A 2-bromo-1-(6-fluoropiridin-2-il)propan-1-ona (1,00 g, 4,31 mmol) en EtOH (30 ml) se le añadió tiourea (335 mg, 4,40 mmol). La mezcla de reacción se calentó a 80 °C durante 16 h. Después de enfriar a temperatura ambiente, el volumen del disolvente se redujo al vacío y la mezcla restante se puso en el congelador durante 2 h. Los sólidos precipitados se retiraron por filtración y se lavaron con Et2O. El sólido se suspendió en agua y se añadió NH4OH. La capa acuosa se extrajo con Et2O. La capa orgánica combinada se lavó con salmuera, se secó (MgSO4), se filtró y se concentró a sequedad para proporcionar 4-(6-fluoropiridin-2-il)-5-metiltiazol-2-amina (760 mg, 84 % de rendimiento) en forma de un sólido de color blanquecino. RMN 1H (500 m Hz , DMSO-d6) 87,98 (c, J = 8,3 Hz, 1H), 7,78 (dd, J = 7,6, 2,4 Hz, 1H), 6,99 (dd, J = 8,1, 2,6 Hz, 1H), 6,89 (s, 2H), 2,61 (s, 3H). LCMS m/z = 209.7 (M+H).+
El ejemplo 13 (3,4 mg, 5,05 % de rendimiento) se obtuvo mediante acoplamiento de BOP como se describe en el ejemplo 1, sustituyendo 4-(6-fluoropiridin-2-il)-5-metiltiazol-2-amina (23,8 mg, 0,114 mmol) por 7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-amina. RMN 1H (500 MHz, DMSO-d6) 8 12,45 (s a, 1H), 8,24-8,17 (m, 2H), 8,08 (c, J = 8,2 Hz, 1H), 7,89 (d a, J = 7,6 Hz, 1H), 7,83 (s a, 1H), 7,81-7,77 (m, 1H), 7,44-7,31 (m, 2H), 7,27-7,21 (m, 2H), 7,10 (dd, J = 8,1,2,0 Hz, 1H), 3,86 (s, 2H), 2,73 (s, 3H). LCMS m/z = 482,0 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,72 min. [Método B]
Ejemplo 15. 2-(4-(2-(benzo[d]tiazol-2-ilamino)-2-oxoetil)-2-fluorofenoxi)nicotinamida

El ejemplo 15 (0,6 mg, 2,2 % de rendimiento) se fabricó a partir del intermedio 8 mediante deshalogenación durante una reacción de Suzuki que implicó 1,3-dimetil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol. RMN 1H (500 MHz, DMSO-da) 8 8,25-8,15 (m, 2H), 7,88 (d a, J = 7,6 Hz, 1H), 7,83 (s a, 1H), 7,79 (s a, 1H), 7,66 (d a, J = 8,2 Hz, 1H), 7,45-7,31 (m, 3H), 7,27-7,14 (m, 3H), 3,82 (s, 2H). LCMS m/z = 422,8 (M+H).+ Pureza por HPLC >97 % con un tiempo de retención de 1,48 min. [Método A]
Ejemplo 17. 2-(2-fluoro-4-(2-((4-(1-metil-2-oxo-1,2-dihidroμmdm-4-N)tiazol-2-N)ammo)-2-oxoetil)fenoxi)nicotinamida.

Etapa 1. A una suspensión del intermedio 1 (1,0 g, 3,7 mmol) en DCE (10 ml) se le añadió SOCh (2,7 ml, 37 mmol) y la mezcla de reacción se calentó a 50 °C. Después de 1 h, los disolventes se eliminaron a presión reducida y se añadieron THF (10 ml), 4-bromotiazol-2-amina (0,70 g, 4,0 mmol) y DIEA (2 ml, 11 mmol). Después de 24 h, los disolventes se eliminaron a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice para proporcionar (1,1 g, 71 % de rendimiento) de N-(4-bromotiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida en forma de un sólido de color pardo. LCMS (ESI) m/z: 432,7-434,7 (M+H).+ r Mn 1H (400 MHz, CD3OD) 88,32 (dd, J = 5,0, 1,9 Hz, 1H), 8,27 (dd, J = 7,6, 1,9 Hz, 1H), 7,39-7,25 (m, 4H), 7,08 (s, 1H), 3,90 (s, 2H).
Etapa 2. Se oxidó N-(4-bromotiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida (1,0 g, 2,3 mmol) de una manera similar a la del intermedio 5 para proporcionar 2-(4-(2-((4-bromotiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida, HCl (0,98 g, 87% de rendimiento). LCMS(ESI) (ESI) m/z: 450,9-452,9 (M+H).+ RMN 1H (400 MHz, DMSO-d6) 812,72 (s, 1H), 8,24-8,17 (m, 2H), 7,87-7,81 (m, 1H), 7,80-7,73 (m, 1H), 7,45-7,30 (m, 3H), 7,25 (dd, J = 7,4, 5,0 Hz, 1H), 7,21 (dd, J = 8,8, 1,1 Hz, 1H), 3,86 (s, 2H)
Etapa 3. El ejemplo 17 (2,4 mg, 4,0 % de rendimiento) se preparó a partir del producto de la etapa 2 mediante un procedimiento de acoplamiento cruzado de Suzuki como se ha descrito anteriormente para el ejemplo 4. RMN 1H (500 MHz, DMSO-d6) 88,26-8,11 (m, 2H), 7,92 (s, 1H), 7,83 (s a, 1H), 7,79 (s a, 1H), 7,74 (d, J = 7,3 Hz, 1H), 7,43 7,31 (m, 2H), 7,27-7,18 (m, 2H), 6,91 (s, 1H), 6,74 (d a, J = 7,0 Hz, 1H), 3,84 (s, 2H), 3,41 (s, 3H). LCMS m/z = 480,1 (M+H).+ Pureza por HPLC 94 % con un tiempo de retención de 1,10 min. [Método B]
Ejemplo 18. 2-(2-fluoro-4-(2-oxo-2-((4-(2-oxo-1,2-dihidropiridin-4-il)tiazol-2-il)amino)etil)fenoxi)nicotinamida

Etapa 1. A 4-bromopiridin-2(1H)-ona (0,90 g, 5,2 mmol) en dioxano (10 ml) se le añadió 5,5,5',5'-tetrametil-2,2'-bi(1,3,2-dioxaborinano) (1,3 g, 5,7 mmol), acetato potásico (1,5 g, 16 mmol). La mezcla de reacción se desgasificó con N2 durante 15 min y se añadió PdCh(dppf)DCM (0,21 g, 0,26 mmol) y la mezcla de reacción se calentó a 90 °C durante 24 h. La mezcla de reacción se repartió con agua (20 ml) y acetato de etilo (50 ml). La capa acuosa se extrajo con acetato de etilo (2 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4), se filtraron y se concentraron a presión reducida para proporcionar 4-(5,5-dimetil-1,3,2-dioxaborinan-2-il)piridin-2(1H)-ona en forma de un aceite oscuro LCMS m/z = 208 (M+H).+
Etapa 2. El ejemplo 18 (3,7 mg, 6,0 % de rendimiento) se preparó a partir del producto de la etapa 2 de la misma manera que se ha descrito para el ejemplo 17, usando 4-(5,5-dimetil-1,3,2-dioxaborinan-2-il)piridin-2(1H)-ona. RMN 1H (500 MHz, DMSO-da) 88,29-8,13 (m, 2H), 7,85 (s, 1H), 7,71 (s a, 1H), 7,66 (s a, 1H), 7,46-7,28 (m, 3H), 7,27-7,04 (m, 2H), 6,84 (s, 1H), 6,68 (d a, J = 6,7 Hz, 1H), 3,83 (s, 2H), 2,99 (s, 1H). LCMS m/z = 465,9 (M+H).+ Pureza por HPLC 96 % con un tiempo de retención de 1,04 min. [Método A]
Ejemplo 19. (4-(2-(2-(4-((3-carbamoilpiridm-2-il)oxi)-3-fluorofeml)acetamido)-5-metiltiazol-4-il)piridm-2-il)carbamato de metilo

Etapa 1. Al intermedio 1 (0,3 g, 1 mmol) en THF (2 ml) se le añadió 4-bromo-5-metiltiazol-2-amina (0,2 g, 1 mmol) y piridina (0,3 ml, 3 mmol) y la mezcla de reacción se agitó durante 18 h. La mezcla de reacción se concentró a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice para proporcionar N-(4-bromo-5-metiltiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida (0,46 g, 99 % de rendimiento). LCMS (ESI) m/z: 444-446,8 (M+H).+ RMN 1H (400 MHz, DMSO-d6) 812,54 (s, 1H), 8,47 (dd, J = 7,5, 2,0 Hz, 1H), 8,43-8,36 (m, 1H), 7,45-7,32 (m, 3H), 7,23 (dd, J = 8,3, 1,0 Hz, 1H), 3,85 (s, 2H), 2,27 (s, 3H).
Etapa 2. Una mezcla de 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo (0,13 g, 0,27 mmol), KOAc (4 g, 40 mmol), precatalizador xphos de 2a generación (0,1 g, 0,1 mmol), (4-cloropiridin-2-il)carbamato de metilo (2,5 g, 13 mmol) y ácido hipodibórico (1,8 g, 20 mmol) en EtOH (130 ml) se desgasificó tres veces mediane un ciclo de vaciado/llenado con N2. La mezcla de reacción se calentó a 80 °C durante 3 h. El disolvente se eliminó mediante vacío y el sólido se suspendió con una mezcla de metanol y DCM. La suspensión se filtró y el filtrado se concentró para dar ácido (2-((metoxicarbonil)amino)piridin-4-il)borónico en forma de un sólido de color amarillo. LCMS (ESI) m/z: 197,1 (M+H).+
Etapa 3. El ejemplo 19 (1 mg, 3 % de rendimiento) se preparó a partir del producto de la etapa 2 de una manera similar a la del ejemplo 4 usando N-(4-bromo-5-metiltiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida y ácido (2-((metoxicarbonil)amino)piridin-4-il)borónico. RMN 1H (500 MHz, DMSO-d6) 88,30 (d, J = 5,2 Hz, 1H), 8,21 (d a, J = 4,0 Hz, 2H), 8,19 (s, 1H), 7,83 (s a, 1H), 7,79 (s a, 1H), 7,39-7,31 (m, 3H), 7,27-7,23 (m, 1H), 7,21 (d a, J = 8,2 Hz, 1H), 3,91 (s, 1H), 3,77 (s a, 2H), 3,70 (s, 3H), 2,56 (s, 3H), 1,65 (s, 1H). LCMS m/z = 537,1 (M+H).+ Pureza por HPLC 98 % con un tiempo de retención de 1,46 min. [Método A]
Ejemplo 20. 2-(2-fluoro-4-(2-((5-metil-4-(1-metil-6-oxo-1,6-dihidroμmdm-3-N)tiazol-2-N)ammo)-2-oxoetil)fenoxi)nicotinamida

Etapa 1. A 5-bromo-1-metilpiridin-2(1H)-ona (0,75 g, 3,9 mmol) en dioxano se le añadió 5,5,5',5'-tetrametil-2,2'-bi(1,3,2-dioxaborinano) (1,0 g, 4,4 mmol), KOAc (1,2 g, 12 mmol). La mezcla de reacción se desgasificó con N2, después se añadió PdCh(dppf)-DCM (0,16 g, 0,19 mmol) y la mezcla de reacción se calentó a 90 °C durante 18 h. La mezcla de reacción se dejó enfriar, se filtró y se repartió con agua (20 ml) y EtOAc (50 ml). La capa acuosa se extrajo con EtOAc (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4), se filtraron y se concentraron a presión reducida para proporcionar 5-(5,5-dimetil-1,3,2-dioxaborinan-2-il)-1-metilpiridin-2(1H)-ona (0,67 g, 76 % de rendimiento) en forma de un aceite oscuro). LCMS m/z = 222 (M+H).+
Etapa 2. El ejemplo 20 (1,8 mg, 5,0 % de rendimiento) se preparó a partir del producto de la etapa 1 de una manera similar a la usada para el ejemplo 4, usando 5-(5,5-dimetil-1,3,2-dioxaborinan-2-il)-1-metilpiridin-2(1H)-ona y N-(4-bromo-5-metiltiazol-2-il)-2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetamida. RMN 1H (500 Mh z , DMSO-d6) 88,21 (d a, J = 7,5 Hz, 1H), 8,18 (d a, J = 2,9 Hz, 1H), 7,89 (s a, 1H), 7,73 (dd a, J = 9,3, 2,3 Hz, 2H), 7,63 (s a, 1H), 7,41-7,29 (m, 2H), 7,24 (dd, J = 7,3, 5,0 Hz, 1H), 7,21 (d a, J = 8,2 Hz, 1H), 6,48 (d, J = 9,3 Hz, 1H), 3,81 (s, 2H), 3,51 (s, 3H), 2,42 (s, 3H), 1,25 (s a, 1H). LCMS m/z = 494,2 (M+H).+ Pureza por HPLC 92 % con un tiempo de retención de 1,20 min. [Método A]
Ejemplo 21. 2-(4-(2-((4-(1-(difluorometil)-6-oxo-1,6-dihidropiridin-3-il)-5-metiltiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida
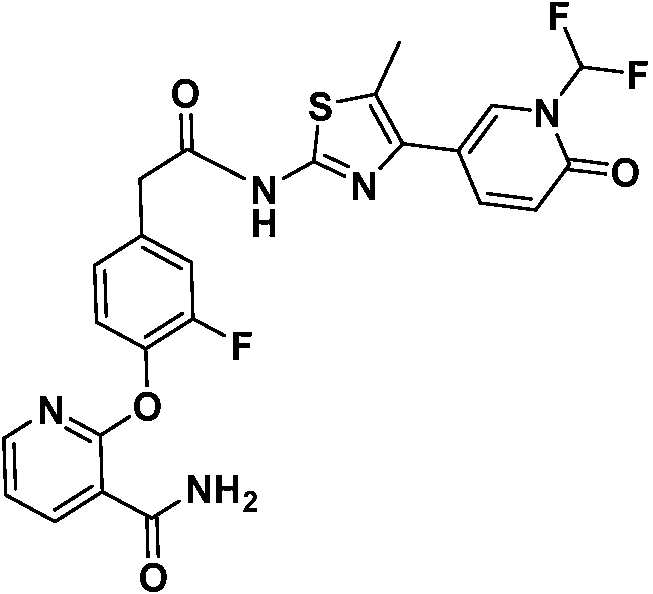
El ejemplo 21 (11 mg, 31 % de rendimiento) se preparó de una manera similar a la del ejemplo 20 sustituyendo 5-bromo-1-(difluorometil)piridin-2(1H)-ona por 5-bromo-1-metilpiridin-2(1H)-ona. RMN 1H (500 MHz, DMSO-d6) 812,27 (s a, 1H), 8,28-8,13 (m, 2H), 7,94-7,85 (m, 3H), 7,71 (s a, 1H), 7,65 (s a, 1H), 7,41-7,30 (m, 2H), 7,28-7,14 (m, 2H), 6,63 (d, J = 9,3 Hz, 1H), 3,83 (s, 2H), 2,45 (s, 3H). LCMS m/z = 529,9 (M+H).+ Pureza por HPLC 100% con un tiempo de retención de 1,42 min. [Método B]
Ejemplo 22. 2-(2-cloro-4-(2-((5-metil-4-(6-metilpiridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

Etapa 1. A 1-(6-metilpiridin-2-il)propan-1-ona (0,3 g, 2 mmol) y AcOH (3 ml), calentados a 90 °C, se les añadió Br2 (0,1 ml, 2 mmol) en AcOH (1 ml). Después de 1 h, los disolventes se eliminaron a presión reducida y al residuo se le añadió EtOH (15 ml) y tiourea (0,15 g, 2,0 mmol) y la mezcla de reacción se calentó a 60 °C durante 72 h. Los disolventes se eliminaron a presión reducida y el residuo se repartió con NH4OH dil. (20 ml) y EtOAc (50 ml). La capa acuosa se extrajo con acetato de etilo (2 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4) para proporcionar 5-metil-4-(6-metilpiridin-2-il)tiazol-2-amina (0,3 g, 70 % de rendimiento) de un sólido de color castaño. RMN 1H (400 MHz, CDCh) 87,68-7,51 (m, 2H), 7,05 (dd, J = 7,3, 1,1 Hz, 1H), 4,91 (s a, 2H), 2,68 (s, 3H), 2,61 (s, 3H). LCMS (ESI) m/z: 206,0 (M+H).+
Etapa 3. El ejemplo 22 (2 mg, 3 % de rendimiento) se obtuvo de una manera similar a la del ejemplo 2 usando el intermedio 7 y 5-metil-4-(6-metilpiridin-2-il)tiazol-2-amina. RMN 1H (500 MHz, DMSO-d6) 812,39 (s, 1H), 8,26-8,20 (m, 1H), 8,20-8,15 (m, 1H), 7,81 (s a, 1H), 7,78-7,72 (m, 3H), 7,57 (s, 1H), 7,31-7,21 (m, 2H), 7,19-7,09 (m, 2H), 3,84 (s, 2H), 2,75 (s, 3H), 2,52 (s, 3H), LCMS m/z = 494,0 (M+H).+ Pureza por HPLC 96 % con un tiempo de retención de 1,57 min. [Método B]
Ejemplo 23. 2-(2-doro-4-(2-((4-ddobutM-5-(pindm-2-il)tiazol-2-il)ammo)-2-oxoetil)fenoxi)mcotmamida

Etapa 1. 1-ciclobutil-2-(piridin-2-il)etanona (2,40 g, 13,7 mmol) y NaOAc (1,24 g, 15,1 mmol) se combinaron y se disolvieron en AcOH (75 ml). Se añadió Br2 (0,5 ml, 9 mmol) a la mezcla de reacción en una porción. La mezcla de reacción se agitó a ta durante 20 min, después se concentró a presión reducida. Al residuo se le añadió agua y se extrajo con EtOAc (2 x 100 ml). Los extractos orgánicos combinados se lavaron dos veces con NaHCO3 ac., salmuera, se secaron (MgSO4), se filtraron y después se concentraron a sequedad. El residuo se purificó por cromatografía sobre gel de sílice usando hexanos/EtOAc. Se obtuvo 2 -bromo-1 -ciclobutil-2 -(piridin-2 -il)etanona (2 g, 60 % de rendimiento) en forma de un sólido de color amarillo claro. RMN 1H (500 MHz, CDCla) 88,52 (d, J = 4,88 Hz, 1H) 7,72 (td, J = 7,71, 1,68 Hz, 1H) 7,56 (d, J = 7,93 Hz, 1H) 7,23 (dd, J = 7,48, 4,73 Hz, 1H) 5,53 (s, 1H) 3,64 (quint., J = 8,47 Hz, 1H) 2,18 2,40 (m, 2H) 2,00-2,18 (m, 2H) 1,87- 2,00 (m, 1 H) 1,74-1,87 (m, 1 H). LCMS m/z = 254,0, 256,0 (M+H).+
Etapa 2. A 2-bromo-1-ciclobutil-2-(piridin-2-il)etanona (2 g, 8 mmol) en EtOH (50 ml), se le añadió K2CO3 (1,1 g, 8,1 mmol) seguido de tiourea (0,6 g mg, 8 mmol). La mezcla de reacción se calentó a 60 °C durante 18 h, se enfrió y la mezcla de reacción se filtró. El filtrado se concentró a presión reducida. Al residuo se le añadió agua y se extrajo con EtOAc (2 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y después se concentraron a sequedad. El residuo se trituró en DCM y hexanos. Los sólidos se eliminaron por filtración y se lavaron con hexanos para proporcionar 4-ciclobutil-5-(piridin-2-il)tiazol-2-amina (1,3 g, 74 % de rendimiento), que se obtuvo en forma de un sólido de color castaño. RMN 1H (500 MHz, DMSO-cfe) 8 8,65 (s, 0,5H) 8,34-8,56 (m, 1H) 7,59-7,83 (m, 1H) 7,31-7,44 (m, 1H) 7,20 (s, 1,5H) 7,02-7,16 (m, 1H) 4,01 (quint., J = 8,62 Hz, 0,4H) 3,91 (quint., J =
8,55 Hz, 0,6H) 2,30-2,46 (m, 2H) 2,10-2,30 (m, 2H) 1,94 (spt, J = 9,41 Hz, 1H) 1,70-1,87 (m, 1H). LCMS m/z = 232.0 (M+H).+
Etapa 3. El ejemplo 23 (5 mg, 7 % de rendimiento) se obtuvo de manera similar a la descrita para el ejemplo 2 usando el intermedio 7 y 4-ciclobutil-5-(piridin-2-il)tiazol-2-amina RMN 1H (500 MHz, DMSO-da) 88,55 (d a, J = 4,3 Hz, 1H), 8,23 (d a, J = 7,4 Hz, 1H), 8,16 (d a, J = 3,4 Hz, 1H), 7,83 (t, J = 7,7 Hz, 1H), 7,69 (s a, 1H), 7,64 (s a, 1H), 7,55 (s, 1H), 7,50 (d, J = 8,0 Hz, 1H), 7,36 (c, J = 8,3 Hz, 2H), 7,25 (td, J = 8,0, 5,0 Hz, 2H), 4,00 (quint., J = 8,3 Hz, 1H), 3,85 (s, 2H), 2,43-2,24 (m, 4H), 2,00 (sxt, J = 9,3 Hz, 1H), 1,93-1,81 (m, 1H) LCMS m/z = 520,2 (M+H).+ Pureza por HPLC 95 % con un tiempo de retención de 1,91 min. [Método A]
Los siguientes ejemplos 24-48, se prepararon como se ha descrito siguiendo el procedimiento general usado para el ejemplo 4







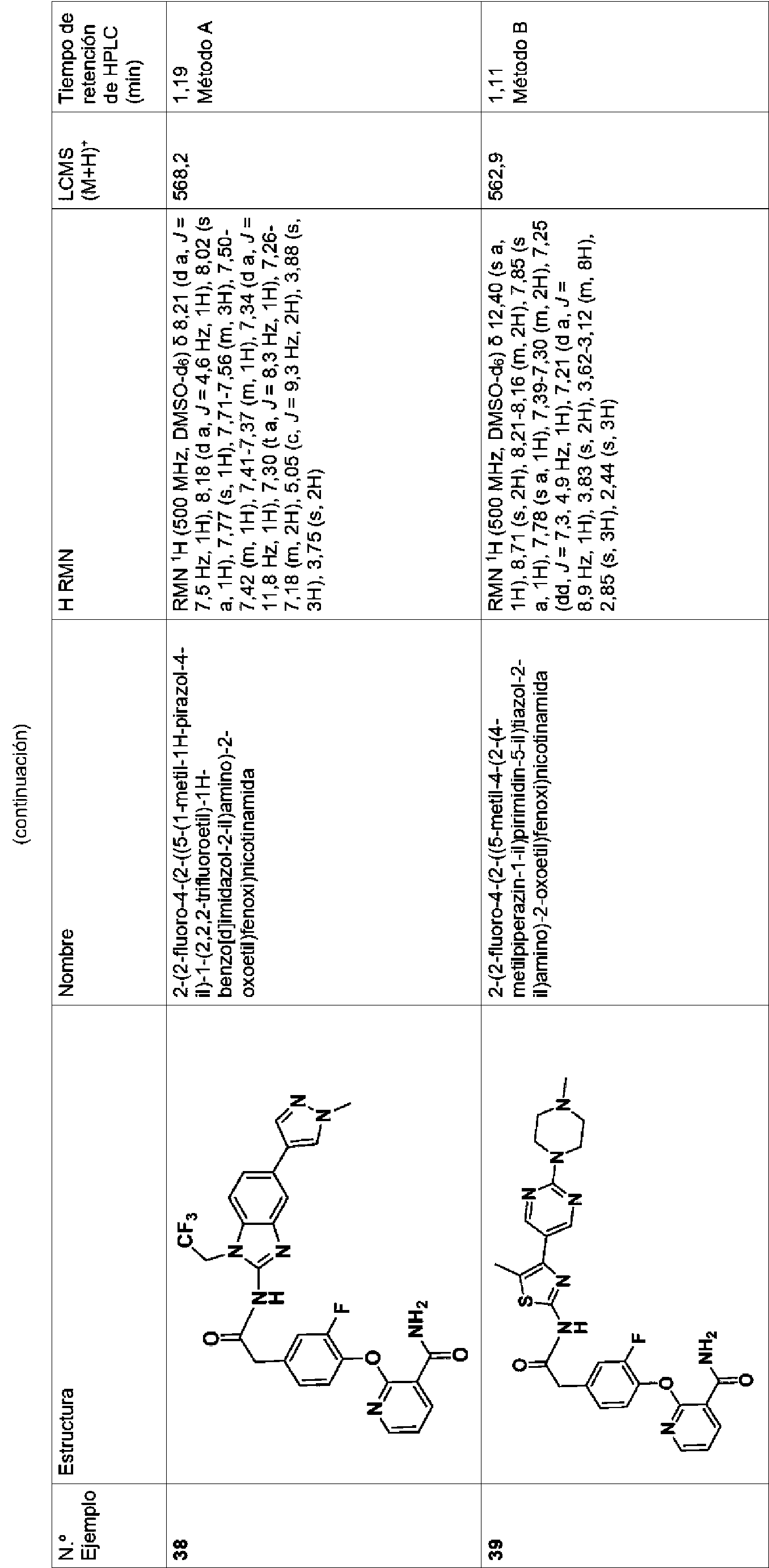



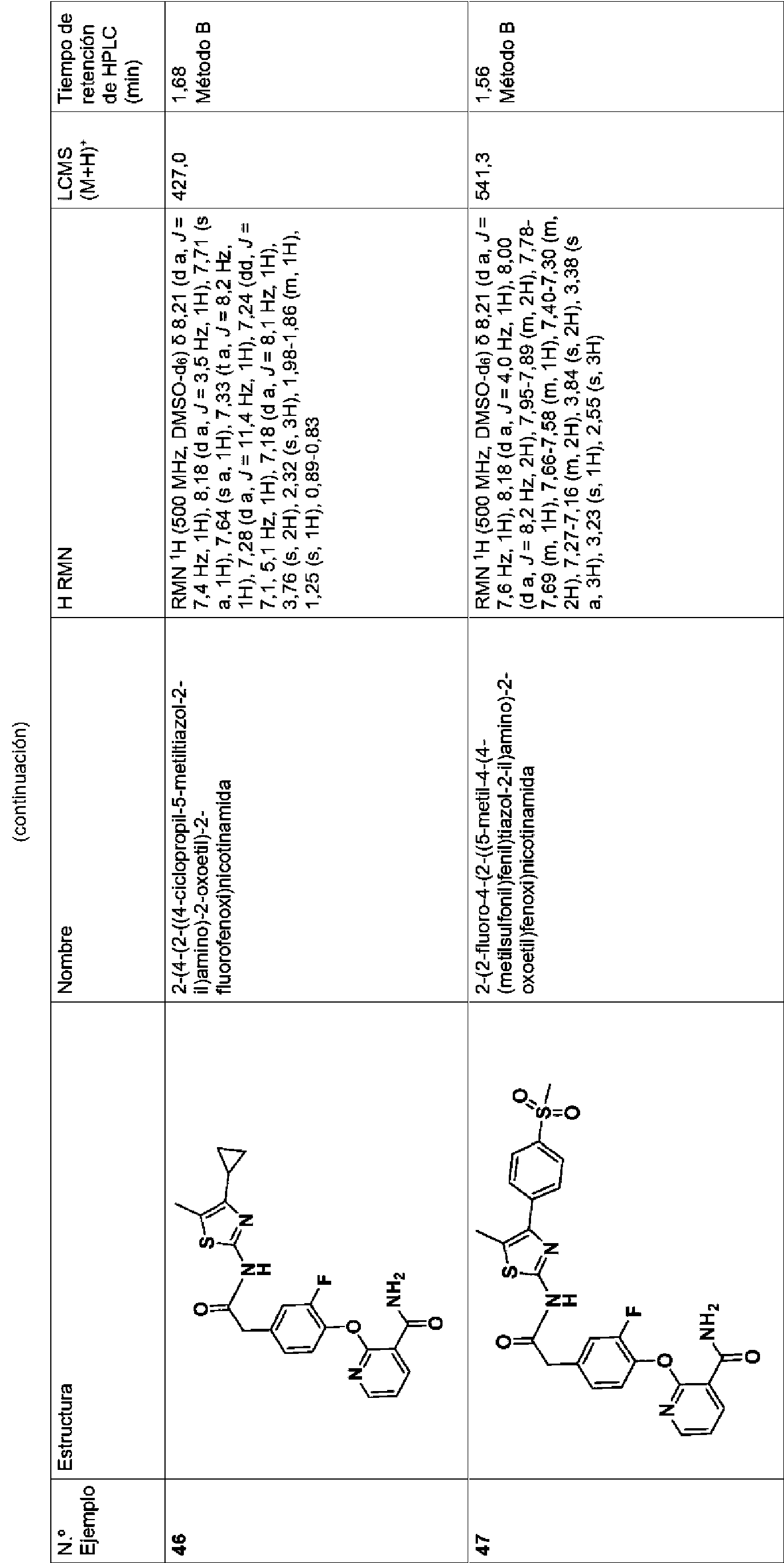

Los ejemplos 49-105 se prepararon siguiendo el procedimiento general usado para el ejemplo 2































Ejemplo 106. 2-(4-(1-((7-metoxy-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-1-oxopropan-2-il)fenoxi)nicotinamida.

E tap a 1. A 2-(4-hidroxifenil)acetato de metilo (2,23 g, 13,4 mmol) en DMSO (10 ml) se le añadió 2-(4-hidroxifenil)acetato de metilo (2,23 g, 13,4 mmol) y K2CO3 (4,08 g, 29,5 mmol) y la mezcla de reacción se calentó a 60 °C durante 24 h. Se añadió agua (25 ml) y la mezcla de reacción se extrajo con acetato de etilo (3 x 30 ml). Las capas orgánicas combinadas se lavaron con agua (25 ml), después salmuera (25 ml) y se secaron (MgSO4), se filtraron y se concentraron a presión reducida para proporcionar 2-(4-((3-cianopiridin-2-il)oxi)fenil)acetato de metilo (2,9 g, 81 % de rendimiento) en forma de un sólido de color castaño; RMN 1H (400 MHz, CDCh) 88,34 (dd, J = 5,1, 2,0 Hz, 1H), 8,04 (dd, J = 7,5, 2,0 Hz, 1H), 7,48-7,37 (m, 2H), 7,23-7,15 (m, 2H), 7,12 (dd, J = 7,5, 5,1 Hz, 1H), 3,74 (s, 3H), 3,69 (s, 2H). LCMS (ESI) m /z : 268,9 (M+H).+
E tap a 2.
A 2-(4-((3-cianopiridin-2-il)oxi)fenil)acetato de metilo (0,20 g, 0,74 mmol) y yodometano (0,046 ml, 0,74 mmol) en DMF (3 ml), enfriados a 0 °C, se les añadió NaH (0,066 g, 1,6 mmol)(suspensión al 60 % en aceite). Después de 24 h, la mezcla de reacción se inactivó mediante la adición de agua (10 ml) y se extrajo con acetato de etilo (3 x 10 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4). Se obtuvo 2-(4-((3-cianopiridin-2-il)oxi)fenil)propanoato de metilo (0,149 g) en forma de un aceite de color amarillo y se usó sin más purificación en la etapa siguiente. LCMS (ESI)
m /z :
282,9 (M+H).+
E ta p a 3. Se obtuvo ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)propanoico (0,2 g) a partir de la oxidación en H 2O2 de 2-(4-((3-cianopiridin-2-il)oxi)fenil)propanoato de metilo según se describe para el intermedio 5, seguido de la adición de LiOH (0,2 g). Después de 24 h, la mezcla de reacción se acidificó a pH = 5 mediante la adición de HCl 1 N y se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4), se filtraron y se concentraron a presión reducida para proporcionar un sólido de color blanco. LCMS (ESI) m /z : 286,9 (M+H).+
El ejemplo 106 (1,4 mg, 4,5% ) se preparó mediante el acoplamiento de ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)propanoico y 7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-amina de una manera similar a como se describe para el ejemplo 1. RMN 1H (500 MHz, DMSO-d6) 88,15 (s a, 2H), 7,94-7,81 (m, 1H), 7,80-7,70 (m, 1H), 7,67-7,51 (m, 1H), 7,42 (d, J = 7,7 Hz, 2H), 7,24-7,15 (m, 1H), 7,14 (d, J = 7,7 Hz, 2H), 6,85 (s a, 2H), 4,41-4,30 (m, 1H), 4,08 - 3,95 (m, 1H), 2,92 (d, J = 7,3 Hz, 2H), 2,90-2,80 (m, 2H), 1,45 (d, J = 6,6 Hz, 3H) un Me bajo el disolvente. LCMS m/z = 500,9 (M+H).+ Pureza por HPLC 95,9 % con un tiempo de retención de 1,91 min. [Método A]
Ejemplo 107. 2-(4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

El ejemplo 107 (3,3 mg, 6,8 % de rendimiento) se preparó de una manera similar a la del ejemplo 3 sustituyendo el intermedio 6 por el intermedio 5. RMN 1H (500 MHz, DMSO-da) 58,23-8,11 (m, 2H), 7,82 (d, J = 8 ,5 Hz, 2H), 7,78 (s a, 1H), 7,40 (d, J = 8,2 Hz, 2H), 7,27 (s, 1H), 7,25-7,19 (m, 1H), 7,16 (d, J = 8,2 Hz, 2H), 6,96 (d, J = 8,6 Hz, 1H), 4,69 (s a, 1H), 3,85 (s, 2H), 3,78 (s, 2H), 1,23 (s, 6H). LCMS m/z = 493,0 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,39 min. [Método B]
Ejemplo 108. 2-(4-(2-((7-(2-hidroxi-2-metilpropoxi)-4,5-dihidronafto[1,2-d]tiazol-2-N)ammo)-2-oxoetil)fenoxi)nicotinamida

E tap a 1. A 7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-amina, bromhidrato (0,22 g, 0,70 mmol) en agitación en DCM (5 ml) se le añadió BBr3 (0,702 ml, 0,702 mmol). Después de 72 h,
La mezcla de reacción se enfrió a 0 °C, se inactivó mediante la adición de K2CO3 acuoso y MeOH, se filtró y se concentró a presión reducida y se purificó por cromatografía sobre gel de sílice usando DCM/MeOH al 0-10 % como eluyentes para proporcionar 2-amino-4,5-dihidronafto[1,2-d]tiazol-7-ol (0,22 g) en forma de un sólido de color pardo claro. LCMS (ESI) m /z : 218,8 (M+H).+
E ta p a 2.
Se preparó 1-((2-amino-4,5-dihidronafto[1,2-d]tiazol-7-il)oxi)-2-metilpropan-2-ol (0,12 g, 61 %) a partir de 2-amino-4,5-dihidronafto[1,2-d]tiazol-7-ol de una manera similar a la descrita en el ejemplo 3. RMN 1H (400 MHz, CDCh) 5 7,66-7,62 (m, 1H), 6,87-6,79 (m, 2H), 4,91 (s a, 2H), 3,84 (s, 2H), 3,07-2,97 (m, 2H), 2,90-2,82 (m, 2H), 2,44-2,24 (m, 1H), 1,38 (s, 6H), LCMS (ESI)
m /z :
290,9 (M+H).+
El ejemplo 108 (4,3 mg, 8,4%) se obtuvo mediante el acoplamiento de 1-((2-amino-4,5-dihidronafto[1,2-d]tiazol-7-il)oxi)-2-metilpropan-2-ol y ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acético como se describe en el ejemplo 2. RMN 1H (500 MHz, DMSO-d6) 58,20-8,10 (m, 2H), 7,84 (s a, 1H), 7,76 (s a, 1H), 7,56 (d,
J
= 8,3 Hz, 1H), 7,38 (d,
J
= 8,2 Hz, 2H), 7,25-7,18 (m, 1H), 7,14 (d,
J
= 8,2 Hz, 2H), 6,90-6,83 (m, 1H), 6,86-6,78 (m, 1H), 4,78 (s a, 1H), 3,00-2,91 (m, 2H), 2,90-2,81 (m, 2H), 1,27-1,16 (m, 7H), picos ocultos bajo el pico de agua. LCMS m/z = 545,1 (M+H).+ Pureza por HPLC 99 % con un tiempo de retención de 1,59 min. [Método B]
Ejemplo 109. 2-(4-(2-((6-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

El ejemplo 109 (5,2 mg, 11 %) se preparó como se ha descrito anteriormente para el ejemplo 3 sustituyendo el intermedio 6 por el intermedio 5. RMN 1H (500 MHz, DMSO-d6) 58,23-8,12 (m, 2H), 7,79 (s a, 1H), 7,75 (s a, 1H), 7,63 (d,
J
= 8,8 Hz, 1H), 7,54 (s, 1H), 7,39 (d,
J
= 8,2 Hz, 2H), 7,21 (s a, 1H), 7,20-7,11 (m, 2H), 7,04 (d,
J
= 8,8 Hz, 1H), 3,83 (s, 2H), 3,75 (s, 2H), 1,27-1,15 (m, 8H); LCMS m/z = 493,0 (M+H),+ pureza por HPLC 97 % con un tiempo de retención de 1,32 min. [Método B]
Ejemplo 110. 2-(2-fluoro-4-(2-((7-(2-hidroxi-2-metilpropoxi)-[1,2,4]triazolo[1,5-a] piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

E tap a 1.
A 7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-amina (0,23 g, 1,4 mmol) en AcOH (3 ml) se le añadió HBr al 48
%
(1,5 ml) y la mezcla de reacción se calentó a 100 °C durante 48 h. Los disolventes se eliminaron a presión reducida y el residuo se destiló azeotrópicamente con tolueno (2 x 10 ml) para proporcionar amino-[1,2,4]triazolo[1,5-a]piridin-7-ol, bromhidrato (0,30 g, 36 % de rendimiento) sólido de color pardo. Lc Ms m/z = 151.1 (M+H).+
E ta p a 2. Se preparó 1-((2-amino-[1,2,4]triazolo[1,5-a]piridin-7-il)oxi)-2-metilpropan-2-ol (23 mg, 23 % de rendimiento) a partir de bromhidrato de amino-[1,2,4]triazolo[1,5-a]piridin-7-ol de una manera similar a la descrita para el ejemplo 3. RMN 1H (400 MHz, CD3OD) 88,55 (d, J = 7,5 Hz, 1H), 7,19 (d, J = 2,6 Hz, 1H), 7,12 (dd, J = 7,4, 2,8 Hz, 1H), 4,04 (s, 2H), 3,34 (dt, J = 3,3, 1,7 Hz, 1H), 1,37 (s, 6H). LCMS m/z = 223.1 (M+H).+
E ta p a 3.
El ejemplo 110 (7,0 mg, 12 % de rendimiento) se obtuvo de una manera similar a la descrita para el ejemplo 8 usando el intermedio 2 y 1-((2-amino-[1,2,4]triazolo[1,5-a]piridin-7-il)oxi)-2-metilpropan-2-ol. RMN 1H (500 m Hz , DMSO-da) 810,95 (s a, 1H), 8,65 (s a, 1H), 8,27-8,14 (m, 2H), 7,82 (s a, 1H), 7,78 (s a, 1H), 7,44-7,29 (m, 2H), 7,28 7,17 (m, 2H), 7,06 (s a, 1H), 6,78 (d a,
J
= 7,3 Hz, 1H), 4,75 (s, 1H), 3,86 (s, 2H), 3,39 (s, 2H), 1,23 (s, 6H). LCMS m/z = 495,3 (M+H).+ Pureza por HPLC 99 % con un tiempo de retención de 1,05 min. [Método A]
Ejemplo 111. 2-(4-(2-((1 -(2,2-difluoroetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida

E tap a 1.
Al intermedio 5 (0,8 g, 3 mmol) en MeOH (25 ml) se le añadió exceso de cloruro de tionilo (1,5 ml, 21 mmol). Después de 24 h, la mezcla de reacción se concentró a presión reducida y se purificó por cromatografía sobre gel de sílice usando hexano/EtOAc como eluyentes para proporcionar (0,30 g, 35 %) 2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acetato de metilo LCMS (ESI)
m /z :
304,8 (M+H).+
E tap a 2.
A 1-(2,2-difluoroetil)-1H-benzo[d]imidazol-2-amina (15 mg, 0,076 mmol) y 2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acetato de metilo (28 mg, 0,091 mmol) en tolueno (2 ml) se le añadió trimetilaluminio (0,11 ml, 0,23 mmol). Después de 5 minutos, la mezcla de reacción se calentó con irradiación con microondas a 120 °C durante 30 min. La mezcla de reacción se concentró y se inactivó mediante la adición de TFA y se purificó por HPLC de fase inversa para proporcionar (1,9 mg, 4,1 %) del ejemplo 111.
RMN 1H (500 MHz, DMSO-d6) 88,18 (d,
J
= 7,2 Hz, 2H), 7,83 (d,
J
= 11,2 Hz, 2H), 7,66-7,44 (m, 2H), 7,44-7,12 (m, 6H), 4,69-4,55 (m, 2H), 3,71 (s a, 2H). LCMS m/z = 470,1 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,19 min. [Método B]
Ejemplo 112. 2-(2-cloro-4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

El ejemplo 112 (4 mg, 7
%
de rendimiento) se fabricó de una manera similar a la del ejemplo 108 sustituyendo el intermedio 4 por el intermedio 5. RMN 1H (500 MHz, DMSO-da) 88,39-8,07 (m, 2H), 7,79 (d a,
J
= 12,8 Hz, 2H), 7,64 (d a,
J
= 8,9 Hz, 1H), 7,56 (d a,
J
= 13,1 Hz, 1H), 7,38 (s, 2H), 7,24 (dd a,
J
= 7,2, 5,0 Hz, 1H), 7,05 (d a,
J
= 8,5 Hz, 1H), 4,03-3,84 (m, 2H), 3,76 (s, 2H), 1,21 (s, 6H), LCMS m/z = 526,9 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,45 min. [Método A]
Ejemplo 113. 2-(2-doro-4-(2-((7-(2-hidroxi-2-metilpropoxi)-4,5-dihidronafto[1,2-d]tiazol-2-N)ammo)-2-oxoetil)fenoxi)nicotinamida

El ejemplo 113 (2 mg, 3 % de rendimiento) se fabricó de una manera similar a la del ejemplo 108 sustituyendo 1-((2-amino-4,5-dihidronafto[1,2-d]tiazol-7-il)oxi)-2-metilpropan-2-ol por 1-((2-aminobenzo[d]tiazol-5-il)oxi)-2-metilpropan-2-ol. RMN 1H (500 MHz, DMSO-d6) 88,39-8,07 (m, 2H), 7,79 (d a,
J
= 12,8 Hz, 2H), 7,64 (d a,
J
= 8,9 Hz, 1H), 7,56 (d a,
J
= 13,1 Hz, 1H), 7,38 (s, 2H), 7,24 (dd a,
J
= 7,2, 5,0 Hz, 1H), 7,05 (d a,
J
= 8,5 Hz, 1H), 4,03-3,84 (m, 2H), 3,76 (s, 2H), 1,21 (s, 6H), LCMS m/z = 579,2 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,69 min. [Método A]
Ejemplo 114. 2-(2-fluoro-4-(2-(6-(2-hidroxi-2-metilpropoxi)indolin-1-il)-2-oxoetil)fenoxi)nicotinamida (no de acuerdo con la invención)
E tap a 1. Se obtuvo 6-(2-hidroxi-2-metilpropoxi)indolin-1-carboxilato de tere-butilo (0,12 g, 0,40 mmol, 94% de rendimiento) en forma de un aceite transparente de una manera similar a la descrita en el ejemplo 3, sustituyendo 6-hidroxiindolin-1-carboxilato de tere-butilo por 2-aminobenzo[d]tiazol-5-ol). RMN 1H (400 MHz, CDCh) 87,65-7,45 (m, 1H), 7,04 (d, J = 8,1 Hz, 1H), 6,52 (dd, J = 8,1, 2,4 Hz, 1H), 4,07-3,94 (m, 2H), 3,82 (s, 2H), 3,04 (t, J = 8,6 Hz, 2H), 2,16 (s a, 1H), 1,59 (s a, 9H), 1,35 (s, 6 H), LCMS (ESI) m /z : 308,1 (M+H).+
E tap a 2.
A 6-(2-hidroxi-2-metilpropoxi)indolin-1-carboxilato de tere-butilo (0,12 g, 0,40 mmol) en DCM se le añadió exceso de HCl 4 N en dioxano (5 ml) y se dejó en agitación 24 h. La mezcla de reacción se concentró a presión reducida para proporcionar 1-(indolin-6-iloxi)-2-metilpropan-2-ol, HCl (98 mg, 100 % de rendimiento) en forma de una película oscura. LCMS (ESI)
m /z:
208,1 (M+H).+
E tap a 3. 1-(indolin-6-iloxi)-2-metilpropan-2-ol, Se acopló HCl a ácido 2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acético, HCl como se describe en el ejemplo 3 para proporcionar el ejemplo 114 (4 mg, 11 % de rendimiento). RMN 1H (500 MHz, DMSO-d6) 88,23-8,13 (m, 2H), 7,88 (s a, 1H), 7,79 (s a, 1H), 7,71 (s, 1H), 7,33 (t, J = 8,2 Hz, 1H), 7,29-7,20 (m, 2H), 7,13 (dd, J = 19,1, 8,2 Hz, 2H), 6,57 (dd, J = 8,2, 2,1 Hz, 1H), 4,20 (t, J = 8,4 Hz, 2H), 3,95-3,86 (m, 2H), 3,21-3,03 (m, 2H), 1,17 (s, 6 H). LCMS m/z = 480,3 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,48 min. [Método A]
Ejemplo 115. 2-(2-cloro-4-(2-((5-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

Al intermedio 4 (28 mg g, 0,090 mmol) y 5-metoxibenzo[d]tiazol-2-amina (17 mg, 0,090 mmol) se le añadió 1 ml de DCM seguido de piridina (7,8 μl, 0,090 mmol) y POCh (9 μl, 0,09 mmol). Después de agitar durante 24 h, los disolventes se eliminaron a presión reducida y el residuo se repartió con NaHCO3 sat. (2 ml) y EtOAc (3 ml). La capa acuosa se extrajo con EtOAc (2 ml). Las capas orgánicas combinadas se concentraron a presión reducida y al residuo se le añadió exceso de K2CO3 (54 mg, 0,38 mmol), algunas escamas de MgO y DMSo (0,3 ml). A la mezcla de reacción se le añadió H 2O2 (0,040 ml, 0,38 mmol). Después de agitar durante 24 h, la mezcla de reacción se inactivó mediante la adición de bisulfito sódico ac. (2 ml) y se extrajo con EtOAc (3 x 2 ml). Los disolventes se eliminaron a presión reducida y el residuo se purificó por HPLC de fase inversa para proporcionar (3,1 mg, 5,0 % de rendimiento) del ejemplo 115. RMN 1H (500 MHz, DMSO-d6) 88,22 (d, J = 7,6 Hz, 1H), 8,16 (d, J = 5,3 Hz, 1H), 7,87-7,73 (m, 3H), 7,57 (s, 1H), 7,38 (s, 2H), 7,33-7,20 (m, 2H), 6,94 (dd, J = 8,7, 2,3 Hz, 1H), 3,89 (d, J = 3,1 Hz, 3H), 3,82 (s, 2H). LCMS m/z = 496,1 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,55 min. [Método B]
Ejemplo 116. 2-(2-cloro-4-(2-((7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

El ejemplo 116 (4,3 mg, 7,2 % de rendimiento) se preparó de una manera similar a la descrita para el ejemplo 115 sustituyendo 7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-amina, HBr por 5-metoxibenzo[d]tiazol-2-amina. RMN 1H (500 MHz, DMSO-da) 88,28-8,12 (m, 2H), 7,87-7,73 (m, 2H), 7,60-7,50 (m, 2H), 7,41-7,28 (m, 2H), 7,28-7,15 (m, 1H), 6,96-6,60 (m, 2H), 3,84 (s, 2H), 3,70 (s, 3H), 3,06-2,93 (m, 2H), 2,93-2,82 (m, 2H). LCMS m/z = 521,1 (M+H).+ Pureza por HPLC 100 % con un tiempo de retención de 1,87 min. [Método B]
Ejemplo 117. 2-(2-metoxi-4-(2-((5-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

E ta p a 1 A 2-cloronicotinonitrilo (0,31 g, 2,2 mmol) en DMSO (2 ml) se le añadió 2-(4-hidroxi-3-metoxifenil)acetato de etilo (0,50 g, 2,2 mmol) y K2CO3 (0,70 g, 4,9 mmol) y la mezcla de reacción se calentó a 60 °C durante 24 h. La mezcla de reacción enfriada se diluyó con agua y el sólido resultante se recogió y secó al vacío para proporcionar 2-(4-((3-cianopiridin-2-il)oxi)-3-metoxifenil)acetato de etilo (55 mg, 79 % de rendimiento) en forma de un sólido de color pardo. LCMS (ESI) m/z: 313,1 (M+H).+ RMN 1H (400 MHz, CDCla) 88,30 (dd, J = 5,1, 2,0 Hz, 1H), 8,01 (dd, J = 7,5, 2,0 Hz, 1H), 7,16 (d, J = 8,1 Hz, 1H), 7,08 (dd, J = 7,6, 5,0 Hz, 1H), 7,01 (d, J = 1,8 Hz, 1H), 6,96 (dd, J = 7,9, 2,0 Hz, 1H), 4,21 (c, J = 7,1 Hz, 2H), 3,77 (s, 3H), 3,65 (s, 2H), 1,38-1,27 (m, 3H).
E ta p a 2. A 2-(4-((3-cianopiridin-2-il)oxi)-3-metoxifenil)acetato de etilo (50 mg, 0,16 mmol) y 5-metoxibenzo[d]tiazol-2-amina (0,032 g, 0,18 mmol) en tolueno (2 ml) se le añadió Me3Al (0,24 ml, 0,48 mmol). Después de 5 minutos, la mezcla de reacción se calentó con irradiación con microondas a 120 °C durante 30 min. La mezcla de reacción se inactivó mediante la adición de HCl 1 N (2 ml), se extrajo con EtOAc (3 x 2 ml) y las capas orgánicas se lavaron con NaHCO3 sat. y se concentraron a presión reducida para proporcionar 2-(4-((3-cianopiridin-2-il)oxi)-3-metoxifenil)-N-(5-metoxibenzo[d]tiazol-2-il)acetamida LCMS (ESI) m /z : 447,1(m H).+
E tap a 3. 2-(4-((3-cianopiridin-2-il)oxi)-3-metoxifenil)-N-(5-metoxibenzo[d]tiazol-2-il)acetamida se convirtió en la carboxamida 117 como se describe en el ejemplo 115 y se purificó por HPLC de fase inversa para proporcionar ejemplo 117 (2,3 mg, 2,3% de rendimiento). RMN 1H (500 MHz, DMSO-d6) 88,20 (dd, J = 7,6, 1,8 Hz, 1H), 8,14 (d, J = 5,4 Hz, 1H), 7,82 (d, J = 8,5 Hz, 1H), 7,80-7,77 (m, 1H), 7,70 (s a, 1H), 7,28 (d, J = 2,1 Hz, 1H), 7,22-7,13 (m, 3H), 7,04-6,88 (m, 2H), 3,88-3,78 (m, 5H), 3,68 (s, 3H) LCMS m/z = 465,1 (M+H).+ Pureza por HPLC 95 % con un tiempo de retención de 1,49 min. [Método A]
Ejemplo 118. 2-(4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[dltiazol-2-il)amino)-2-oxoetil)-2-metoxifenoxi)nicotinamida

El ejemplo 118 (2,6 mg, 4,0 % de rendimiento) se preparó de una manera similar a la descrita para el ejemplo 115 sustituyendo 1-((2-aminobenzo[d]tiazol-5-il)oxi)-2-metilpropan-2-ol (ejemplo 3, etapa 1) por 5-metoxibenzo[d]tiazol-2-amina. RMN 1H (500 MHz, DMSO-d6) 58,20 (d a, J = 7,4 Hz, 1H), 8,14 (d a, J = 3,0 Hz, 1H), 7,82 (s a, 1H), 7,71 (s a, 1H), 7,64 (d, J = 8 ,8 Hz, 1H), 7,55 (d, J = 2 ,2 Hz, 1H), 7,25 (s a, 1H), 7,23-7,13 (m, 3H), 7,05 (dd a, J = 8,7, 2 ,4 Hz, 1H), 6,97 (d a, J = 8,0 Hz, 1H), 3,83 (s, 2H), 3,75 (s, 2H), 3,68 (s, 3H), 3,57 (s a, 1H), 3,17 (s, 1H), 1,21 (s, 6H). LCMS m/z = 522,9 (M+H).+ Pureza por Hp Lc 95 % con un tiempo de retención de 1,42 min. [Método B]
Ejemplo 119. 2-(2-bromo-4-(2-((7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

E tap a 1. A 2-cloronicotinonitrilo (3,4 g, 24 mmol) en DMSO (20 ml) se le añadió ácido 2-(3-bromo-4-hidroxifenil)acético (5,6 g, 24 mmol) y K2CO3 (7,4 g, 53 mmol) y la mezcla de reacción se calentó a 80 °C durante 24 h. La mezcla de reacción enfriada se diluyó con agua, se acidificó con HCl, se filtró para proporcionar ácido 2-(3-bromo-4-((3-cianopiridin-2-il)oxi)fenil)acético (7,8 g, 96% de rendimiento) en forma de un sólido de color castaño. RMN 1H (400 MHz, CDCla) 88,32 (dd, J = 5,0, 1,9 Hz, 1H), 8,07 (dd, J = 7,5, 2,0 Hz, 1H), 7,66 (d, J = 2,0 Hz, 1H), 7,38 (dd, J = 8,3, 2,1 Hz, 1H), 7,26 (d, J = 8,1 Hz, 1H), 7,15 (dd, J = 7,6, 5,0 Hz, 1H), 3,71 (s, 2H). LCMS (ESI) m/z: 333-334,9 (M+H).+
E tap a 2.
A una suspensión de ácido 2-(3-bromo-4-((3-cianopiridin-2-il)oxi)fenil)acético (0,8 g, 2,5 mmol) en DCE (5 ml) se le añadió cloruro de tionilo (1,8 ml, 24 mmol) y la mezcla de reacción se calentó a 50 °C durante 3 h. La mezcla de reacción se concentró a presión reducida para proporcionar cloruro de 2-(3-bromo-4-((3-cianopiridin-2-il)oxi)fenil)acetilo (0,88 g, 100 % de rendimiento) en forma de un sólido de color castaño. LCMS (ESI) m/z: 346-348,9 (M+H)+ para el éster metílico.
E tap a 3. El ejemplo 119 (1,8 mg, 3,0 % de rendimiento) se preparó de una manera similar a la descrita para el ejemplo 8 usando cloruro de 2-(3-bromo-4-((3-cianopiridin-2-il)oxi)fenil)acetilo y 7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-amina. RMN 1H (500 MHz, DMSO-d6) 8 11,06-10,88 (m, 1H), 8,62 (d a, J = 6,7 Hz, 1H), 8,23 (dd, J = 7,5, 1,7 Hz, 1H), 8,16 (dd, J = 4,6, 1,5 Hz, 1H), 7,81 (s a, 1H), 7,75 (s a, 1H), 7,69 (s, 1H), 7,48-7,38 (m, 1H), 7,33 (d, J = 8,2 Hz, 1H), 7,24 (dd, J = 7,3, 4,9 Hz, 1H), 7,06 (s a, 1H), 6,77 (d a, J = 7,0 Hz, 1H), 3,88 (s, 2H), 3,56 (s, 3H), LCMS m/z = 496,7 (M+H).+ Pureza por HPLC 98 % con un tiempo de retención de 1,03 min. [Método A]
Ejemplo 120. 2-(4-(2-((1-(2,2-difluoroetil)-5-metoxMH-benzo[c/]imidazol-2-N)ammo)-2-oxoetil)-2-fluorofenoxi)nicotinamida

E tap a 1.
A 5-metoxi-1H-benzo[d]imidazol-2-amina (0,34 g, 2,1 mmol) y 1,1-difluoro-2-yodoetano (0,4 g, 2 mmol) en DMF (5 ml), enfriados a 0 °C, se les añadió NaH (0,17 g, 4,2 mmol) en aceite y la mezcla de reacción se agitó a ta. Después de 72 h, la mezcla de reacción se inactivó mediante la adición de agua (20 ml) y se extrajo con acetato de etilo (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4). El residuo se purificó por cromatografía sobre gel de sílice, eluyendo con DCM/MeOH al 0-10 % para proporcionar 0,2 g de una espuma de color pardo en forma de una mezcla de isómeros. LCMS (ESI)
m /z :
228,0 (M+H).+ La purificación de una muestra de 160 mg de esta mezcla de isómeros mediante SFC proporcionó 1-(2,2-difluoroetil)-5-metoxi-1H-benzo[d]imidazol-2-amina (27 mg, 25% de rendimiento) y 1-(2,2-difluoroetil)-6-metoxi-1H-benzo[d]imidazol-2-amina (58 mg, 51 % de rendimiento).
E ta p a 2. A una suspensión del intermedio 1 (2,2 g, 8,1 mmol) en DCE (16 ml) se le añadió SOCI2 (5,9 ml, 81 mmol) y la mezcla de reacción se calentó a 50 °C. La reacción se hizo transparente y se agitó durante 1 h. Los disolventes se eliminaron a presión reducida para proporcionar cloruro de 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetilo (2,3 g, 98 % de rendimiento) en forma de un sólido de color blanco.
E tap a 3.
A cloruro de 2-(4-((3-cianopiridin-2-il)oxi)-3-fluorofenil)acetilo (33 mg, 0,11 mmol) en THF (2 ml) y piridina (28 μl, 0,34 mmol) se le añadió 1-(2,2-difluoroetil)-5-metoxi-1H-benzo[d]imidazol-2-amina (26 mg, 0,11 mmol) y la mezcla de reacción se agitó a ta durante 24 h. La mezcla de reacción se concentró a presión reducida y el residuo se oxidó como se describe en el ejemplo 115 para proporcionar el ejemplo 120 (2,8 mg, 3,8 % de rendimiento); RMN 1H (500 MHz, DMSO-da) 88,29-8,13 (m, 2H), 7,79-7,69 (m, 1H), 7,67-7,58 (m, 1H), 7,48-7,38 (m, 1H), 7,36-7,29 (m, 2H), 7,27-7,19 (m, 2H), 7,16 (s a, 1H), 6,86 (d a,
J
= 8,8 Hz, 1H), 6,55-6,19 (m, 2H), 4,64-4,56 (m, 2H), 3,81 (s, 3H), 3,78 (s, 2H). LCMS m/z = 500,1 (M+H).+ Pureza por HPLC 95 % con un tiempo de retención de 1,18 min. [Método B]
Los ejemplos 121-131 se prepararon como se ha descrito siguiendo el procedimiento general para el ejemplo 8 o el ejemplo 9 y el intermedio 8







Ejemplo 134. 2-(4-(2-((7-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida

Se fabricó 2-(4-(2-((7-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida 134 (0,17 g, 30% ) mediante el acoplamiento del intermedio 2 con 7-bromo-[1,2,4]triazolo[1,5-a]piridin-2-amina, seguido de oxidación con H 2O2 según se describe para el intermedio 8. RMN 1H (400 MHz, DMSO-d6) 811,18 (s a, 1H), 8,96-8,69 (m, 1H), 8,40-8,15 (m, 3H), 8,07 (d, J = 2,0 Hz, 1H), 7,82 (s a, 1H), 7,80 (s a, 1H), 7,35-7,31 (m, 2H), 7,27-7,23 (m, 2h ), 3,83 (s a, 2H), LCm S m/z = 487,1 (M+H).+ Pureza por HPlC 96 % con un tiempo de retención de 1,12 min.
[Método A]
Los ejemplos 135-137 se prepararon siguiendo el procedimiento general usado para el ejemplo 4


Ejemplo 138. 2-(2-fluoro-4-(2-((6-(1 -metil-1H-pirazol-5-il)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

E tap a 1.
Se fabricó 2-(4-(2-((6-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida (28 mg, 7,0
%
de rendimiento) usando un procedimiento similar al del ejemplo 134 sustituyendo 6-bromo-[1,2,4]triazolo[1,5-a]piridin-2-amina por 7-bromo-[1,2,4]triazolo[1,5-a]piridin-2-amina. LCMS m/z = 485-488,1 (M+H).+
E ta p a 2.
El ejemplo 138 (1,4 mg, 4,0 % de rendimiento) se fabricó usando una reacción de Suzuki con 2-(4-(2-((6-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida y 1-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol como se describe en el ejemplo 4. RMN 1H (500 MHz, DMSO-da) 89,15 (s, 1H), 8,27-8,15 (m, 2H), 7,87 (s, 1H), 7,85-7,81 (m, 3H), 7,54 (d,
J
= 1,5 Hz, 1H), 7,42-7,31 (m, 2H), 7,28-7,15 (m, 2H), 6,57 (d,
J
= 1,5 Hz, 1H), 3,93 (s, 3H), 3,91 (s, 2H), 2,56 (s, 1H). LCMS m/z = 486,9 (M+H).+ Pureza por HPLC 96 % con un tiempo de retención de 0,98 min. [Método A]
Ejemplo 139. 2-(4-(2-((8-(difluorometoxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida

E ta p a 1.
A 3-(difluorometoxi)piridin-2-amina (0,20 g, 1,2 mmol) en dioxano (2 ml) se le añadió carbonisotiocianatidato de O-etilo (0,16 ml, 1,4 mmol), después de 2 h, se añadieron EtOH (3 ml), clorhidrato de hidroxilamina (0,4 g, 6 mmol) y DIEA (0,7 ml, 4 mmol) y la mezcla de reacción se calentó a 60 °C. Después de 48 h, un sólido de color blanco se eliminó por filtración y el filtrado se concentró, se extrajo con EtOAc, se lavó con salmuera y se secó (MgSO4). La 8-(difluorometoxi)-[1,2,4]triazolo[1,5-a]piridin-2-amina se recogió (0,25 g) y se usó sin más purificación. RMN 1H (400 MHz, DMSO-d6) 89,43 (s a, 2H), 8,48 (dd,
J
= 6,7, 1,0 Hz, 1H), 7,72-7,40 (m, 1H), 7,38-7,22 (m, 1H), 6,87 (dd,
J
= 7,9, 6,6 Hz, 1H).
E tap a 2.
El ejemplo 139 (5,6 mg, 9,5 % de rendimiento) se preparó mediante acoplamiento de 8-(difluorometoxi)-[1,2,4]triazolo[1,5-a]piridin-2-amina y el intermedio 2, seguido de oxidación como se describe en el intermedio 8. RMN 1H (500 MHz, DMSO-d6) 811,30 (s a, 1H), 8,78 (d,
J
= 6,7 Hz, 1H), 8,25-8,13 (m, 2H), 7,84 (s a, 1H), 7,79 (s a, 1H), 7,54-7,46 (m, 2H), 7,43-7,32 (m, 2H), 7,28-7,20 (m, 2H), 7,15 (t,
J
= 7,3 Hz, 1H), 3,83 (s a, 2H). LCMS m/z = 473,0 (M+H).+ Pureza por HPLC 99 % con un tiempo de retención de 1,08 min. [Método B]
Ejemplo 140. 2-(2-fluoro-4-(2-oxo-2-((6-(trifluorometoxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)etil)fenoxi)nicotinamida

El ejemplo 140 (2,2 mg, 3,5
%
de rendimiento) se preparó usando el mismo procedimiento del ejemplo 139 sustituyendo 5-(trifluorometoxi)piridin-2-amina en la etapa 1 por 3-(difluorometoxi)piridin-2-amina. RMN 1H (500 MHz, DMSO-da) 89,29 (s a, 1H), 8,21 (d a,
J
= 7,4 Hz, 1H), 8,18 (d a,
J
= 3,6 Hz, 1H), 7,87-7,72 (m, 3H), 7,71 (s a, 1H), 7,63 (s a, 1H), 7,36-7,30 (m, 2H), 7,26-7,20 (m, 2H), 3,85 (s a, 2H). LCMS m/z = 491,0 (M+H).+ Pureza por HPLC 97 % con un tiempo de retención de 1,40 min. [Método A]
Ejemplo 141 2-(2-fluoro-4-(2-oxo-2-((8-(t
N fl
uorometil)-[1,2,4]tnazolo[1,5-a]μmdm-2-il)amino)etil)fenoxi)nicotinamida

El ejemplo 141 (3,5 mg, 5,7 % de rendimiento) se preparó usando el mismo procedimiento del ejemplo 139 sustituyendo 3-(trifluorometil)piridin-2-amina en la etapa 1 por 3-(difluorometoxi)piridin-2-amina. RMN 1H (500 MHz, DMSO-d6) 811,50 (s a, 1H), 9,17 (d a,
J
= 6,4 Hz, 1H), 8,33-8,16 (m, 2H), 8,14 (d a,
J
= 7,0 Hz, 1H), 7,85 (s a, 1H), 7,81 (s a, 1H), 7,48-7,34 (m, 2H), 7,31 (t a,
J
= 7,0 Hz, 1H), 7,26 (d a,
J
= 7,0 Hz, 2H), 3,93 (s a, 2H). LCMS m/z = 475,0 (M+H).+ Pureza por HPLC 96 % con un tiempo de retención de 1,17 min. [Método B]
Ejemplo 142. 2-(2-fluoro-4-(2-((7-fluoro-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

El ejemplo 142 (0,6 mg, 1 % de rendimiento) se preparó usando el mismo procedimiento del ejemplo 139 sustituyendo la 4-fluoropiridin-2-amina en la etapa 1 por 3-(difluorometoxi)piridin-2-amina. RMN 1H (500 MHz, DMSO-d6) 8 10,01 (s, 1H), 9,04-8,79 (m, 1H), 8,21 (d a,
J
= 7,5 Hz, 1H), 8,19 (d a,
J
= 4,6 Hz, 1H), 7,70 (s a, 1H), 7,64 (s a, 1H), 7,57 (dd,
J
= 9,1, 2,3 Hz, 1H), 7,38-7,29 (m, 2H), 7,28-7,18 (m, 2H), 7,14 (td,
J
= 7,7, 2,5 Hz, 1H), 3,85 (s a, 2H). LCMS m/z = 425,1 (M+H).+ Pureza por HPlC 97 % con un tiempo de retención de 0,93 min. [Método A]
Ejemplo 143. 2-(4-(2-((7-(benciloxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida

El ejemplo 143 (1,3 mg, 2% de rendimiento) se preparó mediante un procedimiento similar al del ejemplo 139 sustituyendo 4-(benciloxi)piridin-2-amina en la etapa 1 por 3-(difluorometoxi)piridin-2-amina. RMN 1H (500 MHz, DMSO-da) 88,70 (d a,
J
= 7,3 Hz, 1H), 8,26-8,16 (m, 2H), 7,84 (s a, 1H), 7,81 (s a, 1H), 7,53 (d a,
J
= 7,3 Hz, 2H), 7,45 (t a,
J
= 7,3 Hz, 2H), 7,44-7,31 (m, 4H), 7,30-7,17 (m, 3H), 6,86 (dd a,
J
= 7,0, 2,1 Hz, 1H), 5,27 (s, 2H), 3,93 (s, 2H). LCMS m/z = 513,2 (M+H).+ Pureza por HPLC 94 % con un tiempo de retención de 1,45 min. [Método B]
Ejemplo 144. 2-(4-(2-((1-(ddopropNmetil)-6-(1-metiMH-pirazol-4-il)-1H-benzo[d]imidazol-2-N)ammo)-2-oxoetil)-2-fluorofenoxi)nicotinamida
E ta p a 1.
A 4-bromo-2-fluoro-1-nitrobenceno (0,60 g, 2,5 mmol) en THF (2 ml), enfriado a 0 °C, se le añadió ciclopropilmetanamina (0,19 g, 2,7 mmol) y DIEA (0,5 ml, 3,0 mmol).
La mezcla de reacción se agitó a ta durante 18 h. La mezcla de reacción se concentró a presión reducida y el residuo se disolvió en acetona (3 ml)/agua (1,5 ml), se enfrió a 0 °C y se añadió lentamente NH4Cl (0,70 g, 12 mmol) y cinc (1,6 g, 25 mmol). El cinc se eliminó por filtración, la acetona se concentró a presión reducida. La capa acuosa se repartió con EtOAc (50 ml). La capa acuosa se extrajo con EtOAc (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (Na2SO4). El residuo se purificó por cromatografía sobre gel de sílice y se eluyó con DCM/MeOH al 0-10 % para proporcionar 5-bromo-N1-(ciclopropilmetil)bencen-1,2-diamina (0,40 g, 67 % de rendimiento) en forma de un aceite oscuro. RMN 1H (400 MHz, CDCh) 86,88-6,71 (m, 1H), 6,60 (d, J = 8,1 Hz, 1H), 6,33 (d a, J = 7,7 Hz, 1H), 3,79-3,40 (m, 1H), 3,08-2,84 (m, 2H), 1,13-0,99 (m, 1H), 0,71-0,54 (m, 2H), 0,40-0,12 (m, 2H).
E ta p a 2.
5-bromo-N1-(ciclopropilmetil)bencen-1,2-diamina (0,40 g, 1,7 mmol) y bromuro ciánico (0,26 g, 2,5 mmol) se agitaron en MeOH (6,6 ml)/H2O (1,6 ml) durante 18 h. Los disolventes se redujeron a presión reducida y después se inactivaron mediante la adición de hidróxido de amonio. Después de diluir con agua, se eliminó un sólido de color pardo oscuro por filtración para proporcionar 6-bromo-1-(ciclopropilmetil)-1H-benzo[d]imidazol-2-amina (0,36 g, 83 % de rendimiento). LCMS (ESI)
m /z :
265-267,9 (M+H).+ RMN 1H (400 MHz, DMSO-d6) 87,41 (t,
J
= 1,1 Hz, 1H), 7,05 (d,
J
= 1,1 Hz, 2H), 6,53 (s, 2H), 3,92 (d,
J
= 7,0 Hz, 2H), 1,32-1,12 (m, 1H), 0,63-0,25 (m, 4H).
E tap a 3.
El intermedio 5 se acopló con 6-bromo-1-(ciclopropilmetil)-1H-benzo[d]imidazol-2-amina como se describe en el ejemplo 2 para proporcionar 2-(4-(2-((6-bromo-1-(ciclopropilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida (72 mg, 36 % de rendimiento) en forma de un sólido de color naranja. LCMS (ESI)
m /z:
537 539,9 (M+H).+
E ta p a 4.
El ejemplo 144 (4,0 mg, 21 % de rendimiento) se fabricó de una manera similar a la del ejemplo 4. RMN 1H (500 MHz, DMSO-d6) 88,39-8,08 (m, 4H), 7,91 (s, 1H), 7,80 (s a, 2H), 7,72 (s a, 1H), 7,42 (s a, 2H), 7,37-7,27 (m, 2H), 7,23 (d a,
J
= 8,5 Hz, 2H), 4,03 (d a,
J
= 7,6 Hz, 2H), 3,88 (s, 3H), 3,69 (s, 2H), 1,33 (m, 1H), 0,47 (s a, 4H). LCMS m/z = 540,1 (M+H).+ Pureza por HPLC 99 % con un tiempo de retención de 1,19 min. [Método B]
Ejemplo 145. 2-(4-(2-((1-(ddopropNmetil)-6-(dimetilfosforM)-1H-benzo[d]imidazol-2-N)ammo)-2-oxoetil)-2-fluorofenoxi)nicotinamida

2-(4-(2-((6-bromo-1-(cidopropilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida (32 mg, 0,060 mmol), óxido de dimetilfosfina (46. mg, 0,60 mmol) y TEA (25 μl, 0,18 mmol) se combinaron en acetonitrilo (2,3 ml) y se desgasificaron con una corriente de N2. Después de purgar durante 5 min, se añadió (Ph3P)4Pd (0) (7 mg, 6 μmol) y la mezcla de reacción se calentó con irradiación con microondas a 120 °C durante 40 min. La mezcla de reacción se dejó enfriar, se filtró y se purificó por HPLC de fase inversa para proporcionar el ejemplo 145 (14 mg, 35 % de rendimiento). RMN 1H (500 MHz, DIVISOR) 88,21 (d a, J = 7,4 Hz, 1H), 8,18 (s a, 1H), 7,86 (d a, J = 10,7 Hz, 1H), 7,70 (s a, 1H), 7,66 (s a, 1H), 7,59 (d a, J = 5,8 Hz, 2H), 7,44-7,28 (m, 2H), 7,23 (d a, J = 5,4 Hz, 2H), 4,05 (s a, 2H), 3,96-3,64 (m, 2H), 2,55 (s, 1H), 1,71 (s, 3H), 1,69 (s, 3H), 1,30 (s a, 1H), 0,47 (d a, J = 7,6 Hz, 3H), 0,32 (s a, 1H), LCMS m/z = 536,1 (M+H).+ Pureza por HPlC 100 % con un tiempo de retención de 1,09 min. [Método A]
Los ejemplos 146-150 a continuación se prepararon siguiendo el procedimiento general usado para el ejemplo 145



Ejemplo 151. 2-(2-fluoro-4-(2-((5-metil-4-(6-metilpiridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida

E ta p a 1. A 1-(6-metilpiridin-2-il)propan-1-ona (0,30 g, 2,0 mmol) y AcOH (3 ml), calentados a 90 °C, se les añadió Br2 (0,10 ml, 2,0 mmol) en AcOH (1 ml). Después de 1 h, los disolventes se eliminaron a presión reducida y se añadieron EtOH (15 ml) y tiourea (0,15 g, 2,01 mmol) y la mezcla de reacción se calentó a 60 °C durante 72 h. Los disolventes se eliminaron a presión reducida y el residuo se repartió con NH4OH 0,1 M (20 ml) y EtOAc (50 ml). La capa acuosa se extrajo con EtOAc (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (15 ml) y se secaron (MgSO4), se filtraron y se concentraron para proporcionar 5-metil-4-(6-metilpiridin-2-il)tiazol-2-amina (0,30 g, 74 % de rendimiento) en forma de un sólido de color castaño. RMN 1H (400 MHz, CDCh) 87,68-7,51 (m, 2H), 7,05 (dd, J = 7,3, 1,1 Hz, 1H), 4,91 (s a, 2H), 2,68 (s, 3H), 2,61 (s, 3H). LCMS (ESI) m /z : 206,0 (M+H).+
E ta p a 2. El ejemplo 151 (6 mg, 8 % de rendimiento) se fabricó usando BOP para acoplar el intermedio 5 y 5-metil-4-(6-metilpiridin-2-il)tiazol-2-amina como se describe en el ejemplo 2. RMN 1H (500 MHz, DMSO-d6) 8 12,38 (s, 1H), 8,28-8,13 (m, 2H), 7,83 (s a, 1H), 7,79 (s a, 1H), 7,77-7,66 (m, 2H), 7,48-7,31 (m, 2H), 7,30-7,20 (m, 2H), 7,16 (d a, J = 6,7 Hz, 1H), 2,84 (s, 2H), 2,75 (s, 3H), 2,50 (s, 3H), LCMS m/z = 478,1 (M+H).+Pureza por HPLC 95 % con un tiempo de retención de 1,62 min. [Método A]
Ejemplo 152. Preparación 2-(4-(2-((1-(cidobutilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida

E ta p a 1. Se añadieron KOH (105 mg, 1,88 mmol) y (bromometil)ciclobutano (0,21 ml, 1,9 mmol) a una solución de 2-aminobenzoimidazol (250 mg, 1,87 mmol) en acetona (10 ml) y se calentó a 60 °C durante 8 h y después se dejó enfriar a ta. Después de agitar durante 48 h, el disolvente se evaporó a presión reducida. El residuo se disolvió en EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía de fase normal (DCM al 100 % a MeOH al 10 %/DCM al 90 %) para dar 1 -(ciclobutilmetil)-1H-benzo[d]imidazol-2-amina. RMN 1H (400 MHz, DMSO-de) 87,17-7,05 (m, 2H), 6,93-6,81 (m, 2H), 6,33 (s, 2H), 4,00 (d, J = 7,3 Hz, 2H), 2,80-2,66 (m, 1H), 1,92-1,75 (m, 6 H). LCMS m/z = 202.0 (M+H).+
E ta p a 2. El intermedio 2 (72 mg, 0,25 mmol) se añadió a una solución de 1-(ciclobutilmetil)-1H-benzo[d]imidazol-2-amina (50 mg, 0,20 mmol) y piridina (60 μl, 0,70 mmol) en THF a 0 °C. La mezcla de reacción se dejó calentar a ta. Después de agitar durante 18 h, la mezcla de reacción se diluyó con EtOAc y se lavó con agua y salmuera, se secó sobre sulfato sódico, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía de fase normal (DCM al 100 % a MeOH al 10 %/DCM al 90 %) para dar un sólido de color castaño. El nitrilo se hidrolizó a la amida primaria de manera similar a la descrita en el ejemplo 8 para proporcionar el ejemplo 152 (25 mg, 17 %) en forma de un sólido de color blanco. RMN 1H (500 MHz, DMSO-cfe) 8 8,21-8,15 (m, 2H), 7,84-7,76 (m, 2H), 7,72-7,62 (m, 1H), 7,61-7,55 (m, 1H), 7,42-7,29 (m, 4H), 7,24 (dd, J = 7,4, 5,0 Hz, 2H), 4,29 (s a, 2H), 3,89 (s a, 2H), 2,80 (dt, J = 15,1, 7,5 Hz, 1H), 1,95-1,88 (m, 2H), 1,87-1,77 (m, 4H). LCMS m/z = 474,0 (M+H).+ Pureza por HPLC: 98 % con un tiempo de retención de 8,37 min. [Método C].
Ejemplo 153. 2-(2-fluoro-4-(2-((1-isopropil-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-fenoxi)nicotinamida

E tap a 1.
Se preparó 1-isopropil-1H-benzo[d]imidazol-2-amina (110 mg, 34 %) de una manera similar a la descrita para la etapa 1 del ejemplo 152 sustituyendo (bromometil)ciclobutano por 2-bromopropano. LCMS m/z = 176.1 (M+H).+
E ta p a 2.
Se preparó 2-(2-fluoro-4-(2-((1-isopropil-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-fenoxi)nicotinamida 153 (49 mg, 13 %) de una manera similar a la del ejemplo 152 sustituyendo 1-(ciclobutilmetil)-1H-benzo[d]imidazol-2-amina por 1-isopropil-1H-benzo[d]imidazol-2-amina. RMN 1H (500 MHz, DMSO-da) 88,20-8,16 (m, 2H), 7,84-7,77 (m, 3H), 7,62 (s a, 1H), 7,39-7,30 (m, 4H), 7,26-7,22 (m, 2H), 5,04-4,96 (m, 1H), 3,96-3,88 (m, 2H), 1,59 (s, 3H), 1,58 (s, 3H). LCMS m/z = 448,0 (M+H).+ Pureza por HPlC: 93 % con un tiempo de retención de 7,49 min. [Método C].
Los ejemplos 154-177 se prepararon siguiendo el procedimiento general usado para el ejemplo 152.












Ejemplo 178. 2-(4-(2-((1-(cidopropilmetil)-6-(2-hidroxi-2-metilpropoxi)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2 -fluorofenoxi)nicotinamida
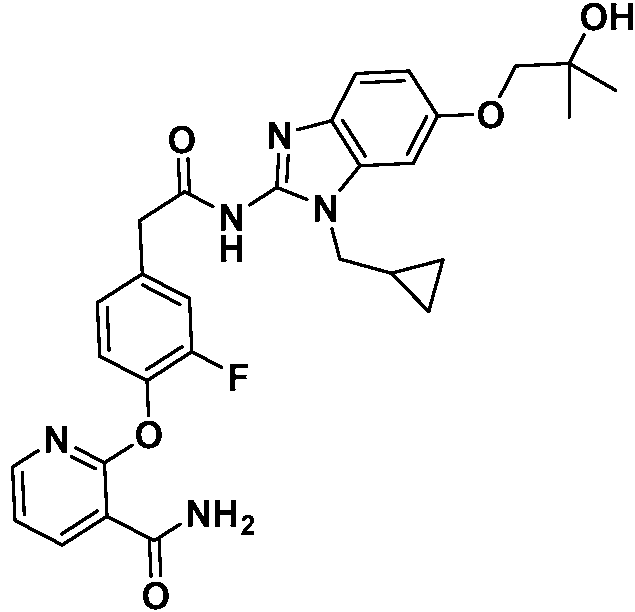
E ta p a 1. Una mezcla de 3-fluoro-4-nitrofenol (1,0 g, 6,4 mmol), (bromometil)benceno (0,80 ml, 7,0 mmol), K2CO3 (2,6 g, 19 mmol) y TBAI (16 mg, 0,050 mmol) se añadió a DMF (15 ml) y se agitó a ta. Después de agitar durante 18 h, se añadió agua (75 ml) y los sólidos resultantes se recogieron mediante filtración al vacío, se lavaron con agua y se secaron para dar 4-(benciloxi)-2-fluoro-1-nitrobenceno en forma de un sólido de color amarillo (1,5 g, 95 %). RMN 1H (500 MHz, DMSO-da) 88,16 (t, J = 9,1 Hz, 1H), 7,51-7,45 (m, 2H), 7,45-7,40 (m, 2H), 7,39-7,35 (m, 1H), 7,28 (dd, J = 13,6, 2,6 Hz, 1H), 7,08-7,03 (m, 1H), 5,30-5,26 (m, 2H). LCMS m/z = 248.1 (M+H).+
E ta p a 2. 4-(benciloxi)-2-fluoro-1-nitrobenceno (869 mg, 3,52 mmol), ciclopropilmetanamina (250 mg, 3,52 mmol) y Cs2COa (1,1 g, 3,5 mmol) se añadieron a ACN (15 ml) y se irradiaron a 120 °C durante 15 min. La mezcla de reacción se concentró a presión reducida y el residuo se repartió entre EtOAc y agua. La capa orgánica se lavó con salmuera, se secó sobre sulfato sódico, se filtró y se concentró. El intermedio se disolvió en EtOH (30 ml) y se trató con NH4Cl (0,90 g, 18 mmol) en agua (15 ml). La solución se calentó a 60 °C antes de añadir Zn (3,4 g, 53 mmol). Después de 2 h, la mezcla se dejó enfriar a ta, se filtró a través de un lecho de Celite y se lavó con EtOAc. La capa orgánica se lavó con agua y salmuera y se secó sobre Na2SO4. El residuo se purificó por cromatografía de fase normal para dar el producto deseado (396 mg, 42 %). LCMS m/z = 269.1 (M+H).+
E tap a 3. A una solución de 5-(benciloxi)-N1-(ciclopropilmetil)bencen-1,2-diamina (0,40 g, 1,5 mmol) en MeOH (6 ml) y agua (1,5 ml), se le añadió bromuro ciánico (0,171 g, 1,62 mmol) y se agitó a ta. Después de 18 h, la mezcla de reacción se ajustó a pH = 9 mediante la adición de NH4OH conc. y se diluyó con agua (75 ml). El sólido resultante se recogió y se secó mediante filtración al vacío para dar 6-(benciloxi)-1-(ciclopropilmetil)-1H-benzo[d]imidazol-2-amina en forma de un sólido de color rojizo-anaranjado. LCMS m/z = 294.0 (M+H).+
E ta p a 4. A una solución de 6-(benciloxi)-1-(ciclopropilmetil)-1H-benzo[d]imidazol-2-amina (200 mg, 0,682 mmol) en THF (15 ml), se le añadió TEA (0,095 ml, 0,68 mmol) y BOC-anhídrido (0,24 ml, 1,0 mmol). Después de agitar durante 18 h, la reacción se diluyó con agua y se extrajo con EtOAc (3x). La capa orgánica combinada se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se disolvió en EtOH, se trató con Pd/C (145 mg) y se sometió a atmósfera de hidrógeno (379,212 kPa (55 psi)). Después de 2 h, la suspensión se filtró a través de un lecho de Celite y el filtrado se concentró para dar (1-(ciclopropilmetil)-6-hidroxi-1H-benzo[d]imidazol-2-il)carbamato de tere-butilo (150 mg, 71 %). LCMS m/z = 304.1 (M+H).+
E ta p a 5.
Se fabricó (1-(ciclopropilmetil)-6-(2-hidroxi-2-metilpropoxi)-1H-benzo[d]imidazol-2-il)carbamato de tere-butilo de una manera similar a la de la etapa 1 del ejemplo 3 a partir de (1-(ciclopropilmetil)-6-hidroxi-1H-benzo[d]imidazol-2-il)carbamato de tere-butilo. El material se llevó a la reacción siguiente sin más purificación. LCMS m/z = 376.1 (M+H).+
E tap a 6. Se disolvió (1-(ciclopropilmetil)-6-(2-hidroxi-2-metilpropoxi)-1H-benzo[d]imidazol-2-il)carbamato de tere-butilo (24 mg, 0,060 mmol) en HCl (4,0 M en dioxano) (320 μl, 1,28 mmol) y se agitó a ta. Después de 1 h, se añadió más HCl (4,0 M en dioxano) (320 μl, 1,28 mmol) y una gota de HCl (conc.) y la agitación continuó. Después de agitar durante un total de 6 h, la mezcla de reacción se concentró a presión reducida y se colocó a alto vacío durante 14 h. La sal HCl de 1-((2-amino-1-(ciclopropilmetil)-1H-benzo[d]imidazol-6-il)oxi)-2-metilpropan-2-ol se llevó a la reacción siguiente reacción sin más purificación. LCMS m/z = 276.1 (M+H).+
E ta p a 7. Se preparó la 2-(4-(2-((1-(ciclopropilmetil)-6-(2-hidroxi-2-metilpropoxi)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida del ejemplo 178 (4,9 mg, 11 %) de una manera similar a la del ejemplo 152 reemplazando 1-(ciclobutilmetil)-1H-benzo[d]imidazol-2-amina por sal HCl de 1-((2-amino-1-(ciclopropilmetil)-1H-benzo[d]imidazol-6-il)oxi)-2-metilpropan-2-ol. RMN 1H (500 MHz, DMSO-cfe) 8 8,18-8,12 (m, 2H), 7,87-7,82 (m, 1H), 7,78-7,74 (m, 1H), 7,49 (d, J = 8,9 Hz, 1H), 7,36-7,31 (m, 2H), 7,29-7,26 (m, 1H), 7,25-7,21 (m, 2H), 6,96 (d a, J = 8,9 Hz, 1H), 4,19-4,12 (m, 2H), 3,91-3,87 (m, 1H), 3,80-3,76 (m, 1H), 1,29-1,16 (m, 8H), 0,52-0,39 (m, 4H). LCMS m/z = 548,3 (M+H).+ Pureza por HPLC: 96 % con un tiempo de retención de 1,54 min. [Método A].
Claims (14)
1. Un compuesto que tiene la fórmula (II):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona independientemente entre halógeno y alquilo C-i-a;
R5 es un heterociclo de 4-15 miembros que comprende átomos de carbono y 1-4 heteroátomos seleccionados entre N, NR8, O y S(O)p, en donde dicho heterociclo está sustituido con 1-4 R7;
R7, en cada caso, se selecciona independientemente de H, =O, NO2, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2V C O 2H, -(CH2)n-CO2(alquilo C1.4), -(CH2)n-NR8R8, -NHCO(alquilo C1.4), -NHCOCF3, -NHCO2(alquilo C1.
4), -NHCO2(CH2) 2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2) 2OH, -NHCO2(CH2)2NH2, -NHCO2(CH2)2N(alquilo ^ .4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1.4), -NHC(O)NR8R8, -NHSO2(alquilo C1.4), -SO2NH2, -SO2NH(alquilo C1-4), -SO2N(alquilo C1-4)2, -SO2NH(CH2) 2OH, -SO2NH(CH2) 2O(alquilo C1-4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, -NHCO-carbociclo, -NHCO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dichos alquilo, alquenilo, alquinilo, alcoxi, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, C(O)alquilo C1-4, -(CH2)nC(O)NRaRa, C(O)O-alquilo, SO2alquilo, SO2NRaRa, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dichos alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CF3, alquilo C1-4, alcoxi C1.4, CH2OH, CO2H, CO2(alquilo C1.4), CONH2, -(CH2)nNRaRa, -(CH2)nCONRaRa, -O(CH2)nheterociclo, -O(CH2)(2-4)NRaRa, -(CH2)n-heterociclo de 4-10 miembros, en donde dichos alquilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 Rb;
Ra, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)nOH, CO(alquilo C1.4), COCF3, CO2(alquilo C1.4), -CONH2, -CONH-alquilen C1-4-CO2(alquilo C1-4), alquilen C1-4-CO2(alquilo C1.4), Rc, CO2Rc y CONHRc; como alternativa, Ra y Ra se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros, en donde dichos alquilo, alquileno y heterociclo están sustituidos por 0-4 Rb; Rb, en cada caso, se selecciona independientemente de =O, halógeno, alquilo C1-4, alcoxi C1-4, OCF3, NH2, NO2, N(alquilo C1-4)2, CO(alquilo C1-4), CO(haloalquilo C1-4), CO2(alquilo C1-4), CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, -CONH-alquilen C1-4-O(alquilo C1.4), -CONH-alquilen C1-4-N(alquilo C1-4)2, -CONH-alquilen C1-4-N(alquilo C1.
4)2, -alquilen C1-4-O-P(O)(OH)2, -NHCO2(alquilo C1.4), -Rc, CORc, CO2Rc y CONHRc;
Rc, en cada caso, se selecciona independientemente de -(CH2)n-cicloalquilo C3-6, -(CH2)n-fenilo y -(CH2)nheterociclo de 5 a 6 miembros que contienen átomos de carbono y 1-4 heteroátomos seleccionados entre el grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p; en donde cada resto del anillo está sustituido por 0-2 Rd; Rd, en cada caso, se selecciona independientemente de =O, halógeno, -OH, alquilo C1-4, NH2, NH(alquilo C1-4), N(alquilo C1-4)2, alcoxi C1.4 y -NHCO(alquilo C1.4) y heterociclo que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p;
n, en cada caso, se selecciona independientemente de 0 , 1, 2 , 3 y 4;
p, en cada caso, se selecciona independientemente entre 0 , 1 y 2 ;
o
un compuesto que tiene la fórmula (III):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona entre F, Cl, Br y Oalquilo C1-4;
R4 se selecciona entre H y alquilo C1-4;
R5 se selecciona entre

R7, en cada caso, se selecciona independientemente de H, =O, NO2, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2)n-CO(alquilo C1-4), -(CH2)n-CO2H, -(CH2)n-CO2(alquilo C1-4), -(CH2)n-NR8R8, -NHCO(alquilo C1-4), -NHCOCF3, -NHCO2(alquilo C1.4), -NHCO2(CH2) 2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2) 2OH, -NHCO2(CH2)2NH2, -NHCO2(CH2)2N(alquilo C1-4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1-4), -NHC(O)NR8R8, -NHSO2(alquilo C1.4), -P(=O)(alquilo C1-3)2, -SO2alquilo C1.4, -SO2NH2, -SO2NH(alquilo C1.4), -SO2N(alquilo C1-4)2, -SO2NH(CH2) 2OH, -SO2NH(CH2) 2O(alquilo C1.4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, - NHCO-carbociclo, -NHCO-heterociclo, -CO-carbociclo, -CO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dichos alquilo, alquenilo, alquinilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, -(CH2)n-C(O)alquilo C1.4, -(CH2)n-C(O)carbociclo, -(CH2)n-C(O)heterociclo, -(CH2)n-C(O)NRaRa, -(CH2)n-C(O)O-alquilo, -(CH2)n-C(O)O-carbociclo, -(CH2)n-C(O)O-heterociclo, -(CH2)n-SO2alquilo, -(CH2)n-SO2carbociclo, -(CH2)n-SO2heterociclo, -(CH2)n-SO2NRaRa, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dichos alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
como alternativa, R8 y R8 se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros sustituido con 0-4 R9;
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CN, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO(alquilo C1-4), CO2H, CO2(alquilo C1-4), -(CH2)nNRaRa, -(CH2)nCONRaRa, -O(CH2)ncarbociclo, -O(CH2)nheterociclo, -O(CH2)nNRaRa, NRaCO2(alquilo C1.4), S(O)palquilo C1.4, -(CR10R10)n-carbociclo, -(CR10R10)nheterociclo de 4-10 miembros, en donde dichos alquilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 Rb;
R10 se selecciona entre H y alquilo C1-4;
Ra, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)nOH, CO(alquilo C1.4), COCF3, CO2(alquilo C1.4), -CONH2, -CONH-alquilen C1-4-CO2(alquilo C1.4), alquilen C1-4-CO2(alquilo C1.4), Rc, CO2Rc y CONHRc; como alternativa, Ra y Ra se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros, en donde dichos alquilo, alquileno y heterociclo están sustituidos por 0-4 Rb; Rb, en cada caso, se selecciona independientemente de =O, OH, halógeno, alquilo C1-4, alcoxi C1-4, OCF3, NH2, NO2, N(alquilo C1-4)2, CO(alquilo C1.4), CO(haloalquilo C1-4), CO2(alquilo C1.4), CONH2, -CONH(alquilo C1.4), -CON(alquilo C1-4)2, -CONH-alquilen C1-4-O(alquilo C1.4), -CONH-alquilen C1-4-N(alquilo C1-4)2, -CONH-alquilen C1.
4-N(alquilo ^ .4)2, -alquilen C1-4-O-P(O)(OH)2, -NHCO2(alquilo C1.4), -Rc, CORc, CO2Rc y CONHRc;
Rc, en cada caso, se selecciona independientemente de -(CH2)n-cicloalquilo C3-6, -(CH2)n-fenilo y -(CH2)nheterociclo de 5 a 6 miembros que contienen átomos de carbono y 1-4 heteroátomos seleccionados entre el grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p; en donde cada resto del anillo está sustituido por 0-2 Rd; Rd, en cada caso, se selecciona independientemente de =O, halógeno, -OH, alquilo C1-4, NH2, NH(alquilo C1.4), N(alquilo C1-4)2, alcoxi C1.4 y -NHCO(alquilo C1.4) y heterociclo que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p;
n, en cada caso, se selecciona independientemente de 0 , 1, 2 , 3 y 4; y
p, en cada caso, se selecciona independientemente de 0 , 1 y 2.
2. El compuesto (II) de la reivindicación 1, o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona independientemente entre F, Cl y Br;
R5 se selecciona entre



R7, en cada caso, se selecciona independientemente de H, =O, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2)n-CO2H, -(CH2)n-CO2(alquilo C1-4), -(CH2)n-NR8R8, -NHCO(alquilo C1-4), -NHCOCF3, -NHCO2(alquilo C1-4), -NHCO2(CH2) 2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2) 2OH, -NHCO2(CH2)2NH2, -NHCO2(CH2)2N(alquilo ^ .4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1.4), -NHC(O)NR8R8, -NHSO2(alquilo C1.4), -SO2NH2, -SO2NH(alquilo C1-4), -SO2N(alquilo C1-4)2, -SO2NH(CH2) 2OH, -SO2NH(CH2) 2O(alquilo C1-4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, -NHCO-carbociclo, -NHCO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados entre N, NR8, O y S(O)p, en donde dichos alquilo, alquenilo, alquinilo, alcoxi, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dichos alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9; y
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CF3, alquilo C1.4, alcoxi C1-4, CH2OH, CO2H, CO2(alquilo C1.4), CONH2, -(CH2)nNH2, -(CH2)nCONH2, -O(CH2)nheterociclo, -O(CH2)(2-4)NH2, -(CH2)n-heterociclo de 4-10 miembros.
3. El compuesto (II) de la reivindicación 2, o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona independientemente entre F y Cl;
R5 se selecciona entre


R7, en cada caso, se selecciona independientemente de H, halógeno, alquilo C1-4, alcoxi C1-4, un carbociclo y un heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dichos alquilo, alquenilo, alquinilo, alcoxi, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4 y -(CH2)n-cicloalquilo C3-6; y
R9, en cada caso, se selecciona independientemente de halógeno, OH, CHF2, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO2H, CO2(alquilo C1-4) y CONH2.
4. El compuesto (II) de la reivindicación 3, o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde R7, en cada caso, se selecciona independientemente de H, F, Cl, Br, Me, Et, OMe, OEt, OCH2C(CH2)2OH,

y
R9, en cada caso, se selecciona independientemente de F, Cl, Br, OH, CHF2, CF3, alquilo C1-4 y alcoxi C1-4.
5. El compuesto de acuerdo con la reivindicación 1 que tiene la fórmula (III):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona entre F, Cl, Br y Oalquilo C1-4;
R4 se selecciona entre H y alquilo C1-4;
R5 se selecciona entre


y
R7, en cada caso, se selecciona independientemente de H, =O, NO2, halógeno, alquilo C1-4, alcoxi C1-4, CN, OH, CF3, -(CH2)n-CO(alquilo C1-4), -(CH2)n-CO2H, -(CH2)n-CO2(alquilo C1-4), -(CH2)n-NR8R8, -NHCO(alquilo C1-4), -NHCOCF3, -NHCO2(alquilo C1.4), -NHCO2(CH2) 2O(alquilo C1.4), -NHCO2(CH2)3O(alquilo C1.4), -NHCO2(CH2) 2OH, -NHCO2(CH2)2NH2, -NHCO2(CH2)2N(alquilo C1-4)2, -NHCO2CH2CO2H, -CH2NHCO2(alquilo C1-4), -NHC(O)NR8R8, -NHSO2(alquilo C1.4), -P(=O)(alquilo C1-3)2, -SO2alquilo C1.4, -SO2NH2, -SO2NH(alquilo C1.4), -SO2N(alquilo C1-4)2, -SO2NH(CH2) 2OH, -SO2NH(CH2) 2O(alquilo C1.4), -(CH2)n-CONR8R8, -O(CH2)n-carbociclo, -O(CH2)n-heterociclo, -NHCO-carbociclo, -NHCO-heterociclo, -CO-carbociclo, -CO-heterociclo, -(CH2)n-carbociclo y -(CH2)n-heterociclo que comprende átomos de carbono y 1-4 heteroátomos seleccionados de N, NR8, O y S(O)p, en donde dichos alquilo, alquenilo, alquinilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
R8, en cada caso, se selecciona independientemente de H, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, -(CH2)n-C(O)alquilo C1.4, -(CH2)n-C(O)carbociclo, -(CH2)n-C(O)heterociclo, -(CH2)n-C(O)NRaRa, -(CH2)n-C(O)O-alquilo, -(CH2)n-C(O)O-carbociclo, -(CH2)n-C(O)O-heterociclo, -(CH2)n-SO2alquilo, -(CH2)n-SO2carbociclo, -(CH2)n-SO2heterociclo, -(CH2)n-SO2NRaRa, -(CH2)n-carbociclo y -(CH2)n-heterociclo, en donde dichos alquilo, carbociclo y heterociclo están sustituidos con 0-4 R9;
como alternativa, R8 y R8 se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros sustituido con 0-4 R9;
R9, en cada caso, se selecciona independientemente de halógeno, OH, NO2, CHF2, CN, CF3, alquilo C1-4, alcoxi C1-4, CH2OH, CO(alquilo C1-4), CO2H, CO2(alquilo C1-4), -(CH2)nNRaRa, -(CH2)nCONRaRa, -O(CH2)ncarbociclo, -O(CH2)nheterociclo, -O(CH2)nNRaRa, NRaCO2(alquilo C1.4), S(O)palquilo C1.4, -(CR10R10)n-carbociclo, -(CR10R10)nheterociclo de 4-10 miembros, en donde dichos alquilo, alcoxilo, carbociclo y heterociclo están sustituidos con 0-4 Rb;
R10 se selecciona entre H y alquilo C1-4;
Ra, en cada caso, se selecciona independientemente de H, alquilo C1-4, -(CH2)nOH, CO(alquilo C1.4), COCF3, CO2(alquilo C1.4), -CONH2, -CONH-alquilen C1-4-CO2(alquilo C1.4), alquilen C1-4-CO2(alquilo C1.4), Rc, CO2Rc y CONHRc; como alternativa, Ra y Ra se toman junto con el átomo de nitrógeno al que están unidos para formar un heterociclo de 4 a 10 miembros, en donde dichos alquilo, alquileno y heterociclo están sustituidos por 0-4 Rb; Rb, en cada caso, se selecciona independientemente de =O, OH, halógeno, alquilo C1-4, alcoxi C1-4, OCF3, NH2, NO2, N(alquilo C1-4)2, CO(alquilo C1.4), CO(haloalquilo C1-4), CO2(alquilo C1.4), CONH2, -CONH(alquilo C1.4), -CON(alquilo C1-4)2, -CONH-alquilen C1-4-O(alquilo C1.4), -CONH-alquilen C1-4-N(alquilo C1-4)2, -CONH-alquilen C1.
4-N(alquilo ^ .4)2, -alquilen C1-4-O-P(O)(OH)2, -NHCO2(alquilo C1.4), -Rc, CORc, CO2Rc y CONHRc;
Rc, en cada caso, se selecciona independientemente de -(CH2)n-cicloalquilo C3-6, -(CH2)n-fenilo y -(CH2)nheterociclo de 5 a 6 miembros que contienen átomos de carbono y 1-4 heteroátomos seleccionados entre el grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p; en donde cada resto del anillo está sustituido por 0-2 Rd; Rd, en cada caso, se selecciona independientemente de =O, halógeno, -OH, alquilo C1-4, NH2, NH(alquilo C1-4), N(alquilo C1-4)2, alcoxi C1-4 y -NHCO(alquilo C1-4) y heterociclo que contiene átomos de carbono y 1-4 heteroátomos seleccionados del grupo que consiste en: N, NH, N(alquilo C1-4), O y S(O)p;
n, en cada caso, se selecciona independientemente de 0 , 1, 2 , 3 y 4; y
p, en cada caso, se selecciona independientemente de 0 , 1 y 2.
6. El compuesto de la reivindicación 5 o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona entre F, Cl y OCH3;
R5 se selecciona entre

R7 se selecciona entre H, alquilo C1-3 sustituido con halógeno,

y

R8 se selecciona entre H y alquilo C1-3 opcionalmente sustituido con halógeno;
R9 se selecciona entre F, Cl, Cn , alquilo C1-4 sustituido con 0-2 Rb, alcoxi C1-3, cicloalquilo C3-6, NHCOOalquilo C1.
4, SO2alquilo C1-3 y

y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
7. El compuesto de la reivindicación 5 o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona independientemente entre F, Cl y OCH3;
R5 se selecciona entre

R7 se selecciona entre H, F, Cl, Br, alquilo C1-3 sustituido con 0-4 R9, alcoxi C1-3 sustituido con 0-4 R9, CN, CO2alquilo C1-4, P(=O)(alquilo C1-3)2, SO2alquilo C1-4,

R8 se selecciona entre H y alquilo C1-3 sustituido con 0-4 R9;
R9 se selecciona entre halógeno, OH, CN, alquilo C1-3 sustituido con 0-4 Rb, alcoxi C1-3 sustituido con 0-4 Rb, CO2(alquilo C1.4), CONH2, NHCO2(alquilo C1-3), cicloalquilo C3-6, fenilo y heterociclo; y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
8. El compuesto de la reivindicación 5 o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona independientemente entre F, Cl y OCH3;
R5 se selecciona entre

y

R7 se selecciona entre H, CN, alquilo C1-3 sustituido con 0-4 R9, alcoxi C1-3 sustituido con 0-4 R9, CO2alquilo C1-4, CONH2, P(=O)(alquilo C1-3)2, SO2alquilo C1.3,

R8 se selecciona entre H y alquilo C1-3 sustituido con 0-4 R9;
R9 se selecciona entre halógeno, OH, CN, alquilo C1-3 sustituido con 0-4 Rb; alcoxi C1-3 sustituido con 0-4 Rb, CO2(alquilo C1.4), CONH2, NHCO2(alquilo C1.3), cicloalquilo C3-6, fenilo y heterociclo; y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
9. El compuesto de la reivindicación 5 o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde
R2 se selecciona entre F, Cl y OCH3;
R5 es

R7 se selecciona entre H, F, Cl, Br, CN, alquilo C1-3 sustituido con 0-4 R9, alcoxi C1-3 sustituido con 0-4 R9, CO2alquilo C1.4, CONH2, P(=O)(alquilo C1-3)2, SO2alquilo C1-3y

R8 se selecciona entre H y alquilo C1-3 sustituido con 0-4 R9;
R9 se selecciona entre halógeno, OH, CN, alquilo C1-3 sustituido con 0-4 Rb; alcoxi C1-3 sustituido con 0-4 Rb, CO2(alquilo C1.4), CONH2, NHCO2(alquilo C1-3), cicloalquilo C3-6, fenilo y heterociclo; y
Rb, en cada caso, se selecciona independientemente entre OH y halógeno.
10. Una composición farmacéutica que comprende uno o más compuestos de acuerdo con una cualquiera de las reivindicaciones 1-9 y un vehículo o un diluyente farmacéuticamente aceptables.
11. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-9 para su uso en terapia.
12. Un compuesto de acuerdo con la reivindicación 1, en donde el compuesto se selecciona de:
2-(2-fluoro-4-(2-((7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-(1,3-dimetil-1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((5-(3,5-dimetil-1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-(1-metil-3-(trifluorometil)-1H-pirazol-5-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-cloro-4-(2-((1-(ciclopropilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((6-bromoimidazo[1,2-a]piridin-2-il)amino)-2-oxoetil)-2-clorofenoxi)nicotinamida;
2-(2-cloro-4-(2-((1-metil-1H-pirazolo[3,4-b]piridin-3-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(pirazin-2-il)-1,3,4-tiadiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-metil-5-(piridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-(5-fluoropiridin-2-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-metil-4-feniltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-(6-fluoropiridin-2-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-(benzo[d]tiazol-2-ilamino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-(1-metil-2-oxo-1,2-dihidropiridin-4-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((4-(2-oxo-1,2-dihidropiridin-4-il)tiazol-2-il)amino)etil)fenoxi)nicotinamida;
(4-(2-(2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acetamido)-5-metiltiazol-4-il)piridin-2-il)carbamato de metilo; 2-(2-fluoro-4-(2-((5-metil-4-(1-metil-6-oxo-1,6-dihidropiridin-3-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(4-(2-((4-(1-(difluorometil)-6-oxo-1,6-dihidropiridin-3-il)-5-metiltiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-cloro-4-(2-((5-metil-4-(6-metilpiridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-cloro-4-(2-((4-ciclobutil-5-(piridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(2-oxo-1,2-dihidropiridin-4-il)benzo[d]tiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-(1-metil-2-oxo-1,2-dihidropiridin-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(4-(2-((5-(3-ciclopropil-1-metil-1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(2-fluoro-4-(2-oxo-2-((5-(1,3,4-trimetil-1H-pirazol-5-il)benzo[d]tiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((5-(1,3-dimetil-1H-pirazol-5-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((5-(1-(difluorometil)-1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(2-fluoro-4-(2-((5-(1-metil-1H-pirazol-5-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-(1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(3-(trifluorometil)-1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)etil)fenoxi)nicotinamida; 2-(2-fluoro-4-(2-((5-(1-metil-1H-pirazol-3-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-(l-(metil-d3)-1H-pirazol-3-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-(1-metil-1H-pirazol-4-il)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((6-(1-metil-1H-pirazol-4-il)imidazo[1,2-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-cloro-4-(2-((6-(1-metil-1H-pirazol-4-il)imidazo[1,2-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-(1-metil-1H-pirazol-4-il)-1-(2,2,2-trifluoroetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(2-(4-metilpiperazin-1-il)pirimidin-5-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(2-fluoro-4-(2-((4-(2-metoxipirimidin-5-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(pirimidin-5-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-(2-(2-hidroxipropan-2-il)pirimidin-5-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(2-fluoro-4-(2-((4-(6-metoxipiridin-2-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(piridin-4-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((4-(6-cianopiridin-2-il)-5-metiltiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((4-ciclopropil-5-metiltiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(4-(metilsulfonil)fenil)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-(4-(isopropilsulfonil)fenil)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-(benzo[d]tiazol-2-ilamino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((4-(3-cloro-4-metilfenil)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((4-(3,4-dimetoxifenil)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-oxo-2-((4-feniltiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-(benzo[d]oxazol-2-ilamino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-metil-1,3,4-tiadiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((4-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-oxo-2-((5-fenil-1,3,4-tiadiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((5-metil-4-feniltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-oxo-2-((4-(2-oxo-2H-cromen-5-il)tiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-oxo-2-((4-(3-(trifluorometoxi)fenil)tiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((4-(3-cianofenil)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((4-(3-fluorofenil)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acetamido)benzo[d]tiazol-5-carboxilato de metilo;
2-(2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acetamido)benzo[d]tiazol-6-carboxamida;
2-(4-(2-((6-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-metil-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-oxo-2-((5-(propilsulfonil)-1H-benzo[d]imidazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acetamido)benzo[d]tiazol-6-carboxilato de metilo;
2-(4-(2-((5-ciano-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-benzoil-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((7-cloro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-(4-((3-carbamoilpiridin-2-il)oxi)fenil)acetamido)-1-metil-1H-benzo[d]imidazol-5-carboxilato de metilo;
2-(4-(2-((1-metil-1H-indazol-3-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((6-(metilsulfonil)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-oxo-2-((4-(m-tolilamino)quinazolin-6-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((4-(4-fluorofenil)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((1-bencil-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-clorofenoxi)nicotinamida;
2-(2-cloro-4-(2-((4-(2-fluorofenil)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-cloro-4-(2-((4-(6-metilimidazo[2,1-b]tiazol-5-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((8-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((8-metoxi-[1,2,4|triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4,5,6,7-tetrahidrotiazolo[4,5-c]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((4-(6-fluoropiridin-3-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(1-metil-1H-pirazol-5-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(piridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(2-metilpirimidin-4-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(piridin-3-il)-1,3,4-tiadiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(trifluorometil)-1,3,4-tiadiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((5-cidopropil-1,3,4-tiadiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((5-(3,4-dimetoxibencil)-1,3,4-tiadiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((8-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((4-((1-metilpiperidin-4-il)oxi)fenil)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(piridin-4-il)-1,3,4-tiadiazol-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((4,5-dimetiltiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((1-(2,2,2-trifluoroetil)-1H-pirazol-3-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((1-(5-(trifluorometil)piridin-2-il)-1H-pirazol-3-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((3-cidopropil-1,2,4-tiadiazol-5-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-doro-4-(2-((4-(2-fluoropiridin-3-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((4-(6-fluoropiridin-3-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((5-metil-4-(1-metil-1H-pirazol-4-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((4-(6-doropiridin-3-il)-5-metiltiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(1-((7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-1-oxopropan-2-il)fenoxi)nicotinamida;
2-(4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((7-(2-hidroxi-2-metilpropoxi)-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((6-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((7-(2-hidroxi-2-metilpropoxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(4-(2-((1-(2,2-difluoroetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-doro-4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((7-(2-hidroxi-2-metilpropoxi)-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(2-doro-4-(2-((5-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((7-metoxi-4,5-dihidronafto[1,2-d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-metoxi-4-(2-((5-metoxibenzo[d]tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((5-(2-hidroxi-2-metilpropoxi)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-metoxifenoxi)nicotinamida;
2-(2-bromo-4-(2-((7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((1-(2,2-difluoroetil)-5-iTietoxMH-benzo[d]iiTiidazol-2-il)aiTiino)-2-oxoetil)-2-fluorofenoxi)mcotmaiTiida; 2-(2-doro-4-(2-((5-(difluorometoxi)tiazolo[5,4-b]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((1H-indazol-3-il)amino)-2-oxoetil)-2-dorofenoxi)nicotinamida;
2-(4-(2-((1-(2,2-difluoroetil)-6-metoxi-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((7-metoxi-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((5-(trifluorometil)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((8-fluoro-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((6-fluoro-[l,2,4]triazolo[l,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((6-(trifluorometil)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((6,7-dimetil-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((5,6-dimetil-[l,2,4]triazolo[l,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((7-(trifluorometil)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((6-(trifluorometil)imidazo[1,2-a]piridin-2-il)amino)etil)fenoxi)nicotinamida;
2-(4-(2-((7-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((7-(1-(difluoroiTietil)-1H-pirazol-4-ilH1,2,4]tnazolo[1,5-a]pindin-2-il)aiTiino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((7-(1,3-dimetil-1H-pirazol-4-il)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((7-(1-metil-1H-pirazol-5-il)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(2-fluoro-4-(2-((6-(l-metil-1H-pirazol-5-il)-[l,2,4]triazolo[l,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; 2-(4-(2-((8-(difluorometoxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((6-(trifluorometoxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((8-(trifluorometil)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((7-fluoro-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((7-(benciloxi)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((l-(ddopropNiTietil)-6-(1-iTietiMH-pirazol-4-il)-1H-benzo[d]iiTiidazol-2-il)aiTiino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((1-(ddopropilmetil)-6-(dimetilfosforil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-
fluorofenoxi)nicotinamida;
2-(4-(2-((5-(dimetilfosforil)benzo[d]tiazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((5-(dimetilfosforil)-[1,2,4]triazolo[1,5-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((5-(dimetilfosforil)-1-(2,2,2-trifluoroetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((6-(dimetilfosforil)imidazo[1,2-a]piridin-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((1-(2,2-difluoroetil)-6-(dimetilfosforil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((5-metil-4-(6-metilpiridin-2-il)tiazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((1-(cidobutilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(2-fluoro-4-(2-((1-isopropil-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-fenoxi)nicotinamida;
2-(2-fluoro-4-(2-oxo-2-((1-(3,3,3-trifluoropropil)-1H-benzo[d]-imidazol-2-il)-amino)etil)fenoxi)nicotinamida;
2-(2-fluoro-4-(2-((1-(2-metoxietil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-fenoxi)nicotinamida;
2-(2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acetamido)-1-(2-metoxietil)-1H-benzo[d]imidazol-5-carboxamida; 2-(2-fluoro-4-(2-((6-metoxi-1-(2-metoxietil)-1H-benzo[d]-imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-(4-((3-carbamoilpiridin-2-il)oxi)-3-fluorofenil)acetamido)-1-(2-metoxietil)-1H-benzo[d]-imidazol-6-carboxamida; 2-(2-fluoro-4-(2-oxo-2-((1-((tetrahidro-2H-piran-4-il)metil)-1H-benzo[d]imidazol-2-il)amino)etil)-fenoxi)nicotinamida; 2-(4-(2-((1-(cidopropilmetil)-6-metoxi-1H-benzo[d]-imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(cidopropilmetil)-4-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(cidopropilmetil)-5-metoxi-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(cidopropilmetil)-4-metoxi-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(cidopropilmetil)-7-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(cidopropilmetil)-5-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(2,2-difluoroetil)-6-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((1-(cidopropilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((1-(cidopropilmetil)-6-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(4-(2-((1-(2,2-difluoroetil)-4-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
2-(4-(2-((6-(benciloxi)-1-(cidopropilmetil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida; 2-(2-doro-4-(2-((1-(cidopropilmetil)-4-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((1-(cidopropilmetil)-6-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((1-(2,2-difluoroetil)-4-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-doro-4-(2-((1-(cidopropilmetil)-5-fluoro-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(4-(2-((1-(ciclopropilmetil)-6-(trifluorometil)-1H-benzo[d]imidazol-2-ii)amino)-2-oxoetii)-2-fluorofenoxi)nicotinamida;
2-(2-cloro-4-(2-((1-(ciclopropilmetil)-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida;
2-(2-cloro-4-(2-((1-(ciclopropilmetil)-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)fenoxi)nicotinamida; o
2-(4-(2-((1-(cidopropilmetil)-6-(2-hidroxi-2-metilpropoxi)-1H-benzo[d]imidazol-2-il)amino)-2-oxoetil)-2-fluorofenoxi)nicotinamida;
o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo.
13. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-9 para su uso en la profilaxis y/o el tratamiento de trastornos asociados a la actividad aberrante de Rho cinasa, en donde dicho trastorno se selecciona del grupo que consiste en un trastorno cardiovascular, un trastorno relacionado con el músculo liso, una enfermedad fibrótica, una enfermedad inflamatoria, trastornos neuropáticos, trastornos oncológicos y un trastorno autoinmunitario.
14. El compuesto para su uso de acuerdo con la reivindicación 13, en donde dicho trastorno cardiovascular se selecciona del grupo que consiste en angina de pecho, ateroesclerosis, ictus, enfermedad cerebrovascular, insuficiencia cardíaca, arteriopatía coronaria, infarto de miocardio, vasculopatía periférica, estenosis, vasoespasmo, hipertensión e hipertensión pulmonar.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762531624P | 2017-07-12 | 2017-07-12 | |
| PCT/US2018/041559 WO2019014300A1 (en) | 2017-07-12 | 2018-07-11 | PHENYLACETAMIDES AS ROCK INHIBITORS |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2951212T3 true ES2951212T3 (es) | 2023-10-18 |
Family
ID=63036481
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18746509T Active ES2951212T3 (es) | 2017-07-12 | 2018-07-11 | Fenilacetamidas como inhibidores de ROCK |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11306081B2 (es) |
| EP (1) | EP3652164B1 (es) |
| JP (1) | JP7206253B2 (es) |
| KR (1) | KR102680164B1 (es) |
| CN (1) | CN110869360B (es) |
| ES (1) | ES2951212T3 (es) |
| WO (1) | WO2019014300A1 (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102491994B1 (ko) | 2016-07-07 | 2023-01-25 | 브리스톨-마이어스 스큅 컴퍼니 | Rock의 억제제로서의 스피로락탐 |
| ES2829400T3 (es) | 2016-11-30 | 2021-05-31 | Bristol Myers Squibb Co | Inhibidores tricíclicos de Rho cinasa |
| TW201908293A (zh) | 2017-07-12 | 2019-03-01 | 美商必治妥美雅史谷比公司 | 作為rock抑制劑之5員及雙環雜環醯胺 |
| CN110869361B (zh) | 2017-07-12 | 2023-06-02 | 百时美施贵宝公司 | 用于治疗心力衰竭的rock的五元氨基杂环和5,6元或6,6元双环氨基杂环抑制剂 |
| EP3652167B1 (en) | 2017-07-12 | 2021-06-02 | Bristol-Myers Squibb Company | Spiroheptanyl hydantoins as rock inhibitors |
| US12060341B2 (en) | 2017-07-12 | 2024-08-13 | Bristol-Myers Squibb Company | Spiroheptanyl hydantoins as ROCK inhibitors |
| CN111278825B (zh) | 2017-11-03 | 2023-05-02 | 百时美施贵宝公司 | 二氮杂螺rock抑制剂 |
| BR112021008883A2 (pt) | 2018-11-06 | 2021-08-10 | Cervello Therapeutics, Llc | inibidores de rock quinase |
| US11248004B2 (en) | 2018-11-06 | 2022-02-15 | Cervello Therapeutics, Llc. | Substituted isoquinolines as rock kinase inhibitors |
| KR20220097434A (ko) * | 2019-11-01 | 2022-07-07 | 브리스톨-마이어스 스큅 컴퍼니 | 톨 수용체 억제제로서의 치환된 피라졸 화합물 |
| CN118434843A (zh) | 2021-11-11 | 2024-08-02 | 学校法人同志社 | 角膜内皮细胞的冷冻保存制剂及其制造方法 |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW200306819A (en) * | 2002-01-25 | 2003-12-01 | Vertex Pharma | Indazole compounds useful as protein kinase inhibitors |
| NZ540161A (en) | 2002-10-30 | 2008-03-28 | Vertex Pharma | Compositions useful as inhibitors of rock and other protein kinases |
| WO2008036540A2 (en) | 2006-09-20 | 2008-03-27 | Boehringer Ingelheim International Gmbh | Rho kinase inhibitors |
| CL2007003874A1 (es) | 2007-01-03 | 2008-05-16 | Boehringer Ingelheim Int | Compuestos derivados de benzamida; composicion farmaceutica que comprende a dichos compuestos; y su uso para tratar enfermedades cardiovasculares, hipertension, aterosclerosis, reestenosis, ictus, insuficiencia cardiaca, lesion isquemica, hipertensio |
| EP2331507A2 (en) | 2008-09-18 | 2011-06-15 | Astellas Pharma Inc. | Heterocyclic carboxamide compounds |
| US9302989B2 (en) | 2010-11-15 | 2016-04-05 | Abbvie Inc. | NAMPT and rock inhibitors |
| TW201443023A (zh) | 2013-01-18 | 2014-11-16 | 必治妥美雅史谷比公司 | 作爲rock抑制劑之酞□酮及異喹啉酮 |
| AR094929A1 (es) | 2013-02-28 | 2015-09-09 | Bristol Myers Squibb Co | Derivados de fenilpirazol como inhibidores potentes de rock1 y rock2 |
| EP2961746B1 (en) | 2013-02-28 | 2018-01-03 | Bristol-Myers Squibb Company | Phenylpyrazole derivatives as potent rock1 and rock2 inhibitors |
| US9663529B2 (en) | 2013-07-02 | 2017-05-30 | Bristol-Myers Squibb Company | Tricyclic pyrido-carboxamide derivatives as rock inhibitors |
| ES2633987T3 (es) | 2013-07-02 | 2017-09-26 | Bristol-Myers Squibb Company | Derivados de pirido-carboxamidas tricíclicas como inhibidores de ROCK |
| US9902702B2 (en) | 2014-07-15 | 2018-02-27 | Bristol-Myers Squibb Company | Spirocycloheptanes as inhibitors of rock |
| WO2016028971A1 (en) | 2014-08-21 | 2016-02-25 | Bristol-Myers Squibb Company | Tied-back benzamide derivatives as potent rock inhibitors |
| AR103990A1 (es) | 2015-01-09 | 2017-06-21 | Bristol Myers Squibb Co | Ureas cíclicas como inhibidoras de rock |
| US10112929B2 (en) | 2015-03-09 | 2018-10-30 | Bristol-Myers Squibb Company | Lactams as inhibitors of rock |
| AR107354A1 (es) | 2016-01-13 | 2018-04-18 | Bristol Myers Squibb Co | Salicilamidas espiroheptanos y compuestos relacionados como inhibidores de rock |
| US10562887B2 (en) | 2016-05-27 | 2020-02-18 | Bristol-Myers Squibb Company | Triazolones and tetrazolones as inhibitors of ROCK |
| KR102491994B1 (ko) | 2016-07-07 | 2023-01-25 | 브리스톨-마이어스 스큅 컴퍼니 | Rock의 억제제로서의 스피로락탐 |
| KR102473481B1 (ko) | 2016-07-07 | 2022-12-01 | 브리스톨-마이어스 스큅 컴퍼니 | 강력하고 선택적인 rock 억제제로서의 락탐, 시클릭 우레아 및 카르바메이트, 및 트리아졸론 유도체 |
| KR102449652B1 (ko) | 2016-07-07 | 2022-09-29 | 브리스톨-마이어스 스큅 컴퍼니 | Rock의 억제제로서의 스피로-융합 시클릭 우레아 |
| ES2829400T3 (es) | 2016-11-30 | 2021-05-31 | Bristol Myers Squibb Co | Inhibidores tricíclicos de Rho cinasa |
| CN110869361B (zh) | 2017-07-12 | 2023-06-02 | 百时美施贵宝公司 | 用于治疗心力衰竭的rock的五元氨基杂环和5,6元或6,6元双环氨基杂环抑制剂 |
| EP3652167B1 (en) | 2017-07-12 | 2021-06-02 | Bristol-Myers Squibb Company | Spiroheptanyl hydantoins as rock inhibitors |
| TW201908293A (zh) | 2017-07-12 | 2019-03-01 | 美商必治妥美雅史谷比公司 | 作為rock抑制劑之5員及雙環雜環醯胺 |
| CN111278825B (zh) | 2017-11-03 | 2023-05-02 | 百时美施贵宝公司 | 二氮杂螺rock抑制剂 |
-
2018
- 2018-07-11 US US16/629,736 patent/US11306081B2/en active Active
- 2018-07-11 JP JP2020501553A patent/JP7206253B2/ja active Active
- 2018-07-11 CN CN201880045811.XA patent/CN110869360B/zh active Active
- 2018-07-11 WO PCT/US2018/041559 patent/WO2019014300A1/en unknown
- 2018-07-11 EP EP18746509.1A patent/EP3652164B1/en active Active
- 2018-07-11 ES ES18746509T patent/ES2951212T3/es active Active
- 2018-07-11 KR KR1020207003732A patent/KR102680164B1/ko active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019014300A1 (en) | 2019-01-17 |
| EP3652164A1 (en) | 2020-05-20 |
| EP3652164B1 (en) | 2023-06-21 |
| US11306081B2 (en) | 2022-04-19 |
| JP2020527560A (ja) | 2020-09-10 |
| US20210147407A1 (en) | 2021-05-20 |
| KR20200028424A (ko) | 2020-03-16 |
| JP7206253B2 (ja) | 2023-01-17 |
| CN110869360B (zh) | 2023-12-15 |
| KR102680164B1 (ko) | 2024-07-01 |
| CN110869360A (zh) | 2020-03-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2951212T3 (es) | Fenilacetamidas como inhibidores de ROCK | |
| ES2882432T3 (es) | Amidas de 5 miembros y heterocíclicas bicíclicas como inhibidores de ROCK | |
| ES2814325T3 (es) | Derivados de lactama, urea cíclica y carbamato y de triazolona como inhibidores potentes y selectivos de ROCK | |
| ES2829550T3 (es) | Ureas cíclicas espiro-condensadas como inhibidores de ROCK | |
| ES2898698T3 (es) | Salicilamidas de espiroheptanos y compuestos relacionados como inhibidores de cinasa Rho (ROCK) | |
| ES2684776T3 (es) | Ftalazinonas e isoquinolinonas como inhibidores de ROCK | |
| ES2829400T3 (es) | Inhibidores tricíclicos de Rho cinasa | |
| ES2894128T3 (es) | Inhibidores aminoheterocíclicos de cinco miembros y aminoheterocíclicos bicíclicos de 5,6 o 6,6 miembros de ROCK para el tratamiento de la insuficiencia cardíaca | |
| ES2838573T3 (es) | Derivados de benzamida ligados como inhibidores potentes de ROCK | |
| ES2634628T3 (es) | Derivados tricíclicos de pirido-carboxamida como inhibidores ROCK | |
| ES2937039T3 (es) | Inhibidores diazaespiro de ROCK | |
| ES2730112T3 (es) | Lactamas como inhibidores de ROCK | |
| ES2815681T3 (es) | Ureas cíclicas como inhibidores de ROCK | |
| AU2018379321B2 (en) | 1,2,4-oxadiazole derivatives as histone deacetylase 6 inhibitors | |
| EP2961746A1 (en) | Phenylpyrazole derivatives as potent rock1 and rock2 inhibitors | |
| KR102716960B1 (ko) | 디아자스피로 rock 억제제 | |
| US20160090364A1 (en) | Phthalazinones and isoquinolinones as rock inhibitors |