ES2947464T3 - Inhibidores de la proteína tirosina fosfatasa - Google Patents
Inhibidores de la proteína tirosina fosfatasa Download PDFInfo
- Publication number
- ES2947464T3 ES2947464T3 ES19801996T ES19801996T ES2947464T3 ES 2947464 T3 ES2947464 T3 ES 2947464T3 ES 19801996 T ES19801996 T ES 19801996T ES 19801996 T ES19801996 T ES 19801996T ES 2947464 T3 ES2947464 T3 ES 2947464T3
- Authority
- ES
- Spain
- Prior art keywords
- amine
- pyrazin
- thio
- amino
- pyrido
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Enzymes And Modification Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Se proporcionan compuestos de Fórmula Ia o un estereoisómero, tautómero o sal farmacéuticamente aceptable de los mismos, que son útiles para el tratamiento de enfermedades hiperproliferativas en vista de su capacidad para inhibir SHP2. Se describen métodos para usar compuestos de fórmula I o un estereoisómero, tautómero o sal farmacéuticamente aceptable de los mismos, para diagnóstico, prevención o tratamiento in vitro, in situ e in vivo de tales trastornos en células de mamíferos, o condiciones patológicas asociadas. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Inhibidores de la proteína tirosina fosfatasa
ANTECEDENTES DE LA INVENCIÓN
CAMPO DE LA INVENCIÓN
La presente invención se refiere a compuestos que inhiben SHP2 y son útiles para tratar enfermedades hiperproliferativas y neoplásicas. La presente invención se refiere además a compuestos de la presente invención para su uso en el tratamiento del cáncer o de enfermedades hiperproliferativas.
DESCRIPCIÓN DEL ESTADO DE LA TÉCNICA
SHP2 es una proteína triosina fosfatasa (PTP) que contiene dominios Src Homología 2 (SH2) codificada por el gen PTPN11. SHP2 contribuye a múltiples funciones celulares, incluyendo la proliferación, la diferenciación, el mantenimiento del ciclo celular y la migración. SHP2 es necesaria para la plena activación de la vía RasZERKl/2, una cascada de señalización clave en la biología del cáncer que se encuentra corriente abajo de una amplia gama de receptores tirosina quinasas y otros transductores de señales. También se ha demostrado que SHP2 promueve la señalización PI3K/AKT, JAK/STAT, JNK y NF-KΒ, que también están asociadas con diversos cánceres humanos. SHP2 es una oncoproteína. Véase Frankson, Rochelle, et al. “Therapeutic Targeting of Oncogenic Tyrosine Phosphatases.” Cancer Research. Vol. 77, No. 21 (2017): pp. 5701-5705. Fedele, Carmine, et al. “SHP2 Inhibition Prevents Adaptive Resistance to MEK inhibitors in Multiple Cancer Models.” Cancer Discovery. Vol. 8, No. 10 (2018): pp. 1237-49. Nichols, Robert J., et al. “Efficacy of SHP2 phosphatase inhibition in cancers with nucleotide- cycling oncogenic RAS, RAS-GTP dependent oncogenic BrAf and NF 1 loss.” bioRxiv 188730; doi: https://doi.org/10.1101/188730.
Por lo tanto, los inhibidores de pequeña molécula de SHP2 serían útiles para tratar un amplio espectro de cánceres, tales como, por ejemplo, melanoma, leucemias mielomonocíticas juveniles, neuroblastoma, mieloide crónico positivo con cromosoma Filadelfia, leucemias linfoblásticas agudas con cromosoma Filadelfia positivo, leucemias mieloides agudas, neoplasias mieloproliferativas (tales como policitemia vera, trombocitemia esencial y mielofibrosis primaria), cáncer de mama, cáncer de pulmón, cáncer de hígado, cáncer colorrectal, cáncer de esófago, cáncer gástrico, carcinoma de células escamosas de cabeza y cuello, glioblastoma, linfoma anaplásico de células grandes, carcinoma de tiroides, neoplasias spitzoides, así como, Neurofibramatosis y Síndrome de Noonan.
Los inhibidores de SHP2 son conocidos, véase, por ejemplo, los documentos WO 2015/107493; WO 2015/107494; WO 2015/107495; WO 2016/203404; WO 2016/203405; WO 2016/203406; WO 2017/210134; WO 2017/211303; WO 2017/216706; WO 2018/013597; WO 2018/057884; WO 2018/081091; WO 2018/136264; WO 2018/136265; y WO 2018/172984. Sin embargo, es bien sabido que hay dificultades para desarrollar un compuesto y convertirlo en un medicamento aprobado. DiMasi, Joseph A. “Success rates for new drugs entering clinical testing in the United States.” Clinical Pharmacology & Therapeutics. Vol. 58, no. 1 (1995): pp. 1-14. Scannell, JW, Bosley J. “When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis.” PloS ONE 11(2) (2016): e0147215. doi: 10.1371/joumal.pone.0147215.
SUMARIO DE LA INVENCIÓN
Existe una necesidad continua de agentes terapéuticos nuevos y novedosos que puedan utilizarse para el cáncer y las afecciones hiperproliferativas. El diseño y desarrollo de nuevos compuestos farmacéuticos es esencial.
La invención proporciona un compuesto seleccionado de la Fórmula Ila:

o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que:
Li es S;
R1 se selecciona de fenilo, heteroarilo, arilo bicíclico, heterociclilo bicíclico y heteroarilo bicíclico,
en el que fenilo, heteroarilo, arilo bicíclico, heterociclo bicíclico y heteroarilo bicíclico están opcionalmente sustituidos con uno o más grupos seleccionados del grupo que consiste en halógeno, OH, oxo, ciano, alquilo opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y un heterociclo opcionalmente sustituido con halógeno, ciano, OH o alquilo opcionalmente sustituido con OH u oxo; R2 es:

X11 se selecciona de CR13R14, SiR13R14, NH y O;
X12 se selecciona de CHR15 y NH, en donde uno o ambos de X11 y X12 debe ser carbono;
R10 se selecciona de hidrógeno y alquilo;
R11 se selecciona de hidrógeno, OH y CH2NH2;
R12, R16 y R17 son hidrógeno;
R13 se selecciona de hidrógeno, OH y (alquilo de C0-C3)NRbRc;
R14 se selecciona de hidrógeno, OH, alquilo opcionalmente sustituido con halógeno, OH, metilo, OCH3 y un heteroarilo;
R15 se selecciona de hidrógeno o NH2;
o uno de los siguientes grupos pueden unirse:
R10 y R11 pueden unirse como CH2NHCH2 para formar un bicíclico fusionado,
R10 y R15 pueden unirse como alquilo para formar un bicíclico puenteado,
R11 y R12 pueden unirse como alquilo sustituido con NH2 para formar un espirociclo,
R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico, en donde el cicloalquilo, heterociclo, carbociclo y heterociclo se sustituyen opcionalmente con F, Cl, OH, OCH3, CN, metilo o NH2 para formar un espirociclo,
R10 y R16 pueden unirse como alquilo, O o NH para formar un bicíclico puenteado,
R11 y R15 pueden unirse como alquilo para formar un bicíclico puenteado,
R11 y R16 pueden unirse como alquilo u O para formar un bicíclico puenteado,
R11 y R17 pueden unirse como alquilo para formar un bicíclico puenteado, o
R13 y R15 pueden unirse como NHCH2 o cicloalquilo en donde el cicloalquilo se sustituye con NH2 para formar un bicíclico fusionado;
R48 se selecciona de hidrógeno y metilo;
Ra es hidrógeno, alquilo opcionalmente sustituido con OH, metoxi, halógeno o ciano o ciclopropilo;
Rb y Rc se seleccionan independientemente de hidrógeno, alquilo y un grupo Boc; y
a, b, c y d se seleccionan de 0 y 1.
Otro aspecto proporciona un compuesto de acuerdo con la Fórmula Ila, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, a un paciente que lo necesite para su uso en el tratamiento de un trastorno hiperproliferativo. El compuesto puede administrarse solo o junto con al menos otro compuesto antihiperproliferativo o quimioterapéutico.
Otro aspecto proporciona un compuesto de Fórmula Ila, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, solo o en combinación con uno o más compuestos adicionales que tienen propiedades anticancerígenas para su uso en el tratamiento del cáncer.
Otro aspecto proporciona una composición farmacéutica que comprende un compuesto de Fórmula Ila, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, y un portador, diluyente o excipiente farmacéuticamente aceptable.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
A continuación, se hará referencia en detalle a determinadas realizaciones, cuyos ejemplos se ilustran en las estructuras y fórmulas adjuntas. Aunque se describirán las realizaciones enumeradas, se entenderá que no pretenden limitar la invención a dichas realizaciones. Por el contrario, la invención pretende abarcar todas las alternativas, modificaciones y equivalentes que puedan incluirse en el ámbito de la presente invención, tal como se define en las reivindicaciones. Un experto en la técnica reconocerá muchos métodos y materiales similares o equivalentes a los descritos en el presente documento, que podrían utilizarse en la práctica de la presente invención. La presente invención no se limita en modo alguno a los métodos y materiales descritos. En el caso de que una o más de las publicaciones incorporadas y materiales similares difieran o contradigan esta solicitud, incluyendo, pero no limitándose a términos definidos, uso de términos, técnicas descritas o similares, esta solicitud prevalecerá.
DEFINICIONES
La frase "una" o "un" entidad, tal como se utiliza en el presente documento, se refiere a una o más de esa entidad; por ejemplo, un compuesto se refiere a uno o más compuestos o al menos a un compuesto. Como tales, los términos "un" (o "una"), "uno o más" y "al menos uno" pueden utilizarse indistintamente en el presente documento.
La frase "tal como se define en el presente documento" se refiere a la definición más amplia para cada grupo tal como se proporciona en la Descripción Detallada de la Invención o en la reivindicación más amplia. En todas las demás realizaciones proporcionadas a continuación, los sustituyentes que pueden estar presentes en cada realización, y que no se definen explícitamente, conservan la definición más amplia proporcionada en la Descripción Detallada de la Invención.
Tal como se utilizan en esta memoria descriptiva, ya sea en una frase transitoria o en el cuerpo de la reivindicación, los términos "comprende(n)" y "que comprende(n)" deben interpretarse con un significado abierto. Es decir, los términos deben interpretarse como sinónimos de las frases "que tiene al menos" o "que incluye al menos". Cuando se utiliza en el contexto de un proceso, el término "que comprende" significa que el proceso incluye al menos los pasos mencionados, pero puede incluir pasos adicionales. Cuando se utiliza en el contexto de un compuesto o composición, el término "que comprende" significa que el compuesto o composición incluye al menos las características o componentes mencionados, pero también puede incluir características o componentes adicionales. Además, las palabras "incluir", "que incluye" e "incluye", cuando se utilizan en esta memoria descriptiva y en las reivindicaciones siguientes, pretenden especificar la presencia de características, números enteros, componentes o pasos indicados, pero no excluyen la presencia o adición de una o más características, números enteros, componentes, pasos o grupos de los mismos.
El término "independientemente" se utiliza en el presente documento para indicar que una variable se aplica en cualquier caso sin tener en cuenta la presencia o ausencia de una variable que tenga esa misma definición o una diferente dentro del mismo compuesto. Así, en un compuesto en el que R" aparece dos veces y se define como "independientemente carbono o nitrógeno", ambos R" pueden ser carbono, ambos R" pueden ser nitrógeno, o un R" puede ser carbono y el otro nitrógeno.
Cuando cualquier variable (por ejemplo, R1, R4a, Ar, X1 o Het) aparece más de una vez en cualquier fracción o fórmula que represente y describa compuestos empleados o reivindicados en la presente invención, su definición en cada aparición es independiente de su definición en cualquier otra aparición. Asimismo, las combinaciones de sustituyentes y/o variables sólo son admisibles si tales dan lugar a compuestos estables.
El término "opcional" u "opcionalmente" como se usa en el presente documento significa que un evento o circunstancia descrito posteriormente puede ocurrir, pero no necesariamente, y que la descripción incluye instancias en las que ocurre el evento o circunstancia y las instancias en las que no ocurre. Por ejemplo, "opcionalmente sustituida" significa que la fracción opcionalmente sustituida puede incorporar un hidrógeno o un sustituyente.
El término "aproximadamente" se utiliza en el presente documento para significar aproximadamente, en la región de, más o menos, o alrededor de. Cuando el término "aproximadamente" se utiliza junto con un intervalo numérico, modifica dicho intervalo ampliando los límites por encima y por debajo de los valores numéricos establecidos. En general, el término "aproximadamente" se utiliza en el presente documento para modificar un valor numérico por encima y por debajo del valor indicado en una variación del 20 %.
Tal y como se utiliza en el presente documento, la mención de un intervalo numérico para una variable pretende transmitir que la invención puede practicarse con la variable igual a cualquiera de los valores dentro de ese intervalo. Así, para una variable que es intrínsecamente discreta, la variable puede ser igual a cualquier valor entero del intervalo numérico, incluyendo los puntos finales del intervalo. Del mismo modo, para una variable que es intrínsecamente continua, la variable puede ser igual a cualquier valor real del intervalo numérico, incluyendo los puntos finales del intervalo. Por ejemplo, una variable que se describe con valores entre 0 y 2 puede ser 0, 1 o 2 para las variables que son intrínsecamente discretas, y puede ser 0,0, 0,1, 0,01, 0,001 o cualquier otro valor real para las variables que son intrínsecamente continuas.
Los compuestos de la invención presentan tautomerismo. Los compuestos tautoméricos pueden existir como dos o más especies interconvertibles. Los tautómeros prototrópicos son el resultado de la migración de un átomo de hidrógeno unido covalentemente entre dos átomos. Los tautómeros existen generalmente en equilibrio y los intentos de aislar un tautómero individual suelen producir una mezcla cuyas propiedades químicas y físicas son consistentes con una mezcla de compuestos. La posición del equilibrio depende de las características químicas de la molécula. Por ejemplo, en muchos aldehídos alifáticos y cetonas, tal como el acetaldehído, predomina la forma ceto, mientras que en los fenoles predomina la forma enol. Tautómeros prototrópicos comunes incluyen tautómeros de ceto/enol (-C(=O)-CH2- ^ -C(-OH)=CH-), amida/ácido imídico (-C(=O)-NH- ^ -C(-OH)=N-) y amidina (-C(=NR)-NH- ^ -C(-NHR)=N-). Los dos últimos son particularmente comunes en los anillos heteroarilo y heterocíclico, y la presente invención abarca todas las formas tautoméricas de los compuestos.
El experto en la técnica apreciará que algunos de los compuestos de la invención pueden contener uno o más centros quirales y, por lo tanto, existir en dos o más formas estereoisoméricas. Los racematos de estos isómeros, los isómeros individuales y las mezclas enriquecidas en un enantiómero, así como diastereómeros cuando hay dos centros quirales, y las mezclas parcialmente enriquecidas con diastereómeros específicos están dentro del alcance de la presente invención. La presente invención incluye todos los estereoisómeros individuales (por ejemplo, enantiómeros), mezclas racémicas o mezclas parcialmente resueltas de los compuestos de la invención y, en su caso, las formas tautoméricas individuales de los mismos.
Los compuestos de la invención pueden contener un centro básico y se forman sales de adición ácida adecuadas a partir de ácidos que forman sales no tóxicas. Ejemplos de sales de ácidos inorgánicos incluyenclorhidrato, bromhidrato, yodhidrato, cloruro, bromuro, yoduro, sulfato, bisulfato, nitrato, fosfato y fosfato de hidrógeno. Ejemplos de sales de ácidos orgánicos incluyen acetato, fumarato, pamoato, aspartato, besilato, carbonato, bicarbonato, camsilato, D y L-lactato, D y L-tartrato, esilato, mesilato, malonato, orotato, gluceptato, metilsulfato, estearato, glucuronato, 2-napsilato, tosilato, hibenzato, nicotinato, isetionato, malato, maleato, citrato, gluconato, succinato, sacarato, benzoato, esilato y sales de pamoato. Para una revisión de las sales adecuadas, véase Berge, Stephen M., et al. “Pharmaceutical salts.” J, Pharm, Sci. Vol. 66, No. 1 (1977): 1-19, and Paulekuhn, G. Steffen, et al. “Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database.” J. Med. Chern. Vol. 50, No. 26 (2007): 6665-6672.
Los términos técnicos y científicos utilizados en el presente documento tienen el significado comúnmente entendido por un experto en la técnica a la que pertenece la presente invención, a menos que se defina de otro modo.
En el presente documento se hace referencia a diversas metodologías y materiales conocidos por los expertos en la técnica. Una obra de referencia estándar que expone los principios generales de la farmacología incluye Hardman, Joel Griffith, et al. Goodman & Gilman's The Pharmacological Basis of Therapeutics. Nueva York: McGraw-Hill Professional, 2001. Los materiales de partida y reactivos utilizados en la preparación de estos compuestos generalmente están disponibles de proveedores comerciales, tales como Sigma-Aldrich (St. Louis, MO), o se preparan por métodos conocidos por los expertos en la técnica siguiendo los procedimientos establecidos en las referencias. Los materiales, reactivos y similares a los que se hace referencia en la siguiente descripción y ejemplos pueden obtenerse de fuentes comerciales, a menos que se indique lo contrario. Los procedimientos sintéticos generales se han descrito en tratados, como Louis F. Fieser y Mary Fieser, Reagents for Organic Synthesis, v. 1-23, Nueva York: Wiley 1967-2006 ed. (también disponible en el sitio web de Wiley InterScience®); LaRock, Richard C., Comprehensive Organic Transformations: A Guide to Functional Group Preparations. Nueva York: Wiley-VCH, 1999; B. Trost e I. Fleming, eds. Comprehensive Organic Synthesis, v. 1-9, Oxford: Pergamon 1991; A. R. Katritzky y C. W. Rees, eds. Comprehensive Heterocyclic Chemistry. Oxford: Pergamon 1984; A. R. Katritzky y C. W. Rees, eds. Comprehensive Heterocyclic Chemistry II. Oxford: Pergamon 1996; y Paquette, Leo A., ed. Organic Reactions, v. 1-40, Nueva York: Wiley & Sons 1991; y resultará familiar a los expertos en la técnica.
El término “alquilo” incluye radicales de cadena lineal o ramificada de átomos de carbono. Se han abreviado algunas porciones alquilo, por ejemplo, metilo (“Me”), etilo (“Et”), propilo (“Pr”) y butilo (“Bu”) y se utilizan abreviaturas adicionales para designar isómeros específicos de los compuestos, por ejemplo, 1 -propilo o n-propilo (“n-Pr”), 2-propilo o isopropilo (“ i-Pr”), 1 -butilo o n-butilo (“n-Bu”), 2-metil-1 -propilo o isobutilo (“/-Bu”), 1 -metilpropilo o s-butilo (“s-Bu”), 1,1 -dimetiletilo o f-butilo (“f-Bu”) y similares. Las abreviaturas se utilizan algunas veces junto con abreviaturas elementales y estructuras químicas, por ejemplo, metanol (“MeOH”) o etanol (“EtOH”). En ciertas modalidades, alquilo es alquilo de C1-10. En ciertas modalidades, alquilo es alquilo de C1-6.
El término “Boc” significa ter-butiloxicarbonilo. Abreviaturas adicionales utilizadas en toda la solicitud incluyen, por ejemplo, bencilo (“Bn”), fenilo (“Ph”), acetato (“Ac”) y mesilato (“Ms”).
Los términos "heterociclo" y "heterocíclico" incluyen anillos saturados o parcialmente insaturados de cuatro a siete miembros que contienen uno, dos o tres heteroátomos seleccionados del grupo que consiste en O, N, S, S(=O) y S(=O)2. En ciertos casos, estos términos pueden estar específicamente más limitados, tales como, "heterocíclico de cinco a seis miembros" que solo incluye anillos de cinco y seis miembros.
El término "heteroarilo" incluye anillos aromáticos de cinco a seis miembros que contienen uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en O, N y S. En ciertos casos, estos términos pueden estar específicamente más limitados, tal como heteroarilo de cinco a seis miembros, en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno. Como bien saben los expertos en la técnica, los anillos heteroarilo tienen menos carácter aromático que sus homólogos totalmente carbonados. Así, a efectos de la invención, un grupo heteroarilo sólo necesita tener cierto grado de carácter aromático.
Un enlace dibujado en un sistema de anillo (en oposición a conectado en un vértice distinto) indica que el enlace puede estar unido a cualquiera de los átomos de anillo adecuados. Un enlace con una línea ondulada indica el punto de unión.
Los términos "tratar" o "tratamiento" se refieren a medidas terapéuticas, profilácticas, paliativas o preventivas. Los resultados clínicos beneficiosos o deseados incluyen, pero no se limitan a, alivio de los síntomas, disminución de la extensión de la enfermedad, estabilización (es decir, no empeoramiento) del estado de la enfermedad, retraso o ralentización de la progresión de la enfermedad, mejora o paliación del estado de la enfermedad, y remisión (parcial o total), detectable o indetectable. "Tratamiento" también puede significar prolongar la supervivencia en comparación con la supervivencia esperada si no se recibe tratamiento. Entre las personas que necesitan tratamiento se incluyen las que ya padecen la enfermedad o trastorno, así como las propensas a padecerla o aquellas en las que se desea prevenirla.
Las frases "cantidad terapéuticamente eficaz" o "cantidad eficaz" significan una cantidad de un compuesto descrito en el presente documento que, cuando se administra a un mamífero que necesita dicho tratamiento, es suficiente para (i) tratar o prevenir la enfermedad, afección o trastorno particular, (ii) atenuar, mejorar o eliminar uno o más síntomas de la enfermedad, afección o trastorno particular, o (iii) prevenir o retrasar la aparición de uno o más síntomas de la enfermedad, afección o trastorno particular descrito en el presente documento. La cantidad de un compuesto que corresponderá a dicha cantidad variará en función de factores tales como el compuesto concreto, la afección de la enfermedad y su gravedad, la identidad (por ejemplo, peso) del mamífero que necesita tratamiento, pero puede determinarse de forma rutinaria por un experto en la técnica.
Los términos "cáncer" y "canceroso" se refieren o describen la condición fisiológica en mamíferos que se caracteriza típicamente por un crecimiento celular anormal o no regulado. Un "tumor" comprende una o más células cancerosas. Ejemplos de cáncer incluyen, entre otros, carcinoma, linfoma, blastoma, sarcoma y leucemia o neoplasias linfoides. Ejemplos más particulares de tales cánceres incluyen el cáncer de células escamosas (. por ejemplo, cáncer epitelial de células escamosas), cáncer de pulmón, incluyendo cáncer de pulmón de células pequeñas, cáncer de pulmón de células no pequeñas ("NSCLC"), adenocarcinoma de pulmón y carcinoma escamoso de pulmón, cáncer de peritoneo, cáncer hepatocelular, cáncer gástrico o de estómago, incluyendo cáncer gastrointestinal, cáncer de páncreas, glioblastoma, cáncer de cuello uterino, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama, cáncer de colon, cáncer de recto, cáncer colorrectal, carcinoma de endometrio o de útero, carcinoma de glándulas salivales, cáncer de riñón o renal, cáncer de próstata, cáncer de vulva, cáncer de tiroides, carcinoma hepático, carcinoma anal, carcinoma de pene, cáncer de piel, incluyendo melanoma, así como cáncer de cabeza y cuello.
La frase "farmacéuticamente aceptable" indica que la sustancia o composición es compatible química y/o toxicológicamente con los demás ingredientes que componen una formulación y/o con el mamífero tratado con ella.
La frase "sal farmacéuticamente aceptable", tal como se utiliza en el presente documento, se refiere a sales orgánicas o inorgánicas farmacéuticamente aceptables de un compuesto descrito en el presente documento.
Los compuestos descritos en el presente documento también incluyen otras sales de dichos compuestos que no son necesariamente sales farmacéuticamente aceptables, y que pueden ser útiles como intermedios para preparar y/o
purificar compuestos descritos en el presente documento y/o para separar enantiómeros de 8 compuestos descritos en el presente documento.
El término "mamífero" hace referencia a un animal de sangre caliente que tiene o corre el riesgo de desarrollar una enfermedad descrita en el presente documento e incluye, pero no se limitan a, cobayas, perros, gatos, ratas, ratones, hámsteres y primates, incluyendo humanos.
INHIBIDORES DE SHP2
En el presente documento se proporcionan compuestos y formulaciones farmacéuticas de los mismos que son potencialmente útiles en el tratamiento de enfermedades, afecciones y/o trastornos modulados por SHP2.
La invención proporciona un compuesto seleccionado de la Fórmula Ila:

o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que:
L1 es S;
R1 se selecciona de fenilo, heteroarilo, arilo bicíclico, heterociclilo bicíclico y heteroarilo bicíclico, en el que fenilo, heteroarilo, arilo bicíclico, heterociclo bicíclico y heteroarilo bicíclico están opcionalmente sustituidos con uno o más grupos seleccionados del grupo que consiste en halógeno, OH, oxo, ciano, alquilo opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo) opcionalmente sustituido con halógeno, ciano u O h , NHRa, y un heterociclo opcionalmente sustituido con halógeno, ciano, OH o alquilo opcionalmente sustituido con OH u oxo;
R2 es:

X11 se selecciona de CR13R14, SiR13R14, NH y O;
X12 se selecciona de CHR15 y NH, en el que uno o ambos de X11 y X12 deben ser carbono;
R10 se selecciona de hidrógeno y alquilo;
R11 se selecciona de hidrógeno, OH y CH2NH2;
R12, R16 y R17 son hidrógeno;
R13 se selecciona de hidrógeno, OH y (alquilo C0-C3 )NRbRc;
R14 se selecciona de hidrógeno, OH, alquilo opcionalmente sustituido con halógeno, OH, metilo, OCH3 y un heteroarilo;
R15 se selecciona de hidrógeno o NH2;
o puede unirse uno de los siguientes grupos:
R10 y R11 pueden unirse como CH2NHCH2 para formar un bicíclico fusionado,
R10 y R15 pueden unirse como alquilo para formar un bicíclico con puente,
R11 y R12 pueden unirse como alquilo sustituido con NH2 para formar un espirociclo,
R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico, en el que el cicloalquilo, heterociclo, carbociclo y heterociclo están opcionalmente sustituidos con F, Cl, OH, OCH3, CN, metilo o NH2 para formar un espirociclo,
R10 y R16 pueden unirse como alquilo, O o NH para formar un bicíclico con puente,
R11 y R15 pueden unirse como alquilo para formar un bicíclico con puente,
R11 y R16 pueden unirse como alquilo u O para formar un bicíclico con puente,
R11 y R17 pueden unirse como alquilo para formar un bicíclico con puente, o
R13 y R15 pueden unirse como NHCH2, o cicloalquilo en el que el cicloalquilo está sustituido con NH2 para formar un bicíclico fusionado;
R48 se selecciona de hidrógeno y metilo;
Ra es hidrógeno, alquilo opcionalmente sustituido con OH, metoxi, halógeno o ciano, o ciclopropilo;
Rb y Rc se seleccionan independientemente de hidrógeno, alquilo y un grupo Boc; y a, b, c y d se seleccionan de 0 y 1.
En otra realización, se proporcionan compuestos de la invención o un estereoisómero o sal farmacéuticamente aceptable de los mismos.
En otra realización, se proporcionan compuestos de la invención o un tautómero o sal farmacéuticamente aceptable de los mismos.
En otra realización, se proporcionan compuestos de la invención o un estereoisómero o tautómero de los mismos. En otra realización, se proporcionan compuestos de la invención o un estereoisómero de los mismos.
En otra realización, se proporcionan compuestos de la invención o un tautómero de los mismos.
En otra realización, se proporcionan compuestos de la invención o una sal farmacéuticamente aceptable de los mismos.
En ciertas realizaciones, R1 se selecciona de fenilo, un heteroarilo de 5 a 6 miembros en el que el heteroarilo contiene de uno a cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, un arilo bicíclico de 10 miembros, un heterociclilo bicíclico de 9-10 miembros en el que el heterociclilo contiene uno a tres heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, y un heteroarilo bicíclico de 9-10 miembros en el que el heteroarilo bicíclico contiene uno a tres heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el fenilo, heteroarilo, arilo bicíclico, heterociclilo bicíclico y heteroarilo bicíclico están opcionalmente sustituidos con uno a tres grupos seleccionados del grupo que consiste en halógeno; oxo; ciano; alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH; -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano, OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno, azufre y SO2. En ciertas realizaciones, R1 se selecciona de fenilo, un heteroarilo de 5 a 6 miembros en el que el heteroarilo contiene uno a cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, un arilo bicíclico de 10 miembros, un heterociclilo bicíclico de 9-10 miembros en el que el heterociclilo contiene uno a tres heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, y un heteroarilo bicíclico de 9-10 miembros en el que el heteroarilo bicíclico contiene uno a tres heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el fenilo, el heteroarilo, el arilo bicíclico, el heterociclo bicíclico y el heteroarilo bicíclico están opcionalmente sustituidos con uno a tres grupos seleccionados del grupo que consiste en halógeno; ciano; alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH; -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano, OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno, azufre y SO2. En ciertas realizaciones, R1 se selecciona del grupo que consiste en: (a) fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con
halógeno, y ciano; (b) un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno a tres seleccionados del halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno, azufre y SO2; en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre; (c) un arilo bicíclico de 10 miembros opcionalmente sustituido con halógeno o metilo; (d) un heterociclilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, oxo, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, o ciano, en el que el heterociclilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno; y (e) un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, o ciano, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en: (a) fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno y ciano; (b) un heteroarilo de 6 miembros, opcionalmente sustituido con uno a tres grupos seleccionados del halógeno; alquilo C1-C3opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2; en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno; (c) un arilo bicíclico de 10 miembros; (d) un heterociclo bicíclico de 9-10 miembros opcionalmente sustituido con halógeno u oxo, en el que el heterociclo bicíclico contiene uno o dos heteroátomos de nitrógeno; y (e) un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con halógeno, ciano o alquilo C1-C3 opcionalmente sustituido con halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en: (a) fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, y ciano; (b) heteroarilo de 6 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2; en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno; (c) un arilo bicíclico de 10 miembros; (d) un heterociclilo bicíclico de 9-10 miembros, en el que el heterociclilo bicíclico contiene uno o dos heteroátomos de nitrógeno; y (e) un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con halógeno, ciano o alquilo C1-C3 opcionalmente sustituido con halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en fenilo, 2,3-diclorofenilo, 3-clorofenilo, 4-fluorofenilo, 3-cloro-2-trifluorometilfenilo, 2-cloro-3-metoxifenilo, 3-cloro-2-fluorofenilo, 2-cloro-6-fluoro-3-metoxifenilo, 2,3-dicloro-4-metoxifenilo, 2-cloro-3-cianofenilo, 2-cloro-3-fluorofenilo, 2-amino-3-cloropiridin-4-ilo, 3-cloro-2-(pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-metoxipiridin-4-ilo, 3-cloro-2-(metilamino)piridin-4-ilo), 3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((2-hidroxietil)amino)piridin-4-ilo, 3-cloro-2-metilpiridin-4-ilo, 6-amino-2,3-dicloropiridin-4-ilo, 3-cloro-2-(2-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 2-amino-3-metilpiridin-4-ilo, 3-cloro-2-(1,1-dioxidotiomorfolino)piridin-4-ilo, 3-cloro-2-(4-hidroxipiperidin-1-il)piridin-4-ilo, 3-cloro-2-morfolinopiridin-4-ilo, 2-(4-acetilpiperazin-1-il)-3-cloropiridin-4-ilo, 3-cloro-2-((S)-3-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((S)-3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-(hidroximetil)piridin-4-ilo, 3-cloro-2-metilpiridin-4-ilo, 2-aminopiridin-3-ilo, 6-cloro-2-metilpiridin-3-ilo, 6-amino-2-cloropiridin-3-ilo, 2-cloro-6-metilpiridin-3-ilo, 3-cloro-2-((R)-3-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 2,3-dimetilpiridin-4-ilo, 2-amino-3-fluoropiridin-4-ilo, 6-amino-2-(trifluorometil)piridin-3-ilo, 2-amino-5-cloropiridin-4-ilo, 6-amino-4,5-dicloropiridin-3-ilo, 6-amino-3-cloro-2-metoxipiridin-4-ilo, 2-amino-3-metoxipiridin-4-ilo, 6-amino-5-cloropirimidin-4-ilo, 2- (trifluorometil)piridin-3-ilo, 3-cloro-2-((2-metoxietil)amino)piridin-4-ilo, 3-cloro-2-(ciclopropilamino)piridin-4-ilo, 3-fluoropiridin-4-ilo, 3-cloropiridin-4-ilo, 3-(trifluorometil)piridin-4-ilo, 3-cloro-2-((2-ciano-2-metilpropil)amino)piridin-4-ilo, 3- cloro-2-((3-metoxipropil)amino)piridin-4-ilo, 2-amino-3-(trifluorometil)piridin-4-ilo, 2-(trifluorometil)piridin-4-ilo, 3,4-dihidroquinolin-1(2H)-ilo, 3,4-dihidroquinoxalin-1(2H)-ilo, 2,3-dihidro-1 H-pirrol[2,3-b]piridin-4-ilo, 3,4-dihidro-1,5-naftiridin-1(2H)-ilo, 3,3-difluoro-2-oxo-2,3-dihidro-1H-pirrol[2,3-b]piridin-4-ilo, 7-hidroxi-6,7-dihidro-5H-ciclopenta[b]piridin-4-ilo, naftalen-1-ilo, 1-metil-1H-indazol-7-ilo, pirazol[1,5-a]piridin-4-ilo, pirazol[1,5-a]pirazin-4-ilo, isoquinolin-8-ilo, 3H-imidazo[4,5-b]piridin-7-ilo, 6-cloroimidazo[1,2-a]piridin-3-ilo, 6-cianoimidazo[1,2-a]piridin-3-ilo, 1H-pirazol[3,4-b]piridin-4-ilo, 1,8-naftiridin-4-ilo, 1 H-pirrol[2,3-b]piridin-4-ilo, 1 -metil-1 H-pirrol[2,3-b]piridin-4-ilo, 5-cloro-1 H-pirrol[2,3-b]piridin-4-ilo, 5-(trifluorometil)-1 H-pirrol[2,3-b]piridin-4-ilo, 3-metil-1 H-pirrol[2,3-b]piridin-4-ilo, 6-amino-1 H-pirrol[2,3-b]piridin-4-ilo y 5-fluoro-1 H-pirrol[2,3-b]piridin-4-ilo.
En ciertas realizaciones, R1 se selecciona de fenilo, un heteroarilo de 5 a 6 miembros en el que el heteroarilo contiene uno a cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, un arilo bicíclico de 10 miembros, y un heteroarilo bicíclico de 9-10 miembros en el que el heteroarilo bicíclico contiene uno a tres heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el fenilo, heteroarilo, arilo bicíclico y heteroarilo bicíclico están opcionalmente sustituidos con uno a tres grupos seleccionados del grupo que consiste en halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, - O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 se selecciona del grupo que consiste en: (a) fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, y ciano; (b) un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre; (c) un arilo bicíclico de 10 miembros opcionalmente sustituido con halógeno o metilo; y (d) un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres 25 grupos seleccionados de halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, o ciano, en el que el heteroarilo bicíclico contiene de uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en: (a) fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, y ciano; (b) un heteroarilo de 6 miembros, opcionalmente sustituido con uno o dos grupos seleccionados entre halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene un heteroátomo de nitrógeno, en el que el heteroarilo contiene un heteroátomo de nitrógeno; (c) un arilo bicíclico de 10 miembros; y (d) un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con alquilo C1-C3, en el que el heteroarilo bicíclico contiene de uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 2,3-diclorofenilo, 3-clorofenilo, 3-cloro-2- trifluorometilfenilo, 2-cloro-3-metoxifenilo, 3-cloro-2-fluorofenilo, 2-cloro-6-fluoro-3-metoxifenilo, 2,3-dicloro-4-metoxifenilo, 2-cloro-3-cianofenilo, 2-cloro-3-fluorofenilo, 2-amino-3-cloropiridin-4-ilo, 3-cloro-2-(pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-metoxipiridin-4-ilo, 3-cloro-2-(metilamino)piridin-4-ilo), 3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((2-hidroxietil)amino)piridin-4-ilo, naftalen-1-ilo, i-metil-1H-indazol-7-¡lo, pirazol[1,5-a]piridin-4-ilo, pirazol[1,5-a]pirazin-4-ilo e isoquinolin-8-ilo.
En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno a tres grupos seleccionados del grupo que consiste en halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u O h , NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, y ciano. En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, trifluorometilo, metoxi y ciano. En ciertas realizaciones, R1 se selecciona del grupo que consiste en fenilo, 2,3-diclorofenilo, 3-clorofenilo, 4-fluorofenilo, 3- cloro-2-trifluorometilfenilo, 2-cloro-3-metoxifenilo, 3-cloro-2-fluorofenilo, 2-cloro-6-fluoro-3-metoxifenilo, 2,3-dicloro-4- metoxifenilo, 2-cloro-3-cianofenilo y 2-cloro-3-fluorofenilo.
En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno o más grupos seleccionados del grupo que consiste en halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno a tres grupos seleccionados del grupo que consiste en halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u O h , NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, y ciano. En ciertas realizaciones, R1 es fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados del grupo que consiste en halógeno, trifluorometilo, metoxi y ciano. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 2,3-diclorofenilo, 3-clorofenilo, 3-cloro-2-trifluorometilfenilo, 2-cloro-3-metoxifenilo, 3-cloro-2-fluorofenilo, 2-cloro-6-fluoro-3-metoxifenilo, 2,3-dicloro-4-metoxifenilo, 2-cloro-3-cianofenilo y 2-cloro-3-fluorofenilo.
En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno; ciano; alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH; -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano, OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno, azufre y SO2; en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; ciano; alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH; - O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH; NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano, OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno, azufre y SO2; en el que el heteroarilo contiene uno, dos, tres o
cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno a tres seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno, azufre y SO2; en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno a tres seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con Oh u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2; en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, donde el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno, azufre y SO2; donde el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno, azufre y SO2; en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2; en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 5 o 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2; en el que el heteroarilo contiene uno o dos heteroátomos de nitrógeno. En ciertas realizaciones, R1 es piridinilo o pirimidinilo opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno, azufre y SO2. En ciertas realizaciones, R1 es piridinilo o pirimidinilo opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2. En ciertas realizaciones, R1 es piridinilo o pirimidinilo opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 5 o 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2. En ciertas realizaciones, R1 es piridinilo o pirimidinilo opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y heterociclo de 5 o 6 miembros opcionalmente sustituido con OH, CH2OH o C(=O)CH3, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2. En ciertas realizaciones, R1 es piridinilo o pirimidinilo opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa; y un heterociclo de 5 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 2-amino-3-cloropiridin-4-ilo, 3-cloro-2-(pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-metoxipiridin-4-ilo, 3-cloro-2-(metilamino)piridin-4-ilo), 3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((2-hidroxietil)amino)piridin-4-ilo, 3-cloro-2-metilpiridin-4-ilo, 6-amino-2,3-dicloropiridin-4-ilo, 3-cloro-2-(2-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 2-amino-3-metilpiridin-4-ilo, 3-cloro-2-(1,1-dioxidotiomorfolino)piridin-4-ilo, 3-cloro-2-(4-hidroxipiperidin-1-il)piridin-4-ilo, 3-cloro-2-morfolinopiridin-4-ilo, 2-(4-acetilpiperazin-1-il)-3-cloropiridin-4-ilo, 3-cloro-2-((S)-3-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((S)-3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-(3-hidroxipirrolidin-1 -il)piridin-4-ilo, 3-cloro-2-(hidroximetil)piridin-4-ilo, 3-cloro-2-metilpiridin-4-ilo, 2-aminopiridin-3-ilo, 6-cloro-2-metilpiridin-3-ilo, 6-amino-2-cloropiridin-3-ilo, 2-cloro-6-metilpiridin-3-ilo, 3-cloro-2-(R)-3-(hidroximetil)pirrolidin-1- il)piridin-4-ilo, 2,3-dimetilpiridin-4-ilo, 2-amino-3-fluoropiridin-4-ilo, 6-amino-2-(trifluorometil)piridin-3-ilo, 2-amino-5-cloropiridin-4-ilo, 6-amino-4,5-dicloropiridin-3-ilo, 6-amino-3-cloro-2-metoxipiridin-4-ilo, 2-amino-3-metoxipiridin-4-ilo, 6-amino-5-cloropirimidin-4-ilo, 2-(trifluorometil)piridin-3-ilo, 3-cloro-2-((2-metoxietil)amino)piridin-4-ilo, 3-cloro-2-(ciclopropilamino)piridin-4-ilo, 3-fluoropiridin-4-ilo, 3-cloropiridin-4-ilo, 3-(trifluorometil)piridin-4-ilo, 3-cloro-2-((2-ciano-2- metilpropil)amino)piridin-4-ilo, 3-cloro-2-((3-metoxipropil)amino)piridin-4-ilo, 2-amino-3-(trifluorometil)piridin-4-ilo y 2-(trifluorometil)piridin-4-ilo.
En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente
sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un heteroarilo de 5 a 6 miembros opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene un heteroátomo de nitrógeno, en el que el heteroarilo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene uno, dos, tres o cuatro heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene un heteroátomo de nitrógeno, en el que el heteroarilo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R1 es heteroarilo de 6 miembros, opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 5 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene un heteroátomo de nitrógeno, en el que el heteroarilo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R1 es piridinilo opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es piridinilo opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R1 es piridinilo opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa y heterociclo de 5 miembros opcionalmente sustituido con OH, en el que el heterociclo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R1 es piridinilo opcionalmente sustituido con uno o dos grupos seleccionados de halógeno, metoxi, NHRa y pirrolidinilo opcionalmente sustituido con OH. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 2-amino-3-cloropiridin-4-ilo, 3-cloro-2-(pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-metoxipiridin-4-ilo, 3-cloro-2-(metilamino)piridin-4-ilo), 3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-ilo y 3-cloro-2-((2-hidroxietil)amino)piridin-4-ilo.
En ciertas realizaciones, R1 es un arilo bicíclico de 10 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un arilo bicíclico de 10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R1 es un arilo bicíclico de 10 miembros opcionalmente sustituido con halógeno o metilo. En ciertas realizaciones, R1 es un arilo bicíclico de 10 miembros. En ciertas realizaciones, R1 es naftalen-1-ilo.
En ciertas realizaciones, R1 es un heterociclo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno, OH, oxo, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heterociclo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heterociclo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, OH, oxo, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u O h , NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heterociclo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heterociclo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno, OH, oxo, ciano, alquilo C1-C3
opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heterociclo bicíclico contiene uno o dos heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heterociclo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, OH y oxo, en el que el heterociclo bicíclico contiene uno o dos heteroátomos de nitrógeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 3,4-dihidroquinolin-1(2H)-ilo, 3,4-dihidroquinoxalin-1(2H)-ilo, 2,3-dihidro-1H-pirrolo[2.3-6]piridin-4-ilo y 3,4-dihidro-1,5-naftiridin-1(2H)-ilo, 3,3-difluoro-2-oxo-2,3-dihidro-1H-pirrolo[2.3-6]piridin-4-ilo y 7-hidroxi-6,7-dihidro-5H-ciclopenta[b]piridin-4-ilo.
En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heteroarilo bicíclico contiene de uno a tres nitrógenos, azufres u oxígenos heteroátomos. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con: halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno; -O(alquilo C1-C3) opcionalmente sustituido con halógeno; ciano; o NHRa; en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de: halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno; -O(alquilo C1-C3) opcionalmente sustituido con halógeno; ciano; o NHRa; en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con: halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno; -O(alquilo C1-C3) opcionalmente sustituido con halógeno; ciano; o NHRa; en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, o NHRa, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, o NHRa, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con metilo, trifluorometilo, ciano, halógeno o amino, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos metilo, metoxi, trifluorometilo, ciano, halógeno o amino, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos metilo, trifluorometilo, ciano, halógeno o amino, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con metilo, trifluorometilo, ciano, halógeno o amino, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 1-metil-1H-indazol-7-ilo, pirazol[1,5-a]piridin-4-ilo, pirazol[1,5-a]pirazin-4-ilo, isoquinolin-8-ilo, 3H-imidazo[4,5-b]piridin-7-ilo, 6-cloroimidazo[1,2-a]piridin-3-ilo, 6-cianoimidazo[1,2-a]piridin-3-ilo, 1 H-pirazol[3,4-b]piridin-4-ilo, 1,8-naftiridin-4-ilo, 1H-pirrol[2,3-b]piridin-4-ilo, 1 -metil-1 H-pirrol[2,3-b]piridin-4-ilo, 5-cloro-1 H-pirrol[2,3-b]piridin-4-ilo, 5-(trifluorometil)-1 H-pirrol[2,3-b]piridin-4-ilo, 3-metil-1 H-pirrol[2,3-b]piridin-4-ilo, 6-amino-1 H-pirrol[2,3-b]piridin-4-ilo y 5-fluoro-1 H-pirrol[2,3-b]piridin-4-ilo.
En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno o más grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heteroarilo contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9 a 10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, o ciano, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos seleccionados de halógeno, alquilo C1-C3 opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, o ciano, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente
sustituido con halógeno, alquilo Ci -C3opcionalmente sustituido con halógeno, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, o ciano, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con alquilo C1-C3 o halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno, azufre u oxígeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con alquilo C1-C3 o halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con metilo o halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos metilo, metoxi, trifluorometilo o halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9-10 miembros opcionalmente sustituido con uno a tres grupos metilo o halógeno, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 es un heteroarilo bicíclico de 9 10 miembros opcionalmente sustituido con metilo, en el que el heteroarilo bicíclico contiene uno a tres heteroátomos de nitrógeno. En ciertas realizaciones, R1 se selecciona del grupo que consiste en 1-metil-1H-indazol-7-ilo, pirazol[1,5-a]piridin-4-ilo, pirazol[1,5-a]pirazin-4-ilo e isoquinolin-8-ilo.
En ciertas realizaciones, Ra es hidrógeno; alquilo C1-C4 opcionalmente sustituido con OH, metoxi, halógeno o ciano; o ciclopropilo. En ciertas realizaciones, Ra es hidrógeno, alquilo C1-C4 opcionalmente sustituido con OH, metoxi o ciano, o ciclopropilo. En ciertas realizaciones, Ra es hidrógeno, metilo, 2-hidroxietilo, 2-metoxietilo, ciclopropilamino o 2-ciano-2-metilpropilo.
En ciertas realizaciones, Ra es hidrógeno o alquilo C1-C3 opcionalmente sustituido con OH, metoxi, halógeno o ciano. En ciertas realizaciones, Ra es hidrógeno o alquilo C1-C3 opcionalmente sustituido con OH. En ciertas realizaciones, Ra es hidrógeno o alquilo C1-C3 opcionalmente sustituido con OH. En ciertas realizaciones, Ra es hidrógeno, metilo o 2-hidroxietilo.
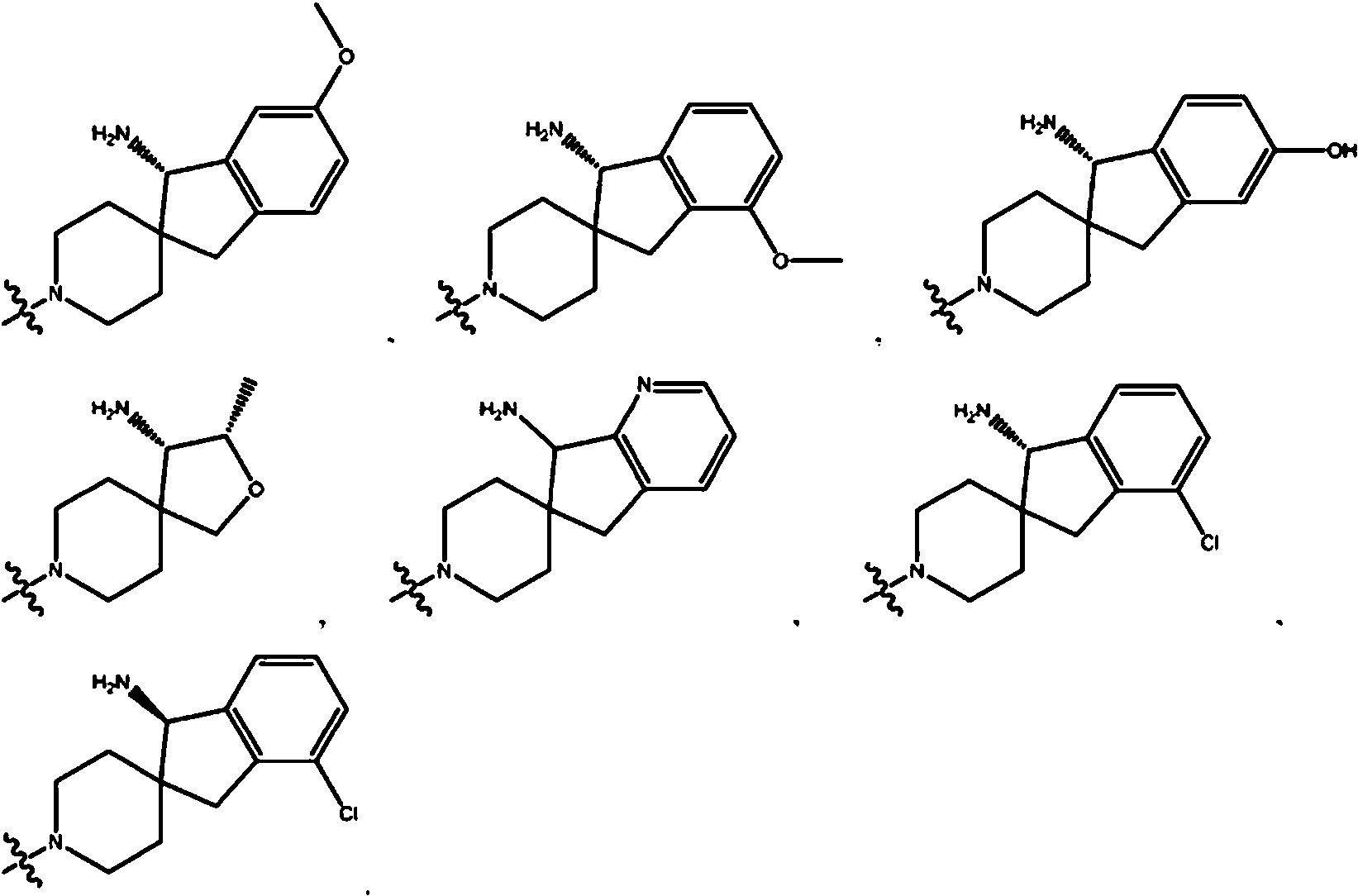
En ciertas realizaciones, R2 se selecciona del grupo que consiste en:



En ciertas realizaciones, X11 se selecciona de CR13R14, SiR13R14, NH y O. En ciertas realizaciones, X11 se selecciona de CR13R14, NH y O. En ciertas realizaciones, X11 se selecciona de CR13R14 y SiR13R14 En ciertas realizaciones, X11 se selecciona de NH y O. En ciertas realizaciones, X11 es CR13R14 En ciertas realizaciones, X11 es SiR13R14 En ciertas realizaciones, X11 es NH. En ciertas realizaciones, X11 es O.
En ciertas realizaciones, X12 se selecciona de CHR15 y NH. En ciertas realizaciones, X12 es CHR15 En ciertas realizaciones, X12 es NH.
En ciertas realizaciones, X11 se selecciona de CR13R14, SiR13R14, NH y O; y X12 se selecciona de CHR15 y NH, en el que uno o ambos de X11 y X12 debe ser carbono. En ciertas realizaciones, X11 es CR13R14; y X12 se selecciona de CHR15 y NH o X11 se selecciona de CR13R14, SiR13R14, NH y O; y X12 es CHR15. En ciertas realizaciones, X11 es CR13R14; y X12 se selecciona de CHR15 y NH. En ciertas realizaciones, X11 se selecciona de CR13R14, SiR13R14, NH y O; y X12 es CHR15. En ciertas realizaciones, X11 es CR13R14; y X12 es CHR15. En ciertas realizaciones, X11 es CR13R14; y X12 es NH. En ciertas realizaciones, X11 es SiR13R14; y X12 es CHR15. En ciertas realizaciones, X11 es NH; y X12 es CHR15. En ciertas realizaciones, X11 es O; y X12 es CHR15.
En ciertas realizaciones, R10 se selecciona de hidrógeno y alquilo. En ciertas realizaciones, R10 se selecciona de hidrógeno y alquilo de C1-C3. En ciertas realizaciones, R10 se selecciona de hidrógeno y metilo. En ciertas realizaciones, R10 es hidrógeno. En ciertas realizaciones, R10 es metilo.
En ciertas realizaciones, R11 se selecciona de hidrógeno, OH y CH2NH2. En ciertas realizaciones, R11 se selecciona de hidrógeno, OH y CH2NH2. En ciertas realizaciones, R11 es hidrógeno. En ciertas realizaciones, R11 es OH. En ciertas realizaciones, R11 es CH2NH2.
En ciertas realizaciones, R12, R16 y R17 son hidrógeno.
En ciertas realizaciones, R13 se selecciona de hidrógeno, OH y (alquilo de C0-C3)NRbRc. En ciertas realizaciones, R13 se selecciona de hidrógeno, OH, CH2NH2, NH2, NH(CH3), N(CH3)2, C(NH2)(CH3)2 o NHBoc. En ciertas realizaciones, R13 es hidrógeno. En ciertas realizaciones, R13 es OH. En ciertas realizaciones, R13 es (alquilo de C0-C3)NRbRc. En ciertas realizaciones, R13 es CH2NH2. En ciertas realizaciones, R13 es NH2. En ciertas realizaciones, R13 es NH(CH3). En ciertas realizaciones, R13 es N(CH3)2. En ciertas realizaciones, R13 es C(NH2)(CH3)2. En ciertas realizaciones, R13 es NHBoc.
En ciertas realizaciones, Rb y Rc se seleccionan independientemente de hidrógeno, alquilo y un grupo Boc. En ciertas realizaciones, Rb y Rc se seleccionan independientemente de hidrógeno, alquilo de C1-C3 y un grupo Boc. En ciertas realizaciones, Rb y Rc se seleccionan independientemente de hidrógeno, metilo y un grupo Boc. En ciertas realizaciones, Rb se selecciona de hidrógeno, metilo y un grupo Boc y Rc es hidrógeno o metilo. En ciertas
realizaciones, Rb se selecciona de hidrógeno, metilo y un grupo Boc y Rc es hidrógeno. En ciertas realizaciones, Rb y Rc se seleccionan independientemente de hidrógeno y metilo. En ciertas realizaciones, Rb es metilo y Rc es hidrógeno. En ciertas realizaciones, Rb y Rc son hidrógeno. En ciertas realizaciones, Rb es un grupo Boc, Rc es hidrógeno.
En ciertas realizaciones, R14 se selecciona de hidrógeno, OH, alquilo opcionalmente sustituido con halógeno, OH, metilo, OCH3 y un heteroarilo. En ciertas realizaciones, R14 se selecciona de hidrógeno, OH, alquilo de C1-C3 opcionalmente sustituido con halógeno, OH, metilo, OCH3 y un heteroarilo de 5 a 6 miembros en el que el heteroarilo contiene de uno a tres heteroátomos seleccionados de nitrógeno, oxígeno y azufre. En ciertas realizaciones, R14 se selecciona de hidrógeno, OH, alquilo de C1-C3 opcionalmente sustituido con halógeno, OH, metilo, OCH3 y un heteroarilo de 6 miembros en el que el heteroarilo contiene un heteroátomo de nitrógeno. En ciertas realizaciones, R14 se selecciona de hidrógeno, OH, metilo, etilo, propilo, CF3, CH2OH, CH2CH2OH, CH2C(CH3)2OH, CH2OCH3, CH2CH2OCH3 y -(CH2)piridin-2-ilo.
En ciertas realizaciones, R15 se selecciona de hidrógeno o NH2. En ciertas realizaciones, R15 es hidrógeno. En ciertas realizaciones, R15 es NH2.
En ciertas realizaciones, dos grupos R en R2 pueden unirse para formar un biciclo puenteado, espirocíclico o fusionado. Sólo un conjunto de dos grupos R dentro R2 pueden unirse para formar el biciclo puenteado, espirocíclico o fusionado, seleccionado de: R10 y R11 pueden unirse para formar un bicíclico fusionado, R10 y R15 pueden unirse para formar un bicíclico puenteado, R11 y R12 pueden unirse para formar un espirocíclico, R13 y R14 pueden unirse para formar un espirocíclico, R10 y R16 pueden unirse para formar un bicíclico puenteado, R11 y R15 pueden unirse para formar un bicíclico puenteado, R11 y R16 pueden unirse para formar un bicíclico puenteado, R11 y R17 pueden unirse para formar un bicíclico puenteado o R13 y R15 pueden unirse para formar un bicíclico fusionado.
En ciertas realizaciones, R10 y R11 pueden unirse como CH2NHCH2 para formar un bicíclico fusionado.
En ciertas realizaciones, R10 y R15 pueden unirse como alquilo para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R15 pueden unirse como alquilo de C1-C4 para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R15 pueden unirse como etilo o propilo para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R15 pueden unirse como etilo para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R15 pueden unirse como propilo para formar un bicíclico puenteado.
En ciertas realizaciones, R11 y R12 pueden unirse como alquilo sustituido con NH2 para formar un espirociclo. En ciertas realizaciones, R11 y R12 pueden unirse como alquilo de C1-C4 sustituido con NH2 para formar un espirociclo. En ciertas realizaciones, R11 y R12 pueden unirse como ciclobutano sustituido con NH2 para formar un espirociclo.
En ciertas realizaciones, R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico, en donde el cicloalquilo, heterociclo, carbociclo y heterociclo se sustituyen opcionalmente con F, Cl, OH, OCH3, CN, metilo o NH2, para formar un espirociclo. En ciertas realizaciones, R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo de C3-C6, heterociclo de 4 a 6 miembros en donde el heterociclo contiene de uno a tres heteroátomos seleccionados del grupo que consiste de nitrógeno, oxígeno y azufre, carbociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado y un heterociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado en donde el heterociclo contiene de uno a tres heteroátomos seleccionados del grupo que consiste de nitrógeno, oxígeno y azufre, en donde el cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico se sustituyen opcionalmente con F, Cl, OH, OCH3, CN, metilo o NH2, para formar un espirociclo. En ciertas realizaciones, R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo de C3-C6, heterociclo de 4 a 6 miembros en donde el heterociclo contiene de uno a tres heteroátomos seleccionados del grupo que consiste de nitrógeno y oxígeno, carbociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado y un heterociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado en donde el heterociclo contiene de uno a tres heteroátomos seleccionados del grupo que consiste de nitrógeno y oxígeno, en donde el cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico se sustituyen opcionalmente con F, Cl, OH, OCH3, CN, metilo o NH2, para formar un espirociclo. En ciertas realizaciones, R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo de C3-C6, heterociclo de 4 a 6 miembros en donde el heterociclo contiene un heteroátomo seleccionado del grupo que consiste de nitrógeno y oxígeno, carbociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado y un heterociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado en donde el heterociclo contiene un heteroátomo seleccionado del grupo que consiste de nitrógeno y oxígeno, en donde el cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico se sustituyen opcionalmente con F, Cl, OH, OCH3, CN, metilo o NH2, para formar un espirociclo. En ciertas realizaciones, R13 y R14 pueden unirse como un grupo seleccionado de ciclopentilo, tetrahidrofurano, azetidina, 2,3-dihidro-1H-indeno, 6,7-dihidro-5H-ciclopenta[6]piridina, 2,3-dihidrobenzofurano o biciclo[4.2.0]octa-1(6),2,4-trieno, opcionalmente sustituido con F, Cl, OH, OCH3, CN, metilo o NH2, para formar un espirociclo.
En ciertas realizaciones, R10 y R16 pueden unirse como alquilo, O o NH para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R16 pueden unirse como alquilo C1-C4, O o NH para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R16 pueden unirse como alquilo C1-C3, O o NH para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R16 pueden unirse como alquilo C1-C3 para formar un bicíclico puenteado. En ciertas realizaciones,
R10 y R16 pueden unirse como O para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R16 pueden unirse como NH para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R16 pueden unirse como metilo, etilo, propilo, O o NH, para formar un bicíclico puenteado. En ciertas realizaciones, R10 y R16 pueden unirse como metilo, etilo o propilo para formar un bicíclico puenteado.
En ciertas realizaciones, R11 y R15 pueden unirse como alquilo para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R15 pueden unirse como alquilo C1-C4 para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R15 pueden unirse como alquilo C1-C3 para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R15 pueden unirse como metilo o etilo para formar un bicíclico puenteado.
En ciertas realizaciones, R11 y R16 pueden unirse como alquilo o para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R16 pueden unirse como alquilo C1-C4 o O para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R16 pueden unirse como alquilo C1-C3 o O para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R16 pueden unirse como alquilo C1-C3 para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R16 pueden unirse como metilo, etilo o O, para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R16 pueden unirse como metilo o etilo para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R16 pueden unirse como O para formar un bicíclico puenteado.
En ciertas realizaciones, R11 y R17 pueden unirse como alquilo para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R17 pueden unirse como alquilo C1-C4 para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R17 pueden unirse como alquilo C1-C3 para formar un bicíclico puenteado. En ciertas realizaciones, R11 y R17 pueden unirse como etilo para formar un bicíclico puenteado.
En ciertas realizaciones, R13 y R15 pueden unirse como NHCH2 o cicloalquilo sustituido con NH2 para formar un bicíclico fusionado. En ciertas realizaciones, R13 y R15 pueden unirse como NHCH2 o cicloalquilo de C3-C6 sustituido con NH2 para formar un bicíclico fusionado. En ciertas realizaciones, R13 y R15 pueden unirse como NHCH2 o ciclopentilo o ciclohexilo en donde el ciclopentilo o ciclohexilo se sustituyen con NH2 para formar un bicíclico fusionado. En ciertas realizaciones, R13 y R15 pueden unirse como NHCH2 para formar un bicíclico fusionado. En ciertas realizaciones, R13 y R15 pueden unirse como ciclopentilo o ciclohexilo en donde el ciclopentilo o ciclohexilo se sustituyen con NH2 para formar un bicíclico fusionado.
En ciertas realizaciones, a, b, c y d se seleccionan de 0 y 1.
En ciertas realizaciones, a, b, c y d son 1, en la que R2 es:

En ciertas realizaciones, a es 0, y b, c, y d son 1, en la que R2 es:

En ciertas realizaciones, a y b son 0 y c y d son 1, en la que R2 es:

En ciertas realizaciones, a, b y c son 0 y d es 1, en la que R2 es:

En ciertas realizaciones, a, b y d son 0 y c es 1, en la que R2:

En ciertas realizaciones, a, b, c y d son 0, en la que R2 es:

En ciertas realizaciones, R48 se selecciona de hidrógeno y metilo. En ciertas realizaciones, R48 es hidrógeno. En ciertas realizaciones, R48 es metilo.
Se apreciará que ciertos compuestos descritos en el presente documento pueden contener centros asimétricos o quirales, y por lo tanto existir en diferentes formas estereoisoméricas. Se pretende que todas las formas estereoisoméricas de los compuestos descritos en el presente documento, incluyendo, pero no limitándose a, diastereómeros, enantiómeros y atropisómeros, así como mezclas de los mismos tales como mezclas racémicas, formen parte de los presentes compuestos.
En las estructuras mostradas en el presente documento, donde la estereoquímica de cualquier átomo quiral particular no se especifica, entonces todos los estereoisómeros se contemplan e incluyen como los compuestos descritos en el presente documento. Cuando la estereoquímica se especifica mediante una cuña sólida o una línea discontinua que representa una configuración particular, entonces ese estereoisómero está así especificado y definido.
También se apreciará que ciertos compuestos descritos en el presente documento pueden utilizarse como intermedios para otros compuestos descritos en el presente documento.
Se apreciará además que los compuestos descritos en el presente documento pueden existir en formas no solvatadas, así como solvatadas con solventes farmacéuticamente aceptables, tales como agua, etanol y similares, y se pretende que los compuestos abarquen tanto formas solvatadas como no solvatadas.
SÍNTESIS DE LOS COMPUESTOS
Los compuestos descritos en el presente documento pueden sintetizarse por rutas sintéticas que incluyen procesos análogos a los bien conocidos en las técnicas químicas, particularmente a la luz de la descripción contenida en el presente documento. Los materiales de partida suelen estar disponibles en fuentes comerciales tales como Sigma-Aldrich (St. Louis, MO), Alfa Aesar (Ward Hill, MA) o TCI (Portland, OR), o se preparan fácilmente mediante métodos bien conocidos por los expertos en la técnica (por ejemplo, se preparan mediante métodos generalmente descritos en Louis F. Fieser y Mary Fieser, Reagents for Organic Synthesis, v. 1-23, Nueva York: Wiley 1967-2006 ed. (también disponible a través del sitio web de Wiley InterScience®), o Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlín, incluyendo los suplementos (también disponible a través de la base de datos en línea de Beilstein)).
Con fines ilustrativos, los Esquemas 1-3 muestran métodos generales para preparar los compuestos descritos en el presente documento, así como intermedios clave. Para una descripción más detallada de cada uno de los pasos de la reacción, véase la sección Ejemplos. Los expertos en la técnica apreciarán que pueden utilizarse otras rutas sintéticas para sintetizar los compuestos. Aunque en los Esquemas se representan y discuten a continuación materiales de partida y reactivos específicos, pueden sustituirse fácilmente otros materiales de partida y reactivos para proporcionar una variedad de derivados y/o condiciones de reacción. Además, muchos de los compuestos preparados por los métodos descritos a continuación pueden modificarse aún más a la luz de esta divulgación utilizando química convencional bien conocida por los expertos en la técnica.

El Esquema 1 muestra un esquema general para la síntesis del compuesto 1.4. Se puede hacer reaccionar 6-cloropirido[2,3-£))]pirazin-2(1H)-ona 1.1 con POCl o anhídrido tríflico ("Tf2O") para dar piridopirazina 1.2, donde R1 es Cl o triflato ("OTf"). La piridopirazina 1.2 puede someterse a una reacción SNAR para proporcionar piridopirazina 1.3, donde R2 se define en el presente documento. Otra reacción SNAR de la piridopirazina 1.3 da piridopirazina 1.4, donde La es S, CH, O o NH, y R1 es como se define en el presente documento. Alternativamente, puede utilizarse un acoplamiento Negisi o cobre para proporcionar también piridopirazina 1.4.

El Esquema 2 muestra un esquema general para preparar piridopirazina 2.1, en el que R1 y R2son como se definen en el presente documento. La piridopirazina 1.3 puede someterse a una reacción de Suzuki para obtener piridopirazina 2.1.

El Esquema 3 muestra un esquema general para preparar el compuesto 3.4, en el que Rj es I, Br o Cl, Rk1 se selecciona de halógeno y metilo; Rk2 se selecciona de ciano, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo está enlazado con nitrógeno y contiene uno o dos heteroátomos de nitrógeno; y Rm es hidrógeno, Na o K. El compuesto 3.1, donde X es C o N, puede someterse a una reacción SNAR para proporcionar el compuesto 3.2. El compuesto 3.2 puede someterse a un acoplamiento cruzado de Buchwald para obtener el compuesto 3.3. El compuesto 3.3 se hace reaccionar con etóxido sódico para obtener el compuesto 3.4.
MÉTODOS DE SEPARACIÓN
Puede ser ventajoso separar los productos de reacción entre sí y/o de los materiales de partida. Los productos deseados de cada paso o serie de pasos se separan y/o purifican (en lo sucesivo, se separan) hasta el grado deseado de homogeneidad mediante las técnicas habituales en la técnica. Normalmente, estas separaciones implican la extracción multifásica, la cristalización a partir de un disolvente o una mezcla de disolventes, la destilación, la sublimación o la cromatografía. La cromatografía puede implicar cualquier número de métodos incluyendo, por ejemplo: fase inversa y fase normal; exclusión por tamaño; intercambio iónico; métodos y aparatos de cromatografía líquida de alta, media y baja presión; analítica a pequeña escala; cromatografía de lecho móvil simulado ("SMB") y preparativa de capa fina o gruesa, así como técnicas de cromatografía de capa fina a pequeña escala y cromatografía instantánea. Un experto en la técnica aplicará las técnicas más adecuadas para conseguir la separación deseada.
Las mezclas diastereoméricas pueden separarse en sus diastereómeros individuales sobre la base de sus diferencias fisicoquímicas por métodos bien conocidos por los expertos en la técnica, tal como por cromatografía y/o cristalización fraccionada. Los enantiómeros pueden separarse convirtiendo la mezcla enantiomérica en una mezcla diastereomérica por reacción con un compuesto ópticamente activo apropiado (por ejemplo, un auxiliar quiral tal como un alcohol quiral o cloruro ácido de Mosher), separando los diastereómeros y convirtiendo (por ejemplo, hidrolizando) los diastereoisómeros individuales en los correspondientes enantiómeros puros. Los enantiómeros también pueden separarse utilizando una columna HPLC quiral.
Un solo estereoisómero, por ejemplo, un enantiómero, sustancialmente libre de su estereoisómero puede obtenerse por resolución de la mezcla racémica utilizando un método tal como la formación de diastereómeros utilizando agentes de resolución ópticamente activos (Eliel, E. y Wilen, S. Stereochemistry of Organic Compounds. Nueva York: John Wiley & Sons, Inc., 1994; Lochmuller, C. H., et al. “Chromatographic resolution of enantiomers: Selective review.” J, Chromatogr, Vol. 113, No. 3 (1975): pp. 283-302). Las mezclas racémicas de los compuestos quirales descritos en el presente documento pueden separarse y aislarse por cualquier método adecuado, incluyendo: (1) formación de sales iónicas diastereoméricas con compuestos quirales y separación por cristalización fraccionada u otros métodos, (2) formación de compuestos diastereoméricos con reactivos derivatizantes quirales, separación de los diastereómeros y conversión a los estereoisómeros puros, y (3) separación de los estereoisómeros sustancialmente puros o enriquecidos directamente bajo condiciones quirales. Véase: Wainer, Irving W., ed. Drug Stereochemistry: Analytical Methods and Pharmacology. Nueva York: Marcel Dekker, Inc., 1993.
Según el método (1), pueden formarse sales diastereoméricas por reacción de bases quirales enantioméricamente puras tales como brucina, quinina, efedrina, estricnina, a-metil-p-feniletilamina (anfetamina) y similares con compuestos asimétricos con funcionalidad ácida, tales como ácido carboxílico y ácido sulfónico. Se puede inducir la separación de las sales diastereoméricas mediante cristalización fraccionada o cromatografía iónica. Para la separación de los isómeros ópticos de los compuestos aminados, la adición de ácidos carboxílicos o sulfónicos quirales, tales como ácido canforsulfónico, ácido tartárico, ácido mandélico o ácido láctico, puede dar lugar a la formación de las sales diastereoméricas.
Alternativamente, por el método (2), el sustrato que se va a resolver se hace reaccionar con un enantiómero de un compuesto quiral para formar un par diastereomérico (Eliel, E. y Wilen, S. Stereochemistry of Organic Compounds. Nueva York: John Wiley & Sons, Inc., 1994, p. 322). Los compuestos diastereoméricos pueden formarse haciendo reaccionar compuestos asimétricos con reactivos derivatizantes quirales enantioméricamente puros, tales como los derivados mentílicos, seguido de la separación de los diastereómeros y la hidrólisis para producir el enantiómero puro o enriquecido. Un método para determinar la pureza óptica consiste en fabricar ésteres quirales, tales como un éster mentílico, por ejemplo, (-) cloroformato mentílico en presencia de base, o éster Mosher, acetato de a-metoxi-a-(trifluorometil)fenilo (Jacob III, Peyton. “Resolution of (±)-5-Bromonomicotine. Synthesis of (R)- and (S)-Nomicotine of High Enantiomeric Purity.” J. Org, Chern, Vol. 47, No. 21 (1982): pp. 4165-4167), de la mezcla racémica, y analizando el espectro de 'H RMN para detectar la presencia de los dos enantiómeros atropisoméricos o diastereómeros. Los diastereómeros estables de los compuestos atropisoméricos pueden separarse y aislarse mediante cromatografía en fase normal y en fase inversa siguiendo los métodos para la separación de naftil-isoquinolinas atropisoméricas (WO 96/15111).
Por el método (3), una mezcla racémica de dos enantiómeros se puede separar por cromatografía utilizando una fase estacionaria quiral (Lough, W.J., ed., Chiral Liquid Chromatography. Nueva York: Chapman y Hall, 1989; Okamoto, Yoshio, et al. “Optical resolution of dihydropyridine enantiomers by high-performance liquid chromatography using phenylcarbamates of polysaccharides as a chiral stationary phase.” J. de Chromatogr. Vol. 513 (1990): pp. 375-378). Los enantiómeros enriquecidos o purificados pueden distinguirse mediante métodos utilizados para distinguir otras moléculas quirales con átomos de carbono asimétricos, tal como la rotación óptica y el dicrαsmo circular.
EVALUACIÓN BIOLÓGICA
La determinación de la actividad de SHP2 de un compuesto de Fórmula I es posible mediante una serie de métodos de detección directos e indirectos. Ciertos compuestos ejemplares descritos en el presente documento se ensayaron para su ensayo de inhibición de SHP2 (Ejemplo Biológico 1). Se utilizó un ensayo celular (Ejemplo Biológico 2) para determinar el efecto de los inhibidores de SHP2 sobre la señalización corriente abajo mediante el ensayo de fosforilación de ERK1/2.
ADMINISTRACIÓN Y FORMULACIONES FARMACÉUTICAS
Los compuestos descritos en el presente documento pueden administrarse por cualquier vía conveniente y adecuada a la afección que se va a tratar. Las vías adecuadas incluyen oral, parenteral (incluyendo subcutánea, intramuscular, intravenosa, intraarterial, intradérmica, intratecal y epidural), transdérmica, rectal, nasal, tópica (incluyendo bucal y sublingual), vaginal, intraperitoneal, intrapulmonar e intranasal.
Los compuestos pueden administrarse en cualquier forma administrativa conveniente, por ejemplo, comprimidos, polvos, cápsulas, soluciones, dispersiones, suspensiones, jarabes, aerosoles, supositorios, geles, emulsiones, parches, etc. Dichas composiciones pueden contener componentes convencionales en las preparaciones farmacéuticas, por ejemplo, diluyentes, portadores, modificadores del pH, edulcorantes, agentes de carga y otros agentes activos. Si se desea la administración parenteral, las composiciones serán estériles y en forma de solución o suspensión inyectable o para perfusión.
Una formulación típica se prepara mezclando un compuesto descrito en el presente documento y un portador, diluyente o excipiente. Los portadores, diluyentes y excipientes adecuados son bien conocidos por los expertos en la técnica y se describen en detalle en, por ejemplo, Ansel, Howard C., et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Filadelfia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Filadelfia: Lippincott, Williams & Wilkins, 2000; y Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Las formulaciones también pueden incluir uno o más tampones, agentes estabilizadores, tensioactivos, agentes humectantes, agentes lubricantes, emulsionantes, agentes de suspensión, conservantes, antioxidantes, agentes opacificantes, deslizantes, auxiliares de procesamiento, colorantes, edulcorantes, agentes perfumantes, agentes aromatizantes, diluyentes y otros aditivos conocidos para proporcionar una presentación elegante del fármaco ( es decir, un compuesto descrito en el presente documento o una composición farmacéutica del mismo) o ayudar en la fabricación del producto farmacéutico (es decir, medicamento).
Una realización incluye una composición farmacéutica que comprende un compuesto de Fórmula Ila, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo. Otra realización proporciona una composición farmacéutica que comprende un compuesto de Fórmula Ila, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, junto con un portador, diluyente o excipiente farmacéuticamente aceptable.
MÉTODOS DE TRATAMIENTO CON COMPUESTOS DE LA INVENCIÓN
También se proporcionan uno o más compuestos descritos en el presente documento, o un estereoisómero, tautómero o sal farmacéuticamente aceptable de los mismos para su uso en tratar o prevenir una enfermedad o afección. En una
realización, se proporciona un compuesto de la invención, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad hiperproliferativa en un mamífero.
Otra realización proporciona un compuesto de la invención - o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso en tratar o prevenir el cáncer en un mamífero.
Otra realización proporciona un compuesto de la invención, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso en tratar o prevenir el dolor en un mamífero.
Otra realización proporciona un compuesto de la invención, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento o prevención de un trastorno inflamatorio en un mamífero.
En ciertas realizaciones, la enfermedad hiperproliferativa es cáncer. En determinadas realizaciones, el cáncer puede seleccionarse de melanoma, leucemias mielomonocíticas juveniles, neuroblastoma, leucemias mieloides crónicas con cromosoma Filadelfia positivo, leucemias linfoblásticas agudas con cromosoma Filadelfia positivo, leucemias mieloides agudas, neoplasias mieloproliferativas (tales como policitemia vera, trombocitemia esencial y mielofibrosis primaria), cáncer de mama, cáncer de pulmón, cáncer de hígado, cáncer colorrectal, cáncer de esófago, cáncer gástrico, carcinoma de células escamosas de cabeza y cuello, glioblastoma, linfoma anaplásico de células grandes, carcinoma de tiroides y neoplasias spitzoides. En ciertas realizaciones, el cáncer es un melanoma. En algunas realizaciones, el cáncer es una leucemia mielomonocítica juvenil. En ciertas realizaciones, el cáncer es neuroblastoma. En ciertas realizaciones, el cáncer es mieloide crónico con cromosoma Filadelfia positivo. En ciertas realizaciones, el cáncer es una leucemia linfoblástica aguda con cromosoma Filadelfia positivo. En ciertas realizaciones, el cáncer es una leucemia mieloide aguda. En ciertas realizaciones, el cáncer es una neoplasia mieloproliferativa, tal como policitemia vera, trombocitemia esencial y mielofibrosis primaria. En ciertas realizaciones, el cáncer se selecciona del grupo que consiste en policitemia vera, trombocitemia esencial y mielofibrosis primaria. En ciertas realizaciones, el cáncer es olicitemia vera. En ciertas realizaciones, el cáncer es trombocitemia esencial. En ciertas realizaciones, el cáncer es pielofibrosis Primaria. En ciertas realizaciones, el cáncer es de mama. En ciertas realizaciones, el cáncer es de pulmón. En ciertas realizaciones, el cáncer es de hígado. En ciertas realizaciones, el cáncer es colorrectal. En ciertas realizaciones, el cáncer es de esófago. En ciertas realizaciones, el cáncer es gástrico. En ciertas realizaciones, el cáncer es un carcinoma de células escamosas de cabeza y cuello. En ciertas realizaciones, el cáncer es un glioblastoma. En ciertas realizaciones, el cáncer es un linfoma anaplásico de células grandes. En ciertas realizaciones, el cáncer es un carcinoma de tiroides. En ciertas realizaciones, el cáncer es una neoplasia espitzoide.
En ciertas realizaciones, el cáncer se selecciona del grupo que consiste en NSCLC, un cáncer de colon, un cáncer esofágico, un cáncer rectal, JMML, cáncer de mama, melanoma, y un cáncer pancreático.
En ciertas realizaciones, la enfermedad o trastorno puede seleccionarse de neurofibramatosis y síndrome de Noonan. En ciertas realizaciones, la enfermedad o trastorno es la neurofibramatosis. En ciertas realizaciones, la enfermedad o trastorno es el síndrome de Noonan.
En ciertas realizaciones, la enfermedad o trastorno es la schwannomatosis.
En ciertas realizaciones, la enfermedad o trastorno que comprende una célula que contiene una mutación que codifica la variante KRASG12C. Véase el documento WO 2019/051084.
En ciertas realizaciones, la enfermedad hiperproliferativa es una enfermedad o trastorno que comprende una célula con una mutación que codifica una variante de pérdida de función NF1 (“NF1LOF”). En ciertas realizaciones, la mutación NF 1 es una mutación de pérdida de función. En ciertas realizaciones, la enfermedad o trastorno es un tumor que comprende células con una mutación de pérdida de función NF1. En ciertas realizaciones, el tumor es un NSCLC o un melanoma. En ciertas realizaciones, la enfermedad se selecciona de neurofibromatosis tipo I, neurofibromatosis tipo II, schwannomatosis y síndrome de Watson.
En ciertas realizaciones, la enfermedad o trastorno asociado con una mutación de la vía RAS en una célula del sujeto que hace que la célula dependa, al menos parcialmente, del flujo de señalización a través de SHP2. En ciertas realizaciones, la mutación de la vía RAS es una mutación RAS seleccionada de una mutación KRAS, una mutación NRAS, una mutación SOS, una mutación BRAF de clase III, una mutación MEK1 de clase I, una mutación MEK1 de clase II y una mutación Fl. En ciertas realizaciones, la mutación KRAS se selecciona de una mutación KRASG12A, una mutación KRASG12C, una mutaciónKRASG12D, una mutación KRASG12F, una mutación KRASG121, una mutación KRASG12L, una mutación KRASG12R, una mutación KRASG12S, una mutación KRASG12V y una mutación KRASG12Y. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12A. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12C. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12D. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12F. En ciertas realizaciones, la mutación KRAS es una mutación KRASG121. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12L. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12R. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12S. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12V. En ciertas realizaciones, la mutación KRAS es una mutación KRASG12Y. En ciertas realizaciones, la mutación de clase III de BRAF se selecciona de una o más de las siguientes sustituciones de aminoácidos en BRAF humano: D287H; P367R; V459L; G466V; G466E; G466A;
S467L; G469E; N581S; N581I; D594N; D594G; D594A; D594H; F595L; G596D; G596R y A762E. En ciertas realizaciones, la mutación MEK1 de clase I se selecciona de una o más de las siguientes sustituciones de aminoácidos en MEK1 humana: D67N; P124L; P124S; y L177V. En ciertas realizaciones, la mutación MEK1 de clase II se selecciona de una o más de las siguientes sustituciones de aminoácidos en MEK1 humana: AE51-Q58; AF53-Q58; E203K; L177M; C121S; F53L; K57E; Q56P; y K57N.
TERAPIA COMBINADA
Los compuestos descritos en el presente documento y los estereoisómeros, tautómeros y sales farmacéuticamente aceptables de los mismos pueden emplearse solos o en combinación con otros agentes terapéuticos para el tratamiento. Los compuestos descritos en el presente documento pueden utilizarse en combinación con uno o más fármacos adicionales, por ejemplo, un agente antihiperproliferativo (o anticancerígeno) que actúe sobre una proteína objetivo diferente. El segundo compuesto de la formulación de combinación farmacéutica o régimen de dosificación preferiblemente tiene actividades complementarias al compuesto descrito en el presente documento, de tal manera que no se afectan adversamente entre sí. Dichas moléculas están convenientemente presentes en combinación en cantidades que son eficaces para el fin previsto. Los compuestos pueden administrarse juntos en una composición farmacéutica unitaria o por separado y, cuando se administran por separado, esto puede ocurrir simultánea o secuencialmente en cualquier orden. Dicha administración secuencial puede ser próxima en el tiempo o remota en el tiempo.
En ciertas realizaciones, el compuesto de la invención se administra en combinación con un inhibidor de la vía RAS. En ciertas realizaciones, el inhibidor de la vía RAS es un inhibidor de MEK o de ERK. En ciertas realizaciones, el inhibidor de la vía Ras se selecciona de uno o más de trametinib, binimetinib, selumetinib, cobimetinib, LErafAON (NeoPharm), ISIS 5132; vemurafenib, pimasertib, TAK733, R04987655 (CH4987655); CI-1040; PD-0325901; CH5126766; MAP855; AZD6244; Refametinib (RDEA 119/BAY 86-9766); GDC-0973/XL581; AZD8330 (ARRY-424704/ARRY-704); R05126766; ARS-853; LY3214996; BVD523; GSK1 120212; Ulixertinib, y Abemaciclib.
EJEMPLOS
Se incluyen los siguientes Ejemplos. Los expertos en la técnica reconocerán que las reacciones químicas descritas pueden adaptarse fácilmente para preparar una serie de otros compuestos descritos en el presente documento, y se considera que los métodos alternativos para preparar los compuestos están dentro del alcance de esta invención. Por ejemplo, la síntesis de compuestos no ejemplificados puede realizarse con éxito mediante modificaciones evidentes para los expertos en la técnica, por ejemplo, protegiendo adecuadamente los grupos interferentes, utilizando otros reactivos adecuados conocidos en la técnica distintos de los descritos, y/o realizando modificaciones rutinarias de las condiciones de reacción. Alternativamente, se reconocerá que otras reacciones divulgadas en el presente documento o conocidas en la técnica tienen aplicabilidad para preparar otros compuestos descritos en el presente documento.
En los Ejemplos descritos a continuación, a menos que se indique lo contrario, todas las temperaturas se expresan en grados Celsius. Los reactivos se adquirieron de proveedores comerciales tales como Sigma- Aldrich, Alfa Aesar o TCI, y se utilizaron sin más purificación a menos que se indicara lo contrario.
Las reacciones expuestas a continuación se realizaron generalmente bajo una presión positiva de nitrógeno o argón 0 con un tubo de secado (a menos que se indique lo contrario) en disolventes anhidros, y los matraces de reacción se equiparon típicamente con septos de goma para la introducción de sustratos y reactivos a través de jeringa. El material de vidrio se secó al horno y/o al calor.
La cromatografía en columna se realizó en un sistema Biotage (Fabricante: Dyax Corporation) con una columna de gel de sílice o en un cartucho SepPak de sílice (Waters) (salvo que se indique lo contrario). *Los espectros de 1H RMN registraron en un instrumento Varian que funcionaba a 400 MHz. Los espectros 1H-RMN se obtuvieron como soluciones CDCh, CD3OD, D2O, (CD3)2SO, (CD3)2CO, C6D6, CD3CN (informados en ppm), utilizando tetrametilsilano (0,00 ppm) o disolvente residual (CDCh: 7,26 ppm; CD3OD: 3,31 ppm; D2O: 4,79 ppm; (Cd 3)2SO: 2,50 ppm; (CD3)2CO: 2,05 ppm; C6D6: 7,16 ppm; CD3CN: 1,94 ppm) como patrón de referencia. Cuando se comunican las multiplicidades de los picos se utilizan las siguientes abreviaturas: s (singlete), d (doblete), t (triplete), q (cuarteto), m (multiplete), br (ensanchado), dd (doblete de dobletes), dt (doblete de tripletes). Cuando se indican, las constantes de acoplamiento se expresan en hercios (Hz).
Ejemplo 1 Biológico
Ensayo enzimático SHP2
Se configuró un ensayo cinético de intensidad de fluorescencia para SHP2 de longitud completa que monitoriza la cantidad de 6,8-difluoro-7-hidroxi-4-metilcumarina ("DiFMU") formada tras la hidrólisis de 6,8-difluoro-4-metilumbeliferil fosfato ("DiFMUP") por SHP2. Las mezclas de ensayo consistieron en 25 mM K+HEPES, pH 7,4, 0.01 % Triton X-100, 1 mM DTT, 50 mM KCl, 100 μg/ml de Y-globulina bovina, 50 μM DiFMUP, 1 μM péptido activador de SHP2
(LN(pY)IDLDLV(dPEG8)LST(pY)ASINFQK-amida), 1 μM SHP2 de longitud completa (SHP2(2-527) marcado con His6, expresado recombinantemente en E. coli y purificada internamente) y 2 % de dimetilsulfóxido ("DMSO") (del compuesto). Los compuestos se diluyeron normalmente en DMSO en un intervalo de dosificación de 10 puntos creado mediante un protocolo de dilución en serie de 3 veces a una dosis máxima de 20 μM. El ensayo se realizó en placas de microtitulación de 384 pocillos, de poliestireno, de bajo volumen, no tratadas y de color negro (Costar 4511), en un volumen final de 20 μl. Los pocillos de control bajo carecían de enzima. Los ensayos se iniciaron mediante la adición de una mezcla de SHP2 y el péptido activador y, tras una mezcla de 15 segundos en un agitador orbital, se leyeron en modo cinético durante 15 minutos (30 segundos/ciclo) a temperatura ambiente en un lector de microplacas PerkinElmer EnVision (AEx = 355 nm, AEm = 460 nm). Las velocidades iniciales (pendientes de las tangentes en t = 0) se estimaron a partir de ajustes exponenciales a las curvas de progreso ligeramente no lineales y luego se convirtieron en porcentaje de control ("POC") mediante la siguiente ecuación:

Se ajustó un modelo logístico de 4 parámetros a los datos POC de cada compuesto. A partir de ese ajuste, se estimó el IC50, que se define como la concentración de compuesto a la que la curva cruza 50 POC.
La Tabla 1 contiene datos representativos de los Ejemplos divulgados en el presente documento. El IC50 indicado en la Tabla 1 puede proceder de un único ensayo o de la media de varios ensayos. Los Ejemplos 1-51 se probaron en el ensayo anterior y resultaron tener un IC50 inferior a 10 μM. Los Ejemplos 1-51 se probaron en el ensayo anterior y resultaron tener un IC50 inferior a 9,5 μM. Los Ejemplos 1-6, 8- 20, 22-46, 48, 49 y 51 se probaron en el ensayo anterior y resultaron tener un IC50 inferior a 5 μM. Los Ejemplos y Ejemplos 1-4, 6, 8-20, 22-33, 35-45, 48, 49 y 51 de Referencia se probaron en el ensayo anterior y resultaron tener una IC50 de 2,5 μM o menos. Los Ejemplos y Ejemplos 1, 2, 4, 6, 8, 9, 12-18, 20, 22, 24-33, 35, 36, 38-45, 48, 49 y 51 de Referencia se probaron en el ensayo anterior y resultaron tener un IC50 de 1 μM o menos.
La Tabla 1 contiene los Ejemplos probados en el ensayo anterior:
TABLA 1

(continuación)

(continuación)

(continuación)

Ejemplo 2 Biológico
Ensayo celular de fosfo-p44/42 MAPK (Erkl/2) (Thr202/Tyr204)
La inhibición de la fosforilación de ERK1/2 (Thr202/Tyr204) se determinó mediante el siguiente ensayo celular, que consiste en incubar las células con un compuesto durante 1 hora y cuantificar la señal de pERK mediante Western In-Cell en células fijadas y normalizar con respecto a la señal de GAPDH. Las células KYSE520 se obtuvieron de DSMZ y se cultivaron en RPM i suplementado con 10 % de suero bovino fetal, penicilina/estreptomicina, 2 mM de dipéptido L-alanil-L-glutamina en 0,85 % de NaCl (Glutamax™), aminoácidos no esenciales y piruvato sódico. Las células se sembraron en placas de 96 pocillos a 30.000 células/pocillo y se dejaron adherir durante la noche a 37 °C/5 % de CO2. Las células se trataron con compuestos preparados como una serie de diluciones de 10 puntos, 1:3 (intervalo: 20 μM - 1 nM), con una concentración final de DMSO del 0,5 %. Tras 1 hora de incubación, las células se fijaron en formaldehído al 3,7 % en solución salina tamponada con fosfato de Dulbecco ("dPBS") a temperatura ambiente durante 20 minutos. A continuación, las células se lavaron con dPBS y se permeabilizaron en MeOH al 100 % a temperatura ambiente durante 10 minutos. Tras la permeabilización, las células se lavaron en dPBS y se incubaron en LI-COR Blocking Buffer (LI-COR Biosciences, Cat#927-40000) durante 1 hora o más. A continuación, se incubaron las placas con un anticuerpo específico para los sitios de fosforilación de ERK1/2 dependientes de MEK, treonina 202 y tirosina 204 (Cell Signaling Technologies; Cat# 9101), corriente abajo de SHP2 en la vía de transducción de señales de la MAP cinasa, así como GAPDH (Millipore; Cat# MAB374). El anticuerpo pErkl/2 (Thr202/Tyr204) se diluyó en tampón de bloqueo LI-COR que contenía 0,05 % de polisorbato-20 (Tween-20) a 1:250; GAPDH se diluyó a 1:2.500. Las placas se incubaron durante la noche a 4 °C. Tras lavarlas con PBS/0,05 % de Tween-20, las células se incubaron con anticuerpos secundarios marcados con fluorescencia (Anti-conejo-Alexa Flour680, Invitrogen Cat#A21109; Antiratón-IRDye800CW, Li-cor Bioscieces Cat#926-32210, ambos en dilución 1:1000) durante 1 hora. A continuación, se lavaron las células, como se ha indicado anteriormente, y se analizó la fluorescencia a longitudes de onda de 680 nm y 800 nm utilizando el Aerius Infrared Imaging System (LI-COR Biosciences, modelo 9250). La señal de Erkl/2 fosforilada (Thr202/Tyr204) se normalizó con respecto a la señal de GAPDH para cada pocillo. Los valores IC50 se calcularon a partir de los valores normalizados utilizando un ajuste de 4 parámetros en el software BioAssay. La Tabla 2 contiene datos representativos de los Ejemplos divulgados en el presente documento. El IC50 indicado en la Tabla 2 puede proceder de un único ensayo o de la media de varios ensayos.
La Tabla 2 contiene Ejemplos seleccionados probados en el ensayo anterior:
TABLA 2

Ejemplo A Intermedio

trifluorometanosulfonato de 6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡lo
Se añad¡ó N-et¡l-N-¡soprop¡lpropan-2-am¡na (3.0 ml, 16 mmol) a una pasta de 6-clorop¡r¡do[2.3-b1p¡raz¡n-2(H)-ona (2.0 g. 11 mmol) en d¡clorometano ("DCM") (110 ml. 11 mmol) enfr¡ado a 0 °C. segu¡do de Tf2O (2.1 ml. l3 mmol). La reacc¡ón se mantuvo a 0 °C durante 30 m¡nutos. La reacc¡ón se concentró y se cromatograf¡ó d¡rectamente usando acetato de et¡lo ("EtOAc")/hexanos al 5-50 % para dar tr¡fluorometanosulfonato de 6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡lo (2.6 g. 8.2 mmol. rend¡m¡ento del 75 %). 1H RMN (400 MHz. (CDCh) 88.98 (s. 1H). 8.38 (d. 1H. J=8.6 Hz). 7.84 (d. 1H. J=8.6Hz).
Ejemplo B Intermedio

Paso A: Se añad¡ó éster 2-et¡lhexíl¡co del ác¡do 3-mercaptoprop¡ón¡co (2.3 ml. 22 mmol) y la base de Hun¡g (6.9 ml.
39 mmol) a una mezcla de 3-cloro-4-yodop¡r¡d¡n-2-am¡na (5.0 g. 20 mmol). Pd(OAc)2 (0.22 g. 0.98 mmol) y xantfos (1.1 g. 2.0 mmol) en d¡oxano (65 ml. 20 mmol) bajo gas Ar. La reacc¡ón se calentó a 100 °C bajo argón durante 18 horas. La reacc¡ón se d¡luyó en EtOAc y se f¡ltró a través de síl¡ce d¡atomácea (Cel¡te®). El f¡ltrado se concentró para proporc¡onar 3-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo (4.3 g. 17 mmol. 88 % de rend¡m¡ento).
Paso B: Se añad¡ó NaOEt (7.1 ml. 19 mmol) a 3-((2-am¡no-3- clorop¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo (4.3 g. 17 mmol)
en tetrahidrofurano ("THF") (87 ml, 17 mmol) y se agitó bajo N2 durante 1 hora a temperatura ambiente. Se añadió DCM (20 ml), y esta mezcla se agitó durante 5 minutos. La reacción se concentró y el sólido se tituló con DCM, se filtró y se secó. Los sólidos se introdujeron en agua (pasta) y se añadió HCl 1N para llevar el pH a 6 aproximadamente. Los sólidos se filtraron y se lavaron con agua para proporcionar 2-amino-3-cloropiridina-4-tiol (1,4 g, 9,0 mmol, 52 % de rendimiento). 1H RMN(400 MHz, (CDa^SO) 811,4 (br, 1H), 7,06 (d, 1H, J= 6,8 Hz), 6,70 (br, 2H), 6,65 (d, 1H, J=7,0 Hz); m/z (esi/APCI) M+1 = 161,0.
Ejemplo C Intermedio

2.6-d¡cloropir¡do[2.3-61p¡raz¡na
Se colocó 6-cloropirido[2,3-8]pirazin-2(H)-ona (0,95 g, 5,2 mmol) en tolueno (5 ml). Se añadió POCh (5,3 ml, 57 mmol) y la reacción se sometió a reflujo durante 22 horas. La reacción se enfrió a 0 °C y se añadió agua seguida de bicarbonato saturado. La mezcla se extrajo con DCM (3 X 50 ml), los orgánicos se combinaron, se secaron, se filtraron y se concentraron para proporcionar 2,6-dicloropirido[2,3-8]pirazina (0,90 g, 4,5 mmol, 86 % de rendimiento). El intermedio se utilizó tal cual.
Ejemplo D Intermedio

3-cloro-2-metox¡p¡r¡d¡na-4-t¡ol
Se preparó 3-cloro-2-metoxipiridina-4-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 3-cloro-4-yodo-2-metoxipiridina por 3-cloro-4-yodopiridin-2-amina en el Paso A. m/z (esi/APCI) M+l = 176,0.
Ejemplo E Intermedio

3-cloro-2-(pirrolidin-1-il)piridin-4-tiol\
Paso B: Se preparó 3-cloro-2-(pirrolidin-1-il)piridin-4-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 3-cloro-4-yodo-2-(pirrolidin-1-il)piridina por 3-cloro-4-yodopiridin-2-amina en el Paso A. 1H RMN(400 MHz, (CD3)2SO) 811,28 (br, 1H), 7,06 (br, 1H), 6,82 (d, 1H, 6,7 Hz), 3,56 (m, 4H), 1,88 (m, 4H); m/z (esi/APCI) M+1 = 215,0.
Ejemplo F Intermedio

Diclorhidrato de(R)-8-azaespiro[4.51decan-1-amina
Paso A: Se agregaron tetraetoxititanio (5,0 g, 22 mimóles) y (R)-2-metilpropan-2-sulfinaimida (1,4 g, 12 mimóles) a una solución de 8-boc-1-oxo-8-aza-espiro[4.5]decano (1,4 g, 5,5 mmoles) en THF (37 ml, 5,5 mmoles). La reacción se calentó a 65 °C durante la noche. La solución se enfrió a 0 °C y se agregó MeOH (20 ml) seguido por la adición por goteo de LiBH4 (5,5 ml, 11 mmoles). La reacción se agitó a 0 °C durante una hora. La reacción se vertió después en NH4Cl y la suspensión se filtró a través de Celite®. El Celite® se lavó con EtOAc. La mezcla se separó y la capa acuosa se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El material se sometió a cromatografía utilizando un gradiente de EtOAc/hexanos al 10-70 % para dar (Rj-1-(((Rj-ferf-butilsulfinil)amino)-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (0,7 g, 2,0 mmoles, 35 % de rendimiento).
Paso B: Se colocó (Rj-1-(((Rj-ferf-butilsulfinil)amino)-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (1,6 g, 4,3 mmoles) en MeOH (10 ml) y se trató con HCl (11 ml, 43 mmoles). La reacción se agitó durante una hora a temperatura ambiente. La reacción se concentró y el sólido resultante se tituló con metilo ferf-butiléter (“MTBE”) (30 ml) y se filtró para proporcionar diclorhidrato de (Rj-8-azaespiro[4.5]decan-1-amina (0,88 g, 3,9 mmoles, 89 % de rendimiento). m/z (esi/APCI) M+1 = 155,2.
Ejemplo G Intermedio

Se preparó 2-cloro-3-metoxibencenotiol de acuerdo con el Ejemplo B Intermedio, sustituyendo l-bromo-2-cloro-3-metoxibenceno por 3-cloro-4-yodopiridin-2-amina en el Paso A. m/z (esi/APCI) M+l = 175,0.
Ejemplo intermedio H

2-cloro-3-mercaptobenzonitrilo
Se preparó 2-cloro-3-mercaptobenzonitrilo de acuerdo con el Ejemplo B Intermedio, sustituyendo 3 bromo-2-clorobenzonitrilo por 3-cloro-4-yodopiridin-2-amina en el paso A. m/z (esi/APCI) M+1 = 170,0.
Ejemplo I Intermedio

Se preparó 1-metil-1H-indazol-7-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 7-broimo-1-imetil-1H-indazol por 3-cloro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 165,1.
E je m p lo J In te rm e d io

p¡razol[1.5-a1p¡rid¡n-4-t¡ol
Se preparó p¡razol[1,5-a]pir¡d¡n-4-t¡ol de acuerdo con el Ejemplo B Intermedio. sustituyendo 4-bromo-pirazol(1,5-a)piridina por 3-doro-4-yodopirid¡n-2-am¡na en la Paso A. m/z (esi/APCI) M+1 = 151,1.
Ejemplo K Intermedio

p¡razol[1,5-a1p¡raz¡n-4-t¡ol
Se preparó pirazol[1,5-a]piraz¡n-4-t¡ol de acuerdo con el Ejemplo B Intermedio, al sustituir 4-clorop¡razol[1,5-a]piraz¡na por 3-cloro-4-yodopir¡d¡n-2-am¡na en la Paso A. m/z (esi/APCI) M+1 = 152,1.
Ejemplo L Intermedio

Se preparó 3-cloro-2-(met¡lam¡no)p¡r¡din-4-t¡ol de acuerdo con el Ejemplo Intermedio E, sustituyendo metilamina por pirolidina en la Paso A. m/z (esi/ApCI) M+1 = 175,0.
Ejemplo M Intermedio

isoquinolin-8-tiol
Se preparó isoquinolin-8-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 8-bromoisoquinolina por 3-cloro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 321,1.
E je m p lo N In te rm e d io

3-cloro-2-metilp¡r¡d¡n-4-t¡olato de sodio
Paso A: Se agregaron 2-etilhexiléster del ácido 3-mercaptopropiónico (0,47 ml, 4,3 mmoles) y base de Hunig (1,4 ml, 7,9 mmoles) a una mezcla de 3-cloro-4-yodo-2-met¡lp¡r¡d¡na (1,0 g, 3,945 mmoles), Pd(OAc)2 (0,044 g, 0,20 mmoles) y xantfos (0,230 g, 0,40 mmoles) en dioxano (13 ml, 4,0 mmoles) bajo gas Ar. La reacción se calentó a 100 °C bajo argón durante 18 horas. La reacción se diluyó en EtOAc (60 ml) y se filtró a través de Celite®. El filtrado se concentró y el residuo resultante se purificó mediante gel de sílice (EtOAc en hexanos al 5-60 %) para proporcionar 3-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)tio)propanoato de metilo (962 mg, 3,9 mmoles, 99 % de rendimiento).
Paso B: Se agregó NaOEt (1,6 ml, 4,3 mmoles) a 3-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)propanoato de metilo (962 mg, 3,9 mmoles) en THF (20 ml, 3,9 mmoles) y se agitó bajo N2 durante una hora a temperatura ambiente. La mezcla de reacción se vertió sobre éter y los sólidos se filtraron y lavaron con éter para proporcionar 3-cloro-2-met¡lp¡r¡d¡n-4-tiolato de sodio (701 mg, 3,9 mmoles, 99 % de rendimiento).
Ejemplo O Intermedio

(ft)-N-((1R3ft)-3-h¡drox¡-8-azaesp¡ro[4.5ldecan-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da
Paso A: Una mezcla de 4-form¡lp¡per¡d¡n-1-carbox¡lato de ferf-butilo (30 g, 140,84 mmoles), ferf-butóxido de litio (13,4 g, 169 mmoles) y bromuro de alilo (10,2 ml, 161,96 mmoles) en Dm F (288 ml) se agitó durante 15 minutos a 0 °C. La mezcla se vertió en un embudo de separación que contenía NH4Cl:H2O saturado acuoso (1:1, 160 ml) y se extrajo con EtOAc (5 X 60 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y concentraron bajo presión reducida. El residuo obtenido se purificó mediante cromatografía instantánea (gradiente: EtOAc/Hexanoal 0-10 %) para dar 4-form¡l-4-(prop-2-en-1-¡l)p¡per¡d¡n-1-carbox¡lato de ferf-butilo (24 g, 67 %) como un aceite incoloro. LCMS: 254.1 (M+H).
Paso B: Se agregó bromuro de vinilmagnesio (1 M en THF, 102 ml, 102,92 mmoles) a una solución agitada de 4-form¡l-4-(prop-2-en-1-¡l)p¡per¡d¡n-1-carbox¡lato de ferf-butilo (21 g, 83,00 mmoles) en THF (260 ml) a -78 °C bajo una atmósfera de N2. La solución resultante se calentó gradualmente a temperatura ambiente en el plazo de 1 hora. La mezcla se vertió en un embudo de separación que contenía la solución acuosa saturada de NH4Cl (60 ml) y se extrajo con EtOAc (3 X 100 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y secaron bajo presión reducida para dar el producto deseado sin purificar de 4-(1-h¡drox¡prop-2-en-1-¡l)-4-(prop-2-en-1-¡l)p¡per¡d¡n-1-carboxilato de ferf-butilo (21 g) como un aceite. Este compuesto se utilizó para el siguiente paso sin ninguna purificación adicional. LCMS: 282,4 (M+H).
Paso C: Se agregó periodinano de Dess-Martin (54g, 128,11 mmoles) a una solución agitada de 4-(1-hidroxiprop-2-en-1-¡l)-4-(prop-2-en-1-¡l)p¡per¡d¡n-1-carbox¡lato de ferf-butilo (18 g, 64,05 mmoles) en DCM (240 ml) y se agitó durante una hora a temperatura ambiente bajo una atmósfera de nitrógeno. La mezcla se filtró a través de una almohadilla de Celite®, el filtrado se vertió en un embudo de separación que contenía una solución saturada de NaHCO3 y se extrajo con DCM. Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y concentraron bajo presión reducida para proporcionar un semi-sólido amarillo (20 g). Este semi-sólido se suspendió en hexano (100 ml) y se sometió a ultrasonido durante 5 minutos. La suspensión blanca se filtró a través de una almohadilla de Celite® y los compuestos volátiles se retiraron bajo presión reducida para dar un aceite amarillo sin purificar. El residuo obtenido se combinó con el producto sin purificar de otro lote (tamaño del lote: 3 g de 4-(1-h¡drox¡prop-2-en-1-¡l)-4-(prop-2-en-1-¡l)p¡per¡d¡n-1-carboxilato de ferf-butilo) y se purificó mediante cromatografía instantánea (gradiente: EtOAc/hexano al 0-10 %) para dar 4-(prop-2-en-1-¡l)-4-(prop-2-eno¡l)p¡per¡d¡n-1-carbox¡lato de ferf-butilo (15 g, 65 %, 2 pasos) como un aceite. LCMS: 280.1 (M+H).
Paso D: Se agregó Grubbs II (1,3 g, 1,61 mmoles) a una solución agitada de 4-(prop-2-en-1-il)-4-(prop-2-eno¡l)p¡per¡d¡n-1-carbox¡lato de ferf-butilo (15 g, 53,76 mmoles) en tolueno (desgasificado, 540 ml) y la mezcla resultante se agitó durante 45 minutos a 85 °C. El solvente se concentró y el residuo resultante (17,9 g) se purificó
mediante cromatografía en columna de sílice (gradiente: EtOAc/hexano al 0-50 %) para dar 1-oxo-8-azaespiro[4.5]dec-2- en-8-carboxilato de ferf-butilo (10 g, 74 %) como un sólido. LCMS: 252,0 (M+h ).
Paso E: Una mezcla de CuCl (532 mg, 5,37 mmoles), (S)-Tol-BINAP (3,6 g, 5,37 mmoles) y ferf-butóxido de sodio (517 mg, 5,37 mmoles) en THF (45 ml) se agitó durante 30 minutos a temperatura ambiente. Se agregó bis(pinacolato)diboro (11,8 g, 46,61 mmoles) en THF (15 ml) y la mezcla de reacción de dejo agitar a temperatura ambiente durante 10 minutos. Se agregó una solución de 1-oxo-8-azaespiro[4.5]dec-2-en-8-carboxilato de ferf-butilo (9 g, 35,85 mmoles) en THF (36 ml), seguida por MeOH (2,7 ml). La mezcla resultante se agitó durante 16 horas a temperatura ambiente. Se agregó agua (110 ml), seguida por perborato de sodio (17,9 g, 179,28 mmoles) y la mezcla resultante se agitó vigorosamente durante 10 minutos a 0 °C. La suspensión verde resultante se filtró a través de una almohadilla de Celite®, se vertió en un embudo de separación que contenía una solución saturada de NaHCO3 y se extrajo con EtOAc (3 X 50 ml). Las fases orgánicas combinadas se secaron, filtraron y concentraron bajo presión reducida. El residuo obtenido se purificó mediante cromatografía en columna de sílice (gradiente: EtOAc/hexano al 0 60 %) para dar el producto deseado de (3R)-3-hidroxi-1-oxo-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (5,1 g, 53 %) como un sólido.
Paso F: Se agregó cloruro de ferf-butildimetilsililo (1,9 g, 13,0 mmoles) a la solución agitada de imidazol (817 mg, 12,0 mmoles) en DMF seco (27 ml) a 0 °C y se agitó durante 15 minutos a temperatura ambiente. Se agregó (3R)-3-hidroxi-1-oxo-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (2,7 g, 10,0 mmoles) a la mezcla de reacción y se dejó agitar a temperatura ambiente durante 4 horas. La mezcla de reacción se diluyó mediante la adición de agua fría y se extrajo con EtOAc (2 X 30 ml). Entonces la capa orgánica combinada se secó sobre sulfato de sodio y se concentró bajo presión reducida. El residuo resultante se purificó mediante cromatografía instantánea (gradiente: EtOAc/hexano al 0 30 %) para dar (3R)-3-[(ferf-butildimetilsilil)oxi]-1-oxo-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (3,1 g, 81 %) como un sólido. Lc Ms : 384,3 (M+H).
Paso G: Una solución de (3R)-3-[(ferf-butildimetilsilil)oxi]-1-oxo-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (7,4 g, 27,3 mmoles), etóxido de titanio (IV) (22,8 ml, 109,2 mmoles) y (R)-(+)-2-metil-2-propansulfinamida (6,6 g, 54,6 mmoles) en THF (80 ml) se agitó durante 4 horas a 65 °C. A la mezcla de la reacción se agregó agua, se agitó durante 15 minutos y se filtró a través de una almohadilla de Celite®. El filtrado se lavó con EtOAc (2 X 60 ml), se secó y concentró para dar el producto sin purificar. El residuo obtenido se purificó mediante cromatografía instantánea (gradiente: EtOAc/hexano al 0-40 %) para dar (1E,3R)-3-[(tert-butildimetilsilil)oxi]-1-{[(R)-2-metilpropan-2-sulfinil]imino}-8azaespiro[4.5]decan-8-carboxilato de ferf-butilo (3,5g, 53 %). LCMS: 487,6 (M+H).
Paso H: Una solución de (1E,3R)-3-[(tert-butildimetilsilil)oxi]-1-{[(R)-2-metilpropan-2-sulfinil]imino}-8 azaespiro[4.5]decan-8-carboxilato de ferf-butilo (745 mg, 1,53 mmoles) en t Hf (7 ml) se enfrió a -78 °C seguida por la adición de MeOH (0,07 ml). Se agregó borohidruro de litio (84 mg, 3,83 mmoles) y la mezcla de reacción resultante se agitó durante 3 horas a -78 °C. Al término, la solución acuosa saturada de NH4Cl (10 ml) se agregó lentamente para extinguir el exceso de borohidruro y la mezcla de reacción se diluyó con EtOAc (20 ml). La mezcla resultante se agitó vigorosamente durante 15 minutos a temperatura ambiente y después se extrajo con EtOAc (2 X 20 ml). La mezcla de reacción se concentró para dar el producto sin purificar de (1R,3R)-3-[(ferf-butildimetilsilil)oxi]-1-{[(R)-2-metilpropan-2-sulfinil]amino}-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (745 mg) como una masa pegajosa incolora, que procedió al siguiente paso sin ninguna purificación. LCMS: 489,4 (M+H).
Paso I: Se agregó una solución de TBAF (1,0M en THF, 2,3 ml, 2,29 mmoles) a una solución agitada de (1R,3R)-3-[(ferf-butildimetilsilil)oxi]-1-{[(R)-2-metilpropan-2-sulfinil]amino}-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (745 mg, 1,52 mmoles) en THF (7 ml) a temperatura ambiente y se dejó agitar durante 2 horas a la misma temperatura. Al término, la mezcla de reacción se trató con una solución saturada de NaHCO3 y se extrajo con EtOAc (2 X 20 ml). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y concentraron. El residuo resultante se purificó mediante cromatografía instantánea (gradiente: MeOH/DCM al 0-10 %) para dar (1R,3R)-3-hidroxi-1-{[(R)-2-metilpropan-2-sulfinil]amino}-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (315 mg, 55 %, 2 pasos) como un sólido.
Paso J: Se agregó (1R,3R)-3-hidroxi-1-{[(R)-2-metilpropan-2-sulfinil]amino}-8-azaespiro[4.5]decan-8-carboxilato de ferf-butilo (200 mg, 0,53 mmoles) a una solución de TFNDCM (2 ml, 1:10) a 0 °C y se agitó durante 2 horas a temperatura ambiente. Después del término, la mezcla de reacción se concentró. El residuo obtenido se disolvió en MeOH (0,5 ml) y se agregó NaHCO3 sólido a la mezcla de reacción. La mezcla de reacción resultante se agitó durante 15 minutos a temperatura ambiente, se filtró y el filtrado se concentró bajo presión reducida. El residuo obtenido se trituró con una mezcla de MeOH al 10 % en DCM y el residuo sólido se filtró. El filtrado se concentró para dar el producto sin purificar como un sólido pegajoso marrón claro. Este se purificó mediante cromatografía de columna que utiliza alúmina neutra como fase estacionaria y MeOH al 0-15 % en DCM como la fase móvil para dar (R)-N-[(1R,3R)-3- hidroxi-8-azaespiro[4.5]decan-1-il]-2-metilpropan-2-sulfinamida (79,2 mg, 32 %) como un sólido.
E je m p lo P In te rm e d io

6-((ferf-butoxicarboml)am¡no)-2,3-d¡dorop¡rid¡n-4-tiolato de sodio
Paso A: Se agregó por goteo LiHMDS (1 M en THF; 26 ml, 26,99 mmoles) a una solución agitada de 5,6-dicloropiridin-2-amina (2,0 g, 12,27 mmoles) en THF (64 ml) a 0 °C y la mezcla de reacción se agitó durante 15 minutos a la misma temperatura. Se agregó Boc2O (3,1 ml, 13,49 mmoles) y se agitó durante 15 minutos a esta temperatura. La mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante otros 30 minutos. Después del término (monitorizado mediante TLC), la mezcla de reacción se extinguió con agua enfriada con hielo (20 ml) y se extrajo con acetato de etilo (3 X 30 ml). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y el filtrado se retiró bajo presión reducida para dar el producto sin purificar, el cual se purificó mediante cromatografía combiflash en gel de sílice utilizando acetato de etilo al 5 % en hexano como un eluyente para producir ^^-dicloropiridin^-i^carbamato de tert-butilo (2,8 g, 86 %) como un sólido. LCMS: 207 (M+H-56).
Paso B: Se agregó n-BuLi (1,9M in THF; 4 ml, 7,63 mmoles) a una solución agitada de diisopropilamina (1,1 ml, 7,63 mmoles) en THF (8 ml) a -78 °C y se agitó durante una hora. Después se agregó una solución de (5,6-dicloropiridin-2- il)carbamato de tert-butilo (1,0 g, 3,82 mmoles) en THF (6 ml) a la misma temperatura y se agitó a -78 °C durante otras 2 horas. Se agregó una solución de yodo (1,2 g, 4,58 mmoles) en THF (6 ml) a -78 °C y se agitó durante 30 minutos adicionales. Después del término (monitorizado mediante TLC), la mezcla de reacción se diluyó con una solución saturada de cloruro de amonio (10 ml) y la mezcla de reacción se extrajo con acetato de etilo (2 X 20 ml). La parte orgánica se secó sobre Na2SO4 anhidro, se filtró y concentró bajo presión reducida para dar el producto sin purificar, el cual se purificó mediante cromatografía combiflash en gel de sílice utilizando acetato de etilo en hexano al 3- 5 % como un eluyente para producir ^^-dicloro^-yodopiridin^-i^carbamato de tert-butilo (0,85 g, 57 %) como un sólido. LCMS: 332,8 (M+H-56).
Paso C: Se agregó DIPEA (9,0 ml, 51,54 mmoles) a una solución agitada de ^^-dicloro^-yodopiridin^-i^carbamato de tert-butilo (10,0 g, 25,77 mmoles) y metiléster del ácido 3-mercapto-propiónico (3,2 ml, 28,35 mmoles) en dioxano (150 ml) y se desgasificó con argón durante 15 minutos. Se agregaron xantfos (0,75 g, 1,28 mmoles) y Pd(OAc)2 (0,35 g, 1,54 mmoles) y se desgasificaron durante otros 10 minutos con argón. La mezcla de reacción se agitó en un baño de aceite precalentado a 100 °C durante 4 horas. Después del término (monitorizado mediante TLC), la mezcla de reacción se filtró a través de una almohadilla de Celite® y se lavó con acetato de etilo (2 X 80 ml). El solvente se evaporó y el producto sin purificar se purificó a través de cromatografía de columna instantánea en gel de sílice utilizando acetato de etilo al 20 % en hexano como un eluyente para producir 3-((6-((ferf-butoxicarbonil)amino)-2,3-dicloropiridin^-il^io^ropanoato de metilo (8,1 g, 82 %). Lc Ms : 381,0 (M+H).
Paso D: Se agregó NaOEt (21 % en peso en EtOH; 4,7 ml, 14,47 mmoles) a una solución agitada de 3-((6-((tert-butoxicarboni^amino^^-dicloropiridin^-il)tio^ropanoato de metilo (5,0 g, 13,16 mmoles) en THF (50 ml) a 0 °C y se agitó durante 40 minutos a la misma temperatura. Después del término (monitorizado mediante TLC), la mezcla de reacción se concentró y el producto sin purificar se trituró con DCM (20 ml), éter dietílico (20 ml) y pentano (40 ml) a 0 °C y el sólido se filtró y secó al vacío para producir el 6-((terf-butox¡carbon¡l)am¡no)-2,3-d¡clorop¡r¡d¡n-4-t¡olato de sodio deseado (3,98 g, 94 %) como un sólido. UPLC: 293,0 (M+H).
Ejemplo Q Intermedio

(1 ft^ ft^S ^^ -o xa ^ -a za b ic ic lo re ^ .n o c ta n ^ -a m in a
Paso A: Se diluyó ácido Rel-(1S*,5R*,6R*)-3-(tert-butox¡carbon¡l)-8-oxa-3-azab¡c¡clo[3.2.1]octan-6-carboxíl¡co (365 mg, 1,42 mmoles) con tert-butanol (7 ml), seguido por la adición de trietilamina (395 μl, 2,84 mmoles) y la adición por goteo de difenilfosforilazida (336 μl, 1,56 mmoles). Después de agitar durante 30 minutos, la reacción se calentó a 93 °C y se agitó durante 12 horas. La reacción se dejó enfriar y se diluyó con DCM y carbonato de sodio al 10 %. Las capas se separaron y el DCM se secó sobre MgSO4, se filtró y concentró. El material se purificó en gel de sílice eludido
con acetato de etilo/hexanos al 10-100 % para producir (1S*,5S*,6R*)-6-((tert-butoxicarbonil)amino)-8-oxa-3-azabiciclo[3.2.1]octan-3-carboxilato de tert-butilo (144 mg, 0,44 mmoles, 31 % de rendimiento).
Paso B: Se diluyó (1S*,5S*,6R*)-6-((tert-butoxicarbonil)amino)-8-oxa-3-azabiciclo[3.2.1]octan-3-carboxilato de tert-butilo (146 mg, 0,45 mmoles) con DCM (2 ml), seguido por la adición de TFA (2 ml). Después de agitar durante 3 horas, la reacción se concentró para producir (1R*,5R*,6S*)-8-oxa-3-azabiciclo[3.2.1]octan-6-amina (57 mg, 0,45 mmoles, 100 % de rendimiento).
Ejemplo R Intermedio

Se diluyó 4-(2-aminopropan-2-il)piperidin-1-carboxilato de tert-butilo (260 mg, 1,07 mmoles) con DCM (3 ml), seguido por la adición de TFA (3 ml). Después de agitar durante 2 horas, la reacción se concentró para producir 2-(piperidin-4-il)propan-2-amina (140 mg, 0,98 mmoles, 92 % de rendimiento).
Ejemplo S Intermedio

(S)-(1-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-2-¡l)metanol
Paso A: Se agregó (S)-pirrolidin-2-ilmetanol l (0,79 g, 7,8 mmoles) a una solución de 3-cloro-2-fluoro-4-yodopiridina (1,0 g, 3,9 mmoles) en DMSO (4 ml) y la reacción se calentó a 80 °C durante 4 horas. La reacción se vertió en agua y se extrajo con MTBE. Las fases orgánicas se lavaron con agua, salmuera, se secaron sobre MgSO4 y se concentraron in vacuo para dar (S)-(1-(3-cloro-4-yodopiridin-2-il)pirrolidin-2-il)metanol (1,2 g, 91 % de rendimiento). m/z (esi/APCI) M+1 = 339,0.
Paso B: Se agregaron xantfos (0,21 g, 0,35 mmoles), etantioato de potasio (0,40 g, 3,5 mmoles) y Pd2(dba)3 (0,19 g, 0,21 mmoles) a una solución de (S)-(1-(3-cloro-4-yodopiridin-2-il)pirrolidin-2-il)metanol (1,2 g, 3,5 mmoles) en dioxanos (15 ml). La reacción se roció con argón durante 10 minutos y se calentó a 80 °C durante 2 horas. La reacción se filtró a través de papel GHF y se concentró in vacuo. El residuo se sometió a cromatografía utilizando EtOAc/DCM al 0 100 % como eluyente para dar (S)-S-(3-cloro-2-(2-(hidroximetil)pirrolidin-1-il)piridin-4-il)etantioato (0,90 g, 89 % de rendimiento). m/z (esi/APCI) M+1 = 287,1.
Paso C: Se agregó hidróxido de amonio (0,70 ml, 13 mmoles) a una solución de (S)-S-(3-cloro-2-(2-(hidroximetil)pirrolidin-1-il)piridin-4-il)etantioato (0,90 g, 3,1 mmoles) en THF (15 ml) y la reacción se agitó a temperatura ambiente durante 2 horas. La reacción se concentró in vacuo y se agregó agua al residuo. El agua se acidificó aproximadamente a un pH de 5 utilizando HCl 1 N. La capa acuosa se extrajo con EtOAc. Las fases orgánicas se separaron, se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron in vacuo para dar (S)-(1-(3-cloro-4-mercaptopiridin-2-il)pirrolidin-2-il)metanol (0,44g, 57 %). m/z (esi/APCI) M+1 = 245,1.
Ejemplo T Intermedio

1,1-dióxido de 4-(3-cloro-4-mercaptopir¡d¡n-2-¡l)t¡omorfol¡na
Se preparó 1,1-d¡óx¡do de 4-(3-cloro-4-mercaptop¡n d¡n-2-¡l)t¡oiT ioif:ol¡na de acuerdo con el Ejemplo Intermed¡o S, sust¡tuyendo el 1,1 d¡óx¡do de t¡omorfol¡na por (S)-pirrolidin-2-ilTetanol en la Paso A.
Ejemplo U Intermedio

1-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡per¡d¡n-4-ol
Paso A: Se agregó p¡per¡d¡n-4-ol (0,39 g, 3,9 mmoles) a una soluc¡ón de 3-cloro-2-fluoro-4-yodop¡r¡d¡na (0,50 g, 1,9 mmoles) en DMSO (2 ml) y la reacc¡ón se calentó a 80 °C durante la noche. La reacc¡ón se vert¡ó en agua y se extrajo con EtOAc. Las fases orgán¡cas se lavaron con agua (3 X), salmuera, se secaron sobre MgSO4 y se concentraron ¡n vacuo para dar 1-(3-cloro-4-yodop¡r¡d¡n-2-¡l)p¡pend¡n-4-ol. m/z (es¡/APCI) M+1 = 339,0.
Paso B: Se agregaron xantfos (0,062 g, 0,11 mmoles), base de Hun¡g (0,37 ml, 2,1 mmoles), 3-mercaptopropanoato de 2-et¡lhex¡lo (0,23 g, 1,1 mmoles) y Pd2(dba)3 (0,058 g, 0,064 mmoles) a una soluc¡ón de 1-(3-cloro-4-yodop¡r¡d¡n-2-¡l)p¡per¡d¡n-4-ol (0,36 g, 1,1 mmoles) en d¡oxanos (2 ml). La reacc¡ón se roc¡ó con argón durante 10 m¡nutos y se calentó a 100 °C durante 2 horas. La reacc¡ón se enfr¡ó, se f¡ltró a través de papel GHF y se concentró ¡n vacuo. Se agregaron THF (5 ml) y etanolato de sod¡o (0,34 g, 1,1 mmoles) al res¡duo y la reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se d¡luyó con DCM, se ag¡tó 10 m¡nutos y se concentró ¡n vacuo. El mater¡al se extrajo en agua, se h¡zo bás¡co con NaOH y la capa acuosa se lavó con EtOAc. Las capas se separaron. La capa acuosa se ac¡d¡f¡có con HCl 1 M y se extrajo dos veces con EtOAc. Las fases orgán¡cas comb¡nadas se lavaron con salmuera, se secaron sobre MgSO4 y concentraron ¡n vacuo para dar 1-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡pend¡n-4-ol (0,10 g, 38 %). m/z (es¡/APCI) M+1 = 245,1.
Ejemplo V Intermedio

1-(4-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)etan-1-ona
Se preparó 1-(4-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)etan-1-ona de acuerdo con el Ejemplo Intermed¡o U, sust¡tuyendo 1-(p¡peraz¡n-1-¡l)etan-1-ona por p¡per¡d¡n-4-ol en la Paso A.
E je m p lo W In te rm e d io

3-cloro-2-morfol¡nop¡r¡din-4-t¡olato de sodio
Paso A: Se agregó morfolina (0,34 g, 3,9 mmoles) a una solución de 3-cloro-2-fluoro-4-yodopirid¡na (0,50 g, 1,9 mmoles) en DMSO (2 ml) y la reacción se calentó a 80 °C durante la noche. La reacción se vertió en agua y se extrajo con EtOAc. Las capas se separaron. Las fases orgánicas se lavaron con agua (3 X), salmuera, se secaron sobre MgSO4 y se concentraron in vacuo para dar 1-(3-cloro-4-yodopir¡d¡n-2-¡l)p¡perid¡n-4-ol (0,5g, 79 %). m/z (esi/APCI) M+1 = 325,0.
Paso B: Se agregaron Xantfos (0,089 g, 0,15 mmoles), base de Hunig (0,54 ml, 3,1 mmoles), 3-mercaptopropanoato de 2-etilhexilo (0,34 g, 1,5 mmoles) y Pd2(dba)3 (0,085 g, 0,092 mmoles) a una solución de 4-(3-cloro-4-yodopirid¡n-2-¡l)morfolina (0,50 g, 1,5 mmoles) en dioxanos (2 ml). La reacción se roció con argón durante 10 minutos y se calentó a 100 °C durante 2 horas. La reacción se enfrió, se filtró a través de papel GHF y se concentró in vacuo. Se agregaron THF (5 ml) y etanolato de sodio (0,50 g, 1,5 mmoles) al residuo y la reacción se agitó a temperatura ambiente durante una hora. La reacción después se diluyó con DCM, se agitó 10 minutos y se concentró in vacuo. El residuo se dividió de nuevo en DCM. La suspensión que se formó se filtró y el sólido se lavó con DCM para dar el producto sin purificar de 3-cloro-2-morfolinopir¡d¡n-4-t¡olato de sodio. m/z (esi/APCI) M+1 = 231,1.
Ejemplo X Intermedio

1-(3-cloro-4-mercaptopir¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol
Se preparó 1-(3-cloro-4-mercaptopir¡d¡n-2-¡l)p¡rrolid¡n-3-ol de acuerdo con el Ejemplo Intermedio W, sustituyendo 3-pirrolidinol (0,32 ml, 3,9 mmoles) por morfolina en la Paso A. m/z (esi/APCI) M+1 = 231,1.
Ejemplo Y Intermedio

3-cloro-2-(h¡droximet¡l)p¡r¡d¡n-4-t¡olato de sodio
Paso A: Se agregaron xantfos (0,13 g, 0,22 mmoles), Pd2(dba)3 (0,12 g, 0,13 mmoles), N-etil-N-isopropilpropan-2-amina (0,79 ml, 4,4 mmoles) y 3-mercaptopropanoato de 2-etilhexilo (0,48 g, 2,2 mmoles) a una solución de (3,4-dicloropir¡d¡n-2-¡l)metanol (0,39 g, 2,2 mmoles) en dioxanos (4 ml). La reacción se roció con argón durante 10 minutos y se calentó a 100 °C durante la noche. La reacción se enfrió, se filtró a través de papel GHF y se concentró in vacuo. El residuo se sometió a cromatografía utilizando EtOAc/Hexanos al 20-80 % como eluyente para dar 3-((3-cloro-2-(hidrox¡met¡l)p¡r¡d¡n-4-¡l)tio)propanoato de 2-etilhexilo (0,43 g, 1,2 mmoles, 55 % de rendimiento). m/z (esi/APCI) M+1 = 360,2.
Paso B: Se agregó etanolato de sodio (0,426 g, 1,31 mmoles) a una solución de 3-((3-cloro-2-(hidrox¡met¡l)pir¡d¡n-4-il)tio)propanoato de 2-etilhexilo (0,43 g, 1,19 mmoles) en THF (10 ml) y la reacción se agitó a temperatura ambiente durante una hora. La reacción se concentró y los sólidos se suspendieron en MTBE y se filtraron. Los sólidos se
lavaron además con MTBE para dar 3-cloro-2-(hidroximetil)piridin-4-tiolato de sodio (0,158 g, 66,9 % de rendimiento). m/z (esi/APCI) M+1 = 176,1.
Ejemplo Z Intermedio

(S)-3-cloro-2-(3-(h¡drox¡met¡l)p¡rrolid¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sodio
Paso A: Se agregó (S)-pirrolidin-3-ilmetanol (0,39 g, 3,9 mmoles) a una solución de 3-cloro-2-fluoro-4-yodopiridina (0,50 g, 1,9 mmoles) en DMSO (2 ml) y la reacción se calentó a 80 °C durante la noche. La reacción se vertió en agua y se extrajo con MTBE. Las fases orgánicas se lavaron con agua, salmuera, se secaron sobre MgSO4 y se concentraron in vacuo para dar (S)-(1-(3-cloro-4-yodopiridin-2-il)pirrolidin-3-il)metanol (0,51 g, 78 % de rendimiento). m/z (esi/APCI) M+1 = 339,0.
Paso B: Se agregaron xantfos (0,087 g, 0,15 mmoles), Pd2(dba)3 (0,083 g, 0,090 mmoles) y 3-mercaptopropanoato de 2-etilhexilo (0,33 g, 1,5 mmoles) a una solución de (S)-(1-(3-cloro-4-yodopiridin-2-il)pirrolidin-3-il)metanol (0,51 g, 1,5 mmoles) en dioxanos (4 ml). La reacción se roció con argón durante 10 minutos y se calentó a 100 °C durante 2 horas. La reacción se enfrió, se filtró a través de papel GHF y se concentró in vacuo. El residuo se sometió a cromatografía utilizando EtOAc/Hexanos al 20-80 % como eluyente para dar 3-((3-cloro-2-((S)-3-(hidroximetil)pirrolidin-1-il)piridin-4-il)tio)propanoato de 2-etilhexilo (0,52 g, 80 % de rendimiento). m/z (esi/APCI) M+1 = 429,2.
Paso C: Se agregó etanolato de sodio (0,39 g, 1,2 mmoles) a una solución de 3-((3-cloro-2-((S)-3-(hidroximetil)pirrolidin-1-il)piridin-4-il)tio)propanoato de 2-etilhexilo (0,52 g, 1,2 mmoles) en THF (10 ml) y la reacción se agitó a temperatura ambiente durante una hora. La reacción se filtró y los sólidos se lavaron con MTBE para dar (S)-3-cloro-2-(3-(hidroximetil)pirrolidin-1-il)piridin-4-tiolato de sodio (0,10 g, 34 % de rendimiento). m/z (esi/APCI) M+1 = 245,1.
Ejemplo AB Intermedio

(S)-3-cloro-2-(3-h¡drox¡p¡rrolid¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sodio
Se preparó (S)-3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-tiolato de sodio de acuerdo con el Ejemplo Intermedio Z, sustituyendo (S)-(-)-3-hidroxipirrolidina por (S)-pirrolidin-3-ilmetanol en la Paso A. m/z (esi/APCI) M+1 = 231,1.
Ejemplo AC Intermedio

Se preparó 3-cloro-2-metilpiridin-4-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 3,4-didoro-2-metilpiridina por 3-doro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 159,6.
Ejemplo AD Intermedio

Se preparó 2-aminopiridin-3-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 3-bromo-2-piridinamina por 3-cloro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 127,1.
Ejemplo AE Intermedio

6-cloro-2-met¡lp¡r¡d¡n-3-t¡ol
Se preparó 6-cloro-2-metilpiridin-3-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 3-bromo-6-cloro-2-metilpiridina por 3-doro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 160,1.
Ejemplo AF Intermedio

Se preparó 6-amino-2-doropiridin-3-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 2-amino-5-bromo-6-cloropiridina por 3-doro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 161,1.
Ejemplo AG Intermedio

Se preparó 2-cloro-6-metilpiridin-3-tiol de acuerdo con el Ejemplo B Intermedio, sustituyendo 3-bromo-2-cloro-6-metilpiridina por 3-doro-4-yodopiridin-2-amina en la Paso A. m/z (esi/APCI) M+1 = 160,1.
Ejemplo AH Intermedio

(E)-(1-(3-cloro-4-mercaptopindin-2-il)pirrolidin-3-il)metanol
Se preparó (R)-(1-(3-cloro-4-mercaptopindin-2-il)pirrolidin-3-il)metanol de acuerdo con el Ejemplo E Intermedio, sustituyendo (R)-3-pirrolidin-3-il-metanol por pirrolidina en la Paso A. m/z (esi/APCI) M+1 = 245,1.
Ejemplo AI Intermedio

(E)-1-(3-cloro-4-mercaptopindin-2-il)pirrolidin-3-ol
Se preparó (R)-(1-(3-cloro-4-mercaptopiridin-2-il)pirrolidin-3-il)metanol de acuerdo con el Ejemplo E Intermedio, sustituyendo (R)-(+)-3-pirrolidinol por pirrolidina en la Paso A. m/z (esi/APCI) M+1 = 231,1.
Ejemplo AJ Intermedio

2,2.2-trifluoroacetato de (E)-N-((S)-1,3-dihidroespiro[inden-2,4'-pipendinl-1-il)-2-metilpropan-2-sulfinamida
Paso A: Se colocaron 1-oxo-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-butilo (1,0 g, 3,32 mmoles) y (R)-2-metilpropan-2-sulfinamida (1,21 g, 10,0 mmoles) en THF (10 ml). Se agregó Ti(OEt)4 (4,87 ml, 23,23 mmoles) y la reacción se calentó a 65 °C durante 2 días. La reacción se enfrió y se agregó EtOAc, seguido por agua. Los sólidos se filtraron y las capas se separaron. La capa orgánica se secó, filtró y concentró para proporcionar el material sin purificar que se utilizó en el siguiente paso.
Paso B: Se colocó (R,E)-1-((terf-but¡lsulf¡n¡l)¡m¡no)-1,3-d¡h¡droesp¡ro[¡nden-2,4’-p¡per¡d¡n]-1’-carbox¡lato de ferf-butilo (1,32 g, 3,26 mmoles) en Th F (15 ml) y se enfrió a -45 °C. Se agregó NaBH4 (0,19 g, 4,89 mmoles) y la reacción se dejó calentar lentamente a temperatura ambiente y se agitó durante 18 horas. Se agregó agua y la mezcla se extrajo con DCM (3 X 25 ml). Los extractos se combinaron y concentraron y el residuo resultante se purificó mediante gel de sílice (MeOH en DCM al 0-5 % con NH4OH al 2 %). El primer pico de elución se recolectó para proporcionar (S)-1
(((R)-ferf-but¡lsulf¡n¡l)am¡no)-1,3-d¡h¡droesp¡ro[¡nden-2,4’-piper¡d¡n]-1’-carbox¡lato de ferf-butilo (500 mg, 1,23 mimóles, 38 % de rend¡m¡ento).
Paso C: Se colocó (S)-1-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-1,3-d¡h¡droesp¡ro[¡nden-2,4’-piper¡d¡n]-1’-carbox¡lato de ferf-butilo (500 mg, 1,23 mmoles) en DCM (1 ml) y se enfr¡ó a 0 °C. Se agregó TFA (1 ml) y la reacc¡ón se ag¡tó durante 45 m¡nutos. La reacc¡ón se concentró y el mater¡al se ut¡l¡zó tal como está. m/z (es¡/APCl) M+1 = 307,1.
Ejemplo AK Intermedio

6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l 4-nitrobencensulfonato
Se disolvió parcialmente 6-cloropir¡do[2,3-b]p¡raz¡n-2-ol (20 g, 110 mmoles) en DMF (200 ml) y se agregó trietilamina (18,4 ml, 132 mmoles). Después se agregó cloruro de 4-nitrobencensulfonilo (24,4 g, 110 mmoles). Después de 20 minutos, la reacción se vertió en agua (1,5 L), se agitó vigorosamente durante 15 minutos, se filtró y secó en un horno al vacío para producir 4-nitrobencensulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (37,4 g, 102 mmoles, 93 % de rendimiento) como un sólido. Espectro de masas: m/z = 367,0 (M+H).
Ejemplo AL Intermedio

(ft)-1’-(((ft)-ferf-but¡lsulf¡n¡l)am¡no)-1’,3’-d¡h¡droesp¡ro[azet¡d¡n-3,2’-¡nden]-1-carbox¡lato de ferf-butilo
Paso A: Se disolvió 3-cianoazet¡d¡n-1-carbox¡lato de ferf-butilo (1 g, 5,49 mmoles) en THF (15 ml) y se enfrió a -78 °C. Se agregó por goteo N-litiohexametildisilazano (6,86 ml, 6,86 mmoles) a la solución. Se agregó una solución de 1-(bromometil)-2-yodobenceno (1,79 g, 6,04 mmoles) en THF (2 ml) a la mezcla mediante una jeringa bajo nitrógeno. Después de 3 horas, la reacción se extinguió con NH4Cl saturado y se extrajo con EtOAc (2 x 15 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron. El residuo resultante se purificó utilizando una columna de gel de sílice de 80 g y EtOAc/hexanos al 10-70 % como el sistema de solvente que produce 3-ciano-3-(2-yodobencil)azet¡d¡n-1-carbox¡lato de ferf-butilo (1,7 g, 4,27 mmoles, 78 % de rendimiento) como un aceite. m/z (esi/APCI) M+1 =399,1.
Paso B: Se disolvió 3-ciano-3-(2-yodobenc¡l)azet¡d¡n-1-carbox¡lato de ferf-butilo (1,6 g, 4,0 mmoles) en THF (40 ml) y la solución se enfrió a -78 °C. Se agregó una solución de n-butil-litio (1,9 ml, 4,8 mmoles) en hexano a la reacción bajo nitrógeno por goteo durante 10 minutos. La reacción se mantuvo a -78 °C durante 2 horas, se llevó a temperatura ambiente y se extinguió con una solución saturada de NH4CL El producto se extrajo con EtOAc y la capa orgánica se secó y evaporó. El residuo resultante se purificó utilizando una columna de gel de sílice de 40 g (EtOAc/hexanos al 10-60 %) produciendo 1’-oxo-1’,3’-d¡h¡droespiro[azet¡d¡n-3,2’-¡nden]-1-carbox¡lato de ferf-butilo (0,73 g, 2,7 mmoles, 66 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =274,1.
Paso C: Se suspendió 1’-oxo-1’,3’-d¡h¡droespiro[azet¡d¡n-3,2’-¡nden]-1-carbox¡lato de ferf-butilo (0,72 g, 2,63 mmoles) en tetraetoxititanio (2,50 ml, 18,4 mmoles) y (ft)-2-met¡lpropan-2-sulfinam¡da (0,958 g, 7,90 mmoles) se agregó a la mezcla. La reacción se calentó en un baño de aceite hasta 90 °C durante 18 horas. La mezcla de reacción se enfrió a temperatura ambiente y se diluyó con EtOAc (20 ml). Se agregó salmuera (50 ml) a la mezcla y la reacción se agitó vigorosamente durante 10 minutos. El sólido formado se filtró a través de una almohadilla de Celite® y la capa orgánica se separó. La capa acuosa se extrajo una vez con EtOAc (30 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron para dar un residuo. El residuo resultante se purificó utilizando una columna de gel de sílice de 40 g (EtOAc/hexanos al 20-80 %) produciendo (R,E)-1’-((ferf-but¡lsulfin¡l)¡m¡no)-1’,3’-d¡h¡droespiro[azet¡d¡n-3,2’-¡nden]-1-carbox¡lato de ferf-butilo (0,82 g, 2,18 mmoles, 83 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =377,2.
Paso D: Se disolvió (R,E)-1’-((tert-but¡lsulf¡n¡l)¡m¡no)-1’,3’-dih¡droesp¡ro[azet¡d¡n-3,2’-¡nden]-1-carbox¡lato de ferf-butilo (0,10 g, 0,27 mmoles) en THF (3 ml) y se enfrió a -78 °C. Se agregó por porciones NaBH4 (0,030 g, 0,80 mmoles) a la mezcla y la reacción se calentó hasta temperatura ambiente durante 3 horas. La reacción se extinguió con NH4Cl saturado y se extrajo con EtOAc (2 x 10 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron. El residuo resultante se purificó utilizando una columna de gel de sílice de 12 g (mezcla de EtOAc/hexanos al 10-80 % ) recolectando el 1er pico para proporcionar (R)-1’-(((R)-tert-but¡lsulf¡n¡l)am¡no)-1’,3’-dih¡droesp¡ro[azet¡d¡n-3,2’-¡nden]-1-carboxilato de ferf-butilo (0,032 g, 0,085 mmoles, 32 % de rendimiento) y el 2o pico para dar (S)-1’-(((R)-tert-but¡lsulf¡n¡l)am¡no)-1’,3’-dih¡droesp¡ro[azet¡d¡n-3,2’-¡nden]-1-carbox¡lato de ferf-butilo (0,025 g, 0,066 mmoles, 25 % de rendimiento). m/z (esi/APCI) M+1 =379,2.
Ejemplo AM Intermedio

(S)-1'-(((R)-tert-but¡lsulf¡n¡l)am¡no)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden1-1-carbox¡lato de ferf-butilo
Este compuesto se preparó utilizando procedimientos similares a los descritos según el Ejemplo AL Intermedio, recolectando el segundo pico eludido en la Paso D. m/z (esi/APCI) M+1 =379,2.
Ejemplo AN Intermedio

(S)-8-(((R)-tert-but¡lsulf¡n¡l)am¡no)esp¡ro[b¡c¡clo[4.2.01octan-7.4'-p¡per¡d¡n1-1.3.5-tr¡eno-1'-carbox¡lato de ferf-butilo
Paso A: Se disolvió 2-(2-yodofenil)acetonitrilo (1,3 ml, 10 mmoles) en DMF (20 ml) y la solución se enfrió a 0 °C. Se agregó una suspensión de NaH (1,0 g, 26 mmoles) al 60 % en aceite mineral a la solución en porciones y la mezcla se calentó a 60 °C durante 1,5 horas. Se agregó bis(2-cloroetil)carbamato de ferf-butilo (3,0 g, 12 mmoles) a la reacción y la mezcla se agitó a 60 °C durante 2 horas. La reacción se enfrió a temperatura ambiente, se agregó salmuera (25 ml) y la mezcla se extrajo con EtOAc (3 X 100 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron para dar un residuo, el cual se purificó con cromatografía instantánea (EtOAc/hex 5-70 %). El segundo pico eludido se recolectó para proporcionar 4-(2-yodofenil)-4-c¡anop¡perid¡n-1-carbox¡lato de ferf-butilo (1,8 g, 4,9 mmoles, 48 % de rendimiento) como un aceite. m/z (esi/APCI) M+1 =413.
Paso B: Se agregó una solución 1 M de hidruro de diisobutilaluminio (“DIBAL-H”; 2,4 ml, 2,4 mmoles) en DCM a una solución de 4-ciano-4-(2-yodofen¡l)p¡perid¡n-1-carbox¡lato de ferf-butilo (0,9 g, 2,2 mmoles) enfriada a -78 °C. La reacción se agitó a -78 °C, después se calentó a temperatura ambiente y se agitó durante la noche. Se agregó DIBAL-H adicional y la reacción se agitó durante 2 horas. La reacción se extinguió con una solución de HCl 2 M y la mezcla se extrajo con EtOAc (3 x 15 ml). Las capas orgánicas se combinaron, se lavaron con una solución saturada de NaHCO3, se secaron y evaporaron para dar un residuo el cual se purificó utilizando una columna de gel de sílice de 40 g (EtOAc/hexanos al 5-60 %) para proporcionar 4-formil-4-(2-yodofen¡l)p¡perid¡n-1-carbox¡lato de ferf-butilo (0,35 g, 0,84 mmoles, 39 % de rendimiento) m/z (esi/APCI) M+1 =416,0.
Paso C: Se disolvió 4-formil-4-(2-yodofen¡l)p¡perid¡n-1-carbox¡lato de ferf-butilo (0,088 g, 0,21 mmoles) en THF (3 ml) y se agregaron (R)-2-metilpropan-2-sulfinamida (0,027 g, 0,22 mmoles) y tetraetoxititanio (0,058 ml, 0,42 mmoles) a la mezcla. La reacción se agitó a 60 °C durante la noche. Se agregó salmuera (10 ml) y la mezcla se agitó vigorosamente durante 5 minutos. Se agregó EtOAc (10 ml) a la mezcla y el precipitado formado se retiró mediante
filtración. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (10 ml). Las capas orgánicas combinadas se secaron y evaporaron. El residuo resultante se purificó utilizando una columna de gel de sílice (12 gm) (EtOAc/hexanos al 0-60 %) produciendo (R,Z)-4-(((tert-butilsulfinil)imino)metil)-4-(2-yodofenil)piperidin-1-carboxilato de ferf-butilo (0,067 g, 0,13 mmoles, 61,0 % de rendimiento) como un aceite. m/z (esi/APCI) M+1 =519,1.
Paso D: Se disolvió (R,Z)-4-(((tert-butilsulfinil)imino)metil)-4-(2-yodofenil)piperidin-1-carboxilato de ferf-butilo (0,067 g, 0,13 mmoles) en THF (2 ml) y se enfrió a -78 °C. Se agregó una solución 1,4 M de sec-butil-litio (0,18 ml, 0,26 mmoles) en ciclohexano a la solución mediante una jeringa. La mezcla se agitó a -78 °C durante 2 horas y se extinguió con NH4Cl saturado. La mezcla se extrajo con EtOAc (2 x 5 ml) y las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron. El residuo resultante se purificó utilizando una columna de gel de sílice de 12 g (EtOAc/hexanos al 10-70 %) recolectando el 1er pico que produjo (S)-8-(((R)-tertbutilsulfinil)amino)espiro[biciclo[4.2.0]octan-7,4'-piperidin]-1,3,5-trieno-1'-carboxilato de ferf-butilo (0,021 g, 71 %) como un sólido. m/z (esi/APCI) M+1 =393,1.
Ejemplo AO Intermedio

(R)-8-(((R)-tert-but¡lsulf¡n¡l)am¡no)esp¡ro[b¡c¡clo[4.2.01octan-7.4'-p¡per¡d¡n1-1.3.5-tr¡eno-1'-carboxilato de ferf-butilo
Se preparó (R)-8-(((R)-tert-but¡lsulf¡n¡l)am¡no)esp¡ro[b¡c¡clo[4.2.0]octan-7,4'-p¡perid¡n]-1,3,5-tr¡eno-1'-carbox¡lato de ferf-butilo siguiendo el Ejemplo AN Intermedio al recolectar el 2o pico de la Paso D. m/z (esi/APCI) M+1 =393,1.
Ejemplo AP Intermedio

Sal de tri-HCl de (S)-5,7-dihidroespiro[ciclopenta[¿)1piridin-6,4'-piperidin1-5-amina
Paso A: Se disolvió 2-cloropiridina (458 ml, 4,84 mmoles) en THF seco (15 ml) y la solución se enfrió a -70 °C en IPNbaño de hielo seco. Se agregó por goteo diisopropilamida de litio (“LDA”; 2,75 ml, 5,50 mmoles) a la mezcla y la reacción se calentó a -60 °C y se agitó a esa temperatura durante 1,5 horas. Se agregó 4-formil-4-metilpiperidin-1-carboxilato de ferf-butilo (1 g, 4,40 mmoles) en THF (3 ml) a la mezcla y se agitó a -60 °C durante una hora. La reacción se extinguió con agua y se dividió entre EtOAc y agua. La capa orgánica se separó, se secó y concentró. El residuo resultante se purificó utilizando una columna de gel de sílice de 40 g (EtOAc/hexanos al 10-80 %) produciendo 4-((2-cloropiridin-3-il)(hidroxi)metil)-4-metilpiperidin-1-carboxilato de ferf-butilo (1,06 g, 3,11 mmoles, 71 % de rendimiento).
Paso B: Se disolvió 4-((2-cloropiridin-3-il)(hidroxi)metil)-4-metilpiperidin-1-carboxilato de ferf-butilo (1,06 g, 3,11 mmoles) en DCM (8 ml) y se agregó DMP (2,64 g, 6,22 mmoles) a la solución. La mezcla se agitó a temperatura ambiente durante una hora y se extinguió con una solución de bisulfito de sodio al 10 %. La capa orgánica se separó y lavó con una solución saturada de bicarbonato de sodio y salmuera, se secó y evaporó. El residuo resultante se purificó utilizando cromatografía instantánea en gel de sílice (EtOAc/hexanos al 10-100 %) proporcionando 4-(2-cloronicotinoil)-4-metilpiperidin-1-carboxilato de ferf-butilo (0,31 g, 0,92 mmoles, 29 % de rendimiento) como un aceite.
Paso C: Se disolvió 4-(2-cloronicotinoil)-4-metilpiperidin-1-carboxilato de ferf-butilo (8,13 g, 24,0 mmoles) en mesitileno (70 ml) en un tubo de presión. Se agregaron tetrafluoroborato de triciclohexilfosfonio (0,884 g, 2,40 mmoles), diacetoxipaladio (0,269 g, 1,20 mmoles), ácido piválico (0,735 g, 7,20 mmoles) y carbonato de cesio (15,6 g, 48,0 mmoles) a la mezcla de reacción. Se burbujeó gas nitrógeno durante 5 minutos en la mezcla de reacción y el tubo se selló y calentó a 140 °C durante 72 horas. La reacción se enfrió a temperatura ambiente y se diluyó con EtOAc (50 ml). La mezcla se filtró utilizando una almohadilla de Celite® y se lavó varias veces con EtOAc. El filtrado se evaporó
bajo vacío para dar un residuo. El residuo se purificó utilizando una columna de sílice de 330 g (EtOAc/hexanos al 20 80 %) para dar 5-oxo-5,7-dihidroespiro[ciclopenta[£)]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo (2,23 g, 7,37 mmoles, 31 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =303,1.
Paso D: Se suspendió 5-oxo-5,7-dihidroespiro[ciclopenta[6]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo (2,23 g, 7,375 mmoles) en tetraetoxititanio (6,98 ml, 51,62 mmoles) y se agregó (R)-2-metilpropan-2-sulfinamida (2,68 g, 22,12 mmoles) a la mezcla. La reacción se calentó a 90 °C y se agitó durante 18 horas. La reacción se enfrió a temperatura ambiente y se agregó EtOAc (250 ml), seguido por salmuera (200 ml). La mezcla se agitó vigorosamente durante 10 minutos y después se filtró para retirar el precipitado formado. La capa de EtOAc se separó y lavó dos veces con salmuera, se secó y evaporó. El residuo resultante se purificó utilizando una columna de gel de sílice de 120 g (EtOAc/Hexanos al 10-100 %). La recolección del segundo pico eludido proporcionó (R,Z)-5-((tert-butilsulfinil)imino)-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo (2,8 g, 6,90 mmoles, 94 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =406,2.
Paso E: Se disolvió (R,Z)-5-((tert-butilsulfinil)imino)-5,7-dihidroespiro[ciclopenta[£)]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo (0,15 g, 0,37 mmoles) en THF (2 ml) en un vial. La solución se enfrió a -78 °C y se agregó en una porción L¡BH4 (0,28 ml, 0,55 mmoles) 2 M en THF. La reacción se mantuvo a -78 °C durante 1 hora. La reacción se calentó lentamente a temperatura ambiente y se agitó durante 18 horas. La reacción se extinguió con NH4Cl saturado seguido por la extracción con EtOAc (3 X 10 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron. El residuo resultante se purificó utilizando cromatografía de columna que utiliza una columna de sílice de 24 g (EtOAc/Hexanos al 10-80 %) dio (S)-5-(((R)-ferf-butilsulfinil)amino)-5,7-dihidroespiro[ciclopenta[£)]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo (0,11 g, 0,27 mmoles, 73 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =408,2.
Paso F: Se disolvió (S)-5-(((R)-ferf-butilsulfinil)amino)-5,7-dihidroespiro[ciclopenta[£)]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo (3,45 g, 8,46 mmoles) en DCM (10 ml) y a esa solución (4 ml) se agregó una solución de HCl 4 M en dioxano. La mezcla de reacción se agitó a temperatura ambiente durante la noche. Se agregó éter (100 ml) a la mezcla y el precipitado formado se filtró para dar (S)-5,7-dihidroespiro[ciclopenta[£)]piridin-6,4'-piperidin]-5-amina (1,62 g, 7,97 mmoles, 94 % de rendimiento) como un sólido como la sal de tri-HCl. m/z (esi/APCI) M+1 =204,1.
Ejemplo AQ Intermedio

(R)-5-(((R)-ferf-butilsulfinil)amino)-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo
Se preparo (R)-5-(((R)-ferf-butilsulfinil)amino)-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de ferf-butilo de acuerdo con el procedimiento descrito bajo el Ejemplo AP Intermedio, que recolectó el 2o pico eludido en la Paso E. m/z (esi/APCI) M+1 =408,2.
Ejemplo AR Intermedio

(S)-(1'-(6-mercaptopirido[2,3-;b]pirazin-2-il)-1,3-dihidroespironnden-2,4'-piperidin]-1-il)carbamato de ferf-butilo
Paso A: Se suspendió diclorhidrato de (S)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina (3 g, 10,9 mmoles) en 1,4-dioxano (36,3 ml, 10,9 mmoles) y se agregó trietilamina (4,56 ml, 32,7 mmoles) a la mezcla. Después de 20 minutos de agitar a temperatura ambiente, se agregó trifluorometansulfonato de cloropirido[2,3-b]pirazin-2-ilo (3,42 g, 10,9
mimóles) y la mezcla se agitó a temperatura ambiente durante 2 horas. Se agregaron 3-mercaptopropanoato de metilo (1,31 g, 10,9 mmoles), Pd(OAc)2 (0,245 g, 1,09 mmoles) y xantfos (1,26 g, 2,18 mmoles) a la reacción y se burbujeó nitrógeno en la mezcla de reacción durante 2 minutos. La reacción se calentó a 90 °C durante la noche y se enfrió a temperatura ambiente, se mezcló con EtOAc (30 ml) y se filtró. El filtrado se evaporó y el residuo se purificó con una columna de gel de sílice (80 g) (MeOH/DCM al 2-20 %) para proporcionar (S)-3-((2-(1-amino-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)propanoato de metilo (1,6 g, 3,56 mmoles, 33 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =450,2.
Paso B: Se disolvió (S)-3-((2-(1-amino-1,3-dihidroespiro[inden-2,4'-piperidin]-T-il)pirido[2,3-b]pirazin-6-il)tio)propanoato de metilo (1,6 g, 3,6 mmoles) en DCM (25 ml). Se agregaron trietilamina (“TEA”; 0,99 ml, 7,1 mmoles), 4-dimetilaminopiridina (“DMAP”; 0,043 g, 0,36 mmoles) y anhídrido de BOC (0,91 ml, 3,9 mmoles) a la mezcla y se agitaron a temperatura ambiente durante la noche. La reacción se extinguió con agua (30 ml) y se extrajo con DCM (30 ml). Las capas orgánicas combinadas se secaron y evaporaron. El residuo resultante se purificó utilizando una columna de gel de sílice (80 g) (EtOAc/hexanos 10-80 %) produciendo (S)-3-((2-(1-((ferf-butoxicarbonil)amino)-1,3-dihidroespiro[inden-2,4'-piperidin]-T-il)pirido[2,3-£)]pirazin-6-il)tio)propanoato de metilo (0,48 g, 0,87 mmoles, 25 % de rendimiento) como un sólido. m/z (esi/APCI) M+1 =550,2.
Paso C: Se disolvió (S)-3-((2-(1-((ferf-butoxicarbonil)amino)-1,3-dihidroespiro[inden-2,4'-piperidin]-T-il)pirido[2,3-6]pirazin-6-il)tio)propanoato de metilo (0,48 g, 0,87 mmoles) en una solución de THF (10 ml) y etanolato de sodio (0,65 ml, 1,75 mmoles) al 21 % en etanol y se agregó lentamente a la solución. La reacción se agitó a temperatura ambiente durante una hora y se extinguió con NH4Cl saturado (10 ml). La capa acuosa se extrajo con EtOAc (2 x 15 ml) y las capas orgánicas combinadas se lavaron con salmuera, se secaron y evaporaron. El residuo resultante se purificó utilizando una columna de gel de sílice (40 g) (EtOAc/hexanos 10-100 %) produciendo (S)-(1'-(6-mercaptopirido[2,3-6]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)carbamato de ferf-butilo (0,321 g, 0,69 mmoles, 79 % de rendimiento) como un sólido. m/z (esi/APCl) M+1 =464,2.
Ejemplo AS Intermedio

3-cloro-2-((2-h¡drox¡et¡l)am¡no)p¡r¡d¡n-4-t¡olato de sodio
Paso A: A una solución agitada de 3-cloro-2-fluoro-4-yodopiridina (1,0 g, 3,89 mmoles) en DMSO (5 ml) se agregó 2-aminoetan-1-ol (0,47 ml, 7,78 mmoles) y se agitó a 70 °C durante 16 horas. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se secó (Na2SO4), se concentró y el residuo resultante se purificó mediante cromatografía de columna en gel de sílice (EtOAc-Hexanos al 40 %) para producir 2-((3-cloro-4-yodopiridin-2-il)amino)etan-1-ol (820 mg, 70 % de rendimiento) como un sólido. m/z (esi) M+1 = 298,8.
Paso B: A una solución agitada de 2-((3-cloro-4-yodopiridin-2-il)amino)etan-1-ol (500 mg, 1,67 mmoles) y 3-mercaptopropanoato de metilo (0,2 ml, 1,84 mmoles) en dioxano (5 ml) se agregó DIPEA (0,6 ml, 3,35 mmoles) y se desgasificó con argón durante 10 minutos. Se agregaron Xantfos (48 mg, 0,08 mmoles) y Pd(OAc)2 (23 mg, 0,10 mmoles) y se desgasificaron durante otros 10 minutos. La mezcla de reacción se agitó en un baño de aceite precalentado en un tubo sellado a 100 °C durante 4 horas. La mezcla de reacción se filtró a través de una almohadilla de Celite® y se lavó con acetato de etilo. Se evaporó el disolvente y el material sin purificar se purificó mediante cromatografía de columna en gel de sílice (EtOAc/hexano al 60 %) para producir 3-((3-cloro-2-((2-hidroxietil)amino)piridin-4-il)tio)propanoato de metilo (460 mg, 94 % de rendimiento) como un sólido. m/z (esi) M+1 = 290,9.
Paso C: A una solución agitada de 3-((3-cloro-2-((2-hidroxietil)amino)piridin-4-il)tio)propanoato (500 mg, 1,72 mmoles) en THF (10 ml) se agregó NaOEt (21 % en peso de EtOH) (1,5 ml, 2,06 mmoles) a 0 °C y se agitó durante 30 minutos a 0 °C. La mezcla de reacción se concentró y el producto sin purificar se trituró con DCM y el precipitado sólido se filtró para producir 3-cloro-2-((2-hidroxietil)amino)piridin-4-tiolato de sodio (350 mg, 90 % de rendimiento) como un sólido. m/z (esi) M+1 = 205,1.
Ejemplo AT Intermedio

Diclorohidrato de 1-am¡no-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-5-carbon¡tr¡lo
Paso A: A una soluc¡ón ag¡tada de 5-cloro-2,3-d¡h¡dro-1H-¡nden-1-ona (5.0 g. 30.12 mmoles) en DMF (50 ml) se agregó NaH (60 % en peso de paraf¡na) (3.61 g. 90.36 mmoles) a 0 °C y se ag¡tó a temperatura amb¡ente durante 30 m¡nutos. Se agregó por porc¡ones N-benc¡l-2-cloro-N-(2-cloroet¡l)etan-1-am¡na (8.89 g. 33.13 mmoles) a 0 °C y se ag¡tó a temperatura amb¡ente durante otras 5 horas. La mezcla de reacc¡ón se ext¡ngu¡ó con agua helada y se extrajo con acetato de et¡lo. La parte orgán¡ca se secó (Na2SO4). se f¡ltró. concentró y el producto s¡n purificar se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc-hexano al 25 %) para produc¡r 1'-benc¡l-5-clorosp¡ro[¡nden-2.4'-p¡per¡d¡n1-1(3H)-ona (2 g. 20 % de rend¡m¡ento) como un líqu¡do. m/z (es¡) M+1 = 325.9.
Paso B: A una soluc¡ón ag¡tada de 1'-benc¡l-5-clorosp¡ro[¡nden-2.4'-p¡per¡d¡n1-1(3H)-ona (2.0 g. 6.15 mmoles) en DCE (20 ml) se agregó cloroform¡ato de 1-cloroet¡lo (3.49 g. 24.61 mmoles) a 0 °C y se ag¡tó durante 10 m¡nutos. La mezcla de reacc¡ón se ag¡tó a 80 °C durante 16 horas. Después. la mezcla de reacc¡ón se concentró y el mater¡al s¡n pur¡f¡car se d¡solv¡ó en MeOH (20 ml) y se ag¡tó de nuevo a 80 °C durante 1 hora. La mezcla de reacc¡ón se concentró para produc¡r clorh¡drato de 5-clorosp¡ro[¡nden-2.4'-p¡per¡d¡n1-1(3H)-ona (1.45 g de producto s¡n pur¡f¡car) como un líqu¡do gomoso. que se ut¡l¡zó para el s¡gu¡ente paso s¡n pur¡f¡cac¡ón ad¡c¡onal. m/z (es¡) M+1 = 236.1.
Paso C: A una soluc¡ón ag¡tada de clorh¡drato de 5-cloroesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1(3H)-ona (1.45 g. 6.17 mmoles) en DCM (15 ml) se agregó tr¡et¡lam¡na (3.43 ml. 24.68 mmoles) y anhídr¡do de Boc (2.12 ml. 9.25 mmoles) a 0 °C y la mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante 16 horas. La mezcla de reacc¡ón se concentró y pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc-hexano al 30 %) para produc¡r 5-cloro-1-oxo-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de ferf-but¡lo (400 mg. 19 % de rend¡m¡ento. 2 Pasos) como un sól¡do. m/z (es¡) M+1 = 336.3.
Paso D: A una soluc¡ón ag¡tada de 5-cloro-1-oxo-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de terf-but¡lo (1.4 g. 4.14 mmoles) en DMF (10 ml) se agregaron Zn(CN)2 (982.0 mg. 8.35 mmoles) y polvo de z¡nc (55.0 mg. 0.83 mmoles) y se ag¡tó durante 10 m¡nutos. La mezcla de reacc¡ón se desgas¡f¡có con argón. después se agregaron tr¡x¡efos (383.0 mg. 0.41 mmoles) segu¡do por Pd(OAc)2 (232.0 mg. 0.41 mmoles). La mezcla de reacc¡ón se ag¡tó a 120 °C durante 16 horas. La mezcla de reacc¡ón se d¡luyó con agua y se extrajo con acetato de et¡lo. La parte orgán¡ca se secó (Na2SO4). se f¡ltró y concentró y el mater¡al s¡n pur¡f¡car se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc-hexano al 30 %) para produc¡r 5-c¡ano-1-oxo-1.3-d¡h¡droesp¡ro [¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de terf-but¡lo (250 mg. 18 % de rend¡m¡ento) como un sól¡do pegajoso. m/z (es¡) M+1 = 326.3.
Paso E: A una soluc¡ón ag¡tada de 5-c¡ano-1-oxo-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de terf-but¡lo (500 mg. 1.53 mmoles) en etóx¡do de t¡tan¡o (IV) (1.62 ml. 7.66 mmoles) se agregó (R)-2-met¡lpropan-2-sulf¡nam¡da (204.5 mg. 1.68 mmoles) y se ag¡tó a 90 °C durante una hora. La mezcla de reacc¡ón se vert¡ó sobre EtOAc y salmuera. se ag¡tó durante 15 m¡nutos. El prec¡p¡tado sól¡do se f¡ltró. La capa orgán¡ca se lavó con salmuera. se secó (Na2SO4) y concentró para produc¡r 1-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-5-c¡ano-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de ferf-but¡lo (600 mg de producto s¡n pur¡f¡car) como un líqu¡do gomoso. que se ut¡l¡zó para el s¡gu¡ente paso s¡n pur¡f¡cac¡ón ad¡c¡onal. m/z (es¡) M+1 = 430.3.
Paso F: A una soluc¡ón ag¡tada de 1-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-5-c¡ano-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de ferf-butMo (600.0 mg. 1.39 mmoles) en MeOH (10 ml) se agregó NaBH4 (105.8 mg. 2.79 mmoles) a -10 °C y se ag¡tó a temperatura amb¡ente durante 2 horas. La mezcla de reacc¡ón se concentró. se d¡luyó con acetato de et¡lo y se lavó con agua. La capa orgán¡ca se secó (Na2SO4). se f¡ltró. concentró y pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc-hexano al 40 %) para produc¡r 1-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-5-c¡ano-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de ferf-but¡lo (430 mg. 71 % de rend¡m¡ento) como un sól¡do. m/z (es¡) M+1 = 432.1.
Paso G: A una soluc¡ón ag¡tada 1-(((R)-ferf-but¡lsulf¡n¡l) am¡no)-5-c¡ano-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de ferf-but¡lo (430.0 mg. 0.99 mmoles) en MeOH (3 ml) a 0 °C se agregó 4M HCl en d¡oxano (3 ml) y se ag¡tó a 0 °C durante 2 horas. La mezcla de reacc¡ón se concentró y el producto s¡n pur¡f¡car se tr¡turó con éter d¡etíl¡co para produc¡r 3-am¡no-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-6-carbon¡tr¡lo d¡clorh¡drato (250 mg. 84% de rend¡m¡ento) como un sól¡do. m/z (es¡) M+1 = 228.4.
Ejemplo AU Intermedio

(S)-5-(((ft)-terf-but¡lsulf¡n¡l)am¡no)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[¿)lp¡r¡d¡n-6.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-butilo
Paso A: Una mezcla de c¡clopentan-1,3-d¡ona (20.0 g. 204.08 mmoles). but-3-en-2-ona (26.78 ml. 306.12 mmoles). tam¡ces moleculares 4Á (100 g) y NH4OAc (31.42 g. 408.16 mmoles) en tolueno (800 ml) se ag¡tó hasta reflujo durante 24 horas. Después del térm¡no. la mezcla de reacc¡ón se f¡ltró a través de un lecho de Cel¡te®. se concentró y el res¡duo resultante se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 50 %) para produc¡r 2-met¡l-6.7-d¡h¡dro-5H-c¡clopenta[6lp¡r¡d¡n-5-ona (6.0 g. 20 % de rend¡m¡ento) como un líquido. m/z (es¡) M+1 = 148.3.
Paso B: A una soluc¡ón ag¡tada de 2-met¡l-6.7-d¡h¡dro-5H-c¡clopenta[6lp¡r¡d¡n-5-ona (5.0 g. 34.01 mmoles) en DMF (60 ml) se agregó NaH (60 % en peso en paraf¡na) (4.0 g. 102.04 mmoles) a 0 °C y se agitó durante 30 m¡n a temperatura amb¡ente. Se agregó por porc¡ones clorh¡drato de N-benc¡l-2-cloro-N-(2-cloroet¡l)etan-1-am¡na (7.31 g. 27.21 mmoles) a 0 °C y se ag¡tó durante otras 16 horas a temperatura amb¡ente. La mezcla de reacc¡ón se ext¡ngu¡ó con agua helada y se extrajo con acetato de et¡lo. La parte orgán¡ca se secó (Na2SO4). se f¡ltró. concentró y el mater¡al de producto s¡n pur¡f¡car se pur¡f¡có med¡ante gel de síl¡ce (MeOH en DCM al 0-15 %) para produc¡r 1'-benc¡l-2-met¡lesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-5(7H)-ona (1.2 g. 11 % de rend¡m¡ento) como un líquido. m/z (es¡) M+1 = 306.9.
Paso C: A una soluc¡ón ag¡tada de 1'-benc¡l-2-met¡lesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-5(7H)-ona (400.0 mg. 1.31 mmoles) en etanol (15 ml) se agregaron form¡ato de amon¡o (247.14 mg. 3.92 mmoles) segu¡do por Pd/C (200 mg) y la mezcla de reacc¡ón se purgó con argón durante 10 m¡nutos. Después. la mezcla de reacc¡ón se somet¡ó a reflujo a 80 °C durante 16 horas. La mezcla de reacc¡ón se concentró para produc¡r 2-met¡lesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-5(7H)-ona. el cual se ut¡l¡zó para el s¡gu¡ente paso s¡n pur¡f¡cac¡ón ad¡c¡onal. m/z (es¡) M+1 = 217.2.
Paso D: A una soluc¡ón ag¡tada de 2-met¡lesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-5(7H)-ona (280.0 mg. 1.29 mmoles) en DCM (10 ml) se agregó tr¡et¡lam¡na (0.72 ml. 5.18 mmoles) a 0 °C. segu¡do por anh¡druro de Boc (0.45 ml. 1.94 mmoles) y la mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La mezcla de reacc¡ón se concentró y pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 30 %) para produc¡r 2-met¡l-5-oxo-5.7-d¡h¡droesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-butílo (200 mg. 48 % de rend¡m¡ento. 2 Pasos) como un sól¡do. m/z (es¡) M+1 = 317.2.
Paso E: Se agregaron 2-met¡l-5-oxo-5.7-d¡h¡droesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-but¡lo (400 mg. 1.26 mmoles) y (R)-2-met¡lpropan-2-sulf¡nam¡da (460.21 mg. 3.80 mmoles) al etóx¡do de t¡tan¡o (IV) (866.20 mg. 3.79 mmoles) a 90 °C y se ag¡tó a 90 °C durante 5 horas. La mezcla de reacc¡ón se vert¡ó sobre acetato de et¡lo y salmuera. Después de ag¡tar durante 15 m¡nutos. el sól¡do prec¡p¡tado se f¡ltró y la parte líqu¡da se separó. La capa orgán¡ca se lavó con salmuera. se secó (Na2SO4). se f¡ltró y concentró bajo pres¡ón reduc¡da. El res¡duo resultante se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce para produc¡r (R.£)-1-((tert-but¡lsulf¡n¡l)¡m¡no)-5-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-but¡lo (410 mg. 77 % de rend¡m¡ento) como un líqu¡do gomoso. m/z (es¡) M+1 = 420.2.
Paso F: A una soluc¡ón de (R.£)-1-((tert-but¡lsulf¡n¡l)¡m¡no)-5-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-but¡lo (410 mg. 0.98 mmoles) en MeOH (10 ml) a 0 °C se agregó NaBH4 (185 mg. 4.89 mmoles) y se ag¡tó a temperatura amb¡ente durante 4 horas. La mezcla de reacc¡ón se ext¡ngu¡ó con el agua helada y se extrajo con EtOAc. La capa orgán¡ca comb¡nada se secó (Na2SO4). se f¡ltró y concentró bajo pres¡ón reduc¡da. El res¡duo resultante se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (MeOH-DCM al 1 %) y después con HPLC preparat¡va (Ch¡ralpak IG (21.0 x 250 mm). n-Hexano 5 p/EtOH/IPA : 80/20/0.1. 21.0 ml/m¡n. 20 m¡n. 276 nm. MeOH) para produc¡r (R)-5-(((R)-tert-but¡lsulf¡n¡l)am¡no)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[6lp¡r¡d¡n-6.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-but¡lo (40 mg. 10 % de rend¡m¡ento). m/z (es¡) M+1 = 422.4.
Ejemplo AV Intermedio

(ffl-5-(((ffl-tert-but¡lsulf¡n¡l)am¡no)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[¿)lp¡r¡d¡n-6.4'-p¡per¡d¡nl-1'-carbox¡lato de tert-butilo
Se preparó (R)-5-(((R)-tert-but¡lsulf¡n¡l)am¡no)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-but¡lo de acuerdo con el Ejemplo AU Intermed¡o, al recolectar el segundo p¡co en la Paso F. m/z (es¡) M+1 = 422,5.
Ejemplo AW Intermedio

Clorh¡drato de 3-metox¡-5.7-d¡h¡droesp¡ro[c¡clopenta[¿)lp¡r¡d¡n-6.4'-p¡per¡d¡nl-5-am¡na
Paso A: A una soluc¡ón ag¡tada de 5-bromo-6-clorop¡r¡d¡n-3-ol (5,0 g, 24,16 mmoles) y K2CO3 (5,0 g, 36,25 mmoles) en ACN (30 ml) se agregó Mel (1,65 ml, 26,58 mmoles) y la mezcla se agitó a temperatura amb¡ente durante 16 horas. La mezcla de reacc¡ón se extrajo con EtOAc. La fase orgán¡ca se secó (sobre Na2SO4), se f¡ltró y concentró bajo pres¡ón reduc¡da. El res¡duo se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce ut¡l¡zando una soluc¡ón de EtOAc/hexano (EtOAc/hexano al 0-10 %) para produc¡r 3-bromo-2-cloro-5-metox¡p¡r¡d¡na (3,48 g, 65 % de rend¡m¡ento) como un sól¡do. m/z (es¡) M+1 = 223,9.
Paso B: A una soluc¡ón ag¡tada de 4-form¡l-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-but¡lo (3,89 g, 17,12 mmoles) en THF (55 ml) se agregó por goteo ¡PrMgBr (19,76 ml, 1,3M ¡n THF, 25,68 mmoles) a 0 °C y se ag¡tó a temperatura amb¡ente durante 2 horas. Se agregó por goteo 3-bromo-2-cloro-5-metox¡p¡r¡d¡na (5,68 g, 25,68 mmoles) en THF (10 ml) a 0 °C y se ag¡tó a temperatura amb¡ente durante 30 m¡nutos. La mezcla de reacc¡ón se ext¡ngu¡ó con una soluc¡ón saturada de NH4Cl y se extrajo con EtOAc. La parte orgán¡ca se secó (sobre Na2SO4), se f¡ltró, concentró y pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 15-20 %) para produc¡r 4-((2-cloro-5-metox¡p¡r¡d¡n-3-¡l)(h¡drox¡)met¡l)-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-butílo (2,157 g, 83 % de rend¡m¡ento) como un sólido pegajoso. m/z (es¡) M+1 = 371,4.
Paso C: A una soluc¡ón ag¡tada de 4-((2-cloro-5-metox¡p¡r¡d¡n-3-¡l)(h¡drox¡)met¡l)-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-but¡lo (1,1 g, 2,97 mmoles) en DCM (16 ml) se agregó peryod¡nano de Dess-Mart¡n (1,89 g, 4,45 mmoles) a 0 °C y se ag¡tó a temperatura amb¡ente durante 3 horas bajo una atmósfera de n¡trógeno. La mezcla de reacc¡ón se ext¡ngu¡ó con una soluc¡ón saturada de t¡osulfato de sod¡o y se extrajo con DCM. La fase orgán¡ca se lavó con una soluc¡ón de NaOH 1 N, se concentró y pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 15-20 %) para produc¡r 4-(2-cloro-5-metox¡n¡cot¡no¡l)-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-but¡lo (860 mg, 78 % de rend¡m¡ento) como un ace¡te. m/z (es¡) M+1 = 369,1.
Paso D: A un tubo sellado secado a la flama bajo argón se agregó tetrafluoroborato de tr¡c¡clohex¡lfosfon¡o (145 mg, 0,39 mmoles), acetato de palad¡o (II) (44 mg, 0,19 mmoles), ác¡do p¡vál¡co (121 mg, 1,18 mmoles), carbonato de ces¡o (1,54 g, 4,72 mmoles) y 4-(2-cloro-5-metox¡n¡cot¡no¡l)-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-but¡lo (1,45 g, 3,94 mmoles) en mes¡t¡leno (20 ml). La mezcla se desgas¡f¡có con argón durante 10 m¡nutos y se calentó a 140 °C durante 48 horas. La mezcla de reacc¡ón se enfr¡ó a temperatura amb¡ente y se f¡ltró a través de un lecho de Cel¡te® y se lavó con EtOAc. La fase orgán¡ca se lavó con salmuera, se secó sobre Na2SO4, se f¡ltró y concentró. El res¡duo se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 0-10 %) para dar 3-metox¡-5-oxo-5,7-d¡h¡droesp¡ro[c¡clopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-but¡lo (0,84 g, 64 % de rend¡m¡ento) como un sól¡do, m/z (es¡) M+1 = 333,2.
Paso E: A una solución agitada de 3-metox¡-5-oxo-5,7-d¡h¡droesp¡ro[c¡clopenta[£)]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-but¡lo (575 mg, 1,73 mmoles) en etóx¡do de t¡tan¡o (IV) (2,2 ml, 10,38 mmoles), se agregó (R)-(+)-2-met¡l-2-propansulf¡nam¡da (629 mg, 5,19 mmoles) y se ag¡tó a 110 °C durante 4 horas. La mezcla de reacc¡ón se dejó enfr¡ar a temperatura amb¡ente y la mezcla de reacc¡ón se d¡luyó con EtOAc y agua. La mezcla resultante se ag¡tó v¡gorosamente durante 15 m¡nutos a temperatura amb¡ente y luego se f¡ltró a través de una almohad¡lla de Cel¡te®. El f¡ltrado se extrajo con EtOAc, se lavó con salmuera, se secó sobre Na2SO4 y se f¡ltró. La fase orgán¡ca se concentró y el res¡duo resultante se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 40-50 %) para proporc¡onar (R,Z)-5-((ferf-but¡lsulf¡n¡l)¡m¡no)-3-metox¡-5,7-d¡h¡droesp¡ro[c¡dopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de íerí-butílo como un sem¡sól¡do (0,64 g, 84 % de rend¡m¡ento). m/z (es¡) M+1 = 437,2.
Paso F: A una soluc¡ón ag¡tada de (R,Z)-5-((ferf-but¡lsulf¡n¡l)¡m¡no)-3-metox¡-5,7-d¡h¡droesp¡ro[c¡dopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de íerí-butílo (590 mg, 1,35 mmoles) en MeOH (10 ml), se agregó boroh¡druro de sod¡o (256 mg, 6,77 mmoles) a 0 °C y se ag¡tó durante 3 horas. Después del térm¡no de la reacc¡ón, se dejó que la mezcla ¡ncrementara la temperatura a temperatura amb¡ente. La soluc¡ón acuosa saturada de NH4Cl se agregó lentamente para ext¡ngu¡r la reacc¡ón, el MeOH se evaporó y la mezcla de reacc¡ón se extrajo con EtOAc. La fase orgán¡ca se lavó con salmuera, se secó sobre Na2SO4, se f¡ltró y concentró. El res¡duo resultante se pur¡f¡có med¡ante cromatografía de columna en gel de sílice (MeOH/DCM al 0-4 %) para dar 5-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-3-metox¡-5,7-d¡h¡droesp¡ro[c¡dopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-but¡lo (0,58 g, 97 % de rend¡m¡ento) como un sól¡do. m/z (es¡) M+1 = 437,9.
Paso G: A una soluc¡ón ag¡tada de 5-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-3-metox¡-5,7-d¡h¡droesp¡ro[c¡dopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-but¡lo (600 mg, 1,37 mmoles) en MeOH (12 ml) se agregó d¡oxano-HCl (4M; 12 ml) a 0 °C y se ag¡tó durante 2 horas. La mezcla de reacc¡ón se concentró y el mater¡al crudo se trituró con éter d¡etíl¡co para produc¡r clorh¡drato de 3-metox¡-5,7-d¡h¡droesp¡ro[c¡dopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na (418 mg, 99 % de rend¡m¡ento) como un sól¡do. El mater¡al crudo se ut¡l¡zó en el s¡gu¡ente paso s¡n pur¡f¡cac¡ón. m/z (es¡) M+1 = 234,3.
Ejemplo AX Intermedio
(^)-1-(((S)-ferf-but¡lsulf¡n¡l)am¡no)-6-cloro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de terf-but¡lo
Paso A: A una soluc¡ón ag¡tada de 6-cloro-2,3-d¡h¡dro-1H-¡nden-1-ona (2,0 g, 12,05 mmoles) en DMF (40 ml) se agregó NaH (60 % en ace¡te m¡neral) (1,45 g, 36,14 mmoles) a 0 °C y la mezcla se ag¡tó durante 30 m¡nutos a 0-5 °C. Se agregó por porc¡ones clorh¡drato de N-benc¡l-2-cloro-N-(2-cloroet¡l)etan-1-am¡na (3,06 g, 13,25 mmoles) y la mezcla se agitó a 60 °C durante 16 horas. La reacc¡ón se ext¡ngu¡ó con una soluc¡ón de salmuera y se extrajo con EtOAc. Las capas orgán¡cas comb¡nadas se lavaron con salmuera, se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y concentraron bajo pres¡ón reduc¡da para dar el producto s¡n pur¡f¡car, el cual se pur¡f¡có med¡ante una soluc¡ón en gel de síl¡ce (EtOAc/hexano) para dar 1'-benc¡l-6-cloroesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona (1,55 g, 40 % de rend¡m¡ento) como un ace¡te. m/z (es¡) M+1: 325,9.
Paso B: A una soluc¡ón ag¡tada de 1'-benc¡l-6-cloroesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona (1,55 g, 4,78 mmoles) en d¡cloroetano (“DCE”; 24 ml) se agregó cloroform¡ato de 1-cloroet¡lo (1,55 ml, 14,34 mmoles) y se somet¡ó a reflujo durante una hora. Después, el DCE se evaporó bajo pres¡ón reduc¡da y se agregó MeOH (24 ml) y se somet¡ó a reflujo durante una hora. Se evaporó MeOH hasta sequedad para dar 6-cloroesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona (1,13 g, crudo) como un sól¡do. m/z (es¡) M+1: 236,2.
Paso C: A una soluc¡ón ag¡tada de 6-cloroesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona (1,13 g, 4,79 mmoles) en DCM (20 ml), se agregó TEA (2,66 ml, 19,16 mmoles) segu¡da por anh¡druro de BOC (1,65 ml, 7,19 mmoles) a 0 ° C y se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se evaporó hasta sequedad y se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (EtOAc/hexano al 15-20 %) para produc¡r 6-cloro-1-oxo-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-but¡lo (1,3 g, 81 %, rend¡m¡ento 2 Pasos) como un sól¡do. m/z (es¡) M+1: 336,3.
Paso D: Una soluc¡ón de 6-cloro-1-oxo-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-but¡lo (800 mg, 2,38 mmoles), etóx¡do de t¡tan¡o (IV) (1,99 ml, 9,5 mmoles) y (R)-(+)-2-met¡propan-2-sulf¡nam¡da (577,5 mg, 4,76 mmoles) en THF (15 ml) se agitó a 90 °C durante 12 horas. La reacc¡ón se enfr¡ó a temperatura amb¡ente y se ext¡ngu¡ó con agua. El compuesto se extrajo con EtOAc. Las fases orgán¡cas comb¡nadas se lavaron con salmuera, se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y concentraron bajo pres¡ón reduc¡da para dar el crudo, el cual se pur¡f¡có med¡ante
cromatografía de columna en gel de sílice (EtOAc/hexano al 20-25 %) para dar (R,E)-1-((ferf-but¡lsulflml)im¡no)-6-doro-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de terf-butilo (861 mg, 94 % de rendimiento) como un sólido. m/z (esi) M+1: 438,8.
Paso E: A una solución agitada de (R^-l-^tert-butilsulflm O im ino^-cloro-l^-d ih idroespiropnden^^'-piperid in^l'-carboxilato de tert-butilo (1 g, 2,28 mmoles) en MeOH (20 ml) se agregó borohidruro de sodio (43 mg, 11,42 mmoles) a 0 °C y se agitó a 25 °C durante 3 horas. La solución acuosa saturada de NH4Cl se agregó lentamente para extinguir el exceso de borohidruro y la mezcla de reacción se diluyó con EtOAc. La mezcla resultante se agitó vigorosamente durante 15 minutos a temperatura ambiente y después se extrajo con EtOAc. Las partes orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y concentraron bajo presión reducida para dar el crudo, el cual se purificó mediante purificación de HPLC preparativa normal (Chiralpak IC (20,0 x 250 mm), hexano 5 |j/EtOH/iPrNH280/20/0,1, 1,0 ml/min) se recolectó el pico 1 para proporcionar (É)-1-(((S)-terf-butMsulflnM)amino)-6-cloro-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de terf-butilo, (528 mg, 51 % de rendimiento). m/z (esi) M+1: 441,2.
Ejemplo AY Intermedio

(S)-1-(((S)-terf-but¡lsulf¡n¡l)am¡no)-6-cloro-1.3-d¡h¡droesp¡ro^nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de tert-butilo
Se preparó de acuerdo con el Ejemplo AX Intermedio, se recolectó el pico 2 en la Paso E para proporcionar (S)-1-(((S)-tert-but¡lsulf¡n¡l)am¡no)-6-cloro-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡pe^d¡n]-1'-carbox¡lato de terf-butilo. m/z (esi) M+1: 441,2.
Ejemplo AZ Intermedio

(S)-1-(((E)-ferf-but¡lsulf¡n¡l)am¡no)-5-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de tert-butilo
Paso A: A una solución agitada de 5-met¡l-2,3-d¡h¡dro-1H-¡nden-1-ona (4,0 g, 27,39 mmoles) en DMF (80 ml) se agregó por porciones NaH (60 % de dispersión en aceite mineral, 1,97 g, 82,19 mmoles) a 0 °C. La mezcla se agitó a 0 °C durante 30 minutos. Se agregó por porciones clorhidrato de N-Bencil-2-cloro-N-(2-cloroetil)etan-1-amina (8,09 g, 30,13 mmoles) a la mezcla de reacción y se agitó a temperatura ambiente durante 16 horas. La reacción se diluyó con salmuera y se extrajo con EtOAc. Las partes orgánicas se combinaron y lavaron con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y concentraron bajo presión reducida. El residuo se purificó mediante cromatografía de columna en gel de sílice (EtOAc/hexano al 25 %) para producir 1'-benc¡l-5-met¡lesp¡ro[¡nden-2,4'-p¡pe^d¡n]-1(3H)-ona (4,0 g, 48 % de rendimiento) como un sólido. m/z (esi) M+1 = 305,6.
Paso B: A una solución agitada de 1'-benc¡l-5-met¡lesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona (5,0 g, 16,37 mmoles) en DCE (100 ml) se agregó cloroformiato de cloroetilo (6,97 g, 49,11 mmoles) a 0 °C y se agitó durante 10 minutos. La mezcla de reacción se agitó a 80 °C durante una hora. La mezcla de reacción se concentró hasta sequedad y se agregó MeOH (100 ml) y agitó a 75 °C durante una hora. La mezcla de reacción se concentró bajo presión reducida para producir 5-met¡lesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona (3,5 g de producto sin purificar) como un líquido, que se utilizó para el siguiente paso sin purificación adicional. m/z (esi) M+1 = 215,9.
Paso C: A una solución agitada de 5-met¡lesp¡ro[¡nden-2,4'-p¡pe^d¡n]-1(3H)-ona (3,5 g, 16,27 mmoles) en DCM (35 ml) se agregó trietilamina (9,07 ml, 65,11 mmoles) a 0 °C. Se agregó anhidruro de Boc (5,61 ml, 24,42 mmoles) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. La mezcla de reacción se concentró bajo presión reducida y se purificó mediante cromatografía de columna en gel de sílice (EtOAc/hexano al 10 %) para producir 5-met¡l-1-oxo-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-butilo (520 mg, 68 % de rendimiento, 2 Pasos) como un sólido. m/z (esi) M+1 = 316,2.
Paso D: Se agregaron 5-metil-1-oxo-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de ferf-butilo (1,9 gm, 6,03 mimóles) y (R)-(+)-2-metilpropan-2-sulflnam¡da (2,19 g, 18,09 mmoles) en etóxido de titanio (IV) (4,12 g, 18,09 mmoles) caliente (100 °C) y se agitó a 100 °C durante 19 horas. La mezcla de reacción se vertió en EtOAc y salmuera y la mezcla se agitó durante 15 minutos. Los sólidos se filtraron y la parte líquida se separó. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida. El residuo resultante se purificó mediante cromatografía de columna en gel de sílice (MeOH/DCM al 1 %) para producir (R,E)-1-((tert-butilsulf¡n¡l)¡m¡no)-5-met¡l-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de ferf-butilo (1,25 g, 49 % de rendimiento) como un sólido. m/z (esi) M+1 = 418,9.
Paso E: A una solución de (R,E)-1-((ferf-but¡lsulf¡n¡l)¡m¡no)-5-met¡l-1,3-d¡h¡dro-esp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carboxilato de ferf-butilo (1,3 g, 3,10 mmoles) a 0 °C en MeoH (30 ml) se agregó borohidruro de sodio (470 mg, 12,42 mmoles) y se agitó a temperatura ambiente durante 4 horas. La mezcla de reacción se extinguió con agua helada y se extrajo con EtOAc. Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y concentraron bajo presión reducida. El residuo resultante se purificó mediante cromatografía de columna en gel de sílice (EtOAc/hexano al 30 %) para producir (S)-1-(((R)-ferf-but¡lsulf¡n¡l)am¡no)-5-met¡l-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carboxilato de ferf-butilo (310 mg, 24 % de rendimiento) (m/z (esi) M-1 = 419,3).
Ejemplo BA Intermedio
(R)-1-(((R)-tert-but¡lsulf¡n¡l)am¡no)-5-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de tert-butilo
Se preparó (R)-1-(((R)-tert-but¡lsulf¡n¡l)am¡no)-5-met¡l-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-butilo se preparó de acuerdo con el Ejemplo AZ Intermedio al recolectar el segundo pico en la Paso E. (m/z (esi) M-1 = 419,3).
Ejemplo BC Intermedio

Diclorhidrato de 3-fluoro-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-5-am¡na
Paso A: A una solución agitada de 3-bromo-2-cloro-5-fluoropiridina (5,0 g, 22,02 mmoles) en THF (100 ml) se agregó por goteo cloruro de isopropilmagnesio (1,5 M en THF) (15 ml) a 0 °C y se agitó a temperatura ambiente durante 2 h. Se agregó por goteo 4-form¡l-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-butilo (9,2 ml, 44,05 mmoles) en THF (50 ml) a 0 °C y se agitó a temperatura ambiente durante 30 min. La reacción se extinguió con una solución saturada de cloruro de amonio y se extrajo con EtOAc. La parte orgánica se secó, filtró, concentró y purificó mediante cromatografía de columna para producir el 4-((2-cloro-5-fluoropir¡din-3-¡l)(h¡droxi)metil)-4-met¡lp¡peridin-1 -carboxilato de tert-butilo deseado (7,0 g, 89 % de rendimiento) como un sólido pegajoso amarillo claro. m/z (esi) M+1 = 359,3.
Paso B: A una solución agitada de 4-((2-cloro-5-fluorop¡r¡d¡n-3-¡l)(h¡drox¡)met¡l)-4-met¡lp¡per¡d¡n-1-carbox¡lato de tertbutilo (2,7 g, 6,14 mmoles) en DCM (25 ml) se agregó DMP (4,7 g, 11,31 mmoles) a 0 °C y se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se extinguió con tiosulfato de sodio (30 ml) y se extrajo con DCM. La parte orgánica se lavó con una solución de NaOH 2 M, se secó (Na2SO4), se filtró y concentró. El residuo resultante se purificó mediante cromatografía instantánea en gel de sílice (EtOAc-hexano al 20 %) para producir 4-(2-cloro-5-fluoron¡cot¡no¡l)-4-met¡lp¡per¡d¡n-1-carbox¡lato de tert-butilo (1,8 g, 67 % de rendimiento) como un sólido marrón. m/z (esi) M+1 = 357,2.
Paso C: A una solución agitada de 4-(2-doro-5-fluoronicotinoil)-4-metilpiperidin-1-carboxilato de tert-butilo (2 g, 5,61 mimóles) en 1,3,5 mesitileno (10 ml) se agregaron PCy3H.BF4 (210 mg, 0,56 mmoles), tBuCOOH (172 mg, 1,68 mmoles) y Cs2CO3 (2,2 g, 6,74 mmoles) a temperatura ambiente y se desgasificaron con un balón de N2 durante 5 min. Se agregó Pd(OAc)2 (70 mg, 0,28 mmoles) a la mezcla de reacción y se desgasificó de nuevo con un globo de N2. La mezcla de reacción se agitó a 140 °C durante 72 h. La reacción se enfrió y purificó mediante cromatografía instantánea en gel de sílice (EtOAc/hexano al 50 %) para producir 3-fluoro-5-oxo-5,7-dihidroespiro[ciclopenta[b]pindin-6,4'-piperidin]-1'-carboxilato de tert-butilo (1 g, 56 % de rendimiento) como un sólido marrón. m/z (esi) M+1 = 321,2.
Paso D: A una solución agitada de 3-fluoro-5-oxo-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de tert-butilo (1 g, 3,12 mmoles) en Ti(OEt)4 (2,5 ml, 9,37 mmoles) se agregó R-(+)-2-metil-2-propansulfinamida (0,8 g, 6,25 mmoles) a temperatura ambiente y la mezcla de reacción se agitó a 100 °C durante 16 h. La mezcla de reacción se extinguió con agua y se agregó un exceso de acetato de etilo. La solución se filtró mediante un embudo sinterizado y la capa orgánica se separó. La capa orgánica se secó (Na2SO4), se filtró, concentró y purificó mediante cromatografía instantánea en gel de sílice (EtOAc/hexano al 20 %) para producir (R,Z)-5-((tert-butilsulfinil)imino)-3-fluoro-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de tert-butilo (1 g, 76 % de rendimiento) como un sólido amarillo. m/z (esi) M+1 = 424,0.
Paso E: A una solución agitada de (R,Z)-5-((tert-butilsulfinil)imino)-3-fluoro-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de tert-butilo (1,1 g, 2,6 mmoles) en metanol (50 ml) se agregó NaBH4 (400 mg, 10,4 mmoles) a 0 °C y se agitó durante 10 min. La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 3 h. La mezcla de reacción se extinguió con hielo, se evaporó y extrajo con EtoAc. La capa orgánica se concentró y purificó mediante cromatografía instantánea en gel de sílice (EtOAc/hexano al 30 %) para producir 5-(((R)-tertbutilsulfinil)amino)-3-fluoro-5,7-dihidroespiro [ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de tert-butilo (800 mg, 72 % de rendimiento) de sólido blanquizco. m/z (esi) M+1 = 426,1.
Paso F: A una solución agitada de 5-(((R)-tert-butilsulfinil)amino)-3-fluoro-5,7-dihidroespiro [ciclopenta[b]piridin-6,4'-piperidin]-1'-carboxilato de tert-butilo (700 mg, 1,6 mmoles) en MeOH (4 ml) se agregó HCl (4 M en 1,4-dioxano) (5 ml) a 0 ° C y se agitó durante 1,5 h. La mezcla de reacción se concentró para producir diclorhidrato de 3-fluoro-5,7-dihidroespiro[ciclopenta[b]piridin-6,4' piperidin]-5-amina (480 mg, 99 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 222,1.
Ejemplo BD Intermedio

((4-met¡l-1,4-azas¡l¡nan-4-¡l)met¡l)carbamato de tert-butilo
Paso A: En un matraz de dos cuellos de 500mL que contiene una solución de bromuro de vinilmagnesio (1,0M en THF)(107 ml, 107 mmoles) en THF (50 ml) a 0 °C, se agregó lentamente dicloro(clorometil)(metil)silano (5 g, 30,58 mmoles) y la mezcla de reacción se agitó a temperatura ambiente durante 16 hr. Después del término de la reacción, la mezcla de reacción se extinguió lentamente con una cantidad en exceso de una solución saturada acuosa de cloruro de amonio y se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se extrajo con acetato de etilo (2 x 200 ml) y la capa orgánica combinada se secó sobre sulfato de sodio anhidro, se filtró y concentró bajo presión reducida para dar (clorometil)(metil)divinilsilano (2,6 g el producto sin purificar) como un aceite amarillo claro. El producto sin purificar se utilizó en el siguiente paso sin purificación adicional.
Paso B: Una suspensión de (clorometil)(metil)divinilsilano (2) (2,5 g, 17,48 mmoles), ftalimida de potasio (3,5 g, 19,2 moles) y yoduro de potasio (0,29 g, 1,74 mmoles) en DMF (15 ml) se calentó a 80 °C durante 16 hr bajo una atmósfera de N2. La mezcla de reacción se extinguió lentamente con agua y se extrajo con acetato de etilo (2 x 100 ml), se lavó con salmuera (100mL) y la capa orgánica combinada se secó sobre sulfato de sodio anhidro, se filtró y concentró bajo presión reducida para dar el producto sin purificar, el cual se purificó mediante normal cromatografía en columna de sílice (eludido en acetato de etilo en hexano al 5-8 %) para producir 2-((metildivinilsilil) metil) isoindolin-1, 3-diona como un aceite amarillo claro (2,0 g, 25 %; 2 Pasos).
Paso C: Una solución de 9-BBN (0,5M en THF) (54,6 ml, 27,3 mmoles) y 2-((metildivinilsilil) metil) isoindolin-1,3-diona (2 g, 7,81 mmoles) en THF (1 ml) se agitó durante 16 h a temperatura ambiente. La mezcla de reacción se trató posteriormente con H2O (2 ml) y después con una solución acuosa de NaOH (3M) (6 ml) a temperatura ambiente. Se agregó por goteo una solución acuosa de H2O2 (30 %) (6 ml) a los 15 min a 0 °C y la solución se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se enfrió a temperatura ambiente, se agregó agua, la capa orgánica se separó y la capa acuosa se lavó con DCM. La capa orgánica después se secó sobre sulfato de sodio anhidro y se
evaporó bajo presión reducida. El residuo resultante se purificó mediante normal cromatografía en columna de sílice (eludido en acetato de etilo en hexano al 75-80 %) para producir 2-((bis(2-hidroxietil)(metil)silil) metil)isoindolin-1,3-diona como un sólido blanco (240 mg, 11 %).
Paso D: Se agregaron en secuencia cloruro de metansulfonilo (0,4 ml, 5,98 mmoles) y TEA (1,37 ml, 9,52 mmoles) a -20 °C en una sola porción a una solución agitada de 2-((bis(2-hidroxietil)(metil)silil)metil)isoindolin-1,3-diona (800 mg, 2,72 mmoles) en DCM (500 ml) y la mezcla se agitó a -20 °C durante 3 h. Se agregó alilamina (en exceso) a -20 °C en una sola porción a la mezcla y después se calentó a 20 °C y se agitó a 20 °C durante 16 h. El disolvente y el exceso de alilamina se retiraron de la mezcla de reacción bajo presión reducida, seguido por la adición en secuencia de acetato de etilo y una solución saturada de NaHCO3 a 20 °C. La capa orgánica se separó, la capa acuosa se extrajo con acetato de etilo y los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron bajo presión reducida para dar el crudo que se purificó mediante cromatografía en columna de sílice normal (eludido en metanol en DCM al 5 %) para producir 2-((1-alil-4-metil-1,4-azasilinan-4-il)metil)isoindolin-1,3-diona como un aceite amarillo claro (122 mg, 14 % en 2 Pasos).
Paso E: Una mezcla de 2-((1-alil-4-metil-1,4-azasilinan-4-il)metil)isoindolin-1,3-diona (450 mg, 1.43 mmoles) y metilhidrazina (40 %. Sol, acuosa) (0,12 ml, 2,15 mmoles) en EtOH-THF (10mL) se sometió a reflujo durante 2 hr. El acetato de etilo se evaporó in vacuo, se agregó DCM a la mezcla de reacción y se filtró. La parte sólida se retiró mediante filtración y el filtrado se evaporó bajo presión reducida para dar (1-alil-4-metil-1,4-azasilinan-4-il) metanamina (7) (255 mg de producto sin purificar) como un aceite amarillo claro. El crudo se utilizó en el siguiente paso sin purificación adicional.
Paso F: ((1-alil-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo
A una solución agitada de (1-alil-4-metil-1,4-azasilinan-4-il)metanamina (7) (170 mg, 0,92 mmoles) en DCM (10 ml) se agregó anhidruro de Boc (0,33 ml, 1,38 mmoles) y la mezcla de reacción se agitó a temperatura ambiente durante 4hr. Después del término del material de partida, se agregó agua y la capa acuosa se extrajo con acetato de etilo (2 x 50 ml). Las capas orgánicas se secaron sobre sulfato de sodio anhidro, se filtraron y evaporaron bajo presión reducida para dar el crudo, el cual se purificó mediante cromatografía en columna de sílice normal (eludido en metanol en DCM al 2 %) para producir ((1-alil-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo como un aceite amarillo claro (130 mg, 2 pasos, 32 %).
Paso G: A una solución de ((1-alil-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo (165 mg, 0,64 mmoles) en DCM (10 ml), se agregó ácido 1,3-dimetilbarbitúrico (198,3 mg, 0,70 mmoles) seguido por la adición de (PPh3)4Pd (73,1 mg, 0,06 mmoles). La mezcla de reacción se purgó después con N2 y se calentó en un baño de aceite a 40 °C durante 3 hr. Después del término de la reacción, la mezcla de reacción se evaporó bajo presión reducida para dar el crudo, el cual se purificó mediante amina cromatografía en columna de sílice (eludido en metanol en DCM al 3 %) para producir ((4-metil-1,4-azasilinan-4-il) metil)carbamato de tert-butilo (91 mg, 60 %) como un sólido amarillo claro.
Ejemplo BE Intermedio

4-(pir¡d¡n-2-¡lmet¡l)p¡per¡d¡n-4-am¡na
Se preparó 4-(piridin-2-ilmetil)piperidin-4-amina de acuerdo con el Ejemplo R Intermedio, al sustituir 4-(2-aminopropan-2-il)piperidin-1-carboxilato de tert-butilo por 4-amino-4-(piridin-2-ilmetil)piperidin-1 -carboxilato de tert-butilo en el Paso A. m/z (esi/APCI) M+1 = 192,2.
E je m p lo BF In te rm e d io
Se preparó 4-bencilpiperidin-4-amina de acuerdo con el Ejemplo R Intermedio, al sustituir 4-(2-aminopropan-2-il)piperidin-1 -carboxilato de ferf-butilo por 4-amino-4-bencilpiperidin-1-carboxilato de tert-butilo en el Paso A. m/z (esi/APCI) M+1 = 191,1.
Ejemplo BG Intermedio

4-(2-metoxiet¡l)p¡per¡d¡n-4-am¡na
Se preparó 4-(2-metoxietil)piperidin-4-amina de acuerdo con el Ejemplo R Intermedio, al sustituir 4-(2-aminopropan-2-il)piperidin-1-carboxilato de ferf-butilo por 4-amino-4-(2-metoxietil)piperidin-1-carboxilato de tert-butilo en el Paso A. m/z (esi/APCI) M+1 = 159,2.
Ejemplo BH Intermedio

3-((2-am¡no-3-fluoropir¡d¡n-4-¡l)t¡o)propanoato de metilo
Se disolvió 2-amino-3-fluoro-4-yodopiridina (1,1 g, 4,4 mmoles) en 1,4-dioxano (10 ml, 4,4 mmoles). Se agregaron 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (0,26 g, 0,45 mmoles) y 3-((2-amino-3-fluoropiridin-4-il)tio)propanoato de metilo (0,95 g, 4,1 mmoles) a la solución de la reacción. La reacción se colocó bajo nitrógeno. Se agregó N,N-diisopropiletilamina (1,5 ml, 8,9 mmoles) y la solución resultante se agitó a 100 °C . El crudo se purificó mediante gel de sílice (EtOAc:hexano al 0-100 %) para proporcionar 3-((2-amino-3-fluoropiridin-4-il)tio)propanoato de metilo (0,95 g, 92 % de rendimiento). m/z (esi/APCI) M+1 = 231,1.
Ejemplo BI Intermedio
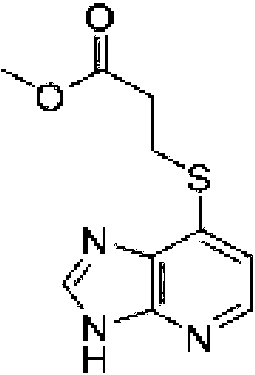
3-((3H-¡m¡dazo[4.5-b1p¡r¡din-7-¡l)t¡o)propanoato de metilo
Se preparó 3-((3H-¡midazo[4,5-b]p¡r¡d¡n-7-¡l)t¡o)propanoato de metilo de acuerdo con el Ejemplo BH Intermedio, al sustituir 2-amino-3-fluoro-4-yodopiridina por 7-bromo-3H-imidazo[4,5-b]piridina en el Paso A. m/z (esi/APCI) M+1 = 238,1.
Ejemplo BJ Intermedio

3-((2.3-d¡met¡lpir¡d¡n-4-¡l)t¡o)propanoato de metilo
Se preparó 3-((2,3-d¡met¡lp¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo de acuerdo con el Ejemplo BH Intermed¡o, al sust¡tu¡r 2-am¡no-3-fluoro-4-yodop¡r¡d¡na por 4-bromo-2,3-d¡met¡lp¡r¡d¡na en el Paso A. m/z (es¡/APCI) M+1 = 226.1.
Ejemplo BK Intermedio

3-((6-am¡no-2-(tr¡fluoromet¡l)p¡r¡d¡n-3-¡l)t¡o)propanoato de met¡lo
Se preparó 3-((6-am¡no-2-(tr¡fluoromet¡l)p¡r¡d¡n-3-¡l)t¡o)propanoato de met¡lo de acuerdo con el Ejemplo BH Intermed¡o, al sustituir 2-am¡no-3-fluoro-4-yodop¡r¡d¡na por 5-bromo-6-tr¡fluoromet¡lp¡r¡d¡n-2-¡lam¡na en el Paso A. m/z (es¡/APCI) M+1 = 281.
Ejemplo BL Intermedio

3-((2.3-d¡clorop¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo
Se preparó 3-((2,3-d¡dorop¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo de acuerdo con el Ejemplo BH Intermed¡o, al sust¡tu¡r 2-am¡no-3-fluoro-4-yodop¡r¡d¡na por 2,3-d¡cloro-4-yodop¡r¡d¡na en el Paso A. m/z (es¡/APCI) M+1 = 281.
E je m p lo BM In te rm e d io

2-am¡no-5-clorop¡r¡d¡n-4-t¡ol
El 2-amino-5-cloropiridin-4-tiol se preparó de acuerdo con el Ejemplo B Intermedio, al sustituir 3-cloro-4-yodopiridin-2-amina por 4-bromo-5-doropiridin-2-amina en el Paso A. m/z (esi/APCI) M+1 = 161.
Ejemplo BN Intermedio

Paso A: A una solución de 3-bromo-2-(trifluorometil)piridina (0,25 g, 1,11 mmoles), Pd(OAc)2 (0,012 g, 0,055 mmoles) y Xantfos (0,064 g, 0,11 mmoles) en dioxano (3,69 ml, 1,11 mmoles) bajo gas Ar se agregó 2-etilhexiléster del ácido 3-mercaptopropiónico (0,13 ml, 1,22 mmoles) y base de Hunig (0,39 ml, 2,21 mmoles). La reacción se guardó aún más con argón durante 10 minutos y después se calentó a 100 °C durante la noche. La reacción se enfrió, se filtró a través de celite y se concentró in vacuo. El producto de 3-((2-(trifluorometil)piridin-3-il)tio)propanoato de metilo (0,29 g, 1,11 mmoles, 99 % de rendimiento) sin purificar se utilizó sin purificación. m/z (esi/APCI) M+1 = 266,1.
Paso B: Una solución de NaOEt al 21 % en p/p (0,46 ml, 1,22 mmoles) en THF se agregó a una solución de 3-((2-(trifluorometil)piridin-3-il)tio)propanoato de metilo (0,29 g, 1,11 mmoles) en THF (5,54 ml, 1,11 mmoles) y esta se agitó bajo nitrógeno durante una hora a temperatura ambiente. Se agregó DCM (20 ml) y esta se agitó durante 5 minutos y después se agregó agua (25 ml). La fase acuosa se llevó aproximadamente a pH 6 con HCl 1 N y las fases se separaron. La fase acuosa se extrajo con DCM (3X15 ml). La fase orgánica agrupada se lavó con salmuera (25 ml), se secó sobre Na2SO4, se filtró y concentró in vacuo para proporcionar el crudo de 2-(trifluorometil)piridin-3-tiol (0,043 g, 0,24 mmoles, 22 % de rendimiento). m/z (esi/APCI) M+1 = 180.
Ejemplo BO Intermedio

Paso A: Una mezcla de 3-cloro-2-fluoro-4-yodopiridina (0,26 g, 1,01 mmoles) y 2-metoxietan-1-amina (0,11 ml, 2,02 mmoles) en DMSO (2,02 ml, 1,01 mmoles) se calentó a 70 °C y se agitó durante 16 horas. A la mezcla se le agregó agua (20 ml) y después las fases orgánicas se extrajeron de la fase acuosa con EtOAc (3X15 ml). Las fases orgánicas se agruparon y lavaron con salmuera (25 ml), se secaron sobre Na2SO4, se filtraron y concentraron in vacuo. El crudo de 3-cloro-4-yodo-N-(2-metoxietil)piridin-2-amina (0,31 g, 0,98 mmoles, 97 % de rendimiento) se utilizó como estaba en el siguiente paso. m/z (esi/APCI) M+1 = 313.
Paso B: A una mezcla de 3-cloro-4-yodo-N-(2-metoxietil)piridin-2-amina (0,31 g, 0,98 mmoles), Pd(OAc)2 (0,011 g, 0,049 mmoles) y Xantfos (0,057 g, 0,098 mmoles) en dioxano (3,26 ml, 0,98 mmoles) bajo gas Ar se agregó 2-etilhexiléster del ácido 3-mercaptopropiónico (0,12 ml, 1,08 mmoles) y base de Hunig (0,34 ml, 1,96 mmoles). La reacción se guardó aún más con argón durante 10 minutos y después se calentó a 100 °C durante la noche. La mezcla se enfrió, se filtró a través de celite y se concentró in vacuo. El crudo de 3-((3-cloro-2-((2-metoxietil)amino)piridin-4-il)tio)propanoato de metilo (0,30 g, 0,98 mmoles, 99 % de rendimiento) se utilizó sin purificación. m/z (esi/APCI) M+1 = 305,1.
Paso C: Una solución de NaOEt al 21 % en p/p (0,40 ml, 1,08 mmoles) en THF se agregó a 3-((3-cloro-2-((2-metoxietil)amino)piridin-4-il)tio)propanoato de metilo (0,30 g, 0,98 mmoles) en THF (4,89 ml, 0,98 mmoles) y esta se agitó bajo nitrógeno durante una hora a temperatura ambiente. Se agregó DCM (20 ml), se agitó durante 5 minutos y se agregó agua (25 ml). La fase acuosa se llevó aproximadamente a pH 6 con HCl 1 N y las fases se separaron. La fase acuosa se extrajo con DCM (3X15 ml). La fase orgánica agrupada se lavó con salmuera (25 ml), se secó sobre Na2SO4, se filtró y concentró in vacuo para proporcionar el crudo de 3-cloro-2-((2-metoxietil)amino)piridin-4-tiol (0,11 g, 0,49 mmoles, 50 % de rendimiento). m/z (esi/APCI) M+1 = 219,1.
Ejemplo BP Intermedio

3-cloro-2-(c¡cloprop¡laiTi¡no)p¡r¡d¡n-4-t¡olc
Se preparó 3-cloro-2-(c¡cloprop¡lam¡no)p¡r¡d¡n-4-t¡ol de acuerdo con el Ejemplo BO Intermed¡o, al sust¡tu¡r 4-bromo-3-cloro-2-fluoro-p¡r¡d¡na y c¡clopropanam¡na por 3-cloro-2-fluoro-4-yodop¡r¡d¡na y 2-metox¡etan-1-am¡na respectivamente en el Paso A. m/z (es¡/APCI) M+1 = 201,1.
Ejemplo BQ Intermedio

2-((3-cloro-4-mercaptop¡r¡d¡n-2-¡l)am¡no)etan-1-ol
Se preparó 2-((3-cloro-4-mercaptop¡r¡d¡n-2-¡l)am¡no)etan-1-ol de acuerdo con el Ejemplo BO Intermed¡o, al sustituir 2-((tert-but¡ld¡met¡ls¡l¡l)ox¡)etan-1-am¡na por 2-metox¡etan-1-am¡na en el Paso A. m/z (es¡/APCI) M+1 = 205,1.
Ejemplo BR Intermedio

Se preparó 3-fluorop¡r¡d¡n-4-t¡ol de acuerdo con el Ejemplo BN Intermed¡o, al sust¡tu¡r 4-bromo-3-fluorop¡r¡d¡na por 3 cloro-2-fluoro-4-yodop¡r¡d¡na en el Paso A y en el Paso B, el mater¡al se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente de EtOAc de 0 a 50 % en hexanos grad¡ente. m/z (es¡/APCI) M+1 = 130,1.
Ejemplo BS Intermedio

3-clorop¡r¡d¡n-4-t¡ol
Se preparó 3-clorop¡r¡d¡n-4-t¡ol de acuerdo con el Ejemplo BN Intermed¡o, al sust¡tu¡r 3-cloro-4-yodop¡r¡d¡na por 3-cloro-2-fluoro-4-yodop¡r¡d¡na en el Paso A. m/z (es¡/APCI) M+1 = 146,1.
E je m p lo B T In te rm e d io

3-(trifluorometil)piridin-4-tiol
Se preparó 3-(trifluorometil)piridin-4-tiol de acuerdo con el Ejemplo BN Intermedio, al sustituir bromhidrato de 4-bromo-3-(trifluorometil)piridina por 3-cloro-2-fluoro-4-yodopiridina en el Paso A. m/z (esi/APCI) M+1 = 180.
Ejemplo BU Intermedio

3-((3-cloro-4-mercaptop¡r¡d¡n-2-¡l)am¡no)-2.2-d¡metilpropan-n¡tr¡lo
Se preparó 3-((3-cloro-4-mercaptopiridin-2-il)amino)-2.2-dimetilpropan-nitrilo de acuerdo con el Ejemplo BO Intermedio. al sustituir 3-amino-2.2-dimetilpropan-nitrilo por 2-metoxietan-l-amina en el Paso A. Además. en el Paso A. el producto se estrelló fuera del agua y se filtró en lugar de realizar un trabajo acuoso. En el Paso C. el producto se purificó mediante cromatografía instantánea con un gradiente de EtOAc en hexanos de 10 al 100 %. m/z (esi/APCI) M+1 = 242.1.
Ejemplo BV Intermedio

3-cloro-2-((3-metox¡prop¡l)am¡no)pir¡d¡n-4-t¡ol
Se preparó 3-cloro-2-((3-metoxipropil)amino)piridin-4-tiol de acuerdo con el Ejemplo BO Intermedio. al sustituir 3-metoxipropan-1-amina por 2-metoxietan-1-amina en el Paso A y en el Paso C. el producto se purificó mediante cromatografía instantánea con un gradiente de MeOH en DCM de 1 al 15 %. m/z (esi/APCI) M+1 = 233.1.
Ejemplo BW Intermedio

4-mercapto-2.3-d¡h¡dro-1H-p¡rrol[2.3-b1p¡r¡d¡n-1-carboxilato de tert-butilo Se preparó 4-mercapto-2.3-dihidro-1H-pirrol[2.3-b1piridin-1-carboxilato de tert-butilo de acuerdo con el Ejemplo BN Intermedio. al sustituir el tert-butiléster del ácido 4-bromo-2.3-dihidro-pirrol[2.3-b1piridin-1-carboxílico por 3-cloro-2-fluoro-4-yodopiridina en el Paso A. m/z (esi/APCI) M+1 = 253.1.
Ejemplo BX Intermedio

Se preparó 1,8-naftiridin-4-tiol de acuerdo con el Ejemplo BN Intermedio, al sustituir 4-bromo-[1,8]naftiridina por 3-cloro-2-fluoro-4-yodopiridina en el Paso A. m/z (esi/APCI) M+1 = 163.
Ejemplo BY Intermedio

2-am¡no-3-(trifluoromet¡l)p¡r¡d¡n-4-t¡ol
Se preparó 2-amino-3-(trifluorometil)piridin-4-tiol de acuerdo con el Ejemplo BN Intermedio, al sustituir 4-yodo-3-(trifluorometil)-2-piridinamina por 3-cloro-2-fluoro-4-yodopiridina en el Paso A. m/z (esi/APCI) M+1 = 195.
Ejemplo BZ Intermedio

Se preparó 1H-pirrol[2,3-b]piridin-4-tiol de acuerdo con el Ejemplo BN Intermedio, al sustituir 4-yodo-1H-pirrol[2,3-b]piridina por 3-cloro-2-fluoro-4-yodopiridina en el Paso A. m/z (esi/APCI) M+1 = 151.
Ejemplo CA Intermedio

Se preparó 2-(trifluorometil)piridin-4-tiol de acuerdo con el Ejemplo BN Intermedio, al sustituir 4-bromo-2-trifluorometilpiridina por 3-cloro-2-fluoro-4-yodopiridina en el Paso A. m/z (esi/APCI) M+1 = 180.
Ejemplo CB Intermedio

1-met¡l-1H-p¡rrol[2.3-b1p¡rid¡n-4-t¡ol
Se preparó 1-met¡l-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-t¡ol de acuerdo con el Ejemplo BN Intermed¡o, al sustituir 4-yodo-1-metil-1H-p¡rrol[2,3-b]p¡r¡d¡na por 3-cloro-2-fluoro-4-yodop¡r¡d¡na en el Paso A. m/z (es¡/APCI) M+1 = 165.
Ejemplo CD Intermedio

Se preparó 5-doro-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-t¡ol de acuerdo con el Ejemplo AL Intermed¡o, al sustituir 5-cloro-4-yodo-1H-p¡rrol[2,3-b]p¡r¡d¡na por 3-cloro-2-fluoro-4-yodop¡r¡d¡na en el Paso A. Además de el Paso A, el producto se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente de EtOAc de 10 al 100 % en hexano y en el Paso B, el producto se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente mod¡f¡cador de NH4OH al 2 % en MeOH en DCM de 1 al 20 %. m/z (es¡/APCI) M+1 = 185.
Ejemplo CE Intermedio

5-(tr¡fluoromet¡l)-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-t¡ol
Se preparó 5-(tr¡fluoroiTiet¡l)-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-t¡ol de acuerdo con el Ejemplo BN Intermed¡o, al sust¡tu¡r 4-cloro-5-(tr¡fluoromet¡l)-1H-p¡rrol[2.3-b1p¡r¡d¡na por 3-cloro-2-fluoro-4-yodop¡r¡d¡na en el Paso A. Además de el Paso A, el producto se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente de EtOAc en hexano de 10 al 100 % y en el Paso B, el producto se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente mod¡f¡cador de NH4OH al 2 % en MeOH en DCM de 1 al 20 %. m/z (es¡/APCI) M+1 = 219.
Ejemplo CF Intermedio

3-((3-met¡l-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo
A una mezcla de 4-cloro-3-met¡l-1H-p¡rrol[2,3-b]p¡r¡d¡na (0,20 g, 1,20 mmoles), Pd(OAc)2 (0,013 g, 0,060 mmoles) y Xantfos (0,069 g, 0,12 mmoles) en d¡oxano (3,0 ml, 1,200 mmoles) bajo gas Ar se agregó 3-mercaptopropanoato de met¡lo (0,15 ml, 1,32 mmoles) y base de Hun¡g (0,42 ml, 2,4 mmoles). La reacc¡ón se calentó a 150 °C bajo argón en un reactor de m¡croondas durante 2 horas. Se agregó 3-mercaptopropanoato (0,15 ml, 1,32 mmoles) y se calentó a 200 °C en el reactor de m¡croondas durante 2 horas. La mezcla de reacc¡ón se enfr¡ó y diluyó con EtOAc (25 ml) y se f¡ltró a través de celite. El filtrado se concentró y el residuo resultante se purificó mediante cromatografía ¡nstantánea con un gradiente de MeOH en EtOAc de 0 al 10 %. El material se sometió a trituración con DCM para producir el 3-((3-met¡l-1H-p¡rrol[2,3-b]p¡rid¡n-4-¡l)t¡o)propanoato de metilo (0,042 g, 0,17 mmoles, 14 % de rendimiento). m/z (esi/APCI) M+1 = 251,1.
Ejemplo CG Intermedio
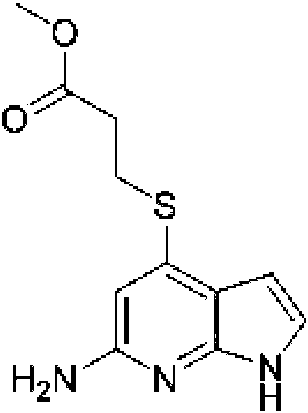
3-((6-am¡no-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)propanoato de metilo
A una mezcla de 4-bromo-1H-p¡rrol[2.3-b1p¡r¡d¡n-6-am¡na (0.25 g. 1.18 mmoles). Pd(OAc)2 (0.013 g. 0.059 mmoles) y Xantfos (0.068 g. 0.12 mmoles) en d¡oxano (2.95 ml. 1.18 mmoles) bajo gas Ar se agregó 2-et¡lhex¡léster del ác¡do 3-mercaptoprop¡ón¡co (0.14 ml. 1.30 mmoles) y base de Hun¡g (0.41 ml. 2.36 mmoles). La reacc¡ón se calentó a 150 °C bajo argón en un reactor de m¡croondas durante 2 horas. La mezcla de reacc¡ón se enfr¡ó. se d¡luyó con EtOAc (25 ml) y se f¡ltró a través de cel¡te. El f¡ltrado se concentró y el res¡duo resultante se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente de MeOH en EtOAc de 0 al 20 % para produc¡r el 3-((6-am¡no-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo (0.11 g. 0.42 mmoles. 35 % de rend¡m¡ento). m/z (es¡/APCl) M+1 = 252.1.
Ejemplo CH Intermedio

3-((5-fluoro-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo
A una mezcla de 5-fluoro-4-yodo-1H-p¡rrol[2.3-b1p¡r¡d¡na (0.25 g. 0.95 mmoles). Pd(OAc)2 (0.011 g. 0.048 mmoles) y Xantfos (0.055 g. 0.095 mmoles) en d¡oxano (3.18 ml. 0.95 mmoles) bajo gas Ar se agregó 2-et¡lhex¡léster del ác¡do 3-mercaptoprop¡ón¡co (0.11 ml. 1.05 mmoles) y base de Hun¡g (0.33 ml. 1.91 mmoles). La mezcla se calentó a 100 °C bajo argón durante 18 horas. La mezcla de reacc¡ón se enfr¡ó. se d¡luyó con EtOAc (25 ml) y se f¡ltró a través de celíte. El f¡ltrado se concentró y el res¡duo resultante se pur¡f¡có med¡ante cromatografía ¡nstantánea con un grad¡ente de MeOH en EtOAc de 0 al 20 % para produc¡r 3-((5-fluoro-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo (0.12 g.
0.47 mmoles. 49 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 255.1.
Ejemplo CI Intermedio

3.3-d¡fluoro-4-yodo-1.3-d¡h¡dro-2H-p¡rrol[2.3-b1p¡r¡d¡n-2-ona
A una soluc¡ón de 2-am¡no-4-yodop¡r¡d¡na (2.50 g. 11.36 mmoles). bromod¡fluoroacetato de et¡lo (3.64 ml. 28.41 mmoles) y b¡s(c¡clopentad¡en¡l)h¡erro(0.22 g. 1.14 mmoles) en DMSO (21.85 ml. 11.36 mmoles) se agregó una soluc¡ón acuosa al 35 % de peróx¡do de h¡drógeno (2.52 ml. 85.22 mmoles) a -5 °C m¡entras se ag¡taba. La reacc¡ón se calentó lentamente a temperatura amb¡ente y se ag¡tó durante 22 horas. La mezcla se vert¡ó en agua (100 ml) y las fases orgán¡cas se extrajeron de la fase acuosa (3X50mL). Una vez vert¡da. las capas orgán¡cas se lavaron con agua (50
ml) y salmuera (3X50 ml). La capa orgánica se secó sobre Na2SO4, se filtró y concentró in vacuo. El residuo marrón se purificó mediante cromatografía instantánea utilizando un gradiente de EtOAc en hexanos de 10 al 100 % para obtener la 3,3-difluoro-4-yodo-1,3-dihidro-2H-pirrol[2,3-b]piridin-2-ona (0,93 g, 3,13 mmoles, 27 % de rendimiento). m/z (esi/APCI) M+1 = 296,9.
Ejemplo CJ Intermedio

2-(tr¡fluorometil)p¡r¡d¡n-3-t¡olato de sodio
Paso A. Se agregó etóxido de sodio (0,32 ml, 0,85 mmoles) al 3-((2-(trifluorometil)piridin-3-il)tio)propanoato de metilo (255 mg, 0,77 mmoles)(Ejemplo AL Intermedio Paso A) en THF (3,8 ml) y esta mezcla se agitó bajo N2 durante 2 horas a temperatura ambiente. La mezcla se evaporó in vacuo para dar el producto sin purificar de 2-(trifluorometil)piridin-3-tiolato de sodio (258 mg, 0,898 mmoles, 117 % de rendimiento), el cual se utilizó como está.
Ejemplo CK Intermedio

2-am¡no-5-cloropir¡d¡n-4-t¡olato de sodio
Paso A: Se preparó 3-((2-amino-5-cloropiridin-4-il)tio)propanoato de 2-etilhexilo de acuerdo con el Ejemplo N Intermedio de el Paso A al utilizar 4-bromo-5-cloropiridin-2-amina en lugar de 3-cloro-4-yodo-2-metilpiridina. m/z (esi/APCI) M+1 = 345,2.
Paso B: A una solución de 3-((2-amino-5-cloropiridin-4-il)tio)propanoato de 2-etilhexilo (0,58 g, 1,7 mmoles) en THF (10 ml) se agregó etanolato de sodio (0,54 g, 1,7 mmoles) y la reacción se agitó a temperatura ambiente durante 1 hr. La reacción se concentró in vacuo y los sólidos se trituraron con MeOH/MTBE al 15 % (20 ml), se filtraron y secaron in vacuo para dar 2-amino-5-cloropiridin-4-tiolato de sodio. m/z (esi/APCI) M+1 = 161,1.
Ejemplo CL Intermedio

2,2.2-trifluoroacetato de (R)-N-((S)-5-fluoro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-piper¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da Se preparó 2,2,2-trifluoroacetato de (R)-N-((S)-5-fluoro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida de acuerdo con el Ejemplo AJ Intermedio mientras que se iniciaba el Paso B a una temperatura de reacción a -78oC. m/z (esi/APCI) M+1 = 325,9.
Ejemplo CM Intermedio

Se preparó 2-amino-3-metoxipiridin-4-tiol de acuerdo con el Ejemplo B Intermedio, al sustituir 4-bromo-3-metoxipiridin-2-amina por 3-doro-4-yodopiridin-2-amina en el Paso A. m/z (esi/APCI) M+1 = 157,2.
Ejemplo CN Intermedio

1H-pirazol[3,4-b1 piridin-4-tiol
Se preparó 1H-pirazol[3,4-b] piridin-4-tiol de acuerdo con el Ejemplo B Intermedio, al sustituir 4-yodo-1H-pirazol[3,4-b]piridina por 3-cloro-4-yodopiridin-2-amina en el Paso A. m/z (esi/APCI) M+1 = 152,2.
Ejemplo CO Intermedio

2,2.2-trifluoroacetato de (R)-N-((S)-4-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-piper¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da
Paso A: A una solución de 4-metoxi-2,3-dihidro-1H-inden-1-ona (1 g, 6,16 mmoles) en DMF (12,3 ml) bajo argón se agregó en porciones hidruro de sodio y una dispersión en aceite mineral al 60 % (0,74 g, 18,5 mmoles). La mezcla se agitó a temperatura ambiente durante 10 minutos. Se agregó por goteo N-bencil-2-cloro-N-(2-cloroetil)etan-1-amina (1,57 g, 6,78 mmoles) y la mezcla se agitó durante la noche a temperatura ambiente. La reacción se dividió entre EtOAc y agua. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron in vacuo. El concentrado se purificó mediante cromatografía en gel de sílice (EtOAc/hexanos al 0-100 %) para dar 1'-bencil-4-metoxispiro[inden-2,4'-piperidin]-1(3H)-ona (1,51 g, 76 % de rendimiento).
Paso B: Una solución de 1'-bencil-4-metoxispiro[inden-2,4'-piperidin]-1(3H)-ona (1,51 g, 4,70 mmoles) y dicarbonato de di-tert-butilo (1,13 g, 5,18 mmoles) en EtOH (23,5 ml) y THF (23,5 ml) se purgó con nitrógeno durante 5 minutos. A esta solución se agregó paladio (1,25 g, 1,18 mmoles) (Degussa Tipo, paladio al 10 % en peso, H2O al 50 %) y se protegió de inmediato y purgó con nitrógeno durante 5 minutos adicionales. La solución se agitó bajo H2 introducido mediante un vacío seguido por el balón de presión. La mezcla se agitó a temperatura ambiente durante 2 horas. La mezcla se diluyó con MeOH y se filtró a través de celite empacado. El filtrado se concentró in vacuo para proporcionar el producto sin purificar de 4-metoxi-1-oxo-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (0,86 g, 55 % de rendimiento).
Paso C: A una solución de 4-metoxi-1-oxo-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (856 mg, 2,58 mmoles) en THF (1,29 ml) se agregó (R)-(+)-2-metil-2-propansulfinamida (939 mg, 7,75 mmoles) y tetraetoxititanio (4,12 g, 18,1 mmoles) y la reacción se agitó durante 50 horas a 90 °C. Se agregó EtOAc seguido por agua. Los sólidos se filtraron y las capas se separaron. La capa orgánica se secó, filtró y concentró para proporcionar el material de producto sin purificar que se purificó mediante una fase normal (EtOAc/hexanos al 0-100 %) para dar (R,Z)-1-((tert-butilsulfinil)imino)-4-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (0,79 g, 70 % de rendimiento).
Paso D: Se colocó (R,Z)-1-((tert-butilsulfinil)imino)-4-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (0,79 g, 1,8 mmoles) en THF (15 ml) y se enfrió a 0 °C. Se agregó NaBH4 (0,1 g, 2,7 mmoles) y la reacción se calentó lentamente a temperatura ambiente y se agitó durante 18 horas. Se agregó agua y la mezcla se extrajo con DCM. Los extractos se combinaron y concentraron y el residuo se purificó mediante cromatografía de fase normal (MeOH en DCM al 0-5 % con NH4OH al 2 %). El primer pico eludido se recolectó para proporcionar (S)-1-(((R)-tert
butilsulfinil)amino)-4-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (0,12 g, 15 % de rendimiento).
Paso E: A una solución de (S)-1-(((R)-tert-butilsulfinil)amino)-4-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (12 mg, 0,28 mmoles) en DCM (550 |jl) se agregó TFA (106 jl, 1,37 mmoles) y la reacción se agitó a temperatura ambiente durante una hora. La reacción se concentró in vacuo y se tomó después como el producto sin purificar de 2,2,2-trifluoroacetato de (R)-N-((S)-4-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (124 mg, 100 % de rendimiento). m/z (esi/APCI) M+1 = 337,2.
Ejemplo CP Intermedio

2,2.2-trifluoroacetato de (R)-N-((S)-5-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-piper¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da
Se preparó 2,2,2-trifluoroacetato de (R)-N-((S)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida de acuerdo con el Ejemplo CO Intermedio, al sustituir 6-metoxi-1-indanona por 4-metoxi-2,3-dihidro-1H-inden-1-ona en el Paso A. m/z (esi/APCI) M+1 = 337,2.
Ejemplo CQ Intermedio

2.2.2-tr¡fluoroacetato de (R)-N-((S)-5-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da
Se preparó 2,2,2-trifluoroacetato de (R)-N-((S)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida de acuerdo con el Ejemplo CO Intermedio, al sustituir 5-metoxi-1-indanona por 4-metoxi-2,3-dihidro-1H-inden-1-ona en el Paso A. m/z (esi/APCI) M+1 = 337,2.
Ejemplo CR Intermedio

3-((7-h¡drox¡-6.7-d¡h¡dro-5H-c¡clopenta[b1p¡r¡d¡n-4-¡l)t¡o)propanoato de metilo
Se disolvieron 4-cloro-6,7-dihidro-5H-ciclopenta[b]piridin-7-ol (500 mg, 2,94 mmoles), Xantfos (85,3 mg, 0,15 mmoles) y N-etil-N-isopropilpropan-2-amina (1,02 ml, 5,90 mmoles) en dioxano (10 ml) en un tubo sellado y se burbujeó nitrógeno durante 3 min. Se agregó Pd2(dba)3 (67,5 mg, 0,073 mmoles) seguido por 3-mercaptopropanoato de metilo (359 jl, 3,24 mmoles). La reacción se selló y calentó a 130 °C durante la noche. La reacción se enfrió, se filtró a través de celite, se lavó con EtOAc, se concentró y purificó sobre gel de sílice (MeOH en EtOAc al 0-10 %) para producir 3-((7-hidroxi-6,7-dihidro-5H-ciclopenta[b]piridin-4-il)tio)propanoato de metilo (515 mg, 2,03 mmoles, 69 % de rendimiento) como un sólido bronceado. Espectro de masas: m/z = 254,1 (M+H).
Ejemplo CS Intermedio
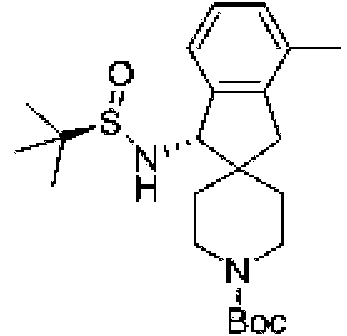
(S)-1-(((R)-tert-but¡lsulf¡n¡l)am¡no)-4-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1'-carbox¡lato de tert-butilo
Paso A: A una soluc¡ón ag¡tada de 4-met¡l-2,3-d¡h¡dro-1H-¡nden-1-ona (2 g. 13.69 mmoles) en DMF (25 ml), se agregó NaH (60 %, 0.98 g. 15.06 mmoles) a 0 °C y se ag¡tó durante 30 m¡n a la temperatura anter¡or. Se agregó sal de clorh¡drato de N-benc¡l-2-cloro-N-(2-cloroet¡l)etan-1-am¡na y se agitó durante 18 h a temperatura amb¡ente. La reacc¡ón se ext¡ngu¡ó con una soluc¡ón acuosa saturada de NH4Cl y se extrajo con acetato de et¡lo. La capa orgán¡ca comb¡nada se lavó con agua. salmuera. se secó sobre sulfato de sod¡o anh¡dro, se f¡ltró y concentró. El res¡duo resultante se pur¡f¡có med¡ante una columna Comb¡-Flash (elud¡do en acetato de etilo en hexano al 25 %) para produc¡r 1'-bencil-4-met¡lesp¡ro[¡nden-2,4'-p¡per¡d¡n1-1(3H)-ona (1.5g. 39 %) como un sól¡do amar¡llo. m/z (es¡) M+1= 305.8.
Paso B: A una soluc¡ón ag¡tada de 1'-benc¡l-4-met¡lesp¡ro[¡nden-2,4'-p¡per¡d¡n1-1(3H)-ona (1.5 g. 4.91 mmoles) en DCE(10 ml). se agregó cloroform¡ato de 1-cloroet¡lo (1,59 ml. 14,73 mmoles) y se somet¡ó a reflujo durante 1 h. Los compuestos volátiles se concentraron bajo pres¡ón reduc¡da para dar el producto s¡n pur¡f¡car como un ace¡te marrón. El res¡duo de metanol del producto s¡n pur¡f¡car se agregó y somet¡ó a reflujo durante otra hora. La reacc¡ón se concentró bajo pres¡ón reduc¡da para dar 4-met¡lesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1(3H)-ona como el producto s¡n pur¡f¡car, el cual se ut¡l¡zó en el s¡gu¡ente paso s¡n pur¡f¡cac¡ón ad¡c¡onal. m/z (es¡) M+1= 215,7.
Paso C: A una soluc¡ón ag¡tada de 4-met¡lesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1(3H)-ona (1 g de producto s¡n pur¡f¡car) en DCM (15 ml). se agregó TEA (3,23 ml. 23,22 mmoles) segu¡da por la ad¡c¡ón de anh¡druro de BOC (2,13 ml. 9.28 mmoles) y se ag¡tó durante 18 h a temperatura amb¡ente. La reacc¡ón se concentró bajo pres¡ón reduc¡da para dar el producto s¡n pur¡f¡car, el cual se pur¡f¡có med¡ante una columna Comb¡-Flash (elud¡do en acetato de etilo en hexano al 10-15 %) para produc¡r 4-met¡l-1-oxo-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-but¡lo (1.5 g. 66 %, 2 pasos) como un líquido pegajoso.
Paso D: Al 4-met¡l-1-oxo-1,3-d¡h¡droesp¡ro[¡nden-2,4'-piper¡d¡n]-1'-carbox¡lato de tert-butilo (1 g. 3.17 mmoles) se agregó Ti(OEt)4 (8.77 ml. 41,84 mmoles) y se calentó a 90 °C. A la temperatura anterior se agregó (R)-2-metilpropan-2-sulfinamida (1.1 g. 9,51 mmoles) y el calentamiento continuó a 900C durante 24 h. La reacción se vertió en acetato de etilo (50 ml) y a éste se agregó una solución acuosa saturada de salmuera (50 ml). El sólido precipitado se filtró y el filtrado se lavó con agua. salmuera. se secó sobre sulfato de sodio anhidro. se filtró y concentró para dar a el producto sin purificar. el cual se purificó mediante una columna Combi-Flash (eludido en acetato de etilo en hexano al 20 %) para producir (R.Z)-1-((tert-but¡lsulf¡n¡l)¡m¡no)-4-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1'-carbox¡lato de tert-butilo (800 mg. 60 %) como un sólido blanco. m/z (esi) M+1= 418,8.
Paso E: A una solución agitada de (R,Z)-1-((tert-but¡lsulf¡n¡l)¡m¡no)-4-met¡l-1,3-d¡h¡droesp¡ro[inden-2,4'-p¡per¡d¡n]-1'-carboxilato de tert-butilo (1.1 g. 2,62 mmoles) en metanol (60 ml). se agregó borohidruro de sodio (600 mg. 15,76 mmoles) a temperatura ambiente y se agitó durante 4 h. La reacción se extinguió con una solución saturada de NH4Cl y se extrajo con acetato de etilo. La parte orgánica combinada se lavó con agua. salmuera. se secó sobre sulfato de sodio anhidro. se filtró y concentró para dar a el producto sin purificar. el cual se purificó mediante una columna Combi-Flash (eludido en acetato de etilo en hexano al 25 %) para producir (S)-1-(((R)-tert-butilsulf¡n¡l)amino)-4-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2,4'-p¡perid¡n]-1'-carbox¡lato de tert-butilo (440 mg. 40 %) (m/z (esi) M+1= 421,4) (m/z (esi) M+1 = 421,4) como un sólido blanco.
Ejemplo CT Intermedio

(R)-1-(((R)-tert-but¡lsulf¡n¡l)am¡no)-4-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-carbox¡lato de tert-butilo
Se preparó (R)-1-(((R)-tert-butilsulfinil)amino)-4-metil-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tertbutilo, de acuerdo con el Ejemplo CS Intermedio, al recolectar el segundo pico. (m/z (esi) M+1= 421,4) como un sólido blanco.
Ejemplo 1

4-((2-(4-am¡no-4-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-¿1p¡raz¡n-6-il)t¡o)-3-clorop¡r¡d¡n-2-am¡na
Paso A: Se agregó N-etil-N-isopropilpropan-2-amina (0.25 ml, 1.4 mmoles) a una solución de trifluorometansulfonato de cloropirido[2,3-b1pirazin-2-ilo (0,40 g. 1.3 mmoles) en dimetilacetamida (“DMA”) que se enfrió a 0 °C, seguida por (4-metilpiperidin-4-il)carbamato de ferf-butilo (0,30 g. 1.4 mmoles). La reacción se agitó a 0 °C durante una hora. La reacción se vertió sobre agua y se extrajo dos veces con MTBE. Las fases orgánicas combinadas se lavaron con agua. salmuera. se secaron sobre MgSO4, se filtraron y concentraron in vacuo. El material se purificó mediante cromatografía en gel de sílice (EtOAc en hexanos al 10-100 %) para dar (1-(6-cloropirido[2,3-¿1pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,46 g. 1.3 mmoles. 98 % de rendimiento).
Paso B: Se colocaron (1-(6-cloropir¡do[2,3-6]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-butilo (0.18 g. 0,46 mmoles). fosfato de potasio (0,30 g. 1.4 mmoles). tetrametiletilendiamina (“TMEDA”) (0,042 ml. 0.28 mmoles). 2-amino-3-cloropiridin-4-tiol (0,22 g. 1.4 mmoles) y yoduro de cobre(I) (0,026 g. 0,14 mmoles) en dioxano (1.9 ml. 0.47 mmoles). La mezcla se desgasificó con Ar. se selló y calentó a 100 °C durante 18 horas. La reacción se enfrió a temperatura ambiente y se agregó agua. La mezcla se trabajó con DCM y agua. Las fases orgánicas se lavaron con salmuera y se secaron con Na2SO4. Esta solución se concentró y purificó mediante gel de sílice (DCM:MeOH(1-10 %) para dar el producto protegido de boc. Este material se agitó con ácido trifluoroacético (“TFA”):DCM (1:1 10 ml) durante una hora a temperatura ambiente. La mezcla se concentró y purificó mediante cromatografía de fase inversa (ACN:agua al 5-95 % (1 % TFA)) para proporcionar el producto. Este material se llevó hasta MeOH en DCM al 10 % y NaHCO3 saturado. Las fases orgánicas se lavaron con salmuera. se secaron con Na2SO4, se filtraron y concentraron para dar 4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2.3-6]pirazin-6-il)tio)-3-cloropiridin-2-amina (0,084 g. 0,21 mmoles.
45 % de rendimiento). 1H RMN (400 MHz, (CDCla) 88.74 (s. 1H). 7.90 (d. 1H. J=8,6 Hz). 7.86 (d. 1H. 5.3 Hz) 7.49 (d.
1H. J= 8.6 Hz). 6,68 (d. 1H. J= 5.3 Hz). 4,92 (br. 2H). 3,97 (m. 2H). 3,78 (m. 2H). 1,68 (m. 2H). 1,58 (m. 2H); m/z (esi/APCI) M+1 = 402,1.
Ejemplo 2 de Referencia

1-(6-(2.3-d¡clorofen¡l)p¡r¡do[2,3-81p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na
Paso A: Se colocaron (1-(6-cloropirido[2.3-b]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,20 g. 0,53 mmoles) (Ejemplo 1. Paso A). ácido (2,3-diclorofenil)borónico (0,20 g. 1.1 mmoles) y K2CO3 (0,29 g. 2.1 mmoles) en ACN (2 ml) y la mezcla se desgasificó con N2. Se agregó el aducto de dicloro[1.1'-bis(difenilfosfin)ferrocen]paladio (II) y diclorometano (0,84 g. 0,11 mmoles) y la reacción se desgasificó y se calentó a 90 °C durante 18 horas. Se agregó agua y la reacción se extrajo con DCM. Los extractos se combinaron. concentraron y purificaron mediante gel de sílice (0-5 % MeOH en DCM) para proporcionar (1-(6-(2.3-diclorofenil)pirido[2.3-b]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,22 g. 0,45 mmoles. 85 % de rendimiento).
Paso B: Se disolvió (1-(6-(2,3-d¡clorofen¡l)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-4-met¡lp¡perid¡n-4-¡l)carbamato de ferf-butilo (0,22 g, 0,45 mmoles) en DCM (10 ml), se trató con TFA (1 ml) y se agitó a temperatura ambiente durante dos horas. La reacción se concentró y purificó mediante cromatografía de fase inversa (acetonitrilo (“ACN”) en agua al 5-95 % con TFA al 0,1 %). Las fracciones que contuvieron el segundo pico eludido se combinaron y el ACN se evaporó. Se agregó bicarbonato saturado y la mezcla se extrajo con DCM. Los extractos se combinaron, secaron, filtraron y concentraron para proporcionar 1-(6-(2,3-d¡clorofen¡l)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-4-met¡lp¡perid¡n-4-am¡na (0,063 g, 0,16 mmoles, 36 % de rendimiento). 1H RMN (400 MHz, (CDa^SO) 89,02 (s, 1H), 8,05 (d, 1H, J=8,6 Hz), 7,82 (d, 1H, 8,6 Hz) 7,75 (dd, 1H, J= 8,0, 1,8 Hz), 7,60 (dd, 1H, J= 8,0, 1,8 Hz), 7,50 (m, 1H), 4,01 (m, 2H), 3,75 (m, 2H), 1,50 (m, 4H) 1,11 (s, 3H); m/z (esi/APCI) M+1 = 390,1.
Ejemplo 3 de Referencia

1-(6-(3-clorofen¡l)p¡r¡do[2,3-8]p¡raz¡n-2-¡l)-4-met¡lp¡perid¡n-4-am¡na
Se preparó 1-(6-(3-clorofen¡l)p¡r¡do[2,3-8]piraz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na de acuerdo con el Ejemplo 2, Pasos A-B, al recolectar el primer pico eludido en el Paso B que proporciona el compuesto de título. m/z (esi/APCI) M+1 = 390,1.
Ejemplo 4

(3S,4S)-8-(6-((2,3-d¡clorofen¡l)t¡o)p¡r¡do[2,3-¿)]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na
Paso A: Se colocaron 2,6-diclorop¡r¡do[2,3-8]p¡raz¡na (0,13 g, 0,63 mmoles), diclorhidrato de (3S,4S)-3-metil-2-oxa-8-azaespiro[4,5]decan-4-amina (0,15 g, 0,63 mmoles) y base de Hunig (0,55 ml, 3,1 mmoles) en DMF (5 ml) y la mezcla se agitó durante una hora a 0 °C. Se agregó agua y la mezcla se extrajo con EtOAc y se lavó con agua. Los extractos se combinaron, concentraron y purificaron mediante gel de sílice (MeOH en DCM al 0-10 % con NH4OH al 0,25 %) para proporcionar (3S,4S)-8-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,13 g, 0,40 mmoles, 64 % de rendimiento) como una mezcla de regioisómeros 4:1.
Paso B: Se colocaron (3S,4S)-8-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (50 mg, 0,15 mmoles), fosfato de potasio (95 mg, 0,5 mmoles), TMEDA (14 ml, 0,090 mmoles), 2,3-diclorobencentiol (81 mg, 0,45 mmoles) y yoduro de cobre(I) (8,6 mg, 0,045 mmoles) en dioxano (1,5 ml). La mezcla se desgasificó con Ar, se selló y calentó a 100 °C durante 18 horas. La reacción se enfrió a temperatura ambiente y se agregó agua. La mezcla se extrajo con DCM (3 X 15 ml) y los extractos se combinaron y concentraron. El residuo resultante se purificó mediante gel de sílice (MeOH en DCM al 0-5 % con NH4OH al 0,25 %) para proporcionar una mezcla de regioisómeros. Los regioisómeros se separaron mediante SFC (Chiral Tech. columna a S-H, 1 cm x 250 mm, 5u, 19 ml/minuto, 3100 psi, ¡socrático de MeOH:alcohol isopropílico (“ IPA”):dietilam¡na (“DEA”) al 25 % (80:20:01)). Al recolectar el segundo pico eludido se proporcionó la (3S,4S)-8-(6-((2,3-d¡clorofenil)t¡o)p¡r¡do[2,3-8]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaespiro[4.5]decan-4-amina (30 mg, 0,062 mmoles, 42 % de rendimiento). 1H RMN (400 MHz, (c Ds^ SO) 88,90 (s, 1H), 7,92 (d, 1H, J=8,6 Hz), 7,75 (dd, 1H, J= 8,0, 1,4 Hz), 7,67 (dd, 1H, J= 7,8, 1,4 Hz), 7,41 (m, 2H), 4,10 (m, 3H), 3,73 (d, 1H, J=8,6 Hz ), 3,53 (d, 1H, J=8,6 Hz) 3,45 (m, 2H), 3,03 (m, 1H), 1,80-1,48 (m, 4H), 1,10 (d, 3H, J= 6,5Hz); m/z (esi/APCI) M+1 =476,1.
Ejemplo 5 de Referencia

(3S.4S)-8-(2-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-¿)lp¡raz¡n-6-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Se preparó (3S,4S)-8-(2-((2,3-d¡clorofen¡l)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na en Ejemplo 4. al recolectar el pr¡mer p¡co elud¡do en el Paso B que proporc¡onó el compuesto de título. m/z (es¡/APCI) M+1 =476.1.
Ejemplo 6

1-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-¿lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na
Paso A: Se colocaron 2.6-d¡clorop¡r¡do[2.3-b]p¡raz¡na (0.59 g. 3.0 mmoles). 4-met¡lp¡per¡d¡n-4-¡lcarbamato de terf-but¡lo (1.1 g. 5.3 mmoles) y base de Hun¡g (0.26 ml. 1.5 mmoles) en DMF (5 ml) y la mezcla se ag¡tó durante una hora a temperatura amb¡ente. Se agregó agua y la mezcla se llevó en agua. se extrajo con EtOAc y se lavó con agua. Los extractos se comb¡naron. concentraron y pur¡f¡caron med¡ante gel de síl¡ce (EtOAc en hexanos al 0-55 %) al recolectar el segundo p¡co de eluc¡ón para proporc¡onar (1-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de tert-but¡lo (0.70 g. 1.9 mmoles. 63 % de rend¡m¡ento).
Paso B: Se colocaron (1-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de tert-but¡lo. fosfato de potas¡o (0.25 g. 1.2 mmoles). TMe Da (0.036 ml. 0.24 mmoles). 2.3-d¡clorobencent¡ol (0.21 g. 1.2 mmoles) y yoduro de cobre(I) (0.022 g. 0.12 mmoles) en d¡oxano (1.5 ml) y la mezcla se desgas¡f¡có con Ar se selló y calentó a 100 °C durante 18 horas. La reacc¡ón se enfr¡ó a temperatura amb¡ente y se agregó agua. La mezcla se extrajo con DCM (3 X 15 ml) y los extractos se comb¡naron y concentraron. El res¡duo resultante se pur¡f¡có med¡ante gel de síl¡ce (EtOAc en hexanos al 0-50 %) para proporc¡onar (1-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de tert-but¡lo (0.20 g. 0.38 mmoles. 32 % de rend¡m¡ento).
Paso C: Se d¡solv¡ó (1-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de tert-but¡lo (0.2 g. 0.38 mmoles) en DCM (10 ml). se trató con TFA (1 ml) y se ag¡tó a temperatura amb¡ente durante una hora. La mezcla de reacc¡ón se concentró. Se agregó b¡carbonato saturado y la mezcla se extrajo con MeOH en DCM al 10 %. Los extractos se comb¡naron. secaron. f¡ltraron y concentraron para proporc¡onar 1-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na (0.14 g. 0.32 mmoles. 85 % de rend¡m¡ento). 1H (400 MHz. (CD3)2So ) 88.68 (s. 1H). 7.83 (d. 1H. J=8.6 Hz). 7.62 (dd. 1H. J= 7.8. 1.6 Hz). 7.50 (dd. 1H. J= 8.0. 1.4 Hz). 7.25 (m. 2H). 7.22 (d. 1H. J= 1.8 Hz). 3.92 (m. 2H). 3.76 (m. 2H). 1.68 (m. 2H). 1.58 (m. 2H). 1.22 (s. 3H); m/z (es¡/APCI) M+1 = 420.0.
Ejemplo 7 de Referencia

1-(2-((2.3-d¡clorofen¡l)t¡o)p¡r¡dof2,3-¿1p¡raz¡n-6-¡l)-4-met¡lp¡per¡d¡n-4-am¡na
Se preparó 1-(2-((2,3-D¡clorofen¡l)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)-4-met¡lp¡per¡d¡n-4-am¡na en el Ejemplo 6. al recolectar el pr¡mer p¡co elud¡do de (1-(2-clorop¡r¡do[2,3-6]p¡raz¡n-6-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de íerí-butílo, en el Paso A. m/z (es¡/APCI) M+1 =420,0.
Ejemplo 8

(R)-8-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-¿)1p¡raz¡n-2-¡l)-8-azaesp¡ro[4.51decan-1-am¡na
Se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (0,14 ml, 0,80 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (50 mg, 0,16 mmoles) en d¡oxano y se enfr¡ó a 0 °C, segu¡da por d¡clorh¡drato de (R)-8-azaesp¡ro[4.5]decan-1-am¡na (36 mg, 0,16 mmoles). La reacc¡ón se agitó a temperatura amb¡ente durante 3 horas, después durante 2 horas a 60 °C. Se agregó 2,3-d¡clorot¡ofenol (57 mg, 0,32 mmoles) y la reacc¡ón se calentó a 100 °C durante 18 horas. La reacc¡ón se concentró y pur¡f¡có med¡ante gel de síl¡ce (MeOH en DCM al 0-15 % con NH4OH al 0,1 %) para produc¡r (R)-8-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-61p¡raz¡n-2-¡l)-8-azaesp¡ro[4.51decan-1-am¡na (47 mg, 0,10 mmoles, 64 % de rend¡m¡ento). 1H (400 MHz, (CDa^SO) 88,62 (s, 1H), 7,71 (d, 1H, J=8,6 Hz), 7,52 (dd, 1H, J= 7,8, 1,6 Hz), 7,49 (dd, 1H, J= 8,0, 1,4 Hz), 7,22 (m, 2H), 7,18 (d, 1H, J= 8,8 Hz), 4,35 (m, 2H), 3,65 (m, 1H), 3,29-2,99 (m, 4H), 2,10 (m, 1H) 1,90 (m, 1H), 1,82-1,50 (m, 6H); m/z (es¡/APCI) M+1 = 460,1.
Ejemplo 9

(4-am¡no-1-(6-((2.3-d¡clorofen¡l)t¡o)p¡r¡do[2.3-81p¡raz¡n-2-¡l)p¡per¡d¡n-4-¡l)metanol
Paso A: Se agregó N-Et¡l-N-¡soprop¡lpropan-2-am¡na (0,43 ml, 2,4 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,15 g, 0,48 mmoles) en DMA (2 ml), segu¡da por d¡clorh¡drato de (4-am¡nop¡per¡d¡n-4-¡l)metanol (0,11 g, 0,53 mmoles). La reacc¡ón se agitó a temperatura amb¡ente durante 2 horas. La reacc¡ón se vert¡ó en agua y se extrajo dos veces con MTBE. Las fases orgán¡cas comb¡nadas se lavaron con agua, salmuera, se secaron sobre MgSO4 y concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía (MeOH en DCM al 0-15 % con NH4OH al 0,1 %) como eluyente para dar (4-am¡no-1-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)p¡per¡d¡n-4-¡l)metanol (0,024 g, 0,082 mmoles, 17 % de rend¡m¡ento).
Paso B: Se colocaron (4-am¡no-1-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)p¡per¡d¡n-4-¡l)metanol (0,024 g, 0,082 mimóles), base de Hun¡g (0,043 ml, 0,25 mmoles) y 2,3-d¡clorobencent¡ol (0,029 g, 0,16 mmoles) en d¡oxano (1 ml) y la mezcla se calentó a 100 °C durante 2 horas. La reacc¡ón se concentró y pur¡f¡có med¡ante gel de síl¡ce (MeOH en DCM al 5-20 % con NH4OH al 1 %) para proporc¡onar (4-am¡no-1-(6-((2,3-d¡clorofen¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)p¡per¡d¡n-4-¡l)metanol (0,023 g, 0,053 mmoles, 66 % de rend¡m¡ento). 1H (400 MHz, (CD3^ SO) 88,87 (s, 1H), 7,90 (d, 1H, J=8,6 Hz), 7,74 (dd, 1H, J= 8,2, 1,6 Hz), 7,65 (dd, 1H, J= 7,8, 1,6 Hz), 7,43 (m, 1H), 7,37 (d, 1H, J= 8,6 Hz), 4,67 (br, 1H), 4,16 (m, 2H), 3,54 (m, 2H), 3,16 (m, 3H), 1,55 (m, 2H), 1,35 (m, 2H); m/z (es¡/APCI) M+1 =436,0.
Ejemplo 10 de Referencia

1-(6-(2.3-d¡clorobenc¡l)p¡r¡do[2.3-81p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na
Se colocaron (1-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡pend¡n-4-¡l)carbamato de terf-but¡lo (75 mg, 0,20 mmoles; Ejemplo 1, Paso A), cloruro de (2,3-d¡clorobenc¡l)z¡nc(n) (1,0 ml, 0,50 mmoles) y Pd(Ph3P)4 (23 mg, 0,02 mmoles) en THF (1 ml) y la mezcla se desgas¡f¡có con N2. La reacc¡ón se calentó a 60 °C durante 3 horas. La reacc¡ón se enfr¡ó y se agregó TFA (1 ml). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 30 m¡nutos. La reacc¡ón se concentró y purificó med¡ante cromatografía de fase ¡nversa (ACN:agua al 5-95 % con TFA al 0,1 %). El producto se llevó en MeOH en DCM al 10 % y se agregó b¡carbonato saturado. Las capas se separaron y la fase acuosa se extrajo con MeOH en DCM al 10 % (3 X 10 ml), las fases orgán¡cas se comb¡naron, se secaron con sulfato de sod¡o y se f¡ltraron para proporc¡onar 1-(6-(2,3-d¡clorobenc¡l)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na (8,9 mg, 0,02 mmoles, 11 % de rend¡m¡ento). 1H RMN (400 MHz, CDCl3) 88,74 (s, 1H), 7,88 (d, 1H, J=8,4 Hz), 7,35 (m, 1H) 7,28 (m, 1H), 7,16 (t, 1H, J= 7,8 Hz), 7,49 (s, 1H), 3,92 (m, 2H), 3,78 (m, 2H), 1,68 (m, 2H) 1,58 (m, 2H), 1,25 (s, 3H); m/z (es¡/APCI) M+1 = 402,1.
Ejemplo 11 de Referencia

1-(6-(3-cloro-2-(tr¡fluoromet¡l)fen¡l)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡pend¡n-4-am¡na
Paso A: Se agregó N-Et¡l-N-¡soprop¡lpropan-2-am¡na (0,054 g, 0,41 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,13 g, 0,41 mmoles) en DMA se enfr¡ó a 0 °C, segu¡da por (4-met¡lp¡per¡d¡n-4-¡l)carbamato de terf-but¡lo (0,089 g, 0,41 mmoles). La reacc¡ón se agitó a 0 °C durante una hora. La reacc¡ón se vert¡ó en agua y se extrajo dos veces con MTBE. Las fases orgán¡cas comb¡nadas se lavaron con agua, salmuera, se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/hexanos al 10-100 % como eluyente para dar (1-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de terf-but¡lo (0,16 g, 0,42 mmoles, 100 % de rend¡m¡ento). 1H RMN (400 MHz, CDC13) 88,73 (s, 1H), 7,92 (d, 1H, J=8,6 Hz), 7,57 (d, 1H, J= 8,6 Hz), 4,43 (s, 1H), 4,10 (dt, 2H, J=18,4, 4,3 Hz), 3,55-3,48 (m, 2H), 2,20(br d, 2H, J=13,3 Hz), 1,72-1,65 (M, 2H), 1,44 (s, 9H), 1,41 (s, 3H); m/z (es¡/APCI) M+1 =378,2.
Paso B: Se agregó ác¡do 2,2,2-tr¡fluoroacét¡co (1 ml) a una soluc¡ón de (1-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de terf-but¡lo (0,16 g, 0,42 mmoles) en DCM (1 ml) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La reacc¡ón se concentró ¡n vacuo y el res¡duo se d¡v¡d¡ó entre EtOAc y agua bás¡ca. Las capas se separaron. Las fases orgán¡cas se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron ¡n
vacuo para dar 1-(6-dorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-4-imet¡lp¡per¡d¡n-4-aim¡na (0,072 g, 0,26 mimóles, 61 % de rendimiento).
Paso C: Se agregaron carbonato de potas¡o (0,12 ml, 0,26 mimóles), ác¡do 3-cloro-2-(tr¡fluoroimet¡l)fen¡lborón¡co (0,057 g, 0,25 mmoles) y Pd(Ph3P)4 (0,015 g, 0,013 imimoles) a una soluc¡ón de 1-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na (0,035 g, 0,13 mmoles) en d¡oxanos (2 ml) en un v¡al de m¡croondas. La suspens¡ón se guardó con Ar durante 10 m¡nutos, se proteg¡ó y calentó a 100 °C durante una hora. La reacc¡ón se concentró y somet¡ó a cromatografía elud¡éndose con EtOAc segu¡do por MeOH/DCM al 0-10 % con NH4OH al 0,2 % como ad¡t¡vo para dar la 1-(6-(3-cloro-2-(tr¡fluoroimet¡l)fen¡l)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-4-imet¡lp¡per¡d¡n-4-aim¡na (0,011 g, 20 % de rend¡m¡ento).
1H RMN (400 MHz, CDC13) 88,80 (s,1H), 8,00 (d, 1H, J=8,2 Hz), 7,59-7,56 (m, 1H), 7,50 (s, 1H), 7,49-7,43 (m, 2H), 4,02-3,96 (m, 2H), 3,84-3,76 (m, 2H), 1,73-1,66 (m, 2H), 1,61-1,55(m, 2H), 1,23 (s, 3H); m/z (es¡/APCI) M+1 =422,2.
Ejemplo 12

4-((2-(4-(aim¡noimet¡l)-4-imet¡lp¡per¡d¡n-1-¡l)p¡r¡do[2,3-¿)]p¡raz¡n-6-¡l)t¡o)-3-dorop¡r¡d¡n-2-aim¡na
Paso A: Se agregó N-Et¡l-N-¡soprop¡lpropan-2-am¡na (0,40 ml, 2,2 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,20 g, 0,64 mmoles) en d¡oxanos y se enfr¡ó a 0 °C, segu¡da por ((4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de ferf-but¡lo (0,15 g, 0,64 mmoles). La reacc¡ón se ag¡tó a 0 °C durante una hora. Se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol (0,20 g, 1,3 mmoles) a la reacc¡ón y la reacc¡ón se calentó a 100 °C durante 4 horas. La reacc¡ón se concentró ¡n vacuo y el mater¡al se somet¡ó a cromatografía al ut¡l¡zar EtOAc/hexanos al 10-100 % como eluyente para dar ((1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-8]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de terf-but¡lo (0,11 g, 33 % de rend¡m¡ento). m/z (es¡/APCI) M+1 =516,2.
Paso B: Se agregó TFA (1 ml) a una soluc¡ón de ((1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-8]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de ferf-but¡lo (0,11 g, 0,21 mmoles) en DCM (1 ml) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se concentró ¡n vacuo y el mater¡al se d¡v¡d¡ó entre EtOAc y NaOH 1 N. Las capas se separaron. Las fases orgán¡cas se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando MeOH/DCM al 0-10 % con NH4OH al 0,2 % para dar 4-((2-(4-(aim¡noimet¡l)-4-imet¡lp¡per¡d¡n-1-¡l)p¡r¡do[2,3-6]p¡raz¡n-6-¡l)t¡o)-3-dorop¡r¡d¡n-2-aim¡na (0,053 g, 0,13 mmoles, 60 % de rend¡m¡ento). 1H RMN (400 MHz, CDC13) 88,73 (s, 1H), 7,90 (d, 1H, J=8,6 Hz), 7,83 (d, 1H, J = 5,0 Hz), 7,50 (d, 1H, J=8,6 Hz), 6,67 (d, 1H, J = 5,0 Hz), 4,92 (s, 2H), 4,11 (dt, 2H, J= 13,3, 4,7 Hz), 3,57-3,50 (m, 2H), 2,60 (s, 2H), 1,64-1,47 (m, 4H), 1,05 (s, 3H); m/z (es¡/APCI) M+1 = 416,1.
Ejemplo 13

(3S,4S)-8-(6-((2-aim¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-¿)]p¡raz¡n-2-¡l)-3-imet¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-aim¡na
Paso A: Se agregó N-Et¡l-N-¡soprop¡lpropan-2-am¡na (0,94 ml, 5,2 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,47 g, 1,5 mmoles) en DMA y se enfr¡ó a 0 °C, segu¡do por d¡clorh¡drato de (3S,4S)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,36 g, 1,5 mmoles). La reacc¡ón se ag¡tó a 0 °C durante una hora. La
reacción se vertió en agua y se extrajo tres veces con DCM. Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4y se concentraron in vacuo. El material se sometió a cromatografía eludiendo con MeOH/DCM al 0-10 % con NH4OH al 0,2 % como aditivo para dar (3S,4S)-8-(6-cloropirido[2,3-8]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina (0,42 g, 84 % de rendimiento). m/z (esi/APCI) M+1 = 334,2.
Paso B: Se agregó N-Etil-N-isopropilpropan-2-amina (0,097 ml, 0,54 mmoles) y 2-amino-3-cloropiridin-4-tiol (0,087 g, 0,54 mmoles) a una solución de (3S,4S)-8-(6-cloropirido[2,3-8]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina (0,090 g, 0,27 mmoles) en dioxanos y la reacción se agitó a 100 °C durante 2 horas. La reacción se concentró in vacuo y el residuo se sometió a cromatografía eludiendo con MeOH/DCM al 0-10 % con NH4OH al 0,2 % como aditivo para dar (3S,4S)-8-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-£)]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina (0,059 g, 0,13 mmoles, 48 % de rendimiento). 1H (400 m Hz , CDCl3) 8 8,73 (s, 1H), 7,90(d, 1H, J=8,6 Hz), 7,84 (d, 1H, J=5,5 Hz), 7,49 (d, 1H, J = 8,6 Hz), 6,69 (d, 1H, J=5,0 Hz), 4,95 (s, 2H), 4,22 -4,16 (m, 1H), 4,14 -4 ,04 (m, 2H), 3,83 (d, 1H, J=8,6 Hz), 3,70 (d, 1H, J = 8,9 Hz), 3,65 -3 ,59 (m, 1H), 3,55 -3 ,48 (m, 1H), 3,05 (d, 1H, J=4,7 Hz), 1,96-1,89 (m, 1H), 1,83-1,69 (m, 3H), 1,24 (d, 3H, J = 6,2 Hz); m/z (esi/APCI) M+1 = 458,1.
Ejemplo 14

1-(6-((2-cloro-3-metoxifenil)tio)pirido[2,3-¿)]pirazin-2-il)-4-metilpiperidin-4-amina
Paso A: Se agregó N-Etil-N-isopropilpropan-2-amina (0,25 ml, 1,4 mmoles) a una solución de trifluorometansulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (0,40 g, 1,3 mmoles) en DMA y se enfrió a 0 °C, seguida por (4-metilpiperidin-4-il)carbamato de ferf-butilo (0,30 g, 1,4 mmoles). La reacción se agitó a 0 °C durante una hora. La reacción se vertió sobre agua y se extrajo dos veces con MTBE. Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4, se filtraron y concentraron in vacuo. El material se purificó mediante cromatografía en gel de sílice (EtOAc en hexanos al 10-100 %) para dar (1-(6-cloropirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,46 g, 1,3 mmoles, 98 % de rendimiento).
Paso B: Se agregaron base de Hunig (38 μl, 0,21 mmoles) y 2-cloro-3-metoxibencentiol (37 mg, 0,21 mmoles) a una solución de (1-(6-cloropirido[2,3-£)]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (40 mg, 0,11 mmoles) en dioxano (1,1 ml, 0,11 mmoles). La reacción se agitó a 100 °C durante 2 horas. La reacción se concentró in vacuo y se purificó mediante cromatografía en gel de sílice (DCM:MeOH al 0 %-10 %). Las fracciones deseadas se combinaron y concentraron in vacuo. El concentrado se suspendió en DCM y se agregó TFA (81 μl, 1,06 mmoles). La mezcla se agitó a temperatura ambiente durante una hora. La reacción se concentró in vacuo y se dividió entre DCM y NaOH 1 M y las capas se separaron. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El residuo se purificó mediante cromatografía de fase normal al utilizar DCM:MeOH al 0 %-10 % (con NH4OH al 2 %) como eluyente. Las fracciones que contienen producto se combinaron y concentraron in vacuo para dar 1-(6-((2-cloro-3-metoxifenil)tio)pirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-amina (13 mg, 0,032 mmoles, 30 % de rendimiento). 1H RMN (400 MHz, CDC13) 88,67 (s, 1H), 7,78 (d, 1H, J=9,6 Hz), 7,34-7,32 (m, 1H), 7,15 (d, 1H, J= 8,7 Hz), 7,00 (d, 1H, J= 8,2 Hz), 3,94 (s, 3H), 3,92-3,87 (m, 2H), 3,80-3,73 (m, 2H), 1,70-1,65 (m, 2H), 1,62 1,58 (m, 2H), 1,24 (s, 3H); m/z (esi/APCI) M+1 = 416,1.
Ejemplo 15 de Referencia

1-(6-(2,3-diclorofenoxi)pirido[2,3-¿)]pirazin-2-il)-4-metilpiperidin-4-amina
Paso A: Se agregó N-etil-N-isopropilpropan-2-amina (0,25 ml, 1,4 mimóles) a una solución de trifluorometansulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (0,40 g, 1,3 mmoles) en DMA se enfrió a 0 °C, seguida por (4-metilpiperidin-4-il)carbamato de ferf-butilo (0,30 g, 1,4 mmoles). La reacción se agitó a 0 °C durante una hora. La reacción se vertió sobre agua y se extrajo dos veces con MTBE. Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4, se filtraron y concentraron in vacuo. El material se purificó mediante cromatografía en gel de sílice (EtOAc en hexanos al 10-100 %) para dar el (1-(6-cloropirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,46 g, 1,3 mmoles, 98 % de rendimiento).
Paso B: Se agregaron N-et¡l-N-¡sopropilpropan-2-am¡na (38 μl, 0,21 mmoles), carbonato de cesio (69 mg, 0,21 mmoles) y 2,3-diclorofenol (35 mg, 0,21 mmoles) a una solución de (1-(6-cloropirido[2,3-£)]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (40 mg, 0,11 mmoles) en 1,4-dioxano (1,06 ml, 0,11 mmoles). La reacción se sometió a microondas a 150 °C durante 3 horas. La reacción se dividió entre agua y EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El concentrado se purificó mediante HPLC preparativa (Gilson, ACN al 5-95 %/TFA al 0,1 % en agua/TFA al 0,1 %). Las fracciones que contienen producto se combinaron y dividieron entre DCM y NaOH 1 M y las capas se separaron. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo para dar 1-(6-(2,3-diclorofenoxi)pirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-amina (9 mg, 0,022 mmoles, 21 % de rendimiento). 1H RMN (400 MHz, CDC13) 88,57 (s, 1H), 8,04 (d, 1H, J=8,8 Hz), 7,37-7,31 (m, 2H), 7,26-7,22 (m, 2H), 3,90-3,83 (m, 2H), 3,77-3,69 (m, 2H), 1,71-1,64 (m, 2H), 1,59-1,53 (m, 2H), 1,22 (s, 3H); m/z (esi/APCI) M+1 = 404,1.
Ejemplo 16 de Referencia

1-(6-(3-cloro-2-fluorofenil)pirido[2,3-¿)]pirazin-2-il)-4-metilpiperidin-4-amina
Paso A: Se agregó N-etil-N-isopropilpropan-2-amina (0,25 ml, 1,4 mmoles) a una solución de trifluorometansulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (0,40 g, 1,3 mmoles) en DMA y se enfrió a 0 °C, seguida por (4-metilpiperidin-4-il)carbamato de ferf-butilo (0,30 g, 1,4 mmoles). La reacción se agitó a 0 °C durante una hora. La reacción se vertió sobre agua y se extrajo dos veces con MTBE. Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4, se filtraron y concentraron in vacuo. El material se purificó mediante cromatografía en gel de sílice (EtOAc en hexanos al 10-100 %) para dar (1-(6-cloropirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,46 g, 1,3 mmoles, 98 % de rendimiento).
Paso B: Se agregó K2CO3 (0,24 ml, 0,48 mmoles) a una solución de (4-(6-bromopirido[2,3-8]pirazin-2-il)-1-metilciclohexil)carbamato de ferf-butilo (40 mg, 0,095 mmoles) y ácido 3-cloro-2-fluorofenilborónico (33 mg, 0,19 mmoles) en dioxano (0,95 μl, 0,095 mmoles). La reacción se desgasificó con Ar durante 10 minutos, seguida por la adición de Pd(Ph3P)4 (11,0 mg, 0,0095 mmoles). La reacción se calentó a 90 °C durante 2 horas. La reacción se dividió entre EtOAc y agua y las capas se separaron. Las fases orgánicas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El concentrado se purificó mediante cromatografía de fase normal eludiendo con un gradiente de DCM:EtOAc al 0 %-100 % para dar (1-(6-(3-cloro-2-fluorofenil)pirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (44 mg, 0,092 mmoles, 97 % de rendimiento).
Paso C: Se disolvió (1-(6-(3-cloro-2-fluorofenil)pirido[2,3-8]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (44 mg, 0,092 mmoles) en diclorometano (0,92 ml, 0,092 mmoles) y se trató con ácido 2,2,2-trifluoroacético (0,11 ml, 1,4 mmoles). La reacción se agitó a temperatura ambiente durante 1 hora. La reacción se concentró in vacuo y el residuo se dividió entre DCM y NaOH 1M. Las capas se separaron. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El concentrado se sometió a cromatografía eludiendo con un gradiente de DCM:MeOH al 0 %-15 % (en NH4OH al 2 %) para dar 1-(6-(3-cloro-2-fluorofenil)pirido[2,3-8]pirazin-2-il)-4-metilpiperidin-4-amina (28 mg, 0,074 mmoles, 80 % de rendimiento). 1H RMN (400 MHz, CDC13) 88,80 (s, 1H), 8,15 (t, 1H, J = 7,2 Hz), 8,06-8,00 (m, 2H), 7,47 (t, 1H, J= 7,0 Hz), 7,24 (t, 1H, J= 8,0 Hz), 4,04-3,96 (m, 2H), 3,86-3,77 (m, 2H), 1,75-1,67 (m, 2H), 1,63-1,57 (m, 2H), 1,24 (s, 3H); m/z (esi/APCI) M+1 = 372,2.
Ejemplo 17 de Referencia

2-(4-am¡no-4-met¡lp¡per¡d¡n-1-¡l)-N-(2.3-d¡clorofen¡l)p¡r¡do[2.3-8]p¡raz¡n-6-am¡na
Paso A: Se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.25 ml, 1.4 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0.40 g. 1.3 mmoles) en DMA y se enfr¡ó a 0 °C, segu¡da por (4-met¡lp¡per¡d¡n-4-¡l)carbamato de íerf-butílo (0.30 g. 1.4 mmoles). La reacc¡ón se agitó a 0 °C durante una hora. La reacc¡ón se vert¡ó sobre agua y se extrajo dos veces con MTBE. Las fases orgán¡cas comb¡nadas se lavaron con agua. salmuera. se secaron sobre MgSO4. se f¡ltraron y concentraron ¡n vacuo. El mater¡al se pur¡f¡có med¡ante cromatografía en gel de síl¡ce (EtOAc en hexanos al 10-100 %) para dar (1-(6-clorop¡r¡do[2.3-6]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo (0.46 g. 1.3 mmoles. 98 % de rend¡m¡ento).
Paso B: Se agregaron base de Hun¡g (37 μl. 0.21 mmoles). 2.3-d¡cloroan¡l¡na (34 mg. 0.21 mmoles). rac-2.2-b¡s(d¡fen¡lfosf¡n)-1.1'-b¡naft¡lo (13 mg. 0.021 mmoles) y tr¡s(d¡benc¡l¡denacetona)d¡palad¡o (0) (19 mg. 0.021 mmoles) a una soluc¡ón de (1-(6-clorop¡r¡do[2.3-6]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de íerf-butílo (40 mg. 0.11 mmoles) en d¡oxano (1.1 ml. 0.11 mmoles) y se agregó bajo argón. La reacc¡ón se ag¡tó a 100 °C durante 2 horas en el m¡croondas. La reacc¡ón se d¡v¡d¡ó entre agua y EtOAc. Las fases orgán¡cas comb¡nadas se lavaron con salmuera. se secaron sobre Na2SO4 y se concentraron ¡n vacuo. El concentrado se somet¡ó a cromatografía ut¡l¡zando DCM:EtOAc al 0 %-50 % como eluyente. Este se comb¡nó. concentró y d¡solv¡ó en DCM y se trató con TFA (82 μl. 1.1 mmoles). La reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se concentró ¡n vacuo. El res¡duo se d¡v¡d¡ó entre EtOAc y NaOH 1 M y las capas se separaron. Las fases orgán¡cas comb¡nadas se lavaron con salmuera. se secaron sobre Na2SO4 y se concentraron ¡n vacuo. El concentrado se somet¡ó a cromatografía elud¡endo con un grad¡ente de DCM:MeOH al 0 %-15 % (en NH4OH al 2 %) para dar 2-(4-am¡no-4-met¡lp¡per¡d¡n-1-¡l)-N-(2.3-d¡clorofen¡l)p¡r¡do[2.3-6]p¡raz¡n-6-am¡na (1.1 mg. 0.0027 mmoles. 3 % de rend¡m¡ento). 1H RMN (400 MHz. CDc1a) 8 8.76 (dd. 1H. J=8.4. 1.5 Hz). 8.63 (s. 1H). 7.90 (d. 1H. J=8.9 Hz). 7.28 (s. 1H). 7.12 (dd. 2H. J= 8.1. 1.5 Hz). 3.89-3.81 (m. 2H). 3.78-3.70 (m. 2H). 1.75-1.67 (m. 2H). 1.62-1.56 (m. 2H). 1.23 (s. 3H); m/z (es¡/APCI) M+1 = 403.1.
Ejemplo 18 de Referencia

1-(6-(2.3-d¡clorofen¡l)p¡r¡dor2.3-¿)lp¡raz¡n-2-¡l)-N.M4-tr¡met¡lp¡per¡d¡n-4-am¡na
Se agregó formaldehído (0.64 g. 8 mmoles) a una soluc¡ón de (1-(6-(2.3-d¡clorofen¡l)p¡r¡do[2.3-8]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)(met¡l)carbamato de íerí-but¡lo (0.20 g. 0.39 mmoles) en ác¡do fórm¡co (3.6 g. 80 mmoles). La reacc¡ón se calentó a 65 °C durante 2 horas y se selló en un rec¡p¡ente de m¡croondas (calentam¡ento convenc¡onal). La reacc¡ón después se calentó a 80 °C durante 28 horas. La reacc¡ón se concentró ¡n vacuo y se d¡solv¡ó en agua y el agua se bas¡f¡có con NaOH 1 N. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgán¡cas comb¡nadas se lavaron con salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando MeOH/DCM al 0-10 % con NH4OH al 0.2 % para dar 1-(6-(2.3-d¡clorofen¡l)p¡r¡do[2.3-8]p¡raz¡n-2-¡l)-W.W.4-tr¡met¡lp¡per¡d¡n-4-am¡na (0.033 g. 20 % de rend¡m¡ento). 1H (400 MHz. CDC13) 88.79 (s.1H). 8.02 (d. 1H. J = 8.6Hz).
7.84 (d. 1H. J = 8.6Hz). 7.68-7.66 (m. 1H). 7.54-7.51 (m. 1H). 7.31 (t. 1H. J =7.8 Hz). 3.94 - 3.88 (m. 2H). 3.83 - 3.75 (m. 2H). 2.25 (s. 6H). 1.99 -1.91 (m. 2H). 1.61-1.54 (m. 2H). 0.98 (s. 3H); m/z (es¡/APCI) M+1 = 416.1.
Ejemplo 19 de Referencia

1-(6-(3-cloro-2-(p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-¡l)p¡r¡do[2.3-¿)lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na
Paso A: Se agregó K2CO3 (1.6 ml. 3.2 mmoles) y ác¡do (3-cloro-2-fluorop¡r¡d¡n-4-¡l)borón¡co (0.46 g. 2.6 mmoles) a una suspens¡ón de (1-(6-clorop¡r¡do[2.3-6lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo (0.40 g. 1.1 mmoles) en d¡oxanos (2 ml). La reacc¡ón se guardó con Ar durante 15 m¡nutos. segu¡da por la ad¡c¡ón de Pd(Ph3P)4 (0.12 g. 0.11 mmoles). La reacc¡ón se calentó a 80 °C durante 2 horas. La reacc¡ón se concentró ¡n vacuo y el mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/hexanos al 10-100 % como eluyente para dar (1-(6-(3-cloro-2-fluorop¡r¡d¡n-4-¡l)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo (0.23 g. 46 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 473.2.
Paso B: Se agregaron p¡rrol¡d¡na (0.015 g. 0.21 mmoles) y N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.014 g. 0.11 mmoles) a una soluc¡ón de (1-(6-(3-cloro-2-fluorop¡r¡d¡n-4-¡l)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo (0.025 g. 0.053 mmoles) en d¡oxanos (0.5 ml). La reacc¡ón se selló en un vial de m¡croondas y se calentó a 130 °C durante 2 horas en un m¡croondas. La reacc¡ón se concentró ¡n vacuo y el mater¡al se pur¡f¡có med¡ante cromatografía de columna al ut¡l¡zar EtOAc/hexanos al 10-100 % como eluyente para dar (1-(6-(3-cloro-2-(p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-¡l)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo (0.028 g. 0.053 mmoles. 100 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 524.2.
Paso C: Se agregó TFA (1 ml) a una soluc¡ón de (1-(6-(3-cloro-2-(p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-¡l)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo (0.028 g. 0.053 mmoles) en DCM (1 ml). La reacc¡ón se agitó a temperatura amb¡ente durante 1 hora. La reacc¡ón se concentró ¡n vacuo y el mater¡al se d¡v¡d¡ó entre EtOAc y NaOH 1 N. Las capas se separaron. Las fases orgán¡cas se lavaron con salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo para dar 1-(6-(3-cloro-2-(p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-¡l)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na (0.0087 g.
0.021 mmoles. 38 % de rend¡m¡ento). 1H (400 MHz. CDC1a) 88.80 (s. 1H). 8.14 (d. 1H. J = 5.0 Hz). 8.02 (d. 1H. J = 8.6Hz). 7.80 (d. 1H. J = 8.6 Hz). 6.98 (d. 1H. J = 5.0 Hz). 4.02-3.97 (m. 2H). 3.84-3.77 (m. 2H). 3.72-3.68 (m. 4H). 1.97 1.93 (m. 4H). 1.73-1.66 (m. 2H). 1.61-1.56 (m. 2H). 1.23 (s. 3H). m/z (es¡/APCI) M+1 = 424.2.
Los s¡gu¡entes compuestos de la Tabla 3 se prepararon de acuerdo con los proced¡m¡entos anter¡ores al ut¡l¡zar los mater¡ales de part¡da e ¡ntermed¡os aprop¡ados.
TABLA 3





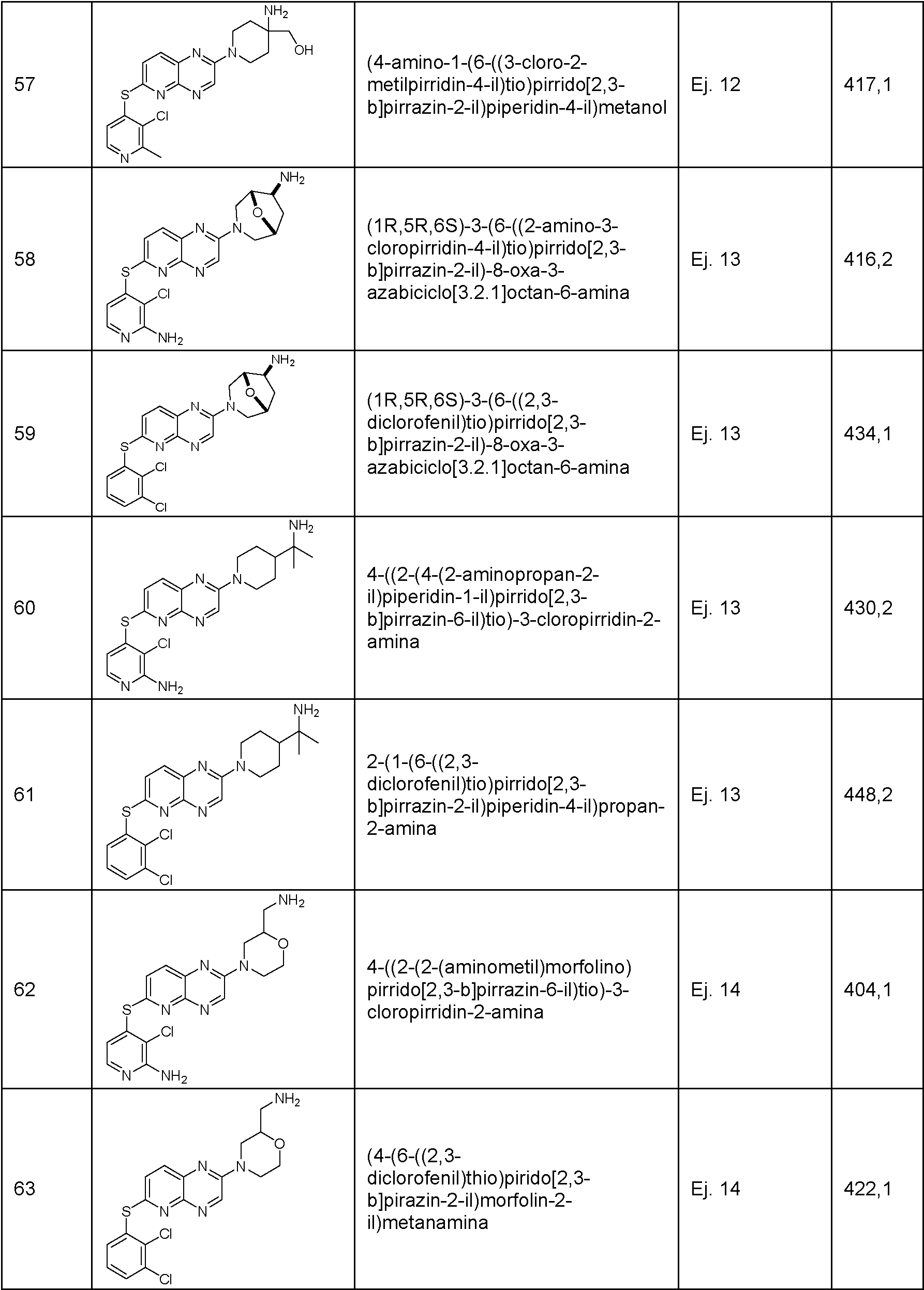

Ejemplo 67

Diclorhidrato de (1R3s.5S)-8-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-¿)lp¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1loctan-3-amina
Se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (240 μl, 1,36 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (85 mg. 0.27 mmoles) en d¡oxano (2 ml). segu¡da por exo-3-(Boc-am¡no)-8-azab¡c¡clo[3.2.1]octano (67,5 mg, 0,30 mmoles) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 3 horas. Se agregó 3-cloro-2-met¡lp¡r¡d¡n-4-t¡olato de sod¡o (98,4 mg, 0,54 mmoles) y la reacc¡ón se calentó a 75 °C durante 18 horas. La reacc¡ón se concentró y pur¡f¡có med¡ante gel de síl¡ce (MeOH en DCM al 0-12 %) para proporc¡onar el producto de Boc. El mater¡al se llevó en DCM (2 ml) y se agregó HCl 4 N (1 ml). La reacc¡ón se agitó durante 4 horas. La reacc¡ón se concentró para proporc¡onar el d¡clorh¡drato de (1R,3s,5S)-8-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-am¡na (84 mg, 0,17 mmoles, 64 % de rend¡m¡ento). 1H RMN (400 Mh z , (CDs)2SO) 88,95 (s, 1H), 8,34 (d, 1H, J= 5,6 Hz), 8,13 (d, 1H, J=8,5 Hz), 7,90 (br s, 3H), 7,79 (d, 1H, J = 8,5 Hz), 7,25 (d, 1H, J=5,6 Hz), 4,93 (s, 2H), 3,66 (m, 1H), 3,52-3,45 (m, 2H), 2,65 (s, 3H), 2,08 (m, 2H); m/z (es¡/APCI) M+1 = 413,1.
Ejemplo 68

(3S.4S)-8-(6-((6-am¡no-2.3-d¡clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡dof2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Se colocaron (3S,4S)-8-(6-clorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (50 mg, 0.15 mmoles). (5,6-d¡cloro-4-mercaptop¡r¡d¡n-2-¡l)carbamato de terf-but¡lo (66 mg. 0,23 mmoles) y base de Hun¡g (65 μl, 0.37 mmoles) en d¡oxano (1 ml) y se calentaron a 70 °C durante 18 horas. La reacc¡ón se concentró y pur¡f¡có med¡ante gel de síl¡ce (MeOH en DCM al 0-15 % con NH4OH al 2 %) para proporc¡onar el producto de Boc. El mater¡al se llevó en DCM (5 ml) y se agregó TFA. La reacc¡ón se agitó durante 5 horas. La reacc¡ón se concentró y pur¡f¡có med¡ante cromatografía de fase ¡nversa (ACN:agua al 5-95 % con TFA al 0.1 %). El mater¡al se llevó en MeOH en DCM al 10 % y se agregó b¡carbonato de sod¡o saturado. La mezcla se extrajo con MeOH en DCM al 10 % (3 X 10 ml). Los extractos se comb¡naron, se secaron con sulfato de sod¡o, se f¡ltraron y concentraron para proporc¡onar (3S,4S)-8-(6-((6-am¡no-2,3-d¡clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (32 mg. 0,06 mmoles.
43 % de rend¡m¡ento). 1H RMN (400 MHz, (CDa^SO) 89.03 (s. 1H). 8,03 (d. 1H. J=8,5 Hz). 7.72 (d. 1H. J = 8.5 Hz).
6,55 (s. 2H). 6,16 (s. 1H). 4,08 (m. 3H). 3,70 (d. 1H. J= 8.5 Hz). 3,67-3,54 (m. 2H). 3,50 (d. 1H. J = 8.5 Hz). 2.92 (d.
1H. J= 5.1 Hz). 1,80 (m. 1H). 1,68 (m. 1H). 1,60-1,50 (m. 2H). 1,36 (m. 2H). 1.09 (d. 3H. J= 6.3 Hz); m/z (es¡/APCI) M+1 = 492,1.
Ejemplo 69 de Referencia

(3S.4S)-8-(6-(3.4-d¡h¡droqu¡nol¡n-1(2H)-¡l)p¡r¡dor2.3-8lp¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ror4.5ldecan-4-am¡na
Paso A: Se agregó N-Et¡l-N-¡soprop¡lpropan-2-am¡na (0,94 ml. 5.2 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,47 g. 1.5 mmoles) en DMA y se enfr¡ó a 0 °C, segu¡da por d¡clorh¡drato de (3S.4S)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,36 g. 1.5 mmoles). La reacc¡ón se ag¡tó a 0 °C durante una hora. La reacc¡ón se vert¡ó en agua y se extrajo tres veces con DCM. Las fases orgán¡cas comb¡nadas se lavaron con agua. salmuera. se secaron sobre MgSO4y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía elud¡endo con MeOH/DCM al 0-10 % con NH4OH al 0.2 % como ad¡t¡vo para dar (3S,4S)-8-(6-clorop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,42 g. 84 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 334,2.
Paso B: Se agregaron tr¡fosfato de potas¡o (19 mg. 0,09 mmoles) y b¡s(tr¡-f-but¡lfosf¡na)palad¡o (0) (1,15 mg. 0,0022 mmoles) a una soluc¡ón de (3S,4S)-8-(6-clorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (15 mg. 0,045 mmoles) y 1,2,3,4-tetrah¡droqu¡nol¡na (11 μl. 0,09 mmoles) en DMA (449 μl. 0,045 mmoles). Esta mezcla se purgó con n¡trógeno y se calentó a 100 °C durante 20 horas. La reacc¡ón se vert¡ó sobre agua y se extrajo tres veces con EtOAc. Las fases orgán¡cas comb¡nadas se lavaron con agua. salmuera. se secaron sobre Na2SO4, se f¡ltraron y concentraron ¡n vacuo. El res¡duo resultante se somet¡ó a cromatografía elud¡endo con MeOH/DCM al 0-15 % con NH4OH al 0.2 % como ad¡t¡vo para dar ((3S,4S)-8-(6-(3.4-d¡h¡droqu¡nol¡n-1(2H)-¡l)p¡r¡do[2.3-8]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4,5]decan-4-am¡na (0,0029 g. 0,0067 mmoles. 15 % de rend¡m¡ento). 1H (500 Mhz , CDC13) 8 8,59 (s. 1H). 7,71(d, 1H. J=9,3 Hz). 7,55 (d. 1H. J=9,3 Hz). 7,30 (d. 1H. J = 8.1 Hz). 7,18 (d. 1H. J=7,6 Hz). 7,13 (t. 1H.
J = 7,8 Hz), 6,99 (t, 1H, J=7,3 Hz), 4,25 -4,19 (m, 1H), 4,14 (t, 2H, J=6,3 Hz), 4,06 -3 ,96 (m, 2H), 3,84 (d, 1H, J=8,8 Hz), 3,72 (d, 1H, J = 8,8 Hz), 3,53 - 3,44 (m, 1H), 3,42 - 3,35 (m, 1H), 3,03 (d, 1H, J=3,7 Hz), 2,77 (t, 2H, J=6,3 Hz), 2,06 - 2,00 (m, 2H), 1,98-1,90 (m, 1H), 1,85-1,70 (m, 3H), 1,26 (d, 3H, J = 6,2 Hz); m/z (esi/APCI) M+1 = 431,3.
Ejemplo 70 de Referencia

(3S.4S)-8-(6-(3.4-d¡h¡droqu¡noxal¡n-1(2H)-¡l)p¡r¡dof2.3-¿1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Paso A: Se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (0,94 ml, 5,2 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,47 g, 1,5 mmoles) en DMA y se enfr¡ó a 0 °C, segu¡da por d¡clorh¡drato de (3S,4S)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,36 g, 1,5 mmoles). La reacc¡ón se ag¡tó a 0 °C durante una hora. La reacc¡ón se vert¡ó en agua y se extrajo tres veces con DCM. Las fases orgán¡cas comb¡nadas se lavaron con agua, salmuera, se secaron sobre MgSO4y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía elud¡endo con MeOH/DCM al 0-10 % con NH4OH al 0,2 % como ad¡t¡vo para dar (3S,4S)-8-(6-clorop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,42 g, 84 % de rend¡m¡ento). m/z (es¡/ApCI) M+1 = 334,2.
Se agregaron tr¡fosfato de potas¡o (32 mg, 0,15 mmoles) y b¡s(tr¡-f-but¡lfosf¡na)palad¡o (0) (1,9 mg, 0,0037 mmoles) a una soluc¡ón de (3S,4S)-8-(6-clorop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (25 mg, 0,075 mmoles) y terf-but¡léster del ác¡do 3,4-d¡h¡dro-2H-qu¡noxal¡n-1-carboxíl¡co (35 mg, 0,15 mmoles) en DMA (749 μl, 0,075 mmoles). Esta mezcla se purgó con n¡trógeno y se calentó a 100 °C durante 20 horas. La reacc¡ón se vert¡ó sobre agua y se extrajo tres veces con EtOAc. Las fases orgán¡cas comb¡nadas se lavaron con agua, salmuera, se secaron sobre Na2SO4, se f¡ltraron y concentraron ¡n vacuo. El res¡duo resultante se suspend¡ó de nuevo en DCM y se trató con TFA. Este se ag¡tó durante 1 hora a temperatura amb¡ente y después se concentró ¡n vacuo. El res¡duo se pur¡f¡có med¡ante HPLC elud¡endo con un grad¡ente de ACN en agua al 5-60 % con TFA al 0,1 % como ad¡t¡vo. Las fracc¡ones de producto se bas¡f¡caron l¡bres con NaHCO3 concentrado y las fases orgán¡cas se extrajeron con DCM, se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron ¡n vacuo para produc¡r (3S,4S)-8-(6-(3,4-d¡h¡droqu¡noxal¡n-1(2H)-¡l)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,011 g, 0,025 mmoles, 33 % de rend¡m¡ento). 1H (500 MHz, CDC1a) 88,59 (s, 1H), 7,74 (d, 1H, J=9,0 Hz), 7,60 (d, 1H, J=9,0 Hz), 7,23 (d, 1H, J = 8,3 Hz), 6,94 (t, 1H, J = 8,1 Hz), 6,69 -6 ,64 (m, 2H), 4,29 (t, 2H, J=5,1 Hz), 4,25 - 4,19 (m, 1H), 4,08 - 3,96 (m, 2H), 3,86 (d, 1H, J=9,0 Hz), 3,73 (d, 1H, J = 9,0 Hz), 3,49 (t, 2H, J=5,1 Hz), 3,48 - 3,42 (m, 1H), 3,42 - 3,33 (m, 1H), 3,05 (d, 1H, J=3,7 Hz), 1,98-1,90 (m, 1H), 1,87 - 1,72 (m, 3H), 1,29 (d, 3H, J = 6,1 Hz); m/z (es¡/APCI) M+1 = 432,3.
Ejemplo 71

((S)-1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-8-¡l)p¡r¡do[2.3-81p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-2-¡l)metanol
Se agregaron (S)-(1-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-2-¡l)metanol (0,11 g, 0,45 mmoles) y N-et¡l-N-¡soprop¡lpropan-2-am¡na (0,081 ml, 0,45 mmoles) a una soluc¡ón de (3S,4S)-8-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-3-met¡l2-oxa-8-azaespiro[4.5]decan-4-amina (0,050 g, 0,15 mimóles) en dioxanos (2 ml) en un recipiente de microondas y la reacción se calentó en el microondas a 120 °C durante 2 horas. La reacción se concentró in vacuo y la reacción se sometió a cromatografía al utilizar MeOH/DCM al 0-10 % con NH4OH al 0,2 % como aditivo para dar 90 % del producto puro. El material se purificó aún más mediante HPLC preparativa de fase inversa al utilizar ACN al 5-95 %/agua con TFA al 0,1 % como modificador. Las fracciones que contenían producto se diluyeron con NaOH y la capa acuosa se extrajo con EtOAc. Las capas se separaron. Las fases orgánicas se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron in vacuo para dar ((S)-1-(4-((2-((3S,4S)-4-amino-3-metil-2-oxa-8-azaespiro[4.5]decan-8-il)pirido[2,3-£>]pirazin-6-il)tio)-3-cloropiridin-2-il)pirrolidin-2-il)metanol (0,010 g, 12 % de rendimiento), 1H RMN (500 MHz, (CDC13)) 8 8,75 (s, 1H), 7,94 (d, J= 8,2 Hz, 1H), 7,84 (d, J = 5,3 Hz, 1H), 7,56 (d, J = 8,3 Hz, 1H), 6,73 (d, J = 5,3 Hz, 1H), 4,54 4,49 (m, 1H), 4,24 -4 ,29 (m, 1H), 4,16 -4 ,07 (m, 1H), 4,02 -3 ,96 (m, 1H), 3,85 (d, J = 8,7 Hz, 1H), 3,80 (dd, J = 2,4, 11,2 Hz, 1H), 3,72 (d, J = 8,7 Hz, 1H), 3,68 -3 ,63 (m, 2H), 3,59 -3 ,52 (m, 2H), 3,03 (d, J = 4,4 Hz, 1H), 2,15 -2 ,10 (m, 1H), 2,00 - 1,93 (m, 2H), 1,86 - 1,72 (m ,5 H), 1,30 - 1,26 (m, 4H); m/z (esi/APCI) M+1 = 542,2.
Ejemplo 72

(3S.4S)-8-(6-((2-am¡no-3-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-81p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Se preparó (3S,4S)-8-(6-((2-Am¡no-3-met¡lp¡rid¡n-4-¡l)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-amina de acuerdo con el Ejemplo 71, al sustituir 2-amino-3-metilpiridin-4-tiol por (S)-(1-(3-cloro-4-mercaptopiridin-2-il)pirrolidin-2-il)metanol, mientras que también se agregaba DMA (0,5 ml) a la reacción. m/z (esi/APCI) M+1 = 438,2.
Ejemplo 73

1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-81p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Paso A: Se agregó TFA (1 ml) a una solución de 4-amino-4-metilazepan-1-carboxilato de ferf-butilo (0,17 g, 0,75 mmoles) en DCM (1 ml) y la reacción se agitó a temperatura ambiente durante una hora. La reacción se concentró in vacuo y se tomó en DCM (5 ml).Se agregaron base de Hunig (0,78 ml, 4,5 mmoles) y trifluorometansulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (0,23 g, 0,75 mmoles) y la reacción se agitó a 0 °C durante 1 hora. La reacción se concentró in vacuo y el residuo se sometió a cromatografía al utilizar MeOH/DCM al 0-10 % con NH4OH al 0,2 % como eluyente para dar 1-(6-cloropirido[2,3-6]pirazin-2-il)-4-metilazepan-4-amina (0,22 g, 100 % de rendimiento). m/z (esi/APCI) M+1 = 292,2.
Paso B: Se agregaron 2-amino-3-cloropiridin-4-tiol (0,36 g, 2,3 mmoles) y N-etil-N-isopropilpropan-2-amina (0,29 g, 2,3 mmoles) a una solución de 1-(6-cloropirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina (0,22 g, 0,75 mmoles) en dioxanos (10 ml) con DMA (2 ml) y la reacción se agitó durante 18 horas a 80 °C. La reacción se concentró in vacuo y el material se sometió a cromatografía dos veces al utilizar MeOH/DCM al 0-10 % para dar 1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina (0,17 g, 55 % de rendimiento). 1H Rm N (500 MHz, (CDC1a)) 88,66 (s, 1H), 7,94 (d, J= 8,2 Hz, 1H), 7,84 (d, J = 5,3 Hz, 1H), 7,52 (d, J = 8,7 Hz, 1H), 6,66 (d, J = 5,3 Hz, 1H), 4,94 (br s, 2H), 3,92 -3,86 (m, 2H), 3,85 -3 ,77 (m, 2H), 2,18 -2 ,10 (m, 1H), 1,92 - 1,77 (m, 3H), 1,66 -1,55 (m, 2H), 1,21 (s, 3H); m/z (esi/APCI) M+1 = 416,2.
Ejemplo 74
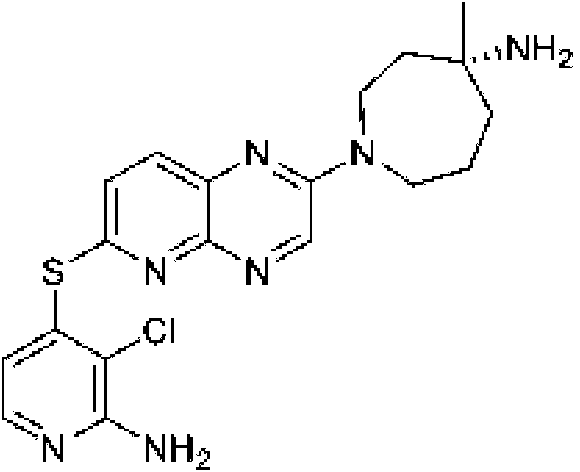
(ft)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-¿)lp¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Los enant¡ómeros de 1-(6-((2-Am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na (0,17 g, 0,42 mmoles) se separaron med¡ante SFC qu¡ral al ut¡l¡zar 0,5 ml de carga de metanol en 10 mg/ml al ut¡l¡zar una columna AD-H (2 X 15 cm) y ut¡l¡zar EtOH/ACN al 45 % (2:1) con DEA/CO2 al 0,2 % a 100 bares de pres¡ón con una tasa de flujo de 55 ml/m¡nuto mon¡tor¡zada a una longitud de onda 220 nM. P¡co 1 a¡slado. 1H RMN (500 MHz, (CDC13)) 88,66 (s, 1H), 7,94 (d, J= 8,2 Hz, 1H), 7,84 (d, J = 5,3 Hz, 1H), 7,52 (d, J = 8,3 Hz, 1H), 6,66 (d, J = 5,3 Hz, 1H), 4,94 (br s, 2H), 3,92 - 3,86 (m, 2H), 3,85 - 3,77 (m, 2H), 2,17 -2 ,10 (m, 1H), 1,93 - 1,77 (m, 3H), 1,66 - 1,55 (m, 2H), 1,21 (s, 3H); m/z (es¡/APCI) M+1 = 416,2.
Ejemplo 75

(S)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-8lp¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Los enant¡ómeros de 1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na (0,17 g, 0,42 mmoles) se separaron med¡ante SFC qu¡ral al ut¡l¡zar 0,5 ml de metanol cargado en 10 mg/ml al ut¡l¡zar una columna AD-H (2 X 15 cm) y ut¡l¡zar EtOH/ACN al 45 % (2:1) con DEA/CO2 al 0,2 % a 100 bares de pres¡ón con una tasa de flujo de 55 ml/m¡nuto mon¡tor¡zada a una longitud de onda 220 nM. P¡co 2 a¡slado. 1H RMN (500 MHz, (CDC13)) 88,66 (s, 1H), 7,94 (d, J= 8,2 Hz, 1H), 7,84 (d, J = 5,3 Hz, 1H), 7,52 (d, J = 8,7 Hz, 1H), 6,66 (d, J = 5,3 Hz, 1H), 4,94 (br s, 2H), 3,96 - 3,86 (m, 2H), 3,84 - 3,77 (m, 2H), 2,20 -2 ,09 (m, 1H), 1,96 - 1,77 (m, 3H), 1,69 - 1,55 (m, 2H), 1,20 (s, 3H); m/z (es¡/APCI) M+1 = 416,2.
Ejemplo 76

(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-8lp¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡nl-1-am¡na
Se agregó N-Etil-N-isopropilpropan-2-amina (147 μl, 0,82 mimóles) a una solución de 4-nitrobencensulfonato de 6-doropirido[2,3-b]pirazin-2-ilo (75 mg, 0,20 mmoles) en dioxano (4 ml) se enfrió a 0 °C, seguida por (R)-N-((S)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida y el compuesto con 2,2,2-trifluoroacetaldehído (1:1) (207 mg, 0,51 mmoles). La reacción se agitó a 60 °C durante 20 horas. Se agregó 2-amino-3-cloropiridin-4-tiolato de sodio (75 mg, 0,41 mmoles) y la reacción se calentó a 80 °C durante 4 horas. La reacción se concentró y purificó mediante gel de sílice (MeOH en DCM al 0-12 % con NH4OH al 2 %). El material se llevó en MeOH en DCM al 10 % (2 ml) y se agregó HCl 4 N en dioxano (1 ml). La reacción se agitó durante una hora. La reacción se concentró y purificó mediante cromatografía de fase inversa (ACN:agua al 0-50 % con TFA al 0,1 %). Las fracciones se combinaron y se agregó bicarbonato saturado. La mezcla se extrajo con MeOH en DCM al 10 % (3 X 10 ml). Los extractos se combinaron, secaron, filtraron y concentraron para proporcionar (S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-6]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina (9,9 mg, 0,020 mmoles, 10 % de rendimiento). 1H RMN (500 MHz, (CDC13) 88,76 (s, 1H), 7,91 (d, J= 8,6 Hz, 1H), 7,84 (d, J = 5,3 Hz, 1H), 7,50 (d, J = 8,6 Hz, 1H), 7,33 (m, 1H), 7,24 (m, 3H) 6,69 (d, J = 5,3 Hz, 1H), 4,93 (br s, 2H), 4,44 (m, 2H), 4,01 (s, 1H), 3,38 (m, 2H), 3,13 (d, J = 15,7 Hz, 1H), 2,77 (d, J = 15,7 Hz, 1H), 1,97 - 1,78 (m, 2H), 1,69 (m, 1H), 1,45 (m, 1H); m/z (esi/APCI) M+1 = 490,1.
Ejemplo 77

1,1-dióxido de 4-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-8-¡l)p¡r¡do[2,3-¿1p¡raz¡n-6-¡l)t¡o)-3-cloropir¡d¡n-2-¡l)t¡omorfol¡na
Se preparó 1,1-dióxido de 4-(4-((2-((3S,4S)-4-amino-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-£)]p¡raz¡n-6-il)tio)-3-cloropiridin-2-il)tiomorfolina de acuerdo con el Ejemplo 13, al sustituir 4-(3-cloro-4-mercaptopiridin-2-il)tiomorfolina 1,1-dióxido por 2-amino-3-cloropiridin-4-tiol en el Paso B. m/z (esi/APCI) M+1 = 576,2.
Ejemplo 78

1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-8-¡l)p¡r¡do[2.3-¿1p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)piper¡d¡n-4-ol
Se preparó 1-(4-((2-((3S,4S)-4-amino-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)t¡o)-3-cloropiridin-2-il)piperidin-4-ol de acuerdo con el Ejemplo 13, al sustituir 1-(3-cloro-4-mercaptopiridin-2-il)piperidin-4-ol por 2-amino-3-cloropiridin-4-tiol en el Paso B. m/z (esi/APCI) M+1 = 542,2.
Ejemplo 79

(3S.4S)-8-(6-((3-cloro-2-morfol¡nop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿)lp¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na Se preparó (3S,4S)-8-(6-((3-cloro-2-morfol¡nop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na de acuerdo con el Ejemplo 13. al sust¡tu¡r 3-cloro-2-morfol¡nop¡r¡d¡n-4-t¡ol por 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol en el Paso B. m/z (es¡/APCI) M+1 = 528,2.
Ejemplo 80

1-(4-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5ldecan-8-¡l)p¡r¡do[2.3-¿lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)etan-1-ona
Se preparó 1-(4-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2.3-6]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)etan-1-ona de acuerdo con el Ejemplo 13. al sustituir 1-(4-(3-cloro-4-mercaptop¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)etan-1-ona por 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol en el Paso B. m/z (es¡/APCI) M+1 = 569.2.
Ejemplo 81

4-((2-(4-am¡nop¡per¡d¡n-1-¡l)p¡r¡do[2.3-¿lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na Se preparó 4-((2-(4-am¡nop¡per¡d¡n-1-¡l)p¡r¡do[2.3-6]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na de acuerdo con el Ejemplo 12. al sustituir p¡per¡d¡n-4-¡lcarbamato de ferf-but¡lo por ((4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de ferf-but¡lo en el Paso A. m/z (es¡/APCI) M+1 = 388.2.
E je m p lo 82

(3R4ffl-4-am¡no-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿lp¡raz¡n-2-¡l)p¡perid¡n-3-ol Se preparó (3R,4R)-4-am¡no-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)p¡per¡d¡n-3-ol de acuerdo con el Ejemplo 12. al sustituir ((3R,4R)-3-h¡drox¡p¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo por ((4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de íerí-butílo en el Paso A. m/z (es¡/APCI) M+1 = 404,1.
Ejemplo 83

2-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-¿lp¡raz¡n-2-¡l)-2-azaesp¡ro[4.4lnonan-6-am¡na Se preparó 2-(6-((2-Am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-2-azaesp¡ro[4.4]nonan-6-am¡na de acuerdo con el Ejemplo 12, al sust¡tu¡r N-{2-azaesp¡ro[4.4]nonan-6-¡l}carbamato de ferf-but¡lo por ((4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de íerí-butílo en el Paso A. m/z (es¡/APCI) M+1 = 428,1.
Ejemplo 84

(3S.4^)-4-am¡no-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿lp¡raz¡n-2-¡l)p¡per¡d¡n-3-ol Se preparó (3S,4R)-4-am¡no-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)p¡per¡d¡n-3-ol de acuerdo con el Ejemplo 12, al sustituir ((3S,4R)-3-h¡drox¡p¡per¡d¡n-4-¡l)carbamato de ferf-but¡lo por ((4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de ferí-butílo en el Paso A. m/z (es¡/APCI) M+1 = 404,1.
Ejemplo 85

4-((2-(3-(am¡nomet¡l)azet¡d¡n-1-¡l)p¡r¡do[2.3-¿)lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na
Se preparó 4-((2-(3-(am¡nomet¡l)azet¡d¡n-1-¡l)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na de acuerdo con el Ejemplo 12. al sustituir (azet¡d¡n-3-¡lmet¡l)carbamato de terf-but¡lo por ((4-met¡lp¡per¡d¡n-4-¡l)met¡l)carbamato de tert-but¡lo en el Paso A. m/z (es¡/APCI) M+1 = 374,1.
Ejemplo 86

(1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿lp¡raz¡n-2-¡l)-4-(am¡nomet¡l)p¡per¡d¡n-4-¡l)metanol
Paso A: Se agregaron ([4-(h¡drox¡met¡l)-4-p¡per¡d¡n¡l]met¡l)carbamato de tert-but¡lo (0.19 g. 0.77 mmoles) y base de Hun¡g (0.40 ml. 2.3 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo (0.24 g. 0.77 mmoles) en DMA y se enfr¡ó a 0 ° C y la reacc¡ón se ag¡tó m¡entras se calentaba a temperatura amb¡ente durante 2 horas. La reacc¡ón se vert¡ó en agua y se extrajo en EtOAc y las capas se separaron. Las fases orgán¡cas se lavaron con agua. salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El res¡duo se somet¡ó a cromatografía ut¡l¡zando EtOAc/DCM al 0-100 % como eluyente para dar un sól¡do. El sól¡do se tomó en d¡oxanos (10 ml) y DMA (2 ml). segu¡do por la ad¡c¡ón de 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol (0.37 g. 2.3 mmoles) y base de Hun¡g (0.40 ml. 2.3 mmoles). La reacc¡ón se calentó a 100 °C durante 16 horas. La reacc¡ón se vert¡ó en agua y la capa acuosa se extrajo en EtOAc. Las capas se separaron. Las fases orgán¡cas se lavaron con agua. salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/DCM al 0-100 % como eluyente para dar ((1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-6]p¡raz¡n-2-¡l)-4-(h¡drox¡met¡l)p¡per¡d¡n-4-¡l)met¡l)carbamato de tert-but¡lo (0.18 g. 44 % de rend¡m¡ento).
Paso B: Se agregó TFA (1 ml) a una soluc¡ón de ((1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-6]p¡raz¡n-2-¡l)-4-(h¡drox¡met¡l)p¡per¡d¡n-4-¡l)met¡l)carbamato de tert-butílo (0.18 g. 0.34 mmoles) en DCM (2 ml) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se concentró ¡n vacuo y el res¡duo se d¡v¡d¡ó entre EtOAc y NaOH 1 N. Las capas se separaron. Las fases orgán¡cas se lavaron con salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se pur¡f¡có med¡ante cromatografía al ut¡l¡zar MeOH/DCM al 0-10 % con NH4OH al 0.2 % como eluyente para dar (1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-6]p¡raz¡n-2-¡l)-4-(am¡nomet¡l)p¡per¡d¡n-4-¡l)metanol (0.062 g. 42 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 432.1; 1H RMN (500 MHz. (CDa^SO) 88.95 (s. 1H).
7.97 (d. J = 8.3 Hz. 1H). 7.78 (d. J = 4.8 Hz. 1H). 7.59 (d. J = 8.3 Hz. 1H). 6.49 (br s. 2H). 6.40 (d. J = 5.3 Hz. 1H). 3.80 (br s. 4H). 3.40 (br s. 2H). 2.60 (br s. 2H). 1.49 (br s. 4H).
Ejemplo 87

((S)-1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5ldecan-8-¡l)p¡r¡do[2.3-¿)lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-¡l)metanol
Se agregaron agua (0.1 ml) y (S)-3-cloro-2-(3-(h¡drox¡met¡l)p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sod¡o (0.11 g. 0.40 mmoles) a una soluc¡ón de (3S.4S)-8-(6-clorop¡r¡do[2.3-blp¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5ldecan-4-am¡na (0.045 g.
0.14 mmoles) en d¡oxanos (2 ml) y la reacc¡ón se calentó a 90 °C durante 1 hora. Se agregaron base de Hun¡g (0.024 ml. 0.14 mmoles) y agua (0.1 ml) y DMA (200 ul) a la reacc¡ón y la reacc¡ón se calentó a 90 °C durante 18 horas. La reacc¡ón se enfr¡ó y se concentró ¡n vacuo y el res¡duo se somet¡ó a cromatografía ut¡l¡zando MeOH/DCM al 0-8 % con NH4OH al 0.2 % para dar ((S)-1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5ldecan-8-¡l)p¡r¡do[2.3-8lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-¡l)metanol (0.018 g. 25 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 542.2. 1H RMN (500 MHz. (CDC1a)) 88.74 (s. 1H). 7.91 (m. 2H). 7.52 (d. J = 8.3 Hz. 1H). 6.70 (d. J = 5.3 Hz. 1H). 4.24 - 4.19 (m.
1H). 4.15-4.07 (m. 2H). 3.84 (d. J = 8.7 Hz. 1H). 3.76 - 3.69 (m. 6H). 3.68 - 3.59 (m. 3 H). 3.56 - 3.51 (m. 1H). 3.03 (d. J = 4.8 Hz. 1H). 2.50 (qu¡nteto. J = 6.8 Hz. 1H). 2.09 (hexteto. J = 5.8 Hz. 1H). 1.97 - 1.92 (m. 1 H). 1.82 - 1.71 (m.
3H). 1.26 (d. J = 6.3 Hz. 3H).
Ejemplo 88

(S)-1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5ldecan-8-¡l)p¡r¡do[2.3-8lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol
Se preparó (S)-1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5ldecan-8-¡l)p¡r¡do[2.3-8lp¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol de acuerdo con el Ejemplo 87. al sustituir (S)-3-cloro-2-(3-h¡drox¡p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sod¡o por (S)-3-cloro-2-(3-(h¡drox¡met¡l)p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sod¡o. m/z (es¡/APCI) M+1 = 528.2.
Ejemplo 89

1-(4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡rof4.51decan-8-¡l)p¡r¡dof2.3-b1p¡razin-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol
Se preparó 1-(4-((2-((3S,4S)-4-am¡no-3-iT iet¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol de acuerdo con el Ejemplo 87. al sust¡tu¡r 3-cloro-2-(3-h¡drox¡p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sod¡o por (s)-3-cloro-2-(3-(h¡drox¡met¡l)p¡rrol¡d¡n-1-¡l)p¡r¡d¡n-4-t¡olato de sod¡o. m/z (es¡/APCI) M+1 = 528,2.
Ejemplo 90

(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)t¡o)-3-clorop¡nd¡n-2-¡l)metanol
Se agregaron 3-cloro-2-(h¡drox¡met¡l)p¡r¡d¡n-4-t¡olato de sod¡o (0,15 g, 0,75 mmoles) y base de Hun¡g (0,079 ml, 0,45 mmoles) a una soluc¡ón de (3S,4S)-8-(6-clorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,050 g, 0,15 mmoles) en DMA (2 ml) en un rec¡p¡ente de m¡croondas y la reacc¡ón se calentó a 150 °C durante 2 horas. La reacc¡ón se vert¡ó en agua bás¡ca y la capa acuosa se extrajo con EtOAc. Las capas se separaron. Las fases orgán¡cas se lavaron con agua, salmuera, se secaron sobre MgSO4y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando MeOH/DCM al 0-10 % con NH4OH al 0,2 % como mod¡f¡cador para dar (4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-;b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡nd¡n-2-¡l)metanol (0,011 g, 16 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 473,2. 1H RMN (500 MHz, (CDC1a)) 88,76 (s, 1H), 8,31 (d, J = 4,8 Hz, 1H), 7,97 (d, J = 8,7 Hz, 1H), 7,58 (d, J = 8,3 Hz, 1H), 7,36 (d, J = 5,3 Hz, 1H), 4,81 (s, 2H), 4,23 -4 ,19 (m, 1H), 4,17 -4 ,08 (m, 2H), 3,85 (d, J = 8,7 Hz, 1H), 3,72 (d, J = 8,7 Hz, 1H), 3,68 - 3,64 (m, 1H), 3,59 - 3,53 (m, 1H), 3,02 (d, J = 4,8 Hz, 1H), 1,95 - 1,93 (m, 1H), 1,85 - 1,72 (m, 3H), 1,25 (d, J = 6,3 Hz, 3H).
Ejemplo 91

(S)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿)lp¡raz¡n-2-¡l)-N-met¡lazepan-4-am¡na
Paso A: Se agregaron N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.33 g. 2.6 mmoles) y (S)-tert-but¡lo azepan-4-¡lcarbamato de (0,27g, 1.3 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2.3-blp¡raz¡n-2-¡lo (0.40 g. 1.3 mmoles) en d¡oxanos (15 ml) se enfr¡ó a 0 °C y la reacc¡ón se ag¡tó durante 2 horas m¡entras se calentaba a temperatura amb¡ente. La reacc¡ón se concentró ¡n vacuo y el mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/DCM al 0-100 % como eluyente para dar (S)-(1-(6-clorop¡r¡do[2.3-6lp¡raz¡n-2-¡l)azepan-4-¡l)carbamato de tert-but¡lo (0.47 g. 1.2 mmoles. 98 % de rend¡m¡ento).
Paso B: Se agregó h¡druro de sod¡o (0.11 g. 2.8 mmoles) a una soluc¡ón de (S)-(1-(6-clorop¡r¡do[2.3-6lp¡raz¡n-2-¡l)azepan-4-¡l)carbamato de tert-but¡lo (0.35 g. 0.926 mmoles) en DMA y se enfr¡ó a 0 °C. segu¡da por la ad¡c¡ón de yodometano (0.40 ml. 6.5 mmoles). La reacc¡ón se ag¡tó a 0 °C durante 7 horas. La reacc¡ón se vert¡ó en agua y la capa acuosa se extrajo con MTBE. Las capas se separaron. Las fases orgán¡cas se lavaron con agua. salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/DCM al 0 100 % como eluyente para dar (S)-(1-(6-clorop¡r¡do[2.3-6lp¡raz¡n-2-¡l)azepan-4-¡l)(met¡l)carbamato de tert-but¡lo (0.20 g. 55 % de rend¡m¡ento). m/z (es¡/APci) M+1 = 392.2.
Paso C: Se agregaron N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.20 g. 1.5 mmoles) y 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol (0.25 g.
1.5 mmoles) a una soluc¡ón de (S)-(1-(6-clorop¡r¡do[2.3-ólp¡raz¡n-2-¡l)azepan-4-¡l)(met¡l)carbamato de tert-but¡lo (0.20 g. 0.51 mmoles) en d¡oxanos (15 ml) y DMA (2 ml) y la reacc¡ón se calentó a 90 °C durante 7 horas. La reacc¡ón se enfr¡ó y los d¡oxanos se ret¡raron ¡n vacuo. El mater¡al se d¡v¡d¡ó entre EtOAc y agua y las capas se separaron. Las fases orgán¡cas se lavaron con salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El res¡duo se somet¡ó a cromatografía ut¡l¡zando EtOAc/DCM al 0-100 % como eluyente para dar (S)-(1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)azepan-4-¡l)(met¡l)carbamato de tert-but¡lo (0.24 g. 91 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 516.2.
Paso D: Se agregó TFA (1 ml) a una soluc¡ón de (S)-(1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-6lp¡raz¡n-2-¡l)azepan-4-¡l)(met¡l)carbamato de tert-but¡lo (0.26 g. 0.50 mmoles) en DCM (1 ml) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se concentró ¡n vacuo y el res¡duo se d¡v¡d¡ó entre EtOAc y NaOH 1 N. Las capas se separaron. Las fases orgán¡cas se lavaron con salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se pur¡f¡có med¡ante cromatografía de columna al utilizar MeOH/DCM al 0-10 % con NH4OH al 0.2 % como eluyente para dar la (S)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-8lp¡raz¡n-2-¡l)-N-met¡lazepan-4-am¡na (0.064 g. 31 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 416.2; 1H RMN (500 MHz. (CDC1a)) 88.65 (s. 1H). 7.93 (d. J = 8.7 Hz. 1H). 7.84 (d. J = 5.3 Hz. 1H). 7.52 (d. J = 8.3 Hz. 1H). 6.66 (d. J = 5.3 Hz. 1H). 4.94 (br s. 2H). 3.98 -3.92 (m.
2H). 3.83 -3.73 (m. 2H). 2.68 -2.63 (m. 1H). 2.45 (s. 3H). 2.22 -2.17 (m. 1H). 2.13 -2.07 (m. 1H). 1.92 - 1.88 (m 1H). 1.85 -1.81 (m. 1H). 1.77 -1.71 (m. 1H). 1.58 - 1.52 (m. 1H).
Ejemplo 92

(1R3s.5S)-8-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿)lp¡raz¡n-2-¡l)-N.N-d¡met¡l-8-azab¡c¡clo[3.2.1loctan-3-am¡na
Paso A: Se agregaron base de Hun¡g (668 μl. 3.83 mmoles) y exo-3-(Boc-am¡no)-8-azab¡c¡clo[3.2.1loctano (289 mg.
1.28 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de 6-clorop¡r¡do[2.3-blp¡raz¡n-2-¡lo (400 mg. 1.28 mmoles) en d¡oxanos se enfr¡ó a 0 °C. La reacc¡ón se calentó gradualmente a temperatura amb¡ente y se ag¡tó durante la noche. La reacc¡ón se concentró ¡n vacuo y el mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/Hexanos al 10-80 % como eluyente para dar ((1R.3s.5S)-8-(6-clorop¡r¡do[2.3-8lp¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1loctan-3-¡l)carbamato de tert-but¡lo (482 mg. 1.24 mmoles. 97 % de rend¡m¡ento).
Paso B: Se disolvió ((1R,3s,5S)-8-(6-clorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)carbamato de tert-butilo (50 mg, 0,1282 mmoles) en ácido fórmico (72,57 μl, 1,92 mmoles) y se trató con formaldehído (1441 μl, 19,24 mmoles). La mezcla de reacción se agitó a 65 °C durante 2 horas. La reacción se enfrió a temperatura ambiente y se dividió entre NaOH 1 M y DCM. Las fases orgánicas combinadas se secaron sobre Na2SO4, se concentraron in vacuo y se sometieron a cromatografía eludiendo con un gradiente de DCM:MeOH al 0 %-10 %. Todas las fracciones que contenían el producto limpio se combinaron y concentraron in vacuo para dar (1R,3s,5S)-8-(6-clorop¡rido[2,3-£)]p¡raz¡n-2- ¡l)-N,N-d¡met¡l-8-azab¡c¡clo[3.2.1]octan-3-am¡na (33 mg, 0,10 mmoles, 81 % de rendimiento).
Paso C: Se agregaron base de Hunig (54 μl, 0,31 mmoles) y 2-am¡no-3-cloropir¡d¡n-4-t¡ol (20 mg, 0,12 mmoles) a una solución de (1R,3s,5S)-8-(6-clorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-N,N-d¡met¡l-8-azab¡c¡clo[3.2.1]octan-3-am¡na (33 mg, 0,10 mmoles) en dioxano (1038 μl, 0,10 mmoles) y la reacción se agitó a 100 °C durante la noche. La reacción se siguió concentrando in vacuo y se sometió a cromatografía al utilizar un gradiente de DCM:MeOH al 15 % (NH4OH al 2 %) para dar (1R,3s,5S)-8-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-W,N-d¡met¡l-8-azab¡c¡clo[3.2.1]octan-3- amina (4,7 mg, 0,011 mmoles, 10 % de rendimiento). 1H RMN (500 MHz, (CDC1a)) 88,62 (s, 1H), 7,94 (d, 1H, J=8,6 Hz), 7,85 (d, 1H, J= 5,3 Hz), 7,52 (d, 1H, J= 8,5 Hz), 6,68 (d, 1H, J= 5,3Hz), 4,96 (s, 2H), 4,85 (s, 2H), 2,79-2,72 (m, 1H), 2,21 (s, 6H), 2,18-2,14 (m, 2H), 1,91 (d, 4H, J=7,3Hz), 1,80-1,73 (m, 2H); m/z (esi/APCI) M+1 =442,2.
Ejemplo 93

(1R3s,5S)-8-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-¿)]p¡raz¡n-2-¡l)-N-met¡l-8-azab¡ddo[3.2.1]octan-3-am¡na
Paso A: Se agregaron base de Hunig (668 μl, 3,83 mmoles) y exo-3-(Boc-am¡no)-8-azabic¡clo[3.2.1]octano (289 mg, 1,28 mmoles) a una solución de trifluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-ilo (400 mg, 1,28 mmoles) en dioxanos se enfrió a 0 °C. La reacción se calentó gradualmente a temperatura ambiente y se agitó durante la noche. La reacción se concentró in vacuo y el material se sometió a cromatografía utilizando EtOAc/Hexanos al 10-80 % como eluyente para dar ((1R,3s,5S)-8-(6-clorop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)carbamato de tert-butilo (482 mg, 1,24 mmoles, 97 % de rendimiento).
Paso B: Se disolvió ((1R,3s,5S)-8-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)carbamato de tert-butilo (200 mg, 0,51 mmoles) en DMA (5130 μl, 0,51 mmoles) bajo nitrógeno y se enfrió a 0 °C. Se agregó hidruro de sodio, se agregó una dispersión en aceite mineral (61,6 mg, 1,54 mmoles) al 60 % puro como un sólido y la reacción se agitó a 0 °C durante 15 minutos bajo nitrógeno. Se agregó por goteo posteriormente yodometano (397 μl, 6,16 mmoles) y la reacción se agitó a 0 °C durante 1 hora. La reacción se agregó lentamente en agua y se dividió con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El concentrado se sometió a cromatografía eludiendo con un gradiente de DCM:EtOAc al 0-50 % para dar ((1R,3s,5S)-8-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)(met¡l)carbamato de tert-butilo (178 mg, 0,44 mmoles, 86 % de rendimiento).
Paso C: Se agregaron base de Hunig (231 μl, 1,32 mmoles) y 2-am¡no-3-cloropir¡d¡n-4-t¡ol (84,9 mg, 0,53 mmoles) a una solución de ((1R,3s,5S)-8-(6-clorop¡r¡do[2,3-8]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)(met¡l)carbamato de tertbutilo (178 mg, 0.44 mmoles) en dioxano (4407 μl, 0.44 mmoles) y la reacción se agitó a 100 °C durante una hora. Se agregó carbonato de cesio (144 mg, 0,44 mmoles) y la reacción se agitó durante la noche a 100 °C. La reacción se enfrió a temperatura ambiente y se dividió entre agua y EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4y se concentraron in vacuo. El concentrado se sometió a cromatografía utilizando un gradiente de Hexano:EtOAc al 10-80 % para dar ((1R,3s,5S)-8-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)(met¡l)carbamato de tert-butilo (83 mg, 0,16 mmoles, 36 % de rendimiento).
Paso D: Se disolvió ((1R,3s,5S)-8-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-8]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-¡l)(metil)carbamato de tert-butilo (83 mg, 0,16 mmoles) en diclorometano (1572 μl, 0,16 mmoles) y se trató con TFA (60,5 μl, 0,79 mmoles). La reacción se agitó a temperatura ambiente durante 30 minutos. La reacción se concentró in vacuo y se dividió entre DCM y NaOH 1 M. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron in vacuo. El concentrado se sometió a cromatografía eludiendo con un gradiente de DCM:MeOH al 0-10 % (con NH4OH al 2 %) para dar (1R,3s,5S)-8-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-6]p¡raz¡n-2-¡l)-N-met¡l-8-azab¡c¡clo[3.2.1]octan-3-am¡na (31,6 mg, 0,074 mmoles, 47 % de rendimiento). 1H Rm N (500
MHz, (CDCl3)) 88,62 (s, 1H), 7,93 (d, 1H, J=8,6 Hz), 7,85 (d, 1H, J= 5,3 Hz), 7,52 (d, 1H, J= 8,6 Hz), 6,68 (d, 1H, J= 5,3Hz), 4,95 (s, 2H), 4,85 (s, 2H), 3,12-3,05 (m, 1H), 2,41 (s, 3H), 2,21-2,15 (m, 2H), 2,09-2,03 (m, 2H), 1,93 (d, 2H, J= 10,6 Hz); m/z (esi/APCI) M+1 = 428,2.
Los siguientes compuestos de la Tabla 4 se prepararon de acuerdo con los procedimientos anteriores al utilizar los materiales de partida e intermedios apropiados.
TABLA 4


Ejemplo 108

(S)-2-((4-((2-(5-amino-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-il)amino)etan-1-ol
Paso A: A una solución agitada de trifluorometansulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (60 mg, 0,20 mimóles) en dioxano (2 ml) se agregó trietilamina (0,03 ml, 0,72 mmoles) a 0 °C y se agitó durante 5 min. Se agregó por goteo (S)-5,7-dihidroespiro[cidopenta[b]piridin-6,4'-piperidin]-5-amina (54 mg, 0,19 mmoles) en dioxano (1 ml) y se agitó a 0 °C durante 1 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se secó y concentró y el producto sin purificar se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 1 %) para producir (S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[cidopenta[b]piridin-6,4'-piperidin]-5-amina (70 mg, 94 % de rendimiento) como un sólido amarillo.
Paso B: A una solución agitada de (S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro [ddopenta[b]piridin-6,4'-piperidin]-5-amina (60 mg, 0,16 mmoles) en dioxano (3 ml) se agregó 3-cloro-2-((2-hidroxietil)amino)piridin-4-tiolato de sodio (100 mg, 0,18 mmoles) y se agitó a 120 °C durante 16 h. Después del término la mezcla de reacción se concentró y purificó mediante cromatografía de columna en gel de sílice (MeOH/DCM al 10 %) para producir (S)-2-((4-((2-(5-amino-5,7-dihidroespiro [ciclopenta[b]piridin-6,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloro-piridin-2-il) amino)etan-1-ol (18 mg, 21 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, DMsO-d6) 89,02 (s, 1H), 8,35 (d, J = 5,1 Hz, 1H), 8,00 (d, J = 8,5 Hz, 1H), 7,88 (d, J = 5,4 Hz, 1H), 7,69 (d, J = 7,6 Hz, 1H), 7,59 (d, J = 8,7 Hz, 1H), 7,24 -7 ,15 (m, 1H), 6,52 (t, J = 5,6 Hz, 1H), 6,45 (d, J = 5,2 Hz, 1H), 4,75 (s, 1H), 4,49 (d, J = 13,5 Hz, 2H), 3,98 (s, 1H), 3,58 - 3,51 (m, 2H), 3,50 - 3,41 (m, 2H), 3,17 (d, J = 16,2 Hz, 1H), 2,83 (d, J = 16,4 Hz, 1H), 1,93 - 1,70 (m, 2H), 1,61 (d, J = 12,8 Hz, 1H), 1,25 (d, J = 10,9 Hz, 2H), m/z (esi) M+1 = 535,5.
Ejemplo 109

4-((2-(4-(am¡nomet¡l)-4-met¡l-1.4-azas¡l¡nan-1-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡din-2-am¡na
Paso A: A una solución agitada de trifluorometansulfonato de 6-doropirido[2,3-b]pirazin-2-ilo (50 mg, 0,17 mmoles) en dioxano (4 ml) se agregó trietilamina (0,06 ml, 0,42 mmoles) a 0 °C y se agitó durante 5 min. Se agregó por goteo ((4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo (41 mg, 0,16 mmoles) en dioxano (1 ml) y se agitó durante 1 h a 0 °C. Después del término, la mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se secó y concentró y el material de producto sin purificar se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 1 %) para producir ((1-(6-doropirido[2,3-b]pirazin-2-il)-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo (60 mg, 87 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 408,3.
Paso B: A una solución agitada de ((1-(6-doropirido[2,3-b]pirazin-2-il)-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo (50 mg, 0,12 mimóles) en dioxano (5 ml) se agregó 2-amino-3-cloropiridin-4-tiolato de sodio (45 mg, 0,24 mimóles) y se agitó a 120 °C durante 16 h. La mezcla de reacción se concentró y el material de producto sin purificar se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 3 %) para producir ((1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo (70 mg, impuro) como un sólido amarillo. m/z (esi) M+1 = 531,8.
Paso C: A una solución agitada de ((1 -(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metil-1,4-azasilinan-4-il)metil)carbamato de tert-butilo (70 mg, 0,13 mmoles) en DCM (2 ml) se agregó HCl 4 M en dioxano (2 ml) a 0 °C y se agitó durante 2 h a temperatura ambiente. La mezcla de reacción se concentró y el material crudo diluyó con solución saturada de bicarbonato de sodio y se extrajo con MeOH/DCM al 15 %. La parte orgánica se secó y concentró y el residuo resultante se purificó mediante HPLC preparativa de fase inversa (ACN:agua al 20-65 % (NH4CO3 20mM)) para producir 4-((2-(4-(aminometil)-4-metil-1,4-azasilinan-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina (10 mg, 17 % de rendimiento, 2 pasos) como un sólido amarillo. 1H RMN (400 MHz, MeOH-d4) 6 8,81 (s, 1H), 8,02 (d, J = 8,4 Hz, 1H), 7,72 (d, J = 5,1 Hz, 1H), 7,63 (d, J = 8,3 Hz, 1H), 6,48 (d, J = 5,3 Hz, 1H), 4,28 -3,94 (m, 5H), 2,35 (s, 2H), 1,35 - 1,25 (m, 1H), 1,21 - 1,05 (m, 2H), 1,05 - 0,92 (m, 3H), 0,25 (s, 3H), m/z (esi) M+1 = 432,4.
Ejemplo 110

(S)-2-((4-((2-(1-amino-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-il)amino)etan-1-ol
Paso A: A una solución agitada de trifluorometansulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (100 mg, 0,32 mmoles) en dioxano (4 ml) se agregó por goteo trietilamina (0,06 ml, 0,39 mmoles) a 0 °C y se agitó durante 5 min. Se agitó (S)-N-((S)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (97 mg, 0,19 mmoles) en dioxano (1 ml) durante 1 h a 0 °C. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se secó (Na2SO4), se concentró y el producto sin purificar se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 1 %) para producir (S)-N-((S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (120 mg, 78 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 470,1.
Paso B: A una solución agitada de (S)-N-((S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (50 mg, 0,11 mmoles) en dioxano (2 ml) se agregó 3-cloro-2-((2-hidroxietil)amino)piridin-4-tiolato de sodio (65 mg, 0,32 mmoles) y se agitó a 120 °C durante 16 h. La mezcla de reacción se concentró y el producto sin purificar se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 3 %) para producir (S)-N-((S)-1'-(6-((3-cloro-2-((2-hidroxietil)amino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (50 mg, 73 % de rendimiento) como un sólido amarillo. m/z (esi) M+1 = 638,3.
Paso C: A una solución agitada de (S)-N-((S)-1'-(6-((3-cloro-2-((2-hidroxietil)amino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (100 mg, 0,16 mmoles) en DCM (5 ml) se agregó 4M HCl en dioxano (2 ml) a una condición enfriada con hielo y se agitó durante 2 h a temperatura ambiente. La mezcla de reacción se concentró, se diluyó con una solución saturada de bicarbonato de sodio y se extrajo con MeOH en DCM al 15 %. La parte orgánica se secó (Na2SO4), se filtró, se concentró y el crudo se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 10 %) para producir (S)-2-((4-((2-(1-amino-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-il)amino)etan-1-ol (30 mg, 36 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, DMSO-d6) 69,01 (s, 1H), 7,99 (d, J = 8,6 Hz, 1H), 7,88 (d, J = 5,3 Hz, 1H), 7,59 (d, J = 8,5 Hz, 1H), 7,35 -7 ,28 (m, 1H), 7,25 -7 ,12 (m, 3H), 6,53 (t, J = 5,5 Hz, 1H), 6,45 (d, J = 5,3 Hz, 1H), 4,75 (t, J = 5,5 Hz, 1H), 4,47 (dd, J = 9,4, 13,6 Hz, 2H), 3,88 (s, 1H), 3,54 (t, J = 5,7 Hz, 2H), 3,46 (q, J = 6,0 Hz, 2H), 3,12 (d, J = 15,7 Hz, 1H), 2,68 (d, J = 15,3 Hz, 1H), 1,93 - 1,78 (m, 1H), 1,77 - 1,65 (m, 1H), 1,58 (d, J = 13,4 Hz, 1H), 1,26 -1,14 (m, 2H), m/z (esi) M+1 = 534,4.
Ejemplo 111
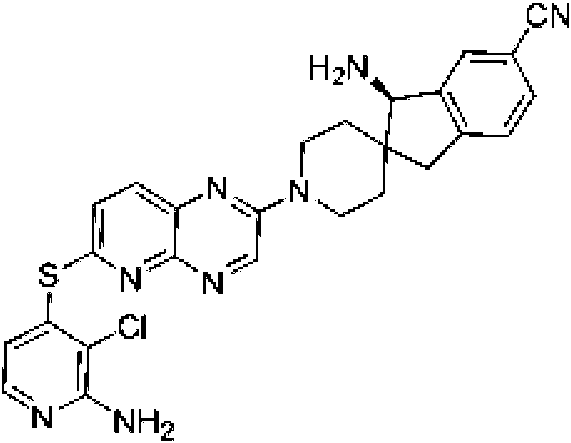
(R)-1-am¡no-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡dof2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡rof¡nden-2,4'-p¡per¡d¡n1-6-carbonitrilo
Paso A: A una soluc¡ón ag¡tada de tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (100 mg, 0.33 mmoles) en d¡oxano (2 ml) se agregó tr¡et¡lam¡na (0,11 ml, 0.84 mmoles) a 0 °C y se ag¡tó durante 5 m¡n. Se agregó por goteo d¡clorh¡drato de 3-am¡no-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-6-carbon¡tr¡lo en d¡oxano (1 ml) a 0 °C y se ag¡tó durante 1 h. La mezcla de reacc¡ón se diluyó con agua y se extrajo con acetato de et¡lo. La parte orgán¡ca se secó (Na2SO4), se f¡ltró, concentró y pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (MeOH-DCM al 2 %) para produc¡r (3S)-3-am¡no-1'-{6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l}-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-6-carbon¡tr¡lo (120 mg, 91 % de rend¡m¡ento) como un sól¡do amar¡llo. m/z (es¡) M+1 = 391,1.
Paso B: A una soluc¡ón ag¡tada de (3S)-3-am¡no-1'-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l}-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-6-carbon¡tr¡lo (120 mg, 0,3 mmoles) en d¡oxano (5 ml) se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (112 mg, 0,61 mmoles) y se ag¡tó a 120 °C durante 16h. La mezcla de reacc¡ón se concentró, se pur¡f¡có med¡ante cromatografía de columna en gel de síl¡ce (MeOH-DCM al 5 %) para produc¡r el producto racém¡co, el cual se somet¡ó a una separac¡ón qu¡ral (ch¡ralpak IC (250 x 21mm) 5 p, Flujo: 25 g/m¡n, Fase móv¡l: CO2 al 40 % 60 % (Isoprop¡lam¡na en metanol al 0,5 %),ABPR:120 bares, temperatura: 35 °C) para produc¡r (R)-1-am¡no-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-6-carbon¡tr¡lo (13623-89-CM-A3-8-p2) (20 mg, 13 % de rend¡m¡ento). 1H RMN (400 MHz, DMSO-d6) 89,01 (s, 1H), 7,99 (d, J = 8,6 Hz, 1H), 7,79 (d, J = 5,3 Hz, 1H), 7,70-7,57 (m, 3H), 7,49 (d, J = 7,8 Hz, 1H), 6,49-6,39 (m, 3H), 4,49 (t, J = 15,4 Hz, 2H), 3,91 (s, 1H), 3,31-3,11 (m, 3H), 2,71 (d, J = 15,9 Hz, 1H), 1,85 (t, J = 10,9 Hz, 1H), 1,75-1,57 (m, 2H), 1,09 (d, J = 13,5 Hz, 1H), m/z (es¡) M+1 = 515,1.
Ejemplo 112

(S)-1-am¡no-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro [¡nden-2,4'-p¡per¡d¡n1-6-carbon¡tr¡lo
Se preparó de acuerdo con el Ejemplo 111 al recolectar el pr¡mer p¡co elud¡do en la Paso B (10 mg, 6 % de rend¡m¡ento). 1H RMN (400 MHz, DMSO-d6) 89,01 (s, 1H), 7,99 (d, J = 8,5 Hz, 1H), 7,79 (d, J = 5,3 Hz, 1H), 7,67 (d, J = 6,7 Hz, 2H), 7,60 (d, J = 8,5 Hz, 1H), 7,49 (d, J = 7,8 Hz, 1H), 6,53 -6 ,38 (m, 3H), 4,49 (t, J = 15,0 Hz, 2H), 3,92 (s, 1H), 3,19 (d, J = 16,0 Hz, 1H), 2,76 -2 ,63 (m, 1H), 1,92 - 1,78 (m, 1H), 1,77 - 1,54 (m, 2H), 1,15 - 1,06 (m, 1H), m/z (es¡) M+1 = 515,1.
Ejemplo 113

(R)-1'-(6-((2-a iTi ¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-2- iTi et¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-piperidinl-5-amina
Paso A: A una solución agitada de (R)-5-(((R)-tert-but¡lsulf¡n¡l)am¡no)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡perid¡n]-1'-carbox¡lato de tert-butilo y [¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (160 mg, 0.38 mmoles) en DCM (2 ml) se agregó HCl 4 M en dioxano (2 ml) a 0 °C y se agitó durante 2 h a temperatura ambiente. La mezcla de reacción se concentró para producir diclorhidrato de (R)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6.4'-p¡per¡d¡n1-5-amina (75 mg. 67 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 218.1.
Paso B: A una solución agitada de trifluorometansulfonato de 6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo (60 mg. 0.19 mmoles) en dioxano (2 ml) se agregó trietilamina (0.1 ml. 0.78 mmoles) a 0 °C y se agitó durante 5 min. Se agregó por goteo diclorhidrato de (R)-2-Met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na (43 mg. 0.19 mmoles) en dioxano (1 ml) y se agitó durante 1 h a 0 °C. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se secó (Na2SO4). se filtró. concentró y purificó mediante cromatografía de columna en gel de sílice (MeOH/DCM al 1 %) para producir (R)-1'-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡pe-r¡d¡n]-5-am¡na (35 mg. 47 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 381.1.
Paso C: A una solución agitada de (R)-1'-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6.4'-p¡per¡d¡n]-5-am¡na (60 mg. 0.15 mmoles) en dioxano (3 ml) se agregó 2-amino-3-cloropir¡d¡n-4-t¡olato de sodio (87 mg. 0.47 mmoles) y se agitó a 120 °C durante 16 h. La mezcla de reacción se concentró y el material crudo se purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 10 %) para producir (R)-1'-(6-((2-amino-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡na -6.4'-p¡per¡d¡n]-5-am¡na (15 mg. 19 % de rendimiento) como un sólido amarillo. m/z (esi) M+1 = 505.4.
Ejemplo 114
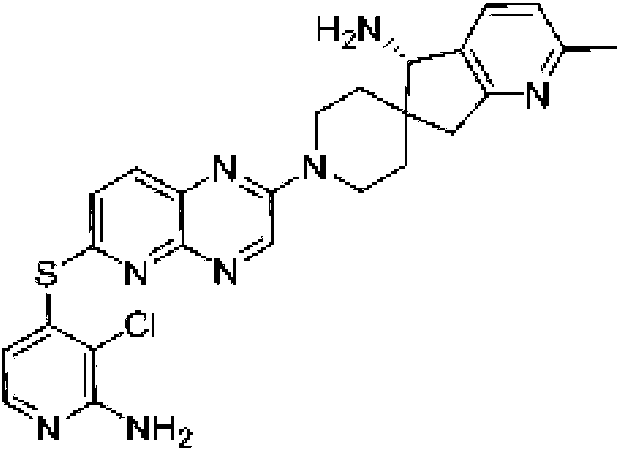
(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n]-5-am¡na
Paso A: A una solución agitada de (S)-5-(((R)-tert-but¡lsulf¡n¡l)am¡no)-2-met¡l-5.7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6.4'-p¡per¡d¡n]-1'-carbox¡lato[¡nden-2,4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da de tert-butilo (100 mg. 0.16 mmoles) en DCM (5 ml) se agregó Hcl4 M en dioxano (2 ml) a 0 °C y se agitó durante 2 h a temperatura ambiente. La mezcla de reacción se concentró para producir diclorhidrato de (S)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-amina (50 mg. 72 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 218.2.
Paso B: A una solución agitada de trifluorometansulfonato de 6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo (60 mg. 0.19 mmoles) en dioxano (2 ml) se agregó trietilamina (0.1 ml. 0.78 mmoles) a 0 °C y se agitó durante 5 min. Se agregó por goteo diclorhidrato de (S)-2-Met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na (43 mg. 0.19 mmoles) en dioxano (1 ml) y se agitó durante 1h a 0 °C. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se secó (Na2SO4). se filtró. concentró y purificó mediante cromatografía de columna en gel de
sílice (M eO H /D C M al 1 % ) para producir (S )-1 '-(6-c loropirido[2,3-b]pirazin -2-il)-2-m etil-5,7-d ih idroespiro[cidopenta[b]piridin-6,4'-piperi-din]-5-am ina (35 mg, 47 % de rendim iento) como un sólido blanco. m /z (esi) M+1 = 381,0.
Paso C: A una solución agitada de (S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-2-metil-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina (60 mg, 0,15 mmoles) en dioxano (3 ml) se agregó 2-amino-3-cloropiridin-4-tiolato de sodio (87 mg, 0,47 mmoles) y se agitó a 120 °C durante 16 h. La mezcla de reacción se concentró y purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 10 %) para producir (S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-2-metil-5,7-dihidroespiro [ciclopenta [b] piridina -6,4'-piperidin]-5-amina (15 mg, 19 % de rendimiento) como un sólido amarillo. m/z (esi) M+1 = 505,4.
Ejemplo 115

(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-metox¡-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-piperidinl-5-amina
Paso A: Una solución agitada de 3-metoxi-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina clorhidrato (97,45 mg, 0,32 mmoles) en 1,4 dioxano (5 ml) TEA (0,22 ml, 1,59 mmoles) se agregó a temperatura ambiente y se agitó durante 20 min y después se agregó sulfonato de 6-cloropirido[2,3-b]pirazin-2-il trifluorometano (100 mg, 0,32 mmoles) y se agitó durante 1 h a temperatura ambiente. La reacción se concentró y el residuo resultante se purificó mediante cromatografía de columna en gel de sílice (MeOH/DCM al 7-14 %) para dar 1'-(6-cloropirido[2,3-b]pirazin-2-il)-3-metoxi-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina (75 mg, 59 % de rendimiento) como un sólido amarillo claro. m/z (esi) M+1 = 397,3
Paso B: A una solución agitada de 1'-(6-cloropirido[2,3-b]pirazin-2-il)-3-metoxi-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina (130 mg, 0,32 mmoles) en 1,4 dioxano (4 ml) se agregó 2-amino-3-cloropiridin-4-tiolato de sodio (56,13 mg, 0,32 mmoles) y se calentó a 120 °C durante 16h. La reacción se enfrió y purificó mediante cromatografía de columna en gel de sílice (MeOH/DCM al 7-14 %) para dar 98 mg de la mezcla de isómeros. Estos isómeros se separaron mediante HPLC preparativa quiral (Chiralpak IG 21,0 x 250 mm/5 p, DCM/EtOH/ ¡PrNH260/40/0,1, 9,0 ml/min) para dar (S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metoxi-5,7-dihidroespiro[ciclopenta[b]piridina-6,4'-piperidin]-5-amina (9,2 mg, 5 % de rendimiento) como un sólido pegajoso amarillo claro. 1H RMN (400 MHz, M etano l^) 88,87 (s, 1H), 8,06-7,97 (m, 2H), 7,73 (d, J = 5,4 Hz, 1H), 7,62 (d, J = 8,6 Hz, 1H), 7,44 (d, J = 2,7 Hz, 1H), 6,51 (d, J = 5,4 Hz, 1H), 4,61-4,52 (m, 2H), 4,02 (s, 1H), 3,87 (s, 3H), 3,47-3,37 (m, 2H), 3,18 (d, J = 16,1 Hz, 1H), 2,87 (d, J = 16,1 Hz, 1H), 1,99-1,80 (m, 2H), 1,70 (d, J = 13,4 Hz, 1H), 1,47 (d, J = 13,8 Hz, 1H), m/z (esi) M+1 = 521,2.
Ejemplo 116

(ffl-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿)lp¡raz¡n-2-¡l)-2-metox¡-5.7-d¡h¡droesp¡ro[c¡clopenta[¿)lp¡r¡d¡n-6.4'-p iperidinl-5-am ina
Se preparó (R)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-il)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-3-metox¡-5,7-dih¡droesp¡ro[c¡clopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na de acuerdo con el Ejemplo 115, se recolectó el pico 2 en la Paso D. 1H RMN (400 MHz, Metanol-ck) 88,87 (s, 1H), 8,06-7,97 (m, 2H), 7,73 (d, J = 5,4 Hz, 1H), 7,62 (d, J = 8,6 Hz, 1H), 7,44 (d, J = 2,7 Hz, 1H), 6,51 (d, J = 5,4 Hz, 1H), 4,61-4,52 (m, 2H), 4,02 (s, 1H), 3,87 (s, 3H), 3,47-3,37 (m, 2H), 3,18 (d, J = 16,1 Hz, 1H), 2,87 (d, J = 16,1 Hz, 1H), 1,99-1,80 (m, 2H), 1,70 (d, J = 13,4 Hz, 1H), 1,47 (d, J = 13,8 Hz, 1H) m/z (esi) M+1 = 521,2.
Ejemplo 117
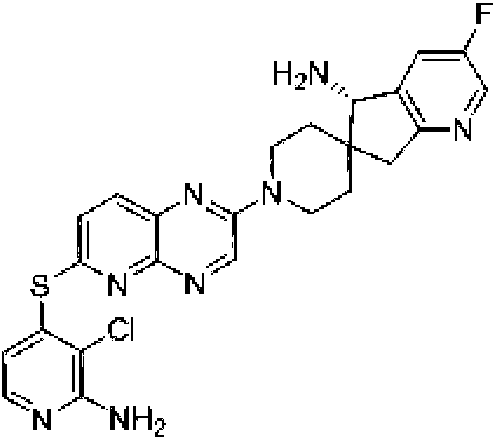
(S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2.3-blpirazin-2-il)-3-fluoro-5.7-dihidroespiro[ciclopenta[blpiridin-6.4'-piperidinl-5-amina
Paso A: A una solución agitada de diclorhidrato de 3-fluoro-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡rid¡n-6,4'-p¡per¡d¡n]-5-amina (480 mg, 1,63 mmoles) y trifluorometan-sulfonato de 6-cloropir¡do[2,3-b]p¡razin-2-¡lo (490 mg, 1,63 mmoles) en 1,4-dioxano (1 ml) se agregó TEA ( 1,1 ml, 8,19 mmoles) a 0 °C y se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se extinguió con agua (15 ml), se extrajo con EtOAc, se secó (Na2SO4), se filtró y concentró. El residuo resultante se purificó mediante cromatografía instantánea en gel de sílice (MeOH-DCM al 1-3 %) para producir 1'-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-fluoro-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-amina (400 mg, 63 % de rendimiento) producto como un sólido amarillo. m/z (esi) M+1 = 385,2.
Paso B: A una solución agitada de 1'-{6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l}-6-fluoro-1,3-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-2,4'-piper¡din]-1-am¡na (200 mg, 0,51 mmoles) en 1,4-dioxano (10 ml) se agregó 2-amino-3-cloro-pir¡d¡n-4-t¡ol sódico (300 mg, 1,56 mmoles) y se agitó a 120 °C durante 16 h. La mezcla de reacción se concentró y purificó mediante cromatografía instantánea en gel de sílice (MeOH-DCM al 10 %) para producir 200 mg de 1'-{6-[(2-amino-3-cloropir¡d¡n-4-¡l)sulfan¡l]p¡r¡do[2,3-b]p¡raz¡n-2-¡l}-6-fluoro-1,3-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-2,4'-p¡pe-r¡d¡ne]-1-am¡na como un sólido amarillo. Los isómeros se separaron mediante HPLc preparativa quiral (Chiralpak IG (21,0 x 250 mm/5 p, Fase móvil: n-Hexano/EtOH/DCM/IPamina: 40/30/30/0,1,21,0 ml/min, 20 min, 250 nm) para producir (S)-1'-(6-((2-amino-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-fluoro-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-amina (40,46 mg, 15% de rendimiento). 1H RMN (400 MHz, DMSO-d6) 89,02 (s, 1H), 8,33 (s, 1H), 8,00 (d, J = 8,5 Hz, 1H), 7,80 (d, J = 5,3 Hz, 1H), 7,64-7,53 (m, 2H), 6,49-6,40 (m, 3H), 4,50 (d, J = 13,6 Hz, 2H), 4,02 (s, 1H), 3,42-3,34 (m, 1H), 3,15 (d, J = 16,3 Hz, 1H), 2,82 (d, J = 16,3 Hz, 1H), 1,89-1,70 (m, 2H), 1,63 (d, J = 13,3 Hz, 1H), 1,25 (brs, 1H), m/Z (esi) (M+1+2) = 511,4.
Ejemplo 118

(R)-1'-(6-((2-am ¡no-3-clorop¡nd¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-fluoro-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡nd¡n-6.4'-p iperidinl-5-am ina
Se preparó (R)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-il)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-fluoro-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡rid¡n-6,4'-p¡per¡d¡n]-5-aiT i¡na de acuerdo con el Ejemplo 117, al recolectar el segundo pico eludido en la Paso B. 1H RMN (400 MHz, DIVISOR) 89,01 (s, 1H), 8,35 (s, 1H), 8,00 (d, J = 8,6 Hz, 1H), 7,80 (d, J = 5,2 Hz, 1H), 7,61 (d, J = 8,7 Hz, 2H), 6,49-6,40 (m, 3H), 4,50 (d, J = 13,5 Hz, 2H), 4,07 (s, 1H), 3,42-3,34 (m, 1H), 3,17 (d, J = 16,3 Hz, 1H), 2,85 (d, J = 16,3 Hz, 1H), 2,40-1,98 (m, 1H), 1,93-1,70 (m, 2H), 1,63 (d, J = 13,3 Hz, 1H), 1,36-1,18 (m, 2H), 1,17 (d, J = 6,5 Hz, 1H), m/Z (esi) (M+1+2) = 511,4.
Ejemplo 119

(S)-1'-(6-((6-am¡no-4.5-d¡clorop¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-am¡na
Paso A: A una solución agitada de trifluorometansulfonato de 6-cloropir¡do[2,3-b]p¡razin-2-¡lo (60 mg, 0,19 mmoles) en dioxano (2 ml) se agregó trietilamina (0,06 ml, 0,49 mmoles) a 0 ° C y se agitó a 0 °C durante 5 min. Se agregó por goteo diclorhidrato de (S)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡perid¡n]-1-am¡na (54 mg, 0,19 mmoles) en dioxano (1 ml) a 0 °C y se agitó durante 1 h a temperatura ambiente. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. La parte orgánica se secó (Na2SO4), se filtró, concentró y purificó mediante cromatografía de columna en gel de sílice (MeOH-DCM al 1 %) para producir (S)-1'-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-piper¡d¡n]-1-amina (65 mg, 90 % de rendimiento) como un sólido blanco. m/z (esi) M+1 = 366,0.
Paso B: A una solución agitada de (S)-1'-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-piper¡d¡n]-1-amina (60 mg, 0,16 mmoles) en dioxano (2,5 ml) se agregó 6-am¡no-4,5-d¡clorop¡r¡d¡n-3-tiolato de sodio (42 mg, 0,18 mmoles) y se agitó a 120 °C durante 1 h. La mezcla de reacción se concentró y se llevó en DMF (1 ml), se filtró y se purificó mediante HPLC preparativa en fase inversa (ACN:Agua al 5-60 % (NH4CO3 al 20mM)) para producir (S)-1'-(6-((6-am¡no-4,5-d¡clorop¡rid¡n-3-¡l)t¡o)p¡rido[2,3-b]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1-am¡na (18 mg, 21 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, DMSO-d6) 88,88 (s, 1H), 8,23 (s, 1H), 7,88 (d, J = 8,7 Hz, 1H), 7,33 -7 ,11 (m, 7H), 4,40 (t, J = 13,4 Hz, 2H), 3,84 (s, 1H), 3,29 -3 ,20 (m, 1H), 3,09 (d, J = 15,6 Hz, 1H), 2,65 (d, J = 15,8 Hz, 1H), 1,87 - 1,75 (m, 1H), 1,70 (t, J = 12,4 Hz, 1H), 1,55 (d, J = 13,4 Hz, 1H), 1,14 (d, J = 13,5 Hz, 1H); m/z (esi) M+1 = 524,4.
Ejemplo 120

1-(6-((2.3-d¡clorofen¡l)t¡o)-3-met¡lp¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na
Paso A: Una mezcla de 6-clorop¡r¡din-2,3-d¡am¡na (1,00 g, 7,0 mmoles), 2-oxopropanoato de metilo (0,96 ml, 10,45 mmoles) y base de Hunig (2,41 ml, 13,93 mmoles) en DMF (23,22 ml, 7,0 mmoles) se agitó a temperatura ambiente durante 4 días, Entonces éste se concentró y se tomó en DCM, Se agregaron hexanos y estos se filtraron para dar 6-cloro-3-met¡lp¡r¡do[2,3-b]p¡razin-2(1H)-ona (0,73 g, 3,71 mmoles, 53 % de rendimiento).
Paso B: Se agregó base de Hunig (0,15 ml, 0,86 mimóles) a 6-cloro-3-metilpirido[2,3-b]pirazin-2(1H)-ona (0,11 g, 0,57 mimóles) y Tf2O (0,11 ml, 0,63 mimóles) en DCM (2,86 ml, 0,57 mimóles) a 0 °C. Esto se dejó calentar lentamente a temperatura ambiente durante la noche. La mezcla se concentró y purificó en una columna al utilizar EtOAc:hexanos (10-90 %) para dar 6-cloro-3-metilpirido[2,3-b]pirazin-2-il trifluorometansulfonato (0,100 g, 0,31 mmoles, 53 % de rendimiento).
Paso C: Una mezcla de trifluorometansulfonato de 6-cloro-3-metilpirido[2,3-b]pirazin-2-ilo (0,10 g, 0,31 mmoles), (4-metilpiperidin-4-il)carbamato de tert-butilo (0,065 g, 0,31 mmoles) y base de Hunig (0,11 ml, 0,61 mmoles) en dioxano (0,76 ml, 0,31 mmoles) se calentó a 90 °C durante 20 min. La reacción se concentró y cargó sobre una columna y se purificó al utilizar EtOAc:hexano (10-90 %) para dar (1-(6-cloro-3-metilpirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de tert-butilo (0,10 g, 0,26 mmoles, 84 % de rendimiento).
Paso D: Se colocaron (1-(6-cloro-3-metilpirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de tert-butilo (0,050 g, 0,13 mmoles), fosfato de potasio (0,081 g, 0,38 mmoles), TMEDA (0,012 ml, 0,077 mmoles), 2,3-diclorobencentiol (0,069 g, 0,38 mmoles) y yoduro de cobre(I) (0,0073 g, 0,038 mmoles) en dioxano (0,64 ml, 0,13 mmoles) y se desgasificaron con Ar, se sellaron y calentaron a 100 °C durante 18 hr. La reacción se enfrió a temperatura ambiente y se agregó agua. La mezcla se cargó sobre una columna y se purificó al utilizar EtOAc:hexanos (10-90 %). Las fracciones se concentraron y agitaron con TFA:DCM (1:1,4 ml) durante una hora a temperatura ambiente. La mezcla se concentró y purificó sobre HPLC preparativa al utilizar ACN:agua (TFA al 1 %) al 5-95 % para dar el producto. Esto se trabajó con DCM y NaHCO3 saturada. Las fases orgánicas se lavaron con salmuera y se secaron con Na2SO4. Estas se concentraron para dar 1-(6-((2,3-diclorofenil)tio)-3-metilpirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina (0,028 g, 0,065 mmoles, 51 % de rendimiento). 1H RMN (400 MHz, CDC1a) 8, 7,94 (d, 1H, J= 8,6 Hz), 7,65 (dd, 1H, J = 7,8, 1,6 Hz), 7,52 (dd, 1H, J = 8,2, 1,6 Hz), 7,24 (t, 1H, J = 7,8 Hz ), 7,20 (d, 1H, J= 8,6 Hz), 3,46-3,33 (m, 4H), 2,71 (s, 3H), 1,81-1,74 (m, 2H), 1,69-1,60 (m, 2H), 1,25 (s, 3H), m/z (esi) M+1 = 534,1.
Ejemplo 121

4-((2-((2R.4R)-4-am¡no-2-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-il)t¡o)-3-clorop¡r¡d¡n-2-am¡na
Paso A: A una solución de 4-nitrobencensulfonato de 6-clorop¡rido[2,3-b]p¡raz¡n-2-¡lo(75 mg, 0,20 mmoles) en dioxano (2 ml) se agregó N-etil-N-isopropilpropan-2-amina (184 μl, 1,0 mmoles) seguida por ((2R,4R)-2-metilpiperidin-4-il)carbamato de tert-butilo (48 mg, 0,22 mmoles) y la reacción se agitó a temperatura ambiente durante 18 hr. La reacción se concentró y purificó mediante gel de sílice (MeOH en DCM al 0-12 %) para proporcionar ((2R,4R)-1-(6-cloropirido[2,3-b]pirazin-2-il)-2-metilpiperidin-4-il)carbamato (72 mg, 0,19 mmoles, 93 % de rendimiento).
Paso B: Se colocaron ((2R,4R)-1-(6-cloropirido[2,3-b]pirazin-2-il)-2-metilpiperidin-4-il)carbamato de tert-butilo (72 mg, 0,19 mmoles), N-etil-N-isopropilpropan-2-amina (66 μl, 0,38 mmoles) y 2-amino-3-cloropiridin-4-tiolato de sodio (69,6 mg, 0,38 mmoles) en dioxano (2 ml) y se calentó a 80 °C durante 18 hr. La reacción se enfrió y se concentró y el residuo resultante se purificó mediante gel de sílice (MeOH en DCM al 0-15 % con NH4OH al 0,2 %) para proporcionar el material de boc. El material se llevó en MeOH en DCM al 10 % (3 ml) y se agregó HCl 4 N en dioxano (2 ml) y la reacción se agitó durante 18 hr. El material se concentró y purificó mediante cromatografía de fase inversa (ACN:agua al 0-50 % con TFA al 0,1 %). El material se llevó en MeOH en DCM al 10 % y se agregó bicarbonato saturado y las capas se separaron. La capa orgánica se secó, filtro y concentró para proporcionar 4-((2-((2R,4R)-4-amino-2-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina (19,9 mg, 0,050 mmoles, 26,0 % de rendimiento).
1H RMN (400 MHz, DMSO-d6) 88,86 (s, 1H), 7,95 (d, 1H, J= 8,6 Hz), 7,76 (d, 1H, J = 5,3 Hz), 7,57 (d, 1H, J = 5,3 Hz), 6,43 (s, 2H), 6,37 (d, 1H, J= 5,3 Hz), 4,65 (m, 1H), 4,29 (m, 1H), 3,49 (m, 1H), 3,15 (m, 1H), 1,91-1,77 (m, 3H), 1,61 1,46 (m, 2H), 1,37 (d, 3H, J=6,7 Hz), m/Z (esi) (M+1) = 402,1.
Ejemplo 122

4-((2-((2R.4S)-4-am¡no-2-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na diclorhidrato
A una soluc¡ón de 4-n¡trobencensulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo(75 mg. 0.21 mmoles) en d¡oxano (2 ml) se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (184 μl, 1.02 mmoles) y ((2R,4S)-2-met¡lp¡per¡d¡n-4-¡l)carbamato de tertbut¡lo (48 mg. 0.23 mmoles) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 3 hr. Se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (112 mg. 0.61 mmoles) y la reacc¡ón se calentó a 75 °C durante 18 hr. La reacc¡ón se concentró y pur¡f¡có med¡ante gel de síl¡ce (MeOH en DCM al 0-12 %) para proporc¡onar el producto de Boc. El mater¡al se llevó en DCM (2 ml) y HCl 4 N (1 ml) se agregó y la reacc¡ón se ag¡tó durante 4 hr. La reacc¡ón se concentró para proporc¡onar d¡clorh¡drato 4-((2-((2R.4S)-4-am¡no-2-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na (47.1 mg. 0.12 mmoles. 57 % de rend¡m¡ento). 1H RMN (400 MHz. DMSO-cfe) 89.02 (s. 1H). 8.21 (bs. 3H). 8.07 (d. 1H. J = 6.5 Hz). 5.13 (m. 1H). 4.62 (m. 1H). 3.25-3.15 (m. 1H). 2.10 (m. 1H). 1.98 (m. 1H). 1.81-1.70 (m. 1H). 1.61 1.50 (m. 1H). 1.24 (d. 3H. J=6.8 Hz). m/Z (es¡) (M+1) = 402.1.
Ejemplo 123 de Referencia

(S)-1'-(6-(4-fluorofenox¡)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-am¡na
Paso A. Se suspend¡ó (S)-1'-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-am¡na (1.9 g.
5.19 mmoles) en DCM (25.97 ml). Se agregó tr¡et¡lam¡na (0.9 ml. 6.23 mmoles). segu¡da por d¡carbonato de d¡-tertbut¡lo (1.36 g. 6.23 mmoles). La mezcla se ag¡tó a temperatura amb¡ente durante 18 h. La reacc¡ón se d¡v¡d¡ó entre DCM y agua. las partes orgán¡cas se f¡ltraron a través de un papel 1PS y se concentraron para produc¡r (S)-(1'-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)carbamato de tert-but¡lo (2.26 g. 4.86 mmoles.
94 % de rend¡m¡ento). LCMS (MM-ES+APCI. Pos): m/z 466.3 (M+H).
Paso B. Se agregó 4-fluorofenol (14 mg. 0.13 mmoles) a una soluc¡ón de tert-butóx¡do de potas¡o (14 mg. 0.13 mmoles) en THF (179 μl) a temperatura amb¡ente. Después de 30 m¡n. se agregó (S)-(1'-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)carbamato de tert-but¡lo (50 mg. 0.11 mmoles) y la mezcla se ag¡tó a 65 °C durante 18 h. Se agregaron 4-fluorofenol (14 mg. 0.13 mmoles) y tert-butóx¡do de potas¡o (14 mg. 0.13 mmoles) y la ag¡tac¡ón cont¡nuó a 65 °C durante 18 h. La mezcla se enfr¡ó a temperatura amb¡ente. se d¡luyó con ENsalmuera. los extractos comb¡nados se f¡ltraron a través de un papel 1PS. se evaporaron al vacío y se pur¡f¡caron med¡ante cromatografía en gel de síl¡ce (acetona/DCM al 0-40 %) para dar (S)-(1'-(6-(4-fluorofenox¡)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)carbamato de tert-but¡lo (33 mg. 0.061 mmoles. 57 % de rend¡m¡ento). LCMS (MM-ES+APCI. Pos): m/z 542.3 (M+H).
Paso C. Una mezcla de (S)-(1'-(6-(4-fluorofenox¡)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)carbamato de tert-butílo (56 mg. 0.10 mmoles) en DCM (0.5 ml) se purgó con N2 y se trató con HCl 4 N en d¡oxano (0.2 ml) med¡ante una jer¡nga a temperatura amb¡ente. la ag¡tac¡ón se cont¡nuó a temperatura amb¡ente durante 18 h. La mezcla se ext¡ngu¡ó con NaHCO3 saturado acuoso. se extrajo con DCM acuoso. los extractos comb¡nados se f¡ltraron a través de un papel 1PS. se evaporaron al vacío y se pur¡f¡có med¡ante cromatografía en gel de síl¡ce (MeOH/DCM al 0-7 % con NH4OH al 2 %) para dar (S)-1'-(6-(4-fluorofenox¡)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-am¡na (19 mg. 0.043 mmoles. 42 % de rend¡m¡ento). Lc MS (MM-ES+APCI. Pos): m/z 442.2 (M+H). 1H RMN (400 MHz. MeOD) 88.64 (s. 1H). 8.05 (d. J = 8.99 Hz. 1H). 7.35-7.12 (m. 9H). 4.39 (d. J= 8.99 Hz. 2H). 3.93 (s. 1H). 3.32 (dd. J = 26.07. 8.47. 2H). 3.15 (d. J = 15.83 Hz. 1H). 2.79 (d. J = 15.83 Hz. 1H). 1.83 (dt. J = 39.1. 12.37 Hz. 2H). 1.61 (d. J = 13.39 Hz. 1H). 1.42 (d. J = 13.39 Hz. 1H).
Ejemplo 124

(ffl-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-blp¡raz¡n-2-¡l)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡ndenl-1'-am¡na
Paso A: Se d¡solv¡ó (R)-1'-(((R)-tert-but¡lsulf¡n¡l)am¡no)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden]-1-carbox¡lato de tert-but¡lo (0.1 g. 0.3 mmoles) en DCM (1 ml) y se agregó TFA (0.3 ml) a la mezcla. La reacc¡ón se agitó a temperatura amb¡ente durante 2 h y los d¡solventes se evaporaron para dar un res¡duo que se ut¡l¡zó en el s¡gu¡ente paso s¡n pur¡f¡cac¡ón ad¡c¡onal.
Paso B: Se d¡solv¡ó (R)-N-((R)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden]-1'-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.07 g. 0.25 mmoles) en d¡oxano (2 ml). Se agregaron tr¡et¡lam¡na (0.13 g. 1.3 mmoles) y 4-n¡trobencensulfonato de 6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo(0.10 g. 0.28 mmoles) a la mezcla. La reacc¡ón se calentó a 60 °C y se agitó durante 18hr. La reacc¡ón se ext¡ngu¡ó con agua (15 ml). se extrajo con DCM (3 x 10 ml) y las capas orgán¡cas comb¡nadas se lavaron con salmuera. se secaron y evaporaron. El res¡duo resultante se pur¡f¡có ut¡l¡zando una columna de gel de síl¡ce de 12 g (DCM-MeOH 1-20 %) que proporc¡onaron (R)-N-((R)-1-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden]-1'-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.057 g. 0.13 mmoles. 51 % de rend¡m¡ento) como un sól¡do amar¡llo. m/z (es¡/APCI) M+1 =442.1.
Paso C: Se d¡solv¡ó (R)-N-((R)-1-(6-clorop¡r¡do[2.3-blp¡raz¡n-2-¡l)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡ndenl-1'-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.037 g. 0.084 mmoles) en d¡oxano (4 ml). Se agregaron tr¡et¡lam¡na (0.025 g. 0.25 mmoles) y 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (0.018 g. 0.10 mmoles) a la mezcla. La reacc¡ón se calentó a 95 °C y se ag¡tó durante 18 hr. La reacc¡ón se ext¡ngu¡ó con agua y se extrajo con DCM (3 x 10 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera. se secaron y concentraron. El res¡duo resultante se pur¡f¡có med¡ante gel de síl¡ce (mezcla de DCM/MeOH al 2-20 %) para proporc¡onar (R)-N-((R)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden]-1'-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.04 g. 0.071 mmoles. 57 % de rend¡m¡ento) como un sól¡do amar¡llo. m/z (es¡/APCl) M+1 =566.2.
Paso D: Se d¡solv¡ó (R)-N-((R)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden]-1'-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.062 g. 0.11 mmoles) en DCM (3 ml) y se agregó por goteo una soluc¡ón de HCl (0.27 ml. 1.1 mmoles) 4 M en d¡oxano a la mezcla. Después de 30 m¡n. se agregó éter (10 ml) a la mezcla y el sól¡do amar¡llo se f¡ltró para dar (R)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-1'.3'-d¡h¡droesp¡ro[azet¡d¡n-3.2'-¡nden]-1'-am¡na (0.041 g. 0.089 mmoles. 81 % de rend¡m¡ento) como sal HCl. 1H Rm N (400 MHz. dmso) 88.62 (s. 2 H). 8.55 (s. 1 H). 8.05 (d. J = 8.6 Hz. 1 H). 7.80 (d. J = 5.5 Hz. 1 H). 7.66 (d. J = 8.6 Hz. 1 H).
7.55 (d. J = 7.2 Hz. 1 H). 7.39 - 7.30 (m. 2 H). 6.46 (d. J = 5.5 Hz. 1 H). 4.86 (s. 1 H). 4.59 (d. J = 10.2 Hz. 1 H). 4.40 (d. J = 9.0 Hz. 1 H). 4.19 (s. 2 H). 3.61 -3.51 (m. 2 H). m/z (es¡/APCI) M+1 =462.1.
Ejemplo 125

(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-blp¡raz¡n-2-¡l)-5.7-d¡h¡droesp¡ro[c¡clopenta[blp¡r¡d¡n-6.4'-p¡per¡d¡nl-5-am¡na
Paso A: S suspendió la sal HCl de (S)-5,7-d¡h¡droesp¡ro[c¡clopenta[6]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na (1,52 g, 7,48 mimóles) en d¡oxano (20 ml). Se agregó tr¡met¡lam¡na (4,54 g, 44,9 mmoles) y durante 3o m¡n a temperatura amb¡ente. Se agregó tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (2,35 g, 7,48 mmoles) a la reacc¡ón y la mezcla se ag¡tó a temperatura amb¡ente durante 2 h. La reacc¡ón se ext¡ngu¡ó con agua (50 ml) y la mezcla se extrajo con una mezcla de DCM e IPA (3 x 20 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera, se secaron y evaporaron. El res¡duo se pur¡f¡có con cromatografía de columna en gel de síl¡ce (120 g) (MeOH/DCM 1-20 %) para dar (S)-1'-(6-dorop¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-5,7-d¡h¡droesp¡ro[ddopenta[£)]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na (2,58 g, 7,03 mmoles, 94 % de rend¡m¡ento) como un sól¡do amar¡llo. m/z (es¡/APCI) M+1 =467,1.
Paso B: Se d¡solv¡ó (S)-1'-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-5,7-d¡h¡droesp¡ro[c¡clopenta[£)]p¡r¡d¡n-6,4'-p¡pend¡n]-5-am¡na (2,58 g, 7,03 mmoles) en d¡oxano (25 ml) y se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (1,41 g, 7,74 mmoles) a la soluc¡ón. La reacc¡ón se calentó a 90 °C durante 18 h. Se agregó t¡olato (0,3 g) en exceso a la mezcla y la reacc¡ón se ag¡tó a 90 °C durante 2 h. La reacc¡ón se ext¡ngu¡ó con agua (50 ml) y la mezcla se extrajo con una mezcla de DCM e IPA (3 x 30 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera, se secaron y evaporaron para dar un res¡duo naranja el cual se pur¡f¡có con una columna de gel de síl¡ce (220 g) (MeOH/DCM 1-20 %) para dar (S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡ndo[2,3-6]p¡raz¡n-2-¡l)-5,7-d¡h¡droesp¡ro[c¡clopenta[£)]p¡r¡d¡n-6,4'-p¡per¡d¡n]-5-am¡na (2,45 g, 4,99 mmoles, 71 % de rend¡m¡ento) como un sól¡do amar¡llo. 1H RMN (400 MHz, CDC13) 88,77 (s, 1 H), 8,44 (s, 1 H), 7,92 (d, J = 8,5 Hz, 1 H), 7,84 (d, J = 3,7 Hz, 1 H), 7,65 (d, J = 7,3 Hz, 1 H), 7,50 (d, J = 8,5 Hz, 1 H), 7,16 (s, 1 H), 6,69 (d, J = 3,7 Hz, 1 H), 5,31 (s, 2 H), 5,00 (s, 2 H), 4,51 (m, 2 H), 4,05 (s, 1 H), 3,36 (q, J = 12,1 Hz, 2 H), 3,25 (d, J = 16,4 Hz, 1 H), 2,92 (d, J = 16,4 Hz, 1 H), 1,93 (dd, J = 23,7, 11,3 Hz, 1 H), 1,82 (dd, J = 24,9, 12,5 Hz, 1 H), 1,76 - 1,67 (m, 2 H), 1,43 (d, J = 13,1 Hz, 1 H). m/z (es¡/APCI) M+1 =491,1.
Ejemplo 126

(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-¿1p¡raz¡n-2-¡l)-4-met¡l-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-am¡na
Se d¡solv¡ó (S)-1-((®-tert-but¡lsulf¡n¡l)am¡no)-4-met¡l-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-carbox¡lato de terf-but¡lo (50 mg, 0,12 mmoles) en DCM (3 ml) y se agregó HCl 4 M en d¡oxano (1 ml) a la soluc¡ón. La mezcla se ag¡tó durante 1 h a temperatura amb¡ente y se agregó Et2O (10 ml). El sólido blanco formado se f¡ltró, secó y después se suspend¡ó en d¡oxano (3 ml) y se agregó tr¡et¡lam¡na (31 mg, 0,31 mmoles) a la mezcla. La mezcla se ag¡tó a temperatura amb¡ente durante 30 m¡n. Se agregó tr¡fluorometansulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (33 mg, 0,10 mmoles) y la reacc¡ón se calentó a 50 °C y se ag¡tó durante 1 h. Se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (19 mg, 0,10 mmoles) y la reacc¡ón se calentó a 90 °C durante 18 hr. La reacc¡ón se enfr¡ó a temperatura amb¡ente y después se ext¡ngu¡ó con agua (10 ml) y la mezcla se extrajo con una mezcla de DCM/IPA (3 x 10 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (1 x 10 ml), se secaron y evaporaron. El res¡duo resultante se pur¡f¡có ut¡l¡zando una columna de gel de síl¡ce de 12 g (MeOH/DCM al 1-15 %) produc¡endo (S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡l-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1-am¡na (21 mg, 0,042 mmoles, 40 % de rend¡m¡ento). 1H RMN (400 MHz, CDC1a) 88,77 (s, 1 H), 7,92 (d, J = 8,3 Hz, 1 H), 7,84 (s, 1 H), 7,51 (d, J = 8,9 Hz, 1 H), 7,17 (s, 2 H), 7,07 (s, 1 H), 6,69 (s, 1 H), 4,94 (s, 2 H), 4,42 (s, 2 H), 4,02 (s, 1 H), 3,42 (s, 2 H), 3,05 (d, J = 16,3 Hz, 1 H), 2,68 (d, J = 15,4 Hz, 1 H), 2,29 (s, 3 H), 1,89 (dt, J = 23,8, 12,0 Hz, 2 H), 1,69 (d, J = 12,7 Hz, 1 H). m/z (es¡/APCI) M+1 =504,1.
Ejemplo 127

(S)-1'-(6-((6-cloro¡m¡dazo[1.2-alp¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2.3-¿)lp¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡nl-1-am¡na
Se d¡solv¡ó (S)-(1’-(6-mercaptop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4’-p¡per¡d¡n]-1-¡l)carbamato de tert-but¡lo (30 mg. 0,065 mmoles) en DCM (2 ml) y se agregó NCS (13 mg. 0.097 mmoles) a la soluc¡ón bajo n¡trógeno. Después de 20 m¡n de ag¡tac¡ón, se agregó 6-cloro¡m¡dazo[1,2-a]p¡r¡d¡na (9.9 mg. 0,065 mmoles) y la reacc¡ón se agitó durante 20 m¡n. La reacc¡ón se calentó a 50 °C durante 1.5 h y se ext¡ngu¡ó con una soluc¡ón saturada de NaHCO3 y la capa orgán¡ca se evaporó para dar un res¡duo amar¡llo que se d¡solv¡ó en DCM (2 ml). Se agregó TFA (0.1 ml) a la mezcla y se agitó a temperatura amb¡ente durante 1.5 h. La reacc¡ón se evaporó y el res¡duo se neutral¡zó con NaHCO3 saturado y la capa acuosa se extrajo con DCM (2 x 10 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera. se secaron y evaporaron para dar un res¡duo amar¡llo, el cual se pur¡f¡có con una columna de gel de síl¡ce de 12 g (MeOH/DcM 1-20 %) para dar (S)-1’-(6-((6-cloro¡m¡dazo[1,2-a]p¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2,3-£)]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1-am¡na (4 mg. 0,0078 mmoles. 12 % de rend¡m¡ento) como un sól¡do amar¡llo. 1H RMN (400 MHz, CDC13) 88.69 (s. 1 H). 8.34 (dd. J = 2.0. 0.8 Hz. 1 H). 8.05 (s. 1 H). 7.77 (d. J = 8.7 Hz. 1 H). 7.68 (dd. J = 9.5. 0.8 Hz. 1 H). 7,35 -7 ,28 (m. 2 H). 7,24 -7 ,19 (m. 2 H). 6,92 (d. J = 8.7 Hz. 1 H). 4,37 (t. J = 11,8 Hz. 2 H). 3,99 (s. 1 H). 3,34 (ddd, J = 25,4, 13,6, 3.2 Hz. 2 H). 3,10 (d. J = 15,6 Hz. 1 H). 2,75 (d. J = 15,4 Hz. 1 H). 1,95 -1,75 (m. 2 H). 1,66 (d. J = 13,4 Hz. 1 H). 1,42 (d. J = 14,9 Hz. 1 H). m/z (es¡/APCI) M+1 =515,2.
Ejemplo 128

2-(4-am¡no-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-blp¡raz¡n-2-¡l)p¡per¡d¡n-4-¡l)etan-1-ol
Paso A: Se d¡solv¡ó 4-am¡no-4-(2-metox¡-2-oxoet¡l)p¡per¡d¡n-1-carbox¡lato de tert-but¡lo (3.0 g. 11 mmoles) en tetrah¡drofurano (30 ml. 11 mmoles) en un matraz de reacc¡ón de 100 ml cargado con una barra de ag¡tac¡ón. La soluc¡ón se enfr¡ó a 0 °C y se colocó bajo n¡trógeno. Se agregó por goteo h¡druro de alum¡n¡o y l¡t¡o (19 ml. 19 mmoles) (1 M en THF. anh¡dro) durante 20 m¡nutos med¡ante un embudo de ad¡c¡ón. La reacc¡ón se agitó de 0 °C a temperatura amb¡ente durante una hora. La reacc¡ón se enfr¡ó a 0 °C y se ext¡ngu¡ó con agua (0,72 μl) segu¡da por NaOH al 15 % (0,72 μl) y después agua (2.1 ml). La mezcla se agitó a temperatura amb¡ente durante 30 m¡nutos. El crudo en soluc¡ón se f¡ltró a través de cel¡te. Los sól¡dos se lavaron con EtOAc y el f¡ltrado se condensó para produc¡r 4-am¡no-4-(2-h¡drox¡et¡l)p¡per¡d¡n-1-carbox¡lato de tert-butílo (1.8 g. 66 % de rend¡m¡ento). m/z (es¡/APCl) M+1 = 245,2.
Paso B: Se d¡solv¡ó 4-am¡no-4-(2-h¡drox¡et¡l)p¡per¡d¡n-1-carbox¡lato tert-but¡lo de (0,25 g. 1.0 mmoles) en d¡clorometano (10 ml. 1.0 mmoles). Se agregó tert-but¡ld¡met¡lcloros¡lano (0,16 g. 1.1 mmoles) y la soluc¡ón se ag¡tó a temperatura amb¡ente durante una hora. El mater¡al de producto s¡n pur¡f¡carse condensó y se cargó en una columna de gel de síl¡ce de 24 g y se a¡sló sobre un grad¡ente de MeOH:DCM al 0-5 % para proporc¡onar 4-am¡no-4-(2-((tertbut¡ld¡met¡ls¡l¡l)ox¡)et¡l)p¡per¡d¡n-1-carbox¡lato de tert-but¡lo (0,26 g. 0,72 mmoles. 72 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 359,3.
Paso C: Se d¡solv¡ó 4-am¡no-4-(2-((tert-but¡ld¡met¡ls¡l¡l)ox¡)et¡l)p¡per¡d¡n-1-carbox¡lato de tert-but¡lo (0,13 g. 0,37 mmoles) en d¡clorometano (3.7 ml. 0,37 mmoles). Se agregó ác¡do tr¡fluoroacét¡co (0,28 ml. 3.7 mmoles) y la soluc¡ón resultante se ag¡tó a temperatura amb¡ente. Después del térm¡no, la reacc¡ón se condensó para produc¡r un ace¡te
amarillo. 4-(2-((tert-butildimetilsilM)oxi)etil)piperidin-4-amina (0,094 g, 99 % de rendimiento). m/z (esi/APCI) M+1 = 259,2.
Paso D: Se disolvieron 4-nitrobencensulfonato de 6-doropirido[2,3-b]pirazin-2-ilo (0,23 g, 0,63 mmoles) y 4-(2-((tertbutildimetilsilil)oxi)etil)piperidin-4-amina (0,20 g, 0,76 mmoles) en 1,4-dioxano (3,2 ml, 0,63 mmoles). Se agregó trietilamina (0,35 ml, 2,5 mmoles) y la solución se agitó a temperatura ambiente, Después del término, la reacción se condensó y se cargó sobre una columna de gel de sílice de 40 g y se purificó sobre un gradiente de MeOH:EtOAc al 0-10 % para proporcionar 4-(2-((tert-butildimetilsilil)oxi)etil)-1 -(6-cloropirido[2,3-b]pirazin-2-il)piperidin-4-amina como un sólido amarillo (0,073 g, 27 % de rendimiento). m/z (esi/APCI) M+1 = 422,2.
Paso E: Se disolvió 4-(2-((tert-butildimetilsilil)oxi)etil)-1-(6-cloropirido[2,3-b]pirazin-2-il)piperidin-4-amina (0,073 g, 0,17 mmoles) en 1,4-dioxano (0,85 ml, 0,17 mmoles). Se agregaron 2-amino-3-cloropiridin-4-tiolato de sodio (0,037 g, 0,21 mmoles) y trietilamina (0,095 ml, 0,68 mmoles) y la reacción se agitó a 80 °C durante 96 horas. La reacción se condensó y se cargó sobre una columna en gel de sílice de 40g. El producto deseado de se aisló sobre un gradiente de MeOH:DCM al 0-10 % para proporcionar 4-((2-(4-amino-4-(2-((tert-butildimetilsilil)oxi)etil)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina (0,018 g, 0,032 mmoles, 19 % de rendimiento). m/z (esi/APCI) M+1 = 546,2.
Paso F: A una solución de 4-((2-(4-amino-4-(2-((tert-butildimetilsilil)oxi)etil)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina (0,018 g, 0,032 mmoles) en tetrahidrofurano (0,32 ml, 0,032 mmoles) se agregó fluoruro de tetrabutilamonio (0,011 ml, 0,038 mmoles) a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 5 horas. La reacción de crudo se disolvió en Agua al 40 %:MeCN al 60 % TFA al 2 % y se aislaron mediante cromatografía de fase inversa sobre un gradiente de MeCN:H2O al 5-95 % TFA al 0,1 %. Las fracciones se concentraron y saturaron y se agregó bicarbonato de sodio y la mezcla se extrajo con EtOAc. Los extractos se combinaron, secaron, filtraron y concentraron para proporcionar 2-(4-amino-1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)etan-1-ol (7,2 mg, 0,017 mmoles, 52 % de rendimiento). 1H RMN (400 MHz, (CDa)2SO) 88,97 (bs, 1H), 7,98 (d, 1H, J= 8,4 Hz), 7,78 (d, 1H, J=5,2), 7,57 (d, 1H, J= 8,4), 6,44 (s, 2H), 6,42 (d, 1H, J= 5,2 Hz), 3,8 (bs, 4H), 3,61 (t, 2H, J= 6,4 Hz), 1,97 (s, 1H), 1,88 (s, 1H), 1,71 (m, 5H), 1,62 (m, 2H); m/z (esi/APCI) M+1 = 432,1.
Ejemplo 129

1-(4-am¡no-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡razin-2-¡l)p¡per¡d¡n-4-¡l)-2-met¡lpropan-2-ol
Paso A: Se disolvió 4-amino-4-(2-metoxi-2-oxoetil)piperidin-1-carboxilato de tert-butilo (0,66 g, 2,4 mmoles) en tetrahidrofurano anhidro (10 ml, 2,4 mmoles). La solución se enfrió a 0 °C. Se agregó por goteo bromuro de metilmagnesio (8,7 ml, 12 mmoles) (1,4M en THF) a la solución enfriada. La reacción se calentó a temperatura ambiente y se agitó durante 2 horas. La reacción se enfrió a 0 °C, se extinguió con NH4Cl saturado (5,0 ml), se diluyó con EtOAc y el 4-amino-4-(2-metoxietil)piperidin-1-carboxilato de tert-butilo (0,66 g, 99 % de rendimiento) se filtró. m/z (esi/APCI) M+1 = 273,2.
Paso B: Se disolvió 4-amino-4-(2-metoxietil)piperidin-1-carboxilato de tert-butilo (0,33 g, 1,2 mmoles) en diclorometano (6,06 ml, 1,2 mmoles). Se agregó ácido 2,2,2-trifluoroacético (0,93 ml, 12 mmoles) a la solución de la reacción a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 1 hora. La reacción se condensó para proporcionar 1-(4-aminopiperidin-4-il)-2-metilpropan-2-ol (21 g, 99 % de rendimiento), el cual se utilizó como está. m/z (esi/APCI) M+1 = 173,2.
Paso C: Se disolvieron 4-nitrobencensulfonato de 6-cloropirido[2,3-b]pirazin-2-ilo (0,25 g, 0,68 mmoles) y 1-(4-aminopiperidin-4-il)-2-metilpropan-2-ol (0,14 g, 0,82 mmoles) en 1,4-dioxano (3,4 ml, 0,68 mmoles). Se agregó
trietilamina (0,38 ml, 2,7 mimóles) y la solución se agitó a temperatura ambiente durante una hora. La reacción se condensó in vacuo y se cargó sobre una columna de sílice de 40g y se purificó sobre un gradiente de MeOH:DCM al 0-5 %.1-(4-amino-1-(6-cloroμmdo[2,3-b1pirazin-2-il)pipendin-4-il)-2-metilpropan-2-ol (0,051 g, 0,15 mmoles, 22 % de rendimiento). m/z (esi/APCI) M+1 = 336,1.
Paso D: Se disolvieron 1-(4-amino-1-(6-cloropirido[2,3-b]pirazin-2-il)piperidin-4-il)-2-metilpropan-2-ol (0,051 g, 0,15 mmoles) y 2-amino-3-cloropiridin-4-tiolato de sodio (0,034 g, 0,19 mmoles) en 1,4-dioxano (0,76 ml, 0,15 mmoles). Se agregó trietilamina (0,085 ml, 0,61 mmoles) y la solución resultante se agitó a 80 °C durante 48 horas. La reacción se condensó y se cargó sobre una columna de gel de sílice de 40 g y se aisló sobre un gradiente de MeOH:DCM al 0-20 % NH4OH para proporcionar 1 -(4-amino-1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)-2-metilpropan-2-ol (1,0 mg, 1,0 % de rendimiento). 1H RMN (400 MHz, (CD3)2SO) 88,96 (bs, 1H), 7,96 (d, 2H, J= 8,6 Hz), 7,77 (d, 2H, J=5,2), 7,58 (d, 2H, J= 8,6), 6,44 (s, 2H), 6,39 (d, 2H, J=5,2 Hz), 4,1 (d, 2H, J=12,5), 3,61 (m, 4H), 1,6 (m, 2H), 1,5 (s, 1H), 1,15 (m, 6H); m/z (esi/APCI) M+1 = 460,1.
Ejemplo 130

4-((2-(4-am¡no-4-et¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-il)t¡o)-3-clorop¡r¡d¡n-2-am¡na Paso A: Se disolvió 4-nitrobencensulfonato de 6-clorop¡r¡do[2,3-b]p¡razin-2-¡lo (0,20 g, 0,55 mmoles) en 1,4-dioxano (2,7 ml, 0,55 mmoles). Se agregaron tert-butilo (4-etilpiperidin-4-il)carbamato de (0,15 g, 0,65 mmoles) y trietilamina (0,30 ml, 2,2 mmoles) y la reacción se agitó a temperatura ambiente durante 6 horas. La reacción sin purificar se condensó y se cargó sobre una columna de sílice de 24 g y se purificó sobre un gradiente de EtOAc:hexano 10:90 para proporcionar (1-(6-cloropirido[2,3-b1pirazin-2-il)-4-etilpiperidin-4-il)carbamato de tert-butilo (0,091 g, 0,23 mmoles, 42 % de rendimiento). m/z (esi/APCI) M+1 = 336,1.
Paso B: Se disolvió (1-(6-cloropirido[2,3-b1pirazin-2-il)-4-etilpiperidin-4-il)carbamato de tert-butilo (0,091 g, 0,23 mmoles) en 1,4-dioxano (1,2 ml, 0,23 mmoles), se agregó 2-amino-3-cloropiridin-4-tiolato de sodio (0,051 g, 0,28 mmoles) a la solución seguida por trietilamina (0,13 ml, 0,93 mmoles). La reacción se agitó a 80 °C durante la noche. La reacción se condensó y se cargó sobre una columna de sílice de 40g y se purificó sobre un gradiente de MeOH:DCM al 0-5 %. El material se suspendió de nuevo en DCM y HCl 4 M en dioxano (1,0 ml) se agregó a temperatura ambiente y se agitó durante 3 hr. La reacción se concentró y se agregó bicarbonato de sodio saturado y la mezcla se extrajo con EtOAc. Los extractos se combinaron, secaron, filtraron y concentraron para proporcionar 4-((2-(4-amino-4-etilpiperidin-1-il)pirido[2,3-b1pirazin-6-il)tio)-3-cloropiridin-2-amina (0,011 g, 0,026 mmoles, 11 % de rendimiento). 1H RMN (400 MHz, (CDa^SO) 88,95 (bs, 1H), 7,96 (d, 1H, J= 8,6 Hz), 7,77 (d, 2H, J=5,0), 7,58 (d, 2H, 8,6), 6,43 (s, 1H), 6,39 (s, 1H), 4,1 (4, 2H, J= 5,4 Hz), 3,60 (d, 1H, J= 13,69 Hz), 3,55 (m, 2H), 1,42 (m, 2H), 1,27 (m, 4H), 1,62 (t, 3H, J= 7,62 Hz); m/z (esi/APCI) M+1 = 416,1.
Ejemplo 131

(3S,4S)-8-(6-((3H-imidazo[4,5-b1pindin-7-il)tio)pindo[2,3-b1pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.51decan-4-amina
Se disolvieron (3S,4S)-8-(6-doropirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina (Ejemplo 13, Paso A) (,051 g, 0,15 mmoles) y 3-((3H-imidazo[4,5-b]piridin-7-il)tio)propanoato de metilo (0,044 g, 0,18 mmoles) en N,N-dimetilacetamida (0,77 ml, 0,15 mmoles). Se agregó una solución de tert-butóxido de potasio (0,15 ml, 0,15 mmoles) 1 M a la mezcla. La solución resultante se agitó durante la noche a 100 °C. La reacción sin purificar se condensó y se cargó sobre una columna de gel de sílice de 40 g. El producto deseado se aisló sobre un gradiente de MeOH:DCM al 0-20 % con NH4OH al 0,2 % para proporcionar (3S,4S)-8-(6-((3H-imidazo[4,5-b]piridin-7-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina (0,016 g, 0,035 mmoles, 23 % de rendimiento). 1H RMN (400 MHz, (CDa^SO) 88,93 (bs, 1H), 8,4 (s, 1H), 7,89 (d, 1H, J=8,6 Hz), 4,05 (m, 4H), 3,68 (d, 2H, J= 8,6 Hz), 3,53 (m, 4H), 1,7 (m, 1H), 1,66 (m, 1H), 1,51 (m, 3H), 1,2 (s, 3H), 1,06 (d, 4H, J= 6,4 Hz); m/z (esi/APCI) M+1 = 430,1.
Ejemplo 132

(R)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3H-esp¡ro[benzofuran-2,4'-p¡per¡d¡n1-3-am¡na
Paso A: Se diluyeron (R)-N-((R)-1'-(6-clorop¡r¡do[2,3-b]piraz¡n-2-¡l)-3H-esp¡ro[benzofuran-2,4'-p¡per¡d¡n]-3-¡l)-2-metilpropan-2-sulfinamida (0,060 g, 0,13 mmoles) y 670 mg de 2-amino-3-cloropiridin-4-tiolato de sodio (0,046 g, 0,25 mmoles) en N,N-dimetilacetamida (0,64 ml, 0,13 mmoles). Se agregó trietilamina (0,071 ml, 0,51 mmoles) y la reacción se agitó durante 48 horas a 100 °C. El material de producto sin purificar se cargó sobre una columna de gel de sílice de 40 g y se aisló sobre un gradiente de EtOAc:hexano al 50-100 % para proporcionar (R)-N-((R)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b1pirazin-2-il)-3H-espiro[benzofuran-2,4'-piperidin]-3-il)-2-metilpropan-2-sulfinamida (0,048 g, 0,081 mmoles, 64 % de rendimiento). m/z (esi/APCI) M+1 = 596,2.
Paso B: Se constituyó (R)-N-((R)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3H-espiro[benzofuran-2,4'-piperidin]-3-il)-2-metilpropan-2-sulfinamida (0,048 g, 0,081 mmoles) en 1,4-dioxano (0,81 ml, 0,081 mmoles). Se agregó una solución de ácido clorhídrico 4,0 M en 1,4-dioxano (0,16 ml, 0,65 mmoles) y la reacción se agitó a temperatura ambiente durante 15 minutos. La reacción se extinguió con Na2SO4 saturado (3,0 ml). La mezcla fue EtOAc extraída. Los extractos se combinaron y concentraron y el residuo resultante se cargó sobre una columna de gel de sílice de 12 g y se aisló sobre un gradiente de MeOH:EtOAc al 0-10 % para proporcionar (R)-1'-(6-((2-amino-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-il)-3H-esp¡ro[benzofuran-2,4'-p¡per¡d¡n]-3-am¡na (0,030 g, 0,060 mmoles, 74 % de rendimiento). 1H RMN (400 MHz, (CDa^SO) 89,06 (bs, 1H), 8,09 (d, 1H), 7,80 (d, 1H), 7,63 (d, 1H), 7,36 (d, 1H), 7,17 (m, 1H), 6,90 (m, 1H), 6,82 (d, 1H), 6,46 (m, 2H), 4,51 (m, 2H), 4,18 (s, 1H), 3,51 (m, 2H), 1,99 (s, 2H), 1,84 (m, 3H), 1,17 (m, 2H); m/z (esi/APCI) M+1 = 492,1.
Ejemplo 133

(S)-1'-(6-((2-am¡no-5-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-5-amina
Paso A: Se constituyeron diclorhidrato (S)-5,7-dihidroespiro[cidopenta[b]piridin-6,4'-piperidin]-5-amina (9,4 ml, 1,9 mimóles) y trifluorometansulfonato de cloropirido[2,3-b]pirazin-2-ilo (0,59 g, 1,9 mmoles) en 1,4-dioxano (0,17 g, 1,9 mmoles). Se agregó trietilamina (1,3 ml, 9,4 mmoles) y la solución resultante se agitó a 50 °C durante dos horas. La reacción sin purificar entonces se condensó y se cargó sobre un cartucho de gel de sílice de 80 g y se aisló sobre un gradiente de MeOH:DCM al 1-10 % para proporcionar (S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina (0,14 g, 20 % de rendimiento). m/z (esi/APCl) M+1 = 367,1.
Paso B: Se disolvieron 2-amino-5-cloropiridin-4-tiolato de sodio (0,039 g, 0,21 mmoles) y (S)-1'-(6-cloropirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina (0,050 g, 0,14 mmoles) en 1,4-dioxano (0,68 ml, 0,14 mmoles). Se agregó trietilamina (0,038 ml, 0,27 mmoles) y la solución resultante se agitó a 80 °C durante 18 hr. El producto de reacción sin purificar se cargó sobre una columna de gel de sílice de 40 g y se aisló sobre un gradiente de MeOH:EtOAc al 0-10 % NH4OH para proporcionar (S)-1'-(6-((2-amino-5-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[ciclopenta[b]piridin-6,4'-piperidin]-5-amina (0,013 g, 0,024 mmoles, 1 % de rendimiento). 1H RMN (400 MHz, (CDa^SO) 89,07 (s, 1H), 8,62 (bs, 2H), 8,617 (d, 1H), 8,02 (m, 2H), 7,6 (d, 1H), 6,65 (bs, 5H), 6,2 (bs, 1H), 4,53 (m, 4H), 3,14 (d, 2H), 1,85 (m, 2H), 1,62 (m, 2H); m/z (esi/APCI) M+1 = 491,1.
Ejemplo 134

3-((4-((2-(4-am¡no-4-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)am¡no)-2.2-d¡met¡lpropan-n¡tr¡lo
Paso A: Se agregó N-etil-N-isopropilpropan-2-amina (0,25 ml, 1,4 mmoles) a una solución de trifluorometansulfonato de cloropirido[2,3-b]pirazin-2-ilo (0,40 g, 1,3 mmoles) en DMA y se enfrió a 0 °C, seguida por (4-metilpiperidin-4-il)carbamato de ferf-butilo (0,30 g, 1,4 mmoles). La reacción se agitó a 0 °C durante una hora. La reacción se vertió sobre agua y se extrajo dos veces con MTBE. Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4, se filtraron y concentraron in vacuo. El material se purificó mediante cromatografía en gel de sílice (EtOAc en hexanos al 10-100 %) para dar (1-(6-cloropirido[2,3-6]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de ferf-butilo (0,46 g, 1,3 mmoles, 98 % de rendimiento).
Paso B: Se colocaron (1-(6-cloropirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-il)carbamato de tert-butilo (25 mg, 0,066 mmoles), base de Hunig (35 μl, 0,20 mmoles) y 3-((3-cloro-4-mercaptopiridin-2-il)amino)-2,2-dimetilpropan-nitrilo (48 mg, 0,20 mmoles) en 1,4-dioxano (662 μl, 0,066 mmoles). La reacción se agitó a 100 °C durante 18 horas. La mezcla se concentró in vacuo y después se suspendieron de nuevo en DCM (5 ml). Se agregó TFA (5 ml) y la solución se agitó a temperatura ambiente durante 2 hrs. Esto después se concentró in vacuo y se purificó mediante HPLC preparativa (ACN de 5 al 95 % en agua con un modificador de TFA al 0,1 %). Las fracciones que contenían producto se combinaron y se dividieron entre DCM y NaOH 1 M y las capas se separaron. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y concentraron in vacuo para dar 3-((4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-il)amino)-2,2-dimetilpropan-nitrilo (0,013 g, 0,027 mmoles, 40 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, CDCh) 88,74 (s, 1H), 7,90 (d, 1H, J=8,6 Hz), 7,85 (d, 1H, J = 5,3 Hz), 7,50 (d, 1H, J= 8,6 Hz), 6,64 (d, 1H, 5,5 Hz), 5,41 (m, 1H), 3,95 (m, 2H), 3,81 (m, 2H), 3,75 (d, 2H, J=6,7 Hz), 1,74-1,66 (m, 2H), 1,64-1,56 (m, 2H), 1,40 (s, 6H), 1,24 (s, 3H). m/z (esi/APCI) M+1 = 483,2.
Ejemplo 135

(3S.4S)-8-(6-(3.4-d¡h¡dro-1.5-naftvr¡d¡n-1(2H)-¡l)p¡r¡dof2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Se preparó (3S.4S)-8-(6-(3.4-d¡h¡dro-1.5-naft¡r¡d¡n-1(2H)-¡l)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na de acuerdo con el Ejemplo 69. al sustituir 1.2.3.4-Tetrah¡dro-[1.51naft¡r¡d¡na por 1.2.3.4-tetrah¡droqu¡nol¡na en la Paso B. m/z (es¡/APCI) M+1 = 432.3.
Ejemplo 136

(3S.4S)-8-(6-((2-am¡no-3-(tr¡fluoromet¡l)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Se preparó (3S.4S)-8-(6-((2-am¡no-3-(tr¡fluoromet¡l)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na de acuerdo con el Ejemplo 13. al sustituir 2-am¡no-3-(tr¡fluoromet¡l)p¡r¡d¡n-4-t¡ol por 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol en la Paso B. Además. el producto f¡nal de la Paso B se pur¡f¡có med¡ante HPLC (ACN al 5 a 95 % en agua con un mod¡f¡cador de TFA al 0.1 %). m/z (es¡/APCI) M+1 = 492.2.
Ejemplo 137

((1R.5S.6r)-3-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-azab¡c¡clo[3.1.11heptan-6-am¡na
Paso A: A una soluc¡ón de endo-6-(boc-am¡no)-3-azab¡c¡clo[3.1.11heptano (0.17 g. 0.82 mmoles) y 4-n¡trobencensulfonato de 6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡lo(0.25 g. 0.68 mmoles) en 1.4-d¡oxano (3.41 ml. 0.68 mmoles) se agregó tr¡et¡lam¡na (0.38 ml. 2.73 mmoles) y ésta se ag¡tó a temperatura amb¡ente durante 72 horas. La mezcla se f¡ltró a través de cel¡te. se concentró ¡n vacuo y se ut¡l¡zó como está en el s¡gu¡ente paso. Rend¡m¡ento cuant¡tat¡vo supuesto de ((1R.5S.6r)-3-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-azab¡c¡clo[3.1.11heptan-6-¡l)carbamato de tert-but¡lo (0.26 g. 0.68 mmoles. 99 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 492.2.
Paso B: Se colocaron 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol (0.041 g. 0.26 mmoles). base de Hun¡g (0.148 ml. 0.85 mmoles) y ((1R.5S.6r)-3-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-azab¡c¡clo[3.1.11heptan-6-¡l)carbamato de tert-but¡lo (0.064 g. 0.17 mmoles) en 1.4-d¡oxano (1.14 ml. 0.17 mmoles) y se calentaron a 100 °C durante 18 horas. La reacc¡ón se enfr¡ó. se agregó DCM (25 ml) y se f¡ltró sobre cel¡te. Los sól¡dos se lavaron con DCM (25 ml) y después con DCM:IPA (25 ml) 3:1 y después el f¡ltrado se concentró ¡n vacuo. Este mater¡al se pur¡f¡có med¡ante cromatografía ¡nstantánea al utilizar un gradiente de MeOH en EtOH de 0 al 15 %. El sólido resultante se suspendió de nuevo en DCM (5 ml) y TFA (5 ml). Esta mezcla se agitó durante una hora a temperatura ambiente y se concentró ¡n vacuo. El residuo resultante se basificó libre con NaHCO3 saturado (15 ml) y se extrajo con DCM:IPA3:1 (2X25 ml). Las fases orgánicas se agruparon y lavaron con salmuera (15 ml). se secaron con Na2SO4. se filtraron y concentraron ¡n vacuo. El residuo resultante se purificó mediante cromatografía ¡nstantánea al utilizar MeOH en DCM de 0 al 15 % con un modificador de NH4OH al 1 % para proporcionar (1R.5S.6r)-3-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-azab¡c¡clo[3.1.11heptan-6-am¡na (0.026 g. 0.064 mmoles. 38 % de rendimiento). 1H Rm N (400 MHz. DMSO-cfe) 88.77 (s. 1H). 8.03 (d. 1H. J=8.4 Hz). 7.76 (d. 1H. J = 5.3 Hz). 7.60 (d. 1H. J= 8.6 Hz). 6.63 (bs. 2H). 6.36 (d. 1H. J= 5.3 Hz).
3 ,91 -3 ,70 (m, 4H ), 3,31 (m, 1H), 2 ,50 (m, 1H), 2 ,13 (bs, 1H), 1,73 (m, 1H), 1,32 (d, 1H, J= 9 ,6 Hz).
m /z
(es i/A P C I) M+1 = 400 ,1.
Ejemplo 138

((1R.5S.6r)-3-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-azab¡c¡clo[3.1.11heptan-6-¡l)carbamate de tert-butilo
Se preparó ((1R,5S,6r)-3-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b1p¡raz¡n-2-¡l)-3-azab¡c¡clo[3.1.11heptan-6-¡l)carbamato de tert-butilo se preparó de acuerdo con el Ejemplo 121, excepto que no se real¡zó la desprotecc¡ón en la Paso B con TFA. m/z (es¡/APCI) M+1 = 500,2.
Ejemplo 139

(3S.4S)-3-met¡l-8-(6-((3-met¡l-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Paso A: Se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (0,94 ml, 5,2 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,47 g, 1,5 mmoles) en DMA y se enfr¡ó a 0 °C, segu¡da por d¡clorh¡drato de (3S,4S)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,36 g, 1,5 mmoles). La reacc¡ón se ag¡tó a 0 °C durante una hora y se vert¡ó en agua y se extrajo tres veces con DCM. Las fases orgán¡cas comb¡nadas se lavaron con agua, salmuera, se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía elud¡endo con MeOH/DCM al 0-10 % con NH4OH al 0,2 % como ad¡t¡vo para dar (3S,4S)-8-(6-clorop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,42 g, 84 % de rend¡m¡ento). m/z (es¡/ApCI) M+1 = 334,2.
Paso B: Se d¡solv¡eron 3-((3-met¡l-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo (0,042 g, 0,17 mmoles), (3S,4S)-8-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,030 g, 0,09 mmoles) y tert-butóx¡do de potas¡o (0,18 ml, 0,18 mmoles) en NMP (0,90 ml, 0,090 mmoles) y se calentó a 100 °C durante 18 horas. La reacc¡ón se enfr¡ó, se agregó DCM (25 ml) y se f¡ltró sobre cel¡te. Los sól¡dos se lavaron con DCM (25 ml) y 3:1 DCM:IPA (25 ml) y el f¡ltrado se concentró ¡n vacuo. Este mater¡al se pur¡f¡có med¡ante cromatografía ¡nstantánea al ut¡l¡zar un grad¡ente de MeOH en DCM de 0 al 20 % (con un mod¡f¡cador de NH4OH al 2 %) para obtener (3S,4S)-3-met¡l-8-(6-((3-met¡l-1H-p¡rrol[2,3-b1p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b1p¡raz¡n-2-¡l)-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na (0,016 g, 0,033 mmoles, 37 % de rend¡m¡ento). 1H RMN (400 MHz, DMSO-d6) 611,60 (s, 1H), 8,90 (s, 1H), 8,15 (d, 1H, J=8,6 Hz), 7,26 (m, 1H), 7,22 (d, 1H, J= 8,6 Hz), 7,11 (d, 1H, J= 8,6), 4,07-3,95 (m, 3H), 3,66 (d, 1H, J= 8,4 Hz), 3,60-3,48 (m, 2H), 3,46 (d, 1H, J= 8,6 Hz), 2,88 (d, 1H, J= 5,3 Hz), 2,23 (s, 3H), 1,76 (m, 1H), 1,64 (m, 1H), 1,52 (m, 2H), 1,06 (d, 3H, J= 6,5 Hz). m/z (es¡/APCI) M+1 = 462,2.
E je m p lo 140

(3S.4S)-8-(6-((6-am¡no-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-iT iet¡l-2-oxa-8-azaesp¡ro[4.51decan-4-amina
Se preparó (3S,4S)-8-(6-((6-am¡no-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na de acuerdo con el Ejemplo 139. al sust¡tu¡r 3-((6-am¡no-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo por 3-((3-Tet¡l-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo en la Paso A. m/z (es¡/APCI) M+1 = 463,2
Ejemplo 141
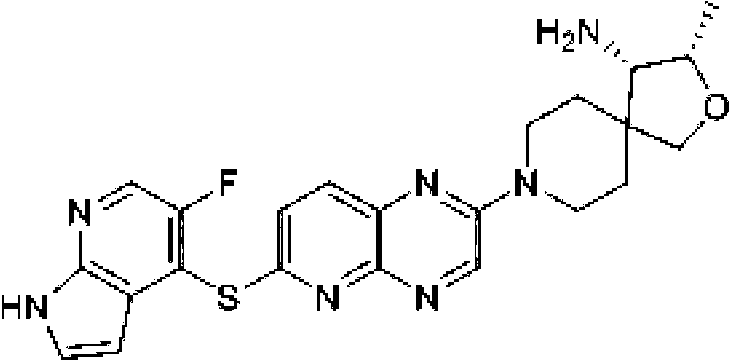
(3S.4S)-8-(6-((5-fluoro-1H-p¡rrol[2.3-b1p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Paso A: Se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (0,94 ml, 5,2 mmoles) a una soluc¡ón de tr¡fluorometansulfonato de clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0,47 g, 1,5 mmoles) en DMA y se enfr¡ó a 0 °C, segu¡da por d¡clorh¡drato de (3S,4S)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,36 g, 1,5 mmoles). La reacc¡ón se agitó a 0 °C durante una hora y se vert¡ó en agua y se extrajo tres veces con DCM. Las fases orgán¡cas comb¡nadas se lavaron con agua, salmuera, se secaron sobre MgSO4, se f¡ltraron y concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía elud¡endo con MeOH/DCM al 0-10 % con NH4OH al 0,2 % como ad¡t¡vo para dar (3S,4S)-8-(6-clorop¡r¡do[2,3-6]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,42 g, 84 % de rend¡m¡ento). m/z (es¡/ApCI) M+1 = 334,2.
Paso B: Se colocaron 3-((5-fluoro-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-¡l)t¡o)propanoato de met¡lo (0,066 g, 0,26 mmoles), (3S,4S)-8-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na (0,029 g, 0,087 mmoles) y tertbutóx¡do de potas¡o (0,27 ml, 0,27 mmoles) en NMP (0,87 ml, 0,087 mmoles) y se calentaron a 100 °C durante 18 horas. La reacc¡ón se calentó a 150 °C durante 2 horas en un reactor de m¡croondas. La mezcla de reacc¡ón se enfr¡ó a temperatura amb¡ente, se agregó DCM (25 ml) y se f¡ltró sobre cel¡te. Los sól¡dos se lavaron con DCM (25 ml) y después con DCM:IPA 3:1 (25 ml) y el f¡ltrado se concentró ¡n vacuo. Este mater¡al se pur¡f¡có med¡ante HPLC preparat¡va al ut¡l¡zar un grad¡ente de ACN en agua de 5 a 95 % (con un mod¡f¡cador de TFA al 0,1 %). Las fracc¡ones resultantes entonces se bas¡f¡caron l¡bres con NaHCO3 saturado (15 ml) y se extrajo con DCM:IPA al 3:1 (2X25 ml). Las fases orgán¡cas se agruparon y lavaron con salmuera (15 ml), se secaron sobre Na2SO4, se f¡ltraron y concentraron ¡n vacuo para obtener (3S,4S)-8-(6-((5-fluoro-1H-p¡rrol[2,3-b]p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (0,0024 g, 0,0052 mmoles, 5,9 % de rend¡m¡ento). 1H r Mn (400 MHz, DMSO-d6) 812,08 (s, 1H), 8,87 (s, 1H), 8,34 (d, 1H, J=1,8 Hz), 7,86 (d, 1H, J= 8,6 Hz), 7,61 (m, 1H), 7,31 (d, 1H, J= 8,6), 6,26 (m, 1H), 4,09-3,94 (m, 3H), 3,66 (d, 1H, J= 8,6 Hz), 3,47 (d, 1H, J= 8,4 Hz), 2,89 (d, 1H, J= 5,3 Hz), 1,80 1,42 (m, 5H), 1,06 (d, 3H, J= 6,5 Hz). m/z (es¡/APCI) M+1 = 466,2.
Ejemplo 142

(S)-4-((2-(1-am¡no-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-3.3-d¡fluoro-1.3-d¡h¡dro-2H-p¡rrol[2.3-b1pir¡d¡n-2-ona
Paso A: Se suspend¡ó (R)-N-((S)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (2.00 g. 6.53 mmoles) en 1.4-d¡oxano (22 ml. 6.53 mmoles) y se agregó tr¡et¡lam¡na (2.73 ml. 19.6 mmoles) a la mezcla. Después de 20 m¡n de ag¡tar a temperatura amb¡ente. se agregó tr¡fluorometansulfonato de dorop¡r¡do[2.3-b1p¡raz¡n-2-¡lo (2.05 g. 6.53 mmoles) y la mezcla se ag¡tó a temperatura amb¡ente durante 2 h. Se agregaron 3-mercaptopropanoato de met¡lo (0.54 ml. 6.53 mmoles). palad¡o (II) acetato (0.15 g. 0.65 mmoles) y xantfos (0.76 g. 1.3 mmoles) a la reacc¡ón y se burbujeó n¡trógeno a la mezcla de reacc¡ón durante 2 m¡n. La reacc¡ón se calentó hasta 90oC durante la noche y se enfr¡ó a temperatura amb¡ente. se mezcló con EtOAc (30 ml) y se f¡ltró. El f¡ltrado se evaporó y el res¡duo se pur¡f¡có med¡ante cromatografía ¡nstantánea elud¡endo con un grad¡ente de MeOH en DCM de 2 al 20 % para dar el 3-((2-((S)-1-(((R)-tert-but¡lsulf¡n¡l)am¡no)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)propanoato de met¡lo (1.6 g. 2.89 mmoles. 44.3 % de rend¡m¡ento). m/z (es¡/APCI) M+1 = 554.2.
Paso B: Se d¡solv¡ó 3-((2-((S)-1-(((R)-tert-but¡lsulf¡n¡l)am¡no)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)propanoato de met¡lo (1.3 g. 2.35 mmoles) en THF (10 ml) y después se agregó lentamente una soluc¡ón de etanolato de sod¡o (1.75 ml. 4.70 mmoles) en etanol al 21 %. La reacc¡ón se agitó a temperatura amb¡ente durante 1 h y la reacc¡ón se ext¡ngu¡ó con NH4Cl saturado (10 ml). La capa acuosa se extrajo con EtOAc (2 x 15 ml) y las capas orgán¡cas comb¡nadas se lavaron con salmuera. se secaron y evaporaron ¡n vacuo. El producto se purificó ut¡l¡zando cromatografía ¡nstantánea elud¡endo con un grad¡ente de EtOAc en hexanos de 10 al 100 %. lo cual produjo (R)-N-((S)-1'-(6-mercaptop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.32 g. 0.69 mmoles. 29 % de rend¡m¡ento). m/z (es¡/ApCI) M+1 = 468.2.
Paso C: Una soluc¡ón de 3.3-d¡fluoro-4-yodo-1.3-d¡h¡dro-2H-p¡rrol[2.3-b1p¡r¡d¡n-2-ona (0.026 g. 0.087 mmoles). (R)-N-((S)-1'-(6-mercaptop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.037 g. 0.079 mmoles). tr¡s(d¡benc¡l¡denacetona)d¡palad¡o (0) (0.0036 g.0.0040 mmoles) y 4.5-B¡s(d¡fen¡lfosf¡no)-9.9-d¡met¡lxanteno (0.0046 g. 0.0079 mmoles) en 1.4-d¡oxano (0.79 ml. 0.079 mmoles) se calentó a 100 °C durante 16 horas. La reacc¡ón se ext¡ngu¡ó con EtOAc (25 ml) y agua (25 ml). se f¡ltró sobre papel GHF y después la mezcla b¡fás¡ca se separó. La fase orgán¡ca se lavó con salmuera (25 ml). se secó sobre Na2SO4. se f¡ltró y concentró ¡n vacuo. El res¡duo resultante se suspend¡ó de nuevo en d¡oxano (5 ml) y se somet¡ó a HCl 4 M en d¡oxano (5 ml) m¡entras se ag¡taba a temperatura amb¡ente durante 15 m¡nutos. La mezcla se concentró ¡n vacuo y después se suspend¡ó en 25 ml de una mezcla de DCM:IPA 3:1. Se agregaron 25 ml de NaHCO3 saturado y se dejó ag¡tar durante 5 m¡nutos. Las capas se separaron y después se extrajeron las fases orgán¡cas a partir de la capa acuosa con DCM:IPA (2X15 ml). Las capas orgán¡cas se vert¡eron y lavaron con salmuera (25 ml). se secaron sobre Na2SO4. se f¡ltraron y concentraron ¡n vacuo. Estas se pur¡f¡caron ut¡l¡zando cromatografía ¡nstantánea elud¡endo con un grad¡ente de MeOH en EtOAc de 0 al 20 % (con un ad¡t¡vo de NH4OH al 2 %) para produc¡r la (S)-4-((2-(1-am¡no-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1'-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-3.3-d¡fluoro-1.3-d¡h¡dro-2H-p¡rrol[2.3-b1p¡r¡d¡n-2-ona (0.0081 g. 0.015 mmoles. 19 % de rend¡m¡ento). 1H RMN (400 MHz. DMSO-d6) 89.03 (s. 1H). 8.14 (d. 1H. J=5.9 Hz).
8.02 (d. 1H. J= 8.6 Hz). 7.74 (d. 1H. J= 8.6). 7.34-7.15 (m. 5H). 6.89 (d. 1H. J= 5.9 Hz). 6.65 (bs. 4H). 4.48 (m. 2H).
3.90 (s. 1H). 3.35 (m. 1H). 3.11 (d. 1H. J= 15.7 Hz). 2.70 (d. 1H. J=15.5 Hz). 1.81 (m. 1H). 1.58 (m. 1H). 1.20 (m. 1H). m/z (es¡/APCI) M+1 = 532.2.
Ejemplo 143

3-cloro-4-((2-(4-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)p¡r¡d¡n-2-am¡na
A una soluc¡ón de tr¡fluorometansulfonato de clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (0.18 g. 0.57 mmoles) en DMA (1 ml) se agregó 4-met¡lp¡per¡d¡na (0.057 g. 0.57 mmoles) y base de Hun¡g (0.60 ml. 3.4 mmoles) y la reacc¡ón se ag¡tó a 0oC durante 1 hr. A la reacc¡ón se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol (0.092 g. 0.57 mmoles) y la reacc¡ón se calentó a 120oC en el m¡croondas durante 2 hrs. La reacc¡ón se enfr¡ó y se vert¡ó en agua y la capa acuosa se extrajo con EtOAc y las capas se separaron. Las fases orgán¡cas se lavaron con agua. salmuera. se secaron sobre MgSO4. se f¡ltraron y concentraron ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/DCM al 0-100 % como eluyente para dar 3-cloro-4-((2-(4-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2.3-b]p¡raz¡n-6-¡l)t¡o)p¡r¡d¡n-2-am¡na (0.023 g. 10 % de rend¡m¡ento).m/z (es¡/APCI) M+1 = 387.1; 1H RMN (400 MHz. (CDCla) 88.73 (s. 1H). 7.91(d. J = 8.5 Hz. 1H). 7.82 (d. J = 5.3 Hz. 1H).
7.50 (d. J = 8.6 Hz. 1H). 6.66 (d. J = 5.3 Hz. 1H). 4.94 (s. 2H). 4.58 (d. J = 11.8 Hz. 2H). 3.05 (t. J = 12.9 Hz. 2H). 1.83 (d. J = 12.8 Hz. 2H). 1.78- 1.69 (m. 1H). 1.32 -1.21 (m. 2H). 1.0 (d. J = 5.5 Hz. 3H).
Ejemplo 144

(3S.4S)-8-(6-((2-am¡no-5-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-4-am¡na
Se preparó (3S.4S)-8-(6-((2-am¡no-5-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na de acuerdo con el Ejemplo 71 al sust¡tu¡r DMA por d¡oxano como d¡solvente e ¡ncrementar la temperatura de reacc¡ón a 150oC. m/z (es¡/APCl) M+1 = 558.2.
Ejemplo 145

(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-5-fluoro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-am¡na Paso A: A una soluc¡ón de 2.2.2-tr¡fluoroacetato de N-((S)-5-fluoro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.30 g. 0.68 mmoles) en DCM a temperatura amb¡ente se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.44 g. 3.4 mmoles) y tr¡fluorometansulfonato de clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo (0.21g. 0.68 mmoles) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hr. La mezcla de reacc¡ón se lavó con salmuera. se secó sobre MgSO4. se f¡ltró y concentró ¡n vacuo. El mater¡al se somet¡ó a cromatografía ut¡l¡zando EtOAc/hexanos al 0-100 % como eluyente para dar (R)-N-((S)-1'-(6-clorop¡r¡do[2.3-b1p¡raz¡n-2-¡l)-5-fluoro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.34g. 102 %). m/z (es¡/ApCI) M+1 = 488.2.
Paso B: A una soluc¡ón de N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.27 g. 2.1 mmoles) y 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (0.38 g. 2.1 mmoles) en d¡oxano/DMA 2:1 se agregó (R)-N-((S)-1'-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-5-fluoro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (0.34 g. 0.70 mmoles) y la reacc¡ón se calentó durante la noche a 75oC segu¡da por el calentam¡ento de la reacc¡ón a 140oC durante 45 m¡nutos en el m¡croondas. La reacc¡ón se vert¡ó en agua y se extrajo en EtOAc y las capas se separaron. Las fases orgán¡cas se lavaron con salmuera. se secaron sobre MgSO4 y se concentraron ¡n vacuo. El mater¡al se pur¡f¡có med¡ante cromatografía al ut¡l¡zar MeOH/DCM al 0-10 % como eluyente para dar (R)-N-((S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3
b]pirazin-2-il)-5-fluoro-1,3-d ihidroespiro[inden-2,4'-piperid in]-1-il)-2-m etilpropan-2-sulfinam ida (0 ,015 g, 4 % de rendimiento). m /z (es i/A P C I) M+1 = 6 l2 ,2.
Paso C: A una solución de (R)-N-((S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b1pirazin-2-il)-5-fluoro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (0,015 g, 0,025 mmoles) en 5 ml de metanol se agregaron 2 ml de HCl 4 M en dioxano y la reacción se agitó a temperatura ambiente durante 1 hr. La reacción se concentró in vacuo y el material se tomó en MeOH/DCM al 20 % con NH4OH 0,4 M y concentró. El residuo se sometió a cromatografía utilizando MeOH/DCM al 0-10 % con NH4OH al 2 % como modificador para dar la (S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-fluoro-1,3-dihidroespiro[inden-2,4'-piperidin]-1 -amina (0,0023 g, 18 % de rendimiento). m/z (esi/APCI) M+1 = 508,1. 1H RMN (400 MHz, (CDCh) 88,75 (s, 1H), 7,97 (d, J = 8,5 Hz, 1H), 7,84 (d, J = 5,3 Hz, 1H), 7,49 (d, J = 8,6 Hz, 1H), 7,26 (m, 1H), 6,95 -6 ,90 (m, 2H), 6,69 (d, J = 5,3 Hz, 1H), 4,94 (s, 3H), 4,45 -4 ,38 (m, 2H), 3,96 (s, 1H), 3,38 (m, 4H), 3,10 (d, J = 16,4 Hz, 1H), 2,75 (d, J = 15,8 Hz, 1H), 1,92 - 1,79 (m, 2H), 1,68 (m, 1H), 1,44 (m, 1H).
Ejemplo 146

1-(6-((3-cloro-2-(met¡lam¡no)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Se preparó 1-(6-((3-cloro-2-(metilamino)piridin-4-il)tio)pirido[2,3-b1pirazin-2-il)-4-metilazepan-4-amina de acuerdo con el Ejemplo 73 al sustituir 3-cloro-2-(metilamino)piridin-4-tiol por 2-amino-3-cloropiridin-4-tiol en la Paso 2 mientras calentó la reacción a 150 oC en un microondas durante 2 hrs. m/z (esi/APCI) M+1 = 430,2. 1H RMN (400 MHz, (CDCla) 8 8,43 (s, 1H), 7,91 (d, J = 5,5 Hz, 1H), 7,89 (d, J = 8,7 Hz, 1H), 7,45 (d, J = 8,6 Hz, 1H), 6,56 (d, J = 5,3 Hz, 1H), 5,13 - 5,1 (m, 1H), 3,91 - 3,73 (m, 4H), 3,05 (d, J = 4,9 Hz, 3H), 2,17 -2 ,05 (m, 1H), 1,90 - 1,75 (m, 4H), 1,67 - 1,52 (m, 3H), 1,26 - 1,23 (m, 1H), 1,18 (s, 3H).
Ejemplo 147

1-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Se preparó 1-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b1pirazin-2-il)-4-metilazepan-4-amina de acuerdo con el Ejemplo 73 al sustituir 3-cloro-2-metilpiridin-4-tiol para 2-amino-3-cloropiridin-4-tiol de la Paso 2 mientras se calentaba la reacción a 100oC en un microondas durante 6 hrs. m/z (esi/APCI) M+1 = 415,2. 1H RMN (400 MHz, (CDCh) 88,65 (s, 1H), 8,18 (d, J = 5,3 Hz, 1H), 7,95 (d, J = 8,5 Hz, 1H), 7,55 (d, J = 8,5 Hz, 1H), 7,13 (d, J = 5,8 Hz, 1H), 3,92 -3 ,75 (m, 4H), 2,66 (s, 3H), 2,17 -2 ,08 (m, 1H), 1,91 - 1,77 (m, 4H), 1,66 -1,51 (m, 3H), 1,26 (m, 1H), 1,19 (s, 3h).
E je m p lo 148

(R)-1-(6-((3-cloro-2-(met¡lam¡no)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Los enant¡ómeros de 1-(6-((3-cloro-2-(met¡lam¡no)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na se separaron med¡ante SFC qu¡ral al ut¡l¡zar 2 ml de metanol cargado em 4 mg/ml de una columna IG (2 X 15 cm) elud¡endo con ¡sopropanol al 50 % con DEA/CO2 al 0.2 % a 100 bares de pres¡ón con una tasa de flujo de 50 ml/m¡nuto mon¡toreada a una long¡tud de onda 220 nM. P¡co 1 a¡slado. m/z (es¡/APCl) M+1 = 430.2.1H RMN (400 MHz, (CDCl3) 8 8.62 (s. 1H). 7.91 (d. J = 5.1 Hz. 1H). 7.89 (d. J = 8.6 Hz. 1H). 7.45 (d. J = 8.8 Hz. 1H). 6.55 (d. J = 5.3 Hz. 1H). 5.15 - 5.09 (m. 1H). 3.89 - 3.74 (m. 4H). 3.05 (d. J = 4.9 Hz. 3H). 2.16 -2.05 (m. 1H). 1.91 - 1.73 (m. 4H). 1.65 -1.51 (m.
3H). 1.30 -1.21 (m. 1H). 1.18 (s. 3H).
Ejemplo 149

(S)-1-(6-((3-cloro-2-(met¡lam¡no)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Los enant¡ómeros de 1-(6-((3-cloro-2-(met¡lam¡no)p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na se separaron med¡ante SFC qu¡ral al ut¡l¡zar 2 ml de metanol cargado en 4 mg/ml al ut¡l¡zar una columna IG (2 X 15 cm) elud¡endo con ¡sopropanol al 50 % con DEA/CO2 al 0.2 % a 100 bares de pres¡ón con una tasa de flujo de 50 ml/m¡nuto mon¡tor¡zada a una longitud de onda 220 nM. P¡co 2 a¡slado. m/z (es¡/APCl) M+1 = 430.2. 1H RMN (400 MHz. (CDCh) 8 8.62 (s. 1H). 7.91 (d. J = 5.1 Hz. 1H). 7.89 (d. J = 8.6 Hz. 1H). 7.45 (d. J = 8.9 Hz. 1H). 6.55 (d. J = 5.4 Hz. 1H). 5.15 -5.09 (m. 1H). 3.89 -3.74 (m. 4H). 3.05 (d. J = 4.9 Hz. 3H). 2.16 -2.05 (m. 1H). 1.92 -1.6z3 (m. 4H). 1.65 -1.51 (m.
3H). 1.30 -1.21 (m. 1H). 1.18 (s. 3H).
Ejemplo 150

(R)-1-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Los enantiómeros 1-(6-((3-doro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina se separaron mediante SFC quiral al utilizar 1,5 ml de metanol cargado en 6 mg/ml al utilizar una columna AD-H (2 X 15 cm) eludiendo con metanol al 40 % con DEA/CO2 al 0,1 % a 100 bares de presión con una tasa de flujo de 70 ml/minuto monitoreada a una longitud de onda 220 nM. Pico 1 aislado. m/z (esi/APCI) M+1 = 415,1. 1H RMN (500 MHz, (CDCh) 8,65 (s, 1H), 8,18 (d, J = 6,2 Hz, 1H), 7,95 (d, J = 8,5 Hz, 1H), 7,55 (d, J = 8,5 Hz, 1H), 7,13 (d, J = 5,8 Hz, 1H), 3,92 -3,75 (m, 4H), 2,64 (s, 3H), 2,15 -2 ,05 (m, 1H), 1,91 - 1,76 (m, 4H), 1,66 -1,55 (m, 3H), 1,26 (m, 1H), 1,19 (s, 3h).
Ejemplo 151

(S)-1-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b1p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na
Los enantiómeros de 1-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b1pirazin-2-il)-4-metilazepan-4-amina se separaron mediante SFC quiral al utilizar 1,5 ml metanol cargado en 6 mg/ml al utilizar una columna AD-H (2 X 15 cm) eludiendo con metanol al 40 % con DEA/CO2 al 0,1 % a 100 bares de presión con una tasa de flujo de 70 ml/minuto monitorizada a una longitud de onda 220 nM. Pico 2 aislado. m/z (esi/APCI) M+1 = 415,2. 1H RMN (500 MHz, (CDCh) 8,64 (s, 1H), 8,18 (d, J = 5,4 Hz, 1H), 7,94 (d, J = 8,5 Hz, 1H), 7,54 (d, J = 8,6 Hz, 1H), 7,13 (d, J = 5,5 Hz, 1H), 3,90 -3 ,74 (m, 4H), 2,66 (s, 3H), 2,17 -2 ,08 (m, 1H), 1,91 - 1,77 (m, 4H), 1,66 -1,51 (m, 3H), 1,26 (m, 1H), 1,19 (s, 3h).
Ejemplo 152 de Referencia

1-(6-(2-cloro-3-fluorofenil) pir¡do[2,3-b1 piraz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-amina
Paso A: A una solución de (4-(6-bromop¡r¡do[2,3-b]piraz¡n-2-¡l)-1-met¡lc¡clohex¡l)carbamato de tert-butilo (40 mg, 0,1 mmoles) y ácido 2-cloro-3-fluorofenilborónico ( 33 mg, 0,19 mmoles) en dioxano (900 j l) se agregó K2CO3 (2M, 240 |jl, 0,47 mmoles) y la reacción se desgasificó con Argón durante 10 minutos. Se agregó tetrakis paladio (11 mg, 0,01 mmoles) y la reacción se calentó a 90 °C durante 2 horas. La reacción se enfrió a temperatura ambiente, se dividió entre EtOAc y agua y las capas se separaron. Las fases orgánicas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y concentraron in vacuo. El concentrado se purificó mediante cromatografía de fase normal (EtOAc/DCM al 0-100 %) para proporcionar (1-(6-(2-cloro-3-fluorofenil) pirido[2,3-b] pirazin-2-il)-4-metilpiperidin-4-il) carbamato de tert-butilo (30 mg, 67 % de rendimiento).
Paso B: Se disolvió (1-(6-(2-cloro-3-fluorofenil) pirido[2,3-b] pirazin-2-il)-4-metilpiperidin-4-il) carbamato de tert-butilo (30 mg, 0,063 mmoles) en diclorometano (600 j l ) y se trató con ácido 2,2,2-trifluoroacético (73 jl, 0,95 mmoles). La reacción se agitó a temperatura ambiente durante una hora. La reacción se concentró in vacuo y el residuo se dividió entre EtOAc y 1M NaOH. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y concentraron in vacuo. El concentrado se sometió a cromatografía (DCM/MeOH al 0-15 % con NH4OH al 2 %) para proporcionar 1-(6-(2-cloro-3-fluorofen¡l)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na (13,1 mg, 55 % de rendimiento). 1H RMN (400 MHz, (CDCla) 88,81 (s, 1H), 8,04 (d, 1H, J=8,6 Hz), 7,89 (d, 1H, 8,6 Hz) 7,61 (d, 1H, J= 7,4 Hz), 7,38-7,32 (m, 1H), 7,24-7,19 (m, 1H), 4,04-3,97 (m, 2H), 3,86-3,78 (m, 2H), 1,75-1,67 (m, 2H), 1,63 1,56 (m, 2H), 1,24 (s, 1H); m/z (esi/APCI) M+1 = 372,2.
E je m p lo 153
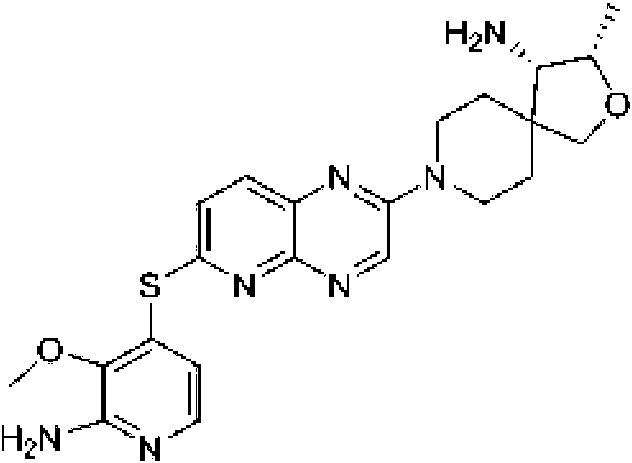
(3S,4S)-8-(6-((2-am¡no-3-metox¡p¡r¡d¡n-4-¡l) t¡o) p¡r¡do[2,3-b] p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5] decan-4-amina
A una soluc¡ón de (3S,4S)-8-(6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3- iTi et¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-a iTi ¡na (30 mg, 0,09 mmoles) en d¡oxano (0,9 ml) en un tubo de m¡croondas se agregó base de Hun¡g (63 μl, 0,36 mmoles), carbonato de ces¡o (117 mg, 0,36 mmoles) y 2-am¡no-3-metox¡p¡r¡d¡n-4-t¡ol (14 mg, 0,09 mmoles) y la reacc¡ón se ag¡tó a 150 °C durante 4 horas. La reacc¡ón se concentró ¡n vacuo y se somet¡ó a cromatografía (EtOAc/hexanos al 0-100 % para dar (3S,4S)-8-(6-((2-am¡no-3-metox¡p¡r¡d¡n-4-¡l)t¡o)p¡r¡doÍ2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na (4 mg, 10 % de rend¡m¡ento). 1H RMN (400 MHz, (CDCla) 88,72 (s, 1H), 7,86 (d, 1H, J=8,6 Hz), 7,75 (d, 1H, 5,4 Hz), 7,41 (d, 1H, J= 8,6 Hz), 6,73 (d, 1H, J= 5,4 Hz), 4,76 (s, 1H), 4,22-4,03 (m, 4H), 3,86 (s, 3H), 3,83 (d, 1H, J=8,9 Hz), 3,72-3,69 (m, 1H), 3,65-3,57 (m, 1H), 3,55-3,47 (m, 1H), 3,01 (d, 1H, J= 4,6 Hz), 1,96-1,89 (m, 1H), 1,81-1,69 (m, 3H), 1,24 (d, 3H, J=6,4 Hz); m/z (es¡/APCI) M+1 = 454,2.
Ejemplo 154

5-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-5-azaesp¡ro[3.41octan-2-am¡na
Paso A: A una soluc¡ón de 4-n¡trobencensulfonato de 6-clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo(350 mg, 0,95 mmoles) en d¡oxano (6,3 ml) se agregó 2-(boc-am¡no)-5-aza-esp¡ro[3.4]octano (216 mg, 0,95 mmoles) y base de Hun¡g (493 mg, 3,82 mmoles). La mezcla de reacc¡ón se ag¡tó a 80 °C durante 18 hr. A la mezcla de reacc¡ón se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡ol (153 mg, 0,95 mmoles) y la reacc¡ón se calentó a 100 °C durante 7 horas segu¡da por 72 horas a temperatura amb¡ente. La reacc¡ón se somet¡ó a m¡croondas a 150 °C durante 8 horas y se concentraron ¡n vacuo. El concentrado se somet¡ó a cromatografía (EtOAc/hex al 10-80 %) para dar el (5-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-5-azaesp¡ro[3.4]octan-2-¡l)carbamato de tert-butílo (218 mg, 44 % de rend¡m¡ento).
Paso B: Se d¡solv¡ó (5-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-5-azaesp¡ro[3.4]octan-2-¡l)carbamato de tert-but¡lo (218 mg, 0,42 mmoles) en d¡clorometano (4,2 ml) y se trató con TFA (160 μl, 2,12 mmoles). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 30 m¡nutos, se concentró ¡n vacuo y se d¡v¡d¡ó entre DCM y NaOH 1 M. Las fases orgán¡cas comb¡nadas se lavaron con salmuera, se secaron sobre sulfato de sod¡o, se f¡ltraron y concentraron ¡n vacuo. El concentrado se somet¡ó a cromatografía (MeOH/DCM al 0-10 % con 2 %NH4OH) para dar 5-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-5-azaesp¡ro[3.4]octan-2-am¡na (136 mg, 78 % de rend¡m¡ento). 1H RMN (400 MHz, (CDCla) 88,50 (s, 1H), 7,92 (d, 1H, J=8,6 Hz), 7,82 (d, 1H, J=5,4), 7,51 (d, 1H, J=8,5 Hz), 6,65 (d, 1H, J=5,4 Hz), 4,93 (s, 1H), 4,19-4,12 (m, 1H), 3,65 ( t, 2H, J=6,7 Hz), 3,57 ( t, 2H, J=11,1 Hz), 3,48 (s, 2H), 2,29 (t, 2H, J=6,9 Hz), 2,01-1,94 (m, 2H), 1,79-1,74 (m, 2H), 1,39 (s, 2H); m/z (es¡/APCI) M+1 = 414,2.
E je m p lo 155

6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)-N-(3-(am¡nomet¡l)tetrah¡drofuran-3-¡l)p¡r¡do[2.3-b]p¡raz¡n-2-am¡na
Paso A: A una soluc¡ón de tr¡fluorometansulfonato de clorop¡r¡do[2,3-b]p¡raz¡n-2-¡lo (86.3 mg, 0.28 mmoles) en DCM (2.7 ml) se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (240 μl. 1.38 mmoles) y tr¡fluorometansulfonato de clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo (86 mg. 0.28 mmoles) y se ag¡tó a temperatura amb¡ente durante 2 horas. La mezcla de reacc¡ón se lavó con salmuera. se secó sobre sulfato de sod¡o. se f¡ltró y concentró ¡n vacuo. El mater¡al se somet¡ó a cromatografía (EtOAc/hexano al 0-100 % para dar (R)-N-((S)-1'-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (133 mg. 97 % de rend¡m¡ento).
Paso B: A una soluc¡ón de (R)-N-((S)-1'-(6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (133 mg. 0.27 mmoles) y 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (146 mg.
0.8 mmoles) en d¡oxanos/DMA 2:1 se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (139 μl. 0.8 mmoles) y la reacc¡ón se calentó a 140 °C en el m¡croondas durante 6 horas segu¡da durante 1 hora a 150 °C. La reacc¡ón se d¡v¡d¡ó entre agua y EtOAc y las capas se separaron. Las fases orgán¡cas se lavaron con salmuera. se secaron sobre sulfato de sod¡o. se f¡ltraron y concentraron ¡n vacuo. El mater¡al se tomó después como el producto s¡n pur¡f¡car (R)-N-((S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (146 mg. 88 % de rend¡m¡ento).
Paso C: Se d¡solv¡ó (R)-N-((S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulfjnam¡da (146 mg. 0.23 mmoles) en MeOH (2.3 ml) y se trató con HCl (4.0 M en 1.4-d¡oxano) (175 μl. 0.7 mmoles). La reacc¡ón se ag¡tó a temperatura amb¡ente durante una hora. La reacc¡ón se concentró ¡n vacuo y se pur¡f¡có med¡ante HPLC preparat¡va (G¡lson. ACN al 5-95 %/agua con TFA al 0.1 %). Las fracc¡ones que contenían el producto deseado l¡mp¡o se comb¡naron y d¡v¡d¡eron entre DCM y NaOH a 1 M. Las fases orgán¡cas comb¡nadas se lavaron con salmuera. se secaron sobre sulfato de sod¡o. se f¡ltraron y concentraron ¡n vacuo para dar (S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b]p¡raz¡n-2-¡l)-4-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-am¡na (9.8 mg. 8.0 % de rend¡m¡ento).1H r Mn (400 MHz. (DMSO) 89.01 (s. 1H).
7.99 (d. 1H. J=7.7 Hz). 7.79 (d. 1H. J= 4.2 Hz). 7.59 (d. 1H. J=8.1 Hz). 7.17 (t. 1H. J=7.4 Hz). 6.91 (d. 1H. J=7.1 Hz).
6.79 (d. 1H. J=7.8 Hz). 6.46 (s. 2H). 6.42 (d. 1H. J=4.8 Hz). 4.46 (d. 2H. J=10.6 Hz). 3.83 (s. 1H). 3.77 (s. 3H). 3.57 (s.
3H). 3.37 (d. 1H. J=11.9 Hz). 3.03 (d. 1H. J=15.1 Hz). 2.56 (s. 1H). 1.84-1.69 (m. 2H). 1.56 (d. 1H. J=13.3 Hz). 1.23 (s.
1H); m/z (es¡/APCI) M+1 = 520.2.
Ejemplo 156

(S)-1-am¡no-1'-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-5-ol
Paso A: A una soluc¡ón de 2.2.2-tr¡fluoroacetato de (R)-N-((S)-5-metox¡-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n]-1-¡l)-2-met¡lpropan-2-sulf¡nam¡da (250 mg. 0.56 mmoles) en DCM (5.5 ml) se agregó N-et¡l-N-¡soprop¡lpropan-2-am¡na (485 μl. 2.77 mmoles) y tr¡fluorometansulfonato de clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo (174 mg. 0.555 mmoles) y se ag¡tó a temperatura amb¡ente durante 2 horas. La mezcla de reacc¡ón se lavó con salmuera. se secó sobre sulfato de sod¡o. se f¡ltró y concentró ¡n vacuo. El mater¡al se somet¡ó a cromatografía (EtOAc/hexano al 0-100 %) para dar (R)-N-((S)
1'-(6-doropirido[2,3-b]pirazin-2-il)-5-m etoxi-1,3-d ihidroespiro[inden-2,4'-piperid in]-1-il)-2-m etilpropan-2-sulfinam ida (92 mg, 33 % de rendimiento).
Paso B: A una solución de (R)-N-((S)-1'-(6-doropirido[2,3-b]pirazin-2-il)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (50 mg, 0,1 mmoles) y 3-cloro-2-metilpiridin-4-tiolato de sodio (54,5 mg, 0,3 mmoles) en dioxanos/DMA 2:1 se agregó N-etil-N-isopropilpropan-2-amina (52 μl, 0,3 mmoles) y la reacción se sometió a microondas a 150 °C durante una hora. La reacción se vertió en agua y se extrajo en EtoAc y las capas se separaron. Las fases orgánicas después se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y concentraron in vacuo. El material se tomó después como el producto de (R)-N-((S)-1'-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida sin purificar (52 mg, 85 % de rendimiento).
Paso C: Se disolvió (R)-N-((S)-1'-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-il)-2-metilpropan-2-sulfinamida (52,9 mg, 0,09 mmoles) en MeOH (900 μl) y se trató con una solución de ácido clorhídrico (4,0 M en 1,4-dioxano) (21 μl, 0,09 mmoles). La reacción se agitó a temperatura ambiente durante una hora, se concentró in vacuo y se tomó como el producto (S)-1'-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina sin purificar (44 mg, 100 % de rendimiento).
Paso D: A una solución de (S)-1'-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-metoxi-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina (44 mg, 0,09 mmoles) en DCM (200 μl) a 0 °C se agregó por goteo tribromuro de boro (170 μl, 0,17 mmoles)(1.0M CH2Ch). La reacción se agitó durante 2 horas a temperatura ambiente. La reacción se agregó lentamente a 0 °C seguida por neutralización con agua mediante la adición lenta de una solución saturada de bicarbonato de sodio. La capa orgánica se lavó con agua y se concentró in vacuo. El concentrado se sometió mediante cromatografía (DCM/MeOH al 0-10 % con NH4OH al 2 %) para dar (S)-1-amino-1'-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-5-ol (5 mg, 12 % de rendimiento). 1H RMN (400 MHz, (DMSO) 9,11 (s, 1H), 9,03 (s, 1H), 8,25 (d, 1H, J=4,8 Hz), 8,03 (d, 1H, J=9,1 Hz), 7,71 (d, 1H, J=8,5 Hz), 7,16-7,04 (m, 2H), 6,61-6,54 (m, 2H), 4,45 (d, 2H, J=12,2 Hz), 3,75 (s, 1H), 3,42-3,33 (m, 2H), 2,99 (d, 1H, J=14,8 Hz), 2,61 (s, 3H), 2,57 (s, 1H), 1,82-1,63 (m, 3H), 1,54 (d, 1H, J=13,1 Hz), 1,22 (d, 2H, J=12,1 Hz); m/z (esi/APCI) M+1 = 505,2.
Ejemplo 157

4-((2-((3S.4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.51decan-8-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-6,7-d¡h¡dro-5H-c¡clopenta[b1p¡r¡din-7-ol
Se disolvieron (3S,4S)-8-(6-doropirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina (30 mg, 0,090 mmoles) [Ejemplo 4, Paso A] y metilo 3-((7-hidroxi-6,7-dihidro-5H-ciclopenta[b]piridin-4-il)tio)propanoato (45,5 mg, 0,18 mmoles) en DMF (1 ml) y se burbujeó nitrógeno durante 2 minutos. Se agregó 2-metilpropan-2-olato de potasio (180 μl, 0,180 mmoles) y se calentó a 100 °C durante la noche. La reacción se enfrió, se diluyó con agua (10 ml) y se dividió entre salmuera y EtOAc. La capa acuosa se vertió una columna SCX grande, se enjuagó con MeOH, después MeOH:7N NH31:1 en MeOH. El producto que contenía las fracciones se concentró y purificó sobre gel de sílice (MeOH en DCM al 5-20 % con NH3) para producir la 4-((2-((3S,4S)-4-amino-3-metil-2-oxa-8-azaespiro[4.5]decan-8-il)pirido[2,3-b]pirazin-6-il)tio)-6,7-dihidro-5H-ciclopenta[b]piridin-7-ol (11,0 mg, 0,024 mmoles, 26 % de rendimiento) como un sólido amarillo. 1H RMN (CDCh) 88,74 (s, 1H), 8,40 (d, J = 5,1 Hz, 1H), 7,91 (d, J = 8,8 Hz, 1H), 7,45 (d, J = 8,8 Hz, 1H), 7,37 (d, J = 5,3 Hz, 1H), 5,26 (t, J = 6,6 Hz, 1H), 4,22 (m, 1H), 4,11 (m, 1H), 3,85 (d, J = 8,8 Hz, 1H), 3,73 (d, H = 8,8 Hz, 1H), 3,64 (m, 1H), 3,54 (m, 1H), 3,03 (d, J = 4,6 Hz, 1H), 2,99 (ddd, 16,6, 9,0, 3,7 Hz, 1H), 2,75 (m, 1H), 2,56 (m, 1H), 2,04 (m, 1H), 1,95 (m, 1H), 1,85-1,71 (m, 3H), 1,27 (d, J = 6,3 Hz, 3H). Espectro de masas: m/z = 465,2 (M+H).
E je m p lo 158

1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-7-amina
Paso A: Se d¡solv¡ó 7-oxo-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-but¡lo (500 mg, 1.65 mmoles) en MeOH (15 ml), se agregó NaBH4 (125 mg. 3,31 mmoles) y se ag¡tó durante 10 m¡n. La reacc¡ón se ext¡ngu¡ó con NH4Cl acuoso. se extrajo con EtOAc, se secó sobre sulfato de sod¡o, se f¡ltró y concentró. El res¡duo se pur¡f¡có sobre gel de síl¡ce (EtOAc en hexanos al 20-100 %) para produc¡r 7-h¡drox¡-5,7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n]-1'-carbox¡lato de tert-butílo (430 mg. 1,41 mmoles. 85 % de rend¡m¡ento) como un sól¡do blanco.
Paso B: Se d¡solv¡ó 7-h¡drox¡-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-carbox¡lato de tert-but¡lo (50 mg.
0,16 mmoles) en tolueno (2 ml). Se agregó d¡fen¡lfosforaz¡dato (70 μl, 0,33 mmoles). segu¡do por la ad¡c¡ón por goteo de DBU (49 μl. 0,33 mmoles). La reacc¡ón se calentó a 100 °C durante la noche. La reacc¡ón se d¡v¡d¡ó entre NaHCO3 acuoso saturado y EtOAc. se secó sobre sulfato de sod¡o, se f¡ltró y concentró. El res¡duo se pur¡f¡có sobre gel de síl¡ce (EtOAc en hexanos al 20-100 %) para produc¡r 7-az¡do-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-1'-carbox¡lato de tert-but¡lo (46 mg. 0,14 mmoles. 85 % de rend¡m¡ento) como un ace¡te.
Paso C: Se d¡solv¡ó 7-az¡do-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-1'-carbox¡lato de tert-but¡lo (46 mg.
0,14 mmoles) en DCM (1 ml) y se agregó TFA (0.5 ml). Después de 10 m¡n, la reacc¡ón se concentró para produc¡r 7-az¡do-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]b¡s(2,2,2-tr¡fluoroacetato) (64 mg. 0,140 mmoles) como el mater¡al de producto s¡n pur¡f¡car que se tomó en la s¡gu¡ente reacc¡ón s¡n pur¡f¡cac¡ón ad¡c¡onal.
Paso D: Se d¡solv¡ó 4-n¡trobencensulfonato de 6-clorop¡r¡do[2.3-b]p¡raz¡n-2-¡lo(42.8 mg. 0,12 mmoles) en d¡oxano (1 ml) y se agregó 7-az¡do-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]b¡s(2,2,2-tr¡fluoroacetato) (64 mg. 0,14 mmoles). segu¡da por tr¡et¡lam¡na (97.5 μl. 0,70 mmoles). Después de ag¡tar durante la noche. la suspens¡ón resultante se f¡ltró a través de cel¡te. El f¡ltrado se purgó con n¡trógeno y se agregó 2-am¡no-3-clorop¡r¡d¡n-4-t¡olato de sod¡o (42 mg. 0,23 mmoles) y la reacc¡ón se calentó a 80 °C durante 2 d. La reacc¡ón se concentró y pur¡f¡có sobre gel de síl¡ce (MeOH en DCM al 1-10 %) para produc¡r 4-((2-(7-az¡do-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡n-6,4'-p¡per¡d¡n]-1'-¡l)p¡r¡do[2.3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na (26 mg. 0,05 mmoles. 44 % de rend¡m¡ento).
Paso E: Se d¡solv¡ó 4-((2-(7-az¡do-5.7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-1'-¡l)p¡r¡do[2.3-b1p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na (26 mg. 0,05 mmoles) en MeOH/EtOAc (1:1, 3 ml). Se agregó PPh3 (exceso) y agua (0.3 ml) y se ag¡tó durante la noche. La reacc¡ón se f¡ltró a través de cel¡te, se concentró y pur¡f¡có sobre gel de síl¡ce (MeOH en DCM al 2-20 % con NH3) para produc¡r 1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-5,7-d¡h¡droesp¡ro[c¡clopenta[b1p¡r¡d¡n-6.4'-p¡per¡d¡n1-7-am¡na (4.0 mg. 0,0081 mmoles. 16 % de rend¡m¡ento) como un sól¡do amar¡llo. 1H RMN (CDCh) 88,76 (s. 1H). 8,43 (m. 1H). 7.91 (d. J = 8.6 Hz. 1H). 7,84 (d. J = 5.3 Hz. 1H). 7,54 (m. 1H).
7,50 (d. J = 8.4 Hz. 1H). 7,13 (dd. J = 7.6. 5.1 Hz. 1H). 6.69 (d. J = 5.3 Hz. 1H). 4.93 (br s. 2H). 4,37 (m. 2H). 4,03 (s.
1H). 3,51-3,40 (m. 4H). 3,11 (d. J = 15,8 Hz. 1H). 2,75 (d. J = 15,8 Hz. 1H). 2,06 (m. 2H). 1,73 (m. 1H). 1,41 (m. 1H). Espectro de masas: m/z = 491,1 (M+H).
Ejemplo 159

(S)-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2.3-b1p¡raz¡n-2-¡l)-4-cloro-1.3-d¡h¡droesp¡ro[¡nden-2.4'-p¡per¡d¡n1-1-am¡na
Paso A: Se disolvió (S)-1-(((R)-tert-butilsulfinil)amino)-4-doro-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-carboxilato de tert-butilo (100 mg, 0,23 mimóles) en unas pocas gotas de DCM y después se agregó HCl 4 N en dioxano (0,5 ml) y se agitó durante 30 min. La reacción se diluyó con Et2O (5 ml) y se filtró. El sólido pegajoso se secó en un horno al vacío para producir diclorhidrato de (S)-4-cloro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina (70 mg, 99 % de rendimiento).
Paso B: Se suspendió (S)-4-cloro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina diclorhidrato (35 mg, 0,113 mmoles) en dioxano (1 ml) y se agregó trifluorometansulfonato de cloropirido[2,3-b]pirazin-2-ilo (35,4 mg, 0,11 mmoles), seguida por trietilamina (78,8 μl, 0,57 mmoles). La reacción se calentó a 50 °C durante 20 min. Se agregó 3-amino-2-clorobencentiolato de sodio (24,6 mg, 0,136 mmoles) y la reacción se calentó a 90 °C durante 30 min. La reacción se enfrió a temperatura ambiente, se purgó con nitrógeno durante 1 min, se agregó 3-amino-2-clorobencentiolato de sodio (24,6 mg, 0,14 mmoles) y se calentó a 90 °C bajo nitrógeno durante 3 hr. La reacción se enfrió y se agregó directamente a una columna de 12 g (MeOH en EtOAc al 0-10 % con NH4OH al 1 %) para producir (R)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-cloro-1,3-dihidroespiro[inden-2,4'-piperidin]-1 -amina (24,8 mg, 0,047 mmoles, 42 % de rendimiento) como un sólido amarillo. 1H RMN (CDCla) 88,76 (s, 1H), 7,92 (d, J = 8,4 Hz, 1H), 7,83 (d, J = 5,3 Hz, 1H), 7,50 (d, J = 8,6 Hz, 1H), 7,25-7,17 (m, 3H), 6,69 (d, J = 5,3 Hz, 1H), 4,97 (br s, 2H), 4,45 (m, 2H), 4,04 (s, 1H), 3,40 (m, 2H), 3,19 (d, J = 16,2 Hz, 1H), 2,78 (d, J = 16,2 Hz, 1H), 1,97-1,79 (m, 3H), 1,70 (m, 2H), 1,43 (m, 1H). Espectro de masas: m/z = 524,1 (M+H).
Los siguientes compuestos de la Tabla 5 se prepararon de acuerdo con los procedimientos anteriores al utilizar los materiales de partida e intermedios apropiados.
TABLA 5









Claims (24)
1. Un compuesto seleccionado de la Fórmula Ila:

o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que:
L1 es S;
R1 se selecciona de fenilo, heteroarilo, arilo bicíclico, heterociclilo bicíclico y heteroarilo bicíclico,
en el que fenilo, heteroarilo, arilo bicíclico, heterociclo bicíclico y heteroarilo bicíclico están opcionalmente sustituidos con uno o más grupos seleccionados del grupo que consiste en halógeno, OH, oxo, ciano, alquilo opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo) opcionalmente sustituido con halógeno, ciano u OH, NHRa, y un heterociclo opcionalmente sustituido con halógeno, ciano, OH o alquilo opcionalmente sustituido con OH u oxo; R2 es:

X11 se selecciona de CR13R14, SiR13R14, Nh y O;
X12 se selecciona de CHR15 y NH, en donde uno o ambos de X11 y X12 debe ser carbono;
R10 se selecciona de hidrógeno y alquilo;
R11 se selecciona de hidrógeno, OH y CH2NH2;
R12, R16 y R17 son hidrógeno;
R13 se selecciona de hidrógeno, OH y (alquilo de C0-C3)NRbRc;
R14 se selecciona de hidrógeno, OH, alquilo opcionalmente sustituido con halógeno, OH, metilo, OCH3 y un heteroarilo;
R15 se selecciona de hidrógeno o NH2;
o uno de los siguientes grupos pueden unirse:
R10 y R11 pueden unirse como CH2NHCH2 para formar un bicíclico fusionado,
R10 y R15 pueden unirse como alquilo para formar un bicíclico puenteado,
R11 y R12 pueden unirse como alquilo sustituido con NH2 para formar un espirociclo,
R13 y R14 pueden unirse como un grupo seleccionado de cicloalquilo, heterociclo, carbociclo bicíclico y heterociclo bicíclico, en donde el cicloalquilo, heterociclo, carbociclo y heterociclo se sustituyen opcionalmente con F, Cl, OH, OCH3, CN, metilo o NH2 para formar un espirociclo,
R10 y R16 pueden unirse como alquilo, O o NH para formar un bicíclico puenteado,
R11 y R15 pueden unirse como alquilo para formar un bicíclico puenteado,
R11 y R16 pueden unirse como alquilo u O para formar un bicíclico puenteado,
R11 y R17 pueden unirse como alquilo para formar un bicíclico puenteado, o
R13 y R15 pueden unirse como NHCH2 o cicloalquilo en donde el cicloalquilo se sustituye con NH2 para formar un bicíclico fusionado;
R48 se selecciona de hidrógeno y metilo;
Ra es hidrógeno, alquilo opcionalmente sustituido con OH, metoxi, halógeno o ciano o ciclopropilo;
Rb y Rc se seleccionan independientemente de hidrógeno, alquilo y un grupo Boc; y
a, b, c y d se seleccionan de 0 y 1.
2. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que R1 es fenilo opcionalmente sustituido con uno a tres grupos seleccionados del grupo que consiste en halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con halógeno, ciano u OH, -O(alquilo C1-C3) opcionalmente sustituido con halógeno, ciano u OH, NHRa y heterociclo de 3 a 6 miembros opcionalmente sustituido con halógeno, ciano u OH, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y azufre.
3. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que R1 es un heteroarilo de 6 miembros, opcionalmente sustituido con uno a tres grupos seleccionados de halógeno; alquilo C1-C3 opcionalmente sustituido con halógeno u OH; metoxi; NHRa y heterociclo de 3 a 6 miembros opcionalmente sustituido con OH o alquilo C1-C3 opcionalmente sustituido con OH u oxo, en el que el heterociclo contiene uno o dos heteroátomos seleccionados de nitrógeno, oxígeno y SO2; donde el heteroarilo contiene uno o dos heteroátomos de nitrógeno.
4. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que R1 se selecciona del grupo que consiste en fenilo, 2,3-diclorofenilo, 3-clorofenilo, 4-fluorofenilo, 3-cloro-2-trifluorometilfenilo, 2-cloro-3-metoxifenilo, 3-cloro-2-fluorofenilo, 2-cloro-6-fluoro-3-metoxifenilo, 2,3-dicloro-4-metoxifenilo, 2-cloro-3-cianofenilo, 2-cloro-3-fluorofenilo, 2-amino-3-cloropiridin-4-ilo, 3-cloro-2-(pirrolidina-1-il)piridina-4-ilo, 3-cloro-2-metoxipiridin-4-ilo, 3-cloro-2-(metilamino)piridina-4-il), 3-cloro-2-(3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((2-hidroxietil)amino)piridin-4-ilo, 3-cloro-2-metilpiridin-4-ilo, 6-amino-2,3-dicloropiridin-4-ilo, 3-cloro-2-(2-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 2-amino-3-metilpiridin-4-ilo, 3-cloro-2-(1,1-dioxidotiomorfolino)piridin-4-ilo, 3-cloro-2-(4-hidroxipiperidin-1-il)piridin-4-ilo,3-cloro-2-morfolinopiridin-4-ilo,2-(4-acetilpiperazin-1-il)-3-cloropiridin-4-ilo, 3-cloro-2-((S)-3-(hidroximetil)pirrolidin-1-il)piridin-4-ilo, 3-cloro-2-((S)-3-hidroxipirrolidin-1-il)piridin-4-ilo, 3-cloro-2-(3-hidroxipirrolidin-1 -il)piridin-4-ilo, 3-cloro-2-(hidroximetil)piridin-4-ilo, 3-cloro-2-metilpiridin-4-ilo, 2-aminopiridin-3-ilo, 6-cloro-2-metilpiridin-3-ilo, 6-amino-2-cloropiridin-3-ilo,2-cloro-6-metilpiridin-3-ilo,3-cloro-2-((R)-3-(hidroximetil)pirrolidin-1- il)piridin-4-ilo, 2,3-dimetilpiridin-4-ilo, 2-amino-3-fluoropiridin-4-ilo, 6-amino-2-(trifluorometil)piridin-3-ilo, 2-amino-5-cloropiridin-4-ilo, 6-amino-4,5-dicloropiridin-3-ilo, 6-amino-3-cloro-2-metoxipiridin-4-ilo, 2-amino-3-metoxipiridin-4-ilo, 6-amino-5-cloropirimidin-4-ilo, 2-(trifluorometil)piridin-3-ilo, 3-cloro-2-((2-metoxietil)amino)piridin-4-ilo, 3-cloro-2-(ciclopropilamino)piridin-4-ilo, 3-fluoropiridin-4-ilo, 3-cloropiridin-4-ilo, 3-(trifluorometil)piridin-4-ilo, 3-cloro-2-((2-ciano-2- metilpropil)amino)piridin-4-ilo, 3-cloro-2-((3-metoxipropil)amino)piridin-4-ilo, 2-amino-3-(trifluorometil)piridin-4-ilo,2-(trifluorometil)piridin-4-ilo,3,4-dihidroquinolin-1(2H)-ilo, 3,4-dihidroquinoxalin-1(2H)-ilo, 2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-ilo, 3,4-dihidro-1,5-naftiridin-1(2H)-ilo, 3,3-difluoro-2-oxo-2,3-dihidro-1H- pirrolo[2,3-b]piridin-4-ilo, 7-hidroxi-6, 7-dihidro-SH-ciclopenta[£)]piridin-4-ilo, naftalen-1-ilo, 1-metil-1H-indazol-7-ilo, pirazolo[l,5-a]piridina-4-ilo, pirazolo[l,5-a]pirazin-4-ilo, isoquinolin-8-ilo, 3H-imidazo[4,5-6]piridin-7-ilo, 6-cloroimidazo[l,2-a]piridin-3-ilo, 6-cianoimidazo[l,2-a]piridin-3-ilo, 1H-pirazolo[3,4-b]piridin-4-ilo, 1,8-naftiridin-4-ilo, 1H-pirrolo[2,3-b]piridin-4-ilo, 1-metil-1H-pirrolo[2,3-b]piridin-4-ilo, 5-cloro-1H-pirrolo[2,3-6]piridin-4-ilo,5-(trifluorometil)-1H-pirrolo[2,3-bípiridin-4-ilo, 3-metil-1H-pirrolo[2,3- £)]piridin-4-ilo,6-amino-1H-pirrolo[2,3-b]piridin-4-ily5-fluoro-1H-pirrolo[2,3-6]piridin-4-ilo.
5. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que a y b son O y d son 1, de modo que R2 es

6. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que R13 y R14 se unen para formar un grupo seleccionado de cicloalquilo C3-C6, heterociclo de 4 a 6 miembros en el que el heterociclo contiene un heteroátomo seleccionado del grupo que consiste en nitrógeno y oxígeno, carbociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado y un heterociclo bicíclico de 8 a 10 miembros saturado o parcialmente insaturado en el que el heterociclo contiene un heteroátomo seleccionado del grupo que consiste en nitrógeno y oxígeno, en el que el cicloalquilo, el heterociclo, el carbociclo bicíclico y el heterociclo bicíclico están opcionalmente sustituidos con F, Cl, OH, OCH3 , CN, metilo o NH2.
7. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que R2 se selecciona del grupo que consiste en:
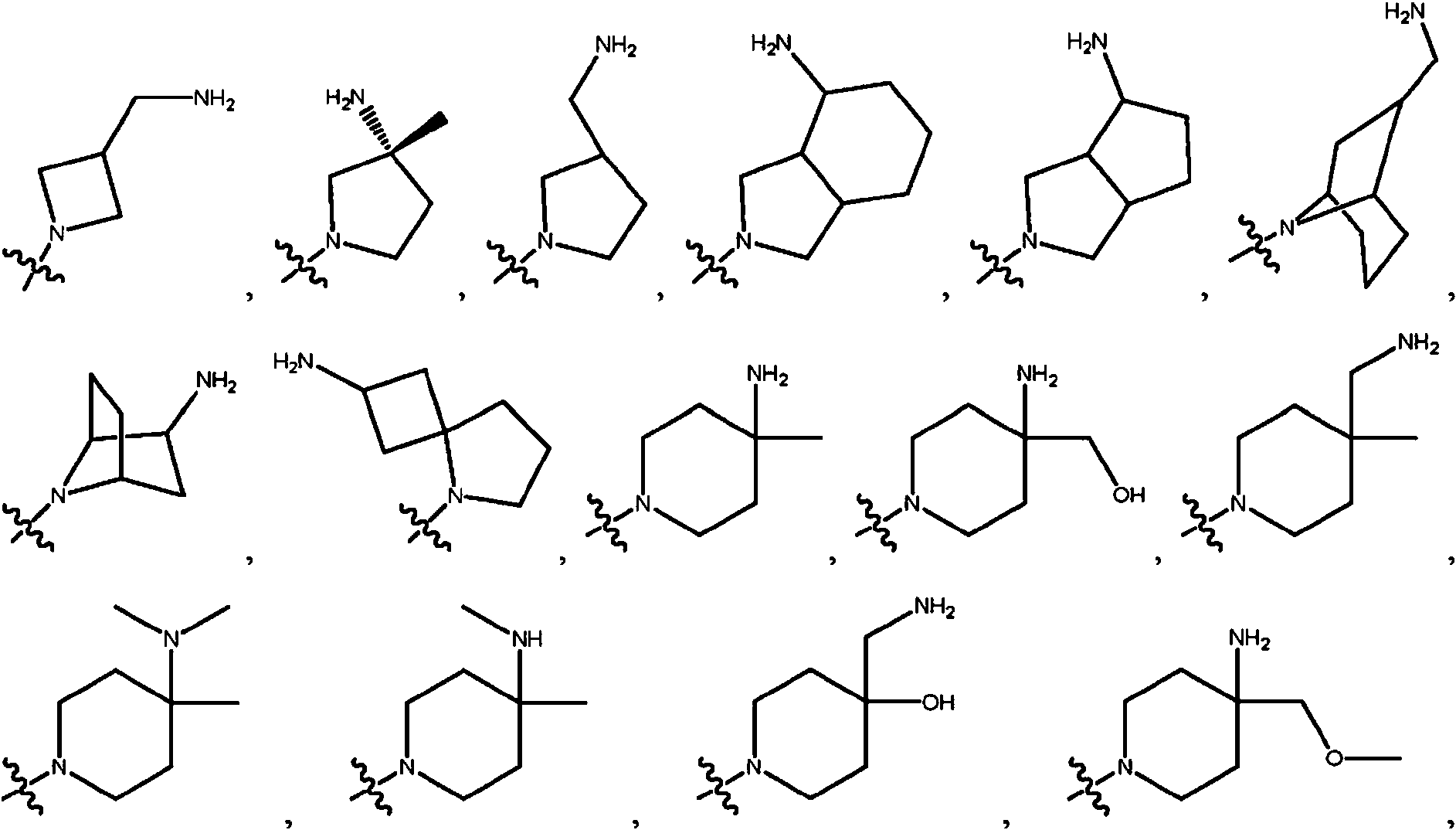



8. El compuesto de la reivindicación 1, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, en el que R48 es hidrógeno.
9. El compuesto de la reivindicación 1, en el que el compuesto se selecciona del grupo que consiste en:
4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
(35.45) -8-(6-((2,3-diclorofenil)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
1-(6-( (2,3-diclorofenil)tio)pirido[2,3-b]pirazin-2-il)-4-metil piperidin-4-amina;
(R)-8-(6-((2,3-diclorofenil)tio)pirido[2,3-b]pirazin-2-il)-8-azaespiro[4.S]decan-1-amina;
(4-amino-1-(6-((2,3-diclorofenil)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)metanol;
4-((2-(4-(aminometil)-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
(35.45) -8-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
1-(6-((2-cloro-3-metoxifenil)tio)pirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina;
1-(6-((3-cloro-2-metoxipiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina;
(R)-4-((2-(3-amino-3-metilpirrolidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
(35.45) -8-(6-((3-cloro-2-(pirrolidin-1-il)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
cloro-4-((2-(4-metil-4-(metilamino)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)piridin-2-amina;
(35.45) -8-(6-((3-cloro-2-(metilamino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
3-metil-1-(6-((1-metil-1H-indazol-7-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-amina;
3- ((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-2-clorobenzonitrilo;
4- metil-1-(6-(pirazolo[1,5-a]piridin-4-iltio)pirido[2,3-b]pirazin-2-il)piperidin-4-amina
4-metil-1-(6-(pirazolo[1,5-a]pirazin-4-iltio)pirido[2,3-b]pirazin-2-il)piperidin-4-amina;
(R) -1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)azepan-4-amina;
1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-(aminometil)piperidin-4-ol;
(4-amino-1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)metanol;
(4-amino-1-(6-((3-cloro-2-metoxipiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)metanol;
(4-amino-1-(6-((3-cloro-2-(pirrolidin-1-il)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)metanol;
1-(6-(isoquinolin-8-iltio)pirido[2,3-b]pirazin-2-il-4-metilpiperidin-4-amina;
1-(6-((3-cloro-2-(pirrolidin-1-il)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina;
(S) -1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)azepan-4-amina;
(R,3s, SS)-8-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-8-azabiciclo[3.2.1]octan-3-amina;
4-((2-(4-amino-4-(metoximetil)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
4-((2-(2,7-diazaespiro[3.5]nonan-7-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
4-((2-(2,7-diazaespiro[3.5]nonan-7-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
(1R,5S)-9-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l-9-azab¡ddo[3 .3.1]nonan-3-amina;
(4-amino-1-(6-((3-doro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)metanol;
(1R,5R,6S)-3-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-8-oxa-3-azab¡ddo[3.2.1]odan-6-am¡na; (1R,5R,6S)-3-(6-((2,3-d¡dorofeml)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-8-oxa-3-azab¡ddo[3 .2.1]octan-6-am¡na;
4-((2-(4-(2-am¡nopropan-2-¡l)p¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
2-(1-(6-((2,3-d¡dorofen¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)p¡per¡d¡n-4-¡l)propan-2-am¡na;
4-((2-(2-(am¡nomet¡l)morfol¡no)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-4-am¡na;
(4-(6-((2,3-d¡dorofeml)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)morfol¡n-2-¡l)metanam¡na;
D¡clorh¡drato de (1R,3s,5S)-8-(6-((3-doro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-8-azab¡c¡clo[3.2.1]octan-3-am¡na;
(35.45) -8-(6-((6-am¡no-2,3-d¡dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na;
((S)-1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-2-¡l)metanol;
(35.45) -8-(6-((2-am¡no-3-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na; 1-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na;
(R) -1-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na;
(S) -1-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lazepan-4-am¡na;
(S)-1'-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1-am¡na; 4-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-6]p¡raz¡n-6-¡l)t¡o)-3-dorop¡r¡d¡n-2-¡l)t¡omorfol¡na1,1-d¡óx¡do;1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-6]p¡raz¡n-6-¡lt¡o)-3-clorop¡r¡d¡n-2-¡l)p¡per¡d¡n-4-ol;
(35.45) -8-(6-((3-doro-2-morfol¡nop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na;
1- (4-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)t¡o)-3-dorop¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)etan-1-ona;
4-((2-(4-am¡nop¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
(3R,4R)-4-am¡no-1-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)p¡per¡d¡n-3-ol;
2- (6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-2-azaesp¡ro[4.4]nonan-6-am¡na;
(3S,4R)-4-am¡no-1-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)p¡per¡d¡n-3-ol;
4-((2-(3-(am¡nomet¡l)azet¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
(1-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-(am¡nomet¡l)p¡per¡d¡n-4-¡l)metanol;
((S)-1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-6]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-¡l)metanol;
(S)-1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-6]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol;
1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-6]p¡raz¡n-6-¡l)t¡o)-3-dorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-ol;
(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.S]decan-8-¡l)p¡ndo[2,3-£i]p¡raz¡n-6-¡l)t¡o)-3-dorop¡nd¡n-2-il)metanol;
(S)-1-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-N- met¡lazepan-4-am¡na;
(1R,3s,5S)-8-(6-((2-am¡no-3-dorop¡nd¡n-4-¡l)t¡o)p¡ndo[2,3-b]p¡raz¡n-2-¡l)-W,N-d¡met¡l-8-azab¡ddo[3.2.1]odan-3-am¡na;
(1R,3s,SS)-8-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-N- met¡l-8-azab¡c¡clo[3.2.1]octan-3-am¡na; 1-(6-((3-cloro-2-met¡lp¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4- met¡lp¡per¡d¡n-4-am¡na;
3- ((2-(4-am¡no-4-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)p¡r¡d¡n-2-am¡na;
1-(6-((6-cloro-2-met¡lp¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na;
5- ((2-(4-am¡no-4-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-6-clorop¡r¡d¡n-2-am¡na;
1- (6-((2-cloro-6-met¡lp¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na;
2- (6-((2-am¡no-3-dorop¡nd¡n-4-¡l)t¡o)p¡rido[2,3-b]p¡raz¡n-2-¡l)odah¡dro-1H-¡so¡ndol-4-am¡na;
2-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)octah¡droc¡clopenta [e]p¡rrol-4-am¡na;
4- ((2-(3-(am¡nomet¡l)-9-azab¡c¡clo[3.3.1]nonan-9-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
4-((2-(3-(am¡nomet¡l)-8-azab¡c¡clo[3.2.1]octan-8-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
4-((2-(3,7-d¡azab¡c¡clo[4.2.0]octan-3-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
4-((2-(4-am¡no-4-(tr¡fluoromet¡l)p¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
(3S,4S)-8-(6-((6-am¡no-2-clorop¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na;
((R)-1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡r¡do[2,3-£)]p¡raz¡n-6-¡l)t¡o)-3-dorop¡nd¡n-2-¡l)p¡rrol¡d¡n-3-¡l)metanol;
(R) -1-(4-((2-((3S,4S)-4-am¡no-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-8-¡l)p¡ndo[2,3-£i]p¡raz¡n-6-¡lt¡o)-3-dorop¡nd¡n-2-¡l)p¡rrol¡d¡n-3-ol;
(S) -2-((4-((2-(5-am¡no-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡na-6,4'-p¡per¡d¡n]-1'-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)am¡no)etan-1-ol;
4-((2-(4-(Am¡nomet¡l)-4-met¡l-1,4-azas¡l¡nan-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
(S)-2-((4-((2-(1-am¡no-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1'-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)am¡no)etan-1-ol;
(R) -1-am¡no-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)- 1, 3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡na]-6- carbon¡tr¡lo;
(S) -1-am¡no-1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡na]-6-carbon¡tr¡lo;
(R) -1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡na-6,4'-p¡per¡d¡n]-5-am¡na;
(S) -1'-(6-((2-am¡no-3-clorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-2-met¡l-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡na-6,4'-p¡per¡d¡n]-5-am¡na;
1- (6-((2-cloro-6-met¡lp¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-met¡lp¡per¡d¡n-4-am¡na;
2- (6-((2-am¡no-3-dorop¡nd¡n-4-¡l)t¡o)p¡rido[2,3-b]p¡raz¡n-2-¡l)odah¡dro-1H-¡so¡ndol-4-am¡na;
2-(6-((2-am ino-3-doropiridin-4-il)tio)p irido[2,3-b]pirazin-2-il)octahidrocidopenta[e]pirro l-4-am ina;
4-((2-(3-(aminometil)-9-azabicido[3.3.1]nonan-9-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3-(aminometil)-8-azabicido[3.2.1]octan-8-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3,7-diazabicido[4.2.0]octan-3-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(4-amino-4-(trifluorometil)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
(3S,4S)-8-(6-((6-amino-2-doropiridin-3-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
((R)-1-(4-((2-((3S,4S)-4-amino-3-irietil-2-oxa-8-azaespiro[4.5]decan-8-N)pindo[2,3-£)]pirazin-6-N)tio)-3-doropindin-2-il)pirrolidin-3-il)metanol;
(R) -1-(4-((2-((3S,4S)-4-amino-3-irietil-2-oxa-8-azaespiro[4.5]decan-8-N)pindo[2,3-£)]pirazin-6-Ntio)-3-doropindin-2-il)pirrolidin-3-ol;
(S) -2-((4-((2-(5-amino-5,7-dihidroespiro[cidopenta[b]piridina-6,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-il)airimo)ethan-1-ol;
4-((2-(4-(Aminometil)-4-metil-1,4-azasilinan-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
(S)-2-((4-((2-(1-amino-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-il)amino)etan-1-ol;
(R) -1-amino-1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidina]-6-carbonitrilo;
(S) -1-amino-1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidine]-6-carbonitrilo;
(R) -1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-2-metil-5,7-dihidroespiro[cidopenta[b]piridina-6,4'-piperidin]-5-amina;
(S) -1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-2-metil-5,7-dihidroespiro[cidopenta[b]piridina-6,4'-piperidin]-5-amina;
(S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metoxi-5,7-dihidroespiro[ciclopenta[b]piridina-6,4'-piperidin]-5-amina;
(R) -1-(6-((2-amino-3-doropindin-4-N)tio)pindo[2,3-b]pirazin-2-N)-2-irietoxi-5,7-dihidroespiro[ddopenta[£)]μmdina-6,4'-piperidin]-5-amina;
(S) -1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-fluoro-5,7-dihidroespiro[ciclopenta[b]piridina-6,4'-piperidin]-5-amina;
(R) -1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-fluoro-5,7-dihidroespiro[ciclopenta[b]piridina-6,4'-piperidin]-5-amina;
(S) -1'-(6-((6-amino-4,5-dicloropiridin-3-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
1-(6-((2,3-diclorofenil)tio)-3-metilpirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina;
4-((2-((2R,4R)-4-amino-2-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
Diclorhidrato de 4-((2-((2R,4S)-4-amino-2-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina; (R) -1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1',3'-dihidroespiro[azetidine-3,2'-inden]-1'-amina; (S) -1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[ciclopenta[b]piridina-6,4'-piperidin]-5-amina;
(S )-1 '-(6-((2 -am ino-3-doropirid in -4-il)tio )p irido[2,3-b]pirazin -2-il)-4-m etil-1, 3-d ihidroespiro[inden-2,4-p iperid in]-1-amina;
(S)-1-(6-((6-doroimidazo[1,2-a]piridin-3-N)tio)pirido[2,3-b]pirazin-2-N)-1,3-dihidroespiro[inden-2,4-piperidin]-1-amina;
2- (4-amino-1-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)etan-1-ol;
1-(4-amino-1-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)piperidin-4-il)-2-metilpropan-2-ol;
4-((2-(4-amino-4-etilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-amina;
(35.45) -8-(6-((3H-imidazo[4,5-b]piridin-7-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
(R) -1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3H- espiro[benzofuran-2,4'-piperidin]-3-amina; (S) -1'-(6-((2-amino-5-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[ciclopenta[b]piridina-6,4'-piperidin]-5-amina;
3- ((4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-cloropiridin-2-il)amino)-2,2-dimetilpropanonitrilo;
(35.45) -8-(6-((2-amino-3-(trifluorometil)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
((1R,5S,6r)-3-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-azabiciclo[3 .1.1]heptan-6-amina;
(tert-butil((lR,5S,6r)-3-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-azabiciclo[3.1.1]heptan-6-il)carbamato;
(35.45) -3-metil-8-(6-((3-metil-1H-pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-2-oxa-8-azaespiro[4.S]decan-4- amina;
(35.45) -8-(6-((6-amino-1H-pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
(35.45) -8-(6-((5-fluoro-1H-pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
(S)-4-((2-(1-amino-1,3-dihidroespiro[inden-2,4'-piperidin]-1'-il)pirido[2,3-b]pirazin-6-il)tio)-3,3-difluoro-1,3-dihidro-2H-pirrolo[2,3-b]piridin-2-ona;
3-cloro-4-((2-(4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)piridin-2-amina;
(35.45) -8-(6-((2-amino-5-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
(S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-fluoro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
1-(6-((3-cloro-2-(metilamino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina;
1-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina;
(R) -1-(6-((3-cloro-2-(metilamino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina;
(S) -1-(6-((3-cloro-2-(metilamino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina;
(R) -1-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina;
(S) -1-(6-((3-cloro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilazepan-4-amina;
(35.45) -8-(6-((2-amino-3-metoxipiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro [4.5]decan-4-amina;
5- (6-((2-amino-3-doropindin-4-N)tio)pirido[2,3-b]pirazin-2-il-5-azaespiro[3. 4]octan-2-amina;
6- ((2-amino-3-doropiridin-4-il)tio)-N-(3-(aminometil)tetrahidrofuran-3-il)pirido[2,3-b]pirazin-2-amina;
(S)-1-amino-1'-(6-((3-doro-2-metilpiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-5-ol; 4-((2-((3S,4S)-4-amino-3-metil-2-oxa-8-azaespiro[4.5]decan-8-ilpirido[2,3-b]pirazin-6-il)tio)-6,7-dihidro-5H-ddopenta[b]piridin-7-ol;
1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5,7-dihidroespiro[cidopenta[b]piridina-6,4'-piperidin]-7- amina;
(S)-1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-doro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
(2R, 4R)-4-amino-8-(6-((2-airiino-3-doropindin-4-N)tio)pirido[2,3-b]pirazin-2-N)-8-azaespiro[4.5]decan-2-ol;
4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doro-N- metilpiridin-2-amina;
(S)-1'-(6-((2-(trifluorometil)piridin-3-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina; (S)-1-(6-((2-(trifluoroiTietil)pindin-3-N)tio)pindo[2,3-b]pirazin-2-N)-5,7-dihidroespiro[ddopenta[;b]piridina-6,4-piperidin]-5-amina;
(S)-1-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1',3'-dihidroespiro[azetidina-3,2'-inden]-1'-amina; (S)-1-(6-((2-amino-3-doropindin-4-N)tio)pindo[2,3-b]pirazin-2-N)espiro[biddo[4.2.0]odano-7,4-piperidina]-1(6),2,4-trien-8-amina;
(R)-1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)espiro[bicido[4.2.0]octano-7,4'-piperidina]-1(6),2,4-trien-8-amina;
(R)-1-(6-((2-amino-3-doropindin-4-N)tio)pindo[2,3-b]pirazin-2-N)-5,7-dihidroespiro[ddopenta[6]piridina-6,4-piperidin]-5-amina;
(R) -1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metil-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
(S) -1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-doro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
(R) -1'-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-5-doro-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
(S) -1'-(6-((6-amino-3-doro-2-metoxipiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
(S)-3-((2-(1-amino-1,3-dihidroespiro[inden-2,4-piperidin]-1-N)pindo[2,3-£)]pirazin-6-N)tio)iiTiidazo[1,2-a]μmdina-6-carbonitrilo;
4-((2-(4-amino-4-propilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
(35.45) -8-(6-((2,3-dimetil piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina; (35.45) -8-(6-((2-amino-3-fluoropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
(35.45) -8-(6-((6-amino-2-(trifluorometil)piridin-3-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
(35.45) -8-(6-((2,3-dicloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina; (S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-6-metil-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina;
(R )-1 '-(6-((2-am ino-3-doropirid in-4-il)tio)p irido[2,3-b]pirazin-2-il)-6-m etil-1,3-d ih idroespiro[inden-2,4 '-p iperid in]-1-amina;
4-((2-(4-amino-4-(piridin-2-ilmetil)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(4-amino-4-(2-metoxietil)piperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
(35.45) -8-(6-((3-doro-2-((2-metoxietil)ammo)pindin-4-il)tio)pindo[2,3-£)]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
(35.45) -8-(6-((3-cloro-2-(ciclopropilamino)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
2-((4-((2-((3S,4S)-4-amino-3-metil-2-oxa-8-azaespiro[4.5]decan-8-il)pirido[2,3-6]pirazin-6-il)tio)-3-doropindin-2-il)amino)etan-1-ol;
(35.45) -8-(6-((3-fluoropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.S]decan-4-amina;
(35.45) -8-(6-((3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
(35.45) -3-metil-8-(6-((3-(trifluorometil)piridin-4-iltio)pirido[2,3-b]pirazin-2-il)-2-oxa-8-azaespiro[4.5]decan-4-amina; (35.45) -8-(6-((1H-pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
(35.45) -3-metil-8-(6-((2-(trifluorometil)piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-2-oxa-8-azaespiro[4.5]decan-4-amina;
(35.45) -3-metil-8-(6-((1-metiMH-pirrolo[2,3-b]pindin-4-il)tio)pindo[2,3-6]pirazin-2-il)-2-oxa-8-azaespiro[4.5]decan-4-amina;
(35.45) -8-(6-((5-doro-1H-pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-3-metil-2-oxa-8-azaespiro[4.5]decan-4-amina;
(35.45) -3-metil-8-(6-((5-(trifluorometil)-1H-pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-2-oxa-8-azaespiro[4.5]decan-4-amina;
4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doro-N-(2-metoxietil)piridin-2-amina;
4-((2-(4-amino-4-metilpiperidin-1-il)pirido[2,3-b]pirazin-6-il)tio)-3-doro-N-(3-metoxipropil)piridin-2-amina;
1-(6-((2,3-dihidro-1H -pirrolo[2,3-b]piridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina;
1-(6-((1,8-naftiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-metilpiperidin-4-amina;
1-(6-((2-amino-3-doropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-4-(hidroximetil)piperidin-4-ol;
(7R,5S,8s)-3-(6-((2-amino-3-doropindin-4-il)tio)pindo[2,3-b]pirazin-2-il)-3-azabiddo[3 .2.1]octan-8-amina;
4-((2-(3,6-diazabicido[3.1.1]heptan-3-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3,8-diazabicido[3.2.1]octan-3-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3,6-diazabicido[3.2.2]nonan-6-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-((1S,4S)-2,5-diazabicido[2.2.1]heptan-2-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3,8-diazabicido[3.2.1]octan-8-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3,8-diazabicido[3.2.1]octan-8-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(9-oxa-3,7-diazabicido[3.3.1]nonan-3-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-(3-oxa-7,9-diazabicido[3.3.1]nonan-7-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
4-((2-((1S,4S)-2,5-diazabicido[2.2.2]octan-2-il)pirido[2,3-b]pirazin-6-il)tio)-3-doropiridin-2-amina;
(S)-1-(4-((2-(5-am¡no-5,7-d¡h¡droesp¡ro[ddopenta[b]p¡nd¡na-6,4'-p¡perid¡n]-1'-¡l)μmdo[2, 3-b]p¡raz¡n-6-¡l)t¡o)-3-doropiridin-2-il)piperidin-4-ol;
((S)-1-(4-((2-((S)-5-am¡no-5,7-d¡h¡droesp¡ro[c¡dopenta[b]p¡r¡d¡na-6,4'-p¡per¡d¡n]-1'-¡l)p¡r¡do[2, 3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-¡l)p¡rrol¡d¡n-3-¡l)metanol;
4-((2-(4-(am¡nomet¡l)-4-fluorop¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
(1-(6-((2,3-d¡clorofen¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-fluorop¡ per¡d¡n-4-¡l)metanam¡na;
(1S,2S,4R)-7-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-7-azab¡c¡clo[2.2.1 ]heptan-2-am¡na; (35.45) -8-(6-((1H-p¡razolo[3,4-b]p¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na;
3- doro-4-((2-((4aR,7aR)-hexah¡drop¡n'olo[3,4-b][1,4]oxaz¡n-4(4aH)-¡l)p¡rido[2,3-6]p¡raz¡n-6-¡l)t¡o)p¡nd¡na-2-am¡na; 4- ((2-((2S,5S)-5-am¡no-2-met¡lp¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
(35.45) -8-(6-((6-am¡no-5-dorop¡r¡m¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3-met¡l-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na;
8-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-3,3-d¡fluoro-8-azaesp¡ro[4.S]decan-1-am¡na;
(S)-1'-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-6-metox¡-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1-am¡na;
(R)-1'-(6-((2-am¡no-3-dorop¡r¡d¡n-4-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-4-cloro-1,3-d¡h¡droesp¡ro[¡nden-2,4'-p¡per¡d¡n]-1-am¡na;
(35.45) -3-met¡l-8-(6-((2-(tr¡fluoromet¡l)p¡r¡d¡n-3-¡l)t¡o)p¡r¡do[2,3-b]p¡raz¡n-2-¡l)-2-oxa-8-azaesp¡ro[4.5]decan-4-am¡na; y
4-((2-(4-am¡no-4-benz¡lp¡per¡d¡n-1-¡l)p¡r¡do[2,3-b]p¡raz¡n-6-¡l)t¡o)-3-clorop¡r¡d¡n-2-am¡na;
o un estereo¡sómero, tautómero o sal farmacéut¡camente aceptable del m¡smo.
10. El compuesto de la Re¡v¡nd¡cac¡ón 1 que es:

o un estereo¡sómero, tautómero o sal farmacéut¡camente aceptable del m¡smo.
11. El compuesto de la re¡v¡nd¡cac¡ón 1 que es (S)-1'-(6-((2-am¡no-3-dorop¡nd¡n-4-¡l)t¡o)p¡ndo[2,3-b]p¡raz¡n-2-¡l)-5,7-d¡h¡droesp¡ro[c¡clopenta[b]p¡r¡d¡na-6,4'-p¡per¡d¡n]-5-am¡na.
12. El compuesto de la Re¡v¡nd¡cac¡ón 1 que es:

o un estereoisómero tautómero o sal farmacéuticamente aceptable del mismo.
13. El compuesto de la reivindicación 1 que es(S)-1'-(6-((2-amino-3-cloropiridin-4-il)tio)pirido[2,3-b]pirazin-2-il)-1,3-dihidroespiro[inden-2,4'-piperidin]-1-amina.
14. Una composición farmacéutica que comprende un compuesto de cualquiera de las reivindicaciones 1 a 13, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo.
15. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1 a 13, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, y al menos un vehículo, excipiente o diluyente farmacéuticamente aceptable.
16. Un compuesto de cualquiera de las reivindicaciones 1 a 13 o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso como un medicamento.
17. Un compuesto de una cualquiera de las reivindicaciones 1 a 13 o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso en terapia.
18. Un compuesto de cualquiera de las reivindicaciones 1 a 13 o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad hiperproliferativa.
19. El compuesto para su uso de acuerdo con la reivindicación 18, en el que la enfermedad hiperproliferativa es cáncer.
20. El compuesto para su uso de acuerdo con la reivindicación 18, en el que la enfermedad hiperproliferativa se selecciona del grupo que consiste en melanoma, leucemias mielomoncíticas juveniles, neuroblastoma, mieloide crónica con cromosoma Filadelfia positivo, leucemias linfoblásticas agudas con cromosoma Filadelfia positivo, leucemias mieloides agudas, neoplasmas mieloproliferativos (tales como policitemia vera, trombocitemia esencial y mielofibrosis primaria), cáncer de mama, cáncer de pulmón, cáncer de hígado, cáncer colorrectal, cáncer de esófago, cáncer gástrico, carcinoma de células escamosas de cabeza y cuello, glioblastoma, linfoma anaplásico de células grandes, carcinoma de tiroides y neoplasias spitzoides.
21. El compuesto para uso de acuerdo con la reivindicación 18, en el que la enfermedad hiperproliferativa se selecciona del grupo que consiste en neurofibromatosis y síndrome de Noonan.
22. El compuesto para usar de acuerdo con una cualquiera de las reivindicaciones 18 a 20, donde dicho compuesto, o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, se administra junto con al menos otro agente quimioterapéutica usado para tratar o mejorar un trastorno hiperproliferativo.
23. El compuesto para uso de acuerdo con la reivindicación 22, en el que al menos otro agente quimioterapéutico es un inhibidor de la vía RAS.
24. El compuesto para uso de acuerdo con la reivindicación 23, en el que al menos otro agente quimioterapéutico se selecciona de uno o más de trametinib, binimetinib, selumetinib, cobimetinib, LErafAON, ISIS 5132, vemurafenib, pimaserib, TAK.733, R04987655, CI-1040 , PD-0325901, CH5126766, MAP855, AZD6244, refametinib, GDC-0973/XL581, AZD8330, R05126766, ARS-853, LY3214996, BVD523, GSKl 120212, ulixertinib y abemaciclib.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862746952P | 2018-10-17 | 2018-10-17 | |
| US201962916119P | 2019-10-16 | 2019-10-16 | |
| PCT/US2019/056786 WO2020081848A1 (en) | 2018-10-17 | 2019-10-17 | Protein tyrosine phosphatase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2947464T3 true ES2947464T3 (es) | 2023-08-09 |
Family
ID=68536896
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19801996T Active ES2947464T3 (es) | 2018-10-17 | 2019-10-17 | Inhibidores de la proteína tirosina fosfatasa |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US20210380582A1 (es) |
| EP (1) | EP3867238B1 (es) |
| JP (1) | JP7449282B2 (es) |
| KR (1) | KR102649419B1 (es) |
| CN (1) | CN114341124A (es) |
| AU (1) | AU2019359885B2 (es) |
| BR (1) | BR112021005733A2 (es) |
| CA (1) | CA3116561C (es) |
| CL (1) | CL2021000972A1 (es) |
| CO (1) | CO2021004966A2 (es) |
| CR (1) | CR20210189A (es) |
| CU (1) | CU20210029A7 (es) |
| CY (1) | CY1126122T1 (es) |
| DO (1) | DOP2021000070A (es) |
| EC (1) | ECSP21027049A (es) |
| ES (1) | ES2947464T3 (es) |
| HU (1) | HUE062404T2 (es) |
| IL (1) | IL282179A (es) |
| MA (1) | MA53921A (es) |
| MX (1) | MX2021004410A (es) |
| NI (1) | NI202100027A (es) |
| PE (1) | PE20211465A1 (es) |
| PH (1) | PH12021550674A1 (es) |
| PL (1) | PL3867238T3 (es) |
| SG (1) | SG11202102985YA (es) |
| WO (1) | WO2020081848A1 (es) |
| ZA (1) | ZA202101960B (es) |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3515916B1 (en) | 2016-09-22 | 2023-06-07 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of use thereof |
| TW201819386A (zh) | 2016-10-24 | 2018-06-01 | 美商傳達治療有限公司 | Shp2磷酸酶抑制劑及其使用方法 |
| EP3630770A1 (en) | 2017-05-26 | 2020-04-08 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazine derivatives as shp2 phosphatase inhibitors |
| US11701354B2 (en) | 2017-09-29 | 2023-07-18 | D. E. Shaw Research, Llc | Pyrazolo[3,4-b]pyrazine derivatives as SHP2 phosphatase inhibitors |
| TW202003471A (zh) | 2018-03-21 | 2020-01-16 | 美商傳達治療有限公司 | Shp2磷酸酶抑制劑及其使用方法 |
| WO2020072656A1 (en) | 2018-10-03 | 2020-04-09 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| GEP20237561B (en) | 2019-04-02 | 2023-10-25 | Array Biopharma Inc | Protein tyrosine phosphatase inhibitors |
| EP4034539A1 (en) | 2019-09-24 | 2022-08-03 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of making and using the same |
| CN114901662A (zh) | 2019-11-08 | 2022-08-12 | 锐新医药公司 | 双环杂芳基化合物及其用途 |
| IL299131A (en) | 2020-06-18 | 2023-02-01 | Revolution Medicines Inc | Methods for delaying, preventing and treating acquired resistance to RAS inhibitors |
| EP4208261A1 (en) | 2020-09-03 | 2023-07-12 | Revolution Medicines, Inc. | Use of sos1 inhibitors to treat malignancies with shp2 mutations |
| EP4214209A1 (en) | 2020-09-15 | 2023-07-26 | Revolution Medicines, Inc. | Indole derivatives as ras inhibitors in the treatment of cancer |
| TWI825637B (zh) | 2021-03-31 | 2023-12-11 | 美商輝瑞股份有限公司 | 啶-1,6(2h,7h)-二酮 |
| EP4334321A1 (en) | 2021-05-05 | 2024-03-13 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2022235866A1 (en) | 2021-05-05 | 2022-11-10 | Revolution Medicines, Inc. | Covalent ras inhibitors and uses thereof |
| CN117616031A (zh) | 2021-05-05 | 2024-02-27 | 锐新医药公司 | 用于治疗癌症的ras抑制剂 |
| CN115960109A (zh) * | 2021-05-31 | 2023-04-14 | 药雅科技(上海)有限公司 | 稠环类shp2磷酸酶抑制剂的制备及其应用 |
| CN115340561A (zh) * | 2021-05-14 | 2022-11-15 | 药雅科技(上海)有限公司 | Shp2磷酸酶稠环类抑制剂的制备及其应用 |
| TW202244049A (zh) * | 2021-05-12 | 2022-11-16 | 大陸商藥雅科技(上海)有限公司 | Shp2磷酸酶抑制劑的製備及其應用 |
| WO2022259157A1 (en) | 2021-06-09 | 2022-12-15 | Novartis Ag | A triple pharmaceutical combination comprising dabrafenib, trametinib and a shp2 inhibitor |
| TW202317100A (zh) | 2021-06-23 | 2023-05-01 | 瑞士商諾華公司 | 包含kras g12c抑制劑的藥物組合及其用於治療癌症之用途 |
| KR20230011245A (ko) | 2021-07-09 | 2023-01-20 | 주식회사 카나프테라퓨틱스 | Shp2 억제제 및 이의 용도 |
| KR20240055778A (ko) | 2021-09-01 | 2024-04-29 | 노파르티스 아게 | Tead 억제제를 포함하는 제약 조합물 및 암의 치료를 위한 이의 용도 |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| TW202346305A (zh) * | 2022-01-05 | 2023-12-01 | 美商雷密克斯醫療公司 | 用於調節剪切之化合物及方法 |
| WO2023172940A1 (en) | 2022-03-08 | 2023-09-14 | Revolution Medicines, Inc. | Methods for treating immune refractory lung cancer |
| CN114621186A (zh) * | 2022-05-12 | 2022-06-14 | 上海维申医药有限公司 | 作为ras信号通路调控剂的杂环化合物 |
| WO2023230205A1 (en) | 2022-05-25 | 2023-11-30 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5543523A (en) | 1994-11-15 | 1996-08-06 | Regents Of The University Of Minnesota | Method and intermediates for the synthesis of korupensamines |
| JPWO2007029847A1 (ja) * | 2005-09-07 | 2009-03-19 | 萬有製薬株式会社 | 二環性芳香族置換ピリドン誘導体 |
| US9481675B2 (en) * | 2009-09-11 | 2016-11-01 | Merck Sharp & Dohme Corp. | Gyrase inhibitors |
| CA2804304C (en) * | 2010-05-24 | 2020-02-25 | Intellikine, Llc | Heterocyclic compounds and uses thereof |
| JO3517B1 (ar) | 2014-01-17 | 2020-07-05 | Novartis Ag | ان-ازاسبيرو الكان حلقي كبديل مركبات اريل-ان مغايرة وتركيبات لتثبيط نشاط shp2 |
| US9815813B2 (en) | 2014-01-17 | 2017-11-14 | Novartis Ag | 1-(triazin-3-yl/pyridazin-3-yl)-piper(-azine)idine derivatives and compositions therefor for inhibiting the activity of SHP2 |
| ES2699351T3 (es) | 2014-01-17 | 2019-02-08 | Novartis Ag | Derivados de 1-piridazin/triazin-3-il-piper(-azina)/idina/pirolidina y composiciones de las mismas para inhibir la actividad de SHP2 |
| CN112625028A (zh) | 2015-06-19 | 2021-04-09 | 诺华股份有限公司 | 用于抑制shp2活性的化合物和组合物 |
| EP3310771B1 (en) | 2015-06-19 | 2020-07-22 | Novartis AG | Compounds and compositions for inhibiting the activity of shp2 |
| ES2741746T3 (es) | 2015-06-19 | 2020-02-12 | Novartis Ag | Compuestos y composiciones para inhibir la actividad de SHP2 |
| WO2017210134A1 (en) | 2016-05-31 | 2017-12-07 | Board Of Regents, University Of Texas System | Heterocyclic inhibitors of ptpn11 |
| CA3026784A1 (en) | 2016-06-07 | 2017-12-14 | Jacobio Pharmaceuticals Co., Ltd. | Heterocyclic pyrazine derivatives useful as shp2 inhibitors |
| MX2018015625A (es) | 2016-06-14 | 2019-03-06 | Novartis Ag | Compuestos y composiciones para inhibir la actividad de shp2. |
| SG11201900157RA (en) | 2016-07-12 | 2019-02-27 | Revolution Medicines Inc | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric shp2 inhibitors |
| EP3515916B1 (en) | 2016-09-22 | 2023-06-07 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of use thereof |
| TW201819386A (zh) * | 2016-10-24 | 2018-06-01 | 美商傳達治療有限公司 | Shp2磷酸酶抑制劑及其使用方法 |
| EP3571189B1 (en) | 2017-01-23 | 2023-03-29 | Revolution Medicines, Inc. | Pyridine compounds as allosteric shp2 inhibitors |
| JP7240319B2 (ja) | 2017-01-23 | 2023-03-15 | レヴォリューション・メディスンズ,インコーポレイテッド | アロステリックshp2阻害剤としての二環式化合物 |
| EP3601239A4 (en) | 2017-03-23 | 2020-05-13 | Jacobio Pharmaceuticals Co., Ltd. | INNOVATIVE HETEROCYCLIC DERIVATIVES USEFUL AS SHP2 INHIBITORS |
| WO2019051084A1 (en) | 2017-09-07 | 2019-03-14 | Revolution Medicines, Inc. | SHP2 INHIBITOR COMPOSITIONS AND METHODS OF TREATING CANCER |
-
2019
- 2019-10-17 BR BR112021005733-1A patent/BR112021005733A2/pt unknown
- 2019-10-17 HU HUE19801996A patent/HUE062404T2/hu unknown
- 2019-10-17 CR CR20210189A patent/CR20210189A/es unknown
- 2019-10-17 JP JP2021520556A patent/JP7449282B2/ja active Active
- 2019-10-17 MX MX2021004410A patent/MX2021004410A/es unknown
- 2019-10-17 CU CU2021000029A patent/CU20210029A7/es unknown
- 2019-10-17 US US17/285,997 patent/US20210380582A1/en active Pending
- 2019-10-17 WO PCT/US2019/056786 patent/WO2020081848A1/en active Application Filing
- 2019-10-17 KR KR1020217014574A patent/KR102649419B1/ko active IP Right Grant
- 2019-10-17 SG SG11202102985YA patent/SG11202102985YA/en unknown
- 2019-10-17 AU AU2019359885A patent/AU2019359885B2/en active Active
- 2019-10-17 EP EP19801996.0A patent/EP3867238B1/en active Active
- 2019-10-17 PE PE2021000527A patent/PE20211465A1/es unknown
- 2019-10-17 ES ES19801996T patent/ES2947464T3/es active Active
- 2019-10-17 CA CA3116561A patent/CA3116561C/en active Active
- 2019-10-17 MA MA053921A patent/MA53921A/fr unknown
- 2019-10-17 PL PL19801996.0T patent/PL3867238T3/pl unknown
- 2019-10-17 CN CN201980068343.2A patent/CN114341124A/zh active Pending
-
2021
- 2021-03-24 ZA ZA2021/01960A patent/ZA202101960B/en unknown
- 2021-03-25 PH PH12021550674A patent/PH12021550674A1/en unknown
- 2021-04-08 IL IL282179A patent/IL282179A/en unknown
- 2021-04-16 NI NI202100027A patent/NI202100027A/es unknown
- 2021-04-16 CL CL2021000972A patent/CL2021000972A1/es unknown
- 2021-04-16 EC ECSENADI202127049A patent/ECSP21027049A/es unknown
- 2021-04-16 DO DO2021000070A patent/DOP2021000070A/es unknown
- 2021-04-19 CO CONC2021/0004966A patent/CO2021004966A2/es unknown
-
2023
- 2023-08-01 CY CY20231100379T patent/CY1126122T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CL2021000972A1 (es) | 2021-10-15 |
| CU20210029A7 (es) | 2021-11-04 |
| NI202100027A (es) | 2021-08-24 |
| PL3867238T3 (pl) | 2023-09-11 |
| SG11202102985YA (en) | 2021-05-28 |
| MX2021004410A (es) | 2021-07-06 |
| AU2019359885A1 (en) | 2021-04-15 |
| DOP2021000070A (es) | 2021-05-16 |
| ZA202101960B (en) | 2023-11-29 |
| US20210380582A1 (en) | 2021-12-09 |
| EP3867238C0 (en) | 2023-06-07 |
| KR20210076098A (ko) | 2021-06-23 |
| JP7449282B2 (ja) | 2024-03-13 |
| CN114341124A (zh) | 2022-04-12 |
| ECSP21027049A (es) | 2021-05-31 |
| WO2020081848A1 (en) | 2020-04-23 |
| AU2019359885B2 (en) | 2024-01-18 |
| EP3867238B1 (en) | 2023-06-07 |
| KR102649419B1 (ko) | 2024-03-21 |
| IL282179A (en) | 2021-05-31 |
| CR20210189A (es) | 2021-09-14 |
| BR112021005733A2 (pt) | 2021-07-27 |
| PE20211465A1 (es) | 2021-08-05 |
| PH12021550674A1 (en) | 2021-12-13 |
| MA53921A (fr) | 2022-01-26 |
| CA3116561A1 (en) | 2020-04-23 |
| HUE062404T2 (hu) | 2023-10-28 |
| CA3116561C (en) | 2023-09-12 |
| CO2021004966A2 (es) | 2021-04-30 |
| EP3867238A1 (en) | 2021-08-25 |
| JP2022504936A (ja) | 2022-01-13 |
| CY1126122T1 (el) | 2023-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2947464T3 (es) | Inhibidores de la proteína tirosina fosfatasa | |
| JP7284830B2 (ja) | タンパク質チロシンホスファターゼ阻害剤 | |
| US10934302B1 (en) | SHP2 phosphatase inhibitors and methods of use thereof | |
| AU2020202707B2 (en) | Heteroaryl pyridone and aza-pyridone compounds as inhibitors of Btk activity | |
| CN111566104B (zh) | 一种螺芳环化合物及其应用 | |
| JP6545863B2 (ja) | Fgfr阻害剤としての置換三環式化合物 | |
| KR101961500B1 (ko) | 세린/트레오닌 키나제 억제제 | |
| WO2018218133A1 (en) | Pyrazolo[3,4-b]pyrazine derivatives as shp2 phosphatase inhibitors | |
| CN105732637B (zh) | 杂芳化合物及其在药物中的应用 | |
| BRPI0708615A2 (pt) | compostos de pirazol heterobicìclicos e métodos de uso | |
| CA2790847A1 (en) | Fused bicyclic kinase inhibitors | |
| TW202330529A (zh) | 雜環化合物及使用方法 | |
| TW202317566A (zh) | 雜環化合物及使用方法 | |
| EP3390404A1 (en) | Tricyclic compounds and compositions as kinase inhibitors | |
| RU2780532C1 (ru) | Ингибиторы протеинтирозинфосфатазы | |
| WO2024020419A1 (en) | Aza-quinazoline compounds and methods of use | |
| TW202400612A (zh) | 作為HPK1抑制劑之四氫吡啶并[3,4-d]嘧啶化物 |