ES2923771T3 - Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue - Google Patents
Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue Download PDFInfo
- Publication number
- ES2923771T3 ES2923771T3 ES17714475T ES17714475T ES2923771T3 ES 2923771 T3 ES2923771 T3 ES 2923771T3 ES 17714475 T ES17714475 T ES 17714475T ES 17714475 T ES17714475 T ES 17714475T ES 2923771 T3 ES2923771 T3 ES 2923771T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- compound
- mixture
- methoxyphenyl
- enantiomer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D209/26—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with an acyl radical attached to the ring nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/405—Indole-alkanecarboxylic acids; Derivatives thereof, e.g. tryptophan, indomethacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Abstract
La presente invención se refiere a compuestos de indolina sustituidos, métodos para prevenir o tratar infecciones virales del dengue mediante el uso de dichos compuestos y también se refiere a dichos compuestos para uso como medicamento, más preferiblemente para uso como medicamento para tratar o prevenir infecciones virales del dengue. La presente invención además se refiere a composiciones farmacéuticas o preparaciones combinadas de los compuestos, a las composiciones o preparaciones para usar como medicina, más preferiblemente para la prevención o tratamiento de infecciones virales del dengue. La invención también se refiere a procesos para la preparación de los compuestos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue
La presente invención se refiere a derivados de indolina sustituidos, a dichos compuestos para su uso como medicamento, más preferentemente, para su uso como medicamento para tratar o prevenir infecciones virales por dengue. La presente invención se refiere además a composiciones farmacéuticas o preparados combinados de los compuestos, a las composiciones o preparados para su uso como un medicamento, más preferentemente para la prevención o el tratamiento de infecciones virales por dengue. La invención también se refiere a procesos para la preparación de los compuestos.
ANTECEDENTES DE LA INVENCIÓN
Los flavivirus, los cuales son transmitidos por mosquitos o garrapatas, provocan infecciones potencialmente letales para el hombre, tales como la encefalitis y la fiebre hemorrágica. Se conocen cuatro serotipos distintos, pero estrechamente relacionados, del flavivirus dengue, denominados DENV-1, -2, -3 y -4. El dengue es endémico en la mayoría de las regiones tropicales y subtropicales en todo el mundo, predominantemente en áreas urbanas y semiurbanas. Según la Organización Mundial de la Salud (OMS), 2500 millones de personas, de las cuales mil millones son niños, corren el riesgo de sufrir una infección por DENV (OMS, 2002). Se estima que en todo el mundo se producen cada año de 50 a 100 millones de casos de fiebre por dengue [FD], medio millón de casos de enfermedad por dengue grave (es decir, fiebre hemorrágica por dengue [FHD] y síndrome de choque por dengue [SCD]) y más de 20,000 muertes. La FHD se ha convertido en una de las principales causas de hospitalización y fallecimiento entre los niños en regiones endémicas. Por todo ello, el dengue representa la causa más común de enfermedad arbovírica. Debido a grandes brotes recientes en países situados en América Latina, sudeste asiático y Pacífico Occidental que incluye Brasil, Puerto Rico, Venezuela, Camboya, Indonesia, Vietnam, Tailandia), el número de casos de dengue ha aumentado drásticamente en los últimos años. No es solo el hecho de que el número de casos de dengue aumente a medida que la enfermedad se propaga a nuevas áreas, sino que los brotes tienden a ser más graves.
Aunque se han hecho progresos en el desarrollo de vacunas contra el dengue con la disponibilidad de la vacuna Dengvaxia®, se han encontrado muchas dificultades. Estas incluyen la existencia de un fenómeno denominado potenciación dependiente de anticuerpos (PDA). La recuperación de una infección provocada por un serotipo proporciona inmunidad de por vida frente a ese serotipo pero únicamente confiere una protección parcial y transitoria frente a una infección posterior provocada por uno de los otros tres serotipos.
Después de la infección con otro serotipo, los anticuerpos heterólogos preexistentes forman complejos con el serotipo de virus del dengue que provoca la nueva infección, pero no neutralizan el patógeno. En su lugar, se cree que se facilita la entrada del virus en las células, lo cual da como resultado una replicación incontrolada del virus y títulos virales máximos mayores. Tanto en infecciones primarias como secundarias, unos títulos virales mayores están asociados con una enfermedad del dengue más grave. Puesto que los anticuerpos maternos pueden transmitirse fácilmente a los niños por la lactancia materna, esta podría ser una de las razones por las que los niños se ven más afectados que los adultos por la enfermedad grave por dengue.
En lugares en los que circulan simultáneamente dos o más serotipos, que también se denominan regiones hiperendémicas, el riesgo de enfermedad severa por dengue es significativamente superior debido a que existe un riesgo mayor de experimentar una infección secundaria más grave. Además, en una situación de hiperendemia, la probabilidad de que emerjan cepas más virulentas aumenta, lo cual a su vez aumenta la probabilidad de fiebre hemorrágica por dengue (FHD) o síndrome de choque por dengue.
Los mosquitos portadores del dengue, que incluyen Aedes aegypti y Aedes albopictus (mosquito tigre), se están desplazando hacia el norte del globo. Según los Centros para el Control y la Prevención de Enfermedades (CCPE) de los Estados Unidos (EE. UU.), en la actualidad ambos mosquitos son omnipresentes en el sur de Texas. La propagación hacia el norte de los mosquitos portadores del dengue no está confinada a los EE. UU., sino que también se ha observado en Europa.
Dengvaxia®, la vacuna para el dengue producida por Sanofi Pasteur se aprobó en primer lugar en México y mientras tanto, ha sido aprobada en más países. Sin embargo, la vacuna se puede mejorar considerablemente debido a que presenta una eficacia limitada, especialmente contra DENV-1 y -2, una eficacia baja en sujetos no expuestos previamente al flavivirus y una pauta posológica prolongada.
Pese a estas limitaciones, la vacuna supone un punto de inflexión en las zonas endémicas puesto que ofrecerá protección a gran parte de la población, aunque probablemente no a bebés muy pequeños, quienes se ven muy afectados por el dengue. Además, la pauta posológica y la eficacia muy limitada en sujetos no expuestos previamente al flavivirus hacen que sea inadecuada y que probablemente no merezca la pena/no sea rentable para personas que viajen de zonas no endémicas a zonas endémicas de dengue. Las limitaciones mencionadas anteriormente de las vacunas contra el dengue son la razón por la que se necesita un agente antiviral contra el dengue que sea profiláctico y se pueda aplicar previamente a la exposición.
Además, a día de hoy, no se dispone de fármacos antivirales específicos para el tratamiento o la prevención de una infección por el virus de la fiebre del dengue. Claramente, sigue existiendo una gran necesidad médica no cubierta en lo que se refiere a productos terapéuticos para la prevención o el tratamiento de infecciones virales en animales, más en particular en seres humanos, y especialmente de infecciones virales provocadas por Flavivirus, más en particular el virus del Dengue. Es indispensable disponer de compuestos con una potencia antiviral satisfactoria, niveles bajos o nulos de efectos secundarios, una actividad de amplio espectro contra múltiples serotipos del virus del Dengue, una baja toxicidad y/o unas propiedades farmacocinéticas o farmacodinámicas satisfactorias.
El documento WO-2010/021878 divulga derivados de 2-fenilpirrolidina e indolina como antagonistas del receptor de mentol frío para el tratamiento de enfermedades inflamatorias y centrales. El documento WO-2013/045516 divulga derivados de indol o indolina para su uso en el tratamiento de infecciones virales por dengue.
A continuación, la presente invención proporciona compuestos, derivados de indolina sustituidos, que muestran una elevada y potente actividad contra los cuatro (4) serotipos del virus del dengue.
COMPENDIO DE LA INVENCIÓN
La presente invención se basa en el descubrimiento inesperado de que al menos uno de los problemas mencionados anteriormente se puede resolver mediante los compuestos actuales de la invención.
La presente invención proporciona compuestos que se ha observado que poseen una potente actividad antiviral contra los cuatro (4) serotipos conocidos actualmente. La presente invención demuestra además que estos compuestos inhiben eficazmente la proliferación del virus del Dengue (DENV). Por lo tanto, estos compuestos constituyen una clase útil de compuestos potentes que se pueden utilizar en el tratamiento y/o la prevención de infecciones virales en animales, mamíferos y seres humanos, más específicamente para el tratamiento y/o la prevención de infecciones con virus del Dengue.
La presente invención se refiere además al uso de tales compuestos como medicamentos y a su uso para la elaboración de medicamentos para tratar y/o prevenir infecciones virales, en particular con virus que pertenecen a la familia de los virus del Dengue en animales o mamíferos, más en particular en seres humanos. La invención también se refiere a métodos para preparar todos estos compuestos y a composiciones farmacéuticas que los comprenden en una cantidad eficaz.
La presente invención también se refiere a los compuestos para su uso en el tratamiento o la prevención de infecciones virales por dengue en seres humanos mediante la administración de una cantidad eficaz de uno o más de tales compuestos, o una de sus sales farmacéuticamente aceptables, opcionalmente combinado con uno o más medicamentos diferentes, como otro agente antivírico, a un paciente que lo necesite.
Un aspecto de la invención es la provisión de compuestos que tienen la fórmula (I), una forma estereoisomérica, una sal, solvato o polimorfo farmacéuticamente aceptable de los mismos:



En una realización alternativa, la presente invención se refiere a un compuesto que tienen la fórmula (I)

una forma estereoisomérica, una sal , solvato o polimorfo farmacéuticamente aceptable del mismo, donde
R1 es trifluorometilo, R2 es hidrógeno, R3 es hidrógeno, y R es metoxi; o
R1 es trifluorometilo, R2 es hidrógeno, R3 es hidrógeno, y R es flúor; o
R1 es trifluorometoxi, R2 es hidrógeno, R3 es hidrógeno, y R es hidrógeno; o
R1 es trifluorometilo, R2 es hidrógeno, R3 es hidrógeno, y R es hidrógeno; o
R1 es trifluorometoxi, R2 es hidrógeno, R3 es hidrógeno, y R es metoxi; o
R1 es trifluorometoxi, R2 es hidrógeno, R3 es hidrógeno, y R es flúor; o
R1 es trifluorometilo, R2 es metoxi, R3 es hidrógeno, y R es hidrógeno; o
R1 es trifluorometilo, R2 es metoxi, R3 es hidrógeno, y R es flúor; o
R1 es trifluorometoxi, R2 es hidrógeno, R3 es metilo, y R es hidrógeno; o
R1 es trifluorometilo, R2 es hidrógeno, R3 es metilo, y R es hidrógeno; o
R1 es trifluorometilo, R2 es metoxi, R3 es hidrógeno, y R es metoxi; o
R1 es trifluorometilo, R2 es flúor, R3 es hidrógeno, y R es hidrógeno; o
R1 es trifluorometoxi, R2 es metoxi, R3 es hidrógeno, y R es hidrógeno.
Parte de la invención actual es también una composición farmacéutica que comprende un compuesto mencionado anteriormente o una forma estereoisomérica, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, junto con uno o más excipientes, diluyentes o portadores farmacéuticamente aceptables.
Las sales farmacéuticamente aceptables de dichos compuestos incluyen las sales de adición de ácidos y de bases de los mismos. Las sales de adición de ácido adecuadas se forman a partir de ácidos que forman sales atóxicas. Las sales de adición de base adecuadas se forman a partir de bases que forman sales atóxicas.
Los compuestos de la invención también pueden existir en formas sin solvatar y solvatadas. El término "solvato" se utiliza en la presente para describir un complejo molecular que comprende el compuesto de la invención y una o más moléculas de un disolvente farmacéuticamente aceptables, por ejemplo, etanol.
El término "polimorfo" se refiere a la capacidad del compuesto de la invención de existir en más de una forma o estructura cristalina.
Los compuestos de la presente invención se pueden administrar como productos amorfos o cristalinos. Se pueden obtener, por ejemplo, como masas compactas sólidas, polvos o películas mediante métodos tales como la precipitación, cristalización, liofilización, secado por pulverización o secado por evaporación. Se pueden administrar solos o combinados con uno o más compuestos de la invención diferentes o combinados con uno o más fármacos diferentes. En general, se administrarán como una formulación asociados con uno o más excipientes farmacéuticamente aceptables. El término "excipiente" se utiliza en la presente para describir cualquier ingrediente que no sea el o los compuestos de la invención. La selección del excipiente depende en gran medida de factores tales como el modo particular de administración, el efecto del excipiente sobre la solubilidad y estabilidad, y la naturaleza de la forma farmacéutica.
Los compuestos de la presente invención o cualquiera de sus subgrupos se pueden formular en varias formas farmacéuticas con el fin de poderlos administrar. Como composiciones adecuadas, se pueden citar todas las composiciones empleadas normalmente para administrar fármacos por vía sistémica. Para preparar las composiciones farmacéuticas de esta invención, se combina una cantidad eficaz del compuesto particular, opcionalmente en forma de sal de adición, como principio activo, en mezcla íntima con un portador farmacéuticamente aceptable, pudiendo adoptar dicho portador una gran variedad de formas dependiendo de la forma del preparado que se desee para la administración. Estas composiciones farmacéuticas se encuentran deseablemente en una forma farmacéutica unitaria adecuada, por ejemplo, para administración oral o rectal. Por ejemplo, en la preparación de las composiciones en una forma farmacéutica oral, se puede emplear cualquiera de los medios farmacéuticos habituales tales como, por ejemplo, agua, glicoles, aceites, alcoholes y similares en el caso de preparados líquidos orales tales como suspensiones, jarabes, elixires, emulsiones y soluciones; o portadores sólidos tales como almidones, azúcares, caolín, diluyentes, lubricantes, aglutinantes, agentes desintegrantes y similares en el caso de polvos, pastillas, cápsulas y comprimidos. Debido a su fácil administración, los comprimidos y las cápsulas representan las formas farmacéuticas unitarias orales más convenientes, en cuyo caso se emplean obviamente portadores farmacéuticos sólidos. También se incluyen los preparados en forma sólida que se pueden convertir, poco antes de su uso, en formas líquidas.
Es especialmente ventajoso formular las composiciones farmacéuticas mencionadas anteriormente en forma farmacéutica unitaria por su facilidad de administración y uniformidad de dosificación. La expresión "forma farmacéutica unitaria", tal como se utiliza en la presente, se refiere a unidades físicamente discretas adecuadas como dosis unitarias, donde cada unidad contiene una cantidad predeterminada de principio activo calculada para producir el efecto terapéutico deseado asociado con el portador farmacéutico requerido. Ejemplos de formas de dosificación unitaria de este tipo son comprimidos (incluyendo comprimidos con muesca o revestidos), cápsulas, píldoras, paquetes de polvos, obleas, supositorios, disoluciones o suspensiones inyectables y similares, y múltiples segregados de las mismas.
Los expertos en el tratamiento de enfermedades infecciosas serán capaces de determinar la cantidad eficaz a partir de los resultados de las pruebas que se presentan posteriormente en la presente. En general, se considera que una cantidad diaria eficaz sería de 0.01 mg/kg a 50 mg/kg de peso corporal, más preferentemente de 0.1 mg/kg a 10 mg/kg de peso corporal. Puede resultar apropiado administrar la dosis requerida como dos, tres, cuatro o más subdosis en intervalos adecuados a lo largo del día. Dichas subdosis se pueden formular como formas farmacéuticas unitarias, por ejemplo, que contengan de 1 a 1000 mg y, en particular, de 5 a 200 mg de principio activo por forma farmacéutica unitaria.
La dosis exacta y la frecuencia de administración dependen del compuesto particular de la invención utilizado, la afección particular que se esté tratando, la gravedad de la afección que se esté tratando, la edad, el peso y el estado físico general del paciente particular, así como también de otra medicación que pueda estar tomando el individuo, como bien sabrán los expertos en la técnica. Además, es obvio que la cantidad eficaz se puede reducir o incrementar dependiendo de la respuesta del sujeto tratado y/o dependiendo de la evaluación del médico que prescriba los compuestos de la presente invención. Por consiguiente, los intervalos de la cantidad eficaz que se han mencionado anteriormente son solamente orientativos y no se pretende que limiten el alcance ni el uso de la invención de ningún modo.
También se pretende que la presente exposición incluya cualquiera de los isótopos de los átomos presentes en los compuestos de la invención. Por ejemplo, los isótopos de hidrógeno incluyen tritio y deuterio, y los isótopos de carbono incluyen C-13 y C-14.
Los presentes compuestos utilizados en la presente invención también pueden existir en su forma estereoquímicamente isomérica, que define todos los posibles compuestos constituidos por los mismos átomos enlazados mediante la misma secuencia de enlaces pero que tienen estructuras tridimensionales diferentes, que no son intercambiables. A menos que se mencione o indique lo contrario, la designación química de los compuestos engloba la mezcla de todas las formas estereoquímicamente isoméricas posibles que dichos compuestos puedan poseer.
Dicha mezcla puede contener todos los diastereómeros y/o enantiómeros de la estructura molecular básica de dicho compuesto. Se pretende que todas las formas estereoquímicamente isoméricas de los compuestos utilizados en la presente invención, tanto en forma pura como mezcladas con las otras, queden englobadas dentro del alcance de la presente invención, incluyendo cualquiera de las mezclas racémicas o racematos.
Las formas estereoisoméricas puras de los compuestos y los intermedios que se mencionan en la presente se definen como isómeros sustancialmente exentos de otras formas enantioméricas o diastereoméricas de la misma estructura molecular básica de dichos compuestos o intermedios. En particular, la expresión "estereoisoméricamente puro" se refiere a compuestos o intermedios que tienen un exceso estereoisomérico de al menos un 80 % (es decir, un mínimo de un 90 % de un isómero y un máximo de un 10 % de los otros isómeros posibles) y hasta un exceso estereoisomérico de un 100 % (es decir, un 100 % de un isómero y nada de los demás), más concretamente, compuestos o intermedios que tienen un exceso estereoisomérico desde un 90 % hasta un 100 %, aún más concretamente que tienen un exceso estereoisomérico desde un 94 % hasta un 100 % y, aún más concretamente, que tienen un exceso estereoisomérico desde un 97 % hasta un 100 %. Las expresiones "enantioméricamente puro" y "diastereoméricamente puro" se deben interpretar de un modo similar, pero entonces haciendo referencia al exceso enantiomérico y el exceso diastereomérico de la mezcla en cuestión, respectivamente.
Pueden obtenerse formas estereoisoméricas puras de compuestos e intermedios utilizados en esta invención mediante la aplicación de procedimientos conocidos en la técnica. Por ejemplo, los enantiómeros se pueden separar unos de otros mediante la cristalización selectiva de sus sales diastereoméricas con ácidos o bases ópticamente activos. Algunos ejemplos de estos son el ácido tartárico, ácido dibenzoiltartárico, ácido ditoluoiltartárico y ácido alcanforsulfónico. Como alternativa, los enantiómeros se pueden separar mediante técnicas cromatográficas utilizando fases estacionarias quirales. Dichas formas estereoquímicamente isoméricas puras también pueden derivarse de las formas estereoquímicamente isoméricas puras correspondientes de los materiales de partida apropiados, siempre que la reacción se produzca de manera estereoespecífica. Preferentemente, si se desea obtener un estereoisómero específico, dicho compuesto se sintetizará mediante métodos de preparación estereoespecíficos. Estos métodos emplearán ventajosamente materiales de partida enantioméricamente puros.
Los compuestos de fórmula (I) de la presente invención tienen todos al menos un átomo de carbono quiral como se indica en la figura siguiente mediante el átomo de carbono marcado con *:

Debido a la presencia de dicho átomo de carbono quiral, un "compuesto de fórmula (I)" puede ser el enantiómero (R), el enantiómero (S), la forma racémica, o cualquier combinación posible de los dos enantiómeros individuales en cualquier proporción. Cuando la configuración absoluta (R) o (S) de un enantiómero no es conocida, este enantiómero también pude identificarse indicando si el enantiómero es dextrógiro (+) o levógiro(-) después de medir la rotación óptica específica de dicho enantiómero particular.
En un aspecto, la presente invención se refiere a un primer grupo de un compuesto de fórmula (I) donde los compuestos de fórmula (I) tienen la rotación específica (+).
En un aspecto adicional, la presente invención se refiere a un segundo grupo de compuestos de fórmula (I) donde los compuestos de fórmula (I) tienen la rotación específica (-).
Ejemplos
Métodos de LC/MS
La medición por cromatografía líquida de alto rendimiento (HPLC) se realizó utilizando una bomba de LC, un haz de diodos (DAD) o un detector de UV y una columna, según se especifique en los métodos respectivos. Cuando fue necesario, se incluyeron detectores adicionales (véase la tabla de métodos más adelante).
El flujo procedente de la columna se introdujo en un espectrómetro de masas (MS), el cual se configuró con una fuente de iones a presión atmosférica. Es parte del conocimiento de un experto en la materia ajustar los parámetros ajustables (por ejemplo, intervalo de barrido, tiempo de permanencia, etc.) con el fin de obtener iones que permitan la identificación del peso molecular (PM) monoisotópico nominal del compuesto. La adquisición de datos se realizó con el software adecuado.
Los compuestos se describen según sus tiempos de retención (Tr) experimentales e iones. Si no se especifica de otro modo en la tabla de datos, el ion molecular descrito corresponde a [M+H]+ (molécula protonada) y/o [M-H]-(molécula desprotonada). En caso de que el compuesto no se pueda ionizar directamente, se especifica el tipo de aducto (es
decir, [M+NH4]+, [M+HCOO]', etc...). Para moléculas con patrones isotópicos múltiples (Br, Cl), el valor indicado es el obtenido para la masa isotópica más baja. Todos los resultados se obtuvieron con incertidumbres experimentales que están habitualmente asociadas al método usado.
En lo sucesivo en la presente, "SQD" significa detector de cuadrupolo único, "MSD" detector selectivo de masas, "TA" temperatura ambiente, "BEH" híbrido con puente de etilsiloxano/sílice, "DAD" detector de haz de diodos, "HSS" sílice de alta resistencia.
Códigos del método de LC/MS (el flujo se expresa en ml/min; la temperatura de la columna (T) en °C; el tiempo de análisis en minutos)

Métodos de SFC/MS
La medición de SFC se llevó a cabo utilizando un sistema de cromatografía analítica de fluidos supercríticos (SFC) compuesto por una bomba binaria para suministrar dióxido de carbono (CO2) y un modificador, un automuestreador, un horno para la columna, un detector de haz de diodos dotado de una celda de flujo de alta presión que resiste una presión de hasta 400 bar. Si se configura con un espectrómetro de masas (MS) el flujo desde la columna se lleva al (MS). Es parte del conocimiento de un experto en la materia ajustar los parámetros ajustables (por ejemplo, intervalo de barrido, tiempo de permanencia, etc.) con el fin de obtener iones que permitan la identificación del peso molecular (PM) monoisotópico nominal del compuesto. La adquisición de datos se realizó con el software adecuado.
Métodos analíticos de SFC/MS (flujo expresado en ml/min; temperatura de la columna (T) en °C; tiempo de análisis en minutos, contrapresión (BPR) en bar).


Puntos de fusión
Los valores son valores máximos o intervalos de fusión, y se obtienen con incertidumbres experimentales que están normalmente asociadas con este método analítico.
DSC823e (indicado como DSC)
Para varios compuestos, los puntos de fusión se determinaron con un DSC823e (Mettler-Toledo). Se midieron los puntos de fusión con un gradiente de temperatura de 10 °C/minuto. La temperatura máxima fue 300 °C.
Rotaciones ópticas:
Las rotaciones ópticas se midieron en un polarímetro Perkin-Elmer 341 con una lámpara de sodio y se expresaron tal como se indica a continuación: [a]° (A, c g/100 ml, disolvente, T°C).
[a]AT = (100a)/(/x c): donde l es la longitud del recorrido en dm y c es la concentración en g/100 ml para una muestra a una temperatura T (°C) y una longitud de onda A (en nm). Si la longitud de onda de la luz utilizada es de 589 nm (la línea D del sodio), entonces podría utilizarse el símbolo D en su lugar. El signo de la rotación (+ o -) debería proporcionarse siempre. Cuando se utiliza esta ecuación, la concentración y el disolvente siempre se proporcionan entre paréntesis después de la rotación. La rotación se indica utilizando grados y no se proporcionan unidades de concentración (se asume que son g/100 ml).
Ejemplo 1 : Síntesis de 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometil)indolin-1-il)etanona (Compuesto 1) y separación quiral en los Enantiómeros 1A y 1B.

Síntesis del intermedio 1a:
Una mezcla de ácido 2-(4-cloro-2-metoxifenil)acético [CAS 170737-95-8] (1.55 g, 7.75 mmol) y 6-(trifluorometil)indolina [CAS 181513-29-1] (1.45 g, 7.75 mmol), HOBt (2.78 g, 11.6 mmol), EDCI (2.23 g, 11.6 mmol) y trietilamina (2.15 ml, 15.5 mmol) en CH2Cl2 (40 ml) se agitó a temperatura ambiente durante 12 h. Se añadió agua y las capas se separaron. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. La purificación se llevó a cabo mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 120 g, heptano/EtOAc, 80/20). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-metoxifenil)-1-(6-(trifluorometil)-indolin-1-il)etanona 1a (1.66 g).
Síntesis del intermedio 1b:
A -78 °C, en un flujo de N2, se añadió gota a gota LiHMDS 1 M en THF (8.98 ml, 8.98 mmol) a una mezcla de 2-(4-cloro-2-metoxifenil)-1-(6-(trifluorometil)-indolin-1-il)etanona 1a (1.66 g, 4.49 mmol) en THF (18 ml). La mezcla se agitó durante 15 min a -78 °C y se añadió gota a gota una solución de NBS (879 mg, 4.94 mmol) en THF (7 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió por la adición de una solución acuosa saturada de NH4CL La mezcla se extrajo con EtOAc, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. La purificación se llevó a cabo mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, heptano/EtOAc, 80/20). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-bromo-2-(4-cloro-2-metoxifenil)-1-(6-(trifluorometil)indolin-1-il)etanona 1b (1.23 g).
Síntesis del Compuesto 1 y separación quiral en los Enantiómeros 1A y 1B:
Una mezcla de 2-bromo-2-(4-cloro-2-metoxifenil)-1-(6-(trifluorometil)indolin-1-il)etanona 1b (1.2 g, 2.68 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1] (735 mg, 4.01 mmol) y trietilamina (558 pl, 4.01 mmol) en CH3CN (50 ml) se agitó a 70 °C durante 6 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con HCl 1 N. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 40 g, CH2Cl2/heptano: 97.5/2.5). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometil)-indolin-1-il)etanona racémica (Compuesto 1, 900 mg) después de la cristalización en CH3CN. Este lotes se combinó con 2 lotes más (cantidad total: 1.84 g). Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralcel® OD-H 5 pm 250 x 30 mm, Fase móvil: 55 % de CO2, 45 % de MeOH). El primer enantiómero eluido se purificó adicionalmente por cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 40 g, CH2Cb/MeOH, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad para dar, después de la solidificación en éter de petróleo/CHaCN/éter diisopropílico, Enantiómero 1A (540 mg). El segundo enantiómero eluido se purificó adicionalmente por cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 40 g, CH2Cl2/MeOH, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad para dar, después de la solidificación en éter de petróleo/CH3CN/éter diisopropílico, Enantiómero 1B (560 mg).
Compuesto 1:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.24 (m, 2 H) 3.59 - 3.67 (m, 5 H) 3.78 - 3.87 (m, 2 H) 3.90 (s, 3 H) 3.98 - 4.07 (m, 1 H) 4.33 - 4.42 (m, 1 H) 4.79 (t a, J=4.7 Hz, 1 H) 5.60 (d, J=8.5 Hz, 1 H) 5.76 (s, 1 H) 5.87 (s a, 2 H) 6.44 (d a, J=8.5 Hz, 1 H) 7.03 (dd, J=8.2, 1.9 Hz, 1 H) 7.15 (d, J=1.6 Hz, 1 H) 7.32 (d, J=8.5 Hz, 1 H) 7.39 (d, J=7.9 Hz, 1 H) 7.46 (d, J=7.9 Hz, 1 H) 8.36 (s, 1 H)
LC/MS (método LC-A): TR 3.35 min, MH+ 551
Punto de fusión: 194°C
Enantiómero 1A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.16 - 3.30 (m, 2 H) 3.57 - 3.67 (m, 5 H) 3.79 - 3.88 (m, 2 H) 3.90 (s, 3 H) 4.03 (td, J=10.3, 7.1 Hz, 1 H) 4.37 (td, J=10.1,6.6 Hz, 1 H) 4.79 (t, J=5.4 Hz, 1 H) 5.61 (d, J=8.5 Hz, 1 H) 5.77 (t, J=1.9 Hz, 1 H) 5.87 (s a, 2 H) 6.44 (d, J=8.8 Hz, 1 H) 7.03 (dd, J=8.2, 2.2 Hz, 1 H) 7.15 (d, J=1.9 Hz, 1 H) 7.32 (d, J=8.2 Hz, 1 H) 7.39 (d, J=7.9 Hz, 1 H) 7.46 (d, J=7.6 Hz, 1 H) 8.37 (s, 1 H) LC/MS (método LC-A): TR 3.34 min, MH+ 551
[a]
d20: -26.5° (c 0.3091, DMF)
SFC quiral (método SFC-A): tR 1.45 min, MH+ 551, pureza quiral 100 %.
Enantiómero 1B:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.20 - 3.30 (m, 2 H) 3.58 - 3.68 (m, 5 H) 3.80 - 3.88 (m, 2 H) 3.91 (s, 3 H) 4.03 (td, J=10.2, 7.1 Hz, 1 H) 4.38 (td, J=10.2, 6.6 Hz, 1 H) 4.80 (t, J=5.5 Hz, 1 H) 5.61 (d, J=8.8 Hz, 1 H) 5.77 (t, J=2.0 Hz, 1 H) 5.88 (s a, 2 H) 6.45 (d, J=8.5 Hz, 1 H) 7.04 (dd, J=8.2, 1.9 Hz, 1 H) 7.15 (d, J=1.9 Hz, 1 H) 7.32 (d, J=8.2 Hz, 1 H) 7.39 (d, J=7.6 Hz, 1 H) 7.47 (d, J=7.9 Hz, 1 H) 8.37 (s, 1 H) LC/MS (método LC-A): TR 3.34 min, MH+ 551
[a]
d20: 28.8° (c 0.2845, DMF)
SFC quiral (método SFC-A): tR 3.64 min, MH+ 551, pureza quiral 100 %.
Ejemplo 2.1 : síntesis de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometil)indolin-1-il)etanona (Compuesto 2).

Síntesis del intermedio 2a:
Una mezcla de ácido 2-(4-cloro-2-fluorofenil)acético [CAS 194240-75-0] (504 mg, 2.67 mmol) y 6-(trifluorometil)indolina [CAS 181513-29-1] (500 mg, 2.67 mmol), HOBt (541 mg, 4 mmol), EDCI (768 mg, 4 mmol) y trietilamina (743 ql, 5.34 mmol) en CH2Cl2 (6 ml) se agitó a temperatura ambiente durante 12 h. Se añadió agua y las capas se separaron. La capa orgánica se lavó con agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida para dar 2-(4-cloro-2-fluorofenil)-1-(6-(trifluorometil)indolin-1-il)etanona 2a (1.04 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del intermedio 2b:
A -78 °C, en un flujo de N2, se añadió gota a gota LiHMDS 1 M en THF (5.5 ml, 5.5 mmol) a una solución de 2-(4-cloro-2-fluorofenil)-1 -(6-(trifluorometil)indolin-1 -il)etanona 2a (980 mg, 2.74 mmol) en THF (8 ml). La mezcla de reacción se agitó durante 15 min a -78 °C y se añadió gota a gota una solución de NBS (536 mg, 3.01 mmol) en THF (5 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió por la adición de una solución acuosa saturada de NH4CL La mezcla se extrajo con EtOAc, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida para dar 2-bromo-2-(4-cloro-2-fluorofenil)-1 -(6-(trifluorometil)indolin-1 -il)etanona 2b (1.3 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 2:
Una mezcla de 2-bromo-2-(4-cloro-2-fluorofenil)-1 -(6-(trifluorometil)-indolin-1 -il)etanona 2b (500 mg, 1.15 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1] (315 mg, 1.72 mmol) y trietilamina (296 ql, 1.72 mmol) en CH3CN (5 ml) se agitó a 50 °C durante 24 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con HCl 1 N. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El residuo se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 qm, 24 g, gradiente de heptano/EtOAc de 70/30 a 50/50). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1 -(6-(trifluorometil)-indolin-1 -il)etanona (Compuesto 2 , 136 mg) después de la cristalización en Et2O.
Compuesto 2:
1H RMN (400 MHz, DMSO-afe) 5 ppm 3.20 - 3.28 (m, 2 H) 3.57 - 3.69 (m, 5 H) 3.79 - 3.91 (m, 2 H) 4.03 - 4.13 (m, 1 H) 4.39 - 4.49 (m, 1 H) 4.78 (s a, 1 H) 5.71 (d, J=9.1 Hz, 1 H) 5.77 - 5.82 (m, 1 H) 5.93 (d, J=2.0 Hz, 2 H) 6.59 (d, J=9.1 Hz, 1 H) 7.32 (dd, J=8.3, 1.8 Hz, 1 H) 7.38 - 7.51 (m, 4 H) 8.36 (s, 1 H)
LC/MS (método LC-A): TR 3.36 min, MH+ 539
Punto de fusión: 168°C
Ejemplo 2.2 : síntesis de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometil)indolin-1 -il)etanona (Compuesto 2) y separación quiral en los Enantiómeros 2A y 2B .

A -78 °C, en un flujo N2, se añadió gota a gota LiHMDS 1 M en THF (38.5 ml, 38.5 mmol) a una mezcla de 2-(4-cloro-2-fluorofenil)acetato de metilo [CAS 917023-04-2] (3.9 g, 19.3 mmol) en t Hf (120 ml). Se añadió gota a gota una solución de TMSCl (3.9 ml, 30.8 mmol) en THF (30 ml). La mezcla se agitó durante 15 min a -78 °C y se añadió gota a gota una solución de NBS (3.77 g, 21.2 mmol) en THF (50 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió por la adición de una solución acuosa saturada de NH4CL La mezcla se extrajo con EtOAc, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida para dar 2-bromo-2-(4-cloro-2-fluorofenil)acetato de metilo 2c (5.4 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del intermedio 2d:
Una mezcla de 2-bromo-2-(4-cloro-2-fluorofenil)acetato de metilo 2c (4 g, 12.8 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1] (2.1 g, 11.6 mmol), trietilamina (2.4 ml, 17.4 mmol) en CH3CN (80 ml) se agitó a 50 °C durante 12 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con agua. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El compuesto se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, CH2Cl2/heptano, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acetato de metilo 2d (2.1 g).
Síntesis del intermedio 2e:
Una mezcla de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acetato de metilo 2d (1 g, 2.6 mmol) y LiOH (330 mg, 7.8 mmol) en THF/agua (1/1) (40 ml) se agitó a temperatura ambiente durante 1 h. La mezcla se diluyó con agua. Se acidificó lentamente la fase acuosa con HCl 3 N y se extrajo con EtOAc. Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron, y el disolvente se evaporó a presión reducida para dar ácido 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 2e (2.7 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 2 y separación quiral en los Enantiómeros 2A y 2B:
Se añadió HATU (1.85 g, 4.87 mmol) a una mezcla de 6-(trifluorometil)indolina [CAS 181513-29-1] (607 mg, 3.24 mmol), ácido 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 2e (1.2 g, 3.24 mmol) y diisopropiletilamina (1.6 ml, 9.74 mmol) en DMF (35 ml). La mezcla resultante se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua y el precipitado se eliminó por filtración. Los sólidos se lavaron con agua y se recogieron en EtOAc. La solución orgánica se lavó con una al 10 % de K2CO3 en agua y salmuera, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, CH2Cl2/heptano: 99.5/0.5). Se realizó una segunda purificación mediante SFC aquiral (fase estacionaria: 2-Etilpiridina 5 gm 150 x 30 mm, Fase móvil: CO2 al 70 %, MeOH al 30 %) para proporcionar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1 -(6-(trifluorometil)indolin-1 -il)-etanona (Compuesto 2 , 550 mg) en forma de mezcla racémica. Este lote se combinó con otro lote (cantidad total: 950 mg). Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 gm 250 x 20 mm, Fase móvil: 70 % de CO2, 30 % de iPrOH (+ 0.3 % de iPrNH2)) para dar, después de la solidificación en éter de petróleo/éter diisopropílico, el primer Enantiómero eluido 2A (384 mg) y el segundo Enantiómero eluido 2B (375 mg).
Enantiómero 2A:
1H RMN (500 MHz, DMSO-ds) 5 ppm 3.20 - 3.29 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.85 (dq, J=10.6, 5.2 Hz, 2 H) 4.08 (td, J=10.1,7.3 Hz, 1 H) 4.40 - 4.48 (m, 1 H) 4.80 (t, J=5.5 Hz, 1 H) 5.71 (d, J=8.8 Hz, 1 H) 5.80 (t, J=2.0 Hz, 1 H) 5.93 (d, J=1.9 Hz, 2 H) 6.61 (d, J=8.8 Hz, 1 H) 7.33 (dd, J=8.2, 1.9 Hz, 1 H) 7.38 - 7.51 (m, 4 H) 8.36 (s, 1 H)
LC/MS (método LC-A): TR 3.34 min, MH+ 539
[a]D20: -26.4° (c 0.2691, DMF)
SFC quiral (método SFC-B): tR 0.83 min, MH+ 539, pureza quiral 100 %.
Enantiómero 2B:
1H RMN (500 MHz, DMSO-ds) 5 ppm 3.21 - 3.29 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.85 (tq, J=10.4, 5.0 Hz, 2 H) 4.04 - 4.12 (m, 1 H) 4.40 - 4.48 (m, 1 H) 4.80 (t, J=5.5 Hz, 1 H) 5.71 (d, J=8.8 Hz, 1 H) 5.78 - 5.81 (m, 1 H) 5.93 (d, J=1.6 Hz, 2 H) 6.61 (d, J=8.8 Hz, 1 H) 7.33 (dd, J=8.5, 1.9 Hz, 1 H) 7.39 - 7.52 (m, 4 H) 8.36 (s, 1 H) LC/MS (método LC-A): TR 3.34 min, MH+ 539
[a]
d20: 27.3° (c 0.2564, DMF)
SFC quiral (método SFC-B): tR 1.69 min, MH+ 539, pureza quiral 99.07 %.
Ejemplo 3.1 : síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 3) y separación quiral en los Enantiómeros 3A y 3B .

Se añadió peróxido de benzoílo (5 mg) a una mezcla de 2-(4-clorofenil)-acetato de metilo [CAS 52449-43-1] (5.0 g, 29.7 mmol) y NBS (4.82 g, 27.1 mmol) en CH3CN (80 ml). La mezcla se calentó a reflujo durante 48 h y el disolvente se evaporó a presión reducida. La mezcla se recogió en 80/20 de ciclohexano/EtOAc y el precipitado se eliminó por filtración y se desechó (succinimida). El filtrado se concentró a presión reducida para dar 2-bromo-2-(4-clorofenil)acetato de metilo 3a (7.2 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del intermedio 3b:
Una mezcla de 2-bromo-2-(4-clorofenil)acetato de metilo 3a (6 g, 3.80 mmol), 3-(2-(ferc-butoxi)etoxi)-5-metoxianilina [CAS 1428973-39-0] (4.63, 4.05 mmol), trietilamina (4.04 ml, 29.0 mmol) en CH3CN (30 ml) se agitó a 50 °C durante 12 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con agua. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El compuesto se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 40 g, 90/10 de heptano/EtOAc). Las fracciones puras se
combinaron y se evaporaron a sequedad para dar 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-acetato de metilo 3b (4.8 g).
Síntesis del intermedio 3c:
Una mezcla de 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)acetato de metilo 3b (4.8 g, 11.4 mmol) y LiOH (1.43 mg, 34.1 mmol) en THF/agua (1/1) (50 ml) se agitó a temperatura ambiente durante 1 h. La mezcla se diluyó con agua. Se acidificó lentamente la fase acuosa con HCl 3 N y se extrajo con EtOAc. Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron, y el disolvente se evaporó a presión reducida para dar ácido 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)acético 3c (4.6 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del intermedio 3d:
Se añadió HATU (2.10 g, 5.52 mmol) a una mezcla de 6-(trifluorometoxi)indolina [CAS 953906-76-8] (747 mg, 3.68 mmol), ácido 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)acético 3c (1.5 g, 3.68 mmol) y diisopropiletilamina (1.82 ml, 11.03 mmol) en DMF (30 ml). La mezcla resultante se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. La purificación se llevó a cabo mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, heptano/EtOAc, 85/15). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-1-(6-(trifluorometoxi)indolin-1-il)etanona 3d (1.33 g).
Síntesis de la separación quiral del Compuesto 3 en los Enantiómeros 3A y 3B:
Una mezcla de 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-1 -(6-(trifluorometoxi)indolin-1 -il)etanona 3d (1.33 g, 2.24 mmol) en HCl 4 M en dioxano (25 ml) se agitó a temperatura ambiente durante 18 h. La mezcla se diluyó con agua y se basificó con K2CO3. Se extrajo la fase acuosa con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, CH2Cl2/MeOH/NH4OH, 99/1/0.1). Las fracciones puras se combinaron y se evaporaron a sequedad para proporcionar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona 3 (1 g) en forma de una mezcla racémica. Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 gm 250 x 20 mm, Fase móvil: 55 % de CO2, 40 % de EtOH (+ 0.3 % de iP rN ^), 5 % de CH2CL). Los enantiómeros se separaron adicionalmente a través de SFC preparativa quiral (Fase estacionaria: Chiralcel® OD-H 5 gm 250 x 30 mm, Fase móvil: 50 % de CO2, 50 % de EtOH (+ 0.3 % de iPrNH2)) para dar, después de la solidificación en éter de petróleo/éter diisopropílico, el primer Enantiómero eluido 3A (294 mg) y el segundo Enantiómero eluido 3B (244 mg).
Compuesto 3:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.08 - 3.25 (m, 2 H) 3.58 - 3.68 (m, 5 H) 3.79 - 3.89 (m, 2 H) 4.00 - 4.11 (m, 1 H) 4.47 - 4.57 (m, 1 H) 4.79 (t, J=5.4 Hz, 1 H) 5.56 (d a, J=8.8 Hz, 1 H) 5.76 (s, 1 H) 5.94 (s, 2 H) 6.47 (d a, J=8.5 Hz, 1 H) 7.01 (d a, J=7.9 Hz, 1 H) 7.34 (d, J=8.2 Hz, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.55 (d a, J=8.2 Hz, 2 H) 8.03 (s a, 1 H)
LC/MS (método LC-A): TR 3.36 min, MH+ 537
Punto de fusión: 162°C
Enantiómero 3A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.08 - 3.25 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.79 - 3.89 (m, 2 H) 4.05 (td, J=10.4, 7.3 Hz, 1 H) 4.52 (td, J=10.4, 6.3 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.56 (d, J=8.8 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.94 (d, J=1.9 Hz, 2 H) 6.47 (d, J=8.5 Hz, 1 H) 7.01 (dd, J=8.0, 1.7 Hz, 1 H) 7.33 (d, J=8.2 Hz, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.03 (s, 1 H)
LC/MS (método LC-A): TR 3.36 min, MH+ 537
[a]D20: 51.9° (c 0.2736, DMF)
SFC quiral (método SFC-C): tR 2.55 min, MH+ 537, pureza quiral 100 %.
Enantiómero 3B:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.08 - 3.26 (m, 2 H) 3.59 - 3.68 (m, 5 H) 3.80 - 3.90 (m, 2 H) 4.05 (td, J=10.4, 7.3 Hz, 1 H) 4.52 (td, J=10.4, 6.3 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.56 (d, J=8.8 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.94 (d, J=1.6 Hz, 2 H) 6.47 (d, J=8.5 Hz, 1 H) 7.01 (dd, J=8.2, 1.6 Hz, 1 H) 7.34 (d, J=8.2 Hz, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.03 (s, 1 H)
LC/MS (método LC-A): TR 3.36 min, MH+ 537
[ajo20: -51.1° (c 0.2973, DMF)
SFC quiral (método SFC-C): ír 3.56 min, MH+ 537, pureza quiral 99.58 %.
Ejemplo 3.2 : síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 3) y separación quiral en los Enantiómeros 3A y 3B .

Una mezcla de 2-bromo-2-(4-clorofenil)acetato de metilo 3a (19.2 g, 72.9 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1 j (13.4 g, 72.9 mmol) y trietilamina (15.2 ml, 109.3 mmol) en CH3CN (115 ml) se calentó a reflujo durante 3 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con HCl 1 N. La fase orgánica se separó, se separó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida para dar el intermedio en bruto 3e (30 g). Esta fracción se combinó con otro lote del intermedio en bruto 3e (cantidad total: 37 g) y se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 400 g, heptano/EtOAc, 60/40). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-clorofenil)-2-((3-(2-hidroxi-etoxi)-5-metoxifenil)amino)acetato de metilo 3e (26 g).
Síntesis del intermedio 3f:
Una mezcla de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)-amino)acetato de metilo 3e (10 g, 27.3 mmol) y LiOH (3.44 g, 82.0 mmol) en THF/agua (1/1) (200 ml) se agitó a temperatura ambiente durante 2 h. La mezcla se diluyó con agua. Se acidificó lentamente la fase acuosa con HCl 3 N y se extrajo con EtOAc. Las capas orgánicas se lavaron con agua, se separaron, se secaron sobre MgSO4, se filtraron, y el disolvente se evaporó a presión reducida para dar ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 3f (9.5 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 3 y separación quiral en los Enantiómeros 3A y 3B:
En flujo N2 a 5 °C, se añadió gota a gota anhídrido propilfosfónico (2.56 ml, 4.26 mmol) a una mezcla de 6-(trifluorometoxi)indolina [CAS 953906-76-8] (577 mg, 2.84 mmol), ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 3f (1.3 g, 3.70 mmol) y diisopropiletilamina (1.03 ml, 6.25 mmol) en 2-Me-THF (30 ml). La mezcla se agitó a temperatura ambiente durante 4 h. Se añadió agua y la mezcla se extrajo con EtOAc. La capa orgánica se lavó con una solución al 10 % de K2CO3 en agua y después con agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice, (15-40 gm, 40 g, CH2CL/MeOH, 99/1). Las fracciones puras se combinaron y se evaporaron a sequedad, para dar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 3, 800 mg) en forma de una mezcla racémica. Esta fracción se combinó con otro lote (cantidad total: 1.4 g) y se cristalizó en éter diisopropílico para dar 1.03 g del Compuesto 3. Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 gm 250 x 20 mm, Fase móvil: 55 % de CO2, 45 % de EtOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido se purificó adicionalmente a través de cromatografía de fase inversa (fase estacionaria: X-bridge-C-18 10 gm 30 x 150 mm, fase móvil: gradiente de 60/40 a 0/100 de NH4HCO3 al 0.2 %/CH3CN) para dar el Enantiómero 3A (312 mg). El segundo Enantiómero eluido 3B (436 mg) no se purificó adicionalmente.
Ejemplo 4 : síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometil)indolin-1-il)etanona (Compuesto 4) y separación quiral en los Enantiómeros 4A y 4B .

Síntesis del intermedio 4a:
Se añadió HATU (2.24 g, 5.88 mmol) a una mezcla de 6-(trifluorometil)indolina [CAS 181513-29-1] (734 mg, 3.92 mmol), ácido 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)acético 3c (1.6 g, 3.92 mmol) y diisopropiletilamina (1.95 ml, 11.8 mmol) en DMF (30 ml). La mezcla resultante se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua y EtOAc. La capa orgánica se separó, se lavó con una solución al 10 % de K2CO3 en agua, se lavó con salmuera, se secó sobre MgSO4, se filtró, y el disolvente se concentró a presión reducida. El residuo en bruto se purificó por cromatografía en columna sobre gel de sílice (15-40 gm, 40 g, 85/15 de heptano/EtOAc). Las fracciones puras se combinaron y el disolvente se concentró a presión reducida para dar 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-1 -(6-(trifluorometil)indolin-1 -il)etanona 4a (1.38 g).
Síntesis del Compuesto 4 y separación quiral en los Enantiómeros 4A y 4B:
Se mezcló 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-1 -(6-(trifluorometil)indolin-1 -il)etanona 4a (1.38 g, 2.39 mmol) con HCl 4 M en dioxano (25 ml) agitado a temperatura ambiente durante 18 h. La mezcla se diluyó con agua y se basificó con K2CO3. Se extrajo la fase acuosa con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, CH2Cl2/MeOH/NH4OH, 99/1/0.1). Las fracciones puras se combinaron y se evaporaron a sequedad para proporcionar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometil)indolin-1-il)etanona (Compuesto 4 , 1.08 g) en forma de una mezcla racémica. Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralcel® OJ-H 5 gm 250 x 20 mm, Fase móvil: 60 % de CO2, 40 % de EtOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (413 mg) se solidificó en heptano/ para dar el Enantiómero 4A (327 mg). El segundo enantiómero eluido (410 mg) se solidificó en heptano/éter diisopropílico para dar el Enantiómero 4B (330 mg).
Compuesto 4:
1H RMN (500 MHz, DMSO-da) 5 ppm 3.16 - 3.32 (m, 2 H) 3.59 - 3.68 (m, 5 H) 3.79 - 3.90 (m, 2 H) 4.03 (td, J=10.4, 6.9 Hz, 1 H) 4.53 (td, J=10.3, 6.5 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.58 (d, J=8.5 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.95 (s, 2 H) 6.44 (d, J=8.5 Hz, 1 H) 7.37 - 7.41 (m, 1 H) 7.43 - 7.49 (m, 3 H) 7.56 (d, J=8.5 Hz, 2 H) 8.38 (s, 1 H)
LC/MS (método LC-A): TR 3.31 min, MH+ 521
Punto de fusión: 160°C
Enantiómero 4A:
1H RMN (400 MHz, DMSO-da) 5 ppm 3.15 - 3.29 (m, 2 H) 3.58 - 3.70 (m, 5 H) 3.79 - 3.89 (m, 2 H) 4.03 (td, J=10.4, 7.6 Hz, 1 H) 4.53 (td, J=10.2, 6.3 Hz, 1 H) 4.78 (s a, 1 H) 5.57 (d, J=9.1 Hz, 1 H) 5.74 - 5.80 (m, 1 H) 5.95 (d, J=1.0 Hz, 2 H) 6.43 (d a, J=8.6 Hz, 1 H) 7.36 - 7.41 (m, 1 H) 7.42 - 7.49 (m, 3 H) 7.56 (d, J=8.6 Hz, 2 H) 8.38 (s, 1 H)
LC/MS (método LC-B): TR 3.03 min, MH+ 521
[a]D20: 52.0° (c 0.3036, DMF)
SFC quiral (método SFC-D): ír 1.82 min, MH+ 521, pureza quiral 100 %.
Enantiómero 4B:
1H RMN (400 MHz, DMSO-cfe) 5 ppm 3.18 - 3.29 (m, 2 H) 3.59 - 3.70 (m, 5 H) 3.84 (m, 2 H) 3.97 - 4.10 (m, 1 H) 4.46 - 4.59 (m, 1 H) 4.78 (s a, 1 H) 5.57 (d a, J=8.6 Hz, 1 H) 5.76 (s, 1 H) 5.95 (s, 2 H) 6.43 (d a, J=8.6 Hz, 1 H) 7.35 - 7.50 (m, 4 H) 7.56 (d a, J=8.6 Hz, 2 H) 8.38 (s, 1 H)
LC/MS método LC-B): TR 3.03 min, MH+ 521
[a]D20: -51.8° (c 0.3418, DMF)
SFC quiral (método SFC-D): ír 3.17 min, MH+ 521, pureza quiral 99.56 %.
Ejemplo 5 : síntesis de 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 5) y separación quiral en los Enantiómeros 5A y 5B .

Síntesis del intermedio 5a:
Una mezcla de 6-(trifluorometoxi)indolina [CAS 953906-76-8] (1 g, 4.92 mmol), ácido 2-(4-cloro-2-metoxifenil)acético [CAS 170737-95-8] (1.09 g, 5.41 mmol), HATU (2.81 g, 7.38 mmol) y diisopropiletilamina (2.44 ml, 14.8 mmol) en DMF (10 ml) se agitó a temperatura ambiente durante 12 h. Se añadieron agua y EtOAc y las capas se separaron. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El residuo se purificó mediante cromatografía sobre gel de sílice (15-40 gm, 80 g, 85/15 de heptano/EtOAc). Las fracciones puras se combinaron, y el disolvente se concentró a presión reducida para dar, después de la cristalización en CHaCN/heptano, 2-(4-cloro-2-metoxifenil)-1 -(6-(trifluorometoxi)indolin-1 -il)etanona 5a (1.53 g).
Síntesis del intermedio 5b:
A -78 °C, en un flujo de N2, se añadió gota a gota LiHMDS 1 M en THF (7.93 ml, 7.93 mmol) a una mezcla de 2-(4-cloro-2-metoxifenil)-1-(6-(trifluorometoxi)-indolin-1-il)etanona 5a (1.53 g, 3.97 mmol) en THF (12 ml). La mezcla se agitó durante 15 min a -78 °C y se añadió gota a gota una solución de NBS (776 mg, 4.36 mmol) en THF (10 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió por la adición de una solución acuosa saturada de NH4CL La mezcla se extrajo con EtOAc, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida para dar 2-bromo-2-(4-cloro-2-metoxifenil)-1 -(6-(trifluorometoxi)indolin-1 -il)etanona 5b (1.70 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 5 y separación quiral en los Enantiómeros 5A y 5B:
Una mezcla de 2-bromo-2-(4-cloro-2-metoxifenil)-1-(6-(trifluorometoxi)indolin-1-il)etanona 5b (1.37 g, 2.95 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1] (810 mg, 4.42 mmol) y diisopropiletilamina (762 gl, 4.42 mmol) en CH3CN (20 ml) se agitó a 50 °C durante 8 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con HCl 1 N. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El compuesto se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, CH2Cl2/heptano, 98.5/1.5). Las fracciones puras se combinaron y se evaporaron a sequedad para dar, después de la cristalización en CH3CN, 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1 -(6
(trifluorometoxi)indolin-1-il)etanona (Compuesto 5 , 500 mg) en forma de una mezcla racémica. Este lote se combinó con otro lote (cantidad total: 903 mg). Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IC 5 gm 250 x 30 mm, Fase móvil: 65 % de CO2, 35 % de iPrOH (+ 0.3 % de iPrNhb)). El primer enantiómero eluido (453 mg) se solidificó en éter de petróleo/éter diisopropílico para dar el Enantiómero 5A (355 mg). El segundo enantiómero eluido (436 mg) se solidificó en éter de petróleo/éter diisopropílico para dar el Enantiómero 5B (342 mg).
Compuesto 5:
1H RMN (500 MHz, DMSO-ds) 5 ppm 3.11 - 3.23 (m, 2 H) 3.59 - 3.67 (m, 5 H) 3.79 - 3.87 (m, 2 H) 3.90 (s, 3 H) 4.01 -4.09 (m, 1 H) 4.32 - 4.41 (m, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.59 (d, J=8.5 Hz, 1 H) 5.76 (s, 1 H) 5.87 (s a, 2 H) 6.46 (d a, J=8.8 Hz, 1 H) 6.98 - 7.06 (m, 2 H) 7.14 (d, J=1.6 Hz, 1 H) 7.30 - 7.35 (m, 2 H) 8.02 (s, 1 H)
LC/MS (método LC-A): TR 3.38 min, MH+ 567
Punto de fusión: 162°C
Enantiómero 5A:
1H RMN (500 MHz, DMSO-da) 5 ppm 3.10 - 3.25 (m, 2 H) 3.59 - 3.67 (m, 5 H) 3.78 - 3.88 (m, 2 H) 3.90 (s, 3 H) 4.04 (td, J=10.3, 7.1 Hz, 1 H) 4.37 (td, J=10.2, 6.8 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.59 (d, J=8.5 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.84 - 5.89 (m, 2 H) 6.46 (d, J=8.5 Hz, 1 H) 6.99 - 7.05 (m, 2 H) 7.14 (d, J=2.2 Hz, 1 H) 7.31 (d, J=8.2 Hz, 1 H) 7.34 (d, J=8.2 Hz, 1 H) 8.02 (s, 1 H)
LC/MS (método LC-A): TR 3.39 min, MH+ 567
[a]D20: 31.1° (c 0.2736, DMF)
SFC quiral (método SFC-E): tR 2.02 min, MH+ 567, pureza quiral 100 %.
Enantiómero 5B:
1H RMN (500 MHz, DMSO-da) 5 ppm 3.09 - 3.25 (m, 2 H) 3.60 - 3.67 (m, 5 H) 3.78 - 3.87 (m, 2 H) 3.90 (s, 3 H) 4.04 (td, J=10.2, 6.9 Hz, 1 H) 4.37 (td, J=10.2, 6.8 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.59 (d, J=8.5 Hz, 1 H) 5.76 (t, J=1.9 Hz, 1 H) 5.87 (s a, 2 H) 6.47 (d, J=8.5 Hz, 1 H) 6.99 - 7.05 (m, 2 H) 7.14 (d, J=1.9 Hz, 1 H) 7.31 (d, J=8.2 Hz, 1 H) 7.34 (d, J=8.2 Hz, 1 H) 8.02 (s, 1 H)
LC/MS (método LC-A): TR 3.39 min, MH+ 567
[a]D20: -31.0° (c 0.2773, DMF)
SFC quiral (método SFC-E): tR 3.00 min, MH+ 567, pureza quiral 100 %.
Ejemplo 6.1 : síntesis de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 6)

Síntesis del Compuesto 6:
Se añadió HATU (1.54 g, 4.06 mmol) a una mezcla de 6-(trifluorometoxi)indolina [CAS 953906-76-8] (550 mg, 2.70 mmol), ácido 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 2e (1 g, 2.70 mmol) y diisopropiletilamina (1.34 ml, 8.11 mmol) en DMF (30 ml). La mezcla resultante se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua. El precipitado se eliminó por filtración y se lavó con agua. El sólido se disolvió en EtOAc, se lavó con una solución al 10 % de K2CO3 en agua y después con salmuera. La capa orgánica se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El residuo en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, CH2Cl2/MeOH, 99.5/0.5) para proporcionar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 6 , 500 mg). Una muestra analítica del Compuesto 6 se obtuvo por cristalización en éter diisopropílico.
Compuesto 6:
1H RMN (500 MHz, DMSO-ds) 5 ppm 3.14 - 3.22 (m, 2 H) 3.58 - 3.68 (m, 5 H) 3.80 - 3.90 (m, 2 H) 4.05 - 4.15 (m, 1 H) 4.38 - 4.47 (m, 1 H) 4.80 (t, J=5.5 Hz, 1 H) 5.70 (d, J=9.1 Hz, 1 H) 5.79 (t, J=1.9 Hz, 1 H) 5.93 (d, J=1.9 Hz, 2 H) 6.63 (d, J=9.1 Hz, 1 H) 7.03 (dd, J=8.2, 1.6 Hz, 1 H) 7.31 - 7.37 (m, 2 H) 7.42 - 7.51 (m, 2 H) 8.02 (s, 1 H)
LC/MS (método LC-A): TR 3.39 min, MH+ 555
Punto de fusión: 166°C
Ejemplo 6.2 : síntesis de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 6) y separación quiral en los Enantiómeros 6A y 6B .

Síntesis del intermedio 6a:
Se añadió HATU (7.02 g, 18.5 mmol) a una mezcla de 6-(trifluorometoxi)indolina [CAS 953906-76-8] (2.5 g, 12.31 mmol), ácido 2-(4-cloro-2-fluorofenil)acético [CAS 194240-75-0] (2.32 g, 12.3 mmol) y diisopropiletilamina (6.1 ml, 36.9 mmol) en DMF (100 ml). La mezcla resultante se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua, y el precipitado se eliminó por filtración y se lavó con agua. El residuo se recogió con EtOAc y la capa orgánica se lavó con una solución al 10 % de K2CO3 en agua, se lavó con salmuera, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El producto en bruto se cristalizó en éter diisopropílico para dar 2-(4-cloro-2-fluorofenil)-1 -(6-(trifluorometoxi)indolin-1 -il)etanona 6a (4 g).
Síntesis del intermedio 6b:
A -78 °C, en un flujo de N2, se añadió gota a gota LiHMDS 1 M en THF (21.4 ml, 21.4 mmol) a una mezcla de 2-(4-cloro-2-fluorofenil)-1-(6-(trifluorometoxi)indolin-1-il)etanona 6a (4 g, 10.7 mmol) en THF (60 ml). La mezcla se agitó durante 15 min a -78 °C y se añadió gota a gota una solución de NBS (2.1 g, 11.8 mmol) en THF (40 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió con una solución acuosa saturada de NH4CL La mezcla se extrajo con EtOAc, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida para dar 2-bromo-2-(4-cloro-2-fluorofenil)-1 -(6-(trifluorometoxi)indolin-1 -il)etanona 6b (4.8 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 6 y separación quiral en los Enantiómeros 6A y 6B:
Una mezcla de 2-bromo-2-(4-cloro-2-fluorofenil)-1-(6-(trifluorometoxi)indolin-1-il)etanona 6b (4.8 g, 10.6 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1] (2.3 g, 12.7 mmol) y diisopropiletilamina (2.2 ml, 12.7 mmol) en CH3CN (200 ml) se agitó a 70 °C durante 72 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con HCl 1 N y agua. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El compuesto se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 80 g, CH2Cl2/heptano, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1 -(6-(trifluorometoxi)indolin-1 -il)etanona racémica (Compuesto 6 , 3 g) después de la cristalización en CH3CN/éter diisopropílico. Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralcel® OD-H 5 pm 250 x 30 mm, Fase móvil: 60 % de CO2, 40 % de EtOH (+ 0.3 %
de ¡PrNhh)). El primer enantiómero eluido (1.45 g) se solidificó por trituración con MeOH/agua para dar el Enantiómero 6A (1.409 g). El segundo enantiómero eluido (1.41 g) se solidificó por trituración con MeOH/agua para dar el Enantiómero 6B (1.37 g).
Enantiómero 6A:
1H RMN (400 MHz, DMSO-afe) 5 ppm 3.13 - 3.21 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.78 - 3.91 (m, 2 H) 4.04 - 4.14 (m, 1 H) 4.37 - 4.48 (m, 1 H) 4.77 (t, J=5.6 Hz, 1 H) 5.69 (d, J=9.1 Hz, 1 H) 5.80 (s, 1 H) 5.93 (d, J=1.5 Hz, 2 H) 6.60 (d a, J=9.1 Hz, 1 H) 7.02 (d a, J=8.1 Hz, 1 H) 7.30 - 7.38 (m, 2 H) 7.41 - 7.51 (m, 2 H) 8.02 (s, 1 H) LC/MS (método LC-A): TR 3.41 min, MH+ 555
[a]
D20: -25.9° (c 0.27, DMF)
SFC quiral (método SFC-F): tR 4.08 min, MH+ 555, pureza quiral 100 %.
Enantiómero 6B:
1H RMN (400 MHz, DMSO-afe) 5 ppm 3.13 - 3.20 (m, 2 H) 3.59 - 3.68 (m, 5 H) 3.79 - 3.91 (m, 2 H) 4.04 - 4.14 (m, 1 H) 4.38 - 4.49 (m, 1 H) 4.77 (t, J=5.6 Hz, 1 H) 5.69 (d, J=9.1 Hz, 1 H) 5.80 (s, 1 H) 5.93 (d, J=2.0 Hz, 2 H) 6.60 (d, J=8.6 Hz, 1 H) 7.02 (d a, J=9.1 Hz, 1 H) 7.29 - 7.38 (m, 2 H) 7.42 - 7.50 (m, 2 H) 8.02 (s, 1 H) LC/MS (método LC-A): TR 3.41 min, MH+ 555
[a]
D20: 23.3° (c 0.27, DMF)
SFC quiral (método SFC-F): tR 2.25 min, MH+ 555, pureza quiral 99.42 %.
Ejemplo 7.1 : síntesis de 2-(4-clorofen¡l)-2-((3-(2-h¡drox¡etox¡)-5-metox¡fen¡l)am¡no)-1-(5-metox¡-6-(tr¡fluoromet¡l)¡ndol¡n-1-¡l)etanona (Compuesto 7)

Síntesis del intermedio 7a:
Una mezcla de 1-metox¡-4-n¡tro-2-(tr¡fluoromet¡l)benceno [CAS 654-76-2] (24.5 g, 110.8 mmol) y 4-clorofenoxiacetonitrilo [CAS 3598-13-8] (20.4 g, 121.9 mmol) en DMF (100 ml) se añadió gota a gota durante 30 min a una solución agitada de íBuOK (27.4 g, 243.7 mmol) en DMF (100 ml) a -10 °C. Después de la adición, la solución de color púrpura se mantuvo a -10 °C durante 1 h. Se añadieron hielo-agua (500 ml) y HCl 6 N (500 ml) y el precipitado se eliminó por filtración, se lavó con agua y se secó al vacío para proporcionar 2-(5-metox¡-2-n¡tro-4-(tr¡fluoromet¡l)-fenil)acetonitrilo 7a (40.4 g) que se usó tal cual en la siguiente etapa.
Síntesis del intermedio 7b:
Una solución de 2-(5-metoxi-2-nitro-4-(trifluorometil)fenil)acetonitrilo 7a (26 g, 99.9 mmol) en etanol/agua (9/1) (500 ml) y AcOH (5.2 ml) se hidrogenó durante 1 h a presión (3.5 Bar) con Pd al 10 %/C (15.3 g) como el catalizador. La mezcla de reacción se filtró a través de un lecho de celite® y la torta de filtro se lavó con una mezcla de CH2Cl2 y CH3OH. Los filtrados combinados se concentraron a presión reducida. El residuo en bruto se filtró a través de un lecho de sílice (60-200 gm) usando 80/20 de heptano/EtOAc como el eluyente. Las fracciones que contenían el compuesto esperado se combinaron y el disolvente se concentró a presión reducida para dar 5-metoxi-6-(trifluorometil)-1 H-indol 7b (15.6 g).
Síntesis del intermedio 7c:
A 0 °C, se añadió gota a gota BH3-Piridina (23.5 ml, 232.4 mmol) a una solución de 5-metoxi-6-(trifluorometil)-1 H-indol 7b (10 g, 46.5 mmol) en EtOH (60 ml). Se añadió lentamente HCl 6 N (140 ml) mientras se mantuvo la temperatura de reacción por debajo de 10 °C. La mezcla se agitó a 0 °C durante 2 h. Se añadió agua (200 ml) y la mezcla se basificó hasta pH 8-9 con una solución concentrada de NaOH en agua, mientras se mantuvo la temperatura de reacción por debajo de 20 °C. El precipitado se eliminó por filtración, se lavó con agua (dos veces) y se co-evaporó a presión reducida con tolueno para dar 5-metoxi-6-(trifluorometil)indolina 7c (9 g).
Síntesis del intermedio 7d:
Se añadió HATU (0.84 g, 2.21 mmol) a una mezcla de 5-metoxi-6-(trifluorometil)indolina 7c (320 mg, 1.47 mmol), ácido 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)acético 3c (631 mg, 1.55 mmol) y diisopropiletilamina (731 gl, 4.42 mmol) en DMF (18 ml). La mezcla de reacción se agitó a temperatura ambiente durante 12 h. La reacción se diluyó con agua y EtOAc. La capa orgánica se separó, se lavó con una solución al 10 % de K2CO3 en agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El residuo en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, CH2Cl2/MeOH, 99.5/0.5) para dar 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-1 -(5-metoxi-6-(trifluorometil)-indolin-1 -il)etanona 7d (839 mg).
Síntesis del Compuesto 7:
Se añadió 2-((3-(2-(ferc-butoxi)etoxi)-5-metoxifenil)amino)-2-(4-clorofenil)-1 -(5-metoxi-6-(trifluorometil)indolin-1 -il)etanona 7d (1.15 g, 1.89 mmol) a HCl 4 M en dioxano (20 ml) y la mezcla se agitó a temperatura ambiente durante 18 h. La mezcla se diluyó con agua y se basificó con K2CO3. Se extrajo la fase acuosa con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, cH2Cl2/MeOH/NH4OH, 99/1/0.1). Las fracciones puras se combinaron y se evaporaron a sequedad para proporcionar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1 -(5-metoxi-6-(trifluorometil)-indolin-1 -il)etanona (Compuesto 7 , 915 mg). Se obtuvo una muestra analítica del Compuesto 7 por cristalización en CH3CN.
Compuesto 7:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.15 - 3.29 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.79 - 3.90 (m, 5 H) 3.96 - 4.07 (m, 1 H) 4.51 (td, J=10.4, 6.0 Hz, 1 H) 4.78 (t, J=5.0 Hz, 1 H) 5.54 (d, J=8.5 Hz, 1 H) 5.76 (s, 1 H) 5.95 (s, 2 H) 6.40 (d a, J=8.5 Hz, 1 H) 7.23 (s, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.56 (d, J=8.5 Hz, 2 H) 8.34 (s, 1 H)
LC/MS (método LC-A): TR 3.21 min, MH+ 551
Punto de fusión: 188°C
Ejemplo 7.2 : síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona (Compuesto 7) y separación quiral en los Enantiómeros 7A y 7B .

En un flujo de N2, a 5 °C, se añadió gota a gota anhídrido propilfosfónico (4.15 ml, 6.91 mmol) a una mezcla de 5-metoxi-6-(trifluorometil)indolina 7c (1 g, 4.60 mmol), ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 3f (1.94 g, 5.53 mmol) y diisopropiletilamina (1.67 ml, 10.1 mmol) en DMF (20 ml). La mezcla se agitó a temperatura ambiente durante 7 h. Se añadió agua y la mezcla se extrajo con EtOAc. La capa orgánica se
lavó con una solución al 10 % de K2CO3 en agua, y después con agua. La fase orgánica se secó con MgSÜ4, se filtró y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice, (15-40 gm, 90 g, ChbCL/MeOH, 99/1). Las fracciones puras se combinaron y se evaporaron a sequedad, para dar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona (Compuesto 7 , 2.17 g) en forma de una mezcla racémica. Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 gm 250 x 20 mm, Fase móvil: 45 % de CO2, 55 % de iPrOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (980 mg) se cristalizó en MeOH para proporcionar el Enantiómero 7A (711 mg). El segundo enantiómero eluido (1.08 g) se purificó adicionalmente por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, CH2Cl2/MeOH, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad (950 mg) para proporcionar, después de la cristalización en MeOH, el Enantiómero 7B (770 mg).
Enantiómero 7A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.15 - 3.31 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.79-3.90 (m, 5 H) 3.95 - 4.04 (m, 1 H) 4.51 (td, J=10.4, 6.3 Hz, 1 H) 4.80 (t, J=5.4 Hz, 1 H) 5.54 (d, J=8.5 Hz, 1 H) 5.76 (s, 1 H) 5.95 (s, 2 H) 6.41 (d, J=8.5 Hz, 1 H) 7.24 (s, 1 H) 7.44 (d, J=8.2 Hz, 2 H) 7.56 (d, J=8.5 Hz, 2 H) 8.34 (s, 1 H)
LC/MS (método LC-A): TR 3.22 min, MH+ 551
[a]
D20: -45.2° (c 0.314, DMF)
SFC quiral (método SFC-G): tR 2.35 min, MH+ 551, pureza quiral 100 %.
Punto de fusión: 112°C
Enantiómero 7B:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.15 - 3.31 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.79 - 3.90 (m, 5 H) 3.95 - 4.05 (m, 1 H) 4.51 (td, J=10.3, 6.5 Hz, 1 H) 4.80 (t a, J=5.0 Hz, 1 H) 5.54 (d, J=8.8 Hz, 1 H) 5.76 (s, 1 H) 5.95 (s, 2 H) 6.41 (d, J=8.8 Hz, 1 H) 7.24 (s, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.56 (d, J=8.5 Hz, 2 H) 8.34 (s, 1 H) LC/MS (método LC-A): TR 3.21 min, MH+ 551
[a]
D20: 43.8° (c 0.27, DMF)
SFC quiral (método SFC-G): tR 3.84 min, MH+ 551, pureza quiral 100 %.
Punto de fusión: 112°C
Ejemplo 8.1 : síntesis de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona (Compuesto 8).

Se añadió HATU (308 mg, 0.81 mmol) a una mezcla de 5-metoxi-6-(trifluorometil)indolina 7c (117 mg, 0.54 mmol), ácido 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 2e (200 mg, 0.54 mmol) y diisopropiletilamina (0.267 ml, 1.61 mmol) en DMF (10 ml). La mezcla de reacción se agitó a temperatura ambiente durante 12 h. La reacción se diluyó con agua, causando precipitación. El precipitado se separó por filtración y se lavó con agua. Se disolvió el sólido en EtOAc. La capa orgánica se lavó con una solución al 10 % de K2CO3 en agua y con salmuera, se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El residuo en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, CH2Cl2/MeOH, 99.5/0.5). El residuo se cristalizó en Et2O/éter diisopropílico para dar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona (Compuesto 8 , 35 mg).
Compuesto 8:
1H RMN (500 MHz, DMSO-db) 5 ppm 3.19 - 3.28 (m, 2 H) 3.59 - 3.69 (m, 5 H) 3.85 (m, 5 H) 3.99 - 4.08 (m, 1 H) 4.37 - 4.47 (m, 1 H) 4.80 (t, J=5.4 Hz, 1 H) 5.68 (d a, J=9.1 Hz, 1 H) 5.79 (s, 1 H) 5.93 (s, 2 H) 6.58 (d a, J=9.1 Hz, 1 H) 7.26 (s, 1 H) 7.33 (d a, J=7.6 Hz, 1 H) 7.42 - 7.52 (m, 2 H) 8.33 (s, 1 H)
LC/MS (método LC-A): TR 3.27 min, MH+ 569
Punto de fusión: 176°C
Ejemplo 8.2 : síntesis de 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona (Compuesto 8) y separación quiral en los Enantiómeros 8A y 8B .

Síntesis del intermedio 8a:
Se añadió HATU (2.9 g, 7.6 mmol) a una mezcla de 5-metoxi-6-(trifluorometil)-indolina 7c (1.1 g, 5.06 mmol), ácido 2-(4-cloro-2-fluorofenil)acético [CAS 194240-75-0] (1.05 g, 5.57 mmol) y diisopropiletilamina (2.51 ml, 15.2 mmol) en DMF (30 ml). La mezcla de reacción se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua, causando precipitación. El precipitado se separó por filtración y se lavó con agua. El residuo se recogió con EtOAc y la solución orgánica se lavó con una solución al 10 % de K2CO3 en agua y después con salmuera. La solución orgánica se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. El residuo en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 80 g, heptano/EtOAc, 90/10 a 60/40). Las fracciones puras se combinaron y el disolvente se evaporó a presión reducida para dar 2-(4-cloro-2-fluorofenil)-1-(5-metoxi-6-(trifluorometil)-indolin-1 -il)etanona 8a (1.8 g).
Síntesis del intermedio 8b:
A -78 °C, en un flujo de N2, se añadió gota a gota LiHMDS 1 M en THF (9.3 ml, 9.3 mmol) a una mezcla de 2-(4-cloro-2-fluorofenil)-1 -(5-metoxi-6-(trifluorometil)indolin-1 -il)etanona 8a (1.8 g, 4.65 mmol) en THF (25 ml). Se añadió gota a gota TMSCl (0.7 ml, 0.86 mmol). La mezcla se agitó durante 15 min a -78 °C y se añadió gota a gota una solución de NBS (1 g, 5.57 mmol) en THF (15 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió por la adición de una solución acuosa saturada de NH4CL La mezcla se extrajo con EtOAc. La capa orgánica se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida para dar 2-bromo-2-(4-cloro-2-fluorofenil)-1-(5-metoxi-6-(trifluorometil)indolin-1 -il)etanona 8b (2.1 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 8 y separación quiral en los Enantiómeros 8A y 8B:
Una mezcla de 2-bromo-2-(4-cloro-2-fluorofenil)-1-(5-metoxi-6-(trifluorometil)-indolin-1-il)etanona 8b (2.1 g, 4.5 mmol), 2-(3-amino-5-metoxifenoxi)-etanol [CAS 725237-16-1] (0.99 g, 5.4 mmol) y diisopropiletilamina (1.16 ml, 6.75 mmol) en CH3CN (80 ml) se agitó a 70 °C durante 72 h. La mezcla se concentró a presión reducida, se diluyó con EtOAc y se lavó con HCl 1 N y agua. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El compuesto se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 80 g, CH2Cl2). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-fluorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona racémica (Compuesto 8 , 850 mg) después de la cristalización en CH3CN. Esta fracción se combinó con otro lote (cantidad total: 1.3 g). Los enantiómeros
se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 pm 250 x 20 mm, Fase móvil: 70 % de CO2, 30 % de EtOH (+ 0.3 % de iPrNFb)). El primer enantiómero eluido (680 mg) se solidificó por trituración con CH3CN para dar el Enantiómero 8A (590 mg). El segundo enantiómero eluido (630 mg) se solidificó por trituración con CH3CN para dar el Enantiómero 8B (569 mg).
Enantiómero 8A:
1H RMN (400 MHz, DMSO-ds) 5 ppm 3.19 - 3.27 (m, 2 H) 3.59 - 3.69 (m, 5 H) 3.78 - 3.91 (m, 5 H) 3.98 - 4.08 (m, 1 H) 4.35 - 4.47 (m, 1 H) 4.77 (t, J=5.6 Hz, 1 H) 5.67 (d, J=9.1 Hz, 1 H) 5.79 (t, J=1.8 Hz, 1 H) 5.93 (d, J=2.0 Hz, 2 H) 6.55 (d, J=9.1 Hz, 1 H) 7.25 (s, 1 H) 7.31 (dd, J=8.3, 1.8 Hz, 1 H) 7.42 - 7.50 (m, 2 H) 8.32 (s, 1 H)
LC/MS (método LC-A): TR 3.26 min, MH+ 569
[a]D20: -28.9° (c 0.225, DMF)
SFC quiral (método SFC-H): tR 4.51 min, MH+ 569, pureza quiral 100 %.
Enantiómero 8B:
1H RMN (400 MHz, DMSO-dfe) 5 ppm 3.19- 3.27 (m, 2 H) 3.59 - 3.68 (m, 5 H) 3.80 - 3.90 (m, 5 H) 3.98 - 4.09 (m, 1 H) 4.35 - 4.47 (m, 1 H) 4.77 (t, J=5.3 Hz, 1 H) 5.67 (d, J=9.1 Hz, 1 H) 5.79 (t, J=1.8 Hz, 1 H) 5.93 (d, J=1.5 Hz, 2 H) 6.54 (d, J=8.6 Hz, 1 H) 7.25 (s, 1 H) 7.31 (dd, J=8.6, 2.0 Hz, 1 H) 7.42 - 7.50 (m, 2 H) 8.32 (s, 1 H)
LC/MS (método LC-A): TR 3.26 min, MH+ 569
[a]D20: 25.7° (c 0.2333, DMF)
SFC quiral (método SFC-H): tR 5.81 min, MH+ 569, pureza quiral 100 %.
Ejemplo 9 : síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(4-metil-6-(trifluorometoxi)indolin-1 -il)etanona (Compuesto 9) y separación quiral en los Enantiómeros 9A y 9B .

A una solución de 2-metil-4-(trifluorometoxi)anilina [CAS 86256-59-9] (10.0 g, 52.3 mmol) en dioxano (20 ml) se le añadió anhídrido trifluoroacético (8 ml, 57.2 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 h. Se concentró la solución a presión reducida. El residuo se repartió entre EtOAc y HCl 1 N. Se separaron las fases. Se lavó la fase orgánica con disolución saturada de NaHCO3 en agua, H2O y salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida para permitir la obtención de 14.7 g de 2,2,2-trifluoro-W-(2-metil-4(trifluorometoxi)fenil)acetamida 9a en forma de polvo blanco. Se usó el compuesto en la siguiente etapa sin purificación adicional.
Síntesis del intermedio 9c:
A anhídrido acético (11.4 ml, 61.1 mmol), enfriado a 0 °C, se le añadió gota a gota ácido nítrico al 70 % (3.9 ml). Se añadió gota a gota 2,2,2-trifluoro-W-(2-metil-4-(trifluorometoxi)fenil)-acetamida 9a (5 g, 17.4 mmol) y se calentó la mezcla de reacción a 55 °C durante 12 h. Tras el enfriamiento a temperatura ambiente, se diluyó la mezcla con EtOAc y se lavó con H2O. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se disolvió en metanol (46 ml). Se añadió K2CO32 M (23 ml, 46 mmol) y se calentó la mezcla de reacción a 70 °C durante 4 h. Se añadió más K2CO32 M (10 ml, 20 mmol) y se calentó la mezcla de reacción a 70 °C durante 12 h. Se concentró parcialmente la mezcla de reacción a presión reducida pare retirar el metanol. Se extrajo el residuo con EtOAc. La fase orgánica se lavó con H2O y salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice utilizando un gradiente de EtOAc (de 20 % a 50 %) en heptano para obtener 3.6 g de 2-metil-6-nitro-4-(trifluorometoxi)anilina 9c en forma de sólido amarillo.
Síntesis del intermedio 9d:
A una solución de 2-metil-6-nitro-4-(trifluorometoxi)anilina 9c (1.8 g, 7.69 mmol) en ácido acético (10.9 ml), se añadió gota a gota una solución de nitrito de sodio (0.806 g, 11.7 mmol) en H2SO4/H2O (2 ml, 1/1). La mezcla de reacción se agitó a temperatura ambiente durante 30 min. Se añadieron H2O (22 ml) y urea (0.802 g, 13.4 mmol). Después de 10 min a temperatura ambiente, se añadió gota a gota una solución de yoduro potásico (1.7 g, 10.2 mmol) en H2O (11 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 30 minutos. Se filtró el sólido amarillo, se lavó con H2O y se secó para proporcionar 2.4 g de 2-yodo-1-metil-3-nitro-5-(trifluorometoxi)benceno 9d.
Síntesis del intermedio 9e:
A una solución de 2-yodo-1 -metil-3-nitro-5-(trifluorometoxi)benceno 9d (3.5 g, 10.0 mmol) en EtOH (30 ml), se le añadió una solución de NH4Cl (2.7 g, 49.9 mmol) en H2O (30 ml). La mezcla de reacción se calentó a 50 °C. Se añadió hierro (2.6 g, 46.9 mmol) y la mezcla de reacción se calentó a reflujo durante 40 min. Después del enfriamiento a temperatura ambiente, la mezcla de reacción se filtró a través de Celite®. Se lavaron los sólidos con EtOH. Se concentró el filtrado parcialmente a presión reducida para retirar EtOH. El residuo se repartió entre EtOAc y una solución saturada de NaHCO3 en agua. Se separaron las fases. La fase orgánica se lavó con H2O y salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice utilizando un gradiente de EtOAc (de 0 % a 25 %) en heptano para obtener 2.9 g de 2-yodo-3-metil-5-(trifluorometoxi)anilina 9e en forma de aceite amarillo.
Síntesis del intermedio 9f:
Una disolución de 2-yodo-3-metil-5-(trifluorometoxi)anilina 9e (2.9 g, 9.1 mmol) en trietilamina (23 ml), se desgasificó con argón durante 15 minutos. Se añadieron diclorobis(trifenilfosfina)paladio(II) (0.327 g, 0.47 mmol), yoduro de cobre(I) (0.164 g, 0.86 mmol) y trimetilsililacetileno (1.8 ml, 13.1 mmol). La mezcla de reacción se calentó a 65 °C durante 12 h. Después del enfriamiento a temperatura ambiente, la mezcla de reacción se diluyó con H2O y se extrajo con EtOAc (3x). Las fases orgánicas se combinaron, se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. Se purificó el residuo por medio de cromatografía sobre gel de sílice usando un gradiente de EtOAc (de 0 % a 20 %) en heptano para permitir 2.6 g de 3-metil-5-(trifluorometoxi)-2-((trimetilsilil)-etinil)anilina 9f en forma de aceite naranja.
Síntesis del intermedio 9g:
A una solución de 3 -metil-5-(trifluorometoxi)-2-((trimetilsil)etinil)anilina 9f (2.7 g, 9.3 mmol) en NMP (27 ml), se le añadió íBuOK (3.1 g, 27.8 mmol). La mezcla de reacción se calentó a 80 °C durante 4 h. Después del enfriamiento a temperatura ambiente, la mezcla de reacción se diluyó con H2O y se extrajo con EtOAc (2x). Las fases orgánicas se combinaron, se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. Se purificó el residuo por medio de cromatografía sobre gel de sílice usando un gradiente de EtOAc (de 0 % a 20 %) en heptano para permitir la obtención de 1.7 g de 4-metil-6-(trifluorometoxi)-1 H-indol 9g en forma de aceite naranja.
Síntesis del intermedio 9h:
A 0 °C, se añadió gota a gota BH3-Piridina (1.2 ml, 11.6 mmol) a una disolución de 4-metil-6-(trifluorometoxi)-1 H-indol 9g (0.5 g, 2.32 mmol) en EtOH (3 ml). Se añadió lentamente HCl 6 N (6 ml) mientras se mantenía la temperatura de reacción por debajo de 10 °C. La mezcla se agitó a 0 °C durante 3 h. Se añadió agua (12 ml) y la mezcla se basificó hasta pH 8-9 con una disolución concentrada de NaOH en agua (la temperatura de reacción se mantuvo por debajo de 20 °C). La mezcla se extrajo con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. Se añadió tolueno y la disolución se concentró a presión reducida para dar 450 g de 4-metil-6-(trifluorometoxi)indolina 9h.
Síntesis del Compuesto 9 y separación quiral en los Enantiómeros 9A y 9B:
En un flujo de N2 a 5 °C, se añadió anhídrido propilfosfónico (1.87 ml, 3.11 mmol) a una mezcla de 4-metil-6-(trifluorometoxi)indolina 9h (450 mg, 2.07 mmol), ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)-amino)acético 3f (729 mg, 2.07 mmol) y diisopropiletilamina (753 pl, 4.56 mmol) en DMF (20 ml). La mezcla se agitó a temperatura ambiente durante 7 h. Se añadió agua y la mezcla se extrajo con EtOAc. La capa orgánica se lavó con una solución de K2CO3 al 10 % en agua, después con agua, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice, (15-40 pm, 24 g, CH2Cl2/MeOH, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad, para dar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1 -(4-metil-6-(trifluorometoxi)indolin-1 -il)etanona racémica (Compuesto 9 , 411 mg). Los enantiómeros del Compuesto 9 se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 pm 250 x 20 mm, Fase móvil: 50 % de CO2, 50 % de iPrOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (180 mg) se solidificó en heptano/éter diisopropílico para proporcionar el Enantiómero 9A (121 mg). El segundo enantiómero eluido (180 mg) se solidificó en heptano/éter diisopropílico para proporcionar el Enantiómero 9B (132 mg).
Enantiómero 9A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 2.20 (s, 3 H) 2.98 - 3.17 (m, 2 H) 3.59 - 3.69 (m, 5 H) 3.79 - 3.90 (m, 2 H) 4.05 (td, J=10.6, 6.9 Hz, 1 H) 4.52 (td, J=10.5, 6.1 Hz, 1 H) 4.77 (t, J=5.5 Hz, 1 H) 5.55 (d, J=8.8 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.94 (d, J=1.9 Hz, 2 H) 6.43 (d, J=8.8 Hz, 1 H) 6.88 (s, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 7.88 (s, 1 H)
LC/MS (método LC-A): TR 3.48 min, MH+ 551
[a]D20: -40.6° (c 0.2067, DMF)
SFC quiral (método SFC-G): tR 2.08 min, MH+ 551, pureza quiral 100 %.
Enantiómero 9B:
1H RMN (500 MHz, DMSO-afe) 5 ppm 2.20 (s, 3 H) 2.98 - 3.17 (m, 2 H) 3.59 - 3.67 (m, 5 H) 3.79 - 3.90 (m, 2 H) 4.05 (td, J=10.6, 6.9 Hz, 1 H) 4.52 (td, J=10.5, 6.1 Hz, 1 H) 4.77 (t, J=5.5 Hz, 1 H) 5.55 (d, J=8.5 Hz, 1 H) 5.76 (t, J=1.9 Hz, 1 H) 5.94 (d, J=1.9 Hz, 2 H) 6.43 (d, J=8.8 Hz, 1 H) 6.88 (s, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 7.88 (s, 1 H)
LC/MS (método LC-A): TR 3.48 min, MH+ 551
[a]D20 42.6° (c 0.2392, DMF)
SFC quiral (método SFC-G): tR 3.34 min, MH+ 551, pureza quiral 99.7 %.
Ejemplo 10 : síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(4-metil-6-(trifluorometil)indolin-1-il)etanona (Compuesto 10) y separación quiral en los Enantiómeros 10A y 10B.

Síntesis del intermedio 10a:
Se añadió Pd/C (10 %) (1.18 g) a una solución de 1-bencil-4-metil-6-(trifluorometil)indolina [CAS 1156512-79-6] (11.8 g, 40.5 mmol) en AcOH (11.8 ml) y MeOH (118 ml). La reacción se agitó a temperatura ambiente durante 12 h en una atmósfera de H2. La mezcla se filtró a través de un lecho de Celite® y se concentró a presión reducida. El residuo se
recogió con CH2CI2, se lavó con agua, salmuera, se secó sobre MgSÜ4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida sobre gel de sílice (heptano/EtOAc, 9/1). Las fracciones puras se combinaron y el disolvente se evaporó a sequedad para dar 8.2 g de 4-metil-6-(trifluorometil)indolina 10a.
Síntesis del Compuesto 10 y separación quiral en los Enantiómeros 10A y 10B:
Una mezcla de 4-metil-6-(trifluorometil)indolina 10a (515 mg, 2.56 mmol), ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 3f (900 mg, 2.56 mmol), diisopropiletilamina (1.27 ml, 7.67 mmol) y HATU (1.46 g, 3.84 mmol) en DMF (7 ml) se agitó a temperatura ambiente durante 18 h. Se añadió agua y la mezcla se extrajo con EtOAc. La capa orgánica se lavó con agua (varias veces), se secó sobre MgSO4, se filtró, y el disolvente se evaporó a presión reducida. La purificación se llevó a cabo mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, 50/50 de heptano/EtOAc). Esta fracción se combinó con otro lote (cantidad total: 640 mg) y se purificó adicionalmente por cromatografía de fase inversa (fase estacionaria: X-Bridge® C18 10 gm 30 x 150 mm, Fase móvil: gradiente del 60 % de NH4HCO3 al 0.2 %, CH3CN al 40 % al 0 % de NH4HCO3 al 0.2 %, CH3CN al 100 %). Las fracciones puras se combinaron y se evaporaron a sequedad, para dar 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(4-metil-6-(trifluorometil)indolin-1-il)-etanona racémica (Compuesto 10, 425 mg). Los enantiómeros del Compuesto 10 se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 gm 250 x 20 mm, Fase móvil: 50 % de CO2, 50 % de EtOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (180 mg) se solidificó en heptano/éter diisopropílico para proporcionar el Enantiómero 10A (145 mg). El segundo enantiómero eluido (170 mg) se solidificó en heptano/éter diisopropílico para proporcionar el Enantiómero 10B (113 mg).
Enantiómero 10A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 2.25 (s, 3 H) 3.05 - 3.24 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.80 - 3.90 (m, 2 H) 4.04 (td, J=10.6, 6.9 Hz, 1 H) 4.54 (td, J=10.5, 6.1 Hz, 1 H) 4.78 (t, J=5.5 Hz, 1 H) 5.57 (d, J=8.5 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.95 (d, J=0.9 Hz, 2 H) 6.42 (d, J=8.5 Hz, 1 H) 7.25 (s, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.56 (d, J=8.5 Hz, 2 H) 8.23 (s, 1 H)
LC/MS (método LC-A): TR 3.43 min, MH+ 535
[a]D20: -46.2° (c 0.2275, DMF)
SFC quiral (método SFC-I): tR 2.26 min, MH+ 535, pureza quiral 100 %.
Enantiómero 10B:
1H RMN (500 MHz, DMSO-ds) 5 ppm 2.25 (s, 3 H) 3.05 - 3.24 (m, 2 H) 3.59 - 3.68 (m, 5 H) 3.80 - 3.91 (m, 2 H) 4.04 (td, J=10.5, 7.1 Hz, 1 H) 4.54 (td, J=10.4, 6.3 Hz, 1 H) 4.78 (t, J=5.5 Hz, 1 H) 5.57 (d, J=8.5 Hz, 1 H) 5.76 (t, J=2.0 Hz, 1 H) 5.95 (d, J=0.9 Hz, 2 H) 6.42 (d, J=8.8 Hz, 1 H) 7.25 (s, 1 H) 7.44 (d, J=8.5 Hz, 2 H) 7.56 (d, J=8.2 Hz, 2 H) 8.23 (s, 1 H)
LC/MS (método LC-A): TR 3.43 min, MH+ 535
[a]D20: 43.0° (c 0.2092, DMF)
SFC quiral (método SFC-I): tR 3.61 min, MH+ 535, pureza quiral 100 %.
Ejemplo 11 : síntesis de 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona (Compuesto 11) y separación quiral en los Enantiómeros 11A y 11B.

A -78 °C, en un flujo de N2, se añadió gota a gota LiHMDS 1.5 M en THF (6.2 ml, 9.32 mmol) a una mezcla de 2-(4-cloro-2-metoxifenil)acetato de metilo [CAS 193290-23-2] (1 g, 4.66 mmol) en THF (30 ml). Se añadió gota a gota una solución de TMSCl (0.95 ml, 7.45 mmol) en THF (10 ml). La mezcla se agitó durante 15 min a -78 °C y se añadió gota a gota NBS (0.912 g, 5.13 mmol) en THF (10 ml). Después de agitar durante 2 h a -78 °C, la reacción se interrumpió con una solución saturada de NH4CL La mezcla se extrajo con EtOAc, se secó sobre MgSCU, se filtró, y el disolvente se evaporó a presión reducida para dar 2-bromo-2-(4-cloro-2-metoxifenil)acetato de metilo 11a (1.4 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del intermedio 11b:
Una mezcla de 2-bromo-2-(4-cloro-2-metoxifenil)acetato de metilo 11a (0.5 g, 1.71 mmol), 2-(3-amino-5-metoxifenoxi)etanol [CAS 725237-16-1] (328 mg, 1.79 mmol), trimetilamina (355 pl, 2.56 mmol) en CH3CN (10 ml) se agitó a 50 °C durante 12 h. La mezcla se diluyó con EtOAc y se lavó con agua. La fase orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 40 g, CH2Cl2/heptano: 99/1). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acetato de metilo 11b (510 mg).
Síntesis del intermedio 11c:
Se agitó 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acetato de metilo 11b (1.2 g, 3.03 mmol) y LiOH (382 mg, 9.10 mmol) en THF/agua (1/1) (20 ml) se agitó a temperatura ambiente durante 1 h. La mezcla se diluyó con agua. Se acidificó lentamente la fase acuosa con HCl 3 N y se extrajo con EtOAc. Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y el disolvente se evaporó a presión reducida para dar ácido 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)-amino)acético 11c (1.12 g). El compuesto se usó como tal en la siguiente etapa.
Síntesis del Compuesto 11 y separación quiral en los Enantiómeros 11A y 11B:
Se añadió HATU (0.692 g, 1.82 mmol) a una mezcla de 5-metoxi-6-(trifluorometil)indolina 7c (264 mg, 1.21 mmol), ácido 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 11c (520 mg, 1.36 mmol) y diisopropiletilamina (0.602 ml, 3.64 mmol) en DMF (14 ml). La mezcla resultante se agitó a temperatura ambiente durante 12 h. La mezcla se diluyó con agua y EtOAc. La capa orgánica se separó, se lavó con una solución al 10 %
de K2CO3 en agua y después con salmuera. La fase orgánica se secó con MgSÜ4, se filtró y el disolvente se evaporó a presión reducida. El producto en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 40 g, CH2Cl2/MeOH, 99.5/0.5). La fracción pura se combinaron y se evaporaron a sequedad para proporcionar 2-(4-cloro-2-metoxifenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometil)indolin-1-il)etanona racémica (Compuesto 11,398 mg). Este lote se combinó con otro lote (cantidad total: 535 mg). Los enantiómeros se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IA 5 pm 250 x 20 mm, Fase móvil: 70 % de CO2, 30 % de iPrOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (240 mg) se cristalizó en CHaCN/éter diisopropílico para dar el Enantiómero 11A (194 mg). El segundo enantiómero eluido (240 mg) se cristalizó en CH3CN/éter diisopropílico para dar el Enantiómero 11B (189 mg).
Compuesto 11:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.19 - 3.29 (m, 2 H) 3.59 - 3.69 (m, 5 H) 3.78 - 3.88 (m, 5 H) 3.91 (s, 3 H) 3.95 -4.04 (m, 1 H) 4.31 - 4.41 (m, 1 H) 4.80 (t, J=5.4 Hz, 1 H) 5.59 (d a, J=8.5 Hz, 1 H) 5.73 - 5.78 (m, 1 H) 5.87 (s a, 2 H) 6.41 (d a, J=8.5 Hz, 1 H) 7.03 (dd, J=8.2, 1.3 Hz, 1 H) 7.15 (d, J=1.3 Hz, 1 H) 7.25 (s, 1 H) 7.33 (d, J=8.2 Hz, 1 H) 8.33 (s, 1 H)
LC/MS (método LC-A): TR 3.22 min, MH+ 581
Punto de fusión: 224 °C
Enantiómero 11A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.16 - 3.30 (m, 2 H) 3.60 - 3.68 (m, 5 H) 3.77 - 3.88 (m, 5 H) 3.90 (s, 3 H) 3.99 (td, J=10.2, 7.3 Hz, 1 H) 4.35 (td, J=10.2, 6.6 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.58 (d, J=8.5 Hz, 1 H) 5.73 - 5.78 (m, 1 H) 5.87 (s, 2 H) 6.41 (d, J=8.8 Hz, 1 H) 7.02 (dd, J=8.2, 1.9 Hz, 1 H) 7.14 (d, J=1.9 Hz, 1 H) 7.24 (s, 1 H) 7.32 (d, J=8.5 Hz, 1 H) 8.33 (s, 1 H)
LC/MS (método LC-A): TR 3.25 min, MH+ 581
[a]D20: -31.4° (c 0.274, DMF)
SFC quiral (método SFC-H): tR 3.60 min, MH+ 581, pureza quiral 100 %.
Punto de fusión: 175°C
Enantiómero 11B:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.17 - 3.30 (m, 2 H) 3.59 - 3.68 (m, 5 H) 3.78 - 3.88 (m, 5 H) 3.91 (s, 3 H) 3.99 (td, J=10.2, 7.3 Hz, 1 H) 4.35 (td, J=10.2, 6.6 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.58 (d, J=8.5 Hz, 1 H) 5.73 - 5.78 (m, 1 H) 5.87 (s, 2 H) 6.41 (d, J=8.8 Hz, 1 H) 7.02 (dd, J=8.2, 1.9 Hz, 1 H) 7.14 (d, J=1.9 Hz, 1 H) 7.24 (s, 1 H) 7.32 (d, J=8.2 Hz, 1 H) 8.33 (s, 1 H)
LC/MS (método LC-A): TR 3.25 min, MH+ 581
[a]D20: 29.4° (c 0.272, DMF)
SFC quiral (método SFC-H): tR 4.96 min, MH+ 581, pureza quiral 100 %.
Punto de fusión: 175°C
Ejemplo 12 : síntesis de 2-(4-clorofenil)-1-(5-fluoro-6-(trifluorometil)indolin-1-il)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)etanona (Compuesto 12) y separación quiral en los Enantiómeros 12A y 12B.

Síntesis del intermedio 12a:
A 0 °C, se añadió gota a gota BH3-Piridina (10.5 ml, 103.5 mmol) a una disolución de 5-fluoro-6-(trifluorometil)-1 H-indol [CAS 1493800-10-4](7 g, 34.5 mmol) en EtOH (45 ml). Se añadió lentamente HCl 6 N (150 ml) mientras se mantenía la temperatura de reacción por debajo de 10 °C. La mezcla se agitó a 0 °C durante 3 h. Se añadió agua (210 ml) y la mezcla se basificó hasta pH 8-9 con una disolución concentrada de NaOH en agua (la temperatura de reacción se mantuvo por debajo de 20 °C). La mezcla se extrajo con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. Se añadió tolueno y la solución se concentró a presión reducida. La purificación se llevó a cabo mediante cromatografía ultrarrápida sobre gel de sílice (15-40 pm, 120 g, 85/15 de heptano/EtOAc). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 3.5 g de 5-fluoro-6-(trifluorometil)indolina 12a.
Síntesis del Compuesto 12 y separación quiral en los Enantiómeros 12A y 12B:
En un flujo N2, a 5 °C, se añadió gota a gota anhídrido propilfosfónico (1.76 ml, 2.93 mmol) a una mezcla de 5-fluoro-6-(trifluorometil)indolina 12a (400 mg, 1.95 mmol), ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)-amino)acético 3f (823 mg, 2.34 mmol) y diisopropiletilamina (709 pl, 4.29 mmol) en DMF (30 ml). La mezcla se agitó a temperatura ambiente durante 5 h. Se añadió agua y el precipitado se eliminó por filtración y se lavó con una solución al 10 % de K2CO3 en agua. El sólido se recogió con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice, (15-40 pm, 40 g, CH2Cl2/MeOH, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad, para dar 2-(4-clorofenil)-1 -(5-fluoro-6-(trifluorometil)indolin-1 -il)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)etanona racémica (Compuesto 12, 825 mg). La cristalización en CH3CN/éter diisopropílico proporcionó el Compuesto 12 (448 mg) en forma de una fracción cristalina. Los enantiómeros del Compuesto 12 se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralcel® OJ-H 5 pm 250 x 20 mm, Fase móvil: 60 % de CO2, 40 % de EtOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (193 mg) se solidificó en heptano/éter diisopropílico para dar el Enantiómero 12A (164 mg). El segundo enantiómero eluido (190 mg) se solidificó en heptano/éter diisopropílico para dar el Enantiómero 12B (131 mg).
Compuesto 12:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.18 - 3.31 (m, 2 H) 3.62 (s, 3 H) 3.63- 3.67 (m, 2 H) 3.80 - 3.89 (m, 2 H) 4.03 (td, J=10.3, 7.4 Hz, 1 H) 4.54 (td, J=10.2, 6.3 Hz, 1 H) 4.79 (t a, J=5.0 Hz, 1 H) 5.57 (d, J=8.5 Hz, 1 H) 5.76 (s, 1 H) 5.94 (s, 2 H) 6.44 (d, J=8.8 Hz, 1 H) 7.42 - 7.48 (m, 3 H) 7.55 (d, J=8.2 Hz, 2 H) 8.39 (d, J=6.6 Hz, 1 H)
LC/MS (método LC-A): TR 3.37 min, MH+ 539
Punto de fusión: 130°C
Enantiómero 12A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.17 - 3.32 (m, 2 H) 3.62 (s, 3 H) 3.64 (c, J=5.6 Hz, 2 H) 3.79 - 3.90 (m, 2 H) 4.03 (td, J=10.2, 7.3 Hz, 1 H) 4.54 (td, J=10.4, 6.3 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.57 (d, J=8.5 Hz, 1 H) 5.76 (s, 1 H) 5.94 (s, 2 H) 6.44 (d, J=8.8 Hz, 1 H) 7.41 - 7.48 (m, 3 H) 7.55 (d, J=8.5 Hz, 2 H) 8.39 (d, J=6.3 Hz, 1 H)
LC/MS (método LC-A): TR 3.37 min, MH+ 539
[a]D20: 50.2° (c 0.299, DMF)
SFC quiral (método SFC-D): tR 1.91 min, MH+ 539, pureza quiral 100 %.
Enantiómero 12B:
1H RMN (500 MHz, DMSO-ds) 5 ppm 3.18 - 3.32 (m, 2 H) 3.62 (s, 3 H) 3.65 (c, J=5.3 Hz, 2 H) 3.78 - 3.90 (m, 2 H) 4.03 (td, J=10.2, 7.3 Hz, 1 H) 4.54 (td, J=10.2, 6.3 Hz, 1 H) 4.79 (t, J=5.4 Hz, 1 H) 5.57 (d, J=8.8 Hz, 1 H) 5.76 (s, 1 H) 5.94 (s, 2 H) 6.44 (d, J=8.5 Hz, 1 H) 7.41 - 7.49 (m, 3 H) 7.55 (d, J=8.2 Hz, 2 H) 8.39 (d, J=6.6 Hz, 1 H)
LC/MS (método LC-A): TR 3.38 min, MH+ 539
[a]D20: -51.0° (c 0.3, DMF)
SFC quiral (método SFC-D): tR 3.93 min, MH+ 539, pureza quiral 99.52 %.
Ejemplo_____13: síntesis de 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometoxi)indolin-1-il)etanona (Compuesto 13) y separación quiral en los Enantiómeros 13A y 13B.

Una solución de 4-metoxi-3-(trifluorometoxi)anilina [CAS 647855-21-8] (3.1 g, 15.0 mmol) en tolueno (50 ml) se trató con W-bromosuccinimida (2.8 g, 15.7 mmol) a 5 °C y la mezcla resultante se agitó a 5-10 °C durante 2 h. La mezcla se diluyó con agua y se extrajo con EtOAc. Los extractos orgánicos combinados se secaron sobre MgSO4, se filtraron y se evaporaron a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 24 g, heptano/EtOAc, 95/5 a 90/10) Las fracciones puras se combinaron y se evaporaron a sequedad para dar 2-bromo-4-metoxi-5-(trifluorometoxi)anilina 13a (2.5 g).
Síntesis del intermedio 13b:
Una solución de 2-bromo-4-metoxi-5-(trifluorometoxi)anilina 13a (2.72 g, 9.51 mmol) en DMF (30 ml) se desgasificó con N2 durante 15 min. Se añadieron diclorobis(trifenilfosfina)paladio (II) (667 mg, 0.95 mmol), yoduro de cobre (I) (362 mg, 1.90 mmol), trietilamina (3.96 ml, 28.53 mmol) y trimetilsililacetileno (3.95 ml, 28.5 mmol). La mezcla de reacción se calentó a 70 °C durante 12 h en un flujo de N2. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con H2O y se extrajo con EtOAc. Las fases orgánicas se combinaron, se secaron sobre MgSO4, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 80 g, heptano/EtOAc, 85/15). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 4-metoxi-5-(trifluorometoxi)-2-((trimetilsilil)etinil)anilina 13b (1.4 g).
Síntesis del intermedio 13c:
A una solución de 4-metoxi-5-(trifluorometoxi)-2-((trimetilsilil)etinil)anilina 13b (1.2 g, 3.96 mmol) en NMP (11 ml) en un flujo de N2, se le añadió en una porción fBuOK (1.33 g, 11.9 mmol). La mezcla de reacción se calentó a 80 °C durante 4 h, después se vertió en hielo/agua y se acidificó con HCl 3 N hasta pH 4-5. Se extrajo la mezcla de reacción con EtOAc. Las fases orgánicas se combinaron, se lavaron con H2O, se secaron sobre MgSO4, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice (15-40 gm, 40 g, heptano/EtOAc, 85/15). Las fracciones puras se combinaron y se evaporaron a sequedad para dar 5-metoxi-6-(trifluorometoxi)-1 H-indol 13c (490 mg).
Síntesis del intermedio 13d:
A 0 °C, se añadió gota a gota BH3-Piridina (10.5 ml, 103.82 mmol) a una disolución de 5-metoxi-6-(trifluorometoxi)-1 H-indol 13c (8 g, 34.6 mmol) en EtOH (45 ml). Se añadió lentamente HCl 6 N (6 ml) mientras se mantenía la temperatura por debajo de 10 °C. La mezcla se agitó a 0 °C durante 3 h. Se añadió agua (210 ml) y la mezcla se basificó hasta pH 8-9 con una disolución concentrada de NaOH en agua (la temperatura de reacción se mantuvo por debajo de 20 °C). La mezcla se extrajo con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSO4, se filtró y el disolvente se evaporó a presión reducida. Se añadió tolueno y la solución se concentró a presión reducida para dar 7.5 g de 5-metoxi-6-(trifluorometoxi)indolina 13d.
Síntesis del Compuesto 13 y separación quiral en los Enantiómeros 13A y 13B:
En un flujo de N2 a 5 °C, se añadió gota a gota anhídrido propilfosfónico (2.1 ml, 3.35 mmol) a una mezcla de 5-metoxi-6-(trifluorometoxi)indolina 13d (552 mg, 2.37 mmol), ácido 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)acético 3f (1 g, 2.84 mmol) y diisopropiletilamina (861 gl, 5.21 mmol) en DMF (40 ml). La mezcla se agitó a temperatura ambiente durante 5 h. Se añadió agua y el precipitado se eliminó por filtración y se lavó con una
solución al 10 % de K2CO3 en agua. El sólido se recogió con EtOAc. La fase orgánica se lavó con agua, se secó sobre MgSÜ4, se filtró y el disolvente se evaporó a presión reducida. La purificación se realizó por cromatografía ultrarrápida sobre gel de sílice, (15-40 gm, 40 g, ChbCL/MeOH, 99.5/0.5). Las fracciones puras se combinaron y se evaporaron a sequedad, para dar, después de la cristalización en éter diisopropílico, 2-(4-clorofenil)-2-((3-(2-hidroxietoxi)-5-metoxifenil)amino)-1-(5-metoxi-6-(trifluorometoxi)indolin-1-il)etanona racémica (Compuesto 13, 970 mg). Los enantiómeros del Compuesto 13 se separaron mediante SFC quiral preparativa (Fase estacionaria: Chiralpak® IC 5 gm 250 x 30 mm, Fase móvil: 55 % de CO2, 45 % de EtOH (+ 0.3 % de iPrNH2)). El primer enantiómero eluido (400 mg) se solidificó en heptano/éter diisopropílico para dar el Enantiómero 13A (332 mg). El segundo enantiómero eluido (397 mg) se solidificó en heptano/éter diisopropílico para dar el Enantiómero 13B (344 mg).
Compuesto 13:
1H RMN (500 MHz, ODMSO-afe) 5 ppm 3.10 - 3.26 (m, 2 H) 3.62 (s, 3 H) 3.65 (c, J=5.4 Hz, 2 H) 3.81 (s, 3 H) 3.82 -3.88 (m, 2 H) 4.03 (td, J=10.3, 7.1 Hz, 1 H) 4.49 (td, J=10.3, 6.5 Hz, 1 H) 4.80 (t, J=5.4 Hz, 1 H) 5.53 (d, J=8.8 Hz, 1 H) 5.75 (s, 1 H) 5.94 (d, J=1.6 Hz, 2 H) 6.45 (d, J=8.5 Hz, 1 H) 7.20 (s, 1 H) 7.44 (d, J=8.2 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.06 (s, 1 H)
LC/MS (método LC-A): TR 3.30 min, MH+ 567
Punto de fusión: 165°C
Enantiómero 13A:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.10 - 3.26 (m, 2 H) 3.62 (s, 3 H) 3.65 (c, J=5.4 Hz, 2 H) 3.81 (s, 3 H) 3.82 - 3.88 (m, 2 H) 4.03 (td, J=10.3, 7.1 Hz, 1 H) 4.49 (td, J=10.3, 6.5 Hz, 1 H) 4.80 (t, J=5.4 Hz, 1 H) 5.53 (d, J=8.8 Hz, 1 H) 5.75 (s, 1 H) 5.94 (d, J=1.6 Hz, 2 H) 6.45 (d, J=8.5 Hz, 1 H) 7.20 (s, 1 H) 7.44 (d, J=8.2 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.06 (s, 1 H)
LC/MS (método LC-A): TR 3.31 min, MH+ 567
[a]D20: 43.2° (c 0.285, DMF)
SFC quiral(método SFC-J): tR 1.82 min, MH+ 567, pureza quiral 100 %.
Enantiómero 13B:
1H RMN (500 MHz, DMSO-afe) 5 ppm 3.09 - 3.26 (m, 2 H) 3.61 (s, 3 H) 3.64 (c, J=5.3 Hz, 2 H) 3.81 (s, 3 H) 3.82 - 3.88 (m, 2 H) 4.02 (td, J=10.4, 6.9 Hz, 1 H) 4.48 (td, J=10.3, 6.1 Hz, 1 H) 4.79 (t, J=5.5 Hz, 1 H) 5.53 (d, J=8.5 Hz, 1 H) 5.75 (s, 1 H) 5.94 (d, J=1.6 Hz, 2 H) 6.44 (d, J=8.5 Hz, 1 H) 7.20 (s, 1 H) 7.43 (d, J=8.2 Hz, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.06 (s, 1 H)
LC/MS (método LC-A): TR 3.31 min, MH+ 567
[a]D20: -45.9° (c 0.301, DMF)
SFC quiral(método SFC-J): tR 3.15 min, MH+ 567, pureza quiral 98.96 %.
Tabla: compuestos preparados como se ha descrito anteriormente
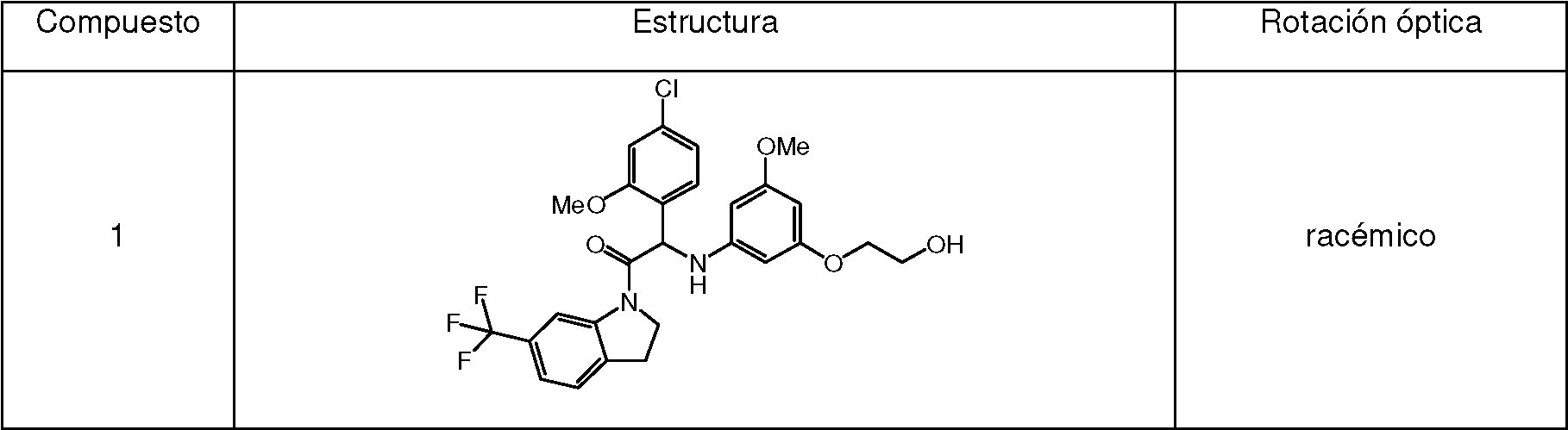



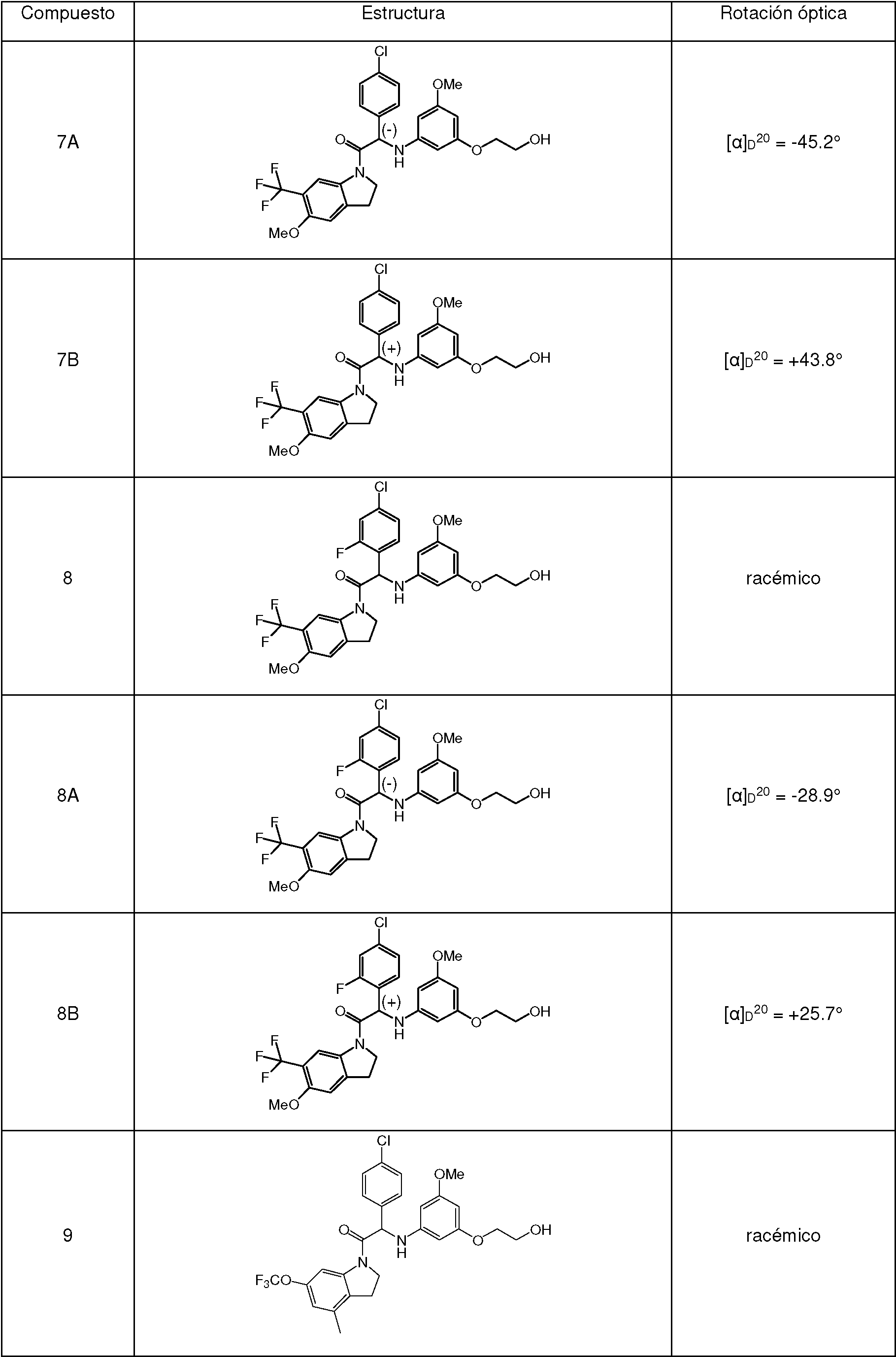



ACTIVIDAD ANTIVIRAL DE LOS COMPUESTOS DE LA INVENCIÓN
Ensayo an tiv ira l de l D EN V-2
Se evaluó la actividad antiviral de todos los compuestos de la invención frente a la cepa 16681 del DENV-2, la cual se marcó con la proteína fluorescente verde potenciada (eGPF). El medio de cultivo consiste en un medio esencial mínimo complementado con un 2 % de suero bovino fetal inactivado térmicamente, un 0.04 % de gentamicina (50 mg/ml) y L-glutamina 2 mM. Se suspendieron células Vero, obtenidas de ECACC, en el medio de cultivo y se añadieron 25 pl a placas de 384 pocillos (2500 células/pocillo), que ya contenían los compuestos antivirales. Habitualmente, estas placas contienen diluciones en serie con un factor de 5, de 9 etapas de dilución del compuesto de prueba con 200 veces la concentración final en DMSO al 100 % (200 nl). Además, cada concentración del compuesto se evalúa por cuadruplicado (intervalo de concentración final: 25 pM - 0.000064 pM o 2.5 pM - 0.0000064 pM para los compuestos más activos). Finalmente, cada placa contiene pocillos que se han asignado como controles del virus (que contienen células y virus en ausencia del compuesto), controles de las células (que contienen células en ausencia de virus y compuesto) y controles del medio (que contiene medio en ausencia de células, virus y compuestos). A los pocillos asignados como control del medio, se añadieron 25 pl de medio de cultivo en lugar de células Vero. Una vez que se añadieron las células a las placas, las placas se incubaron durante 30 minutos a temperatura ambiente para permitir que las células se distribuyeran de manera homogénea dentro de los pocillos. A continuación, las placas se incubaron en una incubadora totalmente humidificada (37 °C, 5 % de CO2) hasta el día siguiente. Después, se añadió la cepa 16681 del DENV-2, marcada con eGFP, con una multiplicidad de infección (MDI) de 0.5. Por lo tanto, se añadieron 15 pl de suspensión de virus a todos los pocillos que contenían compuesto de prueba y a los pocillos asignados como control del virus. En paralelo, se añadieron 15 pl de medio de cultivo a los controles del medio y de las células. A continuación, las placas se incubaron durante 3 días en una incubadora totalmente humidificada (37 °C, 5 % de CO2). El día de la lectura, se midió la fluorescencia de eGFP utilizando un microscopio de fluorescencia automático a 488 nm (láser azul). Utilizando un sistema de LIMS propio, se calcularon las curvas de respuesta a la dosis de inhibición para cada compuesto y se determinó la concentración eficaz que produce la mitad del efecto máximo (CE50). Por lo tanto, la inhibición porcentual (I) para cada concentración de prueba se calcula utilizando la siguiente fórmula: I = 100*(St-Scc)/(Svc-Scc); St, Scc y Svc son las cantidades de señal de eGFP en los pocillos del compuesto de prueba, control de las células y control del virus, respectivamente. La CE50 representa la concentración de un compuesto con la que se inhibe la replicación del virus en un 50 %, que se mide como una reducción del 50 % de la intensidad de fluorescencia de eGFP en comparación con el control del virus. La CE50 se calcula utilizando una interpolación lineal (Tabla 1).
En paralelo, se evaluó la toxicidad de los compuestos en las mismas placas. Una vez que se realizó la lectura de la señal de eGFP, se añadieron 40 pl de ATPlite, un tinte de viabilidad celular, a todos los pocillos de las placas de 384 pocillos. El ATP está presente en todas las células metabólicamente activas y su concentración se reduce muy rápidamente cuando las células experimentan necrosis o apoptosis. El sistema de ensayo ATPLite se basa en la producción de luz provocada por la reacción de ATP con luciferasa y D-luciferina añadidas. Las placas se incubaron durante 10 minutos a temperatura ambiente. A continuación, las placas se midieron en un ViewLux. También se determinó la concentración que produce la mitad del efecto citotóxico máximo (CC50), definida como la concentración requerida para reducir la señal de luminiscencia en un 50 % en comparación con la de los pocillos de control de las células. Finalmente, se determinó el índice de selectividad (IS) para los compuestos, que se calculó de la siguiente manera: IS = CC50/CE50.
Tabla 1: CEso. CCso e IS para los compuestos de la invención en el ensayo antiviral del DENV-2


Ensayo de PCR cuantitativa con retrotranscriptasa tetravalente (RT-qPCR)
Se evaluó la actividad antiviral de los compuestos de la invención frente a la cepa TC974#666 del DENV-1 (NCPV), la cepa 16681 del DENV-2, la cepa H87 del DENV-3 (NCPV) y la cepa H241 (NCPV) del DENV-4 en un ensayo de Rt -qPCR. Por lo tanto, se infectaron células Vero con el DENV-1 o -2 o -3 o -4 en presencia o ausencia de compuestos de prueba. El día 3 después de la infección, las células se sometieron a lisis y los lisados celulares se utilizaron para preparar ADNc de tanto una diana viral (la 3’UTR del DENV; Tabla 2) como un gen de referencia celular (p-actina, Tabla 2). Posteriormente, se realizó una PCR doble a tiempo real en un instrumento Lightcycler480. El valor de Cp generado es inversamente proporcional a la cantidad de expresión de ARN de estas dianas. La inhibición de la replicación del DENV por un compuesto de prueba da como resultado un desplazamiento de Cp para el gen 3’UTR. Por otra parte, si un compuesto de prueba es tóxico para las células, se observará un efecto similar en la expresión de p-actina. El método de AACp comparativo se utiliza para calcular CE50, que se basa en la expresión génica relativa del gel diana (3’UTR) normalizada con el gen constitutivo celular (p-actina). Además, los valores de CC50 se determinan basándose en los valores de Cp adquiridos para el gen constitutivo de p-actina.
Tabla 2 : Cebadores y sondas utilizados para la RT-PCR cuantitativa a tiempo real.

El medio de cultivo consistió en un medio esencial mínimo complementado con un 2 % de suero bovino fetal inactivado térmicamente, un 0.04 % de gentamicina (50 mg/ml) y L-glutamina 2 mM. Se suspendieron células Vero, obtenidas de ECACC, en el medio de cultivo y se añadieron 75 pl/pocillo en placas de 96 pocillos (10 000 células/pocillo), que ya contenían los compuestos antivirales. Habitualmente, estas placas contienen una dilución en serie con un factor de 5, de 9 etapas de dilución del compuesto de prueba con 200 veces la concentración final en DMSO al 100 % (500 nl; intervalo de concentración final: 25 pM - 0.000064 pM o 2.5 pM - 0.0000064 pM para los compuestos más activos). Además, cada placa contiene pocillos que se han asignado como controles del virus (que contienen células y virus en ausencia de compuesto) y controles de las células (que contienen células en ausencia de virus y compuesto). Una vez se añadieron las células a las placas, las placas se incubaron en una incubadora totalmente humidificada (37 °C, 5 % de CO2) hasta el día siguiente. Se diluyeron los serotipos 1, 2, 3 y 4 de los virus del dengue con el fin de obtener un Cp de ~22-24 en el ensayo. Por lo tanto, se añadieron 25 pl de suspensión de virus a todos los pocillos que contenían
compuesto de prueba y a los pocilios asignados como control del virus. En paralelo, se añadieron 25 gl de medio de cultivo a los controles de las células. A continuación, las placas se incubaron durante 3 días en una incubadora totalmente humidificada (37 °C, 5 % de CO2). Después de 3 días, se retiró el sobrenadante de los pocillos y las células se lavaron dos veces con PBS enfriado con hielo (~ 100 gl). Los sedimentos de células dentro de las placas de 96 pocillos se almacenaron a -80 °C durante al menos 1 día. A continuación, se extrajo el ARN utilizando el kit de lisis Cells-to-CTTM, de acuerdo con las directrices del fabricante (Life Technologies). Los lisados celulares pueden almacenarse a -80 °C o utilizarse inmediatamente en la etapa de retrotranscripción.
En la preparación de la etapa de retrotranscripción, se preparó la mezcla A (tabla 3A) y se dispensaron 7.57 gl/pocillo en una placa de 96 pocillos. Después de la adición de 5 gl de los lisados celulares, se realizó una etapa de desnaturalización de cinco minutos a 75 °C (tabla 3B). Posteriormente, se añadieron 7.43 gl de mezcla B (tabla 3C) y se inició la etapa de retrotranscripción (tabla 3D) para generar ADNc.
Finalmente, se preparó una mezcla de RT-qPCR, mezcla C (tabla 4A), y se dispensaron 22.02 gl/pocillo en placas LightCycler qPCR de 96 pocillos en las que se añadieron 3 gl de ADNc y la qPCR se realizó de acuerdo con las condiciones de la tabla 4B en un LightCycler 480.
Utilizando el software LightCycler y un sistema LIMS propio, se calcularon las curvas de respuesta a la dosis para cada compuesto y se determinaron la concentración eficaz que produce la mitad del efecto máximo (CE50) y la concentración que produce la mitad del efecto citotóxico máximo (CC50) (Tablas 5-8).
Tabla 3: Síntesis de ADNc usando la Mezcla A, desnaturalización, la Mezcla B y retrotranscripción.
A Mezcla A

Volumen mezcla/pocillo (gl) 7.57


Etapa de
B desnaturalización:

C Mezcla B


Volumen total de la mezcla
7.43
(pl)
D Protocolo de síntesis de ADNc

Tabla 4: Protocolo y mezcla de qPCR.

Volumen de mezcla/tubo (pl) 22.02

Protocolo qPCR3



Tabla 6: CEso. CC50 e IS para los compuestos frente al serotipo 2 en los ensayos de RT-qPCR


Tabla 7: CE50. CC50 e IS para los compuestos frente al serotipo 3 en los ensayos de RT-qPCR

Tabla 8: CEso. CCso e IS para los compuestos frente al serotipo 4 en los ensayos de RT-qPCR

Ejemplos de la técnica anterior.
Se han ensayado los compuestos (56) y (170) divulgados en el documento WO-2013/045516 en un ensayo antiviral del DENV-2 análogo a los compuestos de la presente invención y su actividad obtenida se enuncia a continuación.

Claims (14)
1. Un compuesto que tiene la fórmula (I)

compuestos se seleccionan del grupo que comprende:


2. Una composición farmacéutica que comprende un compuesto de acuerdo con la reivindicación 1 o su forma estereoisomérica, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo junto con uno o más excipientes, diluyentes o portadores farmacéuticamente aceptables.
3. Un compuesto de acuerdo con la reivindicación 1 o su forma estereoisomérica, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo o una composición farmacéutica de acuerdo con la reivindicación 2 para su uso como medicamento.
4. Un compuesto de acuerdo con la reivindicación 1 o su forma estereoisomérica, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo o una composición farmacéutica de acuerdo con la reivindicación 2 para su uso en el tratamiento del dengue.
5. Uso de un compuesto representado por cualquiera de las fórmulas estructurales de la reivindicación 1, una forma estereoisomérica, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo que comprende un grupo indol mono o di-sustituido para su uso en la inhibición de la replicación del virus (o los virus) del dengue en una muestra biológica o en un paciente.
6. El compuesto para su uso de acuerdo con la reivindicación 5 que comprende además coadministrar un agente terapéutico adicional.
7. El compuesto para su uso de la reivindicación 6, en el que dicho agente terapéutico adicional se selecciona entre un agente antivírico o una vacuna contra el dengue, o ambos.
8. El compuesto según la reivindicación 1, en el que el compuesto se selecciona de


o una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, donde la rotación óptica se mide a 20 °C usando una longitud de onda de 589 nm en DMF como disolvente.
9. Una composición farmacéutica que comprende un compuesto de acuerdo con la reivindicación 8, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, junto con uno o más excipientes, diluyentes o portadores farmacéuticamente aceptables.
10. Un compuesto de acuerdo con la reivindicación 8, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo o una composición farmacéutica de acuerdo con la reivindicación 9 para su uso como medicamento.
11. Un compuesto de acuerdo con la reivindicación 8, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo o una composición farmacéutica de acuerdo con la reivindicación 9 para su uso en el tratamiento del dengue.
12. Un compuesto representado por cualquiera de las fórmulas estructurales de la reivindicación 8, una sal, solvato o polimorfo farmacéuticamente aceptable del mismo que comprende un grupo indol mono o di-sustituido para su uso en la inhibición de la replicación del virus (o los virus) del dengue en una muestra biológica o en un paciente.
13. Los compuestos para su uso de acuerdo con la reivindicación 12, que comprende además coadministrar un agente terapéutico adicional.
14. Los compuestos para su uso de acuerdo con la reivindicación 13, donde dicho agente terapéutico adicional se selecciona entre un agente antivírico o una vacuna contra el dengue, o ambos.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16163281 | 2016-03-31 | ||
| PCT/EP2017/057663 WO2017167953A1 (en) | 2016-03-31 | 2017-03-31 | Substituted indoline derivatives as dengue viral replication inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2923771T3 true ES2923771T3 (es) | 2022-09-30 |
Family
ID=55646444
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17714475T Active ES2923771T3 (es) | 2016-03-31 | 2017-03-31 | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US10913716B2 (es) |
| EP (1) | EP3436436B1 (es) |
| JP (1) | JP6931357B2 (es) |
| KR (1) | KR102359735B1 (es) |
| CN (1) | CN109195949B (es) |
| AU (1) | AU2017240077A1 (es) |
| BR (1) | BR112018069601A2 (es) |
| CA (1) | CA3013407A1 (es) |
| CL (1) | CL2018002718A1 (es) |
| CO (1) | CO2018008421A2 (es) |
| CR (1) | CR20180496A (es) |
| EA (1) | EA201892216A1 (es) |
| EC (1) | ECSP18073366A (es) |
| ES (1) | ES2923771T3 (es) |
| IL (1) | IL261943A (es) |
| MA (1) | MA44498A (es) |
| MX (1) | MX2018011788A (es) |
| PH (1) | PH12018502016A1 (es) |
| SG (1) | SG11201808270PA (es) |
| WO (1) | WO2017167953A1 (es) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201116559D0 (en) | 2011-09-26 | 2011-11-09 | Univ Leuven Kath | Novel viral replication inhibitors |
| GB201305376D0 (en) | 2013-03-25 | 2013-05-08 | Univ Leuven Kath | Novel viral replication inhibitors |
| JOP20160086B1 (ar) | 2015-05-08 | 2021-08-17 | 2 Katholieke Univ Leuven Ku Leuven Research And Development | مشتقات اندول مستبدلة احاديا او ثنائيا بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JO3633B1 (ar) | 2015-09-16 | 2020-08-27 | Katholieke Univ Leuven Ku Leuven Research & Development | مشتقات اندول مستبدلة احاديا او ثنائيا بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JOP20160198B1 (ar) | 2015-09-16 | 2022-03-14 | Janssen Pharmaceuticals Inc | مشتقات اندول مستبدلة احاديا او ثنائيا بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| MX2018011784A (es) | 2016-03-31 | 2019-02-13 | Janssen Pharmaceuticals Inc | Derivados de indol sustituidos como inhibidores de la replicacion virica del dengue. |
| WO2017167953A1 (en) | 2016-03-31 | 2017-10-05 | Janssen Pharmaceuticals, Inc. | Substituted indoline derivatives as dengue viral replication inhibitors |
| BR112018068956A2 (pt) | 2016-04-01 | 2019-01-22 | Janssen Pharmaceuticals Inc | derivados do composto indol substituídos como inibidores da replicação viral da dengue |
| JOP20170069B1 (ar) | 2016-04-01 | 2021-08-17 | 1 Janssen Pharmaceuticals Inc | مشتقات اندولين مستبدلة بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JOP20180025B1 (ar) | 2017-03-31 | 2021-08-17 | Janssen Pharmaceuticals Inc | مشتقات اندولين مستبدلة بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JOP20180026A1 (ar) * | 2017-03-31 | 2019-01-30 | Univ Leuven Kath | مشتقات اندولين مستبدلة بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| ES2929667T3 (es) | 2017-05-22 | 2022-11-30 | Janssen Pharmaceuticals Inc | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue |
| PE20200342A1 (es) | 2017-05-22 | 2020-02-14 | Janssen Pharmaceuticals Inc | Derivados de indolina sustituidos como inhibidores de la replicacion virica de dengue |
| CN109485596A (zh) * | 2018-12-19 | 2019-03-19 | 山东理工职业学院 | 一种含吲哚啉的登革病毒抑制剂的制备方法 |
Family Cites Families (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2308443A1 (en) | 1997-10-27 | 1999-05-06 | Michael Lyle Denney | Morpholino-n-ethyl ester prodrugs of indole spla2 inhibitors |
| GB0110832D0 (en) | 2001-05-03 | 2001-06-27 | Virogen Ltd | Antiviral compounds |
| WO2003050295A2 (en) | 2001-12-12 | 2003-06-19 | Conforma Therapeutics Corporation | Assays and implements for determining and modulating hsp90 binding activity |
| GB0215293D0 (en) | 2002-07-03 | 2002-08-14 | Rega Foundation | Viral inhibitors |
| JP2008526980A (ja) | 2005-01-14 | 2008-07-24 | ジェネラブズ テクノロジーズ インコーポレーティッド | ウイルス感染症を治療するためのインドール誘導体 |
| US20060194835A1 (en) | 2005-02-09 | 2006-08-31 | Migenix Inc. | Compositions and methods for treating or preventing flaviviridae infections |
| CA2726985A1 (en) | 2008-06-03 | 2009-12-10 | Siga Technologies, Inc. | Small molecule inhibitors for the treatment or prevention of dengue virus infection |
| TWI532497B (zh) * | 2008-08-11 | 2016-05-11 | 曼凱公司 | 超快起作用胰島素之用途 |
| US8143259B2 (en) | 2008-08-19 | 2012-03-27 | Janssen Pharmaceutica, Nv | Cold menthol receptor antagonists |
| TWI453207B (zh) | 2008-09-08 | 2014-09-21 | Signal Pharm Llc | 胺基三唑并吡啶,其組合物及使用其之治療方法 |
| WO2010091413A1 (en) | 2009-02-09 | 2010-08-12 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| GB0910003D0 (en) | 2009-06-11 | 2009-07-22 | Univ Leuven Kath | Novel compounds for the treatment of neurodegenerative diseases |
| US8993604B2 (en) | 2009-06-30 | 2015-03-31 | Siga Technologies, Inc. | Treatment and prevention of dengue virus infections |
| PT2523951E (pt) | 2010-01-15 | 2015-09-14 | Gilead Sciences Inc | Inibidores de vírus flaviviridae |
| EP2552211A4 (en) | 2010-03-26 | 2013-10-23 | Glaxo Group Ltd | INDAZOLYL-PYRIMIDINE AS KINASEHEMMER |
| JP5716205B2 (ja) | 2011-03-29 | 2015-05-13 | 学校法人日本大学 | グルコシダーゼ活性阻害用組成物及びそのスクリーニング方法 |
| GB201116559D0 (en) * | 2011-09-26 | 2011-11-09 | Univ Leuven Kath | Novel viral replication inhibitors |
| GB201305376D0 (en) | 2013-03-25 | 2013-05-08 | Univ Leuven Kath | Novel viral replication inhibitors |
| MX368158B (es) | 2013-10-23 | 2019-09-20 | Janssen Sciences Ireland Uc | Derivados de carboxamida y su uso como medicamentos para el tratamiento de la hepatitis b. |
| JP6464176B2 (ja) | 2014-01-31 | 2019-02-06 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | 芳香族p2’基を有する第xia因子阻害剤としてのマクロ環 |
| ME03344B (me) | 2014-10-01 | 2019-10-20 | Janssen Pharmaceuticals Inc | Mono- ili di-supstituisani indoli kao inнibitori replikacije denga virusa |
| NO2721243T3 (es) | 2014-10-01 | 2018-10-20 | ||
| SG11201702463YA (en) | 2014-10-01 | 2017-04-27 | Janssen Pharmaceuticals Inc | Mono- or di-substituted indole derivatives as dengue viral replication inhibitors |
| JOP20150335B1 (ar) | 2015-01-16 | 2022-03-14 | Janssen Pharmaceuticals Inc | مشتقات اندول بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| AR103680A1 (es) | 2015-02-23 | 2017-05-24 | Lilly Co Eli | Inhibidores selectivos de bace1 |
| JOP20160086B1 (ar) | 2015-05-08 | 2021-08-17 | 2 Katholieke Univ Leuven Ku Leuven Research And Development | مشتقات اندول مستبدلة احاديا او ثنائيا بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JOP20160198B1 (ar) | 2015-09-16 | 2022-03-14 | Janssen Pharmaceuticals Inc | مشتقات اندول مستبدلة احاديا او ثنائيا بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JO3633B1 (ar) | 2015-09-16 | 2020-08-27 | Katholieke Univ Leuven Ku Leuven Research & Development | مشتقات اندول مستبدلة احاديا او ثنائيا بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| EP3370698B1 (en) | 2015-11-03 | 2022-01-26 | Zoetis Services LLC | Sol-gel polymer composites and uses thereof |
| MX2018011784A (es) * | 2016-03-31 | 2019-02-13 | Janssen Pharmaceuticals Inc | Derivados de indol sustituidos como inhibidores de la replicacion virica del dengue. |
| WO2017167953A1 (en) | 2016-03-31 | 2017-10-05 | Janssen Pharmaceuticals, Inc. | Substituted indoline derivatives as dengue viral replication inhibitors |
| BR112018068538B1 (pt) | 2016-03-31 | 2023-12-12 | Takeda Pharmaceutical Company Limited | Composto, medicamento, e, uso de um composto |
| CN113150170A (zh) | 2016-04-01 | 2021-07-23 | 凯德药业股份有限公司 | 嵌合受体及其方法和用途 |
| IL296966A (en) | 2016-04-01 | 2022-12-01 | Amgen Inc | Chimeric flt-3 receptors and methods of using them |
| EA039392B1 (ru) | 2016-04-01 | 2022-01-21 | СИГНАЛ ФАРМАСЬЮТИКАЛЗ, ЭлЭлСи | Способ лечения рака с применением замещенного аминопуринового соединения |
| UA123912C2 (uk) | 2016-04-01 | 2021-06-23 | Басф Се | Біциклічні сполуки |
| TW202313671A (zh) | 2016-04-01 | 2023-04-01 | 美商凱特製藥公司 | 嵌合抗原和t細胞受體及使用方法 |
| BR112018068956A2 (pt) | 2016-04-01 | 2019-01-22 | Janssen Pharmaceuticals Inc | derivados do composto indol substituídos como inibidores da replicação viral da dengue |
| JOP20170069B1 (ar) * | 2016-04-01 | 2021-08-17 | 1 Janssen Pharmaceuticals Inc | مشتقات اندولين مستبدلة بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JOP20180025B1 (ar) | 2017-03-31 | 2021-08-17 | Janssen Pharmaceuticals Inc | مشتقات اندولين مستبدلة بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| JOP20180026A1 (ar) | 2017-03-31 | 2019-01-30 | Univ Leuven Kath | مشتقات اندولين مستبدلة بصفتها مانعات للتكاثر الفيروسي لحمى الفنك |
| PE20200342A1 (es) | 2017-05-22 | 2020-02-14 | Janssen Pharmaceuticals Inc | Derivados de indolina sustituidos como inhibidores de la replicacion virica de dengue |
| ES2929667T3 (es) | 2017-05-22 | 2022-11-30 | Janssen Pharmaceuticals Inc | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue |
| CN109485596A (zh) * | 2018-12-19 | 2019-03-19 | 山东理工职业学院 | 一种含吲哚啉的登革病毒抑制剂的制备方法 |
-
2017
- 2017-03-31 WO PCT/EP2017/057663 patent/WO2017167953A1/en active Application Filing
- 2017-03-31 EP EP17714475.5A patent/EP3436436B1/en active Active
- 2017-03-31 CA CA3013407A patent/CA3013407A1/en not_active Abandoned
- 2017-03-31 SG SG11201808270PA patent/SG11201808270PA/en unknown
- 2017-03-31 CN CN201780021312.2A patent/CN109195949B/zh active Active
- 2017-03-31 AU AU2017240077A patent/AU2017240077A1/en not_active Abandoned
- 2017-03-31 MA MA044498A patent/MA44498A/fr unknown
- 2017-03-31 KR KR1020187026918A patent/KR102359735B1/ko active IP Right Grant
- 2017-03-31 CR CR20180496A patent/CR20180496A/es unknown
- 2017-03-31 US US16/088,459 patent/US10913716B2/en active Active
- 2017-03-31 EA EA201892216A patent/EA201892216A1/ru unknown
- 2017-03-31 BR BR112018069601A patent/BR112018069601A2/pt not_active Application Discontinuation
- 2017-03-31 MX MX2018011788A patent/MX2018011788A/es unknown
- 2017-03-31 JP JP2018550333A patent/JP6931357B2/ja active Active
- 2017-03-31 ES ES17714475T patent/ES2923771T3/es active Active
-
2018
- 2018-08-10 CO CONC2018/0008421A patent/CO2018008421A2/es unknown
- 2018-09-20 PH PH12018502016A patent/PH12018502016A1/en unknown
- 2018-09-25 CL CL2018002718A patent/CL2018002718A1/es unknown
- 2018-09-26 IL IL261943A patent/IL261943A/en unknown
- 2018-09-28 EC ECSENADI201873366A patent/ECSP18073366A/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| US20200339511A1 (en) | 2020-10-29 |
| WO2017167953A1 (en) | 2017-10-05 |
| BR112018069601A2 (pt) | 2019-01-29 |
| EP3436436A1 (en) | 2019-02-06 |
| MX2018011788A (es) | 2019-05-20 |
| US10913716B2 (en) | 2021-02-09 |
| PH12018502016A1 (en) | 2019-07-15 |
| KR20180132056A (ko) | 2018-12-11 |
| AU2017240077A1 (en) | 2018-08-09 |
| CN109195949A (zh) | 2019-01-11 |
| ECSP18073366A (es) | 2018-10-31 |
| EA201892216A1 (ru) | 2019-03-29 |
| KR102359735B1 (ko) | 2022-02-08 |
| JP2019510027A (ja) | 2019-04-11 |
| EP3436436B1 (en) | 2022-05-11 |
| SG11201808270PA (en) | 2018-10-30 |
| IL261943A (en) | 2018-10-31 |
| CR20180496A (es) | 2018-12-06 |
| MA44498A (fr) | 2019-02-06 |
| CO2018008421A2 (es) | 2018-10-22 |
| JP6931357B2 (ja) | 2021-09-01 |
| CN109195949B (zh) | 2021-09-17 |
| CL2018002718A1 (es) | 2018-12-14 |
| CA3013407A1 (en) | 2017-10-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2923771T3 (es) | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue | |
| ES2884067T3 (es) | Derivados de indolina sustituidos como inhibidores de la replicación viral del dengue | |
| ES2884151T3 (es) | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue | |
| ES2893298T3 (es) | Derivados de indol mono- o disustituidos como inhibidores de la replicación viral del dengue | |
| ES2884157T3 (es) | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue | |
| ES2887673T3 (es) | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue | |
| ES2882488T3 (es) | Derivados de indol sustituidos como inhibidores de la replicación vírica del dengue | |
| ES2778431T3 (es) | Derivados de indol mono- o disustituidos como inhibidores de la replicación viral del dengue | |
| ES2877404T3 (es) | Derivados de indol mono o disustituidos como inhibidores de la replicación viral del dengue | |
| ES2714074T3 (es) | Derivados de indol mono- o disustituidos como inhibidores de la replicación del virus del dengue | |
| ES2929667T3 (es) | Derivados de indolina sustituidos como inhibidores de la replicación vírica de dengue | |
| OA18876A (en) | Substituted indoline derivatives as dengue viral replication inhibitors |