ES2897422T3 - Compuestos pirroles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos - Google Patents
Compuestos pirroles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos Download PDFInfo
- Publication number
- ES2897422T3 ES2897422T3 ES19199330T ES19199330T ES2897422T3 ES 2897422 T3 ES2897422 T3 ES 2897422T3 ES 19199330 T ES19199330 T ES 19199330T ES 19199330 T ES19199330 T ES 19199330T ES 2897422 T3 ES2897422 T3 ES 2897422T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- compounds
- optionally substituted
- aliphatic
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 174
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 title description 30
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 title description 16
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 title description 4
- 239000008194 pharmaceutical composition Substances 0.000 title description 4
- 239000003909 protein kinase inhibitor Substances 0.000 title description 4
- 150000003233 pyrroles Chemical class 0.000 title description 2
- 239000000203 mixture Substances 0.000 claims description 100
- 150000003839 salts Chemical class 0.000 claims description 38
- 239000003981 vehicle Substances 0.000 claims description 13
- 239000003937 drug carrier Substances 0.000 claims description 12
- 239000002671 adjuvant Substances 0.000 claims description 11
- 229940125773 compound 10 Drugs 0.000 claims description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 claims description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 claims 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 claims 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 claims 1
- 229940125797 compound 12 Drugs 0.000 claims 1
- 229940126543 compound 14 Drugs 0.000 claims 1
- 229940126142 compound 16 Drugs 0.000 claims 1
- 229940125782 compound 2 Drugs 0.000 claims 1
- 125000001931 aliphatic group Chemical group 0.000 description 68
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 49
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- 238000000034 method Methods 0.000 description 38
- 239000000243 solution Substances 0.000 description 38
- 239000002904 solvent Substances 0.000 description 37
- 239000003814 drug Substances 0.000 description 35
- 229910052739 hydrogen Inorganic materials 0.000 description 34
- 235000002639 sodium chloride Nutrition 0.000 description 34
- 239000001257 hydrogen Substances 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- -1 nucleoside triphosphate Chemical class 0.000 description 30
- 239000007787 solid Substances 0.000 description 30
- 101000876610 Dictyostelium discoideum Extracellular signal-regulated kinase 2 Proteins 0.000 description 29
- 101001052493 Homo sapiens Mitogen-activated protein kinase 1 Proteins 0.000 description 29
- 201000010099 disease Diseases 0.000 description 28
- 238000006243 chemical reaction Methods 0.000 description 27
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- 206010028980 Neoplasm Diseases 0.000 description 24
- 229940079593 drug Drugs 0.000 description 24
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 21
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 21
- 208000035475 disorder Diseases 0.000 description 21
- 239000003112 inhibitor Substances 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 19
- 230000000694 effects Effects 0.000 description 19
- 201000011510 cancer Diseases 0.000 description 18
- 125000001424 substituent group Chemical group 0.000 description 17
- 239000000460 chlorine Substances 0.000 description 16
- 229910052801 chlorine Inorganic materials 0.000 description 16
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- 125000006239 protecting group Chemical group 0.000 description 16
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- 239000011541 reaction mixture Substances 0.000 description 15
- 0 *c(cc1)cnc1I Chemical compound *c(cc1)cnc1I 0.000 description 14
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 102000001253 Protein Kinase Human genes 0.000 description 14
- 210000004027 cell Anatomy 0.000 description 14
- 108060006633 protein kinase Proteins 0.000 description 14
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 13
- 238000000576 coating method Methods 0.000 description 13
- 150000002431 hydrogen Chemical class 0.000 description 13
- 229910052757 nitrogen Inorganic materials 0.000 description 13
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 12
- 229910052731 fluorine Inorganic materials 0.000 description 12
- 229940002612 prodrug Drugs 0.000 description 12
- 239000000651 prodrug Substances 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 239000011737 fluorine Substances 0.000 description 11
- 229940124597 therapeutic agent Drugs 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 230000004913 activation Effects 0.000 description 10
- 229910052736 halogen Inorganic materials 0.000 description 10
- 150000002367 halogens Chemical class 0.000 description 10
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- 229910052938 sodium sulfate Inorganic materials 0.000 description 10
- 235000011152 sodium sulphate Nutrition 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 125000000217 alkyl group Chemical group 0.000 description 9
- 150000007942 carboxylates Chemical group 0.000 description 9
- 239000012043 crude product Substances 0.000 description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 9
- 238000011065 in-situ storage Methods 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 229910019142 PO4 Inorganic materials 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 201000001441 melanoma Diseases 0.000 description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 8
- 230000035772 mutation Effects 0.000 description 8
- 235000021317 phosphate Nutrition 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- 208000012902 Nervous system disease Diseases 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 238000002483 medication Methods 0.000 description 7
- 201000006417 multiple sclerosis Diseases 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- 239000010452 phosphate Substances 0.000 description 7
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 7
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 208000023275 Autoimmune disease Diseases 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 102000043136 MAP kinase family Human genes 0.000 description 6
- 108091054455 MAP kinase family Proteins 0.000 description 6
- 108091000080 Phosphotransferase Proteins 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 230000003213 activating effect Effects 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 239000011248 coating agent Substances 0.000 description 6
- 125000000753 cycloalkyl group Chemical group 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- 235000019441 ethanol Nutrition 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 125000001072 heteroaryl group Chemical group 0.000 description 6
- 125000005842 heteroatom Chemical group 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 239000001301 oxygen Chemical group 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- 102000020233 phosphotransferase Human genes 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 229910052717 sulfur Chemical group 0.000 description 6
- 239000011593 sulfur Chemical group 0.000 description 6
- GDSLUYKCPYECNN-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-[(4-fluorophenyl)methyl]benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCC2=CC=C(C=C2)F)C=CC=1 GDSLUYKCPYECNN-UHFFFAOYSA-N 0.000 description 5
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 5
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 5
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 5
- 208000006673 asthma Diseases 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 5
- 239000012045 crude solution Substances 0.000 description 5
- 239000008101 lactose Substances 0.000 description 5
- 208000019423 liver disease Diseases 0.000 description 5
- 208000015122 neurodegenerative disease Diseases 0.000 description 5
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 102000016914 ras Proteins Human genes 0.000 description 5
- 108010014186 ras Proteins Proteins 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 238000004007 reversed phase HPLC Methods 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 239000001993 wax Substances 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- 208000024827 Alzheimer disease Diseases 0.000 description 4
- 208000020084 Bone disease Diseases 0.000 description 4
- 208000026310 Breast neoplasm Diseases 0.000 description 4
- 208000020446 Cardiac disease Diseases 0.000 description 4
- 208000024172 Cardiovascular disease Diseases 0.000 description 4
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 108010050904 Interferons Proteins 0.000 description 4
- 102000014150 Interferons Human genes 0.000 description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 4
- 208000025966 Neurological disease Diseases 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 239000012472 biological sample Substances 0.000 description 4
- 125000003865 brosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)S(*)(=O)=O 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- 239000003246 corticosteroid Substances 0.000 description 4
- 229960001334 corticosteroids Drugs 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 229960004397 cyclophosphamide Drugs 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- 208000019622 heart disease Diseases 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- 229940047124 interferons Drugs 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 125000001736 nosyl group Chemical group S(=O)(=O)(C1=CC=C([N+](=O)[O-])C=C1)* 0.000 description 4
- 210000003800 pharynx Anatomy 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000003586 protic polar solvent Substances 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 201000000980 schizophrenia Diseases 0.000 description 4
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 230000019491 signal transduction Effects 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 235000017550 sodium carbonate Nutrition 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 4
- 102000003390 tumor necrosis factor Human genes 0.000 description 4
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 3
- NCBIFXNQFURYON-UHFFFAOYSA-N 4-[5-chloro-2-(propan-2-ylamino)pyridin-4-yl]-1h-pyrrole-2-carboxylic acid Chemical compound C1=NC(NC(C)C)=CC(C=2C=C(NC=2)C(O)=O)=C1Cl NCBIFXNQFURYON-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 102000000589 Interleukin-1 Human genes 0.000 description 3
- 108010002352 Interleukin-1 Proteins 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 3
- 150000001204 N-oxides Chemical class 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- 108091023040 Transcription factor Proteins 0.000 description 3
- 102000040945 Transcription factor Human genes 0.000 description 3
- FXXACINHVKSMDR-UHFFFAOYSA-N acetyl bromide Chemical compound CC(Br)=O FXXACINHVKSMDR-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000005137 alkenylsulfonyl group Chemical group 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 3
- 125000001118 alkylidene group Chemical group 0.000 description 3
- 230000000172 allergic effect Effects 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 230000000118 anti-neoplastic effect Effects 0.000 description 3
- 229940034982 antineoplastic agent Drugs 0.000 description 3
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 208000010668 atopic eczema Diseases 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 238000013270 controlled release Methods 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- 238000007429 general method Methods 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- 230000003054 hormonal effect Effects 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 230000000116 mitigating effect Effects 0.000 description 3
- KSERXGMCDHOLSS-UHFFFAOYSA-N n-[1-(3-chlorophenyl)-2-hydroxyethyl]-4-[5-chloro-2-(propan-2-ylamino)pyridin-4-yl]-1h-pyrrole-2-carboxamide Chemical compound C1=NC(NC(C)C)=CC(C=2C=C(NC=2)C(=O)NC(CO)C=2C=C(Cl)C=CC=2)=C1Cl KSERXGMCDHOLSS-UHFFFAOYSA-N 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 239000006174 pH buffer Substances 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- 210000000664 rectum Anatomy 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 208000037803 restenosis Diseases 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 229960004295 valine Drugs 0.000 description 3
- YHPDPZLDOQQYER-YUOVYVCQSA-N (2S,3S)-2-amino-5-(3-chlorophenyl)-5-[[4-[5-chloro-2-(propan-2-ylamino)pyridin-4-yl]-1H-pyrrole-2-carbonyl]amino]-2,3-dimethylpentanoic acid Chemical compound C[C@@H](CC(C1=CC(=CC=C1)Cl)NC(=O)C2=CC(=CN2)C3=CC(=NC=C3Cl)NC(C)C)[C@@](C)(C(=O)O)N YHPDPZLDOQQYER-YUOVYVCQSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- PAVKKFAIAQPCRG-UHFFFAOYSA-N 2,2,2-trichloro-1-(4-iodo-1h-pyrrol-2-yl)ethanone Chemical compound ClC(Cl)(Cl)C(=O)C1=CC(I)=CN1 PAVKKFAIAQPCRG-UHFFFAOYSA-N 0.000 description 2
- XXLFCBBWDXLUOK-UHFFFAOYSA-N 2,5-dichloro-4-nitro-1-oxidopyridin-1-ium Chemical compound [O-][N+](=O)C1=CC(Cl)=[N+]([O-])C=C1Cl XXLFCBBWDXLUOK-UHFFFAOYSA-N 0.000 description 2
- XGPMALPFZKLBRM-UHFFFAOYSA-N 2-[(4-bromo-5-methylpyridin-2-yl)amino]butyl acetate Chemical compound CC(=O)OCC(CC)NC1=CC(Br)=C(C)C=N1 XGPMALPFZKLBRM-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- DXJQKWNJVPTPSK-UHFFFAOYSA-N 2-chloro-5-methyl-4-nitro-1-oxidopyridin-1-ium Chemical compound CC1=C[N+]([O-])=C(Cl)C=C1[N+]([O-])=O DXJQKWNJVPTPSK-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- BPGYVHYGKOBDSX-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,4,6-trimethylphenyl)sulfonylpyrrole-2-carboxylic acid Chemical compound CC1(OB(OC1(C)C)C=1C=C(N(C1)S(=O)(=O)C1=C(C=C(C=C1C)C)C)C(=O)O)C BPGYVHYGKOBDSX-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- MAQBAWYSDZIKIC-UHFFFAOYSA-N 5-chloro-2-fluoro-4-iodopyridine Chemical compound FC1=CC(I)=C(Cl)C=N1 MAQBAWYSDZIKIC-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 2
- 208000003200 Adenoma Diseases 0.000 description 2
- 206010001233 Adenoma benign Diseases 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 206010004593 Bile duct cancer Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- FRDOMQZQFJXSJN-UHFFFAOYSA-N CC1(C)BOOC1(C)C Chemical group CC1(C)BOOC1(C)C FRDOMQZQFJXSJN-UHFFFAOYSA-N 0.000 description 2
- DRPCQWCZXNNVJK-QMMMGPOBSA-N CC[C@@H](CO)NC1=CC([N+]([O-])=O)=C(C)C=[N+]1[O-] Chemical compound CC[C@@H](CO)NC1=CC([N+]([O-])=O)=C(C)C=[N+]1[O-] DRPCQWCZXNNVJK-QMMMGPOBSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 208000010667 Carcinoma of liver and intrahepatic biliary tract Diseases 0.000 description 2
- 208000006029 Cardiomegaly Diseases 0.000 description 2
- 201000003883 Cystic fibrosis Diseases 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 2
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 2
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 2
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 2
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 206010073069 Hepatic cancer Diseases 0.000 description 2
- 206010019842 Hepatomegaly Diseases 0.000 description 2
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 108010005716 Interferon beta-1a Proteins 0.000 description 2
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 2
- 206010023347 Keratoacanthoma Diseases 0.000 description 2
- 102000003855 L-lactate dehydrogenase Human genes 0.000 description 2
- 108700023483 L-lactate dehydrogenases Proteins 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- 229940124647 MEK inhibitor Drugs 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 2
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 102000013009 Pyruvate Kinase Human genes 0.000 description 2
- 108020005115 Pyruvate Kinase Proteins 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- 201000010208 Seminoma Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 239000001099 ammonium carbonate Substances 0.000 description 2
- 235000012501 ammonium carbonate Nutrition 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 239000000010 aprotic solvent Substances 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 2
- 229960002170 azathioprine Drugs 0.000 description 2
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 201000007180 bile duct carcinoma Diseases 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 2
- 201000001531 bladder carcinoma Diseases 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 125000005621 boronate group Chemical class 0.000 description 2
- 210000000133 brain stem Anatomy 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 230000006552 constitutive activation Effects 0.000 description 2
- 239000002285 corn oil Substances 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 239000002385 cottonseed oil Substances 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- 239000010779 crude oil Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-O diazynium Chemical group [NH+]#N IJGRMHOSHXDMSA-UHFFFAOYSA-O 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 229940043264 dodecyl sulfate Drugs 0.000 description 2
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 2
- 229940126864 fibroblast growth factor Drugs 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000019634 flavors Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 208000005017 glioblastoma Diseases 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 235000001727 glucose Nutrition 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000003906 humectant Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000000099 in vitro assay Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 208000003849 large cell carcinoma Diseases 0.000 description 2
- 210000002429 large intestine Anatomy 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 210000000088 lip Anatomy 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 201000002250 liver carcinoma Diseases 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 201000005249 lung adenocarcinoma Diseases 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- YIJKTFQVBNGYKP-UHFFFAOYSA-N methyl 4-[5-chloro-2-(propan-2-ylamino)pyridin-4-yl]-1-(2,4,6-trimethylphenyl)sulfonylpyrrole-2-carboxylate Chemical compound COC(=O)C1=CC(C=2C(=CN=C(NC(C)C)C=2)Cl)=CN1S(=O)(=O)C1=C(C)C=C(C)C=C1C YIJKTFQVBNGYKP-UHFFFAOYSA-N 0.000 description 2
- VZYVEPCGYYFESE-UHFFFAOYSA-N methyl 4-[5-chloro-2-(propan-2-ylamino)pyridin-4-yl]-1-(4-methylphenyl)sulfonylpyrrole-2-carboxylate Chemical compound COC(=O)C1=CC(C=2C(=CN=C(NC(C)C)C=2)Cl)=CN1S(=O)(=O)C1=CC=C(C)C=C1 VZYVEPCGYYFESE-UHFFFAOYSA-N 0.000 description 2
- XUMRTTYVSXENOU-UHFFFAOYSA-N methyl 4-iodo-1-(4-methylphenyl)sulfonylpyrrole-2-carboxylate Chemical compound COC(=O)C1=CC(I)=CN1S(=O)(=O)C1=CC=C(C)C=C1 XUMRTTYVSXENOU-UHFFFAOYSA-N 0.000 description 2
- KRNGDJYQOKCQCD-UHFFFAOYSA-N methyl 4-iodo-1h-pyrrole-2-carboxylate Chemical compound COC(=O)C1=CC(I)=CN1 KRNGDJYQOKCQCD-UHFFFAOYSA-N 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 210000000214 mouth Anatomy 0.000 description 2
- 230000000626 neurodegenerative effect Effects 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 2
- 239000012454 non-polar solvent Substances 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 239000003605 opacifier Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 2
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 201000010198 papillary carcinoma Diseases 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000002304 perfume Substances 0.000 description 2
- 229930029653 phosphoenolpyruvate Natural products 0.000 description 2
- DTBNBXWJWCWCIK-UHFFFAOYSA-N phosphoenolpyruvic acid Chemical compound OC(=O)C(=C)OP(O)(O)=O DTBNBXWJWCWCIK-UHFFFAOYSA-N 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 229920000747 poly(lactic acid) Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 238000005956 quaternization reaction Methods 0.000 description 2
- 239000012429 reaction media Substances 0.000 description 2
- 201000010174 renal carcinoma Diseases 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 230000035939 shock Effects 0.000 description 2
- 208000000649 small cell carcinoma Diseases 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 206010041823 squamous cell carcinoma Diseases 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 2
- 229960001940 sulfasalazine Drugs 0.000 description 2
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 210000001685 thyroid gland Anatomy 0.000 description 2
- 208000030901 thyroid gland follicular carcinoma Diseases 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 210000002105 tongue Anatomy 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- 208000010576 undifferentiated carcinoma Diseases 0.000 description 2
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- JCBPETKZIGVZRE-BYPYZUCNSA-N (2s)-2-aminobutan-1-ol Chemical compound CC[C@H](N)CO JCBPETKZIGVZRE-BYPYZUCNSA-N 0.000 description 1
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 1
- 125000006569 (C5-C6) heterocyclic group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- SMWFQHUDVVMMMC-UHFFFAOYSA-N 1-(4-methylphenyl)sulfonyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrrole-2-carboxylic acid Chemical compound CC1(OB(OC1(C)C)C=1C=C(N(C=1)S(=O)(=O)C1=CC=C(C)C=C1)C(=O)O)C SMWFQHUDVVMMMC-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- BBFDGMDENAEMKF-UHFFFAOYSA-N 2,2,2-trichloro-1-(1h-pyrrol-2-yl)ethanone Chemical compound ClC(Cl)(Cl)C(=O)C1=CC=CN1 BBFDGMDENAEMKF-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- GCTFDMFLLBCLPF-UHFFFAOYSA-N 2,5-dichloropyridine Chemical compound ClC1=CC=C(Cl)N=C1 GCTFDMFLLBCLPF-UHFFFAOYSA-N 0.000 description 1
- ATOGAUQNUOCZIH-UHFFFAOYSA-N 2-(chloroamino)-2-phenylethanol Chemical compound OCC(NCl)C1=CC=CC=C1 ATOGAUQNUOCZIH-UHFFFAOYSA-N 0.000 description 1
- VRHJBWUIWQOFLF-WLHGVMLRSA-N 2-[2-(4-benzo[b][1,4]benzothiazepin-6-ylpiperazin-1-yl)ethoxy]ethanol;(e)-but-2-enedioic acid Chemical compound OC(=O)\C=C\C(O)=O.C1CN(CCOCCO)CCN1C1=NC2=CC=CC=C2SC2=CC=CC=C12 VRHJBWUIWQOFLF-WLHGVMLRSA-N 0.000 description 1
- VXLYOURCUVQYLN-UHFFFAOYSA-N 2-chloro-5-methylpyridine Chemical compound CC1=CC=C(Cl)N=C1 VXLYOURCUVQYLN-UHFFFAOYSA-N 0.000 description 1
- KIFPIAKBYOIOCS-UHFFFAOYSA-N 2-methyl-2-(trioxidanyl)propane Chemical compound CC(C)(C)OOO KIFPIAKBYOIOCS-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 108091006112 ATPases Proteins 0.000 description 1
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- ZFWRXWWFQAMUCQ-HLIVEPPXSA-N CC[C@@H](CO)NC(N=CC1C)=CC1c1c[nH]c(C(N[C@H](CO)c2cccc(Cl)c2)=O)c1 Chemical compound CC[C@@H](CO)NC(N=CC1C)=CC1c1c[nH]c(C(N[C@H](CO)c2cccc(Cl)c2)=O)c1 ZFWRXWWFQAMUCQ-HLIVEPPXSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 102100023033 Cyclic AMP-dependent transcription factor ATF-2 Human genes 0.000 description 1
- 108010009392 Cyclin-Dependent Kinase Inhibitor p16 Proteins 0.000 description 1
- 102100024458 Cyclin-dependent kinase inhibitor 2A Human genes 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 239000012824 ERK inhibitor Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 108050009340 Endothelin Proteins 0.000 description 1
- 102000002045 Endothelin Human genes 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 102100039788 GTPase NRas Human genes 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- 108010072051 Glatiramer Acetate Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 101000974934 Homo sapiens Cyclic AMP-dependent transcription factor ATF-2 Proteins 0.000 description 1
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 1
- 101000997829 Homo sapiens Glial cell line-derived neurotrophic factor Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101001050288 Homo sapiens Transcription factor Jun Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020843 Hyperthermia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- QZRGKCOWNLSUDK-UHFFFAOYSA-N Iodochlorine Chemical compound ICl QZRGKCOWNLSUDK-UHFFFAOYSA-N 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 description 1
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 108010071563 Proto-Oncogene Proteins c-fos Proteins 0.000 description 1
- 102000007568 Proto-Oncogene Proteins c-fos Human genes 0.000 description 1
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 description 1
- 101710141955 RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- FTALBRSUTCGOEG-UHFFFAOYSA-N Riluzole Chemical compound C1=C(OC(F)(F)F)C=C2SC(N)=NC2=C1 FTALBRSUTCGOEG-UHFFFAOYSA-N 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 231100000632 Spindle poison Toxicity 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102100023132 Transcription factor Jun Human genes 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000719 anti-leukaemic effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000002001 anti-metastasis Effects 0.000 description 1
- 230000000648 anti-parkinson Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 239000001961 anticonvulsive agent Substances 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229940082992 antihypertensives mao inhibitors Drugs 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000000939 antiparkinson agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 229940039856 aricept Drugs 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 230000003305 autocrine Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 125000005620 boronic acid group Chemical class 0.000 description 1
- 238000002725 brachytherapy Methods 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 description 1
- 229960002802 bromocriptine Drugs 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- AIUWGJYWSXNNHX-UHFFFAOYSA-N calcium;2,3-dihydroxypropyl octadecanoate Chemical compound [Ca].CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO AIUWGJYWSXNNHX-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- 229960004205 carbidopa Drugs 0.000 description 1
- TZFNLOMSOLWIDK-JTQLQIEISA-N carbidopa (anhydrous) Chemical compound NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 TZFNLOMSOLWIDK-JTQLQIEISA-N 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 239000000544 cholinesterase inhibitor Substances 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000010835 comparative analysis Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 229940038717 copaxone Drugs 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000000315 cryotherapy Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- USVZFSNDGFNNJT-UHFFFAOYSA-N cyclopenta-1,4-dien-1-yl(diphenyl)phosphane (2,3-dichlorocyclopenta-1,4-dien-1-yl)-diphenylphosphane iron(2+) Chemical compound [Fe++].c1cc[c-](c1)P(c1ccccc1)c1ccccc1.Clc1c(cc[c-]1Cl)P(c1ccccc1)c1ccccc1 USVZFSNDGFNNJT-UHFFFAOYSA-N 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical compound CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- BSHICDXRSZQYBP-UHFFFAOYSA-N dichloromethane;palladium(2+) Chemical compound [Pd+2].ClCCl BSHICDXRSZQYBP-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- FPAFDBFIGPHWGO-UHFFFAOYSA-N dioxosilane;oxomagnesium;hydrate Chemical compound O.[Mg]=O.[Mg]=O.[Mg]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O FPAFDBFIGPHWGO-UHFFFAOYSA-N 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000010894 electron beam technology Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- JRURYQJSLYLRLN-BJMVGYQFSA-N entacapone Chemical compound CCN(CC)C(=O)C(\C#N)=C\C1=CC(O)=C(O)C([N+]([O-])=O)=C1 JRURYQJSLYLRLN-BJMVGYQFSA-N 0.000 description 1
- 229960003337 entacapone Drugs 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 102000036444 extracellular matrix enzymes Human genes 0.000 description 1
- 108091007167 extracellular matrix enzymes Proteins 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 230000036433 growing body Effects 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000000262 haloalkenyl group Chemical group 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 229960003878 haloperidol Drugs 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000004475 heteroaralkyl group Chemical group 0.000 description 1
- 125000005114 heteroarylalkoxy group Chemical group 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 230000036031 hyperthermia Effects 0.000 description 1
- 238000009217 hyperthermia therapy Methods 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 239000000367 immunologic factor Substances 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910001410 inorganic ion Inorganic materials 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 239000008274 jelly Substances 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 210000000867 larynx Anatomy 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960001786 megestrol Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000008883 metastatic behaviour Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- QXXXFVPRHBSWAZ-UHFFFAOYSA-N methyl 1-(4-methylphenyl)sulfonyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrrole-2-carboxylate Chemical compound COC(=O)C1=CC(B2OC(C)(C)C(C)(C)O2)=CN1S(=O)(=O)C1=CC=C(C)C=C1 QXXXFVPRHBSWAZ-UHFFFAOYSA-N 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000002899 monoamine oxidase inhibitor Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 230000004118 muscle contraction Effects 0.000 description 1
- 229960004866 mycophenolate mofetil Drugs 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 239000003900 neurotrophic factor Substances 0.000 description 1
- 238000009203 neutron therapy Methods 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 230000006548 oncogenic transformation Effects 0.000 description 1
- 102000027450 oncoproteins Human genes 0.000 description 1
- 108091008819 oncoproteins Proteins 0.000 description 1
- 229940041678 oral spray Drugs 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- WXHIJDCHNDBCNY-UHFFFAOYSA-N palladium dihydride Chemical compound [PdH2] WXHIJDCHNDBCNY-UHFFFAOYSA-N 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- YEHCICAEULNIGD-MZMPZRCHSA-N pergolide Chemical compound C1=CC([C@H]2C[C@@H](CSC)CN([C@@H]2C2)CCC)=C3C2=CNC3=C1 YEHCICAEULNIGD-MZMPZRCHSA-N 0.000 description 1
- 229960004851 pergolide Drugs 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 230000004983 pleiotropic effect Effects 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000004632 polycaprolactone Substances 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- FASDKYOPVNHBLU-ZETCQYMHSA-N pramipexole Chemical compound C1[C@@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-ZETCQYMHSA-N 0.000 description 1
- 229960003089 pramipexole Drugs 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 238000002661 proton therapy Methods 0.000 description 1
- 239000000649 purine antagonist Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000003790 pyrimidine antagonist Substances 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000000163 radioactive labelling Methods 0.000 description 1
- 239000002287 radioligand Substances 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 229940038850 rebif Drugs 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 229960004181 riluzole Drugs 0.000 description 1
- 229940106887 risperdal Drugs 0.000 description 1
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 229940035004 seroquel Drugs 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 101150006137 sir gene Proteins 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 210000001138 tear Anatomy 0.000 description 1
- IRDFFAPCSABAGK-UHFFFAOYSA-N tert-butyl dihydrogen phosphate Chemical compound CC(C)(C)OP(O)(O)=O IRDFFAPCSABAGK-UHFFFAOYSA-N 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 210000002229 urogenital system Anatomy 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000010497 wheat germ oil Substances 0.000 description 1
- 235000021119 whey protein Nutrition 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 229940039925 zyprexa Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Cardiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Oncology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Gastroenterology & Hepatology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Communicable Diseases (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Vascular Medicine (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Virology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
El compuesto **(Ver fórmula)**
Description
DESCRIPCIÓN
Compuestos pirróles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos
Campo técnico de la invención
La presente invención se refiere a compuestos útiles como inhibidores de las proteínas cinasas y sales farmacéuticamente aceptables de los mismos. La invención también proporciona composiciones farmacéuticamente aceptables que contienen los compuestos de la invención.
Antecedentes de la invención
La búsqueda de nuevos agentes terapéuticos se ha visto auxiliada en gran medida en los últimos años por una mejor comprensión de la estructura de las enzimas y otras biomoléculas asociadas a enfermedades que son el objetivo. Una clase importante de enzimas que ha sido objeto de un extenso estudio es la de las proteínas cinasas.
Las proteínas cinasas constituyen una gran familia de enzimas relacionadas estructuralmente que son responsables del control de diversos procesos de transducción de la señal dentro de la célula. (Véase, Hardie, G. y Hanks, S. (1995) The Protein Kinase Facts Book, I y II, Academic Press, San Diego, CA). Se cree que las proteínas cinasas han evolucionado de un gen ancestral común debido a la conservación de su estructura y función catalítica. Casi todas las cinasas tienen un dominio catalítico similar de 250 a 300 aminoácidos. Las cinasas se pueden categorizar en familias por los sustratos a los que fosforilan (por ejemplo, proteína-tirosina, proteína-serina/treonina, lípidos, etc.). Se han identificado motivos de secuencia que corresponden generalmente a los de cada una de esas familias de cinasas (véase, por ejemplo, Hanks, S.K., Hunter, T., FASEB J., 9:576-596 (1995); Knighton et al., Science, 253:407-414 (1991); Hiles et al., Cell, 70:419-429 (1992); Kunz et al., Cell,73:585-596 (1993); Garcia-Bustos et al., EMBO J., 13:2352-2361 (1994)).
En general, las proteínas cinasas actúan de mediadores en la señalización intracelular al efectuar una transferencia de fosforilo desde un nucleósido trifosfato a un aceptor de proteína que está implicado en una vía de señalización. Estas fosforilaciones actúan como interruptores moleculares de encendido/apagado que pueden modular o regular la función biológica de la proteína diana. Estas fosforilaciones son en última instancia activadas en respuesta a diversos estímulos extracelulares y otros. Los ejemplos de dichos estímulos incluyen señales de estrés ambiental y químico (por ejemplo, shock osmótico, shock de calor, radiación ultravioleta, endotoxina bacteriana y H2O2), citocinas (por ejemplo, interleucina-1 (IL-1) y factor de necrosis tumoral a (TNF-a)), y factores de crecimiento (por ejemplo, factor estimulante de las colonias de granulocitos y macrófagos (GM-CSF) y factor de crecimiento de fibroblastos (FGF)). Un estímulo extracelular puede afectar a una o más respuestas celulares relacionadas con la multiplicación celular, la migración, la diferenciación, la secreción de hormonas, la activación de los factores de transcripción, la contracción muscular, el metabolismo de la glucosa, el control de la síntesis de proteínas y la regulación del ciclo celular.
Muchas enfermedades están asociadas a respuestas celulares anómalas activadas por acontecimientos mediados por proteínas cinasas. Estas enfermedades incluyen enfermedades autoinmunitarias, enfermedades inflamatorias, osteopatías, enfermedades metabólicas, neuropatías y enfermedades neurodegenerativas, cáncer, enfermedades cardiovasculares, alergias y asma, enfermedad de Alzheimer y enfermedades hormonales. En consecuencia, se ha hecho un esfuerzo importante en la química medicinal por encontrar inhibidores de las proteínas cinasas que sean eficaces como agentes terapéuticos. No obstante, considerando la ausencia de opciones de tratamiento disponibles en la actualidad para la mayoría de las afecciones asociadas a la proteínas cinasas, existe todavía una gran necesidad de nuevos agentes terapéuticos que inhiban estas proteínas diana.
Las células de mamíferos responden a estímulos extracelulares activando cascadas de señalización que son mediadas por miembros de la familia de las proteínas cinasas activadas por mitógenos (MAP), que incluyen las cinasas reguladas por señal extracelular (ERK), las cinasas p38 MAP y las cinasas c-Jun N-terminal (JNK). Las cinasas MAP (MAPK) son activadas por diversas señales que incluyen factores de crecimiento, citocinas, radiación UV e inductores de estrés. Las MAPK son serina/treonina cinasas y su activación se produce por fosforilación dual de treonina y tirosina en el segmento Thr-X-Tyr del bucle de activación. Las MAPK fosforilan varios sustratos incluidos factores de transcripción, que a su vez regulan la expresión de conjuntos de genes específicos y así median una respuesta específica al estímulo.
ERK2 es una proteína cinasa ampliamente distribuida que alcanza su máxima actividad cuando tanto Thr183 como Tyr185 son fosforiladas por la MAP cinasa cinasa, MEK1 (Anderson et al., 1990, Nature 343, 651; Crews et al., 1992, Science 258, 478). Luego de la activación, ERK2 fosforila muchas proteínas reguladoras, incluidas las proteínas cinasas Rsk90 (Bjorbaek et al., 1995, J. Biol. Chem. 270, 18848) y MAPKAP2 (Rouse et al., 1994, Cell 78, 1027), y factores de transcripción, tales como ATF2 (Raingeaud et al., 1996, Mol. Cell Biol. 16, 1247), Elk-1 (Raingeaud et al.
1996), c-Fos (Chen et al., 1993 Proc. Natl. Acad. Sci. USA 90, 10952) y c-Myc (Oliver et al., 1995, Proc. Soc. Exp. Biol. Med. 210, 162). ERK2 también es un objetivo posterior de las rutas dependientes de Ras/Raf (Moodie et al., 1993, Science 260, 1658) y trasmite las señales de estas proteínas potencialmente oncógenas. Se ha demostrado
que ERK2 desempeña un papel fundamental en el control negativo de la multiplicación de las células del cáncer de mama (Frey y Mulder, 1997, Cancer Res. 57, 628) y se ha informado de hiperexpresión de ERK2 en cáncer de mama humano (Sivaraman et al., 1997, J Clin. Invest. 99, 1478). También se ha implicado a ERK2 activada en la proliferación de células de músculo liso estimuladas por endotelina en las vías respiratorias, lo que sugiere un papel de esta cinasa en el asma (Whelchel et al., 1997, Am. J. Respir. Cell Mol. Biol. 16, 589).
La sobreexpresión de receptores tirosina cinasas, tales como EGFR y ErbB2 (Arteaga CL, 2002, Semin Oncol. 29, 3 9; Eccles SA, 2001, J Mammary Gland Biol Neoplasia 6:393-406; Mendelsolm J & Baselga J, 2000, Oncogene 19, 6550-65), así como mutaciones activantes en las proteínas Ras GTPasas (Nottage M & Siu LL, 2002, Curr Pharm Des 8, 2231-42; Adjei AA, 2001, J Natl Cancer Inst 93, 1062-74) o los mutantes B-Raf (Davies H. et al., 2002, Nature 417, 949-54; Brose et al., 2002, Cancer Res 62, 6997-7000) son importantes contribuyentes al cáncer humano. Estas alteraciones genéticas se correlacionan con un mal pronóstico clínico y resultan en la activación de la cascada de transducción de señal Raf-1/2/3 - MEK1/2 - ERK1/2 en un amplio grupo de tumores humanos. ERK activada (es decir ERK1 y/o ERK2) es una molécula de señalización central que ha sido asociada al control de la proliferación, la diferenciación, la supervivencia celular independiente del anclaje y la angiogénesis, contribuyendo a varios procesos que son importantes para la formación y el avance de los tumores malignos. Estos datos sugieren que un inhibidor de ERK1/2 ejercerá actividad pleiotrópica, por ejemplo efectos preapoptóticos, antiproliferativos, antimetastásicos y antiangiogénicos, y ofrecerá una oportunidad terapéutica contra un amplio grupo de tumores humanos.
Existe un creciente cuerpo de evidencia que implica la activación constitutiva de la ruta ERK MAPK en el comportamiento oncógeno de determinados cánceres. Se encuentran mutaciones activantes de Ras en ~30 % de todos los cánceres, con algunos, tales como cáncer pancreático (90 %) y de colon (50 %), que ostentan tasas particularmente altas de mutación (ref). También se han identificado mutaciones de Ras en 9-15 % de los melanomas, pero las mutaciones somáticas de sentido erróneo en B-Raf que confieren activación constitutiva son más frecuentes y se encuentran en 60-66 % de los melanomas malignos. Las mutaciones activantes de Ras, Raf y MEK son capaces de transformar oncógenamente los fibroblastos in vitro, y las mutaciones de Ras o Raf conjuntamente con la pérdida de un gen supresor de tumores (por ejemplo p16INK4A) pueden causar un desarrollo espontáneo del tumor in vivo. Se ha demostrado una mayor actividad de ERK en estos modelos y también ha sido comunicada ampliamente en tumores humanos apropiados. En el melanoma, la elevada actividad basal de ERK resultante de las mutaciones B-Raf o N-Ras o la activación del factor de crecimiento autocrino está bien documentada y se ha asociado al crecimiento tumoral rápido, a mayor supervivencia celular y a resistencia a la apoptosis. Además, la activación de ERK se considera una fuerza conductora importante detrás del comportamiento altamente metastásico del melanoma asociado a una mayor expresión tanto de proteasas degradantes de la matriz extracelular como de integrinas que promueven la invasión así como de la disminución de las moléculas de adhesión E-cadherina que normalmente median las interacciones de los queratinocitos para controlar el crecimiento de los melanocitos. Estos datos considerados en su conjunto, señalan a ERK como un objetivo terapéutico promisorio para el tratamiento del melanoma, una enfermedad intratable en la actualidad.
Sumario de la invención
Se ha encontrado en la actualidad que los compuestos de esta invención y composiciones farmacéuticamente aceptables de los mismos, son eficaces como inhibidores de la proteína cinasa ERK. Estos compuestos tienen las fórmulas:

o sales farmacéuticamente aceptables de los mismos.
Estos compuestos, sales y composiciones farmacéuticamente aceptables de los mismos, son útiles para tratar o atenuar la gravedad de diversos trastornos, especialmente trastornos proliferativos, tales como el cáncer.
Los compuestos provistos por esta invención también son útiles para el estudio de las cinasas en fenómenos biológicos y patológicos y el estudio de las vías de transducción intracelular de señales mediadas por dichas cinasas, y la evaluación comparativa de nuevos inhibidores de las cinasas.
Descripción detallada de ciertas realizaciones de la invención
1. Descripción general de los compuestos de la invención:
La presente invención se refiere a compuestos de fórmulas:

o sales farmacéuticamente aceptables de los mismos.
2. Compuestos y definiciones:
Los compuestos de esta invención incluyen los descritos precedentemente en general, y se ilustran además mediante las clases, subclases y especies dadas a conocer en el presente documento. Como se usa en el presente documento,
se aplicarán las definiciones siguientes a menos que se indique lo contrario. Para los propósitos de esta invención, los elementos químicos se identifican de conformidad con la tabla periódica de los elementos, versión CAS, Handbook of Chemistry and Physics, 75a Ed. Además, otros principios generales de química orgánica se describen en "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999, y "March's Advanced Organic Chemistry", 5a Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, Nueva York: 2o0l.
Como se usa en el presente documento, el término "profármaco" se refiere a un derivado de una molécula de fármaco precursora que requiere transformación dentro del organismo para liberar el principio activo, y que tiene mejores propiedades físicas y/o de liberación respecto a la molécula de fármaco precursora. Los profármacos se diseñan para mejorar propiedades farmacéuticas y/o farmacocinéticas asociadas a la molécula de fármaco precursora. La ventaja de un profármaco se basa en sus propiedades físicas, como mayor solubilidad en agua para la administración parenteral a pH fisiológico en comparación con el fármaco precursor, o que aumenta la absorción en el tubo digestivo, o que puede aumentar la estabilidad del fármaco en el almacenamiento a largo plazo. En los últimos años se han explorado varios tipos de derivados biorreversibles para utilizar en el diseño de profármacos. El uso de ésteres como un tipo de profármaco para fármacos que contienen una función carboxilo o hidroxilo es conocido en la técnica, como se describe por ejemplo en "The Organic Chemistry of Drug Design and Drug Interaction" Richard Silverman, publicado por Academic Press (1992).
Como se describe en el presente documento, los compuestos de la invención pueden estar opcionalmente sustituidos con uno o más sustituyentes, tales como los que se ilustran en general antes, o como se ejemplifican mediante clases, subclases y especies particulares de la invención. Se apreciará que la frase "opcionalmente sustituido" se usa indistintamente con la frase "sustituido o sin sustituir". En general, el término "sustituido", ya sea precedido por el término "opcionalmente", o no, alude al reemplazo de los radicales hidrógeno de una determinada estructura por el radical de un sustituyente especificado. A menos que se indique lo contrario, un grupo opcionalmente sustituido puede tener un sustituyente en cada posición sustituible del grupo, y cuando más de una posición en cualquier estructura determinada pueda ser sustituida con más de un sustituyente elegido de un grupo especificado, el sustituyente puede ser el mismo o diferente en cada posición.
Las combinaciones de sustituyentes previstas por esta invención son preferentemente las que resultan en la formación de compuestos estables o químicamente factibles. El término "estable", como se usa en el presente documento, se refiere a compuestos que no se alteran sustancialmente cuando son sometidos a las condiciones para permitir su producción, detección y preferentemente su recuperación, purificación y uso para uno o más de los propósitos dados a conocer en el presente documento. En algunas realizaciones, un compuesto estable o un compuesto químicamente factible es aquel que no se altera sustancialmente cuando se mantiene a una temperatura de 40 °C o menor, en ausencia de humedad u otras condiciones químicamente reactivas, durante al menos una semana.
El término "alifático" o la expresión "grupo alifático" como se usan en el presente documento, significan una cadena lineal (es decir no ramificada) o ramificada, una cadena de hidrocarburo sustituida o sin sustituir que está completamente saturada o que contiene una o más unidades de insaturación, o un hidrocarburo monocíclico que está completamente saturado o que contiene una o más unidades de insaturación, pero que no es aromático (también denominado en el presente documento como "carbociclo", "cicloalifático" o "cicloalquilo"), que tiene un solo punto de unión al resto de la molécula. En ciertas realizaciones, los grupos alifáticos contienen de 1 a 6 átomos de carbono alifáticos, y aún en otras realizaciones los grupos alifáticos contienen de 1 a 4 átomos de carbono alifáticos. En algunas realizaciones, "cicloalifático" (o "carbociclo" o "cicloalquilo") se refiere a un hidrocarburo C3-C6 monocíclico completamente saturado o que contiene una o más unidades de insaturación, pero que no es aromático, que tiene un único punto de unión al resto de la molécula. Los grupos alifáticos adecuados incluyen, pero sin limitarse a, grupos alquilo, alquenilo o alquinilo, lineales o ramificados, sustituidos o sin sustituir e híbridos de los mismos, tales como (cicloalquil)alquilo, (cicloalquenil)alquilo o (cicloalquil)alquenilo.
El término "insaturado", como se usa en el presente documento, significa que un resto tiene una o más unidades de insaturación.
Los términos "haloalquilo", "haloalquenilo" y "haloalcoxi" significan alquilo, alquenilo o alcoxi, según el caso, sustituido con uno o más átomos de halógeno. El término "halógeno" significa F, Cl, Br o I.
El término "arilo" empleado solo o como parte de un resto más grande como un "aralquilo", "aralcoxi" o "ariloxialquilo", se refiere a sistemas de anillo monocíclicos, bicíclicos y tricíclicos que tienen un total de cinco a catorce miembros en el anillo, en donde al menos un anillo del sistema es aromático y en donde cada anillo del sistema contiene de 3 a 7 miembros. El término "arilo" se puede usar indistintamente con la expresión "anillo arilo".
Un grupo arilo (incluidos aralquilo, aralcoxi, ariloxialquilo y similares) o heteroarilo (incluidos heteroaralquilo y heteroarilalcoxi y similares) puede contener uno o más sustituyentes. Los sustituyentes adecuados en el átomo de carbono insaturado de un grupo arilo o heteroarilo se eligen entre halógeno; Ro; ORo; SRo; 1,2-metilenodioxi, 1,2-etilenodioxi; fenilo (Ph) opcionalmente sustituido con Ro ;-O(Ph) opcionalmente sustituido con Ro; (CH2)1-2(Ph), opcionalmente sustituido con Ro ; CH=CH(Ph), opcionalmente sustituido con Ro ; NO2 ; CN; N(R° )2 ; NR° C(O)R° ; NR° C(O)N(R° )2 ; NRoCO2Ro ; -NRoNRoC(O)Ro; NRoNRoC(O)N(Ro)2; NRoNRoCO2Ro; C(O)C(O)Ro ; C(O)CH2C(O)Ro;
CO2Ro ; C(O)Ro ; C(O)N(Ro)2; OC(O)N(Ro)2 ; S(O)2Ro ; SO2N(Ro)2 ; S(O)Ro; NR° SO2N(R° )2; NR° SO2R° ; C(=S)N(Ro)2; C(=NH)-N(r °)2; o (CH2)o-2NHC(O)R° donde en cada aparición independiente de Ro se elige entre hidrógeno, alifático C1-6 opcionalmente sustituido, un anillo heteroarilo o heterocíclico de 5-6 miembros sin sustituir, fenilo, O(Ph) o CH2(Ph), o, no obstante la definición anterior, dos apariciones independientes de R° , en el mismo sustituyente o sustituyentes diferentes, tomados juntos con el átomo o los átomos a los cuales el grupo R° está unido, forman un anillo cicloalquilo, heterociclilo, arilo o heteroarilo de 3 a 8 miembros que tiene 0-4 heteroátomos elegidos independientemente entre nitrógeno, oxígeno o azufre. Los sustituyentes opcionales en el grupo alifático de R° se eligen entre NH2, NH(alifático C1-4), N(alifático Ci -4)2, halógeno, alifático C1-4, OH, O(alifático C1-4), NO2, CN, CO2H, CO2(alifático C1-4), O(haloalifático C1-4) o haloalifático C1-4 , en donde cada uno de los grupos alifáticos C1-4 precedentes de Ro no está sustituido.
Un grupo alifático o heteroalifático o un anillo heterocíclico no aromático puede contener uno o más sustituyentes. Los ejemplos de sustituyentes adecuados en el carbono saturado de un grupo alifático o heteroalifático, o de un anillo heterocíclico no aromático se eligen entre los enumerados antes para el carbono insaturado de un grupo arilo o heteroarilo e incluyen además los siguientes: =O, =S, =NNHR*, =NN(R*)2 , =NNHC(O)R*, =NNHCO2(alquilo), =NNHSO2(alquilo) o =NR*, donde cada R* se elige independientemente entre hidrógeno o un alifático C1-6 opcionalmente sustituido. Los sustituyentes opcionales en el grupo alifático de R* se eligen entre NH2, NH(alifático C1-4), N(alifático C1-4)2 , halógeno, alifático C1-4, Oh , O(alifático C1-4), NO2 , CN, CO2H, CO2(alifático C1-4), O(haloalifático C1-4) o haloalifático C1-4 , en donde cada uno de los grupos alifáticos C1-4 precedentes de R* no está sustituido.
Los sustituyentes opcionales en el nitrógeno de un anillo heterocíclico no aromático se eligen entre R+, N(R+)2 , C(O)R+ , CO2R+, C(O)C(O)R+ , C(O)CH2C(O)R+, SO2R+ , SO2N(R+)2 , C(=S)N(R+)2, C(=NH)-N(R+)2 o NR+SO2R+ ; en donde R+ es hidrógeno, un alifático C1-6 opcionalmente sustituido, fenilo opcionalmente sustituido, O(Ph) opcionalmente sustituido, CH2(Ph) opcionalmente sustituido, (CH2)1-2(Ph) opcionalmente sustituido; CH=CH(Ph) opcionalmente sustituido; o un anillo heteroarilo o heterocíclico de 5-6 miembros sin sustituir que tiene uno a cuatro heteroátomos elegidos independientemente entre oxígeno, nitrógeno o azufre, o, no obstante la definición anterior, dos apariciones independientes de R+ , en el mismo sustituyente o sustituyentes diferentes, tomados junto con el átomo o los átomos a los cuales cada grupo R+ está unido, forman un anillo cicloalquilo, heterociclilo, arilo o heteroarilo de 3-8 miembros que tiene 0-4 heteroátomos elegidos independientemente entre nitrógeno, oxígeno o azufre. Los sustituyentes opcionales en el grupo alifático o el anillo fenilo de R+ se eligen entre NH2, NH(alifático C1-4), N(alifático C1-4)2, halógeno, alifático C1-4 , OH, O(alifático C1-4), NO2 , CN, CO2H, CO2(alifático C1-4), O(haloalifático C1-4) o halo (alifático C1-4), en donde cada uno de los grupos alifáticos C1-4 precedentes de R+ no está sustituido.
Salvo indicación en contrario, las estructuras descritas también están destinadas a incluir todos los isómeros (por ejemplo, enantiómeros, diastereoisómeros e isómeros geométricos (o conformacionales)) de la estructura; por ejemplo, las configuraciones R y S para cada centro asimétrico, los isómeros de doble enlace (Z) y (E), y los isómeros conformacionales (Z) y (E). Por consiguiente, los isómeros estereoquímicos individuales así como las mezclas de enantiómeros, diastereoisómeros e isómeros geométricos (o conformacionales) de los compuestos de la presente invención están comprendidos por el alcance de la misma. A menos que se indique lo contrario, todos los tautómeros de los compuestos de la invención están comprendidos por el alcance de ésta. Además, a menos que se indique lo contrario, las estructuras descritas en el presente documento también pretenden incluir los compuestos que difieren únicamente en la presencia de uno o más átomos enriquecidos isotópicamente. Por ejemplo, los compuestos que tienen las estructuras de la presente, excepto por el remplazo de un átomo de hidrógeno por un deuterio o tritio, o el remplazo de un átomo de carbono por un carbono enriquecido en 13C o 14C, están comprendidos por el alcance de la invención. Dichos compuestos son útiles, por ejemplo, como herramientas analíticas o sondas en ensayos biológicos.
2. Métodos generales para proporcionar los presentes compuestos:
Los compuestos de esta invención se pueden preparar o aislar en general mediante métodos de síntesis y/o pseudo síntesis conocidos por los expertos en la materia para compuestos análogos y como se ilustra mediante los esquemas generales I, II y III a continuación y los ejemplos preparativos siguientes.
Esquema I

Reactivos y condiciones: (a) i ICI, CH2CI2, ii NaOMe, MeOH; (b) PG-Cl, DMAP, trietilamina; (c) Pd(dppf); (d) R1-NH2; (e) Pd(PPh3)4, 4; (f) desprotección/saponificación; (g) condiciones de acoplamiento.
El Esquema general I anterior muestra un método general para preparar los compuestos de la presente invención. En el paso (a), el compuesto pirrol 1 es yodado y esterificado para formar 2. En el paso (b), el resto pirrol se protege opcionalmente en el -NH- con un grupo protector de amino adecuado para formar 3. Los grupos protectores de amino son bien conocidos en la técnica y se describen en detalle en Protecting Groups in Organic Synthesis, Theodora W. Greene y Peter G. M. Wuts, 1991, publicado por John Wiley and Sons. El resto de yodo del compuesto 3 es desplazado por un ácido o éster borónico apropiado. Como se describe más adelante, se utiliza bis(pinacolato)diborano para formar el compuesto 4 sin embargo se pueden emplear otros ésteres o ácidos borónicos para esta reacción que serán evidentes para los expertos en la materia.
Debido a que los compuestos de la presente están relacionados con un resto de piridina multisustituida, se considera el orden de reacción y se utilizan métodos para activar las posiciones en la piridina para dirigir la regioquímica. En el paso (d) anterior, el primer grupo saliente L1 puede ser desplazado por un alcohol, amina o tiol según se desee. Un experto en la materia comprenderá que se pueden usar diversos grupos salientes L1 para esta reacción. Los ejemplos de tales grupos incluyen, pero no se limitan a, halógeno y éteres activados. Esta reacción puede ser seguida, en el paso (e), por el remplazo de un segundo grupo saliente L2 a través de una reacción de acoplamiento catalizada por un metal o un desplazamiento nucleofílico para formar el compuesto 7. Un experto en la materia comprenderá que se pueden usar diversos grupos salientes L2 para esta reacción. Los ejemplos de dichos grupos incluyen, pero no se limitan a, halógeno, éteres activados, ácido borónico o ésteres borónicos.
En el paso (f), el grupo protector del resto de pirrol se retira por métodos adecuados para retirar el grupo protector de amino usado. Dependiendo de qué grupo protector de amino se use, las condiciones adecuadas para eliminarlo pueden saponificar simultáneamente o de lo contrario proporcionar el resto carboxilato como se describió antes para el compuesto 8. Si las condiciones adecuadas para retirar el grupo protector de amino no son adecuadas para proporcionar el compuesto carboxilato 8, entonces se debe emplear otro paso. Los compuestos de fórmula I se prepararon a partir de 8 acoplando el carboxilato resultante con una amina deseada como se describe en el paso (g). Un experto en la materia comprenderá que diversas condiciones son útiles para dicha reacción de acoplamiento y pueden incluir el paso de activación del resto carboxilato del compuesto 8 antes de, o simultáneamente con, el tratamiento con la amina deseada. Tales condiciones incluyen, pero no se limitan a, las descritas en detalle en la sección de ejemplos siguiente.
Esquema II

Reactivos y condiciones: (a) AC2O/H2O2; (b) HNO3/H2SO4 ; (c) R1-NH2; (d) L2.
El esquema II anterior describe una ruta alternativa para preparar el compuesto intermedio 6 útil para la preparación de los compuestos de la presente invención. En el paso (a), el N-óxido del compuesto 9 se prepara mediante tratamiento con peróxido. El N-óxido compuesto 10 se trata después con ácido nítrico para formar el compuesto nitro 11. El grupo L1 de 11 es desplazado con la amina deseada R1-NH2 para formar 12 y después se introduce el grupo L2 en el paso (d) para obtener el compuesto intermedio 6. El compuesto 6 se puede utilizar después para preparar los compuestos de la presente invención según el esquema general I anterior y los ejemplos que se proporcionan más adelante.
Esquema III
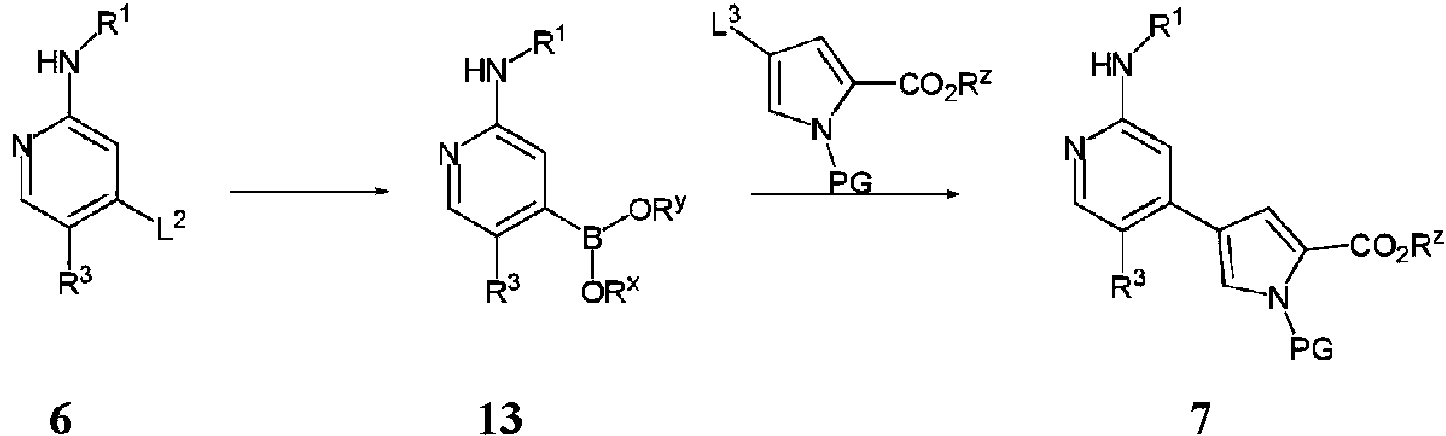
El esquema III anterior muestra un método alternativo para preparar el compuesto 7 a partir de 6. En este método, el grupo L2 del compuesto piridinilo 6 es desplazado por un ácido borónico o éster derivado para formar 13, en donde Rx y Ry tienen los significados definidos posteriormente para los compuestos de fórmula A. Este resto boronato es desplazado después por el grupo saliente L3 del pirrol descrito anteriormente, en donde que L3 es un grupo saliente adecuado, para formar el compuesto 7. El compuesto 7 se usa después para preparar los compuestos de la presente invención por los métodos expuestos antes en los esquemas I y II, por los descritos en la sección de ejemplos y por métodos conocidos por los expertos en la materia.
Un experto en la materia comprenderá que se pueden preparar diversos compuestos de la presente invención según el método general de los esquemas I, II y III, y los Ejemplos de síntesis expuestos a continuación.
De acuerdo con otro caso, la presente solicitud se refiere a un compuesto de fórmula A:

o una sal del mismo, en donde:
PG es un grupo protector de amino adecuado;
Rz es un grupo protector de carboxilato adecuado; y
Rx y Ry son independientemente hidrógeno o alifático C1-6 opcionalmente sustituido, o:
Rx y Ry se toman juntos para formar un anillo de 5-7 miembros opcionalmente sustituido.
Los grupos protectores de amino adecuados son bien conocidos en la técnica e incluyen los descritos en detalle en Protecting Groups in Organic Synthesis, Theodora W. Greene y Peter G. M. Wuts, 1991, publicado por John Wiley and Sons. En ciertas realizaciones, el grupo PG de A es un resto alquilo o arilsulfonilo. Los ejemplos de tales grupos incluyen mesilo, tosilo, nosilo, brosilo y 2,4,6-trimetilbencenosulfónico ("Mts"). Otros ejemplos de tales grupos son Bn,
PMB, Ms, Ts, SiRs, MOM, BOM, Tr, Ac, CO2R, CH2OCH2CH2Si(CHs)3.
Los grupos protectores de carboxilato adecuados son bien conocidos en la técnica y se describen en detalle en Protecting Groups in Organic Synthesis, Theodora W. Greene y Peter G. M. Wuts, 3a edición, 1999, publicado por John Wiley and Sons. En cierta realizaciones, el grupo Rz de A es un grupo alifático C1-6 opcionalmente sustituido o un grupo arilo opcionalmente sustituido. Los ejemplos de grupos Rz adecuados incluyen metilo, etilo, propilo, isopropilo, butilo, isobutilo, bencilo y fenilo en donde cada grupo está opcionalmente sustituido.
En ciertos casos, uno o ambos de Rx y Ry son hidrógeno.
En otros casos, Rx y Ry se toman juntos para formar un anillo de 5-6 miembros opcionalmente sustituido. Aún en otras realizaciones, Rx y Ry se toman juntos para formar un resto 4,4,5,5-tetrametildioxaborolano. Otros derivados boronato adecuados contemplados incluyen ácido borónico, B(O-alifático C1-10)2 y B(O-arilo)2.
De acuerdo con otro caso más, la presente solicitud proporciona un compuesto de fórmula B:

o una sal del mismo, en donde:
R1 es un grupo alifático C1-6, en donde R1 está opcionalmente sustituido con hasta 2 grupos elegidos independientemente entre -OR o -haloalquilo C1-3;
cada R es independientemente hidrógeno o alifático C1-4 ;
R3 es hidrógeno, alifático C1-3, flúor o cloro; y
L2 es un grupo saliente adecuado.
En ciertos casos, la presente solicitud proporciona un compuesto de fórmula B, como se definió en general y en las clases y subclases descritas antes y en esta sección, en donde L2 no es yodo cuando R3 es cloro y R1 es isopropilo.
Un grupo saliente adecuado es un grupo químico que es desplazado fácilmente por un resto químico entrante deseado. Por lo tanto, la elección de un grupo saliente adecuado específico depende de su capacidad para desplazar fácilmente al resto químico entrante de fórmula A. Los grupos salientes adecuados son bien conocidos en la técnica, por ejemplo, véase, "Advanced Organic Chemistry," Jerry March, 5a Ed., pág. 351-357, John Wiley and Sons, N.Y. Tales grupos salientes incluyen, pero no se limitan a, halógeno, grupos alcoxi, sulfoniloxi, alquilsulfonilo opcionalmente sustituido, alquenilsulfonilo opcionalmente sustituido, arilsulfonilo opcionalmente sustituido y diazonio. Los ejemplos de grupos salientes adecuados incluyen cloro, yodo, bromo, flúor, metanosulfonilo (mesilo), tosilo, triflato, nitrofenilsulfonilo (nosilo) y bromofenilsulfonilo (brosilo). En ciertos casos, el grupo L2 de B es yodo.
De acuerdo con un caso alternativo, el grupo saliente adecuado se puede generar in situ dentro del medio de reacción. Por ejemplo, L2 en un compuesto de fórmula B se puede generar in situ a partir de un precursor de ese compuesto de fórmula B, en donde dicho precursor contiene un grupo que es fácilmente reemplazado por L2 in situ. En una ilustración específica de un reemplazo de ese tipo, dicho precursor de un compuesto de fórmula B contiene un grupo (por ejemplo, un grupo cloro o un grupo hidroxilo) que es reemplazado in situ por L2 , tales como un grupo yodo. La fuente del grupo yodo puede ser, por ejemplo, yoduro de sodio. La generación in situ de un grupo saliente adecuado de ese tipo es bien conocida en la técnica, por ejemplo, véase, "Advanced Organic Chemistry," Jerry March, pág. 430-431, 5a Ed., John Wiley and Sons, N.Y.
De acuerdo con ciertos casos, el resto R1 de fórmula B, es un grupo alifático C1-4 opcionalmente sustituido con -OR o -haloalquilo C1-3. En ciertas realizaciones, el resto R1 de fórmula B es un grupo alifático C1-4 opcionalmente sustituido con -OH, -CH2 F, -CHF2 o -CF3. En otras realizaciones, el resto R1 de fórmula B es un grupo alifático C1-4 opcionalmente sustituido con -OH. Aún en otras realizaciones, R1 no está sustituido.
De acuerdo con otro caso, el resto R1 de fórmula B es isopropilo, 2-butilo, ciclopropilo o etilo, en donde cada resto está opcionalmente sustituido con -OH o -CF3.
En otros casos, el resto R3 de fórmula B es hidrógeno, metilo o cloro.
Un compuesto de fórmula B se puede preparar a partir de un compuesto de fórmula B':
o una sal del mismo, en donde:
R3 es hidrógeno, alifático C-i-3, flúor o cloro; y
L1 y L2 son cada uno independientemente un grupo saliente adecuado.
En ciertos casos, la presente solicitud proporciona un compuesto de fórmula B', como se definió en general y en las clases y subclases descritas antes y en esta sección, en donde L2 no es un resto boronato cuando R3 es cloro y R1 es flúor.
En ciertos casos, se proporciona un compuesto de B' en donde L2 es -B(ORx)(ORy). En otras realizaciones, uno o ambos de Rx y Ry son hidrógeno. En otras realizaciones, Rx y Ry se toman juntos para formar un anillo de 5-6 miembros opcionalmente sustituido. Aún en otras realizaciones, Rx y Ry se toman juntos para formar un resto 4,4,5,5-tetrametildioxaborolano.
Como se ha descrito anteriormente, un grupo saliente adecuado es un grupo químico que es desplazado fácilmente por un resto químico entrante deseado. Los grupos salientes adecuados son bien conocidos en la técnica, por ejemplo, véase, "Advanced Organic Chemistry," Jerry March, 5a Ed., pág. 351-357, John Wiley and Sons, N.Y. Dichos grupos salientes incluyen. pero no se limitan a, halógeno, grupos alcoxi, sulfoniloxi, alquilsulfonilo opcionalmente sustituido, alquenilsulfonilo opcionalmente sustituido, arilsulfonilo opcionalmente sustituido y diazonio. Los ejemplos de grupos salientes adecuados incluyen cloro, yodo, bromo, flúor, metanosulfonilo (mesilo), tosilo, triflato, nitrofenilsulfonilo (nosilo) y bromofenilsulfonilo (brosilo). En ciertas realizaciones, el resto L1 de B' es halógeno. En otra realización, el resto L1 de B' es un grupo alquilsulfonilo opcionalmente sustituido, alquenilsulfonilo opcionalmente sustituido o arilsulfonilo opcionalmente sustituido. En otras realizaciones, el resto L1 de B' es flúor.
De acuerdo con un caso alternativo, el grupo saliente adecuado se puede generar in situ dentro del medio de reacción. Por ejemplo, los grupos L1 o L2 en un compuesto de fórmula B' se pueden generar in situ a partir de un precursor de ese compuesto de fórmula B', en donde dicho precursor contiene un grupo que es fácilmente reemplazado por L1 o L2 in situ. La generación in situ de un grupo saliente adecuado de ese tipo es bien conocida en la técnica, por ejemplo, véase, "Advanced Organic Chemistry," Jerry March, pág. 430-431,5a Ed., John Wiley and Sons, N.Y.
De acuerdo con otro caso, la presente solicitud proporciona un método para preparar un compuesto de fórmula B:

o una sal del mismo, que comprende el paso de hacer reaccionar un compuesto de fórmula B':

o una sal del mismo, con un compuesto de fórmula R1-NH2 en donde dicha reacción se realiza en un medio adecuado y en donde:
R1 es un grupo alifático C1-6, en donde R1 está opcionalmente sustituido con hasta 2 grupos elegidos independientemente entre -OR o -haloalquilo C1-3;
R es hidrógeno o alifático C1-4;
R3 es hidrógeno, alifático C1-3, flúor o cloro; y
L1 y L2 son cada uno independientemente un grupo saliente adecuado.
En ciertos casos, dicha reacción se lleva a cabo opcionalmente en presencia de una base adecuada. Un experto en la materia comprenderá que el desplazamiento de un grupo saliente por un resto amino se logra con o sin la presencia de una base adecuada. Tales bases adecuadas son bien conocidas en la técnica e incluyen bases orgánicas e inorgánicas.
Un medio adecuado es un disolvente o una mezcla de disolventes que, en combinación con los compuestos combinados, puede facilitar el avance de la reacción entre ellos. El disolvente adecuado puede solubilizar uno o más de los componentes de la reacción, o alternativamente, el disolvente adecuado puede facilitar la agitación de una suspensión de uno o más de los componentes de la reacción. Los ejemplos de disolventes adecuados útiles en la presente invención son un disolvente prótico, un hidrocarburo halogenado, un éter, un hidrocarburo aromático, un disolvente polar o no polar aprótico, o cualquiera de las mezclas de los mismos. Tales mezclas incluyen, por ejemplo, mezclas de disolvente próticos y apróticos, tales como benceno/metanol/agua; benceno/agua; DME/agua y similares. Estos y otros disolventes adecuados son bien conocidos en la técnica, por ejemplo, véase, "Advanced Organic Chemistry," Jerry March, 5a edición, John Wiley and Sons, N.Y.
De acuerdo con otro caso más, uno o más reactivos pueden actuar como el disolvente adecuado. Por ejemplo, una base orgánica, tales como trietilamina o diisopropiletilamina, si se utiliza en dicha reacción, puede servir como el disolvente además de su rol como reactivo de basificación.
En ciertos casos, la presente solicitud proporciona un compuesto de fórmula B' en donde R1 y R3 son los definidos en general y en las clases y subclases descritas antes y en esta sección.
De acuerdo con otro aspecto, la presente solicitud proporciona un compuesto de fórmula C:

o una sal del mismo, en donde:
PG es un grupo protector de amino adecuado;
Rz es un grupo protector de carboxilato adecuado;
R1 es un grupo alifático C1-6, en donde R1 está opcionalmente sustituido con hasta 2 grupos elegidos independientemente entre -OR o -haloalquilo C1-3;
cada R es independientemente hidrógeno o alifático C1-4; y
R3 es hidrógeno, alifático C1-3, flúor o cloro.
Como se ha indicado anteriormente, los grupos protectores de amino adecuados son bien conocidos en la técnica e incluyen los descritos en detalle en Protecting Groups in Organic Synthesis, Theodora W. Greene y Peter G. M. Wuts, 1991, publicado por John Wiley and Sons. En ciertas realizaciones, el grupo PG de C es un resto alquilo o arilsulfonilo. Los ejemplos de dichos grupos incluyen mesilo, tosilo, nosilo, brosilo y 2,4,6-trimetilbencenosulfónico ("Mts").
Los grupos protectores de carboxilato adecuados son bien conocidos en la técnica y se describen en detalle en Protecting Groups in Organic Synthesis, Theodora W. Greene y Peter G. M. Wuts, 1991, publicado por John Wiley and Sons. En ciertas realizaciones, el grupo Rz de C es un grupo alifático C1-6 opcionalmente sustituido o un grupo arilo opcionalmente sustituido. Los ejemplos de grupos Rz adecuados incluyen metilo, etilo, propilo, isopropilo, butilo, isobutilo, bencilo y fenilo, en donde cada grupo está opcionalmente sustituido.
De acuerdo con ciertos casos, el resto R1 de fórmula C, es un grupo alifático C1-4 opcionalmente sustituido con -OR o -haloalquilo C1-3. En ciertas realizaciones, el resto R1 de fórmula C es un grupo alifático C1-4 opcionalmente sustituido con -OH, -CH2F, -CHF2 o -CF3. En otras realizaciones, el resto R1 de fórmula C es un grupo alifático C1-4 opcionalmente sustituido con -OH. Aún en otras realizaciones, R1 no está sustituido.
De acuerdo con otro caso, el resto R1 de fórmula C es isopropilo, 2-butilo, ciclopropilo o etilo, en donde cada resto está opcionalmente sustituido con -OH o -CF3.
En otros casos, el resto R3 de fórmula C es hidrógeno, metilo o cloro.
Aún en otro caso de la presente solicitud se refiere a un método para preparar un compuesto de fórmula C:

o una sal del mismo, que comprende el paso de hacer reaccionar un compuesto de fórmula A:

o una sal del mismo, con un compuesto de fórmula B:
o una sal del mismo, en donde dicha reacción se lleva a cabo en un medio adecuado y en donde:
PG es un grupo protector de amino adecuado;
L2 es un grupo saliente adecuado.
Rz es un grupo protector de carboxilato adecuado;
Rx y Ry son independientemente hidrógeno o alifático C1-6 opcionalmente sustituido, o:
Rx y Ry se toman juntos para formar un anillo de 5-7 miembros opcionalmente sustituido;
R1 es un grupo alifático C1-6, en donde R1 está opcionalmente sustituido con hasta 2 grupos elegidos independientemente entre -OR o -haloalquilo C1-3;
cada R es independientemente hidrógeno o alifático C1-4 ; y
R3 es hidrógeno, alifático C1-3, flúor o cloro.
En ciertos casos, dicha reacción se lleva a cabo en presencia de Ni (II), Pd (0) o Pd(II) donde cada catalizador puede estar asociado a un ligando, tales como ligandos a base de ferroceno o fosfina. En otras realizaciones, dicha reacción se lleva a cabo en presencia de Pd(PPh3)4.
Un medio adecuado es un disolvente o una mezcla de disolventes que, en combinación con los compuestos combinados, puede facilitar el avance de la reacción entre ellos. El disolvente adecuado puede solubilizar uno o más de los componentes de la reacción, o alternativamente, el disolvente adecuado puede facilitar la agitación de una suspensión de uno o más de los componentes de la reacción. Los ejemplos de disolventes adecuados útiles en la presente invención son un disolvente prótico, un hidrocarburo halogenado, un éter, un hidrocarburo aromático, un disolvente polar o no polar aprótico, o cualquiera de las mezclas de los mismos. Tales mezclas se incluyen, por ejemplo, mezclas de disolvente próticos y apróticos, tales como benceno/metanol/agua; benceno/agua; DME/agua y similares.
Estos y otros disolventes adecuados son bien conocidos en la técnica, por ejemplo, véase, "Advanced Organic Chemistry," Jerry March, 5a edición, John Wiley and Sons, N.Y.
En ciertos casos, la reacción entre los compuestos A y B para formar C se lleva a cabo en una mezcla de DME y agua.
En ciertos casos, la reacción entre los compuestos A y B para formar C se lleva a cabo a una temperatura que varía de entre aproximadamente 20 °Ca 150 °C. En otras realizaciones, la reacción entre los compuestos A y B para formar C se lleva a cabo con irradiación de microondas a una temperatura que varía de entre aproximadamente 100 °C a 250 °C.
Aún en otros casos, la reacción entre los compuestos A y B para formar C se lleva a cabo a un pH algo básico. De acuerdo con otro caso, la presente solicitud proporciona un profármaco de un compuesto de fórmula I en donde dicho profármaco tiene la fórmula II:

o una sal farmacéuticamente aceptable del mismo, en donde:
R1 es un grupo alifático C1-6, en donde R1 está opcionalmente sustituido con hasta 2 grupos elegidos independientemente entre -OR, -OR4 o -haloalquilo C1-3;
cada R es independientemente hidrógeno o alifático C1-6 ;
cada R2 es independientemente R, flúor o cloro;
m es 0, 1 o 2;
R3 es hidrógeno, alifático C1-3 , flúor o cloro;
cada R4 es independientemente hidrógeno, -C(R)2O-R5 o R5, siempre que al menos un grupo R4 o R8 no sea hidrógeno;
cada R5 es independientemente -C(O)R6 , -C(O)OR6 , -C(O)-Q-R6 , -C(O)-(CH2)n-C(O)OR6, -C(O)-(CH2)n-C(O)N(R7)2 , -C(O)-(CH2)n-CH(R6)N(R7)2, -P(O)(OR6)2 ;
cada R6 es independientemente hidrógeno, un grupo alifático C1-6 opcionalmente sustituido o un anillo de 5-8 miembros saturado, parcialmente insaturado o totalmente insaturado, opcionalmente sustituido, que tiene 0-4 heteroátomos elegidos independientemente entre nitrógeno, oxígeno o azufre;
cada R7 es independientemente hidrógeno, -C(O)R6 , -C(O)OR6 , -S(O)2 R6, -OR6 , un grupo alifático C1-6 opcionalmente sustituido o un anillo de 5-8 miembros saturado, parcialmente insaturado o totalmente insaturado, opcionalmente sustituido, que tiene 0-4 heteroátomos elegidos independientemente entre nitrógeno, oxígeno o azufre, o:
dos R6 en el mismo átomo de nitrógeno tomados juntos con el átomo de nitrógeno al cual están unidos forman un anillo de 4-7 miembros saturado, parcialmente insaturado o totalmente insaturado que tiene 1-3 heteroátomos además del átomo de nitrógeno, elegidos independientemente entre nitrógeno, oxígeno o azufre;
cada n es 0-6;
Q es una cadena alquilideno C1-10 opcionalmente sustituida en donde de cero a cuatro unidades de metileno de Q están reemplazadas independientemente por -O-, -N(R)-, -S-, -S(O)-, -S(O)2- o - C(O)-; y
R8 es hidrógeno o -C(R)2O-R5.
De acuerdo con ciertos casos, el resto R1 de fórmula II es un grupo alifático C1-4 opcionalmente sustituido con -OR o -haloalquilo C1-3. En ciertas realizaciones, el resto R1 de fórmula Ii es un grupo alifático C1-4 opcionalmente sustituido con -OH, -CH2F, -CHF2 o -CF3. En otras realizaciones casos, el resto R1 de fórmula II es un grupo alifático C1-4 opcionalmente sustituido con -OH. Aún en otras realizaciones, R1 no está sustituido.
De acuerdo con otros casos, el resto R1 de fórmula II es isopropilo, 2-butilo, ciclopropilo o etilo, en donde cada resto está opcionalmente sustituido con -OH, -CHF2 , -CH2F o -CF3. De acuerdo con otra realización, el resto R1 de fórmula II es isopropilo, 2-butilo, ciclopropilo o etilo, en donde cada resto está opcionalmente sustituido con -OH o -CF3. Otro aspecto de la presente divulgación se refiere a un compuesto de fórmula II en donde cada R2 es independientemente hidrógeno, alifático C1-3 o cloro. De acuerdo con otro aspecto más, la presente solicitud se refiere a un compuesto de fórmula II en donde R2 es cloro y m es 1.
En otros casos, el resto R3 de fórmula II es hidrógeno, metilo o cloro.
En ciertos casos, R4 es C(O)-Q-R6. Aún otras realizaciones casos se refieren a un compuesto de fórmula II en donde R4 es C(O)-Q-R6 y Q es una cadena alquilideno C1-8 opcionalmente sustituida en donde de cero a cuatro unidades de metileno de Q están reemplazadas independientemente por -O-, -N(R)-, -S-, -S(O)-, -S(O)2- o -C(O)- y R6 es el definido
en general y en las clases y subclases descritas antes y en esta sección. De acuerdo con otra realización, Q es una cadena alquilideno C1-8 opcionalmente sustituida en donde de dos a cuatro unidades de metileno de Q están reemplazadas independientemente por -O-. Dichos grupos Q incluyen -CH2OCH2CH2O-, -CH2OCH2CH2OCH2CH2O-y similares.
Aún otro aspecto de la presente divulgación proporciona un compuesto de fórmula II en donde R4 es C(O)-(CH2)n-CH(R6)N(R7)2. En ciertas realizaciones, n es 0-2. En otras realizaciones, R6 es un grupo alifático C1-6 opcionalmente sustituido. Los ejemplos de tales grupos R6 incluyen metilo, bencilo, etilo, isopropilo, t-butilo y similares. Los grupos R7 del C(O)-(CH2)n-CH(R6)N(R7)2 de fórmula II incluyen hidrógeno y un grupo alifático C1-6 opcionalmente sustituido. Los ejemplos de tales grupos incluyen metilo, bencilo, etilo, isopropilo, t-butilo y similares.
De acuerdo con un aspecto, la presente divulgación proporciona un compuesto de fórmula II en donde R8 es hidrógeno. De acuerdo con un caso, R4 es un éster de L-valina.
En ciertas realizaciones de la presente solicitud, el grupo R4 de fórmula II es -P(O)(OR6)2. En otras realizaciones, cada R6 es independientemente hidrógeno o un grupo alifático C1-6 opcionalmente sustituido. Los ejemplos de dichos grupos R6 incluyen metilo, bencilo, etilo, isopropilo, t-butilo y similares. Aún en otras realizaciones, el grupo R4 de fórmula II es -P(O)(OH)2.
En ciertos casos, un compuesto de fórmula II proporciona una mejora con respecto a una o más características físicas o fisiológicas. En otros casos, un compuesto de fórmula II imparte una mejora con respecto a una o más características físicas o fisiológicas.
Los compuestos representativos de la fórmula II se exponen en la Tabla 2 a continuación.
Tabla 2
Los métodos para preparar dichos profármacos incluyen los expuestos en detalle en la sección de Ejemplos posterior y los métodos conocidos por los expertos en la materia.
3. Usos, formulación y administración
Composiciones farmacéuticamente aceptables
Como se ha tratado anteriormente, la presente invención proporciona compuestos que son inhibidores de las proteínas cinasas, y por lo tanto los compuestos de la presente son útiles para el tratamiento de enfermedades, trastornos y afecciones incluidos, pero no se limitan a, cáncer, trastornos autoinmunitarios, trastornos neurodegenerativos y neurológicos, esquizofrenia, trastornos óseos, enfermedad hepática y trastornos cardíacos. En consecuencia, en otro aspecto de la presente invención, se proporcionan composiciones farmacéuticamente aceptables, que contienen cualquiera de los compuestos descritos en el presente documento, y opcionalmente contienen un portador, adyuvante o vehículo farmacéuticamente aceptable. En ciertas realizaciones, estas composiciones pueden contener opcionalmente uno o más agentes terapéuticos adicionales.
Se apreciará también que algunos de los compuestos de la presente invención pueden existir en forma libre para el tratamiento, o cuando proceda, como un derivado farmacéuticamente aceptable de los mismos. De acuerdo con la presente divulgación, un derivado farmacéuticamente aceptable incluye, pero sin limitarse a, sales, ésteres, sales de dichos ésteres, farmacéuticamente aceptables, o cualquier otro aducto o derivado que una vez administrado a un paciente que lo necesita sea capaz de proporcionar, directa o indirectamente, un compuesto como los descritos en el presente documento, o un metabolito o residuo de los mismos.
Como se usa en el presente documento, la expresión "sal farmacéuticamente aceptable" se refiere a las sales que son, al juicio razonable del médico, adecuadas para usar en contacto con los tejidos de los seres humanos y animales inferiores sin toxicidad, irritación, respuesta alérgica ni similares indebidas, y que están de acuerdo con una relación riesgo/beneficio razonable. Una "sal farmacéuticamente aceptable" significa cualquier sal o sal de un éster de un compuesto de esta invención, atóxica, que luego de la administración a un receptor, es capaz de proporcionar, directa o indirectamente, un compuesto de esta invención o un metabolito o un residuo del mismo, activo como inhibidor. Como se usa en el presente documento, la expresión "metabolito o un residuo del mismo, activo como inhibidor" significa que un metabolito o un residuo del mismo es también un inhibidor de la proteína cinasa ERK2.
Las sales farmacéuticamente aceptables son bien conocidas en la técnica. Por ejemplo, S. M. Berge et al., describen sales farmacéuticamente aceptables en detalle en J. Pharmaceutical Sciences, 1977, 66, 1-19. Las sales farmacéuticamente aceptables de los compuestos de esta invención incluyen las derivadas de ácidos y bases inorgánicos y orgánicos, adecuados. Son ejemplos de sales de adición de ácido farmacéuticamente aceptables, atóxicas, las sales de un grupo amino formadas con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico o con ácidos orgánicos, tales como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico o mediante otros métodos utilizados en la técnica, tales como el intercambio iónico. Otras sales farmacéuticamente aceptables incluyen, sales de adipato, alginato, ascorbato, aspartato, bencenosulfonato, benzoato, bisulfato, borato, butirato, canforato, canforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, formiato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, yodohidrato, 2-hidroxietanosulfonato, lactobionato, lactato, laurato, laurilsulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, picrato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato, undecanoato, valerato, y similares. Las sales derivadas de bases adecuadas incluyen sales de metales alcalinos, metales alcalinotérreos, amonio y N+( alquilo C-m )4. Esta invención también contempla la cuaternización de todos los grupos que contienen nitrógeno básico de los compuestos dados a conocer en el presente documento. Se pueden obtener productos solubles o dispersables en agua o aceite mediante dicha cuaternización. Las sales de metales alcalinos o alcalinotérreos representativas incluyen sales de sodio, litio, potasio, calcio, magnesio y similares. Otras sales farmacéuticamente aceptables incluyen, cuando sea apropiado, amonio, amonio cuaternario y cationes amina no tóxicos, formadas usando contraiones, tales como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, aril sulfonato y alquil sulfonato inferior.
Como se ha descrito anteriormente, las composiciones farmacéuticamente aceptables de la presente invención contienen además un portador, adyuvante o vehículo farmacéuticamente aceptable, el cual, como se usa en el presente documento, incluye cualquier y todos los disolventes, diluyentes, u otros vehículos líquidos, auxiliares de dispersión o suspensión, tensioactivos, isotónicos, espesantes o emulsionantes, conservantes, aglutinantes sólidos, lubricantes y similares, según se adapte a la forma farmacéutica particular deseada. Remington's Pharmaceutical Sciences, 16a edición, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) da a conocer diversos portadores utilizados en la formulación de composiciones farmacéuticamente aceptables y técnicas conocidas para la preparación de las mismas. Excepto en la medida en que cualquier medio portador convencional sea incompatible con los compuestos de la invención, por ejemplo por producir algún efecto biológico indeseable o de lo contrario interaccionar de manera nociva con otro(s) componente(s) de la composición farmacéuticamente aceptable, su uso está contemplado como comprendido por el alcance de esta invención. Algunos ejemplos de materiales que pueden servir
como portadores farmacéuticamente aceptables incluyen, pero no se limitan a, intercambiadores de iones, alúmina, estearato de aluminio, lecitina, proteínas del suero, como seroalbúmina humana, sustancias amortiguadoras como fosfatos, glicina, ácido sórbico o sorbato de potasio, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, como sulfato de protamina, fosfato ácido disódico, fosfato ácido de potasio, cloruro de sodio, sales de cinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, poliacrilatos, ceras, polímeros de bloque de polietileno-polioxipropileno, grasa de la lana, azúcares como lactosa, glucosa y sacarosa; almidones como almidón de maíz y almidón de papa; celulosa y sus derivados como carboximetilcelulosa sódica, etilcelulosa y acetato de celulosa; tragacanto en polvo; malta; gelatina; talco; también pueden estar presentes en la composición a criterio del formulador, excipientes como manteca de cacao y cera para supositorios; aceites como aceite de cacahuate, aceite de semilla de algodón; aceite de cártamo; aceite de sésamo, aceite de oliva; aceite de maíz y aceite de soja; glicoles; como propilenglicol o polietilenglicol; ésteres como oleato de etilo y laurato de etilo; agar; amortiguadores del pH como hidróxido de magnesio e hidróxido de aluminio; ácido algínico: agua apirógena; solución salina isotónica; solución de Ringer; alcohol etílico y soluciones amortiguadoras de fosfato, así como otros lubricantes compatibles atóxicos como laurilsulfato de sodio y estearato de magnesio, al igual que colorantes, agentes de liberación, agentes de recubrimiento, edulcorantes, saborizantes y perfumes, conservantes y antioxidantes.
Usos de los compuestos y composiciones farmacéuticamente aceptables
También se desvela un compuesto de la presente invención, o una composición farmacéuticamente aceptable que comprende un compuesto de la presente invención, para su uso en el tratamiento o la reducción de la gravedad del cáncer, un trastorno inmunitario, un trastorno neurodegenerativo o neurológico, una enfermedad hepática o un trastorno cardíaco. En ciertas realizaciones de la presente invención una "cantidad eficaz" del compuesto o la composición farmacéuticamente aceptables en la cantidad eficaz para tratar o atenuar la gravedad de la enfermedad, la afección o el trastorno seleccionado entre cáncer, un trastorno autoinmunitario, un trastorno neurodegenerativo o neurológico, esquizofrenia, un trastorno óseo, una enfermedad hepática o un trastorno cardíaco. Los compuestos y las composiciones, de acuerdo con la presente invención, se pueden administrar utilizando cualquier cantidad y cualquier vía de administración que sean eficaces para tratar o atenuar la gravedad de un cáncer, un trastorno autoinmunitario, un trastorno neurodegenerativo o neurológico, la esquizofrenia, un trastorno óseo, una enfermedad hepática o un trastorno cardíaco. La cantidad exacta necesaria variará de un sujeto a otro, dependiendo de la especie, la edad y el estado general de salud del sujeto, la gravedad de la infección, el fármaco particular, su modo de administración, y similares. Los compuestos de la invención se formulan preferentemente en formas farmacéuticas unitarias para facilitar la administración y la uniformidad de dosis. La expresión "forma farmacéutica unitaria" como se usa en el presente documento se refiere a una unidad físicamente discreta de un fármaco apropiado para el paciente que se va a tratar. Se entenderá, sin embargo, que el uso diario total de los compuestos y las composiciones de la presente invención será decidido por el médico tratante según su criterio profesional. El nivel de dosis eficaz específico para cualquier paciente u organismo particular dependerá de una serie de factores como el trastorno en tratamiento y la gravedad del mismo; la actividad del compuesto específico empleado; la composición específica empleada; la edad, el peso corporal, el estado general de salud, el género y la dieta del paciente; el tiempo de administración, la vía de administración y la velocidad de eliminación del compuesto específico empleado; la duración del tratamiento; otros fármacos utilizados en combinación o simultáneamente con el compuesto específico empleado y factores semejantes conocidos en la técnica médica. El término "paciente" como se usa en el presente documento, significa un animal, preferentemente un mamífero, y muy preferentemente un humano.
Las composiciones farmacéuticamente aceptables de esta invención se pueden administrar a los humanos y otros animales por vía oral, rectal, parenteral, intracisternal, intravaginal, intraperitoneal, tópica (por ejemplo mediante polvos, pomadas o gotas), bucal como un aerosol oral o nasal, o similares, dependiendo de la gravedad de la infección en tratamiento. En ciertas realizaciones, los compuestos de la invención se pueden administrar por vía oral o parenteral en dosis entre aproximadamente 0,01 mg/kg y aproximadamente 50 mg/kg y preferentemente entre aproximadamente 1 mg/kg y aproximadamente 25 mg/kg de peso corporal del sujeto por día, una o más veces al día, para obtener el efecto terapéutico deseado.
Las formas de dosificación líquidas para administración oral incluyen, pero no se limitan a, emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables. Además de los principios activos, las formas farmacéuticas líquidas pueden contener diluyentes inertes comúnmente utilizados en la técnica como, por ejemplo, agua u otros disolventes, solubilizantes y emulsionantes, como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, dimetilformamida, aceites (en particular, aceites de semilla de algodón, maní, maíz, germen de trigo, oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurfurílico, polietilenglicoles y ésteres de sorbitán de ácidos grasos, y mezclas de los mismos. Además de diluyentes inertes, las composiciones orales también pueden contener adyuvantes como humectantes, emulsionantes y suspendentes, edulcorantes, saborizantes y perfumes.
Las preparaciones inyectables, por ejemplo, las suspensiones estériles inyectables acuosas u oleosas se pueden formular de acuerdo con las técnicas conocidas empleando dispersantes o humectantes y suspendentes adecuados. La preparación estéril inyectable también puede ser una solución, suspensión o emulsión estéril inyectable en un diluyente o disolvente atóxico, aceptable para uso parenteral, por ejemplo, como una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que se pueden emplear se encuentran el agua, la solución de Ringer y la
solución de cloruro de sodio isotónica U.S.P. Además, convencionalmente se emplean aceites fijos estériles como disolvente o medio de suspensión. Para este propósito se puede emplear cualquier aceite fijo blando incluidos los mono o diglicéridos sintéticos. Además, en la preparación de inyectables se usan ácidos grasos como el ácido oleico.
Las formulaciones inyectables se pueden esterilizar, por ejemplo, por filtración a través de un filtro de retención de bacterias, o mediante la incorporación de agentes esterilizantes en forma de composiciones sólidas estériles que se pueden disolver o dispersar en agua estéril o cualquier otro medio inyectable estéril antes de usarlas.
Para prolongar el efecto de un compuesto de la presente invención, a menudo es deseable retardar la absorción del compuesto de la inyección subcutánea o intramuscular. Esto se puede lograr mediante el uso de una suspensión líquida de material cristalino o amorfo con baja solubilidad en agua. La velocidad de absorción del compuesto depende entonces de su velocidad de disolución que, a su vez, puede depender del tamaño del cristal y de la forma cristalina. Alternativamente, la absorción retardada de una forma del compuesto administrada por vía parenteral se logra disolviendo o suspendiendo el compuesto en un vehículo oleoso. Las formas inyectables en depot se elaboran por formación de matrices de microencapsulación del compuesto en polímeros biodegradables como poliláctidopoliglicólido. Dependiendo de la proporción entre compuesto y polímero y de la naturaleza del polímero particular empleado, se puede controlar la velocidad de liberación del compuesto. Son ejemplos de otros polímeros biodegradables los poli(ortoésteres) y poli(anhídridos). También se preparan formulaciones inyectables en depot por atrapamiento del compuesto en liposomas o microemulsiones que sean compatibles con los tejidos corporales.
Las composiciones para administración rectal o vaginal son preferentemente supositorios que se pueden preparar mezclando los compuestos de esta invención con excipientes o portadores no-irritantes adecuados como manteca de cacao, polietilenglicol, o una cera para supositorio, que sean sólidos a temperatura ambiente pero líquidos a la temperatura corporal y por lo tanto, se fundan en el recto o la cavidad vaginal y liberen el principio activo.
Las formas de dosificación sólidas para administración oral incluyen cápsulas, comprimidos, pastillas, polvos y gránulos. En dichas formas farmacéuticas sólidas, el principio activo se mezcla con al menos un excipiente o portador inerte farmacéuticamente aceptable como citrato de sodio o fosfato dicálcico y/o a) rellenos o diluyentes, tales como almidones, lactosa, sacarosa, glucosa, manitol y ácido silícico, b) aglutinantes, tales como por ejemplo carboximetilcelulosa, alginatos, gelatina, polivinilpirrolidona, sacarosa y acacia, c) humectantes, tales como glicerol, d) desintegrantes, tales como agar-agar, carbonato de calcio, almidón de papa o tapioca, ácido algínico, ciertos silicatos y carbonato de sodio; e) retardantes de la solución, tales como parafina; f) aceleradores de la absorción, tales como compuestos de amonio cuaternario, g) humectantes, tales como, por ejemplo, alcohol cetílico y monoestearato de glicerol, h) absorbentes, tales como caolín y bentonita y i) lubricantes, tales como talco, estearato de calcio, estearato de magnesio, polietilenglicoles sólidos, laurilsulfato de sodio y mezclas de los mismos. En el caso de las cápsulas, los comprimidos y las pastillas, las formas farmacéuticas también pueden comprender agentes tamponantes.
También se pueden emplear composiciones sólidas de tipo similar como relleno de cápsulas de gelatina blanda y dura, utilizando excipientes como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, pastillas y gránulos se pueden preparar con recubrimientos y cubiertas, como recubrimientos entéricos y otros recubrimientos conocidos en la técnica de la formulación farmacéutica. Dichas formas pueden contener opcionalmente opacificantes y también pueden ser de una composición tal, que liberen sólo el principio o principios activos, o los liberen preferencialmente en cierta parte del tracto intestinal, opcionalmente, de manera retardada. Los ejemplos de composiciones de inclusión que se pueden utilizar comprenden sustancias poliméricas y ceras. Se pueden emplear composiciones sólidas de tipo similar como rellenos de cápsulas de gelatina blanda y dura, utilizando excipientes como lactosa o azúcar de la leche así como polietilenglicoles de alto peso molecular y similares.
Los compuestos activos también pueden estar en forma microencapsulada con uno o más excipientes como los indicados antes. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, pastillas y gránulos se pueden preparar con recubrimientos y cubiertas, como recubrimientos entéricos, recubrimientos de liberación controlada y otros recubrimientos conocidos en la técnica de la formulación farmacéutica. En dichas formas farmacéuticas sólidas el principio activo se puede mezclar con al menos un diluyente inerte como sacarosa, lactosa o almidón. Dichas formas farmacéuticas también pueden contener, como es la práctica habitual, sustancias adicionales diferentes de los diluyentes inertes, por ejemplo, lubricantes de compresión y otros auxiliares de compresión como estearato de magnesio y celulosa microcristalina. En el caso de las cápsulas, los comprimidos y las pastillas, las formas farmacéuticas también pueden contener amortiguadores del pH. Dichas formas pueden contener opcionalmente opacificantes y también pueden ser de una composición tal, que liberen sólo el principio o principios activos, o los liberen preferencialmente en cierta parte del tracto intestinal, opcionalmente, de manera retardada. Los ejemplos de composiciones de inclusión que se pueden utilizar comprenden sustancias poliméricas y ceras.
Las formas farmacéuticas para administración tópica o transdérmica de un compuesto de esta invención incluyen pomadas, pastas, cremas, lociones, geles, polvos, soluciones, aerosoles, inhalantes o parches. El principio activo se mezcla en condiciones asépticas con un portador farmacéuticamente aceptable y cualquier conservante o amortiguador del pH que sea necesario. También se contemplan la formulación oftálmica, las gotas para los oídos y las gotas oculares como comprendidas por el alcance de esta invención. Además, la presente invención contempla el
uso de parches transdérmicos, que tienen la ventaja adicional de proporcionar una liberación controlada de un compuesto al organismo. Dichas formas farmacéuticas se pueden preparar disolviendo o dispensando el compuesto en el medio apropiado. Se pueden utilizar potenciadores de la absorción para aumentar el flujo del compuesto a través de la piel. La velocidad se puede controlar o bien proporcionando una membrana que controle la velocidad o dispersando el compuesto en una matriz polimérica o un gel.
Como se ha descrito en general anteriormente, los compuestos de la invención son útiles como inhibidores de las proteínas cinasas ERK. En una realización, los compuestos y las composiciones de la invención son inhibidores de una o ambas de las proteínas cinasas ERK1 y ERK2 y por lo tanto, sin querer quedar restringidos por ninguna teoría en particular, los compuestos y las composiciones son particularmente útiles para tratar o atenuar la gravedad de una enfermedad, una afección o un trastorno en los que la activación de una o ambas de las proteínas cinasas ERK1 y ERK2 está implicada en la enfermedad, la afección o el trastorno. Cuando la activación de las proteínas cinasas ERK1 y/o ERK2 está implicada en una enfermedad, una afección o un trastorno particular, la enfermedad, la afección o el trastorno también se pueden denominar "enfermedad, afección o síntoma de enfermedad mediado por ERK1 o ERK2". En consecuencia, en otro caso, la presente solicitud proporciona un método para tratar o atenuar la gravedad de una enfermedad, una afección o un trastorno en los que la activación de una o ambas de las proteínas cinasas ERK1 y ERK2 está implicada en dicha enfermedad, afección o trastorno.
La actividad de un compuesto utilizado en esta invención como un inhibidor de las proteínas cinasas ERK1 y/o ERK2 se puede determinar in vitro, in vivo o en una línea celular. Los ensayos in vitro incluyen ensayos que determinan la inhibición de la actividad de fosforilación o la actividad de ATPasa de las proteínas cinasas ERK1 o ERK2 activadas. Alternativamente los ensayos in vitro cuantifican la capacidad del inhibidor para unirse a las proteínas cinasas ERK1 o ERK2. La unión del inhibidor se puede medir mediante radiomarcado del inhibidor antes de la unión, aislando el complejo inhibidor/ERK1 o inhibidor/ERK2 y determinando la cantidad de radiomarcador unido. Alternativamente, la unión del inhibidor se puede determinar realizando un experimento de competición en el cual se incuban nuevos inhibidores con las proteínas cinasas ERK1 o ERK2 unidas a radioligandos conocidos.
La expresión "inhibe mensurablemente", como se usa en el presente documento significa un cambio mensurable en la actividad de la proteína cinasa ERK1 o ERK2 entre una muestra que contiene dicha composición y una proteína cinasa ERK1 o e RK2 y una muestra equivalente que contiene proteína cinasa ERK1 o ERK2 en ausencia de dicha composición. Dichas mediciones de la actividad de la proteína cinasa son conocidas por los expertos en la materia e incluyen los métodos expuestos más adelante en el presente documento.
De acuerdo con otro caso, la solicitud se refiere a un compuesto de la presente invención o una composición que contenga dicho compuesto para su uso en un método para inhibir la actividad de la proteína quinasa ERK1 o ERK2 en un paciente.
La expresión "afección o enfermedad mediada por ERK", como se usa en el presente documento significa cualquier enfermedad o afección perjudicial en la que se sabe que ERK desempeña un papel. La expresión "afección o" enfermedad" mediada por ERK" también significa las enfermedades o afecciones que se alivian mediante el tratamiento con un inhibidor de ERK. Tales afecciones incluyen, sin limitación, cáncer, accidente cerebrovascular, diabetes, hepatomegalia, enfermedades cardiovasculares inclusive cardiomegalia, enfermedad de Alzheimer, fibrosis quística, virosis, enfermedades autoinmunitarias, aterosclerosis, reestenosis, psoriasis, trastornos alérgicos inclusive asma, inflamación, trastornos neurológicos y enfermedades hormonales. El término "cáncer" incluye, pero no se limita a, los cánceres siguientes: de mama, de ovario, de cuello de útero, de próstata, de testículos, de aparato genitourinario, de esófago, de laringe, glioblastoma, neuroblastoma, de estómago, de piel, queratoacantoma, de pulmón, carcinoma epidermoide, carcinoma de células grandes, carcinoma microcítico, adenocarcinoma pulmonar, óseo, de colon, adenoma, de páncreas, adenocarcinoma, de tiroides, carcinoma folicular, carcinoma indiferenciado, carcinoma papilar, seminoma, melanoma, sarcoma, carcinoma de vejiga, carcinoma de hígado y vías biliares, carcinoma de riñón, trastornos mieloides, trastornos linfoides, enfermedad de Hodgkin, de células pilosas, de cavidad bucal y faringe (oral), de labios, de lengua, de boca, de faringe, de intestino delgado, de colon-recto, de intestino grueso, de recto, de cerebro y sistema nervioso central, y leucemia.
En consecuencia, otro caso de la presente solicitud se refiere a tratar o atenuar la gravedad de una o más enfermedades en las que se sabe que ERK desempeña un papel. Específicamente, la presente solicitud se refiere a una composición según la presente solicitud para su uso en un método para tratar o atenuar la gravedad de la enfermedad o afección seleccionada entre cáncer, accidente cerebrovascular, diabetes, hepatomegalia, enfermedades cardiovasculares inclusive cardiomegalia, enfermedad de Alzheimer, fibrosis quística, virosis, enfermedades autoinmunitarias, aterosclerosis, reestenosis, psoriasis, trastornos alérgicos inclusive asma, inflamación, trastornos neurológicos y enfermedades hormonales.
De acuerdo con otro caso, la presente solicitud se refiere a un método para tratar un cáncer seleccionado entre cáncer de mama, de ovario, de cuello de útero, de próstata, de testículos, de aparato genitourinario, de esófago, de laringe, glioblastoma, neuroblastoma, de estómago, de piel, queratoacantoma, de pulmón, carcinoma epidermoide, carcinoma de células grandes, carcinoma microcítico, adenocarcinoma pulmonar, óseo, de colon, adenoma, de páncreas, adenocarcinoma, de tiroides, carcinoma folicular, carcinoma indiferenciado, carcinoma papilar, seminoma, melanoma,
sarcoma, carcinoma de vejiga, carcinoma de hígado y vías biliares, carcinoma de riñón, trastornos mieloides, trastornos linfoides, enfermedad de Hodgkin, de células pilosas, de cavidad bucal y faringe (oral), de labios, de lengua, de boca, de faringe, de intestino delgado, de colon-recto, de intestino grueso, de recto, de cerebro y sistema nervioso central, y leucemia.
Otra realización se refiere a un método para tratar melanoma, cáncer de mama, cáncer de colon o cáncer pancreático en un paciente que lo necesita.
También se apreciará que los compuestos y las composiciones farmacéuticamente aceptables de la presente invención se pueden emplear en terapias de combinación, es decir, los compuestos y las composiciones farmacéuticamente aceptables se pueden administrar simultáneamente con, antes de, o a continuación de, uno o más de otros medicamentos o procedimientos médicos deseados. La combinación particular de terapias (medicamentos o procedimientos) para emplear en un régimen de combinación tendrá en cuenta la compatibilidad de los medicamentos y/o los procedimientos deseados y el efecto terapéutico buscado que se debe lograr. También se apreciará que las terapias empleadas pueden lograr un efecto deseado para el mismo trastorno (por ejemplo, un compuesto de la invención se puede administrar simultáneamente con otro medicamento utilizado para tratar el mismo trastorno), o pueden lograr efectos diferentes (por ejemplo, control de cualquier efecto adverso). Como se usa en el presente documento, los agentes terapéuticos adicionales que normalmente se administran para tratar o prevenir una enfermedad o afección particular, son conocidos como "apropiados para la enfermedad o afección en tratamiento".
Por ejemplo, se pueden combinar quimioterápicos u otros fármacos antiproliferativos con los compuestos de esta invención para tratar enfermedades proliferativas y cáncer. Los ejemplos de quimioterápicos conocidos, incluyen, pero no se limitan a, por ejemplo otras terapias o antineoplásicos que se pueden utilizar en combinación con los antineoplásicos de la invención, incluyen cirugía, radioterapia (en unos pocos ejemplos, radiación gamma, radioterapia de haz de neutrones, radioterapia de haz de electrones, terapia de protones, braquiterapia e isótopos radiactivos sistémicos, por nombrar unos pocos), hormonoterapia, modificadores de la respuesta biológica (interferones, interleucinas y factor de necrosis tumoral (TNF) por nombrar unos pocos), hipertermia y crioterapia, fármacos para atenuar los efectos adversos (por ejemplo, antieméticos) y otros antineoplásicos aprobados, incluidos, pero no se limitan a, fármacos alquilantes (mecloretamina, clorambucilo, Ciclofosfamida, Melfalán, Ifosfamida), antimetabolitos (Metotrexato), antagonistas de purinas y antagonistas de pirimidinas (6-Mercaptopurina, 5-Fluorouracilo, Citarabilo, Gemcitabina), venenos del huso (Vinblastina, Vincristina, Vinorelbina, Paclitaxel), podofilotoxinas (Etopósido, Irinotecán, Topotecán), antibióticos (Doxorrubicina, Bleomicina, Mitomicina), nitrosoureas (Carmustina, Lomustina), iones inorgánicos (Cisplatino, Carboplatino), enzimas (Asparraginasa) y hormonas (Tamoxifeno, Leuprolida, Flutamida y Megestrol), Gleevec™, adriamicina, dexametasona y ciclofosfamida. Por una discusión completa de tratamientos contra el cáncer actualizados, consulte http://www.nci.nih.gov/, una lista de los antineoplásicos aprobados por la FDA en http://www.fda.gov/cder/cancer/druglistframe.htm y el Manual Merck, 17a Ed. 1999.
Otros ejemplos de medicamentos con los que los inhibidores de esta invención también se pueden combinar incluyen, sin limitación: tratamientos para la enfermedad de Alzheimer como Aricept® y Excelon®; tratamientos para la enfermedad de Parkinson como L-DOPA/carbidopa, entacapona, ropinrol, pramipexol, bromocriptina, pergolida, trihexefendilo y amantadina; medicamentos para tratar la esclerosis múltiple (MS) como beta interferón (por ejemplo, Avonex® y Rebif®), Copaxone® y mitoxantrona; tratamientos para el asma como albuterol y Singulair®; medicamentos para tratar la esquizofrenia como zyprexa, risperdal, seroquel y haloperidol; antiinflamatorios como corticoesteroides, bloqueantes de TNF, IL-1 RA, azatioprina, ciclofosfamida y sulfasalazina; inmunomoduladores e inmunosupresores como ciclosporina, tacrolimus, rapamicina, micofenolato mofetilo, interferones, corticoesteroides, ciclofosfamida, azatioprina y sulfasalazina; factores neurotróficos como inhibidores de la acetilcolinesterasa, inhibidores de MAO, interferones, anticonvulsantes, bloqueantes de los canales iónicos, riluzol y antiparkinsonianos; medicamentos para tratar enfermedades cardiovasculares como betabloqueantes, inhibidores de ACE, diuréticos, nitratos, bloqueantes de los canales de calcio y estatinas; medicamentos para tratar enfermedades hepáticas como los corticoesteroides, colestiramina, interferones y antivirales; medicamentos para tratar trastornos sanguíneos como corticoesteroides, antileucémicos y factores de crecimiento; y medicamentos para tratar trastornos de inmunodeficiencia como gammaglobulina.
La cantidad de agente terapéutico adicional presente en las composiciones de esta invención no será mayor que la cantidad que se administraría normalmente en una composición que contuviera ese agente terapéutico como el único agente activo. Preferentemente, la cantidad de agente terapéutico adicional en las composiciones dadas a conocer en la presente variará entre aproximadamente 50 % y 100 % de la cantidad normalmente presente en una composición que contiene ese fármaco como el único principio terapéuticamente activo.
En un caso alternativo, los métodos de esta solicitud que utiliza composiciones que no contienen un agente terapéutico adicional, comprende el paso adicional de administrar por separado a dicho paciente otro agente terapéutico. Cuando esos agentes terapéuticos adicionales se administran por separado se pueden administrar al paciente antes, secuencialmente con, o luego de la administración de las composiciones de esta invención.
Los compuestos de esta invención o composiciones farmacéuticamente aceptables de los mismos también se pueden incorporar en composiciones para recubrir dispositivos médicos implantables, tales como prótesis, válvulas artificiales,
injertos vasculares, estents y catéteres. Por consiguiente, la presente solicitud, en otro caso, incluye una composición para recubrir un dispositivo implantable que contiene un compuesto de la presente invención como los descritos en general antes y en las clases y subclases de esta parte, y un portador adecuado para recubrir dicho dispositivo implantable. Aún en otro aspecto, la presente invención incluye un dispositivo implantable recubierto con una composición que contiene un compuesto de la presente invención como los descritos en general antes, y en las clases y subclases de esta parte, y un portador adecuado para recubrir dicho dispositivo implantable.
Los estents vasculares, por ejemplo, han sido utilizados para solucionar el problema de reestenosis (reestrechamiento de la pared de los vasos luego de una lesión). Sin embargo, los pacientes que usan estents u otros dispositivos implantables corren el riesgo de formación de coágulos o activación de las plaquetas. Estos efectos indeseados se pueden prevenir o mitigar con un recubrimiento previo del dispositivo con una composición farmacéuticamente aceptable que contenga un inhibidor de cinasas. Los recubrimientos adecuados y la preparación general de los dispositivos implantables recubiertos se describen en las patentes de Estados Unidos 6.099.562; 5.886.026; y 5.304.121. Los recubrimientos son habitualmente materiales poliméricos biocompatibles como un polímero de hidrogel, polimetildisiloxano, policaprolactona, polietilenglicol, ácido poliláctico, etileno vinil acetato y mezclas de los mismos. Los recubrimientos pueden opcionalmente recubrirse con una capa superior adecuada de fluorosilicona, polisacáridos, polietilenglicol, fosfolípidos o combinaciones de los mismos, para impartirle a la composición características de liberación controlada.
Otro caso de la solicitud se refiere a inhibir la actividad de la proteína cinasa ERK1 o ERK2 en una muestra biológica o en un paciente, cuyo método comprende administrar al paciente, o poner en contacto dicha muestra biológica con, un compuesto de la presente invención o una composición que contenga dicho compuesto. La expresión "muestra biológica", como se usa en el presente documento, incluye, sin limitación, cultivos celulares o extractos; material de biopsia obtenido de un mamífero o extractos de los mismos; y sangre, saliva, orina, heces, semen, lágrimas u otros líquidos corporales o extractos de los mismos.
La inhibición de la actividad de la proteína cinasa ERK1 o ERK2 en una muestra biológica es útil para distintos propósitos conocidos por los expertos en la materia. Los ejemplos de dichos propósitos incluyen, pero no se limitan a, transfusión sanguínea, trasplante de órganos, almacenamiento de muestras y ensayos biológicos.
Ejemplos de síntesis
Como se usa en el presente documento, el término "Tr" se refiere al tiempo de retención de HPLC, en minutos, asociado al compuesto. Salvo indicación en contrario, el método HPLC utilizado para obtener el tiempo de retención informado es el siguiente:
Columna: Agilent / ZORBAX SB-C18 / 5 pm / 3,0 x 150 mm / PN 883975-302 / SN USBM001410
Gradiente: 10 a 90 % de MeCN en 8 minutos
Flujo: 1,0 ml/minuto
Detección: 214 nm y 254 nm
A menos que se indique lo contrario, cada RMN 1H se obtuvo a 500 MHz en CDCh y los números de los compuestos corresponden a los números de los compuestos enumerados en la Tabla 1.
Ejemplo de referencia 1

2,2,2-Tricloro-1-(4-yodo-1H-pirrol-2-il)etanona: A una solución en agitación de 50 g (235 mmol, 1,0 equiv.) de 2,2,2-tricloro-1-(1H-pirrol-2-il)-etanona en diclorometano seco (400 ml) en atmósfera de nitrógeno, se le añadió gota a gota una solución de monocloruro de yodo (39 g, 240 mmol, 1,02 equivalentes) en diclorometano (200 ml). La mezcla resultante se agitó a temperatura ambiente durante 2 horas. La solución se lavó con carbonato de potasio al 10 %, agua, tiosulfato de sodio 1,0 M, cloruro de sodio saturado, se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El sólido se recristalizó de hexanos/acetato de metilo para obtener el compuesto del título (68,5 g, 86 %) en forma de un sólido incoloro (86 %). MS FIA: 335,8, 337,8 ES-.
Éster metílico del ácido 4-yodo-1H-pirrol-2-carboxílico: A una solución en agitación de 2,2,2-tricloro-1-(4-yodo-1 H-pirrol-2-il)etanona (68 g, 201 mmol, 1,0 equivalente) en metanol seco (400 ml) en atmósfera de nitrógeno, se le añadió una solución de metóxido de sodio en metanol (4,37 M, 54 ml, 235 mmol, 1,2 equivalentes) en el transcurso de 10 minutos. La mezcla resultante se agitó a temperatura ambiente durante 1 hora. Se retiraron los volátiles a presión reducida y el producto en bruto se repartió después entre agua y ferc-butilmetil éter. La fase orgánica se separó, se lavó dos veces con agua y cloruro de sodio saturado, se secó sobre sulfato de sodio, se filtró y se concentró al vacío para obtener el compuesto del título (48 g, 96 %) en forma de un sólido incoloro que se usó directamente sin purificación adicional.
Éster metílico del ácido 4-yodo-1-(tolueno-4-sulfonil)-1H-pirrol-2-carboxílico: Se disolvió éster metílico del ácido 4-yodo-1H-pirrol-2-carboxílico (24,6 g, 98 mmol, 1,0 equivalente) en diclorometano (150 ml) y trietilamina (30 ml, 215,6 mmol, 2,2 equivalentes). Se añadieron 4-(dimetilamino)piridina (1,2 g, 9,8 mmol, 0,1 equivalente) y cloruro de p-toluenosulfonilo (20,6 g, 107,8 mmol, 1,1 equivalentes) y la mezcla de reacción se agitó durante 16 horas a temperatura ambiente. La reacción se detuvo con HCl 1 M y la capa orgánica se lavó con bicarbonato de sodio acuoso y una solución acuosa saturada de cloruro de sodio. Después de secar sobre sulfato de magnesio, se retiró el disolvente a presión reducida y el residuo se cristalizó de ferc-butilmetil éter, produciendo el compuesto del título en forma de un sólido de color amarillo pálido (30 g, 75 %). Tr(min) 8,259 minutos.
Éster metílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1-(tolueno-4-sulfonil)-1H-pirrol-2-carboxílico: A una solución desgasificada de éster metílico del ácido 4-yodo-1-(tolueno-4-sulfonil)-1H-pirrol-2-carboxílico (20 g, 49,4 mmol, 1,0 equivalente) y bis(pinacolato)diborano (15 g, 65 mmol, 1,3 equivalentes) en DMF (200 ml) en atmósfera de nitrógeno, se le añadió aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio (II) (3,6 g, 4,9 mmol, 0,1 equivalente). Después la mezcla de reacción se agitó a 80 °C durante 18 horas. Después de retirar la DMF a presión reducida, el residuo oleoso espeso resultante se suspendió en éter dietílico (500 ml) e inmediatamente precipitó un sólido. Este sólido se retiró por filtración y el filtrado se lavó con HCl 1 M, agua, solución acuosa saturada de cloruro de sodio y se secó en MgSO4. La concentración produjo el compuesto del título en forma de un sólido de color blanco que se usó sin purificación adicional (10 g, 50 %). CL/EM: Tr(min) 4,6; 406,4 ES+. MS FIA: 406,2 ES+. RMN 1H 5 1,2 (s, 12H), 2,35 (s, 3H), 3,8 (s, 3H), 7,2 (m, 3H), 7,8 (d, 2H), 8,0 (s, 1H).
W,W-2-(5-Cloro-4-yodo-piridil)-isopropilamina:
Método A. (Microondas)
En un tubo para microondas de 10 ml, se disolvió 5-cloro-2-fluoro-4-yodopiridina (1,0 g, 3,9 mmol, 1,0 equivalente) en DMSO (4,0 ml) y después se le añadió isopropilamina (0,99 ml, 11,7 mmol, 3,0 equivalentes). El tubo se cerró herméticamente y se colocó bajo irradiación de microondas durante 600 s a 150 °C. Esta reacción se repitió seis veces. Se combinaron las mezclas de reacción, después se diluyeron en acetato de etilo y se lavaron con agua. Después de secar sobre sulfato de sodio, el disolvente se evaporó para obtener el compuesto del título en forma de un aceite espeso de color pardo (5,6 g, 80 %) que se usó directamente sin purificación adicional. Tr(min) 4,614; MS FIA: 296,9 ES+. RMN 1H 81,25 (d, 6H), 3,65 (m, 1H), 7,15 (s, 1H), 7,75 (s, 1H).
Método B: (Térmico)
Se disolvió 5-cloro-2-fluoro-4-yodopiridina (400 mg, 1,55 mmol, 1,0 equivalente) en etanol (5,0 ml) y después se le añadió isopropilamina (0,66 ml, 7,8 mmol, 5,0 equivalentes). La solución resultante se agitó a 80 °C durante 48 horas. Después la mezcla de reacción se diluyó en acetato de etilo y se lavó con agua. Después de secar sobre sulfato de sodio, el disolvente se evaporó y se obtuvo un aceite espeso de color pardo, que se purificó mediante cromatografía ultrarrápida en gel de sílice eluyendo con mezclas de hexanos/acetato de etilo (desde 99:1 hasta 80:20) para obtener el compuesto del título en forma de un sólido de color amarillo pálido (96 mg, 21 %).
Éster metílico del ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1-(tolueno-4-sulfonil)-1H-pirrol-2-carboxílico: A una solución de W,W-2-(5-cloro-4-yodo-piridil)-isopropilamina (0,53 g, 1,8 mmol, 1,0 equivalente) y éster metílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1-(tolueno-4-sulfonil)-1H-pirrol-2-carboxílico (0,78 g, 1,8 mmol, 1,0 equivalente) en DME (4,0 ml) se le añadió una solución acuosa de carbonato de sodio 2M (1,0 ml) seguido de Pd(PPh3)4 (0,21 mg, 0,18 mmol, 0,1 equivalente). El tubo para microondas se cerró herméticamente y la mezcla de reacción se irradió con microondas durante 1800 s a 170 °C. El producto en bruto de seis reacciones se combinó, se diluyó en acetato de etilo y se lavó con agua. Después de secar la capa orgánica con sulfato de sodio, se retiró el disolvente y el aceite espeso resultante se adsorbió en gel de sílice. Después se purificó el producto en bruto mediante cromatografía ultrarrápida en sílice, eluyendo con mezclas de hexanos/acetato de etilo (desde 99:1 hasta 70:30) para obtener el compuesto del título en forma de un sólido de color amarillo (3,1 g, 61 % en dos pasos). Tr(min) 6,556. MS FIA: 448,1 ES+. RMN 1H 81,45 (d, 6H), 2,5 (s, 3H), 3,81 (s, 3H), 6,8 (s, 1H), 7,35 (s, 1H), 7,4 (d, 2H), 8,0 (m, 3H), 8,3 (s, 1H).
Éster metílico del ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1-(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico: A una solución de W,W-2-(5-cloro-4-yodo-piridil)-isopropilamina (96 mg, 0,32 mmol, 1,0 equivalente) y éster metílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1-(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico (152 mg, 0,35 mmol, 1,1 equivalentes) en DME (2 ml), se le añadió una solución acuosa de carbonato de sodio 2 M (0,2 ml) seguida de Pd(PPh3)4 (37 mg, 0,032 mmol, 0,1 equivalente). La mezcla de reacción se agitó a 80 °C durante 16 horas. El producto en bruto se diluyó en acetato de etilo y se lavó con agua. Después de secar la capa orgánica con sulfato de sodio, se retiró el disolvente y el aceite espeso resultante se adsorbió en gel de sílice. Después se purificó el producto en bruto mediante cromatografía ultrarrápida sobre sílice, eluyendo con mezclas de hexanos/acetato de etilo (desde 99:1 hasta 80:20) para obtener el compuesto del título en forma de un sólido de color amarillo (65 mg, 43 %). Tr(min) 7,290. MS FIA:476,1 ES+.
Ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1H-pirrol-2-carboxílico:
Método A. (Microondas)
Se añadió una solución de éster metílico del ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1-(tolueno-4-sulfonil)-1 H-pirrol-2-carboxílico (3,1 g, 6,9 mmol, 1,0 equivalente) en THF (2,0 ml) a una solución de hidróxido de litio monohidratado (710 mg, 17,3 mmol, 2,5 equivalentes) en agua (3,0 ml). El tubo para microondas se cerró herméticamente y la mezcla de reacción se irradió con microondas durante 1200 s a 150 °C. La solución en bruto se acidificó con HCl acuoso 6 N. El disolvente se evaporó para obtener el compuesto del título que se usó directamente sin purificación adicional. Tr(min): 3,574. FIA MS: 279,9 ES+; 278,2 ES-.
Método B: (Térmico)
Se añadió una solución de éster metílico del ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1 -(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico (0,69 g, 1,4 mmol, 1,0 equivalente) en THF (3,0 ml) a una solución de hidróxido de litio monohidratado (1,19 g, 29 mmol, 20,0 equivalentes) en agua (3,0 ml). Después la mezcla se calentó a reflujo durante 8 horas. La solución en bruto se acidificó con HCl acuoso 6 N hasta que se enturbió, el disolvente orgánico se retiró parcialmente y el producto precipitó. El compuesto del título se aisló por filtración y se lavó con agua y éter dietílico, produciendo un sólido de color blanco (0,38 g, 96 %).
[1-(3-Clorofenil)-2-hidroxietil]amida del ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1H-pirrol-2-carboxílico: A una suspensión de ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1H-pirrol-2-carboxílico (1,93 g, 6,9 mmol, 1,0 equivalente)
en DMF (5,0 ml) se le añadió EDCI (1,45 g, 7,6 mmol, 1,1 equivalentes), HOBt (0,94 g, 6,9 mmol, 1,0 equivalente) y (S)-3-clorofenilgMcinol (1,58 g, 7,6 mmol, 1,1 equivalentes). Después se le añadió diisopropiletilamina (2,7 ml) y la mezcla resultante se agitó a temperatura ambiente durante toda la noche. Después la mezcla se vertió en agua y se extrajo con acetato de etilo. Después de secar sobre sulfato de sodio, el disolvente se retiró y el producto en bruto se adsorbió en gel de sílice. La purificación se efectuó mediante cromatografía ultrarrápida sobre sílice, eluyendo con mezclas de hexanos/acetona (desde 80:20 hasta 60:40) para obtener el compuesto del título en forma de un sólido de color blanco (1,9 g, 64 %). T r(min) 4,981s. FIA MS: 433,1 ES+; 431,2 ES-. RMN 1H (CD3OD) 5 1,31 (d, 6H), 3,85 (m, 3H), 5,15 (t, 1H), 7,01 (s, 1H), 7,25 (m, 3H), 7,4 (s, 1H), 7,45 (s, 1H), 7,7 (s, 1H), 7,95 (s, 1H).
Ejemplo de referencia 2
El compuesto 1-9 se preparó también de acuerdo con el siguiente método alternativo:
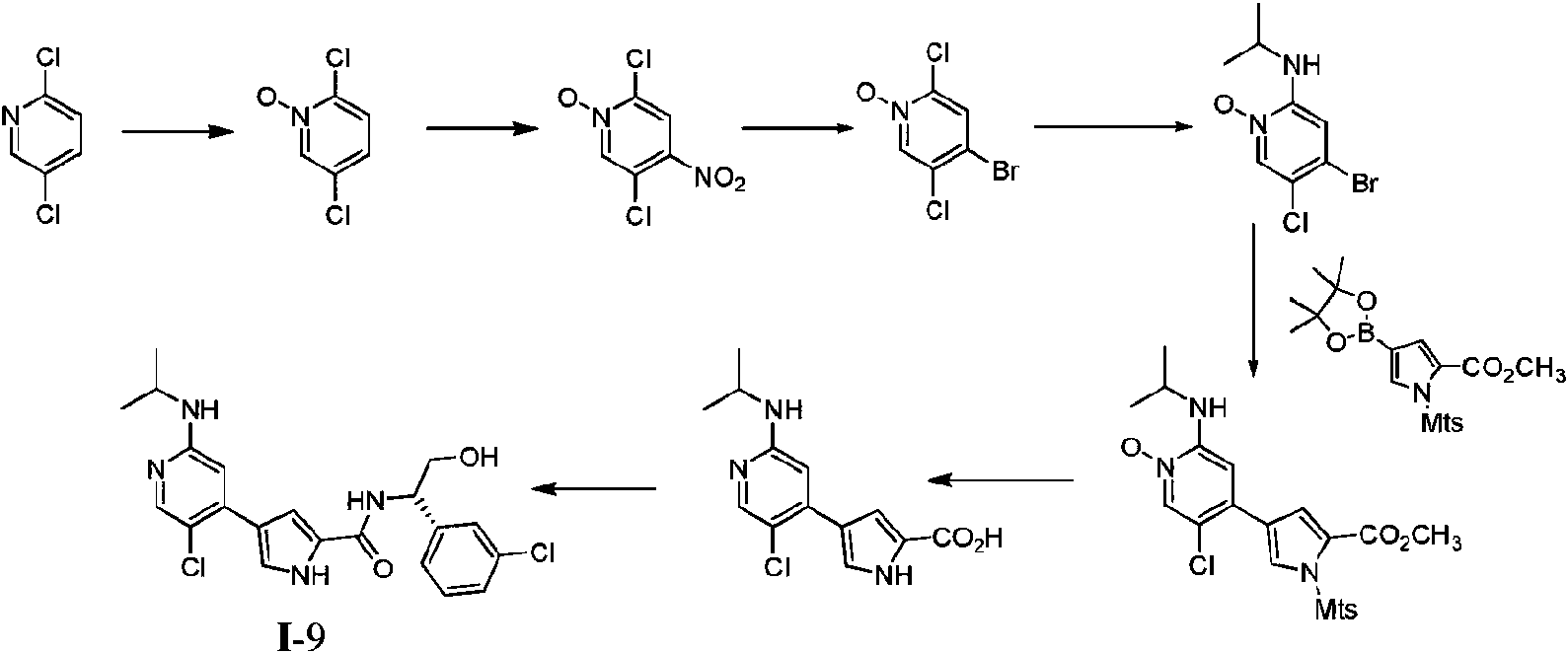
N-óxido de 2,5-dicloro-4-nitropiridina: A una suspensión de 2-cloro-5-cloropiridina (10 g, 0,067 mol) en anhídrido acético (25 ml) se le añadió peróxido de hidrógeno al 30 % (25 ml) en pequeñas porciones. Esta mezcla se agitó a temperatura ambiente durante 24 horas y después se calentó a 60 °C durante 30 horas. Después de retirar el exceso de ácido acético a presión reducida, el residuo se añadió en pequeñas porciones a ácido sulfúrico concentrado (15 ml). La solución resultante se añadió a una mezcla de ácido sulfúrico concentrado (15 ml) y ácido nítrico fumante (25 ml) y después se calentó a 100 °C durante 90 minutos. La mezcla de reacción se vertió sobre hielo, se neutralizó con carbonato de amonio sólido y finalmente con amoníaco acuoso hasta que se obtuvo un pH básico y se formó un precipitado. El precipitado se recogió por filtración para obtener el compuesto del título en forma de un sólido de color amarillo pálido (3,1 g), T r(min) 3,75. MS FIA no muestra picos. RMN 1H (DMSO-d6) 58,78 (s, 1H), 9,15 (s, 1H).
N-óxido de 4-bromo-2-cloro-5-W-isopropilpiridin-2-amina: A 2,5-dicloro-4-nitropiridina N-óxido (400 mg, 1,9 mmol) se le añadió bromuro de acetilo (2 ml) muy lentamente. Después la mezcla de reacción se calentó a 80 °C durante 10 minutos. El disolvente se retiró bajo una corriente de nitrógeno y el producto en bruto se secó en alto vacío. El material en bruto (165 mg, 0,62 mmol) se disolvió en etanol (2 ml), se le añadió /so-propilamina (0,53 ml) y la mezcla resultante se calentó a 80 °C durante 2 horas. La solución en bruto se purificó después por HPLC de fase inversa (acetonitrilo/agua/TFA al 1 %) para obtener el compuesto del título en forma de un sólido de color amarillo pálido (60 mg, 36,6 %). Tr(min) 5,275. MS FIA: 264,8, 266,9 ES+.
[1-(3-Clorofenil)-2-hidroxietil]amida del ácido 4-(5-cloro-2-isopropilaminopiridin-4-il)-1H-pirrol-2-carboxílico (1-9): Se disolvieron N-óxido de 4-bromo-2-cloro-5-W-isopropilpiridin-2-amina (25 mg, 0,094 mmol, 1,0 equivalente) y éster metílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1-(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico (39 mg, 0,094 mmol, 1,0 equivalente) en benceno (5 ml) después se le añadieron Na2CO3 2 M (1 ml) y Pd(PPh3)4 (115,6 mg, 0,1 mmol, 0,2 equivalente) y la suspensión resultante se calentó a reflujo a 80 °C durante 16 horas. La mezcla de reacción se diluyó en acetato de etilo, se lavó con agua y se secó sobre sulfato de sodio anhidro para obtener N-óxido de éster metílico del ácido 4-(5-cloro-2-isopropilamino-piridin-4-il)-1-(2,4,6-trimetil-bencenosulfonil)-1 H-pirrol-2-carboxílico (Tr(min) 6,859. MS FIA: 492,0 ES+) que después se trató con una solución de PCh 2 M en diclorometano (1 ml) a temperatura ambiente. Después de 10 minutos, el disolvente se retiró bajo una corriente de nitrógeno y el aceite en bruto se disolvió en metanol (1 ml) y NaOH acuoso 1 M (1 ml). La mezcla resultante se calentó a reflujo durante 16 horas, después la solución en bruto se acidificó empleando HCl acuoso 1 M y se retiró el disolvente. El ácido 4-(5-cloro-2-isopropilamino-piridin-4-il)-1H-pirrol-2-carboxílico resultante (Tr(min) 3,527. MS FIA: 279,4 ES+; 278,2 Es-) se suspendió en DMF (3 ml) junto con e Dc I (36 mg, 0,19 mmol, 2 equivalentes), HOBt (26 mg, 0,19 mmol, 2 equivalentes), sal de HCl de (S)-3-clorofenilglicinol (59 mg, 0,28 mmol, 3 equivalentes) y DIEA (0,12 ml, 0,75 mmol, 8 equivalentes). La mezcla resultante se agitó a temperatura ambiente durante 16 horas. La mezcla de reacción se diluyó en acetato de etilo, se lavó con agua y se secó sobre sulfato de sodio. Después de retirar el disolvente a presión reducida, el producto en bruto se purificó por HPLC de fase inversa (acetonitrilo/agua/TFA al 1 %) para obtener el compuesto del título en forma de un sólido de color blanco (4,8 mg, 8,1 %).
Ejemplo 3
El compuesto 1-3 se preparó como sigue a continuación:

N-óxido de 2-cloro-5-metil-4-nitropiridina: El compuesto del título se preparó de manera sustancialmente similar a la descrita por Z. Talik, A. Puszko, Roczniki Chemii Ann. Soc. Chim. Polonorum 1976, 50, 2209, como sigue a continuación. A una suspensión de 2-cloro-5-metilpiridina (10 g, 0,078 mol) en anhídrido acético (25 ml) se le añadió peróxido de hidrógeno al 30 % (25 ml) en pequeñas porciones. Esta mezcla se agitó a temperatura ambiente durante 24 horas y después se calentó a 60 °C durante 30 horas. Después de retirar el exceso de ácido acético a presión reducida, el residuo se añadió en pequeñas porciones a ácido sulfúrico concentrado (15 ml). La solución resultante se añadió a una mezcla de ácido sulfúrico concentrado (15 ml) y ácido nítrico fumante (25 ml) y después se calentó a 100 °C durante 90 minutos. La mezcla de reacción se vertió sobre hielo, se neutralizó con carbonato de amonio sólido y finalmente con amoníaco acuoso hasta que se obtuvo un pH básico y se formó un precipitado. Este precipitado se recogió por filtración para obtener el compuesto del título en forma de un sólido de color amarillo pálido (9,4 g, 0,050 mol, Tr(min) 3,272, FIA ES+ 188,9, E S -188,0).
N-óxido de 2-((S)-1-hidroximetilpropilamino)-5-metil-4-nitro-piridina: Se disolvió 2-cloro-5-metil-4-nitropiridina N-óxido (200 mg, 1,06 mmol, 1,0 equivalente) en etanol (1,5 ml). Después se le añadió (S)-2-aminobutanol (284 mg, 3,2 mmol, 3,0 equivalentes) y la mezcla resultante se calentó a reflujo durante 16 horas. La solución en bruto se purificó después por HPLC de fase inversa (acetonitrilo/agua/TFA) para obtener el compuesto del título en forma de un sólido de color pardo (146 mg, 0,61 mmol, T r(min) 3,787; FIA e S+ 241,8, ES- no se observó).
Éster 2-(4-bromo-5-metilpiridin-2-ilamino)-butílico del ácido acético: Se disolvió 2-((S)-1-hidroximetilpropilamino)-5-metil-4-nitro-piridina N-óxido (146 mg, 0,61 mmol) en bromuro de acetilo (1,5 ml). Después la mezcla se calentó a 90 °C durante 3 horas. Se evaporó el bromuro de acetilo bajo una corriente de nitrógeno y el material en bruto se disolvió en una solución 2 M de PCl3 en diclorometano (2 ml). La mezcla resultante se agitó a temperatura ambiente durante 1 hora y la mezcla de reacción se vertió en una solución acuosa saturada de NaHCO3 y se extrajo con acetato de etilo. La capa orgánica se lavó con agua, después el disolvente se secó sobre Na2SO4 y se retiró a presión reducida para obtener el compuesto del título en forma de un aceite de color pardo (149 mg, (Tr(min) 4,146; FIA ES+ 300,9, ES-no se observó).
Éster metílico del ácido 4-[2-(S)-(1-acetoximetilpropilamino)-5-metilpiridin-4-il]-1-(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico: A una solución de éster 2-(4-bromo-5-metilpiridin-2-ilamino)-butílico del ácido acético (149 mg, 0,5 mmol, 1,0 equivalente) y éster metílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1-(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico (215 mg, 0,50 mmol, 1,0 equivalente) en benceno (5 ml) se le añadió Na2CO32 M (1 ml) y Pd(PPh3)4 (115,6 mg, 0,1 mmol, 0,2 equivalente). Después de calentar a reflujo durante 16 horas, la mezcla se vertió en agua y se extrajo con acetato de etilo. El extracto orgánico se secó en Na2SO4 y el disolvente se retiró a presión reducida para obtener el compuesto del título (Tr(min) 6,684; FIA ES+ 528,3, ES- no se observó) que se utilizó en el paso siguiente.
[1 -(S)-(3-Clorofenilglicinol]amida del ácido 4-[2-(S)-hidroximetilpropilamino)-5-metilpiridin-4-il]-1 H-pirrol-2-carboxílico (1-3): El éster metílico del ácido 4-[2-(S)-(1-acetoximetilpropilamino)-5-metilpiridin-4-il]-1-(2,4,6-trimetilbencenosulfonil)-1H-pirrol-2-carboxílico en bruto se disolvió en metanol (1,5 ml) y NaOH acuoso 1 M (2 ml), y la mezcla se calentó a reflujo durante 16 horas. Después de acidificar con HCl acuoso 1 M (2,2 ml), el disolvente se retiró a presión reducida y el producto en bruto se suspendió a continuación en DMF (5 ml). Después de añadir EDCI (192 mg, 1,0 mmol), HOBt (135 mg, 1,0 mmol) y DIeA (0,48 ml, 3,0 mmol), la mezcla se agitó durante 30 minutos a temperatura ambiente antes de añadir sal de HCl de (S)-3-clorofenilglicinol (312 mg, 1,5 mmol) y después la mezcla de reacción se agitó durante 16 horas a temperatura ambiente. La mezcla de reacción se disolvió en acetato de etilo,
se lavó con agua, la capa orgánica se secó en Na2SO4 y el disolvente se retiró a presión reducida. El residuo en bruto se purificó por HPLC de fase inversa (acetonitrilo/agua/TFA) para obtener el compuesto del título como un sólido incoloro (29,3 mg, 0,07 mmol, T r(min) 4,563; FIA ES+ 443,1, ES- 441,5, ES+ 443,1, ES- 441,5; RMN 1H (CD3OD) 8 1,0 (t, 3H), 1,50 (m, 1H), 1,7 (m, 1H), 2,3 (s, 3H), 3,5 (m, 1H), 3,7 (m, 2H), 3,8 (m, 2H), 5,1 (t, 1H), 7,0 (s, 1H), 7,3 (m, 4H), 7,4 (dos s, 2H), 7,6 (s, 1H).
Ejemplo 4
Datos de caracterización
Los compuestos de la presente invención se prepararon por métodos sustancialmente similares a los descritos en los Ejemplos 1 a 3 anteriores y por métodos conocidos por los expertos en la materia. Los datos de caracterización para estos compuestos se resumen en la tabla 3 siguiente que incluye datos de HPLC, MS y RMN 1H. A menos que se indique lo contrario, los datos de RMN 1H se obtuvieron a 500 MHz y todos los desplazamientos químicos se informan en ppm. Los números de los compuestos corresponden a los números de los compuestos indicados en las tablas 1,2 y 3. Como se usa en el presente documento, el término "T r" se refiere al tiempo de retención, en minutos, obtenido para el compuesto designado usando el método de HPLC descrito antes. Cuando se obtuvo más de una medición analítica para un determinado compuesto, como los descritos en el presente documento, sólo se proporciona una única medida de ejemplo.

Ejemplo 5
Síntesis de profármacos
Los profármacos de fórmula II se preparan a partir de compuestos hidroxilo de fórmula I por diversos métodos conocidos por los expertos en la materia. Estos métodos incluyen, pero no se limitan a, acilación mediante un ácido carboxílico deseado o formación de fosfato. Cuando el resto hidroxilo de fórmula I se acila por un aminoácido deseado, el resto amino del aminoácido puede ser opcionalmente protegido por un grupo protector de amino adecuado como los descritos anteriormente en el presente documento.
La preparación del profármaco L-valina del compuesto de referencia 1-9 se describe en detalle a continuación.

(2S)-(S)-2-(4-(5-cloro-2-(isopropilamino)piridina-4-il)-1H-pirrol-2-carboxamido)-2-(3-clorofenil)etil-2-amino-2-metilbutanoato (II-1): A una solución del compuesto 1-9 (1 g, 2,3 mmol, 1,0 equivalente) en diclorometano (50 ml) se le añadieron DIEA (1,1 ml, 6,9 mmol, 3,0 equivalentes) y W-BOC-L-Valina (1,2 g, 5,52 mmol, 2,4 equivalentes). Después se le añadió PyBOP (2,9 g, 5,75 mmol, 2,5 equivalentes) lentamente y la mezcla resultante se agitó a temperatura ambiente durante 48 horas. Después la mezcla se lavó con agua y se secó sobre sulfato de sodio. El sólido en bruto se adsorbió en sílice y después se purificó mediante cromatografía ultrarrápida eluyendo con mezclas de hexano/acetato de etilo (desde 90:10 hasta 50:50), produciendo el compuesto protegido con Boc en forma de un sólido de color blanco (786 mg). Este producto intermedio (761 mg, 1,2 mmol) se disolvió en dioxano (1 ml) y se trató con una solución de HCl 4 M en dioxano. La mezcla resultante se agitó durante 16 horas a temperatura ambiente. Después se retiró el disolvente y se obtuvo la sal 2 x HCl del compuesto del título en forma de un sólido de color blanco (571 mg). HPLC Tr: 4,56 minutos. FIA MS: 531,9 ES+; 529,8 ES-. CL/EM: T r: 2,07 minutos; 532,0 ES+; 530,1 ES-. RMN 1H (CD3OD) 80,9 (dd, 6H), 1,35 (d, 6H), 2,2 (m, 1H), 3,9 (m, 2H), 4,7 (m, 2H), 5,6 (m, 1H), 7,1 (s, 1H), 7,3 (d, 1H), 7,35 (t, 1H), 7,4 (d, 1H), 7,5 (s, 1H), 7,6 (s, 1H), 7,75 (s, 1H), 7,95 (s, 1H).
La preparación de un profármaco fosfato del compuesto de referencia 1-9 se describe en detalle a continuación.

4-(5-Cloro-2-(isopropilamino)piridina-4-il)-A/-((S)-1-(3-clorofenil)-2-hidroxietil)-1H-pirrol-2-carboxamida fosfato de diferc-butilo: Se disolvieron el compuesto 1-9 (1 g, 2,3 mmol, 1,0 equivalente) y tetrazol (241 mg, 3,45 mmol, 1,5 equivalentes) en diclorometano (5 ml) y acetonitrilo (5 ml) en atmósfera de nitrógeno a temperatura ambiente. Se añadió gota a gota diisopropil fosfoamidita de di-ferc-butilo (1,1 ml, 3,45 mmol, 1,5 equivalentes) y la mezcla resultante se agitó durante 16 horas. Después la mezcla de reacción se enfrió en un baño de hielo, se trató con una solución de ferc-butilhidroxiperóxido 6 M (3 ml) y se agitó durante 20 minutos. La solución transparente se diluyó en diclorometano y una pequeña cantidad de metanol, se lavó con Na2S2O3 y agua, y se secó sobre sulfato de sodio. El aceite en bruto se adsorbió en gel de sílice y se purificó primero mediante cromatografía ultrarrápida eluyendo con mezclas de hexano/acetona (desde 90:10 hasta 60:40) y después mediante HPLC de fase inversa (acetonitrilo/agua/TFA al 1 %), produciendo el di-t-butil éter intermedio en forma de un sólido de color blanco (336 mg). HPLC Tr: 6,53 minutos, MS FIA: 625,0 ES+; 623,1ES-.
4-(5-Cloro-2-(isopropilamino)piridina-4-il)-A/-((S)-1-(3-clorofenil)-2-hidroxietil)-1H-pirrol-2-carboxamida fosfato (II-2): El intermedio fosfato de t-butilo (336 mg, 0,54 mmol) se suspendió en dioxano (5 ml) y se le añadió una solución de HCl 4 M en dioxano. La mezcla resultante se agitó a temperatura ambiente durante 1 hora. Después se retiró el disolvente y el fosfato libre se disolvió en una solución de d Ms O (15 ml), metanol (50 ml) y agua (25 ml) y se trató con una solución de Na2CO3 2 M (0,25 ml). El metanol se retiró a presión reducida y la mezcla de agua/DMSO se retiró empleando un liofilizador para obtener el compuesto del título en forma de un sólido de color blanquecino (187 mg).
HPLC T r: 4,24 minutos. FIA MS: 512,9 ES+; 510,9 ES-. CL/EM: Tr 2,39 minutos; 512,9 ES+; 510,9 ES-. RMN 1H (CD3OD) 8 1,2 (d, 6H), 3,9 (m, 1H), 4,1 (dm, 2H), 5,1 (m, 1H), 6,7 (s, 1H), 7,2 (d, 1H), 7,25 (t, 1H), 7,3 (m, 2H), 7,45 (s, 1H), 7,55 (s, 1H), 7,9 (s, 1H).
Ejemplo 6
Ensayo de inhibición de ERK2:
Se ensayaron compuestos para determinar si inhibían a ERK2 mediante un ensayo espectrofotométrico acoplado para la enzima (Fox et al (1998) Protein Sci 7, 2249). En este ensayo, una concentración fija de ERK2 activada (10 nM) se incubó con diversas concentraciones del compuesto en d Ms O (2,5%) durante 10 minutos a 30 °C en solución amortiguadora HEPES 0,1 M, pH 7,5, que contenía MgCh 10 mM, fosfoenolpiruvato 2,5 mM, NADH 200 pM, 150 pg/ml de piruvato cinasa, 50 pg/ml de lactato deshidrogenasa y péptido erktide 200 pM. La reacción se inició por adición de ATP 65 pM. Se monitoreó la velocidad de disminución de la absorbancia a 340 nM. Se evaluó la CI50 a partir de los datos de velocidad como una función de la concentración del inhibidor.
Se encontró que los compuestos de la presente invención son inhibidores de la proteína cinasa ERK2. En ciertas realizaciones, se encontró que los compuestos inhibían la cinasa ERK2 a una concentración <0,1 pM. En otras realizaciones, se encontró que los compuestos inhibían la cinasa ERK2 a una concentración <0,01 pM.
Ejemplo 7
Ensayo de inhibición de ERK1
Se ensayaron compuestos para determinar si inhibían a ERK1 mediante un ensayo espectrofotométrico acoplado para la enzima (Fox et al (1998) Protein Sci 7, 2249). En este ensayo, una concentración fija de ERK1 activada (20 nM) se incubó con diversas concentraciones del compuesto en d Ms O (2,0%) durante 10 minutos a 30 °C en solución amortiguadora HEPES 0,1 M, pH 7,6, que contenía MgCh 10 mM, fosfoenolpiruvato 2,5 mM, NADH 200 pM, 30 pg/ml de piruvato cinasa, 10 pg/ml de lactato deshidrogenasa y péptido erktide 150 pM. La reacción se inició por adición de ATP 140 pM (20 pl). Se monitoreó la velocidad de disminución de la absorbancia a 340 nM. Se evaluó la Ki a partir de los datos de velocidad como una función de la concentración del inhibidor.
Si bien hemos descrito una serie de realizaciones de esta invención, es evidente que nuestros ejemplos básicos pueden ser modificados para proporcionar otras realizaciones que utilicen los compuestos y los métodos de esta invención. Por consiguiente, se apreciará que el alcance de esta invención debe ser definido por las reivindicaciones adjuntas en lugar de por las realizaciones específicas que han sido representadas a modo de ejemplo.
Claims (16)
1. El compuesto

2. Una composición que comprende un compuesto de acuerdo con la reivindicación 1 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
3. El compuesto

4. Una composición que comprende un compuesto de acuerdo con la reivindicación 3 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
5. Una sal farmacéuticamente aceptable del compuesto

6. Una composición que comprende la sal farmacéuticamente aceptable del compuesto de acuerdo con la reivindicación 5 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
7. Una sal farmacéuticamente aceptable del compuesto

8. Una composición que comprende la sal farmacéuticamente aceptable del compuesto de acuerdo con la reivindicación 7 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
9. El compuesto

10. Una composición que comprende un compuesto de acuerdo con la reivindicación 9 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
11. El compuesto

12. Una composición que comprende un compuesto de acuerdo con la reivindicación 11 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
13. Una sal farmacéuticamente aceptable del compuesto
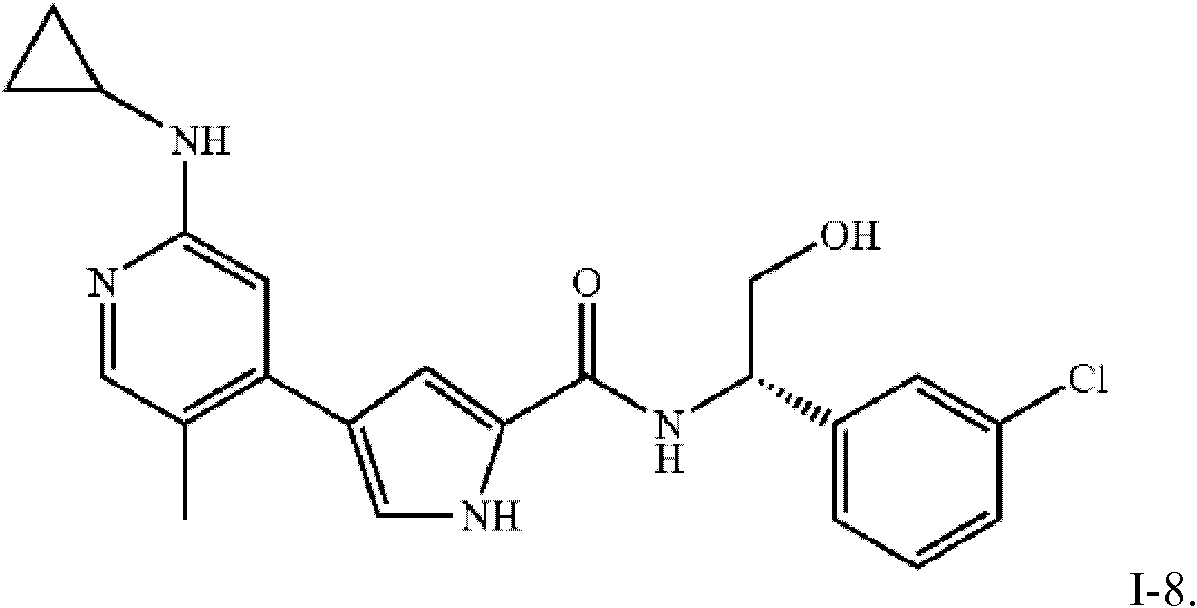
14. Una composición que comprende la sal farmacéuticamente aceptable del compuesto de acuerdo con la reivindicación 13 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
15. Una sal farmacéuticamente aceptable del compuesto

16. Una composición que comprende la sal farmacéuticamente aceptable del compuesto de acuerdo con la reivindicación 15 y un portador, adyuvante o vehículo farmacéuticamente aceptable.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US57130904P | 2004-05-14 | 2004-05-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2897422T3 true ES2897422T3 (es) | 2022-03-01 |
Family
ID=35106760
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19199330T Active ES2897422T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos pirroles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
| ES17184079T Active ES2751761T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos de pirrol como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
| ES14163944.3T Active ES2651439T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos de pirrol como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
| ES05748073.3T Active ES2513965T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos pirroles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17184079T Active ES2751761T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos de pirrol como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
| ES14163944.3T Active ES2651439T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos de pirrol como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
| ES05748073.3T Active ES2513965T3 (es) | 2004-05-14 | 2005-05-13 | Compuestos pirroles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos |
Country Status (26)
| Country | Link |
|---|---|
| US (6) | US7354939B2 (es) |
| EP (5) | EP3305776B1 (es) |
| JP (1) | JP5132305B2 (es) |
| CN (1) | CN1976919A (es) |
| AR (2) | AR051735A1 (es) |
| AU (1) | AU2005245885B2 (es) |
| BR (1) | BRPI0511111A (es) |
| CA (1) | CA2566461C (es) |
| CY (1) | CY1122336T1 (es) |
| DK (1) | DK3305776T3 (es) |
| ES (4) | ES2897422T3 (es) |
| HK (2) | HK1203505A1 (es) |
| HU (1) | HUE047130T2 (es) |
| IL (1) | IL179207A0 (es) |
| LT (1) | LT3305776T (es) |
| MX (1) | MXPA06013209A (es) |
| NO (1) | NO20065727L (es) |
| NZ (1) | NZ551582A (es) |
| PL (1) | PL3305776T3 (es) |
| PT (1) | PT3305776T (es) |
| RU (1) | RU2376299C2 (es) |
| SI (1) | SI3305776T1 (es) |
| TW (1) | TW200607803A (es) |
| UA (1) | UA84930C2 (es) |
| WO (1) | WO2005113541A1 (es) |
| ZA (1) | ZA200609975B (es) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2004221881A1 (en) * | 2003-03-13 | 2004-09-30 | Vertex Pharmaceuticals Incorporated | Compositions useful as protein kinase inhibitors |
| WO2005100342A1 (en) * | 2004-03-26 | 2005-10-27 | Vertex Pharmaceuticals, Incorporated | Pyridine inhibitors of erk2 and uses thereof |
| AU2005245885B2 (en) * | 2004-05-14 | 2011-01-20 | Vertex Pharmaceuticals, Incorporated | Pyrrole compounds as inhibitors of ERK protein kinase, synthesis thereof and intermediates thereto |
| US8546404B2 (en) | 2005-12-13 | 2013-10-01 | Merck Sharp & Dohme | Compounds that are ERK inhibitors |
| EP1984331B1 (en) | 2006-02-16 | 2010-10-20 | Schering Corporation | Pyrrolidine derivatives as erk inhibitors |
| WO2008153858A1 (en) * | 2007-06-05 | 2008-12-18 | Schering Corporation | Polycyclic indazole derivatives and their use as erk inhibitors for the treatment of cancer |
| ES2556353T3 (es) | 2008-02-21 | 2016-01-15 | Merck Sharp & Dohme Corp. | Compuestos que son inhibidores de las ERK |
| CN102146074B (zh) * | 2011-01-26 | 2013-02-06 | 江苏先声药物研究有限公司 | 吡咯衍生物的制备方法及应用 |
| WO2012172093A1 (en) | 2011-06-17 | 2012-12-20 | Merz Pharma Gmbh & Co. Kgaa | Dihydroindolizine derivate as metabotropic glutamate receptor modulators |
| CA2926645C (en) | 2013-10-14 | 2022-10-25 | Indiana University Research And Technology Corporation | Use of acamprosate to modulate erk 1/2 activation in animal models for fxs and asd and individuals diagnosed with fxs and asd |
| JPWO2015060368A1 (ja) | 2013-10-23 | 2017-03-09 | 武田薬品工業株式会社 | 複素環化合物 |
| US10668055B2 (en) | 2013-12-20 | 2020-06-02 | Biomed Valley Discoveries, Inc. | Cancer treatment using combinations of ERK and RAF inhibitors |
| WO2015095835A1 (en) * | 2013-12-20 | 2015-06-25 | Biomed Valley Discoveries, Inc. | Methods of modulating radioiodine uptake for the treatment of radioiodine-refractory cancers |
| WO2015095840A1 (en) | 2013-12-20 | 2015-06-25 | Biomed Valley Discoveries, Inc. | Cancer treatments using combinations of cdk and erk inhibitors |
| WO2015095807A1 (en) * | 2013-12-20 | 2015-06-25 | Biomed Valley Discoveries, Inc. | Cancer treatments using combinations of egfr and erk inhibitors |
| WO2015095831A1 (en) * | 2013-12-20 | 2015-06-25 | Biomed Valley Discoveries, Inc. | Cancer treatments using combinations of mtor and erk inhibitors |
| WO2015095833A1 (en) * | 2013-12-20 | 2015-06-25 | Biomed Valley Discoveries, Inc. | Treatment of hematologic cancers |
| WO2015095842A2 (en) * | 2013-12-20 | 2015-06-25 | Biomed Valley Discoveries, Inc. | Methods and compositions for treating non-erk mapk pathway inhibitor-resistant cancers |
| ES2918375T3 (es) | 2013-12-20 | 2022-07-15 | Biomed Valley Discoveries Inc | Tratamientos contra el cáncer usando combinaciones de inhibidores de la ruta de PI3K/Akt y ERK |
| EP3082779B1 (en) * | 2013-12-20 | 2023-06-07 | Biomed Valley Discoveries, Inc. | Cancer treatments using combinations of type 2 mek and erk inhibitors |
| AU2014368925A1 (en) * | 2013-12-20 | 2016-07-21 | Biomed Valley Discoveries, Inc. | Cancer treatments using combinations of MEK type I and ERK inhibitors |
| AU2016211246B2 (en) * | 2015-01-30 | 2020-08-27 | Biomed Valley Discoveries, Inc. | Crystalline forms of C21H22CI2N4O2 |
| WO2016123581A1 (en) * | 2015-01-30 | 2016-08-04 | Biomed Valley Discoveries, Inc. | Crystalline c21h22c12n4o2 malonate |
| WO2016187028A1 (en) * | 2015-05-15 | 2016-11-24 | Celgene Avilomics Research, Inc. | Heteroaryl compounds, synthesis thereof, and intermediates thereto |
| WO2016192063A1 (en) * | 2015-06-03 | 2016-12-08 | Changzhou Jiekai Pharmatech Co. Ltd | Heterocyclic compounds as erk inhibitors |
| CN106536522B (zh) * | 2015-06-03 | 2020-08-28 | 捷思英达医药技术(上海)有限公司 | 用于治疗银屑病的杂环化合物 |
| EP3365334B1 (en) | 2015-10-21 | 2024-07-17 | Otsuka Pharmaceutical Co., Ltd. | Benzolactam compounds as protein kinase inhibitors |
| EP3170822A1 (en) | 2015-11-18 | 2017-05-24 | AGV Discovery | Azaindole derivatives and their use as erk kinase inhibitors |
| CN109890827A (zh) * | 2016-10-05 | 2019-06-14 | 芝诺罗耶尔蒂里程碑有限责任公司 | 螺环化合物 |
| GB201706327D0 (en) | 2017-04-20 | 2017-06-07 | Otsuka Pharma Co Ltd | A pharmaceutical compound |
| US11406627B2 (en) * | 2017-10-12 | 2022-08-09 | Novartis Ag | Combinations of MDM2 inhibitors with inhibitors of ERK for treating cancers |
| WO2019180141A1 (en) | 2018-03-23 | 2019-09-26 | Bayer Aktiengesellschaft | Combinations of rogaratinib |
| WO2021020841A1 (en) * | 2019-07-29 | 2021-02-04 | Standigm Inc. | Composition for preventing or treating metabolic liver disease |
| EP4212531A1 (en) | 2022-01-14 | 2023-07-19 | AGV Discovery | Azaindole derivatives and their use as erk kinase inhibitors |
| WO2023169480A1 (zh) * | 2022-03-08 | 2023-09-14 | 甘李药业股份有限公司 | 氘代化合物,及其制备方法和应用 |
| AU2023238106A1 (en) * | 2022-03-24 | 2024-09-26 | Biomed Valley Discoveries, Inc. | Deuterated analogs of pyrrole inhibitors of erk, synthesis thereof and intermediates thereto |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5716981A (en) | 1993-07-19 | 1998-02-10 | Angiogenesis Technologies, Inc. | Anti-angiogenic compositions and methods of use |
| US5565413A (en) * | 1994-12-06 | 1996-10-15 | Zeneca Limited | Substituted pyridyl phenyl ketone herbicides |
| US5472966A (en) * | 1995-03-29 | 1995-12-05 | Bristol-Myers Squibb Company | Antidepressant heteroarylaminoalkyl derivatives of naphthyl-monazines |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| CA2383492A1 (en) * | 1999-06-22 | 2000-12-28 | Takeda Chemical Industries, Ltd. | Acylhydrazine derivatives, their production and use |
| MY130778A (en) * | 2001-02-09 | 2007-07-31 | Vertex Pharma | Heterocyclic inhibitiors of erk2 and uses thereof |
| US7304061B2 (en) * | 2002-04-26 | 2007-12-04 | Vertex Pharmaceuticals Incorporated | Heterocyclic inhibitors of ERK2 and uses thereof |
| EP1554269A1 (en) * | 2002-07-09 | 2005-07-20 | Vertex Pharmaceuticals Incorporated | Imidazoles, oxazoles and thiazoles with protein kinase inhibiting activities |
| HUE029020T2 (en) | 2002-07-18 | 2017-02-28 | Janssen Pharmaceutica Nv | Substituted triazine kinase inhibitors |
| GB0217780D0 (en) * | 2002-07-31 | 2002-09-11 | Glaxo Group Ltd | Compounds |
| NZ538715A (en) | 2002-08-14 | 2007-07-27 | Vertex Pharma | Protein kinase inhibitors and uses thereof |
| AU2004221881A1 (en) * | 2003-03-13 | 2004-09-30 | Vertex Pharmaceuticals Incorporated | Compositions useful as protein kinase inhibitors |
| AU2005245885B2 (en) * | 2004-05-14 | 2011-01-20 | Vertex Pharmaceuticals, Incorporated | Pyrrole compounds as inhibitors of ERK protein kinase, synthesis thereof and intermediates thereto |
-
2005
- 2005-05-13 AU AU2005245885A patent/AU2005245885B2/en active Active
- 2005-05-13 UA UAA200613208A patent/UA84930C2/ru unknown
- 2005-05-13 MX MXPA06013209A patent/MXPA06013209A/es active IP Right Grant
- 2005-05-13 BR BRPI0511111-0A patent/BRPI0511111A/pt not_active IP Right Cessation
- 2005-05-13 RU RU2006144445/04A patent/RU2376299C2/ru active
- 2005-05-13 ES ES19199330T patent/ES2897422T3/es active Active
- 2005-05-13 US US11/128,870 patent/US7354939B2/en not_active Ceased
- 2005-05-13 ES ES17184079T patent/ES2751761T3/es active Active
- 2005-05-13 WO PCT/US2005/016902 patent/WO2005113541A1/en active Application Filing
- 2005-05-13 ES ES14163944.3T patent/ES2651439T3/es active Active
- 2005-05-13 EP EP17184079.6A patent/EP3305776B1/en active Active
- 2005-05-13 PT PT171840796T patent/PT3305776T/pt unknown
- 2005-05-13 ES ES05748073.3T patent/ES2513965T3/es active Active
- 2005-05-13 DK DK17184079T patent/DK3305776T3/da active
- 2005-05-13 EP EP14163944.3A patent/EP2799434B1/en active Active
- 2005-05-13 LT LTEP17184079.6T patent/LT3305776T/lt unknown
- 2005-05-13 HU HUE17184079A patent/HUE047130T2/hu unknown
- 2005-05-13 PL PL17184079T patent/PL3305776T3/pl unknown
- 2005-05-13 EP EP19199330.2A patent/EP3608315B1/en active Active
- 2005-05-13 TW TW094115644A patent/TW200607803A/zh unknown
- 2005-05-13 EP EP05748073.3A patent/EP1753738B1/en active Active
- 2005-05-13 CN CNA200580021418XA patent/CN1976919A/zh active Pending
- 2005-05-13 AR ARP050101987A patent/AR051735A1/es not_active Application Discontinuation
- 2005-05-13 CA CA2566461A patent/CA2566461C/en active Active
- 2005-05-13 SI SI200532267T patent/SI3305776T1/sl unknown
- 2005-05-13 EP EP21190432.1A patent/EP3943489A1/en active Pending
- 2005-05-13 JP JP2007513432A patent/JP5132305B2/ja active Active
- 2005-05-13 ZA ZA200609975A patent/ZA200609975B/en unknown
- 2005-05-13 NZ NZ551582A patent/NZ551582A/en unknown
-
2006
- 2006-11-13 IL IL179207A patent/IL179207A0/en unknown
- 2006-12-12 NO NO20065727A patent/NO20065727L/no not_active Application Discontinuation
-
2013
- 2013-08-23 US US14/544,396 patent/USRE46097E1/en active Active
-
2015
- 2015-05-04 HK HK15104226.1A patent/HK1203505A1/xx unknown
-
2016
- 2016-06-28 US US15/195,021 patent/USRE47318E1/en active Active
- 2016-11-18 AR ARP160103545A patent/AR106755A2/es active IP Right Grant
-
2018
- 2018-05-15 HK HK18106295.9A patent/HK1247910A1/zh unknown
-
2019
- 2019-03-25 US US16/363,905 patent/USRE48266E1/en active Active
- 2019-12-03 CY CY20191101265T patent/CY1122336T1/el unknown
-
2020
- 2020-10-15 US US17/071,566 patent/USRE49500E1/en active Active
-
2023
- 2023-02-22 US US18/173,004 patent/US20230183276A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2897422T3 (es) | Compuestos pirroles como inhibidores de proteínas cinasas ERK y composiciones farmacéuticas que contienen esos compuestos | |
| EP1753744B1 (en) | Prodrugs of pyrrolylpyrimidine erk protein kinase inhibitors | |
| JP2007530595A (ja) | Erk2のピリジンインヒビターおよびそれらの使用 | |
| CN106496132B (zh) | N-(4-取代苯基)-2-取代乙酰胺类化合物及其作为sirt2蛋白抑制剂的用途 | |
| KR20020038817A (ko) | 암 치료를 위한 항세포증식제로서의 치환 피롤 | |
| KR20070031934A (ko) | Erk 단백질 키나아제의 억제제로서의 피롤 화합물, 이의합성 및 이의 중간체 | |
| HRP20020513A2 (en) | Substituted bisindolylmaleimides for the inhibition of cell proliferation |