ES2893621T3 - Métodos para el tratamiento de los síntomas residuales de la esquizofrenia - Google Patents
Métodos para el tratamiento de los síntomas residuales de la esquizofrenia Download PDFInfo
- Publication number
- ES2893621T3 ES2893621T3 ES14867061T ES14867061T ES2893621T3 ES 2893621 T3 ES2893621 T3 ES 2893621T3 ES 14867061 T ES14867061 T ES 14867061T ES 14867061 T ES14867061 T ES 14867061T ES 2893621 T3 ES2893621 T3 ES 2893621T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- compounds
- pharmaceutically acceptable
- agents
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 139
- 208000024891 symptom Diseases 0.000 title claims abstract description 85
- 201000000980 schizophrenia Diseases 0.000 title claims abstract description 37
- 150000001875 compounds Chemical class 0.000 claims abstract description 320
- 150000003839 salts Chemical group 0.000 claims abstract description 87
- 238000011282 treatment Methods 0.000 claims abstract description 56
- 239000000164 antipsychotic agent Substances 0.000 claims abstract description 28
- 239000003085 diluting agent Substances 0.000 claims abstract description 12
- 206010041243 Social avoidant behaviour Diseases 0.000 claims abstract description 9
- 206010014557 Emotional poverty Diseases 0.000 claims abstract description 8
- 239000000203 mixture Substances 0.000 claims description 129
- 239000003814 drug Substances 0.000 claims description 119
- 239000004005 microsphere Substances 0.000 claims description 84
- 229940079593 drug Drugs 0.000 claims description 74
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 claims description 56
- -1 Temazapam Chemical compound 0.000 claims description 46
- 238000009472 formulation Methods 0.000 claims description 42
- 229940124597 therapeutic agent Drugs 0.000 claims description 29
- 239000011159 matrix material Substances 0.000 claims description 26
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 claims description 22
- 239000000935 antidepressant agent Substances 0.000 claims description 20
- 230000003111 delayed effect Effects 0.000 claims description 20
- 230000002459 sustained effect Effects 0.000 claims description 18
- 229960001534 risperidone Drugs 0.000 claims description 16
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 claims description 16
- 230000000694 effects Effects 0.000 claims description 15
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 claims description 12
- 239000007972 injectable composition Substances 0.000 claims description 12
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 claims description 9
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 claims description 9
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 claims description 8
- 239000002775 capsule Substances 0.000 claims description 8
- 229940044551 receptor antagonist Drugs 0.000 claims description 8
- 239000002464 receptor antagonist Substances 0.000 claims description 8
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 claims description 8
- 229960002073 sertraline Drugs 0.000 claims description 8
- 229960003692 gamma aminobutyric acid Drugs 0.000 claims description 7
- VSWBSWWIRNCQIJ-GJZGRUSLSA-N (R,R)-asenapine Chemical compound O1C2=CC=CC=C2[C@@H]2CN(C)C[C@H]2C2=CC(Cl)=CC=C21 VSWBSWWIRNCQIJ-GJZGRUSLSA-N 0.000 claims description 6
- HXTGXYRHXAGCFP-OAQYLSRUSA-N (r)-(2,3-dimethoxyphenyl)-[1-[2-(4-fluorophenyl)ethyl]piperidin-4-yl]methanol Chemical compound COC1=CC=CC([C@H](O)C2CCN(CCC=3C=CC(F)=CC=3)CC2)=C1OC HXTGXYRHXAGCFP-OAQYLSRUSA-N 0.000 claims description 6
- PMXMIIMHBWHSKN-UHFFFAOYSA-N 3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-9-hydroxy-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a]pyrimidin-4-one Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCC(O)C4=NC=3C)=NOC2=C1 PMXMIIMHBWHSKN-UHFFFAOYSA-N 0.000 claims description 6
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 claims description 6
- PLDUPXSUYLZYBN-UHFFFAOYSA-N Fluphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 PLDUPXSUYLZYBN-UHFFFAOYSA-N 0.000 claims description 6
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 claims description 6
- KLPWJLBORRMFGK-UHFFFAOYSA-N Molindone Chemical compound O=C1C=2C(CC)=C(C)NC=2CCC1CN1CCOCC1 KLPWJLBORRMFGK-UHFFFAOYSA-N 0.000 claims description 6
- RGCVKNLCSQQDEP-UHFFFAOYSA-N Perphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 RGCVKNLCSQQDEP-UHFFFAOYSA-N 0.000 claims description 6
- ZGUGWUXLJSTTMA-UHFFFAOYSA-N Promazinum Chemical compound C1=CC=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZGUGWUXLJSTTMA-UHFFFAOYSA-N 0.000 claims description 6
- KLBQZWRITKRQQV-UHFFFAOYSA-N Thioridazine Chemical compound C12=CC(SC)=CC=C2SC2=CC=CC=C2N1CCC1CCCCN1C KLBQZWRITKRQQV-UHFFFAOYSA-N 0.000 claims description 6
- GFBKORZTTCHDGY-UWVJOHFNSA-N Thiothixene Chemical compound C12=CC(S(=O)(=O)N(C)C)=CC=C2SC2=CC=CC=C2\C1=C\CCN1CCN(C)CC1 GFBKORZTTCHDGY-UWVJOHFNSA-N 0.000 claims description 6
- NTJOBXMMWNYJFB-UHFFFAOYSA-N amisulpride Chemical compound CCN1CCCC1CNC(=O)C1=CC(S(=O)(=O)CC)=C(N)C=C1OC NTJOBXMMWNYJFB-UHFFFAOYSA-N 0.000 claims description 6
- 229960003036 amisulpride Drugs 0.000 claims description 6
- 229960004372 aripiprazole Drugs 0.000 claims description 6
- 229960005245 asenapine Drugs 0.000 claims description 6
- 229960005123 cariprazine Drugs 0.000 claims description 6
- KPWSJANDNDDRMB-QAQDUYKDSA-N cariprazine Chemical compound C1C[C@@H](NC(=O)N(C)C)CC[C@@H]1CCN1CCN(C=2C(=C(Cl)C=CC=2)Cl)CC1 KPWSJANDNDDRMB-QAQDUYKDSA-N 0.000 claims description 6
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 claims description 6
- 229960001076 chlorpromazine Drugs 0.000 claims description 6
- 229960005426 doxepin Drugs 0.000 claims description 6
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 claims description 6
- RMEDXOLNCUSCGS-UHFFFAOYSA-N droperidol Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC=C(N2C(NC3=CC=CC=C32)=O)CC1 RMEDXOLNCUSCGS-UHFFFAOYSA-N 0.000 claims description 6
- 229960000394 droperidol Drugs 0.000 claims description 6
- 229960002690 fluphenazine Drugs 0.000 claims description 6
- 229960003878 haloperidol Drugs 0.000 claims description 6
- 229960003162 iloperidone Drugs 0.000 claims description 6
- XMXHEBAFVSFQEX-UHFFFAOYSA-N iloperidone Chemical compound COC1=CC(C(C)=O)=CC=C1OCCCN1CCC(C=2C3=CC=C(F)C=C3ON=2)CC1 XMXHEBAFVSFQEX-UHFFFAOYSA-N 0.000 claims description 6
- 229960000423 loxapine Drugs 0.000 claims description 6
- 229960001432 lurasidone Drugs 0.000 claims description 6
- PQXKDMSYBGKCJA-CVTJIBDQSA-N lurasidone Chemical compound C1=CC=C2C(N3CCN(CC3)C[C@@H]3CCCC[C@H]3CN3C(=O)[C@@H]4[C@H]5CC[C@H](C5)[C@@H]4C3=O)=NSC2=C1 PQXKDMSYBGKCJA-CVTJIBDQSA-N 0.000 claims description 6
- SLVMESMUVMCQIY-UHFFFAOYSA-N mesoridazine Chemical compound CN1CCCCC1CCN1C2=CC(S(C)=O)=CC=C2SC2=CC=CC=C21 SLVMESMUVMCQIY-UHFFFAOYSA-N 0.000 claims description 6
- 229960000300 mesoridazine Drugs 0.000 claims description 6
- 229960004938 molindone Drugs 0.000 claims description 6
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 claims description 6
- 229960001800 nefazodone Drugs 0.000 claims description 6
- 229960005017 olanzapine Drugs 0.000 claims description 6
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 claims description 6
- 229960001057 paliperidone Drugs 0.000 claims description 6
- 229960002296 paroxetine Drugs 0.000 claims description 6
- 229960000762 perphenazine Drugs 0.000 claims description 6
- YVUQSNJEYSNKRX-UHFFFAOYSA-N pimozide Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)CCCN1CCC(N2C(NC3=CC=CC=C32)=O)CC1 YVUQSNJEYSNKRX-UHFFFAOYSA-N 0.000 claims description 6
- 229960003634 pimozide Drugs 0.000 claims description 6
- 229960003111 prochlorperazine Drugs 0.000 claims description 6
- WIKYUJGCLQQFNW-UHFFFAOYSA-N prochlorperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 WIKYUJGCLQQFNW-UHFFFAOYSA-N 0.000 claims description 6
- 229960003598 promazine Drugs 0.000 claims description 6
- 229960004431 quetiapine Drugs 0.000 claims description 6
- URKOMYMAXPYINW-UHFFFAOYSA-N quetiapine Chemical compound C1CN(CCOCCO)CCN1C1=NC2=CC=CC=C2SC2=CC=CC=C12 URKOMYMAXPYINW-UHFFFAOYSA-N 0.000 claims description 6
- 102000005962 receptors Human genes 0.000 claims description 6
- 108020003175 receptors Proteins 0.000 claims description 6
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 claims description 6
- 229960000652 sertindole Drugs 0.000 claims description 6
- GZKLJWGUPQBVJQ-UHFFFAOYSA-N sertindole Chemical compound C1=CC(F)=CC=C1N1C2=CC=C(Cl)C=C2C(C2CCN(CCN3C(NCC3)=O)CC2)=C1 GZKLJWGUPQBVJQ-UHFFFAOYSA-N 0.000 claims description 6
- 229960002784 thioridazine Drugs 0.000 claims description 6
- 229960005013 tiotixene Drugs 0.000 claims description 6
- PHLBKPHSAVXXEF-UHFFFAOYSA-N trazodone Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1 PHLBKPHSAVXXEF-UHFFFAOYSA-N 0.000 claims description 6
- 229960003991 trazodone Drugs 0.000 claims description 6
- 229960002324 trifluoperazine Drugs 0.000 claims description 6
- ZEWQUBUPAILYHI-UHFFFAOYSA-N trifluoperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 ZEWQUBUPAILYHI-UHFFFAOYSA-N 0.000 claims description 6
- 229960000607 ziprasidone Drugs 0.000 claims description 6
- MVWVFYHBGMAFLY-UHFFFAOYSA-N ziprasidone Chemical compound C1=CC=C2C(N3CCN(CC3)CCC3=CC=4CC(=O)NC=4C=C3Cl)=NSC2=C1 MVWVFYHBGMAFLY-UHFFFAOYSA-N 0.000 claims description 6
- 229960004496 zotepine Drugs 0.000 claims description 6
- HDOZVRUNCMBHFH-UHFFFAOYSA-N zotepine Chemical compound CN(C)CCOC1=CC2=CC=CC=C2SC2=CC=C(Cl)C=C12 HDOZVRUNCMBHFH-UHFFFAOYSA-N 0.000 claims description 6
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 claims description 5
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 claims description 5
- YLXDSYKOBKBWJQ-LBPRGKRZSA-N N-[2-[(8S)-2,6,7,8-tetrahydro-1H-cyclopenta[e]benzofuran-8-yl]ethyl]propanamide Chemical compound C1=C2OCCC2=C2[C@H](CCNC(=O)CC)CCC2=C1 YLXDSYKOBKBWJQ-LBPRGKRZSA-N 0.000 claims description 5
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 claims description 5
- 229960001653 citalopram Drugs 0.000 claims description 5
- 229960004170 clozapine Drugs 0.000 claims description 5
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 claims description 5
- 230000000295 complement effect Effects 0.000 claims description 5
- 239000002552 dosage form Substances 0.000 claims description 5
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 claims description 5
- 229960002464 fluoxetine Drugs 0.000 claims description 5
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 claims description 4
- 101000831616 Homo sapiens Protachykinin-1 Proteins 0.000 claims description 4
- 102000004310 Ion Channels Human genes 0.000 claims description 4
- 229940123730 Orexin receptor antagonist Drugs 0.000 claims description 4
- 102100024304 Protachykinin-1 Human genes 0.000 claims description 4
- 230000009471 action Effects 0.000 claims description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 4
- 229960002866 duloxetine Drugs 0.000 claims description 4
- 239000003326 hypnotic agent Substances 0.000 claims description 4
- ADNPLDHMAVUMIW-CUZNLEPHSA-N substance P Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 ADNPLDHMAVUMIW-CUZNLEPHSA-N 0.000 claims description 4
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 claims description 4
- 229960004688 venlafaxine Drugs 0.000 claims description 4
- IGLYMJRIWWIQQE-QUOODJBBSA-N (1S,2R)-2-phenylcyclopropan-1-amine (1R,2S)-2-phenylcyclopropan-1-amine Chemical compound N[C@H]1C[C@@H]1C1=CC=CC=C1.N[C@@H]1C[C@H]1C1=CC=CC=C1 IGLYMJRIWWIQQE-QUOODJBBSA-N 0.000 claims description 3
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 claims description 3
- GBBSUAFBMRNDJC-MRXNPFEDSA-N (5R)-zopiclone Chemical compound C1CN(C)CCN1C(=O)O[C@@H]1C2=NC=CN=C2C(=O)N1C1=CC=C(Cl)C=N1 GBBSUAFBMRNDJC-MRXNPFEDSA-N 0.000 claims description 3
- XKFPYPQQHFEXRZ-UHFFFAOYSA-N 5-methyl-N'-(phenylmethyl)-3-isoxazolecarbohydrazide Chemical compound O1C(C)=CC(C(=O)NNCC=2C=CC=CC=2)=N1 XKFPYPQQHFEXRZ-UHFFFAOYSA-N 0.000 claims description 3
- BQBQAWXALAGUIW-GHVWMZMZSA-N 6-[5-[[(2s)-2-(4-fluorophenyl)-4-methylpiperazin-1-yl]methyl]furan-3-yl]-3h-1,3-benzoxazol-2-one;hydrate;hydrochloride Chemical compound O.Cl.C1([C@@H]2N(CC=3OC=C(C=3)C=3C=C4OC(O)=NC4=CC=3)CCN(C2)C)=CC=C(F)C=C1 BQBQAWXALAGUIW-GHVWMZMZSA-N 0.000 claims description 3
- RPUBHMMISKEXSR-MLCLTIQSSA-N 680993-85-5 Chemical compound OC(=O)\C=C/C(O)=O.C1C2=CC=CN=C2N2CCN(C)C[C@@H]2C2=CC=CC=C21 RPUBHMMISKEXSR-MLCLTIQSSA-N 0.000 claims description 3
- VMIYHDSEFNYJSL-UHFFFAOYSA-N Bromazepam Chemical compound C12=CC(Br)=CC=C2NC(=O)CN=C1C1=CC=CC=N1 VMIYHDSEFNYJSL-UHFFFAOYSA-N 0.000 claims description 3
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 claims description 3
- 108010056643 Corticotropin-Releasing Hormone Receptors Proteins 0.000 claims description 3
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 claims description 3
- 229940122880 Estrogen receptor agonist Drugs 0.000 claims description 3
- 229940123935 Galanin receptor agonist Drugs 0.000 claims description 3
- 229940103526 Growth hormone receptor agonist Drugs 0.000 claims description 3
- 102000002265 Human Growth Hormone Human genes 0.000 claims description 3
- 108010000521 Human Growth Hormone Proteins 0.000 claims description 3
- 239000000854 Human Growth Hormone Substances 0.000 claims description 3
- CBIAWPMZSFFRGN-UHFFFAOYSA-N Indiplon Chemical compound CC(=O)N(C)C1=CC=CC(C=2N3N=CC(=C3N=CC=2)C(=O)C=2SC=CC=2)=C1 CBIAWPMZSFFRGN-UHFFFAOYSA-N 0.000 claims description 3
- YJPIGAIKUZMOQA-UHFFFAOYSA-N Melatonin Natural products COC1=CC=C2N(C(C)=O)C=C(CCN)C2=C1 YJPIGAIKUZMOQA-UHFFFAOYSA-N 0.000 claims description 3
- COSPVUFTLGQDQL-UHFFFAOYSA-N Nelotanserin Chemical compound C1=C(C=2N(N=CC=2Br)C)C(OC)=CC=C1NC(=O)NC1=CC=C(F)C=C1F COSPVUFTLGQDQL-UHFFFAOYSA-N 0.000 claims description 3
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 claims description 3
- 102000002512 Orexin Human genes 0.000 claims description 3
- RXBKMJIPNDOHFR-UHFFFAOYSA-N Phenelzine sulfate Chemical compound OS(O)(=O)=O.NNCCC1=CC=CC=C1 RXBKMJIPNDOHFR-UHFFFAOYSA-N 0.000 claims description 3
- AQRLDDAFYYAIJP-UHFFFAOYSA-N Pruvanserin Chemical compound C1=CC(F)=CC=C1CCN1CCN(C(=O)C=2C=3NC=C(C=3C=CC=2)C#N)CC1 AQRLDDAFYYAIJP-UHFFFAOYSA-N 0.000 claims description 3
- PPTYJKAXVCCBDU-UHFFFAOYSA-N Rohypnol Chemical compound N=1CC(=O)N(C)C2=CC=C([N+]([O-])=O)C=C2C=1C1=CC=CC=C1F PPTYJKAXVCCBDU-UHFFFAOYSA-N 0.000 claims description 3
- 229960002629 agomelatine Drugs 0.000 claims description 3
- YJYPHIXNFHFHND-UHFFFAOYSA-N agomelatine Chemical compound C1=CC=C(CCNC(C)=O)C2=CC(OC)=CC=C21 YJYPHIXNFHFHND-UHFFFAOYSA-N 0.000 claims description 3
- 229960004538 alprazolam Drugs 0.000 claims description 3
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 claims description 3
- 229960000836 amitriptyline Drugs 0.000 claims description 3
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 claims description 3
- 229960002519 amoxapine Drugs 0.000 claims description 3
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 claims description 3
- 150000003936 benzamides Chemical class 0.000 claims description 3
- 230000005540 biological transmission Effects 0.000 claims description 3
- 229960002729 bromazepam Drugs 0.000 claims description 3
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 claims description 3
- 229960001058 bupropion Drugs 0.000 claims description 3
- 229960002495 buspirone Drugs 0.000 claims description 3
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 claims description 3
- 150000001733 carboxylic acid esters Chemical group 0.000 claims description 3
- 229960003778 casopitant Drugs 0.000 claims description 3
- XGGTZCKQRWXCHW-WMTVXVAQSA-N casopitant Chemical compound C1([C@H]2C[C@H](CCN2C(=O)N(C)[C@H](C)C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)N2CCN(CC2)C(C)=O)=CC=C(F)C=C1C XGGTZCKQRWXCHW-WMTVXVAQSA-N 0.000 claims description 3
- 229960001403 clobazam Drugs 0.000 claims description 3
- CXOXHMZGEKVPMT-UHFFFAOYSA-N clobazam Chemical compound O=C1CC(=O)N(C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 CXOXHMZGEKVPMT-UHFFFAOYSA-N 0.000 claims description 3
- 229960004606 clomipramine Drugs 0.000 claims description 3
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 claims description 3
- 229960003120 clonazepam Drugs 0.000 claims description 3
- 229960004362 clorazepate Drugs 0.000 claims description 3
- XDDJGVMJFWAHJX-UHFFFAOYSA-N clorazepic acid Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)O)N=C1C1=CC=CC=C1 XDDJGVMJFWAHJX-UHFFFAOYSA-N 0.000 claims description 3
- 229960003914 desipramine Drugs 0.000 claims description 3
- 229960003529 diazepam Drugs 0.000 claims description 3
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 claims description 3
- 229960004341 escitalopram Drugs 0.000 claims description 3
- 229960002336 estazolam Drugs 0.000 claims description 3
- CDCHDCWJMGXXRH-UHFFFAOYSA-N estazolam Chemical compound C=1C(Cl)=CC=C(N2C=NN=C2CN=2)C=1C=2C1=CC=CC=C1 CDCHDCWJMGXXRH-UHFFFAOYSA-N 0.000 claims description 3
- 229940011871 estrogen Drugs 0.000 claims description 3
- 239000000262 estrogen Substances 0.000 claims description 3
- GBBSUAFBMRNDJC-INIZCTEOSA-N eszopiclone Chemical compound C1CN(C)CCN1C(=O)O[C@H]1C2=NC=CN=C2C(=O)N1C1=CC=C(Cl)C=N1 GBBSUAFBMRNDJC-INIZCTEOSA-N 0.000 claims description 3
- 229960001578 eszopiclone Drugs 0.000 claims description 3
- JCYLWUVDHLVGER-UHFFFAOYSA-N evt-201 Chemical compound O1C(CN(C)C)=NC(C2=C3N(C4=CC=CC(Cl)=C4C(=O)N(C)C3)C=N2)=N1 JCYLWUVDHLVGER-UHFFFAOYSA-N 0.000 claims description 3
- 229960002200 flunitrazepam Drugs 0.000 claims description 3
- 229960003528 flurazepam Drugs 0.000 claims description 3
- SAADBVWGJQAEFS-UHFFFAOYSA-N flurazepam Chemical compound N=1CC(=O)N(CCN(CC)CC)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F SAADBVWGJQAEFS-UHFFFAOYSA-N 0.000 claims description 3
- 229960004038 fluvoxamine Drugs 0.000 claims description 3
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 claims description 3
- 229960002870 gabapentin Drugs 0.000 claims description 3
- 239000003382 histamine H3 receptor agonist Substances 0.000 claims description 3
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 claims description 3
- 229960004801 imipramine Drugs 0.000 claims description 3
- 229950003867 indiplon Drugs 0.000 claims description 3
- 229960002672 isocarboxazid Drugs 0.000 claims description 3
- 229960005417 ketanserin Drugs 0.000 claims description 3
- FPCCSQOGAWCVBH-UHFFFAOYSA-N ketanserin Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCN2C(C3=CC=CC=C3NC2=O)=O)CC1 FPCCSQOGAWCVBH-UHFFFAOYSA-N 0.000 claims description 3
- 229960001848 lamotrigine Drugs 0.000 claims description 3
- PYZRQGJRPPTADH-UHFFFAOYSA-N lamotrigine Chemical compound NC1=NC(N)=NN=C1C1=CC=CC(Cl)=C1Cl PYZRQGJRPPTADH-UHFFFAOYSA-N 0.000 claims description 3
- 229960004391 lorazepam Drugs 0.000 claims description 3
- 229960004090 maprotiline Drugs 0.000 claims description 3
- QSLMDECMDJKHMQ-GSXCWMCISA-N maprotiline Chemical compound C12=CC=CC=C2[C@@]2(CCCNC)C3=CC=CC=C3[C@@H]1CC2 QSLMDECMDJKHMQ-GSXCWMCISA-N 0.000 claims description 3
- 229960003987 melatonin Drugs 0.000 claims description 3
- DRLFMBDRBRZALE-UHFFFAOYSA-N melatonin Chemical compound COC1=CC=C2NC=C(CCNC(C)=O)C2=C1 DRLFMBDRBRZALE-UHFFFAOYSA-N 0.000 claims description 3
- 229960003793 midazolam Drugs 0.000 claims description 3
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 claims description 3
- RONZAEMNMFQXRA-UHFFFAOYSA-N mirtazapine Chemical compound C1C2=CC=CN=C2N2CCN(C)CC2C2=CC=CC=C21 RONZAEMNMFQXRA-UHFFFAOYSA-N 0.000 claims description 3
- 229960001785 mirtazapine Drugs 0.000 claims description 3
- DYCKFEBIOUQECE-UHFFFAOYSA-N nefazodone hydrochloride Chemical compound [H+].[Cl-].O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 DYCKFEBIOUQECE-UHFFFAOYSA-N 0.000 claims description 3
- KJONHKAYOJNZEC-UHFFFAOYSA-N nitrazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1 KJONHKAYOJNZEC-UHFFFAOYSA-N 0.000 claims description 3
- 229960001454 nitrazepam Drugs 0.000 claims description 3
- 230000002474 noradrenergic effect Effects 0.000 claims description 3
- 229960001158 nortriptyline Drugs 0.000 claims description 3
- 108060005714 orexin Proteins 0.000 claims description 3
- MEEQBDCQPIZMLY-HNNXBMFYSA-N osemozotan Chemical compound C1=C2OCOC2=CC(OCCCNC[C@@H]2OC3=CC=CC=C3OC2)=C1 MEEQBDCQPIZMLY-HNNXBMFYSA-N 0.000 claims description 3
- 229960004535 oxazepam Drugs 0.000 claims description 3
- ADIMAYPTOBDMTL-UHFFFAOYSA-N oxazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(O)N=C1C1=CC=CC=C1 ADIMAYPTOBDMTL-UHFFFAOYSA-N 0.000 claims description 3
- 229960004790 phenelzine sulfate Drugs 0.000 claims description 3
- AYXYPKUFHZROOJ-ZETCQYMHSA-N pregabalin Chemical compound CC(C)C[C@H](CN)CC(O)=O AYXYPKUFHZROOJ-ZETCQYMHSA-N 0.000 claims description 3
- 229960001233 pregabalin Drugs 0.000 claims description 3
- 229960002601 protriptyline Drugs 0.000 claims description 3
- BWPIARFWQZKAIA-UHFFFAOYSA-N protriptyline Chemical compound C1=CC2=CC=CC=C2C(CCCNC)C2=CC=CC=C21 BWPIARFWQZKAIA-UHFFFAOYSA-N 0.000 claims description 3
- 229950003077 pruvanserin Drugs 0.000 claims description 3
- 229960001150 ramelteon Drugs 0.000 claims description 3
- YGYBFMRFXNDIPO-QGZVFWFLSA-N repinotan Chemical compound O=S1(=O)C2=CC=CC=C2C(=O)N1CCCCNC[C@@H]1OC2=CC=CC=C2CC1 YGYBFMRFXNDIPO-QGZVFWFLSA-N 0.000 claims description 3
- 229950009693 repinotan Drugs 0.000 claims description 3
- 229950009626 ritanserin Drugs 0.000 claims description 3
- JUQLTPCYUFPYKE-UHFFFAOYSA-N ritanserin Chemical compound CC=1N=C2SC=CN2C(=O)C=1CCN(CC1)CCC1=C(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 JUQLTPCYUFPYKE-UHFFFAOYSA-N 0.000 claims description 3
- HKFMQJUJWSFOLY-OAQYLSRUSA-N sarizotan Chemical compound C1=CC(F)=CC=C1C1=CN=CC(CNC[C@@H]2OC3=CC=CC=C3CC2)=C1 HKFMQJUJWSFOLY-OAQYLSRUSA-N 0.000 claims description 3
- 229950007903 sarizotan Drugs 0.000 claims description 3
- 229940076279 serotonin Drugs 0.000 claims description 3
- 239000003772 serotonin uptake inhibitor Substances 0.000 claims description 3
- 229940099190 serzone Drugs 0.000 claims description 3
- 229960000660 tasimelteon Drugs 0.000 claims description 3
- PTOIAAWZLUQTIO-GXFFZTMASA-N tasimelteon Chemical compound CCC(=O)NC[C@@H]1C[C@H]1C1=CC=CC2=C1CCO2 PTOIAAWZLUQTIO-GXFFZTMASA-N 0.000 claims description 3
- PBJUNZJWGZTSKL-MRXNPFEDSA-N tiagabine Chemical compound C1=CSC(C(=CCCN2C[C@@H](CCC2)C(O)=O)C2=C(C=CS2)C)=C1C PBJUNZJWGZTSKL-MRXNPFEDSA-N 0.000 claims description 3
- 229960001918 tiagabine Drugs 0.000 claims description 3
- 229960003741 tranylcypromine Drugs 0.000 claims description 3
- 229960003386 triazolam Drugs 0.000 claims description 3
- JOFWLTCLBGQGBO-UHFFFAOYSA-N triazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl JOFWLTCLBGQGBO-UHFFFAOYSA-N 0.000 claims description 3
- ZSCDBOWYZJWBIY-UHFFFAOYSA-N trimipramine Chemical compound C1CC2=CC=CC=C2N(CC(CN(C)C)C)C2=CC=CC=C21 ZSCDBOWYZJWBIY-UHFFFAOYSA-N 0.000 claims description 3
- 229960002431 trimipramine Drugs 0.000 claims description 3
- 229950002976 volinanserin Drugs 0.000 claims description 3
- HUNXMJYCHXQEGX-UHFFFAOYSA-N zaleplon Chemical compound CCN(C(C)=O)C1=CC=CC(C=2N3N=CC(=C3N=CC=2)C#N)=C1 HUNXMJYCHXQEGX-UHFFFAOYSA-N 0.000 claims description 3
- 229960004010 zaleplon Drugs 0.000 claims description 3
- ZAFYATHCZYHLPB-UHFFFAOYSA-N zolpidem Chemical compound N1=C2C=CC(C)=CN2C(CC(=O)N(C)C)=C1C1=CC=C(C)C=C1 ZAFYATHCZYHLPB-UHFFFAOYSA-N 0.000 claims description 3
- 229960001475 zolpidem Drugs 0.000 claims description 3
- 229960000820 zopiclone Drugs 0.000 claims description 3
- YFGHCGITMMYXAQ-UHFFFAOYSA-N 2-[(diphenylmethyl)sulfinyl]acetamide Chemical compound C=1C=CC=CC=1C(S(=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-UHFFFAOYSA-N 0.000 claims description 2
- 239000003185 4 aminobutyric acid B receptor stimulating agent Substances 0.000 claims description 2
- 229940121723 Melatonin receptor agonist Drugs 0.000 claims description 2
- 229940123445 Tricyclic antidepressant Drugs 0.000 claims description 2
- 229960004823 armodafinil Drugs 0.000 claims description 2
- YFGHCGITMMYXAQ-LJQANCHMSA-N armodafinil Chemical compound C=1C=CC=CC=1C([S@](=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-LJQANCHMSA-N 0.000 claims description 2
- 229960001165 modafinil Drugs 0.000 claims description 2
- 230000002035 prolonged effect Effects 0.000 claims description 2
- 229940071773 rozerem Drugs 0.000 claims description 2
- 239000003215 serotonin 5-HT2 receptor antagonist Substances 0.000 claims description 2
- 239000003029 tricyclic antidepressant agent Substances 0.000 claims description 2
- PJDFLNIOAUIZSL-UHFFFAOYSA-N vigabatrin Chemical compound C=CC(N)CCC(O)=O PJDFLNIOAUIZSL-UHFFFAOYSA-N 0.000 claims description 2
- 229960005318 vigabatrin Drugs 0.000 claims description 2
- XJGVXQDUIWGIRW-UHFFFAOYSA-N loxapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2OC2=CC=C(Cl)C=C12 XJGVXQDUIWGIRW-UHFFFAOYSA-N 0.000 claims 3
- 239000002767 noradrenalin uptake inhibitor Substances 0.000 claims 1
- 229940127221 norepinephrine reuptake inhibitor Drugs 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 description 56
- 229920000642 polymer Polymers 0.000 description 54
- 229940126062 Compound A Drugs 0.000 description 53
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 53
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- 239000012458 free base Substances 0.000 description 33
- 239000003963 antioxidant agent Substances 0.000 description 32
- 235000006708 antioxidants Nutrition 0.000 description 32
- 208000028017 Psychotic disease Diseases 0.000 description 30
- 230000003078 antioxidant effect Effects 0.000 description 29
- 239000002253 acid Substances 0.000 description 26
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 25
- 229920000747 poly(lactic acid) Polymers 0.000 description 25
- 238000002360 preparation method Methods 0.000 description 25
- 239000012071 phase Substances 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 239000004626 polylactic acid Substances 0.000 description 20
- 239000004372 Polyvinyl alcohol Substances 0.000 description 19
- 229920002451 polyvinyl alcohol Polymers 0.000 description 19
- 238000011068 loading method Methods 0.000 description 18
- 239000011859 microparticle Substances 0.000 description 18
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 17
- 230000001154 acute effect Effects 0.000 description 17
- 239000000243 solution Substances 0.000 description 17
- 208000035475 disorder Diseases 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 15
- 239000007903 gelatin capsule Substances 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 239000007864 aqueous solution Substances 0.000 description 12
- 150000002148 esters Chemical class 0.000 description 12
- 208000020925 Bipolar disease Diseases 0.000 description 11
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 11
- 239000000651 prodrug Substances 0.000 description 11
- 229940002612 prodrug Drugs 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 10
- 208000019116 sleep disease Diseases 0.000 description 10
- 230000004888 barrier function Effects 0.000 description 9
- 239000003795 chemical substances by application Substances 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 8
- 229920001577 copolymer Polymers 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 241000700159 Rattus Species 0.000 description 7
- 230000000561 anti-psychotic effect Effects 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 208000019901 Anxiety disease Diseases 0.000 description 6
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 6
- 206010012239 Delusion Diseases 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 229940005513 antidepressants Drugs 0.000 description 6
- 230000036506 anxiety Effects 0.000 description 6
- 230000006399 behavior Effects 0.000 description 6
- 210000004556 brain Anatomy 0.000 description 6
- 239000008367 deionised water Substances 0.000 description 6
- 229910021641 deionized water Inorganic materials 0.000 description 6
- 231100000868 delusion Toxicity 0.000 description 6
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 208000024714 major depressive disease Diseases 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 6
- 230000003204 osmotic effect Effects 0.000 description 6
- 208000022610 schizoaffective disease Diseases 0.000 description 6
- 230000000392 somatic effect Effects 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- 208000004547 Hallucinations Diseases 0.000 description 5
- 241000282414 Homo sapiens Species 0.000 description 5
- 239000013543 active substance Substances 0.000 description 5
- 208000029560 autism spectrum disease Diseases 0.000 description 5
- 230000015556 catabolic process Effects 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- 235000014655 lactic acid Nutrition 0.000 description 5
- 239000004310 lactic acid Substances 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 230000004962 physiological condition Effects 0.000 description 5
- 239000000902 placebo Substances 0.000 description 5
- 229940068196 placebo Drugs 0.000 description 5
- 229920000728 polyester Polymers 0.000 description 5
- 150000003077 polyols Chemical group 0.000 description 5
- 238000009097 single-agent therapy Methods 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 238000000935 solvent evaporation Methods 0.000 description 5
- 238000013268 sustained release Methods 0.000 description 5
- 239000012730 sustained-release form Substances 0.000 description 5
- TUNFSRHWOTWDNC-UHFFFAOYSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 5
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 4
- WIHMBLDNRMIGDW-UHFFFAOYSA-N 1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-3h-2-benzofuran-5-carbonitrile;hydron;bromide Chemical compound [Br-].O1CC2=CC(C#N)=CC=C2C1(CCC[NH+](C)C)C1=CC=C(F)C=C1 WIHMBLDNRMIGDW-UHFFFAOYSA-N 0.000 description 4
- 102000049773 5-HT2A Serotonin Receptor Human genes 0.000 description 4
- 108010072564 5-HT2A Serotonin Receptor Proteins 0.000 description 4
- RDMFHRSPDKWERA-UHFFFAOYSA-N 5H-Pyrido[4,3-b]indole Chemical class C1=NC=C2C3=CC=CC=C3NC2=C1 RDMFHRSPDKWERA-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- LFMYNZPAVPMEGP-PIDGMYBPSA-N Fluvoxamine maleate Chemical compound OC(=O)\C=C/C(O)=O.COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 LFMYNZPAVPMEGP-PIDGMYBPSA-N 0.000 description 4
- JVTAAEKCZFNVCJ-REOHCLBHSA-N L-lactic acid Chemical compound C[C@H](O)C(O)=O JVTAAEKCZFNVCJ-REOHCLBHSA-N 0.000 description 4
- GIYXAJPCNFJEHY-UHFFFAOYSA-N N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]-1-propanamine hydrochloride (1:1) Chemical compound Cl.C=1C=CC=CC=1C(CCNC)OC1=CC=C(C(F)(F)F)C=C1 GIYXAJPCNFJEHY-UHFFFAOYSA-N 0.000 description 4
- 229920000954 Polyglycolide Polymers 0.000 description 4
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- 239000003693 atypical antipsychotic agent Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 208000010877 cognitive disease Diseases 0.000 description 4
- 230000006735 deficit Effects 0.000 description 4
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 230000000848 glutamatergic effect Effects 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 4
- 206010022437 insomnia Diseases 0.000 description 4
- 235000019136 lipoic acid Nutrition 0.000 description 4
- AGBQKNBQESQNJD-UHFFFAOYSA-N lipoic acid Chemical compound OC(=O)CCCCC1CCSS1 AGBQKNBQESQNJD-UHFFFAOYSA-N 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 239000002530 phenolic antioxidant Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 229940044601 receptor agonist Drugs 0.000 description 4
- 239000000018 receptor agonist Substances 0.000 description 4
- 150000004671 saturated fatty acids Chemical class 0.000 description 4
- 230000000698 schizophrenic effect Effects 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 229960002663 thioctic acid Drugs 0.000 description 4
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 4
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 4
- KTGRHKOEFSJQNS-BDQAORGHSA-N (1s)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-3h-2-benzofuran-5-carbonitrile;oxalic acid Chemical compound OC(=O)C(O)=O.C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 KTGRHKOEFSJQNS-BDQAORGHSA-N 0.000 description 3
- RKDVKSZUMVYZHH-UHFFFAOYSA-N 1,4-dioxane-2,5-dione Chemical compound O=C1COC(=O)CO1 RKDVKSZUMVYZHH-UHFFFAOYSA-N 0.000 description 3
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 description 3
- 208000020706 Autistic disease Diseases 0.000 description 3
- GHVNFZFCNZKVNT-UHFFFAOYSA-N Decanoic acid Natural products CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 108090000862 Ion Channels Proteins 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 108010089430 Phosphoproteins Proteins 0.000 description 3
- 102000007982 Phosphoproteins Human genes 0.000 description 3
- 229920001710 Polyorthoester Polymers 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 102000019208 Serotonin Plasma Membrane Transport Proteins Human genes 0.000 description 3
- 108010012996 Serotonin Plasma Membrane Transport Proteins Proteins 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 3
- 230000001430 anti-depressive effect Effects 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 235000010323 ascorbic acid Nutrition 0.000 description 3
- 229960005070 ascorbic acid Drugs 0.000 description 3
- 239000011668 ascorbic acid Substances 0.000 description 3
- 208000028683 bipolar I disease Diseases 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 208000015114 central nervous system disease Diseases 0.000 description 3
- 230000003291 dopaminomimetic effect Effects 0.000 description 3
- 238000004945 emulsification Methods 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 230000003400 hallucinatory effect Effects 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 150000002440 hydroxy compounds Chemical class 0.000 description 3
- 238000010255 intramuscular injection Methods 0.000 description 3
- 239000007927 intramuscular injection Substances 0.000 description 3
- JJTUDXZGHPGLLC-UHFFFAOYSA-N lactide Chemical compound CC1OC(=O)C(C)OC1=O JJTUDXZGHPGLLC-UHFFFAOYSA-N 0.000 description 3
- YQZBAXDVDZTKEQ-UHFFFAOYSA-N loxapine succinate Chemical compound [H+].[H+].[O-]C(=O)CCC([O-])=O.C1CN(C)CCN1C1=NC2=CC=CC=C2OC2=CC=C(Cl)C=C12 YQZBAXDVDZTKEQ-UHFFFAOYSA-N 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 229920005862 polyol Polymers 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000002400 serotonin 2A antagonist Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 238000010254 subcutaneous injection Methods 0.000 description 3
- 239000007929 subcutaneous injection Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 2
- XGCDBGRZEKYHNV-UHFFFAOYSA-N 1,1-bis(diphenylphosphino)methane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CP(C=1C=CC=CC=1)C1=CC=CC=C1 XGCDBGRZEKYHNV-UHFFFAOYSA-N 0.000 description 2
- BGMZUEKZENQUJY-UHFFFAOYSA-N 2-(4-iodo-2,5-dimethoxyphenyl)-1-methylethylamine Chemical compound COC1=CC(CC(C)N)=C(OC)C=C1I BGMZUEKZENQUJY-UHFFFAOYSA-N 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- 206010003805 Autism Diseases 0.000 description 2
- 206010005885 Blunted affect Diseases 0.000 description 2
- 208000028698 Cognitive impairment Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 208000024254 Delusional disease Diseases 0.000 description 2
- 206010012289 Dementia Diseases 0.000 description 2
- 208000020401 Depressive disease Diseases 0.000 description 2
- 208000026331 Disruptive, Impulse Control, and Conduct disease Diseases 0.000 description 2
- DYEFUKCXAQOFHX-UHFFFAOYSA-N Ebselen Chemical compound [se]1C2=CC=CC=C2C(=O)N1C1=CC=CC=C1 DYEFUKCXAQOFHX-UHFFFAOYSA-N 0.000 description 2
- NMYAHEULKSYAPP-UHFFFAOYSA-N Eptapirone Chemical compound O=C1N(C)C(=O)C=NN1CCCCN1CCN(C=2N=CC=CN=2)CC1 NMYAHEULKSYAPP-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102100022630 Glutamate receptor ionotropic, NMDA 2B Human genes 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 208000009668 Neurobehavioral Manifestations Diseases 0.000 description 2
- 0 O[C@](CCCN1C[C@]2c3cccc(*CC4)c3N4[C@]2CC1)c(cc1)ccc1F Chemical compound O[C@](CCCN1C[C@]2c3cccc(*CC4)c3N4[C@]2CC1)c(cc1)ccc1F 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- 229920002732 Polyanhydride Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 208000006289 Rett Syndrome Diseases 0.000 description 2
- 208000036750 Schizophrenia, residual type Diseases 0.000 description 2
- 208000020186 Schizophreniform disease Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 206010042008 Stereotypy Diseases 0.000 description 2
- 229930003427 Vitamin E Natural products 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 238000011374 additional therapy Methods 0.000 description 2
- 238000011360 adjunctive therapy Methods 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 230000016571 aggressive behavior Effects 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 238000006065 biodegradation reaction Methods 0.000 description 2
- 208000022257 bipolar II disease Diseases 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 235000005473 carotenes Nutrition 0.000 description 2
- 150000001746 carotenes Chemical class 0.000 description 2
- 239000005018 casein Substances 0.000 description 2
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 2
- 235000021240 caseins Nutrition 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 150000004292 cyclic ethers Chemical class 0.000 description 2
- USRHYDPUVLEVMC-FQEVSTJZSA-N dapoxetine Chemical compound C1([C@H](CCOC=2C3=CC=CC=C3C=CC=2)N(C)C)=CC=CC=C1 USRHYDPUVLEVMC-FQEVSTJZSA-N 0.000 description 2
- 229960005217 dapoxetine Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 229960003638 dopamine Drugs 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 229950000789 eplivanserin Drugs 0.000 description 2
- VAIOZOCLKVMIMN-PRJWTAEASA-N eplivanserin Chemical compound C=1C=CC=C(F)C=1\C(=N/OCCN(C)C)\C=C\C1=CC=C(O)C=C1 VAIOZOCLKVMIMN-PRJWTAEASA-N 0.000 description 2
- 229950002850 eptapirone Drugs 0.000 description 2
- 229960005086 escitalopram oxalate Drugs 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 230000005713 exacerbation Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 238000010579 first pass effect Methods 0.000 description 2
- 238000009093 first-line therapy Methods 0.000 description 2
- 229960002107 fluvoxamine maleate Drugs 0.000 description 2
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 229920002674 hyaluronan Polymers 0.000 description 2
- 229960003160 hyaluronic acid Drugs 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 230000000147 hypnotic effect Effects 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 230000005032 impulse control Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 208000015046 intermittent explosive disease Diseases 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000036651 mood Effects 0.000 description 2
- 229920005615 natural polymer Polymers 0.000 description 2
- 239000006199 nebulizer Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000002357 osmotic agent Substances 0.000 description 2
- 229940039748 oxalate Drugs 0.000 description 2
- 208000002851 paranoid schizophrenia Diseases 0.000 description 2
- 229920001432 poly(L-lactide) Polymers 0.000 description 2
- 229920001281 polyalkylene Polymers 0.000 description 2
- 229920001610 polycaprolactone Polymers 0.000 description 2
- 239000004632 polycaprolactone Substances 0.000 description 2
- 239000004417 polycarbonate Substances 0.000 description 2
- 229920000515 polycarbonate Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000004633 polyglycolic acid Substances 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 208000020016 psychiatric disease Diseases 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 239000002469 receptor inverse agonist Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000000697 serotonin reuptake Effects 0.000 description 2
- 208000022925 sleep disturbance Diseases 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 208000011117 substance-related disease Diseases 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 229930003799 tocopherol Natural products 0.000 description 2
- 239000011732 tocopherol Substances 0.000 description 2
- 235000019149 tocopherols Nutrition 0.000 description 2
- 238000002604 ultrasonography Methods 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 235000019165 vitamin E Nutrition 0.000 description 2
- 239000011709 vitamin E Substances 0.000 description 2
- 229940046009 vitamin E Drugs 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- QUEDXNHFTDJVIY-UHFFFAOYSA-N γ-tocopherol Chemical class OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1 QUEDXNHFTDJVIY-UHFFFAOYSA-N 0.000 description 2
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 2
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- MQZOATSIFWSKKT-OASXIEIISA-N (3s,4r)-3-(1,3-benzodioxol-5-yloxymethyl)-4-(4-fluorophenyl)piperidine;hydrate;dihydrochloride Chemical compound O.Cl.Cl.C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1.C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 MQZOATSIFWSKKT-OASXIEIISA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- IJVRPNIWWODHHA-UHFFFAOYSA-N 2-cyanoprop-2-enoic acid Chemical class OC(=O)C(=C)C#N IJVRPNIWWODHHA-UHFFFAOYSA-N 0.000 description 1
- XBBVURRQGJPTHH-UHFFFAOYSA-N 2-hydroxyacetic acid;2-hydroxypropanoic acid Chemical compound OCC(O)=O.CC(O)C(O)=O XBBVURRQGJPTHH-UHFFFAOYSA-N 0.000 description 1
- WHBMMWSBFZVSSR-UHFFFAOYSA-N 3-hydroxybutyric acid Chemical compound CC(O)CC(O)=O WHBMMWSBFZVSSR-UHFFFAOYSA-N 0.000 description 1
- 239000003477 4 aminobutyric acid receptor stimulating agent Substances 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 102000056834 5-HT2 Serotonin Receptors Human genes 0.000 description 1
- 108091005479 5-HT2 receptors Proteins 0.000 description 1
- 102100022738 5-hydroxytryptamine receptor 1A Human genes 0.000 description 1
- 101710138638 5-hydroxytryptamine receptor 1A Proteins 0.000 description 1
- 102100036321 5-hydroxytryptamine receptor 2A Human genes 0.000 description 1
- 101710138091 5-hydroxytryptamine receptor 2A Proteins 0.000 description 1
- 206010001540 Akathisia Diseases 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 208000037857 Alzheimer's disease agitation Diseases 0.000 description 1
- 206010002942 Apathy Diseases 0.000 description 1
- 208000036640 Asperger disease Diseases 0.000 description 1
- 201000006062 Asperger syndrome Diseases 0.000 description 1
- 208000021465 Brief psychotic disease Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010011971 Decreased interest Diseases 0.000 description 1
- 206010054089 Depressive symptom Diseases 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 1
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 1
- 108050004812 Dopamine receptor Proteins 0.000 description 1
- 102000015554 Dopamine receptor Human genes 0.000 description 1
- 229940121891 Dopamine receptor antagonist Drugs 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 229940123431 GABA B receptor agonist Drugs 0.000 description 1
- 102000005915 GABA Receptors Human genes 0.000 description 1
- 108010005551 GABA Receptors Proteins 0.000 description 1
- 229940121909 GABA receptor agonist Drugs 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010060891 General symptom Diseases 0.000 description 1
- 208000011688 Generalised anxiety disease Diseases 0.000 description 1
- 101710195187 Glutamate receptor ionotropic, NMDA 2B Proteins 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 206010024642 Listless Diseases 0.000 description 1
- 206010026749 Mania Diseases 0.000 description 1
- 229940123784 Melatonin receptor antagonist Drugs 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 1
- XLBVNMSMFQMKEY-BYPYZUCNSA-N N-methyl-L-glutamic acid Chemical compound CN[C@H](C(O)=O)CCC(O)=O XLBVNMSMFQMKEY-BYPYZUCNSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- KYYIDSXMWOZKMP-UHFFFAOYSA-N O-desmethylvenlafaxine Chemical compound C1CCCCC1(O)C(CN(C)C)C1=CC=C(O)C=C1 KYYIDSXMWOZKMP-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 229920002730 Poly(butyl cyanoacrylate) Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 206010036437 Posturing Diseases 0.000 description 1
- 208000001431 Psychomotor Agitation Diseases 0.000 description 1
- 108010038912 Retinoid X Receptors Proteins 0.000 description 1
- 208000036353 Rett disease Diseases 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- 206010041250 Social phobia Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 206010041349 Somnolence Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 206010042635 Suspiciousness Diseases 0.000 description 1
- 206010064805 Tachyphrenia Diseases 0.000 description 1
- 241001516476 Vanda Species 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 230000009798 acute exacerbation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 229940124604 anti-psychotic medication Drugs 0.000 description 1
- 229940005529 antipsychotics Drugs 0.000 description 1
- 239000003420 antiserotonin agent Substances 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- 229940005530 anxiolytics Drugs 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 230000001174 ascending effect Effects 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 229940127236 atypical antipsychotics Drugs 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 208000025307 bipolar depression Diseases 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 235000021152 breakfast Nutrition 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 229940047493 celexa Drugs 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 208000024825 childhood disintegrative disease Diseases 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000003759 clinical diagnosis Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000006999 cognitive decline Effects 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 235000009508 confectionery Nutrition 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 229960001623 desvenlafaxine Drugs 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000003210 dopamine receptor blocking agent Substances 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 208000029364 generalized anxiety disease Diseases 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 229920000578 graft copolymer Polymers 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 208000031424 hyperprolactinemia Diseases 0.000 description 1
- 229940053999 hypnotics and sedatives melatonin receptor agonists Drugs 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 229960000685 levomilnacipran Drugs 0.000 description 1
- 229940054157 lexapro Drugs 0.000 description 1
- 150000002632 lipids Chemical group 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229940009622 luvox Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 238000011034 membrane dialysis Methods 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- VUZPPFZMUPKLLV-UHFFFAOYSA-N methane;hydrate Chemical compound C.O VUZPPFZMUPKLLV-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 229960000600 milnacipran Drugs 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 231100001223 noncarcinogenic Toxicity 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- GELRVIPPMNMYGS-RVXRQPKJSA-N paroxetine hydrochloride Chemical compound Cl.C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 GELRVIPPMNMYGS-RVXRQPKJSA-N 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- RKEWSXXUOLRFBX-UHFFFAOYSA-N pimavanserin Chemical compound C1=CC(OCC(C)C)=CC=C1CNC(=O)N(C1CCN(C)CC1)CC1=CC=C(F)C=C1 RKEWSXXUOLRFBX-UHFFFAOYSA-N 0.000 description 1
- FIADGNVRKBPQEU-UHFFFAOYSA-N pizotifen Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2CCC2=C1C=CS2 FIADGNVRKBPQEU-UHFFFAOYSA-N 0.000 description 1
- 229960004572 pizotifen Drugs 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 239000002745 poly(ortho ester) Substances 0.000 description 1
- 229920002312 polyamide-imide Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 230000036544 posture Effects 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 210000002442 prefrontal cortex Anatomy 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000003518 presynaptic effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 229940035613 prozac Drugs 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000004627 regenerated cellulose Substances 0.000 description 1
- 230000006833 reintegration Effects 0.000 description 1
- 238000007151 ring opening polymerisation reaction Methods 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229940100992 sarafem Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000000862 serotonergic effect Effects 0.000 description 1
- 239000002399 serotonin 2A agonist Substances 0.000 description 1
- 239000000952 serotonin receptor agonist Substances 0.000 description 1
- BLFQGGGGFNSJKA-XHXSRVRCSA-N sertraline hydrochloride Chemical compound Cl.C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 BLFQGGGGFNSJKA-XHXSRVRCSA-N 0.000 description 1
- 208000012201 sexual and gender identity disease Diseases 0.000 description 1
- 208000015891 sexual disease Diseases 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 208000020685 sleep-wake disease Diseases 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 239000003174 triple reuptake inhibitor Substances 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229940020965 zoloft Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5146—Organic macromolecular compounds; Dendrimers obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyamines, polyanhydrides
- A61K9/5153—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Psychiatry (AREA)
- Dermatology (AREA)
- Optics & Photonics (AREA)
- Nanotechnology (AREA)
- Physics & Mathematics (AREA)
- Anesthesiology (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Inorganic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Saccharide Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Un compuesto de Fórmula I: **(Ver fórmula)** en la que: Fórmula I X es -O-, -NH- o -N(CH3)-; e Y es -O- o -C(O)-; en forma libre o de sal farmacéuticamente aceptable, opcionalmente en mezcla o asociación con un diluyente o un vehículo farmacéuticamente aceptables, para su uso en un método para el tratamiento de los síntomas residuales de la esquizofrenia seleccionados entre retraimiento emocional, retraimiento social pasivo o apático y evitación social activa, que comprende administrar a un paciente que lo necesite una cantidad eficaz del compuesto, en donde el paciente es un paciente que no ha respondido o no ha respondido adecuadamente a un tratamiento con otro agente antipsicótico.
Description
DESCRIPCIÓN
Métodos para el tratamiento de los síntomas residuales de la esquizofrenia
La presente solicitud reivindica prioridad de las solicitudes provisionales de EE. UU. N.° 61/911.416 presentada el 3 de diciembre de 2013; 61/925607 presentada el 9 de enero de 2014; 61/975702 presentada el 4 de abril de 2014; y 62/032.326 presentada el 1 de agosto de 2014.
Campo técnico
La presente invención se refiere al uso de gamma-carbolinas fusionadas con heterociclo sustituido particulares como se describe en el presente documento, en formas libre o de sal farmacéuticamente aceptable, como productos farmacéuticos y composiciones farmacéuticas como terapia primaria o adicional en el tratamiento de las fases agudas y residuales de la esquizofrenia, incluyendo particularmente el tratamiento de los síntomas residuales tales como evitación social activa, retraimiento social pasivo, retraimiento emocional, ansiedad, tensión, pensamiento estereotipado, y preocupaciones somáticas. Los compuestos divulgados en el presente documento pueden usarse para tratar tanto los síntomas agudos como los síntomas residuales que aparecen durante las exacerbaciones agudas pero que definen la fase residual de la enfermedad, p. ej., esquizofrenia, una vez que los síntomas agudos disminuyen. Por lo tanto, los compuestos divulgados en el presente documento pueden usarse individualmente o en combinación con otros medicamentos antipsicóticos, así como con otros agentes activos que tratan trastornos comórbidos tales como depresión y/o trastornos del sueño. Como el perfil de respuesta a través de los síntomas asociados a la esquizofrenia puede ser particularmente beneficioso para mejorar la función social, los compuestos divulgados en el presente documento pueden usarse para mejorar la integración social y la función social. Dicho perfil de respuesta puede ser beneficioso para las fases prodrómica, de exacerbación y residual de la esquizofrenia.
Antecedentes de la invención
La psicosis, particularmente la esquizofrenia, afecta al 1,1 % de la población mundial. Esta enfermedad comprende tres fases: fase prodrómica, fase activa y fase residual. La fase prodrómica es una fase temprana en donde se observan signos y síntomas subclínicos. Estos síntomas pueden incluir pérdida de interés en las actividades habituales, retraimiento respecto de los amigos y miembros de la familia, confusión, problemas de concentración, sensación de desgana y apatía. La fase activa se caracteriza por la exacerbación de los síntomas positivos, tales como delirios, alucinaciones y desconfianza. La fase residual se caracteriza por síntomas negativos tales como retraimiento emocional, retraimiento social pasivo, y pensamiento estereotipado, y síntomas de psicopatología general que incluyen evitación social activa, ansiedad, tensión, y preocupaciones somáticas. Los síntomas de la fase residual suelen ir acompañados de depresión, disfunción cognitiva e insomnio. En conjunto, estos síntomas de la fase residual no son bien tratados por muchos de los fármacos antipsicóticos actualmente disponibles en el mercado y, por lo tanto, son habitualmente observados después de que los síntomas de la fase activa hayan remitido tras el tratamiento. Es en esta fase de la enfermedad cuando los pacientes desearían poder volver a llevar una vida más productiva y satisfactoria, pero como los síntomas negativos residuales y el deterioro cognitivo no se tratan adecuadamente, este objetivo se ve frustrado. Sigue habiendo una necesidad urgente de agentes antipsicóticos, que puedan tratar no solo los síntomas de la fase activa o aguda, sino también los síntomas de la fase residual de la psicosis, p. ej., esquizofrenia.
Se sabe que las gamma-carbolinas fusionadas con heterociclo sustituido son agonistas o antagonistas de los receptores de 5-HT2 , particularmente de los receptores de 5-HT2A y 5-HT2C , en el tratamiento de los trastornos del sistema nervioso central. Estos compuestos se han divulgado en las patentes estadounidenses N.° 6.548.493; 7.238.690; 6.552.017; 6.713.471; U.S. RE39680, U.S. RE39679, U.S. N.° 7.183.282 y 7.071.186, como compuestos novedosos y de uso médico para el tratamiento de trastornos asociados a la modulación del receptor de 5-HT2A , tales como obesidad, ansiedad, depresión, psicosis, esquizofrenia, trastornos del sueño, trastornos sexuales, migraña, autismo, afecciones asociadas al dolor cefálico, fobias sociales y trastornos gastrointestinales, tales como disfunción de la motilidad del tracto gastrointestinal. El documento PCT/US08/03340 y la solicitud de EE. UU. N.° 10/786.935 también divulgan métodos para preparar gamma-carbolinas fusionadas con heterociclo sustituido y usos de estas gamma-carbolinas como agonistas y antagonistas de la serotonina útiles para el control y la prevención de trastornos del sistema nervioso central, tales como comportamiento adictivo y trastornos del sueño.
De manera adicional, los documentos WO 2009/145900, WO 2011/133224, WO 2013/155505, WO 2013/155504 y WO 2013/155506 enseñan compuestos de gamma-carbolina fusionada con heterociclo sustituido adicionales y/o su uso para el tratamiento de uno o más trastornos que implican las rutas de 5-HT2A, transportador de serotonina (SERT) y/o dopamina D1/D2.
Si bien las referencias citadas anteriormente relativas a los compuestos de gamma-carbolina fusionada con heterociclo sustituido enseñan el tratamiento de ciertos trastornos asociados a la psicosis y/o a la depresión, ninguna de estas referencias divulga el tratamiento de los síntomas residuales de la psicosis, particularmente los síntomas residuales de la esquizofrenia.
Sumario de la invención
La presente invención se expone en las reivindicaciones adjuntas. Se ha descubierto que unos compuestos de gamma-carbolina fusionada con heterociclo sustituido particulares (los compuestos descritos en el presente documento a continuación) poseen mecanismos de acción que se cree que tienen el potencial de producir una terapia antipsicótica innovadora. Los Compuestos de Fórmula I combinan un potente antagonismo del receptor de serotonina 5-HT2A, modulación de la fosfoproteína del receptor de dopamina, o DPPM, modulación glutamatérgica e inhibición de la recaptación de serotonina en un único fármaco candidato para el tratamiento de la esquizofrenia aguda y residual. En los receptores de dopamina D2, los Compuestos de Fórmula I tienen propiedades duales y actúan como antagonistas postsinápticos y agonistas parciales presinápticos. Los Compuestos de Fórmula I también estimulan la fosforilación de los receptores glutamatérgicos NMDA NR2B, o GluN2B, manera específica en el mesolímbico. Se cree que esta selectividad regional en las áreas cerebrales que se piensa que median la eficacia de los fármacos antipsicóticos, junto con las interacciones serotoninérgicas, glutamatérgicas, y dopaminérgicas, puede dar lugar a una eficacia antipsicótica para los síntomas positivos, negativos, afectivos y cognitivos asociados a la esquizofrenia. La inhibición de la recaptación de serotonina podría permitir una actividad antidepresiva para el tratamiento del trastorno esquizoafectivo, depresión comórbida, y/o como tratamiento independiente para el trastorno depresivo mayor. Se cree que los Compuestos de Fórmula I también pueden ser útiles para el tratamiento del trastorno bipolar y otros trastornos psiquiátricos y neurodegenerativos, particularmente alteraciones del comportamiento asociadas a la demencia, autismo y otras enfermedades del SNC. Estas características de los Compuestos de Fórmula I pueden ser capaces de mejorar la calidad de vida de los pacientes con esquizofrenia y mejorar la función social para permitirles una mayor integración en sus familias y en su lugar de trabajo. De manera adicional, los Compuestos de Fórmula I pueden demostrar que tratan los trastornos a dosis bajas (p. ej., sueño, agresión y agitación) o a dosis altas (p. ej., esquizofrenia aguda exacerbada y residual, trastornos bipolares y trastornos del estado de ánimo).
Como los compuestos descritos a continuación son eficaces en el tratamiento no solo de los síntomas agudos, sino también de los síntomas residuales de la psicosis, la invención proporciona, por lo tanto, en un aspecto, métodos para usar los compuestos de gamma-carbolina fusionada con heterociclo sustituido particulares (los compuestos descritos en el presente documento a continuación), ya sea individualmente o como terapia adyuvante para el tratamiento de los síntomas residuales de la psicosis, particularmente esquizofrenia. Esta es una utilidad nueva e inesperada.
Por tanto, la presente invención hace referencia a un compuesto de Fórmula I:

en donde:
X es -O-, -NH- o -N(CH3)-; e
Y es -O- o -C(O)-;
en forma libre o de sal farmacéuticamente aceptable, opcionalmente en mezcla o asociación con un diluyente o vehículo farmacéuticamente aceptable, para su uso en un método para el tratamiento de los síntomas residuales de la esquizofrenia seleccionados entre retraimiento emocional, retraimiento social pasivo o apático y evitación social activa, que comprende administrar a un paciente que lo necesite una cantidad eficaz del compuesto, en donde el paciente es un paciente que no ha respondido o no ha respondido adecuadamente a un tratamiento con otro agente antipsicótico.
También se describe en el presente documento un método (Método A) para el tratamiento de los síntomas residuales que comprende administrar a un paciente que lo necesite una cantidad eficaz del Compuesto de Fórmula I:
en donde:
X es -O-, -NH- o -N(CH3)
Y es -O-, -C(R2)(OH) -, -C(R3)(OR1) o -C(O)-;
R1 es alquilo C1-6 (p. ej., metilo) o un acilo fisiológicamente hidrolizable y aceptable, p. ej., seleccionado entre -C(O)-alquilo C1-21 (p. ej., -C(O)-alquilo C1-5, -C(O)-alquilo C6-15 o -C(O)-alquilo C16-21), por ejemplo, en donde R1 es -C(O)-alquilo C6, -C(O)-alquilo C7, -C(O)-alquilo C9 , -C(O)-alquilo C11, -C(O)-alquilo C13, o -C(O)-alquilo C15 p. ej., en donde dicho compuesto se hidroliza para proporcionar un ácido graso natural o no natural, saturado o insaturado de fórmula R1-OH y un Compuesto de Fórmula 1 en donde Y es -C(R2)(OH)-, p. ej., en donde el compuesto se hidroliza para formar el Compuesto hidroxi de Fórmula I en donde Y es -C(R2)(Oh )-, por una parte, y ácido octanoico, ácido decanoico, ácido dodecanoico, ácido tetradecanoico o ácido hexadecanoico, por otra parte);
R2 es H o -alquilo C1-6 (p. ej., metilo); y
R3 es H o -alquilo C1-6 (p. ej., metilo);
p. ej., en donde "alquilo" se refiere a un resto de hidrocarburo de cadena recta, opcionalmente saturada o insaturada y opcionalmente sustituida con uno o más grupos hidroxi o alcoxi C1-22 (p. ej., etoxi);
en forma libre o de sal farmacéuticamente aceptable.
El compuesto de Fórmula I del Método A puede ser un compuesto en donde:
X es -O-, -NH- o -N(CH3) -;
Y es -O-, -C(H)(OH) -, -C(H)(ORi) o -C(O)-; y
R1 es un acilo fisiológicamente hidrolizable y aceptable, p. ej., seleccionado entre -C(O)-alquilo C1-21 (p. ej., -C(O)-alquilo C1-5, -C(O)-alquilo C6-15 o -C(O)-alquilo C16-21), por ejemplo en donde R1 es -C(O)-alquilo C6, -C(O)-alquilo C7 , -C(O)-alquilo C9, -C(O)-alquilo C11, -C(O)-alquilo C13, o -C(O)-alquilo C15 p. ej., en donde dicho compuesto se hidroliza para proporcionar un ácido graso natural o no natural, saturado o insaturado de fórmula R1-OH y un Compuesto de Fórmula I en donde Y es -C(R2)(OH)-, p. ej., en donde el compuesto se hidroliza para formar el Compuesto hidroxi de Fórmula I en donde Y es -C(R2)(OH)-, por una parte, y ácido octanoico, ácido decanoico, ácido dodecanoico, ácido tetradecanoico o ácido hexadecanoico, por otra parte);
p. ej., en donde "alquilo" se refiere a un resto de hidrocarburo de cadena recta, opcionalmente saturada o insaturada y opcionalmente sustituida con uno o más grupos hidroxi o alcoxi C1-22 (p. ej., etoxi);
en forma libre o de sal farmacéuticamente aceptable.
El paciente del Método A puede padecer síntomas residuales de psicosis, por ejemplo, esquizofrenia (por ejemplo, de subtipo residual), trastorno delirante (p. ej., de tipo somático), depresión mayor con psicosis, trastorno bipolar con síntomas psicóticos, trastorno psicótico breve, trastorno esquizofreniforme, trastorno esquizoafectivo o psicosis causada por una afección médica o por consumo de sustancias. Preferentemente, el paciente padece síntomas residuales de la esquizofrenia.
Los síntomas de la fase residual pueden incluir: síntomas negativos, tales como afecto embotado, retraimiento emocional, escasa compenetración, retraimiento social pasivo o apático, dificultad para el pensamiento abstracto, falta de espontaneidad y fluidez en la conversación y pensamiento estereotipado; síntomas de psicopatología general, tales como preocupación somática, ansiedad, sentimientos de culpa, tensión, manerismos y posturas, depresión, retraso motor, falta de cooperación, contenido de pensamientos inusuales, desorientación, atención deficiente, falta de juicio y visión interna, alteración de la volición, control deficiente de los impulsos, preocupación y evitación social activa; deterioro cognitivo y trastornos del sueño (p. ej., insomnio). El Método A puede tratar los síntomas residuales (síntomas negativos, efectivos y cognitivos) asociados a la esquizofrenia.
En una realización, la cantidad eficaz del Compuesto de Fórmula I es de aproximadamente 1 mg a aproximadamente 140 mg por dosis al día, en otra realización, de aproximadamente 2,5 mg a aproximadamente 120 mg, en otra realización, de aproximadamente 10 mg a aproximadamente 120 mg por dosis al día, en otra realización, de aproximadamente 60 mg a aproximadamente 120 mg por dosis al día, en aún otra realización, de aproximadamente 10 mg a aproximadamente 60 mg por dosis al día, en otra realización, de aproximadamente 20 mg a aproximadamente 60 mg por dosis al día, en aún otra realización, aproximadamente 20 mg, aproximadamente 40 mg, o aproximadamente 60 mg por dosis al día. Los compuestos usados de acuerdo con la presente invención son eficaces en el tratamiento tanto de los síntomas positivos, que se producen durante la fase activa o aguda de la psicosis (p. ej., eficaces en el tratamiento de los síntomas positivos tales como delirios y alucinaciones) como de los síntomas negativos y otros síntomas residuales que se observan generalmente en la fase residual. Preferentemente, la cantidad eficaz para el tratamiento de los síntomas agudos y residuales de la esquizofrenia es de aproximadamente 60 mg al día. En una realización particular, las dosificaciones divulgadas en el presente documento para administración oral se basan en la cantidad de los Compuestos de Fórmula I en forma de sal de adición de ácido, particularmente en forma de sal de adición de ácido toluensulfónico.
Por lo tanto, en el presente documento se describen métodos de la siguiente manera:
1.1 Método A que comprende un compuesto de Fórmula I, en donde X es -N(CH3);
1.2 Método A que comprende un compuesto de Fórmula I, en donde X es -NH;
1.3 Método A que comprende un compuesto de Fórmula I, en donde X es O;
1.4 Método A o cualquiera de 1.1-1.3, que comprende un compuesto de Fórmula I, en donde Y es -C(O)-;
1.5 Método A o cualquiera de 1.1-1.3, que comprende un compuesto de Fórmula I, en donde Y es -O-;
1.6 Método A o cualquiera de 1.1-1.3, que comprende un compuesto de Fórmula I, en donde Y es -C(R2)(OH)-, p. ej., -C(H)(OH)-;
1.7 Método A o cualquiera de 1.1-1.3, que comprende un compuesto de Fórmula I, en donde Y es -C(R3)(OR1), p. ej., -C(H)(ORi);
1.8 Método A o fórmula 1.7, en donde R1 es -C(O)-alquilo C1-21 (p. ej., -C(O)-alquilo C1-5, -C(O)-alquilo C6-15 o -C(O)-alquilo C16-21), preferentemente dicho alquilo es una cadena recta, opcionalmente saturada o insaturada y opcionalmente sustituida con uno o más grupos hidroxi o alcoxi C1-22 (p. ej., etoxi), por ejemplo R1 es -C(O)-alquilo C6, -C(O)-alquilo C7 , -C(O)-alquilo C9 , -C(O)-alquilo C11, -C(O)-alquilo C13 o -C(O)-alquilo C15, en donde dicho compuesto se hidroliza para formar el residuo de un ácido graso natural o no natural, saturado o insaturado, p. ej., el compuesto se hidroliza para formar el compuesto hidroxi, por una parte, y ácido octanoico, ácido decanoico, ácido dodecanoico, ácido tetradecanoico o ácido hexadecanoico, por otra parte);
1.9 Método A o fórmula 1.7, en donde R1 es -C(O)-alquilo C6-15, p. ej., -C(O)-alquilo C9 ;
1.10 Método A o fórmula 1.7, en donde R1 es -C(O)-alquilo C1-5 , p. ej., -C(O)-alquilo C3 ;
1.11 Método A o fórmula 1.7, en donde R1 es -alquilo C1-6 (p. ej., metilo);
1.12 Método A o cualquiera de la fórmula 1.1-1.11, en donde R2 es H o -alquilo C1-6 (p. ej., metilo);
1.13 Método A o cualquiera de la fórmula 1.1-1.11, en donde R2 es H;
1.14 Método A o cualquiera de la fórmula 1.1-1.11, en donde R2 es -alquilo C1-6 (p. ej., metilo);
1.15 Método A o cualquiera de la fórmula 1.1 -1.1 1 , en donde R3 es H o -alquilo C1-6 (p. ej., metilo);
1.16 Método A o cualquiera de la fórmula 1.1-1.11, en donde R3 es H;
1.17 Método A o cualquiera de la fórmula 1.1-1.11, en donde R3 es -alquilo C1-6 (p. ej., metilo);
1.18 cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I se selecciona entre el grupo que consiste en un compuesto de Fórmula I en donde:
X es -O- e Y es -C(H)(OH)-,
X es -NH- e Y es -C(H)(OH)-,
X es -N(CHa)- e Y es -C(H)(OH)-,
X es -O- e Y es -C(O)-,
X es -O- e Y es -O-,
X es -N(CHa)- e Y es -C(O)-,
X es -N(CH3)- e Y es -O-,
X es -NH- e Y es -C(O)-,
X es -NH- e Y es -O-,
X es -N(CH3)- e Y es -C(H)(ORi),
X es -NH- e Y es -C(H)(ORi), o
X es -O- e Y es -C(H)(ORi);
X es -O- e Y es -C(CHa)(OH)-,
X es -NH- e Y es C(CH3)(OH)-,
X es -N(CH3)- e Y es C(CH3)(OH)-,
1.19 cualquiera de los métodos anteriores en donde X es -O- e Y es -C(O)- en el Compuesto de Fórmula I;
1.20 cualquiera de los métodos anteriores en donde X es -NH- e Y es -C(H)(OH)- en el Compuesto de Fórmula I;
1.21 cualquiera de los métodos anteriores en donde X es -N(CH3)- e Y es -C(H)(OH)- en el Compuesto de Fórmula I;
1.22 cualquiera de los métodos anteriores en donde X es -O- e Y es -C(O)- en el Compuesto de Fórmula I;
1.23 cualquiera de los métodos anteriores en donde X es -O- e Y es -O- en el Compuesto de Fórmula I;
1.24 cualquiera de los métodos anteriores en donde X es -N(CH3)- e Y es -C(O)- en el Compuesto de Fórmula I; 1.25 cualquiera de los métodos anteriores en donde X es -O- e Y es -C(H)(OH)- en el Compuesto de Fórmula I 1.26 cualquiera de los métodos anteriores en donde X es -NH- e Y es -C(H)(OH)- en el compuesto de Fórmula I;
1.27 cualquiera de los métodos anteriores en donde X es -N(CH3)- e Y es -C(H)(OH)- en el compuesto de Fórmula I;
1.28 cualquiera de los métodos anteriores en donde X es -N(CH3)- e Y es -C(H)(ORi) y R1 es -C(O)-alquilo C1-21 en el compuesto de Fórmula I;
1.29 cualquiera de los métodos anteriores en donde X es -N(H)- e Y es -C(H)(ORi) y R1 es -C(O)-alquilo C1-21 en el compuesto de Fórmula I;
1.30 cualquiera de los métodos anteriores en donde X es -O- e Y es -C(H)(ORi) y R1 es -C(O)-alquilo C1-21 en el compuesto de Fórmula I;
1.31 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula
IA:

1.32 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula IB:

1.33 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula IC:

1.34 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula ID (en ocasiones denominado en el presente documento Compuesto B):

1.35 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula IE (en ocasiones denominado en el presente documento Compuesto A):

1.36 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula IF:

1.37 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula IG:

1.38 Cualquiera de los métodos anteriores, en donde el Compuesto de Fórmula I se administra por vía oral, por ejemplo una vez al día en una dosis única diaria o dos veces al día en una dosis dividida, por ejemplo en forma de comprimido o cápsula.
1.39 Cualquiera de los métodos anteriores en donde la cantidad eficaz del Compuesto de Fórmula I es una dosificación oral diaria de 10-120 mg/día, p. ej., aproximadamente 60 mg de la sal de adición de ácido ptoluensulfónico del Compuesto de Fórmula iE:
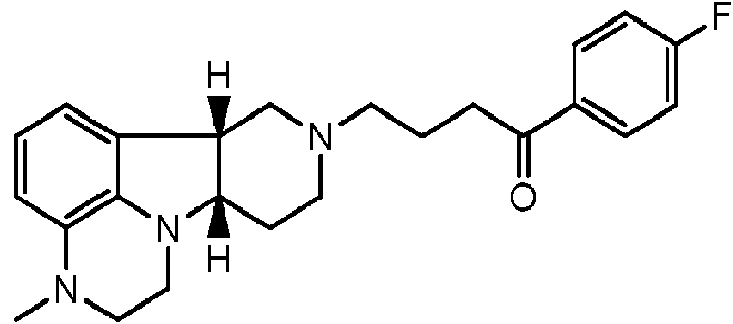
1.40 Cualquiera de los métodos anteriores en donde el Compuesto de Fórmula I se administra como una forma inyectable de liberación sostenida, p. ej., una forma inyectable de liberación lenta, p. ej., administrada una o dos veces al mes, p. ej., en forma de una micropartícula bioerosionable, p. ej., el Compuesto de Fórmula IE:

p. ej., en forma de base libre.
1.41 Cualquiera de los métodos anteriores que comprende administrar una formulación inyectable de acción prolongada de un Compuesto de Fórmula I, p. ej., Composición 2, p. ej., cualquiera de las Composiciones 2.1, et seq., como se establece a continuación.
1.42 Cualquiera de los métodos anteriores que comprende además administrar uno o varios agentes terapéuticos, tales como agentes antipsicóticos adicionales y/o agentes antidepresivos y/o agentes hipnóticos; 1.43 Método 1.38, en donde el uno o varios agentes terapéuticos se seleccionan entre agentes antidepresivos tales como compuestos que modulan la actividad de GAbA (p. ej., mejora la actividad y facilita la transmisión de GABA), un agonista del receptor de GABA-B, un modulador de 5-HT (p. ej., un agonista del receptor de 5-HTm, un antagonista del receptor de 5-HT2A, un agonista inverso del receptor de 5-HT2A, etc.), un agonista del receptor de melatonina, un modulador de los canales iónicos (p. ej., bloqueador), un antagonista de receptor/inhibidor de
la recaptación de serotonina-2, (SARIs), un antagonista del receptor de orexina, un agonista del receptor de H3, un antagonista del receptor noradrenérgico, un agonista del receptor de galanina, un antagonista del receptor de CRH, una hormona del crecimiento humano, un agonista del receptor de la hormona del crecimiento, un estrógeno, un agonista del receptor de estrógenos, un fármaco de neuroquinina-1; y agentes antipsicóticos, p. ej., agentes antipsicóticos atípicos, en forma libre o de sal farmacéuticamente aceptable;
1.44 Método 1.42 o 1.43, en donde el uno o varios agentes terapéuticos son agentes antipsicóticos, p. ej., clorpromazina, haloperidol, droperidol, flufenazina, loxapina, mesoridazina, molindona, perfenazina, pimozida, proclorperazina, promazina, tioridazina, tiotixeno, trifluoperazina, clozapina, aripiprazol, olanzapina, quetiapina, risperidona, ziprasidona, paliperidona, asenapina, lurasidona, iloperidona, cariprazina, amisulprida, zotepina, sertindol, en donde el uno o varios agentes terapéuticos se administran como complemento del compuesto de Fórmula I o el compuesto de Fórmula I es un complemento del uno o varios agentes terapéuticos;
1.45 Cualquiera de los métodos anteriores, en donde la cantidad eficaz es de 1 mg a 120 mg al día o de 10 mg a 120 mg al día, o de 10 mg a 60 mg al día, o de 10 mg a 40 mg al día, o de 1 mg a 10 mg al día, o 10 mg al día, 20 mg al día, 40 mg al día o 60 mg al día; En un método particular, la cantidad eficaz del Compuesto de Fórmula I divulgado en esta fórmula se basa en la cantidad del Compuesto de Fórmula I en una sal de adición de ácido, forma sin profármaco, p. ej., en sal de adición de ácido toluensulfónico, forma sin profármaco.
1.46 Método A o cualquiera de 1.38-1.45, en donde el uno o varios agentes terapéuticos son agentes antidepresivos, p. ej., uno o más antidepresivos seleccionados entre inhibidores selectivos de la recaptación de serotonina (SSRIs) (p. ej., seleccionados entre citalopram, oxalato de escitalopram, fluoxetina, maleato de fluvoxamina, paroxetina, sertralina, dapoxetina), inhibidores de la recaptación de serotonina y norepinefrina (SNRIs) (p. ej., seleccionados entre venlafaxina, desvenlafaxina, duloxetina, milnaciprán, levomilnaciprán, sibutramina) y antidepresivos tricíclicos; inhibidores de triple recaptación, ansiolíticos, busperona, y trazadona; 1.47 Métodos de 1.45 en donde el compuesto de Fórmula I se administra como complemento de uno o varios agentes terapéuticos, tales como agentes antidepresivos SSRI o los agentes antidepresivos SSRI se administran como complemento del compuesto de Fórmula I;
1.48 El método de 1.47, en donde dicho uno o más agentes antidepresivos se seleccionan entre SSRI tales como citalopram (Celexa, Cipramil, Emocal, Sepram, Seropram), oxalato de escitalopram (Lexapro, Cipralex, Esertia), fluoxetina (Prozac, Fontex, Seromex, Seronil, Sarafem, Fluctin (EUR)), maleato de fluvoxamina (Luvox, Faverin), paroxetina (Paxil, Seroxat, Aropax, Deroxat, Paroxat), sertralina (Zoloft, Lustral, Serlain), dapoxetina; en forma libre o de sal farmacéuticamente aceptable;
1.49 Cualquiera de los métodos anteriores, en donde el compuesto de Fórmula I se administra como parte de una composición de microesferas inyectables de acción prolongada;
1.50 El método de 1.49, en donde la composición de microesferas inyectables de acción prolongada es una composición de acuerdo con uno cualquiera de 2.1 a 2.22 del presente documento.
El paciente es un paciente que no ha respondido o no ha respondido adecuadamente al tratamiento con otro agente antipsicótico, p. ej., clorpromazina, haloperidol, droperidol, flufenazina, loxapina, mesoridazina, molindona, perfenazina, pimozida, proclorperazina, promazina, tioridazina, tiotixeno, trifluoperazina, clozapina, aripiprazol, olanzapina, quetiapina, risperidona, ziprasidona, paliperidona, asenapina, lurasidona, iloperidona, cariprazina, amisulprida, zotepina, sertindol. Por lo tanto, el compuesto de Fórmula I puede administrarse como terapia primaria o como terapia adicional, p. ej., como complemento de otro agente antipsicótico.
También se describe en el presente documento un método (Método B) para el tratamiento de cualquiera de los siguientes trastornos: trastorno esquizoafectivo, depresión comórbida, trastorno depresivo mayor, trastorno bipolar (p. ej., trastorno bipolar I y/o bipolar II), trastorno del espectro autista (p. ej., trastorno autista, trastorno de Asperger, trastorno generalizado del desarrollo no especificado (PDD-NOS), trastorno de Rett (síndrome de Rett), trastorno desintegrativo de la infancia) que comprende administrar a un paciente que lo necesite una cantidad eficaz de un Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable. El trastorno del Método B puede ser un trastorno bipolar (p. ej., trastorno bipolar I y/o trastorno bipolar II). El trastorno del Método B puede ser un trastorno del espectro autista. El trastorno del Método B puede ser un trastorno depresivo mayor.
Por lo tanto, dependiendo de la combinación de trastornos a tratar, los Compuestos de Fórmula I pueden usarse estratégicamente. Por ejemplo, a dosis más bajas (por ejemplo, una dosis oral diaria de 1-10 mg, p. ej., 1 mg, 5 mg y 10 mg del Compuesto de Fórmula I en forma de sal de adición de ácido toluensulfónico), los Compuestos de Fórmula I son útiles para el tratamiento del trastorno del sueño, agresión y agitación, enfermedad de Alzheimer y otras demencias, trastorno del espectro autista, enfermedad de Parkinson y trastorno explosivo intermitente (IED). A dosis más altas (por ejemplo, una dosis oral diaria de 60 mg del Compuesto de Fórmula I en forma de sal de adición de ácido toluensulfónico), los Compuestos de Fórmula I son útiles para tratar la esquizofrenia aguda exacerbada y residual, depresión bipolar, trastorno depresivo mayor, trastorno de ansiedad generalizado. A dosis muy altas (por ejemplo, una dosis oral diaria de 120 mg del Compuesto de Fórmula I en forma de sal de adición de ácido toluensulfónico), la administración matutina puede producir somnolencia/sedación. Por lo tanto, en tales dosis diarias más altas, es preferible la administración por la noche.
Los Compuestos de Fórmula I pueden presentarse en forma libre o de sal, p. ej., como sales de adición de ácido. En esta memoria, a menos que el lenguaje indique lo contrario, debe entenderse que como "Compuestos de Fórmula I", "agentes antipsicóticos", "agentes antidepresivos", "otros agentes terapéuticos", y similares, abarcan los compuestos
en cualquier forma, por ejemplo, en forma libre o de sal de adición de ácido, o cuando los compuestos contienen sustituyentes ácidos, en forma de sal de adición de base. Los Compuestos de Fórmula I están destinados a ser usados como productos farmacéuticos, por lo tanto, resultan preferentes las sales farmacéuticamente aceptables. Por lo tanto, las sales que no son adecuadas para usos farmacéuticos y que pueden ser útiles, por ejemplo, para el aislamiento o la purificación de los Compuestos libres de Fórmula I o sus sales farmacéuticamente aceptables, también se incluyen. Las sales farmacéuticamente aceptables incluyen, por ejemplo, las sales de clorhidrato, mesilato y tosilato. Preferentemente, los Compuestos de Fórmula I, particularmente en donde X es -N(CH3)- e Y es -C(O)-, están en forma de sal de tosilato (adición de ácido toluensulfónico). Cuando las cantidades de dosificación de las sales se dan en peso, p. ej., miligramos al día o miligramos por dosis unitaria, la cantidad de dosificación de la sal puede basarse en el peso de la base libre correspondiente o indicarse de otro modo. En una realización particular, la dosificación de los Compuestos de Fórmula I en forma de sal de adición de ácido para la administración oral se basa en el peso de la forma de sal de adición de ácido toluensulfónico, no en la forma de base libre. Por ejemplo, en una realización particular, la cantidad de dosificación de 10 mg, 60 mg, 120 mg del Compuesto de Fórmula I se basa respectivamente, en 10 mg, 60 mg y 120 mg del Compuesto de Fórmula I en forma de sal de adición de ácido toluensulfónico, no se basa en la cantidad de la base libre. Por ejemplo, la dosificación de 60 mg del compuesto para administración oral de los Compuestos de Fórmula I (p. ej., en donde X es -N(CH3)- e Y es -C(O)-) en forma de sal de adición de ácido toluensulfónico se refiere al compuesto en forma de sal de tosilato, que equivale a aproximadamente 41,7 mg de dicho compuesto en forma de base libre.
En el presente documento también se describen los métodos anteriores, p. ej., Método A, p. ej., cualquiera de 1.1 1.50, y el Método B, en donde el Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable se administra en una composición, en donde dicho Compuesto de Fórmula I en forma libre o de sal farmacéuticamente aceptable está en mezcla o en asociación con un diluyente o vehículo farmacéuticamente aceptable.
En el presente documento también se describen los métodos anteriores, p. ej., Método A, p. ej., cualquiera de 1.1 1.50, y el Método B, en donde el Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable se administra en una formulación de liberación inmediata o sostenida o retardada, p. ej., formulación de liberación lenta.
La formulación de liberación sostenida o retardada comprende los Compuestos de Fórmula I divulgados en el presente documento (p. ej., el compuesto de fórmula I o cualquiera de los descritos en cualquiera de las fórmulas 1.1-1.50) en una matriz polimérica. Como alternativa, los Compuestos de Fórmula I están dispersos o disueltos dentro de la matriz polimérica. La matriz polimérica puede comprender polímeros convencionales usados en formulaciones de liberación lenta, tales como polímeros seleccionados entre un poliéster de un ácido graso hidroxi y derivados del mismo, o un polímero de un alfa-cianoacrilato de alquilo, un oxalato de polialquileno, un poli(orto)éster, un policarbonato, un poliorto-carbonato, un poliaminoácido, un éster de ácido hialurónico, y mezclas de los mismos. El polímero puede seleccionarse entre el grupo que consiste en polilactida, poli d, l-lactida, poli glicólido, polímero de PLGA 50:50, PLGA 75:25, PLGA 85:15 y p LgA 90:10. El polímero puede seleccionarse entre poli(ácido glicólico), poli-ácido D,L-láctico, poli-ácido L-láctico, copolímeros de los anteriores, poli(ácidos carboxílicos alifáticos), copolioxalatos, policaprolactona, polidioxonona, poli(ortocarbonatos), poli(acetales), poli(ácido láctico-caprolactona), poliortoésteres, poli(ácido glicólico-caprolactona), polianhídridos, y polímeros naturales entre los que se incluyen albúmina, caseína, y ceras, tales como, mono y diestearato de glicerol, y similares. La matriz polimérica puede comprender poli(d,l-lactida-co-glicólido). El Compuesto de Fórmula I en una matriz polimérica puede estar en mezcla o en asociación con un diluyente o vehículo farmacéuticamente aceptable.
Las formulaciones de liberación sostenida o retardada descritas anteriormente son particularmente útiles para la liberación sostenida o retardada, en donde los compuestos de Fórmula I se liberan tras la degradación de la matriz polimérica. Estas formulaciones pueden liberar los Compuestos de Fórmula I durante un periodo de hasta 180 días, p. ej., de aproximadamente 14 a aproximadamente 30 a aproximadamente 180 días. Por ejemplo, la matriz polimérica puede degradar y liberar los Compuestos de Fórmula I durante un periodo de aproximadamente 30, aproximadamente 60 o aproximadamente 90 días. En otro ejemplo, la matriz polimérica puede degradar y liberar los Compuestos de Fórmula I durante un periodo de aproximadamente 120, o aproximadamente 180 días.
La formulación de liberación sostenida o retardada puede formularse para su administración por inyección.
Por ejemplo, la divulgación proporciona formulaciones inyectables de acción prolongada (IAP) de los Compuestos de Fórmula I. Dichas IAP pueden optimizarse en cuanto a la composición del vehículo, tamaño de partícula, peso molecular del vehículo, carga del principio activo y dosificación, p. ej., como se describe en los ejemplos. Además de la conveniencia de la administración, y de asegurar el cumplimiento por parte del paciente, las formulaciones IAP de los Compuestos de Fórmula I proporcionan sorprendentemente ventajas en cuanto a la farmacocinética y los efectos secundarios. Cuando un Compuesto de Fórmula I se administra mediante una formulación IAP, a diferencia de una forma de dosificación oral, se evita el metabolismo de primer paso en el hígado, lo que significa que una menor proporción del Compuesto de Fórmula I se metaboliza antes de llegar al cerebro. Las formulaciones de liberación sostenida o retardada, como se describen en el presente documento, en general producen menos efectos secundarios extrapiramidales y proporcionan una mejor tolerabilidad y una dosis total reducida que las correspondientes formulaciones de liberación inmediata. Las formulaciones de liberación sostenida o retardada, y particularmente las formulaciones inyectables de acción prolongada, permiten a un paciente alcanzar y mantener
niveles terapéuticamente eficaces de fármaco en el SNC mientras recibe una dosificación total mucho menor que la que se necesitaría para alcanzar el mismo nivel corporal de fármaco usando formulaciones orales de liberación inmediata. En el caso de las formulaciones inyectables de acción prolongada, este efecto se debe en parte a que se evita el metabolismo de primer paso que se produce con los medicamentos orales, incluyendo los medicamentos de liberación sostenida y retardada.
La cantidad eficaz del Compuesto de Fórmula I cuando se administra como una formulación inyectable de acción prolongada es, por lo tanto, mucho menor que la cantidad eficaz cuando se administra por vía oral, p. ej., de aproximadamente 100 mg al mes a aproximadamente 600 mg al mes, y preferentemente de 150 mg al mes a 300 mg al mes.
Por lo tanto, la divulgación proporciona una formulación inyectable de acción prolongada (Composición 2) que comprende microesferas poliméricas, en donde las microesferas comprenden:
una matriz polimérica de poli(D,L-láctido-co-glicólido) (PLGA) y
una cantidad eficaz de un Compuesto de Fórmula I, como se ha descrito anteriormente, en forma libre o de sal farmacéuticamente aceptable, estando el Compuesto de Fórmula I disperso, disuelto o encapsulado en la matriz polimérica.
Por ejemplo, la divulgación proporciona:
2.1 Composición 2, en donde el polímero de PLGA es aproximadamente 75:25 PLA/PLG con grupos terminales de ácido carboxílico o éster carboxílico.
2.2 Composición 2 o 2.1, en donde el polímero de PLGA es aproximadamente 75:25 PLA/PLG con grupos terminales de ácido carboxílico.
2.3 Composición 2, 2.1 o 2.2, en donde el Compuesto de Fórmula I está en forma de base libre.
2.4 Cualquier Composición anterior en donde el Compuesto de Fórmula I se selecciona entre Compuestos de Fórmula IA, IB, IC, ID, IE.
2.5 Cualquier composición anterior en donde el Compuesto de Fórmula I es el Compuesto de Fórmula IE:

p. ej., en forma de base libre.
2.6 Cualquiera de las Composiciones anteriores, en donde el Compuesto de Fórmula I es un compuesto de Fórmula ID:

p. ej., en forma de base libre.
2.7 Cualquier Composición anterior en donde el intervalo de peso molecular promedio del polímero de PLGA es, por ejemplo, de 20 kD a 200 kD, por ejemplo, de 24.000 a 38.000 daltons, o aproximadamente 113.000 daltons o aproximadamente 159.000 daltons.
2.8 Cualquier Composición anterior en donde el plazo de tiempo para la degradación completa de las microesferas y la liberación de los compuestos farmacológicos encapsulados es, p. ej., menos de 6 meses, menos de 4 meses, menos de 3 meses, menos de 2 meses, o menos de 1 mes.
2.9 Cualquiera de las Composiciones anteriores en donde el diámetro de las microesferas, p. ej., el diámetro promedio (o D50), el diámetro del percentil 10 (D10), el diámetro del percentil 25 (D25), el diámetro del percentil 75 (D75), o el diámetro del percentil 90 (D90), es de aproximadamente 10 pm a aproximadamente 200 pm, por ejemplo, de aproximadamente 20 pm a aproximadamente 160 pm, o de aproximadamente 20 pm a aproximadamente 120 pm, o de aproximadamente 20 pm a aproximadamente 100 pm, o de aproximadamente 20 pm a aproximadamente 80 pm, o de aproximadamente 20 pm a aproximadamente 70 pm, o de aproximadamente 20 pm a aproximadamente 60 pm, o de aproximadamente 20 pm a aproximadamente 50 pm, o de aproximadamente 20 pm a aproximadamente 40 pm, o de aproximadamente 20 pm a aproximadamente 30
|jm, o de aproximadamente 25 pm a aproximadamente 70 pm, o de aproximadamente 25 pm a aproximadamente 60 pm, o de aproximadamente 25 pm a aproximadamente 50 pm, o de aproximadamente 25 pm a aproximadamente 40 pm, o de aproximadamente 30 pm a aproximadamente 60 pm, o de aproximadamente 30 a 50 pm, o de aproximadamente 30 pm a aproximadamente 40 pm, o de aproximadamente 30 pm a aproximadamente 120 pm, o de aproximadamente 40 pm a aproximadamente 120 pm, o de aproximadamente 40 pm a aproximadamente 100 pm, o de aproximadamente 40 pm a aproximadamente 80 pm, o de aproximadamente 40 pm a aproximadamente 70 pm, o de aproximadamente 40 pm a aproximadamente 60 pm, o de aproximadamente 40 pm a aproximadamente 50 pm, o de aproximadamente 50 pm a aproximadamente 100 pm, o de aproximadamente 50 pm a aproximadamente 80 pm, o de aproximadamente 50 pm a aproximadamente 70 pm, o de aproximadamente 50 pm a aproximadamente 60 pm, o de aproximadamente 60 pm a aproximadamente 100 pm, o de aproximadamente 60 pm a aproximadamente 90 pm, o de aproximadamente 60 pm a aproximadamente 80 pm, o de aproximadamente 60 pm a aproximadamente 70 pm, o de aproximadamente 70 pm a aproximadamente 100 pm, o de aproximadamente 70 pm a aproximadamente 90 pm, o de aproximadamente 70 pm a aproximadamente 80 pm, o de aproximadamente 75 pm a aproximadamente 110 pm, o de aproximadamente 40 pm a aproximadamente 160 pm, o de aproximadamente 50 pm a aproximadamente 160 pm, o de aproximadamente 50 pm a aproximadamente 120 pm, o aproximadamente 20 pm, aproximadamente 30 pm, aproximadamente 40 pm, aproximadamente 50 pm, aproximadamente 60 pm, aproximadamente 70 pm, aproximadamente 80 pm, aproximadamente 90 pm o aproximadamente 100 pm.
2.10 Cualquiera de las Composiciones anteriores en donde el diámetro de las microesferas, p. ej., el diámetro promedio (o D50), el diámetro del percentil 10 (D10), el diámetro del percentil 25 (D25), el diámetro del percentil 75 (D75), o el diámetro del percentil 90 (D90), es de 10 pm a 160 pm, por ejemplo, de 20 pm a 70 pm, de 25 pm a 70 pm, de 40 a 120 pm, o de 20 pm a 60 pm, o de 20 pm a 50 pm, de 30 pm a 60 pm, de 30 a 50 pm, de 40 pm a 50 pm, o aproximadamente 30 pm, o aproximadamente 40 pm, o aproximadamente 50 pm.
2.11 Cualquier composición anterior en donde la cantidad del Compuesto de Fórmula I disperso, disuelto o encapsulado en cada microesfera, en promedio, es de aproximadamente 5 % en peso a aproximadamente 50 % en peso, por ejemplo, de aproximadamente 10 % en peso a aproximadamente 50 % en peso, o de aproximadamente 20 % en peso a aproximadamente 40 % en peso, o de aproximadamente 30 % en peso a aproximadamente 40 % en peso, o, por ejemplo, aproximadamente 8,5 % en peso, o aproximadamente 16 % en peso, o aproximadamente 30 % en peso, o aproximadamente 35 % en peso, o aproximadamente 40 % en peso.
2.12 Cualquier Composición anterior en donde la viscosidad inherente es de aproximadamente 0,1 a aproximadamente 1, por ejemplo, de aproximadamente 0,3 a aproximadamente 0,4, aproximadamente 0,7, aproximadamente 0,8, aproximadamente 0,9 dl/g.
2.13 Cualquiera de las Composiciones anteriores para su uso en el Método A, por ejemplo, cualquiera de los Métodos 1.1 et seq., o en el Método B, como se ha descrito anteriormente.
2.14 Cualquiera de las Composiciones anteriores para su uso en pacientes que tienen dificultad para adherirse a un régimen de tratamiento regular, tanto de forma intencionada como no intencionadamente.
2.15 Cualquiera de las Composiciones anteriores para su administración a pacientes de forma semanal, quincenal o mensual, o una vez cada 2, 34, 5 o 6 meses.
2.16 Cualquiera de las Composiciones anteriores para inyección intramuscular, intraperitoneal, intratecal, epidural, o subcutánea, por ejemplo, inyección subcutánea o intramuscular, p. ej., inyección intramuscular.
2.17 Cualquiera de las Composiciones anteriores para inyección intramuscular.
2.18 Cualquiera de las Composiciones anteriores, que comprende además un antioxidante, p. ej., en una cantidad eficaz para inhibir o reducir la oxidación del Compuesto de Fórmula 1.
2.19 Cualquiera de las Composiciones anteriores que comprende además un antioxidante, en donde el antioxidante es un antioxidante soluble en agua (p. ej., ácido ascórbico, ácido lipoico), o un antioxidante soluble en lípidos (p. ej., ácido lipoico, vitamina E, tocoferoles, carotenos o antioxidantes fenólicos), o un antioxidante neutro o débilmente básico, o un antioxidante catalítico (por ejemplo, ebseleno), o un antioxidante que contiene metales.
2.20 Cualquiera de las Composiciones anteriores que comprende además un antioxidante, en donde el antioxidante es un antioxidante lipídico o neutro o débilmente básico, p. ej., en donde el polímero comprende grupos terminales carboxi.
2.21 Cualquiera de las Composiciones anteriores, que comprende además un antioxidante, en donde el antioxidante es un antioxidante fenólico (p. ej., butilhidroxianisol (BHA) o butilhidroxitolueno (BHT)).
2.22 Cualquiera de las Composiciones anteriores, que comprende además un antioxidante, en donde el antioxidante es BHT.
También se describen en el presente documento los Métodos A o B descritos anteriormente, en donde el Compuesto de Fórmula I se formula en un sistema de administración oral de liberación controlada osmótica (OROS) para la administración de los Compuestos de Fórmula I, p. ej., análogo a los sistemas descritos en los documentos WO 2000/35419 y EP 1 539 115 (publicación de EE. UU. N.° 2009/020263). En el presente documento, se describen, por lo tanto, los Métodos A o B descritos anteriormente, en donde el Compuesto de Fórmula I se formula en un dispositivo que comprende (a) una cápsula de gelatina que contiene el Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable, como se ha descrito anteriormente; (b) una pared multicapa superpuesta en la cápsula de gelatina que comprende, en orden ascendente de la cápsula: (i) una capa de barrera, (ii) una capa expandible, y (iii) una capa semipermeable; y (c) un orificio formado o que puede formarse a través de la pared.
(Composición P.1)
También se describen en el presente documento los Métodos A o B descritos anteriormente, en donde el compuesto de Fórmula I se formula en una composición que comprende una cápsula de gelatina que contiene un líquido, los Compuestos de Fórmula I, en forma libre o de sal farmacéuticamente aceptable, como se ha descrito anteriormente, estando la cápsula de gelatina rodeada por una pared compuesta que comprende una capa de barrera que está en contacto con la superficie externa de la cápsula de gelatina, una capa expandible que está en contacto con la capa de barrera, una capa semipermeable que abarca la capa expandible, y un orificio de salida formado o que puede formarse en la pared. (Composición P.2)
También se describen en el presente documento los Métodos A o B descritos anteriormente, en donde el compuesto de Fórmula I se formula en una composición que comprende una cápsula de gelatina que contiene un líquido, el Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable, como se ha descrito anteriormente, estando la cápsula de gelatina rodeada por una pared compuesta que comprende una capa de barrera que está en contacto con la superficie externa de la cápsula de gelatina, una capa expandible que está en contacto con la capa de barrera, una capa semipermeable que abarca la capa expandible, y un orificio de salida formado o que puede formarse en la pared, en donde la capa de barrera forma un sello entre la capa expandible y el entorno en el orificio de salida.
(Composición P.3)
También se describe en el presente documento una composición que comprende una cápsula de gelatina que contiene un líquido, el Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable, como se ha descrito anteriormente, estando la cápsula de gelatina rodeada por una capa de barrera que está en contacto con la superficie externa de la cápsula de gelatina, una capa expandible que está en contacto con una porción de la capa de barrera, una capa semipermeable que abarca al menos la capa expandible, y un orificio de salida formado o que puede formarse en la forma de dosificación que se extiende de la superficie externa de la cápsula de gelatina al entorno de uso. (Composición P.4). La capa expansible puede formarse en una o más secciones discretas, tales como por ejemplo, dos secciones situadas en lados o extremos opuestos de la cápsula de gelatina.
Los Compuestos de Fórmula I en el sistema de administración oral de liberación osmótica controlada (es decir, en la Composición P.1-P.4) como se ha descrito anteriormente pueden estar en una formulación líquida, cuya formulación puede ser pura, un agente activo líquido, un agente activo líquido en una solución, suspensión, una emulsión o una composición autoemulsionante o similares.
En el documento WO 2000/35419 puede encontrarse más información sobre las composiciones del sistema de administración oral de liberación osmótica controlada que incluyen las características de la cápsula de gelatina, capa de barrera, capa expansible, capa semipermeable y orificio. Otros sistemas de administración oral de liberación osmótica controlada pueden encontrarse en el documento EP 1539115 (publicación de EE. UU. N.° 2009/0202631). También se describen en el presente documento los Métodos A o B descritos anteriormente, en donde el compuesto de fórmula I se formula en un dispositivo que comprende (a) dos o más capas, dichas dos o más capas que comprenden una primera capa y una segunda capa, comprendiendo dicha primera capa el Compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable como se ha descrito anteriormente, comprendiendo dicha segunda capa un polímero; (b) una pared exterior que rodea dichas dos o más capas; y (c) un orificio en dicha pared exterior. (Composición P.5)
La Composición P.5 utiliza preferentemente una membrana semipermeable que rodea el núcleo de tres capas descrito en el presente documento: en dichas composiciones, la primera capa se denomina primera capa de fármaco y contiene bajas cantidades de fármaco (p. ej., los Compuestos de Fórmula I) y un agente osmótico tal como una sal, la capa intermedia denominada segunda capa de fármaco contiene mayores cantidades de fármaco, excipientes y ninguna cantidad de sal; y la tercera capa denominada capa de empuje contiene agentes osmóticos y ningún fármaco. Se perfora al menos un orificio a través de la membrana en el extremo de la primera capa de fármaco del comprimido en forma de cápsula. (Composición P.6)
La Composición P.5 o P.6 puede comprender una membrana que define un compartimento, rodeando la membrana una subcapa protectora interior, al menos un orificio de salida formado o que puede formarse en ella y al menos una porción de la membrana que es semipermeable; una capa expandible situada dentro del compartimento remoto del orificio de salida y en comunicación fluida con la porción semipermeable de la membrana; una primera capa de fármaco situada adyacente al orificio de salida; y una segunda capa de fármaco situada dentro del compartimento entre la primera capa de fármaco y la capa expandible, comprendiendo las capas de fármaco el Compuesto de Fórmula I en forma libre o de sal farmacéuticamente aceptable de la misma. Dependiendo de la viscosidad relativa de la primera capa de fármaco y de la segunda capa de fármaco, se obtienen diferentes perfiles de liberación. Es indispensable identificar la viscosidad óptima para cada capa. En la presente invención, la viscosidad se modula mediante la adición de una sal, p. ej., cloruro de sodio. El perfil de administración del núcleo depende del peso, de la
formulación y del espesor de cada una de las capas de fármaco. (Composición P.7)
La Composición P.7 puede comprender la primera capa de fármaco que comprende sal y la segunda capa de fármaco que no contiene sal. La Composición P.5-P.7 puede comprender opcionalmente una capa promotora del flujo entre la membrana y las capas de fármaco. Las Composiciones P.1-P.7 se denominarán generalmente composición de sistema de administración oral de liberación osmótica controlada.
También se describe en el presente documento una composición farmacéutica como la descrita anteriormente que comprende un Compuesto de Fórmula I en forma libre o de sal farmacéuticamente aceptable, p. ej., como se describe en cualquiera de los Métodos A o 1.1-1.50, o el Método B en mezcla con un diluyente o vehículo farmacéuticamente aceptable, p. ej., en una formulación de liberación inmediata o sostenida o retardada, incluyendo una formulación inyectable de acción prolongada, para su uso en el tratamiento de los síntomas residuales como se describe en cualquiera de los Métodos A, o 1.1-1.50, o para su uso en el tratamiento de los trastornos como se describe en el Método B.
También se describe en el presente documento el uso del Compuesto de Fórmula I o la composición farmacéutica que comprende el compuesto de Fórmula I en forma libre o de sal farmacéuticamente aceptable como se describe en los Métodos A o 1.1-1.50, formulado en una formulación de liberación inmediata o sostenida o retardada, incluyendo una formulación inyectable de acción prolongada, como se ha descrito anteriormente, en la preparación de un medicamento) para el tratamiento de los síntomas residuales como se describe en cualquiera de los Métodos A o 1.1-1.50, o para el tratamiento de los trastornos como se describe en el Método B.
También se describe el uso del Compuesto de Fórmula I o de cualquier composición farmacéutica como se ha descrito anteriormente, (p. ej., las Composiciones P.1 a P.7 o la Composición 2) que comprende el Compuesto de Fórmula I en forma libre o de sal farmacéuticamente aceptable como se describe en los Métodos A o 1.1-1.50, en donde el compuesto está en mezcla o asociación con un antioxidante. Sin quedar ligado a teoría alguna particular, se cree que la presencia de un antioxidante dentro de la composición estabilizará el compuesto de Fórmula I. En una realización preferida, las microesferas inyectables de acción prolongada contienen un antioxidante, en donde se cree que el antioxidante estabilizará el compuesto de Fórmula I durante la liberación de la microesfera.
Descripción detallada de la invención
La presente invención proporciona una necesidad no satisfecha en el tratamiento no solo de los síntomas agudos, sino también de los síntomas residuales de la psicosis, particularmente esquizofrenia. Los pacientes que padecen esquizofrenia son tratados actualmente con agentes antipsicóticos convencionales o atípicos. Estos agentes, que pueden ser eficaces en el tratamiento de los síntomas positivos de la psicosis, en general son inadecuados para tratar los síntomas residuales. Por tanto, se necesita un tratamiento diferente o adicional para mejorar los resultados. En una realización, el método definido en la reivindicación 1 usa un compuesto de Fórmula I solo, como se ha descrito anteriormente, o se usa en una combinación del compuesto de Fórmula I y uno o más agentes antipsicóticos diferentes para el tratamiento de los síntomas residuales mencionados en la reivindicación 1 y los síntomas agudos de la psicosis.
Como se usa en el presente documento, "síntomas residuales" incluyen síntomas negativos y síntomas generales de psicopatología descritos en la Escala de Síntomas Positivos y Negativos (PANSS) para la esquizofrenia descrita en Kay et al., Schizophr. Bull. (1987) 13(2):261-276. Los síntomas negativos incluyen: afecto embotado, retraimiento emocional, escasa compenetración, retraimiento social pasivo/apático, dificultad para el pensamiento abstracto, falta de espontaneidad y fluidez en la conversación y pensamiento estereotipado. Los síntomas de psicopatología general incluyen: preocupación somática, ansiedad, sentimientos de culpa, tensión, manerismos y posturas, depresión, retraso motor, falta de cooperación, contenido de pensamientos inusuales, desorientación, atención deficiente, falta de juicio y visión interna, alteración de la volición, control deficiente de los impulsos, preocupación y evitación social activa. Los síntomas residuales también pueden incluir depresión, deterioro cognitivo y trastornos del sueño (p. ej., insomnio). De estos síntomas residuales, los Compuestos de la Fórmula I son particularmente útiles para el tratamiento del retraimiento social pasivo, pensamiento estereotipado, preocupaciones somáticas, ansiedad, tensión, evitación social activa y depresión. La mayoría de los pacientes con esquizofrenia presentan déficits en la función social que impiden una reintegración satisfactoria en la sociedad. Los déficits en la función social pueden medirse usando el factor prosocial derivado de la PANSS. El factor prosocial está compuesto por elementos de las subescalas de Psicopatología Positiva, Negativa y General, tales como evitación social activa, retraimiento emocional, retraimiento social pasivo, pensamiento estereotipado, conducta alucinatoria y desconfianza. Este factor ha demostrado ser sensible al cambio en el entorno del ensayo clínico. Dado que los Compuestos de Fórmula I, particularmente el Compuesto A definido en el Ejemplo 1, reducen los síntomas negativos y también tratan otros campos sintomáticos, se cree que los Compuestos de Fórmula I pueden ser útiles para tratar los déficits de la función social. Por lo tanto, los compuestos descritos en el presente documento son particularmente útiles para mejorar la integración social y la función social en pacientes que padecen esquizofrenia. La función social es la capacidad de reconocer, comprender, procesar y usar referencias externas para resolver problemas, mantener el rendimiento laboral y llevar a cabo relaciones interpersonales.
El tratamiento de estos síntomas residuales también es particularmente eficaz en los pacientes esquizofrénicos que también padecen depresión. 60 mg del Compuesto de Fórmula I, particularmente el Compuesto A definido en el Ejemplo 1 en forma de sal de adición de ácido toluensulfónico, administrado una vez al día por la mañana mejora los síntomas asociados a la esquizofrenia medidos por una disminución estadísticamente significativa y clínicamente significativa de la puntuación total de la PANSS tras 28 días de tratamiento. Este compuesto A tampoco causa hiperprolactinemia, EPS/acatisia, aumento de peso o problemas de seguridad cardiovascular. A la misma dosificación, este compuesto A también mejora de forma estadísticamente significativa la subescala de síntomas positivos y la subescala de psicopatología general de la PANSS. Además, el Compuesto de Fórmula I, particularmente el Compuesto A definido en el Ejemplo 1 en forma de sal de adición de ácido toluensulfónico a 60 mg mejora los síntomas negativos en un subgrupo de pacientes con síntomas negativos prominentes al inicio del estudio. De manera significativa, también mejora ciertos elementos de las subescalas de síntomas negativos y psicopatología general acordes con la mejora de la función social.
La expresión "síntomas agudos" de psicosis o esquizofrenia se refiere a los síntomas positivos de la PANSS, tales como delirios, desorganización conceptual, conducta alucinatoria y desconfianza.
El término "psicosis" se refiere a enfermedades tales como esquizofrenia, trastorno delirante, depresión mayor con psicosis, trastorno bipolar con síntomas psicóticos, trastorno psicótico breve, trastorno esquizofreniforme, trastorno esquizoafectivo o psicosis causada por una afección médica o por consumo de sustancias. Preferentemente, el paciente que padece síntomas residuales de psicosis es un paciente que padece síntomas residuales de esquizofrenia.
La expresión "trastorno bipolar" hace referencia a un trastorno caracterizado por cambios extremos en el estado de ánimo. Los individuos con trastorno bipolar pueden experimentar sentimientos intensos de sobreexcitación, irritabilidad e impulsividad con creencias grandiosas y pensamientos acelerados, denominado episodio maníaco. Los síntomas de la depresión pueden incluir sensación de cansancio, desesperanza y tristeza, con dificultad para concentrarse y pensamientos suicidas. Algunas personas experimentan ambos tipos de síntomas en el mismo episodio "mixto". Los síntomas graves del trastorno bipolar pueden ir asociados a alucinaciones o delirios, mencionado de otro modo como psicosis.
Los términos "tratamiento" y "tratar" deben entenderse, en consecuencia, como la profilaxis y el tratamiento o la mejora de los síntomas de la enfermedad y/o el tratamiento de la causa de la enfermedad.
El término "paciente" puede incluir un paciente humano o no humano.
Si no se especifica lo contrario o se deduce claramente del contexto, los siguientes términos tienen el siguiente significado en el presente documento:
(i) "Alquilo", como se usa en el presente documento, es un resto de hidrocarburo saturado o insaturado, p. ej., de uno a veintiún átomos de carbono de longitud, que puede ser lineal o ramificado (por ejemplo, n-butilo o terc-butilo), preferentemente linear, a menos que se especifique lo contrario. Por ejemplo, "alquilo C1-21" representa alquilo que tiene de 1 a 21 átomos de carbono. El alquilo puede estar opcionalmente sustituido con uno o más grupos hidroxi o alcoxi C1-22 (p. ej., etoxi). Como alternativa, el alquilo contiene de 1 a 21 átomos de carbono, preferentemente de cadena recta y opcionalmente saturado o insaturado, por ejemplo, R 1 es una cadena de alquilo que contiene de 1 a 21 átomos de carbono, preferentemente 6-15 átomos de carbono, 16-21 átomos de carbono, p. ej., para que junto con el -C(O)- al que se une, p. ej., cuando se escinde del compuesto de Fórmula I, forme el residuo de un ácido graso natural o no natural, saturado o insaturado.
Métodos de preparación de los compuestos de fórmula I
Los compuestos de la fórmula I y sus sales farmacéuticamente aceptables pueden hacerse usando los métodos descritos y ejemplificados en cualquiera de las siguientes patentes o solicitudes: patente de EE. UU. N.° 6.548.493; 7.238.690; 6.552.017; 6.713.471; U.S. RE39680; U.S. RE39679; PCT /US08/03340; solicitud de EE. UU. con n.° de serie 10/786.935; WO 2011/133224 A1, WO 2009/114181 y WO 2013/155505. Si no están disponibles comercialmente, los materiales de partida para estos procesos pueden realizarse por procedimientos, que se seleccionan entre la técnica química usando técnicas que son similares o análogas a la síntesis de compuestos conocidos.
Los compuestos de Fórmula I descritos en el presente documento incluyen:

en forma libre o de sal farmacéuticamente aceptable. Los Compuestos de Fórmula I descritos en el presente documento también incluyen otros compuestos específicos en donde Y es -C(H)(OH)- o -C(H)(ORi) en donde R1 se ha definido previamente y X es -O-, -N(H)- o -N(CH3)-. Estos se incluyen, por ejemplo:

en forma libre o de sal farmacéuticamente aceptable. Un Compuesto de Fórmula I específico también incluye el Compuesto A que es el compuesto de Fórmula I en donde X es -N(CH3)- e Y es -C(O)-. Las expresiones "Compuestos de Fórmula I" y "Compuestos de Fórmula I" pueden usarse indistintamente y pueden usarse como único agente terapéutico, o también pueden usarse en combinación o para la coadministración con otros agentes activos. Asimismo, en los métodos de la presente invención, la expresión "un compuesto de Fórmula I" puede incluir más de uno de los compuestos de Fórmula I.
Los Compuestos de Fórmula I pueden, en algunos casos, existir también en forma de profármaco. Una forma de profármaco es un compuesto que se convierte en el cuerpo en un Compuesto de Fórmula I activo. El término "profármaco" es un término reconocido en la técnica y se refiere a un precursor de fármaco antes de la administración que genera o libera el metabolito activo in vivo tras la administración, a través de algún proceso químico o fisiológico. Por ejemplo, cuando los Compuestos de Fórmula I contienen sustituyentes hidroxi o carboxi, estos sustituyentes pueden formar ésteres fisiológicamente hidrolizables y aceptables. Como se usa en el presente documento, "éster fisiológicamente hidrolizable y aceptable" significa ésteres de Compuestos de Fórmula I que son hidrolizables en condiciones fisiológicas para producir ácidos (en el caso de los Compuestos de Fórmula I que tienen sustituyentes hidroxi) o alcoholes (en el caso de los Compuestos de Fórmula I que tienen sustituyentes carboxi) que son, a su vez, fisiológicamente tolerables a las dosis que se van a administrar. Por ejemplo, cuando Y del compuesto de Fórmula I es -C(H)(OR1), y R1 es un acilo fisiológicamente hidrolizable y aceptable, por ejemplo -C(O)-alquilo C1
21, p. ej., -C(O)-alquilo C3 o -C(O)-alquilo C9 , estos compuestos pueden hidrolizarse en condiciones fisiológicas para producir un compuesto de Fórmula I en donde Y es-C(H)(OH), por una parte, y alquilo C1-21-C(O)OH, p. ej., alquilo C3-C(O)OH o alquilo C9-C(O)OH, por otra parte. En particular, los Compuestos de Fórmula I, en donde Y es -O-,-C(R2)(OH)-, -C(R3)(OR1) o -C(O)-; R1 es alquilo C1-6 (p. ej., metilo) y R2 y R3 son H o alquilo C1-6 (p. ej., metilo) son restos activos. En contraposición, los Compuestos de Fórmula I, cuando R1 es -C(O)- alquilo C1-21 es un resto fisiológicamente lábil, tendrán una actividad débil o nula, pero en condiciones fisiológicas se someterán a hidrólisis para producir los Compuestos de Fórmula I, R1 se escinde para dejar -C(R2)(OH) o -C(R3)(OH) y el otro producto de hidrólisis no es tóxico en concentraciones relevantes, p. ej., en concentraciones que serían proporcionadas por la hidrólisis in vivo de una dosificación del compuesto profármaco. En algunas condiciones fisiológicas, los Compuestos de Fórmula I, en donde R1 es alquilo C1-6 (p. ej., metilo), también pueden someterse a una conversión in vivo en el compuesto más activo en donde R1 es H, y por lo tanto, estos compuestos pueden considerarse tanto restos activos como profármacos.
Como se apreciará, el término profármaco abarca así pues las formas farmacéuticas convencionales de profármacos. Cuando se usa un profármaco (p. ej., el compuesto de fórmula (I) en donde R1 es -C(O)- alquilo C1-21), la cantidad de dosificación se calcula en función de la cantidad del Compuesto de Fórmula I activo, p. ej., Y es -O-, -C(R2)(OH)-, -C(R3)(OR1) o -C(O)-; R1 es alquilo C1-6 (p. ej., metilo), R2 y R3 son H o alquilo C1-6 (p. ej., metilo), particularmente el compuesto de fórmula (I) en donde Y es C(=O) o Y es C(H)(OH) en forma de base libre o en forma de sal, p. ej., en forma de sal de adición de ácido toluensulfónico.
Los Compuestos de Fórmula I pueden comprender uno o más átomos de carbono quirales. Los compuestos existen así en formas isoméricas individuales, p. ej., formas enantioméricas o diastereoméricas, o como mezclas de formas individuales, p. ej., mezclas racémicas/diastereoméricas. Puede estar presente cualquier isómero en el que el centro asimétrico esté en la configuración (R)-, (S)-, o (R,S). La invención debe entenderse que abarca tanto los isómeros individuales ópticamente activos como las mezclas (p. ej., mezclas racémicas/diastereoméricas) de los mismos. Por consiguiente, los Compuestos de Fórmula I pueden ser una mezcla racémica o pueden estar predominantemente, p. ej., en forma isomérica pura, o sustancialmente pura, p. ej., superior al 70 % de exceso enantiomérico/diaestereomérico ("ee"), preferentemente superior al 80 % de ee, más preferentemente superior al 90 % de ee, lo más preferentemente superior al 95 % de ee. La purificación de dichos isómeros y la separación de dichas mezclas isoméricas puede realizarse mediante técnicas convencionales conocidas en la técnica (p. ej., cromatografía en columna, TLC preparativa, HPLC preparativa, lecho móvil simulado y similares).
Los isómeros geométricos por naturaleza de los sustituyentes alrededor de un doble enlace o de un anillo pueden estar presentes en forma cis (Z) o trans (E), y ambas formas isoméricas se engloban dentro del alcance de esta invención.
Los Compuestos de Fórmula I pueden incluirse como una formulación de liberación sostenida o retardada, p. ej., formulación de liberación lenta, p. ej., dispersando, disolviendo o encapsulando los Compuestos de Fórmula I en una matriz polimérica como se ha descrito anteriormente en P.1-P.7, de manera que el Compuesto se libera de forma continua a medida que el polímero se degrada con el tiempo. La liberación de los Compuestos de Fórmula I de la matriz polimérica proporciona la liberación controlada y/o retardada y/o sostenida de los Compuestos, p. ej., de la composición de liberación lenta farmacéutica, en un sujeto, por ejemplo un animal de sangre caliente tal como el hombre, al que se le administra la liberación lenta farmacéutica. Por tanto, la liberación lenta farmacéutica administra los Compuestos de Fórmula I al sujeto en concentraciones eficaces para el tratamiento de la enfermedad o afección médica particular durante un periodo de tiempo sostenido, p. ej., 14-180 días, preferentemente aproximadamente 30, aproximadamente 60 o aproximadamente 90 días.
Los polímeros útiles para la matriz polimérica en las composiciones descritas en el presente documento (p. ej., composiciones de liberación lenta) pueden incluir un poliéster de un ácido graso hidroxi y derivados del mismo u otros agentes tales como ácido poliláctico, ácido poliglicólico, ácido policítrico, ácido polimálico, ácido poli-betahidroxibutírico, polímero de apertura de anillo de épsilon-capro-lactona, copolímero de ácido láctico-ácido glicólico, copolímero de ácido 2-hidroxibutírico-ácido glicólico, copolímero de ácido poliláctico-polietilenglicol o copolímero de ácido poliglicólico-polietilenglicol), un polímero de un alfa-cianoacrilato de alquilo (por ejemplo, poli(butil 2-cianoacrilato)), un oxalato de polialquileno (por ejemplo, oxalato de politrimetileno u oxalato de politetrametileno), un poliortoéster, un policarbonato (por ejemplo, carbonato de polietileno o carbonato de polietilpropileno), un poliortocarbonato, un poliaminoácido (por ejemplo, poli-gamma-L-alanina, poli-gamma-bencil-L-ácido glutámico o poli-metil-L-ácido glutámico), un éster de ácido hialurónico, y similares, pudiendo usarse uno o varios de estos polímeros.
Si los polímeros son copolímeros, pueden ser cualquiera de los copolímeros aleatorios, en bloque y/o de injerto. Cuando los ácidos alfa-hidroxicarboxílicos, los ácidos hidroxidicarboxílicos y los ácidos hidroxicarboxílicos mencionados anteriormente tienen actividad óptica en sus moléculas, puede utilizarse uno cualquiera de los isómeros D, isómeros L y/o isómeros DL. Entre otros, puede usarse el polímero de ácido alfa-hidroxicarboxílico (preferentemente el polímero de ácido láctico-ácido glicólico), su éster, ésteres de ácido poli-alfa-cianoacrílico, etc., y se prefiere el copolímero de ácido láctico-ácido glicólico (también denominado poli(lactida-alfa-glicólido) o poli(ácido láctico-co-glicólico), y en lo sucesivo denominado PLGA). Por tanto, en un aspecto, el polímero útil para la matriz polimérica es PLGA. Como se usa en el presente documento, el término PLGA incluye polímeros de ácido láctico
(también denominados polilactida, poli(ácido láctico) o PLA). Lo más preferentemente, el polímero es el polímero biodegradable de poli(d,l-lactida-co-glicólido).
Cuando está presente, la matriz polimérica es un material polimérico biocompatible y biodegradable. El término "biocompatible" se define como un material polimérico que es no tóxico, no es cancerígeno y no induce significativamente inflamación en los tejidos del cuerpo. El material de la matriz debe ser biodegradable, en donde el material polimérico debe degradarse mediante procesos corporales a productos fácilmente desechables por el cuerpo y no debe acumularse en el cuerpo. Los productos de la biodegradación también deben ser biocompatibles con el cuerpo en el sentido de que la matriz polimérica es biocompatible con el cuerpo. Algunos ejemplos útiles de materiales de matriz polimérica incluyen poli(ácido glicólico), poli-ácido D,L-láctico, poli-ácido L-láctico, copolímeros de los anteriores, poli(ácidos carboxílicos alifáticos), copolioxalatos, policaprolactona, polidioxonona, poli(ortocarbonatos), poli(acetales), poli(ácido láctico-caprolactona), poliortoésteres, poli(ácido glicólicocaprolactona), polianhídridos, y polímeros naturales entre los que se incluyen albúmina, caseína, y ceras, tales como, mono y diestearato de glicerol, y similares. El polímero preferido para su uso en la práctica de esta invención es dl-(polilactida-co-glicólido). Se prefiere que la relación molar de lactida a glicólido en dicho copolímero esté en el intervalo de aproximadamente 75:25 a 50:50.
Los polímeros de PLGA útiles pueden tener un peso molecular promedio de aproximadamente 5.000 a 500.000 daltons, p. ej., de aproximadamente 150.000 daltons, o de 20.000 a 200.000 daltons, por ejemplo, de 24.000 a 38.000 daltons, o aproximadamente 113.000 daltons o aproximadamente 159.000 daltons. Por ejemplo, el polímero de PLGA tiene un peso molecular promedio de 24.000 a 38.000 daltons. Dependiendo de la velocidad de degradación que se quiera conseguir, se pueden usar diferentes pesos moleculares de polímeros. Para un mecanismo difusional de liberación de fármacos, el polímero debe permanecer intacto hasta que todo el fármaco se libere de la matriz polimérica y luego se degrade. El fármaco también puede liberarse de la matriz polimérica a medida que el excipiente polimérico se bioerosiona.
El PLGA puede prepararse por métodos convencionales, o puede estar disponible comercialmente. Por ejemplo, el PLGA puede producirse por polimerización de apertura de anillo con un catalizador adecuado a partir de lactida cíclica, glicólido, etc. (véase el documento EP-0058481B2; Effects of polymerization variables on PLGA properties: molecular weight, composition and chain structure).
Se cree que el PLGA es biodegradable por medio de la degradación de toda la composición polimérica sólida, debido a la ruptura de los enlaces éster hidrolizables y escindibles enzimáticamente en condiciones biológicas (por ejemplo, en presencia de agua y enzimas biológicas que se encuentran en los tejidos de animales de sangre caliente, tales como los humanos) para formar ácido láctico y ácido glicólico. Tanto el ácido láctico como el ácido glicólico son productos solubles en agua, y no tóxicos del metabolismo normal, que pueden seguir biodegradándose para formar dióxido de carbono y agua. En otras palabras, se cree que el PLGA se degrada mediante la hidrólisis de sus grupos éster en presencia de agua, por ejemplo en el cuerpo de un animal de sangre caliente tal como el hombre, para producir ácido láctico y ácido glicólico y crear el microclima ácido. El ácido láctico y el ácido glicólico son subproductos de diversas rutas metabólicas en el cuerpo de un animal de sangre caliente, tal como el hombre, en condiciones fisiológicas normales y, por lo tanto, son bien tolerados y producen una toxicidad sistémica mínima.
La matriz polimérica útil para la invención puede comprender un polímero de estrella en donde la estructura del poliéster tiene forma de estrella. Estos poliésteres tienen un único residuo de poliol como resto central rodeado de cadenas de residuos ácidos. El resto de poliol puede ser, p. ej., glucosa o, p. ej., manitol. Estos ésteres son conocidos y se describen en el documento Gb 2.145.422 y en la patente de EE. UU. N.° 5.538.739.
Los polímeros en estrella pueden prepararse usando compuestos polihidrixo, p. ej., poliol, p. ej., glucosa o manitol como iniciador. El poliol contiene al menos 3 grupos hidroxi y tiene un peso molecular de hasta aproximadamente 20.000 daltons, con al menos 1, preferentemente al menos 2, p. ej., como media 3 de los grupos hidroxi del poliol en forma de grupos éster, que contienen cadenas de polilactida o copolilactida. Los poliésteres ramificados, p. ej., poli (d, 1-lactida-co-glicólido) tienen un resto central de glucosa que tiene líneas rectas de cadenas lineales de polilactida.
La formulación de liberación sostenida o retardada, como se ha descrito anteriormente, puede comprender el polímero en forma de micropartículas (p. ej., microesferas) o nanopartículas, o en forma líquida, con los Compuestos de Fórmula I dispersos o encapsulados en su interior. Por "micropartículas" se entienden las partículas sólidas que contienen los Compuestos de Fórmula I en solución o en forma sólida, en donde dicho compuesto está disperso o disuelto dentro del polímero que sirve de matriz de la partícula. Mediante una selección adecuada de los materiales poliméricos, se puede realizar una formulación de micropartículas en la que las micropartículas resultantes presenten tanto propiedades de liberación difusional como de liberación por biodegradación.
Cuando el polímero está en forma de micropartículas, las micropartículas pueden prepararse usando cualquier método apropiado, tal como por ejemplo un método de evaporación de disolventes o de extracción de disolventes. Por ejemplo, en el método de evaporación de disolventes, los Compuestos de Fórmula I y el polímero pueden disolverse en un disolvente orgánico volátil (por ejemplo, una cetona tal como acetona, un hidrocarburo halogenado
tal como cloroformo o cloruro de metileno, un hidrocarburo aromático halogenado, un éter cíclico tal como dioxano, un éster tal como acetato de etilo, un nitrilo tal como acetonitrilo, o un alcohol tal como etanol) y se dispersa en una fase acuosa que contiene un estabilizador de emulsión adecuado (por ejemplo, alcohol polivinílico, PVA). A continuación, el disolvente orgánico se evapora para proporcionar micropartículas con los Compuestos de Fórmula I encapsulados en ellas. En el método de extracción con disolventes, los Compuestos de Fórmula I y el polímero pueden disolverse en un disolvente polar (tal como acetonitrilo, diclorometano, metanol, acetato de etilo o formiato de metilo) y luego dispersarse en una fase acuosa (tal como una solución de agua/PVA). Se produce una emulsión para proporcionar micropartículas con los Compuestos de Fórmula I encapsulados en su interior. El secado por pulverización es una técnica de preparación alternativa para preparar las micropartículas.
Otro método para preparar las micropartículas descritas en el presente documento también se describe en la patente de EE. UU. N.° 4.389.330 y en la patente de EE.UU. N.° 4.530.840.
La micropartícula descrita en el presente documento puede prepararse mediante cualquier método capaz de producir micropartículas en un intervalo de tamaño aceptable para su uso en una composición inyectable. Un método preferido de preparación es el descrito en la patente de EE. UU. N.° 4.389.330. En este método, el agente activo se disuelve o dispersa en un disolvente apropiado. Al medio que contiene el agente se añade el material de matriz polimérica en una cantidad relativa al principio activo que proporciona un producto que tiene la carga deseada de agente activo. Opcionalmente, todos los ingredientes del producto de micropartículas pueden mezclarse en conjunto en el medio disolvente.
Los disolventes para los Compuestos de Fórmula I y el material de matriz polimérica que pueden emplearse incluyen disolventes orgánicos, tales como acetona; hidrocarburos halogenados, tales como cloroformo, cloruro de metileno y similares; compuestos de hidrocarburos aromáticos; compuestos de hidrocarburos aromáticos halogenados; éteres cíclicos; alcoholes, tales como, alcohol bencílico; acetato de etilo; y similares. El disolvente puede ser una mezcla de alcohol bencílico y acetato de etilo. Puede encontrarse más información para la preparación de micropartículas útiles para la invención en la publicación de patente de EE. UU. número 2008/0069885, cuyo contenido se incorpora en el presente documento por referencia en su totalidad.
La cantidad de los Compuestos de Fórmula I incorporados en las micropartículas varía habitualmente de aproximadamente 1 % en peso a aproximadamente 90 % en peso, preferentemente de 30 a 50 % en peso, más preferentemente de 35 a 40 % en peso. Por % en peso se entiende partes de los Compuestos de Fórmula I por peso total de micropartícula.
La formulación de liberación sostenida o retardada puede comprender un diluyente o vehículo farmacéuticamente aceptable, tal como un diluyente o vehículo miscible con agua.
La formulación de liberación sostenida o retardada puede ser una formulación inyectable de acción prolongada. Como alternativa, el inyectable de acción prolongada puede formularse usando microesferas poliméricas, p. ej., microesferas que comprenden una matriz de PLGA con Compuestos de Fórmula I en forma libre o de sal farmacéuticamente aceptable dispersos, disueltos o encapsulados en su interior. Preferentemente, el polímero de PLGA es aproximadamente 75:25 PLA/PLG (lactida:glicólido) con grupos terminales de ácido carboxílico o éster carboxílico. Preferentemente, el Compuesto de Fórmula I está presente en la microesfera como su base libre. Las microesferas pueden prepararse usando métodos conocidos en la técnica, por ejemplo, mediante un método de evaporación de disolventes por emulsificación sin microtamizado, o un método de evaporación de disolventes por emulsificación con microtamizado en seco, o un método de evaporación de disolventes por emulsificación con microtamizado en húmedo.
La velocidad a la que se degrada las microesferas de PLGA depende en gran medida del intervalo de peso molecular elegido para las moléculas de polímero y del tamaño de las microesferas. El intervalo de peso molecular promedio del polímero de PLGA puede ser de 24.000 a 38.000 daltons. El polímero de PLGA puede tener un peso molecular promedio de aproximadamente 113.000 daltons. Como alternativa, el polímero de PLGA puede tener un peso molecular promedio de aproximadamente 159.000 daltons. El plazo de tiempo para la degradación completa de las microesferas y la liberación de los compuestos farmacológicos encapsulados puede ser, p. ej., menos de 6 meses, menos de 4 meses, menos de 3 meses, menos de 2 meses, o menos de 1 mes.
El diámetro de las microesferas, p. ej., el diámetro promedio (o D50), el diámetro del percentil 10 (D10), el diámetro del percentil 25 (D25), el diámetro del percentil 75 (D75), o el diámetro del percentil 90 (D90),puede ser de aproximadamente 1 pm a aproximadamente 100 pm, o de aproximadamente 2 pm a aproximadamente 80 pm, o de aproximadamente 2 pm a aproximadamente 60 pm, o de aproximadamente 2 pm a aproximadamente 50 pm, o de aproximadamente 2 pm a aproximadamente 40 pm, o de aproximadamente 2 pm a aproximadamente 30 pm, o de aproximadamente 5 pm a aproximadamente 35 pm, o de aproximadamente 5 pm a aproximadamente 25 pm, o de aproximadamente 5 pm a aproximadamente 20 pm, o de aproximadamente 10 pm a aproximadamente 20 pm, o de aproximadamente 10 pm a aproximadamente 2 0 0 pm, de aproximadamente 2 0 pm a aproximadamente 160 pm, o de aproximadamente 2 0 pm a aproximadamente 12 0 pm, o de aproximadamente 2 0 pm a aproximadamente 100 pm, o de aproximadamente 20 pm a aproximadamente 80 pm, o de aproximadamente 20 pm a aproximadamente 70
adamente adamente adamente adamente
adamente  adamente
adamente
pm, aproximadamente 30 pm, aproximadamente 40 pm, aproximadamente 50 pm, aproximadamente 60 pm,
aproximadamente 70 pm, aproximadamente 80 pm, aproximadamente 90 pm o aproximadamente 100 pm. Dichas
mediciones del tamaño de partícula pueden realizarse, por ejemplo, mediante fotomicroscopía, microscopía
electrónica de barrido, difracción láser, dispersión de la luz y otras técnicas conocidas por los expertos en la materia.
La cantidad de fármaco encapsulado en cada microesfera, en promedio, puede ser de aproximadamente 5 % en
peso a aproximadamente 50 % en peso, por ejemplo, de 10 % en peso a aproximadamente 50 % en peso, o de
aproximadamente 20 % en peso a aproximadamente 40 % en peso, o de aproximadamente 30 % en peso a
aproximadamente 35 % en peso, o aproximadamente 8,5 % en peso, o aproximadamente 16 % en peso, o
aproximadamente 30 % en peso, o aproximadamente 35 % en peso, o aproximadamente 40 % en peso. En una
realización preferida, la cantidad de fármaco encapsulado en cada microesfera es de aproximadamente 8,5 % en
peso o aproximadamente 16 % en peso.
Los detalles de la composición del sistema de administración oral de liberación osmótica pueden encontrarse en el
documento EP 1539115 (publicación de EE. UU. N.° 2009/0202631) y WO 2000/35419.
Una "cantidad terapéuticamente eficaz" es cualquier cantidad de los Compuestos de Fórmula I que, cuando se
administra a un sujeto que padece una enfermedad o trastorno, es eficaz en causar una reducción, remisión o
regresión de la enfermedad o trastorno durante el periodo de tiempo previsto para el tratamiento.
Se estima que la depresión está presente en el 50 % de los pacientes esquizofrénicos. Los pacientes
esquizofrénicos con depresión comórbida, en comparación con los pacientes que solo padecen esquizofrenia, tienen
una peor salud mental y física en general, una menor calidad de vida, un mayor deterioro de las relaciones sociales,
una menor satisfacción con el sueño, capacidad de realizar actividades cotidianas, capacidad de trabajo, transporte,
apoyo social y autoestima. Por lo tanto, los síntomas depresivos exacerban los déficits en el funcionamiento
psicosocial y aumentan el riesgo de recaída psicótica. A diferencia de los antagonistas del receptor de dopamina, los
Compuestos de Fórmula I, particularmente la Fórmula A definida en el Ejemplo 1, normalizan la actividad
dopaminérgica cerebral, particularmente en la corteza prefrontal. Los Compuestos de Fórmula I, particularmente la
Fórmula A definida en el Ejemplo 1, se unen a los receptores de 5-HT2A y D2 de la dopamina. De manera adicional,
los compuestos de Fórmula I también modulan la fosfoproteína glutamatérgica (D1/GIuN2b) y la fosfoproteína
dopaminérgica (DPPM). Los Compuestos de Fórmula I también presentan una afinidad de unión nanomolar para el
transportador de serotonina en comparación con los antidepresivos conocidos. Por lo tanto, además de tratar los
síntomas agudos (p. ej., alucinaciones y delirios) y los síntomas residuales, los compuestos de Fórmula I también
son útiles para el tratamiento de la depresión y/o los trastornos del sueño. Para los pacientes esquizofrénicos que
también padecen depresión, los Compuestos de Fórmula I son particularmente útiles para mejorar la PANSS total y
los síntomas negativos. Si bien uno de los fármacos antipsicóticos atípicos, risperidona, también es útil para tratar
algunos síntomas negativos, este compuesto es menos eficaz en comparación con el subgrupo de pacientes con
depresión en el inicio del estudio tratados con el Compuesto de Fórmula I (véanse las Figuras 2 y 3 y el Ejemplo 1).
Por lo tanto, algunos de los métodos descritos en el presente documento son particularmente útiles para el
tratamiento de la depresión y/o los trastornos del sueño en pacientes que padecen síntomas agudos y/o residuales
de la psicosis, así como para el tratamiento de los síntomas agudos y/o residuales de la psicosis en pacientes que
padecen depresión y/o trastornos del sueño.
También se describe en el presente documento un método para tratar síntomas residuales mediante la
administración del compuesto de Fórmula I en combinación con uno o más agentes terapéuticos tales como
agente(s) antipsicótico(s) y/o agente(s) antidepresivo(s) y/o agentes hipnóticos. En dichos métodos, los agentes
antipsicóticos y/o antidepresivos y/o hipnóticos pueden ser un complemento del compuesto de Fórmula I o el
compuesto de Fórmula I puede ser un complemento del agente antipsicótico y/o agente antidepresivo y/o agente
hipnótico. Como se usa en el presente documento, el término "adjunto/a" o "complemento" se refiere a cualquier
tratamiento que se usa junto con otro para aumentar la posibilidad de curación, o para aumentar la eficacia del primer tratamiento. En otras palabras, la terapia adjunta actúa como una ayuda al tratamiento principal. Algunas veces, el compuesto de fórmula I se usa como monoterapia para tratar los síntomas agudos y/o los síntomas residuales de la esquizofrenia, así como la depresión y/o los trastornos del sueño (p. ej., insomnio) en pacientes que padecen esquizofrenia.
Otros agentes terapéuticos que pueden administrarse opcionalmente a un paciente que los necesite incluyen compuestos que modulan la actividad de GABA (p. ej., potencian la actividad y facilitan la transmisión de GABA), un agonista del receptor de GABA-B, un modulador de 5-HT (p. ej., un agonista del receptor de 5-HTi a , un antagonista del receptor de 5-HT2A, un agonista inverso del receptor de 5-HT2A, etc.), un agonista del receptor de melatonina, un modulador de los canales iónicos (p. ej., bloqueador), un antagonista de receptor/inhibidor de la recaptación de serotonina-2, (SARIs), un antagonista del receptor de orexina, un agonista del receptor de H3, un antagonista del receptor noradrenérgico, un agonista del receptor de galanina, un antagonista del receptor de CRH, una hormona del crecimiento humano, un agonista del receptor de la hormona del crecimiento, un estrógeno, un agonista del receptor de estrógenos, un fármaco de la neuroquinina-1 y agentes antipsicóticos, p. ej., agentes antipsicóticos atípicos, en forma libre o de sal farmacéuticamente aceptable.
El término "GABA" se refiere al ácido gamma-aminobutírico. Los compuestos de GABA son compuestos que se unen al receptor de GABA, e incluyen, pero no se limitan a uno o más de doxepina, alprazolam, bromazepam, clobazam, clonazepam, clorazepato, diazepam, flunitrazepam, flurazepam, lorazepam, midazolam, nitrazepam, oxazepam, temazapam, triazolam, indiplon, zopiclona, eszopiclona, zaleplón, Zolpidem, gabaxadol, vigabatrina, tiagabina, EVT 201 (Evotec Pharmaceuticals) o estazolam.
Otros agentes terapéuticos opcionales son los antagonistas del receptor 5HT2A, tales como ketanserina, risperidona, eplivanserina, volinanserina (Sanofi-Aventis, Francia), pruvanserina, pimavanserina (ACP-103), MDL 100907 (Sanofi-Aventis, Francia), HY10275 (Eli Lilly), APD125 (Arena Pharmaceuticals, San Diego, CA), o AVE8488 (Sanofi-Aventis, Francia).
Otros agentes terapéuticos opcionales aún son pizotifeno.
Otros agentes terapéuticos opcionales son los agonistas del receptor de 5HT1A, tales como repinotán, sarizotán, eptapirona, buspirona o MN-305 (MediciNova, San Diego, CA).
Otros compuestos opcionales son los agonistas del receptor de melatonina, tales como melatonina, ramelteón (ROZERe M®, Takeda Pharmaceuticals, Japón), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Descubrimiento, Fase II o agomelatina.
Otros agentes terapéuticos opcionales son los bloqueadores de los canales iónicos, tales como lamotrigina, gabapentina o pregabalina.
Otros agentes terapéuticos opcionales son los antagonistas del receptor de orexina, tales como orexina, una 1,3-biarilurea, SB-334867-a (GlaxoSmithKline, R.U.), GW649868 (GlaxoSmithKline) o un derivado de benzamida, por ejemplo.
Otros agentes terapéuticos opcionales son los antagonistas de receptores/inhibidores de la recaptación de la serotonina-2 (SARI), tales como Org 50081 (Organon - Países Bajos), ritanserina, nefazodona, serzone o trazodona. Otros agentes terapéuticos opcionales son los fármacos de neuroquinina-1, tales como Casopitant (GlaxoSmithKline).
Los ejemplos específicos de agentes terapéuticos adicionales incluyen modafinilo, armodafinilo, doxepina, alprazolam, bromazepam, clobazam, clonazepam, clorazepato, diazepam, flunitrazepam, flurazepam, lorazepam, midazolam, nitrazepam, oxazepam, temazapam, triazolam, indiplon, zopiclona, eszopiclona, zaleplón, zolpidem, gabaxadol, vigabatrina, tiagabina, EVT 201 (Evotec Pharmaceuticals), estazolam, ketanserina, risperidona, eplivanserina, volinanserina (Sanofi-Aventis, Francia), pruvanserina, MDL 100907 (Sanofi-Aventis, Francia), HY10275 (Eli Lilly), APD125 (Arena Pharmaceuticals, San Diego, CA), AVE8488 (Sanofi-Aventis, Francia), repinotán, sarizotán, eptapirona, buspirona, MN-305 (MediciNova, San Diego, CA), melatonina, ramelteón (ROZEREM®, Takeda Pharmaceuticals, Japón), VEC-162 (Evotec Pharmaceuticals, Rockville, MD), PD-6735 (Descubrimiento, Fase II), agomelatina, lamotrigina, gabapentina, pregabalina, orexina, una 1,3-biarilurea, SB-334867-a (GlaxoSmithKline, R.U.), GW649868 (GlaxoSmithKline), un derivado de benzamida, Org 50081 (Organon -Países Bajos), ritanserina, nefazodona, serzone, trazodona, Casopitant (GlaxoSmithKline), amitriptilina, amoxapina, bupropión, citalopram, clomipramina, desipramina, doxepina, duloxetina, escitalopram, fluoxetina, fluvoxamina, imipramina, isocarboxazida, maprotilina, mirtazapina, nefazodona, nortriptilina, paroxetina, sulfato de fenelzina, protriptilina, sertralina, tranilcipromina, trazodona, trimipramina, venlafaxina, clorpromazina, haloperidol, droperidol, flufenazina, loxapina, mesoridazina, molindona, perfenazina, pimozida, proclorperazina, promazina, tioridazina, tiotixeno, trifluoperazina, clozapina, aripiprazol, olanzapina, quetiapina, risperidona, ziprasidona, paliperidona,
asenapina, lurasidona, iloperidona, cariprazina, amisulprida, zotepina, sertindol, en forma libre o de sal farmacéuticamente aceptable.
Las composiciones de combinación descritas en el presente documento pueden incluir mezclas de los fármacos combinados, así como dos o más composiciones separadas de los fármacos, cuyas composiciones individuales pueden ser, por ejemplo, coadministradas juntas a un paciente en el mismo o en diferentes momentos.
También se describe en el presente documento el uso del Compuesto de Fórmula I o de cualquiera de las composiciones farmacéuticas descritas en el presente documento (Composiciones P.1-P.7 y Composiciones 2, y 2.1-2.17) que comprenden el Compuesto de Fórmula I en forma libre o de sal farmacéuticamente aceptable como se describe en los Métodos A o 1.1-1.50, en donde el compuesto está en mezcla con un antioxidante. Preferentemente, las formulaciones de microesferas inyectables de acción prolongada descritas en el presente documento contienen un antioxidante. Algunas veces, el antioxidante es un antioxidante soluble en agua (p. ej., ácido ascórbico, ácido lipoico), aunque en otros casos, el antioxidante es un antioxidante soluble en lípidos (p. ej., ácido lipoico, vitamina E, tocoferoles, carotenos y antioxidantes fenólicos). Algunas veces, el antioxidante es un antioxidante neutro o débilmente básico. Otros posibles antioxidantes incluyen antioxidantes catalíticos (p. ej., ebseleno) y los antioxidantes que contienen metales. Preferentemente, el antioxidante es un antioxidante fenólico, tal como butilhidroxitolueno (BHT) o butilhidroxianisol (BHA). Más preferentemente, el antioxidante es BHT.
Las dosificaciones empleadas en la práctica de la presente invención variarán, por supuesto, por ejemplo, de la enfermedad o afección particular a tratar, el Compuesto de Fórmula I particular usado, el modo de administración y la terapia deseada. A menos que se indique otra cosa, una cantidad del Compuesto de Fórmula I para su administración (ya sea administrado como base libre, profármaco o como forma de sal) se refiere o se basa en la cantidad del Compuesto de Fórmula I en forma de base libre (es decir el cálculo de la cantidad se basa en la cantidad de base libre). En una realización particular, sin embargo, la dosificación del Compuesto de Fórmula I se administra basándose en la cantidad de la forma de sal, p. ej., la forma de sal de adición de ácido toluensulfónico. Los compuestos de Fórmula I pueden administrarse por cualquier vía adecuada, entre las que se incluyen vía oral, vía parenteral o vía transdérmica, pero se administran preferentemente por vía oral. Como monoterapia, el compuesto de Fórmula I puede administrarse a aproximadamente de 1 mg a 120 mg al día o de 10 mg a 120 mg al día, o de 10 mg a 60 mg al día, o de 10 mg a 40 mg al día, o de 1 mg a 10 mg al día, o de 10 mg al día, 20 mg al día, 40 mg al día o 60 mg al día, preferentemente aproximadamente 60 mg del compuesto en forma de sal de adición de ácido toluensulfónico al día. Cuando se administran 120 mg, se administran preferentemente por la noche.
Como formulación de microesferas inyectables de acción prolongada, la dosificación del Compuesto de Fórmula I depende tanto de la carga promedio del fármaco dentro de las microesferas (expresada en % p/p) como de la dosificación de microesferas administradas (mg/kg). Como monoterapia, el Compuesto de Fórmula I puede administrarse en forma de microesferas para proporcionar una dosificación de 1 a 50 mg/kg del Compuesto de Fórmula I, por ejemplo, de 5 a 25 mg/kg, preferentemente 5-10 mg/kg, por ejemplo aproximadamente 5 mg/kg. Puede proporcionarse una dosis de aproximadamente 5 mg/kg del compuesto de Fórmula I, por ejemplo, empleando una dosis de 60 mg/kg de microesferas, en donde cada microesfera contiene en promedio una carga de aproximadamente 8,5 % p/p del Compuesto de Fórmula I.
Las dosificaciones de un compuesto de Fórmula I y/o del otro agente antipsicótico y/o antidepresivo del Método A pueden ser iguales o inferiores a la dosificación aprobada para el fármaco, a la dosificación de prueba clínica o bibliográfica o a la dosificación usada para el fármaco como monoterapia. Por ejemplo, la dosificación diaria del compuesto de Fórmula I que se administrará en combinación con otro agente antipsicótico y/o un agente antidepresivo es de aproximadamente 1 mg a aproximadamente 140 mg, en otra realización de aproximadamente 1 mg a aproximadamente 120 mg, en otra realización de aproximadamente 10 mg a aproximadamente 120 mg, en otra realización de aproximadamente 10 mg a aproximadamente 60 mg, en otra realización de aproximadamente 10 mg a aproximadamente 40 mg, en otra realización de aproximadamente 20 mg a aproximadamente 40 mg, en otra realización de aproximadamente 1 mg a aproximadamente 10 mg, y en aún otra realización, aproximadamente 60 mg del compuesto en forma de base libre o de sal de adición de ácido toluensulfónico. La cantidad de agente antipsicótico a administrar en combinación con el compuesto de Fórmula I es de aproximadamente 0,01 mg a aproximadamente 1000 mg, en otra realización, de aproximadamente 0,1 mg a aproximadamente 600 mg, p. ej., de aproximadamente 1 mg a aproximadamente 200 mg, p. ej., de aproximadamente 1 mg a aproximadamente 50 mg, p. ej., de aproximadamente 1 mg a aproximadamente 15 mg, p. ej., aproximadamente 4 mg. La cantidad de agente antidepresivo a administrar en combinación con el compuesto de Fórmula I es de aproximadamente 0,01 mg a aproximadamente 2000 mg, en otra realización de aproximadamente 0,1 mg a aproximadamente 200 mg, en otra realización de aproximadamente 10 mg a aproximadamente 200 mg. En una realización particular, el segundo agente terapéutico es el agente antipsicótico risperidona a una dosis diaria de aproximadamente 2 mg a aproximadamente 4 mg y el antidepresivo es sertralina y la dosificación diaria de sertralina está entre aproximadamente 20 mg y 100 mg.
Las dosificaciones de un compuesto de Fórmula I y/o del segundo (o tercer) agente terapéutico del Método A pueden ser menores que cuando se utilizan en monoterapia. Por lo tanto, la dosificación diaria de un compuesto de
Fórmula I puede ser inferior a 100 mg una vez al día, o menos de 60 mg, o menos de 40 mg, o menos de 30 mg, o menos de 20 mg, o menos de 10 mg. También se prefiere que las dosificaciones tanto del Compuesto de Fórmula I como del agente antipsicótico y/o del agente antidepresivo del Método A sean menores que las dosificaciones usadas para los fármacos individuales como monoterapia. Por lo tanto, por ejemplo, el Método A puede comprender administrar (1) un Compuesto de Fórmula I a una dosificación inferior a 100 mg una vez al día, p. ej., menos de 60 mg o menos de 40 mg; y/o (2) un antidepresivo, por ejemplo un SSRI, tal como sertralina, a una dosificación diaria de menos de 50 mg, más preferentemente, menos de 20 mg, aún más preferentemente, menos de 10 mg, más preferentemente menos de 6 mg; y/o (3) un agente antipsicótico, por ejemplo risperidona, a una dosificación diaria de menos de 4 mg, en forma libre o de sal farmacéuticamente aceptable.
Para el tratamiento de los trastornos descritos en el presente documento, en donde la formulación de liberación sostenida o retardada se usa para lograr una mayor duración de la acción, las dosificaciones serán más altas en relación con la composición de acción más corta, p. ej., más de 1-100 mg, p. ej., 25 mg, 50 mg, 100 mg, 500 mg, 1000 mg, o más de 1000 mg. El régimen de dosificación para la formulación de liberación sostenida o retardada puede incluir una dosificación inicial oral de liberación inmediata junto con la liberación de suministro lento para proporcionar un nivel sanguíneo estable del fármaco. La duración de la acción de los Compuestos de Fórmula I puede controlarse mediante la manipulación de la composición polimérica, es decir, la relación polímero:fármaco y el tamaño de las micropartículas. Cuando las formulaciones descritas en el presente documento son una formulación de liberación lenta, se prefiere la administración por inyección. En una realización preferida, la formulación es una formulación de microesferas inyectables de acción prolongada, como se describe anteriormente en el presente documento.
Los compuestos que se administran en los métodos descritos en el presente documento pueden estar en forma de ácido libre o base libre o como sales farmacéuticamente aceptables. La expresión "sales farmacéuticamente aceptables" se refiere a los derivados de los compuestos divulgados anteriormente, en donde el compuesto original se modifica mediante la preparación de sales de ácido o de base. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero sin limitación, sales minerales u orgánicas de residuos básicos tales como aminas; sales alcalinas u orgánicas de residuos ácidos tales como ácidos carboxílicos; y similares. Las sales farmacéuticamente aceptables incluyen las sales convencionales no tóxicas o las sales de amonio cuaternario del compuesto original formado, por ejemplo, a partir de ácidos inorgánicos u orgánicos no tóxicos. Por ejemplo, dichas sales convencionales no tóxicas incluyen las derivadas de ácidos inorgánicos tales como clorhídrico, bromhídrico, sulfúrico, sulfámico, fosfórico, nítrico y similares; y las sales preparadas a partir de ácidos orgánicos, tales como acético, propiónico, succínico, glicólico, esteárico, láctico, málico, tartárico, cítrico, ascórbico, palmoico, maleico, hidroximaleico, fenilacético, glutámico, benzoico, salicílico, sulfanílico, 2-acetoxibenzoico, fumárico, toluensulfónico, metansulfónico, etandisulfónico, oxálico, isetiónico y similares. Preferentemente, los Compuestos de Fórmula I están en forma de sal de adición de ácido toluensulfónico.
Las sales farmacéuticamente aceptables de los compuestos que se usarán en los métodos descritos en el presente documento pueden sintetizarse a partir del compuesto original que contiene un resto básico o ácido mediante métodos químicos convencionales. En general, dichas sales pueden prepararse haciendo reaccionar las formas de base libre de estos compuestos con una cantidad estequiométrica del ácido apropiado en agua o en un disolvente orgánico, o en una mezcla de ambos; en general, se prefieren medios no acuosos tales como éter dietílico, acetato de etilo, etanol, isopropanol o acetonitrilo. Pueden encontrarse más detalles acerca de la preparación de estas sales, p. ej., sales de ácido toluensulfónico en forma amorfa o cristalina, en los documentos PCT/US08/03340 (WO 2008/112280) y/o WO 2009/114181 y WO 2011/133224.
Las composiciones farmacéuticas que comprenden los Compuestos de Fórmula I pueden prepararse usando diluyentes o excipientes convencionales y técnicas conocidas en la técnica galénica. Por ejemplo, los compuestos pueden administrarse en una amplia variedad de formas de dosificación diferentes, es decir, pueden combinarse con diversos vehículos inertes farmacéuticamente aceptables en forma de comprimidos, cápsulas, pastillas para chupar, tabletas, caramelos duros, polvos, pulverizadores, suspensión acuosa, soluciones inyectables, elixires, jarabes y similares.
Para administración oral, las composiciones farmacéuticas pueden adoptar la forma de, por ejemplo, comprimidos o cápsulas preparados por medios convencionales con excipientes farmacéuticamente aceptables, tales como agentes aglutinantes (p. ej., almidón de maíz pregelatinizado, polivinilpirrolidona o hidroxipropilmetilcelulosa); cargas (p. ej., lactosa, celulosa microcristalina o fosfato de calcio); lubricantes (p. ej., estearato de magnesio, talco o sílice); desintegrantes (p. ej., almidón de patata o glicolato de almidón de sodio); o agentes humectantes (p. ej., lauril sulfato de sodio). Los comprimidos pueden estar recubiertos por métodos bien conocidos en la técnica. Las preparaciones líquidas para administración oral pueden adoptar la forma de, por ejemplo, soluciones, jarabes o suspensiones, o pueden presentarse como un producto seco para su constitución con agua u otro vehículo adecuado antes de su uso. Dichas preparaciones líquidas pueden prepararse por medios convencionales con aditivos farmacéuticamente aceptables, tales como agentes de suspensión (p. ej., jarabe de sorbitol, metilcelulosa o grasas comestibles hidrogenadas); agentes emulsionantes (p. ej., lecitina o acacia); vehículos no acuosos (p. ej., aceite de almendras, ésteres oleosos o alcohol etílico); y conservantes (p. ej., p-hidroxibenzoatos de metilo o propilo o ácido sórbico).
Para la formulación de liberación inmediata, el compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable como se ha descrito anteriormente, puede formularse con un diluyente o vehículo farmacéuticamente aceptable. Para la formulación de liberación sostenida o retardada, el compuesto del compuesto de Fórmula I, en forma libre o de sal farmacéuticamente aceptable como se ha descrito anteriormente, puede formularse como se ha descrito anteriormente en cualquiera de las Composiciones P.1-P.7.
En una realización particular, el Compuesto de Fórmula I se formula en una cápsula como se indica a continuación:

Para la administración bucal, la composición puede adoptar la forma de comprimidos o pastillas para chupar formuladas de manera convencional.
Los Compuestos de Fórmula I pueden formularse para su administración parenteral por inyección, incluyendo el uso de técnicas de cateterización convencionales o infusión. Las formulaciones para inyección pueden presentarse en forma de dosificación unitaria, por ejemplo, en ampollas o en envases multidosis, con un conservante añadido. Las composiciones pueden adoptar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilizantes y/o dispersantes. Como alternativa, el principio activo puede estar en forma de polvo para su reconstitución con un vehículo adecuado, p. ej., agua estéril libre de pirógenos, antes de su uso.
Los Compuestos de Fórmula I también pueden formularse en composiciones rectales tales como supositorios o enemas de retención, p. ej., que contengan bases para supositorio convencionales, tales como manteca de cacao u otros glicéridos.
Para la administración intranasal o la administración por inhalación, los Compuestos de Fórmula I se administran convenientemente en forma de solución o suspensión de un recipiente de pulverización de bomba que es apretado o bombeado por el paciente o como una presentación de pulverización en aerosol de un recipiente o nebulizador presurizado, con el uso de un propulsor adecuado, p. ej., diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado. En el caso de un aerosol presurizado, la unidad de dosificación puede determinarse dotándolo de una válvula para suministrar una cantidad medida. El recipiente o nebulizador presurizado puede contener una solución o suspensión del compuesto activo. Las cápsulas y los cartuchos (hechos, por ejemplo, de gelatina) para su uso en un inhalador o insuflador pueden formularse conteniendo una mezcla de polvo de un compuesto activo y una base de polvo adecuada, tal como lactosa o almidón. Cuando se desean suspensiones y/o elixires acuosos para administración oral, el(los) principios(s) activo(s) esencial(es) puede combinarse con varios agentes edulcorantes o aromatizantes, materia colorante o tintes y, si así se desea, también agentes emulsionantes y/o de suspensión, junto con diluyentes tales como agua, etanol, propilenglicol, glicerina y diversas combinaciones similares de los mismos.
Todas las referencias citadas anteriormente se incorporan por la presente en su totalidad.
El siguiente ejemplo es para ilustrar la invención.
EJEMPLO 1: Tratamiento de los síntomas agudos así como residuales de la esquizofrenia
A efectos de este ejemplo, el Compuesto A se refiere al compuesto de Fórmula I en donde X es -N(CH3)- e Y es -C(O)-; sal de tosilato a menos que se indique lo contrario. Se realiza un ensayo aleatorizado, doble ciego, controlado con placebo, en pacientes ingresados. Se realizan cuatro brazos de tratamiento, con una duración de 4 semanas, con dosificación QAM de la siguiente manera: 60 mg de Compuesto A; 120 mg de Compuesto A; control positivo (4 mg de Risperidona); y placebo. El resultado primario se mide por el cambio del inicio del estudio en la puntuación total de la Escala de Síndromes Positivos y Negativos (PANSS) en el día 28. Las mediciones secundarias exploran las características diferenciadoras clave.
El brazo de tratamiento con 60 mg de Compuesto A recibe 60 mg de Compuesto A una vez al día durante 28 días. El brazo de tratamiento con 120 mg de Compuesto A recibe 60 mg de compuesto A el día 1 del estudio, seguido de 120 mg de Compuesto A una vez al día durante 27 días. El brazo de tratamiento con risperidona recibe 2 mg de risperidona el día 1 del estudio, seguido de 4 mg de risperidona una vez al día durante 27 días. El brazo de tratamiento con placebo recibe placebo una vez al día durante 28 días. Todas las dosis se administran por la mañana con el desayuno.
Criterios clave de inclusión: Diagnóstico clínico de esquizofrenia de acuerdo con DSM-IV-TR confirmado por SCID-CT modificado. Puntuación de la Escala Breve de Evaluación Psiquiátrica (BPRS) de 40 o más (18 elementos puntuados de 1 a 7 cada uno). Puntuación mínima de 4 o superior en al menos dos de los siguientes elementos positivos: desconfianza, desorganización conceptual, conducta alucinatoria, contenido de pensamientos inusuales. El episodio exacerbado actual no dura más de 4 semanas. Una historial suficiente y/o un informante independiente deben verificar que el estado actual es un estado exacerbado para el individuo. Respuesta previa a la terapia antipsicótica en los últimos 5 años, definida como una disminución clínicamente significativa y documentada de los delirios y/o alucinaciones durante un episodio exacerbado documentado (y al menos 3 meses de exposición previa a la terapia antipsicótica).
El ensayo tiene una alta tasa de finalización de los sujetos (74 %) en comparación con una media del 62 % de finalización en otros ensayos de antipsicóticos de 4 semanas. El 19 % interrumpe el tratamiento del estudio (durante el día 1-28). El 7 % completa el tratamiento del estudio hasta el día 28, pero se pierde el seguimiento. A 60 mg, el ensayo muestra que el Compuesto A demuestra eficacia antipsicótica en el cambio total de la PANSS en el criterio de valoración principal respecto del inicio del estudio en el día 28:
Tabla 1

Para los pacientes con síntomas negativos prominentes al inicio del estudio (p. ej., pacientes con una puntuación de 4 o más en al menos 3 elementos de síntomas negativos al inicio del estudio), 60 mg del Compuesto A mejoran los síntomas negativos cualitativamente (véase la Figura 1).
En un subgrupo de pacientes con síntomas de depresión secundarios a la esquizofrenia medidos por la puntuación de corte para la depresión (Escala de Depresión de Calgary para la Esquizofrenia, CDSS, puntuación > 6) al inicio del estudio (que representa aproximadamente el 16 % de los pacientes del estudio), 60 mg de Compuesto A reduce significativamente la depresión medida por la CDSS (p = 0,044). El Compuesto A a 60 mg también mejora de forma contundente la PANSS total (véase la Figura 2) y los síntomas negativos (véase la Figura 3).
A 60 mg, el Compuesto A también demuestra mejorar significativamente el cambio del Factor Prosocial de la PANSS del inicio del estudio en comparación con el placebo (Véase la Figura 4).
EJEMPLOS 2-10: Preparación de formulaciones de microesferas inyectables de acción prolongada
Ejemplo 2: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,32-0,44 dl/g) de base libre del Compuesto A (Lote A)
A efectos de los Ejemplos 2-10, el Compuesto A se refiere al compuesto de Fórmula I donde X es -N(CH3) - e Y es -C(O) - en forma de base libre.
2 g de poli(alcohol vinílico) (PVA, 87-89 % hidrolizado, peso molecular típico 13.000-23.000) en 400 ml de agua desionizada se someten a ultrasonidos en un baño de ultrasonidos durante 5-10 min y luego se filtran para dar una solución acuosa de PVA al 0,5 %. El filtrado se transfiere a un matraz de fondo plano de 500 ml. Se disuelven 0,80 g de Compuesto A y 1,2 g de polímero de PLGA (PLA/PLG = 75/25, 0,32-0,44 dl/g, grupos terminales ácidos) en 15 ml de diclorometano. Esta solución se añade a la solución acuosa de PVA al 0,5 % gota a gota con agitación vigorosa. Se utiliza un agitador mecánico vertical y la velocidad de agitación es de unas 700 ppm durante la adición. Una vez completada la adición, la mezcla se agita a esta velocidad durante una hora y luego se agita a aproximadamente 550 ppm durante 4 horas. Se aplica una corriente de argón durante todo el proceso para promover la evaporación del diclorometano de la solución acuosa. Se apila un microtamiz con un tamaño de 75 pm sobre la parte superior de un microtamiz de 20 pm. La suspensión se vierte lentamente sobre los microtamices apilados y luego se lava con agua al menos cinco veces. Las microesferas recogidas en el microtamiz de 20 pm se transfieren a un tubo falcon
de 50 ml con agua desionizada y luego se liofilizan para obtener 1,43 g de microesferas de PLGA del Compuesto A con una distribución de tamaños de 20-60 pm. El contenido de fármaco en las microesferas obtenidas es de 37,5 %, según lo determinado por HPLC. La carga inicial de esta preparación es del 40 % (calculada sobre la base de 0,8 g de base libre del Compuesto A en 1,2 g del polímero de PLGA). La eficiencia de atrapamiento del fármaco es del 93,8 %.
Ejemplo 3: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,32-0,44 dl/g) de base libre del Compuesto A (Lote B)
Este lote de microesferas de PLGA del Compuesto A se prepara usando un procedimiento similar al descrito en el Ejemplo 2, salvo que se utiliza agua esterilizada en todo el proceso en lugar de agua desionizada y se esteriliza todos los instrumentos de laboratorio que están en contacto con las microesferas. Se obtienen 1,49 g de microesferas de PLGA del Compuesto A con una distribución de tamaño de 20-50 pm. El contenido de fármaco en las microesferas obtenidas es de 38 %, según lo determinado por HPLC. La carga inicial de fármaco para este lote es del 40 % y la eficiencia de atrapamiento del fármaco es del 85,8 %.
Ejemplo 4: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,89 dl/g) de base libre del Compuesto A (Lote C)
2 g de poli(alcohol vinílico) (PVA, 87-89 % hidrolizado, peso molecular típico 13000-23000) en 400 ml de agua esterilizada se someten a ultrasonidos en un baño de ultrasonidos durante 5-10 min y luego se filtran para dar una solución acuosa de PVA al 0,5 %. El filtrado se transfiere a un matraz de fondo plano de 500 ml. Se disuelven 0,80 g de Compuesto A y 1,2 g de polímero de PLGA (PLA/PLG = 75/25; 0,89 dl/g; PM: 159.000 d; grupos terminales éster) en 16 ml de diclorometano con tratamiento con ultrasonidos. Esta solución se añade a la solución acuosa de PVA al 0,5 % gota a gota con agitación vigorosa. Se utiliza un agitador mecánico vertical y la velocidad de agitación es ~ 700 ppm durante la adición. Una vez completada la adición, la mezcla se agita a esta velocidad durante una hora y luego se agita a aproximadamente 550 ppm durante 4 horas. Se aplica una corriente de argón durante todo el proceso para promover la evaporación del diclorometano de la solución acuosa. Se apila un microtamiz con un tamaño de 75 pm sobre la parte superior de un microtamiz de 20 pm. La suspensión se vierte lentamente sobre los microtamices apilados y luego se lava con agua al menos cinco veces. Las microesferas recogidas en el microtamiz de 20 pm se transfieren a un tubo falcon de 50 ml con agua desionizada y luego se liofilizan para obtener 0,78 g de microesferas de PLGA del Compuesto A con una distribución de tamaños de 20-70 pm. El contenido de fármaco en las microesferas obtenidas es de 36 %, según lo determinado por HPLC. La carga inicial de esta preparación es del 40 % (calculada sobre la base de 0,8 g de base libre del Compuesto A en 1,2 g del polímero de p LgA). La eficiencia de atrapamiento del fármaco es del 90 %.
Ejemplo 5: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,68 dl/g) de base libre del Compuesto A (Lote D)
Este lote de microesferas de PLGA del Compuesto A se prepara usando un procedimiento similar al descrito en el Ejemplo 4, excepto que se usa un nuevo tipo de polímero de PLGA (PLA/p Lg = 75/25; 0,68 dl/g; PM: 113.000 d; grupos terminales ácidos) en la preparación de las microesferas. Se obtienen 1,11 g de microesferas de PLGA del Compuesto A con una distribución de tamaño de 25-75 pm. El contenido de fármaco en las microesferas obtenidas es de 36 %, según lo determinado por HPLC. La carga inicial de fármaco para este lote es del 40 % y la eficiencia de atrapamiento del fármaco es del 90 %.
Ejemplo 6: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,68 dl/g) de base libre del Compuesto A (Lote E)
Este lote de microesferas de PLGA se prepara usando un procedimiento similar al descrito en el Ejemplo 4, salvo que en la preparación de las microesferas se usa un tipo diferente de polímero de PLGA (PLA/PLG = 75/25; 0,68 dl/g; PM: 113.000 d; grupos terminales ácidos) y un microtamiz con un tamaño de poro de 75 pm en la preparación de microesferas. Se obtienen 0,25 g de microesferas de PLGA del Compuesto A con una distribución de tamaño de 75-110 pm. El contenido de fármaco en las microesferas obtenidas es de 37 %, según lo determinado por HPLC. La carga inicial de fármaco para este lote es del 40 % y la eficiencia de atrapamiento del fármaco es del 93 %.
Ejemplo 7: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,68 dl/g) de base libre del Compuesto A (Lote F)
Este lote de microesferas de PLGA del Compuesto A se prepara usando un procedimiento similar al descrito en el Ejemplo 4, excepto que se usa un nuevo tipo de polímero de PLGA (PLA/p Lg = 75/25; 0,68 dl/g; PM: 113.000 d; grupos terminales ácidos) y microtamices con tamaños de poro de 53 pm y 106 pm, respectivamente, en la preparación de microesferas. Se obtienen 1,22 g de microesferas de PLGA con una distribución de tamaños de 52 101 pm. El contenido de fármaco en las microesferas obtenidas es de 37 %, según lo determinado por HPLC. La carga inicial de fármaco para este lote es del 40 % y la eficiencia de atrapamiento del fármaco es del 93 %.
Ejemplo 7-A: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,32-0,44 dl/g) de base libre del Compuesto A (Lote F)
2 g de poli(alcohol vinílico) (PVA, 87-89 % hidrolizado, peso molecular típico 13000-23000) en 400 ml de agua esterilizada se someten a ultrasonidos en un baño de ultrasonidos durante 5-10 min y luego se filtran para dar una solución acuosa de PVA al 0,5 %. El filtrado se transfiere a un matraz de fondo plano de 500 ml. Se disuelven 0,40 g de Compuesto A y 2,1 g de polímero de PLGA (PLA/PLG = 75/25; 0,32-0,44 dl/g; grupos terminales ácidos) en 21 ml de diclorometano con tratamiento con ultrasonidos. Esta solución se añade a la solución acuosa de PVA al 0,5 % gota a gota con agitación vigorosa. Se utiliza un agitador mecánico vertical y la velocidad de agitación es ~ 500 ppm durante la adición. Una vez completada la adición, la mezcla se agita a aproximadamente 500 ppm durante 5 horas. Se aplica una corriente de argón durante todo el proceso para promover la evaporación del diclorometano de la solución acuosa. Se apila un microtamiz con un tamaño de 75 pm sobre la parte superior de un microtamiz de 30 pm. La suspensión se vierte lentamente sobre los microtamices apilados y luego se lava con agua al menos cinco veces. Las microesferas recogidas en el microtamiz de 30 pm se transfieren a un vial de vidrio de 7,78 g (% oz) y luego se secan al vacío para obtener 1,43 g de microesferas de PLGA de base libre del Compuesto A con una distribución de tamaños de 25-70 pm. El contenido de fármaco en las microesferas obtenidas es de 8,5 %, según lo determinado por HPLC. La carga inicial de esta preparación es del 16 % (calculada sobre la base de 0,4 g de base libre del Compuesto A en 2,1 g del polímero de PLGA). La eficiencia de atrapamiento del fármaco es del 53 %.
Ejemplo 8: Preparación de las microesferas de PLGA (PLA/PLG = 75/25, 0,68 dl/g) de base libre del Compuesto B (Lote G)
A efectos de este ejemplo, el Compuesto B se refiere al compuesto de Fórmula I en donde X es -N(CH3) - e Y es -CH(OH)-. 2 g de poli(alcohol vinílico) (PVA, 87-89 % hidrolizado, peso molecular típico 13000-23000) en 400 ml de agua desionizada se someten a ultrasonidos en un baño de ultrasonidos durante 5-10 min y luego se filtran para dar una solución acuosa de PVA al 0,5 %. El filtrado se transfiere a un matraz de fondo plano de 500 ml. Se disuelven 0,80 g de Compuesto B (base libre) y 1,2 g de polímero de PLGA (PLA/PLG = 75/25, 0,32-0,44 dl/g, grupos terminales ácidos) en 15 ml de diclorometano. Esta solución se añade a la solución acuosa de PVA al 0,5 % gota a gota con agitación vigorosa. Se utiliza un agitador mecánico vertical y la velocidad de agitación es de unas 700 ppm durante la adición. Una vez completada la adición, la mezcla se agita a esta velocidad durante una hora y luego se agita a aproximadamente 550 ppm durante 4 horas. Se aplica una corriente de argón durante todo el proceso para promover la evaporación del diclorometano de la solución acuosa. Se apila un microtamiz con un tamaño de 75 pm sobre la parte superior de un microtamiz de 20 pm. La suspensión se vierte lentamente sobre los microtamices apilados y luego se lava con agua al menos cinco veces. Las microesferas recogidas en el microtamiz de 20 pm se transfieren a un tubo falcon de 50 ml con agua desionizada y luego se liofilizan para obtener 1,36 g de microesferas de PLGA del Compuesto B con una distribución de tamaños de 13-60 pm. El contenido de fármaco en las microesferas obtenidas es de 29 %, según lo determinado por HPLC. La carga inicial de esta preparación es del 40 % (calculada sobre la base de 0,8 g de base libre del Compuesto B en 1,2 g del polímero de PLGA). La eficiencia de atrapamiento del fármaco es del 72,5 %.
Ejemplo 9: Determinación de la carga de microesferas inyectables de acción prolongada
Se disuelven aproximadamente 5 mg de microesferas en 10 ml de una mezcla 1:2 v/v de diclorometano/acetonitrilo. Se transfiere 1 ml de la solución a un microtubo de 1,5 ml y se elimina el disolvente usando Savant Speed Vac; luego se reconstituye el residuo en 95 % de acetonitrilo/5 % de agua. Esta solución se filtra (filtro de jeringa de nylon de 0,2 pm de Waters) y las mediciones se llevan a cabo por triplicado a través de UPLC Waters Acquity con un detector de absorbancia UV PDA ajustado a 314 nm. La fase móvil es un gradiente acetonitrilo-agua con 0,1 % v/v de ácido fórmico. Se utiliza la columna Waters Acquity UPLC HSS T3 (2,1 X 50 mm) con un caudal de 0,3 ml/min. Se prepara una curva convencional para incluir aproximadamente 0,1-0,7 mg/ml de la concentración esperada del Compuesto A.
La carga de fármaco se determina como: porcentaje de carga de fármaco =100 x (10 x concentración de la solución filtrada)/peso de las microesferas utilizadas. Los resultados se presentan como la media ±DT.
Ejemplo 10: Determinación de la liberación del fármaco de microesferas inyectables de acción prolongada
Los experimentos de liberación de fármacos de microesferas se llevan a cabo en tampón fosfato 0,1 M (pH 7,4) que contiene ácido ascórbico 10 mM. Las suspensiones de microesferas que contienen cantidades conocidas de fármaco (aproximadamente 30 mg) se colocan en un dispositivo de diálisis de membrana de celulosa regenerada (Float-a-Lyzer). El Float-a-Lyzer se coloca en un tubo falcon de 45 ml que contiene 40 ml de tampón, el medio de liberación se mantiene a 37 °C y se agita horizontalmente a 100 rpm en un horno. El tampón se sustituye con una solución fresca a intervalos de tiempo predeterminados. El contenido de fármaco del medio de liberación se determina mediante UPLC utilizando la absorbancia UV-vis a 314 nm con una curva convencional construida con concentraciones conocidas de ITI07 (3~30 pg/ml).
Ejemplo 11: Farmacocinética de las microesferas inyectables de acción prolongada
Para la caracterización in vivo, se determina la liberación del fármaco in vivo (estudio farmacocinético). La farmacocinética de la formulación se estudia en ratas macho Sprague-Dawley adultas. A las ratas (N=12/experimento) se les inyecta por vía subcutánea en la región intraescapular una suspensión (2 ml/kg) de base libre del Compuesto A formulado en microesferas de PLGA biodegradables (aproximadamente 60 mg/rata) suspendidas en una solución de carboximetilcelulosa de baja viscosidad al 0,5 % en solución salina estéril que contiene 0,1 % de Tween-20. En momentos específicos después de la inyección (es decir, de 24 horas a 28 días), las ratas (N=2 o 3 por punto de tiempo) se someten a pruebas de comportamiento de sacudidas de cabeza inducido por el agonista de 5-HT2A como indicador funcional de la actividad del antagonista de 5-HT2A debido a los niveles circulantes del Compuesto A. Para estas mediciones, a las ratas se les inyecta por vía intraperitoneal 2, 5-dimetoxi-4-yodoanfetamina (DOI) (2,5 mg/kg) en un volumen de 2 ml/kg. Cinco minutos más tarde, se observa a las ratas en busca de comportamientos estereotípicos de sacudidas de cabeza, que se cuentan y registran manualmente. Las ratas se matan por decapitación en puntos de tiempo especificados (es decir, 24 h-28 días después de la inyección de la formulación) y se recoge sangre del tronco y tejido cerebral para analizar los niveles del Compuesto A y sus metabolitos conocidos (Compuesto B), usando un método HPLC-MS/MS.
Usando un procedimiento como el descrito o similarmente descrito anteriormente, se analizan varias formulaciones de microesferas inyectables de acción prolongada para su perfil de liberación in vivo del compuesto de Fórmula I en donde X es -N(c H3)- e Y es -C(O)-(base libre del Compuesto A). Los resultados se resumen en las siguientes tablas. La tabla 2 muestra las características físicas de las formulaciones de microesferas IAP 2-7 y 7A. La distribución del tamaño de las partículas se determina mediante fotomicroscopía.
Tabla 2

La carga real del fármaco describe el % peso/peso del Compuesto A en las microesferas. Las microesferas se dosifican a los animales a 60 mg/kg o a 30 mg/kg. La dosis de Compuesto A, en la Tabla 2, representa la cantidad de fármaco que se administra al animal (carga real de fármaco x dosis de microesferas). Los niveles plasmáticos y cerebrales del Compuesto A y del metabolito principal del Compuesto A (Compuesto B), se miden el día 1, día 3, día 7, día 10, día 14 y día 21 después de la inyección subcutánea en ratas. Los resultados se muestran en la Tabla 3.
Tabla 3

continuación

Se observa, inesperadamente, que se obtiene un perfil de liberación superior (liberación prolongada) usando una carga de microesferas más baja. La formulación de microesferas con una carga del 8,5 % en peso (Ej. 7-A) produce un perfil de liberación con un nivel cerebral máximo de fármaco en el día 7, con niveles medibles de fármaco hasta el día 21 del estudio (11 ng/ml de nivel cerebral en el día 21). En contraposición, las microesferas con mayor carga de fármaco dan lugar a una liberación máxima más temprana del fármaco, sin niveles medibles o con niveles bajos de fármaco que se mantienen más allá del día 10 del estudio.
Claims (18)
1. Un compuesto de Fórmula I:

en la que:
X es -O-, -NH- o -N(CH3)-; e
Y es -O- o -C(O)-;
en forma libre o de sal farmacéuticamente aceptable, opcionalmente en mezcla o asociación con un diluyente o un vehículo farmacéuticamente aceptables, para su uso en un método para el tratamiento de los síntomas residuales de la esquizofrenia seleccionados entre retraimiento emocional, retraimiento social pasivo o apático y evitación social activa, que comprende administrar a un paciente que lo necesite una cantidad eficaz del compuesto,
en donde el paciente es un paciente que no ha respondido o no ha respondido adecuadamente a un tratamiento con otro agente antipsicótico.
2. El compuesto para el uso de acuerdo con la reivindicación 1, en donde el compuesto de Fórmula I es un compuesto en donde:
X es -O-, -NH- o -N(CH3)-; e
Y es -C(O)-;
en forma libre o de sal farmacéuticamente aceptable.
3. El compuesto para el uso de acuerdo con la reivindicación 1, en donde el compuesto de Fórmula I se selecciona entre un grupo que consiste en compuestos de fórmula I en donde:
X es -N(CH3)- e Y es -C(O)-,
X es -N(CH3)- e Y es -O-,
X es -NH- e Y es -C(O)-,
X es -NH- e Y es -O-.
4. El compuesto para el uso de la reivindicación 1, en donde X es -N(CH3)- e Y es -C(O)- o -C(H)(OH).
5. El compuesto para el uso de cualquiera de las reivindicaciones anteriores, que comprende además la administración de uno o varios agentes terapéuticos, tales como agentes antipsicóticos y/o agentes antidepresivos y/o agentes hipnóticos adicionales.
6. El compuesto para el uso de cualquiera de las reivindicaciones 1-4, en donde la administración del compuesto de Fórmula I es complementaria a la administración del uno o varios agentes terapéuticos; o en donde la administración del uno o varios agentes terapéuticos es complementaria a la administración del compuesto de Fórmula I.
7. El compuesto para el uso de la reivindicación 5 o de la reivindicación 6, en donde los agentes terapéuticos se seleccionan entre compuestos que potencian la actividad de GABA y facilitan la transmisión de GABA, un agonista de GABA-B, un modulador de 5-HT, un agonista del receptor de melatonina, un modulador de canales iónicos, un antagonista del receptor/inhibidor de la recaptación de serotonina-2 (SARI), un antagonista del receptor de orexina, un agonista del receptor de H3, un antagonista del receptor noradrenérgico, un agonista del receptor de galanina, un antagonista del receptor de CRH, una hormona del crecimiento humano, un agonista del receptor de la hormona del crecimiento, un estrógeno, un agonista del receptor de estrógenos, un fármaco de neuroquinina-1; y agentes antipsicóticos, en forma libre o de sal farmacéuticamente aceptable.
8. El compuesto para el uso de la reivindicación 5 o de la reivindicación 6, en donde el uno o varios agentes terapéuticos se seleccionan entre el grupo que consiste en modafinilo, armodafinilo, doxepina, alprazolam, bromazepam, clobazam, clonazepam, clorazepato, diazepam, flunitrazepam, flurazepam, lorazepam, midazolam, nitrazepam, oxazepam, temazapam, triazolam, indiplon, zopiclona, eszopiclona, zaleplón, Zolpidem, gabaxadol, vigabatrina, tiagabina, EVT 201 (Evotec Pharmaceuticals), estazolam, ketanserina, risperidona, eplivanserina, volinanserina (Sanofi-Aventis, Francia), pruvanserina, MDL 100907 (Sanofi-Aventis, Francia), HY10275 (Eli Lilly), APD125 (Arena Pharmaceuticals, San Diego, CA), AVE8488 (Sanofi-Aventis, Francia), repinotán, sarizotán,
eptapirona, buspirona, MN-305 (MediciNova, San Diego, CA), melatonina, ramelteón (ROZEREM®, Takeda Pharmaceuticals, Japón), VEC-162 (Evotec Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery), agomelatina, lamotrigina, gabapentina, pregabalina, orexina, una 1,3-biarilurea, SB-334867-a (GlaxoSmithKline, R.U.), GW649868 (GlaxoSmithKline), un derivado de benzamida, Org 50081 (Organon - Países Bajos), ritanserina, nefazodona, serzone, trazodona, Casopitant (GlaxoSmithKline), amitriptilina, amoxapina, bupropión, citalopram, clomipramina, desipramina, doxepina, duloxetina, escitalopram, fluoxetina, fluvoxamina, imipramina, isocarboxazida, maprotilina, mirtazapina, nefazodona, nortriptilina, paroxetina, sulfato de fenelzina, protriptilina, sertralina, tranilcipromina, trazodona, trimipramina, venlafaxina, clorpromazina, haloperidol, droperidol, flufenazina, loxapina, mesoridazina, molindona, perfenazina, pimozida, proclorperazina, promazina, tioridazina, tiotixeno, trifluoperazina, clozapina, aripiprazol, olanzapina, quetiapina, risperidona, ziprasidona, paliperidona, asenapina, lurasidona, iloperidona, cariprazina, amisulprida, zotepina, sertindol, en forma libre o de sal farmacéuticamente aceptable.
9. El compuesto para el uso de la reivindicación 8, en donde el uno o varios agentes terapéuticos son agentes antipsicóticos seleccionados entre clorpromazina, haloperidol, droperidol, flufenazina, loxapina, mesoridazina, molindona, perfenazina, pimozida, proclorperazina, promazina, tioridazina, tiotixeno, trifluoperazina, clozapina, aripiprazol, olanzapina, quetiapina, risperidona, ziprasidona, paliperidona, asenapina, lurasidona, iloperidona, cariprazina, amisulprida, zotepina, sertindol, en forma libre o de sal farmacéuticamente aceptable.
10. El compuesto para el uso de la reivindicación 8, en donde el uno o varios agentes terapéuticos son agentes antidepresivos seleccionados entre uno o más de amitriptilina, amoxapina, bupropión, citalopram, clomipramina, desipramina, doxepina, duloxetina, escitalopram, fluoxetina, fluvoxamina, imipramina, isocarboxazida, maprotilina, mirtazapina, nefazodona, nortriptilina, paroxetina, sulfato de fenelzina, protriptilina, sertralina, tranilcipromina, trazodona, trimipramina, y venlafaxina.
11. El compuesto para el uso de la reivindicación 5, en donde el uno o varios agentes terapéuticos son agentes antidepresivos seleccionados entre inhibidores selectivos de la recaptación de serotonina (SSRIs), inhibidores de la recaptación de serotonina y de norepinefrina (SNRIs), y antidepresivos tricíclicos.
12. El compuesto para el uso de cualquiera de las reivindicaciones anteriores, en donde la cantidad eficaz del compuesto de Fórmula I es una dosis diaria de aproximadamente 1 mg a aproximadamente 140 mg.
13. El compuesto para el uso de cualquiera de las reivindicaciones anteriores, en donde el paciente no ha respondido adecuadamente a un tratamiento con otro agente antipsicótico o una combinación de agentes antipsicóticos seleccionados entre clorpromazina, haloperidol, droperidol, flufenazina, loxapina, mesoridazina, molindona, perfenazina, pimozida, proclorperazina, promazina, tioridazina, tiotixeno, trifluoperazina, clozapina, aripiprazol, olanzapina, quetiapina, risperidona, ziprasidona, paliperidona, asenapina, lurasidona, iloperidona, cariprazina, amisulprida, zotepina, sertindol.
14. El compuesto para el uso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde el compuesto de Fórmula I se administra por vía oral como una composición que comprende un diluyente o un vehículo farmacéuticamente aceptables como una forma de dosis unitaria oral que es un comprimido o una cápsula.
15. El compuesto para el uso de acuerdo con una cualquiera de las reivindicaciones anteriores, en donde el compuesto de Fórmula I está formulado en una formulación de liberación sostenida o retardada.
16. El compuesto para el uso de acuerdo con una cualquiera de las reivindicaciones 1 a 13, en donde el compuesto de Fórmula I está formulado en una formulación de liberación sostenida o retardada, que es una formulación de microesferas inyectables de acción prolongada y/o una composición inyectable de acción prolongada que comprende una matriz de PLGA con una relación PLA/PLG de 75:25 con grupos terminales de ácido carboxílico o éster carboxílico.
17. El compuesto para el uso de la reivindicación 15 o de la reivindicación 16, en donde el compuesto de Fórmula I se administra una o dos veces al mes como una formulación inyectable de acción prolongada, en donde la cantidad eficaz es de 100 mg al mes a 600 mg al mes.
18. El compuesto para el uso de la reivindicación 17, en donde la cantidad eficaz es de 150 mg al mes a 300 mg al mes.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361911416P | 2013-12-03 | 2013-12-03 | |
| US201461925607P | 2014-01-09 | 2014-01-09 | |
| US201461975702P | 2014-04-04 | 2014-04-04 | |
| US201462032326P | 2014-08-01 | 2014-08-01 | |
| PCT/US2014/068443 WO2015085004A1 (en) | 2013-12-03 | 2014-12-03 | Novel methods |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2893621T3 true ES2893621T3 (es) | 2022-02-09 |
Family
ID=53274093
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14867061T Active ES2893621T3 (es) | 2013-12-03 | 2014-12-03 | Métodos para el tratamiento de los síntomas residuales de la esquizofrenia |
Country Status (17)
| Country | Link |
|---|---|
| US (7) | US9956227B2 (es) |
| EP (2) | EP3076967B1 (es) |
| JP (4) | JP6475727B2 (es) |
| KR (8) | KR102372145B1 (es) |
| CN (2) | CN105939712A (es) |
| AU (4) | AU2014360452C1 (es) |
| BR (1) | BR112016012781A2 (es) |
| CA (4) | CA2932610C (es) |
| DK (1) | DK3076967T3 (es) |
| ES (1) | ES2893621T3 (es) |
| HU (1) | HUE056496T2 (es) |
| IL (3) | IL305990A (es) |
| MX (3) | MX2016007219A (es) |
| PL (1) | PL3076967T3 (es) |
| PT (1) | PT3076967T (es) |
| RU (1) | RU2682658C1 (es) |
| WO (1) | WO2015085004A1 (es) |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101999300B1 (ko) | 2007-03-12 | 2019-07-12 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | 치환된 헤테로환 융합 감마-카르볼린 합성 |
| US20110021538A1 (en) | 2008-04-02 | 2011-01-27 | Arena Pharmaceuticals, Inc. | Processes for the preparation of pyrazole derivatives useful as modulators of the 5-ht2a serotonin receptor |
| USRE48948E1 (en) | 2008-04-18 | 2022-03-01 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable polymer |
| USRE48839E1 (en) * | 2008-05-27 | 2021-12-07 | Intra-Cellular Therapies, Inc | Methods and compositions for sleep disorders and other disorders |
| EP2364142B1 (en) | 2008-10-28 | 2018-01-17 | Arena Pharmaceuticals, Inc. | Compositions of a 5-ht2a serotonin receptor modulator useful for the treatment of disorders related thereto |
| TR201910327T4 (tr) | 2009-10-23 | 2019-07-22 | Janssen Pharmaceutica Nv | Oreksin reseptör modülatörleri olarak di-ornatılmış oktahidropirrolo [3,4-c]pirroller. |
| RU2591194C2 (ru) | 2010-04-22 | 2016-07-10 | Интра-Селлулар Терапиз, Инк. | Органические соединения |
| WO2013155506A1 (en) | 2012-04-14 | 2013-10-17 | Intra-Cellular Therapies, Inc. | Novel compositions and methods |
| ES2846823T3 (es) | 2013-03-15 | 2021-07-29 | Intra Cellular Therapies Inc | Compuestos orgánicos |
| AU2014360452C1 (en) | 2013-12-03 | 2019-05-16 | Intra-Cellular Therapies, Inc. | Novel methods |
| BR112016023162B1 (pt) | 2014-04-04 | 2022-11-29 | Intra-Cellular Therapies, Inc | Compostos orgânicos gama-carbolinas, composições farmacêuticas compreendendo os ditos compostos e uso dos mesmos no tratamento ou na profilaxia de transtornos de sistema nervoso central |
| CN106456638A (zh) | 2014-04-04 | 2017-02-22 | 细胞内治疗公司 | 有机化合物 |
| CA3013771C (en) | 2015-02-27 | 2024-01-02 | Kindred Biosciences, Inc. | Stimulation of appetite, management of weight loss, and treatment of anorexia in dogs and cats |
| RU2018103338A (ru) * | 2015-07-15 | 2019-08-15 | Аксовант Сайенсиз Гмбх | Производные диарил- и арилгетероарилмочевины для профилактики и лечения галлюцинаций, ассоциированных с нейродегенеративным заболеванием |
| WO2017117514A1 (en) * | 2015-12-31 | 2017-07-06 | Tung Roger D | Deuterated iti-007 |
| CN105560192A (zh) * | 2016-01-07 | 2016-05-11 | 万全万特制药江苏有限公司 | 一种棕榈酸帕利哌酮长效微球注射液的制备方法 |
| PL3838274T3 (pl) | 2016-01-26 | 2024-04-02 | Intra-Cellular Therapies, Inc. | Pochodna pirydo[3',4':4,5]pirolo[1,2,3-DE]chinoksaliny do zastosowania w leczeniu zaburzeń OUN |
| SI3426251T1 (sl) | 2016-03-10 | 2022-07-29 | Janssen Pharmaceutica Nv | Postopki za zdravljenje depresije z antagonisti receptorja oreksina-2 |
| EP3888656A1 (en) | 2016-03-25 | 2021-10-06 | Intra-Cellular Therapies, Inc. | Deuterated heterocyclic gamma-carboline compounds and their use in the treatment or prophylaxis of a central nervous system disorder |
| JP2019513143A (ja) | 2016-03-28 | 2019-05-23 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | 新規塩類および結晶 |
| JP2019510039A (ja) * | 2016-03-28 | 2019-04-11 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | 新規組成物および方法 |
| JP6997718B2 (ja) * | 2016-03-28 | 2022-01-18 | イントラ-セルラー・セラピーズ・インコーポレイテッド | 新規共結晶 |
| CN110072518B (zh) | 2016-10-12 | 2021-10-26 | 细胞内治疗公司 | 无定形固体分散体 |
| TW201827051A (zh) * | 2016-12-02 | 2018-08-01 | 美商神經性分泌生物科學公司 | 戊苯那嗪(valbenazine)於治療精神分裂症或情感性精神分裂症之用途 |
| PL3338768T3 (pl) | 2016-12-20 | 2020-05-18 | Lts Lohmann Therapie-Systeme Ag | Transdermalny system terapeutyczny zawierający asenapinę |
| KR102614709B1 (ko) | 2016-12-20 | 2023-12-18 | 에르테에스 로만 테라피-시스테메 아게 | 아세나핀 및 폴리실록산 또는 폴리이소부틸렌을 함유하는 경피흡수 치료 시스템 |
| JP6987868B2 (ja) | 2016-12-29 | 2022-01-05 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | 有機化合物 |
| WO2018126140A1 (en) | 2016-12-29 | 2018-07-05 | Intra-Cellular Therapies, Inc. | Organic compounds |
| MX2019011329A (es) | 2017-03-24 | 2019-12-02 | Intra Celular Therapies Inc | Composiciones novedosas y metodos. |
| EP3644973B1 (en) | 2017-06-26 | 2021-03-24 | LTS LOHMANN Therapie-Systeme AG | Transdermal therapeutic system containing asenapine and silicone acrylic hybrid polymer |
| AU2018307479B2 (en) | 2017-07-26 | 2024-06-20 | Intra-Cellular Therapies, Inc. | Organic compounds |
| IL272252B2 (en) * | 2017-07-26 | 2024-03-01 | Intra Cellular Therapies Inc | Transformed histories of gamma-carbolines fused with heterocyclics, pharmaceutical preparations containing them and their use for treatment |
| US11440911B2 (en) | 2017-09-26 | 2022-09-13 | Intra-Cellular Therapies, Inc. | Salts and crystals |
| JP2021502959A (ja) | 2017-10-10 | 2021-02-04 | ニューロクライン バイオサイエンシーズ,インコーポレイテッド | 特定のvmat2インヒビターを投与するための方法 |
| KR102089737B1 (ko) | 2017-11-01 | 2020-03-16 | 한국화학연구원 | 에씨탈로프람을 함유한 미립구형 서방출 주사제 및 그의 제조방법 |
| EP3720435B1 (en) | 2017-12-05 | 2024-03-06 | Sunovion Pharmaceuticals Inc. | Crystal forms and production methods thereof |
| WO2019113079A1 (en) | 2017-12-05 | 2019-06-13 | Sunovion Pharmaceuticals Inc. | Nonracemic mixtures and uses thereof |
| US11980617B2 (en) * | 2018-03-16 | 2024-05-14 | Intra-Cellular Therapies, Inc. | Methods of treating acute depression and/or acute anxiety |
| CA3102948A1 (en) | 2018-06-08 | 2019-12-12 | Intra-Cellular Therapies, Inc. | Novel methods |
| KR20210031455A (ko) | 2018-06-11 | 2021-03-19 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | 치환된 헤테로환 융합 감마-카르볼린 합성 |
| CA3101420A1 (en) | 2018-06-20 | 2019-12-26 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system containing asenapine |
| BR112021001738A2 (pt) | 2018-06-21 | 2021-06-29 | Aquestive Therapeutics, Inc. | sistema e método para fabricação de doses unitárias individuais personalizadas contendo ativos farmacêuticos |
| AU2019328528A1 (en) * | 2018-08-31 | 2021-03-18 | Intra-Cellular Therapies, Inc. | Novel methods |
| MX2021002321A (es) | 2018-08-31 | 2021-04-28 | Intra Cellular Therapies Inc | Nuevos metodos. |
| EP3846778A1 (en) | 2018-09-07 | 2021-07-14 | Aquestive Therapeutics, Inc. | Oral film compositions and dosage forms having precise active dissolution profiles |
| KR20200087594A (ko) | 2019-01-11 | 2020-07-21 | 한국화학연구원 | 레티노익산을 포함하는 미립구형 서방출 제제 및 그의 제조방법 |
| US11160758B2 (en) | 2019-06-04 | 2021-11-02 | Sunovion Pharmaceuticals Inc. | Modified release formulations and uses thereof |
| KR20220029744A (ko) * | 2019-07-07 | 2022-03-08 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | 신규 방법 |
| BR112022008294A2 (pt) | 2019-11-01 | 2022-07-26 | Aquestive Therapeutics Inc | Composições de profármaco e métodos de tratamento |
| KR20220126287A (ko) | 2019-11-14 | 2022-09-15 | 어퀘스티브 테라퓨틱스, 아이엔씨. | 멀티모달 조성물 및 치료 방법 |
| EP4072554A4 (en) * | 2019-12-11 | 2023-12-20 | Intra-Cellular Therapies, Inc. | ORGANIC COMPOUND |
| US11344503B2 (en) * | 2019-12-13 | 2022-05-31 | Halo Science LLC | Cariprazine release formulations |
| KR102383448B1 (ko) | 2020-04-20 | 2022-04-06 | 한국화학연구원 | 락트산 또는 글리콜산을 포함하는 미립구형 서방출 제제 및 그의 제조방법 |
| JP2024505429A (ja) | 2021-01-15 | 2024-02-06 | アクエスティブ セラピューティクス インコーポレイテッド | プロドラッグ組成物及び治療の方法 |
| AU2022226584A1 (en) * | 2021-02-24 | 2023-08-17 | Oakwood Laboratories, Llc | Microsphere formulations comprising lurasidone and methods for making and using the same |
| WO2022192476A1 (en) | 2021-03-09 | 2022-09-15 | Aquestive Therapeutics, Inc. | Dosage forms having equivalent biocomparable profiles |
| AU2023204432A1 (en) * | 2022-01-03 | 2024-07-18 | Engrail Therapeutics, Inc. | Deuterated organic compounds and uses thereof |
| WO2023225620A1 (en) | 2022-05-18 | 2023-11-23 | Intra-Cellular Therapies, Inc. | Novel methods |
Family Cites Families (120)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2490813A (en) | 1944-11-29 | 1949-12-13 | Standard Oil Co | Continuous process for making aryl amines |
| US3299078A (en) | 1962-10-01 | 1967-01-17 | Smith Kline French Lab | Pyrido [3', 4': 4, 5] pyrrolo [3, 2, 1-hi] indoles and-[3, 2, 1-ij] quinolines |
| US3813392A (en) | 1969-06-09 | 1974-05-28 | J Sellstedt | Pyrrolo(1,2,3-alpha epsilon)quinoxalin-2(3h)-ones and related compounds |
| US4238607A (en) | 1972-06-19 | 1980-12-09 | Endo Laboratories Inc. | Pyridopyrrolo benzheterocycles |
| US4183936A (en) | 1972-06-19 | 1980-01-15 | Endo Laboratories, Inc. | Pyridopyrrolobenzheterocycles |
| US4115577A (en) | 1972-06-19 | 1978-09-19 | Endo Laboratories, Inc. | Pyridopyrrolobenzheterocycles |
| US3914421A (en) | 1972-06-19 | 1975-10-21 | Endo Lab | Pyridopyrrolobenzheterocycles for combatting depression |
| IE41352B1 (en) | 1974-04-01 | 1979-12-19 | Pfizer | 5-aryl-1,2,3,4-tetrahydro- -carbolines |
| US4001263A (en) | 1974-04-01 | 1977-01-04 | Pfizer Inc. | 5-Aryl-1,2,3,4-tetrahydro-γ-carbolines |
| US4219550A (en) | 1978-11-09 | 1980-08-26 | E. I. Du Pont De Nemours And Company | Cis- and trans- octahydropyridopyrrolobenzheterocycles |
| US4389330A (en) | 1980-10-06 | 1983-06-21 | Stolle Research And Development Corporation | Microencapsulation process |
| IE52535B1 (en) | 1981-02-16 | 1987-12-09 | Ici Plc | Continuous release pharmaceutical compositions |
| US4530840A (en) | 1982-07-29 | 1985-07-23 | The Stolle Research And Development Corporation | Injectable, long-acting microparticle formulation for the delivery of anti-inflammatory agents |
| US4522944A (en) | 1982-12-23 | 1985-06-11 | Erba Farmitalia | Carboxamido-derivatives of 5H-1,3,4-thiadiazolo[3,2-a]pyrimidines, compositions and use |
| CH656884A5 (de) | 1983-08-26 | 1986-07-31 | Sandoz Ag | Polyolester, deren herstellung und verwendung. |
| ZA871987B (en) | 1986-03-19 | 1988-05-25 | Kumiai Chemical Industry Co | 5h-1,3,4-thiadiazole(3,2-a)pyrimidin-5-one derivatives and fungicidal compositions containing the same |
| EP0242690B1 (en) | 1986-04-07 | 1993-06-30 | Kumiai Chemical Industry Co., Ltd. | 5H-1,3,4-Thiadiazolo[3,2-a]pyrimidin-5-one derivatives and agricultural-horticultural fungicide composition containing the same |
| HU208484B (en) | 1988-08-17 | 1993-11-29 | Chinoin Gyogyszer Es Vegyeszet | Process for producing pharmaceutical composition containing acid additional salt of selegilin as active component for treating schisofrenia |
| US5114976A (en) | 1989-01-06 | 1992-05-19 | Norden Michael J | Method for treating certain psychiatric disorders and certain psychiatric symptoms |
| US5538739A (en) | 1989-07-07 | 1996-07-23 | Sandoz Ltd. | Sustained release formulations of water soluble peptides |
| IT1271352B (it) | 1993-04-08 | 1997-05-27 | Boehringer Ingelheim Italia | Derivati dell'indolo utili nel trattamento dei disturbi del sistema nervoso centrale |
| JP3645906B2 (ja) | 1993-11-19 | 2005-05-11 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | マイクロカプセル封入された3−ピペリジニル−置換1,2−ベンズイソオキサゾール類及び1,2−ベンズイソチアゾール類 |
| ATE372966T1 (de) | 1994-03-25 | 2007-09-15 | Isotechnika Inc | Verbesserung der effektivität von arzneimitteln duren deuterierung |
| US5576460A (en) | 1994-07-27 | 1996-11-19 | Massachusetts Institute Of Technology | Preparation of arylamines |
| US5654482A (en) | 1996-02-29 | 1997-08-05 | Xerox Corporation | Triarylamine processes |
| US5648539A (en) | 1996-02-29 | 1997-07-15 | Xerox Corporation | Low temperature arylamine processes |
| US5648542A (en) | 1996-02-29 | 1997-07-15 | Xerox Corporation | Arylamine processes |
| US5847166A (en) | 1996-10-10 | 1998-12-08 | Massachusetts Institute Of Technology | Synthesis of aryl ethers |
| US5705697A (en) | 1997-01-30 | 1998-01-06 | Xerox Corporation | Arylamine processes |
| US5723669A (en) | 1997-01-30 | 1998-03-03 | Xerox Corporation | Arylamine processes |
| US5723671A (en) | 1997-01-30 | 1998-03-03 | Xerox Corporation | Arylamine processes |
| TWI242011B (en) | 1997-03-31 | 2005-10-21 | Eisai Co Ltd | 1,4-substituted cyclic amine derivatives |
| US6323366B1 (en) | 1997-07-29 | 2001-11-27 | Massachusetts Institute Of Technology | Arylamine synthesis |
| GB2328686B (en) | 1997-08-25 | 2001-09-26 | Sankio Chemical Co Ltd | Method for producing arylamine |
| US6884429B2 (en) | 1997-09-05 | 2005-04-26 | Isotechnika International Inc. | Medical devices incorporating deuterated rapamycin for controlled delivery thereof |
| US6395939B1 (en) | 1997-10-06 | 2002-05-28 | Massachusetts Institute Of Technology | Diaryl ether condensation reactions |
| CA2322194C (en) | 1998-02-26 | 2011-04-26 | Massachusetts Institute Of Technology | Metal-catalyzed arylations and vinylations of hydrazines, hydrazones, hydroxylamines and oximes |
| US6235936B1 (en) * | 1998-02-26 | 2001-05-22 | Massachusetts Institute Of Technology | Metal-catalyzed arylations of hydrazines, hydrazones, and related substrates |
| US5902901A (en) | 1998-05-07 | 1999-05-11 | Xerox Corporation | Arylamine processes |
| US6395916B1 (en) | 1998-07-10 | 2002-05-28 | Massachusetts Institute Of Technology | Ligands for metals and improved metal-catalyzed processes based thereon |
| US6307087B1 (en) | 1998-07-10 | 2001-10-23 | Massachusetts Institute Of Technology | Ligands for metals and improved metal-catalyzed processes based thereon |
| US7223879B2 (en) | 1998-07-10 | 2007-05-29 | Massachusetts Institute Of Technology | Ligands for metals and improved metal-catalyzed processes based thereon |
| DE69929609T2 (de) | 1998-07-10 | 2006-09-28 | Massachusetts Institute Of Technology, Cambridge | Liganden für metalle und metall-katalysiertes verfahren |
| US20010008942A1 (en) | 1998-12-08 | 2001-07-19 | Buchwald Stephen L. | Synthesis of aryl ethers |
| CN1161101C (zh) | 1998-12-17 | 2004-08-11 | 阿尔扎有限公司 | 用多层包衣将填充液体的明胶胶囊转变成控制释放的系统 |
| NO309305B1 (no) | 1999-02-19 | 2001-01-15 | Norsk Hydro As | Anvendelse av benzaldehydderivater ved fremstilling av farmasöytiske preparater for forebygging og/eller behandling av kreft, samt visse nye benzaldehydderivater |
| US6407092B1 (en) | 1999-04-23 | 2002-06-18 | Pharmacia & Upjohn Company | Tetracyclic azepinoindole compounds |
| AR023574A1 (es) | 1999-04-23 | 2002-09-04 | Pharmacia & Upjohn Co Llc | Compuestos de azepinindol tetraciclico,composiciones farmaceuticas y el uso de dichos compuestos para preparar un medicamento, e intermediarios |
| US7071186B2 (en) | 1999-06-15 | 2006-07-04 | Bristol-Myers Squibb Pharma Co. | Substituted heterocycle fused gamma-carbolines |
| US6713471B1 (en) | 1999-06-15 | 2004-03-30 | Bristol-Myers Squibb Pharma Company | Substituted heterocycle fused gamma-carbolines |
| MXPA01012969A (es) | 1999-06-15 | 2003-10-14 | Bristol Myers Squibb Pharma Co | Gamma-carbolinas fusionadas de heterociclo sustituido. |
| US6541639B2 (en) | 2000-07-26 | 2003-04-01 | Bristol-Myers Squibb Pharma Company | Efficient ligand-mediated Ullmann coupling of anilines and azoles |
| US6849619B2 (en) | 2000-12-20 | 2005-02-01 | Bristol-Myers Squibb Company | Substituted pyridoindoles as serotonin agonists and antagonists |
| US6699852B2 (en) | 2000-12-20 | 2004-03-02 | Bristol-Myers Squibb Pharma Company | Substituted pyridoindoles as serotonin agonists and antagonists |
| US6759554B2 (en) | 2001-04-24 | 2004-07-06 | Massachusetts Institute Of Technology | Copper-catalyzed formation of carbon-heteroatom and carbon-carbon bonds |
| DE10123129A1 (de) | 2001-05-02 | 2002-11-14 | Berolina Drug Dev Ab Svedala | Deuterierte 3-Piperidinopropiophenone sowie diese Verbindungen enthaltende Arzneimittel |
| WO2003014118A1 (en) | 2001-08-08 | 2003-02-20 | Pharmacia & Upjohn Company | THERAPEUTIC 1H-PYRIDO[4,3-b]INDOLES |
| EP1314554A1 (fr) | 2001-11-23 | 2003-05-28 | Kba-Giori S.A. | Dispositif de décollage d'éléments de sécurité |
| DE10162121A1 (de) | 2001-12-12 | 2003-06-18 | Berolina Drug Dev Ab Svedala | Deuterierte substituierte Pyrazolyl-Benzolsulfonamide sowie diese Verbindungen enthaltende Arzneimittel |
| US20050232995A1 (en) | 2002-07-29 | 2005-10-20 | Yam Nyomi V | Methods and dosage forms for controlled delivery of paliperidone and risperidone |
| RU2321391C2 (ru) | 2002-07-29 | 2008-04-10 | Алза Корпорейшн | Способы и лекарственные формы для контролируемой доставки палиперидона |
| KR20050032107A (ko) | 2002-08-02 | 2005-04-06 | 메사추세츠 인스티튜트 오브 테크놀로지 | 구리를 촉매로 한 탄소-헤테로원자 결합 및 탄소-탄소결합 형성방법 |
| US7223870B2 (en) | 2002-11-01 | 2007-05-29 | Pfizer Inc. | Methods for preparing N-arylated oxazolidinones via a copper catalyzed cross coupling reaction |
| US20040142970A1 (en) | 2002-11-01 | 2004-07-22 | Kathryn Chung | Treatment of hyperkinetic movement disorder with donepezil |
| EP1581221B1 (en) | 2002-12-19 | 2011-05-18 | Bristol-Myers Squibb Company | Substituted tricyclic gamma-carbolines as serotonin receptor agonists and antagonists |
| US7601740B2 (en) * | 2003-01-16 | 2009-10-13 | Acadia Pharmaceuticals, Inc. | Selective serotonin 2A/2C receptor inverse agonists as therapeutics for neurodegenerative diseases |
| AU2004259741A1 (en) | 2003-07-21 | 2005-02-03 | Smithkline Beecham Corporation | (2S,4S)-4-fluoro-1-[4-fluoro-beta-(4-fluorophenyl)-L-phenylalanyl]-2-pyrrolidinecarbonitrile p-toluenesulfonic acid salt and anhydrous crystalline forms thereof |
| SA04250304B1 (ar) | 2003-09-26 | 2009-03-11 | سولفاي فارماسوتيكالز بي . في | مشتقات هكسا - واوكتاهيدرو - بيريدو {1،2-a} بيرازين ذات نشاط مضاد لـ nk1 |
| JP2005259113A (ja) | 2004-02-12 | 2005-09-22 | Ricoh Co Ltd | プロセス編集装置、プロセス管理装置、プロセス編集プログラム、プロセス管理プログラム、記録媒体、プロセス編集方法及びプロセス管理方法 |
| WO2005095396A1 (en) | 2004-03-05 | 2005-10-13 | Pharma C S.A. | 8-phenoxy-ϝ carboline derivatives |
| US20050222209A1 (en) | 2004-04-01 | 2005-10-06 | Zeldis Jerome B | Methods and compositions for the treatment, prevention or management of dysfunctional sleep and dysfunctional sleep associated with disease |
| US7592454B2 (en) | 2004-04-14 | 2009-09-22 | Bristol-Myers Squibb Company | Substituted hexahydro-pyridoindole derivatives as serotonin receptor agonists and antagonists |
| JP5289765B2 (ja) | 2004-09-20 | 2013-09-11 | マウント シナイ スクール オブ メディシン | 自閉症、強迫神経症、および衝動性の治療のためのメマンチン(ナメンダ)の使用 |
| BRPI0515528A (pt) | 2004-09-21 | 2008-07-29 | Pfizer Prod Inc | n-metil hidroxietilamina úteis no tratamento de condições do sistema nervoso central |
| US7614727B2 (en) | 2004-09-30 | 2009-11-10 | Fujifilm Corporation | Liquid ejection head, manufacturing method thereof, and image forming apparatus |
| JP4709849B2 (ja) | 2004-12-15 | 2011-06-29 | エフ.ホフマン−ラ ロシュ アーゲー | アルツハイマー病治療のためのグリシントランスポーターi(glyt−1)インヒビターとしての二環式及び三環式置換フェニルメタノン類 |
| CA2595607C (en) * | 2005-01-25 | 2014-07-15 | Epix Delaware, Inc. | Substituted arylamine compounds and their use as 5-ht6 modulators |
| WO2006081251A2 (en) | 2005-01-25 | 2006-08-03 | Celgene Corporation | Methods and compositions using 4-amino-2-(3-methyl-2,6-dioxopiperidin-3-yl)-isoindole-1-3-dione |
| US20090104274A1 (en) * | 2005-03-01 | 2009-04-23 | Ajay Khopade | Process of making microspheres |
| EP1919287A4 (en) | 2005-08-23 | 2010-04-28 | Intra Cellular Therapies Inc | ORGANIC COMPOUNDS FOR TREATING A REDUCED DOPAMINE RECEPTOR SIGNALING ACTIVITY |
| CN101309917B (zh) | 2005-10-06 | 2013-09-11 | 奥斯拜客斯制药有限公司 | 具有增强治疗性质的胃h+,k+-atp酶氘代抑制剂 |
| KR20080114688A (ko) | 2006-01-13 | 2008-12-31 | 와이어쓰 | 5-히드록시트립타민 수용체에 대한 리간드로서의 술포닐 치환된 1h-인돌 |
| US7750168B2 (en) | 2006-02-10 | 2010-07-06 | Sigma-Aldrich Co. | Stabilized deuteroborane-tetrahydrofuran complex |
| AU2007292848A1 (en) | 2006-09-08 | 2008-03-13 | Braincells, Inc. | Combinations containing a 4-acylaminopyridine derivative |
| KR101999300B1 (ko) | 2007-03-12 | 2019-07-12 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | 치환된 헤테로환 융합 감마-카르볼린 합성 |
| US20110269777A1 (en) | 2007-08-01 | 2011-11-03 | Medivation Neurology, Inc. | Methods and compositions for treating schizophrenia using antipsychotic combination therapy |
| ES2421237T7 (es) | 2007-08-15 | 2013-09-30 | Arena Pharmaceuticals, Inc. | Derivados de imidazo[1,2-a]piridin como moduladores del receptor serotoninérgico 5ht2a en el tratamiento de trastornos relacionados con el mismo |
| US20090209608A1 (en) | 2007-08-29 | 2009-08-20 | Protia, Llc | Deuterium-enriched asenapine |
| US20090076159A1 (en) | 2007-09-19 | 2009-03-19 | Protia, Llc | Deuterium-enriched eplivanserin |
| EP2262502A4 (en) | 2008-02-05 | 2011-12-28 | Clera Inc | COMPOSITIONS AND METHODS FOR RELIEVING DEPRESSION OR ENHANCING COGNITIVE PROCESSES |
| CN101986784A (zh) | 2008-02-07 | 2011-03-16 | 先灵公司 | 基因工程tslpr抗体 |
| US8648077B2 (en) * | 2008-03-12 | 2014-02-11 | Intra-Cellular Therapies, Inc. | 4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)-1-(4-fluorophenyl)-1-butanone toluenesulfonic acid addition salt and salt crystals |
| USRE48839E1 (en) * | 2008-05-27 | 2021-12-07 | Intra-Cellular Therapies, Inc | Methods and compositions for sleep disorders and other disorders |
| US8309772B2 (en) | 2008-07-31 | 2012-11-13 | Celanese International Corporation | Tunable catalyst gas phase hydrogenation of carboxylic acids |
| US20100159033A1 (en) | 2008-09-29 | 2010-06-24 | Auspex Pharmaceuticals, Inc. | Benzisoxazole modulators of d2 receptor, and/or 5-ht2a receptor |
| WO2010037142A1 (en) * | 2008-09-29 | 2010-04-01 | The Corporation Of Mercer University | Nanospheres encapsulating bioactive material and method for formulation of nanospheres |
| CN102421418A (zh) * | 2009-03-09 | 2012-04-18 | 得克萨斯系统大学评议会 | 在药物递送中有用的中空金纳米球(HAuNSs)和装载HAuNSs的微球体 |
| RU2591194C2 (ru) * | 2010-04-22 | 2016-07-10 | Интра-Селлулар Терапиз, Инк. | Органические соединения |
| CA3115378A1 (en) | 2010-12-16 | 2012-06-21 | Sunovion Pharmaceuticals Inc. | Sublingual films comprising apomorphine and an organic base |
| WO2013155506A1 (en) | 2012-04-14 | 2013-10-17 | Intra-Cellular Therapies, Inc. | Novel compositions and methods |
| BR112015005743A2 (pt) | 2012-09-14 | 2017-07-04 | Abbvie Deutschland | derivados tricíclicos de quinoxalina e quinolina |
| ES2846823T3 (es) * | 2013-03-15 | 2021-07-29 | Intra Cellular Therapies Inc | Compuestos orgánicos |
| AU2014360452C1 (en) * | 2013-12-03 | 2019-05-16 | Intra-Cellular Therapies, Inc. | Novel methods |
| CN106456638A (zh) | 2014-04-04 | 2017-02-22 | 细胞内治疗公司 | 有机化合物 |
| BR112016023162B1 (pt) * | 2014-04-04 | 2022-11-29 | Intra-Cellular Therapies, Inc | Compostos orgânicos gama-carbolinas, composições farmacêuticas compreendendo os ditos compostos e uso dos mesmos no tratamento ou na profilaxia de transtornos de sistema nervoso central |
| US10179776B2 (en) | 2014-06-09 | 2019-01-15 | Intra-Cellular Therapies, Inc. | Compounds and methods of use to treat schizophrenia |
| AU2016272089B2 (en) | 2015-06-03 | 2021-02-18 | Triastek, Inc. | Dosage forms and use thereof |
| PL3838274T3 (pl) | 2016-01-26 | 2024-04-02 | Intra-Cellular Therapies, Inc. | Pochodna pirydo[3',4':4,5]pirolo[1,2,3-DE]chinoksaliny do zastosowania w leczeniu zaburzeń OUN |
| EP3888656A1 (en) | 2016-03-25 | 2021-10-06 | Intra-Cellular Therapies, Inc. | Deuterated heterocyclic gamma-carboline compounds and their use in the treatment or prophylaxis of a central nervous system disorder |
| WO2017165755A1 (en) | 2016-03-25 | 2017-09-28 | Intra-Cellular Therapies, Inc. | Organic compounds |
| JP6997718B2 (ja) * | 2016-03-28 | 2022-01-18 | イントラ-セルラー・セラピーズ・インコーポレイテッド | 新規共結晶 |
| WO2018106916A1 (en) | 2016-12-07 | 2018-06-14 | Concert Pharmaceuticals, Inc. | Deuterated quinoxaline compounds |
| MX2019011329A (es) * | 2017-03-24 | 2019-12-02 | Intra Celular Therapies Inc | Composiciones novedosas y metodos. |
| EP3717484A4 (en) | 2017-11-27 | 2021-08-04 | Egis Gyógyszergyár Zrt. | PROCESS FOR THE PRODUCTION OF LUMATEPERON AND ITS SALT |
| EP3746081A4 (en) * | 2018-01-31 | 2021-10-27 | Intra-Cellular Therapies, Inc. | INNOVATIVE USES |
| US11980617B2 (en) * | 2018-03-16 | 2024-05-14 | Intra-Cellular Therapies, Inc. | Methods of treating acute depression and/or acute anxiety |
| US20210009592A1 (en) * | 2018-03-23 | 2021-01-14 | Intra-Cellular Therapies, Inc. | Organic compounds |
| JP2022500362A (ja) * | 2018-08-29 | 2022-01-04 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | 新規組成物および方法 |
| US20220362241A1 (en) * | 2019-09-25 | 2022-11-17 | Intra-Cellular Therapies, Inc. | Novel methods |
| WO2023225620A1 (en) * | 2022-05-18 | 2023-11-23 | Intra-Cellular Therapies, Inc. | Novel methods |
-
2014
- 2014-12-03 AU AU2014360452A patent/AU2014360452C1/en active Active
- 2014-12-03 DK DK14867061.5T patent/DK3076967T3/da active
- 2014-12-03 MX MX2016007219A patent/MX2016007219A/es unknown
- 2014-12-03 EP EP14867061.5A patent/EP3076967B1/en not_active Revoked
- 2014-12-03 CA CA2932610A patent/CA2932610C/en active Active
- 2014-12-03 KR KR1020217012502A patent/KR102372145B1/ko active IP Right Grant
- 2014-12-03 CN CN201480074647.7A patent/CN105939712A/zh active Pending
- 2014-12-03 PL PL14867061T patent/PL3076967T3/pl unknown
- 2014-12-03 KR KR1020237014108A patent/KR20230058736A/ko active Application Filing
- 2014-12-03 MX MX2021007665A patent/MX2021007665A/es unknown
- 2014-12-03 IL IL305990A patent/IL305990A/en unknown
- 2014-12-03 KR KR1020247025920A patent/KR20240119197A/ko active Search and Examination
- 2014-12-03 KR KR1020237003673A patent/KR102692169B1/ko active IP Right Grant
- 2014-12-03 CA CA3148303A patent/CA3148303C/en active Active
- 2014-12-03 JP JP2016536205A patent/JP6475727B2/ja active Active
- 2014-12-03 BR BR112016012781A patent/BR112016012781A2/pt not_active Application Discontinuation
- 2014-12-03 KR KR1020227023990A patent/KR102495941B1/ko active IP Right Grant
- 2014-12-03 KR KR1020217024479A patent/KR102421656B1/ko active IP Right Grant
- 2014-12-03 ES ES14867061T patent/ES2893621T3/es active Active
- 2014-12-03 EP EP20153731.3A patent/EP3666271A1/en active Pending
- 2014-12-03 US US15/101,874 patent/US9956227B2/en active Active
- 2014-12-03 CA CA3148326A patent/CA3148326A1/en active Pending
- 2014-12-03 CN CN202210062907.5A patent/CN114366741A/zh active Pending
- 2014-12-03 PT PT14867061T patent/PT3076967T/pt unknown
- 2014-12-03 KR KR1020167017585A patent/KR102287288B1/ko active IP Right Grant
- 2014-12-03 RU RU2016126445A patent/RU2682658C1/ru active
- 2014-12-03 CA CA3205777A patent/CA3205777A1/en active Pending
- 2014-12-03 KR KR1020217012501A patent/KR102373288B1/ko active IP Right Grant
- 2014-12-03 WO PCT/US2014/068443 patent/WO2015085004A1/en active Application Filing
- 2014-12-03 HU HUE14867061A patent/HUE056496T2/hu unknown
- 2014-12-03 IL IL293523A patent/IL293523B2/en unknown
- 2014-12-03 IL IL246002A patent/IL246002B/en unknown
-
2016
- 2016-06-02 MX MX2022011140A patent/MX2022011140A/es unknown
-
2018
- 2018-03-12 US US15/918,955 patent/US10322134B2/en active Active
-
2019
- 2019-01-17 AU AU2019200301A patent/AU2019200301B2/en active Active
- 2019-02-01 JP JP2019016947A patent/JP6874028B2/ja active Active
- 2019-04-23 US US16/392,409 patent/US10960009B2/en active Active
- 2019-06-17 US US16/443,240 patent/US10960010B2/en active Active
- 2019-07-10 US US16/507,956 patent/US11026951B2/en active Active
-
2020
- 2020-04-03 AU AU2020202386A patent/AU2020202386C1/en active Active
-
2021
- 2021-02-12 JP JP2021021096A patent/JP7262495B2/ja active Active
- 2021-04-30 US US17/245,939 patent/US20210322433A1/en active Pending
- 2021-12-22 AU AU2021290277A patent/AU2021290277B2/en active Active
-
2023
- 2023-04-11 JP JP2023064176A patent/JP2023089113A/ja active Pending
- 2023-10-04 US US18/481,168 patent/US20240024334A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2893621T3 (es) | Métodos para el tratamiento de los síntomas residuales de la esquizofrenia |