ES2864035T3 - Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus - Google Patents
Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus Download PDFInfo
- Publication number
- ES2864035T3 ES2864035T3 ES20171633T ES20171633T ES2864035T3 ES 2864035 T3 ES2864035 T3 ES 2864035T3 ES 20171633 T ES20171633 T ES 20171633T ES 20171633 T ES20171633 T ES 20171633T ES 2864035 T3 ES2864035 T3 ES 2864035T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- ethyl
- pharmaceutically acceptable
- formula
- imidazo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 **(*)OC(*(CC1)CC(*=C2C(c(c3c4)ccc4-c4cc(*)c(*)cc4*)=**3I)=C1*2I)=O Chemical compound **(*)OC(*(CC1)CC(*=C2C(c(c3c4)ccc4-c4cc(*)c(*)cc4*)=**3I)=C1*2I)=O 0.000 description 3
- CFDDUWYBUWFMRA-UHFFFAOYSA-N CCc(c(-c(cc1)cc2c1c(C(CC1)=CC(C3)=C1CCN3C(C1=NC=CNC1)=O)n[nH]2)c1)cc(O)c1F Chemical compound CCc(c(-c(cc1)cc2c1c(C(CC1)=CC(C3)=C1CCN3C(C1=NC=CNC1)=O)n[nH]2)c1)cc(O)c1F CFDDUWYBUWFMRA-UHFFFAOYSA-N 0.000 description 1
- BHJGVQFPHZIMFE-UHFFFAOYSA-N CCc(cc(c(F)c1)N=O)c1-c(cc1)cc2c1c(-c1nc(CNCC3)c3[nH]1)n[nH]2 Chemical compound CCc(cc(c(F)c1)N=O)c1-c(cc1)cc2c1c(-c1nc(CNCC3)c3[nH]1)n[nH]2 BHJGVQFPHZIMFE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto de fórmula: **(Ver fórmula)** o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo.
Description
DESCRIPCIÓN
Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus
[0001] Esta invención se refiere, entre otras cosas, a un compuesto que es un agente antiinflamatorio (p. ej., a través de la inhibición de uno o más de los miembros de la familia de la quinasa Janus (JAK)). La invención también se refiere al uso de dicho compuesto en terapia, incluso en terapias mono y combinadas, especialmente en el tratamiento de enfermedades inflamatorias, incluidas las enfermedades inflamatorias del pulmón (como el asma y la enfermedad pulmonar obstructiva crónica (EPOC)), ojo (como uveítis o enfermedad del ojo seco (EOS, también conocida como queratoconjuntivitis seca y xeroftalmia)) y tracto gastrointestinal (como enfermedad de Crohn (EC) y colitis ulcerosa (CU)).
[0002] La lista o discusión de un documento aparentemente publicado anteriormente en esta especificación no debe tomarse necesariamente como un reconocimiento de que el documento es parte del estado de la técnica o es de conocimiento general común.
[0003] Las JAK son una familia de tirosina quinasas intracelulares que juegan un papel esencial en la señalización de numerosas citoquinas implicadas en la patogénesis de enfermedades inflamatorias y son críticos para inmunidades tanto innata como adaptativa (Clark, JD; Flanagan, ME; Telliez, J.-BJ Med. Chem. 2014, 57, 5023-5038). La familia comprende cuatro miembros, JAK1, JAK2 y TYK2, todos los cuales se expresan de manera ubicua, y JAK3, que se encuentra solo en células hematopoyéticas. Estas enzimas muestran una alta homología de secuencia y están unidas constitutivamente a la cola citoplasmática de los receptores de citocinas. Cuando una citocina se une a su receptor, se produce la multimerización (dimerización o complejos de orden superior) de las subunidades del receptor, llevando las enzimas JAK asociadas con cada subunidad próximas entre sí. Luego, los miembros de la familia JAK se auto y/o trans-fosforilan, lo que desencadena una serie de eventos de fosforilación que finalmente dan como resultado la fosforilación y activación de los transductores de señal y los activadores de proteínas de la transcripción (STAT). Un dímero STAT fosforilado luego se transloca al núcleo de la célula donde se une a los genes diana para modular su expresión y alterar la función celular. Es importante destacar que no existen vías compensatorias conocidas alrededor de la señalización JAK/STAT y, como tal, las enzimas JAK son esenciales en la regulación de las citoquinas que emiten señales a través de estas vías. Como resultado de su papel fundamental en la señalización de citocinas, las enzimas JAK se han convertido en objetivos para el descubrimiento de fármacos y los esfuerzos de desarrollo que han dado lugar a dos productos comercializados, tofacitinib y ruxolitinib, así como a varios compuestos en desarrollo (Norman, P. Expert Opin. Investig. Drugs 2014, 23, 1067-1077). De los dos productos comercializados, el inhibidor de pan-JAK tofacitinib (Flanagan, ME, et al. J. Med. Chem.
2010, 53, 8468-8484) es más relevante para las enfermedades inflamatorias, ya que se comercializa para el tratamiento de la artritis reumatoide (Yamaoka, K.; Tanaka, Y. Expert Opin. Pharmacother.2014, 15, 103-113) e investigado en la clínica para enfermedades inflamatorias del intestino (EII), como UC (Sandborn, WJ, et al. New Engl. J. Med. 2012, 367, 616-624; Vuitton, L.; Koch, S.; Peyrin-Biroulet, L. Curr. Drug Targ.2013, 14, 1385-1391; Panes, J., et al. BMC Gastroenterol. 2015, 15, 14), mientras que el inhibidor de JAK1/JAK2 ruxolitinib se comercializa para el tratamiento de la mielofibrosis (Mesa, RA; Yasothan, U.; Kirkpatrick, P. Nat. Rev. Drug Discov. 2012, 11, 103-104). El tofacitinib y otros inhibidores de JAK se han propuesto como posibles terapias para otros trastornos inmunológicos, incluida la EPOC (Barnes, PJ Nat. Rev. Drug Discov. 2013, 12, 543-559; Fenwick, PS, et al. PLoS ONE 2015, 10 (6), e0128757), DED (Beals, CR; Woldemussie, E. Publicación de solicitud de patente estadounidense US 2010/0267751, 21 de octubre de 2010; Liew, SH, et al. Ophthalmology 2012, 119, 1328-1335; Huang, J.-F., et al. Ophthalmology 2012, 119, e43-e50) y uveítis (Huang, J.-F.; Zhang, Y.; Hirakawa, B., 2013 Association for Research in Vision and Ophthalmology Annual Meeting, Seattle, EE. UU., 05-09 de mayo de 2013, Resumen 2536).
[0004] Las quinasas JAK funcionan como dímeros homo o heterodímeros que son específicos de las subunidades del receptor de citocinas. Por ejemplo, los heterodímeros JAK1-JAK3 se asocian con la cadena común y de receptores para controlar la señalización asociada con IL2, IL4, IL7, IL9, IL15 e IL21, citocinas predominantemente asociadas con funciones inmunes adaptativas. Sin embargo, JAK1 también funciona como un heterodímero con JAK2 y TYK2 para regular la señalización a través de una amplia gama de receptores de citocinas. De esta manera, JAK1 modula la señalización de varias citocinas proinflamatorias asociadas con la respuesta inmune innata, como la IL6 y los interferones de tipo I. JAK2 es el único miembro de la familia JAK que puede funcionar como homodímero. En esta combinación, JAK2 controla la señalización de diversas citocinas y factores de crecimiento, como IL3, IL5, factor estimulante de colonias de granulocitos y macrófagos, eritropoyetina y trombopoyetina.
[0005] La importancia potencial de JAK1 a EII se destaca por el hecho de que dos inhibidores de JAK1 selectivos, filgotinib (GLPG0634) y ABT-494, están en fase 2 de ensayos clínicos para CD (Galien, R., et al. Gastroenterology 2014, 146 (Supl 1), S-49, Resumen 188). El filgotinib es de particular interés para el tratamiento de la EII, dado que, cuando se trataron biopsias de colon de pacientes con EII con este compuesto, se observó la regulación de la expresión de IL6 y MX1 y una relación con la fosforilación de STAT3 (Dupont, S., et al. Inflamm. Res. 2015, 64 (Suppl 2), S202, Resumen B252). Además, filgotinib ha demostrado eficacia en el modelo de colitis inducida por sulfato de sodio de dextrano en ratón, estando asociada la eficacia con la inhibición de la fosforilación de STAT3 en el colon inflamado (Merciris, D., et al. 9th Congress of the European Crohn's and Colitis Organisation, Copenhague, Dinamarca, 20-22 de febrero de 2014, Resumen P072). JAK2 está implicado en que los polimorfismos del gen correspondiente son factores de riesgo tanto para la EC como para la CU, especialmente en caucásicos (Zhang, JX, et al. Inflammation 2014, 37, 793-800). Además, muchos
componentes de la vía IL23, incluidos JAK2 y TYK2, son verdaderos genes de susceptibilidad a la EII, lo que sugiere un papel fundamental de esta vía en el mantenimiento de la homeostasis inmunitaria intestinal (Lees, CW, et al. Gut 2011, 60, 1739-1753; Khor, B.; Gardet, A.; Xavier, RJ Nature 2011,474, 307-317). También debe tenerse en cuenta que, cuando se administra por vía oral, el inhibidor de JAK1/2 AZD1480 inhibe el cáncer de colon asociado a colitis, un modelo de tumorigénesis inducida por inflamación (Stuart, E. y col., Mol. Cancer Ther. 2014, 13, 468-474). Esto es importante, dado que los pacientes con Cu y EC del colon tienen un mayor riesgo de desarrollar cáncer colorrectal (Farraye, FA, et al. Gastroenterology 2010, 138, 738-745). La importancia de JAK3 en la homeostasis inmune ha sido subrayada por las observaciones de que las mutaciones con pérdida de función en humanos dan como resultado un fenotipo de inmunodeficiencia combinada grave (Casanova, J.-L.; Holland, SM; Notarangelo, LD Immunity 2012, 36, 515-528). Además, el inhibidor selectivo de JAK3 JANEX1 demostró actividad en modelos preclínicos de colitis (Uckun, FM y col. Bioorg. Med. Chem. 2008, 16, 1287-1298). No obstante, se ha indicado que la inhibición de JAK3 por sí sola no es suficiente para lograr la máxima eficacia antiinflamatoria y que es necesaria una inhibición adicional de JAK1 para mejorar la actividad celular (Thoma, G.; Drückes, P.; Zerwes, H.-G. Bioorg. Med. Chem. Lett. 2014, 24, 4617-4621).
[0006] Como se ha indicado anteriormente, todas las enzimas JAK están asociadas con EII en una forma u otra. Como tal, los inhibidores de pan-JAK pueden ofrecer la mejor posibilidad de lograr eficacia en los pacientes. En este sentido, el inhibidor de pan-JAK tofacitinib ha demostrado (Boland, BS; Sandborn, WJ; Chang, JT Gastroenterol. Clin. N. Am. 2014, 43, 603-617) eficacia dependiente de la dosis en ensayos clínicos de fase 2 para CU, aunque a dosis superiores a las aprobadas para el tratamiento de la artritis reumatoide (5 mg dos veces al día). Actualmente se están realizando estudios de fase 3 para evaluar la eficacia de este compuesto (10 mg dos veces al día) como terapia de inducción en pacientes con CU activa, y a 5 y 10 mg dos veces al día como terapia de mantenimiento en pacientes con CU que han respondido a la terapia de inducción de tofacitinib. Sin embargo, como resultado de la inhibición sistémica de JAK, la terapia con tofacitinib se asocia con efectos secundarios que limitan la dosis, en particular, infecciones oportunistas, anomalías lipídicas relacionadas con la dosis y supresión de la médula ósea, que está mediada por la inhibición de JAK2 y produce anemia, trombocitopenia y neutropenia. Por tanto, aunque tofacitinib ha demostrado eficacia en pacientes con CU, existe la necesidad de agentes eficaces que presenten un perfil de efectos secundarios mejorado.
[0007] Cuando se administra por inhalación, el inhibidor PF06263276 clínico pan-JAK (Compuesto de Referencia; Coe, J.W., et al. WO2013/014567, 31 de enero de 2013) se ha informado para atenuar IL6 estimulada por inducción pSTAT en ratón de pulmón. Además, después de la administración tópica, este compuesto redujo la hinchazón de la oreja del ratón inducida por una inyección de IL23 (Jones, P. Frontiers in Medicinal Chemistry 2015, Amberes, Bélgica, 14-16 de septiembre de 2015, Resumen IL23). La estructura cristalina de PF06263276 unida a JAK2 revela que este compuesto interactúa con la quinasa empleando el modo de unión de Tipo 1.5 (Foloppe, N., et al. Bioorg. Med. Chem. 2006, 14, 1792-1804; Zuccotto, F., et al. J. Med. Chem. 2010, 53, 2681-2694; Wang, T., et al. Bioorg. Med. Chem. Lett. 2010, 20, 153-156) de manera que su extremo fenólico "Noroeste" actúa como donante de enlace de hidrógeno y aceptor de enlace de hidrógeno, uniéndolo profundamente dentro de un bolsillo hidrofóbico. El modo de unión observado da como resultado que PF06263276 exhiba una cinética de desplazamiento lento y una duración de acción celular extendida.

[0008] Ahora hemos descubierto, sorprendentemente, que compuestos que llevan ciertos sustituyentes aminoheteroarilo inhiben una o más enzimas JAK y por lo tanto poseen buenas propiedades anti-inflamatorias.
[0009] La invención proporciona un compuesto de la fórmula:

o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo.
[0010] La invención también proporciona una formulación farmacéutica que comprende el compuesto, o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, en mezcla con un adyuvante farmacéuticamente aceptable, diluyente o portador.
[0011] La invención también proporciona un producto de combinación que comprende
(A) el compuesto, o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, y
(B) otro agente terapéutico,
en el que cada uno de los componentes (A) y (B) se formula en mezcla con un adyuvante, diluyente o portador farmacéuticamente aceptable.
[0012] La invención también proporciona el compuesto, o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o la formulación farmacéutica o el producto de combinación, para uso en el tratamiento o prevención de una enfermedad inflamatoria.
[0013] También se describe un compuesto de fórmula I,

en el que:
X representa halo;
Ak representa C1-6 alquilo opcionalmente sustituido con uno o más átomos de flúor;
s representa 1 o 2;
L representa C(O), S(O)2, CH2 o un enlace;
Q representa un anillo heteroaromático de 6 miembros que contiene 1, 2 o 3 átomos de N;
R1 representa R3;
R2 representa H, R3, C1-6 alquilo o C3-7 cicloalquilo, cuyos dos últimos grupos están opcionalmente sustituidos con uno o más sustituyentes seleccionados entre C1-4 alquilo, halo, hidroxi y oxo; o R1 y R2 , junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 4 a 7 miembros que está completamente saturado o parcialmente insaturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que R1 y R2 están unidos) y en el que
(a) el grupo heterocíclico contiene un heteroátomo adicional que es S y está opcionalmente sustituido por uno o más sustituyentes seleccionados de halo, hidroxi, oxo, C1-4 alquilo, C1-4 alcoxi y C1-4 hidroxialquilo o
(b) el grupo heterocíclico contiene opcionalmente uno o más heteroátomos adicionales seleccionados entre N, o y S, está sustituido por uno o más sustituyentes seleccionados de C1-4 hidroxialquilo, C1-4 carboxialquilo y -(C1-4 alquileno)-NRaRb, y está opcionalmente sustituido además por uno o más sustituyentes seleccionados de halo, hidroxi, oxo, CO2H, C1-4 alquilo y C1-4 alcoxi o
(c) el grupo heterocíclico está fusionado con un anillo heterocíclico de 5 o 6 miembros completamente saturado o parcialmente insaturado, cuyo anillo heterocíclico contiene s uno o más heteroátomos seleccionados entre N, O y S y está opcionalmente sustituido con uno o más sustituyentes seleccionados entre oxo y C1-4 alquilo;
R3 representa
Hetx o
C1-6 alquilo sustituido por
CO2H,
-(OCH2CH2)o-4ORc,
Hety,
Hetz,
NRdC(O)Re o
S (o) 1-2 R f,
cuyo grupo C1-6 alquilo está opcionalmente sustituido adicionalmente por uno o más sustituyentes seleccionados de halo, hidroxi y oxo;
Ra, Rb, Rc, Rd y Re representan independientemente H o C1-4 alquilo,
o Ra y Rb, junto con el átomo de N al que están unidos, forman un grupo heterocíclico 4 a 7 miembros que está completamente saturado, parcialmente insaturado o totalmente aromático y grupo heterocíclico que contiene un átomo de N (el átomo al que Ra y Rb están unidos) y, opcionalmente, uno o más heteroátomos adicionales seleccionados de O, S y N, y grupo heterocíclico el cual está opcionalmente sustituido por uno o más sustituyentes seleccionados de halo, hidroxi, oxo, C1-4 alquilo, C1-4 alcoxi y C1-4 hidroxialquilo;
Rf representa C1-6 alquilo o C3-7 cicloalquilo;
Hetx representa, independientemente de cada aparición, un grupo heterocíclico de 4 a 7 miembros que está completamente saturado, parcialmente insaturado o completamente aromático, cuyo grupo contiene uno o más heteroátomos seleccionados entre N, O y S, y cuyo grupo está opcionalmente sustituido por uno o más sustituyentes seleccionados entre C1-4 alquilo, halo, hidroxi y oxo;
Hety representa, independientemente de cada aparición, un grupo heterocíclico de 5 a 10 miembros que es completamente aromático, cuyo grupo contiene uno o más heteroátomos seleccionados entre N, O y S, y cuyo grupo está opcionalmente sustituido con uno o más sustituyentes seleccionados de C1-4 alquilo, halo, hidroxi y oxo; y
Hetz representa, independientemente en cada aparición, un grupo heterocíclico de 4 a 7 miembros que está completamente saturado o parcialmente insaturado, cuyo grupo contiene uno o más heteroátomos seleccionados de N, O y S, y cuyo grupo está sustituido por oxo o hidroxi, y cuyo grupo está opcionalmente sustituido adicionalmente con uno o más sustituyentes seleccionados entre C1-4 alquilo, halo, hidroxi y oxo,
o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo.
[0014] Las sales farmacéuticamente aceptables que se pueden mencionar incluyen sales de adición de ácido y sales de adición de base. Dichas sales pueden formarse por medios convencionales, por ejemplo, mediante la reacción de un ácido libre o una forma de base libre de un compuesto de fórmula I con uno o más equivalentes de un ácido o base apropiada, opcionalmente en un disolvente, o en un medio en cuya sal es insoluble, seguido de la eliminación de dicho disolvente, o dicho medio, utilizando técnicas estándar (p. ej., al vacío, por liofilización o por filtración). Las sales también se pueden preparar intercambiando un contraión de un compuesto de fórmula I en forma de sal con otro contraión, por ejemplo usando una resina de intercambio iónico adecuada.
[0015] Los ejemplos de sales farmacéuticamente aceptables incluyen sales de adición de ácido derivadas de ácidos minerales y ácidos orgánicos, y sales derivadas de metales.
[0016] Para evitar dudas, los compuestos de fórmula I pueden contener los átomos indicados en cualquiera de sus formas isotópicas naturales o no naturales. A este respecto, las realizaciones de la divulgación que pueden mencionarse incluyen aquellas en las que:
(a) el compuesto de fórmula I no está enriquecido isotópicamente ni marcado con respecto a ningún átomo del compuesto; y
(b) el compuesto de fórmula I está enriquecido o marcado isotópicamente con respecto a uno o más átomos del compuesto.
[0017] Las referencias en el presente documento a un "derivado isotópico" se refieren a la segunda de estas dos realizaciones. En realizaciones particulares de la divulgación, el compuesto de fórmula I está enriquecido o marcado isotópicamente (con respecto a uno o más átomos del compuesto) con uno o más isótopos estables. Por tanto, los
compuestos de la divulgación que pueden mencionarse incluyen, por ejemplo, compuestos de fórmula I que están enriquecidos isotópicamente o marcados con uno o más átomos tales como deuterio o similares.
[0018] Los compuestos de fórmula I puede exhibir tautomerismo. Se describen todas las formas tautoméricas y mezclas de las mismas.
[0019] A menos que se especifique lo contrario, grupos alquilo y grupos alcoxi como se ha definido en este documento pueden ser de cadena lineal o, cuando hay un número suficiente (es decir, un mínimo de tres) de átomos de carbono, ser ramificados. Los grupos alquilo particulares que pueden mencionarse incluyen, por ejemplo, metilo, etilo, n-propilo, isopropilo, butilo, n-butilo y terc-butilo. Los grupos alcoxi particulares que pueden mencionarse incluyen, por ejemplo, metoxi, etoxi, propoxi y butoxi.
[0020] A menos que se especifique lo contrario, los grupos cicloalquilo como se definen en el presente documento pueden, cuando hay un número suficiente (es decir, un mínimo de cuatro) de átomos de carbono, ser parcialmente cíclicos/acíclicos.
[0021] A menos que se especifique lo contrario, los grupos alquileno tal como se definen en el presente documento pueden ser de cadena lineal o, cuando hay un suficiente número (es decir, un mínimo de dos) de átomos de carbono, ser ramificados.
[0022] A menos que se indique lo contrario, el punto de unión de los grupos arilo puede ser por medio de cualquier átomo del sistema de anillos. Sin embargo, cuando los grupos arilo son bicíclicos o tricíclicos, se unen al resto de la molécula a través de un anillo aromático. Los grupos Ca-14 arilo incluyen fenilo, naftilo y similares.
[0023] Para evitar la duda, los sustituyentes oxo que pueden estar presentes en los grupos heterocíclicos representados por Hetx, Hety, Hetz, N(R1)R2 y N(Ra)Rb puede estar unido a cualesquiera átomos apropiados en el anillo heterocíclico que incluyen, cuando las valencias lo permitan, átomos C, N y/o S dentro del anillo (formando así grupos ceto, N-óxido, S(O) y/o S(O)2).
[0024] Los valores de Hetx que pueden mencionarse incluyen piperidinilo (p. ej., piperidin-4-ilo).
[0025] Los valores de Het y que pueden mencionarse incluyen piridinilo (p. ej., piridin-2-ilo).
[0026] Los valores de Hetz que pueden mencionarse incluyen isotiazolidinilo (p. ej., isotiazolidin-2-ilo), oxazolidinilo (p. ej., oxazolidin-3-ilo) o, particularmente, piperidinilo (p. ej., piperidin-1-ilo o piperidin-4-ilo), pirrolidinilo (p. ej., pirrolidin-1-ilo) y tiomorfolinilo (p. ej., tiomorfolin-4-ilo).
[0027] Los ejemplos de grupos heterocíclicos que pueden estar formados por R1, R2 y el átomo de N al que están unidos incluyen, pero no se limitan a, azetidin-1-ilo, hexahidropirrolo[3,4-c]pirrol-2(lH)-ilo, piperazin-1-ilo, piperidin-1-ilo, pirrolidin-1-ilo y tiomorfolin-4-ilo.
[0028] A menos que se especifique lo contrario, el término "halo" incluye referencias a fluoro, cloro, bromo o yodo, en particular, a fluoro, cloro o bromo, especialmente flúor o cloro.
[0029] Las realizaciones de la divulgación que se pueden mencionar incluyen compuestos de fórmula I en los que:
R1 representa R3;
R2 representa H, R3, Ci-a alquilo o C3-7 cicloalquilo, cuyos dos últimos grupos están opcionalmente sustituidos con uno o más sustituyentes seleccionados entre C1-4 alquilo, halo, hidroxi y oxo;
o R1 y R2, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 4 a 7 miembros que está completamente saturado o parcialmente insaturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que R1 y R2 están unidos) y en donde o bien
(a) el grupo heterocíclico contiene un heteroátomo adicional que es S y está opcionalmente sustituido por uno o más sustituyentes seleccionados de halo, hidroxi, oxo, C1-4 alquilo, C1-4 alcoxi y C1-4 hidroxialquilo o
(b) el grupo heterocíclico contiene opcionalmente uno o más heteroátomos adicionales seleccionados de N, O y S, es sustituido por uno o más sustituyentes seleccionados de C1-4 hidroxialquilo, C1-4 carboxialquilo y -(C1-4 alquileno)-NRaRb, y está opcionalmente sustituido además por uno o más sustituyentes seleccionados de halo, hidroxi, oxo, CO2H, C1-4 alquilo y C1-4 alcoxi; y
R3 representa
Hetx o
C1-a alquilo sustituido por
CO2H,
-(OCH2CH2)o-4ORc,
Hety o
Hetz,
cuyo grupo C i-6 alquilo está opcionalmente sustituido adicionalmente por uno o más sustituyentes seleccionados entre halo, hidroxi y oxo.
[0030] Las realizaciones alternativas de la descripción que se pueden mencionar incluyen aquellas en las que una o más de las siguientes definiciones se aplican a los compuestos de fórmula I:
(i) R1 y R2, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 4 a 7 miembros que está completamente saturado o parcialmente insaturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que están unidos R1 y R2), y
en donde el grupo heterocíclico está fusionado a un anillo heterocíclico de 5 o 6 miembros completamente saturado o parcialmente insaturado, cuyo anillo heterocíclico contiene uno o más heteroátomos seleccionados entre N, O y S y está opcionalmente sustituido con uno o más sustituyentes seleccionados entre oxo y C1-4 alquilo; (ii) R3 representa C1-6 alquilo sustituido por
NRdC(O)Re o
S(O)1-2Rf,
cuyo grupo C1-6 alquilo está opcionalmente sustituido adicionalmente con uno o más sustituyentes seleccionados entre halo, hidroxi y oxo.
[0031] Las realizaciones de la divulgación que pueden mencionarse incluyen compuestos de fórmula I en relación con los cuales se aplican uno o más de los siguientes:
(i) X representa cloro o, particularmente, flúor;
(ii) Ak representa C1-4 alquilo opcionalmente sustituido con uno o más átomos de flúor;
(iii) s representa 1;
(iv) L representa C(O);
(v) Q representa un anillo heteroaromático de 6 miembros que contiene 2 átomos de N, como pirazinilo;
(vi) R1 representa R3;
(vii) R2 representa H, C1-3 alquilo o R3;
(viii) R1 y R2, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 4 a 6 miembros que está completamente saturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que R1 y R2 están unidos) y en donde
(a) el grupo heterocíclico contiene un heteroátomo adicional que es S y está opcionalmente sustituido por uno o más sustituyentes seleccionados de halo, hidroxi, oxo, C1-2 alquilo, C1-2 alcoxi y C1-2 hidroxialquilo, o
(b) el grupo heterocíclico contiene opcionalmente un heteroátomo adicional seleccionado de N, O y S, está sustituido por un sustituyente seleccionado de C1-3 hidroxialquilo, C1-3 carboxialquilo y -(C1-3 alquileno)-NRaRb, y está opcionalmente sustituido además por uno o más hidroxi o C1-2 grupos alquilo, o
(c) el grupo heterocíclico está condensado con un anillo totalmente saturado de 5 ó 6 miembros de anillo heterocíclico, cuyo anillo heterocíclico contiene uno o dos heteroátomos seleccionados entre N, O y S y está opcionalmente sustituido con uno a tres sustituyentes sele formado a partir de oxo y C1-2 alquilo
(p. ej., R1 y R2, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 5 o 6 miembros que está completamente saturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que están unidos R1 y R2) y en donde
(a) el grupo heterocíclico contiene un heteroátomo adicional que es S y está opcionalmente sustituido con uno o más sustituyentes seleccionados entre halo, hidroxi, oxo, C1-2 alquilo, C1-2 alcoxi y C1-2 hidroxialquilo o
(b) el grupo heterocíclico contiene opcionalmente un heteroátomo adicional seleccionado de N, O y S, está sustituido por un sustituyente seleccionado de C1-3 hidroxialquilo, C1-3 carboxialquilo y -(C1-3 alquileno)-NR a Rb, y está opcionalmente sustituido adicionalmente con C1-2 alquilo);
(ix) R3 representa
Hetx o
C1-4 alquilo sustituido con (p. ej., C2-3 n-alquilo terminado en)
CO2H,
-(OCH2CH2)0-3ORc,
Hety,
Hetz,
NRdC(O)Re o
S(O)-,.2Rf,
(p. ej., R3 representa
Hetx o
C1.4 alquilo sustituido por
CO2H,
-(OCH2CH2)0-1ORc,
Hety o
Hetz);
(x) Ra y Rb representan H o C1.2 alquilo, o Ra y Rb, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 5 o 6 miembros que está completamente saturado, parcialmente insaturado o totalmente aromático y cuyo grupo heterocíclico contiene un átomo de N (el átomo al que están unidos Ra y Rb) y, opcionalmente, uno o dos otros heteroátomos seleccionados de O, S y N, y cuyo grupo heterocíclico está opcionalmente sustituido por uno o más sustituyentes seleccionados de hidroxi, oxo, C1-2 alquilo, C1-2 alcoxi y C1-2 hidroxialquilo;
(xi) Rc representa H o C1-2 alquilo;
(xii) Rd y Re representan independientemente H o, particularmente, C1-2 alquilo;
(xiii) Rf representa C1.2 alquilo;
(xiv) Hetx representa, independientemente en cada aparición, un grupo heterocíclico de 5 ó 6 miembros que está completamente saturado, cuyo grupo contiene uno o dos heteroátomos seleccionados de N, O y S, y cuyo grupo está opcionalmente sustituido por uno a tres sustituyentes seleccionados entre C1-2 alquilo, halo, hidroxi y oxo; (xv) Hety representa, independientemente en cada aparición, un grupo heterocíclico de 5 o 6 miembros que es completamente aromático, cuyo grupo contiene de uno a tres heteroátomos seleccionados entre N, O y S y cuyo grupo está opcionalmente sustituido con uno o más sustituyentes seleccionados entre C1-2 alquilo, halo, hidroxi y oxo;
(xvi) Hetz representa, independientemente en cada aparición, un grupo heterocíclico de 5 ó 6 miembros que está completamente saturado, cuyo grupo contiene uno o dos heteroátomos seleccionados de N, O y S, y cuyo grupo está sustituido por oxo o hidroxi, y cuyo grupo está opcionalmente sustituido adicionalmente con uno o más sustituyentes seleccionados entre C1-2 alquilo, halo, hidroxi y oxo.
[0032] Las realizaciones de la divulgación que pueden mencionarse incluyen aquellas en las que el compuesto de fórmula I es un compuesto de fórmula Ia o Ib,

en donde X, Ak, L, Q, R1 y R2 son como se definieron anteriormente.
[0033] Las realizaciones de la divulgación que pueden mencionarse incluyen aquellas en las que una o más de las siguientes definiciones se aplican a los compuestos de fórmula I, Ia o Ib:
(i) X representa flúor;
(ii) Ak representa C1-3 alquilo, tal como etilo;
(iii) L representa C(O);
(iv) Q representa pirazinilo;
(v) R1 representa R3;
(vi) R2 representa H, metilo o R3;
(vii) R1 y R2, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 5 o 6 miembros que es totalmente saturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que están unidos R1 y R2) y en donde
(a) el grupo heterocíclico contiene un heteroátomo adicional que es S y está opcionalmente sustituido con uno o más sustituyentes seleccionados de hidroxi, oxo y metilo o
(b) el grupo heterocíclico contiene opcionalmente un heteroátomo adicional seleccionado entre N, O y S, está sustituido con un sustituyente seleccionado entre C1-2 hidroxialquilo (p. ej., hidroximetilo) y -(C1-3 alquileno)-NRaRb, y está opcionalmente sustituido adicionalmente con metilo;
(viii) R3 representa
Hetx o
C1-4 alquilo sustituido con
CO2H (p. ej., para formar-(CH2)3-CO2H),
-(OCH2CH2)0-2ORc (p. ej., para formar-(CH2)2-(OCH2CH2)2ORc o -(CH2)2-ORc),
Hety (p. ej. para formar-(CH2)1-2-Hety),
Hetz (p. ej. para formar-(CH2)1-3-Hetz) o
NRdC(O)Re (p. ej. para formar-(CH2)2-NRdC(O)Re)
(p. ej., R3 representa
Hetx o
C1-4 alquilo sustituido por
CO2H (p. ej. para formar-(CH2)3-CO2H),
-ORc (p. ej. para formar -(CH2)2-ORc),
Hety (p. ej., para formar -(CH2^-2-Hety) o
Hetz (p. ej., para formar -(CH2^-3-Hetz));
(ix) Ra y Rb representan H o metilo, o Ra y Rb, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 5 o 6 miembros que es totalmente aromático y cuyo grupo heterocíclico contiene un átomo de N (la átomo al que Ra y Rb están unidos) y, opcionalmente, uno o dos heteroátomos adicionales seleccionados de O, S y N, y grupo heterocíclico el cual está opcionalmente sustituido por uno o más sustituyentes seleccionados de metilo, metoxi e hidroximetilo;
(x) Rc representa H o metilo;
(xi) Rd y Re representan independientemente H o, particularmente, metilo;
(xii) Hetx representa, independientemente en cada aparición, un grupo heterocíclico de 5 ó 6 miembros que está totalmente saturado, cuyo grupo contiene uno o dos heteroátomos seleccionados de N, O y S, y cuyo grupo está opcionalmente sustituido por uno o dos sustituyentes seleccionados entre metilo, hidroxi y oxo;
(xiii) Hety representa, independientemente en cada aparición, un grupo heterocíclico de 5 o 6 miembros que es completamente aromático, cuyo grupo contiene uno o dos heteroátomos seleccionados entre N, O y S y cuyo grupo está opcionalmente sustituido con uno o dos sustituyentes metilo;
(xiv) Hetz representa, independientemente en cada aparición, un grupo heterocíclico de 5 ó 6 miembros que está totalmente saturado, cuyo grupo contiene uno o dos heteroátomos seleccionados de N, O y S, y cuyo grupo está sustituido por oxo o hidroxi, y cuyo grupo está opcionalmente sustituido adicionalmente con uno o más sustituyentes seleccionados entre metilo, hidroxi y oxo.
[0034] Otras realizaciones de la divulgación que se pueden mencionar incluyen aquellas en las que el compuesto de fórmula I es un compuesto de fórmula Ic,

en donde X, Ak, R1 y R2 son como se definieron anteriormente.
[0035] Las realizaciones de la divulgación que pueden mencionarse incluyen aquellas en las que una o más de las siguientes definiciones se aplican a los compuestos de fórmula I, la, Ib o Ic:
(i) X representa flúor;
(ii) Ak representa etilo;
(iii) R1 representa R3;
(iv) R2 representa H, metilo o R3 (p. ej., R2 representa metilo o R3);
(v) R1 y R2, junto con el átomo de N al que están unidos, forman un grupo heterocíclico de 5 o 6 miembros que está completamente saturado, cuyo grupo heterocíclico contiene un átomo de N (el átomo al que R1 y R2 están unidos) y en donde o bien
(a) el grupo heterocíclico contiene un heteroátomo adicional que es S y el grupo heterocíclico está opcionalmente sustituido por oxo o
(b) el grupo heterocíclico está sustituido con C1-2 hidroxialquilo (p. ej., hidroximetilo);
(vi) R3 representa alquilo C1-3 (p. ej., etilo) sustituido con
-(OCH2CH2)0-2ORc (por ejemplo para formar -(CH2)2-(OCH2CH2)2ORc o -(CH2)2-ORc), Hetz (p. ej. para formar -(CH2)2-3-Hetz) o
NCH3C(O)CH3 (p. ej. para formar-(CH2)2-NCH3C(O)CH3
(p. ej., R3 representa C1-3 alquilo (p. ej., etilo) sustituido por
-ORc (p. ej., para formar -(c H2)2-Orc) o
Hetz (p. ej., para formar-(CH2)2-3-Hetz));
(vii) Rc representa metilo;
(viii) Hetz representa un grupo heterocíclico de 5 o 6 miembros que está completamente saturado, cuyo grupo contiene uno o dos heteroátomos seleccionados de N, O y S, y cuyo grupo está sustituido con oxo y está opcionalmente sustituido adicionalmente por un sustituyente seleccionado de metilo y oxo (p. ej., Hetz representa pirrolidinilo o isotiazolidinilo, tales como pirrolidin-1-ilo sustituido por oxo (p. ej., en la posición 2) o isotiazolidin-2-ilo disustituido con oxo en la posición 1) (p. ej., Hetz representa un grupo heterocíclico de 5 o 6 miembros que está completamente saturado, cuyo grupo contiene uno o dos heteroátomos (p. ej., un heteroátomo) seleccionado de N, O y S, y cuyo grupo está sustituido con oxo (p. ej., Hetz representa pirrolidinilo, tal como pirrolidin-1-ilo, sustituido por oxo (p. ej., en la posición 2))).
[0036] Otros compuestos de fórmula I, Ia, Ib y Ic que pueden ser mencionados incluyen los compuestos de los ejemplos descritos a continuación. Por tanto, las realizaciones de la divulgación que pueden mencionarse incluyen aquellas en las que el compuesto de fórmula I, la, Ib e Ic es un compuesto seleccionado de la lista:
ácido 4-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il) (metil)amino)butanoico;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-((2-(4-hidroxipiperidin-1-il)etil)amino)pirazin-2-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(metil(piridin-2-ilmetil)amino)pirazin-2-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-((2-(1-oxidotiomorfolino)etil)amino)pirazin-2-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(1-oxidotiomorfolino)pirazin-2-il)metanona;
(5-(4-(2-(dimetilamino)etil) piperazin-1-il)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridina-5-il)(5-((2-(4-hidroxi-1-metilpiperidin-4-il)etil)amino)pirazin-2-il)metanona;
(S)-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(2-(hidroximetilo) pirrolidin-1-il)pirazin-2-il)metanona;
(5-(bis(2-metoxietil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)metanona;
(5-(4-(2-(1H-imidazol-1-il)etil)piperazin-1-il)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenilo))-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)metanona;
1-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil) pirrolidin-2-ona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-(4-(2-hidroxietil)piperazin-1 -il)pirazin-2-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((2-(2-(2-metoxietoxi) etoxi)etil)amino)pirazin-2-il)metanona;
N-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)-N-metilacetamida;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-(3-(hidroximetilo) azetidin-1 -il)pirazin-2-il)metanona;
(5-(bis(2-hidroxietil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((2-(metilsulfonil)etil)amino)pirazin-2-il)metanona;
1-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)amino)etil) pirrolidin-2-ona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((2R,3R,4R,5S)-3,4,5-trihidroxi-2-(hidroximetilo)piperidin-1-il)pirazin-2-il)metanona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((3aR, 6aS)-5-metilhexahidropirrolo[3,4-c]pirrol-2(1H)-il)pirazin-2-il)metanona;
3-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)oxazolidin-2-ona;
(5-((2-(1,1-dioxidoisotiazolidin-2-il)etil)(metil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)metanona; y
(3aR,6aS)-5-(5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)tetrahidropirrolo[3,4-c]pirrol-1,3(2H,3aH)-diona,
o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo.
[0037] Otras realizaciones de la divulgación que se pueden mencionar incluyen aquellos en los que o bien
(i) el compuesto de fórmula I, Ia, Ib o Ic representa, o
(ii) el compuesto de fórmula I, Ia, Ib o Ic es tan definido anteriormente, siempre que no represente
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(1-oxidotiomorfolino)pirazin-2-il)metanona;
1-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)pirrolidin-2-ona;
(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((2-(2-(2-metoxietoxi) etoxi)etil)amino)pirazin-2-il)metanona;
N-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)-N-metilacetamida; y/o
(5-((2-(1,1-dioxidoisotiazolidin-2-il)etil)(metil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4)-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)metanona;
o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo
(p. ej., (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(1-oxidotiomorfolino)pirazin-2-il)metanona; y/o 1-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)pirrolidin-2-ona, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo).
[0038] Los ejemplos de sales de compuestos de fórmula I, la, Ib e Ic incluyen todas las sales farmacéuticamente aceptables, tales como, sin limitación, sales de adición de ácidos de ácidos minerales fuertes como HCl, H2SO4 y sales HBr (p. ej., HCl o sales HBr) y sales de adición de ácidos orgánicos fuertes tales como ácido metanosulfónico.
[0039] Las referencias en el presente documento a un compuesto de la divulgación (un compuesto de fórmula I, Ia, Ib o Ic) pretenden incluir referencias al compuesto y a todas las sales, solvatos y/o tautómeros farmacéuticamente aceptables
de dicho compuesto, a menos que el contexto indica específicamente lo contrario. A este respecto, los solvatos que se pueden mencionar incluyen hidratos.
[0040] Los compuestos de la descripción (compuestos de fórmula I, Ia, Ib y Ic) son inhibidores de la quinasa JAK y son por lo tanto útiles en medicina, en particular para el tratamiento de enfermedades inflamatorias. Por lo tanto, otros aspectos de la divulgación que pueden mencionarse incluyen los siguientes.
(a) Una formulación farmacéutica que comprende un compuesto de fórmula I, la, Ib o Ic, como se define aquí anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, mezclado con un adyuvante, diluyente o vehículo farmacéuticamente aceptable.
(b) Un producto de combinación que comprende
(A) un compuesto de fórmula I, la, Ib o Ic, como se definió anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, y
(B) otro agente terapéutico,
en donde cada uno de los componentes (A) y (B) se formulan mezclados con un adyuvante, diluyente o vehículo farmacéuticamente aceptable.
En este aspecto, el producto de combinación puede ser una formulación farmacéutica única (combinación) o un kit de partes.
Por tanto, este aspecto abarca una formulación farmacéutica que incluye un compuesto de fórmula I, la, Ib o Ic, como se definió anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, y otro agente terapéutico, en mezcla con un adyuvante, diluyente farmacéuticamente aceptable. o vehículo (cuya formulación se denominará en lo sucesivo "preparación combinada"). También incluye un kit de partes que comprende componentes:
(i) una formulación farmacéutica que incluye un compuesto de fórmula I, la, Ib o Ic, como se definió anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, en mezcla con un compuesto farmacéuticamente aceptable. adyuvante, diluyente o portador; y
(ii) una formulación farmacéutica que incluye otro agente terapéutico, mezclado con un adyuvante, diluyente o vehículo farmacéuticamente aceptable,
cuyos componentes (i) y (ii) se proporcionan cada uno en una forma adecuada para la administración junto con el otro. Por tanto, el componente (i) del kit de piezas es el componente (A) anterior mezclado con un adyuvante, diluyente o vehículo farmacéuticamente aceptable. De manera similar, el componente (ii) es el componente (B) anterior mezclado con un adyuvante, diluyente o vehículo farmacéuticamente aceptable.
(c) Un proceso para preparar la formulación farmacéutica del aspecto (a) anterior, comprendiendo dicho proceso la etapa de mezclar el compuesto de fórmula I, la, Ib o Ic, como se define anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, con un adyuvante, diluyente o portador farmacéuticamente aceptable. Las realizaciones de este aspecto que pueden mencionarse incluyen aquellas en las que el adyuvante, diluyente o portador farmacéuticamente aceptable es un adyuvante, diluyente o portador tópicamente aceptable (y/o donde el proceso es para preparar una formulación farmacéutica tópica, es decir, una formulación farmacéutica que es adaptado para administración tópica).
(d) Un compuesto de fórmula I, la, Ib o Ic, como se define aquí anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, para uso en medicina (o para uso como medicamento o como producto farmacéutico).
(e) Un compuesto de fórmula I, la, Ib o Ic, como se define aquí anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o una formulación farmacéutica o producto de combinación como se define en relación con el aspecto (a) o (b), para su uso en el tratamiento o prevención de una enfermedad inflamatoria.
(f) El uso de
un compuesto de fórmula I, la, Ib o Ic, como se definió anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o
una formulación farmacéutica o un producto de combinación como se define en relación con el aspecto (a) o (b),
para la preparación de un medicamento para el tratamiento o la prevención de una enfermedad inflamatoria.
(g) Un método para tratar o prevenir una enfermedad inflamatoria, comprendiendo dicho método administrar a un sujeto una cantidad eficaz de
un compuesto de fórmula I, la, Ib o Ic, como se ha definido anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o
una formulación farmacéutica o un producto de combinación como se define en relación con el aspecto (a) o (b).
(h) Un método para sensibilizar a un sujeto a los efectos antiinflamatorios de un corticosteroide, comprendiendo dicho método administrar al sujeto una cantidad eficaz de
un compuesto de fórmula I, la, Ib o Ic, como se ha definido anteriormente, o una sal farmacéuticamente aceptable., solvato o derivado isotópico del mismo, o
una formulación farmacéutica o producto de combinación como se define en relación con el aspecto (a) o (b).
[0041] Las realizaciones de este aspecto que pueden mencionarse incluyen aquellas en las que el sujeto es alguien que se ha vuelto refractario a los efectos antiinflamatorios de un corticosteroide.
[0042] Las referencias en el presente documento a "prevenir una enfermedad inflamatoria" incluyen referencias a prevenir (o reducir la probabilidad de) la recurrencia de una enfermedad inflamatoria en un sujeto que previamente ha padecido dicha enfermedad (p. ej., un sujeto que ha recibido previamente tratamiento para esa enfermedad, por ejemplo el tratamiento de acuerdo con el método descrito en (g) anterior).
[0043] Por lo tanto, todavía aspectos adicionales de la divulgación que se pueden mencionar incluyen los siguientes.
(i) Un compuesto de fórmula I, la, Ib o Ic, como se define aquí anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o una formulación farmacéutica o producto de combinación como se define en relación con el aspecto (a) o (b), para su uso en la reducción de la probabilidad de recurrencia de una enfermedad inflamatoria en un sujeto que ha recibido previamente tratamiento para esa enfermedad (p. ej., tratamiento con un compuesto de fórmula I, la, Ib o Ic, como se define anteriormente en la presente, o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o una formulación farmacéutica o producto de combinación como se define en relación con el aspecto (a) o (b)).
(j) El uso de
un compuesto de fórmula I, la, Ib o Ic, como se define aquí anteriormente, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o
una formulación farmacéutica o producto de combinación como se define en relación con el aspecto (a) o (b),
para la preparación de un medicamento para reducir la probabilidad de recurrencia de una enfermedad inflamatoria en un sujeto que ha recibido previamente tratamiento para esa enfermedad (p. ej., tratamiento con un compuesto de fórmula I, la, Ib o Ic, como se describe anteriormente o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o una formulación farmacéutica o un producto de combinación como se define en relación con el aspecto (a) o (b)).
(k) Un método para reducir la probabilidad de recurrencia de una enfermedad inflamatoria en un sujeto que ha recibido previamente tratamiento para esa enfermedad (p. ej., tratamiento con un compuesto de fórmula I, la, Ib o Ic, como se define anteriormente en la presente, o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o una formulación farmacéutica o producto de combinación como se define en relación con el aspecto (a) o (b)), comprendiendo dicho método administrar a dicho sujeto una cantidad eficaz de
un compuesto de fórmula I, la, Ib o Ic, como se define aquí anteriormente, o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o
una formulación farmacéutica o producto de combinación como se define en conexión con el aspecto (a) o (b).
Formulaciones
[0044] En relación con los aspectos (a) y (b) anterior, diluyentes y vehículos que pueden ser mencionados incluyen los adecuados para la administración parenteral, oral, tópica, mucosal y rectal.
[0045] Las formulaciones farmacéuticas y productos de combinación de aspectos (a) y (b) arriba se pueden preparar por ejemplo, para administración parenteral, subcutánea, intramuscular, intravenosa, intra-articular, intravítrea, periocular, retrobulbar, subconjuntival, sub-Tenon, tópica ocular o periarticular, particularmente en forma de soluciones, emulsiones o suspensiones líquidas; para la administración oral, particularmente en forma de comprimidos o cápsulas, y que involucran especialmente tecnologías destinadas a proporcionar la liberación de fármacos dirigida al colon (Patel, M. M. Expert Opin. Drug Deliv. 2011, 8 (10), 1247-1258); para administración tópica, por ejemplo pulmonar o intranasal, particularmente en forma de polvos, gotas nasales o aerosoles y administración transdérmica; para administración ocular
tópica, particularmente en forma de soluciones, emulsiones, suspensiones, ungüentos, implantes/insertos, geles, jaleas o formulaciones de micropartículas liposomales (Ghate, D.; Edelhauser, HF Expert Opin. Drug Deliv.2006, 3 (2), 275-287); para administración ocular, particularmente en forma de implantes, liposomas y nanopartículas biodegradables y no biodegradables (Thrimawithana, TR et al. Drug Discov. Today 2011, 16 (5/6), 270-277); para la administración mucosa, por ejemplo, a la mucosa bucal, sublingual o vaginal, y para la administración rectal, por ejemplo, en forma de supositorio o enema.
[0046] Las formulaciones farmacéuticas y productos de combinación de aspectos (a) y (b) arriba pueden ser convenientemente administrados en forma de dosificación unitaria y pueden prepararse por cualquiera de los métodos bien conocidos en la técnica farmacéutica, por ejemplo como se describe en Remington de Pharmaceutical Sciences, 17a ed., Mack Publishing Company, Easton, PA., (1985). Las formulaciones para administración parenteral pueden contener como excipientes agua estéril o solución salina, alquilenglicoles tales como propilenglicol, polialquilenglicoles tales como polietilenglicol, aceites de origen vegetal, naftalenos hidrogenados y similares. Las formulaciones para administración nasal pueden ser sólidas y pueden contener excipientes, por ejemplo, lactosa o dextrano, o pueden ser soluciones acuosas u oleosas para su uso en forma de gotas nasales o pulverizadores medidos. Para la administración bucal, los excipientes típicos incluyen azúcares, estearato de calcio, estearato de magnesio, almidón pregelatinizado y similares.
[0047] Las formulaciones farmacéuticas y productos de combinación adecuados para la administración oral pueden comprender uno o más vehículos fisiológicamente y/o excipientes compatibles y puede estar en forma sólida o líquida. Los comprimidos y cápsulas se pueden preparar con agentes aglutinantes, por ejemplo, jarabe, goma arábiga, gelatina, sorbitol, tragacanto o polivinilpirrolidona; cargas, tales como lactosa, sacarosa, almidón de maíz, fosfato cálcico, sorbitol o glicina; lubricantes, tales como estearato de magnesio, talco, polietilenglicol o sílice; y tensioactivos, como lauril sulfato de sodio. Las composiciones líquidas pueden contener aditivos convencionales tales como agentes de suspensión, por ejemplo, jarabe de sorbitol, metilcelulosa, jarabe de azúcar, gelatina, carboximetilcelulosa o grasas comestibles; agentes emulsionantes como lecitina o goma arábiga; aceites vegetales como aceite de almendras, aceite de coco, aceite de hígado de bacalao o aceite de cacahuete; conservantes como el hidroxianisol butilado (BHA) y el hidroxitolueno butilado (BHT). Las composiciones líquidas pueden encapsularse, por ejemplo, en gelatina para proporcionar una forma de dosificación unitaria.
[0048] Las formas de dosificación orales sólidas incluyen comprimidos, cápsulas de dos piezas de cubierta dura y cápsulas blandas de gelatina elástica (SEG). Tales cápsulas de cubierta dura de dos piezas pueden estar hechas, por ejemplo, de gelatina o hidroxilpropilmetilcelulosa (HPMC).
[0049] Una formulación de cubierta seca comprende típicamente de aproximadamente 40% a 60% p/p de concentración de gelatina, alrededor de un 20% a la concentración de 30% de plastificante (tal como glicerina, sorbitol o propilenglicol) y alrededor de un 30% a 40% de concentración de agua. También pueden estar presentes otros materiales tales como conservantes, colorantes, opacificantes y aromatizantes. El material de relleno líquido comprende un fármaco sólido que se ha disuelto, solubilizado o dispersado (con agentes de suspensión como cera de abejas, aceite de ricino hidrogenado o polietilenglicol 4000) o un fármaco líquido en vehículos o combinaciones de vehículos como aceite mineral, aceites vegetales, triglicéridos, glicoles, polioles y agentes tensioactivos.
[0050] Un compuesto de la invención se puede administrar tópicamente (por ejemplo al pulmón, ojo o intestinos). Por tanto, las realizaciones de los aspectos (a) y (b) anteriores que pueden mencionarse incluyen formulaciones farmacéuticas y productos de combinación que están adaptados para la administración tópica. Tales formulaciones incluyen aquellas en las que los excipientes (incluido cualquier adyuvante, diluyente y/o vehículo) son tópicamente aceptables.
[0051] La administración tópica al pulmón se puede lograr mediante el uso de una formulación de aerosol. Las formulaciones en aerosol comprenden típicamente el ingrediente activo suspendido o disuelto en un propulsor de aerosol adecuado, tal como un clorofluorocarbono (CFC) o un hidrofluorocarbono (HFC). Los propulsores de CFC adecuados incluyen tricloromonofluorometano (propulsor 11), diclorotetrafluoroetano (propulsor 114) y diclorodifluorometano (propulsor 12). Los propulsores HFC adecuados incluyen tetrafluoroetano (HFC-134a) y heptafluoropropano (HFC-227). El propulsor comprende típicamente del 40% al 99,5%, por ejemplo, del 40% al 90% en peso de la composición total para inhalación. La formulación puede comprender excipientes que incluyen codisolventes (p. ej., etanol) y tensioactivos (p. ej., lecitina, trioleato de sorbitán y similares). Otros posibles excipientes incluyen polietilenglicol, polivinilpirrolidona, glicerina y similares. Las formulaciones en aerosol se envasan en botes y se administra una dosis adecuada por medio de una válvula dosificadora (p. ej., la suministrada por Bespak, Valois o 3M o alternativamente por Aptar, Coster o Vari).
[0052] La administración tópica al pulmón puede también lograrse mediante el uso de una formulación no presurizada tal como una solución o suspensión acuosa. Esto se puede administrar por medio de un nebulizador, por ejemplo, uno que puede ser de mano y portátil o para uso doméstico u hospitalario (es decir, no portátil). La formulación puede comprender excipientes tales como agua, tampones, agentes de ajuste de la tonicidad, agentes de ajuste del pH, tensioactivos y codisolventes. Líquido de suspensión y formulaciones de aerosol (si presurizadas o no presurizadas) contendrán típicamente el compuesto de la invención en forma finamente dividida, por ejemplo con un D50 de 0,5-10 pm por ejemplo, alrededor de 1-5 pm. Las distribuciones de tamaño de partícula se pueden representar usando valores D10, D50 y D90. El valor mediano D50 de las distribuciones de tamaño de partícula se define como el tamaño de partícula en micrones que divide la distribución a la mitad. La medición derivada de difracción de láser se describe con más precisión como una
distribución de volumen, y en consecuencia el valor D50 obtenido utilizando este procedimiento se denomina de manera más significativa como valor Dv50 (mediana para una distribución de volumen). Como se usa en el presente documento, los valores Dv se refieren a distribuciones de tamaño de partículas medidas usando difracción láser. De manera similar, los valores D10 y D90, utilizados en el contexto de la difracción láser, se toman como valores de Dv10 y Dvg0 y se refieren al tamaño de partícula en donde el 10% de la distribución se encuentra por debajo del valor D10, y el 90% de la distribución se encuentra por debajo del valor D90, respectivamente.
[0053] La administración tópica al pulmón puede conseguirse también mediante el uso de una formulación de polvo seco. Una formulación de polvo seco contendrá el compuesto de la divulgación en forma finamente dividida, típicamente con un diámetro aerodinámico de masa media (MMAD) de 1-10 pm o una D50 de 0,5-10 pm por ejemplo, alrededor de 1-5 pm. Los polvos del compuesto de la invención en forma finamente dividida se pueden preparar mediante un proceso de micronización o un proceso de reducción de tamaño similar. La micronización se puede realizar utilizando un molino de chorro como los fabricados por Hosokawa Alpine. La distribución de tamaño de partícula resultante puede medirse usando difracción láser (p. ej., con un instrumento Malvern Mastersizer 2000S). La formulación típicamente contener un diluyente tópicamente aceptable, tal como lactosa, glucosa o manitol (preferiblemente lactosa), por lo general de partícula grande tamaño, por ejemplo, un MMAD de 50 pm o más, por ejemplo 100 pm o más o una D50 de 40-150 pm. Como se usa en este documento, el término "lactosa" se refiere a un componente que contiene lactosa, que incluye a-lactosa monohidrato, p-lactosa monohidrato, a-lactosa anhidra, p-lactosa anhidra y lactosa amorfa. Los componentes de lactosa pueden procesarse mediante micronización, tamizado, molienda, compresión, aglomeración o secado por pulverización. También se incluyen las formas disponibles comercialmente de lactosa en diversas formas, por ejemplo, Lactohale® (lactosa de grado de inhalación; DFE Pharma), InhaLac®70 (lactosa tamizada para inhalador de polvo seco; Meggle), Pharmatose® (DFE Pharma) y Respitose® (tamizado lactosa de grado de inhalación; DFE Pharma). En una realización, el componente de lactosa se selecciona del grupo que consiste en a-lactosa monohidrato, a-lactosa anhidra y lactosa amorfa. Preferiblemente, la lactosa es a-lactosa monohidrato.
[0054] Las formulaciones de polvo seco pueden contener también otros excipientes tales como estearato de sodio, estearato de calcio o estearato de magnesio.
[0055] Una formulación de polvo seco es típicamente administrada usando un dispositivo de inhalador de polvo seco (DPI). Ejemplos de sistemas de suministro de polvo seco incluyen SPINHALER, DlSKHALER, TURBOHALe R, DISKUS y CLICKHALER. Otros ejemplos de sistemas de administración de polvo seco incluyen ECLIPSE, NEXT, ROTAHALER, HANDIHALER, AEROLISER, CYCLOHALER, BREEZHALER/NEOHALER, MONODOSIS, FLOWCAPS, TWINCAPS, X-CAPS, TURBOSPIN, ELPENHALER, MIATHERLER, NOVOLISTHALER, TWISTHALER inhalador de polvo, MICRODOSIS, PULVINAL, EASYHALER, ULTRAHALER, TAIFUN, PULMOJET, OMNIHALER, GYROHALER, TAPER, CONIX, XCELOVAIR y PROHALER.
[0056] En una realización se proporciona un compuesto de la presente invención en una formulación de polvo seco micronizado, por ejemplo que comprende además lactosa de un grado adecuado, opcionalmente junto con estearato de magnesio, se envasa en un dispositivo de dosis única tal como AEROLISER o llenado en un dispositivo de dosificación múltiple como DISKUS.
[0057] Los compuestos de la presente invención se pueden administrar también por vía rectal, por ejemplo en forma de supositorios o enemas, que incluyen soluciones acuosas u oleosas, así como suspensiones y emulsiones. Dichas composiciones se preparan siguiendo procedimientos estándar, bien conocidos por los expertos en la técnica. Por ejemplo, se pueden preparar supositorios mezclando el ingrediente activo con una base de supositorio convencional como manteca de cacao u otros glicéridos, por ejemplo, Suppocire. En este caso, el fármaco se mezcla con un excipiente no irritante adecuado que es sólido a temperaturas ordinarias pero líquido a la temperatura rectal y, por lo tanto, se derretirá en el recto para liberar el fármaco. Tales materiales son manteca de cacao y polietilenglicoles.
[0058] En general, para composiciones destinadas a ser administradas por vía tópica al ojo en forma de gotas para los ojos o los ojos ungüentos, la cantidad total del inhibidor será de aproximadamente 0,0001 a menos de 4,0% (p/p).
[0059] Preferentemente, para la administración ocular tópica, las composiciones administradas según la presente invención se formularán como soluciones, suspensiones, emulsiones y otras formas de dosificación. Generalmente se prefieren las soluciones acuosas, basándose en la facilidad de formulación, así como en la capacidad del paciente para administrar tales composiciones fácilmente mediante la instilación de una o dos gotas de las soluciones en los ojos afectados. Sin embargo, las composiciones también pueden ser suspensiones, geles viscosos o semiviscosos u otros tipos de composiciones sólidas o semisólidas. Se pueden preferir las suspensiones para compuestos que son escasamente solubles en agua.
[0060] Las composiciones administradas según la presente invención pueden incluir también otros diversos ingredientes, incluyendo, pero no limitado a agentes de tonicidad, tampones, agentes tensioactivos, polímero estabilizador, conservantes, co-disolventes y agentes de construcción de viscosidad. Las composiciones farmacéuticas preferidas de la presente invención incluyen el inhibidor con un agente de tonicidad y un tampón. Las composiciones farmacéuticas de la presente invención pueden incluir además opcionalmente un tensioactivo y/o un agente paliativo y/o un polímero estabilizador.
[0061] Pueden emplearse varios agentes de tonicidad para ajustar la tonicidad de la composición, preferiblemente a la de las lágrimas naturales para composiciones oftálmicas. Por ejemplo, cloruro de sodio, cloruro de potasio, cloruro de magnesio, cloruro de calcio, azúcares simples, como dextrosa, fructosa, galactosa y/o simplemente polioles, como los alcoholes de azúcar manitol, sorbitol, xilitol, lactitol, isomaltitol, maltitol y pueden añadirse hidrolizados de almidón hidrogenado a la composición de tonicidad fisiológica aproximada. Dicha cantidad de agente de tonicidad variará, dependiendo del agente particular que se añada. En general, sin embargo, las composiciones tendrán un agente de tonicidad en una cantidad suficiente para hacer que la composición final tenga una osmolalidad oftálmicamente aceptable (generalmente aproximadamente 150-450 mOsm, preferiblemente 250-350 mOsm y lo más preferiblemente a aproximadamente 290 mOsm). En general, los agentes de tonicidad de la invención se presentarán en el intervalo de 2 a 5% p/p. Los agentes de tonicidad preferidos de la invención incluyen los azúcares simples o los alcoholes de azúcar, tales como D-manitol.
[0062] Un sistema de tampón apropiado (p. ej., fosfato de sodio, acetato de sodio, citrato de sodio, borato de sodio o ácido bórico) se puede añadir a las composiciones para prevenir la deriva del pH en condiciones de almacenamiento. La concentración particular variará, dependiendo del agente empleado. Sin embargo, preferiblemente, el tampón será escogido para mantener un pH diana dentro del intervalo de pH 5 a 8.
[0063] Los tensioactivos opcionalmente pueden emplearse para suministrar mayores concentraciones de inhibidor. Los tensioactivos funcionan para solubilizar el inhibidor y estabilizar la dispersión coloidal, tal como solución micelar, microemulsión, emulsión y suspensión. Los ejemplos de tensioactivos que pueden usarse opcionalmente incluyen polisorbato, poloxámero, estearato de polioxil 40, aceite de ricino polioxílico, tiloxapol, tritón y monolaurato de sorbitán. Los tensioactivos preferidos para ser empleados en la invención tienen un "HLB" hidrófilo/lipófilo/balance en el intervalo de 12,4 a 13,2 y son aceptables para uso oftálmico, tales como TritonX114 y tiloxapol.
[0064] Los agentes adicionales que pueden añadirse a las composiciones oftálmicas de la presente invención son demulcentes que funcionan como un polímero estabilizante. El polímero estabilizador debe ser un ejemplo iónico/cargado con precedencia para uso ocular tópico, más específicamente, un polímero que lleva carga negativa en su superficie que puede exhibir un potencial zeta de (-)10-50 mV para la estabilidad física y capaz de hacer una dispersión en agua (es decir, soluble en agua). Un polímero estabilizador preferido de la invención sería polielectrolito, o polielectrolitos si hay más de uno, de la familia de poliacrilatos reticulados, tales como carbómeros, policarbofilo y Pemulen®, específicamente Carbómero 974p (ácido poliacrílico), en 0,1-0,5% p/p.
[0065] Otros compuestos también se pueden añadir a las composiciones oftálmicas de la presente invención para aumentar la viscosidad del vehículo. Los ejemplos de agentes potenciadores de la viscosidad incluyen, pero no se limitan a: polisacáridos, tales como ácido hialurónico y sus sales, condroitín sulfato y sus sales, dextranos, varios polímeros de la familia de la celulosa, polímeros vinílicos y polímeros de ácido acrílico.
[0066] Los productos oftálmicos tópicos se envasan típicamente en forma de multidosis. Por tanto, se requieren conservantes para evitar la contaminación microbiana durante el uso. Los conservantes adecuados incluyen: cloruro de benzalconio, clorobutanol, bromuro de benzododecinio, metil parabeno, propil parabeno, alcohol feniletílico, edentado disódico, ácido sórbico, policuaternio-1 u otros agentes conocidos por los expertos en la técnica. Dichos conservantes se emplean típicamente a un nivel de 0,001 a 1,0% p/v. Las composiciones de dosis unitarias de la presente invención serán estériles, pero típicamente no conservadas. Por lo tanto, dichas composiciones generalmente no contendrán conservantes.
[0067] El médico, u otra persona experta, será capaz de determinar una dosificación adecuada para los compuestos de la invención, y por tanto la cantidad del compuesto de la invención que debe ser incluido en cualquier particular, formulación farmacéutica (ya sea en la unidad forma de dosificación o de otro modo).
[0068] Las realizaciones de la invención que pueden mencionarse en relación con los productos de combinación descritos en (b) anteriormente incluyen aquellas en las que el otro agente terapéutico es uno o más agentes terapéuticos que los expertos en la técnica saben que son adecuados para tratar enfermedades inflamatorias (p. ej., las enfermedades específicas que se mencionan a continuación).
[0069] Por ejemplo, para el tratamiento de trastornos respiratorios (tales como COPD o asma), el otro agente terapéutico es uno o más agentes seleccionados de entre la lista que comprende:
- esteroides (p. ej., budesonida, dipropionato de beclometasona, propionato de fluticasona, furoato de mometasona, furoato de fluticasona; otro ejemplo es ciclesonida);
- agonistas beta, particularmente agonistas beta2 (p. ej., terbutalina, salbutamol, salmeterol, formoterol; otros ejemplos son vilanterol, olodaterol, reproterol y fenoterol); y
- xantinas (p. ej., teofilina).
[0070] Por ejemplo, para el tratamiento de trastornos respiratorios (tales como COPD o asma), el otro agente terapéutico es uno o más agentes seleccionados de entre la lista que comprende:
- antagonistas muscarínicos (p. ej., el tiotropio, umeclidinio, glicopirronio, aclidinio y daratropio, cualquiera de estos, por ejemplo, como la sal de bromuro); e
- inhibidores de la fosfodiesterasa.
[0071] Además, para el tratamiento de trastornos gastrointestinales (tales como enfermedad de Crohn o colitis ulcerosa), el otro agente terapéutico puede ser, por ejemplo, uno o más agentes seleccionados de la lista que comprende:
- ácido 5-aminosalicílico, o un profármaco del mismo (tal como sulfasalazina, olsalazina o balsalazida);
- corticosteroides (p. ej., prednisolona, metilprednisolona o budesonida);
- inmunosupresores (p. ej. ciclosporina, tacrolimus, metotrexato, azatioprina o 6-mercaptopurina);
- anticuerpos anti-TNFa (p. ej., infliximab, adalimumab, certolizumab pegol o golimumab);
- anticuerpos anti-IL12/IL23 (p. ej., ustekinumab) o inhibidores de IL12/IL23 de molécula pequeña (p. ej., apilimod); - anticuerpos anti-a4p7 (p. ej., vedolizumab);
- bloqueadores del receptor de tipo toll (TLR) (p. ej., BL-7040; Avecia (Cambridge, Reino Unido));
- bloqueadores MAdCAM-1 (p. ej., PF-00547659);
- anticuerpos contra la molécula de adhesión celular a4-integrina (p. ej., natalizumab);
- anticuerpos contra la subunidad a del receptor de IL2 (p. ej., daclizumab o basiliximab);
- anticuerpos anti-Smad7 (p. ej., mongersen (GED0301; al-P-ambo-2'-desoxi-P-tioguanilil-(3' ^ 5')-P-tiotimidilil-(3' ^ 5')-2'-desoxi-5-metil-P-tiocitidilil-(3' ^ 5')-2'-desoxi-P-tioguanilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-P-tiotimidilil-(3' ^ 5')-P-tiotimidilil-(3' ^ 5')-2'-desoxi-Ptiocitidilil-(3' ^ 5')-P-tiotimidil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-5-metil-P-tiocitidilil-(3' ^ 5')-2'-desoxi-Ptioguanilil-(3' ^ 5')-2'-desoxi-P-tiocitidilil-(3' ^ 5')-2'-desoxi-Ptioadenilil-(3' ^ 5')-2'-desoxi-Ptioguanilil-(3' ^ 5')-2'-desoxicitidina));
- moduladores del receptor de esfingosina 1-fosfato 1 (S1P1) (p. ej., ozanimod ((S)-5-(3-(1-((2-hidroxietil)amino)-2,3-dihidro-1H-inden-4-ilo)-1,2,4-oxadiazol-5-il)-2-isopropoxibenzonitrilo), amiselimod (MT1303;
- 2-amino-2-{2-[4-(heptiloxi)-3-(trifluorometil)fenil]etil}propano-1,3-diol) o APD334 (ácido 2-[7-[4-ciclopentil-3-(trifluorometil)benciloxi]-1,2,3,4-tetrahidrociclopenta[b]indol-3(R)-il]acético));
- inhibidores de STAT3 (p. ej., TAK-114; (3E)-1-metil-3-(2-oxo-lH-indol-3-iliden)indol-2-ona);
- Inhibidores de quinasas de espectro estrecho (p. ej., TOP1288);
- inhibidores de la quinasa de proteína 1 que interactúa con el receptor (RIP1) (p. ej., GSK2982772);
- inhibidores de Syk y profármacos de los mismos (p. ej., fostamatinib y R-406);
- Inhibidores de la fosfodiesterasa-4 (p. ej., tetomilast);
- HMPL-004;
- probióticos;
- moduladores de microbioma (p. ej., SGM1019);
- Dersalazina;
- semapimod/CPSI-2364; e
- inhibidores de la proteína quinasa C (p. ej., AEB-071).
[0072] Para el tratamiento de trastornos oculares (tales como uveítis y queratoconjuntivitis seca (ojo seco)), el otro agente terapéutico puede ser, por ejemplo, uno o más agentes seleccionados de la lista, que comprenden:
- corticosteroides (p. ej., dexametasona, prednisolona, acetónido de triamcinolona, difluprednato o acetónido de fluocinolona);
- agonistas de glucocorticoides (p. ej., mapracorat);
- inmunosupresores (p. ej., ciclosporina, voclosporina, azatioprina, metotrexato, micofenolato de mofetilo o tacrolimus); - anticuerpos anti-TNFa (p. ej., infliximab, adalimumab, certolizumab pegol, ESBA-105 o golimumab);
- anticuerpos anti-IL-17A (p. ej., secukinumab);
- inhibidores de mTOR (p. ej., sirolimus);
- VGX-1027;
- agonistas del receptor de adenosina A3 (p. ej., CF-101);
- lifitegrast;
- inhibidores de quinasas de espectro estrecho (p. ej., TOP1630);
- bloqueadores de IL1 (p. ej., EBI-005; Hou et al. PNAS 2013, 110 (10), 3913-3918);
- RGN-259 (timosina p 4); - SI-614; - OTX-101;
- inhibidores de JNK (p. ej., XG-104);
- inhibidores de la señalización de MAP quinasa (p. ej., DA-6034; ácido {[2-(3,4-dimetoxifenil)-5-metoxi-4-oxocromen-7-il]oxi}acético);
- estimuladores de mucina (p. ej., rebamipida; ácido 2 -[(4-clorobenzoil)amino]-3-(2-oxo-1H-quinolin-4-il)propanoico); - MIM-D3 (Tavilermide; ver, por ejemplo, US 2013/0345395); e
- inhibidores de la proteína quinasa C (p. ej., AEB-071).
Usos médicos
[0073] El compuesto de la invención puede ser utilizado como una monoterapia para enfermedades inflamatorias, o en terapias de combinación para tales enfermedades.
[0074] Por lo tanto, las realizaciones de los aspectos (e) a (g) anteriores que se pueden mencionar incluyen aquellos en los que el compuesto de fórmula I, Ia, Ib o Ic (o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo) es el único ingrediente farmacológicamente activo utilizado en el tratamiento.
[0075] Sin embargo, en otras realizaciones de los aspectos (e) a (g) arriba, el compuesto de fórmula I, Ia, Ib o Ic (o sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo) se administra a un sujeto al que también se le administra uno o más de otros agentes terapéuticos (p. ej., en los que uno o más de otros agentes terapéuticos son como se definieron anteriormente en relación con productos de combinación).
[0076] Cuando se utiliza aquí, el término "enfermedad inflamatoria" incluye específicamente las referencias a una cualquiera o más de las siguientes:
(i) enfermedades pulmonares o trastornos que tienen un componente inflamatorio, tales como la fibrosis quística, hipertensión pulmonar, sarcoidosis pulmonar, fibrosis idiopática pulmonar o, particularmente, EPOC (incluyendo bronquitis crónica y enfisema), asma o asma pediátrica;
(ii) enfermedades o trastornos de la piel que tienen un componente inflamatorio, como dermatitis atópica, dermatitis alérgica, dermatitis de contacto o psoriasis;
(iii) enfermedades o trastornos nasales que tienen un componente inflamatorio, como rinitis alérgica, rinitis o sinusitis;
(iv) enfermedades oculares o trastornos que tienen un componente inflamatorio, como conjuntivitis, conjuntivitis alérgica, glaucoma, retinopatía diabética, edema macular (incluido el edema macular diabético), oclusión de la vena central de la retina (OVCR), degeneración macular seca y/o húmeda relacionada con la edad (DMAE), inflamación de cataratas posoperatoria o, en particular, queratoconjuntivitis seca (ojo seco, también conocido como xeroftalmia), uveítis (que incluye uveítis posterior, anterior y panuveítis), injerto de córnea y rechazo de trasplante de células del limbo; y
(v) enfermedades o trastornos gastrointestinales que tienen un componente inflamatorio, tales como enteropatía sensible al gluten (enfermedad celíaca), esofagitis eosinofílica, enfermedad de injerto contra huésped intestinal o, particularmente, enfermedad de Crohn o colitis ulcerosa.
[0077] Las referencias en el presente documento a enfermedades que tienen un componente inflamatorio incluyen referencias a enfermedades que implican inflamación, haya o no otros síntomas (no inflamatorios) o consecuencias de la enfermedad.
[0078] De acuerdo con un aspecto adicional de la descripción, se proporciona un proceso para la preparación de un compuesto de fórmula I, cuyo procedimiento comprende:
(a) reacción de un compuesto de fórmula II,

en donde X, Ak, S, L y Q son como se definieron anteriormente y LG representa un grupo saliente (p. ej., halógeno, metanosulfonilo o triflato), con un compuesto de fórmula III,
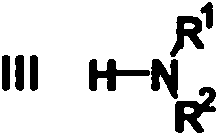
en donde R1 y R2 son como se definieron anteriormente, por ejemplo en condiciones conocidas por los expertos en la técnica., como a través de (i) una reacción típica de SNAr en presencia de un disolvente orgánico adecuado (p. ej., un disolvente aprótico polar como DMSO, DMF, THF o mezclas de los mismos) y una base (p. ej., DIPEA
o TEA) o (ii) mediante un acoplamiento Buchwald-Hartwig (Lundgren, RJ; Stradiotto, M. Aldrichim. Acta 2012, 45 (3), 59-65) empleando un catalizador de metal de transición, por ejemplo, paladio y un ligando adecuado, por ejemplo, BrettPhos (J. Am. Chem. Soc. 2008, 130, 13552-13554);
(b) para compuestos de fórmula I en donde L representa C(O), reacción de un compuesto de fórmula IV,

en donde X, Ak y s son como se definieron anteriormente, con un compuesto de fórmula VIII,

en donde Q, R1 y R2 son como se definen anteriormente, por ejemplo, en condiciones conocidas por los expertos en la técnica, como la reacción con el compuesto de fórmula VIII en condiciones estándar de formación de enlaces amida (p. ej., que implica un agente de acoplamiento, como T3P o HATU (documento WO2013/014567), opcionalmente en presencia de una base, como DIPEA, en un disolvente aprótico polar, como DMF);
(c) reacción de un compuesto de fórmula IV, como se define aquí anteriormente, con un compuesto de fórmula IX,

donde LG1 representa un grupo saliente (p. ej., halógeno o sulfonato) y L, Q, R1 y R2 son como se definen anteriormente, por ejemplo, en condiciones conocidas por los expertos en la técnica, como en presencia de una base, como DIPEA, y un disolvente inerte adecuado (p. ej., DMF), opcionalmente a temperaturas elevadas de hasta 50°C;
(d) para compuestos de fórmula I en donde L representa CH2, reacción de un compuesto de fórmula IV, como se define anteriormente, con un compuesto de fórmula X,
O 1
H
X
^Q -N ,R , X
'r 2
en donde Q, R1 y R2 son como se definen anteriormente, por ejemplo en condiciones conocidas por los expertos en la técnica, tales como condiciones de alquilación reductora estándar en presencia de un agente reductor (p. ej., triacetoxiborohidruro de sodio: J. Am. Chem. Soc. 1996, 61, 3849-3862) y un disolvente inerte adecuado (p. ej., 1,2-dicloroetano, THF o DMF), opcionalmente en presencia de ácido acético; o
(e) desprotección de un derivado protegido de un compuesto de fórmula I, en condiciones conocidas por los expertos en la técnica, en donde el derivado protegido lleva un grupo protector en un átomo de O o N del compuesto de fórmula I (y, para evitar la duda, un derivado protegido de un compuesto de fórmula I puede o puede no representar otro compuesto de fórmula I).
[0079] Los ejemplos de derivados protegidos de compuestos de fórmula I incluyen aquellos en los que:
- un átomo de O está protegido con un grupo bencilo, cuyo grupo bencilo puede eliminarse mediante hidrogenación, por ejemplo en presencia de un catalizador de paladio (tal como Pd/C);
- un átomo de O de un ácido (p. ej., un ácido carboxílico) está protegido con un grupo alquilo (como metilo, etilo o terc-butilo), cuyo grupo alquilo puede eliminarse mediante hidrólisis básica (p. ej., para grupos metilo o etilo, por una reacción de hidrólisis usando un hidróxido de metal alcalino tal como hidróxido de sodio) o hidrólisis ácida (p. ej., para un grupo terc-butilo, por una reacción de hidrólisis usando un ácido tal como ácido trifluoroacético);
- un átomo de N de una amina está protegido con un grupo carbamato, tal como un bencilo o terc-butil carbamato, cuyos grupos pueden eliminarse en condiciones similares a los utilizados para eliminar grupos de bencilo o terc-butilo de átomos de O.
[0080] El compuesto de fórmula II puede prepararse de acuerdo con procedimientos similares a los descritos en WO2013/014567. Por tanto, el compuesto de fórmula II se puede producir (esquema 1) condensando una amina de fórmula IV con (i) un ácido de fórmula V en condiciones estándar de formación de enlace amida (p. ej., que implica un agente de acoplamiento, como T3P o HATU, opcionalmente en presencia de una base, como DIPEA, en un disolvente aprótico polar, como DMF), (ii) un electrófilo de fórmula VI, es decir, un cloruro de ácido, cloruro de sulfonilo, haluro de alquilo o haluro de arilo activado, donde LG1 representa un grupo saliente (p. ej., halógeno o sulfonato) desplazado preferentemente antes de LG, en presencia de una base, como DIPEA, y un disolvente inerte adecuado (p. ej., DMF), opcionalmente a temperaturas elevadas de hasta 50°C, o (iii) un aldehído de fórmula VII en presencia de un agente reductor (p. ej., triacetoxiborohidruro de sodio: J. Am. Chem. Soc. 1996, 61, 3849-3862) y un disolvente inerte adecuado (p. ej., 1,2- dicloroetano, THF o DMF), opcionalmente en presencia de ácido acético.

[0081] Los compuestos de fórmula VIII, IX y X se pueden preparar por reacción de un compuesto de fórmula III, como se ha definido, con un compuesto de fórmula V, VI o VII, respectivamente, a través de cualquiera de los acoplamientos SNAr o Buchwald-Hartwig, como se describe arriba.
[0082] Los compuestos de fórmula III, V, VI y VII son o bien comercialmente disponibles, o se pueden obtener usando los procedimientos citados, o pueden prepararse fácilmente por métodos convencionales por los expertos en la técnica.
[0083] Los aspectos de la invención descritos en este documento (p. ej., los compuestos, combinaciones, métodos y usos mencionados anteriormente) pueden tener la ventaja de que, en el tratamiento de las condiciones descritas en el presente documento, que pueden ser más convenientes para el médico y/o paciente que, ser más eficaces que, ser menos tóxicos que, tener mejor selectividad, tener un rango de actividad más amplio que, ser más potentes que, producir menos efectos secundarios que, tener un mejor perfil farmacocinético y/o farmacodinámico que, tener más morfología de estado sólido adecuada que, tener mejor estabilidad a largo plazo que, o pueden tener otras propiedades farmacológicas útiles sobre compuestos, combinaciones, métodos (tratamientos) o usos similares conocidos en la técnica anterior para su uso en el tratamiento de esas afecciones o de otro modo.
[0084] El compuesto de la invención puede además (o alternativamente):
- exhibir una larga duración de acción y/o persistencia de la acción (p. ej., en comparación con el compuesto de referencia);
- ser más potente en los ensayos de enzimas JAK bioquímicos (p. ej., en comparación con el compuesto de referencia); - mostrar valores menores CI50 en ensayos celulares que evalúan la liberación de citoquinas (p. ej., en comparación con el compuesto de referencia);
- producir concentraciones sistémicas más bajas después de la dosificación oral (p. ej., en comparación con el compuesto de referencia);
- mantener una concentración de fármaco local relativamente alta entre dosis (p. ej., una concentración local alta en relación con otros inhibidores de JAK descritos anteriormente);
- exhiben propiedades que son particularmente adecuadas para la administración tópica/local (p. ej., después de la administración tópica/local, la generación de concentraciones tisulares altas pero concentraciones plasmáticas bajas de los compuestos de fórmula (I) y/o una rápida eliminación de los compuestos de fórmula (I) del plasma, por ejemplo como resultado de una elevada extracción renal o hepática);
- mostrar citotoxicidades reducidas (p. ej., en comparación con el compuesto de referencia); y/o
- exhiben solubilidades mejoradas (p. ej., en comparación con el compuesto de referencia) en medios biológicamente relevantes, por ejemplo, en líquido colónico simulado en estado de ayuno (FaSSCoF; Vertzoni, M., et al. Pharm. Res.
2010, 27, 2187-2196).
SECCIÓN EXPERIMENTAL
[0085] Se definen a continuación las abreviaturas utilizadas en la presente memoria. Cualquier abreviatura no definida pretende transmitir su significado generalmente aceptado.
Ejemplos de química
Procedimientos generales
[0086] Todos los materiales de partida y disolventes se obtuvieron de fuentes comerciales o se prepararon de acuerdo con la cita de la literatura. A menos que se indique lo contrario, se agitaron todas las reacciones. Las soluciones orgánicas se secaron de forma rutinaria sobre sulfato de magnesio anhidro.
Cromatografía líquida de alta eficacia de fase inversa preparativa
[0087] Realizada usando detección UV a 210-400 nm con una columna Waters X-Bridge Prep-C18, 5 pm, columna 19x50 mm eluyendo con un gradiente de H2O-MeCN que contiene 0,1% de bicarbonato de amonio durante 10 min.
Métodos analíticos
Cromatografía líquida de alta eficacia de fase inversa
[0088] Método 1: Agilent Infinity, X-Select, Waters X-Select C18, 2,5 pm (4,6 x 30 mm) a 40°C; velocidad de flujo 2,5 a 4,5 ml min'1 eluyó con un gradiente H2O-MeCN que contiene 0,1% v/v de ácido fórmico durante 4 min empleando detección UV a 254 nm. Información del gradiente: 0-3,00 min, aumento de 95% de H2O-5% de MeCN a 5% de H2O-95% de MeCN; 3,00-3,01 min, mantenido a 5% de H2O-95% de MeCN, la velocidad de flujo aumentó a 4,5 ml min-1; 3,01-3,50 min, mantenido a 5% de H2O-95% de MeCN; 3,50-3,60 min, volvió a H2O al 95% - MeCN al 5%, el caudal se redujo a 3,50 ml min-1; 3,60-3,90 min, mantenido a 95% de H2O-5% de MeCN; 3,90-4,00 min, mantenido a 95% H2O-5% MeCN, velocidad de flujo reducida a 2,5 ml min-1.
[0089] Método 2: Waters Acquity UPLC C18, 1,7 pm (2,1 x 50 mm) a 40°C; inyectar volumen 2 pL; velocidad de flujo 0,77 ml min-1 eluyó con un gradiente de H2O-MeCN que contiene bicarbonato de amonio 10 mM durante 3 min empleando detección UV a 210-400 nm. Información del gradiente: 0-0,11 min, mantenido a 95% de H2O-5% de MeCN; 0,11-2,15 min, rampa de 95% H2O-5% de MeCN al 5% H2O-95% de MeCN; 2,15-2,49 min, mantenido a 5% de H2O-95% de MeCN; 2,49-2,56 min, devuelto a 95% de H2O-5% de MeCN; 2,56-3,00 min, mantenido a 95% de H2O-5% de MeCN.
Espectroscopia 1H RMN
[0090] Los espectros 1H RMN se adquirieron en un espectrómetro de BrukerAvance III a 400 MHz usando un disolvente no deuterado residual como referencia y a menos que se especifique de otro modo se realizaron en DMSO-d6.
Preparación del compuesto de la invención
Ejemplo 1
(2-(6-(2-Etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazor4,5-c1piridin-5-il)(5-(metil(1-met¡lp¡perid¡n-4-¡l)am¡no)p¡raz¡n-2-¡l)metanona
[0091]

(i) edio
edio 5-Clorop¡raz¡n-2-¡lW2-í6-í2-et¡l-5-fluoro-4-h¡drox¡fen¡l)-1H-¡ndazol-3-ilo)-1.4.6.7-tetrah¡dro-5H-¡m¡dazor4.5-clp¡r¡d¡n-5-il)metanona
5-Clorop¡raz¡n-2-¡lW2-í6-í2-et¡l-5-fluoro-4-h¡drox¡fen¡l)-1H-¡ndazol-3-ilo)-1.4.6.7-tetrah¡dro-5H-¡m¡dazor4.5-clp¡r¡d¡n-5-il)metanona
[0092]

[0093] A una solución agitada de 5-et¡l-2-fluoro-4-(3-(4,5,6,7-tetrah¡dro-1H-¡m¡dazo[4,5-c]p¡rid¡n-2-¡l)-1H-¡ndazol-6-il)fenol.2HCl (Coe, J.W., et al. WO2013/014567, 31 de enero de 2013; 500 mg, 1,11 mmol), ácido 5-cloropirazina-2-carboxílico (176 mg, 1,11 mmol) y DIPEA (970 ml, 5,55 mmol) en DMF (10 ml) se añadió T3P (50% en EtOAc) (1,90 ml, 3,33 mmol) y la solución amarilla resultante se dejó agitar a ta durante 2 h. La mezcla de reacción se diluyó con agua (10 ml) para formar un precipitado. El precipitado se recogió por filtración y luego se lavó con agua (10 ml) y éter dietílico (10 ml) para dar un sólido de color amarillo pálido. El sólido se disolvió en MeOH (15 ml) y se concentró al vacío para producir el compuesto del subtítulo como un sólido amarillo (521 mg).
1H RMN (400 MHz, DMSO-d6) 8: 13,30-13,19 (m, 1H), 12,58 (s, 1H), 8,88 (m, 1H), 8,78 (m, 1H), 8,34 y 8,25 (2 x d, 1H), 7,39 y 7,37 (2 x s, 1H), 7,15-6,98 (m, 2H), 6,96-6,87 (m, 1H), 4,76 y 4,59 (2x s, 2H), 4,04 (t, 1H), 3,73 (t, 1H), 2,89 (s, 1H), 2,84-2,71 (m, 2H), 2,49-2,43 (m, 1H), 1,30-1,20 (m, 1H), 1,01 (m, 3H). CLEM m/z 518 (M+H)+ (ES+)
(ji)________ (2-(6-(2-et¡l-5-fluoro-4-h¡drox¡fen¡l)-1H-¡ndazol-3-¡l)-1.4.6.7-tetrah¡dro-5H-¡m¡dazo[4.5-clp¡r¡d¡n-5-¡l)(5-(met¡l(1-met¡lp¡perid¡n-4-¡l)am¡no)p¡raz¡n-2-¡l)metanona
[0094]

[0095] Se añadió una solución del producto de la etapa (i) (Intermedio A; 75,0 mg, 0,145 mmol) en DMSO (2 ml) a N, 1-dimetilpiperidin-4-amina (4,35 mg, 0,290 mmol), seguido de DIPEA (101 pL, 0,579 mmol). La solución se agitó a ta durante 18 h. La mezcla de reacción se filtró y luego se purificó mediante HPLC preparativa (Waters, Basic (bicarbonato de amonio al 0,1%), Basic, Waters X-Bridge Prep-C18, 5 pm, columna de 19x50 mm, MeCN al 10-40% en agua) para producir el compuesto del título como un sólido blanco (16,3 mg).
1H RMN (400 MHz, DMSO-d6) 8: 13,22 (s, 1H), 12,52 y 12,27 (2 x s, 1H), 9,86 (s, 1H), 8,43-8,38 (m, 1H), 8,37-8,21 (m, 1H), 8,16 (s, 1H), 7,37 (s, 1H), 7,16-6,97 (m, 2H), 6,91 (d, 1H), 4,83-4,62 (m, 2H), 4,48-4,36 (m, 1H), 4,01-3,86 (m, 2H), 3,30 (s, 2H), 2,99 (s, 3H), 2,90-2,73 (m, 4H), 2,18 (s, 3H), 2,02 (t, 2H), 1,90-1,75 (m, 2H), 1,57 (d, 2H), 1,02 (t, 3H). CLEM m/z 610,5 (M+H)+ (ES+)
Ejemplo de referencia 2
[0096] Los siguientes compuestos de la descripción se prepararon haciendo reaccionar la amina apropiada con el Intermedio A empleando métodos análogos a los descritos anteriormente para el Ejemplo 1 (p. ej., a temperaturas de 20-90°C). La única excepción fue el Ejemplo 2(a), que se preparó haciendo reaccionar el Intermedio A con 4-(metilamino)butanoato de metilo, luego el éster metílico resultante se aisló y saponificó con LiOH, antes de purificarse por RP-HPLC, para producir el correspondiente ácido.
(a) 4-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)butanoico
[0097]

[0098] 1H RMN (400 MHz, DMSO-de) 5: 13,22 (s, 1H), 12,51 (s, 1H), 9,95 (s, 1H), 8,47-8,23 (m, 2H), 8,17 (s, 1H), 7,37 (s, 1H), 7,16-6,97 (m, 2H), 6,92 (d, 1H), 4,91-4,60 (m, 2H), 3,94 (s, 3H), 3,60 (t, 2H), 3,12 (d, 3H), 2,79 (s, 4H), 2,25-2,17 (m, 2H)), 1,84-1,72 (m, 2H), 1,02 (t, 3H). CLEM m/z 599,5 (M+H)+ (ES+)
(b) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-((2-(4-hidroxipiperidin-1-il)etil)amino)pirazin-2-il)metanona
[0099]

[0100] 1H RMN (400 MHz, DMSO-cfe) 5: 13,22 (s, 1H), 12,52 y 12,26 (2 x s, 1H), 9,81 (s, 1H), 8,43-8,20 (m, 2H), 7,95 (d, 1H), 7,51 (s, 1H), 7,37 (s, 1H), 7,17-6,97 (m, 2H), 6,91 (d, 1H), 4,86-4,59 (m, 2H), 4,54 (t, 1H), 3,91 (s, 2H), 3,49-3,37 (m, 3H), 2,85-2,69 (m, 4H), 2,49-2,41 (m, 4H), 2,06 (t, 2H), 1,76-1,63 (m, 2H), 1,45-1,31 (m, 2H), 1,02 (t, 3H). CLEM m/z 626,5 (M+H)+ (ES+)
(c) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(metil(piridin-2-ilmetil)amino)pirazin-2-il)metanona
[0101]

[0102] 1H RMN (400 MHz, DMSO-d6) 5: 13,23 (s, 1H), 12,53 y 12,28 (2 x s, 1H), 9,79 (s, 1H), 8,57-8,49 (m, 1H), 8,45 8,17 (m, 3H), 7,81-7,71 (m, 1H), 7,38 (s, 1H), 7,33-7,23 (m, 2H), 7,16- 6,99 (m, 2H), 6,92 (d, 1H), 4,96 (s, 2H), 4,84-4,62 (m, 2H), 3,93 (s, 2H), 3,26 (s, 3H), 2,79 (s, 2H), 2,50-2,44 (m, 2H), 1,02 (t, 3H). CLEM m/z 604,5 (M+H)+ (ES+)
(d) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-((2-(1-oxidotiomorfolino)etil)amino)pirazin-2-il)metanona
[0103]

[0104] 1H RMN (400 MHz, m etano l^) 5: 8,39 (d, 1H), 8,28 (s, 1H), 7,96 (d, 1H), 7,41 (s, 1H), 7,16 (s, 1H), 6,92 (dd, 2H), 4,95-4,89 (asumiendo 2H, oscurecido por solvente), 4,06 (s, 2H), 3,59 (t, 2 H), 3,38-3,34 (asume 4H, oscurecido por solvente), 3,18-2,81 (m, 10H), 2,73 (t, 2H), 2,55 (q, 2H), 1,07 (t, 3H). CLEM m/z 644,5 (M+H)+ (ES+)
(e) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(1 -oxidotiomorfolino)pirazin-2-il)metanona
[0105]

[0106] 1H RMN (400 MHz, DMSO-d6) 5: 13,21 (s, 1H), 12,52 y 12,29 (2 x s, 1H), 9,80 (s, 1H), 8,52-8,41 (m, 2H), 8,41 8,19 (m, 1H), 7,38 (s, 1H), 7,18-6,98 (m, 2H), 6,91 (d, 1H), 4,91-4,59 (m, 2H), 4,44-4,25 (m, 2H), 4.08-3,82 (m, 4H), 2,97 (ddd, 2H), 2,88-2,70 (m, 4H), 2,49-2,43 (asumiendo 2H, oscurecido por disolvente), 1,02 (t, 3H). CLEM m/z 601,4 (M+H)+ (ES+)
(f) (5-(4-(2-(dimetilamino)etil)piperazin-1-il)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)metanona
[0107]

[0108] 1H RMN (400 MHz, DMSO-d6) 5: 13,21 (s, 1H), 12,51 y 12,26 (2 x s, 1H), 9,85 (s, 1H), 8,45-8,20 (m, 3H), 7,37 (s, 1H), 7,18-6,96 (m, 2H), 6,91 (d, 1H), 4,83-4,60 (m, 2H), 4.03-3,82 (m, 2H), 3,72-3,59 (m, 4H), 2,78 (s, 2H), 2,54-2,51 (suponga 4H, oscurecido por disolvente), 2,48-2,34 (m, 6H), 2,15 (s, 6 H), 1,01 (t, 3H). CLEM m/z 639,6 (M+H)+ (ES+)
(g) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-((2-(4-hidroxi-1-metilpiperidin-4-il)etil)amino)pirazin-2-il)metanona
[0109]

[0110] 1H RMN (400 MHz, DMSO-cfe) 5: 13,22 (s, 1H), 12,52 y 12,27 (2 x s, 1H), 9,68 (s, 1H), 8,43-8,21 (m, 2H), 7,89 (d, 1H), 7,53 (s, 1H), 7,38 (s, 1H), 7,17-6,97 (m, 2H), 6,92 (d, 1H), 4,85-4,59 (m, 2H), 4,18 (d, 1H), 3,92 (s, 2H), 3,46-3,36 (m, 2H), 2,86-2,71 (m, 2H), 2,49-2,43 (asumiendo 2H, oscurecido por solvente), 2,42-2,20 (m, 4H), 2,14 (s, 3H), 1,74-1,62 (m, 2H), 1,58-1,44 (m, 4H), 1,02 (t, 3H). CLEM m/z 640,5 (M+H)+ (ES+)
(h) (S)-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-ilo)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)(5-(2-(hidroximetilo)pirrolidin-1-il)pirazin-2-il)metanona
[0111]

[0112] 1H RMN (400 MHz, DMSO-d6) 5: 13,22 (s, 1H), 12,52 y 12,29 (2 x s, 1H), 9,83 (s, 1H), 8,49-8,20 (m, 2H), 8,04 (s, 1H), 7,38 (s, 1H), 7,20-6,96 (m, 2H), 6,92 (d, 1H), 4,95-4,58 (m, 3H), 4,24-4,11 (m, 1H), 4,08-3,86 (m, 2H), 3,67-3,51 (m, 2H), 3,46-3,36 (asumiendo 2H, oscurecido por solvente), 2,80 (s, 2H), 2,49-2,41 (asumiendo 2H, oscurecido por solvente), 2,13-1,86 (m, 4H), 1,02 (t, 3H). CLEM m/z 583,5 (M+H)+ (ES+)
(i) (5-(bis(2-metoxietil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro)-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridin-5-il)metanona
[0113]

[0114] 1H RMN (400 MHz, DMSO-d6) 5: 13,22 (s, 1H), 12,52 y 12,28 (2 x s, 1H), 9,83 (s, 1H), 8,44-8,12 (m, 3H), 7,37 (s, 1H), 7,19-6,96 (m, 2H), 6,91 (d, 1H), 4,88-4,55 (m, 2H), 4,05-3,84 (m, 2H), 3,78 (t, 4H), 3,55 (t, 4H), 3,26 (s, 6H), 2,79 (s, 2H), 2,49-2,43 (asumiendo 2H, oscurecido por el disolvente), 1,01 (t, 3H). CLEM m/z 615,5 (M+H)+ (ES+)
(j) (5-(4-(2-( 1H-imidazol-1-il)etil)piperazin-1-il)pirazin-2-ilo)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-1,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridina-5-il)metanona
[0115]

[0116] 1H RMN (400 MHz, DMSO-d6) 5: 13,22 (s, 1H), 12,53 y 12,28 (2 x s, 1H), 9,87 (s, 1H), 8,49-8,19 (m, 3H), 7,66 (s, 1H), 7,38 (s, 1H), 7,21 (s, 1H), 7,18-6,97 (m, 2H), 6,97-6,83 (m, 2H), 4,87-4,57 (m, 2H), 4,13 (t, 2H), 4,05-3,82 (m, 2H), 3,73-3,61 (m, 4H), 2,79 (s, 2H), 2,69 (t, 2H), 2,60-2,53 (m, 4H), 2,50-2,42 (asumiendo 2H, oscurecido por el disolvente), 1,02 (t, 3H). CLEM m/z 662,5 (M+H)+ (ES+)
(k) 1-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)pirrolidin-2-ona
[0117]

[0118] 1H RMN (400 MHz, DMSO-da) 8: 13,22 (s, 1H), 12,52 y 12,31 (2 x s, 1H), 9,83 (s, 1H), 8,46-8,21 (m, 2H), 8,12 (s, 1H), 7,38 (s, 1H), 7,23-6,97 (m, 2H), 6,92 (d, 1H), 4,93-4,60 (m, 2H), 4,07-3,86 (m, 2H), 3,79 (t, 2H), 3,46-3,37 (m, 4H), 3,12 (s, 3H), 2,80 (s, 2H), 2,48-2,44 (asumiendo 2H, oscurecido por solvente), 2,05 (t, 2H), 1,88-1,73 (m, 2H), 1,02 (t, 3H). CLEM m/z 624,6 (M+H)+ (ES+)
(I) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-(4-(2-hidroxietil)piperazin-1-il)pirazin-2-il)metanona
[0119]

[0120] 1H RMN (400 MHz, DMSO-d6) 8: 13,23 (s, 1H), (2 x s, 12,53 y 12,28, 1H), 9,87 (s, 1H), 8,45-8,38 (m, 1H), 8,37 8,22 (m, 2H), 7,41-7,34 (m, 1H), 7,15-6,98 (m, 2H), 6,91 (d, 1H), 4,81-4,61 (m, 2H), 4,47 (t, 2H), 4.02-3,80 (m, 2H), 3,70 3,61 (m, 4H), 3,55 (q, 2H), 2,84-2,72 (m, 2H), 1,01 (t, 3H). 7H por debajo del pico DMSO. CLEM m/z 612 (M+H)+ (ES+)
(m) (2-(6-(2-etN-5-fluoro-4-hidroxifeml)-1H-indazol-3-N)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-N)(5-((2-(2-(2-metoxietoxi) etoxi)etil)amino)pirazin-2-il)metanona
[0121]

[0122] 1H RMN (400 MHz, DMSO-d6) 8: 13,23 (s, 1H), (2 x s, 12,53 y 12,28, 1H), 9,87 (s, 1H), 8,45-8,38 (m, 1H), 8,37 8,22 (m, 2H), 7,41-7,34 (m, 1H), 7,15-6,98 (m, 2H), 6,91 (d, 1H), 4,81-4,61 (m, 2H), 4,47 (t, 2H), 4.02-3,80 (m, 2H), 3,70 3,61 (m, 4H), 3,55 (q, 2H), 2,84-2,72 (m, 2H), 1,01 (t, 3H). 7H por debajo del pico DMSO. CLEM m/z 645 (M+H)+ (ES+)
(n) N-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)-N-metilacetamida

[0124] 1H RMN (400 MHz, DMSO-da) 8: 13,23 (s, 1H), (2 x s, 12,53 y 12,31, 1H), 9,88 (s, 1H), 8,40 (dd, 1H), 8,38-8,17 (m, 1H), 8,15 (d, 1H), 7,37 (s, 1H), 7,10 (d, 1H), 7.03 (d, 1H), 6,91 (d, 1H), 4,82-4,66 (m, 2H), 3,99-3,86 (m, 2H), 3,76 (dt, 2H), 3,50 (q, 2H), (2 x s, 3,17 y 3,11, 3H), 2,97 (s, 2H), 2,84 (s, 1H), 2,84-2,70 (m, 2H), (2 x s, 1,92 y 1,96, 3H), 1,01 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 612 (M+H)+ (ES+)
(o) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-(3-(hidroximetilo) azetidin-1-il)pirazin-2-il)metanona
[0125]

[0126] 1H RMN (400 MHz, DMSO-d6) 8: 13,23 (s, 1H), (2 x s, 12,54 y 12,29, 1H), 9,87 (s, 1H), 8,40-8,22 (m, 2H), 7,86 (s, 1H), 7,38 (s, 1H), 7,11 (d, 1H), 7,03 (d, 1H), 6,92 (d, 1H)), 4,87-4,79 (m, 1H), 4,74-4,66 (m, 2H), 4,14 (t, 2H), 3,96 (s, 1H), 3,88 (t, 3H), 3,61 (t, 2H), 2,96- 2,73 (m, 3H), 1,02 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 569 (M+H)+ (ES+) (p) (5-(bis(2-hidroxietil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro)-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)metanona
[0127]

[0128] 1H RMN (400 MHz, DMSO-d6) 8: 13,23 (s, 1H), (2 x s, 12,53 y 12,28, 1H), 9,89 (s, 1H), 8,42-8,27 (m, 2H), 8,20 (s, 1H)), 7,37 (s, 1H), 7,10 (d, 1H), 7,02 (d, 1H), 6,92 (d, 1H), 4,87-4,68 (m, 2H), 4,77-4,58 (m, 2H), 4,00- 3,86 (m, 2H), 3,73 3,57 (m, 7 H), 2,86-2,70 (m, 2H), 1,01 (t, 3H). 3H por debajo del pico DMSO. CLEM m/z 587 (M+H)+ (ES+)
(q) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((2-(metilsulfonil)etil)amino)pirazin-2-il)metanona
[0129]
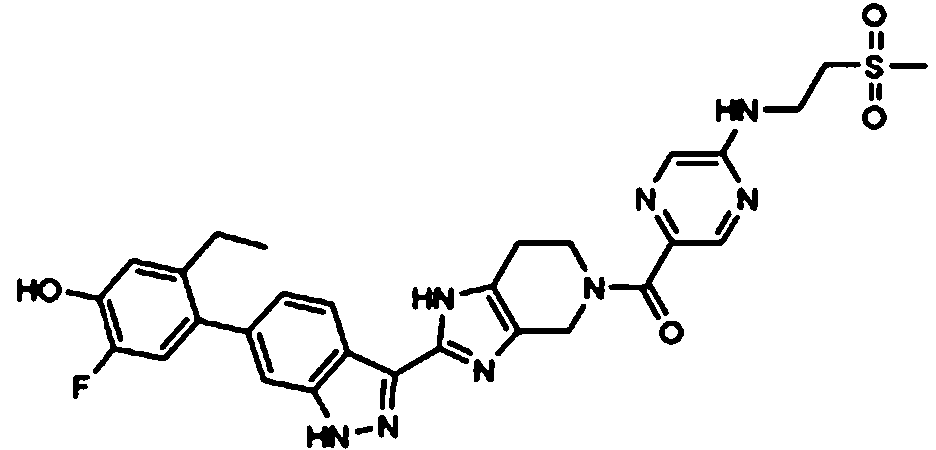
[0130] 1H RMN (400 MHz, DMSO-da) 5: 13,23 (s, 1H), (2 x s, 12,53 y 12,27, 1H), 9,88 (s, 1H), 8,81-8,19 (m, 2H), 7,98 (s, 1H), 7,94-7,85 (m, 1H), 7,38 (s, 1H), 7,11 (d, 1H), 7,02 (d, 1H), 6,92 (d, 1H), 4,84-4,60 (m, 2H), 4,00-3,83 (m, 2H), 3,76 (q, 2H), 3,41 (t, 2H), 3,05 (s, 3H), 2,85-2,61 (m, 2H), 1,02 (t, 3H). 2H por debajo del pico DMSO.
CLEM m/z 605 (M+H)+ (ES+)
(r) 1-(2-((5-(2-(6-(2-etN-5-fluoro-4-hidroxifeml)-1H-indazol-3-N)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]pindin-5-carbonil)pirazin-2-il)amino)etil)pirrolidin-2-ona
[0131]

[0132] 1H RMN (400 MHz, DMSO-d6) 5: 13,22 (s, 1H), (2 x s, 12,53 y 12,27, 1H), 9,87 (s, 1H), 8,38-8,25 (m, 2H), 7,89 (s, 1H), 7,75-7,52 (m, 1H), 7,37 (s, 1H), 7,10 (d, 1H), 7.03 (d, 1H), 6,92 (d, 1H), 4,81-4,57 (m, 2H), 3,99-3,80 (m, 2H), 3,52 3,43 (m, 2H), 3,42-3,33 (m, 4H), 23,75-2,71 (m, 2H), 2,17 (t, 2H), 1,89 (p, 2H), 1,01 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 610 (M+H)+ (ES+)
(s) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((2R,3R,4R,5S)-3,4,5-trihidroxi-2-(hidroximetilo)piperidina-1-il)pirazin-2-il)metanona
[0133]

[0134] 1H RMN (400 MHz, DMSO-d6) 5: 13,23 (s, 1H), (2 x s, 12,53 y 12,27, 1H), 9,88 (s, 1H), 8,41-8,22 (m, 3H), 7,38 (s, 1H), 7,18-7,07 (m, 1H), 7,01 (d, 1H), 6,91 (d, 1H), 5,42-4,93 (m, 3H), 4,91-4,58 (m, 3H), 4,39-4,32 (m, 1H), 4,27 (d, 1H), 4.03-3,87 (m, 2H), 3,83-3,64 (m, 4H), 3,56-3,51 (m, 1H), 3,43 (dd, 1H), 2,87-2,72 (m, 2H), 1,02 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 645 (M+H)+ (ES+)
(t) (2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)(5-((3aR,6aS)-5-metilhexahidropirrolo[3,4-c]pirrol-2(1H)-il)pirazin-2-il)metanona
[0135]

[0136] 1H RMN (400 MHz, DMSO-da) 5: 13,24 (s, 1H), (2 x s, 12,53 y 12,29, 1H), 9,91 (s, 1H), 8,49-8,27 (m, 2H), 8,00 (s, 1H), 7,38 (s, 1H), 7,11 (d, 1H), 7,03 (d, 1H), 6,92 (d, 1H), 4,81-4,67 (m, 2H), 4,00-3,84 (m, 2H), 3,76-3,71 (m, 2H), 3,44 (dd, 2H), 3,02-2,89 (m, 2H), 2,85-2,72 (m, 2H), 2,22 (s, 3H), 1,02 (t, 3H). 2H bajo pico de agua. 2H por debajo del pico DMSO. CLEM m/z 608 (M+H)+ (ES+)
(u) 3-(2-((5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonil)pirazin-2-il)(metil)amino)etil)oxazolidin-2-ona
[0137]

[0138] 1H RMN (400 MHz, DMSO-d6) 5: 13,23 (s, 1H), (2 x s, 12,54and 12,30, 1H), 9,87 (s, 1H), 8,45-8,40 (m, 1H), 8,37 8,24 (m, 1H), 8,15 (s, 1H), 7,38 (s, 1H), 7,07 (dd, 2H), 6,92 (d, 1H), 4,85-4,64 (m, 2H), 4,15 (t, 2H), 4,02-3,89 (m, 2H), 3,82 (t, 2H), 3,63 (t, 2H), 3,42 (t, 2H), 3,14 (s, 3H), 2,88-2,74 (m, 2H), 1,02 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 626 (M+H)+ (ES+)
(v) (5-((2-( 1,1 -dioxidoisotiazolidin-2-il)etil)(metil)amino)pirazin-2-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-6,7-dihidro-1H-imidazo[4,5-c]piridin-5(4H)-il)metanona
[0139]

[0140] 1H RMN (400 MHz, DMSO-d6) 5: 13,23 (s, 1H), (2 x s, 12,53 y 12,30, 1H), 9,88 (s, 1H), 8,45-8,39 (m, 1H), 8,36 8,22 (m, 1H), 8,16 (s, 1H), 7,38 (s, 1H), 7,11 (d, 1H), 7,02 (d, 1H), 6,92 (d, 1H), 4,83-4,67 (m, 2H), 4,01-3,87 (m, 2H), 3,81 (t, 2H), 3,30 (t, 2H), 3,20-3,11 (m, 7H), 2,85-2,73 (m, 2H), 2,18 (p, 2H), 1,02 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 660 (M+H)+ (ES+)
(w) (3aR,6aS)-5-(5-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-5-carbonilo)pirazin-2-il)tetrahidropirrolo[3,4-c]pirrol-1,3(2H,3aH)-diona
[0141]

[0142] 1H RMN (400 MHz, DMSO-da) 8: 13,23 (s, 1H), (2 x s, 12,53 y 12,26, 1H), 11,41 (s, 1H), 9,87 (s, 1H), 8,44 (s, 1H), 8,38-8,23 (m, 1H), 8,18 (s, 1H)), 7,43-7,35 (m, 1H), 7,15-6,99 (m, 2H), 6,92 (d, 1H), 4,83-4,64 (m, 2H), 4,07 (d, 2H), 4,00 3,83 (m, 2H)), 3,69-3,55 (m, 4H), 2,83-2,61 (m, 2H), 1,02 (t, 3H). 2H por debajo del pico DMSO. CLEM m/z 622 (M+H)+ (ES+)
Prueba biológica: Métodos experimentales
Ensayos de unión de enzimas (Kinomescan)
[0143] Actividades de unión de enzima de quinasa de los compuestos descritos en el presente documento se pueden determinar usando un ensayo patentado que mide la unión de competición dirigida al sitio activo a un ligando inmovilizado (Fabian, MA, et al., Nature Biotechnol. 2005, 23, 329-336). Estos ensayos pueden ser realizados por DiscoveRx (anteriormente Ambit; San Diego, CA). El porcentaje de inhibición producido por la incubación con un compuesto de prueba puede calcularse en relación con el control no inhibido.
Ensayos celulares
[0144] Los compuestos de la invención se estudiaron usando uno o más de los siguientes ensayos.
(a) Liberación de IFNv de células PBMC estimuladas CD3/IL2
[0145] PBMCs de sujetos sanos se separan de la sangre entera usando un gradiente de densidad (Lymphoprep, Axis-Shield Healthcare). Las células se añaden a una placa de 96 pocillos y el compuesto a la concentración deseada se añade 2 horas antes de la estimulación con una mezcla de anticuerpo monoclonal para CD3 (1 pg/ml, eBioscience) e IL2 recombinante humana (10 ng/ml, Peprotech). Después de 48 horas de incubación en condiciones normales de cultivo de tejidos, se recogen los sobrenadantes y se determina la liberación de IFNy mediante ELISA Sandwich (Duo-set, R&D System). El CI50 se determina a partir de la curva de respuesta a la dosis.
(b) Liberación de IL-2 e IFNv en células LPMC estimuladas por CD3/CD28 de los pacientes con EII
[0146] Las células mononucleares de lamina propria (LPMCs) se aíslan y purifican a partir de mucosa inflamada EII de muestras quirúrgicas o de mucosa normal de muestras quirúrgicas como a continuación: Se extrae la mucosa de las capas más profundas de las piezas quirúrgicas con un bisturí y se corta en fragmentos de 3-4 mm de tamaño. El epitelio se elimina lavando los fragmentos de tejido tres veces con EDTA 1 mM (Sigma-Aldrich, Poole, Reino Unido) en HBSS (Sigma-Aldrich) con agitación utilizando un agitador magnético, descartando el sobrenadante después de cada lavado. Posteriormente, la muestra se trata con colagenasa tipo 1A (1 mg/mL; Sigma-Aldrich) durante 1 h con agitación a 37°C. La suspensión celular resultante se filtra luego usando un colador celular de 100 pm, se lava dos veces, se resuspende en medio RPMI-1640 (Sigma-Aldrich) que contiene suero de ternero fetal al 10%, 100 U/mL de penicilina y 100 pg/mL de estreptomicina, y utilizado para cultivo celular.
[0147] Las LPMC recién aisladas (2 X 105 células/pocillo) se estimulan con 1 pg/ml de a-CD3/a-CD28 durante 48 h en presencia de control de DMSO o concentraciones apropiadas de compuesto. Después de 48 h, se retira el sobrenadante y se analiza la presencia de TNFa e IFNy mediante R&D ELISA. El porcentaje de inhibición de la liberación de citocinas por los compuestos de prueba se calcula en relación con la liberación de citocinas determinada para el control de DMSO (100%).
(c) Ensayo de citotoxicidad celular
[0148] Se añaden 1 x 105 células Jurkat (linfocitos T humanos inmortalizados) al número apropiado de pocilios de una placa de 96 pocillos en 100 j l de medio (RPMI suplementado con suero bovino fetal al 10%). Se añade 1 j l de control DMSO (concentración final 1,0% v/v) o compuesto de prueba (concentración final 20, 5 o 1 jg/m l) a los pocillos y se incuban a 37°C, 5% CO2. Después de 24 horas, la placa se centrifuga a 1200 rpm durante 3 minutos y se descarta el sobrenadante. Las células se resuspendieron luego en 150 j l (concentración final 7,5 jg/m l) de yoduro de propidio (PI) en PBS y se incubaron a 37°C, 5% de CO2 durante 15 minutos. Después de 15 minutos, las células se analizan mediante citometría de flujo (BD accuri) usando la ventana FL3. El % de viabilidad se calcula como el % de células que son PI negativas en los pocillos de prueba normalizados al control DMSO.
Cribado in vivo: farmacodinámica y actividad anti-inflamatoria
(i) La colitis inducida por DSS en ratones
[0149] Los ratones BDF1 macho no en ayunas de 10-12 semanas de edad se dosifican por sonda oral dos veces al día con vehículo, el artículo de referencia (5-ASA) o compuesto de prueba un día antes (Día -1) de la estimulación de la respuesta inflamatoria mediante tratamiento con dextrano sulfato sódico (DSS). El día 0 del estudio, se administra DSS (5% p/v) en el agua de bebida seguido de la dosificación BID del vehículo (5 ml/kg), referencia (100 mg/kg) o compuesto de prueba (5 mg/kg) durante 7 días. El agua potable con DSS se repone cada 3 días. Durante el estudio, los animales se pesan todos los días y se realizan observaciones de las heces y se registran como una puntuación, según la consistencia de las heces. En el momento del sacrificio del Día 6, se extrae el intestino grueso y se registran la longitud y el peso. Se toman secciones del colon para análisis de MPO, para determinar la infiltración de neutrófilos, o para puntuación histopatológica para determinar la gravedad de la enfermedad.
(ii) Colitis inducida por TNBS en ratones
[0150] Los ratones BDF1 macho no en ayunas de 10-12 semanas de edad se dosifican por sonda oral dos veces al día con vehículo (5 ml/kg), elemento de referencia (budesonida 2,5 mg/kg) o compuesto de prueba (1 o 5 mg/kg) un día antes (Día -1) de la estimulación de la respuesta inflamatoria por tratamiento con ácido 2,4,6-trinitrobencenosulfónico (TNBS) (15 mg/ml en etanol al 50%/50% de solución salina). El día 0 del estudio se administra TNBS (200 j l) por vía intracolónica a través de un catéter de plástico con la dosificación BID del vehículo, la referencia o el compuesto de prueba continuando durante 2 o 4 días. Durante el estudio, los animales se pesan todos los días y se realizan observaciones de las heces y se registran como una puntuación, según la consistencia de las heces. En el momento del sacrificio en el día 2 (o día 4), se extrae el intestino grueso y se registra la longitud y el peso. Se toman secciones del colon para la puntuación de histopatología para determinar la gravedad de la enfermedad.
(iii) Transferencia adoptiva en ratones
[0151] En el día de estudio 0, ratones hembra Balb/C se terminan y los bazos obtenidos para CD45RBalta de aislamiento de células (Usando protocolo de separación de células EII SCID). A continuación, se inyectan por vía intraperitoneal aproximadamente 4 x 105 células/ml de células CD45RBalta (100 jl/ratón) en animales SCID hembra. El día 14 del estudio, los ratones se pesan y se asignan al azar en grupos de tratamiento según el peso corporal. El día 14, los compuestos se administran dos veces al día, mediante sonda oral, en un volumen de dosis de 5 ml/kg. El tratamiento continúa hasta el día 49 del estudio, momento en donde se realiza la necropsia de los animales 4 horas después de la administración matutina. La longitud y el peso del colon se registran y se utilizan como criterio de valoración secundario en el estudio como medida del edema de colon. A continuación, el colon se divide en seis secciones transversales, cuatro de las cuales se utilizan para la puntuación de histopatología (criterio de valoración principal) y dos se homogeneizan para el análisis de citocinas. Los datos se dan como el % de inhibición de la ventana de inducción entre animales ingenuos y animales vehículo, donde una mayor inhibición implica más cerca del fenotipo no enfermo ingenuo.
(iv) Uveítis inducida por endotoxinas en ratas
[0152] Se colocan ratas machos Lewis (6-8 semanas de edad, Charles River UK Limited) en jaulas de 3 a 19-21°C con un ciclo de luz/oscuridad de 12 h (07:00/19:00) y se les alimentó con una dieta estándar de comida para roedores y agua ad libitum. Las ratas que no están en ayunas se pesan, se identifican individualmente en la cola con un marcador permanente y reciben una única administración intravítrea en el humor vítreo derecho (volumen de dosis de 5 j l ) de 100 ng/animal de LPS (Escherichia coli 0111:B4 preparado en PBS, Sigma Aldrich, Reino Unido) utilizando una aguja de calibre 32. Se inyecta PBS a ratas no tratadas. El compuesto de prueba o vehículo (4% de polioxil 40 estearato, 4% de manitol en PBS (pH 7,4)) se administra por vía tópica en el ojo derecho (10 j l ) de los animales 1 hora antes de LPS, en el momento de la administración de LPS y 1,2 y 4 horas después de la administración de LPS. Antes de la administración, la solución a administrar es sonicada para asegurar una solución clara. 6 horas después de la dosificación de LPS, los animales se sacrifican por sobredosis con pentobarbitona (mediante punción cardíaca). Inmediatamente después de la eutanasia, se recogen 10 j l de humor acuoso del ojo derecho de las ratas mediante la punción de la cámara anterior con una aguja de calibre 32 bajo un microscopio quirúrgico. El humor acuoso se diluye en 20 j l de PBS y los recuentos celulares totales se miden inmediatamente usando un contador celular automático Countess (Invitrogen). Después de la recolección del humor acuoso, el ojo derecho de cada animal se enuclea y se diseca en secciones frontal (anterior) y posterior (posterior) alrededor del cristalino. Cada sección se pesa y homogeneiza en 500 j l de PBS estéril seguido de
una centrifugación de 20 minutos a 12000 rpm a 4°C. El sobrenadante resultante se divide en 3 alícuotas y se almacena a -80°C hasta el posterior análisis de citocinas mediante R&D DuoSet ELISA.
Resumen de resultados de cribado In Vitro e In Vivo
[0153] El compuesto del Ejemplo 2(k) es sustancialmente más potente que el Compuesto de Referencia, se presentan más bajas constantes de disociación en los ensayos de unión de enzimas de la familia JAK bioquímica llevados a cabo a DiscoveRx (Tabla 1).
Tabla 1: Constantes de disociación para quinasas seleccionadas determinadas por LeadHunter Discover Services (DiscoveRx Corporation, Fremont, CA), utilizando la tecnología KINOMEscan™.
Constante de disociación (nM)
Compuesto de prueba JAK1 JAK2 JAK3 TYK2
Compuesto de referencia 2,7 0,78 1,9 1,9
Ejemplo 2(k) 1,0 0,047 0,092 0,13
[0154] De manera similar, los compuestos de los ejemplos de la presente invención pueden ser sustancialmente más potentes que el Compuesto de Referencia en la inhibición de liberación de IFNy de células PBMC estimuladas por CD3/IL2 (ensayo (a) anterior) y/o puede mostrar viabilidades mejoradas en el ensayo de citotoxicidad celular (c) anterior (Tabla 2).
Tabla 2: Efectos de los compuestos de la invención sobre la inhibición de la liberación de IFNy a partir de células PBMC estimuladas (ensayo (a) anterior) y la viabilidad celular en células Jurkat (ensayo (c) anterior).
Compuesto de ensayo PBMC IFNy CI 50 (nM) % viabilidad a 1 pg/mL Compuesto de referencia 81,3 49
Ejemplo 1 22,6 14
Ejemplo de referencia 2(a) 731,5 -Ejemplo de referencia 2(b) 87,0 -Ejemplo de referencia 2(c) 37,0 -Ejemplo de referencia 2(d) 269,9 -Ejemplo de referencia 2(e) 57,6 96
Ejemplo de referencia 2(f) 49,3 16
Ejemplo de referencia 2(g) 270,8 -Ejemplo de referencia 2(h) 12,1 56
Ejemplo de referencia 2(i) 14,5 53
Ejemplo de referencia 2(j) 5,3 12
Ejemplo de referencia 2(k) 9,9 93
Ejemplo de referencia 2(l) 9,4 10
Ejemplo de referencia 2(m) 15,2 88
Ejemplo de referencia 2(n) 29,2 87
Ejemplo de referencia 2(o) 22,5 62
Ejemplo de referencia 2(p) 153,7 -Ejemplo de referencia 2(q) 113,2 94
Ejemplo de referencia 2(r) 90,8 -Ejemplo de referencia 2(s) 693,6 -Ejemplo de referencia 2(t) 15,2 18
Ejemplo de referencia 2(u) 73,8 -Ejemplo de referencia 2(v) 7,5 82
Ejemplo de referencia 2(w) 941,5 -
Resumen de estudios adicionales
Determinación de solubilidades en líquido colónico simulado en estado de ayuno (FaSSCoF)
[0155] Las solubilidades de los compuestos de la invención en FaSSCoF a pH 6,5 se determinan usando una modificación de un procedimiento informado previamente (Vertzoni, M., et al. Pharm. Res. 2010, 27, 2187-2196). En lugar del extracto de sal biliar empleado en el procedimiento original (cuyo extracto ya no está disponible), el procedimiento modificado utiliza una mezcla de tauroclorato de sodio (0,15 g), ácido glicocólico (0,15 g), ácido ursodesoxicólico (0,05 g), ácido cólico (0,05 g) y ácido glicodesoxicólico (0,05 g). Estos cinco ácidos biliares se muelen junto con un mortero y una mano para producir un polvo blanco fino que se incorpora al FaSSCoF, como se describe a continuación.
[0156] Medio FaSSCoF: se disuelven Tris(hidroximetilo)aminometano (Tris; 0,275 g) y ácido maleico (0,44 g) en agua (35 ml) para dar una solución cuyo pH se ajusta a 6,5 mediante tratamiento con NaOH 0,5 M (aprox.,12 ml). A continuación, la solución se completa hasta 50 ml con agua. Se añade una porción de esta solución tampón de Tris/maleato (aprox. 25 ml) a un matraz de fondo redondo de 0,5 L, antes de ser tratado con 0,00565 g de la mezcla de ácidos biliares descrita anteriormente. Se agregan soluciones de fosfatidilcolina (0,0111 g) en DCM (0,15 mL) y ácido palmítico (0,0013 g) en DCM (0,15 mL), luego se evapora el solvente orgánico bajo presión reducida a 40°C hasta una solución transparente, sin que se consigue un olor DCM perceptible. El volumen de la solución evaporada se ajusta a 50 mL mediante la adición del resto de tampón Tris/maleato, luego se agrega BSA (0,115 g), antes de disolverse mediante agitación suave.
[0157] Determinación de la solubilidad: Los compuestos de prueba se suspenden en el medio FaSSCoF a pH 6,5 para dar una concentración final máxima de 2-10 mg/ml. Las suspensiones se equilibran a 25°C durante 24 h, antes de filtrarse a través de un filtro C de fibra de vidrio. A continuación, los filtrados se diluyen según sea apropiado para inyección y cuantificación por HPLC con referencia a un estándar. Se inyectan diferentes volúmenes de las soluciones de muestra estándar, diluidas y sin diluir y se calculan las solubilidades usando las áreas de pico determinadas por integración del pico encontrado en el mismo tiempo de retención que el pico principal en la inyección estándar.
[0158] Las solubilidades FaSSCoF se muestran en la Tabla 3 a continuación, lo que revela que, a diferencia del compuesto de referencia, los compuestos de los Ejemplos exhiben solubilidades en el medio FaSSCoF a pH 6,5 de 0,01 mg/ml o mayor.
Tabla 3: Solubilidades medidas para ciertos compuestos de los Ejemplos de la presente invención en FaSSCoF a pH
6,5.
Solubilidad de FaSSCoF pH 6,5 (mg/mL)
Compuesto de ensayo Ejecución 1 Ejecución 2
Compuesto de referencia 0,006 0,006
Ejemplo 1 0,010 0,010
Ejemplo de referencia 2(h) 0,030 0,040
Ejemplo de referencia 2(k) 0,010 -
Determinación de los parámetros farmacocinéticos
[0159] Se realizaron estudios de Sai Life Sciences (Hinjewadi, Pune, India) para investigar la farmacocinética sistémica y distribución en los tejidos de colon total de compuestos de la invención después de una sola administración oral en ratones machos C57BL/6.
[0160] Se les administró a un grupo de veinticuatro ratones macho una solución del compuesto de ensayo formulado a una dosis de 10 mg/10 ml/kg en una solución acuosa de hidroxipropil-6-ciclodextrina (20% p/v en agua). Se recogieron muestras de sangre (aproximadamente 60 pl) del plexo retroorbital de cada ratón bajo anestesia ligera con isoflurano, de modo que las muestras se obtuvieron a las 0,5, 1, 2, 4,6, 8, 12 y 24 h. Las muestras de sangre se recogieron de un conjunto de tres ratones en cada punto de tiempo en un tubo de microcentrífuga etiquetado que contenía K2EDTA como anticoagulante. Las muestras de plasma se separaron por centrifugación a 4000 rpm durante 10 min y se almacenaron por debajo de -70°C hasta el bioanálisis. Después de la recolección de la muestra de sangre, los animales fueron sacrificados humanitariamente mediante asfixia con dióxido de carbono para recolectar el tejido total del colon. Los dos puntos se lavaron abundantemente con PBS frío (pH 7,4) para eliminar el contenido. Los tejidos de colon totales se homogeneizaron con PBS frío (pH 7,4) de dos veces el peso del tejido de colon y se almacenaron por debajo de -70°C. El volumen total fue tres veces el peso total del tejido del colon. Todas las muestras se procesaron para su análisis mediante precipitación de proteínas usando acetonitrilo y se analizaron con un método LC-MS/MS adaptado al propósito. Los parámetros farmacocinéticos se calcularon utilizando la herramienta de análisis no compartimental de Phoenix WinNonlin® (Versión 6,3).
[0161] Los datos catalogados en la Tabla 4 revelan que, después de la administración oral, el compuesto del Ejemplo 2 (k) logra concentraciones colónicas sustanciales, mientras que, en contraste, la exposición sistémica de plasma era muy baja. Además, el Ejemplo 2(k) demuestra proporciones de colon a plasma mucho más altas en comparación con el Compuesto de Referencia, como se revela en la Tabla 5. En las Tablas 4 y 5, "P" se refiere a plasma y "C" se refiere a colon total.
Tabla 4: Las concentraciones plasmáticas medias (ng/ml) o los niveles totales de colon (ng/g) obtenidos después de la administración oral del compuesto de referencia y del Ejemplo 2(k) a ratones a 10 mg/kg en solución en 20% de hidroxipropil-p-ciclodextrina acuosa (volumen de dosis = 10 ml/kg).
Tiempo (h)
Compuesto Matriz 0,5 1 2 4 6 8 12 24 Referencia P 5,2 12,8 2,2 14,4 114 95,0 0,0 0,0 Compuesto C 344 102 2,366 2,972 201 2,555 3,229 16,6 Referencia P 2,4 2,6 1,9 1,1 3,4 4,8 1,2 0,0 Ejemplo 2(k) C 431 1,155 5,447 7,232 4,815 15,217 2,224 44,8
Tabla 5: Proporciones totales de colon a plasma obtenidas tras la administración oral de soluciones del Compuesto de Referencia y del Ejemplo 2(k) a 10 mg/kg a ratones.
Cmax (ng/g o ng/mL) AUCo-24h (hng/g o
hng/mL)
Compuesto C P Relación C P Relación Cmax AUCo-24h Compuesto de 3,229 114 28,2 43,739 368 119 referencia
Ejemplo de 15,217 4,8 3,170 97,062 20 4,853 referencia 2(k)
Abreviaturas
[0162]
aq acuoso
5-ASA ácido 5-aminosalicílico
ATP adenosina-5'-trifosfato
BID bis in die (dos veces al día)
Boc ferc-butoxicarbonil
Br amplio
BSA albúmina de suero bovino
CD Enfermedad de Crohn
COPD enfermedad pulmonar obstructiva crónica
d doblete
8 desplazamiento químico
DCM diclorometano
DIPEA diisopropiletilamina
DMEM medio Eagle modificado de Dulbecco
DMF N,N-dimetilformamida
DMSO dimetil sulfóxido
EDTA ácido etilendiaminotetraacético
ELISA ensayo inmunoabsorbente ligado a enzimas
(ES-) ionización por electrospray, modo negativo
(ES+) ionización por electropulverización, modo positivo
Et etilo
EtaN trietilamina
EtOAc acetato de etilo
EtOH etanol
FaSSCoF fluido colónico simulado en estado de ayunas
FBS suero fetal bovino
FCS suero de ternero fetal
HATU 2-(1H-7-azabenzotriazol-1-il)-1,1,3,3-tetrametiluronio hexafluorofosfato
HBSS solución salina equilibrada de Hank
HPLC cromatografía líquida de alta resolución
HPMC hidroxipropilmetilcelulosa
h, hr o hrs hora(s)
HRP peroxidación de rábano picante
EII Enfermedad inflamatoria intestinal
IFNy interferón-y
IL interleucina
(Continuación)
IPA alcohol isopropílico
JAK Janus quinasa
JNK c-Jun quinasa N-terminal
LC cromatografía líquida
LPMC células mononucleares de lámina propia
LPS lipopolisacárido
m multiplete
(M+H)+ ion molecular protonado
(M-H)- ion molecular desprotonado
Me metilo
MeCN acetonitrilo
MeOH metanol
MHz megahertz
min o mins minuto(s)
MMAD diámetro aerodinámico masa mediano
MPO mieloperoxidasa
MS espectrometría de masas
m/z relación masa/carga
NMP N-metil-2-pirrolidona
RMN resonancia magnética nuclear (espectroscopia)
OD densidad óptica
PBMC células mononucleares de sangre periférica
PBS solución salina tamponada con fosfato
Ph fenilo
PHA fitohemaglutinina
PI yoduro de propidio
PMA acetato de miristato de forbol
q cuarteto
ta o TA temperatura ambiente
RP HPLC cromatografía líquida de alta eficacia de fase inversa
rpm revoluciones por minuto
RPMI Roswell Park Memorial Institute
s singlete
sat o satd saturado
SCID inmunodeficiencia combinada grave
SCX intercambio catiónico soportado sólido (resina)
SDS sulfato de dodecilo de sodio
SNAr sustitución aromática nucleofílica
Syk quinasa de tirosina de bazo
t triplete
‘Bu tere-butilo
T3P ácido 1-propanofosfónico anhídrido cíclico
TEA trietilamina
THF tetrahidrofurano
TFA ácido trifluoroacético
TGFp factor de crecimiento transformante beta
TNFa factor de necrosis tumoral alfa
Tris tris(hidroximetilo)aminometano
TYK2 tirosina quinasa 2
UC colitis ulcerosa
UPLC cromatografía líquida de ultra eficacia
UV ultravioleta
[0163] Los prefijos n-, s -, i-, t- y tere- tienen sus significados habituales: normal, secundario, iso y terciario.
Claims (7)
1. Un compuesto de fórmula:

o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo.
2. Una formulación farmacéutica que comprende un compuesto como se define en la reivindicación 1, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, mezclado con un adyuvante, diluyente o vehículo farmacéuticamente aceptable.
3. Un producto de combinación que comprende
(A) un compuesto como se define en la reivindicación 1, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, y
(B) otro agente terapéutico,
en donde cada uno de los componentes (A) y (B) está formulado en mezcla con un adyuvante, diluyente o portador farmacéuticamente aceptable.
4. Un compuesto como se define en la reivindicación 1, o una sal farmacéuticamente aceptable, solvato o derivado isotópico del mismo, o una formulación farmacéutica como se define en la reivindicación 2 o un producto de combinación como se define en la reivindicación 3, para uso en el tratamiento o prevención de una enfermedad inflamatoria..
5. Un compuesto, formulación o producto de combinación para su uso según la reivindicación 4, en donde la enfermedad inflamatoria se selecciona de la lista que comprende fibrosis quística, hipertensión pulmonar, sarcoidosis pulmonar, fibrosis pulmonar idiopática, EPOC, asma, asma pediátrica, dermatitis atópica, alergia. dermatitis, dermatitis de contacto o psoriasis, rinitis alérgica, rinitis, sinusitis, conjuntivitis, conjuntivitis alérgica, queratoconjuntivitis seca (ojo seco o xeroftalmia), glaucoma, retinopatía diabética, edema macular, oclusión de la vena central de la retina (OVCR), degeneración macular relacionada con la edad seca y/o húmeda (DMAE), inflamación de cataratas posoperatoria, uveítis, rechazo de injerto de córnea y trasplante de células limbares, enteropatía sensible al gluten (enfermedad celíaca), esofagitis eosinofílica, enfermedad de injerto contra huésped intestinal, enfermedad de Crohn y colitis ulcerosa.
6. Un compuesto, formulación o producto de combinación para su uso según la reivindicación 5, en donde la enfermedad inflamatoria es uveítis, queratoconjuntivitis seca (ojo seco o xeroftalmia), enfermedad de Crohn, colitis ulcerosa, asma o EPOC.
7. Un compuesto, formulación o producto de combinación para su uso según la reivindicación 5, en donde:
dicha EPOC es bronquitis crónica o enfisema; y/o
dicho edema macular es edema macular diabético; y/o
dicho uveítis es posterior, anterior o panuveítis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1519381.6A GB201519381D0 (en) | 2015-11-03 | 2015-11-03 | Kinase inhibitors |
| GB201606968 | 2016-04-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2864035T3 true ES2864035T3 (es) | 2021-10-13 |
Family
ID=57256356
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20171633T Active ES2864035T3 (es) | 2015-11-03 | 2016-11-02 | Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10851097B2 (es) |
| EP (3) | EP3712152B1 (es) |
| CA (1) | CA3037243A1 (es) |
| DK (1) | DK3712152T3 (es) |
| ES (1) | ES2864035T3 (es) |
| PL (1) | PL3712152T3 (es) |
| PT (1) | PT3712152T (es) |
| WO (1) | WO2017077283A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA123633C2 (uk) | 2015-11-03 | 2021-05-05 | Тереванс Байофарма Ар Енд Ді Айпі, Елелсі | Сполуки інгібітору jak-кінази для лікування респіраторного захворювання |
| EP3371185B1 (en) | 2015-11-03 | 2020-09-30 | Topivert Pharma Limited | 4,5,6,7-tetrahydro-1h-imidazo[4,5-c]pyridine and 1,4,5,6,7,8-hexahydroimidazo[4,5-d]azepine derivatives as janus kinase inhibitors |
| SG11201907840RA (en) | 2017-03-09 | 2019-09-27 | Theravance Biopharma R&D Ip Llc | Fused imidazo-piperidine jak inhibitors |
| TW201900170A (zh) | 2017-05-01 | 2019-01-01 | 美商施萬生物製藥研發 Ip有限責任公司 | 使用jak抑制劑化合物之治療方法 |
| AR111495A1 (es) | 2017-05-01 | 2019-07-17 | Theravance Biopharma R&D Ip Llc | Compuestos de imidazo-piperidina fusionada como inhibidores de jak |
| JP7096268B2 (ja) | 2017-05-01 | 2022-07-05 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | Jak阻害剤化合物の結晶形態 |
| UA125318C2 (uk) | 2017-08-01 | 2022-02-16 | Тереванс Байофарма Ар Енд Ді Айпі, Елелсі | Піразоло- і триазолобіциклічні сполуки як інгібітори jak-кінази |
| SG11202101751XA (en) | 2018-09-04 | 2021-03-30 | Theravance Biopharma R&D Ip Llc | 5 to 7 membered heterocyclic amides as jak inhibitors |
| TWI808250B (zh) | 2018-09-04 | 2023-07-11 | 美商施萬生物製藥研發 Ip有限責任公司 | 用於製備jak抑制劑之方法及其中間體 |
| AU2019336167B2 (en) | 2018-09-04 | 2024-08-29 | Theravance Biopharma R&D Ip, Llc | Dimethyl amino azetidine amides as JAK inhibitors |
| MX2021004582A (es) | 2018-10-29 | 2021-06-15 | Theravance Biopharma R&D Ip Llc | Compuesto 2-azabiciclo hexano inhibidor de jak. |
| CA3125039A1 (en) | 2019-01-23 | 2020-07-30 | Theravance Biopharma R&D Ip, Llc | Imidazo[1,5-a]pyridine, 1,2,4-triazolo[4,3-a]pyridine and imidazo[1,5-a]pyrazine as jak inhibitors |
| CN114075199A (zh) * | 2020-08-14 | 2022-02-22 | 河南迈英诺医药科技有限公司 | Jak抑制剂化合物及其用途 |
| CN114075200A (zh) * | 2020-08-14 | 2022-02-22 | 河南迈英诺医药科技有限公司 | 用于治疗重症肺炎的jak抑制剂化合物 |
| JP7514025B2 (ja) * | 2019-02-25 | 2024-07-10 | ホーナン メディノ ファーマシューティカル テクノロジー カンパニー リミテッド | Jak阻害剤化合物及びその使用 |
| TW202144343A (zh) | 2020-03-02 | 2021-12-01 | 美商施萬生物製藥研發 Ip有限責任公司 | Jak抑制劑化合物之結晶水合物 |
| CN115043844B (zh) * | 2021-06-21 | 2024-05-17 | 河南迈英诺医药科技有限公司 | Trk激酶抑制剂化合物及其用途 |
| CN117343063A (zh) * | 2022-07-04 | 2024-01-05 | 河南迈英诺医药科技有限公司 | 作为trk抑制剂和/或ret抑制剂的化合物及其用途 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009007839A1 (en) | 2007-07-11 | 2009-01-15 | Pfizer Inc. | Pharmaceutical compositions and methods of treating dry eye disorders |
| US8575336B2 (en) | 2011-07-27 | 2013-11-05 | Pfizer Limited | Indazoles |
| FR2981917B1 (fr) | 2011-10-26 | 2013-11-15 | Cadorit Ag | Recipient pour emballer un premier fluide et un deuxieme fluide |
| JP6248103B2 (ja) | 2012-06-22 | 2017-12-13 | ミメトゲン ファーマシュウティカルズ インコーポレイテッドMimetogen Pharmaceuticals, Inc. | β−ターン環式ペプチド模倣化合物の合成 |
| EP3143021B1 (en) | 2014-05-14 | 2019-06-12 | Pfizer Inc | Pyrazolopyridines and pyrazolopyrimidines |
| MX2020011774A (es) | 2015-05-28 | 2021-06-01 | Theravance Biopharma R&D Ip Llc | Compuestos de naftiridina como inhibidores de quinasa jak. |
| UA123633C2 (uk) | 2015-11-03 | 2021-05-05 | Тереванс Байофарма Ар Енд Ді Айпі, Елелсі | Сполуки інгібітору jak-кінази для лікування респіраторного захворювання |
| EP3371185B1 (en) | 2015-11-03 | 2020-09-30 | Topivert Pharma Limited | 4,5,6,7-tetrahydro-1h-imidazo[4,5-c]pyridine and 1,4,5,6,7,8-hexahydroimidazo[4,5-d]azepine derivatives as janus kinase inhibitors |
| RS60237B1 (sr) | 2015-11-24 | 2020-06-30 | Theravance Biopharma R&D Ip Llc | Prolekovi jak inhibitornih jedinjenja za lečenje gastorintestinalne inflamatorne bolesti |
| TWI726094B (zh) | 2016-04-28 | 2021-05-01 | 美商施萬生物製藥研發Ip有限責任公司 | 作為jak激酶抑制劑之嘧啶化合物 |
-
2016
- 2016-11-02 WO PCT/GB2016/053387 patent/WO2017077283A1/en active Application Filing
- 2016-11-02 EP EP20171633.9A patent/EP3712152B1/en not_active Not-in-force
- 2016-11-02 PL PL20171633T patent/PL3712152T3/pl unknown
- 2016-11-02 ES ES20171633T patent/ES2864035T3/es active Active
- 2016-11-02 DK DK20171633.9T patent/DK3712152T3/da active
- 2016-11-02 PT PT201716339T patent/PT3712152T/pt unknown
- 2016-11-02 EP EP16793973.5A patent/EP3371184A1/en not_active Withdrawn
- 2016-11-02 EP EP21151168.8A patent/EP3865481A1/en not_active Withdrawn
- 2016-11-02 US US15/772,861 patent/US10851097B2/en active Active
- 2016-11-02 CA CA3037243A patent/CA3037243A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| EP3865481A1 (en) | 2021-08-18 |
| CA3037243A1 (en) | 2017-05-11 |
| US20180319791A1 (en) | 2018-11-08 |
| WO2017077283A1 (en) | 2017-05-11 |
| DK3712152T3 (da) | 2021-03-22 |
| EP3712152A1 (en) | 2020-09-23 |
| US10851097B2 (en) | 2020-12-01 |
| PT3712152T (pt) | 2021-04-15 |
| EP3712152B1 (en) | 2021-01-13 |
| EP3371184A1 (en) | 2018-09-12 |
| PL3712152T3 (pl) | 2021-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2864035T3 (es) | Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus | |
| ES2840650T3 (es) | Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus | |
| US9822076B2 (en) | Kinase inhibitor | |
| ES2856902T3 (es) | Derivados de urea útiles como inhibidores de quinasa | |
| US20190233395A1 (en) | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs | |
| WO2016051186A1 (en) | N-phenyl-3-quinazolin-6-yl-benzamide derivatives as p38 kinase inhibitors | |
| KR20180128390A (ko) | 인돌리논 화합물과 섬유성 질환의 치료에서 이의 용도 | |
| JP6995774B2 (ja) | キナーゼ阻害剤 | |
| KR20180102590A (ko) | Cftr 조절제 및 이의 사용방법 |