ES2853375T3 - Sales novedosas y composiciones farmacéuticas de las mismas para el tratamiento de trastornos inflamatorios - Google Patents
Sales novedosas y composiciones farmacéuticas de las mismas para el tratamiento de trastornos inflamatorios Download PDFInfo
- Publication number
- ES2853375T3 ES2853375T3 ES15704974T ES15704974T ES2853375T3 ES 2853375 T3 ES2853375 T3 ES 2853375T3 ES 15704974 T ES15704974 T ES 15704974T ES 15704974 T ES15704974 T ES 15704974T ES 2853375 T3 ES2853375 T3 ES 2853375T3
- Authority
- ES
- Spain
- Prior art keywords
- salt
- compound
- disease
- hcl
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04L—TRANSMISSION OF DIGITAL INFORMATION, e.g. TELEGRAPHIC COMMUNICATION
- H04L63/00—Network architectures or network communication protocols for network security
- H04L63/04—Network architectures or network communication protocols for network security for providing a confidential data exchange among entities communicating through data packet networks
- H04L63/0428—Network architectures or network communication protocols for network security for providing a confidential data exchange among entities communicating through data packet networks wherein the data content is protected, e.g. by encrypting or encapsulating the payload
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04L—TRANSMISSION OF DIGITAL INFORMATION, e.g. TELEGRAPHIC COMMUNICATION
- H04L63/00—Network architectures or network communication protocols for network security
- H04L63/06—Network architectures or network communication protocols for network security for supporting key management in a packet data network
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04L—TRANSMISSION OF DIGITAL INFORMATION, e.g. TELEGRAPHIC COMMUNICATION
- H04L63/00—Network architectures or network communication protocols for network security
- H04L63/08—Network architectures or network communication protocols for network security for authentication of entities
- H04L63/0861—Network architectures or network communication protocols for network security for authentication of entities using biometrical features, e.g. fingerprint, retina-scan
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04L—TRANSMISSION OF DIGITAL INFORMATION, e.g. TELEGRAPHIC COMMUNICATION
- H04L63/00—Network architectures or network communication protocols for network security
- H04L63/20—Network architectures or network communication protocols for network security for managing network security; network security policies in general
- H04L63/205—Network architectures or network communication protocols for network security for managing network security; network security policies in general involving negotiation or determination of the one or more network security mechanisms to be used, e.g. by negotiation between the client and the server or between peers or by selection according to the capabilities of the entities involved
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04L—TRANSMISSION OF DIGITAL INFORMATION, e.g. TELEGRAPHIC COMMUNICATION
- H04L67/00—Network arrangements or protocols for supporting network services or applications
- H04L67/01—Protocols
- H04L67/10—Protocols in which an application is distributed across nodes in the network
- H04L67/104—Peer-to-peer [P2P] networks
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04L—TRANSMISSION OF DIGITAL INFORMATION, e.g. TELEGRAPHIC COMMUNICATION
- H04L67/00—Network arrangements or protocols for supporting network services or applications
- H04L67/50—Network services
- H04L67/51—Discovery or management thereof, e.g. service location protocol [SLP] or web services
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04W—WIRELESS COMMUNICATION NETWORKS
- H04W4/00—Services specially adapted for wireless communication networks; Facilities therefor
- H04W4/80—Services using short range communication, e.g. near-field communication [NFC], radio-frequency identification [RFID] or low energy communication
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04W—WIRELESS COMMUNICATION NETWORKS
- H04W8/00—Network data management
- H04W8/22—Processing or transfer of terminal data, e.g. status or physical capabilities
- H04W8/24—Transfer of terminal data
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- H—ELECTRICITY
- H04—ELECTRIC COMMUNICATION TECHNIQUE
- H04W—WIRELESS COMMUNICATION NETWORKS
- H04W92/00—Interfaces specially adapted for wireless communication networks
- H04W92/16—Interfaces between hierarchically similar devices
- H04W92/18—Interfaces between hierarchically similar devices between terminal devices
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Signal Processing (AREA)
- Computer Networks & Wireless Communication (AREA)
- Computer Security & Cryptography (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Computing Systems (AREA)
- Computer Hardware Design (AREA)
- General Engineering & Computer Science (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Biomedical Technology (AREA)
- Databases & Information Systems (AREA)
- Transplantation (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Peptides Or Proteins (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Una sal aceptable para uso farmacéutico de un compuesto de acuerdo con la Fórmula (I): **(Ver fórmula)** en la que la sal es un aducto Base Libre/HCl/ H2O 1:1:3.
Description
DESCRIPCIÓN
Sales novedosas y composiciones farmacéuticas de las mismas para el tratamiento de trastornos inflamatorios
Campo de la invención
La presente invención se refiere a las formas salinas y cristalinas de un compuesto de acuerdo con la Fórmula I, útiles en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones. En particular, la sal de la invención inhibe JAK, una familia de quinasas de la tirosina, y más particularmente JAK1. La presente invención también proporciona composiciones farmacéuticas que comprenden la sal de la invención.
Antecedentes de la invención
Las terapias actuales para tratar afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones, en particular artritis reumatoide, están lejos de lo satisfactorio y aún permanece vigente una necesidad de identificar nuevos agentes terapéuticos que pueden ser de uso en su tratamiento. Estas afecciones son afecciones crónicas que requieren terapia prolongada, y la ingesta repetida del fármaco. El tratamiento prolongado puede ser una carga pesada sobre el paciente y el médico por igual, ya que el paciente puede ser, o puede convertirse en, intolerante al fármaco, y además, la posología elevada, o la frecuencia posológica alta pueden resultar en efectos colaterales incómodos, y/o bajo cumplimiento del paciente, en los que el paciente puede, ocasionalmente, deliberadamente o accidentalmente, perder una dosis. El impacto de la falta de adherencia varía entre enfermedades crónicas y oscila de mínima a muy significativa (Ingersoll & Cohen, 2008). Por consiguiente, existe una necesidad de identificar nuevos agentes para reforzar el arsenal del médico, y compuestos con régimen de posología de frecuencia baja para mejorar la vida de los pacientes.
Las quinasas Janus (JAK) son quinasas de tirosina citoplasmáticas que transducen la señalización de citosina de los receptores de membrana a los factores de transcripción de STAT. Se describen cuatro miembros de la familia JAK, JAK1, JAK2, JAK3 y TYK2. Tras la unión de la citosina a su receptor, los miembros de la familia JAK se auto- y/o transforsoforilan entre sí, seguido de fosforilación de STAT que luego migran al núcleo para modular la transcripción. La transducción de la señal intracelular de JAK-STAT sirve a los interferones, la mayoría de las interleucinas, así como una variedad de citosinas y factores endócrinos tales como EPO, TPO, GH, OSM, LIF, CNTF, GM-c Sf y PRL (Vainchenker, Dusa, & Constantinescu, 2008).
La combinación de modelos genéticos y la investigación del inhibidor JAK de molécula pequeña revelaron el potencial terapéutico de varias JAK.
JAK1 es una diana en el área de enfermedad inmuno-inflamatoria. JAK1 se heterodimeriza con otros JAK para transducir la señalización proinflamatoria accionada por citosinas. Por consiguiente, la inhibición de JAK1 es de interés para enfermedades inmunoinflamatorias con citosinas asociadas con una patología que usan la señalización de JAK1, tal como IL-2, IL-6, IL-4, IL-5, IL-13, o IFNgamma, así como otras enfermedades accionadas por transducción de la señal mediada por JAK.
En los roles de los miembros de la familia JAK, existe cierta superposición, ya que la mayoría de las vías de señalización incluyen más de un JAK, sin embargo, para algunos factores de crecimiento tales como eritropoyetina y trombopoyetina, solo está involucrado JAK2.
JAK3 cumple una función principal en el bloqueo de la función inmune mediante la transmisión de señales generadas por interleucina (IL)-2.
Por otro lado, TYK2 funcionaría en combinación con JAK2 con el fin de transducir la señalización de citosinas tales como IL-12 y IL-23.
La función de las enzimas JAK se ha estudiado principalmente usando ratones en los que cada uno de los miembros de la familia JAK se ha eliminado. Los ratones knockout de JAK1 exhiben un fenotipo letal perinatal y también tienen desarrollo y función linfoide defectuosos como resultado de la señalización defectuosa de las citosinas a través de JAK1. La deficiencia de JAK2 resulta en letalidad embrionaria el día 12 como resultado de una insuficiencia en la eritropoyesis definitiva. Los ratones con deficiencia de JAK3 tienen fenotipo de inmununodeficiencia combinado (SCID) pero no tienen defectos inmunes (Vertovsek, 2009).
Según se ha observado con los inhibidores JAK compatibles, la inhibición no selectiva puede estar relacionada con efectos colaterales tal como anemia, una tasa aumentada de infecciones, recuentos más bajos de neutrófilos y linfocitos, una reducción en la hemoglobina, y niveles elevados de colesterol (Dolgin, 2011).
Por consiguiente, el desarrollo de un inhibidor de JAK selectivo sería beneficioso para minimizar dichos efectos colaterales.
La degeneración del cartílago es un hito de varias enfermedades, entre las que la artritis reumatoide y la osteoartritis son las más prominentes. La artritis reumatoide (RA) es una enfermedad degenerativa crónica de las articulaciones, caracterizada por la inflamación y destrucción de las estructuras articulares. Cuando la enfermedad no se trata, puede conducir a discapacidad y dolor sustancial debido a la pérdida de la función de las articulaciones y resultar en menor expectativa de vida. El objetivo de una terapia de RA, por lo tanto, no solo es ralentizar la enfermedad sino lograr la remisión para detener la destrucción de las articulaciones y mejorar la calidad de vida. Además de la severidad de la evolución de la enfermedad, la alta prevalencia de RA (~ 0,8% de los adultos están afectados a nivel mundial) significa un alto impacto socio-económico (Smolen & Steiner, 2003) (O'Dell, 2004). JAK1 está implicada en una transducción de señal intracelular para muchas citosinas y hormonas. Las patologías asociadas con cualquiera de estas citosinas y hormonas pueden aliviarse mediante inhibidores de JAK1. Por consiguiente, varis trastornos alérgicos, inflamatorios y autoinmunes podrían beneficiarse a partir del tratamiento con compuestos descritos en esta invención incluyendo artritis reumatoide, lupus eritematoso sistémico, artritis idiopática juvenil, osteoartritis, asma, enfermedad pulmonar obstructiva crónica (EPOC), fibrosis de tejidos, inflamación eosinofílica, esofagitis, enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, colitis ulcerativa), trasplante, enfermedad de injerto contra huésped, psoriasis, miositis, artritis psoriásica, espondilitis anquilosante, artritis idiopática juvenil y esclerosis múltiple. (Kopf, Bachmann, & Marsland, 2010)
La psoriasis es una enfermedad que puede afectar la piel. La causa de psoriasis no se comprende totalmente pero se presume que es una enfermedad relacionada mediada de manera inmune relacionada con la liberación de las citosinas, en particular el TNFa, que produce inflamación y rápida reproducción de las células cutáneas. Esta hipótesis se ha corroborado mediante la observación de que la medicación inmunosupresora puede aclarar las placas de psoriasis. (Zenz, et al., 2005) La psoriasis también puede causar inflamación de las articulaciones, lo que se conoce como artritis psoriásica. Entre el 10-30% de todas las personas con psoriasis también tienen artritis psoriásica. ((CHMP), 18 de noviembre de 2004). Debido a su naturaleza recurrente crónica, la psoriasis es un desafío para tratar. Se ha demostrado recientemente que la inhibición de JAK podría resultar en la mejora exitosa de la condición psoriásica (Punwani, et al., 2012).
La enfermedad inflamatoria intestinal (IBD) es un grupo de afecciones inflamatorias del colon y el intestino delgado. Los tipos principales de IBD son enfermedad de Crohn y colitis ulcerativa. Recientemente, se ha encontrado a través de estudios de asociación de amplitud del genoma (GWAS) que la fosfatasa de tirosina la proteína de células T (TCPTP) es un JAK/STAT y la fosfatasa del receptor del factor de crecimiento que se haya relacionado con la patogénesis de la diabetes tipo 1, artritis reumatoide, y enfermedad de Crohn por GWAS. (Zikherman & Weiss, 2011). Por consiguiente, la inhibición de la vía de JAK puede proporcionar un modo de tratamiento de la IBD.
Los miembros de la familia JAK se han implicado en afecciones adicionales incluyendo trastornos mieloproliferativos (O’Sullivan, Liongue, Lewis, Stephenson, & Ward, 2007), en los que se han identificado mutaciones en JAK2. Esto indica que los inhibidores de JAK en particular JAK2 también pueden ser de utilidad en el tratamiento de trastornos mieloproliferativos. Además, la familia JAK, en particular JAK1, JAK2 y JAK3, se ha relacionado con los cánceres, en particular leucemias (por ej., leucemia mieloide aguda (O’Sullivan, Liongue, Lewis, Stephenson, & Ward, 2007) (Xiang, et al., 2008) y leucemia linfoblástica aguda (Mullighan, 2009)), linfoma de células T cutáneas (Zhang, 1996) o tumores sólidos, por ej., leiomiosarcoma uterino (Constantinescu, Girardot, & Pecquet, 2007), cáncer de próstata (Tam, McGlynn, Traynor, Mukherjee, Bartlett, & Edwards, 2007) y cáncer de mama (Berishaj, et al., 2007). Estos resultados indican que los inhibidores de JAK, en particular JAK1 también pueden tener utilidad en el tratamiento de cánceres (leucemias y tumores sólidos, por ej., liomiosarcoma uterino, cáncer de próstata, cánceres pancreáticos).
Además, la enfermedad de Castleman, mieloma múltiple, glomerulonefritis proliferativo mesangial, psoriasis y sarcoma de Kaposi probablemente se deben a la hipersecreción de la citosina IL-6, cuyos efectos biológicos están mediados por señalización de JAK-STAT intracelular (Naka, Nishimoto, & Kishimoto, 2002). Este resultado muestra que los inhibidores de JAK, también pueden encontrar utilidad en el tratamiento de dichas enfermedades.
De este modo, los compuestos que son potentes inhibidores de JAK ofrecerían el potencial de tratamiento de una amplia variedad de enfermedades y afecciones descritas con anterioridad.
El compuesto {5-[4-(1,1-dioxo-tiomorfolin-4-ilmetil)-fenil]-[1,2,4]triazolo[1,5-a]piridin-2-il}-amida del ácido ciclopropancarboxílico (Compuesto I), que tiene la estructura química:
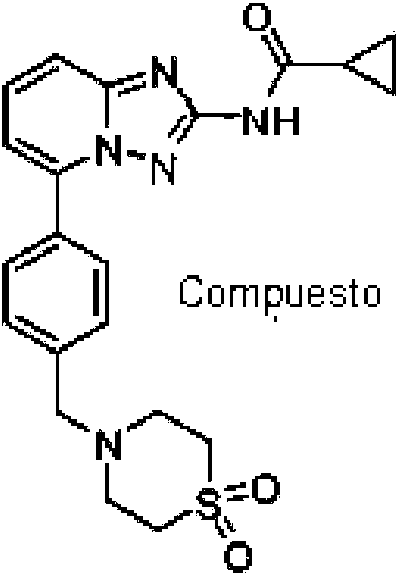
se describe en nuestra solicitud anterior WO 2010/149769 (Menet C. J., 2010) como un inhibidor de JAK y útil en el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativa, alergia, rechazo al trasplante, enfermedades que involucran deterioro de la producción de cartílago, malformaciones congénitas de cartílagos, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones. En adelante este compuesto se denomina Compuesto I. Los datos presentados en el documento WO 2010/149769 demuestran que a pesar de las actividades in vitro similares, el Compuesto I tiene potencia in vivo inesperadamente alta comparado con compuestos estructuralmente similares.
Una característica importante de varias sustancias bioactivas (por ejemplo pero sin limitación, productos farmacéuticos, medicinas y biocidas, normalmente referidos como fármacos) es su ''biodisponibilidad'' o concentración activa en una forma que puede ser absorbida y utilizada por un órgano u organismo diana. En muchos casos, la biodisponibilidad se relaciona con la solubilidad del fármaco en agua.
Para ser de utilidad como agente terapéutico, el fármaco debe ser soluble en un intervalo de concentración adecuado para el período de tiempo requerido. Se encuentran disponibles varias opciones para lograr estas propiedades, incluyendo formulación del fármaco como una píldora, cápsulas, soluciones, u otras formulaciones similares. Son de particular interés los fármacos de "liberación de orden cero", en los que la tasa de liberación del fármaco es constante. Sin embargo, desarrollar estos sistemas puede ser complicado y costoso.
Con frecuencia, los fármacos en su forma de base libre son escasamente solubles en agua, pero la presencia de sitios ácidos (por ejemplo, ácidos carboxílicos, fenoles, ácidos sulfónicos) o sitios alcalinos (por ejemplo grupos amino, centros alcalinos de nitrógeno) pueden usarse ventajosamente para producir las sales del fármaco. Los compuestos iónicos resultantes se tornan mucho más solubles en agua en virtud de su carácter iónico y mejor energía de disolución, y de este modo mejoran la biodisponibilidad. Una pauta de 50 pg/ml para solubilidad acuosa se proporciona en el texto de Lipinsky etal. (Lipinski, Lombardo, Dominy, & Feeney, 2001).
Los agentes formadores de sales están disponibles en un número extenso, y la selección de sales debe diseñarse cuidadosamente. El objetivo de la selección de sales es identificar la mejor forma de sal adecuada para desarrollo, y se basa principalmente en cuatro criterios principales: solubilidad acuosa a varios pH, alto grado de cristalinidad, baja higroscopicidad y estabilidad química óptima (Handbook of Pharmaceutical Salts: Properties, Selection and Use, Stahl, P.H. and Wermuth, C.G. Eds. Wiley-VCH, Weinheim, Germany, 2002).
Si una sal adecuada de un fármaco puede identificarse, se requieren investigaciones adicionales para identificar si hay formas cristalinas alternativas. La disponibilidad de dichas formas alternativas es altamente impredecible y puede requerir una combinación de intuición, diseño empírico cuidadoso, perseverancia y casualidad. En la parte más alta de los desafíos asociados con incluso hallar una o más formas cristalinas definidas, las propiedades de cualquier forma cristalina descubierta de este modo necesita evaluarse cuidadosamente para ver si una o más de ellas es realmente adecuada para desarrollo farmacéutico. De hecho, en un primer aspecto, la cristalinidad de los fármacos afecta, entre otras propiedades físicas y mecánicas, la solubilidad, la tasa de disolución, la fluidez, la dureza, la capacidad de compresión y el punto de fusión. En un segundo aspecto, una forma cristalina puede tener ventajas sobre la forma amorfa, por ejemplo, la purificación al alto grado de pureza requerida por la mayoría de las autoridades regulatorias es más eficiente y por consiguiente cuesta menos para que se forme la forma cristalina que el sólido amorfo. Además, la manipulación de la forma cristalina se mejora sobre la forma amorfa, la cual tiende a ser oleosa, o pegajosa, y en la práctica, el secado de un material cristalino que tiene una temperatura de secado y desolvación bien definida es más fácilmente controlada, que para el sólido amorfo, que tiene una mayor afinidad por los solventes orgánicos y las temperaturas de secado variables. Finalmente el procesamiento corriente abajo del fármaco cristalino permite el control de proceso mejorado. En un tercer aspecto, la estabilidad física y química, y por consiguiente la vida útil también mejoró para las formas cristalinas por sobre las formas amorfas.
Finalmente, las propiedades farmacocinéticas y farmacodinámicas de un fármaco pueden relacionarse con una forma estructural cristalina particular, y es fundamental para producir y retener la misma forma desde la producción hasta la administración al paciente. Por consiguiente, la obtención de sales y/o formas cristalinas por sobre materiales amorfos
es altamente deseable (Hilfiker, Blatter, & von Raumer, 2006).
De este modo, el objetivo de esta invención es divulgar formas de sales y polimorfos de las sales de la invención, que tienen propiedades farmacológicas deseables, y que muestren mejoras en su perfil farmacéutico comparado con la forma de base libre y/o la forma amorfa de la sal de la invención, en particular exposición mejorada in vivo.
Descripción de los dibujos
La Figura 1 muestra el difractograma de XRPD del Compuesto I patrón 1.
La Figura 2 muestra el difractograma de XRPD del Compuesto I patrón 3.
La Figura 3 muestra el difractograma de XRPD del Compuesto I patrón 4.
La Figura 4 (comparativa) muestra el difractograma de XRPD del Compuesto I. HCl.
La Figura 5 muestra el difractograma de XRPD del Compuesto I. HCl.3H2O.
La Figura 6 muestra el análisis de DVS del Compuesto I. HCl.3H2O.
La Figura 7 muestra oligoelementos de DSC del Compuesto I. HCl.3H2O.
La Figura 8 (comparativa) muestra el difractograma de XRPD del Compuesto I. HCl.MeOH.
La Figura 9 muestra el difractograma de XRPD del Compuesto I.HCl.1,5HCO2H.
La Figura 10 (incluyendo los ejemplos comparativos) muestra la exposición del Compuesto I indistintamente como base libre o como una sal de HCl.3H2O tras dosificación po diaria.
La Figura 11 muestra la estructura de cristal del Compuesto I. HCl.3H2O.
Sumario de la invención
La presente invención se basa en la identificación de sales y formas cristalinas novedosas del Compuesto I, útiles en el tratamiento y/o la profilaxis de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones. En particular, las sales de la invención pueden actuar como inhibidores de JAK, y más particularmente de JAK1. La presente invención proporciona también procedimientos para la producción de estas sales y composiciones farmacéuticas que comprenden estas sales.
Por consiguiente, en un aspecto la presente invención proporciona una sal de un compuesto de acuerdo con la Fórmula (I) que sigue (en adelante también denominada Compuesto I):

en la que dicha sal es 1:1:3 de sal de Base Libre/HCl/ H2O.
En otro aspecto de la invención, la sal de la invención es un aducto [Compuesto I.HCL3H2O] en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en cualquiera de una o más de las siguientes posiciones: 7,3, 8,4, 8,8, 10,7, 12,0, 12,2, 13,2, 13,7, 14,5, 16,3, 16,7, 17,6, 19,3, 20,2, 20,6, 21,0, 21,4, 21,8, 22,8, 23,4, 23,9, 24,5, 25,2, 25,7, 25,9, 26,4, 27,2, 27,7, 28,3, 28,6, 28,9, 29,2, 29,6, 7 y 32,7° 20± 0,2° 20. En un aspecto particular de la invención, la sal de la invención exhibe solubilidad y exposición mejoradas por sobre la base libre, lo que puede resultar en una eficacia mejorada y una dosis más baja de fármaco administrada. A su vez, la reducción del nivel de posología de la dosis puede potencialmente reducir la toxicidad que puede ocurrir a través
de la interacción medicamentosa.
En un aspecto particular, las sales de la invención se proporcionan para uso en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
En un aspecto adicional, la presente invención proporciona composiciones farmacéuticas que comprenden una sal de la invención, y un portador, excipiente o diluyente farmacéuticos. En un aspecto particular, la composición farmacéutica puede comprender, además, principios terapéuticamente activos adicionales, adecuados para uso en combinación con las sales de la invención. En un aspecto más particular, el principio terapéuticamente activo es un agente para el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
Más aún, las sales de la invención, útiles en las composiciones farmacéuticas y en los procedimientos de tratamiento divulgados en la presente memoria, son aceptables para uso farmacéutico como se preparan y se usan.
En un aspecto adicional de la invención, esta invención proporciona un procedimiento de tratamiento de un mamífero, en particular seres humanos, afectados con una condición seleccionada de entre las mencionadas en la presente memoria, y particularmente afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo a los trasplantes, enfermedades que implican degradación y/o interrupción de la homeóstasis de cartílagos, malformaciones congénitas de cartílagos, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones, cuyo procedimiento comprende administrar una cantidad efectiva de la composición farmacéutica o las sales de la invención como se describe en la presente memoria.
La presente invención proporciona también composiciones farmacéuticas que comprenden una sal de la invención, y un portador, excipiente o diluyente farmacéutico adecuado para uso en medicina. En un aspecto particular, la composición farmacéutica es para uso en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
En aspectos adicionales, la invención proporciona procedimientos para sintetizar la sal de la invención, con protocolos de síntesis representativos y vías divulgadas más adelante en la presente memoria.
Otros objetivos y otras ventajas resultarán evidentes para aquellos expertos en la técnica a partir de una consideración de la descripción detallada resultante.
Se apreciará que las sales de la invención pueden metabolizarse para producir metabolitos biológicamente activos.
Descripción detallada de la invención
Definiciones
Se pretende que los siguientes términos tengan los significados presentados con estos a continuación y sean útiles en la comprensión de la descripción y el alcance buscado de la presente invención.
Cuando se describe la invención, que puede incluir compuestos, composiciones farmacéuticas que contienen dichos compuestos y procedimientos de uso de dichos compuestos y dichas composiciones, los siguientes términos, si están presentes, tienen los siguientes significados, a menos que se indique de otro modo. Se deberá comprender también que cuando se describe en la presente memoria, cualquiera de las fracciones definidas a continuación puede ser sustituida con una variedad de sustituyentes, y que las definiciones respectivas pretenden incluir dichas fracciones sustituidas dentro de su alcance como se establece a continuación. A menos que se indique de otro modo, el término 'sustituido' se deberá definir como se indica a continuación. Además, debe comprenderse que los términos 'grupos' y 'radicales' pueden considerarse sinónimos cuando se usan en la presente memoria.
Los artículos "un" y "una" pueden usarse en la presente memoria para referir a uno o a más de uno (es decir al menos a uno) de los objetos gramaticales del artículo. A modo de ejemplo, 'un análogo' significa un análogo o más de un análogo.
“Sales de la invención”, y expresiones equivalentes, pretenden abarcar las sales del compuesto de acuerdo con la Fórmula (I) (Compuesto I) como se describe en la presente memoria, cuya expresión incluye las sales aceptables para uso farmacéutico, y los solvatos, por ej., hidratos, y los solvatos de las sales aceptables para uso farmacéutico cuando lo permita el contexto.
“Aceptable para uso farmacéutico” significa aprobado o susceptible de aprobarse por una agencia regulatoria del gobierno federal o estatal o la agencia correspondiente en países fuera de los Estados Unidos, o que aparece en la lista de la Farmacopea de los EE. UU. u otra farmacopea generalmente reconocida para uso en animales, y más
particularmente, en seres humanos.
“Vehículo aceptable para uso farmacéutico” se refiere a un diluyente, adyuvante, excipiente o portador con el que se administra una sal de la invención.
“Solvato” se refiere a formas del compuesto que están asociadas con un disolvente, normalmente por una reacción de solvólisis. Esta asociación física incluye unión de hidrógeno. Los solventes convencionales incluyen agua, etanol, ácido acético y similares. Las sales de la invención pueden prepararse, por ej., en forma cristalina y pueden ser solvatadas o hidratadas. Los solvatos adecuados incluyen solvatos aceptables para uso farmacéutico, tales como hidratos, e incluyen, además, tanto solvatos estequimétricos como solvatos no estequimétricos. En ciertas instancias, el solvato será capaz de aislamiento, por ejemplo, cuando una o más moléculas de disolvente se incorporan en el retículo de cristal del sólido cristalino. “Solvato” abarca tanto los solvatos en fase de solución como aislables. Los solvatos representativos incluyen hidratos, etanolatos y metanolatos.
“Individuo” incluye seres humanos. Los términos “ser humano”, “paciente” e “individuo” se utilizan en la presente memoria como sinónimos.
“Cantidad efectiva” significa la cantidad de una sal de la invención que, cuando se administra a un individuo para tratar una enfermedad, es suficiente para efectuar dicho tratamiento para la enfermedad. La “cantidad efectiva” puede variar dependiendo del compuesto, la enfermedad y su severidad, y la edad, el peso, etc., del individuo que se tratará.
“Prevenir” o “prevención” se refiere a una reducción en el riesgo de adquirir o desarrollar una enfermedad o un trastorno (es decir, causar que al menos uno de los síntomas clínicos de la enfermedad no se desarrolle en un individuo que pueda estar expuesto a un agente causante de la enfermedad, o predispuesto a la enfermedad antes de la aparición de la enfermedad.
El término “profilaxis” se relaciona con “prevención”, y se refiere a una medida o procedimiento cuya finalidad es prevenir, en lugar de tratar o curar una enfermedad. Ejemplos no limitativos de medidas profilácticas pueden incluir la administración de vacunas; la administración de heparina de bajo peso molecular a pacientes hospitalarios en riesgo de trombosis debido a, por ejemplo, inmovilización; y la administración de un agente anti malaria tal como cloroquina, antes de una visita a una región geográfica donde la malaria es endémica o el riesgo de contraer malaria es alto.
“Tratar” o “tratamiento” de cualquier enfermedad o trastorno se refiere, en una realización, a aliviar la enfermedad o el trastorno (es decir, detener la enfermedad o reducir la manifestación, extensión o severidad de al menos uno de sus síntomas clínicos). En otra realización “tratar” o “tratamiento” se refieren a aliviar al menos un parámetro físico, que puede no ser discernible para el individuo. Incluso en otra realización, “tratar” o “tratamiento” se refiere a modular la enfermedad o el trastorno, ya sea físicamente (por ej., estabilización de un síntoma discernible), fisiológicamente (por ej., estabilización de un parámetro físico), o ambos. En una realización adicional, “tratar” o “tratamiento” se refiere a hacer más lenta la progresión de la enfermedad.
Como se usa en la presente memoria, el término “enfermedades inflamatorias” se refiere al grupo de afecciones que incluyen, artritis reumatoide, osteoartritis, artritis idiopática juvenil, psoriasis, artritis psoriásica, enfermedad alérgica de las vías aéreas (por ej. asma, rinitis), enfermedad pulmonar obstructiva crónica (EPOC), enfermedades inflamatorias intestinales (por ej. enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis), estados patológicos accionados por endotoxinas (por ej. complicaciones posteriores a una cirugía de bypass o estados crónicos de endotoxinas que contribuyen por ej. a insuficiencia cardíaca crónica), y enfermedades relacionadas que incluyen los cartílagos, tales como las de las articulaciones. Particularmente, el término se refiere a artritis reumatoide, osteoartritis, enfermedad alérgica de las vías aéreas (por ej., asma) enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades inflamatorias intestinales. Más particularmente, el término se refiere a artritis reumatoide, y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis).
Como se usa en la presente memoria, el término “enfermedad(es) autoinmune(s)” se refiere al grupo de enfermedades que incluyen enfermedad obstructiva de las vías aéreas, incluyendo afecciones tal como EPOC, asma (por ej., asma intrínseco, asma extrínseco, asma por el polvo, asma infantil) particularmente asma crónico o asma habitual (por ejemplo, asma tardío e híper respuesta de las vías aéreas), bronquitis, incluyendo asma bronquial, lupus eritematoso sistémico (SLE), lupus eritematoso cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, esclerosis múltiple, psoriasis, enfermedad del ojo seco, diabetes mellitus tipo I, y complicaciones asociadas con ésta, eczema atópico (dermatitis atópica) tiroiditis (tiroiditis de Hashimoto y autoinmune), dermatitis de contacto y dermatitis eczematosa adicional, enfermedad inflamatoria intestinal (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis), ateroesclerosis y esclerosis lateral amiotrófica. Particularmente el término se refiere a EPOC, asma, lupus eritematoso sistémico, diabetes mellitus tipo I y enfermedad inflamatoria intestinal.
Como se usa en la presente memoria, el término “enfermedad(es) proliferativa(s)” se refiere a afecciones tales como el cáncer (por ej., leiomiosarcoma o cáncer de próstata), trastornos mieloproliferativos (por ej., policitemia vera, trombocitosis esencial y mielofibrosis), leucemia (por ej., leucemia mieloide aguda, leucemia linfoblástica aguda y crónica), mieloma múltiple, psoriasis, restenosis, escleroderma o fibrosis. En particular el término se refiere a cáncer, leucemia, mieloma múltiple y psoriasis.
Como se usa en la presente memoria, el término “cáncer” se refiere a un crecimiento maligno o benigno de células en la piel o en los órganos corporales, por ejemplo, pero sin limitación, mama, próstata, pulmón, riñón, páncreas, estómago o intestino. Un cáncer tiende a infiltrarse en el tejido adyacente y esparcirse (hacer metástasis) a órganos distantes, por ejemplo, a los huesos, al hígado, a los pulmones o al cerebro. Como se usa en la presente memoria, el término cáncer incluye tanto los tipos de células tumorales con metástasis (tales como, pero sin limitación, melanoma, linfoma, leucemia, fibrosarcoma, rabdomiosarcoma y mastocitoma) como los tipos de carcinoma de tejidos (tales como, pero sin limitación, cáncer colorectal, cáncer de próstata, cáncer de pulmón de células pequeñas y cáncer de pulmón de células no pequeñas, cáncer de mama, cáncer de páncreas, cáncer de vejiga, cáncer renal, cáncer gástrico, glioblastoma, cáncer primario de hígado, cáncer de ovario, cáncer de próstata y leiomiosarcoma uterino). En particular, el término “cáncer” se refiere a leucemia linfoblástica aguda, leucemia mielodie, carcinoma adrenocortical, cáncer anal, cáncer de apéndice, astrocitomas, tumor teratoide/rabdoide atípico, carcinoma de células basales, cáncer del conducto biliar, cáncer de vejiga, cáncer de huesos (osteosarcoma e histiocitoma fibroso maligno), glioma del tronco encefálico, tumores de cerebro, tumores del cerebro y la médula espinal, leucemia linfocítica crónica, leucemia mielógena crónica, linfoma de células T cutáneas, tumores embrionarios, cáncer de endometrio, ependimoblastoma, ependimoma, cáncer de esófago, familia de tumores del sarcoma de Ewing, cáncer de ojo, retinoblastoma, cáncer de vesícula biliar, cáncer gástrico (estómago), tumor carcinoide gastrointestinal, tumor estromal gastrointestinal (GIST), tumor de células estromales gastrointestinales, tumor de células germinales, glioma, leucemia de células capilares, cáncer de cabeza y cuello, cáncer hepatocelular (hígado), cáncer hipofaríngeo, melanoma intraocular, tumores de células islote (páncreas endócrino), sarcoma de Kaposi, cáncer de riñón, histiocitosis de células de Langerhans, cáncer laríngeo, leucemia, leucemia linfoblástica aguda, leucemia mieloide aguda, leucemia linfocítica crónica, leucemia mielógena crónica, leucemia de células capilares, cáncer de hígado, cáncer de pulmón de células no pequeñas, cáncer de pulmón de células pequeñas, linfoma de Burkitt, linfoma de células T cutáneas, linfoma de Hodgkin, linfoma no de Hodgkin, linfoma, macroglobulinemia de Waldenstrom, meduloblastoma, meduloepitelioma, melanoma, mesotelioma, cáncer de boca, leucemia mielógena crónica, leucemia mieloide, mieloma múltiple, cáncer asofaríngeo, neuroblastoma, cáncer de pulmón de células no pequeñas, cáncer bucal, cáncer orofaríngeo, osteosarcoma, histocitoma fibroso maligno de huesos, cáncer de ovario, cáncer epitelial de ovario, tumor de células germinales de ovario, tumor potencial maligno bajo de ovario, cáncer de páncreas, papilomatosis, cáncer de paratiroides, cáncer de pene, cáncer de faringe, tumores parenquimales pineales de diferenciación intermedia, pineoblastoma y tumores neuroectodérmicos primitivos supratentoriales, tumor de la glándula pituitaria, mieloma múltiple/neoplasma de células plasmáticas, blastoma pleuropulmonar, linfoma del sistema nervioso central primario, cáncer de próstata, cáncer rectal, cáncer de (riñón) de célula renal, retinoblastoma, rabdomiosarcoma, cáncer de glándulas salivales, sarcoma, familia de tumores de sarcoma de Ewing, sarcoma, síndrome de Sezary, cáncer de piel, cáncer de pulmón de células pequeñas, cáncer de intestino delgado, sarcoma de tejido blando, carcinoma de células escamosas, cáncer de estómago (gástrico), tumores neuroectodérmicos primitivos supratentoriales, linfoma de células T, cáncer testicular, cáncer de garganta, carcinoma de timoma y tímico, cáncer de tiroides, cáncer de uretra, cáncer de útero, sarcoma uterino, cáncer vaginal, cáncer vulvar, macroglobulinemia de Waldenstrom, y tumor de Wilms. Más particularmente, el cáncer se selecciona de cáncer de mama, cáncer de endometrio y cervical, cáncer de pulmón, cáncer de ovario, cáncer de próstata, cáncer hepático, y cáncer pancreático.
Como se usa en la presente memoria, el término “leucemia” se refiere a enfermedades neoplásicas de la sangre y órganos que forman la sangre. Dichas enfermedades pueden causar disfunción de la médula ósea y del sistema inmune, lo cual torna al huésped altamente sensible a infección y sangrado. En particular, el término leucemia se refiere a leucemia mieloide aguda (AML), y leucemia linfoblástica aguda (ALL) y leucemia linfoblástica aguda (CLL).
Como se usa en la presente memoria, el término “alergia” se refiere al grupo de afecciones caracterizadas por un trastorno de hipersensibilidad del sistema inmune que incluye enfermedad alérgica de las vías respiratorias (por ej., asma, rinitis), sinusitis, eczema y urticaria, así como alergias a los alimentos o alergias al veneno de los insectos.
Como se usa en la presente memoria, el término “asma” como se usa en la presente memoria se refiere a cualquier trastorno de los pulmones caracterizado por variaciones en el flujo de gases pulmonares asociados con constricción de las vías aéreas de cualquier causa (intrínseca, extrínseca, o ambas; alérgica o no alérgica). El término asma puede usarse con uno o más adjetivos para indicar la causa.
Como se usa en la presente memoria, el término “rechazo a trasplantes” se refiere a rechazo agudo o crónico a alo- o xenoinjertos de células, tejido, u órganos sólidos de (por ej., islotes pancreáticos, mastocitos, médula ósea, piel, músculo, tejido corneal, tejido neuronal, corazón, pulmón, corazón-pulmón combinados, riñón, hígado, intestino, páncreas, tráquea o esófago, o enfermedades de injerto contra huésped.
Como se usa en la presente memoria, el término “enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos” incluye afecciones tal como osteoartritis, artritis psoriásica, artritis reumatoide juvenil, artritis por gota, artritis séptica o infecciosa, artritis reactiva, distrofia simpática refleja, algositrofia, acondroplasia, enfermedad de Paget, síndrome de Tietze o condritis costal, fibromialgia, osteocondritis, artritis neurogénica o neuropática, artropatía, sarcoidosis, amilosis, hidartrosis, enfermedad periódica, espondilitis reumatoide, formas endémicas de artritis como osteoartritis deformante endémica, enfermedad de Mseleni y enfermedad de Handigodu; degeneración resultante de fibromialgia, lupus eritematoso sistémico, escleroderma y espondilitis anquilosante.
Como se usa en la presente memoria, “malformaciones congénitas de cartílagos” incluye afecciones tales como condrólisis hereditaria, condrodisplasias y pseudocondrodisplasias, en particular, pero sin limitación, microtia, anotia, condrodisplasia metafisaria y trastornos relacionados.
Como se usa en la presente memoria, el término “enfermedades asociadas con la hipersecreción de IL6” incluye afecciones tal como la enfermedad de Castelman, mieloma múltiple, psoriasis, sarcoma de Kaposi y/o glomerulonefritis proliferativa mesangial.
Como se usa en la presente memoria, el término “enfermedades asociadas con la hipersecreción de interferones” incluye afecciones tal como lupus eritematoso sistémico y cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, psoriasis, artritis reumatoide.
Cuando se hace referencia en la presente memoria a intervalos, por ejemplo, pero sin limitación, alquilo Ci-8, la mención de un intervalo debe considerarse una representación de cada miembro de dicho intervalo.
Como se usa en la presente memoria, el término “variante isotópica” se refiere a un compuesto que contiene proporciones no naturales de isotopos en uno o más de los átomos que constituyen dicho compuesto. Por ejemplo, una “variante isotópica” de un compuesto puede contener uno o más isotopos no radiactivos, tal como, por ejemplo, deuterio (2H o D), carbono-13 (13C), nitrógeno-15 (15N), o similares. Se comprenderá que, en un compuesto en el que se realiza dicha sustitución isotópica, los siguientes átomos, donde están presentes, pueden variar, de modo tal que, por ejemplo, cualquier hidrógeno puede ser 2H/D, cualquier carbono puede ser 13C, o cualquier hidrógeno puede ser 15N, y que la presencia y la colocación de dichos átomos puede ser determinada dentro de las habilidades en la técnica. Del mismo modo, la invención puede incluir la preparación de variantes isotópicas con radioisótopos, en la instancia, por ejemplo, en la que los compuestos resultantes pueden usarse para estudios de distribución de tejidos en sustrato y/o fármaco. Los isotopos radiactivos tritio, es decir, 3H, y carbono 14, es decir, 14C, son particularmente útiles para este fin en vista de la facilidad de incorporación y los medios fáciles de detección. Además, pueden prepararse compuestos que estén sustituidos con isotopos de emisión de positrones, tal como 11C, 18F, 15O y 13N, y serían útiles en los estudios de Topografía por Emisión de Positrones (PET) para examinar la ocupación del receptor en el sustrato.
Todas las variantes isotópicas de los compuestos proporcionados en la presente memoria, ya sean radiactivos o no, pretenden estar abarcados dentro del alcance de la invención.
“Tautómeros” se refiere a los compuestos que son formas intercambiables de una estructura de compuesto particular, y que varían en el desplazamiento de los átomos de hidrógeno y electrones. De este modo, dos estructuras pueden estar en equilibrio a través del movimiento de electrones n y un átomo (normalmente H). Por ejemplo, los enoles y las cetonas son tautómeros porque se interconvierten rápidamente por tratamiento indistintamente con ácido o base. Otro ejemplo de tautomerismo es las formas ácido y nitro de fenilnitrometano, que se forman del mismo modo por tratamiento con ácido o base.
Las formas tautoméricas pueden ser relevantes para el logro de la reactividad química óptima y la actividad biológica de un compuesto de interés.
Se apreciará que las sales de la invención pueden metabolizarse para producir metabolitos biológicamente activos.
La invención
La presente invención se refiere a una sal 1:1:3 de Base Libre/HCl/H2O del compuesto {5-[4-(1,1-dioxo-tiomorfolin-4-ilmetil)-fenil]-[1,2,4]triazolo[1,5-a]piridin-2-il}-amida del ácido ciclopropancarboxílico (Compuesto I), y procedimientos para la preparación de la sal, que es útil en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones. En particular, las sales de la invención inhiben JAK, una familia de quinasas de la tirosina, y más particularmente JAK1.
La presente invención también proporciona procedimientos para la profilaxis y/o el tratamiento de enfermedades que incluyen afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones por administración de una sal de la invención, o una composición farmacéutica que contiene dicha sal.
De esta manera, en un aspecto la presente invención proporciona una sal de un compuesto de acuerdo con la Fórmula (I) (Compuesto I) a continuación:

en la que dicha sal es sal 1:1:3 de Base Libre/HCl/hbO.
En una realización, la sal de la invención es aquella formada con un agente formador de sal seleccionado de ácido clorhídrico.
En una realización, la sal de la invención es un solvato. En una realización particular, la sal de la invención es un trisolvato. En una realización mucho más particular, la sal de la invención es un trisolvato.
En una realización, la sal de la invención es un hidrato. En una realización más particular, la sal de la invención es un trihidrato. En una realización mucho más particular, la sal de la invención es un trihidrato.
En una realización, la sal de la divulgación es un aducto [Compuesto I:agente formador de sal:disolvente]. En una realización particular, la sal de la divulgación es un aducto [Compuesto I:agente formador de sal:disolvente] de 1:1:0 a 1:1:4. En una realización más particular, la sal de la divulgación es un aducto [Compuesto I:agente formador de sal:disolvente] 1:1:0, 1:1:1, 1:1:1.5, 1:1:2, o 1:1:3. En una realización mucho más particular, la sal de la invención es un aducto [Compuesto I:agente formador de sal:disolvente] 1:1:3.
En otra realización particular, la sal de la divulgación es un aducto [Compuesto I:HCl:disolvente]. En una realización particular, la sal de la divulgación es un aducto [Compuesto I:HCl:disolvente] de 1:1:0 a 1:1:4. En una realización más particular, la sal de la invención es un aducto [Compuesto I:HCl:disolvente] 1:1:0, 1:1:1, 1:1:1.5, 1:1:2, o 1:1:3. En una realización mucho más particular, la sal de la invención es un aducto [Compuesto I:HCl:disolvente] 1:1:3. En una realización mucho más particular adicional, el disolvente se selecciona de H2O
En otra realización, la sal de la divulgación es un aducto [Compuesto I:HCl:H2O]. En una realización particular, la sal de la invención es un aducto [Compuesto I:HCl:H2O] de 1:1:0 a 1:1:4. En una realización más particular, la sal de la divulgación es un aducto [Compuesto I:HCl:H2O] 1:1:0, 1:1:1, 1:1:1.5, 1:1:2, o 1:1:3. En una realización mucho más particular, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:3.
En una realización, la sal de la invención exhibe picos en un espectro de XRPD.
En una realización, la sal de la invención está en una forma cristalina.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:0 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en cualquiera de una o más de las siguientes posiciones: 7,4, 8,9, 12,4, 14,8, 15,1, 16,9, 17,6, 19,4, 20,7, 21,1, 22,8, 24,9, 26,0, 28,6, 29,8, y 32,6° 20 ± 0,2° 20.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:0 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo al menos en 5, 10, 15, o más de las siguientes posiciones: 7,4, 8,9, 12,4, 14,8, 15,1, 16,9, 17,6, 19,4, 20,7, 21,1, 22,8, 24,9, 26,0, 28,6, 29,8, y 32,6° 20 ± 0,2° 20.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:0 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en todas las siguientes posiciones: 7,4, 8,9, 12,4, 14,8, 15,1, 16,9, 17,6, 19,4, 20,7, 21,1, 22,8, 24,9, 26,0, 28,6, 29,8, y 32,6° 20 ± 0,2° 20. En una realización particular, la sal de la invención está caracterizada por el Patrón XRPD expresado en términos de ángulos 2 theta según lo mostrado en la Figura 4.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:3 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en cualquiera de una o más de las siguientes posiciones: 7,3, 8,4, 8,8, 10,7, 12,0, 12,2, 13,2, 13,7, 14,5, 16,3, 16,7, 17,6, 19,3, 20,2, 20,6, 21,0, 21,4, 21,8, 22,8, 23,4, 23,9, 24,5, 25,2, 25,7, 25,9, 26,4, 27,2, 27,7, 28,3, 28,6, 28,9, 29,2, 29,6, y 32,7° 20 ± 0,2° 20.
En una realización particular, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:3 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo al menos en 5, 10, 15, 20, 25, 30 o más de las siguientes posiciones: 7,3, 8,4, 8,8, 10,7, 12,0, 12,2, 13,2, 13,7, 14,5, 16,3, 16,7, 17,6, 19.3, 20,2, 20,6, 21,0, 21,4, 21,8, 22,8, 23,4, 23,9, 24,5, 25,2, 25,7, 25,9, 26,4, 27,2, 27,7, 28,3, 28,6, 28,9, 29,2, 29,6, y 32,7° 20 ± 0,2° 20.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:H2O] 1:1:3 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en todas las siguientes posiciones: 7.3, 8,4, 8,8, 10,7, 12,0, 12,2, 13,2, 13,7, 14,5, 16,3, 16,7, 17,6, 19,3, 20,2, 20,6, 21,0, 21,4, 21,8, 22,8, 23,4, 23,9, 24,5, 25,2, 25,7, 25,9, 26,4, 27,2, 27,7, 28,3, 28,6, 28,9, 29,2, 29,6, y 32,7° 20 ± 0,2° 20. En una realización particular, la sal de la invención está caracterizada por el Patrón XRPD expresado en términos de ángulos 2 theta según lo mostrado en la Figura 5.
En una realización, el aducto [Compuesto I:HCl:H2O] 1:1:3 en una forma cristalina sólida tiene un tamaño de las partículas de menos de 1000 pM, según lo medido por difracción del láser (Tabla II). En una realización particular, el aducto [Compuesto I:HCl:H2O] 1:1:3 en una forma sólida cristalina tiene un tamaño de partícula entre 50 pm y 800 pm. En una realización más particular, el aducto [Compuesto I:HCl:H2O] 1:1:3 en una forma sólida cristalina tiene un tamaño de partícula entre 200 pm y 600 pm.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:MeOH] 1:1:1 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en cualquiera de una o más de las siguientes posiciones: 7,1, 14,4, 16,6, 17,3, 18,9, 23,4, 24,8, y 29,0° 20 ± 0,2° 20.
En una realización particular, la sal de la invención es un aducto [Compuesto I:HCl:MeOH] 1:1:1 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo al menos en 3, 5, 7 o más de las siguientes posiciones: 7,1, 14,4, 16,6, 17,3, 18,9, 23,4, 24,8, y 29,0° 20 ± 0,2° 20.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:MeOH] 1:1:1 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en todas las siguientes posiciones: 7,1, 14,4, 16,6, 17,3, 18,9, 23,4, 24,8, y 29,0° 20 ± 0,2° 20. En una realización particular, la sal de la invención está caracterizada por el Patrón XRPD expresado en términos de ángulos 2 theta según lo mostrado en la Figura 8.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:HCO2H] 1:1:1,5 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en cualquiera de una o más de las siguientes posiciones: 7,1, 14,4, 14,8, 16,4, 17,4, 18,6, 20,8, 23,4, 24,5, 24,9, y 29,0° 20 ± 0,2° 20.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:HCO2H] 1:1:1,5 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo al menos en 3, 5, 7, 9 o más de las siguientes posiciones: 7,1, 14,4, 14,8, 16,4, 17,4, 18,6, 20,8, 23,4, 24,5, 24,9, y 29,0° 20 ± 0,2° 20.
En una realización, la sal de la invención es un aducto [Compuesto I:HCl:HCO2H] 1:1:1,5 en forma cristalina sólida, en la que la forma cristalina se caracteriza por un pico de difracción por rayos X del polvo en todas las siguientes posiciones: 7,1, 14,4, 14,8, 16,4, 17,4, 18,6, 20,8, 23,4, 24,5, 24,9, y 29,0° 20 ± 0,2° 20. En una realización particular, la sal de la invención está caracterizada por el Patrón XRPD expresado en términos de ángulos 2 theta según lo mostrado en la Figura 9.
En una realización, una sal de la invención se obtiene por combinación del Compuesto I, con un ácido seleccionado de ácido bromhídrico, ácido sulfúrico, ácido toluensulfónico, ácido bencensulfónico, ácido oxálico, ácido maleico, ácido naftalen-2-sulfónico, ácido naftalen-1,5-disulfónico, ácido 1-2-etan disulfónico, ácido metansulfónico, ácido 2-hidroxi etansulfónico, ácido fosfórico, ácido etansulfónico, ácido malónico, ácido 2-5-dihidroxibenzoico, y ácido L-tartárico, en un disolvente inerte, y la precipitación de dicha sal de dicho disolvente. En una realización particular, la sal de la invención se obtiene por el agregado del Compuesto I y un agente formador de sal en un disolvente adecuado para lograr la disolución completa, seguido de una evaporación controlada del disolvente, para lograr la supersaturación, y de este modo la cristalización de la correspondiente sal.
En una realización, la sal de la invención se obtiene mediante la mezcla del Compuesto I, y un ácido en una relación molar de entre 5:1 y 1:5 del Compuesto I: ácido. En una realización particular, la sal de la invención se obtiene mediante la mezcla del Compuesto I, y un ácido en una relación molar de entre 2:1 y 1:2 del Compuesto I: ácido. En una realización, la sal de la invención se obtiene mediante la mezcla de un Compuesto I y un ácido en una relación molar de 1:1.
En otra realización particular, el disolvente para la preparación de la sal de la invención se selecciona de cetonas, alcoholes, ésteres, alquilo C1-10, cicloalquilo C3-7, arilo C6-10 monocíclico o fusionado, sulfóxido, alquilnitrilo C1-10, éteres C1-10 lineales, ramificados o cíclicos, y haloalquilo C1-10. En una realización particular, el disolvente para la preparación de la sal de la invención se selecciona de acetona, anisol, butanol, acetato de butilo, TBME, DMSO, etanol, acetato de etilo, heptano, acetato de isopropilo, MEK, acetato de isopropilo, MeCN, ciclohexano, DCM, dioxano, metanol, nitrometano, THF, THF de metilo, tolueno, agua, acetona acuosa al 10%, THF acuoso al 10%, y metanol al 10%. En una realización particular, el disolvente para la preparación de la sal de la invención se selecciona de dioxano, THF,
acetona, DCM, y MeOH.
En otro aspecto se proporciona un procedimiento de preparación de la sal de la invención que comprende las etapas de:
i) hacer reaccionar el Compuesto I, con un ácido seleccionado de ácido clorhídrico,
en un disolvente inerte; y
ii) precipitar la sal mencionada a partir del disolvente mencionado.
En un aspecto adicional, se proporciona una sal de la invención que se obtiene por, o se obtiene mediante, el procedimiento mencionado con anterioridad.
En una realización, con respecto a la preparación de la sal de la invención, el disolvente inerte se selecciona de dioxano, THF, acetona, Dc M, y MeOH.
En una realización, con respecto a la preparación de la sal de la invención, el disolvente inerte es DCM.
En una realización, con respecto a la preparación de la sal de la invención, el disolvente inerte se selecciona de iPrOH/agua, iPrOH, iBuOH, y tBuOH.
En una realización se proporciona un procedimiento de preparación de un aducto [Compuesto I: HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) Agitar el Compuesto I con agua,
ii) Añadir HCl acuoso,
iii) Agitar, además, la mezcla obtenida en la etapa ii),
iv) Enfriar la mezcla de la etapa iii) hasta 15°C,
v) Continuar agitando durante un máximo de 24h a 15°C,
vi) Separar el sólido resultante por filtración obtenida en la etapa v) previo, y
vii) Secar bajo nitrógeno durante al menos 4h dicho sólido resultante obtenido en la etapa previa vi). En una realización particular se proporciona un procedimiento de preparación de un aducto [Compuesto I: HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) Agitar el Compuesto I con agua a 50°C,
ii) Añadir HCl acuoso,
iii) Agitar la mezcla obtenida en la etapa ii) en forma adicional a 50°C durante 15 min,
iv) Enfriar la mezcla de la etapa iii) hasta 15°C,
v) Continuar agitando durante 12h a 24h a 15°C,
vi) Separar el sólido resultante por filtración obtenida en la etapa previa v), y
vii) Secar bajo nitrógeno durante al menos 4h dicho sólido resultante obtenido en la etapa previa vi). En otra realización se proporciona un procedimiento de preparación de un aducto [Compuesto I: HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) Mezclar el Compuesto I suspendido en DCM, con MeOH,
ii) Añadir agua bajo agitación,
iii) Separar la fase orgánica,
iv) Añadir una solución de HCl a la fase orgánica obtenida en la etapa previa iii),
v) Separar el sólido resultante por filtración obtenida en la etapa previa iv),
vi) Secar dicho sólido resultante obtenido en la etapa previa v),
vii) Añadir el sólido obtenido en la etapa previa vi) a una solución de ácido fórmico/agua, bajo agitación,
viii) Añadir agua a la solución obtenida en la etapa previa vii),
ix) Separar por filtración el sólido resultante obtenido en la etapa previa viii), y
x) Secar el sólido resultante obtenido en la etapa previa ix),
En otra realización se proporciona un procedimiento de preparación de un aducto [Compuesto I: HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) Mezclar el Compuesto I suspendido en DCM, con MeOH a 35°C,
ii) Añadir agua bajo agitación a 35°C durante al menos 15 min,
iii) Separar la fase orgánica,
iv) Añadir una solución p/p al 10% de HCl en MeOH a la fase orgánica obtenida en la etapa previa iii), v) Separar el sólido resultante por filtración obtenida en la etapa previq iv),
vi) Secar dicho sólido resultante obtenido en la etapa previq v),
vii) Añadir el sólido obtenido en la etapa previa vi) a una solución 1,6/0,4 de ácido fórmico/agua, bajo agitación a 55°C
viii) Añadir agua a la solución obtenida en la etapa previq vii),
ix) Separar por filtración el sólido resultante obtenido en la etapa previq viii), y
x) Secar el sólido resultante obtenido en la etapa previq ix),
En otra realización se proporciona un procedimiento de preparación de un aducto [Compuesto I: HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) Hacer reaccionar el Compuesto I suspendido en DCM, con MeOH y trimercaptotriazina de trisodio, ii) Filtrar la suspensión resultante,
iii) Añadir agua bajo agitación,
iv) Separar la fase orgánica,
v) Añadir una solución de HCl a la fase orgánica obtenida en la etapa iv),
vi) Separar el sólido resultante por filtración obtenida en la etapa v),
vii) Secar dicho sólido resultante obtenido en la etapa vi),
viii) Añadir el sólido obtenido en la etapa vii) a una solución de ácido fórmico/agua, bajo agitación, ix) Añadir agua a la solución de la etapa viii),
x) Separar por filtración el sólido resultante obtenido en la etapa ix), y
xi) Secar el sólido resultante obtenido en la etapa x),
En otro aspecto se proporciona un procedimiento de preparación de un aducto [Compuesto I: HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) Hacer reaccionar el Compuesto I suspendido en DCM, con MeOH y trimercaptotriazina de trisodio a 35°C durante al menos 5h,
ii) Filtrar la suspensión resultante,
iii) Añadir agua bajo agitación a 35°C durante al menos 15 min,
iv) Separar la fase orgánica,
v) Añadir una solución p/p al 10% de HCl en MeOH a la fase orgánica obtenida en la etapa iv), vi) Separar el sólido resultante por filtración obtenida en la etapa v),
vii) Secar dicho sólido resultante obtenido en la etapa vi),
viii) Añadir el sólido obtenido en la etapa vii) a una solución 1,6/0,4 de ácido fórmico/agua, bajo agitación a 55°C
ix) Añadir agua a la solución de la etapa viii),
x) Separar por filtración el sólido resultante obtenido en la etapa ix), y
xi) Secar el sólido resultante obtenido en la etapa x),
Alternativamente, la ejecución de una o más de las variables especificadas de un grupo o una realización, o combinaciones de los anteriores también se contemplan en la presente invención.
Composiciones farmacéuticas
Cuando se emplea como producto farmacéutico, la sal de la invención se administra normalmente en la forma de una composición farmacéutica. Dichas composiciones pueden prepararse en un modo muy conocido en la técnica farmacéutica y comprenden al menos una sal activa de la invención.
Generalmente, una sal de la invención se administra en una cantidad farmacéuticamente efectiva. Un médico normalmente determinará la cantidad de la sal de la invención realmente administrada, en vista de las circunstancias relevantes, que incluyen la afección a tratar, la vía de administración elegida, la sal de la invención administrada en realidad, la edad, el peso y la respuesta del paciente individual, la severidad de los síntomas del paciente y similares.
La composición farmacéutica de esta invención puede administrarse por una variedad de vías que incluyen la vía oral, rectal, transdérmica, subcutánea, intraarticular, intravenosa, intramuscular e intranasal. Dependiendo de la vía de administración final, una sal de la invención se formula preferentemente como composiciones inyectables u orales o como ungüentos, lociones, o como parches, todos para administración transdérmica.
Las composiciones para administración oral pueden tener la forma de soluciones o suspensiones líquidas a granel, o polvos a granel. Más comúnmente, sin embargo, las composiciones se presentan en formas farmacéuticas unitarias para facilitar la administración exacta de la dosis. El término "formas farmacéuticas unitarias" se refiere a unidades físicamente separables adecuadas como formas farmacéuticas unitarias para seres humanos y otros mamíferos, en las que cada unidad contiene una cantidad predeterminada de material activo calculada para producir el efecto terapéutico deseado en asociación con un excipiente farmacéutico, vehículo o portador adecuados. Las formas farmacéuticas unitarias típicas incluyen las ampollas o jeringas pre llenadas, pre medidas de composiciones líquidas, o las píldoras, los comprimidos, las cápsulas o similares, en el caso de las composiciones sólidas. En dichas composiciones, la sal de la invención normalmente es un componente menor (de aproximadamente 0,1 a aproximadamente 50% en peso o preferentemente de aproximadamente 1 a aproximadamente 40% en peso) en la que el resto son varios vehículos o portadores y auxiliares de procesamiento útiles para formar la forma farmacéutica deseada.
Las formas líquidas adecuadas para administración oral pueden incluir un vehículo acuoso o no acuoso adecuado con tampones, agentes de suspensión y dispersión, colorantes, sabores y similares. Las formas sólidas incluyen, por ejemplo, cualquiera de los siguientes ingredientes, o sales de la invención de una naturaleza similar: un aglutinante tal como celulosa microcristalina, goma tragacanto o gelatina; un excipiente tal como almidón o lactosa, un agente desintegrante tal como ácido algínico, Primogel o almidón de maíz; un lubricante tal como estearato de magnesio; un deslizante tal como dióxido de silicio coloidal; un agente edulcorante tal como sacarosa o sacarina; o un agente saborizante tal como saborizante de menta o naranja.
Las composiciones inyectables normalmente se basan en solución salina estéril inyectable o solución salina tamponada con fosfato u otros portadores inyectables conocidos en la técnica. Como antes, la sal activa de la invención de acuerdo con la Fórmula I en dichas composiciones es normalmente un componente menor, con frecuencia de aproximadamente 0,05 a 10% en peso en el que el resto es el portador inyectable y similares.
Las composiciones transdérmicas están formuladas normalmente como ungüento o crema tópicos que contienen el(los) principio(s) activo(s), generalmente en una cantidad que oscila de aproximadamente 0,01 a aproximadamente 20% en peso, preferentemente de aproximadamente 0,1 a aproximadamente 20% en peso, preferentemente de aproximadamente 0,1 a aproximadamente 10% en peso, y más preferentemente de aproximadamente 0,5 a aproximadamente 15% en peso. Cuando se formulan como ungüento, los principios activos normalmente se combinan con una base indistintamente parafínica o un ungüento soluble en agua. Alternativamente, los principios activos pueden estar formulados en una crema con, por ejemplo, una base de crema de aceite en agua. Dichas formulaciones transdérmicas son muy conocidas en la técnica y generalmente incluyen ingredientes adicionales para mejorar la penetración dérmica de estabilidad de los principios activos o la formulación. Todos dichos ingredientes y formulaciones transdérmicas conocidos se incluyen dentro del alcance de la invención.
Una sal de la invención puede ser administrada por un dispositivo transdérmico. De esta manera, la administración transdérmica puede lograrse usando un parche, indistintamente del tipo reservorio o membrana porosa, o de una variedad de matriz sólida.
Los componentes descritos más arriba para las composiciones para administración oral, inyectable o tópica son simplemente representativos. Otros materiales, además de las técnicas de procesamiento y similares, se establecen en la Parte 8 de Remington’s Pharmaceutical Sciences, 17th edition, 1985, Mack Publishing Company, Easton, Pennsylvania, la cual se incorpora a la presente a modo de referencia.
Una sal de la invención también pueden administrarse en formas de liberación sostenida o a partir de sistemas de administración de liberación sostenida de fármacos. Una descripción de los materiales de liberación sostenida representativos pueden encontrarse en Remington’s Pharmaceutical Sciences.
Los siguientes ejemplos de formulación incluyen composiciones farmacéuticas representativas que pueden prepararse de acuerdo con esta invención. La presente invención, sin embargo, no está limitada a las siguientes composiciones farmacéuticas.
Formulación 1 - Comprimidos
Una sal de la invención puede mezclarse como polvo seco con un aglutinante de gelatina seca en una relación en peso de aproximadamente 1:2. Una cantidad menor de estearato de magnesio puede añadirse como lubricante. La mezcla puede formarse en comprimidos de 240-270 mg (80-90 mg de sal activa de la invención por comprimido) en una prensa para comprimidos.
Formulación 2 - Cápsulas
Una sal de la invención puede mezclarse como polvo seco con un diluyente de almidón en una relación en peso de aproximadamente 1:1. La mezcla puede llenarse en cápsulas de 250 mg (125 mg de sal activa de la invención por cápsula).
Formulación 3 - Líquido
Una sal de la invención (125 mg), puede mezclarse con sacarosa (1,75 g) y goma xántica (4 mg) y la mezcla resultante puede mezclarse, pasarse a través de un tamiz U.S. malla Nro. 10, y luego mezclarse con una solución realizada previamente de celulosa microcristalina y carboximetilcelulosa de sodio (11:89, 50 mg) en agua. Pueden diluirse benzoato de sodio (10 mg), sabor y color con agua y añadirse con agitación. Luego puede añadirse suficiente agua con agitación. Además, luego puede añadirse suficiente agua para producir un volumen total de 5 ml.
Formulación 4 - Comprimidos
Una sal de la invención puede mezclarse como polvo seco con un aglutinante de gelatina seca en una relación en peso de aproximadamente 1:2. Una cantidad menor de lubritab puede añadirse como lubricante. La mezcla puede formarse en comprimidos de 25-900 mg (8-300 mg de sal activa de la invención) en una prensa para comprimidos.
Formulación 5 - Inyección
Una sal de la invención puede disolverse o suspenderse en un medio acuoso inyectable de solución salina tamponada con fosfato hasta una concentración de aproximadamente 5 mg/ml.
Formulación 6 - Tópica
Alcohol estearílico (250 g) y una vaselina (250 g) pueden fundirse a aproximadamente 75°C y luego una mezcla de una sal de la invención (50 g) metil parabeno (0,25 g), propilparabeno (0,15 g), lauril sulfato de sodio (10 g), y propilenglicol (120 g) disuelto en agua (aproximadamente 370 g) puede añadirse y la mezcla resultante puede agitarse hasta que se congela.
Procedimientos de tratamiento
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en medicina. En una realización particular, la presente invención proporciona sales de la invención o composiciones farmacéuticas que comprenden una sal de la invención para uso en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención en medicina. En una realización particular, la presente invención proporciona sales de la invención o composiciones farmacéuticas que comprenden una sal de la invención para uso en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
En otra realización, la presente invención proporciona sales de la invención o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que implican la degradación y/o interrupción de la homeóstasis de cartílagos, malformaciones congénitas de cartílagos, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición.
En una realización, la presente invención proporciona composiciones farmacéuticas que comprenden una sal de la invención, y otro agente terapéutico. En una realización particular, otro agente terapéutico es un agente para el tratamiento de afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo al trasplante, enfermedades que incluyen degradación y/o interrupción de la homeostasis de cartílago, malformaciones congénitas de cartílago, y/o enfermedades asociadas con la hipersecreción de IL6 o interferones.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de enfermedades inflamatorias. En una realización particular, la enfermedad inflamatoria se selecciona de artritis reumatoide, osteoartritis, enfermedad alérgica de las vías respiratorias (por ej., asma) enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica, o colitis). Más particularmente, la enfermedad inflamatoria se selecciona de artritis reumatoide, y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis).
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de enfermedades inflamatorias. En una realización particular, la enfermedad inflamatoria se selecciona de artritis reumatoide, osteoartritis, enfermedad alérgica de las vías respiratorias (por ej., asma) enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica, o colitis). Más particularmente, la enfermedad inflamatoria se selecciona de artritis reumatoide, y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis).
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de enfermedades inflamatorias. En una realización particular, la enfermedad inflamatoria se selecciona de artritis reumatoide, osteoartritis, enfermedad alérgica de las vías respiratorias (por ej., asma) enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica, o colitis). Más particularmente, la enfermedad inflamatoria se selecciona de artritis reumatoide, y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis).
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre enfermedades inflamatorias, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, la enfermedad inflamatoria se selecciona de artritis reumatoide, osteoartritis, enfermedad alérgica de las vías respiratorias (por ej., asma) enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica, o colitis). Más particularmente, la enfermedad inflamatoria se selecciona de artritis reumatoide, y enfermedades inflamatorias intestinales (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis).
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de enfermedades autoinmunes. En una realización particular, la enfermedad autoinmune se selecciona de EPOC, asma (por ej., asma intrínseco, asma extrínseco, asma por el polvo, asma infantil) particularmente asma crónico o asma habitual (por ejemplo, asma tardío e híper respuesta de las vías aéreas), bronquitis, incluyendo asma bronquial, lupus eritematoso sistémico (SLE), lupus eritematoso cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, esclerosis múltiple, psoriasis, enfermedad del ojo seco, diabetes mellitus tipo I, y complicaciones asociadas con ésta, eczema atópico (dermatitis atópica) tiroiditis (tiroiditis de Hashimoto y autoinmune), dermatitis de contacto y dermatitis eczematosa adicional, enfermedad inflamatoria intestinal (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis), ateroesclerosis y esclerosis lateral amiotrófica. Particularmente, la enfermedad autoinmune se selecciona de EPOC, asma, lupus eritematoso sistémico, diabetes mellitus tipo I y enfermedad inflamatoria intestinal.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de enfermedades autoinmunes. En una realización particular, la enfermedad autoinmune se selecciona de EPOC, asma (por ej., asma intrínseco, asma extrínseco, asma por el polvo, asma infantil) particularmente asma crónico o asma habitual (por ejemplo, asma tardío e híper respuesta de las vías aéreas), bronquitis, incluyendo asma bronquial, lupus eritematoso sistémico (SLE), lupus eritematoso cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, esclerosis múltiple, psoriasis, enfermedad del ojo seco, diabetes mellitus tipo I, y complicaciones asociadas con ésta, eczema atópico (dermatitis atópica) tiroiditis (tiroiditis de Hashimoto y autoinmune), dermatitis de contacto y dermatitis eczematosa adicional, enfermedad inflamatoria intestinal (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis), ateroesclerosis y esclerosis lateral amiotrófica. Particularmente, la enfermedad autoinmune se selecciona de EPOC, asma, lupus eritematoso sistémico, diabetes mellitus tipo I y enfermedad inflamatoria intestinal.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de enfermedades autoinmunes. En una realización particular, la enfermedad autoinmune se selecciona de EPOC, asma (por ej., asma intrínseco, asma extrínseco, asma por el polvo, asma infantil) particularmente asma crónico o asma habitual (por ejemplo, asma tardío e híper respuesta de las vías aéreas), bronquitis, incluyendo asma bronquial, lupus eritematoso sistémico (SLE), lupus eritematoso cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, esclerosis múltiple, psoriasis, enfermedad del ojo seco, diabetes mellitus tipo I, y complicaciones asociadas con ésta, eczema atópico (dermatitis atópica) tiroiditis (tiroiditis de Hashimoto y autoinmune), dermatitis de contacto y dermatitis eczematosa adicional, enfermedad inflamatoria intestinal (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis), ateroesclerosis y esclerosis lateral amiotrófica. Particularmente, la enfermedad autoinmune se selecciona de EPOC, asma, lupus eritematoso sistémico, diabetes mellitus tipo I y enfermedad inflamatoria intestinal.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre enfermedades autoinmunes, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, la enfermedad autoinmune se selecciona de EPOC, asma (por ej., asma intrínseco, asma extrínseco, asma por el polvo, asma infantil) particularmente asma crónico o asma habitual (por ejemplo, asma tardío e híper respuesta de las vías aéreas), bronquitis, incluyendo asma bronquial, lupus eritematoso sistémico (SLE), lupus eritematoso cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, esclerosis múltiple, psoriasis, enfermedad del ojo seco, diabetes mellitus tipo I, y complicaciones asociadas con ésta, eczema atópico (dermatitis atópica) tiroiditis (tiroiditis de Hashimoto y autoinmune), dermatitis de contacto y dermatitis eczematosa adicional, enfermedad inflamatoria intestinal (por ej., enfermedad de Crohn, Whipple, colitis ulcerativa crónica o colitis), ateroesclerosis y esclerosis lateral amiotrófica. Particularmente, la enfermedad autoinmune se selecciona de EPOC, asma, lupus eritematoso sistémico, diabetes mellitus tipo I y enfermedad inflamatoria intestinal.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de enfermedades proliferativas. En una realización particular, la enfermedad proliferativa se selecciona de cáncer y leucemia. En una realización más particular, la enfermedad proliferativa se selecciona de cáncer de mama, cáncer de endometrio y cervical, cáncer de pulmón, cáncer de ovario, cáncer de próstata, cáncer hepático, y cáncer pancreático.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de enfermedades proliferativas. En una realización particular, la enfermedad proliferativa se selecciona de cáncer y leucemia. En una realización más particular, la enfermedad proliferativa se selecciona de cáncer de mama, cáncer de endometrio y cervical, cáncer de pulmón, cáncer de ovario, cáncer de próstata, cáncer hepático, y cáncer pancreático.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de enfermedades proliferativas. En una realización particular, la enfermedad proliferativa se selecciona de cáncer y leucemia. En una realización más particular, la enfermedad proliferativa se selecciona de cáncer de mama, cáncer de endometrio y cervical, cáncer de pulmón, cáncer de ovario, cáncer de próstata, cáncer hepático, y cáncer pancreático.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre enfermedades proliferativas, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, la enfermedad proliferativa se selecciona de cáncer y leucemia. En una realización más particular, la enfermedad proliferativa se selecciona de cáncer de mama, cáncer de endometrio y cervical, cáncer de pulmón, cáncer de ovario, cáncer de próstata, cáncer hepático, y cáncer pancreático.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de alergias. En una realización particular, la alergia se selecciona de enfermedad alérgica de las vías respiratorias (por ej., asma, rinitis), sinusitis, eczema y urticaria, así como alergias a los alimentos o alergias al veneno de insectos.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de la alergia. En una realización particular, la alergia se selecciona de enfermedad alérgica de las vías respiratorias (por ej., asma, rinitis), sinusitis, eczema y urticaria, así como alergias a los alimentos o alergias al veneno de insectos.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de la alergia. En una realización particular, la alergia se selecciona de enfermedad alérgica de las vías respiratorias (por ej., asma, rinitis), sinusitis, eczema y urticaria, así como alergias a los alimentos o alergias al veneno de insectos.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre alergia, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, la alergia se selecciona de enfermedad alérgica de las vías respiratorias (por ej., asma, rinitis), sinusitis, eczema y urticaria, así como alergias a los alimentos o alergias al veneno de insectos.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de rechazo a los trasplantes.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de la rechazo a los trasplantes.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento del rechazo a los trasplantes.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre rechazo a los trasplantes, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de enfermedades que implican degradación y/o interrupción de la homeóstasis de cartílagos. En una realización particular, las enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos se selecciona de osteoartritis, artritis psoriásica, artritis reumatoide juvenil, artritis por gota, artritis séptica o infecciosa, artritis reactiva, distrofia simpática refleja, algositrofia, acondroplasia, enfermedad de Paget, síndrome de Tietze o condritis costal, fibromialgia, osteocondritis, artritis neurogénica o neuropática, artropatía, sarcoidosis, amilosis, hidartrosis, enfermedad periódica, espondilitis reumatoide, formas endémicas de artritis como osteoartritis deformante endémica, enfermedad de Mseleni y enfermedad de Handigodu; degeneración resultante de fibromialgia, lupus eritematoso sistémico, escleroderma y espondilitis anquilosante.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos. En una realización particular, las enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos se selecciona de osteoartritis, artritis psoriásica, artritis reumatoide juvenil, artritis por gota, artritis séptica o infecciosa, artritis reactiva, distrofia simpática refleja, algositrofia, acondroplasia, enfermedad de Paget, síndrome de Tietze o condritis costal, fibromialgia, osteocondritis, artritis neurogénica o neuropática, artropatía, sarcoidosis, amilosis, hidartrosis, enfermedad periódica, espondilitis reumatoide, formas endémicas de artritis como osteoartritis deformante endémica, enfermedad de Mseleni y enfermedad de Handigodu; degeneración resultante de fibromialgia, lupus eritematoso sistémico, escleroderma y espondilitis anquilosante.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de enfermedades que implican degradación y/o interrupción de la homeóstasis de cartílagos. En una realización particular, las enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos se selecciona de osteoartritis, artritis psoriásica, artritis reumatoide juvenil, artritis por gota, artritis séptica o infecciosa, artritis reactiva, distrofia simpática refleja, algositrofia, acondroplasia, enfermedad de Paget, síndrome de Tietze o condritis costal, fibromialgia, osteocondritis, artritis neurogénica o neuropática, artropatía, sarcoidosis, amilosis, hidartrosis, enfermedad periódica, espondilitis reumatoide, formas endémicas de artritis como osteoartritis deformante
endémica, enfermedad de Mseleni y enfermedad de Handigodu; degeneración resultante de fibromialgia, lupus eritematoso sistémico, escleroderma y espondilitis anquilosante.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, las enfermedades que incluyen la degradación y/o interrupción de la homeóstasis de cartílagos se selecciona de osteoartritis, artritis psoriásica, artritis reumatoide juvenil, artritis por gota, artritis séptica o infecciosa, artritis reactiva, distrofia simpática refleja, algositrofia, acondroplasia, enfermedad de Paget, síndrome de Tietze o condritis costal, fibromialgia, osteocondritis, artritis neurogénica o neuropática, artropatía, sarcoidosis, amilosis, hidartrosis, enfermedad periódica, espondilitis reumatoide, formas endémicas de artritis como osteoartritis deformante endémica, enfermedad de Mseleni y enfermedad de Handigodu; degeneración resultante de fibromialgia, lupus eritematoso sistémico, escleroderma y espondilitis anquilosante.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de malformación(es) congénitas de cartílagos. En una realización particular, la(s) malformaciones congénitas de cartílagos se seleccionan de condrólisis hereditaria, condrodisplasias y pseudocondrodisplasias, en particular, pero sin limitación, microtia, anotia, condrodisplasia metafisaria y trastornos relacionados.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de malformación(es) congénitas de cartílagos. En una realización particular, la(s) malformaciones congénitas de cartílagos se seleccionan de condrólisis hereditaria, condrodisplasias y pseudocondrodisplasias, en particular, pero sin limitación, microtia, anotia, condrodisplasia metafisaria y trastornos relacionados.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de malformación(es) congénitas de cartílagos. En una realización particular, la(s) malformaciones congénitas de cartílagos se seleccionan de condrólisis hereditaria, condrodisplasias y pseudocondrodisplasias, en particular, pero sin limitación, microtia, anotia, condrodisplasia metafisaria y trastornos relacionados.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre malformación(es) congénitas de cartílagos, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, la(s) malformaciones congénitas de cartílagos se seleccionan de condrólisis hereditaria, condrodisplasias y pseudocondrodisplasias, en particular, pero sin limitación, microtia, anotia, condrodisplasia metafisaria y trastornos relacionados.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de enfermedades asociadas con la hipersecreción de IL6. En una realización particular, las enfermedades asociadas con la hipersecreción de IL6” se selecciona(n) de la enfermedad de Castelman, mieloma múltiple, psoriasis, sarcoma de Kaposi y/o glomerulonefritis proliferativa mesangial.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de enfermedades asociadas con la hipersecreción de IL6. En una realización particular, las enfermedades asociadas con la hipersecreción de IL6” se selecciona(n) de la enfermedad de Castelman, mieloma múltiple, psoriasis, sarcoma de Kaposi y/o glomerulonefritis proliferativa mesangial.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de enfermedades asociadas con la hipersecreción de IL6. En una realización particular, las enfermedades asociadas con la hipersecreción de IL6” se selecciona(n) de la enfermedad de Castelman, mieloma múltiple, psoriasis, sarcoma de Kaposi y/o glomerulonefritis proliferativa mesangial.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre enfermedades asociadas con la hipersecreción de IL6, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, las enfermedades asociadas con la hipersecreción de IL6” se selecciona(n) de la enfermedad de Castelman, mieloma múltiple, psoriasis, sarcoma de Kaposi y/o glomerulonefritis proliferativa mesangial.
En una realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, para uso en la profilaxis y/o el tratamiento de enfermedades asociadas con la
hipersecreción de interferones. En una realización particular, la enfermedad asociada con hipersecreción de interferones se selecciona de lupus eritematoso sistémico y cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, psoriasis y artritis reumatoide.
En una realización, la presente invención proporciona el uso de una sal de la invención, o composiciones farmacéuticas que comprenden una sal de la invención, en la profilaxis y/o el tratamiento de enfermedades asociadas con la hipersecreción de interferones. En una realización particular, la enfermedad asociada con hipersecreción de interferones se selecciona de lupus eritematoso sistémico y cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, psoriasis y artritis reumatoide.
En otra realización, la presente invención proporciona sales de la invención, o composiciones farmacéuticas que comprenden una sal de la invención para uso en la fabricación de un medicamento para uso en la profilaxis y/o el tratamiento de enfermedades asociadas con la hipersecreción de interferones. En una realización particular, la enfermedad asociada con hipersecreción de interferones se selecciona de lupus eritematoso sistémico y cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, psoriasis y artritis reumatoide.
En un procedimiento adicional de aspectos de tratamiento esta invención proporciona procedimientos de profilaxis y/o tratamiento de un mamífero que sufre enfermedades asociadas con la hipersecreción de interferones, cuyos procedimientos comprenden la administración de una cantidad efectiva de una sal de la invención o una o más de las composiciones farmacéuticas descritas en la presente memoria para el tratamiento o la profilaxis de dicha condición. En una realización particular, la enfermedad asociada con hipersecreción de interferones se selecciona de lupus eritematoso sistémico y cutáneo, nefritis por lupus, dermatomiositis, síndrome de Sjogren, psoriasis y artritis reumatoide.
Un régimen particular de la presente invención comprende la administración a un individuo que sufre afecciones inflamatorias, enfermedades autoinmunes, enfermedades proliferativas, alergia, rechazo a los trasplantes, enfermedades que implican la degradación y/o interrupción de la homeóstasis de cartílagos, malformaciones congénitas y/o enfermedades asociadas con la hipersecreción de IL6 o interferones, de una cantidad efectiva de una sal de la invención durante un período de tiempo suficiente para reducir el nivel de las enfermedades antes mencionadas en el individuo, y preferentemente terminar los procesos responsables para dichos procesos.
Por el uso de estos patrones de dosificación, cada dosis proporciona de aproximadamente 1 a aproximadamente 500 mg de la sal de la invención, en las que cada dosis particular proporciona de aproximadamente 10 a aproximadamente 300 mg, más particularmente aproximadamente 25 a aproximadamente 250 mg, y especialmente 10 mg, 20 mg, 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg o 300 mg.
Los niveles de dosis para inyección oscilan de aproximadamente 0,1 mg/kg/h a al menos 10 mg/kg/h, en total durante de aproximadamente 1 a aproximadamente 120 h y especialmente 24 a 96 h. También puede administrarse un bolo de precarga de aproximadamente 0,1 mg/kg a aproximadamente 10 mg/kg o más para lograr niveles adecuados en estado estacionario. No se espera que la dosis máxima total exceda aproximadamente 1 g/día para un paciente humano de 40 a 80 kg.
Para la profilaxis y/o el tratamiento de afecciones prolongadas, tal como las afecciones degenerativas, el régimen para tratamiento normalmente se prolonga durante muchos meses o años de modo tal que se prefiere la administración oral para conveniencia y tolerancia del paciente. Con la administración oral, de una a cuatro (1 -4) dosis regulares por día, especialmente de una a tres (1-3) dosis regulares por día, normalmente una a dos (1-2) dosis regulares por día, y mucho más normalmente una (1) dosis regular por día son regímenes representativos. Alternativamente para fármacos de efecto prolongado, con la administración oral, una vez cada semana por medio, una vez por semana y una vez por día son regímenes representativos. En particular, el régimen de administración puede ser cada 1 -14 días, más particularmente, 1-10 días, incluso más particularmente 1-7, y mucho más particularmente 1-3 días.
Por el uso de estos patrones de dosificación, cada dosis proporciona de aproximadamente 1 a aproximadamente 500 mg de la sal de la invención, en las que cada dosis particular proporciona de aproximadamente 10 a aproximadamente 300 mg, más particularmente aproximadamente 25 a aproximadamente 250 mg, y especialmente 10 mg, 20 mg, 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg o 300 mg.
Las dosis transdérmicas generalmente se seleccionan para proporcionar niveles en sangre similares o más bajos que los logrados usando dosis inyectables.
Cuando se usa para prevenir la aparición de una condición, una sal de la invención se administrará a un paciente que presente riesgo de desarrollar la afección, normalmente tras el consejo y bajo la supervisión de un médico, a los niveles posológicos descritos más arriba. Los pacientes en riesgo de desarrollar una afección particular generalmente incluyen aquellos que tienen antecedentes familiares de la afección, o aquellos que según la identificación realizada por análisis o determinación genética sean particularmente susceptibles a desarrollar la afección.
Una sal de la invención puede administrarse como principio activo solo o puede administrarse en combinación con otros agentes terapéuticos, incluyendo otras sales de la invención que demuestran la misma actividad terapéutica, o una similar, y que se determine que son seguras y eficaces para dicha administración combinada. En una realización
específica, la administración concomitante de dos (o más) agentes permite el uso de dosis significativamente más bajas, reduciendo de esta manera los efectos colaterales observados.
En una realización, una sal de la invención, o una composición farmacéutica que comprende una sal de la invención se administra como medicamento. En una realización específica, dicha composición farmacéutica comprende, además, un principio activo adicional.
En una realización, una sal de la invención se administra de manera concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de una enfermedad que implica inflamación, los agentes particulares incluyen, pero sin limitación, agentes inmuno reguladores, por ej., azatioprina, corticosteroides (por ej., prednisolona o dexametasona), ciclofosfamida, ciclosporina A, tacrolimo, micofenolato, mofetilo, muromonab-CD3 (OKT3, por ej., Orthocolone®), ATG, aspirina, paracetamol, ibuprofeno, naproxeno y piroxicam.
En una realización, una sal de la invención se administra en forma concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de la artritis (por ej., artritis reumatoide), los agentes particulares incluyen, pero sin limitación, analgésicos, fármacos antiinflamatorios no esteroides (AINE), esteroides DMARDS sintéticos (por ejemplo, pero sin limitación metotrexato, leflunomida, sulfasalazina, auranofina, aurotiomalato de sodio, penicilamina, cloroquina, hidroxicloroquina, azatioprina, tofacitinib, baricitinib, fostamatinib, y ciclosporina), y DMARDS biológicos (por ejemplo, pero sin limitación infliximab, etanercept, adalimumab, rituximab y abatacept).
En una realización, una sal de la invención se administra de manera concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de trastornos proliferativos, los agentes particulares incluyen, pero sin limitación: metotrexato, leukovorina, adriamicina, prednisona, bleomicina, ciclofosfamida, 5- fluorouracilo, paclitaxel, docetaxel, vincristina, vinblastina, vinorelbina, doxorubicina, tamoxifeno, toremifeno, acetato de megestrol, anastrozol, goserelina, anticuerpo monoclonal anti-HER2 (por ej., Herceptin™), capecitabina, clorhidrato de raloxifeno, inhibidores de EGFR (por ej., lressa®, Tarceva™, Erbitux™), inhibidores de VEGF (por ej., Avastin™), inhibidores de proteasoma (por ej., Velcade™), Glivec® e inhibidores de hsp90 (por ej., 17-AAG). Además, la sal de la invención de acuerdo con la Fórmula I puede administrarse en combinación con otras terapias que incluyen, pero sin limitación, radioterapia o cirugía. En una realización específica, el trastorno proliferativo se selecciona de cáncer, enfermedad mieloproliferativa o leucemia.
En una realización, una sal de la invención se administra de manera concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de enfermedades autoinmunes, los agentes particulares incluyen, pero sin limitación: glucocorticoides, agentes citostáticos (por ej., análogos de purina), agentes de alquilación, (por ej., mostazas de nitrógeno (ciclofosfamida), nitrosoureas, sales de platino de la invención y otros), antimetabolitos (porej., metotrexato, azatioprina y mercaptopurina), antibióticos citotóxicos (por ej., dactinomicina antraciclinas, mitomicina C, bleomicina y mitramicina), anticuerpos (por ej., anticuerpos monoclonales anti-CD20, anti-CD25 o anti-CD3 (OTK3), Atgam® y Thymoglobuline®), ciclosporina, tacrolimo, rapamicina (sirolimo), interferones (por ej., IFN-P), proteínas que se unen al TNF (porej., infliximab, etanercept, o adalimumab), micofenolato, fingolimod y miriocina.
En una realización, una sal de la invención se administra de manera concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de rechazo a los trasplantes, los agentes particulares incluyen, pero sin limitación: inhibidores de calcineurina (por ej., ciclosporina o tacrolimo (FK506)), inhibidores de mTOR (por ej., sirolimo, everolimo), anti-proliferativos (por ej., azatioprina, ácido micofenólico), corticosteroides (por ej., prednisolona, hidrocortisona), anticuerpos (por ej., anticuerpos monoclonales del receptor anti-IL-2Ra, basiliximab, daclizumab), anticuerpos policlonales anti-células T (porej., globulina anti-timocito (ATG), globulina anti-linfocito (ALG)).
En una realización, una sal de la invención se administra en forma concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de asma y/o rinitis y/o EPOC, los agentes particulares incluyen, pero sin limitación: agonistas adrecoceptores beta2 (por ej., salbutamol, levalbuterol, terbtalina y bitolterol), epinefrina (inhalada o comprimidos), anticolinérgicos (por ej., bromuro de ipratropio), glucocorticoides (orales o inhalados). Los agonistas p2 de acción prolongada (por ej., salmeterol, formoterol, bambuterol, y albuterol oral de liberación prolongada), combinaciones de esteroides inhalados y broncodilatadores de acción prolongada (por ej., fluticasona/salmeterol, budesonida/formoterol), antagonistas e inhibidores de la síntesis de leucotrieno (por ej., montelukast, zafirlukast y zileuton), inhibidores de liberación de mediadores (por ej., cromoglicato y cetotifeno), reguladores biológicos de la respuesta de IgE (porej., omalizumab), antihistaminas (porej., ceterizina, cinarizina, fexofenadina) y vasoconstrictores (por ej., oximetazolina, xilometazolina, nafazolina y tramazolina).
Además, una sal de la invención puede administrarse en combinación con las terapias de emergencia para el asma y/o EPOC, dichas terapias incluyen administración de oxígeno o helio, salbutamol o terbutalina nebulizados (opcionalmente combinados con un anticolinérgico (por ej., ipratropio), esteroides sistémicos (orales o intravenosos, por ej., prednisona, prednisolona, metilprednisolona, dexametasona o hidrocortisona), salbutamol intravenoso, beta agonistas no específicos, metaproterenol), anticolinérgicos (IV o negulizados, por ej., glicopirrolato, atropina, ipratropio), metilxantinas (teofilina, aminofilina, bamifilina), anestésicos por inhalación que tienen un efecto broncodilatador (por ej., isoflurano, halotano, enflurano), cetamina y sulfato de magnesio intravenoso.
En una realización, una sal de la invención se administra en forma concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de enfermedad inflamatoria de intestino (IBD), los agentes particulares incluyen, pero sin limitación: glucocorticoides (por ej., prednisona, budesonida) agentes inmunomoduladores, sintéticos, modificadores de la enfermedad (por ej., metotrexato, leflunomida, sulfasalazina, mesalazina, azatioprina, 6- mercaptopurina y ciclosporina) agentes inmunomoduladores, biológicos, modificadores de la enfermedad (infliximab, adalimumab, rituximab, y abatacept).
En una realización, una sal de la invención se administra en forma concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de SLE, los agentes particulares incluyen, pero sin limitación: anticuerpos monoclonales humanos (belimumab (Benlysta)), fármacos antirreumáticos modificadores de la enfermedad (DMARD) tal como antimaláricos (por ej., plaquenil, hidroxicloroquina), inmunosupresores (por ej., metotrexato y azatioprina), ciclofosfamida y ácido micofenólico, fármacos inmunosupresores y analgésicos, tal como fármacos antiinflamatorios no esteroides, opiatos (por ej., dextropropoxifeno y co-codamol), opioides (porej., hidrocodona, oxicodona, MS Contin, o metadona) y el parche transdérmico duragésico de fentanilo.
En una realización, una sal de la invención se administra en forma concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de la psoriasis, los agentes particulares incluyen, pero sin limitación: tratamientos tópicos tal como soluciones para el baño, humectantes, cremas y ungüentos medicados que contienen alquitrán de carbón, ditranol (antralina), corticosteroides como desoximetasona (Topicort™), fluocinonida, análogos de vitamina D3 (por ejemplo calcipotriol), aceite de argán y retinoides (etretinato, acitretina, tazaroteno), tratamientos sistémicos tal como metotrexato, ciclosporina, retinoides, tioguanina, hidroxiurea, sulfasalazina, micofenolato mofetilo, azatioprina, tacrolimo, ésteres de ácido fumárico o biologías tal como Amevive™, Enbrel™, Humira™, Remicade™, Raptiva™ y ustekinumab (un bloqueador de IL 12 y IL-23). Además, una sal de la invención puede administrarse en combinación con otras terapias que incluyen, pero sin limitación, fototerapia o fotoquimioterapia (por ej., psoralen y fototerapia ultravioleta A (PUVA)).
En una realización, una sal de la invención se administra en forma concomitante con otro agente terapéutico para el tratamiento y/o la profilaxis de una reacción alérgica, los agentes particulares incluyen, pero sin limitación: antihistaminas (por ej., cetirizina, difenhidramina, fexofenadina, levocetirizina), glucocorticoides (por ej., prednisona, betametasona, beclometasona, dexametasona), epinefrina, teofilina o antileucotrienos (por ej., montelukast o zafirlukast), anti-colinérgicos y descongestivos.
Por administración concomitante se incluye cualquier medio de administración de dos o más agentes terapéuticos al paciente como parte del mismo régimen de tratamiento, como será evidente para los expertos en la técnica. Mientras que dos o más agentes pueden administrarse simultáneamente en una sola formulación, es decir, como una composición farmacéutica individual, esto no es esencial. Los agentes pueden administrarse en diferentes formulaciones y en diferentes momentos.
Procedimientos de síntesis química
General
El compuesto de acuerdo con la Fórmula I, y la sal de 1:1:3 Base Libre/HCl/H2O de la invención pueden prepararse a partir de materiales de partida ampliamente disponibles con el uso de los siguientes procedimientos y procedimientos generales. Se apreciará que donde se presenten las afecciones de los procesos preferidos o típicos (es decir, temperaturas de reacción, tiempos, proporciones en moles de reactivos, disolventes, presiones, etc.), también pueden usarse otras afecciones de proceso a menos que se indique de otro modo. Las afecciones de reacción óptimas pueden variar con los reactivos particulares o el disolvente usado, pero una persona experta en la técnica puede determinar dichas afecciones mediante procedimientos de optimización de rutina.
Además, como resultará evidente para aquellas personas con experiencia en la técnica, puede ser necesario el uso de grupos protectores convencionales para evitar que ciertos grupos funcionales sufran reacciones no deseadas. La elección de un grupo protector adecuado para un grupo funcional particular así como las afecciones adecuadas para la protección y desprotección son muy conocidas en la técnica (Greene, T W; Wuts, P G M;, 1991).
Los siguientes procedimientos se presentan con detalles respecto de la preparación de una sal de la invención como se define anteriormente en la presente memoria y en los ejemplos comparativos. Una sal de la invención puede prepararse a partir de materiales de partida y reactivos comercialmente disponibles o conocidos por una persona capacitada en la técnica de la síntesis orgánica.
Todos los reactivos son de grado comercial y se usan como se recibe sin purificación adicional, a menos que se indique de otro modo. Los solventes anhidros disponibles en el mercado se usan para reacciones conducidas bajo atmósfera inerte. Los solventes de grado reactivo se usan en todos los otros casos, a menos que se especifique de otro modo. La cromatografía en columna se realiza sobre gel de sílice 60 (35-70 gm). La cromatografía de fase delgada se lleva a cabo con el uso de placas F-254 de gel de sílice pre-revestidas (espesor 0,25 mm). Los espectros 1H RMN se registran en un espectrómetro Broker DPX 400 RMN (400 MHz o un espectrómetro Broker Advance 300 RMN (300 MHz). Las desviaciones químicas (5) para los espectros de 1H RMN se informan en partes por millón (ppm) con relación a tetrametilsilano (5 0.00) o el pico del disolvente residual apropiado, es decir CHCl3 (5 7,27), como
referencia interna. Las multiplicidades se expresan como singulete (s), duplete (d), triplete (t), cuarteto (q), quintuplete (quin), multiplete (m) y amplio (br). Los espectros de electroaspersión MS se obtienen sobre u espectrómetro LC/MS de la plataforma Waters o con UPLC Waters Acquity H-Class acoplado a un espetrómetro 3100 detector de Masa Waters. Columnas usadas: Waters Acquity UPLC BEH C18 1,7 gm, 2,1mm ID x 50mm L, Waters Acquity UPLC BEH C18 1,7 gm, 2,1mm ID x 30 mm L, o Waters Xterra MS 5gm C18, 100 x 4,6mm. Los procedimientos utilizan indistintamente gradientes MeCN/H2O (H2O contiene cualquiera de 0,1% TFA o 0,1% NH3) o gradientes MeOH /H2O (H2O contiene 0,05% TFA). El calentamiento en microondas se realiza con un iniciador Biotage.
Tabla I. Lista de abreviaturas usadas en la sección experimental:


Tabla II. Aparato del estudio de sal



Preparación sintética de la sal de la invención
Ejemplo 1. Preparación del Compuesto I
1.1. Vía 1
1.1.1. 4-[4-(4,4,5,5-Tetrametil-[1,3,2]dioxaborolan-2-il)-bencil]-tiomorfolin-1,1-dióxido

Se disuelven 2-(4-Bromometil-fenil)-4,4,5,5-tetrametil-[1,3,2]dioxaborolano (1 eq.) y DIPEA (2 eq.) en DCM/MeOH (5:1 v:v) bajo N2 y 1, 1 -dióxido de tiomorfolino (2 eq.) en porciones. La solución resultante se agitó a temperatura ambiente durante 16h. Después de este tiempo, la reacción está completa. El disolvente se evapora. El compuesto se extrae con EtOAc y agua, se lava con salmuera y se seca sobre MgSO4. Las capas orgánicas se filtran y se evaporan. El compuesto final se aísla sin purificación adicional.
1.1.2. (5-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)-amida del ácido ciclopropanocarboxílico

1.1.2.1. Etapa i): 1-(6-Bromo-piridin-2-il)-3-carboetoxi-tiourea
A una solución de 2-amino-6-bromopiridina (1) (253,8 g, 1,467 mol) en DCM (2,5 L) enfriada hasta 5°C se añade isotiocianato de etoxicarbonilo (173,0 ml, 1,467 mol) gota a gota durante 15 min. La mezcla de reacción se deja entibiar hasta la temperatura ambiente. (20 °C) y se agita durante 16 h. La evaporación al vacío da un sólido que puede recolectarse por filtración, lavarse minuciosamente con petróleo (3 x 600 ml) y secarse al aire para producir el producto deseado. La tiourea puede usarse como tal para la próxima etapa sin ninguna purificación. 1H (400 MHz, CDCh) 5 12,03 (1H, br s), 8,81 (1H, d), 8,15 (1H, br s), 7,60 (1H, t), 7,32 (1H, dd), 4,31 (2H, q), 1,35 (3H, t).
1.1.2.2. Etapa ii): 5-Bromo-[1,2,4]t
riazolo[1,5-a]piridin-2-ilamina
A una suspensión de clorhidrato de hidroxilamina (101,8 g, 1,465 mol) en EtOH/MeOH (1:1, 900 ml) se añade N,N-diisopropilctilamina (145,3 ml, 0,879 mol) y la mezcla se agita a temperatura ambiente (20 °C) durante 1 h. 1-(6-Bromopiridin-2-il)-3-carboetoxi-tiourea (2) (89,0 g, 0,293 mol) se añade luego y la mezcla se calienta lentamente hasta reflujo (Nota: se requiere rascador blanqueador para desactivar el H2S evolucionado). Después de 3h a reflujo, la mezcla se deja enfriar y se filtra para recolectar el sólido precipitado. El producto adicional se recolecta por evaporación al vacío del filtrado, incorporación de H2O (250 ml) y filtración. Los sólidos combinados se lavan consecutivamente con H2O (250 ml), EtOH/MeOH (1:1,250 ml) y Et2O (250 ml) luego se seca al vacío para producir el derivado tiazolopiridina (3) como un sólido. El compuesto puede usarse como tal para la próxima etapa sin ninguna purificación. 1H (400 MHz, DMSO-d6) 57,43-7,34 (2H, m, 2 x H aromático), 7,24 (1H, dd, J 6,8 y 1,8 Hz, H aromático), 6,30 (2H, br, NH2); m/z 213/215 (1:1, M+H+ , 100%).
1.1.2.3. Etapa (iii): (5-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)-amida del ácido ciclopropancarboxílico
A una solución de la 2-amino-triazolopiridina obtenida en la etapa previa (7,10 g, 33,3 mmol) en MeCN anhidro (150 ml) a 5°C se añade Et3N (11,6 ml, 83,3 mmol) seguido de cloruro de ciclopropancarboxílico (83,3 mmol). La mezcla de reacción se deja enfriar luego hasta la temperatura ambiente y se agita hasta que se consume todo el material de partida. Si se requiere, se añade Et3N (4,64 ml, 33,3 mmol) y cloruro de ciclopropancarbonilo (33,3 mmol) adicional para asegurar la reacción completa. Después de la evaporación del disolvente al vacío el residuo resultante se trata con solución de amoníaco metanólico 7 N (50 ml) y se agita a temperatura ambiente (durante 1-16 h) para hidrolizar cualquier producto bis-acilado. El aislamiento del producto se realiza por remoción de los volátiles al vacío seguido de trituración con Et2O (50 ml). Los sólidos se recolectan por filtración, se lavan con H2O (2x50ml), acetona (50 ml) y Et2O (50 ml), luego se secan al vacío para dar el compuesto deseado.
1.1.3. Compuesto I

Se añade 4-[4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-benzil]-tiomorfolin-1,1-dióxido (1,1 eq.) a una solución de (5-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)-amida del ácido ciclopropanocarboxílico en 1,4-dioxano/agua (4:1). K2CO3 (2 eq.) y PdCbdppf (0,03 eq.) se añaden a la solución. La mezcla resultante se calienta luego en un baño de aceite a 90°C durante 16h bajo N2. Se añade agua y la solución se extrae con acetato de etilo. Las fases orgánicas se secan sobre MgSO4 anhidro y se evaporan al vacío.
El compuesto final se obtiene después de purificación por cromatografía flash.
Alternativamente, una vez completa la reacción, un depurador de paladio, tal como 1,2-bis(difenilfosfin)etano, se añade, la mezcla de reacción se deja enfriar y se realiza una filtración. La torta del filtro se reemulsiona en un disolvente adecuado (por ej., acetona), el sólido se separa por filtración, se lava con más acetona, y se seca. El sólido resultante se resuspende en agua, se añade HCl acuoso y después de agitar a temperatura ambiente la solución resultante se filtra sobre celite (Celpure P300). Luego se añade NaOH acuoso al filtrado y la suspensión resultante se agita a temperatura ambiente, el sólido se separa por filtración, se lava con agua y se seca por succión. Finalmente, la torta se solubiliza nuevamente en una mezcla de THF/H2O, se trata con un depurador de paladio (por ej., SMOPEX 234) a 50°C, la suspensión se filtra, los solventes orgánicos se retiran por evaporación y la emulsión resultante se lava con agua y metanol, se seca y se tamiza, para obtener el compuesto deseado como la base libre.
1.2. Vía 2
1.2.1. Etapa 1: [5-(4-hidroximetil-fenil)-[1,2,4-triazolo[1,5-a]-piridin-2-il]-amida del ácido ciclopropancarboxílico

Se añade ácido 4-(hidroximetil)fenilborónico (1,1 eq.) a una solución de (5-bromo-[1,2,4]triazolo[1,5-a]piridin-2-il)-amida del ácido ciclopropanocarboxílico en 1,4-dioxano/agua (4:1). K2CO3 (2 eq.) y PdChdppf (0,03 eq.) se añaden a la solución. La mezcla resultante se calienta luego en un baño de aceite a 90°C durante 16h bajo N2. Se añade agua y la solución se extrae con acetato de etilo. Las fases orgánicas se secan sobre MgSO4 anhidro y se evaporan al vacío. La mezcla resultante se usa sin purificación adicional.
1.2.2. Etapa 2: [5-(4-bromometil-fenil)-[1,2,4]triazolo[1,5-a]piridin-2-il]-amida del ácido cidopropancarboxílico
A una solución de [5-(4-hidroximeti-fenil)-[1,2,4]-triazolo-[1,5-a]piridin-2-il]amida del ácido ciclopropancarboxílico (1,0 eq.) en cloroformo se añade lentamente tribromuro de fósforo (1,0 eq.). La mezcla de reacción se agita a temperatura ambiente durante 20 horas, se desactiva con hielo y agua (20 ml) y se extrae con diclorometano. La fase orgánica se seca sobre MgSO4 anhidro, se filtra y se concentra hasta la sequedad. El residuo blanco resultante se tritura con diclorometano/éter dietílico 2:1 para producir el producto deseado.
1.2.3. Etapa 3:

Se disuelven [5-(4-bromometil-fenil)-[1,2,4]-triazolo-[1,5-a]piridin-2-il]amida del ácido ciclopropancarboxílico (1 eq.) y DIPEA (2 eq.) en DCM/MeOH (5:1 v:v) bajo N2 y se añade gota a gota 1,1- dióxido de tiomorfolina (1,1 eq.). La solución resultante se agitó a temperatura ambiente durante 16h. Después de este tiempo, la reacción está completa. El disolvente se evapora. El compuesto se disuelve en DCM, se lava con agua y se seca sobre MgSO4 anhidro. Las fases orgánicas se filtran y se evaporan. El compuesto final se aísla por cromatografía en columna usando EtOAc para producir el producto deseado.
Ejemplo 2. Preparación de las sales del Compuesto I.
2.1. Protocolo 1
El disolvente se añade a las muestras del Compuesto I (aproximadamente 5 mg) en porciones relativas en volumen de 10, 15 y 20 volúmenes, y después de cada incorporación las muestras se agitan a 50°C en un agitador durante 20 min para alentar la disolución, antes de enfriar hasta la temperatura ambiente para la siguiente incorporación de disolvente. El disolvente se selecciona de isopropanol, etanol, metanol, acetato de isopropilo, THF, TBME, acetona, metil etil cetona, DCM y MeCN.
En estas condiciones, casi la disolución completa del Compuesto I solo ocurre en DCM.
Después de la incorporación de 20 volúmenes relativos de disolvente, se añade HCl (solución 1 M en THF) (12 gl ~ 1 eq.) a cada muestra y se realizan observaciones. Las muestras se almacenan en un agitador sobre un ciclo de calor -frío (50°C / temperatura ambiente, 4 h a cada temperatura) durante 16 h. Los sólidos resultantes se aíslan por filtración al vacío, se secan bajo succión y se analizan por XRPD.
La formación de sal de HCl se observa solamente en etanol, metanol, THF, acetona, metil etil cetona y MeCN.
2.2. Protocolo 2 (incluyendo ejemplos comparativos)
El Compuesto I (~20mg) se trata con 400 gl (20 volúmenes) de disolvente (THF, acetona) y se trata con soluciones de ácido acuosas a temperatura ambiente (véase Tabla III a continuación). Los experimentos se colocan en una cámara de maduración que se cicla entre la temperatura ambiente y 50°C con cuatro horas transcurridas bajo cada condición. Después de tres días los sólidos se aíslan por filtración y cualquier solución se deja evaporar. Cualquier sólido formado se aísla, o los sólidos remanentes se tratan con EtOAc y se colocan nuevamente en la cámara de maduración durante cuatro días. Cualquier sólido adicional se aísla por filtración.
Los sólidos se analizan por XRPD.
Cuando se somete a este protocolo, las sales solo se forman en ácido sulfúrico, pTSA, ácido 1,2-etan disulfónico, ácido 2-hidroxietansulfónico, ácido naftalen2-sulfónico, y ácido maleico.
Tabla III Ácidos usados en el Protocolo 2

2.3. Protocolo 3 (incluyendo los ejemplos comparativos)
A una suspensión del Compuesto I (~25 mg) y disolvente (MeOH, THF, Acetona o DCM) (20 volúmenes relativos) a aproximadamente 40°C se añade 1,05 o 2,1 eq. del ácido (HCl, HBr, H2SO4 , PH-SO3H, ácido oxálico, ácido maleico, o pTSA.H2O), y se realizan observaciones visuales. Las muestras se almacenan en un agitador a 26°C durante 14 h. Se toma una alícuota y el sólido se aísla por filtración al vacío, se seca bajo succión y se analiza por XRPD. Luego las mezclas se almacenan en un agitador a 26°C durante un adicional de 31 h. El sólido remanente de aquellas muestras que exhiben patrones de XRPD cristalinos diferentes a la base libre se aíslan por filtración al vacío y se realiza análisis adicional (DSC, RMN, Estabilidad).
El resto de cada muestra se almacena en un agitador sobre un ciclo de calor / frío (50°C / temperatura ambiente, 4 h a cada temperatura) durante 136 h. Nuevamente, una alícuota de cada muestra se toma y el sólido se aísla por filtración al vacío, se secan bajo succión y se analizan por XRPD. El sólido remanente de aquellas muestras que exhiben patrones de XRPD cristalinos diferentes a la base libre se aíslan por filtración al vacío y se emprende análisis adicional (DSC, RMN, Estabilidad).
Las muestras que permanecen en aislamiento se dejan evaporar lentamente en condiciones ambiente hasta que el disolvente se ha evaporado, y cualquier sólido formado se aísla y se analiza por XRPD.
Cuando se somete a este protocolo, las sales solo se forman en HBr, HCl, ácido sulfúrico, pTSA y ácido maleico.
2.4 Protocolo 4 (incluyendo los ejemplos comparativos)
2.4.1. HCl
HCl (solución 1 M en THF) (655 pL, 0,65 mmol, 1,1 eq.) se añade a una suspensión agitada de Compuesto I (252,6 mg, 0,59 mmol, 1 eq.) y metanol (5,05 ml, 20 vol.) a 50°C. La mezcla se enfría hasta 25°C a 1 °C/min y se agita a 25°C durante 22 h.
El sólido se aísla por filtración al vacío y se seca bajo succión.
El análisis de XRPD confirmó la formación de una sal no higroscópica estable, que contiene 4-5% de agua, que tiene una solubilidad acuosa medida a 1,9 mg/ml.
2.4.2. Ácido maleico (comparativo)
El Compuesto I (246,4 mg, 0,58 mmol, 1 eq.) se suspende en 5 % agua en acetona (4,95 ml, 20 vol.) a 25°C y la muestra se entibia hasta 50°C. Se añade ácido maleico (solución 1 M en THF) (1,2 ml, 1,22 mmol, 2,1 eq.) a la suspensión agitada a 50°C. La mezcla se enfría hasta 25°C a 1°C/min y se agita a 25°C durante 22 h.
El sólido se aisló por filtración al vacío y se seca bajo succión.
El análisis de XRPD confirmó la formación de una sal no higroscópica estable, que tiene una estabilidad acuosa medida a 0,4 mg/ml.
2.4.3. pTSA (comparativo)
pTSA (solución 1 M en EtOH) (1,3 ml, 1,3 mmol, 2,2 eq.) se añade a una suspensión agitada de Compuesto I (250,7 mg, 0,59 mmol, 1 eq.) y THF (5 ml, 20 vol.) a 50°C. La mezcla se enfría hasta 25°C a 1 °C/min y se agita a 25°C durante 22 h.
El sólido se aísla por filtración al vacío y se seca bajo succión.
El análisis de XRPD confirmó la formación de una sal, que resultó ser inestable.
2.5. Análisis de sal complementaria
Los sólidos recuperados de este protocolo también se someten a evaluación de pureza, equivalente de sal (IC), RMN (disolvente residual), DSC, TGA (solvatos) y estabilidad (1 semana a 40°C / Humedad relativa 75%).
2.5.1. Determinación de pKa
Los datos se recolectan en un instrumento Siriuis GIpKa con un accesorio D-PAS. Se realizan las mediciones a 25 °C en solución acuosa por UV y en mezclas de agua y metanol por potenciometría. El medio de titulación fue ajustado en su potencia iónica con KCl 0,15 M (ac.). Los valores encontrados en las mezclas de metanol y agua se corrigen a 0% de co-disolvente por medio de extrapolación de Yasuda-Shedlovsky.
Los datos se refinan usando el software Refinement Pro.
Después de este procedimiento, se obtienen los siguientes valores: 1,58 ± 0,01,3,68 ± 0,02, y 12,02 ±0,01.
2.5.2. Determinación del pH
Las mediciones de pH se realizan sobre el material experimental como emulsión. Normalmente, 1 ml de agua desmineralizada se añade hasta aproximadamente 300 mg del material analizado y la suspensión luego se somete a vórtice. Si se encuentra disponible sólido insuficiente o la viscosidad es demasiado alta para la medición del pH con el uso de un electrodo de pH, la cantidad de agua aumenta hasta que es posible realizar una medición en incrementos de 1 ml. El electrodo de pH se calibró con el uso de una calibración de tres puntos.
Finalmente, el pYH se registra al inicio (= después de 2 min para permitir una medición relativamente estable del pH).
2.5.3. Estudio de solubilidad.
2.5.3.1. Protocolo
La solubilidad de un compuesto se determina en diferentes solventes por equilibrio de un exceso de producto durante al menos 24 h a 20°C en un agitador giratorio. La solución súper saturada luego se filtra y la concentración del producto en el filtrado se determina con el uso de espectrometría UV.
En agua, la influencia del pH se ajusta hasta un pH 2, pH 7 y pH 9. Se usan HCl y NaOH para ajustar el pH.
2.5.3.2. Resultados
Por ejemplo, cuando se somete a este protocolo, la solubilidad de la base libre y la correspondiente medición de sal de HCl se informan en la tabla IV a continuación. Inesperadamente, ir del Compuesto I al Compuesto I. HCl.3H2O no resulta sistemáticamente en una solubilidad mejorada.
Tabla IV Solubilidad del Compuesto I como base libre y la correspondiente sal de HCl en varios solventes

2.5.4. Conclusiones
Como se demostró con anterioridad, la solubilidad del Compuesto I varía en gran medida dependiendo del disolvente usado, y la formación de sal no ocurre inevitablemente con cada ácido, ilustrando la dificultad en seleccionar una combinación satisfactoria de disolvente y ácido, lo que es específico para el Compuesto I.
Ejemplo 3. Estudio de polimorfismo
3.1. Compuesto I
3.1.1. Formación del Compuesto I amorfo
El Compuesto I (38 mg) se calienta en un instrumento de DSC bajo nitrógeno de acuerdo con el siguiente procedimiento: 1) Se calentó desde 30°C hasta 250°C a 10°C/min; 2) Se mantuvo isotérmico durante 1 min; 3) Se enfrió de 250°C a 30°C a 100°C/min; y 4) Se mantuvo isotérmico durante 4 min.
El sólido resultante se confirma como amorfo por análisis de XRPD.
3.1.2. Estudio de polimorfismo
El compuesto amorfo se sometió a cuarenta solventes diferentes (formiato de etilo, TBME, acetona, acetato de metilo, metanol, tetrahidrofurano, éter diisopropílico, acetato de etilo, etanol, metil etil cetona, acetonitrilo, t-BuOH, 2-Propanol, 1-2-Dimetoxietano, Acetato de isopropilo, 1-Propanol, 2-Butanol, nitrometano, 1-4-Dioxano, acetato de propilo, 2-Pentanona, 2-Metil-1-Propanol, Tolueno, Acetato de isobutilo, Metilisobutil cetona, 1-Butanol, 2-Metoxietanol, Acetato de butilo, metil butil cetona, 3-Metil-1-Butanol, 2-Etoxietanol, 1-Pentanol, Anisol, Benzonitrilo, nitrobenceno, IPA 5% agua, EtOH 5% agua, MeCN 5% agua, THF 5% agua, y acetona 5% agua) y se cicla térmicamente entre la temperatura ambiente y 50°C con cuatro horas transcurridas entre cada condición. Después de cinco días, los sólidos se recolectan por filtración y se analizan inicialmente por XRPD.
3.1.3. Resultados de polimorfismo del Compuesto I
Cuando se somete a este protocolo, los sólidos obtenidos se analizaron por XRPD, y los siguientes patrones más estables se informan en la Tabla V, Tabla VI, y Tabla VII a continuación.
Tabla V. Compuesto I Patrón 1 Patrón XRPD (Figura 1)

Tabla VI Compuesto I Patrón 3 Patrón XRPD (Figura 2)

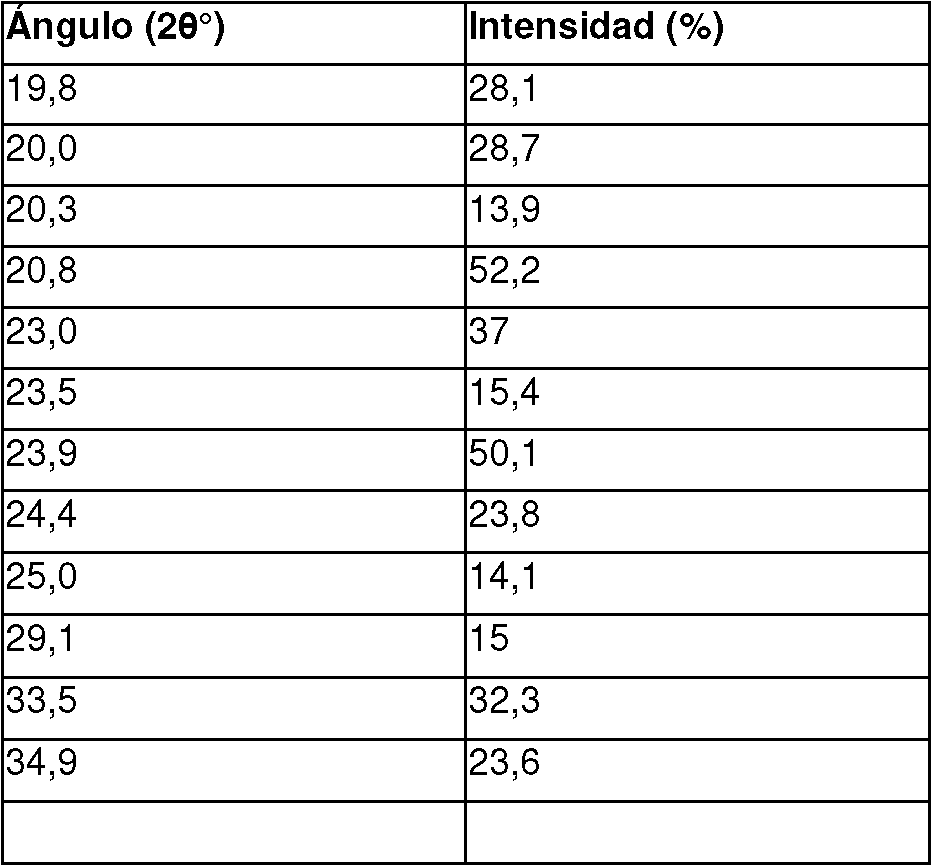
Tabla VII Compuesto I Patrón 4 Patrón XRPD (Figura 3)


3.2 Polimorfismo de las sales de HCI del Compuesto I y sus solvatos
3.2.1 Formación de sales de HCl amorfas del Compuesto I
El Compuesto I (25,47 mg) se mezcla con agua (70 ml, -2750 volúmenes relativos) y se agita a temperatura ambiente durante 30 min. La mezcla resultante se filtra a través de un filtro de jeringa con membrana de PVDF de 0,45 mm y se congela en un baño a -78 °C. El disolvente se remueve usando un liofilizador para producir la sal de mono-HCl amorfa del Compuesto I (confirmada por XRPD, transición del vidrio a 149°C (análisis de DSC modulado))
3.2.2. Protocolos de estudio
Los volúmenes de 25 solventes diferentes (Acetona, anisol, butanol, acetato de butilo, TBME, DMSO, etanol, acetato de etilo, heptano, acetato de isopropilo, MEK, acetato de isopropilo, MeCN, ciclohexano, DCM, dioxano, metanol, nitrometano, THF, THF de metilo, tolueno, agua, acetona acuosa al 10%, THF acuoso al 10% y metanol al 10%), se añaden a la sal de mono-HCl amorfa del Compuesto I, dichos volúmenes oscilan de 125 pL (5 vol rel) a 1 ml (40 vol. Rel.), o hasta que se observa una suspensión móvil o disolución casi completa. La muestra se almacena en un agitador en un ciclo de calor / frío (50°C / temperatura ambiente, 4h) durante 23h.
Después de 23 h, donde se ha formado el sólido, se toma una alícuota y el sólido se aísla por filtración al vacío, mientras que la suspensión remanente se mantiene en un agitador en un ciclo de calor / frío (50°C / temperatura ambiente, 4 h). El sólido de la alícuota se seca bajo succión durante 30 min y se analiza por XRPD, luego se seca de manera adicional en un horno de vacío a 30 °C y 0,0005 Mpa (5 mbar) durante 34 h, y se analiza nuevamente por XRPD. Finalmente, la muestra secada se almacena a 40°C / 75 % HR durante 72 h y se analiza nuevamente por XRPD.
Después de 47 h, donde se ha formado el sólido en la suspensión, se toma una alícuota y el sólido se aísla por filtración al vacío, mientras que la suspensión remanente se mantiene en un agitador en un ciclo de calor / frío (50°C / temperatura ambiente, 4 h). El sólido de la alícuota se almacena bajo condiciones ambiente durante 72 h y 14 d con análisis por XRPD en cada momento de medición. El sólido, que se ha almacenado bajo condiciones ambiente durante 14 d, se almacena a 40 °C / 75 % HR durante 43 h y se analiza nuevamente por XRPD.
Después de 143h, donde se ha formado el sólido, se toma una alícuota de la suspensión remanente analizada en un agitador en un ciclo de calor / frío (50°C / temperatura ambiente, 4 h) y el sólido se aísla por filtración al vacío, se seca bajo succión durante 2 h, luego se analiza por XRPD. El sólido aislado se seca en un horno de vacío a 30°C y 0,0005 Mpa (5 mbar) durante 20 h, y se analiza nuevamente por XRPD. La muestra secada se almacena a 40°C / 75 % HR durante 114 h y se analiza nuevamente por XRPD.
3.2.3. Resultados
Además de este estudio, se identifican dos formas estables.
3.2.3.1. Compuesto I. HCl (comparativo)
HCl (solución 3,6 M en dioxano) (2,5 ml, 9,05 mmol 1,1 eq.) se añade en porciones a una suspensión agitada de Compuesto I amorfo (3,499 g, 8,22 mmol, 1 eq.) y metanol (70 ml, 20 volúmenes relativos) a 50 °C durante un período de 2 min. La mezcla se enfría hasta 20°C a 0,1°/min y se agita a 20°C durante un adicional de 10 h. La mezcla se enfría hasta 15°C a 0,5°C/min y se agita durante 30 min. El sólido resultante se aísla por filtración al vacío, se lava con
metanol (2 x 3,5 ml, 1x7 ml) y se seca bajo succión para producir el Compuesto l. HCl.
3.2.3.2. Compuesto I. HCI.3 H2O
HCl (solución 1 M en THF) (250 pL, 0,25 mmol, 1,05 eq.) se añade a una suspensión de Compuesto I amorfo (100,37 mg, 0,24 mmol, 1 eq.) y DCM (2 ml, 20 volúmenes relativos) a 40°C. La mezcla se almacena en un agitador en un ciclo de calor / frío (40°C / temperatura ambiente, 4 h) durante 70 h. El sólido se aísla por filtración al vacío y se seca bajo succión para producir el Compuesto l. HCL3H2O.
3.2.3.3. Análisis
Los patrones de difracción por rayos X se divulgan en la Tabla VIII y en la Tabla IX a continuación.
Tabla VIII Compuesto I. Patrón XRPD de HCl (Figura 4)

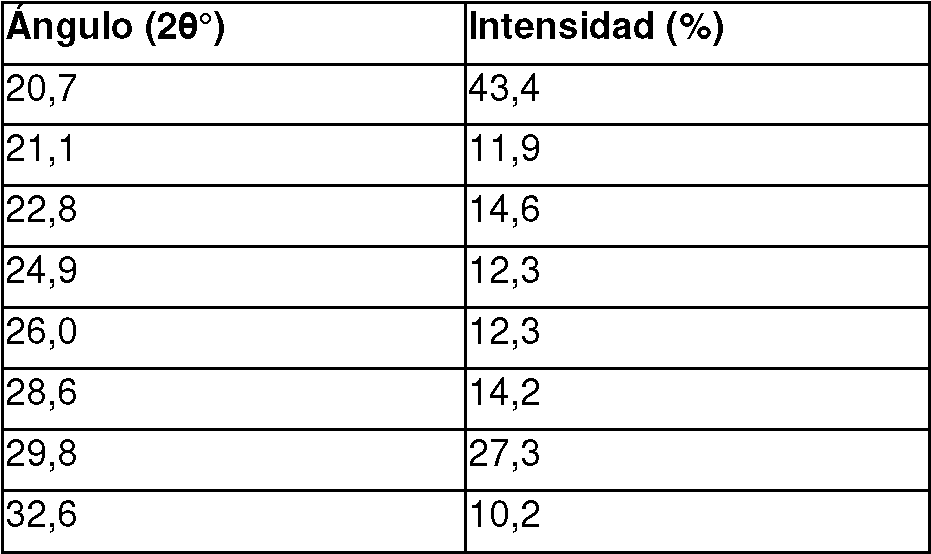
Tabla IX Compuesto I. Patrón HRPD de HCL3H2O (Figura 5)


3.2.3.4. Difracción por rayos X del cristal simple del Compuesto I. HCI.3 H2O (Figura 11)
El Compuesto I. HCL3H2O se recristaliza a partir de acetona:agua (1:1). Los resultados se muestran en la Tabla X a continuación.
Tabla X. Estructura individual de cristal del Compuesto I. HCI.3H2O

El procedimiento de refinamiento se basa en los menos cuadrados de la matriz completa sobre F2. R [F2 > 2o(F2)] = 0,0377 y wR(F2) = 0,0837. Bondad de ajuste (S) = 1,109. El procedimiento de refinamiento usó 5649 reflejos, 339 parámetros y 0 limitaciones. Todas las posiciones de hidrógeno se identificaron usando el mapa de diferencia y aquellas unidas a átomos C y átomos N se colocaron luego en posiciones calculadas y se refinaron usando un modelo de conducción. Aquellos hidrógenos unidos al oxígeno y nitrógeno del agua se refinaron libremente. El Apmax final = 0,314 y Á y Apmin = -0,368 e Á.
La estructura de cristal del Compuesto I. HCl.3H2Ü (Figura 11) muestra la inclusión inesperada de las moléculas de agua en el retículo de cristal, lo que puede proporcionar estabilización adicional del sistema.
Ejemplo 4. Formación a gran escala del Compuesto I . HCI.3 H2O
4.1. Protocolo 1
Al Compuesto I (44 kg, 1,0 eq.) bajo atmósfera inerte, se añade agua (15 vol. rel., 1000 L), y la mezcla se agita a 50°C. Se añaden 3,5 eq. de HCl ac. (5 vol. rel.) durante 10-15 min, a una temperatura máxima de 55°C. Tras la compleción de la adición, se continúa con la agitación a 50°C durante 15 min, y la reacción luego se enfría hasta 15°C y se agita a esa temperatura durante al menos 12h aunque no más de 24h.
El sólido resultante se separa por filtración, y la torta se lava con agua (2,0 vol. rel.) y la torta se seca bajo nitrógeno durante al menos 4h para producir el producto deseado.
4.2. Protocolo 2
Al Compuesto I (45g, 106 mmol, 1 eq.) bajo atmósfera inerte se añade DCM (675 ml) y metanol (225 ml). La suspensión resultante se calienta hasta 35°C bajo agitación, y se añade sal de trisodio de trimercaptotriazina 15% en agua (22,5 g, 14 mmol, 0,13 eq.), y la solución resultante se agita durante 5h, después de lo cual la solución se filtra en un papel de 0,45 pm bajo presión de nitrógeno.
Al filtrado se añade agua (50 ml), y la mezcla bifásica resultante se agita a 35°C durante 15 min, después de cuyo período las fases se separan, y la fase orgánica se deja enfriar hasta 20°C, y se lava dos veces con 50 ml de agua.
La fase orgánica se enfría hasta 15-20°C, luego se añade HCl 10% en metanol (42,4 g, 116mmol, 1,10 eq.) durante 30 min, causando la precipitación de un sólido. La suspensión se agita de manera adicional a 20°C durante 3h, luego el precipitado se aísla por filtración, la torta se lava con metanol (2x50 ml) para producir el compuesto deseado, que se seca al vacío a 45°C durante 3 h. La torta luego se resuspende en agua (220 ml) y se agita durante 6 h a 50°C, y luego se enfría hasta 15-20°C. El sólido resultante se separa por filtración y la torta se lava con agua (2 x 30 ml), y se seca a 45°C durante 3h para producir el producto deseado.
4.3. Protocolo 3
4.3.1. Etapa 1: Compuesto I. HCl.MeOH
Al Compuesto I (100g, 235 mmol, 1 eq.) suspendido en DCM (1,5 L), se añade MeOH (0,5 L), y la solución resultante se calienta hasta 35°C. Se añade trimercaptotriazina de trisodio 85% (8,7 g, 3 mmol, 0,13 eq.) en agua (42 ml) y la mezcla resultante se agita a 35°C durante al menos 5h. La solución luego se filtra sobre un filtro de papel de 0,45 gm bajo presión de nitrógeno.
A la solución resultante se añade agua (150 g), se agita a 35°C durante 15 a 30 min, y se separa la mezcla bifásica. La fase orgánica se lava nuevamente con agua (2 x 150 g).
Finalmente, se añade una solución de HCl en MeOH (10% p/p) (141 g), y la suspensión se agita a 20°C durante 3h, y el sólido resultante se separa por filtración, la torta se lava con MeOH (2 x 118g), se seca al vacío durante 3h a 45°C, para producir el Compuesto l. HCl.MeOH que se analiza por XRPD. (Tabla XI).
4.3.2. Etapa 2: Compuesto I. HCl.3H2O
A ácido fórmico (200g, 1,6 eq.) en agua (36g, 0,4 eq.) se añade Compuesto l. HCl.MeOH (100 g, 1 eq.) obtenido en la etapa 1 anterior. La mezcla resultante se calienta hasta 55°C bajo agitación y la solución se filtra a través de un cartucho de filtro de 0,45 gm. Se añade ácido fórmico 85% ac. (200 g), y la mezcla se enfría hasta 28-32°C bajo suave agitación.
Luego se añade agua (100g) seguido de Compuesto I. HCl.3H2O (1g) causando la precipitación del Compuesto I.HCI.1,5HCO2H, que se analiza por XRPD (Tabla XII).
Bajo agitación a 28-32°C, se añade agua (2L) en porciones en 8 porciones de 100ml, 1 porción de 200 ml, y 2 porciones de 500 ml.
La suspensión acuosa luego se filtra, la torta se lava con agua (2x100 ml) y se seca a 30-35°C para producir el Compuesto l. HCl.3H2O, que se analiza por XRPD y DSC. (Figura 5 y Figura 7)
Tabla XI Compuesto I. Patrón XRPD de HCl:MeOH (Figura 8)

Tabla XII Compuesto I. Patrón XRPD de HCl.HCOOH (Figura 9)

Ejemplo 5. Compuesto I. Estudio de estabilidad de HCL3 H2O.
5.1. Estudio de estabilidad acelerada
5.1.1. Protocolo
Muestras del Compuesto I. HCL3H2O se almacenan en afecciones para evaluar la estabilidad química y la estabilidad física como se describe en la Tabla XIII.
Tabla XIII Condiciones de estabilidad

Las muestras se toman a T0, luego cada mes hasta 3 meses, y se analizan por FT-IR, DSC, y XRPD.
5.2. Estudio de Estabilidad extendida
5.2.1. Protocolo
Las muestras de los compuestos de prueba se almacenan en afecciones para evaluar la estabilidad química y la estabilidad física como se describe en la Tabla XIV que sigue:
Tabla XIV Condiciones de estabilidad

Se toman muestras en el T0, luego cada mes hasta 12 meses, luego a los 18, 24 y 36 meses; y cada una de las alícuotas luego se analiza por HPLC, titulación por Karl-Fisher, FT-IR, DSC, y XRPD.
5.3. Análisis de GVS del Compuesto l. HCI.3 H2O
El análisis de GVS se llevó a cabo para evaluar la estabilidad del Compuesto I. HCL3H2O y se presenta en la Figura 6. La evolución del material se sigue por medio de XPRD en varios momentos de medición.
No se observó cambio en la forma a HR elevada. El Compuesto I. HCL3H2O se deshidrata al Compuesto I.HCl anhidro tras calentar por encima de 60°C o a menos de 10 % HR. La rehidratación del Compuesto I. HCL3H2O ocurre tras enfriar hasta la temperatura ambiente o por exposición al 20 % o más de humedad relativa.
5.3.1 Conclusiones
Inesperadamente, el Compuesto I. HCL3H2O puede deshidratarse en el Compuesto I.HCl, pero se convierte nuevamente en la forma de trihidrato más estable del Compuesto l. HCL3H2O en afecciones normales. Esto no pudo haber sido previsto por los expertos en la técnica.
Ejemplos biológicos
Ejemplo 6. Ensayos in vitro
6.1. Ensayo de inhibición de JAK 1
El dominio catalítico de JAK1 humana recombinante (aminoácidos 850-1154; número de catálogo 08-144) se adquiere de Carna Biosciences. 10 gg de JAK1 se incuba con 12,5 gg de sustrato polyGT (número de catálogo de Sigma P0275) en tampón de reacción de quinasa (15 mM Tris-HCl pH 7,5, 1 mM Dt T, 0,01% Tween-20, 10 mM MgCl2, 2 gM ATP no radiactivo, 0,25 gCi 33P-gamma-ATP (GE Healthcare, número de catálogo AH9968) concentraciones finales) con o sin 5gL que contiene compuesto de prueba o vehículo (DMSO, 1 % concentración final), en un volumen total de 25 gL, en una placa de polipropileno de 96 pocillos (Greiner, base V). Después de 45 min a 30°C, las reacciones se detienen por el agregado de 25 gl/pocillo de ácido fosfórico 150 mM. La totalidad de la reacción de quinasa terminada se transfiere a placas de filtro de 96 pocillos previamente lavadas (ácido fosfórico 75 mM) (número de catálogo de Perkin Elmer 6005177) usando un cosechador de células (Perkin Elmer). Las placas se lavan 6 veces con 300 gl por pocillo de una solución 75 mM de ácido fosfórico y la base de las placas se sella. Se añaden 40 gl/pocillo de Microscint-20,
la parte superior de las placas se sella y se realiza la lectura usando el Topcount (Perkin Elmer). La actividad de quinasa se calcula restando recuentos por minuto (cpm) obtenidos en presencia de un inhibidor de control positivo (10 pM esturosporina) de cpm obtenido en presencia de vehículo. La capacidad que tiene un compuesto de prueba de inhibir su actividad se determina como:
Porcentaje de inhibición = ((cpm determinado para la muestra con el compuesto de prueba presente - cpm determinado para la muestra con inhibidor de control positivo) dividido por (cpm determinado en presencia de vehículo - cpm determinado para la muestra con inhibidor de control positivo)) * 100.
Las series de dilución de la dosis se preparan para los compuestos, permitiendo el análisis de los efectos de respuesta a la dosis en el ensayo de JAK1 y el cálculo de la IC50 para cada compuesto. Cada compuesto se analiza como rutina a la concentración de 20pM seguido de una dilución serial 1/3, 8 puntos (20pM - 6,67pM - 2,22pM - 740nM - 247nM -82nM - 27nM - 9nM) en una concentración final de 1% DMSO. Cuando la potencia de la serie de compuestos aumenta, se preparan más diluciones y/o la concentración superior se reduce (por ej., 5 pM, 1 pM).
Los siguientes compuestos se han analizado para determinar su actividad contra JAK1 y los valores de IC50, según lo determinado usando los ensayos descritos en la presente memoria, se dan a continuación en la Tabla XV.
Tabla XV Valores de los compuestos JAK1 IC50

6.2. Ensayo de determinación de Ki de JAK1
Para la determinación de Ki, se mezclan diferentes cantidades de compuesto con la enzima y la reacción enzimática se sigue como función de la concentración de ATP. La Ki se determina por medio de gráfico doble recíproco de Km contra concentración del compuesto (gráfico de Lineweaver-Burk). 1 pg de JAK1 (Invitrogen, PV4774) se usa en este ensayo. El sustrato es 50nM Ulight-jAK-1 (Tyrl023) Péptido (Perkin Elmer, TRF0121) La reacción se realiza en 25mM MOPS pH 6,8, 0,01%, 2 mM DTT, 5 mM MgCl2 Brij-35 con concentraciones variables de ATP y compuesto. El sustrato fosforilado se mide usando un anticuerpo anti-fosfotirosina marcado Eu PT66 (Perkin Elmer, AD0068). La lectura se realiza en el envision (Perkin Elmer) con excitación a 320 nm y emisión seguido a 615 nm y 665 nm. Por ejemplo, cuando el Compuesto I se analiza en este ensayo, se mide un valor de Ki de 39 nM.
6.3. Ensayo de inhibición de JAK2
El dominio catalítico de JAK2 humana recombinante (aminoácidos 808-1132; número de catálogo PV4210) se adquiere de Invitrogen. 0.025mU de JAK2 se incuba con 2,5 pg de sustrato polyGT (número de catálogo de Sigma P0275) en tampón de reacción de quinasa (5 mM MOPS pH 7,5, 9 mM MgAc, 0.3mM EDTA, 0,06% Brij y 0,6 mM DTT, 1 pM ATP no radiactivo, 0,25 pCi 33P-gamma-ATP (GE Healthcare, número de catálogo AH9968) concentraciones finales) con o sin 5pL que contiene compuesto de prueba o vehículo (DMSO, 1 % concentración final), en un volumen total de 25 pL, en una placa de polipropileno de 96 pocillos (Greiner, base V). Después de 90 min a 30°C, las reacciones se detienen por el agregado de 25 pl/pocillo de ácido fosfórico 150 mM. La totalidad de la reacción de quinasa terminada se transfiere a placas de filtro de 96 pocillos previamente lavadas (ácido fosfórico 75 mM) (número de catálogo de Perkin Elmer 6005177) usando un cosechador de células (Perkin Elmer). Las placas se lavan 6 veces con 300 pl por pocillo de una solución 75 mM de ácido fosfórico y la base de las placas se sella. Se añaden 40 pl/pocillo de Microscint-20, la parte superior de las placas se sella y se realiza la lectura usando el Topcount (Perkin Elmer). La actividad de quinasa se calcula restando recuentos por minuto (cpm) obtenidos en presencia de un inhibidor de control positivo (10 pM esturosporina) de cpm obtenido en presencia de vehículo. La capacidad que tiene un compuesto de prueba de inhibir su actividad se determina como:
Porcentaje de inhibición = ((cpm determinado para la muestra con el compuesto de prueba presente - cpm determinado para la muestra con inhibidor de control positivo) dividido por (cpm determinado en presencia de vehículo - cpm determinado para la muestra con inhibidor de control positivo)) * 100.
Las series de dilución de la dosis se preparan para los compuestos, permitiendo el análisis de los efectos de respuesta a la dosis en el ensayo de JAK2 y el cálculo de la IC50 para cada compuesto. Cada compuesto se analiza como rutina a la concentración de 20pM seguido de una dilución serial 1/3, 8 puntos (20 pM - 6.67 pM - 2.22 pM - 740 nM - 247 nM - 82 nM - 27 nM - 9 nM) en una concentración final de 1% DMSO. Cuando la potencia de la serie de compuestos aumenta, se preparan más diluciones y/o la concentración superior se reduce (por ej., 5 pM, 1 pM).
Los siguientes compuestos se han analizado para determinar su actividad contra JAK2 y los valores de IC50, según lo determinado usando los ensayos descritos en la presente memoria, se dan a continuación en la Tabla XVI.
Tabla XVI Valores de los compuestos JAK2 IC50

6.4. Ensayo de determinación de Kd de JAK2
JAK2 (Invitrogen, PV4210) se usa a una concentración final de 5 nM. El experimento de unión se realiza en 50mM Hepes pH 7,5, 0,01% Brij-35, 10mM MgCl2, 1 mM EGTA usando 25nM rastreador de quinasa 236 (Invitrogen, PV5592) y 2 nM Eu-anti-GST (Invitrogen, PV5594) con concentraciones variadas de compuesto. La detección del rastreador se realiza de acuerdo con el procedimiento de los fabricantes.
Por ejemplo, cuando el Compuesto I se analiza en este ensayo, se mide un valor de Kd de 205 nM.
6.5. Ensayo de inhibición de JAK3
El dominio catalítico de JAK3 humana recombinante (aminoácidos 781-1124; número de catálogo PV3855) se adquiere de Invitrogen. 0.025mU de JAK3 se incuba con 2,5 gg de sustrato polyGT (número de catálogo de Sigma P0275) en tampón de reacción de quinasa (25 mM Tris pH 7,5, 0,5 mM EGTA, 0,5 mM Na3VO4, 5 mM b-glicerolfosfato, 0,01% Triton X-100, 1 pM ATP no radiactivo, 0,25 gCi 33P-gamma-ATP (GE Healthcare, número de catálogo AH9968) concentraciones finales) con o sin 5pL que contiene compuesto de prueba o vehículo (DMSO, 1 % concentración final), en un volumen total de 25 pL, en una placa de polipropileno de 96 pocillos (Greiner, base V). Después de 105 min a 30°C, las reacciones se detienen por el agregado de 25 pl/pocillo de ácido fosfórico 150 mM. La totalidad de la reacción de quinasa terminada se transfiere a placas de filtro de 96 pocillos previamente lavadas (ácido fosfórico 75 mM) (número de catálogo de Perkin Elmer 6005177) usando un cosechador de células (Perkin Elmer). Las placas se lavan 6 veces con 300 pl por pocillo de una solución 75 mM de ácido fosfórico y la base de las placas se sella. Se añaden 40 pl/pocillo de Microscint-20, la parte superior de las placas se sella y se realiza la lectura usando el Topcount (Perkin Elmer). La actividad de quinasa se calcula restando recuentos por minuto (cpm) obtenidos en presencia de un inhibidor de control positivo (10 pM esturosporina) de cpm obtenido en presencia de vehículo. La capacidad que tiene un compuesto de prueba de inhibir su actividad se determina como:
Porcentaje de inhibición = ((cpm determinado para la muestra con el compuesto de prueba presente - cpm determinado para la muestra con inhibidor de control positivo) dividido por (cpm determinado en presencia de vehículo - cpm determinado para la muestra con inhibidor de control positivo)) * 100.
Las series de dilución de la dosis se preparan para los compuestos, permitiendo el análisis de los efectos de respuesta a la dosis en el ensayo de JAK3 y el cálculo de la IC50 para cada compuesto. Cada compuesto se analiza como rutina a la concentración de 20pM seguido de una dilución serial 1/3, 8 puntos (20pM - 6,67pM - 2,22pM - 740nM - 247nM -82nM - 27nM - 9nM) en una concentración final de 1% DMSO. Cuando la potencia de la serie de compuestos aumenta, se preparan más diluciones y/o la concentración superior se reduce (por ej., 5 pM, 1 pM).
Los siguientes compuestos se han analizado para determinar su actividad contra JAK3 y los valores de IC50, según lo determinado usando los ensayos descritos en la presente memoria, se dan a continuación en la Tabla XVII.
Tabla XVII Valores de los compuestos JAK3 IC50

6.6. Ensayo de determinación de Ki de JAK3
Para la determinación de Ki, se mezclan diferentes cantidades de compuesto con la enzima y la reacción enzimática se sigue como función de la concentración de ATP. La Ki se determina por medio de gráfico doble recíproco de Km contra concentración del compuesto (gráfico de Lineweaver-Burk). JAK3 (Carna Biosciences, 09CBS-0625B) se usa a una concentración final de 10 ng/ml. El sustrato es sal de sodio Poly(Glu,Tyr) (4:1) , MW 20000 - 50 000 (Sigma, P0275) La reacción se realiza en 25mM Tris pH 7,5,0,01% Triton X-100, 0,5mM Eg Ta , 2,5mM DTT, 0,5mM Na3VO4, 5mM b- glicerolfosfato, 10mM MgCl2 con concentraciones variadas de ATP y compuesto y se detiene por la incorporación de 150 mM ácido fosfórico. La medición de fosfato incorporado en el sustrato polyGT se realiza por la carga de las muestras en una placa de filtro (usando un cosechador, Perkin Elmer) y posterior lavado. El 33P incorporado en polyGT se mide en un contador de centelleo Topcount después de la incorporación de líquido para centelleo a las placas del filtro (Perkin Elmer).
Por ejemplo, cuando el Compuesto I se analiza en este ensayo, se mide un valor de Ki de 353 nM.
6.7. Ensayo de inhibición de TYK2
El dominio catalítico de TYK2 humana recombinante (aminoácidos 871-1187; número de catálogo 08-147) se adquiere de Carna Biosciences. 5 ng de TYK2 se incuba con 12,5 gg de sustrato polyGT (número de catálogo de Sigma P0275) en tampón de reacción de quinasa (25 mM Hepes pH 7,5, 100 mM NaCl, 0,2 mM Na3VÜ4, 0,1% NP-40, 0,1 gM ATP no radiactivo, 0,125 gCi 33P-gamma-ATP (GE Healthcare, número de catálogo AH9968) concentraciones finales) con o sin 5gL que contiene compuesto de prueba o vehículo (DMSO, 1% concentración final), en un volumen total de 25 gL, en una placa de polipropileno de 96 pocillos (Greiner, base V). Después de 90 min a 30°C, las reacciones se detienen por el agregado de 25 gl/pocillo de ácido fosfórico 150 mM. La totalidad de la reacción de quinasa terminada se transfiere a placas de filtro de 96 pocillos previamente lavadas (ácido fosfórico 75 mM) (número de catálogo de Perkin Elmer 6005177) usando un cosechador de células (Perkin Elmer). Las placas se lavan 6 veces con 300 gl por pocillo de una solución 75 mM de ácido fosfórico y la base de las placas se sella. Se añaden 40 gl/pocillo de Microscint-20, la parte superior de las placas se sella y se realiza la lectura usando el Topcount (Perkin Elmer). La actividad de quinasa se calcula restando recuentos por minuto (cpm) obtenidos en presencia de un inhibidor de control positivo (10 gM esturosporina) de cpm obtenido en presencia de vehículo. La capacidad que tiene un compuesto de prueba de inhibir su actividad se determina de la siguiente manera:
Porcentaje de inhibición = ((cpm determinado para la muestra con el compuesto de prueba presente - cpm determinado para la muestra con inhibidor de control positivo) dividido por (cpm determinado en presencia de vehículo - cpm determinado para la muestra con inhibidor de control positivo)) * 100.
Las series de dilución de la dosis se preparan para los compuestos, permitiendo el análisis de los efectos de respuesta a la dosis en el ensayo de TYK2 y el cálculo de la IC50 para cada compuesto. Cada compuesto se analiza como rutina a la concentración de 20gM seguido de una dilución serial 1/3, 8 puntos (20gM - 6,67gM - 2,22gM - 740nM - 247nM -82nM - 27nM - 9nM) en una concentración final de 1% DMSO. Cuando la potencia de la serie de compuestos aumenta, se preparan más diluciones y/o la concentración superior se reduce (por ej., 5 gM, 1 gM).
Los siguientes compuestos se han analizado para determinar su actividad contra TYK2 y los valores de IC50, según lo determinado usando los ensayos descritos en la presente memoria, se dan a continuación en la Tabla XVIII.
Tabla XVIII Valores de los compuestos TYK2 IC50

6.8. Ensayo de determinación de Kd de TYK2
TYK2 (Carna Biosciences, 09CBS-0983D) se usa a una concentración final de 5 nM. El experimento de unión se realiza en 50mM Hepes pH 7,5, 0,01% Brij-35, 10mM MgCl2, 1mM EGTA usando 50nM rastreador de quinasa 236 (Invitrogen, PV5592) y 2 nM Eu-anti-GST (Invitrogen, PV5594) con concentraciones variadas de compuesto. La detección del rastreador se realiza de acuerdo con el procedimiento de los fabricantes.
Por ejemplo, cuando el Compuesto I se analiza en este ensayo, se mide un valor de Kd de 376 nM.
Ejemplo 7. Ensayos celulares
7.1. Ensayo de señalización de JAK-STAT
Se mantienen células HeLa en medio de Eagle Modificado de Dulbecco (DMEM) que contiene 10% suero de ternero fetal inactivado por calor, 100 U/ml de penicilina y 100 gg/ml de estreptomicina. Se usan células HeLa al 70 % de confluencia para transfección. 20.000 células en 87 gl de medio de cultivo se transfectan transitoriamente con 40 gg de informante pSTAT 1 (2)-luciferasa (Panomics), 8 gg de informante LacZ como informante de control interno y 52 gg de pBSK usando 0,32 gL de Jet-PEI (Polyplus) como reactivo de transfección por en formato de placa de 96 pocillos. Después de la incubación durante la noche, a 37°C, 10% de CO2, se retira el medio de transfección. Se añaden 75 gl de DMEM 1,5% de suero de ternero fetal inactivado con calor. 15 gl de compuesto a concentración 6,7 x se añade durante 60 min y luego 10 gl de OSM humano (Peprotech) a 33 gg/ml de concentración final.
Todos los compuestos se analizan por duplicado comenzando con 20 gM seguido de una dilución serial 1/3, 8 dosis en total (20 gM - 6.6 gM - 2.2 gM - 740 nM - 250 nM - 82 nM - 27 nM - 9 nM) en una concentración final de 0,2% DMSO.
Después de la incubación durante la noche a 37°C, 10% células de CO2 se lisan en 100 gl de tampón de lisis/pocillo (PBS, 0,9 mM CaCl2, 0,5 mM MgCl2, 5% Trehalosa, 0,025% Tergitol NP9, 0,15% BSA). 40 gL de lisado celular se usa para leer la actividad de p-galactosidasa mediante el agregado de 180 gl de solución de p-Gal (30gl de ONPG 4mg/ml 150 gl de tampón de p-Galactosidasa (0,06 M Na2HPO4, 0,04 M NaH2PO4, 1 mM MgCl2)) durante 20 min. La reacción se detiene por la adición de 50 gl de Na2CO31 M. La absorbancia se lee a 405 nm.
La actividad de luciferasa se mide usando 40 pl de lisado celular más 40 pl de Steadylite® como se describe por el fabricante (Perkin Elmer), en el Envision (Perkin Elmer). 10 pM de un inhibidor de pan-JAK se usa como control positivo (100% de inhibición). Como control negativo se usa 0,5% DMSO (0% de inhibición). Los controles positivo y negativo se usan para calcular los valores de z” y “porcentaje de inhibición” (PIN).
Porcentaje de inhibición = ((fluorescencia determinada en presencia de vehículo - fluorescencia determinada para la muestra con compuesto experimental presente) dividido por (fluorescencia determinada en presencia de vehículo -fluorescencia determinada para la muestra sin activación)) * 100.
Los valores PIN se grafican para compuestos analizados en dosis-respuesta, los valores de EC50 se derivan y se divulgan en la Tabla XIX que sigue.
Tabla XIX Valores de JAK-STAT EC50

7.2. Ensayo de señalización de OSM/IL-W
Se muestra que OSM e IL-1 p regulan de manera ascendente sinergísticamente los niveles de MMP13 en la línea celular de condrosarcoma humana SW1353. Las células se siembran en placas de 96 pocillos a 15.000 células/pocillo en un volumen de 120 pL de DMEM (Invitrogen) que contiene 10% (v/v) FBS y 1% penicilina/estreptomicina (InVitrogen) incubada a 37°C / 5% CO2. Las células se incuban previamente con 15 pl de compuesto en medio M199 con 2% DMSO 1 hr antes de activar con 15 pl OSM e IL-1 p para alcanzar 25 pg/ml de OSM and 1 pg/ml de IL-1 p, y los niveles de MMP13 se miden en medio acondicionado 48 h después de activar. La actividad de MMP13 se mide usando un ensayo de captura de anticuerpos. Para este fin, placas de 384 pocillos (NUNC, 460518, MaxiSorb negras) se recubren con 35 pl de 1,5 pg/ml solución de anticuerpo MMP13 anti-humano (R&D Systems, MAB511) durante 24 hrs a 4°C. Después de lavar los pocillos 2 veces con PBS 0,05% Tween, los sitios de unión remanentes se bloquean con 100 pL de leche descremada en polvo al 5% (Santa Cruz, sc-2325, Blotto) en PBS durante 24 hr a 4°C. Luego, los pocillos se lavan dos veces con PBS 0,05% Tween y 35 pL de dilución 1/10 de sobrenadante de cultivo que contiene MMP13 en tampón de bloqueo diluido 100 veces se añade y se incuba durante 4 hr a temperatura ambiente. A continuación los pocillos se lavan dos veces con PBS 0,05% Tween seguido de activación de MMP13 por la adición de 35 pl de una solución 1,5 mM de acetato 4-aminofenilmercúrico (APMA) (Sigma, A9563) e incubación a 37°C durante 1 hr. Los pocillos se lavan nuevamente con PBS 0,05% Tween y se añade 35 pl de sustrato MMP13 (Biomol, P-126, OmniMMP sustrato fluorogénico). Después de la incubación durante 24 hrs a 37°C la fluorescencia del sustrato convertido se mide en un lector Perkin Elmer Wallac EnVision 2102 Multilabel (excitación de longitud de onda: 320 nm, emisión de longitud de onda: 405 nm).
Porcentaje de inhibición = ((fluorescencia determinada en presencia de vehículo - fluorescencia determinada para la muestra con compuesto experimental presente) dividido por (fluorescencia determinada en presencia de vehículo -fluorescencia determinada para la muestra sin activación)) * 100.
Por ejemplo, cuando el Compuesto I se analiza en este ensayo, se mide un valor de EC50 de 2242,5 (± 1098,5) nM.
7.3. Ensayo de proliferación de PBL
Linfocitos de sangre periférica humana PBL) se estimulan con IL-2 y la proliferación se mide usando un ensayo de incorporación. Los PBL se estimulan primero durante 72 hrs con PHA para inducir el receptor de IL-2, luego se dejan en ayuno durante 24 hrs para detener la proliferación celular seguido de estimulación de IL-2 durante otras 72 hrs (incluyendo 24hr de marcado de BrdU). Las células se preincuban con compuestos de prueba 1 hora antes de la adición de IL-2. Las células se cultivan en RPMI 1640 que contiene 10% (v/v) FBS.
Ejemplo 8. Ensayos in vivo
8.1. Estudio de PK/PD
8.1.1. Estudio de biodisponibilidad en perros
8.1.1.1. Ajuste experimental
El objetivo de este experimento es comparar la PK en perros beagle macho sanos (3 perros por grupo) después de una sola administración oral de Compuesto I o como Compuesto I. HCL3H2O formulado como cápsulas de dos potencias diferentes (25 y 100 mg).
Los perros no se ponen en ayuno antes de la administración, y tienen libre acceso a agua. Cada día del tratamiento, se proporciona media ración de alimento después de la toma de muestra de sangre T0, 8 a 17 min antes del tratamiento, y la segunda media ración se administra inmediatamente después de la administración o 1h después del
tratamiento para el período 2. Un período de eliminación de 3 días se asegura entre tratamientos.
Compuesto I o Compuesto I. HCL3H2O se administra a una dosis diana de 10 mg/kg en cápsulas (indistintamente 4 x 25 mg, o 1 cápsula x 100 mg). La composición de la cápsula se describe a continuación en la Tabla XX.
Tabla XX Composición de las cápsulas de 25 y 100 mg

Las cápsulas se administran oralmente con agua (5-10 ml), para proporcionar buen tránsito esofágico. Cada animal se controla al menos una vez por día.
Se recolecta sangre de la vena yugular en tubos heparinizados de litio en el T0 (antes de la administración de comida) y luego a las 1 h, 2h, 4h, 6h, 8h, 10h, y 24h después del tratamiento.
Luego se obtiene el plasma de sangre por centrifugado (2500 g durante 10 min a 4°C), y se almacena a -20°C hasta el análisis.
8.1.1.2, Análisis de plasma
Las alícuotas representativas de plasma se diluyen con plasma de perro de control según la necesidad para asegurar que las concentraciones presentes están dentro del intervalo de la curva de calibración, y se extraen por precipitación de proteínas con 2 volúmenes de acetonitrilo acidificado (con 0,1% ácido fórmico) que contiene Compuesto I deuterado como estándar interno (a 150 gg/ml).
Después de mezclado en vórtice y centrifugado a 4°C, los sobrenadantes se diluyen con un volumen de 0,5 de agua grado HPLC en una placa midi-eppendorf de 96 pocillos. La placa se sella y se agita para asegurar la homogeneidad de la muestra antes del análisis. Las muestras se analizan para el Compuesto I por LC-MS/MS usando un espectrómetro de masa Waters TQS, contra una serie de estándares de calibración coincidentes y control de calidad. El procedimiento de Waters TQS tiene un intervalo de curva estándar de 1,00 gg/ml (límite inferior de cuantificación para muestras no diluidas), hasta un máximo de 4000 gg/ml para el Compuesto I.
El análisis de farmacocinética se realiza usando el software WinNonlin™ versión 5.3, usando concentraciones de animales individuales. El análisis no compartimental se aplica para determinar los parámetros de PK (Cmax, Tmax„ ABCü.iast, t1/2, etc...)
Las concentraciones por debajo del límite de detección se ajustan a cero para la estadística descriptiva y los cálculos de parámetros de PK.
Las dosis reales de Compuesto I administrado a cada perro se usan para normalización de la dosis de los parámetros de PK (Cmax y ABC). Los resultados se presentan en la Tabla a continuación y en la Figura 10.
Tabla XXI Resultados del estudio de biodisponibilidad en perros

El Compuesto I (como base libre) se toma por vía oral y por consiguiente pasa a través de la vía gástrica ácida que contiene HCl, en la que el Compuesto I.HCl debe formarse. La persona experta debe esperar, por consiguiente no ver diferencia entre las dos formas administradas.
Sin embargo, como se ilustra en la Figura 10, en promedio y en las 2 potencias de cápsula, el Compuesto I. HCI.3H2O se absorbe más rápidamente, y muestra exposición mejorada in vivo sobre el Compuesto I, lo que puede resultar en el régimen de administración inferior, y por consiguiente cumplimiento mejorado del paciente, y potencialmente menor toxicidad, o problemas de interacción medicamentosa.
Comentarios finales
Los expertos en la técnica apreciarán que las descripciones que anteceden son ejemplificativas y explicativas en su naturaleza, y pretenden ilustrar la invención y sus realizaciones preferidas. A través de experimentación de rutina, un experto reconocerá modificaciones y variaciones evidentes que pueden realizarse sin alejarse de la invención. Todas dichas modificaciones que entran dentro del alcance de las reivindicaciones anexas pretenden incluirse en ellas. De este modo, la invención tiene como finalidad definirse no por la descripción que antecede, sino por las siguientes realizaciones y sus equivalentes.
Se deberá comprender que factores tal como la capacidad diferencial de penetración celular de los varios compuestos puede contribuir a discrepancias entre la actividad de los compuestos en los ensayos celulares y bioquímicos in vitro.
Al menos algunos de los nombres químicos de la sal de la invención según se dan y se establecen en esta solicitud, pueden generase en una base automatizada por el uso de un programa de software de nomenclatura química disponible en el mercado, y no se han verificado independientemente. Los programas representativos que realizan esta función incluyen la herramienta de nomenclatura Lexichem vendida por Open Eye Software, Inc. y la herramienta Autonom Software comercializada por MDL, Inc. En la instancia en la que el nombre químico indicado y la estructura representada difieran, regirá la estructura representada.
Referencias
(CHMP), C. f. (18 November 2004). Guideline on Clinical Investigation of Medicinal Products indicated for the treatment of Psoriasis.
Berishaj, M., Gao, S. P., Ahmed, S., Leslie, K., Al-Ahmadie, H., Gerald, W. L., et al. (2007). Stat3 is tyrosinephosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Research, 9(3), 1-9.
Constantinescu, S. N., Girardot, M., & Pecquet, C. (2007). Mining for JAK-STAT mutations in cancer. Trends in Biochemical Sciences, 33(3), 122-131.
Dolgin, E. (2011). Companies hope for kinase inhibitor JAKpot. Nat Rev Drug Discov, 10(10), 717-8.
Greene, T W; Wuts, P G M;. (1991). Protecting Groups in Organic Synthesis, Second Edition. New York: Wiley. Hilfiker, R., Blatter, F., & von Raumer, M. (2006). Relevance of Solid-state Properties. In Polymorphism: in the Pharmaceutical Industry (pp. 1-20). Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA.
Ingersoll, K. S., & Cohen, J. (2008). The impact of medication regimen factors on adherence to chronic. J Behav Med, 31(3), 213-224.
Kopf, M., Bachmann, M. F., & Marsland, B. J. (2010). Averting inflammation by targeting the cytokine environment. Nat Rev: Drug Disc, 9, 703-718.
Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (2001). Experimental and computational approaches to estimate. Advanced Drug Delivery Reviews solubility andpermeability in drug discovery and development settings, 46, 3-26.
Menet, C. J. (2010). Patent No. WO2010/149769. PCT.
Mullighan, C. G. (2009). JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl. Acad. Sci., 106(23), 9414-9418.
Naka, T., Nishimoto, N., & Kishimoto, T. (2002). The paradigm of IL-6: from basic science to medicine. Arthritis Res, 4 (suppl 3), S233-S242.
O'Sullivan, L. A., Liongue, C., Lewis, R. S., Stephenson, S. E., & Ward, A. C. (2007). Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immuno, 44(10), 2497-506.
O'Dell, J. R. (2004). Therapeutic Strategies for Rheumatoid Arthritis. N Engl J Med, 350, 2591-602.
Punwani, N., Scherle, P., Flores, R., Shi, J., Liang, J., Yeleswaram, S., et al. (2012). Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol, 67, 658- 64.
Smolen, J. S., & Steiner, G. (2003). Therapeutic strategies for rheumatoid arthritis. Nat Rev: Drug Disc, 2, 473-488. Tam, L., McGlynn, L. M., Traynor, P., Mukheijee, R., Bartlett, J. M., & Edwards, J. (2007). Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. British Journal of Cancer, 97, 378 - 383.
Vainchenker, W., Dusa, A., & Constantinescu, S. N. (2008). JAKs in pathology: Role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Seminars in Cell & Developmental Biology, 19, 385-393.
Verstovsek, S. (2009). Therapeutic potential of JAK2 inhibitors. Hematology Am Soc Hematol Educ Program, 636-42. Xiang, Z., Zhao, Y., Mitaksov, V., Fremont, D. FL, Kasai, Y., Molitoris, A., et al. (2008). Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood, 111, 4809-4812.
Zenz, R., Eferl, R., Florin, L., Hummerich, L., Mehic, D., Scheuch, H., et al. (2005). Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature, 437(7057), 369-75.
Zhang, Q. (1996). Activation of JAK/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proc. Natl. Acad. Sci., 93, 9148-9153.
Zikherman, j., & Weiss, A. (2011). Unraveling the functional implications of GWAS: how T cell protein tyrosine phosphatase drives autoimmune disease. J Clin Invest, 121 (12), 4618-21.
Claims (5)
1. Una sal aceptable para uso farmacéutico de un compuesto de acuerdo con la Fórmula (I):

en la que la sal es un aducto Base Libre/HCl/ H2O 1:1:3.
2. Una sal aceptable para uso farmacéutico o hidrato aceptable para uso farmacéutico de una sal de acuerdo con la reivindicación 1, en la que el hidrato de la sal está en forma cristalina.
3. Un hidrato aceptable para uso farmacéutico de una sal de acuerdo con la reivindicación 2,
en el que el hidrato Base Libre/HCl/H2O 1:1:3 de la sal se caracteriza al menos por un pico de difracción de rayos X en polvo en una cualquiera o más de las siguientes posiciones: 7,25, 8,31, 8,78, 10,69, 11,96, 12,25, 13,17, 13,74, 16,23, 16,71,19,26, 19,64, 20,15, 20,57, 20,95, 21,38, 21,69, 22,77, 23,37, 25,14, 25,71, 25,89, 27,66, 28,57, 28,81, 29,17, 29,58, 30,63, 30,94, 32,06, 32,63, 34,14, 36,22, 36,67 y 37,12° 20 ± 0,2° 20.
4. Un hidrato aceptable para uso farmacéutico de una sal de acuerdo con la reivindicación 2, en el que el hidrato Base Libre/HCl/H2O 1:1:3 de la sal se caracteriza por los siguientes parámetros de difracción:


5. Un procedimiento de preparación de un aducto [Compuesto de acuerdo con la Fórmula I:HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) mezclar el compuesto de Fórmula I suspendido en DCM, con MeOH a 35°C,
ii) añadir agua bajo agitación a 35°C durante al menos 15 min,
iii) separar la fase orgánica,
iv) añadir una solución p/p al 10% de HCl en MeOH a la fase orgánica obtenida en la etapa previa iii), v) separar el sólido resultante por filtración obtenida en la etapa previa iv),
vi) secar dicho sólido resultante obtenido en la etapa previa v),
vii) añadir el sólido obtenido en la etapa previa vi) a una solución 1,6/0,4 de ácido fórmico/agua, bajo agitación a 55°C,
viii) añadir agua a la solución obtenida en la etapa previa vii),
ix) separar por filtración el sólido resultante obtenido en la etapa previa viii), y
secar el sólido resultante obtenido en la etapa previa ix).

5. Un procedimiento de preparación de un aducto [Compuesto de acuerdo con la Fórmula I:HCl:3H2O] en una forma sólida cristalina que comprende las etapas de:
i) mezclar el compuesto de Fórmula I suspendido en DCM, con MeOH a 35°C,
ii) añadir agua bajo agitación a 35°C durante al menos 15 min,
iii) separar la fase orgánica,
iv) añadir una solución p/p al 10% de HCl en MeOH a la fase orgánica obtenida en la etapa previa iii), v) separar el sólido resultante por filtración obtenida en la etapa previa iv),
vi) secar dicho sólido resultante obtenido en la etapa previa v),
vii) añadir el sólido obtenido en la etapa previa vi) a una solución 1,6/0,4 de ácido fórmico/agua, bajo agitación a 55°C,
viii) añadir agua a la solución obtenida en la etapa previa vii),
ix) separar por filtración el sólido resultante obtenido en la etapa previa viii), y
secar el sólido resultante obtenido en la etapa previa ix).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1402071.3A GB201402071D0 (en) | 2014-02-07 | 2014-02-07 | Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| PCT/EP2015/052242 WO2015117981A1 (en) | 2014-02-07 | 2015-02-04 | Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2853375T3 true ES2853375T3 (es) | 2021-09-15 |
Family
ID=50390572
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15704974T Active ES2853375T3 (es) | 2014-02-07 | 2015-02-04 | Sales novedosas y composiciones farmacéuticas de las mismas para el tratamiento de trastornos inflamatorios |
Country Status (27)
| Country | Link |
|---|---|
| US (6) | US9382247B2 (es) |
| EP (2) | EP3102575B1 (es) |
| JP (2) | JP2017505329A (es) |
| KR (1) | KR102478614B1 (es) |
| CN (2) | CN105960407A (es) |
| AR (1) | AR099307A1 (es) |
| AU (4) | AU2015215045B2 (es) |
| CA (1) | CA2938219C (es) |
| CY (1) | CY1124349T1 (es) |
| DK (1) | DK3102575T3 (es) |
| EA (2) | EA201990330A1 (es) |
| ES (1) | ES2853375T3 (es) |
| GB (1) | GB201402071D0 (es) |
| HR (1) | HRP20210194T1 (es) |
| HU (1) | HUE053309T2 (es) |
| IL (1) | IL246833B (es) |
| LT (1) | LT3102575T (es) |
| MX (2) | MX2016010068A (es) |
| NZ (1) | NZ722603A (es) |
| PL (1) | PL3102575T3 (es) |
| PT (1) | PT3102575T (es) |
| RS (1) | RS61450B1 (es) |
| SG (2) | SG10202013187PA (es) |
| SI (1) | SI3102575T1 (es) |
| TW (2) | TWI792593B (es) |
| UY (1) | UY35984A (es) |
| WO (1) | WO2015117981A1 (es) |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201402071D0 (en) | 2014-02-07 | 2014-03-26 | Galapagos Nv | Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| GB201402070D0 (en) * | 2014-02-07 | 2014-03-26 | Galapagos Nv | Pharmaceutical compositions for the treatment of inflammatory disorders |
| KR20170134751A (ko) * | 2015-04-13 | 2017-12-06 | 갈라파고스 엔.브이. | 심혈관 질환의 치료 방법 |
| US20180200257A1 (en) * | 2015-04-13 | 2018-07-19 | Galapagos Nv | Methods for the treatment of inflammatory disorders |
| CN105198878B (zh) * | 2015-09-18 | 2017-07-25 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物f晶型及其制备方法 |
| CN105198880B (zh) * | 2015-09-18 | 2017-07-25 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物a晶型及其制备方法 |
| CN105198879B (zh) * | 2015-09-18 | 2017-09-22 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物c晶型及其制备方法 |
| CN105198876B (zh) * | 2015-09-18 | 2017-09-29 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物h晶型及其制备方法 |
| CN105198877B (zh) * | 2015-09-18 | 2017-09-22 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物g晶型及其制备方法 |
| CN105111207B (zh) * | 2015-09-18 | 2017-07-25 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物d晶型及其制备方法 |
| CN105218539B (zh) * | 2015-09-18 | 2017-07-25 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物b晶型及其制备方法 |
| CN105111206B (zh) * | 2015-09-18 | 2017-07-25 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物e晶型及其制备方法 |
| US20170174788A1 (en) | 2015-12-17 | 2017-06-22 | Gilead Sciences, Inc. | Combination of a jak inhibitor and an mmp9 binding protein for treating inflammatory disorders |
| WO2017106564A1 (en) | 2015-12-17 | 2017-06-22 | Gilead Sciences, Inc. | Combination of a jak inhibitor and a syk inhibitor for treating cancers and inflammatory disorders |
| WO2017106568A1 (en) | 2015-12-17 | 2017-06-22 | Gilead Sciences, Inc. | Combination of a jak inhibitor and a bromodomain inhibitor for treating cancer |
| CN105669669A (zh) * | 2016-03-04 | 2016-06-15 | 上海宣创生物科技有限公司 | 一种环丙烷甲酰胺衍生物的a晶型、b晶型、d晶型、g晶型和m晶型及其制备方法 |
| KR20180120273A (ko) * | 2016-03-21 | 2018-11-05 | 크리스탈 파마테크 씨오., 엘티디. | Jak 관련 질병 예방 또는 치료용 약물의 염산 결정성 형태 및 이의 제조 방법 |
| EP3495364A4 (en) | 2016-08-03 | 2019-09-04 | Crystal Pharmaceutical (Suzhou) Co., Ltd. | NOVEL CRYSTAL FORM OF A JAK1 SELECTIVE INHIBITOR AND MANUFACTURING METHOD AND APPLICATION THEREOF |
| JP2019534304A (ja) | 2016-11-10 | 2019-11-28 | ガラパゴス・ナムローゼ・フェンノートシャップGalapagos N.V. | 炎症性疾患の治療のための化合物及びその医薬組成物 |
| GB201702603D0 (en) | 2017-02-17 | 2017-04-05 | Galápagos Nv | Novel compositions and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| JOP20180018A1 (ar) * | 2017-03-14 | 2019-01-30 | Gilead Sciences Inc | تركيبات صيدلية تتضمن مثبط jak |
| GB201808575D0 (en) | 2018-05-24 | 2018-07-11 | Galapagos Nv | Methods for the treatment of psoriatic arthrits |
| US10815227B2 (en) | 2018-08-27 | 2020-10-27 | Cadila Healthcare Limited | Processes for the preparation of filgotinib |
| US11882438B2 (en) * | 2018-10-29 | 2024-01-23 | Zorday IP, LLC | Network-enabled electronic cigarette |
| WO2020177705A1 (zh) * | 2019-03-05 | 2020-09-10 | 苏州科睿思制药有限公司 | Filgotinib的马来酸盐晶型CSI及其制备方法和用途 |
| WO2021044327A1 (en) * | 2019-09-03 | 2021-03-11 | Dr. Reddy's Laboratories Limited | Solid forms of filgotinib maleate and processes thereof |
| CN113773322B (zh) * | 2021-11-10 | 2022-02-11 | 奥锐特药业(天津)有限公司 | 一种Filgotinib的制备方法 |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2549632B2 (ja) * | 1986-08-20 | 1996-10-30 | 三井石油化学工業株式会社 | オレフインの重合方法 |
| ES2228451T3 (es) | 1999-01-29 | 2005-04-16 | Chugai Seiyaku Kabushiki Kaisha | Promotores de condrogenesis y derivados de indolin-2-ona. |
| EP1391211A1 (en) | 2001-04-27 | 2004-02-25 | Chugai Seiyaku Kabushiki Kaisha | Chondrogenesis promoters |
| US6514989B1 (en) | 2001-07-20 | 2003-02-04 | Hoffmann-La Roche Inc. | Aromatic and heteroaromatic substituted 1,2,4-triazolo pyridine derivatives |
| WO2004039816A1 (ja) * | 2002-10-29 | 2004-05-13 | Shionogi & Co., Ltd. | セフェム化合物の無機酸塩の結晶 |
| MXPA05008568A (es) | 2003-02-14 | 2005-11-04 | Pfizer Prod Inc | Nuevos compuestos anti-inflamatorios triazolo-piridinas. |
| US20050222171A1 (en) | 2004-01-22 | 2005-10-06 | Guido Bold | Organic compounds |
| ES2427046T3 (es) | 2004-06-21 | 2013-10-28 | Galapagos N.V. | Métodos y medios para el tratamiento de la osteoartritis |
| EP1786811A2 (en) | 2004-08-18 | 2007-05-23 | Pharmacia & Upjohn Company LLC | Triazolopyridine compounds |
| JP2008515874A (ja) | 2004-10-07 | 2008-05-15 | ワーナー−ランバート カンパニー リミテッド ライアビリティー カンパニー | 抗菌薬 |
| GB0515026D0 (en) | 2005-07-21 | 2005-08-31 | Novartis Ag | Organic compounds |
| AU2007291190A1 (en) | 2006-08-30 | 2008-03-06 | Cellzome Limited | Triazole derivatives as kinase inhibitors |
| WO2008150015A1 (en) | 2007-06-05 | 2008-12-11 | Takeda Pharmaceutical Company Limited | Heterobicyclic compounds as kinase inhibitors |
| CN101801966A (zh) | 2007-07-18 | 2010-08-11 | 诺瓦提斯公司 | 双环杂芳基化合物和它们作为激酶抑制剂的用途 |
| WO2009017954A1 (en) | 2007-08-01 | 2009-02-05 | Phenomix Corporation | Inhibitors of jak2 kinase |
| AU2008291075A1 (en) | 2007-08-31 | 2009-03-05 | Merck Serono S.A. | Triazolopyridine compounds and their use as ask inhibitors |
| GB0719803D0 (en) | 2007-10-10 | 2007-11-21 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| PE20110063A1 (es) | 2008-06-20 | 2011-02-16 | Genentech Inc | DERIVADOS DE [1, 2, 4]TRIAZOLO[1, 5-a]PIRIDINA COMO INHIBIDORES DE JAK |
| WO2010010187A1 (en) | 2008-07-25 | 2010-01-28 | Galapagos Nv | Novel compounds useful for the treatment of degenerative and inflammatory diseases |
| WO2010010184A1 (en) | 2008-07-25 | 2010-01-28 | Galapagos Nv | [1, 2, 4] triazolo [1, 5-a] pyridines as jak inhibitors |
| WO2010010189A1 (en) | 2008-07-25 | 2010-01-28 | Galapagos Nv | Novel compounds useful for the treatment of degenerative and inflammatory diseases |
| JO3041B1 (ar) | 2008-07-25 | 2016-09-05 | Galapagos Nv | مركبات جديدة مفيدة لمعالجة الأمراض التنكسية والالتهابية |
| WO2010010188A1 (en) | 2008-07-25 | 2010-01-28 | Galapagos Nv | Novel compounds useful for the treatment of degenerative and inflammatory diseases. |
| WO2010010186A1 (en) | 2008-07-25 | 2010-01-28 | Galapagos Nv | Novel compounds useful for the treatment of degenerative and inflammatory diseases |
| EP2305673A4 (en) | 2008-07-31 | 2011-11-02 | Daiichi Sankyo Co Ltd | CRYSTAL OF A THIAZOLIDINE CONNECTION AND METHOD FOR THE PRODUCTION THEREOF |
| WO2010141796A2 (en) | 2009-06-05 | 2010-12-09 | Cephalon, Inc. | PREPARATION AND USES OF 1,2,4-TRIAZOLO [1,5a] PYRIDINE DERIVATIVES |
| TWI462920B (zh) * | 2009-06-26 | 2014-12-01 | 葛萊伯格有限公司 | 用於治療退化性及發炎疾病之新穎化合物 |
| JO3030B1 (ar) | 2009-06-26 | 2016-09-05 | Galapagos Nv | مركب جديد مفيد لمعالجة الامراض التنكسية والالتهابات |
| CA2775027A1 (en) | 2009-09-25 | 2011-03-31 | Kyorin Pharmaceutical Co., Ltd. | Maleic acid salt and crystal thereof |
| CN102146082A (zh) | 2010-02-04 | 2011-08-10 | 江苏恒瑞医药股份有限公司 | 吡咯并n杂环类衍生物的可药用的盐、其制备方法及其在医药上的应用 |
| JP2013049632A (ja) * | 2011-08-30 | 2013-03-14 | Kowa Co | ベンゾチアジン化合物のモノマレイン酸塩を有効成分とするアレルギー疾患の予防及び/又は治療剤 |
| SA112330992B1 (ar) * | 2011-11-10 | 2015-09-13 | كيورين فارماسوتيكال كو.، ليمتد | صورة متبلرة من 7-{(s4، s3)-3-[(سيكلو بروبيل أمينو) ميثيل]-4-فلورو بيروليدين-1-يل}-6-فلورو-1-(2-فلورو إيثيل)- 8-ميثوكسي-4-أوكسو-1، 4-داي هيدرو كينولين-3- حمض كربوكسيلي |
| US20130310340A1 (en) | 2012-05-16 | 2013-11-21 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| SG11201408123SA (en) | 2012-06-22 | 2015-01-29 | Galapagos Nv | Aminotriazolopyridine for use in the treatment of inflammation, and pharmaceutical compositions thereof |
| GB201402071D0 (en) * | 2014-02-07 | 2014-03-26 | Galapagos Nv | Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
-
2014
- 2014-02-07 GB GBGB1402071.3A patent/GB201402071D0/en not_active Ceased
-
2015
- 2015-02-04 AU AU2015215045A patent/AU2015215045B2/en active Active
- 2015-02-04 RS RS20210134A patent/RS61450B1/sr unknown
- 2015-02-04 CN CN201580006893.3A patent/CN105960407A/zh active Pending
- 2015-02-04 DK DK15704974.3T patent/DK3102575T3/da active
- 2015-02-04 KR KR1020167023807A patent/KR102478614B1/ko active IP Right Grant
- 2015-02-04 EA EA201990330A patent/EA201990330A1/ru unknown
- 2015-02-04 EA EA201691584A patent/EA032596B1/ru unknown
- 2015-02-04 JP JP2016550633A patent/JP2017505329A/ja active Pending
- 2015-02-04 HU HUE15704974A patent/HUE053309T2/hu unknown
- 2015-02-04 EP EP15704974.3A patent/EP3102575B1/en active Active
- 2015-02-04 IL IL246833A patent/IL246833B/en unknown
- 2015-02-04 WO PCT/EP2015/052242 patent/WO2015117981A1/en active Application Filing
- 2015-02-04 PT PT157049743T patent/PT3102575T/pt unknown
- 2015-02-04 CN CN202010324636.7A patent/CN111499630A/zh active Pending
- 2015-02-04 LT LTEP15704974.3T patent/LT3102575T/lt unknown
- 2015-02-04 PL PL15704974T patent/PL3102575T3/pl unknown
- 2015-02-04 SG SG10202013187PA patent/SG10202013187PA/en unknown
- 2015-02-04 SI SI201531491T patent/SI3102575T1/sl unknown
- 2015-02-04 NZ NZ722603A patent/NZ722603A/en unknown
- 2015-02-04 SG SG11201606433PA patent/SG11201606433PA/en unknown
- 2015-02-04 EP EP20211405.4A patent/EP3842432A1/en not_active Withdrawn
- 2015-02-04 US US14/614,396 patent/US9382247B2/en active Active
- 2015-02-04 MX MX2016010068A patent/MX2016010068A/es active IP Right Grant
- 2015-02-04 CA CA2938219A patent/CA2938219C/en active Active
- 2015-02-04 ES ES15704974T patent/ES2853375T3/es active Active
- 2015-02-06 TW TW110136599A patent/TWI792593B/zh active
- 2015-02-06 UY UY0001035984A patent/UY35984A/es not_active Application Discontinuation
- 2015-02-06 TW TW104104160A patent/TWI767878B/zh active
- 2015-02-06 AR ARP150100351A patent/AR099307A1/es unknown
-
2016
- 2016-07-01 US US15/200,228 patent/US20160376269A1/en not_active Abandoned
- 2016-08-03 MX MX2020003368A patent/MX2020003368A/es unknown
-
2017
- 2017-07-10 US US15/645,308 patent/US10708263B2/en active Active
-
2019
- 2019-04-08 AU AU2019202441A patent/AU2019202441B2/en active Active
- 2019-09-30 US US16/588,207 patent/US10919890B2/en active Active
-
2020
- 2020-01-28 JP JP2020011555A patent/JP2020097595A/ja active Pending
- 2020-10-26 AU AU2020260391A patent/AU2020260391B2/en active Active
-
2021
- 2021-01-08 US US17/144,788 patent/US11667633B2/en active Active
- 2021-02-01 CY CY20211100080T patent/CY1124349T1/el unknown
- 2021-02-03 HR HRP20210194TT patent/HRP20210194T1/hr unknown
-
2022
- 2022-09-23 AU AU2022235624A patent/AU2022235624B2/en active Active
-
2023
- 2023-04-25 US US18/306,867 patent/US20230406857A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2853375T3 (es) | Sales novedosas y composiciones farmacéuticas de las mismas para el tratamiento de trastornos inflamatorios | |
| ES2788671T3 (es) | Composiciones farmacéuticas para el tratamiento de afecciones inflamatorias | |
| JP5591925B2 (ja) | JAK阻害剤としての5−フェニル−[1,2,4]トリアゾロ[1,5−a]ピリジン−2−イルカルボキサミド | |
| JP2018048185A (ja) | 炎症の治療に使用するためのアミノトリアゾロピリジン及びその医薬組成物 | |
| PT2346864E (pt) | Novos compostos úteis para o tratamento de doenças degenerativas e inflamatórias | |
| ES2693019T3 (es) | Derivados de bencimidazol y composiciones farmacéuticas de los mismos para el tratamiento de enfermedades inflamatorias | |
| WO2017133423A1 (zh) | 一种取代的吡啶酰胺类化合物及其应用 | |
| EA041895B1 (ru) | Новые соли и их фармацевтические композиции для лечения воспалительных заболеваний |