ES2817248T3 - Forma farmacéutica para la liberación prolongada de principios activos - Google Patents
Forma farmacéutica para la liberación prolongada de principios activos Download PDFInfo
- Publication number
- ES2817248T3 ES2817248T3 ES13730598T ES13730598T ES2817248T3 ES 2817248 T3 ES2817248 T3 ES 2817248T3 ES 13730598 T ES13730598 T ES 13730598T ES 13730598 T ES13730598 T ES 13730598T ES 2817248 T3 ES2817248 T3 ES 2817248T3
- Authority
- ES
- Spain
- Prior art keywords
- active ingredient
- emulsifier
- compartment
- dosage form
- active
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0002—Galenical forms characterised by the drug release technique; Application systems commanded by energy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/145—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/146—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2031—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Forma farmacéutica con al menos un primer principio activo y al menos un emulsionante, en donde el emulsionante son glicéridos de polietilenglicol que se preparan mediante una reacción de monoglicéridos, diglicéridos y/o triglicéridos con polietilenglicol, en donde la cadena de polietilenglicol es la parte hidrófila del emulsionante y está conectada con una parte lipófila, en donde el emulsionante tiene un valor HLB de al menos 1 y como máximo 16, en donde la relación en masa entre principio activo y emulsionante es de al menos 1 a 30 y como máximo de 1 a 1, y en donde la forma farmacéutica es un comprimido en capas que comprende al menos dos capas, en donde la primera capa es un primer compartimento y la segunda capa es un segundo compartimento, en donde el primer compartimento comprende al menos el primer principio activo y al menos el emulsionante, y el emulsionante no está presente en el segundo compartimento, en donde el primer principio activo es cinarizina o una sal de cinarizina.
Description
DESCRIPCIÓN
Forma farmacéutica para la liberación prolongada de principios activos
Esta invención se refiere a formas farmacéuticas para la liberación prolongada de principios activos, en particular de principios activos que son poco solubles a valores de pH elevados. Una realización particular se refiere a una forma farmacéutica de este tipo, que no solo libera de forma prolongada un primer principio activo, que es poco soluble a valores de pH elevados, sino que también libera un segundo principio activo de manera retardada, en particular un segundo principio activo que tiene mejor solubilidad a valores de pH elevados que el primer principio activo. Para este fin, las realizaciones de esta invención comprenden varios compartimentos, en donde cada uno de los cuales puede contener un principio activo. Una forma farmacéutica de este tipo de acuerdo con la invención se puede dise ñar, por ejemplo, como un comprimido en capas.
Muchos principios activos farmacéuticos son bases orgánicas, es decir, tienen grupos funcionales que presentan un valor de pKb inferior a 7, en particular inferior a 5. Esos principios activos se disuelven mal en el medio intestinal. La razón es que en el intestino predomina un valor de pH alcalino. Por lo tanto, las bases orgánicas ahí no están proto nadas y, por lo tanto, no están cargadas. De este modo, esas moléculas son poco solubles. El intestino, especial mente el intestino delgado, es el lugar en donde normalmente tiene lugar y debe tener lugar la absorción de los prin cipios activos.
Para que un principio activo se pueda absorber, primero se tiene que disolver. Si no se disuelve, no se liberará de su forma farmacéutica.
A las bases orgánicas mencionadas anteriormente se aplica que normalmente se disuelven mejor en un medio áci do. La razón es que el grupo básico en la molécula respectiva está protonado en un medio ácido, por lo que la base orgánica es portadora de una carga positiva. Un medio ácido de ese tipo predomina en un estómago humano en ayunas.
Las circunstancias descritas anteriormente, una mejor solubilidad en el estómago en ayunas por un lado y una peor solubilidad en el intestino por otro lado, conducen a problemas en la formulación de las formas farmacéuticas, en particular si se desea una liberación prolongada.
Hay que tener en cuenta que el pH del estómago humano no es siempre muy ácido. Después de la ingestión de una comida, el valor del pH alcanza valores neutros, generalmente valores entre pH 4 y 6. Solamente de forma gradual el valor del pH vuelve a bajar. En general, un valor de pH < 2 solo se vuelve a alcanzar después de 90 a 120 minu tos. Para el principio activo con las propiedades descritas anteriormente, esto significa que si se toma después de una comida, inicialmente tiene poca solubilidad en el estómago, la cual mejora con el tiempo porque el valor del pH en el estómago vuelve a ser más ácido.
Es particularmente difícil lograr una liberación prolongada de tales principios activos poco solubles. Un enfoque para abordar el problema descrito es la formulación de formas farmacéuticas con liberación prolongada. Esto a menudo se logra proporcionando un recubrimiento a la forma farmacéutica que libera lentamente el principio activo, o disper sando el principio activo en una matriz que se erosiona solo lentamente.
En este caso, sin embargo, reaparece el problema de que el principio activo se disuelve mal en el medio alcalino del intestino. Para una liberación prolongada, en lo posible de forma lineal, es necesario que el principio activo se di suelva de forma continua. En la práctica, esto significa que las formas farmacéuticas que tienen que liberar un prin cipio activo básico con mala solubilidad intestinal durante un período más largo, inicialmente cumplen con este re quisito (en el estómago), pero tan pronto como la forma farmacéutica llega al intestino, la liberación es tan deficiente que el uso no es conveniente.
Por ejemplo, el documento CN 101049309 describe la mejora de la biodisponibilidad de un principio activo que es poco soluble a un valor de pH básico. Sin embargo, el principio activo se proporciona en una forma de administra ción que se disuelve debajo de la lengua y libera el principio activo. Esta forma de administración conduce a una rápida liberación en grandes cantidades, por lo que el principio activo se absorbe a través de la mucosa oral y se evita una absorción por vía gastrointestinal.
Otro problema surge cuando dos principios activos con diferente comportamiento en disolución deben ser liberadas del mismo modo desde una forma farmacéutica. En el caso de principios activos más solubles, en particular aquellas con una carga permanente, surge el problema de que esos principios activos más solubles solo se pueden liberar de forma retardada. Con respecto al primer principio activo, se debe conseguir favorecer la liberación en cualquier caso en el intestino y con respecto al segundo principio activo, se debe conseguir un retardo.
Por ejemplo, el documento WO 2006/099865 describe la liberación de dos principios activos. Sin embargo, la mayo ría de los principios activos se liberan después de unas pocas horas. Sería deseable una liberación prolongada y, al mismo tiempo, una coordinación óptima mejorada de la liberación de los principios activos con un comportamiento en disolución diferente.
Es un objetivo de acuerdo con la invención proporcionar formas farmacéuticas que permiten una liberación prolon gada de un principio activo que es poco soluble a valores de pH elevados. Ciertas realizaciones también logran el objetivo de liberar un principio activo que es poco soluble a valores de pH elevados y un principio activo con mayor solubilidad, de una manera igualmente prolongada. En este contexto, "liberar" significa que el principio activo está disponible en el lugar de la absorción, es decir, en el intestino delgado. Las formas farmacéuticas de acuerdo con la invención tienen un tiempo de residencia prolongado en el estómago, de modo que están un tiempo lo suficiente mente largo en el estómago como para que se pueda disolver una proporción significativa del primer principio activo. Los objetivos descritos anteriormente se logran mediante el objeto de las reivindicaciones. La forma farmacéutica de esta invención contiene al menos un primer principio activo y al menos un emulsionante, ambos tal y como se defi nen en la reivindicación 1, siendo la relación en masa entre el principio activo y el emulsionante de al menos 1 a 30 y como máximo de 1 a 1.
La forma farmacéutica puede comprender solo un compartimento con el primer principio activo o tener varios com partimentos que pueden contener otros principios activos. De acuerdo con la invención, la forma farmacéutica com prende al menos un primer compartimento y un segundo compartimento. De acuerdo con la invención, el primer compartimento comprende al menos el primer principio activo. En realizaciones muy particularmente preferidas, la forma farmacéutica de acuerdo con la invención se compone del primer compartimento que contiene principio activo y el segundo compartimento que contiene principio activo.
La forma farmacéutica de esta invención es un comprimido en capas. La forma farmacéutica de acuerdo con la in vención se diseña preferiblemente de tal manera que no exceda una masa de 1750 mg, preferiblemente 1500 mg, más preferiblemente 1400 mg, incluso más preferiblemente 1200 mg y particularmente de forma preferible 1000 mg. Debido a la formulación, su masa es preferiblemente de al menos 700 mg, más preferiblemente de al menos 800 mg. La masa del segundo compartimento, si está presente, es preferiblemente de al menos 150 mg, más preferible mente al menos 200 mg y en particular como máximo 500 mg, preferiblemente como máximo 400 mg y más preferi blemente como máximo 350 mg.
La forma farmacéutica de acuerdo con la invención es un comprimido en capas que tiene al menos dos capas, sien do la primera capa el primer compartimento descrito anteriormente; y la segunda capa el segundo compartimento descrito anteriormente. Un comprimido en capas de este tipo también puede contener capas adicionales. De forma particularmente preferida, la forma farmacéutica de acuerdo con la invención es un comprimido en capas que consis te en dos capas, siendo una capa el primer compartimento y la otra capa el segundo compartimento.
En el caso del comprimido en capas también puede ser un comprimido de núcleo con recubrimiento. Una capa está dispuesta en el núcleo y otra capa forma el recubrimiento. En otras palabras, el comprimido en capas se puede di señar de manera que una capa rodee completamente a la otra capa. Sin embargo, se prefiere una realización en la que las capas son adyacentes, pero una capa no rodea completamente a la otra. Una capa "no rodea completamen te" a la otra capa cuando preferiblemente no más del 70%, más preferiblemente un máximo del 62% y de forma muy particularmente preferible un máximo del 52% de la superficie exterior total de una capa está recubierta por la otra capa.
En realizaciones particularmente preferidas, la forma farmacéutica descrita en esta memoria es una forma con largo tiempo de residencia en el estómago. Esto significa que la forma farmacéutica no se disuelve tanto en el estómago como para que pueda salir rápidamente del estómago a través del esfínter pilórico. La forma farmacéutica de acuer do con la invención permanece preferiblemente en el estómago del paciente durante al menos 2 horas, más preferi blemente al menos 3 horas, antes de pasar al intestino. Esto se consigue de acuerdo con la invención porque la forma farmacéutica está diseñada de tal manera que solo se disuelve muy lentamente. Como resultado, mantiene durante un largo período de tiempo un tamaño que no le permite salir del estómago.
La forma farmacéutica tiene preferiblemente al menos en un punto, un diámetro de más de 2 mm, en particular más de 4 mm y de manera particularmente preferida al menos 5 mm. Esto asegura que la forma farmacéutica no pueda salir inmediatamente del estómago a través del esfínter pilórico, sino que permanece en el estómago todavía duran te algún tiempo.
El estómago no se vacía inmediatamente después de ingerir alimentos, sino que solo libera gradualmente el bolo alimenticio (quimo) en el intestino delgado. La forma farmacéutica de acuerdo con la invención puede hacer uso de este hecho fisiológico. Como se ha descrito anteriormente, el primer principio activo es preferiblemente un principio activo que se disuelve mejor a un valor de pH bajo, es decir, en un medio ácido que en un medio alcalino.
En el estómago predomina un pH bajo. Sin embargo, después de comer, el pH aumenta a valores más altos, p. ej., aproximadamente a pH 5. El pH disminuye a valores más bajos solo de forma gradual. Esto significa que el primer principio activo inicialmente se disolvería mal después de la ingestión de alimentos porque el valor del pH es dema siado alto. Sin embargo, la forma farmacéutica de acuerdo con la invención permite entonces que parte del primer principio activo se disuelva mediante las medidas descritas en este documento.
A través de los movimientos del estómago, el primer principio activo liberado se mezcla con el quimo y luego se entrega gradualmente al intestino delgado. A continuación, el principio activo se absorbe en el intestino delgado
proximal, es decir, poco después de la salida desde el estómago. Por tanto, el estómago sirve como agente retar dante para el primer principio activo. Al mismo tiempo, si está presente, también se disuelve el segundo principio activo, que preferiblemente tiene una solubilidad más alta que el primer principio activo. El segundo principio activo tiene preferiblemente una mayor solubilidad cuando su solubilidad es al menos una potencia de diez más alta que la solubilidad del primer principio activo.
Tan pronto como el pH en el estómago alcanza un valor más bajo, p. ej., aproximadamente pH 3, el primer principio activo se disuelve mejor. Para que no se libere a la vez todo el contenido restante del primer principio activo, el emulsionante se selecciona preferiblemente de modo que pueda ralentizar la liberación del primer principio activo. Opcionalmente, esto se facilita con al menos un tensioactivo, al menos un triglicérido y/o al menos un agente retar dante.
En el segundo compartimento, no se necesita un emulsionante, por lo que no hay ninguno. En cambio, se encuentra preferiblemente al menos un agente retardante que es adecuado para ralentizar la liberación del segundo principio activo, de modo que aquí se puede lograr una liberación prolongada.
Dado que la forma farmacéutica solo se disuelve muy lentamente, preferiblemente permanece en el estómago hasta que se ha vaciado en gran medida. Una gran parte del bolo alimenticio habrá llegado después al intestino delgado a través del esfínter pilórico. En ese momento, el estómago se vacía mediante las llamadas contracciones de mante nimiento. De esta manera, también llegan cuerpos, que normalmente no pasan por el esfínter pilórico, hasta el intes tino delgado.
La forma farmacéutica continuará liberando entonces los principios activos en el intestino delgado. Esto permite una liberación casi lineal de los dos principios activos.
El primer principio activo es un principio activo que es poco soluble en un medio básico. Esto significa en particular que el primer principio activo en agua con un pH de 7, tiene una solubilidad menor de 0,5 mol/l, en particular menor de 0,1 mol/l, así como preferiblemente menor de 0,01 mol/l. A menos que se especifique lo contrario, la solubilidad se determina en condiciones estándar (DIN 1343), esto también se aplica a todos los demás parámetros de esta descripción.
Esto se aplica regularmente a la cinarizina y a sus sales.
De acuerdo con la invención, "fácilmente soluble" o "buena solubilidad" significa que la sustancia caracterizada de ese modo, se disuelve en el medio o el disolvente correspondiente en una cantidad superior a 0,5 mol/l. "Poco solu ble" o "escasa solubilidad" significa que la sustancia caracterizada de ese modo se disuelve en el medio o el disol vente correspondiente en una cantidad de como máximo 0,5 mol/l, en particular como máximo 0,1 mol/l y preferible mente como máximo 0,01 mol/l.
Como se ha mencionado anteriormente, los principios activos lipófilos se benefician en particular de la matriz de principio activo de esta invención. La ventaja para esos principios activos también es particularmente elevada porque la absorción de principios activos lipófilos generalmente funciona muy bien, tan pronto como se disuelven esas sus tancias. En otras palabras, la disolución de esos principios activos procedentes de una forma farmacéutica es el paso que determina la velocidad durante la absorción del principio activo en el organismo.
No obstante, existen también principios activos cuya lipofilicidad es tan pronunciada que la matriz de principio activo propuesta en esta memoria es insuficiente para asegurar una biodisponibilidad suficiente. Los principios activos que se utilizan de acuerdo con la invención tienen por tanto coeficientes de distribución de n-octanol-agua (logPow) a 20°C, de preferentemente como máximo 100, más preferentemente como máximo 50, en particular como máximo 30, de forma especialmente preferida como máximo 15 y en particular como máximo 7.
Para mejorar la solubilidad del primer principio activo después de una aplicación, se prefiere de acuerdo con la in vención usar el primer principio activo con un tamaño de partícula pequeño. Por lo tanto, el primer principio activo en la forma farmacéutica de esta invención tiene preferiblemente tamaños medios de partícula de como máximo 1000 nm, más preferiblemente como máximo 600 nm y de forma particularmente preferible como máximo 300 nm. De acuerdo con la invención, el tamaño medio de partícula se determina preferiblemente usando el método de análisis de dispersión de luz dinámica.
El tamaño de partícula pequeño es particularmente importante cuando el principio activo no se disuelve o apenas se disuelve en el emulsionante. Esto se aplica normalmente cuando el emulsionante tiene un componente hidrófilo particularmente grande, como muchos poloxámeros, y/o el principio activo es particularmente lipófilo. Se prefiere de acuerdo con la invención que el principio activo se disuelva en el emulsionante casi completamente, de forma prefe rible, completamente. Esto da como resultado una liberación mejorada. Este efecto se puede lograr simplemente adaptando la porción lipófila del emulsionante a la lipofilicidad del principio activo. Cómo se puede lograr esto, se desprende de las realizaciones siguientes sobre el emulsionante y su estructura molecular.
La forma farmacéutica de acuerdo con la invención es especialmente adecuada para una administración de princi pios activos que tienen al menos un grupo amino. Es ventajoso que estos principios activos no presenten grupos
hidroxilo y/o carboxilo. En cualquier caso, esos principios activos se benefician particularmente del diseño de la for ma farmacéutica de acuerdo con la invención. Por tanto, el primer principio activo preferiblemente no tiene ningún grupo hidroxilo ni grupo carboxilo.
De acuerdo con la invención, el primer principio activo es cinarizina o una sal de cinarizina. El primer principio activo se usa en la forma farmacéutica preferiblemente en una cantidad de al menos 10 mg y más preferiblemente al me nos 20 mg o de forma particularmente preferible al menos 40 mg. Las cantidades del primer principio activo preferi blemente no deben exceder un valor de 200 mg, más preferiblemente 100 mg y de forma particularmente preferible 80 mg.
El emulsionante favorece la liberación del primer principio activo. Esto es particularmente importante para que al menos una parte del primer principio activo se disuelva lo más rápida y uniformemente posible ya en el estómago. Como se ha descrito anteriormente, el valor del pH del estómago desciende lentamente después de la ingesta de una comida. Sin embargo, es necesario que el primer principio activo se disuelva ya en el estómago para que pueda mezclarse con el bolo alimenticio. La mezcla se lleva a cabo mediante el movimiento del estómago. El uso de un emulsionante de acuerdo con la invención en las formas farmacéuticas de acuerdo con la invención permite, en particular, una mejora de la solubilidad del primer principio activo a valores de pH más altos. El emulsionante de acuerdo con la invención contribuye a que el primer principio activo idealmente ya se libera en forma disuelta y a continuación se puede absorber.
Para que la mejora de la liberación sea particularmente pronunciada, el emulsionante debe tener preferiblemente un valor de HLB de al menos 1 y como máximo 16, en particular al menos 9 y como máximo 14. El emulsionante es preferiblemente no iónico.
Se ha mostrado que los emulsionantes que tienen un determinado motivo estructural consiguen resultados espe cialmente ventajosos. Ese motivo estructural es una cadena de polietilenglicol. Se ha encontrado que los emulsio nantes con ese motivo estructural poseen una función doble. Por un lado, a valores de pH elevados de aproximada mente pH 4, conducen a una mejora de la solubilidad del primer principio activo. Por otro lado, a valores de pH ba jos, es decir, cuando el primer principio activo es más soluble, provocan un retraso de la liberación. Este es un com portamiento deseable de acuerdo con la invención, ya que la liberación del primer principio activo debería tener lugar de la forma más continua posible durante un período de tiempo largo.
La cadena de polietilenglicol representa la parte hidrófila del emulsionante y está ligada con una parte lipófila. La parte lipófila puede ser ventajosamente un residuo de glicérido u otra cadena de poli(óxido de alquileno).
Como residuos de glicéridos se toman en consideración los residuos de monoglicéridos y diglicéridos, prefiriéndose los residuos de diglicéridos. Los residuos de monoglicéridos pueden no ser lo suficientemente lipófilos, dependiendo de la longitud de la cadena de los residuos de ácidos grasos implicados.
Cadenas de poli(óxido de alquileno) particularmente adecuadas son poli(óxidos de alquileno) que tienen cadenas de carbono ininterrumpidas de al menos 3 átomos de carbono. Un ejemplo preferido de una cadena de poli(óxido de alquileno) en un emulsionante de esta invención, es el poli(óxido de propileno).
Esos emulsionantes se pueden obtener mediante procedimientos conocidos por el experto en la materia. Los glicéridos de polietilenglicol de acuerdo con la invención se preparan haciendo reaccionar los glicéridos correspondientes con polietilenglicol. Los glicéridos de polietilenglicol disponibles comercialmente son, por ejemplo, Gelucire® 43/01 y Gelucire® 50/13.
Los emulsionantes descritos en esta memoria se pueden preparar mediante una reacción, en particular una alcoholisis, de monoglicéridos, diglicéridos y/o triglicéridos con poli(óxidos de alquileno). Esos materiales de partida están disponibles comercialmente. Los emulsionantes de acuerdo con la invención son preferiblemente mezclas de los productos de reacción de las sustancias mencionadas.
En realizaciones preferidas, el emulsionante se selecciona entre Gelucire® 50/13, Gelucire® 43/01, Poloxamero 407 y mezclas de los mismos.
Se logra una buena solubilización cuando la relación entre las masas de principio activo y emulsionante es de al menos 1 a 30, más preferiblemente de al menos 1 a 20 y de forma particularmente preferible de al menos 1 a 10 e incluso más preferiblemente de al menos 1 a 8. La relación indicada es como máximo de 1 a 1, preferiblemente de menos de 1 a 1, más preferiblemente como máximo de 1 a 2, incluso más preferiblemente como máximo de 1 a 3 e incluso más preferiblemente como máximo de 1 a 4 y de forma particularmente preferible, como máximo de 1 a 6. En general se aplica que una mayor cantidad de emulsionante permite una mejor solubilización. Sin embargo, debe tenerse en cuenta que una forma farmacéutica no debe exceder ciertos volúmenes máximos para que la ingesta no sea innecesariamente desagradable. Además, una cantidad demasiado grande de emulsionante también puede provocar que se impida la liberación del primer principio activo. Se ha observado que es particularmente adecuada una relación en masa entre principio activo y emulsionante de al menos 1 a 10 y como máximo de 1 a 2, idealmente una relación en masa de al menos 1 a 8 y como máximo de 1 a 2. En el caso de conservar esa relación en masa, se observó una aceleración ventajosa de la liberación, con un tamaño especialmente adecuado de la forma farmacéuti
ca.
De acuerdo con la invención, el primer compartimento comprende el emulsionante. La forma farmacéutica o el pri mer compartimento contiene el emulsionante preferiblemente en una proporción de al menos 7% en peso, preferi blemente al menos 10% en peso, más preferiblemente al menos 15% en peso y de manera particularmente preferida al menos 20% en peso. La proporción de emulsionante no debe exceder preferiblemente una proporción del 50% en peso, más preferiblemente del 40% en peso y de manera particularmente preferida del 32% en peso. Al intentar determinar la relación óptima entre solubilización y volumen de la forma farmacéutica, estas cantidades han mostra do ser ventajosas.
En realizaciones particularmente preferidas, además de las sustancias mencionadas anteriormente, se emplea al menos un fosfolípido. El fosfolípido se selecciona preferiblemente a partir de fosfatidilcolinas (lecitinas), fosfatidiletanolaminas (cefalinas), fosfatidilserinas, esfingomielinas y mezclas de las mismas. Se prefieren particularmente las fosfatidilcolinas (lecitinas). Sorprendentemente, se ha encontrado que los fosfolípidos a su vez, producen los efectos ventajosos descritos anteriormente, como los emulsionantes mencionados anteriormente.
El emulsionante adicional también puede ser un fosfolípido. El fosfolípido potencia los efectos beneficiosos de las sustancias mencionadas anteriormente.
En realizaciones preferidas, la forma farmacéutica también contiene al menos un tensioactivo. El tensioactivo sirve para reforzar la solubilización mediada por el emulsionante. Si se utiliza una mezcla de emulsionante y tensioactivo, estas dos sustancias actúan de forma sinérgica. Esto permite reducir la masa total de emulsionante y tensioactivo en relación con el principio activo. Como se ha mencionado anteriormente, para lograr la mejor solución posible, puede ser necesario elegir una cantidad muy grande de emulsionante en relación con el principio activo. Esta cantidad elevada se puede reducir utilizando un tensioactivo.
Los tensioactivos preferidos son tensioactivos iónicos, especialmente tensioactivos aniónicos. Los jabones metálicos han mostrado ser especialmente ventajosos. Por jabones metálicos se entiende preferentemente sustancias que contienen un ácido orgánico como componente aniónico y un catión metálico como componente catiónico. El catión metálico se selecciona preferiblemente a partir de iones de metales alcalinos y alcalinotérreos. Se prefieren particu larmente los iones de metales alcalinos y especialmente sodio y potasio. Entre los iones de metales alcalinotérreos, se prefieren particularmente magnesio y calcio.
Los ácidos orgánicos preferidos que pueden aparecer como componentes aniónicos en los tensioactivos, son ácidos con al menos 6 átomos de carbono. Se ha observado que los ácidos orgánicos con cadenas de carbono de longitud más corta son menos eficaces. Sin embargo, los ácidos orgánicos preferiblemente no deben exceder longitudes de cadena de 22 átomos de carbono. En caso de cadenas de carbono más largas, el efecto sinérgico en la interacción con el emulsionante no es muy fuerte. Se da especial preferencia a los ácidos orgánicos con longitudes de cadena de los tensioactivos de al menos 10 y como máximo 18, más preferiblemente al menos 12 y como máximo 16 áto mos de carbono.
Los ácidos orgánicos pueden tener cadenas de carbono ramificadas o no ramificadas, preferiblemente las cadenas de carbono no están ramificadas. Un tensioactivo particularmente preferido es el miristato de sodio.
Los tensioactivos de acuerdo con la invención también tienen la función de ralentizar el paso gastrointestinal del bolo alimenticio y, por tanto, la absorción del primer principio activo. Curiosamente, se ha descubierto que esto se puede lograr en particular mediante los jabones metálicos descritos en esta memoria y que estos pueden tener un efecto directo sobre el control del paso gastrointestinal, con el resultado de que en general se puede absorber más del primer principio activo.
La proporción de tensioactivo en la forma farmacéutica de acuerdo con la invención debe ser preferiblemente de al menos 2% en peso, preferiblemente de al menos 7% en peso, más preferiblemente de al menos 12% en peso y de forma particularmente preferible de al menos 17% en peso. Se ha mostrado que los efectos del tensioactivo son particularmente pronunciados. Sin embargo, no se debe exceder una cantidad de preferiblemente 35% en peso, más preferiblemente 25% en peso y de forma particularmente preferible 20% en peso. Esto dificultaría demasiado la liberación del primer principio activo. La masa absoluta del tensioactivo en la forma farmacéutica no debe exceder un valor de 300 mg, más preferiblemente 250 mg y de forma particularmente preferible 200 mg. La cantidad mínima de tensioactivo preferiblemente no debe ser inferior a un valor de 10 mg, más preferiblemente 50 mg y de forma particu larmente preferible 100 mg.
La cantidad de tensioactivo en la relación en masa con el primer principio activo no debe ser inferior a 1 a 1, preferi blemente 1,5 a 1 y de forma particularmente preferible 2 a 1. Preferiblemente, esa relación no debe superar un valor de 5 a 1, más preferiblemente de 4 a 1 y en particular de 3 a 1. Si se tienen en cuenta estos valores, el efecto venta joso descrito anteriormente se puede utilizar de manera óptima.
En una realización preferida, la forma farmacéutica de esta invención tiene al menos un triglicérido. El triglicérido se usa para ralentizar el paso gastrointestinal de la forma farmacéutica. El triglicérido también se puede usar en combi nación con el tensioactivo para ralentizar aún más la liberación del primer principio activo.
El triglicérido comprende preferiblemente un residuo de ácido graso que tiene al menos 10 átomos de carbono, más preferiblemente al menos 12 átomos de carbono y de forma particularmente preferible al menos 16 átomos de car bono. Esta longitud de cadena mínima es deseable porque prolonga el tiempo de permanencia de la forma farma céutica en el estómago de forma particularmente pronunciada y, por lo tanto, la liberación del primer principio activo se puede favorecer de manera particularmente eficiente. Sin embargo, la longitud de la cadena no debería exceder preferiblemente un valor de 22 átomos de carbono, más preferiblemente 20 átomos de carbono. En realizaciones particularmente preferidas, al menos dos y de forma muy particularmente preferible, todos los residuos de ácidos grasos en el triglicérido cumplen esos requisitos.
El triglicérido se puede usar preferiblemente en una proporción de más del 5% en peso, preferiblemente al menos 7% en peso, más preferiblemente al menos 10% en peso, incluso más preferiblemente al menos 15% en peso y de forma particularmente preferible al menos 20% en peso, en la forma farmacéutica de acuerdo con la invención. Sin embargo, no se debe exceder una proporción máxima de preferiblemente 40% en peso, más preferiblemente 30% en peso y de forma particularmente preferible 25% en peso, porque de lo contrario la liberación del primer principio activo se restringiría demasiado.
La masa absoluta del triglicérido en la forma farmacéutica no debe exceder un valor de 500 mg, más preferiblemente 400 mg y de forma particularmente preferible 300 mg. La cantidad mínima de triglicérido preferiblemente no debe ser inferior a un valor de 50 mg, más preferiblemente 100 mg y de forma particularmente preferible 150 mg.
El contenido en triglicéridos en la relación en masa con el primer principio activo es preferiblemente superior a 1 a 1. El contenido en triglicéridos en la relación en masa con el primer principio activo, preferiblemente no debe ser inferior a una proporción de 2 a 1, incluso más preferiblemente de 3 a 1 y de forma particularmente preferible de 3,5 a 1. Preferiblemente, esa relación no debe superar un valor de 20 a 1, más preferiblemente de 10 a 1, incluso más prefe riblemente de 7 a 1 y en particular de 6 a 1. Si se tienen en cuenta esos valores, el efecto ventajoso descrito ante riormente se puede utilizar de manera óptima.
El primer compartimento puede comprender un coadyuvante de prensado. El coadyuvante de prensado se seleccio na preferiblemente entre lactosa y almidón de maíz. El coadyuvante de prensado es más preferiblemente almidón de maíz. En otra realización, la lactosa se utiliza como coadyuvante de prensado. El coadyuvante de prensado se usa, entre otras cosas, para elevar el punto de fusión de una mezcla de principio activo y emulsionante durante la prepa ración, de modo que sea posible un procesamiento adicional de esa mezcla.
La proporción de ese coadyuvante de prensado en la forma farmacéutica, o cuando la forma farmacéutica tiene varios compartimentos, en el primer compartimento debe ser preferiblemente de al menos 10% en peso, más prefe riblemente al menos 20% en peso y de forma particularmente preferible al menos 30% en peso. Si se usa una canti dad demasiado escasa de coadyuvante de prensado, no se logrará el efecto deseado. Una cantidad demasiado elevada de coadyuvante de prensado agranda la forma farmacéutica y, por tanto, no es deseable. Por tanto, la pro porción de coadyuvante de prensado en la forma farmacéutica, o cuando la forma farmacéutica tiene varios compar timentos, en el primer compartimento, no debería exceder un valor del 65% en peso, preferiblemente del 60% en peso, más preferiblemente del 50% en peso y de forma particularmente preferible del 40% en peso.
La forma farmacéutica de esta invención también puede contener un agente retardante. El agente retardante se selecciona preferiblemente a partir de sustancias orgánicas poliméricas, en particular aquellas que son hinchables. Los agentes retardantes preferidos son polímeros de acrilato, polímeros de metacrilato y sus copolímeros así como mezclas de los mismos. Otros agentes retardantes preferidos son celulosas, especialmente las celulosas de viscosi dad elevada. El agente retardante de la forma farmacéutica de esta invención es preferiblemente un polímero, espe cialmente un polisacárido. Se prefieren celulosa, derivados de celulosa, alginatos y mezclas de los mismos, así co mo sus hidratos, pero también son adecuados los poliacrilatos, polimetacrilatos y sus copolímeros y mezclas. Los derivados de celulosa particularmente preferidos son metilcelulosa e hidroxipropilmetilcelulosa.
Se utiliza preferentemente un agente retardante que, en una solución acuosa en una proporción del 2% en peso, a una temperatura de 20°C y una presión de 101,325 kPa, tiene una viscosidad de al menos 1500 mPas. La viscosi dad se mide con un viscosímetro de caída de bola (DIN 53015). Dependiendo de la prolongación deseada de la liberación del primer principio activo, puede ser deseable elegir también viscosidades más altas. Cuanto mayor sea la viscosidad del agente retardante, mayor será la prolongación de la liberación. En realizaciones particularmente preferidas, la viscosidad del agente retardante es de incluso a al menos 2500 mPas y de manera particularmente preferida al menos 3500 mPas. Sin embargo, si la viscosidad es demasiado alta, el primer principio activo se liberará de forma demasiado lenta. Esto podría conducir a que el primer principio activo no se libere por completo durante su paso por el tracto gastrointestinal. Por lo tanto, la viscosidad debería limitarse a un máximo de 200.000 mPas, más preferiblemente a un máximo de 120.000 mPas.
La proporción de este agente retardante en la forma farmacéutica, o cuando la forma farmacéutica tiene varios com partimentos, en el primer compartimento, debe ser preferiblemente de al menos 10% en peso, más preferiblemente al menos 20% en peso y de manera particularmente preferida al menos 30% en peso. Si se usa una cantidad dema siado reducida del agente retardante, no se logrará el efecto deseado. Si las cantidades son demasiado elevadas, el primer principio activo no se liberará con la suficiente rapidez. Por tanto, la proporción de agente retardante en la
forma farmacéutica, o cuando la forma farmacéutica tiene varios compartimentos, en el primer compartimento, no debe exceder un valor del 60% en peso, más preferiblemente del 50% en peso y de forma particularmente preferible del 40% en peso. En la relación en masa con el primer principio activo, el contenido en agente retardante debería ser preferiblemente de al menos 3 a 1, más preferiblemente de al menos 5 a 1 y de forma particularmente preferible de al menos 7 a 1. Sin embargo, esa relación no debe exceder un valor de 20 a 1, más preferiblemente de 15 a 1 y de forma particularmente preferible de 10 a 1.
Además, se puede añadir un alcohol graso al primer y/o al segundo compartimento. El alcohol graso es preferible mente un alcohol con una longitud de cadena de al menos 10 átomos de carbono, más preferiblemente de al menos 12 átomos de carbono y de forma particularmente preferible de al menos 14 átomos de carbono. Sin embargo, la longitud de la cadena no debe exceder un valor de preferiblemente 20 átomos de carbono, más preferiblemente 18 átomos de carbono y en particular 16 átomos de carbono. Se ha mostrado que la adición de un alcohol graso es adecuada para retrasar la liberación de los principios activos, en particular del segundo principio activo.
La proporción en peso del alcohol graso en la forma farmacéutica o en el primer compartimento (si la forma farma céutica tiene varios compartimentos) es preferiblemente de al menos 5% en peso, más preferiblemente de al menos 10% en peso y de forma particularmente preferible de al menos 18% en peso. En caso de tener una proporción de alcohol graso demasiado baja en la forma farmacéutica o en el primer compartimento (si la forma farmacéutica tiene varios compartimentos), el efecto de acuerdo con la invención puede verse afectado. En caso de que el contenido sea demasiado alto, la identidad estructural de la forma farmacéutica puede verse afectada. Por lo tanto, el conteni do en alcohol graso en la forma farmacéutica o en el primer compartimento (si la forma farmacéutica tiene varios compartimentos) es preferiblemente como máximo del 40% en peso, más preferiblemente como máximo del 35% en peso y de forma particularmente preferible como máximo del 30% en peso.
La forma farmacéutica de esta invención puede comprender otros coadyuvantes farmacéuticos. Estos coadyuvantes se pueden seleccionar preferiblemente a partir de agentes lubricantes, agentes deslizantes, agentes aglutinantes, agentes desintegrantes, antioxidantes, agentes formadores de complejos, agentes de recubrimiento, agentes de flujo, conservantes, cargas, plastificantes, pigmentos y mezclas de esas sustancias.
Un coadyuvante preferido en la forma farmacéutica es un vehículo farmacéuticamente aceptable. Los vehículos preferidos son polisacáridos, en particular celulosa y/o lactosa. El vehículo confiere a la forma farmacéutica una estabilidad suficiente. Finalmente, la forma farmacéutica contiene una gran proporción de emulsionante. El emulsio nante tiene solo una contribución muy limitada a la integridad estructural de la forma farmacéutica. La forma farma céutica contiene preferiblemente el vehículo en una proporción en peso de al menos un 10% en peso, más preferi blemente de al menos un 20% en peso y de manera particularmente preferida de al menos un 30% en peso.
En resumen, la forma farmacéutica, o el primer compartimento, si la forma farmacéutica tiene varios compartimentos, puede contener, entre otros elementos:
a. un primer principio activo,
b. un emulsionante para mejorar la solubilidad del primer principio activo con valores de pH altos y para ralenti zar la disolución del primer principio activo con valores de pH bajos;
c. opcionalmente, un tensioactivo para mejorar la solubilidad del primer principio activo y ralentizar el transporte del primer principio activo desde el estómago al intestino;
d. opcionalmente, un triglicérido también para mejorar la solubilidad del primer principio activo y para ralentizar el transporte del primer principio activo desde el estómago al intestino;
e. opcionalmente, un agente retardante para ralentizar la disolución del primer principio activo con valores de pH bajos;
f. opcionalmente, un coadyuvante de prensado para favorecer el procesamiento de una mezcla del primer prin cipio activo y el emulsionante durante la preparación;
g. opcionalmente un alcohol graso para ralentizar la disolución del primer principio activo con valores de pH ba jos;
h. opcionalmente otros coadyuvantes.
La forma farmacéutica de acuerdo con la invención tiene al menos dos compartimentos que preferiblemente contie nen diferentes principios activos. El primer compartimento tiene las sustancias mencionadas anteriormente, a saber, el primer principio activo y el emulsionante así como, opcionalmente, un tensioactivo, un triglicérido, un agente retar dante, un coadyuvante de prensado, un alcohol graso y otros coadyuvantes opcionales. La forma farmacéutica está compuesta de forma muy especialmente preferida por dos compartimentos que contienen principio activo con princi pios activos diferentes, teniendo el primer compartimento las sustancias mencionadas anteriormente. De acuerdo con la invención, "principios activos diferentes" significa que los principios activos de los compartimentos difieren, es
decir, que el mismo principio activo no está contenido en diferentes compartimentos.
El primer compartimento tiene al menos el primer principio activo, el segundo compartimento tiene al menos un se gundo principio activo. Además del primer y el segundo principio activo, la forma farmacéutica podría contener más principios activos. Por tanto, el primer compartimento y al menos el segundo compartimento pueden contener otros principios activos además del primer o el segundo principio activo. Sin embargo, de manera muy particularmente preferida, la proporción de principio activo de la forma farmacéutica consiste en el primer y el segundo principio acti vo.
La proporción en peso del primer compartimento en la forma farmacéutica de acuerdo con la invención, es preferi blemente del 45 al 100% en peso, más preferiblemente del 55 al 87,5% en peso. La razón de ello es que el primer compartimento no solo tiene que facilitar la liberación prolongada, sino también mediar en la solubilización del primer principio activo, que es difícil de formular.
Un segundo compartimento de la forma farmacéutica contiene al menos el segundo principio activo. El segundo principio activo es preferiblemente un principio activo con mayor solubilidad en agua que el primer principio activo, que por tanto se disuelve mejor que el primer principio activo tanto en el estómago como en el intestino. Por tanto, el segundo principio activo tiene preferiblemente una solubilidad que es al menos una potencia de diez más alta que la del primer principio activo, en particular a pH 7.
El segundo compartimento puede contener en particular:
a. un segundo principio activo,
b. opcionalmente un agente retardante para ralentizar la liberación del segundo principio activo,
c. opcionalmente un alcohol de cadena larga para ralentizar la liberación del segundo principio activo, d. opcionalmente, un tensioactivo como se ha descrito anteriormente, para ralentizar el transporte del principio activo desde el estómago al intestino;
e. opcionalmente un triglicérido como se ha descrito anteriormente, para ralentizar el transporte del principio ac tivo desde el estómago al intestino;
f. opcionalmente otros coadyuvantes.
En la técnica anterior, no se presenta ninguna forma farmacéutica convincente que permita liberar tanto un principio activo poco soluble (en esta memoria: el primer principio activo) como un segundo principio activo, en particular si este tiene una solubilidad más alta que el primer principio activo, en una forma farmacéutica, cada uno retardado. Finalmente, se debe favorecer la liberación del principio activo poco soluble y, en el caso de un segundo principio activo más soluble, se debe impedir al mismo tiempo la liberación del segundo principio activo. Esta invención ofrece una solución a este problema proporcionando una forma farmacéutica de acuerdo con la invención que comprende preferiblemente dos compartimentos, en donde el primer compartimento contiene el primer principio activo y el se gundo compartimento contiene el segundo principio activo.
En el contexto de la forma farmacéutica descrita en esta memoria, es un desafío retardar la liberación del segundo principio activo. Una razón es que se requiere una cantidad comparativamente elevada de emulsionante para disol ver el primer principio activo. Esto conduce al hecho de que el volumen de la forma farmacéutica ya está casi agota do debido a la medida necesaria que mejora la solubilidad, en relación con el primer principio activo. Por tanto, la medida para la liberación retardada del segundo principio activo debe ser posible con un volumen mínimo.
Tampoco se prefiere un recubrimiento simple de la forma farmacéutica completa, porque ese recubrimiento evitaría la liberación de ambos principios activos. Por tanto, de acuerdo con la invención, la forma farmacéutica comprende dos compartimentos, comprendiendo el primer compartimento al menos el primer principio activo y al menos un emulsionante. El segundo compartimento contiene el segundo principio activo. Para que el segundo principio activo se libere durante un período más largo, el segundo compartimento contiene preferiblemente un agente retardante. En una realización preferida de esta invención, la forma farmacéutica comprende al menos dos compartimentos en forma de capas. Para ello, una de las capas es preferiblemente el primer compartimento y otra de las capas es el segundo compartimento. La forma farmacéutica también puede incluir otras capas, en particular una tercera capa. En una realización de esta invención, la forma farmacéutica de esta invención comprende una cubierta y un núcleo. Para ello, el núcleo puede ser un compartimento de esta invención, en particular el primer compartimento. La cubier ta puede ser otro compartimento, en particular el segundo compartimento. Preferiblemente, la cubierta rodea com pletamente el núcleo. En otras palabras, el núcleo se encuentra completamente dentro de la cubierta.
En principio, todos los principios activos se pueden beneficiar de la administración en la forma farmacéutica de acuerdo con la invención, que se deben administrar durante un período de tiempo prolongado. Ejemplos de princi pios activos y clases de principios activos que se pueden usar ventajosamente como segundo principio activo en el
contexto de la presente invención, son:
Fármacos para el tratamiento del dolor y para la terapia del dolor con analgésicos de acción periférica, analgésicos de acción central y no analgésicos adyuvantes. Se trata preferiblemente en este caso de los siguientes analgésicos y sustancias adyuvantes:
ácido acetilsalicílico, ibuprofeno, diclofenaco, indometacina, naproxeno, piroxicam, paracetamol, metamizol, celecoxib, parecoxib, tramadol, petidina, codeína, dihidrocodeína, piritramida, tilidina, morfina, hidromorfona, oxicodona, levometadona, fentanilo, sufentanilo, buprenorfina, pentazocina, naloxona, flupirtina, carbamazepina, metoprolol, metoclopramida, amitriptilina, doxepina, clomipramina, mianserina, maprotilina, triptano tal como, p. ej., naratriptán, rizatriptán, sumatriptán, zolmitriptán, antagonistas de calcio tales como: flunarizina, topiramat, ácido valproico, fenitoína, baclofeno, otros agentes tales como: toxina botulínica, ergotamina, lisurida, metisergida, pizotifeno, oxcarbazepina, gabapentina y lamotrigina, dexametasona, metilprednisolona, prednisolona, triamcinolona, diazepam, tetrazepam, tizanidina, butilescopolamina y/o una combinación de tilidina y naloxona.
Fármacos para el tratamiento del sistema nervioso, solos o en combinación, p. ej., trastornos convulsivos (especial mente clonazepam, diazepam, lorazepam, midazolam, clobazam, fenitoína, clometiazol, ácido valproico, fenobarbital, gabapentina, lamotrigina, oxcarbazepina, pregabalina, topiramato, etosuximida, levetrazetam, mesuximida, primidona, nitrazepam y/o vigabitrina), síndrome de Parkinson (en particular levodopa, con benserazida/carbidopa, bromocriptina, cabergolina, dihidroergocriptina, lisurida, mesilato de pergolida, pramipexol, ropinirol, apomorfina, biperideno, clorhidrato de metixeno, trihexfenidilo, entacapona, amantadina, budidina, selegilina y/o apomorfina), ictus (en particular ácido acetilsalicílico, clopidogrel, dipiridamol, ticlopidina, heparina, fenprocumona, warfarina, protamina, fitomenadiona, nimodipina, paracetamol, tramadol y/o buprenorfina), presión intracraneal (especialmente furosemida y/o manitol), temblor (especialmente propranolol, clozapina, alprazidolam y/o primidona).
Fármacos para el tratamiento de enfermedades psiquiátricas, tales como trastornos de ansiedad (en particular, alprazolam, diazepam, fluoxetina, paroxetina, clorprotixeno, levomepromazina, tioridazina, flupentixol y/o fluespirileno), depresiones (en particular, imipramina, amitriptilina, desipramina, maprotilina, minaserina, citalopram, fluoxeti na, paroxetina, trazodona, moclobemid y/o miratazepam), psicosis y esquizofrenias (en particular, sulpirida, promazina, melperona, tioridazina, clorprotixeno, perazina, pimozida, flufenazina, olanzapina y/o risperidona), trastornos del sueño (en particular, triazolam, brotizolam, oxazepam, flurazepam, nitrazepam, temazepam, tartrato de zolpidem, zopiclona, prometazina, clorprotixeno, pipamperona, tioridazina y/o hidrato de cloral), inquietud y confusión (en parti cular, alprazolam, oxazepam, doxepina, clomipramina, imipramina, tioridazina y/o perazina), demencia de tipo Alzheimer (en particular, donepezil, rivastigmina, tacrina, memantina, nimodipina y/o seleginina).
Fármacos para el tratamiento de enfermedades cardiovasculares, tales como cardiopatía coronaria/angina de pecho (en particular, ácido acetilsalicílico, clopidrogel, ticlopidina, dinitrato de isosorbida, mononitrato de isosorbida, nitro glicerina, molsidomina, trapidil, metoprolol, bisoprolol, atenolol, acebutolol, carvedilol, nitrendipina, nifedipina, diltiazem, verapamilo, benazepril, lisinopril, ramipril, fosinopril y/o enalapril), infarto de miocardio e insuficiencia cardía ca (en particular, mononitrato y dinitrato de isosorbida, clopidogrel, ticlopidina, captoprol, ramipril, lisinopril, candesartán, eprosartán, irbesatán, losartán, clortalidona, xipamida, hidroclorotiazida, furosemida, piretanida, triametereno, glicósidos de digitalis, carvedilol, metoprolol y/o prazosina), arritmias cardíacas (en particular, ajmalina, quinidina, disopiramida, flecainida, propafenona, propranolol, carvedilol, amiodarona, verapamilo y/o diltiazem), hipertensión (en particular, metoprolol, atenolol, urapidil, clonidina, dihidralazina, clortalidona, hidroclorotiazida, furosemida, felodipina, israpidina, lacidipina, diltiazem, captopril, enalapril, fosinopril, lisinopril, ramnipril, verapamilo, candesartán, eprosartán, irbesatán, losartán, doxazosina, bunazosina, prazosina, terazosina, moxonidina, dihidralazina y/o minoxidil).
Fármacos, solos o en combinación, para el tratamiento de las vías respiratorias y los pulmones (en particular, teofilina, metilprednisolona, flucortona, dexametasona, montekulast, roxitromicina, eritromicina, azitromicina, ciprofloxacina, claritromicina, levofloxacina, ofloxacina, doxiciclina, ampicilina y sulbactam, amoxicilina, cefuroxima, clindamicina, cefotiam, cefuroxima, ceftazidima, ceftriaxona, piperacilina y/o moxifloxacina).
Fármacos para el tratamiento del tracto gastrointestinal y el páncreas (en particular, fluconazol, mesalazina, sulfasalazina, budenosida, azatioprina, prednisona, metronidazol, infliximab, loperamida, cotrimoxazol, ciprofloxacina, metronidazol, vancomicina, esomeprazol, lansoprazol, pantoprazol, rabeprazol, cimetidina, famotidina, ranitidina, nizatidina, sucralfato, misoprostol, metoclopramida, pirenzepina, bisacodilo, domperidona, sulpirida, alizaprida, dimenhidrinato, cinarizina, flunarizina, levomeprazina, ondansetrón, betahistina y/o aprepitant).
Fármacos para protegerse de una infección con antibióticos o sustancias antivirales eficaces (en particular, aciclovir, amantadina, azitromicina, bacampicilina, cefaclor, cefazolina, cefixima, cefprozilo, ceftriaxona, cloroquina, ciprofloxa cina, clotrimazol, dicloxacilina, doxiciclina, econazol, eritromicina, etambutol, fosfomicina, flucloxacilina, fluconazol, ácido fusídico, gramicidina, idoxuridina, indinavir, interferón, itraconazol, isoniazida, josamicina, ketoconazol, lamivudina, lomefloxacina, mafenida, mebendazol, mesalazina, mezlozilina, mupirocina, miconazol, naftifina, ácido nalidíxico, norfloxacina, ofloxacina, oxacilina, oxitetraciclina, piperacilina, praziquantel, primaquim, proguanil, ribavirina, rifabutina, rimantadina, roxotromicina, saquinavir, espectinomicina, espriramicina, estavudina, sulbactam, teicoplanina, terbinafina, tetraciclina, tetroxoprim, ticarcilina, tinidazol, tromantadina, tolnaftato, vancomicina, zidovudina y/o
zalcitabina).
Fármacos para el tratamiento de la disfunción eréctil (especialmente sildenafilo, tadalafilo, vardenafilo, teobromina, cafeína y/o teofilina).
En realizaciones preferidas, la forma farmacéutica de acuerdo con la invención no comprende antibióticos en una cantidad farmacéuticamente eficaz, en particular ningún representante de la clase de principios activos de las tetraciclinas. Los antibióticos afectan a la flora intestinal al destruir bacterias importantes en el intestino. En la forma far macéutica de esta invención, este efecto daría lugar a fuertes efectos secundarios porque la forma farmacéutica en parte solo se disuelve muy tarde.
En realizaciones preferidas, la forma farmacéutica de acuerdo con la invención no incluye inhibidores de la bomba de protones. Los inhibidores de la bomba de protones aumentan el valor del pH en el estómago y, por tanto, interfie ren en la disolución de muchos fármacos de la primera unidad que contiene principio activo, en particular los princi pios activos débilmente básicos.
El segundo principio activo está contenido preferiblemente en el segundo compartimento en forma de pequeñas partículas. Esto significa que el tamaño medio de partícula del segundo principio activo preferiblemente no excede de 1000 nm. Más preferiblemente, el tamaño medio de partícula del segundo principio activo no es superior a 300 nm. El tamaño de partícula del principio activo se determina mediante difracción láser.
El segundo principio activo puede ser un principio activo que sea fácilmente soluble en agua. El segundo principio activo es en particular un principio activo que tiene al menos un grupo hidroxilo, al menos un grupo carboxilo y/o al menos una carga permanente. Un principio activo preferido es el dimenhidrinato.
El segundo principio activo se usa preferiblemente en una cantidad de al menos 50 mg y más preferiblemente de al menos 80 mg o, de forma particularmente preferible, de al menos 100 mg, en la forma farmacéutica. Las cantidades del segundo principio activo no deben exceder preferiblemente un valor de 400 mg, más preferiblemente 300 mg y de forma particularmente preferible 200 mg.
El agente retardante en el segundo compartimento de la forma farmacéutica de esta invención es preferiblemente un polímero, en particular un polisacárido. Se prefieren celulosas, derivados de celulosa, alginatos o mezclas de los mismos, pero también son posibles poliacrilatos, polimetacrilatos y sus copolímeros y mezclas. Los derivados de celulosa particularmente preferidos son metilcelulosa e hidroxipropilmetilcelulosa. Esos derivados de celulosa permi ten una liberación particularmente ventajosa del segundo principio activo y no muestran ningún comportamiento desfavorable dependiente del pH.
Se utiliza preferentemente un agente retardante el cual, en solución acuosa en una proporción de 2% en peso, a una temperatura de 20°C y una presión de 101,325 kPa, tiene una viscosidad de al menos 1500 mPas. La viscosidad se mide con un viscosímetro de caída de bola (DIN 53015). Dependiendo del retardo deseado en la liberación del se gundo principio activo, puede ser deseable también elegir viscosidades más altas. Cuanto mayor sea la viscosidad del agente retardante, mayor será el retardo de la liberación. En realizaciones particularmente preferidas, la viscosi dad del agente retardante es incluso de al menos 2500 mPas y de manera particularmente preferida de al menos 3500 mPas. Sin embargo, si la viscosidad es demasiado alta, el segundo principio activo se liberará demasiado lentamente. Esto conduciría a que el segundo principio activo podría no liberarse completamente durante su paso por el tracto gastrointestinal. Por lo tanto, la viscosidad debería limitarse a un máximo de 200.000 mPas, más prefe riblemente a un máximo de 120.000 mPas.
La proporción de ese agente retardante en el segundo compartimento debería ser preferiblemente de al menos 10% en peso, más preferiblemente de al menos 20% en peso y de manera particularmente preferida de al menos 30% en peso. Si se emplea una cantidad demasiado baja del agente retardante, no se consigue el efecto de acuerdo con la invención. Si las cantidades son demasiado elevadas, el segundo principio activo no se libera con la suficiente rapi dez. Por lo tanto, la proporción de agente retardante en el segundo compartimento no debe exceder un valor de 60% en peso, más preferiblemente de 50% en peso y de forma particularmente preferible de 40% en peso.
Además, se puede añadir un alcohol de cadena larga al primer y/o al segundo compartimento. "Cadena larga" indica en este caso un alcohol con una longitud de cadena de al menos 10 átomos de carbono, más preferiblemente al menos 12 átomos de carbono y de forma particularmente preferible, al menos 14 átomos de carbono. Sin embargo, la longitud de la cadena no debe exceder un valor de preferiblemente 20 átomos de carbono, más preferiblemente 18 y en particular 16 átomos de carbono. Se ha mostrado que la adición de un alcohol de cadena larga es adecuada para retrasar la liberación de los principios activos, en particular del segundo principio activo.
La proporción en peso del alcohol de cadena larga en el segundo compartimento de esta invención es preferible mente de al menos 5% en peso, más preferiblemente de al menos 10% en peso y de forma particularmente preferi ble, de al menos 18% en peso. En caso de una proporción de alcohol de cadena larga en el segundo compartimento demasiado baja, no se logra el efecto deseado. En caso de un contenido demasiado alto, la integridad estructural de la forma farmacéutica puede verse afectada. Por tanto, el contenido en alcohol de cadena larga en el segundo com partimento es preferiblemente como máximo de 40% en peso, más preferiblemente como máximo de 35% en peso y
de forma particularmente preferible, como máximo de 30% en peso.
El segundo compartimento en la forma farmacéutica de acuerdo con la invención también puede contener otros coadyuvantes. Esos coadyuvantes se pueden seleccionar preferiblemente a partir de agentes lubricantes, agentes deslizantes, agentes aglutinantes, agentes desintegrantes, antioxidantes, agentes formadores de complejos, agentes de revestimiento, agentes de flujo, conservantes, cargas, plastificantes, pigmentos y mezclas de esas sustancias. Una realización particularmente preferida de la forma farmacéutica de acuerdo con la invención comprende cinarizina como primer principio activo, se prefiere de forma muy particular que el primer principio activo sea cinarizina y el segundo principio activo sea dimenhidrinato. La forma farmacéutica es adecuada para un uso en la terapia del vérti go de cualquier origen. Por tanto, el uso de esta forma farmacéutica para el tratamiento del vértigo de cualquier origen también está de acuerdo con la invención.
Una gran ventaja de la forma farmacéutica de acuerdo con la invención es que se puede preparar de forma econó mica.
El procedimiento para preparar la forma farmacéutica comprende preferiblemente las etapas:
a. Mezclar los componentes de la forma farmacéutica,
b. Granular la mezcla y
c. Preparar una forma farmacéutica monolítica a partir del granulado.
La mezcla de los componentes de la forma farmacéutica tiene lugar en realizaciones en las que la forma farmacéuti ca comprende al menos dos compartimentos, preferiblemente mezclando los componentes de cada compartimento por separado para obtener al menos dos mezclas de compartimentos. En realizaciones en las que la forma farma céutica comprende al menos dos compartimentos, la granulación de la mezcla comprende preferiblemente granular las al menos dos mezclas de compartimentos para obtener al menos dos granulados de compartimentos. La prepa ración de una forma farmacéutica monolítica a partir del granulado comprende, en realizaciones en las que la forma farmacéutica comprende al menos dos compartimentos, preferiblemente la compresión del granulado de al menos dos compartimentos.
Los ingredientes de cada compartimento se mezclan preferiblemente y se someten a una granulación en estado fundido. La forma farmacéutica de acuerdo con la invención preferiblemente no tiene ningún recubrimiento. En reali zaciones alternativas, la forma farmacéutica tiene un recubrimiento que se disuelve fácilmente.
En la preparación de los dos compartimentos, los componentes del primer compartimento se mezclan preferiblemen te entre sí y a partir de ellos se produce un granulado en estado fundido. Lo mismo se hace preferiblemente con los componentes del segundo compartimento. Sin embargo, el granulado también se puede producir con la ayuda de un proceso de pulverización.
A continuación se prepara una forma farmacéutica a partir de los granulados. En el caso de un comprimido en ca pas, el granulado se comprime en una prensa para comprimidos.
En realizaciones preferidas del procedimiento de preparación, el primer principio activo se mezcla con el emulsio nante para preparar el primer compartimento. El principio activo se disuelve preferiblemente en el emulsionante. Opcionalmente, se puede utilizar un disolvente. Después se mezcla el principio activo, el emulsionante y el disolven te. Dependiendo de la naturaleza exacta del principio activo y el emulsionante, son posibles diferentes disolventes. Se da preferencia al uso de coadyuvantes habituales en la tecnología farmacéutica, en particular alcoholes, ésteres y/o cetonas, en particular acetona.
A continuación, esta mezcla se pulveriza preferiblemente sobre un vehículo. El vehículo puede ser el coadyuvante para el prensado descrito anteriormente y/o el agente retardante. La pulverización puede tener lugar preferiblemente en un sistema de lecho fluidizado.
En una realización alternativa, el principio activo se mezcla con el emulsionante, en particular en una mezcladora de circulación forzada, y se funde. Esta mezcla se aplica luego sobre el vehículo, que puede ser el coadyuvante para el prensado y/o el agente retardante.
Los granulados preparados por el procedimiento de fusión o el procedimiento alternativo de pulverización, antes de la compresión se pueden mezclar con coadyuvantes tales como agentes lubricantes y auxiliares de flujo y después comprimirlos directamente para formar los compartimentos deseados.
Si es necesario, el principio activo se muele previamente, en particular en un nano molino. El polvo obtenido de ese modo se incorpora preferiblemente en el emulsionante. Para la incorporación se utiliza un disolvente en el que no se disuelva el principio activo, en particular agua. La incorporación evita una aglomeración de nuevo de las partículas del principio activo.
El segundo compartimento se puede preparar de la misma forma. Preferiblemente, se produce un granulado de principio activo y agente retardante, por ejemplo, por medio de una granulación en húmedo, y ese granulado se comprime luego opcionalmente con coadyuvantes para formar el segundo compartimento.
Ejemplos
Ejemplo 1
Se prepararon formas farmacéuticas con las siguientes composiciones (cantidades en mg en cada caso):

Los ingredientes especificados se mezclaron y luego se procesaron en un granulado por medio de una granulación en estado fundido convencional y después se prensaron para formar comprimidos. La liberación se determinó a pH 5,5 usando un aparato con paletas.
La Figura 1 muestra la influencia de la cantidad relativa de emulsionante en la forma farmacéutica sobre el compor tamiento en disolución. La ordenada muestra la cantidad de principio activo liberado en porcentaje, la abscisa los minutos transcurridos. Se puede observar que la cantidad de emulsionante también aumenta la cantidad de principio activo liberado.
Ejemplo 2
Se preparó una forma farmacéutica que tenía la siguiente composición:

En primer lugar, los ingredientes del primer compartimento se mezclaron entre sí y se procesaron para formar un granulado mediante granulación en estado fundido. A continuación, los ingredientes del segundo compartimento se procesaron también de la misma forma para formar un granulado en estado fundido. A continuación, los dos granu lados se comprimieron juntos en una prensa para comprimidos para formar una forma farmacéutica con dos compar timentos.
Ejemplo 3
El siguiente ejemplo muestra la influencia de un triglicérido sobre la liberación de principio activo del principio activo poco soluble. El experimento se llevó a cabo con un valor de pH de 5,5 en un aparato con paletas. Las cantidades de los componentes se expresan en mg.
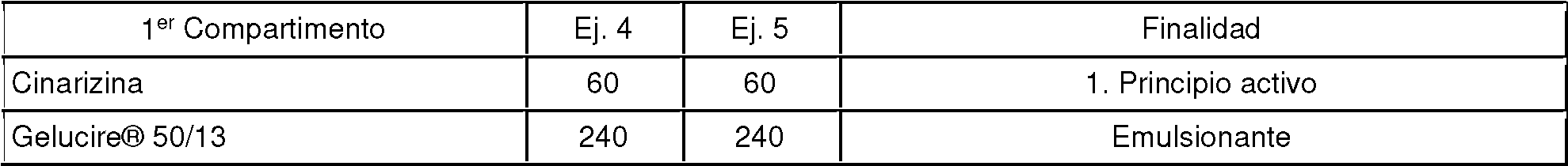

La Figura 2 muestra la influencia del triglicérido en la forma farmacéutica sobre el comportamiento en disolución del primer principio activo. Se puede observar que el triglicérido aumenta la liberación de cinarizina. Según el Ejemplo 5, 23,6 mg de cinarizina ya estaban en solución después de 480 min. Esto es sorprendente y ventajoso ya que la cinarizina solo se disuelve con un valor de pH de 5,5 (tampón acuoso) hasta un máximo de 1 a 2 mg. La ordenada muestra la cantidad de principio activo liberado en porcentaje, la abscisa los minutos transcurridos.
Ejemplo 4
El siguiente ejemplo muestra experimentos adicionales para determinar la influencia del emulsionante sobre la canti dad de principio activo poco soluble disuelta. Para ello, las siguientes composiciones se comprimieron en forma de comprimidos (cantidades en mg en cada caso). La prueba se llevó a cabo a pH 5,5 en un sistema Bio-Dis (30 minu tos con 15 inmersiones/minuto). El recipiente se cambió cada 30 minutos. Las soluciones se evaluaron fotométricamente.
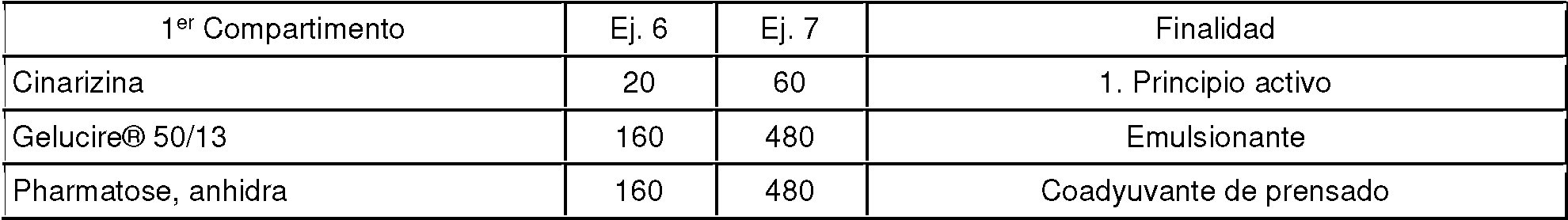
Se obtuvieron los siguientes valores para la cantidad de cinarizina disuelta:

Es sorprendente que cuando se aumenta la cantidad total de principio activo, también aumenta la cantidad absoluta de principio activo que se disuelve. Esto deja en claro que la cantidad de cinarizina liberada está influenciada por el emulsionante y no está restringida por una concentración límite.
Ejemplo 5
El siguiente ejemplo muestra experimentos para formular un segundo compartimento de la forma farmacéutica de acuerdo con la invención. Para ello, se prepararon comprimidos en capas con dimensiones de 18 x 8 mm, mediante el procedimiento de preparación de acuerdo con la invención, con las siguientes composiciones (cantidades en cada caso en mg), utilizando una mezcla de placebo para el 1. Compartimento.

La Figura 3 muestra la liberación de dimenhidrinato desde la forma farmacéutica. La ordenada muestra la cantidad de principio activo liberado en porcentaje, la abscisa los minutos transcurridos. Se puede observar que el dimenhi
drinato se libera inicialmente de forma rápida desde el segundo compartimiento en 60 minutos, después el principio activo se libera más lentamente y casi de forma lineal. Se logró un retardo del principio activo en el segundo compar timento de 10 horas.
Claims (10)
1. Forma farmacéutica con al menos un primer principio activo y al menos un emulsionante, en donde el emulsionan te son glicéridos de polietilenglicol que se preparan mediante una reacción de monoglicéridos, diglicéridos y/o triglicéridos con polietilenglicol, en donde la cadena de polietilenglicol es la parte hidrófila del emulsionante y está conec tada con una parte lipófila, en donde el emulsionante tiene un valor HLB de al menos 1 y como máximo 16, en donde la relación en masa entre principio activo y emulsionante es de al menos 1 a 30 y como máximo de 1 a 1, y en donde la forma farmacéutica es un comprimido en capas que comprende al menos dos capas, en donde la prime ra capa es un primer compartimento y la segunda capa es un segundo compartimento,
en donde el primer compartimento comprende al menos el primer principio activo y al menos el emulsionante, y el emulsionante no está presente en el segundo compartimento,
en donde el primer principio activo es cinarizina o una sal de cinarizina.
2. Forma farmacéutica según la reivindicación 1, en la que la relación en masa entre principio activo y emulsionante es inferior a 1 a 1.
3. Forma farmacéutica según la reivindicación 1, en la que los compartimentos contienen diferentes principios acti vos.
4. Forma farmacéutica según al menos una de las reivindicaciones precedentes, en donde la forma farmacéutica contiene un segundo principio activo en el segundo compartimento.
5. Forma farmacéutica según la reivindicación 4, en la que el segundo principio activo se disuelve en agua en una cantidad superior a 0,5 mol/l.
6. Forma farmacéutica según la reivindicación 5, en la que el segundo compartimento contiene un agente retardante.
7. Forma farmacéutica según la reivindicación 5 o 6, en la que el segundo principio activo es dimenhidrinato.
8. Forma farmacéutica según al menos una de las reivindicaciones precedentes, en donde la forma farmacéutica es un comprimido en capas en forma de un comprimido de núcleo con recubrimiento.
9. Procedimiento para la preparación de una forma farmacéutica según al menos una de las reivindicaciones prece dentes, con las etapas de:
a. mezclar los componentes de la forma farmacéutica,
b. granular la mezcla y
c. preparar una forma farmacéutica monolítica a partir del granulado.
10. Forma farmacéutica según una de las reivindicaciones 1 a 8, para uso en un procedimiento terapéutico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102012105512.2A DE102012105512A1 (de) | 2012-06-25 | 2012-06-25 | Arzneiform zur verlängerten Freisetzung von Wirkstoffen |
| PCT/EP2013/063163 WO2014001268A1 (de) | 2012-06-25 | 2013-06-24 | Arzneiform zur verlängerten freisetzung von wirkstoffen |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2817248T3 true ES2817248T3 (es) | 2021-04-06 |
Family
ID=48670585
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13730598T Active ES2817248T3 (es) | 2012-06-25 | 2013-06-24 | Forma farmacéutica para la liberación prolongada de principios activos |
Country Status (22)
| Country | Link |
|---|---|
| EP (1) | EP2863891B1 (es) |
| KR (1) | KR102160837B1 (es) |
| AU (1) | AU2013283474B2 (es) |
| BR (1) | BR112014032583B1 (es) |
| CA (1) | CA2876294C (es) |
| CO (1) | CO7240370A2 (es) |
| DE (1) | DE102012105512A1 (es) |
| EA (2) | EA028064B1 (es) |
| ES (1) | ES2817248T3 (es) |
| HR (1) | HRP20201534T1 (es) |
| HU (1) | HUE050945T2 (es) |
| IL (1) | IL236465B (es) |
| IN (1) | IN2014MN02511A (es) |
| LT (1) | LT2863891T (es) |
| MX (1) | MX371143B (es) |
| PH (1) | PH12014502834A1 (es) |
| PL (1) | PL2863891T3 (es) |
| PT (1) | PT2863891T (es) |
| RS (1) | RS60872B1 (es) |
| SI (1) | SI2863891T1 (es) |
| UA (1) | UA116776C2 (es) |
| WO (1) | WO2014001268A1 (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| HUE043399T2 (hu) * | 2014-06-26 | 2019-08-28 | Hennig Arzneimittel Gmbh&Co Kg | Gyógyszer, különbözõ okokból eredõ szédülés kezelésére |
| JP6371463B2 (ja) | 2014-07-17 | 2018-08-08 | ファーマシューティカル マニュファクチュアリング リサーチ サービシズ,インコーポレーテッド | 即時放出性乱用抑止性液体充填剤形 |
| EP3209282A4 (en) | 2014-10-20 | 2018-05-23 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| KR101625344B1 (ko) * | 2015-12-21 | 2016-06-08 | 주식회사 유영제약 | 세레콕시브 및 둘록세틴을 함유하는 약제학적 조성물 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9405304D0 (en) * | 1994-03-16 | 1994-04-27 | Scherer Ltd R P | Delivery systems for hydrophobic drugs |
| IE80467B1 (en) * | 1995-07-03 | 1998-07-29 | Elan Corp Plc | Controlled release formulations for poorly soluble drugs |
| JP2001520984A (ja) * | 1997-10-27 | 2001-11-06 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | 水難溶性薬剤の固態溶剤及び固体分散体 |
| US6368622B2 (en) * | 1999-01-29 | 2002-04-09 | Abbott Laboratories | Process for preparing solid formulations of lipid regulating agents with enhanced dissolution and absorption |
| US7374779B2 (en) * | 1999-02-26 | 2008-05-20 | Lipocine, Inc. | Pharmaceutical formulations and systems for improved absorption and multistage release of active agents |
| CA2551254A1 (en) * | 2003-12-31 | 2005-07-21 | Pfizer Products Inc. | Stabilized pharmaceutical solid compositions of low-solubility drugs, poloxamers, and stabilizing polymers |
| US20050281876A1 (en) * | 2004-06-18 | 2005-12-22 | Shun-Por Li | Solid dosage form for acid-labile active ingredient |
| DE102005014141B4 (de) * | 2005-03-23 | 2006-12-21 | Hennig Arzneimittel Gmbh & Co. Kg | Tablettenförmige Retardzubereitung gegen Schwindel |
| EP1942888A2 (en) | 2005-11-02 | 2008-07-16 | Cytokinetics, Inc. | Certain chemical entities, compositions, and methods |
| WO2007086078A2 (en) * | 2006-01-30 | 2007-08-02 | Panacea Biotec Ltd. | Novel pharmaceutical compositions and process of preparation thereof |
| CN101049309A (zh) * | 2006-04-03 | 2007-10-10 | 陈茜 | 桂利嗪滴丸及其制备方法 |
| WO2009040818A1 (en) * | 2007-09-25 | 2009-04-02 | Solubest Ltd | Compositions comprising lipophilic active compounds and method for their preparation |
| JP5646340B2 (ja) * | 2007-12-12 | 2014-12-24 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | ポリマー性対イオンと活性成分との塩 |
| PE20120858A1 (es) * | 2009-06-11 | 2012-08-01 | Photocure Asa | Composiciones solidas que comprenden acido 5-aminolevulinico |
| WO2011024029A1 (en) * | 2009-08-24 | 2011-03-03 | Abdi Ibrahim Ilac Sanayi Ve Ticaret Anonim Sirketi | Fast disintegrating dosage forms of cinnarizine and dimenhydrinate combination |
| DE102011051304A1 (de) * | 2011-06-24 | 2012-12-27 | Hennig Arzneimittel Gmbh & Co. Kg | Wirkstoffmatrix |
| DE102011051308A1 (de) * | 2011-06-24 | 2012-12-27 | Hennig Arzneimittel Gmbh & Co. Kg | Herstellungsverfahren und Arzneiform |
| DE102011053068A1 (de) * | 2011-08-29 | 2013-02-28 | Hennig Arzneimittel Gmbh & Co. Kg | Darreichungsform mit stabilisierten Wirkstoffpartikeln |
-
2012
- 2012-06-25 DE DE102012105512.2A patent/DE102012105512A1/de not_active Ceased
-
2013
- 2013-06-24 HU HUE13730598A patent/HUE050945T2/hu unknown
- 2013-06-24 KR KR1020147036365A patent/KR102160837B1/ko active IP Right Grant
- 2013-06-24 IN IN2511MUN2014 patent/IN2014MN02511A/en unknown
- 2013-06-24 EP EP13730598.3A patent/EP2863891B1/de active Active
- 2013-06-24 PT PT137305983T patent/PT2863891T/pt unknown
- 2013-06-24 CA CA2876294A patent/CA2876294C/en active Active
- 2013-06-24 AU AU2013283474A patent/AU2013283474B2/en active Active
- 2013-06-24 LT LTEP13730598.3T patent/LT2863891T/lt unknown
- 2013-06-24 SI SI201331778T patent/SI2863891T1/sl unknown
- 2013-06-24 EA EA201500043A patent/EA028064B1/ru not_active IP Right Cessation
- 2013-06-24 EA EA201792143A patent/EA035815B1/ru unknown
- 2013-06-24 UA UAA201500503A patent/UA116776C2/uk unknown
- 2013-06-24 PL PL13730598T patent/PL2863891T3/pl unknown
- 2013-06-24 BR BR112014032583-9A patent/BR112014032583B1/pt active IP Right Grant
- 2013-06-24 MX MX2014015642A patent/MX371143B/es active IP Right Grant
- 2013-06-24 ES ES13730598T patent/ES2817248T3/es active Active
- 2013-06-24 RS RS20201085A patent/RS60872B1/sr unknown
- 2013-06-24 WO PCT/EP2013/063163 patent/WO2014001268A1/de active Application Filing
-
2014
- 2014-12-19 PH PH12014502834A patent/PH12014502834A1/en unknown
- 2014-12-25 IL IL236465A patent/IL236465B/en active IP Right Grant
-
2015
- 2015-01-15 CO CO15007539A patent/CO7240370A2/es unknown
-
2020
- 2020-09-25 HR HRP20201534TT patent/HRP20201534T1/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| BR112014032583A2 (pt) | 2017-06-27 |
| WO2014001268A1 (de) | 2014-01-03 |
| EP2863891B1 (de) | 2020-08-26 |
| PT2863891T (pt) | 2020-09-25 |
| IL236465A0 (en) | 2015-02-26 |
| HRP20201534T1 (hr) | 2021-02-19 |
| UA116776C2 (uk) | 2018-05-10 |
| PH12014502834B1 (en) | 2015-02-02 |
| LT2863891T (lt) | 2020-12-10 |
| HUE050945T2 (hu) | 2021-01-28 |
| DE102012105512A1 (de) | 2014-04-24 |
| BR112014032583A8 (pt) | 2021-06-22 |
| EP2863891A1 (de) | 2015-04-29 |
| AU2013283474B2 (en) | 2017-12-14 |
| AU2013283474A1 (en) | 2015-01-29 |
| PH12014502834A1 (en) | 2015-02-02 |
| CA2876294A1 (en) | 2014-01-03 |
| PL2863891T3 (pl) | 2021-01-25 |
| EA201500043A1 (ru) | 2015-09-30 |
| IL236465B (en) | 2020-06-30 |
| CO7240370A2 (es) | 2015-04-17 |
| RS60872B1 (sr) | 2020-11-30 |
| KR102160837B1 (ko) | 2020-09-29 |
| MX371143B (es) | 2020-01-20 |
| EA035815B1 (ru) | 2020-08-14 |
| SI2863891T1 (sl) | 2020-11-30 |
| MX2014015642A (es) | 2015-08-05 |
| EA201792143A1 (ru) | 2018-05-31 |
| CA2876294C (en) | 2020-09-08 |
| KR20150041612A (ko) | 2015-04-16 |
| BR112014032583B1 (pt) | 2022-05-17 |
| IN2014MN02511A (es) | 2015-07-17 |
| EA028064B1 (ru) | 2017-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2817248T3 (es) | Forma farmacéutica para la liberación prolongada de principios activos | |
| Gopalakrishnan et al. | Floating drug delivery systems: A Review | |
| ES2922387T3 (es) | Microparticulas recubiertas de liberacion modificada de al menos un principio activo y forma galenica oral que las comprende | |
| ES2692450T3 (es) | Formas farmacéuticas de multimicropartículas que resisten a la descarga inmediata del principio activo en presencia de alcohol | |
| KR102546742B1 (ko) | 붕해가 개선된 경구용 고형 제제 조성물 및 이의 제조 방법 | |
| US20130217777A1 (en) | Process for making multiparticulate gastroretentive dosage forms | |
| JPWO2011019045A1 (ja) | 崩壊性粒子組成物及び口腔内速崩壊錠 | |
| WO2007106960A1 (en) | Controlled-release floating dosage forms | |
| JP2011157348A (ja) | 崩壊性高強度球状粒子組成物 | |
| ES2566507T3 (es) | Comprimidos multiparticulados y procedimiento para su fabricación | |
| Kumari | Recent Development inFloating Drug Delivery System: A Review | |
| ES2870562T3 (es) | Composiciones y procedimientos para el tratamiento del crecimiento celular anormal | |
| KR102220130B1 (ko) | 유효 성분 조합물의 변형 방출을 위한 모놀리식 투여형 | |
| ES2755075T3 (es) | Forma farmacológica para la liberación dirigida de principios activos | |
| Kandwal et al. | Floating drug delivery system: A novel approach | |
| Dey et al. | A Complete Sojourn of Current Trends in Gastro-retentive Drug Delivery System: Recent Advances and Patent Survey | |
| Khanam et al. | A review on novel approaches incorporated in the formulation of gastro retentive drug delivery system | |
| WO2012175737A1 (de) | Wirkstoffmatrix | |
| WO2012175747A1 (de) | Herstellungsverfahren und arzneiform | |
| AU2018297656A1 (en) | Novel pharmaceutical composition | |
| Shilpa et al. | A brief review on floating drug delivery system | |
| Mistry et al. | TIME AND SITE SPECIFIC GASTRO-RETENTIVE FLOATING PULSATILE DRUG DELIVERY SYSTEM: A REVIEW | |
| Kumar et al. | A REVIEW ON FLOATING TABLET | |
| Khan et al. | RECENT TRENDS IN GASTRORETENTIVE FLOATING DRUG DELIVERY SYSTEM-A REVIEW | |
| Shah et al. | FLOATING MICROSPHERES AS GASTRORETENTIVE DRUG DELIVERY SYSTEMS: A REVIEW |