ES2811342T3 - Antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar glomeruloesclerosis y nefropatía inducida por IgA - Google Patents
Antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar glomeruloesclerosis y nefropatía inducida por IgA Download PDFInfo
- Publication number
- ES2811342T3 ES2811342T3 ES17157697T ES17157697T ES2811342T3 ES 2811342 T3 ES2811342 T3 ES 2811342T3 ES 17157697 T ES17157697 T ES 17157697T ES 17157697 T ES17157697 T ES 17157697T ES 2811342 T3 ES2811342 T3 ES 2811342T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- formulation
- day
- tablets
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC1=C(C)C(N(COC)*C2=CCCC=C=C2C)=C**1 Chemical compound CC1=C(C)C(N(COC)*C2=CCCC=C=C2C)=C**1 0.000 description 5
- HGCSZGGMKXHVGU-UHFFFAOYSA-N C=NC1(CCCC1)C(N)=O Chemical compound C=NC1(CCCC1)C(N)=O HGCSZGGMKXHVGU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2031—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Hospice & Palliative Care (AREA)
- Psychology (AREA)
Abstract
Un compuesto de Fórmula I: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de un trastorno seleccionado entre el grupo que consiste en glomeruloesclerosis y nefropatía inducida por IgA, en el que la cantidad administrada del compuesto de Fórmula I, o la sal farmacéuticamente aceptable del mismo, es de 200 mg a 800 mg.
Description
DESCRIPCIÓN
Antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar glomeruloesclerosis y nefropatía inducida por IgA
ANTECEDENTES DE LA INVENCIÓN
Campo de la invención
La presente invención se refiere a un compuesto de bifenilsulfonamida que es un antagonista doble de receptores de angiotensina y endotelina, a composiciones farmacéuticas que contienen tal compuesto, a métodos de fabricación de las formulaciones farmacéuticas que contienen tal compuesto, y a métodos de uso de tal compuesto en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II y otras enfermedades. Descripción de la técnica relacionada
La angiotensina II (AngII) y la endotelina-1 (ET-1) son dos de los péptidos vasoactivos endógenos más potentes conocidos en la actualidad, y se cree que desempeñan un papel en el control del tono vascular y la remodelación tisular patológica asociada a una diversidad de enfermedades, que incluyen nefropatía diabética, insuficiencia cardiaca, e hipertensión arterial crónica o persistente. En la actualidad, los bloqueantes de receptores de angiotensina (ARB), que bloquean la actividad de AngII, se usan de manera generalizada como tratamiento para nefropatía diabética, insuficiencia cardiaca, e hipertensión arterial crónica o persistente. Además, existe un conjunto de datos creciente que demuestra los beneficios terapéuticos potenciales de los antagonistas de receptores de ET (ERA) en el bloqueo de la actividad de ET-1.
También se sabe que AngII y ET-1 funcionan en conjunto en el control de la tensión arterial y la remodelación tisular patológica. Por ejemplo, los ARB no solamente bloquean la acción de AngII en su receptor, sino que también limitan la producción de ET-1. De forma similar, los ERA bloquean la actividad de ET-1 e inhiben la producción de AngII. Por lo tanto, el bloqueo simultáneo de las actividades de AngII y ET-1 puede ofrecer una mejor eficacia que el bloqueo de cada sustancia por sí sola.
En modelos en ratas bien validados de hipertensión arterial humana crónica o persistente, la combinación de un ARB y un ERA da como resultado un efecto sinérgico. Además, aunque los ARB son el tratamiento de referencia para los pacientes con nefropatía diabética, se ha informado de una eficacia mejorada con la coadministración de un ERA en el desarrollo clínico de Fase 2.
Existen datos preclínicos y clínicos iniciales que sugieren que en comparación con cada mecanismo por sí solo, el bloqueo simultáneo de angiotensina II y endotelina 1 en sus receptores respectivos, ATI y ETA, puede proporcionar una opción de tratamiento mejorada para varias enfermedades cardiovasculares.
Los documentos de Patente US 2002/143024 y WO 00/01389 se refieren a compuestos de bifenilsulfonamida que son antagonistas combinados de receptores de angiotensina y endotelina, y a métodos de uso de tales compuestos en el tratamiento de afecciones tales como hipertensión y otras enfermedades, así como a composiciones farmacéuticas que contienen tales compuestos.
SUMARIO DE LA INVENCIÓN
La invención es como se define en las reivindicaciones, específicamente un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de glomeruloesclerosis y nefropatía inducida por IgA en el que la cantidad administrada del compuesto de Fórmula I, o sal farmacéuticamente aceptable
del mismo, es de 200 mg a 800 mg.
La invención también proporciona el uso de un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo en la fabricación de un medicamento para el tratamiento de un trastorno seleccionado entre el grupo que consiste en glomeruloesclerosis y nefropatía inducida por IgA en el que la cantidad administrada del compuesto de Fórmula I, o sal farmacéuticamente aceptable del mismo, es de 200 mg a 800 mg.
En la medida en que en el presente documento se desvelan otros usos del compuesto o la sal farmacéuticamente aceptable del mismo, estos se incluyen meramente con fines descriptivos.
Se desvelan métodos de administración, formas farmacéuticas, formulaciones farmacéuticas, y regímenes de tratamiento de un compuesto de bifenilsulfonamida de la siguiente fórmula I, enantiómeros (incluyendo atropisómeros), diastereómeros, sales y metabolitos de los mismos, métodos de fabricación de formulaciones farmacéuticas, y métodos de uso de las formulaciones:

Algunas divulgaciones proporcionan un método para tratar un trastorno dependiente de endotelina o dependiente de angiotensina II en un sujeto que lo necesita, que comprende administrar un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para ello. En algunas otras divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II es nefropatía diabética. En algunas divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II es hipertensión arterial crónica. En algunas divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II es hipertensión arterial persistente. En algunas divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II es hipertensión.
En algunas divulgaciones, el método comprende administrar un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo en una cantidad de aproximadamente 50 mg/día a aproximadamente 1000 mg/día. En el contexto de algunas realizaciones de la invención, la cantidad del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, administrada al sujeto humano es de aproximadamente 200 mg/día a 800 mg/día, más preferentemente aproximadamente 400 mg/día, lo más preferentemente aproximadamente 800 mg/día. En algunas realizaciones de la invención, la cantidad del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, administrada al sujeto humano puede ser 200 mg/día, aproximadamente 400 mg/día, o 800 mg/día. En otras divulgaciones, la cantidad del compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, administrada al sujeto humano puede ser aproximadamente 100 mg/día.
En algunas realizaciones, la tensión arterial sistólica del sujeto humano disminuye debajo de al menos 160 mmHg,
debajo de al menos 140 mmHg, debajo de al menos 140 mmHg, debajo de al menos 130 mmHg, o debajo de al menos 120 mmHg. En algunas realizaciones, la tensión arterial diastólica del sujeto humano disminuye debajo de al menos 120 mmHg, debajo de al menos 110 mmHg, debajo de al menos 100 mmHg, o debajo de al menos 90 mmHg. En algunas realizaciones, la tensión arterial sistólica o diastólica del sujeto humano disminuye en al menos aproximadamente 5 mmHg, al menos aproximadamente 8 mmHg, aproximadamente 10 mmHg, al menos aproximadamente 12 mmHg, o aproximadamente 14 mmHg en comparación con la tensión arterial sistólica o diastólica antes del tratamiento.
En algunas realizaciones, el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, se administra con una frecuencia no mayor de cuatro veces, dos veces, o una vez al día. En algunas realizaciones, el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, se administra cuatro veces, dos veces, o una vez al día.
En algunas divulgaciones, la tensión arterial sistólica de menos de 140 mmHg o la tensión arterial diastólica de menos de 90 mmHg se alcanza en 16 semanas, en 14 semanas, en 12 semanas, en 10 semanas, o en 8 semanas de administración del compuesto I, o una sal farmacéuticamente aceptable del mismo.
Algunas divulgaciones proporcionan una composición farmacéutica que comprende un compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de un trastorno dependiente de endotelina o dependiente de angiotensina II en un sujeto que lo necesita. En algunas divulgaciones, la cantidad del compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo puede ser de aproximadamente 50 mg a aproximadamente 1000 mg. En algunas divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II puede ser hipertensión arterial persistente. En algunas divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II puede ser nefropatía diabética. En algunas divulgaciones, el trastorno dependiente de endotelina o dependiente de angiotensina II puede ser hipertensión. En el contexto de la invención, la cantidad de compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, es de 200 mg a 800 mg. En algunas realizaciones de la invención, la cantidad de compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, es 200 mg, aproximadamente 400 mg, u 800 mg. En otra divulgación, la cantidad del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, es aproximadamente 100 mg.
El uso de la composición farmacéutica que comprende un compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, en el tratamiento del trastorno dependiente de endotelina o dependiente de angiotensina II puede disminuir la tensión arterial sistólica del sujeto humano de forma que esté debajo de al menos 160 mmHg, debajo de al menos 150 mmHg, debajo de al menos 140 mmHg, debajo de al menos 130 mmHg, o debajo de al menos 120 mmHg. El uso de la composición farmacéutica que comprende un compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, en el tratamiento del trastorno dependiente de endotelina o dependiente de angiotensina II puede disminuir la tensión arterial diastólica del sujeto humano de forma que esté debajo de al menos 120 mmHg, debajo de al menos 110 mmHg, debajo de al menos 100 mmHg, o debajo de al menos 90 mmHg. El uso de la composición farmacéutica que comprende un compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, en el tratamiento del trastorno dependiente de endotelina o dependiente de angiotensina II puede disminuir la tensión arterial sistólica o diastólica del sujeto humano en al menos aproximadamente 5 mmHg, al menos aproximadamente 8 mmHg, al menos aproximadamente 10 mmHg, al menos aproximadamente 12 mmHg, o al menos aproximadamente 14 mmHg.
En algunas realizaciones, la composición farmacéutica del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, se administra con una frecuencia no mayor de cuatro veces, dos veces, o una vez al día. En algunas realizaciones, la composición farmacéutica del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, se administra cuatro veces, dos veces, o una vez al día.
En algunas realizaciones, la tensión arterial sistólica de menos de 140 mmHg o la tensión arterial diastólica de menos de 90 mmHg se alcanza en 16 semanas, en 14 semanas, en 12 semanas, en 10 semanas, o en 8 semanas de la administración de la composición farmacéutica del compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo.
0.
En algunas realizaciones, el compuesto de fórmula I se puede proporcionar en formas farmacéuticas de disolución rápida. En algunas realizaciones, las formulaciones pueden tener una o más de: friabilidad, compresión, disolución, uniformidad, solubilidad, palatabilidad mejoradas, y similares. Además, en algunas realizaciones las formulaciones pueden permitir al menos uno o más de: inicio rápido, niveles plasmáticos mayores y/o más rápidos, y similares.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo, y al menos un excipiente, en el que el compuesto de fórmula I comprende más de aproximadamente un 35 % p/p de la composición y los excipientes comprenden de aproximadamente un 5 % a aproximadamente un 65 % p/p de la composición. Cada excipiente se puede seleccionar individualmente entre el grupo que consiste en celulosa microcristalina, hidroxipropilcelulosa, poloxámero 188, almidón glicolato sódico, y croscarmelosa sódica. El compuesto de fórmula I se puede proporcionar
en una cantidad de aproximadamente un 40 % a aproximadamente un 65 % p/p de la composición, o aproximadamente un 50 % a aproximadamente un 60 % p/p de la composición. Un excipiente puede ser celulosa microcristalina. La celulosa microcristalina puede comprender de aproximadamente un 5 % a aproximadamente un 65 % p/p, de aproximadamente un 15 % a aproximadamente un 50 % p/p, de aproximadamente un 20 % a aproximadamente un 40 % p/p, de aproximadamente un 25 % a aproximadamente un 35 % p/p, o aproximadamente un 28 % a aproximadamente un 30 % p/p de la composición. La celulosa microcristalina puede ser celulosa microcristalina silicificada. Un excipiente es poloxámero 188. El poloxámero 188 puede comprender de aproximadamente un 0,1 % a aproximadamente un 10 % p/p, o de aproximadamente un 1 % a aproximadamente un 8 % p/p de la composición. El poloxámero 188 puede comprender aproximadamente un 5 % p/p de la composición.
La composición puede comprender además uno o más lubricantes, en la que los uno o más lubricantes pueden comprender hasta aproximadamente un 5 % p/p de la composición. Los uno o más lubricantes se pueden seleccionar cada uno individualmente entre el grupo que consiste en lauril sulfato sódico, estearato magnésico, estearato cálcico, estearil fumarato sódico, ácido esteárico, aceite vegetal hidrogenado, behenato de glicerilo, y polietilenglicol. Un lubricante puede ser estearato magnésico. El estearato magnésico puede comprender de aproximadamente un 0,1 % a aproximadamente un 1,5 % p/p de la composición. El estearato magnésico puede comprender aproximadamente un 0,5 % p/p o aproximadamente un 1,0 % p/p de la composición. Un lubricante puede ser lauril sulfato sódico. El lauril sulfato sódico puede comprender de aproximadamente un 0,1 % a aproximadamente un 5 % p/p, o de aproximadamente un 0,3 % a aproximadamente un 2 % p/p de la composición. El lauril sulfato sódico puede comprender aproximadamente un 1,0 % p/p de la composición. La composición puede comprender además un deslizante. El deslizante puede ser dióxido de silicio coloidal. El dióxido de silicio coloidal puede comprender de aproximadamente un 0,01 % a aproximadamente un 1,5 % p/p de la composición. El dióxido de silicio coloidal puede comprender aproximadamente un 0,1 % p/p de la composición. El compuesto de fórmula I se puede proporcionar en una cantidad de aproximadamente 100 mg, o aproximadamente 200 mg, o aproximadamente 400 mg, o aproximadamente 800 mg.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y al menos un excipiente. El excipiente se puede seleccionar entre el grupo que consiste en celulosa microcristalina, monohidrato de lactosa, croscarmelosa sódica, almidón glicolato sódico, hidroxipropilcelulosa, y poloxámero 188. Un excipiente puede ser celulosa microcristalina, que puede comprender de aproximadamente un 20 % a aproximadamente un 50 % p/p, o de aproximadamente un 20 % a aproximadamente un 40 % p/p, o de aproximadamente un 28 % a aproximadamente un 30 % p/p de la composición. La composición puede comprender además un deslizante. La composición puede comprender además uno o más lubricantes. La composición puede comprender además uno o más tensioactivos.
El compuesto de fórmula I se puede proporcionar en una cantidad de aproximadamente 100 mg, o aproximadamente 200 mg, o aproximadamente 400 mg, o aproximadamente 800 mg.
La composición puede tener un peso total de aproximadamente 50 mg a aproximadamente 1500 mg. La composición puede tener un peso total de aproximadamente 50 mg, aproximadamente 75 mg, aproximadamente 100 mg, aproximadamente 150 mg, aproximadamente 175 mg, aproximadamente 200 mg, aproximadamente 250 mg, aproximadamente 300 mg, aproximadamente 350 mg, aproximadamente 400 mg, aproximadamente 450 mg, aproximadamente 500 mg, aproximadamente 550 mg, aproximadamente 600 mg, aproximadamente 700 mg, aproximadamente 750 mg, aproximadamente 800 mg, aproximadamente 900 mg, aproximadamente 1000 mg, aproximadamente 1100 mg, aproximadamente 1200 mg, aproximadamente 1300 mg, aproximadamente 1400 mg, o aproximadamente 1500 mg.
La composición puede estar en forma de un comprimido, un comprimido revestido de película, una cápsula, una cápsula de gelatina, un comprimido oblongo, una miniesfera, o una microesfera. La composición puede estar en forma de un comprimido revestido de película.
Un comprimido puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo, teniendo los comprimidos una dureza de al menos aproximadamente 4 Kp. El comprimido puede tener una dureza de al menos aproximadamente 6 Kp, o al menos aproximadamente 8 Kp, o al menos aproximadamente 10 Kp, o al menos aproximadamente 12 Kp, o al menos aproximadamente 14 Kp. El comprimido puede tener una dureza de aproximadamente 4 Kp, aproximadamente 6 Kp, aproximadamente 8 Kp, aproximadamente 10 Kp, aproximadamente 12 Kp, aproximadamente 14 Kp, o aproximadamente 16 Kp.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, en la que la composición tiene una liberación de al menos un 85 por ciento del compuesto de fórmula I, o la sal del compuesto de fórmula I, en 45 minutos con el uso del aparato de disolución I de tipo II de la Farmacopea de EE.UU. (USP) a 50 rpm o 60 rpm en HCl 0,1 N. La composición puede tener al menos una velocidad de liberación de un 85 por ciento a los 30 minutos, o a los 20 minutos, o a los 15 minutos. La composición puede tener al menos una velocidad de liberación de un 90 por ciento a los 30 minutos. La composición puede tener al menos una velocidad de liberación de un 95 por ciento a los
30 minutos.
Algunas divulgaciones proporcionan un método para tratar hipertensión arterial crónica o persistente, que comprende identificar un individuo que necesita tal tratamiento, y administrar las composiciones farmacéuticas del compuesto de fórmula I.
Algunas divulgaciones proporcionan un método de producción de una composición farmacéutica que comprende mezclar de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I y uno o más excipientes, en el que los uno o más excipientes comprenden de aproximadamente un 5 % a aproximadamente un 65 % p/p de la composición. Un excipiente desvelado es celulosa microcristalina. En algunas divulgaciones, la celulosa microcristalina comprende de aproximadamente un 20 % a aproximadamente un 50 % p/p, o de aproximadamente un 28 % a aproximadamente un 30 % p/p de la composición. En algunas divulgaciones, el método comprende además añadir de aproximadamente un 0,5 % a aproximadamente un 1,0 % p/p de estearato magnésico. En algunas divulgaciones, el método comprende además añadir aproximadamente un 5 % p/p de poloxámero 188. En algunas divulgaciones, el método comprende además añadir aproximadamente un 1,0 % p/p de lauril sulfato sódico. En algunas divulgaciones, el método comprende además añadir aproximadamente un 0,1 % p/p de dióxido de silicio coloidal. En algunas divulgaciones, el compuesto de fórmula I se proporciona en una cantidad de aproximadamente 100 mg, o aproximadamente 200 mg, o aproximadamente 400 mg, o aproximadamente 800 mg. Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y celulosa microcristalina que comprende aproximadamente un 29 % p/p de la composición, croscarmelosa sódica que comprende aproximadamente un 5 % p/p de la composición, hidroxipropilcelulosa que comprende aproximadamente un 3 % p/p de la composición, poloxámero 188 que comprende aproximadamente un 5 % p/p de la composición, dióxido de silicio coloidal que comprende aproximadamente un 0,1 % p/p de la composición, y estearato magnésico que comprende aproximadamente un 0,5 % p/p de la composición.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y celulosa microcristalina que comprende aproximadamente un 22 % p/p de la composición, monohidrato de lactosa que comprende aproximadamente un 11 % p/p de la composición, lauril sulfato sódico que comprende aproximadamente un 1 % p/p de la composición, croscarmelosa sódica que comprende aproximadamente un 5 % p/p de la composición, hidroxipropilcelulosa que comprende aproximadamente un 3,0 % p/p de la composición, dióxido de silicio coloidal que comprende aproximadamente un 0,1 % p/p de la composición, y estearato magnésico que comprende aproximadamente un 1 % p/p de la composición.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y celulosa microcristalina que comprende aproximadamente un 29 % p/p de la composición, croscarmelosa sódica que comprende aproximadamente un 5 % p/p de la composición, hidroxipropilcelulosa que comprende aproximadamente un 3 % p/p de la composición, poloxámero 188 que comprende aproximadamente un 5 % p/p de la composición, dióxido de silicio coloidal que comprende aproximadamente un 0,1 % p/p de la composición, y estearato magnésico que comprende aproximadamente un 1 % p/p de la composición.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y celulosa microcristalina que comprende aproximadamente un 33 % p/p de la composición, lauril sulfato sódico que comprende aproximadamente un 1 % p/p de la composición, croscarmelosa sódica que comprende aproximadamente un 5 % p/p de la composición, hidroxipropilcelulosa que comprende aproximadamente un 3 % p/p de la composición, dióxido de silicio coloidal que comprende aproximadamente un 0,1 % p/p de la composición, y estearato magnésico que comprende aproximadamente un 1 % p/p de la composición.
Una composición farmacéutica puede comprender de aproximadamente 100 mg a aproximadamente 800 mg del compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y celulosa microcristalina que comprende aproximadamente un 29 % p/p de la composición, hidroxipropilcelulosa que comprende aproximadamente un 3 % p/p de la composición, poloxámero 188 que comprende aproximadamente un 5 % p/p de la composición, almidón glicolato sódico que comprende aproximadamente un 5 % p/p de la composición, dióxido de silicio coloidal que comprende aproximadamente un 0,1 % p/p de la composición, y estearato magnésico que comprende aproximadamente un 1 % p/p de la composición.
DESCRIPCIÓN DETALLADA DE LA REALIZACIÓN PREFERENTE
Lo siguiente son definiciones de los términos usados en la presente memoria descriptiva. La definición inicial proporcionada para un grupo o término de la presente memoria se aplica a ese grupo o término a lo largo de la presente memoria descriptiva, individualmente o como parte de otro grupo, a menos que se indique de otra manera.
El compuesto de fórmula I puede formar sales. Se entiende que la referencia a un compuesto de fórmula I en la presente memoria incluye la referencia a las sales del mismo, a menos que se indique de otra manera. El término "sal(es)", como se emplea en la presente memoria, indica sales ácidas y/o básicas formadas con ácidos y bases orgánicas y/o inorgánicas. Además, cuando el compuesto de fórmula I contiene un resto básico y un resto ácido, se pueden formar iones dipolares ("sales internas"), y se incluyen dentro del término "sal(es)", tal como se usa en la presente memoria. Son preferentes las sales farmacéuticamente aceptables (es decir, no tóxicas, fisiológicamente aceptables), aunque también son útiles otras sales, por ejemplo, en las etapas de aislamiento o purificación que se pueden emplear durante la preparación. Las sales del compuesto de fórmula I se pueden formar, por ejemplo, haciendo reaccionar el compuesto de fórmula I con una cantidad de ácido o base, tal como una cantidad equivalente, en un medio tal como uno en el que la sal precipita, o en un medio acuoso seguido de liofilización.
El compuesto de fórmula I que contiene un resto básico puede formar sales con una diversidad de ácidos inorgánicos y orgánicos. Las sales de adición de ácido ejemplares incluyen acetatos (tales como los formados con ácido acético o ácido trihaloacético, por ejemplo, ácido trifluoroacético), adipatos, alginatos, ascorbatos, aspartatos, benzoatos, bencenosulfonatos, bisulfatos, boratos, butiratos, citratos, alcanforatos, alcanforsulfonatos, ciclopentanopropionatos, digluconatos, dodecilsulfatos, etanosulfonatos, fumaratos, glucoheptanoatos, glicerofosfatos, hemisulfatos, heptanoatos, hexanoatos, clorhidratos (formados con ácido clorhídrico), bromhidratos (formados con bromuro de hidrógeno), yodhidratos, 2-hidroxietanosulfonatos, lactatos, maleatos (formados con ácido maleico), metanosulfonatos (formados con ácido metanosulfónico), 2-naftalenosulfonatos, nicotinatos, nitratos, oxalatos, pectinatos, persulfatos, 3-fenilpropionatos, fosfatos, picratos, pivalatos, propionatos, salicilatos, succinatos, sulfatos (tales como los formados con ácido sulfúrico), sulfonatos (tales como los mencionados en la presente memoria), tartratos, tiocianatos, toluenosulfonatos tales como tosilatos, undecanoatos, y similares.
El compuesto de fórmula I que contiene un resto ácido puede formar sales con una diversidad de bases inorgánicas y orgánicas. Las sales básicas ejemplares incluyen sales de amonio, sales de metales alcalinos tales como sales de sodio, litio, y potasio, sales de metales alcalinotérreos tales como sales de calcio y magnesio, sales con bases orgánicas (por ejemplo, aminas orgánicas) tales como benzatinas, diciclohexilaminas, hidrabaminas (formadas con N,N-bis(deshidroabietil)etilendiamina), N-metil-D-glucaminas, N-metil-D-glucamidas, t-butilaminas, y sales con aminoácidos tales como arginina, lisina y similares. Los grupos que contienen nitrógeno básico se pueden hacer cuaternarios con agentes tales como haluros de alquilo inferior (por ejemplo, cloruros, bromuros, y yoduros de metilo, etilo, propilo, y butilo), sulfatos de dialquilo (por ejemplo, sulfatos de dimetilo, dietilo, dibutilo, y diamilo), haluros de cadena larga (por ejemplo, cloruros, bromuros, y yoduros de decilo, laurilo, miristilo y estearilo), haluros de aralquilo (por ejemplo, bromuro de bencilo y fenetilo), y otros.
También se contemplan profármacos y solvatos del compuesto de fórmula I. El término "profármaco" indica un compuesto que, tras la administración a un sujeto, experimenta una conversión química mediante procesos metabólicos o químicos para producir un compuesto de fórmula I, o una sal y/o solvato del mismo. Los solvatos del compuesto de fórmula I son preferentemente hidratos. También se contempla cualquier tautómero.
Se contemplan todos los estereoisómeros del compuesto de fórmula I, tales como los que pueden existir debido a carbonos asimétricos en los sustituyentes R, incluyendo formas enantioméricas (que pueden existir incluso en ausencia de carbonos asimétricos, por ejemplo, atropisómeros) y formas diastereoméricas. Los estereoisómeros individuales del compuesto de fórmula I pueden estar sustancialmente exentos, por ejemplo, de otros isómeros, o pueden estar mezclados, por ejemplo, en forma de racematos o con todos los demás estereoisómeros, o con otros estereoisómeros seleccionados. Los centros quirales del compuesto de fórmula I pueden tener configuración S o R.
Métodos de Preparación
El compuesto de fórmula I se puede preparar mediante métodos tales como los ilustrados en los siguientes Esquemas I a II. El experto en la materia puede seleccionar disolventes, temperaturas, presiones, y otras condiciones de reacción.

En una divulgación, el compuesto de fórmula I se puede sintetizar mediante el método descrito en el Esquema I. El 4-bromobenzonitrilo sustituido de fórmula I-1 (X es bromo o mesilato) se puede tratar con etóxido sódico en DMF, por ejemplo se puede tratar alcohol etílico en DMF con hidruro sódico, para proporcionar 4-bromo-3-(etoximetil)benzonitrilo. El 4-bromo-3-(etoximetil)benzonitrilo se puede convertir en 4-bromo-3-(etoximetil)benzaldehído mediante reducción del nitrilo a un aldehído. Por ejemplo, se puede tratar 4-bromo-3-(etoximetil)benzonitrilo con DIBAL-H seguido de metanol y ácido clorhídrico para proporcionar 4-bromo-3-(etoximetil)benzaldehído. El 4-bromo-3-(etoximetil)benzaldehído se puede acoplar después con un compuesto de fórmula I-2 en presencia de un catalizador de paladio, por ejemplo tetraquis(trifenilfosfina)paladio(0), en las condiciones adecuadas para proporcionar un compuesto de fórmula I-3 (R es SEM o MEM). El aldehído del compuesto de fórmula I-3 se puede reducir hasta un alcohol, por lo que se proporciona un compuesto de fórmula I-4.
Por ejemplo, el compuesto de fórmula I-3 se puede tratar con borohidruro sódico en alcohol etílico o alcohol metílico para proporcionar el compuesto de fórmula I-4 (R es SEM o MEM). El alcohol bencílico de fórmula I-4 se puede convertir después en bromuro de bencilo, por lo que se proporciona un compuesto de fórmula I-5 (R es SEM o MEM). Por ejemplo, el alcohol bencílico de fórmula I-4 en DMF en presencia de tetrabromuro de carbono se puede tratar con trifenilfosfina para proporcionar el bromuro de bencilo de fórmula I-5. El bromuro de bencilo de fórmula I-5 se puede tratar con clorhidrato de 2-N-butil-1,3-diazaespiro[4.4]non-1-en-4-ona para proporcionar el compuesto de fórmula I-6 (R es SEM o MEM). Por ejemplo, el clorhidrato de 2-N-butil-1,3-diazaespiro[4.4]non-1-en-4-ona se puede tratar con hidruro sódico en DMF seguido de la adición del bromuro de bencilo de fórmula I-5 para proporcionar el compuesto de fórmula I-6. El compuesto de fórmula I-6 se puede desproteger en condiciones ácidas, por ejemplo se puede tratar el compuesto de fórmula I-6 en alcohol etílico con ácido clorhídrico 6 N, por lo que se proporciona el compuesto de fórmula I, 4-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2-(etoximetil)[1,1'-bifenil]-2-sulfonamida.

En otra divulgación, el compuesto de fórmula I se puede sintetizar mediante el método descrito en el Esquema II. Se puede tratar 4-bromo-3-metilbenzoato de etilo (110 g, 450 mmol) con NBS para proporcionar 4-bromo-3-metilbenzoato de etilo. El 4-bromo-3-metilbenzoato de etilo se puede tratar con etóxido sódico en alcohol etílico, por ejemplo una disolución al 21 % de etóxido sódico en etanol, para proporcionar 4-bromo-3-(etoximetil)benzoato de etilo. El 4-bromo-3-(etoximetil)benzoato de etilo se puede acoplar después con ácido 2-[[N-(4,5-dimetil-3-isoxazolil)-N-(metoximetil)amino]sulfonil]-fenilborónico en presencia de un catalizador de paladio, por ejemplo tetraquis(trifenilfosfina)paladio(0), en las condiciones adecuadas para proporcionar N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. La N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil) [1,1'-bifenil]-2-sulfonamida se puede reducir a un alcohol, por lo que se proporciona N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. Por ejemplo, la N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida en THF se puede tratar con una disolución de DIBAL-H en tolueno para proporcionar N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. La N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida se puede convertir después en 4'-(bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. Por ejemplo, la N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida en DMF en presencia de
tetrabromuro de carbono se puede tratar con trifenilfosfina para proporcionar 4'-(bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. La 4'-(bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida se puede tratar con clorhidrato de 2-N-butil-1,3-diazaespiro[4.4]non-1-en-4-ona para proporcionar 4'-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil-N-](4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. Por ejemplo, se puede tratar clorhidrato de 2-N-butil-1,3-diazaespiro[4.4]non-1-en-4-ona con hidruro sódico en DMF seguido de la adición de 4'-(bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida para proporcionar 4'-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil-N-](4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida. La 4'-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil-N-](4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida se puede desproteger en condiciones ácidas, por lo que se proporciona el compuesto de fórmula I, 4'-[(2-butil-4-oxo-1,3-diazaespiro[4,4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)[1,1'-bifenil]-2-sulfonamida. Por ejemplo, se puede tratar 4'-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil-N-](4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida en alcohol etílico con ácido clorhídrico 6 N para proporcionar el compuesto de fórmula I, 4'-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)[1,1'-bifenil]-2-sulfonamida.
El compuesto de fórmula I y las sales del mismo son antagonistas de los receptores de endotelina (en especial, ET-1) y de angiotensina II (en especial, el subtipo AT1) ("antagonistas dobles de receptores de angiotensina y endotelina"), y son útiles en el tratamiento de afecciones asociadas a niveles incrementados de ET y/o niveles incrementados de angiotensina II, y de todos los trastornos dependientes de endotelina o dependientes de angiotensina II. Así, son útiles como agentes antihipertensivos. Mediante la administración de una composición que tiene el compuesto de fórmula I, se reduce la tensión arterial de un hospedador mamífero hipertenso (por ejemplo, un ser humano). También son útiles en la hipertensión portal crónica o persistente, hipertensión arterial crónica o persistente secundaria a tratamiento con eritropoyetina, hipertensión arterial crónica o persistente con renina baja, e hipertensión arterial crónica o persistente.
El compuesto de fórmula I también es útil en el tratamiento de los trastornos relacionados con la función de las células renales, glomerulares y mesangiales, incluyendo insuficiencia renal aguda (tal como isquémica, nefrotóxica, o glomerulonefritis) y crónica (tal como diabética, hipertensiva o mediada por el sistema inmunitario), nefropatía diabética, lesión glomerular, daño renal secundario a la vejez o relacionado con diálisis, nefroesclerosis (en especial nefroesclerosis hipertensiva), nefrotoxicidad (incluyendo nefrotoxicidad relacionada con técnicas de formación de imágenes y agentes de contraste y con ciclosporina), isquemia renal, reflujo vesicoureteral primario, glomeruloesclerosis y similares. El compuesto de fórmula I también es útil en el tratamiento de trastornos relacionados con la función paracrina y endocrina. El compuesto de fórmula I también es útil en el tratamiento de nefropatía diabética, nefropatía inducida por IgA, y nefropatía inducida por hipertensión. La expresión "nefropatía diabética", tal como se usa en la presente memoria, se entenderá que incluye las etapas incipientes y manifiestas de la nefropatía diabética, diagnosticada o no, pero más en general la diagnosticada por un médico.
El compuesto de fórmula I también es útil en el tratamiento de la endotoxemia o choque por endotoxinas, así como en choque hemorrágico. El compuesto de fórmula I también es útil en la mitigación del dolor asociado al cáncer, tal como dolor asociado al cáncer de próstata, y dolor óseo asociado al cáncer óseo. El compuesto de fórmula I es útil además en la prevención y/o la reducción de lesión de órganos asociada a los efectos proliferativos celulares de la endotelina.
El compuesto de fórmula I también es útil en enfermedad hipóxica e isquémica, y como agente anti-isquémico para el tratamiento, por ejemplo, de isquemia cardiaca, renal y cerebral, y reperfusión (tal como la que se da tras cirugía con circulación extracorpórea), vasoespasmo coronario y cerebral, y similares.
Además, el compuesto de fórmula I también es útil como agente antiarrítmico; agente antianginoso; agente antifibrilador; agente antiasmático; agente antiateroesclerótico y antiarterioesclerótico (lo que incluye agente antiarterioesclerótico en trasplantes); aditivo para soluciones cardioplégicas para circulación extracorpórea; agente auxiliar para terapia trombolítica; y agente antidiarreico. El compuesto de fórmula I puede ser útil en terapia del infarto de miocardio; terapia para enfermedad vascular periférica (por ejemplo, enfermedad de Raynaud, claudicación intermitente y enfermedad de Takayasu); tratamiento de hipertrofia cardiaca (por ejemplo, cardiomiopatía hipertrófica); tratamiento de hipertensión pulmonar primaria (por ejemplo, plexogénica, embólica) en adultos y en el recién nacido, e hipertensión pulmonar secundaria a insuficiencia cardiaca, lesión por radioterapia y quimioterapia, u otro traumatismo; tratamiento de trastornos vasculares del sistema nervioso central, tales como ictus, migraña y hemorragia subaracnoidea; tratamiento de trastornos conductuales del sistema nervioso central; tratamiento de enfermedades gastrointestinales tales como colitis ulcerosa, enfermedad de Crohn, lesión de la mucosa gástrica, úlcera, enfermedad inflamatoria intestinal y enfermedad isquémica intestinal; tratamiento de enfermedades relacionadas con la vesícula biliar o las vías biliares, tal como colangitis; tratamiento de pancreatitis; regulación del crecimiento celular; tratamiento de hipertrofia prostática benigna; reestenosis tras angioplastia o tras cualquier procedimiento que incluya un trasplante y colocación de mallas intravasculares; terapia de insuficiencia cardiaca congestiva, incluyendo inhibición de la fibrosis; inhibición de dilatación, remodelación y disfunción ventricular izquierda; y tratamiento de hepatotoxicidad y de muerte súbita. El compuesto de fórmula I es útil en el tratamiento de anemia de células falciformes, incluyendo el inicio y/o la evolución de las crisis dolorosas de esta
enfermedad; tratamiento de las consecuencias perjudiciales de tumores productores de ET tales como hipertensión arterial crónica o persistente resultante de un hemangiopericitoma; tratamiento de enfermedad y lesión hepática temprana y avanzada, incluyendo las complicaciones concomitantes (por ejemplo, hepatotoxicidad, fibrosis y cirrosis); tratamiento de enfermedades espásticas del tracto urinario y/o la vejiga; tratamiento de síndrome hepatorrenal; tratamiento de enfermedades inmunológicas que implican vasculitis, tales como lupus, esclerosis sistémica, crioglobulinemia mixta; y tratamiento de fibrosis asociada a disfunción renal y hepatotoxicidad. El compuesto de fórmula I es útil en terapia de trastornos metabólicos y neurológicos; cáncer; diabetes mellitus insulinodependiente y no insulinodependiente; neuropatía; retinopatía; epilepsia; ictus hemorrágico e isquémico; remodelación ósea; psoriasis; y enfermedades inflamatorias crónicas tales como artritis, artritis reumatoide, osteoartritis, sarcoidosis y dermatitis eccematosa (todos los tipos de dermatitis).
El compuesto de fórmula I además es útil en el tratamiento de trastornos que implican broncoconstricción y trastornos de inflamación pulmonar crónica o aguda, tales como enfermedad pulmonar obstructiva crónica (COPD) y síndrome de dificultad respiratoria del adulto (ARDS).
El compuesto de fórmula I también es útil en el tratamiento de disfunción sexual en hombres (disfunción eréctil, por ejemplo, debida a diabetes mellitus, lesión de la médula espinal, prostatectomía radical, etiología psicógena o cualquier otra causa) y en mujeres por mejora del flujo sanguíneo en los genitales, en especial, el cuerpo cavernoso.
El compuesto de fórmula I también es útil en el tratamiento de demencia, incluyendo demencia por Alzheimer, demencia senil y demencia vascular. Además, el compuesto de fórmula I además es útil en la reducción de la morbilidad general y/o mortalidad como resultado de las utilidades anteriores. Por lo tanto, se describen métodos para el tratamiento de todos los trastornos dependientes de endotelina o dependientes de angiotensina II, que comprenden la etapa de administrar a un sujeto que lo necesita el compuesto de fórmula I en una cantidad eficaz. Se pueden emplear otros agentes terapéuticos tales como los descritos más adelante con el compuesto de fórmula I en los métodos desvelados. En los métodos, se puede(n) administrar tal(es) otro(s) agente(s) terapéutico(s) antes, simultáneamente o después de la administración del compuesto de fórmula I de la presente descripción.
Composiciones Farmacéuticas
Una composición farmacéutica puede comprender agentes tensioactivos, vehículos, diluyentes, excipientes, agentes suavizantes, agentes de suspensión, sustancias formadoras de películas, y asistentes de revestimiento fisiológicamente aceptables, o una combinación de los mismos; y un compuesto descrito en la presente memoria. Los vehículos o diluyentes aceptables para el uso terapéutico se conocen bien en la técnica farmacéutica, y se describen, por ejemplo, en Remington's Pharmaceutical Sciences, 18a Ed., Mack Publishing Co., Easton, PA (1990). Se pueden proporcionar conservantes, estabilizantes, colorantes, edulcorantes, perfumes, agentes aromatizantes, y similares en la composición farmacéutica. Por ejemplo, se puede añadir benzoato sódico, ácido ascórbico y ésteres de ácido p-hidroxibenzoico como conservantes. Además, se pueden usar antioxidantes y agentes de suspensión. Se pueden usar alcoholes, ésteres, alcoholes alifáticos sulfatados y similares como agentes tensioactivos; se pueden usar sacarosa, glucosa, lactosa, almidón, celulosa cristalizada, manitol, silicato anhidro ligero, aluminato magnésico, metasilicato aluminato de magnesio, silicato de aluminio sintético, carbonato cálcico, bicarbonato sódico, fosfato dicálcico, carboximetilcelulosa cálcica, y similares como excipientes; se pueden usar estearato magnésico, talco, aceite hidrogenado y similares como agentes suavizantes; se pueden usar aceite de coco, aceite de oliva, aceite de sésamo, aceite de cacahuete, soja como agentes de suspensión o lubricantes; se puede usar acetato ftalato de celulosa como derivado de un carbohidrato tal como celulosa o azúcar, o copolímero de acetato de metilometacrilato como derivado de polivinilo como agentes de suspensión; y se pueden usar plastificantes tales como ésteres de ftalato y similares como agentes de suspensión.
La expresión "composición farmacéutica" se refiere a una mezcla de un compuesto descrito en la presente memoria con otros componentes químicos, tales como diluyentes o vehículos. La composición farmacéutica facilita la administración del compuesto a un organismo. Existen múltiples métodos para administrar un compuesto en la técnica, que incluyen, pero sin limitación, administración oral, mediante inyección, aerosol, parenteral, y tópica. Las composiciones farmacéuticas también se pueden obtener haciendo reaccionar los compuestos con ácidos orgánicos o inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico, y similares.
El término "vehículo" define un compuesto químico que facilita la incorporación de un compuesto a las células o tejidos. Por ejemplo, el dimetilsulfóxido (DMSO) es un vehículo utilizado habitualmente, ya que facilita la absorción de muchos compuestos orgánicos en las células o tejidos de un organismo.
El término "diluyente" define compuestos químicos diluidos en agua que disolverán el compuesto de interés, así como estabilizarán la forma biológicamente activa del compuesto. Las sales disueltas en soluciones tamponadas se utilizan como diluyentes en la técnica. Una solución tamponada usada habitualmente es solución salina tamponada con fosfato, ya que imita las condiciones salinas de la sangre humana. Debido a que las sales tamponadoras pueden controlar el pH de una solución a concentraciones bajas, un diluyente tamponado raramente modifica la actividad biológica de un compuesto.
La expresión "fisiológicamente aceptable" define un vehículo o diluyente que no elimina la actividad biológica y las propiedades del compuesto.
Las composiciones farmacéuticas descritas en la presente memoria se pueden administrar a un paciente humano por sí mismas, o en composiciones farmacéuticas en las que se mezclan con otros ingredientes activos, como en una terapia de combinación, o con vehículos o excipiente(s) adecuados. Las técnicas para la formulación y administración del compuesto de fórmula I se pueden hallar en "Remington's Pharmaceutical Sciences", Mack Publishing Co., Easton, PA, 18a edición, 1990.
Se puede proporcionar una dosis baja del compuesto de fórmula I en formas farmacéuticas de comprimidos, comprimidos revestidos de película, cápsulas, comprimidos oblongos, píldoras, cápsulas de gelatina, miniesferas, microesferas, o grageas. Preferentemente, las formulaciones descritas en la presente memoria pueden proporcionar cualidades favorables de procesamiento de fármacos, que incluyen, por ejemplo, pero sin limitación, velocidades rápidas de prensado de comprimidos, fuerza de compresión reducida, fuerzas de expulsión reducidas, uniformidad de la mezcla, uniformidad del contenido, dispersión uniforme del color, tiempo de disgregación acelerado, disolución rápida, friabilidad baja (preferente para el procesamiento posterior tal como envasado, envío, recogida y embalaje, etc.) y características físicas de la forma farmacéutica (por ejemplo, peso, dureza, grosor, friabilidad) con poca variación.
La formulación puede producir una forma farmacéutica de disolución rápida, para la que al menos un 85 % de la cantidad etiquetada de la sustancia farmacológica se disuelve en 45 minutos, mediante el uso del aparato de disolución de tipo II de la Farmacopea de EE.UU. (USP) mediante la utilización de HCl 0,1 N a 37 °C con una velocidad de paleta de 50 rpm. La formulación puede producir una forma farmacéutica de disolución rápida, para la que al menos un 85 % de la cantidad etiquetada de la sustancia farmacológica se disuelve en 45 minutos, mediante el uso del aparato de disolución de tipo II de la Farmacopea de EE.UU. (USP) mediante la utilización de HCl 0,1 N a 37 °C con una velocidad de paleta de 60 rpm. La formulación puede producir una forma farmacéutica de disolución rápida, para la que al menos un 85 % de la cantidad etiquetada de la sustancia farmacológica se disuelve en 30 minutos, mediante el uso del aparato de disolución de tipo II de la Farmacopea de EE.UU. (USP) mediante la utilización de HCl 0,1 N a 37 °C con una velocidad de paleta de 50 rpm. La formulación puede producir una forma farmacéutica de disolución rápida, para la que al menos un 85 % de la cantidad etiquetada de la sustancia farmacológica se disuelve en 30 minutos, mediante el uso del aparato de disolución de tipo II de la Farmacopea de EE.UU. (USP) mediante la utilización de HCl 0,1 N a 37 °C con una velocidad de paleta de 50 rpm. La formulación puede producir una forma farmacéutica de disolución rápida, para la que al menos un 85 % de la cantidad etiquetada de la sustancia farmacológica se disuelve en 20 minutos, mediante el uso del aparato de disolución de tipo II de la Farmacopea de EE.UU. (USP) mediante la utilización de HCl 0,1 N a 37 °C con una velocidad de paleta de 50 rpm. La formulación puede producir una forma farmacéutica de disolución rápida, para la que al menos un 85 % de la cantidad etiquetada de la sustancia farmacológica se disuelve en 20 minutos, mediante el uso del aparato de disolución de tipo II de la Farmacopea de EE.UU. (USP) mediante la utilización de HCl 0,1 N a 37 °C con una velocidad de paleta de 50 rpm.
Las formulaciones pueden requerir fuerzas mínimas de compresión de comprimidos para conseguir una dureza de aproximadamente 2 kp a aproximadamente 25 kp. La formulación puede requerir fuerzas de compresión para conseguir una dureza, por ejemplo, de al menos aproximadamente 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, o 22 kp. Tales fuerzas mínimas de compresión pueden posibilitar que los comprimidos permanezcan relativamente porosos y se disgreguen rápidamente con un desgaste mínimo de las herramientas de compresión y de la prensa de comprimidos.
Las formulaciones pueden producir comprimidos, incluyendo comprimidos revestidos de película, que tienen un valor de friabilidad de un 1 % o menos. Así, el valor de friabilidad puede ser de aproximadamente un 0,9 %, 0,8 %, 0,75 %, 0,6 %, 0,5 %, 0,4 %, 0,3 %, 0,25 %, 0,2 %, 0,15 %, 0,1 %, 0,08 %, 0,06 %, 0,04 %, 0,02 % o menos.
El compuesto de fórmula I se puede formular fácilmente, por ejemplo, combinando la sustancia farmacológica con cualquier excipiente(s) farmacéuticamente aceptable(s) adecuado(s), por ejemplo, pero sin limitación, aglutinantes, diluyentes, disgregantes, lubricantes, cargas, vehículos, revestimientos, deslizantes, aromatizantes, aditivos colorantes, y similares, como se expone más adelante. Tales composiciones se pueden preparar para el almacenamiento y procesamiento posteriores.
Excipientes
Los excipientes aceptables para el uso terapéutico son muy conocidos en la técnica farmacéutica, y se describen, por ejemplo, en Handbook of Pharmaceutical Excipients, 5a edición (Raymond C Rowe, Paul J Sheskey y Sián C Owen, eds. 2005), y Remington: The Science and Practice of Pharmacy, 21a edición (Lippincott Williams & Wilkins, 2005). El término "vehículo" o "excipiente" puede significar en la presente memoria cualquier sustancia, no en sí misma un agente terapéutico, usada como vehículo y/o diluyente y/o adyuvante para la administración de un agente terapéutico a un sujeto o añadida a una composición farmacéutica para mejorar sus propiedades de manipulación o
de almacenamiento o para permitir o facilitar la formación de una unidad de dosis de la composición en un artículo discreto tal como una cápsula, comprimido, comprimido revestido de película, comprimido oblongo, cápsula de gelatina, píldora, miniesfera, microesfera, y similares, adecuada para la administración oral. Los excipientes pueden incluir, a modo de ilustración y no de limitación, diluyentes, disgregantes, agentes aglutinantes, agentes humectantes, polímeros, lubricantes, deslizantes, revestimientos, edulcorantes, agentes solubilizantes añadidos para enmascarar o contrarrestar un sabor u olor desagradable, aromatizantes, colorantes, perfumes, y sustancias añadidas para mejorar el aspecto de la composición.
Los excipientes aceptables incluyen, por ejemplo, pero sin limitación, celulosa microcristalina, lactosa, sacarosa, polvo de almidón, almidón de maíz o derivados del mismo, ésteres de ácidos alcanoicos de celulosa, ésteres de alquilo de celulosa, talco, ácido esteárico, estearato magnésico, óxido magnésico, sales de sodio y calcio de ácido fosfórico y sulfúrico, gelatina, goma arábiga, alginato sódico, polivinilpirrolidona, y/o poli(alcohol vinílico), solución salina, dextrosa, manitol, monohidrato de lactosa, lecitina, albúmina, glutamato sódico, clorhidrato de cisteína, croscarmelosa sódica, almidón glicolato sódico, hidroxipropilcelulosa, poloxámero (por ejemplo, los poloxámeros 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, 407, y benzoato de poloxámero 105, dibenzoato de poloxámero 182407, y similares), lauril sulfato sódico, dióxido de silicio coloidal y similares. Los ejemplos de excipientes adecuados para los comprimidos y las cápsulas incluyen, pero sin limitación, celulosa microcristalina, celulosa microcristalina silicificada, monohidrato de lactosa, croscarmelosa sódica, almidón sódico, hidroxipropilcelulosa, poloxámero 188, lauril sulfato sódico, dióxido de silicio coloidal, estearato magnésico. Los ejemplos de excipientes adecuados para las cápsulas de gelatina blandas incluyen aceites vegetales, ceras, grasas, polioles semisólidos y líquidos. Los excipientes adecuados para la preparación de las soluciones y los jarabes incluyen, sin limitación, agua, polioles, sacarosa, azúcar invertido y glucosa. El compuesto también se puede producir en forma microencapsulada. Si se desea, se pueden utilizar preparaciones potenciadoras de la absorción (por ejemplo, liposomas).
Las composiciones y formulaciones pueden incluir cualquier otro agente que proporcione una mejor transferencia, administración, tolerancia, y similar. Estas composiciones y formulaciones pueden incluir, por ejemplo, polvos, pastas, gelatinas, ceras, aceites, lípidos, vesículas que contienen lípidos (catiónicos o aniónicos) (tales como Lipofectin™), conjugados de ADN, pastas de absorción anhidras, emulsiones de aceite en agua y agua en aceite, emulsiones de Carbowax (polietilenglicoles de diversos pesos moleculares), geles semisólidos, y mezclas semisólidas que contienen Carbowax.
Cualquiera de las mezclas anteriores puede ser apropiada en tratamientos y terapias de acuerdo con la descripción de la presente memoria, con tal de que el ingrediente activo de la formulación no sea inactivado por la formulación, y que la formulación sea fisiológicamente compatible y tolerable con la vía de administración. Véase también Baldrick P. "Pharmaceutical excipient development: the need for preclinical guidance." Regul. Toxicol. Pharmacol. 32(2):210-8 (2000), Charman WN "Lipids, lipophilic drugs, and oral drug delivery-some emerging concepts". J. Pharm. Sci.
89(8):967-78 (2000), y las citas de ese documento para una información adicional relacionada con formulaciones, excipientes y vehículos bien conocidos por los químicos farmacéuticos.
Uno o más, o cualquier combinación, de los excipientes enumerados se puede incluir o excluir de manera específica de las formulaciones y/o métodos descritos en la presente memoria.
Como entenderán los expertos en la materia, las cantidades de los excipientes estarán determinadas por las dosis de fármaco y el tamaño de la forma farmacéutica. El tamaño de la forma farmacéutica puede ser de aproximadamente 175 mg. El tamaño de la forma farmacéutica puede ser de aproximadamente 350 mg. El tamaño de la forma farmacéutica puede ser de aproximadamente 700 mg. Este peso de la forma farmacéutica es arbitrario, y un experto en la técnica sabrá que se puede preparar un intervalo de pesos. El intervalo preferente de la forma farmacéutica es de aproximadamente 50 mg a aproximadamente 1500 mg, más habitualmente de aproximadamente 100 mg a aproximadamente 1000 mg, más habitualmente de aproximadamente 175 mg a aproximadamente 700 mg, siendo el peso preferente de la forma habitual de aproximadamente 175 mg, aproximadamente 350 mg, o aproximadamente 700 mg.
Lubricantes
Se pueden emplear lubricantes en la fabricación de ciertas formas farmacéuticas. Por ejemplo, a menudo se empleará un lubricante al producir comprimidos. Se puede añadir un lubricante justo antes de la etapa de compresión, y se puede mezclar con la formulación durante un periodo mínimo de tiempo para obtener una buena dispersión. Se pueden usar uno o más lubricantes. Los ejemplos de lubricantes adecuados incluyen, pero sin limitación, estearato magnésico, estearato cálcico, estearato de zinc, ácido esteárico, talco, behenato de glicerilo, polietilenglicol, polímeros de poli(óxido de etileno) (por ejemplo, disponibles con las marcas comerciales registradas Carbowax® para polietilenglicol y Polyox® para poli(óxido de etileno) de Dow Chemical Company, Midland, Mich.), lauril sulfato sódico, lauril sulfato magnésico, oleato sódico, estearil fumarato sódico, DL-leucina, sílice coloidal, y otros como se conoce en la técnica. Los lubricantes habituales son estearato magnésico, estearato cálcico, estearato de zinc y mezclas de estearato magnésico con lauril sulfato sódico. Los lubricantes pueden comprender de aproximadamente un 0,25 % a aproximadamente un 50 % del peso del comprimido, habitualmente de
aproximadamente un 1 % a aproximadamente un 40 %, más habitualmente de aproximadamente un 5 % a aproximadamente un 30 %, más habitualmente de un 20 % a un 30 %. Se puede añadir estearato magnésico como lubricante, por ejemplo, para mejorar el flujo del polvo, prevenir que la mezcla se adhiera al equipo de compresión y a las superficies de los punzones y proporcionar lubricación para permitir que los comprimidos se expulsen limpiamente de las matrices de comprimidos. Se puede añadir estearato magnésico a las formulaciones farmacéuticas habitualmente a concentraciones que oscilan de aproximadamente un 0,1 % a aproximadamente un 5,0 % p/p, o de aproximadamente un 0,25 % a aproximadamente un 4 % p/p, o de aproximadamente un 0,5 % a aproximadamente un 3 % p/p, o de aproximadamente un 0,75 % a aproximadamente un 2 % p/p, o de aproximadamente un 0,8 % a aproximadamente un 1,5 % p/p, o de aproximadamente un 0,85 % a aproximadamente un 1,25 % p/p, o de aproximadamente un 0,9 % a aproximadamente un 1,20 % p/p, o de aproximadamente un 0,85 % a aproximadamente un 1,15 % p/p, o de aproximadamente un 0,90 % a aproximadamente un 1,1 % p/p, o de aproximadamente un 0,95 % a aproximadamente un 1,05 % p/p, o de aproximadamente un 0,95 % a aproximadamente un 1 % p/p. Los intervalos anteriores son ejemplos de intervalos habituales. El experto en la materia reconocerá otros lubricantes y/o cantidades que se pueden usar en las formulaciones descritas en la presente memoria. Como reconocerá el experto en la materia, cuando se incorporan a las formulaciones desveladas en la presente memoria, las cantidades de la carga o cargas principales y/u otros excipientes se pueden reducir en consecuencia para acomodar la cantidad del lubricante o lubricantes añadidos para mantener inalterado el peso unitario total del comprimido.
Aditivos Colorantes
También se pueden incluir aditivos colorantes. Los colorantes se pueden usar en cantidades suficientes para distinguir las dosis de las formas farmacéuticas. Preferentemente, se añaden aditivos colorantes aprobados para el uso en fármacos (21 CFR 74) a las formulaciones comerciales para diferenciar las dosis de los comprimidos. Se pueden hacer uso de otros colorantes farmacéuticamente aceptables y combinaciones.
Aglutinantes
Se pueden usar aglutinantes, por ejemplo, para conferir cualidades cohesivas a una formulación, y así asegurar que la forma farmacéutica resultante permanezca intacta tras la compactación. Los materiales aglutinantes adecuados incluyen, pero sin limitación, celulosa microcristalina, gelatina, azúcares (que incluyen, por ejemplo, sacarosa, glucosa, dextrosa y maltodextrina), polietilenglicol, ceras, gomas naturales y sintéticas, polivinilpirrolidona, almidón pregelatinizado, povidona, polímeros celulósicos (que incluyen, por ejemplo, hidroxipropilcelulosa (HPC), hidroxipropilmetilcelulosa (HPMC), metilcelulosa, hidroxietilcelulosa, y similares), hidroxipropilcelulosa (HPC), y similares. Por lo tanto, en algunas realizaciones, las formulaciones desveladas en la presente memoria pueden incluir al menos un aglutinante para aumentar la compresibilidad del excipiente o excipientes principales. Por ejemplo, la formulación puede incluir al menos uno de los siguientes aglutinantes en los intervalos preferentes siguientes: de aproximadamente un 2 % a aproximadamente un 6 % p/p de hidroxipropilcelulosa (Klucel), de aproximadamente un 2 % a aproximadamente un 5 % p/p de polivinilpirrolidona (PVP), de aproximadamente un 1 % a aproximadamente un 5 % p/p de metilcelulosa, de aproximadamente un 2 % a aproximadamente un 5 % de hidroxipropilmetilcelulosa, de aproximadamente un 1 % a aproximadamente un 5 % p/p de etilcelulosa, de aproximadamente un 1 % a aproximadamente un 5 % p/p de carboximetilcelulosa sódica, y similares. Los intervalos anteriores son intervalos preferentes ejemplares. El experto en la materia conocerá otros aglutinantes y/o cantidades que se pueden usar en las formulaciones descritas en la presente memoria. Como reconocerá el experto en la materia, cuando se incorporan a las formulaciones desveladas en la presente memoria, las cantidades de la carga o cargas principales y/u otros excipientes se pueden reducir en consecuencia para acomodar la cantidad del aglutinante añadido para mantener inalterado el peso unitario total del comprimido. El aglutinante o aglutinantes se pueden pulverizar desde una solución, por ejemplo mediante granulación húmeda, para incrementar la actividad de aglutinación.
Disgregantes
Se pueden usar disgregantes, por ejemplo, para facilitar la disgregación de los comprimidos tras la administración, y en general son almidones, arcillas, celulosas, alginatos, gomas, o polímeros reticulados. Los disgregantes adecuados incluyen, pero sin limitación, polivinilpirrolidona reticulada (PVP-XL), almidón glicolato sódico, ácido algínico, ácido metacrílico DYB, celulosa microcristalina, crospovidona, polacrilina potásica, almidón glicolato sódico, almidón, almidón pregelatinizado, croscarmelosa sódica, y similares. Si se desea, la formulación farmacéutica puede contener además cantidades menores de sustancias auxiliares no tóxicas tales como agentes humectantes o emulsionantes, agentes tamponadores del pH y similares, por ejemplo, acetato sódico, monolaurato de sorbitán, trietanolamina-acetato sódico, trietanolamina-oleato, lauril sulfato sódico, dioctil sulfosuccinato sódico, ésteres de ácidos grasos de polioxietileno sorbitán, etc., y similares.
Los intervalos anteriores son ejemplos de intervalos preferentes. El experto en la materia conocerá otros disgregantes y/o cantidades de disgregantes que se pueden usar en las formulaciones descritas en la presente memoria. Como reconocerá el experto en la materia, cuando se incorporan a las formulaciones descritas en la presente memoria, las cantidades de la carga o cargas principales y/u otros excipientes se pueden reducir en
consecuencia para acomodar la cantidad del disgregante añadido para mantener inalterado el peso unitario total del comprimido.
Revestimientos
Las formulaciones pueden incluir un revestimiento, por ejemplo, un revestimiento de película. Cuando implican revestimientos de película, las preparaciones de revestimiento pueden incluir, por ejemplo, un polímero formador de películas, un plastificante, o similares. Además, los revestimientos pueden incluir pigmentos y/u opacificantes. Los ejemplos no limitantes de polímeros formadores de películas incluyen hidroxipropilmetilcelulosa, hidroxipropilcelulosa, metilcelulosa, polivinilpirrolidina, y almidones. Los ejemplos no limitantes de plastificantes incluyen polietilenglicol, citrato de tributilo, sebacato de dibutilo, aceite de ricino, y monoglicérido acetilado. Además, los ejemplos no limitantes de pigmentos y opacificantes incluyen óxidos de hierro de diversos colores, tintes de laca de muchos colores, dióxido de titanio, y similares.
Diluyentes
Se pueden usar diluyentes, y se seleccionan en general de uno o más de los compuestos sacarosa, fructosa, glucosa, galactosa, lactosa, maltosa, azúcar invertido, carbonato cálcico, lactosa, almidón, celulosa microcristalina, monohidrato de lactosa, fosfato dicálcico, fosfato dicálcico anhidro, un poliol farmacéuticamente aceptable tal como xilitol, sorbitol, maltitol, manitol, isomaltitol y glicerol, polidextrosa, almidón, o similares, o cualquier mezcla de los mismos.
Tensioactivos
Se pueden usar tensioactivos. El uso de tensioactivos como agentes humectantes en las formas farmacológicas orales se describe en la bibliografía, por ejemplo en H. Sucker, P. Fuchs, P. Speiser, Pharmazeutische Technologie, 2a edición, Thieme 1989, página 260. Se sabe de otros artículos, tales como los publicados en Advanced Drug Delivery Reviews (1997), 23, páginas 163-183, que además es posible usar tensioactivos, entre otros, para mejorar la permeabilidad y la biodisponibilidad de los compuestos activos farmacéuticos. Los ejemplos de tensioactivos incluyen, pero sin limitación, tensioactivos aniónicos, tensioactivos no iónicos, tensioactivos dipolares y una mezcla de los mismos. Preferentemente, los tensioactivos se seleccionan entre el grupo que consiste en éster de ácidos grasos de poli(oxietileno) sorbitán, estearato de poli(oxietileno), poli(oxietileno) alquil éter, glicérido poliglicolado, poli(oxietileno)-aceite de ricino, éster de ácidos grasos de sorbitán, poloxámero, sal de ácidos grasos, sales biliares, sulfato de alquilo, lecitina, micelas mixtas de sales biliares y lecitina, éster de glucosa de vitamina E TPGS (succinato de D-a-tocoferilo polietilenglicol 1000), lauril sulfato sódico, y similares, y una mezcla de los mismos.
Deslizantes
Se pueden usar deslizantes. Los ejemplos de deslizantes que se pueden usar incluyen, pero sin limitación, dióxido de silicio coloidal, trisilicato magnésico, celulosa en polvo, almidón, talco y fosfato cálcico, o similares, y mezclas de los mismos.
Los excipientes anteriores pueden estar presentes en una cantidad de hasta aproximadamente un 95 % del peso total de la composición, o hasta aproximadamente un 85 % del peso total de la composición, o hasta aproximadamente un 75 % del peso total de la composición, o hasta aproximadamente un 65 % del peso total de la composición, o hasta aproximadamente un 55 % del peso total de la composición, o hasta aproximadamente un 45 % del peso total de la composición, o hasta aproximadamente un 43 % del peso total de la composición, o hasta aproximadamente un 40 % del peso total de la composición, o hasta aproximadamente un 35 % del peso total de la composición, o hasta aproximadamente un 30 % del peso total de la composición, o hasta aproximadamente un 25 % del peso total de la composición, o hasta aproximadamente un 20 % del peso total de la composición, o hasta aproximadamente un 15 % del peso total de la composición, o hasta aproximadamente un 10 % del peso total de la composición, o menos.
Las vías adecuadas de administración pueden incluir, por ejemplo, administración oral, rectal, transmucosa, tópica, o intestinal; administración parenteral, que incluye inyección intramuscular, subcutánea, intravenosa, intramedular, así como inyección intratecal, intraventricular directa, intraperitoneal, intranasal, o intraocular. El compuesto de fórmula I se puede administrar además en formas farmacéuticas de liberación sostenida o controlada, que incluyen inyecciones de liberación lenta, bombas osmóticas, píldoras, parches transdérmicos (que incluyen electrotransporte), y similares, para la administración prolongada y/o controlada en el tiempo, pulsátil a una velocidad predeterminada. Las composiciones farmacéuticas se pueden fabricar de una manera que se conozca en sí misma, por ejemplo, por medio de procesos convencionales de mezcla, solución, granulación, fabricación de grageas, levigación, emulsión, encapsulación, atrapamiento o compresión.
De ese modo se pueden formular composiciones farmacéuticas para uso de manera convencional mediante el uso de uno o más vehículos fisiológicamente aceptables, que comprenden excipientes y agentes auxiliares que facilitan
el procesamiento de los compuestos activos en preparaciones que se puedan usar farmacéuticamente. La formulación adecuada depende de la vía de administración elegida. Se puede usar cualquiera de los métodos, vehículos, y excipientes muy conocidos como sea adecuado y se entienda en la técnica; por ejemplo, en Remington's Pharmaceutical Sciences, mencionado anteriormente.
Se pueden preparar inyectables en formas convencionales, como soluciones o suspensiones líquidas, formas sólidas adecuadas para la solución o suspensión en líquidos antes de la inyección, o como emulsiones. Los excipientes adecuados son, por ejemplo, agua, solución salina, dextrosa, manitol, lactosa, lecitina, albúmina, glutamato sódico, clorhidrato de cisteína, y similares. Además, si se desea, las composiciones farmacéuticas inyectables pueden contener cantidades menores de sustancias auxiliares no tóxicas, tales como agentes humectantes, agentes tamponadores del pH, y similares. Los tampones fisiológicamente compatibles incluyen, pero sin limitación, solución de Hank, solución de Ringer, o tampón de solución salina fisiológica. Si se desea, se pueden utilizar preparaciones potenciadoras de la absorción (por ejemplo, liposomas).
Para la administración transmucosa, se pueden usar agentes penetrantes adecuados para la barrera a penetrar en la formulación.
Las formulaciones farmacéuticas para la administración parenteral, por ejemplo, mediante inyección rápida o infusión continua, incluyen soluciones acuosas de los compuestos activos en una forma hidrosoluble. Además, las suspensiones de los compuestos activos se pueden preparar en forma de suspensiones oleosas adecuadas para inyección. Los disolventes o vehículos lipófilos adecuados incluyen aceites grasos tales como aceite de sésamo, u otros aceites orgánicos tales como aceites de soja, pomelo o almendra, o ésteres de ácidos grasos sintéticos, tales como oleato de etilo o triglicéridos, o liposomas. Las suspensiones acuosas pueden contener sustancias que aumentan la viscosidad de la suspensión, tales como carboximetilcelulosa sódica, sorbitol, o dextrano. Opcionalmente, la suspensión puede contener también estabilizantes adecuados o agentes que aumentan la solubilidad de los compuestos para permitir la preparación de soluciones muy concentradas. Las formulaciones para inyección se pueden presentar en una forma farmacéutica unitaria, por ejemplo, en ampollas o recipientes de múltiples dosis, con un conservante añadido. Las composiciones pueden tomar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilizantes y/o dispersantes. Alternativamente, el ingrediente activo puede estar en forma de polvo para reconstitución con un vehículo adecuado, por ejemplo agua estéril apirógena, antes del uso.
Para la administración oral, el compuesto de fórmula I se puede formular fácilmente combinando el compuesto activo con vehículos farmacéuticamente aceptables muy conocidos en la técnica. Tales vehículos posibilitan que el compuesto de fórmula I se formule en forma de comprimidos, comprimidos revestidos de película, píldoras, grageas, cápsulas, líquidos, geles, cápsulas de gelatina, miniesferas, microesferas, jarabes, suspensiones espesas, suspensiones y similares, para la ingestión oral por parte de un paciente a tratar. Las preparaciones farmacéuticas para uso oral se pueden obtener combinando el compuesto activo con un excipiente sólido, opcionalmente moliendo la mezcla resultante, y procesando la mezcla de gránulos, tras la adición de agentes auxiliares adecuados, si se desea, para obtener núcleos de grageas o comprimidos. Los excipientes adecuados son, en particular, cargas tales como azúcares, que incluyen lactosa, sacarosa, manitol, o sorbitol; preparaciones de celulosa tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma de tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa sódica, y/o polivinilpirrolidona (PVP). Si se desea, se pueden añadir agentes disgregantes, tales como polivinilpirrolidona reticulada, agar, o ácido algínico o una sal del mismo tal como alginato sódico. Se proporcionan núcleos de grageas con revestimientos adecuados. Para este fin se pueden usar soluciones concentradas de azúcares, que pueden contener opcionalmente goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, soluciones de laca y disolventes orgánicos adecuados o mezclas de disolventes. Se pueden añadir colorantes o pigmentos a los revestimientos de comprimidos 0 grageas para la identificación o para caracterizar las diferentes combinaciones de dosis del compuesto activo. Para este fin se pueden usar soluciones concentradas de azúcares, que pueden contener opcionalmente goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, soluciones de laca y disolventes orgánicos adecuados o mezclas de disolventes. Se pueden añadir colorantes o pigmentos a los comprimidos o revestimientos de grageas para la identificación o para caracterizar las diferentes combinaciones de dosis del compuesto activo. Además, se pueden añadir estabilizantes. Todas las formulaciones para administración oral deberían estar en las dosis adecuadas para tal administración. Se desvelan formulaciones del compuesto de fórmula 1 con un perfil aceptable de disolución de liberación inmediata y un método de fabricación sólido y escalable.
Las preparaciones farmacéuticas que se pueden usar oralmente incluyen cápsulas duras hechas de gelatina, así como cápsulas selladas blandas hechas de gelatina y un plastificante tal como glicerol o sorbitol. Las cápsulas duras pueden contener los ingredientes activos en mezcla con una carga tal como lactosa, aglutinantes tales como almidones, y/o lubricantes tales como talco o estearato magnésico y, opcionalmente, estabilizantes. En las cápsulas blandas, los compuestos activos pueden estar disueltos o suspendidos en líquidos adecuados, tales como aceites grasos, parafina líquida o polietilenglicoles líquidos. Además, se pueden añadir estabilizantes. Todas las formulaciones para administración oral deben estar en dosis adecuadas para tal administración.
Para administración bucal, las composiciones pueden tomar la forma de comprimidos o pastillas para chupar
formulados de una manera convencional.
Para la administración mediante inhalación, el compuesto de fórmula I se administra de manera conveniente en forma de una presentación en aerosol desde envases presurizados o un nebulizador, con el uso de un propelente adecuado, por ejemplo, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado. En el caso de un aerosol presurizado la unidad posológica se puede determinar proporcionando una válvula para liberar una cantidad medida. Las cápsulas y cartuchos, por ejemplo, de gelatina para el uso en un inhalador o insuflador se pueden formular para que contengan una mezcla en polvo del compuesto y una base en polvo adecuada, tal como lactosa o almidón.
En la presente memoria se describen además diversas composiciones farmacéuticas muy conocidas en la técnica farmacéutica para aplicaciones que incluyen administración intraocular, intranasal, e intraauricular. Los agentes penetrantes adecuados para estas aplicaciones se conocen generalmente en la técnica. Las composiciones farmacéuticas para la administración intraocular incluyen soluciones oftálmicas acuosas de los compuestos activos en forma hidrosoluble, tales como gotas oculares, o en goma de gelano (Shedden et al., Clin. Ther., 23(3):440-50 (2001)) o hidrogeles (Mayer et al., Ophthalmologica, 210(2):101-3 (1996)); pomadas oftálmicas; suspensiones oftálmicas, tales como microparticulados, partículas poliméricas pequeñas que contienen fármacos que se suspenden en un vehículo líquido (Joshi, A., J. Ocul. Pharmacol., 10(1):29-45 (1994)), formulaciones liposolubles (Alm et al., Prog. Clin. Biol. Res., 312:447-58 (1989)), y microesferas (Mordenti, Toxicol. Sci., 52(1 ):101 -6 (1999)); e insertos oculares. Tales formulaciones farmacéuticas adecuadas se formulan muy a menudo y preferentemente para que sean estériles, isotónicas y tamponadas para proporcionar estabilidad y comodidad. Las composiciones farmacéuticas para administración intranasal pueden incluir además gotas y aerosoles preparados a menudo para simular en muchos aspectos las secreciones nasales, para asegurar el mantenimiento de la acción ciliar normal. Como se describe en Remington's Pharmaceutical Sciences, 18a Ed., Mack Publishing Co., Easton, PA (1990), y conocen muy bien los expertos en la materia, las formulaciones adecuadas son con mucha frecuencia y preferentemente isotónicas, ligeramente tamponadas para mantener un pH de 5,5 a 6,5, y con mucha frecuencia y preferentemente incluyen conservantes antimicrobianos y estabilizantes farmacológicos adecuados. Las formulaciones farmacéuticas para administración intraauricular incluyen suspensiones y pomadas para la aplicación tópica en el oído. Los disolventes comunes para tales formulaciones auditivas incluyen glicerina y agua.
El compuesto de fórmula I también se puede formular en composiciones rectales tales como supositorios o enemas de retención, por ejemplo, que contienen bases de supositorio convencionales tales como manteca de cacao u otros glicéridos.
Además de las formulaciones descritas previamente, el compuesto de fórmula I también se puede formular en forma de una preparación de liberación lenta. Tales formulaciones de acción prolongada se pueden administrar mediante implantación (por ejemplo, de manera subcutánea o intramuscular) o mediante inyección intramuscular. Así, por ejemplo, el compuesto de fórmula I se puede formular con materiales poliméricos o hidrófobos adecuados (por ejemplo, en forma de una emulsión en un aceite aceptable) o resinas de intercambio iónico, o en forma de derivados poco solubles, por ejemplo, en forma de una sal poco soluble.
Para los compuestos hidrófobos, un vehículo farmacéutico adecuado puede ser un sistema de codisolventes que comprende alcohol bencílico, un tensioactivo apolar, un polímero orgánico miscible con agua, y una fase acuosa. Un sistema de codisolventes habitual usado es el sistema de codisolventes VPD, que es una solución de un 3 % p/v de alcohol bencílico, un 8 % p/v del tensioactivo apolar Polisorbato 80™, y un 65 % p/v de polietilenglicol 300, llevado hasta el volumen total con etanol absoluto. Naturalmente, las proporciones de un sistema de codisolventes se pueden variar considerablemente sin destruir sus características de solubilidad y toxicidad. Además, se puede variar la identidad de los componentes de los codisolventes: por ejemplo, se pueden usar otros tensioactivos apolares de toxicidad baja en vez de POLISORBATO 80™; se puede variar el tamaño de la fracción de polietilenglicol; otros polímeros biocompatibles pueden sustituir al polietilenglicol, por ejemplo, polivinilpirrolidona; y otros azúcares o polisacáridos pueden sustituir a la dextrosa.
Alternativamente, se pueden emplear otros sistemas de administración para los compuestos farmacéuticos hidrófobos. Los liposomas y las emulsiones son ejemplos bien conocidos de vehículos de administración para fármacos hidrófobos. También se pueden emplear ciertos disolventes orgánicos tales como dimetilsulfóxido, aunque normalmente a costa de una toxicidad mayor. Además, los compuestos se pueden administrar mediante el uso de un sistema de liberación sostenida, tal como matrices semipermeables de polímeros hidrófobos sólidos que contienen el agente terapéutico. Se han establecido diferentes materiales de liberación sostenida y son muy conocidos por los expertos en la materia. Dependiendo de su naturaleza química, las cápsulas de liberación sostenida pueden liberar los compuestos durante un periodo de unas semanas a más de 100 días. Dependiendo de la naturaleza química y la estabilidad biológica del reactivo terapéutico, se pueden emplear estrategias adicionales para estabilización de proteínas.
Los agentes destinados a administrarse de forma intracelular se pueden administrar mediante el uso de técnicas muy conocidas para los expertos en la materia. Por ejemplo, tales agentes se pueden encapsular en liposomas. Todas las moléculas presentes en una solución acuosa en el momento de la formación de los liposomas se
incorporan al interior acuoso. El contenido liposómico se protege del microentorno y, debido a que los liposomas se fusionan con las membranas celulares, se transportan de manera eficaz al citoplasma celular. El liposoma se puede revestir con un anticuerpo específico de tejido. Los liposomas se transportarán y se absorberán selectivamente en el órgano deseado. Alternativamente, se pueden administrar directamente de manera intracelular moléculas orgánicas hidrófobas pequeñas.
Se pueden incorporar agentes terapéuticos o de diagnóstico adicionales a las composiciones farmacéuticas. Además, o alternativamente, las composiciones farmacéuticas se pueden combinar con otras composiciones que contienen otros agentes terapéuticos o de diagnóstico.
Métodos de Administración
El compuesto de fórmula I o las composiciones farmacéuticas se pueden administrar al paciente mediante cualquier medio adecuado. Los ejemplos no limitantes de los métodos de administración incluyen, entre otros, (a) administración a través de vías orales, incluyendo administración en cápsulas, comprimidos, gránulos, aerosoles, jarabes, u otras formas similares; (b) administración a través de vías no orales tales como rectal, vaginal, intrauretral, intraocular, intranasal, o intraauricular, incluyendo administración en forma de una suspensión acuosa, una preparación oleosa o similares, o en forma de un gotero, aerosol, supositorio, bálsamo, pomada o similares; (c) administración por medio de inyección, por vía subcutánea, intraperitoneal, intravenosa, intramuscular, intradérmica, intraorbital, intracapsular, intraespinal, intraesternal, o similar, incluyendo administración con una bomba de infusión; (d) administración local tal como mediante inyección directamente en el área renal o cardiaca, por ejemplo, mediante implante de liberación sostenida; así como (e) administración por vía tópica; como consideren adecuado los expertos en la materia para poner en contacto el compuesto de fórmula I con el tejido vivo.
Las composiciones farmacéuticas adecuadas para administración incluyen composiciones en las que el compuesto de fórmula I está contenido en una cantidad eficaz para conseguir el propósito deseado. La cantidad terapéuticamente eficaz del compuesto de fórmula I desvelado en la presente memoria necesaria como dosis dependerá de la vía de administración, el tipo de animal, incluyendo un ser humano, a tratar, y las características físicas del animal específico en cuestión. La dosis se puede adaptar para conseguir un efecto deseado, pero dependerá de factores tales como el peso, la dieta, la medicación concurrente y otros factores que reconocerán los expertos en las técnicas médicas. De manera más específica, una cantidad terapéuticamente eficaz significa una cantidad de compuesto eficaz para prevenir, mitigar o mejorar los síntomas de la enfermedad, o para prolongar la supervivencia del sujeto a tratar. La determinación de una cantidad terapéuticamente eficaz se halla dentro de la capacidad de los expertos en la materia.
Como será fácilmente evidente para un experto en la materia, la posología in vivo útil a administrar y el modo particular de administración variarán dependiendo de la edad, el peso y la especie de mamífero tratado, y el uso específico para el que se emplea el compuesto de fórmula I. La determinación de los niveles posológicos eficaces, que son los niveles posológicos necesarios para conseguir el resultado deseado, la puede llevar a cabo un experto en la materia mediante el uso de métodos farmacológicos rutinarios. En general, las aplicaciones clínicas humanas de productos comienzan a niveles de dosis inferiores, y se incrementa el nivel de dosis hasta que se alcanza el efecto deseado. Alternativamente, se pueden usar estudios in vitro aceptables para establecer las dosis útiles y las vías de administración de las composiciones identificadas mediante los presentes métodos con el uso de métodos farmacológicos establecidos.
En estudios con animales no humanos, las aplicaciones de los productos potenciales comienzan a niveles de dosis mayores, y las dosis disminuyen hasta que ya no se alcanza el efecto deseado o hasta que desaparecen los efectos secundarios adversos. La dosis puede oscilar ampliamente, dependiendo de los efectos deseados y la indicación terapéutica. Por lo general, las dosis pueden estar entre aproximadamente 10 microgramos/kg y 100 mg/kg de peso corporal, preferentemente entre aproximadamente 100 microgramos/kg y 10 mg/kg de peso corporal. Alternativamente, las dosis se pueden basar en y calcular con el área superficial del paciente, como entienden los expertos en la materia.
Dependiendo de la gravedad y sensibilidad de la afección a tratar, la posología también puede ser una única administración de una composición de liberación lenta, y el curso de tratamiento dura de varios días a varias semanas o hasta conseguir la curación o hasta alcanzar una disminución del estado patológico. La cantidad de una composición a administrar dependerá, por supuesto, de muchos factores que incluyen el sujeto a tratar, la gravedad de la afección, la manera de administración, el juicio del médico que prescribe el tratamiento. El compuesto de fórmula I se puede administrar por vía oral o por medio de inyección a una dosis de 0,001 a 2500 mg/kg de peso corporal del paciente al día. El intervalo de dosis para seres humanos adultos es en general de 0,01 mg a 10 g/día. Los comprimidos u otras formas de presentación proporcionadas en unidades discretas pueden contener de manera conveniente una cantidad del compuesto de fórmula I que es eficaz a tal dosis o como múltiplo de la misma, por ejemplo, unidades que contienen de 5 mg a 1000 mg, normalmente de aproximadamente 100 mg a aproximadamente 800 mg. La cantidad precisa de compuesto administrada a un paciente será responsabilidad del médico que lo atiende. Sin embargo, la dosis empleada dependerá de varios factores, que incluyen la edad y el sexo del paciente, el trastorno preciso a tratar, y su gravedad. Además, la vía de administración puede variar dependiendo
de la afección y su gravedad.
La formulación, vía de administración y dosis exactas para las composiciones farmacéuticas del compuesto de fórmula I las puede elegir el médico individual en vista de la afección del paciente. (Véase, por ejemplo, Fingl et al.
1975, en "The Pharmacological Basis of Therapeutics" con referencia particular al cap. 1). En general, el intervalo de dosis de la composición administrada al paciente puede ser de aproximadamente 0,01 a aproximadamente 1000 mg/kg de peso corporal del paciente. La dosis puede ser una única dosis o una serie de dos o más dosis administradas en el curso de uno o más días, según lo necesite el paciente. En los casos en los que se han establecido dosis para seres humanos de los compuestos para al menos cierta afección, se pueden usar las mismas dosis, o dosis que estén entre aproximadamente un 0,1 % y aproximadamente un 500 %, más preferentemente entre aproximadamente un 25 % y aproximadamente un 250 % de la dosis establecida para seres humanos. Cuando no está establecida una dosis para seres humanos, como será el caso para compuestos farmacéuticos recién descubiertos, se puede deducir una dosis adecuada para seres humanos a partir de los valores de DE50 o DI50, u otros valores adecuados obtenidos de estudios in vitro o in vivo, tal como se determina mediante estudios de toxicidad y estudios de eficacia en animales.
Se debería indicar que el médico que aplica el tratamiento sabrá cómo y cuándo terminar, interrumpir, o ajustar la administración debido a la toxicidad o a disfunciones de órganos. A la inversa, el médico que aplica el tratamiento además sabrá ajustar el tratamiento a niveles mayores si la respuesta clínica no fuera adecuada (excluyendo la toxicidad). La magnitud de una dosis administrada en el tratamiento del trastorno de interés variará con la gravedad de la afección a tratar y la vía de administración. La gravedad de la afección se puede determinar, por ejemplo, en parte mediante métodos habituales de determinación del pronóstico. Además, la dosis y quizás la frecuencia de la dosis variarán también según la edad, el peso corporal, y la respuesta del paciente individual. Se puede usar un programa comparable al discutido anteriormente en medicina veterinaria.
En el contexto de la presente invención, la cantidad del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo es de 200 mg a 800 mg. En la medida en que se mencionan otras cantidades en la presente memoria, se incluyen únicamente con fines de referencia. Aunque la dosis exacta se determinará para cada fármaco, en la mayoría de los casos, se pueden hacer ciertas generalizaciones con respecto a la dosis. La pauta posológica diaria para un paciente humano adulto puede ser, por ejemplo, una dosis oral de entre 0,1 mg y 2000 mg de cada ingrediente activo, preferentemente entre 1 mg y 1500 mg, por ejemplo, de 5 a 1000 mg. Una dosis oral de cada ingrediente activo puede estar entre 1 mg y 1000 mg, preferentemente entre 50 mg y 900 mg, por ejemplo, de 100 a 800 mg. En los casos de administración de una sal farmacéuticamente aceptable, las dosis se pueden calcular como la base libre. La composición se puede administrar de 1 a 4 veces al día. Alternativamente, las composiciones del compuesto de fórmula I se pueden administrar mediante infusión intravenosa continua, preferentemente a una dosis de cada ingrediente activo de hasta 1000 mg al día. Como entenderán los expertos en la materia, en ciertas situaciones puede ser necesario administrar el compuesto desvelado en la presente memoria en cantidades que superan, o incluso superan en gran medida, el intervalo posológico preferente indicado anteriormente, para tratar de manera eficaz y agresiva enfermedades o infecciones especialmente agresivas. En algunas realizaciones, el compuesto de fórmula I se administrará durante un periodo de terapia continua, por ejemplo durante una semana o más, o durante meses o años.
En algunas realizaciones, la pauta posológica del compuesto de fórmula I se administra durante un periodo de tiempo, periodo de tiempo que puede ser, por ejemplo, de al menos aproximadamente 4 semanas a al menos aproximadamente 8 semanas, de al menos aproximadamente 4 semanas a al menos aproximadamente 12 semanas, de al menos aproximadamente 4 semanas a al menos aproximadamente 16 semanas, o mayor. La pauta posológica del compuesto de fórmula I se puede administrar tres veces al día, dos veces al día, a diario, cada dos días, tres veces a la semana, cada dos semanas, tres veces al mes, una vez al mes, de forma sustancialmente continua o continuamente.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 10 mg a aproximadamente 1000 mg, de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método para usar una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 100 mg a aproximadamente 1000 mg, de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 100 mg a aproximadamente 900 mg, de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 100 mg de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento. Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 200 mg de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento. Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 400 mg de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento. Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 800 mg de fármaco por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento. Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 0,01 mg a aproximadamente 1000 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 0,1 mg a aproximadamente 500 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 0,1 mg a aproximadamente 100 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 0,5 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la
duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 1 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 5 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 25 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
Algunas divulgaciones proporcionan un método de uso de una cantidad eficaz del compuesto de fórmula I en el tratamiento de trastornos dependientes de endotelina o dependientes de angiotensina II en un paciente, que comprende administrar al paciente una posología del compuesto de fórmula I que contiene una cantidad de aproximadamente 50 mg de fármaco por kilogramo de peso corporal por dosis del compuesto de fórmula I, por vía oral, tres veces al mes, una vez al mes, una vez a la semana, una vez cada tres días, una vez cada dos días, una vez al día, dos veces al día, o tres veces al día de forma sustancialmente continua o continuamente, durante la duración deseada del tratamiento.
La cantidad y el intervalo de dosis se pueden ajustar individualmente para proporcionar niveles plasmáticos del resto activo que sean suficientes para mantener los efectos de modulación, o una concentración eficaz mínima (CEM). La CEM variará para cada compuesto, pero se puede estimar a partir de datos in vitro. Las dosis necesarias para alcanzar la CEM dependerán de las características del individuo y de la vía de administración. Sin embargo, se pueden usar ensayos de HPLC o bioensayos para determinar las concentraciones plasmáticas.
Los intervalos posológicos también se pueden determinar usando el valor de CEM. Las composiciones se pueden administrar usando un régimen que mantiene los niveles plasmáticos por encima de la CEM durante un 10-90 % del tiempo, por ejemplo, entre un 15-30 %, 20-45 %, 25-50 %, 30-55 %, 35-60 %, 40-65 %, 45-70 %, 50-75 %, 55-80 %, 60-90 %, 65-75 %, 70-80 %, 75-85 %, 15-90 %, 20-90 %, 25-90 %, 30-90 %, 35-90 %, 40-90 %, 45-90 %, 50-90 %, 55-90 %, 60-90 %, 65-90 %, 70-90 %, 75-90 %, o 80-90 %. Las composiciones se pueden administrar mediante el uso de un régimen que mantiene los niveles plasmáticos por encima de la CEM durante un 20-90 % del tiempo. Las composiciones se pueden administrar mediante el uso de un régimen que mantiene los niveles plasmáticos por encima de la CEM durante un 30-90 % del tiempo, entre un 40-90 % y más en general entre un 50-90 %.
En los casos de administración local o absorción selectiva, la concentración local eficaz del fármaco puede no estar relacionada con la concentración plasmática.
La cantidad de la composición administrada puede depender del sujeto a tratar, del peso del sujeto, la gravedad de la aflicción, la forma de administración y el criterio del médico que prescribe el tratamiento.
El compuesto de fórmula I desvelado en la presente memoria se puede estudiar con respecto a su eficacia y toxicidad usando métodos conocidos. Por ejemplo, se puede establecer la toxicología del compuesto de fórmula I determinando la toxicidad in vitro hacia una línea celular, tal como una línea celular mamífera, y preferentemente humana. Los resultados de tales estudios a menudo son predictivos de la toxicidad en animales, tales como mamíferos, o de manera más específica, en seres humanos. Alternativamente, se puede determinar la toxicidad del compuesto de fórmula I en un modelo animal, tal como en ratones, ratas, conejos, o monos, usando métodos conocidos. La eficacia del compuesto de fórmula I se puede establecer usando varios métodos reconocidos, tales como métodos in vitro, modelos animales, o ensayos clínicos en seres humanos. Existen modelos in vitro reconocidos para prácticamente cada clase de afección, que incluyen, pero sin limitación, cáncer, enfermedad cardiovascular, y diversas disfunciones inmunitarias. De forma similar, se pueden usar modelos animales aceptables para establecer la eficacia de los productos químicos para tratar tales afecciones. Cuando se selecciona un modelo
para determinar la eficacia, el experto en la materia se puede guiar por el estado de la técnica para elegir un modelo, dosis, y vía de administración, y régimen adecuados. Por supuesto, también se pueden usar ensayos clínicos en seres humanos para determinar la eficacia de un compuesto en seres humanos.
Las composiciones se pueden presentar, si se desea, en un envase o dispositivo dispensador que puede contener una o más formas farmacéuticas unitarias que contienen el ingrediente activo. El envase puede comprender, por ejemplo, una lámina de metal o plástico, tal como un blíster. El envase o dispositivo dispensador puede ir acompañado de instrucciones para la administración. El envase o dispensador también puede ir acompañado de una nota asociada al recipiente en la forma prescrita por un organismo estatal que regula la fabricación, el uso, o la comercialización de productos farmacéuticos, nota que refleja la aprobación por parte del organismo estatal de la forma farmacéutica para la administración humana o veterinaria. Tal nota, por ejemplo, puede ser el etiquetado aprobado por la Administración de Alimentos y Medicamentos de EE.UU. para fármacos de prescripción, o el prospecto del producto aprobado. También se pueden preparar composiciones que comprenden el compuesto de fórmula I formulado en un vehículo farmacéutico compatible, colocarlas en un recipiente adecuado, y etiquetarlas para el tratamiento de una afección indicada.
El experto en la materia puede determinar una cantidad eficaz del compuesto de fórmula I, e incluye cantidades ejemplares de dosis para un ser humano de aproximadamente 0,1 a aproximadamente 100 mg/kg, preferentemente de aproximadamente 0,2 a aproximadamente 50 mg/kg y más preferentemente de aproximadamente 0,5 a aproximadamente 25 mg/kg de peso corporal (o de aproximadamente 1 a aproximadamente 2500 mg, preferentemente de aproximadamente 100 a aproximadamente 800 mg) de compuesto activo al día, que se puede administrar en una única dosis o en forma de dosis divididas individuales, tal como de 1 a 4 veces al día. Se entenderá que el nivel específico de la dosis y la frecuencia de la dosis para cualquier sujeto particular puede variar, y dependerá de una diversidad de factores que incluyen la actividad del compuesto específico empleado, estabilidad metabólica y duración de acción de ese compuesto, especie, edad, peso corporal, salud general, sexo y dieta del sujeto, modo y momento de la administración, velocidad de excreción, combinación de fármacos, y gravedad de la afección particular. Los sujetos preferentes para el tratamiento incluyen animales, lo más preferentemente especies mamíferas tales como seres humanos, y animales domésticos tales como perros, gatos y similares, sometidos a trastornos dependientes de endotelina o dependientes de angiotensina II.
También se desvelan composiciones farmacéuticas que comprenden el compuesto de fórmula I capaces de tratar un trastorno dependiente de endotelina o dependiente de angiotensina II en una cantidad eficaz para los mismos, y un vehículo o diluyente farmacéuticamente aceptable. Las composiciones pueden contener otros agentes terapéuticos como se describe más adelante, y se pueden formular, por ejemplo, empleando vehículos o diluyentes sólidos o líquidos convencionales, así como aditivos farmacéuticos de un tipo adecuado para el modo de administración deseado (por ejemplo, excipientes, aglutinantes, conservantes, estabilizantes, aromas, etc.) según métodos tales como los conocidos en la técnica de formulación farmacéutica o requeridos por la práctica farmacéutica aceptada.
El compuesto de fórmula I se puede administrar mediante cualquier medio adecuado, por ejemplo, por vía oral, tal como en forma de comprimidos, cápsulas, gránulos o polvos; sublingual; bucal; parenteral, tal como mediante inyección subcutánea, intravenosa, intramuscular, o intraesternal o mediante técnicas de infusión (por ejemplo, en forma de soluciones o suspensiones acuosas o no acuosas inyectables estériles); nasal, tal como mediante pulverización por inhalación; tópica, tal como en forma de una crema o pomada; o rectal, tal como en forma de supositorios; en formas farmacéuticas unitarias que contienen vehículos o diluyentes no tóxicos farmacéuticamente aceptables. En algunas realizaciones, la cantidad del compuesto de fórmula I administrada en la formulación puede ser de 800 mg por dosis unitaria. La cantidad del compuesto de fórmula I administrada al día puede ser de 800 mg. Por ejemplo, se pueden administrar aproximadamente 400 mg del compuesto en una formulación dos veces al día, se pueden administrar 200 mg del compuesto en una formulación cuatro veces al día, se pueden administrar 100 mg del compuesto en una formulación ocho veces al día.
El compuesto de fórmula I, por ejemplo, se administra en una forma adecuada para liberación inmediata o liberación prolongada. La liberación inmediata o liberación prolongada se puede conseguir mediante el uso de composiciones farmacéuticas adecuadas que comprenden el compuesto de fórmula I, o, en particular en el caso de liberación prolongada, mediante el uso de dispositivos tales como implantes subcutáneos o bombas osmóticas. La cantidad del compuesto de fórmula I, en la formulación de liberación inmediata, puede ser de aproximadamente 800 mg por unidad posológica. La cantidad del compuesto de fórmula I, en la formulación de liberación inmediata, administrada al día puede ser de aproximadamente 800 mg. Por ejemplo, se pueden administrar aproximadamente 400 mg del compuesto en una formulación de liberación inmediata dos veces al día, se pueden administrar aproximadamente 200 mg del compuesto en una formulación de liberación inmediata cuatro veces al día, se pueden administrar aproximadamente 100 mg del compuesto en una formulación de liberación inmediata ocho veces al día.
El compuesto de fórmula I se puede administrar también por vía liposómica. Por ejemplo, la sustancia activa se puede utilizar en una composición tal como un comprimido, cápsula, solución o suspensión que contiene de aproximadamente 5 mg a aproximadamente 1000 mg por dosis unitaria del compuesto de fórmula I o en forma tópica para la curación de heridas (0,01 a 5 % en peso del compuesto de fórmula I, 1 a 5 tratamientos al día). La cantidad del compuesto de fórmula I, administrada por vía liposómica, puede ser de aproximadamente 800 mg por
unidad posológica. La cantidad del compuesto de fórmula I, en la formulación de liposomas, administrada al día puede ser de aproximadamente 800 mg. Por ejemplo, se pueden administrar aproximadamente 400 mg del compuesto en una formulación de liposomas dos veces al día, se pueden administrar aproximadamente 200 mg del compuesto en una formulación de liposomas cuatro veces al día, se pueden administrar aproximadamente 100 mg del compuesto en una formulación de liposomas ocho veces al día.
El compuesto de fórmula I se puede componer de una manera convencional con un vehículo, excipiente, aglutinante, conservante, estabilizante, aromatizante, etc., fisiológicamente aceptable o con un vehículo tópico. La cantidad del compuesto de fórmula I puede ser de aproximadamente 800 mg por unidad posológica. La cantidad del compuesto de fórmula I administrada al día puede ser de aproximadamente 800 mg. Por ejemplo, se pueden administrar aproximadamente 400 mg del compuesto dos veces al día, se pueden administrar aproximadamente 200 mg del compuesto cuatro veces al día, se pueden administrar aproximadamente 100 mg del compuesto ocho veces al día.
El compuesto de fórmula I también se puede formular en composiciones tales como soluciones o suspensiones estériles para administración parenteral. Se pueden componer de aproximadamente 0,1 a aproximadamente 800 miligramos del compuesto de fórmula I, habitualmente 200 miligramos, más habitualmente aproximadamente 400 miligramos, lo más habitualmente aproximadamente 800 miligramos, con un vehículo, excipiente, aglutinante, conservante, estabilizante, etc., fisiológicamente aceptable en una forma farmacéutica unitaria como requiere la práctica farmacéutica aceptada. La cantidad de la sustancia activa en estas composiciones o preparaciones es preferentemente tal que se obtiene una dosis adecuada en el intervalo indicado. La cantidad del compuesto de fórmula I, administrada por vía parenteral, puede ser de aproximadamente 800 mg por dosis unitaria. La cantidad del compuesto de fórmula I administrada al día puede ser de aproximadamente 800 mg. Por ejemplo, se pueden administrar aproximadamente 400 mg del compuesto dos veces al día, se pueden administrar aproximadamente 200 mg del compuesto cuatro veces al día, o se pueden administrar aproximadamente 100 mg del compuesto ocho veces al día.
Las composiciones ejemplares para la administración oral incluyen suspensiones que pueden contener, por ejemplo, celulosa microcristalina para conferir volumen, ácido algínico o alginato sódico como agente de suspensión, metilcelulosa como potenciador de la viscosidad, y agentes edulcorantes o aromatizantes tales como los conocidos en la técnica; y comprimidos de liberación inmediata que pueden contener, por ejemplo, celulosa microcristalina, fosfato dicálcico, almidón, estearato magnésico y/o lactosa y/u otros excipientes, aglutinantes, extensores, disgregantes, diluyentes y lubricantes tales como los conocidos en la técnica. Los comprimidos moldeados, los comprimidos prensados o los comprimidos liofilizados son formas ejemplares que se pueden usar. Las composiciones ejemplares incluyen las que formulan el compuesto de fórmula I con diluyentes de disolución rápida tales como manitol, lactosa, sacarosa y/o ciclodextrinas. También se pueden incluir en tales formulaciones excipientes de peso molecular elevado tales como celulosas (Avicel) o polietilenglicoles (PEG). Tales formulaciones también pueden incluir un excipiente para ayudar en la adhesión mucosa, tal como hidroxipropilcelulosa (HPC), hidroxipropilmetilcelulosa (HPMC), carboximetilcelulosa sódica (SCMC), copolímero de anhídrido maleico (por ejemplo, Gantrez), y agentes para controlar la liberación, tales como copolímero poliacrílico (por ejemplo, Carbopol 934). También se pueden añadir agentes lubricantes, deslizantes, aromatizantes, colorantes y estabilizantes para facilitar la fabricación y el uso.
Las composiciones ejemplares para la administración mediante aerosol o inhalación nasal incluyen soluciones en solución salina que pueden contener, por ejemplo, alcohol bencílico u otros conservantes adecuados, promotores de la absorción para aumentar la biodisponibilidad, y/u otros agentes solubilizantes o dispersantes tales como los conocidos en la técnica.
Las composiciones ejemplares para la administración parenteral incluyen soluciones o suspensiones inyectables que pueden contener, por ejemplo, diluyentes o disolventes parenteralmente aceptables no tóxicos adecuados, tales como manitol, 1,3-butanodiol, agua, solución de Ringer, una solución isotónica de cloruro sódico, u otro dispersante o humectante adecuado y agentes de suspensión, que incluyen mono o diglicéridos sintéticos, y ácidos grasos, que incluyen ácido oleico.
Las composiciones ejemplares para la administración rectal incluyen supositorios que pueden contener, por ejemplo, un excipiente no irritante adecuado, tal como manteca de cacao, ésteres de glicéridos sintéticos o polietilenglicoles, que son sólidos a las temperaturas habituales, pero se licúan y/o se disuelven en la cavidad rectal para liberar el fármaco.
Las composiciones ejemplares para la administración tópica incluyen un vehículo tópico tal como Plastibase (aceite mineral gelificado con polietileno). Por ejemplo, el compuesto de fórmula I se puede administrar de manera tópica para tratar enfermedades vasculares periféricas, y como tal se puede formular en forma de una crema o pomada.
El compuesto de fórmula I se puede emplear solo o en combinación con otros agentes terapéuticos adecuados útiles en el tratamiento de los trastornos dependientes de endotelina o dependientes de angiotensina II. Por ejemplo, el compuesto de fórmula I se puede formular en combinación con inhibidores de la enzima conversora de endotelina (ECE), tales como fosforamidón; antagonistas de receptores de tromboxano, tales como ifetrobán; activadores de los
canales de potasio; inhibidores de trombina (por ejemplo, hirudina y similares); inhibidores de factores de crecimiento, tales como moduladores de la actividad de PDGF; antagonistas del factor activador de plaquetas (PAF); agentes antiplaquetarios tales como bloqueantes de GPIIb/IIIa (por ejemplo, abciximab, eptifibatida, y tirofibán), antagonistas de P2Y(AC) (por ejemplo, clopidogrel, ticlopidina y CS-747), y aspirina; anticoagulantes tales como warfarina, heparinas de peso molecular bajo tales como enoxaparina, inhibidores del Factor VIIa, e inhibidores del Factor Xa tales como los descritos en la patente de EE.UU. n.° 6.297.233, expedida el 2 de octubre de 2001; inhibidores de renina; inhibidores de la enzima conversora de angiotensina (ACE) tales como captopril, zofenopril, fosinopril, ceranapril, alacepril, enalapril, delapril, pentopril, quinapril, ramipril, lisinopril y las sales de tales compuestos; inhibidores de endopeptidasa neutra (NEP); inhibidores de vasopeptidasa (inhibidores dobles NEP-ACE), tales como omapatrilat y gemopatrilat; inhibidores de HMG CoA reductasa, tales como pravastatina, lovastatina, atorvastatina, simvastatina, NK-104 (también conocido como itavastatina, o nisvatatina o nisbatatina) y ZD-4522 (también conocido como rosuvastatina, o atavastatina o visastatina); inhibidores de escualeno sintetasa; fibratos; secuestrantes de ácidos biliares tales como questrán; niacina; agentes antiateroescleróticos tales como inhibidores de ACAT; inhibidores de MTP tales como los descritos en el documento de EE.UU. con n.° de serie 09/007.938, presentado el 16 de enero de 1998; bloqueantes de los canales de calcio, tales como besilato de amlodipina; activadores de los canales de potasio; agentes alfa-adrenérgicos, agentes beta-adrenérgicos tales como carvedilol y metoprolol; agentes antiarrítmicos; diuréticos, tales como clorotiazida, hidroclorotiazida, flumetiazida, hidroflumetiazida, bendroflumetiazida, metilclorotiazida, triclorometiazida, politiazida o benzotiazida, así como ácido etacrínico, tricrinafeno, clortalidona, furosemida, musolimina, bumetanida, triamtereno, amilorida y espironolactona, y las sales de tales compuestos; agentes trombolíticos tales como el activador del plasminógeno tisular (tPA), tPA recombinante, estreptoquinasa, uroquinasa, prouroquinasa y complejo activador de estreptoquinasa-plasminógeno anisoilado (APSAC); agentes antidiabéticos tales como biguanidas (por ejemplo metformina), inhibidores de glucosidasa (por ejemplo, acarbosa), insulinas, meglitinidas (por ejemplo, repaglinida), sulfonilureas (por ejemplo, glimepirida, gliburida, y glipizida), combinaciones biguanida/gliburida tales como las descritas en la patente de EE.UU. n.° 6.586.438, expedida el 1 de julio de 2003 y la patente de EE.UU. n.° 7.598.262, expedida el 6 de octubre de 2009; tiozolidindionas (por ejemplo troglitazona, rosiglitazona y pioglitazona), y agonistas de PPAR-gamma; antagonistas de receptores mineralocorticoides, tales como espironolactona y eplerenona; secretagogos de la hormona del crecimiento, tales como los descritos en la patente de EE.UU. n.° 6.380.184, expedida el 30 de abril de 2002, y el documento de EE.UU. con n.° de serie 6.518.292, presentado el 11 de febrero de 2003; inhibidores de aP2, tales como los descritos en el documento de EE.UU. con n.° de serie 7.390.824, presentado el 24 de junio de 2008, y el documento de EE.UU. con n.° de serie 09/390.275, presentado el 7 de septiembre de 1999; glucósidos digitálicos; ouabaína; fármacos antiinflamatorios no esteroideos (AINE), tales como aspirina e ibuprofeno; inhibidores de fosfodiesterasa, tales como inhibidores de PDE III (por ejemplo, cilostazol) e inhibidores de PDE V (por ejemplo, sildenafil); inhibidores de proteína tirosina quinasa; antiinflamatorios; antiproliferativos tales como metotrexato, FK506 (tacrólimus, Prograf), micofenolato y mofetilo; agentes quimioterápicos; inmunosupresores; agentes antineoplásicos y agentes citotóxicos (por ejemplo, agentes alquilantes, tales como mostazas nitrogenadas, sulfonatos de alquilo, nitrosoureas, etileniminas, y triazenos); antimetabolitos tales como antagonistas de folato, análogos de purina, y análogos de pirimidina; antibióticos, tales como antraciclinas, bleomicinas, mitomicina, dactinomicina, y plicamicina; enzimas, tales como L-asparaginasa; inhibidores de farnesil-proteína transferasa; agentes hormonales, tales como glucocorticoides (por ejemplo, cortisona), estrógenos/antiestrógenos, andrógenos/antiandrógenos, progestinas, y antagonistas de la hormona liberadora de la hormona luteinizante, acetato de octreótido; agentes de alteración de microtúbulos, tales como ecteinascidinas o sus análogos y derivados; agentes estabilizantes de microtúbulos, tales como paclitaxel (Taxol®), docetaxel (Taxotere®), y epotilonas A-F o sus análogos o derivados; productos derivados de plantas, tal como alcaloides de la vinca, epipodofilotoxinas, taxanos; e inhibidores de topoisomerasa; inhibidores de prenil-proteína transferasa; y agentes diversos, tales como hidroxiurea, procarbazina, mitotano, hexametilmelamina, complejos de coordinación de platino, tales como cisplatino y carboplatino); ciclosporinas; esteroides tales como prednisona o dexametasona; compuestos de oro; fármacos citotóxicos tales como azatiprina y ciclofosfamida; inhibidores de TNF-alfa tales como tenidap; anticuerpos anti-TNF o receptor de TNF soluble, tales como etanercept (Enbrel) rapamicina (sirolimus o Rapamune), leflunimida (Arava); e inhibidores de ciclooxigenasa-2 (COX-2) tales como celecoxib (Celebrex) y rofecoxib (Vioxx).
Si se formula en forma de una dosis fija, tales productos de combinación emplean el compuesto de fórmula I dentro del intervalo posológico descrito más adelante, y el otro agente farmacéuticamente activo dentro de su intervalo posológico aprobado. El compuesto de fórmula I también se puede formular con, o puede ser útil junto con, agentes antifúngicos e inmunosupresores tales como anfotericina B, ciclosporinas y similares para contrarrestar la contracción glomerular y la nefrotoxicidad secundarias a tales compuestos. El compuesto de fórmula I se puede usar también junto con la hemodiálisis.
Los otros agentes terapéuticos anteriores, cuando se emplean en combinación con el compuesto de fórmula I, se pueden usar, por ejemplo, en las cantidades indicadas en Physicians' Desk Reference (PDR) o como determine de otra manera el experto en la materia.
Se pueden emplear los siguientes ensayos para determinar el grado de actividad de un compuesto ("fármaco") como antagonista de receptores de endotelina y angiotensina II. Se ha probado el compuesto de fórmula I descrito en los Ejemplos siguientes en estos ensayos, y ha mostrado actividad.
Ensayo de Unión de Células Adheridas de ETa/b
Se cultivaron células CHO-K1 que expresaban el receptor humano de endotelina A o endotelina B en medio F12 de Ham (Gibco/BRL, Grand Island, N.Y.) con suero bovino fetal al 10 % (Hyclone), complementado con 300 pg/ml de Geneticina (G-418 Gibco BRL Products, Grand Island, N.Y.), y se mantuvieron a 37 °C con CO2 al 5 % en un incubador humidificado. Veinticuatro horas antes del ensayo, las células se trataron con tripsina-EDTA al 0,25 % y se sembraron en placas Falcon de cultivo de tejidos de 96 pocillos a una densidad de 1,8 x 104 células/pocillo (la monocapa debía alcanzar un 80-90 % de confluencia en el día del ensayo).
En el ensayo con células adheridas, se aspiraron los medios de cultivo de cada pocillo y las monocapas se lavaron con 50 pl de PBS (sin Mg++, Ca++). El ensayo de unión se llevó a cabo en un volumen total de 125 pl que consistió en tampón de ensayo (Tris 50 mM, pH 7,4, que incluyó BSA al 1 %, y fosforamidón 2 pM), y 25 pl de ET-1 500 nM (para definir la unión inespecífica) o fármaco competitivo. La reacción se inició con la adición de 25 pl de [125I]-ET-1 0,25 nM (New England Nuclear). La incubación se llevó a cabo con agitación orbital suave, a 4 °C, y se alcanzó el equilibrio a las 4 horas. La reacción se terminó mediante la aspiración del tampón de reacción y dos lavados posteriores con PBS frío (sin Mg++, Ca++). Las células se disociaron mediante la adición de 100 pl de NaOH 0,5 N, seguido de incubación durante 40 minutos. Las muestras se transfirieron después desde el formato de 96 pocillos a tubos para realizar un recuento en un contador gamma Cobra (Packard). Los datos se analizaron con el programa informático de ajuste de curvas SigmaPlot.
Ensayo de Unión de RASMC
Los ensayos se llevaron a cabo en un volumen total de 250 pl en placas de microtitulación de 96 pocillos. La mezcla de incubación contuvo 50 pl de [125]I-Sar-Ile-Angiotensina II (0,2 nM), 25 pl de fármaco disuelto en DMSO, o angiotensina II (1 pM) para definir la unión inespecífica. La unión a células musculares lisas aórticas de rata (RASMC) se llevó a cabo en medio RPMI (Gibco BRL Products, Grand Island, N.Y.) que contuvo BSA al 0,1 % durante 2 horas a temperatura ambiente con agitación continua. El radioligando sin unir se retiró mediante lavado de los pocillos. Las RASMC con radioligando unido se lisan con Triton X al 1 % y BSA al 0,1 % en agua destilada durante 15 minutos a temperatura ambiente con agitación continua. La solución de cada pocillo se transfirió a tubos y se colocaron en un contador gamma.
Los siguientes Ejemplos ilustran las realizaciones de la presente divulgación, y no pretenden limitar el alcance de las reivindicaciones. En la medida en que los Ejemplos contengan materia objeto que se halle fuera del alcance de las reivindicaciones, se incluyen simplemente con fines de referencia. Las abreviaturas empleadas en la presente memoria se definen a continuación.
Abreviaturas
Ac = acetilo
BOC = terc-butoxicarbonilo
n-Bu = n-butilo
BSA = albúmina de suero bovino
p.e. = punto de ebullición
CDI = 1,1 '-carbonildiimidazol
d = días
DIBAL-H = hidruro de diisobutilaluminio
DMF = W,W-dimetilformamida
DMSO = dimetilsulfóxido
EDTA = ácido etilendiamintetraacético
eq = equivalentes
Et = etilo
ET = endotelina
ET-1 = endotelina-1
EtOAc = acetato de etilo
EtOH = etanol
g = gramos
h = horas
kg = kilogramos
Me = metilo
MEM = metoxietoximetilo
MeOH = metanol
m2/g = metros cuadrados por gramo, se usa como medida del área superficial de las partículas
min = minutos
ml = mililitros
mmol = milimoles
mm Hg = milímetros de mercurio
HR = humedad relativa
MOM = metoximetilo
p.f. = punto de fusión
Ms = metanosulfonilo
NBS = N-bromosuccinimida
°C = grados Celsius
°F = grados Fahrenheit
PBS = solución salina tamponada con fosfato
Ph = fenilo
n-Pr = n-propilo
|jl = microlitros
|jg = microgramos
SEM = 2-(trimetilsiloxi)etoximetilo
t.a. = temperatura ambiente
TFA = ácido trifluoroacético
THF = tetrahidrofurano
Métodos Generales
Se emplearon los siguientes Métodos Generales en las Preparaciones y Ejemplos.
Método General 1: Alquilación de Heterociclos o Alcoholes Alifáticos
RCH2X ^ RCH2-OCH2CH3 o RCH?-heterociclo
X = Br o MsO
Se añadió hidruro sódico (dispersión al 60 % en aceite mineral, 1,2 eq) a 0 °C a una solución 1,0 M o suspensión de un heterociclo adecuado o alcohol etílico (1,5 eq) en DMF. La mezcla se dejó calentar a t.a., se agitó durante 20 min, y se volvió a enfriar después a 0 °C. A la mezcla de heterociclo se añadió una solución del bromuro de alquilo o metanosulfonato de alquilo adecuado (1,0 eq) en una cantidad mínima de DMF. La mezcla resultante se dejó calentar a t.a. y se agitó durante 16-24 h. La mezcla de reacción se diluyó con EtOAc y se lavó con agua y salmuera. La fase orgánica se secó sobre sulfato sódico y se concentró, y el residuo se sometió a cromatografía sobre gel de sílice con hexanos/acetato de etilo como eluyente para proporcionar el producto de alquilación.
Método General 2: Reducción de un Nitrilo Aromático a un Aldehido Aromático Usando DIBAL-H
ArCN ^ ArCHO
Se añadió DIBAL-H (solución 1,5 M en tolueno, 1,5 eq) gota a gota a 0 °C a una solución 0,5 M de un nitrilo aromático (1,0 eq) en tolueno o tolueno/diclorometano 9:1. La solución se agitó a 0 °C durante 1-4 h, y se trató después con metanol en exceso. Después de 15 min, se añadió ácido clorhídrico 2 N y la mezcla se agitó enérgicamente durante otros 15 min. Las fases se separaron, y la fase acuosa se extrajo con acetato de etilo. Las fases orgánicas combinadas se secaron sobre sulfato sódico y se concentraron para proporcionar el aldehído en bruto, que se utilizó en bruto o purificado por medio de cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente.
Método General 3: Acoplamiento de Suzuki de Bromuros de Arilo con Ácidos Arilborónicos
ArBr Ar'BÍORb ^ Ar-Ar'
R = H o alquilo
En una solución de 1,0 eq de un ácido (o éster) arilborónico y el bromuro de arilo adecuado (1,0 eq) en tolueno:etanol 2:1 (concentración 0,1 M para cada reactivo) se introdujo nitrógeno durante 15 minutos. Se añadieron tetraquis(trifenilfosfina)paladio(0) (0,05 eq) y carbonato sódico acuoso 2 M (3 eq), y la mezcla se calentó a 85 °C durante 3 h en atmósfera de nitrógeno. La mezcla se enfrió y se añadió acetato de etilo y agua. La fase orgánica se lavó una vez con carbonato sódico acuoso saturado, se secó sobre sulfato sódico, y se concentró. El residuo se sometió a cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente para proporcionar el producto de biarilo.
Ejemplos no limitantes de ácidos arilborónicos usados: ácido [2-[[(4,5-dimetil-3-isoxazolil)[(2-metoxietoxi)metil]amino]-sulfonil]fenil]borónico (preparado como se describe en la patente de EE.UU. n.° 5.612.359 y la solicitud de patente de EE.UU. con n.° de serie 09/013.952, presentada el 27 de enero de 1998); ácido 2-[[N-(4,5-dimetil-3-isoxazolil)-N-(metoximetil)amino]sulfonil]-fenilborónico.
Método General 4: Reducción de Arilaldehídos a Alcoholes Bencílicos Usando Borohidruro Sódico ArCHO ^ ArCH2-OH
Se añadió borohidruro sódico (0,5 eq) a 0 °C a una solución 0,2 M de un aldehído aromático en etanol o metanol absoluto. La mezcla se dejó calentar a t.a. y se agitó durante 1-2 h. Se añadió una solución acuosa de fosfato monopotásico (o ácido clorhídrico diluido) y la mezcla se agitó durante otros 15 min. La mezcla se concentró parcialmente y el residuo se repartió entre acetato de etilo y agua. La fase acuosa se extrajo dos veces con acetato de etilo y los extractos orgánicos combinados se secaron sobre sulfato sódico y se concentraron. El alcohol bencílico se usó en bruto directamente o se purificó mediante cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente.
Método General 5: Conversión de Alcoholes Bencílicos en Bromuros de Bencilo
RCH2OH ^ RCH2Br
A una solución 0,2 M del alcohol bencílico en DMF a 0 °C se añadió tetrabromuro de carbono (1,5 eq) seguido de trifenilfosfina (1,5 eq). La mezcla se agitó a 0 °C durante 4 h, se diluyó con 10 partes de hexanos/acetato de etilo 2:1, y se lavó con agua y salmuera. La solución se secó sobre sulfato sódico y se concentró, y el residuo se sometió a cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente para producir el producto de bromuro de bencilo.
Método General 6: Hidrólisis de los Grupos Protectores de Sulfonamida SEM o MEM Usando Ácido Clorhídrico/Etanol

R = MEM o SEM
A una solución 0,1 M de una N-heteroarilsulfonamida protegida con SEM o MEM en un volumen de EtOH al 95 % se añadió un volumen igual de HCl acuoso 6 N, y la solución resultante se calentó a reflujo durante 1 h. La mezcla de reacción se concentró y el pH de la solución se ajustó a pH 8 mediante el uso de una solución acuosa de bicarbonato sódico. Después se reacidificó a pH 5 con ácido acético glacial. La mezcla se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se lavaron con agua y salmuera, se secaron sobre sulfato sódico, y se concentraron. El residuo se purificó mediante HPLC preparativa de fase inversa, o mediante cromatografía en gel de sílice usando cloroformo/metanol o hexanos/acetona como eluyentes.
Procedimiento General: Purificación mediante Cromatografía de Intercambio Aniónico
Se llevó a cabo cromatografía de intercambio aniónico en cartuchos SAX de Varian (forma de acetato, 1,5-3 g) o cartuchos CUQAX13M6-AC de United Chemical Technologies (forma de acetato, 3 g). Tras un lavado con metanol, el cartucho se cargó con una solución en diclorometano del producto en bruto. La elución de las impurezas con diclorometano, seguida de la elución del producto deseado con al TFA 1-3 % en diclorometano o diclorometano/metanol, proporcionó el producto purificado.
Procedimiento General: Purificación mediante HPLC Preparativa en Fase Inversa
Se llevó a cabo HPLC preparativa en fase inversa con cromatógrafos líquidos Shimadzu 8A usando columnas YMC S5 ODS (20 x 100, 20 x 250, o 30 x 250 mm). La elución en gradiente se llevó a cabo con mezclas de metanol/agua en presencia de TFA al 0,1 %. En ciertos casos, un producto que eluyó en forma de una sal de TFA se convirtió posteriormente en la base libre correspondiente mediante extracción con solución acuosa de bicarbonato sódico o carbonato sódico.
Métodos de HPLC Analítica Empleados en la Caracterización de los Ejemplos
Se llevó a cabo HPLC analítica en cromatógrafos líquidos Shimadzu LC10AS usando los siguientes métodos: A. Gradiente lineal del 0 al 100 % de disolvente B a lo largo de 4 min, con 1 min de mantenimiento al 100 % de B; Visualización UV a 220 nm; Columna: YMC S5 ODS Ballistic de 4,6 x 50 mm; Caudal: 4 ml/min; Disolvente A: 0,1 % de ácido trifluoroacético, 90 % de agua, 10 % de metanol; Disolvente B: 0,1 % de ácido trifluoroacético, 90 % de metanol, 10 % de agua; B. Gradiente lineal del 0 al 100 % de disolvente B a lo largo de 30 min, con 5 min de mantenimiento al 100 % de B; Visualización UV a 254 nm; Columna: YMC S3 ODS de 6 x 150 mm; Caudal: 1,5 ml/min; Disolvente A: 0,2 % de ácido fosfórico, 90 % de agua, 10 % de metanol; Disolvente B: 0,2 % de ácido fosfórico, 90 % de metanol, 10 % de agua; C. Gradiente lineal del 0 al 100 % de disolvente B a lo largo de 4 min, con 1 min de mantenimiento al 100 % de B; Visualización UV a 220 nm; Columna: YMC S5 ODS Ballistic de 4,6 x
50 mm; Caudal: 4 ml/min; Disolvente A: 0,2 % de ácido fosfórico, 90 % de agua, 10 % de metanol; Disolvente B: 0,2 % de ácido fosfórico, 90 % de metanol, 10 % de agua; D. Gradiente lineal del 45 al 100 % de disolvente B a lo largo de 2 min, con 1 min de mantenimiento al 100 % de B; Visualización UV a 220 nm; Columna: Phenomenex Primesphere de 4,6 x 30 mm; Caudal: 5 ml/min; Disolvente A: 0,2 % de ácido fosfórico, 90 % de agua, 10 % de metanol; Disolvente B: 0,2 % de ácido fosfórico, 90 % de metanol, 10 % de agua; E. Mismas condiciones que en (B), pero con un gradiente lineal del 40 al 100 % de disolvente B a lo largo de 30 min, con 5 min de mantenimiento al 100 % de B; F. Mismas condiciones que en (B), pero con un gradiente lineal del 70 al 100 % de disolvente B a lo largo de 30 min, con 5 min de mantenimiento al 100 % de B; G. Mismas condiciones que en (D), pero con un gradiente lineal del 40 al 100 % de disolvente B a lo largo de 2 min, con 1 min de mantenimiento al 100 % de B; H. Gradiente lineal del 0 al 100 % de disolvente B a lo largo de 2 min, con 1 min de mantenimiento al 100 % de B; Visualización UV a 220 nm; Columna: Phenomenex Primesphere de 4,6 x 30 mm; Caudal: 5 ml/min; Disolvente A: 0,1 % de ácido trifluoroacético, 90 % de agua, 10 % de metanol; Disolvente B: 0,1 % de ácido trifluoroacético, 90 % de metanol, 10 % de agua; I. Mismas condiciones que en (B), pero con un gradiente lineal del 50 al 100 % de disolvente B a lo largo de 30 min, con 5 min de mantenimiento al 100 % de B; J. Mismas condiciones que en (C), pero con un gradiente lineal del 0 al 100 % de disolvente B a lo largo de 8 min, con 1 min de mantenimiento al 100 % de B; K. Mismas condiciones que en (D), pero con un gradiente lineal del 0 al 100 % de disolvente B a lo largo de 2 min, con 1 minuto de mantenimiento al 100 % de B.
Habiéndose descrito en este momento la invención en términos generales, se entenderá con mayor facilidad por referencia a los siguientes ejemplos, que se incluyen con fines meramente ilustrativos.
EJEMPLOS
Ejemplo 1

Nombre: 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-2'-formil-N-(4,5-dimetil-3-isoxazolil)-[[1,1'-bifenil]-2-sulfonamida]
A. 4,-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-2,-formil-N-(4,5-dimetil-3-isoxazolil)-N-(2-metoxietoximetil)[1,1'-bifenil]-2-sulfonamida
Se llevó a cabo acoplamiento de Suzuki catalizado con paladio de 4-bromo-3-(etoximetil)benzaldehído y ácido [2-[[(4,5-dimetil-3-isoxazolil)[(2-metoxietoxi)metil]amino]sulfonil]fenil]borónico según el Método General 3 para proporcionar N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(formil)-N-((metoxietoxi)metil)[1,1'-bifenil]-2-sulfonamida (81 %) tras cromatografía sobre gel de sílice.
B. 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-2'-formil-N-(4,5-dimetil-3-isoxazolil)-[1,1'-bifenil]-2-sulfonamida
El tratamiento de N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(formil)-N-((metoxietoxi)metil)[1,1'-bifenil]-2-sulfonamida con ácido clorhídrico acuoso 6 N según el Método General 6 para retirar el grupo protector MEM proporcionó el compuesto del título (85 %): fR = 0,38, MeOH al 5 % en cloruro de metileno.
Ejemplo 2
Se sintetizó 4'-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-[1,1'-bifenil]-2-sulfonamida mediante combinaciones de los Métodos Generales.

Nombre: 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-[1,1'-bifenil]-2-sulfonamida

Material de Partida: X- Br u OMs
Métodos Generales Aplicados (rendimiento, %): Método General 1, EtOH (77); Método General 2 (80); Método General 3 (70); Método General 4 (98); Método General 5 (80); Método General 1 (83); Método General 6 (86) M/z (MH)+: 593
Pureza en % mediante HPLC: >98
Tiempo de retención en HPLC, min (método de HPLC): 18,75 (E)
Ejemplo 3
4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil) [1,1'-bifenil]-2-sulfonamida [cristalina]

Síntesis Alternativa de 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil) [1,1 '-bifenil]-2-sulfonamida
Etapa A. 4-Bromo-3-(bromometil)benzoato de etilo
Se trató 4-bromo-3-metilbenzoato de etilo (110 g, 450 mmol) con NBS según el procedimiento del Ejemplo 5. La cromatografía sobre gel de sílice con hexanos/acetato de etilo como eluyente proporcionó 4-bromo-3-(bromometil)benzoato de etilo (91 g, 62 %) en forma de un sólido blanco.

Etapa B. 4-Bromo-3-(etoximetil)benzoato de etilo
Se trató una solución de 4-bromo-3-(bromometil)benzoato de etilo (89 g, 280 mmol) en una mezcla de etanol (300 ml) y DMF (50 ml) a 0 °C con etóxido sódico (135 ml de una solución al 21 % en etanol). La mezcla se dejó calentar a t.a. y se agitó durante 16 h. El etanol se evaporó a presión reducida. Se añadió acetato de etilo al residuo y la mezcla se lavó con agua y salmuera. La fase orgánica se secó sobre sulfato sódico y se concentró, y el residuo se sometió a cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente para proporcionar 4-bromo-3-(etoximetil)benzoato de etilo (67 g, 84 %) en forma de un aceite ligeramente amarillo.

Etapa C. N-(4,5-Dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida 4-Bromo-3-(etoximetil)benzoato de etilo (32 g, 100 mmol) se sometió a acoplamiento de Suzuki con ácido 2-[[N-(4,5-dimetil-3-isoxazolil)-N-(metoximetil)amino]sulfonil]-fenilborónico según el Método General 3. La cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente proporcionó N-(4,5-Dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida (52 g) en forma de un aceite amarillo, contaminado con productos secundarios procedentes del ácido borónico.

Etapa D. N-(4,5-Dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida Se trató N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida (muestra completa) con DIBAL-H según el siguiente procedimiento:
Se trata una solución de N-(4,5-dimetil-3-isoxazolil)-4'-(etoxicarbonil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida (0,3 mmol) en THF (5 ml) con DIBAL-H (0,53 ml de una solución 1,5 M en tolueno, 0,8 mmol) a -78 °C. La temperatura se dejó elevar a -25 °C y la mezcla se agitó durante 2 h. Se añade cloruro amónico acuoso saturado a la mezcla de reacción enfriada, seguido de extracción con acetato de etilo. Las fases orgánicas combinadas se secan sobre sulfato sódico y se concentran para proporcionar N-(4,5-Dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida en forma de un aceite amarillo crudo.

Etapa E. 4'-(Bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil) [1,1'-bifenil]-2-sulfonamida Se convirtió N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-4'-(hidroximetil)-N-(metoximetil)[1,1 '-bifenil]-2-sulfonamida (muestra completa) en el bromuro correspondiente según el Método General 5. La cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente proporcionó 4'-(Bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida] (38 g, pureza estimada del 83 % mediante RMN 1H) en forma de un aceite amarillo claro.
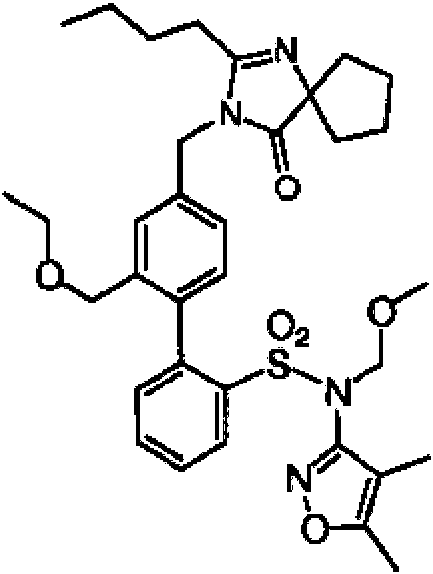
Etapa F. 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil) [1,1 '-bifenil]-2-sulfonamida
Se usó 4'-(bromometil)-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida] (muestra completa) para alquilar 2-butil-1,3-diazaespiro[4.4]non-1-en-4-ona según el Método General 1. El residuo en bruto se sometió a cromatografía sobre gel de sílice usando hexanos/acetato de etilo/trietilamina como eluyente para proporcionar 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil-N-](4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil)[1,1'-bifenil]-2-sulfonamida (32 g, 53 % a partir de la Etapa B) en forma de un aceite ligeramente amarillo.

Etapa G. 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)[1,1'-bifenil]-2-sulfonamida
El grupo protector MOM de 4'-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2'-(etoximetil)-N-(metoximetil) [1,1'-bifenil]-2-sulfonamida (32 g, 53 mmol) se retiró según el Método General 6. El producto en bruto se purificó mediante cromatografía en gel de sílice usando hexanos/acetato de etilo/ácido acético
como eluyente para proporcionar el compuesto del título (26 g, 88 %) en forma de una espuma amorfa: MS n/e 593 (modo ESI+); Tiempo de retención de HPLC 18,75 min (método de h PlC E); Pureza de hPlC > 96 %.
Ejemplo 4

Etapa H. 4-[(2-Butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2-(etoximetil)[1,1-bifenil]-2-sulfonamida
Cristalización
La 4-[(2-butil-4-oxo-1,3-diazaespiro[4.4]non-1-en-3-il)metil]-N-(4,5-dimetil-3-isoxazolil)-2-(etoximetil)[1,1-bifenil]-2-sulfonamida amorfa (1 g) se disolvió en 5 ml de isopropanol, y después se añadieron 5 ml de agua a la mezcla gota a gota y la mezcla se calentó hasta 40 °C para proporcionar una solución clara. La solución se dejó reposar a temperatura ambiente y los cristales blancos así obtenidos se filtraron y se lavaron con una cantidad pequeña de mezcla 2:1 de isopropanol/agua y se secaron para proporcionar 0,87 g de un sólido cristalino blanco. p.f.: 148 °C. Ejemplo 5
Método General 1: Bromación Bencílica Usando N-Bromosuccinimida

A una solución 0,4 M de un compuesto aromático sustituido con metilo, tal como 4-bromo-3-metilbenzoato de etilo en tetracloruro de carbono, se añadió N-bromosuccinimida (1,05 eq) y peróxido de benzoílo (0,03 eq), y la mezcla se calentó a reflujo durante 8-16 h. La mezcla se enfrió y se filtró, y el filtrado se concentró. El residuo se purificó mediante trituración con hexanos/acetato de etilo 3:1, o mediante cromatografía sobre gel de sílice usando hexanos/acetato de etilo como eluyente para proporcionar el producto monobromado, tal como 4-bromo-3-(bromometil)benzoato de etilo.
Ejemplo 6
Comprimidos de dosis de 200 mg y 400 mg para Administración Oral Usando Granulación de Bajo Cizallamiento Se procesaron dos formulaciones utilizando celulosa microcristalina frente a una mezcla de celulosa microcristalina y lactosa usando granulación de bajo cizallamiento. Las composiciones de la Formulación B y la Formulación C se muestran en las Tablas 1 y 2. Se produjeron comprimidos de dosis de 200 mg y 400 mg usando un método de fabricación por granulación húmeda de bajo cizallamiento con una carga de fármaco de un 57 %.


Las formulaciones se granularon con un mezclador de tipo planetario Kitchen Aid usando agua mili-Q como líquido de granulación. La Formulación C se granuló con un total de 84 gramos de agua a una velocidad de adición de 14 gramos por minuto. La Formulación B se granuló con un total de 94 gramos de agua con una velocidad de adición de 14 gramos por minuto. Ambas granulaciones se secaron en bandejas en un horno ventilado a 60 °C durante aproximadamente 15 horas. Las granulaciones secas se molieron usando un aparato 197S Quadro Comil equipado con una criba de rejilla de 0,050 pulgadas, un impulsor redondo, y un espaciador de 0,175 pulgadas a una velocidad del 20 % del aparato Comil. Los excipientes extragranulares se ajustaron basándose en el contenido en porcentaje. Cada formulación se mezcló en bolsas durante 5 minutos antes de añadir la lubricación, y se mezcló durante 1 minuto más después de añadir la lubricación.
Se determinaron las propiedades físicas de las mezclas (densidad aparente y contenido de humedad LOD). La mezcla se comprimió en un equipo de prensado de comprimidos Piccola BD con una estructura de alimentación por
gravedad y tolva a una velocidad de 36 rpm. Se monitorizaron las fuerzas de precompresión, compresión principal y expulsión usando el programa informático "The Director", escrito por SMI Inc. Se seleccionaron pesos de comprimidos de 350 mg y 700 mg para producir comprimidos de dosis de 200 y 400 mg, respectivamente, de cada formulación. Los comprimidos de 350 mg (dosis de 200 mg) se comprimieron con herramientas de compresión cóncavas estándar con forma de cápsula de 0,2500 pulgadas x 0,5000 pulgadas, y los comprimidos de 700 mg (dosis de 400 mg) se comprimieron con herramientas de compresión cóncavas estándar ovaladas de 0,3300 x 0,7100. Se seleccionó un intervalo de durezas de comprimidos para determinar la solidez de las formulaciones, y se registró el grosor de los comprimidos. Se monitorizó el peso, la dureza, el grosor y la friabilidad de los comprimidos durante el proceso.
El proceso de granulación de bajo cizallamiento consiguió densificar moderadamente las formulaciones, y el secado en bandejas llevó la granulación húmeda hasta un contenido de humedad aceptablemente bajo, tal como se indica mediante los resultados de la densidad y LOD como se muestra en la Tabla 3.

Ambas dosis de comprimido de la Formulación C y de la Formulación B se comportaron bien en la prensa rotativa de comprimidos con respecto al flujo y la compresibilidad, lo que dio como resultado pesos estables de comprimidos y durezas aceptables de comprimidos, y una friabilidad baja (Tablas 4 y 5). Ambas formulaciones de comprimidos se disgregaron rápidamente en HCl 0,1 N, pero la Formulación B se disgregó más rápidamente que la Formulación C (Tabla 5). La Formulación C experimentó problemas de adherencia de comprimidos durante los ensayos de compresión. Además, se estudió la adición de estearato magnésico.


Se llevó a cabo un análisis de HPLC final en las muestras de disolución. Los resultados de los ensayos de disolución indicaron que la Formulación C tuvo una velocidad de disolución sustancialmente más rápida que la Formulación B en los medios de disolución de HCl 0,1 N a las dos velocidades de paleta de 50 y 60 rpm (Tabla 6).

Los resultados de los ensayos y de sustancias relacionadas para la Formulación C y la Formulación B se muestran en la Tabla 7.
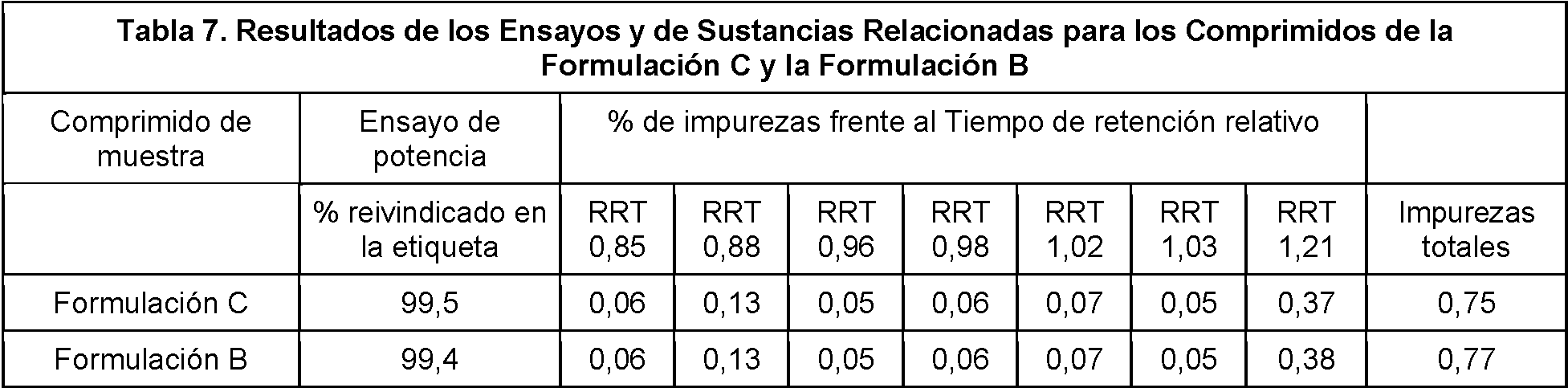
Ejemplo 7
Comprimidos de dosis de 200 mg y 400 mg para Administración Oral mediante el Uso de Granulación Húmeda de Alto Cizallamiento
En el Ejemplo 6, se observó que la Formulación C de una composición basada en celulosa microcristalina que incluía Poloxámero 188 tenía un perfil de liberación de fármaco sustancialmente más rápido comparado con la Formulación B de una formulación de combinación de celulosa microcristalina/lactosa (Tabla 6). La Formulación C necesitó un incremento de lubricante debido a problemas de adherencia observados en los ensayos de compresión. En este ejemplo, se incrementó la cantidad de estearato magnésico en la Formulación C para proporcionar la Formulación D. Además, se creó un nuevo diseño experimental para analizar el superdisgregante almidón glicolato sódico además de croscarmelosa sódica para proporcionar la Formulación E, por lo que se incrementaron las velocidades de disgregación/disolución. La granulación húmeda de alto cizallamiento se sustituyó por granulación de bajo cizallamiento para mejorar la velocidad de disolución y proporcionar un método de fabricación más moderno y adecuado para el escalado futuro de la fabricación.
La Formulación D es una formulación basada en celulosa microcristalina con Poloxámero 188 que incluye un incremento de un 0,5 % de la lubricación y adición de croscarmelosa sódica. La Formulación E es una formulación similar que usa un disgregante de almidón glicolato sódico en vez de croscarmelosa sódica. La Formulación A es una formulación adicional que incluye celulosa microcristalina, croscarmelosa sódica con lauril sulfato sódico en lugar de Poloxámero 188. Las composiciones de las Formulaciones D, E, y A se muestran en las Tablas 8 y 9. Se prepararon comprimidos de dosis a 200 mg y 400 mg proporcionales a la dosis con los ingredientes en la cantidad mostrada en las Tablas 8 y 9.
Las formulaciones se granularon usando granulación húmeda de alto cizallamiento en el granulador de alto cizallamiento Niro PP 1 de 10 litros equipado con un inserto de 7,5 litros. Se usó agua Mili-Q como solución aglutinante. El tamaño de lote para cada formulación fue 750 gramos. Los materiales intragranulares se mezclaron en el granulador durante 3 minutos a una velocidad de impulsor de 300 rpm antes de la granulación. La granulación de la Formulación D necesitó 251 gramos de agua a una velocidad de pulverización de 41,8 g/minuto. La granulación de la Formulación A necesitó 322 gramos de agua a una velocidad de pulverización de 41,8 g/minuto. La Formulación E necesitó 311 gramos de agua a una velocidad de pulverización de 52 g/minuto. La velocidad del impulsor se ajustó a 305 rpm, y el cortador se ajustó a alta velocidad durante la granulación. Se determinó de
manera cualitativa el punto final de la granulación mediante el método "bola de nieve" (primer ensayo), y las granulaciones se mezclaron 5 minutos más a 300 rpm sin cortador. Cada formulación se secó con el secador de lecho fluidizado Niro Aeromatic MP-1 a una temperatura de aire de entrada de 65 °C hasta un contenido de humedad LOD menor de un 2,0 %. Las granulaciones secas se molieron usando un aparato 197S Quadro Comil equipado con una criba de rejilla de 0,050 pulgadas, un impulsor redondo, y un espaciador de 0,175 pulgadas a una velocidad de un 20 % del aparato Comil.


Los excipientes extragranulares se ajustaron con respecto al contenido y se añadieron a cada granulación de ejemplo. La mezcla se mezcló en bolsas durante 5 minutos. Se extendió estearato magnésico en la mezcla, y se mezcló durante otros 3 minutos. Las mezclas finales se comprimieron en una prensa de comprimidos Piccola ED usando la estructura de alimentación con paletas a una velocidad de prensa de 36 rpm. Los comprimidos de 350 mg (dosis de 200 mg) se comprimieron con herramientas de compresión cóncavas estándar con forma de cápsula de 0,2500 pulgadas x 0,5000 pulgadas, y los comprimidos de 700 mg (dosis de 400 mg) se comprimieron con herramientas de compresión cóncavas estándar ovaladas de 0,3300 x 0,7100. Se seleccionó como objetivo una dureza de comprimido de 15 kp para las formulaciones.
Las propiedades físicas de las mezclas finales para la Formulación D y la Formulación A, mostraron pocas diferencias con respecto a la densidad aparente, densidad compactada, ángulo de reposo, compresibilidad o distribución de tamaño de partícula (Tablas 10 y 11). Todos los ejemplos exhibieron un flujo aceptable, tal como se indica mediante el ángulo de reposo, compresibilidad y valores del instrumento Flodex (Tabla 10). Todas las formulaciones exhibieron una baja humedad, tal como se muestra en la Tabla 11. La Formulación E fue sustancialmente más densa que las otras dos Formulaciones, y tuvo una distribución de tamaño de partícula sustancialmente mayor tal como se muestra en la Tabla 11. El proceso de granulación húmeda de la Formulación E produjo gránulos grandes, bien definidos. Los gránulos duros y grandes producidos durante la granulación dieron como resultado tiempos de molienda significativamente mayores que los observados en los otros ejemplos. Algunas esferas, gránulos y partículas secas no se pudieron moler y se descartaron.



Los resultados de LOD para las Formulaciones A, D, E, G y F se muestran en la Tabla 12.

Todas las mezclas de formulaciones se comportaron bien en la prensa de comprimidos (Tabla 13) y produjeron comprimidos duros, consistentes y de calidad elevada con una friabilidad baja. El lubricante incrementado en la Formulación D eliminó los problemas de adherencia de los comprimidos que se observaron antes en la Formulación C. El tiempo de disgregación de los comprimidos a los 49,70 minutos para la Formulación E fue significativamente mayor que el tiempo de disgregación de los comprimidos observado en los otros ejemplos (Tabla 14).



El tensioactivo de Poloxámero 188 (Formulación D) superó al tensioactivo de lauril sulfato sódico (Formulación A) con respecto a la disolución. Tanto la Formulación A como la Formulación D superaron a la Formulación E. El perfil de disolución para el comprimido de dosis de 400 mg de la Formulación A mostró una liberación de fármaco de un 77 % en 30 minutos y una liberación de fármaco de un 89 % en 60 minutos, mientras el perfil de disolución para el comprimido de dosis de 400 mg de la Formulación D mostró una liberación de fármaco de un 92 % en 30 minutos y una liberación de fármaco de un 96 % en 60 minutos (Tabla 15).

Los comprimidos de Formulación A de dosis de 200 mg tuvieron una liberación en disolución más rápida que los comprimidos de dosis de 400 mg como se muestra en la Tabla 16.

Tanto la Formulación D como la Formulación A demostraron tener un grado elevado de actividad con sustancias relacionadas bajas. Se llevó a cabo una determinación de la estabilidad a corto plazo con los comprimidos de Formulación D y Formulación A de dosis de 200 mg en condiciones de envasado y de recipiente abierto. Los comprimidos envasados se colocaron en botellas de HDPE con cierres sellados por inducción y se sometieron a condiciones de estabilidad aceleradas a 50 °C (humedad ambiental). Los comprimidos de recipiente abierto se colocaron en botellas abiertas a 40 °C/75 % de HR (humedad relativa). Los comprimidos se ensayaron a las 2 y 4 semanas. El estudio de estabilidad a corto plazo indicó que tanto la Formulación D como la Formulación A fueron formulaciones químicamente estables. Los comprimidos de dosis de 200 mg para la Formulación D y la Formulación A siguieron siendo activos y sin un incremento significativo de las sustancias relacionadas durante al menos 4 semanas a 40 °C/75 % de Hr en los recipientes abiertos. Los comprimidos de los recipientes cerrados a 50 °C fueron estables durante 4 semanas, con solamente cierta degradación menor observada en la muestra de la Formulación D a las 4 semanas. No se observó un incremento de la degradación en la Formulación A.

A partir de la experimentación llevada a cabo, en particular la disolución, se observó que el Poloxámero 188 fue un buen tensioactivo y que la croscarmelosa sódica fue un buen disgregante. La granulación húmeda de alto cizallamiento demostró ser un método de fabricación eficaz. Las formulaciones que usan granulación húmeda de bajo cizallamiento tuvieron menos ventajas en comparación con las formulaciones fabricadas con la granulación húmeda de alto cizallamiento. El ensayo de disolución proporcionó algunas de tales evidencias de las diferencias entre los dos métodos.
Ejemplo 8
Estudios de Compresión para Evaluar la Formulación A y la Formulación C
Se llevaron a cabo estudios de compresión de comprimidos con la Formulación D y la Formulación A. La compresión se llevó a cabo en la prensa de comprimidos Piccola BD a una velocidad de 36 rpm. Se seleccionaron cinco fuerzas de compresión principales que oscilaron de 3 a 10 kN para someter a tensión a la formulación. Se midió el peso de los comprimidos (mg) y el grosor de los bordes de los comprimidos (pulgadas).
Los estudios de compresión mostraron que todas las formulaciones fueron muy comprimibles. La Formulación A comenzó a alcanzar su dureza máxima de aproximadamente 21 kp a 10,0 kN, mientras la Formulación D alcanzó su dureza máxima de aproximadamente 16,3 kp a 9,9 kN. También se observó en los estudios de compresión que la Formulación A necesitó más fuerza que la Formulación D para conseguir el mismo grosor de comprimidos. La
Formulación D tuvo fuerzas de expulsión ligeramente inferiores. La Formulación A proporcionó una dureza mayor en comparación con la Formulación D.
Ejemplo 9
Optimización de la Granulación de Alto Cizallamiento
El análisis de cribado de la mezcla final de la Formulación D indicó un nivel elevado de partículas finas en la granulación (~50 %). Para optimizar adicionalmente la Formulación D y reducir el nivel de partículas finas en la granulación, se emprendió una experimentación para mejorar posiblemente el proceso de granulación de alto cizallamiento incrementando la cantidad de fluido de granulación de un 25 % p/p (Formulación F) a un 30 % p/p (Formulación G). Las formulaciones usadas en la experimentación de optimización se presentan en la Tabla 18.

Se mezclaron ambos lotes de excipientes de pre-granulación usando un mezclador 1 Quart V. El compuesto de fórmula I (IFA) se envolvió entre los excipientes mezclados en el mezclador Niro PP-1 durante 5 minutos con el cortador apagado. Las formulaciones se granularon mediante granulación de alto cizallamiento con el uso del aparato Niro PP1, equipado con un inserto de 7,5 litros. Se usó agua Mili-Q como disolución de granulación. El tamaño del lote fue de 1000 gramos para la Formulación G y 950 g para la Formulación F debido al IFA limitado. La Formulación G se granuló usando 413 gramos de agua a una velocidad de pulverización de 68,6 g/minuto, y la Formulación F se granuló mediante el uso de 305 gramos de agua a una velocidad de pulverización de 50,9 g/minuto. La granulación seca se molió mediante el uso de un aparato 197S Quadro Comil equipado con una criba de rejilla de 0,050 pulgadas, un impulsor redondo, y un espaciador de 0,175 pulgadas a una velocidad del 20 % del aparato Comil. Los excipientes extragranulares se ajustaron basándose en el contenido en porcentaje de la granulación molida final.
La granulación se mezcló en un mezclador 2 Quart V durante 5 minutos tras la adición de la croscarmelosa y el dióxido de silicio coloidal. La mezcla se mezcló otros 3 minutos tras la adición del estearato magnésico. La mezcla se comprimió en una prensa Piccola BD mediante el uso de la estructura de alimentación de paletas y tolva a una velocidad de 36 rpm. Los comprimidos de 350 mg se comprimieron con herramientas de compresión cóncavas estándar con forma de cápsula de 0,2500 pulgadas x 0,5000 pulgadas y los comprimidos de 700 mg se comprimieron con herramientas de compresión cóncavas estándar ovaladas de 0,3300 x 0,7100.
Los comprimidos de dosis de 200 mg procesados con un 25 % de fluido de granulación (Formulación F) se envasaron a 30 comprimidos por botella en botellas de HDPE de 50 cm3 con cierres sellados por inducción y se
colocaron en condiciones de estabilidad a 25 °C/25 % de HR y 40 °C/75 % de HR durante hasta 3 meses junto con los comprimidos de Formulación A de dosis de 200 mg.
La granulación de la Formulación F tuvo un aspecto pulverulento más fino que la Formulación G, y formó gránulos bien definidos. Las partículas finas se redujeron drásticamente en la Formulación G en la que se usó un 30 % de fluido de granulación durante el procesamiento en comparación con la Formulación F en la que se usó un 25 % de fluido de granulación durante el procesamiento, en el análisis con criba de las mezclas finales, y la distribución de tamaño de partícula de la granulación del 30 % (Formulación G) fue sustancialmente mayor que en la granulación del 25 % (Formulación F) (Tabla 11). Las mezclas finales de ambas granulaciones fueron capaces de producir comprimidos robustos, de buena calidad con una dureza, peso y grosor coherentes, y friabilidad baja (Tabla 12). Los ensayos de actividad y de sustancias relacionadas no indicaron problemas con la actividad o las impurezas. El tiempo de disgregación de los comprimidos se dobló aproximadamente con aumento del 30 % de granulación de ~15 minutos a ~30 minutos (Tabla 14). La disolución también fue notablemente más lenta con los comprimidos de granulación del 30 % en comparación con los comprimidos de granulación del 25 % (Tabla 19).

Ejemplo 10
Estudio de Estabilidad de la Formulación F y A
Se envasaron y se colocaron en condiciones de estabilidad comprimidos de dosis de 200 mg de Formulación F y Formulación A. Los comprimidos se envasaron a 30 comprimidos por botella en botellas de HDPE de 50 cm3 con cierres sellados por inducción.
Tanto la Formulación F como la Formulación A demostraron ser estables durante al menos tres meses a 25 °C/60 % de HR y 40 °C/75 % de HR con respecto al aspecto, actividad, sustancias relacionadas, disolución, y contenido de humedad. No hubo indicación de inestabilidad (Tabla 20).



Ejemplo 11
Desarrollo de Comprimidos de Formulación D de dosis de 100 mg y Evaluación del Efecto del Tamaño de Partícula de IFA
La Formulación D de granulación de alto cizallamiento se usó para explorar el comprimido de dosis de 100 mg de dosis proporcional. Se empleó el procedimiento de granulación de alto cizallamiento que utiliza un 25 % de fluido de granulación. Además, se determinó el efecto de la utilización de un IFA (el compuesto de fórmula I) con un tamaño de partícula mayor mediante la fabricación de comprimidos de dosis de 400 mg así como los comprimidos de dosis de 100 mg a partir de una mezcla común mediante el uso de un IFA con un tamaño de partícula mayor para compararlo con los lotes previos de comprimidos de dosis de 400 mg que usaron el IFA con un tamaño de partícula menor.
Se hicieron comprimidos de dosis de 100 mg usando un procedimiento de granulación de alto cizallamiento utilizando un 25 % de fluido de granulación con un 57 % de carga de fármaco. Se seleccionaron como objetivo pesos de comprimidos de 175 mg para producir comprimidos de dosis de 100 mg de la Formulación D usando el IFA con mayor tamaño de partícula (Tabla 22).


El valor de actividad usado para determinar el peso ajustado del compuesto de Fórmula I (IFA) fue de un 98,80 %. La cantidad total de compuesto de Fórmula I calculada para el lote fue 578,4 gramos para un tamaño de lote de 1 kg. Los excipientes se mezclaron en bolsas y después se usaron para envolver el compuesto de Fórmula I en el granulador de alto cizallamiento PP-1. Los materiales se cargaron en el aparato PP-1 y se premezclaron con el impulsor ajustado a 306 rpm sin cortador durante 3 minutos. La granulación se llevó a cabo con la velocidad del impulsor ajustada a 306 rpm, y la velocidad del cortador ajustada a alta velocidad. Tras el análisis de la granulación, se determinó que era necesario añadir agua adicional. Se añadió un total de 57,8 g de agua adicional a la granulación para conseguir una adición total de agua de 378,8 g a una velocidad de 53,5 g/min o un 28 % de adición de fluido de granulación. La granulación se secó hasta una pérdida por desecación de un 0,7 % en lecho fluido MP-1 con una temperatura del aire de entrada de 65 °C. La granulación se molió usando un aparato 197S Quadro Comil equipado con una criba de 0,050 pulgadas con el uso de un espaciador de 150 a un 20 % de velocidad. El rendimiento de la granulación molida resultante fue de un 80,1 %. Los excipientes extragranulares se ajustaron y se mezclaron en bolsas. La mezcla final resultante se comprimió mediante el uso de una estructura de alimentación por gravedad y tolva a aproximadamente 42 rpm en una prensa de comprimidos Korsch PHI 03 equipada con herramientas de compresión de comprimidos planos ovalados de 0,2000 x 0,4000 (Hob# 21971) para los comprimidos de dosis de 100 mg, y de comprimidos ovalados modificados de 0,3360 x 0,6720 (Hob# 1907) para los comprimidos de dosis de 400 mg. Se seleccionó como objetivo una dureza de 10 kp y un peso de comprimido de 175 mg para los comprimidos de dosis de 100 mg con el uso de la Formulación D (mayor tamaño de partícula de IFA), y se seleccionó como objetivo una dureza de 15 kp y un peso de 700 mg para los comprimidos de dosis de 400 mg con el uso de la Formulación D (mayor tamaño de partícula de IFA).
Evaluación de comprimidos de dosis de 100 mg y 400 mg usando la Formulación D (mayor tamaño de partícula de IFA)
Las propiedades físicas de la mezcla final se compararon con los lotes previos de la Formulación D (Tabla 13). Se descubrió que la densidad aparente y las propiedades de flujo, tal como se miden mediante la compresibilidad y el ángulo de reposo, fueron muy similares. Se descubrió que la densidad compactada de la mezcla final fue algo menor que en los lotes previos, y se descubrió que la cantidad de partículas finas en el análisis con criba fue sustancialmente mayor que en las granulaciones de formulaciones previas que utilizaron un 25 % de fluido de granulación, aunque la granulación utilizó un 28 % de fluido de granulación. El IFA de mayor tamaño pareció afectar a las propiedades de la granulación. Sin embargo, la mezcla final se comportó bien en la prensa de comprimidos, y se produjeron comprimidos coherentes que cumplieron los objetivos de peso y dureza de los comprimidos con una friabilidad baja para los comprimidos de dosis de 100 mg y 400 mg. Las propiedades de los comprimidos se presentan en la Tabla 13. El análisis de la actividad se llevó a cabo con los comprimidos de dosis de 100 mg, y, de manera similar a todas las formulaciones previas, la actividad fue elevada (97,7 %) y las sustancias relacionadas fueron bajas (1,03 % en total). El ensayo de disolución en los comprimidos de dosis de 100 mg y 400 mg indicó una velocidad rápida de liberación que cumple fácilmente los criterios para un comprimido de liberación inmediata. El perfil de disolución se presenta en la Tabla 23.

Comprimidos de dosis de 400 mg de Formulación F  100 102
100 102
Por lo tanto, el uso del IFA con mayor tamaño de partícula no afectó negativamente a la disolución de los comprimidos o a su fabricación. El perfil de disolución de los comprimidos de dosis de 400 mg de Formulación D mediante el uso del IFA de mayor tamaño de partícula fue prácticamente idéntico al lote de estabilidad de la Formulación F. Sin embargo, el mayor tamaño de partícula sí requirió el uso de más fluido de granulación en el proceso de fabricación.
Ejemplo 12
Utilizando los procedimientos descritos en los ejemplos 6 a 11, los comprimidos y cápsulas pueden contener el compuesto de fórmula I en cantidades de aproximadamente 10 mg a aproximadamente 800 mg. El compuesto de fórmula I y los ingredientes inertes están presentes en las cantidades en porcentaje en peso descritas, que el experto en la materia determina fácilmente a partir de los ejemplos previos. Por ejemplo, el experto en la materia, siguiendo los procedimientos de los ejemplos 1 a 12 de una manera análoga, puede preparar comprimidos y cápsulas, además de los ya expuestos, en los que el compuesto de fórmula I está presente en cantidades de aproximadamente 10 mg, aproximadamente 20 mg, aproximadamente 30 mg, aproximadamente 40 mg, aproximadamente 50 mg, aproximadamente 60 mg, aproximadamente 70 mg, aproximadamente 80 mg, aproximadamente 90 mg, aproximadamente 100 mg, aproximadamente 110 mg, aproximadamente 120 mg, aproximadamente 130 mg, aproximadamente 140 mg, aproximadamente 150 mg, aproximadamente 160 mg, aproximadamente 170 mg, aproximadamente 180 mg, aproximadamente 190 mg, aproximadamente 200 mg, aproximadamente 210 mg, aproximadamente 220 mg, aproximadamente 230 mg, aproximadamente 240 mg, aproximadamente 250 mg, aproximadamente 260 mg, aproximadamente 270 mg, aproximadamente 280 mg, aproximadamente 290 mg, aproximadamente 300 mg, aproximadamente 310 mg, aproximadamente 320 mg, aproximadamente 330 mg, aproximadamente 340 mg, aproximadamente 350 mg, aproximadamente 360 mg, aproximadamente 370 mg, aproximadamente 380 mg, aproximadamente 390 mg, aproximadamente 400 mg, aproximadamente 410 mg, aproximadamente 420 mg, aproximadamente 430 mg, aproximadamente 440 mg, aproximadamente 450 mg, aproximadamente 460 mg, aproximadamente 470 mg, aproximadamente 480 mg, aproximadamente 490 mg, aproximadamente 500 mg, aproximadamente 510 mg, aproximadamente 520 mg, aproximadamente 530 mg, aproximadamente 540 mg, aproximadamente 550 mg, aproximadamente 560 mg, aproximadamente 570 mg, aproximadamente 580 mg, aproximadamente 590 mg, aproximadamente 600 mg, aproximadamente 610 mg, aproximadamente 620 mg, aproximadamente 630 mg, aproximadamente 640 mg, aproximadamente 650 mg, aproximadamente 660 mg, aproximadamente 670 mg, aproximadamente 680 mg, aproximadamente 690 mg, aproximadamente 700 mg, aproximadamente 710 mg, aproximadamente 720 mg, aproximadamente 730 mg, aproximadamente 740 mg, aproximadamente 750 mg, aproximadamente 760 mg, aproximadamente 770 mg, aproximadamente 780 mg, aproximadamente 790 mg, o aproximadamente 800 mg.
Ejemplo 13
El ensayo de fase IIa se inició con un periodo de preinclusión con placebo de 3-4 semanas. Se aleatorizaron 141 pacientes hipertensos de estadio I y II cualificados para tomar 200 mg, 500 mg del compuesto de fórmula I, o placebo, diariamente durante 4 semanas. Se estudiaron criterios de valoración de eficacia claves de la monitorización ambulatoria de la tensión arterial (ABPM) sistólica media de 24 horas, ABPM diastólica media de 24 horas, tensión arterial sistólica (SBP) y tensión arterial diastólica (DBP) en reposo media, SBP y DBP diurna media, SBP y DBP nocturna media, y SBP y DBP durante las 2 horas finales de la posología. 93 pacientes (población de eficacia) estuvieron disponibles para el análisis de ABPM.
El compuesto de fórmula I a 200 mg o 500 mg una vez al día produjo una mayor reducción estadísticamente significativa de la tensión arterial sistólica y diastólica en pacientes hipertensos de estadio I y II en comparación con el placebo (Tabla 24). Se demostró que el fármaco fue seguro y bien tolerado por los pacientes. La mayoría de los eventos adversos informados fueron de gravedad leve o moderada, e incluyeron cefaleas y dolencias musculoesqueléticas y respiratorias menores (Tabla 25). No se observaron cambios notables en la función del hígado y en los análisis hematológicos (Tabla 26).




Ejemplo 14
El ensayo de Fase IIb se inició con un periodo de preinclusión con placebo de 4 semanas, y se aleatorizaron 261 pacientes cualificados con SBP/DBP dentro del intervalo de 140-179/90-109 mmHg con el compuesto de fórmula I a 200 mg, 400 mg, 800 mg, irbesartán a 300 mg, o placebo, tomados a diario por la mañana durante 12 semanas. Se estudiaron los criterios clave de valoración de la eficacia de SBP, DBP en reposo y % de pacientes con control de BP (<140/90 mmHg), y todos los parámetros de seguridad habituales. Los resultados mostraron que cada dosis del compuesto de fórmula I exhibió una disminución de BP mayor estadísticamente dependiente de la dosis en SBP y DBP (p < 0,001), y en control de BP (p < 0,0013) que con el placebo. La eficacia del compuesto de fórmula I 200 mg fue similar a irbesartán, 400 mg alcanzaron un control mejor de BP que irbesartán (p < 0,05), y a 800 mg fue estadísticamente superior a irbesartán en todos los criterios de valoración como se demuestra en la Tabla 27. El inicio de la acción del compuesto de fórmula I fue rápido en 4 horas y la eficacia se sostuvo a lo largo de 12 semanas.
Tabla 27. Criterios de Valoración de Eficacia en la Semana 12 (Final del Tratamiento)
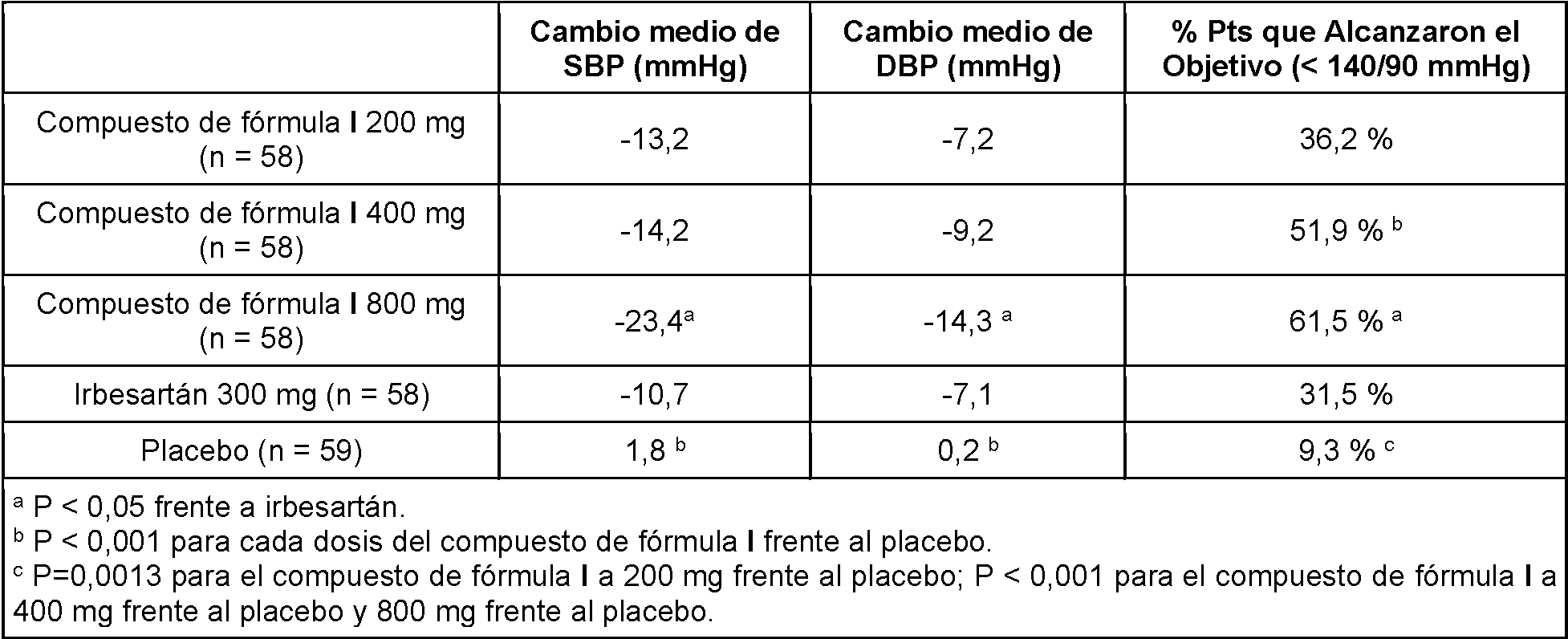
No se informaron eventos adversos graves (SAE) durante el tratamiento activo (Tabla 28). No hubo cambios notables observados en los ensayos de la función hepática (Tabla 29), creatinina, nitrógeno ureico y signos vitales. En conclusión, el compuesto de fórmula I es un agente nuevo, extremadamente potente, con un mecanismo de acción doble, que promete ser un tratamiento importante de nefropatía diabética, hipertensión arterial crónica o persistente, hipertensión y otras enfermedades.


Ejemplo 15
El estudio comienza con pacientes cualificados con nefropatía diabética. Se pueden llevar a cabo análisis y determinaciones de cribado a lo largo de un periodo de no más de 2 semanas. Tras el cribado, todos los pacientes
elegibles se someterán a un periodo de preinclusión con placebo de 4 semanas para asegurar que la tensión arterial inicial permanece estable y continúan cumpliendo los criterios de elegibilidad para la aleatorización. Los pacientes elegibles se aleatorizan para asignarlos al compuesto de fórmula I: 200 mg, 400 mg, o 800 mg; irbesartán 300 mg; o placebo, tomados a diario durante 12 semanas. Se estudian los criterios clave de valoración de la eficacia de BP sistólica, BP diastólica en reposo y % de pacientes con BP de control (<140/90 mmHg), y todos los parámetros de seguridad habituales. Se registran los cambios desde el valor inicial hasta la medida final en el curso del tratamiento. Se espera que el tratamiento con cada dosis del compuesto de fórmula I de un sujeto humano, por ejemplo un sujeto humano que tiene nefropatía diabética, exhiba una mayor disminución de BP estadísticamente dependiente de la dosis de SBP y DBP, y en control de BP que con el placebo e irbesartán.
Claims (15)
1. Un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de un trastorno seleccionado entre el grupo que consiste en glomeruloesclerosis y nefropatía inducida por IgA, en el que la cantidad administrada del compuesto de Fórmula I, o la sal farmacéuticamente aceptable del mismo, es de 200 mg a 800 mg.
2. El compuesto para el uso de acuerdo con la reivindicación 1, en el que la cantidad administrada del compuesto de Fórmula I, o la sal farmacéuticamente aceptable del mismo, es 200 mg, 400 mg, u 800 mg.
3. El compuesto para el uso de acuerdo con la reivindicación 2, en el que la cantidad administrada del compuesto de Fórmula I, o la sal farmacéuticamente aceptable del mismo, es 200 mg.
4. El compuesto para el uso de acuerdo con la reivindicación 2, en el que la cantidad administrada del compuesto de Fórmula I, o la sal farmacéuticamente aceptable del mismo, es 400 mg.
5. El compuesto para el uso de acuerdo con la reivindicación 2, en el que la cantidad administrada del compuesto de Fórmula I, o la sal farmacéuticamente aceptable del mismo, es 800 mg.
6. El compuesto para el uso de acuerdo con la reivindicación 1, en el que el trastorno es glomeruloesclerosis.
7. El compuesto para el uso de acuerdo con la reivindicación 1, en el que el trastorno es nefropatía inducida por IgA.
8. Uso de un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo, en la fabricación de un medicamento para el tratamiento de un trastorno seleccionado entre el grupo que consiste en glomeruloesclerosis y nefropatía inducida por IgA, en el que la cantidad del compuesto de Fórmula I o la sal farmacéuticamente aceptable del mismo es de 200 mg a 800 mg.
9. El uso de acuerdo con la reivindicación 8, en el que la cantidad del compuesto de Fórmula I o la sal farmacéuticamente aceptable del mismo es 200 mg, 400 mg, u 800 mg.
10. El uso de acuerdo con la reivindicación 9, en el que la cantidad del compuesto de Fórmula I o la sal farmacéuticamente aceptable del mismo es 200 mg.
11. El uso de acuerdo con la reivindicación 9, en el que la cantidad del compuesto de Fórmula I o la sal farmacéuticamente aceptable del mismo es 400 mg.
12. El uso de acuerdo con la reivindicación 9, en el que la cantidad del compuesto de Fórmula I o la sal farmacéuticamente aceptable del mismo es 800 mg.
13. El uso de acuerdo con la reivindicación 8, en el que el trastorno es glomeruloesclerosis.
14. El uso de acuerdo con la reivindicación 8, en el que el trastorno es nefropatía inducida por IgA.
15. Una forma farmacéutica unitaria que comprende 800 mg de un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16541909P | 2009-03-31 | 2009-03-31 | |
| US16544709P | 2009-03-31 | 2009-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2811342T3 true ES2811342T3 (es) | 2021-03-11 |
Family
ID=42828649
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17157697T Active ES2811342T3 (es) | 2009-03-31 | 2010-03-29 | Antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar glomeruloesclerosis y nefropatía inducida por IgA |
| ES14155459.2T Active ES2636087T3 (es) | 2009-03-31 | 2010-03-29 | Un antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar la glomeruloesclerosis |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14155459.2T Active ES2636087T3 (es) | 2009-03-31 | 2010-03-29 | Un antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar la glomeruloesclerosis |
Country Status (18)
| Country | Link |
|---|---|
| US (4) | US20140142149A1 (es) |
| EP (4) | EP3708163A1 (es) |
| JP (3) | JP2012522058A (es) |
| CN (2) | CN102421434A (es) |
| CY (3) | CY1119232T1 (es) |
| DK (2) | DK2732818T3 (es) |
| ES (2) | ES2811342T3 (es) |
| FI (1) | FIC20240031I1 (es) |
| FR (1) | FR24C1038I2 (es) |
| HR (2) | HRP20171197T1 (es) |
| HU (3) | HUE049450T2 (es) |
| LT (3) | LT2732818T (es) |
| NL (1) | NL301291I2 (es) |
| PL (2) | PL2732818T3 (es) |
| PT (2) | PT2732818T (es) |
| SI (2) | SI3222277T1 (es) |
| SM (2) | SMT201700416T1 (es) |
| WO (1) | WO2010114801A1 (es) |
Families Citing this family (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG187592A1 (en) | 2010-07-23 | 2013-03-28 | Univ Boston | Anti-despr inhibitors as therapeutics for inhibition of pathological angiogenesis and tumor cell invasiveness and for molecular imaging and targeted delivery |
| AR083417A1 (es) | 2010-10-14 | 2013-02-21 | Novartis Ag | Composiciones farmaceuticas que contienen un dgat1 inhibidor |
| US20130022551A1 (en) * | 2011-07-22 | 2013-01-24 | Trustees Of Boston University | DEspR ANTAGONISTS AND AGONISTS AS THERAPEUTICS |
| SG11201401583YA (en) * | 2011-10-17 | 2014-09-26 | Lexicon Pharmaceuticals Inc | Solid dosage forms of (s)-ethyl 2-amino-3-(4-(2-amino-6-((r)-1-(4-chloro-2-(3-methyl-1h-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate |
| ES2643403T3 (es) | 2011-12-28 | 2017-11-22 | Global Blood Therapeutics, Inc. | Compuestos de benzaldehído sustituidos y métodos para su uso en el aumento de la oxigenación tisular |
| HK1203412A1 (en) | 2011-12-28 | 2015-10-30 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| EP2875359A4 (en) | 2012-03-30 | 2015-08-19 | Charles R Drew University Of Medicine And Science | COMPOSITIONS AND METHODS FOR TREATING OR PREVENTING METABOLISM SYNDROME DISEASES |
| US20140271923A1 (en) | 2013-03-14 | 2014-09-18 | Christopher Brian Reid | Compositions & formulations for preventing and treating chronic diseases that cluster in patients such as cardiovascular disease, diabetes, obesity, polycystic ovary syndrome, hyperlipidemia and hypertension, as well as for preventing and treating other diseases and conditions |
| US8952171B2 (en) | 2013-03-15 | 2015-02-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9458139B2 (en) | 2013-03-15 | 2016-10-04 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| CN105073728A (zh) | 2013-03-15 | 2015-11-18 | 全球血液疗法股份有限公司 | 化合物及其用于调节血红蛋白的用途 |
| WO2014150258A1 (en) | 2013-03-15 | 2014-09-25 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| MX379235B (es) | 2013-03-15 | 2025-03-11 | Global Blood Therapeutics Inc | Compuestos y sus usos para modular la hemoglobina. |
| SG10201910773VA (en) | 2013-06-13 | 2020-01-30 | Akebia Therapeutics Inc | Compositions and methods for treating anemia |
| EA201992707A1 (ru) | 2013-11-18 | 2020-06-30 | Глобал Блад Терапьютикс, Инк. | Соединения и их применения для модуляции гемоглобина |
| US12168004B2 (en) | 2014-02-05 | 2024-12-17 | Merck Sharp & Dohme Llc | Treatment of migraine |
| JP6491669B2 (ja) | 2014-02-05 | 2019-03-27 | メルク・シャープ・アンド・ドーム・コーポレーションMerck Sharp & Dohme Corp. | Cgrp活性化合物の錠剤製剤 |
| CN114181195A (zh) | 2014-02-07 | 2022-03-15 | 全球血液疗法股份有限公司 | 一种化合物的结晶多晶型物 |
| CN104367576A (zh) * | 2014-10-24 | 2015-02-25 | 南方医科大学南方医院 | 化合物icg-001在制备治疗高血压的药物中的用途 |
| CA2995716A1 (en) | 2015-08-24 | 2017-03-02 | Trustees Of Boston University | Anti-despr monoclonal antibody targeted therapy and imaging for cancer and stroke |
| SG11201804647TA (en) | 2015-12-04 | 2018-06-28 | Global Blood Therapeutics Inc | Dosing regimens for 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| TWI663160B (zh) | 2016-05-12 | 2019-06-21 | 全球血液治療公司 | 用於合成2-羥基-6-((2-(1-異丙基-1h-吡唑-5-基)-吡啶-3-基)甲氧基)苯甲醛之方法 |
| TW202332423A (zh) * | 2016-10-12 | 2023-08-16 | 美商全球血液治療公司 | 包含2-羥基-6-((2-(1-異丙基-1h-吡唑-5-基)吡啶-3-基)甲氧基)-苯甲醛之片劑 |
| JP2019530713A (ja) | 2016-10-13 | 2019-10-24 | レトロフィン, インコーポレイテッド | 腎臓疾患または障害を処置するためのビフェニルスルホンアミド化合物 |
| KR102568472B1 (ko) | 2017-02-27 | 2023-08-18 | 이도르시아 파마슈티컬스 리미티드 | 엔도텔린 관련 질환의 치료를 위한 4-피리미딘술파미드 유도체와 활성 성분의 조합물 |
| WO2018200691A2 (en) * | 2017-04-25 | 2018-11-01 | Proteus Digital Health, Inc. | Lisinopril compositions with an ingestible event marker |
| JP7404230B2 (ja) | 2017-09-18 | 2023-12-25 | トラスティーズ オブ ボストン ユニバーシティ | ネトーシスおよび好中球活性化を処置するための方法 |
| US12383545B1 (en) | 2018-06-08 | 2025-08-12 | Allergan Pharmaceuticals International Limited | Treatment of migraine |
| WO2019241609A1 (en) | 2018-06-15 | 2019-12-19 | Trustees Of Boston University | Polypeptide compositions and methods for site-specific targeting of therapeutic agents |
| US11014884B2 (en) | 2018-10-01 | 2021-05-25 | Global Blood Therapeutics, Inc. | Modulators of hemoglobin |
| KR102902972B1 (ko) | 2018-10-04 | 2025-12-22 | 트래버르 테라퓨틱스, 인코포레이티드 | Iv형 콜라겐 질환의 치료를 위한 비페닐 설폰아미드 화합물 |
| CA3124127A1 (en) * | 2018-12-21 | 2020-06-25 | Travere Therapeutics, Inc. | Amorphous sparsentan compositions |
| ES3048296T3 (en) * | 2018-12-28 | 2025-12-09 | Gnt Pharma Co Ltd | Compositions comprising 5-benzylaminosalicylic acid compounds for use in treating neurodegenerative disorders in a companion animal |
| EP3925957A4 (en) | 2019-02-15 | 2022-11-02 | Tohoku University | 1,3-DIOXOLANE DERIVATIVE |
| KR20230015874A (ko) | 2019-12-17 | 2023-01-31 | 치누크 세라퓨틱스, 인크. | 아트라센탄에 의해 iga 신장병증을 치료하는 방법 |
| CN116390712A (zh) | 2020-07-29 | 2023-07-04 | 阿勒根制药国际有限公司 | 治疗偏头痛 |
| WO2022081792A1 (en) | 2020-10-15 | 2022-04-21 | Trustees Of Boston University | Antibody therapies and methods for treating coronavirus infection |
| CA3206184A1 (en) | 2020-12-22 | 2022-06-30 | Allergan Pharmaceuticals International Limited | Treatment of migraine |
| WO2022246200A1 (en) | 2021-05-21 | 2022-11-24 | Travere Therapeutics, Inc. | Methods of treating kidney diseases or disorders |
| JP7458085B2 (ja) * | 2021-07-28 | 2024-03-29 | 株式会社三協 | 錠剤用崩壊剤及びこれを利用した錠剤 |
| CN117836294A (zh) | 2021-08-26 | 2024-04-05 | 上海翰森生物医药科技有限公司 | 含芳环类生物拮抗剂、其制备方法和应用 |
| WO2023049920A1 (en) | 2021-09-27 | 2023-03-30 | Allergan Pharmaceuticals International Limited | Combination comprising atogepant for treating migraine |
| IL312723A (en) * | 2021-12-28 | 2024-07-01 | Alchemedicine Inc | Compound, angiotensin ii type 1 receptor antagonist, and pharmaceutical composition |
| WO2024073672A1 (en) | 2022-09-30 | 2024-04-04 | Travere Therapeutics, Inc. | Combination therapy with sparsentan and a sglt2 inhibitor for treating kidney diseases or disorders |
| CN115636799A (zh) * | 2022-11-04 | 2023-01-24 | 苏州莱安医药化学技术有限公司 | 一种双效内皮素-血管紧张素受体拮抗剂的制备方法 |
| CN120752231A (zh) | 2023-02-24 | 2025-10-03 | 江苏豪森药业集团有限公司 | 含芳环类衍生物拮抗剂的盐、其制备方法和应用 |
| US20240382461A1 (en) * | 2023-05-15 | 2024-11-21 | Pharmazz, Inc. | Pharmaceutical composition and method for prevention and treatment of hearing loss |
| WO2024249509A1 (en) | 2023-05-31 | 2024-12-05 | Travere Therapeutics, Inc. | Sparsentan for use in a method of treating iga-mediated diseases |
| IT202300018276A1 (it) * | 2023-09-06 | 2025-03-06 | Procos Spa | Processo per la preparazione di sparsentan |
| WO2025064688A1 (en) | 2023-09-20 | 2025-03-27 | Travere Therapeutics, Inc. | Sparsentan for treating immunoglobulin a nephropathy (igan) |
| WO2025094214A1 (en) * | 2023-11-02 | 2025-05-08 | Msn Laboratories Private Limited, R&D Center | Process for the preparation of 2-[4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-(ethoxymethyl)phenyl]-N-(4,5-dimethyl-1,2-oxazol-3-yl) benzenesulfonamide and its intermediates thereof |
| IT202400000246A1 (it) * | 2024-01-09 | 2025-07-09 | Procos Spa | Processo per la preparazione di sparsentan |
| CN120289447A (zh) * | 2025-06-11 | 2025-07-11 | 成都臻拓医药科技有限公司 | 一种司帕生坦的晶型及其制备方法、药物组合物 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US793898A (en) | 1905-03-01 | 1905-07-04 | William Mcintosh | Locomotive-tender frame. |
| US1395298A (en) | 1919-11-20 | 1921-11-01 | Walter H Richman | Container |
| US5612359A (en) | 1994-08-26 | 1997-03-18 | Bristol-Myers Squibb Company | Substituted biphenyl isoxazole sulfonamides |
| US6638937B2 (en) * | 1998-07-06 | 2003-10-28 | Bristol-Myers Squibb Co. | Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists |
| WO2000001389A1 (en) * | 1998-07-06 | 2000-01-13 | Bristol-Myers Squibb Co. | Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists |
| US6380184B1 (en) | 1998-10-28 | 2002-04-30 | Bristol-Myers Squibb Co. | Benzoazepines and analogs thereof useful as growth hormone secretagogues |
| EP1156803A4 (en) | 1999-02-09 | 2004-03-17 | Bristol Myers Squibb Co | FXA LACTAM INHIBITORS AND METHOD |
| US6518292B1 (en) | 1999-03-12 | 2003-02-11 | Bristol-Myers Squibb Co. | Heterocyclic aromatic compounds usefuls as growth hormone secretagogues |
| US7390824B1 (en) | 1999-09-07 | 2008-06-24 | Bristol-Myers Squibb Company | Method for treating diabetes employing an aP2 inhibitor and combination |
| US6586438B2 (en) | 1999-11-03 | 2003-07-01 | Bristol-Myers Squibb Co. | Antidiabetic formulation and method |
| DE60030764T2 (de) * | 1999-12-15 | 2007-09-13 | Bristol-Myers Squibb Co. | Biphenyl - sulfonamide als duale angiotensin - endothelin - rezeptor - antagonisten |
-
2010
- 2010-03-29 EP EP20152292.7A patent/EP3708163A1/en active Pending
- 2010-03-29 LT LTEP14155459.2T patent/LT2732818T/lt unknown
- 2010-03-29 DK DK14155459.2T patent/DK2732818T3/en active
- 2010-03-29 ES ES17157697T patent/ES2811342T3/es active Active
- 2010-03-29 EP EP10759268A patent/EP2413930A4/en not_active Withdrawn
- 2010-03-29 SI SI201032000T patent/SI3222277T1/sl unknown
- 2010-03-29 PL PL14155459T patent/PL2732818T3/pl unknown
- 2010-03-29 EP EP14155459.2A patent/EP2732818B1/en active Active
- 2010-03-29 PT PT141554592T patent/PT2732818T/pt unknown
- 2010-03-29 ES ES14155459.2T patent/ES2636087T3/es active Active
- 2010-03-29 SM SM20170416T patent/SMT201700416T1/it unknown
- 2010-03-29 SM SM20200269T patent/SMT202000269T1/it unknown
- 2010-03-29 SI SI201031505T patent/SI2732818T1/sl unknown
- 2010-03-29 WO PCT/US2010/029093 patent/WO2010114801A1/en not_active Ceased
- 2010-03-29 JP JP2012503575A patent/JP2012522058A/ja active Pending
- 2010-03-29 HU HUE17157697A patent/HUE049450T2/hu unknown
- 2010-03-29 PL PL17157697T patent/PL3222277T3/pl unknown
- 2010-03-29 LT LTEP17157697.8T patent/LT3222277T/lt unknown
- 2010-03-29 PT PT171576978T patent/PT3222277T/pt unknown
- 2010-03-29 CN CN2010800188487A patent/CN102421434A/zh active Pending
- 2010-03-29 HU HUE14155459A patent/HUE033494T2/en unknown
- 2010-03-29 EP EP17157697.8A patent/EP3222277B1/en active Active
- 2010-03-29 CN CN201410045095.9A patent/CN104107176A/zh active Pending
- 2010-03-29 DK DK17157697.8T patent/DK3222277T3/da active
-
2012
- 2012-12-19 US US13/720,452 patent/US20140142149A1/en not_active Abandoned
-
2014
- 2014-11-12 JP JP2014229994A patent/JP2015052001A/ja active Pending
-
2015
- 2015-02-25 US US14/631,768 patent/US9662312B2/en active Active
-
2016
- 2016-08-16 JP JP2016159420A patent/JP2017008081A/ja active Pending
-
2017
- 2017-07-28 CY CY20171100811T patent/CY1119232T1/el unknown
- 2017-08-03 HR HRP20171197TT patent/HRP20171197T1/hr unknown
- 2017-08-25 US US15/687,407 patent/US9993461B2/en active Active
-
2018
- 2018-03-28 US US15/938,956 patent/US20180344705A1/en not_active Abandoned
-
2020
- 2020-04-24 HR HRP20200665TT patent/HRP20200665T1/hr unknown
- 2020-05-08 CY CY20201100424T patent/CY1123017T1/el unknown
-
2024
- 2024-09-30 HU HUS2400033C patent/HUS2400033I1/hu unknown
- 2024-10-01 FI FIC20240031C patent/FIC20240031I1/fi unknown
- 2024-10-07 FR FR24C1038C patent/FR24C1038I2/fr active Active
- 2024-10-07 NL NL301291C patent/NL301291I2/nl unknown
- 2024-10-08 LT LTPA2024527C patent/LTPA2024527I1/lt unknown
- 2024-10-14 CY CY2024028C patent/CY2024028I1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2811342T3 (es) | Antagonista de receptores de endotelina y angiotensina II de bifenilsulfonamida para tratar glomeruloesclerosis y nefropatía inducida por IgA | |
| EP3525784B1 (en) | Sparsentan for use in the treatment of alport syndrome | |
| TW200530227A (en) | Benzimidazole derivative and use thereof | |
| CN101890028A (zh) | 稳定化药物组合物 | |
| CN109982687A (zh) | 联合疗法 | |
| CN111225917A (zh) | 取代咪唑并吡啶酰胺及其用途 | |
| JP2022514569A (ja) | 非晶質スパルセンタン組成物 | |
| WO2002049630A2 (en) | Method for preventing or treating pain by administering an endothelin antagonist | |
| US8124640B2 (en) | Pharmaceutical composition based on idazoxan, salts, hydrates or polymorphs thereof | |
| WO2007106468A2 (en) | Formulations of sitaxsentan sodium | |
| CN112839651A (zh) | 联合疗法 | |
| CN110167535A (zh) | 包含噁嗪衍生物的药物组合物及其在治疗或预防阿尔茨海默病中的用途 | |
| HK1198010B (en) | A biphenylsulfonamide endothelin and angiotensin ii receptor antagonist to treat glomerulosclerosis | |
| HK1244687B (en) | A biphenylsulfonamide endothelin and angiotensin ii receptor antagonist to treat glomerulosclerosis and iga-induced nephropathy | |
| HK1198010A (en) | A biphenylsulfonamide endothelin and angiotensin ii receptor antagonist to treat glomerulosclerosis | |
| CN109721557A (zh) | 来曲唑晶ii型固体物质及制备方法和其药物组合物与用途 | |
| US8476307B2 (en) | Pharmaceutical composition based on idazoxan, salts, hydrates or polymorphs thereof | |
| US7595335B2 (en) | Pharmaceutical composition based on idazoxan, salts, hydrates or polymorphs thereof | |
| EA040851B1 (ru) | Комбинированная терапия | |
| BR122025008185A2 (pt) | Usos terapêuticos de compostos de bifenil sulfonamida para a fabricação de um medicamento para tratar doenças ou transtornos renais em um indivíduo |