ES2758506T3 - Péptidos citotóxicos y conjugados de estos - Google Patents
Péptidos citotóxicos y conjugados de estos Download PDFInfo
- Publication number
- ES2758506T3 ES2758506T3 ES14835606T ES14835606T ES2758506T3 ES 2758506 T3 ES2758506 T3 ES 2758506T3 ES 14835606 T ES14835606 T ES 14835606T ES 14835606 T ES14835606 T ES 14835606T ES 2758506 T3 ES2758506 T3 ES 2758506T3
- Authority
- ES
- Spain
- Prior art keywords
- methyl
- methoxy
- mmol
- amino
- pyrrolidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title description 95
- 230000001472 cytotoxic effect Effects 0.000 title description 68
- 231100000433 cytotoxic Toxicity 0.000 title description 67
- 102000004196 processed proteins & peptides Human genes 0.000 title description 65
- 150000001875 compounds Chemical class 0.000 claims abstract description 184
- 150000003839 salts Chemical class 0.000 claims abstract description 46
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 32
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims abstract description 32
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 28
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 22
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 21
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 15
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 13
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 11
- 125000001715 oxadiazolyl group Chemical group 0.000 claims abstract description 5
- 125000003226 pyrazolyl group Chemical group 0.000 claims abstract description 5
- 125000000714 pyrimidinyl group Chemical group 0.000 claims abstract description 5
- 125000003831 tetrazolyl group Chemical group 0.000 claims abstract description 5
- -1 leukein Chemical compound 0.000 claims description 405
- 230000027455 binding Effects 0.000 claims description 96
- 238000009739 binding Methods 0.000 claims description 93
- 108091007433 antigens Proteins 0.000 claims description 87
- 102000036639 antigens Human genes 0.000 claims description 87
- 239000000427 antigen Substances 0.000 claims description 84
- 229940127121 immunoconjugate Drugs 0.000 claims description 81
- 229910052739 hydrogen Inorganic materials 0.000 claims description 62
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 59
- 229910052799 carbon Inorganic materials 0.000 claims description 49
- 206010028980 Neoplasm Diseases 0.000 claims description 46
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 30
- 201000011510 cancer Diseases 0.000 claims description 28
- 229940024606 amino acid Drugs 0.000 claims description 22
- 235000001014 amino acid Nutrition 0.000 claims description 22
- 150000001413 amino acids Chemical class 0.000 claims description 22
- 125000000217 alkyl group Chemical group 0.000 claims description 21
- 239000003814 drug Substances 0.000 claims description 21
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 claims description 18
- 239000003795 chemical substances by application Substances 0.000 claims description 15
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 15
- 238000011282 treatment Methods 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 13
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 9
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims description 7
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 7
- 235000018417 cysteine Nutrition 0.000 claims description 7
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 7
- 239000004472 Lysine Substances 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 235000018977 lysine Nutrition 0.000 claims description 6
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 claims description 5
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 5
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 5
- 229930182817 methionine Natural products 0.000 claims description 5
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 4
- 239000004475 Arginine Substances 0.000 claims description 4
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 4
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 4
- 239000004471 Glycine Substances 0.000 claims description 4
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 4
- ZGUNAGUHMKGQNY-ZETCQYMHSA-N L-alpha-phenylglycine zwitterion Chemical compound OC(=O)[C@@H](N)C1=CC=CC=C1 ZGUNAGUHMKGQNY-ZETCQYMHSA-N 0.000 claims description 4
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 claims description 4
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 4
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims description 4
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 claims description 4
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 claims description 4
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 4
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 claims description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 4
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 4
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 4
- 239000004473 Threonine Substances 0.000 claims description 4
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 4
- 235000004279 alanine Nutrition 0.000 claims description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims description 4
- 235000009697 arginine Nutrition 0.000 claims description 4
- 235000009582 asparagine Nutrition 0.000 claims description 4
- 229960001230 asparagine Drugs 0.000 claims description 4
- 235000013922 glutamic acid Nutrition 0.000 claims description 4
- 239000004220 glutamic acid Substances 0.000 claims description 4
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 4
- 235000004554 glutamine Nutrition 0.000 claims description 4
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims description 4
- 229960000310 isoleucine Drugs 0.000 claims description 4
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 4
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 4
- 229960005190 phenylalanine Drugs 0.000 claims description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 4
- 239000004474 valine Substances 0.000 claims description 4
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 2
- 102200001405 rs377584435 Human genes 0.000 claims 1
- 125000002883 imidazolyl group Chemical group 0.000 abstract description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 7
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 abstract 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 353
- 239000011541 reaction mixture Substances 0.000 description 219
- 230000014759 maintenance of location Effects 0.000 description 210
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 209
- 238000006243 chemical reaction Methods 0.000 description 158
- 239000000047 product Substances 0.000 description 152
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 150
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 131
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 127
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 119
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 118
- 239000000243 solution Substances 0.000 description 103
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 97
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 86
- 239000002253 acid Substances 0.000 description 86
- 210000004027 cell Anatomy 0.000 description 84
- 238000000034 method Methods 0.000 description 83
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 79
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 73
- 239000007821 HATU Substances 0.000 description 66
- 238000004007 reversed phase HPLC Methods 0.000 description 59
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 58
- 125000005647 linker group Chemical group 0.000 description 54
- 239000000203 mixture Substances 0.000 description 54
- 238000002953 preparative HPLC Methods 0.000 description 51
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 49
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 45
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 42
- 239000000611 antibody drug conjugate Substances 0.000 description 41
- 229940049595 antibody-drug conjugate Drugs 0.000 description 41
- 239000012634 fragment Substances 0.000 description 36
- 235000019439 ethyl acetate Nutrition 0.000 description 33
- 239000002904 solvent Substances 0.000 description 33
- 238000005160 1H NMR spectroscopy Methods 0.000 description 30
- 239000007787 solid Substances 0.000 description 26
- 230000015572 biosynthetic process Effects 0.000 description 25
- 150000003840 hydrochlorides Chemical class 0.000 description 25
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 24
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 23
- 108090000623 proteins and genes Proteins 0.000 description 23
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 22
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 22
- WMFOQBRAJBCJND-UHFFFAOYSA-M lithium hydroxide Inorganic materials [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 22
- 239000012074 organic phase Substances 0.000 description 21
- 238000000746 purification Methods 0.000 description 21
- 230000002829 reductive effect Effects 0.000 description 21
- 239000000562 conjugate Substances 0.000 description 19
- 238000003786 synthesis reaction Methods 0.000 description 19
- 239000013598 vector Substances 0.000 description 19
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 18
- 235000018102 proteins Nutrition 0.000 description 18
- 102000004169 proteins and genes Human genes 0.000 description 18
- 125000001424 substituent group Chemical group 0.000 description 18
- 229940079593 drug Drugs 0.000 description 17
- 230000014509 gene expression Effects 0.000 description 17
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 16
- 238000004458 analytical method Methods 0.000 description 16
- 229910052805 deuterium Inorganic materials 0.000 description 16
- 238000004128 high performance liquid chromatography Methods 0.000 description 16
- 238000000338 in vitro Methods 0.000 description 16
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 229920001184 polypeptide Polymers 0.000 description 15
- 229920006395 saturated elastomer Polymers 0.000 description 15
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 125000003275 alpha amino acid group Chemical group 0.000 description 14
- 201000010099 disease Diseases 0.000 description 14
- 239000013604 expression vector Substances 0.000 description 14
- 101100383064 Bacillus subtilis (strain 168) cdaR gene Proteins 0.000 description 13
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 13
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- 239000001257 hydrogen Substances 0.000 description 13
- 238000001727 in vivo Methods 0.000 description 13
- 230000035772 mutation Effects 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 239000007832 Na2SO4 Substances 0.000 description 12
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- 125000006580 bicyclic heterocycloalkyl group Chemical group 0.000 description 12
- 239000003112 inhibitor Substances 0.000 description 12
- 229910052938 sodium sulfate Inorganic materials 0.000 description 12
- 235000011152 sodium sulphate Nutrition 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- GMORVOQOIHISPT-UHFFFAOYSA-N 2-ethylhexanamide Chemical compound CCCCC(CC)C(N)=O GMORVOQOIHISPT-UHFFFAOYSA-N 0.000 description 11
- 206010025323 Lymphomas Diseases 0.000 description 11
- 229930040373 Paraformaldehyde Natural products 0.000 description 11
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 239000000945 filler Substances 0.000 description 11
- 229920002866 paraformaldehyde Polymers 0.000 description 11
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 10
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 10
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 10
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 10
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 10
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 208000035475 disorder Diseases 0.000 description 10
- 230000002496 gastric effect Effects 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 9
- 125000002947 alkylene group Chemical group 0.000 description 9
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 9
- 239000000460 chlorine Chemical group 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 230000021615 conjugation Effects 0.000 description 9
- 229940088598 enzyme Drugs 0.000 description 9
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 9
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 9
- 235000019341 magnesium sulphate Nutrition 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- 102000040430 polynucleotide Human genes 0.000 description 9
- 108091033319 polynucleotide Proteins 0.000 description 9
- 239000002157 polynucleotide Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 239000011734 sodium Substances 0.000 description 9
- ZREGAIWSPAJLPD-HNNXBMFYSA-N (1s)-2-phenyl-1-(5-phenyl-1h-imidazol-2-yl)ethanamine Chemical compound C([C@H](N)C=1NC=C(N=1)C=1C=CC=CC=1)C1=CC=CC=C1 ZREGAIWSPAJLPD-HNNXBMFYSA-N 0.000 description 8
- NKBWMBRPILTCRD-UHFFFAOYSA-N 2-Methylheptanoic acid Chemical compound CCCCCC(C)C(O)=O NKBWMBRPILTCRD-UHFFFAOYSA-N 0.000 description 8
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 8
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 239000000872 buffer Substances 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 8
- 239000003921 oil Substances 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- PQRNDCLOGHHYLO-VIFPVBQESA-N (2r)-n-[3-(2-azidoethylamino)-3-oxopropyl]-2,4-dihydroxy-3,3-dimethylbutanamide Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCN=[N+]=[N-] PQRNDCLOGHHYLO-VIFPVBQESA-N 0.000 description 7
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 7
- 241000588724 Escherichia coli Species 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 125000001314 canonical amino-acid group Chemical group 0.000 description 7
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 7
- 239000005516 coenzyme A Substances 0.000 description 7
- 229940093530 coenzyme a Drugs 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 208000032839 leukemia Diseases 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 6
- 206010035226 Plasma cell myeloma Diseases 0.000 description 6
- 206010039491 Sarcoma Diseases 0.000 description 6
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 230000002927 anti-mitotic effect Effects 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- ILKFHZRKLTWFKR-UHFFFAOYSA-N benzyl n-benzylcarbamate Chemical compound C=1C=CC=CC=1COC(=O)NCC1=CC=CC=C1 ILKFHZRKLTWFKR-UHFFFAOYSA-N 0.000 description 6
- 239000011230 binding agent Substances 0.000 description 6
- 208000035269 cancer or benign tumor Diseases 0.000 description 6
- 230000004663 cell proliferation Effects 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 230000008878 coupling Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- 238000010511 deprotection reaction Methods 0.000 description 6
- 239000003623 enhancer Substances 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 150000002430 hydrocarbons Chemical class 0.000 description 6
- 230000001939 inductive effect Effects 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- 208000003747 lymphoid leukemia Diseases 0.000 description 6
- 201000000050 myeloid neoplasm Diseases 0.000 description 6
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 6
- 230000003287 optical effect Effects 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- AKLOSWXBWKPISE-JTQLQIEISA-N (1S)-2-phenyl-1-(1H-pyrazol-5-yl)ethanamine Chemical compound C1(=CC=CC=C1)C[C@H](N)C1=NNC=C1 AKLOSWXBWKPISE-JTQLQIEISA-N 0.000 description 5
- OCXHPHOVIMNVPQ-UTHKHBGESA-N (2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(O)=O OCXHPHOVIMNVPQ-UTHKHBGESA-N 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 5
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 5
- 206010006187 Breast cancer Diseases 0.000 description 5
- 208000026310 Breast neoplasm Diseases 0.000 description 5
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 5
- 108020004414 DNA Proteins 0.000 description 5
- 206010061968 Gastric neoplasm Diseases 0.000 description 5
- 208000032612 Glial tumor Diseases 0.000 description 5
- 206010018338 Glioma Diseases 0.000 description 5
- 208000017604 Hodgkin disease Diseases 0.000 description 5
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 5
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 5
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 5
- 208000034578 Multiple myelomas Diseases 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 5
- 206010029260 Neuroblastoma Diseases 0.000 description 5
- 206010033128 Ovarian cancer Diseases 0.000 description 5
- 206010061535 Ovarian neoplasm Diseases 0.000 description 5
- 208000024770 Thyroid neoplasm Diseases 0.000 description 5
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 239000003242 anti bacterial agent Substances 0.000 description 5
- 230000000890 antigenic effect Effects 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 5
- 238000001516 cell proliferation assay Methods 0.000 description 5
- 239000007795 chemical reaction product Substances 0.000 description 5
- 210000001072 colon Anatomy 0.000 description 5
- 208000029742 colonic neoplasm Diseases 0.000 description 5
- 125000004093 cyano group Chemical group *C#N 0.000 description 5
- 231100000599 cytotoxic agent Toxicity 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 230000002255 enzymatic effect Effects 0.000 description 5
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 5
- 150000002431 hydrogen Chemical class 0.000 description 5
- 238000003018 immunoassay Methods 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 229940090044 injection Drugs 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 201000005202 lung cancer Diseases 0.000 description 5
- 208000020816 lung neoplasm Diseases 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 201000001441 melanoma Diseases 0.000 description 5
- 208000025113 myeloid leukemia Diseases 0.000 description 5
- 208000025402 neoplasm of esophagus Diseases 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- 208000023958 prostate neoplasm Diseases 0.000 description 5
- 150000003254 radicals Chemical class 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 201000002510 thyroid cancer Diseases 0.000 description 5
- OVJXUELLYTXSHZ-AWEZNQCLSA-N (1s)-2-phenyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)ethanamine Chemical compound C([C@H](N)C=1OC(=NN=1)C=1C=CC=CC=1)C1=CC=CC=C1 OVJXUELLYTXSHZ-AWEZNQCLSA-N 0.000 description 4
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- AUDYZXNUHIIGRB-UHFFFAOYSA-N 3-thiophen-2-ylpyrrole-2,5-dione Chemical compound O=C1NC(=O)C(C=2SC=CC=2)=C1 AUDYZXNUHIIGRB-UHFFFAOYSA-N 0.000 description 4
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 4
- WOJKKJKETHYEAC-UHFFFAOYSA-N 6-Maleimidocaproic acid Chemical compound OC(=O)CCCCCN1C(=O)C=CC1=O WOJKKJKETHYEAC-UHFFFAOYSA-N 0.000 description 4
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 4
- 206010009944 Colon cancer Diseases 0.000 description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- ZFOMKMMPBOQKMC-KXUCPTDWSA-N L-pyrrolysine Chemical compound C[C@@H]1CC=N[C@H]1C(=O)NCCCC[C@H]([NH3+])C([O-])=O ZFOMKMMPBOQKMC-KXUCPTDWSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 4
- 229920002684 Sepharose Polymers 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- HYSPJPGXSALJRR-DHIFEGFHSA-N [4-[[(2s)-5-(carbamoylamino)-2-[[(2s)-2-[6-(2,5-dioxopyrrol-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl (4-nitrophenyl) carbonate Chemical compound N([C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NC=1C=CC(COC(=O)OC=2C=CC(=CC=2)[N+]([O-])=O)=CC=1)C(=O)CCCCCN1C(=O)C=CC1=O HYSPJPGXSALJRR-DHIFEGFHSA-N 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 125000000539 amino acid group Chemical group 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- CERDIVAFXRCORQ-UHFFFAOYSA-O butoxy-hydroxy-oxophosphanium Chemical compound CCCCO[P+](O)=O CERDIVAFXRCORQ-UHFFFAOYSA-O 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 238000003780 insertion Methods 0.000 description 4
- 230000037431 insertion Effects 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- 230000000155 isotopic effect Effects 0.000 description 4
- 210000003734 kidney Anatomy 0.000 description 4
- HNGRJOCEIWGCTB-LURJTMIESA-N methyl (2s)-3-amino-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound COC(=O)[C@H](CN)NC(=O)OC(C)(C)C HNGRJOCEIWGCTB-LURJTMIESA-N 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000013612 plasmid Substances 0.000 description 4
- 230000036515 potency Effects 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 235000017550 sodium carbonate Nutrition 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 239000003053 toxin Substances 0.000 description 4
- 231100000765 toxin Toxicity 0.000 description 4
- 108700012359 toxins Proteins 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 238000000108 ultra-filtration Methods 0.000 description 4
- 230000003612 virological effect Effects 0.000 description 4
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 4
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 3
- SVMIVBMALOISKL-UHFFFAOYSA-N 1-(6-hydroxyhexyl)pyrrole-2,5-dione Chemical compound OCCCCCCN1C(=O)C=CC1=O SVMIVBMALOISKL-UHFFFAOYSA-N 0.000 description 3
- STVVMTBJNDTZBF-UHFFFAOYSA-N 2-amino-3-phenylpropan-1-ol Chemical compound OCC(N)CC1=CC=CC=C1 STVVMTBJNDTZBF-UHFFFAOYSA-N 0.000 description 3
- YTZFUAXOTCJZHP-UHFFFAOYSA-N 3-[2-(2,5-dioxopyrrol-1-yl)ethoxy]propanoic acid Chemical compound OC(=O)CCOCCN1C(=O)C=CC1=O YTZFUAXOTCJZHP-UHFFFAOYSA-N 0.000 description 3
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 3
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 3
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical group FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 108060003951 Immunoglobulin Proteins 0.000 description 3
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 3
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 3
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 108010076504 Protein Sorting Signals Proteins 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 3
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- PUJDIJCNWFYVJX-UHFFFAOYSA-N benzyl carbamate Chemical compound NC(=O)OCC1=CC=CC=C1 PUJDIJCNWFYVJX-UHFFFAOYSA-N 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 230000022534 cell killing Effects 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 229910052801 chlorine Chemical group 0.000 description 3
- 235000015165 citric acid Nutrition 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229940125807 compound 37 Drugs 0.000 description 3
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 3
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 239000002254 cytotoxic agent Substances 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 3
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000011737 fluorine Chemical group 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 102000018358 immunoglobulin Human genes 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 230000036210 malignancy Effects 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- TWXDDNPPQUTEOV-FVGYRXGTSA-N methamphetamine hydrochloride Chemical compound Cl.CN[C@@H](C)CC1=CC=CC=C1 TWXDDNPPQUTEOV-FVGYRXGTSA-N 0.000 description 3
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 3
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 3
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 150000007523 nucleic acids Chemical class 0.000 description 3
- 239000001301 oxygen Chemical group 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 230000028327 secretion Effects 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical group O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- DLNKOYKMWOXYQA-VXNVDRBHSA-N (+)-norephedrine Chemical compound C[C@@H](N)[C@@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-VXNVDRBHSA-N 0.000 description 2
- NXQBRADSLPEODL-QMMMGPOBSA-N (1s)-2-phenyl-1-(2h-tetrazol-5-yl)ethanamine Chemical compound C([C@H](N)C=1NN=NN=1)C1=CC=CC=C1 NXQBRADSLPEODL-QMMMGPOBSA-N 0.000 description 2
- IFAMSTPTNRJBRG-YIZRAAEISA-N (1s,2s,4r)-3-[(2-methylpropan-2-yl)oxycarbonyl]-3-azabicyclo[2.2.1]heptane-2-carboxylic acid Chemical compound C1C[C@@H]2[C@@H](C(O)=O)N(C(=O)OC(C)(C)C)[C@H]1C2 IFAMSTPTNRJBRG-YIZRAAEISA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 2
- HYUJTBXBZQMMDQ-VIFPVBQESA-N (2S)-1-methoxy-3-(3-nitrophenyl)propan-2-amine Chemical compound COC[C@H](CC1=CC(=CC=C1)[N+](=O)[O-])N HYUJTBXBZQMMDQ-VIFPVBQESA-N 0.000 description 2
- VGBXXXWWAFSRIA-VIFPVBQESA-N (2S)-2-amino-3-phenylpropane-1-sulfonamide Chemical compound N[C@H](CS(=O)(=O)N)CC1=CC=CC=C1 VGBXXXWWAFSRIA-VIFPVBQESA-N 0.000 description 2
- AIQKYLNXJKSLHP-NSHDSACASA-N (2S)-2-amino-N-(3-azidopropylsulfonyl)-3-phenylpropanamide Chemical compound N[C@H](C(=O)NS(=O)(=O)CCCN=[N+]=[N-])CC1=CC=CC=C1 AIQKYLNXJKSLHP-NSHDSACASA-N 0.000 description 2
- JDSSVJPEXKWINW-INIZCTEOSA-N (2S)-2-amino-N-[2-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxy]ethyl]-3-phenylpropanamide Chemical compound N[C@H](C(=O)NCCOCCOCCN1C(C=CC1=O)=O)CC1=CC=CC=C1 JDSSVJPEXKWINW-INIZCTEOSA-N 0.000 description 2
- VVGOTPOKECMNAT-INIZCTEOSA-N (2S)-2-amino-N-[2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethyl]-3-phenylpropanamide Chemical compound N[C@H](C(=O)NCCOCCOCCOCCN=[N+]=[N-])CC1=CC=CC=C1 VVGOTPOKECMNAT-INIZCTEOSA-N 0.000 description 2
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 2
- UZYSMEFHEZAPDI-VIFPVBQESA-N (2s)-2-amino-3-(3-aminophenyl)propan-1-ol Chemical compound OC[C@@H](N)CC1=CC=CC(N)=C1 UZYSMEFHEZAPDI-VIFPVBQESA-N 0.000 description 2
- SNRJFODLMSOJIX-QMMMGPOBSA-N (2s)-2-amino-3-(3-nitrophenyl)propan-1-ol Chemical compound OC[C@@H](N)CC1=CC=CC([N+]([O-])=O)=C1 SNRJFODLMSOJIX-QMMMGPOBSA-N 0.000 description 2
- GMTRSZXBNADBMK-VIFPVBQESA-N (2s)-2-amino-3-(4-aminophenyl)propan-1-ol Chemical compound OC[C@@H](N)CC1=CC=C(N)C=C1 GMTRSZXBNADBMK-VIFPVBQESA-N 0.000 description 2
- PCLGMKWXPXAGEO-QMMMGPOBSA-N (2s)-2-amino-3-(4-nitrophenyl)propan-1-ol Chemical compound OC[C@@H](N)CC1=CC=C([N+]([O-])=O)C=C1 PCLGMKWXPXAGEO-QMMMGPOBSA-N 0.000 description 2
- KRJLRVZLNABMAT-YFKPBYRVSA-N (2s)-3-amino-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](CN)C(O)=O KRJLRVZLNABMAT-YFKPBYRVSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- MQLACMBJVPINKE-UHFFFAOYSA-N 10-[(3-hydroxy-4-methoxyphenyl)methylidene]anthracen-9-one Chemical compound C1=C(O)C(OC)=CC=C1C=C1C2=CC=CC=C2C(=O)C2=CC=CC=C21 MQLACMBJVPINKE-UHFFFAOYSA-N 0.000 description 2
- GVVWCRQQBUQLMZ-UHFFFAOYSA-N 2-(2-methyl-1,3-dioxolan-2-yl)ethanamine Chemical compound NCCC1(C)OCCO1 GVVWCRQQBUQLMZ-UHFFFAOYSA-N 0.000 description 2
- AZYDCZDQGPLAQS-UHFFFAOYSA-N 2-(4-methoxyphenyl)-5,5-dimethyl-1,3-dioxane-4-carbaldehyde Chemical compound C1=CC(OC)=CC=C1C1OC(C=O)C(C)(C)CO1 AZYDCZDQGPLAQS-UHFFFAOYSA-N 0.000 description 2
- LRWPHTXGTSBOQS-UHFFFAOYSA-N 2-(4-methoxyphenyl)-5,5-dimethyl-1,3-dioxane-4-carboxylic acid Chemical compound C1=CC(OC)=CC=C1C1OC(C(O)=O)C(C)(C)CO1 LRWPHTXGTSBOQS-UHFFFAOYSA-N 0.000 description 2
- SDUPGQBSWSABRG-UHFFFAOYSA-N 2-phenyl-1-pyrimidin-2-ylethanamine Chemical compound N=1C=CC=NC=1C(N)CC1=CC=CC=C1 SDUPGQBSWSABRG-UHFFFAOYSA-N 0.000 description 2
- SJTKQIPAACOOGS-UHFFFAOYSA-N 3,3-dimethylbutane-1,2,4-triol Chemical compound OCC(C)(C)C(O)CO SJTKQIPAACOOGS-UHFFFAOYSA-N 0.000 description 2
- YGSGZOINLXDDAU-JTQLQIEISA-N 3-[(2S)-2-amino-3-methoxypropyl]aniline Chemical compound N[C@@H](CC=1C=C(N)C=CC1)COC YGSGZOINLXDDAU-JTQLQIEISA-N 0.000 description 2
- TXMDOUPVKVGPIS-UHFFFAOYSA-N 3-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxy]propanoic acid Chemical compound OC(=O)CCOCCOCCN1C(=O)C=CC1=O TXMDOUPVKVGPIS-UHFFFAOYSA-N 0.000 description 2
- WUIABRMSWOKTOF-OYALTWQYSA-N 3-[[2-[2-[2-[[(2s,3r)-2-[[(2s,3s,4r)-4-[[(2s,3r)-2-[[6-amino-2-[(1s)-3-amino-1-[[(2s)-2,3-diamino-3-oxopropyl]amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2r,3s,4s,5r,6r)-4-carbamoyloxy-3,5-dihydroxy-6-(hydroxymethyl)ox Chemical compound OS([O-])(=O)=O.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C WUIABRMSWOKTOF-OYALTWQYSA-N 0.000 description 2
- DBRIOBHILRGDPP-UHFFFAOYSA-N 3-azidopropane-1-sulfonic acid Chemical compound OS(=O)(=O)CCCN=[N+]=[N-] DBRIOBHILRGDPP-UHFFFAOYSA-N 0.000 description 2
- FVRXLSOOPLSSNW-UHFFFAOYSA-N 3-azidopropane-1-sulfonyl chloride Chemical compound ClS(=O)(=O)CCCN=[N+]=[N-] FVRXLSOOPLSSNW-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical class OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- WEAMRAGCXUIACZ-UHFFFAOYSA-N 6-(2,5-dioxopyrrol-1-yl)-N-propylsulfonylhexanamide Chemical compound O=C1N(C(C=C1)=O)CCCCCC(=O)NS(=O)(=O)CCC WEAMRAGCXUIACZ-UHFFFAOYSA-N 0.000 description 2
- ZWIGHFZUSGHZEZ-UHFFFAOYSA-N 6-(2,5-dioxopyrrol-1-yl)hexanal Chemical compound O=CCCCCCN1C(=O)C=CC1=O ZWIGHFZUSGHZEZ-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 101710117290 Aldo-keto reductase family 1 member C4 Proteins 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 108010024976 Asparaginase Proteins 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- 101710205625 Capsid protein p24 Proteins 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 2
- 238000001712 DNA sequencing Methods 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- 108010016626 Dipeptides Proteins 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 2
- 102000003792 Metallothionein Human genes 0.000 description 2
- 108090000157 Metallothionein Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 101710181812 Methionine aminopeptidase Proteins 0.000 description 2
- 101710160066 Mitochondrial holo-[acyl-carrier-protein] synthase Proteins 0.000 description 2
- 241000713333 Mouse mammary tumor virus Species 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 101710177166 Phosphoprotein Proteins 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 241000710961 Semliki Forest virus Species 0.000 description 2
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 2
- 101710149279 Small delta antigen Proteins 0.000 description 2
- 241000191940 Staphylococcus Species 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- 102100022563 Tubulin polymerization-promoting protein Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- CHKFLBOLYREYDO-SHYZEUOFSA-N [[(2s,4r,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-4-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)C[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 CHKFLBOLYREYDO-SHYZEUOFSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 2
- 230000002494 anti-cea effect Effects 0.000 description 2
- 229940046836 anti-estrogen Drugs 0.000 description 2
- 230000001833 anti-estrogenic effect Effects 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 230000003127 anti-melanomic effect Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000003418 antiprogestin Substances 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- FUSUHKVFWTUUBE-UHFFFAOYSA-N buten-2-one Chemical compound CC(=O)C=C FUSUHKVFWTUUBE-UHFFFAOYSA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229960002436 cladribine Drugs 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125877 compound 31 Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 239000002619 cytotoxin Substances 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- VSPMNQQOQWDCQX-UHFFFAOYSA-N diazonio(3-sulfamoylpropyl)azanide Chemical compound NS(=O)(=O)CCCN=[N+]=[N-] VSPMNQQOQWDCQX-UHFFFAOYSA-N 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 150000002019 disulfides Chemical class 0.000 description 2
- AFOSIXZFDONLBT-UHFFFAOYSA-N divinyl sulfone Chemical compound C=CS(=O)(=O)C=C AFOSIXZFDONLBT-UHFFFAOYSA-N 0.000 description 2
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000000328 estrogen antagonist Substances 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- 150000007857 hydrazones Chemical class 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-M isobutyrate Chemical compound CC(C)C([O-])=O KQNPFQTWMSNSAP-UHFFFAOYSA-M 0.000 description 2
- 125000005921 isopentoxy group Chemical group 0.000 description 2
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000004574 piperidin-2-yl group Chemical group N1C(CCCC1)* 0.000 description 2
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 2
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 239000013615 primer Substances 0.000 description 2
- 230000003623 progesteronic effect Effects 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 239000013557 residual solvent Substances 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 239000006152 selective media Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000019187 sodium-L-ascorbate Nutrition 0.000 description 2
- 239000011755 sodium-L-ascorbate Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- FQZYTYWMLGAPFJ-OQKDUQJOSA-N tamoxifen citrate Chemical compound [H+].[H+].[H+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 FQZYTYWMLGAPFJ-OQKDUQJOSA-N 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 102000013498 tau Proteins Human genes 0.000 description 2
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 2
- FQJRZOYTUCCBQR-UHFFFAOYSA-N tert-butyl 2-methylheptanoate Chemical compound CCCCCC(C)C(=O)OC(C)(C)C FQJRZOYTUCCBQR-UHFFFAOYSA-N 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- ORYDPOVDJJZGHQ-UHFFFAOYSA-N tirapazamine Chemical compound C1=CC=CC2=[N+]([O-])C(N)=N[N+]([O-])=C21 ORYDPOVDJJZGHQ-UHFFFAOYSA-N 0.000 description 2
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 241001529453 unidentified herpesvirus Species 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 239000013603 viral vector Substances 0.000 description 2
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- ZJIFDEVVTPEXDL-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) hydrogen carbonate Chemical compound OC(=O)ON1C(=O)CCC1=O ZJIFDEVVTPEXDL-UHFFFAOYSA-N 0.000 description 1
- OMEXBARTDSXSIO-VIFPVBQESA-N (2S)-2-amino-N-methylsulfonyl-3-phenylpropanamide Chemical compound N[C@H](C(=O)NS(=O)(=O)C)CC1=CC=CC=C1 OMEXBARTDSXSIO-VIFPVBQESA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- FDKWRPBBCBCIGA-REOHCLBHSA-N (2r)-2-azaniumyl-3-$l^{1}-selanylpropanoate Chemical compound [Se]C[C@H](N)C(O)=O FDKWRPBBCBCIGA-REOHCLBHSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- FAZIHZPDHJBQKN-JTQLQIEISA-N (2s)-1-methoxy-3-phenylpropan-2-amine Chemical compound COC[C@@H](N)CC1=CC=CC=C1 FAZIHZPDHJBQKN-JTQLQIEISA-N 0.000 description 1
- BXSIPZGNIBVSLK-AJZOCDQUSA-N (2s)-1-tritylaziridine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 BXSIPZGNIBVSLK-AJZOCDQUSA-N 0.000 description 1
- ORFNVPGICPYLJV-YTVPMEHESA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-[6-(2,5-dioxopyrrol-1-yl)hexanoyl-methylamino]-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-methoxy-2-methylpropanoyl]amino]-3-phenylpropan Chemical compound C([C@H](NC(=O)[C@H](C)[C@@H](OC)[C@@H]1CCCN1C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCCCN1C(C=CC1=O)=O)C(C)C)OC)C(O)=O)C1=CC=CC=C1 ORFNVPGICPYLJV-YTVPMEHESA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- WHXSMMKQMYFTQS-BJUDXGSMSA-N (6Li)Lithium Chemical compound [6Li] WHXSMMKQMYFTQS-BJUDXGSMSA-N 0.000 description 1
- VNTHYLVDGVBPOU-QQYBVWGSSA-N (7s,9s)-9-acetyl-7-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 VNTHYLVDGVBPOU-QQYBVWGSSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- HNWOYAYJUYWRGD-UHFFFAOYSA-N 1-(6-aminohexyl)pyrrole-2,5-dione Chemical compound NCCCCCCN1C(=O)C=CC1=O HNWOYAYJUYWRGD-UHFFFAOYSA-N 0.000 description 1
- NNHYAHOTXLASEA-UHFFFAOYSA-N 1-(dimethoxymethyl)-4-methoxybenzene Chemical compound COC(OC)C1=CC=C(OC)C=C1 NNHYAHOTXLASEA-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- SHDYDABQVJGHLD-UHFFFAOYSA-N 1-[2-[2-(2-aminoethoxy)ethoxy]ethyl]pyrrole-2,5-dione Chemical compound NCCOCCOCCN1C(=O)C=CC1=O SHDYDABQVJGHLD-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- AFFLGGQVNFXPEV-UHFFFAOYSA-N 1-decene Chemical group CCCCCCCCC=C AFFLGGQVNFXPEV-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- ZNUKSRFJVSGOTL-UHFFFAOYSA-N 1-nitro-4-(trichloromethyl)benzene Chemical compound [O-][N+](=O)C1=CC=C(C(Cl)(Cl)Cl)C=C1 ZNUKSRFJVSGOTL-UHFFFAOYSA-N 0.000 description 1
- GVPDKCZQAZOFJX-UHFFFAOYSA-N 1-pyrimidin-2-ylethanamine Chemical compound CC(N)C1=NC=CC=N1 GVPDKCZQAZOFJX-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- OBETXYAYXDNJHR-UHFFFAOYSA-N 2-Ethylhexanoic acid Chemical compound CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- FPVCVHVTMPCZTH-UHFFFAOYSA-N 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethanamine Chemical compound NCCOCCOCCOCCN=[N+]=[N-] FPVCVHVTMPCZTH-UHFFFAOYSA-N 0.000 description 1
- UPMDUJBOYVOHRH-UHFFFAOYSA-N 2-[4-[2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethoxy]phenyl]-5-methylsulfonyl-1,3,4-oxadiazole Chemical compound O1C(S(=O)(=O)C)=NN=C1C1=CC=C(OCCOCCOCCOCCN=[N+]=[N-])C=C1 UPMDUJBOYVOHRH-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- FZDFGHZZPBUTGP-UHFFFAOYSA-N 2-[[2-[bis(carboxymethyl)amino]-3-(4-isothiocyanatophenyl)propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)C(C)CN(CC(O)=O)CC(N(CC(O)=O)CC(O)=O)CC1=CC=C(N=C=S)C=C1 FZDFGHZZPBUTGP-UHFFFAOYSA-N 0.000 description 1
- WXHLLJAMBQLULT-UHFFFAOYSA-N 2-[[6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl]amino]-n-(2-methyl-6-sulfanylphenyl)-1,3-thiazole-5-carboxamide;hydrate Chemical compound O.C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1S WXHLLJAMBQLULT-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- DJQYYYCQOZMCRC-UHFFFAOYSA-N 2-aminopropane-1,3-dithiol Chemical compound SCC(N)CS DJQYYYCQOZMCRC-UHFFFAOYSA-N 0.000 description 1
- CSTIZSQKHUSKHU-UHFFFAOYSA-N 2-azidoethanamine Chemical compound NCCN=[N+]=[N-] CSTIZSQKHUSKHU-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- LSTRKXWIZZZYAS-UHFFFAOYSA-N 2-bromoacetyl bromide Chemical compound BrCC(Br)=O LSTRKXWIZZZYAS-UHFFFAOYSA-N 0.000 description 1
- HTWIZMNMTWYQRN-UHFFFAOYSA-N 2-methyl-1,3-dioxolane Chemical compound CC1OCCO1 HTWIZMNMTWYQRN-UHFFFAOYSA-N 0.000 description 1
- CVKMFSAVYPAZTQ-UHFFFAOYSA-N 2-methylhexanoic acid Chemical compound CCCCC(C)C(O)=O CVKMFSAVYPAZTQ-UHFFFAOYSA-N 0.000 description 1
- UXSCTIIPGJOZNA-UHFFFAOYSA-N 2-nitro-4-(6-oxohexoxy)benzaldehyde Chemical compound [N+](=O)([O-])C1=C(C=O)C=CC(=C1)OCCCCCC=O UXSCTIIPGJOZNA-UHFFFAOYSA-N 0.000 description 1
- VSMANSLTTPNSMB-UHFFFAOYSA-N 3-(4-nitrophenyl)propan-1-ol Chemical compound OCCCC1=CC=C([N+]([O-])=O)C=C1 VSMANSLTTPNSMB-UHFFFAOYSA-N 0.000 description 1
- IEHDSRSXQAOLJT-UHFFFAOYSA-N 3-[2-(2-azidoethoxy)ethoxy]propanoic acid Chemical compound OC(=O)CCOCCOCCN=[N+]=[N-] IEHDSRSXQAOLJT-UHFFFAOYSA-N 0.000 description 1
- YSFOTVPQFVYTQX-UHFFFAOYSA-N 3-[2-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound OC(=O)CCOCCOCCOCCN1C(=O)C=CC1=O YSFOTVPQFVYTQX-UHFFFAOYSA-N 0.000 description 1
- UQWLGZFJGVEDCQ-UHFFFAOYSA-N 3-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound OCCOCCOCCOCCOCCC(O)=O UQWLGZFJGVEDCQ-UHFFFAOYSA-N 0.000 description 1
- BTFZSOVKVDCKQD-UHFFFAOYSA-N 3-[2-[2-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound OC(=O)CCOCCOCCOCCOCCN1C(=O)C=CC1=O BTFZSOVKVDCKQD-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- JVYNJRBSXBYXQB-UHFFFAOYSA-N 4-[3-(4-carboxyphenoxy)propoxy]benzoic acid;decanedioic acid Chemical compound OC(=O)CCCCCCCCC(O)=O.C1=CC(C(=O)O)=CC=C1OCCCOC1=CC=C(C(O)=O)C=C1 JVYNJRBSXBYXQB-UHFFFAOYSA-N 0.000 description 1
- MMBZCFJKAQZVNI-VPENINKCSA-N 4-amino-5,6-difluoro-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound FC1=C(F)C(N)=NC(=O)N1[C@@H]1O[C@H](CO)[C@@H](O)C1 MMBZCFJKAQZVNI-VPENINKCSA-N 0.000 description 1
- WAGMYTXJRVPMGW-UHFFFAOYSA-N 4-azidobutanoic acid Chemical compound OC(=O)CCCN=[N+]=[N-] WAGMYTXJRVPMGW-UHFFFAOYSA-N 0.000 description 1
- SUTWPJHCRAITLU-UHFFFAOYSA-N 6-aminohexan-1-ol Chemical compound NCCCCCCO SUTWPJHCRAITLU-UHFFFAOYSA-N 0.000 description 1
- FPFTWHJPEMPAGE-UHFFFAOYSA-N 6-hydroxy caproaldehyde Chemical compound OCCCCCC=O FPFTWHJPEMPAGE-UHFFFAOYSA-N 0.000 description 1
- IWHLYPDWHHPVAA-UHFFFAOYSA-N 6-hydroxyhexanoic acid Chemical compound OCCCCCC(O)=O IWHLYPDWHHPVAA-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 102100022749 Aminopeptidase N Human genes 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 241000132092 Aster Species 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 241000304886 Bacilli Species 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- NEHKHPQFTUZMMX-UHFFFAOYSA-N C1(CCC(N1)=O)=O.C1(CCCCC1)C(=O)O Chemical compound C1(CCC(N1)=O)=O.C1(CCCCC1)C(=O)O NEHKHPQFTUZMMX-UHFFFAOYSA-N 0.000 description 1
- WCJLOEFXANROAO-UHFFFAOYSA-N CCCCOP(O)=O Chemical compound CCCCOP(O)=O WCJLOEFXANROAO-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 1
- 102100036239 Cyclin-dependent kinase 2 Human genes 0.000 description 1
- 102100024457 Cyclin-dependent kinase 9 Human genes 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- FDKWRPBBCBCIGA-UWTATZPHSA-N D-Selenocysteine Natural products [Se]C[C@@H](N)C(O)=O FDKWRPBBCBCIGA-UWTATZPHSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 108020001019 DNA Primers Proteins 0.000 description 1
- 239000012624 DNA alkylating agent Substances 0.000 description 1
- 229940126161 DNA alkylating agent Drugs 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 1
- 101100326341 Drosophila melanogaster brun gene Proteins 0.000 description 1
- 102000001301 EGF receptor Human genes 0.000 description 1
- 108060006698 EGF receptor Proteins 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000305071 Enterobacterales Species 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000701959 Escherichia virus Lambda Species 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 102100029095 Exportin-1 Human genes 0.000 description 1
- 108010014172 Factor V Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102100037362 Fibronectin Human genes 0.000 description 1
- 102000002090 Fibronectin type III Human genes 0.000 description 1
- 108050009401 Fibronectin type III Proteins 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 101000690301 Homo sapiens Aldo-keto reductase family 1 member C4 Proteins 0.000 description 1
- 101000757160 Homo sapiens Aminopeptidase N Proteins 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000980930 Homo sapiens Cyclin-dependent kinase 9 Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101000684208 Homo sapiens Prolyl endopeptidase FAP Proteins 0.000 description 1
- 101001116548 Homo sapiens Protein CBFA2T1 Proteins 0.000 description 1
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 102100024319 Intestinal-type alkaline phosphatase Human genes 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- 229940124179 Kinesin inhibitor Drugs 0.000 description 1
- 102100027629 Kinesin-like protein KIF11 Human genes 0.000 description 1
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- 125000000998 L-alanino group Chemical group [H]N([*])[C@](C([H])([H])[H])([H])C(=O)O[H] 0.000 description 1
- 239000002211 L-ascorbic acid Substances 0.000 description 1
- 235000000069 L-ascorbic acid Nutrition 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- FFFHZYDWPBMWHY-VKHMYHEASA-N L-homocysteine Chemical compound OC(=O)[C@@H](N)CCS FFFHZYDWPBMWHY-VKHMYHEASA-N 0.000 description 1
- QEFRNWWLZKMPFJ-ZXPFJRLXSA-N L-methionine (R)-S-oxide Chemical compound C[S@@](=O)CC[C@H]([NH3+])C([O-])=O QEFRNWWLZKMPFJ-ZXPFJRLXSA-N 0.000 description 1
- QEFRNWWLZKMPFJ-UHFFFAOYSA-N L-methionine sulphoxide Natural products CS(=O)CCC(N)C(O)=O QEFRNWWLZKMPFJ-UHFFFAOYSA-N 0.000 description 1
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 description 1
- 125000000510 L-tryptophano group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C(C([H])([H])[C@@]([H])(C(O[H])=O)N([H])[*])C2=C1[H] 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 229940119336 Microtubule stabilizer Drugs 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- LKJPYSCBVHEWIU-UHFFFAOYSA-N N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methylpropanamide Chemical compound C=1C=C(C#N)C(C(F)(F)F)=CC=1NC(=O)C(O)(C)CS(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-UHFFFAOYSA-N 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- HFVPBQOSFYXKQZ-DTWKUNHWSA-N N6-[(2R)-3,4-Dihydro-2H-pyrrol-2-ylcarbonyl]-L-lysine Chemical compound [O-]C(=O)[C@@H]([NH3+])CCCCNC(=O)[C@H]1CCC=N1 HFVPBQOSFYXKQZ-DTWKUNHWSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 108091061960 Naked DNA Proteins 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 241000233805 Phoenix Species 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102100023832 Prolyl endopeptidase FAP Human genes 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 108020005067 RNA Splice Sites Proteins 0.000 description 1
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000607720 Serratia Species 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101710192266 Tegument protein VP22 Proteins 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 102000004357 Transferases Human genes 0.000 description 1
- 108090000992 Transferases Proteins 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- AKDLZRSCZBAIQF-UHFFFAOYSA-N [N].C1(C=CC(N1)=O)=O Chemical compound [N].C1(C=CC(N1)=O)=O AKDLZRSCZBAIQF-UHFFFAOYSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- SWPYNTWPIAZGLT-UHFFFAOYSA-N [amino(ethoxy)phosphanyl]oxyethane Chemical compound CCOP(N)OCC SWPYNTWPIAZGLT-UHFFFAOYSA-N 0.000 description 1
- PDKXJKWLFFZPPF-UHFFFAOYSA-N [dimethylamino-(3-oxidotriazolo[4,5-b]pyridin-3-ium-1-yl)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(C(N(C)C)=[N+](C)C)N=[N+]([O-])C2=N1 PDKXJKWLFFZPPF-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000370 acceptor Substances 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 229940064305 adrucil Drugs 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 229940098174 alkeran Drugs 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical group C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- 150000001371 alpha-amino acids Chemical class 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 229940124691 antibody therapeutics Drugs 0.000 description 1
- 238000011230 antibody-based therapy Methods 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000003080 antimitotic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003096 antiparasitic agent Substances 0.000 description 1
- 229940125687 antiparasitic agent Drugs 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 229940078010 arimidex Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 108010044540 auristatin Proteins 0.000 description 1
- CREXVNNSNOKDHW-UHFFFAOYSA-N azaniumylideneazanide Chemical group N[N] CREXVNNSNOKDHW-UHFFFAOYSA-N 0.000 description 1
- 125000004320 azepan-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WARCRYXKINZHGQ-UHFFFAOYSA-N benzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1 WARCRYXKINZHGQ-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- YILCBSAIBMIFQA-UHFFFAOYSA-N benzyl n-[(4-aminophenyl)methyl]carbamate Chemical compound C1=CC(N)=CC=C1CNC(=O)OCC1=CC=CC=C1 YILCBSAIBMIFQA-UHFFFAOYSA-N 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 229950004221 besilate Drugs 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- 230000004193 beta-amyloid degradation Effects 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 102000006635 beta-lactamase Human genes 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 229940108502 bicnu Drugs 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- 229960004395 bleomycin sulfate Drugs 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 229940111214 busulfan injection Drugs 0.000 description 1
- 229940112133 busulfex Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- KVUAALJSMIVURS-ZEDZUCNESA-L calcium folinate Chemical compound [Ca+2].C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 KVUAALJSMIVURS-ZEDZUCNESA-L 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 229940097647 casodex Drugs 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 238000003181 co-melting Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000004154 complement system Effects 0.000 description 1
- 239000000306 component Substances 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000001120 cytoprotective effect Effects 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 229940052372 daunorubicin citrate liposome Drugs 0.000 description 1
- 229940107841 daunoxome Drugs 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 229940070968 depocyt Drugs 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- BLEBFDYUDVZRFG-UHFFFAOYSA-N dichloromethane;propan-2-ol Chemical compound ClCCl.CC(C)O BLEBFDYUDVZRFG-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- GMLFPSKPTROTFV-UHFFFAOYSA-N dimethylborane Chemical compound CBC GMLFPSKPTROTFV-UHFFFAOYSA-N 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 1
- 229930188854 dolastatin Natural products 0.000 description 1
- 229960002918 doxorubicin hydrochloride Drugs 0.000 description 1
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 229940099302 efudex Drugs 0.000 description 1
- 229940073038 elspar Drugs 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethanedisulfonate group Chemical group C(CS(=O)(=O)[O-])S(=O)(=O)[O-] AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- ZPQNOPCJUCMNTL-UHFFFAOYSA-N ethyl 4-azidobutanoate Chemical compound CCOC(=O)CCCN=[N+]=[N-] ZPQNOPCJUCMNTL-UHFFFAOYSA-N 0.000 description 1
- XBPOBCXHALHJFP-UHFFFAOYSA-N ethyl 4-bromobutanoate Chemical compound CCOC(=O)CCCBr XBPOBCXHALHJFP-UHFFFAOYSA-N 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 108700002148 exportin 1 Proteins 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 1
- 239000003193 general anesthetic agent Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229940084910 gliadel Drugs 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N glutaric acid Chemical compound OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 102000054751 human RUNX1T1 Human genes 0.000 description 1
- 210000000688 human artificial chromosome Anatomy 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229940096120 hydrea Drugs 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229940090411 ifex Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 101150066555 lacZ gene Proteins 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-M lactobionate Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-M 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000000370 laser capture micro-dissection Methods 0.000 description 1
- 229940063725 leukeran Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- SCEZYJKGDJPHQO-UHFFFAOYSA-M magnesium;methanidylbenzene;chloride Chemical compound [Mg+2].[Cl-].[CH2-]C1=CC=CC=C1 SCEZYJKGDJPHQO-UHFFFAOYSA-M 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Chemical class 0.000 description 1
- 239000002184 metal Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- WRVLBJXFSHALRZ-FUVGGWJZSA-N methyl (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoate Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 WRVLBJXFSHALRZ-FUVGGWJZSA-N 0.000 description 1
- SWVMLNPDTIFDDY-FVGYRXGTSA-N methyl (2s)-2-amino-3-phenylpropanoate;hydrochloride Chemical class Cl.COC(=O)[C@@H](N)CC1=CC=CC=C1 SWVMLNPDTIFDDY-FVGYRXGTSA-N 0.000 description 1
- LLAZQXZGAVBLRX-UHFFFAOYSA-N methyl 2,5-dioxopyrrole-1-carboxylate Chemical compound COC(=O)N1C(=O)C=CC1=O LLAZQXZGAVBLRX-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- LSDPWZHWYPCBBB-UHFFFAOYSA-O methylsulfide anion Chemical compound [SH2+]C LSDPWZHWYPCBBB-UHFFFAOYSA-O 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 230000001459 mortal effect Effects 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 229940090009 myleran Drugs 0.000 description 1
- CZFNISFYDPIDNM-UHFFFAOYSA-N n,n-dimethylformamide;oxolane Chemical compound CN(C)C=O.C1CCOC1 CZFNISFYDPIDNM-UHFFFAOYSA-N 0.000 description 1
- NWKYZYGOSPOKDY-UHFFFAOYSA-N n,n-dimethylformamide;pyridine Chemical compound CN(C)C=O.C1=CC=NC=C1 NWKYZYGOSPOKDY-UHFFFAOYSA-N 0.000 description 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000003935 n-pentoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 1
- 229950006327 napsilate Drugs 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229940085033 nolvadex Drugs 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 230000017111 nuclear migration Effects 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-M octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC([O-])=O QIQXTHQIDYTFRH-UHFFFAOYSA-M 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 102000027450 oncoproteins Human genes 0.000 description 1
- 108091008819 oncoproteins Proteins 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229940115458 pantolactone Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Chemical group 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229960005141 piperazine Drugs 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 238000013492 plasmid preparation Methods 0.000 description 1
- 229940063179 platinol Drugs 0.000 description 1
- 229940098901 polifeprosan 20 Drugs 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- XTUSEBKMEQERQV-UHFFFAOYSA-N propan-2-ol;hydrate Chemical compound O.CC(C)O XTUSEBKMEQERQV-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 238000005086 pumping Methods 0.000 description 1
- 229940117820 purinethol Drugs 0.000 description 1
- OENLEHTYJXMVBG-UHFFFAOYSA-N pyridine;hydrate Chemical compound [OH-].C1=CC=[NH+]C=C1 OENLEHTYJXMVBG-UHFFFAOYSA-N 0.000 description 1
- IIHQNAXFIODVDU-UHFFFAOYSA-N pyrimidine-2-carbonitrile Chemical compound N#CC1=NC=CC=N1 IIHQNAXFIODVDU-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000003439 radiotherapeutic effect Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000022983 regulation of cell cycle Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- MVQFLINMDQDTKB-UHFFFAOYSA-N s-(6-oxohexyl) ethanethioate Chemical compound CC(=O)SCCCCCC=O MVQFLINMDQDTKB-UHFFFAOYSA-N 0.000 description 1
- MOODSJOROWROTO-UHFFFAOYSA-N salicylsulfuric acid Chemical compound OC(=O)C1=CC=CC=C1OS(O)(=O)=O MOODSJOROWROTO-UHFFFAOYSA-N 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 235000016491 selenocysteine Nutrition 0.000 description 1
- 229940055619 selenocysteine Drugs 0.000 description 1
- ZKZBPNGNEQAJSX-UHFFFAOYSA-N selenocysteine Natural products [SeH]CC(N)C(O)=O ZKZBPNGNEQAJSX-UHFFFAOYSA-N 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000010183 spectrum analysis Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 230000010473 stable expression Effects 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- DIORMHZUUKOISG-UHFFFAOYSA-N sulfoformic acid Chemical compound OC(=O)S(O)(=O)=O DIORMHZUUKOISG-UHFFFAOYSA-N 0.000 description 1
- 229940071103 sulfosalicylate Drugs 0.000 description 1
- DHCDFWKWKRSZHF-UHFFFAOYSA-N sulfurothioic S-acid Chemical compound OS(O)(=O)=S DHCDFWKWKRSZHF-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960003454 tamoxifen citrate Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical compound O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- GINSRDSEEGBTJO-UHFFFAOYSA-N thietane 1-oxide Chemical compound O=S1CCC1 GINSRDSEEGBTJO-UHFFFAOYSA-N 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 229950002376 tirapazamine Drugs 0.000 description 1
- 238000012090 tissue culture technique Methods 0.000 description 1
- 230000025366 tissue development Effects 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 229960002190 topotecan hydrochloride Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000006276 transfer reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 230000004862 vasculogenesis Effects 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 210000002845 virion Anatomy 0.000 description 1
- 239000000277 virosome Substances 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229940102001 zinc bromide Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0205—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-(X)3-C(=0)-, e.g. statine or derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
- A61K47/6817—Toxins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K19/00—Hybrid peptides, i.e. peptides covalently bound to nucleic acids, or non-covalently bound protein-protein complexes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/02—Linear peptides containing at least one abnormal peptide link
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Abstract
Un compuesto o estereoisómero de este que tiene la estructura de Fórmula (I)**Fórmula** donde:**Fórmula** R1 es o R2 es metilo, etilo, isopropilo o sec-butilo;**Fórmula** R3 es o ; R4 es -OH, alcoxi C1-C6, -N(R14)2, -R16, -NR12(CH2)mN(R14)2, -NR12(CH2)mR16, -NHS(O)2R11, o ;**Fórmula** R5 es alquilo C1-C6, -C(=O)R11, -(CH2)mOH, -C(=O)(CH2)mOH, -C(=O)((CH2)mO)nR12, -((CH2)mO)nR12 o alquilo C1-C6 que se sustituye opcionalmente con -CN, -C(=O)NH2 o de 1 a 5 hidroxilos; R8 es H; cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos; cada R12 se selecciona independientemente a partir de H y alquilo C1-C6; R13 es tetrazolilo, imidazolilo sustituido con fenilo, oxadiazolilo sustituido con fenilo, pirazolilo, pirimidinilo**Fórmula** o o -CH2S(=O)2NH2; cada R14 se selecciona independientemente a partir de H y alquilo C1-C6; R16 es un heterocicloalquilo de 4-8 miembros sin sustituir conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O; R19 es H o alquilo C1-C6; cada m se selecciona independientemente entre 1, 2, 3, 4, 5, 6, 7, 8, 9 y 10, y cada n se selecciona independientemente entre 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 y 18, o una sal farmacéuticamente aceptable de este.
Description
DESCRIPCIÓN
Péptidos citotóxicos y conjugados de estos
Campo de la invención
La invención proporciona compuestos que son péptidos citotóxicos antimitóticos, y son útiles para tratar trastornos proliferativos celulares. La invención también incluye conjugados que comprenden dichos péptidos citotóxicos conectados a un resto de unión a antígeno y a composiciones farmacéuticas que contienen estos conjugados. También se incluyen métodos para utilizar estos compuestos y conjugados para tratar trastornos de proliferación celular, incluidos cánceres.
Antecedentes
El uso de conjugados anticuerpo-fármaco (ADC) para el suministro dirigido de inhibidores de la proliferación celular y/o agentes citotóxicos para células específicas ha sido el foco de una investigación considerable. Antibody-Drug Conjúgate, Methods in Molecular Biology, Vol. 1045, Editor L. Ducry, Humana Press (2013). Los ADC incluyen un anticuerpo seleccionado por su capacidad para unirse a una célula utilizada como diana para la intervención terapéutica, conectado a un fármaco seleccionado por su actividad citostática o citotóxica. La unión del anticuerpo a la célula utilizada como diana suministra de este modo el fármaco al sitio en el que se requiere el efecto terapéutico.
Muchos anticuerpos que reconocen y se unen selectivamente a las células utilizadas como diana, como las células cancerosas, se han divulgado para su uso en ADC, y también se han descrito muchos métodos para unir compuestos de carga (fármaco) tales como citotoxinas a los anticuerpos. Sin embargo, a pesar del trabajo exhaustivo sobre ADC, solamente unas pocas clases de inhibidores de la proliferación celular se han utilizado extensamente como cargas de ADC. Aunque el primer ADC aprobado para el uso en seres humanos en los EE. UU. se lanzó en el 2000 (y más tarde se retiró del mercado), una década más tarde, solamente unas pocas clases químicas de compuestos farmacológicos (maitansinoides, auristatinas, calicheamicinas y duocarmicinas) han alcanzado los ensayos clínicos como cargas para ADC. Antibody-Drug Conjugates: the Next Generation of Moving Parts, A. Lash, Start-Up, Dic. de 2011, 1-6. Dado el ampliamente reconocido valor de los ADC como productos terapéuticos, particularmente para tratar el cáncer, continúa habiendo por tanto una necesidad de péptidos citotóxicos con propiedades mejoradas para su uso como cargas en los ADC.
Compendio de la invención
La invención proporcionada en la presente incluye péptidos citotóxicos y métodos para utilizar dichos péptidos citotóxicos como el componente farmacológico de un conjugado anticuerpo-fármaco (ADC). La presente invención incluye péptidos citotóxicos novedosos y el uso de dichos péptidos citotóxicos novedosos como cargas para ADC. La invención incluye además métodos e intermedios útiles para incorporar dichos péptidos citotóxicos novedosos en ADC, y métodos para utilizar los compuestos y conjugados novedosos para tratar trastornos de proliferación celular. Dichos péptidos citotóxicos son agentes antimitóticos que inhiben la división celular bloqueando la polimerización de la tubulina y, de este modo, bloqueando la migración nuclear y la división celular y nuclear.
En un aspecto de la invención hay péptidos citotóxicos, o estereoisómeros de estos, y sales farmacéuticamente aceptables de estos, que tienen la estructura de Fórmula (I)

R2 es metilo, etilo, isopropilo o sec-butilo;
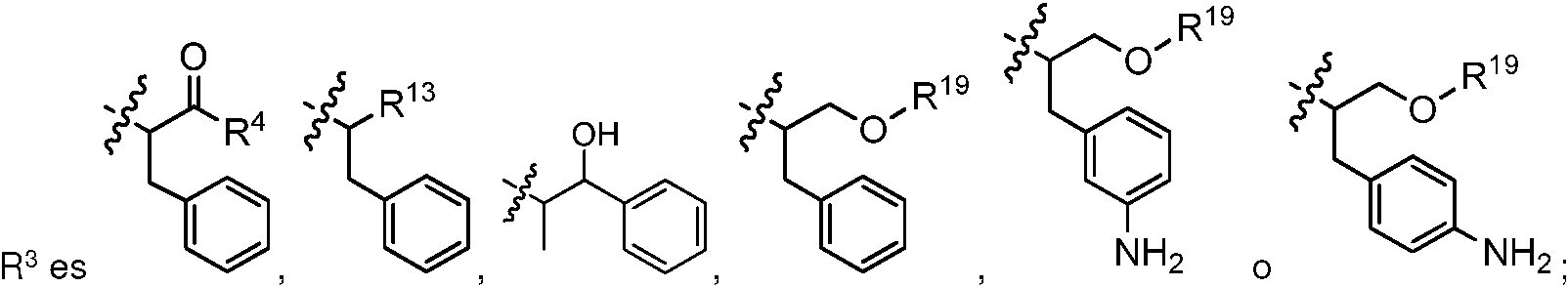
R4 es -OH, alcoxi C1-C6 , -N(R14)2, R 16 -NR12(CH2)mN(R14)2 , -NR12(CH2)mR16, -NHS(O)2 Rn,

R5 es alquilo Ci-C6, -C(=0)R11, -(CH2)mOH, -C(=0)(CH2)mOH, -C(=0)((CH2)m0 )nR12, -((CH2)mO)nR12 o alquilo Ci-Ce que se sustituye opcionalmente con -CN, -C(=0)NH2 o de 1 a 5 hidroxilos,
R8 es H;
cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente entre H y alquilo C1-C6 ;
O
<, 11 -1 -P -O H R13 es tetrazolilo, ¡midazolilo sustituido con fenilo, oxadiazolilo sustituido con fenilo, pirazolilo, pirimidinilo, f H ,

cada R14 se selecciona independientemente entre H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros sin sustituir conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
R19 es H o alquilo C1-C6 ;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 y 18.
En la presente se divulgan péptidos citotóxicos, o un estereoisómero de estos, y sales farmacéuticamente aceptables de estos, que tienen la estructura de Fórmula (I)

donde:
R1 es un heterocicloalquilo de 6 miembros conectado en C que contiene 1-2 heteroátomos de N y un puente alquileno C1-C2 o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros conectado en C que contiene 1 -2 heteroátomos de N, donde cada uno está sin sustituir o cada uno está sustituido con un R7 y de 0 a 3 sustituyentes seleccionados independientemente entre R5 y R6, o cada uno está sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
R2 es alquilo C1-C6 ;


R5 es alquilo Ci-C6, -C (=0)R 11, -(CH2)mOH, -C(=0)(CH2)mOH, -C(=0)((CH2)m0)nR12, -((CH2)mO)nR12, o alquilo Ci-Ce que está sustituido opcionalmente con -CN, -C(=O)NH2 o de 1 a 5 hidroxilos;
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2 , -R16 y -N R 12C(=O)R11;
R7 es LR9;
R8 es H o LR9;
cada L se selecciona independientemente entre -L1L2L3L4L5L6-, -L6L5L4L3L2 L1-, -L1L2L3L4L5-, -L5L4L3L2L1-,-L1L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la presente;

cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente entre H y alquilo C1-C6 ;
O
«, 11 - f -P -O H R13 es tetrazolilo, imidazolilo sustituido con fenilo, oxadiazolilo sustituido con fenilo, pirazolilo, pirimidinilo, ? H ,

cada R14 se selecciona independientemente entre H y alquilo C1-C6 ;
R15 es 2-piridilo o 4-piridilo;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O, que está sin sustituir o sustituido con -LR9;
cada
X3 es
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10; y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
En la presente se divulgan también compuestos de fórmula (I), donde,
R1 es un heterocicloalquilo de 6 miembros conectado en C que contiene 1-2 heteroátomos de N y un puente alquileno C1-C2 o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros conectado en C que contiene 1 -2 heteroátomos de N, donde cada uno está sin sustituir o cada uno está sustituido con un R7 y de 0 a 3 sustituyentes seleccionados independientemente entre R5 y R6, o cada uno está sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
R2 es alquilo C1-C6 ;


cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente entre H y alquilo C1-C6 ;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R15 es 2-piridilo o 4-piridilo;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O, que está sin sustituir o sustituido con -LR9;

X 3 e s
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10; y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
En determinadas realizaciones de este aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I), R1 es un heterocicloalquilo de 6 miembros conectado en C que contiene 1-2 heteroátomos de N y un puente alquileno C1-C2 o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros conectado en C que contiene 1 -2 heteroátomos de N, donde cada uno está sin sustituir o cada uno está sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
R2 es alquilo C1-C6 ;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos, -C(=0)R11, -(CH2)mOH, -C(=0)(CH2)mOH, -C(=0)((CH2)m0)nR12, o -((CH2)mO)nR12;
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2, -R16 y -N R 12C(=O)R11;
R8 es H;
cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros sin sustituir conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
En determinadas realizaciones de este aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I), son péptidos citotóxicos que tienen la estructura de Fórmula (Ia):

En otras realizaciones del aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I) o Fórmula (Ia), son péptidos citotóxicos que tienen la estructura de Fórmula (Ib):

En determinadas realizaciones del aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I), son péptidos citotóxicos que tienen la estructura de Fórmula (Ic):

En otras realizaciones del aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I) o Fórmula (Ic), son péptidos citotóxicos que tienen la estructura de Fórmula (Id):

En determinadas realizaciones del aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I), son péptidos citotóxicos que tienen la estructura de Fórmula (Ie):

En otras realizaciones del aspecto de los péptidos citotóxicos que tienen la estructura de Fórmula (I) o Fórmula (Ie), son péptidos citotóxicos que tienen la estructura de Fórmula (If):

La presente invención proporciona inmunoconjugados, también denominados en la presente ADC, que contienen péptidos citotóxicos conectados a un resto de unión a antígeno tal como un anticuerpo o fragmento de anticuerpo. Estos conjugados que comprenden péptidos citotóxicos son útiles para tratar trastornos de proliferación celular, particularmente cuando el péptido citotóxico está conectado con un anticuerpo que reconoce células cancerosas y, por tanto, promueve el suministro de los péptidos citotóxicos a una célula utilizada como diana para el ataque. Los inmunoconjugados son especialmente útiles para tratar determinados tipos de cáncer tal como se detalla adicionalmente en la presente. Los datos proporcionados en la presente demuestran que estos inmunoconjugados son inhibidores eficaces de la proliferación celular; sin ceñirse a ninguna teoría, se cree que su actividad se debe a la inhibición de la polimerización de la tubulina en las células.
En un aspecto de los inmunoconjugados de la invención incluyen inmunoconjugados de Fórmula (II):

Ab representa un resto de unión a antígeno;
L se selecciona entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -LsL4L3L2L1-,-L1L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la presente;
y es un número entero de 1 a 16;
R101 es un radical divalente heterocicloalquílico de 6 miembros que contiene 1-2 heteroátomos de N y un puente
y r v alquileno C1-C2 , donde el radical divalente heterocicloalquílico de 6 miembros está conectado en C al grupo O y está conectado en N a L1, o está conectado en C a L1, y el radical divalente heterocicloalquílico de 6 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
o R101 es un radical divalente heterocicloalquílico bicíclico fusionado de 5-8 miembros que contiene 1-2 heteroátomos de N, donde el radical divalente heterocicloalquílico bicíclico fusionado de 5-8 miembros está conectado en C al grupo
y r v
O y está conectado en N a Li, o está conectado en C a L1, y el radical divalente heterocicloalquílico bicíclico fusionado de 5-8 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
R2 es alquilo C1-C6 ;

R5 es alquilo Ci-C6, -C(=0)R11, -(CH2)mOH, -C(=0)(CH2)m0H, -C(=0)((CH2)m0 )nR12, -((CH2)mO)nR12, o alquilo Ci-C6 que está sustituido opcionalmente con -CN, -C(=0)NH2 o de 1 a 5 hidroxilos,
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2 , -R16 y -N R 12C(=O)R11;
R11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R13 es tetrazolilo, imidazolilo sustituido con fenilo, oxadiazolilo sustituido con fenilo, pirazolilo, pirimidinilo,

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
R19 es H o alquilo C1-C6 ;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10,
y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
En un aspecto de los inmunoconjugados de la invención incluyen inmunoconjugados de Fórmula (II):

Ab representa un resto de unión a antígeno;
L se selecciona entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2L1-,-L1L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la presente;
es un número entero de 1 a 16;

R2 es metilo, etilo, isopropilo o sec-butilo;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos, -C(=0)R11, -(CH2)mOH, -C(=0)(CH2)mOH, -C(=0)((CH2)m0)nR12, o -((CH2)mO)nR12;
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2 , -R16 y -N R 12C(=O)R11;
R11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H alquilo C1-C6 ;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
También se divul an en la presente inmunoconu ados que tienen la estructura de Fórmula (111):

donde:
Ab representa un resto de unión a antígeno;
L se selecciona entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2L1-,-L1L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la presente;
y es un número entero de 1 a 16;
R1 es un heterocicloalquilo de 6 miembros que contiene 1 -2 heteroátomos de N y un puente alquileno C1-C2 , donde el heterocicloalquilo de 6 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros que contiene 1-2 heteroátomos de N, donde el heterocicloalquilo bicíclico fusionado de 5-8 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos, -C(=0)R11, -(CH2)mOH, -C(=0)(CH2)mOH, -C(=0)((CH2)m0)nR12, o -((CH2)mO)nR12;
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2 , -R16 y -N R 12C(=O)R11;
R11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;
cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
R17 es un enlace, -NH NHS=O2-,  o
o

cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10,
y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
También se divulgan en la presente inmunoconjugados que tienen la estructura de Fórmula (III):

donde:
Ab representa un resto de unión a antígeno;
L se selecciona entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2L1-,-L1L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la presente;
y es un número entero de 1 a 16;
R1 es un heterocicloalquilo de 6 miembros que contiene 1 -2 heteroátomos de N y un puente alquileno C1-C2 , donde el heterocicloalquilo de 6 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros que contiene 1-2 heteroátomos de N, donde el heterocicloalquilo bicíclico fusionado de 5-8 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
R2 es alquilo C1-C6 ;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos C(=O)R11, -(CH2)mOH, C(=O)(CH2)mOH, -C(=O)((CH2)mO)nR12, o -((CH2)mO)nR12;
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2 , -R16 y -N R 12C(=O)R11;
R11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;
cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;

cada m se selecciona independientemente entre 1 2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
La invención proporciona métodos para preparar dichos ADC utilizando péptidos citotóxicos de Fórmula (I) como la carga (fármaco) a suministrar. En dichos péptidos citotóxicos, el extremo N o el extremo C del péptido citotóxico se ha modificado para que tenga un grupo funcional reactivo, y opcionalmente uno o más componentes conectores, para facilitar la conexión del péptido citotóxico ya sea directa o indirectamente al anticuerpo o fragmento de unión a antígeno, por ejemplo, los aspectos descritos anteriormente de los péptidos citotóxicos de Fórmula (I). Además, la invención proporciona métodos para utilizar estos ADC con el fin de tratar trastornos de proliferación celular.
En otro aspecto, la invención proporciona composiciones farmacéuticas que comprenden un inmunoconjugado de Fórmula (II) o subfórmula de esta, mezclado con al menos un portador o excipiente farmacéuticamente aceptable, opcionalmente mezclado con dos o más portadores o excipientes farmacéuticamente aceptables, y métodos para utilizar estas composiciones con el fin de tratar trastornos de proliferación celular.
En otro aspecto, la invención proporciona un método para tratar una afección caracterizada por una proliferación celular excesiva o indeseada, que comprende administrar a un sujeto que necesite dicho tratamiento una cantidad eficaz de un inmunoconjugado de Fórmula (II). El sujeto para el tratamiento puede ser un mamífero y es preferentemente un ser humano. Las afecciones que se pueden tratar con los inmunoconjugados y métodos descritos en la presente incluyen varias formas de cáncer, tales como tumores gástricos, mieloides, de colon, nasofaríngeos, esofágicos y de próstata, glioma, neuroblastoma, cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer colorrectal, cáncer de tiroides, leucemia (por ejemplo, leucemia mielógena, leucemia linfocítica, leucemia mielógena aguda (LMA), leucemia mieloide crónica (LMC), leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de linaje T o LLA-T, leucemia linfocítica crónica (LLC), síndrome mielodisplásico (SMD), tricoleucemia), linfoma (linfoma de Hodgkin (LH), linfoma no Hodgkin (LNH)), mieloma múltiple, cáncer de vejiga, renal, gástrico (por ejemplo, tumores del estroma gastrointestinal (TEGI)), de hígado, melanoma y pancreático, y sarcoma. Otros trastornos de proliferación celular que se pueden tratar con estos métodos y composiciones incluyen retinopatía diabética, fibrosis hepática y pulmonar, síndrome de Sjogren y lupus eritematoso.
La invención incluye composiciones de las Fórmulas (I)-(II) y las subfórmulas de estas tal como se describen en la presente, y todos los estereoisómeros (incluidos diastereoisómeros y enantiómeros), tautómeros y versiones isotópicamente enriquecidas de estos (que incluyen sustituciones con deuterio), así como sales farmacéuticamente aceptables de estos compuestos. La presente invención también comprende polimorfos de Fórmula (I) (o subfórmulas de estos) y sales, particularmente sales farmacéuticamente aceptables, de estos.
Breve descripción de los dibujos
Figura 1. Ensayos de proliferación celular in vitro de ADC anti-Her2 con mutación Cys (A) y etiqueta A1/ybbR (B) con el clon 40 de MDA-MB-231, ADC anti-Her2 con mutación Cys (C) y etiqueta A1/ybbR (D) con el clon 16 de MDA-MB-231, ADC anti-Her2 con mutación Cys (E) y etiqueta A1/ybbR (F) con HCC1954, y ADC anti-Her2 con mutación Cys (G) y etiqueta A1/ybbR (H) con células JimT-1.
Figura 2. Ensayos de proliferación celular in vitro de ADC de anticuerpo 20507-HC-E152C-10 (A), anticuerpo 20507-HC-S375C-47 (B), anticuerpo 20507-HC-ins388-A1-20 (C) y anticuerpo 20507-HC-ins388-A1-49-22 (D). Figura 3. Estudios farmacocinéticos de anti-Her2-LC-S159C-10 (A), anti-Her2-LC-S159C-47 (B), anti-Her2-LC-S159C-77 (C), anti-Her2-LC-S159C-80 (D), anti-Her2-LC-S159C-79 (E), anti-Her2-LC-S159C-78 (F), anti-Her2-LC-S159C-14 (G), anti-Her2-HC-E152C-S375C-10 (H) y anti-Her2-10 (I) en ratones que no han recibido tratamiento.
Figura 4. Estudios farmacocinéticos de ADC de anticuerpo 20507-HC-E152C-10 (A), anticuerpo 20507-LC-K107C-47 (B), anticuerpo 20507-HC-ins388-A1-20 (C), anticuerpo 20507-HC-E152C-S375C-10 (D), anticuerpo 20507-10 (E), anti-Her2-HC-ins388-ybbR-20 (G), anticuerpo 20507-HC-ins388-A1-49-22 (H), anti-Her2-HC-ins388-A1-49-22 (I), y anti-Her2-HC-ins388-ybbR-(i-11)-75 (J) en ratones que no han recibido tratamiento con una dosis de 1 mg/kg. El estudio farmacocinético del anticuerpo 20507-HC-ins388-A1-20 (F) se llevó a cabo en ratones que portaban tumores con una dosis de 10 mg/kg.
Figura 5. Estudios de estabilidad in vitro de los ADC. Cronología de la retención de carga de anti-Her2-LC-S159C-10 incubado en tampones con diferente pH a 37 °C (A), Cronología de la hidrólisis del anillo de succinimida de anti-Her2-LC-S159C-10 incubado en tampones con diferente pH a 37 °C (B), Cronología de la retención de carga de anti-Her2-LC-S159C-47 incubada en tampones con diferente pH a 37 °C (C), Cronología de la hidrólisis del anillo de succinimida de anti-Her2-LC-S159C-47 incubado en tampones con diferente pH a 37 °C (D), tasa de retención de carga de anti-Her2-LC-S159C-10, anti-Her2-LC-S159C-77, anti-Her2-LC-S159C-80, anti-Her2-LC-S159C-79, anti-Her2-LC-S159C-78 y anti-Her2-LC-S159C-14 incubados en tampones a pH 8,5 a 37°C durante 24 horas (E), Extensión de la hidrólisis del anillo de succinimida de anti-Her2-LC-S159C-10, anti-Her2-LC-S159C-77, anti-Her2-LC-S159C-80, anti-Her2-LC-S159C-79, anti-Her2-LC-S159C-78 y anti-Her2-LC-S159C-14 incubados en tampones a pH 8,5 a 37 °C durante 24 horas (F). Figura 6. Estudio de eficacia in vivo de ADC de anticuerpo 20507 en el modelo tumora1H526. (A) ADC de anticuerpo 20507-HC-E152C-10 y anticuerpo 20507-HC-E152C-MMAF. (B) ADC de anticuerpo 20507-HC-E152C-10, anticuerpo 20507-HC-ins388-A1-20 y anticuerpo 20507-LC-S159C-MMAF. (c ) Anticuerpo 20507-HC-E152C-S375C-10 y anticuerpo 20507-10. (D) a Dc de anticuerpo 20507-HC-E152C-10, anticuerpo 20507-HC-ins388-A1 -49-22 y anti-Her2-HC-ins388-A1-49-22.
Descripción detallada
Las siguientes definiciones se aplican a menos que se proporcionen otras expresamente.
El término «aminoácido» se refiere a aminoácidos canónicos, sintéticos y artificiales, así como también a análogos de aminoácidos y miméticos de aminoácidos que funcionan de manera similar a los aminoácidos canónicos. Los aminoácidos canónicos son aminoácidos proteinógenos codificados por el código genético e incluyen alanina, arginina, asparagina, ácido aspártico, cisteína, glutamina, ácido glutámico, glicina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina, así como selenocisteína, pirrolisina y pirrolinacarboxilisina. Los análogos de aminoácidos se refieren a compuestos que tienen la misma estructura química básica que un aminoácido canónico, es decir, un carbono a que está unido a un hidrógeno, un grupo carboxilo, un grupo amino y un grupo R, p. ej., homoserina, norleucina, sulfóxido de metionina, metionina metil sulfonio. Tales análogos tienen grupos R modificados (p. ej., norleucina) o esqueletos peptídicos modificados, pero conservan la misma estructura química básica que un aminoácido canónico.
La expresión «resto de unión a antígeno», tal como se utiliza en la presente, se refiere a un resto capaz de unirse específicamente a un antígeno, e incluye anticuerpos y fragmentos de anticuerpos.
El término «anticuerpo», tal como se utiliza en la presente se refiere a un polipéptido de la familia de las inmunoglobulinas que es capaz de unirse a un antígeno correspondiente de forma no covalente, reversible y de manera específica. Por ejemplo, un anticuerpo IgG presente en la naturaleza es un tetrámero que comprende al menos dos cadenas pesadas (H) y dos cadenas ligeras (L) interconectadas por enlaces disulfuro. Cada cadena pesada comprende una región variable de cadena pesada (abreviada en la presente como Vh) y una región constante de cadena pesada. La región constante de cadena pesada está constituida por tres dominios: CH1, CH2 y CH3. Cada cadena ligera comprende una región variable de cadena ligera (abreviada en la presente como Vl) y una región
constante de cadena ligera. La región constante de cadena ligera comprende un dominio, Cl. Las regiones Vh y Vl se pueden subdividir además en regiones de hipervariabilidad, denominadas regiones determinantes de la complementariedad (CDR), intercaladas con regiones que están más conservadas, denominadas regiones de marco (FR). Cada Vh y Vl está compuesta por tres CDR y cuatro FR dispuestas desde el extremo amínico hasta el extremo carboxílico en el siguiente orden: FR1, CDR1, FR2, CDR2, FR3, CDR3 y FR4. Las regiones variables de las cadenas pesada y ligera contienen un dominio de unión que interacciona con un antígeno. Las regiones constantes de los anticuerpos pueden mediar la unión de la inmunoglobulina a factores o tejidos hospedadores, que incluyen varias células del sistema inmunitario (p. ej., células efectoras) y el primer componente (Clq) del sistema del complemento clásico.
El término «anticuerpo» incluye anticuerpos monoclonales, anticuerpos humanos, anticuerpos humanizados, anticuerpos de camélido, anticuerpos quiméricos y anticuerpos anti-idiotípicos (anti-Id) (que incluyen, por ejemplo, de anticuerpos anti-Id a anticuerpos). Los anticuerpos pueden ser de cualquier isotipo/clase (por ejemplo, IgG, IgE, IgM, IgD, IgA e IgY), o subclase (por ejemplo, IgG1, IgG2, IgG3, IgG4, IgA1 e IgA2).
Tanto las cadenas ligeras como las pesadas se dividen en regiones de homología estructural y funcional. Los términos «constante» y «variable» se utilizan funcionalmente. A este respecto, se apreciará que los dominios variables de tanto las porciones de cadena ligera (Vl) como pesada (Vh) determinan el reconocimiento y la especificidad antigénicos. En cambio, los dominios constantes de la cadena ligera (Cl) y la cadena pesada (CH1, CH2 o CH3) confieren propiedades biológicas importantes tales como secreción, movilidad a través de la placenta, unión a receptores de Fc y unión al complemento. Por convención, la numeración de los dominios de región constante aumenta a medida que pasan a ser más distales del sitio de unión a antígeno o extremo amino del anticuerpo. El extremo N es una región variable y en el extremo C hay una región constante; los dominios CH3 y Cl comprenden en realidad los dominios carboxi terminales de la cadena pesada y ligera, respectivamente.
La expresión «fragmento de unión a antígeno», tal como se utiliza en la presente, se refiere a una o más partes de un anticuerpo que retienen la capacidad para interactuar de forma específica con (por ejemplo, mediante unión, impedimento estérico, estabilización/desestabilización, distribución espacial) un epítopo de un antígeno. Los ejemplos de fragmentos de unión incluyen Fv monocatenarios (scFv), Fv conectados mediante disulfuro (sdFv), fragmentos Fab, fragmentos F(ab’), un fragmento monovalente constituido por los dominios VL, VH, CL y CH1; un fragmento F(ab)2, un fragmento bivalente que comprende dos fragmentos Fab unidos por un puente disulfuro en la región bisagra; un fragmento Fd constituido por los dominios VH y CH1; un fragmento Fv constituido por los dominios VL y VH de un único brazo de un anticuerpo; un fragmento dAb (Ward et al., Nature 341:544-546, 1989), que está constituido por un dominio VH; y una región determinante de la complementariedad (CDR) aislada, u otros fragmentos de unión a epítopo de un anticuerpo.
Además, aunque los dos dominios del fragmento Fv, VL y VH estén codificados por genes separados, se pueden unir, utilizando métodos recombinantes, a través de un conector sintético que permite que sean preparados como una única cadena proteica en la que las regiones VL y VH se aparean para formar moléculas monovalentes (conocidas como Fv monocatenario («scFv»); remítase, p. ej., a Bird et al., Science 242:423-426, 1988; y Huston et al., Proc. Natl. Acad. Sci. 85:5879-5883, 1988). También se pretende que dichos anticuerpos monocatenarios estén englobados en la expresión «fragmento de unión a antígeno». Estos fragmentos de unión a antígeno se obtienen utilizando técnicas convencionales conocidas para los expertos en la técnica y los fragmentos se criban para determinar su utilidad de la misma manera que para los anticuerpos intactos.
Los fragmentos de unión a antígeno también se pueden incorporar en anticuerpos de dominio único, maxicuerpos, minicuerpos, nanocuerpos, intracuerpos, diacuerpos, triacuerpos, tetracuerpos, v-NAR y bis-scFv (remítase, por ejemplo, a Hollinger y Hudson, Nature Biotechnology 23:1126-1136, 2005). Se pueden injertar fragmentos de unión a antígeno en matrices basados en polipéptidos tales como fibronectina tipo III (Fn3) (remítase a la Pat. de EE. UU. N.° 6 703 199, que describe monocuerpos de polipéptidos de fibronectina).
Se pueden incorporar fragmentos de unión a antígeno en moléculas monocatenarias que comprenden un par de segmentos Fv tándem (VH-CH1-VH-CH1) que, junto con polipéptidos de cadena ligera complementarios, forman un par de regiones de unión a antígeno (Zapata et al., Protein Eng. 8:1057-1062, 1995; y la Pat. de EE. UU. N.° 5641 870).
La expresión «anticuerpo monoclonal» o «composición de anticuerpos monoclonales», tal como se utiliza en la presente, se refiere a polipéptidos, que incluyen anticuerpos y fragmentos de unión a antígeno que tienen una secuencia de aminoácidos sustancialmente idéntica o proceden de la misma fuente genética. Esta expresión también incluye preparados de moléculas de anticuerpo de composición monomolecular. Una composición de anticuerpo monoclonal presenta una única especificidad de unión y afinidad por un epítopo particular.
La expresión «anticuerpo humano», tal como se utiliza en la presente, incluye anticuerpos con regiones variables en las que tanto las regiones de marco como las CDR proceden de secuencias de origen humano. Además, si el anticuerpo contiene una región constante, la región constante también procede de dichas secuencias humanas, p. ej.,
secuencias de la línea germinal humana, o versiones mutadas de secuencias de la línea germinal humana o un anticuerpo que contiene secuencias de marco consenso derivadas del análisis de secuencias de marco humanas, por ejemplo, tal como se describe en Knappik, et al. J. Mol. Biol. 296:57-86, 2000).
Los anticuerpos humanos incluyen residuos de aminoácidos no codificados por secuencias humanas (p. ej., mutaciones introducidas mediante mutagénesis aleatoria o con especificidad por un sitio in vitro, o mediante mutación somática in vivo, o una sustituición para promover la estabilidad o la producción).
El término anticuerpo «humanizado», tal como se utiliza en la presente, se refiere a un anticuerpo que conserva la reactividad de un anticuerpo no humano a la vez que es menos inmunogénico en seres humanos. Esto se puede conseguir, por ejemplo, conservando las regiones CDR no humanas y reemplazando las partes restantes del anticuerpo con sus homólogos humanos. Remítase, p. ej., a Morrison et al., Proc. Natl. Acad. Sci. USA, 81:6851-6855 (1984); Morrison y Oi, Adv. Immunol., 44:65-92 (1988); Verhoeyen et al., Science, 239:1534-1536 (1988); Padlan, Molec. Immun., 28:489-498 (1991); Padlan, Molec. Immun., 31 (3): 169-217 (1994).
La expresión «se une específicamente» o «se une selectivamente», cuando se utiliza en el contexto de describir la interacción entre un antígeno (p. ej., una proteína o un glicano) y un anticuerpo, fragmento de anticuerpo o agente de unión derivado de anticuerpo, se refiere a una reacción de unión que determina la presencia del antígeno en una población heterogénea de proteínas y otros agentes biológicos, p. ej., en una muestra biológica, p. ej., una muestra de sangre, suero, plasma o tejido. De este modo, en determinadas condiciones de inmunoensayo designadas, los anticuerpos o agentes de unión con una especificidad de unión particular se unen a un antígeno particular en una cantidad que es al menos dos veces la de referencia y no se unen sustancialmente en ninguna cantidad significativa a otros antígenos presentes en la muestra. En una realización, en las condiciones de inmunoensayo designadas, el anticuerpo o agentes de unión con una especificidad de unión particular se unen a un antígeno particular en una cantidad que es al menos diez (10) veces la de referencia y no se unen sustancialmente en ninguna cantidad significativa a otros antígenos presentes en la muestra. La unión específica a un anticuerpo o agente de unión en tales condiciones puede requerir que el anticuerpo o agente hayan sido seleccionados por su especificidad por una proteína particular. Según corresponda o se desee, esta selección se puede conseguir sustrayendo anticuerpos que reaccionen de forma cruzada con moléculas de otras especies (p. ej., de ratón o rata) u otros subtipos. Como alternativa, en algunas realizaciones, se seleccionan anticuerpos o fragmentos de anticuerpo que reaccionan de forma cruzada con determinadas moléculas deseadas.
Se pueden utilizar varios formatos de inmunoensayo para seleccionar anticuerpos específicamente inmunorreactivos con una proteína particular. Por ejemplo, se utilizan inmunoensayos ELISA en fase sólida de forma rutinaria para seleccionar anticuerpos específicamente inmunorreactivos con una proteína (remítase, por ejemplo, a Harlow & Lane, Using Antibodies, A Laboratory Manual (1998), para consultar una descripción de formatos y condiciones de inmunoensayo que se pueden utilizar para determinar una inmunorreactividad específica). Habitualmente, una reacción de unión específica o selectiva producirá una señal de al menos el doble por encima de la señal de referencia y, más habitualmente, al menos de 10 a 100 veces por encima de la referencia.
El término «afinidad», tal como se utiliza en la presente, se refiere a la fuerza de interacción entre el anticuerpo y el antígeno en sitios antigénicos individuales. Dentro de cada sitio antigénico, la región variable del «brazo» del anticuerpo interacciona a través de fuerzas no covalentes débiles con el antígeno en numerosos sitios; cuantas más interacciones, más fuerte es la afinidad.
La expresión «anticuerpo aislado» se refiere a un anticuerpo que está sustancialmente exento de otros anticuerpos que tienen diferentes especificidades antigénicas. Un anticuerpo aislado que se une específicamente a un antígeno puede, sin embargo, tener reactividad cruzada respecto a otros antígenos. Además, un anticuerpo aislado puede estar sustancialmente exento de otro material celular y/o productos químicos.
Los términos «polipéptido» y «proteína» se utilizan indistintamente en la presente para referirse a un polímero de residuos de aminoácidos. Los términos se aplican a polímeros de aminoácidos canónicos, así como a polímeros de aminoácidos no canónicos. A menos que se indique lo contrario, una secuencia polipeptídica particular también engloba de forma implícita variantes modificadas de esta.
El término «inmunoconjugado» o la expresión «conjugado de anticuerpo-fármaco», tal como se utiliza en la presente, se refiere a la conexión de un resto de unión a antígeno tal como un anticuerpo o un fragmento de unión a antígeno de este con un péptido citotóxico de Fórmula (I). La conexión puede ser enlaces covalentes, o interacciones no covalentes, y puede incluir quelación. Se pueden emplear varios conectores, conocidos en la técnica, para formar el inmunoconjugado.
El término «citotoxina» o «agente citotóxico», tal como se utiliza en la presente, se refiere a cualquier agente que es perjudicial para el crecimiento y la proliferación de las células y puede actuar para reducir, inhibir o destruir una célula o neoplasia maligna.
La expresión «agente anticanceroso», tal como se utiliza en la presente, se refiere a cualquier agente que se puede utilizar para tratar un trastorno proliferativo celular tal como el cáncer, que incluye agentes citotóxicos, agentes quimioterapéuticos, radioterapia y agentes radioterapéuticos, agentes anticancerosos dirigidos y agentes inmunoterapéuticos.
La expresión «resto farmacológico» o «carga», tal como se utiliza en la presente, se refiere a un resto químico que se conjuga o se puede conjugar con un anticuerpo o fragmento de unión a antígeno para formar un inmunoconjugado, y puede incluir cualquier resto que sea útil unir al anticuerpo o fragmento de unión a antígeno. Por ejemplo, «resto farmacológico» o «carga» incluye los péptidos citotóxicos descritos en la presente. Los inmunoconjugados de la invención comprenden uno o más péptidos citotóxicos descritos en la presente como una carga, pero también pueden incluir una o más cargas diferentes. Otras cargas incluyen, por ejemplo, una carga o resto farmacológico puede ser un agente anticanceroso, un agente antiinflamatorio, un agente antifúngico, un agente antibacteriano, un agente antiparasitario, un agente antiviral o un agente anestésico. En determinadas realizaciones, se selecciona un resto farmacológico entre un inhibidor de Eg5, un inhibidor de HSP90, un inhibidor de IAP, un inhibidor de mTor, un estabilizador de microtúbulos, un desestabilizador de microtúbulos, una auristatina, una dolastatina, un maitansitoide, un MetAP (metionina-aminopeptidasa), un inhibidor de la exportación nuclear de proteínas CRM1, un inhibidor de DPPIV, un inhibidor de reacciones de transferencia de fosforilo en las mitocondrias, un inhibidor de la síntesis de proteínas, un inhibidor de cinasas, un inhibidor de CDK2, un inhibidor de CDK9, un inhibidor del proteasoma, un inhibidor de cinesina, un inhibidor de HDAC, un agente que daña el ADN, un agente alquilante de ADN, un intercalante de ADN, un agente de unión al surco menor del ADN y un inhibidor de DHFR. Los ejemplos adecuados incluyen calicheamicinas tales como gamma-calicheamicina; y maitansinoides tales como DM1, DM3 y DM4. Los métodos para unir cada uno de estos a un conector compatible con los anticuerpos y el método de la invención son conocidos en la técnica. Remítase, por ejemplo, a Singh et al., (2009) Therapeutic Antibodies: Methods and Protocols, vol. 525, 445 457.
El término «tumor» se refiere a la proliferación y el crecimiento celular neoplásico, ya sea maligno o benigno, y todas las células y tejidos precancerosos y cancerosos.
La expresión «actividad antitumoral» se refiere a una reducción en la tasa de proliferación, viabilidad o actividad metastásica de células tumorales. Una posible vía para mostrar actividad antitumoral es mostrar una disminución en la velocidad de crecimiento de células anormales que surge durante la terapia o una estabilidad o reducción del tamaño tumoral. Dicha actividad se puede evaluar utilizando modelos tumorales in vitro o in vivo aceptados, que incluyen modelos de xenoinjerto, modelos de aloinjerto, modelos de MMTV y otros conocidos modelos conocidos en la técnica para investigar la actividad antitumoral.
La expresión «neoplasia maligna» se refiere a un tumor no benigno o un cáncer. Tal como se utiliza en la presente, el término «cáncer», incluye una neoplasia maligna caracterizada por un crecimiento celular desregulado o incontrolado. Los tipos de cáncer ilustrativos incluyen: carcinomas, sarcomas, leucemias y linfomas.
El término «cáncer» incluye tumores malignos primarios (por ejemplo, aquellos cuyas células no hayan migrado a sitios en el cuerpo del sujeto aparte del sitio del tumor original) y tumores malignos secundarios (por ejemplo, aquellos que surgen de la metástasis, la migración de células tumorales a sitios secundarios que son diferentes del sitio del tumor original).
Tal como se utiliza en la presente, la expresión «portador farmacéuticamente aceptable» incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, tensioactivos, antioxidantes, conservantes (p. ej., agentes antibacterianos, agentes antifúngicos), agentes isotónicos, agentes retardantes de la absorción, sales, conservantes, estabilizantes farmacológicos, aglutinantes, excipientes, agentes disgregantes, lubricantes, agentes edulcorantes, agentes aromatizantes, colorantes y combinaciones de estos, tal como serían conocidos para los expertos en la técnica (remítase, por ejemplo, a Remington’s Pharmaceutical Sciences, 18.a ed. Mack Printing Company, 1990, págs.
1289-1329). Salvo en lo que concierne a cualquier portador convencional que sea incompatible con el principio activo, se contempla su uso en las composiciones terapéuticas o farmacéuticas.
La expresión «una cantidad terapéuticamente eficaz» de un compuesto de la presente invención se refiere a una cantidad del compuesto de la presente invención que provocará la respuesta biológica o médica de un sujeto, por ejemplo, la reducción o inhibición de una enzima o una actividad proteica, o mejorará los síntomas, aliviará las afecciones, ralentizará o retrasará la progresión de la enfermedad, o prevendrá una enfermedad, etc. En una realización, la expresión «una cantidad terapéuticamente eficaz» se refiere a la cantidad del compuesto de la presente invención que, cuando se administra a un sujeto, es eficaz para, al menos en parte, aliviar, inhibir, prevenir y/o mejorar una afección o un trastorno o una enfermedad.
El término «sujeto», tal como se utiliza en la presente, se refiere a un animal. Habitualmente el animal es un mamífero. Un sujeto también se refiere, por ejemplo, a primates (p. ej., seres humanos, hombres o mujeres), vacas, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, peces y pájaros. En ciertas realizaciones, el sujeto es un primate. En realizaciones específicas, el sujeto es un ser humano.
Tal como se utiliza en la presente, el término «inhibir», «inhibición» o la expresión «que inhibe» se refiere a la reducción o supresión de una determinada afección, síntoma o trastorno, o enfermedad o una reducción significativa en la actividad basal de una actividad o proceso biológico.
Tal como se utiliza en la presente, el término «tratar», la expresión «que trata» o el término «tratamiento» de cualquier enfermedad o trastorno se refiere en una realización, a mejorar la enfermedad o trastorno (es decir, ralentizar o detener o reducir el desarrollo de la enfermedad o al menos uno de los síntomas clínicos de esta). En otra realización, «tratar», «que trata» o «tratamiento» se refiere a mitigar o mejorar al menos un parámetro físico que incluye los que pueden no ser perceptibles por el paciente. En otra realización más, «tratar», «que trata» o «tratamiento» se refiere a modular la enfermedad o trastorno, ya sea físicamente, (p. ej., estabilización de un síntoma perceptible), fisiológicamente, (p. ej., estabilización de un parámetro físico), o ambos. En otra realización más, «tratar», «que trata» o «tratamiento» se refiere a prevenir o retrasar la progresión de la enfermedad o trastorno.
Tal como se utiliza en la presente, un sujeto «necesita» un tratamiento si dicho sujeto se beneficiaría biológicamente, médicamente, o en su calidad de vida, de dicho tratamiento.
Tal como se utiliza en la presente, se debe interpretar que el término «un», «una», «el/la» y términos similares utilizados en el contexto de la presente invención (especialmente en el contexto de las reivindicaciones) cubren tanto el singular como el plural a menos que se indique lo contrario en la presente o se contradiga claramente por el contexto.
En determinadas realizaciones, los inmunoconjugados modificados de la invención se describen según una proporción «péptido citotóxico respecto a anticuerpo» de, por ejemplo, 1, 2, 3, 4, 5, 6, 7, u 8, o 12 o 16; esta proporción corresponde a "y" en la Fórmula (II) y la Fórmula (III). Aunque esta proporción tiene un valor entero para una molécula de conjugado específica, se sobreentiende que habitualmente se utiliza un valor promedio para describir una muestra que contiene muchas moléculas, debido a algún grado de inhomogeneidad dentro de una mezcla de un inmunoconjugado. A la carga promedio para una muestra de un inmunoconjugado se hace referencia en la presente como «proporción de fármaco respecto a anticuerpo» o DAR. En algunas realizaciones, la DAR se encuentra entre aproximadamente 1 y aproximadamente 16, y habitualmente es de aproximadamente 1,2, 3, 4, 5, 6, 7, u 8. En algunas realizaciones, al menos un 50% de una muestra en peso es un compuesto que tiene la DAR promedio más o menos 2, y preferentemente al menos un 50% de la muestra es un producto que contiene la DAR promedio más o menos 1.5. Las realizaciones preferidas incluyen inmunoconjugados en los que la DAR es de aproximadamente 2 a aproximadamente 8, por ejemplo, aproximadamente 2, aproximadamente 3, aproximadamente 4, aproximadamente 5, aproximadamente 6, aproximadamente 7 o aproximadamente 8. En estas realizaciones, una DAR de «aproximadamente q» significa que el valor medido para DAR se encuentra dentro de un ±20% de q, o preferentemente dentro de un ±10% de q.
Tal como se utiliza en la presente, la expresión «un isómero óptico» o «un estereoisómero» se refiere a cualquiera de las diversas configuraciones estereoisoméricas que pueden existir para un compuesto determinado de la presente invención e incluye isómeros geométricos. Se sobreentiende que un sustituyente puede estar unido a un centro quiral de un átomo de carbono. El término «quiral» se refiere a moléculas que tienen la propiedad de no superponerse a su imagen especular, mientras que el término «aquiral» se refiere a moléculas que se pueden superponer a su imagen especular. Por lo tanto, la invención incluye enantiómeros, diastereómeros o racematos del compuesto. Los «enantiómeros» son un par de estereoisómeros cuyas imágenes especulares no se pueden superponer entre sí. Una mezcla 1:1 de un par de enantiómeros es una mezcla «racémica». El término se utiliza para designar una mezcla racémica cuando sea apropiado. Los «diastereoisómeros» son estereoisómeros que tienen al menos dos átomos asimétricos, pero que no son imágenes especulares entre sí. La estereoquímica absoluta se especifica de acuerdo con el sistema R-S de Cahn-Ingold-Prelog. Cuando un compuesto es un enantiómero puro, la estereoquímica en cada uno de los carbonos quirales se puede especificar ya sea como R o S. Los compuestos resueltos, cuya configuración absoluta es desconocida, se pueden designar (+) o (-) dependiendo de la dirección (dextrógira o levógira) en la que desvían el plano de luz polarizada a la longitud de onda de la línea D del sodio. Ciertos compuestos descritos en la presente contienen uno o más centros o ejes asimétricos y, por tanto, pueden dar lugar a enantiómeros, diastereómeros y otras formas estereoisoméricas que pueden definirse, en términos de su estereoquímica absoluta, como (R) o (S).
Dependiendo de la elección de los materiales de partida y los procedimientos, los compuestos se pueden encontrar presentes en forma de uno de los posibles isómeros o como mezclas de estos, por ejemplo, como isómeros ópticos puros, o como mezclas de isómeros tales como mezclas de racematos y diastereómeros, dependiendo del número de átomos de carbono asimétricos. Se pretende que la presente invención incluya todos los isómeros posibles de este tipo, incluidas mezclas racémicas, mezclas diastereoméricas y formas ópticamente puras, a menos que se indique de otro modo, por ejemplo, cuando se identifica un isómero específico. Se pueden preparar isómeros (R) y (S) ópticamente activos utilizando sintones quirales o reactivos quirales, o se pueden resolver utilizando técnicas convencionales. Si el compuesto contiene un doble enlace, el sustituyente puede tener la configuración E o Z. Si el compuesto contiene un cicloalquilo disustituido, el sustituyente del cicloalquilo puede tener una configuración cis o trans. También se pretende que todas las formas tautoméricas estén incluidas.
Tal como se utilizan en la presente, los términos «sal» o «sales» se refieren a una sal de adición de ácido o base de un compuesto de la invención. El término «sales» incluye en particular «sales farmacéuticamente aceptables». La expresión «sales farmacéuticamente aceptables» se refiere a sales que conservan la efectividad biológica y las propiedades de los compuestos de esta invención y que habitualmente no son indeseadas desde un punto de vista biológico o de otra forma. En muchos casos, los compuestos de la presente invención son capaces de formar sales de ácidos y/o bases en virtud de la presencia de grupos amino y/o carboxilo o grupos similares a estos.
Se pueden formar sales por adición de ácidos farmacéuticamente aceptables con ácidos inorgánicos y ácidos orgánicos, p. ej., sales acetato, aspartato, benzoato, besilato, bromuro/hidrobromuro, bicarbonato/carbonato, bisulfato/sulfato, canfosulfonato, cloruro/hidrocloruro, cloroteofilonato, citrato, etandisulfonato, fumarato, gluceptato, gluconato, glucuronato, hipurato, hidroyoduro/yoduro, isetionato, lactato, lactobionato, laurilsulfato, malato, maleato, malonato, mandelato, mesilato, metilsulfato, naftoato, napsilato, nicotinato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/hidrógeno-fosfato/dihidrógeno-fosfato, poligalacturonato, propionato, estearato, succinato, sulfosalicilato, tartrato, tosilato y trifluoroacetato.
Los ácidos inorgánicos a partir de los cuales pueden derivarse sales incluyen, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico y ácido fosfórico.
Los ácidos orgánicos a partir de los cuales se pueden derivar sales incluyen, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido toluenosulfónico y ácido sulfosalicílico. Se pueden formar sales de adición de bases farmacéuticamente aceptables con bases inorgánicas y orgánicas.
Las bases inorgánicas a partir de las cuales se pueden derivar las sales incluyen, por ejemplo, sales de amonio y metales de las columnas I a XII de la tabla periódica. En determinadas realizaciones, las sales se obtienen a partir de sodio, potasio, amonio, calcio, magnesio, hierro, plata, zinc y cobre; algunas sales particularmente adecuadas incluyen sales de amonio, potasio, sodio, calcio y magnesio.
Las bases orgánicas a partir de las cuales se pueden derivar las sales incluyen, por ejemplo, aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas de origen natural, aminas cíclicas y resinas de intercambio iónico básicas. Ciertas aminas orgánicas incluyen isopropilamina, benzatina, colinato, dietanolamina, dietilamina, lisina, meglumina, piperazina y trometamina.
Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar a partir de un resto básico o ácido, por métodos químicos convencionales. Generalmente, este tipo de sales se pueden preparar haciendo reaccionar formas ácidas libres de estos compuestos con una cantidad estequiométrica de la base apropiada (tal como hidróxido, carbonato o bicarbonato de Na, Ca, Mg o K), o haciendo reaccionar formas de bases libres de estos compuestos con una cantidad estequiométrica del ácido apropiado. Las reacciones de este tipo normalmente se llevan a cabo en agua o en un disolvente orgánico, o en una mezcla de los dos. Generalmente, cuando sea posible, es deseable el uso de medios no acuosos tales como éter, acetato de etilo, etanol, isopropanol o acetonitrilo. Se pueden encontrar listas de sales adecuadas adicionales, p. ej., en "Remington's Pharmaceutical Sciences", 20.a ed., Mack Publishing Company, Easton, Pa., (1985); y en "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" de Stahl y Wermuth (Wiley-VCH, Weinheim, Alemania, 2002).
También se pretende que cualquier fórmula proporcionada en la presente represente formas no marcadas, así como formas marcadas isotópicamente de los compuestos. Los compuestos marcados isotópicamente tienen estructuras representadas por las fórmulas dadas en la presente, salvo por que uno o más átomos se reemplazan por un átomo que tiene una masa atómica o un número másico seleccionado. Algunos ejemplos de isótopos que se pueden incorporar en compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro tales como 2H, 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36Cl, 125I respectivamente. La invención incluye diversos compuestos marcados isotópicamente tal como se definen en la presente, por ejemplo, aquellos en los que están presentes isótopos radiactivos tales como 3H y 14C, o aquellos en los que están presentes isótopos no radiactivos tales como 2H y 13C. Los compuestos marcados isotópicamente de este tipo son útiles en estudios metabólicos (con 14C), estudios de reacción cinéticos (con, por ejemplo, 2H o 3H), técnicas de detección o formación de imágenes, tales como la tomografía por emisión de positrones (PET) o la tomografía computarizada por emisión de fotón único (SPECT), incluidos los ensayos de distribución tisular de sustratos o fármacos, o en el tratamiento radiactivo de pacientes. En particular, un 18F o compuesto marcado puede ser particularmente deseable para estudios de PET o SPECT. Generalmente se pueden preparar compuestos de fórmula (I) marcados isotópicamente mediante técnicas convencionales conocidas por los expertos en la técnica o mediante procesos análogos a los descritos en los Ejemplos y Preparaciones adjuntos utilizando un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado empleado previamente.
Además, la sustitución con isótopos más pesados, particularmente deuterio (es decir, 2H o D), puede proporcionar ciertas ventajas terapéuticas como resultado de una mayor estabilidad metabólica, por ejemplo, una mayor semivida in vivo o requisitos de dosificación reducida o una mejora en el índice terapéutico. La concentración de un isótopo más pesado de este tipo, específicamente deuterio, se puede definir con el factor de enriquecimiento isotópico. El término «factor de enriquecimiento isotópico», tal como se utiliza en la presente, se refiere a la relación entre la abundancia isotópica y la abundancia natural de un isótopo específico. Si un sustituyente en un compuesto de esta invención se denomina deuterio, dicho compuesto tendrá un factor de enriquecimiento isotópico para cada átomo de deuterio designado de al menos 3500 (52,5% de incorporación de deuterio en cada átomo de deuterio designado), al menos 4000 (60% de incorporación de deuterio), al menos 4500 (67,5% de incorporación de deuterio), al menos 5000 (75% de incorporación de deuterio), al menos 5500 (82,5% de incorporación de deuterio), al menos 6000 (90% de incorporación de deuterio), al menos 6333,3 (95% de incorporación de deuterio), al menos 6466,7 (97% de incorporación de deuterio), al menos 6600 (99% de incorporación de deuterio) o al menos 6633,3 (99,5% de incorporación de deuterio).
Algunos solvatos farmacéuticamente aceptables de acuerdo con la invención incluyen aquellos en los que el disolvente de cristalización puede estar sustituido con isótopos, p. ej., D2O, d6-acetona, d6-DMSO, así como solvatos con disolventes no enriquecidos.
Algunos compuestos de la invención, es decir, los compuestos de fórmula (I) que contienen grupos capaces de actuar como dadores y/o aceptores de enlaces de hidrógeno pueden ser capaces de formar cocristales con formadores de cocristales adecuados. Estos cocristales se pueden preparar a partir de compuestos de fórmula (I) mediante procedimientos de formación de cocristales conocidos. Tales procedimientos incluyen moler, calentar, cosublimar, cofundir o poner en contacto en disolución los compuestos de fórmula (I) con el formador de cocristales en condiciones de cristalización y aislar los cocristales formados de este modo. Los formadores de cocristales adecuados incluyen los descritos en el documento WO 2004/078163. Por lo tanto, la invención proporciona, además, cocristales que comprenden un compuesto de fórmula (I).
Cualquier átomo asimétrico (p. ej., carbono) del compuesto o compuestos de la presente invención puede estar presente en forma racémica o enriquecida enantioméricamente, por ejemplo, la configuración (R)-, (S)- o (R,S). En determinadas realizaciones, cada átomo asimétrico tiene al menos un 50% de exceso enantiomérico, al menos un 60% de exceso enantiomérico, al menos un 70% de exceso enantiomérico, al menos un 80% de exceso enantiomérico, al menos un 90% de exceso enantiomérico, al menos un 95% de exceso enantiomérico o al menos un 99% de exceso enantiomérico de la configuración (R)- o (S)-, es decir, para compuestos ópticamente activos, a menudo se prefiere utilizar un enantiómero hasta la exclusión sustancial del otro enantiómero. Los sustituyentes en átomos con dobles enlaces insaturados pueden estar presentes, si es posible, en forma cis (Z) o trans (E).
Por consiguiente, tal como se utiliza en la presente, un compuesto de la presente invención puede estar en forma de uno de los posibles isómeros, rotámeros, atropisómeros, tautómeros o mezclas de estos, por ejemplo, como isómeros geométricos (cis o trans), diastereómeros, isómeros ópticos (enantiómeros), racematos o mezclas de estos sustancialmente puros. La expresión «sustancialmente puro» o «sustancialmente exento de otros isómeros», tal como se utiliza en la presente, se refiere a que el producto contiene menos de un 5%, y preferentemente menos de un 2% de otros isómeros en relación con la cantidad del isómero preferido, en peso.
Cualesquiera mezclas resultantes de isómeros se pueden separar, basándose en las diferencias fisicoquímicas de los constituyentes, en los racematos, diastereómeros, isómeros geométricos u ópticos puros o sustancialmente puros, por ejemplo, mediante cromatografía y/o cristalización fraccionada.
Cualesquiera racematos resultantes de productos finales o intermedios se pueden resolver en los enantiómeros ópticos mediante métodos conocidos, p. ej., mediante la separación de las sales diastereoméricas de estos, obtenidas con una base o ácido ópticamente activo, y la liberación del compuesto ácido o básico ópticamente activo. En particular, se puede emplear de este modo un resto básico para resolver los compuestos de la presente invención en sus enantiómeros ópticos, por ejemplo, mediante la cristalización fraccionada de una sal formada con un ácido ópticamente activo, por ejemplo, ácido tartárico, ácido dibenzoiltartárico, ácido diacetiltartárico, ácido di-O,O'-ptoluoiltartárico, ácido mandélico, ácido málico o ácido canfor-10-sulfónico. Los productos racémicos también se pueden resolver por cromatografía quiral, por ejemplo, cromatografía líquida de alta resolución (HPLC) utilizando un adsorbente quiral.
Además, los compuestos de la presente invención, incluidas sus sales, también se pueden obtener en forma de sus hidratos o incluir otros disolventes utilizados para su cristalización. Los compuestos de la presente invención pueden formar solvatos de forma inherente o intencional con disolventes farmacéuticamente aceptables (que incluyen agua); por lo tanto, se pretende que la invención englobe tanto formas solvatadas como no solvatadas. El término «solvato» se refiere a un complejo molecular de un compuesto de la presente invención (incluidas las sales farmacéuticamente aceptables de este) con una o más moléculas de disolvente. Dichas moléculas de disolvente son aquellas utilizadas comúnmente en la técnica farmacéutica, de las que se tiene constancia que son inocuas para el receptor, p. ej., agua y etanol. El término «hidrato» se refiere al complejo donde la molécula de disolvente es agua.
Los compuestos de Fórmula (I) de la presente invención, incluidas sales, hidratos y solvatos de estos, pueden formar polimorfos de forma inherente o intencional.
El término «tiol-maleimida», tal como se utiliza en la presente, se refiere a un grupo formado mediante la reacción de un tiol con maleimida, que tiene esta fórmula general

, donde Y y Z son grupos que se han de conectar a través de la conexión tiol-maleimida y pueden comprender componentes de conectores, anticuerpos o cargas.
El término «escindible», tal como se utiliza en la presente, se refiere a un conector o componente de conector que conecta dos restos mediante conexiones covalentes, pero que se descompone para cortar la conexión covalente entre los restos en condiciones fisiológicamente relevantes, habitualmente un conector escindible se corta in vivo más rápidamente en un ambiente intracelular que cuando se encuentra fuera de una célula, lo que provoca que la liberación de la carga tenga lugar preferentemente dentro de una célula diana. La escisión puede ser enzimática o no enzimática, pero generalmente libera una carga de un anticuerpo sin degradar el anticuerpo. La escisión puede dejar alguna parte de un conector o componente de un conector unida a la carga, o puede liberar la carga sin ninguna parte o componente residual del conector.
El término «Pcl», tal como se utiliza en la presente, se refiere a pirrolinocarboxilisina, por ejemplo,

El compuesto correspondiente, donde R20 es metilo es pirrolisina.
La expresión «no escindible», tal como se utiliza en la presente, se refiere a un conector o componente de un conector que no es especialmente susceptible de descomposición en condiciones fisiológicas, por ejemplo, es al menos tan estable como el anticuerpo o la parte de fragmento de unión a antígeno del inmunoconjugado. Dichos conectores a menudo se denominan «estables», lo que significa que son suficientemente resistentes a la degradación para mantener la carga conectada al resto de unión a antígeno Ab hasta que el Ab mismo se degrade al menos parcialmente, es decir, la degradación del Ab precede a la escisión del conector in vivo. La degradación de la porción de anticuerpo de un ADC que tiene un conector estable o no escindible pueden dejar algo o todo el conector, y uno o más grupos aminoácidos de un anticuerpo, unidos al resto de fármaco o carga que se suministra in vivo.
Los términos «alquilo C1-C3 », «alquilo C2-C3 », «alquilo C1-C4 », «alquilo C1-C5 », «alquilo C1-C6 » y «alquilo C2-C6», tal como se utilizan en la presente, se refieren a un hidrocarburo de cadena lineal o ramificada totalmente saturado que contiene 1 -3 átomos de carbono, 2-3 átomos de carbono, 1 -4 átomos de carbono, 1 -5 átomos de carbono, 1 -6 átomos de carbono o 2-6 átomos de carbono, respectivamente. Algunos ejemplos de grupos «alquilo C1-C3 » incluyen metilo, etilo, n-propilo e isopropilo. Algunos ejemplos de grupos «alquilo C2-C3 » incluyen etilo, n-propilo e isopropilo. Algunos ejemplos de grupos «alquilo C1-C4 » incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo y ferf-butilo. Algunos ejemplos de grupos «alquilo C1-C5 » incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, ferf-butilo, n-pentilo e isopentilo. Algunos ejemplos de grupos «alquilo C1-C6» incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, ferf-butilo, n-pentilo, isopentilo y hexilo. Algunos ejemplos de grupos «alquilo C2-C6» incluyen etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, ferf-butilo, n-pentilo, isopentilo y hexilo.
Tal como se utiliza en la presente, el término «alquileno» se refiere a un grupo alquilo divalente que tiene de 1 a 10 átomos de carbono, y dos valencias abiertas para unirse a otros rasgos. A menos que se indique lo contrario, alquileno se refiere a restos que tienen de 1 a 10 átomos de carbono, de 1 a 6 átomos de carbono o de 1 a 4 átomos de carbono. Algunos ejemplos representativos de alquileno incluyen metileno, etileno, n-propileno, isopropileno, n-butileno, sec-
butileno, isobutileno, ferf-butileno, n-pentileno, isopentileno, neopentileno, n-hexileno, 3-metilhexileno, 2,2-dimetilpentileno, 2,3-dimetilpentileno, n-heptileno, n-octileno, n-nonileno y n-decileno.
Los términos «alcoxi C1-C3 », «alcoxi C2-C3 », «alcoxi C1-C4», «alcoxi C1-C5 », «alcoxi C1-C6 » y «alcoxi C2-C6 », tal como se utilizan en la presente, se refieren a los grupos -O-alquilo C1-C3 , -O-alquilo C2-C3 , -O-alquilo C1-C4 , -O-alquilo C1-C5 , -O-alquilo C1-C6 y -O-alquilo C2-C6 respectivamente, donde los grupos «alquilo C1-C3 », «alquilo C2-C3 », «alquilo C1-C4 », «alquilo C1-C5 », «alquilo C1-C6 » y «alquilo C2-C6 » son como se definen en la presente. Los ejemplos de grupos «alcoxi C1-C3 » incluyen metoxi, etoxi, n-propoxi e isopropoxi. Los ejemplos de grupos «alcoxi C2-C3 » incluyen etoxi, n-propoxi e isopropoxi. Los ejemplos de grupos «alcoxi C1-C4 » incluyen metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, sec-butoxi y ferf-butoxi. Los ejemplos de grupos «alcoxi C1-C5 » incluyen metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, sec-butoxi, ferf-butoxi, n-pentiloxi e isopentiloxi. Los ejemplos de grupos «alcoxi C1-C6 » incluyen metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, sec-butoxi, ferf-butoxi, n-pentiloxi, isopentiloxi y hexiloxi. Los ejemplos de grupos «alcoxi C2-C6 » incluyen etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, sec-butoxi, ferf-butoxi, npentiloxi, isopentiloxi y hexiloxi.
Tal como se utiliza en la presente, el término «halógeno» (o halo) se refiere a flúor, bromo, cloro o yodo, en particular, flúor o cloro. Los grupos y restos sustituidos con halógeno tales como alquilo sustituido con halógeno (haloalquilo) pueden estar mono-, poli- o perhalogenados.
Tal como se utiliza en la presente, el término «heteroátomos» se refiere a átomos de nitrógeno (N), oxígeno (O) o azufre (S), en particular nitrógeno u oxígeno, a menos que se indique lo contrario.
La expresión «heterocicloalquilo de 4-8 miembros», tal como se utiliza en la presente se refiere a una estructura anular hidrocarbonada monocíclica de 4-8 miembros saturada, donde de uno a dos de los carbonos anulares de la estructura anular hidrocarbonada se reemplazan por de uno a dos grupos NR, donde R es hidrógeno, un enlace, un grupo R5 tal como se define en la presente o un grupo R7 tal como se define en la presente. Los ejemplos de grupos heterocicloalquílicos de 4-8 miembros, tal como se utilizan en la presente, incluyen azetadinilo, azetadin-1-ilo, azetadin-2-ilo, azetadin-3-ilo, pirrolidinilo, pirrolidin-1-ilo, pirrolidin-2-ilo, pirrolidin-3-ilo, pirrolidin-4-ilo, pirrolidin-5-ilo, piperidinilo, piperidin-1-ilo, piperidin-2-ilo, piperidin-3-ilo, piperidin-4-ilo, piperidin-5-ilo, piperidin-6-ilo, piperazinilo, piperazin-1-ilo, piperazin-2-ilo, piperazin-3-ilo, piperazin-4-ilo, piperazin-5-ilo, piperazin-6-ilo, azepanilo, azepan-1-ilo, azepan-2-ilo, azepan-3-ilo, azepan-4-ilo, azepan-5-ilo, azepan-6-ilo y azepan-7-ilo.
La expresión «heterocicloalquilo de 6 miembros», tal como se utiliza en la presente se refiere a una estructura anular hidrocarbonada monocíclica de 6 miembros saturada, donde de uno a dos de los carbonos anulares de la estructura anular hidrocarbonada se reemplazan por de uno a dos grupos NR, donde R es hidrógeno, un enlace, un grupo R5 tal como se define en la presente o un grupo R7 tal como se define en la presente. Los ejemplos de grupos heterocicloalquílicos de 6 miembros, tal como se utilizan en la presente, incluyen piperidinilo, piperidin-1-ilo, piperidin-2- ilo, piperidin-3-ilo, piperidin-4-ilo, piperidin-5-ilo, piperidin-6-ilo, piperazinilo, piperazin-1-ilo, piperazin-2-ilo, piperazin-3- ilo, piperazin-4-ilo, piperazin-5-ilo y piperazin-6-ilo.
La expresión «heterocicloalquilo de 5-8 miembros», tal como se utiliza en la presente se refiere a una estructura anular hidrocarbonada bicíclica fuisionada de 5-8 miembros saturada, donde de uno a dos de los carbonos anulares de la estructura anular hidrocarbonada se reemplazan por de uno a dos grupos NR, R es hidrógeno, un enlace, un grupo R5 tal como se define en la presente o un grupo R7 tal como se define en la presente. Los ejemplos de grupos heterocicloalquílicos bicíclicos fusionados de 5-8 miembros, tal como se utilizan en la presente, incluyen 3-azabiciclo[3.1.0]hexanilo y 3-azabiciclo[4.1.0]heptanilo.
La convención de nomenclatura de inmunoconjugados utilizada en la presente es el Número de compuesto-anticuerpo, donde Número de compuesto se refiere al compuesto de Fórmula (I) utilizado para la conjugación al anticuerpo particular.
Conectores
Los péptidos citotóxicos proporcionados en la presente para su uso como cargas de ADC se pueden unir a un conector, L, o directamente a un resto de unión a antígeno. Los conectores adecuados para su uso en dichos ADC son muy conocidos en la técnica, y se pueden utilizar en los conjugados de la invención. El conector, L, se puede unir al resto de unión a antígeno en cualquier posición disponible adecuada en el resto de unión a antígeno: habitualmente, L se une a un átomo de nitrógeno de amino disponible (es decir, una amina primaria o secundaria, en lugar de una amida) o un átomo de oxígeno hidroxílico, o a un sulfihidrilo disponible tal como en una cisteína. La unión del conector, L, a los péptidos citotóxicos proporcionados en la presente puede ser en el extremo N del péptido citotóxico o en el extremo c del péptido citotóxico. Se conoce una amplia variedad de conectores para su uso en ADC (remítase, por ejemplo, a Lash, Anfibody-Drug Conjugales: fhe Nexf Generafion of Moving Parfs, Start-Up, Dic. 2011, 1 -6), y se pueden utilizar en conjugados dentro del alcance de la invención.
El conector, L, en la Fórmula (I), Fórmula (II) y Fórmula (III) es un resto conector que comprende uno o más componentes de conector L1, L2 , L3 , L4 , L5 , L6, etc. En determinadas realizaciones, un componente de conector puede
representar un enlace que conecta los grupos que lo flanquean. En determinadas realizaciones, L es -*Li L2L3L4L5L6-, donde el * denota el sitio de unión al péptido citotóxico. En determinadas realizaciones, un componente de conector puede representar un enlace que conecta los grupos que lo flanquean. En determinadas realizaciones, L es -*L1 L2L3L4L5-, donde el * denota el sitio de unión al péptido citotóxico. En determinadas realizaciones, un componente de conector puede representar un enlace que conecta los grupos que lo flanquean. En determinadas realizaciones, L es -*L1 L2L3L4-, donde el * denota el sitio de unión al citotóxico. En determinadas realizaciones, un componente de conector puede representar un enlace que conecta los grupos que lo flanquean. En determinadas realizaciones, L es -*L1 L2L3-, donde el * denota el sitio de unión al péptido citotóxico. En una realización preferida, L es - T 1L2-, donde el * denota el sitio de unión al péptido citotóxico. En una realización determinada, L es -L1-. Algunos conectores y componentes de conector preferidos se representan en la presente.
El conector, L, en la Fórmula (I), Fórmula (II) y Fórmula (III) puede ser divalente, lo que significa que se puede utilizar para conectar solamente una carga por conector a un resto de unión a antígeno, o puede ser trivalente y es capaz de conectar dos cargas por conector a un resto de unión a antígeno o puede ser polivalente. Se pueden utilizar conectores trivalentes, tetravalentes y polivalentes para aumentar la carga de una carga (fármaco) en un resto de unión a antígeno (por ejemplo, un anticuerpo), con lo cual aumenta la proporción de fármaco respecto a anticuerpo (DAR) sin necesidad de sitios adicionales en el anticuerpo para unir múltiples conectores. Algunos ejemplos de dichos conectores proporcionados en Bioconjugate Chem, 1999 Marzo-Abril;10(2):279-88; US6638499; Clin Cáncer Res Octubre 15, 2004 10; 7063; y el documento WO2012/113847A1.
Un conector, L, para su uso en los compuestos de Fórmula (I) y los inmunoconjugados de Fórmula (II) y Fórmula (III) puede ser escindible o no escindible. Los conectores escindibles tales como aquellos que contienen una hidrazona, un disulfuro, el dipéptido Val-Cit, y los que contienen un resto p-aminobenciloxicarbonilo escindible con glucuronidasa, son muy conocidos en la técnica, y se pueden utilizar. Remítase, por ejemplo, a Ducry, et al., Bioconjugate Chem., vol.
21, 5-13 (2010). Para los inmunoconjugados de que comprenden un conector escindible, el conector es sustancialmente estable in vivo hasta que el inmunoconjugado se une o entra a la célula, momento en el cual las enzimas intracelulares o las condiciones químicas intracelulares (pH, capacidad de reducción) escinden el conector para liberar el péptido citotóxico.
Como alternativa, se pueden utilizar conectores no escindibles en los compuestos de Fórmula (I) y los inmunoconjugados de Fórmula (II) y Fórmula (III). Los conectores no escindibles carecen de componentes estructurales diseñados para degradarse en las células, y por tanto, sus estructuras pueden variar de forma sustancial. Remítase, por ejemplo, a Ducry, et al., Bioconjugate Chem., vol. 21,5-13 (2010). Se cree que estos inmunoconjugados entran en una célula diana y experimentan degradación proteolítica del anticuerpo en lugar de descomposición del conector, por lo que al menos una parte del conector, o todo, e incluso algo del anticuerpo o fragmento de anticuerpo puede permanecer unido a la carga.
El conector, L, en los compuestos de Fórmula (I) y los inmunoconjugados de Fórmula (II) y Fórmula (III) contiene normalmente habitualmente dos o más componentes de conector, que se pueden seleccionar por conveniencia en ensamblaje del conjugado, o se pueden seleccionar de modo que influyan en las propiedades del conjugado. Los componentes de conector adecuados para formar un conector, L, son conocidos en la técnica, al igual que los métodos para construir el conector L. Los componentes de conector pueden incluir los grupos utilizados normalmente para unir un grupo a un aminoácido, espaciadores tales como grupos alquileno y oligómeros de óxido de etileno, aminoácidos y péptidos cortos hasta aproximadamente 4 aminoácidos en longitud; un enlace; y conexiones de carbonilo, carbamato, carbonato, urea, éster y amida. Los componentes de conector pueden comprende grupos tiol-maleimida, tioéteres, amidas y ésteres; grupos que se escinden fácilmente in vivo en condiciones que se encuentran en las células diana o alrededor de ellas, tales como disulfuros, hidrazonas, dipéptidos como Val-Cit y grupos benciloxicarbonilo sustituidos; espaciadores para orientar la carga en una posición adecuada respecto al resto de unión a antígeno tal como anillos fenilo, heteroarilo, cicloalquilo o heterociclilo, y cadenas alquilénicas; y/o grupos que potencian las propiedades farmacocinéticas, tales como alquileno sustituido con uno o más grupos polares (carboxi, sulfonato, hidroxilo, amina, aminoácido, sacárido), y cadenas alquilénicas que contienen uno o más -NH- u -O - en lugar de uno o varios grupos metileno tales como éteres glicólicos (-CH2CH2O-)p donde p es 1-10, que puede potenciar la solubilidad o reducir la agregación intermolecular, por ejemplo.
Además, los componentes de conector pueden comprender restos químicos que se forman fácilmente mediante reacción entre dos grupos reactivos. Algunos ejemplos de dichos restos químicos se proporcionan en la Tabla 1.
Tabla 1





donde: R32 en la Tabla 1 es H, alquilo C1-4, fenilo, pirimidina o piridina; R35 en la Tabla 1 es H, alquilo Ci-6, fenilo o alquilo C1-4 sustituido con de 1 a 3 grupos -OH; cada R36 en la Tabla 1 se selecciona independientemente entre H, alquilo C1-6, fluoro, benciloxi sustituido con -C(=O)OH, bencilo sustituido con -C(=O)OH, alcoxi C1-4 sustituido con -C(=O)OH y alquilo C1-4sustituido con -C(=O)OH; R37 en la Tabla 1 se selecciona independientemente entre H, fenilo y piridina, cada R5 en la Tabla 1 se selecciona independientemente entre H o alquilo C1-6; R12 en la Tabla 1 es H, -CH3 o fenilo; R50 en la Tabla 1 es H o nitro.
En algunas realizaciones, un componente de conector de un conector, L, de los inmunoconjugados de Fórmula (II) es un grupo formado tras la reacción de un grupo funcional reactivo con una de las cadenas laterales de aminoácidos utilizadas normalmente para la conjugación, por ejemplo, el tiol de la cisteína, o el -N H 2 libre de lisina, o un grupo Pcl o Pyl introducido mediante modificación en un anticuerpo. Remítase, por ejemplo, a Ou, et al., PNAS 108(26), 10437 42 (2011). Los componentes de conector formados mediante reacción con un residuo de cisteína del resto de unión a

componentes de conector formados mediante reacción con el -N H 2 de un residuo de Usina del resto de unión a antígeno, donde cada p es 1-10 y cada R es independientemente H o alquilo C1-4 (preferentemente metilo) incluyen


, donde R20 es H o Me, y R30 es H, Me o Fenilo, para la conexión, donde el grupo acilo mostrado se une a la parte de lisina de un Pcl o Pyl en un anticuerpo modificado. Un componente de conector formado
tras la reacción de un puente disulfuro de Ab, , y un compuesto de Fórmula (I) que contiene una
, y un compuesto de Fórmula (I) que contiene una
hidroxilamina componente de conector formado tras la reacción de un puente disulfuro de Ab, «AAAAAAA
componente de conector formado tras la reacción de un puente disulfuro de Ab, «AAAAAAA
,S 
, .
En algunas realizaciones, un componente de conector de un conector, L, de los inmunoconjugados de Fórmula (II) y Fórmula (III) incluyen, por ejemplo, grupos alquileno -(CH2)n- (donde n es habitualmente 1-10 o 1-6), unidades de etilenglicol (-CH2CH2O-V (donde n es 1-20, habitualmente 1-10 o 1-6), -O-, -S-, carbonilo (-C(=O)-), amidas -C(=O)-NH- o -NH-C(=O)-, ásteres -C(=O)-O- o -O-C(=O)-, sistemas anulares que tienen dos puntos disponibles de unión tales como un anillo divalente seleccionado entre fenilo (incluidos fenilos 1,2- 1,3- y 1,4- disustituidos), heteroarilo C5-6, cicloalquilo C3-8 que incluye ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo 1,1 -disustituido, y ciclohexilo 1,4-disustituido y anillos heterocíclicos C4-8, y ejemplos específicos representados a continuación; aminoácidos -NH-CHR*-C=O- o -C(=O)-CHR*-NH-, o grupos derivados de aminoácidos que se unen a N de una estructura adyacente (por ejemplo, a un nitrógeno de maleimida) que tiene la fórmula [N]-CHR*-C(=O)- donde R* es la cadena lateral de un aminoácido conocido (frecuentemente uno de los aminoácidos canónicos, por ejemplo, trp, ala, asp, lys y gly, pero que incluyen también, por ejemplo, norvalina, norleucina, homoserina, homocisteína, fenilglicina, citrulina y otros denominados normalmente alfa-aminoácidos), polipáptidos de aminoácidos conocidos (por ejemplo, dipáptidos, tripáptidos, tetrapáptidos, etc.), uniones tiol-maleimida (a partir de la adición de -SH a maleimida), -S-CR2- y otros éteres de tiol tales como -S-CR2-C(=O)- o -C(=O)-CR2-S- donde R es independientemente en cada caso H o alquilo C1-4, -CH2-C(=O)-, y disulfuros (-S-S-), así como combinaciones de cualquiera de estos con otros componentes de conector descritos a continuación, por ejemplo, un enlace, un conector no escindible enzimáticamente, un conector no escindible, un conector escindible enzimáticamente, un conector fotoestable, un conector fotoescindible o un conector que comprende un espaciador autodestructivo.
En determinadas realizaciones, el conector, L, de los inmunoconjugados de Fórmula (II) es - T 1L2L3L4L5L6-, donde el * denota el sitio de unión al páptido citotóxico. En determinadas realizaciones, el conector, L, de los inmunoconjugados de Fórmula (II) es -*L1L2L3L4L5-, donde el * denota el sitio de unión al páptido citotóxico. En determinadas realizaciones, el conector, L, de los compuestos de los inmunoconjugados de Fórmula (II) es - T 1L2L3L4-, donde el * denota el sitio de unión al páptido citotóxico. En determinadas realizaciones, el conector, L, de los inmunoconjugados de Fórmula (II) es - T 1L2L3-, donde el * denota el sitio de unión al páptido citotóxico. En una realización preferida, el conector, L, de los inmunoconjugados de Fórmula (II) es - T 1L2-, donde el * denota el sitio de unión al páptido citotóxico.
El componente de conector L1 de los inmunoconjugados de Fórmula (II) se selecciona entre -(CH2)m-, -C(=O)(CH2)m-, -C=O X1X2C=O CH2 m-, -C =O X1X2C =O CH2 mNR12C=O CH2 m- -C=OX1X2C=O CH2 mX3 CH2 m- -


cada R25 se selecciona independientemente entre H o alquilo C1-4 ;
Raa es una cadena lateral de un aminoácido seleccionado entre glicina, alanina, triptófano, tirosina, fenilalanina, leucina, isoleucina, valina, asparagina, ácido glutámico, glutamina, ácido aspático, histidina, arginina, lisina, cisteína, metionina, serina, treonina, fenilglicina y f-butilglicina;
R32 se selecciona independientemente entre H, alquilo C1-4, fenilo, pirimidina y piridina;

R33 se selecciona independientemente entre

R34 se selecciona independientemente entre H, alquilo C1-4, y haloalquilo C1-6;



donde:
R20 es H o Me, y R30 es H, -CH3 o fenilo

cada R25 se selecciona independientemente entre H y alquilo C1-4 ;
Raa es una cadena lateral de un aminoácido seleccionado entre glicina, alanina, triptófano, tirosina, fenilalanina, leucina, isoleucina, valina, asparagina, ácido glutámico, glutamina, ácido aspático, histidina, arginina, lisina, cisteína, metionina, serina, treonina, fenilglicina y f-butilglicina;
R32 se selecciona independientemente entre H, alquilo C1-4, fenilo, pirimidina y piridina;

R33 se selecciona independientemente entre

R3
X
1



cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
Péptidos citotóxicos
Los péptidos citotóxicos divulgados en la presente, o un estereoisómero de estos, y sales farmacéuticamente aceptables de estos, son compuestos que tienen la estructura de Fórmula (I)


cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R15 es 2-piridilo o 4-piridilo;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O, que está sin sustituir o sustituido con -LR9;
cada R19 es H o alquilo C1-C6 ;
X3 es
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6 , 7, 8 , 9 y 10; y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6 , 7, 8 , 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
También se divulgan en la presente péptidos citotóxicos, o un estereoisómero de estos, y sales farmacéuticamente aceptables de estos, que tienen la estructura de Fórmula (I)

donde:
R1 es

R2 es metilo, etilo, isopropilo o sec-butilo;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos, -C(=0)R11, -(CH2)mOH, C(=0)(CH2)mOH, -C(=0)((CH2)m0)nR12, o -((CH2)mO)nR12;
R7 es LR9;
R8 es H o LR9;
cada L es un conector seleccionado independientemente entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2Li -,-Li L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la presente;

cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R15 es 2-piridilo o 4-piridilo;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O, que está sin sustituir o sustituido con -LR9;
X3 e s 
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6 , 7, 8 , 9 y 10;
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6 , 7, 8 , 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
Métodos sintéticos
Todos los materiales de partida, componentes, reactivos, ácidos, bases, agentes deshidratantes, disolventes y catalizadores utilizados para sintetizar los compuestos de la presente invención están comercializados o se pueden producir mediante métodos de síntesis orgánica conocidos por los expertos en la técnica (remítase, por ejemplo, a Houben-Weyl 4.a Ed. 1952, Methods of Organic Synthesis, Thieme, Volumen 21). Además, los compuestos de la presente invención se pueden producir mediante métodos de síntesis orgánica conocidos para un experto en la técnica, habida cuenta de los siguientes ejemplos.
Se proporcionan ejemplos ilustrativos de las estrategias sintéticas para el compuesto de Fórmula (I) y subfórmulas de esta, en los siguientes Esquemas generales. En los siguientes esquemas, R1, R2, R3, R4, R5, R6 y L son como se definen en la presente. Aunque los esquemas generales muestran reactivos específicos utilizados para varios pasos sintéticos, se sobreentiende que se pueden utilizar otros reactivos conocidos para lograr dichos pasos sintéticos.
A continuación, en los Esquemas 1 a 3 se muestran estrategias sintéticas para los compuestos de Fórmula (I) y subfórmulas de esta, donde R1 es un heterocicloalquilo de 6 miembros conectado en C que contiene 1 -2 heteroátomos de N y un puente alquileno C1-C2 o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros conectado en C que contiene 1-2 heteroátomos de N, donde cada uno está sin sustituir o cada uno está sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6.
Esquema 1

En el Esquema 1, R3 se acopla con un péptido corto mediante formación de un enlace amida seguida por un paso de desprotección con un acoplamiento posterior de R1 mediante formación de un enlace amida. En el Esquema 1, a modo O

r 1^ 0 H
de ejemplo, K puede ser Boc que posteriormente se desprotege después del acoplamiento. En
r 
el Esquema 1, a modo de ejemplo, t '
Esquema 2

El Esquema 2 ilustra una estrategia sintética ilustrativa para los compuestos de Fórmula (I), y subfórmulas de esta,
donde  Esquema 2, R3 se acopla con un péptido corto mediante formación de un enlace amida seguida por un paso de desprotección con un acoplamiento posterior de R1 mediante formación
Esquema 2, R3 se acopla con un péptido corto mediante formación de un enlace amida seguida por un paso de desprotección con un acoplamiento posterior de R1 mediante formación

de un enlace amida. En el Esquema 2, a modo de ejemplo, R puede ser Boc que O
. K 'OH posteriormente se desprotege después del acoplamiento. En el Esquema 1, a modo de ejemplo, R1 también

amida seguida por un paso de desprotección con un acoplamiento posterior de R1 mediante formación de un enlace
O 
OH
amida. En el Esquema 3, a modo de ejemplo, R1 puede ser Boc , que posteriormente se O
r ^ O H
desprotege después del acoplamiento. En el Esquema 1, a modo de ejemplo, K también puede ser

A continuación, en los Esquemas 4 a 6 se muestran estrategias sintéticas para los compuestos conectados al extremo
N terminal de Fórmula (I), y subfórmulas de esta, donde R1 es .
.
Esquema 4

En el Esquema 4, R3 se acopla con un péptido corto mediante formación de un enlace amida seguida por un paso de desprotección con un acoplamiento posterior de R1 mediante formación de un enlace amida. En el Esquema 3, a modo

En el Esquema 5, R1 es inicialmente ^—NH ' o r5 (remítase a los Esquemas 1 a 3) y a continuación se une

donde cada uno se
sustituye con un R7. Se muestran ejemplos ilustrativos en el esquema a continuación:


C1-C2 o donde cada uno se sustituye con un R7, se obtienen mediante un proceso de dos pasos tal como se observa en el Esquema 6.
donde cada uno se sustituye con un R7, se obtienen mediante un proceso de dos pasos tal como se observa en el Esquema 6.
donde RGi y RG2 son grupos reactivos tales como los que se proporcionan en la Tabla 1, y L' es uno o más componentes conectores.
Se muestran ejemplos ilustrativos en el esquema a continuación:

A continuación, en el Esquema 7 se muestra una estrategia sintética para los compuestos conectados al extremo C

terminal de Fórmula (I), y subfórmulas de esta, donde R1 es C N HH 0 R
Esquema 7

A continuación, en el Esquema 8 se muestra una estrategia sintética alternativa para los compuestos conectados al
extremo C terminal de Fórmula (I), y subfórmulas de esta, donde R1 es .
.

A continuación, en el Esquema 9 se muestra una estrategia sintética alternativa para los compuestos conectados al 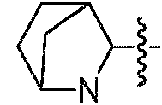
extremo C terminal de Fórmula (I), y subfórmulas de esta, donde R1 es CH
Esquema 9

A continuación, en el Esquema 10 se muestra una estrategia sintética alternativa para los compuestos conectados al extremo C terminal de Fórmula (I), y subfórmulas de esta, donde R1es
Esquema 10

A continuación, en el Esquema 11 se muestra una estrategia sintética alternativa para los compuestos conectados al
extremo C terminal de Fórmula (I), y subfórmulas de esta, donde R1 es o
o 

A continuación, en el Esquema 12 se muestra una estrategia sintética alternativa para los compuestos conectados al 
C H
extremo C terminal de Fórmula (I), y subfórmulas de esta, donde R1 es NH 0 R
Esquema 12

La invención incluye además cualquier variante de los procesos de la presente, en la que se utilice un producto intermedio que se pueda obtener en cualquier etapa de estos como material de partida y se lleven a cabo los pasos restantes o en la que los materiales de partida se formen in situ en las condiciones de reacción, o en la que los componentes de reacción se utilicen en forma de sus sales o material ópticamente puro.
Los ejemplos siguientes pretenden ilustrar la invención y no se debe interpretar que suponen limitaciones de la misma. Las temperaturas se indican en grados Celsius. Si no se menciona lo contrario, todas las evaporaciones se llevan a cabo a presión reducida, habitualmente entre aproximadamente 15 mm de Hg y 100 mm de Hg (= 20-133 mbar). Las abreviaturas utilizadas son las convencionales en la técnica. Todas las reacciones se llevaron a cabo en nitrógeno utilizando disolventes de grado comercial sin ninguna destilación adicional. Los reactivos se utilizaron como grado comercial sin purificación adicional. La cromatografía en capa fina se llevó a cabo utilizando placas de gel de sílice CCF. La cromatografía en columna se llevó a cabo utilizando un sistema ISCO Combiflash Companion, utilizando columnas Redisep® preempaquetadas de grado flash.
La HPLC preparativa se llevó a cabo en un sistema Waters Autopurification utilizando las siguientes condiciones: Columna Sunfire C18 30 x 100 mm, 5 pm, elución en gradiente con CH3CN en agua 0.05%de TFA-CH3CN a 30 mL/min.
Métodos analíticos
A menos que se indique lo contrario, se utilizaron los siguientes métodos de HPLC y HPLC/MS en la preparación de los Intermedios y los Ejemplos.
El análisis LC/MS se llevó a cabo en un sistema Agilent 1200sl/6140.
Columna: Waters Acquity HSS T3 C18, 50x2,0, 1,8gm
Fase móvil: A) H2 O 0,05% de TFA; B: acetonitrilo 0,035% de TFA
Método de bombeo:

Detección: Matriz de diodos UV a 190 nm - 400 nm
Análisis MS: 200 - 1350 uma
ELSD:60 °C
Parámetros de MS:

Procedimiento sintético para los intermedios
Síntesis de (S)-2-fenil-1-(5-fenil-1,3,4-oxadiazol-2-il)etanamina (i-1)
n - n
y —Ph
Ptv
n h 2 ° <M >
Paso 1: Se añadió ácido (S)-2-((f-butoxicarbonil)amino)-3-fenilpropanoico (200 mg, 0,754 mmol) a diclorometano (5,5 mL) a 0 °C, seguido de carbonildiimidazol (128 mg, 0,792 mmol). Después de agitar a 0 °C durante 30 min, se añadió benzohidrazida (103 mg, 0,754 mmol). Después de 45 min adicionales a 0 °C, se añadieron tetrabromuro de carbono (497 mg, 1,5 mmol) y trifenilfosfina (198 mg, 0,754 mmol). La mezcla se agitó durante 2 h a 0 °C y, a continuación, a t.a. durante 16 h. Se añadió agua a la mezcla y se extrajo con DCM (5 mL X3). Las fases orgánicas se combinaron, se secaron con Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante una columna en gel de sílice (20-40% de acetato de etilo en hexanos) para obtener [(1 S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-il)etil]carbamato de f-butilo. MS m/z 366 (M+H). Tiempo de retención 1,351 min. 1H RMN (400 MHz, Cloroformo-d) 58,03 - 7,85 (m, 2H), 7,62 - 7,38 (m, 3H), 7,33 - 7,16 (m, 3H), 7,18 - 7,04 (m, 2H), 5,35 (d, J = 7,9 Hz, 1H), 5,15 (d, J = 9,1 Hz, 1H), 3,28 (d, J = 6,6 Hz, 2H), 1,54 (s, 9H).
Paso 2: Al compuesto obtenido en el paso 1, (548 mg, 1,5 mmol) en DCM (5 mL) se añadió TFA (1,5 mL). La solución resultante se agitó a temperatura ambiente durante 18 h y a continuación se concentró para obtener la sal de TFA de (S)-2-fenil-1-(5-fenil-1,3,4-oxadiazol-2-il)etanamina (i-1). Se utilizó sin purificación adicional. MS m/z 266 (M+H). Tiempo de retención 0,858 min.
Síntesis de 2-fenil-1-(pirimidin-2-il)etanamina (i-2).
Se añadió gota a gota cloruro de bencilmagnesio (1,2 mL, 2,4 mmol) (2 M en THF) a 2-cianopirimidina (210 mg, 2,00 mmol) en tolueno (10 mL) a 0 °C. La reacción se agitó a esta temperatura durante 1 h. A continuación, se añadió 2-butanol (10 mL), seguido de borohidruro sódico (106 mg. 2,80 mmol). La reacción se agitó a t.a. durante 1 h y, a continuación, se desactivó con MeOH (3 mL) y agua. La mezcla se extrajo con EtOAc (2 X 30 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró. El producto crudo se purificó mediante HPLC preparativa (10-30% de acetonitrilo en agua con un 0,05% de TFA) para obtener 2-fenil-1 -(pirimidin-2-il)etanamina (i-2). MS m/z 200,2 (M+H). Tiempo de retención 0,637 min.1H RMN (400 MHz, Acetonitrilo-d3) 58,75 (d, J = 5,0 Hz, 2H), 7,41 (t, J = 4,9 Hz, 1 h ), 7,27 (m, 3H), 7,14 - 7,05 (m, 2H), 4,84 (t, J = 6,7 Hz, 1H), 3,45 (dd, J = 14,1, 6,3 Hz, 1H), 3,33 (dd, J = 14,1,7,1 Hz, 1H).
Síntesis de (S)-2-fenil-1 -(1 H-pirazol-3-il)etanamina (i-3)

Paso 1: Se añadió monohidrato de hidrazina (0,034 mL, 0,69 mmol) a (S,£)-(5-(dietilamino)-3-oxo-1-fenilpent-4-en-2-il)carbamato de f-butilo (60 mg, 0,17 mmol) en MeOH (5 mL). La reacción se calentó a 70 °C durante 2 h y a continuación 50 °C durante 3 días. La mezcla de reacción se concentró, se recuperó en agua y se extrajo con DCM (5 mL X 2). Las fases de DCM se combinaron, se secaron con Na2SO4, se filtraron y se concentraron. El residuo se purificó mediante CCF preparativa en gel de sílice (4% de MeOH en DCM) para obtener (S)-(2-fenil-1-(1H-pirazol-3-il)etil)carbamato de f-butilo. MS m/z 288,2 (M+H). Tiempo de retención 1,310 min. 1H RMN (400 MHz, Cloroformo-d) 5 7,53 - 7,35 (m, 1H), 7,28 - 6,90 (m, 5H), 6,01 (s, 1H), 5,47 - 5,25 (m, 0,3 H), 5,15 - 4,84 (m, 0,7H), 3,40 (s, 1H), 3,09 (d, J = 8,0 Hz, 2H), 1,34 (d, J = 31,2 Hz, 9H).
Paso 2: Una solución del compuesto obtenido en el paso 1 (38 mg, 0,13 mmol) en DCM (2 mL) se trató con TFA (0,5 mL) a t.a. durante 2 h y a continuación se concentró para proporcionar la sal de TFA de (S)-2-fenil-1-(1 H-pirazol-3-il)etanamina (i-3). El producto se utilizó en el paso siguiente sin purificación adicional. MS m/z 188,2 (M+H). Tiempo de retención 0,616 min.
Síntesis de (S)-(3-(2-amino-3-hidroxipropil)fenil)carbamato de f-butilo (i-4)

Paso 1: Se añadió BH3 en THF (1M, 10 mL) a ácido (S)-2-((f-butoxicarbonil)amino)-3-(3-nitrofenil)propanoico (562 mg, 1,81 mmol) en THF (10 mL) con agitación a 0 °C. A continuación, la reacción se agitó a 50 °C durante 1 h. La mezcla de reacción se enfrió a 0 °C, se desactivó con agua, se diluyó con EtOAc y se lavó con K2CO3 acuoso al 10%, se secó con MgSO4, se filtró y se concentró. El crudo se purificó mediante una columna en gel de sílice (30-70% de EtOAchexanos) para obtener (S)-(1-hidroxi-3-(3-nitrofenil)propan-2-il)carbamato de f-butilo como un sólido blanco. MS m/z 319,1 (M+Na). Tiempo de retención 1,183 minutos. 1H RMN (600 MHz, Cloroformo-d) 58,13 - 8,04 (m, 2H), 7,57 (d, J = 7,7 Hz, 1H), 7,46 (dd, J = 8,9, 7,6 Hz, 1H), 4,76 (s, 1H), 3,87 (dc, J = 8,0, 4,6, 4,1 Hz, 1H), 3,69 (dd, J = 10,9, 3,9 Hz, 1H), 3,58 (dd, J = 10,8, 4,7 Hz, 1H), 2,97 (td, J = 13,1, 12,5, 7,3 Hz, 2H), 1,37 (s, 9H).
Paso 2: A (S)-(1-hidroxi-3-(3-nitrofenil)propan-2-il)carbamato de f-butilo (0,31 g, 1,0 mmol) en acetonitrilo (5 mL) se añadió ácido clorhídrico al 10% (5 mL). La mezcla de reacción se agitó a t.a. durante 48 h y a continuación se concentró para proporcionar (S)-2-amino-3-(3-nitrofenil)propan-1-ol en forma de sal de HCl. MS m/z 197,2 (M+H). Tiempo de retención 0,775 min.
Paso 3: La sal de HCl del (S)-2-amino-3-(3-nitrofenil)propan-1-ol (0,243 g, 1,046 mmol) se disolvió en MeOH (10 mL) y se añadió un 10% de paladio sobre carbono (50 mg, 0,047 mmol). Se acopló un globo de hidrógeno de 2 L. La reacción se purgó con H2 tres veces y a continuación se agitó a t.a. durante 1 h. El LCMS indicó que la reacción era completa. La reacción se filtró a través de un lecho de celite y se concentró para proporcionar (S)-2-amino-3-(3-aminofenil)propan-1-ol como una sal de HCl. MS m/z 167,2 (M+H). Tiempo de retención 0,373 min.
Paso 4: Se combinaron la sal de HCl del (S)-2-amino-3-(3-aminofenil)propan-1 -ol (0,212 g, 1,046 mmol) y Boc2 O (228 mg, 1,05 mmol) y dioxano-agua-AcOH (10:9:1, 20mL) y se agitaron a t.a. durante 3 días. El LCMS indicó que la reacción estaba completa en un 75%. Se añadió Boc2O adicional (150 mg) y la reacción se continuó agitando durante 6 h. A continuación, la mezcla de reacción se concentró y se purificó con HPLC preparativa (10-40% de acetonitrilo en agua con un 0,05% de TFA) para proporcionar (S)-(3-(2-amino-3-hidroxipropil)fenil)carbamato de f-butilo (i-4) como un aceite. MS m/z 267,2 (M+H). Tiempo de retención 1,011 min.
Síntesis de (S)-(4-(2-amino-3-hidroxipropil)fenil)carbamato de f-butilo (i-5)
Bocv
Paso 1: A ácido (S)-2-((f-butoxicarbonil)amino)-3-(4-nitrofenil)propanoico (0,80 g, 2,58 mmol) en THF (10 mL) se añadió complejo de sulfuro de dimetilo y borano (1,00 mL, 10,5 mmol) a 0 °C. La reacción se agitó durante 10 min a 0 °C y, a continuación, a t.a. durante 5 h. A continuación, la reacción se desactivó con agua a 0 °C. La mezcla desactivada se repartió entre DCM y Na2CO3 acuoso 1 M. La fase de DCM se separó, se secó con Na2SO4, se filtró y se concentró para proporcionar (S)-(1-hidroxi-3-(4-nitrofenil)propan-2-il)carbamato de f-butilo como un sólido blanco.
MS m/z 319,1(M+Na). Tiempo de retención 1,031 min. 1H RMN (400 MHz, Cloroformo-d) 58,10 (d, J = 8,8 Hz, 2H), 7,33 (d, J = 8,7 Hz, 2H), 4,73 (s, 1H), 3,83 (s, 1H), 3,70 - 3,56 (m, 1H), 3,50 (d, J = 4,6 Hz, 1H), 2,91 (d, J = 7,1 Hz, 2H), 1,32 (s, 9H).
Paso 2: Se agitó el (S)-(1-hidroxi-3-(4-nitrofenil)propan-2-il)carbamato de f-butilo (300 mg, 1,01 mmol) en acetonitrilo (5 mL) y ácido clorhídrico al 10% (5 mL) a t.a. durante 4 h y a continuación se concentró. El residuo se trató con Na2 CO3 acuoso saturado, y se extrajo con DCM-iPrOH (10:1, 10 mL X3). Las fases orgánicas se combinaron, se secaron y se concentraron para proporcionar (S)-2-amino-3-(4-nitrofenil)propan-1 -ol. MS m/z 197,2 (M+H). Tiempo de retención 0,512 min. 1H RMN (400 MHz, Cloroformo-d) 58,32 - 7,92 (m, 2H), 7,41 - 7,21 (m, 2H), 4,18 - 4,00 (m, 1H), 3,66 - 3,49 (m, 2H), 3,49-3,36 (m, 1H), 3,25-3,00 (m, 1H), 3,01 - 2,74 (m, 2H), 2,70-2,65 (m, 1H).
Paso 3: El (S)-2-amino-3-(4-nitrofenil)propan-1 -ol (200 mg, 1,019 mmol) se disolvió en MeOH (10 mL) y se añadió un 10% de Pd/C (50 mg). Se acopló un globo de hidrógeno de 2 L. La reacción se purgó con H2 tres veces y a continuación se agitó a t.a. durante 3 h. La mezcla de reacción se filtró a través de un lecho de celite y, a continuación, se concentró para proporcionar (S)-2-amino-3-(4-aminofenil)propan-1-ol. MS m/z 167,2 (M+H). Tiempo de retención 0,240 min.
Paso 4: El (S)-2-amino-3-(4-aminofenil)propan-1 -ol (168 mg, 1,012 mmol) se disolvió en dioxano (10mL)-agua (9 mL)-AcOH (1 mL) y dicarbonato de f-butilo (0,28 g, 1,28 mmol) se combinaron y se agitaron a t.a. durante 16 h. La mezcla de reacción se concentró entonces y se purificó con ISCO utilizando una columna C18, se eluyó con un 10-40% de acetonitrilo en agua con un 0,05% de TFA para proporcionar TFA de (S)-(4-(2-amino-3-hidroxipropil)fenil)carbamato de f-butilo (i-5). MS m/z 267,2 (M+H). Tiempo de retención 0,764 min. 1H RMN (400 MHz, Acetonitrilo-d3) 57,59 (s, 1H), 7,36 (d, J = 8,5 Hz, 2H), 7,16 (d, J = 8,5 Hz, 2H), 6,14 (s, 3H), 3,69 (dd, J = 11,7, 3,2 Hz, 1H), 3,57 - 3,36 (m, 2H), 2,86 (d, J = 7,1 Hz, 2H), 1,47 (s, 9H).
Síntesis de (S)-(3-(2-amino-3-metoxipropil)fenil)carbamato de f-butilo (i-6)

Paso 1: Se añadió complejo de sulfuro de dimetilo y borano (3,00 mL, 31,6 mmol) a ácido (S)-2-((fbutoxicarbonil)amino)-3-(3-nitrofenil)propanoico (1,5 g, 4,83 mmol) en THF (10 mL) a 0 °C. La reacción se agitó durante 10 min a 0 °C y, a continuación, a t.a. durante 6 h. A continuación, la reacción se desactivó con agua a 0 °C. La mezcla de reacción desactivada se repartió entre DCM y Na2CO3 acuoso 1 M. La fase de DCM se separó, se secó con Na2SO4, se filtró y se concentró para proporcionar (S)-(1 -hidroxi-3-(3-nitrofenil)propan-2-il)carbamato de f-butilo como un sólido blanco. MS m/z 319,1 (M+Na). Tiempo de retención 1,031 min. 1H RMN (400 MHz, Cloroformo-d) 58,14 - 7,97 (m, 2H), 7,57 (dt, J = 7,7, 1,4 Hz, 1H), 7,46 (dd, J = 8,8, 7,6 Hz, 1H), 4,77 (d, J = 14,5 Hz, 1H), 3,87 (s, 1H), 3,69 (dd, J = 10,9, 3,8 Hz, 1H), 3,58 (dd, J = 10,9, 4,7 Hz, 1H), 2,95 (t, J = 6,8 Hz, 2H), 1,37 (s, 9H).
Paso 2: A (S)-(1-hidroxi-3-(3-nitrofenil)propan-2-il)carbamato de f-butilo (0,200 g, 0,675 mmol) en THF/DMF 4:1 (10 mL) a 0°C se añadió NaH (60% en aceite mineral, 0,048 g, 1,2 mmol) lentamente, seguido de yoduro de metilo (0,19 g, 1,3 mmol). La mezcla resultante se agitó a t.a. durante 1 h. La reacción se desactivó cuidadosamente mediante la adición lenta de agua hasta que no se observó burbujeo (H2). El producto crudo se extrajo con EtOAc (10 mL X3). Las fases orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron. El residuo se purificó con ISCO utilizando una columna C18 y se eluyó con un 30-67% de ACN en agua con un 0,05% de TFA para obtener (S)-(1-metoxi-3-(3-nitrofenil)propan-2-il)carbamato de f-butilo como un sólido blanco. MS m/z 333,1(M+Na). Tiempo de retención 1,205 min. 1H Rm N (400 MHz, Cloroformo-d) d 8,10 (dd, J = 4,5, 2,5 Hz, 2H), 7,59 (d, J = 7,5 Hz, 1H), 7,48 (dd, J = 8,8, 7,6 Hz, 1H), 4,96 (d, J = 8,7 Hz, 1H), 4,07 - 3,88 (m, 1H), 3,43 - 3,28 (m, 5H), 2,98 (d, J = 7,2 Hz, 2H), 1,40 (s, 9H).
Paso 3: Se agitó el (S)-(1-metoxi-3-(3-nitrofenil)propan-2-il)carbamato de f-butilo (124 mg, 0,400 mmol) en acetonitrilo (3 mL) y ácido clorhídrico al 10% (3 mL) a t.a. durante 4 h y a continuación se concentró. Se añadió Na2CO3 acuoso saturado al residuo y la mezcla resultante se extrajo con DCM-iPrOH (10:1, 10 mL X3). Las fases orgánicas se combinaron, se secaron y se concentraron para proporcionar (S)-1-metoxi-3-(3-nitrofenil)propan-2-amina. MS m/z 211,2 (M+H). Tiempo de retención 0,622 min.
Paso 4: La (S)-1 -metoxi-3-(3-nitrofenil)propan-2-amina se disolvió en MeOH (10 mL) y se añadió un 10% de Pd/C (50 mg). Se acopló un globo de hidrógeno de 2 L. La reacción se purgó con H2 tres veces y a continuación se agitó a t.a. durante 3 h. La mezcla de reacción se filtró a través de un lecho de celite y a continuación se concentró para proporcionar (S)-3-(2-amino-3-metoxipropil)anilina. MS m/z 181,2 (M+H). Tiempo de retención 0,282 min.
Paso 5: La (S)-3-(2-amino-3-metoxipropil)anilina (62,6 mg, 0,347 mmol) en dioxano (3 mL)-agua (3 mL)-AcOH (0,6 mL) y dicarbonato de f-butilo (0,093 mL, 0,4 mmol) se combinaron y se agitaron a t.a. durante 16 h. La mezcla de reacción se concentró entonces y se purificó con ISCO utilizando una columna C18, se eluyó con un 10-40% de acetonitrilo en agua con un 0,05% de TFA para proporcionar la sal de TFA del (S)-(3-(2-amino-3metoxipropil)fenil)carbamato de f-butilo (i-6). MS m/z 281,2 (M+H). Tiempo de retención 0,856 min. 1H RMN (400 MHz, Acetonitrilo-d3) d 7,61 (s, 1H), 7,40 - 7,12 (m, 3H), 6,89 (dt, J = 7,4, 1,5 Hz, 1H), 6,78 (s, 3H), 3,59 (m, 1H), 3,50 (dd, J = 10,6, 3,4 Hz, 1H), 3,38 (dd, J = 10,6, 6,9 Hz, 1H), 3,33 (s, 3H), 2,90 (d, J=7,5 Hz, 3H), 1,48 (s, 9H).
Síntesis de ácido (3R,4S,5S)-4-((S)-W,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoico (i-7)

Paso 1: Sal de HCl de Dil-OfBu (388 mg, 0,982 mmol), ácido (1 R,3S,4S)-2-(f-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (287 mg, 1,19 mmol), HATU (411 mg, 1,08 mmol) y DieA (0,42 mL, 2,38 mmol) y DMF (5 mL) se combinaron y se agitaron a t.a. durante 30 min. La mezcla de reacción se diluyó con agua (10 mL) y se purificó con RP-C18 ISCO para proporcionar (3R,4S,5S)-4-((S)-W,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoato de f-butilo. MS (m+1) = 582,5, TR de pico de HPLC = 1,542 min
Paso 2: El producto obtenido en el paso 1 (540 mg, 0,93 mmol) en HCl 4 M en 1,4-dioxano (10 mL) se agitó a t.a. durante la noche. La mezcla de reacción se concentró en para proporcionar ácido (3R,4S,5S)-4-((S)-W,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoico. MS (m+1) = 426,2, TR de pico de HPLC = 0,736 min
Paso 3: El producto obtenido en el paso 2 (430 mg, 0,93 mmol), solución de formaldehído al 37% (0,38 mL, 4,7 mmol), ácido acético (0,27 mL, 4,65 mmol), NaBHaCN (585 mg, 9,31 mmol) y MeOH (10 mL) se combinaron y agitaron a t.a. durante 30 min y después se concentraron. El residuo se purificó con RP-C18 ISCO para proporcionar 450 mg de ácido (3R,4S,5S)-4-((s)-W,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoico como una sal de TFA. La sal de TFA se trató con 10 mL de solución de HCl 12 N y se concentró dos veces para proporcionar la sal de HCl del ácido (3R,4S,5S)-4-((S)-W,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoico (i-7). MS (m+1) = 440,2, TR de pico de HPLC = 0,754 min
Síntesis de Boc-Dap-OMe: ((S)-2-((1 R,2R)-1.3-dimetoxi-2-metil-3-oxopropil)pirrolidino-1-carboxilato de ferf-butilo) (i-8)

Se combinaron Boc-Dap-OH (Small Molecules Inc., 3,11 g, 10,8 mmol), K2CO3 (2,99 g, 21,6 mmol), yodometano (2,95 g) y acetona (55 mL). La reacción se agitó a 20 °C durante 2 h. Se añadió un yoduro de metilo adicional (2,28 g) a la reacción y la reacción se agitó a 40 °C durante 3 h. La mezcla de reacción se concentró. El residuo se repartió entre 200 mL de EtOAc y 100 mL de H2O. La fase orgánica se separó, se lavó con 50 mL de NaCl ac. saturado, se secó con MgSO4, se filtró y se concentró, lo que proporcionó Boc-Dap-OMe (i-8) como un aceite amarillo. MS (ESI+) m/z calc 324,2, encontrado 324,2 (M+23). Tiempo de retención 1,245 min.
Síntesis de Dap-OMe: ((2fl,3fiV3-metoxi-2-metil-3-((S)-pirrolidin-2-il)propanoato de metilo) (i-9)

Se combinó Boc-Dap-OMe (3,107 g, 10,3 mmol) con HCl en éter dietílico (2 M, 10 mL) y se concentró. Esta operación se repitió. La reacción fue completa después del 7° tratamiento. Se obtuvo la sal de HCl de Dap-OMe (i-9) como un sólido blanco después de concentrarla. MS (ESI+) m/z calc 202,1, found 202,2 (M+1). Tiempo de retención 0,486 min.
1H RMN (400 MHz, CDCla): 54,065-4,041 (m, 1H), 3,732 (a.s, 1H), 3,706 (s, 3H), 3,615 (s, 3H), 3,368 (a.s, 1H), 3,314 (a.s, 1H), 2,795 (c, 1H, J=6,8Hz), 2,085-1,900 (m, 4H), 1,287 (d, 3H, J=7,2Hz).
Síntesis de (R)-2,4-dihidroxi-3,3-dimetil-W-(3-oxo-3-((2-oxopropii)amino)propil)butanamida (i-10)

Paso 1: Se disolvió ácido pantoteico (50 mg, 0,23 mmol) en DMF (5 mL) y se añadieron azida de difenilfosforilo (98 pL, 0,46 mmol) y 2-(2-metil-1,3-dioxolan-2-il)etanamina (40 mg, 0,34 mmol). La mezcla de reacción se enfrió hasta 0 °C y se añadió trietilamina (79 pL, 0,57 mmol). La mezcla de reacción se agitó a 0 °C durante 10 min, y a continuación se agitó a t.a. durante 24 h. Se añadió EtOAc (50 mL) y se lavó con solución de HCl 0,1 N (20 mL), solución de NaOH 0,1 N (20 mL), salmuera (20 mL), se secó con Na2SO4 y se filtró. El filtrado se concentró al vacío y el residuo se purificó mediante HPLC y se liofilizó para proporcionar (R)-2,4-dihidroxi-3,3-dimetil-N-(3-(((2-metil-1,3-dioxolan-2-il)metil)amino)-3-oxopropil)butanamida. MS (m+1) = 319,2, t R de pico de Hp Lc = 0,466 min
Paso 2: La (R)-2,4-dihidroxi-3,3-dimetil-N-(3-(((2-metil-1,3-dioxolan-2-il)metil)amino)-3-oxopropil)butanamida (46 mg, 0,14 mmol) se disolvió en THF (5 mL) y solución de HCl 3N (3mL) y se agitó a t.a. durante 4 h. Después de enfriar hasta 0 °C, la mezcla de reacción se neutralizó con solución de NaOH 1 N y se concentró a la mitad del volumen al vacío. La mezcla de reacción se purificó con ISCO RP-C18 y se liofilizó para proporcionar (R)-2,4-dihidroxi-3,3-dimetil-N-(3-oxo-3-((2-oxopropil)amino)propil)butanamida (i-10). MS (m+1) = 275,2, TR de pico de HPLC = 0,337 min, 1H-RMN (MeOD, 400 MHz) 53,99 (s, 2H), 3,84 (s, 1H), 3,42~4,47 (m, 2H), 3,42 (d, 1H, J = 11,2 Hz), 3,34 (d, 1H, J = 11,2 Hz), 2,45 (t, 2H, J = 6,8 Hz), 2,10 (s, 3H), 0,87 (s, 6H).
Síntesis de (R)-N-(3-((2-azidoetil)amino)-3-oxopropil)-2,4-dihidroxi-3,3-dimetilbutanamida (i-11)

Se disolvió ácido pantoteico (50 mg, 0,23 mmol) en DMF (5 mL) y se añadieron azida de difenilfosforilo (98 pL, 0,46 mmol) y 2-azidoetanamina (30 mg, 0,34 mmol). La mezcla de reacción se enfrió hasta 0 °C y se añadió trietilamina (79 pL, 0,57 mmol). La mezcla de reacción se agitó a 0 °C durante 10 min, y a continuación se agitó a t.a. durante 24 h. Se añadió EtOAc (50 mL) y se lavó con solución de HCl 0,1 N (20 mL), solución de NaOH 0,1 N (20 mL), salmuera (20 mL), se secó con Na2SO4 y se filtró. El filtrado se concentró al vacío y el residuo se purificó mediante HPLC y se liofilizó para proporcionar (R)-N-(3-((2-azidoetil)amino)-3-oxopropil)-2,4-dihidroxi-3,3-dimetilbutanamida (i-11). MS (m+1) = 288,2, TR de pico de HPLC = 0,504 min, 1H-RMN (MeOD, 400 MHz) 5 3,84 (s, 1H), 3,41~4,47 (m, 3H), 3,31 ~3,35 (m, 5H), 2,40 (t, 2H, J = 6,8 Hz), 0,87 (s, 6H).
Síntesis de (R)-2,4-dihidroxi-3,3-dimetil-N-(3-oxo-3-((3-oxobutil)amino)propil)butanamida (i-12)
OH OH| Y H H
> < ^ o rN' ^ r o N^ Y
(i-12)
Paso 1: Se disolvió sal hemicálcica del ácido pantoteico (100 mg, 0,390 mmol) en CH3 CN (10 mL) y se intercambió por ácido pantoteico utilizando una resina de ácido sulfúrico. Se disolvió ácido pantoteico (10 mg, 0,046 mmol) en DMF (2 mL) y se añadieron azida de difenilfosforilo (20 pL, 0,091 mmol) y 2-(2-metil-1,3-dioxolan-2-il)etanamina (7 mg, 0,005 mmol). La mezcla de reacción se enfrió hasta 0 °C y se añadió trietilamina (16 pL, 0,114 mmol). La mezcla de reacción se agitó a 0 °C durante 10 min, y a continuación se agitó a t.a. durante 24 h. Se añadió EtOAc (50 mL) y se lavó con solución de HCl 0,1 N (20 mL), solución de NaOH 0,1 N (20 mL), salmuera (20 mL), se secó con Na2SO4 y se filtró. El filtrado se concentró al vacío y el residuo se purificó mediante HPLC y se liofilizó para proporcionar (R)-2,4-dihidroxi-3,3-dimetil-N-(3-((2-(2-metil-1,3-dioxolan-2-il)etil)amino)-3-oxopropil)butanamida. Ms (m+1) = 333,2, TR de pico de HPLC = 0,512 min
Paso 2: La (R)-2,4-dihidroxi-3,3-dimetil-N-(3-((2-(2-metil-1,3-dioxolan-2-il)etil)amino)-3-oxopropil)butanamida (6 mg, 0,018 mmol) se disolvió en THF (2 mL) y solución de HCl 3N (1mL) y se agitó a t.a. durante 4 h. Después de enfriar hasta 0 °C, la mezcla de reacción se neutralizó con solución de NaOH 1 N y se concentró a la mitad del volumen al
vacío. La mezcla de reacción se purificó con ISCO RP-C18 y se liofilizó para proporcionar (R)-2,4-dihidroxi-3,3-dimetil-W-(3-oxo-3-((3-oxobutil)amino)propil)butanamida (i-12). MS (m+1) = 289,2, TR de pico de h PlC = 0,362 min, 1H-RMN (MeOD, 400 MHz) 53,83 (s, 1H), 3,37~4,45 (m, 3H), 3,34 (d, 2H, J = 7,2 Hz), 3,32 (d, 1H, J = 3,2 Hz), 2,65 (t, 2H, J = 6,4Hz), 2,34 (t, 2H, J = 6,8 Hz), 2,10 (s, 3H), 0,87 (s, 6H).
Síntesis de 2,4-dihidroxi-3,3-dimetil-W-(3-oxobutil)butanamida (i-13)

Paso 1: Se disolvió hidruro de litio y aluminio (583 mg, 15 mmol) en THF (100 mL) y se enfrió hasta 02C. Se añadió una solución de (±)-pantolactona (1g, 8 mmol) en THF (50 mL) a 0 °C y se agitó a t.a. durante 4h. Se añadió a la mezcla de reacción sulfato sódico anhidro lentamente, seguido de EtOAc (50 mL). La mezcla de reacción se filtró sobre un lecho corto de celite y el filtrado se concentró. El residuo se purificó con ISCO (de un 5% a un 20% de MeOH en CH2 Cl2 ) para proporcionar 3,3-dimetilbutano-1,2,4-triol. 1H-RMN (CDCl3, 400 MHz) 53,71~3,74 (m, 1H), 3,65 (dd, 1H, J = 4,8 y 7,6 Hz), 3,57 (dd, 1H, J = 2,4 y 4,8 Hz), 3,54 (d, 1H, J = 7,2 Hz), 3,48 (d, 1H, J = 7,2 Hz), 0,95 (s, 3H), 0,93 (s, 3H).
Paso 2: Se disolvió 3,3-dimetilbutano-1,2,4-triol (570 mg, 4 mmol) y 1-(dimetoximetil)-4-metoxibenceno (1,16 g, 6 mmol) en CH2 Cl2 (50 mL) y se añadió ácido (7,7-dimetil-2-oxobiciclo[2.2.1]heptan-1-il)metanosulfónico (99 mg, 0,4 mmol). La mezcla de reacción se agitó a t.a. durante 2 h y se añadió trietilamina (0,29 mL, 2 mmol). Después de la concentración, el residuo se purificó con ISCO (de un 0% a un 30% de EtOAc en n-hexano) para proporcionar 2-(4-metoxifenil)-5,5-dimetil-1,3-dioxan-4-il)metanol. 1H-RMN (CDCla, 400 MHz) 57,44 (d, 2H, J = 6,0 Hz), 6,91 (d, 2H, J = 6,0 Hz), 5,47 (s, 1H), 3,90 (s, 1H), 3,81 (s, 3H), 3,59~3,70 (m, 5H), 1,14 (s, 3H), 0,84 (s, 3H).
Paso 3: Se disolvió DMSO (0,27 mL, 4 mmol) en CH2 Cl2 anhidro (20 mL) y se añadió cloruro de oxalilo (0,25 mL, 3 mmol) a -78 °C. La mezcla de reacción se agitó durante 15 min a -78 °C y se añadió lentamente una solución de 2-(4-metoxifenil)-5,5-dimetil-1,3-dioxan-4-il)metanol (485 mg, 2 mmol) en CH2 Cl2 anhidro (1 mL). La mezcla de reacción se agitó a -78 °C durante 30 min y se añadió trietilamina (1,34 mL, 10 mmol). Se dejó que la mezcla de reacción se calentase hasta t.a. y se agitó durante 1 h. La mezcla de reacción se repartió entre agua (50 mL) y CH2 Cl2 (100 mL), y la fase orgánica se lavó con NaHCO3 sat. (50 mL) y salmuera (50 mL), se secó con Na2SO4, se filtró y se concentró. El residuo se purificó con ISCO (de un 20% a un 50% de EtOAc en n-hexano) para proporcionar 2-(4-metoxifenil)-5,5-dimetil-1,3-dioxano-4-carbaldehído. MS (m+1) = 251,2, TR de pico de HPLC = 1,105 min.
Paso 4: Se disolvió 2-(4-metoxifenil)-5,5-dimetil-1,3-dioxano-4-carbaldehído (289 mg, 1 mmol) en acetona/CH2 Cl2 (3:1, 20 mL) y se añadió una solución recién preparada de NaH2PO4.H2O (1593 mg, 12 mmol) y NaCLO (528 mg, 6 mmol) en agua (5 mL) a t.a. La mezcla de reacción se agitó durante 30 min a t.a. y se concentró. El residuo se purificó con ISCO (C18) para proporcionar ácido 2-(4-metoxifenil)-5,5-dimetil-1,3-dioxano-4-carboxílico. MS (m+1) = 267,2, TR de pico de HPLC = 0,957 min.
Paso 5: Se disolvió ácido 2-(4-metoxifenil)-5,5-dimetil-1,3-dioxano-4-carboxílico (40 mg, 0,2 mmol) en DMF (3 mL) y se añadieron HATU (39 mg, 0,2 mmol) y DIEA (0,05 mL, 0,3 mmol). La mezcla de reacción se agitó durante 10 min a t.a. y se añadió 2-(2-metil-1,3-dioxolan-2-il)etanamina (40 mg, 0,3 mmol). La mezcla de reacción se agitó a t.a. durante 1 h y se purificó mediante HPLC preparativa para proporcionar 2-(4-metoxifenil)-5,5-dimetil-W-(2-(2-metil-1,3-dioxolan-2-il)etil)-1,3-dioxano-4-carboxamida. MS (m+1) = 380,2, TR de pico de HPLC = 1,102 min, 1H-r Mn (CDCl3, 400 MHz) 5 7,44 (d, 2H, J = 5,6 Hz), 7,33 (s a, 1H), 6,90 (d, 2H, J = 5,2 Hz), 5,46 (s, 1H), 4,08 (s, 1H), 3,82~3,88 (m, 2H), 3,81 (s, 3H), 3,75 (m, 1H), 3,68 (dd, 2H, J = 7,6 y 16,0 Hz),3,38 (m, 2H), 1,86 (m, 4H), 1,31 (s, 3H), 1,11 (s, 3H), 1,09 (s, 3H).
Paso 5: Se disolvió 2-(4-metoxifenil)-5,5-dimetil-W-(2-(2-metil-1,3-dioxolan-2-il)etil)-1,3-dioxano-4-carboxamida (10 mg, 0,03 mmol) en HCl 3 M en MeOH (1 mL) y se añadió agua (0,1 mL). La mezcla de reacción se concentró al vacío y se purificó con ISCO (C18) para proporcionar 2,4-dihidroxi-3,3-dimetil-W-(3-oxobutil)butanamida (i-13). MS (m+1) = 218,2, TR de pico de HPLC = 0,400 min, 1H-RMN (MeOD-d4, 400 MHz) 53,84 (s, 1H), 3,31~3,44 (m, 4H), 2,70 (t, 2H, J = 4,0 Hz),2,12 (s, 3H), 0,88 (s, 3H).
Procedimiento sintético para los péptidos no conectados
Ejemplo 1: (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1)

Paso 1: A una solución de BocVal-Dil-Dap-OH (1,00 g, 1,75 mmol) en W,W-dimetilformamida (DMF, 20,0 mL) a 0°C se añadieron W,W-diisopropiletilamina (DIEA, 0,677 g, 5,25 mmol) y hexafluorofosfato de 1-[bis(dimetilamino)metileno]-1 H-1,2,3-triazolo[4,5-b]piridinio-3-óxido (HATU)(0,731 g, 1,93 mmol). A continuación, la solución resultante se agitó durante 5 minutos y se añadió a una solución de sal de HCl del éster metílico de L-fenilalanina (0,377 g, 1,75 mmol) y DIEA (0,226 g, 1,75 mmol) en DMF (5,0 mL) a 0 °C. La mezcla de reacción se calentó hasta temperatura ambiente, se agitó durante 30 minutos adicionales y después se concentró. El residuo se purificó mediante HPLC en fase inversa utilizando el sistema ISCO, columna C18, se eluyó con un 20-90% de acetonitrilo-agua para obtener BocVal-Dil-Dap-PheOMe: MS m/z 733,4 (M+1); tiempo de retención 1,47 minutos.
Paso 2: A una solución de BocVal-Dil-Dap-PheOMe (0,683 g, 0,932 mmol) obtenida en el paso 1 en metanol (20 mL) se añadió HCl (4 N en 1,4-dioxano, 16 mL). La mezcla de reacción se agitó a temperatura ambiente durante 7 horas y se concentró. El residuo se disolvió en dioxano y se liofilizó para obtener la sal de HCl de Val-Dil-Dap-PheOMe: MS m/z 633,4 (M+1); tiempo de retención 0,96 minutos.
Paso 3: Se disolvió ácido (1 R,3S,4S)-W-boc-2-azabiciclo[2.2.1]heptano-3-carboxílico (12,6 mg, 0,052 mmol) en DMF (1 mL) en un matraz de fondo redondo de 15 mL. Se añadieron DlEA (12,3 mg, 0,095 mmol) y HATU (19 mg, 0,050 mmol). La mezcla de reacción se agitó durante 10 minutos y se añadió la sal de HCl de Val-Dil-Dap-PheOMe (30 mg, 0,090 mmol) en DMF (1,0 mL). La mezcla de reacción se agitó durante 1 hora. El análisis LCMS indicó que la reacción estaba completa. El crudo se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 20-90% de acetonitrilo-H2O que contenía un 0,05% de ácido trifluoroacético (TFA). Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1 -metoxi-3-(((S)-1 -metoxi-1 -oxo-3-fenilpropan-2-il)amino)-2-metil-3-oxopropil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo: MS m/z 856,6 (M+1); tiempo de retención 1,67 minutos.
Paso 4: El producto obtenido en el paso 3 se disolvió en diclorometano (DCM) (2,0 mL) y se trató con TFA (0,5 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. El análisis LCMS mostró que la reacción estaba completa. La mezcla de reacción se concentró con un evaporador rotatorio para proporcionar el compuesto 1 como una sal de TFA: MS m/z 756,6 (M+1); tiempo de retención 1,22 minutos.
Ejemplo 2: Ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.2]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2)

2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1) (38,4 mg, 0,044 mmol), LiOH monohidratado (50,0 mg, 1,19 mmol) y una mezcla de disolventes de MeOH-H2O (2:1, 4,0 mL). La mezcla se agitó a temperatura ambiente durante 60 horas. El análisis LC-MS indicó que la reacción estaba completa. La mezcla de reacción se concentró y se purificó mediante HPLC en fase inversa, columna C18, se eluyó con acetonitrilo-H2O (10-70%) que contenía un 0,05% de TFA.
Las fracciones que contenían el producto deseado se combinaron y se concentraron para proporcionar el compuesto 2 como una sal de TFA, MS m /z742,5 (M+1). Tiempo de retención 1,15 minutos.
Ejemplo 3: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (3)

Paso 1: A una solución de Boc-Val-Dil-Dap-OH (20,0 mg, 0,035 mmol) en DMF (1,0 mL) en un matraz de fondo redondo de 15 mL se añadió DIEA (9,0 mg, 0,070 mmol), seguida de hexafluorofosfato de N,N,N',N'-tetrametil-0-(1 H-benzotriazol-1-il)uronio (HBTU) (13,3 mg, 0,035 mmol). La mezcla de reacción se agitó durante 10 minutos antes de añadir (1 S,2R)-2-amino-1-fenilpropan-1-ol (6,4 mg, 0,042 mmol) en DMF (1,0 mL) a la mezcla de reacción. La reacción se agitó durante 1 hora. El análisis LCMS indicó que la reacción estaba completa. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener ((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2r )-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamato de ferf-butilo, MS m/z 705,4 (M+1). Tiempo de retención 1,39 minutos.
Paso 2: A una solución de ((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((1 S,2R)-1-hidroxi-1-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamato de ferf-butilo (24,7 mg, 0,035 mmol) en DCM (2,0 mL) se añadió TFA (1,0 mL). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas y se concentró para obtener una mezcla de (S)-2-amino-N-((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)-N,3-dimetilbutanamida (MS m/z 605,4 (M+1) Tiempo de retención 0,96 minutos y el éster de TFA de este (MS m/z 701,4 (M+1)), Tiempo de retención 1,17 minutos. La mezcla se utilizó en el paso siguiente sin purificación adicional.
Paso 3: A una solución de ácido (1 R,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (8,4 mg, 0,035 mmol) en DMF (1,0 mL) se añadieron DIEA (0,024 ml, 0,14 mmol) y HBTU (13,3 mg, 0,035 mmol). La mezcla de reacción se agitó durante 10 minutos y se añadió a una solución de la mezcla de productos obtenida en el paso 2 (25,2 mg, 0,035 mmol) (que contenía el éster de TFA) en DMF (0,5 mL). La mezcla de reacción se mantuvo a temperatura ambiente durante 18 horas y a continuación el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-90% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían los productos deseados se concentraron para obtener una mezcla de (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo (MS m/z 828,5 (M+1)) Tiempo de retención 1,42 minutos y el éster de TFA de este (MS m/z 924,4 (M+1)) Tiempo de retención 1,61 minutos.
Paso 4: A una solución de la mezcla obtenida en el paso 3 en DCM (1,5 mL) se añadió TFA (1,0 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y a continuación se concentró para obtener una mezcla de (1 R,3S,4S)-N-((S)-1 -(((3r ,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (MS m/z 728,4 (M+1)), tiempo de retención 0,99 minutos y 2,2,2-trifluoroacetato de (1 S,2R)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-1-fenilpropilo (MS m/z 824,5 (M+1)), tiempo de retención 1,31 minutos. Esta mezcla se utilizó en el paso siguiente sin purificación adicional.
Paso 5: A una solución de la mezcla obtenida en el paso 4 en MeOH-H2O (1:1, 3,0 mL) se añadió LiOH (10,0 mg, 0,418 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 18 horas y, a continuación, se concentró hasta un volumen total de aproximadamente 1 mL. La mezcla cruda se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-35% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el compuesto 3, MS m/z 728,4 (M+1). Tiempo de retención 0,99 minutos.
Ejemplo 4: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(metilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (4)

Paso 1: Se disolvió ácido (S)-2-((ferí-butoxicarbonil)amino)-3-fenilpropanoico (132,5 mg, 0,50 mmol) en DMF (4 mL). Se añadieron DIEA (0,523 mL, 3,0 mmol) y HATU (475 mg, 1,25 mmol). Después de 15 minutos, se añadió metanosulfonamida (143 mg) y la mezcla de reacción se agitó durante 2 horas. El análisis LC/MS indicó que la reacción se había completado. El producto se purificó mediante Prep-HPLC, columna C18, se eluyó con un 20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 243,1 (M+1). Tiempo de retención 1,023 minutos. El producto se disolvió en DCM (2 mL). Se añadió TFA (2 mL) y se agitó durante 1 hora a temperatura ambiente. El análisis LC/MS indicó que la reacción estaba completa. El producto desprotegido también se purificó mediante Prep-HPLC, se eluyó con un 10-40% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 243,1 (M+1). Tiempo de retención 0,403 minutos. RMN (400 MHz, CD3OD): 57,41-7,30 (m, 5H), 4,10-4,06 (m, 1H), 3,32-3,25 (m, 1H), 3,19 (s, 3H), 3,12-3,07 (m, 1H).
Paso 2: Se disolvió Boc-Val-Dil-Dap (65,5 mg, 0,115 mmol) en DMF (2mL). Se añadieron DIEA (59,2 mg, 80 uL) y HATU (27,7 mg, 0,099 mmol). Después de 10 minutos, se añadió (S)-2-amino-N-(metilsulfonil)-3-fenilpropanamida (18,5 mg, 0,076 mmol) y la mezcla de reacción se agitó durante 1 hora a temperatura ambiente. El análisis LC/MS indicó que la reacción se había completado. El producto se purificó mediante Prep-HPLC, columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 796,4 (M+1). Tiempo de retención 1,388 minutos. El producto se disolvió en HCl en MeOH (3 M, 3 mL). El disolvente se eliminó lentamente. El análisis LC/MS indicó que la reacción se había completado. MS m/z 696,3 (M+1). Tiempo de retención 1,046 minutos.
Paso 3: Se disolvió ácido (1 R,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (14,23 mg, 0,059 mmol) en DMF (2mL). Se añadieron DIEA (22,9 mg, 0,177 mmol) y HATU (20,19 mg, 0,053 mmol). Después de 10 minutos, se añadió el producto del paso previo (21,6 mg, 0,029 mmol) y la mezcla de reacción se agitó durante 2 horas a temperatura ambiente. El análisis LC/MS indicó que la reacción se había completado. El producto se purificó mediante Prep-HPLC, columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de t Fa . Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 919,5 (M+1). Tiempo de retención 1,370 minutos. El producto se disolvió en HCl en MeOH (3 M, 3 mL). El disolvente
se eliminó lentamente. El análisis LC/MS indicó que la reacción se había completado. MS m/z 819,5 (M+1). Tiempo de retención 1,096 minutos.
Ejemplo 5: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(metilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (5)

El compuesto 4 (5 mg, 0,00584 mmol) se disolvió en MeOH (2,0 mL). Se añadieron paraformaldehído (5,97 mg, 0,199 mmol) y ácido acético (6,0 uL). Se añadió cianoborohidruro sódico (12,5 mg, 0,199 mmol) y la mezcla de reacción se calentó hasta 50 °C y se agitó durante 1 hora. El análisis LC/MS indicó que la reacción se había completado. El producto se purificó mediante Prep-HPLC, columna C18, se eluyó con un 10-90% de acetonitrilo-H2 O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 833,5 (M+1). Tiempo de retención 0,983 minutos.
Ejemplo 6: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1H-tetrazol-5-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (6)

Paso 1: Se disolvieron N-Boc-aminonitrilo (0,5 g, 2,03 mmol), azida sódica (0,264 g, 4,06 mmol) y bromuro de zinc (0,229 g, 1,02 mmol) en una mezcla de mezcla de disolventes 2-propanol-agua (1:1, 60 mL) y la mezcla de reacción se agitó a reflujo durante 16 horas. Después de que la reacción se completase, se añadieron 5 mL de ácido cítrico al 10% y 30 mL de acetato de etilo y la agitación se continuó hasta que no quedó sólido. La fase acuosa se extrajo dos veces con acetato de etilo. La fase orgánica combinada se lavó con agua y se secó con Na2SO4 anhidro. El disolvente se eliminó y el residuo se purificó mediante columna en gel de sílice, se eluyó con metanol al 10% en DCM. Las fracciones que contenían el producto deseado se concentraron, se redisolvieron en acetato de etilo, se lavaron con salmuera, se secaron y se concentraron para proporcionar (S)-(2-fenil-1-(2H-tetrazol-5-il)etil)carbamato de ferf-butilo MS m/z 290,2 (M+1). 1H RMN (400 MHz, CDCla) 57,40 - 7,24 (m, 3H), 7,22 - 7,12 (m, 2H), 5,22 - 5,02 (m, 2H), 3,49 -3,24 (m, 2H), 1,40 (s, 9H).
Paso 2: En un matraz de fondo redondo de 15 mL se añadió (S)-(2-fenil-1-(2H-tetrazol-5-il)etil)carbamato de ferf-butilo (30 mg, 0,104 mmol), TFA (2 mL) y DCM (4 mL) para proporcionar una solución transparente que se agitó a temperatura ambiente durante 1 hora. El LCMS mostró que el grupo Boc se había escindido. La solución se concentró para obtener (S)-2-fenil-1-(2H-tetrazol-5-il)etanamina crudo en forma de sal de TFA (M+1 190,2), que se utilizó sin purificación adicional en el siguiente paso.
Paso 3: En un matraz de fondo redondo de 15 mL se añadió Boc-Val-Dil-Dap-OH (59,3 mg, 0,104 mmol) y DIEA (0.072 ml, 0,415 mmol) en DMF (2 mL) proporciona una solución transparente. Se añadió HATU (43,4 mg, 0,114 mmol) y la mezcla de reacción se agitó entonces durante 5 minutos y a continuación se añadió la sal de TFA de (S)-2-fenil1- (2H-tetrazol-5-il)etanamina obtenida en el paso 2 (0,104 mmol). La solución se agitó a temperatura ambiente durante 72 horas. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamato de tert-butilo MS m/z 743,5 (M+1). Tiempo de retención 1,325 minutos.
Paso 4: En un matraz de fondo redondo de 15 mL se añadió ((S)-1 -(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1 -metoxi-2- metil-3-oxo-3-(((S)-2-fenil-1-(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3- metil-1 -oxobutan-2-il)carbamato de tert-butilo (46 mg, 0,056mmol), TFA (2 mL) y DCM (4 mL) para proporcionar una solución transparente que se agitó a temperatura ambiente durante 1 hora. El LCMs mostró que el grupo Boc se había escindido. La solución se concentró para obtener la sal de TFA de (S)-2-amino-W-((3R,4s,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4- il)-W,3-dimetilbutanamida cruda. MS m/z 643,5 (M+1). Tiempo de retención 0,947 minutos, que se utilizó en el paso siguiente sin purificación adicional.
Paso 5: A una solución de ácido (1 R,3S,4S)-2-(tert-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (7,6 mg, 0,032 mmol) en DMF (1 mL) se añadió DiEa (0,014 mL, 0,079 mmol) y HATU (12 mg, 0,032 mmol), que a continuación se añadió a una solución de la sal de TFA de (S)-2-amino-W-((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)-W,3-dimetilbutanamida (20 mg, 0,026 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas y a continuación el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-70% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de tert-butilo como sal de TFA MS m/z 866,6 (M+1). Tiempo de retención 1,407 minutos.
Paso 6: A una solución de la sal de TFA de (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1-(1H-tetrazol-5-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de tert-butilo (10,2 mg, 0,012 mmol) en DCM (2 mL) se añadió TFA (1 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y a continuación se concentró para obtener (1 R,3S,4S)-W-((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (6) como sal de TFA. MS m/z 766,6 (M+1). Tiempo de retención 0,985 minutos.
Ejemplo 7: Ácido (fí)-1-((-2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico. (7)

Paso 1: El ácido ((fí)-1-(((benciloxi)carbonil)amino)-2-feniletil)fosfónico (100 mg, 0,313 mmol), (sintetizado por el siguiente procedimiento descrito en J. Chem. Soc. Perkin Trans. I 1984, 2845) se disolvió en piridina (5 mL) y se añadió n-BuOH (35 mg, 0,46 mmol), seguido de cloruro de pivaloílo (70 mg, 0,58 mmol). El LCMS indicó que la reacción estaba incompleta, por lo tanto, se añadieron tres porciones más de n-BuOH y cloruro de pivaloílo hasta que se consumió todo el ácido fosfínico. A continuación, se añadió una solución de yodo (160 mg, 0,630 mmol) en 2 mL de piridina-H2O (10% de agua) y la mezcla de reacción se agitó durante 20 minutos. El LCMS mostró que la reacción estaba completa. La piridina se eliminó al vacío. Se añadió solución de tiosulfato acuosa y la mezcla de reacción se extrajo con EtOAc. A continuación, la fase de EtOAc se secó, se concentró y se purificó con ISCO (columna C18 de 5,5 g), se eluyó con un 10%-60% de acetonitrilo en agua con un 0,5% de TFA para obtener ((1R)-1-(butoxi(hidroxi)fosforil)-2-feniletil)carbamato de bencilo como un sólido blanco. MS m/z 392,1 (M+1). Tiempo de retención 1,179 minutos. 1H RMN (400 MHz, CD3CN) d 7,42 - 7,18 (m, 8H), 7,18 - 7,00 (m, 2H), 6,10 (s, 1H), 5,07 -4,59 (m, 2H), 4,20-4,35 (m, 1H), 4,13 - 3,93 (m, 2H), 3,15-3,30 (m, 1H), 2,85-2,75 (s, 1H), 1,71 - 1,47 (m, 2H), 1,47 -1,23 (m, 2H), 0,89 (t, J = 7,3 Hz, 3H).
Paso 2: A una solución de ((1R)-1-(butoxi(hidroxi)fosforil)-2-feniletil)carbamato de bencilo (84,7 mg, 0,216 mmol) en MeOH (5 mL) se añadió un 10% de Pd/C (26 mg). Se acopló un globo de hidrógeno y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas. El catalizador se eliminó mediante filtración a través de Celite, y los filtrados se evaporaron hasta sequedad para proporcionar ((R)-1-amino-2-feniletil)fosfonato de hidrógeno y butilo. MS m/z 258,1 (M+1). Tiempo de retención 0,789 minutos, que se utilizó en el paso siguiente sin purificación.
Paso 3: En un matraz de fondo redondo de 15 mL se añadió Boc-Val-Dip-Dap-OH (80 mg, 0,140 mmol) y DIEA (62,9 mg, 0,487 mmol) en DMF (2 mL) para obtener una solución transparente. Se añadió HATU (53 mg, 0,139 mmol) y la mezcla de reacción se agitó durante 5 minutos y a continuación se añadió ((R)-1-amino-2-feniletil)fosfonato de hidrógeno y butilo (41,9 mg, 0,163 mmol). La solución se agitó a temperatura ambiente durante 18 horas. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 40-60% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ((2S)-1 -(((3R,4S,5S)-1 -((2S)-2-((1 R,2R)-3-(((1 R)-1 -(butoxi(hidroxi)fosforil)-2-feniletil)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3
metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamato de ferf-butilo. MS m/z 811,4 (M+1). Tiempo de retención 1,376 minutos.
Paso 4: A una solución de ((2S)-1-(((3fí,4S,5S)-1-((2S)-2-((1fí,2fí)-3-(((1fí)-1-(butoxi(hidroxi)fosforil)-2-feniletil)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamato de ferf-butilo (106 mg, 0,131 mmol) en DCM (3 mL) se añadió TFA (1 mL), y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora y a continuación se concentró. Aproximadamente 2/3 se convirtieron al ácido fosfónico ácido (1-((2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico. MS m/z 655,3 (M+1). Tiempo de retención 0,957 minutos. El otro 1/3 era (1-((2R,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfonato de hidrógeno y butilo. MS m/z 711,4 (M+1). Tiempo de retención 1,038 minutos. La mezcla se utilizó en el paso siguiente sin separación.
Paso 5: A una solución de ácido (1 fí,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (3,8 mg, 0,016 mmol) en DMF (1 mL) se añadió DIEA (6,1 mg, 0,047 mmol) y después HATU (5,9 mg, 0,016 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 10 minutos y a continuación se añadió a una mezcla de la amina del paso 4 (12 mg, 0,016 mmol) que contenía principalmente el ácido fosfónico. La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. El crudo se purificó con ISCO utilizando una columna C18, 4,5 g, se eluyó con un 10-70% de acetonitrilo en agua con un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ácido ((fí)-1-((2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1fí,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico Ms m/z 878,5 (M+1). Tiempo de retención 1,307 minutos, y (1 R,3S,4S)-3-(((2S)-1 -(((3R,4S,5s )-1 -((2S)-2-((1 R,2R)-3-(((1 R)-1 -(butoxi(hidroxi)fosforil)-2-feniletil)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo MS m/z 934,5 (M+1). Tiempo de retención 1,447 minutos.
Paso 6: A una solución de ácido ((fí)-1-((2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1fí,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico (11,0 mg, 0,012 mmol) en DCM (2 mL) se añadió TFA (1 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y a continuación se concentró para obtener ácido ((R)-1-((2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico (7). MS m/z 778,4 (M+1). Tiempo de retención 0,973 minutos.
Ejemplo 8: Ácido (fí)-1-((2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1fí,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (8)

Paso 1: En un matraz de fondo redondo de 15 mL se añadió Boc-Val-Dip-Dap-OH (50 mg, 0,087 mmol) y DIEA (33,9 mg, 0,262 mmol) en DMF (2 mL) para obtener una solución transparente. Se añadió HATU (33,3 mg, 0,087 mmol) y la mezcla de reacción se agitó durante 5 minutos y a continuación se añadió a ácido ((fí)-1-amino-2-feniletil)fosfínico (41 mg, 0,154 mmol), (sintetizado siguiendo el procedimiento descrito en J. Chem. Soc. Perkin Trans. I 1984, 2845). La solución se agitó a temperatura ambiente durante 18 horas. El LCMS indicó la formación del producto deseado. El
crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-50% de acetonitrilo-hbO que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((tert-butoxicarbonil)amino)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico. MS m/z 739,4 (M+1). Tiempo de retención 1,248 minutos.
Paso 2: A una solución de ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((tert-butoxicarbonil)amino)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (69,1 mg, 0,094 mmol) en DCM (2 mL) se añadió TFA (1 mL) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora y a continuación se concentró para obtener ácido ((R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-amino-N, 3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (MS m/z 639,3 (M+1); tiempo de retención 0,851 minutos) que se utilizó sin purificación adicional en el paso siguiente.
Paso 3: A una solución de ácido (1 R,3S,4S)-2-(terí-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (11,3 mg, O, 047 mmol) en DMF (1 mL) se añadió DIEA (0,033 mL, 0,188 mmol), seguido de HATU (17,9 mg, 0,047 mmol). La mezcla de reacción se agitó durante 10 minutos y a continuación se añadió a una solución de ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (35,4 mg, 0,047 mmol) en DMF (1 mL). El LCMS indicó que la reacción se completó en 10 minutos. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-55% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(tert-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico. MS m/z 862,5 (M+1). Tiempo de retención 1,372 minutos.
Paso 4: A una solución de ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(terí-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (60 mg, 0,070 mmol) en DCM (2 mL) se añadió TFA (1 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y a continuación se concentró para proporcionar ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (8). MS m/z 762,5 (M+1). Tiempo de retención 1,220 minutos.
Ejemplo 9: Ácido (R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (9)

A una solución de ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (8) (20 mg, 0,023 mmol) en MeOH (2 mL) se añadió paraformaldehído (10 mg, 0,33 mmol) y ácido acético (0,019 mL, 0,333 mmol), seguido de cianoborohidruro sódico (20 mg, 0,32 mmol). La mezcla de reacción se agitó a 50 °C durante 1 hora y después a temperatura ambiente durante 2 días. El LCMS mostró que la reacción estaba completa. La mezcla de reacción se filtró a través de Celite para eliminar el residuo insoluble y el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-50% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ácido ((R)-1 -((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (9). MS m/z 776,4 (M+1). Tiempo de retención 0,944 minutos.
Ejemplo 10: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (50)

Paso 1: Se añadieron DIEA (0,013 mL, 0,075 mmol) y HATU (18,5 mg, 0,049 mmol) a ácido (1 R,3S,4S)-2-(tertbutoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (9,8 mg, 0,040 mmol) en DMF (1 mL). La mezcla de reacción se agitó durante 5 min y después se añadió a Val-Dil-Dap-OH (17,7 mg, 0,038 mmol) en DMF. La reacción se agitó a t.a. durante 16 h. A continuación, el crudo se purificó mediante HPLC preparativa (30-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(t-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2- metilpropanoico. MS m/z 695,4 (M+H). Tiempo de retención 1,376 min.
Paso 2: Al producto obtenido en el paso 1 (5,9 mg, 0,008 mmol) en DMF (1 mL) se añadieron DIEA (1,1 mg, 0,008 mmol) y HATU (3,8 mg, 0,010 mmol). Después de agitar la reacción durante 5 min, se añadió (S)-2-(S)-2-amino-3-fenilpropan-1-ol (1,9 mg, 0,013 mmol) en DMF. La reacción se agitó a t.a. durante 1 h. El crudo se purificó mediante HPLC preparativa (20-90% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3- metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de t-butilo. MS m/z 828,5 (M+H). Tiempo de retención 1,388 min.
Paso 3: El producto obtenido en el paso 2 (4 mg, 0,005 mmol) en DCM (3 mL) se trató con TFA (1 mL) a t.a. durante 1 h y a continuación se concentró para proporcionar el compuesto (50) como sal de TFA. MS m/z 728,5 (M+H). Tiempo de retención 1,008 min.
Ejemplo 11: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1-hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (51)

Se combinaron (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (50) (6,1 mg, 0,008 mmol), MeOH (2 mL), ácido acético (0,005 mL, 0,09 mmol), paraformaldehído (3 mg, 0,1 mmol), y cianoborohidruro sódico (5 mg, 0,08 mmol) a t.a. y a continuación se agitaron a 50 °C durante 1 h. La mezcla de reacción se enfrió entonces hasta t.a., se filtró y se purificó mediante HPLC preparativa (20-40% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (51) como sal de TFA. MS m/z 742,5 (M+H). Tiempo de retención 1,008 min.
Ejemplo 12: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -(3-aminofenil)-3-hidroxipropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (52)

Paso 1: Se añadieron DIEA (0,105 mL, 0,60 mmol) y HATU (45,5 mg, 0,12 mmol) a ácido (3fí,4S,5S)-4-((S)-/V,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoico (i-7) (57 mg, 0,12 mmol) en DMF (2 mL). La mezcla de reacción se agitó a t.a. durante 5 min y después se añadió DapOMe (i-9) (28,5 mg, 0,12 mmol) en DMF (1 mL). La mezcla de reacción se agitó a t.a. durante 1 h y después se purificó mediante HPLC preparativa (10-50% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoato de metilo. MS m/z 623,5 (M+H). Tiempo de retención 1,225 min.
Paso 2: Se añadió LiOH (30 mg, 1,25 mmol) al producto obtenido en el paso 1 (43,2 mg, 0,059 mmol) en MeOH-H2O (1:1, 4 mL). La mezcla de reacción se agitó a TA durante 18 h, se concentró y se acidificó con HCl (1 N, 1 mL). El crudo se purificó mediante HPLC preparativa (10-38% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (2R,-3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico como sal de TFA. MS m/z 609,5 (M+H). Tiempo de retención 0,962 min.
Paso 3: Al producto obtenido en el paso 2 (45,7 mg, 0,063 mmol) en DMF (1 mL) se añadieron DIEA (0,055 mL, 0,32 mmol) y HATU (24,0 mg, 0,063 mmol). La mezcla de reacción se agitó a t.a. durante 10 min y a continuación se añadió a la sal de TFA del (S)-(3-(2-amino-3-hidroxipropil)fenil)carbamato de f-butilo (i-4) (24,1 mg, 0,063 mmol) en DMF (1 mL). La mezcla de reacción se agitó a t.a. durante 1 h y después se concentró. El crudo se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (3-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-hidroxipropil)fenil)carbamato de f-butilo como sal de TFA. MS m/z 857,5 (M+H). Tiempo de retención 1,145 min.
Paso 4: Una solución del producto obtenido en el paso 3 (61,4 mg, 0,063 mmol) en acetonitrilo-agua (1:1,4 mL) con un 5% de HCl se agitó a t.a. durante 24 h. A continuación, la mezcla de reacción se concentró y se purificó mediante HPLC preparativa (10-30% de acetonitrilo-H2O que contenía un 0,05% de TFA) para proporcionar el compuesto (52) como sal de TFA. Ms m/z 757,5 (M+H). Tiempo de retención 0,744 min.
Ejemplo 13: (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (53)

A la sal de TFA de (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1) (55,4 mg, 0,064 mmol) en MeOH (5 mL) se añadieron ácido acético (0,009 mL, 0,2 mmol), paraformaldehído (24 mg, 0,79 mmol) y a continuación cianoborohidruro sódico (25 mg, 0,40 mmol). La mezcla de reacción se agitó a 40 °C durante 16 h, se filtró, se concentró y se purificó mediante HPLC preparativa (10-45% de acetonitrilo-agua con un 0,05% de TFA) para obtener el compuesto (53) como sal de TFA. MS m/z 770,3 (M+H). Tiempo de retención 1,100 min.
Ejemplo 14: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (9d)

Se disolvió la sal de TFA del compuesto (53) (50,8 mg, 0,057 mmol) en MeOH-H2O (1:1,5 mL) y se añadió LiOH (20 mg, 0,835 mmol). La reacción se agitó a 40 °C durante 1 h. Se eliminó el MeOH mediante evaporación. Se añadió agua al residuo y se acidificó con AcOH (0,040 mL). El crudo se purificó mediante HPLC preparativa (27-33% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (54) como sal de TFA. MS m/z 756,5 (M+H). Tiempo de retención 0,985 min.
Ejemplo 15: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (55)

Se añadieron DIEA (10,2 mg, 0,014 mL) y HATU (7,7 mg, 0,020 mmol) a la sal de TFA del ácido (2R, 3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (Paso 2, Ejemplo 12) (12,3 mg, 0,017 mmol) en DMF (1 mL). La reacción se agitó durante 15 min, y posteriormente se añadió (S)-2-amino-3-fenilpropano-1-sulfonamida (4,3 mg, 0,020 mmol) en DMF (0,5 mL). La reacción se agitó a t.a. durante 1 h adicional. El crudo se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA para obtener el compuesto (55). MS m/z 805,5 (M+1). Tiempo de retención 0,965 min.
Ejemplo 16: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (56)

Paso 1: A Boc-Dap-OH (21,6 mg, 0,075 mmol) en DMF (2 mL) se añadieron DIEA (48,5 mg, 0,066 mL) y HATU (26,2 mg, 0,069 mmol). La reacción se agitó durante 15 min, y posteriormente se añadió (S)-2-amino-3-fenilpropano-1-sulfonamida (13,4 mg, 0,063 mmol). La mezcla de reacción se agitó a t.a. durante 2 h y después se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1-fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-carboxilato de f-butilo. MS m/z 484,2 (M+1). Tiempo de retención 1,130 min.
Paso 2: Se disolvió (S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-carboxilato de f-butilo (28,5 mg, 0,059 mmol) en HCl metanólico (3 M, 3 mL). El disolvente se eliminó lentamente bajo una corriente de N2 seguida de presión reducida durante la noche para proporcionar (2R,3R)-3-metoxi-2-metil-N-((S)-1-fenil-3-sulfamoilpropan-2-il)-3-((S)-pirrolidin-2-il)propanamida como sal de HCl. MS m/z 384,2 (M+1). Tiempo de retención 0,630 min.
Paso 3: A Cbz-Val-Dil-OH (28,7 mg, 0,066 mmol) en DMF (1 mL) se añadieron DIEA (0,048 mL) y HATU (22,9 mg, 0,060 mmol). La reacción se agitó durante 15 min, y posteriormente se añadió (2R,3R)-3-metoxi-2-metil-N-((S)-1-fenil-3-sulfamoilpropan-2-il)-3-((S)-pirrolidin-2-il)propanamida (23 mg, 0,055 mmol) en DMF (1mL). La mezcla de reacción se agitó a t.a. durante 2 h y se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-1-fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamato de bencilo. MS m/z 802,4 (M+1). Tiempo de retención 1,298 min.
Paso 4: El producto obtenido en el paso 3 (24,6 mg, 0,031 mmol), 10% de Pd-C (32,7 mg) y EtOAc (3 mL) se combinaron y se agitaron en hidrógeno durante 8 h a t.a. La mezcla de reacción se filtró y se concentró para proporcionar (S)-2-amino-N-((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)-N,3-dimetilbutanamida. MS m/z 668,4 (M+1). Tiempo de retención 0,888 min.
Paso 5: Se combinaron ácido (1 R,3S,4S)-2-(f-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (7,0 mg, 0,029 mmol), DMF (1 mL), DIEA (0,021 mL) y HATU (10,1 mg, 0,027 mmol) y se agitaron a t.a. durante 15 min, y a continuación se añadió el producto obtenido en el paso 4 (16,2 mg, 0,024 mmol) en DMF (1 mL). La mezcla de reacción se agitó a t.a. durante 2 h y se purificó mediante HPLC preparativa (30-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-1-fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de f-butilo. MS m/z 891,5 (M+1). Tiempo de retención 1,319 min.
Paso 6: El producto obtenido en el paso 5 (13,2 mg, 0,015 mmol) se disolvió en HCl metanólico (3 M, 3 mL). El disolvente se eliminó lentamente bajo una corriente de N2 seguido de presión reducida durante la noche para proporcionar (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (56) en forma de sal de HCl. MS m/z 791,5 (M+1). Tiempo de retención 0,923 min.
Ejemplo 17: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1 H-imidazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (57)

Se añadieron DIEA (33 mg, 0,26 mmol) y HATU (19 mg, 0,051 mmol) al ácido (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(f-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5
metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (40 mg, 0,043 mmol) en DMF (2 mL). La reacción se agitó a t.a. durante 15 min, y posteriormente se añadió (S)-2-fenil-1-(5-fenil-1 H-imidazol-2-il)etanamina (22,4 mg, 0,085 mmol). La reacción se agitó a t.a. durante 1 h. El crudo se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1 H-imidazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de f-butilo. MS m/z 940,5 (M+1). Tiempo de retención 1,333 min. Este producto (13,9 mg, 0,015 mmol) se disolvió en HCl metanólico (3 M, 3 mL). El disolvente se eliminó lentamente bajo una corriente de N2 seguido de presión reducida durante la noche para proporcionar el compuesto (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1 H-imidazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (57) en forma de sal de HCl. MS m/z 840,5 (M+1). Tiempo de retención 0,936 min.
Ejemplo 18: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-3-(((S)-1 -metoxi-3-fenilpropan-2-il)amino)-2-metil-3-oxopropil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (58)

El compuesto (58) se preparó mediante el procedimiento descrito para el compuesto (57) utilizando la sal de HCl de la (S)-1-metoxi-3-fenilpropan-2-amina en lugar de (S)-2-fenil-1-(5-fenil-1 H-imidazol-2-il)etanamina. MS m/z 742,5 (M+1). Tiempo de retención 0,997 min.
Ejemplo 19: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-pirazol-3-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (59)

El compuesto (59) se preparó mediante el procedimiento descrito para el compuesto (57) utilizando la sal de HCl de la (S)-2-fenil-1-(1H-pirazol-3-il)etanamina en lugar de (S)-2-fenil-1-(5-fenil-1H-imidazol-2-il)etanamina. MS m/z 764,5 (M+1). Tiempo de retención 0,959 min.
Ejemplo 20: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-Metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(pirimidin-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (60), y (1 R,3S,4s )-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((R)-2-fenil-1-(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (61)

fenil-1 -(pirimidin-2-il)etanamina en lugar de (S)-2-fenil-1-(5-fenil-1H-imidazol-2-il)etanamina. Los compuestos (60) y (61) protegidos con Boc se separaron en HPLC preparativa (30-65% de acetonitrilo-H2O que contenía un 0,05% de TfA). La eliminación del grupo Boc de los compuestos (60) y (61) protegidos con Boc proporcionó ((1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (60) y (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((R)-2-fenil-1-(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (61) en forma de sal de HCl, respectivamente. MS m/z 776,5 (M+1). Tiempo de retención 1,001 min (60) y 1,016 min (61).
Ejemplo 21: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (62)

El compuesto (62) se preparó en el procedimiento descrito para el compuesto (57) utilizando la (S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-il)etanamina en lugar de (S)-2-fenil-1 -(5-fenil-1 H-imidazol-2-il)etanamina con en el paso 1. Después de la eliminación del grupo Boc, se obtuvo el compuesto (62) como sal de HCl. MS m/z 842,5 (M+1). Tiempo de retención 1,112 min.
Ejemplo 22: (1 R,3S,4S)-2-Cianometil)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (63)

Paso 1: Se añadieron DIEA (104 mg, 0,80 mmol) y HATU (122 mg, 0,32 mmol) a una solución de ácido (1R,3S,4S)-2- (ferí-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (78 mg, 0,32 mmol) en DMF (3 mL). La mezcla de reacción se agitó a t.a. durante 5 min y después se añadió a Val-Dil-Dap-OMe (130 mg, 0,27 mmol) en DMF (2 mL). A continuación, la mezcla de reacción se agitó a t.a. durante 1 h y se concentró. Se añadió solución de bicarbonato sódico saturada (5 mL) al residuo y el producto se extrajo con DCM (10 mL X 3). Las fases orgánicas se combinaron, se secaron y se concentraron para obtener (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-1,3-dimetoxi-2-metil-3- oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferí-butilo. El producto se utilizó en el paso siguiente sin purificación adicional. MS m/z 710,5 (M+H). Tiempo de retención 1,440 min.
Paso 2: El producto obtenido en el paso 1 (190 mg, 0,27 mmol) en DCM (10 mL) se trató con TFA (2 mL) a t.a. durante 3 h, y a continuación se concentró para proporcionar (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoato de metilo en forma de sal de TFA. MS m/z 610,5 (M+H). Tiempo de retención 1,003 min. El producto se utilizó en el paso siguiente sin purificación adicional.
Paso 3: Al producto obtenido en el paso 2 (193 mg, 0,27 mmol) en MeOH (10 mL) se añadieron ácido acético (0,015 mL, 0,27 mmol), paraformaldehído (40 mg, 1,3 mmol) y cianoborohidruro sódico (84 mg, 1,4 mmol). La reacción se agitó a 50 °C durante 16 h. El LCMS indicó que aproximadamente un 90% se había convertido en el compuesto cianometilado y aproximadamente un 10% se convirtió en el compuesto metilado. La mezcla de reacción se filtró y se purificó mediante HPLC preparativa (20-60% de acetonitrilo-H2O que contenía un 0,05% de TFA). Las fracciones que contenían el aducto ciano se recogieron y se concentraron para proporcionar (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(cianometil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoato de metilo en forma de sal de TFA. MS m/z 648,5 (M+H). Tiempo de retención 1,261 min.
Paso 4: Al producto (0,12 g, 0,16 mmol) obtenido en el paso 3 en MeOH-H2O (1:1,5 mL) se añadió LiOH (50 mg, 2,09 mmol). La mezcla de reacción se agitó a t.a. durante 16 h y después se acidificó con 0,2 mL de HCl al 10%. El grupo ciano se hidrolizó parcialmente para formar un producto carbamoilmetilado además del producto cianometilo. La reacción se concentró y los dos productos se aislaron mediante HPLC preparativa (20-50% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (2R,3R)-3-((S)-1-((3R,4s,5S)-4-((S)-2-((1R,3S,4S)-2-(cianometil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico, MS m/z 634,4 (M+H), tiempo de retención 1,138 min, y ácido (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(2-amino-2-oxoetil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico en forma de sales de TFA. MS m/z 652,4 (M+H). Tiempo de retención 0,888 min.
Paso 5: A la sal de TFA del ácido (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1R,3S,4S)-2-(cianometil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (6 mg, 0,008 mmol) en DMF se añadió DIEA (3,1 mg, 0,024 mmol) y HATU (3,7 mg, 0,0096 mmol). La reacción se agitó a t.a. durante 5 min, y posteriormente se añadió (S)-2-amino-3-fenilpropan-1-ol (2,4 mg, 0,016
mmol). La reacción se agitó a t.a. durante 1 h. El crudo se purificó mediante HPLC preparativa (10-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (63) en forma de sal de TFA. MS m/z 767,5 (M+H). Tiempo de retención 1,189 min.
Ejemplo 23: (1 R,3S,4S)-2-(2-amino-2-oxoetil-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (64)

A la sal de TFA del ácido (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(2-amino-2-oxoetil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (paso 4, Ejemplo 22) (6,1 mg, 0,008 mmol) en DMF se añadieron DIEa (3,1 mg, 0,024 mmol) y HATU (3,7 mg, 0,0096 mmol). La reacción se agitó a t.a. durante 5 min, y posteriormente se añadió (S)-2-amino-3-fenilpropan-1-ol (2,4 mg, 0,016 mmol). La reacción se agitó a t.a. durante 1 h. El crudo se purificó mediante HPLC preparativa (10-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (64) en forma de sal de TFA. MS m/z 785,5 (M+H). Tiempo de retención 0,951 min.
Ejemplo 24: (1 R,3S,4S)-2-Acetil)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (65)

Paso 1: A la sal de TFA del (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoato de metilo (paso 2, Ejemplo 63) (13 mg, 0,021 mmol) en DCM (2 mL) se añadieron DIEA (0,014 mL, 0,082 mmol) y anhídrido acético (0,0039 mL, 0,041 mmol). La reacción se agitó a t.a. durante 1 h. Se añadió Na2CO3 acuoso (2 M) y la mezcla de reacción se extrajo con DCM (5 mL X 3). Las fases orgánicas se combinaron, se secaron con Na2SO4, se filtraron y a continuación se concentraron para obtener (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-acetil-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoato de metilo. El producto se utilizó en el paso siguiente sin purificación adicional. MS m/z 651,5 (M+H). Tiempo de retención 1,188 min.
Paso 2: Al producto obtenido en el paso 1 en MeOH:H2O (1:1,2 mL) se añadió LiOH (10 mg, 0,42 mmol). La reacción se llevó a cabo a t.a. durante 16 h. La mezcla de reacción se concentró y se añadieron 0,040 mL de HOAc. El crudo se purificó mediante HPLC preparativa (10-50% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (2R,-3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-acetil-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico. MS m/z 637,4 (M+H). Tiempo de retención 1,158 min.
Paso 3: A una solución del producto obtenido en el paso 2 (5 mg, 0,008 mmol) en DMF (1 mL) se añadieron DIEA (2,7 mg, 0,021 mmol) y HATU (3,9 mg, 0,010 mmol). La reacción se agitó a t.a. durante 5 min, y posteriormente se añadió (S)-2-amino-3-fenilpropan-1 -ol (1,6 mg, 0,010 mmol). La reacción se agitó a t.a. durante 1 h y a continuación el crudo se purificó mediante HPLC preparativa (10-60% de acetonitrilo-H2O que contenía un 0,05% de TFA para obtener el compuesto (65). MS m/z 770,5 (M+H). Tiempo de retención 1,121 min.
Ejemplo 25: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,4S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(4-fenil-1 H-imidazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (66)

Se añadieron DIEA (0,0097 mL) y HATU (3,2 mg, 0,0083 mmol) a la sal de TFA del ácido (2R, 3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (Paso 2, Ejemplo 12) (4,0 mg, 0,0055 mmol) en DMF (0,5 mL). La reacción se agitó durante 15 min a t.a., y se añadió (S)-2-fenil-1 -(4-fenil-1 H-imidazol-2-il)etanamina (2,9 mg, 0,011 mmol) en DMF (0,5 mL). La mezcla de reacción se agitó durante 2 h a t.a. y a continuación se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (66). MS m/z 854,5 (M+1). Tiempo de retención 0,980 min.
Ejemplo 26: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-3-(((S)-1 -metoxi-3-fenilpropan-2-il)amino)-2-metil-3-oxopropil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (67)
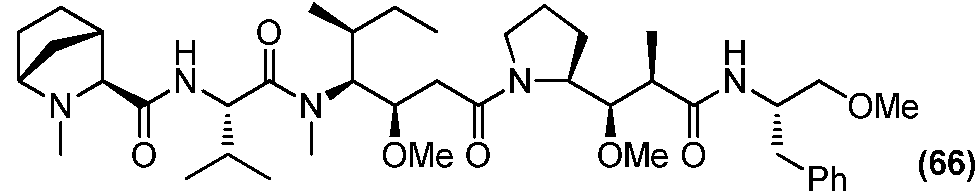
El compuesto (67) se obtuvo mediante el método descrito para el compuesto (66) utilizando la sal de HCI de la (S)-1-metoxi-3-fen¡lpropan-2-amina en lugar de (S)-2-fenil-1-(5-fen¡l-1H-¡m¡dazol-2-¡l)etanam¡na. MS m/z 756,5 (M+1). Tiempo de retención 1,046 min.
Ejemplo 27: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-pirazol-3-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (68)

El compuesto (68) se preparó mediante el método descrito para el compuesto (66) utilizando la sal de HCI de la (S)-2-fenil-1-(1H-pirazol-3-il)etanamina en lugar de (S)-2-fenil-1-(5-fenil-1H-imidazol-2-il)etanamina. MS m/z 778,5 (M+1). Tiempo de retención 0,998 min.
Ejemplo 28: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-Metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1- (pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2- azabiciclo[2.2.1]heptano-3-carboxamida (69), y (1 R,3S,4s )-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((R)-2-fenil-1-(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (70)

(pirimidin-2-il)etanamina en lugar de (S)-2-fenil-1-(5-fenil-1H-imidazol-2-il)etanamina después de una separación por HPLC preparativa (30-55% de acetonitrilo-H2O que contenía un 0,05% de TFA) de los dos diastereoisómeros. MS m/z 790,5 (M+1). Tiempo de retención 1,016 min y 1,043 min para los compuestos (69) y (70), respectivamente.
Ejemplo 29: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (71)

El compuesto (71) se obtuvo mediante el método descrito para el compuesto (66) utilizando la (S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-¡l)etanamina en lugar de (S)-2-fen¡l-1-(5-fenil-1H-¡m¡dazol-2-¡l)etanam¡na. MS m/z 856,5 (M+1). Tiempo de retención 1,120 min.
Ejemplo 30: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -(3-aminofenil)-3-metoxipropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (72)

Se añadieron DIEA (0,012 mL, 0,069 mmol) y HATU (7,89 mg, 0,021 mmol) al ácido (2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (Paso 2, Ejemplo 12) (10 mg, 0,014 mmol) en DMF (2 mL). La reacción se agitó a t.a. durante 5 min y a continuación se añadió la sal de TFA del (S)-(3-(2-amino-3-metoxipropil)fenil)carbamato de f-butilo (10,9 mg, 0,028 mmol). La reacción se agitó a t.a. durante 1 h y después el crudo se purificó mediante HPLC preparativa (20-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (3-((S)-2-((2R,-3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-metoxipropil)fenil)carbamato de f-butilo como sal de TFA. MS m/z 871,5 (M+H). Tiempo de retención 1,157 min. A este producto (13,6 mg, 0,014 mmol) en acetonitrilo (2 mL) se añadió ácido clorhídrico al 10% (2 mL). La mezcla de reacción se agitó a t.a. durante 2 h y a continuación se concentró para obtener (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -(3-aminofenil)-3-metoxipropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (72) como sal de HCl. MS m/z 771,5 (M+H). Tiempo de retención 0,883 min.
Ejemplo 31: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1-(4-aminofenil)-3-hidroxipropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (73)

(2-amino-3-hidroxipropil)fenil)carbamato de f-butilo en lugar de la sal de TFA del (S)-(3-(2-amino-3-metoxipropil)fenil)carbamato de f-butilo. MS m/z 757,5 (M+H). Tiempo de retención 0,787 min.
Ejemplo 32: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (74)
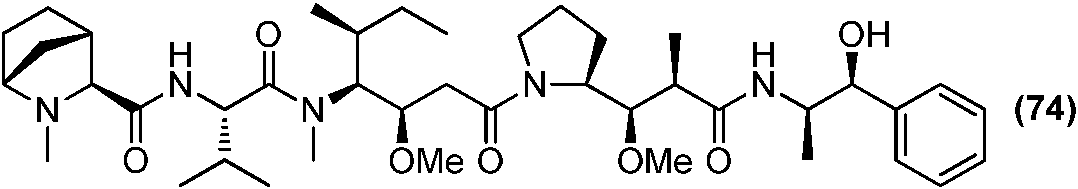
Se añadieron DIEA (0,006 mL, 0,035 mmol) y HATU (4,0 mg, 0,010 mmol) al ácido (2R, 3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (5 mg, 0,007 mmol) en DMF (1 mL). La reacción se agitó a t.a. durante 5 min, y posteriormente se añadió (1 S,2R)-(+)-norefedrina (3 mg, 0,02 mmol). La mezcla de reacción se agitó a t.a. durante 1 h y después se purificó mediante HPLC preparativa (20-50% de acetonitrilo-H2O que contenía un 0,05% de TFA). Las fracciones que contenían el producto deseado se combinaron y se añadió ácido clorhídrico al 10%. La concentración proporcionó el compuesto (74) en forma de sal de HCl. MS m/z 742,5 (M+H). Tiempo de retención 1,005 min.
Procedimiento sintético para los compuestos de Fórmula (I) conectados en el extremo N term inal ejemplo de Carga
Ejemplo de carga 33: Ácido (S)-2-((-2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (10)

A una solución de ácido 6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanoico (EMCA)(1,2 mg, 0,0058 mmol) en DMF (1,0 mL) en un matraz de fondo redondo de 15 mL se añadió DIEA (3,0 mg, 0,023 mmol), seguida de HATU (2,7 mg, 0,0070 mmol). La mezcla de reacción se agitó durante 10 minutos antes de añadir una solución de la sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2) (5,0 mg, 0,0058 mmol) en DMF (1,0 mL) a la mezcla de reacción. La mezcla de reacción se agitó durante 1 hora. El análisis LCMS indicó que la reacción estaba completa. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-80% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto se concentraron para obtener el compuesto 10, MS m/z 935,6 (M+1). Tiempo de retención 1,17 minutos.
Ejemplo de carga 34: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (11)

Paso 1: A un matraz de fondo redondo de 100 mL se añadió 6-amino-1 -hexanol (1,00 g, 6,44 mmol) en una solución de NaHCO3 acuoso saturado (12,0 mL). La mezcla se enfrió a 0 °C y se añadió W-metoxicarbonilmaleimida (0,750 g, 6,44 mmol). La mezcla de reacción se agitó a 0 °C durante 1,5 horas. Después, la mezcla de reacción se acidificó a 0 °C con HCl 2 M hasta pH 1. La mezcla de reacción acidificada se extrajo con acetato de etilo (AcOEt). La fase orgánica se concentró. El residuo se disolvió en DCM, se cargó en una columna de gel de sílice, y se eluyó con MeOH/DCM (0-4%) para obtener 1-(6-hidroxihexil)-1H-pirrol-2,5-diona como un sólido blanco, MS m/z 198,2 (M+1). 1H RMN (400 MHz, CDCla): 56,68 (s, 2H), 3,63 (t, d = 6,4 Hz, 2H), 3,52 (t, d = 7,2 Hz, 2H), 1,63-1,52 (m, 4H), 1,43-1,28 (m, 4H).
Paso 2: Se agregó reactivo de Dess-Martin (618 mg, 1,44 mmol) a 1-(6-hidroxihexil)-1 H-pirrol-2,5-diona (237 mg, 1,20 mmol) en DCM (10,0 mL). Después de 1 hora a temperatura ambiente, la mezcla de reacción se diluyó con DCM (10 mL) y se filtró. El filtrado se concentró y se purificó mediante ISCO (gel de sílice, 0-20% de EtOAc/Hexano) para proporcionar 6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanal como un aceite incoloro, MS m/z 196,2 (M+1). 1H RMN (400 MHz, CDCla): 59,76 (t, J = 1,6 Hz, 1H), 6,69 (s, 2H), 3,52 (t, J= 7,2 Hz, 2H), 2,43 (td, J = 7,2 Hz, 1,6 Hz, 2H), 1,70-1,56 (m, 4H), 1,36-1,28 (m, 2H).
Paso 3: El compuesto 2 (5,0 mg, 0,0067 mmol) y 6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanal (6,6 mg, 0,034mmol) se disolvieron en MeOH (1,0 mL). Se añadió cianoborohidruro sódico (4,2 mg, 0,067 mmol). La mezcla de reacción se agitó durante 3 horas a temperatura ambiente. El análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico 11, MS m/z 921,6 (M+1). Tiempo de retención 1,07 minutos.
Ejemplo de carga 35: Ácido ((S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(((4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencil)oxi)carbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (12)

En un matraz de fondo redondo de 15 mL a temperatura ambiente se añadieron MC-Val-Cit-PABC-PNP (5,2 mg, 0,0070 mmol), sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2) (5,0 mg, 0,0058 mmol) y 1-hidroxi-7-azabenzotriazol (HOAT) (0,6 mg, 0,005 mmol), seguido de piridina-DMF (1:4, 1,25 mL). A la solución resultante se añadió DIEA (2,3 mg, 0,018 mmol). La mezcla de reacción se agitó durante 72 horas, momento en el que el compuesto 2 ya se había consumido. La mezcla de reacción se concentró y el residuo se purificó mediante HPLc en fase inversa, columna C18, se eluyó con un 20-80% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener el compuesto 12, MS m/z 1340,7 (M+1). Tiempo de retención 1,15 minutos.
Ejemplo de carga 36: Ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-(2-(2-azidoetoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (13)

Paso 1: Al ácido 3-(2-(2-azidoetoxi)etoxi)propanoico (6,6 mg, 0,033 mmol) en DMF (2 mL) se añadieron DIEA (0,011 mL, 0,065 mmol) y HATU (10,3 mg, 0,027 mmol). Después de 15 minutos, se añadió el compuesto 1 (8,2 mg, 0,010 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. El análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1R,3S,4S)-2-(3-(2-(2-azidoetoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (MS m/z 941,3 (M+1). Tiempo de retención 1,30 minutos.
Paso 2: El producto éster del paso 1 se disolvió en acetonitrilo (0,3 mL) y H2O (0,2 mL). Se añadió NaOH acuoso (1,0 N, 0,15 mL). La mezcla de reacción se agitó durante 30 minutos. El crudo se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 13 ácido (S)
2-((-2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1fí,3S,4S)-2-(3-(2-(2-azidoetoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (MS m/z 927,5 (M+1). Tiempo de retención 1,21 minutos.
Ejemplo de carga 37: Ácido (S)-2-((2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-(2-(2-(4-((2,5-dioxo-2,5-dihidro-1 AV-pirrol-1 -il)metil)-1 H-1,2,3-triazol-1-il)etoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (14)

azidoetoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (13)(5,4 mg, 0,058 mmol),1-(prop-2-in-1-il)-1 H-pirrol-2,5-diona (1,6 mg, 0,012 mmol) y CuSÜ4 (0,7 mg, 0,005 mmol) en DMF (1,2 mL) y H2O (0.3 mL) se trató con la sal sódica del ácido L-ascórbico (2,6 mg, 0,015 mmol) y se agitó a temperatura ambiente durante 2 horas. El análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-(2-(2-(4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)metil)-1 H-1,2,3-triazol-1 -il)etoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico 14, MS m/z 1062,5 (M+1). Tiempo de retención 1,15 minutos.
Ejemplo de carga 38: Ácido (S)-2-((2R,2fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-(((2-(2-(2-(vinilsulfonil)etoxi)etoxi)etil)sulfonil)etil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (15)

Paso 1: se añadió f-BuOK (119 mg, 1,10 mmol) a una solución de divinilsulfona (1,60 g, 13,5 mmol) y etilenglicol (330 mg, 5,32 mmol) en THF (100 mL). La mezcla de reacción se agitó a temperatura ambiente durante 18 horas. El disolvente se eliminó a presión reducida para proporcionar un crudo que se purificó mediante cromatografía en columna de gel de sílice (EtOAc-Hexanos 2:1 a 3:1) para obtener ((2-(2-(2-vinilsulfoniletoxi)etoxi)etil)sulfonil)eteno como un jarabe incoloro. 1H RMN (400 MHz, CDCh) d 6,75 (dd, J = 9,9 Hz, 16,6 Hz, 2H), 6,39 (d, J = 16,6 Hz, 2H), 6,09 (d, J = 9,9 Hz, 2H), 3,88 (t, J = 5,7 Hz, 4H), 3,61 (s, 4H), 3,24 (t, J = 5,7 Hz, 4H).
Paso 2: A una solución de ((2-(2-(2-vinilsulfoniletoxi)etoxi)etil)sulfonil)eteno (13,3 mg, 0,045 mmol) en DCM-i-PrOH (2:1) se añadieron la sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2) (10,0 mg, 0,012 mmol) y DIEA (0,0020 mL, 0,012 mmol). La mezcla de reacción se calentó hasta 80 °C durante 18 horas, momento en el que el análisis LCMS indicó que la reacción estaba completa en un 70-80%. La mezcla de reacción se concentró y el residuo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-50% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para proporcionar el compuesto 15 como una sal de TFA, MS m/z 1040,4 (M+1). Tiempo de retención 1,03 minutos.
Ejemplo de carga 39: Ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-(3-(metil(2-(vinilsulfonil)etil)amino)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (16)

Paso 1: A una solución de ácido 3-((terf-butoxicarbonil)(metil)amino)propanoico (5,4 mg, 0,027 mmol) en DMF (1,0 mL) se añadieron DIEA (0,0070 mL, 0,040 mmol) y HATU (9,1 mg, 0,024 mmol). La mezcla de reacción se agitó durante 5 minutos y se añadió sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2) (11,4 mg, 0,013 mmol). La reacción se completó en 1 hora a juzgar por el análisis LCMS. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el ácido (S)-2-((-2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-((tertbutoxicarbonil)(metil)amino)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico MS m/z 927,5 (M+1). Tiempo de retención 1,28 minutos.
Paso 2: A una solución del producto obtenido en el paso 1 (6,4 mg, 0,0069 mmol) en DCM (2,0 mL) se añadió TFA (1,0 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y se concentró para obtener la sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)(3-(metilamino)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico. MS m/z 827,4 (M+1). Tiempo de retención 0,99 minutos. Este producto se utilizó en el paso siguiente sin purificación adicional.
Paso 3: A una solución de la sal de TFA del producto obtenido en el paso 2 (6,5 mg, 0,0069 mmol) en i-PrOH (2,0 mL) se añadieron divinilsulfona (20,0 mg, 0,169 mmol) y DIEA (0,010 mL, 0,057 mmol). La mezcla de reacción se agitó a 80 °C durante 1 hora, momento en el que la reacción estaba completa a juzgar por el análisis LCMS y la mezcla de reacción se concentró. El residuo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-60% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el compuesto 16, MS m/z 945,4 (M+1). Tiempo de retención 0,99 minutos.
Ejemplo de carga 40: (1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanoil)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1 -fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (17)
Este compuesto se sintetizó utilizando el mismo método tal como se ha descrito para el compuesto (4) (en el Ejemplo 4) a partir de EMCA (5,5 mg, 0,026 mmol), DIEA (10,0 mg, 0,078 mmol), HBTU (9,8 mg, 0,026 mmol) y la sal de TFA de la (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((1 S,2R)-1 -hidroxi-1-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (3) (21,8 mg, 0,026 mmol). El compuesto 17 se obtuvo después de la purificación mediante HPLC en fase inversa, MS m/z 921,5 (M+1). Tiempo de retención 1,25 minutos.
Ejemplo de carga 41: Ácido (S)-2-((-2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-mercaptohexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (18)

Paso 1: A una solución de etanotioato de S-(6-oxohexilo) (4,28 mg, 0,025 mmol) y sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2) (7,0 mg, 0,0082 mmol) en MeOH (2,0 mL) se añadió ácido acético (0,0050 mL, 0,083 mmol) y cianoborohidruro sódico (2,57 mg, 0,041 mmol). La mezcla de reacción se calentó a 50 °C durante 2 horas y el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-70% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron, lo que proporcionó ácido (S)-2-((-2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(acetiltio)hexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico MS m/z 900,5 (M+1), tiempo de retención 1,17 minutos.
Paso 2: El producto obtenido en el paso 1 se disolvió en MeOH:H2O (2:1, 3,0 mL). A la solución se añadió hidróxido de litio (5,0 mg, 0,21 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas y, a continuación, se concentró hasta aproximadamente 1,5 mL. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-60% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el compuesto 18, MS m/z 858,5 (M+1). Tiempo de retención 1,16 minutos.
Ejemplo de carga 42: Ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(3-amino-4-formilfenoxi)hexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (19)

Paso 1: A una solución de 2-nitro-4-((6-oxohexil)oxi)benzaldehído (20,1 mg, 0,076 mmol) y (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1) (16,5 mg, 0,019 mmol) en DMF (2,0 mL) se añadió ácido acético (0,0076 mL, 0,13 mmol) y cianoborohidruro sódico (11,9 mg, 0,190 mmol). La mezcla de reacción se calentó a 50 °C durante 2 horas y el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-70% de acetonitrilo-H2Ü, que contenía un 0,05% de TFA. Las fracciones que contienen el aldehido deseado (MS m/z 1005,5 (M+1), tiempo de retención 1,27 minutos) y los intermedios del alcohol deseado (MS m/z 1007,5 (M+1), tiempo de retención 1,21 minutos) se combinaron y se concentraron y utilizaron en el paso siguiente.
Paso 2: La mezcla obtenida del paso 1 que contenía el aldehido y el alcohol se disolvió en DCM (2,0 mL) y se añadió peryodinano de Dess-Martin (4,0 mg, 0,0095 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se lavó entonces con solución de Na2S2Ü3 acuosa y se extrajo con DCM. La fase de DCM se secó con MgSÜ4 anhidro, se filtró y se concentró. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-70% de acetonitrilo-H2Ü que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para proporcionar la sal de TFA de (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(4-formil-3-nitrofenoxi)hexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo, MS m/z 1005,5 (M+1). Tiempo de retención 1,27 minutos. El producto también contenía algo de ácido hidrolizado, MS m/z 991,5 (M+1). Tiempo de retención 1,22 minutos.
Paso 3: A una solución del producto obtenido en el paso 2 (16,9 mg, 0,015 mmol) en EtOH al 70% en agua se añadieron hierro en polvo (0,8 mg, 0,02 mmol) y HCl (0,1 N, 0,15 mL, 0,015 mmol). La mezcla de reacción se agitó vigorosamente a temperatura ambiente durante 18 horas. Se formó un precipitado marrón. La mezcla se filtró a través de un lecho de Celite y el filtrado se concentró. El crudo se purificó con ISCO, columna C18, se eluyó con un 30-100% de acetonitrilo-H2O. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener (S)-2-((2R,3R)-3-((S)-1-((3fí,3S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(3-amino-4-formilfenoxi)hexil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo, MS m/z 975,5 (M+1). Tiempo de retención 1,23 minutos.
Paso 4: A una solución del producto obtenido en el paso 3 (4,6 mg, 0,0047 mmol) en MeOH-H2O (1,5:1, 2,5 mL) se añadió hidróxido de litio (10,0 mg, 0,435 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 4 horas. La mezcla de reacción se concentró hasta aproximadamente un 50% del volumen y se acidificó con HCl 1N hasta pH 5. El crudo se purificó con ISCO, columna C18, se eluyó con un 30-75% de acetonitrilo-H2O. Las fracciones que contenían el producto deseado se combinaron, se neutralizaron con 0,3 mg de LiOH, y se liofilizaron para obtener el compuesto 19, MS m/z 961,5 (M+1). Tiempo de retención 1,15 minutos.
Ejemplo de carga 43: Aducto con coenzima A del ácido (S)-2-((-2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-AÍ,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (20)

A una solución de sal de trilitio de la coenzima A (CoA) (7,6 mg, 0,0096 mmol) en tampón de fosfato 100 mM que contiene EDTA 5 mM a pH 7,5 se añadió una solución de ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (17) (9,0 mg, 0,0096 mmol) en DMSO (0,048 mL). La mezcla de reacción se dejó reposar a temperatura ambiente durante 1 hora, momento en el que la reacción estaba completa a juzgar por el análisis LCMS. La muestra se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-60% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el aducto de CoA 20, MS m/z 852 (M/2+1). Tiempo de retención 0,98 minutos.
Ejemplo de carga 44: Ácido (S)-2-((2R,3fí)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2-bromoacetamido)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (21)

Paso 1: A una solución de ácido 6-((ferí-butoxicarbonil)amino)hexanoico (12 mg, 0,051 mmol) en DMF (2,0 mL) se añadieron DIEA (18 mg, 0,14 mmol) y HATU (18 mg, 0,047 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 10 minutos antes de añadir ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (2) (40 mg, 0,047 mmol). La reacción se completó en media hora. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-((ferf-butoxicarbonil)amino)hexanoil)-2
azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico, MS m/z 955,5 (M+1). Tiempo de retención 1,32 minutos.
Paso 2: A una solución del compuesto obtenido en el paso 1 (15,6 mg, 0,016 mmol) en DCM (2,0 mL) se añadió TFA (1,0 mL). La mezcla de reacción se agitó a temperatura ambiente durante 30 minutos y se concentró para obtener la sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1R,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico, MS m/z 855,5 (M+1). Tiempo de retención 1,01 minutos.
Paso 3: La sal de TFA del ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1R,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (20 mg, 0,021 mmol) se disolvió en DCM y se trató con DIEA (12 mg, 0,093 mmol). La mezcla de reacción se enfrió hasta 0 °C. A continuación, se añadió a la mezcla de reacción una solución de bromuro de 2-bromoacetilo (9,0 mg, 0,045 mmol) en DCM (0,2 mL) con agitación. La mezcla de reacción se agitó a 0 °C durante 10 min y el análisis LCMS mostró que el material de partida de amina se había consumido. Se añadió NaHCÜ3 acuoso saturado para desactivar la reacción. La mezcla de reacción se extrajo con DCM (5 mL X 3). Las fases orgánicas se combinaron y se concentraron. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-45% de acetonitrilo-H2Ü que contenía un 0,05% de TFA. Las fracciones se combinaron y se concentraron para obtener el compuesto 21, MS m/z 975,3 (M+1). Tiempo de retención 1,19 minutos.
Ejemplo de carga 45: Ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(aminooxi)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (22)

Paso 1: A una solución de 6-(((1-etoxietilideno)amino)oxi)hexanoato de litio (6,3 mg, 0,028 mmol) en DMF (1,0 mL) se añadió HATU (8,9 mg, 0,023 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 20 minutos antes de añadir la mezcla de reacción completa a una solución de la sal de TFA de (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1) (20 mg, 0,021 mmol) y DIEA (6,0 mg, 0,047 mmol) en DMF (1,0 mL). Después de agitar a temperatura ambiente durante 2 horas, la mezcla de reacción se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 40-80% de acetonitrilo-H2Ü, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron. El análisis LCMS reveló que el grupo protector en el resto alcoxilamina se eliminó para proporcionar la sal de TFA de (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(aminooxi)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo, MS m/z 885,5 (M+1). Tiempo de retención 1,10 minutos.
Paso 2: A una solución del compuesto obtenido en el Paso 1 (24,3 mg, 0,023 mmol) en MeOH-H2O (1:1, 2,0 mL) se añadió hidróxido de litio (20 mg, 0,84 mmol). La reacción se monitorizó por LCMS. Tras la finalización, el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-40% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener la sal de TFA del compuesto 22, MS m/z 871,5 (M+1). Tiempo de retención 1,03 minutos.
Ejemplo de carga 46: Ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-((S)-aziridina-2-caboxamido)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (23)

Paso 1: En un vial de 7 mL, se disolvió ácido 6-((ferf-butoxicarbonil)amino)hexanoico (16 mg, 0,069 mmol) en DMF anhidra (2 mL). Se añadieron DIEA (0,036 mL, 0,21 mmol) y HATU (24 mg, 0,062 mmol). La mezcla de reacción se agitó durante 10 min antes de añadir (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1, sal de HCl, 30 mg, 0,034 mmol). La mezcla de reacción se agitó durante 2 horas adicionales a temperatura ambiente. El LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de ACN-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (S)-2-((2R,3R)-3-((S)-1-((3R,4s ,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-((ferí-butoxicarbonil)amino)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo. MS m/z 969,6 (M+1). Tiempo de retención 1,42 minutos. El producto así obtenido (21 mg, 0,022 mmol) se disolvió en HCl en MeOH (3 M, 2 mL). El disolvente se eliminó lentamente a presión reducida. El análisis LCMS del residuo indicó la eliminación completa del grupo Boc. El residuo se recuperó en acetonitrilo y agua y se liofilizó para obtener (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo como sal de HCl. MS m/z 869,5 (M+1). Tiempo de retención 1,01 minutos.
Paso 2: El producto del paso previo (10 mg, 0,012 mmol) se disolvió en THF (0,8 mL), MeOH (0,1 mL) y H2O (0,1 mL). Se añadió hidróxido de litio monohidratado (4,83 mg, 0,115 mmol). La mezcla de reacción se agitó durante 4 horas a temperatura ambiente. El LCMS indicó que la reacción se había completado. Los disolventes se eliminaron a presión reducida. El residuo se neutralizó utilizando ácido clorhídrico 0,1 N, se recuperó en acetonitrilo y H2O, y se liofilizó para obtener ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico que contenía algo de LiCl. MS m/z 855,6 (M+1). Tiempo de retención 0,98 minutos.
Paso 3: En un vial de 7 mL, se disolvió ácido (S)-1-tritilaziridina-2-carboxílico(7,6 mg, 0,023 mmol) en DMF anhidra (2 mL). Se añadieron DIEA (0,010 mL, 0,021 mmol) y HATU (7,9 mg, 0,021 mmol). La mezcla de reacción se agitó durante 10 minutos antes de ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (10 mg, 0,012 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas más. El LCMS indicó que la reacción se había completado. El disolvente se eliminó a presión reducida. El crudo se purificó con ISCO en fase inversa, utilizando una columna C18ac. (5,5 g), se eluyó con un 10-100% de acetonitrilo-H2O. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para proporcionar ácido (S)-2-((-2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-(6-((S)-1 -tritilaziridina-2-carboxamido)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico. MS m/z 1166,5 (M+1). Tiempo de retención 1,49 minutos.
Paso 4: El producto del Paso 3 (4,0 mg, 0,0034 mmol) se disolvió en MeOH/CHCl3 (1:1, 1 mL) y se enfrió hasta 0 °C. Se añadió TFA (0,0040 mL, 0,051 mmol) gota a gota. La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 1 hora. El LCMS indicó que la reacción estaba completa en aproximadamente un 60%. Se añadió de nuevo TFA (0,0040 mL, 0,051 mmol). Después de otra 1 hora a temperatura ambiente, el LCMS indicó que la reacción estaba completa. Los disolventes se evaporaron a presión reducida. El residuo se disolvió en MeOH y se purificó con ISCO en fase inversa, utilizando una columna C18ac. (5,5 g), se eluyó con un 10-100% de acetonitrilo
H2O. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para proporcionar ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-((S)-aziridina-2-carboxamido)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (23). MS m/z 924,6 (M+1). Tiempo de retención 1,012 minutos.
Ejemplo de carga 47: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(4-(4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)metil)-1 H-1,2,3-triazol-1-il)butanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (24)

Paso 1: A una solución de 4-bromobutanoato de etilo (3,4 g, 0,0174 mol) en DMF (100mL) se añadió azida sódica (1,7 g, 0,0262 mol). La mezcla se calentó hasta 80 °C y se agitó durante la noche. La mezcla de reacción se diluyó con agua y se extrajo 3 veces con éter. La fase orgánica se lavó con agua 3 veces, se secó con MgSO4, se filtró y se concentró para proporcionar el producto crudo que se utilizó directamente en el paso siguiente sin purificación adicional.
Paso 2: Se disolvió 4-azidobutanoato de etilo (157 mg, 1,0 mmol) en THF (4 mL), MeOH (0,5mL) y agua (0,5 mL). A continuación, se añadió LiOH.H2O(168 mg, 4,0 mmol) y la mezcla de reacción se agitó durante 2 horas a temperatura ambiente. El LCMS indicó que la reacción se había completado. La reacción se detuvo, el pH se ajustó a 2-3 utilizando HCl 1 N y la mezcla de reacción se extrajo con EtOAc. La fase orgánica combinada se secó con MgSO4, se concentró para proporcionar el producto crudo que se utilizó directamente en el paso siguiente sin purificación adicional. 1H RMN (400 MHz, CD3OD): 53,36 (t, J=6,8 Hz, 2H), 2,39 (t, J=7,2 Hz, 2H), 1,89-1,82 (m, 2H).
Paso 3: Una solución de ácido 4-azidobutanoico (19 mg, 0,147 mmol), 1-(prop-2-in-1-il)-1H-pirrol-2,5-diona (39,8 mg, 0,294 mmol) y CuSO4 (17,62 mg, 0,11 mmol) en DMF (3,0 mL) y H2O (0,75mL) se trató son sal sódica del ácido L-ascórbico (72,9 mg, 0,368 mmol) y se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se purificó mediante Prep-HPLC, columna C18, se eluyó con un 20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 265,1 (M+1 ). Tiempo de retención 0,642 minutos. 1H RMN (400 MHz, CD3OD): 57,94 (s, 1H), 6,86 (s, 2H), 4,77 (s, 2H), 4,43 (t, J=7,0 Hz, 2H), 2,31 (t, J=7,2 Hz, 2H), 2,17-2,13 (m, 2H).
Paso 4: Se disolvió ácido 4-(4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)metil)-1 H-1,2,3-triazol-1 -il)butanoico (4,5 mg, 0,017 mmol) en DMF (1 mL). Se añadieron DIEA (9,9 uL, 0,057 mmol) y HATU (5,61 mg, 0,015 mmol) y la mezcla se agitó durante 10 minutos antes de la adición de 2 (9,72 mg, 0,011 mmol). La mezcla de reacción se agitó entonces durante 1 hora a temperatura ambiente. El análisis LC/MS indicó que la reacción se había completado. El producto se purificó mediante Prep-HPLC, columna C18, se eluyó con un 20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado 24 se combinaron y se liofilizaron para obtener un sólido blanco. MS m/z 988,5.1 (M+1). Tiempo de retención 1,074 minutos.
Ejemplo de carga 48: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(metilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamida (25)

Se disolvió EMCA (4,1 mg, 0,019 mmol) en DMF (2 mL). Se añadieron DIEA (8,31 mg, 0,064 mmol) y HATU (5,87 mg, 0,015 mmol) y después de 10 minutos se añadió el compuesto 4 (11 mg, 0,013 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. El análisis LC/MS indicó que la reacción se había completado. El producto se purificó mediante Prep-HPLC, columna C18, se eluyó con un 30-50% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el producto deseado 25 como un sólido blanco. MS m/z 1012,5 (M+1). Tiempo de retención 1,222 minutos.
Ejemplo de carga 49: (1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanoil)-A/-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (26)

A una solución de EMCA (2,5 mg, 0,012 mmol) en DMF (1 mL) se añadió DIEA (6,2 uL, 0,035 mmol) y después HATU (4,5 mg, 0,012 mmol). La mezcla de reacción se agitó durante 5 minutos y a continuación se añadió a la sal de TFA de (1 R,3S,4S)-W-((s )-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-tetrazol-5-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (6,7 mg, 0,0076 mmol). La mezcla de reacción se mantuvo a temperatura ambiente durante 1 hora y a continuación el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-60% de acetonitrilo-H2O, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener el compuesto 26, MS m/z 959,5 (M+1). Tiempo de retención 1,220 minutos.
Ejemplo de carga 50: Ácido (R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico (27)

A una solución de EMCA (3,3 mg, 0,016 mmol) en DMF (1 mL) se añadió DIEA (2,7 uL, 0,016 mL) y después HATU (5,93 mg, 0,016 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 10 minutos y a continuación se añadió a una solución de ácido ((fí)-1-((2R,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico 7 (10 mg, 0,011 mmol) en DMF. La mezcla de reacción se agitó a temperatura ambiente durante 1 hora.
El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-60% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ácido (R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfónico 27. MS m/z 971,5 (M+1). Tiempo de retención 1,181 minutos.
Ejemplo de carga 51: Ácido (R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (28)

A una solución de EMCA (2,4 mg, 0,011 mmol) en DMF (1 mL) se añadió DIEA (6,6 uL, 0,038 mmol) y HATU (4,0 mg, 10,42 gmol). La mezcla de reacción se agitó durante 5 minutos y a continuación se añadió a una solución de ácido ((fí)-1-((2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico 8 (8,3 mg, 0,0095 mmol) en DMF (1 mL). La mezcla de reacción se completó en 10 minutos y el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-55% de acetonitrilo-H2Ü, que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico 28. MS m/z 955,5 (M+1). Tiempo de retención 1,151 minutos.
Ejemplo de carga 52: ((R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfonato de butilhidrógeno (29)

A una solución de ácido ((R)-1-((1fí,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1fí,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (8) (4,2 mg, 0,0044 mmol) en piridina (1 mL) se añadió n-BuOH (3,3 mg, 0,044 mmol) y después cloruro de pivaloílo (5,3 mg, 0,044 mmol). La reacción se monitorizó mediante LCMS hasta que todo el ácido fosfórico se convirtió en el éster. A continuación, se añadió una solución de yodo recién preparada (11 mg, 0,044 mmol) en piridina húmeda-agua (10:1, 1 mL). La reacción se monitorizó mediante LCMS hasta su finalización. La piridina se eliminó al vacío y el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 30-55% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener ((fí)-1-((2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfonato de butilhidrógeno 29. MS m/z 1027,5 (M+1). Tiempo de retención 1,300 minutos. El éster es proclive a la hidrólisis en condiciones ácidas.
Ejemplo de carga 53: Ácido (S)-2-((2fí,3fí)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-((((1 R,8S,9s)-biciclo[6.1.0]non-4-in-9-ilmetoxi)carbonil)amino)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (75)

Al ácido (S)-2-((2R,3R)-3-((S)-1-((3R,2S,5S)-4-((S)-2-((1 fí,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (paso 2, Ejemplo 44) (5 mg, 0,005 mmol) en DMF-THF (1:1, 2 mL) se añadió (2,5-dioxopirrolidin-1 -il) carbonato de (1R,8S,9s)-biciclo[6.1.0]non-4-in-9-ilmetilo (1.5 mg, 0,005 mmol) y DiEA (0,0025 mL, 0,014 mmol). La mezcla de reacción se agitó a t.a. durante 30 min y después se purificó mediante HPLC preparativa (40-65% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (75). MS m/z 1031,6 (M+H). Tiempo de retención 1,337 min.
Ejemplo de carga 54: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -hidroxi-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (76)

A una solución de MC-Val-Cit-PAB-PNP (1,9 mg, 0,0026 mmol), sal de TFA del compuesto (50) (1,8 mg, 0,002 mmol) en DMF (1 mL) se añadieron piridina (0,25 mL), HOAT (0,29 mg, 0,002 mmol) y DIEA (0,0054 mL, 0,031 mmol). La reacción se agitó a 40 °C durante 2 h y después a 30 °C durante 18 h. La mezcla de reacción se concentró y se purificó mediante HPLC preparativa (20-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (76). MS m/z 664,0 (M/2+H). Tiempo de retención 1,165 min.
Ejemplo de carga 55: Ácido (S)-2-((2R,3fí)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-AÍ,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (77)

A una solución de ácido 3-(2-(maleimido)etoxi)propanoico (2,2 mg, 0,010 mmol) en DMF (1 mL) se añadieron HATU (3,7 mg, 0,0098 mmol) y DIEA (3,6 mg, 0,028 mmol). La reacción se agitó durante 5 minutos y después se añadió ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (8 mg, 0,0093 mmol) en DMF (0,5 mL). La mezcla de reacción se agitó a t.a. durante 1 h y después se concentró. El crudo se purificó mediante HPLC preparativa (10-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (77). MS m/z 937,5 (M+H). Tiempo de retención 1,138 min.
Ejemplo de carga 56: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-(2-(2-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)etoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (78)
El compuesto (78) se preparó mediante el método descrito para el compuesto (77) utilizando ácido 3-(2-(2-(maleimido)etoxi)etoxi)propanoico (2,6 mg, 0,010 mmol) en lugar de ácido 3-(2-(maleimido)etoxi)propanoico. MS m/z 981,5 (M+H). Tiempo de retención 1,140 min.
Ejemplo de carga 57: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(3-(2-(2-(2-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)etoxi)etoxi)etoxi)propanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (79)

El compuesto (79) se preparó mediante el método descrito para el compuesto (77) utilizando ácido 3-(2-(2-(2-(maleimido)etoxi)etoxi)etoxi)propanoico (3,1 mg, 0,010 mmol) en lugar de ácido 3-(2-(maleimido)etoxi)propanoico. MS m/z 1025,5 (M+H). Tiempo de retención 1,143 min.
Ejemplo de carga 58: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(1-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)-3,6,9,12-tetraoxapentadecan-15-oil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (80)

El compuesto (80) se preparó mediante el método descrito para el compuesto (77) utilizando ácido 1-(maleimido)-3,6,9,12-tetraoxapentadecan-15-oico (3,6 mg, 0,010 mmol) en lugar de ácido 3-(2-(maleimido)etoxi)propanoico. MS m/z 1069,5 (M+H). Tiempo de retención 1,144 min.
Ejemplo de carga 59: Ácido (S)-2-((2R,2R)-3-((S)-1-((3R,4S,5S)-4-((S)-W,3-dimetil-2-((1 R,3S,4S)-2-(4-(1-(2-(2-(2-(2-(4-(5-(metilsulfonil)-1,3,4-oxadiazol-2-il)fenoxi)etoxi)etoxi)etoxi)etil)-1 H-1,2,3-triazol-4-il)butanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (81)

Paso 1: Se disolvió (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoato de metilo (1) (63 mg, 0,080 mmol) en ACN (0,75 mL) y agua (0,5 mL). Se añadió NaOH (1 M, 0,35 mL). La reacción se agitó 2 h a t.a. Después de neutralizarla con HCl 1 N hasta aproximadamente pH 5, la mezcla de reacción se diluyó con agua y se liofilizó para proporcionar ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico crudo. MS m/z 742,4 (M+1). Tiempo de retención 1,010 min. El producto se utilizó en el paso siguiente sin purificación adicional.
Paso 2: A una solución de ácido hex-5-inoico (5,4 mg, 0,049 mmol) en DMF (2 mL) se añadió DIEA (26,1 mg, 0,35 mmol) y HATU (16,9 mg, 0,044 mmol). La reacción se agitó a t.a. durante 15 min. A continuación, se añadió el producto
obtenido en el paso 1 (30 mg, 0,040 mmol). La reacción se agitó a t.a. durante 2 h. El crudo se purificó mediante HPLC preparativa (10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (S)-2-(2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(hex-5-inoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico. MS m/z 836,5 (M+1). Tiempo de retención 1,224 min.
Paso 3: Se suspendieron ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(hex-5-inoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (7 mg, 0,0084 mmol) y 2-(4-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etoxi)fenil)-5-(metilsulfonil)-1,3,4-oxadiazol (3.7 mg, 0.0084 mmol) en f-BuOH (1 mL) y agua (1 mL). Se añadieron L-ascorbato sódico (1,7 mg, 0,0084 mmol) en 0,3 mL de H2O y CuSO4 (0,3 mg, 0,0017 mmol) en 0,3 mL de H2O secuencialmente utilizando una jeringa y la reacción se agitó a t.a. durante 3 h. La mezcla de reacción se purificó mediante preparativa (10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (81) como un sólido blanco. MS m/z 639,4 (M/2+1). Tiempo de retención 1,196 min.
Ejemplo de carga 60: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-Metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (82)

Se combinaron (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida ((56), 8,7 mg, 0,011 mmol), MC-Val-Cit-PABC-PNP (9,7 mg, 0,013 mmol), HOAT (1,7 mg, 0,011 mmol) y DIEA (0,013 ml, 0,077 mmol) en piridina (0,5 mL) y DMF (2 mL). La reacción se agitó durante 4 h a t.a. La mezcla de reacción se purificó mediante HPLC preparativa (10-60% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (82) como un sólido blanco. MS m/z 695,5 (M/2+1). Tiempo de retención 1,139 min.
Ejemplo de carga 61: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-Metox¡-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(4-fenil-1 H-imidazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (83)

El compuesto (83) se preparó mediante el método descrito para el compuesto (82) utilizando (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metox¡-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1 H-imidazol-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (57) en lugar del compuesto (56). MS m/z 720,0 (M/2+1). Tiempo de retención 1,169 min.
Ejemplo de carga 62: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-metox¡-1 -((S)-2-((1 R,2R)-1 -metoxi-3-(((S)-1 -metoxi-3-fenilpropan-2-il)amino)-2-metil-3-oxopropil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (84)

El compuesto (84) se preparó mediante el método descrito para el compuesto (82) utilizando (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-3-(((S)-1 -metoxi-3-fenilpropan-2-il)amino)-2-metil-3-oxopropil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (58) en lugar del compuesto (56). MS m/z 671,0 (M/2+1). Tiempo de retención 1,236 min.
Ejemplo de carga 63: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-pirazol-3-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (85)

El compuesto (85) se preparó mediante el método descrito para el compuesto (82) utilizando (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(1 H-pirazol-3-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (59) en lugar del compuesto (56). MS m/z 682,1(M/2+1). Tiempo de retención 1,172 min.
Ejemplo de carga 64: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (86)

El compuesto (86) se preparó mediante el método descrito para el compuesto (83) utilizando (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(5-fenil-1,3,4-oxadiazol-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (62) en lugar del compuesto (56). MS m /z721, 1 (M/2+1). Tiempo de retención 1,280 min.
Ejemplo de carga 65: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1-(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (87)

El compuesto (87) se preparó mediante el método descrito para el compuesto (82) utilizando (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-2-fenil-1 -(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (60) en lugar del compuesto (56). Tiempo de retención 1,204 min.
Ejemplo de carga 66: (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-Metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((R)-2-fenil-1 -(pirimidin-2-il)etil)amino)propil)pirrolidin-1 -il)-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (88)

El compuesto (88) se preparó mediante el método descrito para el compuesto (82) utilizando (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((R)-2-fenil-1 -(pirimidin-2-il)etil)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (61) en lugar del compuesto (56). MS m /z688,0 (M/2+1). Tiempo de retención 1,221 min.
Ejemplo de carga 67: Ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(5-((2,5-dioxopirrolidin-1-il)oxi)-5-oxopentanamido)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (89)
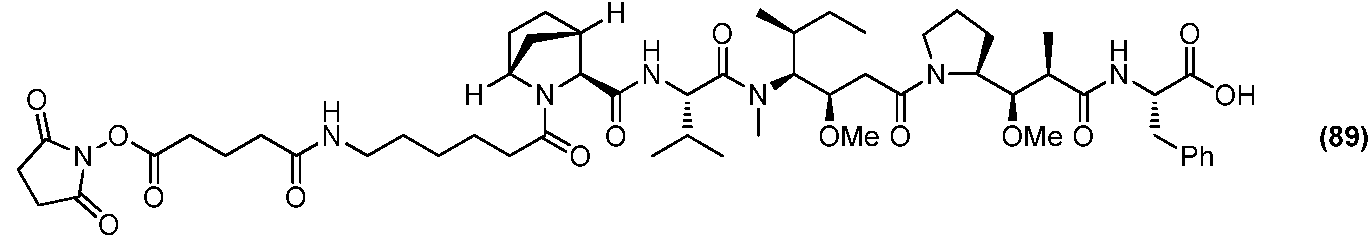
Una solución de ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-aminohexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico ((paso 2, Ejemplo 44) 20 mg, 0,021 mmol) y DIEA (0,018 mL, 0,10 mmol) en DMF (1 mL) se añadió a glutarato de bis(2,5-dioxopirrolidin-1-ilo) (10,1 mg, 0,031 mmol) y DIEA (0,018 mL) en Dm F (1 mL). La reacción se agitó durante 2 h a t.a. El crudo se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener ácido (S)-2-((2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(5-((2,5-dioxopirrolidin-1-il)oxi)-5-oxopentanamido)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (89). MS m/z 1066,5 (M+1). Tiempo de retención 1,103 min.
Procedimiento sintético para los compuestos de Fórmula (I) conectados en el extremo C term inal ejemplo de Carga
Ejemplo de carga 68: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1-((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (30)

Paso 1: En un matraz de fondo redondo de 15 mL, se añadieron ácido (1 R,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (12 mg, 0,050 mmol) y DIEA (0,032 mL, 0,18 mmol) en d Mf (2,0 mL), seguidos de HATU (19 mg, 0,050 mmol). La solución resultante se agitó durante 5 minutos. A continuación, se añadió la sal de TFA de N-(4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5
metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencil)-6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamida (48,5 mg, 0,047 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-70% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferí-butilo, MS m/z 1139,6 (M+1). Tiempo de retención 1,39 minutos.
Paso 2: En un matraz de fondo redondo de 15 mL se añadieron el producto obtenido en el paso 1 (42,6 mg, 0,037 mmol), TFA (2,0 mL) y DCM (4,0 mL), que dan como resultado una solución transparente. La mezcla de reacción se agitó a temperatura ambiente durante 1 hora, momento en el cual un análisis LCMS mostró que el Boc se había eliminado completamente. La mezcla de reacción se concentró para obtener el compuesto 30 como sal de TFA, MS m/z 1039,6 (M+1). Tiempo de retención 1,06 minutos.
Ejemplo de carga 69: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (31)

En un matraz de fondo redondo de 15 mL, se añadieron la sal de TFA de (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (30) (20 mg, 0,017 mmol), paraformaldehído (5,9 mg, 0,21 mmol) y ácido acético (0,0029 mL, 0,050 mmol) en MeOH (2,0 mL). A la suspensión resultante se añadió NaCNBH3 (6,6 mg, 0,11 mmol).
La mezcla de reacción se agitó a temperatura ambiente durante 18 horas. Se añadieron formaldehído y NaCNBH3 adicionales y la mezcla de reacción se calentó hasta 50 °C durante 1 hora hasta completar la reacción. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-50% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el compuesto 31 como sal de TFA, MS m/z 1053,7 (M+1)). Tiempo de retención 1,07 minutos.
Ejemplo de carga 70: (1 R,3S,4S)-2-Acetil-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (32)

En un matraz de fondo redondo de 15 mL, se añadieron ácido acético (0,79 mg, 0,013 mmol), DIEA (1,7 mg, 0,013 mmol) y DMF (1,0 mL), seguidos de HBTU (2,2 mg, 0,0058 mmol). La mezcla de reacción se agitó durante 5 minutos antes de añadir la sal de TFA de (2S,3S)-N-((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1-((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-3-metilpirrolidin-2-carboxamida (30) (5,5 mg, 0,0048 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-50% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el compuesto 32, MS m/z 1081,3 (M+1). Tiempo de retención 1,22 minutos.
Ejemplo de carga 71: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(6-hidroxihexil)-2-azabiciclo[2.2.1]heptano-3-carboxamida (33)

Al compuesto 30 (3,0 mg, 0,0029 mmol) en MeOH (1,0 mL) se añadió 6-hidroxihexanal (6,7 mg, 0,058 mmol), seguido de NaBHaCN (9,1 mg, 0,14 mmol). Después de 30 minutos, se añadió NaBHaCN (9,1 mg, 0,14 mmol) adicional. Después de otros 30 minutos, el análisis LCMS indicó que la reacción se había completado. La mezcla de reacción se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(6-hidroxihexil)-2-azabiciclo[2.2.1]heptano-3-carboxamida 33, MS m/z 1139,6 (M+1). Tiempo de retención 1,10 minutos.
Ejemplo de carga 72: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(6-hidroxihexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamida (34)

Al ácido 6-hidroxihexanoico (3,8 mg, 0,029 mmol) en DMF (1 mL) se añadieron DIEA (7,5 mg, 0,058 mmol) y HBTU (9,1 mg, 0,024 mmol). Después de 10 minutos, se añadió el compuesto 30 (10 mg, 0,0096 mmol). La mezcla de reacción se agitó durante 1 hora, momento en el que el análisis LCMS indicó que la reacción estaba completa. La mezcla de reacción se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 34, MS m/z 1153,5 (M+1). Tiempo de retención 1,20 minutos.
Ejemplo de carga 73: (2S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-3-azabiciclo[3.1.0]hexano-2-carboxamida (35)

butoxicarbonil)-3-azabiciclo[3.1.0]hexano-2-carboxílico (12,5 mg, 0,055 mmol), DIEA (28,5 mg, 0,22 mmol), HATU (21 mg, 0,055 mmol) y la sal de TFA de N-(4-((S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencil)-6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamida (11 mg, 0,011 mmol). Después de la purificación, se obtuvo el intermedio protegido con Boc, MS m/z 1125,5 (M+1). Tiempo de retención 1,34 minutos. El producto así obtenido (10 mg, 0,0089 mmol) se trató con TFA (2,0 mL) en DCM (4,0 mL). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y el análisis LCMS mostró que el Boc se había eliminado completamente. La solución se concentró para obtener el compuesto 35 como sal de TFA, MS m/z 1025,5 (M+1). Tiempo de retención 1,06 minutos.
Ejemplo de carga 74: (2S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-3-metil-3-azabiciclo[3.1.0]hexano-2-carboxamida (36)

il)hexanamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-3-azabiciclo[3.1.0]hexano-2-carboxamida (35) (7,9 mg, 0,0062 mmol) y paraformaldehído (2,7 mg, 0,089 mmol) en MeOH (2,0 mL) se añadió NaCNBH3 (11 mg, 0,018 mmol). La mezcla de reacción se agitó a 50 °C durante 2 horas. El crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 20-50% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se concentraron para obtener el compuesto 36 como sal de TFA, MS m/z 1039,5 (M+1). Tiempo de retención 1,07 minutos.
Ejemplo de carga 75: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1-((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(2,5,8,11,14,17,20,23,26,29,32,35-dodecaoxaoctatriacontan-38-oil)-2-azabiciclo[2.2.1]heptano-3-carboxamida (37)
Al ácido 2,5,8,11,14,17,20,23,26,29,32,35-dodecaoxaoctatriacontan-38-oico (17,0 mg, 0,029 mmol) en DMF (1,5 mL) se añadieron DIEA (7,5 mg, 0,058 mmol) y HATU (9,2 mg, 0,024 mmol). Después de 10 minutos, se añadió el compuesto 30 (10,0 mg, 0,0096 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. La mezcla de reacción se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 37, MS m/z 805,6 ((M+2)/2). Tiempo de retención 1,25 minutos.
Ejemplo de carga 76: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1-((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(2,5,8,11,14,17,20,23-octaoxahexacosan-26-oil)-2-azabiciclo[2.2.1]heptano-3-carboxamida (38)
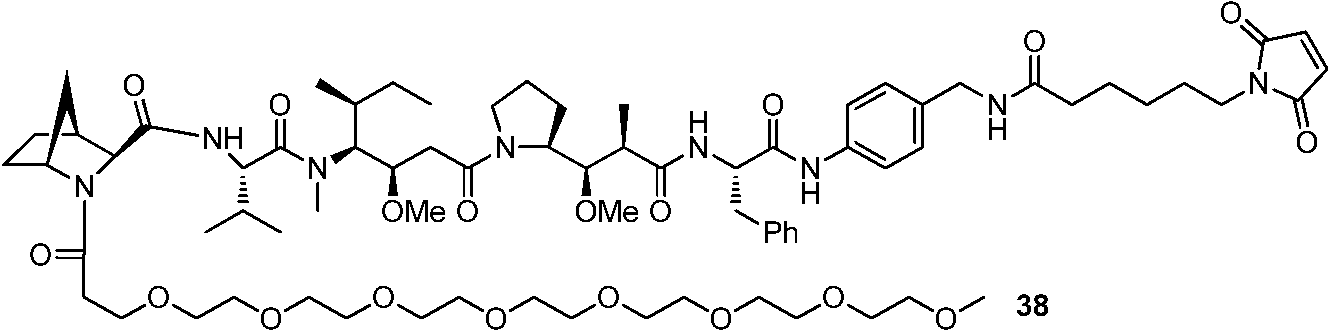
El compuesto 38 se sintetizó mediante el mismo método tal como se ha descrito para el compuesto 37 utilizando el compuesto 30 (10 mg, 0.0096 mmol) y ácido 2,5,8,11,14,17,20,23-octaoxahexacosan-26-oico (11,91 mg, 0,029 mmol), MS m/z 717,5 ((M+2)/2). Tiempo de retención 1,25 minutos.
Ejemplo de carga 77: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1-((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(1-hidroxi-3,6,9,12-tetraoxapentadecan-15-oil)-2-azabiciclo[2.2.1]heptano-3-carboxamida (39)

El compuesto 39 se sintetizó mediante el mismo método descrito para el compuesto 37 utilizando el compuesto 30 (10 mg, 0,0096 mmol), ácido 1-hidroxi-3,6,9,12-tetraoxapentadecan-15-oico (7,7 mg, 0,029 mmol), DIEA (7,46 mg, 0,058 mmol) y HBTU (9,12 mg, 0,024 mmol) en DMF (1,5 mL), MS m/z 1287,6 (M+1)). Tiempo de retención 1,18 minutos.
Ejemplo de carga 78: (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1-((4-((4-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)metil)ciclohexanocarboxamido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (40)

Paso 1: Se disolvió ácido (S)-2-((ferí-butoxicarbonil)amino)-3-fenilpropanoico (964 mg, 3,63 mmol) en DMF (10 mL). Se añadieron DIEA (1,27 g, 9,84 mmol) y HATU (1,13 g, 3,03 mmol). Después de 10 minutos, se añadió 4-aminobencilcarbamato de bencilo (388 mg, 1,51 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente, momento en el que el análisis LCMS indicó que la reacción se había completado. Se agregó EtOAc (60 mL) a la reacción. A continuación, la mezcla de reacción se lavó con NaHCO3 acuoso saturado. La fase acuosa se extrajo con EtOAc (2 x 30 mL). Las fases orgánicas combinadas se lavaron con H2O (5 x 10 mL), se secaron con MgSO4, se filtraron y se concentraron para proporcionar el producto crudo. El producto crudo se disolvió en DCM (5,0 mL) y se trató con TFA (5,0 mL). Después de 1 hora a temperatura ambiente, el análisis LCMS indicó que la reacción se había completado. Los disolventes se eliminaron a presión reducida. El residuo se purificó con ISCO utilizando un 0-8% de MeOH con amoniaco 2 M en DCM para obtener (S)-4-(2-amino-3-fenilpropanamido)bencilcarbamato de bencilo como un sólido blanco, MS m/z 404,2(M+1)). 1H RMN (400 MHz, CD3OD): 57,44-7,23 (m, 14H), 5,10 (s, 2H), 4,26 (s, 2H), 4,12 (d, J = 7,4 Hz, 1H), 3,28-3,22 (m, 1H), 3,15-3,10 (m, 1H).
Paso 2: Se disolvieron (S)-4-(2-amino-3-fenilpropanamido)bencilcarbamato de bencilo (201,7 mg, 0,50 mmol) y ácido (2R, 3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-(fert-butoxicarbonil)amino)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (429 mg, 0,75 mmol) en DMF (6 mL). A continuación, se añadieron DIEA (323 mg, 2,50 mmol) y HATU (342 mg, 0,90 mmol). La mezcla de reacción se agitó durante 1 hora a temperatura ambiente. La mezcla de reacción se purificó mediante HPLC en fase inversa para proporcionar 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-amino-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo, MS m/z 957,5 (M+1), Tiempo de retención 1,54 minutos. El producto protegido con Boc (393 mg, 0,41 mmol) se disolvió en HCl metanólico (3 M, 15 mL). El disolvente se evaporó lentamente a presión reducida. El análisis LCMS indicó que la reacción de desprotección se había completado. Se añadieron acetonitrilo y agua y la solución resultante se liofilizó para obtener 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-amino-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3
metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo como una sal de HCl, MS m/z 857,5 (M+1). Tiempo de retención 1,16 minutos.
Paso 3: Se disolvió ácido (1 R,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (190mg, 0,788 mmol) en DMF (5,0 mL). Se añadieron DIEA (254 mg, 1,97 mmol) y HATU (270 mg, 1,71 mmol). La mezcla de reacción se agitó durante 15 minutos y se añadió 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo (336 mg, 0,394 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente, momento en el que el análisis LCMS indicó que la reacción estaba completa. La mezcla de reacción se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (1 R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1-((4-((((benciloxi)carbonil)amino)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo, MS m/z 1080,5 (M+1), Tiempo de retención 1,56 minutos.
El producto protegido con Boc (88 mg, 0,081 mmol) se disolvió en HCl metanólico (3 M, 6,0 mL). El disolvente se evaporó lentamente a presión reducida. El análisis LCMS indicó que la reacción de desprotección se había completado. Se añadieron acetonitrilo y agua y la solución resultante se liofilizó para obtener 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo como una sal de HCl, m S m /z980,5 (M+1).
Paso 4: Se disolvió 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-W,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo (65,8 mg, 0,067 mmol) en MeOH (4 mL). Se añadieron paraformaldehído (22,8 mg, 0,76 mmol) y ácido acético (0,023 mL, 0,40 mmol), seguidos de cianoborohidruro sódico (47,7 mg, 0,76 mmol). La mezcla de reacción se calentó hasta 50 °C durante 1 hora. El análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-^O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N-3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo MS m/z 994,5 (M+1). Tiempo de retención 1,21 minutos.
Paso 5: Se disolvió 4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetilo-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanamido)bencilcarbamato de bencilo (48 mg, 0,048 mmol) en MeOH (5,0 mL) y se volatilizó en N2. Se añadió Pd/C (20,5 mg, 10% de Pd). El recipiente de reacción se evacuó y se rellenó de nuevo con H2. Esta operación se repitió cinco veces para reemplazar la atmósfera de la reacción con H2. La mezcla de reacción se agitó durante 2 horas a temperatura ambiente en H2. El análisis LCMS indicó que la reacción se había completado. La mezcla de reacción se filtró y se concentró para obtener (1 R,3S,4S)-A/-((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1 -((4-(aminometil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida, MS m/z 860,5 (M+1). Tiempo de retención 0,86 minutos. El producto así obtenido se utilizó en el paso siguiente sin purificación adicional.
Paso 6: Se disolvieron (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1-((4-(aminometil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (12 mg, 0,014 mmol) y 4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)metil)ciclohexanocarboxilato de 2,5-dioxopirrolidin-1-ilo (5.6 mg, 0.017 mmol) en DMF (1 mL), y se añadió DIEA (10,8 mg, 0,084 mmol). La mezcla de reacción se agitó durante 1 hora a temperatura ambiente. El análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 40, MS m/z 1079,5 (M+1). Tiempo de retención 1,12 minutos.
Ejemplo de^ carga 79: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((3-(2-(2-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)etoxi)etoxi)propanamido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (41)

Al ácido 3-(2-(2-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)etoxi)etoxi)propanoico (7,2 mg, 0,028 mmol) en DMF (1,5 mL) se añadieron DiEA (10,8 mg, 0,084 mmol) y HATU (8,0 mg, 0,021 mmol). Después de 10 minutos, se añadió (1 R,3S,4s )-N-((S)-1 -(((3R,4S,5S)-1 -((s )-2-((1 R,2R)-3-(((S)-1 -((4-(aminometil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (12 mg, 0,014 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 41, MS m/z 1099,5 (M+1). Tiempo de retención 1,07 minutos.
Ejemplo de carga 80: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (42)

A NaHCOa acuoso saturado (3,0 mL) se añadió (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1-((4-(aminometil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (20,0 mg, 0,023 mmol). La suspensión resultante se enfrió hasta 0 °C y se añadió 2,5-dioxo-2,5-dihidro-1H-pirrol-1-carboxilato de metilo (14,4 mg, 0,093 mmol) La mezcla de reacción se agitó a 0 °C durante 1,5 horas. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 42, MS m/z 940,5 (M+1). Tiempo de retención 1,13 minutos.
Ejemplo de^ carga 81: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((3-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexil)ureido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (43)

Paso 1: Se disolvió (1fí,3S,4S)-3-(((S)-1-(((3fí,4S,5S)-1-((S)-2-((1fí,2fí)-3-(((S)-1-((4-((((benciloxi)carbonil)amino)-metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo (211 mg, 0,195 mmol) en MeOH (10 mL). Se añadió Pd/C (41,6 mg, 10% de Pd). El recipiente de reacción se evacuó y se rellenó de nuevo con H2. Esta operación se repitió cinco veces para reemplazar la atmósfera de la reacción con H2. La mezcla de reacción se agitó durante 2 horas a temperatura ambiente en H2. El análisis LCMS indicó que la reacción se había completado. La mezcla de reacción se filtró y se concentró para proporcionar (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-(aminometil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo, MS m/z 946,6 (M+1), que se utilizó en el paso siguiente sin purificación.
Paso 2: Se disolvió (1fí,3S,4S)-3-(((S)-1-(((3fí,4S,5S)-1-((S)-2-((1fí,2fí)-3-(((S)-1-((4-(aminometil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo (30 mg, 0,032 mmol) en DMF (3 mL) y THF (3 mL). A continuación, se añadieron DIEA (20,5 mg, 0,16 mmol) y carbonocloridrato de 4-nitrofenilo (12,8 mg, 0,063 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. El análisis LC/MS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (1R,3S,4S)-3-(((S)-1-(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-(((S)-1 -((4-((((4-nitrofenoxi)carbonil)amino)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-3-oxopropil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo, MS m/z 1111,5 (M+1). Tiempo de retención 1,54 minutos.
Paso 3: A (1 R,3S,4S)-3-(((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-(((S)-1 -((4-((((4-nitrofenoxi)carbonil)amino)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-3-oxopropil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo (13,7 mg, 0,012 mmol) disuelto en DMF (1,0 mL) y THF (1,0 mL) se añadieron 1-(6-aminohexil)-1H-pirrol-2,5-diona (14,5 mg, 0,074 mmol) y DIEA (31,9 mg, 0,25 mmol). La mezcla de reacción se agitó durante 4 horas a temperatura ambiente. El análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (1 R,3S,4S)-3-(((S)-1-(((3fí,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((3-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexil)ureido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4
il)(metil)amino)-3-metil-1-oxobutan-2-il)carbamoil)-2-azabiciclo[2.2.1]heptano-2-carboxilato de ferf-butilo, MS m/z 1168,6 (M+1). El producto protegido con Boc así obtenido se disolvió en HCl metanólico (3 M, 2,0 mL). El disolvente se eliminó lentamente a presión reducida. El análisis LCMS indicó que la reacción se había completado. El residuo se disolvió en acetonitrilo y agua y se liofilizó para proporcionar (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-1-((S)-2-((1 R,2R)-3-(((S)-1 -((4-((3-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexil)ureido)metil)fenil)amino)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida como sal de HCl, MS m/z 1068,6 (M+1). Tiempo de retención 1,09 minutos.
Paso 4: Se disolvió (1fí,3S,4S)-N-((S)-1-(((3fí,4S,5S)-1-((S)-2-((1fí,2fí)-3-(((S)-1-((4-((3-(6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexil)ureido)metil)fenil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (8,4 mg, 0,0079 mmol) en MeOH (1,5 mL). Se añadieron paraformaldehído (2,7 mg, 0,089 mmol) y ácido acético (0,0027 mL, 0,046 mmol), seguidos de cianoborohidruro sódico (5,6 mg, 0,089 mmol). La mezcla de reacción se calentó hasta 50 °C durante 1 hora con agitación. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 43, MS m/z 1082,6 (M+1). Tiempo de retención 1,11 minutos.
Ejemplo de carga 82: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -((4-((6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)metil)fenil)(metil)amino)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (44)

Paso 1: Se disolvió ácido 6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoico (349 mg, 1,65 mmol) en DMF (10 mL). A continuación, se añadieron DIEA (820 mg, 6,35 mmol) y HATU (579 mg, 1,52 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 10 minutos. Se añadió entonces (4-(aminometil)fenil)(metil)carbamato de ferf-butilo (300 mg, 1,27 mmol). La mezcla de reacción se agitó durante 1 hora a temperatura ambiente. Se agregó EtOAc (30 mL) a la reacción. A continuación, la mezcla de reacción se lavó con NaHCO3 acuoso saturado. La fase acuosa se extrajo con EtOAc (2 x 30 mL). La fase orgánica combinada se lavó con H2O (5 x 10 mL), se secó con MgSO4, se concentró y se purificó con ISCo (EtOAc/Hexano 0-80%). El producto deseado, MS m/z 374,2 (M+1-tBu), tiempo de retención 1,156 minutos, se obtuvo como un aceite amarillo. El producto se disolvió en DCM (3 mL) y se trató con TFA (1 mL). Después de 1 hora a temperatura ambiente, los disolventes se eliminaron a presión reducida. El residuo se recuperó en acetonitrilo y H2O y se liofilizó para obtener 6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)-N-(4-(metilamino)bencil)hexanamida como un sólido amarillo (MS m/z 330,2 (M+1), Tiempo de retención 0,61 minutos).
Paso 2: A ácido (S)-2-((ferf-butoxicarbonil)amino)-3-fenilpropanoico (219 mg, 0,827 mmol) disuelto en DMF (5 mL) se añadieron DIEA (356 mg, 2,76 mmol) y HATU (288 mg, 0,758 mmol). Después de agitar durante 10 minutos a temperatura ambiente, se añadió 6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)-N-(4-(metilamino)bencil)hexanamida (227 mg, 0,689 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. Se agregó EtOAc (20 mL) a la reacción. A continuación, la mezcla de reacción se lavó con NaHCO3 acuoso saturado. La fase acuosa se extrajo con EtOAc(2 x 20 mL). La fase orgánica combinada se lavó con H2O (5 x 10 mL), se secó con MgSO4 anhidro, se concentró y se purificó con ISCO (EtOAc/Hexano 0-75%), lo que proporcionó el producto deseado. MS m/z 577,3 (M+1). Tiempo de retención 1,19 minutos. 1H RMN (400 MHz, DMSO-d6): 510,00 (s, 1H), 8,24 (t, J = 6,0 Hz, 1h), 7,52 (d, j = 8,4 Hz, 2H), 7,32-7,09 (m, 7H), 7,01 (s, 2H), 4,31 (m, 1H), 4,19 (d, J = 6,0 Hz, 2H), 3,38 (t, J = 7,0 Hz, 2H), 3,17 (d, J= 7,2 Hz, 2H), 3,00 (m, 1H), 2,85 (m, 1H), 2,10 (t, J = 7,4 Hz, 2H), 1,54-1,44 (m, 4H), 1,31 (s, 9H), 1,22-1,15 (m, 4H). El producto se disolvió en HCl 3 M en MeOH (5 mL). Los disolventes se eliminaron lentamente a presión reducida. El residuo se recuperó en acetonitrilo y H2O y se liofilizó para obtener (S)-N-(4-(2-amino-N-metil-3-fenilpropanamido)bencil)-6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamida en forma de sal de HCl. MS m/z 477,2 (M+1). Tiempo de retención 0,83 minutos.
Paso 3: A Boc-Val-Dil-Dap-OH (347 mg, 0,607 mmol) disuelto en DMF (4 mL) se añadieron DIEA (261 mg, 2,02 mmol) y HATU (282 mg, 0,49 mmol). Después de agitar durante 15 minutos a temperatura ambiente, se añadió (S)-N-(4-(2-amino-N-metil-3-fenilpropanamido)bencil)-6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamida (193 mg, 0,404 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. La mezcla de reacción se purificó mediante HPLC en fase inversa para proporcionar el producto deseado N-(4-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-N-metil-3-fenilpropanamido)bencilo)-6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamida. MS m/z 1030,5 (M+1). Tiempo de retención 1,430 minutos. El producto se disolvió en HCl metanólico 3 M (3 mL). Los disolventes se eliminaron a presión reducida. El residuo se recuperó en acetonitrilo y H2O y se liofilizó para obtener el producto deseado N-(4-((S)-2-((2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-2-amino-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-N-metil-3-fenilpropanamido)bencilo)-6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamida en forma de sal de HCl. MS m/z 930,5 (M+1). Tiempo de retención 1,07 minutos.
Pasos 4-5: Siguiendo el mismo procedimiento descrito para la preparación del compuesto 30 y el compuesto 31, se obtuvo el compuesto 44. MS m/z 1067,6 (M+1). Tiempo de retención 1,10 minutos.
Ejemplo de carga 83: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(3-azidopropilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (45)

Paso 1: A una solución agitada de azida sódica (3,50 g, 53,8 mmol) en agua (25 mL) se añadió una solución de 1,3-propanosulfona (6,10 g, 50,0 mmol) en acetona (25 mL). La mezcla de reacción se agitó a temperatura ambiente durante 24 horas y se concentró a sequedad. El sólido resultante se suspendió en éter dietílico (100 mL) y se agitó a reflujo durante 1 hora. La suspensión se enfrió hasta temperatura ambiente y el sólido se recogió por filtración, se lavó con acetona y éter dietílico y se secó al vacío, lo que proporcionó ácido 3-azido-1-propanosulfónico. MS m/z 188,1 (M+1). 1H RMN (400 MHz, CD3OD): 53,47 (t, J = 6,8 Hz, 2H), 2,87 (t, J = 7,6 Hz, 2H), 2,07-2,00 (m, 2H).
Paso 2: Se suspendió ácido 3-azido-1-propanosulfónico (2,07 g, 13,0 mmol) en tolueno. Se añadió PCls (2,61 g, 13,0 mmol). La mezcla se calentó a reflujo durante 3 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y se filtró para eliminar componentes insolubles. La masa retenida sobre el filtro se lavó con DCM. Los filtrados
combinados se concentraron para proporcionar cloruro de 3-azidopropano-1-sulfonilo como un aceite amarillo oscuro, que se utilizó en el paso siguiente sin purificación adicional.
Paso 3: A NH4OH (5 mL) enfriado a 0°C se añadió cloruro de 3-azidopropano-1 -sulfonilo (1,75 g, 9,53 mmol). Después de 10 minutos, la mezcla de reacción se calentó hasta temperatura ambiente y se agitó a la misma temperatura durante 3 horas. La mezcla oleosa se volvió transparente. La mezcla de reacción se extrajo con EtOAc tres veces. La fase orgánica se lavó con salmuera, se secó con MgSO4 anhidro y se concentró. El disolvente residual se eliminó además bajo alto vacío durante 18 horas para proporcionar 3-azidopropano-1-sulfonamida. MS m/z 187,1 (M+1). 1H RMN (400 MHz, CDCla): 54,83 (s, 2H), 3,51 (t, J = 6,4 Hz, 2H), 3,23 (t, J = 7,6 Hz, 2H), 2,17-2,10 (m, 2H).
Paso 4: Se disolvió ácido (S)-2-((ferf-butoxicarbonil)amino)-3-fenilpropanoico (100 mg, 0,38 mmol) en DMF (4 mL) seguido de la adición de DIEA (0,395 mL, 2,26 mmol) y HATU (358 mg, 0,940 mmol). Después de 15 minutos, se añadió 3-azidopropano-1-sulfonamida (186 mg, 1,13 mmol). La mezcla de reacción se agitó durante 2 horas, momento en el que el análisis LCMS indicó que la reacción estaba completa. El crudo se purificó mediante HPLC en fase inversa utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (S)-(1-(3-azidopropilsulfonamido)-1-oxo-3-fenilpropan-2-il)carbamato de ferf-butilo. MS m/z 312,1 (M+1-Boc). Tiempo de retención 1,15 minutos. El producto así obtenido (72,4 mg, 0,176 mmol) se disolvió en HCl metanólico 3 M (5 mL). El disolvente se eliminó a presión reducida. El residuo se recuperó en acetonitrilo y H2O y se liofilizó para proporcionar (S)-2-amino-N-((3-azidopropil)sulfonil)-3-fenilpropanamida como un sólido rosado amarillento. MS m/z 312,1 (M+1).
1H RMN (400 MHz, CD3OD): 57,42-7,31 (m, 5H), 4,16-4,13 (m, 1H), 3,51-3,47 (m, 4H), 3,32-3,26 (m, 1H), 3,13-3,08 (m, 1H), 2,00-1,94 (m, 2H).
Paso 5: A Boc-Val-Dil-Dap-OH (195 mg, 0,34 mmol) disuelto en DMF (4 mL) se añadieron DIEA (132 mg, 1,02 mmol) y HATU (108 mg, 0,28 mmol). La mezcla de reacción se agitó durante 15 minutos a temperatura ambiente antes de añadir (S)-2-amino-N-((3-azidopropil)sulfonil)-3-fenilpropanamida (59,2 mg, 0,17 mmol). La mezcla de reacción se agitó durante 2 horas más a temperatura ambiente. El crudo se purificó mediante HPLC en fase inversa para proporcionar el producto deseado (95 mg, 65% de rendimiento, MS m/z 865,4 (M+1), Tiempo de retención 1,43 minutos). El producto se disolvió en HCl 3 M en MeOH (3 mL). Los disolventes se eliminaron al vacío. A continuación, se añadieron acetonitrilo y H2O al residuo y la solución se liofilizó para obtener el producto deseado, (S)-1-(((3R,4S,5S)-1 -((S)-2-((1 fí,2fí)-3-(((S)-N-1 -(3-azidopropilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-2-amino-3-metil-1 -oxobutano. MS m/z 765,4 (M+1). Tiempo de retención 1,04 minutos.
Paso 6: A ácido (1 R,3S,4S)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxílico (16,5 mg, 0,068 mmol) en DMF (2,0 mL) se añadieron DIEA (17,6 mg, 0,137 mmol) y HATU (21,6 mg, 0,057 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 10 minutos antes de añadir (S)-1-(((3R,4S,5S)-1-(((S)-2-((1 R,2R)-3-(((S)-N-1-(3-azidopropilsulfonamido)-1-oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-3-oxoheptan-4-il)(metil)amino)-2-amino-3-metil-1-oxobutano (20 mg, sal de TFA, 0,023 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente, momento en el que el análisis LCMS indicó que la reacción estaba completa.
El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de ACN-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(3-azidopropilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-(ferf-butoxicarbonil)-2-azabiciclo[2.2.1]heptano-3-carboxamida. MS m/z 988,5 (M+1). Tiempo de retención 1,51 minutos. El producto así obtenido (9,4 mg, 0,0095 mmol) se disolvió en HCl metanólico (3 M, 2,0 mL). El disolvente se eliminó lentamente a presión reducida. El residuo se disolvió en acetonitrilo y H2O y se liofilizó para proporcionar el compuesto 45 como una sal de HCl. MS m/z 888,5 (M+1). Tiempo de retención 1,10 minutos.
Ejemplo de carga 84: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(3-azidopropilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (46)

il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1 -oxobutan-2-il)-2-azabiciclo[2.2.1]heptano-3-carboxamida (45) (8,8 mg, 0,0099 mmol) se disolvió en MeOH (2,0 mL). Se añadieron paraformaldehído (10,1 mg, 0,337 mmol) y ácido acético (0,0102 mL), seguidos de cianoborohidruro
sódico (21,2 mg, 0,337 mmol). La mezcla de reacción se calentó a 50 °C con agitación durante 1 hora. Se añadieron paraformaldehído (10,1 mg, 0,337 mmol), ácido acético (0,0102 mL) y cianoborohidruro sódico (21.2 mg, 0,337 mmol). Después de 1 hora a 50 °C, el análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de ACN-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 46. MS m/z 902,5 (M+1). Tiempo de retención 1,12 minutos.
Ejemplo de carga 85: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-N-1 -(3-(4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)metil)-1 H-1,2,3-triazol-1 -il)propilsulfonamido)-1 -oxo-3-fenilpropan-2-il)amino)-1 -metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (47)

fenilpropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (46)(5,2 mg, 0,0058 mmol), 1-(prop-2-in-1-il)-1 H-pirrol-2,5-diona (1,56 mg, 0,012 mmol) y CuSO4 (0,7 mg, 0,004 mmol) en DMF (2,0 mL) y H2O (0,5 mL) se trató con sal sódica del ácido L-ascórbico (2,5 mg, 0,014 mmol) y se agitó a temperatura ambiente durante 2 horas. Se añadieron CuSO4 (0,7 mg, 0,004 mmol) y sal sódica del ácido L-ascórbico (2,5 mg, 0,014 mmol) adicionales. Después de 2 horas adicionales a temperatura ambiente, el análisis LCMS indicó que la reacción se había completado. El crudo se purificó mediante HPLC en fase inversa, utilizando una columna C18, se eluyó con un 10-90% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron para obtener el compuesto 47. MS m/z 1037,4 (M+1). Tiempo de retención 1,00 minutos.
Ejemplo de carga 86: ((R)-1 -((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfonato de 6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexilhidrógeno (48).

A ácido ((R)-1-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-2-feniletil)fosfínico (9) (10,2 mg, 0,011 mmol) en piridina (2 mL) se añadió 1 -(6-hidroxihexil)-1 H-pirrol-2,5-diona (13,5 mg, 0,068 mmol) y después cloruro de pivaloílo (40 mg, 0,332 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas y la reacción se monitorizó mediante LCMS hasta que desapareció un 90% del ácido fosfínico. A continuación, se añadió una solución de I2 recién preparada en un 5% de H2O en piridina. Una vez se completó el paso de oxidación, la piridina se eliminó mediante alto vacío. El crudo se disolvió en acetonitrilo y el crudo se purificó mediante HPLC en fase inversa, columna C18, se eluyó con un 10-60% de acetonitrilo-H2O que contenía un 0,05% de TFA. Las fracciones que contenían el producto deseado se concentraron para obtener el compuesto 48. MS m/z 971,5 (M+1). Tiempo de retención 1,038 minutos.
Ejemplo de carga 87: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((1 R,2R)-3-(((S)-1 -(3-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanamido)fenil)-3-hidroxipropan-2-il)amino)-1-metoxi-2-metil-3-oxopropil)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (90)
Se trató EMCA (1,3 mg, 0,006 mmol) en DMF (0,5 mL) con DIEA (0,006 mL, 0,03 mmol) y HATU (2,3 mg, 0,006 mmol) a t.a. durante 10 min, y a continuación se añadió la sal de TFA del compuesto (52) (6 mg, 0,006 mmol) en DMF (0,5 mL). La reacción se agitó a t.a. durante 16 h. El crudo se purificó mediante HPLC preparativa (10-45% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (90) en forma de sal de TFA. MS m/z 950,6 (M+H). Tiempo de retención 0,934 min.
Ejemplo de carga 88: (3-((S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-hidroxipropil)fenil)carbamato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (91)

Se añadió piridina (0,25 mL) a la sal de TFA del compuesto (52) (6 mg, 0,006 mmol), MC-Val-Cit-PAB-PNP (13 mg, 0,018 mmol) en d MF (1 mL), seguida de HOAT (0,8 mg, 0,006 mmol) y DlEA (13 mg, 0,098 mmol). La reacción se agitó a 40 °C durante 48 h. El crudo se purificó mediante HPLC preparativa (25-40% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (91) en forma de sal de TFA. MS m/z 678,6 (M/2+H). Tiempo de retención 0,959 min.
Ejemplo de carga 89: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-3-metoxi-1 -((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-(N-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)hexanoil)-sulfamoilpropan)-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (92)
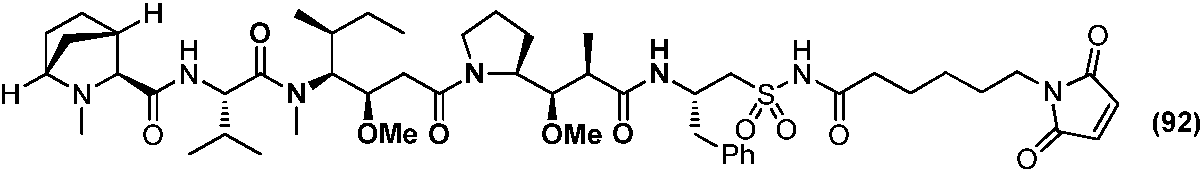
Se disolvió EMCA (12,1 mg, 0,057 mmol) en DMF (1 mL). Se añadieron DIEA (0,0024 mL) y HATU (19,7 mg, 0,052 mmol). A continuación, se añadió (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1-metoxi-2-metil-3-oxo-3-(((S)-1-fenil-3-sulfamoilpropan-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida ((55), 26,4 mg, 0,029 mmol) en DMF (2 mL). La reacción se agitó a t.a. durante 2 h. A continuación, se añadieron EMCA (12,1 mg, 0,057 mmol), DIEA (0,0024 mL) y HATU (19,7 mg, 0,052 mmol) adicionales. Después de 2 h, se añadieron de nuevo EMCA (12,1 mg, 0,057 mmol), DIEA (0,0024 mL) y HATU (19,7 mg, 0,052 mmol). A continuación, la reacción se calentó a 50 °C durante 2 h. La reacción se enfrió y se añadieron e Mc A (12,1 mg, 0,057mmol), DIEA (0,0024 mL) y HATU (19,7 mg, 0,052 mmol) adicionales. La reacción se agitó durante 16 h a t.a. El LCMS indicó que aproximadamente un 20% del compuesto (55) se había convertido en el producto. La mezcla de reacción se purificó mediante HPLC preparativa (30-50% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (1 R,3S,4S)-N-((S)-1-(((3R,4S,5S)-3-metoxi-1-((S)-2-((1 R,2R)-1 -metoxi-2-metil-3-oxo-3-(((S)-1 -fenil-3-(N-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)hexanoil)-sulfamoilpropan)-2-il)amino)propil)pirrolidin-1-il)-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (92). MS m/z 998,5 (M+1). Tiempo de retención 1,041 min.
Ejemplo de carga 90: (1 R,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((3R,4R,7S)-7-bencil-20-(4-((2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)metil)-1 H-1,2,3-triazol-1 -il)-4-metil-5,8-dioxo-2,12,15,18-tetraoxa-6,9-diazaicosan-3-il)pirrolidin-1 -il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (93)

Paso 1: Se trató ácido (S)-2-((f-butoxicarbonil)amino)-3-fenilpropanoico (175 mg, 0,66 mmol) en DMF (4 mL) con DIEA (0,48 mL, 2,75 mmol) y HATU (230 mg, 0,605 mmol) durante 15 min, seguidos de la adición de 2-(2-(2-(2azidoetoxi)etoxi)etoxi)etanamina (120 mg, 0,55 mmol). La mezcla de reacción se agitó durante la noche. El crudo se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (S)-(1-azido-13-oxo-15-fenil-3,6,9-trioxa-12-azapentadecan-14-il)carbamato de f-butilo. MS m/z 466,3 (M+1). Tiempo de retención 1,170 min.
Paso 2: Se disolvió (S)-(1-azido-13-oxo-15-fenil-3,6,9-trioxa-12-azapentadecan-14-il)carbamato de f-butilo (117 mg, 0,251 mmol) en HCl metanólico (3 M, 5 mL). El disolvente se eliminó lentamente mediante evaporación, lo que dio como resultado la eliminación completa del grupo Boc. El disolvente residual se eliminó adicionalmente a presión reducida durante la noche para obtener (S)-2-amino-N-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etil)-3-fenilpropanamida como sal de HCl. MS m/z 366,1 (M+1). Tiempo de retención 0,858 min.
Paso 3: Se trató (2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (Paso 2, Ejemplo 12, 8 mg, 0,01 mmol) en DMF (1 mL) con DIEA (0,011 mL, 0,066 mmol) y HATU (4,63 mg, 0,012 mmol) durante 15 min, seguidos de la adición de (S)-2-amino-N-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etil)-3-fenilpropanamida (5,3 mg, 0,013 mmol) en DMF (1 mL). La reacción se agitó durante 2 h a t.a. El crudo se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (1 R,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((3R,4R,7S)-20-azido-7-bencil-4-metil-5,8-dioxo-2,12,15,18-tetraoxa-6,9-diazaicosan-3-il)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida. MS m/z 956,5 (M+1). Tiempo de retención 1,051 min.
Paso 4: Al producto obtenido en el paso 3 (6,2 mg, 0,0058 mmol) y 1 -(prop-2-in-1 -il)-1 H-pirrol-2,5-diona (1,6 mg, 0,012 mmol) en f-BuOH (1 mL) y agua (1 mL) se añadieron L-ascorbato sódico (1,1 mg, 0,0058 mmol) en 0,2 mL de H2O y se añadieron CuSO4 (0,2 mg, 0,001 mmol) en 0,1 mL de agua. La mezcla de reacción se agitó a t.a. durante 4 h y después se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener el compuesto (93). MS m/z 1091,6 (M+1). Tiempo de retención 0,980 min.
Ejemplo de carga 91: (1 R,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((3R,4R,7S)-7-bencil-17-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)-4-metil-5,8-dioxo-2,12,15-trioxa-6,9-diazaheptadecan-3-il)pirrolidin-1-il)-3-metoxi-5-metil-1-oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (94)
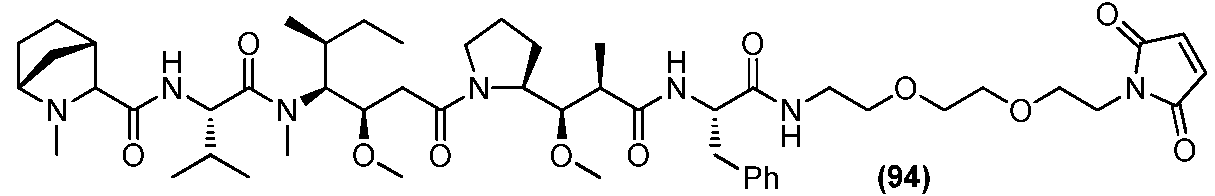
Paso 1: Se combinaron (2-(2-(2-aminoetoxi)etoxi)etil)carbamato de f-butilo (250 mg, 1,0 mmol) y 2,5-dioxo-2,5-dihidro-1H-pirrol-1-carboxilato de metilo (156 mg, 1,0 mmol) en NaHCO3 acuoso saturado (10 mL) y se agitó durante 1,5 h a 0 °C. La mezcla de reacción se acidificó hasta pH 2 con ácido clorhídrico (2 M) y se extrajo con EtOAc. La fase orgánica se lavó con salmuera, se secó con MgSO4 y se concentró. El crudo se purificó con ISCO utilizando un 0-4% de MeOH/DCM para proporcionar (2-(2-(2-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)etoxi)etoxi)etil)carbamato de f-butilo como un aceite incoloro. m S m/z 229,2 (M+1-Boc). Tiempo de retención 0,963 min. 1H RMN (400 MHz, Cloroformo-d) 56,71 (s, 2H), 5,04 (s a, 1H), 3,74 (t, J = 5,4 Hz, 2H), 3,64 (t, J = 5,4 Hz, 2H), 3,61-3,59 (m, 2H), 3,56-3,54 (m, 2H), 3,50 (t, J = 5,2 Hz, 2H), 3,31 -3,26(m, 2H), 1,44 (s, 9H).
Paso 2: Se trató (2-(2-(2-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)etoxi)etoxi)etil)carbamato de f-butilo (184 mg, 0,56 mmol) en DCM (2 mL) con TFA (0,4 mL) a 0 °C durante 30 min y después a t.a. durante 2 h. La mezcla de reacción se concentró para proporcionar 1-(2-(2-(2-aminoetoxi)etoxi)etil)-1H-pirrol-2,5- en forma de sal de TFA. MS m/z 229,2 (M+1). Tiempo de retención 0,353 min.
Paso 3: Se activó Boc-L-Phe-OH (30 mg, 0,113 mmol) en DMF (1 mL) con DIEA (88 mg) y HATU (43 mg, 0,113 mmol) durante 15 min, y se añadió sal de TFA de 1-(2-(2-(2-aminoetoxi)etoxi)etil)-1H-pirrol-2,5-diona (46,4 mg) en DMF (1 mL). La mezcla de reacción se agitó a t.a. durante 2 h y después se purificó mediante HPLC preparativa (20-70% de acetonitrilo-H2O que contenía un 0,05% de TFA) para obtener (S)-(1-((2-(2-(2-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)etoxi)etoxi)etil)amino)-1-oxo-3-fenilpropan-2-il)carbamato de f-butilo. MS m/z 476,2 (M+1). Tiempo de retención 1,091 min. Este producto (31 mg, 0,065 mmol) en DCM (2 mL) se trató con TFA (0,2 mL) a 0 °C durante 30 min y después a t.a. durante 2 h. La mezcla de reacción se concentró para proporcionar (S)-2-amino-N-(2-(2-(2-(2,5-dioxo-2.5- dihidro-1H-pirrol-1-il)etoxi)etoxi)etil)-3-fenilpropanamida como sal de TFA. MS m/z 376,2 (M+1). Tiempo de retención 0,649 min.
Paso 4: A la sal de TFA del ácido (2R,3R)-3-((S)-1-((3fí,4S,5S)-4-((S)-N,3-dimetil-2-((1 R,3S,4S)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamido)butanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanoico (Paso 2, Ejemplo 12) (7,2 mg, 0,010 mmol) en DMF (1 mL) se añadieron DIEA (7,7 mg) y HATU (4,18 mg, 0,011 mmol). La reacción se agitó durante 15 min, y se añadió la sal de TFA de (S)-2-amino-N-(2-(2-(2-(2,5-dioxo-2.5- dihidro-1H-pirrol-1-il)etoxi)etoxi)etil)-3-fenilpropanamida (6,3 mg, 0,013 mmol) en DMF (1 mL). La mezcla de
reacción se agitó a t.a. durante 2 h y se purificó mediante HPLC preparativa (20-70% de acetonitrilo-hbO que contenía un 0,05% de TFA) para obtener el compuesto (93) en forma de sal de TFA. MS m/z 966,5 (M+1). Tiempo de retención 1,016 min.
Ejemplo de carga 92: (1 R,3S,4S)-N-((S)-1 -(((3R,4S,5S)-1 -((S)-2-((3R,4R,7S)-7-bencil-14-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)-4-metil-5,8-dioxo-2,12-dioxa-6,9-diazatetradecan-3-il)pirrolidin-1 -il)-3-metoxi-5-metil-1 -oxoheptan-4-il)(metil)amino)-3-metil-1-oxobutan-2-il)-2-metil-2-azabiciclo[2.2.1]heptano-3-carboxamida (95)

El compuesto (95) se preparó mediante el método descrito para el compuesto (94) utilizando (2-(2-aminoetoxi)etil)carbamato de f-butilo en lugar de (2-(2-(2-aminoetoxi)etoxi)etil)carbamato de f-butilo. MS m/z 922,5 (M+1). Tiempo de retención 1,044 min.
Procedimiento sintético para los análogos de la Coenzima A
Ejemplo de carga 93: Aducto con 3-buten-2-ona de Coenzima A (49)

Se disolvió sal de trilitio de la Coenzima A (259 mg, Sigma, ensayo >93%) en 2,0 mL de tampón de fosfato con EDTA (fosfato 100 mM, EDTA 5 mM, pH 7,5). A la mezcla de reacción se añadió 3-buten-2-ona (29,0 pL, Aldrich, 99%), y la mezcla de reacción se dejó reposar a 20 °C durante 75 minutos. La mezcla de reacción completa se cargó en una columna ISCO C18Aq Gold de 15,5 g que se preequilibró con un 100% de H2O. El producto deseado se eluyó con un 100% de H2O. Las fracciones que contenían el producto deseado puro se combinaron y se liofilizaron, lo que proporcionó el compuesto 49 como un sólido cristalino. MS (ESI+) m/z 838,2 (M+1). H-RMN (400MHz, D2O) 58,525 (s, 1H), 8,235 (s, 1H), 6,140 (d, 1H, J=7,2Hz), 4,746 (m, 1H), 4,546 (s a, 1H), 4,195 (s a, 1H), 3,979 (s, 1H), 3,786 (dd, 1H, J= 4,8, 9,6Hz), 3,510 (dd, 1H, J=4,8, 9,6Hz), 3,429 (t, 2H, J= 6,6Hz), 3,294S (t, 2H, J=6,6Hz), 2,812 (t, 2H, J=6,8Hz), 2,676 (t, 2H, J=6,8Hz), 2,604 (t, 2H, J=6,8Hz), 2,420 (t, 2H, J=6,6Hz), 2,168 (s, 3H), 0,842 (s, 3H), 0,711 (s, 3H) (nota: algunos picos que solapan con D2O no se indican).
Ejemplo de carga 94: Análogo de cetona-Coenzima A (CoA-(i-10))
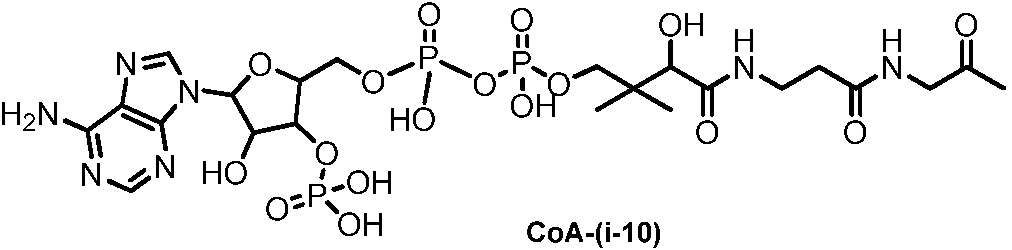
El compuesto (i-10) se convirtió en el análogo de CoA funcionalizada con cetona CoA-(i-10) haciendo reaccionar una concentración 5 mM del compuesto (i-10) con una concentración 25 mM de ATP en presencia de CoAA de Sfaphylococcus aureus 10 pM, CoAD de Escherichia coli 25 pM y CoAE de Escherichia coli 20 pM durante aproximadamente 14 h a 37 °C en tampón HEPES 50 mM (pH 8,0) que contenía 20 MgCl2. La enzima soluble se separó por ultrafiltración a través de un filtro centrífugo Amicon Ultra con un límite de 10 kDa. La conversión enzimática del compuesto (i-10) en el análogo de CoA CoA-(i-10) se verificó mediante la formación del ADC de anti-Her2-HC-ins388-ybbR-(i-10)-22 (remítase a la Tabla 9 y a la Tabla 10).
Ejemplo de carga 95: Análogo de azida-Coenzima A (CoA-(i-11))

El compuesto (i-11) se convirtió en el análogo de CoA funcionalizado con azida CoA-(i-11) haciendo reaccionar una concentración 5 mM del compuesto (i-11) con una concentración 25 mM de ATP en presencia de CoAA de
Staphylococcus aureus 10 pM, CoAD de Escheríchia coli 25 pM y CoAE de Escheríchia coli 20 pM durante aproximadamente 14 h a 37 °C en tampón HEPES 50 mM (pH 8,0) que contenía 20 MgCÍ2. La enzima soluble se separó por ultrafiltración a través de un filtro centrífugo Amicon Ultra con un límite de 10 kDa. La conversión enzimática del compuesto (i-11) en el análogo de CoA CoA-(i-11) se verificó mediante análisis LC-MS después de la reacción de una pequeña fracción del ultrafiltrado con un exceso molar en un factor cinco del compuesto (75). La reacción de química click sin cobre se llevó a cabo durante 3 h a 23 °C en un 50% (v/V) de DMSO/H2O, y la mezcla de reacción se separó en una columna Acquity UPLC HSS T3 en fase inversa (100 Á, 2.1 mm x 50 mm, Waters) utilizando un gradiente de elución de 1,35 min de un 10 a un 100% de acetonitrilo en agua que contenía un 0,05% de TFA con un caudal de 0,9 mL/min. El análisis espectral de masas reveló la presencia del aducto deseado con lo cual se confirmó la conversión del compuesto (i-11) con las tres enzimas biosintéticas de CoA en el análogo de CoA CoA-(i-11). MS m/z 904,6 ((M+2/2). Tiempo de retención 0,94 minutos.
Ejemplo de carga 96: Análogo de cetona-Coenzima A (CoA-(i-12))

El compuesto (i-12) se convirtió en el análogo de CoA funcionalizado con cetona CoA-(i-12) haciendo reaccionar una concentración 5 mM del compuesto (i-12) con una concentración 25 mM de ATP en presencia de CoAA de Staphylococcus aureus 10 pM, CoAD de Escherichia coli 25 pM y CoAE de Escherichia coli 20 pM durante aproximadamente 16 h a 37 °C en tampón HEPES 50 mM (pH 8,0) que contenía 20 MgCl2. Después de la centrifugación de la mezcla de reacción a 20817 x g durante 2 min, la enzima soluble se separó mediante ultrafiltración a través de un filtro centrífugo Amicon Ultra con un límite de 10 kDa (15 min; 14000 x g). La conversión enzimática del compuesto (i-12) en el análogo de CoA CoA-(i-12) se verificó mediante la formación del ADC de anti-Her2-HC-ins388-ybbR-(i-12)-22 (remítase a la Tabla 10).
Ejemplo de carga 97: Análogo de cetona-Coenzima A (CoA-(i-13))

El compuesto (i-13) se convirtió en el análogo de CoA funcionalizado con cetona CoA-(i-13) haciendo reaccionar una concentración 10 mM del compuesto (i-13) con una concentración 25 mM de ATP en presencia de CoAA de Staphylococcus aureus 10 pM, CoAD de Escherichia coli 25 pM y CoAE de Escherichia coli 20 pM durante aproximadamente 48 h a 37 °C en tampón HEPES 50 mM (pH 8,0) que contenía MgCl2 20 mM. Después de la centrifugación de la mezcla de reacción a 20817 x g durante 2 min, la enzima soluble se separó mediante ultrafiltración a través de un filtro centrífugo Amicon Ultra con un límite de 10 kDa (15 min; 14000 x g).
Procedimiento sintético del péptido comparativo
Síntesis del ácido (S)-2-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)-W-met¡lhexanam¡do)-3-met¡lbutanam¡do)-W,3-d¡met¡lbutanam¡do)-3-metoxi-5-met¡lheptano¡l)p¡rrol¡d¡n-2-¡l)-3-metox¡-2-metilpropanamido)-3-fenilpropanoico (MC-MMAF)

Se disolvió MMAF-OMe (135 mg, Concortis Biosystems) en CH3CN (10 mL). A la solución transparente resultante se añadieron 5 mL de agua, seguidos de 0,375 mL de hidróxido sódico acuoso 1 N (certificado, Fisher Scientific). La mezcla de reacción se agitó magnéticamente a 21 °C durante 18 horas, momento en el que el análisis LCMS indicó una reacción completa. La mezcla de reacción se congeló y se liofilizó, lo que proporcionó la sal sódica de MMAF. Tiempo de retención en LCMS 0,911 minutos. MS (ESI+) m/z 732,5 (M+1 ). La sal sódica de MMAF completa así obtenida en la reacción previa se disolvió en 10mL de DMSO. En un recipiente de reacción separado, se trató ácido 6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoico (95 mg) con HATU (165 mg) y DIEA (0,126 mL) en 3,0 mL de DMSO a 21 °C durante 25 min. La mezcla de reacción completa del éster activado se añadió a la solución de sal sódica de MMAF, y la mezcla de reacción se agitó a la misma temperatura durante 3 horas. La mezcla de reacción se repartió entre 40 mL de EtOAc y 20 mL de ácido cítrico acuoso al 5%. La fase orgánica se separó y la fase acuosa se extrajo con 20 mL de EtOAc. Las fases orgánicas combinadas se lavaron con 10 mL de NaCl acuoso saturado, se secaron con MgSO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó en un instrumento ISCO CombiFlash utilizando una columna ISCO C18gold de 15,5 g. El material deseado se eluyó con un 50% de CH3CN en H2O. Las fracciones que contenían el producto deseado se combinaron y se liofilizaron, lo que proporcionó el compuesto como un sólido blanco. LCMS tiempo de retención 1,392 minutos. MS (ESI+) m/z 925,6 (M+1).
Ejemplo 98: Ensayo de destrucción de células in vitro de las cargas
Para la evaluación de la potencia de destrucción celular de los péptidos citotóxicos de Fórmula (I) in vitro, se llevaron a cabo ensayos de proliferación celular en paralelo con 8 líneas celulares: Clon 16 de MDA-MB231, clon 40, células JimT1, HCC1954, H526, KU812, CMK11-5 y células Jurkat. Además, se llevaron a cabo ensayos de proliferación celular, in vitro, utilizando 3 líneas celulares diferentes: A375, SKBR3 y NCI-N87. Todas las líneas celulares se describen en más detalle en el Ejemplo 106 y también se utilizaron para evaluar la potencia in vitro de los inmunoconjugados de la invención. Los ensayos de proliferación celular se llevaron a cabo con Cell-Titer-Glo™ (Promega) cinco días después de que las células se incubasen con varias concentraciones de ADC (Riss etal., (2004) Assay Drug Dev Technol. 2:51 -62). En algunos estudios, los ensayos basados en células son de alto rendimiento y se llevaron a cabo en un sistema automatizado (Melnick et al., (2006) Proc Natl Acad Sci U S A. 103:3153-3158). Las potencias de destrucción de células in vitro obtenidas para determinados ejemplos de péptidos citotóxicos de Fórmula (I) se proporcionan en la Tabla 2a y la Tabla 2b.
T l 2 . D r i n l l in vi r I nM rmin i i xi F rm l I

Tabla 2b. Destrucción de células in vitro (CI50 (nM)) de determinados péptidos citotóxicos de Fórmula (I)

n. d., no determinado
Restos de unión a antígeno
El resto de unión a antígeno en la Fórmula (II) o (III) puede ser cualquier resto que se una selectivamente a un marcador de la superficie celular que se encuentra en un tipo de células diana. En algunos aspectos, Ab es un anticuerpo o un fragmento de anticuerpo (por ejemplo, fragmento de unión a antígeno de un anticuerpo) que se une específicamente a un antígeno que se encuentra predominante o preferencialmente en la superficie de las células cancerosas, por ejemplo, un antígeno asociado a tumores. En algunos aspectos, Ab es un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmento de unión a antígeno) que se une específicamente a una proteína receptora de la superficie celular u otras moléculas de la superficie celular, un factor regulador de la supervivencia celular, un factor
regulador de la proliferación celular, una molécula asociada con (por ejemplo, de la que se sabe o se sospecha que contribuye funcionalmente a) el desarrollo o la diferenciación tisular, una linfocina, una citocina, una molécula implicada en la regulación del ciclo celular, una molécula implicada en la vasculogénesis o una molécula asociada con (por ejemplo, de la que se sabe o se sospecha que contribuye funcionalmente a) la angiogénesis. Un antígeno asociado con un tumor puede ser un factor de diferenciación de tipo clúster (es decir, una proteína CD). En algunos aspectos de la invención, el resto de unión a antígeno de la invención se une específicamente a un antígeno. En algunos aspectos de la invención, el resto de unión a antígeno de la invención se une específicamente a dos o más antígenos descritos en la presente, por ejemplo, el resto de unión a antígeno de la invención es un anticuerpo biespecífico o multiespecífico o un fragmento de unión a antígeno de este.
Los anticuerpos o fragmentos de unión a antígeno ilustrativos incluyen anticuerpo anti-receptor de estrógenos, anticuerpo anti-receptor de progesterona, anticuerpo anti-p53, anticuerpo anti-HER-2, anticuerpo anti-EGFR, anticuerpo anti-catepsina D, anticuerpo anti-Bcl-2, anticuerpo anti-cadherina E, anticuerpo anti-CA125, anticuerpo anti-CA15-3, anticuerpo anti-CA19-9, anticuerpo anti-c-erbB-2, anticuerpo antiglicoproteína P, anticuerpo anti-CEA, anticuerpo antiproteína del retinoblastoma, anticuerpo antioncoproteína ras, anticuerpo anti-Lewis X, anticuerpo anti-Ki-67, anticuerpo anti-PCNA, anticuerpo anti-CD3, anticuerpo anti-CD4, anticuerpo anti-CD5, anticuerpo anti-CD7, anticuerpo anti-CD8, anticuerpo anti-CD9/p24, anticuerpo anti-CD1, anticuerpo anti-CD11c, anticuerpo anti-CD13, anticuerpo anti-CD14, anticuerpo anti-CD15, anticuerpo anti-CD19, anticuerpo anti-CD20, anticuerpo anti-CD22, anticuerpo anti-CD23, anticuerpo anti-CD30, anticuerpo anti-CD31, anticuerpo anti-CD33, anticuerpo anti-CD34, anticuerpo anti-CD35, anticuerpo anti-CD38, anticuerpo anti-CD39, anticuerpo anti-CD41, anticuerpo anti-LCA/CD45, anticuerpo anti-CD45RO, anticuerpo anti-CD45RA, anticuerpo anti-CD71, anticuerpo anti-CD95/Fas, anticuerpo anti-CD99, anticuerpo anti-CD100, anticuerpo anti-S-100, anticuerpo anti-CD106, anticuerpo antiubiquitina, anticuerpo anti-c-myc, anticuerpo anticitoqueratina, anticuerpo anticadenas ligeras lambda, anticuerpo antimelanosomas, anticuerpo antiantígeno específico de la próstata, anticuerpo antiantígeno tau, anticuerpo antifibrina, anticuerpo antiqueratinas y anticuerpo antiantígeno Tn.
En una realización, el resto de unión a antígeno de los conjugados de anticuerpo-fármaco (ADC) de Fórmula (II) se une específicamente a un receptor codificado por un gen ErbB. El resto de unión a antígeno se puede unir específicamente a un receptor de ErbB seleccionado entre EGFR, HER2, HER3 y HER4. El resto de unión a antígeno puede ser un anticuerpo que se unirá específicamente al dominio extracelular (ECD) del receptor HER2 e inhibirá el crecimiento de las células tumorales que sobreexpresen el receptor HER2. El anticuerpo puede ser un anticuerpo monoclonal, por ejemplo, un anticuerpo monoclonal murino, un anticuerpo quimérico o un anticuerpo humanizado. Un anticuerpo humanizado puede ser huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 o huMAb4D5-8 (trastuzumab). El anticuerpo puede ser un fragmento de anticuerpo, por ejemplo, un fragmento Fab.
Los restos de unión a antígeno en la Fórmula (II) o (III) incluyen anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión a antígeno) contra receptores de la superficie celular y antígenos asociados a tumores. Dichos antígenos asociados con tumores son conocidos en la técnica y se pueden preparar para su uso en la generación de anticuerpos utilizando métodos e información que son muy conocidos en la técnica. En los intentos por descubrir dianas celulares eficaces para el diagnóstico y la terapia del cáncer, los investigadores han tratado de identificar polipéptidos transmembrana o asociados con tumores de otro modo que se expresan específicamente en la superficie de uno o más tipos particulares de células cancerosas, en comparación con una o más células no cancerosas normales. A menudo, dichos polipéptidos asociados con tumores se expresan de forma más abundante sobre la superficie de las células cancerosas, en comparación con la superficie de las células no cancerosas. La identificación de dichos polipéptidos antigénicos de la superficie celular asociados con tumores ha dado lugar a la capacidad de utilizar específicamente como diana células cancerosas para su destrucción a través de terapias basadas en anticuerpos. Los anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmento de unión a antígeno) útiles para los inmunoconjugados de la invención incluyen anticuerpos modificados o diseñados tales como un anticuerpo modificado para introducir un residuo de cisteína (Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y etal.; NatBiotechnol2008, 26:925-932; documento WO2013093809), u otro aminoácido reactivo, que incluye Pcl, pirrolisina, etiquetas peptídicas (tales como etiquetas S6, A1 e ybbR), y aminoácidos artificiales, en lugar de al menos un aminoácido de la secuencia nativa, con lo cual se proporciona así un sitio reactivo en el anticuerpo o fragmento de unión a antígeno para su conjugación a un péptido citotóxico de Fórmula (I) o una subfórmula de esta. Por ejemplo, los anticuerpos o fragmentos de anticuerpo se pueden modificar para que incorporen Pcl o pirrolisina (W. Ou et al. (2011) PNAS 108 (26), 10437-10442) o aminoácidos artificiales (J.Y. Axup, K.M. Bajjuri, M. Ritland, B.M. Hutchins, C.H. Kim, S.A. Kazane, R. Halder, J.S. Forsyth, A.F. Santidrian, K. Stafin, Y. Lu et al. Proc Natl Acad Sci U S A, 109 (2012), pp. 16101-16106; para consultar una revisión, remítase a C.C. Liu y P.G. Schultz (2010) Annu Rev Biochem 79, 413-444; C.H. Kim, J.Y. Axup, P.G. Schultz (2013) Curr Opin Chem Biol. 17, 412-419) como sitios para la conjugación a un fármaco. De forma similar, se pueden introducir en un anticuerpo etiquetas peptídicas para los métodos de conjugación enzimática (Strop P. et al. Chem Biol. 2013, 20(2):161-7; Rabuka D., Curr Opin Chem Biol. 2010 Dec;14(6):790-6; Rabuka D,et al., Nat Protoc. 2012, 7(6):1052-67). Otro ejemplo es el uso de 4’-fosfopanteteinil-transferasas (PPTasa) para la conjugación de análogos de Co-enzima A (documento WO2013184514). Los métodos para conjugar dichos anticuerpos alterados o modificados con cargas o combinaciones de conector-carga son conocidos en la técnica.
Los restos de unión a antígeno (por ejemplo, anticuerpos y fragmentos de unión a antígeno) útiles en la invención pueden tener también otras modificaciones o estar conjugados con otros restos tales como etiquetas de polietilenglicol, albúmina y otro polipéptido de fusión.
Los anticuerpos utilizados en los ejemplos de la presente tienen las secuencias de cadena ligera y cadena pesada enumeradas en la Tabla 3. Estos anticuerpos se diseñaron para que contuviesen residuos de cisteína o etiquetas enzimáticas PPTasa para la conjugación con especificidad por un sitio con péptidos citotóxicos de la invención. Los ejemplos de la presente ilustran que estos anticuerpos diseñados son un anticuerpo adecuado para su uso en los inmunoconjugados de Fórmula (II) o (III). Además, también se pueden utilizar anticuerpos no diseñados para la preparación de los inmunoconjugados de Fórmula (II) o (III) mediante métodos tradicionales (Carter PJ, Senter PD, Antibody-drug conjugates forcáncer therapy, Cancer J. 2008, 14(3): 154-69; J.E. Stefano, M. Busch, L. Hou, A. Park, y D.A. Gianolio, págs. 145-171, y M.-P. Brun y L. Gauzy-Lazo, págs. 173-187 en Antibody-Drug Conjúgate, Methods in Molecular Biology, Vol. 1045, Editor L. Ducry, Humana Press (2013).
Tabla 3. Secuencias de aminoácidos de los anticuer os de eem lo


La SEQ ID NO: 1 y la SEQ ID NO: 2 son la secuencia de aminoácidos completa de la cadena pesada (HC) y la cadena ligera (LC) del anticuerpo anti-Her2 de origen natural, respectivamente. Las regiones CDR están subrayadas. La SEQ ID NO: 3 es la secuencia de aminoácidos de la región constante de LC de anti-Her2 LC-S159C y del anticuerpo mutante anticuerpo 20507-LC-S159C. La SEQ ID NO: 4, SEQ ID NO: 5 y SEQ ID NO: 6 son las secuencias de aminoácidos de las regiones constantes para la cadena pesada de los anticuerpos mutantes HC-E152C y HC-S375C y HC-E152C-S375C, respectivamente. La SEQ ID NO:7 es la mutación LC-K107C de cadena ligera del anticuerpo 20507. La SEQ ID NO: 8 es la secuencia de aminoácidos de la región constante de la cadena pesada mutante para anti-Her2 HC-ins388-ybbR donde la etiqueta ybbR se inserta después del residuo Glu388 de HC. La SEQ ID NO: 9 es la secuencia de aminoácidos de la región constante de la cadena pesada mutante para ambos anticuerpos anti-Her2 y anticuerpo 20507 HC-ins388-A1, donde la etiqueta A1 se inserta después del residuo Glu388 de HC. El residuo Cys mutante y las etiquetas de secuencias insertadas de ybbR y A1 se muestran en negrita y están subrayadas en las secuencias de las cadenas mutantes correspondientes. La SEQ ID NO: 10 y la SEQ ID NO: 11 son las secuencias de aminoácidos de las regiones constantes de la cadena pesada y la cadena ligera del anticuerpo 20507 de origen natural, respectivamente.
Producción del anticuerpo
Los anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión a anticuerpo) de la invención se pueden producir por cualquier medio conocido en la técnica, que incluye expresión recombinante, síntesis química y digestión enzimática de tetrámeros de anticuerpos, mientras que los anticuerpos monoclonales completos se pueden obtener, por ejemplo, mediante producción de hibridomas o recombinante. La expresión recombinante puede ser a partir de cualesquiera células hospedadoras apropiadas conocidas en la técnica, por ejemplo, células hospedadoras de mamífero, células hospedadoras bacterianas, células hospedadoras de levadura, células hospedadoras de insecto, etc.
La invención proporciona además polinucleótidos que codifican los anticuerpos descritos en la presente, p. ej., polinucleótidos que codifican regiones variables de cadena ligera o pesada o segmentos que comprenden las regiones determinantes de la complementariedad según se describen en la presente.
Las secuencias de polinucleótidos se pueden producir por síntesis de novo de ADN en fase sólida o por mutagénesis por PCR de una secuencia existente (por ejemplo, secuencias como las descritas en los ejemplos siguientes) que codifica un anticuerpo o su fragmento de unión. La síntesis química directa de ácidos nucleicos se puede lograr mediante métodos conocidos en la técnica tales como el método del fosfotriéster de Narang et al., Meth. Enzymol.
68:90, 1979; el método del fosfodiéster de Brown et al., Meth. Enzymol. 68:109, 1979; el método de la dietilfosforamidita de Beaucage et al., Tetra. Lett., 22:1859, 1981; y el método del soporte sólido de la patente de EE. UU. N.° 4458 066. La introducción de mutaciones en una secuencia de polinucleótidos por PCR se puede realizar como se describe, por ejemplo, en PCR Technology: Principies and Applications for DNA Amplification, H.A. Erlich (Ed.), Freeman Press, Ny , NY, 1992; PCR Pr o t o c o l s : A Guide to Methods and Applications, Innis et al. (Ed.), Academic Press, San Diego, CA, 1990; Mattila et al., Nucleic Acids Res. 19:967, 1991; y Eckert et al., PCR Methods and Applications 1:17, 1991.
También se proporcionan en la invención vectores de expresión y células hospedadoras para producir los anticuerpos o fragmentos de anticuerpos descritos anteriormente. Se pueden emplear diversos vectores de expresión para expresar los polinucleótidos que codifican las cadenas o fragmentos de unión de los anticuerpos de la invención. Tanto los vectores de expresión virales como los no virales se pueden utilizar para producir los anticuerpos en una célula hospedadora de mamífero. Los vectores y sistemas no virales incluyen plásmidos, vectores episómicos, habitualmente con un casete de expresión para expresar una proteína o ARN, y cromosomas artificiales humanos (remítase, por ejemplo, a Harrington et al., Nat Genet 15:345, 1997). Por ejemplo, los vectores no virales útiles para la expresión de los polinucleótidos y polipéptidos en células de mamíferos (por ejemplo, humanas) incluyen pThioHis A, B y C, pcDNA3.1/His, pEBVHis A, B y C, (Invitrogen, San Diego, CA), vectores MPSV y muchos otros vectores conocidos en la técnica por expresar otras proteínas. Los vectores virales útiles incluyen vectores basados en retrovirus, adenovirus, virus adenoasociados, virus del herpes, vectores basados en SV40, virus del papiloma, virus de Epstein Barr HBP, vectores del virus vaccinia y virus Semliki Forest (SFV). Remítase a, Smith, Annu. Rev. Microbiol. 49:807, 1995; y Rosenfeld et al., Cej] 68:143, 1992.
La elección del vector de expresión depende de las células hospedadoras previstas en las que se va a expresar el vector. Habitualmente, los vectores de expresión contienen un promotor y otras secuencias reguladoras (por ejemplo, potenciadores) que están unidas operativamente a los polinucleótidos que codifican una cadena o un fragmento de anticuerpo de la invención. En algunas realizaciones, se emplea un promotor inducible para evitar la expresión de secuencias insertadas excepto en condiciones inductoras. Los promotores inducibles incluyen, por ejemplo, arabinosa, lacZ, promotor de metalotioneína o un promotor de choque térmico. Los cultivos de organismos transformados se pueden expandir en condiciones no inductoras sin sesgar a la población para codificar secuencias cuyos productos de expresión sean mejor tolerados por las células hospedadoras. Además de los promotores, también pueden ser necesarios o deseables otros elementos reguladores para la expresión eficiente de una cadena o fragmento de anticuerpo de la invención. Estos elementos incluyen habitualmente un codón de iniciación ATG y un sitio de unión al ribosoma adyacente u otras secuencias. Además, la eficiencia de la expresión se puede mejorar mediante la inclusión de potenciadores apropiados para el sistema celular en uso (remítase, por ejemplo, a Scharf et al., Results Probl. Cell Differ. 20:125, 1994; y Bittner et al., Meth. Enzvmol.. 153:516, 1987). Por ejemplo, el potenciador s V40 o el potenciador CMV se pueden utilizar para aumentar la expresión en células hospedadoras de mamífero.
Los vectores de expresión también pueden proporcionar una posición de secuencia de señal de secreción para formar una proteína de fusión con polipéptidos codificados por secuencias de anticuerpos insertadas. Más a menudo, las secuencias de anticuerpos insertadas se conectan a secuencias señal antes de su inclusión en el vector. Los vectores a utilizar para recibir secuencias que codifican dominios variables de cadena ligera y pesada de anticuerpos en ocasiones también codifican regiones constantes o partes de ellas. Dichos vectores permiten la expresión de las regiones variables como proteínas de fusión con las regiones constantes, lo que conduce a la producción de anticuerpos intactos o fragmentos de los mismos. Generalmente, dichas regiones constantes son humanas.
Las células hospedadoras para albergar y expresar las cadenas de anticuerpos de la invención pueden ser procariotas o eucariotas. La Escherichia coli es un hospedador procariota útil para clonar y expresar los polinucleótidos de la presente invención. Otros hospedadores microbianos adecuados para su uso incluyen bacilos tales como Bacillus subtilis, y otras enterobacterias tales como Salmonella, Serratia y varias especies de Pseudomonas. En estos hospedadores procariotas, también se pueden producir vectores de expresión, que habitualmente contienen secuencias de control de la expresión compatibles con la célula hospedadora (por ejemplo, un origen de replicación). Además, puede estar presente cualquier número de una serie de promotores conocidos tales como el sistema promotor de lactosa, un sistema promotor de triptófano (trp), un sistema promotor de beta-lactamasa o un sistema promotor de fagos lambda. Los promotores habitualmente controlan la expresión, opcionalmente con una secuencia operadora, y tienen secuencias de sitio de unión a ribosomas, para iniciar y completar la transcripción y la traducción. También se pueden emplear otros microbios tales como levaduras, para expresar los anticuerpos o fragmentos de anticuerpo de la invención. También se pueden utilizar células de insectos combinadas con vectores de baculovirus.
En un aspecto, se utilizan células hospedadoras de mamífero para expresar y producir los anticuerpos y fragmentos de anticuerpo de la presente invención. Por ejemplo, pueden ser una línea celular del hibridoma que expresa genes de inmunoglobulina endógenos o una línea celular de mamífero que alberga un vector de expresión exógeno. Estas incluyen cualquier célula animal o humana normal mortal o normal o anormal inmortal. Por ejemplo, se han desarrollado varias líneas celulares hospedadoras adecuadas capaces de secretar inmunoglobulinas intactas, incluidas las líneas celulares CHO, varias líneas celulares Cos, células HeLa, líneas celulares de mieloma, linfocitos B transformados e hibridomas. El uso del cultivo de células de tejidos de mamíferos para expresar polipéptidos se trata en general en, por ejemplo, Winnacker, From Genes to Clones, VCH Publishers, N.Y., N.Y., 1987. Los vectores de expresión para células hospedadoras de mamíferos pueden incluir secuencias de control de la expresión tales como un origen de replicación, un promotor y un potenciador (remítase, por ejemplo, a Queen et al., Immunol. Rev.
89:49-68, 1986), y los sitios de información de procesamiento necesarios tales como sitios de unión a ribosomas, sitios de corte y empalme de ARN, sitios de poliadenilación y secuencias de terminación de la transcripción. Estos vectores de expresión contienen generalmente promotores derivados de genes de mamíferos o de virus de mamíferos. Los promotores adecuados pueden ser constitutivos, específicos del tipo de célula, específicos del estadio y/o modulables o regulables. Los promotores útiles incluyen el promotor de metalotioneína, el promotor constitutivo tardío mayor del
adenovirus, el promotor de MMTV inducible por dexametasona, el promotor de SV40, el promotor de MRP polIII, el promotor constitutivo de MPSV, el promotor de CMV inducible por tetraciclina (tales como el promotor de CMV temprano inmediato humano), el promotor de CMV constitutivo y las combinaciones de promotor-potenciador conocidas en la técnica.
Los métodos para introducir vectores de expresión que contienen las secuencias de polinucleótidos de interés varían dependiendo del tipo de hospedador celular. Por ejemplo, la transfección con cloruro de calcio se utiliza comúnmente para las células procariotas, mientras que el tratamiento con fosfato de calcio o la electroporación se pueden utilizar para otros hospedadores celulares (remítase de forma general a Sambrook et al., supra). Otros métodos incluyen, por ejemplo, electroporación, tratamiento con fosfato de calcio, transformación mediada por liposomas, inyección y microinyección, métodos balísticos, virosomas, inmunoliposomas, conjugados de policatión:ácido nucleico, ADN desnudo, viriones artificiales, fusión a la proteína estructural VP22 del virus del herpes (Elliot y O'Hare, Cell 88:223, 1997), captación de ADN potenciada por el agente y transducción ex vivo. Para la producción a largo plazo de alto rendimiento de proteínas recombinantes, a menudo se deseará una expresión estable. Por ejemplo, las líneas celulares que expresan de forma estable cadenas o fragmentos de unión de anticuerpos se pueden preparar utilizando vectores de expresión de la invención que contienen orígenes virales de replicación o elementos de expresión endógena y un gen marcador seleccionable. Después de la introducción del vector, se puede permitir que las células se multipliquen durante 1-2 días en un medio enriquecido antes de cambiar al medio selectivo. El propósito del marcador seleccionable es conferir resistencia a la selección, y su presencia permite la multiplicación de células que expresan con éxito las secuencias introducidas en medios selectivos. Las células resistentes, transfectadas de forma estable, pueden proliferar utilizando técnicas de cultivo tisular apropiadas para el tipo de célula.
Ejemplo 99: Clonación de anticuerpos anti-Her2 y anticuerpo 20507 con mutación etiqueta A1/ybbR y Cys para estudios de conjugación
Oligonucleótidos de ADN que codifican regiones variables de cadenas pesadas y ligeras de un anticuerpo anti-Her2 (Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. (1992) Proc. Nati. Acad. Sci. USA, 89, 4285-4289. Humanization of an anti-p185her2 antibody for human Cáncer therapy) se sintetizaron químicamente y se clonaron en dos vectores de expresión de mamíferos, pOG-HC y pOG-LC que contienen las regiones constantes de IgG1 humano y cadena ligera kappa humana, lo que dio como resultado dos constructos de origen natural, HC de anticuerpo pOG-anti-Her2 y LC de anticuerpo pOG-anti-Her2, respectivamente. En estos vectores, la expresión de cadena pesada y ligera de anticuerpos en células de mamífero está impulsada por un promotor de CMV. Los vectores codifican una secuencia señal de 24 aminoácidos sintéticos, MKTFILLLWVLLLWVIFLLPGATA (SEQ ID NO:12), en el extremo N terminal de la cadena pesada y la cadena ligera para guiar su secreción a partir de células de mamífero. La secuencia señal se ha validado para que sea eficiente para controlar la secreción proteica en cientos de proteínas de mamífero en células 293 Freestyle™ (Gonzalez R, Jennings LL, Knuth M, Orth AP, Klock HE, Ou W, Feuerhelm J, Hull MV, Koesema E, Wang Y, Zhang J, Wu C, Cho CY, Su AI, Batalov S, Chen H, Johnson K, Laffitte B, Nguyen DG, Snyder EY, Schultz Pg , Harris JL, Lesley SA. (2010) Proc Nati Acad Sci U S A. 107:3552-7). Se empleó la mutagénesis dirigida por oligonucleótidos para preparar la mutación LC-S159C del anticuerpo anti-Her2. Los cebadores sentido y antisentido (Tabla 4) que corresponden al sitio de mutación LC-S159C en las regiones constantes de la cadena ligera kappa humana se sintetizaron químicamente. Las reacciones de PCR se llevaron a cabo utilizando ADN polimerasa PfuUltra II Fusion HS (Stratagene) con HC de anticuerpo pOG-anti-Her2 y LC de anticuerpo pOG-anti-Her2 como moldes. Los productos de PCR se confirmaron en geles de agarosa y se trataron con DPN I y después se transformaron en células DH5a (Klock et al., (2009) Methods Mol Biol. 498:91-103).
La secuencia del constructo con mutación LC-S159C se confirmó mediante secuenciación de ADN. La secuencia de aminoácidos de la cadena pesada del anticuerpo anti-Her2 de origen natural se muestra como SEQ ID NO: 1 y la de la cadena ligera se muestra como SEQ ID NO: 2. La secuencia de aminoácidos del anticuerpo con mutación LC-S159C se muestra en la Tabla 3 con C159 en negrita y subrayada. Los residuos de aminoácidos en la cadena ligera kappa humana y la cadena pesada de IgG 1 humana se numeran de acuerdo con el sistema de numeración Eu (Edelman et al, (1969) Proc Nati Acad Sci USA, 63:78-85). El anticuerpo anti-Her2-LC-S159C se clonó además en vectores que contenían marcadores de selección antibióticos para la selección de clones de células transfectados de forma estable en medios que contenían los correspondientes antibióticos.
De forma similar, un mutante con Cys doble, HC-E152C-S375C, del anticuerpo anti-Her2 y cuatro mutantes con Cys (HC-E152C, HC-S375C, LC-K107C, LC-S159C, HC-E152C-S375C) de un segundo anticuerpo, el anticuerpo 20507, se clonaron utilizando los cebadores de ADN enumerados en la Tabla 4 y los procedimientos anteriores. El anticuerpo 20507 contiene una cadena pesada de IgG1 y una cadena ligera kappa humana. Las partes constantes de la cadena pesada y ligera del anticuerpo 20507 son idénticas en la secuencia de aminoácidos a aquellas en el anticuerpo anti-Her2. Las secuencias de aminoácidos de las regiones constantes de los mutantes con Cys del anticuerpo anti-Her2 y del anticuerpo 20507 se muestran en la Tabla 3 con Cys mutada en negrita y subrayadas.
Inserción de las secuencias peptídicas ybbR y A1 en la región constante de la cadena pesada del anticuerpo anti-Her2 se logró mediante métodos de biología molecular estándar. El vector HC del anticuerpo pOG-anti-Her2 sirvió como
molde de PCR para obtener los mutantes de inserción HC-ins388-ybbR y HC-ins388-A1 correspondientes. La Tabla 4 enumera los cebadores sentido y antisentido que se utilizaron para la clonación de estos constructos. El vector que codifica el mutante de inserción anticuerpo 20507-HC-ins388-A1 se preparó amplificando la región variable de la cadena pesada del anticuerpo 20507. El fragmento de ADN amplificado se movió después al vector existente que codifica el mutante de inserción anti-Her2-HC-ins388-A1. Todos los vectores de expresión resultantes que codifican cadenas pesadas con etiquetas peptídicas se confirmaron mediante secuenciación de ADN. Los anticuerpos anti-Her2 que contenían las inserciones HC-ins388-ybbR y HC-ins388-A1 se clonaron adicionalmente en vectores con marcadores de selección de antibióticos, con lo cual permite así el posterior aislamiento de los clones que expresan de forma estable los respectivos constructos con etiquetas peptídicas. Las secuencias de aminoácidos de las regiones constantes de los mutantes con la inserción A1/ybbR del anticuerpo anti-Her2 y del anticuerpo 20507 se muestran en la Tabla 3. La etiqueta peptídica insertada se muestra en negrita y está subrayada.
Los anticuerpos anti-Her2 y anticuerpo 20507 con mutación etiqueta A1 y Cys se prepararon como se describe en el Ejemplo 100, y se conjugaron con un péptido citotóxico ilustrativo de fórmula (I) tal como se describe en los Ejemplos 101, 102, 103, 104 y 105.
T l 4. n i ADN l r m i n iliz r l n r ni r m n .


Ejemplo 100: Preparación de anticuerpos anti-Her2 y anticuerpo 20507 con mutación de Cys y de etiqueta A1/ybbR.
El mutante de anti-Her2 y anticuerpo 20507 con Cys, etiqueta A1 y etiqueta ybbR se expresó en células 293 Freestyle™ cotransfectando plásmidos de cadena pesada y cadena ligera utilizando un método de transfección transitoria tal como se ha descrito previamente (Meissner, etal., BiotechnolBioeng. 75:197-203 (2001)). Los plásmidos de ADN utilizados en la cotransfección se prepararon utilizando un kit de preparación de plásmidos Qiagen conforme al protocolo del fabricante. Se cultivaron células 293 Freestyle™ en suspensión en medio de expresión Freestyle™ (Invitrogen) a 37 °C en un 5% de CO2. El día antes de la transfección, las células se dividieron hasta 0,7 x 106 células /mL en medio fresco. En el día de la transfección, la densidad celular alcanzó típicamente 1,5 x 106 células/mL. Las células se transfectaron con una mezcla de plásmidos de cadena pesada y cadena ligera con la proporción de 1:1 utilizando el método PEI (Meissner et al., 2001 supra). Las células transfectadas se cultivaron adicionalmente durante cinco días. El medio del cultivo se recogió por centrifugación del cultivo a 2000x g durante 20 min y se filtró a través de filtros de 0,2 micrómetros. Los anticuerpos expresados se purificaron a partir del medio filtrado utilizando Protein A-Sepharose™ (GE Healthcare Life Sciences). Los anticuerpos IgG se eluyeron de la columna Protein A-Sepharose™ utilizando un tampón de elución a pH 3,0. Las soluciones de IgG eluidas se neutralizaron inmediatamente con Tris-HCl 1 M (pH 8,0) seguido de un intercambio de tampón a PBS.
Los constructos de expresión para los anticuerpos anti-Her2-LC-S159C, anti-Her2-HC-ins388-ybbR y anti-Her2-HC-ins388-A1 también se transfectaron en células CHO. Siguiendo protocolos estándar, las células que expresaban de forma estable estos anticuerpos se seleccionaron entonces utilizando antibióticos. Todos los constructos de anticuerpos anti-Her2 expresados en los clones de células CHO seleccionados se purificaron mediante cromatografía Protein A-Sepharose tal como se ha descrito anteriormente.
En un estudio separado, el anticuerpo anti-Her2-LC-S159C se expresó de forma estable en la célula CHO y después se realizó una selección mediante selección con antibióticos. El anticuerpo anti-Her2-LC-S159C expresado en el clon de células CHO establecido se purificó mediante procedimientos con columna Protein A-Sepharose tal como se ha descrito anteriormente.
Inmunoconjugados
Los inmunoconjugados de la invención que comprenden dichos péptidos citotóxicos como una carga (fármaco) incluyen conjugados de Fórmula (II):

donde:
Ab representa un resto de unión a antígeno;
L es un conector seleccionado entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2L1 - ,-L1 L2L3L4 -, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1, donde -L1, L2, L3 , tal c Lo4m , o se d Le5 ,finen en y la prese Ln6te; son es un número entero de 1 a 16;

R2 es metilo, etilo, isopropilo o sec-butilo;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos, -C(=0)R11, -(CH2)mOH, -C(=0)(CH2)mOH, -C(=0)((CH2)m0)nR12, o -((CH2)mO)nR12;
R11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
Otros inmunoconjugados divulgados en la presente que comprenden dichos péptidos citotóxicos como una carga (fármaco) incluyen conjugados de Fórmula (III):

donde:
Ab representa un resto de unión a antígeno;
L se selecciona entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2L1-,-L1L2L3L4-; -L4L3L2L1-,-L1L2L3-, -L3L2L1-L1L2-, -L2L1- y -L1, donde -L1, L2 , L3 , L4 , L5 , y L6 son como se definen en la pres y es un número entero de 1 a 16;
R1 es un heterocicloalquilo de 6 miembros que contiene 1 -2 heteroátomos de N y un puente alquileno C1-C2 , donde el heterocicloalquilo de 6 miembors está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
o R1 es un heterocicloalquilo bicíclico fusionado de 5-8 miembros que contiene 1-2 heteroátomos de N, donde el heterocicloalquilo bicíclico fusionado de 5-8 miembros está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionados independientemente entre R5 y R6;
R2 es alquilo C1-C6 ;

R5 es alquilo C1-C6 , alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos, -C(=O)R11, -(CH2)mOH, -C(=O)(CH2)mOH, -C(=O)((CH2)mO)nR12, o -((CH2)mO)nR12;
R6 es halo, oxo, OH, alquilo C1-C6 , -N(R14)2 , -R16 y -N R 12C(=O)R11;
R11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;
cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros unido en N que contiene 1-2 heteroátomos seleccionados independientemente entre N
R17 es un enlace, -NH

cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
Se proporcionan otros inmunoconjugados de Fórmula (II) de la invención en las realizaciones enumeradas más adelante.
La invención proporciona inmunoconjugados que comprenden uno o más péptidos citotóxicos antimitóticos conectados a un resto de unión a antígeno tal como un anticuerpo o fragmento de anticuerpo. Los inmunoconjugados preferidos de la invención son los de Fórmula (II) tal como se describen en la presente. Los métodos para producir dichos inmunoconjugados son muy conocidos en la técnica. Los inmunoconjugados preferidos incluyen los divulgados en la Tabla 5, y variaciones de estos que tienen otro resto de unión a antígeno en lugar del anticuerpo anti-Her2, particularmente conjugados de este tipo donde el anticuerpo anti-Her2 se reemplaza por un anticuerpo seleccionado de la siguiente lista: anticuerpo antirreceptor de estrógenos, anticuerpo antirreceptor de progesterona, anticuerpo antip53, anticuerpo anti-EGFR, anticuerpo anticatepsina D, anticuerpo anti-Bcl-2, anticuerpo anti-cadherina E, anticuerpo anti-CA125, anticuerpo anti-CA15-3, anticuerpo anti-CA19-9, anticuerpo anti-c-erbB 2, anticuerpo antiglicoproteína P, anticuerpo anti-CEA, anticuerpo antiproteína del retinoblastoma, anticuerpo anti-oncoproteína ras, anticuerpo anti-Lewis X, anticuerpo anti-Ki-67, anticuerpo anti-PCNA, anticuerpo anti-CD3, anticuerpo anti-CD4, anticuerpo anti-CD5, anticuerpo anti-CD7, anticuerpo anti-CD8, anticuerpo anti-CD9/p24, anticuerpo anti-CD1, anticuerpo anti-CD11c, anticuerpo anti-CD13, anticuerpo anti-CD14, anticuerpo anti-CD15, anticuerpo anti-CD19, anticuerpo anti-CD20, anticuerpo anti-CD22, anticuerpo anti-CD23, anticuerpo anti-CD30, anticuerpo anti-CD31, anticuerpo anti-CD33, anticuerpo anti-CD34, anticuerpo anti-CD35, anticuerpo anti-CD38, anticuerpo anti-CD39, anticuerpo anti-CD41, anticuerpo anti-LCA/CD45, anticuerpo anti-CD45RO, anticuerpo anti-CD45RA, anticuerpo anti-CD71, anticuerpo anti-CD95/Fas, anticuerpo anti-CD99, anticuerpo anti-CD100, anticuerpo anti S-100, anticuerpo anti-CD106, anticuerpo antiubiquitina, anticuerpo anti-myc-c, anticuerpo anticitoqueratina, anticuerpo anticadenas ligeras lambda, anticuerpo antimelanosomas, anticuerpo antiantígeno específico de próstata, anticuerpo antiantígeno tau, anticuerpo antifibrina, anticuerpo antiqueratinas y anticuerpo antiantígeno Tn.
En algunas realizaciones, un inmunoconjugado de Fórmula (II), o subfórmula de este, que comprende un anticuerpo o fragmento de anticuerpo Ab que tiene actividad de unión a antígenos, donde el conector L se une al Ab en un átomo de azufre de cisteína del Ab. Se proporcionan grupos reactivos habituales para la reacción con un grupo con azufre de cisteína y el grupo resultante formado en la Tabla 1. Algunos ejemplos de componentes conectores formados

mediante reacción con un residuo de cisteína del resto de unión a antígeno incluyen O

En otras realizaciones, un inmunoconjugado de Fórmula (II) que comprende Ab, un anticuerpo o fragmento de anticuerpo que tiene actividad de unión a antígeno, donde el conector se une al Ab mediante un puente disulfuro en

el Ab. En dichas realizaciones, se forma un resto mediante reacción de  y un compuesto de Fórmula (I) que contiene una hidroxilamina.
y un compuesto de Fórmula (I) que contiene una hidroxilamina.
En algunas realizaciones, un inmunoconjugado de Fórmula (II), o subfórmula de este, comprende un anticuerpo o fragmento de anticuerpo Ab que tiene actividad de unión a antígenos, donde el conector L se une al Ab en un -NH2 libre de lisina. Los componentes conectores formados mediante reacción con el -N H 2 de un residuo de lisina del resto de unión a antígeno, donde cada p es 1-10 y cada R es independientemente H o alquilo C1-4 (preferentemente metilo)
incluyen
En algunas realizaciones, un inmunoconjugado de Fórmula (II), o subfórmula de esta, comprende un anticuerpo o fragmento de anticuerpo Ab que tiene actividad de unión a antígenos, donde el conector L se une al Ab en un grupo Pcl o Pyl introducido mediante modificación en un anticuerpo. Remítase, por ejemplo, a Ou, et al., PNAS 108(26), 10437-42 (2011). Los componentes conectores formados mediante reacción con un grupo Pcl o Pyl incluyen

anticuerpo modificado.
En algunas realizaciones, un inmunoconjugado de Fórmula (II), o subfórmula de esta, comprende un anticuerpo o fragmento de anticuerpo Ab que tiene actividad de unión a antígenos, donde el conector L se une al Ab en un residuo de serina en un péptido S6, ybbR o A1 introducido mediante modificación en un anticuerpo. Los componentes conectores formados mediante reacción con dichos residuos de serina incluyen

A modo de ejemplo, un esquema de reacción general para la formación de inmunoconjugados de Fórmula (II) se muestra en el Esquema 13 a continuación:
Esquema 13

donde RG1 es un grupo reactivo 1 de la Tabla 1 y RG2 es un grupo reactivo 1 de la Tabla 1 y el producto de reacción de los respectivos grupos (tal como se observa en la Tabla 1) es un componente conector del conector L. R101, R2, R3, L y Ab son como se definen en la presente.
Otro esquema de reacción general para la formación de inmunoconjugados de Fórmula (II) se muestra en el Esquema 14 a continuación:
Esquema 14

donde RGi es un grupo reactivo 1 de la Tabla 1 y RG2 es un grupo reactivo 1 de la Tabla 1 y el producto de reacción de los respectivos grupos (tal como se observa en la Tabla 1) es un componente conector del conector L. R101, R2, R3, L y Ab son como se definen en la presente.
Un esquema de reacción general para la formación de inmunoconjugados de Fórmula (III) se muestra en el Esquema 15 a continuación:
Esquema 15

donde RG1 es un grupo reactivo 1 de la Tabla 1 y RG2 es un grupo reactivo 1 de la Tabla 1 y el producto de reacción de los respectivos grupos (tal como se observa en la Tabla 1) es un componente conector del conector L. R1, R2, R3, L y Ab son como se definen en la presente.
Otro esquema de reacción general para la formación de inmunoconjugados de Fórmula (II) se muestra en el Esquema 16 a continuación:
Esquema 16

donde RG1 es un grupo reactivo 1 de la Tabla 1 y RG2 es un grupo reactivo 1 de la Tabla 1 y el producto de reacción de los respectivos grupos (tal como se observa en la Tabla 1) es un componente conector del conector L. R1, R2, R3, L y Ab son como se definen en la presente.
En otro aspecto, la presente invención proporciona una composición farmacéutica que comprende un inmunoconjugado de Fórmula (II) de la presente invención, o subfórmula de este, y al menos un portador farmacéuticamente aceptable. La composición farmacéutica se puede formular para vías de administración particulares tales como administración intravenosa y administración parenteral.
Los inmunoconjugados de la invención se formulan habitualmente como soluciones o suspensiones en tampón acuoso y/o solución acuosa isotónica. Estos se administran habitualmente por vía parenteral, ya sea por inyección o por infusión. Los métodos para su formulación y administración son similares a aquellos para la formulación y administración de otros productos farmacéuticos de base biológica tales como productos terapéuticos de anticuerpos, y son conocidos por los expertos en la técnica.
Determinadas composiciones inyectables son soluciones o suspensiones isotónicas acuosas, y se preparan supositorios convenientemente a partir de emulsiones o suspensiones grasas. Dichas composiciones pueden esterilizarse y/o contener adyuvantes tales como agentes conservantes, estabilizantes, humectantes o emulsionantes, promotores de disolución, sales para regular la presión osmótica y/o tampones. Además, también pueden contener otras sustancias valiosas desde el punto de vista terapéutico. Dichas composiciones se preparan de acuerdo con métodos convencionales de mezcla, granulación o recubrimiento, respectivamente, y contienen aproximadamente un 0,1-75%, o contienen aproximadamente un 1-50%, del principio activo.
La potencia de destrucción de células in vitro proporcionada en la Tabla 2 obtenida para ciertos péptidos citotóxicos de Fórmula (I) muestra que dichos compuestos de fórmula (I) presentan actividades farmacológicas valiosas, y de este modo, estos compuestos se pueden utilizar como la carga de un ADC. Los inmunoconjugados que comprenden un compuesto de fórmula (I), tal como se demuestra en la presente, presentan actividad sustancial sobre células in vitro y sobre tumores in vivo utilizados como diana, tal como se demuestra por la potente inhibición del crecimiento de los tumores xenoinjertados que representan diferentes tipos de cáncer humano. Por tanto, los inmunoconjugados de Fórmula (II) de la invención, que comprenden una carga de Fórmula (I), y una subfórmula de estos, conectados a un resto de unión a antígeno tal como un anticuerpo, también son útiles para tratar tipos de cáncer tales como tumores gástricos, mieloides, de colon, nasofaríngeos, esofágicos y de próstata, glioma, neuroblastoma, cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer colorrectal, cáncer de tiroides, leucemia (por ejemplo, leucemia mielógena, leucemia linfocítica, leucemia mielógena aguda (LMA), leucemia mieloide crónica (LMC), leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de linaje T o LLA-T, leucemia linfocítica crónica (LLC), síndrome mielodisplásico (SMD), tricoleucemia), linfoma (linfoma de Hodgkin (LH), linfoma no Hodgkin (LNH)), mieloma múltiple, cáncer de vejiga, renal, gástrico (por ejemplo, tumores del estroma gastrointestinal (TEGI)), hígado, melanoma y pancreático, y sarcoma.
Una realización de la invención proporciona la conjugación de un compuesto de fórmula (I) y subfórmulas de esta, a un resto de unión a antígeno, y la formación de este modo de un inmunoconjugado de Fórmula (II), tal como se describe en la presente.
Los inmunoconjugados de la invención que comprenden un compuesto de Fórmula (I), o subfórmula de esta, son particularmente útiles para tratar tipos de cáncer de los que se tiene constancia en la técnica que se inhiben con toxinas antimitóticas, y se demostró en la presente que esos tipos de tumores son susceptibles de inhibición con los compuestos y conjugados de la invención. Algunas indicaciones adecuadas para el tratamiento incluyen tumores gástricos, mieloides, de colon, nasofaríngeos, esofágicos y de próstata, glioma, neuroblastoma, cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer colorrectal, cáncer de tiroides, leucemia (por ejemplo, leucemia mielógena, leucemia linfocítica, leucemia mielógena aguda (LMA), leucemia mieloide crónica (LMC), leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de linaje T o LLA-T, leucemia linfocítica crónica (LLC), síndrome mielodisplásico (SMD), tricoleucemia), linfoma (linfoma de Hodgkin (LH), linfoma no Hodgkin (LNH)), mieloma múltiple, cáncer de vejiga, renal, gástrico (por ejemplo, tumores del estroma gastrointestinal (TEGI), hígado, melanoma y pancreático, y sarcoma. Los inmunoconjugados de la invención que comprenden un compuesto de Fórmula (I), o una subfórmula de esta, son particularmente útiles en terapia. En una realización adicional, la terapia es para una enfermedad que se puede tratar con toxinas antimitóticas. En otra realización, los compuestos de la invención son útiles para tratar tipos de cáncer, que incluyen tumores gástricos, mieloides, de colon, nasofaríngeos, esofágicos y de próstata, glioma, neuroblastoma, cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer colorrectal, cáncer de tiroides, leucemia (por ejemplo, leucemia mielógena, leucemia linfocítica, leucemia mielógena aguda (LMA), leucemia mieloide crónica (LMC), leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de linaje T o LLA-T, leucemia linfocítica crónica (LLC), síndrome mielodisplásico (SMD), tricoleucemia), linfoma (linfoma de Hodgkin (LH), linfoma no Hodgkin (LNH)), mieloma múltiple, cáncer de vejiga, renal, gástrico (por ejemplo, tumores del estroma gastrointestinal (TEGI)), hígado, melanoma y pancreático, y sarcoma.
Los métodos habitualmente comprenden administrar una cantidad eficaz de un inmunoconjugado de la invención tal como se describe en la presente o una composición farmacéutica que comprende dichos inmunoconjugados a un sujeto que necesite dicho tratamiento. El inmunoconjugado se puede administrar mediante cualquier método adecuado tal como los descritos en la presente, y la administración se puede repetir a intervalos seleccionados por un facultativo a cargo del tratamiento.
Por tanto, como una realización adicional, la presente invención proporciona el uso de un inmunoconjugado de fórmula (I) o cualquiera de las realizaciones de dichos compuestos descritos en la presente, para la fabricación de un medicamento. En una realización adicional, el medicamento es para el tratamiento de una enfermedad que se puede
tratar con toxinas antimitóticas. En otra realización, la enfermedad se selecciona entre tumores gástricos, mieloides, de colon, nasofaríngeos, esofágicos y de próstata, glioma, neuroblastoma, cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer colorrectal, cáncer de tiroides, leucemia (por ejemplo, leucemia mielógena, leucemia linfocítica, leucemia mielógena aguda (LMA), leucemia mieloide crónica (LMC), leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de linaje T o LLA-T, leucemia linfocítica crónica (LLC), síndrome mielodisplásico (SMD), tricoleucemia), linfoma (linfoma de Hodgkin (LH), linfoma no Hodgkin (LNH)), mieloma múltiple, cáncer de vejiga, renal, gástrico (por ejemplo, tumores del estroma gastrointestinal (TEGI)), hígado, melanoma y pancreático, y sarcoma.
La composición o combinación farmacéutica de la presente invención puede estar en dosis unitarias de aproximadamente 1-1000 mg de uno o varios principios activos para un sujeto de aproximadamente 50-100 kg, o aproximadamente 1-500 mg o aproximadamente 1-250 mg o aproximadamente 1-150 mg o aproximadamente 0,5 100 mg, o aproximadamente 1 -50 mg de principios activos. La dosificación terapéuticamente eficaz de un compuesto, la composición farmacéutica o las combinaciones de estas, depende de la especie del sujeto, del peso corporal, de la edad y del estado individual, del trastorno o enfermedad, o de la gravedad de esta, que se esté tratando. Un facultativo, médico o veterinario experto puede determinar fácilmente la cantidad eficaz de cada uno de los principios activos necesaria para prevenir, tratar o inhibir el progreso del trastorno o la enfermedad.
Las propiedades de dosificación citadas anteriormente son demostrables en pruebas in vitro e in vivo que utilizan convenientemente mamíferos, p. ej., ratones, ratas, perros, monos ,u órganos aislados, tejidos y preparados de estos. Los compuestos de la presente invención se pueden aplicar in vitro en forma de soluciones, p. ej., soluciones acuosas, e in vivo por vía enteral, parenteral, convenientemente por vía intravenosa, p. ej., en forma de una suspensión o en solución acuosa. La dosificación in vitro puede oscilar entre concentraciones de aproximadamente 10-3 molar y 10-12 molar. Una cantidad terapéuticamente eficaz in vivo puede variar en función de la vía de administración, entre aproximadamente 0,1-500 mg/kg, o entre aproximadamente 1-100 mg/kg.
Un inmunoconjugado de Fórmula (II) o Fórmula (III), o subfórmulas de estas, de la presente invención se puede administrar simultáneamente con, o antes o después de, uno o más coagentes terapéuticos diferentes. Un inmunoconjugado de Fórmula (II) o Fórmula (III), o una subfórmula de estas, de la presente invención se puede administrar por separado, por una vía de administración idéntica o diferente, o de forma conjunta en la misma composición farmacéutica que el o los coagentes.
En una realización, la invención proporciona un producto que comprende un compuesto de fórmula (I) de acuerdo con la invención, o una subfórmula de esta, y al menos un coagente terapéutico diferente como una preparación combinada para uso simultáneo, separado o secuencial en terapia. En una realización, la terapia es el tratamiento de una enfermedad o afección tal como cáncer con una toxina antimitótica. Los productos proporcionados como un preparado combinado incluyen una composición que comprende un inmunoconjugado de Fórmula (II) o subfórmula de esta, y el o los otros coagentes terapéuticos juntos en la misma composición farmacéutica, o el inmunoconjugado de Fórmula (II), o subfórmula de esta, y el o los otros coagentes terapéuticos en forma separada, p. ej., en forma de un kit.
En una realización, la invención proporciona una composición farmacéutica que comprende un inmunoconjugado de Fórmula (II), o una subfórmula de esta, y otro u otros coagentes terapéuticos. Opcionalmente, la composición farmacéutica puede comprender un portador farmacéuticamente aceptable, tal como se ha descrito anteriormente.
Los coagentes adecuados para su uso con los compuestos y conjugados de la invención incluyen otros agentes anticancerosos, agentes antialérgicos, agentes antieméticos, analgésicos, agentes antiinflamatorios, agentes citoprotectores y combinaciones de estos.
Los coagentes específicos considerados para su uso en combinación con los compuestos y conjugados divulgados en la presente incluyen anastrozol (Arimidex®), bicalutamida (Casodex®), sulfato de bleomicina (Blenoxane®), busulfán (Myleran®), inyección de busulfán (Busulfex®), capecitabina (Xeloda®), N4-pentoxicarbonil-5-deoxi-5-fluorocitidina, carboplatino (Paraplatin®), carmustina (BiCNU®), clorambucilo (Leukeran®), cisplatino (Platinol®), cladribina (Leustatin®), ciclofosfamida (Cytoxan® o Neosar®), citarabina, arabinósido de citosina (Cytosar-U®), inyección de liposomas de citarabina (DepoCyt®), dacarbazina (DTIC-Dome®), dactinomicina (Actinomicina D, Cosmegan), clorhidrato de daunorubicina (Cerubidine®), inyección de liposomas de citrato de daunorubicina (DaunoXome®), dexametasona, docetaxel (Taxotere®), clorhidrato de doxorubicina (Adriamycin®, Rubex®), etopósido (Vepesid®), fosfato de fludarabina (Fludara®), 5-fluorouracilo (Adrucil®, Efudex®), flutamida (Eulexin®), tezacitibina, Gemcitabine (difluorodeoxicitidina), hidroxiurea (Hydrea®), Idarubicina (Idamicina®), ifosfamida (IFEX®), irinotecán (Camptosar®), L-asparaginasa (ELSPAR®), leucovorina cálcica, melfalán (Alkeran®), 6-mercaptopurina (Purinethol®), metotrexato (Folex®), mitoxantrona (Novantrone®), mylotarg, paclitaxel (Taxol®), phoenix (Ytrio90/MX-DTPA), pentostatina, polifeprosán 20 con implante de carmustina (Gliadel®), citrato de tamoxifeno (Nolvadex®), tenipósido (Vumon®), 6-tioguanina, tiotepa, tirapazamina (Tirazone®), clorhidrato de topotecán para inyección (Hycamptin®), vinblastina (Velban®), vincristina (Oncovin®), y vinorelbina (Navelbine®).
En una realización, la invención proporciona un kit que comprende dos o más composiciones farmacéuticas separadas, al menos una de las cuales contiene un compuesto de Fórmula (II), o subfórmula de esta. En una realización, el kit
comprende un medio para mantener por separado dichas composiciones, tal como un recipiente, frasco dividido o envase de aluminio dividido.
En las terapias combinadas de la invención, el inmunoconjugado de Fórmula (II), o subfórmula de esta, de la invención y el otro coagente terapéutico pueden ser fabricados y/o formulados por el mismo o diferentes fabricantes. Además, el inmunoconjugado de Fórmula (II), o subfórmula de esta, de la invención y el otro agente terapéutico se pueden integrar en una terapia combinada: (i) antes de dispensar el producto combinado a los facultativos (p. ej., en el caso de un kit que comprende el compuesto de la invención y el otro agente terapéutico), (ii) por parte de los mismos facultativos (o bajo la supervisión del facultativo) poco antes de la administración; (iii) en los mismos pacientes, p. ej., durante la administración secuencial del compuesto de la invención y el otro agente terapéutico.
La invención también proporciona un inmunoconjugado de Fórmula (II), o subfórmula de esta, para su uso en un método de tratamiento de una enfermedad o afección con un péptido citotóxico. La invención también proporciona un inmunoconjugado de Fórmula (II), o subfórmula de esta, para su uso en un método de tratamiento de una enfermedad o afección con un péptido citotóxico, donde el inmunoconjugado de Fórmula (II), o subfórmula de esta, se prepara para la administración con otro agente terapéutico. La invención también proporciona otro coagente terapéutico para su uso en un método de tratamiento de una enfermedad o afección con un péptido citotóxico, donde el otro coagente terapéutico se prepara para la administración con un inmunoconjugado de Fórmula (II), o subfórmula de esta. La invención también proporciona un inmunoconjugado de Fórmula (II), o subfórmula de esta, para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica, donde el inmunoconjugado de Fórmula (II), o subfórmula de esta, se administra con otro coagente terapéutico. La invención también proporciona otro coagente terapéutico para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica, donde el otro coagente terapéutico se administra con un inmunoconjugado de Fórmula (II), o subfórmula de esta.
La invención también proporciona el uso de un inmunoconjugado de Fórmula (II), o subfórmula de esta, para el tratamiento de una enfermedad o afección con un péptido citotóxico, donde el paciente ha sido tratado previamente (p. ej., dentro de un periodo de 24 horas) con otro agente terapéutico. La invención también proporciona el uso de otro agente terapéutico para tratar una enfermedad o afección con una toxina antimitótica, donde el paciente ha sido tratado previamente (p. ej., dentro de un periodo de 24 horas) con un inmunoconjugado de Fórmula (II), o subfórmula de esta.
La invención también proporciona un inmunoconjugado de Fórmula (II), o subfórmula de esta, para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica. La invención también proporciona un inmunoconjugado de Fórmula (II), o subfórmula de esta, para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica, donde el inmunoconjugado de Fórmula (II), o subfórmula de esta, se prepara para la administración con otro agente terapéutico. La invención también proporciona otro coagente terapéutico para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica, donde el otro coagente terapéutico se prepara para la administración con un inmunoconjugado de Fórmula (II), o subfórmula de esta. La invención también proporciona un inmunoconjugado de Fórmula (II), o subfórmula de esta, para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica, donde el inmunoconjugado de Fórmula (II), o subfórmula de esta, se administra con otro coagente terapéutico. La invención también proporciona otro coagente terapéutico para su uso en un método de tratamiento de una enfermedad o afección con una toxina antimitótica, donde el otro coagente terapéutico se administra con un inmunoconjugado de Fórmula (II), o subfórmula de esta.
La invención también proporciona el uso de un inmunoconjugado de Fórmula (II), o subfórmula de esta, para el tratamiento de una enfermedad o afección con una toxina antimitótica, donde el paciente ha sido tratado previamente (p. ej., dentro de un periodo de 24 horas) con otro agente terapéutico. La invención también proporciona el uso de otro agente terapéutico para tratar una enfermedad o afección con una toxina antimitótica, donde el paciente ha sido tratado previamente (p. ej., dentro de un periodo de 24 horas) con un inmunoconjugado de Fórmula (II), o subfórmula de esta. Conjugación de Conector-Carga (L-P) con un resto de unión a antígeno
Ejemplo 101: Conjugados de anticuerpo y fármaco (ADC) formados mediante conjugación de anticuerpos anti-Her2 y anticuerpo 20507 con mutación de Cys con los péptidos citotóxicos de Fórmula (I)
Se conocen en la técnica numerosos métodos para conjugar conectores-cargas a un resto de unión a antígeno (revisadas en, por ejemplo: Antibody-Drug Conjúgate, Methods in Molecular Biology, Vol. 1045, Editor L. Ducry, Humana Press (2013)). En este ejemplo, los compuestos de Fórmula (I) de la invención que comprenden un conector se conjugaron con residuos de cisteína introducidos mediante modificación en un anticuerpo utilizando métodos descritos en Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W. (2008) Nature Biotechnology 26:925-932. La conjugación de los péptidos citotóxicos de Fórmula (I) de la invención se ilustra en la presente utilizando un pequeño conjunto de anticuerpos con mutación de Cys, pero se prevé que los péptidos citotóxicos se puedan conjugar a la mayor parte, si no a todos, los mutantes de anticuerpos con Cys posibles. Además,
se espera que los péptidos citotóxicos se puedan conjugar con mutantes con Cys de numerosos anticuerpos, si no todos.
Debido a que las Cys introducidas mediante modificación en los anticuerpos expresados en células de mamíferos son modificadas por aductos (disulfuros) tales como el glutatión (GSH) y/o la cisteína durante su biosíntesis (Chen et al., 2009), la Cys modificada en el producto expresado inicialmente no es reactiva frente a reactivos que reaccionan con tiol tales como grupos maleimido o bromo- o yodoacetamida. Para conjugar la cisteína introducida mediante modificación después de la expresión, es necesario eliminar los aductos de cisteína o glutatión reduciendo estos disulfuros, lo cual generalmente permite reducir todos los disulfuros en la proteína expresada. Esto se puede lograr exponiendo en primer lugar el anticuerpo a un agente reductor tal como ditiotreitol (DTT) seguido por un procedimiento que permita la reoxidación de todos los enlaces disulfuro nativos del anticuerpo para restablecer y/o estabilizar la estructura del anticuerpo funcional. Por consiguiente, con el fin de reducir todos los enlaces disulfuro nativos y el disulfuro unido entre los aductos de GSH o cisteína del residuo de cisteína introducido mediante modificación, se añadió DTT recién preparada a los constructos anti-Her2 o anticuerpo 20507 con mutación de Cys purificados, hasta una concentración final de 20 mM. Después de la incubación con DTT a 37 °C durante 1 hora, las mezclas se dializaron a 4 °C frente a PBS durante tres días con intercambio de tampón diario para eliminar el DTT y reoxidar los enlaces disulfuro nativos. Un método alternativo es eliminar los reactivos reductores a través de una columna de desalinización tal como Sephadex G-25. Una vez que la proteína está totalmente reducida, se añade ascorbato oxidado (ácido deshidroascórbico) 1 mM a las muestras desalinizadas y las incubaciones de reoxidación se llevan a cabo durante 20 horas. Ambos métodos producen resultados similares. Sin embargo, los intentos para seguir los protocolos de reoxidación descritos anteriormente en la bibliografía utilizando CuSÜ4 dieron como resultado la precipitación de las proteínas (Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W. (2008) Nature Biotechnology 26:925). Todos los ejemplos de la presente utilizan el protocolo de diálisis descrito anteriormente. La reoxidación restaura los disulfuros intracatenarios, mientras que la diálisis elimina las cisteínas y los glutationes conectados inicialmente a la cisteína o cisteínas introducidas mediante modificación del anticuerpo.
Después de la reoxidación, el anticuerpo se conjugó con un péptido citotóxico de Fórmula (I), donde el péptido citotóxico de Fórmula (I) comprende un conector y un resto reactivo. A modo de ejemplo, se añadieron péptidos citotóxicos de Fórmula (I) que tenían un resto maleimida conectado (10 equivalentes molares respecto al anticuerpo) a los anticuerpos anti-Her2 o anticuerpo 20507 con mutación de cys reoxidados en tampón PBS (pH 7,2). Las incubaciones se llevaron a cabo durante 1 hora. El proceso de conjugación se monitorizó mediante HPLC en fase inversa, la cual es capaz de separar los anticuerpos conjugados de los no conjugados. Las mezclas de reacción de conjugación se analizaron en una columna PRLP-S (4000 A, 50 mm x 2,1 mm, Agilent), se calentaron hasta 80 °C y la elución de la columna se llevó a cabo con un gradiente lineal de un 30-60% de acetonitrilo en agua que contenía un 0,1% de TFA con un caudal de 1,5 mL/min. La elución de proteínas de la columna se monitorizó a 280 nm, 254 nm y 215 nm.
La eficiencia de conjugación de varios péptidos citotóxicos que tienen una maleimida conectada a un anticuerpo anti-Her2 o anticuerpo 20507 con mutación de Cys varió en función de la solubilidad de los péptidos citotóxicos utilizados, pero la mayoría de las reacciones dieron como resultado más de un 90% de conjugado (Tablas 5 y 6). Para evaluar el estado de agregación, los conjugados resultantes se analizaron en una columna de cromatografía de exclusión de tamaños (GE, Superdex200, 3,2/30) con un caudal de 0,1 mL/min en PBS. Todos los conjugados eran principalmente monoméricos. La mayoría de los conjugados contienen menos de un 3% de material dimérico y oligomérico (Tablas 5 y 6), lo que indica que la conjugación de dichos péptidos citotóxicos que tienen una maleimida conectada al anticuerpo anti-Her2 o anticuerpo 20507 con mutación de Cys no provocó agregación. De forma similar, la conjugación enzimática a través de las etiquetas A1 o ybbR (Ejemplos 104 y 105) también procede con eficiencias de conjugación superiores a un 90% (Tabla 7 y 9) y da como resultado conjugados que son monoméricos con menos de un 3% de agregados detectables (Tablas 7 y 9).
Los conjugados también se caracterizaron en cuanto a la carga promedio de un péptido citotóxico en el resto de unión del anticuerpo, denominada generalmente proporción de fármaco respecto a anticuerpo (DAR). El valor de DAR se extrapola a partir de los datos de LC-MS para las muestras reducidas y desglicosiladas. El LC/MS permite la cuantificación del número promedio de moléculas de carga (fármaco) unidas a un anticuerpo en un ADC. La HPLC separa el anticuerpo en cadenas ligeras y pesadas, y separa la cadena pesada (HC) y cadena ligera (LC) conforme al número de grupos de Conector-Carga por cadena. Los datos espectrales de masas permiten la identificación de las especies que componen la mezcla, por ejemplo, LC, LC+1, LC+2, HC, HC+1, HC+2, etc. A partir de la carga promedio en las cadenas LC y HC, se puede calcular la DAR promedio para un ADC. La DAR para una muestra de conjugado determinada representa el número promedio de moléculas de fármaco (carga) unidas a un anticuerpo tetramérico que contiene dos cadenas ligeras y dos cadenas pesadas. Las Tablas 5 y 6 proporcionan los valores de DAR obtenidos para los conjugados obtenidos con los anticuerpos anti-Her2 o anticuerpo 20507 y determinados péptidos citotóxicos que tienen una maleimida conectada.
Como comparación, el anticuerpo muíante anticuerpo 20507-LC-S159SC se conjugó con maleimidocaproilmonometilauristatina F (MC-MMAF; Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC, Meyer DL, Oflazoglu E, Toki BE, Sanderson RJ, Zabinski RF, Wahl AF, Senter PD. Bioconjug. Chem. 2006 Ene-Feb;17(1):114-24.) siguiendo los mismos protocolos. Las propiedades del ADC del anticuerpo 20507-LC-S159C-MMAF de comparación se enumeran en la Tabla 6.
Ejemplo 102: Preparación de conjugados de anticuerpo y fármaco a través de reducción parcial de enlaces disulfuro nativos de anticuerpos no modificados.
Los fármacos citotóxicos de la invención también se pueden conjugar con residuos de cisteína nativos de anticuerpos no modificados utilizando un procedimiento que conlleva la reducción parcial de los anticuerpos (Doronina, S. O., Toki, B. E., Torgov, M. Y., Mendelsohn, B. A., Cerveny, C. G., Chace, D. F., DeBlanc, R. L., Gearing,R. P., Bovee, T. D., Siegall, C. B., Francisco, J. A., Wahl, A. F., Meyer, D. L., y Senter, P. D. (2003) Development of potent monoclonal antibody auristatin conjugales forcáncer therapy. Nat. Biotechnol. 21, 778-784). En este ejemplo, los enlaces disulfuro inter- e intracatenarios de los anticuerpos anti-Her2 y anticuerpo 20507 con una concentración de 5 a 10 mg/mL se redujeron en primer lugar parcialmente en PBS que contenía EDTA 2 mM añadiendo mercaptoetilamina sólida hasta una concentración final de 50 mM e incubando la mezcla a 37 °C durante 1 hora. Después de una desalinización y adición de un 1 % p/v de detergente PS-20, los anticuerpos parcialmente reducidos (1 -2 mg/mL) se hicieron reaccionar durante la noche a 4 °C con de 0,5 a 1 mg de compuesto de carga 10 por 10 mg de anticuerpo. Los ADC resultantes, anti-Her2-10 y anticuerpo 20507-10, se purificaron mediante cromatografía de Proteína A. Después de un lavado inicial con PBS, los conjugados se eluyeron con citrato 50 mM, a pH 2,7, NaCl 140 mM, se neutralizaron y se esterilizaron por filtración. Se determinó mediante cromatografía de interacción hidrófoba y MS que la DAR promedio de los dos ADC resultantes, anti-Her2-10 y anticuerpo 20507-10 era 4,1 y 4,6, respectivamente. Las propiedades bioquímicas seleccionadas de los dos ADC se mostraron en las Tablas 5 y 6. Además del anticuerpo anti-Her2 y el anticuerpo 20507, se espera que los péptidos citotóxicos de la invención se pueden conjugar con los residuos de cisteína nativos de numerosos anticuerpos, si no todos, utilizando el método anterior (Ejemplo 102).
T l . Pr i v ri AD ni-H r2 n m i n .


a El nombre consiste en una descripción del anticuerpo mutado y el último número que corresponde al compuesto utilizado en el paso de conjugación química.
b La eficiencia de conjugación se midió mediante HPLC en fase inversa y describe el porcentaje de anticuerpo convertido a ADC.
c Proporción de fármaco respecto a anticuerpo según la HPLC en fase inversa.
d La agregación se midió cromatografía de exclusión de tamaños analítica e incluye especies diméricas y oligoméricas.
T l . Pr i v ri AD l ni r 2 7 n m i n .

a El nombre consiste en una descripción del anticuerpo mutado y el último número que corresponde al compuesto utilizado en el paso de conjugación química.
b La eficiencia de conjugación se midió mediante HPLC en fase inversa y describe el porcentaje de anticuerpo convertido a ADC.
c Proporción de fármaco respecto a anticuerpo según la HPLC en fase inversa.
d La agregación se midió cromatografía de exclusión de tamaños analítica e incluye especies diméricas y oligoméricas.
Ejemplo 103: Preparación de conjugados de anticuerpo y fármaco utilizando 1,3-dicloropropan-2-ona para reconectar enlaces disulfuro nativos de anticuerpos no modificados.
La conjugación a los residuos de cisteína nativos de los anticuerpos no modificados utilizando el procedimiento en el Ejemplo 102 tiene la desventaja de que algunos enlaces disulfuro nativos que estabilizan de forma natural el anticuerpo se rompen y permanecen así después de la conjugación del fármaco. En un método alternativo que supera esta desventaja, los enlaces disulfuro inter- e intracatenarios del anticuerpo se reducen en primer lugar y después se reconectan químicamente mediante una reacción con 1,3-dicloropropan-2-ona. En el proceso, los cuatro enlaces disulfuro intercatenarios nativos en un anticuerpo se reemplazan con un «puente cetona» de tres carbonos (Esquema 17). El grupo cetona se puede conjugar entonces específicamente con un fármaco citotóxico en el segundo paso. El ADC resultante tiene hasta cuatro fármacos unidos específicamente en la localización de los cuatro enlaces disulfuro intercatenarios nativos de un anticuerpo. Al contrario que la conjugación tradicional a disulfuros nativos parcialmente reducidos (Ejemplo 102), los ADC preparados en el ejemplo son más estables. En un ejemplo, se preparó anti-Her2 recombinante sin modificar mediante métodos estándar y tal como se ha descrito anteriormente. Después de la purificación, el anti-Her2 modificado se conjugó a un fármaco citotóxico en dos pasos siguiendo el Esquema 17.
Esquema 17

Paso 1: Reducción de puentes disulfuro nativos y nueva formación de puentes utilizando 1,3-dicloropropan-2-ona: Se añadió TCEPHCl (47,2 pg, 0,165 pmol) a una solución de IgG anti-Her2 (2036 pg, 0,014 pmol) y 1,3-dicloropropan-2-ona (220 pg, 1,648 pmol) en tampón Tris (pH 7,4, 0,25 M, 177pL) a 4 °C. La mezcla resultante se mantuvo a 4 °C durante 4 h. A continuación, la mezcla de reacción se desalinizó utilizando una columna Zeba spin 7K MWCO con PBS (pH 7,4) como tampón de elución. La solución resultante se concentró con un filtro Amicon 10K para proporcionar el anti-Her2 modificado). ESI (Eluyente A: agua 0,1% de ácido fórmico, Eluyente B: Acetonitrilo 0,04% de Ácido fórmico. Gradiente: de un 3 a un 80% de B en 2 min - Flujo 1,0 mL/min. Columna: Proswift Monolith 4,6*50mm 40 °C); 145399 Da (después de la desglicosilación con PNGasa F (New England biolab)).
Paso 2: Conjugación del fármaco citotóxico: Una solución de ácido (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((1 R,3S,4S)-2-(6-(aminooxi)hexanoil)-2-azabiciclo[2.2.1]heptano-3-carboxamido)-N,3-dimetilbutanamido)-3-metoxi-5-metilheptanoil)pirrolidin-2-il)-3-metoxi-2-metilpropanamido)-3-fenilpropanoico (22) (77 pg, 0,078 pmol, 0,77 pL en DMSO) y ácido 3,5-diaminobenzoico (355 pg, 2,334 pmol, 1,42 pL en DMSO) se añadieron a una solución del anti-Her2 modificado (577,5 pg, 0,0039 pmol, 35 pL en PBS, pH 7,4) a 23 °C. La mezcla resultante se mantuvo a 23 °C durante 21 h. La mezcla de reacción se desalinizó entonces utilizando una columna Zeba spin 7K MWCO con PBS (pH 7,4) como tampón de elución dos veces para proporcionar el anti-Her2 modificado conjugado con el compuesto (22). MIS (Eluyente A: agua 0,1% de Ácido fórmico, Eluyente B: Acetonitrilo 0,04 % de Ácido fórmico. Gradiente: de un 3 a un 8 0 % de B en 2 min - Flujo 1,0 mL/min. Columna: Proswift Monolith 4,6*50mm 40 °C); 147955 Da (DAR 3), 148807 Da (DAR 4) (después de la desglicosilación mediante el tratamiento con PNGasa F (New England biolab)). Se calculó que la DAR total era DAR 3,7.
Aunque solamente se muestra en la presente para un anticuerpo anti-Her2, se espera que la estrategia de conjugación en este ejemplo sea aplicable a numerosos anticuerpos diferentes, si no todos.
Ejemplo 104: Conjugación de anticuerpos anti-Her2 y anticuerpo 20507 con mutación de etiqueta A1/ybbR con péptidos citotóxicos de Fórmula (I).
La 4’-fosfopanteteinilación posterior a la traducción es un método versátil para el marcaje con especificidad por un sitio de proteínas recombinantes con moléculas de bajo peso molecular estructuralmente diversas (Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT (2005) Proc. Natl. Acad. Sci. U.S.A.
102:15815-15820) (Zhou Z, Cironi P, Lin AJ, Xu Y, Hrvatin S, Golan De , Silver PA, Walsh CT, Yin J (2007) ACS Chem. Biol. 2:337-346). Esta estrategia enzimática, que se basa en la acción catalítica de 4’-fosfopanteteinil-transferasas (PPTasas) prolíficas, se adoptó para la preparación de ADC muy homogéneos (documento WO2013184514). El marcaje enzimático se logra incorporando secuencias peptídicas 11 o 12-mer de S6 , ybbR y A1 en varios sitios de la región constante de un anticuerpo IgG1. Aunque el siguiente ejemplo describe la formación de ADC mediada por PPTasa para solamente un sitio, se espera que la estrategia sea aplicable a numerosos sitios de inserción dentro de la matriz de anticuerpos y se espera que sea aplicable a numerosos anticuerpos.
Un trabajo previo de Burkart y colaboradores reveló que las PPTasas son enzimas versátiles que aceptan una serie de análogos de CoA-indicador como sustratos (La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart m D (2004) Chem. Biol. 11:195-201). Por tanto, para convertir una carga farmacológica en un sustrato adecuado para la catálisis con PPTasas, el péptido citotóxico que contenía maleimida de Fórmula (I) se conjugó con el grupo tiol terminal de la CoA mediante una adición de Michael (remítase al Ejemplo 43). Habiendo unido covalentemente el péptido citotóxico a un elemento de reconocimiento enzimático, el análogo de CoA-péptido citotóxico resultante (Compuesto 20) se conjugó enzimáticamente a la secuencia peptídica incorporada del respectivo anticuerpo. A modo de ejemplo, se conjugó una
concentración 2,5 pM de anticuerpo anti-Her2-HC-ins388-ybbR o anticuerpo 20507-HC-ins388-A1 con una concentración 30 pM de análogo de CoA-péptido citotóxico (Compuesto 20) (12 equivalentes molares respecto al anticuerpo) en presencia de 1 pM de Sfp PPTasa de Bacillus subtilis. Las reacciones de conjugación enzimática se llevaron a cabo a temperatura ambiente durante aproximadamente 16 horas en tampón Tris-HCl 75 mM (pH 8,0) suplementado con NaCl 20 mM y MgCl212,5 mM. Para garantizar el marcaje casi completo del anticuerpo 20507-HC-ins388-A1 con la carga citotóxica, el tiempo de incubación se extendió otros tres días y las concentraciones del análogo de CoA-péptido de (Compuesto 20) y Sfp PPTasa se aumentaron hasta 35 pM (14 equivalentes molares respecto al anticuerpo) y 2 pM, respectivamente. Después de la conjugación, se eliminaron la Sfp PPTasa y el exceso de reactivo mediante cromatografía de afinidad de Proteína A (Protein A-Sepharose™, GE Healthcare Life Sciences). La elución de la columna se llevó a cabo con 0,1 M de tampón de acetato sódico (pH 3,0) seguido de la neutralización inmediata con tampón T ris-HCl 1 M (pH 8,0). Finalmente, se intercambio el tampón de los ADC etiquetados con péptidos en PBS utilizando columnas de desalinización PD-10 (GE Healthcare).
La extensión de la conjugación de la carga se determinó mediante HPLC analítica en una columna PLRP-S (4000 Á, 5 pM, 50 x 4,6 mm, Agilent Technologies) con un gradiente lineal de 6 min de un 25-50% de acetonitrilo en agua que contenía un 0,1% de ácido trifluoroacético. La separación en fase inversa de anticuerpo conjugado y no conjugado se monitorizó a una longitud de onda de 280 nm. Con este fin, la integración de picos de HPLC indicó el marcaje casi completo de los anticuerpos anti-Her2-HC-ins388-ybbR y 20507-HC-ins388-A1 con el análogo de CoA-péptido (Compuesto 20). La identidad de los ADC marcados enzimáticamente se confirmó adicionalmente obteniendo los espectros de ESI-MS deconvolucionados de las muestras reducidas y desglicosiladas correspondientes. Tal como se muestra en la Tabla 8, las masas observadas están en consonancia con los pesos moleculares calculados de las cadenas pesadas marcadas con fármacos de los anticuerpos anti-Her2-HC-ins388-ybbR y 20507-HC-ins388-A1. Finalmente, los ADC marcados enzimáticamente se analizaron mediante cromatografía de exclusión de tamaños analítica (AnSEC) en una columna Shodex PROTEIN KW-803. Ambos conjugados de anticuerpo se eluyeron a los tiempos de retención que correspondían al peso molecular aparente del ADC monomérico (remítase a la Tabla 7). No se detectaron otras especies, lo que indica que la conjugación del análogo de CoA-péptido hidrófilo no promovió la agregación de los anticuerpos.
-

a HC-ins388 se refiere a la inserción de la etiqueta peptídica A1 o ybbR después del residuo Glu388 en la cadena pesada. El último numero corresponde al compuesto utilizado en el paso de conjugación química.
b La eficiencia de conjugación se midió mediante HPLC en fase inversa y describe el porcentaje de anticuerpo convertido a ADC.
c Proporción de fármaco respecto a anticuerpo según la HPLC en fase inversa.
d La agregación se midió cromatografía de exclusión de tamaños analítica e incluye especies diméricas y oligoméricas.
- ^

a HC-ins388 se refiere a la inserción de la etiqueta peptídica A1 o ybbR después del residuo Glu388 en la cadena pesada. El último numero corresponde al compuesto utilizado en el paso de conjugación química.
b Masa en Dalton tal como se detecta en un instrumento Agilent 6520 Q-TOF (Agilent Technologies).
c Masa en Dalton tal como se ha predicho para la cadena pesada conjugada.
c Masa en Dalton tal como se ha predicho para la cadena pesada sin acoplar.
Ejemplo 105: Conjugación de dos pasos de los anticuerpos mutantes anti-Her2 y anticuerpo 20507 etiquetados con A1/ybbR con un péptido citotóxico de Fórmula (I)
El método de dos pasos es una estrategia alternativa para preparar ADC con especificidad por un sitio mediante 4’-fosfopanteteinilación posterior a la traducción (documento WO2013184514). El primer paso de esta estrategia se basa
en el mareaje catalizado por PPTasa de un anticuerpo etiquetado con péptidos con un análogo de CoA que contiene un grupo bioortogonal tal como un resto azido, alqueno, alquino, cetona o aldehído. Después de la purificación por afinidad del anticuerpo marcado bioortogonalmente, el segundo paso del método de dos pasos conlleva la conjugación de una carga citotóxica que comprende un resto reactivo con el grupo bioortogonal. A modo de ejemplo, la siguiente sección describe el método de dos pasos para los anticuerpos mutantes anticuerpo 20507 y anti-Her2 que contienen una inserción de etiqueta A1 o ybbR en un sitio específico dentro de la región constante de la cadena pesada. Además, la siguiente sección describe el método de dos pasos para los anticuerpos mutantes anticuerpo 20507 y anti-Her2 que contienen una inserción de etiqueta A1 o ybbR en un sitio específico dentro de la región constante de la cadena pesada. Sin embargo, se prevé que esta estrategia sea ampliamente aplicable a numerosos sitios de inserción dentro de las regiones constantes de una amplia variedad de anticuerpos. Además, aunque el método de dos pasos se ilustra para el ligamiento de oximas y la química click sin cobre, se puede concebir que esta estrategia se podría extender a otras químicas bioortogonales tales como el ligamiento de Staudinger, la química click basada en isonitrilo y el ligamiento con tetrazina.
El ligamiento con oxima y la química click sin cobre han sido utilizados por varios grupos de investigación como un método eficiente bioortogonal para la preparación de conjugados de proteínas con especificidad por un sitio (Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, Lu Y, Tran H, Seller AJ, Biroc SL, Szydlik A, Pinkstaff JK, Tian F, Sinha SC, Felding-Habermann B, Smider VV, Schultz pG (2012) Proc Natl Acad Sci U S A. 109:16101-16106) (Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi c R (2012) Nat Protoc.
7:1052-1067) (Plass T, Milles S, Koehler C, Schultz C, Lemke eA (2011) Angew Chem Int Ed Engl. 50:3878-3881) (Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carell T (2012) Angew Chem Int Ed Engl. 51:4466-4469). Con el fin de combinar la 4’-fosfopanteteinilación con el ligamiento con oxima, se unió covalentemente un grupo cetona a CoA haciendo reaccionar esta última con metil vinil cetona (remítase al Ejemplo 93). Después, se utilizó la catálisis con PPTasa para conjugar enzimáticamente el grupo cetona bioortogonal con especificidad por un sitio en la etiqueta A1 incorporada de un anticuerpo anti-Her2. Específicamente, se conjugaron 2.5 pM de anticuerpo anti-Her2-HC-ins388-A1 con una concentración 30 pM del análogo de CoA-cetona resultante (Compuesto 49) (12 equivalentes molares respecto al anticuerpo) en presencia de una concentración 1 pM de Sfp PPTasa de Bacillus subtilis. El primer paso del método de dos pasos se llevó a cabo durante aproximadamente 16 horas a temperatura ambiente en tampón T ris-HCl 75 mM (pH 8,0), suplementado con MgCl212,5 mM y NaCl 20 mM. El marcaje casi completo del anticuerpo anti-Her2-HC-ins388-A1 con el análogo de CoA-cetona (Compuesto 49; remítase al Ejemplo 93) se verificó obteniendo los espectros de ESI-MS deconvolucionados de la muestra reducida y desglicosilada. Las condiciones de reacción similares también dieron como resultado una funcionalización con cetona casi cuantitativa del anticuerpo 20507-HC-ins388-A1. Tal como se muestra en la Tabla 10, las masas observadas están en consonancia con los pesos moleculares calculados de las cadenas pesadas funcionalizadas con cetona correspondientes. Después de eliminar la Sfp PPTasa y el exceso de análogo de CoA-cetona mediante cromatografía de afinidad de Proteína A (Protein A-Sepharose™, Ge Healthcare Life Sciences), los anticuerpos activados con cetona, anti-Her2-HC-ins388-A1-49 y anticuerpo 20507-HC-ins388-A1-49, se eluyeron con una concentración 0,1 M de tampón de acetato sódico (pH 3) seguido de la neutralización inmediata con tampón Tris-HCl 1 M (pH 8). Las soluciones de anticuerpos neutralizadas se sometieron a intercambio de tampón en agua y se hicieron pasar a través de filtros con un tamaño de poro de 0,22 pm.
En otro aspecto de esta estrategia de marcaje de dos pasos, se prepararon quimioenzimáticamente análogos de CoA modificados utilizando las enzimas de CoA biosintéticas CoAA, CoAD y CoAE (Worthington AS, Burkart MD (2006) Org Biomol Chem. 4:44-46) (Kosa NM, Haushalter RW, Smith AR, Burkart MD (2012) Nat Methods 9:981-984). Adoptando esta estrategia, se prepararon análogos de CoA funcionalizados con cetona a partir de las moléculas precursoras de pantotenato correspondientes (i-10), (i-12) e (i-13) (remítase a los Ejemplos 94, 96 y 97). Del mismo modo, se sintetizó quimioenzimáticamente un análogo de CoA funcionalizado con azida a partir del respectivo derivado de pantotenato (i-11) (remítase al Ejemplo 95). En esta estrategia, los ultrafiltrados de los Ejemplos 94-97 se utilizaron sin purificación adicional, y los análogos de CoA CoA-(i-10), CoA-(i-11), CoA-(i-12) y CoA-(i-13) se utilizaron para la conjugación a anti-Her2-HC-ins388-ybbR (2,5 pM), con una concentración final de aproximadamente 30 pM. El marcaje de anticuerpos se llevó a cabo en presencia de Sfp PPTasa de B. subtilis 1,5 pM durante de 16 a 72 horas a 23 °C en tampón Tris-HCl 75 mM (pH 8,0), suplementado con MgCl2 12,5 mM y NaCl 20 mM. De forma similar, se conjugó CoA-(i-13) con una concentración final de aproximadamente 25 pM en presencia de enzima Sfp 2 pM.
Se llevó a cabo una cromatografía de afinidad (MabSelect SuRe™, GE Healthcare Life Sciences) de los anticuerpos marcados bioortogonalmente de forma exactamente idéntica a la descrita anteriormente para el análogo ceto-CoA 49. Después de la purificación, las soluciones de anticuerpo neutralizadas se sometieron a intercambio de tampón en PBS. La unión covalente de los restos cetona y azida al anticuerpo etiquetado con ybbR se confirmó mediante análisis de espectrometría de masas siguiendo un tratamiento de muestras con PNGasa F y TCEP (Tabla 10).
La unión con especificidad por un sitio de un grupo cetona permitió el ligamiento con oxima posterior de una carga citotóxica a anti-Her2-HC-ins388-A1 -49, anticuerpo 20507-HC-ins388-A1 -49, anti-Her2-HC-ins388-ybbR-(i-10), y anti-Her2-HC-ins388-ybbR-(i-12) activados con cetona como segundo paso del método de dos pasos. Se hizo reaccionar una concentración de 25 a 67 pM de anticuerpo funcionalizado con cetona con un exceso molar de un factor 7,5 a 40 (0,5 - 1 mM) del análogo aminooxi-péptido (Compuesto 22) en tampón de acetato sódico 50 o 100 mM (pH 4 - 5) que
contiene de un 2,5 a un 5% (v/v) de DMSO. Después de 16 a 36 horas de incubación a 23 o 37 °C, se eliminó el reactivo aminooxi en exceso mediante cromatografía de exclusión de tamaños preparativa en una columna de grado preparativo HiLoad 26/600 Superdex 200 (GE Healthcare). La proporción de fármaco respecto a anticuerpo (DAR) se determinó mediante HPLC analítica en una columna PLRP-S (4000 Á, 5 pM, 50 x 4,6 mm, Agilent Technologies) con un gradiente lineal de 5 min de un 30 a un 60% de acetonitrilo en agua que contenía un 0,1% de ácido trifluoroacético. La traza de HPLC se monitorizó a una longitud de onda de 280 nm y después se integraron los picos del anticuerpo conjugado y no conjugado. Tal como se muestra en la Tabla 9, el método de dos pasos proporcionó un marcaje casi cuantitativo de anti-Her2-HC-ins388-A1 -49 y anticuerpo 20507-HC-ins388-A1 -49 activados con cetona con el análogo aminooxi-péptido (Compuesto 22). Por el contrario, la conjugación con especificidad por un sitio del compuesto 22 al anticuerpo anti-Her2-HC-ins388-ybbR-(i-10) activado con cetona fue menos eficiente, lo que dio lugar a un ADC con una DAR de 1,3. La formación de ADC a través de un ligamiento con oxima se confirmó adicionalmente mediante ESI-MS después de la reducción y desglicosilación de los inmunoconjugados (Tabla 10). Finalmente, la cromatografía de exclusión de tamaños analítica (AnSEC) en columnas Superdex 200 10/300 GL (GE Healthcare) y Protein KW-8035 pm 300 x 8 mm (Shodex) revelaron solamente pequeñas cantidades de ADC agregado, lo que sugiere que se induce poca agregación durante el proceso de conjugación de dos pasos.
La unión con especificidad por un sitio de un resto azida a un anticuerpo modificado permite la posterior conjugación de una carga mediante química click sin cobre. En este ejemplo, la reacción bioortogonal es promovida por la tensión anular del grupo biciclo[6.1.0]nonino (BCN), que se une covalentemente al fármaco-conector. La cicloadición alquinoazida promovida por la tensión se llevó a cabo con el anticuerpo anti-Her2-HC-ins388-ybbR-(i-11) activado con azida en presencia del Compuesto de carga 75 funcionalizado con BCN. Este segundo paso del método de dos pasos se llevó a cabo con una concentración 127 pM de anticuerpo anti-Her2-HC-ins388-ybbR-(i-11) y una concentración 1270 pM del análogo peptídico conectado a BCN (Compuesto 75) en tampón de fosfato sódico 100 mM (pH 7,5) suplementado con NaCl 1 M y un 6% (v/v) de DMSO. Después de aproximadamente 16 horas de incubación a 23 °C, el reactivo de BCN en exceso se eliminó mediante cromatografía de afinidad con Proteína A estándar utilizando MabSelect SuRe™ (GE Healthcare Life Sciences). La elución se llevó a cabo con tampón de elución con IgG (Thermo Scientific), seguida por la neutralización con tampón Tris-HCl 1 M (pH 8) e intercambio de tampón en PBS. Tal como se muestra en la Tabla 10, la conjugación de la carga mediante química click sin cobre se confirmó mediante ESI-MS después de la reducción y la desglicosilación de la muestra de ADC. El mismo método se utilizó para calcular la DAR promedio del inmunoconjugado. En este proceso, se suplementaron 10 pg de ADC con 10 pL de una suspensión densa al 50% de IgG Sepharose 6 Fast Flow (GE Healthcare). La unión a la resina se llevó a cabo con agitación suave durante 1 h a 23 °C. Después de lavar la resina con PBS, el ADC unido por afinidad se desglicosiló mediante adición de 5 pg de PNGasa F y posterior incubación a 37 °C durante 3 horas. La enzima PNGasa F se eliminó lavando la resina de afinidad con PBS. Después, la muestra desglicosilada se eluyó utilizando un 1% de ácido fórmico seguido por la neutralización inmediata con acetato amónico 10 M (pH 5). Para reducir eficazmente el constructo de anticuerpo a las cadenas pesadas y ligeras, se suplementaron 20 pL de eluato con 10 pL de tampón de formiato sódico 100 mM (pH 4,0) que contenía clorhidrato de guanidina 6 M y 5 pL de TCEP 0,66 M en acetato amónico 10 M (pH 5). Después de la incubación durante al menos 30 min a 23 °C, la muestra reducida y desglicosilada se inyectó en un sistema de LC/MS 6550 iFunnel Q-TOF (Agilent Technologies). Se utilizó MassHunter Qualitative Analysis Software (Agilent Technologies) para el procesamiento del registro espectral y la deconvolución espectral. La DAR promedio se calculó como el promedio ponderado del estado de DAR de las alturas de pico relativas a lo largo de una distribución. Tal como se muestra en la Tabla 9, el ADC anti-Her2-HC-ins388-ybbR-(i-11)-75 tiene una DAR promedio de 2,0, lo que sugiere que ambos pasos de conjugación fueron prácticamente cuantitativos. Finalmente, el ADC se examinó por cromatografía de exclusión de tamaños analítica (AnSEC) en una columna Bio SEC-3 (Agilent Technologies) y se observó que era monomérico en un 95%, lo que sugiere que se induce poca agregación en el proceso de conjugación.
Tabla 9.
Propiedades de anticuerpos etiquetados con A1 e ybbR/ADC marcados mediante un proceso de conjugación de dos


a HC-ins388 se refiere a la inserción de la etiqueta peptídica A1 o ybbR después del residuo Glu388 en la cadena pesada. El último número corresponde al compuesto utilizado en el paso de conjugación. Por ejemplo, el anti-Her2-HC-ins388-A1 -49-22 y el anticuerpo 20507-HC-ins388-A1 -49-22 se conjugaron en primer lugar enzimáticamente con el compuesto 49 y después por conjugación química con el compuesto 22.
b La eficiencia de conjugación se midió mediante HPLC en fase inversa y describe el porcentaje de anticuerpo convertido a ADC.
c La eficiencia de conjugación se determinó mediante espectrometría de masas y describe el porcentaje de anticuerpo convertido a ADC.
d Proporción de fármaco respecto a anticuerpo según la HPLC en fase inversa.
e Proporción de fármaco respecto a anticuerpo según la espectrometría de masas.
f La agregación se midió mediante cromatografía de exclusión de tamaños analítica e incluye especies diméricas y oligoméricas.
Tabla 10.
Propiedades de anticuerpos etiquetados con A1 e ybbR/ADC marcados mediante un proceso de conjugación de dos

a HC-ins388 se refiere a la inserción de la etiqueta peptídica A1 o ybbR después del residuo Glu388 en la cadena pesada. El último numero corresponde al compuesto utilizado en el paso de conjugación. Por ejemplo, el anti-Her2-HC-ins388-A1 -49-22 y el anticuerpo 20507-HC-ins388-A1 -49-22 se conjugaron en primer lugar enzimáticamente con el compuesto 49 y después por conjugación química con el compuesto 22.
b Masa en Dalton tal como se detecta en un instrumento Agilent 6520 Q-TOF (Agilent Technologies).
c Masa en Dalton tal como se ha predicho para la cadena pesada conjugada.
c Masa en Dalton tal como se ha predicho para la cadena pesada sin acoplar.
e Pico minoritario de identidad desconocida.
f Cadena no modificada se refiere tanto a las especies no fosfopanteteiniladas como a las funcionalizadas con cetona/azida.
Aunque los inmunoconjugados de Fórmula (II) y Fórmula (III) divulgados en la Tabla 5 y 6 se obtuvieron conjugando los anticuerpos anti-Her2 y anticuerpo 20507 Cys y determinados péptidos citotóxicos de Fórmula (I) que tienen un resto maliemida conectado, también se han utilizado otras combinaciones de conector-carga de la invención. Los inmunoconjugados divulgados en las Tablas 7 a 10 son ejemplos de este tipo. La conjugación a anticuerpos no modificados es otro ejemplo. Esto se puede lograr mediante la conjugación a enlaces disulfuro parcialmente reducidos
tal como se ha descrito en los Ejemplos 102 y 103. Los anticuerpos no modificados también pueden ser lisinas conjugadas utilizando, por ejemplo, el compuesto 89, o una serie de métodos diferentes conocidos en la técnica.
Ejemplo 106: Ensayos de proliferación celular para medir in vitro la potencia de destrucción celular de los ADC de anti-Her2 y anticuerpo 20507 con etiquetas A1/ybbR y Cys.
Se utilizan frecuentemente células que expresan de forma natural antígenos diana o líneas celulares modificadas para que expresen antígenos diana para someter a ensayo la actividad y potencia de los ADC. Para la evaluación de la potencia de destrucción celular de los ADC de anticuerpo anti-Her2 in vitro, se emplearon dos líneas celulares modificadas, clon 16 y clon 40 de MDA-MB231, y cuatro líneas celulares endógenas, células JimT1, HCC1954, NCI-N87 y SKBR3 (Clinchy B, Gazdar A, Rabinovsky R, Yefenof E, Gordon B, Vitetta ES. Breast Cáncer Res Treat. (2000) 61:217-228). Las células del clon 16 de MDA-MB231 expresan de forma estable números de copias elevados (~5x105 copias/célula) de Her2 humano recombinante, mientras que el clon 40 expresa números de copias bajos (~5x103 copias/célula) de Her2 humano. En HCC1954 se expresan de forma endógena niveles elevados de Her2 (~5x105 copias/célula), SKBR-3 (5,4x105 copias/célula) y las líneas celulares NCI-N87 (2,7x105 copias/célula) mientras que las células JimT-1 expresan Her2 humano en un nivel medio (~8x104 copias/célula). Como control negativo, se utilizó la línea celular A375 negativa para Her2. Un efecto citotóxico dependiente de antígeno debería destruir solamente células que expresan suficiente antígeno en la superficie celular y no las céllas que carecen del antígeno. Los ensayos de proliferación celular se llevaron a cabo con Cell-Titer-Glo™ (Promega) cinco días después de que las células se incubasen con varias concentraciones de ADC (Riss et al., (2004) Assay Drug Dev Technol. 2:51-62). En algunos estudios, los ensayos basados en células son de alto rendimiento y se llevaron a cabo en un sistema automatizado (Melnick et al., (2006) Proc Natl Acad Sci U S A. 103:3153-3158).
Todos los ADC anti-Her2 excepto uno (anti-Her2-LC-S159C-44) destruían específicamente líneas celulares con niveles elevados de expresión de Her2, concretamente el clon 16 de MDA-MB231, HCC1954, NCI-N87, SKBR3, pero no mostraban potencia citotóxica hacia células del clon 40 de MDA-MB231, que expresan un nivel bajo de Her2, y tampoco hacia células A375 negativas para Her2 (Figura 1; Tabla 11, Tabla 13). La CI50 de los ADC anti-Her2 en las cuatro líneas celulares que expresan niveles elevados de Her2 oscila de 20 pM a 700 pM (Tabla 11, Tabla 13). En las células JimT-1 que expresan un nivel medio de Her2, las actividades de destrucción celular con los anti-Her2-ADC variaron ampliamente. Algunos ADC fueron activos en células que expresaban niveles elevados de Her2, pero no en células JimT-1. Muchos ADC destruyeron la células JimT-1 con la misma eficacia que las líneas celulares que expresaban niveles elevados de Her2 (Tabla 11, Tabla 13). Los resultados sugieren que los ADC anti-Her2 destruían células Her2+ de forma dependiente de Her2 y que los ADC son activos hacia múltiples tipos de células. De forma similar, los ADC anti-Her2 preparados mediante un método de reducción parcial de anticuerpos de origen natural (Ejemplo 102) y mediante métodos enzimáticos a través de una etiqueta A1 o ybbR (Ejemplos 104 y 105) y mediante una metodología de puentes cetona (Ejemplo 103) también destruían células Her2+ de una forma dependiente de Her2 (Tabla 11), lo que demuestra que los péptidos citotóxicos divulgados en la invención retienen su potencia cuando se conjugan a anticuerpos a través de un método y química de conjugación diferentes.
Para verificar si los péptidos citotóxicos de Fórmula (I) son también activos cuando se conjugan con otros anticuerpos, se conjugaron varios péptidos citotóxicos de Fórmula (I) al anticuerpo 20507, cuyo antígeno se expresa en las células H526, KU812 y CMK11-5, pero no en las células de Jurkat. Tal como se muestra en la Figura 2 y en la Tabla 12, las combinaciones de carga y conector que muestran actividades de destrucción celular en células Her2+ también son activas cuando se conjugan al anticuerpo 20507, al destruir células que expresan el antígeno diana. Los resultados indican que los péptidos citotóxicos de Fórmula (I) descritos en la presente muestran citotoxicidad hacia una amplia gama de tipos de células.
Tabla 11.
Potencia de ADC en un ensayo de destrucción celular in vitro: CI50 de los ADC anti-Her2 en ensayos de proliferación l l r l n 4 MDA-MB2 1 l n 1 MDA-MB2 1 H 1 4 imT-1


a El nombre consiste en una descripción del anticuerpo mutado y el último número que corresponde al compuesto utilizado en el paso de conjugación química. HC-ins388 se refiere a la inserción de la etiqueta peptídica A1 o ybbR después del residuo Glu388 en la cadena pesada.
b La concentración más elevada utilizada en el ensayo fue 6,67E-02 pM. Los valores de CI50 de 6,67E-02 pM se refieren por tanto a la inactividad del ADC en el ensayo.
c El valor es igual a la concentración más elevada utilizada en el ensayo, lo que indica por tanto inactividad del ADC.
Tabla 12
Potencia de ADC en un ensayo de destrucción celular in vitro: CI50 de los ADC de anticuerpo 20507 en ensayos de


a El nombre consiste en una descripción del anticuerpo mutado y el último número que corresponde al compuesto utilizado en el paso de conjugación química. HC-ins388 se refiere a la inserción de la etiqueta peptídica A1 después del residuo Glu388 en la cadena pesada.
b La concentración más elevada utilizada en el ensayo fue 6,67E-02 pM. Los valores de CI50 de 6,67E-02 pM se refieren por tanto a la inactividad del ADC en el ensayo.
Tabla 13
Potencia de ADC en un ensayo de destrucción celular in vitro: CI50 de los ADC anti-Her2 en ensayos de proliferación de células A375 HCC1954 JimT-1 NCI-N87 SKBR3.

a El nombre consiste en una descripción del anticuerpo mutado y el último número que corresponde al compuesto utilizado en el paso de conjugación química.
b La concentración más elevada utilizada en el ensayo fue 6,67E-02 pM. Los valores de CI50 de 6,67E-02 pM se refieren por tanto a la inactividad del ADC en el ensayo.
c N.D.: no determinado
Ejemplo 107: Estudio farmacocinético de ADC de anti-Her2 y anticuerpo 20507.
Se ha demostrado que una semivida en suero prolongada es crítica para una eficacia in vivo elevada de los ADC (Hamblett, et al., "Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate," Clin Cáncer Res., 10:7063-7070 (2004); Alley et al., Bioconjug Chem. 19:759-765 (2008)). La unión de una carga farmacológica hidrófoba a un anticuerpo podría afectar a las propiedades de un anticuerpo, y esto puede dar lugar a una eliminación rápida de los ADC in vivo (Hamblett et al., 2004) y a escasa eficacia in vivo. Para evaluar los efectos de la conjugación de varios péptidos citotóxicos de la invención en la eliminación de los ADC in vivo, se llevaron a cabo estudios farmacocinéticos en ratones que no portaban tumores. Para detectar el péptido citotóxico (es decir, la
parte farmacológica) de los inmunoconjugados de la invención en plasma murino, se generó un anticuerpo anti-MMAF que también reconoce varios péptidos citotóxicos de la invención. Se desarrollaron ELISA para la detección de los inmunoconjugados en una plataforma Gyros™ utilizando un anticuerpo anti-hIgG para capturar moléculas IgG humanas del plasma y un segundo anticuerpo anti-IgG humano y el anticuerpo anti-MMAF para la detección de señales en dos ensayos separados. El anticuerpo anti-MMAF reconoce los péptidos citotóxicos de la invención y, por tanto, se puede utilizar para detectar ADC con los péptidos citotóxicos unidos (ADC «intactos»). Por tanto, los dos ELISA miden la concentración en suero del anticuerpo humano y el ADC «intacto», respectivamente.
Se muestran ejemplos de estudios PK en la Figura 3 y la Figura 4. Se administró a tres ratones por grupo una única dosis de los siguientes ADC con una concentración de 1 mg/kg: ADC de anti-Her2-LC-S159C-10 (Figura 3A), anti-Her2-LC-S159C-47 (Figura 3B), anti-Her2-LC-S159C-77 (Figura 3C), anti-Her2-LC-S159C-80 (Figura 3D), anti-Her2-LC-S159C-79 (Figura 3E), anti-Her2-LC-S159C-78 (Figura 3F), anti-Her2-LC-S159C-14 (Figura 3G), anti-Her2-HC-E152C-S375C-10 (Figura 3H), anti-Her2-10 (Figura 3I), anticuerpo 20507-HC-E152C-10 (Figura 4a ), anticuerpo 20507-LC-K107C-47 (Figura 4B), anticuerpo 20507-HC-ins388-A1-20 (Figura 4C), anticuerpo 20507-HC-E152C-S375C-10 (Figura 4D), anticuerpo 20507-10 (Figura 4E), anti-Her2-HC-ins388-ybbR-20 (Figura 4g ), anticuerpo 20507-HC-ins388-A1 -49-22 (Figura 4H) anti-Her2-HC-ins388-A1-49-22 (Figura 4I), y anti-Her2-HC-ins388-ybbR-(i-11)-75 (Figura 4J). Se administró el anticuerpo 20507-HC-ins388-A1-20 (Figura 4F) con una única dosis de 10 mg/kg. Se recogieron muestras de plasma durante tres semanas y se sometieron a ensayo mediante ELISA utilizando un anticuerpo anti-hIgG para capturar las moléculas IgG que incluían los ADC de anticuerpo 20507 y anti-Her2, así como los respectivos anticuerpos puros. El anticuerpo anti-MMAF y un anticuerpo anti-hIgG se utilizaron después para la detección en dos ensayos separados. El ensayo de anticuerpo anti-MMAF mide la concentración de conjugados de anticuerpos solamente y el anti-hIgG cuantifica tanto conjugados de anticuerpo como anticuerpo que carece de cargas. Se generaron curvas estándar para cada ADC por separado utilizando el mismo material que se inyectó en los ratones. Los ensayos con anti-MMAF y anti-hIgG deberían proporcionar, por tanto, lecturas de concentración idénticas si no tienen lugar cambios en la carga de fármaco del ADC de anticuerpo 20507 o anti-Her2 después de la inyección en los ratones. Para los ADC que pierden parte de la carga, el ensayo con el anticuerpo anti-MMAF medirá una concentración inferior que el ensayo anti-hIgG. Una comparación de las dos lecturas de concentración permite, por tanto, medir la liberación de fármaco a partir de los ADC de anticuerpo 20507 y anti-Her2 durante la incubación in vivo en el ratón.
Tal como se muestran en la Figura 3 y la Figura 4, las concentraciones en plasma obtenidas tanto en ensayos antihIgG como anti-MMAF coinciden bien con la mayoría de ADC con cargas de maleimida que se conjugan con una o más Cys introducidas mediante modificación en ambos anticuerpos anti-Her2 y anticuerpo 20507 (Figura 3A-I, Figura 4D), lo que sugiere que existe una pérdida de fármaco mínima en estos ADC durante el periodo de prueba para estos ADC. Los dos ADC preparados mediante reducción parcial de anticuerpos de origen natural, anti-Her2-10 (Figura 3I) y anticuerpo 20507-10 (Figura 4E), mostraron una pronunciada separación entre las concentraciones determinadas con los ensayos anti-hIgG y ant-MMAF, siendo la primera superior a última, lo que sugiere que hay una pérdida de fármaco significativa durante el periodo de prueba para los dos ADC. Los ADC preparados por métodos mediados por enzimas, ADC de anticuerpo 20507-HC-ins388-A1-20, anti-Her2-HC-ins388-ybbR-20, anticuerpo 20507-HC-ins388-A1-49-22, y anti-Her2-HC-ins388-A1-49-22 mostraron una buena coincidencia entre las concentraciones determinadas mediante ensayos anti-IgG y anti-MMAF (Figura 4F, G, H e I). Por el contrario, el ADC anti-Her2-HC-ins388-ybbR-(i-11)-75 (Figura 4J) presentó una gran separación entre los ensayos anti-hIgG y anti-MMAF, lo que sugiere que el ADC experimenta una desconjugación del fármaco significativa durante el curso del estudio FC.
Tal como se muestra en la Figura 4, las concentraciones plasmáticas obtenidas mediante tanto el ensayo anti-hIgG como el ensayo anti-MMAF coinciden bien con el ADC de anticuerpo 20507-HC-E153C-10, ADC de anticuerpo 20507-LC-K107C-47 y ADC de anticuerpo 20507-HC-ins388-A1 -20, lo que sugiere que hay una pérdida de fármaco mínima en estos ADC durante el periodo de prueba.
Con el fin de determinar la retención de las cargas farmacológicas para los ADC después de tres semanas en la circulación del ratón con más detalle, los ADC se purificaron por afinidad a partir de suero de ratón recogido mediante sangrado terminal y las cargas farmacológicas unidas a los ADC se analizaron mediante análisis MS. En un proceso típico, se diluyeron 200 pL de plasma con una cantidad igual de PBS que contenía EDTA 10 mM. A la dilución, se añadieron 10 pL de resina de afinidad (IgG Select Sepharose 6 Fast flow; GE Healthcare 17-0969-01; suspensión densa al 50%). Se llevó a cabo una incubación de la resina con las muestras de plasma diluidas durante 1 h a temperatura ambiente aplicando agitación suave para evitar que sedimentase la resina. A continuación, la resina se eliminó por filtración y se lavó dos veces con 200 pL de PBS. Para desglicosilar el anticuerpo, se añadieron 10 pL de PNGasa F (1 mg/mL, 1/2x TBS pH 7,4, EDTA 2,5 mM, Glicerol al 50%) diluidos con 10 pL de PBS a la resina y las mezclas se incubaron durante 2-3 h a 37 °C. Después de que se eliminase la PNGasa F lavando la resina de afinidad dos veces con 200 pL de PBS, la muestra se eluyó dos veces de la resina de afinidad añadiendo 20 pL de ácido fórmico al 1% y se eliminó la resina por filtración. Los eluatos combinados se diluyeron con 20 pL de clorhidrato de guanidina 6 M y 5 pL de tampón de reducción (TCEP 0,66 M, acetato amónico 3,3 M, pH 5). Para reducir eficazmente el anticuerpo, las muestras se incubaron durante al menos 30 min a temperatura ambiente antes del análisis. El LCMS se llevó a cabo con un sistema de HPLC Agilent 1260/6550-iFunnel QTOF MS de Agilent Technologies. Se utilizó una cromatografía en fase inversa estándar para desalinizar la muestra con una columna PLRS (8 pm, 2,1 x 50 mm, 1000Á, Agilent) con un caudal de 0,5 mL/min a 80 °C. La elución se llevó a cabo utilizando un gradiente lineal de un 20%- a
un 60%-de acetonitrilo que contenía ácido fórmico al 0,1% en 6 min. Se utilizó Agilent Qualitative Analysis para el procesado del registro espectral y la deconvolución espectral. Para el análisis, el registro espectral se sumó durante el intervalo temporal que cubría la elución de todas las especies relevantes. Los espectros sumados se deconvolucionaron en su estado de carga y se registraron las imágenes de los espectros deconvolucionados. Se extrajeron los valores de intensidad de pico para las especies asignables. Se realizaron asignaciones de estado de DAR y especies fragmentadas basándose en los valores de masa calculada a partir de la secuencia de los anticuerpos analizados y los desplazamientos de masas esperados de los conjugados con las moléculas de fármaco. La DAR promedio se calculó utilizando las alturas de pico relativas de todos los estados de DAR a lo largo de una distribución. Se calculó la DAR de los anticuerpos promedio como la suma de las DAR de 2 cadenas ligeras promedio y 2 cadenas pesadas promedio.
La DAR promedio de los ADC purificados después de tres semanas en la circulación del ratón, medida mediante MS, se comparó a la DAR en los preparados de ADC originales. La «retención de carga» se calculó a partir de la proporción de las dos DAR (DAR de los ADC aislados a partir de plasma de ratón divididas por la DAR del preparado de ADC original), y representa el porcentaje de cargas retenidas en el ADC después de tres semanas en la circulación del ratón. La retención de carga de varios ADC medida mediante MS está en gran medida en consonancia con los resultados obtenidos con el ensayo ELISA mencionado anteriormente utilizando el anticuerpo anti-MMAF (Figuras 3 y 4).
Tal como se muestra en la Tabla 14, la retención de carga osciló ampliamente de un 5% a un 96% dependiendo del compuesto, los sitios de conjugación y el método de conjugación. Para los ADC preparados mediante el método enzimático de PPTasa, la retención de carga (mediante MS) para los compuestos 20 y 22 es superior a un 50%, mientras que para el compuesto 75, la retención de carga es solamente un 5% (Tabla 14). En el último caso, la evaluación de espectrometría de masas del ADC purificado a partir del suero de las sangrías terminales reveló una escisión prácticamente cuantitativa del resto carbamato del fármaco-conector, mientras que la unión mediante química click entre el anticuerpo y la carga citotóxica permaneció estable en circulación.
Para los ADC preparados con péptidos citotóxicos reactivos con Cys que presentan un grupo maleimida, la retención de carga osciló de un 18% a un 96%, dependiendo de la estructura del compuesto y los sitios de conjugación (Tabla 14). Los resultados de MS para los ADC aislados a partir de las muestras de plasma confirman que tenía lugar una desconjugación de las cargas de los ADC entre las cargas de maleimida y el grupo tiol de los residuos de Cys conjugados. No se observaron roturas en las estructuras conectoras. Por lo tanto, la retención de carga para las cargas conectadas con maleimida puede ser inversamente proporcional a la desconjugación mediante adición de Michael, ya que la masa observada del anticuerpo no conjugado recuperado es la del anticuerpo sin modificar.
Se tiene constancia de que la reacción para formar un tioéter entre una maleimida y un grupo tiol libre es reversible (Bioconjugate Chem. 2011,22, 1946-1953). Se cree que la reacción inversa es responsable de la desconjugación de las cargas de fármacos con maleimida de los ADC in vivo e in vitro. Posteriormente, la maleimida de las cargas desconjugadas puede reaccionar con tioles libres en forma de aminoácidos, péptidos o proteínas. (Bioconjugate Chem.
2008, 19, 759-765; Nat. Biotechnol. 2012, 30, 184-189).
Para determinados péptidos citotóxicos de la invención, se observó que la desconjugación de las cargas de los ADC en la circulación sanguínea de los ratones era despreciable (Tabla 14). Después de tres semanas en la circulación de los ratones, se observó que se retenía un 90% de las cargas para los ADC preparados con los compuestos 47, 14, 77, 80, 79 y 78 que se conjugaban a las Cys introducida mediante modificación de los anticuerpos. El análisis MS de estos ADC aislados a partir de muestras de plasma reveló que la masa de los ADC aumentó en 18 dalton para cada carga farmacológica conjugada, lo que sugería que había tenido lugar una reacción de hidrólisis. Se tiene constancia de que el anillo de succinimida resultante de la reacción de un grupo maleimida con una cisteína experimenta una hidrólisis espontánea (Gregory, J. Am. Chem. Soc. 1955, 77, 3922; Knight, P. Biochem. J. 1979, 179, 191-197; Khan M.N. J Pharm Sci. 1984, 73:1767-1771). De hecho, el grupo se puede hidrolizar a propósito exponiendo un conjugado a pH básico (por ejemplo, Biochem. J. 1979179, 191-197; Biochemistry 1976, 15, 2863-8; Chem Commun. 2011; 47: 5452-5454). La apertura de anillo hidrolítica del grupo succinimida producirá una unión tioéter estable que no estará sujeto a la reacción inversa con la maleimida mencionada anteriormente (Bioconjugate Chem. 2011, 22, 1946-1953; Nat. Biotechnol. 2012, 30, 184-189; Nat. Biotechnol. 2014, 32, 1059). Determinados compuestos de la invención, ejemplificados por los compuestos 47, 14, 77, 80, 79 y 78, tienen una hidrólisis del anillo de succinimida mayor cuando se conjugan con residuos de Cys en los anticuerpos y producen así conjugados de anticuerpo y fármaco más estables.
Tabla 14
Retención de carga de ADC seleccionados después de 3 semanas en ratones que no han recibido tratamiento d rmin r IP-M


Ejemplo 108: Estabilidad in vitro de los ADC.
Los efectos de la hidrólisis del anillo de succinimida en la estabilidad de los ADC preparados con los péptidos citotóxicos de la invención se estudiaron adicionalmente in vitro. Los cambios en las masas resultantes de la desconjugación de cargas y la hidrólisis del anillo de succinimida de las cargas de maleimida conjugadas a los anticuerpos se monitorizaron mediante LC-MS. Se ha publicado que la hidrólisis del anillo de succinimida se ve estimulada por determinadas condiciones tales como pH elevado, temperatura elevada o concentraciones de sales elevadas (J. Am. Chem. Soc. 1955, 77, 3922; Biochemistry, 1976, 15, 2 8 3 6 ; Biochem. J. 1979, 179, 191-197; J Pharm Sci. 1984, 73:1767-1771, Bioorg. Med. Chem. Lett. 17 (2007) 6286-6289). Para investigar la estabilidad in vitro de los ADC en función del pH, se incubaron anti-Her2-LC-S159C-10 y anti-Her2-LC-S159C-47 a 37 °C en tampones que oscilaban de pH 7,0 a pH 9,0 y los ADC se analizaron en varios puntos temporales mediante MS para determinar el grado de desconjugación de cargas y de hidrólisis de succinimida. Las poblaciones relativas de los ADC desconjugados, ADC con carga unida con anillo de succinimida hidrolizado y ADC con carga unida con anillo de succinimida intacto se calcularon a partir de las intensidades de MS relativas de las especies de ADC correspondientes. Tal como se muestra en la Figura 5A, el aumento del pH del tampón de incubación aumenta la desconjugación de anti-Her2-LC-S159C-10. En tampón a pH 7,0, el anti-Her2-LC-S159C-10 era estable con aproximadamente un 10% de pérdida de carga después de 10 horas. Sin embargo, la incubación en tampón a pH 8,0 y pH 9,0 durante 10 horas aumentó el grado de desconjugación de carga hasta aproximadamente un 30% y un 60%, respectivamente (Figura 5A). Por el contrario, la incubación de anti-Her2-LC-S159C-47 en tampones con pH 7,0 a pH 9,0 a 37 °C durante 25 horas no dio como resultado más de un 15% de desconjugación (Figura 5C).
En paralelo, el grado de hidrólisis del anillo de succinimida de los dos ADC también se determinó en los distintos puntos temporales (Figura 5B y Figura 5D). Para ambos ADC, el aumento de pH del tampón de incubación estimuló la hidrólisis del anillo de succinimida. Para anti-Her2-LC-S159C-47, la hidrólisis del anillo de succinimida tuvo lugar de forma significativamente más rápida que para anti-Her2-LC-S159C-10. La incubación de anti-Her2-LC-S159C-10 en tampón a pH 7,5 durante 25 horas dio lugar a la desconjugación de un 30% de la carga y se hidrolizó un 20% de la carga que aún estaba unida al anticuerpo (Figura 5A y Figura 5B). En las mismas condiciones de incubación, el anti-Her2-LC-S159C-47 perdió solo aproximadamente un 10% de la carga, mientras que se hidrolizó aproximadamente un 90% de la carga unida (Figura 5C y Figura 5D). Tal como se muestra en la Figura 5E, y F, se analizaron la conjugación de cargas in vitro y la hidrólisis del anillo de succinimida para los ADC anti-Her2-LC-S159C-10, anti-Her2-LC-S159C-77, anti-Her2-LC-S159C-80, anti-Her2-LC-S159C-79, anti-Her2-LC-S159C-78 y anti-Her2-LC-S159C-14 incubados en tampones a pH 8,5 durante 24 horas a 37 °C. Si bien la desconjugación de las cargas no fue detectable para los ADC de anti-Her2-LC-S159C-77, anti-Her2-LC-S159C-80, anti-Her2-LC-S159C-79, anti-Her2-LC-S159C-78 y anti-Her2-LC-S159C-14 (Figura 5E), se observó un grado elevado de hidrólisis del anillo de succinimida para todos los ADC (Figura 5F). Por tanto, determinados compuestos de la invención, ejemplificados por los compuestos 47, 14, 77, 80, 79 y 78, presentan una estabilidad de los ADC mejorada debido a una menor susceptibilidad a la desconjugación a través de la reacción de maleimida inversa y una estabilización adicional mediante la hidrólisis del anillo de succinimida.
Ejemplo 109: Estudios de eficacia in vivo con los ADC de anticuerpo 20507.
Los modelos de xenoinjerto in vivo estimulan la actividad biológica observada en seres humanos y están constituidos por líneas celulares tumorales o tumores primarios humanos bien caracterizados con relevancia para el injerto en ratones atímicos inmunodeficientes. Los estudios sobre el tratamiento de ratones con xenoinjertos tumorales con reactivos anticancerosos han proporcionado una información valiosa sobre la eficacia in vivo de los reactivos evaluados (Sausville y Burger, (2006) Cancer Res. 66:3351-3354). Como las células H526 expresan el antígeno del anticuerpo 20507 en su superficie y son destruidas selectivamente por los ADC de anticuerpo 20507 (Figura 2, Tabla 12), se utilizó la línea celular para generar un modelo de xenoinjerto con el fin de evaluar la actividad in vivo de los ADC de anticuerpo 20507. Todos los estudios animales se llevaron a cabo de conformidad con la Guía para el cuidado y uso de animales de laboratorio (publicación de NIH; National Academy Press, 8.a edición, 2001). Se implantaron células H526 en ratones nu/nu por vía subcutánea (Morton y Houghton, Nat Protoc. 2007;2:247-250). Después de que el tamaño del tumor alcanzase ~200 mm3, se administraron tres ADC de anticuerpo 20507 en el ratón mediante inyección i. v. en una única dosis con una concentración de 3 mg/kg o 10 mg/kg. El crecimiento tumoral se midió periódicamente después de la inyección de ADC. Cada grupo de tratamiento incluía 7 ratones. Un ejemplo de un estudio de eficacia in vivo de este tipo se muestra en la Figura 6A. El tratamiento de los ratones con una concentración de 3 mg/kg de ADC de anticuerpo 20507-HC-E152C-10 provocó una inhibición del crecimiento tumoral, mientras que el tratamiento con una concentración de 10 mg/kg de ADC de anticuerpo 20507-HC-E152C-10 dio lugar a regresión tumoral (Figura 6A). La eficacia del ADC de anticuerpo 20507-HC-E152C-10 es equivalente a la de un ADC de control positivo, anticuerpo 20507-HC-E152C-MMAF conjugado con el compuesto de referencia MC-MMAF. No se observó pérdida de peso asociada con el tratamiento de ADC, lo que sugería una toxicidad sistémica baja. Los resultados confirman que con un tratamiento de una única dosis con una concentración de 10 mg/kg, el ADC de anticuerpo 20507-HC-E152C-10 provocó de forma eficaz una regresión de tumores de H526 sin pérdida de peso significativa. En un segundo estudio, se administraron los ADC de anticuerpo 20507-HC-E152C-10 y anticuerpo 20507-HC-ins388-A1 -20 en ratones que portaban tumores H526 con dosis de 3 mg/kg y 10 mg/kg (Figura 6B). Ambos ADC presentaban una actividad de inhibición tumoral con una concentración de 3 mg/kg y actividad de regresión tumoral con una concentración de 10 mg/kg similares. El ADC de anticuerpo 20507-HC-E152C-10 fue ligeramente más eficaz que el ADC de 20507-HC-ins388-A1 -20.
En otro ejemplo, los inventores comparan la eficacia in vivo de dos ADC de anticuerpo 20507, anticuerpo 20507-HC-E152C-S375C-10 y anticuerpo 20507-10, en el modelo de xenoinjerto H526 (Figura 6C). Los dos ADC se prepararon con la misma carga, conjugando el compuesto 10 con diferentes sitios de Cys utilizando dos métodos diferentes. Se preparó el anticuerpo 20507-HC-E152C-S375C-10 con un anticuerpo con mutación de Cys, tal como se describe en el Ejemplo 101 con el compuesto 10 conjugado a residuos de Cys introducidos mediante modificación, HC-E152C y HC-S375C. El anticuerpo 20507-10 se preparó utilizando el método de reducción parcial del anticuerpo 20507 de origen natural tal como se ha descrito en el Ejemplo 102 con el compuesto 10 conjugado a residuos de Cys nativos. El anticuerpo 20507-10 tiene una DAR ligeramente superior (DAR 4,6) que el anticuerpo 20507-HC-E152C-S375C-10 (DAR 3,9) (Tabla 6). Los estudios farmacocinéticos mostraron que los dos ADC retenían la misma carga en un grado muy diferente durante las tres semanas de cirulación en el ratón (Figura 4D, Figura 4E, Tabla 14): Anticuerpo 20507-HC-E152C-S375C-10 que muestra una retención de carga mucho mejor (56%) que el anticuerpo 20507-10 (20%).
En el modelo de xenoinjerto H526, la misma dosis de anticuerpo 20507-HC-E152C-S375C-10 es más eficaz para inhibir tumores que el anticuerpo 20507-10 (Figura 6C). El anti-Her2-HC-E152C-S375C-10, cuyo antígeno no se expresa en las células H526, no mostró ninguna actividad de inhibición tumoral.
En un tercer estudio, se inyectaron i.v. los ADC de anticuerpo 20507-HC-E152C-10 y anticuerpo 20507-HC-ins388-A1-49-22 en ratones que portaban tumores H526 con una única dosis de 7 mg/kg (Figura 6D). Aunque el ADC de anticuerpo 20507-HC-ins388-A1-49-22 presentaba inhibición tumoral, el ADC de anticuerpo 20507-HC-E152C-10 presentaba regresión tumoral al mismo nivel de dosis, indicativo de una mayor eficacia de este último ADC. No se observó actividad de inhibición tumoral significativa para el ADC de control de isotipo, anti-Her2-HC-ins388-A1-49-22, lo que sugería una destrucción tumoral con especificidad por un antígeno por parte de los ADC de anticuerpo 20507. Juntos, los tres estudios confirman que los péptidos citotóxicos de la invención unidos por una serie de métodos a un anticuerpo pueden provocar la regresión de un cáncer que expresa antígenos en un modelo de xenoinjerto de ratón.
Ejemplo 110: Producción de Sfp 4’-fosfopanteteinil-transferasa (PPTasa)
[0405] La Sfp PPTasa de Bacillus subtilis se clonó en el vector de expresión pET22b utilizando el método PIPE (remítase a Klock et al., Proteins 71:982-994 (2008)). Para permitir la escisión de la etiqueta His6 C terminal, se insertó un sitio de reconocimiento de proteasas de TEV (virus del grabado del tabaco) en dirección 3’ respecto a la secuencia codificadora de Sfp. Todos los cebadores utilizados para la clonación y la secuencia proteica de Sfp se enumeran en la Tabla 15.
Tabla 15.
Cebadores utilizados para la clonación y secuencia proteica de Sfp

Se llevaron a cabo la expresión y purificación de proteínas de acuerdo con Yin et al., (remítase a Nat. Protoc. 1:280-285 (2006)) con algunas modificaciones. En resumen, se inoculó 1 L de medio TB a partir de cultivos saturados durante una noche de células Escherichia coli BL21 (DE3) que albergan el plásmido de expresión pET22b/sfp. El cultivo se agitó a 250 rpm a 37 °C y se indujo mediante la adición de IPTG 1 mM después de alcanzar una densidad óptica (600 nm) de 0,7. La temperatura se redujo hasta 30 °C y el cultivo se agitó a 250 rpm durante aproximadamente 16 horas antes de recolectar las células bacterianas por centrifugación (20 min a 3400 rpm). Antes de su uso, los pélets de células se almacenaron a -20 °C. Para iniciar la purificación de proteínas, los pélets congelados se descongelaron durante aproximadamente 15 minutos en hielo y se resuspendieron en un tampón que contenía Tris/Hcl 20 mM (pH 8), NaCl 0,5 M, imidazol 5 mM y 2 U/mL de ADNasa I (3 mL de tampón por g de peso húmedo de células). Se indujo la lisis celular por sonicación durante un tiempo total de 2 min utilizando pulsos de sonicación de 1 s con lapsos de 1 s intermitentes. Con el fin de eliminar restos celulares insolubles, el lisado resultante se centrifugó a 40000 x g durante 25 min a 4 °C. La enzima Sfp etiquetada con His6 se capturó entonces añadiendo 4 mL de suspensión densa de agarosa Ni-NTA al 50% (Qiagen) al lisado limpiado. Después de agitar durante 1 hora a 4 °C, la mezcla de resina-lisado se vertió en una columna desechable. La resina sedimentada se lavó con 50 volúmenes de columna de Tris/HCl 50 mM (pH 8), NaCl 300 mM e imidazol 20 mM. La elución se llevó a cabo con 6 volúmenes de columna de Tris/HCl 50 mM (pH 8), NaCl 300 mM e imidazol 250 mM. La enzima Sfp se intercambió en tampón de escisión de TEV que contenía Tris/HCl 50 mM (pH 8) y NaCl 50 mM. La eliminación de la etiqueta His6 se llevó a cabo mediante digestión con proteasa de TEV al 7% (p/p) a 23 °C durante 1 hora y después a 4 °C durante aproximadamente 16 horas. La enzima Sfp digerida con TEV se volvió a cargar entonces en columnas de Ni-NTA nuevas preequilibradas con PBS. La enzima escindida se recogió a partir de la fracción que atravesaba la columna y a partir de un paso de lavado que conllevaba 5 volúmenes de columna de Tris/HCl 50 mM (pH 8), NaCl 300 mM e imidazol 20 mM. A continuación, la enzima Sfp purificada se dializó frente a Tris/HCl 10 mM (pH 7,4), EDTA 1 mM y glicerol al 10% utilizando cintas de diálisis Slide-A-Lyzer (Pierce) con un límite de 3,5 kDa. Después de pasar el producto dializado a través de un filtro de 0,22 pm, el rendimiento de la enzima Sfp se cuantificó mediante espectroscopía ultravioleta a 280 nm (ND-1000 UV-Vis Spectrophotometer, NanoDrop Technologies, Wilmington, DE) utilizando un coeficiente de extinción molar de 28620 M-1cm-1. Se obtuvieron 19 mg de enzima Sfp escindida con TEV por cultivo de un litro.
Después, se concentró la Sfp PPTasa hasta 151 pM utilizando unidades de filtración centrífuga Amicon Ultra (Millipore) con un límite de 10 kDa. Se evaluó la pureza de la enzima mediante SDS-PAGE, y la eliminación de la etiqueta His6 se verificó mediante LC-MS. De acuerdo con la cromatografía de exclusión de tamaños analítica, la Sfp PPTasa escindida con TEV es monomérica en un 90%. Se tomaron alícuotas de la enzima concentrada, se ultracongelaron en nitrógeno líquido y se almacenaron a -80 °C.
Claims (11)
1. Un compuesto o estereoisómero de este que tiene la estructura de Fórmula (I)

R2 es metilo, etilo, isopropilo o sec-butilo
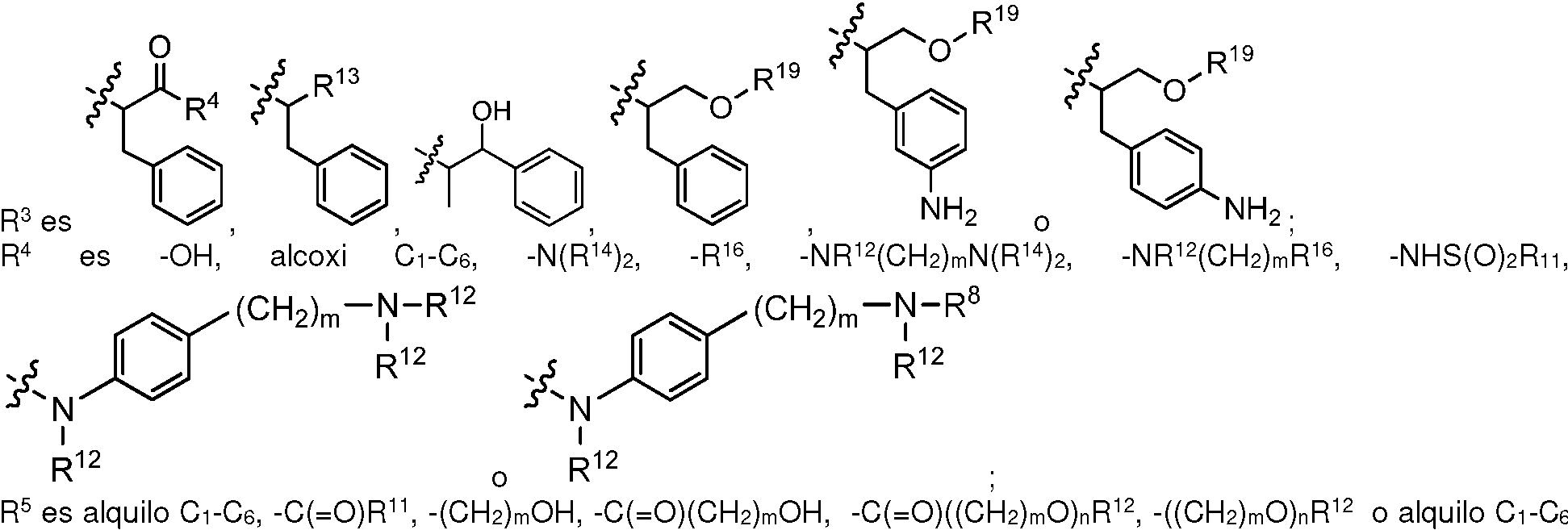
que se sustituye opcionalmente con -CN, -C(=0)NH2 o de 1 a 5 hidroxilos;
R8 es H;
cada R11 se selecciona independientemente entre alquilo C1-C6 y alquilo C1-C6 que se sustituye opcionalmente con de 1 a 5 hidroxilos;

cada R14 se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 es un heterocicloalquilo de 4-8 miembros sin sustituir conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
R19 es H o alquilo C1-C6 ;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10, y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 y 18, o una sal farmacéuticamente aceptable de este.
2. El compuesto de la reivindicación 1, donde el compuesto es un compuesto que tiene la estructura de Fórmula (Ia):

Fórmula (la)
3. El compuesto de la reivindicación 1 seleccionado entre,

4. Un inmunoconjugado de Fórmula (II):

Fórmula (II)
donde:
Ab representa un resto de unión a antígeno;
L se selecciona entre -L1L2L3L4L5L6-, -L6L5L4L3L2L1-, -L1L2L3L4L5-, -L5L4L3L2L1-,-L1L2L3L4-, -L4L3L2L1-,-L1L2L3-, -L3L2L1-,-L1L2-, -L2L1- y -L1,
donde
L1 se selecciona entre -(CH2)m-, -C(=O)(CH2)m-, -C(=O)X1X2C(=O)(CH2)m-, -C(=O)X1X2C(=O)(CH2)mNR12C(=O)(CH2)m-, -C(=O)X1X2C(=O)(CH2)mX3(CH2)m-, -C(=O)X1X2C(=O)((CH2)mO)n(CH2)m-, -C(=O)X1X2C(=O)((CH2)mO)n(CH2)mNR12C(=O)(CH2)m-, -C(=O)X1X2C(=O)((CH2)mO)n(CH2)mNR12C(=O)(CH2)mX3(CH2)m-, -C(=O)X1X2C(=O)((CH2)mO)n(CH2)mX3(CH2)m-, -C(=O)X1X2C(=O)(CH2)mNR12C(=O)((CH2)mO)n(CH2)m-, -C(=O)X1X2C(=O)(CH2)mNR12C(=O)((CH2)mO)n(CH2)mX3(CH2)m-, -C(=O)X1X2(CH2)mX3(CH2)m-, -C(=O)X1X2((CH2)mO)n(CH2)m-, -C(=O)X1X2((CH2)mO)n(CH2)mNR12C(=O)(CH2)m-, -C(=O)X1X2((CH2)mO)n(CH2)mN R12C(=O)(CH2)mX3(CH2)m-, -C(=O)X1X2((CH2)mO)n(CH2)mX3(CH2)m-, -C(=O)X1X2(CH2)mNR12((CH2)mO)n(CH2)m-, -C(=O)X1X2C(=O)(CH2)mNR12((CH2)mO)n(CH2)mX3(CH2)m-, -(CH2)mNR12C(=O)(CH2)m-, -C(=O)((CH2)mO)n(CH2)m-, -(CH2)mS(=O)2((CH2)mO)n(CH2)m-, -C(=O)(CH2)mNR12(CH2)m-, -C(=O)NR12(CH2)m-, -C(=O)NR12(CH2)mX3(CH2)m-, -C(=O)NH(CH2)mNR12C(=O)X1X2C(=O)(CH2)m-, -C(=O)X1C(=O)NR12(CH2)mNR12C(=O)(CH2)m-, -C(=O)X1C(=O)NR12(CH2)mX3(CH2)m-, 


cada R25 se selecciona independientemente entre H y alquilo C1-4 ;
Raa es una cadena lateral de un aminoácido seleccionado entre glicina, alanina, triptófano, tirosina, fenilalanina, leuceina, isoleucina, valina, asparagina, ácido glutámico, glutamina, ácido aspático, histidina, arginina, lisina, cisteína, metionina, serina, treonina, fenilglicina y f-butilglicina;
R32 se selecciona independientemente entre H, alquilo C1-4, fenilo, pirimidina y piridina;

R33 se selecciona independientemente entre

R34 se selecciona independientemente entre H, alquilo C1-4, haloalquilo C1-6;

es un número entero de 1 a 16;

R2 es metilo, etilo, isopropilo o sec-butilo

R6 *11 es alquilo C1-C6 o alquilo C1-C6 que está sustituido opcionalmente con de 1 a 5 hidroxilos;
cada R12 se selecciona independientemente a partir de H y alquilo C1-C6 ;
s O II i -P -O H R13 es tetrazolilo, imidazolilo sustituido con fenilo, oxadiazolilo sustituido con fenilo, pirazolilo, pirimidinilo, 5 H

cada R14 *se selecciona independientemente a partir de H y alquilo C1-C6 ;
R16 * es un heterocicloalquilo de 4-8 miembros conectado en N que contiene 1-2 heteroátomos seleccionados independientemente entre N y O;
R19 es H o alquilo C1-C6 ;
cada m se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9 y 10,
y
cada n se selecciona independientemente entre 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17 y 18.
5. El inmunoconjugado de la reivindicación 4, donde el inmunoconjugado de Fórmula (II) es un inmunoconjugado de Fórmula IIb :

6. El inmunoconjugado de cualquiera de las reivindicaciones 4 o 5, donde:
L1 es -(CH2)mNHC(=O)(CH2)mX3(CH2)m*-, -(CH2)mC(=O)*-, -(CH2)m-, -(CH2)mC(=O)X2X1C(=O)*-, -(CH2)mX2X1C(=O)*--(CH2)m(O(CH2)m)nC(=O)*-, -(CH2)m(O(CH2)m)nC(=O)X2X1C(=O)*-, -(CH2)m(O(CH2)m)nX2X1C(=O)*-, (CH2)m(O(CH2)m)nS(=O)2(CH2)m*-, -(CH2)mNH(CH2)mC(=O)*-, -((CH2)mO)n(CH2)m*-, -(CH2)mX3(CH2)mC(=O)*-, (CH2)mX3(CH2)m-, -(CH2)mX3(CH2)mC(=O)X2X1C(=O)*-, -(CH2)mX3(CH2)mX2X1C(=O)*-, (CH2)mX3(CH2)m(O(CH2)m)nX2X1C(=O)*-, -(CH2)mNHC(=O)*-, -(CH2)mX3(CH2)m(O(CH2)m)nC(=O)*-, (CH2)mX3((CH2)mO)n(CH2)m*-, -(CH2)mC(=O)NH(CH2)m*-, -(CH2)mX3(CH2)mC(=O)NH(CH2)m*-, (CH2)mC(=O)NH(CH2)mNHC(=O)*-, -(CH2)mX3(CH2)mNHC(=O)*-, -(CH2)mC(=O)NH(CH2)mC(=O)*-, (CH2)mX3(CH2)mC(=O)NH(CH2)mNHC(=O)*-, -(CH2)mX3(CH2)mC(=O)NH(CH2)mC(=O)*-, (CH2)mC(=O)NH(CH2)m(O(CH2)m)nC(=O)*-, -(CH2)mX3(CH2)mC(=O)NH(CH2)m(O(CH2)m)nC(=O)*-, (CH2)mC(=O)NH(CH2)mNHC(=O)(CH2)m*-, -(CH2)mC(=O)NH(CH2)mC(=O)NH(CH2)m*-, (CH2)mX3(CH2)mC(=O)NH(CH2)m*-, -(CH2)mS(=O)2*-, -(CH2)mX3(CH2)mS(=O)2*-, - (CH2)mOC(=O)NH(CH2)mC(=O)*-, y (CH2)mOC(=O)NH(CH2)mS(=O)2*-, y donde el * indica el punto de unión a R101;

y 1-3,1-4, 1-5 y 1_6 son un enlace.
8. Una composición farmacéutica que comprende un inmunoconjugado de cualquiera de las reivindicaciones 4 a 7, y uno o más portadores farmacéuticamente aceptables.
9. Una combinación que comprende una cantidad terapéuticamente eficaz de un inmunoconjugado de cualquiera de las reivindicaciones 4 a 7, y uno o más coagentes terapéuticamente activos.
10. Un inmunoconjugado de cualquiera de las reivindicaciones 4 a 7 para su uso como un medicamento.
11. Un inmunoconjugado de cualquiera de las reivindicaciones 4 a 7 para su uso en el tratamiento del cáncer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361917293P | 2013-12-17 | 2013-12-17 | |
| PCT/US2014/070800 WO2015095301A2 (en) | 2013-12-17 | 2014-12-17 | Cytotoxic peptides and conjugates thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2758506T3 true ES2758506T3 (es) | 2020-05-05 |
Family
ID=52463118
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14835606T Active ES2758506T3 (es) | 2013-12-17 | 2014-12-17 | Péptidos citotóxicos y conjugados de estos |
Country Status (16)
| Country | Link |
|---|---|
| US (3) | US9988420B2 (es) |
| EP (2) | EP3753578A1 (es) |
| JP (2) | JP6636925B2 (es) |
| KR (1) | KR20160098257A (es) |
| CN (1) | CN105899526A (es) |
| AU (1) | AU2014364812B2 (es) |
| BR (1) | BR112016012538A2 (es) |
| CA (1) | CA2930614A1 (es) |
| EA (1) | EA201691251A1 (es) |
| ES (1) | ES2758506T3 (es) |
| IL (1) | IL245558B (es) |
| MX (1) | MX2016007865A (es) |
| PL (1) | PL3082877T3 (es) |
| PT (1) | PT3082877T (es) |
| SG (1) | SG11201604877UA (es) |
| WO (1) | WO2015095301A2 (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150115000A (ko) * | 2013-02-08 | 2015-10-13 | 아이알엠 엘엘씨 | 면역접합체의 제조를 위한 항체의 변형에 사용되는 특정 부위 |
| CN105358174B (zh) | 2013-03-15 | 2019-03-15 | 酵活有限公司 | 具细胞毒性和抗有丝分裂的化合物以及其使用方法 |
| US10675355B2 (en) | 2013-12-27 | 2020-06-09 | Var2 Pharmaceuticals Aps | VAR2CSA-drug conjugates |
| AU2014373574B2 (en) | 2013-12-27 | 2020-07-16 | Zymeworks Bc Inc. | Sulfonamide-containing linkage systems for drug conjugates |
| EP3129407A2 (en) * | 2014-03-12 | 2017-02-15 | Novartis Ag | Specific sites for modifying antibodies to make immunoconjugates |
| CN106573956A (zh) * | 2014-06-13 | 2017-04-19 | 诺华股份有限公司 | 澳瑞他汀衍生物及其缀合物 |
| KR102494557B1 (ko) | 2014-09-17 | 2023-02-02 | 자임워크스 비씨 인코포레이티드 | 세포독성 및 항유사분열성 화합물, 그리고 이를 이용하는 방법 |
| US11566082B2 (en) | 2014-11-17 | 2023-01-31 | Cytiva Bioprocess R&D Ab | Mutated immunoglobulin-binding polypeptides |
| US10654887B2 (en) | 2016-05-11 | 2020-05-19 | Ge Healthcare Bio-Process R&D Ab | Separation matrix |
| US10513537B2 (en) | 2016-05-11 | 2019-12-24 | Ge Healthcare Bioprocess R&D Ab | Separation matrix |
| US10889615B2 (en) | 2016-05-11 | 2021-01-12 | Cytiva Bioprocess R&D Ab | Mutated immunoglobulin-binding polypeptides |
| WO2017194592A1 (en) | 2016-05-11 | 2017-11-16 | Ge Healthcare Bioprocess R&D Ab | Method of storing a separation matrix |
| US10730908B2 (en) | 2016-05-11 | 2020-08-04 | Ge Healthcare Bioprocess R&D Ab | Separation method |
| JP6987424B2 (ja) | 2016-05-11 | 2022-01-05 | サイティバ・バイオプロセス・アールアンドディ・アクチボラグ | 分離マトリックスを洗浄および/または消毒する方法 |
| US10703774B2 (en) | 2016-09-30 | 2020-07-07 | Ge Healthcare Bioprocess R&D Ab | Separation method |
| US10517958B2 (en) | 2016-10-04 | 2019-12-31 | Zymeworks Inc. | Compositions and methods for the treatment of platinum-drug resistant cancer |
| JOP20190187A1 (ar) | 2017-02-03 | 2019-08-01 | Novartis Ag | مترافقات عقار جسم مضاد لـ ccr7 |
| UY38265A (es) * | 2018-06-20 | 2020-01-31 | Novartis Ag | Conjugados anticuerpo droga para ablación de células madre hematopoyéticas |
| EP3883914A4 (en) * | 2018-11-23 | 2022-04-06 | Wuxi Biologics Ireland Limited | ORTHOPHTHALALDEHYDE-CONTAINING LINKERS AND METHODS FOR PRODUCTION OF ANTIBODY-DRUG CONJUGATE |
| US20230181756A1 (en) | 2020-04-30 | 2023-06-15 | Novartis Ag | Ccr7 antibody drug conjugates for treating cancer |
| EP4279502A1 (en) * | 2021-01-18 | 2023-11-22 | Ajinomoto Co., Inc. | Compound or salt thereof, and antibody obtained therefrom |
| WO2023061405A1 (zh) * | 2021-10-12 | 2023-04-20 | 成都科岭源医药技术有限公司 | 一种高稳定性的靶向接头-药物偶联物 |
| WO2024023735A1 (en) * | 2022-07-27 | 2024-02-01 | Mediboston Limited | Auristatin derivatives and conjugates thereof |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| DE69230824T2 (de) | 1991-08-09 | 2000-07-27 | Teikoku Hormone Mfg Co Ltd | Neue tetrapeptidderivate |
| ATE282630T1 (de) | 1993-10-01 | 2004-12-15 | Teikoku Hormone Mfg Co Ltd | Dolastatin-derivate |
| US5595756A (en) * | 1993-12-22 | 1997-01-21 | Inex Pharmaceuticals Corporation | Liposomal compositions for enhanced retention of bioactive agents |
| US5641870A (en) | 1995-04-20 | 1997-06-24 | Genentech, Inc. | Low pH hydrophobic interaction chromatography for antibody purification |
| ATE196913T1 (de) | 1995-04-21 | 2000-10-15 | Teikoku Hormone Mfg Co Ltd | Neuartige peptidderivate |
| AU729035B2 (en) | 1997-06-12 | 2001-01-25 | Novartis Ag | Artificial antibody polypeptides |
| US6153655A (en) | 1998-04-17 | 2000-11-28 | Enzon, Inc. | Terminally-branched polymeric linkers and polymeric conjugates containing the same |
| US7803915B2 (en) | 2001-06-20 | 2010-09-28 | Genentech, Inc. | Antibody compositions for the diagnosis and treatment of tumor |
| US20090068178A1 (en) | 2002-05-08 | 2009-03-12 | Genentech, Inc. | Compositions and Methods for the Treatment of Tumor of Hematopoietic Origin |
| JP2007524596A (ja) | 2003-02-28 | 2007-08-30 | トランスフォーム・ファーマシューティカルズ・インコーポレイテッド | 共結晶医薬組成物 |
| SG195524A1 (en) | 2003-11-06 | 2013-12-30 | Seattle Genetics Inc | Monomethylvaline compounds capable of conjugation to ligands |
| JP2008519863A (ja) | 2004-11-12 | 2008-06-12 | シアトル ジェネティクス インコーポレイティッド | N末端にアミノ安息香酸単位を有するオーリスタチン |
| JP2008521828A (ja) | 2004-11-29 | 2008-06-26 | シアトル ジェネティックス, インコーポレイテッド | 操作された抗体およびイムノコンジュゲート |
| WO2007002222A2 (en) | 2005-06-20 | 2007-01-04 | Psma Development Company, Llc | Psma antibody-drug conjugates |
| US7750116B1 (en) | 2006-02-18 | 2010-07-06 | Seattle Genetics, Inc. | Antibody drug conjugate metabolites |
| WO2007103288A2 (en) | 2006-03-02 | 2007-09-13 | Seattle Genetics, Inc. | Engineered antibody drug conjugates |
| AR059900A1 (es) | 2006-03-17 | 2008-05-07 | Genentech Inc | Anticuerpos anti-tat226 e inmunoconjugados |
| PT2211904T (pt) | 2007-10-19 | 2016-11-02 | Seattle Genetics Inc | Agentes de ligação a cd19 e seus usos |
| NO2842575T3 (es) | 2008-03-18 | 2018-02-24 | ||
| JP5933562B2 (ja) | 2010-09-29 | 2016-06-15 | シアトル ジェネティックス, インコーポレイテッド | N−カルボキシアルキル−アウリスタチンおよびその使用 |
| CA2815363C (en) | 2010-10-22 | 2020-07-14 | Seattle Genetics, Inc. | Synergistic effects between auristatin-based antibody drug conjugates and inhibitors of the pi3k-akt mtor pathway |
| PL2678037T3 (pl) | 2011-02-25 | 2015-05-29 | Lonza Ag | Rozgałęziony łącznik do koniugatów białko-lek. |
| US9029406B2 (en) | 2011-03-16 | 2015-05-12 | Seattle Genetics, Inc | N-carboxyalkylauristatins and use thereof |
| CN103826661B (zh) | 2011-04-21 | 2019-03-05 | 西雅图基因公司 | 新的结合剂-药物缀合物(adc)及其用途 |
| KR102030856B1 (ko) | 2011-05-27 | 2019-10-10 | 암브룩스, 인코포레이티드 | 비-천연 아미노산 연결된 돌라스타틴 유도체를 함유하는 조성물, 이를 수반하는 방법, 및 용도 |
| SG11201401452PA (en) | 2011-11-17 | 2014-06-27 | Pfizer | Cytotoxic peptides and antibody drug conjugates thereof |
| EP2794653B1 (en) * | 2011-12-23 | 2019-03-13 | Pfizer Inc | Engineered antibody constant regions for site-specific conjugation and methods and uses therefor |
| EP2855520B1 (en) * | 2012-06-04 | 2018-09-26 | Novartis AG | Site-specific labeling methods and molecules produced thereby |
| KR20150115000A (ko) * | 2013-02-08 | 2015-10-13 | 아이알엠 엘엘씨 | 면역접합체의 제조를 위한 항체의 변형에 사용되는 특정 부위 |
-
2014
- 2014-12-17 EP EP19193628.5A patent/EP3753578A1/en not_active Withdrawn
- 2014-12-17 MX MX2016007865A patent/MX2016007865A/es active IP Right Grant
- 2014-12-17 KR KR1020167015935A patent/KR20160098257A/ko not_active Application Discontinuation
- 2014-12-17 SG SG11201604877UA patent/SG11201604877UA/en unknown
- 2014-12-17 PL PL14835606T patent/PL3082877T3/pl unknown
- 2014-12-17 EA EA201691251A patent/EA201691251A1/ru unknown
- 2014-12-17 US US15/104,727 patent/US9988420B2/en active Active
- 2014-12-17 CN CN201480068696.XA patent/CN105899526A/zh active Pending
- 2014-12-17 ES ES14835606T patent/ES2758506T3/es active Active
- 2014-12-17 PT PT148356066T patent/PT3082877T/pt unknown
- 2014-12-17 AU AU2014364812A patent/AU2014364812B2/en not_active Ceased
- 2014-12-17 EP EP14835606.6A patent/EP3082877B1/en not_active Not-in-force
- 2014-12-17 WO PCT/US2014/070800 patent/WO2015095301A2/en active Application Filing
- 2014-12-17 BR BR112016012538A patent/BR112016012538A2/pt not_active Application Discontinuation
- 2014-12-17 JP JP2016540522A patent/JP6636925B2/ja not_active Expired - Fee Related
- 2014-12-17 CA CA2930614A patent/CA2930614A1/en not_active Abandoned
-
2016
- 2016-05-09 IL IL245558A patent/IL245558B/en not_active IP Right Cessation
-
2018
- 2018-04-24 US US15/961,531 patent/US10392421B2/en active Active
-
2019
- 2019-06-12 US US16/439,204 patent/US10787480B2/en active Active
- 2019-09-27 JP JP2019176561A patent/JP2020023514A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP3082877B1 (en) | 2019-08-28 |
| BR112016012538A2 (pt) | 2017-09-26 |
| US20180244717A1 (en) | 2018-08-30 |
| PL3082877T3 (pl) | 2020-02-28 |
| JP2017503777A (ja) | 2017-02-02 |
| PT3082877T (pt) | 2019-12-03 |
| AU2014364812B2 (en) | 2017-05-25 |
| US20190382441A1 (en) | 2019-12-19 |
| CA2930614A1 (en) | 2015-06-25 |
| IL245558B (en) | 2020-01-30 |
| EP3082877A2 (en) | 2016-10-26 |
| EA201691251A1 (ru) | 2016-11-30 |
| MX2016007865A (es) | 2017-01-11 |
| SG11201604877UA (en) | 2016-07-28 |
| US9988420B2 (en) | 2018-06-05 |
| US10787480B2 (en) | 2020-09-29 |
| WO2015095301A3 (en) | 2015-09-03 |
| EP3753578A1 (en) | 2020-12-23 |
| KR20160098257A (ko) | 2016-08-18 |
| AU2014364812A1 (en) | 2016-07-07 |
| JP6636925B2 (ja) | 2020-01-29 |
| WO2015095301A2 (en) | 2015-06-25 |
| US10392421B2 (en) | 2019-08-27 |
| IL245558A0 (en) | 2016-06-30 |
| CN105899526A (zh) | 2016-08-24 |
| JP2020023514A (ja) | 2020-02-13 |
| US20160311853A1 (en) | 2016-10-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2758506T3 (es) | Péptidos citotóxicos y conjugados de estos | |
| EP3154997B9 (en) | Auristatin derivatives and conjugates thereof | |
| ES2941768T3 (es) | Compuestos de 2H-pirazolo[4,3-d]pirimidina como agonistas del receptor 7 tipo toll (TLR7) y métodos y usos para los mismos | |
| KR102215954B1 (ko) | 튜부리신 화합물, 그의 제조 및 사용 방법 | |
| ES2852052T3 (es) | Péptidos citotóxicos y conjugados de fármaco anticuerpo de los mismos | |
| KR101660146B1 (ko) | 에네디인 화합물, 그의 접합체, 및 그에 대한 용도 및 방법 | |
| KR20170102980A (ko) | 벤조디아제핀 이량체, 그의 접합체, 및 제조 및 사용 방법 | |
| JP2018528161A (ja) | Ksp阻害剤との部位特異的均一複合体 | |
| JP7023933B2 (ja) | セコ-シクロプロパピロロインドール化合物、その抗体-薬物コンジュゲート、ならびに製造および使用方法 | |
| TW202024043A (zh) | 妥布賴森(tubulysins)及其中間體之製備之替代方法 | |
| NZ625745B2 (en) | FGFR antibody drug conjugates (ADCs) and the use thereof |