ES2716748T3 - Procedimientos de preparación de compuestos, tales como 3-arilbutanales, útiles en la síntesis de medetomidina - Google Patents
Procedimientos de preparación de compuestos, tales como 3-arilbutanales, útiles en la síntesis de medetomidina Download PDFInfo
- Publication number
- ES2716748T3 ES2716748T3 ES16706396T ES16706396T ES2716748T3 ES 2716748 T3 ES2716748 T3 ES 2716748T3 ES 16706396 T ES16706396 T ES 16706396T ES 16706396 T ES16706396 T ES 16706396T ES 2716748 T3 ES2716748 T3 ES 2716748T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- reaction
- reacting
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C**(N=C)=CNC Chemical compound C**(N=C)=CNC 0.000 description 2
- ZNGWEEUXTBNKFR-UHFFFAOYSA-O C1COCC[NH2+]C1 Chemical compound C1COCC[NH2+]C1 ZNGWEEUXTBNKFR-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/24—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/02—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of compounds containing imino groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/455—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation with carboxylic acids or their derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/62—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by hydrogenation of carbon-to-carbon double or triple bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/63—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/78—Separation; Purification; Stabilisation; Use of additives
- C07C45/86—Use of additives, e.g. for stabilisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/20—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms
- C07C47/228—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing six-membered aromatic rings, e.g. phenylacetaldehyde
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/20—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms
- C07C47/228—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing six-membered aromatic rings, e.g. phenylacetaldehyde
- C07C47/232—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing six-membered aromatic rings, e.g. phenylacetaldehyde having unsaturation outside the aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
- C07D233/58—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/14—Esters of phosphoric acids containing P(=O)-halide groups
- C07F9/1403—Esters of phosphoric acids containing P(=O)-halide groups containing the structure Hal-P(=O)-O-unsaturated acyclic group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/20—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms
- C07C47/24—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing halogen
Description
DESCRIPCIÓN
Procedimientos de preparación de compuestos, tales como 3-arilbutanales, útiles en la síntesis de medetomidina Campo de la invención
La presente invención se refiere a nuevos procedimientos y a nuevos compuestos químicos usados en, y obtenidos a partir de, dichos procedimientos. En particular, se refiere a nuevos procedimientos de preparación de intermedios, tales como 3-arilbutanales, útiles en la síntesis de la medetomidina.
Antecedentes de la invención
Los compuestos que tienen un núcleo 3-arilbutanal son particularmente útiles como intermedios en la producción de medetomidina, que es un imidazol 4-sustituido que se emplea como el ingrediente activo en productos anti incrustantes como Selektope™ y, en la forma del enantiómero S (conocido como dexmedetomidina), como agente terapéutico.
Sin embargo, la síntesis del núcleo 3-arilbutanal requerido para la producción de medetomidina se ve obstaculizada por la necesidad de la incorporación de un grupo 2,3-dimetil fenilo estéricamente impedido.
La enumeración o la descripción de un documento aparentemente publicado anteriormente en esta memoria descriptiva no debería considerarse necesariamente como un reconocimiento de que el documento forma parte del estado de la técnica o del conocimiento general común.
Los procedimientos para la síntesis de medetomidina usando 3-arilbutanales (específicamente, 3-(2,3-dimetilfenil)butanal) como intermedios sintéticos se describen, por ejemplo, en el documento WO 2013/014428, que describe la síntesis de 3-arilbutanales mediante acoplamiento catalizado por paladio de un boronato de arilo correspondiente y un aldehído crotónico.
El documento WO 2012/120070 describe la síntesis de 3-arilbutenales mediante acoplamiento de Heck catalizado con paladio entre un haluro de arilo correspondiente y alcohol crotílico.
El documento WO 2012/069422 describe la síntesis de 3-arilbutenales mediante una reacción de un reactivo de aril-litio con 4,4-dimetoxi-2-butanona.
Las rutas anteriores a los 3-arilbutanales se han basado también en procedimientos que implican la reacción de derivados de estireno con monóxido de carbono e hidrógeno, en presencia de un catalizador de rodio (véase, por ejemplo, el documento US 4.113.781); y reacciones catalizadas por ácidos de Lewis de bencilacetales con éteres vinílicos (véase, por ejemplo, el documento WO 98/045237).
Dichos procedimientos tienen numerosas desventajas técnicas y económicas, particularmente cuando se utilizan en la fabricación industrial, tales como: la necesidad de condiciones de reacción extremas (tales como temperaturas muy bajas o muy altas y/o presión elevada); la generación de residuos tóxicos; la necesidad de una dilución excesiva de los reactivos; la necesidad de purificación mediante la eliminación de subproductos no deseados; y la necesidad de usar reactivos tóxicos y/o costosos.
Divulgación de la invención
Ahora, los presentes inventores han encontrado que los 3-arilbutanales pueden prepararse usando un procedimiento que comprende la formación altamente eficiente de un 3-aril butenal correspondiente y la reducción selectiva del mismo.
En un primer aspecto de la invención, se proporciona un procedimiento para la preparación de un compuesto de fórmula I

en el que dicho procedimiento comprende hacer reaccionar un compuesto de fórmula II

con uno o más reactivos de Vilsmeier adecuados.
La persona experta en la materia entenderá que todas las referencias en la presente memoria a aspectos particulares de la invención incluyen referencias a todas las realizaciones y combinaciones de una o más realizaciones de ese aspecto de la invención. De esta manera, todas las realizaciones de aspectos particulares de las invenciones pueden combinarse con una o más realizaciones distintas de ese aspecto de la invención para formar realizaciones adicionales sin apartarse de la enseñanza de la invención.
La persona experta en la materia entenderá que la expresión "reactivo de Vilsmeier" se refiere a un reactivo adecuado para realizar la reacción de Vilsmeier-Haack (conocida también como la reacción de Vilsmeier y formilación de Vilsmeier-Haack), cuya reacción y reactivos usados en la presente invención son bien conocidos en la técnica.
En particular, la persona experta en la materia entenderá que un reactivo de Vilsmeier adecuado se formará típicamente mediante una reacción entre una amida adecuada (típicamente una amida N,N-disustituida) y un cloruro de ácido adecuado.
La formación del reactivo de Vilsmeier (es decir, mediante reacción entre la amida y el cloruro de ácido) puede realizarse antes de realizar la reacción de formilación (por ejemplo, la formilación comprendida en el procedimiento del primer aspecto de la invención), en cuyo caso puede hacerse referencia al reactivo como un reactivo de Vilsmeier pre-formado, o puede realizarse in situ como parte de la reacción (por ejemplo, mediante mezclado de una amida adecuada y cloruro de ácido como parte del procedimiento del primer aspecto de la invención).
Las amidas adecuadas usadas en la formación de un reactivo de Vilsmeier incluyen típicamente N-metil formanilida, N,N-dimetil formamida (conocida también como dimetilformamida o DMF). Otras amidas adecuadas que se han usado en la formación de un reactivo de Vilsmeier incluyen N-metil formamida, N-formilpiperidina y N-formilindolina, formamida no sustituida, N,N-dimetil acetamida, N-metil acetamida, N,N-dimetil benzamida, pentametil acetamida y similares.
Las amidas particulares que pueden mencionarse incluyen dimetilformamida.
Los cloruros de ácido adecuados usados en la formación de un reactivo de Vilsmeier incluyen típicamente oxicloruro de fósforo (POCh) y cloruro de oxalilo ((COCl)2). Otros cloruros de ácido adecuados que se han usado en la formación de un reactivo de Vilsmeier incluyen COCl2, SOCl2, CIOCI, CHaCOCl, ArCOCI, ArSO2Cl, PCl5, Me2NSO2Cl y RO2CNHSO2Cl, en la que R representa un grupo alquilo o arilo opcionalmente sustituido adecuado, tal como alquilo C1-6 (por ejemplo, metilo o etilo) o fenilo), y Ar representa arilo, tal como fenilo.
Los cloruros de ácido particulares que pueden mencionarse incluyen cloruro de oxalilo y, más particularmente, oxicloruro de fósforo (POCh).
En una realización particular, el reactivo de Vilsmeier se forma (pre-formado o formado in situ) mediante reacción (es decir, combinación, bajo condiciones adecuadas) de dimetilformamida y oxicloruro de fósforo (POCh).
Tal como se usan en la presente memoria, las referencias a un reactivo o compuesto formado in situ se entenderá que hacen referencia a aquel reactivo o compuesto formado en el recipiente de reacción (es decir, el recipiente en el que se está llevando a cabo el procedimiento definido), en lugar de formado en un recipiente separado y añadido a continuación al recipiente de reacción (al que puede hacerse referencia como el compuesto o reactivo pre formado).
En una realización más particular, el reactivo de Vilsmeier comprende un compuesto de fórmula IIIa y/o un compuesto de fórmula

en las que:
X representa un contraión derivado de la reacción con un cloruro de ácido;
Y representa un sustituyente derivado de la reacción con un cloruro de ácido;
cada Ra y Rb representa independientemente H, alquilo o fenilo C1-3, o Ra y Rb junto con el nitrógeno al que están unidos forman un resto pirolidina, piperidina, morfolina o indolina.
A menos que se especifique lo contrario, las referencias a grupos alquilo en la presente memoria (tal como alquilo C1-6 y alquilo C1-3), puede referirse a grupos de cadena lineal o, cuando hay un número suficiente (es decir, un mínimo de tres) de átomos de carbono, pueden referirse a grupos de cadena ramificada. En particular, pueden referirse a grupos de cadena lineal (por ejemplo, grupos metilo y etilo).
A menos que se especifique lo contrario, las referencias a grupos cicloalquilo usados en la presente memoria pueden referirse a grupos que son completamente cíclicos o, cuando hay un número suficiente (es decir, un mínimo de cuatro) parcialmente cíclicos.
A menos que se especifique lo contrario, las referencias a grupos alcoxi en la presente memoria (por ejemplo, alcoxi C1-6) se entenderá que se refiere a un grupo alquilo similar pero que está unido en el punto de unión al resto de la molécula mediante un átomo de oxígeno (tal como un resto metoxi o etoxi).
La persona experta en la materia entenderá que la referencia a un contraión derivado de la reacción con un cloruro de ácido se referirá al contraión generado durante la formación del reactivo de Vilsmeier mediante la reacción de una amida adecuada (por ejemplo, DMF) con un cloruro de ácido (por ejemplo, POCh). Por ejemplo, cuando el cloruro de ácido adecuado usado en la formación del reactivo de Vilsmeier es POCl3, X representará -PO2Cl2. La persona experta en la materia entenderá que la referencia a un sustituyente derivado de la reacción con un cloruro de ácido se referirá al sustituyente generado durante la formación del reactivo de Vilsmeier mediante la reacción de una amida adecuada (por ejemplo, DMF) con un cloruro de ácido (por ejemplo, POCh). Por ejemplo, cuando el cloruro de ácido adecuado usado en la formación del reactivo de Vilsmeier es POCl3, Y representará POCI2.
Los compuestos particulares de fórmulas IIIa y IIIb que pueden mencionarse incluyen aquellos en los que al menos uno de entre Ra y Rb es distinto de H.
Los compuestos más particulares de fórmulas IIIa y IIIb que pueden mencionarse incluyen aquellos en los que cada Ra y Rb representa independientemente H, metilo o fenilo, o Ra y Rb junto con el nitrógeno al que están unidos forman un resto piperidina o indolina.
Los compuestos todavía más particulares de fórmulas IIIa y IIIb que pueden mencionarse incluyen aquellos en los que cada Ra y Rb representa independientemente H, metilo o fenilo, siempre que al menos uno de entre Ra y Rb sea distinto de H, o Ra y Rb junto con el nitrógeno al que están unidos formen un resto piperidina o indolina.
Los compuestos todavía más particulares de fórmulas IIIa y IIIb que pueden mencionarse incluyen aquellos en los que cada Ra y Rb representa independientemente metilo o fenilo, o Ra y Rb junto con el nitrógeno al que están unidos forman un resto de piperidina o indolina (por ejemplo, un resto de piperidina).
En una realización más particular, el reactivo de Vilsmeier comprende un compuesto de fórmula IIIc y/o un compuesto de fórmula IIId

La persona experta en la materia entenderá que el procedimiento del primer aspecto de la invención puede realizarse en un disolvente adecuado, tal como dimetilformamida (DMF). Cuando se usa DMF como disolvente, puede actuar también como reactivo (es decir, el componente amida) en la formación del reactivo de Vilsmeier requerido.
De esta manera, en una realización más particular, el reactivo de Vilsmeier se forma mediante reacción de dimetilformamida y POCl3, opcionalmente en el que la dimetilformamida actúa también como un disolvente. En dichas realizaciones, la persona experta en la materia entenderá que el reactivo de Vilsmeier puede formarse in situ mediante la adición del cloruro de ácido relevante (por ejemplo, POCh) a un recipiente de reacción que contiene un compuesto de fórmula II según se define en la presente memoria y DMF (que puede actuar también como un disolvente).
La persona experta en la materia entenderá que el procedimiento del primer aspecto de la invención es un procedimiento de múltiples etapas, que tiene una etapa de deshidratación y una etapa de formilación.
De esta manera, en una realización particular del primer aspecto de la invención, el procedimiento puede describirse como comprendiendo las etapas (concertadas) de:
(i) hacer reaccionar el compuesto de fórmula II para formar un compuesto de fórmula IV

y posteriormente
(ii) hacer reaccionar el compuesto de fórmula IV para formar el compuesto de fórmula I.
La persona experta en la materia entenderá además que estas etapas (es decir, las etapas (i) y (ii) indicadas anteriormente) se realizarán de manera concertada in situ, es decir, en un único recipiente de reacción (al que puede hacerse referencia como procedimiento de un solo recipiente), en el que el compuesto de fórmula IV un intermedio transitorio que no está aislado de la mezcla de reacción y no se observa como producto de reacción. En otras palabras, estas etapas pueden ocurrir en una sucesión inmediata.
De esta manera, en una realización más particular del primer aspecto de la invención, la reacción se realiza como un procedimiento de un único recipiente (es decir, en un único recipiente de reacción) y/o (por ejemplo, y) sin aislamiento de un compuesto de fórmula IV.
La persona experta en la materia entenderá que el compuesto de fórmula I puede obtenerse a partir de la mezcla de reacción y opcionalmente puede ser purificado usando técnicas bien conocidas en la técnica, tales como mediante desactivación de la reacción con una solución acuosa (tal como una solución alcalina, por ejemplo, NaOH 4 M), seguido de extracción a partir de la solución acuosa usando un disolvente orgánico adecuado (tal como un éter (por ejemplo, MTBE) o, más particularmente, tolueno).
De esta manera, el procedimiento del primer aspecto de la invención puede describirse como comprendiendo la etapa adicional de aislamiento y opcionalmente purificación del compuesto de fórmula I.
La persona experta en la materia será capaz de ajustar las condiciones de reacción (por ejemplo, la temperatura y/o la presión de la reacción) con el fin de obtener el producto deseado y/o maximizar el rendimiento del mismo. En realizaciones particulares del primer aspecto de la invención:
(I) la reacción (es decir, la reacción de deshidratación y formilación concertada, descrita también como las etapas (i) y (ii) anteriores) puede realizarse a aproximadamente presión atmosférica (es decir, aproximadamente 1 atm); y/o
(II) la formación del reactivo de Vilsmeier (por ejemplo, in situ) puede realizarse a temperatura reducida (es decir, a una temperatura inferior a aproximadamente 20°C), tal como a una temperatura de aproximadamente 0°C a aproximadamente 15°C; y/o
(III) después de la formación del reactivo de Vilsmeier, la reacción puede realizarse a una temperatura elevada (es decir, a una temperatura superior a aproximadamente 20°C), tal como a una temperatura de aproximadamente 70°C a aproximadamente 90°C, por ejemplo, a entre aproximadamente 75°C y aproximadamente 85°C.
Por ejemplo, en una realización particular, la formación del reactivo de Vilsmeier puede realizarse in situ (es decir, en presencia del compuesto de fórmula II y el precursor del reactivo Vilsmeier, tal como dimetilformamida) a temperatura reducida (es decir, a una temperatura inferior a aproximadamente 20°C), tal como a una temperatura de aproximadamente 0°C a aproximadamente 15°C.
En una realización alternativa, la formación del reactivo de Vilsmeier puede realizarse in situ (es decir, en presencia del compuesto de fórmula II y el precursor del reactivo de Vilsmeier, tal como dimetilformamida) a temperatura elevada (es decir, aumentada), por ejemplo, a una temperatura superior a aproximadamente 50°C, tal como a una temperatura de aproximadamente 65°C a aproximadamente 75°C.
Tal como se usa en la presente memoria, el término "aproximadamente" puede hacer referencia a un valor que es ± 10% del valor indicado, y puede eliminarse sin alterar la enseñanza general de la invención.
Tal como se describe en la presente memoria, los presentes inventores han encontrado también un procedimiento para la preparación de 3 aril butanales a partir de 3 aril butenales mediante reducción altamente selectiva.
De esta manera, en un segundo aspecto de la invención, se proporciona un procedimiento para la preparación de un compuesto de fórmula V

en el que el procedimiento comprende hacer reaccionar un compuesto de fórmula I tal como se define en el primer aspecto de la invención con un agente reductor adecuado.
En una realización particular del segundo aspecto de la invención, el procedimiento comprende las etapas de: (i) proporcionar un compuesto de fórmula I usando un procedimiento tal como se describe en el primer aspecto de la invención; y posteriormente
(ii) hacer reaccionar el compuesto de fórmula I con un agente reductor adecuado para formar el compuesto de fórmula V.
En una realización particular, el agente reductor adecuado es una fuente de hidrógeno (es decir, una fuente de átomos de hidrógeno) y la reacción se realiza en presencia de un catalizador adecuado.
En una realización particular, el agente reductor adecuado es una fuente de hidrógeno (es decir, una fuente de átomos de hidrógeno), y la reacción se realiza en presencia de un catalizador o más (por ejemplo, uno) adecuado y un compuesto o más (por ejemplo, uno) adecuado para limitar la actividad (es decir, envenenar) de dicho catalizador.
Las fuentes particulares de hidrógeno que pueden mencionarse incluyen compuestos orgánicos adecuados tal como conocen las personas expertas en la técnica (tales como cicloalquildienos estables (por ejemplo, ciclohexadieno), hidrazina, dihidronaftaleno, dihidroantraceno, isopropanol, ácido fórmico, metanol o ésteres de Hantzsch y similares) y gas hidrógeno (es decir, hidrógeno molecular, H2).
La persona experta en la materia entenderá que la expresión éster de Hantzsch, tal como se usa en la presente memoria, puede hacer referencia a un compuesto que, cuando se oxida, actúa como una fuente de hidrógeno. Por
ejemplo, la expresión puede hacer referencia a un compuesto que, cuando se oxida, actúa como una fuente de hidrógeno; por ejemplo, siguiendo el esquema de reacción mostrado a continuación

La persona experta entenderá que el término catalizador, tal como se usa en la presente memoria, se refiere a una sustancia que aumenta la velocidad de una reacción sin modificar el cambio de energía de Gibbs estándar global en la reacción.
Los catalizadores particulares que pueden mencionarse (por ejemplo, cuando la fuente de hidrógeno es gas hidrógeno) incluyen catalizadores metálicos, tales como paladio (por ejemplo, paladio (Pd) sobre carbono, por ejemplo 1% de Pd sobre carbono, 2% de Pd sobre carbono, 3% de Pd sobre carbono, 4% de Pd sobre carbono, 5% de Pd sobre carbono, 10% de Pd sobre carbono o 20% de Pd sobre carbono (en el que en cada caso el % es en peso, es decir, % en peso), por ejemplo, 3% de Pd sobre carbono), níquel de Raney, catalizador de Wilkinson, catalizador de Crabtree o catalizador de Lindlar. Dichos catalizadores pueden estar presentes en una mezcla de reacción homogénea o heterogénea. Los catalizadores más particulares que pueden mencionarse incluyen paladio sobre carbono (por ejemplo, 3% de Pd sobre carbono).
Los compuestos particulares adecuados para limitar la actividad (es decir, venenos) de dichos catalizadores (por ejemplo, paladio sobre carbón vegetal) que pueden mencionarse incluyen sales básicas (tales como las sales acetato y carbonato, por ejemplo, acetato de potasio y carbonato de potasio), aminas (tales como alquil/aril aminas trisustituidas, por ejemplo, trietanolamina, trietilamina y 4-dimetilaminopiridina (DMAP)), tioles (como los alquil tioles, por ejemplo, dodecil mercaptano) y tiourea.
Los compuestos más particulares adecuados para limitar la actividad (por ejemplo, venenos) de dichos catalizadores (por ejemplo, paladio sobre carbón vegetal) que pueden mencionarse incluyen 4-dimetilaminopiridina (DMAP).
Los compuestos todavía más particulares adecuados para limitar la actividad (es decir, venenos) de dichos catalizadores (por ejemplo, paladio sobre carbono, tal como el 3% de Pd sobre carbono) que pueden mencionarse incluyen una mezcla de DMAP y una amina trisubitituida (por ejemplo, una trialquil amina, tal como trimetilamina). En una realización particular del segundo aspecto de la invención, el procedimiento comprende hacer reaccionar un compuesto de fórmula I tal como se ha definido en el primer aspecto de la invención con una o más fuentes de hidrógeno, uno o más catalizadores adecuados y uno o más compuestos adecuados para limitar la actividad de dicho catalizador o dichos catalizadores.
En una realización más particular del segundo aspecto de la invención, el procedimiento comprende hacer reaccionar un compuesto de fórmula I según se define en el primer aspecto de la invención con hidrógeno molecular (es decir, gas hidrógeno), un catalizador de paladio (tal como paladio sobre carbón vegetal, por ejemplo, 3% en peso de paladio sobre carbón vegetal) y un compuesto o una mezcla de compuestos adecuados para limitar la actividad de dicho catalizador seleccionado de entre:
(i) DMAP; y
(ii) una mezcla de DMAP y una trialquil amina, tal como trietilamina (por ejemplo, una mezcla 1:4 de DMAP:trietilamina).
En una realización particular del segundo aspecto de la invención, el compuesto o la mezcla de compuestos adecuados para limitar la actividad del catalizador puede comprender DMAP, por ejemplo, DMAP en una cantidad que es del 2 a aproximadamente el 8% en moles, tal como aproximadamente del 4 a aproximadamente el 6% en moles (por ejemplo, aproximadamente el 5% en moles, tal como aproximadamente el 4,8% en moles) con relación al compuesto de fórmula I.
La cantidad y el tipo de catalizador y, si está presente, el compuesto usado para envenenar ese catalizador puede seleccionarse con el fin de controlar la selectividad de la reacción de reducción y el rendimiento del producto relevante. Por ejemplo:
- el catalizador (por ejemplo, 3% de Pd sobre carbono) puede ser usado en una cantidad que es menor del 1% en moles (es decir, % de la cantidad molar) del compuesto de fórmula I, tal como aproximadamente del 0,1 a aproximadamente el 0,9% en moles (por ejemplo, de aproximadamente el 0,7 o aproximadamente el 0,3% en moles); y/o
- los uno o más compuestos usados para envenenar ese catalizador pueden usarse en una cantidad que es menor de aproximadamente el 30% en moles del compuesto de fórmula I, que puede comprender menos de aproximadamente el 10% en moles o, particularmente, menos de aproximadamente el 6% en moles de DMAP, tal como aproximadamente el 5% en moles de DMAP (por ejemplo, una mezcla de aproximadamente el 5% en moles de DMAP y aproximadamente el 20% en moles de trietilamina con relación al compuesto de fórmula I).
La persona experta en la materia entenderá que, cuando el agente reductor adecuado es gas hidrógeno, el procedimiento del segundo aspecto de la invención (es decir, la reacción de reducción) puede realizarse a una presión mayor que la presión atmosférica (es decir, de dicho gas hidrógeno), tal como a una presión superior a aproximadamente 1,5 bar (por ejemplo, de aproximadamente 1,5 a aproximadamente 5,5 bar, tal como de aproximadamente 2 bar o aproximadamente 4 a aproximadamente 5 bar). En dichos casos, la reacción puede realizarse en un autoclave.
La persona experta en la materia entenderá que la temperatura a la que se realiza la reacción y la duración durante la cual se mantienen las condiciones de reacción pueden ajustarse para maximizar el rendimiento y la pureza del producto requerido. Por ejemplo, la reacción puede realizarse a temperatura ambiente (es decir, sin calentamiento o enfriamiento) y durante un período superior a aproximadamente 5 horas, por ejemplo, de aproximadamente 5 a aproximadamente 7 horas (por ejemplo, aproximadamente 6 horas) o de aproximadamente 20 a aproximadamente 24 horas (por ejemplo, aproximadamente 22 horas).
La persona experta en la materia entenderá que el procedimiento del segundo aspecto de la invención puede realizarse en presencia de un disolvente adecuado, tal como tolueno, metanol, etanol, dihidronaftaleno, dihidroantraceno, alcohol isopropílico, acetato de isopropilo y/o ácido fórmico. Por ejemplo, el disolvente adecuado puede ser tolueno o acetato de isopropilo.
El compuesto de fórmula V obtenido a partir del procedimiento del segundo aspecto de la invención puede obtenerse con una pureza suficiente de manera que pueda usarse en reacciones posteriores sin purificación. Sin embargo, los presentes inventores han encontrado que las etapas realizadas para purificar el compuesto de fórmula V pueden permitir un almacenamiento más conveniente y una mayor estabilidad de ese compuesto.
De esta manera, en una realización particular del segundo aspecto de la invención, el compuesto de fórmula V se usa en procedimientos adicionales sin purificación adicional (es decir, sin etapas adicionales, realizadas antes de su uso en procedimientos adicionales, que están destinadas a aumentar la pureza de la muestra del compuesto). En una realización más particular del segundo aspecto de la invención, el procedimiento comprende la etapa adicional de aislar y opcionalmente purificar el compuesto de fórmula V.
En particular, los presentes inventores han encontrado que el compuesto de fórmula V puede purificarse y estabilizarse (es decir, para la estabilidad durante el almacenamiento) de una manera altamente eficiente mediante la formación del aducto de bisulfito correspondiente.
De esta manera, en una realización particular del segundo aspecto de la invención, el compuesto de fórmula V se aísla como (y/o purifica vía la formación de) un aducto de bisulfito de fórmula VI.
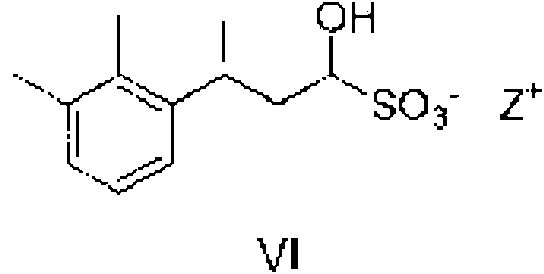
en la que Z representa un metal alcalino.
A menos que se especifique lo contrario, la expresión metal alcalino, cuando se usa en la presente memoria, se refiere a un elemento seleccionado de entre el Grupo I de la tabla periódica de elementos (por ejemplo, sodio, potasio o litio).
Los compuestos de fórmula VI pueden ser novedosos. De esta manera, en un tercer aspecto de la invención, se proporciona un compuesto de fórmula VI según se define en la presente memoria (es decir, en el segundo aspecto de la invención).
Los metales alcalinos particulares que pueden mencionarse incluyen potasio y, más particularmente, sodio. De esta manera, los compuestos particulares de fórmula VI que pueden mencionarse incluyen aquellos en los que Z representa Na.
En una realización particular de los aspectos segundo y tercero de la invención, el compuesto de fórmula VI es un compuesto de fórmula Via

Los compuestos de fórmula VI y VIa pueden representarse también en formas no ionizadas, cuyas formas representarán los mismos compuestos. Por ejemplo, el compuesto de fórmula Via puede representarse como sigue.

Los compuestos de fórmula VI (tales como el compuesto de fórmula Via) pueden prepararse usando técnicas conocidas por las personas expertas en la técnica.
En un cuarto aspecto de la invención, se proporciona un procedimiento para preparar un compuesto de fórmula VI según se define en los aspectos segundo y tercero de la invención, en el que el procedimiento comprende hacer reaccionar un compuesto de fórmula V según se define en el segundo aspecto de la invención con un compuesto que tiene la fórmula ZHSO3, en la que Z es según se define en el compuesto correspondiente de fórmula VI.
De manera alternativa, el cuarto aspecto de la invención puede denominarse como un procedimiento para purificar (es decir, aumentar la pureza de dicho compuesto; por ejemplo, aumentar la pureza en al menos un 10%, por ejemplo, en al menos un 20% o al menos un 30%) un compuesto de fórmula V.
En una realización particular del cuarto aspecto de la invención, el compuesto de fórmula VI es un compuesto de fórmula Via según se define en los aspectos segundo y tercero de la invención.
La persona experta en la materia entenderá que el procedimiento del cuarto aspecto de la invención puede realizarse en presencia de un disolvente adecuado, tal como etanol y/o acetato de etilo.
Los compuestos de fórmula VI (tales como los compuestos de fórmula VIa) pueden purificarse adicionalmente usando técnicas conocidas por las personas expertas en la técnica, tales como recristalización en un disolvente adecuado. De esta manera, en una realización particular del cuarto aspecto de la invención, el procedimiento comprende la etapa adicional de recristalizar el compuesto de fórmula VI.
La persona experta en la materia entenderá que los compuestos de fórmula V pueden obtenerse (es decir, regenerarse) a partir de compuestos de fórmula VI mediante hidrólisis, que puede realizarse bajo condiciones conocidas por las personas expertas en la técnica.
Tal como se ha descrito en la presente memoria, los 3-aril butenales y los 3-aril butanales pueden ser útiles en la síntesis del compuesto comercialmente importante medetomidina.
En un quinto aspecto de la invención, se proporciona un procedimiento para la preparación del compuesto de fórmula VII (es decir, medetomidina)

o una sal adecuada del mismo, en el que el procedimiento comprende las etapas de:
(i) proporcionar un compuesto de fórmula I usando un procedimiento según se ha descrito en el primer aspecto de la invención; y/o
(ii) proporcionar un compuesto de fórmula V usando un procedimiento según se ha descrito en el segundo aspecto de la invención (o, de manera alternativa, mediante hidrólisis de un compuesto de fórmula VI, cuyo compuesto de fórmula VI puede prepararse usando un procedimiento según se ha descrito en el cuarto aspecto de la invención).
Las sales adecuadas que pueden mencionarse incluyen sales farmacéuticamente aceptables conocidas por las personas expertas en la técnica. Las sales más particulares que pueden mencionarse incluyen sales de adición de ácido.
Dichas sales de adición de ácido pueden formarse por medios convencionales, por ejemplo, mediante reacción de una forma de base libre de un compuesto de fórmula I con uno o más equivalentes de un ácido apropiado, opcionalmente en un disolvente o en un medio en el que la sal es insoluble, seguido de la eliminación de dicho disolvente, o dicho medio, usando técnicas estándar (por ejemplo, en vacío mediante liofilización o mediante filtración). Las sales pueden prepararse también intercambiando un contraión de un compuesto de la invención en forma de una sal con otro contraión, por ejemplo, usando una resina de intercambio iónico adecuada.
Las sales de adición de ácido particulares que pueden mencionarse incluyen sales de carboxilato (por ejemplo, sales de formiato, acetato, trifluoroacetato, propionato, isobutirato, heptanoato, decanoato, caprato, caprilato, estearato, acrilato, caproato, propiolato, ascorbato, citrato, glucuronato, glutamato, glicolato, a-hidroxibutirato, lactato, tartrato (por ejemplo, D- o L- tartratos y/o derivados de los mismos, tales como derivados de D- o L-tartrato (por ejemplo, ácido di-p-toluoil-D-tartárico)), fenilacetato, mandelato, fenilpropionato, fenilbutirato, benzoato, clorobenzoato, metilbenzoato, hidroxibenzoato, metoxibenzoato, dinitrobenzoato, o-acetoxibenzoato, salicilato, nicotinato, isonicotinato, cinamato, oxalato, malonato, succinato, suberato, sebacato, fumarato, malato, maleato, hidroximaleato, hipurato, ftalato o tereftalato), sales de haluro (por ejemplo, sales de clorhidrato, bromhidrato o yodohidrato), sales de sulfonato (por ejemplo, sales de bencenosulfonato, metil-, bromo- o clorobencenosulfonato, xilenosulfonato, metanosulfonato, etanosulfonato, propanosulfonato, hidroxietanosulfonato, 1- o 2- naftalenosulfonato o 1,5-naftalenodisulfonato) o sales de sulfato, pirosulfato, bisulfato, sulfito, bisulfito, fosfato, monohidrogenofosfato, dihidrogenofosfato, metafosfato, pirofosfato o nitrato, y similares. Las sales más particulares que pueden mencionarse incluyen la sal de clorhidrato.
En una realización particular, el procedimiento comprende las etapas de:
(I) preparar un compuesto de fórmula V usando un procedimiento según se ha descrito en el segundo aspecto de la invención (incluyendo en el que el compuesto de fórmula V se prepara a partir de un compuesto de fórmula I que se prepara usando un procedimiento según se ha descrito en el primer aspecto de la invención); (II) hacer reaccionar el compuesto de fórmula V para formar un compuesto de fórmula VIII

en la que L1 representa un grupo saliente adecuado; y
(III) hacer reaccionar el compuesto de fórmula VIII en presencia de
(a) una fuente de formamidina, o
(b) formamida,
para formar el compuesto de fórmula VII, o una sal adecuada del mismo.
En una realización alternativa, la etapa (I) en la realización descrita justo anteriormente, muchos requieren por el contrario la preparación de un compuesto de fórmula V mediante hidrólisis de un compuesto de fórmula VI (por ejemplo, un compuesto de fórmula VIa), cuyo compuesto de fórmula VI puede prepararse usando un procedimiento según se describe en el cuarto aspecto de la invención.
En una realización particular, L1 representa un haluro (tal como yodo, cloro o bromo). En una realización más particular, L1 representa bromo.
En una realización particular, la fuente de formamidina es formamidina, acetato de formamidina, hidrohaluro de formamidina, ácido formamidinasulfínico o una mezcla de cloruro de amonio y ácido fórmico.
Los compuestos de fórmula V pueden hacerse reaccionar para proporcionar compuestos de fórmula VIII, y los compuestos de fórmula VIII pueden hacerse reaccionar para proporcionar compuestos de fórmula VII, usando técnicas como las descritas en el documento WO 2013/014428.
En una realización particular, en la que L1 representa bromo, la bromación correspondiente puede realizarse en presencia de un agente de bromación (es decir, cualquier fuente adecuada de iones bromuro), tal como ácido 5,5-dibromo barbitúrico.
De manera alternativa, el agente de bromación puede ser bromo (es decir, Br2). Por ejemplo, la bromación puede realizarse en presencia de bromo y un disolvente adecuado, tal como acetato de isopropilo y, opcionalmente, en presencia de un agente complejante adecuado, tal como 1,4-dioxano.
La persona experta en la materia entenderá que la bromación puede realizarse a una temperatura adecuada, tal como a una temperatura reducida (por ejemplo, a una temperatura de aproximadamente -15 a aproximadamente -25°C, o a una temperatura de aproximadamente -10°C).
En una realización alternativa, la preparación de los compuestos de fórmula VIII puede proceder a través de un intermedio, cuyo intermedio puede ser un derivado del compuesto de fórmula V. Dichos intermedios pueden formarse mediante la adición de componentes adicionales al procedimiento, cuyos componentes pueden ser, en particular, compuestos orgánicos y/o catalíticos (con respecto a la producción de compuestos de fórmula VIII). En particular, el procedimiento para preparar compuestos de fórmula VIII a partir de compuestos de fórmula V puede realizarse en presencia de un compuesto de fórmula IX,
HN(RX)Ry IX
en la que:
RX y Ry, ambos independientemente, representan un grupo alquilo C1-6 opcionalmente sustituido con uno o más sustituyentes seleccionados de entre halo, -CN y alcoxi C1-6 (en el que el último grupo está opcionalmente sustituido con uno o más átomos de flúor), o
RX y Ry pueden tomarse juntos para formar, junto con el átomo de nitrógeno al que están ambos unidos, un grupo heterocicloalquilo de 5 a 7 miembros, que contiene opcionalmente uno o dos grupos adicionales que contienen heteroátomos seleccionados de entre O, S y NRz, y opcionalmente sustituidos con uno o más sustituyentes seleccionados de entre halo, -CN, alquilo C1-6 y alcoxi C1-6 (en el que los dos últimos grupos están opcionalmente sustituidos con uno o más átomos de flúor); y
Rz representa H o un grupo alquilo C1-6, en el que el último grupo está opcionalmente sustituido con uno o más sustituyentes seleccionados de entre halo, -CN y alcoxi C1-6 (en el que el último grupo está opcionalmente sustituido con uno o más átomos de flúor).
Tal como se usa en la presente memoria, el término heterocicloalquilo se refiere a grupos cicloalquilo según se define en la presente memoria pero que contienen al menos un heteroátomo (en particular, al menos un átomo seleccionado de entre O, S y N) como parte de un anillo. En particular, dichos grupos pueden ser monocíclicos. Los compuestos particulares de fórmula IX que pueden mencionarse incluyen aquellos en los que RX y Ry se toman juntos para formar, junto con el átomo de nitrógeno al que están ambos unidos, un grupo heterocicloalquilo
de 5 a 7 miembros (por ejemplo, 6 miembros) que contiene opcionalmente uno o dos (por ejemplo, uno) grupos adicionales que contiene heteroátomos seleccionados de entre O, S y NRz (por ejemplo, un átomo de O).
Los compuestos más particulares de fórmula IX que pueden mencionarse incluyen morfolina.
Cuando el procedimiento para preparar compuestos de fórmula VIII a partir de compuestos de fórmula VIa implica la adición de un compuesto de fórmula IX, la persona experta apreciará que la reacción puede proceder a través de un compuesto de fórmula X

en la que RX y Ry son tal como se ha definido en la presente memoria con respecto a los compuestos de fórmula X.
Los compuestos de fórmula VIII pueden prepararse a partir de compuestos de fórmula X usando técnicas conocidas por las personas expertas en la técnica, tales como bajo condiciones descritas en la presente memoria con respecto a la preparación de compuestos de fórmula VIII a partir de compuestos de fórmula V.
Los compuestos particulares de fórmula X que pueden mencionarse incluyen compuestos de fórmula Xa

En particular, cuando el procedimiento implica la adición de un compuesto de fórmula IX y/o procede a través (o partiendo de) un compuesto de fórmula X (por ejemplo, un compuesto de fórmula Xa), la reacción puede implicar primero la adición de un compuesto de fórmula IX (por ejemplo, morfolina) a un compuesto de fórmula V en presencia de un disolvente adecuado y, opcionalmente, bajo condiciones adecuadas para la eliminación de agua desde la mezcla de reacción (por ejemplo, en el que el disolvente es tolueno y, después de la adición del compuesto de fórmula IX, la reacción se calienta hasta el punto de reflujo del disolvente bajo las denominadas condiciones Dean-Stark (es decir, usando un aparato de destilación Dean-Stark).
Más particularmente, el procedimiento puede implicar la adición de un exceso (es decir, más de un equivalente) del compuesto de fórmula IX con relación al compuesto de fórmula V, tal como de aproximadamente 1,1 a aproximadamente 2 equivalentes (por ejemplo, aproximadamente 1,5 equivalentes).
De manera alternativa, ciertos compuestos de fórmula IX (tal como morfolina) pueden usarse también como el disolvente adecuado (por ejemplo, cuando la reacción se realiza bajo condiciones adecuadas para la eliminación de agua de la mezcla de reacción, tal como bajo las condiciones de Dean-Stark).
Para el procedimiento para la conversión de un compuesto de fórmula V al compuesto de fórmula VIII (en particular, cuando el procedimiento no implica la adición de un compuesto de fórmula IX y/o procede a través de un compuesto de fórmula X), el compuesto de fórmula V puede hacerse reaccionar en presencia de ácido 5,5-dibromo barbitúrico, por ejemplo, de manera que haya al menos un equivalente de iones bromuro en el reactivo empleado (por ejemplo, cuando el reactivo proporciona dos equivalentes del ión haluro, entonces pueden emplearse aproximadamente 0,5 equivalentes, en comparación con el compuesto de fórmula V). El compuesto de fórmula V puede disolverse primero en un disolvente apropiado (por ejemplo, un disolvente aprótico polar, tal como un éter, especialmente tetrahidrofurano (THF)) y puede añadirse un pequeño equivalente molar (es decir, una cantidad sub-estequiométrica) de HCl (por ejemplo, 37% HCl). Esta mezcla puede calentarse primero (por ejemplo, a más de 40°C, tal como a aproximadamente 60°C o hasta el punto de ebullición del disolvente), después de lo
cual puede añadirse el agente de bromación apropiado (por ejemplo, ácido 5,5-dibromo barbitúrico). En particular, puede añadirse en porciones de manera que la temperatura (cuando la reacción se realiza, por ejemplo, en THF) se mantenga por debajo del punto de ebullición (es decir, por debajo de aproximadamente 65°C). El producto deseado puede aislarse mediante un procedimiento de elaboración, por ejemplo, tal como se describe en los ejemplos a continuación.
De manera alternativa, para el procedimiento para la conversión del compuesto de fórmula V en el compuesto de fórmula VIII (en particular, cuando el procedimiento implica la adición de un compuesto de fórmula IX y/o procede a través de (o partiendo de) un compuesto de fórmula X), la reacción puede realizarse en presencia de bromo (es decir, Br2) y, opcionalmente, un disolvente adecuado (tal como acetato de etilo). En particular, dichas reacciones pueden realizarse en presencia de al menos un equivalente de bromo (es decir, un equivalente de Br2) con relación al compuesto de fórmula V (o el compuesto de fórmula X), tal como un ligero exceso de bromo (por ejemplo, de aproximadamente 1,01 a aproximadamente 1,05 equivalentes, tal como aproximadamente 1,04 equivalentes). En una realización particular del quinto aspecto de la invención, la reacción del compuesto de fórmula VIII para formar el compuesto de fórmula VII (es decir, la etapa (III) en la realización descrita anteriormente) puede realizarse en presencia de una fuente de formamidina (por ejemplo, formamidina, o una sal apropiada o un derivado de la misma). Más particularmente, puede realizarse en presencia de acetato de formamidina, aunque también son posibles otros derivados, por ejemplo, otras sales tales como hidrohaluro de formamidina (por ejemplo, HCI), ácido formamidinasulfínico y/u otras sales o derivados que pueden estar disponibles comercialmente). El uso de formamidina y, en particular, acetato de formamidina puede tener la ventaja de que el procedimiento de la invención se mejora, por ejemplo, en términos de rendimiento y pureza.
En una realización más particular, la etapa (III) se realiza en presencia de acetato de formamidina.
En particular, se emplea al menos un equivalente del reactivo que promueve la reacción de formación de anillo de imidazol (por ejemplo, acetato de formamidina) en comparación con el compuesto de fórmula VIII, tal como de aproximadamente 1,1 a aproximadamente 2,5 equivalentes, por ejemplo, aproximadamente 1,5 equivalentes o aproximadamente 2 equivalentes.
En particular, la reacción del compuesto de fórmula VIII para formar el compuesto de fórmula VII puede realizarse en presencia de un disolvente adecuado, tal como un disolvente orgánico polar, por ejemplo, un disolvente alcohólico. Los disolventes particulares que pueden mencionarse son etanol e IPA. La cantidad de disolvente, con relación al compuesto de fórmula VIII, debería ser suficiente para que la reacción proceda de manera eficiente. Por ejemplo, se emplea al menos una relación 1:1 de compuesto de fórmula VIII:disolvente alcohólico (en peso), tal como una relación de al menos 1:2, tal como al menos 1:3. Aunque la relación puede ser de 1:10, las relaciones particulares que pueden mencionarse son de 1:4 a 1:6 (por ejemplo, entre 1:4 y 1:6, tal como aproximadamente 1:5). Más particularmente, la relación puede ser de 1:6 a 1:7 (por ejemplo, entre 1:6 y 1:7, tal como aproximadamente 1:7). Las cantidades más altas de disolvente tienen la desventaja de que la velocidad de reacción puede reducirse debido a la mayor dilución y, además, pueden tener desventajas ambientales/económicas. En particular, el disolvente adecuado puede seleccionarse de entre etanol, alcohol isopropílico (IPA) o una mezcla de etanol o alcohol isopropílico y agua (tal como una mezcla aproximadamente igual) (por ejemplo, alcohol isopropílico (IPA)); por ejemplo, puede emplearse una relación de 1:7 de compuesto de fórmula VIII:IPA (en peso).
Además, la reacción del compuesto de fórmula VIII para formar el compuesto de fórmula VII puede realizarse en presencia de un disolvente ionizante no acuoso (además del disolvente que ya puede estar presente (por ejemplo, el disolvente alcohólico)). A este respecto, los disolventes adicionales particulares que pueden mencionarse son amoniaco líquido o acuoso (por ejemplo, puede emplearse amoníaco acuoso al 25%). En comparación con el compuesto de fórmula VIII, se emplea al menos 1 equivalente molar del disolvente no acuoso (por ejemplo, al menos 5 equivalentes molares, tal como de 5 a 20 equivalentes molares (por ejemplo, entre 5 y 20 equivalentes molares), preferentemente aproximadamente 10 equivalentes molares). En términos de pesos relativos, en comparación con el disolvente primario empleado (por ejemplo, el disolvente alcohólico, tal como IPA), asumiendo que el disolvente adicional es amoniaco acuoso al 25%, la relación de disolvente primario a disolvente adicional es de 1:2 a 10:1 (por ejemplo, entre 1:2 y 10:1), particularmente de 1:1 a 5:1 (por ejemplo, entre 1:1 y 5:1, tal como aproximadamente 1,5:1 o 2:1).
En particular, el disolvente no acuoso puede ser amoníaco líquido; por ejemplo, en comparación con el compuesto de fórmula VIII, pueden usarse de 5 a 20 equivalentes molares de amoníaco líquido (particularmente de aproximadamente 6 a aproximadamente 16, tal como de aproximadamente 8 a aproximadamente 15 (por ejemplo, aproximadamente 9 (tal como aproximadamente 8,9), aproximadamente 12 o 13 (tal como aproximadamente 12,5) o aproximadamente 14 (tal como aproximadamente 14,4) equivalentes molares de amoníaco líquido en comparación con el compuesto de fórmula VIII).
En una realización particular del quinto aspecto de la invención, el disolvente primario usado en la etapa (111) (es decir, en la realización descrita anteriormente) es IPA y el disolvente ionizante no acuoso es amoníaco líquido (es decir, el disolvente usado en la reacción es una mezcla de IPA y amoníaco líquido); por ejemplo, la reacción puede realizarse en un disolvente al menos el 90% (por ejemplo, al menos el 95%, tal como al menos el 99%) del cual consiste en una mezcla de IPA y amoníaco líquido. Más particularmente, en términos de pesos relativos, la relación de IPA a amoníaco líquido puede ser de 6:1 a 12:1, tal como de aproximadamente 8:1 a aproximadamente 11:1 (por ejemplo, aproximadamente 11:1 o aproximadamente 86:10); por ejemplo, la reacción puede realizarse en una mezcla de disolventes que es del 6 al 12% en peso de amoníaco líquido en IPA (por ejemplo, aproximadamente el 8,3% en peso o, particularmente, aproximadamente el 10,4% en peso de amoníaco líquido en IPA).
En una realización particular del quinto aspecto de la invención, la etapa (III) (es decir, en la realización descrita anteriormente) puede realizarse a una temperatura elevada, por ejemplo, por encima de la temperatura ambiente (por ejemplo, a más de 50°C, por ejemplo, a más de 80°C, por ejemplo a aproximadamente 120°C, dependiendo del punto de ebullición del sistema de disolvente que se emplea) durante un período de tiempo (por ejemplo, aproximadamente 2 horas) aunque la temperatura y el tiempo de reacción pueden variarse para maximizar la eficiencia y el rendimiento de la reacción. Por ejemplo, cuando el disolvente es una mezcla de amoníaco líquido e IPA (por ejemplo, 10,4% en peso de amoníaco líquido en IPA), la reacción puede realizarse a una temperatura de aproximadamente 70 a aproximadamente 90°C (por ejemplo, de aproximadamente 75 a aproximadamente 85°C, por ejemplo, de aproximadamente 77 a aproximadamente 83°C). Más particularmente, la reacción puede comprender la etapa de añadir el compuesto de fórmula VIII a una mezcla precalentada que comprende:
(a) parte o la totalidad de los disolventes requeridos (por ejemplo, la mezcla de IPA y amoníaco líquido; por ejemplo, una mezcla que comprende al menos el 50% de los disolventes totales usados en la reacción); y (b) la fuente de formamidina, tal como formamidina (o una sal o un derivado de la misma, tal como acetato de formamidina, hidrohaluro de formamidina y/o ácido formamidinasulfínico) o una mezcla de una sal de amonio (por ejemplo, un haluro de amonio, como cloruro de amonio) y ácido fórmico.
Tal como se usa en la presente memoria, el término precalentado se entenderá como una mezcla que, en el punto de adición del compuesto de fórmula VIII, está a una temperatura elevada, por ejemplo, por encima de la temperatura ambiente (por ejemplo, a más de aproximadamente 50°C, tal como a más de aproximadamente 70°C (por ejemplo, de aproximadamente 70°C a aproximadamente 120°C), dependiendo del punto de ebullición del sistema de disolvente que se emplea y la presión particular a la que se realiza la reacción). Por ejemplo, cuando el disolvente es una mezcla de amoníaco líquido e IPA (por ejemplo, 10,4% en peso de amoníaco líquido en IPA), la mezcla precalentada puede estar a una temperatura de aproximadamente 70°C a aproximadamente 90°C (tal como de aproximadamente 75°C a aproximadamente 85°C, por ejemplo, de aproximadamente 77°C a aproximadamente 83°C).
En una realización más particular, la reacción puede realizarse en un recipiente sellado y, opcionalmente, a presión elevada (es decir, a una presión mayor que la presión atmosférica). En particular, el procedimiento puede realizarse en un recipiente sellado y a una temperatura elevada y/o presión elevada (por ejemplo, en el que el recipiente se sella y a continuación los contenidos se calientan a una temperatura elevada, resultando de esta manera en que la reacción se realice a una presión elevada).
En realizaciones más particulares del quinto aspecto de la invención, una o más de las afirmaciones siguientes pueden aplicarse a la etapa (III) tal como se ha descrito en la realización anterior.
(a) La reacción se realiza en presencia de acetato de formamidina.
(b) La reacción se realiza en un disolvente que es una mezcla de amoníaco líquido IPA, tal como del 6 al 12% en peso de mezcla de amoníaco líquido en IPA (por ejemplo, aproximadamente el 8,3% en peso o, particularmente, aproximadamente el 10,4% en peso). peso del amoníaco líquido en IPA).
(c) La reacción se realiza a una temperatura de aproximadamente 70 a aproximadamente 90°C (tal como de aproximadamente 75 a aproximadamente 85°C, por ejemplo, de aproximadamente 77 a aproximadamente 83°C).
(d) La reacción incluye la etapa de añadir el compuesto de fórmula VIII a una mezcla precalentada de disolvente o disolventes y la formamidina (o una de sus sales o derivados), cloruro de amonio/ácido fórmico o similares, en el que la mezcla precalentada puede estar a una temperatura de aproximadamente 70 a aproximadamente 90°C (tal como de aproximadamente 75 a aproximadamente 85°C, por ejemplo, de aproximadamente 77 a aproximadamente 83°C).
(e) La reacción se realiza en un recipiente sellado y, opcionalmente, a presión elevada.
En una realización particular que puede mencionarse, se aplican cada una de las afirmaciones (b) a (e) anteriores. En una realización más particular, se aplican cada una de las afirmaciones (a) a (e) anteriores.
Tal como se describe en la presente memoria, la persona experta apreciará que el procedimiento del quinto aspecto de la invención (que incluye una cualquiera o más de las realizaciones de la misma) puede comprender además la etapa de hacer reaccionar el compuesto de fórmula VII para formar la sal correspondiente (por ejemplo, la sal de clorhidrato) usando técnicas conocidas por las personas expertas en la técnica.
En una realización particular del quinto aspecto de la invención, el procedimiento comprende la etapa adicional de aislar y opcionalmente purificar el compuesto de fórmula VII, o una sal adecuada del mismo.
El producto de fórmula VII puede aislarse después del procedimiento del quinto aspecto de la invención usando técnicas conocidas por las personas expertas en la técnica. Por ejemplo, el sistema de disolvente (por ejemplo, amoníaco e IPA) puede eliminarse (por ejemplo, preferentemente a presión atmosférica normal, por ejemplo, haciendo hervir el disolvente; aunque el sistema de disolvente puede eliminarse también bajo presión reducida, por ejemplo, mediante destilación a presión reducida) para dejar un residuo. El residuo puede absorberse en una mezcla de agua y disolvente orgánico o, en otra realización, el residuo puede añadirse a agua (en la última realización, el producto crudo puede separarse simplemente de la capa de agua, evitando la necesidad de emplear disolvente orgánico en esa etapa). Cuando el residuo se absorbe en una mezcla de agua y disolvente orgánico, entonces el disolvente orgánico puede ser cualquier disolvente orgánico no soluble en agua que disuelve el producto (por ejemplo, un disolvente orgánico polar tal como acetato de etilo o un disolvente orgánico no polar, tal como un disolvente aromático, por ejemplo, tolueno). En este caso, el pH de la mezcla (el residuo, el agua y el disolvente orgánico) puede ajustarse a 9-10 (empleando, por ejemplo, una base de carbonato, una base de hidróxido, una base de alcóxido o similar, por ejemplo, carbonato de sodio o hidróxido de sodio) y las fases separadas. A continuación, la fase orgánica puede lavarse con ácido clorhídrico diluido de manera que el producto esté sustancialmente en la fase acuosa. Después de separar la fase orgánica, el pH de la fase acuosa (que contiene el producto) puede ajustarse a continuación a pH 9-10 (empleando, por ejemplo, una base de carbonato, una base de hidróxido, una base de alcóxido o similar, por ejemplo, carbonato de sodio o hidróxido de sodio) y el producto se extrajo de nuevo con un disolvente orgánico (por ejemplo, un disolvente orgánico polar, tal como acetato de etilo, o un disolvente orgánico no polar, tal como un disolvente aromático, por ejemplo, tolueno). La fase acuosa se separa y la fase orgánica se concentra para dejar un residuo que contiene el producto deseado.
El producto de fórmula VII puede aislarse después del procedimiento del quinto aspecto de la invención como la base libre. Por ejemplo, la base libre puede aislarse como un sólido (por ejemplo, un sólido cristalino), que puede obtenerse mediante cristalización a partir de un disolvente adecuado (tal como el disolvente orgánico usado en la extracción del producto). De manera alternativa, el producto deseado del compuesto de fórmula VII puede aislarse del residuo de la elaboración como una sal correspondiente (por ejemplo, la sal ácida correspondiente, por ejemplo, la sal HCl), por ejemplo, mediante precipitación. De manera ventajosa, la sal (por ejemplo, la sal HCl) del compuesto de fórmula VII puede ser sólida (o incluso cristalina) y, por lo tanto, puede aislarse fácilmente, por ejemplo, el residuo puede absorberse en un disolvente orgánico (por ejemplo, un disolvente orgánico aprótico polar, tal como acetona) y la precipitación, por ejemplo, de la sal de haluro de hidrógeno puede promoverse mediante la adición del haluro de hidrógeno (por ejemplo, ácido clorhídrico, que puede estar concentrado, por ejemplo, HCl al 37% o puede emplearse HCl gaseoso) a un pH más bajo (por ejemplo, hasta aproximadamente pH 6). El producto puede filtrarse y lavarse con más disolvente (por ejemplo, acetona). Si se desea, un producto adicional o una segunda cosecha de producto (por ejemplo, la sal HCl del compuesto de fórmula VII) puede aislarse mediante la destilación del disolvente desde el licor madre seguido de la adición de acetona libre de agua. Sin embargo, cuando se emplea HCl gaseoso en la etapa de formación de la sal de haluro de hidrógeno, no es necesario realizar el aislamiento de una segunda cosecha de producto (por ejemplo, la sal de HCl del mismo). Si el producto de fórmula VII se aísla como la sal, entonces la sal puede convertirse a la base libre neutralizando la sal. Las condiciones de reacción que pueden emplearse pueden describirse en los ejemplos (por ejemplo, primero la sal se disuelve en agua y a continuación se trata con carbón vegetal), por ejemplo, la neutralización puede realizarse mediante la adición de una base (por ejemplo, un hidróxido tal como hidróxido de sodio), por ejemplo, a temperatura elevada (por ejemplo, aproximadamente 55-60°C) después de lo cual la emulsión resultante puede enfriarse, por ejemplo, a aproximadamente 40°C, y la cristalización puede inducir se mediante la siembra y la base libre puede aislarse posteriormente.
Los procedimientos descritos en la presente memoria pueden operarse como un procedimiento por lotes o pueden operarse como un procedimiento continuo (es decir, flujo), y pueden realizarse a cualquier escala.
A menos que se indique lo contrario, los procedimientos descritos en la presente memoria pueden realizarse con o sin separación (por ejemplo, aislamiento y/o purificación) de cualquier producto intermedio estable.
Las personas expertas en la materia apreciarán que, en los procedimientos descritos en la presente memoria, los grupos funcionales de compuestos intermedios pueden estar, o pueden necesitar ser, protegidos por grupos protectores. La protección y la desprotección de los grupos funcionales puede tener lugar antes o después de cualquiera de las etapas de reacción descritas en la presente memoria.
Los grupos protectores pueden eliminarse según técnicas que son bien conocidas por las personas expertas en la técnica. Por ejemplo, el uso de grupos protectores se describe en "Protective Groups in Organic Chemistry", editado por J.W.F. McOmie, Plenum Press (1973), y "Protective Groups in Organic Synthesis", 3a edición, T.W. Greene & P.G.M. Wutz, Wiley-Interscience (1999).
A menos que se indique lo contrario, el material de partida y los reactivos usados en los procedimientos descritos en la presente memoria pueden estar disponibles comercialmente y/o pueden sintetizarse a partir de materiales de partida disponibles comercialmente usando técnicas conocidas por las personas expertas en la técnica.
En particular, los compuestos de fórmula II pueden prepararse usando materiales de partida disponibles comercialmente y usando técnicas conocidas por las personas expertas en la técnica. Los compuestos de fórmula II pueden prepararse mediante reacción de un compuesto de XI

para formar un intermedio (reactivo de Grignard) de fórmula XII

seguido de la reacción del compuesto de fórmula XII con acetona para formar el compuesto de fórmula II, usando técnicas y condiciones conocidas por las personas expertas en la técnica.
Los compuestos empleados en o producidos por los procedimientos descritos en la presente memoria (es decir, aquellos que implican el procedimiento de la invención) pueden exhibir tautomerismo. Por lo tanto, el procedimiento de la invención abarca el uso o la producción de dichos compuestos en cualquiera de sus formas tautoméricas, o en mezclas de cualquiera de dichas formas.
De manera similar, los compuestos empleados en o producidos por los procedimientos descritos en la presente memoria (es decir, aquellos que implican el procedimiento de la invención) pueden contener también uno o más átomos de carbono asimétricos y, por lo tanto, pueden existir como enantiómeros o diastereoisómeros, y pueden exhibir actividad óptica. De esta manera, el procedimiento de la invención abarca el uso o la producción de dichos compuestos en cualquiera de sus formas ópticas o diastereoisoméricas, o en mezclas (por ejemplo, mezclas racémicas) de cualquiera de dichas formas.
Los compuestos particulares de fórmulas V, VI (incluyendo Via), VII, VIII y X, tal como se describen en la presente memoria que pueden mencionarse incluyen los enantiómeros S de esos compuestos. Por ejemplo, el compuesto de fórmula VII, tal como se ha descrito en la presente memoria, puede proporcionarse en forma de enantiómero S, tal como se muestra a continuación, que puede denominarse dexmedetomidina.

Además, los compuestos empleados en o producidos por los procedimientos descritos en la presente memoria pueden contener dobles enlaces y, de esta manera, pueden existir como isómeros geométricos E (Entgegen) y Z (zusammen) en cada doble enlace individual. La totalidad de dichos isómeros y sus mezclas están incluidos dentro del alcance de la invención.
Los procedimientos descritos en la presente memoria pueden realizarse empleando sales, solvatos o derivados protegidos, produciendo de esta manera compuestos que pueden producirse o no en forma de una sal o solvato correspondiente, o un derivado protegido de los mismos. En particular, los procedimientos descritos en la presente memoria pueden producir compuestos que están en forma de la sal correspondiente (por ejemplo, la sal de clorhidrato), en el que dichos procedimientos pueden comprender además la etapa de hacer reaccionar el producto para formar la sal correspondiente usando técnicas conocidas por las personas expertas en la técnica.
Los procedimientos de la invención pueden tener la ventaja de que son, entre otras cosas, más eficientes (por ejemplo, mayor rendimiento), usan menos energía, usan menos reactivos tóxicos, producen menos subproductos y/o son más económicos de realizar que los procedimientos descritos en la técnica anterior. En particular, los procedimientos tal como se describen en la presente memoria (tal como el procedimiento del primer aspecto de la invención) pueden tener la ventaja de que son más adecuados para su uso en la fabricación industrial a gran escala que los procedimientos descritos en la técnica anterior.
Ejemplos
Los ejemplos siguientes son ejemplos simplemente ilustrativos de los procedimientos de la invención descritos en la presente memoria. Todos los equipos, reactivos y disolventes usados fueron equipos de laboratorio estándar, por ejemplo, objetos de cristal, aparatos de calentamiento y aparatos de HPLC.
Ejemplo 1 - Preparación de alcohol (2-(2,3-xili)-2-propanol) terciario
Se disolvió 2,3-dimetilbromobenceno (150 g, 0,80 mol) en THF (250 ml). A virutas de Mg (20,5 g, 0,84 moles) en THF (300 ml) se añadieron 90 ml de solución de 2,3-dimetilbromobenceno preparada anteriormente. La solución se calentó a 50°C y se añadió 1,2-dibromoetano (1 ml) para iniciar la reacción. El resto de la solución de 2,3-dimetilbromobenceno se añadió durante 50 minutos a 64-65°C. La mezcla se sometió a reflujo durante 1 hora y a continuación 300 ml de THF se separaron mediante destilación a presión atmosférica.
La mezcla se enfrió a 50°C y se añadió tolueno (500 ml) seguido de enfriamiento a temperatura ambiente. A continuación, la solución de acetona (61 g, 1,05 moles) en tolueno (100 ml) se añadió durante 1 hora a 24-29°C. La suspensión obtenida se agitó durante 13 horas a temperatura ambiente.
La suspensión se enfrió a 12°C, se neutralizó con HCl acuoso a 13-15°C. La fase acuosa inferior se cortó y los orgánicos se lavaron con NaHCO3 al 5% (250 ml) y agua (250 ml).
Los disolventes se eliminaron a 40-50°C y a 50-100 mbar. A continuación, se añadió heptano (150 ml) y la solución se enfrió a menos 10°C. Los sólidos se filtraron y se lavaron con heptano frío (100 ml) para obtener la cosecha 1 húmeda (74,4 g) que, después del secado en vacío, dió 73,6 g (55,8% con relación a la teoría) de 2-(2,3-xilil)-2-propanol seco, determinado como puro mediante 1H RMN.
El filtrado se concentró a 52,5 g de residuo. La dilución con heptano (50 ml), la cristalización durante el enfriamiento, la filtración, el lavado con heptano y el secado dieron 2-(2,3-xilil)-2-propanol (cosecha 2, 16,8 g, 12,7% con relación a la teoría) del producto. Rendimiento global del 68,5% a partir de 2,3-dimetilbromobenceno.
Ejemplo 2 - Preparación de aldehído (3-(2,3-xilil)-2-butenal) insaturado
El alcohol 2-(2,3-xilil)-2-propanol terciario (8,83 g, 0,054 moles) se disolvió en DMF (12 ml, 0,16 moles) y la solución se enfrió a 1°C. A continuación, se añadió POCl3 (12,5 ml, 0,13 mol) a <13°C (reacción exotérmica) en 50 minutos. La mezcla se calentó a 75-85°C y se hizo reaccionar durante 2 horas, a continuación, se enfrió a temperatura ambiente y se vertió en agua con hielo. La mezcla se neutralizó con NaOH 4 M, se extrajo con MTBE y el extracto orgánico se lavó con agua.
La eliminación del disolvente dejó 9,06 g de 3-(2,3-xilil)-2-butenal como un aceite con un 92% ensayo mediante RMN (suma de los isómeros E y Z). Rendimiento corregido para el ensayo 89%.
Ejemplo 3 - Preparación de aldehído (3-(2,3-xilil)-butanal) saturado
Se cargó un autoclave con 330 g de una solución al 12,5% de 3-(2,3-dimetilfenil)but-2-enal en acetato de isopropilo (mezcla de isómeros E y Z; 0,24 moles). Se añadieron 4-(dimetilamino)piridina (1,4 g; 12 mmoles; 5% en moles) y catalizador (3% Pd/C; 12,0 g; 1,7 mmoles; 0,7 moles). La hidrogenación se llevó a cabo a 2 bar y a temperatura ambiente durante 6 horas. Un análisis mediante GC de la mezcla de reacción (área%) mostró: 84,9% de 3-(2,3-dimetilfenil)butanal; 0,5% de 3-(2,3-dimetilfenil)but-2-enol; 0,1% de 3-(2,3-dimetilfenil)butanol; 6,8% de aldehido de partida.
Ejemplo 4 - Preparación de aldehido (3-(2,3-xilil)-butanal) saturado
El autoclave se cargó con 20,0 g de solución al 22% de 3-(2,3-dimetilfenil)but-2-enal en tolueno (mezcla de isómeros E y Z; 25,3 mmoles). Se añadió carbón vegetal (1,5 g). La mezcla se agitó durante 1 hora. Se añadieron 4-(dimetilamino)piridina (0,15 g; 1,23 mmoles; 4,8% en moles) y trietilamina (0,50 g; 5 mmoles; 19,7% en moles). Se añadió catalizador (3% Pd/C; 0,55 g; 0,077 mmoles; 0,30% en moles). La hidrogenación se llevó a cabo a 4-5 bar y a temperatura ambiente durante 22 horas. El catalizador se separó mediante filtración y se lavó con tolueno. Después de la evaporación del disolvente, se obtuvieron 26,4 g de un líquido amarillo claro. El % de área GC mostró: 96,9% de 3-(2,3-dimetilfenil)butanal; 0,6% de 3-(2,3-dimetilfenil)but-2-enol; 0,3% de 3-(2,3-dimetilfenil)butanol; 0,9% de aldehido de partida. Ensayo de RMN 15%; Rendimiento 89%.
Ejemplo 5 - Preparación de bromo aldehído (3-(2,3-xilil)-2-bromo-butanal) (1)

3-(2,3-xilil)-butanal, 945,8g, 5,37 mol, se disolvió en 3.880 g de THF. Se añadió ácido clorhídrico al 37%, 31,2 g, 0,32 mol, y la mezcla se calentó a 60°C. Se añadió ácido 5,5-dibromobarbitúrico, 767,6 g, 2,69 mol, en porciones, manteniendo la temperatura por debajo de 65°C. A continuación, la mezcla se agitó durante 30 minutos a 60-65°C. El THF se eliminó bajo presión reducida seguido de la adición de 2.980 g de tolueno. A continuación, el THF residual se destiló bajo presión reducida. La fase de tolueno se lavó con 3 x 3,2 l de agua seguido de 1,6 l de trietanolamina acuosa al 3% y finalmente con 1,6 l de agua. A la fase de tolueno se añadieron 200 mg de trietanolamina y 200 mg de BHT. El tolueno se eliminó a presión reducida dejando 1.150 g, 4,51 moles, 84%, de 3-(2,3-xilil)-2-bromo-butanal (1).
Ejemplo 6 - Síntesis de medetomidina

A un reactor de presión SS se añadió 3-(2,3-xilil)-2-bromo-butanal (1), 1.154 g, 4,53 mol, acetato de formamidina, 939 g, 9,02 mol, etanol, 5.280 g y finalmente amoniaco acuoso al 25%, 3.050 g, 44,9 mol. La mezcla se calentó a 120°C durante 2 horas. El etanol y el amoniaco se eliminaron a presión atmosférica y el residuo se disolvió en 1.200 ml de agua y 700 ml de acetato de etilo. El pH se ajustó a 9-10 con carbonato de sodio y la fase acuosa se separó. El producto se extrajo en agua mediante tres lavados sucesivos con ácido clorhídrico diluido. El pH de la fase acuosa ácida se ajustó a 9-10 con carbonato de sodio y el producto se extrajo en 500 ml de acetato de etilo. La fase acuosa se separó y el acetato de etilo se eliminó a presión reducida. El aceite residual se disolvió en acetona, 4 l, y el producto precipitó como la sal HCl mediante la adición de ácido clorhídrico al 37% a pH 6. La filtración y el lavado con acetona dieron 366 g de Medetomidina x HCl. Se aisló una segunda cosecha de producto, 96 g mediante destilación del disolvente desde el licor madre seguido de la adición de acetona libre de agua. En total, se aislaron 462 g, 1,95 mol, 43% de Medetomidina x HCl pura.
La medetomidina x HCl, 783 g, 3,31 mol, se disolvió en 2,5 l de agua. Se añadió carbón vegetal, 40 g, y la mezcla se agitó durante 30 minutos a 70°C. El carbón vegetal se filtró y se lavó con 0,5 l de agua. El filtrado combinado y
el lavado se diluyeron con 3,1 l de acetona y 0,2 l de agua. La temperatura se ajustó a 55-60°C y se añadió una solución de 132 g, 3,3 mol, de hidróxido de sodio en 0,54 l de agua durante aproximadamente 1 hora. La emulsión resultante se enfrió a aproximadamente 40°C y la cristalización se indujo mediante siembra. La suspensión se enfrió a 0°C, se filtró y la torta del filtro se lavó con 3 x 400 ml de agua. El secado al vacío produjo 590 g, 2,95 mol, 89% de la base libre de medetomidina.
Ejemplo 7 - Preparación de bromo aldehido (3-(2,3-xilil)-2-bromo-butanal)

A 3-(2,3-dimetilfenil)butanal (2) en tolueno (82,2 g, 0,28 moles) se añadió tolueno (93 ml) y morfolina (36 ml, 0,42 moles). La mezcla se calentó a reflujo con una trampa Dean-Stark montada para eliminar el agua formada. Cuando se había eliminado la cantidad teórica de agua y no se destiló más agua, los disolventes se eliminaron mediante destilación hasta alcanzar un volumen residual de 110 ml. El intermedio de enamina resultante (3) se usó directamente.
A un segundo recipiente de reacción se añadió bromo (15 ml, 0,29 mol) y acetato de etilo (566 ml) y la mezcla se enfrió por debajo de -10°C. A esta solución de bromo se añadió la solución de enamina preparada anteriormente a una velocidad que mantenía la temperatura por debajo de -10°C. Después de al menos 10 minutos de agitación, se añadió agua (185 ml) para desactivar la reacción y a continuación la temperatura se incrementó a 25°C. Cuando fue necesario, el pH se ajustó por debajo de 4 añadiendo ácido clorhídrico. La agitación se detuvo y se permitió que las fases se separaran durante 10 minutos. La fase inferior se desechó. Se añadió una solución de bicarbonato de sodio (7,6 g) y tiosulfato de sodio 7,5 g) en agua (132 ml) y la mezcla se agitó durante 10 minutos y a continuación se permitió que las fases se separaran durante 10 minutos. La fase inferior se desechó. Se añadió agua (153 ml) y la mezcla se agitó durante 10 minutos antes de la detención de la agitación y se permitió que las fases se separaran. La fase inferior se desechó. Se cargaron BHT (0,1 g) y trietanolamina (0,1 g). Los disolventes se destilaron con vacío aplicado hasta un volumen residual de 93 ml. La solución del producto crudo que contenía 62,8 g de 2-bromo-3-(2,3-dimetilfenil)butanal (1) se recogió. El rendimiento global a partir del 1-bromo-2,3-dimetilbenceno es del 61%.
Ejemplos 8(a) a (d) - Preparación de medetomidina

Ejemplo 8(a)
La reacción se realizó en una bomba revestida con PTFE. El bromoaldehído (3,4 g; que contenía 2,5 g, 9,8 mmoles de bromoaldehído mediante ensayo de RMN) y 2,04 g (19,6 mmoles) de acetato de formamidina se mezclaron con 30 ml de isopropanol que contenía amoníaco (8,3%; 118 mmol). La mezcla se calentó en un baño
de aceite a 77-80°C durante 2 horas. Después de enfriar a 21°C, un análisis GC mostró un 74,8% de área de Medetomidina y aproximadamente un 11-12% de subproductos de alto punto de ebullición (incluyendo pirazinas). La mezcla de reacción se concentró en rotavapor. Al residuo se añadieron (10,7 g) 15 ml de tolueno, 15 ml de agua y 1 ml de hidróxido de sodio al 30%. La mezcla se calentó a 30°C y las fases se separaron. La fase acuosa inferior (pH-11-12) se desechó. La fase de tolueno se ensayó mediante RMN para dar un rendimiento del 70% de medetomidina.
Ejemplo 8(b)
El acetato de formamidina (0,25 g) se mezcló con 3 ml de solución de amoníaco al 10,4% p/p en 2-propanol. La mezcla se sumergió en un baño de aceite (77°C). El acetato de formamidina se disolvió a aproximadamente 70°C resultando en una solución incolora.
El bromoaldehído (peso de la muestra 0,42 g, calculado para contener 0,31 g de bromoaldehído) se añadió con una microjeringa durante 20 minutos a 77-80°C. La agitación se continuó a una temperatura del baño de 76-81°C durante 2 horas.
Un análisis mediante GC del producto crudo mostró 84,5% de medetomidina y 3,7% de los subproductos de alto punto de ebullición.
Ejemplo 8 (c)
El acetato de formamidina (28,5 g; 0,27 moles) se mezcló con una solución de amoníaco al 10,4% en 2-propanol (260 ml; 1,24 moles de NH3). La mezcla se calentó a 83°C (baño de aceite a 97°C). El bromoaldehído (51 g, 0,137 moles) se bombeó al reactor en 42 minutos. Post reacción a 90-91°C durante 2 horas.
El reactor se enfrió a temperatura ambiente. La mezcla contenía el 80% de medetomidina y el 3,2% de subproductos determinados mediante análisis de % de área GC.
La mezcla de reacción (305,5 g) se concentró a 112,8 g. Al concentrado, se añadieron 150 ml de tolueno, 100 ml de agua y 18 ml de NaOH al 30%. La mezcla se agitó a 50-55°C durante 10 minutos y las fases se separaron. La fase de tolueno se analizó mediante RMN para dar un rendimiento del 81% de medetomidina.
Ejemplo 8 (d)
El acetato de formamidina (21 g; 0,20 moles) se mezcló con una solución de amoníaco al 10,4% en 2-propanol (250 ml; 1,2 moles de NH3). La mezcla se calentó en un baño de aceite a 80°C (baño de aceite a 97°C). El bromoaldehído (50 g; 0,134 moles) se bombeó al reactor en 40 minutos a 85-89°C. Después de 2 horas después de la reacción, la mezcla se enfrió a temperatura ambiente. La mezcla contenía el 80% de medetomidina y el 3,2% de subproductos determinados mediante un análisis del % de área GC.
El aislamiento se llevó a cabo tal como se describe en el Ejemplo 8(c) (directamente arriba).
La fase de tolueno se ensayó mediante RMN para dar un rendimiento del 82,8% de medetomidina.
Ejemplo 9 - Preparación de aldehido insaturado (3-(2,3-xilil)-2-butenal)
El 2-(2,3-xilil)-2-propanol bruto (77,8 g, ensayo 74,1%, 0,352 mol) se disolvió en DMF (66 ml, 0,856 mol). A continuación, se añadió POCl3 (139 g, 907 mmol) durante 4,5 horas a una temperatura que no excedía los 75°C (reacción exotérmica). La mezcla se hizo reaccionar a aproximadamente 70°C durante 4,5 horas. Durante ese período, se añadieron 3 ml adicionales de POCl3. La mezcla se enfrió a temperatura ambiente y se vertió en agua a 0-5°C. La mezcla se neutralizó con NaOH al 25%, se extrajo con tolueno y el extracto orgánico se lavó con agua. La fase orgánica (194,5 g) tenía un ensayo del 28,6% de 3-(2,3-xilil)-2-butenal como suma de isómeros E y Z. El rendimiento corregido del ensayo fue del 91%.
Ejemplo 10
Los productos mencionados en la presente memoria, por ejemplo, obtenidos mediante los procedimientos descritos en la presente memoria (tales como los enumerados en los Ejemplos 8(a) a (d) anteriores), pueden formularse en un producto final adecuado, por ejemplo, en el caso de la síntesis del producto final medetomidina a un agente antiincrustante tal como Selektope™ usando técnicas de formulación estándar. Por ejemplo, la base libre de medetomidina puede disolverse en un disolvente orgánico para preparar el producto final formulado.
De manera alternativa, la medetomidina (por ejemplo, la dexmedetomidina), o una sal farmacéuticamente aceptable de la misma, puede combinarse con uno o más excipientes farmacéuticamente aceptables para proporcionar una formulación farmacéutica. Dichos excipientes farmacéuticamente aceptables serán bien conocidos por las personas expertas en la técnica.
Abreviaturas
Las abreviaturas usadas en la presente memoria serán bien conocidas por las personas expertas en la materia. Por ejemplo, las siguientes abreviaturas pueden tener los significados indicados a continuación.
BHT hidroxitolueno butilado
C Celsius
DMAP 4-(dimetilamino)piridina
DMF dimetilformamida
eq equivalente(s)
h hora(s)
HPLC cromatografía líquida de alto rendimiento
IPA alcohol isopropílico
MTBE metil-tert-butil éter
RMN resonancia magnética nuclear
PTFE politetrafluoroetileno
THF tetrahidrofurano
Claims (15)
1. Procedimiento de preparación de un compuesto de fórmula I

en la que dicho procedimiento comprende hacer reaccionar un compuesto de fórmula II

con uno o más reactivos de Vilsmeier adecuados.
2. Procedimiento según la reivindicación 1, en el que el reactivo de Vilsmeier comprende un compuesto de fórmula IIIa y/o un compuesto de fórmula IIIb

en las que:
X representa un contraión derivado de la reacción con un cloruro de ácido;
Y representa un sustituyente derivado de la reacción con un cloruro de ácido;
cada Ra y Rb independientemente representa H, alquilo o fenilo C1-3, o Ra y Rb tomados junto con el nitrógeno al que están unidos forman un grupo pirolidina, piperidina, morfolina o indolina.
3. Procedimiento según la reivindicación 1 o la reivindicación 2, en el que el reactivo de Vilsmeier se forma mediante reacción de dimetilformamida y POCh, y opcionalmente en el que la dimetilformamida actúa también como un disolvente.
4. Procedimiento según una cualquiera de las reivindicaciones 1 a 3, en el que el procedimiento comprende las etapas de:
(i) hacer reaccionar el compuesto de fórmula II para formar un compuesto de fórmula IV

y posteriormente
(ii) hacer reaccionar el compuesto de fórmula IV para formar el compuesto de fórmula I.
5. Procedimiento según una cualquiera de las reivindicaciones 1 a 4, en el que;
a) el procedimiento se realiza como un procedimiento en un solo recipiente y/o sin aislamiento de un compuesto de fórmula IV y/o;
b) el procedimiento comprende la etapa adicional de aislar y opcionalmente purificar el compuesto de fórmula I.
6. Procedimiento de preparación de un compuesto de fórmula

en la que el procedimiento comprende hacer reaccionar un compuesto de fórmula I tal como se ha definido en la reivindicación 1 con un agente reductor adecuado.
7. Procedimiento según la reivindicación 6, en el que el procedimiento comprende las etapas de:
(i) proporcionar un compuesto de fórmula I usando un procedimiento según una cualquiera de las reivindicaciones 1 a 5; y posteriormente
(ii) hacer reaccionar el compuesto de fórmula I con un agente reductor adecuado para formar el compuesto de fórmula V.
8. Procedimiento según la reivindicación 6 o la reivindicación 7, en el que el agente reductor adecuado es una fuente de hidrógeno y la reacción se realiza en presencia de uno o más catalizadores adecuados, y opcionalmente se realiza en presencia de uno o más compuestos adecuados para limitar la actividad de dicho catalizador.
9. Procedimiento según una cualquiera de las reivindicaciones 6 a 8, en el que;
a) el procedimiento comprende la etapa adicional de aislar y opcionalmente purificar el compuesto de fórmula V y/o;
b) el compuesto de fórmula V se aísla como y/o se purifica a través de la formación de un aducto de bisulfito de fórmula VI

en la que Z representa un metal alcalino.
10. Compuesto de fórmula VI tal como se ha definido en la reivindicación 9.
11. Procedimiento de preparación de un compuesto de fórmula VI como se define en la reivindicación 9, en el que el procedimiento comprende hacer reaccionar un compuesto de fórmula V como se define en la reivindicación 6 con un compuesto que tiene la fórmula ZHSO3, en la que Z es tal como se ha definido en el compuesto de fórmula VI.
12. Procedimiento según la reivindicación 9, un compuesto según la reivindicación 10 o un procedimiento según la reivindicación 11, en los que Z representa Na.
13. Procedimiento de preparación del compuesto de fórmula VII

o una sal adecuada del mismo, en el que el procedimiento comprende las etapas de:
(i) proporcionar un compuesto de fórmula I usando un procedimiento según una cualquiera de las reivindicaciones 1 a 5; o
(ii) proporcionar un compuesto de fórmula V usando un procedimiento según una cualquiera de las reivindicaciones 6 a 9.
14. Procedimiento según la reivindicación 13, en el que el procedimiento comprende las etapas de:
(I) preparar un compuesto de fórmula V usando un procedimiento como se define en una cualquiera de las reivindicaciones 6 a 9;
(II) hacer reaccionar el compuesto de fórmula V para formar un compuesto de fórmula VIII

en la que L1 representa un grupo saliente adecuado; y
(III) hacer reaccionar el compuesto de fórmula VIII en presencia de
(a) una fuente de formamidina, o
(b) formamida,
para formar el compuesto de fórmula VII, o una sal adecuada del mismo.
15. Procedimiento según la reivindicación 13 o la reivindicación 14, en el que el procedimiento comprende la etapa adicional de aislar y opcionalmente purificar el compuesto de fórmula VIII.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1501593.6A GB201501593D0 (en) | 2015-01-30 | 2015-01-30 | New compounds and processes |
| PCT/GB2016/050204 WO2016120635A1 (en) | 2015-01-30 | 2016-01-29 | Processes for the preparation of compounds, such as 3-arylbutanals, useful in the synthesis of medetomidine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2716748T3 true ES2716748T3 (es) | 2019-06-14 |
Family
ID=52705534
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16706396T Active ES2716748T3 (es) | 2015-01-30 | 2016-01-29 | Procedimientos de preparación de compuestos, tales como 3-arilbutanales, útiles en la síntesis de medetomidina |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US10053420B2 (es) |
| EP (1) | EP3250556B1 (es) |
| JP (1) | JP6702623B2 (es) |
| KR (1) | KR102458638B1 (es) |
| CN (1) | CN107428648B (es) |
| DK (1) | DK3250556T3 (es) |
| ES (1) | ES2716748T3 (es) |
| GB (1) | GB201501593D0 (es) |
| HR (1) | HRP20190286T1 (es) |
| HU (1) | HUE042626T2 (es) |
| PL (1) | PL3250556T3 (es) |
| TR (1) | TR201902673T4 (es) |
| WO (1) | WO2016120635A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2023551097A (ja) | 2020-10-21 | 2023-12-07 | アイ - テック エービー | 防汚化合物 |
| WO2023182903A1 (ru) | 2022-03-22 | 2023-09-28 | Общество с ограниченной ответственностью "ВИК-здоровье животных" | Способ получения медетомидина и его производных |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2340812C2 (de) | 1973-08-11 | 1982-05-27 | Basf Ag, 6700 Ludwigshafen | Phenylpropanale sowie ein Verfahren zu deren Herstellung |
| US4416902A (en) | 1982-07-16 | 1983-11-22 | International Flavors & Fragrances Inc. | Flavoring with cyclohexenyl-beta-methyl acrolein derivatives |
| DE19714042A1 (de) | 1997-04-05 | 1998-10-08 | Henkel Kgaa | Verfahren zur Herstellung aromatischer Aldehyde |
| WO2007111630A1 (en) * | 2005-07-29 | 2007-10-04 | Flexitral, Inc. | Compounds which release an odor on a change in ph |
| JP5663791B2 (ja) * | 2009-06-03 | 2015-02-04 | 高砂香料工業株式会社 | 不斉水素化触媒 |
| TW201223934A (en) | 2010-11-26 | 2012-06-16 | Lonza Ag | 3-(2,3-dimethylphenyl)-2-butenal and its use as perfume |
| BR112013018345B1 (pt) * | 2011-01-19 | 2019-02-26 | Dsm Ip Assets B.V. | Hidrogenação de ligação dupla carbono-carbono catalítica |
| WO2012120070A1 (en) | 2011-03-10 | 2012-09-13 | Lonza Ltd | Method for preparation of 3-(2,3-dimethylphenyl)-2-butenal |
| DK2734508T3 (en) | 2011-07-22 | 2018-05-22 | Cambrex Karlskoga Ab | NEW PROCEDURES FOR THE PREPARATION OF 4-SUBSTITUTED IMIDAZOLS |
| WO2012172120A2 (en) | 2012-05-08 | 2012-12-20 | Lonza Ltd | 2-(2,3-dimethylphenyl)-1-propanal and its use as perfume |
-
2015
- 2015-01-30 GB GBGB1501593.6A patent/GB201501593D0/en not_active Ceased
-
2016
- 2016-01-29 DK DK16706396.5T patent/DK3250556T3/en active
- 2016-01-29 HU HUE16706396A patent/HUE042626T2/hu unknown
- 2016-01-29 EP EP16706396.5A patent/EP3250556B1/en active Active
- 2016-01-29 TR TR2019/02673T patent/TR201902673T4/tr unknown
- 2016-01-29 ES ES16706396T patent/ES2716748T3/es active Active
- 2016-01-29 PL PL16706396T patent/PL3250556T3/pl unknown
- 2016-01-29 CN CN201680012547.0A patent/CN107428648B/zh active Active
- 2016-01-29 JP JP2017540562A patent/JP6702623B2/ja active Active
- 2016-01-29 KR KR1020177023707A patent/KR102458638B1/ko active IP Right Grant
- 2016-01-29 US US15/547,288 patent/US10053420B2/en active Active
- 2016-01-29 WO PCT/GB2016/050204 patent/WO2016120635A1/en active Application Filing
-
2019
- 2019-02-12 HR HRP20190286TT patent/HRP20190286T1/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN107428648A (zh) | 2017-12-01 |
| HRP20190286T1 (hr) | 2019-04-05 |
| KR102458638B1 (ko) | 2022-10-24 |
| PL3250556T3 (pl) | 2019-05-31 |
| CN107428648B (zh) | 2021-12-10 |
| KR20170108080A (ko) | 2017-09-26 |
| US10053420B2 (en) | 2018-08-21 |
| EP3250556B1 (en) | 2018-12-05 |
| TR201902673T4 (tr) | 2019-03-21 |
| WO2016120635A1 (en) | 2016-08-04 |
| JP6702623B2 (ja) | 2020-06-03 |
| JP2018505179A (ja) | 2018-02-22 |
| HUE042626T2 (hu) | 2019-07-29 |
| GB201501593D0 (en) | 2015-03-18 |
| DK3250556T3 (en) | 2019-03-11 |
| EP3250556A1 (en) | 2017-12-06 |
| US20180009744A1 (en) | 2018-01-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2816042B1 (en) | 6-oxabicyclo[3,2,0]heptanes for use as intermediates | |
| FI60194B (fi) | Foerfarande foer framstaellning av bensil-monoketaler | |
| US9550716B2 (en) | Process for treprostinil salt preparation | |
| US8163930B2 (en) | Process for preparing pyridinamines and novel polymorphs thereof | |
| US20130211078A1 (en) | Preparation of methionine or selenomethionine from homoserine via a carbamate intermediate | |
| US20080275265A1 (en) | Process for the Preparation of (Aminoalkylamino)Alkyl Halides and Conversion to Amifostine | |
| ES2665093T3 (es) | Nuevos procedimiento de preparación de imidazoles 4-sustituidos | |
| CN102020647A (zh) | 1-(2,2-二氟乙氧基)-6-三氟甲基-n-([1,2,4]三氮唑[1,5-c]嘧啶-2-)苯磺酰胺的制备方法 | |
| CN112266392A (zh) | 制备cenicriviroc及相关类似物的方法 | |
| TWI462913B (zh) | 製備抗壞血酸衍生物之方法 | |
| ES2716748T3 (es) | Procedimientos de preparación de compuestos, tales como 3-arilbutanales, útiles en la síntesis de medetomidina | |
| JP2743461B2 (ja) | 1―メチル―3―アルキル―5―ピラゾールカルボン酸エステル類の製造法 | |
| US7141693B2 (en) | Process for producing β-oxonitrile compound or alkali metal salt thereof | |
| CA3028151A1 (en) | Purified cenicriviroc and purified intermediates for making cenicriviroc | |
| JP3972715B2 (ja) | スルフィド誘導体の製法 | |
| JP3272340B2 (ja) | 1−[(シクロペント−3−エン−1−イル)メチル]−5−エチル−6−(3,5−ジメチルベンゾイル)−2,4−ピリミジンジオンの製造方法 | |
| AU2013200508B2 (en) | Process for preparing pyridinamines and novel polymorphs thereof | |
| JPS5840939B2 (ja) | シクロヘキサンジオン誘導体の製造方法 | |
| JPS58154537A (ja) | オキシ安息香酸低級アルアルキルエステルの製造法 | |
| JP2003137882A (ja) | 2,3−ジメチルチオフェンの製造方法 | |
| JPS591270B2 (ja) | N− ( 1− アダマンチルメチル ) −n’− シンナミルピペラジンノセイホウ | |
| JPH08143538A (ja) | 酸アジド誘導体およびその製造法 | |
| JPS6253947A (ja) | 〔2−(4−アセトキシ−2−イソプロピル−5−メチルフェノキシ)エチル〕−ジメチルアミン塩酸塩の製造方法 | |
| JPS6132312B2 (es) | ||
| JPS59112975A (ja) | 4−置換イソオキサゾ−ル類の製造法 |