ES2703904T3 - Moduladores del receptor de NMDA y usos de los mismos - Google Patents
Moduladores del receptor de NMDA y usos de los mismos Download PDFInfo
- Publication number
- ES2703904T3 ES2703904T3 ES12844456T ES12844456T ES2703904T3 ES 2703904 T3 ES2703904 T3 ES 2703904T3 ES 12844456 T ES12844456 T ES 12844456T ES 12844456 T ES12844456 T ES 12844456T ES 2703904 T3 ES2703904 T3 ES 2703904T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- compound
- stirred
- added
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- DVBUEXCIEIAXPM-PJUQSVSOSA-N C[C@H]([C@@H](C(N(CCC1)[C@@H]1C(N(CCC1)[C@]1(Cc1ccccc1)C(N[C@@H]([C@@H](C)O)C(N)=O)=O)=O)=O)N)O Chemical compound C[C@H]([C@@H](C(N(CCC1)[C@@H]1C(N(CCC1)[C@]1(Cc1ccccc1)C(N[C@@H]([C@@H](C)O)C(N)=O)=O)=O)=O)N)O DVBUEXCIEIAXPM-PJUQSVSOSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1013—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing O or S as heteroatoms, e.g. Cys, Ser
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/397—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having four-membered rings, e.g. azetidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/48—Ergoline derivatives, e.g. lysergic acid, ergotamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/07—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
- C07K5/06165—Dipeptides with the first amino acid being heterocyclic and Pro-amino acid; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0821—Tripeptides with the first amino acid being heterocyclic, e.g. His, Pro, Trp
- C07K5/0823—Tripeptides with the first amino acid being heterocyclic, e.g. His, Pro, Trp and Pro-amino acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Neurology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Zoology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
Compuesto representado por:**Fórmula** en el que: R1 se selecciona del grupo que consiste en hidrógeno, -OH, alquilo C1-C6 y halógeno; R2 y R4 se seleccionan cada uno independientemente del grupo que consiste en alquilo C1-6, hidrógeno, halógeno y -OH; R3 se selecciona del grupo que consiste en -OH, hidrógeno, alquilo C1-C6 y halógeno; R5 es -CH2-fenilo (en el que el fenilo está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados del grupo que consiste en halógeno, alcoxi C1-3 y alquilo C1-3 (opcionalmente sustituido con uno, dos o tres halógenos)); y X se selecciona del grupo que consiste en ORx y NRxRx, en el que RX se selecciona independientemente, para cada caso, del grupo que consiste en hidrógeno y alquilo C1-C6; sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismo.
Description
DESCRIPCIÓN
Moduladores del receptor de NMDA y usos de los mismos
Antecedentes
Un receptor de N-metil-d-aspartato (NMDA) es un receptor postsináptico, ionotrópico que es sensible a, entre otras cosas, los aminoácidos excitadores glutamato y glicina y el compuesto sintético NMDA. El receptor de NMDA controla el flujo de iones tanto divalentes como monovalentes en la célula neuronal postsináptica a través de un canal asociado al receptor (Foster et al., Nature 1987, 329:395-396; Mayer et al., Trends in Pharmacol. Sci. 1990, 11:254-260). El receptor de NMDA se ha implicado durante el desarrollo en la especificación de arquitectura neuronal y conectividad sináptica, y puede estar involucrado en modificaciones sinápticas dependientes de experiencia. Además, también se piensa que los receptores de NMDA están involucrados en trastornos del sistema nervioso central y potenciación a largo plazo.
El receptor de NMDA juega un papel principal en la plasticidad sináptica que subyace a numerosas funciones cognitivas superiores, tales como adquisición de memoria, retención y aprendizaje, así como en determinadas rutas cognitivas y en la percepción de dolor (Collingridge et al., The NMDA Receptor, Oxford University Press, 1994). Además, determinadas propiedades de receptores de NMDA sugieren que estos pueden estar involucrados en el procesamiento de información en el cerebro que subyace a la propia consciencia.
El receptor de NMDA ha atraído un particular interés desde que parece que está involucrado en un amplio espectro de trastornos del SNC. Por ejemplo, durante isquemia cerebral causada por accidente cerebrovascular o lesión traumática, se liberan cantidades excesivas del aminoácido excitador glutamato a partir de neuronas privadas de oxígeno o dañadas. Este exceso de glutamato se une a los receptores de NMDA que abre sus canales iónicos regulados por ligandos; en cambio la entrada de calcio produce un nivel alto de calcio intracelular que activa una cascada bioquímica que da como resultado degradación de proteínas y muerte celular. Este fenómeno, conocido como excitotoxicidad, también se piensa que es responsable del daño neurológico asociado con otros trastornos que oscilan entre hipoglucemia y paro cardíaco y epilepsia. Además, hay informes preliminares que indican participación similar en la neurodegeneración crónica de enfermedades de Huntington, Parkinson y Alzheimer. La activación del receptor de NMDA se ha demostrado que es responsable de convulsiones posteriores a un accidente cerebrovascular, y, en determinados modelos de epilepsia, la activación del receptor de NMDA se ha demostrado que es necesaria para la generación de crisis. La participación neuropsiquiátrica del receptor de NMDA también se ha reconocido ya que el bloqueo del canal de Ca++ receptor de NMDA por el anestésico animal PCP (fenciclidina) produce un estado psicótico en humanos similar a la esquizofrenia (analizado en Johnson, K. y Jones, S., 1990). Además, los receptores de NMDA también se han implicado en determinados tipos de aprendizaje espacial.
El receptor de NMDA se cree que consiste en varias cadenas de proteínas integradas en la membrana postsináptica. Los primeros dos tipos de subunidades descubiertas hasta la fecha forman una región extracelular grande, que probablemente contiene la mayoría de los sitios de unión alostéricos, varias regiones transmembrana enlazadas y plegadas para formar un poro o canal, que es permeable a Ca++ y una región carboxilo terminal. La apertura y cierre del canal está regulada por la unión de diversos ligandos a dominios (sitios alostéricos) de la proteína que reside en la superficie extracelular. La unión de los ligandos se piensa que afecta al cambio conformacional en la estructura global de la proteína que se refleja finalmente en la apertura, apertura parcial, cierre parcial o cierre del canal.
Los compuestos del receptor de NMDA pueden ejercer un efecto dual (agonista/antagonista) en el receptor de NMDA a través de los sitios alostéricos. Estos compuestos se denominan normalmente “agonistas parciales”. En presencia del ligando del sitio principal, un agonista parcial desplazará un poco el ligando y por tanto disminuirá el flujo de Ca++ a través del receptor. En la ausencia de o nivel disminuido del ligando del sitio principal, el agonista parcial actúa para aumentar el flujo de Ca++ a través del canal receptor.
Continúa existiendo una necesidad en la técnica de compuestos nuevos y más específicos/potentes que son capaces de unir el sitio de unión de glicina de receptores de NMDA y proporcionar beneficios farmacéuticos. Además, continúa existiendo una necesidad en la técnica médica de formas de entrega por vía oral de tales compuestos.
Sumario
Se proporcionan en el presente documento, al menos en parte, compuestos que son moduladores de NMDA, por ejemplo, agonistas parciales de NMDA. Por ejemplo, en el presente documento se divulgan compuestos representados por la fórmula:

en la que
R 11, R 12 y R 13 se seleccionan cada uno independientemente del grupo que consiste en H, halógeno, alcoxi C1-3 y alquilo C1-3 (opcionalmente sustituido con uno, dos o tres halógenos);
X1 es OH o NH2,
X2 es H u OH;
y sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismo.
En algunas realizaciones, un halógeno de un compuesto contemplado puede seleccionarse independientemente del grupo que consiste en Cl, Br y F. En algunos casos, X2 en la estructura anterior puede ser OH y X1 en la estructura anterior puede ser NH2.
En algunos casos, cada uno de R , R y R en la estructura anterior pueden ser H. Alternativamente, R y R en la estructura anterior pueden ser H. R12 en la estructura anterior puede seleccionarse del grupo que consiste en H, halógeno, alcoxi C1-3 y alquilo C1-3 (opcionalmente sustituido con uno, dos o tres halógenos). Por ejemplo, R11 y R13 en la estructura anterior pueden ser H y R12 en la estructura anterior puede seleccionarse del grupo que consiste en F, Br, Cl, CH3, CF3 y -OCH3.
En determinadas realizaciones, un compuesto contemplado puede ser sustancialmente más eficaz cuando se administra oralmente comparado con

En otro aspecto, se proporciona un compuesto representado por:

en la que:
R1 se selecciona del grupo que consiste en hidrógeno, -OH, alquilo C1-C6 y halógeno;
R2 y R4 se seleccionan cada uno independientemente del grupo que consiste en alquilo C1-6, hidrógeno, halógeno y -OH;
R3 se selecciona del grupo que consiste en -OH, hidrógeno, alquilo C1-C6 y halógeno;
R5 es -CH2-fenilo (en el que el fenilo está opcionalmente sustituido con uno, dos, o tres sustituyentes seleccionados del grupo que consiste en halógeno, alcoxi C1-3 y alquilo C1-3 (opcionalmente sustituido con uno, dos o tres halógenos)); y
X se selecciona del grupo que consiste en ORx y NRxRx, en el que RX se selecciona independientemente, para cada caso, del grupo que consiste en hidrógeno y alquilo C1-C6; sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismo.
Por ejemplo, un compuesto contemplado puede representarse por:

En otros ejemplos, un compuesto contemplado puede representarse por:

Ċ 

También se proporcionan en el presente documento composiciones farmacéuticamente aceptables que comprenden un compuesto divulgado, y un excipiente farmacéuticamente aceptable. Por ejemplo, tales composiciones pueden ser adecuadas para administración oral a un paciente. En otras realizaciones, tales composiciones pueden ser adecuadas para inyección.
En otro aspecto, se proporciona un compuesto contemplado para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo, que comprende, por ejemplo, administración oral, intravenosa o subcutánea.
Breve descripción de las figuras
La figura 1 muestra un gráfico de la concentración de plasma de CM-4A en función del tiempo tras la administración de la dosis.
La figura 2 muestra gráficos dependientes de la dosis de un ensayo de unión [3H] MK-801 para la evaluación de las propiedades agonísticas de los compuestos contemplados.
La figura 3 muestra gráficos dependientes de la dosis de un ensayo de unión [3H] MK-801 para la evaluación de las propiedades agonísticas de los compuestos contemplados.
La figura 4 muestra un gráfico dependiente de la dosis de un ensayo de unión [3H] MK-801 para la evaluación de las propiedades agonísticas de un compuesto contemplado.
La figura 5 muestra un gráfico que representa la evolución temporal del efecto de CM-4A en corrientes postsinápticas excitadoras de NMDA aislado farmacológicamente provocadas por la vía colateral de Schaffer en un único choque registradas en neuronas piramidales CA1.
La figura 6 muestra un gráfico que representa el efecto de la concentración de CM-4A en la potenciación a largo plazo tras estimulación de alta frecuencia de EPSC de NMDA provocada por la vía colateral de Schaffer registrada en neuronas piramidales CA1.
La figura 7 muestra un gráfico que representa la relación de respuesta a dosis de CM-4A en depresión crónica (LTD).
La figura 8 muestra un gráfico de barras del porcentaje de la potenciación a largo plazo de la pendiente del potencial postsináptico excitador de campo (fEPSP) para los compuestos contemplados.
La figura 9 muestra un gráfico que representa los tiempos de flotación medios de la prueba de Porsolt en función de la concentración de CM-4A.
La figura 10 muestra un gráfico de barras de los tiempos de flotación medios de la prueba de Porsolt para CM-4A y CM-4A en combinación con receptor antagonista AMPA 2,3-dihidroxi-6-nitro-7-sulfamoil-benzo[f]quinoxalina-2,3
diona (NBQX).
Las figuras 11A-11C muestran gráficos de barras que representan los resultados de la prueba de Porsolt para los compuestos contemplados.
La figura 12 muestra un gráfico de barras que representa los resultados de la prueba de Porsolt para los compuestos contemplados después de 1 hora y 24 horas.
La figura 13 muestra un gráfico de barras que representa los resultados de la prueba de hipofagia inducida por novedad (NIH) en ratas para CM-4A.
Las figuras 14A y 14B muestran gráficos de barras que representan los resultados de la prueba de vocalizaciones ultrasónicas (USV) en ratas para CM-4A. La figura 14a cuantifica el aprendizaje emocional positivo, y la figura 14B cuantifica USV hedonistas y aversivas y recompensa.
La figura 15 muestra un gráfico de barras que representa los resultados de la prueba de campo abierto en rata para CM-4A.
La figura 16 muestra un gráfico de barras que representa los resultados de la prueba de cilindro giratorio con aceleración en ratas para CM-4A.
Descripción detallada
Esta divulgación está dirigida generalmente a compuestos que son capaces de modular NMDA, por ejemplo, antagonistas o agonistas parciales de NMDA, y composiciones y/o métodos de uso de los compuestos divulgados. Definiciones
En algunas realizaciones, los compuestos, tal como se describen en el presente documento, pueden sustituirse con cualquier número de sustituyentes o restos funcionales. En general, el término “sustituido” tanto si está precedido por el término “opcionalmente” como si no lo está y sustituyentes contenidos en las fórmulas, se refieren al reemplazo de radicales hidrógeno en una estructura dada con el radical de un sustituyente especificado.
En algunos casos, cuando más de una posición en cualquier estructura dada puede sustituirse con más de un sustituyente seleccionado de un grupo especificado, el sustituyente puede ser o bien el mismo o bien diferente en cada posición.
Tal como se usa en el presente documento, el término “sustituido” se considera que incluye todos los sustituyentes admisibles de compuestos orgánicos. En un aspecto amplio, los sustituyentes admisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y no ramificados, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. En algunas realizaciones, heteroátomos tales como nitrógeno pueden tener sustituyentes hidrógeno y/o cualesquiera sustituyentes admisibles de compuestos orgánicos descritos en el presente documento que satisfagan las valencias de los heteroátomos. Los ejemplos de sustituyentes incluyen acilo; alifático; heteroalifático; arilo; heteroarilo; arilalquilo; heteroarilalquilo; alcoxi; cicloalcoxi; heterociclilalcoxi; heterocicliloxi; heterocicliloxialquilo; alqueniloxi; alquiniloxi; ariloxi; heteroalcoxi; heteroariloxi; alquiltio; ariltio; heteroariltio; oxo; -F;-Cl; -Br; -I; -OH; -NO2; -N3; -CN; -SCN; -SRx; -CF3; -CH2CF3; -CHCh; -CH2OH;-CH2CH2OH; -CH2NH2; -CH2SO2CH3; -ORx, -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2;-C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2 y -C(Rx)3; en los en que en cada caso Rx incluye independientemente hidrógeno, halógeno, acilo, alifático, heteroalifático, arilo, heteroarilo, arilalquilo o heteroarilalquilo, en el que cualquiera de los sustituyentes alifático, heteroalifático, arilalquilo, o heteroarilalquilo descritos anteriormente y en el presente documento pueden ser sustituidos o no sustituidos, ramificados o no ramificados, cíclicos o acíclicos, y en el que cualquiera de los sustituyentes arilo o heteroarilo descritos anteriormente y en el presente documento pueden ser sustituidos o no sustituidos. En algunas realizaciones, las combinaciones de sustituyentes y variables descritos en el presente documento pueden ser preferiblemente aquellas que dan como resultado la formación de compuestos estables. El término “estable”, tal como se usa en el presente documento, se refiere a compuestos que poseen suficiente estabilidad para permitir la fabricación y que mantienen la integridad del compuesto durante un periodo de tiempo suficiente para ser detectado y preferiblemente durante un periodo de tiempo suficiente para ser útil para los propósitos detallado en el presente documento.
El término “alifático”, tal como se usa en el presente documento, incluye tanto hidrocarburos saturados e insaturados, de cadena lineal (es decir, no ramificados), ramificados, acíclicos, cíclicos, o alifáticos policíclicos, que están opcionalmente sustituidos con uno o más grupos funcionales. Como se apreciará por un experto habitual en la técnica, “alifático” se desea en el presente documento para incluir alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, y restos de cicloalquinilo.
Los términos “arilo” y “heteroarilo”, tal como se usan en el presente documento, se refieren a restos insaturados
mono- o policíclicos que tienen preferiblemente de 3-14 átomos de carbono, cada uno de los cuales puede estar sustituido o no sustituido. En determinadas realizaciones, “arilo” se refiere a un sistema de anillo carbocíclico monoo bicíclico que tiene uno o dos anillos aromáticos incluyendo fenilo, naftilo, tetrahidronaftilo, indanilo e indenilo. En determinadas realizaciones, “heteroarilo” se refiere a un sistema de anillo heterocíclico mono- o bicíclico que tiene uno o dos anillos aromáticos en el que uno, dos, o tres átomos del anillo son heteroátomos independientemente seleccionados del grupo que consiste en S, O y N y los átomos del anillo restantes son carbono. Los ejemplos de grupos heteroarilo incluyen piridilo, pirazinilo, pirimidinilo, pirrolilo, pirazolilo, imidazolilo, tiazolilo, oxazolilo, isooxazolilo, tiadiazolilo, oxadiazolilo, tiofenilo, furanilo, quinolinilo e isoquinolinilo.
El término “alquenilo” tal como se usa en el presente documento se refiere a un hidrocarburo lineal o ramificado insaturado que tiene al menos un doble enlace carbono-carbono, tal como un grupo lineal o ramificado de 2-12, 2-10 o 2-6 átomos de carbono, denominados en el presente documento como alquenilo C2-C12, alquenilo C2-C10 y alquenilo C2-C6, respectivamente. A modo de ejemplo los grupos alquenilo incluyen vinilo, alilo, butenilo, pentenilo, hexenilo, butadienilo, pentadienilo, hexadienilo, 2-etilhexenilo, 2-propil-2-butenilo, 4-(2-metil-3-buteno)-pentenilo, etc. El término “alcoxi” tal como se usa en el presente documento se refiere a un grupo alquilo unido a un oxígeno (-O alquil). A modo de ejemplo los grupos alcoxi incluyen grupos con un grupo alquilo de 1-12, 1-8 o 1-6 átomos de carbono, denominados en el presente documento como alcoxi C1-C12, alcoxi C-i-Ca y alcoxi C1-C6, respectivamente. A modo de ejemplo los grupos alcoxi incluyen metoxi, etoxi, etc. De manera similar, a modo de ejemplo los grupos “alquenoxi” incluyen viniloxi, aliloxi, butenoxi, etc.
El término “alcoxicarbonilo” tal como se usa en el presente documento se refiere a un grupo alquilo lineal o ramificado unido a oxígeno, unido a un grupo carbonilo (alquil-O-C(O)-). A modo de ejemplo los grupos alcoxicarbonilo incluyen grupos alcoxicarbonilo de 1-6 átomos de carbono, denominados en el presente documento como alcoxicarbonilo C1-6. A modo de ejemplo los grupos alcoxicarbonilo incluyen metoxicarbonilo, etoxicarbonilo, tbutoxicarbonilo, etc.
El término “alquiniloxi” usado en el presente documento se refiere a un grupo alquinilo lineal o ramificado unido a un oxígeno (alquinil-O)). A modo de ejemplo los grupos alquiniloxi incluyen propiniloxi.
El término “alquilo” tal como se usa en el presente documento se refiere a un hidrocarburo lineal o ramificado saturado, por ejemplo, tal como un grupo lineal o ramificado de 1-6, 1-4 o 1-3 átomos de carbono, denominados en el presente documento como alquilo C1-C6, alquilo C1-C4 y alquilo C1-C3, respectivamente. A modo de ejemplo los grupos alquilo incluyen metilo, etilo, propilo, isopropilo, 2-metil-1 -propilo, 2-metil-2-propilo, 2-metil-1 -butilo, 3-metil-1-butilo, 2-metil-3-butilo, 2,2-dimetil-1-propilo, 2-metil-1 -pentilo, 3-metil-1 -pentilo, 4-metil-1 -pentilo, 2-metil-2-pentilo, 3-metil-2-pentilo, 4-metil-2-pentilo, 2,2-dimetil-1 -butilo, 3,3-dimetil-1 -butilo, 2-etil-1 -butilo, butilo, isobutilo, t-butilo, pentilo, isopentilo, neopentilo, hexilo, heptilo, octilo, etc.
El término “alquilcarbonilo” tal como se usa en el presente documento se refiere a un grupo alquilo lineal o ramificado unido a un grupo carbonilo (alquil-C(O)-). A modo de ejemplo los grupos alquilcarbonilo incluyen grupos alquilcarbonilo de 1-6 átomos, denominados en el presente documento como grupos alquilcarbonilo C1-C6. A modo de ejemplo los grupos alquilcarbonilo incluyen acetilo, propanoilo, isopropanoilo, butanoilo, etc.
El término “alquinilo” tal como se usa en el presente documento se refiere a un hidrocarburo lineal o ramificado insaturado que tiene al menos un triple enlace carbono-carbono, tal como un grupo lineal o ramificado de 2-6 o 3-6 átomos de carbono, denominados en el presente documento como alquinilo C2-6 y alquinilo C3-6, respectivamente. A modo de ejemplo los grupos alquinilo incluyen etinilo, propinilo, butinilo, pentinilo, hexinilo, metilpropinilo, etc.
Los grupos alquilo, alquenilo y alquinilo pueden estar sustituidos opcionalmente, si no se indica lo contrario, con uno o más grupos seleccionados de alcoxilo, alquilo, cicloalquilo, amino, halógeno y -C(O)alquilo. En determinadas realizaciones, los grupos alquilo, alquenilo y alquinilo no están sustituidos, es decir, son no sustituidos.
El término “amida” o “amido” tal como se usa en el presente documento se refiere a un radical de la forma -RaC(O)N(Rb)-, -RaC(O)N(Rb)Rc-, o -C(O)NRbRc, en el que Ra, Rb y Rc se seleccionan cada uno independientemente de alcoxilo, alquilo, alquenilo, alquinilo, amida, amino, arilo, arilalquilo, carbamato, cicloalquilo, éster, éter, formilo, halógeno, haloalquilo, heteroarilo, heterociclilo, hidrógeno, hidroxilo, cetona, y nitro. La amida puede unirse a otro grupo a través del carbono, del nitrógeno, Rb, Rc, o Ra. La amida también puede ser cíclica, por ejemplo Rb y Rc, Ra y Rb, o Ra y Rc pueden unirse para formar un anillo de 3 a 12 miembros, tal como un anillo de 3 a 10 miembros o un anillo de 5 a 6 miembros. El término “carboxamido” se refiere a la estructura -C(O)NRbRc.
El término “amina” o “amino” tal como se usa en el presente documento se refiere a un radical de la forma -NRdRe, en el que Rd y Re se seleccionan independientemente de hidrógeno, alquilo, alquenilo, alquinilo, arilo, arilalquilo, cicloalquilo, haloalquilo, heteroarilo, y heterociclilo. El amino también puede ser cíclico, por ejemplo, Rd y Re se unen juntos con el N para formar un anillo de 3 a 12 miembros, por ejemplo, morfolino o piperidinilo. El término amino también incluye la correspondiente sal de amonio cuaternario de cualquier grupo amino, por ejemplo, -[N(Rd)(Re)(Rf)]+. A modo de ejemplo los grupos amino incluyen grupos aminoalquilo, en los que al menos uno de Rd,
Re, o Rf es un grupo alquilo. En determinada realización, Rd y Re son hidrógeno o alquilo.
El término “cidoalquilo” tal como se usa en el presente documento se refiere a un grupo hidrocarburo saturado o parcialmente insaturado monocíclico de por ejemplo 3-6 o 4-6 carbonos, denominado en el presente documento, por ejemplo, cicloalquilo C3-6 o cicloalquilo C4-6 y derivado de un cicloalcano. A modo de ejemplo los grupos cicloalquilo incluyen ciclohexilo, ciclohexenilo, ciclopentilo, ciclobutilo o ciclopropilo.
Los términos “halo” o “halógeno” o “Hal” tal como se usan en el presente documento se refiere a F, Cl, Br o I. El término “haloalquilo” tal como se usa en el presente documento se refiere a un grupo alquilo sustituido con uno o más átomos de halógeno.
Los términos “heterociclilo” o “grupo heterocíclico” se reconocen en la técnica y se refieren a estructuras de anillo de 3 a 10 miembros saturadas o parcialmente insaturadas, alternativamente anillos de 3 a 7 miembros, cuyas estructuras de anillo incluyen de uno a cuatro heteroátomos, tales como nitrógeno, oxígeno, y azufre. Los heterociclos también pueden ser sistemas de anillos mono, bi, u otros multicíclicos. Un heterociclo puede estar condensado a uno o más anillos saturados, parcialmente insaturados o de arilo. Los grupos heterociclilo incluyen, por ejemplo, biotinilo, cromenilo, dihidrofurilo, dihidroindolilo, dihidropiranilo, dihidrotienilo, ditiazolilo, homopiperidinilo, imidazolidinilo, isoquinolilo, isotiazolidinilo, isoxazolidinilo, morfolinilo, oxolanilo, oxazolidinilo, fenoxantenilo, piperazinilo, piperidinilo, piranilo, pirazolidinilo, pirazolinilo, piridilo, pirimidinilo, pirrolidinilo, pirrolidin-2-onilo, pirrolinilo, tetrahidrofurilo, tetrahidroisoquinolilo, tetrahidropiranilo, tetrahidroquinolilo, tiazolidinilo, tiolanilo, tiomorfolinilo, tiopiranilo, xantenilo, lactonas, lactamas tales como azetidinonas y pirrolidinonas, sultamas y sultonas. El anillo heterocíclico puede estar sustituido en una o más posiciones con sustituyentes tales como alcanoílo, alcoxilo, alquilo, alquenilo, alquinilo, amido, amidino, amino, arilo, arilalquilo, azido, carbamato, carbonato, carboxilo, ciano, cicloalquilo, éster, éter, formilo, halógeno, haloalquilo, heteroarilo, heterociclilo, hidroxilo, imino, cetona, nitro, fosfato, fosfonato, fosfinato, sulfato, sulfuro, sulfonamido, sulfonilo y tiocarbonilo. En determinadas realizaciones, el grupo heterocíclico no está sustituido, es decir, el grupo heterocíclico está no sustituido.
El término “heterocicliloalcoxi” tal como se usa en el presente documento se refiere a un grupo heterociclil-alquil-O-. El término “heterocicliloxi” se refiere a un grupo heterociclil-O-.
El término “heterocicliloxialquilo” se refiere a un grupo heterociclil-O-alquil-.
Los términos “hidroxi” e “hidroxilo” tal como se usa en el presente documento se refieren al radical -OH.
El término “oxo” tal como se usa en el presente documento se refiere al radical =O.
“Farmacéuticamente o farmacológicamente aceptable” incluye entidades moleculares y composiciones que no producen una reacción adversa, alérgica u otra reacción desfavorable cuando se administran a un animal, o un humano, según sea apropiado. “Para administración a humanos, las preparaciones deben satisfacer las normas de esterilidad, pirogenicidad, seguridad general y pureza con arreglo a la oficina de normas biológicas de la FDA.
Tal como se usa en la presente divulgación, el término “agonista del receptor de NMDA parcial” se define como un compuesto que es capaz de unirse a un sitio de unión a glicina de un receptor de NMDA; a bajas concentraciones un agonista del receptor de NMDA actúa sustancialmente como agonista y a altas concentraciones actúa sustancialmente como un antagonista. Estas concentraciones se determinan de manera experimental para cada “agonista parcial.
Tal como se usa en el presente documento “portador farmacéuticamente aceptable” o “excipiente” incluyen todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y que retardan la absorción que son fisiológicamente compatibles. En una realización, el portador es adecuado para administración parenteral. Alternativamente, el portador puede ser adecuado para administración intravenosa, intraperitoneal, intramuscular, sublingual u oral. Portadores farmacéuticamente aceptables incluyen dispersiones o disoluciones acuosas estériles y polvos estériles para la preparación extemporánea de dispersión o disoluciones inyectables estériles. El uso de tales medios y agentes para sustancias farmacéuticamente activas es bien conocido en la técnica. Salvo que cualquiera de los medios convencionales o agentes sean incompatibles con el compuesto activo, se contempla el uso de los mismos en las composiciones farmacéuticas de la invención. También pueden incorporarse compuestos activos adicionales en las composiciones.
El término “sal(es) farmacéuticamente aceptable(s)” tal como se usa en el presente documento se refiere a sales de grupos ácidos o básicos que pueden estar presentes en compuestos usados en las presentes composiciones. Compuestos incluidos en las presentes composiciones que son de naturaleza básica son capaces de formar una amplia variedad de sales con diversos ácidos inorgánicos y orgánicos. Los ácidos que pueden usarse para preparar sales de adición de ácidos farmacéuticamente aceptables de tales compuestos básicos son aquellos que forman sales de adición de ácidos no tóxicas, es decir, sales que contienen aniones farmacológicamente aceptables, incluyendo sales de malato, oxalato, cloruro, bromuro, yoduro, nitrato, sulfato, bisulfato, fosfato, fosfato ácido,
isonicotinato, acetato, lactato, salicilato, citrato, tartrato, oleato, tanato, pantotenato, bitartrato, ascorbato, succinato, maleato, gentisinato, fumarato, gluconato, glucaronato, sacarato, formiato, benzoato, glutamato, metanosulfonato, etanosulfonato, bencenosulfonato, p-toluenosulfonato y pamoato (es decir, 1,1'-metilen-bis-(2-hidroxi-3-naftoato)). Compuestos incluidos en las presentes composiciones que incluyen un resto amino pueden formar sales farmacéuticamente aceptables con diversos aminoácidos, además de los ácidos mencionados anteriormente. Compuestos incluidos en las presentes composiciones que son de naturaleza ácida son capaces de formar sales de bases con diversos cationes farmacológicamente aceptables. Los ejemplos de tales sales incluyen sales de metales alcalinos o alcalinotérreos y, particularmente, sales de calcio, magnesio, sodio, litio, zinc, potasio y hierro.
Los compuestos de la divulgación pueden contener uno o más centros quirales y/o dobles enlaces y, por tanto, existir como estereoisómeros, tales como isómeros geométricos, enantiómeros o diastereómeros. El término “estereoisómeros” cuando se usa en el presente documento consiste en todos los isómeros geométricos, enantiómeros o diastereómeros. Estos compuestos pueden designarse por los símbolos “R” o “S”, dependiendo de la configuración de sustituyentes alrededor del átomo de carbono estereogénico. La presente invención abarca diversos estereoisómeros de estos compuestos y mezclas de los mismos. Los estereoisómeros incluyen enantiómeros y diastereómeros. Las mezclas de enantiómeros o diastereómeros pueden designarse “(+)” en la nomenclatura, pero el experto en la técnica reconocerá que una estructura puede denotar un centro quiral de manera implícita.
Estereoisómeros individuales de compuestos de la presente invención pueden prepararse sintéticamente a partir de materiales de partida comercialmente disponibles que contienen centros asimétricos o estereogénicos, o mediante preparación de mezclas racémicas seguida por métodos de resolución bien conocidos por los expertos habituales en la técnica. Estos métodos de resolución se ejemplifican por (1) unión de una mezcla de enantiómeros a un auxiliar quiral, separación de la mezcla resultante de diastereómeros mediante recristalización o cromatografía y liberación del producto ópticamente puro del auxiliar, (2) formación de sales empleando un agente de resolución ópticamente activo, o (3) separación directa de la mezcla de enantiómeros ópticos en columnas cromatográficas quirales. Las mezclas estereoisoméricas también pueden resolverse en sus estereoisómeros componentes mediante métodos bien conocidos, tales como cromatografía de gases de fase quiral, cromatografía de gases de alta resolución de fase quiral, cristalizar el compuesto como un complejo de sal quiral, o cristalizar el compuesto en un disolvente quiral. Los estereoisómeros también pueden obtenerse a partir de productos intermedios, reactivos y catalizadores estereoméricamente puros, mediante métodos sintéticos asimétricos bien conocidos.
También pueden existir isómeros geométricos en los compuestos de la presente invención. El símbolo = denota un enlace que puede ser un enlace simple, doble o triple tal como se describe en el presente documento. La presente invención abarca los diversos isómeros geométricos y mezclas de los mismos que resultan de la disposición de sustituyentes alrededor de un doble enlace carbono-carbono o la disposición de sustituyentes alrededor de un anillo carbocíclico. Los sustituyentes alrededor de un doble enlace carbono-carbono se designan como configuración “Z” o “E” en la que los términos “Z” y “E” se usan según las normas de la IUPAC. A menos que se especifique lo contrario, las estructuras que representan dobles enlaces abarcan ambos isómeros “E” y “Z”.
Los sustituyentes alrededor de un doble enlace carbono-carbono pueden denominarse alternativamente como “cis” o “trans”, donde “cis” representa sustituyentes en el mismo lado del doble enlace y “trans” representa sustituyentes en lados opuestos del doble enlace. La disposición de sustituyentes alrededor de un anillo carbocíclico se designa como “cis” o “trans”. El término “cis” representa sustituyentes en el mismo lado del plano del anillo y el término “trans” representa sustituyentes en lados opuestos del plano del anillo. Las mezclas de compuestos en las que los sustituyentes se disponen tanto en el mismo lado como en lados opuestos del plano del anillo se designan “cis/trans”.
Los compuestos divulgados en el presente documento pueden existir en formas solvatadas así como en formas no solvatadas con disolventes farmacéuticamente aceptables tales como agua y etanol, y se desea que la invención comprenda tanto formas solvatadas como solvatadas. En una realización, el compuesto es amorfo. En una realización, el compuesto es un polimorfo. En otra realización, el compuesto es una forma cristalina.
La invención también comprende compuestos marcados isotópicamente de la invención que son idénticos a aquellos mencionados en el presente documento, excepto que uno o más átomos están sustituidos con un átomo que tiene una masa atómica o número atómico diferente de la masa atómica o número atómico encontrado habitualmente en la naturaleza. Los ejemplos de isotopos que pueden incorporarse en compuestos de la invención incluyen isotopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, tales como 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente.
Determinados compuestos divulgados marcados isotópicamente (por ejemplo, aquellos marcados con 3H y 14C) son útiles en ensayos de distribución en tejidos de compuesto y/o sustrato. Se prefieren particularmente isotopos tritio (es decir, 3H) y carbono-14 (es decir, 14C) por su facilidad de preparación y detectabilidad. Además, la sustitución con isotopos más pesados tales como deuterio (es decir, 2H) pueden proporcionar determinadas ventajas terapéuticas que dan como resultado una mayor estabilidad metabólica (por ejemplo, semivida in vivo aumentada o requisitos de dosificación reducidos) y por tanto pueden preferirse en algunas circunstancias. Los compuestos
marcados isotópicamente de la invención pueden prepararse generalmente siguiendo procedimientos análogos a aquellos divulgados en los por ejemplo, ejemplos en el presente documento, sustituyendo un reactivo no marcado isotópicamente por un reactivo marcado isotópicamente.
Tal como se usa en la presente divulgación, “NMDA” se define como N-metil-d-aspartato.
En la presente memoria descriptiva, el término “cantidad terapéuticamente eficaz” significa la cantidad del compuesto objeto que provocará la respuesta biológica o médica de un tejido, sistema, animal o humano que es buscado por el investigador, veterinario, médico u otro profesional clínico. Los compuestos de la invención se administran en cantidades terapéuticamente eficaces para tratar una enfermedad. Alternativamente, una cantidad terapéuticamente eficaz de un compuesto es la cantidad necesaria para lograr un efecto terapéutico y/o profiláctico deseado, tal como una cantidad que da como resultado definida como aquella cantidad necesaria para dar la máxima mejora de un comportamiento (por ejemplo, aprendizaje), respuesta fisiológica (por ejemplo, inducción de LTP) o inhibición de dolor neuropático.
Compuestos
Los compuestos de referencia divulgados incluyen aquellos representados por la fórmula:

y sales farmacéuticamente aceptables, estereoisómeros, metabolitos e hidratos de los mismos,
en la que:
R1, R2, R3 y R4 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; grupo alifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; grupo heteroalifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; arilo sustituido o no sustituido; heteroarilo sustituido o no sustituido; -ORx; -NO2; -N3; -CN; -SCN; -SRX; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2;-NRxC(O)ORx; -NRxC(NRx)N(Rx)2; y -C(Rx)3; en los en que en cada caso Rx se selecciona independientemente del grupo que consiste en hidrógeno; halógeno; acilo; grupo alifático opcionalmente sustituido; grupo heteroalifático opcionalmente sustituido; arilo opcionalmente sustituido; y heteroarilo opcionalmente sustituido;
R5 y R6 pueden seleccionarse independientemente del grupo que consiste en -Q-Ar e hidrógeno, siempre y cuando al menos uno de R5 y R6 sea -Q-Ar; en el que Q se selecciona independientemente del grupo que consiste en grupo alifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; grupo heteroalifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; y un enlace; y en el que Ar se selecciona del grupo que consiste en arilo sustituido o no sustituido, y heteroarilo sustituido o no sustituido; o R5 y R6, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros sustituido o no sustituido;
R7 y R8 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; hidroxilo; alquilo C1-C6 sustituido o no sustituido; alcoxilo C1-C6 sustituido o no sustituido; y arilo sustituido o no sustituido; o R7 y R8, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros sustituido o no sustituido;
R9 y R10 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; alquilo C1-C6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo; alquenilo C2-6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo; alquinilo C2-6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo; cicloalquilo C3-6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en alquilo C1-6, halógeno, oxo e hidroxilo; fenilo, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en alquilo C1-6; alcoxilo C1-6; halógeno; hidroxilo; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; y -C(Rx)3;
en los en que en cada caso Rx se selecciona independientemente del grupo que consiste en hidrógeno; halógeno; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; y fenilo; o R9 y R10, junto con N, heterocíclico de 4-6 miembros, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en alquilo C1-6, halógeno, oxo e hidroxilo.
En algunas realizaciones, R1, R2, R3 y R4 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; fenilo; naftilo; heteroarilo; heterociclilo; cicloalquil C3-6-alquil C1-6-; fenil-alquil C1-6-; naftil-alquil C1-6-; heteroaril-alquil C1-6-; y heterociclil-alquil
C1-6-; -ORx; -NO2; -N3; -CN;-SCN; -SRX; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx;-NRxC(NRx)N(Rx)2; y -C(Rx)3;
en el que heteroarilo es un anillo de 5-6 miembros que tiene uno, dos, o tres heteroátomos cada uno independientemente seleccionado de N, O o S; en el que heteroarilo está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rb; en el que heterociclilo es un anillo de 4-7 miembros opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rc; en el que cuando el heterociclilo contiene un resto -NH-, este resto -NH- está opcionalmente sustituido con Rd; en el que alquenilo C2-6 y alquinilo C2-6, están cada uno independientemente opcionalmente sustituidos con uno o más sustituyentes cada uno independientemente seleccionado de Re; en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rf; en el que cicloalquilo C3-6 está independientemente opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rg;
Rb puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; alcoxi C1-6; alqueniloxi C3-6; alquiniloxi C3-6; cicloalcoxi C3-6; alquil C1-6-S(O)w-, en el que w es 0, 1 o 2; alquil C1-6-cicloalquil C3-6-; cicloalquil C3-6-alquil C1-6-; alcoxicarbonil C1-6-N(Ra)-; alquil C1-6-N(Ra)-; alquil C1-6-N(Ra)carbonil-; RaRaN-; RaRaN-carbonil-; RaRaN-carbonil-N(Ra)-; RaRaN-SO2-; y alquil C1-6-carbonil-N(R)-;
Ra y Ra pueden seleccionarse, independientemente para cada caso, del grupo que consiste en hidrógeno y alquilo
C1-6, o Ra y Ra cuando se toman junto con el nitrógeno al que están unidos forman un anillo heterocíclico de 4-6 miembros, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo, y en el que el anillo heterocíclico está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, alquilo, oxo o hidroxilo;
Rc puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; oxo; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; alcoxi C1-6; alqueniloxi C3-6; alquiniloxi
C3-6; cicloalcoxi C3-6; alquil C1-6-S(O)w-, en el que w es 0, 1 o 2; alquil C1-6-cicloalquil C3-6-; cicloalquil C3-6-alquil C1.6-; alcoxicarbonil C1-6-N(Ra)-; alquil C1-6-N(Ra)-; alquil C1-6-N(Ra)carbonil-; RaRaN-; RaRaN-carbonil-; RaRaN-carbonil-N(Ra)-; RaRa'N-SO2-; y alquil C1-6-carbonil-N(Ra)-;
Rd puede seleccionarse, independientemente para cada caso, del grupo que consiste en alquilo C1-6, alquilcarbonilo
C1-6, y alquilsulfonilo C1.6, en el que alquilo C1.6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de halógeno, hidroxilo, y RaRaN-;
Re puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2;
Rf puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2;
Rg puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno, hidroxilo, -NO2; -N3; -CN; -SCN; alquilo C1-6; alcoxi C1.4; alcoxicarbonilo C1.4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2;
Rx puede seleccionarse, independientemente, del grupo que consiste en hidrógeno; halógeno; alquilo C1-6; alquenilo
C2-6; alquinilo C2-6; cicloalquilo C3-6; fenilo; naftilo; heteroarilo; heterociclilo; cicloalquil C3-6- alquil C1.6-; fenil-alquil C1.
6-; naftil-alquil C1.6-; heteroaril-alquil C1-6-; y heterociclil-alquil C1.6-; en el que heteroarilo es un anillo de 5-6 miembros que tiene uno, dos o tres heteroátomos cada uno independientemente seleccionado de N, O o S; en el que heteroarilo está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rb; en el que heterociclilo es un anillo de 4-7 miembros opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rc; en el que cuando heterociclilo contiene un resto -NH-, este resto
-NH- está opcionalmente sustituido con Rd; en el que alquenilo C2-6 y alquinilo C2-6, están cada uno independientemente opcionalmente sustituidos con uno o más sustituyentes cada uno independientemente seleccionado de Re; en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno
independientemente seleccionado de Rf; en el que cicloalquilo C3-6 está independientemente opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rg.
En determinadas realizaciones, al menos uno de R1, R2, R3 y R4 puede ser hidroxilo.
En algunos casos, al menos uno de R1, R2, R3 y R4 puede ser alquilo C1-C6, opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente del grupo que consiste en halógeno, hidroxilo, -NH2 y ciano. En algunas realizaciones, al menos uno de R5 y R6 puede ser -(alquileno C1-C6)-Ar. Al menos uno de R5 y R6 puede ser también -CH2-Ar. En algunos casos, al menos uno de R5 y R6 es -Q-fenil. En determinados ejemplos, uno de R5 y R6 puede ser hidrógeno.
En algunos casos, R7 y R8 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; hidroxilo; alquilo C1-C6; fenilo y naftilo; o R7 y R8, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros; en el que alquilo C1-C6, fenilo, naftilo, el anillo cicloalquilo y el anillo heterocíclico pueden estar cada uno independientemente sustituido por uno o más sustituyentes seleccionados del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2; en el que Ra y Ra pueden seleccionarse, independientemente para cada caso, del grupo que consiste en hidrógeno y alquilo C1-6, o Ra y Ra cuando se toman junto con el nitrógeno al que están unidos forman un anillo heterocíclico de 4-6 miembros, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo, y en el que el anillo heterocíclico está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, alquilo, oxo o hidroxilo.
En algunos casos, R7 y R8 pueden ser hidrógeno.
En una realización a modo de ejemplo, un compuesto puede representarse por:

En otra realización a modo de ejemplo, un compuesto puede representarse por:

En aún otra realización a modo de ejemplo, un compuesto puede representarse por:
Ċ 
En todavía otra realización a modo de ejemplo, un compuesto puede representarse por:


Los compuestos de referencia divulgados también incluyen aquellos representados por la fórmula:

y sales farmacéuticamente aceptables, estereoisómeros, metabolitos e hidratos del mismo, en la que:
R1 y R3 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; grupo alifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; grupo heteroalifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; arilo sustituido o no sustituido; heteroarilo sustituido o no sustituido; -ORx; -NO2; -N3; -CN; -SCN; -SRx; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx;-NRxC(NRx)N(Rx)2; y -C(Rx)3;
en el que en cada caso Rx se selecciona independientemente del grupo que consiste en hidrógeno; halógeno; acilo; grupo alifático opcionalmente sustituido; grupo heteroalifático opcionalmente sustituido; arilo opcionalmente sustituido; y heteroarilo opcionalmente sustituido;
R2 y R4 pueden seleccionarse independientemente del grupo que consiste en hidrógeno y - ORx, siempre que al menos uno de R2 y R4 sea hidrógeno, en el que Rx se selecciona del grupo que consiste en hidrógeno; halógeno; acilo; grupo alifático opcionalmente sustituido; grupo heteroalifático opcionalmente sustituido; arilo opcionalmente sustituido; y heteroarilo opcionalmente sustituido;
R5 y R6 pueden seleccionarse independientemente del grupo que consiste en -Q-Ar e hidrógeno; en el que Q se selecciona independientemente del grupo que consiste en grupo alifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; grupo heteroalifático cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; y un enlace; y en el que Ar se selecciona del grupo que consiste en arilo sustituido o no sustituido, y heteroarilo sustituido o no sustituido; o R5 y R6, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros sustituido o no sustituido;
R7 y R8 se seleccionan independientemente del grupo que consiste en hidrógeno; halógeno; hidroxilo; alquilo C1-C6 sustituido o no sustituido; alcoxi C1-C6 sustituido o no sustituido; y arilo sustituido o no sustituido; o R7 y R8, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros sustituido o no sustituido;
R9 y R10 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; alquilo C1-C6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo; alquenilo C2-6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo; alquinilo C2-6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo; cicloalquilo C3-6, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en alquilo C1-6, halógeno, oxo e hidroxilo; fenilo, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en alquilo C1-6; alcoxi C1-6; halógeno; hidroxilo; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; y -C(Rx)3; en el que en cada caso Rx se selecciona independientemente del grupo que consiste en hidrógeno; halógeno; alquilo
C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; y fenilo; o R9 y R10, junto con N, forman un anillo heterocíclico de
4-6 miembros, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en alquilo C1-6, halógeno, oxo e hidroxilo.
En algunas realizaciones, R1 y R3 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; fenilo; naftilo; heteroarilo; heterociclilo; cicloalquil C3-6-alquil C1-6-; fenil-alquil C1-6-; naftil-alquil C1-6-; heteroaril-alquil C1-6-; y heterociclil-alquil C1-6-; -ORx; -NO2; -N3; -CN; -SCN; -SRX; -C(O)Rx; -CO2(Rx); -C(O)N(Rx)2; -C(NRx)N(Rx)2; -OC(O)Rx; -OCO2Rx; -OC(O)N(Rx)2; -N(Rx)2; -SORx; -S(O)2Rx; -NRxC(O)Rx; -NRxC(O)N(Rx)2; -NRxC(O)ORx; -NRxC(NRx)N(Rx)2; y -C(Rx)3; en el que heteroarilo es un anillo de 5-6 miembros que tiene uno, dos o tres heteroátomos cada uno independientemente seleccionado de N,
O o S; en el que heteroarilo está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rb; en el que heterociclilo es un anillo de 4-7 miembros opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rc; en el que cuando el heterociclilo contiene un resto -NH-, ese resto -NH- está opcionalmente sustituido por Rd; en el que alquenilo C2-6 y alquinilo C2-6, están cada uno independientemente opcionalmente sustituidos con uno o más sustituyentes cada uno independientemente seleccionado de Re; en el que alquilo C1-6 está opcionalmente sustituido con uno o más
sustituyentes cada uno independientemente seleccionado de Rf; en el que cicloalquilo C3-6 está independientemente opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rg;
Rb puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; alcoxi C1-6; alqueniloxi C3-6; alquiniloxi C3-6; cicloalcoxi C3-6; alquil C1-6-S(O)w-, en el que w es 0, 1 o 2; alquil C1-6-cicloalquil C3-6-; cicloalquil C3-6-alquil C1.6-; alcoxicarbonil C1-6-N(Ra)-; alquil C1-6-N(Ra)-; alquil C1-6-N(Ra)carbonil-; RaRaN-; RaRaN-carbonil-; RaRaN-carbonil-N(Ra)-; RaRaN-SO2-; y alquil C1-6-carbonil-N(Ra)-;
Ra y Ra pueden seleccionarse, independientemente para cada caso, del grupo que consiste en hidrógeno y alquilo
C1-6, o Ra y Ra cuando se toman junto con el nitrógeno al que están unidos forman un anillo heterocíclico de 4-6 miembros, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo, y en el que el anillo heterocíclico está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, alquilo, oxo o hidroxilo;
Rc puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; oxo; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; alcoxi C1-6; alqueniloxi C3-6; alquiniloxi
C3-6; cicloalcoxi C3-6; alquil C1-6-S(O)w-, en el que w es 0, 1 o 2; alquil C1-6-cicloalquil C3-6-; cicloalquil C3-6-alquil C1.6-; alcoxicarbonil C1-6-N(Ra)-; alquil C1-6-N(Ra)-; alquil C1-6-N(Ra)carbonil-; RaRaN-; RaRaN-carbonil-; RaRaN-carbonil-N(Ra)-; RaRa'N-SO2-; y alquil C1-6-carbonil-N(Ra)-;
Rd puede seleccionarse, independientemente para cada caso, del grupo que consiste en alquilo C1-6, alquilcarbonilo
C1-6, y alquilsulfonilo C1.6, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de halógeno, hidroxilo, y RaRaN-;
Re puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquilC1-4S(O)w-, en el que w es 0, 1 o 2;
Rf puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2;
Rg puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno, hidroxilo, -NO2; -N3; -CN; -SCN; alquilo C1-6; alcoxi C1.4; alcoxicarbonilo C1.4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y al 4S(O)w-, en el que w es 0, 1 o 2;
Rx puede seleccionarse, independientemente, del grupo que consiste en hidrógeno; halógeno; alquilo C1-6; alquenilo
C2-6; alquinilo C2-6; cicloalquilo C3-6; fenilo; naftilo; heteroarilo; heterociclilo; cicloalquil C3-6-alquil C1-6-; fenil-alquil C1-6-; naftil-alquil C1.6-; heteroaril-alquil C1.6-; y heterociclil-alquil C1-6-; en el que heteroarilo es un anillo de 5-6 miembros que tiene uno, dos o tres heteroátomos cada uno independientemente seleccionado de N, O o S; en el que heteroarilo está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rb; en el que heterociclilo es un anillo de 4-7 miembros opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rc; en el que cuando el heterociclilo contiene un resto -NH-, este resto -NH- está opcionalmente sustituido por Rd; en el que alquenilo C2-6 y alquinilo C2-6, están cada uno independientemente opcionalmente sustituidos con uno o más sustituyentes cada uno independientemente seleccionado de Re; en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rf; en el que cicloalquilo C3-6 está independientemente opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rg.
En algunos casos, R2 y R4 pueden seleccionarse independientemente del grupo que consiste en hidrógeno y -ORx, siempre que al menos uno de R2 y R4 sea hidrógeno, en el que Rx puede seleccionarse del grupo que consiste en hidrógeno; halógeno; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; fenilo; naftilo; heteroarilo; heterociclilo; cicloalquil C3-6-alquil C1-6-; fenil-alquil C1-6-; naftil-alquil C1-6-; heteroaril-alquil C1-6-; y heterociclil-alquil
C1-6-; en el que heteroarilo es un anillo de 5-6 miembros que tiene uno, dos o tres heteroátomos cada uno independientemente seleccionado de N, O o S; en el que heteroarilo está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rb; en el que heterociclilo es un anillo de 4-7 miembros opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rc; en el que cuando heterociclilo contiene un resto -NH-, este resto -NH- está opcionalmente sustituido por Rd; en el que alquenilo C2-6 y alquinilo C2-6, están cada uno independientemente opcionalmente sustituidos con uno o más sustituyentes cada uno independientemente seleccionado de Re; en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rf; en el que cicloalquilo C3-6 está independientemente opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de Rg;
Rb puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; alcoxi C1.6; alqueniloxi cicloalcoxi C3-6; alquil C1-6-S(O)w-, en el que w es 0, 1 o 2; alquil C1-6-cicloalquil C3-6-; cicloalquil C3-6-alquil C1.6-; alcoxicarbonil C1-6-N(Ra)-; alquil C1-6-N(Ra)-; alquil C1-6-N(Ra)carbonil-; RaRaN-; RaRaN-carbonil-; RaRaN-carbonil-N(Ra)-; RaRaN-SO2-; y alquil C1-6-carbonil-N(Ra)-;
Ra y Ra pueden seleccionarse, independientemente para cada caso, del grupo que consiste en hidrógeno y alquilo
C1-6, o Ra y Ra cuando se toman junto con el nitrógeno al que están unidos forman un anillo heterocíclico de 4-6 miembros, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo, y en el que el anillo heterocíclico está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, alquilo, oxo o hidroxilo;
Rc puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; oxo; alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; alcoxi C1-6; alqueniloxi C3-6; alquiniloxi
C3-6; cicloalcoxi C3-6; alquil C1-6-S(O)w-, en el que w es 0, 1 o 2; alquil C1-6-cicloalquil C3-6-; cicloalquil C3-6-alquil C1-6-; alcoxicarbonil C1-6-N(Ra)-; alquil C1-6-N(Ra)-; alquil C1-6-N(Ra)carbonil-; RaRaN-; RaRaN-carbonil-; RaRaN-carbonil-N(Ra)-; RaRa'N-SO2-; y alquil C1-6-carbonil-N(Ra)-;
Rd puede seleccionarse, independientemente para cada caso, del grupo que consiste en alquilo C1-6, alquilcarbonilo
C1-6, y alquilsulfonilo C1.6, en el que alquilo C1.6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de halógeno, hidroxilo, y RaRaN-;
Re puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2;
Rf puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4S(O)w-, en el que w es 0, 1 o 2;
Rg puede seleccionarse, independientemente para cada caso, del grupo que consiste en halógeno, hidroxilo, -NO2; -N3; -CN; -SCN; alquilo C1-6; alcoxi C1.4; alcoxicarbonilo C1.4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y al 4S(O)w-, en el que w es 0, 1 o 2.
En determinadas realizaciones, R5 y R6 pueden seleccionarse independientemente del grupo que consiste en -Q-Ar e hidrógeno; en el que Q se selecciona independientemente del grupo que consiste en alquilo C1-6; alquenilo C2-6; alquinilo C2-6; cicloalquilo C3-6; heterociclilo; cicloalquil C3-6-alquil C1-6-; heterociclil-alquil C1.6-; y un enlace; y en el que Ar se selecciona del grupo que consiste en fenilo, naftilo, y heteroarilo sustituido o no sustituido; o R5 y R6, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros, opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado de halógeno, hidroxilo, -NO2; -N3; -CN; -SCN; alquilo C1-6; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y al 4S(O)w-, en el que w es 0, 1 o 2; y
en el que Ra y Ra pueden seleccionarse, independientemente para cada caso, del grupo que consiste en hidrógeno y alquilo C1-6, o Ra y Ra cuando se toman junto con el nitrógeno al que están unidos forman un anillo heterocíclico de
4-6 miembros, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo, y en el que el anillo heterocíclico está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, alquilo, oxo o hidroxilo.
En determinadas realizaciones, al menos uno de R1, R2, R3 y R4 puede ser hidroxilo.
En algunos casos, al menos uno de R1, R2, R3 y R4 puede ser alquilo C1-C6, opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente del grupo que consiste en halógeno, hidroxilo, -NH2 y ciano.
En algunas realizaciones, al menos uno de R5 y R6 puede ser -(alquileno C1-C6)-Ar, por ejemplo, uno de R5 y R6 también puede ser -CH2-Ar. En algunos casos, al menos uno de R5 y R6 es -Q-fenilo. En determinados ejemplos, uno de R5 y R6 puede ser hidrógeno.
En algunos casos, R7 y R8 pueden seleccionarse independientemente del grupo que consiste en hidrógeno; halógeno; hidroxilo; alquilo C1-C6; fenilo; y naftilo; o R7 y R8, junto con los átomos a los que están unidos, forman un anillo cicloalquilo o heterocíclico de 4-6 miembros; en el que alquilo C1-C6, fenilo, naftilo, el anillo cicloalquilo y el anillo heterocíclico pueden sustituirse cada uno independientemente con uno o más sustituyentes seleccionados del grupo que consiste en halógeno; hidroxilo; -NO2; -N3; -CN; -SCN; alcoxi C1-4; alcoxicarbonilo C1-4; RaRaN-; RaRaN-carbonilo; RaRaN-SO2-; y alquil C1-4-S(O)w-, en el que w es 0, 1 o 2; en el que Ra y Ra pueden seleccionarse,
independientemente para cada caso, del grupo que consiste en hidrógeno y alquilo C1-6, o Ra y Ra cuando se toman junto con el nitrógeno al que están unidos forman un anillo heterocíclico de 4-6 miembros, en el que alquilo C1-6 está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, oxo e hidroxilo, y en el que el anillo heterocíclico está opcionalmente sustituido con uno o más sustituyentes cada uno independientemente seleccionado del grupo que consiste en halógeno, alquilo, oxo o hidroxilo.
En algunos casos, R7 y R8 pueden ser hidrógeno.
En una realización de referencia, un compuesto puede representarse por:

En otra realización de referencia, un compuesto puede representarse por:

En aún otra realización de referencia, un compuesto puede representarse por:

Los compuestos de la presente divulgación y formulaciones de los mismos pueden tener una pluralidad de centros quirales. Cada centro quiral puede ser independientemente R, S, o cualquier mezcla de R y S. Por ejemplo, en algunas realizaciones, un centro quiral puede tener una razón R:S de entre aproximadamente 100:0 y aproximadamente 50:50, de entre aproximadamente 100:0 y aproximadamente 75:25, de entre aproximadamente 100:0 y aproximadamente 85:15, de entre aproximadamente 100:0 y aproximadamente 90:10, de entre aproximadamente 100:0 y aproximadamente 95:5, de entre aproximadamente 100:0 y aproximadamente 98:2, de entre aproximadamente 100:0 y aproximadamente 99:1, de entre aproximadamente 0:100 y 50:50, de entre aproximadamente 0:100 y aproximadamente 25:75, de entre aproximadamente 0:100 y aproximadamente 15:85, de entre aproximadamente 0:100 y aproximadamente 10:90, de entre aproximadamente 0:100 y aproximadamente 5:95, de entre aproximadamente 0:100 y aproximadamente 2:98, de entre aproximadamente 0:100 y aproximadamente 1:99, de entre aproximadamente 75:25 y 25:75, y aproximadamente 50:50. Formulaciones de los compuestos divulgados que comprenden una razón mayor de uno o más isómeros (es decir, R y/o S) pueden poseer característica terapéutica potenciada relativa a formulaciones racémicas de mezcla de compuestos o compuestos divulgados.
Los compuestos divulgados pueden contemplar la apertura eficiente del canal del catión en el receptor de NMDA, por ejemplo pueden unirse o asociarse con el sitio de glutamato del receptor de NMDA para asistir en la apertura del canal del catión. Los compuestos divulgados pueden usarse para regular (abrir o cerrar) el receptor de NMDA a través de la acción como un agonista.
Los compuestos tal como se describen en el presente documento pueden ser agonistas parciales del receptor de NMDA del sitio de glicina. Un agonista parcial tal como se usa en este contexto se entenderá que significa que a baja concentración, el análogo actúa como un agonista y a alta concentración, el análogo actúa como un antagonista. La unión de glicina no está inhibida por glutamato o por inhibidores competitivos de glutamato, y tampoco se une al mismo sitio que el glutamato en el receptor de NMDA. Un segundo y separado sitio de unión para glicina existe en el receptor de NMDA. El canal iónico regulado por ligando del receptor de NMDA está, por tanto, bajo el control de al menos estos dos sitios alostéricos distintos. Los compuestos divulgados pueden ser capaces de unirse o asociarse con el sitio de unión de glicina del receptor de NMDA. En algunas realizaciones, los compuestos divulgados pueden poseer una potencia que es 10 veces o mayor que la actividad de los agonistas parciales del sitio de glicina del receptor de NMDA existentes. Por ejemplo, los compuestos divulgados pueden poseer una potencia mejorada de 10 veces a 20 veces en comparación con GLYX-13. GLYX-13 se representa por:

Por ejemplo, en el presente documento se proporcionan compuestos que pueden ser al menos aproximadamente 20 veces más potentes en comparación con GLYX-13, según se mide por conductancia neuronal única regulada por el receptor de NMDA activado por ráfaga (Inmda) en un cultivo de neuronas piramidales CA1 del hipocampo a una concentración de 50 nM. En otra realización, un compuesto proporcionado puede ser capaz de generar una conductancia neuronal única regulada por el receptor de NMDA provocado por impacto único mejorada (Inmda) en neuronas piramidales CA1 del hipocampo a concentraciones de 100 nM a 1 |iM. Los compuestos divulgados pueden tener potencia mejorada en comparación con GLYX-13 medida por la magnitud de potenciación a largo plazo (LTP) en las sinapsis CA-1-de la vía colateral de Schaffer en cortes de hipocampo in vitro.
Los compuestos divulgados pueden exhibir un alto índice terapéutico. El índice terapéutico, tal como se usa en el presente documento, se refiere a la razón de la dosis que produce una toxicidad en el 50 % de la población (es decir, TD50) con respecto a la mínima dosis eficaz para el 50 % de la población (es decir, ED50). Por tanto, el índice terapéutico = (TD50MED50). En algunas realizaciones, un compuesto divulgado puede tener un índice terapéutico de al menos aproximadamente 10:1, al menos aproximadamente 50:1, al menos aproximadamente 100:1, al menos aproximadamente 200:1, al menos aproximadamente 500:1, o al menos aproximadamente 1000:1.
Composiciones
En otros aspectos, se proporcionan formulaciones y composiciones que comprenden los compuestos divulgados y opcionalmente un excipiente farmacéuticamente aceptable. En algunas realizaciones, una formulación contemplada comprende una mezcla racémica de uno o más de los compuestos divulgados.
Las formulaciones contempladas pueden prepararse en cualquiera de una variedad de formas para su uso. A modo de ejemplo, los compuestos pueden prepararse en una formulación adecuada para administración oral, inyección subcutánea u otros métodos para administrar un agente activo a un animal conocido en las técnicas farmacéuticas.
Las cantidades de un compuesto divulgado tal como se describe en el presente documento en una formulación pueden variar según factores tales como el grado de actividad de la enfermedad, edad, sexo y peso del individuo. Los regímenes de dosificación pueden ajustarse para proporcionar la respuesta terapéutica óptima. Por ejemplo, puede administrarse un bolo simple, pueden administrarse varias dosis divididas a lo largo del tiempo o puede reducirse o aumentarse la dosis de manera proporcional según se indique por las exigencias de la situación terapéutica. Es especialmente ventajoso para formular composiciones parenterales en forma unitaria de dosificación para facilidad de administración y uniformidad de dosificación. Forma unitaria de dosificación tal como se usa en el presente documento se refiere a unidades separadas físicamente adaptadas como dosificaciones unitarias para el mamífero que se va a tratar; cada unidad contiene una cantidad predeterminada de compuesto activo calculada para producir el efecto terapéutico deseado asociado con el portador farmacéutico requerido.
La memoria descriptiva para las formas unitarias de dosificación de la invención está impuesta por y directamente depende de (a) las características únicas del compuesto seleccionado y el efecto terapéutico particular que va a lograrse, y (b) las limitaciones inherentes en la técnica de formación de compuestos de dicho compuesto activo para el tratamiento de sensibilidad en individuos.
Las composiciones terapéuticas normalmente pueden ser estériles y estables en las condiciones de fabricación y almacenamiento. La composición puede formularse como una disolución, microemulsión, liposoma u otra estructura ordenada adecuada para una concentración de fármaco alta. El portador puede ser un disolvente o medio de dispersión que contiene, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol y polietilenglicol
líquido) y mezclas adecuadas de los mismos. La fluidez apropiada puede mantenerse, por ejemplo, mediante el uso de un recubrimiento tal como lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersión y mediante el uso de tensioactivos. En muchos casos, será preferible incluir agentes isotónicos, por ejemplo, azúcares, polialcoholes tales como manitol, sorbitol o cloruro de sodio en la composición. La absorción prolongada de las composiciones inyectables puede facilitarse incluyendo en la composición un agente que retrase la absorción, por ejemplo, sales de monoestearato y gelatina.
Los compuestos pueden administrarse en una formulación diferida, por ejemplo en una composición que incluye un polímero de liberación lenta. Los compuestos pueden prepararse con portadores que protegerán el compuesto frente a la liberación rápida, tales como una formulación de liberación controlada, incluyendo implantes y sistemas de entrega microencapsulados. Pueden usarse polímeros biocompatibles, biodegradables, tales como acetato de etilenvinilo, polianhídridos, ácido poliglicólico, colágeno, poliortoesteres, ácido poliláctico y copolímeros polilácticos, poliglicólicos (PLG). Muchos métodos para la preparación de tales formulaciones se conocen generalmente por los expertos en la técnica.
Pueden prepararse disoluciones inyectables estériles incorporando el compuesto en la cantidad requerida en un disolvente apropiado con un ingrediente o una combinación de ingredientes enumerados anteriormente, según se requiera, seguido por esterilización por filtración. Generalmente, las dispersiones se preparan incorporando el compuesto activo en el interior de un vehículo estéril que contiene un medio básico de dispersión y los otros ingredientes necesarios de aquellos enumerados anteriormente. En el caso de polvos estériles para la preparación de disoluciones inyectables estériles, los métodos de preparación preferidos son secado a vacío y liofilización que produce un polvo del principio activo más cualquier ingrediente deseado adicional a partir de una disolución del mismo esterilizada por filtración previamente.
En algunas realizaciones, determinados compuestos divulgados son capaces de la entrega de una cantidad eficaz de compuesto cuando se administran oralmente a un paciente. Por ejemplo, en determinadas realizaciones, determinados compuestos divulgados son más eficaces cuando se administran oralmente a un paciente en comparación con la administración oral a un paciente de un compuesto de peptidilo representado por:

Según un aspecto alternativo de la invención, un compuesto puede formularse con uno o más compuestos adicionales que mejoran la solubilidad del compuesto.
Métodos
Se proporcionan uno o más de los compuestos divulgados para su uso en métodos para tratamiento de trastornos cognitivos y para mejora del aprendizaje. Tales métodos incluyen administrar una formulación farmacéuticamente aceptable de uno o más de los compuestos divulgados a un paciente necesitado de la misma. También se contemplan métodos de tratamiento de pacientes que padecen de, déficits de memoria asociados con la edad, esquizofrenia, trastornos del aprendizaje especiales, crisis, convulsiones posteriores a un accidente cerebrovascular, isquemia cerebral, hipoglucemia, paro cardíaco, epilepsia, migrañas, así como enfermedad de Huntington, Parkinson y Alzheimer.
Otros métodos contemplados incluyen el tratamiento de isquemia cerebral, accidente cerebrovascular, traumatismo cerebral, tumores cerebrales, dolor neuropático agudo, dolor neuropático crónico, trastornos del sueño, adicción a las drogas, depresión, determinados trastornos de la visión, síndrome de abstinencia del alcohol, ansiedad, dificultades de aprendizaje y de memoria, autismo, epilepsia, demencia asociada a SIDA, atrofia multisistémica, parálisis supranuclear progresiva, ataxia de Friedrich, síndrome de Down, síndrome del cromosoma X frágil, esclerosis tuberosa, atrofia olivopontocerebelosa, parálisis cerebral, neuritis óptica inducida por fármacos, neuropatía periférica, mielopatía, retinopatía isquémica, retinopatía diabética, glaucoma, paro cardíaco, trastornos de la conducta, trastornos del control de impulsos, enfermedad de Alzheimer, pérdidas de memoria que acompañan estadios tempranos de la enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia, mejora de la adición a opiáceos, adicción a la nicotina, adicción al alcohol, traumatismo craneoencefálico, lesión de la médula espinal, síndrome de estrés postraumático y corea de Huntington.
Por ejemplo, se proporciona en el presente documento un compuesto divulgado para su uso en un método de tratamiento de depresión en un paciente necesitado del mismo, que comprende administrar un compuesto divulgado, por ejemplo, administrando de manera aguda un compuesto divulgado. En determinadas realizaciones, el paciente resistente a tratamiento se identifica como uno que ha sido tratado con al menos dos tipos de tratamientos
antidepresivos antes de la administración de un compuesto divulgado. En otras realizaciones, el paciente resistente a tratamiento es uno que se identifica como reacio o incapaz de tolerar un efecto secundario de al menos un tipo de tratamiento antidepresivo.
Los estados de depresión más comunes incluyen trastorno distímico y trastorno depresivo mayor. Otros estados de depresión se desarrollan bajo circunstancias únicas. Tales estados de depresión incluyen depresión psicótica, depresión posparto, trastorno afectivo estacional (TAE), trastorno del estado de ánimo, depresiones causadas por estados médicos crónicos tales como cáncer o dolor crónico, quimioterapia, estrés crónico, trastornos de estrés postraumático, y trastorno bipolar (o trastorno maníaco depresivo).
La depresión resistente se produce en pacientes que sufren de depresión que son resistentes a tratamientos convencionales farmacológicos, incluyendo antidepresivos tricíclicos, IMAO, SSRI y fármacos ansiolíticos y/o inhibidores de captación doble y triple, así como tratamientos no farmacológicos tales como psicoterapia, terapia electroconvulsiva, estimulación del nervio vago y/o estimulación magnética transcraneal. Un paciente resistente a tratamiento puede identificarse como uno que fracasa en experimentar alivio de uno o más síntomas de depresión (por ejemplo, sentimientos tristes o ansiosos persistentes, sentimientos de impotencia, desesperanza, pesimismo) a pesar de someterse a uno o más tratamientos farmacológicos o no farmacológicos convencionales. En determinadas realizaciones, un paciente resistente a tratamiento es uno que fracasa en experimentar alivio de uno o más síntomas de depresión a pesar de someterse a tratamiento con dos fármacos antidepresivos diferentes. En otras realizaciones, un paciente resistente a tratamiento es uno que fracasa en experimentar alivio de uno o más síntomas de depresión a pesar de someterse a tratamiento con cuatro fármacos antidepresivos diferentes. Un paciente resistente a tratamiento también puede identificarse como uno que es reacio o incapaz de tolerar los efectos secundarios de uno o más tratamientos farmacológicos o no farmacológicos convencionales.
En aún otro aspecto, se proporciona un compuesto divulgado para su uso en un método para potenciar el alivio del dolor y para proporcionar analgesia a un animal.
En determinadas realizaciones, se proporciona un compuesto divulgado para su uso en métodos para tratamiento de esquizofrenia. Por ejemplo, puede tratarse esquizofrenia tipo paranoide, esquizofrenia tipo desorganizado (es decir, esquizofrenia hebefrénica), esquizofrenia tipo catatónico, esquizofrenia tipo indiferenciado, esquizofrenia tipo residual, depresión posesquizofrénica y esquizofrenia simple usando los métodos y composiciones contemplados en el presente documento. Los trastornos psicóticos tales como trastornos esquizoafectivos, trastornos delirantes, trastornos psicóticos breves, trastornos psicóticos compartidos y trastornos psicóticos con delirios o alucinaciones también pueden tratarse usando las composiciones contempladas en el presente documento.
La esquizofrenia paranoide puede caracterizarse porque están presentes alucinaciones auditivas o delirios, pero no lo están trastorno del pensamiento, comportamiento desorganizado o aplanamiento afectivo. Los delirios pueden ser persecutorios y/o grandiosos, pero además de estos, pueden estar presentes otros temas tales como celos, religiosidad o somatización.
La esquizofrenia tipo desorganizado puede caracterizarse porque se presentan juntos trastorno del pensamiento y poca afectividad.
La esquizofrenia tipo catatónico puede caracterizarse porque el sujeto puede estar casi inmóvil o mostrar movimiento agitado, sin propósito. Los síntomas pueden incluir estupor catatónico y flexibilidad cérea.
La esquizofrenia tipo indiferenciado puede caracterizarse porque están presentes síntomas psicóticos pero no se cumplen los criterios para los tipos paranoide, desorganizado o catatónico.
La esquizofrenia tipo residual puede caracterizarse porque están presentes síntomas positivos solo a una baja intensidad.
La depresión posesquizofrénica puede caracterizarse porque surge un episodio depresivo después de una enfermedad esquizofrénica porque todavía pueden estar presentes algunos síntomas esquizofrénicos de bajo nivel. La esquizofrenia simple puede caracterizarse por desarrollo progresivo e insidioso de síntomas negativos prominentes sin historial de episodios psicóticos.
En algunas realizaciones, se proporciona un compuesto divulgado para su uso en métodos para el tratamiento de síntomas psicóticos que pueden presentarse en otros trastornos mentales, incluyendo trastorno bipolar, trastorno límite de la personalidad, intoxicación por drogas y psicosis inducida por drogas.
En otra realización, se proporciona un compuesto divulgado para su uso en métodos para el tratamiento de delirios (por ejemplo, “no extravagantes”) que pueden estar presentes en, por ejemplo, trastorno delirante.
También se proporciona un compuesto divulgado para su uso en métodos para el tratamiento de retraimiento social
en condiciones incluyendo trastorno de ansiedad social, trastorno de personalidad evitativo y trastorno de personalidad esquizotípico.
De manera adicional, se proporciona un compuesto divulgado para su uso en métodos para el tratamiento de trastorno obsesivo-compulsivo (TOC).
Ejemplos
Los siguientes ejemplos se proporcionan solamente con propósitos ilustrativos, y no pretenden limitar el alcance de la divulgación.
Ejemplo de referencia 1 - Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((S)-2-amino-3-h¡drox¡propano¡l)p¡rrol¡d¡n-2-carbonil)p¡rrol¡d¡n-2-carboxam¡da (CM-1).
Se usó la siguiente secuencia de reacción (esquema A) para sintetizar (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((S)-2-amino-3-hidroxipropanoil)pirrolidin-2-carbonil)pirrolidin-2-carboxamida (CM-1):
Esquema A. Síntesis de CM-1.

Síntesis de (S)-1-((S)-3-acetoxi-2-(benciloxicarbonilamino)-propanoil)-pirrolidin-2-carboxilato de terc-butilo (2):
Se disolvió ácido (S)-3-acetoxi-2-(benciloxicarbonilamino)-propanoico (1,5 g, 5,33 mmol) en CH2Cl2 (15 ml). Se le añadieron N-metilmorfolina (NMM) (0,64 ml, 5,87 mmol) y cloroformiato de isobutilo (IBCF) (0,72 ml, 6,12 mmol) a -15 °C y se agitó durante 30 minutos bajo atmósfera inerte. Se le añadieron gota a gota una mezcla de (S)-pirrolidin-2-carboxilato de terc-butilo (1) (998 mg, 5,87 mmol) y NMM (0,64 ml, 5,87 mmol) en DMF (5 ml) a la mezcla de reacción y se continuó agitando durante otras 3 h a TA. Se diluyó la mezcla de reacción con DCM (200 ml), se lavó con agua (50 ml), disolución de ácido cítrico (10 ml) y salmuera (10 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 30 % de EtOAc/hexano para proporcionar el compuesto 2 (1,6 g, 69,5 %).
1H-RMN: (200 MHz, DMSO-d6): 87,81-7,76 (d, J = 20,5 Hz, 1H), 7,35-7,30 (m, 5H), 5,03-4,97 (m, 2H), 4,61-4,55 (m, 1H), 4,32-4,16 (m, 2H), 4,08-3,87 (m, 2H), 3,65-3,59 (m, 1H), 2,21-2,11 (m, 2H), 1,98 (s, 3H), 1,91- 1,75 (m, 2H), 1,37 (s, 9H).
Masa m/z: 435,0 [M++1].
Síntesis de ácido (S)-1-((S)-3-acetoxi-2-(benciloxicarbonilamino)-propanoil)-pirrolidin-2-carboxílico (3):
A una disolución del compuesto 2 (1 g, 2,30 mmol) en CH2Cl2 (5 ml), se le añadió TFA-DCM al 20 % (10 ml) y se
agitó a TA durante 2 h. Se diluyó la mezcla de reacción con agua (10 ml) y se extrajo con EtOAc ( 2* 15 ml). Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida para producir el compuesto 3 (800 mg, 92 %).
1H-RMN: (200 MHz, DMSO-da): 812,58 (s a, 1H), 7,81-7,77 (d, J= 8,0 Hz, 1H), 7,35-7,27 (m, 5H), 5,04-4,96 (m, 2H), 4.66- 4,60 (m, 1H), 4,32-4,24 (m, 2H), 4,04-3,86 (m, 1H), 3,66-3,59 (t, J = 12,6 Hz, 2H), 2,17-2,07 (m, 3H), 1,98-1,80 (m, 4H).
Masa m/z: 379,0 [M++1].
Síntesis de (2S,3R)-2-((S)-1-((S)-1-((R)-3-acetoxi-2-(benciloxicarbonilamino)-propanoil)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)-3-hidroxibutanoato de metilo (5):
Se disolvió el compuesto 3 (1,0 g, 2,64 mmol) en CH2Cl2 (10 ml), se le añadieron a la mezcla de reacción NMM (0,32 g, 3,17 mmol) e IBCF (0,41 g, 3,04 mmol) a -15 °C y se agitó durante 30 minutos bajo atmósfera inerte. Se le añadieron gota a gota una mezcla de (2S,3R)-3-hidroxi-2-((S)-pirrolidin-2-carboxamido)-butanoato de metilo (4) (0,73 g, 3,17 mmol) y NMM (0,35 ml) en DMF (3 ml) a la mezcla de reacción a -15 °C y se continuó agitando durante otras 3 h a TA. Se diluyó la mezcla de reacción con DCM (200 ml), se lavó con agua (20 ml), disolución de ácido cítrico (2 * 20 ml) y salmuera (2 * 50 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 5 % de CHaOH/EtOAc para proporcionar el compuesto (5) (0,29 g, 19 %).
1H-RMN: (500 MHz, DMSO-d6): 87,83-7,81 (m, 1H), 7,72-7,70 (m, 1H), 7,36-7,35 (m, 5H), 5,07-5,01 (m, 2H), 4,99 4,93 (m, 1H), 4,58 (s, 1H), 4,50-4,48 (m, 1H), 4,26-4,22 (m, 2H), 4,07-4,00 (m, 2H), 3,89-3,86 (m, 1H), 3,61-3,55 (m, 5H), 3,53 (s, 1H), 3,39 (s, 1H), 2,12 (s, 1H), 1,98 (s, 3H), 1,94-1,83 (m, 4H), 1,81-1,80 (m, 3H), 1,05 (d, J= 6,5 Hz, 3H).
Masa m/z: 591,0 [M++1].
Síntesis de bencil-(R)-1-((S)-2-((S)-2-((2S,3R)-1-(aminooxi)-3-hidroxi-1-oxobutan-2-ilcarbamoil)-pirrolidin-1-carbonil)-pirrolidin-1-il)-3-hidroxi-1-oxopropan-2-ilcarbamato (6):
Se le añadió una disolución de amoníaco metanólico (3 ml) al compuesto 5 (0,28 g, 0,47 mmol) y se agitó a TA durante 18 h. Se evaporaron los compuestos volátiles a presión reducida para proporcionar el compuesto 6 (0,21 g, 82.3 %).
1H-RMN: (500 MHz, DMSO-d6): 87,38-7,31 (m, 5H), 7,26 (s, 1H), 7,10-7,03 (m, 2H), 6,65 (s a, 1H), 5,04-5,01 (m, 2H), 4,98-4,84 (m, 1H), 4,76-4,75 (m, 1H), 4,61 (s, 1H), 4,38-4,31 (m, 2H), 4,02-4,00 (m, 2H), 3,77-3,74 (m, 1H), 3.67- 3,56 (m, 3H), 3,44-3,37 (m, 2H), 2,14-1,86 (m, 8H), 1,01-1,00 (m, 3H).
Masa m/z: 550 [M++1].
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((S)-2-amino-3-hidroxipropanoil)-pirrolidin-2-carbonil)-pirrolidin-2-carboxamida (CM-1):
A una disolución del compuesto 6 (0,21 g, 0,39 mmol) en metanol (5 ml), se le añadió a Pd al 10 %/C (30 mg) y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 2 h. Se filtró la mezcla de reacción sobre Celite, se evaporó el disolvente a vacío, y se trituró el residuo bruto obtenido con éter de dietilo para producir CM-1 (130 mg, 83.3 %).
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 87,39 (d, J = 8,0 Hz, 1H), 7,08-7,03 (m, 2H), 6,65 (s a, 1H), 4,89-4,85 (m, 1H), 1,61-1,59 (m, 1H), 4,39-4,38 (m, 1H), 4,02-4,00 (m, 2H), 3,68-3,52 (m, 4H), 3,43-3,36 (m, 2H), 3,22-3,10 (m, 2H), 2,19-2,13 (m, 1H), 2,07-1,98 (m, 1H), 1,93-1,81 (m, 5H), 1,75 (s, 2H), 1,01-1,00 (m, 3H).
CL-EM m/z: 400,2 [M++1].
Pureza de HPLC: 99,27 %.
Síntesis de ácido (S)-1-(benciloxicarbonil) pirrolidin-2-carboxílico (8):
A una disolución con agitación de ácido (S)-pirrolidin-2-carboxílico (7) (2,0 g, 17,39 mmol) en THF: H2O (20 ml, 1:1), se le añadieron Na2CO3 (2,76 g, 26,08 mmol) y Cbz-Cl (3,54 g, 20,80 mmol) y se agitó a Ta durante 18 h. Se lavó la mezcla de reacción con EtOAc (10 ml) y se acidificó la fase acuosa con HCl 3 N y se extrajo con EtOAc (2 * 20 ml). Se lavó la fase orgánica combinada con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para producir el compuesto 8 (3,0 g, 69,7 %).
1H-RMN: (500 MHz, DMSO-da): 812,62 (s a, 1H), 7,36-7,22 (m, 5H), 5,12-5,00 (m, 2H), 4,24-4,15 (dd, J = 5,0, 36,0 Hz, 1H), 3,46-3,31 (m, 2H), 2,25-2,15 (m, 1H), 1,94-1,79 (m, 3H).
Masa m/z: 250,0 [M++1].
Síntesis de (S)-2-((2S, 3R)-3-hidroxi-1-metoxi-1-oxobutan-2-ilcarbamoil)pirrolidin-1-carboxilato de bencilo (9):
Se disolvió el compuesto 8 (5,0 g, 20,08 mmol) en CH2Cl2 (50 ml), se le añadieron NMM (2,43 ml, 22,08 mmol) e IBCF (2,74 ml, 23,09 mmol) y se agitó a -15 °C durante 30 minutos bajo atmósfera inerte. Se le añadieron gota a gota una mezcla de (2S,3R)-2-amino-3-hidroxibutanoato de metilo (2,93 g, 22,08 mmol) y NMM (2,43 ml, 22,08 mmol) en DMF (15 ml) a -15 °C. Se agitó la mezcla de reacción resultante a TA durante 3 h. Se diluyó con DCM (200 ml) y se lavó la fase orgánica con agua (50 ml), salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 30 % de EtOAc/hexano para proporcionar el compuesto 9 (3,1 g, 42 %).
1H-RMN: (500 MHz, DMSO-d6)(rotámeros): 87,98-7,94 (m, 1H), 7,35-7,27 (m, 5H), 5,09-4,94 (m, 3H), 4,44 (dd, J= 5,5, 8,5 Hz, 1H), 4,29-4,27 (m, 1H), 4,12 (s, 1H), 3,62 (s, 3H), 3,44-3,30 (m, 2H), 2,20-2,08 (m, 1H), 1,87-1,78 (m, 3H), 1,08-0,94 (2d, 3H).
Masa m/z: 365,0 [M++1].
Ejemplo de referencia 2 - Síntesis de (S)-N-((S)-1-amino-3-hidroxi-1-oxopropan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil) p¡rrol¡din-2-carbon¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-2):
Se usó la siguiente secuencia de reacción (esquema B) para sintetizar (S)-N-((S)-1-amino-3-hidroxi-1-oxopropan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)pirrolidin-2-carboxamida (CM-2):
Esquema B. Síntesis de CM-2.

Síntesis de ácido (S)-1-(terc-butoxicarbonil)-pirrolidin-2-carboxílico (2):
A una disolución con agitación enfriada con hielo de ácido (S)-pirrolidin-2-carboxílico (1) (3,0 g, 26,08 mmol) en THF:H2O (60 ml, 1:1) se le añadieron Na2CO3 (5,52 g, 52,16 mmol), Boc2O (6,25 g, 26,69 mmol) y se agitó a TA durante 16 h. se diluyó la mezcla de reacción con agua y se lavó con EtOAc (50 ml). Se acidificó la fase acuosa con HCl 2 N y se extrajo con EtOAc (2 * 100 ml). Se secó la fase orgánica combinada sobre Na2SO4 anhidro y se concentró a presión reducida para producir el ácido (S)-1-(terc-butoxicarbonil)-pirrolidin-2-carboxílico (2) (4,8 g, 86 %).
1H-RMN: (500 MHz, DMSO-d6): 812,49 (s a, 1H), 4,08-4,03 (m, 1H), 3,36-3,24 (m, 2H), 2,22-2,11 (m, 1H), 1,87-1,76 (m, 3H), 1,39 (s, 9H).
Masa m/z: 216,0 [M++1].
Síntesis de (S)-2-((S)-3-hidroxi-1-metoxi-1-oxopropan-2-ilcarbamoil)-pirrolidin-1-carboxilato de terc-butilo (3):
Se disolvió el compuesto 2 (2,0 g, 9,00 mmol) en CH2Cl2 (10 ml) enfriado a -15 °C, se le añadieron NMM (1,12 ml, 10,2 mmol) e IBCF (1,26 ml, 1,15 mmol) y se agitó a 0 °C durante 20 minutos. Se le añadieron gota a gota una mezcla de (S)-2-amino-3-hidroxipropanoato de metilo (1,59 g, 10,2 mmol) y NMM (1,12 ml) en DMF (3 ml) a -15 °C y se agitó la mezcla de reacción resultante a TA durante 1 h. Se diluyó con DCM (200 ml), agua (50 ml) y se lavó con HCl 2 N (20 ml) y salmuera (2 * 50 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo
con el 20 % de EtOAC/hexano para proporcionar el compuesto 3 (2,3 g) como un jarabe.
Masa m/z: 317,0 [M++1].
Síntesis de (S)-3-hidroxi-2-((S)-pirrolidin-2-carboxamido)propionato de metilo (4):
Se disolvió (S)-2-((S)-3-hidroxi-1-metoxi-1-oxopropan-2-ilcarbamoil)-pirrolidin-1-carboxilato de terc-butilo (3) (500 mg,
1,58 mmol) en 1,4-dioxano (3 ml) y se le añadió una disolución de HCl en dioxano (3,16 ml, 3,16 mmol) con agitación a TA durante 4 h. Se evaporaron los compuestos volátiles a presión reducida para proporcionar el compuesto 4 (280 mg) como sólido.
1H-RMN: (200 MHz, DMSO-da): 89,99 (s a, 1H), 9,12-9,08 (m, 1H), 8,53 (s a, 1H), 5,48 (s a, 2H), 4,43-4,22 (m, 2H),
3,82-3,67 (m, 4H), 3,56 (s, 3H), 2,36-2,27 (m, 1H), 1,93-1,86 (m, 3H).
Masa m/z: 217,0 [M++1].
Síntesis de (S)-2-((S)-1-((S)-1-((2R, 3S)-3-acetoxi-2-(benciloxicarbonilamino)-butanoil)-pirrolidin-2-carbonil)-pirrolidin-2-carboxamido)-3-hidroxipropanoato de metilo (6):
Se disolvió ácido (2S)-1-((2R)-3-acetoxi-2-(benciloxicarbonilamino)-butanoil)-pirrolidin-2-carboxílico (5) (1,3 g,
2,62 mmol) en CH2Cl2 (15 ml), se le añadió NMM (0,43 ml) e IBCF (0,51 ml) a -10 °C y se agitó durante 30 minutos bajo atmósfera inerte. Se le añadieron gota a gota una mezcla de (S)-metil-3-hidroxi-2-((S)-pirrolidin-2-carboxamido)-propionato (4) (992 mg, 3,93 mmol) y NMM (0,43 ml) en DMF (5 ml) a la mezcla de reacción y se continuó agitando durante otras 3 h a TA. Se diluyó la mezcla de reacción con DCM (200 ml), se lavó con agua (20 ml), disolución de ácido cítrico (2 * 20 ml) y salmuera (2 * 50 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 5 % de CH3OH/CH2O2 para proporcionar el compuesto 6 (270 mg, 17,5 %).
1H-RMN: (500 MHz, DMSO-d6): 88,13 (d, J = 8,0 Hz, 1H), 7,74 (d, J = 7,5 Hz, 1H), 7,38-7,31 (m, 5H), 5,08-4,96 (m,
3H), 4,85-4,82 (m, 1H), 4,56 (d, J = 8,0 Hz, 1H), 4,44-4,42 (m, 2H), 4,27 (d, J = 7,0 Hz, 1H), 4,10 (d, J = 10,5 Hz,
2H), 3,81-3,78 (m, 1H), 3,72-3,70 (m, 1H), 3,61-3,59 (m, 3H), 3,54-3,50 (m, 2H), 2,16-2,14 (m, 1H), 2,05-2,01 (m,
1H), 1,90 (s, 3H), 1,87-1,86 (m, 3H), 1,85-1,84 (m, 3H), 1,21-1,20 (d, J = 6,0 Hz, 3H).
Masa m/z: 591,0 [M++1].
Síntesis de bencil-(2R,3S)-1-((S)-2-((S)-2-((S)-1-(aminooxi)-3-hidroxi-1-oxopropan-2-ilcarbamoil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-ilcarbamato (7):
A una disolución del compuesto 6 (250 g, 0,42 mmol) en CH3OH (2 ml), se le añadió MeOH-NH3 (10 ml) y se agitó a
TA durante 16 h. Se evaporaron los compuestos volátiles a presión reducida para proporcionar el compuesto 7
(190 mg, 84 %).
1H-RMN: (500 MHz, DMSO-d6): 87,60 (d, J = 7,5 Hz, 1H), 7,35-7,30 (m, 5H), 7,18 (d, J = 7,0 Hz, 1H), 7,11-7,06 (m,
2H), 5,05-4,97 (m, 2H), 4,82-4,81 (m, 1H), 4,60-4,59 (m, 2H), 4,33-4,31 (m, 1H), 4,15-4,08 (m, 2H), 3,81-3,79 (m, 1H), 3,72-3,64 (m, 2H), 3,59-3,53 (m, 4H), 2,14 (s, 1H), 2,03 (d, J = 9,0 Hz, 1H), 1,95-1,85 (m, 5H), 1,75 (s, 1H), 1,10
(d, J = 6,5 Hz, 3H).
Masa m/z: 550,0 [M++1].
Síntesis de (S)-N-((S)-1-amino-3-hidroxi-1-oxopropan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-pirrolidin-2-carboxamida (CM-2):
A una disolución del compuesto 7 (190 mg, 0,35 mmol) en metanol (5 ml), se le añadió Pd al 10 %/C (50 mg) y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 2 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el disolvente a vacío y se purificó el producto bruto mediante cromatografía en columna sobre alúmina básica usando CH3OH al 0-5 % en CH2Ó2 como eluyente para producir CM-2 (130 mg, 73 %).
1H-RMN: (500 MHz, DMSO-d6): 87,65-7,60 (m, 1H), 7,12-7,03 (m, 2H), 4,81 (s a, 1H), 4,58-4,57 (m, 1H), 4,49 (m,
1H), 4,38-4,19 (m, 1H), 4,10-4,06 (m, 1H), 3,69-3,62 (m, 2H), 3,59-3,56 (m, 4H), 3,49-3,45 (m, 2H), 3,37-3,26 (m, 2H), 2,19-2,15 (m, 1H), 2,09-1,99 (m, 1H), 1,95-1,84 (m, 5H), 1,75 (s, 1H), 1,06 (d, J = 13,0 Hz, 3H).
CL-EM m/z: 400,8 [M++1],
Pureza de HPLC: 97,71 %.
Ejemplo de referencia 3 - Síntesis de (S)-N-((S)-1-am¡no-3-hidrox¡-1-oxopropan-2-¡l)-1-((S)-1-((S)-2-am¡no-3hidroxipropanoil)- pirrolidin-2-carbonil)-pirrolidin-2-carboxamida (CM-3):
Se usó la siguiente secuencia de reacción (esquema C) para sintetizar (S)-N-((S)-1-amino-3-hidroxi-1-oxopropan-2-il)-1-((S)-1-((S)-2-amino-3-hidroxi-propanoil)-pirrolidin-2-carbonil)-pirrolidin-2-carboxamida (CM-3):
Esquema C. Síntesis de CM-3.

A una disolución con agitación de ácido (S)-pirrolidin-2-carboxílico (3,0 g, 26,08 mmol) en THF:H2O (60 ml, 1:1) a 0 °C se le añadieron Na2CO3 (5,52 g, 52,16 mmol) y Boc2O (6,25 g, 26,69 mmol) y se agitó a TA durante 16 h. Se diluyó la mezcla de reacción con agua y se lavó con EtOAc (50 ml). Se acidificó la fase acuosa con HCl 2 N y se extrajo con EtOAc (2 * 50 ml). Se secó la fase orgánica combinada sobre Na2SO4 anhidro y se concentró a presión reducida para producir el ácido (S)-1-(terc-butoxicarbonil)-pirrolidin-2-carboxílico 2 (4,8 g, 85,7 %).
1H-RMN: (500 MHz, DMSO-d6): 812,49 (s a, 1H), 4,08-4,03 (m, 1H), 3,36-3,24 (m, 2H), 2,22-2,11 (m, 1H), 1,87-1,76 (m, 3H), 1,39 (s, 9H).
Masa m/z: 216,0 [M++1].
Síntesis de (S)-2-((S)-3-hidroxi-1-metoxi-1-oxopropan-2-ilcarbamoil)pirrolidin-1-carboxilato de terc-butilo (3):
Se disolvió el compuesto 2 (2,0 g, 9,00 mmol) en CH2Cl2 (10 ml) enfriado a -15 °C, se le añadieron NMM (1,12 ml, 10,2 mmol) e IBCF (1,26 ml, 1,15 mmol) y se agitó a 0 °C durante 20 minutos. Se le añadieron gota a gota una mezcla de (S)-2-amino-3-hidroxipropanoato de metilo (1,59 g, 10,2 mmol) y NMM (1,12 ml) en DMF (3 ml) a -15 °C. Se agitó la mezcla de reacción resultante a TA durante 1 h. Se diluyó la mezcla de reacción con DCM (200 ml) y agua (25 ml) y se lavó con HCl 2 N (20 ml) y salmuera (10 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAC/hexano para proporcionar el compuesto 3 (2,3 g) como sólido. Masa m/z: 317,0 [M++1].
Síntesis de (S)-3-hidroxi-2-((5)-pirrolidin-2-carboxamido)propanoato de metilo (4):
A una disolución de (S)-2-((S)-3-hidroxi-1-metoxi-1-oxopropan-2-ilcarbamoil)pirrolidin-1-carboxilato de terc-butilo 3 (500 mg, 1,58 mmol) en 1,4-dioxano (3 ml), se le añadió una disolución de HCl en dioxano (3,16 ml, 3,16 mmol) y se agitó a TA durante 4 h. Se evaporaron los compuestos volátiles a presión reducida para proporcionar el compuesto 4 (280 mg) como sólido.
1H-RMN: (200 MHz, DMSO-d6): 89,99 (s a, 1H), 9,12-9,08 (m, 1H), 8,53 (s a, 1H), 5,48 (s a, 2H), 4,43-4,22 (m, 2H), 3,82-3,67 (m, 4H), 3,56 (s, 3H), 2,36-2,27 (m, 1H), 1,93-1,86 (m, 3H).
Masa m/z: 217,0 [M++1].
Síntesis de (S)-2-((S)-1-((S)-1-((S)-2-(benciloxicarbonilamino)-3-hidroxipropanoil)-pirrolidin-2-carbonil)-pirrolidin-2-carboxamido)-3-hidroxipropanoato de metilo (6):
Se disolvió ácido (S)-1-((S)-3-acetoxi-2-(benciloxicarbonilamino)-propanoil)-pirrolidin-2-carboxílico (5) (400 mg, 1,05 mmol) en CH2Cl2 (2 ml), se le añadieron NMM (0,13 ml) e IBc F (0,14 ml) a -15 °C y se agitó durante 30 minutos bajo atmósfera inerte. Se le añadieron gota a gota una mezcla de clorhidrato de (S)-3-hidroxi-2-((S)-pirrolidin-2-carboxamido)-propanoato de metilo (4) (293 mg, 1,16 mmol) y NMM (0,13 ml) en DMF (2 ml) a la mezcla de reacción y se continuó agitando durante otras 3 h a TA. Se diluyó la mezcla de reacción con DCM (200 ml), se lavó con agua
(20 ml) y salmuera (10 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 5 % de CH3OH/CH2O 2 para proporcionar el compuesto 6 (80 mg, 13 %).
1H-RMN: (500 MHz, DMSO-da): 88,09 (d, J = 7,5 Hz, 1H), 7,71 (d, J = 8,0 Hz, 1H), 7,36-7,31 (m, 6H), 5,07-4,99 (m, 3H), 4,59-4,58 (m, 2H), 4,41-4,40 (m, 1H), 4,29-4,24 (m, 3H), 3,86 (t, J = 9,5 Hz, 1H), 3,72-3,68 (m, 1H), 3,64-3,57 (m, 3H), 3,40-3,38 (m, 3H), 2,14-2,01 (m, 2H), 1,98 (s, 3H), 1,90-1,80 (m, 6H).
Masa m/z: 535,0 [M++1].
Síntesis de bencil-(S)-1-((S)-2-((S)-2-((S)-1-amino-3-hidroxi-1-oxopropan-2-ilcarbamoil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxopropan-2-ilcarbamato (7):
A una disolución de (S)-2-((S)-1-((S)-1-((S)-2-(benciloxicarbonilamino)-3-hidroxipropanoil)-pirrolidin-2-carbonil)-pirrolidin-2-carboxamido)-3-hidroxipropanoato de metilo de (6) (60 mg, 1,04 mmol) en MeOH, se le añadió MeOH-NH3 (3 ml) se agitó a TA durante 16 h. Se evaporaron los compuestos volátiles a presión reducida para proporcionar el compuesto 7 (30 mg, 55 %).
1H-RMN: (500 MHz, DMSO-d6): 87,60 (d, J = 7,5 Hz, 1H), 7,36-7,31 (m, 6H), 7,11-7,06 (m, 2H), 5,04-4,98 (m, 2H), 4,82-4,74 (m, 2H), 4,61-4,59 (m, 1H), 4,36-4,30 (m, 2H), 4,10-4,07 (m, 1H), 3,67-3,65 (m, 2H), 3,59-3,55 (m, 6H), 3,44-3,40 (m, 2H), 1,95-1,92 (m, 6H).
Masa m/z: 520,0 [M++1].
Síntesis de (S)-N-((S)-1-amino-3-hidroxi-1-oxopropan-2-il)-1-((S)-1-((S)-2-amino-3-hidroxi-propanoil)-pirrolidin-2-carbonil)-pirrolidin-2-carboxamida (CM-3):
Se disolvió bencil-(S)-1-((S)-2-((S)-2-((S)-1-amino-3-hidroxi-1-oxopropan-2-ilcarbamoil)pirrolidin-1-carbonil)pirrolidin-1- il)-3-hidroxi-1-oxopropan-2-ilcarbamato 7 (300 mg, 0,57 mmol) en metanol (8 ml), se le añadió Pd al 10%/C (50 mg) y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 2 h. Se filtró la mezcla de reacción y se concentró el filtrado a presión reducida para producir CM-3 (150 mg, 68 %).
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 87,62 (d, J = 8,0 Hz, 1H), 7,24 (s a, 1H), 7,14-7,07 (m, 2H), 4,87-4,82 (m, 2H), 4,59-4,57 (m, 1H), 4,37-4,31 (m, 2H), 4,11-4,07 (m, 2H), 3,70-3,39 (m, 8H), 2,17-2,01 (m, 2H), 1,95-1,79 (m, 6H).
CL-EM m/z: 386,4 [M++1].
Pureza de HPLC: 98,45 %.
Ejemplo 4 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbonil)-2-benc¡lp¡rrol¡d¡n-2-carboxam¡da (CM-4A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)-p¡rrol¡d¡n-2-carbon¡l)-2-benc¡lp¡rrol¡d¡n-2-carboxam¡da (CM-4B):
Se usó la siguiente secuencia de reacción (esquema D) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2- il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM-4A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)-pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM-4B):
Esquema D. Síntesis de CM-4A y CM-4B.
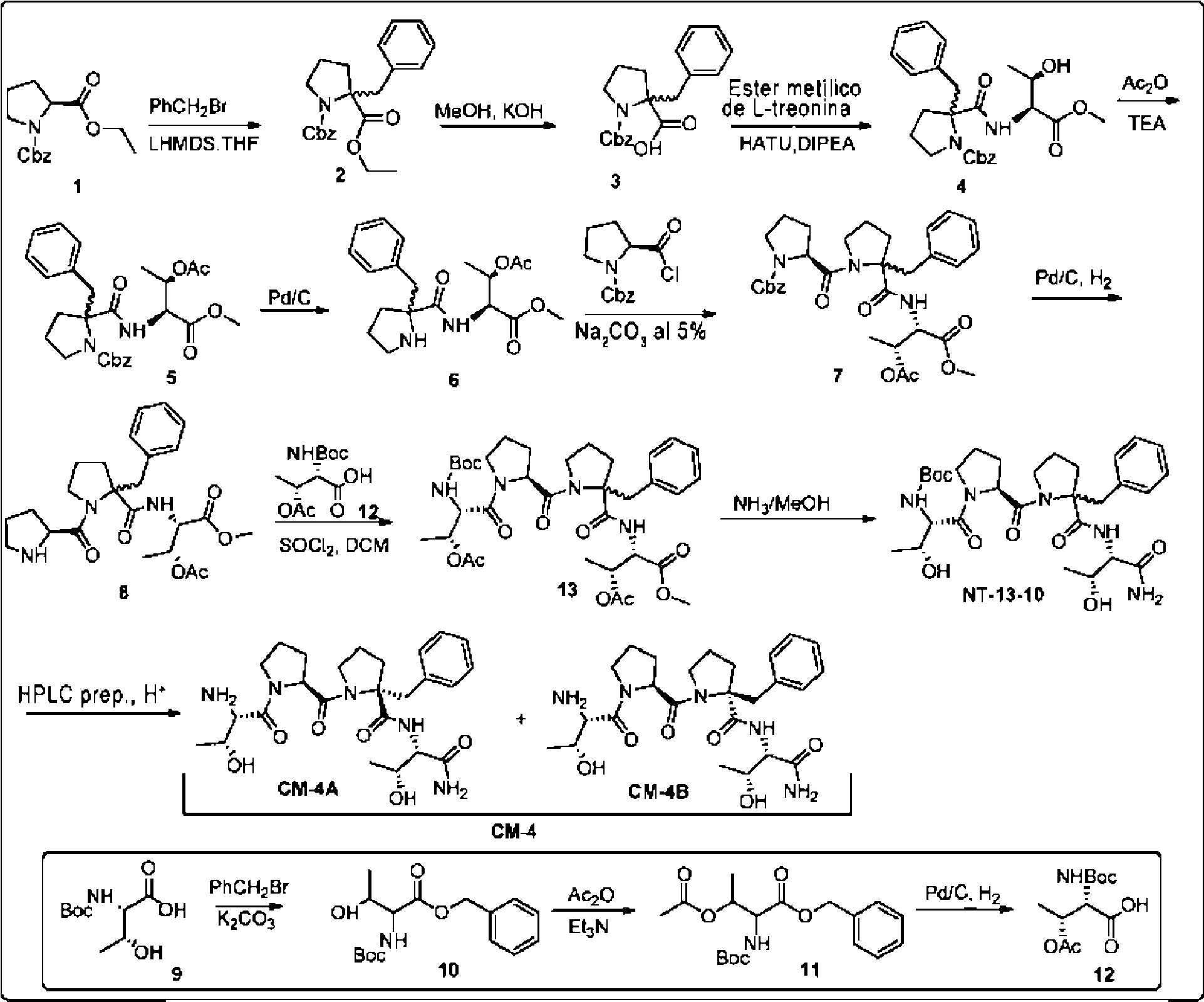
Síntesis de 2-bencilpirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (2):
A una disolución de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo 1 (10 g, 36,10 mmol) en THF (150 ml) bajo atmósfera inerte, se le añadió LiHMDS (1 M en THF) (43,3 ml, 43,3 mmol) a -25 °C y se agitó durante 2 h. Se le añadió gota a gota bromuro de bencilo (5,17 ml, 43,26 mmol) a -25 °C a la mezcla de reacción. Se dejó calentar hasta TA y se agitó durante 2 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 * 200 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 5 % de EtOAc/hexano para proporcionar el compuesto 2 (13 g, 75 %) como líquido.
1H-RMN: (200 MHz, DMSO-da): 87,47-7,32 (m, 5H), 7,27-7,16 (m, 3H), 7,07-7,04 (m, 2H), 5,29-5,06 (m, 2H), 4,16 3,89 (m, 2H), 3,57-3,33 (m, 2H), 3,02-2,78 (m, 2H), 2,13-1,89 (m, 2H), 1,56-1,51 (m, 1H), 1,21-1,04 (m, 3H), 0,93 0,79 (m, 1H).
Masa m/z: 368,2 [M++1].
Síntesis de 2-bencilpirrolidin-1,2-dicarboxilato 1-bencilo y 2-etilo (3):
A una disolución con agitación del compuesto 2 (8,0 g, 21,79 mmol) en CH3OH (20 ml), se le añadió KOH acuoso 2 N (20 ml) y se calentó hasta 100 °C y se agitó durante 16 h. Se evaporaron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando una disolución de HCl y se extrajo con EtOAc (2 * 100 ml). Se secó la fase orgánica combinada sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar el compuesto 3 (6 g, 81 %) como un sólido blanquecino.
1H-RMN: (200 MHz, DMSO-d6): 812,71 (s a, 1H), 7,40-7,30 (m, 5H), 7,25-7,19 (m, 3H), 7,07-7,00 (m, 2H), 5,27-5,02
(m, 2H), 3,59-3,32 (m, 2H), 3,02-2,83 (m, 2H), 2,13-1,91 (m, 2H), 1,58-1,49 (m, 1H), 0,90-0,77 (m, 1H).
Masa m/z: 340,1 [M++1].
Síntesis de 2-bencil-2-((2S,3R)-3-hidroxi-1-metoxi-1-oxobutan-2-ilcarbamoil)pirrolidin-1-carboxilato de bencilo (4): A una suspensión de ácido 2-bencil-1-(benciloxicarbonil)pirrolidin-2-carboxílico (1,0 g, 2,94 mmol), éster metílico de L-treonina (471 mg, 3,53 mmol) en DMF (20 ml), se le añadió HATU (1,12 g, 2,94 mmol) y DIPEA (1,58 ml, 8,84 mmol) a 5 °C. Se agitó la mezcla de reacción a TA durante 16 h. Se diluyó con EtOAc (150 ml) y se lavó con agua (2 * 30 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4, se concentró y se purificó mediante cromatografía en columna de gel de sílice con el 50 % de EtOAc/hexano como eluyente para producir el compuesto 4 (1,0 g, 74%).
1H-RMN: (200 MHz, DMSO-da): 87,62-7,59 (m, 1H), 7,44-7,31 (m, 5H), 7,21-7,18 (m, 3H), 7,06-6,99 (m, 2H), 5,25 5,24 (m, 1H), 5,12-4,94 (m, 2H), 4,30 (s, 1H), 4,15-4,08 (m, 1H), 3,66-3,64 (m, 3H), 3,63-3,49 (m, 2H), 3,14 (s, 1H), 2.89 (s, 1H), 2,09-2,02 (m, 2H), 1,56-1,51 (m, 1H), 1,09-0,98 (m, 4H).
Masa m/z: 455,1 [M++1], 477,3 [M+Na].
Síntesis de 2-((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-ilcarbamoil)-2-bencilpirrolidin-1-carboxilato de bencilo (5) Se disolvió 2-((2S,3R)-3-hidroxi-1-metoxi-1-oxobutan-2-ilcarbamoil)pirrolidin-1-carboxilato de 2-bencilo (3 g, 6,60 mmol) en Th F (30 ml), Et3N (1,11 ml, 7,92 mmol) y Ac2O (742 mg, 7,26 mmol) se le añadieron a TA. Se agitó la mezcla de reacción a TA durante 2 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con CH2O2 y se lavó con HCl dil. Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron a presión reducida. Se purificó el residuo bruto mediante cromatografía en columna usando el 30 % de EtOAc/hexano como eluyente para producir el compuesto 5 (2,5 g, 76 %).
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 88,15-7,71 (m, 1H), 7,42-7,04 (m, 10H), 5,30-5,19 (m, 2H), 5,11-5,09 (m, 1H), 4,99-4,93 (m, 1H), 4,67-4,62 (m, 1H), 3,66-3,64 (m, 3H), 3,55-3,46 (m, 2H), 3,38-3,35 (m, 1H), 2,88-2,69 (m, 1H), 2,17-2,00 (m, 2H), 1,98-1,92 (m, 3H), 1,56-1,46 (m, 1H), 1,23-1,17 (m, 3H), 1,02-0,86 (m, 1H).
CL-EM m/z: 497,4 [M++1].
Síntesis de (2S, 3R)-3-acetoxi-2-(2-bencilpirrolidin-2-carboxamido) butanoato de metilo (6):
A una disolución con agitación del compuesto 5 (4 g, 8,06 mmol) en etanol (50 ml), se le añadió Pd al 10 %/C (1,2 g) y se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 4 h. Se filtró a través de un lecho de Celite y se concentró el filtrado a presión reducida para producir el compuesto 6 (2,2 g, 75 %).
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 88,22-8,17 (m, 1H), 7,24-7,16 (m, 5H), 5,17 (t, J = 11,5 Hz, 1H), 4,48 4,42 (m, 1H), 3,60-3,54 (s, 3H), 3,20 (t, J = 13,5 Hz, 1H), 3,06-2,97 (m, 1H), 2,82-2,68 (m, 3H), 2,08-2,02 (m, 1H), 1.89 (s, 3H), 1,72-1,51 (m, 3H), 1,10 (2d, 3H).
CL-EM m/z: 363 [M++1], 385 [M+Na].
Síntesis de (S)-2-(2-((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-ilcarbamoil)-2-bencilpirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (7):
A una disolución con agitación del compuesto 6 (1 g, 2,76 mmol) y Na2CO3 (732 mg, 6,90 mmol)en CH2Ch:H2O (20 ml, 1:1) se le añadió una disolución de cloruro de ácido [A una disolución de ácido (S)-1-(benciloxicarbonil) pirrolidin-2-carboxílico (825 mg, 3,31 mmol) en CH2Cl2 (20 ml) se le añadió gota a gota SOCl2 (0,60 ml) a 0 °C y se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión reducida para producir (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo] en CH2Cl2 y se agitó la mezcla de reacción a TA durante 2 h. Se evaporaron los compuestos volátiles a presión reducida. Se diluyó el residuo con CH2Cl2 (100 ml), se filtró y el filtrado se concentró a vacío. Se purificó el residuo bruto mediante cromatografía en columna usando el 60 % de EtOAc/hexano como eluyente para proporcionar el compuesto 7 (750 mg, 45 %).
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 87,36-7,23 (m, 8H), 7,15-7,12 (m, 3H), 5,21-5,15 (m, 2H), 5,04-4,92 (m, 1H), 4,57-4,50 (m, 2H), 3,88 (d, J = 14,5 Hz, 1H), 3,65 (s, 3H), 3,54-3,46 (m, 3H), 3,21-3,13 (m, 1H), 3,02-2,90 (m, 2H), 2,19-2,02 (m, 4H), 1,97 (s, 3H), 1,89 (s, 1H), 1,77-1,65 (m, 1H), 1,17 (s, 2H), 1,06 (s, 2H).
Masa m/z: 594,1 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-bencil-1-((S)-pirrolidin-2-carbonil) pirrolidin-2-carboxamido)butanoato de metilo (8):
A una disolución del compuesto 7 (200 mg, 0,336 mmol) en EtOAc (15 ml), se le añadió Pd al 10 %/C (40 mg), se le añadió bajo atmósfera inerte y se agitó durante 12 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida. Se trituró el residuo obtenido con éter (10 ml) para proporcionar el compuesto 8 (125 mg, 81 %) como sólido.
1H-RMN: (500 MHz, CDCla) (rotámeros): 87,88-7,87 (d, 1H, J= 8,5), 7,30-7,26 (m, 2H), 7,24-7,21 (m, 1H), 7,13-7,12 (d, 2H, J = 7), 5,44-5,43 (m, 1H), 4,76-4,74 (m, 1H), 3,94-3,92 (m, 1H), 3,84-3,81 (m, 1H), 3,75 (s, 3H), 3,50 (m, 1H), 3,26-3,12 (m, 3H), 2,90-2,88 (m, 1H), 2,23-2,15 (m, 4H), 2,04 (s, 3H), 1,87-1,77 (m, 5H), 1,27-1,24 (m, 3H).
Masa m/z: 460(M+1).
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (10):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (3,0 g, 13,69 mmol) en DMF (50 ml), se le añadió K2CO3 (3,73 g, 27,39 mmol) y se agitó a TA durante 15min. Se le añadió (bromometil)benceno (2,81 g, 16,43 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (50 ml) y se extrajo con EtOAc (2 x 50 ml). Se lavó la fase orgánica combinada con salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice usando el 20 % de EtOAc/hexano como eluyente para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo 10 (2,8 g, 66 %).
1H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, J = 8,5 Hz, 1H), 5,18-5,08 (m, 2H), 4,76 (d, J = 7 Hz, 1H), 4,08-4,00 (m, 2H), 1,38 (s, 9H), 1,09 (d, J = 6,0 Hz, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)-butanoato de bencilo (11):
A una disolución con agitación de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (2,8 g, 9,06 mmol) en THF (80 ml), se le añadió Ac2O (1,1 g, 10,87 mmol), Et3N (1,51 ml, 10,87 mmol) y DMAP (280 mg) y se agitó a TA durante 15 min. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (150 ml) y se lavó con disolución de Hcl 0,5 N fría (2 x 20 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(tercbutoxicarbonilamino)butanoato 11 (2,8 g, 88 %).
1H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, J = 8,5 Hz, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, J = 3 Hz, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S, 3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (12):
Se disolvió 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo 11 (1,4 g, 3,98 mmol) en EtOAc (40 ml), se le añadió Pd al 10 %/C (600 mg) y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción sobre Celite, se evaporó el disolvente a vacío y se trituró el residuo bruto con hexano para producir ácido (2S, 3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico 12 (0,7 g, 70 %).
1H-RMN: (500 MHz, DMSO-d6): 812,78 (s a, 1H), 6,94 (d, J = 9,5 Hz, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1,39 (s, 9H), 1,10 (d, J = 6,0 Hz, 3H).
Masa m/z: 260,0 [M-1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-1-((2S,3R)-3-acetoxi-2-((terc-butoxicarbonil)-amino)butanoil)pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamido)butanoato de metilo (13):
A una disolución del compuesto ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico 12 (199 mg, 0,76 mmol) en CH2Cl2 (6 ml) que estaba bajo atmósfera inerte, se le añadieron IBCF (125 mg, 0,91 mmol) y NMM (154 mg, 1,52 mmol) a -15 °C y se agitó durante 1 h. Se le añadió una disolución de (2S,3R)-3-acetoxi-2-(2-bencil-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo 8 (350 mg, 0,76 mmol) en DMF (2 ml) a la mezcla de reacción y se agitó durante 1 h a -15 °C. Se dejó calentar la mezcla de reacción resultante hasta TA y se agitó durante 19 h. Se extrajo la mezcla de reacción con EtOAc y se lavó la fase orgánica separada con agua (20 ml), seguido de salmuera (20 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante HPLC preparativa para proporcionar el compuesto 13 (100 mg, 20 %).
1H-RMN: (500 MHz, CD3OD) (rotámeros): 87,30-7,24 (m, 3H), 7,15-7,13 (m, 2H), 4,62-4,55 (m, 2H), 4,29-3,97 (m, 1H), 3,98-3,79 (m, 4H), 3,75 (s, 3H), 3,62-3,22 (m, 2H), 3,23 (d, J = 13,5 Hz, 1H), 3,00-2,95 (q, 1H), 2,37-2,31 (m, 1H), 2,23-2,10 (m, 2H), 2,02-1,88 (m, 3H), 1,46-1,28 (m, 2H), 0,97 (d, J = 7,0 Hz, 6H).
Síntesis de ((2S,3R)-1-((S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-bencilpirrolidin- 1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (NT-13-10).
Se agitó una disolución del compuesto 13 (100 mg, 0,153 mmol) en NH3 metanólico (10 ml) en un tubo sellado a TA durante 72 h. Se concentró la mezcla de reacción a presión reducida. Se lavó el residuo bruto obtenido con éter ( 2 * 2 ml) para proporcionar NT-13-10 (85 mg).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM-4A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM-4B):
Se purificó adicionalmente NT-13-10 (85 mg) mediante HPLC preparativa quiral para proporcionar dos isómeros que con tratamiento con dioxano HCl y evaporación proporcionaron 15 mg de cada uno de CM-4A y CM-4B.
1H-RMN: (500 MHz, CD3OD) (rotámeros): 87,33-7,26 (m, 3H), 7,16 (s, 2H), 4,55-4,54 (m, 1H), 4,39 (s, 1H), 4,14 (s, 1H), 4,01-3,98 (m, 1H), 3,91-3,71 (m, 3H), 3,59 (s, 2H), 3,25-3,16 (m, 1H), 3,04-3,00 (m, 1H), 2,33-2,10 (m, 3H), 2,01-1,91 (m, 2H), 1,86-1,80 (m, 1H), 1,46-1,44 (m, 1H), 1,34-1,29 (m, 1H), 1,25-1,19 (m, 3H), 0,99-0,97 (d, J = 14,0 Hz, 3H).
Masa m/z: 503 [M+]
Pureza de HPLC: 98,1 %.
Síntesis de sal de trifluoroacetato de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)-pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM-4A-TFA):
A una disolución de CM-4A en metanol, se le añadió ácido trifluoroacético en exceso. Se recogió el precipitado resultante mediante filtración, se lavó con metanol, y se secó a vacío para producir CM-4A-TFA.
Ejemplo 5 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-metilbenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-5A) y (S)-N-((2S,3R)-1-amino-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-met¡lbenc¡l)p¡rrol¡d¡n-2-carboxamida (CM-5B):
Se usó la siguiente secuencia de reacción (esquema E) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metilbencil)pirrolidin-2-carboxamida (CM-5A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metilbencil)pirrolidin-2-carboxamida (CM-5B):
Esquema E. Síntesis de CM-5A y CM-5B:

Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución con agitación de L-prolina (SM) (110 g, 956,5 mmol) en etanol, se le añadió cloruro de tionilo (141 ml, 1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TAy se concentró a vacío para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %).
Síntesis de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (2):
A una disolución con agitación del compuesto 1 (170 g, 947 mmol) en CH2Cl2 , se le añadió Et3N (398 ml, 2,83 mol). Tras agitarse durante 30 min, se le añadió Cbz-Cl (1,136 mol) a la mezcla de reacción y se continuó agitando durante otras 12 h a TA. Se lavó la mezcla de reacción con agua y se extrajo con CH2Cl2. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a vacío. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1H-RMN: (200 MHz, DMSO-da): 87,42-7,31 (m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81 (m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1].
Síntesis de 2-(4-metilbencil) pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución con agitación del compuesto 2 (5 g, 0,018 mol) en THF (40 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (36 ml, 0,036 mol) a -78 °C y se agitó durante 30 min. A ésta se le añadió gota a gota bromuro de 4-metilbencilo (5 g, 0,027 mol) a -30 °C y se dejó calentar hasta TA y se agitó durante 2 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 x 100 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtoAc/hexano para proporcionar el compuesto 3 (5,2 g, 76 % ) como líquido.
1H-RMN: (500 MHz, DMSO-d6): 87,47-7,32 (m, 5H), 7,17-7,12 (m, 1H), 7,05-7,01 (m, 1H), 6,95-6,90 (m, 2H), 5,25 5,00 (m, 2H), 4,16-4,12 (m, 1H), 4,00-3,89 (m, 1H), 3,52 (d, 1H), 3,38-3,33 (m, 1H), 2,99-2,89 (m, 2H), 2,23 (s, 3H),
2,12-1,90 (m, 1H), 1,56-1,51 (m, 1H), 1,05-1,01 (m, 3H), 1,00-0,97 (m, 1H), 0,92-0,89 (m, 1H).
Síntesis de ácido 1-((benciloxi) carbonil)-2-(4-metilbencil)pirrolidin-2-carboxílico (4):
A una disolución con agitación del compuesto 3 (5,2 g, 0,0136 mol) en metanol (25 ml) y agua (15 ml), se le añadió NaOH acuoso 2 N (2 g, 0,052 mol) y se calentó hasta 85 °C durante 4 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl 2 N y se extrajo con CH2O 2 (2* 100 ml). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron a presión reducida para proporcionar el compuesto 4 (4,3 g, 89 % ) como un sólido blanquecino.
1H-RMN: (400 MHz, CDCla): 87,45-7,38 (m, 5H), 7,09-6,89 (m, 4H), 5,35-5,26 (m, 2H), 3,67-3,47 (m, 2H), 3,14-2,99 (m, 2H), 2,42-2,39 (m, 1H), 2,29 (s, 3H), 2,22-1,99 (m, 1H), 1,74-1,61 (m, 1H), 1,31-1,22 (m, 1H).
CL-EM (ESI): 354 [M++1].
Síntesis de 2-(((2R, 3S)-3-hidroxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-metilbencil)pirrolidin-1-carboxilato de bencilo (5):
A una disolución con agitación del compuesto 4 (4,3 g, 12,1 mmol) en CH2Cl2 (40 ml), se le añadieron EDCI.HCl (2,5 g, 13,4 mmol) seguido de HOBt (1,82 g, 13,4 mmol) y DIPEA (6,4 ml, 36,5 mmol) a 0 °C. Tras agitarse durante 10 min, se le añadió éster metílico de L-treonina (2,2 g, 13,4 mmol) a la mezcla de reacción y se agitó durante otras 4 h a TA. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con CH2O 2 (150 ml) y se lavó con agua (2 * 30 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4, se concentró y se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 2 % de MeOH/CH2Cl2 para proporcionar el compuesto 5 (4,1 g, 72 %).
1H-RMN: (500 MHz, CDCla): 87,44-7,31 (m, 5H), 7,09-6,85 (m, 5H), 5,32-5,25 (m, 1H), 5,05-4,94 (m, 2H), 4,25-4,20 (m, 1H), 4,15-4,08 (m, 1H), 3,66-3,64 (m, 2H), 3,45-3,41 (m, 2H), 3,14-3,09 (m, 1H), 2,89-2,84 (m, 1H), 2,20 (s, 3H), 2,05-2,02 (m, 2H), 1,55-1,51 (m, 1H), 1,09-0,98 (m, 4H).
CL-EM (ESI): 469 [M++1].
Síntesis de 2-(((2S, 3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-metilbencil)pirrolidin-1-carboxilato de bencilo (6):
A una disolución con agitación del compuesto 5 (4,1 g, 8,75 mmol) en CH2Cl2 (40 ml), se le añadió Et3N (3,0 ml, 21,9 mmol) seguido de Ac2O (1,0 g, 10,5 mmol) a 0 °C y se agitó durante 1 h. A ésta se le añadió DMAP (25 mg, catalítico) y se agitó a TA durante 2 h. Tras el consumo del material de partida (mediante CCF), se diluyó la mezcla de reacción con agua y se extrajo con EtOAc (2 * 100 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar el compuesto 6 (4,2 g, producto bruto). Este fue directamente usado para la próxima etapa sin purificación adicional.
1H-RMN: (500 MHz, CDCh): 87,42-7,30 (m, 5H), 7,09-6,85 (m, 5H), 5,25-5,21 (m, 2H), 5,11-5,09 (m, 1H), 4,67-4,62 (m, 1H), 3,66-3,64 (m, 2H), 3,55-3,46 (m, 3H), 3,15-3,10 (m, 1H), 2,92-2,85 (m, 1H), 2,20 (s, 3H), 2,05-1,95 (m, 5H), 1,56-1,46 (m, 1H), 1,15-1,11 (m, 4H).
CL-EM (ESI): 511 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-(4-metilbencil)pirrolidin-2-carboxamido)butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (4,2 g, 8,22 mmol) en metanol (35 ml), se le añadió Pd al 10 % /C (1,0 g) bajo atmósfera de N2. Se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 3 h. Tras el consumo del material de partida (mediante CCF), se filtró la reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (3,0 g, 100 % ) como jarabe.
1H-RMN: (500 MHz, CDCh): 88,22 (s a, 1H), 7,09-7,06 (m, 4H), 5,23-5,19 (m, 1H), 4,50-4,42 (m, 1H), 3,60 (d, 3H), 3,20 (m, 1H), 3,00-2,97 (m, 1H), 2,75-2,68 (m, 2H), 2,28 (s, 3H), 2,05-2,02 (m, 1H), 1,90 (d, 3H), 1,65-1,51 (m, 3H), 1,10 (dd, 3H).
CL-EM m/z: 377 [M++1].
Síntesis de (2S)-2-(2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-metilbencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (8):
A una disolución con agitación del compuesto 7 (3,0 g, 7,97 mmol) en CH2CI2 (30 ml) y agua (20 ml), se le añadió Na2CO3 (2,1 g, 19,9 mmol) y se agitó a 0 °C durante 5 min. A este cloruro de ácido [A una disolución de ácido (S)-1-(benciloxicarbonil)pirrolidin-2-carboxílico (2,3 g, 9,56 mmol) en CH2Cl2 (10 ml) se le añadió gota a gota SOCl2 (1,4 ml) a 0 °C y se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión reducida] y se agitó la mezcla de reacción a TA durante 2 h. Se concentró la fase orgánica separada a vacío y se separaron los isómeros obtenidos lavando con éter de dietilo (2 * 20 ml) para proporcionar el compuesto 8-F1 (1,6 g) y el compuesto 8-F2 (2,6 g).
Datos analíticos para el compuesto 8-F1:
1H-RMN: (400 MHz, CDCh): 87,39-7,28 (m, 5H), 7,15-6,85 (m, 5H), 5,21-5,05 (m, 2H), 5,04-4,92 (m, 1H), 4,65-4,50 (m, 1H), 4,53-4,45 (m, 1H), 3,65 (s, 3H), 3,54-3,46 (m, 4H), 3,21-3,13 (m, 2H), 2,25-2,16 (m, 4H), 2,05-2,00 (m, 2H), 1,95-1,85 (m, 4H), 1,56-1,51 (m, 2H), 1,15 (dd, 3H).
CL-EM m/z: 608 [M++1].
UPLC (pureza): 97 % .
Datos analíticos para el compuesto 8-F2:
1H-RMN: (400 MHz, CDCla): 88,50 (s a, 1H), 7,36-7,23 (m, 5H), 7,15-6,85 (m, 5H), 5,21-5,05 (m, 2H), 5,04-4,92 (m, 1H), 4,65-4,50 (m, 1H), 4,53-4,45 (m, 1H), 3,65 (s, 3H), 3,54-3,46 (m, 4H), 3,21-3,13 (m, 2H), 2,25-2,16 (m, 4H), 2,05-2,00 (m, 2H), 1,95-1,85 (m, 4H), 1,56-1,51 (m, 2H), 1,15 (dd, 3H).
CL-EM m/z: 608 [M++1]
UPLC (pureza): 96 % .
Síntesis de (2S, 3R)-3-acetoxi-2-((R)-2-(4-metilbencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (9-F1):
A una disolución con agitación del compuesto 8-F1 (1,6 g, 2,63 mmol) en MeOH (20 ml) y EtOAc (20 ml), se le añadió Pd al 10 % /C (0,5 g) bajo atmósfera inerte y se agitó durante 4 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9-F1 (1,1 g, 92 %).
1H-RMN: (400 MHz, CDCla): 88,23 (dd, 1H), 7,20-6,85 (m, 5H), 5,20-5,13 (m, 1H), 4,63-4,59 (m, 1H), 3,85-3,81 (m, 1H), 3,65-3,61 (m, 5H), 3,32-3,25 (m, 2H), 3,12-3,05 (m, 3H), 2,75-2,71 (m, 1H), 2,25 (s, 3H), 2,15-2,13 (m, 2H), 2,00 (d, 3H), 1,77 (m, 3H), 1,27 (dd, 4H).
CL-EM m/z: 474 [M++1].
Síntesis de (2S, 3R)-3-acetoxi-2-((S)-2-(4-metilbencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (9-F2):
A una disolución con agitación del compuesto 8-F2 (2,6 g, 4,20 mmol) en MeOH (25 ml), se le añadió Pd al 10 % /C (1,0 g) bajo atmósfera inerte y se agitó durante 3 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9-F2 (1,1 g, 55 %).
1H-RMN: (400 MHz, CDCls): 88,23 (dd, 1H), 7,20-6,85 (m, 5H), 5,20-5,13 (m, 1H), 4,63-4,59 (m, 1H), 3,85-3,81 (m, 1H), 3,65-3,61 (m, 5H), 3,32-3,25 (m, 2H), 3,12-3,05 (m, 3H), 2,75-2,71 (m, 1H), 2,25 (s, 3H), 2,15-2,13 (m, 2H), 2,00 (d, 3H), 1,77 (m, 3H), 1,27 (dd, 4H).
CL-EM m/z: 474 [M++1].
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (A):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (50 g, 228,3 mmol) en DMF (500 ml), se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15 min. A esta se le añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAc/hexano para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 73 %).
1H-RMN: (500 MHz, DMSO-da): 87,37-7,30 (m, 5H), 6,60 (d, 1H), 5,18-5,08 (m, 2H), 4,76 (d, 1H), 4,08-4,00 (m, 2H), 1.38 (s, 9H), 1,09 (d, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (B):
A una disolución con agitación del compuesto A (52 g, 168,2 mmol) en CH2Cl2 (500 ml), se le añadió Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a TA durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 x 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato B (52 g, 88 %).
1H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, 3H).
Masa m/z: 252 [M++1-des-Boc]
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (C):
A una disolución con agitación del compuesto B (52 g, 148,1 mmol) en MeOH (1 l), se le añadió Pd al 10 % /C bajo atmósfera de N2 y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el filtrado obtenido a vacío y se trituró el residuo bruto con hexano para producir ácido (2S, 3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico C (35 g, 90 %).
1H-RMN: (500 MHz, DMSO-d6): 812,78 (s a, 1H), 6,94 (d, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1.39 (s, 9H), 1,10 (d, 3H).
Masa m/z: 260,0 [M-1].
Síntesis de (2S,3R)-3-acetoxi-2-((R)-1-((S)-1-((2S,3R)-3-acetoxi-2-((terc-butoxicarbonil)amino)butanoil)pirrolidin-2-carbonil)-2-(4-metilbencil)pirrolidin-2-carboxamido)butanoato de metilo (10-F1):
A una disolución con agitación del compuesto C (0,66 g, 2,55 mmol) en CH2Cl2 (15 ml), se le añadieron EDCI (0,49 g, 2,55 mmol), HOBt (0,345 g, 2,55 mmol) seguido de DIPEA (1,2 ml, 6,96 mmol) a 0 °C y se agitó durante 10 min. A esta se le añadió el compuesto 9-F1 (1,1 g, 2,32 mmol) y se agitó durante 15 h. Se extrajo la mezcla de reacción con EtOAc (2 x 75 ml) y se lavó la fase orgánica separada con agua (200 ml), seguido de salmuera (200 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna eluyendo con el 70 % de EtOAc/hexano para proporcionar el compuesto 10-F1 (1,6 g, 100 % ) como jarabe.
1H-RMN: (500 MHz, CDCla): 87,24-6,91 (m, 5H), 5,42-5,01 (m, 2H), 4,73-4,68 (m, 1H), 4,54-4,41 (m, 1H), 3,90-3,85 (m, 1H), 3,67 (s, 3H), 3,65-3,60 (m, 2H), 3,59-3,55 (m, 1H), 3,59-3,55 (m, 1H), 3,30 (s, 3H), 3,12-3,05 (m, 1H), 2,33 (s, 3H), 2,05 (s, 3H), 2,00-1,96 (m, 4H), 1,94-1,85 (m, 4H), 1,43 (s, 9H), 1,25 (d, 3H), 1,15 (d, 3H).
CL-EM (ESI): m/z 717 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-((S)-1-((S)-1-((2S,3R)-3-acetoxi-2-((terc-butoxicarbonil)amino)butanoil)pirrolidin-2-carbonil)-2-(4-metilbencil)pirrolidin-2-carboxamido)butanoato de metilo (10-F2):
A una disolución con agitación del compuesto C (0,66 g, 2,55 mmol) en CH2Cl2 (10 ml), se le añadieron EDCI.HCl (0,48 g, 2,55 mmol), HOBt (0,345 g, 2,55 mmol) seguido de DIPEA (1,2 ml, 6,96 mmol) a 0 °C y se agitó durante 10 min. A esta se le añadió el compuesto 9-F2 (1,1 g, 2,32 mmol) y se agitó durante 15 h. Se extrajo la mezcla de reacción con EtOAc (2 x 75 ml) y se lavó la fase orgánica separada con agua (200 ml), seguido de salmuera (200 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna eluyendo con el 70 % de EtOAc/hexano para proporcionar el compuesto 10-F2 (0,8 g, 50 % ) como jarabe.
1H-RMN: (400 MHz, DMSO-d6): 87,24-6,91 (m, 5H), 5,42-5,01 (m, 2H), 4,73-4,68 (m, 1H), 4,54-4,41 (m, 1H), 3,90 3,85 (m, 1H), 3,67 (s, 3H), 3,65-3,60 (m, 2H), 3,59-3,55 (m, 1H), 3,59-3,55 (m, 1H), 3,30 (s, 3H), 3,12-3,05 (m, 1H), 2,33 (s, 3H), 2,05 (s, 3H), 2,00-1,96 (m, 4H), 1,94-1,85 (m, 4H), 1,43 (s, 9H), 1,25 (d, 3H), 1,15 (d, 3H).
CL-EM (ESI): m/z 717 [M++1].
Síntesis de ((2S,3R)-1-((S)-2-((R)-2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(4-metilbencil)pirrolidin
1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11-F1):
Se llevó una mezcla del compuesto 10-F1 (1,6 g, 2,20 mmol) y NH
3
.metanólico (20 ml) a un autoclave y se calentó hasta 70 °C durante 16 h. Tras el consumo del material de partida (mediante CCF), se concentró la mezcla de reacción a presión reducida para proporcionar el compuesto 11-F1 (0,6 g, 46 %).
1H-RMN: (400 MHz, DIVISOR): 87,32 (s a, 1H), 7,15-7,10 (m, 2H), 7,05-6,91 (m, 3H), 6,65 (s a, 1H), 4,75-4,59 (m, 2H), 4,24-3,45 (m, 8H), 3,35 (s, 1H), 3,05-3,00 (m, 1H), 2,32 (s, 3H), 2,26-1,85 (m, 6H), 1,75 (s, 6H), 1,60-1,55 (m, 1H), 1,47 (s, 9H), 1,15 (d, 3H).
CL-EM (ESI): m/z 618,5 [M++1].
Síntesis de ((2S,3R)-1-((S)-2-((S)-2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(4-metilbencil)pirrolidin-1- carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11-F2):
Se agitó una mezcla de compuesto 10-F2 (0,8 g, 1,11 mmol) y NH
3
.metanólico (20 ml) a TA durante 16 h. Tras el consumo del material de partida (mediante CCF y CL-EM), se concentró la mezcla de reacción a presión reducida para proporcionar el compuesto 11-F2 (0,4 g, 58 %).
1H-RMN: (400 MHz, DMSO-d6): 87,32 (s a, 1H), 7,15-7,10 (m, 2H), 7,05-6,91 (m, 3H), 6,65 (s a, 1H), 4,75-4,59 (m, 2H), 4,24-3,45 (m, 8H), 3,35 (s, 1H), 3,05-3,00 (m, 1H), 2,32 (s, 3H), 2,26-1,85 (m, 6H), 1,75 (s, 6H), 1,60-1,55 (m, 1H), 1,47 (s, 9H), 1,15 (d, 3H).
CL-EM (ESI): m/z 618,5 [M++1].
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2- carbonil)-2-(4-metilbencil) pirrolidin-2-carboxamida (CM-5A):
Una disolución con agitación del compuesto 11-F1 (0,25 g, 0,40 mmol) en HCl 2 N en 1,4-dioxano (7 ml) a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida y se purificó mediante HPLC prep. para proporcionar CM-5A (0,1 g, 44 %) como sal de clorhidrato.
1H-RMN: (400 MHz, D
2
O): 87,35 (t, 2H), 7,25 (d, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,89 (m, 1H), 3,87 3,78 (m, 2H), 3,63 (d, 1H), 3,49-3,40 (m, 1H), 3,25 (d, 1H), 2,56-2,34 (m, 5H), 2,28-2,20 (m, 4H), 1,75-1,71 (m, 1H), 1.56 (d, 3H), 1,32 (d, 3H), 1,15-1,11 (m, 1H).
CL-EM (ESI) m/z: 518 [M++1].
Pureza de UPLC: 99 %.
Rotación óptica [a25o]: 30,51 (C = 1 % en agua).
HPLC-prep.: Rt= 7,56 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metilbencil)pirrolidin-2-carboxamida (CM-5B):
Una disolución con agitación del compuesto 11-F2 (0,4 g, 0,64 mmol) en HCl 2 N en 1,4-dioxano (5 ml) a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para dar producto bruto; que se lavó con EtOAc y npentano para proporcionar CM-5B (0,15 g, 42 %) como sal de clorhidrato.
1H-RMN: (400 MHz, D
2
O): 87,35 (t, 2H), 7,25 (d, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,89 (m, 1H), 3,87 3,78 (m, 2H), 3,63 (d, 1H), 3,49-3,40 (m, 1H), 3,25 (d, 1H), 2,56-2,34 (m, 5H), 2,28-2,20 (m, 4H), 1,75-1,71 (m, 1H), 1.56 (d, 3H), 1,32 (d, 3H), 1,15-1,11 (m, 1H).
CL-EM (ESI) m/z: 518 [M++1].
Pureza de UPLC: 96 %.
Rotación óptica [a25o]: -124,6 (C = 1 % en agua).
HPLC-prep.: Rt = 9,68 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Ejemplo 6 - Síntesis de (R)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-6A) y (S)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-6B):
Se usó la s¡gu¡ente secuenc¡a de reacc¡ón (esquema F) para s¡ntet¡zar (R)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S,3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-6A) y (S)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-6B):
Esquema F. Síntes¡s de CM-6A y CM-6B

A una d¡soluc¡ón con ag¡tac¡ón de L-prol¡na (SM) (110 g. 956.5 mmol) en etanol se le añad¡ó cloruro de t¡on¡lo (141 ml. 1911.3 mmol) y se somet¡ó a reflujo durante 16 h. Se llevó la mezcla de reacc¡ón hasta TAy se concentró a vacío para proporc¡onar el compuesto 1 como sal de clorh¡drato (170 g. 99 %).
Síntes¡s de (S)-1-benc¡lo 2-met¡lo p¡rrol¡d¡n-1.2-d¡carbox¡lato (2):
A una d¡soluc¡ón con ag¡tac¡ón del compuesto 1 (170 g. 947 mmol) en CH2Cl2 se le añad¡ó Et3N (398 ml. 2.83 mol). Tras ag¡tarse durante 30 m¡n. se le añad¡ó Cbz-Cl (1.136 mol) a la mezcla de reacc¡ón y se cont¡nuó ag¡tando durante otras 12 h a TA. Se lavó la mezcla de reacc¡ón con agua y se extrajo con CH2Cl2. Se secó la fase orgán¡ca sobre Na2SO4 anh¡dro y se concentró a vacío. Se pur¡f¡có el producto bruto med¡ante cromatografía en columna para proporc¡onar el compuesto 2 (230 g. 88 %).
1H-RMN: (200 MHz. DMSO-d6): 87.42-731(m. 5H). 5.09 (m. 2H). 4.32-4.23(m. 1H). 4.08-3.98(m. 2H). 3.50-3.38 (m.
2H). 2.30-2.18 (m. 1H). 1.90-1.81(m. 3H). 1.10-1.04 (t.3H).
Masa m/z: 278 [M++1].
Síntesis de 2-(4-fluorobencil) pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución con agitación del compuesto 2 (10 g, 36,0 mmol) en THF seco (100 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (72,1 ml, 72,1 mmol) a -20 °C y se agitó durante 1 h. A esta se le añadió gota a gota bromuro de 4-fluorobencilo (5,34 ml, 43,2 mmol) a -20 °C y se dejó calentar hasta TA y se agitó durante 1 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución acuosa de NH4Cl y se extrajo la fase acuosa con EtOAc (2 x 100 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 30 % de EtOAc/hexano para proporcionar el compuesto 3 (10 g, 72 %).
1H-RMN: (400 MHz, DMSO-da) (rotámeros): 87,46-7,32 (m, 5H), 7,15-7,01 (m, 4H), 5,18-5,09 (m, 2H), 4,16-3,91 (m, 2H), 3,53 (d, 1H), 3,62 (d, 1H), 3,38-3,32 (m, 1H), 2,97-2,81 (m, 1H), 2,14-1,91 (m, 2H), 1,65-1,51 (m, 1H), 1,25 (t, 2H), 1,09 (t, 1H), 0,99-0,85 (m, 1H).
CL-EM (ESI): 386 [M++1].
Síntesis de ácido 1-((benciloxi)carbonil)-2-(4-fluorobencil)pirrolidin-2-carboxílico (4):
A una disolución con agitación del compuesto 3 (10 g, 25,9 mmol) en metanol (50 ml) y agua (50 ml) se le añadió NaOH acuoso 2N (3,11 g, 77,8 mmol) y se calentó hasta 70 °C durante 16 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl acuoso y se extrajo con EtOAc (2 x 100 ml). Se lavaron las fases orgánicas combinadas con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para dar el producto bruto; que se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 2 % de MeOH/CH2Cl2 para proporcionar el compuesto 4 (9 g, 97 %).
Síntesis de 2-(4-fluorobencil)-2-(((2R, 3S)-3-hidroxi-1-metoxi-1-oxobutan-2-il)carbamoil)pirrolidin-1-carboxilato de bencilo (5):
A una disolución con agitación del compuesto 4 (9,5 g, 26,6 mmol) en CH2Cl2 (95 ml) se le añadieron EDCI.HCl (7,61 g, 39,87 mmol) seguido de HOBt (5,42 g, 39,87 mmol) y DIPeA (14,6 ml, 79,7 mmol) a 0 °C bajo atmósfera de N2. Tras agitarse durante 10 min, se le añadió sal de clorhidrato de ester metílico de L-treonina (5,4 g, 31,89 mmol) a la mezcla de reacción y se agitó durante otras 16 h a TA. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con agua (100 ml) y se extrajo con CH2Cl2 (2 x 100 ml). Se secó la fase orgánica sobre Na2SO4 y se concentró para dar producto bruto; que se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 2 % de MeOH/CH2Cl2 para proporcionar el compuesto 5 (13 g, producto bruto).
Síntesis de 2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-fluorobencil)pirrolidin-1-carboxilato de bencilo (6):
A una disolución con agitación del compuesto 5 (13 g, 27,5 mmol) en CH2Cl2 (130 ml) se le añadió Et3N (9,58 ml, 68,7 mmol) seguido de Ac2O (4,13 ml, 41,26 mmol) a 0 °C y se agitó durante 1 h. A esta se le añadió DMAP (0,13 g) y se agitó a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con agua (100 ml) y se extrajo con CH2O 2 (2 x 100 ml). Se secó la fase orgánica sobre Na2SO4 y se concentró para dar producto bruto; que se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 30 % de EtOAc/hexano para proporcionar el compuesto 6 (10 g, 71 %).
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 87,22-7,14 (m, 5H), 7,11-6,98 (m, 4H), 5,26-5,21 (m, 2H), 5,09 (t, 1H), 4,67-4,63 (m, 1H), 3,66-3,64 (m, 3H), 3,69-3,65 (m, 2H), 3,19-3,16 (m, 1H), 2,92-2,85 (m, 1H), 2,10-2,02 (m, 1H), 2,98 (s, 4H), 1,56-1,46 (m, 1H), 1,20-1,14 (m, 3H), 1,12 (t, 1H).
CL-EM (ESI): 515 [M++1].
Masa m/z: 515 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-(4-fluorobencil)pirrolidin-2-carboxamido)butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (10 g, 19,4 mmol) en metanol (100 ml) se le añadió Pd al 10 % /C húmedo (2 g) bajo atmósfera de N2. Se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 4 h. Tras el consumo del material de partida (mediante CCF), se filtró la reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (6,7 g, 91 %).
Síntesis de (2S)-2-(2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-fluorobencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (8):
A una disolución con agitación del compuesto 7 (6,7 g, 17,6 mmol) en CH2CI2 (67 ml) y agua (67 ml) se le añadió Na2CO3 (4,67 g, 44,0 mmol) y se agitó a 0 °C durante 5 min. A este cloruro de ácido [A una disolución de ácido (S)-1-(benciloxicarbonil)pirrolidin-2-carboxílico (20,6 g, 82,8 mmol) en CH2Cl2 (20 ml) se le añadió SOCl2 (3,19 ml, 44,0 mmol) gota a gota a 0 °C y se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión reducida para producir (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo] (5,26 g, 21,15 mmol) y se agitó la mezcla de reacción a TA durante 2 h. Se concentró la fase orgánica separada a vacío y se purificó el material bruto obtenido mediante cromatografía en columna eluyendo con el 2 % de MeOH/CH2Cl2 para proporcionar el compuesto 8 (8,9 g, 83%).
CL-EM m/z: 612 [M++1].
Masa m/z: 612 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-(4-fluorobencil)-1-((S)-pirrolidin-2-carbonil) pirrolidin-2-carboxamido) butanoato de metilo (9):
A una disolución con agitación del compuesto 8 (8,9 g, 14,5 mmol) en MeOH (180 ml) se le añadió Pd al 10 % /C húmedo (1,8 g) bajo atmósfera inerte y se agitó durante 4 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9 (6,9 g, producto bruto).
1 H-RMN: (400 MHz, CD3OD) (rotámeros): 87,22-7,14 (m, 2H), 7,03 (t, 2H), 5,39-5,34 (m, 1H), 4,52 (t, 1H), 3,78-3,72 (m, 2H), 3,74 (d, 3H), 3,35 (s, 2H), 3,29-3,24 (m, 1H), 3,18-3,12 (m, 1H), 2,85-2,83 (m, 1H), 2,31-2,25 (m, 2H), 2,18 2,15 (m, 1H), 2,06 (d, 3H), 1,89-1,75 (m, 3H), 1,71-1,65 (m, 1H), 1,52-1,40 (m, 1H), 1,29 (dd, 3H).
Masa m/z: 478 [M++1].
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (A):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (Boc-Thr) (50 g, 228,3 mmol) en DMF (500 ml) se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15 min. A esta se le añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAc/hexano para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 73 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, 1H), 5,18-5,08 (m, 2H), 4,76 (d, 1H), 4,08-4,00 (m, 2H), 1.38 (s, 9H), 1,09 (d, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (B):
A una disolución con agitación del compuesto A (52 g, 168,2 mmol) en CH2Cl2 (500 ml) se le añadió Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a TA durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 * 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato B (52 g, 88 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (C):
A una disolución con agitación del compuesto B (52 g, 148,1 mmol) en MeOH (1 l) se le añadió Pd al 10 % /C bajo atmósfera de N2 y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el filtrado obtenido a vacío y se trituró el residuo bruto con hexano para producir ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico C (35 g, 90 %).
1 H-RMN: (500 MHz, DMSO-d6): 8 12,78 (s a, 1H), 6,94 (d, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1.39 (s, 9H), 1,10 (d, 3H).
Masa m/z: 260,0 [M+-1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-1-((2S,3R)-3-acetoxi-2-((terc-butoxicarbonil)amino)butanoil)pirrolidin-2-carbonil)-2-(4-fluorobencil)pirrolidin-2-carboxamido)butanoato de metilo (10):
A una disolución con agitación del compuesto C (4,53 g, 17,35 mmol) en CH2CI2 (69 ml) se le añadieron EDCI.HCl (4,14 g, 21,69 mmol), HOBt (2,95 g, 21,7 mmol) seguido de DIPEA (7,99 ml, 43,4 mmol) a 0 ° C y se agitó durante 10 min. A esta se le añadió el compuesto 9 (6,9 g, 14,46 mmol) y se agitó durante 16 h. Se diluyó la mezcla de reacción con agua (75 ml) y se extrajo con EtOAc (2 * 75 ml). Se secó la fase orgánica separada sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna eluyendo con el 2 % de MeOH/cH2Cl2 para proporcionar el compuesto 10 (8,0 g, 77 %).
1 H-RMN: (400 MHz, DMSO-d6) (rotámeros): 87,35-7,29 (m, 1H), 7,23 (m, 1H), 7,13 (d, 2H), 7,08-7,05 (m, 1H), 5,30 5,12 (m, 2H), 4,67-4,34 (m, 3H), 3,98-3,70 (m, 3H), 3,65 (s, 1H), 3,49-3,45 (m, 1H), 3,25-3,10 (m, 1H), 2,32-1,96 (m, 8 H), 1,75-1,62 (m, 2H), 1,49 (d, 9H), 1,32-1,29 (m, 3H), 1,25-1,20 (m, 3H).
CL-EM m/z: 621 [M++1].
Síntesis de ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(4-fluorobencil)pirrolidin-1 - carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11):
Una mezcla con agitación del compuesto 10 (4 g, 5,55 mmol) y NH3 metanólico (20 ml) se llevó a un tubo sellado y se agitó durante 48 h. Se concentró la mezcla de reacción a vacío para dar producto bruto; que se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 2 % de MeOH/C^Ch para proporcionar el compuesto 11 (1,6 g) como mezcla. Se sometió este material bruto a purificación prep. quiral.
HPLC preparativa quiral de isómeros:
Se separaron los isómeros del compuesto 11 (1,6 g, producto bruto) mediante HPLC preparativa quiral para obtener el compuesto 11-F1 (0,6 g) y el compuesto 11-F2 (0,25 g).
Datos analíticos para el compuesto 11-F1:
1 H-RMN: (400 MHz, DMSO-d6): 8 8,15 (s a, 1H), 7,39-7,35 (m, 2H), 7,24 (s, 1H), 7,11 (t, 2H), 7,04-7,00 (m, 1H), 6.79- 6,74 (m, 2H), 4,75 (d, 1H), 4,21 (t, 2H), 4,02-3,95 (m, 1H), 3,81-3,75 (m, 2H), 3,69-3,56 (m, 2H), 3,53-3,49 (m, 1H), 3,27-3,09 (m, 2H), 2,29-2,10 (m, 1H), 2,05-1,91 (m, 5H), 1,69-1,51 (m, 1H), 1,40 (s, 9H), 1,19-1,11 (m, 6 H), 0,92-0,84 (m, 1H).
HPLC: 85 % .
CL-EM (ESI): m/z 622 [M++1].
HPLC quiral: Rt = 10,69 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Datos analíticos para el compuesto 11-F2:
1 H-RMN: (400 MHz, DMSO-d6): 8 8,15 (s a, 1H), 7,39-7,35 (m, 2H), 7,24 (s, 1H), 7,11 (t, 2H), 7,04-7,00 (m, 1H), 6.79- 6,74 (m, 2H), 4,75 (d, 1H), 4,21 (t, 2H), 4,02-3,95 (m, 1H), 3,81-3,75 (m, 2H), 3,69-3,56 (m, 2H), 3,53-3,49 (m, 1H), 3,27-3,09 (m, 2H), 2,29-2,10 (m, 1H), 2,05-1,91 (m, 5H), 1,69-1,51 (m, 1H), 1,40 (s, 9H), 1,19-1,11 (m, 6 H), 0,92-0,84 (m, 1H).
HPLC: 84 % .
CL-EM (ESI): m/z 622 [M++1].
HPLC quiral: Rt= 18,55 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2- carbonil)-2-(4-fluorobencil)pirrolidin-2-carboxamida (CM-6 A):
A una disolución con agitación del compuesto 11-F1 (0,6 g, 0,96 mmol) en CH2Cl2 (12 ml) se le añadió HCl 4 M en 1,4-dioxano ( 6 ml) a 0 °C y se agitó a t A durante 1 h. Se concentró la mezcla de reacción a vacío para dar producto bruto; que se lavó adicionalmente con EtOAc y se secó a vacío para proporcionar CM-6 A (0,28 g, 52 %) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,41-7,32 (m, 2H), 7,21 (t, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,62 (m, 5H), 3.27 (d, 1H), 2,56-2,50 (m, 1H), 2,35-2,29 (m, 1H), 2,14-2,05 (m, 4H), 1,81-1,75 (m, 1H), 1,35 (d, 3H), 1,32 (d, 3H), 1.19- 1,11 (m, 1H).
CL-EM (ESI) m/z: 522 [M++1].
Pureza de UPLC: 96 %.
HPLC quiral: Rt = 7,73 min (Chiralpak IA, 250 x 4 , 6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Rotación óptica [a20D]: 7,15 (C = 1 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-fluorobencil)pirrolidin-2-carboxamida (CM-6B):
A una disolución con agitación del compuesto 11-F2 (0,25 g, 0,40 mmol) en CH2Cl2 (5 ml) se le añadió HCl 4 M en 1,4-dioxano (2,5 ml) a 0 °C y se agitó a TA durante 3 h. Se concentró la mezcla de reacción a vacío para dar producto bruto; que se lavó adicionalmente con EtOAc y se secó a vacío para proporcionar CM-6B (0,1 g, 45 %) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,41-7,32 (m, 2H), 7,21 (t, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,62 (m, 5H), 3.27 (d, 1H), 2,56-2,50 (m, 1H), 2,35-2,29 (m, 1H), 2,14-2,05 (m, 4H), 1,81-1,75 (m, 1H), 1,35 (d, 3H), 1,32 (d, 3H), 1.19- 1,11 (m, 1H).
CL-EM (ESI) m/z: 522 [M++1].
Pureza de UPLC: 98,38 %.
HPLC quiral: Rt = 11,28 min (Chiralpak IA, 250 x 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Rotación óptica [a20D]: -102,77 (C = 1 % en agua).
Ejemplo 7 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-(tr¡fluoromet¡l)bencil)p¡rrol¡d¡n-2-carboxam¡da (CM-7A) y (S)-N-((2S,3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-(tr¡fluorometil)benc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-7B):
Se usó la siguiente secuencia de reacción (esquema G) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamida (Cm -7A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamida (CM-7B):
Esquema G. Síntesis de CM-7A y CM-7B:

Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución con agitación de L-prolina (SM) (110 g, 956,5 mmol) en etanol se le añadió cloruro de tionilo (141 ml, 1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TA y se concentró a vacío para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %).
Síntesis de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-metilo (2):
A una disolución con agitación del compuesto 1 (170 g, 947 mmol) en CH2Cl2 se le añadió Et3N (398 ml, 2,83 mol). Tras agitarse durante 30 min, se le añadió Cbz-Cl (1,136 mol) a la mezcla de reacción y se continuó agitando durante otras 12 h a TA. Se lavó la mezcla de reacción con agua y se extrajo con CH2Cl2. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a vacío. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1H-RMN: (200 MHz, DMSO-da): 87,42-7,31(m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81(m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1],
Síntesis de 2-(4-(trifluorometil)bencil)pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución con agitación del compuesto 2 (5 g, 0,018 mol) en THF (50 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (27 ml, 0,026 mol) a -78 °C y se agitó durante 30 min. A esta se le añadió gota a gota 1-(bromometil)-4-(trifluorometil)benceno (3,3 ml, 0,021 mol) a -40 °C y se dejó calentar hasta TA y se agitó durante 3 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución acuosa de NH4Cl y se extrajo la fase acuosa con EtOAc (2 * 100 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 10 % de EtOAc/hexano para proporcionar el compuesto 3 (6 g, 76 % ) como líquido.
1H-RMN: (500 MHz, DMSO-d6): 87,64-7,58 (m, 2H), 7,44-7,25 (m, 7H), 5,22-5,12 (m, 2H), 4,16-4,12 (m, 1H), 4,05 3,89 (m, 1H), 3,62 (d, 1H), 3,48-3,39 (m, 1H), 3,16-3,10 (m, 1H), 2,99-2,89 (m, 1H), 2,19-1,99 (m, 2H), 1,63-1,58 (m, 1H), 1,19 (t, 2H), 1,05 (t, 1H), 0,92-0,89 (m, 1H).
Síntesis de 1-((benciloxi)carbonil)-2-(4-(trifluorometil) bencil) pirrolidin-2-carboxílico ácido (4):
A una disolución con agitación del compuesto 3 (6 g, 0,014 mol) en metanol (50 ml) y agua (15 ml) se le añadió NaOH acuoso 2 N (1,1 g, 0,027 mol) y se calentó hasta 70 °C durante 3 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl 2 N y se extrajo con EtOAc (2* 100 ml). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron a presión reducida para proporcionar el compuesto 4 (4 g, 71 % ) como un sólido blanquecino.
1H-RMN: (400 MHz, DMSO-da): 87,59-7,22 (m, 10H), 5,24-5,19 (m, 1H), 5,05-5,00 (m, 1H), 3,65 (d, 1H), 3,35-3,20 (m, 1H), 3,19 (t, 1H), 3,01-2,89 (m, 1H), 2,00-1,97 (m, 2H), 1,65-1,61 (m, 1H), 0,85-0,77 (m, 1H).
CL-EM (ESI): 406 [M+-1].
Masa m/z: 406 [M+-1].
Síntesis de 2-(((2R,3S)-3-hidroxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-(trifluorometil)bencil)pirrolidin-1-carboxilato de bencilo (5):
A una disolución con agitación del compuesto 4 (4 g, 9,80 mmol) en CH2Cl2 (40) se le añadieron EDCI (2,8 g, 14.7 mmol) seguido de HOBt (2,6 g, 19,6 mmol) y DlPEA (3,5 ml, 19,6 mmol) a 0 °C. Tras agitarse durante 10 min, se le añadió sal de clorhidrato de ester metílico de L-treonina (2 g, 11,8 mmol) a la mezcla de reacción y se agitó durante otras 16 h a TA. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con CH2Cl2 (150 ml) y se lavó con NaHCO3 acuoso (2 * 30 ml) seguido de ácido cítrico (2 * 30 ml). Se secó la fase orgánica sobre Na2SO4 y se concentró para proporcionar el compuesto 5 (5 g, producto bruto).
Síntesis de 2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-(trifluorometil)bencil)pirrolidin-1-carboxilato de bencilo (6):
A una disolución con agitación del compuesto 5 (5 g, 9,57 mmol) en CH2Cl2 (50 ml) se le añadió Et3N (4 ml, 28.7 mmol) seguido de Ac2O (2,7 ml, 28,7 mmol) a 0 °C y se agitó durante 1 h. A esta se le añadió DMAP (0,1 g) y se agitó a TA durante 16 h. Se evaporaron los compuestos volátiles a presión reducida y se purificó el material bruto obtenido mediante cromatografía en columna eluyendo con el 20 % de EtOAc/hexano para proporcionar el compuesto 6 (3,4 g, 63 % ) como líquido amarillo claro.
1H-RMN: (500 MHz, DMSO-d6) (rotámeros): 87,62-7,50 (m, 2H), 7,43-7,24 (m, 7H), 5,27-5,21 (m, 2H), 5,11-5,09 (m, 1H), 4,67-4,62 (m, 1H), 3,66-3,64 (m, 4H), 3,55-3,46 (m, 2H), 2,92-2,85 (m, 2H), 2,05-1,95 (m, 1H), 1,99 (s, 3H), 1,56-1,46 (m, 1H), 1,15-1,11 (m, 4H).
CL-EM (ESI): 565 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamido)butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (3,4 g, 6,02 mmol) en metanol (20 ml) se le añadió Pd al 10 % /C húmedo (0,34 g) bajo atmósfera de N2. Se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 6 h. Tras el consumo del material de partida (mediante CCF), se filtró la reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (2,1 g, 77 % ) como jarabe espeso.
1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,22 (s a, 1H), 7,62-7,56 (m, 2H), 7,45-7,39 (m, 2H), 5,23-5,19 (m, 1H), 4,50-4,42 (m, 1H), 3,60 (d, 3H), 3,05-3,00 (m, 1H), 2,97-2,85 (m, 3H), 2,18-2,09 (m, 1H), 1,95 (d, 3H), 1,65-1,51 (m, 3H), 1,10 (d, 2H), 0,75 (d, 1H).
CL-EM m/z: 431 [M++1],
Síntesis de (2S)-2-(2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-(trifluorometil)bencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (8):
A una disolución con agitación del compuesto 7 (4,5 g, 10,4 mmol) en CH2Cl2 (60 ml) y agua (40 ml) se le añadió Na2CO3 (2,2 g, 20,9 mmol) y se agitó a 0 °C durante 5 min. Se añadió (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo (3,3 g, 12,5 mmol) y se agitó la mezcla de reacción a TA durante 2 h. Se concentró la fase orgánica separada a vacío y se purificó el material bruto obtenido mediante cromatografía en columna eluyendo con el 50 % de EtOAc/hexano para proporcionar el compuesto 8 (4,5 g, 65 %) como líquido amarillo. Se sintetizó (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo de la siguiente manera. A una disolución de ácido (S)-1-(benciloxicarbonil)pirrolidin-2-carboxílico (20,6 g, 82,8 mmol) en CH2Cl2 (20 ml) se le añadió gota a gota SOCl2 (20,5 g, 172,6 mmol) a 0 °C y se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión
reducida para producir (S)-2-(dorocarbonil)pirrolidin-1-carboxilato de bencilo.
1 H-RMN: (400 MHz, DMSO-da) (rotámeros): 8 8,50 (s a, 1H), 7,79-7,65 (m, 2H), 7,52-7,37 (m, 7H), 5,42-5,05 (m, 3H), 4,79-4,59 (m, 1H), 4,15 (q, 1H), 3,78 (s, 3H), 3,69-3,50 (m, 3H), 3,21-3,13 (m, 1H), 2,45-2,16 (m, 2H), 2,10 (s, 6H), 2,05-2,00 (m, 2H), 1,95-1,85 (m, 1H), 1,44-1,19 (m, 6H).
CL-EM m/z: 662 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-pirrolidin-2-carbonil)-2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamido)butanoato de metilo (9):
A una disolución con agitación del compuesto 8 (4,5 g, 6,8 mmol) en MeOH (45 ml) se le añadió Pd al 10 % /C húmedo (0,45 g) bajo atmósfera inerte y se agitó durante 16 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9 (3,5 g, producto bruto) como semisólido.
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (A):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (Boc-Thr) (50 g, 228,3 mmol) en DMF (500 ml) se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15 min. A esta se le añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAc/hexano para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 73 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, 1H), 5,18-5,08 (m, 2H), 4,76 (d, 1H), 4,08-4,00 (m, 2H), 1.38 (s, 9H), 1,09 (d, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (B):
A una disolución con agitación del compuesto A (52 g, 168,2 mmol) en CH2Cl2 (500 ml) se le añadieron Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a t A durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 * 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato B (52 g, 88 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (C):
A una disolución con agitación del compuesto B (52 g, 148,1 mmol) en MeOH (1 l) se le añadió Pd al 10 % /C bajo atmósfera de N2 y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el filtrado obtenido a vacío y se trituró el residuo bruto con hexano para producir ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico C (35 g, 90 %).
1 H-RMN: (500 MHz, DMSO-d6): 8 12,78 (s a, 1H), 6,94 (d, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1.39 (s, 9H), 1,10 (d, 3H).
Masa m/z: 260,0 [M-1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-1-((2S,3R)-3-acetoxi-2-((terc-butoxicarbonil)amino)butanoil)pirrolidin-2-carbonil)-2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamido)butanoato de metilo (10):
A una disolución con agitación del compuesto C (1,78 g, 6,83 mmol) en CH2Cl2 (30 ml) se le añadieron EDCI (1,63 g, 8,53 mmol), HOBt (1,53 g, 11,38 mmol) seguido de DIPEA (2 ml, 14,2 mmol) a 0 °C y se agitó durante 10 min. A esta se le añadió el compuesto 9 (3 g, 5,69 mmol) y se agitó durante 16 h. Se extrajo la mezcla de reacción con EtOAc (2 * 75 ml) y se lavó la fase orgánica separada con NaHCO3 acuoso (100 ml), ácido cítrico acuoso (100 ml) seguido de salmuera (100 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna eluyendo con el 70 % de EtOAc/hexano para proporcionar el compuesto 10 (2,1 g, 51 % ) como un sólido blanquecino.
1 H-RMN: (400 MHz, DMSO-da) (rotámeros): 87,71-7,55 (m, 3H), 7,34-7,05 (m, 2H), 5,35-5,20 (m, 1H), 5,09-5,01 (m, 1H), 4,78-4,35 (m, 3H), 3,90-3,85 (m, 2H), 3,67 (s, 4H), 3,39-3,25 (m, 1H), 3,25-3,05 (m, 1H), 2,21-1,96 (m, 12H), 1,39 (d, 9H), 1,35-1,25 (m, 8 H), 0,85 (d, 1H).
Síntesis de ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(4-(trifluorometil)bencil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11):
A una disolución con agitación del compuesto 10 (1 g, 1,29 mmol) en MeOH (10 ml) se le añadió NH3 metanólico (20 ml) a 0 °C y se agitó a TA durante 24 h. Se concentró la mezcla de reacción a vacío para proporcionar el compuesto 11 (0,87 g, producto bruto) como un sólido blanquecino.
HPLC preparativa quiral de isómeros:
Se separaron los isómeros del compuesto 11 (0,3 g, producto bruto) mediante HPLC preparativa quiral para obtener el compuesto 11 (0,3 g) y el compuesto 11-F2 (0,15 g).
Datos analíticos para el compuesto 11:
1 H-RMN: (400 MHz, DMSO-da): 87,59-7,51 (m, 4H), 7,24 (s, 1H), 7,02 (s, 1H), 6,79-6,60 (m, 2H), 5,09 (d, 1H), 4,77 4,74 (m, 1H), 4,64-4,61 (m, 1H), 4,21 (t, 2H), 3,98 (dd, 1H), 3,85-3,80 (m, 2H), 3,71-3,60 (m, 3H), 3,25-3,19 (m, 2H), 2.21- 1,85 (m, 5H), 1,75-1,70 (m, 1H), 1,44 (s, 10H), 1,21 (d, 3H), 1,09 (d, 3H), 0,91-0,85 (m, 1H).
HPLC:93 % .
CL-EM (ESI): m/z 671,6 [M++1].
HPLC quiral: Rt= 11,70 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1 , 00 ml/min).
Datos analíticos para el compuesto 11-F2:
1 H-RMN: (400 MHz, DMSO-d6): 87,59-7,51 (m, 4H), 7,24 (s, 1H), 7,02 (s, 1H), 6,79-6,60 (m, 2H), 5,09 (d, 1H), 4,77 4,74 (m, 1H), 4,64-4,61 (m, 1H), 4,21 (t, 2H), 3,98 (dd, 1H), 3,85-3,80 (m, 2H), 3,71-3,60 (m, 3H), 3,25-3,19 (m, 2H), 2.21- 1,85 (m, 5H), 1,75-1,70 (m, 1H), 1,44 (s, 10H), 1,21 (d, 3H), 1,09 (d, 3H), 0,91-0,85 (m, 1H).
HPLC: 91 % .
CL-EM (ESI): m/z 671,6 [M++1].
HPLC quiral: Rt= 19,68 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1 , 00 ml/min).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S, 3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamida (CM-7A):
A una disolución con agitación del compuesto 11-F1 (0,3 g, 0,45 mmol) en CH2Cl2 (5 ml) se le añadió HCl 4 N en 1,4-dioxano (1 ml) a 0 °C y se agitó a t A durante 3 h. Se concentró la mezcla de reacción a vacío para proporcionar CM-7A (0,15 g, 59 % ) como sal de clorhidrato.
1 H-RMN: (400 MHz, D2O): 87,89 (d, 2H), 7,57 (d, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,79 (m, 5H), 3,35 (q, 2H), 2,56-2,50 (m, 1H), 2,35-2,29 (m, 2H), 2,14-2,05 (m, 3H), 1,81-1,75 (m, 1H), 1,35 (d, 3H), 1,32 (d, 3H), 1,19 1,11 (m, 1H).
CL-EM (ESI) m/z: 572,6 [M++1].
Masa m/z: 572,4 [M++1].
Pureza de UPLC: 97 %.
Rotación óptica [a20o]: 14,86 (C = 1 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-(trifluorometil)bencil)pirrolidin-2-carboxamida (CM-7B):
A una disolución con agitación del compuesto 11-F2 (0,15 g, 0,22 mmol) en CH2CI2 (5 ml) se le añadió HCl 4 N en 1,4-dioxano (0,5 ml) a 0 °C y se agitó a TA durante 3 h. Se concentró la mezcla de reacción a vacío para proporcionar CM-7B (140 mg, 59 %) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,89 (d, 2H), 7,57 (d, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,79 (m, 5H), 3,35 (q, 2H), 2,56-2,50 (m, 1H), 2,35-2,29 (m, 2H), 2,14-2,05 (m, 3H), 1,81-1,75 (m, 1H), 1,35 (d, 3H), 1,32 (d, 3H), 1,19 1,11 (m, 1H).
CL-EM (ESI) m/z: 572,8 [M++1].
Masa m/z: 572,4 [M++1].
Pureza de UPLC: 97,67 %.
Rotación óptica [a20D]: -105,51 (C = 1 % en agua).
Ejemplo 8 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbonil)-2-(3-met¡lbenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-8A) y (S)-N-((2S,3R)-1-amino-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-met¡lbenc¡l)p¡rrol¡d¡n-2-carboxamida (CM-8B):
Se usó la siguiente secuencia de reacción (esquema H) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metilbencil)pirrolidin-2-carboxamida (CM-8A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metilbencil)pirrolidin-2-carboxamida (CM-8B):
Esquema H. Síntesis de CM-8A y CM-8B

Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución con agitación de L-prolina (SM) (110 g, 956,5 mmol) en etanol se le añadió cloruro de tionilo (141 ml, 1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TA y se concentró a vacío para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %).
Síntesis de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-metilo (2):
A una disolución con agitación del compuesto 1 (170 g, 947 mmol) en CH2CI2 se le añadió Et3N (398 ml, 2,83 mol). Tras agitarse durante 30 min, se le añadió Cbz-Cl (1,136 mol) a la mezcla de reacción y se continuó agitando durante otras 12 h a TA. Se lavó la mezcla de reacción con agua y se extrajo con CH2Cl2. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a vacío. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1H-RMN: (200 MHz, DMSO-da): 87,42-7,31(m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81(m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1].
Síntesis de 2-(3-metilbencil)pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución con agitación del compuesto 2 (5 g, 0,018 mol) en THF (50 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (37 ml, 0,036 mol) a -78 °C y se agitó durante 30 min. A esta se le añadió gota a gota bromuro de 3-metilbencilo (4 g, 0,021 mol) a -40 °C y se dejó calentar hasta TA y se agitó durante 2 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 x 100 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtoAc/hexano para proporcionar el compuesto 3 (6 g, 87 % ) como líquido.
1H-RMN: (500 MHz, DMSO-da): 87,47-7,32 (m, 5H), 7,17-7,12 (m, 1H), 7,05-7,01 (m, 1H), 6,95-6,90 (m, 2H), 5,25 5,00 (m, 2H), 4,16-4,12 (m, 1H), 4,00-3,89 (m, 1H), 3,52 (d, 1H), 3,38-3,33 (m, 1H), 2,99-2,89 (m, 2H), 2,23 (s, 3H), 2,12-1,90 (m, 1H), 1,56-1,51 (m, 1H), 1,05-1,01 (m, 3H), 1,00-0,97 (m, 1H), 0,92-0,89 (m, 1H).
CL-EM (ESI): 382 [M++1].
HPLC (pureza): 99 % .
Síntesis de ácido 1-((benciloxi)carbonil)-2-(3-metilbencil)pirrolidin-2-carboxílico (4):
A una disolución con agitación del compuesto 3 (6 g, 0,015 mol) en metanol (30 ml) y agua (15 ml) se le añadió NaOH acuoso 2 N (2 g, 0,052 mol) y se calentó hasta 85 °C durante 6 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl 2 N y se extrajo con EtOAc (2 x 100 ml). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron a presión reducida para proporcionar el compuesto 4 (3,3 g, 78 % ) como un sólido blanquecino.
1H-RMN: (400 MHz, DMSO-d6): 812,71 (s a, 1H), 7,40-7,30 (m, 5H), 7,15-7,11 (m, 1H), 7,07-7,00 (m, 1H), 6,97-6,85 (m, 2H), 5,27-5,20 (m, 1H), 5,05-5,00 (m, 1H), 3,65 (d, 1H), 3,35-3,20 (m, 1H), 3,00-2,85 (m, 2H), 2,26 (s, 3H), 2,00 1,97 (m, 2H), 1,65-1,61 (m, 1H), 0,85-0,77 (m, 1H).
HPLC (pureza): 99,78 % .
Síntesis de 2-(((2R,3S)-3-hidroxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(3-metilbencil)pirrolidin-1-carboxilato de bencilo (5):
A una disolución con agitación del compuesto 4 (3,3 g, 9,0 mmol) en CH2Cl2 (30) se le añadieron EDCI.HCl (2,2 g, 11,4 mmol) seguido de HOBt (1,82 g, 13,5 mmol) y DIPEA (6,1 ml, 27,0 mmol) a 0 °C. Tras agitarse durante 10 min, se añadió sal de clorhidrato de ester metílico de L-treonina (1,95 g, 11,2 mmol) a la mezcla de reacción y se agitó durante otras 16 h a TA. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con EtOAc (150 ml) y se lavó con agua (2 x 30 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4, se concentró y se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 2 % de MeOH/CH2Cl2 para proporcionar el compuesto 5 (3,15 g, 73 %).
1H-RMN: (500 MHz, DMSO-d6): 87,64 (s a, 1H), 7,44-7,31 (m, 5H), 7,12-7,00 (m, 2H), 6,95-6,85 (m, 2H), 5,32-5,25 (m, 1H), 5,05-4,94 (m, 2H), 4,25-4,20 (m, 1H), 4,15-4,08 (m, 1H), 3,66-3,64 (m, 2H), 3,45-3,41 (m, 2H), 3,14-3,09 (m, 1H), 2,89-2,84 (m, 1H), 2,20 (s, 3H), 2,05-2,02 (m, 2H), 1,55-1,51 (m, 1H), 1,09-0,98 (m, 4H).
CL-EM (ESI): 469 [M++1].
Síntesis de 2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(3-metilbencil)pirrolidin-1-carboxilato de bencilo (6):
A una disolución con agitación del compuesto 5 (3,1 g, 6,4 mmol) en CH2CI2 (30 ml) se le añadió Et3N (1,4 ml, 5,6 mmol) seguido de Ac2O (0,8 ml, 8,4 mmol) a 0 °C y se agitó durante 1 h. A esta se le añadió DMAP (0,1 g) y se agitó a TA durante 6 h. Se evaporaron los compuestos volátiles a presión reducida y se purificó el material bruto obtenido mediante cromatografía en columna eluyendo con el 20 % de EtOAc/hexano para proporcionar el compuesto 6 (3,15 g, 92,5 %).
1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,25 (s a, 1H), 7,42-7,30 (m, 5H), 7,12-7,00 (m, 2H), 6,98-6,89 (m, 2H), 5,25-5,21 (m, 2H), 5,11-5,09 (m, 1H), 4,67-4,62 (m, 1H), 3,66-3,64 (m, 2H), 3,55-3,46 (m, 3H), 3,15-3,10 (m, 1H), 2,92-2,85 (m, 1H), 2,20 (s, 3H), 2,05-1,95 (m, 5H), 1,56-1,46 (m, 1H), 1,15-1,11 (m, 4H).
CL-EM (ESI): 511 [M++1].
HPLC (pureza): 98 % .
Síntesis de (2S,3R)-3-acetoxi-2-(2-(3-metilbencil)pirrolidin-2-carboxamido)butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (3,15 g, 5,97 mmol) en metanol (30 ml) se le añadió Pd al 10 % /C húmedo (1,0 g) bajo atmósfera de N2. Se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 4 h. Tras el consumo del material de partida (mediante CCF), se filtró la reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (2,35 g, 99,5 %) como jarabe. 1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,22 (s a, 1H), 7,19-7,16 (m, 1H), 7,00-6,95 (m, 3H), 5,23-5,19 (m, 1H), 4,50-4,42 (m, 1H), 3,60 (d, 3H), 3,20 (m, 1H), 3,00-2,97 (m, 1H), 2,75-2,68 (m, 2H), 2,28 (s, 3H), 2,05-2,02 (m, 1H), 1,90 (d, 3H), 1,65-1,51 (m, 3H), 1,10 (dd, 3H).
CL-EM m/z: 377 [M++1].
HPLC (pureza): 98,5 %.
Síntesis de (2S)-2-(2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(3-metilbencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (8):
A una disolución con agitación del compuesto 7 (2,35 g, 6,30 mmol) en CH2Cl2 (60 ml) y agua (40 ml) se le añadió Na2CO3 (2,0 g, 18,8 mmol) y se agitó a 0 °C durante 5 min. Se le añadió (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo (2,12 g, 7,6 mmol) y se agitó la mezcla de reacción a TA durante 2 h. Se concentró la fase orgánica separada a vacío y se purificó el material bruto obtenido mediante cromatografía en columna eluyendo con el 3 % de MeOH/CH2Cl2 para proporcionar el compuesto 8 (2,5 g, 63%). Se sintetizó (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo de la siguiente manera. A una disolución de ácido (S)-1-(benciloxicarbonil)pirrolidin-2-carboxílico (20,6 g, 82,8 mmol) en CH2Cl2 (20 ml) se le añadió SOCl2 (20,5 g, 172,6 mmol) gota a gota a 0 °C y se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión reducida para producir (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo.
1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,50 (s a, 1H), 7,36-7,23 (m, 5H), 7,15-6,85 (m, 5H), 5,21-5,05 (m, 2H), 5,04-4,92 (m, 1H), 4,65-4,50 (m, 1H), 4,53-4,45 (m, 1H), 3,65 (s, 3H), 3,54-3,46 (m, 4H), 3,21-3,13 (m, 2H), 2,25-2,16 (m, 4H), 2,05-2,00 (m, 2H), 1,95-1,85 (m, 4H), 1,56-1,51 (m, 2H), 1,15 (dd, 3H).
CL-EM m/z: 608 [M++1].
HPLC (pureza): 91,3 %.
Síntesis de (2S,3R)-3-acetoxi-2-(2-(3-metilbencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (9):
A una disolución con agitación del compuesto 8 (2,5 g, 4,0 mmol) en MeOH (30 ml) se le añadió Pd al 10 % /C húmedo (0,5 g) bajo atmósfera inerte y se agitó durante 4 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9 (1,56 g, 79,5 %).
1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,23 (dd, 1H), 7,20-6,85 (m, 5H), 5,20-5,13 (m, 1H), 4,63-4,59 (m, 1H), 3,85-3,81 (m, 1H), 3,65-3,61 (m, 5H), 3,32-3,25 (m, 2H), 3,12-3,05 (m, 3H), 2,75-2,71 (m, 1H), 2,25 (s, 3H), 2,15-2,13 (m, 2H), 2,00 (d, 3H), 1,77 (m, 3H), 1,27 (dd, 4H).
CL-EM m/z: 474 [M++1].
HPLC (pureza): 87,3 %.
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (A):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (Boc-Thr) (50 g, 228,3 mmol) en DMF (500 ml) se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15 min. A esta se le añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAc/hexano para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 73 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, 1H), 5,18-5,08 (m, 2H), 4,76 (d, 1H), 4,08-4,00 (m, 2H), 1.38 (s, 9H), 1,09 (d, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (B):
A una disolución con agitación del compuesto A (52 g, 168,2 mmol) en CH2Cl2 (500 ml) se le añadieron Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a t A durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 * 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato B (52 g, 88 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (C):
A una disolución con agitación del compuesto B (52 g, 148,1 mmol) en MeOH (1 l) se le añadió Pd al 10 % /C bajo atmósfera de N2 y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el filtrado obtenido a vacío y se trituró el residuo bruto con hexano para producir ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico C (35 g, 90 %).
1 H-RMN: (500 MHz, DMSO-d6): 8 12,78 (s a, 1H), 6,94 (d, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1.39 (s, 9H), 1,10 (d, 3H).
Masa m/z: 260,0 [M-1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-1-((2S,3R)-2-((terc-butoxicarbonil)amino)-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metilbencil)pirrolidin-2-carboxamido)butanoato de metilo (10):
A una disolución con agitación del compuesto C (10 g, 0,02 mol) en CH2Cl2 (100 ml) se le añadieron EDCI (5 g, 0,02 mol), HOBt (5 g, 0,04 mol) seguido de DIPEA (10,6 ml, 0,06 mol) a 0 °C y se agitó durante 10 min. A esta se le añadió el compuesto 9 (7 g, 0,025 mol) y se agitó durante 15 h. Se extrajo la mezcla de reacción con EtOAc (2 * 75 ml) y se lavó la fase orgánica separada con agua (200 ml), seguido de salmuera (200 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna eluyendo con el 4 % de MeOH/CH2Cl2 para proporcionar el compuesto 10 (10 g, 67 %) como jarabe.
1 H-RMN: (400 MHz, DMSO-d6) (rotámeros): 87,24-6,91 (m, 5H), 5,42-5,01 (m, 2H), 4,73-4,68 (m, 1H), 4,54-4,41 (m, 1H), 3,90-3,85 (m, 1H), 3,67 (s, 3H), 3,65-3,60 (m, 2H), 3,59-3,55 (m, 1H), 3,59-3,55 (m, 1H), 3,30 (s, 3H), 3,12-3,05 (m, 1H), 2,33 (s, 3H), 2,05 (s, 3H), 2,00-1,96 (m, 4H), 1,94-1,85 (m, 4H), 1,43 (s, 9H), 1,25 (d, 3H), 1,15 (d, 3H). CL-EM (ESI): m/z 675 [M++1].
HPLC: 71 % .
Síntesis de ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3-metilbencil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11):
A una disolución con agitación del compuesto 10 (10 g, 0,014 mol) en MeOH (20 ml) se le añadió NH3 metanólico (100 ml) y se llevó a un tubo sellado. Se agitó la mezcla de reacción a TA durante 20 h. Se concentró la mezcla de reacción a presión reducida. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 4 % de MeOH/CH2Cl2 para proporcionar el compuesto 11 (8,0 g, rendimiento del 99 %).
1H-RMN: (400 MHz, DMSO-da) (rotámeros): 87,32 (s a, 2H), 7,15-7,10 (m, 4H), 7,05-6,91 (m, 4H), 6,65 (s a, 2H), 4,75-4,59 (m, 2H), 4,24-3,45 (m, 8H), 3,35 (s, 1H), 3,05-3,00 (m, 1H), 2,32 (s, 3H), 2,26-1,85 (m, 6H), 1,75 (s, 6H), 1,60-1,55 (m, 1H), 1,47 (s, 9H), 1,15 (d, 3H), 1,05 (d, 3H).
CL-EM (ESI): m/z 618 [M++1].
HPLC preparativa quiral de isómeros:
Se separaron los isómeros del compuesto 11 (8,0 g, 12,9 mmol) mediante HPLC preparativa quiral para obtener el compuesto 11-F1 (0,4 g) y el compuesto 11-F2 (0,25 g).
Datos analíticos para el compuesto 11-F1:
HPLC: 91,1 %.
CL-EM (ESI): m/z 618 [M++1].
HPLC quiral: Rt = 9,20 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Datos analíticos para el compuesto 11-F2:
HPLC: 84 % .
CL-EM (ESI): m/z 618 [M++1].
HPLC quiral: Rt = 13,39 min (Chiralpak IA, 250 * 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metilbencil)pirrolidin-2-carboxamida (CM-8A):
A una disolución con agitación del compuesto 12-F1 (0,4 g, 0,60 mmol) en CH2Cl2 (20 ml) se le añadió HCl 2 N en 1.4- dioxano (2 ml) a 0 °C y se agitó a TA durante 3 h. Se concentró la mezcla de reacción a presión reducida y se trituró el material obtenido con EtOAc y se secó a vacío para proporcionar CM-8A (0,22 g, 66 % ) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,35 (t, 1H), 7,25 (s, 1H), 7,21 (d, 1H), 7,15 (d, 1H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,89 (m, 1H), 3,87-3,78 (m, 2H), 3,63 (d, 1H), 3,49-3,40 (m, 1H), 3,25 (d, 1H), 2,56-2,34 (m, 5H), 2,28-2,20 (m, 4H), 1,75-1,71 (m, 1H), 1,56 (d, 3H), 1,32 (d, 3H), 1,15-1,11 (m, 1H).
CL-EM (ESI) m/z: 518 [M++1].
Pureza de HPLC: 97 %.
Rotación óptica [a25o]: 18,8 (C = 1 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metilbencil)pirrolidin-2-carboxamida (CM-8B):
A una disolución con agitación del compuesto 12-F2 (0,25 g, 0,40 mmol) en CH2Cl2 (20 ml) se le añadió HCl 2 N en 1.4- dioxano (2 ml) a 0 °C y se agitó a Ta durante 3 h. Se concentró la mezcla de reacción a presión reducida y se trituró el material obtenido con EtOAc y se secó a vacío para proporcionar CM-8B (130 mg, 62% ) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,35-7,23 (m, 2H), 7,12-7,00 (m, 2H), 4,45-4,40 (m, 1H), 4,32-4,10 (m, 3H), 3,94-3,75 (m, 3H), 3,60 (d, 1H), 3,25 (d, 1H), 3,14-3,11 (m, 1H), 2,56-2,05 (m, 10H), 1,85-1,79 (m, 2H), 1,45 (d, 3H), 1,25 (d, 3H).
CL-EM (ESI) m/z: 518 [M++1].
Pureza de HPLC: 92 %.
Rotación óptica [a25o]: -104,9 (C = 1 % en agua).
Ejemplo 8 - Síntesis de (R)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-9A) y (S)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-9B):
Se usó la s¡gu¡ente secuenc¡a de reacc¡ón (esquema I) para s¡ntet¡zar (R)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-9A) y (S)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-fluorobenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-9B):
Esquema I. Síntes¡s de CM-9A y CM-9B:

Síntes¡s de clorh¡drato de (S)-p¡rrol¡d¡n-2-carbox¡lato de et¡lo (1):
A una d¡soluc¡ón con ag¡tac¡ón de L-prol¡na (SM) (110 g. 956.5 mmol) en etanol se le añad¡ó cloruro de t¡on¡lo (141 ml. 1911.3 mmol) y se somet¡ó a reflujo durante 16 h. Se llevó la mezcla de reacc¡ón hasta TA y se concentró a vacío para proporc¡onar el compuesto 1 como sal de clorh¡drato (170 g. 99 %).
Síntes¡s de (S)-p¡rrol¡d¡n-1.2-d¡carbox¡lato de 1-benc¡lo y 2-met¡lo (2):
A una d¡soluc¡ón con ag¡tac¡ón del compuesto 1 (170 g. 947 mmol) en CH2Cl2 se le añad¡ó Et3N (398 ml. 2.83 mol). Tras ag¡tarse durante 30 m¡n. se le añad¡ó Cbz-Cl (1.136 mol) a la mezcla de reacc¡ón y se cont¡nuó ag¡tando durante otras 12 h a TA. Se lavó la mezcla de reacc¡ón con agua y se extrajo con CH2Cl2. Se secó la fase orgán¡ca sobre Na2SO4 anh¡dro y se concentró a vacío. Se pur¡f¡có el producto bruto med¡ante cromatografía en columna para proporc¡onar el compuesto 2 (230 g. 88 %).
1H-RMN: (200 MHz. DMSO-da): 87.42-7.31(m. 5H). 5.09 (m. 2H). 4.32-4.23 (m. 1H). 4.08-3.98 (m. 2H). 3.50-3.38 (m. 2H). 2.30-2.18 (m. 1H). 1.90-1.81(m. 3H). 1.10-1.04 (t. 3H).
Masa m/z: 278 [M++1].
Síntes¡s de 2-(3-fluorobenc¡l)p¡rrol¡d¡n-1.2-d¡carbox¡lato de 1-benc¡lo y 2-et¡lo (3):
A una d¡soluc¡ón con ag¡tac¡ón del compuesto 2 (10 g. 0.018 mol) en THF (100 ml) bajo atmósfera ¡nerte se le añad¡ó
LiHMDS (1 M en THF) (76 ml, 0,076 mol) a -78 °C y se agitó durante 1 h. A esta se le añadió gota a gota bromuro de 3-fluorobencilo (5,6 ml, 0,045 mol) a -30 °C y se agitó durante 3 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 * 100 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 12 % de EtOAc/hexano para proporcionar el compuesto 3 (11 g, 78 % ) como líquido.
1H-RMN: (400 MHz, DMSO-da): 87,64-7,58 (m, 2H), 7,44-7,25 (m, 7H), 5,22-5,12 (m, 2H), 4,16-4,12 (m, 1H), 4,05 3,89 (m, 1H), 3,62 (d, 1H), 3,48-3,39 (m, 1H), 3,16-3,10 (m, 1H), 2,99-2,89 (m, 1H), 2,19-1,99 (m, 2H), 1,63-1,58 (m, 1H), 1,19 (t, 2H), 1,05 (t, 1H), 0,92-0,89 (m, 1H).
CL-EM (ESI): 372 [M++1].
Síntesis de ácido 1-((benciloxi)carbonil)-2-(3-fluorobencil)pirrolidin-2-carboxílico (4):
A una disolución con agitación del compuesto 3 (11 g, 0,029 mol) en metanol (50 ml) y agua (50 ml) se le añadió NaOH acuoso 2 N (3,5 g, 0,089 mol) y se calentó hasta 60 °C durante 3 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl 2 N y se extrajo con EtOAc (2 * 100 ml). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron a presión reducida para proporcionar el compuesto 4 (9,5 g, 90 % ) como líquido.
1H-RMN: (400 MHz, DMSO-d6): 87,59-7,22 (m, 10H), 5,24-5,19 (m, 1H), 5,05-5,00 (m, 1H), 3,65 (d, 1H), 3,35-3,20 (m, 1H), 3,19 (t, 1H), 3,01-2,89 (m, 1H), 2,00-1,97 (m, 2H), 1,65-1,61 (m, 1H), 0,85-0,77 (m, 1H).
CL-EM (ESI): 358 [M++1].
Síntesis de 2-(3-fluorobencil)-2-(((2R, 3S)-3-hidroxi-1-metoxi-1-oxobutan-2-il)carbamoil)pirrolidin-1-carboxilato de bencilo (5):
A una disolución con agitación del compuesto 4 (9,5 g, 26,6 mmol) en CH2Cl2 (100 ml) se le añadieron EDCI.HCl (6,1 g, 31,9 mmol) seguido de HOBt (5,4 g, 39,9 mmol) y DIPEA (14 ml, 79,8 mmol) a 0 °C. Tras agitarse durante 10 min, se le añadió sal de clorhidrato de ester metílico de L-treonina (4,97 g, 29,2 mmol) a la mezcla de reacción y se agitó durante otras 16 h a TA. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con CH2Cl2 (150 ml), se lavó con salmuera, se secó sobre Na2SO4 y se concentró para proporcionar el compuesto 5 (13 g, producto bruto).
CL-EM (ESI): 473,7 [M++1].
Síntesis de 2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(3-fluorobencil)pirrolidin-1-carboxilato de bencilo (6):
A una disolución con agitación del compuesto 5 (13 g, 27,5 mmol) en CH2Cl2 (100 ml) se le añadió Et3N (5,8 ml, 41,3 mmol) seguido de Ac2O (3,23 ml, 33,0 mmol) a 0 °C y se agitó durante 10 min. A esta se le añadió DMAP (1,2 g) y se agitó a TA durante 2 h. Se lavó la mezcla de reacción con ácido cítrico y se extrajo con CH2Cl2 (2 * 150 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida para conseguir producto bruto; que se purificó mediante cromatografía en columna eluyendo con el 20 % de EtOAc/hexano para proporcionar el compuesto 6 (9,5 g, 67 % ) como líquido amarillo claro.
1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 87,39-7,23 (m, 6H), 7,05 (t, 1H), 6,95-6,78 (m, 2H), 5,27-5,21 (m, 2H), 5,11-5,09 (m, 1H), 4,67-4,62 (m, 1H), 3,66-3,64 (m, 4H), 3,55-3,46 (m, 2H), 2,92-2,85 (m, 2H), 2,05-1,95 (m, 1H), 1,99 (s, 3H), 1,56-1,46 (m, 1H), 1,15-1,11 (m, 4H).
CL-EM (ESI): 515 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-(3-fluorobencil)pirrolidin-2-carboxamido)butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (9,5 g, 18,4 mmol) en metanol (100 ml) se le añadió Pd al 10 % /C húmedo (1,2 g) bajo atmósfera de N2. Se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 3 h. Tras el consumo del material de partida (mediante CCF), se filtró la reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (6,3 g, 90 %) como líquido. 1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,22 (s a, 1H), 7,62-7,56 (m, 2H), 7,45-7,39 (m, 2H), 5,23-5,19 (m, 1H), 4,50-4,42 (m, 1H), 3,60 (d, 3H), 3,05-3,00 (m, 1H), 2,97-2,85 (m, 3H), 2,18-2,09 (m, 1H), 1,95 (d, 3H), 1,65-1,51 (m, 3H), 1,10 (d, 2H), 0,75 (d, 1H).
CL-EM m/z: 381 [M++1].
Síntesis de (2S)-2-(2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(3-fluorobencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (8):
A una disolución con agitación del compuesto 7 (6,2 g, 16,3 mmol) en CH2Cl2 (50 ml) y agua (50 ml) se le añadió Na2CO3 (4,3 g, 40,7 mmol) y se agitó a 0 °C durante 5 min. Se le añadió (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo (17,93 mmol) y se agitó la mezcla de reacción a TA durante 2 h. Se lavó la mezcla de reacción con agua y se extrajo con CH2O 2 (2* 100 ml). Se concentró la fase orgánica separada a vacío y se purificó el material bruto obtenido mediante cromatografía en columna eluyendo con el 30 % de EtOAc/hexano para proporcionar el compuesto 8 (7,3 g, 73 %) como líquido. Se sintetizó (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo de la siguiente manera. A una disolución de ácido (S)-1-(benciloxicarbonil)pirrolidin-2-carboxílico (4,87 g, 19,5 mmol) en CH2Cl2 (20 ml) se le añadió SOCl2 (3,0 ml, 40,7 mmol) gota a gota a 0 °C y se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión reducida para producir (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo.
1 H-RMN: (400 MHz, DMSO-da) (rotámeros): 8 8,50 (s a, 1H), 7,79-7,65 (m, 2H), 7,52-7,37 (m, 7H), 5,42-5,05 (m, 3H), 4,79-4,59 (m, 1H), 4,15 (q, 1H), 3,78 (s, 3H), 3,69-3,50 (m, 2H), 3,21-3,13 (m, 1H), 2,45-2,16 (m, 2H), 2,10 (s, 6H), 2,05-2,00 (m, 2H), 1,95-1,85 (m, 1H), 1,44-1,19 (m, 6H).
CL-EM m/z: 612 [M++1], 613 [M++2].
Síntesis de (2S, 3R)-3-acetoxi-2-(2-(3-fluorobencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (9):
A una disolución con agitación del compuesto 8 (7,2 g, 11,78 mmol) en MeOH (80 ml) se le añadió Pd al 10 % /C húmedo (1,2 g) bajo atmósfera inerte y se agitó durante 16 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9 (5,2 g, 92 % ) como líquido.
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (A):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (Boc-Thr) (50 g, 228,3 mmol) en DMF (500 ml) se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15 min. A esta se le añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAc/hexano para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 73 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, 1H), 5,18-5,08 (m, 2H), 4,76 (d, 1H), 4,08-4,00 (m, 2H), 1,38 (s, 9H), 1,09 (d, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (B):
A una disolución con agitación del compuesto A (52 g, 168,2 mmol) en CH2Cl2 (500 ml) se le añadió Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a TA durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 * 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato B (52 g, 88 %).
1 H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (C):
A una disolución con agitación del compuesto B (52 g, 148,1 mmol) en MeOH (1 l) se le añadió Pd al 10 % /C bajo atmósfera de N2 y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el filtrado obtenido a vacío y se trituró el residuo bruto con hexano para producir ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico C (35 g, 90 %).
1 H-RMN: (500 MHz, DMSO-da): 8 12,78 (s a, 1H), 6,94 (d, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1.39 (s, 9H), 1,10 (d, 3H).
Masa m/z: 260,0 [M-1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-1-((2S,3R)-2-((terc-butoxicarbonil)amino)-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-fluorobencil) pirrolidin-2-carboxamido) butanoato de metilo (10):
A una disolución con agitación del compuesto C (3,41 g, 13,0 mmol) en CH2Cl2 (50 ml) se le añadieron EDCI (3,12 g, 16,3 mmol), HOBt (2,2 g, 16,3 mmol) seguido de DIPEA (5,7 ml, 32,7 mmol) a 0 °C y se agitó durante 10 min. A esta se le añadió el compuesto 9 (5,2 g, 10,9 mmol) y se agitó durante 16 h. Se extrajo la mezcla de reacción con CH2O 2 ( 2 x 75 ml) y se lavó la fase orgánica separada con NaHCO3 acuoso ( 10 0 ml), ácido cítrico acuoso ( 100 ml) seguido de salmuera (100 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna eluyendo con el 2 % de MeOH/CH2Cl2 para proporcionar el compuesto 10 (6,0 g, 77 % ) como líquido.
1 H-RMN: (400 MHz, DMSO-d6) (rotámeros): 87,71-7,55 (m, 3H), 7,34-7,05 (m, 2H), 5,35-5,20 (m, 1H), 5,09-5,01 (m, 1H), 4,78-4,35 (m, 3H), 3,90-3,85 (m, 2H), 3,67 (s, 3H), 3,39-3,25 (m, 1H), 3,25-3,05 (m, 1H), 2,21-1,96 (m, 12H), 1.39 (d, 9H), 1,35-1,25 (m, 8 H), 0,85 (d, 1H).
CL-EM (ESI): m/z 478,4 [M++1], 479,4 [M++2].
Síntesis de ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il) carbamoil)-2-(3-fluorobencil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11):
A disolución del compuesto 10 (3 g, 4,1 mmol) en amoníaco metanólico (50 ml) se agitó a TA durante 48 h. Se concentró la mezcla de reacción a vacío para dar producto bruto; que se purificó mediante cromatografía en columna de gel de sílice eluyendo con el 4 % de MeOH/CH2Cl2 para proporcionar el compuesto 11 (1,7 g, producto bruto) como sólido blanco.
1 H-RMN: (400 MHz, DMSO-d6): 87,59-7,51 (m, 4H), 7,24 (s, 1H), 7,02 (s, 1H), 6,79-6,60 (m, 2H), 5,09 (d, 1H), 4,77 4,74 (m, 1H), 4,64-4,61 (m, 1H), 4,21 (t, 2H), 3,98 (dd, 1H), 3,85-3,80 (m, 2H), 3,71-3,60 (m, 3H), 3,25-3,19 (m, 2H), 2.21- 1,85 (m, 5H), 1,75-1,70 (m, 1H), 1,44 (s, 10H), 1,21 (d, 3H), 1,09 (d, 3H), 0,91-0,85 (m, 1H).
CL-EM (ESI): m/z 622 [M++1].
HPLC preparativa quiral de isómeros:
Se separaron los isómeros del compuesto 11 (1,7 g) mediante HPLC preparativa quiral para obtener el compuesto 11 (0,4 g) y el compuesto 11-F2 (0,3 g).
Datos analíticos para el compuesto 11:
1 H-RMN: (400 MHz, DMSO-d6): 87,59-7,51 (m, 4H), 7,24 (s, 1H), 7,02 (s, 1H), 6,79-6,60 (m, 2H), 5,09 (d, 1H), 4,77 4,74 (m, 1H), 4,64-4,61 (m, 1H), 4,21 (t, 2H), 3,98 (dd, 1H), 3,85-3,80 (m, 2H), 3,71-3,60 (m, 3H), 3,25-3,19 (m, 2H), 2.21- 1,85 (m, 5H), 1,75-1,70 (m, 1H), 1,44 (s, 10H), 1,21 (d, 3H), 1,09 (d, 3H), 0,91-0,85 (m, 1H).
UPLC:87 % .
CL-EM (ESI): m/z 622 [M++1].
HPLC quiral: Rt = 10,32 min (Chiralpak IA, 250 x 4,6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH (4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Datos analíticos para el compuesto 11-F2:
1 H-RMN: (400 MHz, DMSO-d6): 87,59-7,51 (m, 4H), 7,24 (s, 1H), 7,02 (s, 1H), 6,79-6,60 (m, 2H), 5,09 (d, 1H), 4,77 4,74 (m, 1H), 4,64-4,61 (m, 1H), 4,21 (t, 2H), 3,98 (dd, 1H), 3,85-3,80 (m, 2H), 3,71-3,60 (m, 3H), 3,25-3,19 (m, 2H), 2.21- 1,85 (m, 5H), 1,75-1,70 (m, 1 1,44 (s, 10H), 1,21 (d, 3H), 1,09 (d, 3H), 0,91-0,85 (m, 1H).
1,44 (s, 10H), 1,21 (d, 3H), 1,09 (d, 3H), 0,91-0,85 (m, 1H).
UPLC: 91 % .
CL-EM (ESI): m/z 622 [M++1].
HPLC quiral: Rt = 15,37 min (Chiralpak IA, 250 x 4 , 6 mm, 5 |i; fase móvil (A) TFA al 0,1 % en n-hexano (B) EtOH
(4/1): A:B (80:20); velocidad de flujo: 1,00 ml/min).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-fluorobencil)pirrolidin-2-carboxamida (CM-9A):
A una disolución con agitación del compuesto 11-F1 (0,4 g, 0,64 mmol) en CH2Cl2 (5 ml) se le añadió HCl 4 N en 1.4- dioxano (2 ml) a 0 °C y se agitó a TA durante 2 h. Se concentró la mezcla de reacción a vacío. Se lavó el material bruto obtenido con EtOAc (20 ml) seguido de n-pentano (20 ml) y se secó a vacío para proporcionar CM-9A (0,2 g, 59 % ) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,89 (d, 2H), 7,57 (d, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,79 (m, 5H), 3,35 (q, 2H), 2,56-2,50 (m, 1H), 2,35-2,29 (m, 2H), 2,14-2,05 (m, 3H), 1,81-1,75 (m, 1H), 1,35 (d, 3H), 1,32 (d, 3H), 1,19 1,11 (m, 1H).
CL-EM (ESI) m/z: 522 [M++1], 523 [M++2].
Pureza de UPLC: 97 %.
Pureza de HPLC quiral: 99 %.
Rotación óptica [a20D]: 15,6 (C=1 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-fluorobencil)pirrolidin-2-carboxamida (CM-9B):
A una disolución con agitación del compuesto 11-F2 (0,2 g, 0,32 mmol) en CH2Cl2 (5 ml) se le añadió HCl 4 N en 1.4- dioxano (2 ml) a 0 °C y se agitó a TA durante 2 h. Se concentró la mezcla de reacción a vacío. Se lavó el material bruto obtenido con EtOAc (20 ml) seguido de n-pentano (20 ml) y se secó a vacío para proporcionar CM-9B (0,1 g, 59 % ) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,89 (d, 2H), 7,57 (d, 2H), 4,95-4,90 (m, 1H), 4,43-4,35 (m, 4H), 3,94-3,79 (m, 5H), 3,35 (q, 2H), 2,56-2,50 (m, 1H), 2,35-2,29 (m, 2H), 2,14-2,05 (m, 3H), 1,81-1,75 (m, 1H), 1,35 (d, 3H), 1,32 (d, 3H), 1,19 1,11 (m, 1H).
CL-EM (ESI) m/z: 522 [M++1], 523 [M++2].
Pureza de UPLC: 98 %.
Pureza de HPLC quiral: 98 %.
Rotación óptica [a20D]: -101,46 (C = 1 % en agua).
Ejemplo 10 - Síntesis de N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((S)-2-amino-3-h¡drox¡propano¡l)p¡rrol¡d¡n-2-carbonil)-2-benc¡lp¡rrol¡d¡n-2-carboxam¡da (CM-10):
Se usó la siguiente secuencia de reacción (esquema J) para sintetizar N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((S)-2-amino-3-hidroxipropanoil)pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM -10):
Esquema J. Síntesis de CM-10.

Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución con agitación de L-prolina (SM) (110 g, 956,5 mmol) en etanol se le añadió cloruro de tionilo (141 ml, 1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TA y se concentró a vacío para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %).
Síntesis de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-metilo (2):
A una disolución con agitación del compuesto 1 (170 g, 947 mmol) en CH2Cl2 se le añadió TEA (398 ml, 2,83 mol) y tras 30 min se le añadió Cbz-Cl (1,136 mol). Se agitó la mezcla de reacción a TA durante 12 h. Se lavó la mezcla de reacción con agua y se extrajo con CH2O 2. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a vacío. Se purificó el material bruto obtenido mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1H-RMN: (200 MHz, DMSO-da): 87,42-7,31(m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81(m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1],
Síntesis de 2-bencilpirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo 2 (87 g, 314 mmol) en THF (800 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (351 ml, 351 mmol) a -25 °C y se agitó durante 2 h. Se le añadió gota a gota bromuro de bencilo (45 ml, 376 mmol) a -25 °C a la mezcla de reacción. Se dejó calentar hasta TA y se agitó durante 2 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 * 200 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 5 % de EtoAc/hexano para proporcionar el compuesto 3 (80 g, 69 % ) como
líquido.
1 H-RMN: (200 MHz, DMSO-da): 8 7,47-7,32 (m, 5H), 7,27-7,16 (m, 3H), 7,07-7,04 (m, 2H), 5,29-5,06 (m, 2H), 4,16 3,89 (m, 2H), 3,57-3,33 (m, 2H), 3,02-2,78 (m, 2H), 2,13-1,89 (m, 2H), 1,56-1,51 (m, 1H), 1,21-1,04 (m, 3H), 0,93 0,79 (m, 1H).
Masa m/z: 368,2 [M++1].
Síntesis de 2-bencilpirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (4):
A una disolución con agitación del compuesto 3 (125 g, 340 mmol) en CH3OH (700 ml) se le añadió NaOH acuoso 2 N (680 mmol) y se calentó hasta 100 °C durante 16 h. Se evaporaron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl 2 N y se extrajo con EtOAc (2*100ml). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron a presión reducida para proporcionar el compuesto 4 (90 g, 78 % ) como un sólido blanquecino.
1 H-RMN: (200 MHz, DMSO-d6): 8 12,71 (s a, 1H), 7,40-7,30 (m, 5H), 7,25-7,19 (m, 3H), 7,07-7,00 (m, 2H), 5,27-5,02 (m, 2H), 3,59-3,32 (m, 2H), 3,02-2,83 (m, 2H), 2,13-1,91 (m, 2H), 1,58-1,49 (m, 1H), 0,90-0,77 (m, 1H).
Masa m/z: 340,1 [M++1].
Síntesis de 2-bencil-2-((2S,3R)-3-hidroxi-1-metoxi-1-oxobutan-2-ilcarbamoil)pirrolidin-1-carboxilato de bencilo (5): A una suspensión con agitación del compuesto 4 (50 g, 147,9 mmol), ester metílico de L-treonina (25 g, 147,9 mmol) en DCM (500 ml) se le añadió HATU (56,2 g, 147,9 mmol) y DIPEA (64 ml, 36,98 mmol) a 5 °C. Se agitó la mezcla de reacción a TA durante 3 h. Se diluyó con EtOAc (150 ml) y se lavó con agua (2 x 30 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4, se concentró y se purificó mediante cromatografía en columna de gel de sílice con el 50 % de EtOAc/hexano como eluyente para proporcionar el compuesto 5 (49,6 g, 74 %).
1 H-RMN: (200 MHz, DMSO-d6): 8 7,62-7,59 (m, 1H), 7,44-7,31 (m, 5H), 7,21-7,18 (m, 3H), 7,06-6,99 (m, 2H), 5,25 5,24 (m, 1H), 5,12-4,94 (m, 2H), 4,30 (s, 1H), 4,15-4,08 (m, 1H), 3,66-3,64 (m, 3H), 3,63-3,49 (m, 2H), 3,14 (s, 1H), 2,89 (s, 1H), 2,09-2,02 (m, 2H), 1,56-1,51 (m, 1H), 1,09-0,98 (m, 4H).
Masa m/z: 455,1 [M++1], 477,3 [M+Na].
Síntesis de 2-((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-ilcarbamoil)-2-bencilpirrolidin-1-carboxilato de bencilo (6): A una disolución con agitación del compuesto 5 (49 g, 107,9 mmol) en THF (30 ml) se le añadieron Et3N (22,7 ml, 161,8 mmol) y Ac2O (13,2 g, 129,5 mmol) a TA. Se agitó la mezcla de reacción a TA durante 2 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con CH2O 2 y se lavó con HCl dil. Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron a presión reducida. Se purificó el residuo bruto mediante cromatografía en columna usando el 30 % de EtOAc/hexano como eluyente para proporcionar el compuesto 6 (42 g, 80 %).
1 H-RMN: (500 MHz, DMSO-d6) (rotámeros): 8 8,15-7,71 (m, 1H), 7,42-7,04 (m, 10H), 5,30-5,19 (m, 2H), 5,11-5,09 (m, 1H), 4,99-4,93 (m, 1H), 4,67-4,62 (m, 1H), 3,66-3,64 (m, 3H), 3,55-3,46 (m, 2H), 3,38-3,35 (m, 1H), 2,88-2,69 (m, 1H), 2,17-2,00 (m, 2H), 1,98-1,92 (m, 3H), 1,56-1,46 (m, 1H), 1,23-1,17 (m, 3H), 1,02-0,86 (m, 1H).
CL-EM m/z: 497,4 [M++1],
Síntesis de (2S,3R)-3-acetoxi-2-(2-bencilpirrolidin-2-carboxamido)-butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (50 g, 100,9 mmol) en metanol (1,5 l) se le añadió Pd al 10 % /C bajo atmósfera de N2. Se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 4 h. Tras el consumo del material de partida (mediante CCF), se filtró la reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (28 g, 77 %).
1 H-RMN: (500 MHz, DMSO-d6) (rotámeros): 8 8,22-8,17 (m, 1H), 7,24-7,16 (m, 5H), 5,17 (t, 1H), 4,48-4,42 (m, 1H), 3,60-3,54 (s, 3H), 3,20 (t, 1H), 3,06-2,97 (m, 1H), 2,82-2,68 (m, 3H), 2,08-2,02 (m, 1H), 1,89 (s, 3H), 1,72-1,51 (m, 3H), 1,10 (2d, 3H).
CL-EM m/z: 363 [M++1], 385 [M+Na].
Síntesis de (S)-2-(2-((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-ilcarbamoil)-2-bencilpirrolidin-1-carbonil)-pirrolidin-1carboxilato de bencilo (8):
A una mezcla con agitación del compuesto 7 (25 g, 69,1 mmol) y Na2CO3 (18,3 g, 172,6 mmol) en CH2Ch:H2O (200 ml, 1:1) se le añadió una disolución de (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo en CH2Cl2 y se agitó la mezcla de reacción a TA durante 2 h. Se evaporaron los compuestos volátiles a presión reducida. Se diluyó el residuo con CH2Cl2 (100 ml), se filtró y el filtrado se concentró a vacío. Se purificó el residuo bruto mediante cromatografía en columna usando el 60 % de EtOAc/hexano como eluyente para proporcionar el compuesto 8 (30 g, 73 %). Se preparó (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo de la siguiente manera. A una disolución de ácido (S)-1-(benciloxicarbonil)pirrolidin-2-carboxílico (20,6 g, 82,8 mmol) en CH2Cl2 (20 ml) se le añadió SOCl2 (20,5 g, 172,6 mmol) gota a gota a 0 °C y la disolución resultante se sometió a reflujo durante 2 h. Se retiraron los compuestos volátiles a presión reducida para producir (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo.
1 H-RMN: (500 MHz, DIVISOR) (rotámeros): 87,36-7,23 (m, 8H), 7,15-7,12 (m, 3H), 5,21-5,15 (m, 2H), 5,04-4,92 (m, 1H), 4,57-4,50 (m, 2H), 3,88 (d, 1H), 3,65 (s, 3H), 3,54-3,46 (m, 3H), 3,21-3,13 (m, 1H), 3,02-2,90 (m, 2H), 2,19-2,02 (m, 4H), 1,97 (s, 3H), 1,89 (s, 1H), 1,77-1,65 (m, 1H), 1,17 (s, 2H), 1,06 (s, 2H).
Masa m/z: 594,1 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-2-bencil-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (9): A una disolución con agitación del compuesto 8 (30 g, 50,05 mmol) en MeOH (300 ml) se le añadió Pd al 10 %/C, se le añadió bajo atmósfera inerte y se agitó durante 12 h bajo atmósfera de H2 (presión de globo). Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida. Se trituró el residuo obtenido con éter (10 ml) para proporcionar el compuesto 9 (21 g, 90 %) como sólido.
1 H-RMN: (500 MHz, CDCla) (rotámeros): 8 7,88-7,87 (m, 1H), 7,30-7,26 (m, 2H), 7,24-7,21 (m, 1H), 7,13-7,12 (d, 2H), 5,44-5,43 (m, 1H), 4,76-4,74 (m, 1H), 3,94-3,92 (m, 1H), 3,84-3,81 (m, 1H), 3,75 (s, 3H), 3,50 (m, 1H), 3,26-3,12 (m, 3H), 2,90-2,88 (m, 1H), 2,23-2,15 (m, 4H), 2,04 (s, 3H), 1,87-1,77 (m, 5H), 1,27-1,24 (m, 3H).
Masa m/z: 460(M+1).
Síntesis de ácido 2-((terc-butoxicarbonil)amino)butanoico (B):
A una disolución con agitación de ácido 2-aminobutanoico A (50 g, 0,47 mol) en THF: H2O (1 l, 1: 1) se le añadió NaHCO3 (80 g, 0,95 mol) y se enfrió la mezcla de reacción hasta 0 °C. A esta se le añadió anhídrido de Boc (124 g, 0,57 mol) gota a gota y se dejó a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se lavó la reacción con éter y se neutralizó la fase acuosa con HCl acuoso (pH~4-5) y se extrajo con EtOAc (2 * 250 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro, se filtraron y se concentraron a vacío para proporcionar el compuesto B (40 g, 41 %) como jarabe espeso incoloro.
Síntesis de (2S,3R)-3-acetoxi-2-(2-bencil-1-((S)-1-((S)-2-((terc-butoxicarbonil)amino)-3-hidroxipropanoil)pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (10):
A una mezcla con agitación del compuesto 9 (0,6 g, 1,30 mmol) y compuesto B (267 mg, 1,30 mmol) en CH2Cl2 (10 ml) se le añadieron EDCI (248 mg, 1,30 mmol), HOBt (351 mg, 2,60 mmol) seguido de DIPEA (167 mg, 1,30 mmol) bajo atmósfera de argón. Se agitó la mezcla de reacción resultante durante 3 h y además se agitó a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con agua y se extrajo con CH2Cl2 (2 * 150 ml). Se lavó la fase orgánica separada con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 50 % de EtOAc/hexano para proporcionar el compuesto 10 (0,5 g, 59 %) como un sólido blanquecino.
1 H-RMN: (400 MHz, DMSO-d6) (rotámeros): 87,30-7,24 (m, 6H), 7,12-7,10 (m, 1H), 5,29-5,20 (m, 1H), 4,79-4,60 (m, 2H), 4,55 (t, 1H), 4,38-4,30 (m, 2H), 3,79-3,60 (m, 11H), 2,21-1,87 (m, 11H), 1,36 (s, 9H), 1,25-1,15 (m, 4H).
CL-EM (ESI): 647(M+1).
Síntesis de ((2S)-1-((2S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-bencilpirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxopropan-2-il)carbamato de terc-butilo (11):
Se llevó una disolución del compuesto 10 (0,5 g, 0,77 mmol) en NH3 metanólico (5 ml) a un tubo sellado y se agitó durante 24 h. Tras el consumo del material de partida (mediante CCF), se concentró la mezcla de reacción a presión reducida para dar producto bruto. Se purificó el material obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 10 % de Me-OH/CH2Cl2 para proporcionar el compuesto 11 (275 mg, 60 % ) como un sólido blanquecino.
1H-RMN: (400 MHz, DMSO-da) (rotámeros): 87,35-7,20 (m, 6H), 7,15 (d, 1H), 7,01-6,97 (m, 2H), 6,77 (d, 1H), 5,09 (d, 1H), 4,75 (t, 2H), 4,39-4,35 (m, 1H), 4,24-4,20 (m, 1H), 3,92 (dd, 1H), 3,74-3,49 (m, 6H), 3,21-3,11 (m, 2H), 2,23 1,91 (m, 6H), 1,80-1,56 (m, 2H), 1,41 (s, 9H), 1,09 (d, 3H), 0,89-0,80 (m, 1H).
CL-EM (ESI): 590(M+1).
Masa (m/z): 590(M+1).
Síntesis de N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((S)-2-amino-3-hidroxipropanoil)pirrolidin-2-carbonil)-2-bencilpirrolidin-2-carboxamida (CM-10):
A una disolución con agitación del compuesto 11 (175 mg, 0,29 mmol) en dioxano (3 ml) se le añadió HCl 4 N en 1,4-dioxano (2 ml) bajo atmósfera de N2. Se agitó la mezcla de reacción a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida, se diluyó con éter de dietilo y además se agitó durante otros 15 min. Se decantó la fase de éter y se secó a vacío para proporcionar CM-10 (108 mg, 74 % ) como un sólido blanquecino.
1H-RMN: (400 MHz, DMSO-d6) (rotámeros): 88,29 (s a, 3H), 7,39-7,20 (m, 7H), 7,07-7,01 (m, 1H), 5,45-5,40 (m, 1H), 5,06 (d, 1H), 4,79-4,76 (m, 1H), 4,27-4,20 (m, 2H), 4,01 (d, 1H), 3,91-3,85 (m, 4H), 3,69-3,55 (m, 5H), 3,29-3,21 (m, 2H), 2,10-1,87 (m, 6H), 1,08 (s, 3H).
CL-EM (ESI): 490(M+2).
Ejemplo 11 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-metoxibenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-11A) y (S)-N-((2S,3R)-1-amino-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(4-metoxibenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-11B):
Se usó la siguiente secuencia de reacción (esquema K) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metoxibencil)pirrolidin-2-carboxamida (CM-1 1 A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metoxibencil)pirrolidin-2-carboxamida (CM-11B):
Esquema K. Síntesis de CM-11A y CM-11B.

Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución de L-prolina (SM) (110 g, 956,5 mmol) en etanol se le añadió cloruro de tionilo (141 ml,
1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TA y se concentró a presión reducida para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %). Se usó este material directamente para la siguiente etapa sin purificación adicional.
Síntesis de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (2):
A una disolución de 1 (170 g, 947 mmol) en DCM se le añadió TEA (398 ml, 2,83 mol) y tras 30 min se añadió Cbz-Cl (1,136 mol). Se agitó la mezcla de reacción a TA durante 12 h. Se lavó la mezcla de reacción con agua y se extrajo con DCM. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1 H-RMN: (200 MHz, DMSO-da): 8 7,42-7,31(m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81(m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1].
Síntesis de 2-(4-metoxibencil)pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución del compuesto 2 (10 g, 0,038 mol) en THF (100 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (76 ml, 0,076 mol) a -25 °C y se agitó durante 2 h. Se añadió bromuro de 4-metoxibencilo (11,5 g, 0,057 mol) gota a gota a -25 °C, se dejó calentar hasta TA y se agitó durante 2 h. Se enfrió la mezcla de reacción hasta 5 °C, se extinguió con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 * 200 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 10 % de EtOAc/hexano para proporcionar el compuesto 3 (11 g, 75 %).
1 H-RMN: (400 MHz, CDCla): 8 7,44-7,29 (m, 6H), 7,01 (t, 2H), 6,81-6,79 (m, 2H), 5,32 (t, 1H), 5,15 (d, 1H), 3,82 (d, 5H), 3,55 (d, 1H), 3,50 (s, 2H), 3,02 (d, 2H), 2,14-1,99 (m, 2H), 1,65-1,62 (m, 1H), 1,09-0,95 (m, 1H).
CL-EM (m/z):384,3 [M++1].
Síntesis de ácido 1-((benciloxi)carbonil)-2-(4-metoxibencil)pirrolidin-2-carboxílico (4):
A una disolución con agitación del compuesto 3 (1 g, 0,026 mol) en CH3OH (50 ml) se le añadió NaOH acuoso 2 N (25 ml) y se calentó hasta 100 °C durante 16 h. Se evaporaron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con agua enfriada con hielo (50 ml) y se lavó con éter (50 ml). Se acidificó la fase acuosa hasta pH~2 usando HCl 2 N y se extrajo con EtOAc (2 * 100 ml). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron a presión reducida para proporcionar el compuesto 4 (10 g, 94 %).
1 H-RMN: (400 MHz, DMSO-d6): 8 12,55 (s a, 1H), 7,49-7,65 (m, 5H), 6,99 (t, 2H), 6,78-6,75 (m, 2H), 5,25 (d, 1H), 5,11 (t, 1H), 3,72 (s, 3H), 3,51-3,42 (m, 3H), 2,99-2,95 (m, 2H), 2,15-2,09 (m, 2H), 1,62-1,57 (m, 1H), 0,98-0,94 (m, 1H).
CL-EM (m/z):368,3 [M+-1]
HPLC (pureza): 95 % .
Síntesis de 2-(((2R,3S)-3-hidroxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-metoxibencil)pirrolidin-1-carboxilato de bencilo (5):
A una suspensión de compuesto 4 (10 g, 0,027 mol), ester metílico de L-treonina (4,5 g, 0,027 mol) en DCM (100 ml) se le añadieron HATU (10,2 g, 0,027 mol) y DIPEA (11,8 ml, 0,067 mol) a 5 °C. Se agitó la mezcla de reacción a t A durante 2 h. Se diluyó la mezcla de reacción con DCM (150 ml), se lavó con agua (2 * 30 ml), salmuera y se secó sobre Na2SO4, se concentró y se purificó mediante cromatografía en columna de gel de sílice con el 50 % de EtOAc/hexano como eluyente para producir el compuesto 5 (7,8 g, 59 %).
1 H-RMN: (400 MHz, DMSO-d6): 8 7,47-7,42 (m, 6H), 6,99-6,95 (m, 2H), 6,84-6,77 (m, 2H), 5,35-5,24 (m, 1H), 5,12 4,95 (m, 2H), 4,35-4,32 (m, 1H), 4,19-4,15 (m, 1H), 3,74 (s, 3H), 3,65 (d, 2H), 3,59-3,50 (m, 2H), 3,14-3,05 (m, 1H), 3,92-3,85 (m, 1H), 2,71 (s, 5H), 2,19-2,05 (m, 2H), 1,68-1,60 (m, 1H), 1,14-1,05 (m, 3H).
CL-EM (m/z):4858,3 [M++1].
Síntesis de 2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)-carbamoil)-2-(4-metoxibencil)pirrolidin-1-carboxilato de bencilo (6):
A una disolución con agitación del compuesto 5 (7,8 g, 0,016 mol) en DCM (100 ml) se le añadieron Et3N (3,36 ml, 0,024 mol) y Ac2O (1,9 g, 0,019 mol) a TA y se agitó durante 2 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con CH2Cl2 y se lavó con HCl dil. Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron a presión reducida. Se purificó el residuo bruto mediante cromatografía en columna usando el 30 % de EtOAc/n-hexano como eluyente para proporcionar el compuesto 6 (7,6 g, 90 %). 1H-RMN: (400 MHz, DMSO-d6): 87,45-7,40 (m, 5H), 6,99-6,95 (m, 2H), 6,82-6,75 (m, 2H), 5,31-5,25 (m, 2H), 4,62 4,55 (m, 1H), 3,74 (s, 3H), 3,65 (t, 2H), 3,09-3,01 (m, 2H), 3,19-3,10 (m, 1H), 2,92-2,87 (m, 1H), 2,21-1,99 (m, 6 H), 1,59-1,45 (m, 1H), 1,25-1,15 (m, 4H).
CL-EM (m/z):527,4 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(2-bencilpirrolidin-2-carboxamido)butanoato de metilo (7):
A una disolución con agitación del compuesto 6 (7,6 g, 0,014 mol) en metanol (100 ml) se le añadió Pd al 10 % /C (1,5 g) y se agitó la mezcla de reacción bajo atmósfera de H2 (presión de globo) durante 12 h. Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 7 (4,5 g, 80 %).
1H-RMN: (400 MHz, DMSO-d6): 88,25 (s a, 1H), 6,81 (t, 2H), 5,2-5,17 (m, 1H), 4,52 (dd, 1H), 3,73 (d, 3H), 3,64 (d, 3H), 3,05 (s a, 1H), 2,86-2,80 (m, 2H), 2,12 (s a, 1H), 1,95 (d, 3H), 1,71-1,62 (m a, 3H), 1,12 (d, 1H), 0,82 (d, 1H). CL-EM (m/z): 393,3 [M++1].
Síntesis de (2S)-2-(2-(((2S,3R)-3-acetoxi-1-metoxi-1-oxobutan-2-il)carbamoil)-2-(4-metoxibencil)pirrolidin-1-carbonil)pirrolidin-1 -carboxilato de bencilo (8 ):
A una disolución con agitación del compuesto 7 (4,5 g, 11,4 mmol) y Na2CO3 (3,0 g, 28,0 mmol) en CH2Cl2 (54 ml) y H2O (36 ml) se le añadió una disolución de cloruro de ácido (3,4 g, 13,0 mmol) en CH2Cl2 y se agitó la mezcla de reacción a TA durante 2 h. Se evaporaron los compuestos volátiles a presión reducida. Se diluyó el residuo con CH2Cl2 (100 ml), se filtró y se concentró el filtrado a presión reducida. Se purificó el residuo bruto mediante cromatografía en columna usando el 30 % de EtOAc/hexano como eluyente para proporcionar el compuesto 8 (7,0 g, 96 %).
1H-RMN: (400 MHz, DMSO-d6): 87,45-7,36 (m, 4H), 7,22-6,69 (m, 3H), 5,27-5,05 (m, 2H), 4,69-4,55 (m, 1H), 3,75 3,62 (m, 4H), 3,55-3,47 (m, 3H), 3,25-3,01 (m, 1H), 2,31-1,85 (m, 6 H), 1,75-1,45 (m, 1H), 1,22-1,09 (m, 3H).
Síntesis de (2S,3R)-3-acetoxi-2-(2-(4-metoxibencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamido)butanoato de metilo (9):
A una disolución del compuesto 8 (7,0 g, 11,2 mmol) en MeOH (100 ml) se le añadió Pd al 10 % /C (2,0 g) y se agitó bajo atmósfera de H2 (presión de globo) durante 12 h. Se filtró la mezcla de reacción a través de un lecho de Celite y se concentró a presión reducida para proporcionar el compuesto 9 (5,4 g, 99 %).
1H-RMN: (400 MHz, DMSO-d6): 89,25 (m a, 1H), 8,01-7,85 (m, 1H), 7,11-6,99 (m, 2H), 6,85 (d, 2H), 5,29-5,22 (m, 1H), 4,65-4,60 (m, 1H), 4,44-4,22 (m, 1H), 3,72 (s, 3H), 3,55 (s, 3H), 3,49 (d, 3H), 3,39-3,30 (m, 5H), 3,15-3,09 (m, 3H), 2,25-2,20 (m, 1H), 2,02 (s, 9H), 1,65-1,60 (m, 2H), 1,21 (t, 3H).
CL-EM (m/z):490,4 [M++1].
Síntesis de (2S,3R)-3-acetoxi-2-(1-((S)-1-((2S,3R)-3-acetoxi-2-((terc-butoxicarbonil)amino)butanoil)pirrolidin-2-carbonil)-2-(4-metoxibencil)pirrolidin-2-carboxamido)butanoato de metilo (10):
A una disolución del compuesto 9 (5,4 g, 81,1 mmol) en CH2Cl2 (100 ml) bajo atmósfera inerte se le añadieron EDCI HCl (3,1 g, 16,0 mmol), HOBt (2,2 g, 16,0 mmol) seguido de DIPEA (4,2 g, 33,0 mmol) y compuesto C (3,4 g, 13,0 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 12 h. Se extrajo la mezcla de reacción con EtOAc ( 2 x 75 ml) y se lavó la fase orgánica separada con agua ( 200 ml), seguido de salmuera ( 200 ml), se secó sobre Na2SO4 y se concentró a presión reducida. Se purificó el material bruto mediante cromatografía en columna para proporcionar el compuesto 10 (5,8 g, 72 %).
1H-RMN: (400 MHz, DMSO-d6): 87,22-7,01 (m, 2H), 6,85-6,82 (m, 1H), 5,25-5,05 (m, 1H), 4,65-4,20 (m, 2H), 3,85 3,42 (m, 7H), 3,22-3,12 (m, 1H), 2,22-1,85 (m, 8 H), 1,52 (d, 6 H), 1,29-1,10 (m, 5H).
Síntesis de ((2S,3R)-1-((2S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(4-metoxibencil)-pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (11):
Se agitó una disolución del compuesto 10 (4 g, 5,45 mmol) en NH3 metanólico (40 ml) a TA durante 36 h. Se concentró la mezcla de reacción a presión reducida. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice seguido de purificación preparativa para proporcionar el compuesto 11 -Fr-1 (200 mg) y compuesto-11-Fr-2 (150 mg).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metoxibencil)pirrolidin-2-carboxamida (CM-11A):
A una disolución del compuesto 11-Fr-1 (0,2 g, 0,3 mmol) en DCM (3 ml) se le añadió HCl 4 N en 1,4-dioxano (2 ml) y se agitó a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-11A (0,12 g, 66 % ) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,32 (d, 2H), 7,05 (d, 2H), 4,99-4,95 (m, 2H), 4,42 (d, 1H), 4,37 (s, 3H), 3,81 (s, 3H), 3.74 (d, 1H), 3,72-3,65 (m, 1H), 3,62 (d, 1H), 3,35-3,30 (m, 1H), 3,29-3,25 (m, 1H), 3,22 (d, 1H), 2,51-2,50 (m, 1H), 2.35- 2,30 (m, 1H), 2,19-2,05 (m, 4H), 1,75-1,72 (m, 1H), 1,55 (d, 3H), 1,25 (d, 3H), 1,21-1,15 (m, 1H).
CL-EM (m/z):534,5 [M++1].
Pureza de HPLC: 97 %.
Rotación óptica [a20D]: 34,33 (C=1 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(4-metoxibencil)pirrolidin-2-carboxamida (CM-11B):
A una disolución del compuesto 11 -Fr-2 (0,15 g, 0,2 mmol) en DCM (3 ml) se le añadió HCl 4N en 1,4-dioxano (2 ml) y se agitó a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-11B (90 mg, 66 %) como sal de clorhidrato.
1H-RMN: (400 MHz, D2O): 87,32 (d, 2H), 7,05 (d, 2H), 4,99-4,95 (m, 2H), 4,42 (d, 1H), 4,37 (s, 3H), 3,81 (s, 3H), 3.74 (d, 1H), 3,72-3,65 (m, 1H), 3,62 (d, 1H), 3,35-3,30 (m, 1H), 3,29-3,25 (m, 1H), 3,22 (d, 1H), 2,51-2,50 (m, 1H), 2.35- 2,30 (m, 1H), 2,19-2,05 (m, 4H), 1,75-1,72 (m, 1H), 1,55 (d, 3H), 1,25 (d, 3H), 1,21-1,15 (m, 1H).
CL-EM (m/z):534,5 [M++1].
Pureza de HPLC: 92 %.
Rotación óptica [a20o]: -108,63 (C = 1 % en agua).
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (A):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (Boc-Thr) (50 g, 228,3 mmol) en DMF (500 ml) se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15min. Se añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice usando el 20 % de EtOAc/hexano como eluyente para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 73 %).
1H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, J = 8,5 Hz, 1H), 5,18-5,08 (m, 2H), 4,76 (d, J = 7 Hz, 1H), 4,08-4,00 (m, 2H), 1,38 (s, 9H), 1,09 (d, J = 6,0 Hz, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (B):
A una disolución con agitación de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo A (52 g, 168,2 mmol) en DCM (500 ml) se le añadieron Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a TA durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 * 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(tercbutoxicarbonilamino)butanoato B (52 g, 88 %).
1H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, J = 8,5 Hz, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m,
1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, J = 3 Hz, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (C):
Se disolvió 3-acetoxi-2-(terc-butoxicarbonNamino)butanoato de bencilo B (52 g, 148,1 mmol) en MeOH (1 l), se añadió Pd al 10 % /C y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción sobre Celite, se evaporó el disolvente a presión reducida y se trituró el residuo bruto con hexano para producir ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico C (35 g, 90 %).
1H-RMN: (500 MHz, DMSO-da): 812,78 (s a, 1H), 6,94 (d, J = 9,5 Hz, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1,39 (s, 9H), 1,10 (d, J = 6,0 Hz, 3H).
Masa m/z: 260,0 [M-1].
Ejemplo 12 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3.5-d¡bromobencil)p¡rrol¡d¡n-2-carboxam¡da (CM-12A) y (S)-N-((2S,3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3.5-d¡bromobencil)p¡rrol¡d¡n-2-carboxam¡da (CM-12B):
Se usó la siguiente secuencia de reacción (esquema L) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,5-dibromobencil)pirrolidin-2-carboxamida (CM-12A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,5-dibromobencil)pirrolidin-2-carboxamida (CM-12B):
Esquema L. Síntesis de CM-12A y CM-12B:

Síntesis de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución con agitación de ácido pirrolidin-2-carboxílico (SM) (100 g, 0,87 mol) en metanol (800 ml) se le añadió lentamente cloruro de tionilo (76,9 ml, 1,04 mol) a 0 °C. Se calentó la mezcla de reacción a reflujo durante 12 h. Tras el consumo del material de partida (mediante CCF), se concentró la reacción a presión reducida para proporcionar el compuesto 1 (143,9 g, sal de HCl).
1H-RMN: (400 MHz, CDCla) (rotámeros): 83,89 (s, 3H), 3,68-3,62 (m, 2H), 3,59-3,47 (m, 2H), 2,49-2,37 (m, 1H), 2,27-2,05 (m, 3H).
CL-EM (m/z): 166 [M++1].
Síntesis de pirrolidin-1,2-dicarboxilato de 1-terc-butilo y 2-metilo (2):
A una disolución con agitación del compuesto 1 (35 g, 0,22 mol) en CH2Cl2 (175 ml) se le añadieron Et3N (90 ml, 0,65 mol) seguido de Boc-anhídrido (56,9 ml, 0,26 mol) a 0 °C. Se agitó la mezcla de reacción a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con agua (100 ml) y se extrajo con CH2Cl2 ( 2 x 100 ml). Se lavó la fase orgánica con agua, salmuera, se secó sobre Na2SO4 y se concentró. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 30 % de EtOAc/nhexano para proporcionar el compuesto 2 (41 g, 95 %).
1H-RMN: (400 MHz, CDCla) (rotámeros): 84,25-4,21 (m, 1H), 3,75 (s, 3H), 3,57-3,26 (m, 2H), 2,29-2,10 (m, 1H), 1,99-1,75 (m, 3H), 1,45 (s, 9H).
CL-EM (m/z): 130 [(M++1)-Boc].
Síntesis de 2-(3,5-dibromobencil)pirrolidin-1,2-dicarboxilato de 1-terc-butilo y 2-metilo (3):
A una disolución con agitación del compuesto 2 (10 g, 0,043 mol) en THF (50 ml) se le añadió LiHMDS (65,5 ml, 0,065 mol) a -25 °C y se agitó durante 2 h. A esto se le añadió bromuro de 3,5-dibromobencilo (17,1 g, 0,052 mol) gota a gota a -25 °C y se agitó durante 2 h a TA. Tras el consumo del material de partida (mediante CCF), se extinguió la reacción con NH4Cl a 0 °C. Se lavó la fase orgánica separada con agua y se extrajo con EtOAc. Se secó la fase orgánica separada sobre Na2SO4 y se concentró a presión reducida para obtener producto bruto, que se purificó mediante cromatografía en columna de gel de sílice para proporcionar el compuesto 3 (10 g, 50 %).
1H-RMN: (500 MHz, DMSO-d6): 87,72 (s, 1H), 7,41 (d, 2H), 3,69 (d, 3H), 3,37-3,35 (m, 2H), 3,07-3,01 (m, 1H), 2,89 2,81 (m, 1H), 2,08-2,01 (m, 2H), 1,68-1,62 (m, 1H), 1,45 (s, 9H), 1,19-1,12 (m, 1H).
CL-EM (m/z): 419,1 [(M++1)-Boc].
Síntesis de 2-(3,5-dibromobencil)pirrolidin-2-carboxilato de metilo (4):
A una disolución con agitación del compuesto 3 (10 g, 0,016 mol) en metanol (20 ml) se le añadió MeOH.HCl (80 ml) bajo atmósfera de N2 y se agitó durante 8 h a TA. Se evaporaron los compuestos volátiles a presión reducida para proporcionar el compuesto 4 (8 g, 93 %) como un sólido de color blanco.
1H-RMN: (400 MHz, DMSO-d6): 810,95 (s a, 1H), 9,45 (s a, 1H), 7,81 (s, 1H), 7,65 (s, 2H), 3,75 (s, 3H), 3,55 (d, 1H), 3,39 (d, 1H), 3,22 (s a, 1H), 2,43 (t, 1H), 2,15-2,10 (m, 2H), 1,85-1,81 (m, 1H).
CL-EM (m/z): 378 [(M+-HCl].
Síntesis de ácido (S)-1-((aliloxi)carbonil)pirrolidin-2-carboxílico (A):
A una disolución con agitación de ácido (S)-pirrolidin-2-carboxílico (SM) (5 g, 0,043 mol) en 1,4-dioxano (50 ml) y agua (100 ml) se le añadió Na2CO3 (11,5 g, 0,11 mol) seguido de cloroformiato de alilo (6,2 g, 0,052 mol) a 0 °C y se agitó durante 12 h. Se extrajo la mezcla de reacción con EtOAc (2 x 75 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida para dar producto bruto; que se purificó mediante cromatografía en columna de gel de sílice para proporcionar el compuesto A (3,28 g, 37 %).
Síntesis de (S)-2-((R)-2-(3,5-dibromobencil)-2-(metoxicarbonil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de alilo (5): A una disolución con agitación del compuesto 4 (8 g, 0,019 mol) y Na2CO3 (5,1 g, 0,048 mol) en DCM:H2O (150 ml, 3:2) se le añadió una disolución de cloruro de ácido B y se agitó durante 2 h a TA. Se extrajo la fase acuosa con CH2Cl2 (2 x 50 ml). Se lavó la fase orgánica separada con agua (50 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida. Se purificó el material bruto obtenido mediante cromatografía en columna de gel de sílice para proporcionar el compuesto 5 (8 g, 74 %). El cloruro de ácido B se preparó de la siguiente manera. A una disolución del compuesto A (4,6 g, 0,023 mol) en DCM se le añadió SOCl2 (5,7 g, 0,048 mol) gota a gota a 0 °C y se sometió la disolución resultante a reflujo durante 2 h; se eliminó el disolvente de la reacción a presión reducida. 1H-RMN: (400 MHz, DMSO-d6): 87,75-7,42 (m, 2H), 7,37 (s, 1H), 5,95 (s a, 1H), 9,45 (s a, 1H), 7,81 (s, 1H), 7,65 (s, 2H), 3,75 (s, 3H), 3,55 (d, 1H), 3,39 (d, 1H), 3,22 (s a, 1H), 2,43 (t, 1H), 2,15-2,10 (m, 2H), 1,85-1,81 (m, 1H).
CL-EM (m/z): 559,3 [(M++1].
Síntesis de ácido (R)-1-((S)-1-((aliloxi)carbonil)pirrolidin-2-carbonil)-2-(3,5-dibromobencil)pirrolidin-2-carboxílico (6):
A una disolución con agitación del compuesto 5 (8 g, 0,014 mol) en MeOH (50 ml) se le añadió disolución de NaOH 2 N (10 ml) y se calentó a reflujo durante 5 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua y se lavó con éter. Se acidificó la fase acuosa a pH~2 usando disoluciones de HCl 2 N y se extrajo con EtOAc. Se secó la fase orgánica separada sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida para proporcionar el compuesto 6 (7 g, 90 %). Se usó este material directamente para la siguiente etapa sin purificación adicional.
CL-EM (m/z): 543,3 [(M+-1].
Síntesis de (2S,3R)-2-amino-3-hidroxibutanoato de metilo (C):
A una disolución con agitación de ácido (2S,3R)-2-amino-3-hidroxibutanoico (200 g, 1,68 mol) en metanol (1,2 l) se le añadió SOCl2 (244 ml, 3,36 mol) gota a gota a 0 °C y se agitó durante 1 h. Se sometió la mezcla de reacción resultante a reflujo durante 24 h. Tras el consumo del material de partida (mediante CCF), se calentó la mezcla de reacción hasta TA y se concentró a presión reducida y se lavó con n-hexano (2 * 50 ml). Se disolvió el residuo en EtOH (1 l) y se neutralizó con Et3N (471 ml, 3,36 mol) y se agitó de nuevo durante 2 h. Se separó por filtración el sólido precipitado; se concentró el filtrado obtenido a presión reducida para proporcionar el compuesto C (195 g, 80 %).
1 H-RMN: (400 MHz, DMSO-d6): 88,51 (s a, 3H), 4,13-4,10 (m, 1H), 3,91 (s a, 1H), 1,20 (d, 3H).
CL-EM (m/z): 134,1 [M++1].
Síntesis de (2S,3R)-2-amino-3-hidroxibutanamida (D):
Se llevó una disolución del compuesto C (190 g, 1,35 mol) en IPA (2 l) a autoclave y se purgó con gas NH3 (7-8 kg) y se agitó a 35 °C durante 24 h. Después se eliminó el gas NH3 y se concentró la mezcla de reacción a presión reducida y se añadió CH2Cl2 y se filtró. Se sometió el sólido obtenido a reflujo en EtOH durante 1 h a 78 °C. Se filtró la masa de reacción en condición de calentamiento y se añadió n-hexano al filtrado y se agitó de nuevo durante otras 4 h. Se filtró el sólido precipitado obtenido y se secó a presión reducida para proporcionar el compuesto D (160 g, 47 %).
1 H-RMN: (500 MHz, DMSO-d6): 8 7,38 (s a, 1H), 7,02 (s a, 1H), 4,66 (s a, 1H), 3,77-3,70 (m, 1H), 2,93 (d, 1H), 2,72 (m a, 1H), 1,05 (d, 3H).
CL-EM (m/z): 119,1 [M++1].
UPLC (pureza de ELSD): 99,9 % .
Síntesis de (S)-2-((R)-2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3,5-dibromobencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de alilo (7):
A una disolución con agitación del compuesto 6 (5 g, 9,19 mmol) en CH2Cl2 (100 ml) se le añadió HOBt (1,8 g, 13,7 mmol), EDCI.HCl (2,6 g, 13,7 mmol) seguido de DIPEA (3,5 g, 27,0 mmol) y compuesto D (1,3 g, 11,0 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 12 h. Tras el consumo del material de partida (mediante CCF), se extinguió la reacción con agua y se extrajo con CH2O 2 (2 * 100 ml). Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 7 (5 g, 84 %).
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-2-(3,5-dibromobencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2 -carboxamida (8 ):
A una disolución con agitación del compuesto 7 (1 g, 1,35 mmol) en THF (50 ml) se le añadió DABCO (1,3 g, 12,4 mmol) seguido de Pd(PPh3)4 (358 mg, 0,3 mmol) a TA y se agitó durante 30 min. Se diluyó la mezcla de reacción con EtOAc (75 ml) y disolución de NaHCO3 saturada (75 ml). Se secó la fase orgánica separada sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar el compuesto 8 (560 mg, 64 %). Se usó este material directamente para la siguiente etapa sin purificación adicional.
Síntesis de ((2S,3R)-1-((S)-2-((R)-2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3,5-dibromobencil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (9):
A una disolución con agitación del compuesto 8 (1,8 g, 3,20 mmol) en CH2Cl2 (100 ml) se le añadieron EDCI.HCl (0,916 g, 4,80 mmol), HOBt (0,65 g, 4 ,80 mmol) seguido de DIPEA (1,7 ml, 9,60 mmol) y ácido (2S,3R)-2-((tercbutoxicarbonil)amino)-3-hidroxibutanoico (0,84 g, 3,80 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 12 h. Se extinguió la mezcla de reacción con agua (100 ml) y se extrajo con CH2Cl2 (2 * 150 ml). Se lavaron las fases orgánicas separadas con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión
reducida para obtener producto bruto; que se purificó mediante cromatografía en columna de gel de sílice seguido de purificación por HPLC preparativa para proporcionar el compuesto 9-Fr-1 (140 mg) y el compuesto 9-Fr-2 (150 mg).
Datos de HPLC quiral para el compuesto 9-Fr-1:
1H-RMN: (400 MHz, DMSO-da): 87,71 (d, 3H), 7,21 (s, 1H), 6,99-6,75 (m, 2H), 6,59 (d, 1H), 5,01 (d, 1H), 4,65-4,60 (m, 2H), 4,19 (t, 2H), 3,99 (d, 1H), 3,89-3,85 (m, 2H), 3,65-3,61 (m, 2H), 3,44-3,39 (m, 3H), 2,25-2,15 (m, 1H), 2,02 1,95 (m, 5H), 1,75-1,72 (m, 1H), 1,35 (s, 9H), 1,25 (d, 3H), 1,19 (d, 3H), 0,99-0,95 (m, 1H).
HPLC: 99,83 % .
CL-EM (m/z): 762,3 [M++1].
Datos de HPLC quiral para el compuesto 9-Fr-2:
1H-RMN: (400 MHz, DMSO-d6): 87,71 (d, 3H), 7,21 (s, 1H), 6,99-6,75 (m, 2H), 6,59 (d, 1H), 5,01 (d, 1H), 4,65-4,60 (m, 2H), 4,19 (t, 2H), 3,99 (d, 1H), 3,89-3,85 (m, 2H), 3,65-3,61 (m, 2H), 3,44-3,39 (m, 3H), 2,25-2,15 (m, 1H), 2,02 1,95 (m, 5H), 1,75-1,72 (m, 1H), 1,35 (s, 9H), 1,25 (d, 3H), 1,19 (d, 3H), 0,99-0,95 (m, 1H).
HPLC: 92 % .
CL-EM (m/z):762,2 [M++1].
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3S)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,5-dibromobencil)pirrolidin-2-carboxamida (CM-12A):
A una disolución con agitación del compuesto 9-Fr-1 (140 mg, 0,183 mmol) en CH2Cl2 (3 ml) se le añadió dioxano.HCl (2 ml) a 0 °C bajo atmósfera de N2. Se agitó la mezcla de reacción a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-12A (120 mg, 93 %).
1H-RMN: (400 MHz, D2O): 87,84 (s, 1H), 7,61 (s, 2H), 4,39 (d, 4H), 3,92-3,85 (m, 4H), 3,65-3,62 (m, 2H), 3,45 (d, 1H), 2,59-2,55 (m, 1H), 2,48-2,44 (m, 2H), 2,15-2,19 (m, 3H), 1,91-1,87 (m, 1H), 1,60 (s, 3H), 1,25 (s, 3H).
CL-EM (m/z):662 (M+1).
HPLC: 97,55 % .
Rotación óptica [a20o]: 29,21 (C=0,5 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,5-dibromobencil)pirrolidin-2-carboxamida (CM-12B):
A una disolución con agitación del compuesto 9-Fr-2 (150 mg, 0,197 mmol) en CH2Cl2 (3 ml) se le añadió dioxano.HCl (2 ml) a 0 °C bajo atmósfera de N2. Se agitó la mezcla de reacción a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-12B (130 mg, 94 %).
1H-RMN: (400 MHz, D2O): 87,84 (s, 1H), 7,61 (s, 2H), 4,39 (d, 4H), 3,92-3,85 (m, 4H), 3,65-3,62 (m, 2H), 3,45 (d, 1H), 2,59-2,55 (m, 1H), 2,48-2,44 (m, 2H), 2,15-2,19 (m, 3H), 1,91-1,87 (m, 1H), 1,60 (s, 3H), 1,25 (s, 3H).
CL-EM (m/z):662 (M+1).
HPLC: 92,11 % .
Rotación óptica [a20o]: -78,70 (C = 0,5 % en agua).
Ejemplo 13 - Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-metoxibenc¡l)p¡rrol¡d¡n-2-carboxam¡da (CM-13A) y N-((2S,3R)-1-amino-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S.3R)-2-am¡no-3-h¡drox¡butano¡l)p¡rrol¡d¡n-2-carbon¡l)-2-(3-metox¡benc¡l)p¡rrol¡d¡n-2-carboxamida (CM-13B):
Se usó la siguiente secuencia de reacción (esquema M) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metoxibencil)pirrolidin-2-carboxamida (CM-13A) y N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metoxibencil)pirrolidin-2-carboxamida (CM-13B):
Esquema M. Síntesis de CM-13A y CM-13B:

Síntesis de ácido (S)-1-((benciloxi)carbonil pirrolidin-2-carboxílico (A):
A una disolución con agitación de ácido (S)-pirrolidin-2-carboxílico (250 g, 2,17 mol) en agua (1 l) se le añadió Na2CO3 (576 g, 5,43 mol) y se agitó durante 1 h. Tras enfriarse hasta 0 °C, se añadió cloroformiato de bencilo (444 g, 2,61 mol) gota a gota a la mezcla de reacción y se agitó de nuevo durante 1 h. Se calentó la mezcla de reacción resultante hasta TA y se agitó adicionalmente durante 24 h. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con agua (1 l) y éter (1,5 l). Se trató la fase acuosa separada con PhCH3 (1,5 l) y se acidificó usando HCl 6 N. Se extrajo la fase acuosa con EtOAc (3* 1,5 l); se lavaron los extractos orgánicos combinados con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida para proporcionar el compuesto A (450 g, 84 % ) como un jarabe de color amarillo claro.
1 H-RMN: (400 MHz, DMSO-da): 8 12,71 (s a, 1H), 7,40-7,30 (m, 5H), 5,19-5,01 (m, 2H), 4,25 (dd, 1H), 3,51-3,50 (m, 2H), 2,29-2,15 (m, 1H), 1,89-1,80 (m, 3H).
CL-EM (m/z): 250 [M++1].
Síntesis de (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo (B):
A una disolución con agitación del compuesto A (2,5 g, 0,01 mol) en CH2Cl2 (50 ml) se le añadió SOCl2 (2,7 g, 0,02 mol) a 0 °C y se agitó durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar el compuesto B como producto bruto. Se usó este material directamente para la siguiente etapa sin purificación adicional.
Síntesis de ácido (2S,3R)-2-(((benciloxi)carbonil)amino)-3-hidroxibutanoico (E):
A una disolución con agitación de NaHCO3 (529 g, 6,30 mol) en agua (1 l) se le añadió ácido (2S,3R)-2-amino-3-hidroxibutanoico (250 g, 2,10 mol) a TA y se agitó durante 30 min. Se enfrió la mezcla de reacción hasta 0 °C, se añadió Cbz-Cl (850 ml, 2,52 mol, al 50 % en PhCH3) gota a gota a la reacción y se agitó durante 1 h. Se calentó la mezcla de reacción hasta TA y se agitó de nuevo durante 28 h. A esto se le añadió MTBE (1 l) y se agitó durante 20 min. Se agitó la fase acuosa separada con tolueno y se agitó durante 20 min. Se acidificó la fase acuosa con HCl 1 N (pH~1-2) y se extrajo con EtOAc (3 * 1,5 l). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. Se agitó el material bruto con diciclohexilamina (819 ml, 4,20 mol) durante 4 h para obtener un sólido blanco, se filtró y se secó. Se sometió el sólido obtenido a reflujo con EtOAc (1,5 l) durante 1 h y después se filtró. Se disolvió el material sólido en agua (1 l) y se acidificó con H2SO4 dil. y se agitó de nuevo durante 30 min. Se extrajo la fase acuosa con EtOAc (3 * 1 l). Se lavó la fase orgánica separada con salmuera, se secó sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida. Se trituró el material bruto obtenido con n-hexano durante 2 h para obtener un sólido blanco. Se filtró el material sólido obtenido y se secó a
presión reducida para proporcionar el compuesto E (230 g, 43 %).
1H-RMN: (400 MHz, DMSO-da): 87,35-7,32 (m, 5H), 6,91 (d, 1H), 5,06 (s, 2H), 4,13-4,09 (m, 1H), 3,99-3,95 (m, 1H), 1,02 (d, 3H).
CL-EM (m/z):254,1(M+1).
UPLC (pureza de ELSD): 99,82 %.
Síntesis de (2S,3R)-2-amino-3-hidroxibutanoato de metilo (C):
A una disolución con agitación de ácido (2S,3R)-2-amino-3-hidroxibutanoico (200 g, 1,68 mol) en metanol (1,2 l) se le añadió SOCh (244 ml, 3,36 mol) gota a gota a 0 °C y se agitó durante 1 h. Se sometió la mezcla de reacción resultante a reflujo durante 24 h. Tras el consumo del material de partida (mediante CCF), se calentó la mezcla de reacción hasta TA y se concentró a presión reducida y se lavó con n-hexano (2 * 50 ml). Se disolvió el residuo en EtOH (1 l) y se neutralizó con Et3N (471 ml, 3,36 mol) y se agitó de nuevo durante 2 h. Se separó por filtración el sólido precipitado; se concentró el filtrado obtenido a presión reducida para proporcionar el compuesto C (195 g, 80 %).
1H-RMN: (400 MHz, DMSO-d6): 88,51 (s a, 3H), 4,13-4,10 (m, 1H), 3,91 (s a, 1H), 1,20 (d, 3H).
CL-EM (m/z): 134,1 [M++1].
Síntesis de (2S,3R)-2-amino-3-hidroxibutanamida (D):
Se llevó una disolución del compuesto F (190 g, 1,35 mol) en IPA (2 l) a autoclave y se purgó con gas NH3 (7-8 kg) y se agitó a 35 °C durante 24 h. Después se eliminó el gas NH3 y se concentró la mezcla de reacción a presión reducida y se añadió CH2Cl2 y se filtró. Se sometió el sólido obtenido a reflujo en EtOH durante 1 h a 78 °C. Se filtró la masa de reacción en condición de calentamiento y se añadió n-hexano al filtrado y se agitó de nuevo durante otras 4 h. Se filtró el sólido precipitado obtenido y se secó a presión reducida para proporcionar el compuesto D (160 g, 47 %).
1H-RMN: (500 MHz, DMSO-d6): 87,38 (s a, 1H), 7,02 (s a, 1H), 4,66 (s a, 1H), 3,77-3,70 (m, 1H), 2,93 (d, 1H), 2,72 (m a, 1H), 1,05 (d, 3H).
CL-EM (m/z): 119,1 [M++1].
UPLC (pureza de ELSD): 99,9 % .
Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de etilo (1):
A una disolución de L-prolina (SM) (110 g, 956,5 mmol) en etanol se le añadió cloruro de tionilo (141 ml, 1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TA y se concentró a presión reducida para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %).
Síntesis de (S)-pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (2):
A una disolución de 1 (170 g, 947 mmol) en DCM se le añadió TEA (398 ml, 2,83 mol) y tras 30 min se añadió Cbz-Cl (1,136 mol). Se agitó la mezcla de reacción a TA durante 12 h. Se lavó la mezcla de reacción con agua y se extrajo con DCM. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1H-RMN: (200 MHz, DMSO-d6): 87,42-7,31(m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81(m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1].
Síntesis de 2-(3-metoxibencil)pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-etilo (3):
A una disolución del compuesto 2 (10 g, 37,9 mmol) en THF seco (100 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (76 ml, 0,076 mol) a -25 °C y se agitó durante 2 h. Se añadió bromuro de 3-metoxibencilo (9,21 g, 45,59 mmol) gota a gota a -25 °C a la mezcla de reacción, se dejó calentar hasta TA y se agitó durante 30 min. Se extinguió la mezcla de reacción con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 * 200 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 10 % de EtoAc/hexano para proporcionar el compuesto 3 (10 g, 69 %).
1 H-RMN: (400 MHz, CDCI3): 8 7,41-7,32 (m, 5H), 7,19-7,09 (m, 1H), 6,80 (t, 1H), 6,65 (d, 2H), 5,30 (t, 1H), 5,19 (t, 1H), 3,79-3,70 (m, 5H), 3,55-3,52 (m, 2H), 3,10-3,07 (m, 2H), 2,15-2,08 (m, 1H), 1,79-1,72 (m, 1H), 1,10-0,99 (m, 1H).
Síntesis de 2-(3-metoxibencil)pirrolidin-2-carboxilato de metilo (4):
A una disolución con agitación del compuesto 3 (10 g, 26,17 mmol) en CH3OH (100 ml) se le añadió Pd/C (3 g) y se agitó bajo atmósfera de H2 a TA durante 10 h. Tras el consumo del material de partida (mediante CCF), se filtró la mezcla de reacción a través de un lecho de Celite y se lavó con MeOH. Se concentró el filtrado obtenido a presión reducida para proporcionar el compuesto 4 (5,3 g, 82 %). Se usó este material directamente para la siguiente etapa sin purificación adicional.
1 H-RMN: (400 MHz, DMSO-d6): 8 7,15 (t, 1H), 6,59-6,55 (m, 3H), 3,71 (s, 3H), 3,55 (s, 3H), 2,99 (d, 1H), 2,85-2,81 (m, 3H), 2,09-2,05 (m, 1H), 1,75-1,67 (m, 3H).
CL-EM (m/z):250,2 [M++1].
Síntesis de (2S)-2-(2-(3-metoxibencil)-2-(metoxicarbonil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (5): A una suspensión de compuesto 4 (4,3 g, 17,47 mmol) en DCM:H2O (700 ml, 2:5) se le añadió Na2CO3 (4,6 g, 43,69 mmol) seguido de una disolución de Int-B (5,2 g, 20,97 mmol) en DCM a TA y se agitó durante 2 h. Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4, se concentró a presión reducida para proporcionar el compuesto 5 (7 g) como producto bruto. Se usó este material directamente para la siguiente etapa sin purificación adicional.
Síntesis de ácido (S)-1-(1-((benciloxi)carbonil)pirrolidin-2-carbonil)-2-(3-metoxibencil)pirrolidin-2-carboxílico (6 ): A una disolución con agitación del compuesto 5 (7 g, 14,58 mmol) en MeOH (100 ml) se le añadió disolución de NaOH 1 N (100 ml) y se agitó a temperatura de reflujo durante 5 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua y se ajustó a pH~ 2 usando disoluciones de ácido cítrico saturadas. Se extrajo la fase acuosa con EtOAc. Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron a presión reducida para proporcionar el compuesto 6 (7,6 g) como producto bruto.
1 H-RMN: (400 MHz, DMSO-d6): 8 12,45 (s a, 1H), 7,85-7,75 (m, 4H), 6,99-6,82 (m, 2H), 5,34-5,29 (m, 1H), 3,74 (d, 1H), 3,65-3,41 (m, 2H), 3,39-3,34 (m, 1H), 3,01 (t, 1H), 2,25-2,21 (m, 1H), 2,01-1,92 (m, 3H), 1,12 (t, 1H).
CL-EM (m/z):467 [M++1].
Síntesis de (S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3-metoxibencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (7):
A una disolución con agitación del compuesto 6 (2 g, 4,29 mmol) en CH2Cl2 (30 ml) se le añadieron EDCI.HCl (1,23 g, 6,43 mmol), HOBt (869 mg, 6,43 mmol) seguido de DIPEA (1,66 g, 12,87 mmol) y compuesto D (506 mg, 4,29 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 12 h. Tras el consumo del material de partida (mediante CCF), se extinguió la reacción con agua y se extrajo con CH2O 2 (2* 100 ml). Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 7 (1,5 g, 61 %).
1 H-RMN: (400 MHz, DMSO-d6): 8 7,49-7,37 (m, 6 H), 7,11-6,92 (m, 2H), 6,88-6,82 (m, 2H), 5,35-5,01 (m, 2H), 4,89 4,52 (m, 1H), 4,22 (d, 1H), 4,01-3,84 (m, 1H), 3,72 (d, 2H), 3,62-3,55 (m, 4H), 3,22-3,18 (m, 1H), 2,22-1,75 (m, 5H), 1,09 (s a, 3H).
Síntesis de N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-2-(3-metoxibencil)-1-((S)-pirrolidin-2-carbonil)pirrolidin-2-carboxamida (8 ):
A una disolución con agitación del compuesto 7 (1,5 g, 2,65 mmol) en MeOH (20 ml) se le añadió Pd al 10 % /C (0,4 g) y se agitó bajo atmósfera de H2 a TA durante 6 h. Tras el consumo del material de partida (mediante CCF), se filtró la mezcla de reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 8 (1 g, 87 %) como un sólido blanco.
1 H-RMN: (400 MHz, DMSO-d6): 8 7,22-7,18 (m, 2H), 7,08-7,01 (m, 1H), 6,98 (s a, 1H), 6,84 (t, 1H), 6,75-6,71 (m, 2H), 5,05 (d, 1H), 4,23 (t, 1H), 5,09 (s a, 1H), 3,95 (d, 1H), 3,74 (s, 4H), 3,61 (t, 2H), 3,09-3,01 (m, 2H), 2,72-2,68 (m, 1H), 2,17-2,01 (m, 3H), 1,85-1,80 (m, 5H), 1,08 (d, 3H).
CL-EM (m/z): 433 [M++1].
Síntesis de ((2S,3R)-1-((S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3-metoxibencil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de bencilo (9):
A una disolución con agitación del compuesto 8 (2 g, 4,62 mmol) en CH2O 2 (30 ml) se le añadieron HOBt (937 mg, 6,94 mmol), EDCI.HCl (1,32 g, 6,93 mmol) seguido de DIPEA (1,79 g, 13,82 mmol) y compuesto E (1,17 g, 4,62 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 12 h. Tras el consumo del material de partida (mediante CCF), se extinguió la reacción con agua y se extrajo con CH2O 2 (2* 100 ml). Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna seguido de purificación por HPLC preparativa para proporcionar el compuesto 9-Fr-1 (0,5 g) y el compuesto 9-Fr-2 (0,1 g) como un sólido blanco.
Datos de HPLC quiral para el compuesto 9-Fr-1:
1H-RMN: (400 MHz, DMSO-d6): 87,39 (s, 4H), 7,19-7,15 (m, 2H), 7,09-7,02 (m, 2H), 6,75-6,71 (m, 2H), 5,10-4,99 (m, 2H), 4,76-4,71 (m, 1H), 4,29-4,21 (m, 1H), 4,07-3,87 (m, 2H), 3,71 (d, 2H), 3,65-3,62 (m, 1H), 3,45 (d, 1H), 3,21 3,14 (m, 1H), 2,15-2,01 (m, 5H), 1,72 (m a, 1H), 1,22-1,15 (m, 2H), 1,05 (t, 2H).
HPLC: 41,56%.
CL-EM (m/z): 668 [M++1].
Datos de HPLC quiral para el compuesto 9-Fr-2:
1H-RMN: (400 MHz, DMSO-d6): 87,391 (s, 4H), 7,19-7,15 (m, 2H), 7,09-7,02 (m, 2H), 6,75-6,71 (m, 2H), 5,10-4,99 (m, 2H), 4,76-4,71 (m, 1H), 4,29-4,21 (m, 1H), 4,07-3,87 (m, 2H), 3,71 (d, 2H), 3,65-3,62 (m, 1H), 3,45 (d, 1H), 3,21 3,14 (m, 1H), 2,15-2,01 (m, 5H), 1,72 (m a, 1H), 1,22-1,15 (m, 2H), 1,05 (t, 2H).
HPLC: 58,43 % .
CL-EM (m/z): 762,2 [M++1].
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metoxibencil)pirrolidin-2-carboxamida (CM-13A):
A una disolución del compuesto 9-Fr-1 (0,5 g, 0,0.09 mol) en DCM (2 ml) se le añadió HCl de éter (2 ml) y se agitó a TA durante 1 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-13A (45 mg, 84 %) como un sólido blanco.
1H-RMN: (400 MHz, DMSO-d6): 88,01 (s a, 3H), 7,35 (d, 1H), 7,21 (t, 1H), 7,09 (s, 1H), 7,01 (s, 1H), 6,89 (d, 1H), 6.69 (s, 2H), 5,49 (s a, 1H), 4,69 (t, 1H), 4,35-4,31 (m, 1H), 4,15 (d, 1H), 4,09 (m a, 1H), 3,91-3,85 (m, 4H), 3,75-3,71 (m, 1H), 3,49 (d, 2H), 3,19 (s, 2H), 3,12 (d, 1H), 2,21-2,15 (m, 1H), 2,12-2,05 (m, 3H), 1,97-1,92 (m, 3H), 1,75-1,71 (m, 1H), 1,41 (d, 3H), 1,09 (d, 3H).
CL-EM (m/z): 534 [M++1].
Pureza de HPLC: 94 %.
Síntesis de N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3-metoxibencil)pirrolidin-2-carboxamida (CM-13B):
A una disolución del compuesto 9-Fr-2 (0,1 g, 0,187 mmol) en DCM (3 ml) se le añadió HCl de éter (2 ml) y se agitó a TA durante 1 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-13B (80 mg, 75 %) como un sólido blanco.
1H-RMN: (400 MHz, DMSO-d6): 88,01 (s a, 3H), 7,35 (d, 1H), 7,21 (t, 1H), 7,09 (s, 1H), 7,01 (s, 1H), 6,89 (d, 1H), 6.69 (s, 2H), 5,49 (s a, 1H), 4,69 (t, 1H), 4,35-4,31 (m, 1H), 4,15 (d, 1H), 4,09 (m a, 1H), 3,91-3,85 (m, 4H), 3,75-3,71 (m, 1H), 3,49 (d, 2H), 3,19 (s, 2H), 3,12 (d, 1H), 2,21-2,15 (m, 1H), 2,12-2,05 (m, 3H), 1,97-1,92 (m, 3H), 1,75-1,71 (m, 1H), 1,41 (d, 3H), 1,09 (d, 3H).
CL-EM (m/z): 534 [M++1].
Pureza de HPLC: 92 %.
Ejemplo 14 - Síntesis de (R)-N-((2S.3R)-1-am¡no-3-h¡drox¡-1-oxobutan-2-¡l)-1-((S)-1-((2S,3R)-2-am¡no-3
hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (CM-14A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-i r )-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (CM-14B):
Se usó la siguiente secuencia de reacción (esquema N) para sintetizar (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (CM-14A) y (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (CM-14B):
Esquema N. Síntesis de CM-14A y CM-14B:

Síntesis de (S)-1-((benciloxi) carbonil)ácido pirrolidin-2-carboxílico (A):
A una disolución con agitación de ácido (S)-pirrolidin-2-carboxílico (250 g, 2,17 mol) en agua (1 l) se le añadió Na2CO3 (576 g, 5,43 mol) y se agitó durante 1 h. Tras enfriarse hasta 0 °C, se añadió cloroformiato de bencilo (444 g, 2,61 mol) gota a gota a la mezcla de reacción y se agitó de nuevo durante 1 h. Se calentó la mezcla de reacción resultante hasta TA y se agitó adicionalmente durante 24 h. Tras el consumo del material de partida (mediante CCF), se diluyó la reacción con agua (1 l) y éter (1,5 l). Se trató la fase acuosa separada con PhCH3 (1,5 l) y se acidificó usando HCl 6 N. Se extrajo la fase acuosa con EtOAc (3 x 1,5 l). Se lavaron los extractos orgánicos combinados con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida para proporcionar el compuesto A (450 g, 84 %) como un jarabe de color amarillo claro.
1H-RMN: (400 MHz, DMSO-da): 812,71 (s a, 1H), 7,40-7,30 (m, 5H), 5,19-5,01 (m, 2H), 4,25 (dd, 1H), 3,51-3,50 (m, 2H), 2,29-2,15 (m, 1H), 1,89-1,80 (m, 3H).
CL-EM (m/z): 250 [M++1].
Síntesis de (S)-2-(clorocarbonil)pirrolidin-1-carboxilato de bencilo (B):
A una disolución con agitación del compuesto A (2,5 g, 0,01 mol) en CH2Cl2 (50 ml) se le añadió SOCl2 (2,7 g, 0,02 mol) a 0 °C y se agitó durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar el compuesto B como producto bruto. Se usó este material directamente para la siguiente etapa sin purificación adicional.
Síntesis de (2S,3R)-2-amino-3-hidroxibutanoato de metilo (C):
A una disolución con agitación de ácido (2S,3R)-2-amino-3-hidroxibutanoico (200 g, 1,68 mol) en metanol (1,2 l) se le añadió SOCl2 (244 ml, 3,36 mol) gota a gota a 0 °C y se agitó durante 1 h. Se sometió la mezcla de reacción resultante a reflujo durante 24 h. Tras el consumo del material de partida (mediante CCF), se calentó la mezcla de reacción hasta TA y se concentró a presión reducida y se decantó con n-hexano (2 * 50 ml). Se disolvió el residuo en EtOH (1 l) y se neutralizó con Et3N (471 ml, 3,36 mol) y se agitó de nuevo durante 2 h. Se separó por filtración el sólido precipitado; se concentró el filtrado obtenido a presión reducida para proporcionar el compuesto C (195 g, 80 %).
1H-RMN: (400 MHz, DMSO-da): 88,51 (s a, 3H), 4,13-4,10 (m, 1H), 3,91 (s a, 1H), 1,20 (d, 3H).
CL-EM (m/z): 134,1 [M++1].
Síntesis de (2S,3R)-2-amino-3-hidroxibutanamida (D):
Se llevó una disolución del compuesto F (190 g, 1,35 mol) en IPA (2 l) a autoclave y se purgó con gas NH3 (7-8 kg) y se agitó a 35 °C durante 24 h. Después se eliminó el gas NH3 y se concentró la mezcla de reacción a presión reducida y se añadió CH2Cl2 y se filtró. Se sometió el sólido obtenido a reflujo en EtOH durante 1 h a 78 °C. Se filtró la masa de reacción en condición de calentamiento y se añadió n-hexano al filtrado y se agitó de nuevo durante otras 4 h. Se filtró el sólido precipitado obtenido y se secó a presión reducida para proporcionar el compuesto D (160 g, 47 %).
1H-RMN: (500 MHz, DMSO-d6): 87,38 (s a, 1H), 7,02 (s a, 1H), 4,66 (s a, 1H), 3,77-3,70 (m, 1H), 2,93 (d, 1H), 2,72 (m a, 1H), 1,05 (d, 3H).
CL-EM (m/z): 119,1 [M++1].
UPLC (pureza de ELSD): 99,9 % .
Síntesis de 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo (E):
A una disolución de ácido 2-(terc-butoxicarbonilamino)-3-hidroxibutanoico (50 g, 228,3 mmol) en DMF (500 ml) se le añadió K2CO3 (63 g, 456,6 mmol) y se agitó a TA durante 15 min. A esto se le añadió bromuro de bencilo (46,83 g, 273,9 mmol) y se agitó a TA durante 6 h. Se diluyó la mezcla de reacción con agua (500 ml) y se extrajo con EtOAc (2 * 750 ml). Se lavaron las fases orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el material bruto mediante cromatografía en columna de gel de sílice eluyendo con el 20 % de EtOAc/hexano para proporcionar 2-(terc-butoxicarbonilamino)-3-hidroxibutanoato de bencilo E (52 g, 73 %).
1H-RMN: (500 MHz, DMSO-d6): 87,37-7,30 (m, 5H), 6,60 (d, 1H), 5,18-5,08 (m, 2H), 4,76 (d, 1H), 4,08-4,00 (m, 2H), 1,38 (s, 9H), 1,09 (d, 3H).
Masa m/z: 310,0 [M++1], 210 [M+-des-Boc].
Síntesis de 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato de bencilo (F):
A una disolución con agitación del compuesto E (52 g, 168,2 mmol) en CH2Cl2 (500 ml) se le añadieron Ac2O (20,5 g, 201,9 mmol), Et3N (25,4 g, 252,4 mmol) y DMAP (3,5 g) y se agitó a t A durante 2 h. Se retiraron los compuestos volátiles a presión reducida. Se diluyó el residuo obtenido con EtOAc (750 ml) y se lavó con disolución de HCl 0,5 N fría (2 * 200 ml). Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para proporcionar 3-acetoxi-2-(terc-butoxicarbonilamino)butanoato F (52 g, 88 %).
1H-RMN: (500 MHz, DMSO-d6): 87,35-7,34 (m, 5H), 7,27-7,25 (d, 1H), 5,18-5,06 (m, 3H), 4,34-4,32 (m, 1H), 1,90 (s, 3H), 1,39 (s, 9H), 1,16 (d, 3H).
Masa m/z: 252 [M++1-des-Boc].
Síntesis de ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico (G):
A una disolución con agitación del compuesto F (52 g, 148,1 mmol) en MeOH (1 l) se le añadió Pd al 10 % /C bajo atmósfera de N2 y se agitó la mezcla de reacción bajo atmósfera de hidrógeno durante 16 h. Se filtró la mezcla de reacción a través de un lecho de Celite, se evaporó el filtrado obtenido a presión reducida y se trituró el residuo bruto con hexano para producir ácido (2S,3R)-3-acetoxi-2-(terc-butoxicarbonilamino)butanoico G (35 g, 90 %).
1 H-RMN: (500 MHz, DMSO-da): 8 12,78 (s a, 1H), 6,94 (d, 1H), 5,16-5,14 (m, 1H), 4,17-4,15 (m, 1H), 1,95 (s, 3H), 1,39 (s, 9H), 1,10 (d, 3H).
Masa m/z: 260,0 [M-1].
Síntesis de clorhidrato de (S)-pirrolidin-2-carboxilato de metilo (1):
A una disolución de L-prolina (SM) (110 g, 956,5 mmol) en metanol se le añadió cloruro de tionilo (141 ml, 1911,3 mmol) y se sometió a reflujo durante 16 h. Se llevó la mezcla de reacción hasta TA y se concentró a presión reducida para proporcionar el compuesto 1 como sal de clorhidrato (170 g, 99 %).
Síntesis de pirrolidin-1,2-dicarboxilato de (S)-1-bencilo y 2-metilo (2):
A una disolución de 1 (170 g, 947 mmol) en DCM se le añadió TEA (398 ml, 2,83 mol) y tras 30 min se añadió Cbz-Cl (1,136 mol). Se agitó la mezcla de reacción a TA durante 12 h. Se lavó la mezcla de reacción con agua y se extrajo con DCM. Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna para proporcionar el compuesto 2 (230 g, 88 %).
1 H-RMN: (200 MHz, DMSO-d6): 8 7,42-7,31(m, 5H), 5,09 (m, 2H), 4,32-4,23(m, 1H), 4,08-3,98(m, 2H), 3,50-3,38 (m, 2H), 2,30-2,18 (m, 1H), 1,90-1,81(m, 3H), 1,10-1,04 (t, 3H).
Masa m/z: 278 [M++1].
Síntesis de 2-(3,4,5-trimetoxibencil)pirrolidin-1,2-dicarboxilato de 1-bencilo y 2-metilo (3):
A una disolución del compuesto 2 (3,4 g, 12,9 mmol) en THF seco (60 ml) bajo atmósfera inerte se le añadió LiHMDS (1 M en THF) (25 ml, 25,0 mmol) a -25 °C y se agitó durante 2 h. Se añadió 5-(bromometil)-1,2,3-trimetoxibenceno (4 g, 15,5 mmol) gota a gota a -25 °C a la mezcla de reacción. Se dejó calentar hasta TA y se agitó durante 2 h. Se extinguió la mezcla de reacción con disolución de NH4Cl saturada y se extrajo la fase acuosa con EtOAc (2 x 200 ml). Se secaron los extractos orgánicos combinados sobre Na2SO4 anhidro y se concentraron a presión reducida. Se purificó el residuo bruto obtenido mediante cromatografía en columna de gel de sílice eluyendo con el 15 % de EtOAc/hexano para proporcionar el compuesto 3 (4,8 g, 84 %).
CL-EM (m/z):444,4 [M++1].
HPLC: 92 % .
Síntesis de 2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxilato de metilo (4):
A una disolución con agitación del compuesto 3 (4,8 g, 10,8 mmol) en CH3OH (40 ml) se le añadió Pd/C (1,5 g) y se agitó bajo atmósfera de H2 a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se filtró la mezcla de reacción a través de un lecho de Celite y se lavó con MeOH. Se concentró el filtrado obtenido a presión reducida para proporcionar el compuesto 4 (3,0 g, 90 %).
1 H-RMN: (400 MHz, DMSO-d6): 8 6,49 (s, 2H), 3,72 (s, 6H), 3,61 (s, 6H), 2,99 (d, 1H), 2,85-2,81 (m, 2H), 2,78 (d, 1H), 2,12-2,07 (m, 1H), 1,79-1,71 (m, 3H).
CL-EM (m/z): 310,2 [M++1].
Síntesis de (2S)-2-(2-(metoxicarbonil)-2-(3,4,5-trimetoxibencil)-pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (5):
A una suspensión de compuesto 5 (3 g, 9,70 mmol) en DCM:H2O (45 ml, 5:4) se le añadió Na2CO3 (2,5 g, 24,20 mmol) seguido de una disolución de Int-B (3,2 g, 11,60 mmol) en DCM a TA y se agitó durante 2 h. Se lavó la fase orgánica con salmuera, se secó sobre Na2SO4, se concentró a presión reducida para proporcionar el compuesto 5 (4,6 g) como producto bruto. Se usó este material directamente para la siguiente etapa sin purificación adicional.
CL-EM (m/z):541,3 [M++1].
Síntesis de ácido (S)-1-(1-((benciloxi)carbonil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxílico (6): A una disolución con agitación del compuesto 5 (4,6 g, 8,80 mmol) en MeOH (40 ml) se le añadió disolución de NaOH 2 N (10 ml) y se agitó a temperatura de reflujo durante 16 h. Se evaporaron los compuestos volátiles a presión reducida y se diluyó el residuo obtenido con agua y se ajustó a pH~ 2 usando disolución de HCl 1 N. Se extrajo la fase acuosa con EtOAc. Se secaron los extractos orgánicos combinados sobre Na2SO4 y se concentraron
a presión reducida para proporcionar el compuesto 6 (4 g) como producto bruto.
Síntesis de (S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3,4,5-trimetoxibencil)pirrolidin-1-carbonil)pirrolidin-1-carboxilato de bencilo (7):
A una disolución con agitación del compuesto 6 (4 g, 7,60 mmol) en CH2Cl2 (50 ml) se le añadieron HOBt (1,5 g, 11,40 mmol), EDCI.HCl (2,10 g, 11,40 mmol) seguido de DIPEA (4,1 ml, 22,8 mmol) y compuesto D (987 mg, 8.00 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se extinguió la reacción con agua y se extrajo con CH2O 2 (2* 100 ml). Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida para obtener producto bruto, que se purificó mediante cromatografía en columna para proporcionar el compuesto 7 (3 g, 63 %).
CL-EM (m/z):625,6 [M+-1].
Síntesis de N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (8):
A una disolución con agitación del compuesto 7 (3 g, 4,79 mmol) en MeOH (30 ml) se le añadió Pd al 10 % /C (0,7 g) bajo atmósfera de N2. Se agitó la mezcla de reacción a TA durante 16 h bajo atmósfera de H2. Tras el consumo del material de partida (mediante CCF), se filtró la mezcla de reacción a través de un lecho de Celite y se concentró el filtrado a presión reducida para proporcionar el compuesto 8 (2,1 g, 91 % ) como un sólido blanco.
Síntesis de ((2S,3R)-1-((S)-2-(2-(((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)carbamoil)-2-(3,4,5-trimetoxibencil)pirrolidin-1-carbonil)pirrolidin-1-il)-3-hidroxi-1-oxobutan-2-il)carbamato de terc-butilo (9):
A una disolución con agitación del compuesto 8 (2 g, 4,00 mmol) en CH2O 2 (30 ml) se le añadieron HOBt (823 mg, 6.00 mmol), EDCI.HCl (1,16 g, 6,00 mmol) seguido de DIPEA (2,2 ml, 11,6 mmol) y compuesto G (1,068 g, 4,80 mmol) a 0 °C. Se agitó la mezcla de reacción a TA durante 16 h. Tras el consumo del material de partida (mediante CCF), se extinguió la reacción con agua y se extrajo con CH2O 2 (2 * 100 ml). Se secó la fase orgánica sobre Na2SO4 anhidro y se concentró a presión reducida. Se purificó el producto bruto mediante cromatografía en columna seguido de purificación por HPLC preparativa para proporcionar el compuesto 9-Fr-1 (0,12 g) y el compuesto 9-Fr-2 (0,2 g) como un sólido blanco.
Datos de HPLC quiral para el compuesto 9-Fr-1:
CL-EM (m/z): 594,5 [(M++1)-Boc].
HPLC: 94 % .
Datos de HPLC quiral para el compuesto 9-Fr-2:
CL-EM (m/z): 594,5 [(M++1)-Boc].
HPLC: 95 % .
Síntesis de (R)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (CM-14A):
A una disolución del compuesto 9-Fr-1 (0,12 g, 0,17 mmol) en DCM (2,5 ml) se le añadió 1,4-dioxano HCl (2,5 ml) y se agitó a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-14A (80 mg) como sal de HCl.
1H-RMN: (400 MHz, D2O): 86,89 (s, 2H), 4,99 (t, 1H), 4,45-4,40 (m, 4H), 3,98 (s, 6H), 3,85 (s, 4H), 3,71-3,65 (m, 4H), 3,51 (d, 1H), 3,39 (d, 1H), 2,62-2,55 (m, 1H), 2,44-2,37 (m, 1H), 2,24-2,20 (m, 4H), 1,84-1,82 (m, 1H), 1,51 (d, 3H), 1,27 (d, 3H), 1,15-1,09 (m, 1H).
CL-EM (m/z):594,5 [M++1].
Pureza de HPLC: 93 %.
Rotación óptica [a19,9o]: 21,91 (C = 0,5 % en agua).
Síntesis de (S)-N-((2S,3R)-1-amino-3-hidroxi-1-oxobutan-2-il)-1-((S)-1-((2S,3R)-2-amino-3-hidroxibutanoil)pirrolidin-2-carbonil)-2-(3,4,5-trimetoxibencil)pirrolidin-2-carboxamida (CM-14B):
A una disolución del compuesto 9-Fr-2 (0,2 g, 0,20 mmol) en DCM (2,5 ml) se le añadió 1,4-dioxano HCl (2,5 ml) y
se agitó a TA durante 2 h. Se concentró la mezcla de reacción a presión reducida para proporcionar CM-14B (170 mg) como sal de HCl.
1H-RMN: (400 MHz, D2O): 86,89 (s, 2H), 4,99 (t, 1H), 4,45-4,40 (m, 4H), 3,98 (s, 6H), 3,85 (s, 4H), 3,71-3,65 (m, 4H), 3,51 (d, 1H), 3,39 (d, 1H), 2,62-2,55 (m, 1H), 2,44-2,37 (m, 1H), 2,24-2,20 (m, 4H), 1,84-1,82 (m, 1H), 1,51 (d, 3H), 1,27 (d, 3H), 1,15-1,09 (m, 1H).
CL-EM (m/z):594,5 [M++1].
Pureza de HPLC: 95 %.
Rotación óptica [a19,9D]: -92,57(C = 0,5 % en agua).
Ejemplo 15 - Farmacocinética y metabolismo de fármaco
Este ejemplo demuestra las propiedades farmacocinéticas y de metabolismo de fármaco de compuestos de prueba. Se sometieron a prueba los compuestos CM-5A, CM-6A, CM-7A, CM-8A y CM-9A en un ensayo de solubilidad acuosa in vitro, permeabilidad de MDCK y estabilidad metabólica. Los compuestos CM-5A, CM-6A y CM-7A también se sometieron a prueba para determinar la farmacocinética en rata. A continuación se resumen un breve procedimiento, resultados y conclusiones.
Solubilidad acuosa:
Procedimiento. Se evaluó la cinética de solubilidad acuosa (método de plON) usando los siguientes parámetros: • Preparación de disolución madre : DMSO
Tampón Tampón libre de fosfato
1 Condición de pH pH 7,4
1 Concentración de prueba 200 |iM con DMSO al 1 % (n = 3)
1 Tiempo de incubación 16 h
1 Filtración placas de filtro de 0,22 |i
1 Análisis CL-EM/EM
Resultados. A continuación se presentan los resultados del ensayo de solubilidad:

Conclusión. Los resultados sugieren que los compuestos de prueba tienen una buena solubilidad acuosa a pH 7,4. Estabilidad metabólica:
Se evaluó la estabilidad metabólica en microsomas de hígado así como en hepatocitos con unos pocos compuestos. Procedimiento para estabilidad microsomal:
Especie : Rata y humano
Concentración de prueba : 1 |iM (n = 2)
Concentración de proteína : 0,125 mg/ml
• Puntos de tiempo : 0, 5, 15, 30 y 60 minutos
• Controles : Sin NADPH y tampón
• Análisis : Monitorización del compuesto original mediante CL-EM/EM (razones de área de picos)
Procedimiento para estabilidad en hepatocitos:
• Preparación de disolución madre : DMSO
• Tampón Tampón de Krebs Henseleit
• Volumen de incubación total 1000 |il
• Densidad celular 1 millón de células/ml
• Concentración de prueba 10 |iM con DMSO al 0,5 % (n = 2)
• Puntos de tiempo 0 y 60 min
• Análisis CL-EM/EM
Resultados:

(H): Hepatocitos; (M): Microsomas
La clasificación de estabilidad metabólica se basó en los siguientes criterios.

Conclusión. Los resultados sugieren que los compuestos de prueba tienen una estabilidad de moderada a alta en microsomas/hepatocitos.
Permeabilidad de MDCK:
Procedimiento. Se evaluó la permeabilidad de compuestos de prueba usando MDCK (células de riñón canino Madin-Darby) usando los siguientes parámetros.
• Concentración de prueba : 5 |iM (n = 3)
• Tampón : HBSS pH 7,4
• Puntos de tiempo : 0, 15, 30, 60 y 90 min
• Análisis : Monitorización del compuesto original usando CL-EM/EM
Resultados. A continuación se presentan los resultados del ensayo de permeabilidad de MDCK:

Conclusión. Los resultados sugieren que los compuestos de prueba tienen una permeabilidad escasa/baja a través de células MDCK. La clasificación de permeabilidad de MDCK se basó en los siguientes criterios.

Biodisponibilidad de CM-4A
Procedimiento. A ratas Sprague Dawley de 2-3 meses de edad macho (1 mg/kg i.v. y 10mg/kg v.o.) y macho y hembra (0,1 y 1 mg/kg v.o.) se les administraron dosis de CM-4A (1 mg/kg i.v. [vena de la cola] o 0,1, 1 o 10 mg/kg v.o. [sonda gástrica]; n = 5-6 por grupo) en vehículo de solución salina estéril y se extrajeron muestras de sangre repetidas a diversos puntos de tiempo mediante extracciones de la vena de la cola lateral o punción cardiaca usando jeringas tratadas con K2-EDTA. Se combinaron los datos para machos y hembras para la dosis de 1 mg/kg v.o. dado que no se observó ninguna diferencia por sexo. Se separó el plasma mediante centrifugación (1000 * G durante 10 min a 4 °C) y se almacenaron las muestras de plasma a -80 °C hasta el ensayo. Se almacenaron las muestras a -80 °C hasta el ensayo. En el día del ensayo, se descongelaron las muestras y patrones de plasma con adiciones conocidas a 4 °C, se extrajeron en 3 volúmenes de acetonitrilo, se centrifugaron a 25000 * G durante 10 min a 4 °C, y se cargó el sobrenadante en viales de inyector automático (4 °C). Se analizaron las muestras usando un instrumento Agilent QQQ LC/MS/MS en modo de monitorización de reacciones múltiples (504,3 -> 171,1, 199,1, 386,1 m/z) y el tiempo de retención de CM-4A fue de 9,3 min con un pico simétrico a ~10 s.
Tal como se muestra en la figura 1, la biodisponibilidad oral de CM-4A es esencialmente lineal desde 0,1 hasta 10 mg/kg.
Ejemplo 16 - Ensayo de unión a [3H]-MK-801
Este ejemplo demuestra un ensayo de unión a [3H]-MK-801 que puede usarse para evaluar propiedades agonistas y/o antagonistas de moduladores de receptor de n MdA candidatos.
Se prepararon membranas sinápticas en bruto a partir de prosencéfalos de rata tal como se describe en Moskal et al. (2001), “The use of antibody engineering to create novel drugs that target N-methyl-D-aspartate receptors”, Curr. Drug Targets, 2:331-45. Se decapitaron ratas macho de 2-3 meses de edad sin anestesia mediante guillotina, y se extirparon rápidamente los cerebros (~90 s) y se disecaron la corteza y el hipocampo completos en una plataforma helada, se congelaron en hielo seco y se almacenaron a -80 °C. Se homogeneizaron las muestras en 20 volúmenes de Tris-HCl 5 mM helado pH 7,4 mediante un instrumento Brinkman Polytron y se sedimentaron a 48000 * g durante 20 min a 4 °C, y se lavaron 3 veces adicionales tal como se describió anteriormente. Después se resuspendieron las membranas en EDTA 5 mM y Tris-HCl 15 mM pH 7,4 y se incubaron durante 1 h a 37 °C, se sedimentaron las membranas a 48000 * g durante 20 min a 4 °C, se congelaron de manera inmediata en nitrógeno líquido y se almacenaron a -80 °C. El día del experimento, se descongelaron las membranas a temperatura ambiente y se lavaron 7 veces adicionales en Tris-HCl 5 mM helado (pH 7,4) tal como se describió anteriormente. Tras el último lavado, se resuspendieron las membranas en tampón de ensayo (Tris-acetato 5 mM pH 7,4) y se determinó el contenido de proteína mediante el ensayo de BCA.
Se realizaron ensayos de unión a [3H]-MK-801 tal como se describe en Urwiler et al. (2009), “Drug design, in vitro pharmacology, and structure-activity relationships of 3-acylamino-2-aminopropionic acid derivatives, a novel class of partial agonists at the glycine site on the N-methyl-D-aspartate (NMDA) receptor complex”, J. Med. Chem., 52:5093-10. Se incubó proteína de membrana (200 |ig) con concentraciones variables de los compuestos de prueba (10'3-10‘ 17 M) con glutamato 50 |iM durante 15 min a 23 °C. Después se incubaron los tubos de ensayo en condiciones no de equilibrio con [3H]-MK-801 (5 nM; 22,5 Ci/mmol) durante 15 min a 23 °C seguido de filtración a través de filtros Whatman GF/B usando un colector de células Brandel M-24R. Después se lavaron los tubos tres veces con tampón de ensayo (Tris-acetato 5 mM pH 7,4) y se analizaron los filtros mediante centelleo de líquido para calcular las desintegraciones por minuto (DPM). Se determinaron los niveles de cero en ausencia de cualquier ligando de glicina
y en presencia de ácido 5,7-dicloroquinurénico 30 mM (5,7-DCKA). Se midió la estimulación máxima en presencia de glicina 1 mM. Había glutamato 50 uM presente en todas las muestras.
Para cada punto de datos (es decir, una única concentración del compuesto de prueba), se calculó el % de unión a [3H]-MK-801 máxima mediante la siguiente fórmula:
% de unión a [ H]-MK-801 máxima — ((DpM(compuesto de prueba) — DPM5,7-DCKA) / (DPMglicina 1 mM — DPM5,7-DCKA)) x 100 % La eficacia para cada compuesto, expresada como el % de aumento de la unión a [3H]-MK-801, se calcula ajustando los datos a una ecuación de “log(agonista) frente a respuesta (tres parámetros)” usando Graph Pad Prism, y se calcularon la potencia (CE50, expresada en pM) y actividad máxima (% de estimulación máxima) para cada compuesto de prueba, siendo la eficacia para el compuesto de prueba el valor superior de mejor ajuste (figuras 2-4). Tabla 1. Datos de ensayo de unión a [3H]-MK-801.

Ejemplo 17 - Corrientes de receptor de NMDA (NMDAR)
Este ejemplo demuestra un ensayo para determinar el efecto de compuestos de prueba sobre corrientes de NMDAR.
Se llevaron a cabo experimentos con cortes de hipocampo de ratas Sprague-Dawley de 14-18 días de edad tal como se describe en Zhang et al. (2008) “A receptor NMDA glycine site partial agonist, GLYX-13 (“GLYX”), simultaneously enhances LTP and reduces LTD at Schaffer collateral-CA1 synapses in hyppocampus”, Neuropharmacology, 55:1238-50. Se obtuvieron registros de células completas a partir de neuronas piramidales CA1 con voltaje fijado a -60 mV, en cortes sometidos a perfusión con LCRA (líquido cefalorraquídeo artificial) que contenía [Mg2+] 0 mM y [Ca2+] 3 mM, más bicuculina 10 uM y CNQX 20 uM para aislar farmacológicamente corrientes postsinápticas excitadoras (EPSC) dependientes de NMDAR. Se aplicaron en baño diversas concentraciones de compuesto de prueba (de 10 nM a 1 uM) y se estimularon fibras colaterales de Schaffer con pulsos eléctricos individuales (80 us de duración) una vez cada 30 s. Las EPSC de NMDAR se caracterizaron por tiempos de aumento y disminución prolongados, y se bloquearon completamente al final de cada experimento mediante aplicación en baño del antagonista específico de NMDAR, ácido D-2-amino-5-fosfonopentanoico (D-AP5; 50 uM). Se calculó la eficacia de un compuesto de prueba como el % de aumento de la corriente de NMDAR con respecto al nivel de referencia. El nivel de referencia se midió como la corriente de NMDAR antes de aplicarse el compuesto de prueba.
Tabla 2. Datos del ensayo de corriente de NMDAR.

NT — no sometido a prueba.
La figura 5 muestra una evolución temporal del efecto de CM-4A (1 uM) sobre las EPSC de NMDA
farmacológicamente aislado provocadas por vía colateral de Schaffer de un único choque registradas en neuronas piramidales CA1 (n = 5). (Cada punto es la medida ± EEM de la amplitud máxima de EPSC o el potencial postsináptico excitador de campo (fEPSP) de n neuronas piramidales).
Ejemplo 18 - Procedimiento 1 de ensayo de potenciación a largo plazo (LTP)
Este ejemplo demuestra un ensayo para determinar el efecto de compuestos de prueba sobre LTP.
Se transfirieron cortes de hipocampo de ratas Sprague-Dawley de 14-18 días de edad a una cámara de registro de superficie de contacto y se sometieron a perfusión continua a 3 ml/min con LCRA oxigenado a 32 °C ± 0,5 °C. Se fabricaron electrodos de registro de baja resistencia de vidrio de borosilicato de pared delgada (1-2 MQ tras rellenarlos con LCRA) y se insertaron en la región dendrítica apical del campo de terminación colateral de Schaffer en el estrato radiado de la región CA1 para registrar potenciales postsinápticos excitadores de campo (fEPSP). Se colocó un electrodo de estimulación de acero inoxidable bipolar (FHC Co.) en fibras de la comisura colateral de Schaffer en el estrato radiado CA3 y se ajustó la intensidad de estímulos de corriente constante para provocar fEPSP aproximadamente semimáximos una vez cada 30 s (50-100 pA; 100 |is duración). Se midió la pendiente de fEPSP mediante interpolación lineal a partir del 20 %-80 % de la desviación negativa máxima, y se confirmó que las pendientes eran estables dentro de un intervalo de ± 10 % durante al menos 10 min antes de comenzar un experimento. Se indujo potenciación a largo plazo (LTP) mediante un tren de estímulos de alta frecuencia (3 x 100 Hz/500 ms; flecha) en sinapsis CA1 colaterales de Schaffer en control (vehículo), cortes sin tratar o cortes tratados previamente con compuesto de prueba (de 10 nM a 100 |iM). Se registraron señales de potenciación a largo plazo usando un amplificador Multiclamp 700B y se digitalizaron con un instrumento Digidata 1322 (Axon Instruments, Foster City, CA). Se analizaron los datos usando el software pClamp (versión 9, Axon Instruments) en un ordenador personal compatible con IBM. Se calculó la eficacia como el % de aumento en la potenciación a largo plazo medida para cortes tratados previamente con compuesto de prueba en comparación con el vehículo.
Tabla 3. Datos del ensayo de LTP.

NT = no sometido a prueba.
La figura 6 muestra una relación de dosis-respuesta de los efectos sobre LTP para CM-2, CM-1 y CM-4A
La figura 7 muestra una relación de dosis-respuesta de los efectos sobre la depresión a largo plazo (LTD) de GLYX-13 (“GLYX”) frente a CM-4A. 1 |iM tanto de GLYX-13 como de CM-4A redujeron significativamente la magnitud de LTD, mientras que concentraciones inferiores dieron como resultado alteraciones de menor magnitud. (Obsérvese que 0,001 |iM representa nada de adición de fármaco; cada punto es la media ± EEM de 8-10 cortes).
Ejemplo 19 - Procedimiento 2 de ensayo de potenciación a largo plazo (LTP)
Este ejemplo demuestra un ensayo para determinar el efecto de compuestos de prueba sobre LTP.
Se llevaron a cabo ensayos tal como se describe en Zhang et al. (2008). Se sometieron ratas Sprague-Dawley (12 18 días de edad; Taconic Farms) a anestesia profunda con isoflurano y se decapitaron. Se extirparon rápidamente los cerebros, se sumergieron en líquido cefalorraquídeo artificial helado (LCRA, 2-4 °C), que contenía (en mM): NaCl 124, KCl 4, MgSO42, CaCl22, NaH2PO4 1,25, NaHCO326, glucosa 10; a pH 7,4, continuamente gasificado con el 95 % de 0 2 /el 5 % de CO2). Se realizaron hemisecciones de los cerebros, se cortaron los lóbulos frontales y se adhirieron hemisferios individuales usando adhesivo de cianoacrilato sobre una plataforma sumergida en LCRA helado continuamente gasificado con el 95 % de 0 2 /el 5 % de CO2 durante el corte. Se cortaron cortes coronales de 400 |im de grosor usando un vibratomo (Leica VT1200S) y se transfirieron a una cámara de sujeción de superficie de contacto para su incubación a temperatura ambiente durante un mínimo de una h antes de transferirlos a una cámara de registro de superficie de contacto de tipo Haas sometida a perfusión continua a 3 ml/min con LCRA oxigenado a 32 °C ± 0 ,5 °C . Se fabricaron electrodos de registro de baja resistencia de vidrio de borosilicato de pared delgada (1-2 MQ tras rellenarlos con LCRA) y se insertaron en la región dendrítica apical del campo de terminación colateral de Schaffer en el estrato radiado de la región de campo CA1 para registrar potenciales postsinápticos excitadores de campo (fEPSP). Se colocó un electrodo de estimulación de acero inoxidable bipolar (FHC Co.) en fibras de la comisura colateral de Schaffer en el estrato radiado CA3 y se ajustó la intensidad de estímulos de corriente constante para provocar fEPSP aproximadamente semimáximos una vez cada 30 s (50 100 pA; 100 |is de duración). Se midió la pendiente de fEPSP antes y después de la inducción de LTP mediante
interpolación lineal desde el 20 hasta el 80 % de la desviación negativa máxima, y se confirmó que las pendientes eran estables dentro de un intervalo de ± 10 % durante al menos 15 min antes de comenzar un experimento. Se aplicó una aplicación en baño de compuestos de prueba (1 |iM) 30 min antes de la aplicación de trenes de estímulos colaterales de Schaffer para provocar LTP. Se indujo LTP mediante estimulación de axones colaterales de Schaffer con cuatro trenes de estímulos de ráfaga theta de alta frecuencia de 10 * 100 Hz/5 ráfagas de pulsos cada uno, aplicados a un intervalo entre ráfagas de 200 ms. Cada tren tenía 2 s de duración, y se aplicaron trenes a 15 s de separación. Se registraron las señales usando un amplificador Multiclamp 700B y se digitalizaron con un instrumento Digidata 1322 (Axon Instruments, USA). Se analizaron los datos usando el software pClamp (versión 9, Axon Instruments) en un ordenador personal compatible con IBM.
Tal como se muestra en la figura 8, CM-6A, CM-7A, CM-8A y CM-9A (1 |iM) aumentaron la potenciación a largo plazo tras la estimulación de alta frecuencia de EPSC de NMDA provocadas por la vía colateral de Schaffer de rata registradas en neuronas piramidales CA1. * p <0,05.
Ejemplo 20 - Procedimiento 1 de prueba de Porsolt
Este ejemplo demuestra la prueba de Porsolt para evaluar compuestos de prueba para determinar actividad antidepresiva.
Se llevaron a cabo experimentos tal como se describe en Burgdorf et al. (2009) “The effect of selective breeding for differential rates of 50-kHz ultrasonic vocalizations on emotional behavior in rats”, Devel. Psychobiol., 51:34-46. Se administraron dosis a ratas Sprague-Dawley macho (2-3 meses de edad) de compuesto de prueba (de 0,3 a 30 mg/kg; por vía intravenosa mediante inyección en la vena de la cola, o por vía oral mediante sonda gástrica) o vehículo (1 ml/kg de solución salina estéril, o 1 ml/kg de DMSO para 2,5-diazaespiro[3,4]octan-1-ona) de una manera ciega 1 h antes de las pruebas. Se colocaron los animales en un tubo de vidrio transparente de 46 cm de altura * 20 cm de diámetro lleno hasta 30 cm con agua corriente a temperatura ambiente (23 °C ± 0,5 °C) durante 5 min en el día de la prueba. El encargado del experimento secó a todos los animales con toalla tras cada sesión de nado. Se cambió el agua tras cada dos animales. Se grabaron los animales en cinta de vídeo y se cuantificó la duración total (s) del comportamiento de flotación (definido como el mínimo movimiento requerido para mantener la cabeza del animal por encima del agua) por un encargado del experimento de manera ciega.
Tabla 4. Datos del ensayo de Porsolt.

NT = no sometido a prueba.
La figura 9 muestra la media ± EEM del tiempo de flotación en la prueba de Porsolt de ratas en ratas SD macho de 2-3 meses de edad a las que se les administraron dosis de CM-4A (de 0,1 a 10 mg/kg, v.o., sonda gástrica) o vehículo de solución salina estéril (1 ml/kg, v.o.) 1 h antes de la primera sesión de prueba. Los animales recibieron una única sesión de habituación de Porsolt de 15 min en el día antes de la primera prueba de Porsolt de 5 min. N =9-10 por grupo. Los resultados demuestran un efecto antidepresivo dependiente de la dosis y del tiempo.
La figura 10 muestra la media ± EEM del tiempo de flotación en la prueba de Porsolt de ratas en ratas SD macho de 2-3 meses de edad a las que se les administraron dosis del antagonista de receptor de AMPA, 2,3-dihidroxi-6-nitro-7-sulfamoil-benzo[f]quinoxalin-2,3-diona (NBQX) (10 mg/kg, i.p.) o vehículo de agua destilada (1 ml/kg) 10 min antes de administrar la dosis de CM-4A (1 mg/kg, v.o.) o vehículo de solución salina estéril (1 ml/kg, v.o., sonda gástrica) 1 h antes de las pruebas. Los animales recibieron una única sesión de habituación de Porsolt de 15 min en el día antes de la primera prueba de Porsolt de 5 min. N =6 por grupo. * p <0,05 prueba de comparaciones múltiples a posteriori de PLSD de Fisher frente a todos los demás grupos. Los resultados demuestran que el antagonista de AMPA, NBQX, bloquea el efecto de tipo antidepresivo de CM-4A.
Ejemplo 21 - Procedimiento 2 de prueba de Porsolt
Este ejemplo demuestra la prueba de Porsolt para evaluar los compuestos de prueba para determinar actividad antidepresiva.
Se examinaron los efectos de tipo antidepresivo de compuestos de prueba con la prueba de Porsolt de ratas tal como se describe en Page et al. (1999) y Burgdorf et al. (2009). En este estudio se usaron ratas Sprague Dawley macho (2-3 meses de edad). Se administraron a los animales dosis de compuestos de prueba (0,3 mg/kg, i.v.) o
vehículo de solución salina estéril (1 ml/kg, i.v., vena de la cola) 1 h antes de una única sesión de prueba. A un segundo grupo de ratas Sprague Dawley macho (2-3 meses de edad) se les administraron dosis de compuestos de prueba (0,1 mg/kg, v.o.) o vehículo de solución salina estéril (1 ml/kg, v.o., sonda gástrica) 1 h antes de la primera sesión de prueba, y volvieron a someterse a prueba 24 h tras la administración de dosis. Se colocaron los animales en un tubo de plástico transparente de 46 cm de altura * 20 cm de diámetro lleno hasta 30 cm con agua corriente a temperatura ambiente (22 °C-23 °C) durante 15 min 1 día antes de la administración de dosis (habituación) y 5 min en el/los día(s) de prueba posterior(es). Se cambió el agua cada dos animales. Se grabaron los animales en cinta de vídeo y se cuantificó la duración total (s) del comportamiento de flotación por un encargado del experimento de manera ciega. Se sometieron a prueba CM-6A, CM-7A, CM-8A y CM-9A en la prueba de Porsolt. CM-5A no se sometió a prueba en la prueba de Porsolt.
Tal como se muestra en la figura 11A, CM-6A, CM-7A, CM-8A y CM-9A produjeron un efecto de tipo antidepresivo en la prueba de Porsolt tal como se clasifica mediante tiempo de flotación reducido tras una única dosis (0,3 mg/kg, i.v.) en comparación con vehículo 1 h tras la administración de dosis tal como se clasifica mediante un ANOVA de efecto de fármaco significativo (F(2, 24) = 17,5, p <0,0001), seguido de una prueba de comparaciones múltiples a posteriori de PLSD de Fisher significativa para cada compuesto de prueba frente al grupo de vehículo (* todos p <0,0001). N = 5-6 ratas por grupo.
Tal como se muestra en las figuras 11B y 11C, CM-6A, CM-7A, CM-8A y CM-9A produjeron un efecto de tipo antidepresivo en la prueba de Porsolt tal como se clasifica mediante tiempo de flotación reducido tras una única dosis (0,3 mg/kg, i.v.) en comparación con vehículo a 1 h (figura 11B) y 24 h (figura 11C) tras la administración de dosis tal como se clasifica mediante un ANOVA de mediciones repetidas de efecto de fármaco significativo (F(4, 25) = 58,2, p <0,0001), seguido de una prueba de comparaciones múltiples a posteriori de PLSD de Fisher significativa para compuesto de prueba en cada punto de tiempo frente al grupo de vehículo respectivo (* todos p <0,0001). n = 6 ratas por grupo.
Tal como se muestra en la figura 12, se observaron efectos de tipo antidepresivo robustos en la prueba de Porsolt de ratas. La media ± EEM del tiempo de flotación en la prueba de Porsolt de ratas en ratas SD macho de 2-3 meses de edad a las que se les administraron dosis de control positivo CM-4A (1 mg/kg, v.o.), compuestos de prueba (0,1 mg/kg, v.o.) o vehículo de solución salina estéril (1 ml/kg, v.o., sonda gástrica) a 1 h antes de las pruebas y 24 h tras la administración de la dosis. Los animales recibieron una única sesión de habituación de Porsolt de 15 min en el día antes de la primera prueba de Porsolt de 5 min. N = 6 por grupo. * p <0,05, prueba de comparaciones múltiples a posteriori de PLSD de Fisher frente a vehículo.
Ejemplo 22 - Prueba de hipofagia inducida por novedad (NIH) de rata
La prueba de NIH de rata se llevó a cabo de la siguiente manera. Se privaron a los animales de alimento durante la noche antes de las pruebas, y se colocó comida de laboratorio en la cámara central del campo abierto. Tras las pruebas de NIH, se determinó la latencia para comer en la jaula de residencia de los animales como control. Los antidepresivos de acción aguda así como crónica, pero no el tratamiento con SSRI agudo, disminuyeron la latencia para comer en la jaula nueva pero no en la jaula de residencia de los animales. La figura 13 muestra la media ± EEM de la latencia lineal para comer en la prueba de NIH de ratas SD macho de 2-3 meses de edad tratadas con CM-4A (1 mg/kg, v.o.) o vehículo de solución salina estéril (1 ml/kg, v.o.) 1 h antes de una única sesión de prueba de 10 min. N = 12 por grupo. * p <0,05. CM-4A (1 mg/kg, v.o.) produce un efecto de tipo antidepresivo/ansiolítico en la prueba de NIH de rata.
Ejemplo 23 - Prueba de vocalizaciones de ultrasonidos (USV) de ratas
Las figuras 14A y 14B muestran los resultados de la prueba de USV de rata. Se midió el aprendizaje emocional positivo durante los ensayos de estímulos condicionados (CS) precedentes a los ensayos de estímulos no condicionados (UCS) de cosquilleo (figura 14A). Los animales recibieron ensayos de 15 segundos que consistían en 6 ensayos CS y 6 UCS cada uno (3 min en total). También se midió la velocidad de carrera para animales para autoadministrarse cosquilleo al final de la sesión de 3 min. La figura 14B muestra la media ± EEM de USV hedonistas y aversivas en la prueba de USV de rata en ratas SD macho de 2-3 meses de edad tratadas con CM-4A (1 mg/kg, v.o.) o vehículo de solución salina estéril (1 ml/kg, v.o.) 1 h antes de las pruebas. N = 7-8. * p <0,05. Los resultados demuestran que CM-4A aumenta el aprendizaje emocional positivo y disminuye las USV de 20 kHz aversivas en la prueba de USV de rata, indicando así un efecto antidepresivo.
Ejemplo 24 - Prueba de campo abierto de rata
La figura 15 muestra la media ± EEM de cruces de línea, cruces de compartimento central y tiempo en compartimento central en ratas SD macho de 2-3 meses de edad tratadas con CM-4A (1 mg/kg, v.o.) o vehículo de solución salina estéril (1 ml/kg, v.o.) 1 h antes de una única sesión de prueba de 10 min en el campo abierto. N = 7 8. * p <0,05. Los resultados demuestran que CM-4A produce un efecto de tipo ansiolítico en la prueba de campo abierto de rata sin afectar al comportamiento locomotor.
Ejemplo 24 - Prueba de cilindro giratorio de aceleración de rata
La figura 16 muestra la media ± EEM de latencias de caída (s) en la prueba de cilindro giratorio de aceleración (4 40 RPM a lo largo de 300 s) en ratas SD macho de 2-3 meses de edad tratadas previamente con vehículo de solución salina (1 ml/kg, v.o.), CM-4A (1, 10 o 100 mg/kg, v.o.), o el control positivo ketamina (30 mg/kg, s.c.). Se sometieron los animales a prueba inmediatamente antes de la administración de dosis (0 min), 30 min tras la administración de dosis y 60 min tras la administración de dosis usando un diseño dentro de sujetos. Un día antes de las pruebas, los animales recibieron 3 sesiones de habituación a cilindro giratorio con al menos 30 min entre cada sesión, y el rendimiento de cilindro giratorio en la última sesión de habituación no difirió significativamente del punto de tiempo a los 0 min durante las pruebas. N = 5 para vehículo y dosis de CM-4A, N = 3 para ketamina. * p <0,05. Los resultados demuestran que CM-4A (10-100 mg/kg, v.o.) no produce un efecto de tipo sedante/ataxia en la prueba de cilindro giratorio de aceleración de rata.
Claims (1)
- REIVINDICACIONESCompuesto representado por:
 en el que:R1 se selecciona del grupo que consiste en hidrógeno, -OH, alquilo C 1-C 6 y halógeno;R2 y R4 se seleccionan cada uno independientemente del grupo que consiste en alquilo C 1 -6, hidrógeno, halógeno y -OH;R3 se selecciona del grupo que consiste en -OH, hidrógeno, alquilo C 1-C 6 y halógeno;R5 es -CH2-fenilo (en el que el fenilo está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados del grupo que consiste en halógeno, alcoxi C 1-3 y alquilo C 1-3 (opcionalmente sustituido con uno, dos o tres halógenos)); yX se selecciona del grupo que consiste en ORx y NRxRx, en el que RX se selecciona independientemente, para cada caso, del grupo que consiste en hidrógeno y alquilo C 1 -C 6; sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismoCompuesto según la reivindicación 1, en el que el compuesto se representa por:
en el que:R1 se selecciona del grupo que consiste en hidrógeno, -OH, alquilo C 1-C 6 y halógeno;R2 y R4 se seleccionan cada uno independientemente del grupo que consiste en alquilo C 1 -6, hidrógeno, halógeno y -OH;R3 se selecciona del grupo que consiste en -OH, hidrógeno, alquilo C 1-C 6 y halógeno;R5 es -CH2-fenilo (en el que el fenilo está opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados del grupo que consiste en halógeno, alcoxi C 1-3 y alquilo C 1-3 (opcionalmente sustituido con uno, dos o tres halógenos)); yX se selecciona del grupo que consiste en ORx y NRxRx, en el que RX se selecciona independientemente, para cada caso, del grupo que consiste en hidrógeno y alquilo C 1 -C 6; sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismoCompuesto según la reivindicación 1, en el que el compuesto se representa por: en el queR 11, R 12 y R 13 se seleccionan cada uno independientemente del grupo que consiste en H, halógeno, alcoxi C 1-3 y alquilo C 1-3 (opcionalmente sustituido con uno, dos o tres halógenos);X1 es OH o NH2 ,X2 es H u OH;y sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismo.Compuesto según la reivindicación 2, en el que el halógeno, para cada caso, se selecciona independientemente del grupo que consiste en Cl, Bry F.Compuesto según la reivindicación 2 o 3, en el que el compuesto es sustancialmente más eficaz cuando se administra oralmente a un paciente comparado con la administración oral a un paciente de un compuesto de peptidilo representado por:
en el queR 11, R 12 y R 13 se seleccionan cada uno independientemente del grupo que consiste en H, halógeno, alcoxi C 1-3 y alquilo C 1-3 (opcionalmente sustituido con uno, dos o tres halógenos);X1 es OH o NH2 ,X2 es H u OH;y sales farmacéuticamente aceptables, estereoisómeros e hidratos del mismo.Compuesto según la reivindicación 2, en el que el halógeno, para cada caso, se selecciona independientemente del grupo que consiste en Cl, Bry F.Compuesto según la reivindicación 2 o 3, en el que el compuesto es sustancialmente más eficaz cuando se administra oralmente a un paciente comparado con la administración oral a un paciente de un compuesto de peptidilo representado por: 5. Compuesto según una cualquiera de las reivindicaciones 2-4, en el que X2 es OH y X 1 es NH2.6. Compuesto según una cualquiera de las reivindicaciones 2-5, en el que cada uno de R 11 , R 19 y R 19 es H.7. Compuesto según una cualquiera de las reivindicaciones 2-6, en el que R 11 y R 19 es H y R 19 se selecciona del grupo que consiste en F, Br, Cl, CH3, CF3 y -OCH3.8. Compuesto según la reivindicación 1 representado por:
5. Compuesto según una cualquiera de las reivindicaciones 2-4, en el que X2 es OH y X 1 es NH2.6. Compuesto según una cualquiera de las reivindicaciones 2-5, en el que cada uno de R 11 , R 19 y R 19 es H.7. Compuesto según una cualquiera de las reivindicaciones 2-6, en el que R 11 y R 19 es H y R 19 se selecciona del grupo que consiste en F, Br, Cl, CH3, CF3 y -OCH3.8. Compuesto según la reivindicación 1 representado por: 9. Compuesto según la reivindicación 1 representado por:
9. Compuesto según la reivindicación 1 representado por: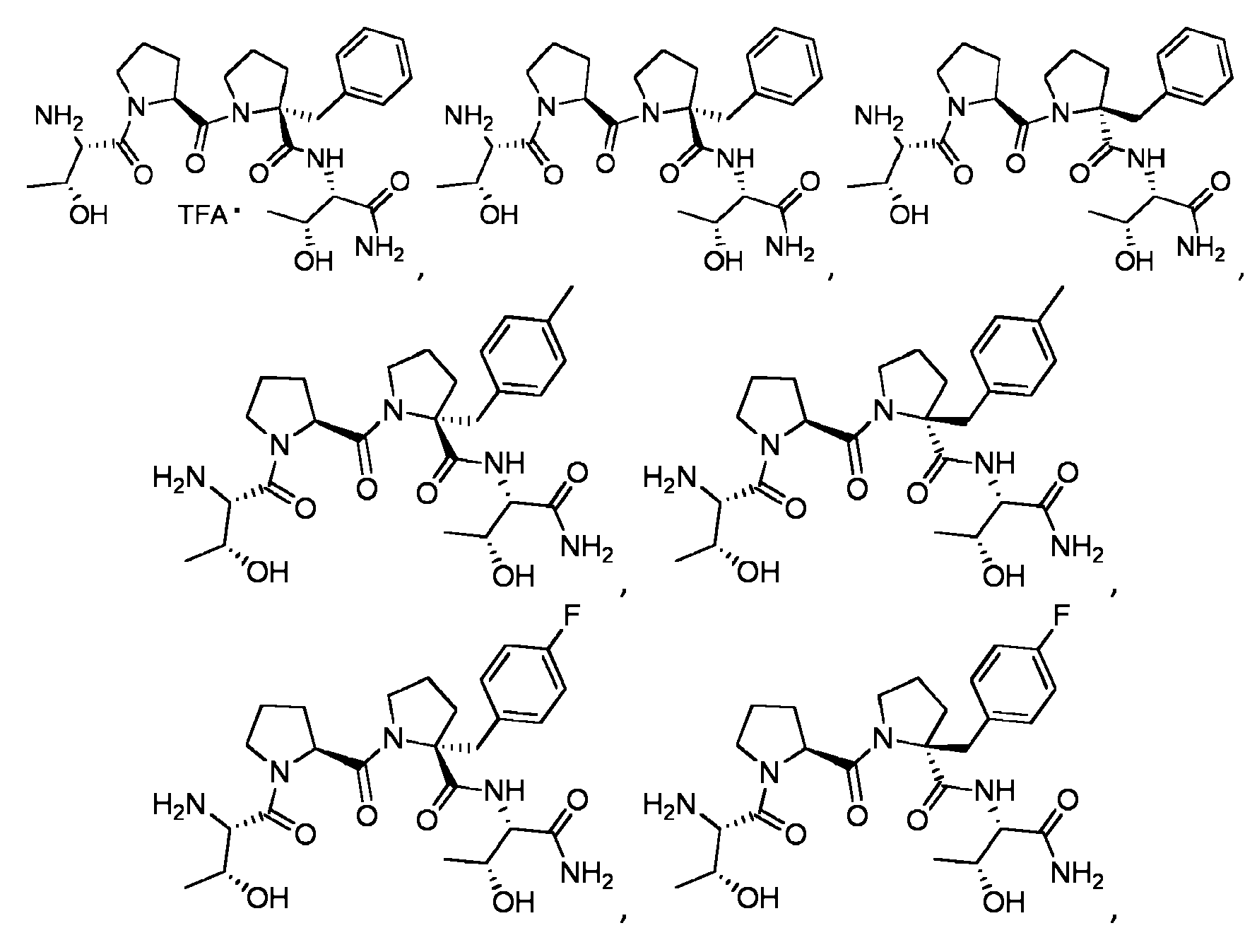
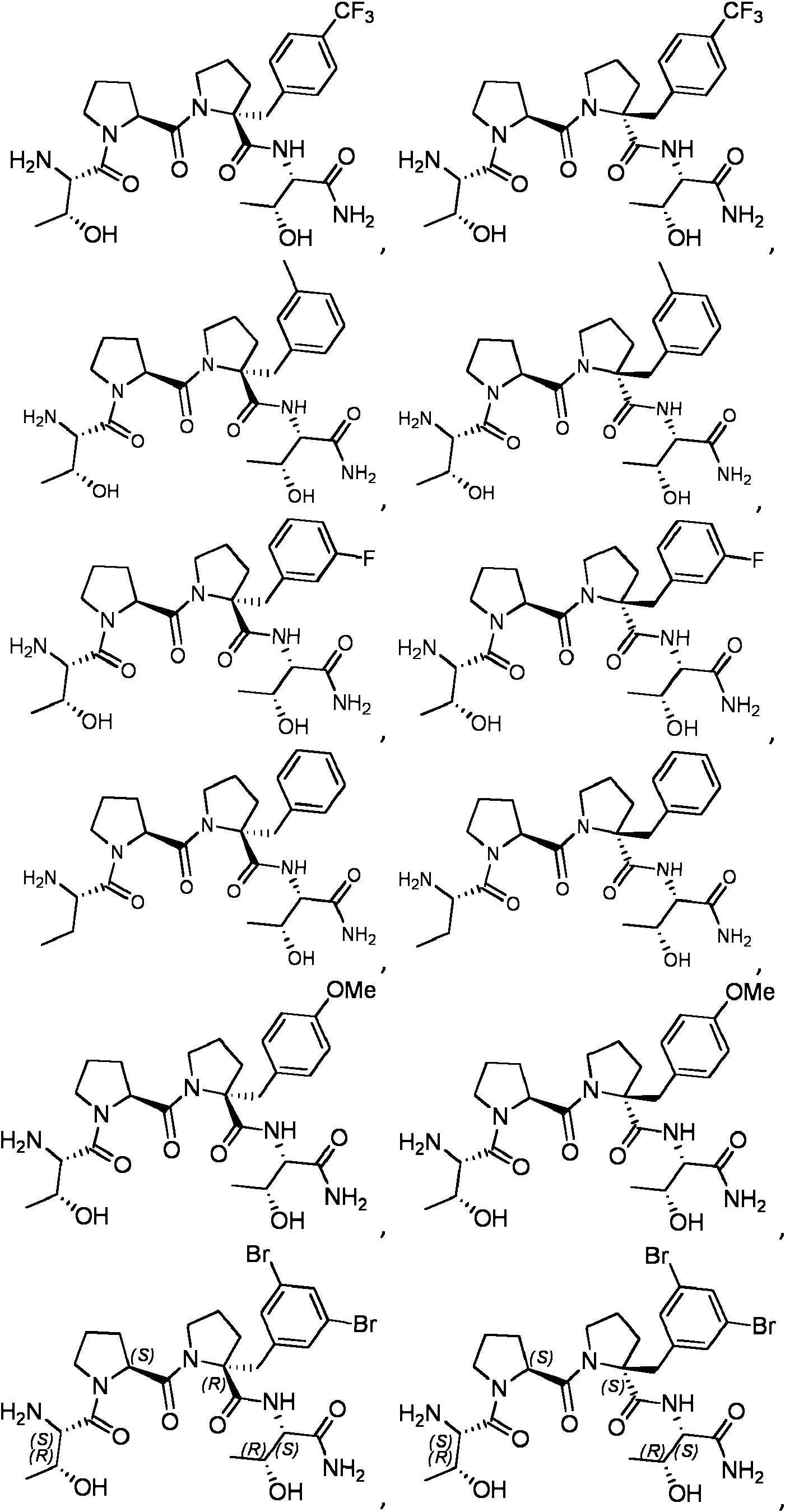
 10. Composición farmacéutica, que comprende:una cantidad terapéuticamente eficaz de un compuesto según una cualquiera de las reivindicaciones 1-9 y un portador farmacéuticamente aceptable, en el que opcionalmente la composición farmacéutica es adecuada para inyección.11. Compuesto según una cualquiera de las reivindicaciones 1-9 para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo.12. Compuesto según una cualquiera de las reivindicaciones 2-7 para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo mediante administración oral.13. Compuesto según una cualquiera de las reivindicaciones 8-9 para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo mediante administración subcutánea o intravenosa.
10. Composición farmacéutica, que comprende:una cantidad terapéuticamente eficaz de un compuesto según una cualquiera de las reivindicaciones 1-9 y un portador farmacéuticamente aceptable, en el que opcionalmente la composición farmacéutica es adecuada para inyección.11. Compuesto según una cualquiera de las reivindicaciones 1-9 para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo.12. Compuesto según una cualquiera de las reivindicaciones 2-7 para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo mediante administración oral.13. Compuesto según una cualquiera de las reivindicaciones 8-9 para su uso en un método de tratamiento de depresión, enfermedad de Alzheimer, trastorno por déficit de atención, TDAH, esquizofrenia o ansiedad, en un paciente necesitado del mismo mediante administración subcutánea o intravenosa.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161550782P | 2011-10-24 | 2011-10-24 | |
| PCT/US2012/061696 WO2013063120A2 (en) | 2011-10-24 | 2012-10-24 | Nmda receptor modulators and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2703904T3 true ES2703904T3 (es) | 2019-03-13 |
Family
ID=48168770
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12844456T Active ES2703904T3 (es) | 2011-10-24 | 2012-10-24 | Moduladores del receptor de NMDA y usos de los mismos |
Country Status (13)
| Country | Link |
|---|---|
| US (6) | US8673843B2 (es) |
| EP (1) | EP2771021B1 (es) |
| JP (1) | JP6210993B2 (es) |
| KR (2) | KR101692275B1 (es) |
| CN (2) | CN104321071A (es) |
| AU (3) | AU2012328927B2 (es) |
| BR (1) | BR112014009851A8 (es) |
| CA (1) | CA2853364C (es) |
| ES (1) | ES2703904T3 (es) |
| HK (1) | HK1206616A1 (es) |
| RU (1) | RU2621049C2 (es) |
| SG (1) | SG11201401776WA (es) |
| WO (1) | WO2013063120A2 (es) |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010033757A1 (en) | 2008-09-18 | 2010-03-25 | Naurex, Inc. | Nmda receptor modulators and uses thereof |
| US8951968B2 (en) | 2009-10-05 | 2015-02-10 | Northwestern University | Methods of treating depression and other related diseases |
| KR101692275B1 (ko) * | 2010-02-11 | 2017-01-04 | 노오쓰웨스턴 유니버시티 | 2차 구조 안정화된 nmda 수용체 조절제 및 그의 용도 |
| SG10201501050RA (en) | 2010-02-11 | 2015-04-29 | Univ Northwestern | Secondary Structure Stabilized NMDA Receptor Modulators And Uses Thereof |
| EP4245369A3 (en) | 2011-09-08 | 2023-11-22 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| JP6531042B2 (ja) | 2013-01-29 | 2019-06-12 | アプティニックス インコーポレイテッド | スピロラクタム系nmda受容体モジュレーターおよびその使用 |
| EA201591404A1 (ru) | 2013-01-29 | 2016-01-29 | Аптиникс Инк. | Спиролактамные модуляторы рецептора nmda и их применение |
| BR112015018092A2 (pt) | 2013-01-29 | 2017-07-18 | Naurex Inc | moduladores de receptor de nmda de espiro-lactama e usos dos mesmos |
| BR112015018094A2 (pt) | 2013-01-29 | 2017-07-18 | Naurex Inc | moduladores de receptor nmda de espiro-lactama e usos dos mesmos |
| JP2016506958A (ja) | 2013-01-29 | 2016-03-07 | ノーレックス, インコーポレイテッドNaurex, Inc. | スピロラクタム系nmda受容体モジュレーターおよびその使用 |
| CN112375115A (zh) | 2013-03-13 | 2021-02-19 | 萨奇治疗股份有限公司 | 神经活性甾类化合物及其使用方法 |
| CN104109189B (zh) * | 2013-04-18 | 2019-05-10 | 中国人民解放军军事医学科学院毒物药物研究所 | Thr-Pro-Pro-Thr四肽的液相合成方法 |
| KR20160077153A (ko) * | 2013-10-28 | 2016-07-01 | 노렉스, 인크. | Nmda 수용체 조절인자 및 이들의 전구약물, 염, 그리고 용도 |
| ES2970222T3 (es) | 2014-06-18 | 2024-05-27 | Sage Therapeutics Inc | Oxiesteroles y procedimientos de uso de los mismos |
| WO2016014982A1 (en) | 2014-07-24 | 2016-01-28 | Naurex, Inc. | N-methyl-d-aspartate receptor modulators and methods of making and using same |
| SG10202011773UA (en) * | 2014-10-07 | 2021-01-28 | Sage Therapeutics Inc | Neuroactive compounds and methods of use thereof |
| BR112018000129B1 (pt) | 2015-07-06 | 2024-01-09 | Sage Therapeutics, Inc | Compostos oxiesteróis, seus usos e composição farmacêutica |
| JP2018519328A (ja) | 2015-07-06 | 2018-07-19 | セージ セラピューティクス, インコーポレイテッド | オキシステロールおよびそれらの使用の方法 |
| MA42409A (fr) | 2015-07-06 | 2018-05-16 | Sage Therapeutics Inc | Oxystérols et leurs procédés d'utilisation |
| EP3362080A1 (en) * | 2015-10-16 | 2018-08-22 | Northwestern University | Pharmaceutical combination of an atypical antipsychotic and an nmda modulator for the treatment of schizophrenia,bipolar disorder, cognitive impairment and major depressive disorder |
| DK3436022T3 (da) | 2016-04-01 | 2022-06-20 | Sage Therapeutics Inc | Oxysteroler og fremgangsmåder til anvendelse heraf |
| WO2017193046A1 (en) | 2016-05-06 | 2017-11-09 | Sage Therapeutics, Inc. | Oxysterols and methods of use thereof |
| WO2017201285A1 (en) | 2016-05-19 | 2017-11-23 | Aptinyx Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| KR102128675B1 (ko) | 2016-05-19 | 2020-06-30 | 앱티닉스 인크. | 스피로-락탐 nmda 수용체 조정제 및 그의 용도 |
| PL3481846T3 (pl) | 2016-07-07 | 2021-12-20 | Sage Therapeutics, Inc. | 11-podstawione 24-hydroksysterole do stosowania w leczeniu stanów związanych z NMDA |
| JP2019527233A (ja) | 2016-08-01 | 2019-09-26 | アプティニックス インコーポレイテッド | スピロ−ラクタムnmda受容体修飾因子及びその使用 |
| KR102503590B1 (ko) | 2016-08-01 | 2023-02-24 | 앱티닉스 인크. | 스피로-락탐 nmda 수용체 조정제 및 그의 용도 |
| EA201990428A1 (ru) | 2016-08-01 | 2019-08-30 | Аптиникс Инк. | Спиролактамовые модуляторы nmda-рецептора и их применение |
| PE20190501A1 (es) | 2016-08-01 | 2019-04-10 | Aptinyx Inc | Moduladores nmda espiro-lactam y metodos de uso de los mismos |
| US11028095B2 (en) | 2016-08-01 | 2021-06-08 | Aptinyx Inc. | Spiro-lactam and bis-spiro-lactam NMDA receptor modulators and uses thereof |
| CN110023323A (zh) | 2016-09-30 | 2019-07-16 | 萨奇治疗股份有限公司 | C7取代的氧固醇及其作为nmda调节剂的方法 |
| MX2019004578A (es) | 2016-10-18 | 2019-10-09 | Sage Therapeutics Inc | Oxisteroles y metodos de uso de los mismos. |
| AU2017345399B2 (en) | 2016-10-18 | 2022-02-24 | Sage Therapeutics, Inc. | Oxysterols and methods of use thereof |
| CN108148113A (zh) * | 2016-12-06 | 2018-06-12 | 深圳先进技术研究院 | 一种nmda受体调控剂四肽衍生物的固相合成方法 |
| CN107474107B (zh) * | 2017-05-18 | 2023-01-17 | 江苏恩华药业股份有限公司 | Glyx-13的制备方法及用于制备glyx-13的化合物 |
| FR3067028B1 (fr) * | 2017-06-06 | 2019-07-12 | Adpuerivitam | Modulateurs de recepteurs nmda, compositions les comprenant et utilisation de ces composes dans le traitement de maladies impliquant le systeme nerveux central |
| EP3658569B1 (en) * | 2017-07-28 | 2024-01-31 | Naurex Inc. | Process and intermediates for synthesis of peptide compounds |
| KR20200115611A (ko) | 2018-01-31 | 2020-10-07 | 앱티닉스 인크. | 스피로-락탐 nmda 수용체 조정제 및 그의 용도 |
| CN110003309B (zh) * | 2019-04-09 | 2021-09-10 | 北京广为医药科技有限公司 | 调节nmda受体活性的化合物、其药物组合物及用途 |
Family Cites Families (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4683221A (en) * | 1986-01-09 | 1987-07-28 | Scripps Clinic And Research Foundation | Lymphocyte-activating polypeptides |
| CA1305177C (en) | 1987-06-30 | 1992-07-14 | Yasufumi Ohfune | Carboxycyclopropylglycine and process for producing the same |
| US4904681A (en) | 1987-12-01 | 1990-02-27 | G. D. Searle & Co. | D-cycloserine and its prodrugs as cognitive enhancers |
| EP0360390A1 (en) | 1988-07-25 | 1990-03-28 | Glaxo Group Limited | Spirolactam derivatives |
| US5061721A (en) | 1989-03-15 | 1991-10-29 | G. D. Searle & Co. | Composition containing d-cycloserine and d-alanine for memory and learning enhancement or treatment of a cognitive or psychotic disorder |
| US5086072A (en) | 1990-06-18 | 1992-02-04 | The United States Of America As Represented By The Department Of Health And Human Services | Treatment of mood disorders with functional antagonists of the glycine/nmda receptor complex |
| CA2058975C (en) | 1990-10-30 | 2000-06-06 | Shuichi Kasai | Antiinflammatory gel preparation |
| JP3318622B2 (ja) * | 1992-05-27 | 2002-08-26 | 独立行政法人産業技術総合研究所 | プロリルエンドペプチダーゼ阻害剤 |
| FR2692268B1 (fr) | 1992-06-15 | 1994-08-19 | Rhone Poulenc Rorer Sa | Nouveaux polypeptides ayant une activité de récepteur NMDA, acides nucléiques codant pour ces polypeptides et utilisations. |
| SE9301667D0 (sv) | 1993-05-14 | 1993-05-14 | Kabi Pharmacia Ab | New use |
| US5523323A (en) | 1993-09-14 | 1996-06-04 | Maccecchini; Maria-Luisa | Use of partial agonists of the NMDA receptor to reduce opiate induced tolerance and dependence |
| US5605911A (en) | 1995-01-31 | 1997-02-25 | Washington University | Use of alpha-2 adrenergic drugs to prevent adverse effects of NMDA receptor hypofunction (NRH) |
| US5741778A (en) | 1996-03-19 | 1998-04-21 | Amgen Inc. | Method for treating Huntington's disease using glial cell line-derived neurotrophic factor (GDNF) protein product |
| US5763393A (en) | 1996-05-17 | 1998-06-09 | Neurotherapeutics L.P. | Neuroactive peptides |
| US6541453B2 (en) | 1996-06-07 | 2003-04-01 | Syngenta Limited | Peptide derivatives |
| WO1998005782A1 (en) | 1996-08-02 | 1998-02-12 | Zymogenetics, Inc. | Testis-specific insulin homolog polypeptides |
| US5902815A (en) | 1996-09-03 | 1999-05-11 | Washington University | Use of 5HT-2A serotonin agonists to prevent adverse effects of NMDA receptor hypofunction |
| JP3955345B2 (ja) | 1996-09-27 | 2007-08-08 | サントリー株式会社 | 新規アミノ酸ダイジハーベイン |
| WO1999002733A1 (en) * | 1997-07-07 | 1999-01-21 | Florida State University | Identification of molecular targets |
| US6897028B1 (en) * | 1997-07-07 | 2005-05-24 | Florida State University | Identification of molecular targets |
| AU1585999A (en) | 1997-11-12 | 1999-05-31 | Neurotherapeutics | Methods for the detection and treatment of disease using a glycosyltransferase |
| US5952389A (en) | 1998-01-13 | 1999-09-14 | Synchroneuron | Methods of treating tardive dyskinesia and other movement disorders |
| US6007841A (en) | 1998-03-13 | 1999-12-28 | Algos Pharmaceutical Corporation | Analgesic composition and method for treating pain |
| US6274314B1 (en) | 1998-04-02 | 2001-08-14 | Nyxis Neurotherapies, Inc. | Diagnostic assay for the modified nucleosides pseudouridine, 7-methyladenosine, or 1-methyladenosine |
| US6197820B1 (en) | 1998-04-06 | 2001-03-06 | Uab Research Foundation | Use of phenylglycine derivatives to decrease neuronal death caused by brain tumors and brain lesions |
| US6025471A (en) | 1998-06-03 | 2000-02-15 | Deghenghi; Romano | Diazaspiro, azepino and azabicyclo therapeutic peptides |
| US6902886B1 (en) * | 1998-11-06 | 2005-06-07 | The Research Foundation Of State University Of New York | Genetic assay for protein nuclear transport |
| AU1724000A (en) | 1998-11-12 | 2000-05-29 | Nyxis, Inc. | Diagnostic assay for cancer |
| US20030064921A1 (en) | 1999-10-27 | 2003-04-03 | The Regents Of The University Of California | Methods and compounds for modulating melanocortin receptor ligand binding and activity |
| WO2001036685A2 (en) | 1999-11-17 | 2001-05-25 | Nyxis Neurotherapies, Inc. | Differential gene expression in cancer |
| EP1255854A2 (en) | 2000-02-01 | 2002-11-13 | Agy Therapeutics, Inc. | Interaction of nmda receptor with protein tyrosine phosphatase |
| AU2001268467A1 (en) | 2000-06-14 | 2001-12-24 | Nyxis Neurotherapies, Inc. | Identification of genes and compounds for treatment of cancer |
| EP1296999A2 (en) | 2000-06-22 | 2003-04-02 | NYXIS NeuroTherapies, Inc. | Neuroactive peptides for treatment of hypoxia and related conditions |
| IL145209A0 (en) | 2000-09-06 | 2002-06-30 | Pfizer Prod Inc | Pharmaceutical combinations for the treatment of stroke and traumatic brain injury |
| EP1186303A3 (en) | 2000-09-06 | 2003-12-10 | Pfizer Products Inc. | Pharmaceutical combinations, for the treatment of stroke and traumatic brain injury, containing a neutrophil inhibiting factor and an selective NMDA-NR2B receptor antagonist |
| UA73619C2 (en) | 2000-12-13 | 2005-08-15 | Pfizer Prod Inc | Stable pharmaceutical compositions of nmda receptor agonist (variants) and method of treatment |
| WO2002047535A2 (en) | 2000-12-14 | 2002-06-20 | Nyxis Neurotherapies, Inc. | High throughput assay to detect inhibitors of the map kinase pathway |
| US20030065138A1 (en) | 2001-03-07 | 2003-04-03 | University Of Utah Research Foundation | Linear gamma-carboxyglutamate rich conotoxins |
| AU2002255705A1 (en) | 2001-03-12 | 2002-09-24 | Nyxis Neurotherapies, Inc | Neuroactive peptides for prevention and/or treatment of hypoxia and neuropathic pain |
| JP2004531731A (ja) * | 2001-05-31 | 2004-10-14 | ザ トラスティーズ オブ プリンストン ユニバーシテイ | Iap結合ペプチドおよびiapに結合する化合物を同定するアッセイ |
| US20030022253A1 (en) * | 2001-07-25 | 2003-01-30 | Nyxis Neurotherapies, Inc. | Method for identifying, isolating and producing neuroactive binding agents and binding agents derived thereby |
| US7273889B2 (en) | 2002-09-25 | 2007-09-25 | Innovative Drug Delivery Systems, Inc. | NMDA receptor antagonist formulation with reduced neurotoxicity |
| WO2005014797A2 (en) | 2003-08-08 | 2005-02-17 | The Burnham Institute | P16 mediated regulation of nmda receptors |
| US7662856B2 (en) | 2003-08-29 | 2010-02-16 | The University Of Houston System | Compositions having antimycrobial activity including a hydroxamate or a hydroxamate and a hydroxlyamine |
| US20050096311A1 (en) | 2003-10-30 | 2005-05-05 | Cns Response | Compositions and methods for treatment of nervous system disorders |
| WO2007025141A2 (en) | 2005-08-26 | 2007-03-01 | Wisconsin Alumni Research Foundation | Poly-beta-peptides from functionalized beta-lactam monomers and antibacterial compositions containing same |
| EP1957093B1 (en) * | 2005-08-29 | 2017-04-12 | SHASHOUA, Victor E. | Neuroprotective and neurorestorative methods and compositions |
| US20070208001A1 (en) | 2006-03-03 | 2007-09-06 | Jincong Zhuo | Modulators of 11- beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| EP2061792A2 (en) | 2006-09-15 | 2009-05-27 | Schering Corporation | Azetidinone derivatives for the treatment of disorders of the lipid metabolism |
| CN101066945B (zh) | 2007-05-25 | 2010-05-19 | 中国科学院上海有机化学研究所 | 一种合成3-位取代内酰胺类化合物的方法 |
| CN101317844B (zh) * | 2007-06-08 | 2010-11-17 | 章劲夫 | 化合物(+)-5-甲基-10,11-二氢-5H-二苯并〔a,d〕环庚烯-5,10-亚胺在制备避孕药物中的应用 |
| CN101125817B (zh) | 2007-08-03 | 2011-09-14 | 中国科学院上海有机化学研究所 | 一种合成高对映选择性醛基取代小环胺类化合物和光学活性3-位取代内酰胺类化合物的方法 |
| WO2009039390A2 (en) | 2007-09-20 | 2009-03-26 | Naurex Inc. | The development of glycobiology-based therapeutics for the treatment of brain tumors |
| KR101647520B1 (ko) | 2008-08-07 | 2016-08-10 | 에프. 호프만-라 로슈 아게 | 거대환식 화합물의 제조 방법 |
| WO2010033757A1 (en) | 2008-09-18 | 2010-03-25 | Naurex, Inc. | Nmda receptor modulators and uses thereof |
| WO2010065709A2 (en) | 2008-12-03 | 2010-06-10 | Amin Khan | Hydroxamic acid derivatives, preparation and therapeutic uses thereof |
| WO2010071132A1 (ja) * | 2008-12-15 | 2010-06-24 | カルピス株式会社 | 皮膚老化抑制ペプチド |
| WO2011003064A2 (en) | 2009-07-02 | 2011-01-06 | Naurex, Inc. | Methods of treating neuropathic pain |
| PT2485751E (pt) | 2009-10-05 | 2014-09-01 | Univ Northwestern | Glyx-13 para uso num método para tratar depressão refratária |
| US8951968B2 (en) | 2009-10-05 | 2015-02-10 | Northwestern University | Methods of treating depression and other related diseases |
| SG10201501050RA (en) | 2010-02-11 | 2015-04-29 | Univ Northwestern | Secondary Structure Stabilized NMDA Receptor Modulators And Uses Thereof |
| KR101692275B1 (ko) | 2010-02-11 | 2017-01-04 | 노오쓰웨스턴 유니버시티 | 2차 구조 안정화된 nmda 수용체 조절제 및 그의 용도 |
| MX357761B (es) | 2011-04-27 | 2018-07-23 | Univ Northwestern | Métodos para tratar la enfermedad de alzheimer, enfermedad de huntington, autismo u otros trastornos. |
| US8795919B2 (en) | 2011-11-29 | 2014-08-05 | Societe BIC & Sanyo Electric Co., Ltd. | Fuel cell layer |
-
2011
- 2011-02-11 KR KR1020127023731A patent/KR101692275B1/ko active IP Right Grant
-
2012
- 2012-06-18 US US13/525,861 patent/US8673843B2/en active Active
- 2012-10-24 WO PCT/US2012/061696 patent/WO2013063120A2/en active Application Filing
- 2012-10-24 CA CA2853364A patent/CA2853364C/en active Active
- 2012-10-24 AU AU2012328927A patent/AU2012328927B2/en active Active
- 2012-10-24 BR BR112014009851A patent/BR112014009851A8/pt not_active Application Discontinuation
- 2012-10-24 EP EP12844456.9A patent/EP2771021B1/en active Active
- 2012-10-24 SG SG11201401776WA patent/SG11201401776WA/en unknown
- 2012-10-24 CN CN201280063834.6A patent/CN104321071A/zh active Pending
- 2012-10-24 CN CN201810978925.1A patent/CN109232713A/zh active Pending
- 2012-10-24 US US14/353,681 patent/US20150051262A1/en not_active Abandoned
- 2012-10-24 RU RU2014121090A patent/RU2621049C2/ru active
- 2012-10-24 KR KR1020147013898A patent/KR102038161B1/ko active IP Right Grant
- 2012-10-24 ES ES12844456T patent/ES2703904T3/es active Active
- 2012-10-24 JP JP2014538931A patent/JP6210993B2/ja active Active
-
2014
- 2014-01-23 US US14/162,328 patent/US20150315237A1/en not_active Abandoned
-
2015
- 2015-07-28 HK HK15107208.6A patent/HK1206616A1/xx unknown
-
2016
- 2016-08-30 US US15/252,132 patent/US20160368947A1/en not_active Abandoned
-
2017
- 2017-08-14 AU AU2017213587A patent/AU2017213587A1/en not_active Abandoned
-
2018
- 2018-02-14 US US15/896,662 patent/US20180237474A1/en not_active Abandoned
- 2018-07-06 US US16/028,664 patent/US20190135862A1/en not_active Abandoned
-
2019
- 2019-06-11 AU AU2019204073A patent/AU2019204073B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2703904T3 (es) | Moduladores del receptor de NMDA y usos de los mismos | |
| US10441571B2 (en) | Spiro-lactam NMDA receptor modulators and uses thereof | |
| ES2706063T3 (es) | Agonistas del receptor de NMDA y usos del mismo | |
| US10195179B2 (en) | Spiro-lactam NMDA receptor modulators and uses thereof | |
| CA2898863A1 (en) | Spiro-lactam nmda receptor modulators and uses thereof |