ES2698511T3 - Nuevos derivados de piridina condensados útiles como inhibidores de c-met tirosina cinasa - Google Patents
Nuevos derivados de piridina condensados útiles como inhibidores de c-met tirosina cinasa Download PDFInfo
- Publication number
- ES2698511T3 ES2698511T3 ES13809182T ES13809182T ES2698511T3 ES 2698511 T3 ES2698511 T3 ES 2698511T3 ES 13809182 T ES13809182 T ES 13809182T ES 13809182 T ES13809182 T ES 13809182T ES 2698511 T3 ES2698511 T3 ES 2698511T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- phenyl
- yloxy
- dihydro
- oxo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003112 inhibitor Substances 0.000 title claims description 7
- 150000003222 pyridines Chemical class 0.000 title description 5
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 title description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 126
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 49
- 239000001257 hydrogen Substances 0.000 claims abstract description 49
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 33
- 150000002367 halogens Chemical group 0.000 claims abstract description 33
- 150000003839 salts Chemical class 0.000 claims abstract description 31
- 150000002431 hydrogen Chemical group 0.000 claims abstract description 21
- 125000001997 phenyl group Chemical class [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 18
- 125000003118 aryl group Chemical group 0.000 claims abstract description 15
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims abstract description 6
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 6
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 6
- 101100516563 Caenorhabditis elegans nhr-6 gene Proteins 0.000 claims abstract description 5
- -1 3-fluoro-4- (3-oxo-3,4-dihydro-2H-pyrido [3,2-b] [1,4] oxazin-8-yloxy) phenyl Chemical group 0.000 claims description 49
- 206010028980 Neoplasm Diseases 0.000 claims description 47
- 239000000203 mixture Substances 0.000 claims description 43
- 201000011510 cancer Diseases 0.000 claims description 30
- 125000000217 alkyl group Chemical group 0.000 claims description 27
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 26
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 25
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 23
- 239000008194 pharmaceutical composition Substances 0.000 claims description 23
- 239000003814 drug Substances 0.000 claims description 19
- IZTVDTIADHKZNM-UHFFFAOYSA-N 5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxylic acid Chemical compound O=C1C(C(=O)O)=CNC=C1C1=CC=C(F)C=C1 IZTVDTIADHKZNM-UHFFFAOYSA-N 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 14
- 201000010099 disease Diseases 0.000 claims description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 10
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 9
- 206010027476 Metastases Diseases 0.000 claims description 8
- 230000009401 metastasis Effects 0.000 claims description 8
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 7
- WLIZYVKCXBYABW-UHFFFAOYSA-N n-[4-(3,4-dihydro-2h-pyrido[3,2-b][1,4]oxazin-8-yloxy)phenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1)=CC=C1OC1=CC=NC2=C1OCCN2 WLIZYVKCXBYABW-UHFFFAOYSA-N 0.000 claims description 7
- 102000001253 Protein Kinase Human genes 0.000 claims description 5
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 5
- 206010017758 gastric cancer Diseases 0.000 claims description 5
- 230000001404 mediated effect Effects 0.000 claims description 5
- 108060006633 protein kinase Proteins 0.000 claims description 5
- 201000011549 stomach cancer Diseases 0.000 claims description 5
- 208000027927 Hereditary papillary renal cell carcinoma Diseases 0.000 claims description 4
- 206010038389 Renal cancer Diseases 0.000 claims description 4
- NAQXSTWFIQVVRY-UHFFFAOYSA-N n-[3-fluoro-4-[(2-oxo-1,3-dihydropyrrolo[2,3-b]pyridin-4-yl)oxy]phenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1CC(=O)N2 NAQXSTWFIQVVRY-UHFFFAOYSA-N 0.000 claims description 4
- 230000002265 prevention Effects 0.000 claims description 4
- BXAOWYGNLKPHSL-UHFFFAOYSA-N 2-(4-fluoroanilino)-3-methyl-5-[5-(2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-9-yloxy)pyridin-2-yl]pyrimidin-4-one Chemical compound N=1C=C(C=2N=CC(OC=3C=4OCCCNC=4N=CC=3)=CC=2)C(=O)N(C)C=1NC1=CC=C(F)C=C1 BXAOWYGNLKPHSL-UHFFFAOYSA-N 0.000 claims description 3
- JBZTXEHTQXPUHW-UHFFFAOYSA-N 2-(4-fluoroanilino)-3-methyl-5-[5-(2,3,4,5-tetrahydropyrido[3,2-b][1,4]thiazepin-9-yloxy)pyridin-2-yl]pyrimidin-4-one Chemical compound N=1C=C(C=2N=CC(OC=3C=4SCCCNC=4N=CC=3)=CC=2)C(=O)N(C)C=1NC1=CC=C(F)C=C1 JBZTXEHTQXPUHW-UHFFFAOYSA-N 0.000 claims description 3
- LDSKYBVNIGKJHA-UHFFFAOYSA-N 2h-1,4-oxazine-2-carboxamide Chemical compound NC(=O)C1OC=CN=C1 LDSKYBVNIGKJHA-UHFFFAOYSA-N 0.000 claims description 3
- IXEGFTIVNVYKIP-UHFFFAOYSA-N 3-fluoro-4-(2,3,4,5-tetrahydropyrido[3,2-b][1,4]thiazepin-9-yloxy)aniline Chemical compound FC1=CC(N)=CC=C1OC1=CC=NC2=C1SCCCN2 IXEGFTIVNVYKIP-UHFFFAOYSA-N 0.000 claims description 3
- WPDIXOUSOUTVMR-UHFFFAOYSA-N 4-(3,4-dihydro-2H-pyrido[3,2-b][1,4]thiazin-8-yloxy)-3-fluoroaniline Chemical compound FC1=CC(N)=CC=C1OC1=CC=NC2=C1SCCN2 WPDIXOUSOUTVMR-UHFFFAOYSA-N 0.000 claims description 3
- LAZWPQZGOUJFPI-UHFFFAOYSA-N 4-(3,4-dihydro-2h-pyrido[3,2-b][1,4]thiazin-8-yloxy)aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=NC2=C1SCCN2 LAZWPQZGOUJFPI-UHFFFAOYSA-N 0.000 claims description 3
- AVJRRINCGCKTCD-UHFFFAOYSA-N 4-[(3,3-difluoro-1,2-dihydropyrrolo[2,3-b]pyridin-4-yl)oxy]-3-fluoroaniline Chemical compound FC1=CC(N)=CC=C1OC1=CC=NC2=C1C(F)(F)CN2 AVJRRINCGCKTCD-UHFFFAOYSA-N 0.000 claims description 3
- JSQCXCALRYVUJY-UHFFFAOYSA-N 4-[(3,3-dimethyl-2,4-dihydropyrido[3,2-b][1,4]oxazin-8-yl)oxy]aniline Chemical compound N1C(C)(C)COC2=C1N=CC=C2OC1=CC=C(N)C=C1 JSQCXCALRYVUJY-UHFFFAOYSA-N 0.000 claims description 3
- IRXOPKSSMIWMLE-UHFFFAOYSA-N 4-[(3,3-dimethyl-2,4-dihydropyrido[3,2-b][1,4]thiazin-8-yl)oxy]-3-fluoroaniline Chemical compound C1=CN=C2NC(C)(C)CSC2=C1OC1=CC=C(N)C=C1F IRXOPKSSMIWMLE-UHFFFAOYSA-N 0.000 claims description 3
- WYZXIMLMXGULAG-UHFFFAOYSA-N 4-[(3,3-dimethyl-2,4-dihydropyrido[3,2-b][1,4]thiazin-8-yl)oxy]aniline Chemical compound N1C(C)(C)CSC2=C1N=CC=C2OC1=CC=C(N)C=C1 WYZXIMLMXGULAG-UHFFFAOYSA-N 0.000 claims description 3
- SIULNZPSBTZEPG-UHFFFAOYSA-N 5-[5-[(3,3-dimethyl-2,4-dihydropyrido[3,2-b][1,4]oxazin-8-yl)oxy]pyridin-2-yl]-2-(4-fluoroanilino)-3-methylpyrimidin-4-one Chemical compound N=1C=C(C=2N=CC(OC=3C=4OCC(C)(C)NC=4N=CC=3)=CC=2)C(=O)N(C)C=1NC1=CC=C(F)C=C1 SIULNZPSBTZEPG-UHFFFAOYSA-N 0.000 claims description 3
- RMIXTFLTXNXKKL-UHFFFAOYSA-N 5-[5-[(3,3-dimethyl-2,4-dihydropyrido[3,2-b][1,4]thiazin-8-yl)oxy]pyridin-2-yl]-2-(4-fluoroanilino)-3-methylpyrimidin-4-one Chemical compound N=1C=C(C=2N=CC(OC=3C=4SCC(C)(C)NC=4N=CC=3)=CC=2)C(=O)N(C)C=1NC1=CC=C(F)C=C1 RMIXTFLTXNXKKL-UHFFFAOYSA-N 0.000 claims description 3
- VPJGESSPJGADBC-UHFFFAOYSA-N 8-[6-[2-(4-fluoroanilino)-1-methyl-6-oxopyrimidin-5-yl]pyridin-3-yl]oxy-4h-pyrido[3,2-b][1,4]oxazin-3-one Chemical compound N=1C=C(C=2N=CC(OC=3C=4OCC(=O)NC=4N=CC=3)=CC=2)C(=O)N(C)C=1NC1=CC=C(F)C=C1 VPJGESSPJGADBC-UHFFFAOYSA-N 0.000 claims description 3
- 206010006187 Breast cancer Diseases 0.000 claims description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 3
- 201000008808 Fibrosarcoma Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 3
- 206010025538 Malignant ascites Diseases 0.000 claims description 3
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 claims description 3
- 206010029260 Neuroblastoma Diseases 0.000 claims description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 3
- 201000000582 Retinoblastoma Diseases 0.000 claims description 3
- 206010039491 Sarcoma Diseases 0.000 claims description 3
- 208000035475 disorder Diseases 0.000 claims description 3
- 208000005017 glioblastoma Diseases 0.000 claims description 3
- 201000010982 kidney cancer Diseases 0.000 claims description 3
- 201000005202 lung cancer Diseases 0.000 claims description 3
- 208000020816 lung neoplasm Diseases 0.000 claims description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 3
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 3
- 201000001441 melanoma Diseases 0.000 claims description 3
- MMYWEXNODBDKCL-UHFFFAOYSA-N n-[3-fluoro-4-[[2-(hydroxymethyl)-3,4-dihydro-2h-pyrido[3,2-b][1,4]oxazin-8-yl]oxy]phenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C=12OC(CO)CNC2=NC=CC=1OC(C(=C1)F)=CC=C1NC(=O)C(C1=O)=CNC=C1C1=CC=C(F)C=C1 MMYWEXNODBDKCL-UHFFFAOYSA-N 0.000 claims description 3
- VGCVRWZIPZWDPL-UHFFFAOYSA-N n-[3-fluoro-4-[[3-(2-hydroxyethyl)-2,3-dihydro-1h-pyrrolo[2,3-b]pyridin-4-yl]oxy]phenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C=12C(CCO)CNC2=NC=CC=1OC(C(=C1)F)=CC=C1NC(=O)C(C1=O)=CNC=C1C1=CC=C(F)C=C1 VGCVRWZIPZWDPL-UHFFFAOYSA-N 0.000 claims description 3
- DGMFNZXEGKFREH-UHFFFAOYSA-N n-[4-(2,3-dihydro-1h-pyrrolo[2,3-b]pyridin-4-yloxy)-3-fluorophenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1CCN2 DGMFNZXEGKFREH-UHFFFAOYSA-N 0.000 claims description 3
- IEWKIQPDCLBITI-UHFFFAOYSA-N n-[4-[[2-(aziridin-1-ylmethyl)-3,4-dihydro-2h-pyrido[3,2-b][1,4]oxazin-8-yl]oxy]-3-fluorophenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1OC(CN1CC1)CN2 IEWKIQPDCLBITI-UHFFFAOYSA-N 0.000 claims description 3
- 208000008798 osteoma Diseases 0.000 claims description 3
- 201000002528 pancreatic cancer Diseases 0.000 claims description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 3
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 3
- 208000001608 teratocarcinoma Diseases 0.000 claims description 3
- BNHKHOQIBAZORZ-UHFFFAOYSA-N 3-fluoro-4-(2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-9-yloxy)aniline Chemical compound FC1=CC(N)=CC=C1OC1=CC=NC2=C1OCCCN2 BNHKHOQIBAZORZ-UHFFFAOYSA-N 0.000 claims description 2
- IXHHCSPUESIWRD-UHFFFAOYSA-N 5-(4-fluorophenyl)-n-(3-fluoro-4-spiro[3,4-dihydropyrido[3,2-b][1,4]oxazine-2,1'-cyclopropane]-8-yloxyphenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1OC1(CC1)CN2 IXHHCSPUESIWRD-UHFFFAOYSA-N 0.000 claims description 2
- 101100481408 Danio rerio tie2 gene Proteins 0.000 claims description 2
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 claims description 2
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 claims description 2
- 101001106413 Homo sapiens Macrophage-stimulating protein receptor Proteins 0.000 claims description 2
- 101000987310 Homo sapiens Serine/threonine-protein kinase PAK 2 Proteins 0.000 claims description 2
- 102100021435 Macrophage-stimulating protein receptor Human genes 0.000 claims description 2
- 101100481410 Mus musculus Tek gene Proteins 0.000 claims description 2
- 101700056750 PAK1 Proteins 0.000 claims description 2
- 102000001332 SRC Human genes 0.000 claims description 2
- 108060006706 SRC Proteins 0.000 claims description 2
- 102100027910 Serine/threonine-protein kinase PAK 1 Human genes 0.000 claims description 2
- 102100027939 Serine/threonine-protein kinase PAK 2 Human genes 0.000 claims description 2
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 claims description 2
- 230000001900 immune effect Effects 0.000 claims description 2
- UWRZHNJKLRWYON-UHFFFAOYSA-N n-[3-fluoro-4-(2-oxospiro[1h-pyrrolo[2,3-b]pyridine-3,1'-cyclopropane]-4-yl)oxyphenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1C1(CC1)C(=O)N2 UWRZHNJKLRWYON-UHFFFAOYSA-N 0.000 claims description 2
- OTCZXXKLOWXXFL-UHFFFAOYSA-N n-[4-[(2,2-difluoro-1,3-dihydropyrrolo[2,3-b]pyridin-4-yl)oxy]-3-fluorophenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1CC(F)(F)N2 OTCZXXKLOWXXFL-UHFFFAOYSA-N 0.000 claims description 2
- XGDZIDIBULRTBN-UHFFFAOYSA-N n-[4-[(3,4-dimethyl-2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-9-yl)oxy]phenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CN=C2NC(C)C(C)COC2=C1OC(C=C1)=CC=C1NC(=O)C(C1=O)=CNC=C1C1=CC=C(F)C=C1 XGDZIDIBULRTBN-UHFFFAOYSA-N 0.000 claims description 2
- 201000002689 pediatric hepatocellular carcinoma Diseases 0.000 claims description 2
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 claims 1
- 208000022873 Ocular disease Diseases 0.000 claims 1
- 230000003394 haemopoietic effect Effects 0.000 claims 1
- 108091008743 testicular receptors 4 Proteins 0.000 claims 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 abstract description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 abstract 1
- 101100073357 Streptomyces halstedii sch2 gene Proteins 0.000 abstract 1
- 239000000047 product Substances 0.000 description 52
- 239000002253 acid Substances 0.000 description 42
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 41
- 238000002360 preparation method Methods 0.000 description 37
- 239000011541 reaction mixture Substances 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- 238000000034 method Methods 0.000 description 24
- 210000004027 cell Anatomy 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 19
- 239000007787 solid Substances 0.000 description 18
- 239000000243 solution Substances 0.000 description 17
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000006243 chemical reaction Methods 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 14
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 235000019441 ethanol Nutrition 0.000 description 11
- 239000007788 liquid Substances 0.000 description 11
- 125000001424 substituent group Chemical group 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 8
- 208000026278 immune system disease Diseases 0.000 description 8
- 239000004615 ingredient Substances 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 208000030533 eye disease Diseases 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 239000011230 binding agent Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 4
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 4
- 229940126543 compound 14 Drugs 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- 230000002354 daily effect Effects 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 125000001072 heteroaryl group Chemical group 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000002674 ointment Substances 0.000 description 4
- 229940124531 pharmaceutical excipient Drugs 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 4
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 4
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 3
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 3
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 3
- MXJQJURZHQZLNN-UHFFFAOYSA-N 4-amino-2-fluorophenol Chemical compound NC1=CC=C(O)C(F)=C1 MXJQJURZHQZLNN-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940125797 compound 12 Drugs 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 229940126086 compound 21 Drugs 0.000 description 3
- 229940125877 compound 31 Drugs 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000003203 everyday effect Effects 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 230000009036 growth inhibition Effects 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 125000002943 quinolinyl group Chemical class N1=C(C=CC2=CC=CC=C12)* 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 2
- YEDUVZXIDQJILE-UHFFFAOYSA-N 4-[(3,3-dimethyl-2,4-dihydropyrido[3,2-b][1,4]oxazin-8-yl)oxy]-3-fluoroaniline Chemical compound C1=CN=C2NC(C)(C)COC2=C1OC1=CC=C(N)C=C1F YEDUVZXIDQJILE-UHFFFAOYSA-N 0.000 description 2
- KRZCOLNOCZKSDF-UHFFFAOYSA-N 4-fluoroaniline Chemical compound NC1=CC=C(F)C=C1 KRZCOLNOCZKSDF-UHFFFAOYSA-N 0.000 description 2
- ZJDWVOKEDWFBCQ-UHFFFAOYSA-N 5-[5-(3,4-dihydro-2h-pyrido[3,2-b][1,4]thiazin-8-yloxy)pyridin-2-yl]-2-(4-fluoroanilino)-3-methylpyrimidin-4-one Chemical compound N=1C=C(C=2N=CC(OC=3C=4SCCNC=4N=CC=3)=CC=2)C(=O)N(C)C=1NC1=CC=C(F)C=C1 ZJDWVOKEDWFBCQ-UHFFFAOYSA-N 0.000 description 2
- HCCNBKFJYUWLEX-UHFFFAOYSA-N 7-(6-methoxypyridin-3-yl)-1-(2-propoxyethyl)-3-(pyrazin-2-ylmethylamino)pyrido[3,4-b]pyrazin-2-one Chemical compound O=C1N(CCOCCC)C2=CC(C=3C=NC(OC)=CC=3)=NC=C2N=C1NCC1=CN=CC=N1 HCCNBKFJYUWLEX-UHFFFAOYSA-N 0.000 description 2
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 125000000129 anionic group Chemical group 0.000 description 2
- 229940111121 antirheumatic drug quinolines Drugs 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 229940126545 compound 53 Drugs 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 150000003246 quinazolines Chemical class 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 239000013074 reference sample Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 1
- ZCZVGQCBSJLDDS-UHFFFAOYSA-N 1,2,3,4-tetrahydro-1,8-naphthyridine Chemical compound C1=CC=C2CCCNC2=N1 ZCZVGQCBSJLDDS-UHFFFAOYSA-N 0.000 description 1
- RUBQQRMAWLSCCJ-UHFFFAOYSA-N 1,2-difluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C(F)=C1 RUBQQRMAWLSCCJ-UHFFFAOYSA-N 0.000 description 1
- MBELGTLJWJJGLF-UHFFFAOYSA-N 1,4-dihydropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CNC=CC1 MBELGTLJWJJGLF-UHFFFAOYSA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- RQMWVVBHJMUJNZ-UHFFFAOYSA-N 4-chloropyridin-2-amine Chemical compound NC1=CC(Cl)=CC=N1 RQMWVVBHJMUJNZ-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- 125000001572 5'-adenylyl group Chemical group C=12N=C([H])N=C(N([H])[H])C=1N=C([H])N2[C@@]1([H])[C@@](O[H])([H])[C@@](O[H])([H])[C@](C(OP(=O)(O[H])[*])([H])[H])([H])O1 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 1
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101100268648 Mus musculus Abl1 gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- LWZFANDGMFTDAV-BURFUSLBSA-N [(2r)-2-[(2r,3r,4s)-3,4-dihydroxyoxolan-2-yl]-2-hydroxyethyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O LWZFANDGMFTDAV-BURFUSLBSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 239000001785 acacia senegal l. willd gum Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000003302 alkenyloxy group Chemical group 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 125000005133 alkynyloxy group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000010410 dusting Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 230000036433 growing body Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 235000011167 hydrochloric acid Nutrition 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 1
- 102000049853 macrophage stimulating protein Human genes 0.000 description 1
- 108010053292 macrophage stimulating protein Proteins 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 125000006216 methylsulfinyl group Chemical group [H]C([H])([H])S(*)=O 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000007758 minimum essential medium Substances 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- ILSBMPQZYHPAIP-UHFFFAOYSA-N n-[3-fluoro-4-[(2-oxo-1,3-dihydropyrrolo[2,3-b]pyridin-4-yl)oxy]phenyl]-1-(4-fluorophenyl)-2-oxopyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1N1C(=O)C(C(=O)NC=2C=C(F)C(OC=3C=4CC(=O)NC=4N=CC=3)=CC=2)=CC=C1 ILSBMPQZYHPAIP-UHFFFAOYSA-N 0.000 description 1
- RDZFFKZMENKECY-UHFFFAOYSA-N n-[3-fluoro-4-[(3-oxo-4h-pyrido[3,2-b][1,4]oxazin-8-yl)oxy]phenyl]-1,5-dimethyl-3-oxo-2-phenylpyrazole-4-carboxamide Chemical compound CN1C(C)=C(C(=O)NC=2C=C(F)C(OC=3C=4OCC(=O)NC=4N=CC=3)=CC=2)C(=O)N1C1=CC=CC=C1 RDZFFKZMENKECY-UHFFFAOYSA-N 0.000 description 1
- JQEXXZKPEDSLNT-UHFFFAOYSA-N n-[4-(2,3-dihydro-1h-pyrrolo[2,3-b]pyridin-4-yloxy)-3-fluorophenyl]-1,5-dimethyl-3-oxo-2-phenylpyrazole-4-carboxamide Chemical compound CN1C(C)=C(C(=O)NC=2C=C(F)C(OC=3C=4CCNC=4N=CC=3)=CC=2)C(=O)N1C1=CC=CC=C1 JQEXXZKPEDSLNT-UHFFFAOYSA-N 0.000 description 1
- KZLUVOZMCBNEOV-UHFFFAOYSA-N n-[4-(2,3-dihydro-1h-pyrrolo[2,3-b]pyridin-4-yloxy)-3-fluorophenyl]-1-(4-fluorophenyl)-2-oxopyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1N1C(=O)C(C(=O)NC=2C=C(F)C(OC=3C=4CCNC=4N=CC=3)=CC=2)=CC=C1 KZLUVOZMCBNEOV-UHFFFAOYSA-N 0.000 description 1
- DGXHQCRALGIMKL-UHFFFAOYSA-N n-[4-(3,4-dihydro-2h-pyrido[3,2-b][1,4]oxazin-8-yloxy)-3-fluorophenyl]-1,5-dimethyl-3-oxo-2-phenylpyrazole-4-carboxamide Chemical compound CN1C(C)=C(C(=O)NC=2C=C(F)C(OC=3C=4OCCNC=4N=CC=3)=CC=2)C(=O)N1C1=CC=CC=C1 DGXHQCRALGIMKL-UHFFFAOYSA-N 0.000 description 1
- RRPJDVRVBLATCY-UHFFFAOYSA-N n-[4-(3,4-dihydro-2h-pyrido[3,2-b][1,4]oxazin-8-yloxy)-3-fluorophenyl]-5-(4-fluorophenyl)-4-oxo-1h-pyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1C(C1=O)=CNC=C1C(=O)NC(C=C1F)=CC=C1OC1=CC=NC2=C1OCCN2 RRPJDVRVBLATCY-UHFFFAOYSA-N 0.000 description 1
- NSPMDZVQNZHIRQ-UHFFFAOYSA-N n-[4-(3,4-dihydro-2h-pyrido[3,2-b][1,4]oxazin-8-yloxy)phenyl]-1-(4-fluorophenyl)-2-oxopyridine-3-carboxamide Chemical compound C1=CC(F)=CC=C1N1C(=O)C(C(=O)NC=2C=CC(OC=3C=4OCCNC=4N=CC=3)=CC=2)=CC=C1 NSPMDZVQNZHIRQ-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 238000010837 poor prognosis Methods 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- JQRYUMGHOUYJFW-UHFFFAOYSA-N pyridine;trihydrobromide Chemical compound [Br-].[Br-].[Br-].C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1 JQRYUMGHOUYJFW-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 201000010174 renal carcinoma Diseases 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 235000020374 simple syrup Nutrition 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 235000011067 sorbitan monolaureate Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- YLTRPTRRNKYFGY-UHFFFAOYSA-N tert-butyl 4-chloro-2,3-dihydropyrrolo[2,3-b]pyridine-1-carboxylate Chemical compound N1=CC=C(Cl)C2=C1N(C(=O)OC(C)(C)C)CC2 YLTRPTRRNKYFGY-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006090 thiamorpholinyl sulfone group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Oncology (AREA)
- Cardiology (AREA)
- Hospice & Palliative Care (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Un compuesto de fórmula I o una de sus sales farmacéuticamente aceptable:**Fórmula** en donde, B no está o es, O, S, OCH2 o SCH2; R1, R2, R3 y R4 son cada uno independientemente hidrógeno, halógeno, alquilo C1-6 sustituido o no sustituido, o carbamoilo, o o R1 y R2 a la vez, o R3 y R4 a la vez, forman =O; A es N o CH; n es 0, 1, 2 o 3; cada R5 es independientemente halógeno, o alquilo C1-6, Z es NHR6 o de fórmula II:**Fórmula** en donde R18 y R19 son cada uno independientemente hidrógeno, alquilo C1-8 sustituido o no sustituido o arilo C6-8 sustituido o no sustituido; R6 tiene la fórmula VI:**Fórmula** en donde: Q3 es hidrógeno; R8 es hidrógeno, o fenil sustituido con halógeno; y R9 es hidrógeno.
Description
DESCRIPCIÓN
Nuevos derivados de piridina condensados útiles como inhibidores de c-Met tirosina cinasa
Campo técnico
Esta invención se refiere a determinados nuevos derivados de piridina fusionados como inhibidores de c-Met, a su síntesis y a su uso para tratar un trastorno mediado por c-Met.
Técnica anterior
El estudio de las vías de transducción de señales en estados normales y patológicos es de considerable interés debido al posible beneficio terapéutico que surge de los nuevos agentes moleculares que se dirigen a algunas de estas vías relacionadas con la enfermedad.
Las tirosina cinasas receptoras (RTK) son enzimas clave en las vías de transducción de señales que catalizan la autofosforilación de restos de tirosina en el dominio citosólico, C-terminal de la proteína. Esto genera sitios de acoplamiento para la captación de proteínas aguas abajo y la posterior propagación de señales involucradas en una serie de episodios celulares incluidos el crecimiento, la proliferación y la supervivencia. La señalización de la cinasa desregulada más generalmente está implicada en una serie diversa de estados patológicos incluidos trastornos inmunológicos e inflamatorios, enfermedades cardiovasculares y neurodegenerativas. Las tirosina cinasas receptoras conocidas abarcan 20 familias y muchos son oncogenes (Blume-Jensen et al. 2001. Nature 411 355-365). c-Met es el miembro prototípico de una subfamilia de RTK que incluye las proteínas Ron relacionadas (receptor de proteínas estimuladoras de macrófagos) y su ortólogo de pollo, Sea. El ligando endógeno es el factor de crecimiento de hepatocitos del factor de crecimiento y motilidad (HGF, también conocido como factor de dispersión). c-Met y HGF se expresan en una variedad de tipos de tejidos, aunque su expresión está normalmente restringida a células de origen epitelial y mesenquimatoso. En cambio, las células tumorales a menudo expresan c-Met activada constitutivamente. En la actualidad existe un conjunto creciente de pruebas convincentes, tanto en estudios con animales como en pacientes con cáncer, en que la señalización de HGF-Met desempeña una función importante en el desarrollo y la evolución del cáncer y está relacionada en particular con fenotipos invasivos. c-Met y HGF se expresan mucho en relación con el tejido circundante en numerosos cánceres y su expresión se correlaciona con un deficiente pronóstico del paciente (Jiang, W. et al. 1999 Crit. Rev. Oncol.-hematol., 29, 209-248.) Las mutaciones puntuales de activación en el dominio de cinasa de c-Met están implicadas en la causa de formas esporádicas y hereditarias de carcinoma renal papilar (Danilkovitch-Miagkova, A. et al. 2002. 1 J. Clin. Invest. 109, 863-867). c-Met es un marcador tanto para el cáncer como para la neoplasia y cabe esperar que los agentes que inhiben la señalización de c-Met-HGF mejoren la evolución de la enfermedad en cánceres relevantes.
El documento WO 2006/116713 A1 describe derivados sustituidos en 5 de 1,2,3,4-tetrahidro-1,8-naftiridina útiles en el tratamiento del cáncer y la angiogenia.
El documento WO 2006/014325 A2 se refiere a quinazolinas y quinolinas que inhiben, regulan y/o modulan las vías de transducción de señales del receptor de cinasa relacionadas con los cambios en actividades celulares. En especial, estos compuestos inhibirán a c-Met, KDR y flt-3.
Compendio de la invención
La presente invención se refiere a nuevos compuestos de piridina fusionados útiles como inhibidores de c-Met y para el tratamiento de afecciones mediadas por c-Met. Los compuestos de la invención tienen la estructura general como fórmula I:
Un compuesto de fórmula I o una de sus sales farmacéuticamente aceptable:

en donde,
B no está o es, O, S, OCH2 o SCH2 ;
R1, R2, R3 y R4 son cada uno independientemente hidrógeno, halógeno, alquilo C1-6 sustituido o no sustituido, o carbamoílo, o
o Ri y R2 a la vez, o R3 y R4 a la vez, forman =O;
A es N oCH;
n es 0 , 1 , 2 ;
cada R5 es independientemente halógeno, o alquilo C1-6,
Z es NHR6 o de fórmula II:

en donde R18 y R19 son cada uno independientemente hidrógeno, alquilo C1-8 sustituido o no sustituido o arilo C6-8 sustituido o no sustituido;
R6 tiene la fórmula VI:

en donde:
Q3 es hidrógeno;
R8 es hidrógeno, o fenil sustituido con halógeno; y
R9 es hidrógeno.
La presente invención proporciona además algunas soluciones técnicas preferidas con respecto al compuesto de fórmula (I).
En algunas realizaciones de fórmula (I), R1, R2 , R3 y R4 son cada uno independientemente hidrógeno, halógeno, alquilo C1-4 sustituido o no sustituido o carbamoílo; o
R1 y R2 a la vez, o R3 y R4 a la vez, forman =O.
En algunas realizaciones de fórmula (I), R1, R2, R3 y R4 son cada uno independientemente hidrógeno, F, o metilo; o R1 y R2 a la vez, o R3 y R4 a la vez, forman =O.
En algunas realizaciones de fórmula (I), R1, R2, R3 y R4 son todos hidrógeno.
En algunas realizaciones de fórmula (I), R1 y R2 a la vez, o R3 y R4 a la vez forman =O.
En algunas realizaciones de fórmula (I), n es 0 o 1.
En algunas realizaciones de fórmula (I), R5 es halógeno o alquilo C1-4.
En algunas realizaciones de fórmula (I), R5 es F o metilo.
En algunas realizaciones de fórmula (I), B falta o es O.
En algunas realizaciones de fórmula (I), A es CH.
En algunas realizaciones de fórmula (I), Z es de fórmula II en donde R18 y R19 son cada uno independientemente hidrógeno, alquilo C1-4 sustituido o no sustituido, en concreto metilo, etilo sustituido o no sustituido, propilo sustituido o no sustituido, arilo C6-8 sustituido o no sustituido.
En algunas realizaciones de fórmula (I), Z es de fórmula II en donde R18 y R19 son cada uno independientemente hidrógeno, metilo o fenilo sustituido con halógeno.
En algunas realizaciones de fórmula (I), Z es de fórmula II en donde R18 es metilo; y Rig es fenilo sustituido con F en la posición para.
En algunas realizaciones, el compuesto de fórmula (I) es de fórmula VIII:

en donde,
Ri, R2 , R3 y R4 son cada uno independientemente hidrógeno, halógeno; o alquilo C1 -3sustituido o no sustituido; o Ri y R2 a la vez, o R3 y R4 a la vez, forman =O;
cada R5 es independientemente halógeno, o alquilo C1-3 sustituido o no sustituido;
n es 0 o 1;
Q3 es hidrógeno; y
R8 es hidrógeno o fenilo sustituido con halógeno; y
Rg es hidrógeno.
La presente invención proporciona además algunas soluciones técnicas preferidas de fórmula VIII:
En algunas realizaciones del compuesto de fórmula VIII, Ri, R2 , R3 y R4 son cada uno independientemente hidrógeno o halógeno; o Ri y R2 a la vez, o R3 y R4 a la vez, forman =O.
En algunas realizaciones del compuesto de fórmula VIII, Ri, R2 , R3 y R4 son cada uno independientemente hidrógeno o halógeno.
En algunas realizaciones del compuesto de fórmula VIII, Ri y R2 son cada uno F; y R3 y R4 son cada uno hidrógeno. En algunas realizaciones del compuesto de fórmula VIII, Ri y R2 son cada uno hidrógeno; y R3 y R4 son cada uno F. En algunas realizaciones del compuesto de fórmula VIII, Ri, R2 , R3 y R4 son cada uno hidrógeno.
En algunas realizaciones del compuesto de fórmula VIII, Ri y R2 a la vez forman =O.
En algunas realizaciones del compuesto de fórmula VIII, R3 y R4 a la vez forman =O.
En algunas realizaciones del compuesto de fórmula VIII, R5 es halógeno.
En algunas realizaciones del compuesto de fórmula VIII, R5 es F.
En algunas realizaciones del compuesto de fórmula VIII, R8 es fenilo sustituido con halógeno en la posición para. En algunas realizaciones del compuesto de fórmula VIII, R8 es fenilo sustituido con flúor en la posición para.
La presente invención proporciona además algunas soluciones técnicas especialmente preferidas con respecto al compuesto de fórmula I. El compuesto es:
N-(4-(2,3-dihidro-iH-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-i,4-dihidropiridin-3-carboxamida; N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirido[3,2-b][i,4]oxazin-8-iloxi) fenil)-5-(4-fluorofenil) )-4-oxo-i,4-dihidropiridin-3-carboxamida;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][i,4]oxazin-8-iloxi)-3-fluorofenil)-5-4-fluorofenil)-4-oxo-i,4-dihidropiridin-3-carboxamida;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][i,4]oxazin-8-iloxi) fenil)-5-(4-fluorofenil)-4 oxo-i,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(2-oxo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
8-(6-(2-(4-fluorofenilamino)-1-metil-6-oxo-1,6-dihidropirimidin-5-il) piridin-3-iloxi)-2H-pirido[3,2-b][ 1,4]oxazin-3 (4H)-ona;
5-(5-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi) piridin-2-il)-2-(4-fluorofenilamino)-3-metilpirimidin-4(3H) )-ona;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1, 4-dihidropiridin-3-carboxamida;
[4-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-feml)-4-oxo-1,4-dihidropiridin-3-carboxílico;
[4-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
5-[5-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-piridin-2-il]-2-(4-fluoro-fenilamino)-3-metil-3H-pirimidin-4-ona;
5-[5-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-piridin-2-il]-2-( 4-fluoro-fenilamino)-3-metil-3H-pirimidin-4- ona;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
5- [5-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-piridin-2-il]-2-(4-fluoro-fenilamino)-3-metil-3H-pirimidin-4-ona;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
[4-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
2-(4-fluoro-fenilamino)-3-metil-5-[5-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-piridin-2-il]-3H-pirimidin-4-ona;
[3-fluoro-4-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
[3-fluoro-4-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
2-(4-fluoro-fenilamino)-3-metil-5-[5-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-piridin-2-il]-3H-pirimidin-4-ona;
4-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
N-(4-(3,4-dimetil-2,3,4,5-tetrahidropirido[3,2-b][1,4]oxazepin-9-iloxi) fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
[4-(3,3-difluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
N-(4-(2,2-difluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(4-(3',4'-dihidrospiro[ciclopropano-1,2'-pirido[3,2-b][1,4]oxazina]-8'-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(2-(hidroximetil)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
8-(2-fluoro-4-(5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxainido)fenoxi)-3,4-dihidro-2H-pirido[3,2-b ][1,4]oxazina-2-carboxamida;
N-(4-(2-(aziridin-1-ilmetil)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(3-(2-hidroxietil)-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-fluorofenilo)-4-oxo-1,4-dihidropiridin-3-carboxamida; o
N-(3-fluoro-4-(2'-oxo-1 ', 2-dihidrospiro[ciclopropano-1,3-pirrolo[2,3-b]piridina]-4-iloxi) fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida.
La presente invención también proporciona una composición farmacéutica que comprende al menos un compuesto descrito en la presente memoria y al menos un excipiente farmacéuticamente aceptable, tal como hidroxipropilmetil celulosa. En la composición, la relación en peso del compuesto al excipiente puede estar comprendida dentro del intervalo, p. ej., de aproximadamente 0,0001 a aproximadamente 10.
La presente invención proporcionó adicionalmente un uso de la composición farmacéutica descrita en la presente memoria para la preparación de un medicamento.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para el tratamiento o prevención del cáncer, metástasis del cáncer, enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para retrasar o prevenir la evolución de la enfermedad en cáncer, metástasis de cáncer, enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para tratar o retrasar la evolución o aparición de cáncer, la metástasis de cáncer, la enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para el tratamiento o prevención del cáncer, la metástasis del cáncer, la enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para retrasar o prevenir la evolución de la enfermedad en cáncer, la metástasis de cáncer, la enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para tratar o retrasar la evolución o aparición de cáncer, la metástasis de cáncer, la enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse como inhibidor de c-Met. Además, la presente invención proporciona al menos un compuesto para uso en el tratamiento del cáncer, la prevención de la metástasis del cáncer o el tratamiento de la enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocular.
En algunas realizaciones, un medicamento preparado de este modo puede usarse para tratar a un paciente que tiene una afección que está mediada por la actividad de la proteína cinasa. Ejemplos de la proteína cinasa incluyen KDR, Tie-2, Flt3, FGFR3, Abl, Aurora A, c-Src, IGF-IR, ALK, c-MET, RON, PAK1, PAK2yTAK1.
En algunas realizaciones, la afección mediada por la actividad de la proteína cinasa es el cáncer.
Los ejemplos de cáncer incluyen un tumor sólido, un sarcoma, un fibrosarcoma, un osteoma, un melanoma, un retinoblastoma, un rabdomiosarcoma, un glioblastoma, un neuroblastoma, un teratocarcinoma, un tumor maligno hematopoyético, una ascitis maligna, cáncer de pulmón, cáncer de mama, cáncer colorrectal, cáncer renal, cáncer pancreático, cáncer de cabeza, cáncer de cuello, carcinoma hereditario de células renales papilares, carcinoma hepatocelular infantil y cáncer gástrico.
También se proporciona al menos un compuesto como se describe en la presente memoria o una de sus sales farmacéuticamente aceptable para uso como medicamento.
Además, se proporciona al menos un compuesto como se describe en la presente memoria o una de sus sales farmacéuticamente aceptable para uso en el tratamiento del cáncer.
Los ejemplos de cáncer incluyen un tumor sólido, un sarcoma, un fibrosarcoma, un osteoma, un melanoma, un retinoblastoma, un rabdomiosarcoma, un glioblastoma, un neuroblastoma, un teratocarcinoma, un tumor maligno hematopoyético, una ascitis maligna, cáncer de pulmón, cáncer de mama, cáncer colorrectal, cáncer renal, cáncer pancreático, cáncer de cabeza, cáncer de cuello, carcinoma hereditario de células renales papilares, carcinoma
hepatocelular infantil y cáncer gástrico. El término “halógeno”, como se emplea en la presente memoria, a menos que se indique de otra manera, significa flúor, cloro, bromo o yodo. Los grupos de halógeno preferidos incluyen F, Cl y Br.
Como se emplea en la presente memoria, a menos que se indique lo contrario, alquilo incluye radicales hidrocarbonados monovalentes saturados que tienen restos lineales, ramificados o cíclicos. Por ejemplo, los radicales alquilo incluyen metilo, etilo, propilo, isopropilo, ciclopropilo, n-butilo, isobutilo, sec-butilo, t-butilo, ciclobutilo, n-pentilo, 3-(2-metil) butilo, 2-pentilo, 2-metilbutilo, neopentilo, ciclopentilo, n-hexilo, 2-hexilo, 2-metilpentilo y ciclohexilo. De manera similar, C1-8 , como en alquilo C1-8, se define para identificar el grupo que tiene 1, 2, 3, 4, 5, 6, 7 u 8 átomos de carbono en una configuración lineal o ramificada.
Los grupos alquenilo y alquinilo incluyen alquenos y alquinos de cadena lineal o ramificada. Del mismo modo, "alquenilo C2-8" y "alquinilo C2-8"se refiere a radicales alquenilo o alquinilo que tienen 2, 3, 4, 5, 6, 7 u 8 átomos de carbono en una configuración lineal o abierta.
Los radicales alcoxilo son éteres oxigenados formados a partir de los grupos alquilo lineales, de cadena ramificada o cíclicos descritos anteriormente.
El término "arilo", como se emplea en esta memoria, a menos que se indique lo contrario, se refiere a un sistema de anillo mono o policíclico no sustituido o sustituido que contiene átomos de anillo de carbono. Los arilos preferidos son sistemas de anillos aromáticos monocíclicos o bicíclicos de 6 a 10 eslabones. Fenilo y naftilo son arilos preferidos. El arilo más preferido es fenilo.
El término "heterociclilo", como se emplea en esta memoria, a menos que se indique lo contrario, representa un sistema de anillo saturado monocíclico no sustituido o sustituido de tres a ocho eslabones que consta de átomos de carbono y de uno a tres heteroátomos seleccionados de N, O o S, y en donde los heteroátomos de nitrógeno o azufre se pueden oxidar opcionalmente, y el heteroátomo de nitrógeno opcionalmente se puede cuaternizar. El grupo heterociclilo puede estar unido a cualquier heteroátomo o átomo de carbono, lo que resulta en la creación de una estructura estable. Los ejemplos de dichos grupos heterociclo incluyen, entre otros, azetidinilo, pirrolidinilo, piperidinilo, piperazinilo, oxopiperazinilo, oxopiperidinilo, oxoazepinilo, azepinilo, tetrahidrofuranilo, dioxolanilo, tetrahidroimidazolilo, tetrahidrotiazolilo, tetrahidrooxazolilo, tetrahidropiranilo, morfolinilo, tiomorfolinilo, sulfóxido de tiamorfolinilo, tiamorfolinil sulfona y oxodiazolilo.
El término "heteroarilo", como se emplea en esta memoria, a menos que se indique lo contrario, representa un sistema de anillo aromático monocíclico no sustituido o sustituido de cinco o seis eslabones o un sistema de anillo heteroaromático benzo-fusionado de nueve o diez eslabones no sustituido o sustituido que consta de átomos de carbono y de uno a cuatro heteroátomos seleccionados de N, O o S y en el que los heteroátomos de nitrógeno o azufre opcionalmente pueden estar oxidados, y el heteroátomo de nitrógeno opcionalmente puede estar cuaternizado. El grupo heteroarilo puede estar unido a cualquier heteroátomo o átomo de carbono, lo que da lugar a la creación de una estructura estable. Los ejemplos de grupos heteroarilo incluyen, entre otros, tienilo, furanilo, imidazolilo, isoxazolilo, oxazolilo, pirazolilo, pirrolilo, tiazolilo, tiadiazolilo, triazolilo, piridilo, piridazinilo, indolilo, azaindolilo, indazolilo, benzimidazolilo, benzofuranilo, benzotienilo, benzisoxazolilo, benzoxazolilo, benzopirazolilo, benzotiazolilo, benzotiadiazolilo, benzotriazolilo, adenililo, quinolinilo o isoquinolino.
El término "carbonilo" se refiere al grupo C(O).
El término "alcoxicarbonilo" se refiere a los ésteres de cadena lineal o ramificada de un derivado de ácido carboxílico de la presente invención del número de átomos de carbono especificado (p. ej., alcoxicarbonilo C1-6), o cualquier número dentro de este intervalo (p. ej., metiloxicarbonilo (MeOCO)-), etiloxicarbonilo o butiloxicarbonilo.
Siempre que el término "alquilo" o "arilo" o cualquiera de sus raíces de prefijo aparezcan en el nombre de un sustituyente (p. ej., aralquilo o dialquilamino) se interpretará que incluye las limitaciones dadas anteriormente para "alquilo" y "arilo". Los números designados de átomos de carbono (p. ej., C1-6) se referirán independientemente al número de átomos de carbono en un resto alquilo o a la porción alquilo de un sustituyente mayor en el que el alquilo aparece como su raíz prefijo.
El término "alquilsulfinilo" se refiere a alquilsulfonas de cadena lineal o ramificada del número de átomos de carbono especificado (p. ej., alquil C1-6 sulfonilo), o cualquier número dentro de este intervalo (p. ej., metilsulfinilo (MeSO-), etilsulfinilo, isopropilsulfinilo).
El término "alquilsulfonilo" se refiere a alquilsulfonas de cadena lineal o ramificada con el número de átomos de carbono especificado (p. ej., alquil C1-6 sulfonilo), o cualquier número dentro de este intervalo p. ej., metilsulfonilo (MeSO2-), etilsulfonilo, isopropilsulfonilo, etc].
El término "alquiltio" se refiere a alquilsulfuros de cadena lineal o ramificada del número de átomos de carbono especificado (p. ej, alquil C ^ tio ), o cualquier número dentro de este intervalo [p. ej., metiltio (MeS-), etiltio, isopropiltio, etc].
El término "alqueniloxi" se refiere al grupo -O-alquenilo, donde el alquenilo se define como anteriormente.
El término "alquiniloxi" se refiere al grupo -O-alquinilo, donde alquinilo se define como anteriormente.
El término "composición", como se emplea en este documento, pretende abarcar un producto que comprende los ingredientes especificados en las cantidades especificadas, así como cualquier producto que resulte, directa o indirectamente, de combinaciones de los ingredientes especificados en las cantidades especificadas. Por consiguiente, las composiciones farmacéuticas que contienen los compuestos de la presente invención como principio activo, así como los métodos para preparar los presentes compuestos, también forman parte de la presente invención. Además, algunas de las formas cristalinas para los compuestos pueden existir como polimorfos y, como tales, pretenden incluirse en la presente invención. Además, algunos de los compuestos pueden formar solvatos con el agua (es decir, hidratos) o disolventes orgánicos comunes y dichos solvatos se desea que estén comprendidos dentro del alcance de esta invención.
Los compuestos de la presente invención también pueden estar presentes en forma de sales farmacéuticamente aceptables. Para su uso en medicina, las sales de los compuestos de esta invención se refieren a "sales farmacéuticamente aceptables" atóxicas. Las formas de sales farmacéuticamente aceptables incluyen sales ácidas/ aniónicas o básicas/catiónicas farmacéuticamente aceptables. La sal ácida/aniónica farmacéuticamente aceptable generalmente toma una forma en la que el nitrógeno básico está protonado con un ácido inorgánico u orgánico. Los ácidos orgánicos o inorgánicos representativos incluyen clorhídrico, bromhídrico, yodhídrico, perclórico, sulfúrico, nítrico, fosfórico, acético, propiónico, glicólico, láctico, succínico, maleico, fumárico, málico, tartárico, cítrico, benzoico, mandélico, metansulfónico, hidroxietansulfónico, bencensulfónico, oxálico, pamoico, 2-naftalensulfónico, ptoluensulfónico, ciclohexansulfámico, salicílico, sacarínico o trifluoroacético. Las sales básicas/catiónicas farmacéuticamente aceptables incluyen, entre otras, las de aluminio, calcio, cloroprocaína, colina, dietanolamina, etilendiamina, litio, magnesio, potasio, sodio y cinc.
Se pretende que la definición de cualquier sustituyente o variable en una posición determinada en una molécula sea independiente de sus definiciones en otra parte de esa molécula. Se entiende que los sustituyentes y los patrones de sustitución en los compuestos de esta invención pueden ser seleccionados por un experto en la técnica para proporcionar compuestos que sean químicamente estables y que puedan sintetizarse fácilmente mediante técnicas conocidas en la técnica, así como los métodos expuestos en la presente memoria.
La presente invención incluye compuestos descritos que pueden contener uno o más centros asimétricos y, por lo tanto, pueden dar lugar a diastereómeros e isómeros ópticos. La presente invención incluye todos los diastereoisómeros posibles así como sus mezclas racémicas, sus enantiómeros resueltos sustancialmente puros, todos los isómeros geométricos posibles y sus sales farmacéuticamente aceptables.
La fórmula I anterior se muestra sin una estereoquímica definitiva en determinadas posiciones. La presente invención incluye todos los estereoisómeros de fórmula I y sus sales farmacéuticamente aceptables. Además, también se incluyen mezclas de estereoisómeros así como estereoisómeros específicos aislados. Durante el curso de los procedimientos sintéticos utilizados para preparar dichos compuestos, o al utilizar procedimientos de racemización o epimerización conocidos por los expertos en la técnica, los productos de dichos procedimientos pueden ser una mezcla de estereoisómeros.
Cuando existe un tautómero del compuesto de fórmula (I), la presente invención incluye cualquier posible tautómero y sus sales farmacéuticamente aceptables, y mezclas de los mismos, excepto cuando se indique específicamente lo contrario.
La expresión "sales farmacéuticamente aceptables" se refiere a sales preparadas a partir de bases o ácidos atóxicos farmacéuticamente aceptables. Cuando el compuesto de la presente invención es ácido, su sal correspondiente puede prepararse convenientemente a partir de bases atóxicas farmacéuticamente aceptables, incluidas bases inorgánicas y bases orgánicas. Las sales derivadas de dichas bases inorgánicas incluyen las de aluminio, amonio, calcio, cobre (ico y oso), férrico, ferroso, litio, magnesio, manganeso (ico y oso), potasio, sodio, cinc y sales similares. Son particularmente preferidas las sales de amonio, calcio, magnesio, potasio y sodio. Las sales derivadas de bases orgánicas atóxicas farmacéuticamente aceptables incluyen sales de aminas primarias, secundarias y terciarias, así como aminas cíclicas y aminas sustituidas, tales como aminas sustituidas naturales y sintetizadas. Otras bases atóxicas orgánicas farmacéuticamente aceptables a partir de las cuales se pueden formar sales incluyen resinas de intercambio iónico tales como, por ejemplo, arginina, betaína, cafeína, colina, N',N'-dibenciletilendiamina, dietilamina, 2-dietilaminoetanol, 2-dimetilaminoetanol, etanolamina, etilendiamina, N-etilmorfolina, N-etilpiperidina, glucamina, glucosamina, histidina, hidrabamina, isopropilamina, lisina, metilglucamina, morfolina, piperazina, piperidina, resinas de poliamina, procaína, purinas, teobromina, trietilamina, trimetilamina, tripropilamina, trometamina y similares.
Cuando el compuesto de la presente invención es básico, su sal correspondiente puede prepararse convenientemente a partir de ácidos atóxicos farmacéuticamente aceptables, incluidos ácidos orgánicos e inorgánicos. Dichos ácidos incluyen, por ejemplo, acético, bencensulfónico, benzoico, alcanforsulfónico, cítrico, etansulfónico, fórmico, fumárico, glucónico, glutámico, bromhídrico, clorhídrico, isetiónico, láctico, maleico, málico, mandélico, metansulfónico, múcico, nítrico, pamoico, pantoténico, fosfórico, succínico, sulfúrico, tartárico, p-toluensulfónico y similares. Los preferidos son los ácidos cítrico, bromhídrico, fórmico, clorhídrico, maleico, fosfórico, sulfúrico y tartárico. Particularmente preferidos son los ácidos fórmico y clorhídrico. Dado que los compuestos de fórmula (I) están destinados a uso farmacéutico, se
proporcionan preferiblemente en forma sustancialmente pura, por ejemplo, al menos 60% puros, más adecuadamente al menos 75% puros, especialmente al menos 98% puros (% son en peso por peso).
Las composiciones farmacéuticas de la presente invención comprenden un compuesto representado por la fórmula I (o una de sus sales farmacéuticamente aceptables) como principio activo, un vehículo farmacéuticamente aceptable y opcionalmente otros ingredientes terapéuticos o adyuvantes. Las composiciones incluyen composiciones adecuadas para administración oral, rectal, tópica y parenteral (incluidas la subcutánea, intramuscular e intravenosa), aunque la ruta más adecuada en cualquier caso dependerá del anfitrión en particular, y de la naturaleza y gravedad de las enfermedades para las cuales se está administrando el principio activo. Las composiciones farmacéuticas pueden presentarse convenientemente en forma galénica unitaria y prepararse por cualquiera de los métodos bien conocidos en la técnica de la farmacia.
En la práctica, los compuestos representados por la fórmula I, o una de sus sales farmacéuticamente aceptables, de esta invención se pueden combinar como el principio activo en mezcla íntima con un vehículo farmacéutico según técnicas de composición farmacéutica convencionales. El excipiente puede tomar una amplia variedad de formas dependiendo de la forma de preparación deseada para administración, p. ej., oral o parenteral (incluida la intravenosa). Por lo tanto, las composiciones farmacéuticas de la presente invención pueden presentarse como unidades discretas adecuadas para la administración oral, tales como cápsulas, sellos o comprimidos, que contienen cada uno una cantidad predeterminada del principio activo. Además, las composiciones pueden presentarse en forma de polvo, de gránulos, de solución, de suspensión en un líquido acuoso, de líquido no acuoso, de emulsión líquida de aceite en agua o de agua en aceite. Además de las formas de dosificación comunes indicadas anteriormente, el compuesto representado por la fórmula I, o una de sus sales farmacéuticamente aceptables, también puede administrarse mediante liberación controlada y/o dispositivos de administración. Las composiciones pueden prepararse por cualquiera de los métodos de farmacia. En general, dichos métodos incluyen una etapa para poner en contacto el principio activo con el principio activo que constituye uno o más ingredientes necesarios. En general, las composiciones se preparan mezclando uniforme e íntimamente el principio activo con excipientes líquidos o excipientes sólidos finamente divididos o ambos. El producto puede entonces moldearse convenientemente en la presentación deseada.
Por lo tanto, las composiciones farmacéuticas de esta invención pueden incluir un excipiente farmacéuticamente aceptable y un compuesto, o una sal farmacéuticamente aceptable, de fórmula I. Los compuestos de fórmula I, o una de sus sales farmacéuticamente aceptables, también pueden incluirse en composiciones farmacéuticas en combinación con uno o más de otros compuestos terapéuticamente activos.
El excipiente farmacéutico empleado puede ser, por ejemplo, un sólido, líquido o gas. Los ejemplos de excipientes sólidos incluyen lactosa, arcilla blanca, sacarosa, talco, gelatina, agar-agar, pectina, goma arábiga, estearato de magnesio y ácido esteárico. Ejemplos de excipientes líquidos son el jarabe de azúcar, el aceite de cacahuete, el aceite de oliva y el agua. Los ejemplos de excipientes gaseosos incluyen dióxido de carbono y nitrógeno. Al preparar las composiciones para la forma galénica oral, se puede emplear cualquier medio farmacéutico conveniente. Por ejemplo, se pueden usar agua, glicoles, aceites, alcoholes, agentes aromatizantes, conservantes, agentes colorantes y similares para formar preparados líquidos orales tales como suspensiones, elixires y soluciones; mientras que los excipientes tales como almidones, azúcares, celulosa microcristalina, diluyentes, agentes de granulación, lubricantes, aglutinantes, agentes disgregadores, y similares pueden usarse para formar preparados sólidos orales tales como polvos, cápsulas y comprimidos. Debido a su facilidad de administración, los comprimidos y las cápsulas son las dosis individuales orales preferidas por las cuales se emplean excipientes farmacéuticos sólidos. Opcionalmente, los comprimidos pueden recubrirse mediante técnicas acuosas o no acuosas habituales.
Un comprimido que contiene la composición de esta invención puede prepararse por compresión o moldeo, opcionalmente con uno o más ingredientes accesorios o adyuvantes. Los comprimidos pueden prepararse comprimiendo, en una máquina adecuada, el principio activo en forma suelta, como en polvo o gránulos, mezclados opcionalmente con un aglutinante, lubricante, diluyente inerte, tensioactivo o agente dispersante. Los comprimidos moldeados pueden fabricarse moldeando en una máquina adecuada, una mezcla del compuesto en polvo humedecido con un diluyente líquido inerte. Cada comprimido contiene preferiblemente de aproximadamente 0,05 mg a aproximadamente 5 g del principio activo y cada sello o cápsula contiene preferiblemente de aproximadamente 0,05 mg a aproximadamente 5 g del principio activo. Por ejemplo, una formulación destinada a administración oral a personas puede contener de aproximadamente 0,5 mg a aproximadamente 5 g de principio activo, compuesto con una cantidad apropiada y conveniente de material excipiente que puede variar desde aproximadamente 5 a aproximadamente 95 por ciento de la composición total. Las formas galénicas unitarias generalmente contendrán entre aproximadamente 1 mg y aproximadamente 2 g del principio activo, generalmente 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg, o 1000 mg.
Las composiciones farmacéuticas de la presente invención adecuadas para administración parenteral pueden prepararse como soluciones o suspensiones de los principios activos en agua. Se puede incluir un tensioactivo adecuado tal como, por ejemplo, hidroxipropilcelulosa. Las dispersiones también se pueden preparar en glicerol, polietilenglicoles líquidos y sus mezclas en aceites. Además, se puede incluir un conservante para evitar el crecimiento perjudicial de microorganismos.
Las composiciones farmacéuticas de la presente invención adecuadas para uso inyectable incluyen soluciones o dispersiones acuosas estériles. Además, las composiciones pueden estar en forma de polvos estériles para la preparación improvisada de dichas soluciones o dispersiones inyectables estériles. En todos los casos, la forma inyectable final debe ser estéril y debe ser efectivamente fluida para una fácil inyectabilidad. Las composiciones farmacéuticas deben ser estables en las condiciones de fabricación y almacenamiento; por lo tanto, preferiblemente debe conservarse contra la acción contaminante de microorganismos tales como bacterias y hongos. El excipiente puede ser un disolvente o medio de dispersión que contenga, por ejemplo, agua, etanol, poliol (p. ej., glicerol, propilenglicol y polietilenglicol líquido), aceites vegetales y una de sus mezclas adecuadas.
Las composiciones farmacéuticas de la presente invención pueden estar en una forma adecuada para uso tópico tal como, por ejemplo, un aerosol, crema, pomada, loción, polvo para espolvorear, o similares. Además, las composiciones pueden estar en una forma adecuada para uso en dispositivos transdérmicos. Estas formulaciones pueden prepararse, utilizando un compuesto representado por la fórmula I de esta invención, o una de sus sales farmacéuticamente aceptables, por métodos de tratamiento convencionales. Como ejemplo, se prepara una crema o ungüento mezclando material hidrófilo y agua, junto con aproximadamente 5% en peso a aproximadamente 10% en peso del compuesto, para producir una crema o pomada que tenga la consistencia deseada.
Las composiciones farmacéuticas de esta invención pueden estar en una forma adecuada para administración rectal en la que el excipiente es un sólido. Es preferible que la mezcla forme supositorios de dosis individual. Los excipientes adecuados incluyen manteca de cacao y otros materiales frecuentemente usados en la técnica. Los supositorios pueden formarse convenientemente mezclando en primer lugar la composición con el (los) soporte(s) ablandado(s) o fundido(s) seguido de enfriamiento y moldeado en moldes.
Además de los ingredientes excipientes mencionados anteriormente, las formulaciones farmacéuticas descritas anteriormente pueden incluir, según corresponda, uno o más ingredientes excipientes adicionales tales como diluyentes, amortiguadores, agentes aromatizantes, aglutinantes, agentes tensioactivos, espesantes, lubricantes, conservantes (incluidos los antioxidantes) y similares. Además, se pueden incluir otros adyuvantes para hacer que la formulación sea isotónica con la sangre del receptor deseado. Las composiciones que contienen un compuesto descrito por la fórmula I, o una de sus sales farmacéuticamente aceptables, también pueden prepararse en forma de polvo o concentrado líquido.
Generalmente, las cantidades de administración del orden de aproximadamente 0,01 mg/kg a aproximadamente 150 mg/kg de peso corporal al día son útiles en el tratamiento de las afecciones indicadas anteriormente, o alternativamente de aproximadamente 0,5 mg a aproximadamente 7 g por paciente al día. Por ejemplo, la inflamación, el cáncer, la psoriasis, la alergia/asma, las enfermedades y las afecciones del sistema inmunológico, las enfermedades y las afecciones del sistema nervioso central (SNC) se pueden tratar de manera eficaz mediante la administración de aproximadamente 0,01 a 50 mg del compuesto por kilogramo de peso corporal al día, o alternativamente, de aproximadamente 0,5 mg a aproximadamente 3,5 g por paciente al día.
Sin embargo, se entiende que el nivel de dosis específico para cualquier paciente en particular dependerá de una variedad de factores incluida la edad, el peso corporal, la salud general, el sexo, la alimentación, el tiempo de administración, la vía de administración, la tasa de excreción, la combinación de fármacos y la gravedad de la enfermedad concreta sometida a tratamiento.
Estos y otros aspectos se harán evidentes a partir de la siguiente descripción escrita de la invención.
Modos de realización de la invención.
La presente invención está dirigida a nuevos compuestos con actividad inhibidora de c-Met. Los compuestos de la invención incluyen los de fórmula I y una de sus sales farmacéuticamente aceptables.

En la fórmula I,
B falta o es O, S, OCH2 o SCH2;
R1, R2, R3 y R4 son cada uno independientemente hidrógeno, halógeno, alquilo C1-6 sustituido o no sustituido, alquenilo C2-8 o carbamoílo; o R1 y R2 a la vez, o R3 y R4 a la vez, forman =O;
A es N oCH;
n es 0, 1, 2 o 3;
cada R5 es independientemente halógeno o alquilo C1-6 ;
cada R50 es independientemente hidrógeno o alquilo C1-6 sustituido o no sustituido;
Z es NHR6 o de fórmula II:

en donde R18 y R19 son cada uno independientemente hidrógeno, alquilo C1-8 sustituido o no sustituido o arilo C6-8 sustituido o no sustituido;
R6 es de fórmula VI:

en donde:
Q3 es hidrógeno;
X 1 es NR8 o CR8 R9;
R8 es hidrógeno o fenil sustituido con halógeno; y
R9 es hidrógeno.
Los siguientes compuestos de la invención se proporcionan para que el lector comprenda los compuestos incluidos en la invención:
N-(4-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida; N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi) fenil)-5-(4-fluorofenilo) )-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-5-4-(fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi) fenil)-5-(4-fluorofenil)-4 oxo-1,4-dihidropiridin-3-carboxamida; N-(3-fluoro-4-(2-oxo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
8-(6-(2-(4-fluorofenilamino)-1-metil-6-oxo-1,6-dihidropirimidin-5-il)piridin-3-iloxi)-2H-pirido[3,2-b][ 1,4]oxazin-3(4H)-ona;
5-(5-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi) piridin-2-il)-2-(4-fluorofenilamino)-3-metilpirimidin-4(3H) )-ona;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1, 4-dihidropiridin-3-carboxamida;
[4-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
[4-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
5-[5-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-piridin-2-il]-2-(4-fluoro-fenilamino)-3-metil-3H-pirimidin-4-ona;
5-[5-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-piridin-2-il]-2-( 4-fluoro-fenilamino)-3-metil-3H-pirimidin-4- ona;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico
5- [5-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-piridin-2-il]-2-(4-fluoro-fenilamino)-3-metil-3H-pirimidin-4-ona;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
[4-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
2-(4-fluoro-fenilamino)-3-metil-5-[5-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-piridin-2-il]-3H-pirimidin-4-ona;
[3-fluoro-4-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
2-(4-fluoro-fenilamino)-3-metil-5-[5-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-piridin-2-il]-3H-pirimidin-4-ona;
[4-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
N-(4-(3,4-dimetil-2,3,4,5-tetrahidrospiro[3,2-b][1,4]oxazepin-9-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
[4-(3,3-difluoro-2,3-dihidro-1 H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
N-(4-(2,2-difluoro-2,3,-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(4-(3',4-dihidrospiro[ciclopropano-1,2-pirido[3,2-b][1,4]oxazina]-8-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(2-(hidroximetil)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
8-(2-fluoro-4-(5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamido)fenoxi)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazina-2-carboxamida;
N-(4-(2-(aziridin-1-ilmetil)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(3-(2-hidroxietil)-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-fluorofenilo)-4-oxo-1,4-dihidropiridin-3-carboxamida; o
N-(3-fluoro-4-(2'-oxo-1', 2'-dihidrospiro[ciclopropano-1,3'-pirrolo[2,3-b]piridina]-4'-iloxi) fenil)-5-(4-fluorofenil)-4-oxo-1.4- dihidropiridin-3-carboxamida.
Como se define en la presente memoria, el término "sujeto" o "paciente" se refiere a un animal, preferiblemente un mamífero, más preferiblemente un ser humano, que ha sido objeto de tratamiento, observación o experimento. La expresión "cantidades terapéuticamente eficaces" empleada en la presente memoria, significa la cantidad de principio activo o agente farmacéutico que provoca la respuesta biológica o medicinal en un sistema hístico, animal o humano que un investigador, veterinario, médico u otro clínico busca, que incluye el alivio de los síntomas de la enfermedad o trastorno que se está tratando.
El término "sustituido", como se define en la presente memoria, incluye múltiples sustituyentes (p. ej., Phe, arilo, heteroarilo), preferiblemente de uno a cinco sustituyentes, más preferiblemente de uno a tres sustituyentes, aún más preferiblemente de uno a dos sustituyentes, seleccionados independientemente de la lista de sustituyentes. Los sustituyentes típicos incluyen, entre otros, -X, -CH3, -C2H5 , -OH, =O, -SH, =S, -NH2 , -CX3 , -CF3, -CN, -NO2, -OS(O2)OH, -C(O)OH, donde cada X es independientemente un halógeno. El término "sustituido" contempla específicamente y permite sustituciones que son frecuentes en la técnica. Sin embargo, los expertos en la técnica entienden generalmente que los sustituyentes deben seleccionarse de manera que no afecten negativamente las características farmacológicas del compuesto o interfieran negativamente en el uso del medicamento.
Los compuestos de la presente invención pueden tener átomos de carbono asimétricos. Dichas mezclas diastereoméricas pueden separarse en sus diastereómeros individuales en base a sus diferencias fisicoquímicas por métodos conocidos por los expertos en la técnica, por ejemplo, por cromatografía o cristalización fraccionada. Los enantiómeros pueden separarse convirtiendo las mezclas enantioméricas en una mezcla diastereomérica por reacción con un compuesto ópticamente activo apropiado (p. ej., alcohol), separando los diastereoisómeros y convirtiendo (p. ej., hidrolizando) los diastereoisómeros individuales en los enantiómeros puros correspondientes. Todos estos isómeros, incluidas las mezclas de diastereómeros y los enantiómeros puros, se consideran parte de la invención.
En general, los compuestos de la presente invención pueden prepararse según el esquema 1 que representa rutas sintéticas generales para compuestos de la invención y no pretenden ser restrictivas. Más específicamente, el esquema 1 representa la síntesis de compuestos de piridina fusionados. Se describen ejemplos específicos posteriormente a estas descripciones sintéticas generales para permitir que un experto en la técnica prepare y use quinazolinas o quinolinas de la invención.
El esquema 1 describe los procedimientos generales que se podrían usar para proporcionar compuestos de la presente invención, por ejemplo, Z es NHR6 en la fórmula I.

Los compuestos de fórmula I de la presente invención, Z es NHR6 en la fórmula I, pueden prepararse a partir de compuestos intermedios 5 y compuestos ácidos adecuados, R6-OH, donde R6 se definió anteriormente, utilizando condiciones de reacción de acoplamiento habituales para la formación de amidas. Compuestos de ácido adecuado, R6-OH, donde R6 se ha definido anteriormente, están disponibles en el mercado, son conocidos en la bibliografía o pueden prepararse convenientemente por una variedad de métodos conocidos por los expertos en la técnica.
Una ruta común para los compuestos intermedios 5 se ilustra en el esquema 1. Los compuestos 1 pueden reaccionar con el compuesto intermedio 2 a temperatura elevada, en presencia de una base adecuada tal como bicarbonato de sodio en un solvente apropiado tal como DMF, seguido de una reducción típica del grupo nitro, para proporciona los compuestos intermedios 5. Una ruta alternativa para la preparación de compuestos intermedios 5 puede ser la reacción de sustitución de los compuestos 1 con el intermedio anilina 3 a temperatura elevada en presencia de una base adecuada, como bicarbonato de sodio, en un disolvente adecuado, como DMF, como se muestra en esquema 1.
El esquema 2 describe los procedimientos generales que se podrían usar para proporcionar compuestos de la presente invención, por ejemplo, Z en grupos en la fórmula II.

Los intermedios adecuados 6 , donde R18 y R19 se definieron anteriormente, están disponibles en el mercado, son conocidos en la bibliografía o pueden prepararse convenientemente por una variedad de métodos conocidos por los expertos en la técnica. Los intermedios 6 pueden reaccionar con los compuestos 1 a temperatura elevada, en presencia de una base adecuada tal como bicarbonato de sodio en un disolvente apropiado como DMF, para proporcionar compuestos de la presente invención, por ejemplo, Z en grupos en la fórmula II.
Para preparar las composiciones farmacéuticas de esta invención, uno o más compuestos de fórmula (I) o una de sus sal como principio activo, se mezclan íntimamente con un excipiente farmacéutico según técnicas de composición farmacéutica convencionales, excipiente que puede tomar una amplia variedad de formas dependiendo de la forma de preparación deseada para su administración (p. ej., oral o parenteral). Los excipientes farmacéuticamente aceptables adecuados son bien conocidos en la técnica. Las descripciones de algunos de estos excipientes farmacéuticamente aceptables se pueden encontrar en The Handbook of Pharmaceutical Excipients, publicado por la American Pharmaceutical Association y la Pharmaceutical Society of Great Britain. Durante cualquiera de los procesos para la preparación de los compuestos de la presente invención, puede ser necesario y/o deseable proteger los grupos sensibles o reactivos en cualquiera de las moléculas en cuestión. Esto puede conseguirse por medio de grupos protectores convencionales, como los descritos en Protective Groups in Organic Chemistry, ed. J. F. W. McOmie, Plenum Press, 1973; y T. W. Greene & P. G. M. Wuts, Protective Groups in síntesis Synthesis, John Wiley & Sons, 1991. Los grupos protectores pueden eliminarse en una etapa posterior conveniente utilizando métodos conocidos en la técnica.
La administración de los compuestos activos puede efectuarse por cualquier método que permita la administración de los compuestos en el punto de actuación (p. ej., células cancerosas). Estos métodos incluyen vías orales, vías rectales, vías intraduodenales, inyección parenteral (incluida la administración intravenosa, subcutánea, intramuscular, intravascular o infusión), administración tópica, etc. La cantidad de compuesto activo administrado dependerá, por supuesto, del sujeto tratado, de la gravedad de la afección, de la forma de administración y del criterio del médico que receta. Sin embargo, una dosis eficaz está comprendida en el intervalo de aproximadamente 0,001 mg a aproximadamente 300 mg (preferiblemente, de aproximadamente 0,01 mg a aproximadamente 100 mg; y, más preferiblemente, de aproximadamente 0,1 mg a aproximadamente 30 mg) y se puede administrar a una dosis de alrededor de 0,001 mg/kg/día a aproximadamente 300 mg/kg/día (preferiblemente, de aproximadamente 0,01 mg/kg/día a aproximadamente 100 mg/kg/día; y, más preferiblemente, de aproximadamente 0,1 mg/kg/día a aproximadamente 30 mg/kg/día).
La composición puede estar, por ejemplo, en una forma adecuada para administración oral tal como un comprimido, cápsula, píldora, polvo, formulación de liberación lenta, solución o suspensión; para inyección parenteral, tal como una solución, suspensión o emulsión estéril; o para administración tópica como una pomada o crema o para administración rectal como supositorio. La composición farmacéutica puede estar en formas galénicas unitarias adecuadas para la administración aislada de dosis precisas. La composición farmacéutica incluirá un vehículo o excipiente farmacéutico convencional y un compuesto según la presente invención como principio activo. Además, puede incluir otros agentes medicinales o farmacéuticos, vehículos, adyuvantes, etc.
Ejemplos de formas de administración parenteral incluyen soluciones o suspensiones de compuestos activos en soluciones acuosas estériles, por ejemplo, soluciones acuosas de propilenglicol o dextrosa. Dichas formas de dosificación pueden amortiguarse adecuadamente, si se desea.
Los vehículos farmacéuticos adecuados incluyen diluyentes o cargas inertes, agua y diversos disolventes orgánicos. Las composiciones farmacéuticas, si se desea, pueden contener ingredientes adicionales tales como aromatizantes, aglutinantes, excipientes y similares. Por lo tanto, para la administración oral, los comprimidos que contienen diversos excipientes, como el ácido cítrico, pueden emplearse junto con varios disgregadores como el almidón, el ácido algínico y determinados silicatos complejos y con agentes aglutinantes como la sacarosa, la gelatina y la goma arábiga. Además, los agentes lubricantes como el estearato de magnesio, el laurilsulfato de sodio y el talco a menudo son útiles para la fabricación de comprimidos.
Las composiciones sólidas de un tipo similar también se pueden emplear en cápsulas de gelatina rellenas blandas y duras. Los materiales preferidos, por lo tanto, incluyen lactosa o azúcar de leche y polietilenglicoles de alto peso molecular. Cuando se desean suspensiones acuosas o elixires para administración oral, el compuesto activo en ellos
puede combinarse con diversos agentes edulcorantes o aromatizantes, materias colorantes o tintes y, si se desea, agentes emulsionantes o agentes de suspensión, junto con diluyentes tales como agua, etanol, propilenglicol, glicerina o una de sus combinaciones.
Ejemplos
Los ejemplos siguientes se proporcionan para ilustrar mejor la presente invención. Los ejemplos 6-15, 18-20, 24-27, 32-35, 39-42, 44-46, 49, 51, 53-55, 58, 61-67 son ejemplos de referencia. Todas las partes y porcentajes son en peso y todas las temperaturas son en grados Celsius, a menos que se indique explícitamente lo contrario. Las siguientes abreviaturas se han utilizado en los ejemplos:
ATP: trifosfato de adenosina;
DIPEA: N,N-diisopropiletilamina;
DMF: N,N-dimetilformamida;
DMA: N,N-dimetiacetamida;
DMAP: 4-N,N-dimetilamiopiridina;
DMSO: Sulfóxido de dimetilo;
DEAD: Azodicarboxilato de dietilo;
HATU: Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio;
DIPEA: N,N-diisopropiletilamina;
EDC: 1-etil-3-(3-dimetilaminopropil)carbodiimida;
TBAB: Bromuro de tetrabutilamonio;
TEA: Trietilamina;
EtOAc: Acetato de etilo;
GSR: Glutatión-S-transferasa;
Crk: CT10 (Retrovirus 10 de tumor de pollo);
min: minutos;
h: horas;
t. a. o T. A.: temperatura ambiente;
SDS: Dodecilsulfato de sodio;
SDS-PAGE: electroforesis en gel de poliacrilamida con dodecilsulfato sódico;
TLC: Cromatografía en capa fina.
Ejemplos
Ejemplo 1
N-(4-(2,3-dihidro-1H-pirrolo[2,3-b]pirídin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropirídin-3-carboxamida (Producto 1)
Etapa 0. Preparación del ácido 1
1. Preparación de A11
Se añadió piridina (70,35 g) a la mezcla de ácido Maxwell (50,32 g) en DCM (450 ml) con agitación, seguido de la adición de A10 (62,28 g) a 0°C. La reacción se continuó a 0°Cdurante 2,5 horas o hasta que se completó la reacción controlada por TLC. La mezcla de reacción se diluyó con agitación con DCM (300 ml) y 750 ml de HCl 1 N. La fase orgánica se separó y se secó al vacío para dar un sólido, A11 (66,70 g).
2. Preparación de A12
Se calentó a 87°C A11 (66,70 g) en etanol (400 ml) y se agitó durante la noche. La mezcla de reacción se enfrió y se concentró al vacío para dar un aceite (55 g). El residuo oleoso se disolvió en DMF/DMA (200 ml), se calentó a 110°Cdurante 3 h, y luego se concentró al vacío, seguido de dilución con acetato de etilo y agitación. El precipitado se recogió por filtración para dar A12 (52 g).
3. Preparación de A13
Se disolvió A12 (27,20g) en etanol (600 ml) al que se añadió NH4CI. La mezcla de reacción se calentó a reflujo durante 2 horas y luego se enfrió a temperatura ambiente. El sólido se recogió por filtración para dar A13 (16,88 g).
4. Preparación de ácido 1
Se añadió solución de NaOH 2 N (120 ml) a A13 (16,88 g) en etanol (50 ml). La mezcla de reacción se agitó y se calentó a 65°Cdurante 2 horas, y luego se enfrió a temperatura ambiente. La mezcla de reacción se añadió en HCl 1,5 N (400 ml) para precipitar el sólido. La filtración fue para recoger el sólido. El sólido se lavó dos veces con agua y se secó al vacío para dar ácido 1 (14,13 g).
Etapa 1. Preparación de 4-doropiridin-2-ilcarbamato de terc-butilo (Compuesto 11)
El compuesto 10 (4-cloropiridin-2-amina, 128 g, 1 mol) se disolvió en DCM (3,0 l), a lo que se añadió TEA (121 g, 1,2 mol) y DMAP (9,76 g, 0,08 mol) con agitación. Después la mezcla se agitó a 15°C-20°C durante varios minutos, a cuya mezcla de reacción se añadió lentamente (Boc)2O (229 g) en DCM (500 ml) a 15°C-20°C. La reacción se continuó por debajo de 25°C hasta que se completó. El precipitado se recogió por filtración y se lavó con DCM (500 ml). El compuesto final 11 se purificó por recristalización con acetato de etilo para dar el compuesto 11 (160 g).
Etapa 2. Preparación de 4-cloro-3-(2-hidroxietil)piridin-2-ilcarbamato de terc-butilo (Compuesto 12)
El compuesto 11 (2,01 g, 8,80 mmol) en THF (15 ml) se enfrió a -78°C, y luego se le añadió gota a gota n-BuLi (12 ml, 19,26 mmol). La mezcla de reacción se calentó a -70°Cy se mantuvo otra media hora. Se añadió óxido de etileno (1,93 g, 43,75mmol) a la mezcla de reacción a -70°Cy se agitó durante la noche para calentar a temperatura ambiente. La reacción se detuvo con agua (1 ml) y se extrajo con acetato de etilo (100 ml) y salmuera (100 ml). Se separó la capa orgánica y se secó. La concentración de la capa orgánica dio el compuesto 12 en forma de sólido amarillo (2,0 g). Etapa 3. Preparación de 4-cloro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-carboxilato de terc-butilo (Compuesto 13) El compuesto 12 (1,37 g, 5,04 mmol) se disolvió en THF (20 ml), al que se añadió Ph3P (1,59g, 6,05 mmol). Se añadió gota a gota DEAD (1,07 g, 6,06 mmol) a la mezcla de reacción a 0°C, y luego se calentó lentamente a temperatura ambiente. Después de concentrar, la mezcla de reacción se purificó por columna de gel de sílice para dar el compuesto 13 en forma de sólido blanco (1,15 g).
Etapa 4. Preparación del compuesto 14
El Compuesto 13 (1,15g, 4,53 mmol) y 4-amino-2-fluoro-fenol (1,15g, 9,06 mmol) se disolvió en DMF (20 ml), al que se añadió a continuación carbonato de cesio (4,43 g, 13,59 mmol). La mezcla de reacción se calentó a 125°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con acetato de etilo. La fase orgánica se recogió y se evaporó al vacío para dar el compuesto 14 en forma de sólido (0,80 g).
Etapa 5. Preparación del compuesto 15
El Compuesto 14 (4,00 g, 11,6 mimóles) y el ácido 1 (2,97 g, 12,8 mimóles) se disolvieron en DCM (60 ml) seguido de la adición de HATU (5,29 g, 13,9 mmoles) y TEA (3,52 g, 34,8 mmoles). La reacción se agitó a temperatura ambiente durante la noche y luego se diluyó con acetato de etilo y agua. La fase orgánica se separó y la capa acuosa se extrajo con acetato de etilo. Toda la capa orgánica se combinó y se secó. Después de la evaporación del disolvente, el residuo se purificó en columna de gel para dar el compuesto l5 en forma de sólido amarillo claro (4,8 g).
Etapa 6. Preparación del producto 1
El Compuesto 15 (3,5 g) se añadió en la mezcla de DCM (15 ml) y ácido trifluoroacético (15 ml), la reacción se continuó durante 3 horas a temperatura ambiente. La mezcla de reacción se evaporó al vacío para dar el producto 1 en forma de sólido rojo claro (2,46 g). MS: 461 (M+H)+ RMN H (DMSO-da): 13,14 (s, 1H), 12,68 (s, 1H), 8,62 (s, 1H), 8,09 (s, 1H), 7,96-8,00 (dd, 1H, J = 9,60Hz 1,80Hz), 7,68-7,72 (m, 2H), 7,61-7,62 (d, 1H, J = 4,50 Hz), 7,39-7,42 (dd, 1H, J = 6,60 Hz 1,20Hz ), 7,20-7,29 (m, 3H), 6,48 (s, 1H), 5,90-5,91 (d, 1H, J = 4,50 Hz), 3,45-3,49 (m, 2H), 2,83-2,89 (m, 2H).
Ejemplo 2
N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi) fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida (Producto 2)

Etapa 1. Preparación del compuesto 21
El compuesto 20 (184 mg, 1,0 mmol), 2-fluoro-4-amino-fenol (190 mg, 1,5 mmol) y carbonato de cesio (652 mg, 2,0 mmol) se mezclaron en N-metil pirrolidona (3 ml). La mezcla de reacción se calentó a 180°C durante la noche bajo N2. A continuación, la mezcla de reacción se añadió en agua (50 ml) y se extrajo con acetato de etilo (100 ml). La fase orgánica se lavó con salmuera, se secó y se concentró, seguido de purificación en columna ultrarrápida para dar el compuesto 21 (105 mg).
Etapa 2. Preparación del producto 2.
HATU (76 mg, 0,2 mmol), DIPEA (26 mg, 0,2 mmol) y ácido 1 (25,6 mg, 0,11 mmol) se mezclaron en DCM (3 ml) a 0°C y se agitaron durante 15 minutos, seguido de la adición del compuesto 21 (27 mg), 0,1mmol). La mezcla de reacción se agitó durante 4 horas a temperatura ambiente. Después de concentrar, la mezcla de reacción se purificó por columna ultrarrapida para dar el producto 2 (16 mg). MS: 491 (M+H)+.
Ejemplo 3
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida (Producto 3)

Etapa 1. Preparación del compuesto 31
El compuesto 30 (1,52 g, 10 mmol), 3,4-difluoronitrobenceno (1,75 g, 1 mmol) y carbonato de cesio (3,60 g, 1 mmol) se mezclaron en DMF (15 ml) y se agitaron a temperatura ambiente durante la noche. Una vez completado el control por TLC, la mezcla de reacción se añadió en agua (400 ml) y se extrajo con acetato de etilo (800 ml). La capa acuosa se extrajo con acetato de etilo. Toda la capa orgánica se combinó, se lavó con salmuera y se secó. Después de la concentración, el residuo se purificó por columna ultrarrápida hasta el compuesto 31 (2,20 g).
Etapa 2. Preparación del compuesto 32
El compuesto 31 (2,20 g) se añadió a la mezcla de Pd/Cal 5% (0,8 g) y metanol (100 ml). La mezcla resultante se agitó bajo H2 hasta que no hubo material de partida. Luego, la mezcla de reacción se filtró para eliminar Pd/C. El filtrado se concentró para dar el compuesto 32 (1,60 g).
Etapa 3. Preparación del producto 3
El producto deseado 3 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2. MS: 477 (M+H)+.
Ejemplo 4
N-(4-(3,4-dihidro-2H-pirído[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropirídin-3-carboxamida (Producto 4)

El compuesto 42 deseado se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 3.
El producto deseado 4 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2. MS: 459 (M+)+.
Ejemplo 5
N-(3-fluoro-4-(2-oxo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-flourofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida (Producto 5)

Etapa 1. Preparación del compuesto 51
El compuesto 50 (3,0 g 20 mmoles) se disolvió en alcohol terc-butílico (200 ml), al que se añadió tribromuro de piridinio (21,5 g 67 mmoles). La mezcla de reacción se agitó a temperatura ambiente durante la noche, seguido de concentración al vacío. El residuo se purificó por extracción y recristalización para dar el compuesto 51 (3,60 g). Etapa 2. Preparación del compuesto 52
El compuesto 51 (3,6 g) se disolvió en alcohol (50 ml), al que se añadió lentamente ácido acético (2,0 ml). La reacción se calentó a reflujo hasta que se completó. La filtración de la mezcla de reacción se empleó para eliminar el sólido y el filtrado se concentró al vacío para dar el compuesto 52 (1,12 g).
Etapa 3. Preparación del compuesto 53
El compuesto 52 (1,12 g, 66 mmol), fluoro-4-aminofenol (930 mg, 73 mmol) y carbonato de cesio (4,30mg, 132 mmol) se mezclaron en DMSO (15 ml) y se calentó a 135°C durante la noche bajo N2. La mezcla de reacción se diluyó con acetato de etilo y agua. La capa orgánica se separó y se secó. La evaporación de la capa orgánica dio el compuesto 53 (660 mg).
Etapa 4. Preparación del producto 5
El producto deseado 5 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2. MS: 475 (M+H)+.
Ejemplo 6
N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-1-(4-fluorofenil)-2-oxo-1,2-dihidropiridin-3-carboxamida (Producto 6)

Etapa 1. Preparación de A21
Se disolvió A20 (3,10 g) en DMF (25 ml) y se enfrió a 0°C, a lo que se añadió 4-fluoroanilina. La reacción se continuó durante 7 horas a 0°C, seguido de la adición de EDC-HCl (5,40 g) y DMAP (0,6 l g) y se agitó durante la noche. La mezcla de reacción se diluyó con agua y acetato de etilo. La capa orgánica se separó y se lavó con salmuera. Después de secar, el disolvente se eliminó por evaporación para dar A21 (2,80 g).
Etapa 2. Preparación de ácido 2
Se mezcló A21 (2,61 g) en solución de NaOH (1,60 g de NaOH y 40 ml de agua) y metanol (8 ml). A continuación, la mezcla de reacción se agitó y se calentó a reflujo durante 1 hora. Después de enfriar a temperatura ambiente, la mezcla de reacción se ajustó a pH = 1. El sólido resultante se recogió por filtración y se lavó con agua, seguido de secado para dar ácido 2 (2,25 g).
Etapa 3. Preparación del producto 6
El producto deseado 6 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2 con ácido 2 en lugar de ácido 1. MS: 491 (M+H)+.
Ejemplo 7
N-(4-(2,3-dihidro-1H-pirrolo[2,3-b]pirídin-4-iloxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-dihidropiridin-3-carboxamida (Producto 7)

El compuesto 71 se obtuvo mediante el compuesto 14 que estaba sin la protección de Boc.
El producto deseado 7 se sintetiza utilizando una secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 2 en lugar de ácido 1. MS: 461 (M+H)+. RMN H (DMSO-d6): 12,10 (s, 1H), 8,57-8,59 (dd, 1H, J = 5,40Hz 1,50Hz), 8,13-8,15 (dd, 1H, J = 5,10Hz 1,80Hz), 7,99-8,03 (dd, 1H, J = 9,60Hz 1,65Hz), 7,60-7,64 (m, 4H), 7,41-7,49 (m, 3H), 7,31-7,36 (t, 1H, J = 13,50Hz), 6,72-6,75 (t, 1H, J = 10,20 Hz), 6,10-6,11 (d, 1H, J = 5,10Hz), 3,62-3,66 (m, 2H), 2,89-2,93 (m, 2H).
Ejemplo 8
N-(4-(3,4-dihidm-2H-pirído[3,2-b][1,4]oxazin-8-iloxi)-3-fluomfenil)-1-(4-fluomfenil)-2-oxo-1,2-dihidmpirídin-3-carboxamida (Producto 8)

El producto deseado 8 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2 con ácido 2 en lugar de ácido 1. MS: 477 (M+H)+. RMN H (DMSO-da): 12,03 (s, 1H), 8,56-8,59 (dd, 1H, J = 5,40Hz 1,65Hz), 8,11-8,13 (dd, 1H, J = 4,80Hz 1,50Hz), 7,93-7,97 (dd, 1H, J = 9,90Hz 1,80Hz), 7,60-7,62 (m, 2H), 7,40-7,46 (m, 4H), 7,12-7,16 (t, 1H, J = 13,80Hz), 6,81 (s, 1H ), 6,70-6,74 (t, 1H, J = 10,50Hz), 6,00 (ms, 1H), 4,11-4,16 (m, 2H), 3,41-3,42 (m, 2H).
Ejemplo 9
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-1-(4-fluorofenil)-2-oxo-1,2-dihidropiridin-3-carboxamida (Producto 9)

El producto deseado 9 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2 con ácido 2 en lugar de ácido 1. MS: 459 (M+H)+. RMN H (DMSO-d6): 11,90 (s, 1H), 8,56-8,58 (dd, 1H, J = 5,40 Hz 1,50 Hz), 8,09-8,11 (dd, 1H, J = 4,8 Hz 1,5 Hz), 7,67-7,71 (d, 2H, J =6,90 Hz), 7,59-7,62 (m, 2H), 7,47-7,48 (d, 1H, J = 4,2 Hz), 7,40-7,44 (t, 2H, J = 13,20 Hz), 6,99-7,01 (d, 2H, J = 6,90 Hz), 6,80 (s, 1H), 6,69-6,73 (t, 1H, J = 10,20 Hz), 6,07-6,08 (d, 1H, J = 4,20 Hz), 4,08-4,10 (m, 2H), 3,40-3,41 (m, 2H).
Ejemplo 10
N-(3-fluoro-4-(2-oxo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-1-(4-fluorofenil)-2-oxo-1,2-dihidropiridin-3-carboxamida (Producto 10)

El producto deseado 10 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2 con ácido 2 en lugar de ácido 1. MS: 475 (M+H)+.
Ejemplo 11
N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirído[3,2-b][1,4]oxazin-8-iloxi)fenil)-N-(4-fluorofenil)ddopropano-1,1-dicarboxamida (Producto 11)

Etapa 1. Preparación de A31
Se añadieron malonato de dietilo (33,01 g), 1,2-dicloroetano (40,38 g), TBAB (2,09 g), benceno (100 ml), carbonato de potasio (72,50 g) y agua (1,00 ml) en un matraz de 500 ml y se calentaron a 115°C durante la noche. La mezcla de reacción se enfrió a temperatura ambiente y se filtró. El filtrado se concentró y se continuó con la purificación de la columna ultrarrápida para dar A31 (24,80 g).
Etapa 2. Preparación de A32
Se mezcló A31 (24,80 g) con etanol (150 ml) y solución de NaOH (5,08 g en 100 H2O) a 0°C. La mezcla de reacción se agitó durante la noche y se calentó lentamente a temperatura ambiente. A continuación, el etanol se evaporó y se lavó con éter para dar A32 (20,45 g).
Etapa 3. Preparación de A33
Se mezclaron A32 (20,45 g) y cloruro de tionilo (150 ml) y se sometieron a reflujo durante 2 horas. A continuación, el cloruro de tionilo se evaporó después de completar la reacción controlada por TLC. El sólido resultante se lavó con DCM para dar A33.
Etapa 4. Preparación de A34
Se añadió lentamente A33 (de la etapa 3) en DCM (100 ml) a la solución de p-fluoroanilina (16,05 g), TEA (40 ml) y DCM (200 ml) a 0°C. La mezcla de reacción se agitó durante la noche y se calentó a temperatura ambiente lentamente. La reacción se detuvo mediante la adición de agua (150 ml). Luego la fase orgánica se lavó dos veces con HCl diluido, se secó con sulfato de sodio anhidro. Por evaporación del disolvente, el residuo se purificó mediante una columna ultrarrápida para dar A34 (20,23 g).
Etapa 5. Preparación de ácido 3
Se mezcló A34 (19,40 g) con etanol (100 ml) y solución de NaOH 2 N (200 ml). La mezcla de reacción se calentó a 65°C y se agitó durante 1 hora. La mezcla de reacción se enfrió por debajo de 10°C y se añadió lentamente a una mezcla de HCl concentrado (50 ml), acetato de etilo (200 ml) y agua (300 ml). La capa orgánica se separó y se lavó dos veces con salmuera y se secó con sulfato de sodio anhidro. La evaporación del disolvente orgánico al vacío dio el ácido 3 (16,21 g).
Etapa 6. Preparación del producto 11
El producto deseado 11 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 3 en lugar de ácido 1. MS: 481 (M+H)+.
Ejemplo 12
N-(4-(2,3-dihidro-1H-pirrolo[2,3-b]pirídin-4-iloxi)-3-fluorofenil)-N-(4-fluorofenil)ddopropropano-1,1-dicarboxamida (Producto 12)

El producto deseado 12 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el Ejemplo 2 con ácido 3 en lugar de ácido 1. MS: 451 (M+H)+. RMN H (DMSO-da): 10,30 (s, 1H), 9,99 (s, 1H), 7,79-7,82 (d, 1H, J = 10,20Hz), 7,56-7,78 (m, 3H), 7,40-7,42 (d, 1H, J = 6,30 Hz), 7,21-7,26 (t, 1H, J = 13,50Hz), 7,12-7,17 (t, 2H, J = 13,20Hz), 6,90-6,95 (t, 1H, J = 13,80Hz), 6,36-6,48 (m, 3H), 5,80-5,86 (dd, 1H, J = 12,00Hz 4,5Hz), 5,39 (s, 1H), 3,41 3,52 (m, 2H), 2,84-2,92 (m, 2H)
Ejemplo 13
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-N-(4-fluorofenil) ddopropano-1,1-dicarboxamida (Producto 13)

El producto deseado 13 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 3 en lugar de ácido 1. MS: 467 (M+H)+.
Ejemplo 14
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-N-(4-fluorofenil) ddopropano-1,1-dicarboxamida (Producto 14)

El producto deseado 14 se sintetiza utilizando la secuencia y las condiciones similares a las descritas para el ejemplo 2 con ácido 3 en lugar de ácido 1. MS: 449 (M+H)+. RMN H (DMSO-d 6 ): 10,04-10,07 (d, 2H, J = 9,30Hz), 7,59-7,64 (m, 4H), 7,46-7,47 (d, 1H, J = 4,20Hz), 7,12-7,16 (t, 2H, J = 13,50 Hz), 6,96-6,98 (d, 2H, J = 6,90Hz), 6,78 (s, 1H), 6,02-6,03 (d, 1H, J = 4,20 Hz), 4,07-4,10 (m, 2H), 3,40-3,43 (m, 2H), 1,44 (s, 4H).
Ejemplo 15
N-(3-fluoro-4-(2-oxo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-N-(4-fluorofenil) cidopropano-1,1-dicarboxamida (Producto 15)

El producto deseado 15 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 3 en lugar de ácido 1. MS: 465 (M+H)+.
Ejemplo 16
8-(6-(2-(4-fluorofenilamino)-1-metil-6-oxo-1,6-dihidropirímidin-5-il)piridin-3-iloxi)-2H-pirído[3,2-b][1,4]oxazin-3 (4H)-ona (Producto 16)
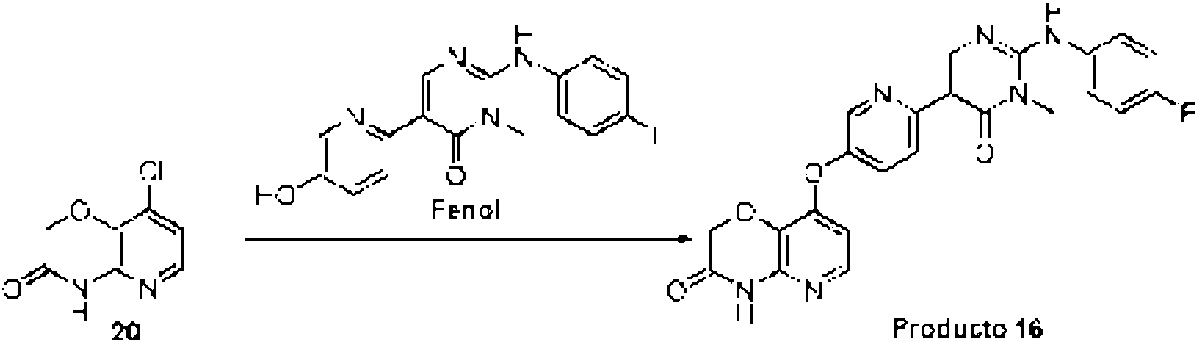
El producto deseado 16 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con fenol en lugar de ácido 1. MS: 461 (M+H)+.
Ejemplo 17
5-(5-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)piridin-2-il)-2-(4-fluorofenilamino)-3-metilpirimidin-4(3H)-ona (Producto 17)

El producto deseado 17 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con fenol en lugar de ácido 1. MS: 431 (M+H)+.
Ejemplo 18
N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-1,5-dimetil-3-oxo-2-fenil-2,3-dihidro-1H-pirazol-4-carboxamida (Producto 18)
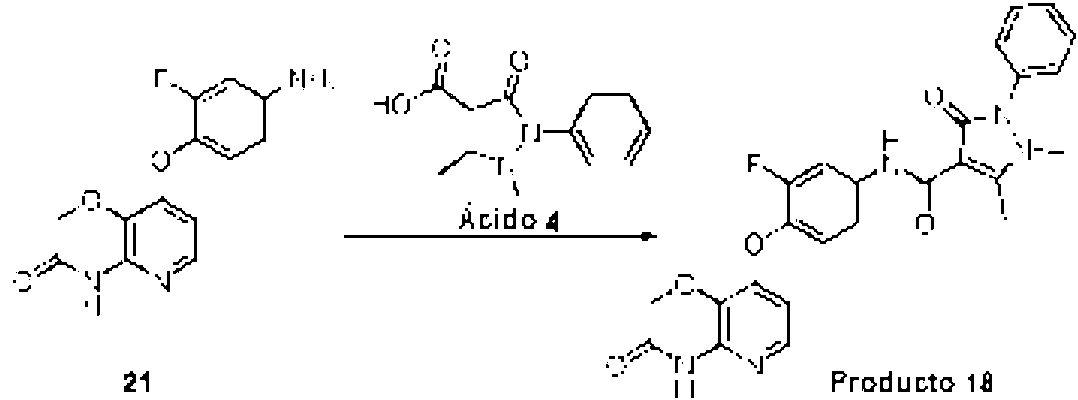
El producto deseado 18 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 4 en lugar de ácido 1. MS: 490 (M+H)+.
Ejemplo 19
N-(4-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-1,5-dimetil-3-oxo-2-fenil-2,3-dihidro-1H-pirazol-4-carboxamida (Producto 19)
El producto deseado 19 se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 4 en lugar de ácido 1. MS: 460 (M+H)+. RMN H (DMSO-da): 10,91 (s, 1H), 7,88-7,95 (dd, 1H, J = 10,50Hz 1,50Hz), 7,60-7,62 (m, 3H), 7,52-7,53 (m, 1H), 7,42-7,44 (m, 2H), 7,22-7,32 (m, 2H), 6,77 (s, 1H), 5,93-5,95 (d, 1H, J = 4,50 Hz), 3,49-3,53 (m, 2H), 3,37 (s, 3H), 2,84-2,89 (m, 2H), 2,70 (s, 3H)
Ejemplo 20
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-1,5-dimetil-3-oxo-2-fenil-2,3-dihidro-1H-pirazol-4-carboxamida (Producto 20)

El producto 20 deseado se sintetiza utilizando la secuencia y condiciones similares a las descritas para el ejemplo 2 con ácido 4 en lugar de ácido 1. MS: 476 (M+H)+. RMN H (DMSO-d6): 10,87 (s, 1H), 7,86-7,90 (dd, 1H, J = 9,60Hz 1,50Hz), 7,42-7,61 (m, 6H), 7,12-7,26 (m, 2H), 6,897 (s, 1H), 5,99-6,00 (d, 1H, J = 4,2Hz), 4,12-4,14 (m, 2H), 3,42 3,43 (m, 2H), 3,36 (s, 3H), 2,70 (s, 3 H )).
Ejemplo 21
N-(4-(3,4-dihidro-2H-pirído[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida (Producto 21)

El producto deseado 21 se sintetiza utilizando la secuencia y condiciones similares descritas para el ejemplo 2. MS: 459 (M+H)+.








Pruebas farmacológicas
Los siguientes ensayos demuestran que determinados compuestos de la presente invención inhiben potentemente la fosforilación de c-Met in vitro, inhiben potentemente c-Met in vivo y tienen actividad antitumoral dependiente de la dosis en determinados modelos de xenoinjerto.
Ensayos biológicos
Se incuba Met (h) con MOPS 8 mM, pH 7,0, EDTA 0,2 mM, KKKSPGEYVNIEFG 250 pM, acetato de Mg 10 mM, [y-33P-ATP] (actividad específica de aprox. 500 cpm/pmol, concentración según sea necesario) y compuesto de prueba 0,2 pM. La reacción se inicia mediante la adición de la mezcla MgATP. Después de la incubación durante 40 minutos a temperatura ambiente, la reacción se detiene mediante la adición de una solución de ácido fosfórico al 3%. Luego se colocan 10 pl de la reacción en una alfombrilla filtrante P30 y se lavan tres veces durante 5 minutos en ácido fosfórico 75 mM y una vez en metanol antes del secado y el recuento de centelleo. Este ensayo se realiza por Millipore. El experimento se lleva a cabo por duplicado. El valor para la muestra de referencia (DMSO) se fijó en 100%, y los valores para las muestras tratadas con compuesto se expresaron como actividad con respecto a la muestra de referencia.
Tabla 1

Análisis de proliferación celular
El análisis de proliferación celular se realizó mediante el protocolo de análisis MTS. En resumen, se cultivarán células U87-MG en medio EMEM, se cultivarán células EBC-1 y células SNU-5 en medio MEM, se cultivarán células MKN45, células NCI-H1975 y células NCI-H1993 en medio RPMI1640, se cultivarán células A549 en medio 5a de McCoy. Todas las células se cultivarán en medio enriquecido con 10% de FBS, a la temperatura de 37°C, 5% de CO2 y 95%
de humedad. Todos los medios de cultivo se comprarán en GIBCO (EE.UU.). Las células se recolectarán respectivamente durante el período de crecimiento logarítmico y se hará el recuento con hemocitómetro. La viabilidad celular es superior al 98% por exclusión con azul tripano. Se ajustan las concentraciones celulares a 2,22 x 105 o 1,11 x 105 o 5,56 x 104 celulas/ml con el medio respectivo. Se añaden 90 pl de suspensiones celulares a las placas de 96 pocillos (por triplicado para cada concentración celular), las densidades celulares finales son 2 x 104 o 1 x 104 o 5 x 103 células/pocillo. La densidad de 5 x 103 células/pocillo se utilizará para la primera prueba. La densidad celular apropiada se determinará y ajustará según los resultados de la primera prueba. Al día siguiente, se disuelve el artículo de prueba o los medicamentos positivos con DMSO o PBS como solución madre. Se dispensan 2 pl de solución de fármaco en 1 ml de medio de cultivo. Se agregan 200 pl de medio de fármaco en placas de 96 pocillos (por triplicado para cada concentración de fármaco) después de desechar el medio anterior. La concentración final del fármaco será 0, 0,03, 0,1, 0,3, 1, 3, 10, 30 o 100 pM. Las placas se cultivarán durante otros 7 días y a continuación se cuantificarán mediante un análisis MTS. Se prepara la solución MTS/PMS inmediatamente antes de su uso, se pipetean 20 pl de la mezcla en cada pocillo de la placa analítica de 96 pocillos que contiene 100 pl de medio de cultivo. (El volumen de reacción final es de 120 pl). Se incuba la placa durante 1-4 horas a 37°C en una atmósfera humidificada y 5% de CO2. Se registra la absorbancia a 490 nm utilizando el espectrofotómetro VictorX5 para microplacas.
Tabla 2

Modelos tumorales de xenoinjerto
Células A549 de cáncer de pulmón no microcítico humano, células MKN45 de cáncer gástrico humano y células EBC-1 de carcinoma escamoso de pulmón humano se expanden en cultivo, se recolectan y se inyectan por vía subcutánea en el flanco derecho de ratones lampiños BALB/c. El compuesto de prueba se prepara en un vehículo apropiado y se administra por sonda oral cuando se comprueban los tumores (6-10 días después del implante). La respuesta del tumor se determina mediante la medición del volumen del tumor realizada dos veces a la semana durante el curso del tratamiento. La inhibición del volumen tumoral (% de inhibición del crecimiento) se calcula comparando los grupos tratados con el grupo de referencia del vehículo. El peso corporal se toma como una medida general de toxicidad. El compuesto del ejemplo 1 demuestra una excelente actividad antitumoral en estos modelos. Por ejemplo, cuando se administra a 60 y 120 mg/kg (cada día x 24), el ejemplo 1 puede causar 76,6% y 96,7% de inhibición del crecimiento de tumores A549, respectivamente. Cuando se administra a 60 y 120 mg/kg (cada día x 22), el ejemplo 1 puede causar una inhibición del crecimiento de 38,9% y 71,9% de los tumores MKN45, respectivamente. Cuando se administra a 40 y 80 mg/kg (cada día x 17), el ejemplo 1 puede causar una inhibición del crecimiento de 56,5% y 100% de los tumores EBC-1, respectivamente.
La sobreexpresión de c-Met en tumores relacionados con c-Met y modelos de xenoinjerto es una característica común de muchos tumores humanos, incluidos los de pulmón, mama, colorrectal, gástrico, renal, pancreático, cabeza y cuello(12). Las mutaciones activadoras de c-Met en el dominio de cinasa están implicadas como la causa de varios tumores, como el carcinoma papilar hereditario de células renales, el carcinoma hepatocelular infantil y el cáncer gástrico(36). Los inhibidores de c-Met de Pfizer demostraron eficacia antitumoral en muchos tumores de xenoinjerto humanos, incluidos U87MG, GTL16, H441, Caki-1 y PC3<7>.
1. Christinsen, J. G., Burrows, J., y Salgia, R. Cáncer Letters 225: 1-26, 2005.
2. Birchmeier, C., Birchmeier, W., Gherardi, E., y Vande Woude, G. F. Nat. Rev. Mol. Cell Biol. 4: 915-925, 2003.
3. Di Renzo, M. F., Olivero, M., Martone, T. et al. Oncogene 19: 1547-1555, 2000.
4. Lee, J. H., Han, S. U., Cho, H. et al. Oncogene 19: 4947-4953, 2000.
5. Ma, P. ej. C., Kijima, T., Maulik, G. et al. Cáncer Res. 63: 6272-6281, 2003. 6. Park, W. S., Dong, S. M., Kim, S. Y. et al. Cáncer Res. 59: 307-310, 1999.
6. Schmidt, L., Duh, F. M., Chen, F., et al. Nat. Genet.16: 68-73, 1997.
7. Zou, H. Y., Li, Qiuhua., Lee, J. H., et al. Cancer Res. 67: 4408-4417, 2007.
Los compuestos de la presente invención se formulan preferiblemente como composiciones farmacéuticas administradas por una variedad de vías. De la manera más preferible, dichas composiciones son para administración oral. Dichas composiciones farmacéuticas y procedimientos para preparar los mismos son bien conocidos en la técnica. Véase, p. ej., REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro, et al., eds., 19a ed., Mack Publishing Co., 1995). Los compuestos de fórmula I son generalmente eficaces en un amplio intervalo de dosis.
Por ejemplo, las dosis diarias normalmente están comprendidas dentro del intervalo de aproximadamente 1 mg a aproximadamente 200 mg de dosis diaria total, preferiblemente de 1 mg a 150 mg de dosis diaria total, más preferiblemente de 1 mg a 50 mg de dosis diaria total. En algunos casos, las cantidades de dosis por debajo del límite inferior del intervalo mencionado anteriormente pueden ser más que adecuados, mientras que en otros casos se pueden emplear dosis aún mayores. El intervalo de dosificación anterior no pretende limitar el alcance de la invención de ninguna manera. Debe entenderse que la cantidad del compuesto realmente administrado la determinará un médico, a la luz de las circunstancias oportunas, incluida la afección a tratar, la vía de administración elegida, el compuesto o compuestos reales administrados, la edad, el peso y la respuesta de cada paciente y la gravedad de los síntomas del paciente.
Claims (34)
1. Un compuesto de fórmula I o una de sus sales farmacéuticamente aceptable:

en donde,
B no está o es, O, S, OCH2 o SCH2 ;
R1, R2, R3 y R4 son cada uno independientemente hidrógeno, halógeno, alquilo C1-6 sustituido o no sustituido, o carbamoilo, o
o R1 y R2 a la vez, o R3 y R4 a la vez, forman =O;
A es NoCH;
n es 0, 1, 2 o 3;
cada R5 es independientemente halógeno, o alquilo C1-6,
Z es NHR6 o de fórmula II:

en donde R18 y R19 son cada uno independientemente hidrógeno, alquilo C1-8 sustituido o no sustituido o arilo C6-8 sustituido o no sustituido;
R6 tiene la fórmula VI:
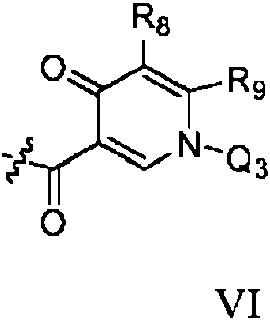
en donde:
Q3 es hidrógeno;
R8 es hidrógeno, o fenil sustituido con halógeno; y
R9 es hidrógeno.
2. El compuesto de la reivindicación 1, en donde R1, R2 , R3 and R4 son cada uno independientemente hidrógeno, halógeno, alquilo C1-4 sustituido o no sustituido or carbamoílo; o
R1 y R2 a la vez, o R3 y R4 a la vez, forman =O.
3. El compuesto de la reivindicación 1 o 2, en donde R1, R2, R3 y R4 son cada uno independientemente hidrógeno, F o metilo; o
R1 y R2 a la vez, o R3 y R4 a la vez, forman =O.
4. El compuesto de cualquiera de las reivindicaciones 1-3, en donde Ri, R2, R3 y R4 forman todos hidrógeno.
5. El compuesto de cualquiera de las reivindicaciones 1-3, en donde R1 y R2 a la vez, o R3 y R4 a la vez, forman =O.
6. El compuesto de cualquiera de las reivindicaciones precedentes, en donde n es 0 o 1.
7. El compuesto de cualquiera de las reivindicaciones precedentes, en donde R5 es halógeno o alquilo C1-4.
8. El compuesto de cualquiera de las reivindicaciones precedentes, en donde R5 es F o metilo.
9. El compuesto de cualquiera de las reivindicaciones precedentes, en donde B falta o es O.
10. El compuesto de cualquiera de las reivindicaciones precedentes, en donde A es CH.
11. El compuesto de cualquiera de las reivindicaciones 1-10, en donde Z tiene la fórmula II; R18 y R19 son cada uno independientemente hidrógeno, alquilo C1-4 sustituido o no sustituido, o arilo C6-8 sustituido o no sustituido.
12. El compuesto de cualquiera de las reivindicaciones 1-11, en donde Z tiene la fórmula II; R18 y R19 son cada uno independientemente hidrógeno, metilo o fenilo sustituido con halógeno.
13. El compuesto de cualquiera de las reivindicaciones 1-12, en donde Z tiene la fórmula II; R18 es metilo y R19 es fenilo sustitutido con F en la posición para.
14. El compuesto de la reivindicación 1, en donde el compuesto tiene la fórmula VIII:

en donde,
R1, R2 , R3 y R4 son cada uno independientemente hidrógeno, halógeno; o alquilo C1-3 sustituido o no sustituido; o R1 y R2 a la vez, o R3 y R4 a la vez, forman =O;
cada R5 es independientemente halógeno, o alquilo C1-3 sustituido o no sustituido;
n es 0 o 1 ;
Q3 es hidrógeno; y
R8 es hidrógeno o fenilo sustituido con halógeno; y
R9 es hidrógeno.
15. El compuesto de la reivindicación 14, en donde R1, R2, R3 y R4 son cada uno independientemente hidrógeno o halógeno; o R1 y R2 a la vez, o R3 y R4 a la vez, forman =O.
16. El compuesto de la reivindicación 14 o 15, en donde R1, R2 , R3 y R4 son cada uno independientemente hidrógeno o halógeno.
17. El compuesto de cualquiera de las reivindicaciones 14-16, en donde R1 y R2 son cada uno F; y R3 y R4 son cada uno hidrógeno.
18. El compuesto de cualquiera de las reivindicaciones 14-16, en donde R1 y R2 son cada uno hidrógeno; y R3 y R4 son cada uno F.
19. El compuesto de cualquiera de las reivindicaciones 14-16, en donde R1, R2 , R3 y R4 son cada uno hidrógeno.
20. El compuesto de la reivindicación 14 o 15, en donde R1 y R2 a la vez forman =O.
21. El compuesto de la reivindicación 14 o 15, en donde R3 y R4 a la vez forman =O.
22. El compuesto de cualquiera de las reivindicaciones 14-21, en donde R5 es halógeno.
23. El compuesto de cualquiera de las reivindicaciones 14-22, en donde R5 es F.
24. El compuesto de cualquiera de las reivindicaciones 14-23, en donde R8 es fenil sustituido con halógeno en la posición para.
25. El compuesto de la reivindicación 24, en donde R8 es fenil sustituido con F en la posición para.
26. El compuesto de la reivindicación 1, en donde el compuesto es
N-(4-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida; N-(3-fluoro-4-(3-oxo-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-5-4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(2-oxo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
8-(6-(2-(4-fluorofenilamino)-1-metil-6-oxo-1,6-dihidropirimidin-5-il)piridin-3-iloxi)-2H-pirido[3,2-b][1,4]oxazin-3(4H)-ona;
5-(5-(2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)piridin-2-il)-2-(4-fluorofenilamino)-3-metilpirimidin-4(3H)-ona;
N-(4-(3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
[4-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
[4-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
5-[5-(3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-piridin-2-il]-2-(4-fluor o-fenilamino)-3-metil-3H-pirimidin-4-ona;
5-[5-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-piridin-2-il]-2-(4-fluoro-fenilamino)-3-metil-3H-pirimidin-4- ona;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-3-fluorofenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]tiazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
5- [5-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-piridin-2-il]-2-(4-fluoro-fenilamino)-3-metil-3H-pirimidin-4- ona;
5- (4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico ácido [4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazm-8-iloxi)-3-fluoro-fenil]-amida del;
[4-(3,3-dimetil-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidropiridin-3-carboxílico;
[4-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
2-(4-fluoro-fenilamino)-3-metil-5-[5-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-piridin-2-il]-3H-pirimidin-4-ona;
[3-fluoro-4-(6,7,8,9-tetrahidro-5-tia-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
[3-fluoro-4-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amina del ácido 5-(4-fluoro-fenil)-4-oxo-1.4- dihidro-piridin-3-carboxílico;
2-(4-fluoro-fenilamino)-3-metil-5-[5-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-piridin-2-il]-3H-pirimidin-4-ona;
[4-(6,7,8,9-tetrahidro-5-oxa-1,9-diaza-benzociclohepten-4-iloxi)-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
N-(4-(3,4-dimetil-2,3,4,5-tetrahidropirido[3,2-b][1,4]oxazepin-9-iloxi)fenil )-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
[4-(3,3-difluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluoro-fenil]-amida del ácido 5-(4-fluoro-fenil)-4-oxo-1,4-dihidro-piridin-3-carboxílico;
N-(4-(2,2-difluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
N-(3-fluoro-4-(2-(hidroximetil)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida;
8-(2-fluoro-4-(5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamido)fenoxi)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-2- carboxamida;
N-(4-(2-(aziridin-1-ilmetil)-3,4-dihidro-2H-pirido[3,2-b][1,4]oxazin-8-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida; o
N-(3-fluoro-4-(3-(2-hidroxietil)-2,3-dihidro-1H-pirrolo[2,3-b]piridin-4-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3- carboxamida.
27. Un compuesto como se muestra a continuación o una de sus sales farmacéuticamente aceptables:
N-(4-(3',4'-dihidrospiro[ciclopropano-1,2'-pirido[3,2-b][1,4]oxazin]-8'-iloxi)-3-fluorofenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida; o
N-(3-fluoro-4-(2'-oxo-1',2'-dihidrospiro[ciclopropano-1,3'-pirrolo[2,3-b]piridin]-4'-iloxi)fenil)-5-(4-fluorofenil)-4-oxo-1,4-dihidropiridin-3-carboxamida.
28. Una composición farmacéutica que comprende un compuesto de cualquiera de las reivindicaciones precedentes y un excipiente farmacéuticamente aceptable.
29. La composición farmacéutica según la reivindicación 28, en donde, dicho compuesto en una relación en peso a dicho excipiente está comprendida en el intervalo entre 0,0001 y 10.
30. El compuesto de cualquiera de las reivindicaciones 1-27 o la composición de la reivindicación 28 o 29 para su uso como medicamento.
31. El compuesto de cualquiera de las reivindicaciones 1-27 o la composición de la reivindicación 28 o 29 para su uso en el tratamiento, prevención o retardo de la aparición o evolución en, el cáncer, metástasis de cáncer, enfermedad cardiovascular, un trastorno inmunológico o un trastorno ocularr.
32. El compuesto de cualquiera de las reivindicaciones 1-27 o la composición de la reivindicación 28 o 29 o para su uso como inhibidor de c-Met en el tratamiento de una enfermedad mediada por una proteína cinasa.
33. El compuesto para uso de la reivindicación 32, en donde dicha proteína cinasa es KDR, Tie-2, Flt3, FGFR3, AbI, Aurora A, c-Src, IGF-IR, ALK, c-MET, RON, PAK1, PAK2 o TAK1.
34. El compuesto para uso de la reivindicación 31, en donde el cáncer es un tumor sólido, un sarcoma, un fibrosarcoma, un osteoma, un melanoma, un retinoblastoma, un rabdomiosarcoma, un glioblastoma, un neuroblastoma, un teratocarcinoma, un tumor maligno hematopoyético, una ascitis maligna, cáncer de pulmón, cáncer de mama, cáncer colorrectal, cáncer renal, cáncer pancreático, cáncer de cabeza, cáncer de cuello, carcinoma hereditario de células renales papilares, carcinoma hepatocelular infantil o cáncer gástrico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2012077924 | 2012-06-29 | ||
| PCT/CN2013/078592 WO2014000713A1 (en) | 2012-06-29 | 2013-07-01 | NOVEL FUSED PYRIDINE DERIVATIVES USEFUL AS c-MET TYROSINE KINASE INHIBITORS |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2698511T3 true ES2698511T3 (es) | 2019-02-05 |
Family
ID=49782283
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13809182T Active ES2698511T3 (es) | 2012-06-29 | 2013-07-01 | Nuevos derivados de piridina condensados útiles como inhibidores de c-met tirosina cinasa |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US9617257B2 (es) |
| EP (1) | EP2867223B1 (es) |
| JP (1) | JP6059342B2 (es) |
| KR (3) | KR101726522B1 (es) |
| AU (1) | AU2013283993B2 (es) |
| BR (1) | BR112014032745B1 (es) |
| CA (1) | CA2878049C (es) |
| ES (1) | ES2698511T3 (es) |
| IN (1) | IN2015DN00372A (es) |
| RU (1) | RU2619130C2 (es) |
| SG (1) | SG11201408750VA (es) |
| TW (1) | TWI520962B (es) |
| WO (1) | WO2014000713A1 (es) |
| ZA (1) | ZA201500344B (es) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106279147A (zh) | 2015-05-21 | 2017-01-04 | 中国科学院上海药物研究所 | 一种吡啶并氮杂环化合物及其制备方法和用途 |
| AU2016297395B2 (en) * | 2015-07-20 | 2019-01-17 | Betta Pharmaceuticals Co., Ltd | Crystalline form of fused pyridine derivative's maleate and uses thereof |
| CN106924260B (zh) * | 2015-12-31 | 2018-05-25 | 北京浦润奥生物科技有限责任公司 | 化合物在制备用于治疗脑胶质瘤的药物中的用途 |
| IL287136B2 (en) | 2016-02-05 | 2023-09-01 | Denali Therapeutics Inc | Receptor inhibitors - interacting with protein kinase 1 |
| HUE058802T2 (hu) | 2016-12-09 | 2022-09-28 | Denali Therapeutics Inc | RIPK1 inhibitorokként alkalmas vegyületek |
| AU2018272986A1 (en) * | 2017-05-26 | 2019-12-12 | Ichnos Sciences SA | Novel inhibitors of MAP4K1 |
| WO2018233527A1 (zh) * | 2017-06-19 | 2018-12-27 | 上海和誉生物医药科技有限公司 | 一种具有csf1r抑制活性的氮杂芳基衍生物、其制备方法和应用 |
| CN109988108B (zh) * | 2017-12-29 | 2022-04-29 | 江苏豪森药业集团有限公司 | 一种卡博替尼的制备方法 |
| SG11202105872RA (en) * | 2018-12-14 | 2021-07-29 | Beta Pharma Inc | Organophosphorus-substituted compounds as c-met inhibitors and therapeutic uses thereof |
| BR112021018168B1 (pt) | 2019-03-21 | 2023-11-28 | Onxeo | Composição farmacêutica, combinação e kit compreendendo uma molécula dbait e um inibidor de quinase para o tratamento de câncer |
| CA3159348A1 (en) | 2019-11-08 | 2021-05-14 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| TWI812301B (zh) * | 2021-06-24 | 2023-08-11 | 南韓商Lg化學股份有限公司 | 作為ron抑制劑之新穎尿素衍生物化合物 |
| US11999750B2 (en) | 2022-01-12 | 2024-06-04 | Denali Therapeutics Inc. | Crystalline forms of (S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydropyrido [3,2-B][1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide |
| JP2025513455A (ja) | 2022-04-22 | 2025-04-24 | バーテックス ファーマシューティカルズ インコーポレイテッド | 疼痛の治療のためのヘテロアリール化合物 |
| EP4289427A1 (en) * | 2022-06-10 | 2023-12-13 | Anagenesis Biotechnologies | Dihydro[1,8]naphthyridin-7-one and pyrido[3,2-b][1,4]oxazin-3-one for use in treating cancer, and metastases in particular. |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4750701B2 (ja) * | 2003-07-07 | 2011-08-17 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | マロンアミド誘導体 |
| DE10357510A1 (de) * | 2003-12-09 | 2005-07-07 | Bayer Healthcare Ag | Heteroarylsubstituierte Benzole |
| US7173031B2 (en) * | 2004-06-28 | 2007-02-06 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| JP5368701B2 (ja) * | 2004-07-02 | 2013-12-18 | エクセリクシス、インコーポレイテッド | c−Metモジュレーター及び使用方法 |
| ES2438017T3 (es) * | 2004-07-30 | 2014-01-15 | Methylgene Inc. | Inhibidores de la señalización del receptor del VEGF y del receptor del HGF |
| JP2008521900A (ja) * | 2004-11-30 | 2008-06-26 | アムジエン・インコーポレーテツド | キノリン及びキナゾリン類似体並びにがん治療のための医薬としてのその使用 |
| JO2787B1 (en) * | 2005-04-27 | 2014-03-15 | امجين إنك, | Alternative amide derivatives and methods of use |
| DK1904504T3 (da) * | 2005-05-20 | 2014-06-23 | Methylgene Inc | Inhibitorer af vegf-receptor- og hgf-receptorsignalering |
| WO2008049855A2 (en) * | 2006-10-27 | 2008-05-02 | Glaxo Group Limited | 7-azaindole derivatives as c-met kinase inhibitors |
| US7960567B2 (en) * | 2007-05-02 | 2011-06-14 | Amgen Inc. | Compounds and methods useful for treating asthma and allergic inflammation |
| KR100961410B1 (ko) * | 2008-10-14 | 2010-06-09 | (주)네오팜 | 단백질 키나제 억제제로서 헤테로사이클릭 화합물 |
| AU2009303602B2 (en) | 2008-10-14 | 2012-06-14 | Sunshine Lake Pharma Co., Ltd. | Compounds and methods of use |
| CN102086211B (zh) * | 2009-12-08 | 2013-09-11 | 广东东阳光药业有限公司 | 作为蛋白激酶抑制剂的芳杂环化合物 |
| CA2805148C (en) * | 2010-07-14 | 2016-01-19 | Zhejiang Beta Pharma Inc. | Novel fused heterocyclic derivatives useful as c-met tyrosine kinase inhibitors |
-
2013
- 2013-06-29 TW TW102123415A patent/TWI520962B/zh not_active IP Right Cessation
- 2013-07-01 AU AU2013283993A patent/AU2013283993B2/en not_active Ceased
- 2013-07-01 KR KR1020157002238A patent/KR101726522B1/ko not_active Expired - Fee Related
- 2013-07-01 US US14/411,515 patent/US9617257B2/en not_active Expired - Fee Related
- 2013-07-01 KR KR1020177003331A patent/KR101770545B1/ko not_active Expired - Fee Related
- 2013-07-01 SG SG11201408750VA patent/SG11201408750VA/en unknown
- 2013-07-01 CA CA2878049A patent/CA2878049C/en not_active Expired - Fee Related
- 2013-07-01 BR BR112014032745-9A patent/BR112014032745B1/pt not_active IP Right Cessation
- 2013-07-01 JP JP2015518816A patent/JP6059342B2/ja not_active Expired - Fee Related
- 2013-07-01 WO PCT/CN2013/078592 patent/WO2014000713A1/en not_active Ceased
- 2013-07-01 RU RU2015102057A patent/RU2619130C2/ru active
- 2013-07-01 KR KR1020177003335A patent/KR101726555B1/ko not_active Expired - Fee Related
- 2013-07-01 EP EP13809182.2A patent/EP2867223B1/en not_active Not-in-force
- 2013-07-01 IN IN372DEN2015 patent/IN2015DN00372A/en unknown
- 2013-07-01 ES ES13809182T patent/ES2698511T3/es active Active
-
2015
- 2015-01-16 ZA ZA2015/00344A patent/ZA201500344B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| RU2015102057A (ru) | 2016-08-20 |
| SG11201408750VA (en) | 2015-01-29 |
| WO2014000713A1 (en) | 2014-01-03 |
| RU2619130C2 (ru) | 2017-05-12 |
| KR101726522B1 (ko) | 2017-04-12 |
| EP2867223B1 (en) | 2018-09-05 |
| JP2015521634A (ja) | 2015-07-30 |
| KR101726555B1 (ko) | 2017-04-12 |
| US9617257B2 (en) | 2017-04-11 |
| BR112014032745A2 (pt) | 2017-06-27 |
| AU2013283993B2 (en) | 2016-07-07 |
| US20150315210A1 (en) | 2015-11-05 |
| TW201418254A (zh) | 2014-05-16 |
| BR112014032745B1 (pt) | 2022-01-04 |
| CA2878049A1 (en) | 2014-01-03 |
| KR20150031320A (ko) | 2015-03-23 |
| ZA201500344B (en) | 2016-08-31 |
| EP2867223A4 (en) | 2016-01-06 |
| TWI520962B (zh) | 2016-02-11 |
| HK1207642A1 (en) | 2016-02-05 |
| EP2867223A1 (en) | 2015-05-06 |
| JP6059342B2 (ja) | 2017-01-11 |
| IN2015DN00372A (es) | 2015-06-12 |
| KR101770545B1 (ko) | 2017-08-22 |
| AU2013283993A1 (en) | 2015-02-05 |
| KR20170017014A (ko) | 2017-02-14 |
| CA2878049C (en) | 2016-12-13 |
| KR20170017015A (ko) | 2017-02-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2698511T3 (es) | Nuevos derivados de piridina condensados útiles como inhibidores de c-met tirosina cinasa | |
| ES2598530T3 (es) | Nuevos derivados heterocíclicos condensados útiles como inhibidores de la tirosina quinasa c-Met | |
| ES2870203T3 (es) | Compuesto derivado tricíclico, método para prepararlo y composición farmacéutica que comprende el mismo | |
| ES2779673T3 (es) | Compuestos de biarilamida como inhibidores de cinasas | |
| ES2797252T3 (es) | Dihidronaftiridinas y compuestos relacionados útiles como inhibidores de quinasas para el tratamiento de enfermedades proliferativas | |
| ES2864839T3 (es) | Macrociclos de diarilo quirales como moduladores de proteína quinasas | |
| ES2729243T3 (es) | Compuestos y composiciones como inhibidores de quinasa | |
| AU2019218186B2 (en) | Urea-substituted aromatic ring-linked dioxinoquinoline compounds, preparation method and uses thereof | |
| CA3083020A1 (en) | Halo-allylamine ssao/vap-1 inhibitor and use thereof | |
| ES2927529T3 (es) | Compuesto heterocíclico condensado | |
| TW202122382A (zh) | 乙內醯脲衍生物 | |
| CN104507930B (zh) | 作为c‑Met酪氨酸激酶抑制剂的新型稠合吡啶衍生物 | |
| KR102436669B1 (ko) | 우레아-치환된 방향족 고리-연결된 디옥시노퀴놀린 화합물 및 이의 제조방법 및 용도 | |
| HK1207642B (en) | Novel fused pyridine derivatives useful as c-met tyrosine kinase inhibitors | |
| WO2025000088A1 (en) | IMIDAZO[1,2-α]PYRIDINE COMPOUNDS FOR USE IN TREATING CANCER AND INFLAMMATORY DISEASES AND METHODS TO PREPARE SAID COMPOUNDS |