EP0659744A1 - Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen - Google Patents
Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen Download PDFInfo
- Publication number
- EP0659744A1 EP0659744A1 EP94120044A EP94120044A EP0659744A1 EP 0659744 A1 EP0659744 A1 EP 0659744A1 EP 94120044 A EP94120044 A EP 94120044A EP 94120044 A EP94120044 A EP 94120044A EP 0659744 A1 EP0659744 A1 EP 0659744A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- acid
- reaction
- benzoyl
- hours
- dicarboxylic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/18—Stationary reactors having moving elements inside
- B01J19/1868—Stationary reactors having moving elements inside resulting in a loop-type movement
- B01J19/1881—Stationary reactors having moving elements inside resulting in a loop-type movement externally, i.e. the mixture leaving the vessel and subsequently re-entering it
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/26—Nozzle-type reactors, i.e. the distribution of the initial reactants within the reactor is effected by their introduction or injection through nozzles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00017—Controlling the temperature
- B01J2208/00106—Controlling the temperature by indirect heat exchange
- B01J2208/00168—Controlling the temperature by indirect heat exchange with heat exchange elements outside the bed of solid particles
- B01J2208/00247—Reflux columns
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
Abstract
Description
- Die vorliegende Erfindung betrifft ein verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen der allgemeinen Formel I

in welcher
X = N-R, CH₂, S, O
Y = N-R, N-CH₃ oder = N-CH₂-CH₂-C₆H₅ und
Z= O, -OH bedeutet wobei in jedem Fall gilt R=H, Alkyl, Cycloalkyl, Aralkyl, Aryl
sowie deren Säureadditionssalze. - Heterocyclische Siebenringketone stellen wichtige Zwischenprodukte dar für die Synthese pharmakologisch aktiver Prinzipien. Hierzu zählen antiasthmatisch und antiallergisch wirksame Strukturen vom Typ des Azelastins (INN) und des Flezelastins (INN) (z.B. DE-OS 3634942 / Drugs Of The Future 12 (3), 283 (1987 / EP 488209), aber auch Strukturen zur Behandlung von Krankheiten des Zentralnervensystems, wie Parkinsonismus, Hyperprolactinämie und Schizophrenie oder zur Behandlung cardiovaskulärer Erkrankungen (z. B. DE-OS 3820775). Auch fungizid wirksame Substanzen mit heterocyclischer Siebenring-Teilstruktur sind bekannt.
Darüberhinaus zeigen Derivate vom Phospholipid-Typ mit heterocyclischer Siebenring-Teilstruktur in der Kopfkomponente antineoplastische Aktivität. - Einen umfassenden Überblick über die Dieckmann-Kondensation erhält man aus Org. Reactions, Vol. 15, 1-203 (1967) sowie der dort zitierten Literatur.
- Bei der Synthese von sieben und höhergliedrigen Ringen werden stets drei Kriterien herausgehoben:
- 1. Hohe Verdünnung zur Favorisierung der intramolekularen gegenüber der intermolekularen Reaktion
- 2. Lange Reaktionszeiten (langsames Zutropfen des Dicarbonsäureesters zur Favorisierung der intramolekularen Reaktion)
- 3. Hohe Überschüsse der verwendeten Base.
- Auffällig sind auch die stark variierenden Ausbeuten, sogar bei der Herstellung identischer Produkte durch unterschiedliche Arbeitsgruppen. Alternative Herstellverfahren wie
- Friedel-Crafts-Acylierung (J. Chem. Soc., Perkin Trans. I 1992, 445)
- Ringerweiterung mit Diazomethan (Bull. Chem. Soc., Japan, 31, 418 (1958); Coll. Czech. Chem. Commun. 51, 2034 (1986))
- Ozonolyse von Cyclohexenon gefolgt von einer reduktiven Aminocyclisierung ( Synth. Commun. 21(7), 881 (1991)) führten nicht zu befriedigenden Ausbeuten an den gewünschten Produkten, abgesehen von der technischen Realisierung der Ozonolyse bzw. der Umgang mit Diazomethan.
- Aufgabe der Erfindung ist die Ausarbeitung eines Verfahrens zur Herstellung von heterocyclischen Siebenringketonen in technischen Dimensionen basierend auf der bekannten Dieckmann-Kondensations-Reaktion.
- Die Erfindung betrifft die Herstellung von Salzen heterocyclischer Siebenringketone mittels Dieckmann-Kondensation.
Hierbei wird so vorgegangen, daß ein in bekannter Weise zugänglicher 1,8-Dicarbonsäureester in inertem Lösungsmittel mit starken Basen umgesetzt wird. Nach saurer Hydrolyse des entstehenden cyclischen β-Enolat-Carbonsäureesters wird der Ester ohne Isolierung verseift und thermisch decarboxyliert. Das gebildete heterocyclische Siebenringketon wird anschließend in Form seines Säureadditionssalzes aus dem Reaktionsgemisch abgetrennt und in reiner Form isoliert. - Überraschenderweise wurde gefunden, daß die Parameter lange Reaktionszeit, hohe Basenüberschüsse und starke Verdünnung nicht (wie bisher angenommen) in besonderer Weise die Höhe der Ausbeute beeinflussen. Bei Reaktionszeiten von 1-6, vorzugsweise 1-3 Stunden und Basenüberschüssen von 0-maximal 20% konnten technische Bedingungen gefunden werden, Verdünnung zu realisieren, ohne auf große Lösungsmittelmengen zurückzugreifen und somit die Raum-Zeit-Ausbeute erheblich zu steigern.
- Hierzu wurden zwei Methoden entwickelt.
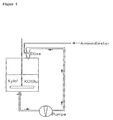
- 1. Die in inertem Lösungsmittel gelöste Base wird im Kreislauf geführt. In diesem Kreislauf befindet sich eine Strahldüse ähnlich dem Prinzip der Wasserstrahlpumpe. In den Seiteneinlaß der Düse wird der Dicarbonsäureester unverdünnt eindosiert (siehe Figur 1).
- 2. Der aufsteigende Dampf längs einer gepackten Füllkörperkolonne wird zur Verdünnung des in unverdünnter Form zudosierten Dicarbonsäureesters, der am Kopf dieser Kolonne dosiert wird, genutzt. Als praktischer Nebeneffekt wird der Diester hierbei bereits auf Reaktionstemperatur gebracht und kann bei Verwendung eines geeigneten Lösungsmittels auch längs der Kolonne absolutiert, bzw. von leichtflüchtigen Nebenprodukten, wie Restlösungsmitteln, befreit werden. Die Konzentration bei den beiden voranstehend beschriebenen Methoden bewegen sich zwischen 0,1 bis 1,5 vorzugsweise zwischen 0,3 bis 0,9 mol/l (Dicarbonsäureester zu anfänglich eingesetzter Lösungsmittelmenge).
Gleichzeitig zur Zugabe des Dicarbonsäureesters wird ein azeotropes Lösungsmittelgemisch am Kopf der Kolonne abgetrennt, und zwar soviel wie der 1-3fachen Volumenmenge an zudosiertem Dicarbonsäureester entspricht. - Als Lösungsmittel kommen in Betracht:
aliphatische Ether wie beispielsweise - Diethylether
- Tetrahydrofuran
- Dioxan
- Benzol
- Toluol
- o-Xylol
- m-Xylol
- p-Xylol
- Xylol (Isomerengemisch)
- Mesitylen
- Alkali- und Erdalkali-Hydride
- Alkali- und Erdalkali-Amide
- Alkali- und Erdalkali-Alkoholate
- Als Alkoholkomponenten für die 1,8-Dicarbonsäureester kommen in Frage
- Alkylalkohole
- Isoalkylalkohole
- Cycloalkylalkohole
- Zu einer im Kreislauf gepumpten Lösung von 123,4 g Kalium-tert.-butylat in 1,3 l siedendem Xylol werden unter Stickstoff im Seitenstrom 245,3 g 4-(2-Carbethoxyethyl-methyl-amino)buttersäure-ethylester binnen 2 Stunden gleichmäßig zudosiert und weitere 0,5 Stunden nachreagieren lassen. Hierbei werden 750 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 300 ml konzentrierter (37%iger) Salzsäure und 500 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 150 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird in 700 ml Isopropanol gelöst, heiß vom ungelösten Kaliumchlorid abfiltriert und bei 0 bis -5°C kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 141,6 g (86,6%) 1-Methyl-perhydroazepin-4-on-HCl.
Schmelzpunkt: 167°C (Zers.) - Zu einer im Kreislauf gepumpten Lösung von 123,4 g Kalium-tert.-butylat in 1,3 l siedendem Xylol werden unter Stickstoff im Seitenstrom 245,3 g 4-(2-Carbethoxyethyl-methyl-amino)buttersäure-ethylester gelöst in 900 ml Xylol binnen 3 Stunden gleichmäßig zudosiert und weitere 1,5 Stunden nachreagieren lassen. Hierbei werden 1500 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 300 ml konzentrierter (37%iger) Salzsäure und 500 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 150 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird in 700 ml Isopropanol gelöst, heiß vom ungelösten Kaliumchlorid abfiltriert und bei 0 bis -5°C kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 145,9 g (89,2%) 1-Methyl-perhydroazepin-4-on-HCl.
Schmelzpunkt: 163-164°C (Zers.) - Zu einer im Kreislauf gepumpten Lösung von 43,6 g Kalium-tert.-butylat in 1,65 l siedendem Xylol werden unter Stickstoff im Seitenstrom 100 g 4-(2-Carbethoxyethyl-benzyl-amino)buttersäure-ethylester gelöst in 780 ml Xylol binnen 3 Stunden gleichmäßig zudosiert und weitere 1,5 Stunden nachreagieren lassen. Hierbei werden 800 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 200 ml konzentrierter (37%iger) Salzsäure und 500 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 100 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird aus Isopropanol erschöpfend extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 60,3 g (80,8%) 1-Benzyl-perhydroazepin-4-on-HCl.
Schmelzpunkt: 191-193°C (Zers.) - Zu einer Lösung von 50,0 kg Kalium-tert.-butylat in 400 l siedendem Xylol werden unter Stickstoff am Kolonnenkopf 90,0 kg 4-(2-Carbethoxyethyl-methyl-amino)buttersäure-ethylester binnen 2-3 Stunden gleichmäßig zudosiert und weitere 1-1,5 Stunden nachreagieren lassen. Hierbei werden ca. 160 l Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 80 l konzentrierter (37%iger) Salzsäure und 100 kg gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 50 l halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird in 400 l Isopropanol gelöst, heiß vom ungelösten Kaliumchlorid abfiltriert und bei 0 - -5°C kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Man erhält 43,2 kg (72,0%) 1-Methyl-perhydroazepin-4-on-HCl.
Schmelzpunkt: 162-165°C (Zers.) - Zu einer Lösung von 61,7 g Kalium-tert.-butylat in 1,5 l siedendem Xylol werden unter Einleiten von Stickstoff am Kopf einer Füllkörperkolonne 168,8 g 4-(2-Carbethoxyethyl-(2-phenylethyl)-amino)buttersäure-ethylester (Gehalt: 94%) binnen 2-3 Stunden gleichmäßig zudosiert und weitere 1-1,5 Stunden nachreagieren lassen. Hierbei werden ca. 500 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 200 ml konzentrierter (37%iger) Salzsäure und 300 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 100 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird in 250 ml Isopropanol heiß suspendiert und in der Kälte kristallisiert. Das Kristallisat wird aus Isopropanol erschöpfend extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 107,7 g (89,7%) 1-(2-Phenylethyl)-perhydroazepin-4-on-HCl.
Schmelzpunkt: 196-198°C (Zers.) - Zu einer Lösung von 30,0 kg Kalium-tert.-butylat in 400 l siedendem Xylol werden unter Stickstoff am Kopf einer Füllkörperkolonne 84,5 kg 4-(2-Carbethoxyethyl-(2-phenylethyl)-amino)buttersäure-ethylester (Gehalt: 92,2%) binnen 2-3 Stunden gleichmäßig zudosiert und weitere 1-1,5 Stunden nachreagieren lassen. Hierbei werden ca. 150 l Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 60 l konzentrierter (37%iger) Salzsäure und 60 kg gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 30 l halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird aus Isopropanol erschöpfend extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichts-konstanz getrocknet. Man erhält 40,0 kg (67,8%) 1-(2-Phenylethyl)-perhydroazepin-4-on-HCl.
Schmelzpunkt: 196-198°C (Zers.)
Elementaranalyse: ber.: C66,26 H7,95 N5,52 gef.: C66,24 H7,94 N5,43 Gehalt (Cl-): 100,28% - Zu einer Lösung von 18,9 g Natriumboranat in 100 ml Wasser setzt man 100 ml 1 N Natronlauge zu. Bei einer Innentemperatur von 0 - 5°C tropft man eine Lösung von 163,6 g 1-Methyl-perhydroazepin-4-on-HCl in 100 ml Wasser zu. Man rührt 2 Stunden bei 0 - 5°C und 2 Stunden bei Raumtemperatur nach. Durch Zugabe von halb-konzentrierter Salzsäure wird der pH-Wert auf 2-3 eingestellt. Man engt im Vakuum zur Trockene ein, nimmt den Rückstand in 600 ml Isopropanol auf, trennt bei 60 - 75°C die anorganischen Salze ab und kristallisiert das Produkt in der Kälte aus. Das Produkt wird abgesaugt und im Vakuum bei erhöhter Temperatur bis zur Gewichtskonstanz ge-trocknet. Man erhält 149 g (90%) 1-Methyl-perhydroazepin-4-ol-HCl.
Schmelzpunkt: 156-158°C. - Zu einer Suspension von 5,0 kg Natriumhydrid (80%ig in Weißöl) in 200 l siedendem Xylol werden unter Stickstoff 34,0 kg 4-(2-Carbethoxy-ethyl-methyl-amino)buttersäure-ethylester gelöst in 400 l Xylol binnen 2-3 Stunden gleichmäßig zudosiert und weitere 0,5 Stunden nachreagieren lassen. Hierbei werden ca. 400 l Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 35 l konzentrierter (37%iger) Salzsäure und 60 kg gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 30 l halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird in 100 l Isopropanol gelöst, heiß vom ungelösten Kaliumchlorid abfiltriert und bei 0 bis -5°C kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Man erhält 11,7 kg (52,0%) 1-Methyl-perhydro-azepin-4-on-HCl.
Schmelzpunkt: 162-164°C. - Zu einer Lösung von 39,0 g Natriumhydrid (80%ig in Weißöl) und 75 ml Ethanol in 1,3 l siedendem Xylol werden unter Einleiten von Stickstoff 245,3 g 4-(2-Carbethoxyethyl-methylamino)buttersäure-ethylester binnen 1,75 Stunden gleichmäßig zudosiert und weitere 0,5 Stunden nachreagieren lassen. Hierbei werden ca. 700 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 80-100°C durch rasche Zugabe in ein Gemisch aus 300 ml konzentrierter (37%iger) Salzsäure und 500 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 150 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird aus 600 ml Isopropanol heiß extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 44,3 g (27,1%) 1-Methyl-perhydroazepin-4-on-HCl.
Schmelzpunkt: 159-161°C (Zers.) - Zu einer Lösung von 61,7 g Kalium-tert.-butylat in 1,5 l siedendem Xylol werden unter Einleiten von Stickstoff am Kopf einer Füllkörperkolonne 181,8 g 4-(2-Carbethoxyethyl-(2-phenylethyl)-amino)buttersäure-n-butylester (Gehalt: 93%) binnen 2-3 Stunden gleichmäßig zudosiert und weitere 1-1,5 Stunden nachreagieren lassen. Hierbei werden ca. 500 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 200 ml konzentrierter (37%iger) Salzsäure und 300 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 100 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird in 250 ml Isopropanol heiß suspendiert und in der Kälte kristallisiert. Das Kristallisat wird aus Isopropanol erschöpfend extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Man erhält 95,4 g (80,8%) 1-(2-Phenylethyl)-perhydroazepin-4-on-HCl.
Schmelzpunkt: 196-197°C (Zers.) - Zu einer Lösung von 61,7 g Kalium-tert.-butylat in 1,5 l siedendem Xylol werden unter Einleiten von Stickstoff am Kopf einer Füllkörperkolonne 153,7 g 4-(2-Carbometh-oxyethyl-(2-phenylethyl)-amino)buttersäure-methylester (Gehalt: 96%) binnen 2-3 Stunden gleichmäßig zudosiert, wobei sich ein schwer rührbarer Niederschlag bildet, der sich bei der Nachreaktionszeit vollständig auflöst und weitere 1-1,5 Stunden nachreagieren lassen. Hierbei werden ca. 500 ml Destillat abgenommen.
- Das Reaktionsgemisch wird bei 50-80°C durch rasche Zugabe in ein Gemisch aus 200 ml konzentrierter (37%iger) Salzsäure und 300 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 100 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird aus Isopropanol erschöpfend extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 105,4 g (86,5%) 1-(2-Phenylethyl)-perhydroazepin-4-on-HCl.
Schmelzpunkt: 196-198°C (Zers.) - Zu einer Lösung von 123,4 g Kalium-tert.-butylat in 1,3 l siedendem Xylol werden unter Einleiten von Stickstoff 245,3 g 4-(2-Carbethoxyethyl-methylamino)buttersäure-ethylester binnen 3 Minuten zugesetzt, wobei rasch 300 ml Lösungsmittel abdestillieren, und weitere 0,5 Stunden nachreagieren lassen. Hierbei werden weitere 350 ml Destillat abgenommen. Das Reaktionsgemisch wird bei 80-100°C durch rasche Zugabe in ein Gemisch aus 300 ml konzentrierter (37%iger) Salzsäure und 500 g gestückeltem Eis hydrolysiert. Die abgetrennte organische Phase wird noch zweimal mit je 150 ml halbkonzentrierter Salzsäure nachgewaschen. Die vereinten wäßrigen Extrakte werden 2 Stunden zum kräftigen Rückfluß erhitzt und anschließend im Vakuum zur Trockene eingeengt. Der Rückstand wird aus 600 ml Isopropanol heiß extrahiert und kristallisiert. Der abfiltrierte Rückstand wird bei erhöhter Temperatur im Vakuum bis zur Gewichtskonstanz getrocknet. Durch Einengen der Mutterlauge auf ein Volumen von ca. 100 ml läßt sich eine Zweitfraktion gewinnen. Man erhält 78,3 g (47,9%) 1-Methyl-perhydroazepin-4-on-HCl.
Schmelzpunkt: 164-165°C (Zers.)
Die vorliegende Erfindung wird durch nachfolgende Beispiele näher erläutert.
Claims (4)
- Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen der allgemeinen Forrmel I

X = N-R, CH₂, S, O
Y = N-R, N-CH₃ oder = N-CH₂-CH₂-C₆H₅ und
Z= O, -OH bedeutet
wobei in jedem Fall gilt R=H, Alkyl, Cycloalkyl, Aralkyl, Aryl sowie deren Salze mittels Dieckmann-Kondensation unter Verwendung starker Basen in inerten Lösungsmitteln gekennzeichnet durch folgende Merkmale:a) daß man einen Überschuß bis zu maximal 20% an starken Basen verwendetb) daß man die Kondensationsreaktion bei Reaktionszeiten von 1 bis 6, vorzugsweise 1-3 Stunden durchführtc) daß man mit Konzentrationen von 0,1 bis 1,5 vorzugsweise 0,3 bis 0,9 mol/l des zu kondensierenden, unverdünnt zulaufenden, Dicarbonsäureesters pro Liter Lösungsmittel arbeitetd) daß man unter Vermeidung der üblichen Verdünnungstechnik entwederdie Reaktion in einer geschlossenen Vorrichtung durchführt, wobei die gelöste Base im Kreislauf geführt und der unverdünnte Dicarbonsäureester in diesen Kreislauf unter Verwendung einer Strahldüse zudosiert wird oder daß das aufsteigende, entstandenen Kondensat gleichzeitig in einer Füllkörperkolonne zur Verdünnung und Erwärmung des unverdünnt am Kopf der Kolonne zudosierten Dicarbonsäureesters dient - Herstellung und Isolierung von Hexahydroazepinolen sowie deren Salze durch Reduktion der Hexahydroazepinone nach Anspruch 1 mittels komplexer Hydride oder Wasserstoff am hydrogenolytisch aktiven Kontakt in Wasser oder Alkoholen.
- Verfahren zur Herstellung von Azelastin-Hydrochlorid dadurch gekennzeichnet, daß man die Verbindung I, worin X= CH₂, Y=N-CH₃ und Z= O bedeutet, in Form ihres Säureadditionssalzes mit Salzsäurea) in organischer Lösung umsetzt mit Benzoylhydrazin und die Zwischenverbindung ohne Isolierung zu einem Benzoyl-azepinyl-hydrazin reduziert undb) diese Verbindung nach saurer Abspaltung des Benzoylrestes in Wasser oder organischem Lösungsmittel mit 2-(4-Chlorphenacetyl)benzoesäure in Azelastin überführt und in Form eines Säureadditionssalzes mit Säure isoliert.
- Verfahren zur Herstellung von Flezelastin-Hydrochlorid dadurch gekennzeichnet, daß man die Verbindung I worin X=CH₂, Y=N-CH₂-CH₂ C₆H₅ und Z=O bedeutet, in Form ihres Säureaddionssalzes mit Säurea) in organischer Lösung umsetzt mit Benzoylhydrazin und die Zwischenverbindung ohne Isolierung zu einem Benzoyl-azepinyl-hydrazin reduziert.b)diese Verbindung nach saurer Abspaltung des Benzoylrestes in Wasser oder organischen Lösungsmitteln mit 2-(4-Fluorphenacetyl)benzoesäure in Flezelastin überführt und in Form eines Säureadditionssalzes mit Säure isoliert.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4343409 | 1993-12-18 | ||
| DE19934343409 DE4343409C2 (de) | 1993-12-18 | 1993-12-18 | Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0659744A1 true EP0659744A1 (de) | 1995-06-28 |
| EP0659744B1 EP0659744B1 (de) | 2001-02-28 |
Family
ID=6505486
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP94120044A Expired - Lifetime EP0659744B1 (de) | 1993-12-18 | 1994-12-17 | Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5760221A (de) |
| EP (1) | EP0659744B1 (de) |
| JP (1) | JP2994220B2 (de) |
| AT (1) | ATE199374T1 (de) |
| CA (1) | CA2138308C (de) |
| DE (2) | DE4345224C2 (de) |
| DK (1) | DK0659744T3 (de) |
| ES (1) | ES2155843T3 (de) |
| GR (1) | GR3035867T3 (de) |
| PT (1) | PT659744E (de) |
| SG (1) | SG46986A1 (de) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100421282B1 (ko) * | 2001-08-22 | 2004-03-09 | 부광약품 주식회사 | 1-메틸헥사히드로-4-아제피논 염산염의 제조방법 |
| KR100426534B1 (ko) * | 2001-09-03 | 2004-04-28 | 한올제약주식회사 | 아젤라스틴의 개선된 합성방법 |
| DE10201689A1 (de) * | 2002-01-17 | 2003-07-31 | Merck Patent Gmbh | Kontinuierliches Verfahren zur Hydrolyse einer lösungsmittelhaltigen organischen Verbindung |
| CN114507184B (zh) * | 2020-11-17 | 2024-02-13 | 好医生药业集团有限公司 | 1-甲基六氢氮杂卓-4-酮盐酸盐的合成方法及其应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3813384A (en) * | 1972-01-17 | 1974-05-28 | Asta Werke Ag Chem Fab | Basically substituted benzyl phthalazone derivatives,acid salts thereof and process for the production thereof |
| DE2206385C2 (de) * | 1972-02-10 | 1983-04-21 | Dr. Karl Thomae Gmbh, 7950 Biberach | 2-Amino-4,5,7,8-tetrahydro-6H-oxazolo-[4,5-d]azepinderivate |
| DE3530793A1 (de) * | 1984-09-14 | 1986-03-27 | Asta-Werke Ag Chemische Fabrik, 4800 Bielefeld | Substituierte benzylphthalazinon-derivate |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH572914A5 (de) * | 1971-01-22 | 1976-02-27 | Asta Werke Ag Chem Fab | |
| DE3634942A1 (de) * | 1985-11-11 | 1987-05-14 | Asta Werke Ag Chem Fab | Neue 4-benzyl-1-(2h)-phthalazinon-derivate |
-
1993
- 1993-12-18 DE DE4345224A patent/DE4345224C2/de not_active Expired - Lifetime
-
1994
- 1994-12-15 JP JP6312189A patent/JP2994220B2/ja not_active Expired - Lifetime
- 1994-12-16 CA CA002138308A patent/CA2138308C/en not_active Expired - Lifetime
- 1994-12-17 AT AT94120044T patent/ATE199374T1/de active
- 1994-12-17 ES ES94120044T patent/ES2155843T3/es not_active Expired - Lifetime
- 1994-12-17 SG SG1996001013A patent/SG46986A1/en unknown
- 1994-12-17 EP EP94120044A patent/EP0659744B1/de not_active Expired - Lifetime
- 1994-12-17 DE DE59409665T patent/DE59409665D1/de not_active Expired - Lifetime
- 1994-12-17 DK DK94120044T patent/DK0659744T3/da active
- 1994-12-17 PT PT94120044T patent/PT659744E/pt unknown
- 1994-12-19 US US08/359,457 patent/US5760221A/en not_active Expired - Lifetime
-
2001
- 2001-05-15 GR GR20010400720T patent/GR3035867T3/el unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3813384A (en) * | 1972-01-17 | 1974-05-28 | Asta Werke Ag Chem Fab | Basically substituted benzyl phthalazone derivatives,acid salts thereof and process for the production thereof |
| DE2206385C2 (de) * | 1972-02-10 | 1983-04-21 | Dr. Karl Thomae Gmbh, 7950 Biberach | 2-Amino-4,5,7,8-tetrahydro-6H-oxazolo-[4,5-d]azepinderivate |
| DE3530793A1 (de) * | 1984-09-14 | 1986-03-27 | Asta-Werke Ag Chemische Fabrik, 4800 Bielefeld | Substituierte benzylphthalazinon-derivate |
Non-Patent Citations (3)
| Title |
|---|
| JOHN P. SCHAEFFER: "The Dieckmann condensation", ORGANIC REACTIONS, vol. 15, 1967, NEW YORK, pages 1 - 203 * |
| SHIRO MOROSAWA: "Studies on Seven-membered Heterocyclic compounds containing Nitrogen. II....", BULLETIN OF THE CHEMICAL SOCIETY OF JAPAN, vol. 31, no. 4, May 1958 (1958-05-01), JAPAN, pages 418 - 422 * |
| ZDENEK POLIVKA ET AL.: "Heterocyclic ethers derived from 6,11-dihydrodibenzo[b,c]thiepin-11-ols and ............", COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, vol. 51, no. 9, 1986, PRAGUE, pages 2034 - 2048 * |
Also Published As
| Publication number | Publication date |
|---|---|
| DE4345224C2 (de) | 1999-07-01 |
| JP2994220B2 (ja) | 1999-12-27 |
| DE59409665D1 (de) | 2001-04-05 |
| CA2138308C (en) | 2002-11-19 |
| DE4345224A1 (de) | 1995-06-22 |
| US5760221A (en) | 1998-06-02 |
| EP0659744B1 (de) | 2001-02-28 |
| SG46986A1 (en) | 1998-03-20 |
| DK0659744T3 (da) | 2001-05-14 |
| JPH0812656A (ja) | 1996-01-16 |
| CA2138308A1 (en) | 1995-06-19 |
| ES2155843T3 (es) | 2001-06-01 |
| PT659744E (pt) | 2001-08-30 |
| ATE199374T1 (de) | 2001-03-15 |
| GR3035867T3 (en) | 2001-08-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE2611690A1 (de) | Cyclische sulfonyloxyimide | |
| DE602005003472T2 (de) | Verfahren zur herstellung von 2-oxo-1-pyrrolidinderivaten durch intramolekulare allylierung | |
| DE4102103A1 (de) | Substituierte benzoxazepine und benzthiazepine, verfahren zu ihrer herstellung und ihre verwendung in arzneimitteln | |
| EP0659744B1 (de) | Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen | |
| EP0057873A1 (de) | Verfahren zur Herstellung von Dimethylsuccinylosuccinat, dessen Dinatriumsalz, Dianilinodihydroterephthalsäuren, deren Dimethylestern und Salzen sowie von Dianilinoterephthalsäuren, deren Dimethylestern und Salzen | |
| DE10201725A1 (de) | Verfahren zur Herstellung und Reinigung von 1,7'-Dimethyl-2'-propyl-2,5'-bi-1H-benzimidazol | |
| DE4343409C2 (de) | Verbessertes Verfahren zur Herstellung von Hexahydroazepinonen und Hexahydroazepinolen | |
| DE3026094C2 (de) | Verfahren zur Herstellung von Cyclopropancarbonsäureamiden | |
| DE1116221B (de) | Verfahren zur Herstellung von ªú¼ª-Pentamethylenbutyrolacton bzw. Salzen der 3,3-Pentamethylen-4-hydroxybuttersaeure | |
| DE2220256C3 (de) | N-(o- bzw. p-nitrobenzoyl)-sulfoximine, verfahren zu ihrer herstellung und verwendung derselben | |
| EP0765878A1 (de) | Heterocyclische Aryl-, Alkyl- und Cycloalkylessigsäureamide als Arzneimittel | |
| CH653996A5 (de) | Tryptaminderivate und verfahren zu deren herstellung. | |
| DE3300522C2 (de) | ||
| EP0003105B1 (de) | Verfahren zur Anilidierung von Carbonsäureestern | |
| DE2653251A1 (de) | Azabicyclo eckige klammer auf 3.1.o eckige klammer zu hexan-derivate, ihre herstellung und verwendung | |
| DE3815046A1 (de) | 3-chlor-2-methylphenethylaminoderivate | |
| EP4301740A1 (de) | Verfahren zur herstellung von alkyl-4-oxotetrahydrofuran-2-carboxylat | |
| DE2720915C2 (de) | N↓1↓-Acyl-2-hydroxy-1,3-diaminopropane und Arzneimittel mit diesen Verbindungen als Wirkstoff | |
| DE880444C (de) | Verfahren zur Herstellung von Chinolinabkoemmlingen | |
| AT250338B (de) | Verfahren zur Herstellung neuer, basischer Derivate von substituierten Benzofuran-2-carbonsäuren und deren Salzen | |
| DE4429978A1 (de) | Verfahren zur Herstellung von Chinazolin-2,4-dionen | |
| AT401931B (de) | Verfahren zur herstellung von (s,s)-(n-(1- ethoxycarbonyl-3-oxo-3-phenylpropyl)-alanin)- (phenylmethyl)ester | |
| CH646167A5 (de) | Verfahren zur herstellung von apovincaminsaeureestern. | |
| DE3231816A1 (de) | Verfahren zur herstellung von cyclopropancarbonsaeure-derivaten | |
| CH368182A (de) | Verfahren zur Herstellung von N-Aminoalkylderivaten von Azepinen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI LU MC NL PT SE |
|
| 17P | Request for examination filed |
Effective date: 19951130 |
|
| 17Q | First examination report despatched |
Effective date: 19960209 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI LU MC NL PT SE |
|
| REF | Corresponds to: |
Ref document number: 199374 Country of ref document: AT Date of ref document: 20010315 Kind code of ref document: T |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: BOVARD AG PATENTANWAELTE |
|
| ITF | It: translation for a ep patent filed |
Owner name: BARZANO' E ZANARDO ROMA S.P.A. |
|
| REF | Corresponds to: |
Ref document number: 59409665 Country of ref document: DE Date of ref document: 20010405 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D Free format text: GERMAN |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 20010503 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2155843 Country of ref document: ES Kind code of ref document: T3 |
|
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PFA Free format text: ASTA MEDICA AKTIENGESELLSCHAFT TRANSFER- VIATRIS GMBH & CO. KG |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: PD4A Free format text: VIATRIS GMBH & CO. KG DE Effective date: 20020909 Ref country code: PT Ref legal event code: PD4A Free format text: ASTA MEDICA GMBH DE Effective date: 20020909 Ref country code: PT Ref legal event code: PC4A Free format text: ASTA MEDICA HEALTH PRODUCTS GMBH & CO. KG DE Effective date: 20020909 |
|
| NLS | Nl: assignments of ep-patents |
Owner name: VIATRIS GMBH & CO. KG |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: TP Ref country code: FR Ref legal event code: CD |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: D3 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: TP Ref country code: FR Ref legal event code: CD Ref country code: FR Ref legal event code: CA |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: GC |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: DG |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PFA Owner name: VIATRIS GMBH & CO. KG Free format text: VIATRIS GMBH & CO. KG#WEISMUELLERSTRASSE 45#60314 FRANKFURT AM MAIN (DE) -TRANSFER TO- VIATRIS GMBH & CO. KG#BENZSTRASSE 1#61352 BAD HOMBURG (DE) |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: TE4A Free format text: VIATRIS GMBH & CO. KG DE Effective date: 20050506 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: CA |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PFA Owner name: MEDA PHARMA GMBH & CO. KG Free format text: VIATRIS GMBH & CO. KG#BENZSTRASSE 1#61352 BAD HOMBURG (DE) -TRANSFER TO- MEDA PHARMA GMBH & CO. KG#BENZSTRASSE 1#61352 BAD HOMBURG (DE) |
|
| NLT1 | Nl: modifications of names registered in virtue of documents presented to the patent office pursuant to art. 16 a, paragraph 1 |
Owner name: MEDA PHARMA GMBH & CO. KG |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: PD4A Owner name: MEDA PHARMA GMBH & CO. KG, DE Effective date: 20070604 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: CD |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PFA Owner name: MEDA PHARMA GMBH & CO. KG Free format text: MEDA PHARMA GMBH & CO. KG#BENZSTRASSE 1#61352 BAD HOMBURG (DE) -TRANSFER TO- MEDA PHARMA GMBH & CO. KG#BENZSTRASSE 1#61352 BAD HOMBURG (DE) |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: MC Payment date: 20131212 Year of fee payment: 20 Ref country code: DK Payment date: 20131219 Year of fee payment: 20 Ref country code: AT Payment date: 20131211 Year of fee payment: 20 Ref country code: SE Payment date: 20131219 Year of fee payment: 20 Ref country code: IE Payment date: 20131230 Year of fee payment: 20 Ref country code: PT Payment date: 20130618 Year of fee payment: 20 Ref country code: CH Payment date: 20131219 Year of fee payment: 20 Ref country code: DE Payment date: 20131220 Year of fee payment: 20 Ref country code: GB Payment date: 20131219 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 20131231 Year of fee payment: 20 Ref country code: NL Payment date: 20131219 Year of fee payment: 20 Ref country code: IT Payment date: 20131217 Year of fee payment: 20 Ref country code: GR Payment date: 20131212 Year of fee payment: 20 Ref country code: ES Payment date: 20131226 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20131219 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20131220 Year of fee payment: 20 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R071 Ref document number: 59409665 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R071 Ref document number: 59409665 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: V4 Effective date: 20141217 Ref country code: PT Ref legal event code: MM4A Free format text: MAXIMUM VALIDITY LIMIT REACHED Effective date: 20141217 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EUP Effective date: 20141217 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: PE20 Expiry date: 20141216 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MK9A |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20141224 Ref country code: GB Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20141216 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: EUG |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK07 Ref document number: 199374 Country of ref document: AT Kind code of ref document: T Effective date: 20141217 |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: MA Ref document number: 20010400720 Country of ref document: GR Effective date: 20141218 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20141217 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20150826 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20141218 |