-
Gebiet der Erfindung
-
Die
vorliegende Erfindung betrifft Perfluoralkyl-modifizierte Polyethylenimine,
insbesondere wasserlösliche
Polyperfluoralkyl(allyloxy/iodpropyloxy)- oder Polyperfluoralkylfluorallyl-substituierte
Polyethylenimine, welche als ölabweisende
Mittel für
Substrate, wie Textilien und Papier, und als Schaumstabilisatoren
in alkoholbeständigen
wässrigen.
Brandbekämpfungsschaumzubereitungen
(AR-AFFF) nützlich sind.
-
Hintergrund
der Erfindung
-
Wässrige Feuer-/Brandbekämpfungsschaum-Zubereitungen
(Aqueous Fire-Fighting
Foam (AFFF) formulations) enthalten wasserlösliche Fluor-Tenside zusammen
mit Kohlenwasserstoff-Tensiden. Sie sind zum Löschen von Feuern nichtpolarer
Lösungsmittel
wirksam. Wenn eine AFFF-Zubereitung mit einem brennenden Kohlenwasserstofftreibmittel
in Kontakt kommt, läuft
das Wasser, welches sowohl Fluor- als auch Kohlenwasserstoff-Tenside
enthält,
aus dem Schaum aus und bildet oben auf dem brennenden Treibstoff
einen dünnen
Film. Dieser Film sinkt nicht ab, allerdings verbreitet er sich
spontan über
die Oberfläche
des brennenden Treibstoffs aufgrund seiner geringen Oberflächenenergie
(< 18 Dynes/cm,
was unter der von Heptan liegt). Dort wirkt er als ein Dampfunterdrücker und
löscht
in Kombination mit dem wässrigen
Schaum das Feuer und verhindert das erneute Entzünden des Treibstoffs. Es ist
bei dieser Anwendung wichtig, dass der Schaum ein langes Schaumleben
auf dem heißen
Treibstoff aufweist; andernfalls kann der Treibstoff sich erneut
entzünden,
eine Erscheinung, welche Rückbrand
(burnback) genannt wird. Ein langes Schaumleben, welches Rückbrand-
bzw. Burnback-Beständigkeit
verleiht, wird durch einen Schaum erreicht, welcher "nass", d.h. hydratisiert,
ist und aus dem Wasser auf die Oberfläche herunter austreten und
die Versiegelung wiederherstellen kann. Bei einem nichtpolaren Treibstoff,
wie Benzin, ist diese Aufgabe einfach, da Wasser und die wasserlöslichen
Tenside in dem Treibstoff nicht löslich sind.
-
Diese
Aufgabe ist deutlich schwieriger bei polaren Treibstoffen, wie Isopropanol
und Aceton. Neben den in konventionellen AFFF-Zubereitungen gefundenen
Fluor- und Kohlenwasserstoff-Tensiden enthält eine alkoholbeständige (Alcohol-Resistant; = AR)
AFFF-Zubereitung einen wasserlöslichen,
allerdings in polaren Lösungsmitteln
unlöslichen
fluorchemischen – ebenso
als "alkophob" bezeichneten – Schaumstabilisator
(wie in der vorliegenden Erfindung beschrieben) zusammen mit einem
Polysaccharid, wie Xanthangummi. Wenn diese zusätzlichen Materialien mit einem
brennenden polaren Treibstofffeuer in Kontakt kommen, fallen sie
aus und bilden eine Membran, welche den Schaum vor dem Auflösen in dem
polaren Lösungsmittel
schützt.
Diese Membran bildet eine Dampfbarriere, welche das Feuer löscht und
ein erneutes Entzünden
des Treibstoffs verhindert, wobei gleichzeitig der Schaum hydratisiert
bleibt. Polysaccharide und/oder hochmolekulargewichtige synthetische
Polymere können
in AR-AFFF-Zubereitungen ohne einen fluorchemischen Schaumstabilisator verwendet
werden und ungefähr
dieselbe Wirksamkeit vermitteln. Das Problem mit einem Schaumkonzentrat, welches
nur Polysaccharide und/oder hochmolekulargewichtige synthetische
Polymere enthält,
liegt in dessen hoher Viskosität
und darin, dass sich das Konzentrat thixotrop verhält. Die
Verwendung eines Schaumkonzentrats mit hoher Viskosität ist schwierig,
da ein Pumpen durch eine Feuerspritze schwer, wenn nicht sogar unmöglich ist.
AR-AFFF-Zubereitungen, welche fluorchemische Schaumstabilisatoren
enthalten, erfordern wesentlich geringere Mengen an Polysacchariden
und/oder hochmolekulargewichtigen synthetischen Polymeren und vermindern
auf diese Weise die Viskosität
des Schaumkonzentrats. Zusätzlich
neigen Schaumkonzentrate, welche fluorchemische Schaumstabilisatoren
in AR-AFFF-Zubereitungen
enthalten, zu einem Newtonschen Verhalten.
-
Feuerbekämpfende
Schaumstabilisatoren, welche mindestens eine Perfluoralkylgruppe
und wassersolubilisierende Funktionalitäten, wie Carboxy- und Amidogruppen
enthalten, werden in den US-Patenten 4,460,480 und 5,218,021 beschrieben.
Die französische
Patentanmeldung 2,637,506 A beschreibt ein alkophobes und oleophobes
Feuerlöschschaumkonzentrat,
welches ein Polyhydroxypolyamin, das mindestens ein quaternäres N-Atom
enthält,
und/oder ein Polysaccharid, welches an hochfluorierte C4-C20-Alkylgruppen chemisch gebunden ist, aufweist,
anstelle das Fluortensid getrennt und das Polysaccharid oder andere
alkophobe Mittel in der konzentrierten Mischung zu enthalten.
-
Alkophobe
Feuerbekämpfungsschaumstabilisatoren,
welche mindestens eine Perfluoralkylgruppe zusammen mit polyquaternären Amino-
und Carboxyfunktionalitäten
enthalten, werden in den internationalen Anmeldungen WO 90/02110
A1 und WO 90/03966 A1 zusammen mit den Publikationen von 5. Szönyi in "Fire Safety Journal", 16, Seiten 353-365
(1990) und in "Progress
in Colloid & Polymer
Science", 81, 136-139
(1990) beschrieben.
-
Da
quaternäre
Ammoniumgruppen eine Inkompatibilität mit in Feuerbekämpfungszubereitungen
verwendeten anionischen Tensiden hervorrufen, wurden weitere Verbesserungen
in der WO 94/18245 beschrieben. Diese Referenz lehrt Verbindungen,
welche eine Kombination von mindestens zwei Perfluoralkylgruppen, sich
von anderen quaternären
Ammoniumgruppen unterscheidende Aminogruppen, Carbonsäuregruppen
und andere wassersolubilisierende Gruppen, welche an Aminogruppen
gebunden sind, enthalten. Beispielsweise offenbart das US-Patent 4,606,973
Aminoethylmetharcrylat-Acrylsäure-Copolymere,
in denen die Aminogruppen mit Perfluoralkylcarbonsäuren umgesetzt
worden sind.
-
S.
Szönyi,
Com. Journ. Com. Esp. Deterg., 22, Seiten 297-304 (1991) beschreibt
einen kommerziellen alkophoben Schaumstabilisator des Standes der
Technik als eine perfluoralkylierte Polyaminosäure.
-
Ein
besonders praktischer Weg, für
die Schaumstabilisierung essentielle Aminogruppen mit Perfluoralkyl-substituierten
Verbindungen zu kombinieren, ist die Verwendung von leicht erhältlichem
Polyethylenimin. Die Nützlichkeit
von Polyethyleniminen in Schaumstabilisatorzubereitungen für Feuer
polarer Lösungsmittel
ist sei einiger Zeit bekannt. Beispielsweise beschreibt die japanische
Patentanmeldung 559-230566 Schaumstabilisatoren, welche für polare
Lösungsmittel
nützlich
sind, die ein anionisches oder amphoteres Fluortensid, Polyethylenimin
mit einem Molekulargewicht von 4.000 bis 100.000 und eine polybasische
Säurekomponente enthalten.
-
Das
US-Patent 3,769,307 beansprucht Perfluoralkyl-substituierte Polyethylenimin-Zusammensetzungen
und deren Herstellung. Dieses Patent beansprucht ebenso die Verwendung
solcher Verbindungen als neue Textilausrüstungen, welche oleophobe Eigenschaften
verleihen. Die deutsche Offenlegungsschrift 2 018 461 beschreibt
oberflächenaktive
Mittel und Schaumstabilisatoren für Polyurethanschäume, welche
durch ein oder mehrere Perfluoralkylgruppen substituierte Polyethylenimine
sowie Perfluoralkyl-substituierte Polyamine, welche bis zu 16 Carboxy-
oder Sulfonsäuregruppen
und/oder hydrophile Amidgruppen enthalten, sind. Obwohl kein Bezug
zu Schaumstabilisatorverbindungen für Feuerbekämpfungsschäume für polare Lösungsmittelbrände hergestellt
wird, wird die Zusammensetzung dieses Patents als in Alkohol-/Wasser-Mischungen äußerst löslich, allerdings
in Alkohol (= "alkophob") und Wasser selbst
nur schlecht löslich
beschrieben, was sie einen Kandidaten für solche Schaumstabilisatoren
werden lässt.
In der Tat beschreibt die oben erwähnte WO 94/18245-Referenz die
Synthese eines Perfluoralkyl- und Carboxy-substituierten Polyethylenimins
aus Tetraethylenpentamin, einem Perfluoralkylacylchlorid und Chloressigsäure.
-
Die
japanische offengelegte Patentanmeldung 59-164073 offenbart Reaktionsprodukte
von Polyethylenimin und anionischen fluorierten Tensiden, welche
die saure und fluorierte Komponente für wirksame Schaumstabilisatoren
bei polaren Lösungsmitteln
zur Verfügung
stellen.
-
Die
internationale Anmeldung WO 96/05889 A1 beschreibt ebenso Schaumstabilisatoren,
welche aus Polyaminen mit Perfluoralkyl-Gruppen, die an das Polyamin
durch Esterbindungen gebunden sind, bestehen und zusätzliche
hydrophile Gruppen sowie wahlweise eine hydrophobe Nicht-Perfluoralkyl-Gruppe
enthalten.
-
Wirksame
Schaumstabilisatoren für
polare Lösungsmittel
müssen
im Wesentlichen unlöslich
in diesen Lösungsmitteln
sein. Sie sind gängigerweise
polyperfluoralkylsubstituierte Polyaminosäureverbindungen, wie die oben
beschriebe nen. Die vorliegende Erfindung offenbart Verbindungen,
welche als Schaumstabilisatoren für Brandbekämpfungsschäume, die bei Bränden polarer
Lösungsmittel
verwendet werden, nützlich
sind, welche Polyethylenimin-Derivate, enthaltend anionische und
nichtionische wassersolubilisierende Gruppen, darstellen und weiterhin
mit einer Mischung von Perfluoralkylallyloxy- und Perfluoralkyliodpropyloxy-Gruppen
oder Polyperfluoralkylfluorallyl-Gruppen substituiert sind.
-
Monoperfluoralkyl-
(= RF) substituierte Aminosäuren sind
seit langem als ausgezeichnete amphotere Tenside bekannt, welche
die Oberflächenspannung
des Wassers auf lediglich 16 Dynes/cm vermindern. Solche Verbindungen,
die durch die Umsetzung eines RF-Ethylthiols,
Maleinsäureanhydrid
und einem Di-, Tri- oder
Tetraamin erhalten werden und bis zu zwei RF-,
Carboxy- und Aminogruppen enthalten, werden beispielsweise in den
US-Patenten 4,069,244 und 4,161,602 beschrieben. Di- und Poly-RF-polyaminosäuren, welche durch Umsetzung
eines Di-RF-diols mit einem Dianhydrid und
einem Diamin erhalten werden und 2-6 RF-Gruppen,
4-10 Carboxy- und zwei tertiäre
Aminogruppen enthalten, sind in dem US-Patent 4,153,590 beschrieben. Es
wurde herausgefunden, dass diese amphoteren Verbindungen nützlich als
oberflächenaktive
Mittel sowie Filmbildner in wässrigen
und Harzzubereitungen sind.
-
Eine
weitere Klasse amphoterer Verbindungen mit ähnlichen Eigenschaften, welche
RF-, Säure-
und Aminogruppen enthalten und welche zum Verleihen von ölabweisenden
Eigenschaften bei Papierprodukten nützlich sind, sind Di-RF-aminosäuren,
welche durch die Umsetzung einer Aminosäure, Allylglycidylether und einem
RF-Iodid, wie in dem US-Patent 5,491,261
beschrieben, erhalten werden. Diese Syntheseroute unter Verwendung
eines RF-Iodids anstelle eines RF-Thiols als Ausgangsmaterial ist kosteneffizienter,
da sie mit höheren
Ausbeuten durchgeführt
werden kann und viel weniger Abfall produziert.
-
Es
wurde nun herausgefunden, dass durch eine ähnliche Route polymere RF-Amine, einschließlich polymerer RF-Aminosäuren diesen
Typs, welche als Schaumstabilisatoren für Schäume zur Bekämpfung von polaren Lösungsmittelbränden (polare
Lösungsmittelbrandbekämpfungsschäume) nützlich sind
und welche eine Vielzahl von RF-Gruppen
sowie Amino- und Carboxy- oder andere hydrophile Gruppen enthalten,
bequem in ähnlich
hohen Ausbeuten und im Wesentlichen ohne Verunreinigungen/Abfall
aus einem Polymer, das eine Vielzahl von primären und/oder sekundären Aminogruppen
und eine Vielzahl von Säuregruppen
enthält,
durch Umsetzung mit Allylglycidylether (= AGE) gefolgt von der Zugabe
von RF-Iodid und teilweiser Dehydrohalogenierung
hergestellt werden können.
Die resultierende Mischung von Polyperfluoralkylallyloxy- und Polyperfluoralkyliodpropyl-substituierten
Polyaminosäuren
ist als Fettschutzmittel für
Papier nützlich,
allerdings wurde herausgefunden, was noch viel wichtiger ist, dass
sie als ausgezeichnete Schaumstabilisatoren für wässrige Feuerbekämp fungsschaumzubereitungen
(AFFF), welche bei polaren Lösungsmittelbränden verwendet
werden, wirken.
-
Ähnliche
Verbindungen, welche Polyperfluoralkylfluorallyl-substituierte Polyaminosäuren sind
und welche ausgezeichnete Schaumstabilisatoren für AR-AFFF-Mittel darstellen, können durch
Umsetzung von Polyethyleniminen mit Perfluoralkylethyliodid gefolgt
von der Umsetzung mit aminoreaktiven Säureverbindungen, wie Chloressigsäure oder
Bernsteinsäureanhydrid,
hergestellt werden. Es wird angenommen, dass diese Reaktion über ein
Perfluoralkylethylen-Intermediat und die anschließende Eliminierung
von HF verläuft,
was in einer 3-Perfluoralkyl-2-fluorallylamin-Struktur resultiert.
Die Additionsreaktion von Perfluoralkylethylenen an primäre und sekundäre Amine
ist in den US-Patenten 3,535,381 und 4,853,141 beschrieben.
-
Es
wurde ebenso herausgefunden, dass die Säurefunktionalität für die Leistungsfähigkeit
der Verbindungen nicht essentiell ist, sondern durch andere hydrophile
Gruppen, wie Amid- und/oder Hydroxygruppen, ersetzt werden kann.
Es wurde weiterhin herausgefunden, dass nichtionisch substituierte
Poly-RF-polyethylenimine eine überragende
Leistung mit Salzwasser ergeben; in ähnlicher Weise wurde herausgefunden,
dass Phosphorsäure-substituierte
Poly-RF-polyethylenimine eine überragende
Leistung mit Salzwasser ergeben. Die Leistungsfähigkeit bei Mischung mit Salzwasser
ist ein Hauptanliegen bei Brandbekämpfungsoperationen an Bord
von Schiffen und in Häfen.
-
Die
Verwendung von nichtionisch substituierten und Phosphorsäure-substituierten
Poly-RF-polyethyleniminen als Schaumstabilisatoren
in Salzwasser-Feuerbekämpfungsschaumzubereitungen
ist somit ein weiteres Ziel der vorliegenden Erfindung.
-
Detaillierte Offenbarung
-
Die
Papierleimchemikalien und Schaumstabilisatoren der vorliegenden
Erfindung sind Perfluoralkylallyloxy- und Perfluoralkyliodpropyloxy-substituierte
Polyaminosäuren
oder Poly-R
F-fluorallyl-substituierte Polyaminosäuren, welche
in statistischer Verteilung enthalten
q Einheiten von A-1,
r Einheiten von A-2, s Einheiten von A-3 und t Einheiten von A-4,
worin
A-1 und A-2 Perfluoralkyl-substituierte Aminogruppen
der Formeln
sind,
A-3
eine hydrophil substituierte Amino- oder Amidogruppe der Formel
darstellt
und
A-4 eine substituierte Amino- oder Amidogruppe der Formel
ist,
worin
T
-CH
2CH(OH)CH
2-O-CH
2- oder eine direkte Bindung darstellt, unter
der Voraussetzung, dass, wenn T -CH
2CH(OH)CH
2-O-CH
2- ist, Q
F den Formeln
-CHI-CH2-RF (QF1) und
-CH=CH-RF (QF2)genügt und aus
5 bis 50 mol% Q
F1 und 50 bis 95 mol% Q
F2 besteht, und wenn T eine direkte Bindung
ist, Q
F -CH2CH=CF-RF' (QF3) darstellt,
q,
r, s und t ganze Zahlen von 0 bis 100 sind, wobei die Summe von
q+r+s+t 5 bis 200 ist, die Summe von q+r gleich oder größer 2 ist
und das Verhältnis
von q+r/s 0,1 bis 2 ist,
R Wasserstoff oder Methyl darstellt,
R
F unabhängig
ein monovalenter perfluorierter Alkyl- oder Alkenyl-, geradkettiger
oder verzweigter organischer Rest mit 4 bis 20 vollständig fluorierten
Kohlenstoffatomen ist,
R
F' unabhängig ein
monovalenter perfluorierter Alkyl- oder Alkenyl-, geradkettiger
oder verzweigter organischer Rest mit 3 bis 19 vollständig fluorierten
Kohlenstoffatomen ist, wobei jeder R
F- und
R
F'-Rest
identisch zu oder unterschiedlich von den anderen R
F-
und R
F'-Resten
ist,
X Wasserstoff, -CH
2CH(OH)CH
2-O-CH
2CH=CH
2 oder -G-Y ist,
G eine direkte Bindung
oder eine Verbindungsgruppe der Formel -CH
2-,
-CH
2CHR-, -CH
2-CH
2C
6H
4-, -CH
2CH
2CH
2-,
-C6H
4-, -CH(-COOH)CH
2-,
-CH
2CH
2CONHCH(OH)-,
-COR
1-, -CH
2CHRCONHC(CH
3)
2(CH
2)- oder
eine Mi schung davon darstellt,
worin R wie oben definiert ist,
R
1 -CH=CH-, -CH
2CH
2- oder -C
6H
4- ist und
Y eine Säuregruppe der Formel -COOH,
-SO
3H, -PO
3H
2 oder -(PO
3H)
3H oder ein Salz davon darstellt,
oder
-CONH
2 oder -CH(OH)CH
2OH
oder eine Mischung dieser Gruppen ist,
R
2 einen
Alkylrest mit 1 bis 20 Kohlenstoffatomen oder -CH
2CH
2CON(CH
3)
2, -CH
2CH
2CONHCH
2OH, -CH
2CH
2CON(CH
2OH)
2, -CH
2CH
2N(R
4)
2, -CH
2CR
1-COOR
4 oder -CH
2CH(OH)CH
2-O-CH
2CH=CH
2 darstellt, worin
R
4 einen Alkylrest mit 1 bis 18 Kohlenstoffatomen
oder -CH
2CH
2-OH
darstellt,
R
3 dasselbe wie R
2 oder Wasserstoff ist und R
1 wie
oben definiert ist.
-
Bevorzugt
sind Verbindungen, wie sie oben beschrieben werden, worin QF aus 10 bis 40% QF1 und
60 bis 90% QF2 besteht oder QF3 ist,
RF gesättigt
ist und 6 bis 12 Kohlenstoffatome enthält, vollständig fluoriert ist und mindestens
eine terminale Perfluormethylgruppe aufweist,
RF' gesättigt ist
und 5 bis 11 Kohlenstoffatome enthält, vollständig fluoriert ist und mindestens
eine terminale Perfluormethylgruppe aufweist,
q+r 2 bis 20
ist,
s 5 bis 80 ist und das Verhältnis von q+r/s 0,05 bis 0,5
beträgt,
t
0 bis 5 ist,
R Wasserstoff ist,
R2 -CH2CH(OH)CH2-O-CH2CH=CH2 ist,
R3 Wasserstoff oder -CH2CH(OH)CH2-O-CH2CH=CH2 ist,
Y wie in Anspruch 1 definiert
ist und
G eine direkte Bindung darstellt oder der Formel -CH2-, -CH2CH2-, -CH2CH2CONHCH(OH)-, -CH2-CH2C6H4-, -CH(-COOH)CH-
oder -COR1-, worin R1 -CH2CH2- ist, genügt.
-
Besonders
bevorzugt sind Verbindungen, wie sie oben beschrieben werden, worin
QF, T, RF, RF',
q, r, s, t, R, R1, R2 und
G wie oben definiert sind und Y -COOH oder -CONH2 ist,
wobei Verbindungen, in denen QF QF1 und QF2 ist und
aus 10 bis 40% QF1 und 60 bis 90% QF2 bestehen, T -CH2CH(OH)CH2-O-CH2- ist und
worin G -CH2- darstellt und Y -COOH ist,
besonders bevorzugt sind.
-
Ebenso
besonders bevorzugt sind Verbindungen, worin QF QF3 darstellt, T eine direkte Bindung ist,
G -CH2- darstellt und Y -COOH ist.
-
Ebenso
besonders bevorzugt sind Verbindungen, wie sie oben beschrieben
sind, worin QF, T, RF,
RF', q,
r, s, t, R, R1 und R2 wie
oben beschrieben sind, G -CH2CH2 oder
-CH2-CH2C6H4- ist und Y -SO3H darstellt.
-
Ebenso
besonders bevorzugt sind Verbindungen, wie sie oben beschrieben
sind, worin QF, T, RF,
RF', q,
r, s, t, R, R1 und R2 wie
oben beschrieben sind, G -CH2CH2-
darstellt und Y -PO3H ist.
-
Ebenso
besonders bevorzugt sind Verbindungen, wie sie oben beschrieben
werden, worin QF, T, RF, RF',
q, r, s, t, R, R1 und R2 wie
oben definiert sind, G eine direkte Bindung darstellt und und Y
-(PO3H)3H, -COOH oder
-CH(OH)CH2OH ist.
-
Die
neuen Poly-RF-(allyloxy/iodpropoxy)polyamine
der vorliegenden Erfindung werden erhalten durch zuerst Umsetzen
eines Allylglycidylethers mit einem Teil der primären oder
sekundären
Aminogruppen eines Vorstufen-Polyaminpolymers; die anschließende Umsetzung
dieses Polyallyloxy-substituierten Polyamino-Präpolymers
mit einer aminoreaktiven organischen oder anorganischen sauren Verbindung
oder einer anderen hydrophilen Verbindung und die anschließende Umsetzung
des Produkts dieser Reaktion mit einem Perfluoralkyliodid.
-
Die
neuen Poly-RF-fluorallyl-substituierten
Polyaminosäuren
werden erhalten durch Umsetzung eines Polyamins mit einem Perfluoralkylethyliodid
entweder vor oder nach der Umsetzung mit einer aminoreaktiven Säure oder
einer anderen hydrophilen Verbindung. Aufgrund der basischen Natur
des Reaktionsmediums wird HI eliminiert, und ein Perfluoralkylethylen
wird als eine Zwischenstufe gebildet, welche an eine Aminogruppe addiert.
Während
dieser Reaktion wird ein Äquivalent
HF eliminiert; daher enthält
die resultierende Perfluoralkylgruppe (= QF3)
eine -CF2-Einheit weniger als die entsprechenden
QF1- und QF2-Gruppen.
-
Die
Reaktion wird in einem hochsiedenden polaren Lösungsmittel, vorzugsweise einem
Glykol, wie Ethylen-, Propylen- oder Hexylenglykol, bei Temperaturen
von 90-120°C
innerhalb einer Zeitdauer von 3 bis 20 Stunden durchgeführt.
-
Nützliche
aminoreaktive saure Verbindungen sind halogenierte Carbonsäuren oder
Sulfonsäuren
oder deren Salze der Formel X'-G-Y,
welche unter Eliminierung von X'-H
reagieren, worin X' Chlor
oder Brom ist und G und Y wie oben definiert sind. Bevorzugte Verbindungen
sind Chloressigsäure,
Chlorpropionsäure
und Chlorsulfonsäure
sowie deren Salze. Ebenso geeignet sind Vinyl-ungesättigte Säuren, welche über eine
Michael-Additionsreaktion reagieren, wie Acrylsäure, Itaconsäure, Vinylsulfonsäure und
Vinylphosphonsäure, 2-Acrylamido-2-methylpropansulfonsäure und
Acrylamidoglykolsäure.
Anhydride, welche unter Amidbildung reagieren, wie Maleinsäure-, Bernsteinsäure- oder
Phthalsäureanhydride,
und Natriummetatriphosphat sind ebenso nützlich.
-
Nützliche
aminoreaktive nichtionische hydrophile Verbindungen sind Oxirane
und Chloracylamide, wie Glycidol und Chloracetamid.
-
Die
Reaktion des Polyallyloxy-Polyamino-Präpolymers mit einer aminoreaktiven
organischen oder anorganischen sauren Verbindung oder einer anderen
hydrophilen Verbindung verläuft
leicht bei Temperaturen von 40 bis 75°C. Die Säuren oder Säuresalze können in einem Lösungsmittel
oder vorzugsweise unverdünnt zugesetzt
werden. Nützliche
Lösungsmittel
sind Wasser und Alkohole, wie n-Propanol, 2-Propanol und Hexylenglykol.
-
Bevorzugte
Reaktanden sind -Halosäuren
und deren Salze, wobei Natriumchloracetat am meisten bevorzugt ist.
Ebenso bevorzugt sind α,β-ungesättigte Säuren, wobei
Acrylsäure
am meisten bevorzugt ist. Ebenso bevorzugt sind Maleinsäure- und
Bernsteinsäureanhydride
sowie cyclisches Natriummetatriphosphat sowie Mischungen von Glycidol
und Chloracetamid.
-
Die
Reaktion wird unter entweder wässrigen
oder wasserfreien Bedingungen durchgeführt, und die Zugabe von einem
Katalysator ist nicht erforderlich.
-
Diese
aminoreaktiven Verbindungen können
alleine oder in Kombination miteinander verwendet werden. Alternativ
können
die aminoreaktiven hydrophilen Verbindungen zu dem Polyamin vor
der Zugabe des Allyglycidylethers zugesetzt werden. In diesem Fall
wird vorteilhafterweise ein Lösungsmittel
verwendet. Wasser ist ein bevorzugtes Lösungsmittel. Der Allylglycidylether
wird anschließend
zugesetzt, vorzugsweise unverdünnt
oder in Lösung
unter Verwendung eines Lösungsmittels,
wie Propanol.
-
Das
fertige Produkt wird durch Umsetzung eines Perfluoralkyliodids mit
dem Präpolymer
in Gegenwart eines Initiators freier Radikale, wie eine Azoverbindung
oder einem Peroxid, bei geeigneten Initüerungstemperaturen, vorzugsweise
bei Temperaturen von zwischen 50 und 80°C, erhalten. Natriummetabisulfit
ist vorzugsweise anwesend, um Iod zu Iodid zu reduzieren.
-
Lösungsmittel
können
vorliegen; beispielsweise Ketone, wie Aceton, Methylethylketon oder
Methylpropylketon, oder Alkohole wie Ethanol, Propanol oder Butanol.
Wenn ein Lösungsmittel
verwendet wird, kann es vor der Verdünnung der Reaktionsmischung
mit Wasser abdestilliert werden. Die Reaktion wird typischerweise
innerhalb von 4 bis 10 Stunden bei 50 bis 80°C unter ausreichendem Rühren durchgeführt. Die
resultierende Produktmischung wird mit ausreichend entionisiertem
Wasser zur Einstellung des Feststoffgehalts auf 15 bis 40 Gew.-%
und des Fluorgehalts auf 4 bis 10% verdünnt.
-
Aufgrund
der basischen Natur des Reaktionsmediums wird ein Großteil des
organischen Iodids während
des Verlaufs der Reaktion eliminiert. Das Präpolymer wird daher als eine
Mischung mit Iodpropoxy- und Allyloxy-Bindungen an die Perfluoralkyl-Einheiten
erhalten. Wenn eine vollständige
Eliminierung des organischen Iodids erwünscht ist, ist die Zugabe einer
starken anorganischen Base, wie Natrium- oder Kaliumhydroxid, oder
einer starken organischen Base, wie 1,8-Diazabicyclo(5.4.0)undec-7-en (DBU),
erforderlich.
-
Es
wurde weiterhin herausgefunden, dass die Addition von RF-I
an die Allyloxy-Gruppen unter Verwendung von Natriumdithionit bei
Temperaturen zwischen 0 und 20°C
durchgeführt
werden kann. Huang (Chin. J. Chem. 4, 350 und 358, (1990); Macromol
Symp. 82, 67, 1994) lehren, dass die Verwendung von einem Äquivalent
Dithionit, basierend auf RF-I, bei der Addition
von RF-I an terminal ungesättigte Verbindungen
notwendig ist. Es wurde weiterhin unerwarteter weise herausgefunden,
dass nur 0,02 bis 0,5 Äquivalente,
vorzugsweise 0,05 bis 0,2 Äquivalente
ausreichend sind, um im Wesentlichen eine vollständige Addition an die Allyloxy-substituierte
Polyaminosäure
zu erreichen. Ein Vorteil dieses Verfahrens liegt darin, dass weniger
Färbung
entsteht und mehr organisch gebundenes Iod verbleibt. Zusätzlich kann
das Verfahren in höheren
wässrigen
Verdünnungen
durchgeführt
werden. Die Durchführung
der Addition von RF-I an terminale Doppelbindungen
in einer wässrigen
Lösung,
enthaltend 4 bis 40 Gew.-% eines wasserlöslichen Lösungsmittels, wie einen C1-C4-Alkohol, ein
Amid, wie Dimethylformamid, oder ein Keton, bei 0 bis 40°C in Gegenwart
von 0,02 bis 0,5 Äquivalenten,
vorzugsweise 0,05 bis 0,2 Äquivalenten,
auf der Basis von RF-I, Dithionitionen ist
folglich ein weiterer Gegenstand der vorliegenden Erfindung.
-
Nützliche
Polyamin-Ausgangsmaterialien besitzen anzahlmittlere Molekulargewichte
im Bereich von ungefähr
200 bis 10.000. Sie sind typischerweise Polyalkylenimine, welche
4 bis 300 primäre,
sekundäre
und tertiäre
Aminogruppen in Verhältnissen
im Bereich von 1:1:0 bis 1:2:1 enthalten. Bevorzugt sind Polyethylenimine
mit Molekulargewichten von 1.000 bis 5.000. Diese Polyamin-Ausgangsmaterialien
sind kommerziell erhältlich.
-
Die
folgenden Beispiele verdeutlichen verschiedene Ausführungsformen
der Erfindung und sollen nicht als den Umfang der angehängten Ansprüche begrenzend
interpretiert werden. In den Beispielen beziehen sich alle Teile
auf das Gewicht, solange nichts anderes angegeben ist. Perfluoralkyliodide
CnF2n+1-I mit n =
4 bis 14 wurden von DuPont unter den Handelsnamen ZONYL® TELA-L
und ZONYL® TELA-N
erhalten. Sie wiesen jeweils die folgenden mittleren Telomer-Verteilungen
auf:
ZONYL® TELA-L:
C4 = 4% Maximum, C6 =
50 ± 3%,
C8 = 29 ± 2%, C10 =
11 ± 2%,
C12 = 4 ± 1%, C14 und
höher =
2% Maximum.
ZONYL® TELA-N:
jeweils C6 = 6% Maximum, C8 =
50 ± 3%,
C10 = 29 ± 2%, C12 = 11 ± 1%, C14 und höher
= 4% Maximum.
-
Die
entsprechenden Perfluoralkylethyliodide, CnF2n+1-CH2CH2I, sind erhältlich von
DuPont unter dem Produktnamen ZONYL® TELB-L
und TELB-N und besitzen im Wesentlichen dieselbe Telomer-Kettenlängen-Verteilung
wie TELA-L und -N.
-
Wenn
die Verbindungen der vorliegenden Erfindung zur Verbesserung der Ölabweisung
von Papier verwendet werden, werden sie auf das Papier oder die
Pappe als eine externe Beschichtung durch ein jedes konventionelles
Verfahren, wie das Aufstreichen oder Aufsprühen, oder in einer Leimpresse
in Mengen zur Absetzung von 0,02 bis 0,5% Fluor, bezogen auf das
Gewicht des Papiers, aufgetragen. Zusätzlich zu der Fluorchemikalie
können
jegliche herkömmliche
Bindemittel, welche in der Papierindustrie verwendet werden – wie polymere
Latexbindemittel, Carboxymethylcellulose und Polyvinylalkohol – und Schlichtemittel,
wie ionische und nichtionische Stärken, wie ethoxylierte und
oxidierte Stärken,
und Wasserschlichtemittel, wie Alkylketen-Dimer (AKD) oder Alkylbernsteinsäureanhydrid
(ASA), eingesetzt werden.
-
In
den folgenden Beispielen wurde die externe Schlichte- bzw. Appreturauftragung
unter Verwendung des folgenden Verfahrens erreicht: die Produkte
wurden auf ein Waterleaf-Rohpapier 34# unter Verwendung einer Werner
Mathis Laboraufstreichvorrichtung (Padder) in dem horizontalen Modus
aufgetragen. Die Proben wurden mit 2% Penford 280 Stärke als
Schlichtemittel und Chel® DPTA 41 (von der Ciba
Specialty Chemicals Corp.) als ein Komplexierungsmittel in einem
Standardverfahren aufgetragen. Das Papier wurde 30 Sekunden lang
auf jeder Seite bei 100°C
unter Verwendung eines photographischen Trockners getrocknet.
-
Die ölabweisende
Eigenschaft einer Oberfläche
wird unter Verwendung des TAPPI UM 557 OIL KIT TESTs bestimmt. Dieses
Testverfahren besteht aus der Auftragung von zwölf verschiedenen Mischungen
aus Rizinusöl/Heptan/Toluol
mit einer Oberflächenspannung
im Bereich von 34,5 bis 22,0 Dynes/cm. Die Beurteilung bzw. Einstufung
basiert auf dem auftretenden Eindringen innerhalb von 15 Sekunden
nach der Auftragung; diese Einstufungen verlaufen von 1 (am niedrigsten)
bis 12.
-
Wie
in Spalte 2 des US-Patents 5,496,475 angegeben ist, dessen Lehre
hierin durch Bezugnahme eingeschlossen ist, werden AFFF- und AR-AFFF-Mittel
im Allgemeinen in der Form flüssiger
Konzentrate vertrieben. Diese Konzentrate, welche eher komplexe
Mischungen sind (siehe Spalte 7, Zeilen 9 bis 36), werden mit Frisch-
oder Salzwasser in Verteilungsvorrichtungen verdünnt und auf eine brennende
Flüssigkeit
als ein Schaum gesprüht.
-
Die
Mittel werden gewöhnlich
als sogenannte "3×6-" und "3×3-" AR-AFFF-Konzentrate, wobei der Trend in der
Industrie zu Letzterem hingeht, vertrieben, worin die Zahlen die
Gewichtsprozente des in der verdünnten
Zubereitung zur Bekämpfung
eines einen jeweils nichtpolaren Treibstoff, wie Benzin, und einen
polaren Treibstoff einschließenden
Feuers enthaltenden Konzentrats anzeigt.
-
Wenn
die erfindungsgemäßen Zusammensetzungen
als Schaumstabilisator in einem AR-AFFF-Mittel verwendet werden,
werden sie zu herkömmlichen
AFFF- und AR-AFFF-Zubereitungen
zugesetzt. Die Menge des typischerweise in 3×3-AR-AFFF-Mitteln verwendeten Schaumstabilisators
liegt im Bereich von 1 Gew.-% bis 4 Gew.-% der wirksamen Bestandteile.
Somit sind von 10 bis 40% des Fluors der fertigen Zubereitung von dem
Schaumstabilisator abgeleitet.
-
Um
die Wirksamkeit der neuen Schaumstabilisatoren zu testen, wurde
die folgende Grund-AR-AFFF-Zubereitung, welche frei von jeglichem
Schaumstabilisator ist, verwendet:
| Lodyne® F-102R,
von der Ciba Specialty Chemicals | 5,6% |
| Lodyne® F-204R,
von der Ciba Specialty Chemicals | 2,4% |
| Mirataine®-H2C-HA
von Rhone-Poulenc | 16% |
| Sipex® OLS
von Alcolac | 1,8% |
| Triton® X-102
von der Rohm & Haas
Comp. | 0,8% |
| Butylcarbitol | 10% |
| Keltrol®BT
von der Kelco Comp. | 1,5% |
-
Diese
Mischung wird in den Beispielen als AR-AFFF-Basis bezeichnet.
-
Die
Messungen des Schaumausdehnungsverhältnisses (Foam Expansion Ratio,
FXR) und der Viertelaustrittszeit (Quarter Drain Time, QDT) wurden
unter Verwendung des folgenden Verfahrens durchgeführt. Es
wurde eine 3%ige Lösung
des AR-AFFF in Meer- oder Leitungswasser hergestellt. Die Testlösung wurde durch
Vakuum in den kalibrierten Flüssigkeitsbehälter gezogen;
siehe 1 unten. Das Volumen der Testlösung wurde
auf 100 ml eingestellt. Die Testlösung wurde auf 40 psi mit Druckstickstoff
gepresst. Druckluft wurde angelegt und auf 33 psi eingestellt. Die
Testlösung
wurde mit Luft an der Mischöffnung
vor dem Schäumen an
der Düse
gemischt. Das Volumen des Schaums wurde in einen 1.000 ml Messzylinder
gemessen. Das Schaumausdehnungsverhältnis des Schaums wurde als
das Verhältnis
des Gesamtschaumvolumens zu dem Volumen der Originaltestlösung bestimmt.
Die Viertelaustrittszeit wurde als die Zeit bestimmt, die es zum
Sammeln von 25 ml austretender Flüssigkeit aus dem Schaum benötigt. Jede
Testmessung wurde zweimal durchgeführt, und der Mittelwert wurde
verzeichnet.
-
Die
Schaumlebensdauer auf heißem
2-Propanol wurde unter Verwendung des folgenden Verfahrens gemessen.
Eine 3%ige Lösung
des AR-AFFF wurde in Meer- oder Leitungswasser hergestellt. Die
Testlösung wurde
in den kalibrierten Flüssigkeitsbehälter unter
Anlegung eines Vakuums gefüllt,
siehe 1 unten. Das Volumen der Testlösung wurde
auf 75 ml eingestellt. Die Testlösung
wurde auf 40 psi mit Druckstickstoff gepresst. Druckluft wurde bei
33 psi angelegt. Die Testlösung
wurde mit Luft an der Mischöffnung
vor dem Schäumen
an der Düse
gemischt. Es wurden zu einer Glas-Pyrex-Pfanne, 6,5 Inches × 10 Inches,
250 ml 2-Propanol bei 70°C
zugegeben. Die Testlösung
wurde als Schaum auf das heiße
2-Propanol ausgegeben und bildete einen diese Oberfläche vollständig bedeckenden Überzug.
Die Schaumlebensdauer wurde als die Zeit gemessen, die für die Kollabierung
bzw. den Zusammenfall von 50% der Schaumoberfläche benötigt wurde. Eine jede Testmessung
wurde zweimal durchgeführt,
und der Mittelwert wurde verzeichnet.
-
-
Analytische Verfahren
-
Der
Fortschritt der Reaktion von Allylglycidylether mit Polyethylenimin
wurde mittels Gaschromatographie verfolgt. Man ließ die Reaktion
so lange fortschreiten, bis Allylglycidylether nicht länger detektiert
werden konnte.
-
Es
wurde ebenso der ZONYL®TELA-L-Verbrauch mittels
Gaschromatographie unter Verwendung eines HP 5890 GC mit einem FID-Detektor
und einer Supelco SPB-1-Säule,
60 mesh/0,53 mm × 3,0
m, durchgeführt.
-
Die
Bestimmung von ionischem Chlorid und Iodid wurde durch Titration
wie nachstehend beschrieben durchgeführt:
Ausstattung: Brinkmann-Auto-Titrator,
Model E436; Fisher Ag/AgCl-Referenzelektrode;
Fisher Silver
Billet Indicating Electrode; Aldrich Standard AgCl.
-
Verfahren:
1) Es werden ungefähr
0,2 g Probe für
Chlorid oder 1,0 g für
Iodid in einem 200 ml-Becher eingewogen und mit 150 ml Wasser verdünnt und
1 ml Eisessig zugesetzt. 2) Es wird mit 0,1023 M AgNO3 bei 750
mv und einer Geschwindigkeit von "2" titriert.
-
-
-
1. Beispiel 1
-
Synthese von Poly(N-2-hydroxy-4-oxa-6,7-enheptyl)polyethylenimin
(= Polyallyloxy-PEI)
-
Es
werden 100,0 g (83,3 mmol) Polyethylenimin, Mn 1200 (Epomin® SP-012
von Nippon Shokubai Co.) und 25,0 g entionisiertes Wasser in einen
Rundbodenkolben, welcher mit einem Rührer, einem Stickstoffeinlass
und einem Thermoregulator augestattet ist, gegeben. Diese Mischung
wird unter Rühren
erwärmt.
Sobald eine Temperatur von 65°C
erreicht wird, werden 28,5 g (250 mmol) Allylglycidylether innerhalb
einer Zeitdauer von einer Stunde zugesetzt. Die Reaktionsmischung
wird anschließend
zwei Stunden lang bei 65°C
gerührt.
Der Verbrauch von Allyglycidylether wird mittels Gaschromatographie
verfolgt. Typischerweise wird dieses Produkt nicht isoliert, sondern
direkt in dem nächsten
Schritt verwendet.
-
B: Synthese von Poly-N-2-hydroxy-4-oxa-[6,7-en-
und -6-iod-]-7-RF-heptyl-N-carboxymethylen-poly(ethylenimin)
(= Poly-RF-PEI-carbonsäure)
-
Es
werden 15,0 g (24,4 mmol) des Präpolymers
aus Beispiel 1A in einen Rundbodenkolben, welcher mit einem Rührer, einem
Stickstoffeinlass und einem Thermoregulator ausgestattet ist, gegeben
und erwärmt. Sobald
die Temperatur 40°C
erreicht, werden 18,0 g (154 mmol) Chloressigsäure-Natriumsalz und 5,0 g entionisiertes
Wasser zu dem Kolben zugegeben. Es wird ein Temperaturanstieg von
40°C auf
100°C beobachtet. Wenn
der Temperaturanstieg abnimmt, wird die Reaktionsmischung bei 75°C drei Stunden
lang unter Rühren gehalten.
Die Vollständigkeit
der Umsetzung wird durch Chloridtitration mit Silbernitrat bestimmt.
Die Temperatur wird anschließend
auf 80°C
erhöht,
und 12,78 g (22,0 mmol) Perfluoralkyliodid (ZONYL TELA-N) und 0,46
g (2,4 mmol) Natriummetabisulfit werden zusammen mit 0,19 g (1 mmol)
2,2'-Azobisisobutyronitril
(AIBN) zugegeben. Nach einer Stunde werden 3, 5 g entionisiertes
Wasser zur Verminderung der Viskosität der Mischung zugesetzt. Das
Rühren
wird fünf
Stunden lang bei 80°C
fortgesetzt. Nach fünf
Stunden wird die Mischung auf Raumtemperatur abgekühlt, und
Wasser wird zum Einstellen des Feststoffgehalts auf 28 Gew.-% und
5,0% F zugesetzt. Der Umsatz von RF-Iodid
beträgt
gemäß gaschromatographischer
Bestimmung 95%.
-
2. Beispiele 2 und 3
-
Die
Produkte werden dem Verfahren des Beispiel 1 folgend mit den Verhältnissen
von Natriumchloracetat und Perfluoralkyliodid, wie sie in Tabelle
1 angegeben sind, synthetisiert.
-
3. Beispiel 4
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 100,0 g (83,3 mmol) Polyethylenimin, Mn 1200 (Epomin® SP-012
von der Aceto Corporation) und 25,0 g entionisiertes Wasser in einen
Rundbodenkolben, ausgestattet mit einem Rührer, einem Stickstoffeinlass
und einem Thermoregulator, gegeben. Die Temperatur der Reaktionsmischung
steigt auf 65°C, und
19,0 g (167 mmol) Allylglycidylether werden innerhalb ungefähr einer
Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden lang bei
65°C gerührt. Nach
dieser Zeit ist der Umsatz von Allylglycidylether vollständig, wie
es mittels Gaschromatographie verfolgt wird. Dieses Produkt wird
nicht isoliert, sondern direkt in dem nächsten Schritt verwendet.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 15,0 g (17,4 mmol) des Präpolymers
aus Beispiel 4A in einen Rundbodenkolben, welcher mit einem Rührer, einem
Stickstoffeinlass und einem Thermoregulator ausgestattet ist, gegeben.
Zu diesem Rundbodenkolben werden 20,2 g (174 mmol) Chloressigsäure-Natriumsalz
und 5,0 g entionisiertes Wasser gegeben. Die Reaktionsmischung wird
auf 75°C
erwärmt
und drei Stunden lang gerührt.
Am Ende der drei Stunden werden 9,09 g (15,6 mmol) ZONYL TELA-N
zu der Reaktionsmischung zusammen mit 0,33 g (1,7 mmol) Natriummetabisulfit
und 0,13 g (0,69 mmol) 2,2'-Azobisisobutyronitril
(AIBN) zugegeben. Die Reaktionsmischung wird unter Stickstoff bei
80°C fünf Stunden
lang gerührt.
Nach fünf
Stunden wird die Mischung auf Raumtemperatur abgekühlt, und
Wasser wird zum Einstellen des Feststoffgehalts auf 27 Gew.-% und
3,5% F zugesetzt. Der Umsatz an RF-Iodid,
bestimmt mittels Gaschromatographie, beträgt 95%.
-
Beispiele 5 und 6
-
Die
Produkte werden dem Verfahren des Beispiels 4 folgend unter Verwendung
der Verhältnisse
von Natriumchloracetat und Perfluoralkyliodid, wie sie in Tabelle
1 angegeben sind, synthetisiert.
-
Beispiel 7
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 100,0 g (83,3 mmol) Polyethylenimin, Mn 1200 (Epomin® SP-012
von der Aceto Corporation) und 20,0 g entionisiertes Wasser in einen
Rundbodenkolben, ausgestattet mit einem Rührer, Stickstoffeinlass und
einem Thermoregulator, gegeben. Die Temperatur der Reaktionsmischung
wird auf 65°C
erhöht, und
9,51 g (83,3 mmol) Allylglycidylether werden innerhalb einer Stunde
zugesetzt. Die Reaktionsmischung wird zwei Stunden lang bei 65°C gerührt. Nach
dieser Zeit ist der Umsatz von Allylglycidylether vollständig, wie es
mittels Gaschromatographie verfolgt wird. Dieses Produkt wird nicht
isoliert, sondern direkt in dem nächsten Schritt verwendet.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 15,0 g (9,6 mmol) des Präpolymers
aus Beispiel 5A in einen Rundbodenkolben, ausgestattet mit einem
Rührer,
einem Stickstoffeinlass und einem Thermoregulator, gegeben. Zu diesem
Rundbodenkolben werden 11,2 g (96 mmol) Chloressigsäure-Natriumsalz
und 5,0 g entionisiertes Wasser gegeben. Die Reaktionsmischung wird
auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dem Rundbodenkolben werden 5,05 g (8,68 mmol) ZONYL TELA-N zusammen
mit 0,18 g (0,9 mmol) Natriummetabisulfit und 0,1 g (0,53 mmol)
2,2'-Azobisisobutyronitril
(AIBN) zugesetzt. Die Reaktionsmischung wird unter Stickstoff bei
80°C fünf Stunden
lang gerührt.
Nach fünf
Stunden wird die Mischung auf Raumtemperatur abgekühlt, und
Wasser wird zum Einstellen des Feststoffgehalts auf 34 Gew.-% und
auf 3,6% F zugesetzt. Der Umsatz von R-Iodid, bestimmt mittels Gaschromatographie,
beträgt
94%.
-
Beispiele 8
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 20,0 g (5 mmol) einer 50 Gew.-%igen wässrigen Lösung von Polyethylenimin mit
Mn 2000 von Aldrich in einen Rundbodenkolben, ausgestattet mit einem
Rührer,
Stickstoffeinlass und einem Thermoregulator, gegeben. Die Temperatur
wird auf 65°C
erhöht,
und 2,85 g (25 mmol) Allylglycidylether werden innerhalb einer Zeitdauer
von einer Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden
lang bei 65°C
gerührt.
Nach dieser Zeit ist der Umsatz an Allylglycidylether vollständig, wie
mittels Gaschromatographie bestimmt wird. Dieses Produkt wird nicht
isoliert, sondern direkt in dem nächsten Schritt verwendet.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 13,4 g (11,1 mmol) des Präpolymers
aus Beispiel 8A in einen Rundbodenkolben, ausgestattet mit einem
Rührer,
Stickstoffeinlass und einem Thermoregulator, gegeben. Dazu werden
3,22 g (27,6 mmol) Chloressigsäure-Natriumsalz
zugesetzt, und die Reaktionsmischung wird auf 75°C erwärmt und drei Stunden lang gerührt. Anschließend werden
5,1 g (10 mmol) ZONYL TELA-L zusammen mit 0,21 g (1,1 mmol) Natriummetabisulfit
und 0,09 g (0,45 mmol) 2,2'-Azobisisobutyronitril
(AIBN) zugesetzt. Die Reaktionsmischung wird unter Stickstoff bei
80°C fünf Stunden
lang gerührt.
Nach fünf
Stunden wird die Mischung auf Raumtemperatur abgekühlt, und
100 g Wasser werden zum Einstellen des Feststoffgehalts auf 20 Gew.-%
und 4,2% F zugesetzt. Der Umsatz an RF-Iodid,
bestimmt mittels Gaschromatographie, beträgt 96%.
-
Beispiel 9
-
Ein
Produkt wird dem Verfahren des Beispiels 8 folgend unter Verwendung
von ZONYL TELA-N mit den RF-Verteilungen,
wie sie anstelle von ZONYL TELA-L gegeben sind, synthetisiert.
-
Beispiele 10
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 60,0 g (85,7 mmol) Polyethylenimin, Mn 700 von der Aldrich
Chemicals und 15 g entionisiertes Wasser in einen Kolben, welcher
mit einem Rührer,
einem Stickstoffeinlass und einem Thermoregulator versehen ist,
gegeben. Die Temperatur der Reaktionsmischung wird auf 65°C erhöht, und
19,56 g (171 mmol) Allylglycidylether werden innerhalb einer Zeitdauer
von einer Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden
lang bei 65°C
gerührt.
Nach dieser Zeit ist der Umsatz von Allylglycidylether vollständig, wie
dies mittels Gaschromatographie verfolgt wird. Dieses Produkt wird
nicht isoliert, sondern direkt in dem nächsten Schritt verwendet.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 30 g (54,4 mmol) des Präpolymers
aus Beispiel 10A in einen Rundbodenkolben, welcher mit einem Rührer, einem
Stickstoffeinlass und einem Thermoregulator versehen ist, gegeben.
Dazu werden 25,3 g (218 mmol) Chloressigsäure-Natriumsalz und 9,5 g entionisiertes
Wasser gegeben. Die Reaktionsmischung wird auf 75°C erwärmt und
drei Stunden lang gerührt.
Anschließend
werden 7, 7 g (13 mmol) ZONYL TELA-N zusammen mit 0, 28 g (1, 5
mmol) Natriummetabisulfit und 0,11 g (0,59 mmol) 2,2'-Azobisisobutyronitril
(AIBN) zugesetzt. Die Reaktionsmischung wird unter Stickstoff bei
80°C fünf Stunden
lang gerührt.
Nach fünf
Stunden wird die Mischung auf Raumtemperatur abgekühlt, und
Wasser wird zum Einstellen des Feststoffgehalts auf 24 Gew.-% und
auf 5,1% F zugegeben. Der Umsatz an RF-Iodid,
bestimmt mittels Gaschromatographie, beträgt 95%.
-
Beispiel 11
-
A: Synthese von Poly(allyloxy-PEI)
-
Es
werden 50,0 g (0,04166 mol) Epomin® SP-012
(Polyethylenimin mit Mn 1200 von der Aceto Corporation) und 12,5
g entionisiertes Wasser in einen Kolben, welcher mit einem Rührer, einem
Stickstoffeinlass und einem Thermoregulator versehen ist, gegeben.
Sobald eine Temperatur von 65°C
erreicht worden ist, werden 23,77 g (0,2083 mol) Allylglycidylether
innerhalb einer Zeitdauer von ungefähr einer Stunde zugesetzt.
Die Reaktionsmischung wird zwei Stunden lang bei 65°C gerührt. Nach
dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 25,0 g (0,06036 mol, auf der Basis eines Doppelbindungs-Äquivalents)
des Produkts aus Beispiel 11A und 28,7 g (0,2414 mol) Chloressigsäure-Natriumsalz zu einem
Kolben, welcher mit einem Rührer,
einem Stickstoffeinlass und einem Thermoregulator versehen ist,
gegeben. Die Reaktionsmischung wird auf 75°C erwärmt und drei Stunden lang gerührt. Zu
dieser Zeit werden 31,61 g (0,0543 mol) (ZONYL TELA-N von DuPont)
zu der Mischung zusammen mit 1,15 g (6,034 mmol) Natriummetabisulfit
und 0,46 g (0,241 mmol) 2,2'-Azobisisobutyronitril
(AIBN) zugesetzt. Die Mischung wird unter Stickstoff bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird. Die Reaktionsmischung wird mit 100 g entionisiertem
Wasser unter Erhalt eines Produkts mit einem Feststoffgehalt von 39,6%
und 9,1% F verdünnt.
-
Beispiel 12
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 100,0 g (0,025 mol) Lupasol® G-35
(50% Polyethylenimin mit Mn 1800 von der BASF) in einen Kolben,
welcher mit einem Rührer,
einem Stickstoffeinlass und einem Thermoregulator ausgestattet ist, gegeben.
Sobald die Temperatur von 65°C
erreicht worden ist, werden 14,3 g (0,125 mol) Allylglycidylether innerhalb
einer Zeitdauer von ungefähr
einer Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden lang
bei 65°C
gerührt.
Nach dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 41,6 g (0,037343 mol) des Produkts aus Beispiel 12A und 17,75
g (0,149 mol) Chloressigsäure,
Natriumsalz in einen Kolben, welcher mit einem Rührer, einem Stickstoffeinlass
und einem Thermoregulator ausgestattet ist, gegeben. Die Reaktionsmischung
wird auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dieser Zeit werden 19,56 g (0,0336 mol) (ZONYL TELA-N von DuPont)
zu der Mischung zusammen mit 0,71 g (3,73 mmol) Natriummetabisulfit
und 0,229 g (1,49 mmol) 2,2'-Azobisisobutyronitril
(AIBN) zugesetzt. Die Mischung wird unter Stickstoff bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 150 g entionisiertem Wasser
unter Erhalt eines Produkts mit einem Feststoffgehalt von 26,7%
mit 5,1% F verdünnt.
-
Beispiel 13
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 65,0 g (0,0361 mol) Lupasol® PR-8515
(Polyethylenimin mit Mn 1800 von der BASF) und 4,0 g entionisiertes
Wasser in einen Kolben, welcher mit einem Rührer, einem Stickstoffeinlass
und einem Thermoregulator versehen ist, gegeben. Die Mischung wird
auf 65°C
erwärmt,
und 18,54 g (0,1625 mol) Allylglycidylether werden innerhalb einer
Zeitdauer von ungefähr
einer Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden
lang bei 65°C
gerührt.
Nach dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird.
-
B: Synthese von Poly-RF-PEI-carbonsäure
-
Es
werden 15,5 g (0,02877 mol) des Produkts aus Beispiel 13A und 23,46
g (0,2013 mol) Chloressigsäure-Natriumsalz
in einen Kolben, welcher mit einem Rührer, einem Stickstoffeinlass
und einem Thermoregulator ausgestattet ist, zusammen mit 8,2 g entionisiertem
Wasser gegeben. Die Reaktionsmischung wird auf 75°C erwärmt und
drei Stunden lang gerührt.
Zu dieser Zeit werden 13,0 g (0,02589 mol) (ZONYL TELA-L von DuPont)
zu der Mischung zusammen mit 0,55 g (2,88 mmol) Natriummetabisulfit
und 0,22 g (1,15 mmol) 2,2'-Azobisisobutyronitril
(AIBN) und 1,0 g 1-Propanol zugesetzt. Die Mischung wird unter Stickstoff
bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird. Die Reaktionsmischung wird mit 70 g entionisiertem
Wasser unter Erhalt eines Produkts mit 28,6% Feststoffen mit 5,68%
F verdünnt.
-
Beispiel 14
-
Synthese einer C6F13-(Allyloxy/Iodpropl-substituierten
Polyaminopolysäure
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 100 g (0,0833 mol) Polyethylenimin mit Mn 1200 (Lupasol® G-20
von der BASF) und 7,0 g entionisiertes Wasser in einen Kolben, welcher
mit einem Rührer,
einem Stickstoffeinlass und einem Thermoregulator versehen ist,
gegeben. Sobald eine Temperatur von 65°C erreicht wird, werden 38,03
g (0,333 mol) Allylglycidylether innerhalb einer Zeitdauer von ungefähr einer
Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden lang bei
65°C gerührt. Nach
dieser Zeit ist die Umsetzung vollständig, wie mittels Gaschromatographie
bestimmt wird.
-
B: Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-perfluorhexylheptyl-N-carboxymethyl-poly(ethylenimin)
-
Es
werden 24,6 g (0,0565 mol) des Produkts aus Beispiel 14A und 34,57
g (0,2968 mol) Chloressigsäure-Natriumsalz
zu einem Kolben, welcher mit einem Rührer, einem Stickstoffeinlass
und einem Thermoregulator ausgestattet ist, zusammen mit 12,0 g
entionisiertem Wasser zugegeben. Die Reaktionsmischung wird auf
75°C erwärmt und
drei Stunden lang gerührt.
Zu dieser Zeit werden 22,69 g (0,051 mol) Perfluorhexyliodid zu
der Mischung zusammen mit 1,07 g (5,65 mmol) Natriummetabisulfit
und 0,43 g (2,26 mmol) 2,2'-Azobisisobutyronitril
(AIBN) und 2,0 g 1-Propanol zugesetzt. Die Mischung wird unter Stickstoff
bei 80°C
fünf Stunden lang
gerührt.
Zu dieser Zeit wird die Umsetzung mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 65,0 g entionisiertem Wasser
und 5,0 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 49,0% Feststoffen mit 7,5% F verdünnt.
-
Die
Zusammensetzungen der Beispiele 1 bis 14 sind nachstehend zusammengefasst.
-
-
5. Beispiel 15
-
Leitungsfähigkeit
von Schaumstabilisatoren mit Meerwasser (0,8% F in der Zubereitung)
-
Die
Wirksamkeit der Verbindungen als Schaumstabilisatoren wurde durch
Herstellung eines AR-AFFF-Konzentrats durch Mischen der vorstehend
erwähnten
AR-AFFF-Basiszubereitung mit einem Anteil von 0,6% F mit den Schaumstabilisatoren
der Beispiele 1 bis 14 in einem Anteil von 0,2% F bestimmt. Das Schaumausdehnungsverhältnis, die
Viertelaustrittszeit und die Schaumlebensdauer auf heißem 2-Propanol wurden
unter Verwendung einer Vormischung, welche 3% des Konzentrats in
Meerwasser enthielt, bestimmt. Eine AR-AFFF-Basisprobe ohne zugesetzten
Schaumstabilisator wurde als Kontrolle (Beispiel 15p) verwendet.
Mehrere Beispiele (1, 4, 8, 10, 11 und 13) zeigten eine ausgezeichnete
Schaumlebensdauer auf heißem 2-Propanol
(> 30 Minuten).
-
-
5. Beispiel 16
-
Proben
von Polyperfluoralkyl-substituierten Poly(aminsäuren) (Beispiele 1, 6, 7 und
9) wurden als externe Papierschlichten unter Verwendung der vorstehend
beschriebenen Leimpressauftragung beurteilt. Die Oil-Kit-Zahlen
wurden bei den angewendeten Fluoranteilen verzeichnet.
-
-
6. Beispiel 17
-
Synthese einer Poly-RF-substituierten Poly(aminsulfonsäure)
-
A: Synthese von Polyallyloxy-PEI
-
Es
werden 404,8 g (0,337 mol) Polyethylenimin mit Mn 1200 (Lupasol® G-20
von der BASF) und 26,68 g entionisiertes Wasser in einen mit einem
Rührer,
Stickstoffeinlass und einem Thermoregulator ausgestatteten Kolben
gegeben. Sobald die Temperatur von 65°C erreicht wird, werden 115,47
g (1,012 mol) Allylglycidylether innerhalb einer Zeitdauer von ungefähr einer
Stunde zugesetzt. Die Reaktionsmischung wird zwei Stunden lang bei
65°C gerührt. Nach
dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird.
-
B: Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyll-N-(2-hydroxy-3-sulfonsäurepropyl)poly(ethylenimin)
-
Es
werden 16,6 g (0,03071 mol) des Produkts aus Beispiel 17A und 40,2
g (0,1944 mol) 3-Chlor-2-hydroxy-1-propansulfonsäure-Natriumsalz in einen mit
einem Rührer,
Stickstoffeinlass und einem Thermoregulator ausgestatteten Kolben
zusammen mit 12,0 g entionisiertem Wasser gegeben. Die Reaktionsmischung wird
auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dieser Zeit werden 14,0 g (0,0276 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,58 g (3,07 mmol)
Natriummetabisulfit und 0,24 g (1,23 mmol) 2,2'-Azobisisobutyronitril (AIBN) zugesetzt.
Die Mischung wird unter Stickstoff bei 80°C fünf Stunden lang gerührt. Zu
dieser Zeit wird die Reaktion mittels Gaschromatographie als vollständig bestimmt.
Die Reaktionsmischung wird anschließend mit 25 g entionisiertem
Wasser und 3,25 g Tripropylenglykolmonomethylether unter Erhalt
eines Produkts mit 59,6% Feststoffen verdünnt.
-
7. Beispiel 18
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-perfluorhexylheptyl)-N-carboxymethyl-N-amidomethylpoly(ethylenimin)
-
Es
werden 12,1 g (0,02239 mol) des Produkts aus Beispiel 17A und 8,68
g (0,07455 mol) Chloressigsäure-Natriumsalz
und 6,97 g (0,07455) 2-Chloracetamid in einen Kolben, welcher mit
einem Rührer,
einem Stickstoffeinlass und einem Thermoregulator ausgestattet ist,
zusammen mit 3,0 g entionisiertem Wasser und 2,1 g (0,027 mol) 50%-igem
Natriumhydroxid zugesetzt. Die Reaktionsmischung wird auf 75°C erwärmt und drei
Stunden lang gerührt.
Zu dieser Zeit werden 8,99 g (0,0201 mol) Perfluorhexyliodid zu
der Mischung zusammen mit 0,48 g (2,01 mmol) Natriummetabisulfit,
0,17 g (0,896 mmol) 2,2'-Azobisisobutyronitril
(AIBN) und 1,3 g 1-Propanol gegeben. Die Mischung wird unter Stickstoff
bei 80°C
fünf Stunden
lang gerührt.
Zu dieser Zeit ist die Reaktion vollständig, wie mittels Gaschromatographie
bestimmt wird. Die Reaktionsmischung wird mit 20,5 g entionisiertem
Wasser und 1,98 g Tripropylenglykolmonomethylether unter Erhalt
eines Produkts mit 56,9% Feststoffen mit 7,5% F verdünnt.
-
8. Beispiel 19
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-perfluorhexylheptyl)-N-carboxymethyl-N-amidomethylpoly(ethvlenimin)
-
Es
werden 11,9 g (0,022 mol) des Produkts aus Beispiel 17A, 12,8 g
(0,110 mol) Chloressigsäure-Natriumsalz
und 3,49 g (0,0365 mol) 2-Chloracetamid in einen mit einem Rührer, Stickstoffeinlass
und einem Thermoregulator ausgestatteten Kolben zusammen mit 4,0
g entionisertem Wasser und 1,0 g (0,0125 mol) 50%-igem Natriumhydroxid
zugegeben. Die Reaktionsmischung wird auf 75°C erwärmt und drei Stunden lang gerührt. Zu
dieser Zeit werden 8,84 g (0,0198 mol) Perfluorhexyliodid (von der
Hoechst AG) zu der Mischung zusammen mit 0,42 g (2,2 mmol) Natriummetabisulfit,
0,17 g (0,88 mmol) 2,2'-Azobisisobutyronitril
(AIBN) und 1,5 g 1-Propanol zugesetzt. Die Mischung wird unter Stickstoff
bei 80°C
fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion durch Gaschromatographie als vollständig bestimmt.
Die Reaktionsmischung wird mit 19,0 g entionisiertem Wasser und
1,95 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 52,9% Feststoffen mit 7,5% F verdünnt.
-
9. Beispiel 20
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-perfluorhexylheptyl-N-carboxymethyl-N-amidomethyl-N-(2-hydroxy-3-trimethylammoniumpropyl)poly(ethylenimin)
-
Es
werden 11,1 g (0,0205 mol) des Produkts aus Beispiel 17A, 7,32 g
Glycidyltrimethylammoniumchlorid (Quab 151 von der Degussa), 7,97
g (0,0685 mol) Chloressigsäure-Natriumsalz
und 3,2 g (0,0342 mol) 2-Chloracetamid in einen mit einem Rührer, Stickstoffeinlass
und einem Thermoregulator versehenen Kolben zusammen mit 3,0 g entionisiertem
Wasser gegeben. Die Reaktionsmischung wird auf 75°C erwärmt und
drei Stunden lang gerührt.
Zu dieser Zeit werden 8,23 g (0,01845 mol) Perfluorhexyliodid zu
der Mischung zusammen mit 0,39 g (2,05 mmol) Natriummetabisulfit,
0,17 g (0,82 mmol) 2,2'-Azobisisobutyronitril
(AIBN) und 0,6 g 1-Propanol gegeben. Die Mischung wird unter Stickstoff
bei 80°C
fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 18,1 g entionisiertem Wasser
und 1,86 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 56,6% Feststoffen mit 7,3% F verdünnt.
-
Beispiel 21
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-perfluorhexylheptyl)-N-carboxymethyl-N-triphosphatpoly(ethylenimin)
-
Es
werden 15,6 g (0,02889 mol) des Produkts aus Beispiel 17A, 14,65
g (0,0479 mol) Natriumtrimetaphosphat und 16,8 g (0,144 mol) Chloressigsäure-Natriumsalz zu einen
mit einem Rührer,
Stickstoffeinlass und einem Thermoregulator ausgestatteten Kolben
zusammen mit 7,0 g entionisiertem Wasser gegeben.
-
Die
Reaktionsmischung wird auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dieser Zeit werden 13,2 g (0,02597 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,55 g (2,89 mmol)
Natriummetabisulfit, 0,22 g (1,1 mmol) 2,2'-Azobisisobutyronitril (AIBN) und 1,5
g 1-Propanol gegeben. Die Mischung wird unter Stickstoff bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 34,78 g entionisiertem Wasser
und 3,2 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 55,9% Feststoffen mit 7,1% F verdünnt.
-
10. Beispiel 22
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl)-N-carbomethyl-N-triphosphatpoly(ethylenimin)
-
Es
werden 16,0 g (0,0296 mol) des Produkts aus Beispiel 17A, 30,18
g (0,0987 mol) Natriumtrimetaphosphat von Monsanto und 11,48 g (0,0987
mol) Chloressigsäure-Natriumsalz
in einen mit einem Rührer, Stickstoffeinlass
und einem Thermoregulator ausgestatteten Kolben zusammen mit 8,0
g entionisiertem Wasser gegeben. Die Reaktionsmischung wird auf
75°C erwärmt und
drei Stunden lang gerührt.
Zu dieser Zeit werden 13,55 g (0,0266 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,56 g (2,96 mmol)
Natriummetabisulfit, 0,23 g (1,1 mmol) 2,2'-Azobisisobutyronitril (AIBN) und 1,5
g 1-Propanol gegeben. Die Mischung wird unter Stickstoff bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 24,7 g entionisiertem Wasser
und 3,28 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 65,0% Festoffen mit 7,18% F verdünnt.
-
11. Beispiel 23
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl)-N-carbomethyl-N-2,3-dihydroxypropyl-poly(ethylenimin)
-
Es
werden 20,6 g (0,0381 mol) des Produkts aus Beispiel 17A, 9,81 g
(0,127 mol) Glycidol und 15,1 g (0,127 mol) Chloressigsäure-Natriumsalz
in einen mit einem Rührer,
Stickstoffeinlass und einem Thermoregulator ausgestatteten Kolben
zusammen mit 7,0 g entionisiertem Wasser gegeben. Die Reaktionsmischung
wird auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dieser Zeit werden 17,4 g (0,0349 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,72 g (3,81 mmol)
Natriummetabisulfit und 0,29 g (1,524 mmol) 2,2'-Azobisisobutyronitril (AIBN) zugesetzt.
Die Mischung wird unter Stickstoff bei 80°C fünf Stunden lang gerührt. Zu
dieser Zeit wird die Reaktion mittels Gaschromatographie als vollständig bestimmt. Die
Reaktionsmischung wird mit 56,1 g entionisiertem Wasser und 3,9
g Tripropylenglykolmonome thylether unter Erhalt eines Produkts mit
47,9% Feststoffen mit 7,7% F verdünnt.
-
Beispiel 24
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl)-N-amidomethyelen-N-triphosphatpoly(ethylenimin)
-
Es
werden 16,2 g (0,03 mol) des Produkts aus Beispiel 17A, 29,0 g (0,0949
mol) Natriumtrimetaphosphat von Monsanto und 8,9 g (0,0949 mol)
2-Chloracetamid
in einen mit einem Rührer,
Stickstoffeinlass und einem Thermoregulator ausgestatteten Kolben
zusammen mit 7,0 g entionisiertem Wasser gegeben. Die Reaktionsmischung
wird auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dieser Zeit werden 13,7 g (0,027 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,57 g (2,99 mmol)
Natriummetabisulfit, 0,23 g (1,12 mmol) 2,2'-Azobisisobutyronitril (AIBN) und 2,0
g 1-Propanol zugegeben. Die Mischung wird unter Stickstoff bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 26,97 g entionisiertem
Wasser und 3,26 g Tripropylenglykolmonomethylether unter Erhalt
eines Produkts mit 58,98% Feststoffen mit 6,98% F verdünnt.
-
12. Beispiel 25
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl)-N-amidomethyl-poly(ethylenimin)
-
Es
werden 15,1 g (0,0279 mol) des Produkts aus Beispiel 17A und 16,5
g (0,177 mol) 2-Chloracetamid in einen mit einem Rührer, Stickstoffeinlass
und einem Thermoregulator ausgestatteten Kolben zusammen mit 6,0
g entionisiertem Wasser gegeben. Die Reaktionsmischung wird auf
75°C erwärmt und
drei Stunden lang gerührt.
Zu dieser Zeit werden 12,8 g (0,0251 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,53 g (2,79 mmol)
Natriummetabisulfit, 0,21 g (1,1 mmol) 2,2'-Azobisisobutyronitril (AIBN) und 1,9
g 1-Propanol zugegeben. Die Mischung wird unter Stickstoff bei 80°C fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 45,4 g entionisiertem Wasser
und 3,0 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 43,7% Feststoffen mit 7,3% F verdünnt.
-
13. Beispiel 26
-
Synthese von Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl)-N-amidomethylen-N-2,3-dihydroxypropyl-poly(ethylenimin)
-
Es
werden 12,7 g (0,02349 mol) des Produkts aus Beispiel 17A, 5,74
g (0,0744 mol) Glycidol und 6,96 g (0,0744 mol) 2-Chloracetamid
in einen mit einem Rührer,
Stickstoffeinlass und einem Thermoregulator ausgestatteten Kolben
zusammen mit 3,0 g entionisiertem Wasser gegeben. Die Reaktionsmischung
wird auf 75°C
erwärmt
und drei Stunden lang gerührt.
Zu dieser Zeit werden 10,7 g (0,021 1 mol) Perfluoralkyliodid (ZONYL
TELA-L von DuPont) zu der Mischung zusammen mit 0,47 g (2,35 mmol)
Natriummetabisulfit, 0,23 g (1,12 mmol) 2,2'-Azobisisobutyronitril
(AIBN) und 1,2 g 1-Propanol zugegeben. Die Mischung wird unter Stickstoff bei
80°C fünf Stunden
lang gerührt.
Zu dieser Zeit wird die Reaktion mittels Gaschromatographie als
vollständig
bestimmt. Die Reaktionsmischung wird mit 41,8 g entionisiertem Wasser
und 2,55 g Tripropylenglykolmonomethylether unter Erhalt eines Produkts
mit 28,8% Feststoffen mit 7,3% F verdünnt.
-
14. Beispiel 17
-
Synthese einer Poly-RF-poly(aminosulfonsäure) Poly-N-(2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl)-N-ethylsulfonsäure-poly(ethylenimin)
-
Beispiel 1: A Reaktion
von Polyethylenimin und Vinylsulfonsäure-Natriumsalz
-
In
einen Rundbodenkolben werden 10 g (7,7 mmol) Polyethylenimin (Luposol® G-20,
wasserfrei, Mn=1200 von der BASF) zusammen mit 49,9 g (92,4 mmol)
Vinylsulfonsäure-Natriumsalz
(50%ige wässrige Lösung) gegeben.
Diese Mischung wird 12 Stunden lang bei 80°C gerührt. Nach 12 Stunden wird das
Wasser mittels Vakuum entfernt, wobei eine Ausbeute von 33,2 g (95%)
erhalten wird.
-
Beispiel 2: B Umsetzung
von AGE mit Polyethylenimin-Vinylsulfonsäure-Natriumsalz-Addukt
-
Eine
wässrige
Lösung
von 9,0 g (3,8 mmol) des Polyethylenimin-Vinylsulfonsäure-Natriumsalz-Addukts
27A, gelöst
in 5,3 g entionisiertem Wasser, wird auf 55°C erwärmt. Zu dieser Lösung werden
0,86 g (7,6 mmol) AGE (Allylglycidylether) tropfenweise unter Verwendung
eines Tropftrichters gegeben. Das Erwärmen wird solange fortgesetzt,
bis sämtliches
AGE verbraucht ist. Der Umsatz von AGE wird mittels Gaschromatographie
verfolgt. Die Produktlösung
wird in einer Ausbeute von 14,9 (98%) erhalten.
-
C Umsetzung des Polyallyloxypolyethylenimin-Polyvinylsulfonsäuresalzes
mit Perfluoralkyliodid
-
Eine
Mischung von 14,9 g (7,4 mmol) Polyallyloxypolyethylenimin-Polyvinylsulfonsäurenatriumsalz aus
Schritt B, 4,25 g (7,2 mmol) Perfluoralkyliodid (ZONYL TELA-L) und
0,75 g n-Propanol werden unter Rühren
auf 85°C
erwärmt.
Gleichzeitig werden 0,14 g (0,74 mmol) Natriummetabisulfit und 57
mg (0,3 mmol) VAZO® 67 zugesetzt. Diese Mischung
wird erwärmt
und über
Nacht bei 85°C
gerührt.
Nach dem Rühren über Nacht
wird die Reaktionsmischung mit 10 ml Wasser verdünnt. Die Ausbeute beträgt 29,1
g (97%).
-
15.Beispiel 28
-
Synthese von Poly-N-2-hydroxy-4-oxa-[6,7-en-
und -6-iod]-7-RF-heptyl-N-carboethylpoly(ethylenimin)
-
A. Umsetzung von Polyethylenimin
mit Acrylsäure
-
In
einen Reaktor, welcher 13,0 g (10 mmol) Polyethylenimin (PEI; MW
= 1200) und 14,0 g entionisiertes Wasser enthält, werden 8,6 g (119 mmol)
Acrylsäure
unter Rühren
bei 40°C
zugegeben. Die Mischung wird auf 75°C erwärmt und dort 14 Stunden lang
gehalten, wobei eine gelbe dickflüssige Lösung erhalten wird. Mittels
Gaschromatographie wird die Abwesenheit von Acrylsäure bestätigt. Der
Umsatz zum Produkt beträgt 91%.
Die Struktur des PEI-Acrylsäure-Addukts
wird mittels 1H NMR (500 mhz, CD3OD) bestätigt:
6 = 3,77, -CH 2COO-,
2H), 2,40 (t, -NR2 CH 2COO-, 2H), 2,6-3,1
(bm, -NR2 CH 2 CH 2NR2-, 4H).
-
B. Umsetzung des PEI-Acrylsäure-Addukts
mit Allylglycidylether
-
Zu
einem Reaktionskolben, welcher eine Mischung von 6,3 g (78,5 mmol)
50-%igem Natriumhydroxid und 23,3 g (6,54 mmol) des PEI-Acrylsäure-Addukts
aus Schritt A enthält,
erwärmt
auf 65°C,
werden mittels einer Spritze 2,2 g (19,6 mmol) Allylglycidylether
(AGE) zugesetzt. Nach 4,5 Stunden zeigt die GC-Analyse auf einer
30 m × 0,53
mm SPB-5-Polysiloxansäule
nur Spuren des restlichen Epoxids. Die Struktur des PEI-Acrylsäure-Allylglycidylether-Addukts
wird mittels 1H NMR verifiziert.
-
C. Umsetzung des PEI-Acrylsäure-AGE-Addukts
mit RF-Iodid
-
Zu
einem Reaktionskolben, welcher 10,2 g (20,8 mmol) PEI-Acrylsäure-Allylglycidylether-Addukt
aus Schritt B enthält,
werden 0,1 g (0,654 mmol) Natriummetabisulfit, 0,05 g (0,26 mmol)
2,2'-Azobis-(2-methylbutyronitril)
(DuPont's VAZO-67),
1,5 g n-Propanol und 10,2 g (20,8 mmol) Perfluoralkyliodid (DuPont's ZONYL TELA-L) mit
einer homologen Verteilung von 47,0% C6F13I, 37,2% C8F17I, 11,8% C10F21I, 3,0% C12F25I, 0,8% C14F29I und 0,2% C16F33I gegeben. Die Mischung wird auf 77 bis
80°C erwärmt, und
nach zwei Stunden erfolgt eine weitere Zugabe von Natriummetabisulfit
und VAZO-67 (jeweils 0,1 g und 0,05 g). Nach vier zusätzlichen Stunden
sind 2,7 Gew.-% des Perfluoralkyliodids auf der Basis der Gaschromatographie
unumgesetzt. Es werden 39,7 g entionisiertes Wasser zu dem Produkt
unter Erhalt einer klaren, leicht bernsteinfarbenen Flüssigkeit (80,0
g; 95% Ausbeute) mit einem pH von 9 zugesetzt.
-
Die
Zusammensetzungen der Beispiele 17 bis 28 sind in Tabelle 4 zusammengefasst. Tabelle
4
- (1)1
- Anzahlmittleres Molekulargewicht
- 2
- Bestimmtes Verhältnis von
primären,
sekundären
und tertiären
Aminogruppen
- 3
- Bestimmter Gesamtrestgehalt
an sekundärem
Amin
- 4
- ZONYL TELA-L
- 5
- Perfluorhexyliodid
- HPS
- = 3-Chlor-2-hydroxy-1-propansulfonsäure
- CA
- = 2-Chloracetamid
- CAC
- = Natriumchloracetat
- QUAB
- = Glycidyltrimethylammoniumchlorid
(Quab 151)
- TMP
- = Natriumtrimetaphosphat
- GLY
- = Glycidol
- VSA
- = Vinylsulfonsäure
- AA
- = Acrylsäure
-
(2) Beispiel 29
-
Leistungsfähigkeit
der Schaumstabilisatoren mit Meerwasser (0,8% F in der Zubereitung)
-
Die
Wirksamkeit der Verbindungen als Schaumstabilisatoren wurde durch
Herstellung eines AR-AFFF-Konzentrats durch Mischen der vorstehend
erwähnten
AR-AFFF-Basiszubereitung mit einem Anteil von 0,6% F mit den Schaumstabilisatoren
der Beispiele 17 bis 28 in einem Anteil von 0,2% F bestimmt. Das Schaumausdehnungsverhältnis, die
Viertelaustrittszeit und die Schaumlebensdauer auf heißem 2-Propanol und
Aceton wurden unter Verwendung einer 3%-igen Vormischung in Salzwasser bestimmt.
Ein kommerzieller Schaumstabilisator, DYNAX 5011 (Dynax Corp., Elmsford,
NY), und eine AR-AFFF-Basisprobe ohne zugesetzten Schaumstabilisator
wurden als Kontrolle (Beispiele 29i und 29p) verwendet. Die Ergebnisse
sind in der folgenden Tabelle zusammengefasst.
-
-
17. Beispiel 30
-
Leistungsfähigkeit
der Schaumstabilisatoren mit Leitungswasser (0,8% F in der Zubereitung)
-
Die
Wirksamkeit der Verbindungen als Schaumstabilisatoren wurde durch
Herstellung eines AR-AFFF-Konzentrats durch Mischen der vorstehend
erwähnten
AR-AFFF-Basiszubereitung mit einem Anteil von 0,6% F mit den Schaumstabilisatoren
der Beispiele 17 bis 28 in einem Anteil von 0,2% F bestimmt. Das Schaumausdehnungsverhältnis, die
Viertelaustrittszeit und die Schaumlebensdauer auf heißem 2-Propanol und
Aceton wurden unter Verwendung einer 3%-igen Vormischung in Leitungswasser bestimmt.
Ein kommerzieller Schaumstabilisator, DYNAX 5011 und eine AR-AFFF-Basisprobe
ohne zugesetzten Schaumsta bilisator wurden als Kontrolle (Beispiele
30i und 30p) verwendet. Die Ergebnisse sind in der folgenden Tabelle
zusammengefasst.
-
-
(i) Beispiel 31
-
Das
folgende Beispiel beschreibt die Synthese eines Schaumstabilisators
durch direkte Addition von RF-Iodid an PEI,
gefolgt von der Umsetzung mit Natriumchloracetat.
-
A: Umsetzung von PEI mit
Perfluorethyliodid
-
Synthese von Poly-(N-1,1,2-trihydro-4-fluor-3-perfluoralkylallyl)ethylenimin
-
Bei
85°C werden
10,0 g (19,0 mmol) Perfluorethyliodid (ZONYL TELB-L) zu einer klaren
Lösung
von 8,2 g (195 mäq)
Polyethylenimin mit Mn 1200 (Lupasol® G-20
von der BASF) und 3,0 g Hexylenglykol gegeben. Die Mischung wird
auf 103°C
unter Rühren
erwärmt
und dort sechs Stunden lang unter Erhalt eines schwarzen dickflüssigen Produkts,
welches wasserlöslich
ist, gehalten. Mittels Gaschromatographie (DB-5-Säule, 30 × 0,53 mm)
verbleiben weniger als 5 mol-% des Perfluorethyliodids, und 95 mol-%
des ionischen Iodids werden erhalten, wie durch Silbernitrattitration
bestimmt wird. Das Produkt wird anschließend in 97%-iger Ausbeute gesammelt.
Spektroskopische Daten: 1H NMR (CD3OD, 500 MHz): 2,6-3,2 (4H, bm, -CH 2CH 2-), 3,5 (bm,
2H, -CH 2-CH-),
6,02 (1H, bm, -CH=CF-); 13C NMR (CD3OD, 300
MHz): δ 47,3
(-CH2-CH-), 47,5 und 52,4 (-CH2CH2-), 110,1 (CH=CF-), 110,2-120,3 (CF), 150,5
(-CF=), 19F (CD3OD,
300 MHz): -83,2 (3F, CF3), -115,4 (2F, F2), -119,6 (2F, F7),
-123,1 bis -125,4 (8F, C3 bis C6),
-129,7 (1F, C8).
-
B: Carboxylierung
-
Synthese von Poly-(N-1,1,2-trihydro-3-fluor-3-perfluoralkylallyl)-(N-carboxymethyl)ethylenimin
-
Natriumchloracetat
(13,3 g; 114,1 mmol) wird zu der oben erhaltenen Produktmischung
zugegeben. Die Reaktionsmischung wird auf 84°C vier bis fünf Stunden lang erwärmt. Zu
dieser Zeit wird eine quantitative Menge des Chlorids auf der Basis
von Silbernitrattitration erhalten. Das wasserlösliche Produkt wird in 98%-iger
Ausbeute gesammelt. Die NMR-Spektroskopie verifiziert die Carboxylierung.
-
Beispiel 32
-
Das
folgende Beispiel zeigt das neue Niedrigtemperaturverfahren für die RF-Iodid-Addition
an allylische ungesättigte
Bindungen unter Verwendung geringer Mengen Dithionit als Initiator
in Gegenwart einer Base.
-
Synthese von Poly-N-2-hydroxy-4-oxa-[6,7-en
und -6-iod-]-7-RF-heptyl-N-carboxymethylenpoly(ethylenimin)(= Poly-RF-PEI-carbonsäure)
-
Es
werden 15,0 g (24,4 mmol) des Präpolymers
aus Beispiel 1A in einen mit einem Rührer, Stickstoffeinlass und
einem Thermoregulator ausgestatteten Rundbodenkolben gegeben und
auf 50°C
erwärmt.
Anschließend
wird eine Lösung
von 18,0 g (154 mmol) Chloressigsäure-Natriumsalz mit 27 g entionisiertem
Wasser innerhalb von zwei Stunden zugesetzt, währenddessen die Temperatur
bei 65°C
gehalten wird. Nach zwei Stunden werden 1,71 g (21,3 mmol) 50%-iges
Natriumhydroxid zugesetzt. Die Vollständigkeit der Umsetzung wird
durch Chloridtitration mit Silbernitrat bestimmt. Die Temperatur
wird anschließend
auf 8°C
vermindert, und 4,84 g Hexylenglykol und 11,11 g (21,9 mmol) Perfluoralkyliodid
(ZONYL TELA-L) werden zusammen mit 0,17 g (0,81 mmol) Natriumdithionit
zugesetzt. Nach zwei Stunden wird die Temperatur auf 15°C erhöht, und
ein Rühren
wird weitere vier Stunden fortgesetzt. Anschließend werden 0,65 g (8,13 mmol)
einer 50%-igen Natriumhydroxid-Lösung
zusammen mit 6 g entionisiertem Wasser zugesetzt. Das Produkt wird
in 97%-igem Umsatz, wie mittels Gaschromatographie bestimmt wird,
als eine 48%-ige Lösung
mit einem pH von 7,0 bis 7,4 und einem Gehalt von 7,6% Fluor erhalten.
-
Beispiel 33
-
Leistungsfähigkeit
der Schaumstabilisatoren in Leitungs- und Meerwasser (0,8% F in
der Zubereitung)
-
Die
Wirksamkeit der Verbindungen der Beispiele 31 und 32 als Schaumstabilisatoren
wurde durch Herstellung einer AR-AFFF-Basiszubereitung mit einem
Anteil von 0,6% F mit den Schaumstabilisatoren wie in Tabelle 10
gezeigt in einem Anteil von 0,2% F bestimmt. Das Schaumausdehnungsverhältnis, die
Viertelaustrittszeit und die Schaumlebensdauer auf heißem 2-Propanol
und Aceton wurden unter Verwendung einer 3%-igen Vormischung in
Leitungs- und Meerwasser bestimmt.
-
-
Die
folgenden Beispiele zeigen die Wirksamkeit des neuen Schaumstabilisators
in Kombination mit kommerziellen AR-AFFF- und AFFF-Mitteln.
-
Beispiel 34
-
Die
Wirksamkeit des Produkts des Beispiels 32 wurde durch Zugabe von
3,2 Gew.-% zu kommerziellen AR-AFFF-Mitteln bestimmt. Das Schaumausdehnungsverhältnis, die
Viertelaustrittszeit und die Schaumlebensdauer auf heißem 2-Propanol
und Aceton wurden unter Verwendung einer 3%-igen Vormischung in Salz- und Leitungswasser
bestimmt. Die Ergebnisse sind in der folgenden Tabelle 8 angegeben.
Tabelle
8
- FXR
- = Schaumausdehnungsverhältnis;
- QDT
- = Viertelaustrittszeit;
- FL
- = Schaumlebensdauer,
alles in Minuten
-
Beispiel 35
-
Ein
3%-iges AFFF-Konzentrat wurde hergestellt, welches Lodyne
® S-152B
von der Ciba Specialty Chemicals Corp., 15 Gew.-%; Butylcarbitol
10% und Wasser 75% enthielt. Zu diesem Konzentrat wurden 3,2 Gew.-%
des Produkts aus Beispiel 32 zugesetzt, und das Schaumausdehnungsverhältnis, die
Viertelaustrittszeit und die Schaumlebensdauer auf heißem 2-Propanol
und Aceton wurden unter Verwendung einer 3%-igen Vormischung in
Salz- und Leitungswasser bestimmt. Das unmodifizierte Konzentrat
wurde als Kontrolle verwendet. Die Ergebnisse sind in Tabelle 9
gezeigt.
Tabelle
9
- FXR
- = Schaumausdehnungsverhältnis;
- QDT
- = Viertelaustrittszeit;
- FL
- = Schaumlebensdauer,
alles in Minuten
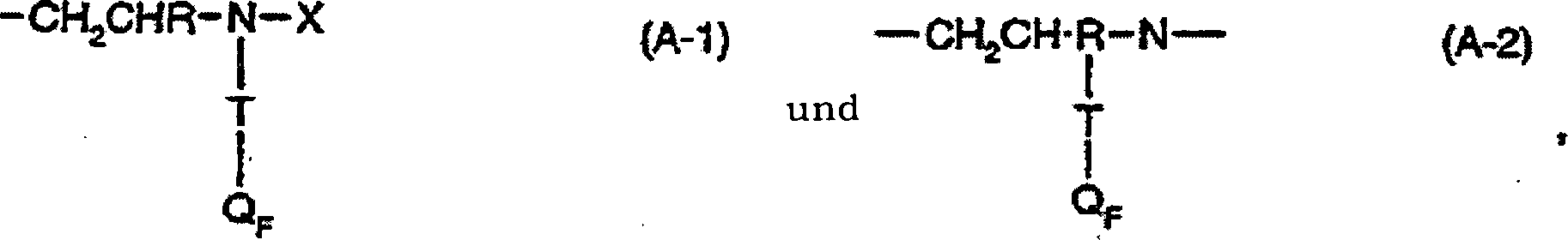


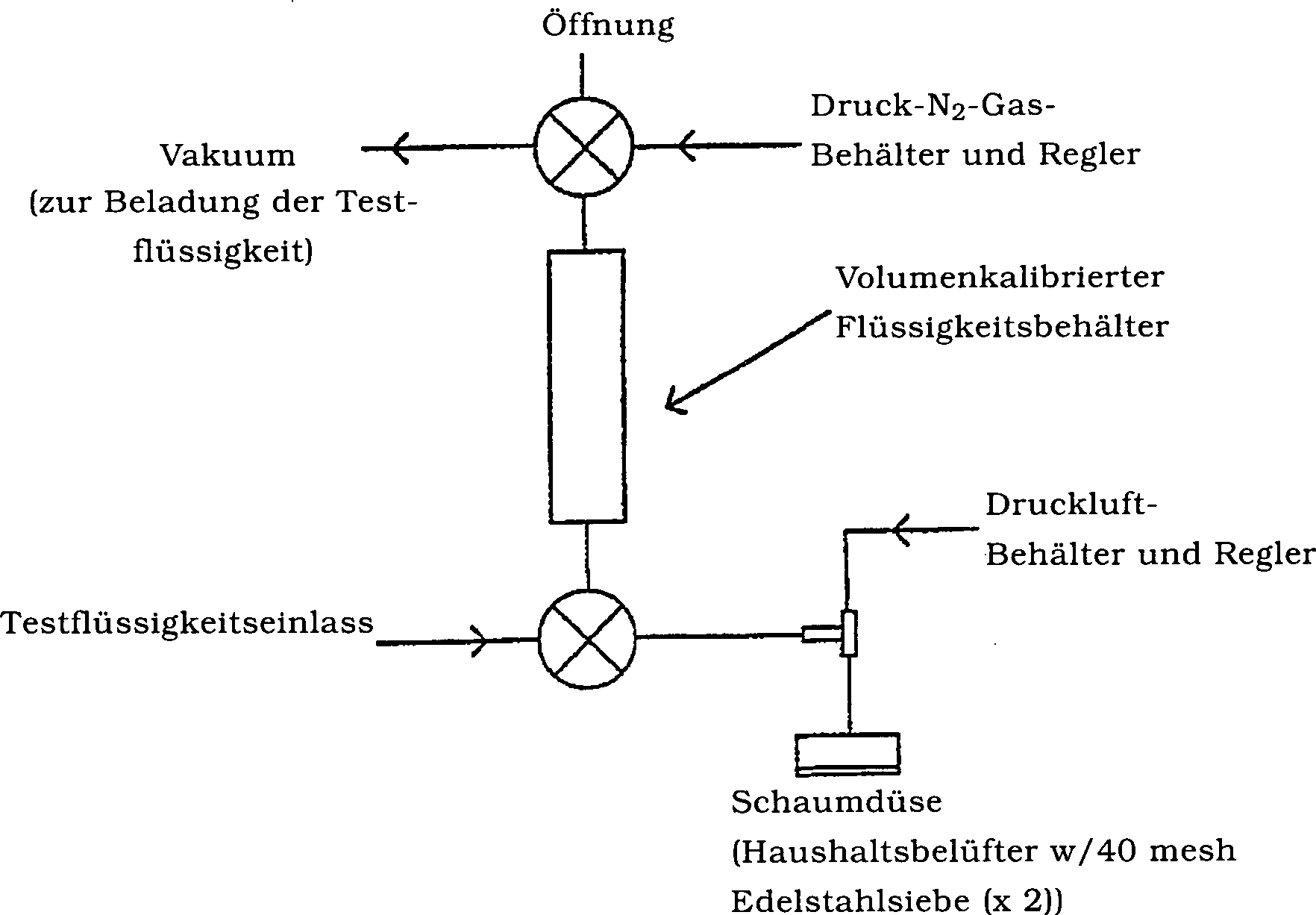


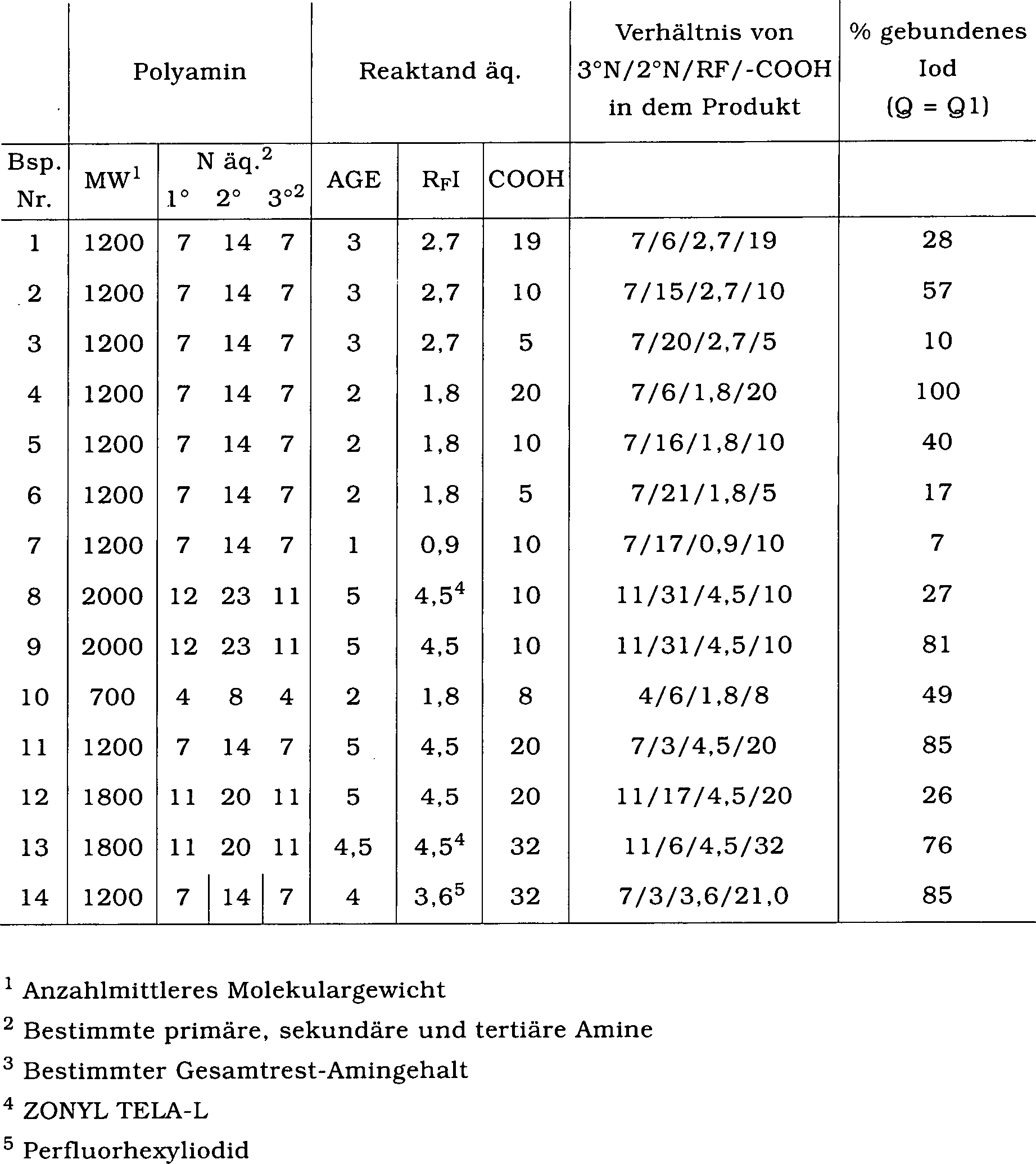
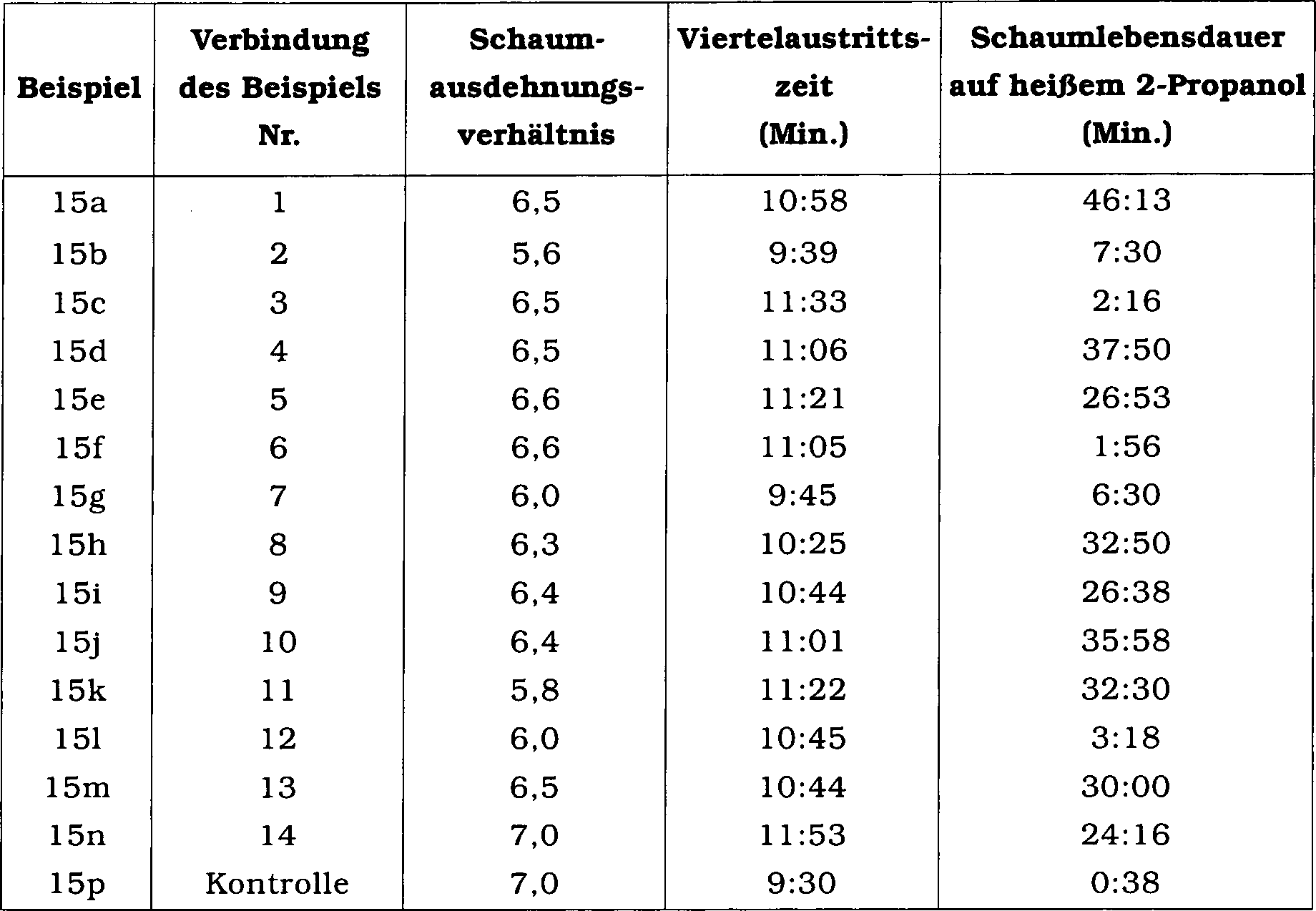

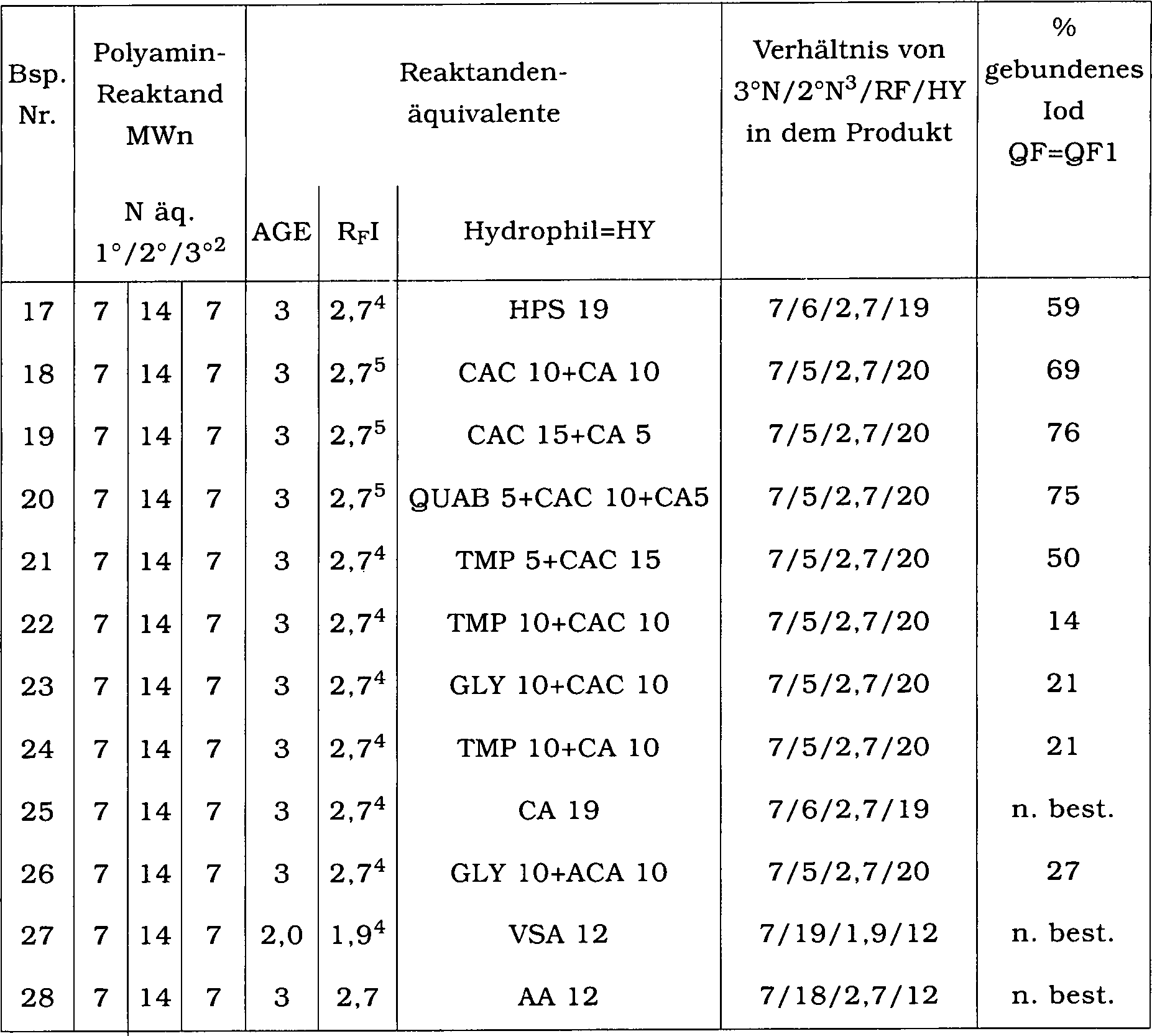



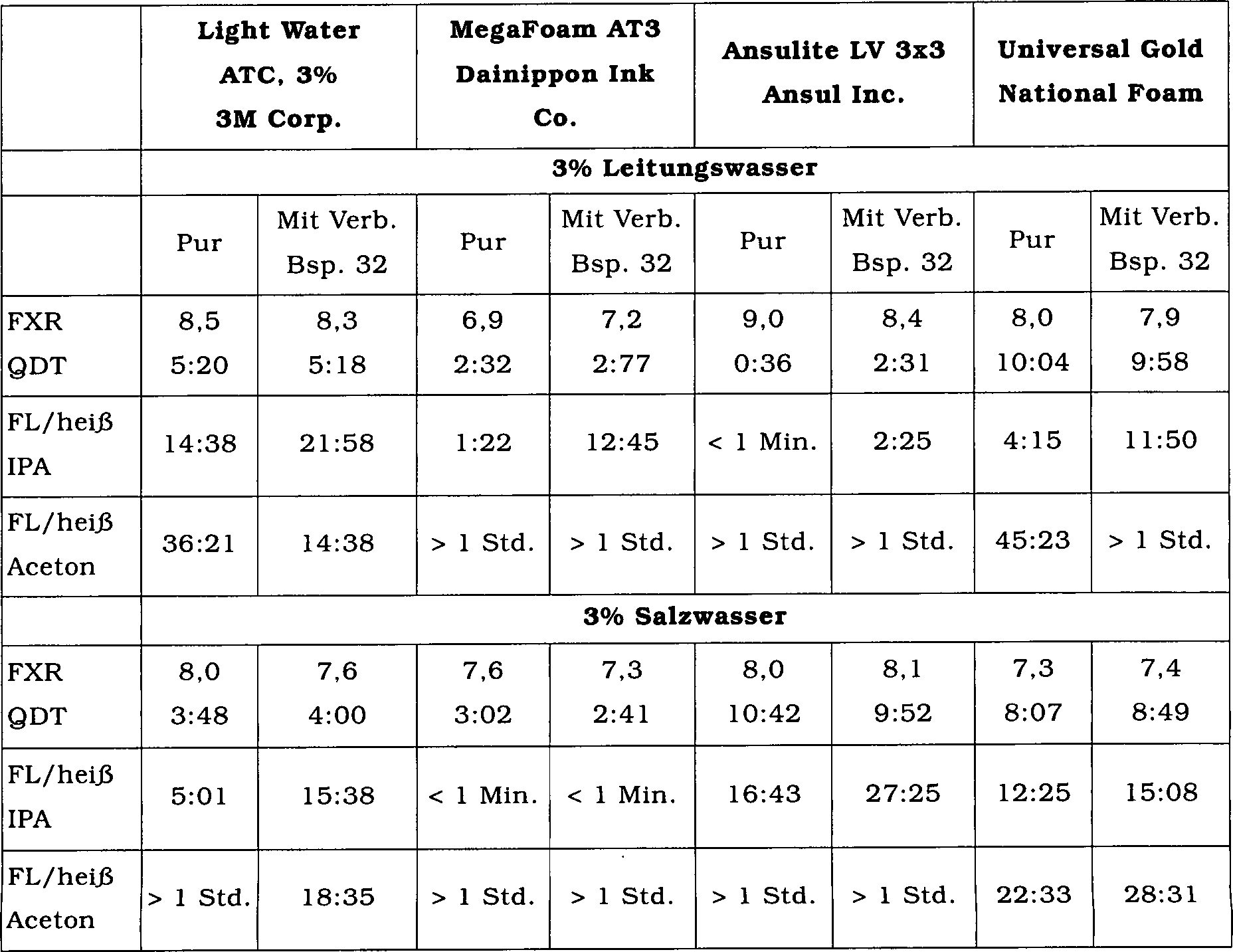

 darstellt und A-4 eine substituierte Amino- oder Amidogruppe der Formel
darstellt und A-4 eine substituierte Amino- oder Amidogruppe der Formel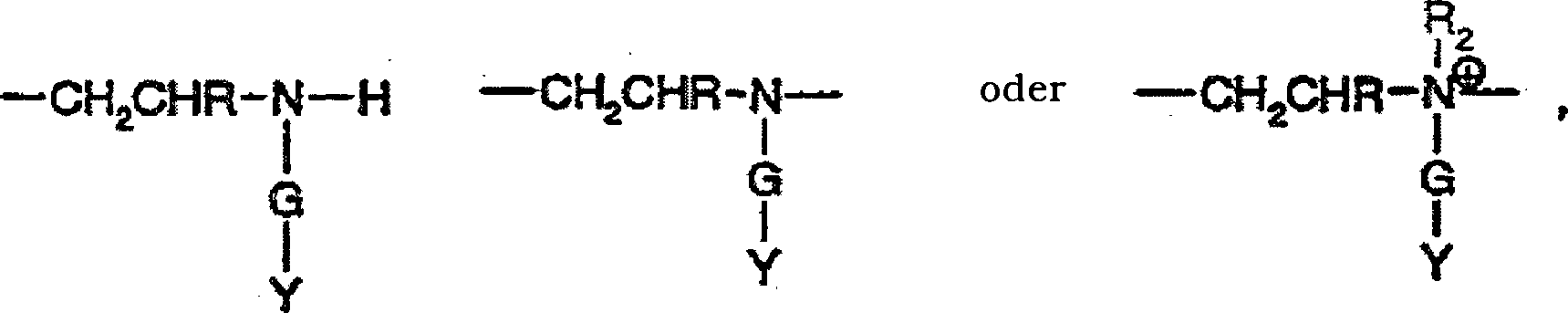 ist, worin T -CH2CH(OH)CH2-O-CH2- oder eine direkte Bindung darstellt, unter der Voraussetzung, dass, wenn T -CH2CH(OH)CH2-O-CH2- ist, QF den Formeln
ist, worin T -CH2CH(OH)CH2-O-CH2- oder eine direkte Bindung darstellt, unter der Voraussetzung, dass, wenn T -CH2CH(OH)CH2-O-CH2- ist, QF den Formeln