DE69918278T2 - Substituierte indolalkansäure - Google Patents
Substituierte indolalkansäure Download PDFInfo
- Publication number
- DE69918278T2 DE69918278T2 DE69918278T DE69918278T DE69918278T2 DE 69918278 T2 DE69918278 T2 DE 69918278T2 DE 69918278 T DE69918278 T DE 69918278T DE 69918278 T DE69918278 T DE 69918278T DE 69918278 T2 DE69918278 T2 DE 69918278T2
- Authority
- DE
- Germany
- Prior art keywords
- methyl
- acetic acid
- trifluorobenzothiazol
- indole
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000002253 acid Substances 0.000 title description 9
- 150000001875 compounds Chemical class 0.000 claims description 70
- -1 nitro, amino Chemical group 0.000 claims description 48
- 229910052739 hydrogen Inorganic materials 0.000 claims description 43
- 239000001257 hydrogen Substances 0.000 claims description 42
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 27
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 25
- 239000011737 fluorine Substances 0.000 claims description 25
- 229910052731 fluorine Inorganic materials 0.000 claims description 25
- 125000000217 alkyl group Chemical group 0.000 claims description 24
- 229910052736 halogen Inorganic materials 0.000 claims description 24
- 150000002367 halogens Chemical group 0.000 claims description 24
- 150000002431 hydrogen Chemical class 0.000 claims description 24
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 18
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 17
- 125000001153 fluoro group Chemical group F* 0.000 claims description 17
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 16
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 15
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical group C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 claims description 14
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 14
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 13
- 239000000460 chlorine Substances 0.000 claims description 12
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 11
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 10
- 239000011734 sodium Substances 0.000 claims description 10
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 9
- 229910052801 chlorine Inorganic materials 0.000 claims description 9
- 229910052757 nitrogen Inorganic materials 0.000 claims description 9
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 8
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 8
- 229910052794 bromium Inorganic materials 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 150000001768 cations Chemical class 0.000 claims description 7
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 claims description 7
- 229910052760 oxygen Inorganic materials 0.000 claims description 7
- 229910052708 sodium Inorganic materials 0.000 claims description 7
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Substances CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 6
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 5
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 5
- 239000011575 calcium Substances 0.000 claims description 5
- 229910052791 calcium Inorganic materials 0.000 claims description 5
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 5
- 239000001301 oxygen Substances 0.000 claims description 5
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 claims description 5
- 125000005034 trifluormethylthio group Chemical group FC(S*)(F)F 0.000 claims description 5
- JCBBUAGGFRHAAR-UHFFFAOYSA-N 2-[2-methyl-5-morpholin-4-yl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C=1C=C2N(CC(O)=O)C(C)=C(C=3SC4=C(F)C=C(F)C(F)=C4N=3)C2=CC=1N1CCOCC1 JCBBUAGGFRHAAR-UHFFFAOYSA-N 0.000 claims description 4
- BUQYFLKPQDSKSS-UHFFFAOYSA-N 2-[2-methyl-5-phenoxy-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C=1C=C2N(CC(O)=O)C(C)=C(C=3SC4=C(F)C=C(F)C(F)=C4N=3)C2=CC=1OC1=CC=CC=C1 BUQYFLKPQDSKSS-UHFFFAOYSA-N 0.000 claims description 4
- KOBFGCSHZBLYOW-UHFFFAOYSA-N 2-[2-methyl-5-phenyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C=1C=C2N(CC(O)=O)C(C)=C(C=3SC4=C(F)C=C(F)C(F)=C4N=3)C2=CC=1C1=CC=CC=C1 KOBFGCSHZBLYOW-UHFFFAOYSA-N 0.000 claims description 4
- VFRYWFLFAVFXIO-UHFFFAOYSA-N 2-[2-methyl-6-morpholin-4-yl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C1=C2N(CC(O)=O)C(C)=C(C=3SC4=C(F)C=C(F)C(F)=C4N=3)C2=CC=C1N1CCOCC1 VFRYWFLFAVFXIO-UHFFFAOYSA-N 0.000 claims description 4
- GFSCGVIGCOFBJX-UHFFFAOYSA-N 2-[2-methyl-6-phenyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C1=C2N(CC(O)=O)C(C)=C(C=3SC4=C(F)C=C(F)C(F)=C4N=3)C2=CC=C1C1=CC=CC=C1 GFSCGVIGCOFBJX-UHFFFAOYSA-N 0.000 claims description 4
- LJTZOVSFODOIIV-UHFFFAOYSA-N 2-[5-chloro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC(Cl)=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 LJTZOVSFODOIIV-UHFFFAOYSA-N 0.000 claims description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 4
- 125000005605 benzo group Chemical group 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 4
- 239000011777 magnesium Substances 0.000 claims description 4
- 229910052749 magnesium Inorganic materials 0.000 claims description 4
- 239000011591 potassium Substances 0.000 claims description 4
- 229910052700 potassium Inorganic materials 0.000 claims description 4
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 claims description 4
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 4
- 125000004738 (C1-C6) alkyl sulfinyl group Chemical group 0.000 claims description 3
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 3
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 3
- 125000005862 (C1-C6)alkanoyl group Chemical group 0.000 claims description 3
- XVWZZDJHMNCSHG-UHFFFAOYSA-N 2-[2,5-dimethyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC(C)=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 XVWZZDJHMNCSHG-UHFFFAOYSA-N 0.000 claims description 3
- HCBULRDWWUHSOC-UHFFFAOYSA-N 2-[2,5-dimethyl-3-[5-(trifluoromethyl)-1,3-benzothiazol-2-yl]indol-1-yl]acetic acid Chemical compound C12=CC(C)=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=CC(C(F)(F)F)=CC=C2S1 HCBULRDWWUHSOC-UHFFFAOYSA-N 0.000 claims description 3
- CKTQUOAACPEOFU-UHFFFAOYSA-N 2-[2,6-dimethyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=C(C)C=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 CKTQUOAACPEOFU-UHFFFAOYSA-N 0.000 claims description 3
- HCEONJPEGLZCMZ-UHFFFAOYSA-N 2-[2,7-dimethyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=CC(C)=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 HCEONJPEGLZCMZ-UHFFFAOYSA-N 0.000 claims description 3
- VBNFGLWTIQTREP-UHFFFAOYSA-N 2-[2-methyl-3-[5-(trifluoromethyl)-1,3-benzothiazol-2-yl]indol-1-yl]acetic acid Chemical compound C12=CC=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=CC(C(F)(F)F)=CC=C2S1 VBNFGLWTIQTREP-UHFFFAOYSA-N 0.000 claims description 3
- UNGJWLWQAXJOEO-UHFFFAOYSA-N 2-[2-methyl-5-phenylmethoxy-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C=1C=C2N(CC(O)=O)C(C)=C(C=3SC4=C(F)C=C(F)C(F)=C4N=3)C2=CC=1OCC1=CC=CC=C1 UNGJWLWQAXJOEO-UHFFFAOYSA-N 0.000 claims description 3
- CUNNSKIRHRLJMS-UHFFFAOYSA-N 2-[4-methyl-2-phenyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound N=1C2=C(F)C(F)=CC(F)=C2SC=1C=1C=2C(C)=CC=CC=2N(CC(O)=O)C=1C1=CC=CC=C1 CUNNSKIRHRLJMS-UHFFFAOYSA-N 0.000 claims description 3
- DFSHWYWKHTUEAJ-UHFFFAOYSA-N 2-[5-fluoro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC(F)=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 DFSHWYWKHTUEAJ-UHFFFAOYSA-N 0.000 claims description 3
- OVRDKEPIWAYWQW-UHFFFAOYSA-N 2-[6-chloro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=C(Cl)C=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 OVRDKEPIWAYWQW-UHFFFAOYSA-N 0.000 claims description 3
- DBRVNNBRSNYWLT-UHFFFAOYSA-N 2-[6-fluoro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=C(F)C=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 DBRVNNBRSNYWLT-UHFFFAOYSA-N 0.000 claims description 3
- LGHOGBGSCCLCQZ-UHFFFAOYSA-N 2-[7-bromo-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=CC(Br)=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 LGHOGBGSCCLCQZ-UHFFFAOYSA-N 0.000 claims description 3
- BKHJELPVVCKDEJ-UHFFFAOYSA-N 2-[7-chloro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=CC(Cl)=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 BKHJELPVVCKDEJ-UHFFFAOYSA-N 0.000 claims description 3
- HMBWWSMTYLJAJP-UHFFFAOYSA-N 2-[7-fluoro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC=CC(F)=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 HMBWWSMTYLJAJP-UHFFFAOYSA-N 0.000 claims description 3
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims description 3
- 239000005977 Ethylene Substances 0.000 claims description 3
- VVZCEUWTIRZKJJ-UHFFFAOYSA-N ethyl 2-[2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetate Chemical compound C(C)OC(CN1C(=C(C2=CC=CC=C12)C=1SC2=C(N=1)C(=C(C=C2F)F)F)C)=O VVZCEUWTIRZKJJ-UHFFFAOYSA-N 0.000 claims description 3
- 125000002346 iodo group Chemical group I* 0.000 claims description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 3
- 125000005004 perfluoroethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- GCNDTOHGLQBVIH-UHFFFAOYSA-N 2-[3-(6-chloro-1,3-benzothiazol-2-yl)-2-methylindol-1-yl]acetic acid Chemical compound C12=CC=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=CC=C(Cl)C=C2S1 GCNDTOHGLQBVIH-UHFFFAOYSA-N 0.000 claims description 2
- SKKDXNWINHMESM-UHFFFAOYSA-N 2-[4-chloro-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=C(Cl)C=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 SKKDXNWINHMESM-UHFFFAOYSA-N 0.000 claims description 2
- WOUZKZPEOGTKMC-UHFFFAOYSA-N 2-[5-bromo-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=CC(Br)=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 WOUZKZPEOGTKMC-UHFFFAOYSA-N 0.000 claims description 2
- NAFDKROMHJGVHR-UHFFFAOYSA-N 2-[5-methoxy-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound FC1=CC(F)=C2SC(C3=C(C)N(CC(O)=O)C4=CC=C(C=C43)OC)=NC2=C1F NAFDKROMHJGVHR-UHFFFAOYSA-N 0.000 claims description 2
- DILGZMIXUSQOLA-UHFFFAOYSA-N 2-[6-bromo-2-methyl-3-[5-(trifluoromethyl)-1,3-benzothiazol-2-yl]indol-1-yl]acetic acid Chemical compound C12=CC=C(Br)C=C2N(CC(O)=O)C(C)=C1C1=NC2=CC(C(F)(F)F)=CC=C2S1 DILGZMIXUSQOLA-UHFFFAOYSA-N 0.000 claims description 2
- HVUIEHYEYLFSFM-UHFFFAOYSA-N 2-[6-methoxy-2-methyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound COC1=CC=C2C(=C(N(C2=C1)CC(=O)O)C)C=1SC2=C(N=1)C(=C(C=C2F)F)F HVUIEHYEYLFSFM-UHFFFAOYSA-N 0.000 claims description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 2
- KYHVTMFADJNSGS-UHFFFAOYSA-N {3-[(4,5,7-trifluoro-1,3-benzothiazol-2-yl)methyl]-1h-indol-1-yl}acetic acid Chemical compound C12=CC=CC=C2N(CC(=O)O)C=C1CC1=NC2=C(F)C(F)=CC(F)=C2S1 KYHVTMFADJNSGS-UHFFFAOYSA-N 0.000 claims description 2
- CJYOVMODZUNUKL-UHFFFAOYSA-N 2-[2,4-dimethyl-3-(4,5,7-trifluoro-1,3-benzothiazol-2-yl)indol-1-yl]acetic acid Chemical compound C12=C(C)C=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=C(F)C(F)=CC(F)=C2S1 CJYOVMODZUNUKL-UHFFFAOYSA-N 0.000 claims 1
- UXEQTQLGTPBEQZ-UHFFFAOYSA-N 2-[3-[(5-fluoro-1,3-benzothiazol-2-yl)methyl]indol-1-yl]acetic acid Chemical compound C12=CC=CC=C2N(CC(=O)O)C=C1CC1=NC2=CC(F)=CC=C2S1 UXEQTQLGTPBEQZ-UHFFFAOYSA-N 0.000 claims 1
- DYNHZTFLLXJYDQ-UHFFFAOYSA-N 2-[3-[(6-fluoro-1,3-benzothiazol-2-yl)methyl]indol-1-yl]acetic acid Chemical compound C12=CC=CC=C2N(CC(=O)O)C=C1CC1=NC2=CC=C(F)C=C2S1 DYNHZTFLLXJYDQ-UHFFFAOYSA-N 0.000 claims 1
- 239000000825 pharmaceutical preparation Substances 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 86
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 60
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 58
- 239000000203 mixture Substances 0.000 description 41
- 238000002360 preparation method Methods 0.000 description 39
- 235000019439 ethyl acetate Nutrition 0.000 description 35
- 238000000034 method Methods 0.000 description 34
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 33
- 239000000243 solution Substances 0.000 description 33
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 27
- 239000000543 intermediate Substances 0.000 description 26
- SIKJAQJRHWYJAI-UHFFFAOYSA-N benzopyrrole Natural products C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 25
- MYTGFBZJLDLWQG-UHFFFAOYSA-N 5-chloro-1h-indole Chemical compound ClC1=CC=C2NC=CC2=C1 MYTGFBZJLDLWQG-UHFFFAOYSA-N 0.000 description 19
- 238000011282 treatment Methods 0.000 description 19
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 17
- 238000002844 melting Methods 0.000 description 17
- 230000008018 melting Effects 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 16
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 16
- 238000003786 synthesis reaction Methods 0.000 description 16
- 108010053754 Aldehyde reductase Proteins 0.000 description 15
- 102100027265 Aldo-keto reductase family 1 member B1 Human genes 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 150000002475 indoles Chemical class 0.000 description 15
- 230000015572 biosynthetic process Effects 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- 125000003118 aryl group Chemical group 0.000 description 13
- 150000003839 salts Chemical class 0.000 description 13
- 238000001816 cooling Methods 0.000 description 12
- 206010012601 diabetes mellitus Diseases 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 11
- 239000000463 material Substances 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 10
- 230000008569 process Effects 0.000 description 10
- 125000006239 protecting group Chemical group 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- 239000004480 active ingredient Substances 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 239000003288 aldose reductase inhibitor Substances 0.000 description 8
- 229940090865 aldose reductase inhibitors used in diabetes Drugs 0.000 description 8
- 125000000623 heterocyclic group Chemical group 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- 231100000252 nontoxic Toxicity 0.000 description 7
- 230000003000 nontoxic effect Effects 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- 235000003599 food sweetener Nutrition 0.000 description 6
- 238000007429 general method Methods 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 239000003765 sweetening agent Substances 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 5
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 5
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 5
- 208000002249 Diabetes Complications Diseases 0.000 description 5
- 206010012655 Diabetic complications Diseases 0.000 description 5
- 101000836540 Homo sapiens Aldo-keto reductase family 1 member B1 Proteins 0.000 description 5
- 229910004298 SiO 2 Inorganic materials 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 125000004190 benzothiazol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N=C(*)SC2=C1[H] 0.000 description 5
- 239000007859 condensation product Substances 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 239000000600 sorbitol Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- JHFOWEGCZWLHNW-UHFFFAOYSA-N 4-fluoro-2-methyl-1-nitrobenzene Chemical compound CC1=CC(F)=CC=C1[N+]([O-])=O JHFOWEGCZWLHNW-UHFFFAOYSA-N 0.000 description 4
- ONYNOPPOVKYGRS-UHFFFAOYSA-N 6-methylindole Natural products CC1=CC=C2C=CNC2=C1 ONYNOPPOVKYGRS-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 4
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 4
- 229910004373 HOAc Inorganic materials 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000000010 aprotic solvent Substances 0.000 description 4
- OCDGBSUVYYVKQZ-UHFFFAOYSA-N beta-dimethylaminomethylindole Natural products C1=CC=C2C(CN(C)C)=CNC2=C1 OCDGBSUVYYVKQZ-UHFFFAOYSA-N 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- DMCPFOBLJMLSNX-UHFFFAOYSA-N indole-3-acetonitrile Chemical compound C1=CC=C2C(CC#N)=CNC2=C1 DMCPFOBLJMLSNX-UHFFFAOYSA-N 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- OTDNCSDKIMJHAS-UHFFFAOYSA-N 1H-indol-2-ylstannane Chemical compound C1=CC=C2NC([SnH3])=CC2=C1 OTDNCSDKIMJHAS-UHFFFAOYSA-N 0.000 description 3
- MGICLRNAZXDKAT-UHFFFAOYSA-N 2-(2-methylindol-1-yl)acetic acid Chemical compound C1=CC=C2N(CC(O)=O)C(C)=CC2=C1 MGICLRNAZXDKAT-UHFFFAOYSA-N 0.000 description 3
- RREODNYNAPILHF-UHFFFAOYSA-N 2-amino-3,4,6-trifluorobenzenethiol;hydrochloride Chemical compound Cl.NC1=C(F)C(F)=CC(F)=C1S RREODNYNAPILHF-UHFFFAOYSA-N 0.000 description 3
- VRVRGVPWCUEOGV-UHFFFAOYSA-N 2-aminothiophenol Chemical class NC1=CC=CC=C1S VRVRGVPWCUEOGV-UHFFFAOYSA-N 0.000 description 3
- PLAZTCDQAHEYBI-UHFFFAOYSA-N 2-nitrotoluene Chemical compound CC1=CC=CC=C1[N+]([O-])=O PLAZTCDQAHEYBI-UHFFFAOYSA-N 0.000 description 3
- MQLYXCLFKUAMQJ-UHFFFAOYSA-N 4,5,7-trifluoro-2-methyl-1,3-benzothiazole Chemical compound FC1=CC(F)=C2SC(C)=NC2=C1F MQLYXCLFKUAMQJ-UHFFFAOYSA-N 0.000 description 3
- SKWTUNAAJNDEIK-UHFFFAOYSA-N 4-fluoro-1-methyl-2-nitrobenzene Chemical compound CC1=CC=C(F)C=C1[N+]([O-])=O SKWTUNAAJNDEIK-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 0 Cc1c(*)c(*)c2[n]c(*)cc2c1* Chemical compound Cc1c(*)c(*)c2[n]c(*)cc2c1* 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 208000017442 Retinal disease Diseases 0.000 description 3
- 206010038923 Retinopathy Diseases 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 3
- 125000001246 bromo group Chemical group Br* 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 125000001072 heteroaryl group Chemical group 0.000 description 3
- 125000001041 indolyl group Chemical group 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 229940057995 liquid paraffin Drugs 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- CLMWKFFAQNVROF-UHFFFAOYSA-N n,n,2-trimethyl-1h-indol-3-amine Chemical compound C1=CC=C2C(N(C)C)=C(C)NC2=C1 CLMWKFFAQNVROF-UHFFFAOYSA-N 0.000 description 3
- 201000001119 neuropathy Diseases 0.000 description 3
- 230000007823 neuropathy Effects 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 208000033808 peripheral neuropathy Diseases 0.000 description 3
- 238000003408 phase transfer catalysis Methods 0.000 description 3
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical class CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- LQQKDSXCDXHLLF-UHFFFAOYSA-N 1,3-dibromopropan-2-one Chemical compound BrCC(=O)CBr LQQKDSXCDXHLLF-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- UHFZUNGNGGEESB-UHFFFAOYSA-N 2-(5-chloro-1h-indol-3-yl)acetonitrile Chemical compound ClC1=CC=C2NC=C(CC#N)C2=C1 UHFZUNGNGGEESB-UHFFFAOYSA-N 0.000 description 2
- QDZFTHLNJVCMSM-UHFFFAOYSA-N 2-[3-(5-fluoro-1,3-benzothiazol-2-yl)-2-methylindol-1-yl]acetic acid Chemical compound C12=CC=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=CC(F)=CC=C2S1 QDZFTHLNJVCMSM-UHFFFAOYSA-N 0.000 description 2
- ZPRQXVPYQGBZON-UHFFFAOYSA-N 2-bromo-1h-indole Chemical class C1=CC=C2NC(Br)=CC2=C1 ZPRQXVPYQGBZON-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- COHVZTXZLIRSTM-UHFFFAOYSA-N 2-methyl-1-nitro-4-phenoxybenzene Chemical compound C1=C([N+]([O-])=O)C(C)=CC(OC=2C=CC=CC=2)=C1 COHVZTXZLIRSTM-UHFFFAOYSA-N 0.000 description 2
- BHNHHSOHWZKFOX-UHFFFAOYSA-N 2-methyl-1H-indole Chemical compound C1=CC=C2NC(C)=CC2=C1 BHNHHSOHWZKFOX-UHFFFAOYSA-N 0.000 description 2
- RGNJKFHZEOSSBO-UHFFFAOYSA-N 4-(1h-indol-5-yl)morpholine Chemical compound C1COCCN1C1=CC=C(NC=C2)C2=C1 RGNJKFHZEOSSBO-UHFFFAOYSA-N 0.000 description 2
- CDGIWYMFRIJCIR-UHFFFAOYSA-N 4-(1h-indol-6-yl)morpholine Chemical compound C1COCCN1C1=CC=C(C=CN2)C2=C1 CDGIWYMFRIJCIR-UHFFFAOYSA-N 0.000 description 2
- IIRHTTDXNXCWHP-UHFFFAOYSA-N 4-(3-methyl-4-nitrophenyl)morpholine Chemical compound C1=C([N+]([O-])=O)C(C)=CC(N2CCOCC2)=C1 IIRHTTDXNXCWHP-UHFFFAOYSA-N 0.000 description 2
- CYMDJHMDVUTLRA-UHFFFAOYSA-N 4-(4-methyl-3-nitrophenyl)morpholine Chemical compound C1=C([N+]([O-])=O)C(C)=CC=C1N1CCOCC1 CYMDJHMDVUTLRA-UHFFFAOYSA-N 0.000 description 2
- YPKBCLZFIYBSHK-UHFFFAOYSA-N 5-methylindole Chemical compound CC1=CC=C2NC=CC2=C1 YPKBCLZFIYBSHK-UHFFFAOYSA-N 0.000 description 2
- YJBIMZVVUOJZSS-UHFFFAOYSA-N 5-phenoxy-1h-indole Chemical compound C=1C=C2NC=CC2=CC=1OC1=CC=CC=C1 YJBIMZVVUOJZSS-UHFFFAOYSA-N 0.000 description 2
- KVVBMAQSZVYTCW-UHFFFAOYSA-N 6-phenyl-1h-indole Chemical compound C1=C2NC=CC2=CC=C1C1=CC=CC=C1 KVVBMAQSZVYTCW-UHFFFAOYSA-N 0.000 description 2
- 235000003911 Arachis Nutrition 0.000 description 2
- 244000105624 Arachis hypogaea Species 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 238000005679 Batcho-Leimgruber synthesis reaction Methods 0.000 description 2
- 208000002177 Cataract Diseases 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- JWDXCWSZNPCJKH-UHFFFAOYSA-N OBO.C1=CC=C2NC=CC2=C1 Chemical class OBO.C1=CC=C2NC=CC2=C1 JWDXCWSZNPCJKH-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 238000006069 Suzuki reaction reaction Methods 0.000 description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- FIAGYDIJZOWVAB-UHFFFAOYSA-N [2-sulfanyl-5-(trifluoromethyl)phenyl]azanium;chloride Chemical compound Cl.NC1=CC(C(F)(F)F)=CC=C1S FIAGYDIJZOWVAB-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 150000004982 aromatic amines Chemical class 0.000 description 2
- 150000001502 aryl halides Chemical class 0.000 description 2
- 125000004540 benzothiazol-5-yl group Chemical group S1C=NC2=C1C=CC(=C2)* 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000011449 brick Substances 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 125000001033 ether group Chemical group 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- PRVMLZZABVMBGR-UHFFFAOYSA-N (4-fluoro-2-sulfanylphenyl)azanium;chloride Chemical compound Cl.NC1=CC=C(F)C=C1S PRVMLZZABVMBGR-UHFFFAOYSA-N 0.000 description 1
- JDSDBDMAPVLZGJ-UHFFFAOYSA-N (5-fluoro-2-sulfanylphenyl)azanium;chloride Chemical compound Cl.NC1=CC(F)=CC=C1S JDSDBDMAPVLZGJ-UHFFFAOYSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical class C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ILGPJIAGKMKHPE-UHFFFAOYSA-N 1-(5-chloro-1h-indol-3-yl)-n,n-dimethylmethanamine Chemical compound C1=C(Cl)C=C2C(CN(C)C)=CNC2=C1 ILGPJIAGKMKHPE-UHFFFAOYSA-N 0.000 description 1
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical compound CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- XNEGMGIEVRYVNY-UHFFFAOYSA-N 2,3,4-trifluorobenzenethiol;hydrochloride Chemical compound Cl.FC1=CC=C(S)C(F)=C1F XNEGMGIEVRYVNY-UHFFFAOYSA-N 0.000 description 1
- SPSWJTZNOXMMMV-UHFFFAOYSA-N 2,3,5,6-tetrafluoroaniline Chemical compound NC1=C(F)C(F)=CC(F)=C1F SPSWJTZNOXMMMV-UHFFFAOYSA-N 0.000 description 1
- JEEDFPVACIKHEU-UHFFFAOYSA-N 2-(2h-indazol-3-yl)acetic acid Chemical class C1=CC=CC2=C(CC(=O)O)NN=C21 JEEDFPVACIKHEU-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- KCYPYKKZHMHYJV-UHFFFAOYSA-N 2-[3-(6-fluoro-1,3-benzothiazol-2-yl)-2-methylindol-1-yl]acetic acid Chemical compound C12=CC=CC=C2N(CC(O)=O)C(C)=C1C1=NC2=CC=C(F)C=C2S1 KCYPYKKZHMHYJV-UHFFFAOYSA-N 0.000 description 1
- ZXLGCODLRRPKAW-UHFFFAOYSA-N 2-[3-(cyanomethyl)indol-1-yl]acetic acid Chemical compound C1=CC=C2N(CC(=O)O)C=C(CC#N)C2=C1 ZXLGCODLRRPKAW-UHFFFAOYSA-N 0.000 description 1
- XDRQQYPIPNJWNA-UHFFFAOYSA-N 2-amino-3,4,6-trifluorobenzenethiol Chemical compound NC1=C(F)C(F)=CC(F)=C1S XDRQQYPIPNJWNA-UHFFFAOYSA-N 0.000 description 1
- KLLLJCACIRKBDT-UHFFFAOYSA-N 2-phenyl-1H-indole Chemical compound N1C2=CC=CC=C2C=C1C1=CC=CC=C1 KLLLJCACIRKBDT-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- UJVBZCCNLAAMOV-UHFFFAOYSA-N 2h-1,2-benzothiazine Chemical class C1=CC=C2C=CNSC2=C1 UJVBZCCNLAAMOV-UHFFFAOYSA-N 0.000 description 1
- ODFFPRGJZRXNHZ-UHFFFAOYSA-N 5-fluoroindole Chemical compound FC1=CC=C2NC=CC2=C1 ODFFPRGJZRXNHZ-UHFFFAOYSA-N 0.000 description 1
- LPYXADUZSWBHCT-UHFFFAOYSA-N 5-phenyl-1h-indole Chemical compound C=1C=C2NC=CC2=CC=1C1=CC=CC=C1 LPYXADUZSWBHCT-UHFFFAOYSA-N 0.000 description 1
- JCQLPDZCNSVBMS-UHFFFAOYSA-N 5-phenylmethoxy-1h-indole Chemical compound C=1C=C2NC=CC2=CC=1OCC1=CC=CC=C1 JCQLPDZCNSVBMS-UHFFFAOYSA-N 0.000 description 1
- FDVBHUXZXNQCCM-UHFFFAOYSA-N 6,6-ditert-butyl-4-methylcyclohexa-2,4-dien-1-ol Chemical compound CC1=CC(C(C)(C)C)(C(C)(C)C)C(O)C=C1 FDVBHUXZXNQCCM-UHFFFAOYSA-N 0.000 description 1
- YTYIMDRWPTUAHP-UHFFFAOYSA-N 6-Chloroindole Chemical compound ClC1=CC=C2C=CNC2=C1 YTYIMDRWPTUAHP-UHFFFAOYSA-N 0.000 description 1
- MAWGHOPSCKCTPA-UHFFFAOYSA-N 6-bromo-1h-indole Chemical compound BrC1=CC=C2C=CNC2=C1 MAWGHOPSCKCTPA-UHFFFAOYSA-N 0.000 description 1
- YYFFEPUCAKVRJX-UHFFFAOYSA-N 6-fluoro-1h-indole Chemical compound FC1=CC=C2C=CNC2=C1 YYFFEPUCAKVRJX-UHFFFAOYSA-N 0.000 description 1
- RDSVSEFWZUWZHW-UHFFFAOYSA-N 7-bromo-1h-indole Chemical compound BrC1=CC=CC2=C1NC=C2 RDSVSEFWZUWZHW-UHFFFAOYSA-N 0.000 description 1
- WMYQAKANKREQLM-UHFFFAOYSA-N 7-chloro-1h-indole Chemical compound ClC1=CC=CC2=C1NC=C2 WMYQAKANKREQLM-UHFFFAOYSA-N 0.000 description 1
- KGWPHCDTOLQQEP-UHFFFAOYSA-N 7-methylindole Chemical compound CC1=CC=CC2=C1NC=C2 KGWPHCDTOLQQEP-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 229920002799 BoPET Polymers 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- SAWVXAKULNDKKH-UHFFFAOYSA-N C(C)#N.ClC=1C=C2C(=CNC2=CC1)O Chemical compound C(C)#N.ClC=1C=C2C(=CNC2=CC1)O SAWVXAKULNDKKH-UHFFFAOYSA-N 0.000 description 1
- BOQUBFRYEZHBJW-UHFFFAOYSA-N CC(C=C(C1SC(Cc2c(-c3ccccc3)[n](CC(O)O)c3c2cccc3)=NC11)F)=C1F Chemical compound CC(C=C(C1SC(Cc2c(-c3ccccc3)[n](CC(O)O)c3c2cccc3)=NC11)F)=C1F BOQUBFRYEZHBJW-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- SDZJYMFUSMVQCU-UHFFFAOYSA-N Cc1c(-c2nc3c(F)c(F)cc(F)c3s2)c2ccccc2n1CC(O)=O.Cc1c(-c2nc3c(F)c(F)cc(F)c3s2)c2cc(Cl)ccc2n1CC(O)=O Chemical compound Cc1c(-c2nc3c(F)c(F)cc(F)c3s2)c2ccccc2n1CC(O)=O.Cc1c(-c2nc3c(F)c(F)cc(F)c3s2)c2cc(Cl)ccc2n1CC(O)=O SDZJYMFUSMVQCU-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- PWVNPKWPLUUGEK-UHFFFAOYSA-N Cl.NC1=C(C(=CC(=C1F)F)F)O Chemical compound Cl.NC1=C(C(=CC(=C1F)F)F)O PWVNPKWPLUUGEK-UHFFFAOYSA-N 0.000 description 1
- YATVUMMGFMFMKY-UHFFFAOYSA-N ClC1=CC=C2C(=C(N(C2=C1)CC(=O)O)C)C=1SC2=C(N1)C(=C(C=C2F)F)F.CC=2C=CC=C1C(=C(N(C21)CC(=O)O)C)C=2SC1=C(N2)C(=C(C=C1F)F)F Chemical compound ClC1=CC=C2C(=C(N(C2=C1)CC(=O)O)C)C=1SC2=C(N1)C(=C(C=C2F)F)F.CC=2C=CC=C1C(=C(N(C21)CC(=O)O)C)C=2SC1=C(N2)C(=C(C=C1F)F)F YATVUMMGFMFMKY-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- CWONLDAQAWGASN-UHFFFAOYSA-N FC1=CC=C2C(=C(N(C2=C1)CC(=O)O)C)C=1SC2=C(N1)C(=C(C=C2F)F)F.C(C2=CC=CC=C2)OC=2C=C1C(=C(N(C1=CC2)CC(=O)O)C)C=2SC1=C(N2)C(=C(C=C1F)F)F Chemical compound FC1=CC=C2C(=C(N(C2=C1)CC(=O)O)C)C=1SC2=C(N1)C(=C(C=C2F)F)F.C(C2=CC=CC=C2)OC=2C=C1C(=C(N(C1=CC2)CC(=O)O)C)C=2SC1=C(N2)C(=C(C=C1F)F)F CWONLDAQAWGASN-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- 235000005135 Micromeria juliana Nutrition 0.000 description 1
- 239000005041 Mylar™ Substances 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- DINBUQZUMRWQSG-UHFFFAOYSA-N OC(CC[n]1c2ccccc2c(Cc2nc(C(F)=C(CC3F)F)c3[s]2)c1)=O Chemical compound OC(CC[n]1c2ccccc2c(Cc2nc(C(F)=C(CC3F)F)c3[s]2)c1)=O DINBUQZUMRWQSG-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 125000000066 S-methyl group Chemical group [H]C([H])([H])S* 0.000 description 1
- 240000002114 Satureja hortensis Species 0.000 description 1
- 235000007315 Satureja hortensis Nutrition 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 238000006619 Stille reaction Methods 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- HKNSIVFWRXBWCK-UHFFFAOYSA-N [N].NC1=CC=CC=C1 Chemical compound [N].NC1=CC=CC=C1 HKNSIVFWRXBWCK-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 150000001243 acetic acids Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 150000001298 alcohols Chemical group 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- NMPVEAUIHMEAQP-UHFFFAOYSA-N alpha-bromo-acetaldehyde Natural products BrCC=O NMPVEAUIHMEAQP-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 150000001543 aryl boronic acids Chemical class 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 125000005620 boronic acid group Chemical class 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- VDDXQSUSMHZCLS-UHFFFAOYSA-N ethenyl trifluoromethanesulfonate Chemical class FC(F)(F)S(=O)(=O)OC=C VDDXQSUSMHZCLS-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- CJDWBNBHHMSCHC-UHFFFAOYSA-N ethyl 2-[3-[(4,5,7-trifluoro-1,3-benzothiazol-2-yl)methyl]indol-1-yl]acetate Chemical compound C12=CC=CC=C2N(CC(=O)OCC)C=C1CC1=NC2=C(F)C(F)=CC(F)=C2S1 CJDWBNBHHMSCHC-UHFFFAOYSA-N 0.000 description 1
- RJTYCUXOHLVMJA-UHFFFAOYSA-N ethyl 2-[5-bromo-3-(cyanomethyl)indol-1-yl]acetate Chemical compound BrC1=CC=C2N(CC(=O)OCC)C=C(CC#N)C2=C1 RJTYCUXOHLVMJA-UHFFFAOYSA-N 0.000 description 1
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- GOERTRUXQHDLHC-UHFFFAOYSA-N gramine Natural products COC1=CC=C2NC=C(CN(C)C)C2=C1 GOERTRUXQHDLHC-UHFFFAOYSA-N 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000007074 heterocyclization reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 150000007975 iminium salts Chemical class 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- SEOVTRFCIGRIMH-UHFFFAOYSA-N indole-3-acetic acid Chemical class C1=CC=C2C(CC(=O)O)=CNC2=C1 SEOVTRFCIGRIMH-UHFFFAOYSA-N 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000006192 iodination reaction Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000006303 iodophenyl group Chemical group 0.000 description 1
- 239000002085 irritant Substances 0.000 description 1
- 231100000021 irritant Toxicity 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- RBXVOQPAMPBADW-UHFFFAOYSA-N nitrous acid;phenol Chemical class ON=O.OC1=CC=CC=C1 RBXVOQPAMPBADW-UHFFFAOYSA-N 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- GYCKQBWUSACYIF-UHFFFAOYSA-N o-hydroxybenzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1O GYCKQBWUSACYIF-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 238000010653 organometallic reaction Methods 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 150000002989 phenols Chemical group 0.000 description 1
- CYQAYERJWZKYML-UHFFFAOYSA-N phosphorus pentasulfide Chemical compound S1P(S2)(=S)SP3(=S)SP1(=S)SP2(=S)S3 CYQAYERJWZKYML-UHFFFAOYSA-N 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000006476 reductive cyclization reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 210000003497 sciatic nerve Anatomy 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 150000003556 thioamides Chemical class 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000007832 transition metal-catalyzed coupling reaction Methods 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Ophthalmology & Optometry (AREA)
- Neurosurgery (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
Description
- Hintergrund der Erfindung
- Die Verwendung von Aldosereduktasehemmern (ARIs) für die Behandlung von diabetischen Komplikationen ist bekannt. Die Komplikationen ergeben sich aus dem erhöhten Spiegel von Glucose in Geweben wie den Nerven, den Nieren, der Netzhaut und der Linse. Diese tritt in den Polyolpfad ein und wird über Aldosereduktase zu Sorbitol umgewandelt. Da Sorbitol Zellmembrane nicht ohne weiteres überquert, sammelt es sich innerhalb gewisser Zellen an, was zu Änderungen des osmotischen Drucks, Veränderungen des Redox-Zustands der Pyridinnucleotide (d.h. einem erhöhten NADH/NAD+ Verhältnis) und einem abgereicherten, intrazellulären Myoinositolspiegel führt. Diese biochemischen Veränderungen, die mit diabetischen Komplikationen in Verbindung gebracht wurden, können durch Hemmer der Aldosereduktase kontrolliert werden.
- Über die Verwendung von Aldosereduktasehemmern für die Behandlung diabetischer Komplikationen wurde bereits ausführlich berichtet, siehe: (a) Textbook of Diabetes, 2. Ausgabe, Pickup, J. C. and Williams, G. (Herausgeber), Blackwell Science, Boston, MA 1997; (b) Larson, E. R.; Lipinski, C. A. and Sarges, R., Medicinal Research Reviews, 1988, 8 (2), 159–198; (c) Dvornik, D. Aldose Reductase Inhibition, Porte, D. (Herausgeber), Biomedical Information Corp., New York, NY, Mc Graw Hill, 1987; (d) Petrash, J. M., Tarle, I., Wilson, D. K., Quiocho. F. A., Perspectives in Diabetes, Aldose Reductase Catalysis and Crystalography: Insights From Recent Advances in Enzyme Structure and Function, Diabetes, 1994, 43, 955; (e) Aotsuka, T., Abe, N., Fukushima, K., Ashizawa, N. und Yoshida, M., Bioorg. & Med. Chem. Letters, 1997, 7, 1677, (f) T., Nagaki, Y., Ishii, A., Konishi, Y., Yago, H., Seishi, S., Okukado, N., Okamoto, K., J. Med. Chem, 1997, 40, 684; (g) Ashizawa, N., Yoshida, M., Sugiyama, Y., Akaike, N., Ohbayashi, S., Aotsuka, T., Abe, N., Fukushima, K., Matsuura, A., Jpn. J. Pharmacol. 1997, 73, 133; (h) Kador, P. F., Sharpless, N. E., Molecular Pharmacology, 1983, 24, 521; (i) Kador, P. F., Kinoshita, J. H., Sharpless, N. E., J. Med. Chem. 1985, 28 (7), 841; (j) Hotta, N., Biomed. & Pharmacother. 1995, 5, 232; (k) Mylar, B., Larson, E. R., Beyer, T. A., Zembrowski, W. J., Aldinger, C. E., Dee, F. D., Siegel, T. W., Singleton, D. H., J. Med. Chem. 1991, 34, 108; (1) Dvornik, D., Croatica Chemica Acta 1996, 69 (2), 613.
- Früher beschriebene Aldosereduktasehemmer, die mit der vorliegenden Erfindung am engsten verwandt sind, umfassen diejenigen, die in (a) US-A-5,700,819: "2-Substituted benzothiazole derivatives useful in the treatment of diabetic complications", (b) US-A-4,868,301: "Processes and intermediates for the preparation of oxophthalazinyl acetic acids having benzothiazole or other heterocyclic side chains", (c) US-A-5,330,997: "1H-indazole-3-acetic acids as aldose reductase inhibitors" und (d) US-A-5,236,945: "1H-indazole-3-acetic acids as aldose reductase inhibitors" erwähnt sind. Obgleich viele Aldosereduktasehemmer in großem Umfang entwickelt wurden, hat keiner bei klinischen Versuchen bei Menschen eine ausreichende Wirksamkeit ohne signifikante unerwünschte Nebenwirkungen gezeigt. So sind gegenwärtig keine Aldosereduktasehemmer als zugelassene, therapeutische Mittel in den Vereinigten Staaten verfügbar, und folglich besteht immer noch ein beträchtlicher Bedarf an neuen, wirksamen und sicheren Medikamenten für die Behandlung diabetischer Komplikationen.
- Zusammenfassung der Erfindung
- Diese Erfindung stellt Verbindungen zur Verfügung, die mit der Aldosereduktase in Wechselwirkung treten und diese hemmen. So stellt die Erfindung unter einem breiten Aspekt Verbindungen der Formel I:oder pharmazeutisch annehmbare Salze derselben zur Verfügung, worin

A eine C1-C4-Alkylengruppe, wahlweise substituiert mit einem C1-C2-Alkyl oder mono- oder disubstituiert mit Halogen, vorzugsweise Fluor oder Chlor ist;
Z eine Bindung, O, S, C(O)NH oder C1-C3-Alkylen, wahlweise substituiert mit C1-C2-Alkyl ist;
R1 Wasserstoff, Alkyl mit 1-6 Kohlenstoffatomen oder Phenyl ist, worin das Phenyl wahlweise mit bis zu drei Gruppen substituiert ist, ausgewählt unter Halogen, Hydroxy, C1-C6-Alkoxy, C1-C6-Alkyl, Nitro, Amino oder Mono- oder Di(C1-C6)-Alkylamino;
R2, R3, R4 und R5 jeweils unabhängig sind Wasserstoff, Halogen, Nitro oder eine Alkylgruppe mit 1-6 Kohlenstoffatomen, wobei die Alkylgruppen mit einem oder mehreren Halogenen substituiert sein können;
OR7, worin jedes R7 unabhängig darstellt Wasserstoff, eine Alkylgruppe mit 1-6 Kohlenstoffatomen, wobei die Alkylgruppe ggfs. mit einem oder mehreren Halogenen substituiert sein kann, oder Benzyl, worin der Phenylanteil wahlweise mit bis zu drei Gruppen substituiert ist, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl oder C1-C6-Alkoxy;
Phenoxy, worin der Phenylanteil wahlweise mit bis zu drei Gruppen substituiert ist, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl oder C1-C6-Alkoxy, Amino und Mono- oder Di(C1-C6)-alkylamino; oder
eine Gruppe der Formelworin
J eine Bindung, CH2, Sauerstoff oder Stickstoff ist; und
jedes r unabhängig 2 oder 3 ist;
R6 Hydroxy bedeutet;
oder O–M+, wobei M+ ein Kation, ausgewählt aus Natrium, Kalium, Ammonium, Magnesium und Calcium, bedeutet;
Ra Wasserstorff C1-C6-Alkyl, Fluor oder Trifluormethyl bedeutet, und
Ar einen Benzothiozolring bedeutet, der ggfs. auf dem Benzoteil durch eines von Iod, Cyano, Nitro, Perfluorethyl, Trifluoracetyl oder (C1-C6)-Alkanoyl, eines oder zwei von Fluor, Chlor, Brom, Hydroxy, (C1-C6)-Alkyl, (C1-C6)-Alkoxy, (C1-C6)-Alkylthio, Trifluormethoxy, Trifluormethylthio, (C1-C6)-Alkylsulfinyl, (C1-C6)-Alkylsulfonyl oder Trifluormethyl, oder zwei Fluor oder zwei Trifluormethyl mit einem Hydroxy und einem (C1-C6)-Alkoxy, oder ein oder zwei Fluor und ein Trifluormethyl, oder drei Fluor substituiert ist. - Die erfindungsgemäßen Verbindungen hemmen die Aldosereduktase. Da die Aldosereduktase kritisch für die Erzeugung eines hohen Sorbitolspiegels bei Menschen mit Diabetes ist, sind Hemmer der Aldosereduktase bei der Vorbeugung und/oder Behandlung verschiedener mit Diabetes verbundener Komplikationen brauchbar. Die erfindungsgemäßen Verbindungen sind deshalb aufgrund ihrer Fähigkeit, die Aldosereduktase zu hemmen, für die Behandlung diabetischer Komplikationen wirksam.
- Somit ermöglicht die Erfindung die Behandlung und/oder Vorbeugung chronischer Komplikationen, die mit Diabetes mellitus verbunden sind, einschließlich beispielsweise Diabetesstar, Retinopathie, Nephropathie und Neuropathie. In einem weiteren Aspekt stellt die Erfindung pharmazeutische Zubereitungen zur Verfügung, die Verbindungen der Formel I enthalten.
- Detaillierte Beschreibung der Erfindung
- Das Numerierungssystem für die Verbindungen der Formel I ist wie folgt:
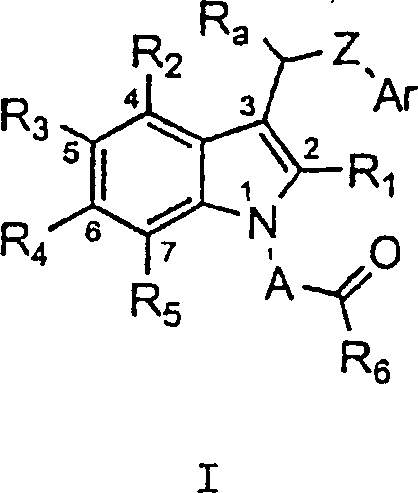
- Wie vorstehend angegeben, stellt die Erfindung neue substituierte Indolalkansäuren zur Verfügung, die bei der Behandlung und/oder der Vorbeugung von Komplikationen brauchbar sind, die mit einem erhöhten Glucosespiegel bei Personen zusammenhängen oder sich daraus ergeben, die an Diabetes mellitus leiden. Diese Verbindungen werden durch die vorstehende Formel I dargestellt.
- In den Verbindungen der Formel I umfassen die durch Ar dargestellten Aryl- und Heteroarylgruppen:
einen Benzothiozolring, der ggfs. auf dem Benzoteil durch eines von Iod, Cyano, Nitro, Perfluorethyl, Trifluoracetyl oder (C1-C6)-Alkanoyl, eines oder zwei von Fluor, Chlor, Brom, Hydroxy, (C1-C6)-Alkyl, (C1-C6)-Alkoxy, (C1-C6)-Alkylthio, Trifluormethoxy, Trifluormethylthio, (C1-C6)-Alkylsulfinyl, (C1-C6)-Alkylsulfonyl oder Trifluormethyl, oder zwei Fluor oder zwei Trifluormethyl mit einem Hydroxy und einem (C1-C6)-Alkoxy, oder ein oder zwei Fluor und ein Trifluormethyl, oder drei Fluor substituiert ist. - Spezifischere Verbindungen der Erfindung sind diejenigen der Formel I, worin Ar ggfs. substituiertes Benzothiazolyl ist. Andere spezifischere Verbindungen sind diejenigen der Formel I, worin Ra Trifluormethyl ist, Z eine kovalente Bindung oder CH2 ist, R6 Hydroxy ist und jedes von R2-R5 unabhängig Wasserstoff, Halogen, stärker bevorzugt Brom oder Chlor ist, C1-C2-Alkyl, Phenoxy, Benzyloxy oder C1-C2-Alkoxy ist, und R1 Wasserstoff oder Methyl ist.
- Bevorzugte Verbindungen der Erfindung sind diejenigen, in denen Z eine kovalente Bindung ist, R6 Hydroxy ist, Ar wahlweise substituiertes Benzothiazol-2-yl, Benzothiazol-5-yl oder Benzoisothiazol-3-yl ist, R2-R5 unabhängig Wasserstoff, Halogen, stärker bevorzugt Brom oder Chlor, C1-C2-Alkyl, Phenoxy, Benzyloxy oder Phenyl sind, worin jeder Phenylanteil wahlweise mit C1-C6-Alkyl, Halogen; oder C1-C6-Alkoxy substituiert ist, Ra Wasserstoff, Fluor oder C1-C2-Alkyl ist, und R1 Wasserstoff oder Methyl ist.
- Andere bevorzugte Verbindungen sind diejenigen, worin sich die Methylenbrücke, die die Indolylgruppe mit Ar verbindet, mit Bezug auf ein Stickstoffatom in Ar α befindet.
- Andere spezifischere Verbindungen der Erfindung sind diejenigen, worin Z eine kovalente Bindung ist, R6 Hydroxy ist, Ra Wasserstoff ist, Ar ggfs. 4,5,6- oder 7-benzo-substituiertes Benzothiazolyl ist oder Ar 2-Benzothiazolyl, substituiert an Benzo mit einem Trifluoracetyl oder Trifluormethylthio oder einem oder zwei von Fluor, Chlor, Brom, Hydroxy, Methyl, Methoxy, Trifluormethyl, Trifluormethoxy, Trifluormethylthio oder einem oder vorzugsweise zwei Fluor und einem Trifluormethyl, oder zwei Fluor oder zwei Trifluormethyl mit einem Methoxy oder drei Fluor, oder mit 6,7-Benzo, und diejenigen, worin eines von R2 und R3 Wasserstoff Fluor, Chlor, Brom oder Methyl ist, und eines von R4 und R5 Wasserstoff oder Chlor, Brom, Methyl, Isopropyl, Methoxy, Nitro oder Trifluormethyl ist; oder R3 und R4 5,6-Difluor sind, Ra Wasserstoff ist; und diejenigen, worin Ar wahlweise substituiertes Benzothiazol-2-yl ist und R3 und R4 jeweils Chlor sind, und R1 Wasserstoff oder Methyl ist.
- Weitere spezifischere Verbindungen sind diejenigen, worin Z eine kovalente Bindung ist, R6 Hydroxy ist, Ar wahlweise substituiertes Benzothiazol-2-yl ist, R3 und R4 Wasserstoff sind, und R5 Methyl ist; diejenigen, worin Z eine kovalente Bindung ist, R6 Hydroxy ist, R3, R4 und R5 Wasserstoff, Chlor, Fluor, Brom oder C1-C2-Alkyl sind, Ra Wasserstoff ist, und Ar wahlweise 4,5,6- oder 7-benzosubstituiertes Benzothiazolyl-2-trifluormethyl ist; und diejenigen, worin Z CH2 ist, R6 Hydroxy ist, Ar ggfs. substituiertes Benzothiazol-2-yl oder Benzothiazol-5-yl ist, und R3, R4 und R5 unabhängig Wasserstoff, Chlor, Fluor, Brom, C1-C2-Alkyl oder Trifluormethyl sind, und Ra Wasserstoff ist.
- Im Allgemeinen ist R1 bei den vorstehend beschriebenen, spezifischen Verbindungen Wasserstoff, Halogen, vorzugsweise Chlor oder Fluor, C1-C6-Alkyl, oder Phenyl, wahlweise substituiert mit bis zu drei Gruppen, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl, C1-C6-Alkoxy, Amino und Mono- oder Di(C1-C6)-alkylamino. Bevorzugte R1-Gruppen sind Wasserstoff und Wasserstoff und Methyl.
- Weitere bevorzugte Verbindungen der Erfindung sind diejenigen, worin Ar ein substituiertes Benzothiazol, d.h. eine Verbindung der Formel III, ist:worin

A eine C1-C4-Alkylengruppe ist, wahlweise substituiert mit C1-C2-Alkyl;
Z eine Bindung oder C1-C3-Alkylen ist, wahlweise substituiert mit C1-C2-Alkyl;
Ra Wasserstoff, C1-C6-Alkyl, Fluor oder Trifluormethyl ist;
R1 Wasserstoff, C1-C6-Alkyl oder Phenyl ist, ggfs. substituiert mit bis zu drei Gruppen, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl, C1-C6-Alkoxy, Amino und Mono- oder Di(C1-C6)-alkylamino;
R2, R3, R4 und R5 jeweils unabhängig Wasserstoff, Halogen, eine Alkylgruppe mit 1-6 Kohlenstoffatomen (die mit einem oder mehreren Halogenen substituiert sein kann), Nitro, OR7;
Phenyl, wobei der Phenylanteil gffs. substituiert ist mit bis zu drei Gruppen, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl oder C1-C6-Alkoxy;
Phenoxy, worin der Phenylanteil wahlweise substituiert ist mit bis zu drei Gruppen, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl oder C1-C6-Alkoxy; oder eine Gruppe der Formelworin
J eine Bindung, CH2, Sauerstoff oder Stickstoff ist; und
jedes r unabhängig 2 oder 3 ist;
R6 Hydroxy oder -O–M+ ist, worin M+ ein Kation ist, das ein pharmazeutisch annehmbares Salz bildet; und
R11, R12, R13 und R14 unabhängig Wasserstoff, Fluor, Clor, Brom, Trifluormethyl oder Nitro sind. - Bei bevorzugten Verbindungen der Formel III stellen die R2, R3, R4 und R5 Substituenten in Kombination eines von Brom, Cyano oder Nitro, eines oder zwei von Fluor, Chlor, Hydroxy, (C1-C6)-Alkyl, (C1-C6)-Alkoxy oder Trifluormethyl oder zwei Fluor oder zwei Methyl mit einem Hydroxy oder ein (C1-C6)-Alkoxy oder ein oder vorzugsweise zwei Fluor und ein Methyl oder drei Fluorgruppen dar. Besonders bevorzugte R2, R3, R4 und R5 Substituenten sind unabhängig Fluor, Chlor, Nitro und Trifluormethyl.
- Bei bevorzugten Verbindungen der Formeln II und III ist A vorzugsweise Methylen, Methylen, substituiert mit einer Methylgruppe, oder Ethylen.
- Bevorzugte Verbindungen gemäß der vorstehenden Formel II umfassen diejenigen, worin R8 Fluor ist, R9 Wasserstoff ist und R10 Brom ist, oder diejenigen, worin R8 und R10 Wasserstoff sind und R9 Nitro ist.
- Bevorzugte Verbindungen der vorstehenden Formel III sind diejenigen, worin die Benzothiazolkomponente mit Nitro, einem, zwei oder drei von Fluor, einem oder zwei von Chlor oder mindestens einer Trifluormethylgruppe substituiert ist. Stärker bevorzugte Verbindungen der Formel II sind diejenigen, worin A Methylen ist, R1 Wasserstoff oder Methyl ist, Z eine Bindung ist und R6 Hydroxy oder C1-C6-Alkoxy ist.
- Besonders bevorzugte Verbindungen der Formel I sind diejenigen, worin R3 und R4 unabhängig Wasserstoff, C1-C6-Alkyl, C1-C6-Alkoxy oder Halogen sind, und Ra Methyl oder Wasserstoff ist, Z eine Bindung ist, A Methylen, mit Methyl substituiertes Methylen oder Ethylen ist, R1 Methyl oder Wasserstoff ist und R11, R12 und R14 Fluor oder Chlor sind.
- Der Begriff "Prodrug-Gruppe" bezeichnet eine Komponente, die in vivo in die Wirkstoffverbindung der Formel I umgewandelt wird, worin R6 Hydroxy ist. Solche Gruppen sind im Allgemeinen im Stand der Technik bekannt und umfassen esterbildende Gruppen zur Bildung einer Ester-Prodrug wie Benzyloxy, Di(C1-C6)-alkylaminoethyloxy, Acetoxymethyl, Pivaloyloxymethyl, Phthalidoyl, Ethoxycarbonyloxyethyl, 5-Methyl-2-oxo-1,3-dioxol-4-yl-methyl und (C1-C6)-Alkoxy, wahlweise substituiert mit N-Morpholino, und amidbildende Gruppen wie Di(C1-C6)-alkylamino. Bevorzugte Prodrug-Gruppen umfassen C1-C6-Alkoxy und O–M+, worin M+ ein Kation darstellt. Bevorzugte Kationen umfassen Natrium, Kalium und Ammonium. Andere Kationen umfassen Magnesium und Calcium. Weitere bevorzugte Prodrug-Gruppen umfassen O+M++, worin M++ ein zweiwertiges Kation wie Magnesium oder Calcium ist.
- In bestimmten Situationen können Verbindungen der Formel I ein oder mehrere asymmetrische Kohlenstoffatome enthalten, so dass die Verbindungen in verschiedenen stereoisomeren Formen existieren können. Diese Verbindungen können beispielsweise Racemate oder optisch aktive Formen sein. In diesen Situationen können die einzelnen Enantiomere, d.h. optisch aktive Formen, durch asymmetrische Synthese oder durch Aufspaltung der Racemate erhalten werden. Die Aufspaltung der Racemate kann beispielsweise durch herkömmliche Verfahren wie Kristallisation in Gegenwart eines Aufspaltungsmittels oder Chromatographie unter Verwendung von beispielsweise einer chiralen HPLC-Säule bewirkt werden.
- Des weiteren kann, falls die Verbindung oder Prodrug der Erfindung als Säureadditionssalz erhalten wird, die freie Base durch Basischstellen einer Lösung des Säuresalzes erhalten werden. Umgekehrt kann, wenn das Produkt eine freie Base ist, ein Additionssalz, insbesondere ein pharmazeutisch annehmbares Additions salz durch Lösen der freien Base in einem geeigneten organischen Lösungsmittel und Behandeln der Lösung mit einer Säure gemäß herkömmlichen Verfahren zur Herstellung von Säureadditionssalzen aus Baseverbindungen hergestellt werden.
- Nichttoxische, pharmazeutische Salze umfassen Salze von Säuren wie Salzsäure, Phosphorsäure, Bromwasserstoffsäure, Schwefelsäure, Sulfinsäure, Ameisensäure, Toluolsulfonsäure, Methansulfonsäure, Salpetersäure, Benzoesäure, Zitronensäure, Weinsäure, Maleinsäure, Hydroiodsäure, Alkansäure, wie Essigsäure, HOOC-(CH2)(CH2)n-ACOOH, worin n 0-4 ist, und dergleichen. Nichttoxische pharmazeutische Baseadditionssalze umfassen Salze von Basen wie Natrium, Kalium, Calcium, Ammonium und dergleichen. Fachleute werden eine große Vielfalt von nichtoxischen, pharmazeutisch annehmbaren Additionssalzen erkennen.
- Wie hier verwendet sind die Begriffe 2-Benzothiazolyl und Benzothiazol-2-yl Synonyme.
- Repräsentative Gruppen der Formelumfassen diejenigen, worin J Sauerstoff ist und jedes r 2(Morpholinyl) ist, J Stickstoff ist und jedes r 2(Piperazinyl) ist oder jedes r 2- und das andere 3(Homopiperazinyl) ist, oder J CH2 ist und jedes r 2(Piperidinyl) ist oder ein r 2- und das andere 3(Homopiperidinyl) ist. Bevorzugte Gruppen dieser Formel sind Morpholinyl und Piperazinyl.
- Die nachfolgenden Verbindungen der Erfindung sind angegeben, um dem Leser ein Verständnis der unter die Erfindung fallenden Verbindungen zu vermitteln.
3-(4,5,7-Trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
5-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
2-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
5-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
7-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
6-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
5-Benzyloxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
6-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
5-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
6-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
3-Methyl-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-2-Propionsäure
3-Methyl-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-3-Propionsäure
3-(5-Trifluormethylbenzothiazol-2-yl)methylindol-N-Essigsäure
5-Methyl-3-(5-trifluormethylbenzothiazol-2-yl)methylindol-N-Essigsäure
3-(3-Nitrophenyl)methylindol-N-Essigsäure - Die vorstehend angegebenen Verbindungen, die in den nachfolgenden Beispielen und der weiteren nachfolgenden Beschreibung der Erfindung näher beschrieben sind, dienen der Veranschaulichung, sollen jedoch den Umfang der in Erwägung gezogenen Verbindungen auf irgendeine Weise beschränken.
- Die Verbindungen der allgemeinen Formel I können oral, topisch, parenteral, durch Inhalieren oder Sprühen oder rektal in Dosierungseinheitsformulierungen verabreicht werden, die herkömmliche, nichttoxische, pharmazeutisch annehmbare Träger, Hilfsstoffe und Vehikel enthalten. Der Begriff parenteral, wie hier verwendet, umfasst subkutane Injektionen, intravenöse, intramuskuläre, intrasternale Injektions- oder Infusionstechniken. Des weiteren wird eine pharmazeutische Formulierung zur Verfügung gestellt, die eine Verbindung der allgemeinen Formel I und einen pharmazeutisch annehmbaren Träger umfasst. Eine oder mehrere Verbindungen der allgemeinen Formel I können zusammen mit einem oder mehreren nichttoxischen, pharmazeutisch annehmbaren Trägern und/oder Verdünnungsmitteln und/oder Hilfsstoffen und falls gewünscht anderen Wirkbestandteilen vorhanden sein. Die pharmazeutischen Zusammensetzungen, die Verbindungen der allgemeinen Formel I enthalten, können in einer Form sein, die für die orale Anwendung geeignet ist, beispielsweise als Tabletten, Pastillen, wässerige oder ölige Suspensionen, dispergierbare Pulver oder Körnchen, Emulsionen, harte oder weiche Kapseln oder Sirupe oder Elixiere.
- Zusammensetzungen, die für die orale Anwendung gedacht sind, können gemäß einem beliebigen im Stand der Technik bekannten Verfahren für die Herstellung von pharmazeutischen Zusammensetzungen hergestellt werden, und solche Zusammensetzungen können eines oder mehrere Mittel, ausgewählt aus der Gruppe, bestehend aus Süßungsmitteln, Aromastoffen, Färbemitteln und Konservierungsmitteln, enthalten, um pharmazeutisch feine und wohlschmeckende Zubereitungen zur Verfügung zu stellen. Tabletten enthalten den Wirkbestandteil in Mischung mit nichttoxischen, pharmazeutisch annehmbaren Arzneimittelträgern, die für die Herstellung von Tabletten geeignet sind. Diese Arzneimittelträger können beispielsweise inerte Verdünnungsmittel wie Calciumcarbonat, Natriumcarbonat, Lactose, Calciumphosphat oder Natriumphosphat, Granulierungs- und Aufschlussmittel, beispielsweise Maisstärke oder Alginsäure, Bindemittel, beispielsweise Stärke, Gelatine oder Akazie, und Gleitmittel, beispielsweise Magnesiumstearat, Stearinsäure oder Talkum sein. Die Tabletten können unbeschichtet sein oder sie können mittels bekannter Techniken beschichtet werden, um die Zersetzung und Absorption im Magendarmtrakt zu verzögern und dadurch für eine Langzeitwirkung während eines längeren Zeitraums zu sorgen. Beispielsweise kann ein zeitlich verzögerndes Material wie Glycerylmonostearat oder Glyceryldistearat verwendet werden.
- Formulierungen zur oralen Anwendung können auch als harte Gelatinekapseln, bei denen der Wirkbestandteil mit einem inerten, festen Verdünnungsmittel, beispielsweise Calciumcarbonat, Calciumphosphat oder Kaolin gemischt wird, oder als weiche Gelatinekapseln dargeboten werden, bei denen der Wirkbestandteil mit Wasser oder einem Ölmedium, beispielsweise Erdnussöl, flüssigem Paraffin oder Olivenöl gemischt wird.
- Wässerige Suspensionen enthalten die Wirkstoffe in Mischung mit Arzneimittelträgern, die für die Herstellung von wässerigen Suspensionen geeignet sind. Solche Arzneimittelträger sind Suspensionsmittel, beispielsweise Natriumcarboxymethylcellulose, Methylcellulose, Hydropropylmethylcellulose, Natriumalginat, Polyvinylpyrrolidon, Tragantgummi und Akaziengummi; Dispergier- oder Benetzungsmittel können ein in der Natur vorkommendes Phosphatid, beispielsweise Lecithin, oder Kondensationsprodukte eines Alkylenoxids mit Fettsäuren, beispielsweise Polyoxyethylenstearat, oder Kondensationsprodukte von Ethylenoxid mit langkettigen, aliphatischen Alkoholen, beispielsweise Heptadecaethylenoxycetanol, oder Kondensationsprodukte von Ethylenoxid mit von Fettsäuren und einem Hexitol abgeleiteten Partialestern, wie Polyoxyethylensorbitonmonooleat, oder Kondensationsprodukte von Ethylenoxid mit von Fettsäuren und Hexitolanhydriden abgeleiteten Partialestern, beispielsweise Polyethylensorbitanmonooleat, sein. Die wässerigen Suspensionen können auch ein oder mehrere Konservie rungsmittel, beispielsweise Ethyl, oder n-Propyl-p-hydroxybenzoat, ein oder mehrere Färbemittel, einen oder mehrere Geschmackstoffe und ein oder mehrere Süßungsmittel, wie Sucrose oder Saccharin, enthalten.
- Ölige Suspensionen können durch Suspendieren der Wirkbestandteile in einem Pflanzenöl, beispielsweise Arachisöl, Olivenöl, Sesamöl oder Kokosnussöl oder in einem Mineralöl, wie flüssigem Paraffin, formuliert werden. Die öligen Suspensionen können ein Dickungsmittel, beispielsweise Bienenwachs, hartes Paraffin oder Cetylalkohol, enthalten. Süßungsmittel, wie diejenigen; die vorstehend angegeben sind, und Aromastoffe können zugegeben werden, um wohlschmeckende, orale Zubereitungen zur Verfügung zu stellen. Diese Zusammensetzungen können durch die Zugabe eines Antioxidationsmittels wie Ascorbinsäure konserviert werden.
- Dispergierbare Pulver und Körnchen, die für die Herstellung einer wässerigen Suspension durch die Zugabe von Wasser geeignet sind, stellen den Wirkbestandteil in Mischung mit einem Dispersions- oder Benetzungsmittel, Suspensionsmittel und einem oder mehreren Konservierungsmitteln zur Verfügung. Geeignete Dispersions- oder Benetzungsmittel und Suspensionsmittel sind durch diejenigen, die bereits vorstehend erwähnt wurden, veranschaulicht. Zusätzliche Arzneimittelträger, beispielsweise Süßungsmittel, Aromastoffe und Färbemittel, können auch vorhanden sein.
- Pharmazeutische Zusammensetzungen der Erfindung können auch in Form von Öl-in-Wasser-Emulsionen vorliegen. Die Ölphase kann ein Pflanzenöl, beispielsweise Olivenöl oder Arachisöl, oder ein Mineralöl, beispielsweise flüssiges Paraffin, oder Mischungen davon sein. Geeignete Emulgatoren können in der Natur vorkommende Gummis, beispielsweise Akaziengummi oder Tragantgummi, in der Natur vorkommende Phosphatide, beispielsweise Sojabohne, Lecithin und Ester oder Partialester, die von Fettsäuren und Hexitol abgeleitet sind, Anhydride, beispielsweise Sorbitanmonooleat, und Kondensationsprodukte dieser Partialester mit Ethylenoxid, beispielsweise Polyoxyethylensorbitanmonooleat, sein. Die Emulsionen können auch Süßungsmittel und Aromastoffe enthalten.
- Sirups und Elixiere können mit Süßungsmitteln, beispielsweise Glycerol, Propylenglycol, Sorbitol oder Sucrose formuliert werden. Solche Formulierungen können auch ein Milderungsmittel, ein Konservierungsmittel und Aromastoffe und Färbemittel enthalten. Die pharmazeutischen Zusammensetzungen können in Form einer sterilen, injizierbaren, wässerigen oder öligen Suspension vorliegen. Diese Suspension kann gemäß dem, was im Stand der Technik bekannt ist, unter Verwendung von denjenigen geeigneten Dispergier- oder Benetzungsmitteln und Suspensionsmitteln, die vorstehend erwähnt wurden, formuliert werden. Die sterile, injizierbare Zubereitung kann auch eine sterile, injizierbare Lösung oder Suspension in einem nichttoxischen, parenteral annehmbaren Verdünnungsmittel oder Lösungsmittel, beispielsweise als Lösung in 1,3-Butandiol, vorliegen. Unter den annehmbaren Trägern und Lösungsmitteln, die verwendet werden können, sind Wasser, die Ringer-Lösung und eine isotonische Natriumchloridlösung. Des weiteren werden sterile, nichtflüssige Öle herkömmlicherweise als Lösungsmittel oder Suspensionsmedium verwendet. Für diesen Zweck kann jedes milde, nichtflüssige Öl verwendet werden, einschließlich synthetischer Mono- oder Diglyceride. Des weiteren können Fettsäuren wie Ölsäure bei der Herstellung der Injektionslösungen Verwendung finden.
- Die Verbindungen der allgemeinen Formel I können auch in der Form von Zäpfchen für die rektale Verabreichung des Arzneimittels verabreicht werden. Diese Zusammensetzungen können durch Mischen des Arzneimittels mit einem geeigneten nichtreizenden Arzneimittelträger, der bei normalen Temperaturen fest, jedoch bei der rektalen Temperatur flüssig ist und deshalb im Rektum zur Freisetzung des Arzneimittels schmilzt, hergestellt werden. Solche Materialien sind Kakaobutter und Polyethylenglycole.
- Verbindungen der allgemeinen Formel I können parenteral in einem sterilen Medium verabreicht werden. Das Arzneimittel kann in Abhängigkeit von dem verwendeten Träger und der verwendeten Konzentration in dem Träger entweder gelöst oder suspendiert werden. Vorteilhafterweise können Hilfsstoffe wie Lokalanästhika, Konservierungsmittel und Puffermittel in dem Träger gelöst werden.
- Ein Dosierungsniveau in der Größenordnung von etwa 0,1 mg bis etwa 140 mg pro kg Körpergewicht pro Tag sind bei der Behandlung der vorstehend indizierten Zustände brauchbar (etwa 0,5 mg bis etwa 7 g pro Patient pro Tag). Die Menge des Wirkbestandteils, der mit den Trägermaterialien zur Erzeugung einer einzigen Dosierungsform kombiniert werden kann, variiert in Abhängigkeit von dem behandelten Wirt und der besonderen Verabreichungsart. Dosierungseinheitsformen enthalten im Allgemeinen etwa 1 mg bis etwa 1000 mg des Wirkbestandteils.
- Es ist jedoch ersichtlich, dass der spezifische Dosisgehalt für einen bestimmten Patienten von einer Vielzahl von Faktoren, einschließlich der Aktivität der verwendeten, spezifischen Verbindung, dem Alter, dem Körpergewicht, dem allgemeinen Gesundheitszustand, dem Geschlecht, der Diät, dem Zeitpunkt der Verabreichung, dem Verabreichungsweg und der Rate der Ausscheidung, der Arzneimittelkombination und der Schwere der bestimmten Krankheit, die der Therapie unterzogen wird, abhängt.
- Die erfindungsgemäßen Verbindungen können unter Verwendung von bekannten chemischen Reaktionen und Verfahren hergestellt werden. Allgemeine Verfahren zur Synthetisierung der Verbindungen sind nachstehend angegeben. Es ist ersichtlich, dass die Art der für die gewünschte Zielverbindung erforderlichen Substituenten oft das bevorzugte Verfahren der Synthese bestimmt. Alle variablen Gruppen dieser Verfahren sind wie in der generischen Beschreibung beschrieben, falls sie nicht spezifisch nachstehend definiert sind. Detailliertere Verfahren für bestimmte Beispiele sind nachstehend in dem experimentellen Abschnitt angegeben.
- Herstellungsverfahren
- Die erfindungsgemäßen Verbindungen, worin Ar Benzothiazolyl ist, können in geeigneterweise aus einer substituierten Indolkomponente unter Verwendung des nachstehend angegebenen Schemas A hergestellt werden.
- Schema A

- Die Behandlung eines Nitrilindols IV mit einer starken Base, wie beispielsweise Natriumydrid, Butyllithium oder Natrium-tert.-butoxid, in einem polaren, aprotischen Lösungsmittel, wie Acetonitril, Tetrahydrofuran oder N,N-Dimethylformamid, gefolgt von einer Behandlung mit einem Alkylierungsmittel, beispielsweise Ethyl oder tert.-Butylbromacetat, stellt das gewünschte N-alkylierte Produkt V zur Verfügung. Alternativ kann eine Phasentransferkatalyse in einem biphasischen Lösungsmittelsystem verwendet werden. Ein allgemeiner Überblick über solche Alkylierungen ist in Sundberg, R. J. Indoles; Kapitel 11, Academic Press Inc., San Diego, CA, 1996, zu finden. Die Kondensierung mit einem geeigneten 2-Aminothiophenolsalz VI stellt das Benzothiazolzwischenprodukt VII zur Verfügung. Diese Reaktionen werden am häufigsten in einem Alkohollösungsmittel bei erhöhten Temperaturen durchgeführt, jedoch können andere Lösungsmittel wie N,N-Dimethylformamid und N-Methylpyrrolidon verwendet werden oder die Re aktionen können in Abwesenheit von Lösungsmitteln insgesamt durchgeführt werden. Der Umfang der Reaktionsbedingungen, die für diese Umwandlung brauchbar sind, wurden früher beschrieben (US-Patent Nr. 5,700,819). Allgemeine Verfahren für die Herstellung verschiedener substituierter 2-Aminothiophenole sind ebenfalls bekannt (J. Med. Chem., 1991, 34, 108, und Chem. Pharm. Bull., 1994, 42, 1264). Im Allgemeinen wird das beste Verfahren der Synthese durch solche Faktoren, wie die Verfügbarkeit der Ausgangsmaterialien und die Einfachheit der Synthese, bestimmt. Die Entschützung der Alkansäurekomponente VII kann durch Verfahren durchgeführt werden, die für Fachleute üblich sind, um zu den Verbindungen der Formel III zu führen. Das bei der Entschützung verwendete Verfahren hängt vom Typ der Schutzgruppe ab. Eine Beschreibung solcher Schutzgruppen und Verfahren für deren Entschützen ist in: Protective Groups in Organic Synthesis, Zweite Auflage, T. W. Green and P. G. M. Wuts, John Wiley and Sons, New York, 1991, zu finden. Wenn ein Methyl- oder Ethylester verwendet wird, wird eine wässerige Natriumhydroxidlösung in Ethanol oder Dimethoxyethan in geeigneter Weise für sein Entfernen verwendet.
- Falls Nitril IV nicht im Handel erhältlich ist, kann es im Wesentlichen wie nachstehend im Schema B beschrieben, hergestellt werden, das die Bildung von mit 3-Acetonitril substituierten Indolen der Formel IV zeigt, worin Z eine Bindung ist. So wird eine Indolkomponente in einer schwachen Säurelösung, beispielsweise Essigsäure in Ethanol, mit wässerigem Formaldehyd und Dimethylamin in einem Alkohollösungsmittel behandelt. Das 3-(Dimethylamino)-methylindolprodukt kann dann mit Natrium- oder Kaliumcyanid in N,N-Dimethylformamid bei erhöhten Temperaturen behandelt werden, um das mit 3-Acetonitril substituierte Indolzwischenprodukt zu liefern. Alternativ kann ein Iminiumsalz wie N,N-Dimethylmethylenammoniumchlorid verwendet werden, um das 3-(Dimethylamino)-methylindolzwischenprodukt herzustellen.
-

- Das 3-(Dimethylamino)-methylindolzwischenprodukt kann auch zu dem mit 3-Acetonitril substituierten Indolzwischenprodukt über das Trimethylammoniumsalz umgewandelt werden. Das Salz kann durch Behandeln des Graminzwischenprodukts mit einem Alkylierungsmittel wie Methyliodid hergestellt werden. Das Trimethylammoniumsalzzwischenprodukt kann dann in das Nitril durch Behandeln mit Natrium- oder Kaliumcyanid in einem Lösungsmittel wie N,N-Dimethylformamid umgewandelt werden. Im Allgemeinen tritt die Umwandlung zu dem Acetonitril unter milderen Bedingungen auf, wenn das Trimethylammoniumsalz verwendet wird.
- Alternativ können andere Verbindungen, wie diejenigen, worin Z-Ar eine große Vielzahl von substituierten Heterocyclen darstellt, unter Verwendung des in Schema C dargestellten, allgemeinen Verfahrens hergestellt werden. Hier werden substituierte Indolzwischenprodukte, worin X eine aktivierende Gruppe wie Hydroxyl, Halogen, Dialkylamino, Trialkylammonium oder Benzotriazol ist, mit Q-Z-Ar Gruppen unter Verwendung von in der Indol-Chemie gut etablierten Verfahren gekoppelt. Beispiele dieser Verfahren, worin Q Na oder H ist und Z Schwefel, Sauerstoff, Stickstoff, Kohlenstoff oder eine Bindung ist, sind beschrieben in (A) Tidwell, J.H., Peat, A. J., Buchwald, S. L., J. Org. Chem. 59, 7164; (B) Bruneau, P., Delvare, C., Edwards, M. P., McMillan, R. M., J. Med. Chem. 1991, 34, 1028; (C) Gan, T., Cook, J. M., Tetrahedron Lett. 1997, 38, 1301; (D) Cerreto, F., Villa, A., Retico, A., Scalzo, M., Eur. J. Med. Chem. 1992, 27, 701; (E) Majchrzak, M. W., Zobel, J. N., Obradovich, D. J., Synth. Commun., 1997, 27, 3201; (F) DeLeon, C. Y., Ganem, B., J. Org. Chem. 1996, 61, 8730; (G) Katritzky, A. R., Toader, D., Xie, L., J. Org. Chem. 1996, 61, 7571.
- Es ist ersichtlich, dass in Abhängigkeit von der verwendeten, spezifischen Chemie eine Schutzgruppe, P, erforderlich sein kann. Im Allgemeinen stellt P Gruppen dar wie Acyloxy, Alkyl, Sulfonyl oder A-COOR. Die Verwendung dieser allgemeinen Verfahren ist dargestellt in Protective Groups in Organic Synthesis, Zweite Ausgabe, T. W. Green und P. G. M. Wuts, John Wiley and Sons, New York, 1991.
- Schema C
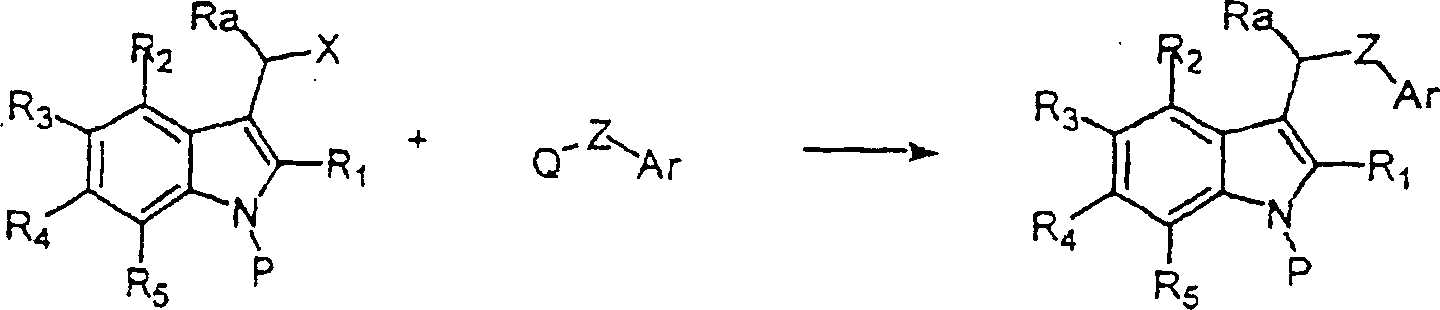
- Im Allgemeinen können die Zwischenverbindungen, worin R2_6 Aryl oder Heteroaryl ist, durch die in dem nachstehenden Reaktionsschema D dargestellte Chemie synthetisiert werden. Beispielsweise stellt die Behandlung des Kaliumsalzes eines gegebenenfalls substituierten Bromindols mit tert.-Butyllithium bei niedriger Temperatur in einem etherischen Lösungsmittel, wie Ether oder Tetrahydrofuran, gefolgt von der Zugabe eines Elektrophils, ein allgemeines Verfahren zum Erhalten von substituierten Indolen dar, wie von Rapoport, H. (J. Org. Chem. 1986, 51, 5106) beschrieben. Bezüglich der Erörterung einer Synthese, worin R Acyl ist, siehe Biorg. Med. Chem. Lett., 1999, 9, 333; worin R Thiomethyl, ist, siehe Heterocycles, 1992, 34, 1169; und worin R Cycloalkyl ist, siehe J. Med. Chem, 1999, 42, 526.
- Insbesondere liefert die Zugabe eines Trialkylborats, gefolgt von einer sauren Behandlung die gewünschten Indolboronsäuren (Heterocycles, 1992, 34, 1169). Indolboronsäuren können in gut etablierten, mit Übergangsmetall katalysierten Kupplungsreaktionen, wie der Suzuki-Reaktion, verwendet werden, um Aryl- und Heteroarylindole zu schaffen. Diese Reaktionen werden am häufigsten in einer Mischung aus etherischen und Alkohollösungsmitteln mit einer wässerigen Base in Gegenwart eines Palladiumkatalysators wie Pd(OAc)2, Pd(OAc)2w/PPh3 oder Pd(PPh3)4 durchgeführt, wie dies in Tetrahedron Lett. 1998, J. Org. Chem., 1999, 64, 1372 und Heterocycles, 1992, 34, 1395 beschrieben ist.
- Alternativ kann ein gegebenenfalls substituiertes Bromoindol mit einer Arylboronsäure und einem Palladiumkatalysator behandelt werden, um Arylindole in großen Mengen zu liefern (Synlett 1994, 93). Ein allgemeiner Überblick über die Suzuki-Kreuzkupplungen zwischen Boronsäuren und Arylhalogeniden ist in Miyaura, N, Suzuki, A., Chem. Rev. 1995, 95, 2457, zu finden.
- Schema D

- Beispielsweise liefert die Behandlung des weiterentwickelten Indolintermediats X mit einer Aryl- oder Heteroarylboronsäure unter Verwendung von mit Pd vermittelten Kupplungsbedingungen das gewünschte Aryl- und Heteroarylindolprodukt XI, wie im Schema (E) gezeigt ist. Im Allgemeinen wird die Nützlichkeit dieses Verfahrens durch die Einfachheit der Synthese der weiterentwickelten Zwischenprodukte des Typs X und die kommerzielle Verfügbarkeit der Aryl- und Heteroarylboronsäuren bestimmt.
- Schema E
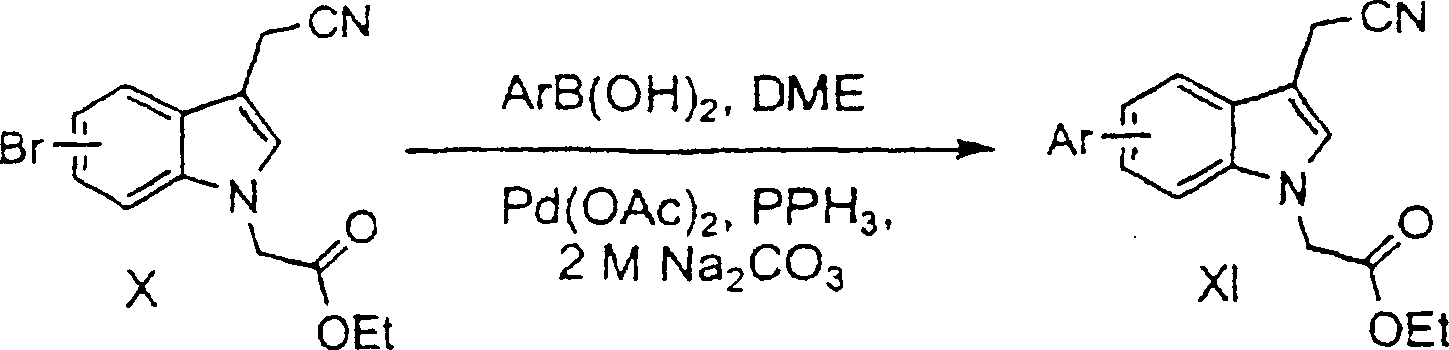
- Des weiteren eliminieren bestimmte organometallische Reaktionen den Bedarf an dem de novo Aufbau des Indolkerns. Beispielsweise dient die Stille-Reaktion als allgemeines Verfahren für die Synthese der regiokontrollierten Substitution von Indolzwischenprodukten, wie von Farina, V., Krishnamurthy, V., Scott, W., Organic Reactions, 1998, 50, 1-652 beschrieben. Wie in dem nachstehenden Schema angegeben, kann das Indol als die Oroganozinnspezies oder das Arylhalogenid dienen. Das Stannylindol (XII), worin P eine geeignete Schutzgruppe, wie 2-(Trimethyl)ethoxymethyl (SEM), oder ein Alkylsubstituent ist, wird mit einer Vielzahl von Partnern (d.h. Vinyl-/Allylhalogeniden, Vinyltriflaten, Aryl-/Heteroarylhalogeniden und Acylhalogeniden) in Gegenwart eines Pd(O)Ln Katalysators behandelt, um die gewünschten Indole (XII) zu liefern (Synnlett 1993, 771, Helv. Chim. Acta 1993, 76,2356, und J. Org. Chem. 1994, 59, 4250). Umgekehrt wird ein Haloindol (XIV) mit einer Vielzahl von Zinnreagenzien unter Stille-Bedingungen behandelt, um die gewünschten substituierten Indole (XV) zu liefern, wie dies in Heterocycles, 1988, 27, 1585 und Synth. Comm., 1992, 22, 1627 beschrieben ist.
-
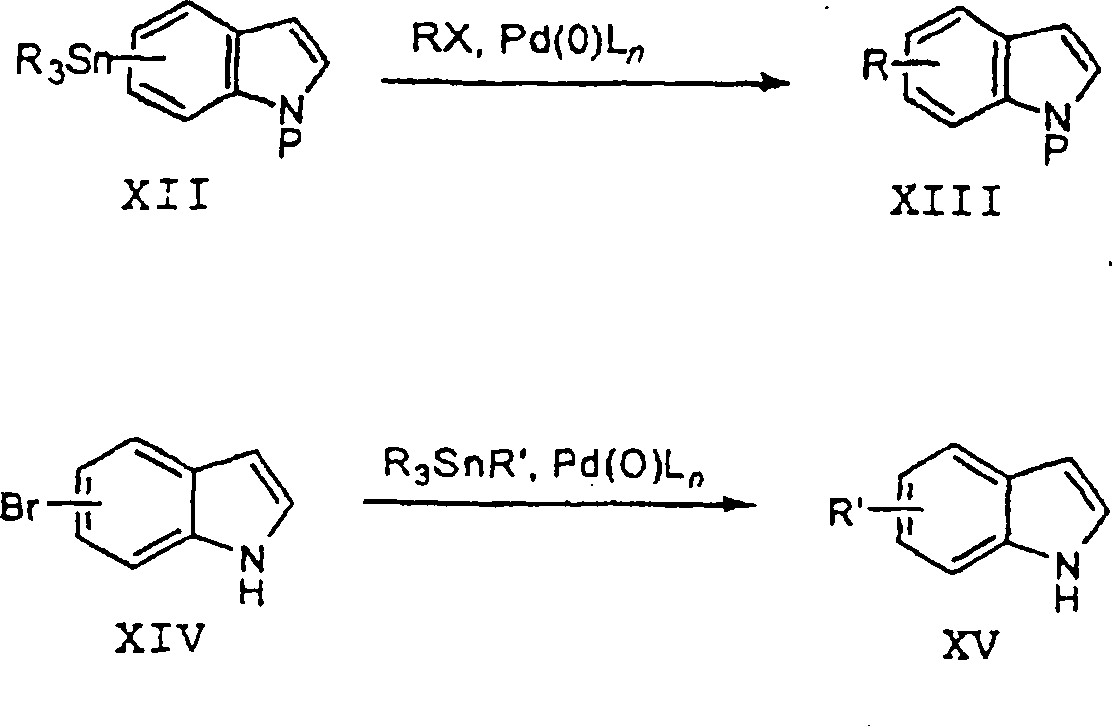
- Ein allgemeines Verfahren für die Synthese von Zwischenverbindungen unter Verwendung von Aminen der Formel NRxRx2(NR1R2 in dem nachstehenden Schema) ist durch das nachstehende Schema F angegeben. In dem Schema F sind Rx und Rx2 gleich oder verschieden und stellen Wasserstoff, C1-C6-Alkyl dar oder Rx und Rx2 stellen zusammen eine Gruppe der Formeldar, worin J und jedes r wie vorstehend für die Formel I definiert sind.
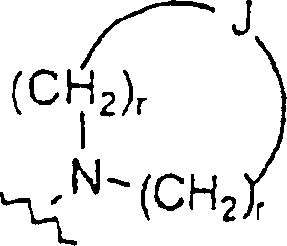
- Wie im Schema F gezeigt, ist die nucleophile Substitution von X (X ist ein Halogen, vorzugsweise Fluor) in einem aromatischen System ein Verfahren, das oft zum Substituieren von aromatischen Ringen mit Amin und Etherfunktionalitäten verwendet wird. Sowohl 4- als auch 5-Fluor-2-nitrotoluol sind ausreichend aktiviert, um einer Substitution mit Aminen in Gegenwart von K2CO3 in einem polaren, aprotischen Lösungsmittel, wie beispielsweise DMSO, unterzogen zu werden, wie dies in J. Med. Chem. 1993, 36, 2716 beschrieben ist. Das Leimgruber-Batcho Zweistufen-Verfahren ist ein allgemeines Verfahren für den Aufbau des Indolringsystems aus dem geeigneten o-Nitrotoluol. Diese Reaktion umfasst die Kondensation eines o-Nitrotoluols mit N,N-Dimethylformamiddimethylacetal, gefolgt von einer reduktiven Cyclisierung unter geeigneten Bedingungen wie Wasserstoff über einem Palladiumkatalysator, oder Zn/HOAc, wie in Sundberg, R. J., Indoles; Kapitel 2, Academic Press Inc., San Diego, CA, 1996, beschrieben ist. Eine repräsentative Beschreibung des Verfahrens ist auch in Organic Synthesis, 1984, 63, 214, zu finden.
- Schema F
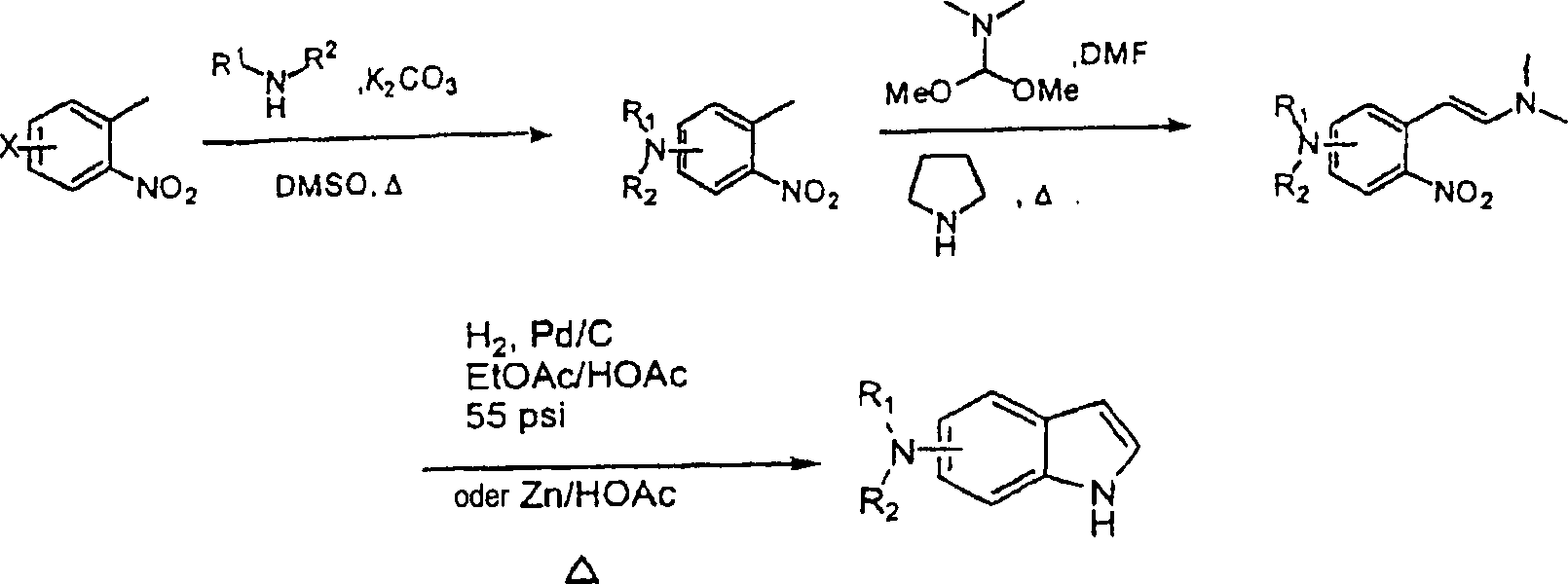
- Ein allgemeines Verfahren für die Synthese der Zwischenverbindungen, worin R eine aromatische, heteroaromatische oder Alkylgruppe ist, ist in dem nachstehenden Schema G angegeben. Wie früher beschrieben, ist die nucleophile Substitution von Halogen, vorzugsweise Fluor, in einem aromatischen System ein Verfahren, das oft verwendet wird, um aromatische Ringe mit Amin und Etherfunktionalitäten zu substituieren. Sowohl das 4- als auch das 5-Fluor-2-nitrotoluol sind ausreichend aktiviert, um einer Substitution mit Alkoholen oder Phenolen in Gegenwart von K2CO3 in einem polaren, aprotischen Lösungsmittel wie DMSO unterzogen zu werden. Ein ähnliches System unter Verwendung von KOH und Phenol ist in J. Med. Chem. 1994, 37, 1955 beschrieben. Alternativ wurden Fest-/Flüssigphasen-Transferkatalyse-(PTC-)Verfahren zur Herstellung von Zwischenethern dieses Typs verwendet, wie dies in Synth. Comm. 1990, 20, 2855 beschrieben ist. Das in geeigneter Weise substituierte o-Nitrotoluol kann dann in das entsprechende Indol durch das bereits beschriebene Leimgruber-Batcho Verfahren umgewandelt werden.
- Schema G
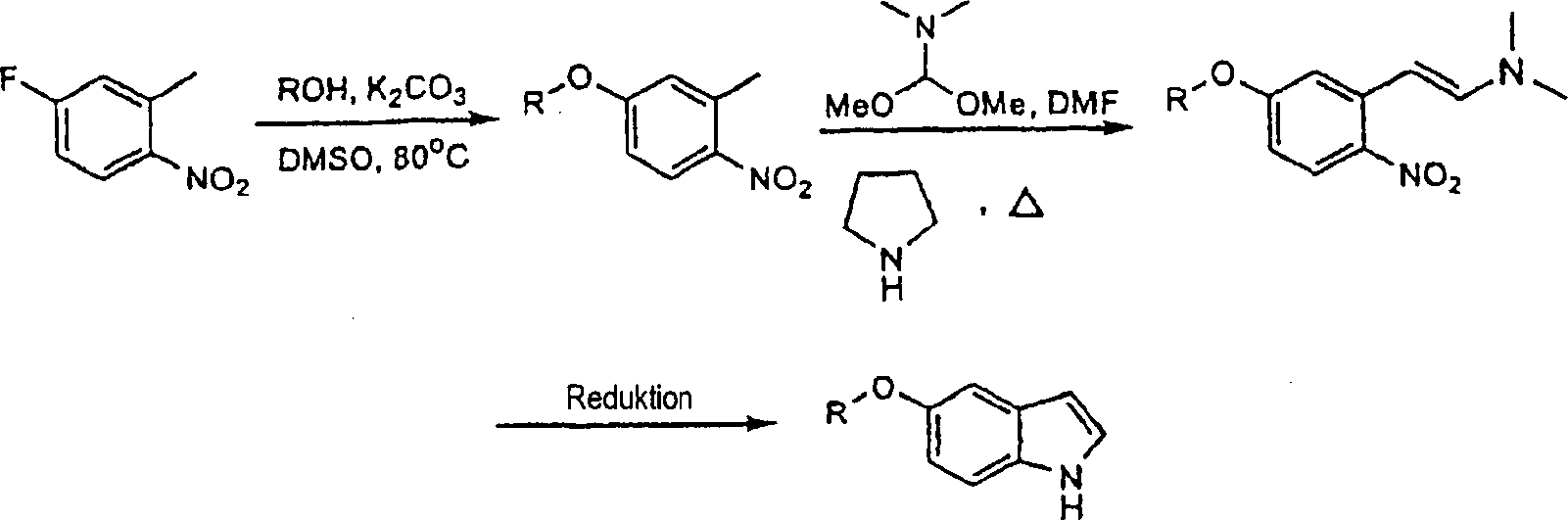
- Die Herstellung der Alkoxyindolzwischenverbindungen, worin R C1-C6-Alkyl ist, ist in dem nachstehenden Schema H angegeben. Im Handel erhältliche Nitrophenole können unter milden Bedingungen mit einer Base, wie beispielsweise K2CO3 oder Cs2CO3, in einem polaren, aprotischen Lösungsmittel, beispielsweise CH3CN, mit einer Vielzahl von geeigneten Alkylhalogeniden alkyliert werden. Siehe Synth. Comm. 1995, 25, 1367. Das Alkoxy-o-nitrotoluol kann dann wie vorstehend erwähnt zu dem gewünschten Indol umgewandelt werden.
- Schema H
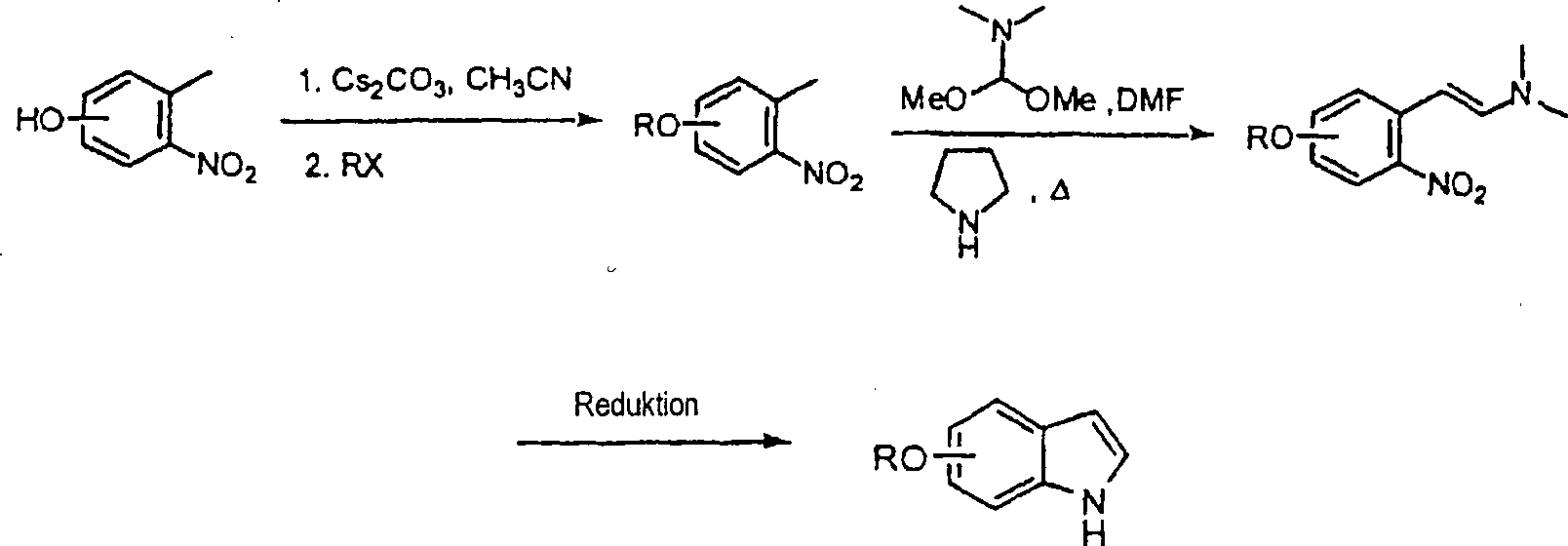
- Alternativ können einige Beispiele der Erfindung, worin Z eine Bindung ist und Ar ein substituierter Heterocyclus wie ein Thiazol ist oder Z Amid ist und Ar ein substituiertes Phenyl ist, in geeigneter Weise aus einem Indol-3-Essigsäurederivat, wie im Schema I dargestellt, hergestellt werden. Unter Verwendung dieses Verfahrens wird die Carbonsäurekomponente aktiviert und mit einem Arylamin gekoppelt. Einige Beispiele von Aktivierungsverfahren, die Fachleuten bekannt sind, umfassen die Bildung von Säurechlorid, Mischanhydriden und Kupplungsreagenzien, wie 1,3-Dicyclohexylcarbodiimid (DCC). Ein Überblick über ein solches Verfahren ist in Bodanszky, M. Principles of Peptide Synthesis; Springer-Verlag: New York, 1984, zu finden. Bei den Beispielen, bei denen Z eine Bindung ist und Ar ein substituiertes Benzothiazol oder Benzoxazol ist, kann das Zwischenamid oder -thiamid zu dem aromatischen Ring cyclisiert werden. Beispiele dieser Typen von Heterocyclen bildenden Reaktionen sind beschrieben in Mylar, B. L. et al., J. Med. Chem. 1991, 34, 108. Des weiteren kann die Carbonsäure zu einem Chlor- oder Brommethylketon umgewandelt und mit Nucleophilen wie Thioamiden oder 2-Aminothiophenolen zur Erzeugung von Thiazol- oder Benzothiazinderivaten kondensiert werden. Beispiele der Verfahren zur Herstellung der Chlor- und Brommethylketone sind in Rotella, D. P., Tetrahedron Lett. 1995, 36, 5453, und Albeck, A., Persky, R., Tetrahedron, 1994, 50, 6333 veran schaulicht. In Abhängigkeit von den Reaktionsbedingungen bei einer gegebenen synthetischen Sequenz kann eine Schutzgruppe erforderlich sein. Es ist auch ersichtlich, dass die spezifische Reihenfolge der bei der Synthese verwendeten Schritte von dem besonderen Beispiel, das hergestellt wird, abhängt. P kann H, A-COOH, niederes A-COO-Alkyl oder eine einfache Schutzgruppe darstellen, die in einer späten Phase der Synthese entfernt werden kann. Wenn eine solche Schutzgruppe verwendet wird, kann die A-CO2R6-Gruppe nahe dem Ende der Synthese eingeführt werden, nachdem die Z-Ar-Gruppe zusammengefügt worden ist. Das Verfahren des Einführens der Z-Ar-Gruppe ist ähnlich denjenigen, die bereits beschrieben wurden.
- Schema I
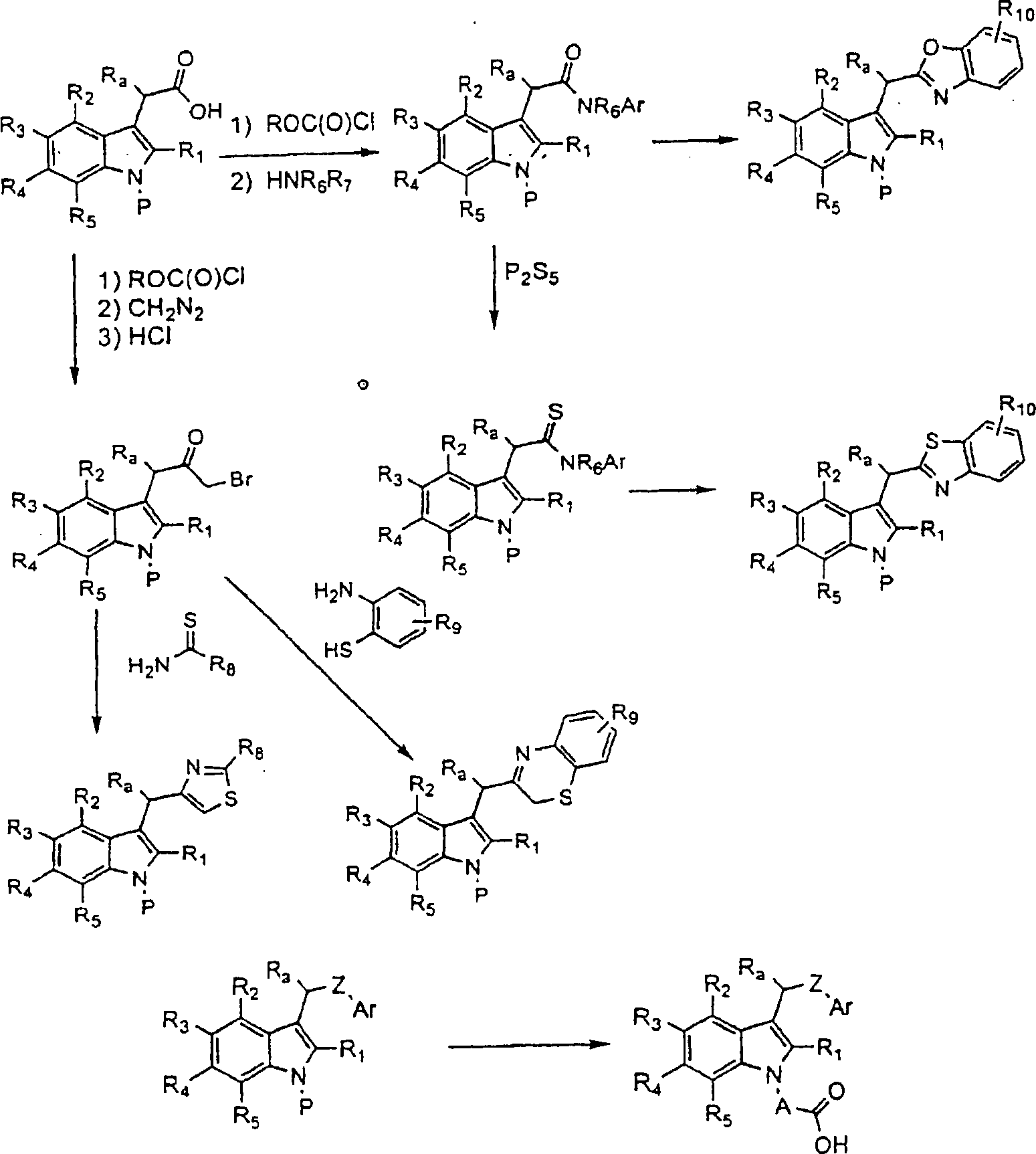
- Eine weitere Strategie umfasst die Synthese von substituierten Indolen über eine intramolekulare Cyclisierung eines Anilinstickstoffs auf ein substituiertes Alkyn, wie im Schema J gezeigt. Typische Ansätze verwenden im Handel erhältliche o-Iodanilinderivate. Wenn diese Zwischenprodukte nicht verfügbar sind, wird die regioselektive ortho-Iodierung der aromatischen Amine verwendet, um das erforderliche Zwischenprodukt zu erzeugen (J. Org. Chem. 1996, 61, 5804). Beispielsweise werden Iodphenyl-Zwischenprodukte mit Trimethylsilylacetylen in Gegenwart eines Pd-Katalysators und einer Cu(I)-Quelle, wie Kupfer(II)-iodid behandelt, um o-Alkynylaniline herzustellen. Siehe Heterocycles, 1996, 43, 2471 und J. Org. Chem. 1997, 62, 6507. Eine weitere Verarbeitung des o-Alkynylanilins zu dem gewünschten Indol kann mittels einer durch Kupfer vermittelten Cyclisierung oder einem baseinduzierten Aminringschluß auf die Alkynfunktionalität erfolgen (J. Med. Chem. 1996, 39, 892). Alternative Modifikationen wurden in den acetylenischen Derivaten durchgeführt, um ausgeklügeltere Indolstrukturen zu erzeugen, wie in J. Am. Chem. Soc. 1991, 113, 6689, Tetrahedron Lett. 1993, 24, 2823 und Tetrahedron Lett. 1993, 34, 6471 beschrieben ist.
- Schema J

- Fachleute werden erkennen, dass die Ausgangsmaterialien variiert und zusätzliche Schritte verwendet werden können, um die Verbindungen, die von der vorliegenden Erfindung umfasst sind, herzustellen, wie dies durch die nachfolgenden Beispiele gezeigt wird. In einigen Fällen kann der Schutz bestimmter reaktiver Funktionalitäten erforderlich sein, um einige der vorstehend angegebenen Umwandlungen zu erzielen. Im Allgemeinen sind der Bedarf an solchen Schutzgruppen sowie die Bedingungen, die zum Befestigen und Entfernen solcher Gruppen notwendig sind, für Fachleute auf dem Gebiet der organischen Synthese ersichtlich.
- Die Herstellung der erfindungsgemäßen Verbindungen wird weiter durch die nachfolgenden Beispiele veranschaulicht, die nicht als die Erfindung in ihrem Umfang auf die spezifischen Verfahren und Verbindungen einschränkend auszulegen sind, die in diesen beschrieben sind.
- Beispiel 1 Herstellung von 2-Methyl-3-(4-,5,7-trifluorbenzothiazol-2-yl)-methylindol-N-Essigsäure

- 2-Methyl-3-(4-,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu dem in Beispiel 2 beschriebenen Verfahren hergestellt mit dem Unterschied, dass man in Teil 1 2-Methylindol anstelle von 5-Chlorindol verwendete: 178-180°C; 1H NMR (DMSO-d6, 300 MHz) δ 7,75-7,62 (m, 1 H), 7,45 (d, J = 9,0 Hz, 1 H), 7,39 (d, J = 9,0 Hz, 1 H), 7,08 (t, J = 9 Hz, 1 H), 6,99 (t, J = 9,0 Hz, 1 H), 5,00 (s, 2 H), 4,60 (s, 2 H), 2,38 (s, 3 H); LRMS berechnet für C19H13F3N2O25: 390,0; gefunden 391,0 (M + 1)+. Anal. berechnet für C19H13F3N2O2S: C, 58,46; H, 3,36; N, 7,18; S, 8,21: Gefunden: C, 58,47; H, 3,29, N, 7,12; S, 8,18.
- Beispiel 2 Herstellung von 5-Chlor-3-(4,5,7-Trifluorbenzothiazol-2-yl)-methylindol-N-Essigsäure
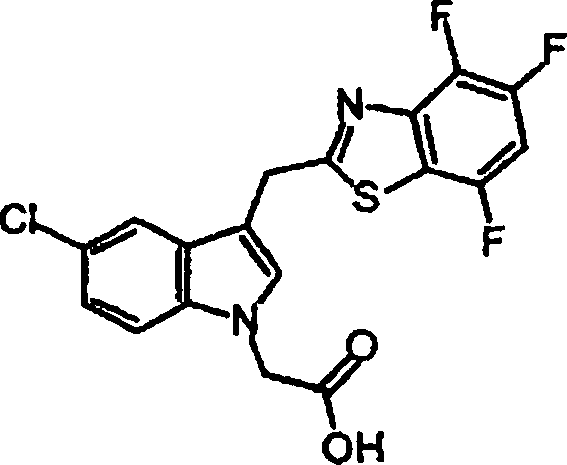
- 5-Chlorindol-3-acetonitril
- Eine Lösung von wässrigem Formaldehyd (37 %, 2,95 ml, 66,0 mMol) und Dimethylamin (40 %, 5,30 ml, 66,0 mMol) in 20 ml EtOH wurde auf 0°C gekühlt. 5-Chlorindol (4,0 g, 26,4 mMol) wurden in einem Gemisch aus HOAc : EtOH (1 : 1, 40 ml) gelöst und tropfenweise zum Reaktionsgemisch gegeben. Nach 2 Stunden Rühren bei dieser Temperatur ließ man das Gemisch sich auf Raumtemperatur erwärmen, wobei über Nacht gerührt wurde. Das Gemisch wurde zu einer gesättigten Lösung von NaHCO3 gegeben und mit 1 N NaOH versetzt, bis der pH-Wert zwischen 9 und 10 lag. Das resultierende Gemisch wurde mit CH2Cl2 (3 mal) extrahiert. Man kombinierte die organischen Anteile und wusch sie mit gesättigtem wässrigen NaCl, trocknete sie über MgSO4, filtrierte und konzentrierte sie im Vakuum. Dabei erhielt man 4,65 g (85 %) 5-Clor-3-[dimethylamino)methyl)]indol als gelbes Pulver. Ohne weitere Reinigung wurde 5-Chlor-3-[(dimethylamino)methyl]indol (4,65 g, 22,4 mMol in Dimethylformamid (80 ml) bei Raumtemperatur unter Rühren gelöst. Dazu gab man KCN (2,18 g, 33,5 mMol) in H2O (10 ml). Das Gemisch wurde auf 140°C erwärmt und 14 Stunden gerührt. Man gab H2O zu und extrahierte das Gemisch mit EtOAc (2 mal). Die organischen Anteile wurden kombiniert und mit gesättigter Salzlösung gewaschen, über MgSO4 filtriert und im Vakuum konzentriert. Der Rückstand wurde durch SiO2-Flash-Chromatographie (3 : 2, Heptan : EtOAc) gereinigt, so dass man 2,65 g (63 %) 5-Chlorindol-3-acetonitril erhielt. 1H NMR: DMSO-d6, 300 MHz) δ 11,30 (brs, 1 H), 7,63 (s, 1 H), 7,42 – 7,38 (m, 2 H), 7,05 (d, J = 6,0 Hz, 1 H), 5,70 (s, 2 H).
- 5-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 5-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 3 (Teile 1 bis 7) hergestellt mit dem Unterschied, dass in Teil 5 5-Chlorindol-3-acetonitril anstelle von 3-Indolylacetonitril verwendet wurde: Schmelzpunkt 188 – 189°C; 1H NMR (DMSO-d6, 300 MHz) δ 7,73-7,68 (m, 1 H), 7,63 (d, J = 1,8 Hz, 1 H), 7,51 (s, 1 H), 7,45 (d, J = 9,0 Hz, 1 H), 7,14 (dd, J1 = 9,0, J2 = 2,4 Hz, 1 H), 5,04 (s, 2 H), 4,65 (s, 2 H); LRMS berechnet für C18H10F3N2O2SCl : 410,0; gefunden 411,0 (M + 1)+. Anal. berechnet für C18H10F3N2O2SCl: C, 52,63; H, 2,45; N, 6,82; S, 7,81. Gefunden: C, 52,56; H, 2,40, N, 6,71, S, 7,72.
- Beispiel 3 Herstellung von 3-(4,5,7-Trifluorbenzothiazo-2-yl)methylindol-N-Essigsäure
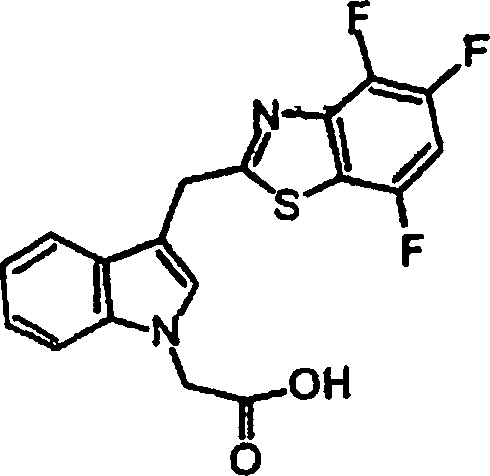
- 2,3,5,6-Tetrafluoracetanilid
- Eine Lösung von 2,3,5,6-Tetrafluoranilin (200 g, 121 Mol) in wasserfreiem Pyridin (103 ml, 1,27 Mol) wurde mit Essigsäureanhydrid (120 ml, 1,27 Mol) behandelt und zwei Stunden auf 120°C erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde die Lösung in eiskaltes Wasser (500 ml) gegossen. Der resultierende Niederschlag wurde filtriert, in Ethylacetat gelöst, über MgSO4 getrocknet, filtriert und konzentriert. Das feste Material wurde mit Heptan (200 ml) gewaschen und getrocknet, so dass 2,3,5,6-Tretrafluoracetanilid als weißer kristalliner Feststoff entstand (206 g, 82 %). Schmelzpunkt: 136-137°C; Rf 0,48 (50 % Ethylacetat in Heptan); 1H NMR (DMSO-d6 300 MHz) δ 10,10 (s, 1 H), 7,87-7,74 m, 1 H), 2,09 (s, 3 H). Anal. berechnet für C8H5F4NO: C, 46,39; H, 2,43; N, 6,67. Gefunden C, 46,35; H, 2,39; N, 6,68.
- 2,3,5,6-Tetrafluorthioacetanilid
- Ein flammengetrockneter 4-Hals-Rundbodenkolben von 5.000 ml Fassungsvermögen wurde mit Phosphorpentasulfid (198 g, 0,45 Mol) beschickt und mit wasserfreiem Benzol (3.000 ml, 0,34 M) verdünnt. 2,3,5,6-Tetrafluoracetanilid (185 g, 0,89 Mol) wurde auf einmal zugesetzt und die hellgelbe Suspension 3 Stunden auf einen leichten Rückfluss erwärmt. Die Lösung wurde auf 0°C gekühlt und filtriert. Das unlösliche Material wurde mit Ether (2 × 250 ml) gewaschen und das kombinierte Filtrat mit 10%igem wässrigem NaOH (750 ml, 500 ml) extrahiert. Nach dem Abkühlen der wässrigen Schicht auf 0°C wurde sie sorgfältig mit konzentriertem HCl (pH 2 bis 3) angesäuert. Das ausgefällte Produkt wurde durch Filtration gesammelt und mit Wasser (500 ml) gewaschen. Das gelborange Material wurde in Ethylacetat (1.000 ml) gelöst, über MgSO4 und Aktivkohle (3 g) getrocknet, durch ein kurzes Siliciumdioxidkissen (50 g) filtriert und konzentriert. Der resultierende Feststoff wurde mit Heptan (500 ml) zerrieben und filtriert, wobei 2,3,5,6-Tetrafluorthioacetanilid (174,9 g, 88 %) entstand. Schmelzpunkt: 103-104°C; Rf 0,67 (50 % Ethylacetat in Heptan); 1H NMR (DMSO-d6; 300 MHz) δ 11,20 (s, 1 H), 8,00-7,88 (m, 1 H), 2,66 (s, 3 H). Anal. berechnet für C8H5F4NS: C, 43,05; H, 2,26; N, 6,28. Gefunden C, 43,10; H, 2,23; N, 6,19.
- 4,5,7-Trifluor-2-methylbenzothiazole
- Ein flammengetrockneter 4-Hals-Rundbodenkolben von 5.000 ml Fassungsvermögen, der mit einem Über-Kopf-Rührer ausgestattet war, wurde mit Natriumhydrid (15,9 g, 0,66 Mol) beschickt und mit wasserfreiem Toluol(3.000 ml, 0,2 M) verdünnt. Die Suspension wurde auf 0°C gekühlt und auf einmal mit 2,3,5,6-Tetrafluorthioacetanilid (134 g, 0,60 Mol) behandelt. Die Lösung wurde über eine Stunde auf Raumtemperatur erwärmt und dann bis zu einem leichten Rückfluss erhitzt. Nach 30 Minuten wurde Dimethylformamid (400 ml) sorgfältig zugegeben und das Gemisch weitere zwei Stunden gerührt. Die Lösung wurde auf 0°C gekühlt und zu Eiswasser (2.000 ml) gegeben. Die Lösung wurde mit Ethylacetat (1.500 ml) extrahiert und mit gesättigtem wässrigem NaCl (1.000 ml) gewaschen. Die organische Schicht wurde bis zur Trockenheit konzentriert, mit Heptan verdünnt und nacheinander mit Wasser (300 ml) und gesättigtem wässrigem NaCl (1.000 ml) gewaschen. Die organische Schicht wurde über MgSO4 getrocknet, filtriert und konzentriert, so dass man 4,5,7-Trifluor-2-methylbenzothiazol (116,8 g, 96 %) als hellgelben Feststoff enthielt. Schmelzpunkt 91 – 92°C, Rf 0,56 (30 % Ethylacetat in Heptan); 1H NMR (DMSO-d6 300 MHz) δ 7,76-7,67 (m, 1 H), 2,87 (s, 3 H). Anal. berechnet für CeH4F3NS: C, 47,29; H, 1,98; N, 6,82; S, 15,78. Gefunden C, 47,56; H, 2,07; N, 6,82; S, 15,59.
- 2-Amino-3,4,6-trifluorthiophenolhydrochlorid
- Eine Lösung von 4,5,7-Trifluor-2-methylbenzothiazol (25,0 g, 123 mMol) in Ethylenglycol (310 ml, 0,4 M) and 30%igem wässrigem NaOH (310 ml, 0,4 M) wurde unter Verwendung eines Stickstoffstroms entgast und dann 3 Stunden bis zu einem leichten Rückfluss erhitzt. Die Lösung wurde auf 0°C abgekühlt und unter Verwendung von konzentriertem HCl (etwa 200 ml) auf einen pH von 3 bis 4 angesäuert. Die Lösung wurde mit Ether (750 ml) extrahiert und mit Wasser (200 ml) gewaschen. Die organische Schicht wurde über Na2SO4 getrocknet, filt riert und mit 2,2-Di-tert-butyl-4-methylphenol (0,135 g, 0,5 Mol-%) behandelt. Nach dem Konzentrieren bis zur Trockenheit wurde das Rohprodukt in wasserfreiem Methanol (200 ml) gelöst und mit einer HCl-Lösung in 1,4-Dioxan (37 ml, 4 N, 148 mMol) behandelt. Das resultierende Gemisch wurde bis zur Trockenheit konzentriert, mit Isopropylether (100 ml) zerrieben und filtriert, so dass man 2-Amino-3,4,6-trifluorphenolhydrochlorid (19,3 g, 73 %) als hellbraunen Feststoff erhielt, der ohne weitere Reinigung verwendet wurde. Schmelzpunkt 121 – 124°C; Rf 0,43 (30 % Ethylacetat in Heptan). Anal. berechnet für C6H5ClF3NS: C, 33,42; H, 2,34; N, 6,50, S, 14,87. Gefunden C, 33,45; H, 2,27; N, 6,48; S, 14,96.
- 3-Cyanomethylindol-N-Essigsäure-Ethlyester:
- Unter einer Stickstoffatmosphäre wurde eine Lösung von 3-Indolylacetonitril (25,0 g, 160 mMol) in trockenem Acetonitril (530 ml, 0,3 M) mit Natriumhydrid (95 %, 4,2 g, 168 mMol) behandelt und 30 Minuten gerührt. Ethylbromacetat (21,3 ml, 192 mMol) wurde über 10 Minuten tropfenweise zugesetzt und die Lösung 16 Stunden bei Raumtemperatur gerührt. Nach dem Konzentrieren unter verringertem Druck wurde der resultierende Rückstand in Ethylacetat gelöst und mit gesättigtem wässrigem NaCl gewaschen. Die organischen Extrakte wurden über MgSO4 getrocknet, filtriert und konzentriert. Das Rohprodukt wurde aus Heptan und Ethylacetat umkristallisiert und ergab die Zielverbindung als weißen kristallinen Feststoff (19 g, 49 %): Schmelzpunkt 98 bis 99°C; Rf 0,29 (30 % Ethylacetat in Heptan); 1H NMR (DMSO-d6 300 MHz) δ 7,59 (dd, J1 = 7,8 Hz, J2 = 0,6 Hz, 1H), 7,40 (dd, J1 = 8,1 Hz, J2 = 0,6 Hz, 1 H), 7,36 (s, 1 H), 7,18 (b t, J = 7,2 Hz, 1 H), 7,10 (b t, J = 7,2 Hz, 1 H), 5,12 (s, 2 H), 4,14 (q, J = 7,2 Hz, 2 H), 4,06, (s, 2 H), 1,20 (t, J = 7,2 Hz, 3 H);); LRMS berechnet für C14H14N2O2: 242,3; Gefunden 243,0 (M + 1)+. Anal. berechnet für C14H14N2O2: C, 69,49; H, 5,82; N, 11,56. Gefunden C, 69,39; H, 5,89; N, 11,59.
- 3-(4,5,7-Trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure-Ethylester:
- Unter einer Stickstoffatmosphäre wurde eine Lösung von 3-Acetonitrilindol-N-Essigsäure-Ethylester (11,0 g, 45,4 mMol) in wasserfreiem Ethanol (90 ml, 0,5 M) mit 2-Amino-3,4,6-trifluorthiophenolhydrochlorid (12,7 g, 59,0 mMol) behandelt und 16 Stunden bis zu einem leichten Rückfluss erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde die Lösung unter verringertem Druck konzent riert, mit Ethylacetat verdünnt und mit 2N HCl und gesättigtem wässrigem NaCl gewaschen. Die organische Schicht wurde über MgSO4 getrocknet, filtriert und konzentriert. Die Reinigung erfolgte durch MPLC (10 – 50 % Ethylacetat in Heptan, 23 ml/min, 150 min) und ergab 3-(4,5,7-Trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure-Ethylester (6,0 g, 36 %) als weißen kristallinen Feststoff: Schmelzpunkt 110-111°C; Rf 0,41 (30 % Ethylacetat in Heptan); 1H NMR (DMSO-d6 300 MHz) δ 7,74-7,66 (m, 1 H), 7,54 (d, J = 7,8 Hz, 1 H), 7,46 (s, 1 H), 7,40 (d, J = 8,1 Hz, 1 H), 7,15 (br t, J = 6,9 Hz, 1 H), 7,04 (br t, J = 7,8 Hz, 1 H), 5,14, s, 2 H), 4,66 (s, 2 H), 4,14 (q, J = 7,2 Hz, 3 H); LRMS berechnet für C20H15F3N2O2S: 404,4; Gefunden 405,0 (M + 1)+. Anal. berechnet für C20H15F3N2O2S: C, 59,40; H, 3,74; N, 6,93; S, 7,93. Gefunden C, 59,52; H, 3,721 N, 6,92; S, 8,04.
- 3-(4,5,7-Trifluorbenzotiahzol-2-yl)methylindol-N-Essigsäure
- Eine Lösung von 3-(4,5,7-Trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure-Ethylester (5,91 g, 14,6 mMol) in 1,2-Dimethoxyethan (73 ml, 0,2 M) wurde auf 0°C gekühlt und 15 Minuten tropfenweise mit wässrigem NaOH 1,25 N, 58 ml, 73,1 mMol behandelt. Nach Abschluss der Zugabe wurde die Lösung weitere 30 Minuten gerührt, mit 2N HCl auf einen pH von 3 angesäuert und unter verringertem Druck konzentriert. Der Rückstand wurde in Ethylacetat (200 ml) gelöst und mit gesättigtem wässrigem NaCl (30 ml) gewaschen. Der organische Extrakt wurde über Na2SO4 getrocknet, filtriert und konzentriert. Das resultierende Material wurde als Suspension in Heptan gerührt, filtriert und getrocknet, wobei man 3-(4,5,7-Trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure (5,38 g, 98 %) als hellgelben Feststoff erhielt: Schmelzpunkt 177 -178°C; Rf 0,44 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,74-7, 65 (m, 1 H), 7,53 (d, J = 7,5 Hz, 1 H), 7,46 (s, 1 H), 7,40 (d, J = 8,1 Hz, 1 H), 7,15 (b t, J = 6,9 Hz, 1 H), 7,03 (b t, J = 7,2 Hz, 1 H), 5,03 (s, 2 H), 4,65 (s, 2, H); LRMS berechnet für C18H11Fe3N2O2S: 376,4; gefunden 375,0 (M – 1)–. Anal. berechnet für C18H11Fe3N2O2S: C, 57,44; H, 2,95; N, 7,44; S, 8,52. Gefunden C, 57,58; H, 2,99; N, 7,38; S, 8,51.
- Beispiel 4 Herstellung von 5-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
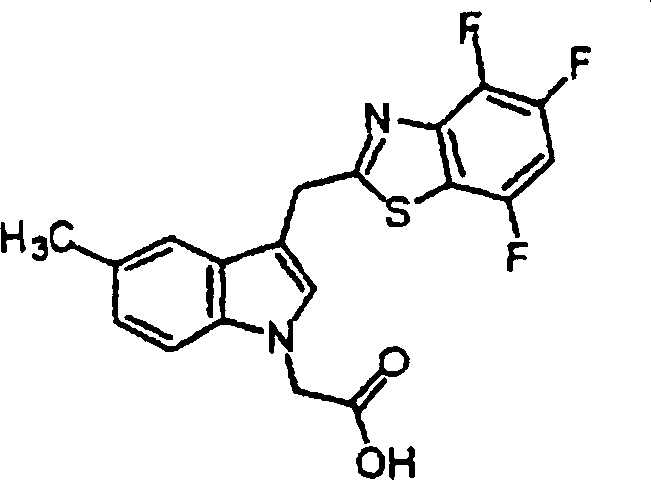
- 5-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 5-Methylindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 131-133°C; 1H NMR (DMSO-d6 300 MHz) δ 7,73-7,62 (m, 1 H), 7,39 (s, 1 H), 7,30 (s, 1 H), 7,27 (d, J = 9,0 Hz, 1 H), 6,96 (dd, J1 = 9,0 Hz, J2 = 2,4 Hz, 1 H), 4,98 (s, 2 H), 4,60 (s, 2 H), 2,32 (s, 3 H); LRMS berechnet für C19H13F3N2O2S: 390,0; Gefunden 391,0 (M + 1)+. Anal. berechnet für C19H13F3N2O2S: C, 58,46; H, 3,36; N, 7,18; S, 8,21. Gefunden: C, 58,36; H, 3,30, N, 7,10, S, 8,20.
- Beispiel 5
- Herstellung von 7-methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 7-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 7-Methylindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 216-218°C; 1H NMR (DMSO-d6 300 MHz) δ 7,73-7,63 (m, 1H), 7,36-7,32 (m, 2 H), 6,92-6,88 (m, 2 H), 5,17 (s, 2 H), 4,60 (s, 2 H), 2,55 (s, 3 H); LRMS berechnet für C19H13F3-N2O2S: 390,0; gefunden 391,0 (M + 1)+. Anal. berechnet für C19H13F3N2O2S: C, 58,46; H, 3,36; N, 7,18; S, 8,21. Gefunden: C, 58,37; H, 3,37; N, 7,11; S, 8,13.
- Beispiel 6
- Herstellung von 6-chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 6-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 6-Chlorindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 194-195°C; 1H NMR (DMSO-d6 300 MHz) δ 7,73-7,63 (m, 1 H), 7,50 (d, J = 8,4 Hz, 1 H), 7,46-7,42 (m, 2 H), 7,00 (dd, J1 = 8,4 Hz, J2 = 2,1 Hz, 1 H), 4,76 (s, 2 H), 4,62 (s, 2 H); LRMS berechnet für C18H10F3N2O2SCl: 410,0; Gefunden 411,0 (M + 1) +, Analyse berechnet für C18H10F3N2O2SCl:: C, 52,63; H, 2,45; N, 6,82; S, 7,81. Gefunden: C, 52,50; H, 2,44, N, 6,74, S, 7,69.
- Beispiel 7 Herstellung von 5-Benzyloxy-3-(4,5,7=trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure

- 5-Benzyloxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 5-Benzyloxyindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 165-168°C; 1H NMR(DMSO-d6 300 MHz) δ 7,73-7,65 (m, 1 H) 7,40-7,30 (m, 3 H), (m, 4 H), 7,28-7,10 (m, 4H), 7,10 (d, J = 2,4 Hz, 1 H), 6,87-6,80 (m, 1H), 5,05 (s, 2 H), 4,95 (s, 2 H), 4,57 (s 2 H); LRMS berechnet für C25H17F3N2O2S: 482,0; Gefunden 483,0 (M + 1)+.
- Beispiel 8
- Herstellung von 6-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 6-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 6-Fluorindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 200-203°C; 1H NMR (DMSO-d6 300 MHz) δ 7,73-7,65 (m, 1 H), 7,53 (dd, J1 = 8,4 Hz, J2 = 3, 3 Hz, 1 H), 7,44 (s, 1 H), 7,34 (dd, J1 = 10,5 Hz, J2 = 2,4 Hz, 1 H), 6,93-6,68 (m, 1 H), 5,11 (s, 2 H), 4,64 (s, 2 H); LRMS berechnet für C18H10F4N2O2S: 394,0; Gefunden 395 (M + 1).
- Beispiel 9 Herstellung von 5-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure

- 5-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zur Beispiel 2 hergestellt, mit dem Unterschied, dass in Teil 1 5-Fluorindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 193-195 C; 1H NMR (DMSO-d6 300 MHz) δ 7,65 (m, 1 H), 7,51 (s, 1 H), 7,42 (br dd, J1 = 9,0 Hz, J2 = 4,8 Hz, 1 H), 7,34 (br dd, J1 = 9,9 Hz, J2 = 2,4 Hz, 1 H), 7,02-6,96 (m, 1 H), 5,03 (s, 2 H), 4,62 (s, 2 H); LRMS berechnet für C18H10F4N2O2S: 394,0. Gefunden 395 (M + 1).
- Beispiel 10
- Herstellung von 6-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 6-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 6-Methylindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 211-213°C, Rf 0,50 (10 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,72-7,63 (m, 1 H), 7,37 (d, J = 7,1 Hz, 1 H), 7,35 (s, 1 H), 7,18 (s, 1 H), 6,85 (d, J = 8,4 Hz, 1 H), 5,08 (s, 2 H), 4,60 (s, 2 H), 2,37 (s, 3 H).
- Beispiel 11
- Herstellung von 3-(5-Trifluormethylbenzothiazol-2-yl)methylindol-N-Essigsäure
- 3-(5-Trifluormethylbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 3 (Teile 5 bis 7) hergestellt mit dem Unterschied, dass in Teil 6 2-Amino-4-(trifluormethyl)benzolthiolhydrochlorid anstelle von 2-Amino-3,4,6-trifluorthiophenolhydrochlorid verwendet wurde: Schmelzpunkt 233-234°C; 1H NMR (DMSO-d6 300 MHz) δ 8,29 (s, 1 H), 8,19 (br d, J = 8,1 Hz, 1 H), 7,68 (br d, J = 9,0 Hz, 1 H), 7,49 (br d, J = 6,9 Hz, 1 H), 7,41 1 H), 7,38 (br d, J = 8,4 Hz, 1 H), 7,12 (br t, J = 6,9 Hz, 1 H), 7,00 (br t, J = 6,9 Hz, 1 H), 5,01 (s, 2 H), 4,60 (s, 2 H).
- Beispiel 12
- Herstellung von 5-Methyl-3-(5-Trifluormethylbenzothiazol-2-yl)methylindol-N-Essigsäure
- 5-Methyl-3-(5-trifluormethylbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt, mit dem Unterschied, dass in Teil 1 5-Methylindol anstelle von 5-Chlorindol und in Teil 2 (Teil 6 von Beispiel 3) 2-Amino-4-(trifluormethyl)benzolthiolhydrochlorid anstelle von 2-Amino-3,4,6-trifluorthiophenolhydrochlorid verwendet wurde: Schmelzpunkt 248-249°C; 1H NMR (DMSO-d6 300 MHz) δ 8,27 (s, 1 H), 8,20 (d, J = 8,4 Hz, 1 H), 7,68 (d, J = 8,4 Hz, 1 H), 7,35 (s, 1 H), 7,27 (s, 1 H), 7,25 (d, J = 8,1 Hz, 1 H), 6,95 (d, J = 8,1 Hz, 1 H), 4,96 (s, 2 H), 4,57 (s, 2 H), 2,31, (s, 3 H); LRMS berechnet für C20H15F3N2O2S: Gefunden 405 (M + H).
- Beispiel 13 Herstellung von 2-Phenyl-3-(4,5,7-trifluorbenzothiazol-2yl)methyl-indol-N-Essigsäure
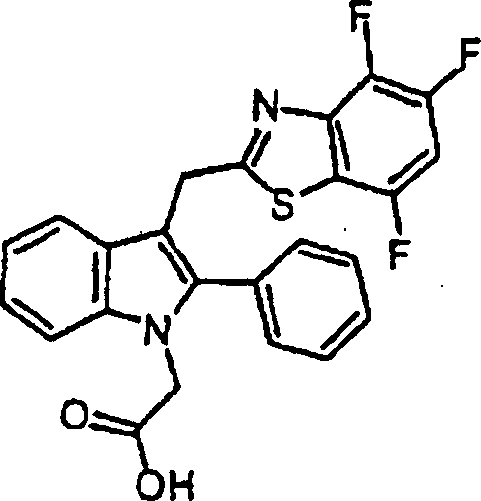
- 2-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 2-Phenylindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 238-239°C; Rf 0,60 (10 % Methanol in Chloroform); 1H NMR (DMSO-d6 300 MHz) δ 7,60-7,70 (m, 1H), 7,39-7,58 (m, 7 H), 7,20 (t, J = 9 Hz, 1 H), 7,07 (t, J = 9 Hz, 1 H), 4,80 (s, 2H), 4,45 (s, 2H); LRMS berechnet für C24H15F3N2O2S: 452,0; Gefunden 453,0 (M + 1)+. Anal. berechnet für C24H15F3N2O2S: C, 63,71; H, 3,34; N, 6,19; S, 7,09. Gefunden: C, 63,46; H, 3,32; N, 6,11; S, 6,96.
- Beispiel 14
- Herstellung von 5-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 3-Cyanomethyl-S-phenylindol-N-Essigsäure-Ethylester
- 5-Brom-3-cyanomethylindol-N-Essigsäure-Ethylester (1,0 g, 3,1 mMol) und Phenylborsäure (0,418 g, 3,4 mMol) wurden bei Raumtemperatur unter einer Stickstoffatmosphäre in wasserfreiem DME gelöst und mit Pd(OAc)2 (2,1 mg, 0,0093 mMol) und PPh3 (7,4 mg, 0,028 mMol) behandelt. Dieses Gemisch wurde bis zum Rückfluss erwärmt und über eine Spritze mit 2 M Na2CO3 (3,11 ml, 6,2 mMol) versetzt. Nach 12 Stunden wurde das Gemisch auf Raumtemperatur gekühlt und zu H2O (50 ml) gegeben. Das resultierende Gemisch wurde mit EtOAc (2 mal, 100 ml) extrahiert. Die organischen Stoffe wurden kombiniert und mit gesättigter NaCl-Lösung gewaschen, über MgSO4 getrocknet, filtriert und im Va kuum konzentriert. Der Rückstand wurde durch SiO2-Flash-Chromatographie gereinigt (Heptan zu 1 : 1 Heptan/EtOAc), um das gewünschte Material als weißen Feststoff zu ergeben (445 mg, 45 %); 1H NMR (DMSO-d6 300 MHz) δ 7,64-7,74 (m, 4 H), 7,39-7,44 (m, 4 H), 7,29-7,34 (m, 1 H), 5,20 (s, 2 H), 4,15 (q, J = 7,2 Hz, 2 H), 4,08 (s, 2 H), 1,20 (t, J = 7,2 Hz, 3 H).
- 5-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 5-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 5-Phenylindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 156-159°C; Rf 0,55 (10 % Methanol in Chloroform): 1H NMR (DMSO-d6 300 MHz) δ 7,66-7,69 (m, 4 H), 7,57-7,60 (m, 1 H), 7,39-7,47 (m, 3 H), 7,29-7,35 (m, 2 H), 5,06 (s, 2 H), 4,66 (s, 2 H); LRMS berechnet für C24H15F3N2O2S: 452,0. Gefunden 453,0 (M + 1)+. Anal. berechnet für C24H15F3N2O2S: C, 63,71; H, 3,34; N, 6,19; S, 7,09. Gefunden: C, 63,54; H, 3,32; N, 6,13; S, 7,01.
- Beispiel 15
- Herstellung von 6-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- Teil 1: 6-Phenylindol
- Eine Lösung von 6-Bromindol (2,0 g, 10,20 mMol) in wasserfreiem Toluol(20 ml) wurde unter einer Stickstoffatmosphäre mit Pd[P(Ph3)]4 (10 % Mol) behandelt. Nachdem man das Gemisch 30 Minuten gerührt hatte, gab man erst Phenylborsäure (1,87 g, 15,30 mMol) in wasserfreiem EtOH (10 ml) und dann gesättigtes NaHCO3 (6 ml) zu. Das zweiphasige Gemisch wurde 24 Stunden bis zum Rückfluss erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde das Gemisch zu einer gesättigten Salzlösung gegeben und mit EtOAc (2 mal) extrahiert. Die organische Schicht wurde über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Flash-Säulenchromatographie (1 : 1 CH2Cl2/Heptan) gereinigt und ergab das gewünschte Material als weißes Pulver (900 mg, 45 %): 1H NMR (DMSO-d6 300 MHz) δ 11,15 (br s, 1 H), 7,58-7,66 (m, 4 H), 7,41-7,47 (m, 2 H), 7,36 (m, 1 H), 7,26-7,31 (m, 2 H), 6,42 (m, 1 H).
- Herstellung von 6-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 6-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 6-Phenylindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 156-159°C; Rf 0,50 (10% Methanol in Chloroform); 1H NMR (DMSO-d6 300 MHz) δ 7,65-7,75 (m, 4 H), 7,57-7,62 (m, 1 H), 7,41-7,50 (m, 3 H), 7,26-7,38 (m, 2 H), 5,12 (s, 2 H), 4,68 (s, 2 H); LRMS berechnet für C24H15F3N2O2S: 452,0. Gefunden 453,0 (M + 1)+. Anal. berechnet für C24H15F3N2O2S: C, 63,71; H, 3,34; N, 6,19; S, 7,09, Gefunden: C, 63,46; H, 3,33; N, 6,10; S, 6,96.
- Beispiel 16 Herstellung von 5-Morpholino-3-(4,5,7-trifluorbenzothiazol-2yl)methylindol-N-Essigsäure
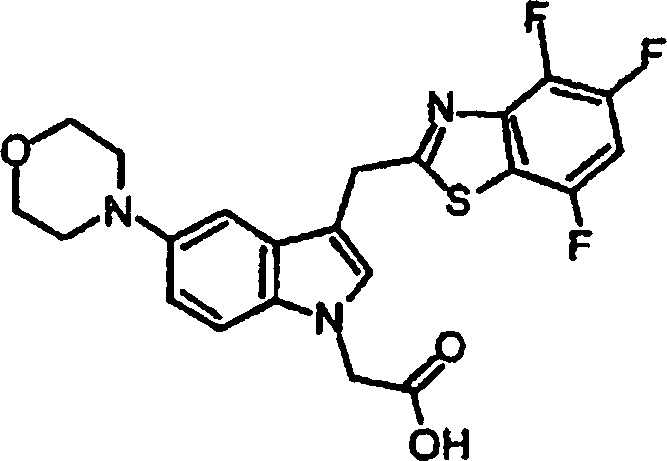
- 5-Morpholino-2-nitrotoluol
- Ein Gemisch aus 5-Fluor-2-nitrotoluol (5,11 g, 32,9 mMol), Morpholin (4,31 ml, 49,4 mMol) und K2CO3 (6,83 g, 49,4 mMol) wurde bei Raumtemperatur unter Rühren in wasserfreiem DMSO (80 ml) verdünnt. Das Gemisch wurde 24 Stunden auf 80°C erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde H2O zugegeben und das resultierende Gemisch mit EtOAc (3 mal, 50 ml) extrahiert. Die organische Schicht wurde mit gesättigtem wässrigen NaCl (100 ml) gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Der verbleibende Feststoff wurde in Heptan (200 ml) zerrieben und filtriert, um das erwünschte Material als gelbes Pulver zu ergeben (7,10 g, 97 %). Rf 0,40 (75 % Heptan/25 % Ethylacetat). 1H NMR (DMSO-d6 300 MHz) δ 7,96 (d, J = 9,9 Hz, 1 H), 8,85- 8,88 (m, 2 H), 3,70 (t, J = 5,0 Hz, 4 H), 3,35 (t, J = 5,0 Hz, 4 H), 2,53 (s, 3 H).
- Herstellung von 5-Morpholinoindol
- Unter einer Stickstoffatmosphäre wurde eine Lösung von 5-Morpholinyl-2-nitrotoluol (7,0 g, 31,5 mMol) in DMF (100 ml) mit Dimethylformamiddimethylacetal (4,81 ml, 36,2 mMol) und Pyrrolidin (2,62 ml, 31,5 ml) behandelt. Das Gemisch wurde auf 100°C erwärmt und 12 Stunden auf dieser Temperatur belassen. Nach dem Abkühlen wurde das Gemisch im Vakuum konzentriert und ergab das gewünschte Intermediat als ziegelroten Feststoff.
- Das Intermediat Enamin wurde in EtOAc (200 ml) gelöst und in eine vorbefüllte Parr-Flasche mit 10 % Pd/C (600 mg) in EtOAc (40 ml) gegeben. Das Gemisch wurde auf einer Parr-Schüttelvorrichtung 2,5 Stunden bei 55 psi hydriert. Der Katalysator wurde durch einen Celite-Pfropfen mit mehreren Wäschen mit EtOAc filtriert und das verbleibende Filtrat im Vakuum konzentriert. Der Rückstand wurde durch SiO2-Flash-Chromatograpie (1 : 1 Hept/EtOAc) gereinigt und ergab 2,0 g (31 % über 2 Teile) des erwünschten Indols als cremefarbenes Pulver: Rf 0,30 (10 % Methanol in Chloroform) 1H NMR (DMSO-d6 300 MHz) δ 10,77 (br s, 1 H), 7,24 (s, 1 H), 7,18-7,20 (m, 1 H), 6,97 (d, J = 1,8 Hz, 1 H), 6,81 (dd, J1 = 8,7 Hz, J2 = 2,1 Hz, 1 H), 6,25 (dd, J1 = 3,0 Hz, J2 = 1,8 Hz, 1 H), 3,7 (t, J = 4,50 Hz, 4 H), 2,96 (t, J = 4,50 Hz, 4 H).
- Herstellung von 5-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 5-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass 5-Morpholinoindol anstelle von 5-Chlorindol verwendet wurde. 1H NMR (DMSO-d6 300 MHz) δ 7,64-7,72 (m, 1 H), 7,34 (s, 1 H), 7,26 (d, J = 9,0 Hz, 1 H), 7,06 (d, J = 2,4 Hz, 1 H), 6,91 (dd, J1 = 9,0 Hz, J2 = 2,4 Hz, 1 H), 4,95 (s, 2 H), 4,60 (s, 2 H), 3,70-3,73 (m, 4 H), 2,37-3,00 (m, 4 H); LRMS berechnet für C22H18F3N3O3S: 461,0; Gefunden 462 (M + 1)+. Anal. berechnet für C22H18F3N3O3S·1H2O: C, 55,11; H, 4,20; N, 8,76; S, 6,69. Gefunden: C, 55,11; H, 4,05; N, 8,57; S, 6,50.
- Beispiel 17
- Herstellung von 6-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- Herstellung von 4-Morpholino-2-nitrotoluol
- Ein Gemisch aus 4-Fluor-2-nitrotoluol (15,34 g, 98,9 mMol), Morpholin (12,94 ml, 49,4 mMol) and K2CO3 (6,83 g, 148,3 mMol) wurde bei Raumtemperatur unter Rühren in wasserfreiem DMSO (250 ml) verdünnt. Das Gemisch wurde 24 Stunden auf 120°C erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde H2O zugegeben und das resultierende Gemisch mit EtOAc (2 mal, 75 ml) extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung (100 ml) gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Der verbleibende Feststoff wurde in Heptan (200 ml) zerrieben und filtriert, um das erwünschte Material als gelbes Pulver zu ergeben (8,00 g, 36,4 %). Rf 0,40 (25 % Ethylacetat in Heptan). 1H NMR (DMSO-d6 300 MHz) δ 7,40 (d, J = 2,7 Hz, 1 H), 7,30 (d, J = 8,7 Hz, 1 H), 7,20 (dd, J1 = 8,7 Hz, J2 = 2,7 Hz, 1 H), 3,70 (t, J = 4,8 Hz, 4 H), 3,35 (t, J = 4,8 Hz, 4 H), 2,36 (s, 3H).
- Herstellung von 6-Morpholinoindol
- Unter einer Stickstoffatmosphäre wurde eine Lösung von 4-Morpholino-2-nitrotoluol (7,1 g, 31,9 mMol) in DMF (100 ml) mit Dimethylformamiddimethylacetal (4,92 ml, 37,1 mMol) und Pyrrolidin (2,67 ml, 31,9 ml) behandelt. Das Gemisch wurde auf 100°C erwärmt und 12 Stunden auf dieser Temperatur belassen. Nach dem Abkühlen wurde das Gemisch im Vakuum konzentriert und ergab das gewünschte Intermediat als ziegelroten Feststoff. Das rohe Intermediat wurde in Eis-HOAc (250 ml) gelöst und auf 85°C erwärmt. Zn (18,17 g, 0,278 Mol) wurde der Lösung über 30 Minuten portionsweise zugesetzt. Das Gemisch wurde 4 Stunden erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde das Gemisch mit gesättigtem NaHCO3 neutralisiert und mit Et2O (3 mal, 300 ml) extrahiert. Die kombinierten organischen Stoffe wurden mit gesättigter Salzlösung gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde durch SiO2-Flash-Chromatographie (Heptan zu 2 : 1 Heptan/EtOAc) gereinigt und ergab das gewünschte Material als weißes kristallines Pulver (1,0 g, 11 % über 2 Teile): Rf 0,50 (2 : 1 Heptan/EtOAc) 1H NMR (DMSO-d6 300 MHz) δ 10,73 (br s, 1 H), 7,35 (d, J = 8,4 Hz, 1 H), 7,11 (d, J = 2,4 Hz, 1 H), 6,80 (s, 1 H), 6,73 (dd, J1 = 8,4 Hz, J2 = 2,4 Hz, 1 H), 6,25 (d, J = 2,4 Hz, 1 H), 3,72 (t, J = 4,8 Hz, 4 H), 3,02 (t, J = 4,8 Hz, 1 H).
- Herstellung von 6-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 6-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 6-Morpholinoindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 178-180°C; 1H NMR(DMSO-d6 300 MHz) δ 7,66-7,72 (m, 1 H), 7,37 (d, J = 8,4 Hz, 1 H), 7,29 (s, 1 H), 7,06 (d, J = 2,4 Hz, 1 H), 6,84 (d, J = 8,4 Hz, 1 H), 4,96 (s, 2 H), 4,58 (s, 2 H), 3,37-3,75 (m, 4 H), 3,09-3,13 (m, 4 H); LRMS berechnet für C22H18F3N3O3S: 46,01; gefunden 462 (M + 1)+. Anal. berechnet für C22H18F3N3O3S: CH2Cl2 0,50H2O: C, 49,74; H, 3,72; N, 7,57; S, 5,77. Gefunden C, 49,73; H, 3,36; N, 7,69; S, 5,58.
- Beispiel 18 Herstellung von 5-Phenoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
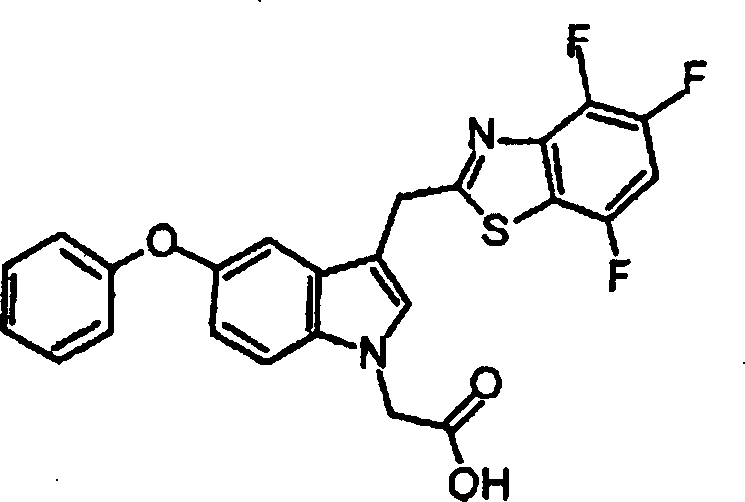
- 5-Phenoxy-2-nitrotoluol
- Eine Lösung von Phenol (12,16 g, 0,129 Mol) in wasserfreiem DMSO wurde mit K2CO3 (17,88 g, 0,129 Mol) behandelt und bei Raumtemperatur 15 Minuten gerührt. 5-Fluor-2-nitrotoluol (13,38 g, 0,086 Mol) wurde mit einer Spritze zur Lösung gegeben. Das resultierende Gemisch wurde 12 Stunden auf 80°C erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde das Gemisch in H2O (100 ml) gegossen. Nach der Extraktion mit EtOAc (2 mal, 100 ml) wurden die organi schen Stoffe kombiniert und mit gesättigter Salzlösung gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Flash-Säulenchromatographie (Heptan zu 8 : 1 Heptan/EtOAc) gereinigt; um das gewünschte Material als gelben kristallinen Feststoff (12,50 g, 63 %) zu ergeben: Rf 0,60 (85 % Heptan/15 % EtOAc); 1H NMR (DMSO-d6 300 MHz) δ 8,05 (d, J = 9,0 Hz, 1 H), 744-7,47 (m, 2 H), 7,23-7,29 (m, 1 H), 7,12-7,16 (m, 2 H), 7,04 (d, J = 2,7 Hz, 1 H), 6,90 (dd, J1 = 9.0 Hz, J2 = 2,7 Hz, 1 H), 2,51 (s, 3 H).
- 5-Phenoxyindol
- Eine Lösung von 5-Phenoxy-2-nitrotoluol (10,03 g, 0,0428 Mol) in wasserfreiem DMF wurde mit N,N-Dimethylformamiddimethyldiacetal (6,73 ml, 0,0508 Mol) und Pyrrolidin (3,63 ml, 0,0438 Mol) behandelt und 2,5 Stunden auf 110°C erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde das Gemisch mit EtOAc (500 ml) verdünnt und mit H2O gewaschen (500 ml). Die organischen Stoffe wurden über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Das rohe Intermediat wurde in Eis-HOAc (250 ml) gelöst und auf 85°C erwärmt. Zn (24,62 g, 0,377 Mol) wurde der Lösung über 30 Minuten portionsweise zugesetzt. Dann wurde das Gemisch 4 Stunden erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde das Gemisch mit gesättigtem NaHCO3 neutralisiert und mit Et2O (3 mal, 300 ml) extrahiert. Die kombinierten organischen Stoffe wurden mit gesättigter Salzlösung gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde durch SiO2-Flash-Chromatographie (Heptan zu 2 : 1 Heptan/EtOAc) gereinigt, um das gewünschte Material als weißes kristallines Pulver zu ergeben (3,1 g, 34 % über 2 Teile): Rf 0,50 (2 : 1 Heptan/EtOAc); 1H NMR(DMSO-d6 300 MHz) δ 11,12 (br s, 1 H), 7,48 (s, 1 H), 7,30-7,38 (m, 1 H), 7,25- 7,29 (m, 2 H), 7,17 (d, J = 2,7 Hz, 1 H), 6,89-7,02 (m, 1 H), 6,86-6,88 (m, 2 H), 6,80 (dd, J1 = 8,7 Hz, J2 = 2,4 Hz, 1 H), 6,37 (m, 1 H).
- Herstellung von 5-Phenoxy-3-(4,5,7-triflurobenzothiazol-2-yl)methylindol-N-Essigsäure
- 5-Phenoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 5-Phenoxyindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 128-130 °C; Rf 0,45 (10 % Methanol in Chloroform); 1H NMR (DMSO-d6 300 MHz) δ 7,65-7,70 (m, 1 H), 7,47 (s, 1 H), 7,42 (d, J = 8,4 Hz, 1 H), 7,21-7,27 (m, 3 H), 6,98 (m, 1 H), 6,83-6,90 (m, 3 H), 5,02 (s, 2 H), 4,60 (s, 2 H); LRMS berechnet für C24H15F3N2O3S: 468,0; Gefunden 467,0 (M – 1)–. Anal. berechnet für C24H15F3N2O3S: C, 55,11; H, 4,20; N, 8,76; S, 6,69. Gefunden: C, 55,11; H, 4,05; N, 8,57; S, 6,50.
- Beispiel 19
- Herstellung von 7-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 7-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 7-Fhorindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 194-196°C; Rf 0,60 (10 Methanol in Chloroform); 1H NMR (DMSO-d6 300 MHz) δ 7,67-7,73 (m, 1 H), 7,46 (s, 1 H), 7,35 (d, J = 7,2 Hz, 1 H), 6,89-6,99 (m, 2 H), 5,06 (s, 2 H), 4,64 (s, 2 H); LRMS berechnet für C18H10F4N2O2S·H2O: C, 50,23; H, 3,28; N, 6,51; S, 7,45. Gefunden C, 50,70; H, 2,52; N, 6,60; S, 7,57. 394,0; Gefunden 395,0 (M + 1)+. Anal. berechnet für C18H10F4N2O2S.
- Beispiel 20
- Herstellung von 7-Brom-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 7-Brom-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 7-Bromindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 228-230°C; Rf 0,40 (10 % Methanol in Chloroform); 1H NMR (DMSO-d6 300 MHz) δ 7,65-7,74 (m, 1 H), 7,57 (d; J = 7,8 Hz, 1 H), 7,49 (s, 1 H), 7,32 (d, J = 7,8 Hz, 1 H), 6,94 (t, J = 7,8 Hz, 1 H), 5,29 (s, 2 H), 4,55 (s, 2 H); LRMS berechnet für C18H10F3N2O2SBr: 454,0 für (79Br und 456,0 für 81Br): Gefunden 453,0 (M – 1)– und 455,0 (M – 1)–. Anal. berechnet für C18H10F3N2O2SBr: C, 47,49; H, 2,21; N, 6,15; S, 7,04, Gefunden: C, 47,65; H, 2,27; N, 6,15; S, 6,98.
- Beispiel 21
- Herstellung von 7-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- 7-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure wurde analog zu Beispiel 2 hergestellt mit dem Unterschied, dass in Teil 1 7-Chlorindol anstelle von 5-Chlorindol verwendet wurde: Schmelzpunkt 228-230°C; Rf 0,38 (10 % Methanol in Chloroform); 1H NMR (DMSO-d6 300 MHz) δ 7,62-7,73 (m, 1 H), 7,52 (d, J = 7,5 Hz, 1 H), 7,49 (s, 1 H), 7,15 (d, J = 7,5 Hz, 1 H), 7,00 (t, J = 7,5 Hz, 1 H), 5,25 (s, 2 H), 4,65 (s, 2 H); LRMS berechnet für C18H10F3N2O2SCl: 410,0. Gefunden 409,0 (M – 1)–. Anal. berechnet für C18H10F3N2O2SCl: C, 52,63; H, 2,45; N, 6,82; S, 7,81. Gefunden: C, 52,60; H, 2,54; N, 6,66; S, 7,59.
- Beispiel 22 Herstellung von 3-[5-Fluorbenzothiazol-2-yl]methylindol-N-Essigsäure
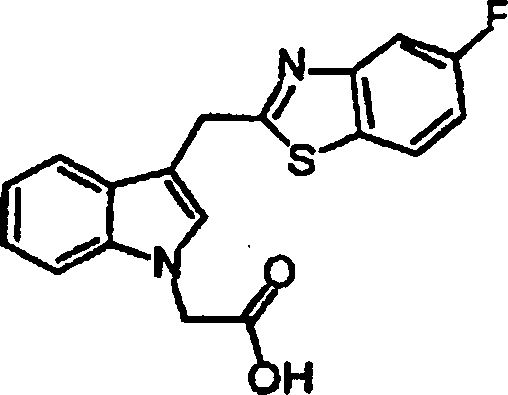
- 3-[5-fluorbenzothiazol-2-yl]methyl-indol-N-Essigsäure wurde analog zu Beispiel 3 hergestellt mit dem Unterschied, dass in Teil 6 2-Amino-4-fluorthiophenolhydrochlorid anstelle von 2-Amino-4,5,7-trifluorthiophenolhydrochlorid verwendet wurde: Schmelzpunkt 208°C (Zersetzung); Rf 0,10 (10 % Methanol in Dichlormethan) 1H NMR (DMSO-d6 300 MHz) δ 12,91 (s, 1 H), 7,98 (dd, J = 8,9, 5,6 Hz: 1 H), 7,78 (dd, J = 10,0, 2,6 Hz, 1 H), 7,50 (d, J = 7,8 Hz, 1 H), 7,40 (s, 1 H), 7,37 (d, J = 7,8 Hz, 1 H), 7,26 (dt, J = 8,9, 2,4 Hz, 1 H), 7,13 (t, J = 7,8 Hz, 1 H), 7,01 (t, J = 7,8 Hz, 1 H), 5,01 (s, 2 H), 4,56 (s, 2 H). LRMS m/z 341,0 (M + 1)+, 339,0 (M – 1). Anal. berechnet für C18H13FN2OS: C, 63,52; H, 3,85; N, 8,23; S, 9,42. Gefunden: C, 63,40; H, 3,80; N, 8,37; S, 9,43.
- Beispiel 23
- 3-[6-Fluorbenzothioazol-2-yl]methylindol-N-Essigsäure
- 3-[6-Fluorbenzothiazol-2-yl]methylindol-N-Essigsäure wurde analog zu Beispiel 3 hergestellt mit dem Unterschied, dass in Teil 6 2-Amino-5-fluorthiophenolhydrochlorid anstelle von 2-Amino-4,5,7-trifluorthiophenolhydrochlorid verwendet wurde. Schmelzpunkt 203°C (Zersetzung), Rf 0,13 (10 % Methanol in Dichlormethan): 1H NMR (DMSO-d6 300 MHz) δ 12,91 (s, 1 H), 7,95 (dd, J = 8,9, 5,0 Hz, 1 H), 7,86 (dd, J = 8,8, 2,8 Hz, 1 H), 7,50 (d, J = 7,5 Hz, 1 H), 7,40-7,35 (m, 2 H), 7,32 (dt, J = 8,9, 2,7 Hz, 1 H), 7,13 (t, J = 7,6 Hz, 1 H), 7,00 (t, J = 7,6 Hz, 1 H), 5,01 (s, 2 H), 4,54 (s, 2 H); LRMS m/z 341,0 (M + 1)+, 339,0 (M–1). Anal. berechnet für C18H13FN2O2S: C, 63,52; H, 3,85; N, 8,23; S, 9,42. Gefunden: C, 63,52; H, 3,86; N, 8,35; S, 9,53.
- Die Verbindungen der Beispiele 24 bis 31 wurden im Wesentlichen nach den in den vorstehenden Beispielen 1 und/oder 2 beschriebenen Verfahren mit entsprechender Substitution der Ausgangsmaterialien hergestellt.
- Beispiel 24 3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-2-propionsäure
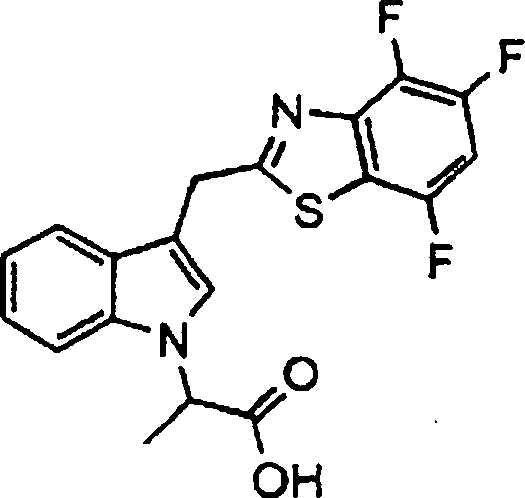
- Schmelzpunkt 176-177°C; Rf 0,34 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,60-7,73 (m, 1 H), 7,60 (s, 1 H), 7,52 (d, J = 8,1 Hz, 1 H), 7,44 (d, J = 8,1 Hz, 1 H), t, J = 7,5 Hz, 1 H), 7,02 (t, J = 7,5 Hz, 1 H), 5,35 (q, J = 8,1 Hz, 1 H), 4,64 (s, 2 H), 1,72 (d, J = 8,1 Hz, 3 H); LRMS berechnet für C19H13F3N2O2S: 390,0. Gefunden 391,0 (M + 1)+. Anal. berechnet für C19H13F3N2O2SH2O : C, 55,88; H, 3,70; N, 6,86; S, 7,85. Gefunden: C, 56,09; H, 3,31; N, 6,89; S, 7,99.
- Beispiel 25 3-(4,5,7-Trifluorbenzothiazol-2-yl)methylindol-N-3-propionsäure
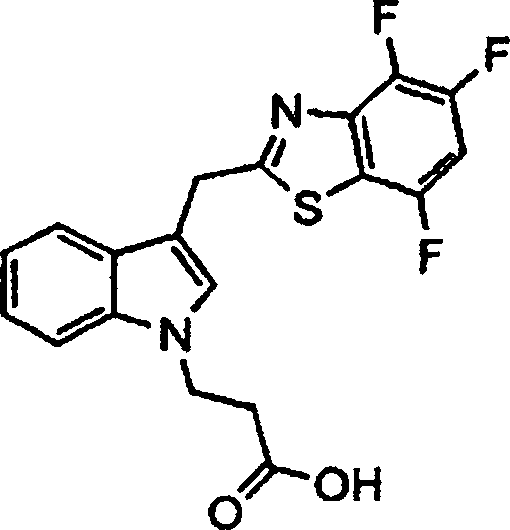
- Schmelzpunkt 200-201°C; Rf 0,50 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 M Hz) δ 7,63-7,71 (m, 1 H), 7,51 (s, 1 H), 7,47 (d, J = 3,0 Hz, 2 H), 7,14 (t, J = 7,5 Hz, 1 H), 7,00 (t, J = 7,5 Hz, 1 H), 4,61 (s, 2 H), 4,39 (t, J = 6,6 Hz, 2 H), 2,75 (t, J = 6,6 Hz, 2 H);. LRMS berechnet für C19H13F3N2O2S 390,0; Gefunden 391,0 (M + 1)+. Anal. berechnet für C19H13F3N2O2S: 58,46; H, 3,36; N, 7,18; S, 8,21. Gefunden: C, 58,63; H, 3,40; N, 7,20; S, 8,30.
- Beispiel 26
- Herstellung von 6-Brom-3-(5-trifluormethylbenzothiazol-2y-l)methylindol-N-Essigsäure
- Schmelzpunkt 265-267°C; Rf 0,19 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 8,28 (s, 1 H), 8,22 (d, J = 8,7 Hz, 1 H), 7,67-7,69 (m, 2 H), 7,43-7,47 (m, 2 H), 7,14 (d, J = 9,0 Hz, 1 H), 5,04 (s, 2 H), 4,61 (s, 2 H); LRMS berechnet für C19H12F3N2O2SBr: 469,0. Gefunden 469,0 (M + 1)+ für Br = 79. Anal. berechnet für C19H12F3N2O2SBr: C, 48,63; H, 2,58; N, 5,97; S, 6,83. Gefunden: C, 48,60; H, 2,63; N, 5,88; S, 6,91.
- Beispiel 27
- 6-Methoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- Schmelzpunkt 118-120°C; Rf 0,27 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,63-7,73 (m, 1 H), 7,39 (s, 1 H), 7,28 (d, J = 8,7 Hz, 1 H), 7,07 (s, 1 H), 6,78 (d, J = 8,7 Hz, 1 H), 4,97 (s, 2 H), 4,61 (s, 2 H); 3,07 (s, 3 H). LRMS berechnet C19H13F3N2O3S: 406,0. Gefunden 407,0 (M +)+. Anal. berechnet für C19H13F3N2O3SH2O:C, 53,77; H, 3,56; N, 6,60; S, 7,56. Gefunden: C, 53,87; H, 3,56; N, 6,67; S, 7,67.
- Beispiel 28 4-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure

- Schmelzpunkt 203-206°C; Rf 0,24 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,63-7,71 (m, 1 H), 7,57 (s, 1 H), 7,33 (d, J = 9,0 Hz, 1 H), 7,12 (dd, J(1) = 9,0, J(2)= 7,8 Hz, 1 H), 7,03 (d, J = 7,8 Hz, 1 H), 5,08 (s, 2 H), 4,78 (s, 2 H). LRMS berechnet für C18H10F3N2O2SCl: 410,0. Gefunden 411,0 (M + 1)+ und 409,0 (M – 1)–.
- Beispiel 29 5-Methoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure

- Schmelzpunkt 165-167°C; Rf 0,37 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,61-7,70 (m, 1 H), 7,35 (d, J = 9,0 Hz, 1 H), 7,26 (s, 1 H), 6,90 (s, 1 H), 6,64 (d, J = 9,0 Hz, 1 H), 4,79 (s, 2 H), 4,56 (s, 2 H), 3,72 (s, 3 H); LRMS berechnet für C10H13F3N2O2S: 406,0. Gefunden 407,0 (M + 1)+ und 405,0 (M – 1)–.
- Beispiel 30
- 5-Brom-3-(4,5,7-trifluorbenzothiazol-2-yl)methylindol-N-Essigsäure
- Schmelzpunkt 209-294°C; Rf 0,18 (20 % Methanol in Dichlormethan); 1H NMR (DMSO-d6 300 MHz) δ 7,78 (d, J = 1,8 Hz, 1 H), 7,65-7,73 (m, 1 H), 7,49 (s, 1 H), 7,61 (d, J = 9,0 Hz, 1 H), 7,25 (dd, J(1) = 9, 0 Hz, J(2) = 1,8 Hz, 1 H), 5,04 (s, 2 H); 4,64 (s, 2 H). LRMS berechnet für C18H10F3N2O2SBr: 455,0. Gefunden 455,0 (M + 1)+ für Br 79 und 457 (M + 1)+ für Br 81.
- Beispiel 31 3-(6-Chlorbenzothiazol-2-yl)methylindol-N-Essigsäure

- Repräsentative erfindungsgemäße Verbindungen wurden auf ihre Stärke, Selektivität und Wirksamkeit als Hemmer der Aldosereduktase beim Menschen untersucht. Der Grad der Hemmwirkung der Verbindungen auf die Aldosereduktase wurde mit ähnlichen Methoden getestet wie von Butera et al. in J. Med. Chem. 1989, 32, 757 beschrieben. Mit diesem Versuch wurden die Konzentrationen bestimmt, die zur Verringerung der Aktivität humaner Aldosereduktase (hALR2) um 50 % (IC50) erforderlich waren.
- In einem zweiten Versuch wurden die gleichen Verbindungen auf ihre Fähigkeit getestet, die Aldehydreduktase (hALR1), ein von der Struktur her verwandtes Enzym, zu hemmen. Die verwendeten Testverfahren waren im Wesentlichen die gleichen wie von Ishii et al., J. Med. Chem. 1996 39: 1924 beschrieben. Mit Hilfe dieses Versuchs wurden die Konzentrationen bestimmt, die zur Verringerung der Aldehydreduktaseaktivität beim Menschen um 50 % (IC50) erforderlich waren.
- Aus diesen Daten wurden die hALRI/hALR2-Verhältnisse ermittelt. Da erwünscht ist, dass die Testverbindungen die Aldosereduktase stark hemmen, sind niedrige hALR2 IC50-Werte erstrebenswert. Andererseits ist eine starke Wirkung der Testverbindungen als Hemmer der Aldehydreduktase unerwünscht, und hohe hALR1 IC50-Werte werden angestrebt. Folglich wird das Verhältnis hALR1/hALR2 dazu verwendet, die Selektivität der Testverbindungen zu bestimmen. Die Bedeutung dieser Selektivität haben Kotani et al., in J. Med. Chem. 40: 684, 1997 beschrieben.
- Die Ergebnisse sämtlicher Tests sind in Tabelle 1 zusammengefasst und veranschaulicht.
-
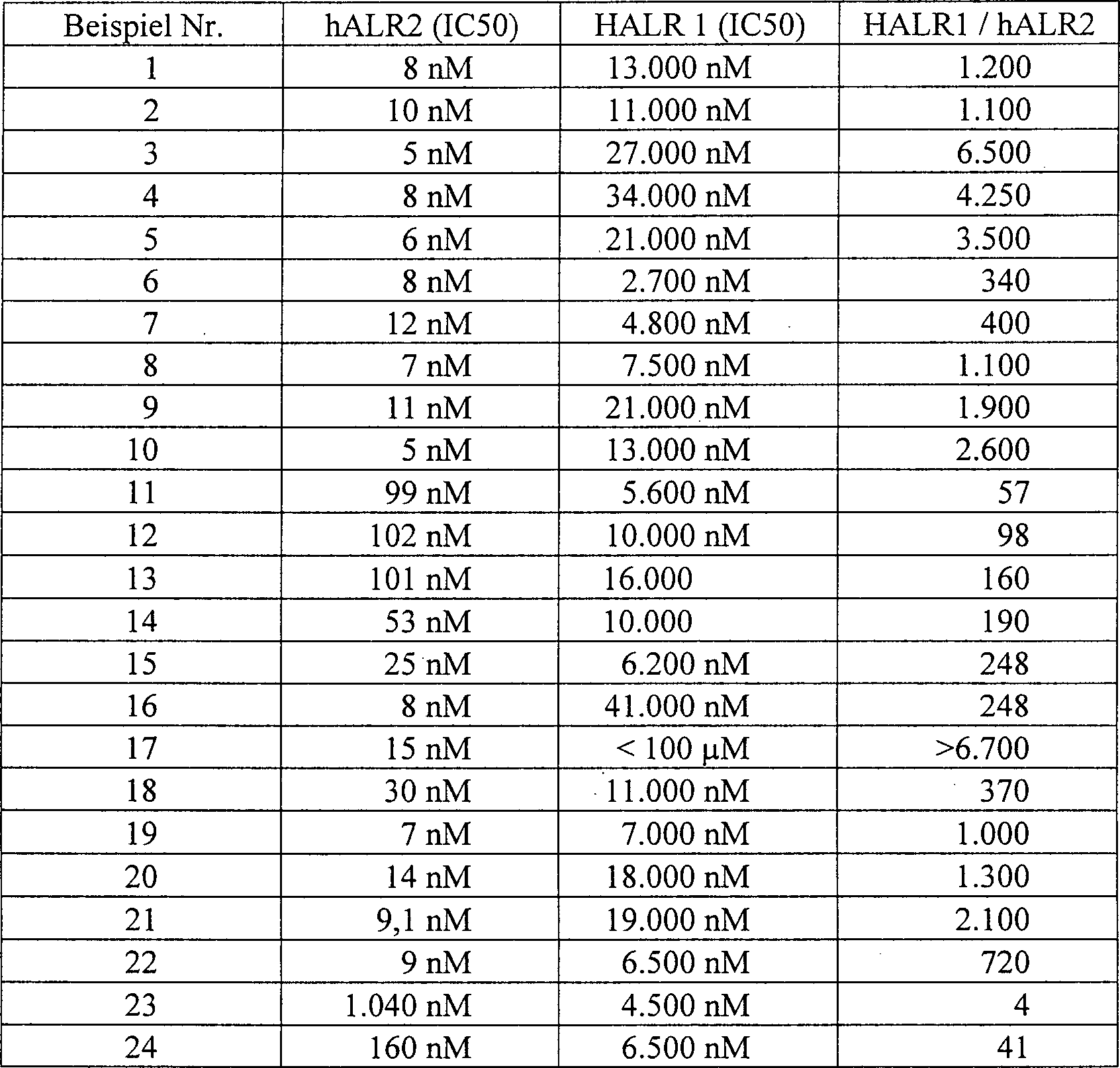
-
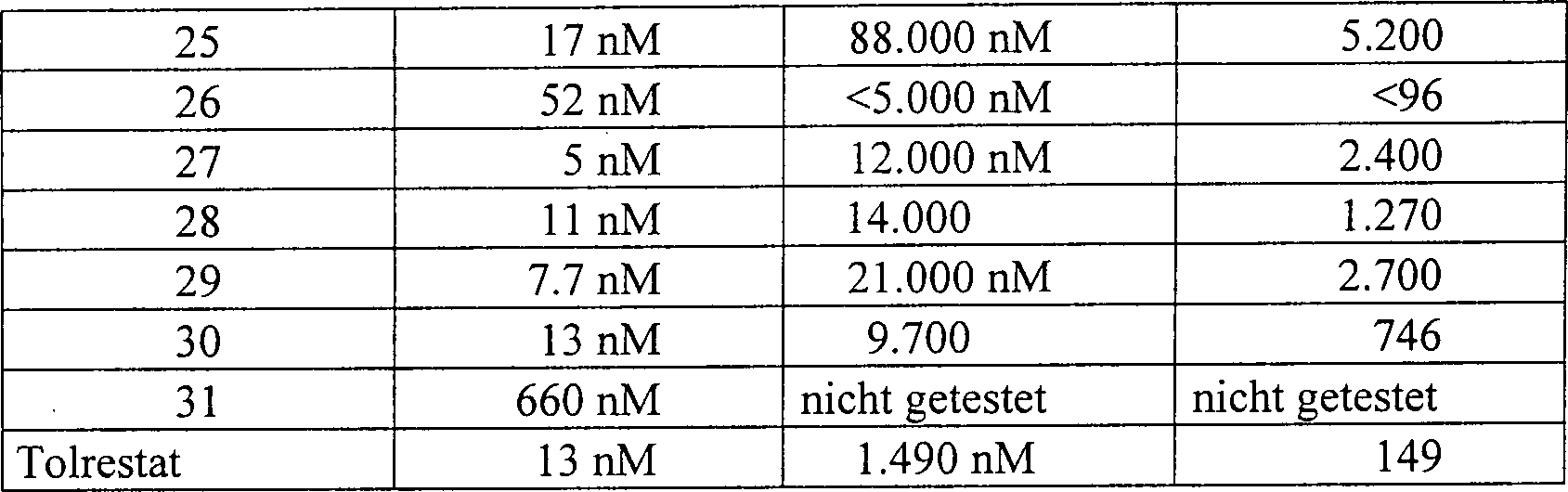
- Diese Ergebnisse zeigen die überlegene Stärke, Selektivität und Wirksamkeit repräsentativer erfindungsgemäßer Verbindungen. Solche Verbindungen eignen sich für die Behandlung chronischer Komplikationen, die eine Folgeerscheinung von Diabetes mellitus sind, z.B. diabetesbedingter Grauer Star, Retinopathie und Neuropathie. Folglich ist ein Aspekt der Erfindung die Behandlung solcher Komplikationen mit den erfindungsgemäßen Verbindungen, wobei Behandlung sowohl Prophylaxe als auch Linderung einschließt. Die Verbindungen eignen sich z.B. für die Behandlung von diabetesbedingtem Grauen Star, der Retinopathie, Nephropathie und Neuropathie.
- In einer dritten bedarfsweise durchgeführten Versuchsreihe können die Verbindungen auf ihre Fähigkeit untersucht werden, Sorbitansammlungen im Ischiasnerv streptozotocin-induzierter diabetischer Ratten zu normalisieren. Die zur Bestimmung der Wirksamkeit verwendeten Testverfahren sind im Wesentlichen die von Mylari et al., J. Med. Chem. 34: 108, 1991.
- Die Erfindung sowie die Art und das Verfahren ihrer Herstellung und Verwendung wurden nun so vollständig, klar, präzise und exakt beschrieben, dass es einem Fachmann auf dem Gebiet, zu dem sie gehört, ermöglicht wird, diese herzustellen und zu verwenden. Es braucht nicht eigens erwähnt zu werden, dass die vorstehenden Erläuterungen bevorzugte Ausführungsformen der Erfindung beschreiben und dass Änderungen vorgenommen werden können, ohne den Rahmen der Erfindung zu verlassen. Um den Gegenstand, der als Erfindung erachtet wird, speziell darzulegen und klar zu beanspruchen, schließen die nachfolgenden Ansprüche diese Patentschrift ab.
Claims (12)
- Verbindung der Formel:worin A eine C1-C4-Alkylengruppe darstellt, die wahlweise mit C1-C2-Alkyl substituiert oder mit Halogen mono- oder disubstituiert ist; Z eine Bindung, O, S, C(O)NH oder C1-C3-Alkylen, wahlweise substituiert mit C1-C2-Alkyl, ist; R1 Wasserstoff Alkyl mit 1-6 Kohlenstoffatomen oder Phenyl darstellt, worin das Phenyl wahlweise mit bis zu drei Gruppen substituiert ist, ausgewählt unter Halogen, Hydroxy, C1-C6-Alkoxy, C1-C6-Alkyl, Nitro, Amino oder Mono- oder Di(C1-C6)-alkylamino; R2, R3, R4 und R5 jeweils unabhängig voneinander sind: – Wasserstoff, Halogen, Nitro oder eine Alkylgruppe mit 1-6 Kohlenstoffatomen, wobei die Alkylgruppe wahlweise mit einem oder mehrerern Halogenen substituiert ist; – OR7, worin jedes R7 unabhängig Wasserstoff eine Alkylgruppe mit 1 bis 6 Kohlenstoffatomen, wobei die Alkylgruppe wahlweise mit einem oder mehreren Halogenen substituiert ist, oder Benzyl darstellt; – Phenyl, worin der Phenylanteil wahlweise substituiert ist mit bis zu drei Gruppen, unabhängig ausgewählt unter Halogen, C1-C6-Alkyl oder C1-C6-Alkoxy; – Phenoxy, worin der Phenylanteil wahlweise substituiert ist mit bis zu drei Gruppen, unabhängig voneinander ausgewählt unter Halogen, C1-C6-Alkyl oder C1-C6-Alkoxy; oder – eine Gruppe der Formel:
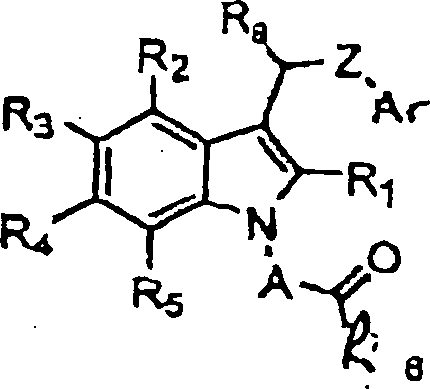 worin J eine Bindung, CH2, Sauerstoff oder Stickstoff ist, und jedes r unabhängig 2 oder 3 ist; R6 Hydroxy oder O–M+ darstellt, worin M+ ein Kation darstellt, ausgewählt unter Natrium, Kalium, Ammonium, Magnesium und Calcium; Ra Wasserstoff, C1-C6-Alkyl, Fluor oder Trifluormethyl darstellt; und Ar darstellt einen Benzothiazolring, wahlweise substituiert auf dem Benzoanteil durch eines von Jod, Cyano, Nitro, Perfluorethyl, Trifluoracetyl oder (C1-C6)Alkanoyl, durch eines oder zwei von Fluor, Chlor, Brom, Hydroxy, (C1-C6)Alkyl, (C1-C6)Alkoxy, (C1-C6)Alkylthio, Trifluormethoxy, Trifluormethylthio, (C1-C6)Alkylsulfinyl, (C1-C6)Alkylsulfonyl oder Trifluormethyl, oder zwei Fluor oder zwei Trifluormethyl mit einem Hydroxy oder einem (C1-C6)Allkoxy oder einem oder zwei Fluor und einem Trifluormethyl oder drei Fluor.
worin J eine Bindung, CH2, Sauerstoff oder Stickstoff ist, und jedes r unabhängig 2 oder 3 ist; R6 Hydroxy oder O–M+ darstellt, worin M+ ein Kation darstellt, ausgewählt unter Natrium, Kalium, Ammonium, Magnesium und Calcium; Ra Wasserstoff, C1-C6-Alkyl, Fluor oder Trifluormethyl darstellt; und Ar darstellt einen Benzothiazolring, wahlweise substituiert auf dem Benzoanteil durch eines von Jod, Cyano, Nitro, Perfluorethyl, Trifluoracetyl oder (C1-C6)Alkanoyl, durch eines oder zwei von Fluor, Chlor, Brom, Hydroxy, (C1-C6)Alkyl, (C1-C6)Alkoxy, (C1-C6)Alkylthio, Trifluormethoxy, Trifluormethylthio, (C1-C6)Alkylsulfinyl, (C1-C6)Alkylsulfonyl oder Trifluormethyl, oder zwei Fluor oder zwei Trifluormethyl mit einem Hydroxy oder einem (C1-C6)Allkoxy oder einem oder zwei Fluor und einem Trifluormethyl oder drei Fluor.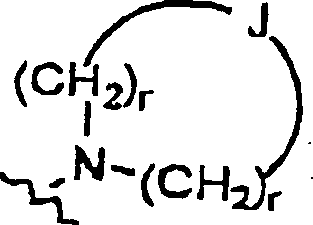
- Verbindung gemäß Anspruch 1, worin Ar ein Benzothiazol der Formel III ist:worin R11, R12, R13 und R14 unabhängig voneinander sind Wasserstoff, Fluor, Chlor, Brom, Trifluormethyl oder Nitro.
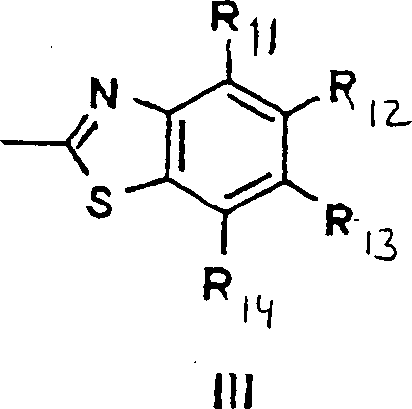
- Verbindung gemäß Anspruch 2, worin A Methylen und Z eine Bindung ist.
- Verbindung gemäß Anspruch 2, worin Ra Wasserstoff und Z eine Bindung ist.
- Verbindung gemäß Anspruch 3, worin A Methylen, Ra Wasserstoff und Z eine Bindung ist.
- Verbindung gemäß Anspruch 5, worin mindestens einer von R11, R12, R13 und R14 Trifluormethyl darstellt.
- Verbindung gemäß Anspruch 6, worin R12 Trifluormethyl ist.
- Verbindung gemäß Anspruch 5, worin R11, R12 und R14 Fluoratome und R13 Wasserstoff ist bzw. sind.
- Verbindung gemäß Anspruch 8, worin R6 Wasserstoff ist.
- Verbindung gemäß Anspruch 1, die ist: 3-(4,5,7- Trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäureethylester; 3-(4,5,7- Trifluorbenzothiazol-2yl)methylindol-N-essigsäure; 5-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 2-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Methyl-3(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 7-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 6-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Benzyloxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 6-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 6-Methyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 3-(5-Trifluormethylbenzothiazol-2-yl)methylindol-N-essigsäure; 5-Methyl-3-(5-trifluormethylbenzothiazol-2-yl)methyl-indol- N-essigsäure; 2-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 6-Phenyl-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 6-Morpholino-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Phenoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 7-Fluor-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 7-Brom-3(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 7-Chlor-3(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 3-[[5-Fluorbenzothiazole-2-yl]methyl]-indol-N-essigsäure; 3-[[6-Fluorbenzothiazole-2-yl]methyl]-indol-N-essigsäure; 3-(4,5,7-Trifluorbenzothiazol-2-yl)methyl-indol-N-2-propionsäure; 3-(4,5,7-Trifluorbenzothiazol-2-yl)methyl-indol-N-3-propionsäure; 6-Brom-3-(5-trifluormethylbenzothiazol-2-yl)methyl-indol-N-essigsäure; 6-Methoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 4-Chlor-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Methoxy-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; 5-Brom-3-(4,5,7-trifluorbenzothiazol-2-yl)methyl-indol-N-essigsäure; oder 3-(6-Chlorbenzothiazol-2-yl)methyl-indol-N-essigsäure.
- Pharmazeutische Zubereitung, umfassend eine wirksame Menge einer Verbindung nach einem der Ansprüche 1 bis 10.
- Verbindung gemäß Anspruch 3, worin Ar ein substituiertes Benzothiazol der Formel III ist, R12 Trifluormethyl ist, A Methylen, mit einer Methylgruppe substituiertes Methylen oder Ethylen ist und R2, R3, R4 und R5 in Kombination darstellen eines von Brom, Cyano oder Nitro, eines oder zwei von Fluor, Chlor, Hydroxy, (C1-C6)Alkyl, (C1-C6)Alkoxy oder Trifluormethyl oder zwei Fluor oder zwei Methyl mit einem Hydroxy oder einem (C1-C6)Alkoxy oder ein oder zwei Fluor und ein Methyl oder drei Fluorgruppen.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8014398P | 1998-03-31 | 1998-03-31 | |
| US80143P | 1998-03-31 | ||
| PCT/US1999/007116 WO1999050268A2 (en) | 1998-03-31 | 1999-03-31 | Substituted indolealkanoic acids |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69918278D1 DE69918278D1 (de) | 2004-07-29 |
| DE69918278T2 true DE69918278T2 (de) | 2005-07-14 |
Family
ID=22155536
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69918278T Expired - Lifetime DE69918278T2 (de) | 1998-03-31 | 1999-03-31 | Substituierte indolalkansäure |
Country Status (29)
| Country | Link |
|---|---|
| US (9) | US6214991B1 (de) |
| EP (1) | EP1066283B1 (de) |
| JP (2) | JP3494990B2 (de) |
| KR (1) | KR100584650B1 (de) |
| CN (1) | CN1205207C (de) |
| AP (1) | AP2000001929A0 (de) |
| AT (1) | ATE269861T1 (de) |
| AU (1) | AU774929B2 (de) |
| BG (1) | BG65446B1 (de) |
| BR (1) | BR9909358A (de) |
| CA (1) | CA2383983C (de) |
| CZ (1) | CZ300706B6 (de) |
| DE (1) | DE69918278T2 (de) |
| EE (1) | EE200000573A (de) |
| ES (1) | ES2224632T3 (de) |
| HU (1) | HUP0101672A3 (de) |
| ID (1) | ID27884A (de) |
| IL (2) | IL138711A0 (de) |
| MX (1) | MXPA02003122A (de) |
| NO (1) | NO20004900L (de) |
| NZ (1) | NZ507172A (de) |
| OA (1) | OA11622A (de) |
| PL (1) | PL201645B1 (de) |
| SK (1) | SK7522001A3 (de) |
| TR (1) | TR200002869T2 (de) |
| TW (1) | TWI243163B (de) |
| WO (1) | WO1999050268A2 (de) |
| YU (1) | YU59600A (de) |
| ZA (1) | ZA200005577B (de) |
Families Citing this family (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6214991B1 (en) * | 1998-03-31 | 2001-04-10 | The Institute For Pharmaceutical Discovery, Inc. | Substituted indolealkanoic acids |
| TNSN99224A1 (fr) * | 1998-12-01 | 2005-11-10 | Inst For Pharm Discovery Inc | Methodes de reduction des niveaux de glucose et triglyceride en serum et pour suppression de l'antigenese utilisant les acides la indolealkanoique |
| WO2001005758A2 (en) * | 1999-07-15 | 2001-01-25 | Nps Allelix Corp. | Indoles and indazoles for the treatment of migraine |
| AU2001238718A1 (en) * | 2000-03-02 | 2001-09-12 | The Institutes For Pharmaceutical Discovery, Llc | Compositions containing a substituted indolealkanoic acid and an angiotensin converting enzyme inhibitor |
| US20020012630A1 (en) * | 2000-05-09 | 2002-01-31 | James Nolan | Methods for testing compounds useful in treating diabetic complications |
| EP1347959A1 (de) | 2000-12-27 | 2003-10-01 | Bayer Aktiengesellschaft | Indol-derivate |
| US7074817B2 (en) | 2001-06-20 | 2006-07-11 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| TWI224101B (en) | 2001-06-20 | 2004-11-21 | Wyeth Corp | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| DE60312474T2 (de) | 2002-01-18 | 2007-12-06 | Aryx Therapeutics, Fremont | 5-ht3-rezeptorantagonisten und verwendungsverfahren |
| CA2493755A1 (en) * | 2002-07-26 | 2004-02-05 | The Institutes For Pharmaceutical Discovery, Llc | Substituted indolealkanoic acids derivative and formulations containing same for use in treatment of diabetic complications |
| CA2395871A1 (en) * | 2002-07-26 | 2004-01-26 | The Institutes For Pharmaceutical Discovery, Llc | Substituted indolealkanoic acids derivatives and formulations containing same for use in treatment of diabetic complications |
| CN100457730C (zh) * | 2002-08-29 | 2009-02-04 | 默克公司 | 具有抗糖尿病活性的吲哚化合物 |
| EP1569900B1 (de) | 2002-12-10 | 2006-06-28 | Wyeth | Substituierte 3-carbonyl-1-yl essigsäure derivate als plasminogen aktivator inhibitor(pai-1) inhibitoren |
| UA80453C2 (en) | 2002-12-10 | 2007-09-25 | Derivatives of substituted dyhydropyranoindol-3,4-dion as inhibitors of plasminogen activator inhibitor-1 (pai-1) | |
| CA2509222A1 (en) | 2002-12-10 | 2004-06-24 | Wyeth | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (pai-1) |
| DE60324183D1 (en) | 2002-12-10 | 2008-11-27 | Wyeth Corp | Aryl-, aryloxy- und alkyloxysubstituierte 1h-indol-3-yl-glyoxylsäurederivateals inhibitoren des plasminogenaktivatorinhibitors-1 (pai-1) |
| CA2509170A1 (en) | 2002-12-10 | 2004-06-24 | Wyeth | Substituted 3-alkyl and 3-arylalkyl 1h-indol-1-yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (pai-1) |
| US7592361B2 (en) | 2003-04-28 | 2009-09-22 | Bayer Pharmaceuticals Corporation | Indole acetic acid derivatives and their use as pharmaceutical agents |
| BRPI0409914A (pt) * | 2003-04-30 | 2006-04-25 | Inst For Pharm Discovery Inc | ácidos carboxìlicos substituìdos |
| JP2007501189A (ja) * | 2003-08-01 | 2007-01-25 | ジェネラブス テクノロジーズ,インコーポレイテッド | フラビウイルス科に対する二環式イミダゾール誘導体 |
| US7163954B2 (en) | 2003-09-25 | 2007-01-16 | Wyeth | Substituted naphthyl benzothiophene acids |
| US7268159B2 (en) | 2003-09-25 | 2007-09-11 | Wyeth | Substituted indoles |
| US7411083B2 (en) | 2003-09-25 | 2008-08-12 | Wyeth | Substituted acetic acid derivatives |
| US7351726B2 (en) | 2003-09-25 | 2008-04-01 | Wyeth | Substituted oxadiazolidinediones |
| US7582773B2 (en) | 2003-09-25 | 2009-09-01 | Wyeth | Substituted phenyl indoles |
| US7446201B2 (en) | 2003-09-25 | 2008-11-04 | Wyeth | Substituted heteroaryl benzofuran acids |
| US7442805B2 (en) | 2003-09-25 | 2008-10-28 | Wyeth | Substituted sulfonamide-indoles |
| US7141592B2 (en) | 2003-09-25 | 2006-11-28 | Wyeth | Substituted oxadiazolidinediones |
| US7332521B2 (en) | 2003-09-25 | 2008-02-19 | Wyeth | Substituted indoles |
| US7265148B2 (en) | 2003-09-25 | 2007-09-04 | Wyeth | Substituted pyrrole-indoles |
| GB0324763D0 (en) | 2003-10-23 | 2003-11-26 | Oxagen Ltd | Use of compounds in therapy |
| EP1805136A1 (de) * | 2004-10-28 | 2007-07-11 | The Institutes for Pharmaceutical Discovery, LLC | Substituierte phenylalkansäuren |
| CN101103026A (zh) * | 2005-01-14 | 2008-01-09 | 健亚生物科技公司 | 用于治疗病毒感染的吲哚衍生物 |
| GB0505048D0 (en) * | 2005-03-11 | 2005-04-20 | Oxagen Ltd | Compounds with PGD antagonist activity |
| US20080051384A1 (en) * | 2006-07-14 | 2008-02-28 | Genelabs Technologies, Inc. | Antiviral agents |
| CA2658496A1 (en) | 2006-07-22 | 2008-01-31 | Oxagen Limited | 2-{5-fluoro-2-methyl-3-[2-(phenylsulfonyl)benzyl]-1h-indol-yl} acetic acid derivatives and esters thereof having crth2 antagonist activity |
| US7750027B2 (en) * | 2008-01-18 | 2010-07-06 | Oxagen Limited | Compounds having CRTH2 antagonist activity |
| CA2712017C (en) | 2008-01-18 | 2016-08-09 | Oxagen Limited | Compounds having crth2 antagonist activity |
| EP2265581A1 (de) * | 2008-01-22 | 2010-12-29 | Oxagen Limited | Verbindungen mit crth2-antagonistischer wirkung |
| JP2011509991A (ja) | 2008-01-22 | 2011-03-31 | オキサジェン リミテッド | Crth2アンタゴニスト活性を有する化合物 |
| GB2457040A (en) * | 2008-01-30 | 2009-08-05 | Oxagen Ltd | 1-Acetic acid indole derivatives with PGD2 activity |
| SG175390A1 (en) | 2009-04-29 | 2011-12-29 | Amarin Corp Plc | Pharmaceutical compositions comprising epa and a cardiovascular agent and methods of using the same |
| HRP20221430T1 (hr) * | 2010-07-16 | 2023-01-06 | The Trustees Of Columbia University In The City Of New York | Inhibitori aldozoreduktaze i njihova uporaba |
| US8916563B2 (en) | 2010-07-16 | 2014-12-23 | The Trustees Of Columbia University In The City Of New York | Aldose reductase inhibitors and uses thereof |
| GB201322273D0 (en) | 2013-12-17 | 2014-01-29 | Atopix Therapeutics Ltd | Process |
| GB201407807D0 (en) | 2014-05-02 | 2014-06-18 | Atopix Therapeutics Ltd | Polymorphic form |
| GB201407820D0 (en) | 2014-05-02 | 2014-06-18 | Atopix Therapeutics Ltd | Polymorphic form |
| RS61239B1 (sr) | 2016-06-21 | 2021-01-29 | Univ Columbia | Inhibitori aldozne reduktaze i postupci korišćenja |
| IL272246B2 (en) | 2017-07-28 | 2026-01-01 | Applied Therapeutics Inc | History of 2-(4-oxo/thioketone/azo-3-((substituted)benzo[d]thiazol-2-yl)methyl)- 3,4-dihydrothieno[3,4-d]pyridazin-1-yl)acetic acid for use as an aldose reductase inhibitor for the treatment of galactosemia or prevention of galactosemia-related complications |
| US11530186B2 (en) | 2018-03-29 | 2022-12-20 | H. Lee Moffitt Cancer Center and Research Center Institute, Inc. | Inhibitors for the β-catenin / T-cell factor protein-protein interaction |
| CA3132136A1 (en) | 2019-04-01 | 2020-10-08 | Andrew Wasmuth | Inhibitors of aldose reductase |
| JP2022531466A (ja) | 2019-05-07 | 2022-07-06 | ユニバーシティ オブ マイアミ | 遺伝性ニューロパチーおよび関連障害の処置および検出 |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3557142A (en) * | 1968-02-20 | 1971-01-19 | Sterling Drug Inc | 4,5,6,7-tetrahydro-indole-lower-alkanoic acids and esters |
| IE47592B1 (en) | 1977-12-29 | 1984-05-02 | Ici Ltd | Enzyme inhibitory phthalazin-4-ylacetic acid derivatives, pharmaceutical compositions thereof,and process for their manufacture |
| DK151884C (da) * | 1979-03-07 | 1988-06-13 | Pfizer | Analogifremgangsmaade til fremstilling af 3-(1-imidazolylalkyl)indolderivater eller farmaceutisk acceptable syreadditionssalte deraf |
| JPS55167282A (en) | 1979-06-12 | 1980-12-26 | Fujisawa Pharmaceut Co Ltd | Piperazine derivative or its salt and its preparation |
| US4283539A (en) | 1979-12-18 | 1981-08-11 | Pfizer Inc. | Isoquinoline acetic acids |
| US4363912A (en) * | 1980-12-15 | 1982-12-14 | Pfizer Inc. | Indole thromboxane synthetase inhibitors |
| US4442118A (en) * | 1981-07-23 | 1984-04-10 | Ayerst, Mckenna & Harrison, Inc. | Aldose reductase inhibition by 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indole-3-acetic acid |
| GB8607294D0 (en) | 1985-04-17 | 1986-04-30 | Ici America Inc | Heterocyclic amide derivatives |
| DE3684410D1 (de) * | 1985-11-07 | 1992-04-23 | Pfizer | Heterocyclische oxophthalazinylessigsaeure. |
| US4939140A (en) | 1985-11-07 | 1990-07-03 | Pfizer Inc. | Heterocyclic oxophthalazinyl acetic acids |
| US4960785A (en) * | 1988-12-16 | 1990-10-02 | Pfizer Inc. | Indolinone derivatives |
| WO1993012786A1 (en) * | 1986-07-10 | 1993-07-08 | Howard Harry R Jr | Indolinone derivatives |
| US4868301A (en) * | 1987-06-09 | 1989-09-19 | Pfizer Inc. | Processes and intermediates for the preparation of oxophthalazinyl acetic acids having benzothiazole or other heterocyclic side chains |
| US5064852A (en) * | 1988-12-16 | 1991-11-12 | Pfizer Inc. | Indolinone derivatives |
| EP0427860A1 (de) | 1989-03-31 | 1991-05-22 | Kyoto Pharmaceutical Industries, Ltd. | Imidazol-derivate, verfahren zur herstellung und verwendung als arzneimittel |
| US4996204A (en) * | 1989-05-11 | 1991-02-26 | Pfizer Inc. | Pyrido[2,3-d]pyridazinones as aldose reductase inhibitors |
| FR2647676A1 (fr) * | 1989-06-05 | 1990-12-07 | Union Pharma Scient Appl | Nouveaux derives de pyridazinone, leurs procedes de preparation, medicaments les contenant, utiles notamment comme inhibiteurs de l'aldose reductase |
| GB8916774D0 (en) * | 1989-07-21 | 1989-09-06 | Bayer Ag | New indole derivatives,a process for their preparation and their use in medicaments |
| FI922308A7 (fi) | 1989-12-15 | 1992-05-21 | Pfizer | Substituoituja oksoftalatsinyylietikkahappoja ja niiden anologeja |
| US5312829A (en) * | 1990-05-21 | 1994-05-17 | Fujisawa Pharmaceutical Co., Ltd. | Indole derivatives |
| GB9011335D0 (en) | 1990-05-21 | 1990-07-11 | Fujisawa Pharmaceutical Co | Indolebutyric acid derivatives and process for preparation thereof |
| US5236945A (en) * | 1990-06-11 | 1993-08-17 | Pfizer Inc. | 1H-indazole-3-acetic acids as aldose reductase inhibitors |
| US5116753A (en) * | 1991-07-30 | 1992-05-26 | The Salk Institute For Biological Studies | Maintenance of pancreatic islets |
| JPH0641071A (ja) * | 1991-09-06 | 1994-02-15 | Taisho Pharmaceut Co Ltd | インドール酢酸エステル誘導体 |
| GB9122590D0 (en) | 1991-10-24 | 1991-12-04 | Lilly Industries Ltd | Pharmaceutical compounds |
| US5391551A (en) | 1993-05-10 | 1995-02-21 | Pfizer Inc. | Method of lowering blood lipid levels |
| GB9317764D0 (en) * | 1993-08-26 | 1993-10-13 | Pfizer Ltd | Therapeutic compound |
| GB9409583D0 (en) | 1994-05-13 | 1994-07-06 | Pfizer Ltd | Indoles |
| US5641800A (en) * | 1994-07-21 | 1997-06-24 | Eli Lilly And Company | 1H-indole-1-functional sPLA2 inhibitors |
| TW401301B (en) | 1994-10-07 | 2000-08-11 | Takeda Chemical Industries Ltd | Antihypertriglyceridemic composition |
| US5700819A (en) * | 1994-11-29 | 1997-12-23 | Grelan Pharmaceutical Co., Ltd. | 2-substituted benzothiazole derivatives and prophylactic and therapeutic agents for the treatment of diabetic complications |
| IL117208A0 (en) * | 1995-02-23 | 1996-06-18 | Nissan Chemical Ind Ltd | Indole type thiazolidines |
| WO1996029075A1 (en) * | 1995-03-20 | 1996-09-26 | Eli Lilly And Company | 5-substituted-3-(1,2,3,6-tetrahydropyridin-4-yl)- and 3-(piperidin-4-yl)-1h-indoles: new 5-ht1f agonists |
| JPH09165371A (ja) | 1995-10-09 | 1997-06-24 | Sankyo Co Ltd | 複素環化合物を含有する医薬 |
| DE19636150A1 (de) * | 1996-09-06 | 1998-03-12 | Asta Medica Ag | N-substituierte Indol-3-glyoxylamide mit antiasthmatischer, antiallergischer und immunsuppressiver/immunmodulierender Wirkung |
| US6214991B1 (en) * | 1998-03-31 | 2001-04-10 | The Institute For Pharmaceutical Discovery, Inc. | Substituted indolealkanoic acids |
| TNSN99224A1 (fr) * | 1998-12-01 | 2005-11-10 | Inst For Pharm Discovery Inc | Methodes de reduction des niveaux de glucose et triglyceride en serum et pour suppression de l'antigenese utilisant les acides la indolealkanoique |
| AU2001238718A1 (en) * | 2000-03-02 | 2001-09-12 | The Institutes For Pharmaceutical Discovery, Llc | Compositions containing a substituted indolealkanoic acid and an angiotensin converting enzyme inhibitor |
-
1999
- 1999-03-31 US US09/282,280 patent/US6214991B1/en not_active Expired - Fee Related
- 1999-03-31 MX MXPA02003122A patent/MXPA02003122A/es active IP Right Grant
- 1999-03-31 YU YU59600A patent/YU59600A/sh unknown
- 1999-03-31 HU HU0101672A patent/HUP0101672A3/hu unknown
- 1999-03-31 CA CA002383983A patent/CA2383983C/en not_active Expired - Fee Related
- 1999-03-31 BR BR9909358-8A patent/BR9909358A/pt not_active Application Discontinuation
- 1999-03-31 OA OA1200000269A patent/OA11622A/en unknown
- 1999-03-31 CN CNB998047732A patent/CN1205207C/zh not_active Expired - Fee Related
- 1999-03-31 ID IDW20002212A patent/ID27884A/id unknown
- 1999-03-31 EP EP99916239A patent/EP1066283B1/de not_active Expired - Lifetime
- 1999-03-31 IL IL13871199A patent/IL138711A0/xx active IP Right Grant
- 1999-03-31 TR TR2000/02869T patent/TR200002869T2/xx unknown
- 1999-03-31 DE DE69918278T patent/DE69918278T2/de not_active Expired - Lifetime
- 1999-03-31 CZ CZ20003607A patent/CZ300706B6/cs not_active IP Right Cessation
- 1999-03-31 ES ES99916239T patent/ES2224632T3/es not_active Expired - Lifetime
- 1999-03-31 AT AT99916239T patent/ATE269861T1/de not_active IP Right Cessation
- 1999-03-31 SK SK752-2001A patent/SK7522001A3/sk not_active Application Discontinuation
- 1999-03-31 AP APAP/P/2000/001929A patent/AP2000001929A0/en unknown
- 1999-03-31 KR KR1020007010884A patent/KR100584650B1/ko not_active Expired - Fee Related
- 1999-03-31 AU AU34595/99A patent/AU774929B2/en not_active Ceased
- 1999-03-31 JP JP2000541171A patent/JP3494990B2/ja not_active Expired - Fee Related
- 1999-03-31 WO PCT/US1999/007116 patent/WO1999050268A2/en not_active Ceased
- 1999-03-31 EE EEP200000573A patent/EE200000573A/xx unknown
- 1999-06-02 TW TW088109147A patent/TWI243163B/zh not_active IP Right Cessation
-
2000
- 2000-09-26 IL IL138711A patent/IL138711A/en not_active IP Right Cessation
- 2000-09-29 BG BG104819A patent/BG65446B1/bg unknown
- 2000-09-29 NO NO20004900A patent/NO20004900L/no not_active Application Discontinuation
- 2000-10-11 ZA ZA200005577A patent/ZA200005577B/en unknown
-
2001
- 2001-03-27 US US09/818,808 patent/US6426344B2/en not_active Expired - Fee Related
-
2002
- 2002-03-01 PL PL348244A patent/PL201645B1/pl not_active IP Right Cessation
- 2002-03-01 NZ NZ507172A patent/NZ507172A/en unknown
- 2002-06-28 US US10/185,863 patent/US6730794B2/en not_active Expired - Fee Related
- 2002-10-02 JP JP2002290240A patent/JP2003155274A/ja active Pending
-
2004
- 2004-04-27 US US10/832,724 patent/US7105514B2/en not_active Expired - Fee Related
-
2006
- 2006-09-12 US US11/531,151 patent/US7304079B2/en not_active Expired - Fee Related
-
2007
- 2007-12-04 US US11/999,524 patent/US7659269B2/en not_active Expired - Fee Related
-
2010
- 2010-02-08 US US12/701,967 patent/US8163932B2/en not_active Expired - Fee Related
- 2010-08-30 US US12/871,293 patent/US20100324039A1/en not_active Abandoned
-
2012
- 2012-06-27 US US13/534,875 patent/US20120270912A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69918278T2 (de) | Substituierte indolalkansäure | |
| DE69916881T2 (de) | Antihypertriglyceridemische, antihyperglykemische, antiangiogene und wundheilende substituierte indolalkansäure | |
| RS59600B1 (sr) | Stabilizovane kompozicije neproteinskog toksina klostridije | |
| DE69118388T2 (de) | Substituierte bizyklische arylderivate mit selektiver leukotrien b4 antagonistischer wirkung | |
| BG61364B2 (bg) | Заместени бета-дикетони | |
| US20010044437A1 (en) | Methods for reducing uric acid levels |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |