-
Hintergrund der Erfindung
-
1. Gebiet der Erfindung
-
Die
vorliegende Erfindung betrifft ein Verfahren zum Steuern des Ladens
eines aktiven Wirkstoffs in Liposomen. Insbesondere betrifft die
vorliegende Erfindung Verfahren zum Modulieren des Ladens von aktiven Wirkstoffen
in multivesikuläre
Liposome.
-
2. Beschreibung des Standes
der Technik
-
Die
optimale Behandlung mit vielen Arzneimitteln erfordert, dass die
Arzneimittelkonzentration auf einem spezifischen Niveau für eine anhaltende
Zeitperiode gehalten wird. Zum Beispiel erfordert die optimale Antikrebs-Behandlung
mit Zellzyklus-spezifischen
Antimetaboliten die Aufrechterhaltung einer zytotoxischen Arzneimittelkonzentration
für eine
anhaltende Zeitperiode. Cytarabin ist ein äußerst Zeitplan-abhängiges Antikrebs-Arzneimittel.
Da dieses Arzneimittel Zellen nur abtötet, wenn diese DNA synthetisieren,
ist eine anhaltende Exposition bei einer therapeutischen Konzentration
des Arzneimittels für
eine optimale therapeutische Wirkung erforderlich. Die therapeutische
Wirksamkeit von solchen Wirkstoffen wird häufig durch die Tatsache verkompliziert,
dass die Halbwertszeit nach einer intravenösen oder subkutanen Dosis so
kurz wie einige wenige Stunden ist. Um eine optimale therapeutische
Wirkung gegen Krebszellen mit einen Zellzyklusphasen-spezifischen Arzneimittel,
wie Cytarabin, zu erreichen, gibt es zwei Hauptanforderungen: erstens
müssen die
Krebszellen einer hohen Arzneimittelkonzentration ausgesetzt werden,
ohne den Wirt signifikant irreversibel zu schädigen; und zweitens muss der
Tumor zu dem Arzneimittel für
eine anhaltende Zeitperiode ausgesetzt werden, um die Anzahl der
Krebszellen zu maximieren, die während
der DNA Synthese, dem anfälligen Teil
des Zyklus der Zellproliferation, kontaktiert werden. Diese Art
von Behandlungsplan erfordert eine hohe Arzneimittelbeladung in
einer Formulierung mit verzögerter
Freisetzung.
-
Bestimmte
andere Arten von Arnzeimitteln sind so toxisch, dass es wichtig
ist, eine niedere Arzneimittelkonzentration über eine verlängerte Zeitperiode
aufrechtzuerhalten. Zum Beispiel ist Amikacin ein Aminoglycosid-Antibiotika
mit einer klinisch-signifikanten Aktivität gegen sowohl Gram-negative
als auch Gram-positive Bakterienstämme. Bei existierenden therapeutischen
Prozeduren wird das Arzneimittel durch intravenöse oder intramuskuläre Weg mit
einem einmal- oder zweimal am Tag-Zeitplan verabreicht. Die am häufigsten
verwendete klinische Dosis ist 15 mg/kg/Tag, welche äquivalent
ist zu einer maximal empfohlenen täglichen Dosis von 1 g pro Tag.
Die Verabreichung des Arzneimittels durch beabstandete Injektionen
führt jedoch
zu der systemischen Exposition des Patienten, und abhängig von
dem Arzneimittel, erhöht
das Risiko von toxischen Nebenwirkungen. Folglich wäre eine
lokale Depot-Zubereitung mit langsamer Freisetzung für die Behandlung
von Infektionen, wie jene, die auf eine lokale Region von Weichgewebe
oder Knochen vorteilhaft ist, beim Erhöhen der lokalen Gewebekonzentrationen
des Arzneimittels vorteilhaft, im Vergleich mit therapeutischen
systemischen Dosen, indem die systemische Toxizität des freien
Arzneimittels reduziert oder vermieden wird. Wenn das Arzneimittel äußerst toxisch
ist oder der Behandlungsplan eine niedere therapeutische Dosis erfordert,
ist eine relativ geringe Arzneimittelbeladung in einer Formulierung
mit langsamer Freisetzung vorteilhaft.
-
Ein
Ansatz, welcher verwendet wurde, um Zusammensetzungen mit kontrollierter
Freisetzung für
die Arzneimittelabgabe bereitzustellen, ist die Liposomeneinkapselung.
Unter den Haupttypen von Lipsomen sind mulitvesikuläre Liposomen
(Kim, et al., Biochim. Biophys. Acta; 728: 339–348, 1983) eindeutig verschieden von
unilamellaren Liposomen (Huang, Biochemistry; 8: 334–352, 1969;
Kim et al., Biochim. Biophys. Acta; 646: 1–10, 1981), multilamellaren
Liposomen (Bangham, et al., J. Mol. Bio., 13: 238–252, 1965),
und stabilen plurilamellaren Liposomen (U.S. Patent Nr. 4.522.803).
Im Gegensatz zu unilamellaren Liposomen, enthalten multivesikuläre Liposome
mehrere wässrige
Kammern. Im Gegensatz zu multilamellaren Liposomen sind die mehreren
wässrigen
Kammern von mulitvesikulären
Liposomen nicht konzentrisch.
-
Der
Stand der Technik beschreibt Verfahren zum Herstellen von multivesikulären Liposomen
(Kim et al., Biochim. Biophys. Acta, 728: 339–348, 1983). Die Einkapselungswirksamkeit
von einigen kleinen Molekülen,
wie Cytosinarabinosid, auch als Cytarabin oder Ara-C bekannt, erwies
sich als relativ gering, und die Freisetzungsrate von eingekapselten
Molekülen
in biologischen Flüssigkeiten
war höher,
als therapeutisch gewünscht
ist.
EP 0 280 503 B1 offenbart
ein Verfahren, welches für
das Steuern der Freisetzungsrate von eingekapselten Molekülen aus
multivesikulären
Liposomen entwickelt wurde, wobei ein Hydrochlorid in den Einkapselungsvorgang
eingeführt
wird, um die Freisetzungsrate in biologischen Fluiden (des aktiven
Wirkstoffs) zu steuern. Weitere Forschung, offenbart in WO 95/13796,
hat gezeigt, dass die Freisetzungsrate von Wirkstoffen aus multivesikulären Liposomen
in menschlichem Plasma durch Einführen einer nicht-Hydrochlorid-Säure in die
wässrige
Lösung,
in welcher der Wirkstoff aus der Bildung des multivesikulären Liposoms
aufgelöst
ist, gesteuert werden.
-
U.S.
Patent Nr. 5.077.056 offenbart Studien, die gezeigt haben, dass
die Freisetzungsrate des eingekapselten biologischen Wirkstoffs
aus Liposomen in eine wässrige
Umgebung durch Einführen
von Protonophoren oder Ionophoren in Liposome moduliert werden können, um
ein Membranpotential zu erzeugen. Zusätzlich ist ein Verfahren bekannt
(U.S. Patent Nr. 5.186.941) für
das Steuern der Freisetzungsrate von Arzneimitteln aus Vesikelzusammensetzungen,
wobei die Liposome einen eingekapselten therapeutischen Wirkstoff
enthalten, in einer Lösung,
die ausreichend gelösten
Stoff enthält,
suspendiert werden, um eine Osmolarität bereitzustellen, die im Wesentlichen
in Bezug auf die Lösung
innerhalb des Vesikels isotonisch ist, und in Bezug auf die physiologische
Kochsalzlösung
hypertonsich ist. In multivesikulären Liposomen ist es bekannt (WO
96/08253), die Freisetzungsrate von aktiven Wirkstoffen durch Einführen eines
osmotischen Spacers in die wässrige
Lösung,
in welcher der Wirkstoff vor der Bildung der multivesikulären Liposomen
aufgelöst
wird, zu steuern.
-
Zusätzlich zu
den biologisch aktiven Wirkstoffen und Säuren oder osmotischen Spacern,
die beabsichtigt sind, die Freisetzungsrate des biologisch aktiven
Wirkstoffs aus den Liposomen zu steuern, ist es gang und gäbe, Verbindungen
miteinzukapseln, die dafür
bestimmt sind, um einer beliebigen einer Reihe von Helferfunktionen
zu dienen. Zum Beispiel bewahren bestimmte biologisch aktive Verbindungen
ihre Aktivität
nur, wenn sie bei einem bestimmten pH gehalten werden. Somit werden
Säuren
und Puffer häufig
notwendigerweise zusätzlich
zu dem aktiven Wirkstoff eingekapselt, um den pH der Arzneimittel-Umgebung
zu steuern. In anderen Fällen
wird ein Gegenion einbezogen, um die Löslichkeit eines biologisch
aktiven Wirkstoffs, der eine geringe Löslichkeit besitzt, zu erhöhen.
-
Diese
Verfahren zum Erzeugen von Liposomenformulierungen mit langsamen
Freisetzungseigenschaften haben sich manchmal mit dem Ziel der Herstellung
von Liposomen, die eine hohe Ladung an aktiven Wirkstoff enthalten,
mit guter Einkapselungswirksamkeit als inkompatibel erwiesen, so
dass wenig des teuren aktiven Wirkstoffs durch Versagen des Einschließens des
Wirkstoffs innerhalb der Liposome verschwendet wird.
-
Somit
existiert der Bedarf an neuen Verfahren zum Herstellen von Liposomen,
zum Beispiel multivesikulären
Liposomen (MVLs), die die Steuerung der Arzneimittelbeladung, entweder
hoch oder niedrig, ermöglichen,
während
die gewünschte
verzögerte
Freisetzung des aktiven Wirkstoffs in den Speicher und biologische
Fluide aufrechterhalten wird. Von speziellem Interesse ist die Entwicklung
von hochbeladenen Formulierungen mit kontrollierter Freisetzung
für Peptide
und Proteine. Es besteht auch ein Bedarf an neuen Verfahren zum
Erreichen dieser Ziele ohne die hohe Einkapselungswirksamkeit zu
opfern, um die Verschwendung von teuren aktiven Wirkstoffen, wie
Arzneimittel und therapeutische Proteine, zu vermeiden.
-
Zusammenfassung der Erfindung
-
Die
vorliegende Erfindung bietet ein Verfahren zum Modulieren des Ladens
eines biologisch aktiven Wirkstoffs in liposomale Formulierungen.
Die Konzentration des biologisch aktiven Wirkstoffs in dem Endprodukt
wird durch Einstellen der Osmolarität der wässrigen Komponente, in welcher
der aktive Wirkstoff für
die Einkapselung aufgelöst
ist, moduliert. Eine umgekehrte Beziehung zwischen Osmolarität und Arzneimittelbeladung
wurde entdeckt, wobei die Beladung mit aktivem Wirkstoff mit abnehmender
Osmolarität
der wässrigen Komponente
zunimmt. Somit können
Liposomen mit entweder hoher Arzneimittelbeladung oder geringer
Arzneimittelbeladung durch Manipulation der Osmolarität der Arzneimittel-enthaltenden
Lösung
vor der Einkapselung erreicht werden. Es wurde außerdem entdeckt,
dass die Modulation der Arzneimittelbeladung, insbesondere um eine
hohe Arzneimittelbeladung zu erreichen, ohne Verlust entweder der
hohen Einkapselungswirksamkeit bei dem Herstellungsverfahren oder
der gewünschten
kontrollierten Freisetzung des Arzneimittels aus dem verwendeten
Endprodukt erreicht werden kann.
-
Durch
das Verfahren dieser Erfindung hergestellte Liposome erreichen stark
verbesserte Ergebnisse, in dem eine gewünschte Menge des aktiven Wirkstoffs
innerhalb eines gegebenen Volumens einer injizierbaren oder implantierbaren
Liposomenformulierung bereitgestellt wird und bieten eine verzögerte Freisetzung des
Arzneimittels bei einer therapeutisch gewünschten Konzentration, wenn
sie an einer in vivo Stelle eingeführt werden. Das gefolgte allgemeine
Prinzip, um das Laden des aktiven Wirkstoffs in liposomale Formulierungen
zu modulieren, wird hierin unter Bezug auf die Herstellung von multivesikulären Liposomen
(MVLs) illustriert. Die Arzneimittelbeladung in Liposomen wird durch
Steuern der Osmolarität
der wässrigen
Lösung,
die während
der Herstellung der Liposome eingekapselt wird, moduliert. Osmolarität ist die
Summe der molaren Konzentrationen von gelösten Stoffen, die in der wässrigen
Lösung
vorhanden sind, einschließlich
der biologisch aktiven Substanz und jeglichen Helfermolekülen, wie
die verwendeten osmotischen Exzipienten, um die Freisetzungsrate
des aktiven Wirkstoffs zu verlangsamen. Wenn der gelöste Stoff
in einer dissoziierten, ionisierten, oder aggregierten Form vorhanden
ist, dann ist die Osmolarität
als die Summe der molaren Konzentrationen der dissoziierten, ionisierten
oder aggregierten Formen definiert. Der Beitrag zu der Osmolarität einer Lösung, der
ein beliebiger gelöster
Stoff in der Lösung
ist, ist etwa äquivalent
zu der Konzentration des gelösten
Stoffes in der Lösung
dividiert durch sein Molekulargewicht. Folglich, als ein allgemeines
Prinzip, je höher das
Molekulargewicht eines gelösten
Stoffes, desto geringer ist die Osmolarität des gelösten Stoffes, und desto kleiner
ist der Beitrag dieses gelösten
Stoffes zu der Gesamtosmolarität
der Lösung.
-
Es
ist weithin bekannt, dass der Grad der Arzneimittelbeladung in Liposome
direkt proportional zu der Konzentration des biologisch aktiven
Wirkstoffs ist. Dementsprechend muss eine hohe Konzentration des
aktiven Wirkstoffs in einer wässrigen
Lösung,
die eingekapselt wird, aufgelöst
sein, um Liposome mit einem hohen Grad an Arzneimittelbeladung zu
erhalten. Die Beladung kann jedoch nicht immer durch Hinzufügen einer weiteren
Konzentration des aktiven Wirkstoffs erhöht werden. Gelöste Stoffe,
mit Ausnahme des biologisch aktiven Wirkstoffs, die in der wässrigen
Lösung
vorhanden sind, die während
der Herstellung der Liposomen verwendet wird, tendieren dazu, die
Menge an biologisch aktiven Wirkstoff zu reduzieren, die in die
Liposomen geladen werden kann. Wenn die Lösung auch osmotische Exzipienten
enthält,
die für
das Regulieren der Löslichkeit
oder Bioaktivität
des aktiven Wirkstoffs notwendig sind, müssen deshalb die vorteilhaften
Wirkungen der osmotischen Helferexzipienten in der Lösung gegenüber ihrer
nachteiligen Wirkung bei der Arzneimittelbeladung ausbalanciert
sein.
-
Um
die Arzneimittelbeladung zu erhöhen,
kann die Osmolarität
der wässrigen
Lösung
verringert werden, ohne die Konzentration des darin aufgelösten aktiven
Wirkstoffes zu verringern, entweder durch Reduzieren der Konzentration
der osmotischen Exzipienten, oder durch Ersetzen eines osmotischen
Exzipienten mit niederem Molekulargewicht durch einen osmotischen
Exzipienten mit hohem Molekulargewicht mit vergleichbarer Funktion
oder Beides. Wenn zum Beispiel der osmotische Exzipient ein Puffer
ist, der verwendet wird, um die Löslichkeit einer bestimmten
Konzentration eines biologisch aktiven Wirkstoffs zu erhalten, wird
ein Puffer mit hohem Molekulargewicht ausgewählt, um eine hohe Ladung an
aktivem Wirkstoff zu erhalten. Im Gegensatz dazu würde, um
die Ladung in einer solchen Situation zu verringern, ein Puffer
mit niederem Molekulargewicht verwendet werden.
-
Obwohl
diese Prinzipien bei der Herstellung von allen Arten von Liposomen
wirksam sind, werden sie hierin bei MVL Formulierungen, die derart
verschiedene aktive Wirkstoffe, wie Cytarabin, Leuprolid, Enkephalin,
Morphin, und Insulin-artiger Wachstumsfaktor I (IGF-1) enthalten,
illustriert. Es wurde in diesen Studien gefunden, dass für eine beliebige
ausgewählte
Konzentration an biologisch aktiven Wirkstoff die Arzneimittelbeladung
in MVLs während
der Herstellung, durch Variieren der Beiträge, die die osmotischen Exzipienten
in der Lösung
zu der Gesamtosmolarität
einer ersten wässrigen
Komponente leisten, effektiv moduliert werden kann. Dieses Prinzip
ist in den Beispielen hierin durch Einstellen der Konzentration
eines osmotischen Modell-Exzipienten, der für gewöhnlich in liposomalen Formulierungen,
entweder Saccharose oder Glycylglycin, verwendet wird, illustriert.
Durch dieses Verfahren können
MVL Formulierungen mit einem großen Bereich an Beladungsniveaus
für einen
beliebigen, gegebenen biologisch aktiven Wirkstoff hergestellt werden.
-
In
dem Verfahren zum Herstellen von multivesikulären Liposomen (MVLs) mit gesteuerter
Arzneimittel-Beladung, wird eine Lipidkomponente, die zumindest
ein amphipathisches Lipid und ein neutrales Lipid enthält, die
in einem oder mehreren organischen Lösungsmitteln aufgelöst sind,
mit einer nicht mischbaren ersten wässrigen Komponente, die einen
oder mehrere biologisch aktiven Wirkstoffe, die einzukapseln sind,
und gegebenenfalls, einen oder mehrere osmotische Exzipienten, wie
ein Helfermolekül,
enthält,
vermischt. Die Ladung des aktiven Wirkstoffs in der Endformulierung
wird von der Gesamtosmolarität
von dieser ersten wässrigen
Komponente abhängen,
welche die Summe der Osmolarität
ist, die durch jeden der in der ersten wässrigen Komponente aufgelösten Feststoffe
beigetragen wird, einschließlich
dem aktiven Wirkstoff und jeglichen osmotischen Exzipienten.
-
Sobald
die Osmolarität
der ersten wässrigen
Komponente eingestellt wurde, um die gewünschte Beladung des aktiven
Wirkstoffs in dem Endprodukt zu erreichen, wird eine Wasser-in-Öl Emulsion
durch Mischen der zwei miteinander nicht mischbaren Komponenten
gebildet. Die Wasser-in-Öl
Emulsion wird dann in eine zweite nicht-mischbare wässrige Komponente
gemischt, um Lösungsmittel-Kügelchen
zu bilden. Das organische Lösungsmittel
wird schlussendlich von den Lösungsmittelkügelchen,
zum Beispiel durch Verdampfung, entfernt, um die Aggregation dieser
in MVLs zu bewirken. In dem letzten Schritt des Verfahrens werden die
MVLs in einem wässrigen
Medium, wie normale Salzlösung
suspendiert. Eine Zusammensetzung, die eine therapeutisch wirksame
Dosis des aktiven Wirkstoffes auf einer Gewicht zu Volumen der Formulierungsbasis enthält, kann
durch Erhöhen
oder Verringern des Volumens des Mediums, in welchem die MVLs, die
den aktiven Wirkstoff enthalten, suspendiert sind, erhalten werden.
-
Um
eine hohe Einkapselungswirksamkeit (oder prozentuelle Ausbeute)
während
der Formulierung der MVLs aufrechtzuerhalten und sicherzustellen,
dass die Freisetzung des aktiven Wirkstoffs in Verwendung bei einer
langsamen therapeutisch-wirksamen Rate erfolgt, enthält die Lipidkomponente
ein oder mehrere amphipathische Lipide mit etwa 13 bis etwa 28,
zum Beispiel, etwa 18 bis 22 Kohlenstoffen in seiner Kohlenstoffkette.
-
Kurze Beschreibung der
Zeichnungen
-
1 ist
ein Diagramm, das den Prozentsatz von IGF-1 zeigt, der in MVLs während der
in vitro Inkubation (Freisetzungsrate) über 7 Tage in Plasma bei 37°C zurückbehalten
wird. Während
der Herstellung des Konzentrats davon wurde Saccharose oder Glycylglycin
als ein osmotischer Exzipient in der wässrigen Komponente variiert,
um die Arzneimittelbeladung zu steuern. ♢ = 80 mg/ml IGF-1
und 2,5 w/v% Saccharose (113,5 mOsm); ∇ = 80 mg/ml IGF-1 und 1 w/v%
Glycylglycin (113,5 mOsm); + = 50 mg/ml IGF-I und 1 w/v% Saccharose
(63,5 mOsm). Die Fehlerbalken repräsentieren die Standardabweichungen.
-
2 ist
ein Diagramm, das die IGF-I Konzentration über 8 Tage in Serum (ng/ml)
von männlichen Ratten
nach einer 10 mg subkutanen Injektion von MVLs, die 80 mg/ml IGF-I
und 2,5 w/v% Saccharose (113,5 mOsm) in der wässrigen Komponente enthalten,
zeigt. Die Daten repräsentieren
die Mittelwerte der Daten für drei
Ratten.
-
Beschreibung der bevorzugten
Ausführungsformen
-
In
dieser Erfindung wird ein Verfahren bereitgestellt, wobei die Menge
von biologisch aktivem Wirkstoff, die pro Einheitsvolumen der Liposomenformulierung
eingekapselt ist, durch Einstellen der Osmolarität der eingekapselten wässrigen
Komponenten, die das Arzneimittel enthält, moduliert wird. In diesem
Verfahren ergibt eine Verringerung der Osmolarität der wässrigen Komponente, in welcher
der aktive Wirkstoff vor der Einkapselung aufgelöst ist, eine erhöhte Konzentration
des aktiven Wirkstoffs in der endgültigen MVL Suspension auf einer
Gewicht zu Volumen-Basis, und umgekehrt.
-
Es
gibt zumindest drei Arten von Liposomen. Der Begriff „multivesikuläre Liposomen
(MVL)", wie in der gesamten
Spezifikation und Ansprüche
verwendet wird, bedeutet künstliche,
mikroskopisch-flüssige
Vesikel, die Lipidmembrane aufweisen, die mehrere nicht-konzentrische
wässrige
Kammern umhüllen.
Im Gegensatz dazu weisen „multilamellare
Liposomen oder Vesikel (MLV)" mehrere „Zwiebelschalen" konzentrische Membranen
auf, zwischen welchen sich schalenartige konzentrische wässrige Kompartimente
befinden sind. Multilamellare Liposomen und multivesikuläre Liposomen
weisen charakteristischerweise einen nach der Länge-gewichteten mittleren Durchmesser
in dem Mikrometerbereich auf, üblicherweise
von 0,5 bis 25 μm.
Der Begriff „unilamellare
Lipsosomen oder Vesikeln (ULV)",
wie hierin verwendet, betrifft liposomale Strukturen mit einer einzelnen
wässrigen
Kammer, üblicherweise
mit einem mittleren Druckmesser im Bereich von etwa 20 bis 500 nm.
-
Multilamellare
und unilamellare Liposomen können
durch mehrere, relativ einfache Verfahren hergestellt werden. Der
Stand der Technik beschreibt eine Reihe von Techniken zum Herstellen
von ULV und MLV (zum Beispiel U.S. Patent Nr. 4.522.803 an Lenk;
4.310.506. an Baldeschweiler; 4.235.871 an Papahadjopoulos; 4.224.179
an Schneider; 4.078.052 an Papahadjopoulos; 4.394.372 an Taylor
4.308.166 an Marchetti; 4.485.054 an Mezei; und 4.508.703 an Redziniak).
-
Im
Gegensatz dazu erfordert die Herstellung von multivesikulären Liposomen
mehrere Verfahrensschritte. In Kürze,
das bevorzugte Verfahren zum Herstellen von MVL ist wie folgt: Im
ersten Schritt wird eine „Wasser-in-Öl" Emulsion durch Auflösen von
zumindest einem amphipathischen Lipid und zumindest einem neutralen
Lipid in einem oder mehreren flüchtigen
organischen Lösungsmittel
für die
Lipidkomponente hergestellt. Zu der Lipidkomponente wird eine nicht-mischbare
erste wässrige
Komponente hinzugefügt,
die einen biologisch aktiven Wirkstoff, der einzukapseln ist, und
ein oder mehrere Helfermoleküle,
d.h. osmotische Exzipienten, enthält, die die MVL mit nützlichen
und vorteilhaften Eigenschaften versehen. Die Mischung wird emulgiert,
und dann mit einer zweiten nicht-mischbaren wässrigen Komponente vermischt,
um einen zweite Emulsion zu bilden. Die zweite Emulsion wird entweder
mechanisch, durch Ultraschallenergie, Verdüsung, und dergleichen, oder
durch Kombinationen davon gemischt, um Lösungsmittelkügelchen
zu bilden, die in der zweiten wässrigen
Komponente suspendiert sind. Die Lösungsmittelkügelchen
enthalten mehrere wässrige Tröpfchen,
wobei der biologisch aktive Wirkstoff, der einzukapseln ist, in
diesen aufgelöst
ist (siehe Kim et al., Biochem. Biophys. Acta, 728: 339–348, 1983).
Für einen
umfangreichen Überblick
von verschiedenen Verfahren zur Herstellung von ULV und MLV sei
auf Szoka et al., Ann. Rev. Biophys. Bioeng. 9: 465–508, 1980
verwiesen.
-
Der
Begriff „Lösungsmittelkügelchen", so wie hierin in
der gesamten Spezifikation und Ansprüchen verwendet wird, bedeutet
ein mikroskopisches sphärisches
Tröpfchen
von organischem Lösungsmittel,
innerhalb dieser sind multiple kleinere Tröpfchen von wässriger
Lösung.
Die Lösungsmittelkügelchen
sind suspendiert und völlig
in einer zweiten wässrigen
Lösung
eingetaucht.
-
Der
Begriff „neutrales
Lipid" bedeutet
ein Öl
oder ein Fett, das selbst keine membranbildende Fähigkeit aufweist
und dem eine hydrophile „Kopfgruppe" fehlt.
-
Der
Begriff „amphipathisches
Lipid" bedeutet
ein Molekül,
das einen hydrophile „Kopfgruppe" und eine hydrophobe „Schwanzgruppe" aufweist und einen
membranbildende Fähigkeit
besitzt.
-
Der
Begriff „zwitterionisches
Lipid" bedeutet
ein amphipathisches Lipid mit einer Nettoladung von Null bei pH
7,4.
-
Der
Begriff „anionisches
Lipid" bedeutet
ein amphipahtisches Lipid mit einer negativen Nettoladung bei pH
7,4.
-
Der
Begriff „kationisches
Lipid" bedeutet
ein amphipathisches Lipid mit einer positiven Nettoladung bei pH
7,4.
-
Für die Herstellung
von multivesikulären
Liposomen ist es notwendig, dass zumindest ein amphipathisches Lipid
und ein neutrales Lipid in der Lipidkomponente eingeschlossen sind.
Die amphiphatischen Lipide können
zwitterionische, anionische oder katonische Lipide sein. Beispiele
von zwitterionischen amphipathischen Lipiden sind Phosphatidylcholine,
Phosphatidylethanolamine, Sphinomyeline etc. Beispiele von anionischen
amphipathischen Lipiden sind Phosphatidylglycerole, Phosphatidylserine,
Phosphatidylinositole, Phosphatidinsäuren, etc. Beispiele von kationischen
amphipatischen Lipiden sind Diacyltrimethylammoniumpropan und Ethylphosphatidylcholin.
Beispiele von neutralen Lipiden umfassen Digylceride, wie Diolein,
Dipalmitolein, und gemischte Caprylin-Caprin-Diglyceride; Triglyceride,
wie Triolein, Tripalmitolein, Trilinolein, Tricaprylin, und Trilaurin;
pflanzliche Öle,
wie Sojabohnenöl;
Sqalen; Tocopherol; und Kombinationen davon. Zusätzlich können Cholesterol oder pflanzliche
Sterole beim Herstellen von multivesikulären Liposomen verwendet werden.
-
So
wie hierin verwendet, umfasst der Begriff „biologisch aktiver Wirkstoff" oder „aktiver
Wirkstoff", wenn
verwendet, um Wirkstoffe zu beschreiben, die in den Kammern des
multivesikulären
Liposoms oder in der wässrigen
Lösung,
die während
der Herstellung von Liposomen verwendet wird, vorhanden sind, Wirkstoffe,
die biologische Aktivität
besitzen, die auf die Behandlung eines bestimmten Krankheitsstadiums
abgezielt ist, entweder in der Form, die aus Vesikel freigesetzt
wird, oder in einer Form, die nach der Freisetzung aus der Vesikelkammer
aktiv wird. Zum Beispiel zählen
zu biologisch aktiven Wirkstoffen Arzneimittel und Pro-Arzneimittel,
die durch die Interaktion mit einem Enzym in einen aktiven Anteil
mit therapeutischer Aktivität
umgewandelt werden. Insektizide, Pestizide und Mittel mit gewünschter
kosmetischer Anwendung sind ebenfalls durch den Begriff „biologisch
aktiver Wirkstoff" umfasst.
-
Der
Begriff „osmotischer
Exzipient" bedeutet
jedes biologisch kompatible gelöste
Feststoffmolekül
in einer wässrigen
Lösung,
das nicht der biologisch aktive Wirkstoff ist. Sowohl Elektrolyte
als auch Nicht-Elektrolyte fungieren als osmotische Exzipienten.
Beim Bestimmen, ob ein bestimmtes Molekül als ein osmotischer Exzipient
agieren wird oder beim Bestimmen der Konzentration des osmotischen
Exzipienten in einer Lösung, zum
Beispiel einem, der innerhalb eines multivesikukären Liposoms eingekapselt ist,
muss Rücksicht
darauf genommen werden, ob, unter den Bedingungen innerhalb der
Lösung
(z.B. pH), das Molekül
vollständig
oder teilweise ionisiert ist. Es sollte auch bestimmt werden, ob
solche Ionen die Lipidmembran durchdringen können (Mahendra K. Jain, van
Nostrand Reinhold Co., The Biomolecular Lipid Bilayer Membrane,
1972, 470 ff). Der Fachmann wird erkennen, dass für die Verwendung
in der vorliegenden Erfindung der osmotische Exzipient so ausgewählt sein
muss, dass jene umgangen werden, die sich für ein Lebewesen, das einer
Therapie unter Verwendung von Liposomen unterzogen wird, als toxisch
oder andererseits schädlich
erweisen würden.
Der Fachmann kann leicht die Eignung eines bestimmten osmotischen
Exzipienten für
die Verwendung in der vorliegenden Erfindung ohne Zurückgreifen
auf unnötige
Experimentierung evaluieren.
-
Bestimmte
osmotische Exzipienten weisen eine inhärente biologische Aktivität auf, und
viele fördern die
biologische Aktivität
des biologisch aktiven Wirkstoffs. Zum Beispiel können Kalziumionen
als ein Gegenion miteingekapselt werden, um die Haltbarkeitsdauer
zu erhöhen
oder um die Bioverfügbarkeit
eines Arzneimittels zu fördern,
sie reichen jedoch nicht aus, um den therapeutischen oder andere
Nutzen der MVL Formulierung zu erreichen. Zusätzlich können verschiedene Stabilisatoren
vorhanden sein. Bestimmte Wirkstoffe, die häufig als Exzipienten klassifiziert
sind, können
sogar eine direkte biologische Aktivität besitzen, und zwar von einer
sehr geringfügigen
bis zu einer ganz signifikanten Aktivität. Zum Beispiel kann der gebräuchliche
Exzipient Mannitol biologisch auch als ein Diuretikum wirken. Sogar
Wasser kann biologisch wirken, um Dehydrierung zu heilen, wenn aber
diese Verbindungen eher als osmotische Exzipienten als als aktive
Wirkstoffe verwendet werden, sind sie mit anderen, die dieselbe
Helferfunktion erfüllen,
relativ untereinander auswechselbar. Osmotische Exzipienten, die
verwendet werden können,
um multivesikuläre
Liposome zu bilden und um die Arzneimittelbeladung des eingekapselten
Wirkstoffes von multivesikulären
Liposomen zu modulieren, umfassen, ohne darauf beschränkt zu sein,
Glucose, Saccharose, Trehalose, Succinat, Glycylglycin, Gluconsäure, Cyclodextrin,
Arginin, Galactose, Mannose, Maltose, Mannitol, Glycin, Lysin, Citrat,
Sorbitol, Dextran, und geeignete Kombinationen davon. Tabelle 1
unter vergleicht die Osmolarität
von Saccharose und Glycylglycin-Lösungen bei unterschiedlichen
Konzentrationen.

- 1 Saccharose Daten aus dem Handbook of Physics
and Chemistry, 67. Ausgabe
- 2 Glycylglycin-Daten wurden basierend
auf der molaren Konzentration berechnet.
-
Fachleute
können
leicht verschiedene Kombinationen von Exzipienten bestimmen und
vorsehen, die in den Vesikeln der Erfindung, ohne auf eine unnötige Experimentierung
zurückzugreifen,
benutzt werden können.
-
Wie
hierin verwendet, bedeutet der Begriff „therapeutisch wirksame Menge
oder Konzentration" die Menge
eines biologisch aktiven Wirkstoffs, die notwendig ist, um eine
gewünschte
pharmakologische Wirkung zu induzieren. Die Menge kann stark variieren,
gemäß der Wirksamkeit
eines bestimmten aktiven Wirkstoffs, dem Alter, dem Gewicht und
Reaktion des individuellen Wirtes, sowie der Natur und Schwere der
Symptome des Wirtes. Dementsprechend gibt es keine obere oder untere
kritische Beschränkung
für die
Menge des aktiven Wirkstoffs. Die therapeutisch wirksame Menge,
die in der vorliegenden Erfindung einzusetzen ist, kann leicht durch
Fachleute bestimmt werden.
-
Wie
hierin verwendet, bedeutet „Arzneimittelbeladung", in einem allgemeinen
quantitativen Sinn, die Menge an biologisch aktivem Wirkstoff, die
in die Produkt-Liposomen-Suspension geladen wird. Es ist deshalb ein
Maß für die Menge
des aktiven Wirkstoffs, die in einer Volumeneinheit der Liposomenformulierung,
die an einen Patienten während
der Verwendung abgegeben wird, verfügbar ist. Spezifischer bedeutet „Arzneimittelbeladung" das Verhältnis von
eingekapseltem Arzneimittel pro Volumeneinheit von Liposomensuspension
zu dem Prozentsatz des eingekapselten Volumens in den Liposomen
selbst. Es etwa gleich zu der Konzentration des, aktiven Wirkstoffs
in der Suspension dividiert durch den Lipokrit der Suspension für das niederprozentige freie
Arzneimittel:
-
So
wie hierin verwendet, bedeutet „prozentuelle Einkapselung
des Arzneimittels, oder anderer Verbindung" das Verhältnis der Menge einer einzukapselnden
Verbindung in der Endsuspension des Liposomen-Herstellungsverfahrens
zu der Gesamtmenge der einzukapselnden Verbindung, die in der ersten
wässrigen
Lösung
des Prozesses verwendet wurde, multipliziert mit 100.
-
-
So
wie hierin verwendet, bedeutet „Lipokrit", welcher in Analogie zu Hämatokrit
definiert ist, das Verhältnis
des durch die Liposomen besetzten Volumens zu dem Gesamtvolumen
der Suspension, multipliziert mit 100.
-
-
So
wie hierin verwendet, bedeutet „prozentuell freies Arzneimittel" das Verhältnis der
Menge des Arzneimittels außerhalb
der Liposomen in der endgültigen
Liposomensuspension, zu der Gesamtmenge an Arzneimittel in der Endsuspension
(das Endprodukt), multipliziert mit 100.
-
-
Die
Verfahren zum Bestimmen dieser Parameter sind in Beispiel 7 dieser
Anmeldung illustriert. Wo immer möglich wird die Verwendung von
osmotischen Exzipienten auf ein Minimum reduziert oder vermieden, um
eine hohe Beladung mit dem biologisch aktiven Wirkstoffs zu erreichen.
In diesem Fall ist die Arzneimittelbeladung direkt von der Konzentration
des aktiven Wirkstoffs in der einzukapselnden Lösung abhängig, da die Osmolarität größtenteils
dem aktiven Wirkstoff zuschreibbar ist. Wenn es nicht möglich ist,
eine erste wässrige Lösung zu
verwenden, die frei von osmotischen Exzipienten ist, kann die Osmolarität der ersten
wässrigen Komponente
durch Austauschen von Exzipienten mit hohem Molekulargewicht gegen
Exzipienten mit niederem Molekulargewicht, wie hoch molekulargewichtige
Puffer oder Stabilisatoren gegen solche mit niederem Molekulargewicht,
verringert werden. Auch durch Auswählen eines negativen Gegenions
für ein
Arzneimittel kann einen Gegenion mit hohem Molekulargewicht durch
eines mit niederem Molekulargewicht ersetzt werden. Zum Beispiel
in Morphinhydrochlorid kann das Chlorid-Ion durch ein Sulfat oder
einem negativen Ion mit noch höherem
Molekulargewicht, wie Phosphat ausgetauscht werden.
-
Falls
es gewünscht
ist, eine Formulierung herzustellen des biologisch aktiven Wirkstoffs
mit niederer Beladung, wie wenn der biologisch aktive Wirkstoff
bei hohen Konzentrationen toxisch ist, kann umgekehrt die Osmolarität der ersten
wässrigen
Lösung
erhöht
werden indem osmotische Exzipienten mit niederem Molekulargewicht
ausgewählt
werden, um ihre Osmolarität
zu erhöhen.
-
Die
untere Grenze für
die Osmolarität
der ersten wässrigen
Komponente kann in der Nähe
von Null sein, wie im Fall, wo der biologisch aktive Wirkstoff ein
hoch molekulargewichtiges Protein oder anderes Makromolekül ist und
keine osmotischen Exzipienten verwendet werden. Andererseits kann
die Osmolarität
der ersten wässrigen
Komponente so hoch wie 1000 mOsm oder höher sein, ohne bei der Verwendung
schädlich oder
toxisch zu sein, da viele der Exzipienten während des Herstellungsverfahrens
aus den Liposomen austreten können.
Im Allgemeinen ist die Osmolarität
der ersten wässrigen
Komponente jedoch im Bereich von etwa 0,01 mOsm bis etwa 1100 mOsm,
zum Beispiel im Bereich von 5 mOsm bis etwa 400 mOsm.
-
Die
Osmolarität
der eingekapselten wässrigen
Komponente in dem endgültigen
liposomalen Produkt ist im Allgemeinen in Bezug auf die wässrige Umgebung
isotonisch, in welcher die MVLs gelagert werden (wie 0,9 Gew.-%
NaCl, oder normale Salzlösung)
oder in welche die MLVs für
die Verwendung eingeführt
werden, wie Serum oder andere physiologich relevante, wässrige Umgebungen.
Jedoch kann die Osmolarität
der wässrigen
Komponente in dem endgültigen
MVL Produkt hypertonisch sein, um eine optimale Abnahme der Freisetzungsrate
des biologisch aktiven Wirkstoffs aus den Liposomen zu bieten. Es
ist deshalb innerhalb des Umfanges dieser Erfindung vorgesehen,
dass die wässrige
Komponente in dem MVL Produkt hypotonisch, isotonisch oder hypertonisch
in Bezug auf das Lagermedium oder die wässrige Umgebung sein kann,
in welche der biologisch aktive Wirkstoff freigesetzt wird.
-
Die
Osmolarität
von normaler Salzlösung
ist ähnlich
zu jener von menschlichem Plasma und anderen in vivo Umgebungen,
wie Cerebrospinalflüssigkeit,
Synovialflüssigkeit,
und subkutane und intramuskuläre Räume. Deshalb
kann Salzlösung
als ein prädiktives
Modell der MVL Arzneimittel-Freisetzung in solchen Umgebungen verwendet
werden. Da die bevorzugte Verwendung der MVLs der Erfindung für die in
vivo Injektion oder Implantation in Gewebe oder Körperhöhlen (z.B.
als Arzneimittel-Depots) ist, werden sie üblicherweise in einem Medium,
wie normaler Salzlösung,
Phosphat-gepufferte Salzlösung,
oder einem anderen osmotisch ähnlichen
Medium gelagert.
-
Die
Freisetzungsrate von aktiven Wirkstoffen aus MVLs wird im Allgemeinen
durch Verringern der Osmolarität
der ersten wässrigen
Komponente, die während
der Herstellung verwendet wird, erhöht. Die Verringerung der Osmolarität der ersten
wässrigen
Komponente kann jedoch eine negative Wirkung auf die verzögerte Freisetzung
und Einkapselungswirksamkeit haben. Diese negative Wirkung kann
durch Verwenden von einem oder mehreren amphipathischen Lipiden
mit etwa 13 bis etwa 28 Kohlenstoffen, zum Beispiel von etwa 18
bis 22 Kohlenstoffen in der Lipidkomponente überwunden werden. Diese allgemeine
Regel gilt, ob die Kohlenstoffkette des amphiphatischen Lipids gesättigt ist,
oder ob sie eine oder mehrere Doppelbindungen enthält. Im Allgemeinen
sollte jedoch beim Auswählen
der Lipide, die beim Formulieren eines multivesikulären Liposoms
zu verwenden sind, beachtet werden, dass es möglich ist, ein organisches
Lösungsmittel
mit einem geringeren Siedepunkt zu verwenden, wenn ein Lipid mit
einer gegebenen Anzahl von Kohlenstoffen in der Kohlenstoffkette
benutzt wird, wenn das Lipid zumindest eine Doppelbindung in der
Kohlenstoffkette enthält.
Die bevorzugten amphipathischen Lipide für die Verwendung beim Herstellen
von multivesikulären
Liposomen dieser Erfindung sind natürlich vorkommende Lipide. Die
vorteilhaften Wirkungen auf die Einkapselungswirksamkeit und verzögerte Freisetzung
des biologisch aktiven Wirkstoffs, die durch Benutzen solcher langkettiger
amphipatihischer Lipide während
der Herstellung von MVLs zu erhalten sind, sind im gleichzeitig
anhängenden U.S.
Patent Anmeldungs Nr. 08/723.583, eingereicht am 1. Oktober, 1996,
mit dem Titel „Methods
for Producing Liposomes With Increased Percent of Compound Encapsulated", die hierin zur
Gänze durch
Bezugnahme aufgenommen ist, offenbart.
-
Eine
repräsentative
Liste von langkettigen amphipathischen Lipiden, die beim Ausführen dieser
Erfindung nützlich
sind, folgt. Diese Liste ist illustrativ und nicht beabsichtigt,
den Umfang der Erfindung auf irgendeine Weise zu beschränken. Ebenfalls
eingeschlossen sind die Abkürzungen,
die verwendetet werden, um die Phospholipide in dieser Anmeldung
und in der wissenschaftlichen Literatur zu bezeichnen.
DOPC
oder DC18:1PC = 1,2-Dioleoyl-sn-glycero-3-phosphocholin
DLPC
oder DC12:0PC = 1,2-Dilauroyl-sn-glycero-3-phosphocholin
DMPC
oder DC14:0PC = 1,2-Dimyristoyl-sn-glycero-3-phosphocholin
DPPC
oder DC16:0PC = 1,2-Dipalmitoyl-sn-glycero-3-phosphocholin
DSPC
oder DC18:0PC = 1,2-Distearoyl-sn-glycero-3-phosphocholin
DAPC
oder DC20:0PC = 1,2-Diarachidoyl-sn-glycero-3-phosphocholin
DBPC
oder DC22:0PC = 1,2-Dibehenoyl-sn-glycero-3-phosphocholin
DC16:1PC
= 1,2-Dipalmitoleoyl-sn-glycero-3-phosphocholin
DC2O:1PC =
1,2-Dieicosenoyl-sn-glycero-3-phosphocholin
DC22:1PC = 1,2-Dierucoyl-sn-glycero-3-phosphocholin
DPPG
= 1,2-Dipalmitoyl-sn-glycero-3-phosphoglycerol
DOPG = 1,2-Dioleoyl-sn-glycero-3-phosphoglycerol
-
Viele
unterschiedliche Arten von flüchtigen
hydrophoben Lösungsmitteln,
wie Ether, Kohlenwasserstoffe, halogenierte Kohlenwasserstoffe,
superkritische Fluide, einschließlich, ohne darauf beschränkt zu sein, CO2, NH3, und Freone,
können
als das Lipidphasen-Lösungsmittel
verwendet werden. Zum Beispiel sind Diethylether, Isopropyl und
andere Ether, Dichlormethan, Chloroform, Tetrahydrofuran, halogenierte
Ether, Ester und Kombinationen davon zufriedenstellend.
-
Therapeutische
biologisch aktive Verbindungen, oder Arzneimittel, für die Einkapselung
in den Verfahren und Zusammensetzungen dieser Erfindung können aus
der allgemeinen Gruppe bestehend aus anti-neoplastischen Mitteln,
antiinfektiöse
Mittel, Hormone, Antidepressiva, entzündungshemmende Wirkstoffe,
antivirale Wirkstoffe, antinociceptive Wirkstoffe, Anxiolytika und
Biologika.
-
Zu
repräsentativen
Beispielen von antineoplastischen Wirkstoffen, die in den Zusammensetzungen und
Verfahren der vorliegenden Erfindung nützlich sind, zählen Methotrexat,
Taxol, Tumornekrosefaktor, Chlorambucil, Interleukine, Etoposid,
Cytarabin, Fluoruracil und Vinblastin.
-
Zu
repräsentativen
Beispielen von antiinfektiösen
Wirkstoffen, die in den Zusammensetzungen und Verfahren der vorliegenden
Erfindung nützlich
sind, zählen
Amikacin, Pentamidin, Metronidazol, Penicillin, Cephalexin, Tetracyclin,
und Chloramphenicol.
-
Zu
repräsentativen
Beispielen von antiviralen Wirkstoffen, die in den Zusammensetzungen
und Verfahren der vorliegenden Erfindung nützlich sind, zählen Dideoxycytidin,
Zidovudin, Acyclovir, Interferone, Didexoyinosin, und Ganciclovir.
-
Zu
repräsentativen
Beispielen von Anxiolytika und Sedativa, die in den Zusammensetzungen
und Verfahren der vorliegenden Erfindung nützlich sind, zählen Benzodiazepine,
wie Diazepam, Bariburate, wie Phenobarbital, und andere Verbindungen,
wie Buspiron und Haloperidol.
-
Zu
repräsentativen
Beispielen von Hormonen, die in den Zusammensetzungen und Verfahren
der vorliegenden Erfindung nützlich
sind, zählen
Estradiol, Prednison, Insulin, Wachstumshormon, Erythropoietin, und
Prostaglandine.
-
Zu
repräsentativen
Beispielen von Antidepressive, die in den Zusammensetzungen und
Verfahren der vorliegenden Erfindung nützlich sind, zählen Fluoxetin,
Trazodon, Imipramin, und Doxepin.
-
Zu
repräsentativen
Beispielen von Antinociceptiva, die in den Zusammensetzungen und
Verfahren der vorliegenden Erfindung nützlich sind, zählen Bupivacain,
Hydromorphin, Oxycodon, Fentanyl, Morphin und Meperidin.
-
Der
Begriff „Biologika" umfasst Nukleinsäuren (DNA
und RNA), Glucosaminoglykane; Protein und Peptide, und umfasst Verbindungen,
wie Cytokine, Hormone (hypophysäre,
adrenale, und pituitäre
Hormone), Wachstumsfaktoren, Vakzine etc. Von besonderem Interesse
sind Interleukin-2, Insulinartiger Wachstumsfaktor-1 (IGF-1), Interferone,
Insulin, Heparin, Leuprolid, Granulocyten-Kolonie-stimulierender
Faktor (G-CSF), Granulozyten-Makrophagen-Kolonie-stimulierender Faktor (GM-CSF),
Tumornekrosefaktor, Inhibin, Tumorwachstumsfaktor Alpha und Beta,
Mullerian initiierende Substanz, Kalcitonin, Hepatitis B Vakzin,
DNA oder RNA Vakzine, DNA für
Gentransfer, und Antisense-Oligonukleotide.
-
Der
biologisch aktive Wirkstoff kann in der vorliegenden Erfindung in
verschiedenen Formen eingesetzt werden, wie molekulare Komplexe
oder biologisch akzeptable Salze. Repräsentative Beispiele von solchen
Salzen sind Succinat, Hydrochlorid, Hydrobromid, Sulfat, Phosphat,
Nitrat, Citrat, Glucuronat, Borat, Acetat, Maleat, Tartrat, Salicylat,
Metallsalze (z.B. Alkali oder Erdalkali), Ammonium oder Aminsalze
(z.B. quarternäres
Ammonium) und dergleichen. Weiters können Derivate der aktiven Wirkstoffe,
wie Ester, Amide, und Ethers davon, die die gewünschten Retentions- und Freisetzungseigenschaften
besitzen, die jedoch bei physiologischem pH oder durch Enzyme leicht
in vivo hydrolysiert werden, ebenfalls als der biologisch aktive
Wirkstoff eingesetzt werden.
-
Die
Konzentration des eingekapselten biologisch aktiven Wirkstoffs kann
von etwa einigen Picomol bis zu mehreren Hundert Millimol variieren.
Die gewünschte
Konzentration des biologisch aktiven Wirkstoffs wird in Abhängigkeit
solcher Eigenschaften, wie der zu behandelnden Krankheit, dem Alter
und Zustand des Patienten, und den besonderen Eigenschaften des
Wirkstoffs variieren. Im Fall wo der Wirkstoff normalerweise mit Nebenwirkungen,
wie Toxizität
assoziiert ist, ist es im Allgemeinen wünschenswert, ein MVL mit geringerer Wirkstoffkonzentration
zu erzeugen und benutzt eine höhere
Konzentration an osmotischen Exzipienten. Der Zusammenhang dieser
verschiedenen Parameter kann leicht durch einen Fachmann beim Auswählen und
Erzeugen einer bestimmten MVL Zusammensetzung, ohne auf unnötige Experimentierung
zurückgreifen
zu müssen,
evaluiert werden.
-
Formulierungen
mit hoher Beladung, die durch das Verfahren dieser Erfindung erhalten
wurden, sind besonders in der pharmazeutischen Industrie für das Reduzieren
der Menge an Liposomen-Formulierung,
die einem Lebewesen verabreicht werden muss (d.h. intramuskulär oder subkutan),
nützlich,
um eine gewünschte therapeutische
Konzentration des Arzneimittels im Blutstrom zu erreichen. Die obere
nützliche
Grenze der Menge an Arzneimittel, die in ein gegebenes Volumen von
Liposomensuspension eingekapselt ist, kann jedoch durch den Lipokrit
der Suspension diktiert werden. Der Fachmann wird erkennen, dass
es schwierig sein kann, eine Liposomen enthaltende Suspension zu
injizieren, wenn der Lipokrit der Suspension zu hoch ist.
-
Der
für die
in vivo Verwendung in Menschen geeignete Dosierungsbereich des biologisch
aktiven Wirkstoffs in multivesikulären Liposomen dieser Erfindung
umfassen den Bereich von 0,001–6,000
mg/m2 Körperoberfläche. Während Dosen
außerhalb
des vorangehenden Dosisbereichs gegeben werden können, umfasst dieser Bereich
die Breite der Verwendung für
praktisch alle der biologisch aktiven Wirkstoffe. Für einen bestimmten
therapeutischen Wirkstoff kann jedoch die bevorzugte Konzentration
leicht, wie zuvor beschrieben ist, bestimmt werden.
-
Die
MVL Formulierungen können
weiters verdünnt
werden, um eine injizierbare Depotformulierung mit verzögerter Freisetzung
von einer beliebigen therapeutisch wirksamen Gesamtdosierung durch
Hinzufügen
eines Suspendiermediums oder eines physiologisch akzeptablen Trägers erreicht
werden. Zu gebräuchlichen geeigneten
Trägern
zählen
wässrige
oder nichtwässrige
Lösungen,
Suspensionen, und Emulsionen. Beispiele von nicht-wässrigen Lösungen sind
Propylenglycol, Polyethylenglycol, pflanzliche Öle, wie Olivenöl, und injizierbare
organische Ester, wie Ethyloleat. Wässrige Träger umfassen Wasser, alkoholische-wässrige Lösungen, Emulsionen oder Suspensionen,
einschließlich
Salzlösung
und gepufferte Medien. Parenterale Vehikel umfassen Natriumchlorid-Lösung, Ringer's Dextrose, Dextrose,
und Ringer-Lactat-Lösung.
Intravenöse
Vehikel umfassen Fluid und Nährstoff-Replenisher,
Elektrolyt-Replenisher (wie jene, die auf der Ringer's Dextrose basieren),
und dergleichen. Konservierungsmittel und andere Zusätze können auch
vorhanden sein, wie antimikrobielle Substantzen, Antioxidantien,
Chelatbildner, und inerte Gase (siehe Remington's Pharmaceutical Science, 16. Ausgabe,
A. Oslo, Hrsg., Mack, Easton, PA. 1980).
-
Die
multivesikulären
Liposome können
durch jede geeignete Route verabreicht werden: zum Beispiel intratumoral,
intra-artikulär
(in Gelenke), intraokular, intramuskulär, intrathecal, intraperitoneal,
subkutan, intravenös,
intralymphatisch, oral und submukosal. Die multivesikulären Liposome
können
unter Verwendung von Verfahren, die im Stand der Technik bekannt
sind, modifiziert werden, indem daran, entweder direkt von indirekt,
mit Hilfe eines Exzipienten-Moleküls oder Peptids Ziel-spezifische
Liganden, wie Antikörper
und andere Rezeptor-spezifische Proteinliganden angeheftet werden,
um eine Organ- oder Zielzell-Spezifität zu vermitteln (Malone, et
al., Proc. Nat'l.
Acad. Sci., U.S.A., 86: 6077, 1989; Gregoriadis, Immunology Today,
11(3): 89, 1990; beide sind durch Bezugnahme hierin aufgenommen).
-
Eine
Reihe von Experimenten wurde ausgeführt, um zu zeigen, dass die
Wirkung der Osmolarität
auf die Arzneimittelbeladung invertiert ist und von anderen Parametern,
die während
des Herstellungsverfahrens verwendet werden, abhängig ist, mit Ausnahme der
Menge des aktiven Wirkstoffs, welche direkt proportional zu der
Menge an aktiven Wirkstoff ist, die in eine liposomale Formulierung
geladen werden kann. Diese zwei Parameter müssen deshalb balanciert sein,
um den gewünschten
Grad an Beladung zu erhalten. Zum Beispiel wurde in Beispiel 1 gezeigt,
dass Cytarabin in MVLs unter Verwendung eines Vortex-Mischers und
einer ersten wässrigen
Komponente, die 40 mg/ml Cytarabin in 20 mM Zitronensäure und
Mengen an Saccharose im Bereich von Null bis 8,0 Gewicht/Volumen-Prozent
(Gew./Vol.-%) enthält,
eingekapselt werden kann. Die entsprechend geschätzte Osmolarität der ersten
wässrigen
Zusammensetzung in diesem Bereich von Formulierungen war 185.9 bis
446.9 mOsm. Der entsprechende Bereich der Arzneimittel-Beladung
(Tabelle 2) war von 61,7 bis 21,0 mg/ml, wobei die % Ausbeute des
Einkapselungsvorgangens relativ konstant bleibt.
-
Durch
Variieren der Konzentration des aktiven Wirkstoffs und der Konzentration
des osmotischen Exzipienten ergibt die Erfindung MVL Formulierungen
mit einem weiten Bereich an Arzneimittelbeladung für einen
bestimmten aktiven Wirkstoff. In Beispiel 2 zum Beispiel wurde Met-Enkephalin
in MVLs unter Verwendung einer ersten wässrigen Komponente, die 40
oder 5 mg/mL Met-Enkephalin in 20 mM Zitronensäure, und 0, 2,5 oder 5 Gew.-/Vol.-%
Saccharose enthält,
eingekapselt, was einen Osmolaritätsbereich der ersten wässrigen Komponente
von 35,5 bis 191,5 mOsm erzeugt. Die Ergebnisse dieser Studien (Tabellen
2 und 3) zeigen, dass das Verringern der Osmolarität in der
ersten wässrigen
Komponente zu einer proportionalen Zunahme in der Arzneimittel-Beladung
führte,
ob die Menge an aktiven Wirkstoff in der ersten wässrigen
Lösung
40 mg/ml oder 5 mg/ml war. Zusätzlich
wurden MVL Formulierungen, die so wenig wie 6,4 mg/mL oder so viel
wie 61,7 mg/mL Arzneimittel enthielten, unter Verwendung des Verfahrens
der Erfindung erhalten. Diese Ergebnisse illustrieren die breite
Anwendbarkeit des Prinzips, das der beanspruchten Erfindung zugrunde
liegt.
-
Weitere
Studien wurden ausgeführt
unter Verwendung von IGF-1 Konzentrationen in der ersten wässrigen
Komponente im Bereich von 10 bis 80 mg/ml, entweder mit oder ohne
100 mM HCl oder 25 mM Zitronensäure
bei konstantem pH. Es wurde gefunden, dass die Löslichkeit und Bioverfügbarkeit
von eingekapseltem IGF-I entsprechend dem pH der ersten wässrigen
Komponente variiert. Studien haben gezeigt, dass bis zu etwa 300
mg/mL IGF-I ist bei einem pH unter 5 löslich sind. Für alle getesteten
IGF-I Konzentrationen variierte die Arzneimittelbeladung, im pH
Bereich, indem das Arzneimittel löslich ist, entsprechend der
Osmolarität. Für IGF-1
Konzentrationen im Bereich von 40 bis 300 mg/ml war die Löslichkeit
im Bereich von 2 bis 4,8 am größten; während der
nützliche
Löslichkeitsbereich
für IGF-I
Konzentrationen im Bereich von etwa 1 mg/ml bis etwa 33 mg/ml von
etwa 1 bis etwa 5 war.
-
Zusätzlich vergleichen
die hergestellten IGF-I Formulierungen die Wirkungen auf die Arzneimittel-Beladung
beim Ersetzten eines Nichtzuckers (Glycylglycin) durch einen Zucker
(Saccharose) als den osmotischen Exzipienten. In einer Reihe von
Formulierungen wurde das langkettige amphipathische Lipid, das verwendet
wurde, um langsame Freissetzungseigenschaften zu vermitteln, von
DEPC zu DOPC geändert,
ohne signifikante Änderung
des Modulierungstrends der Arzneimittelbeladung durch Einstellen
der Osmolarität
der ersten wässrigen
Komponente. Um zu illustrieren, dass das Verfahren dieser Erfindung
unabhängig
von Variablen, wie der Batch-Größe und den
Mischungsverfahren, das während
des Herstellungsverfahren verwendet wurde, ist, wurden die MVL Formulierungen
in unterschiedlichen Batch-Größen und
mit unterschiedlichen Mischertypen hergestellt. Ein Vergleich der
Ergebnisse dieser Tests in den Tabellen 6A bis 6F zeigte, dass die umgekehrte
Beziehung zwischen Osmolarität
und Arzneimittel-Beladung unabhängig
von dem chemischen Charakter eines beliebigen der osmotischen Exzipienten
in der ersten wässrigen
Komponente ist, und dass, für
eine konstante Arzneimittel-Konzentration, der Trend der erhöhten Arzneimittelbeladung
mit verringerte Osmolarität
konsistent ist, obwohl die unterschiedlichen Batch-Größen und
Mischungsverfahren einen etwas unterschiedlichen Beladungsgrad ergeben
können.
-
Die
folgenden Beispiele illustrieren die Art und Weise, in welcher die
Erfindung ausgeführt
werden kann. Es ist jedoch verständlich;
dass die Beispiele für
Illustrationszwecke sind und die Erfindung sollte nicht als Einschränkung auf
eine beliebige der spezifischen Materialien oder Bedingungen hierin
betrachtet werden.
-
Beispiel 1
-
Herstellung
von Liposomen-Formulierungen
-
In
allen den Verfahren zum Herstellen von MVLs, die hierin dargestellt
sind, wurde in einem ersten Schritte eine „Wasser-in-Öl" Emulsion durch Mischen
einer Lipid-Komponente mit einer ersten wässrigen Komponente hergestellt.
Die Lipidkomponente enthielt 0,5–4 ml 13,20 mM DOPC oder DEPC,
19,88 mM Cholesterol, 2,79 mM DPPG, und 2,44 mM Triolein (Avant
Polar Lipids Inc., Alabaster, AL) in Chloroform (Spectrum Chemical
Manufacturing Corp., Gardena, CA) als Lösungsmittel. Ein gleiches Volumen
(0,5–4
mL) einer ersten wässrigen
Lösung,
die Cytarabin, Leuprolid, Morphin, Enkephalin, oder IGF-I und variierende
Konzentrationen eines osmotischen Exzipienten enthielt, wurde mit
der Lipidkomponente unter Verwendung einer Vielzahl von Mischern
gemischt, um die Wirkung der Osmolarität auf die Arzneimittelbeladung
und die prozentuelle Ausbeute der verschiedenen getesteten Kombinationen
zu bestimmen.
-
Herstellung von Cytarabin-enthaltenden
MVL
-
Für Cytarabin
enthielt die Lipidkomponente DEPC, anstelle von DOPC, und vier unterschiedliche
erste wässrige
Lösungen
wurden hergestellt, wobei jede 40 mg/mL Cytarabin (Upjohn Co., Kalamazoo,
MI) in 20 mM Zitronensäure
(Sigma Chemical) und 0, 2,5 oder 8 Gew.-/Vol.-% Saccharose als den
osmotischen Exzipienten-Wirkstoff enthielt. Eine Emulsion des Lipids
und der ersten wässrigen
Komponenten wurde durch Mischen von 0,5 mL der ersten wässrigen
Komponente mit 0,5 ml der Lipidkomponente mittels eines Baxter® Vortexers
bei der maximalen Geschwindigkeit (Einstellung 10) für 6 Minuten
gebildet. Zu der resultierenden ersten Emulsion, wurden 2,5 mL einer
Lösung,
die 4 Gew.-% Glucose bzw. 40 mM Lysin (Spektrum Chemicals) enthielt,
hinzugefügt.
Die resultierende Mischung wurde emulgiert, um eine zweite Emulsion
mit dem Baxter-Vortexer bei der maximalen Geschwindigkeit (Einstellung
10) für
4 Sekunden zu bilden. Die resultierende zweite Emulsion, eine „Wasser-in-Öl-in-Wasser"-Doppelemulsion,
wurde für
die sanfte Verwirbelung in einen 250 ml Erlenmeyer-Kolben, der 10 mL
einer Lösung
von 4 Gewichtsprozent Glukose und 40 mM Lysin enthält, überführt.
-
Um
das organische Lösungsmittel
(Chloroform) von den Partikeln abzudampfen, wurde Stickstoffgas über die
zweite Emulsion bei 37°C
für 20
Minuten unter leichtem Schütteln
geleitet. Die resultierenden multivesikulären Liposomen wurde zweimal
mit 50 mL normaler Salzlösung
durch Zentrifugation bei 600 × g
in einer Tischzentrifuge gewaschen und dann in 0,5–4 ml normaler
Salzlösung
resuspendiert. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 2 unten gezeigt.
-
-
Diese
Ergebnisse zeigen, dass die Arzneimittelbeladung durch Variieren
der Osmolarität
der ersten wässrigen
Lösung
moduliert werden kann, wobei eine verringerte Osmolarität zu einer
erhöhten
Arzneimittelbeladung führt.
Die Zunahme der Arzneimittelbeladung, die durch Verringern der Osmolarität erreicht
wird, führte
zu keiner signifikanten Variation in der prozentuellen Ausbeute
in der MVL Formulierung.
-
Beispiel 2
-
Herstellung von Met-Enkephalin-enthaltenden
multivesikulären
Liposomen-Formulierungen
-
Eine
Lipidkomponente, die DEPC anstelle von DOPC enthielt, wurde wie
in Beispiel 1 hergestellt. Die erste wässrige Komponente enthielt
5 mg/ml Met-Enkephalin (ein Pentapeptid) (Sigma Chemical Co., St.
Louis, MO) in 25 mM Zitronensäure,
und 0, 2,5 oder 5,0 Gew.-/Vol.% Saccharose als osmotischen Exzipienten. Der
Rest der in Beispiel 1 beschriebenen Schritte wurden ausgeführt, um
Met-Enkephalin enthaltende MVLs, die in normaler Salzlösung suspendiert
sind, zu erhalten. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 3 unten gezeigt.
-
-
Die
Daten in Tabelle 3 zeigen wiederum, dass die Beladung von Met-Enkephalin
durch Variieren der Osmolarität
der ersten wässrigen
Lösung
moduliert wird, wobei das Verringern der Osmolarität zu einer
erhöhten
Arzneimittelbeladung führt.
Folglich ist die Wirkung unabhängig
von der Arzneimittelbeladung. Die prozentuelle Ausbeute ist durch
die Abnahme der Osmolarität
nicht signifikant verändert.
Die Wirkung auf die Arzneimittelbeladung, die durch Variieren der
Osmolarität
der ersten wässrigen
Komponente erhalten wird, wird ebenfalls festgestellt, wenn DEPC
durch DOPC in der Lipidkomponente während der Herstellung ersetzt
ist.
-
Beispiel 3
-
Herstellung von Leuprolid-enthaltenden
multivesikulären
Liposomen-Formulierungen
-
Eine
Lipidkomponente, die DOPC anstelle von DEPC enthielt, wurde wie
in Beispiel 1 hergestellt, mit Ausnahme, dass es Leuprolid und eine
3-fach höhere
molare Konzentration von allen vier Lipiden in der Lipidkomponente
enthielt. Die erste wässrige
Komponente enthielt 15 mg/ml Leuprolidacetat (Bachem Bioscience Inc.,
King of Prussia, PA) in 100 mM Phosphorsäure, und 4,0 oder 6,0 Gew.-/Vol.-%
Saccharose als osmotischen Exzipienten. Die Verfahren von Beispiel
1 wurden nachgearbeitet, um die Leuprolid-enthaltenden MVLs zu erhalten,
mit Ausnahme, dass 4 ml der ersten wässrigen Komponente mit 4 mL
der Lipidkomponente unter Verwendung eines TK Autohomogenisators
K bei einer Geschwindigkeit von 9000 U/min für 8 Minuten gemischt wurden,
um die erste Emulsion zu erhalten. Zu der ersten Emulsion wurden
16 mL einer Lösung,
die 4 Gew.-% bzw. 40 mM Lysin (Spekturm Chemicals) enthielt, hinzugefügt. Die
resultierende Mischung wurde emulgiert, um eine zweite Emulsion
mit dem TK Autohomogenisator K bei einer Geschwindigkeit von 4000 U/min
für 1 Minute
zu bilden. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 4 unten gezeigt.
-
-
Wie
in den Beispielen 1 und 2 oben, wird die Beladung von Leuprolid,
einem 9-Aminosäure-Peptid, durch Variieren
der Osmolarität
der ersten wässrigen
Lösung
moduliert, wobei eine Verringerung der Osmolarität zu einer erhöhten Arzneimittelbeladung
führt.
Eine ähnliche
prozentuelle Ausbeute wurde über
den getesteten Osmolaritätsbereich
aufrechterhalten. Es wurde auch gezeigt, dass dieses Ergebnis unabhängig von
der Mixerart ist, die verwendet wurde, um die erste und zweite Emulsion
bei der Herstellung der MVLs zu erzeugen.
-
Beispiel 4
-
Herstellung von Morphin-enthaltenden
multivesikulären
Liposomen-Formulierungen
-
Eine
Lipidkomponente, die DEPC anstelle von DOPC enthielten, wurde wie
in Beispiel 1 hergestellt. Die erste wässrige Komponente enthielt
17 mg/ml Morphinsulfat (Mallinckrodt Chemical Inc., St. Louis, MO)
in 10 mM Salzsäure,
und 0,2, 2,5 oder 5,0 Gew.-(Vol.-% Saccharose als osmotischen Exzipienten.
Die restlichten in Beispiel 1 beschriebenen Schritte wurden ausgeführt, um
Morphinsulphat enthaltende MVLs zu erhalten, die in normaler Salzlösung suspendiert
sind. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 5 unten gezeigt.
-
-
Wiederum
wurde die Beladung von Morphin, einem Lipid-löslichen Arzneimittel, durch
Variieren der Osmolarität
der ersten wässrigen
Lösung
moduliert, wobei eine Verringerung der Osmolarität zu einer erhöhten Arzneimittelbeladung
führte.
Die prozentuelle Ausbeute der MVL Formulierung war im Wesentlichen über den
getesteten Osmolaritätsbereich äquivalent.
-
Beispiel 5
-
Herstellung von IGF-I-enthaltenden
multivesikulären
Liposomen-Formulierungen
-
1. Herstellung von MVLs
in einem 0,5 ml Maßstab
-
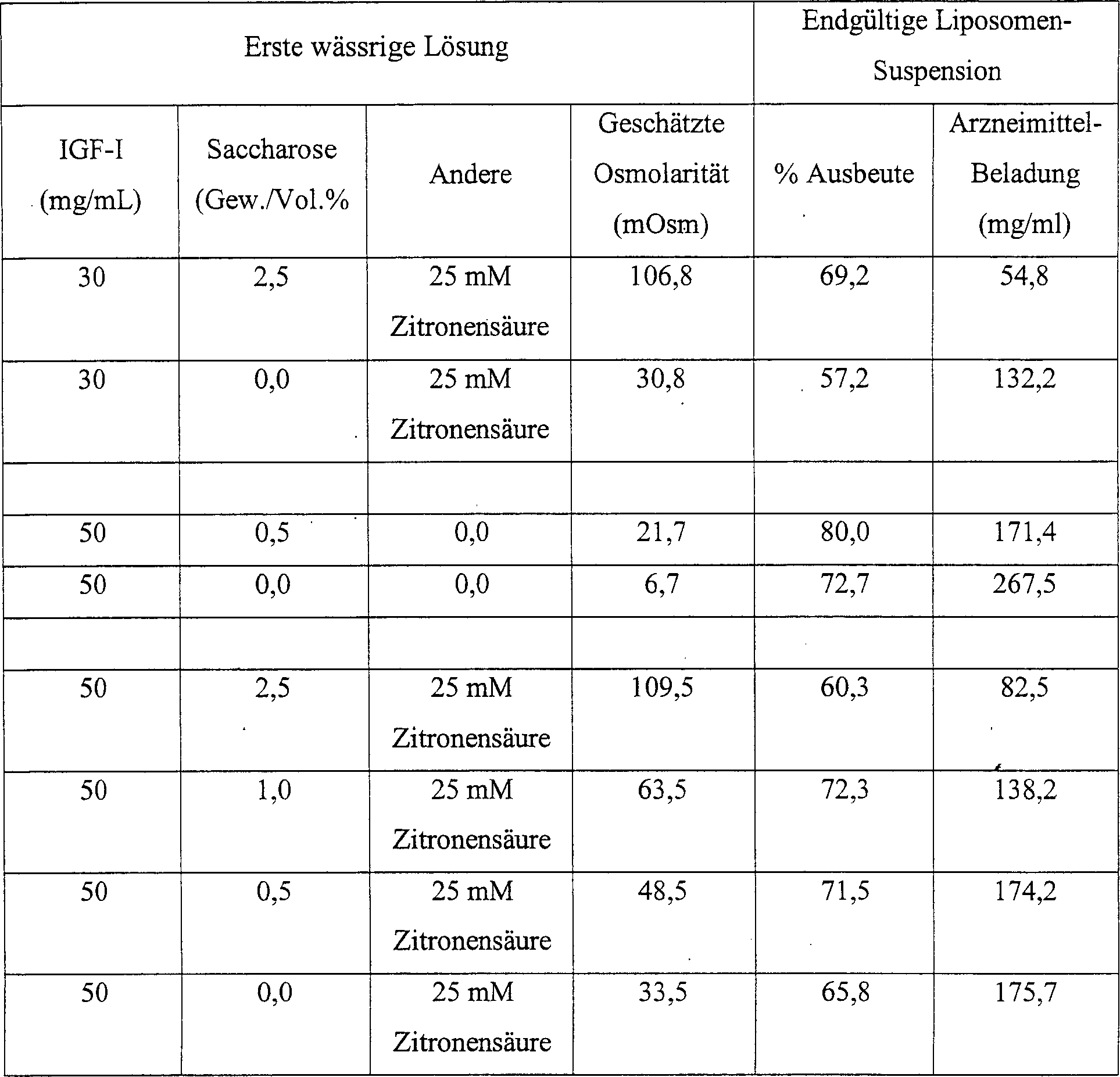
Eine
Lipidkomponente, die DEPC anstelle von DOPC enthielt, wurde wie
in Beispiel 1 hergestellt. Die erste wässrige Komponente enthielt
50 mg/ml IGF-I und 0,25, 0,5, 1,0, 2,5, oder 5,0 Gew./Vol.-% Saccharose als
osmotischen Exzipienten. Die restlichen der in Beispiel 1 beschriebenen
Schritte wurden ausgeführt,
um IGF-I enthaltende MVLs zu erhalten, die in normaler Salzlösung suspendiert
sind. Die geschätzte
Osmolarität (mOsm),
prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 6A unten gezeigt.
-
-
Obwohl
die Beladung von IGF-1 mit den Ergebnissen konsistent ist, die erhalten
wurden, wenn Zitronensäure
als der Puffer verwendet wurde, zeigte IGF-I dann einen gewissen
Abbau in den Studien, die durchgeführt wurden, um das eingekapselte
Protein zu charakterisieren.
-
2. Herstellung von MVLs
in einem 4 ml Maßstab
-
Eine
Lipidkomponente, die DOPC anstelle von DEPC enthielt, wurde wie
in Beispiel 1 hergestellt. Ein erster Satz von Formulierungen verwendete
eine erste wässrige
Komponente, die 20 mg/ml IGF-I (Chiron Corp., Emeryville, CA) in
100 mM Salzsäure
und 2,5, oder 5,0 Gew./Vol.-% Saccharose als osmotischen Exzipienten
enthielt. Ein zweiter Salz verwendete 50 mg/ml IGF-1 in 100 mM Salzsäure und
0 oder 2,5 Gew./Vol.-% Saccharose als osmotischen Exzipienten. Die
Verfahren von Beispiel 1 wurden nachgearbeitet, um die IGF-I enthaltenden
MVLs zu erhalten, mit Ausnahme, dass 4 ml der ersten wässrigen
Komponente mit 4 ml der Lipidkomponente unter Verwendung eines TK
Autohomogenisators K bei einer Geschwindigkeit von 9000 U/min für 8 Minuten
gemischt wurden, um die erste Emulsion zu erhalten. Zu der ersten
Emulsion wurden 16 ml einer Lösung
hinzugefügt,
die 4 Gew.-% Glucose bzw. 40 mM Lysin (Spectrum Chemicals) enthielt.
Die resultierende Mischung wurde emulgiert, um eine zweite Emulsion
mit dem TK Autohomogenisator K bei einer Geschwindigkeit von 4000
U/min für
1 Minute zu bilden. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
unter Verwendung eines Vortex-Mischers und DEPC, einem Lipid mit
einer 22 Kohlenstoffkette, sind in Tabelle 6B unten gezeigt:
-
-
Tabelle
6B zeigt, dass die Beladung, die mit dem TK Mischerverfahren erhalten
wird, ähnlich
zu jenen ist, die erhalten wurde, wenn ein Vortex-Mischer verwendet
wird, um die Emulsionen herzustellen. Die Studien, die durchgeführt wurden,
um das eingekapselte Protein zu charakterisieren, zeigten jedoch
ein verstärktes Vorhandensein
von IGF-I Oligomeren in der Abwesenheit eines Säure-Puffers.
-
3. Herstellung von MVLs
in einem 3 ml Maßstab
-
Eine
Lipidkomponente, die DEPC anstelle von DOPC enthielt, wurde wie
in Beispiel 1 hergestellt. Die erste wässrige Komponente enthielt
eine der drei Formulierungen: (1) 30 mg/ml IGF-I in 25 mM Zitronensäure, und
0 oder 2,5 Gew./Vol.-% Saccharose als osmotischen Exzipienten, (2)
oder 50 mg/mL IGF-I in 25 mM Zitronensäure, und 2,5, 1,0, 0,5 oder
0 Gew./Vol.-% Saccharose, (3) oder 50 mg/ml IGF-I ohne Zitronensäure, und
0 oder 0,5 Gew./Vol.-% Saccharose. Die Verfahren von Beispiel 1
wurden nachgearbeitet, um die MVLs, die IGF-I enthalten, zu erhalten,
mit der Ausnahme, dass 3 mL der ersten wässrigen Komponente mit 3 ml
der Lipidkomponente unter Verwendung eines Omni Mixers ES bei einer
Geschwindigkeit von 10.000 U/min für 12 Minuten gemischt wurden,
um die erste Emulsion zu erhalten. Zu der ersten Emulsion wurden
20 ml einer Lösung
hinzugefügt,
die 4 Gew.-% Glucose bzw. 40 mM Lysin (Spectrum Chemicals) enthielt.
Die resultierende Mischung wurde emulgiert, um eine zweite Emulsion
mit dem Omni Mixer ES bei einer Geschwindigkeit von 4500 U/min für 2 Minute
zu bilden. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 6C unten gezeigt:
-
-
Die
Ergebnisse in Tabelle 6C zeigen eine ähnliche Modulierung der Arzneimittelbeladung
durch die Osmolarität
für jede
der getesteten Arzneimittelkonzentrationen.
-
Beispiel 6
-
Herstellung von eingekapselten
IGF-I in einem 125 ml Maßstab
-
Eine
Standard Lipidkomponente, die DEPC anstelle von DOPC enthielt, wurde
wie für
andere Arzneimittel-Formulierungen hergestellt. Die erste wässrige Komponente
enthielt 15 mg/ml IGF-I, das entweder in einer 5% Saccharose/20
mM Ammoniumcitrat-Lösung,
oder in einer 8% Saccharose/20 mM Ammoniumcitrat-Lösung aufgelöst ist.
125 ml der ersten wässrigen
Lösung wurden
mit 125 ml der Lipidkomponente unter Verwendung eines hochscherenden
Doppelmisch-Gefäßsystems
gemischt, um die erste Emulsion zu erhalten. Dieses Mischsystem
modelliert den Herstellungsmaßstabsprozess,
und wird für
die Maßstabsvergrößerung der
eingekapselten Arzneimittel-Formulierungen verwendet. Die wässrigen
und organischen Komponenten wurden bei einer Geschwindigkeit von
8000 U/min für
30 Minuten in dem ersten Emulsionsgefäß gemischt. Die erste Emulsion
wurde dann bei einer Rate von 167 ml/min in einen Fluidstrom, der
aus 0,04 N Ammoniumhydroxid in 1,5% Glycidlösung besteht, der bei 2400
ml/min fließt,
gepumpt und mittels eines in-line statischen Mixers gemischt, um
die zweite Emulsion zu erhalten. Die Gesamtflussrate durch den statischen Mischer
war 2567 ml/min. Bei dieser Rate war die erste Emulsion nach 90
Sekunden aufgebraucht. Die zweite Emulsion wurde bei Eintritt in
ein Aufnahmegefäß mit einer
Lysin-Lösung
gemischt, und dann unmittelbar mit Stickstoff gespült, um das
organische Lösungsmittel
abzuziehen. Die geschätzte
Osmolarität
(mOsm), prozentuelle Ausbeute, Arzneimittel-Beladung, und % freies
Arzneimittel für
diese Formulierungen sind in Tabelle 7 unten gezeigt.
-
-
Die
Ergebnisse in Tabelle 7 oben zeigen eine ähnliche Zunahme der Arzneimittelbeladung
bei verringerte Saccharosekonzentration, wie für die anderen untersuchten
Arzneimittelformulierungen.
-
Beispiel 7
-
Wirkung des
Austausches von Glycylglycin als den osmotischen Exzipienten
-
Eine
Lipidkomponente, die DEPC anstelle von DOPC enthielt, wurde wie
in Beispiel 1 hergestellt. Die erste wässrige Komponente enthielt
10 mg/ml IGF-I in 25 mM Zitronensäure, und 0, 1,0 oder 2,0 Gew./Vol.-% Glycyclglycin
als osmotischen Exzipienten. Die restlichen der in Beispiel 1 beschriebenen
Schritte wurden ausgeführt,
um IGF-I enthaltende MVLs zu erhalten, die in normaler Salzlösung suspendiert
sind. Die geschätzte Osmolarität (mOsm),
prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 6 D unten gezeigt.
-
-
Eine ähnliche
osmotische Modulierung der Arzneimittelbeladung ist für Formulierungen
unter Verwendung eines Nichtzucker osmotischen Spacers, Glycylglycin,
anstelle von Saccharose gezeigt. Folglich ist die Wirkung der Osmolarität auf die
Arzneimittel-Beladung, gezeigt durch die Daten in der Tabelle 6D,
unabhängig von
der chemischen Struktur des verwendeten osmotischen Exzipienten.
-
Vergleich
der unterschiedlichen osmotischen Exzipienten bei gleicher osmotischer
Stärke
MVLs wurden mit dem Verfahren von Beispiel 1 hergestellt, die IGF-I,
die entweder mit 2,5 Gew./Vol.-% Saccharose oder 1,0 Gew./Vol.-%
Glycylglycin als osmotischen Exzipienten bei ungefähr gleicher
osmotischen Stärke
eingekapselt sind, enthielten. Für
den Vergleich wurde 2,5 Gew./Vol.-% Saccharose oder 1,0 Gew./Vol.-%
Glycylglycin als der osmotische Exzipient in die erste wässrige Komponente
eingeführt,
die 80 mg/ml IGF-I und 25 mM Zitronensäure enthielt.
-
Die
Verfahren von Beispiel 1 wurden nachgearbeitet, um die IGF-I enthaltenden
MVLs zu erhalten, mit Ausnahme, dass 3 ml der ersten wässrigen
Komponente mit 3 mL der Lipidkomponente unter Verwendung eines Omni
Mixers ES bei einer Geschwindigkeit von 10.000 U/min für 20 Minuten
gemischt wurden, um die erste Emulsion zu erhalten. Zu der ersten
Emulsion wurden 20 mL einer Lösung,
die 4 Gew.-% bzw. 40 mM Lysin (Spekturm Chemicals) enthielt, hinzugefügt. Die
resultierende Mischung wurde emulgiert, um einen zweite Emulsion
mit dem Omni Mixer ES bei einer Geschwindigkeit von 4500 U/min für 2 Minute
zu bilden.
-
Um
zu bestimmen, ob die Wirkung auf die Arzneimittel-Beladung nur der
Osmolarität
der ersten wässrigen
Komponente zuzuschreiben ist, wurde einen dritte Formulierung, wie
oben beschrieben, hergestellt, mit Ausnahme, dass sowohl die Konzentration
des osmotischen Exzipienten als auch die des IGF-1s proportional verringert
wurden (von 80 mg/ml IGF-I und 2,5% Saccharose auf 50 mg/ml IGF-I
und 1,0% Saccharose). In dieser Formulierung ersetzte die zweite
wässrige
Komponente 1,5 % Glycin und 40 mM Lysin anstelle von 4% Glucose
und 40 mM Lysin, die in Beispiel 1 verwendet wurden. Tabelle 6E
unten vergleicht die geschätzte
Osmolarität,
% Ausbeute, und Arzneimittelbeladung in der endgültigen Liposomen-Suspension
für diese
drei Formulierungen.
-
Ein
Vergleich der geschätzten
Osmolarität
(mOsm), prozentuelle Ausbeute, und Arzneimittelbeladung dieser Formulierungen
sind in Tabelle 6E unten gezeigt:
-
-
Die
Daten in Tabelle 6E zeigt, dass die Osmolarität des osmotischen Exzipienten
das Ergebnis einer effektiven Variable ist, da zwei unterschiedliche
osmotische Spacer bei etwa gleicher osmotischer Stärke, jedoch
ungleicher molarer Konzentration, eine vergleichbare Wirkung auf
die Arzneimittelbeladung hervorrufen.
-
Beispiel 8
-
Bestimmung der prozentuellen
Einkapselung (oder prozentuellen Ausbeute), Lipokrit, prozentuell
freies Arzneimittel, Partikelgrößen-Verteilung
und Arzneimittelbeladung
-
Tabellen
2 bis 6A–E
zeigen die geschätzte
Osmolarität
(mOsm), % Ausbeute und Arzneimittelbeladung (mg/ml) für die liposomalen
Formulierungen, die in den Beispiel 1–6 oben beschrieben sind. Diese
Parameter wurden wie folgt erhalten:
Die prozentuelle Einkapselung
(oder prozentuelle Ausbeute) des Arzneimittels wurde als der Prozentsatz
der Arzneimittelmenge in der endgültigen Liposomen-Suspension
zu der gesamten Arzneimittelmenge berechnet, die in der ersten wässrigen
Lösung
verwendet wurde. Folglich wurde die prozentuelle Ausbeute des Arzneimittels
als das Verhältnis
der Arzneimittelkonzentration in der endgültigen Suspension mal dem Volumen
der endgültigen
Suspension zu der Arzneimittelkonzentration in der ersten wässrigen
Lösung
mal dem Volumen der ersten wässrigen
Lösung
berechnet. Lipokrit wurde in Analogie zu Hämatokrit berechnet, und zwar
als prozentuelles Verhältnis
des Pelletvolumens zu dem Suspensionsvolumen (siehe Bedingungen
unten, für
das Erhalten des Pelletvolumens).
-
Prozentuell
freies Arzneimittel wurde als das prozentuelle Verhältnis der
Menge des Arzneimittels im Überstand
zu der Arzneimittelmenge in der Endsuspension berechnet. Das prozentuell
freie Arzneimittel kann auch als das prozentuelle Verhältnis der
Arzneimittelkonzentration im Überstand
zu der in der Suspension, mal (1-Lipokirt) berechnet werden. Die
Arzneimittelbeladung, die die Menge an Arzneimittel misst, die in
jeder Einheit des eingekapselten Volumens eingekapselt ist, ist
etwa gleich zu und kann geschätzt
werden (unter der Annahme eines geringen Prozentsatzes an freiem
Arzneimittel) als das Verhältnis
der Arzneimittelkonzentration der endgültigen Liposomensuspension
zu dem Lipokrit. Diese Variablen wurden, wie unten spezifischer beschrieben
ist, bestimmt.
-
Um
den Lipokrit zu bestimmen, wurde etwa 50 μL der multivesikulären Liposomensuspension
in einem Kapillarenröhren
aufgenommen, und das Ende des Röhrchens
wurde versiegelt, während
sichergestellt wurde, dass die Suspension keine Luftblasen enthielt.
Die Suspension wurde in einer Zentrifuge bei 600 × g für 10 Minuten
zentrifugiert, um eine Pelletschicht und eine Überstandsschicht zu erhalten.
Das prozentuelle Verhältnis
der Länge
des Röhrchens,
die durch das Pellet eingenommen wird, zu jener, die durch die Suspension eingenommen
wird, ergibt den Lipokrit.
-
Für die Verwendung
zum Bestimmen der Menge an freiem Arzneimittel in einer Formulierung
wurde Überstand
durch Zentrifugieren von etwa 0,2 mL Suspension für 3 Minuten
bei 600 × g
in einem Eppendorf® Zentrifugenröhrchen erhalten.
Für Cytarabin-
und Morphin-Formulierungen wurde 25–50 μL des Überstandes abgezogen und in
ein Glasröhrchen,
das 1 ml 3:1 Vol./Vol. Isopropanylalkohol:1 N Salzsäure (Fisher
Chemical, Fair Lawn, NJ) pipettiert und kräftig gemischt, um eine klare
Lösung
zu erhalten. Die Absorbanz bei 280 nm für Cytarabin, oder bei 285 nm
für Morphin
wurde mit einem Spektrophotometer (Hitachi® U-2000)
gemessen. Für
Leuprolid-Formulierungen wurden 50 μl des Überstandes abgezogen und in
ein Glasröhrchen,
das 2 ml 1:1 Isopropylalkohol:Wasser, titriert auf pH 10 mittels
0,1 N Ammoniumhydroxid, enthält,
pipettiert, gefolgt von kräftigem
Mischen, um eine klare Lösung
zu erhalten. Die Absorbanz bei 280 nm wurde dann mit einem Spektrophotometer
(Hitachi® U-2000)
gemessen. Für
Enkephalin und IGF-I wurden 25–50 μL des Überstandes
abgezogen und in ein Glassröhrchen,
das 1 ml 3:1 Vol./Vol. Isopropylalkohl:2 N Zitronensäure (Sigma
Chemical) enthält,
pipettiert, gefolgt von kräftigem
Mischen, um eine klare Lösung
zu erhalten. Die Absorbanz bei 275 nm für Enkephalin und IGF-I wurde
mit einem Spektrophotometer (Hitachi® U-2000)
gemessen. Unter Verwendung eines Referenzabsoranzstandards, der
basierend auf den Arzneimittellösungen
von bekannten Konzentrationen in derselben Auflösungslösung etabliert wurde, wurden
die Arzneimittelkonzentrationen in der Suspension und dem Überstand
berechnet.
-
Die
Partikelgrößenverteilung
und der mittlere Partikeldurchmesser wurden durch das Verfahren
der Laserlicht-Diffraktion mittels eines LA-500 oder LA-910 Partikelgrößen-Analysators
von Horiba Inc. (Irvine, CA) bestimmt. Der Volumen-gewichtete mittlere
Partikeldurchmesser für
alle untersuchten Formulierungen war im Allgemeinen im Bereich von
etwa 6 bis 18 μm.
-
Beispiel 9
-
In vitro und In vivo Freisetzung
von hoch Arzneimittel-beladenen, hochprozentige Ausbeute IGF-I Formulierungen
-
Die
physikochemische Integrität
des eingekapselten IGF-I wurde durch SDS-PAGE Untersuchung mittels
Novex® NuPage-Gels
sowie durch RP-HPLC Untersuchung unter Verwendung einer C18 symmetrischen Säule bestätigt. Das
eingekapselte Protein wurde mit 75:25 IPA:2N Zitronensäure extrahiert.
Die Bioaktivität des
eingekapselten IGF-I wurde durch eine mitogene Biountersuchung unter
Verwendung einer MG-63 humanen Osteosarcoma Zelllinie und 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid
(MTT) Farbstoff gemäß dem Verfahren
von W. Lopaczynski et al., Regulatory Peptides, 48: 207–216, 1993
bestätigt.
Die MG-63 Zellen wurden von der American Type Culture Collection
(ATCC# CRL 1427) erhalten, und die Dosis-abhängige mitogene Reaktion von
ruhenden MG-63 Zellen zu hinzugefügtem IGF-I wurde bestimmt.
Die Bioaktivität
von extrahiertem IGF-I wurde als etwa äquivalent zu jener eines nicht
eingekapselten Standards bestätigt.
-
In
vitro Experiment: In Kürze,
ein in vitro Freisetzungsexperiment wurde aufgebaut und wie folgt
durchgeführt:
eine MVL Suspension, die etwa 50 mg/ml IGF-I enthielt, wurde 20
fach in humanes Plasma, das 0,01% NaN3 enthielt,
verdünnt;
eine 0,5 ml Probe in einen Eppendorf-Röhrchen
mit Schraubverschluss wurde für
jeden Zeitpunkt verwendet, und die Proben wurden unter dynamischen/Drehbedingungen
bei 37°C
inkubiert. Zeitpunkt-Proben wurden zu verschiedenen Zeitpunkten
genommen und mit 0,9 ml normaler Salzlösung gewaschen.
-
Partikelpellets
wurden dann durch Zentrifugation in einer Mikrofuge bei 16.000 g
für 4 Minuten
erhalten und bei –20°C bis zur
RP-HPLC Analyse unter Verwendung einer C18 symmetrischen Säule gelagert. 1 zeigt
einige in vitro Plasma-Freisetzungsdaten, die für die drei in Tabelle 6E aufgelisteten,
repräsentativen IGF-I
Formulierungen erhalten wurden. Diese Daten weisen daraufhin, dass
eine verzögerte
Freisetzung von IGF-I über
eine Periode von mehreren Tagen für hoch Arzneimittel-beladene,
hochprozentige Ausbeute IGF-I Formulierungen erreicht wird.
-
In
vivo Experiment: Männliche
Ratten wurden subkutan die drei, in Tabelle 6E gezeigten, MVL-Formulierungen injiziert,
um Informationen über
die in vivo Freisetzungseigenschaften zu erhalten. Jede Ratte erhielt eine
10 mg Dosis IGF-I, und jede der untersuchten Formulierungen wurde
in 3 Ratten injiziert. Blutproben (0,2 ml) wurde zu folgenden Zeitpunkten
gesammelt: Null, und 1, 3, 5, und 7 Tag(e) nach der Injektion und
zwar von der Schwanzvene der Ratten und ermöglicht zu gerinnen. Serum wurde
dann durch Zentrifugation erhalten, und bei –70°C vor der Untersuchung auf die
IGF-I Konzentration mittels eines IGF-I ELISA Satzes DSL-10-5600
(Diagnostic Systems Laborstories, Inc., Webster, TX gemäß den Herstellerangaben)
gelagert.
-
2 zeigt
den zeitlichen Verlauf der durchschnittlichen Serum IGF-I Konzentration
von drei Ratten, die die 10 mg IGF-I der Formulierung A in Tabelle
6E erhielten. Diese Daten weisen darauf hin, dass ein anhaltender
Serumspiegel von IGF-I über
eine Periode von vielen Tagen unter Verwendung von hoch Arzneimittel-beladenen,
hochprozentigen Ausbeute IGF-I Formulierungen erreicht werden kann.