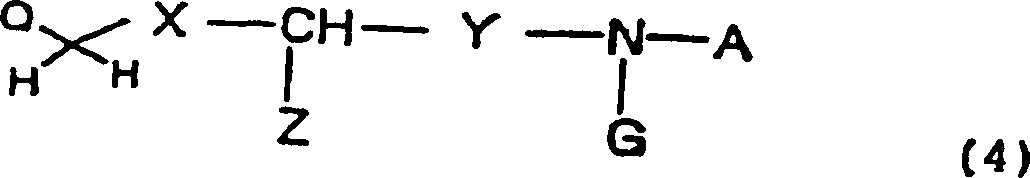-
TECHNISCHES
GEBIET
-
Die
vorliegende Erfindung betrifft neue Naphthyridin-Verbindungen, die
in der Lage sind Antagonismus für
Tachykinin-Rezeptoren, insbesondere Antagonismus für Neurokinin-A-Rezeptoren
(NK2-Rezeptoren) zu zeigen, und deren Salze, Hydrate oder Solvate
sowie pharmazeutische Zusammensetzungen, enthaltend diese Verbindungen.
Die Verbindungen gemäß der vorliegenden
Erfindung sind als prophylaktische oder therapierende Mittel bei
Krankheiten, für
die die Tachykinin-Rezeptoren verantwortlich gemacht werden, wie
Asthma, Bronchitis, Pollakisurie, Harninkontinenz und Colitis geeignet.
-
STAND DER
TECHNIK
-
Der
Begriff „Tachykinine" ist eine allgemeine
Bezeichnung für
eine Gruppe von Peptiden mit einer ähnlichen Struktur, welche Neuropeptide
und im Nervensystem weit verbreitet sind. Tachykinine sind bei der
Geruchswahrnehmung, beim Sehen, beim Hören, bei der Steuerung von
Bewegungen, der Bewegung des Magens und der Kontrolle des Speichelflusses
beteiligt. Zusätzlich
wurde vor kurzem bekannt, dass Tachykinine noch verschiedenste weitere
physiologische Wirkungen wie die Kontraktion der glatten Muskeln
der Atemwege, die Kontraktion der glatten Muskeln der Blase, die
Kontraktion der glatten Muskeln des Darmes, die Auslösung von Überreaktionen
der Atemwege, den Anstieg der Gefäßdurchlässigkeit, die Auslösung von
Husten, die Zufügung
von Schmerzen, das erhöhte
Abscheiden von Schleim, die Auslösung
von Ödemen,
die Erweiterung von Gefäßen, die
Auslösung
von Erbrechen, die Beschleunigung der Ausscheidung von Harn, die
Auslösung
von Angstsymptomen, die Aktivierung von Makrophagen, die Aktivierung
von Mastzellen, etc. zeigen. Aus diesem Grund wurde die Möglichkeit
vorgeschlagen, dass Antagonisten für Tachykinine als therapeutische
Mittel bei Krankheiten, deren krankhafter Zustand eng mit den oben genannten
Wirkungen verknüpft
ist, wie Asthma, Bronchitis, Lungenentzündung, chronisch obstruktive
Lungenerkrankung, Pollakisurie, Harninkontinenz, Colitis, Diabetes,
Zentralkrankheiten, verschiedenste Schmerzen, allergische Krankheiten,
Rheumatoidarthritis, Knochen- und Gelenksentzündungen, verschiedenste Entzündungen,
etc. wirksam werden. Als typische Tachykinine aus Säugetieren
existieren Substanz P, Neurokinin A und Neurokinin B. Es existieren außerdem N-terminale
Erweiterungssubtypen von Neurokinin A.
-
Für diese
drei Haupttachykinine sind mindestens drei Rezeptoren bekannt. Entsprechend
den relativen selektiven Eigenschaften dieser Rezeptoren mit einer
Affinität
für Substanz
P, Neurokinin A oder Neurokinin B werden die Rezeptoren als Neurokinin-1
(NK-1)-Rezeptor, Neurokinin-2 (NK-2)-Rezeptor und Neurokinin-3 (NK-3)-Rezeptor
klassifiziert. Durch diese Rezeptoren weisen Tachykinine verschiedenste
physiologische Wirkungen auf. Es ist bekannt, dass NK2-Rezeptoren
an der Luftstrom-Begrenzung bei Asthma beteiligt sind [Bertrand,
C. et al., Am. J. Physiol. 265, L507-L511 (1993); Perretti, F. et
al., Eur. J. Pharmacol. 273. 129-135 (1995)]. Zusätzlich ist
bekannt, dass NK-2-Rezeptoren auch an Überreaktionen der Atemwege
bei Asthma beteiligt sind und dass Antagonisten für NK-2-Rezeptoren die Überreaktionen
fast vollständig
hemmen [Biochot, E. et al., Br. J. Pharmacol. 114, 259-261 (1995)].
Es ist auch bekannt, dass die Antagonisten bei NK-2-Rezeptoren die
Freisetzung eines chemischen Mediators der Lungen durch Antigen-Belastung
hemmen [Ciabattoni, G. et al., Pharmacodyn. Ther. 328, 357-358 (1994)]
und auch die Ödeme
der Atemwege bei Asthma unterdrücken
[Tousignant C., et al., Br. J. Pharmacol. 108, 383-386 (1993)].
Weiter wurde durch klinische Experimente herausgefunden, dass Antagonisten
für NK-1 – und NK-2-Rezeptoren vor Bronchokonstriktion, hervorgerufen
durch Bradykinin, bei asthmatischen Patienten schützen [Ichinose,
M, et al., Lancet 340, 1248-1251
(1992)].
-
Demzufolge
ist bekannt, dass die Antagonisten für NK-2-Rezeptoren als prophylaktische
oder therapeutische Mittel gegen Asthma geeignet sind. Es ist auch
bekannt, dass die Antagonisten für
NK-2-Rezeptoren und Antagonisten für NK-1-Rezeptoren auch als
Hustenmittel in Fall von Bronchitis, etc geeignet sind [Advenier,
C. et al., Eur. J. Pharmacol. 250, 169-171 (1993); Yasumitsu, R.
et al. Eur. J. Pharmacol. 300, 215-219 (1996)]. Die Antagonisten
für NK-2-Rezeptoren
werden als geeignete prophylaktische oder therapeutische Mittel
gegen Pollakisurie und Harninkontinenz betrachtet [Croci, T. et
al., J. Pharm. Pharmacol. 46, 383-385 (1994); Palea S., et al.,
J. Pharm. Exp. Ther. 277, 700-705 (1996)]. Zusätzlich werden sie als viel
versprechende prophylaktische oder therapeutische Mittel gegen Colitis
betrachtet [Maggi, C.A. et al., Drugs of the Future 18, 155-158
(1993)]. Es ist bekannt, dass NK-2-Rezeptoren bei verschiedensten Schmerzen
[Santucci, V. et al., Eur. J. Pharmacol. 237, 143-146 (1993); Wiesenfeld-Hallin,
Z. et al., Eur. J. Pharmacol. 251, 99-102 (1994)] und Entzündungen
[Lam F.Y. et al., Br. J. Pharmacol. 118, 2107, 2114 (1996)] beteiligt
sind und dass Antagonisten für
NK-2-Rezeptoren die Schmerzen und Entzündungen unterdrücken. Es
ist bekannt, dass NK-2-Rezeptoren auch bei Zentralkrankheiten wie
Angstzuständen
beteiligt sind [S.C. Stratton et al., Br. J. Pharmacol. 112 (supplement)
49p (1993)]. Ferner ist bekannt, dass die Antagonisten für NK-1-Rezeptoren
experimentelles Erbrechen, hervorgerufen durch chemotherapeutische
Medikamente wie zum Beispiel Cisplatin. Schmerzmittel wie Morphin
oder Röntgenbestrahlung,
etc. [Bountra, C. et al., Eur. J. Pharmacol. 249, R3-R4 (1993),
Tatterall, F. D. et al., Eur. J. Pharmacol. 250, R5-R6 (1993)] deutlich
unterdrücken.
-
Verbindungen,
die Antagonisten zu Tachykininen an Tachykinin-Rezeptoren des oben beschriebenen Typs
sind, sind bekannt. Zum Beispiel offenbart die ungeprüfte japanische
Patentveröffentlichung
Nr. 4-261155 Verbindungen, die fähig
sind, Antagonismus für
Neurokinin-Rezeptoren
(insbesondere NK-2-Rezeptoren) zu zeigen. Zusätzlich beschreibt die ungeprüfte japanische
Patentveröffentlichung
Nr. 5-140103 Verbindungen, die
in der Lage sind, Antagonismus für
einen Substanz-P-Rezeptor, Neurokinin-A-Rezeptor oder Neurokinin-B-Rezeptor
zu zeigen. Diese Verbindungen weisen einen einzelnen Ring mit mindestens
einem Stickstoff-Atom auf. Von diesen Verbindungen sind die Verbindungen
gemäß der vorliegenden
Erfindung strukturell und deutlich verschieden, indem sie einen
Naphthyridin-Ring aufwiesen, wie in der chemischen Formel (1), die im
Folgenden beschrieben wird, gezeigt. Auf der anderen Seite sind
auch verschiedene Verbindungen mit einem Naphthyridin-Ring bekannt.
Zum Beispiel offenbart die ungeprüfte japanische Patentveröffentlichung
Nr. 58-57379 Naphthyridin-Verbindungen
mit einem Anti-Schwindel-Effekt. Jedoch wird überhaupt nicht beschrieben,
dass diese Verbindungen Antagonismus für einen Tachykinin-Rezeptor
zeigen.
-
GB-A-2
087 390 offenbart Naphthyridin-Derivate, welche als Arzneimittel
zur Behandlung von Schwindelzuständen
oder zentrales Muskelrelaxans geeignet sind.
-
US-A-3,555,034
offenbart Naphthyridin-Derivate mit einer beruhigenden Wirkung auf
das Nervensystem.
-
BESCHREIBUNG
DER ERFINDUNG
-
Eine
Aufgabe der vorliegenden Erfindung ist, neue Naphthyridin-Verbindungen, ein
Verfahren zu deren Herstellung und pharmazeutische Zusammensetzungen,
umfassend diese Verbindungen als einen wirksamen Bestandteil, bereitstellen.
-
Insbesondere
ist die vorliegende Erfindung dazu gedacht, Naphthyridin-Verbindungen bereitzustellen, die
zur Verhinderung oder Behandlung pathologischer Phänomene oder
Krankheiten, die durch Tachykinine hervorgerufen werden, eingesetzt
werden.
-
Die
Erfinder haben neue Naphthyridin-Derivate, welche durch die allgemeine
Formel (1) dargestellt werden, und pharmakologisch verträgliche Salze
davon gefunden, die Antagonisten für Tachykinine sind, wodurch
die vorliegenden Erfindung erreicht wurde.
-
Die
vorliegende Erfindung betrifft die folgenden Gegenstände (1)
bis (16).
- (1) Ein neues Naphthyridin-Derivat,
dargestellt durch die folgende allgemeine Formel (1): wobei R1,
R2 und R3, X, Y,
Z, A und G die Bedeutung wie in Anspruch 1 definiert haben, oder
ein pharmakologisch verträgliches
Salz davon.
- (2) Eine Verbindung oder ein pharmakologisch verträgliches
Salz davon nach Anspruch 1, wobei R1, R2 und R3 unabhängig ein
Wasserstoffatom, eine Niederalkylgruppe, eine Arylgruppe, eine Aminogruppe,
ein Halogenatom, eine durch NRaCORb dargestellte Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch
1 definiert sind, eine Niederalkoxycarbonylgruppe, eine Carboxylgruppe,
eine Carbamoylgruppe, eine durch NRaCOORb dargestellte Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch
1 definiert sind, oder eine Hydroxyniederalkylgruppe sind, oder
R1 und R2 oder R2 und R3 zusammengenommen
eine C2-C5 Alkylengruppe
oder eine C2-C5 Alkenylengruppe
bilden, vorausgesetzt, dass mindestens einer von R1,
R2 und R3 eine Niederalkylgruppe
ist, wenn keine der Kombinationen einen Ring bildet; Z eine Phenylgruppe
ist, die 1, 2 oder 3 aus Halogenatomen ausgewählte Substituenten aufweisen
kann; A ein Wasserstoffatom, eine C1-C4 Niederalkylgruppe oder eine C1-C4 Niederalkoxygruppe ist; und G eine Benzoylgruppe
ist, die 1, 2 oder 3 aus Halogenatomen und Niederalkoxygruppen ausgewählte Substituenten
aufweisen kann.
- (3) Eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß dem obigen
Gegenstand (1), wobei R1, R2 und
R3 unabhängig
ein Wasserstoffatom, eine Niederalkylgruppe, eine Arylgruppe, eine Aminogruppe,
eine durch NRaCORb dargestellte
Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch 1 definiert sind, eine
Niederalkoxycarbonylgruppe, eine Carbamoylgruppe oder eine durch
NRaCOORb dargestellte
Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch 1 definiert sind, sind,
oder R1 und R2 oder R2 und R3 zusammengenommen
eine C2-C5 Alkylengruppe
oder eine C2-C5 Alkenylengruppe
bilden, vorausgesetzt, dass mindestens einer von R1,
R2 und R3 eine Niederalkylgruppe
ist, wenn keine der Kombinationen einen Ring bildet; Z eine Phenylgruppe
ist, die 1, 2 oder 3 aus Halogenatomen ausgewählte Substituenten aufweisen
kann; A ein Wasserstoffatom, eine C1-C4 Niederalkylgruppe oder eine C1-C4 Niederalkoxygruppe ist; und G eine Benzoylgruppe
ist, die 1, 2 oder 3 aus Halogenatomen und Niederalkoxygruppen ausgewählte Substituenten
aufweisen kann.
- (4) Eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß dem obigen
Gegenstand (1), wobei R1, R2 und
R3 unabhängig
ein Wasserstoffatom, eine Niederalkylgruppe, eine durch NRaCORb dargestellte
Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch 1 definiert sind, oder
eine durch NRaCOORb dargestellte
Aminoschutzgruppe, wobei Ra und Rb wie In Anspruch 1 definiert sind, sind,
oder R1 und R2 oder
R2 und R3 zusammengenommen
eine C2-C5 Alkylengruppe
oder eine C2-C5 Alkenylengruppe bilden,
vorausgesetzt, dass mindestens einer von R1,
R2 und R3 eine Niederalkylgruppe
ist, wenn keine der Kombinationen einen Ring bildet; Z eine Phenylgruppe
ist, die 1, 2 oder 3 aus Halogenatomen ausgewählte Substituenten aufweisen
kann; A ein Wasserstoffatom, eine C1-C4 Niederalkylgruppe oder eine C1-C4 Niederalkoxygruppe
ist; und G eine Benzoylgruppe ist, die 1, 2 oder 3 aus Halogenatomen
und Niederalkoxygruppen ausgewählte
Substituenten aufweisen kann.
- (5) Eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß dem obigen
Gegenstand (1), wobei R1 und R2 zusammengenommen
eine C2-C5 Alkylengruppe
oder eine C2-C5 Alkenylengruppe
bilden; R3 ein Wasserstoffatom, eine Niederalkylgruppe,
eine Arylgruppe, eine Niederalkoxycarbonylgruppe, eine Hydroxyniederalkylgruppe,
eine durch NRaCORb dargestellte
Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch definiert sind, oder eine
durch NRaCOORb dargestellte
Aminoschutzgruppe, wobei Ra und Rb wie in Anspruch 1 definiert sind, sind;
Z eine Phenylgruppe ist, die 1, 2 oder 3 aus Halogenatomen ausgewählte Substituenten
aufweisen kann; A ein Wasserstoffatom, eine C1-C4 Niederalkylgruppe oder eine C1-C4 Niederalkoxygruppe ist; und G eine Benzoylgruppe
ist, die 1, 2 oder 3 aus Halogenatomen und Niederalkoxygruppen ausgewählte Substituenten
aufweisen kann.
- (6) Eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß dem obigen
Gegenstand (1), welche aus den folgenden Verbindungen ausgewählt wird;
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4,6,7,8,8-actahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin-10-carboxamid
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-methoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4,tetrahydro-benzol[b][1,6]naphthyridin-10-carbonsäure oder
ein pharmakologisch verträgliches
Salz davon,
10-Amino-2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[[+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-hydroxymethyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-phenyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-methoxycarbonylamino-1,2,3,4-tetrahydrobenzo[b)[1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-(N-methyl-N-methoxycarbonyl)amino-1,2,3,4-tetrahydrobenzo[b)[1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-chlor-1,2,3,4-tetrahydro-benzo[6][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-benzoylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[/+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-acetylamino-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-difluorophenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo[6][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon, und
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-difluorophenyl)butyl]-10-acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon.
- (7) Eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß dem obigen
Gegenstand (1), welche aus den folgenden Verbindungen ausgewählt wird:
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-((-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin-10-carboxamid
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-methoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4,tetrahydro-benzol[b][1,6]naphthyridin-10-carbonsäure oder
ein pharmakologisch verträgliches
Salz davon,
10-Amino-2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[[-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-hydroxymethyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-phenyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-methoxycarbonylamino-1,2,3,4-tetrahydro-benzo[b)[1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-(N-methyl-N-methoxycarbonyl)amino-1,2,3,4-tetrahydrobenzo[b)[1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-chlor-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-benzoylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-acetylamino-1,2,3,4,6,7,8,9-oerahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-difluorphenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon, und
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-difluorphenyl)butyl]-10-acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon.
- (8) Ein Verfahren zur Herstellung einer Verbindung gemäß einem
der Gegenstände
1 bis 7, welches die Durchführung
einer reduktiven Alkylierung unter Verwendung eines durch die folgende
allgemeine Formel (2) dargestellten 5,6,7,8-Tetrahydro-1,6-naphthyridins wobei R1,
R2 und R3 wie oben
definiert sind, und eines durch die folgende allgemeine Formel allgemeine Formel
(3) dargestellten Aldehyds wobei X, Y, Z, A und G wie
oben definiert sind, umfasst.
- (9) Ein Verfahren zur Herstellung einer Verbindung nach einem
der Gegenstände
1 bis 7, welches die N-Alkylierung einer durch die folgende allgemeine
Formel (2) dargestellten Verbindung: wobei R1,
R2 und R3 wie oben
definiert sind, mit einer durch die folgende allgemeine Formel (4)
dargestellten Verbindung: wobei X, Y, Z, A und G wie
oben definiert sind, Q ein Halogenatom oder eine R4SO2O-Gruppe ist, wobei R4 eine
Niederalkylgruppe, eine Arylgruppe oder eine Trifluormethylgruppe
ist, umfasst.
- (10) Eine pharmazeutische Zusammensetzung, umfassend eine Verbindung
oder ein pharmakologisch verträgliches
Salz davon gemäß einem
der Gegenstände
1 bis 7, und einen pharmazeutisch verträglichen Verdünner oder
Träger.
- (11) Einen Antagonisten für
Tachykinin-Rezeptoren, umfassend eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß einem
der Gegenstände
1 bis 7 als aktiven Bestandteil.
- (12) Einen Antagonisten für
NK-2-Rezeptoren, umfassend eine Verbindung oder ein pharmakologisch
verträgliches
Salz davon gemäß einem
der Gegenstände
1 bis 7 als aktiven Bestandteil.
- (13) Eine pharmazeutische Zusammensetzung zur Prophylaxe oder
Behandlung von Bronchitis, Pollakisurie, Harninkontinenz und Colitis,
umfassend eine Verbindung oder ein pharmakologisch verträgliches
Salz davon gemäß einem
der Gegenstände
1 bis 7 als aktiven Bestandteil.
- (14) Eine pharmazeutische Zusammensetzung zur Prophylaxe oder
Behandlung von Asthma, umfassend eine Verbindung oder ein pharmakologisch
verträgliches
Salz davon gemäß einem
der Gegenstände
1 bis 7 als aktiven Bestandteil.
- (15) Eine Verbindung oder pharmakologisch verträgliches
Salz davon gemäß einem
der Gegenstände
1 bis 7 zur Verwendung als aktiver Bestandteil einer pharmazeutischen
Zusammensetzung.
- (16) Verwendung einer Verbindung oder eines pharmakologisch
verträglichen
Salzes davon gemäß einem der
Gegenstände
1 bis 7 bei der Herstellung eines Antagonisten für Tachykinin-Rezeptoren, umfassend
die Verbindung oder ein Salz davon als aktiven Bestandteil.
-
Die
Verbindungen der vorliegenden Erfindung zeigen einen exzellenten
Antagonismus gegen Tachykinine und sind als prophylaktische oder
therapeutische Mittel für
die folgenden Tachykinin-vermittelten Krankheiten von Säugetieren
wie zum Beispiel Maus, Ratte, Hamster, Hase, Katze, Hund, Vieh,
Schaf, Affe, Mensch, etc. geeignet: Erkrankungen der Atemwege wie
Asthma, Bronchitis, Lungenentzündung,
chronische obstruktive Lungenkrankheit, Bronchokonstriktion, Expektoration,
Husten, etc; Harnerkrankungen, wie Pollakisurie, Harninkontinenz,
Harnblasenentzündung,
Prostataentzündung,
etc.; Zentralkrankheiten wie Angstzustände, Schlaflosigkeit, Depression,
manisch-depressive Psychosen, Gereiztheit, Parkinson'sche Krankheit, psychosomatische
Störung,
mentale Krankheiten, Schizophrenie, etc.; neurodegenerative Krankheiten
wie Demenz in Fall von AIDS, Alzheimerähnliche senile Demenz, Alzheimer
Krankheit, Down Syndrom, Chorea Huntington, etc.; Demyelinationskrankheiten
wie amyotrophe Lateralsklerose, etc.; andere Nervenkrankheiten wie
Nervenkrankheiten aufgrund von Diabetes, AIDS, Chemotherapie oder ähnlichem
und andere periphere Nervenkrankheiten; Neuralgie; Erkrankungen
der Verdauungsorgane wie Krankheiten, hervorgerufen durch Störung des
Viszeralnervs, Reizkolon, Colitis ulcerosa, Crohn-Krankheit, etc.;
Erbrechen wie beispielsweise Erbrechen, hervorgerufen durch Röntgenbestrahlung,
chemotherapeutische Arzneimittel, giftige Substanzen, Toxine, Schwangerschaft,
Funktionsstörungen
des Gleichgewichtsinns, postoperative Krankheiten, Verschluss des
Magens und des Darms, Abnahme der Magen-Darmmotilität, Viszeralschmerz,
Migräne,
Anstieg des Schädelinnendrucks,
Schädelinnenduck-Krankheit
oder Nebenwirkungen durch die Verabreichung verschiedenster Arzneimittel;
Kollagenkrankheit, Sklerodermie; Eosinophilie aufgrund von Infektion
durch Distonatose; Kreislauferkrankungen wie Angina Pectoris, Bluthochdruck,
Herzversagen, Thrombose, Migräne
und Raynaud-Krankheit;
Schmerzen wie Nozizeption, beispielsweise Schmerzen verbunden mit,
zum Beispiel, Krebs, Angina und akuten oder chronischen Entzündungen
sowie neuralgische Schmerzen und Migräne; allergische Krankheiten
wie Rhinitis allergica, Nesselsucht, andere ekzematoide Dermatitis
und Kontaktdermitis; Überempfindlichkeitskrankheiten
wie Überempfindlichkeit
gegenüber
Pflanzen, Augenkrankheiten wie Bindehautentzündung, Frühjahrskonjugativitis, die Zerstörung der
Blut-Kammerwasser-Schranke,
hervorgerufen durch verschiedenste entzündliche Augenkrankheiten, einen
Anstieg des Augeninnendrucks, Pupillenverengung, etc.; entzündliche
Krankheiten wie zum Beispiel Colitis, Schuppenflechte, Bindegewebsentzündung, Rheumatoidarthritis,
Knochen- und Gelenkentzündung,
Nierenentzündung,
Hepatitis, etc.; Osteoporose; Abhängigkeiten wie Alkoholabhängigkeit;
somatische Krankheiten hervorgerufen durch Stress; Algodystrophie-Syndrom wie
beispielsweise Schulter-Hand-Syndrom, Dysthymia; unerwünschte Immunreaktionen
wie zum Beispiel Transplantatabstoßung und Immunoaktivierung;
und Krankheiten verbunden mit Immunodepression wie systemischer
Lupus erythematodes und multiple Sklerose. Weiterhin sind die Verbindungen
der vorliegenden Erfindung bei allen Fällen geeignet, wo Antagonismus
für Tachykinine
gewünscht
ist.
-
BESTE ART UND WEISE, UM
DIE ERFINDUNG DURCHZUFÜHREN
-
In
der vorliegenden Erfindung kann als Beispiel für eine Niederalkylgruppe beispielsweise
eine lineare oder verzweigte Alkylgruppe mit 1 bis 4 Kohlenstoffatomen,
wie zum Beispiel eine Methylgruppe, eine Ethylgruppe, eine n-Propylgruppe,
eine Isopropylgruppe, ein n-Butylgruppe,
eine sec-Butylgruppe, eine tert-Butylgruppe, etc. dienen. Von diesen
können
die Methylgruppe und die Ethylgruppe als bevorzugte Gruppen genannt
werden.
-
Die
Cycloalkylgruppe bezeichnet eine gesättigte cyclische Gruppe von
3 bis 8 Kohlenstoffatomen und bevorzugte Beispiele davon sind die
Cyclopentylgruppe und die Cyclohexylgruppe.
-
In
der vorliegenden Erfindung bezieht sich die Niederalkoxygruppe auf
eine lineare oder verzweigte Alkoxygruppe mit 1 bis 4 Kohlenstoffatomen
und umfasst zum Beispiel eine Methoxygruppe, eine Ethoxygruppe,
eine n-Propoxygruppe, eine Isopropoxygruppe, eine n-Butoxygruppe,
eine Isobutoxygruppe und eine tert-Butoxygruppe. Von diesen Gruppen
können
die Methoxygruppe und die Ethoxygruppe als bevorzugte Gruppen dienen.
-
In
der vorliegenden Erfindung umfasst das Halogenatom ein Fluor-Atom,
ein Brom-Atom und ein Iod-Atom.
-
In
der vorliegenden Erfindung bezeichnet die Arylgruppe eine Arylgruppe
mit 6 bis 14 Kohlenstoffatomen und umfasst zum Beispiel eine Phenylgruppe,
eine Biphenylgruppe, eine Naphthylgruppe, eine Anthrylgruppe und
eine Phenanthrylgruppe. Von diesen Gruppen können die Phenylgruppe und die
Naphthylgruppe als bevorzugte Gruppe dienen.
-
In
der vorliegenden Erfindung bezieht sich die Heteroarylgruppe auf
einen ungesättigten
5- bis 7-gliedrigen Ring enthaltend 1 bis 5 (vorzugsweise 1 oder
2) Heteroatome, vorzugsweise ausgewählt aus der Gruppe Stickstoff-Atom,
Sauerstoff-Atom und Schwefel-Atom. Spezielle Beispiele davon sind
Thienyl, Imidazolyl, Pyridinyl, Pyrimidinyl, Pyridazinyl, etc.
-
Als
die Niederacylgruppe können
beispielsweise Niederacylgruppen mit 1 bis 6 Kohlenstoffatomen wie zum
Beispiel die Formylgruppe, die Acetylgruppe, die Propanoylgruppe,
die Butanoylgruppe, die Pentanoylgruppe, die Hexanoylgruppe, etc.
aufgeführt
werden.
-
Als
Alkenylgruppe können
beispielsweise Alkenylgruppen mit 2 bis 6 Kohlenstoffatomen wie
die Ethenylgruppe, Propenylgruppe, Butenylgruppe, Pentenylgruppe,
Hexenylgruppe, etc aufgeführt
werden. Die C2-C5-Alkylengruppe
umfasst die Ethylengruppe, die Propylengruppe, die Butylengruppe,
die Pentylengruppe, die Hexylengruppe, etc.
-
Die
C2-C5-Alkenylengruppe
umfasst die Ethenylengruppe, die Propenylengruppe, eine Butenylengruppe,
eine Pentenylengruppe, eine Hexenylengruppe, etc.
-
Die
Verbindung der allgemeine Formel (1) kommt als eine optisch aktive
Substanz oder als eine racemische Modifikation vor, da sie ein asymmetrisches
Kohlenstoffatom aufweist. Solch eine Verbindung kann als optisch
aktive Substanz oder als racemische Modifikation isoliert werden.
Es soll klargestellt werden, dass die vorliegende Erfindung alle
solche racemische Modifikationen, optisch aktiven Substanzen oder
Mischungen davon, die Antagonisten für NK-2 sind, umfasst.
-
Die
pharmakologisch verträglichen
Salze der heterocyclischen Verbindungen gemäß der vorliegenden Erfindung
beinhalten zum Beispiel Salze mit Mineralsäuren wie Salzsäure, Schwefelsäure, etc;
und Salze mit organischen Säuren
wie zum Beispiel Essigsäure,
Milchsäure,
Bernsteinsäure,
Fumarsäure,
Maleinsäure,
Zitronensäure,
Benzoesäure,
Methansulfonsäure,
p-Toluolsulfonsäure,
etc. Die Salze können
mittels konventioneller Methoden hergestellt werden.
-
Die
Verbindung, dargestellt durch die allgemeine Formel (1) umfasst
zum Beispiel die folgenden Verbindungen:
- (1)
2-[(+/-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4,6,7,8,9-actahydro-benzo[b][1,6]naphthyridin
oder ein Salz davon,
- (2) 2-{2-[N-(2-Naphthoyl)aminoethyl]}-1,2,3,4,6,7,8,9-octahydrobenzo[b][1,6]naphthyridin
oder ein Salz davon,
- (3) 2-{2-{N-[4-(2-Phenyl)chinolincarbonyl]}-aminoethyl}-1,2,
3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin oder ein Salz davon,
- (4) 2-{3-{N-(1-Naphthoyl)aminopropyl]}-1,2,3,4,6,7,8,9-octahydrobenzo[b][1,6]naphthyridin
oder ein Salz davon,
- (5) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-10-carboxamid
oder ein pharmakologisch verträgliches
Salz davon,
- (6) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl)-10-methoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (7) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-10-carbonsäure oder
ein pharmakologisch verträgliches
Salz davon,
- (8) 10-Amino-2-[(-)-4-(N-benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (9) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-hydroxymethyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (10) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (11) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-phenyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (12) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-methoxycarbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (13) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-(N-methyl-N-methoxycarbonyl)amino-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (14) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-chlor-1 2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (15) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorphenyl)butyl]-10-benzoylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (16) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3, 4-dichlorphenyl)butyl]-10-acetylamino-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon,
- (17) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-difluorphenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon, und
- (18) 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-difluorphenyl)butyl]-10-acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
oder ein pharmakologisch verträgliches
Salz davon.
-
Als
nächstes
wird im Folgenden ein Verfahren zur Herstellung der Verbindungen
gemäß der vorliegenden
Erfindung beschrieben. In jeder chemischen Formel sind R1, R2, R3,
X, Y, Z, A und G wie oben definiert.
-
Die
Naphthyridin-Derivate gemäß der allgemeinen
Formel (1 ) können
durch reduktive Aminierung unter Verwendung einer Verbindung gemäß der allgemeinen
Formel (2) und einem geeigneten Aldehyd gemäß der allgemeinen Formel (3)
hergestellt werden.
-
Die
reduktive Aminierung kann bei –20 °C bis zur
Rückflusstemperatur
in einem Lösungsmittel
wie beispielsweise niedere Alkohole (z.B. Methanol oder Ethanol),
einem Ether (z.B. Tetrahydrofuran) oder Dichlormethan unter Verwendung
eines reduzierenden Mittels wie Natriumcyanotrihydroborat oder Natriumtetrahydroborat
in Gegenwart oder Abwesenheit einer Lewis-Säure wie zum Beispiel Titan(IV)isopropoxid,
Titan(IV)chlorid oder Bortrifluorid-Diethylether-Komplex; einer
Säure wie
zum Beispiel Essigsäure
oder Salzsäure;
oder einem Dehydratsierungsmittel wie einem Molekularsieb durchgeführt werden.
-
Die
Reaktion kann bei 0 °C
bis 100 °C
in einer Wasserstoff-Atmosphäre
in einem Lösungsmittel
wie beispielsweise einem Alkohol (z.B. Methanol oder Ethanol), Ethylacetat
oder Essigsäure
unter Verwendung eines Reduktionskatalysators (z.B. Palladium-Kohlenstoff
oder Raney-Nickel) in der Gegenwart oder Abwesenheit von Essigsäure, Salzsäure oder ähnlichem
durchgeführt
werden.
-
Als
ein alternatives Verfahren kann die Verbindung gemäß der allgemeine
Formel (1) auch durch N-Alkylierung einer Verbindung der allgemeinen
Formel (2) an ihrem Stickstoff-Atom im Ring mit einer Verbindung der
allgemeinen Formel (4) erhalten werden. In der allgemeinen Formel
(4), entspricht Q einer entfernbaren Gruppe, welche allgemein bei
N-Alkylierungen verwendet wird, wie zum Beispiel einem Halogenatom
oder einer R4SO2O-Gruppe,
wobei R4 eine Niederalkylgruppe, eine Arylgruppe,
eine Aralkylgruppe oder ähnliches
ist. Die obige N-Alkylierung kann in einem Lösungsmittel wie Dimethylformamid
oder 2-Butanon oder ohne jegliches Lösungsmittel in der Gegenwart
oder Abwesenheit einer Base wie zum Beispiel Kaliumcarbonat oder Triethanolamin
durchgeführt
werden. Die N-Alkylierung
kann bei 0 °C
bis Rückflusstemperatur
durchgeführt werden.
-
Die
Verbindung der allgemeinen Formel (2) ist ein nützliches Intermediat. Einige
Verbindungen gemäß der allgemeinen
Formel sind wohlbekannt. Im speziellen sind die folgenden Verbindungen
wohlbekannt:
2-Chlor-5,6,7,8-tetrahydro-1,6-naphthyridin-3-ethylester-hydrochlorid,
5,6,7,8-Tetrahydro-9-methyl-thieno[3,2-b][1,6]naphthyridin-dihydrochlorid,
1,2,3,4-Tetrahydro-10-phenyl-benzo[b][1,6]naphthyridin-dihydrochlorid,
6,7,8,9-Tetrahydro-5-phenyl-pyrido[2,3-b][1,6]naphthyridin-dihydrochlorid,
8-Chlor-1,2,3,4-tetrahydro-N-methyl-benzo[b][1,6]naphthyridin-10-amin,
1,2,3,4-Tetrahydro-benzo[b][1,6]naphthyridin-10-amin,
5,6,7,8-Tetrahydro-3-methyl-1,6-naphthyridin,
6,7,8,9-Tetrahydro-5-methyl-pyrido[2,3-b][1,6]naphthyridin,
8-Fluor-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin-10-amin,
5,6,7,8-Tetrahydro-1,6-naphthyridin,
1,2,3,4-Tetrahydro-10-methyl-benzo[b][1,6]naphthyridin-dihydrochlorid,
2,3,5,6,7,8-Hexahydro-9-methyl-thieno[3,2-b][1,6]naphthyridindihydrochlorid,
1,2,3,4-Tetrahydro-8-methoxy-N-methyl-benzo[b][1,6]naphthyridin-10-amin,
5,6,7,8-Tetrahydro-4-hydroxy-1,6-naphthyridin-3-carbonsäureethylester,
6,7,8,9-Tetrahydro-5-phenyl-pyrido[2,3-b][1,6]naphthyridin,
1,2,3,4-Tetrahydro-N-methyl-benzo[b][1,6]naphthyridin-10-amin,
5,6,7,8-Tetrahydro-1,6-naphthyridinhydrochlorid,
2,3,4,6,7,8-Hexahydro-1H-cyclopenta[b][1,6]naphthyridin,
5,6,7,8-Tetrahydro-3-methyl-1,6-naphthyridin-dihydrochlorid,
8-Chlor-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin-10-amin,
5,6,7,8-Tetrahydro-3-nitro-1,6-naphthyridin,
5,6,7,8-Tetrahydro-9-phenyl-thiazolo[4,5-b][1,6]naphthyridin,
8-Chlor-1,2,3,4-tetrahydro-10-phenyl-benzo[b][1,6]naphthyridin,
5,6,7,8-Tetrahydro-2,3-dimethyl-4-phenyl-thieno[2,3-b][1,6]naphthyridin,
8-Fluor-1,2,3,4-tetrahydro-N-methyl-benzo[b][1,6]naphthyridin-10-amin,
1,2,3,4,6,7,8,9-Octahydro-benzo[b][1,6]naphthyridin,
6,7,8,9-Tetrahydro-5-methylpyrido[2,3-b][1,b]naphthyridin-dihydrochlorid,
1,2,3,4-Tetrahydro-8-methoxy-benzo[b][1,6]naphthyridin-10-amin,
5,6,7,8-Tetrahydro-2-methyl-1,6-naphthyridin,
und
1,2,3,4-Tetrahydro-10-phenyl-benzo[b][1,6]naphthyridin.
-
Außer den
wohlbekannten Verbindungen sind die Verbindungen der allgemeinen
Formel (2) neu und werden durch die allgemeine Formel (5) wiedergegeben.
Die wohlbekannten Verbindungen können
zum Beispiel durch die Verfahren, welche in der ungeprüfte japanische
Patentveröffentlichung
Nr. 58-057379, J. Heterocyclic Chem., 33, 1807 (1996), der ungeprüften japanischen
Patentveröffentlichung
Nr. 3-2166, J. Chem. Soc., 708 (1964), J. Org. Chem., 2899 (1966),
und J. Med. Chem., 32, 1 295 (1 989) beschrieben sind, hergestellt
werden. Die neuen Verbindungen der allgemeinen Formel (5) können individuell
durch die folgenden per se wohlbekannten Verfahren (a), (b), (c),
(d), (e), (f), (g), (h) oder (i) hergestellt werden. Herstellungsverfahren
(a)
wobei R
1a ein Wasserstoffatom,
ein Halogenatom oder eine Nitro-Gruppe ist, und R
12 eine
Aminoschutzgruppe (z.B. eine tert-Butoxycarbonylgruppe, eine Benzyloxycarbonylgruppe,
eine 9-Fluorenylmethyloxycarbonylgruppe,
eine Acetylgruppe, eine Formylgruppe oder eine Benzylgruppe) ist.
-
Im
Detail kann eine Verbindung der allgemeinen Formel (7) durch Umsetzung
einer Verbindung gemäß der allgemeinen
Formel (6) mit einem N-geschütztem
Piperidon in einem Lösungsmittel
wie zum Beispiel Dimethylformamid in der Gegenwart von Ammoniumacetat
oder ähnlichem
bei 1 00 – 1
30 °C in
einem Ölbad erhalten
werden. Herstellungsverfahren
(b)
wobei R
12 wie oben definiert
ist. Im Detail kann eine Verbindung der allgemeinen Formel (8) durch
Umsetzung von Anthranilonitril mit einem N-geschützten Piperidon ohne jegliches
Lösungsmittel
oder in einem Lösungsmittel
wie zum Beispiel Dimethylformamid oder Dimethylacetamid in der Gegenwart
von Zinkchlorid oder ähnlichem
bei 90 °C
bis zur Rückflusstemperatur
erhalten werden. Herstellungsverfahren
(c)
wobei R
1b eine Niederalkylgruppe
oder eine Arylgruppe ist, und R
12 wie oben
definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel
(10) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (9)
mit einem N-geschütztem
Piperidon ohne jegliches Lösungsmittel
oder in einem Lösungsmittel
wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol)
oder Dimethylformamid in der Gegenwart einer Säure wie zum Beispiel Schwefelsäure, Essigsäure oder
Salzsäure;
einer Base wie zum Beispiel Kaliumhydroxid oder Natriumhydroxid;
einem Salz wie zum Beispiel Ammoniumacetat oder Piperidinacetat;
oder einer Lewis-Säure
wie zum Beispiel wasserfreiem Ammoniumchlorid oder Titantetrachlorid
bei 130 – 160 °C in einem Ölbad erhalten
werden. Herstellungsverfahren
(d)
wobei R
12 wie oben definiert
ist. Im Detail kann eine Verbindung der allgemeinen Formel (11)
durch Zugabe von Phosphoroxychlorid zu Anthranilsäure und
einem N-geschütztem
Piperidon und Durchführung
der Reaktion unter Erhitzen zum Rückfluss erhalten werden. Herstellungsverfahren
(e)
wobei jedes R
1c und R
1d ein Wasserstoffatom oder eine Niederalkylgruppe
ist, und R
12 wie oben definiert ist. Im
Detail kann eine Verbindung der allgemeinen Formel (13) durch Umsetzung
einer Verbindung gemäß der allgemeinen
Formel (12) mit einem N-geschütztem
Piperidon ohne jegliches Lösungsmittel
oder in einem Lösungsmittel
wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol)
oder Dimethylformamid in der Gegenwart einer Säure wie zum Beispiel Schwefelsäure, Essigsäure oder
Salzsäure;
einer Base wie zum Beispiel Kaliumhydroxid oder Natriumhydroxid;
einem Salz wie zum Beispiel Ammoniumacetat oder Piperidinacetat;
oder einer Lewis-Säure
wie zum Beispiel wasserfreies Ammoniumchlorid oder Titantetrachlorid
bei 130 – 160 °C in einem Ölbad erhalten
werden. Herstellungsverfahren
(f)
wobei R
1e eine Niederalkylgruppe
ist, und R
12 wie oben definiert ist. Im
Detail kann eine Verbindung der allgemeinen Formel (15) durch Umsetzung
einer Verbindung gemäß der allgemeinen
Formel (14) mit einer Niederalkylhalogenalkylgruppe in einem Lösungsmittel
wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol),
Aceton oder Dimethylformamid in der Gegenwart eines basischen Katalysators
wie zum Beispiel Kaliumcarbonat, Kaliumhydroxid oder Natriumhydroxid
oder in der Gegenwart eines sauren Katalysators wie zum Beispiel
Schwefelsäure
oder Salzsäure
bei Raumtemperatur bis Rückflusstemperatur
erhalten werden.
-
Eine
Verbindung der allgemeinen Formel (16) kann durch Reduktion der
Verbindung der allgemeinen Formel (15) erhalten werden. Diese Reaktion
kann bei –78 °C bis Rückflusstemperatur,
beispielsweise durch Verwendung eines Reduktionsmittels wie Lithiumtetrahydroborat,
Calciumtetrahydroborat oder Lithiumaluminiumhydrid und einem Lösungsmittel
wie Wasser oder einem organischen Lösungsmittels wie beispielsweise einem
Alkohol (zum Beispiel Methanol oder Ethanol), einem Ether (z.B.
Tetrahydrofuran, Dimethylether oder Dioxan) oder Toluol durchgeführt werden. Herstellungsverfahren
(g)
wobei R
1e wie oben definiert
ist. Im Detail kann eine Verbindung der allgemeinen Formel (17)
durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (8)
mit einem Säurehalogenid
oder einem Säureanhydrid
erhalten werden. Das Säurehalogenid
umfasst Acetylchlorid, Acetylbromid, Butyrylchlorid, etc. Das Säurehalogenid
umfasst beispielsweise Essigsäureanhydrid,
Trifluoressigsäureanhydrid,
etc. Die Reaktion kann bei –20 °C bis Rückflusstemperatur
durchgeführt
werden. Ein zufrieden stellendes Ergebnis kann erhalten werden, wenn
die Reaktion in Gegenwart eines basischen Katalysators durchgeführt wird.
Der basische Katalysator umfasst Natriumhydroxid, Pyridin, Triethylamin,
4-Dimethylaminopyridin, etc. Was das Lösungsmittel für diese Reaktion
betrifft, so kann kein Lösungsmittel
verwendet werden oder Wasser oder ein Ether (z.B. Tetrahydrofuran,
Dimethylether oder Dioxan) oder ein Halogen-haltiges Lösungsmittel
wie Methylenchlorid oder Chloroform. Herstellungsverfahren
(h)
wobei R
1f ein Wasserstoffatom,
eine Aminocarbonylgruppe, dargestellt durch CONR
aR
b ist, wobei R
a und
R
b wie oben definiert sind, eine Aminogruppe,
eine Niederalkylgruppe, eine Arylgruppe, ein Halogenatom, eine Niederalkoxycarbonylgruppe,
eine Carboxylgruppe, eine Hydroxyniederalkylgruppe, eine Aminoschutzgruppe, dargestellt
durch NR
aCOR
b ist,
wobei R
a und R
b wie
oben definiert sind, oder eine Aminoschutzgruppe, dargestellt durch
NR
aCOOR
b, ist, wobei
R
a und R
b wie oben
definiert sind, und R
12 wie oben definiert
ist.
-
Im
Detail kann eine Verbindung der allgemeinen Formel (19) erhalten
werden, indem eine Verbindung gemäß der allgemeinen Formel (18)
einer Entschützung
durch konventionelle Verfahren wie saure oder alkalische Hydrolyse,
katalytische Reduktion oder ähnlichem
unterworfen wird.
-
Wenn
die Schutzgruppe eine Benzylgruppe ist, kann die Entschützung unter
Wasserstoffatmosphäre bei
Raumtemperatur bis 50 °C
unter Verwendung eines Reduktionskatalysators (z.B. Palladium-Kohlenstoff oder
Raney-Nickel) in Methanol, Ethanol, Wasser, Essigsäure, Trifluoressigsäure oder ähnlichem
durchgeführt werden.
Die Entschützung
kann auch durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (18) unter
Verwendung von Chlorameisensäure-(Chlorethyl)
bei 0 °C
bis Rückflusstemperatur
in einem Lösungsmittel
(z.B. Dichlormethan oder Tetrahydrofuran) und dann Erhitzen des
Rückstandes
unter Rückfluss
in einem alkoholischen Lösungsmittel
(z.B. Methanol oder Ethanol) erfolgen. Herstellungsverfahren
(i)
wobei R
1f wie oben definiert
ist. Im Detail kann eine Verbindung der allgemeinen Formel (20)
erhalten werden, indem eine Verbindung gemäß der allgemeinen Formel (19)
unter Wasserstoff-Atmosphäre
bei Raumtemperatur bis 50 °C
mit Platinoxid oder ähnlichem
in Trifluoressigsäure
umgesetzt wird.
-
Zur
Aufreinigung des Produkts durch Isolierung aus der Reaktionsmischung,
die durch Anwendung jedes der oben genannten Herstellungsverfahren
erhalten werden kann, kann Lösungsmittelextraktion,
Aufkonzentration, Umkristallisation, Chromatographie, etc. jeweils
mittels richtig angewandter, konventioneller Methode verwendet werden.
-
Ein
Salz einer Verbindung gemäß der vorliegenden
Erfindung kann einfach durch konventionelle salzbildende Reaktionen
hergestellt werden.
-
Wenn
die Verbindung gemäß der vorliegenden
Erfindung als Antagonist für
den Tachykinin-Rezeptor verwendet wird, wird sie oral oder parenteral
verabreicht, nachdem sie allein oder in einer Mischung mit einem Excipienten
oder einem Träger
in eine pharmazeutische Zusammensetzung wie beispielsweise eine
Suspension, eine Emulsion, eine Injektion, eine Inhalation, Tabletten,
Pillen, Körnchen,
feine Subtilaes, ein Pulver, Kapseln, eine oralen Lösung, ein
Suppositorium, eine ophthalmische Lösung, eine ophthalmische Salbe,
eine perkutane Lösung,
ein perkutanes Pflaster, eine Salbe, eine Schleimhautlösung, ein
Schleimhautpflaster, ein Spray oder ähnliches konfektioniert wurde.
Als Additiv wie zum Beispiel der Excipient oder der Träger wird
ein pharmazeutisch verträglicher
gewählt
und seine Art und die Menge hängen
von der Applikationsroute und Applikationsmethode ab. Beispielsweise
im Fall einer Injektion sind Natriumchlorid und Zucker wie zum Beispiel Glucose
und Mannitol üblicherweise
bevorzugt. Im Fall von Zusammensetzungen für die orale Verabreichung sind
Stärke,
Lactose, kristalline Cellulose, Magnesiumstearat, etc. bevorzugt.
Falls gewünscht
können
die oben genannten pharmazeutischen Zusammensetzungen Hilfsmittel,
Stabilisatoren, Benetzungsmittel, Emulgatoren, Puffer und andere
konventionelle Additive enthalten.
-
Obwohl
der Gehalt an der Verbindung gemäß der vorliegenden
Erfindung in der pharmazeutischen Zusammensetzung in Abhängigkeit
von der Art der Zusammensetzung variiert wird, beträgt er üblicherweise
0.1 bis 1 00 Gew.-%, vorzugsweise 1 bis 98 Gew.-%. Injektionen zum
Beispiel enthalten den aktiven Bestandteil in einer Menge von üblicherweise
0.1 bis 30 Gew.-%, vorzugsweise 1 bis 10 Gew.-%. Im Fall von Zusammensetzungen
für die
orale Verabreichung wird die Verbindung gemäß der vorliegenden Erfindung
zusammen mit Additiven in Form von Tabletten, Kapseln, einem Pulver,
Körnchen,
einer Lösung,
einem Trockensaft oder ähnlichem
verwendet. Die Kapseln, Tabletten, Körnchen oder Pulver enthalten
den aktiven Bestandteil üblicherweise
in einer Menge von 5 bis 100 Gew.-%, vorzugsweise 25 bis 98 Gew.-%.
-
Die
Dosis wird in Abhängigkeit
von beispielsweise dem Alter, dem Geschlecht, Körpergewicht und den Symptomen
eines Patienten und dem Zweck der Behandlung bestimmt. Zur Behandlung
wird die Verbindung gemäß vorliegenden
Erfindung üblicherweise
in einer Dosis von 0.001 bis 100 mg/kg/Tag im Fall einer parenteralen
Applikation oder 0.01 bis 500 mg/kg/Tag im Fall einer oralen Applikation
in einer oder 2 oder 4 Portionen verabreicht.
-
Die
vorliegende Erfindung wird durch Beschreibung mit nichtlimitierenden
Beispielen im Folgenden veranschaulicht. Das folgende Verfahren
wurde, wenn nicht anders spezifiziert, angewandt.
-
Die
vorliegende Erfindung wird durch die folgenden Beispiele veranschaulicht.
Da die Ausgangsverbindungen, welche zur Herstellung der Verbindungen
gemäß der vorliegenden
Erfindung (1) eingesetzt werden, neue Verbindungen umfassen, werden
auch Beispiele der Herstellung von solchen Ausgangsverbindungen
als Ausführungsbeispiele
beschrieben. Alle in dem Herstellungsverfahren für die Verbindungen gemäß der vorliegenden
Erfindung benötigten
Verbindungen können
mit einem konventionellem Verfahren, nämlich mit demselben Verfahren
wie es in der vorliegenden Beschreibung beschrieben ist, hergestellt
werden.
-
Beispiel 1
-
Synthese von 2-Aminobenzaldehyd
-
Eine
Lösung
von 2-Nitrobenzaldehyd (50 g, 0.33 mol) in Tetrahydrofuran (0.33
Liter) wurde unter Eiskühlung
tropfenweise über
einen Zeitraum von 2 h zu einer wässrigen Lösung (1.2 Liter) von Natriumdithionit (230
g, 1.32 mol) und Natriumcarbonat (1.68 g, 1.59 mol) gegeben, wobei
die Innentemperatur bei 10 °C
oder niedriger gehalten wurde. Nach 30 Minuten wurde die Reaktionslösung mit
Ethylacetat extrahiert, der Extrakt wurde mit Wasser gewaschen und über wasserfreiem
Natriumsulfat getrocknet. Das organische Lösungsmittel wurde mittels Konzentration
bei reduziertem Druck entfernt, um die gewünschte Verbindung (26 g, 64
%) als ein gelbes Öl
zu ergeben.
1H-NMR (200 MHzFT, TMS,
CDCl3)
6.13 (2H, brs),
6.65 (1H,
d, J=8.5Hz),
6.74 (1H, dt, J=1.0Hz, 7.8Hz),
7.31 (1H,
ddd, J=1.6Hz, 7.8Hz, 8.5Hz),
7.47 (1H, dd, J=1.6Hz, 7.8Hz),
9.86
(1H, s).
-
Herstellungsbeispiel 2
-
Synthese von 2-Benzyl-1,2,3,4-Tetrahydrobenzo[b][1,6]-naghthyridin
-
Ein
10%ige ethanolische Lösung
von Kaliumhydroxid (120 ml) wurde über einen Zeitraum von 1 Stunde
tropfenweise zu einer Lösung
von 2-Aminobenzaldehyd
(26 g, 0.21 mol) und 1-Benzyl-4-piperidon (40 g, 0.21 mol) in wasserfreiem
Ethanol (430 ml) gegeben. Nach Rühren über Nacht
wurde die Reaktionslösung
unter reduziertem Druck konzentriert, mit Ethylacetat extrahiert
und die Extraktionslösung
wurde mit Wasser gewaschen und dann über wasserfreiem Natriumsulfat
getrocknet. Das organische Lösungsmittel
wurde mittels Konzentration bei reduziertem Druck entfernt und der
Rückstand
wurde aus Ethylacetat-Isopropanol umkristallisiert, um die gewünschte Verbindung
(37 g, 63 %) als weiße
Kristalle zu ergeben.
1H-NMR (200 MHzFT,
TMS, CDCl3)
2.95 (2H, t, J=6.1 Hz),
3.26
(2H, t, J=6.1 Hz),
3.75 (2H, s),
3.81 (2H, s),
7.24-7.77
(9H, komplex),
7.94-8.03 (1H, m).
-
Herstellungsbeispiel 3
-
Synthese von 1,2,3,4,6,7,8,9-Octahydrobenzo[b][1,6]naphthyridin
-
Platinoxid
(850 mg) wurde zu einer Lösung
von 2-Benzyl-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin (8.5
g, 31 mmol) in Trifluoressigsäure
(155 ml) gegeben und die katalytische Reduktion wurde bei 50 °C für 24 Stunden
durchgeführt.
Der Katalysator wurde durch Filtration entfernt und das Filtrat
wurde bei reduziertem Druck konzentriert. Der Rückstand wurde mit 6N Natriumhydroxid
basisch gestellt und mit Toluol extrahiert. Die Extraktionslösung wurde über wasserfreiem
Natriumsulfat getrocknet. Das organische Lösungsmittel wurde mittels Konzentration
bei reduziertem Druck entfernt und der Rückstand wurde aus Isopropanol
umkristallisiert, um die gewünschte
Verbindung (5.3 g, 90 %) als weiße Kristalle zu ergeben.
1H-NMR (200 MHzFT, TMS, CDCl3)
1.71-1.95
(4H, m),
2.03 (1H, s),
2.70 (2H, brt),
2.87 (4H,
brt),
3.20 (2H, t, J=6.1 Hz),
3.94 (2H, s),
6.99
(1H, s).
-
Herstellungsbeispiel 4
-
Synthese von 1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin
-
2-Benzyl-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin
(4.4 g, 16.04 mmol) wurde in Methanol (88 ml) gelöst. Anschließend wurden
dazu Essigsäure
(1.84 ml, 32.08 mmol) und Palladium-Kohlenstoff (440 mg) gegeben
und die katalytische Reduktion wurde über Nacht bei 50 °C durchgeführt. Der
Katalysator wurde durch Filtration entfernt und das Filtrat wurde
mit Kaliumcarbonat neutralisiert sowie filtriert. Die so erhaltene
organische Phase wurde bei reduziertem Druck konzentriert und der
Rückstand
wurde durch Silicagel-Säulen-Chromatografie
(Methylenchlorid/Methanol = 30/1 bis 10/1 ) gereinigt, um die gewünschte Verbindung
(1.1 g, 37.2 %) als braunen Feststoff zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
3.17 (2H, t,
J=3.0Hz),
3.34 (2H, t, J=5.8Hz),
4.22 (2H, s),
7.42-7.50
(1H, m),
7.64 (1H, ddd, J=1.6, 6.9, 16.9Hz),
7.72 (1H,
brd),
7.78 (1H, brs),
7.99 (1H, d, J=8.1 Hz).
MS
(FAB, m-NBA)
m/z → 185
[M + H]+.
-
Herstellungsbeispiel 5
-
Synthese von 2-t-Butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naghthyridin-10-carboxamid
-
Isatin
(3.99 g, 20 mmol) und 1-t-Butoxycarbonyl-4-piperidon (2.94 g, 20
mmol) wurden in N,N-Dimethylformamid (20 ml) gelöst. Anschließend wurde
dazu Ammoniumacetat (4.63 g, 60 mmol) gegeben und die erhaltene
Mischung wurde bei 1 20 °C
für 3 Stunden
gerührt.
Das Lösungsmittel
wurde mittels Konzentration bei reduziertem Druck entfernt und Aceton
(20 ml) und Wasser (20 ml) wurden zu dem Rückstand gegeben. Die erhaltene
Aufschlämmung
wurde filtriert und der so erhaltene Feststoff wurde durch Suspendieren
in Ethylacetat/Hexan gereinigt, um die gewünschte Verbindung (3.34 g,
50 %) als hellbräunlich-weißen Feststoff
zu ergeben.
1H-NMR (200 MHzFT, TMS,
CDCl3)
1.47 (9H, s),
3.22 (2H,
t, J=6.2Hz),
3.83 (2H, t, J=6.2Hz),
4.83 (2H, s),
6.26
(2H, brs),
7.51-7.61 (1H, m),
7.66-7.77 (1H, m),
7.90-7.97
(1H, m),
8.02 (1H, d, J=8.1 Hz).
MS (FAB, m-NBA)
m/z
328 → [M
+ H]+.
-
Herstellungsbeispiel 6
-
Synthese von 1,2,3, 4-Tetrahydro-benzo[b][1,6]-naphthyridin-10-carboxamid-dihydrochlorid
-
2-t-Butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-10-carboxamid (146 mg,
0.442 mmol) wurde in Dioxan (1 ml) suspendiert. Anschließend wurde
dazu unter Eiskühlung
4N HCl/Dioxan (2.2 ml, 8.83 mmol) gegeben und die erhaltene Mischung
wurde bei Raumtemperatur für
3 Stunden gerührt.
Die Reaktionsmischung wurde konzentriert, um die gewünschte Verbindung
(146 mg, quantitativ) als hellbraunen Feststoff zu ergeben.
1H-NMR (200 MHzFT, TMS, CD3OD)
3.62-3.88
(4H, komplex),
4.69 (2H, s),
7.87-8.01 (1H, m),
8.10-8.26
(3H, komplex).
MS (FAB, m-NBA)
m/z 228 → [M + H]+.
-
Herstellungsbeispiel 7
-
Synthese von 2-t-Butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naohthyridin-10-carbonsäure
-
Isatin
(14.71 g, 100 mmol) und 1-t-Butoxycarbonyl-4-piperidon (19.93 g,
100 mmol) wurden in Ethanol (200 ml) gelöst. Anschließend wurde
dazu eine ethanolische Lösung
(50 ml) von KOH (12.34 g) gegeben und die erhaltene Mischung wurde
bei 70 °C
für 24
Stunden gerührt.
Das Lösungsmittel
wurde mittels Konzentration bei reduziertem Druck entfernt und der
Rückstand
wurde mit Essigsäure
neutralisiert. Das erhaltene unlösliche
Material wurde durch Suspension in Methanol gereinigt, um die gewünschte Verbindung
(9.5 g, 29 %) als hellbräunlich-weißen Feststoff
zu ergeben.
1H-NMR (200 MHzFT, TMS,
DMSO-d6)
1.41 (9H, s),
3.1 4 (2H,
t, J=6.2Hz),
3.76 (2H, t, J=6.2Hz),
4.74 (2H, s),
7.56-7.68
(1H, m),
7.68-7.80 (1H, m),
7.86 (1H, d, J=8.4 Hz),
7.98
(1H, d, J=8.4 Hz).
MS (FAB, m-NBA)
m/z 329 → [M + H]+.
-
Herstellungsbeispiel 8
-
Synthese von 2-t-Butoxycarbonyl-10-methoxycarbonyl-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin
-
2-t-Butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-10-carbonsäure (500
mg, 1.523 mmol) wurde in N,N-Dimethylformamid (3 ml) gelöst. Anschließend wurden
dazu Kaliumcarbonat (315.1 mg, 2.28 mmol) und Methyliodid (0.13
ml, 2.06 mmol) gegeben und die erhaltene Mischung wurde bei Raumtemperatur über Nacht
gerührt.
Wasser wurde zu der Reaktionsmischung gegeben und diese anschließend mit Ethylacetat
extrahiert. Die organische Phase wurde mit gesättigter, wässriger Natriumchlorid-Lösung gewaschen
und über
wasserfreiem Natriumsulfat getrocknet. Die organische Phase wurde
bei reduziertem Druck konzentriert und der erhaltene Rückstand
wurde durch Silicagel-Säulen-Chromatografie
(Hexan/Ethylacetat = 1/1) gereinigt, um die gewünschte Verbindung (487 mg,
93.4 %) als braunes Öl
zu ergeben.
1H-NMR (200 MNzFT, TMS,
CDCl3)
1.50 (9H, s),
3.24 (2H,
t, J=6.1 Hz),
3.84 (2H, t, J=6.2Hz),
4.09 (3H, s),
4.80
(2H, s),
7.50-7.60 (1H, m),
7.67-7.77 (1H, m),
7.78-7.87
(1H, m),
8.03 (1H, d, J=8.1 Hz).
-
Herstellungsbeispiel 9
-
Synthese von 10-Methoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naghthyridin-dihydrochlorid
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten wie
in Herstellungsbeispiel 6 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
3.70-3.87 (4H,
komplex),
4.21 (3H, s),
4.77 (2H, s),
7.92-8.03 (1H,
m),
8.12-8.32 (3H, komplex).
-
Herstellungsbeispiel 10
-
Synthese von 10-Amino-2-t-butoxycarbonyl-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin
-
1-t-Butoxycarbonyl-4-piperidon
(1.69 g, 8.46 mmol), Zinkchlorid (1.50 g, 11.0 mmol) und 2-Aminobenzonitril
(1.0 g, 8.46 mmol) wurden gemischt und bei 90 °C für 1 h umgesetzt. Die Reaktionsmischung
wurde auf Raumtemperatur gekühlt,
um einen Feststoff zu erhalten, der zusammen mit Toluol gemahlen
und filtriert wurde. Der so behandelte Feststoff wurde in Chloroform
suspendiert und anschließend
wurde konzentrierter, wässriger
Ammoniak zugegeben und das Ganze gerührt. Die Chloroform-Phase wurde abgetrennt, über wasserfreiem
Natriumsulfat getrocknet und konzentriert, um die gewünschte Verbindung
(0.54 g, 1.8 mmol, 21 %) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.53 (9H, s),
3.10
(2H, t, J=5.9Hz),
3.80 (2H, t, J=5.8Hz),
4.53 (2H, s),
4.70
(2H, br.s.),
7.35-7.50 (1H, m),
7.55-7.68 (1H, m),
7.73
(1H, d, J=8.3 Hz),
7.91 (1H, d, J=7.7 Hz).
MS (FAB, m-NBA)
m/z → 300 [M
+ H]+,
599 [2M + H]+.
-
Herstellungsbeispiel 11
-
Synthese von 10-Amino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridintrihydrochlorid
-
10-Amino-2-t-butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin (0.538
g, 1.80 mmol) wurde in Dioxan (3 ml) suspendiert. Anschließend wurde
dazu unter Eiskühlung
4N HCl/Dioxan (9 ml) gegeben und die erhaltene Mischung wurde bei
Raumtemperatur für
1 Stunden gerührt.
Die Reaktionsmischung wurde konzentriert, um die gewünschte Verbindung
(0.585 g, quantitativ) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CD 3)
3.35-3.45 (2H,
m),
3.62-3.78 (2H, m),
4.35 (2H, s),
7.66-7.76 (1H,
m),
7.85-7.87 (1H, m),
7.93-8.03 (1H, m),
8.37-8.45
(1H, m).
MS (FAB, m-NBA)
m/z → 200 [M + H]+.
-
Herstellungsbeispiel 12
-
Synthese von 2-t-Butoxycarbonyl-10-hydroxymethyl-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin
-
2-t-Butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-10-methylcarbonsäureester
(1.14 g, 3.05 mmol) wurde in Ethanol (3 ml) gelöst. Anschließend wurden
dazu Natriumtetrahydroborat (138.4 mg, 3.66 mmol) und dann eine
Lösung
von Calciumchlorid (338.5 mg, 3.05 mmol) in Ethanol (3 ml) unter
Eiskühlung gegeben.
Die erhaltene Reaktionsmischung wurde bei Raumtemperatur für 4 Stunden
und dann bei 60 °C
für 3 Stunden
gerührt.
Eine gesättigte,
wässrige
Ammoniumchlorid-Lösung
wurde zu der Reaktionsmischung gegeben und diese wurde anschließend mit
Ethylacetat extrahiert. Die organische Phase wurde mit gesättigter, wässriger
Natriumchlorid-Lösung
gewaschen und über
wasserfreiem Natriumsulfat getrocknet. Die organische Phase wurde
bei reduziertem Druck konzentriert und der erhaltene Rückstand wurde
durch Silicagel-Säulen-Chromatografie
(Hexan/Ethylacetat = 1/1 ) gereinigt, um die gewünschte Verbindung (442 mg,
46 %) als gelbes Öl
zu ergeben.
1H-NMR (200 MHzFT, TMS,
CDCl3)
1.49 (9H, s),
3.21 (2H,
t, J=6.2Hz),
3.81 (2H, t, J=6.2Hz),
4.92 (2H, s),
5.13
(2H, s),
7.50-7.60 (1H, m),
7.63-7.73 (1H, m),
7.97-8.05
(1H, m),
8.14-8.22 (1H, m).
MS (FAB, m-NBA)
m/z 315 → [M + H]+.
-
Herstellungsbeispiel 13
-
Synthese von 10-Hydroxymethyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridindibenzolsulfonat
-
2-t-Butoxycarbonyl-10-hydroxymethyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin (202
mg, 0.643 mmol) wurde in Methanol (1.3 ml) gelöst. Anschließend wurde
dazu Benzolsulfonsäure-Monohydrat
(226.3 mg, 1.285 mmol) gegeben und die erhaltene Mischung wurde
unter Rückfluss
bei 70 °C
für 3 Stunden
erhitzt. Die Reaktionsmischung wurde auf Raumtemperatur gekühlt und
dann konzentriert, um die gewünschte
Verbindung (361.2 mg, quantitativ) als braune Substanz zu ergeben.
1H-NMR (200 MHzFT, TMS, CD3OD)
3.58-3.85
(4H, komplex),
4.97 (2H, s),
5.22 (2H, s),
7.24-7.45
(6H, m),
7.64-7.80 (4H, m),
7.90-8.01 (1H, m),
8.09-8.16
(2H, komplex),
8.59 (1H, d, J=8.bHz),
MS (FAB, m-NBA)
m/z
21 5 → [M
+ H]+.
-
Herstellungsbeispiel 14
-
Synthese von 2-t-Butoxycarbonyl-10-acetylamino-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin
-
10-Amino-2-t-butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin (0.77
g, 2.59 mmol) wurde zu Pyridin (1.0 g, 12.7 mmol) gegeben. Essigsäureanhydrid
(0.53 g, 5.18 mmol) wurde zu der Mischung gegeben und diese anschließend für 7.5 Stunden
unter Rückfluss
erhitzt. Dann wurde Wasser zu der Reaktionsmischung gegeben und
diese wurde anschließend
mit Chloroform extrahiert. Die Chloroform-Phase wurde über wasserfreiem
Natriumsulfat getrocknet und das Lösungsmittel bei reduziertem
Druck abdestilliert. Der erhaltene Rückstand wurde durch Silicagel-Säulen-Chromatografie
gereinigt, um die gewünschte
Verbindung (0.5 g, 1.47 mmol, 46 %) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.49 (9H, s),
2.36
(3H, s),
3.16 (2H, t, J=5.9Hz),
3.82 (2H, t, J=6.2Hz),
4.67
(2H, s),
7.43-7.53 (1H, m),
7.60-7.71 (1H, m),
7.87
(1H, d, J=8.3 Hz),
7.98 (1H, d, J=8.4 Hz).
MS (FAB, m-NBA)
m/z → 342 [M
+ H]+,
683.6 [2M + H]+.
-
Herstellungsbeispiel 15
-
Synthese von 10-Acetylamino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-dihydrochlorid
-
10-Methylcarbonylamino-2-t-butoxycarbonyl-1,2,3,4-tetrahydrobenzol[b][1,6]-naphthyridin
(0.5 g, 1.47 mmol) wurde in einer Mischung aus Dioxan (4.5 ml) und
Methanol (1 ml) suspendiert. Anschließend wurde dazu unter Eiskühlung 4N
HCl/Dioxan (7.35 ml) gegeben und die erhaltene Mischung wurde bei
Raumtemperatur für
1.5 Stunden gerührt.
Die Reaktionsmischung wurde konzentriert, um die gewünschte Verbindung (0.369
g, quantitativ) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
2.44 (3H, s),
3.68-3.87
(4H, m),
4.47 (2H, s),
7.93-8.03 (1H, m),
8.17-8.22
(2H, m),
8.45 (1H, d, J=8.5Hz).
MS (FAB, m-NBA)
m/z → 242 [M
+ H]+.
-
Herstellungsbeispiel 16
-
Synthese von 2-Benzyl-10-phenyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
-
1-t-Butoxycarbonyl-4-piperidon
(1.93 g, 10.2 mmol) und 2-Aminobenzophenon
(2.01 g, 10.2 mmol) wurden in Essigsäure (10 ml) suspendiert. Anschließend wurde
dazu Schwefelsäure
(0.1 ml) gegeben und die Reaktion bei 120 °C für 1 Stunde durchgeführt. Die
Reaktionsmischung wurde in kalte wässrige Ammoniak-Lösung gegossen
und die ausgefallenen Kristalle wurden mit Wasser gewaschen und
aus Ethanol umkristallisiert, um die gewünschte Verbindung (2.60 g,
7.43 mmol) zu ergeben.
1H-NMR (200
MHzFT, TMS, CDCl3)
2.84 (2H, t, J=6.1
Hz),
3.28 (2H, t, J=6.0Hz),
3.56 (2H, s),
3.60 (2H,
s),
7.20-7.37 (10H, m),
7.45-7.55 (2H, m),
7.57-7.67
(1H, m),
8.03 (1H, d, J=8.4Hz),
MS (FAB, m-NBA)
m/z → 351 [M
+ H]+.
-
Herstellungsbeispiel 17
-
Synthese von 10-Phenyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
-
Palladiumschwarz
(125 mg) wurde zu einer Lösung
von 2-Benzyl-10-phenyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
(1.25 g, 3.57 mmol) in Essigsäure
(9 ml) gegeben. Die erhaltene Mischung wurde unter Wasserstoffatmosphäre für 9 Stunden
gerührt.
Der Katalysator wurde mittels Filtration entfernt und das Filtrat wurde
unter reduziertem Druck konzentriert. Der Rückstand wurde mit 1N Natriumhydroxid
basisch gestellt und mit Diethylether extrahiert. Die Extraktionslösung wurde über wasserfreiem
Natriumsulfat getrocknet und bei reduziertem Druck vom Lösungsmittel
befreit. Der Rückstand
wurde mit Diethylether gewaschen, um die gewünschte Verbindung (0.45 g,
1.75 mmol, 49 %) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
3.20-3.36 (4H,
m),
3.86 (2H, s),
7.21-7.28 (3H, m),
7.33-7.38 (2H,
m),
7.45-7.68 (1H, m),
8.04 (1H, d, J=8.4Hz).
MS
(FAB, m-NBA)
m/z → 261
[M + H]+.
-
Herstellungsbeispiel 18
-
Synthese von 2-t-Butoxycarbonyl-10-methoxycarbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
-
2-t-Butoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-10-carboxamid (0.5 g,
1.53 mmol) wurde zu einer Lösung
von Natrium (0.078 g, 3.37mmol) in Methanol (31 ml) gegeben. Anschließend wurde Brom
(0.245 g, 1.53 mmol) tropfenweise dazu gegeben, während die
Temperatur bei 25 °C
oder niedriger gehalten wurde. Nach Beendigung der tropfenweise
Zugabe wurde die erhaltene Mischung unter Rückfluss für 30 Minuten erhitzt. Wasser
wurde zu der Reaktionsmischung gegeben und diese wurde anschließend mit Ethylacetat
extrahiert. Die Ethylacetat-Phase
wurde über
wasserfreiem Natriumsulfat getrocknet und bei reduziertem Druck
konzentriert. Der erhaltene Rückstand
wurde durch Silicagel-Säulen-Chromatografie
(Hexan/Ethylacetat = 1/1 ) gereinigt, um die gewünschte Verbindung (0.46 g,
1.28 mmol, 84 %) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.49 (9H, s),
3.23
(2H, t, J=6.1 Hz),
3.81 (3H, s),
3.84 (2H, t, J=6.3Hz),
4.75
(2H, s),
6.85-7.05 (1H, br.s.),
7.48-7.58 (1H, m),
7.63-7.74
(1H, m),
7.90 (1H, d, J=8.4Hz),
8.02 (1H, d, J=8.4Hz).
MS
(FAB, m-NBA)
m/z → 358
[M + H]+,
715 [2M + H]+.
-
Herstellungsbeispiel 19
-
Synthese von 10-Methoxycarbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin-dihydrochlorid
-
Die
angestrebte Verbindung wurde auf dieselbe Weise erhalten wie in
Herstellungsbeispiel 6 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
3.68-3.87 (4H,
m),
3.92 (3H, s),
4.56 (2H, s),
7.90-8.01 (1H, m),
8.15-8.20
(2H, m),
8.37-8.45 (1H, m).
MS (FAB, m-NBA)
m/z → 258 [M
+ H]+.
-
Herstellungsbeispiel 20
-
Synthese von 2-t-Butoxycarbonyl-10-N-methylmethoxy-carbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
-
Natriumhydrid
(0.038 g, 0.95 mmol) wurde in Dimethylformamid (3 ml) suspendiert
und 2-t-Butoxycarbonyl-10-methoxycarbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
(0.225 g, 0.63 mmol) wurde dazu gegeben. Nach Rühren für 30 Minuten wurde Methyliodid
(0.143 g, 1.01 mmol) dazu gegeben und das Ganze über Nacht gerührt. Wasser
wurde zu der Reaktionsmischung gegeben und diese wurde anschließend mit
Ethylacetat extrahiert. Die Ethylacetat-Phase wurde über wasserfreiem
Natriumsulfat getrocknet und bei reduziertem Druck konzentriert.
Der erhaltene Rückstand
wurde durch Silicagel-Säulen-Chromatografie
(Hexan/Ethylacetat/Methanol = 10 : 10 : 1) gereinigt, um die gewünschte Verbindung
(0.164 g, 0.44 mmol, 70 %) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.50 (9H, s),
3.24
(2H, t, J=6.0Hz),
3.31 (3H, s),
3.61 (2H, s),
3.65-4.14
(3H, m),
4.46-4.90 (2H, komplex),
7.49-7.60 (1H, m),
7.64-7.79
(2H, m),
8.02-8.10 (1H, m).
MS (FAB, m-NBA)
m/z → 358 [M
+ H]+,
715 [2M + H]+.
-
Herstellungsbeispiel 21
-
Synthese von N-Methyl-10-methoxycarbonylamino-1,2,3,4-tetrahydrobenzo[b][1,6]-naphthyridin-dihydrochlorid
-
Die
angestrebte Verbindung wurde auf dieselbe Weise erhalten wie in
Herstellungsbeispiel 6 erhalten.
1H-NMR
(200 MHzFT, TMS, CD3OD)
3.35-3.52 (3H,
m),
3.64-3.96 (7H, komplex),
4.45-4.78 (2H, m),
7.90-8.02
(1H, m),
8.05-8.20 (2H, m),
8.36-8.45 (1H, m).
MS
(FAB, m-NBA)
m/z → 272
[M + H]+.
-
Herstellungsbeispiel 22
-
Synthese von 2-Benzyl-10-chloro-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
-
Anthranilsäure (9.6
g, 70 mmol) und 1-Benzyl-4-piperidon (13.2 g, 70 mmol) wurden in
Phosphoroxychlorid (65.2 ml, 700 mmol) suspendiert und die Suspension
wurde unter Rückfluss
4 Stunden erhitzt. Das überschüssige Phosphoroxychlorid
wurde durch Destillation entfernt und der konzentrierte Rückstand
wurde vorsichtig unter Eiskühlung
zu einer 28%igen wässrigen
Ammoniak-Lösung
gegeben und mit Chloroform extrahiert. Die organische Phase wurde
mit Wasser gewaschen, über
wasserfreiem Natriumsulfat getrocknet und dann bei reduziertem Druck
konzentriert. Der erhaltene Rückstand
wurde durch Silicagel-Säulen-Chromatografie (Hexan/Ethylacetat/Methanol
= 30 : 1 0 : 1) gereinigt, um die gewünschte Verbindung (13 g, 60
%) als hellgelblich-weiße
Substanz zu ergeben.
1H-NMR (200 MHzFT,
TMS, CDCl3)
2.90 (2H, t, J=6.0Hz),
3.24
(2H, t, J=6.0Hz),
3.82 (2H, s),
3.93 (2H, s),
7.27-7.46
(5H, m),
7.50-7.61 (1H, m),
7.64-7.74 (1H, m),
7.96-8.03
(1H, m),
8.12-8.20 (1H, m).
MS (FAB, m-NBA)
m/z 309,
311 → [M
+ H]+.
-
Herstellungsbeispiel 23
-
Synthese von 10-Chloro-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
-
2-Benzyl-10-chloro-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin
(1.54 g, 5 mmol) wurde in Methylenchlorid gelöst. Anschließend wurde
dazu unter Eiskühlung
1-Chloroethylchloroformiat (0.81 ml, 7.5 mmol) gegeben und die Suspension
für 1 Stunde
bei Raumtemperatur gerührt.
Die erhaltene Reaktionsmischung wurde konzentriert und Methanol
(20 ml) wurde hinzugegeben. Die Reaktionsmischung für 1 Stunde
unter Rückfluss auf
80 °C erhitzt.
Die Reaktionsmischung wurde konzentriert und 1N Kaliumhydroxid-Lösung (20
ml) wurde hinzugegeben. Nach Extraktion mit Methylenchlorid wurde
die organische Phase über
wasserfreiem Natriumsulfat getrocknet und dann bei reduziertem Druck
konzentriert. Der erhaltene Rückstand
wurde durch Suspendieren in Diethylether/Hexan gereinigt, um die
gewünschte
Verbindung (656.4 mg, 60 %) als hellgelblich-weiße Substanz zu ergeben.
1H-NMR (200 MHzFT, TMS, CDCl3)
3.08-3.18
(2H, m),
3.23-3.33 (2H, m),
4.23 (2H, s),
7.50-7.60
(1H, m),
7.63-7.73 (1H, m),
7.95-8.02 (1H, m),
8.11-8.18
(1H, m).
MS (FAB, m- NBA)
m/z 219, 221 → [M + H]+.
-
Herstellungsbeispiel 24
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
-
2-Benzyl-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridin
(545 mg, 2,9 mmol) wurde zu einer Lösung von (-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichloro-phenyl)butanol
(675 mg, 1.9 mmol) in Methanol (6 ml) gegeben und für 3 Stunden
gerührt.
Anschließend
wurde eine Lösung
von Natriumcyanotrihydroborat (383 mg, 6.1 mmol) in Tetrahydrofuran
(6 mml) dazugegeben und das ganze über Nacht gerührt. Zu
der Reaktionsmischung wurde 1N Natriumhydroxid hinzugeben und das
Ganze mit Ethylacetat extrahiert. Die organische Phase wurde mit
Wasser und einer gesättigten
Natriumchlorid-Lösung
gewaschen sowie über
wasserfreiem Natriumsulfat getrocknet. Das organische Lösungsmittel
wurde bei reduziertem Druck entfernt und der erhaltene Rückstand wurde
durch Silicagel-Säulen-Chromatografie
(Chloroform/Methanol = 100 : 1 → 30
: 1) gereinigt, um die gewünschte
Verbindung (204 mg, 20 %) zu ergeben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.4-4.2 (22H,
komplex),
6.7-7.5 (9H, komplex).
MS (FAB, m-NBA)
m/z
522, 524 → [M
+ H]+.
-
Herstellungsbeispiel 25
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4,6,7,8,9-octahydrobenzo[b][1,6]naphthyridinfumarat
-
Fumarsäure (45
mg, 0.39 mmol) wurde in Ethanol (0.5 ml) gelöst und anschließend wurde
dazu eine Lösung
von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4,6,7,8,9-octahydrobenzo[b][1,6]naphthyridin
(204 mg, 0.39 mmol) in Ethanol (1 ml) gegeben. Nach 1 Stunde wurde
die Reaktionsmischung bei reduziertem Druck konzentriert und aus
Chloroform-Diethylether umkristallisiert um die gewünschte Verbindung
(87 mg, 35 %) zu ergeben.
1H-NMR (200
MHzFT, TMS, CDCl3)
1.7-4.2 (22H, komplex),
6.68
(2H, s),
6.9-7.7 (8H, komplex).
MS (FAB, m-NBA)
m/z
522, 524, 526 → [M
+ H]+.
-
Herstellungsbeispiel 26
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-methylcarbonylamino-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridin
-
(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butylbenzolsulfonat
(0.57 g, 1.16 mmol) und Triethylamin (0.1 3 g, 1.28 mmol) wurden
zu einer Lösung
von 10-Methylcarbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]-naphthyridin (0.281
g, 1.16 mmol) in einer Mischung aus Tetrahydrofuran (5 ml) und Dimethylsulfoxid
(1 ml) gegeben. Die erhaltene Mischung wurde für 5 Stunden bei Raumtemperatur
und dann für
4 Stunden bei 70 °C
gerührt.
Wasser wurde zu der Reaktionsmischung gegeben und diese wurde anschließend mit
Ethylacetat extrahiert. Die Ethylacetat-Phase wurde über wasserfreiem
Natriumsulfat getrocknet und bei reduziertem Druck konzentriert.
Der erhaltene Rückstand
wurde durch Silicagel-Säulen-Chromatografie (Chloroform – Methanol
= 30 : 1 ) gereinigt, um die gewünschte
Verbindung (0.393 g, 0.683 mmol, 59 %) zu ergeben.
1H-NMR (200 MHzFT, TMS, CDCl3)
1.20-4.10
(19H, komplex),
6.70-7.90 (10H, komplex),
7.99 (1H, br.d.),
8.12
(1H, br.s.).
MS (FAB, m-NBA)
m/z 575, 577 → [M + H]+.
-
Herstellungsbeispiel 27
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4)-dichlorophenyl)butyl]-10-methylcarbonylamino-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridinfumarat
-
Fumarsäure (0.067
g, 0.574 mmol) wurde in Ethanol (1 ml) gelöst und die erhaltene Lösung wurde
zu einer Lösung
von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-methyl-carbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
(0.330 g, 0. 574 mmol) in Ethanol (2 ml) gegeben. Nach 1 Stunde
wurde die Reaktionslösung
bei reduziertem Druck konzentriert, um die gewünschte Verbindung (0.267 g)
zu ergeben.
1H-NMR (200 MHzFT, TMS,
CD3OD)
2.02-4.06 (19H, komplex),
6.72
(2H, s),
6.92-7.23 (2H, komplex),
7.28-7.82 (8H, komplex),
7.87-8.05
(2H, komplex).
MS (FAB, m-NBA)
m/z → 575, 577 [M + H]+.
-
Herstellungsbeispiel 28
-
Synthese von 2-[(-)4-(N-Benzoyl-N-methylamino-3-(3,4-dichloroghenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin-10-carboxamid
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.21-4.10 (16H,
komplex),
6.27-7.58 (11H, komplex),
7.66 (1H, brs),
7.83
(1H, d, J=8.1 Hz),
7.98 (1H, d, J=8.4Hz).
MS (FAB, m-NBA)
m/z
561, 563 → [M
+ H]+.
-
Herstellungsbeispiel 29
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4-tetrahydro-benzo-[b][1,6]naphthyridin-10-carboxamidfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.28-4.18 (16H,
komplex),
6.71 (2H, s),
6.71-8.02 (12H, komplex).
MS
(FAB, m-NBA)
m/z 561, 563 → [M
+ H]+.
-
Herstellungsbeispiel 30
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methylamino-3-(3,4-dichlorophenyl)butyl]-10-methoxycarbonyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.60-4.01 (16H,
komplex),
4.06 (3H, s),
6.72-8.08 (12H, komplex).
MS
(FAB, m-NBA)
m/z 576, 578 → [M
+ H]+,
-
Herstellungsbeispiel 31
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methylamino-3-(3,4-dichlorophenylbutyl]-10-methoxycarbonyl-1,2,3,4-tetrahydro-benzo-[b][1,6]naghthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.60-4.20 (19H,
komplex),
6.72 (2H, s),
6.85-8.05 (12H, komplex).
MS
(FAB, m-NBA)
m/z 576, 578 → [M
+ H]+.
-
Herstellungsbeispiel 32
-
Synthese von 2-[(-1-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4-tetrahydro-benzo-[b][1,6]naphthyridin-10-carbonsäure
-
2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4-tetrahydro-benzo-[b][1,6]naphthyridin-10-methylester
(1 66.6 mg, 0.289 mmol) wurde in Methanol (5 ml) gelöst und anschließend wurde
dazu unter Eiskühlung
1N wässrige
Natriumhydroxid-Lösung
gegeben. Die erhaltene Mischung wurde bei Raumtemperatur für 2 Stunden
und dann bei 70 °C
für 3 Stunden
gerührt.
Nachdem die Reaktionsmischung auf Raumtemperatur abgekühlt war,
wurde dazu eine 2M wässrige
Oxalsäure-Lösung gegeben. Nach Extraktion
mit Methylenchlorid wurde die organische Phase über wasserfreiem Natriumsulfat
getrocknet und bei reduziertem Druck konzentriert, um die gewünschte Verbindung
(97 mg, 58.4 %) als gelben Feststoff zu ergeben.
MS (FAB, m-NBA)
m/z
562, 564 → [M
+ H]+.
-
Herstellungsbeispiel 33
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichloroghenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin-10-carbonsäurefumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.80-4. 60
(16H, komplex),
6.75 (2H, s),
6.70-8.10 (12H, komplex).
MS
(ESI)
m/z 562, 564, 566 → [M
+ H]+.
-
Herstellungsbeispiel 34
-
Synthese von 10-Amino-2-[(-)-4-(N-benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyll-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.25-4.10 (16H,
komplex),
4.58 (2H, br.s.),
7.08-7.50 (9H, komplex),
7.50-7.09
(2H, m),
7.86-8.04 (1H, m).
MS (FAB, m-NBA)
m/z → 533, 535
[M + H]+.
-
Herstellungsbeispiel 35
-
Synthese von 10-Amino-2-[(-)-4-(N-benzoyl-N-methylamino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.70-3.89 (16H,
komplex),
6.71 (2H, s),
6.91-7.20 (2H, komplex),
7.35-7.80
(8H, komplex),
7.82-7.88 (1H, m),
8.34 (1H, br.d.).
MS
(FAB, m-NBA)
m/z → 533,
535 [M + H]+.
-
Herstellungsbeispiel 36
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyllamino-3-(3,4-dichlorophenyl]butyl]-10-hydroxymethyl-1,2,3,4-tetrahydro-benzo-[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.25-4.28 (16H,
komplex),
5.03 (2H, s),
6.71-7.75 (10H, komplex),
7.99
(1H, brd),
8.17 (1H, brd).
MS (ESI)
m/z 548, 550,
552 → [M
+ H]+.
-
Herstellungsbeispiel 37
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-hydroxymethyl-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.70-5.12 (19H,
komplex),
6.72 (2H, s),
6.80-7.90 (10H, komplex).
7.95
(1H, brd),
8.27 (1H, brd).
MS (FAB, m-NBA)
m/z 548,
550 → [M
+ H]+.
-
Herstellungsbeispiel 38
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-phenyl-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.59-3.95 (16H,
komplex),
6.60-7.69 (16H, komplex),
8.03 (1H, br.s.).
MS
(FAB, m-NBA)
m/z → 594,
596 [M + H]+.
-
Herstellungsbeispiel 39
-
Synthese von 2-[(-)-4-(N-Benzovl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-phenyl-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.50-3.85 (16H,
komplex),
6.71 (2H, s),
6.80-7.78 (16H, komplex),
7.99
(1H, br.d.).
MS (FAB, m-NBA)
m/z → 594, 596 [M + H]+.
-
Herstellungsbeispiel 40
-
Synthese von 2-j(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-methoxycarbonylamino-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.18-3.83 (19H,
komplex),
6.72-7.73 (10H, komplex),
7.86 (1H, br.d.),
8.00
(1H, br.d.).
MS (FAB, m-NBA)
m/z → 591, 593 [M + H]+.
-
Herstellungsbeispiel 41
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-methoxycarbonylamino-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
0.84-4.17 (19H,
komplex),
6.72 (2H, s),
6.93-8.08 (12H, komplex).
MS
(FAB, m-NBA)
m/z → 591,
593 [M + H]+.
-
Herstellungsbeispiel 42
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-(N-methyl-N-methoxycarbonylamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridin
-
sDie
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.65-3.90 (22H,
komplex),
7.05-7.67 (11H, komplex),
8.04 (1H, br.d.).
MS
(FAB, m-NBA)
m/z → 605,
607 [M + H]+.
-
Herstellungsbeispiel 43
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-(N-methyl-N-methoxycarbonyllamino-1,2,3,4-tetrahydro-benzo[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
0.82-3.88 (22H,
komplex),
6.74 (2H, s),
6.96-7.86 (11 H, komplex),
8.02
(1H, br.d.).
MS (FAB, m-NBA)
m/z → 605, 607 [M + H]+.
-
Herstellungsbeispiel 44
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methylamino-3-(3,4-dichlorophenyl)butyl]-10-chloro-1,2,3,4-tetrahydrobenzo[b],[b]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.60-4.10 (16H,
komplex),
6.72-7.50 (8H, komplex),
7.55-7.64 (1H,
m),
7.65-7.80 (1H, m),
8.00 (1H, d, J=8.0Hz),
8.18
(1H, dd, J=1.2, 8.4Hz).
MS (FAB, m-NBA)
m/z 552, 554,
556 → [M
+ H]+.
-
Herstellungsbeispiel 45
-
Synthese von 2-[(-)-4-(N-Benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-10-chloro-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.50-4.12 (16H,
komplex),
6.73 (2H, s),
6.80-8.35 (12H, komplex).
MS
(FAB, m-NBA)
m/z 552, 554, 556 → [M + H]+.
-
Herstellungsbeispiel 46
-
Synthese von 10-Benzoylamino-2-[(-)-4-(N-benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4-tetrahydro-benzo-[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MNzFT, TMS, CDCl3)
1.40-4.10 (16H,
komplex),
6.68-8.60 (18H, komplex).
MS (FAB, m-NBA)
m/z → 637, 639
[M + H]+.
-
Herstellungsbeispiel 47
-
Synthese von 10-Benzoylamino-2-[(-)-4-(N-benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4-tetrahydro-benzo-[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.50-4.10 (16H,
komplex),
6.71 (2H, s),
6.80-8.18 (17H, komplex).
MS
(FAB, m-NBA)
m/z 637, 639 → [M
+ H]+.
-
Herstellungsbeispiel 48
-
Synthese von 10-Acetylamino-2-[(-)-4-(N-benzoyl-N-methyl)amino-3-(3,4-dichlorophenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo[b][1,6]naphthyridin
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 26 beschrieben.
1H-NMR
(200 MHzFT, TMS, CDCl3)
1.60-4.15 (27H,
komplex),
6.70-7.50 (9H, komplex).
MS (FAB, m-NBA)
m/z
579, 581 → [M
+ H]+.
-
Herstellungsbeispiel 49
-
Synthese von 10-Acetylamino-2-((-)-4-(N-benzovl-N-methyllamino-3-(3,4-dichloroghenyl)butyl]-1,2,3,4,6,7,8,9-octahydro-benzo-[b][1,6]naphthyridinfumarat
-
Die
im Titel genannte Verbindung wurde auf dieselbe Weise erhalten,
wie in Herstellungsbeispiel 27 beschrieben.
1H-NMR
(200 MHzFT, TMS, CD3OD)
1.65-2.00 (4H,
m),
2.02-4.00 (23H, komplex),
6.67 (2H, s),
6.90-7.60
(8H, komplex).
MS (FAB, m-NBA)
m/z 579, 581 → [M + H]+.
-
Herstellungsbeispiel 50
-
Herstellung von Injektionen
-
Gereinigtes
Wasser wurde zu 30 Gew.-Teilen jeder Verbindung gemäß der vorliegenden
Erfindung und 18 Gew.-Teilen Natriumchlorid (100 Gew.-Teile an Glukose)
gegeben, so dass die Gesamtmenge 2,000 Gew.-Teile sein konnte. Nachdem
Auflösung
erzielt wurde, wurde die Lösung
mittels Filtration durch einen Millipore-Filter GS-Typ® sterilisiert.
2 Gramm des Filtrats wurden in Ampullen dispensiert und jede Ampulle
wurde mit einem Stopfen versehen und mit einem Band abgedichtet,
indem das Band fest um die Ampulle gewickelt wurde. Dadurch wurden
Ampullen erhalten, die jeweils 30 mg der Verbindung gemäß der vorliegenden
Erfindung enthielten.
-
Herstellungsbeispiel 51
-
Herstellung von Tabletten
-
10
Gew.-Teile von jeder Verbindung gemäß der vorliegenden Erfindung,
30 Gew.-Teile Kartoffelstärke, 150
Gew.-Teile kristalline Laktose, 108 Gew.-Teile kristalline Cellulose
und 2 Gew.-Teile Magnesiumstearat wurden in einem Zwillingszylinder-Mischer
gemischt und in Tabletten überführt, die
jeweils 60 mg wogen und jeweils 2 mg der Verbindung gemäß der vorliegenden
Erfindung enthielten.
-
Die
physiologische Aktivität
der Verbindungen gemäß der vorliegenden
Erfindung ist konkret unter Verweis auf die folgenden Testbeispiele
erklärt.
-
Testbeispiel 1
-
Bindung an NK-2 Rezeptoren
-
Duodenuma,
welche aus männlichen
Ratten des Fischer-Stamms isoliert wurden, wurden in 50 mN Tris-HCl-Puffer
(pH 7.4) enthaltend Saccharose (100 mg/l) und Ethylendiamintetraessigsäure (1 mM)
homogenisiert und das erhaltene Homogenat wurde bei 48,000 g und
4 °C für 20 Minuten
zentrifugiert. Die so erhaltenen Pellets wurden dreimal mit der
10-fachen Menge (bezogen auf das Volumen des Kügelchens) 50 mN Tris-HCl-Puffer
(pH 7.4), enthaltend KCl (300 mM) und Ethylendiamintetraessigsäure (10
mM), bezogen auf das Volumen der Pellets, gewaschen, und die erhaltene
Membranzubereitung wurde in der 10-fachen Menge (bezogen auf das
Volumen der Membranzubereitung) 50 mN Tris-HCl-Puffer (pH 7.4) suspendiert
und bei –80 °C bis zur
Verwendung gelagert. Die Membranzubereitung (20 mg/ml) wurde zusammen
mit einem radioaktiven Liganden 125-l-Neurokinin
A (1 × 1
0–9M)
bei Raumtemperatur für
90 Minuten in der Gegenwart von jedem Wirkstoff oder einem Lösungsmittel
dafür in
50 mN Tris-HCl-Puffer (pH 7.4) enthaltend Rinderserumalbumin (10
mg/ml), Bacitracin (40 μg/ml),
Leupeptin (4 μg/ml),
Chymostatin (50 μg/ml),
Antipain (1 × 10–4 M)
und MnCl2 (1 mM) inkubiert. Die Reaktionsmischung
wurde mittels Absaugens durch einen GE/B-Filter, der mit Polyethylenimin
(0.1 %) vorbehandelt war, filtriert und der Filter wurde mit 3 ml
an 50 mN Tris-HCl-Puffer (pH 7.4) enthaltend MnCl2(1
mM), gewaschen. Dann wurde die Radioaktivität im Filter mit einem Gamma-Zähler gemessen.
Die 50 % Hemmkonzentration (IC50) des Arzneimittels
wurde durch Betrachtung der Radioaktivität in der Gegenwart von Nle10
Neurokinin A (4-10) (3 × 10–6M)
aufgrund der nichtspezifischen Bindung des Wirkstoffs mit Hilfe
einer Ausgleichsgerade und durch Messung der Hemmung der spezifischen
Bindung durch den Wirkstoff mittels eines Kontrollexperiments nur
mit dem Lösungsmittel
bestimmt.
-
Die
Ergebnisse sind in Tabelle 1 gezeigt.
-
Tabelle
1 : IC
50 von Wirkstoffen bei Bindung von
radioaktivem Neurokinin A
-
Es
wurde gefunden, wie in Tabelle 1 deutlich wird, dass die Verbindungen
gemäß der vorliegenden Erfindung
die Bindung von Neurokinin A an NK-2 Rezeptoren bei einer niedrigen
Konzentration hemmen. Es tritt nämlich
ein merklich hemmender Effekt bezüglich der Bindung von intrinsisch
stimulierenden Verbindungen an NK-2 Rezeptoren auf.
-
Testbeispiel 2
-
Antagonismus für NK-2 Rezeptoren
-
Männliche
Meerschweinchen des Hartley-Stamms wurden durch einen Schlag auf
den Kopf getötet und
ausgeblutet. Die Tracheen wurden isoliert. Proben, jede zusammengesetzt
aus 5 Schnitten der Tracheen, wurden vorbereitet und bei einer Spannung
von 0.5 gw in einem Magunus-Bad mit einem inneren Volumen von 15
ml, umfassend eine Krebs-Henseleit-Lösung, bei 37 °C suspendiert,
während
eine Gasmischung aus 95% O2 und 5% CO2 in das Bad eingeleitet wurde. In der Krebs-Henseleit-Lösung wurde
enthaltenes Indomethacin (5 × 10–6M)
verwendet, um eine Teilnahme der intrinsischen Prostaglandine zu
verhindern. Nachdem ein maximaler Grad an Kontraktion jeder Probe
durch Verwendung von Methacholin verifiziert wurde, wurde ein Wirkstoff
oder eine Lösung
dafür zu
der Lösung
gegeben. Es folgte eine Inkubation für 1 10 Minuten und Neurokinin A
(1 × 10–9M)
wurde mit den Proben umgesetzt. Phosphoramidon (1 × 10–5M)
zur Verhinderung des Abbaus von Neurokinin A und (±)CP-96345
(3 × 10–7M)
zur Verhinderung der Teilnahme von NK-1 Rezeptoren wurden 30 Minuten
bzw. 20 Minuten vor der Zugabe des Neurokinin A zugegeben. Der Grad
an Kontraktion aufgrund von Neurokinin A wurde in Prozenten basierend
auf dem maximalen Grad an Kontraktion der Proben umgerechnet und
dann wurde die Rate der Hemmung durch den Wirkstoff relativ zu der
prozentualen Kontraktion von anderen Proben, bei denen ein Lösungsmittel
zugegeben wurde, bestimmt.
-
Die
Ergebnisse sind in Tabelle 2 gezeigt.
-
Tabelle
2: Hemmung der Neurokin A-induzierter Kontraktion durch einen Wirkstoff
-
Es
wurde gefunden, wie in Tabelle 2 deutlich wird, dass die Verbindung
gemäß der vorliegenden
Erfindung einen deutlichen, hemmenden Effekt auf die Kontraktion,
hervorgerufen durch Neurokinin A, bei einer sehr geringen Konzentration
aufweist. Die Verbindung zeigt einen sehr starken Antagonismus für NK-2 Rezeptoren.
-
Testbeispiel 3
-
Antagonismus für NK-1 Rezeptoren
-
Männliche
Meerschweinchen des Hartley-Stamms wurden durch einen Schlag auf
den Kopf getötet und
ausgeblutet, und die Ilea isoliert. Eine Probe von etwa 3 cm Länge wurde
vorbereitet und bei einer Spannung von 0.5 gw in einem Magunus-Bad
mit einem inneren Volumen von 1 5 ml. umfassend eine Krebs-Henseleit-Lösung, bei
37 °C suspendiert,
während
eine Gasmischung aus 95% O2 und 5% CO2 in das Bad eingeleitet wurde. Bezüglich der
Krebs-Henseleit-Lösung
wurde enthaltenes Atropin (1 × 10–6M)
verwendet, um eine Teilnahme von intrinsischem Acetylcholin und
der intrinsischen Prostaglandine zu verhindern. Methylsubstanz P (1 × 10–9M),
ein spezifisches stimulierendes Mittel für NK-1 Rezeptoren, wurde mehrfach
auf die Probe in Intervallen von 40 Minuten einwirken gelassen.
Nachdem die prozentuale Kontraktion aufgrund von Methylsubstanz
P konstant geworden war, wurde ein Wirkstoff zu der Lösung gegeben.
Es folgte eine Inkubation für
35 Minuten, und Methylsubstanz P wurde erneut auf die Probe einwirken
gelassen. Der Grad an Hemmung durch den Wirkstoff wurde relativ
zur prozentualen Kontraktion unmittelbar vor der Zugabe des Arzneimittels
bestimmt. Die 50 % Hemmkonzentration (IC50)
des Wirkstoffs wurde mit Hilfe einer Ausgleichsgeraden bestimmt.
-
Tabelle
3: IC
50 eines Werkstoffs auf eine Methylsubstanz
P-induzierte Kontraktion
-
Es
wurde gefunden, wie in Tabelle 3 deutlich wird, dass die Verbindung
gemäß der vorliegenden
Erfindung nicht nur Antagonismus für NK-2 Rezeptoren, sondern
auch Antagonismus für
NK-1 Rezeptoren zeigt.
-
Testbeispiel 4
-
Antagonismus für NK-3 Rezeptoren
-
Männliche
Meerschweinchen des Hartley-Stamms wurden durch einen Schlag auf
den Kopf getötet und
ausgeblutet, und die Ilea isoliert. Eine Probe von etwa 3 cm Länge wurde
vorbereitet und bei einer Spannung von 0.5 gw in einem Magunus-Bad
mit einem inneren Volumen von 1 5 ml, umfassend eine Krebs-Henseleit-Lösung bei
37 °C suspendiert,
während
eine Gasmischung aus 95% O2 und 5% CO2 in das Bad eingeleitet wurde. In der Krebs-Henseleit-Lösung enthaltenes
Atropin (1 × 10–6M)
und Indomethacin (5 × 10–6 M)
wurde verwendet, um eine Teilnahme von intrinsischem Acetylcholin
und der intrinsischen Prostaglandine zu verhindern. Senctid (1 × 10–9M),
ein spezifisches stimulierendes Mittel für NK-3 Rezeptoren, wurde mehrfach
auf die Probe in Intervallen von 50 Minuten einwirken gelassen.
Nachdem die prozentuale Kontraktion aufgrund von Senctid konstant
geworden war, wurde ein Wirkstoff zu der Lösung gegeben. Es folgte eine
Inkubation für
35 Minuten und Senctid wurde ernut auf die Probe einwirken gelassen.
Der Grad an Hemmung durch den Wirkstoff wurde relativ zur prozentualen
Kontraktion unmittelbar vor der Zugabe des Arzneimittels bestimmt.
Die Hemmkonzentration bei 50 % (IC50) des
Wirkstoffs wurde mit Hilfe einer Ausgleichsgeraden bestimmt.
-
Die
Ergebnisse sind in Tabelle 4 gezeigt.
-
Tabelle
3: IC
50 eines Wirkstoffs auf eine Senctid-induzierte
Kontraktion
-
-
Es
wurde gefunden, wie in Tabelle 4 deutlich wird, dass die Verbindung
gemäß der vorliegenden
Erfindung auch Antagonismus für
NK-3 Rezeptoren zeigt.
-
Testbeispiel 5
-
Hemmender Effekt auf die
Bronchokonstriktion
-
Es
wurden nicht behandelte männliche
Meerschweinchen des Hartley-Stamms
und männliche
Meerschweinchen des Hartley-Stamms, die 14 bis 21 Tage zuvor eine
aktive Sensibilisierung durch eine subkutane Verabreichung von 30mg/kg
an Ovalbumin erhalten hatten, eingesetzt. Die Meerschweinchen wurden
mit Pentobarbital anästhesiert
und ein Schlauch wurde in die Trachean jedes Meerschweinchens eingeführt. Die
Meerschweinchen wurden einer Überdruckbeatmung
von 10 ml/kg und 60 Mal/Minute mittels eines Apparates zur künstlichen
Beatmung unterworfen. Der Druck am Rand des Schlauches in den Tracheen
wurde als Indikation der Bronchokonstriktion bestimmt. Spontane
Atmung wurde durch Zugabe von Succinylcholin unterbunden. Den nicht
behandelten Meerschweinchen wurde wiederholt und intravenös 2 nmol/kg
an Nle Neurokinin A (4-10), ein spezifisches stimulierendes Mittel
für NK-2
Rezeptoren, in Intervallen von 10 Minuten injiziert. Nachdem die
dadurch verursachte Bronchokonstriktion stabil wurde, wurde jeder
Wirkstoff intravenös,
oral oder duodenal verabreicht. Nle Neurokinin A (4-10) wurde erneut
3 Minuten nach der intravenösen
Verabreichung oder 50 Minuten nach der oralen oder duodenalen Verabreichung
intravenös
injiziert. Der Grad an Hemmung durch das Arzneimittel wurde relativ
zu der Bronchokonstriktion vor der Verabreichung des Arzneimittels
bestimmt. Den Meerschweinchen, die eine aktive Sensibilisierung
erhalten hatten, wurde 4.6 μmol/kg
Phosphoramidon intravenös
verabreicht und 1 5 Minuten nach der Verabreichung ließ man die
Meerschweinchen für
2 Minuten ein Aerosol aus einer Ovalbumin-Antigen-Lösung (2
mg/ml), hergestellt durch einen Ultraschall-Zerstäuber, inhalieren,
um eine Bronchokonstriktion hervorzurufen. Ein Wirkstoff oder ein
Lösungsmittel dafür wurde
50 Minuten vor der Antigen-Inhalation oral verabreicht. Der Grad
an Hemmung der Wirkstoff-behandelten Meerschweinchen wurde relativ
zu der Bronchokonstriktion, gemessen 8 Minuten nach der Antigen-Inhalation, der Lösungsmittel-behandelten
Gruppe bestimmt.
-
Die
Ergebnisse sind in Tabelle 5 gezeigt.
-
-
-
Es
wurde gefunden, wie in Tabelle 5 deutlich wird, dass die Verbindungen
gemäß der vorliegenden Erfindung
bei niedrigen Dosen die Bronchokonstriktion hemmen. Sie weisen einen
deutlichen antiasthmatischen Effekt auf.
-
Testbeispiel 6
-
Toxischer
Effekt bei Mäusen
-
Die
Toxizität
der Verbindungen gemäß der vorliegenden
Erfindung wurde untersucht.
-
Tabelle
6: Toxizität
der intravenös
verabreichten Wirkstoffs
-
Es
wurde gefunden, wie in Tabelle 6 deutlich wird, dass die Verbindungen
gemäß der vorliegenden Erfindung
solch geringe Nebenwirkungen aufweisen, dass sie nicht toxisch sind,
wenn sie in hohen Dosen verabreicht wurden.
-
Wie
oben beschrieben zeigen die Verbindungen gemäß der vorliegenden Erfindung
einen exzellenten Antagonismus für
Tachykinin-Rezeptoren
und sind gänzlich
zufriedenstellend in Sachen Sicherheit. Es wurde auch bestätigt, dass,
zusätzlich
zu diesen Effekten, die Verbindungen gemäß der vorliegenden Erfindung
einen sehr unmittelbaren Effekt ausüben und so exzellent in Sachen
Sicherheit sind, dass sie nicht die höhergeordneten Strukturen eines
Organs (zum Beispiel Bronchie) modifizieren.
-
INDUSTRIELLE ANWENDBARKEIT
-
Mit
der vorliegenden Erfindung werden neue Naphthyridin-Derivate zur
Verhinderung oder Behandlung verschiedener Symptome, in denen Tachykinine
teilnehmen, und aller Tachykinin-abhängiger Krankheiten wie Krankheiten
der Atmungsorgane bereitgestellt. Zusätzlich wird mit der vorliegenden
Erfindung ein Verfahren zur Herstellung dieser neuen Naphthyridin-Derivate
bereitgestellt. Außerdem
wird mit der vorliegenden Erfindung eine pharmazeutischen Zusammensetzung
für Säugetiere,
umfassend die neuen Naphthyridin-Derivate gemäß der vorliegenden Erfindung,
bereitgestellt, die wirksam gegen die oben genannten Symptome und Krankheiten
ist.

 wobei X, Y, Z, A und G wie oben definiert sind, umfasst.
wobei X, Y, Z, A und G wie oben definiert sind, umfasst.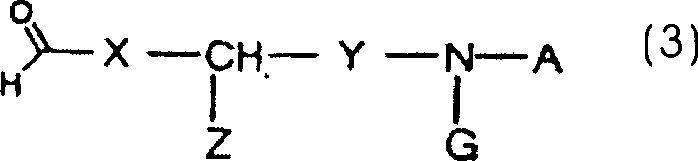
 wobei X, Y, Z, A und G wie oben definiert sind, Q ein Halogenatom oder eine R4SO2O-Gruppe ist, wobei R4 eine Niederalkylgruppe, eine Arylgruppe oder eine Trifluormethylgruppe ist, umfasst.
wobei X, Y, Z, A und G wie oben definiert sind, Q ein Halogenatom oder eine R4SO2O-Gruppe ist, wobei R4 eine Niederalkylgruppe, eine Arylgruppe oder eine Trifluormethylgruppe ist, umfasst.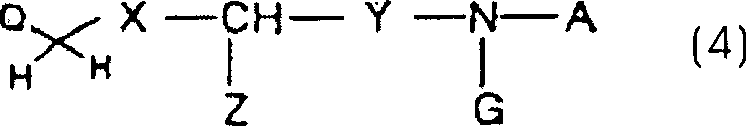

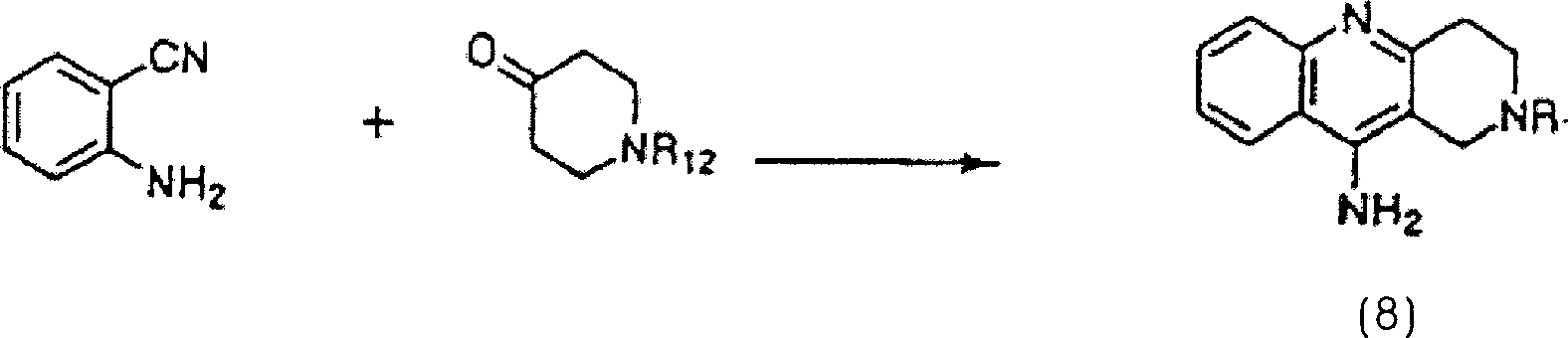 wobei R1b eine Niederalkylgruppe oder eine Arylgruppe ist, und R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (10) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (9) mit einem N-geschütztem Piperidon ohne jegliches Lösungsmittel oder in einem Lösungsmittel wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol) oder Dimethylformamid in der Gegenwart einer Säure wie zum Beispiel Schwefelsäure, Essigsäure oder Salzsäure; einer Base wie zum Beispiel Kaliumhydroxid oder Natriumhydroxid; einem Salz wie zum Beispiel Ammoniumacetat oder Piperidinacetat; oder einer Lewis-Säure wie zum Beispiel wasserfreiem Ammoniumchlorid oder Titantetrachlorid bei 130 – 160 °C in einem Ölbad erhalten werden. Herstellungsverfahren (d)
wobei R1b eine Niederalkylgruppe oder eine Arylgruppe ist, und R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (10) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (9) mit einem N-geschütztem Piperidon ohne jegliches Lösungsmittel oder in einem Lösungsmittel wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol) oder Dimethylformamid in der Gegenwart einer Säure wie zum Beispiel Schwefelsäure, Essigsäure oder Salzsäure; einer Base wie zum Beispiel Kaliumhydroxid oder Natriumhydroxid; einem Salz wie zum Beispiel Ammoniumacetat oder Piperidinacetat; oder einer Lewis-Säure wie zum Beispiel wasserfreiem Ammoniumchlorid oder Titantetrachlorid bei 130 – 160 °C in einem Ölbad erhalten werden. Herstellungsverfahren (d)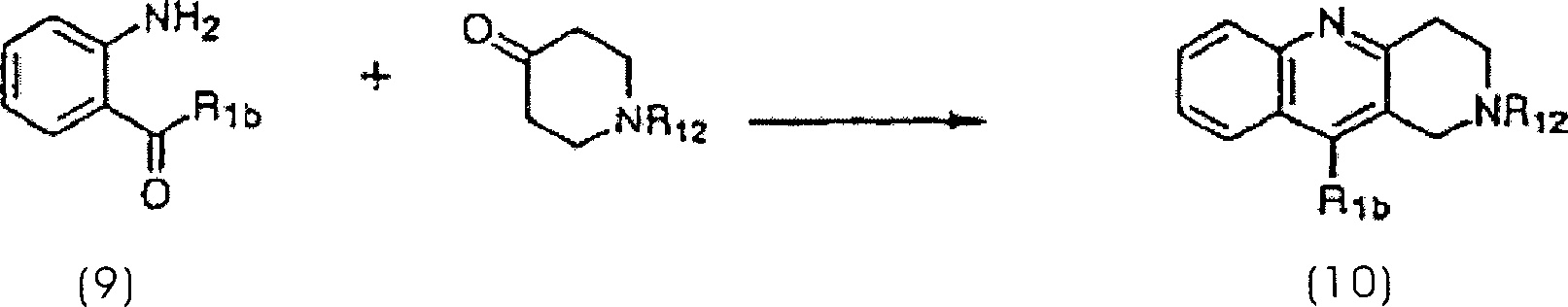 wobei R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (11) durch Zugabe von Phosphoroxychlorid zu Anthranilsäure und einem N-geschütztem Piperidon und Durchführung der Reaktion unter Erhitzen zum Rückfluss erhalten werden. Herstellungsverfahren (e)
wobei R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (11) durch Zugabe von Phosphoroxychlorid zu Anthranilsäure und einem N-geschütztem Piperidon und Durchführung der Reaktion unter Erhitzen zum Rückfluss erhalten werden. Herstellungsverfahren (e) wobei jedes R1c und R1d ein Wasserstoffatom oder eine Niederalkylgruppe ist, und R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (13) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (12) mit einem N-geschütztem Piperidon ohne jegliches Lösungsmittel oder in einem Lösungsmittel wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol) oder Dimethylformamid in der Gegenwart einer Säure wie zum Beispiel Schwefelsäure, Essigsäure oder Salzsäure; einer Base wie zum Beispiel Kaliumhydroxid oder Natriumhydroxid; einem Salz wie zum Beispiel Ammoniumacetat oder Piperidinacetat; oder einer Lewis-Säure wie zum Beispiel wasserfreies Ammoniumchlorid oder Titantetrachlorid bei 130 – 160 °C in einem Ölbad erhalten werden. Herstellungsverfahren (f)
wobei jedes R1c und R1d ein Wasserstoffatom oder eine Niederalkylgruppe ist, und R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (13) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (12) mit einem N-geschütztem Piperidon ohne jegliches Lösungsmittel oder in einem Lösungsmittel wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol) oder Dimethylformamid in der Gegenwart einer Säure wie zum Beispiel Schwefelsäure, Essigsäure oder Salzsäure; einer Base wie zum Beispiel Kaliumhydroxid oder Natriumhydroxid; einem Salz wie zum Beispiel Ammoniumacetat oder Piperidinacetat; oder einer Lewis-Säure wie zum Beispiel wasserfreies Ammoniumchlorid oder Titantetrachlorid bei 130 – 160 °C in einem Ölbad erhalten werden. Herstellungsverfahren (f) wobei R1e eine Niederalkylgruppe ist, und R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (15) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (14) mit einer Niederalkylhalogenalkylgruppe in einem Lösungsmittel wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol), Aceton oder Dimethylformamid in der Gegenwart eines basischen Katalysators wie zum Beispiel Kaliumcarbonat, Kaliumhydroxid oder Natriumhydroxid oder in der Gegenwart eines sauren Katalysators wie zum Beispiel Schwefelsäure oder Salzsäure bei Raumtemperatur bis Rückflusstemperatur erhalten werden.
wobei R1e eine Niederalkylgruppe ist, und R12 wie oben definiert ist. Im Detail kann eine Verbindung der allgemeinen Formel (15) durch Umsetzung einer Verbindung gemäß der allgemeinen Formel (14) mit einer Niederalkylhalogenalkylgruppe in einem Lösungsmittel wie zum Beispiel einem Alkohol (zum Beispiel Methanol oder Ethanol), Aceton oder Dimethylformamid in der Gegenwart eines basischen Katalysators wie zum Beispiel Kaliumcarbonat, Kaliumhydroxid oder Natriumhydroxid oder in der Gegenwart eines sauren Katalysators wie zum Beispiel Schwefelsäure oder Salzsäure bei Raumtemperatur bis Rückflusstemperatur erhalten werden.
 wobei R1f ein Wasserstoffatom, eine Aminocarbonylgruppe, dargestellt durch CONRaRb ist, wobei Ra und Rb wie oben definiert sind, eine Aminogruppe, eine Niederalkylgruppe, eine Arylgruppe, ein Halogenatom, eine Niederalkoxycarbonylgruppe, eine Carboxylgruppe, eine Hydroxyniederalkylgruppe, eine Aminoschutzgruppe, dargestellt durch NRaCORb ist, wobei Ra und Rb wie oben definiert sind, oder eine Aminoschutzgruppe, dargestellt durch NRaCOORb, ist, wobei Ra und Rb wie oben definiert sind, und R12 wie oben definiert ist.
wobei R1f ein Wasserstoffatom, eine Aminocarbonylgruppe, dargestellt durch CONRaRb ist, wobei Ra und Rb wie oben definiert sind, eine Aminogruppe, eine Niederalkylgruppe, eine Arylgruppe, ein Halogenatom, eine Niederalkoxycarbonylgruppe, eine Carboxylgruppe, eine Hydroxyniederalkylgruppe, eine Aminoschutzgruppe, dargestellt durch NRaCORb ist, wobei Ra und Rb wie oben definiert sind, oder eine Aminoschutzgruppe, dargestellt durch NRaCOORb, ist, wobei Ra und Rb wie oben definiert sind, und R12 wie oben definiert ist.





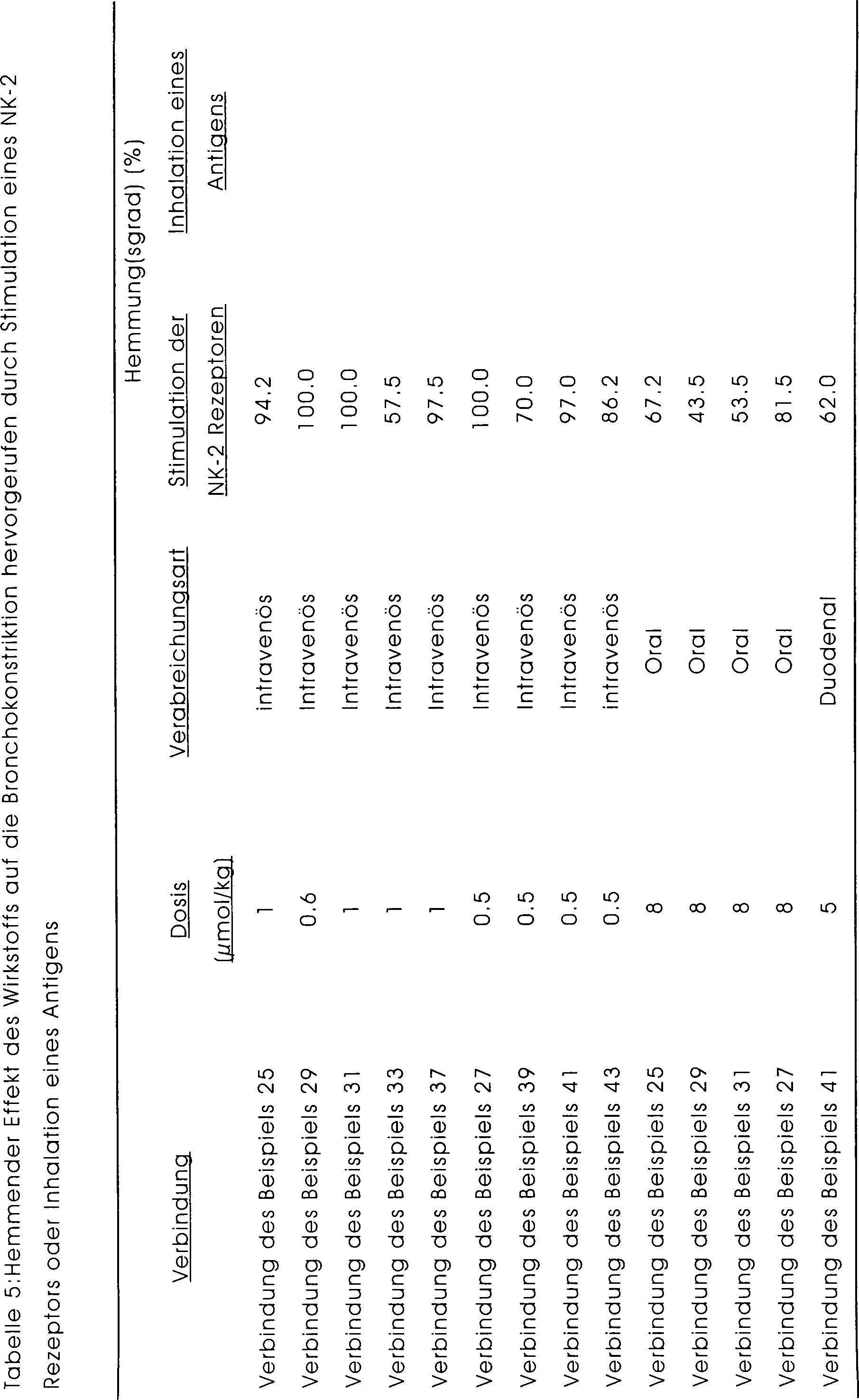

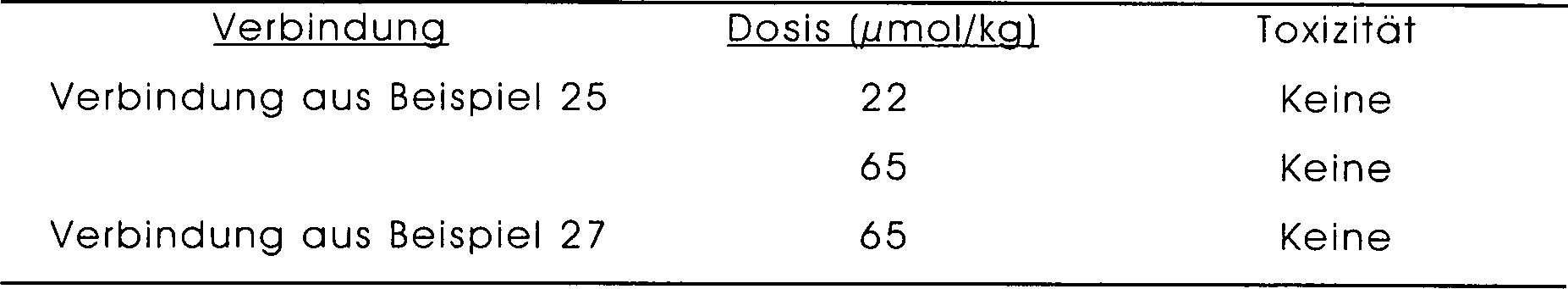
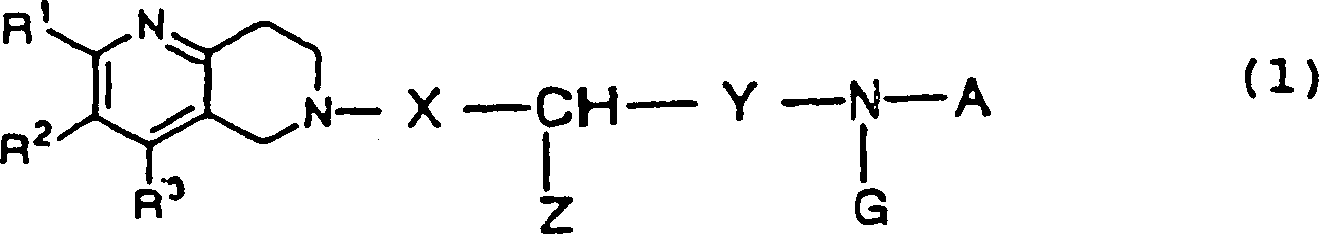
 wobei X, Y, Z, A und G wie oben definiert sind umfasst.
wobei X, Y, Z, A und G wie oben definiert sind umfasst.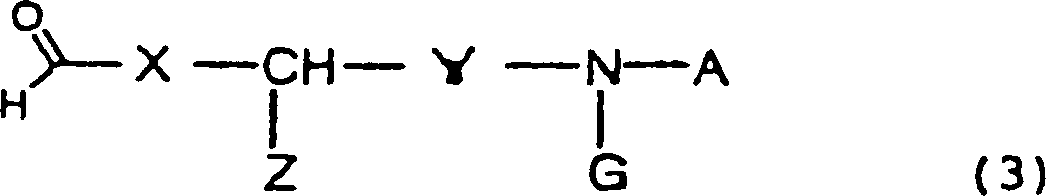
 wobei X, Y, Z, A und G wie oben definiert sind, 62 ein Halogenatom oder eine R4SO2O-Gruppe ist, wobei R4 eine Niederalkylgruppe, eine Arylgruppe oder eine Trifluoromethylgruppe ist, umfasst.
wobei X, Y, Z, A und G wie oben definiert sind, 62 ein Halogenatom oder eine R4SO2O-Gruppe ist, wobei R4 eine Niederalkylgruppe, eine Arylgruppe oder eine Trifluoromethylgruppe ist, umfasst.