-
Die
Erfindung betrifft die Konformationseinschränkung (erzwungene Konformation)
von Peptiden. Insbesondere betrifft die Erfindung die Einschränkung von
Peptiden auf eine α-helikale
Konformation. Außerdem betrifft
diese Erfindung die rationale Entwicklung und Herstellung von HIV-Vakzinen,
die auf HIV-gp41-Polypeptidsequenzen basieren. Weiters betrifft
diese Erfindung verbesserte Verfahren zur Diagnose von HIV-Infektionen
und Immunogene, die in Diagnoseverfahren zweckdienliche Antikörper induzieren.
-
HINTERGRUND
DER ERFINDUNG
-
Verschiedene
Verfahren zur Stabilisierung von α-helikaien
Peptiden wurden bereits beschrieben. Zusatz von Trifluorethanol
oder Hexafluorisopropanol wurde häufig eingesetzt, um α-Helices
in wässriger
Lösung zu
stabilisieren. Die Dimerisierung von α-Helices an hydrophoben Grenzflächen hat
ebenfalls zu einer exogenen Stabilisierung geführt. Kurze α-helikale Peptide wurden stabilisiert,
indem Gruppen an den Termini eingebaut wurden, um den immanenten
Helixdipol zu stabilisieren. Natürlich
vorkommende Capping-Motive sowie organische Matrizen wurden eingesetzt,
um α-Helices
durch Endnucleation zu stabilisieren. Verschiedene Einschränkungen
für nichtkovalente
Seitenketten wurden in Bezug auf die α-Helix-Stabilisierung untersucht,
einschließlich
hydrophaber Wechselwirkungen, Salzbrücken und Metallionenchelatierung
durch sowohl natürliche
als auch nicht-natürliche
Aminosäuren.
-
Schließlich wurden α-Helices
durch kovalente Seitenketten-„Fesseln" stabilisiert. Chorev
et al., Biochemistry 30, 5968–5974
(1991), Osapay et al., J. Am. Chem. Soc. 112, 6046–6051 (1990),
Osapay et al., J. Am. Chem. Soc. 114: 6966–6973 (1990), Bracken et al.,
J. Am. Chem. Soc. 116, 6431–6432
(1994) und Houston et al., J. Peptide Science 1, 274–282 (1995),
beschrieben die Stabilisierung von α-Helices durch Seitienketten-an-Seitenketten-Lactamisierung.
Ravi et al., J. Am. Chem. Soc. 105, 105–109 (1983) und Jackson et al.,
J. Am. Chem. Soc. 113, 9391–9392
(1991), beschrieben die Einschränkung
von Peptiden durch Disulfidbindungen zwischen Resten. Das natürlich vorkommende
Peptid Apamin wurde als Gerüst
zur Präsentation von α-helikalen
Peptidsequenzen verwendet, deren Helixkonformation durch Disulfidbindungen
an Gerüst-Cysteinreste
eingeschränkt
war.
-
AIDS
(Acquired Immune Deficiency Syndrome = erworbenes Immunschwächesyndrom)
wird durch ein Retrovirus ausgelöst,
das als HIV (Human Immunodeficiency Virus = menschliches Immunschwächevirus) bezeichnet
wird. Es wurden große
Anstrengungen unternommen, eine Vakzine zu entwickeln, die eine
schützende
Immunantwort induziert, und zwar ausgehend von der Induktion von
Antikörpern
oder zellulären
Reaktionen. Neuere Bemühungen
nutzten Untereinheitenvakzinen, worin aus Sicherheitsgründen ein
HIV-Protein und kein abgeschwächtes
oder abgetötetes
Virus als Immunogen in der Vakzine eingesetzt wird. Untereinheitenvakzinen
enthaften im Allgemeinen gp120, den Abschnitt des HIV-Hüllproteins,
der sich auf der Oberfläche des
Virus befindet.
-
Das
HIV-Hüllprotein
wurde umfassend beschrieben, und für die HIV-Hülle kodierenden Aminosäure- und
Nucleinsäuresequenzen
von verschiedenen HIV-Stämmen
sind bekannt (G. Myers et al., Human Retroviruses and Aids. A Compilation
and Analysis of Nucleic Acid and Amino Acid Sequences, Los Alamos
National Laboratory, Los Alamos, New Mexico, USA (1992)). Das HIV-Hüllprotein
ist ein Glykoprotein mit etwa 160 kd (gp160), das in der Membrandoppelschicht
in der carboxylterminalen Region verankert ist. Das N-terminale Segment,
gp120, dringt in die wässrige
Umgebung ein, die das Virion umgibt, und das C-terminale Segment, gp41,
durchdringt die Membran. Durch einen wirtszellvermittelten Prozess
wird gp160 gespalten, um gp120 und das komplette Membranprotein
gp41 zu bilden. Da keine kovalente Bindung zwischen gp120 und gp41 vorhanden
ist, wird manchmal freies gp120 von der Oberfläche von Virionen und infizierten
Zellen freigesetzt.
-
gp120
war als Vakzinenkandidat für
Untereinheitenvakzinen Gegenstand intensiver Untersuchungen, da
es das Virusprotein ist, das am leichtesten für einen Immunangriff zugänglich ist.
Klinische Studien am gp120-MN-Stamm sind derzeit am Laufen.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
Erfindung stellt ein Verfahren zur Konstruktion eines eingeschränkten helikalen
Peptids bereit, das folgende Schritte umfasst: (1) das Synthetisieren
eines Peptids, worin das Peptid eine Sequenz aus acht Aminosäureresten
umfasst, worin die Sequenz aus acht Aminosäureresten einen ersten terminalen
Rest und einen zweiten terminalen Rest aufweist, worin der erste
terminate Rest und der zweite terminate Rest eine interne Sequenz
aus sechs Aminosäureresten
flankieren und worin der erste terminate Rest eine Seitenkette aufweist,
die einen eine Amidbindung bildenden Substituenten enthält, und
der zweite terminate Rest eine Seitenkette aufweist, die einen eine
Amidbindung bildenden Substituenten enthält; (2) das Bereitstellen eines
difunktionellen Linkers mit einer ersten funktionellen Gruppe, die
zur Bildung einer Amidbindung mit dem eine Amidbindung bildenden
Seitenketten-Substituenten des ersten terminalen Rests in der Lage
ist, und einer zweiten funktionellen Gruppe, die zur Bildung einer
Amidbindung mit dem eine Amidbindung bildenden Seitenketten-Substituenten
des zweiten terminalen Rests in der Lage ist; und (3) das Zyklisieren
des Peptids durch Umsetzen des eine Amidbindung bildenden Seitenketten-Substituenten des
ersten terminalen Rests mit der ersten funktionellen Gruppe des
difunktionellen Linkers, um eine Amidbindung zu bilden, und das
Umsetzen des eine Amidbindung bildenden Seitenketten-Substituenten
des zweiten terminalen Rests mit der zweiten funktionellen Gruppe
des difunktionellen Linkers, um eine Amidbindung zu bilden, was
ein eingeschränktes
helikales Peptid ergibt.
-
Die
Erfindung stellt außerdem
ein Verfahren zur Konstruktion eines eingeschränkten helikalen Peptids bereit,
das folgende Schritte umfasst: (1) das Synthetisieren eines Peptids,
worin das Peptid eine Sequenz aus acht Aminosäureresten umfasst, worin die
Sequenz aus acht Aminosäureresten
einen ersten terminalen Rest und einen zweiten terminalen Rest aufweist,
worin der erste terminale Rest und der zweite terminale Rest eine interne
Sequenz aus sechs Aminosäureresten
flankieren, worin der erste terminale Rest eine Seitenkette aufweist,
die einen eine Amidbindung bildenden Substituenten enthält, und
der zweite terminate Rest eine Seitenkette aufweist, die einen eine
Amidbindung bildenden Substituenten enthält, und worin der eine A midbindung
bildende Seitenketten-Substituent des ersten terminalen Rests mit
einer solchen ersten Schutzgruppe und der eine Amidbindung bildende
Seitenketten-Substituent
des zweiten terminalen Rests mit einer solchen zweiten Schutzgruppe
geschützt
werden, sodass die erste Schutzgruppe und die zweite Schutzgruppe
unterschiedlich entfernbar sind; (2) das Entfernen der ersten Schutzgruppe,
sodass der eine Amidbindung bildende Seitenketten-Substituent des
ersten terminalen Rests entschützt
wird und der eine Amidbindung bildende Seitenketten-Substituent
des zweiten terminalen Rests nicht entschützt wird; (3) das Bereitstellen
eines difunktionellen Linkers mit einer ersten funktionellen Gruppe,
die zur Bildung einer Amidbindung mit dem eine Amidbindung bildenden
Seitenketten-Substituenten des ersten terminalen Rests in der Lage
ist, und einer zweiten funktionellen Gruppe, die zur Bildung einer
Amidbindung mit dem eine Amidbindung bildenden Seitenketten-Substituenten des
zweiten terminalen Rests in der Lage ist; (4) das Umsetzen des Peptids
mit dem difunktionellen Linker, um eine Amidbindung zwischen der
ersten funktionellen Gruppe des difunktionellen Linkers und dem
eine Amidbindung bildenden Seitenketten-Substituenten des ersten
terminalen Rests zu bilden; (5) das Entfernen der zweiten Schutzgruppe,
um den eine Amidbindung bildenden Seitenketten-Substituenten des zweiten terminalen
Rests zu entschützen;
und (6) das Zyklisieren des Peptids durch intramolekulares Umsetzen
des eine Amidbindung bildenden Seitenketten-Substituenten des zweiten
terminalen Rests mit der zweiten funktionellen Gruppe des difunktionellen
Linkers, um eine Amidbindung zu bilden und ein eingeschränktes helikales
Peptid zu erhalten.
-
Weiters
stellt die Erfindung ein Verfahren zur Konstruktion eines eingeschränkten helikalen
Peptids bereit, das folgende Schritte umfasst: (a) das Synthetisieren
eines Peptids, worin das Peptid eine Sequenz aus acht Aminosäureresten
umfasst, worin die Sequenz aus acht Aminosäureresten einen ersten terminalen
Rest und einen zweiten terminalen Rest aufweist, worin der erste
terminale Rest und der zweite terminale Rest eine interne Sequenz
aus sechs Aminosäureresten
flankieren, worin der erste terminale Rest eine Seitenkette aufweist,
die einen eine Amidbindung bildenden Substituenten enthält, und
der zweite terminale Rest eine Seitenkette aufweist, die einen eine
Amidbindung bildenden Substituenten enthält, worin der erste termina le
Rest an einen difunktionellen Linker gebunden ist, der eine erste
funktionelle Gruppe und eine zweite funktionelle Gruppe aufweist,
worin die erste funktionelle Gruppe in einer Amidbindung mit dem
eine Amidbindung bildenden Seitenketten-Substituenten des ersten terminalen
Rests vorliegt und worin die zweite funktionelle Gruppe des difunktionellen
Linkers zur Bildung einer Amidbindung mit dem eine Amidbindung bildenden
Seitenketten-Substituenten des zweiten terminalen Rests in der Lage
ist; und (b) das Zyklisieren des Peptids durch intramolekulares
Umsetzen des eine Amidbindung bildenden Seitenketten-Substituenten
des zweiten terminalen Rests mit der zweiten funktionellen Gruppe
des difunktionellen Linkers, um eine Amidbindung zu bilden und ein
eingeschränktes
helikales Peptid zu erhalten.
-
Außerdem stellt
die Erfindung ein Verfahren zur Konstruktion eines eingeschränkten helikalen
Peptids bereit, das folgende Schritte umfasst: (1) das Synthetisieren
eines Peptids, worin das Peptid eine Sequenz aus acht Aminosäureresten
umfasst, und worin die Sequenz aus acht Aminosäureresten einen ersten terminalen Rest
und einen zweiten terminalen Rest aufweist, worin der erste terminate
Rest und der zweite terminale Rest unabhängig voneinander aus Asp und
Glu ausgewählt
sind; (2) das Bereitstellen eines Diaminlinkers mit einer ersten
Aminogruppe, die zur Bildung einer Amidbindung mit der Carboxyseitenkette
des ersten terminalen Rests in der Lage ist, und einer zweiten Aminogruppe,
die zur Bildung einer Amidbindung mit der Carboxyseitenkette des
zweiten terminalen Rests in der Lage ist; und (3) das Zyklisieren
des Peptids durch Umsetzen der ersten Aminogruppe des Diaminlinkers
mit der Carboxyseitenkette der ersten terminalen Rests, um eine
Amidbindung zu bilden, und das Umsetzen der zweiten Aminogruppe
des Diaminlinkers mit der Carboxyseitenkette des zweiten terminalen
Rests, um eine Amidbindung zu bilden, was ein eingeschränktes helikales
Peptid ergibt.
-
Die
Erfindung umfasst auch ein Verfahren zur Konstruktion eines eingeschränkten helikalen
Peptids, das folgende Schritte umfasst: (1) das Synthetisieren eines
Peptids, worin das Peptid eine Sequenz aus acht Aminosäureresten
umfasst, worin die Sequenz aus acht Aminosäureresten einen ersten terminalen
Rest und einen zweiten terminalen Rest aufweist, worin der erste
terminale Rest und der zweite terminale Rest eine interne Sequenz
aus sechs Aminosäureresten
flankieren, worin der erste terminale Rest und der zweite terminale
Rest unabhängig
voneinander aus Asp und Glu ausgewählt sind und worin die Carboxyseitenkette
des ersten terminalen Rests mit einer solchen ersten Schutzgruppe
und die Carboxyseitenkette des zweiten terminalen Rests mit einer
solchen zweiten Schutzgruppe geschützt sind, dass die erste Schutzgruppe
und die zweite Schutzgruppe unterschiedlich entfernbar sind; (2)
das Entfernen der ersten Schutzgruppe, sodass die Carboxyseitenkette
des ersten terminalen Rests entschützt wird und die Carboxyseitenkette
des zweiten terminalen Rests nicht entschützt wird; (3) das Umsetzen
des Peptids mit einem Diaminlinker, der eine erste Aminogruppe und
eine zweite Aminogruppe aufweist, um eine Amidbindung zwischen der
entschützten
Carboxyseitenkette des ersten terminalen Rests und der ersten Aminogruppe
des Diaminlinkers zu bilden; (4) das Entfernen der zweiten Schutzgruppe,
um die Carboxyseitenkette des zweiten terminalen Rests zu entschützen; und
(5) das Zyklisieren des Peptids durch intramolekulares Umsetzen
der entschützten
Carboxyseitenkette des zweiten terminalen Rests mit der zweiten
Aminogruppe des Diaminlinkers, um eine Amidbindung zu bilden und
ein eingeschränktes
helikales Peptid zu erhalten.
-
Die
Erfindung umfasst weiters ein Verfahren zur Konstruktion eines eingeschränkten helikalen
Peptids, das folgende Schritte umfasst: (1) das Synthetisieren eines
Peptids, worin das Peptid eine Sequenz aus acht Aminosäureresten
umfasst, worin die Sequenz aus acht Aminosäureresten einen ersten terminalen
Rest und einen zweiten terminalen Rest aufweist, worin der erste
terminale Rest und der zweite terminale Rest eine interne Sequenz
aus sechs Aminosäureresten
flankieren, worin der erste terminale Rest und der zweite terminale
Rest unabhängig
voneinander aus Asp und Glu ausgewählt sind, und worin die Carboxyseitenkette
des ersten terminalen Rests an einen Diaminlinker mit einer ersten
Aminogruppe und einer zweiten Aminogruppe verbunden ist, sodass
die Carboxyseitenkette des ersten terminalen Rests in einer Amidbindung
mit der ersten Aminogruppe des Diaminlinkers vorliegt; und (2) das
Zyklisieren des Peptids durch intramolekulares Umsetzen der entschützten Carboxyseitenkette
des zweiten terminalen Rests mit der zweiten Aminogruppe des Diamin linkers,
um eine Amidbindung zu bilden und ein eingeschränktes helikales Peptid zu erhalten.
-
Die
Erfindung umfasst außerdem
eine Verbindung, die aus der aus Folgendem bestehenden Gruppe ausgewählt ist:
Verbindungen
der Formel (1):
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze Zahl
im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist;
Verbindungen der Formel (6):
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren ist;
q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt ist,
s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist,
mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist;
Verbindungen der Formel (11):
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist; und
Verbindungen der Formel (16):
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze Zahl
im Bereich von (7-(w + y)) bis (9 – (w + y)), Grenzen eingeschlossen,
ist, mit der Maßgabe,
dass x größer als
oder gleich 0 ist.
-
In
einer bevorzugten Ausführungsform
wird eine Verbindung bereitgestellt, die ein eingeschränktes helikales
Peptid enthält,
das wiederum ein Peptid aus einer Sequenz von acht Aminosäureresten
umfasst, worin die Sequenz von acht Aminosäureresten einen ersten terminalen
Rest und eine zweiten terminalen Rest aufweist, die eine interne
Sequenz aus sechs Aminosäureresten
flankieren und die Seitenketten aufweisen, die aneinander gebunden
sind und so eine Arretiergruppierung bilden, um ein eingeschränktes helikales
Peptid zu bilden. Die interne Sequenz aus sechs Aminosäuren weist
die Form gabcde, defgab oder cdefga auf und ist aus einer Gruppe
von Sequenzen ausgewählt,
die eine Sequenz aus sechs zusammenhängenden Aminosäuren in
der HIV-1LAI-Stamm-gp41-Aminosäuresequenz
633 bis 678, in deren homologer Sequenz von einem anderen HIV-Stamm,
in einer Consensus-Sequenz derer homologen Sequenzen von einer beliebigen
HIV-Clade oder einer Aminosäure-Substitutionsvariante
davon umfasst, worin der Aminosäure
633 oder ihrer entsprechenden Aminosäure in der homologen Sequenz,
Consensus-Sequenz oder Sequenzvariante die Position a einer Grundaufstellung
abcdefg für
die Sequenz 633–678
zugeordnet ist (wie in 18 dargestellt).
In diesen Verbindungen liegt die Arretiergruppierung oder Fessel
zwischen benachbarten f-Positionen vor, wenn die interne Sequenz
die Form gabcde aufweist, zwischen benachbarten c-Positionen, wenn
die interne Sequenz die Form defgab aufweist, oder zwischen benachbarten
b-Positionen, wenn
die interne Sequenz die Form cdefga aufweist. Besonders bevorzugt
befindet sich die Arretierung zwischen benachbarten f-Positionen. 18 zeigt die Anordnung der Grundaufstellung abcdefg
mit den Aminosäuren
in der 633-678-Region.
In einer bevorzugten Ausführungsform
weist die interne Sequenz aus sechs Aminosäuren die Form gabcde auf. Die
Verbindungen weisen vorzugsweise antifusogene oder antinfektiöse HIV-Aktivität auf.
-
Bevorzugte
Verbindungen sind solche, die aus der aus eingeschränkten helikalen
Peptiden mit allen möglichen
Sequenzen bestehenden Gruppe ausgewählt sind, die eine oder jede
beliebige Kombination von Aminosäuresubstitutionen,
die in der in 23A und 23B dargestellten
Serie der eingeschränkten
helikalen Peptide I bis XII angeführt sind, in Kombination mit
einer oder jeder beliebigen Kombination von Aminosäuretrunkierungen,
die in der in 23A und 23B dargestellten
Serie der eingeschränkten
helikalen Peptide I bis XII angeführt sind, aufweist. Die Peptide
HIV24 und HIV31 sind besonders bevorzugte Verbindungen dieser Art.
-
In
einer anderen Ausführungsform
werden die Verbindungen der Erfindung, vorzugsweise an Träger gebunden,
als Haptene eingesetzt, und zwar zur Verwendung als Immunogen zur
Bildung von Antikörpern
für diagnostische
Zwecke oder als Vakzine zur prophylaktischen oder therapeutischen
Behandlung von Patienten mit einem Risiko für eine HIV-Infektion. Beispiele
für solche
prophylaktische Anwendungen der Peptide können die Prävention der Übertragung
eines Virus von der Mutter auf das Kind und in anderen Situationen,
in denen die Gefahr einer HIV-Übertragung
besteht, wie beispielsweise bei Unfällen im Gesundheitsbereich,
wo Mitarbeiter mit HIV-hältigen Blutprodukten
in Kontakt kommen, umfassen, sind jedoch nicht darauf beschränkt. Die eingeschränkten Peptide
der Erfindung können
als prophylaktische Vakzine dienen, wobei der Wirt Antikörper gegen
das Peptid der Erfindung bildet, die dann zur Neutralisierung von
HIV-Viren durch beispielsweise Hemmung einer weiteren HIV-Infektion
dienen.
-
KURZBESCHREIBUNG
DER ZEICHNUNGEN
-
1 ist
eine Darstellung, welche die Synthese von Peptid 1b und 1c zeigt.
Reagens a ist 20%igem Piperidin in DMA; Reagens b ist H2NCH2CH2CH2NHR
(R=H oder BOC), BOP, DIPEA, CH2Cl2; Reagens c ist Pd(PPh3)4, 20%igem Piperidin
in DMA, (R=BOC) TFA/CH2Cl2/Anisol/1,2-Ethandithiol
45:45:5:5 Vol./Vol.; Reagens d ist BOP, DIPEA, CH2Cl2; Reagens e ist HF/Anisol/EtSMe 20:2:1 Vol./Vol.,
0°C; und
Reagens f ist CH3NH2,
BOP, CH2Cl2.
-
2 ist
eine Darstellung, welche die Synthese von N-Fmoc-S-Acm-D-thiolysin
(Verbindung 7) zeigt. Reagens a ist nBuLi,
THF, –78°C; Br(CH2)4Br; Reagens b
ist 4-McOBnSN, KOtBu, THF; Reagens c ist
0,25 M HCl, THF/H2O; Reagens d ist Hg/OAc)2, TFA, H2S; Reagens
e ist Acetamidomethanol, TFA; Reagens f ist LiOH, THF/H2O;
und Reagens g ist Fmoc-OSu, Dioxan, NaHCO3.
-
3a und 3b sind
graphische Darstellungen des HN-Hα-
bzw. HN-HN-Abschnitts
des ROESY-Spektrums von Peptid 1c. Das Spektrum wurde bei 280 K,
pH 5,0, 500 MHz und einer Peptidkonzentration von 1,5 mM mit einem
4,5-kHz-Spinlock-Mischimpuls mit einer Dauer von 200 ms aufgenommen.
ROEs sind durch Linien verbunden, anhand derer sequentielle Zuweisungen
vorgenommen wurden. Rechteckige, ovale und rautenförmige Kästchen kennzeichnen
Korrelationen innerhalb von Resten, Sequenz- bzw. (I,I+3)-Korrelationen.
-
4 ist
eine graphische Darstellung von ROE- und 3JHN-Hα-Daten
für die
Peptide 1c und 1b. Für
die dNN- und dαN-Reihe
ist die Beobachtung des sequentiellen ROE durch einen Balken dargestellt,
der zwei Reste verbindet, wobei die Dicke des Balkens die relative
Intensität
des ROE anzeigt. Die nach unten weisenden Pfeile zeigen eine 3JHN–Hα von unter 6,0 Hz an.
Die beobachteten mittleren ROEs (Hα-HN I,I+3 und Hα-Hβ I,I+3) sind
durch die Linien im unteren Teil der Figur angegeben; gestrichelte
Linien und Sterne kennzeichnen ROEs, die aufgrund von Degeneration
der chemischen Verschiebung nicht eindeutig identifiziert werden
konnten. Das Schleifenmotiv über
der Primärsequenz
zeigt die Region an, von der aus den NMR-Daten gefolgert wurde, dass
sie die Helixstruktur aufweist; die gestrichelten Schleifen zeigen
Abschnitte eines Peptids an, in denen nur ein Teil der NMR-Daten
auf einen helikalen Charakter hinweist.
-
5 ist
ein Molekülmodell,
das eine Gruppe von 20 rMD-Strukturen darstellt, die mithilfe von NMR-Daten
für Peptid
1c berechnet wurden. Die Strukturen wurden unter Verwendung der
N-, Cα-
und C-Atome der Reste Thr1 bis Gln10 überlagert. Schwere Rückgrat-
und Seitenkettenatome sind durch durchgehende bzw. gestri chelte
Linien dargestellt. Die Seitenketten von Arg8 und Arg9 sind an Cγ trunkiert,
und alle Seitenkettenatome von Gln11 und Gln12 sind aus Gründen der Übersichtlichkeit
weggelassen.
-
6 ist
eine graphische Darstellung, die das CD-Spektrum von Peptid 1c bei
280, 310, 330, 350 und 370 K zeigt.
-
7 ist
eine graphische Darstellung, die das CD-Spektrum der Peptide 1 und
3 (Apamin-basierte Sequenzen) bei 280 K zeigt.
-
8 ist
eine graphische Darstellung, die das CD-Spektrum der Peptide 2 und
4 (C-Peptid-basierte Sequenzen)
bei 280 K zeigt.
-
9 ist
eine graphische Darstellung, die das Hitzedenaturierungsprofil von
Peptid 1c zeigt, das durch CD-Spektren vor, während und nach dem Erhitzen
auf 87°C
für 1 Tag
erhalten wurden. Kreise kennzeichnen das Anfangsspektrum, das vor
dem Erhitzen von einer Probe erhalten wurde; Quadrats kennzeichnen
das Spektrum, das von einer Probe während einer Inkubation bei
87°C erhalten
wurde; Dreiecke kennzeichnen das Spektrum, das von einer Probe nach
dem Abkühlen
auf 7°C
mit 0,2°C/min
erhalten wurde.
-
10 ist eine graphische Darstellung, die einen
Abschnitt des TOCSY-Spektrums von Peptid 1c zeigt. Die Daten wurden
bei 280 K, pH 5,0, 500 MHz und einer Peptidkonzentration von 1,5
mM mit einer Mischdauer von 90 ms gewonnen. Die durchgehenden Linien
verbinden Kreuzpeaks zwischen Rückgratamid- und
Seitenketten-Protonen;
Zuordnungen sind über
den einzelnen Linien angegeben. Gestrichelte Linien verbinden Kreuzpeaks
zwischen den Seitenkettenamid-Protonen von Gln3 und Gln10 und den
Methylenlinker-Resonanzen.
-
11 ist eine Darstellung, welche die Synthese einer
arretierten Helixspezies des Peptids Asn-Met-Glu-Gln-Gln-Arg-Arg-Phe-Tyr-Glu-Ala-Leu-His
zeigt, worin die Car boxyseitenketten der Glu-Reste kovalent mit
einem 1,5-Pentandiamin-Linker verbunden sind.
-
12 zeigt Sequenzen und schematische Darstellungen
der arretierten Helixpeptid-Ausführungsformen
der Erfindung. Die Zylinder stellen α-Helices dar, wobei die getüpfelten
Flächen
der hydrophoben 4,3-Wiederholung entsprechen. Kovalente Einschränkungen,
die Seitenketten bei I und I+7 verbinden, sind als dunkle Linien
dargestellt.
-
13 ist ein Zirkulardichroismus-Spektrum der Peptide
HIV24 (weiße
Quadrate), HIV30 (weiße
Kreise), HIV31 (schwarze Kreise) und HIV35 (schwarze Quadrate).
Die Spektren wurden bei 7°C
in 10 mM Tris-HCl, pH 7,5 aufgenommen (21).
-
Die 14A und 14B sind
Diagramme, welche die Wirkung von inhibitarischen Peptiden in Primärinfektiositätstests
unter Einsatz von PBMCs mit dem Virus JRCSF, einem NSI-Stamm (14A), und BZ167, einem SI-Stamm (14B), zeigen (22). HIV24 (schwarze Dreiecke),
HIV31 (schwarze Kreise); HIV35 (schwarze Quadrate); DP178 (weiße Quadrate).
-
15 ist eine schematische Darstellung eines Vorschlags
für einen
Mechanismus zur Erzeugung des fusogenen Zustands von gp41 (oben)
und die Hemmung durch eingeschränkte
Peptide (unten).
-
Die 16A bis 16M zeigen
Aminosäuresequenzen
von gp41 von bekannten NIV-Virusstämmen und
ihre Consensus-Seguenzen, basierend auf der statistischen Aminosäurehäufigkeit.
Aminosäuren
sind im herkömmlichen
Einbuchstabencode anangeben. Die Stämme innerhalb der einzelnen
HIV-Claden sind angegeben. Ein „-" in einer Sequenz kennzeichnet die entsprechende
Position in Virussequenzen innerhalb dieser Clade. Eine groß geschriebene
Aminosäure
in einer Consensus-Sequenz zeigt an, dass nur diese Aminosäure an der
entsprechenden Position in Virussequenzen innerhalb dieser Clade
gefunden wurde. Stammbezeichnungen ohne Se quenzinformationen zeigen
an, dass nicht die gesamte gp41-Sequenz bestimmt wurde.
-
17 ist eine Zusammenfassung von Consensus-Sequenzen
von bekannten Stämmen.
Die Peptidsequenz von DP178 ist hervorgehoben. Die Nomenklatur ist
dieselbe wie in den 16A bis 16G.
-
18 ist eine schematische Darstellung von Sequenzanordnungen
von Consensus-Sequenzen
der Claden A, B, C, D und E, der Peptide DP178 und HIV35 sowie des
Neurath-Peptids, worin die Heptaden-Grundaufstellung abcdefg, wie
sie hierin gelehrt wird, bereitgestellt wird und die Positionen
einiger einschränkender
Arretierungen angezeigt sind. Den Aminosäuren in der Sequenz ESQNQQ
von DP178 sind zum Zwecke der vorliegenden Erfindung beispielsweise
die Positionen g, a, b, c, d bzw. e zugeordnet, sodass es die Form
gabcde aufweist. Diese Sequenz ist die interne Sequenz aus sechs
Aminosäuren,
die im Peptid HIF24 vorhanden ist, das eine einfach arretierte Form
der HIV35-Sequenz ist. Positionen von internen Sequenzen der Erfindung
sind jene, die sich zwischen Arretierungsresten befinden, deren
Positionen durch das Symbol „|" angezeigt sind und
denen in diesem Beispiel jeweils die Position f zugeordnet ist.
Positionen zur Platzierung von entweder einer, zwei oder drei Arretierungen
in den repräsentativen
gezeigten Sequenzen sind angegeben. Die Figur stellt fünf helikale
Abschnitte der Form gabcde zur Arretierung dar, wenn Arretierungen an
benachbarten f-Positionen erfolgen. Außerdem sind die Positionen
von helikalen Abschnitten der Form gabcde dargestellt, wenn eine,
zwei oder drei i-bis-i+7-Arretierungen
in einer 633-678-Sequenz oder Variante davon vorhanden sind. Die
zweifach arretierten Varianten sind als (II), (III), HIV31, (VI)
und (VII) bezeichnet. und die einfach arretierten Varianten sind
(VIII), (IX), HIV24, (XI) und (XII). Die dreifach arretierte Variante
ist als (I) bezeichnet.
-
19 ist eine helikale Raddarstellung der repräsentativen
gp42-Fusionsproteinsequenz aus dem HIV-1-LAI-Stamm, weiche das „abcdefg"-Heptaden-Leseraster
und das Heptaden-Wiederholungsmuster, wie es hierin zum Zwecke der
vorliegenden Erfindung zugeordnet ist (siehe 18),
zeigt.
-
20 ist eine schematische Darstellung, welche die
Verwendung der Verbindungen der Erfindung als Haptene zur Immunisierung
darstellt und das gp41-Kerntrimer, seine DP178-Bindungsfurche und
die 633-678-Region, die diese Gruppe bindet, Hapten a präsentiert
die 4,3-Wiederholungsoberfläche
aus HIV24 in einer eingeschränkten
Helix. Diese Fläche
würde vermutlich
niemals dem Immunsystem exponiert werden, da sie wahrscheinlich
nicht gebildet würde,
bis sie der Trimer-„Furchenbindungsstelle" ausgesetzt wird.
Ein dagegen gebildeter Antikörper
wäre jedoch
im Wesentlichen ein antiidiotypischer Antikörper gegen die Trimerfurchen.
Wenn gp41 im Ruhezustand diesem Antikörper ausgesetzt wird, würde es den
C-Terminus von gp41 dazu bringen, eine Helix in einem nichtproduktiven
Zustand, d.h. an den Antikörper
gebunden, zu bilden, wodurch das Protein aus dem Fusionsweg heraus
maskiert wird. Von Antikörpern
gegen Hapten a wird erwartet, dass sie an die angegebene Region
binden und diese Region veranlassen, eine Helix in einer nichtproduktiven Orientierung
zu bilden, wodurch eine letztendliche Anordnung des fusogenen helikalen
Bündels
verhindert wird.
-
21 ist eine schematische Darstellung, die einen
Vorschlag für
den Mechanismus einer Antikörper-Intervention
in HIV-Virusinfektiosität
zeigt.
-
22 zeigt eine Consensus-Sequenz der HIV-gp41-Sequenzen
aus 17 mit allen erlaubten Aminosäuresubstitutionen
jeder einzelnen angeführten
Position. An der fünften
Aminosäureposition
(an der N-terminalen Aminosäure
(linkes Ende) beginnend) sind beispielsweise die Aminosäuren E (Glutaminsäure), D
(Asparaginsäure)
und K (Lysin) erlaubt, ohne dass die H-Bindung gestört wird
und somit ohne dass die Helizität gestört wird
oder die Wechselwirkung des Peptids mit dem Kern-Superhelix-Trimer von gp41 wesentlich
beeinträchtigt
wird. „X" bezeichnet Positionen,
die mit einer beliebigen die Helix nicht brechenden Aminosäure substituiert
werden kann. Die wiederkehrende abcdefg-Heptaden-Grundzuordnung
für die
einzelnen Aminosäurepositionen
in der Sequenz 633 bis 678 zum Zwecke der vorliegenden Erfindung
ist dargestellt. „*" kennzeichnet b-,
c- und f-Positionen, die, wenn sie nicht zur Arretierung der Helix
genutzt werden, durch eine nicht-helixbrechende Aminosäure ersetzt
werden können,
ohne dass die H-Bindung, die Helizität und die Trimerfurchenbindung
wesentlich beeinträchtigt
werden.
-
23 zeigt eine Kurzschrift-Notation spezifischer
Peptide in der Peptidreihe I bis XII (siehe 18), wobei
jeweils Arretierungspositionen, Aminosäuresubstitions-Peptidvarianten und
Trunkierungs-Peptidvarianten angegeben sind. „X" kennzeichnet eine Position, die mit
einer beliebigen nicht-helixbrechenden Aminosäure substituiert werden kann,
vorzugsweise jedoch mit einer Aminosäure, die in einer der in 16 dargestellten bekannten HIV-Sequenzenan
dieser Position vorhanden ist. „B" kennzeichnet eine Position, die zur Überbrückung (oder
Fesselung oder Arretierung) von Resten genutzt wird. Bevorzugte
f-Positionen zur Arretierung sind angegeben; in weniger bevorzugten
Ausführungsformen
können
jedoch auch die c- und einige b-Positionen
zur Arretierung verwendet werden. Wie in 18 sind
Positionen von internen Sequenzen, die für die Erfindung relevant sind,
jene, die sich zwischen Arretierungsresten, deren Positionen durch
das Symbol „|" angezeigt sind,
befinden und in diesem Beispiel der f zugeordneten Position entsprechen.
Positionen zur Platzierung von entweder einer, zwei oder drei Arretierungen
in den repräsentativen
dargestellten Sequenzen sind angezeigt. In der Figur sind fünf helikale
Abschnitte der Form gabcde dargestellt, die zur Arretierung geeignet sind,
wenn Arretierungen an benachbarten f-Positionen erfolgen. „." kennzeichnet Positionen,
die in der letztendlichen eingeschränkten helikalen Peptidverbindung
mitunter fehlen können,
ohne dass die Helixeigenschaften und Furchenbindungseigenschaften
des fertigen eingeschränkten
helikalen Peptids wesentlich beeinträchtigt werden. Einem auf Peptid
I basierenden Peptid, dessen drei Arretierungen wie angegeben platziert sind,
können
beispielsweise eine oder alle fünf
N-terminale Aminosäuren
WXXWE fehlen, die durch „." markiert sind. Außerdem ist
in der Figur eine weitere Reihe von trunkierten Varianten – C-terminal
trunkierten Varianten – dargestellt,
da die fünf
C-terminalen Reste (LWNWF), die mit „." markiert sind, fehlen können. Wenn die
Arretierung zentraler in der 633-678-Sequenz platziert ist, wie
in der Peptidreihe II dargestellt ist, können Peptiden in dieser Reihe
zusätzliche
Aminosäuren
am C-terminalen Ende fehlen, wie durch die mit „." markierten Positionen angezeigt ist.
-
DETAILLIERTE
BESCHREIBUNG DER BEVORZUGTEN AUSFÜHRUNGSFORMEN
-
A. DEFINITIONEN
-
Die
hierin beschriebenen Aminosäuren
und Aminosäurereste
sind nach dem in der nachstehenden Tabelle angeführten, allgemein bekannten
Ein- oder Dreibuchstabencode bezeichnet. Sofern nicht anders angegeben,
liegen diese Aminosäuren
oder Reste in der natürlich
vorkommenden L-Stereoisomerform vor.
-
-
Im
Allgemeinen basieren, sofern nicht anders angegeben, die zur Bezeichnung
von Aminosäuren
und dafür
eingesetzten Schutzgruppen verwendeten Abkürzungen auf den Empfehlungen
der IUPAC-IUB Commision of Biochemical Nomenclature (Biochemistry
11, 1726–1732
(1972)).
-
Die
Bezeichnung -(CH2)n-
wird hierin zur Bezeichnung eines unverzweigten Alkylsubstituenten
mit einer Länge
von n Kohlenstoffen verwendet, worin -(CH2)0- als chemische Bindung definiert ist, d.h.
angibt, dass kein Alkylsubstituent vorhanden ist, -(C2)1- als Methylsubstituent definiert ist, -(CH2)2- als Ethylsubstituent
definiert ist usw.
-
Die
Bezeichnung „C1-C6-Alkyl" steht hierin für einen
gesättigten
aliphatischen Kohlenwasserstoffsubstituenten mit der angegebenen
Anzahl an Kohlenstoffatomen. C1-C6-Alkyl
umfasst zyklische und geradlinige Kohlenwasserstoffe, unverzweigte
und verzweigte Kohlenwasserstoffe, substituierte und unsubstituierte
Kohlenwasserstoffe sowie primäre,
sekundäre
und tertiäre
Kohlenwasserstoffsubstituenten. Repräsentative Beispiele für diese
Alkylsubstituenten umfassen Methyl, Fluorenylmethyl, Ethyl, n-Propyl,
Isopropyl, n-Butyl, Isabutyl, sec-Butyl, tert-Butyl, n-Pentyl, 2-Methylbutyl,
2,2-Dimethylpropyl, n-Hexyl, 2-Methylpentyl, 2,2-Dimethylbutyl,
Cyclohexyl und dergleichen. Die Bezeichnungen „Niederalkyl", „einfaches
Alkyl" und „C1-C6-Alkyl" sind Synonyme und
austauschbar.
-
Die
Bezeichnungen „Peptid", „Polypeptid" und „Protein" sind hierin Synonyme
und beziehen sich auf jegliche proteinartige Verbindung, die eine
Aminosäuresequenz
aus zwei oder mehr Aminosäureresten
umfasst.
-
Ein „eine Amidbindung
bildender Substituent in einer Aminosäure-Seitenkette", ein „eine Amidbindung bildender
Seitenketten-Substituent" und
grammatikalische Varianten davon sind hierin definiert als solche,
die Folgendes umfassen: (1) einen beliebigen Carboxysubstituenten
in der Seitenkette („R"-Gruppe) einer Aminosäure, worin
der Carboxysubstituent zur Bildung einer Amidbindung mit einer Aminogruppe
in einem anderen Molekül
in der Lage ist, d.h. der Carboxysubstituent reagiert mit einer Aminogruppe
in einem anderen Molekül, um
eine Amidbindung zu bilden; und (2) einen beliebigen Aminosubstituenten
in der Seitenkette („R"-Gruppe") einer Aminosäure, worin
der Aminosubstituent zur Bildung einer Amidbindung mit einer Carboxygruppe
in einem anderen Molekül
in der Lage ist, d.h. der Aminosubstituent reagiert mit einer Carboxygruppe
in einem anderen Molekül,
um eine Amidbindung zu bilden.
-
„Unterschiedlich
entfernbare" Schutzgruppen
sind hierin als jedes beliebige Paar von Schutzgruppen definiert,
die in der Lage sind, einen ersten eine Amidbindung bildenden Substituenten
und einen zweiten eine Amidbindung bildenden Substituenten zu schützen, worin
der erste eine Amidbindung bildende Substituent, der mit einem Element
des Paars geschützt
ist, unter Bedingungen entschützt
werden kann, unter denen der zweite eine Amidbindung bildende Substituent,
der mit dem anderen Element des Paars geschützt ist, nicht entschützt wird.
Unterschiedlich entfernbare Schutzgruppen werden hierin auch als „orthogonale" Schutzgruppen bezeichnet,
und der durch solche Schutzgruppen verliehene, unterschiedlich entfernbare
Schutz wird hierin als „orthogonaler" Schutz bezeichnet.
-
Der
Begriff „Epitop" bezeichnet hierin
die Strukturkomponente eines Moleküls, die für spezifische Wechselwirkungen
mit entsprechenden Antikörper-
(Immunglobulin-) Molekülen
verantwortlich ist, die durch das gleiche oder ein verwandtes Antigen
hervorgerufen werden. Allgemeiner gesehen bezeichnet der Begriff ein
Peptid mit den gleichen oder ähnlichen
Immunreaktionseigenschaften, beispielsweise spezifische Antikörperbindungsaffinität, wie das
antigene Protein oder Peptid, das zur Erzeugung des Antikörpers eingesetzt
wird. Somit ist ein Epitop, das von einer spezifischen Peptidsequenz
gebildet wird, im Allgemeinen jedes beliebige Peptid, das mit gegen
die spezifischen Sequenz gerichteten Antikörpern reaktiv ist.
-
Der
Begriff „Antigen" bezeichnet hierin
ein Molekül,
das zur Induktion der Produktion von Antikörpern dient. Alternativ dazu
dient der Begriff zur Bezeichnung eines Moleküls, das mit einem spezifischen
Antikörper reaktiv
ist.
-
Die
Bezeichnung „Immunogen" beschreibt hierin
eine Einheit, die Antikörperproduktion
in einem Wirtstier induziert. In manchen Fällen sind das Antigen und das
Immunogen die gleiche Einheit, während
in anderen Fällen
die beiden Einheiten unterschiedlich sind.
-
Der
Begriff „Untereinheitenvakzine" wird hierin, wie
auf dem Gebiet der Erfindung, zur Bezeichnung einer Virusvakzine
verwendet, die kein Virus, aber ein oder mehrere Virusproteine oder
Fragmente von Virusproteinen enthält. Die Bezeichnung „mehrwertig" bedeutet hierin,
dass die Vakzine ein oder mehrere eingeschränkte helikale Peptide mit einer
auf gp41 basierenden Sequenz aus zumindest zwei HIV-Isolaten mit unterschiedlichen
Aminosäuresequenzen
enthält.
-
Die
Bezeichnung „Breakthrough-Isalat" oder „Breakthrough-Virus" wird hierin, wie
auch auf dem Gebiet der Erfindung, für ein Virus verwendet, das
aus einer Vakzine isoliert ist.
-
B. ALLGEMEINE VERFAHREN
-
Allgemein
stellt die Erfindung ein Verfahren zur Entfernung von Elementen
einer α-hefikalen Sekundärstruktur
aus dem Gefüge
eines Proteins bereit, ohne dass die wohldefinierte Struktur innerhalb
der α-Helix des
Proteins verloren geht. In einem Aspekt dient das Verfahren zur
künstlichen
Rekonstruktion und Charakterisierung der Bindungsdeterminanten,
die innerhalb einer α-helikalen
Bindungsdomäne
eines Proteins von Interesse vorhanden sind. Der Entwurf von Molekülen, die
an einem Proteinbindungsabschnitt zur kompetitiven Bindung in der
Lage sind, erfordert die Fähigkeit,
die übergeordnete
Struktur des natürlichen
Liganden nachzuahmen. Wenn die Struktur des Liganden an der Stelle
des Proteinbindungsabschnitts mit einem kurzen Peptid nachgeahmt
werden kann, dann kann das Peptid dazu verwendet werden, um zu bestimmen,
ob es mäglich
ist, kleine Moleküle
zu entwerfen, die kompetitiv an den Prateinbindungsabschnitt binden.
Die Fähigkeit
eines kurzen Peptids, mit dem natürlichen Liganden um die Bindung
an den Proteinbindungsabschnitt zu konkurrieren, würde darauf
hinweisen, dass die Struktur des Liganden am Kontaktpunkt mit dem
Proteinbindungsabschnitt so aussieht, dass das kurze Peptid als
Modell zum Entwurf von kleinen Molekülen verwendet werden könne, die
mit dem natürlichen
Liganden um die Bindung an den Proteinbindungsabschnitt konkurrieren.
-
In
einem anderen Aspekt werden die Verfahren der Erfindung dazu verwendet,
die Konformationsstruktur eines Proteins oder Peptids zu stabilisieren.
Die vorliegenden Verfahren können
eingesetzt werden, um eine (oder mehrere) α-helikale Determinanten von
Interesse in einem Protein oder Peptid an ihrer Position zu fixieren,
sodass das Protein (oder Peptid) in Umgebungen oder unter Bedingungen,
welche die α-helikale Sekundärstruktur
einer nichteingeschränkten
Protein- oder Peptidspezies destabilisieren oder verschlechtern würden, eine α-helikale
Konformation beibehält.
-
Die
Verfahren der Erfindung sind auch zur Replikation einer Proteinfunktion
ohne intaktes Protein oder intakte funktionelle Domäne zweckdienlich.
Die Replikation der Bindungsaktivität eines Proteins durch ein
eingeschränktes
helikales Peptid der Erfindung würde
die Anwendung von Affinitätsreinigungsverfahren
für den Liganden
des Proteins ermöglichen,
ohne dass ein intaktes Protein oder große Fragmente davon bereitgestellt werden
müssten.
Somit könnte
ein eingeschränktes
helikales Peptid, dass die Bindungsaktivität eines bestimmten Proteins
aufweist, die Verwendung des Proteins in der Affinitätsreinigung
verhindernde Liefer- oder Kostenprobleme lösen. In einem weiteren Beispiel
könnte
ein eingeschränktes
helikales Peptid, das die Konformationsstruktur an der Stelle von
Interesse in einem bestimmten Protein aufweist, zur Isolierung eines
Konformationsepitops vom Rest des Proteins zur Bildung von Antikörpern gegen
das einzelne Epitop von Interesse ohne Störung von den anderen antigenen
Stellen, die im intakten Protein vorhanden sind, verwendet werden.
-
Besonders
bevorzugt ist die Verwendung der Verbindungen der Erfindung, die
eingeschränkte
helikale Peptide mit internen Aminosäuresequenzen aus der HIV-Isolat-LAI-gp41-Aminosäuresequenz
633–678
und Homologen davon aufweisen, als Haptene, Vakzinen und in der
Diagnostik.
-
In
einem weiteren Aspekt können
die Verfahren und Peptide der Erfindung zur Schaffung von kombinatorischen
Bibliotheken von eingeschränkten
helikalen Peptiden verwendet werden, die für chemische Selektionssysteme
geeignet sind.
-
I. Arretiere
Helixpeptide und Anwendungsmöglichkeiten
-
Die
Erfindung stellt arretierte Helixpeptide der Formel (1) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze Zahl
im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist;
-
In
einer anderen Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäurese quenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, und s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist; und r eine beliebige ganze Zahl
im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8) bereit:
worin S nicht vorhanden ist
oder ein Makromalekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist; und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze Zahl
im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17) bereit:
worin S nicht vorhanden
ist oder ein Makromalekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (19) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist; und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (1), Formel (2),
Formel (3), Formel (4), Formel (5), Formel (6), Formel (7), Formel
(8), Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16), Formel (17), Formel (18),
Formel (19) und Formel (20) bereit, worin X, Y und Z gemeinsam bis
zu oder etwa 35 Aminosäuren
enthalten (d.h. arretierte Helixpeptide der Formeln (1), (2), (3),
(4), (5), (6), (7), (8), (9), (10), (11), (12), (13), (14), (15),
(16), (17), (18), (19) und (20), die jeweils insgesamt nicht mehr
als oder etwa 35 Aminosäurereste
enthalten).
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1), Formel (2), Formel
(3), Formel (4), Formel (5), Formel (6), Formel (7), Formel (8),
Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16), Formel (17), Formel (18),
Formel (19) und Formel (24) bereitgestellt, worin X und/oder Y bis
zu oder etwa 30 Aminosäurereste
enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1), Formel (2),
Formel (3), Formel (4), Formel (5), Formel (6), Formel (7), Formel
(8), Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16), Formel (17), Formel (18),
Formel (19) und Formel (20) bereitgestellt, worin X und/oder Y bis
zu oder etwa 25 Aminosäurereste
enthält/enthalten.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1), Formel (2), Formel
(3), Formel (4), Formel (5), Formel (6), Formel (7), Formel (8),
Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16); Formel (17), Formel (18),
Formel (19) und Formel (20) bereitgestellt, worin X und/oder Y bis
zu oder etwa 20 Aminosäurereste
enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1), Formel (2),
Formel (3), Formel (4), Formel (5), Formel (6), Formel (7), Formel
(8), Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16), Formel (17), Formel (18),
Formel (19) und Formel (20) bereitgestellt, worin X und/oder Y bis
zu oder etwa 15 Aminosäurereste
enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1), Formel (2),
Formel (3), Formel (4), Formel (5), Formel (6), Formel (7), Formel
(8), Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16), Formel (17), Formel (18),
Formel (19) und Formel (20) bereitgestellt, worin X und/oder Y bis
zu oder etwa 10 Aminosäurereste
enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1), Formel (2),
Formel (3), Formel (4), Formel (5), Formel (6), Formel (7), Formel
(8), Formel (9), Formel (10), Formel (11), Formel (12), Formel (13),
Formel (14), Formel (15), Formel (16), Formel (17), Formel (18),
Formel (19) und Formel (20) bereitgestellt, worin X und/oder Y bis
zu oder etwa 5 Aminosäurereste
enthält
enthalten.
-
Auch
innerhalb des Schutzumfangs der Erfindung liegen arretierte Helixpeptide
der Formel (1), Formel (2), Formel (3), Formel (4), Formel (5),
Formel (6), Formel (7), Formel (8), Formel (9), Formel (10), Formel
(11), Formel (12), Formel (13), Formel (14), Formel (15), Formel
(16), Formel (17), Formel (18), Formel (19) und Formel (20), worin
X und/oder Y bis zu oder etwa 3 Aminosäurereste enthält/enthalten.
-
Die
Erfindung stellt auch arretierte Helixpeptide der Formel (1a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze Zahl
im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlassen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Forme (12a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminasäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze Zahl
im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (19a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20a) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Y Hydroxyl oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (1 a), Formel (2a),
Formel (3a), Formel (4a), Formel (5a), Formel (6a), Formel (7a),
Formel (8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a),
Formel (13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a),
Formel (18a), Formel (19a) und Formel (20a) bereit, worin X, Y und
Z gemeinsam bis zu oder etwa 12 Aminosäuren enthalten (d.h. arretierte
Helixpeptide der Formeln (1a), (2a), (3a), (4a), (5a), (6a), (7a),
(8a), (9a), (10a), (11a), (12a), (13a), (14a), (15a), (16a), (17a),
(18a), (19a) und (20a), die jeweils insgesamt nicht mehr als oder
etwa 12 Aminosäurereste
enthalten).
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1a), Formel (2a), Formel
(3a), Formel (4a), Formel (5a), Formel (6a), Formel (7a), Formel
(8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a), Formel
(13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a), Formel
(18a), Formel (19a) und Formel (20a) bereitgestellt, worin X und/oder
Y bis zu oder etwa 30 Aminosäurereste
enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1a), Formel (2a),
Formel (3a), Formel (4a), Formel (5a), Formel (6a), Formel (7a),
Formel (8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a),
Formel (13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a),
Formel (18a), Formel (19a) und Formel (20a) bereitgestellt, worin
X und/oder Y bis zu oder etwa 25 Aminosäurereste enthält/enthalten.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1a), Formel (2a), Formel
(3a), Formel (4a), Formel (5a), Formel (6a), Formel (7a), Formel
(8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a), Formel
(13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a), Formel
(18a), Formel (19a) und Formel (20a) bereitgestellt, worin X und/oder
Y bis zu oder etwa 20 Aminosäurereste
enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1a), Formel (2a),
Formel (3a), Formel (4a), Formel (5a), Formel (8a), Formel (7a),
Formel (8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a),
Formel (13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a),
Formel (18a), Formel (19a) und Formel (20a) bereitgestellt, worin
X und/oder Y bis zu oder etwa 15 Aminosäurereste enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1a), Formel (2a),
Formel (3a), Formel (4a), Formel (5a), Formel (6a), Formel (7a),
Formel (8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a),
Formel (13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a),
Formel (18a), Formel (19a) und Formel (20a) bereitgestellt, worin
X und/oder Y bis zu oder etwa 10 Aminosäurereste enthält/enthalten.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1a), Formel (2a),
Formel (3a), Formel (4a), Formel (5a), Formel (6a), Formel (7a),
Formel (8a), Formel (9a), Formel (10a), Formel (11a), Formel (12a),
Formel (13a), Formel (14a), Formel (15a), Formel (16a), Formel (17a),
Formel (18a), Formel (19a) und Formel (20a) bereitgestellt, worin
X und/oder Y bis zu oder etwa 5 Aminosäurereste enthält/enthalten.
-
Auch
innerhalb des Schutzumfangs der Erfindung liegen arretierte Helixpeptide
der Formel (1a), Formel (2a), Formel (3a), Formel (4a), Formel (5a),
Formel (6a), Formel (7a), Formel (8a), Formel (9a), Formel (10a),
Formel (11a), Formel (12a), Formel (13a), Formel (14a), Formel (15a),
Formel (16a), Formel (17a), Formel (18a), Formel (19a) und Formel
(20a), worin X und/oder Y bis zu oder etwa 3 Aminosäurereste
enthält/enthalten.
-
Die
Erfindung stellt auch arretierte Helixpeptide der Formel (1b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze
Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als aller gleich 7 ist, und r eine beliebige
ganze Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Amionsäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminasäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (19b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20b) bereit:
worin Y Hydroxyl oder eine
beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1b), Formel (2b), Formel
(3b), Formel (4b), Formel (5b), Formel (6b), Formel (7b), Formel
(8b), Formel (9b), Formel (10b), Formel (11b), Formel (12b), Formel
(13b), Formel (14b), Formel (15b), Formel (16b), Formel (17b), Formel
(18b), Formel (19b) und Formel (20b) bereitgestellt, worin Y bis
zu oder etwa 30 Aminosäurereste
enthält.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1b), Formel (2b), Formel
(3b), Formel (4b), Formel (5b), Formel (6b), Formel (7b), Formel
(8b), Formel (9b), Formel (10b), Formel (11b), Formel (12b), Formel
(13b), Formel (14b), Formel (15b), Formel (16b), Formel (17b), Formel
(18b), Formel (19b) und Formel (20b) bereitgestellt, worin Y bis
zu oder etwa 25 Aminosäurereste
enthält.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1b), Formel (2b), Formel
(3b), Formel (4b), Formel (5b), Formel (6b), Formel (7b), Formel
(8b), Formel (9b), Formel (10b), Formel (11b), Formel (12b), Formel
(13b), Formel (14b), Formel (15b), Formel (16b), Formel (17b), Formel
(18b), Formel (19b) und Formel (20b) bereitgestellt, worin Y bis
zu oder etwa 20 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1b), Formel (2b),
Formel (3b), Formel (4b), Formel (5b), Formel (6b), Formel (7b),
Formel (8b), Formel (9b), Formel (10b), Formel (11b), Formel (12b),
Formel (13b), Formel (14b), Formel (15b), Formel (16b), Formel (17b),
Formel (18b), Formel (19b) und Formel (20b) bereitgestellt, worin
Y bis zu oder etwa 15 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1b), Formel (2b),
Formel (3b), Formel (4b), Formel (5b), Formel (6b), Formel (7b),
Formel (8b), Formel (9b), Formel (10b), Formel (11b), Formel (12b),
Formel (13b), Formel (14b), Formel (15b), Formel (16b), Formel (17b),
Formel (18b), Formel (19b) und Formel (20b) bereitgestellt, worin
Y bis zu oder etwa 10 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1b), Formel (2b),
Formel (3b), Formel (4b), Formel (5b), Formel (6b), Formel (7b),
Formel (8b), Formel (9b), Formel (10b), Formel (11b), Formel (12b),
Formel (13b), Formel (14b), Formel (15b), Formel (16b), Formel (17b),
Formel (18b), Formel (19b) und Formel (20b) bereitgestellt, worin
Y bis zu oder etwa 5 Aminosäurereste
enthält.
-
Auch
innerhalb des Schutzumfangs der Erfindung liegen arretierte Helixpeptide
der Formel (1b), Formel (2b), Formel (3b), Formel (4b), Formel (5b),
Formel (6b), Formel (7b), Formel (8b), Formel (9b), Formel (10b),
Formel (11b), Formel (12b), Formel (13b), Formel (14b), Formel (15b),
Formel (16b), Formel (17b), Formel (18b), Formel (19b) und Formel
(20b), worin Y bis zu oder etwa 3 Aminosäurereste enthält.
-
Die
Erfindung stellt auch arretierte Helixpeptide der Formel (1c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze
Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäureseqenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausfürhrungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminasäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretiere Helixpeptide der Formel (19c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20c) bereit:
worin X Wasserstoff oder
eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1c), Formel (2c), Formel
(3c), Formel (4c), Formel (5c), Formel (6c), Formel (7c), Formel
(8c), Formel (9c), Formel (10c), Formel (11c), Formel (12c), Formel
(13c), Formel (14c), Formel (15c), Formel (16c), Formel (17c), Formel
(18c), Formel (19c) und Formel (20c) bereitgestellt, worin X bis
zu oder etwa 30 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1c), Formel (2c),
Formel (3c), Formel (4c), Formel (5c), Formel (6c), Formel (7c),
Formel (8c), Formel (9c), Formel (10c), Formel (11c), Formel (12c),
Formel (13c), Formel (14c), Formel (15c), Formel (16c), Formel (17c),
Formel (18c), Formel (19c) und Formel (20c) bereitgestellt, worin
X bis zu oder etwa 25 Aminosäurereste
enthält.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1c), Formel (2c), Formel
(3c), Formel (4c), Formel (5c), Formel (6c), Formel (7c), Formel
(8c), Formel (9c), Formel (10c), Formel (11c), Formel (12c), Formel
(13c), Formel (14c), Formel (15c), Formel (16c), Formel (17c), Formel
(18c), Formel (19c) und Formel (20c) bereitgestellt, worin X bis
zu oder etwa 20 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1c), Formel (2c),
Formel (3c), Formel (4c), Formel (5c), Formel (6c), Formel (7c),
Formel (8c), Formel (9c), Formel (10c), Formel (11c), Formel (12c),
Formel (13c), Formel (14c), Formel (15c), Formel (16c), Formel (17c),
Formel (18c), Formel (19c) und Formel (20c) bereitgestellt, worin
X bis zu oder etwa 15 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1c), Formel (2c),
Formel (3c), Formel (4c), Formel (5c), Formel (6c), Formel (7c),
Formel (8c), Formel (9c), Formel (10c), Formel (11c), Formel (12c),
Formel (13c), Formel (14c), Formel (15c), Formel (16c), Formel (17c),
Formel (18c), Formel (19c) und Formel (20c) bereitgestellt, worin
X bis zu oder etwa 10 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1c), Formel (2c),
Formel (3c), Formel (4c), Formel (5c), Formel (6c), Formel (7c),
Formel (8c), Formel (9c), Formel (10c), Formel (11c), Formel (12c),
Formel (13c), Formel (14c), Formel (15c), Formel (16c), Formel (17c),
Formel (18c), Formel (19c) und Formel (20c) bereitgestellt, worin
X bis zu oder etwa 5 Aminosäurereste
enthält.
-
Auch
innerhalb des Schutzumfangs der Erfindung liegen arretierte Helixpeptide
der Formel (1c), Formel (2c), Formel (3c), Formel (4c), Formel (5c),
Formel (6c), Formel (7c), Formel (8c), Formel (9c), Formel (10c),
Formel (11c), Formel (12c), Formel (13c), Formel (14c), Formel (15c),
Formel (16c), Formel (17c), Formel (18c), Formel (19c) und Formel
(20c), worin X bis zu oder etwa 3 Aminosäurereste enthält.
-
In
einer weiteren Ausführungsform
stellt die Erfindung auch arretierte Helixpeptide der Formel (1d)
bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe, dass
m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze
Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze Zahl
im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7d) bereit:
worin Z eine beliebige Aminasäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe, dass
w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Bereich von (7 – (w
+ y)) bis (9 – (w +
y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17d) bereit:
worin Z eine beliebige Aminasäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (19d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20d) bereit:
worin Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
Die
Erfindung stellt auch arretierte Helixpeptide der Formel (1e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze
Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2e) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt ist,
s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist,
mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eins beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist
und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13e) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15e) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16e) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18e) bereit:
worin S nicht vorhanden
ist oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs A minosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen- eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Form (19e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20e) bereit:
worin S nicht vorhanden ist
oder ein Makromolekül
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs A minasäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1e), Formel (2e), Formel
(3e), Formel (4e), Formel (5e), Formel (6e), Formel (7e), Formel
(8e), Formel (9e), Formel (10e), Formel (11e), Formel (12e), Formel
(13e), Formel (14e), Formel (15e), Formel (16e), Formel (17e), Formel
(18e), Formel (19e) und Formel (20e) bereitgestellt, worin Y bis
zu oder etwa 30 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1e), Formel (2e),
Formel (3e), Formel (4e), Formel (5e), Formel (6e), Formel (7e),
Formel (8e), Formel (9e), Formel (10e), Formel (11e), Formel (12e),
Formel (13e), Formel (14e), Formel (15e), Formel (16e), Formel (17e),
Formel (18e), Formel (19e) und Formel (20e) bereitgestellt, worin
Y bis zu oder etwa 25 Aminosäurereste
enthält.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1e), Formel (2e), Formel
(3e), Formel (4e), Formel (5e), Formel (6e), Formel (7e), Formel
(8e), Formel (9e), Formel (10e), Formel (11e), Formel (12e), Formel
(13e), Formel (14e), Formel (15e), Formel (16e), Formel (17e), Formel
(18e), Formel (19e) und Formel (20e) bereitgestellt, worin Y bis
zu oder etwa 20 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1e), Formel (2e),
Formel (3e), Formel (4e), Formel (5e), Formel (6e), Formel (7e),
Formel (8e), Formel (9e), Formel (10e), Formel (11e), Formel (12e),
Formel (13e), Formel (14e), Formel (15e), Formel (16e), Formel (17e),
Formel (18e), Formel (19e) und Formel (20e) bereitgestellt, worin
Y bis zu oder etwa 15 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1e), Formel (2e),
Formel (3e), Formel (4e), Formel (5e), Formel (6e), Formel (7e),
Formel (8e), Formel (9e), Formel (10e), Formel (11e), Formel (12e),
Formel (13e), Formel (14e), Formel (15e), Formel (16e), Formel (17e),
Formel (18e), Formel (19e) und Formel (20e) bereitgestellt, worin
Y bis zu oder etwa 10 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1e), Formel (2e),
Formel (3e), Formel (4e), Formel (5e), Formel (6e), Formel (7e),
Formel (8e), Formel (9e), Formel (10e), Formel (11e), Formel (12e),
Formel (13e), Formel (14e), Formel (15e), Formel (16e), Formel (17e),
Formel (18e), Formel (19e) und Formel (20e) bereitgestellt, worin
Y bis zu oder etwa 5 Aminosäurereste
enthält.
-
Auch
innerhalb des Schutzumfangs der Erfindung liegen arretierte Helixpeptide
der Formel (1e), Formel (2e), Formel (3e), Formel (4e), Formel (5e),
Formel (6e), Formel (7e), Formel (8e), Formel (9e), Formel (10e),
Formel (11e), Formel (12e), Formel (13e), Formel (14e), Formel (15e),
Formel (16e), Formel (17e), Formel (18e), Formel (19e) und Formel
(20e), worin Y bis zu oder etwa 3 Aminosäurereste enthält.
-
Die
Erfindung stellt auch arretierte Helixpeptide der Formel (1f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze
Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, 2 eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5f) beriet:
worin S Hydroxyl oder ein
Makramolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
beste hend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
sind, mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (19f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20f) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1f), Formel (2f), Formel
(3f), Formel (4f), Formel (5f), Formel (6f), Formel (7f), Formel
(8f), Formel (9f), Formel (10f), Formel (11f), Formel (12f), Formel (13f),
Formel (14f), Formel (15f), Formel (16f), Formel (17f), Formel (18f),
Formel (19f) und Formel (20f) bereitgestellt, worin X bis zu oder
etwa 30 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1f), Formel (2f),
Formel (3f), Formel (4f), Formel (5f), Formel (6f), Formel (7f),
Formel (8f), Formel (9f), Formel (10f), Formel (11f), Formel (12f),
Formel (13f), Formel (14f), Formel (15f), Formel (16f), Formel (17f),
Formel (18f), Formel (19f) und Formel (20f) bereitgestellt, worin
X bis zu oder etwa 25 Aminosäurereste
enthält.
-
Außerdem werden
hierin arretierte Helixpeptide der Formel (1f), Formel (2f), Formel
(3f), Formel (4f), Formel (5f), Formel (6f), Formel (7f), Formel
(8f), Formel (9f), Formel (10f), Formel (11f), Formel (12f), Formel (13f),
Formel (14f), Formel (15f), Formel (16f), Formel (17f), Formel (18f),
Formel (19f) und Formel (20f) bereitgestellt, worin X bis zu oder
etwa 20 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1f), Formel (2f),
Formel (3f), Formel (4f), Formel (5f), Formel (6f), Formel (7f),
Formel (8f), Formel (9f), Formel (10f), Formel (11f), Formel (12f),
Formel (13f), Formel (14f), Formel (15f), Formel (16f), Formel (17f),
Formel (18f), Formel (19f) und Formel (20f) bereitgestellt, worin
X bis zu oder etwa 15 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1f), Formel (2f),
Formel (3f), Formel (4f), Formel (5f), Formel (6f), Formel (7f),
Formel (8f), Formel (9f), Formel (10f), Formel (11f), Formel (12f),
Formel (13f), Formel (14f), Formel (15f), Formel (16f), Formel (17f),
Formel (18f), Formel (19f) und Formel (20f) bereitgestellt, worin
X bis zu oder etwa 10 Aminosäurereste
enthält.
-
Weiters
werden hierin arretierte Helixpeptide der Formel (1f), Formel (2f),
Formel (3f), Formel (4f), Formel (5f), Formel (6f), Formel (7f),
Formel (8f), Formel (9f), Formel (10f), Formel (11f), Formel (12f),
Formel (13f), Formel (14f), Formel (15f), Formel (16f), Formel (17f),
Formel (18f), Formel (19f) und Formel (20f) bereitgestellt, worin
X bis zu oder etwa 5 Aminosäurereste
enthält.
-
Auch
innerhalb des Schutzumfangs der Erfindung liegen arretierte Helixpeptide
der Formel (1f), Formel (2f), Formel (3f), Formel (4f), Formel (5f),
Formel (6f), Formel (7f), Formel (8f), Formel (9f), Formel (10f), Formel
(11f), Formel (12f), Formel (13f), Formel (14f), Formel (15f), Formel
(16f), Formel (17f), Formel (18f), Formel (19f) und Formel (20f),
worin X bis zu oder etwa 3 Aminosäurereste enthält.
-
Die
Erfindung stellt auch arretierte Helixpeptide der Formel (1g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, m und p unabhängig
voneinander aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt sind,
mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und n eine beliebige ganze
Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (2g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (3g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (4g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (5g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (6g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (7g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (8g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (9g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (10g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (11g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (12g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (13g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (14g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (15g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (16g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, w und y unabhängig
voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt sind,
mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Be reich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als oder
gleich 0 ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (17g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (18g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 4 bis 6, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (19g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 5 bis 7, Grenzen eingeschlossen,
ist.
-
In
einer weiteren Ausführungsform
stellt die Erfindung arretierte Helixpeptide der Formel (20g) bereit:
worin S Hydroxyl oder ein
Makromolekül
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, und n eine beliebige ganze Zahl von 3 bis 5, Grenzen eingeschlossen,
ist.
-
Für arretierte
Helixpeptide der Formel (1), (2), (3), (4), (5), (6), (7), (8),
(9), (10), (11), (12), (13), (14), (15), (16), (17), (18), (19),
(20), (1e), (2e), (3e), (4e), (5e), (6e), (7e), (8e), (9e), (10e),
(11e), (12e), (13e), (14e), (15e), (16e), (17e), (18e), (19e), (20e),
(1f), (2f), (3f), (4f), (5f), (6f), (7f), (8f), (9f), (10f), (11f),
(12f), (13f), (14f), (15f), (16f), (17f), (18f), (19f), (20f), (1g),
(2g), (3g), (4g), (5g), (6g), (7g), (8g), (9g), (10g), (11g), (12g),
(13g), (14g), (15g), (16g), (17g), (18g), (19g) oder (20g), die
an ein Makromolekül
gebunden sind, kommt gemäß der Erfindung
jedes beliebige Makromolekül
in Betracht, das als Anker für
den C-Terminus des arretierten Helixpeptids dienen kann. Typischerweise
dient das Makromolekül
als fester Träger.
Der feste Träger
ist im Allgemeinen eine inerte Matrix, wie z.B. ein polymeres Gel,
das ein(e) dreidimensionale(s) Struktur, Gitter oder Netzwerk eines
Materials aufweist. Nahezu jedes beliebige Makromolekül, synthetisch
oder natürlich,
kann ein Gel in einer geeigneten Flüssigkeit bilden, wenn es auf
geeignete Weise mit einem difunktionellen Reagens vernetzt ist.
In einer Ausführungsform
ist das ausgewählte
Makromolekül
zur Verwendung für
die Affinitätschromatographie
geeignet. Die meisten chromatographischen Matrices, die für die Affinitätschromatographie
verwendet werden, sind Xerogele. Solche Gele schrumpfen beim Trocknen
zu einem kompakten Feststoff, der nur die Gelmatrix umfasst. Wenn
das getrocknete Xerogel in der Flüssigkeit resuspendiert wird,
nimmt die Gelmatrix Flüssigkeit
auf, quillt und kehrt wieder in den Gelzustand zurück. Zur
Verwendung hierin geeignete Xerogele umfassen Polymergele, wie z.B.
Cellulose, vernetzte Dextrane (z.B. Sepharose), Agarose, vernetzte Agarose,
Polyacrylamid-Gele und Polyacrylamid-Agarose-Gele.
-
Die
hierin bereitgestellten arretierten Helixpeptide können nach
den in den nachstehenden Abschnitten II und III beschriebenen Verfahren
der Erfindung konstruiert werden.
-
In
einer Ausführungsform
sind die Peptide der Erfindung so entworfen, dass sie die Bindungsdeterminanten
aus α-helikalen
Bindungsdomänen
von bekannten Proteinen isolieren. Solche Peptide sind für eine Reihe
von Anwendungen geeignet, einschließlich der Bestimmung, ob eine
Bindungsdeterminante in einer α-helikalen
Bindungsdomäne
eines bekannten Proteins als Strukturmodell für den Entwurf von Peptidmimetika oder
kleinen Molekülen
geeignet ist, die zur Nachahmung oder Wirkung gegen die Bindungsaktivität des intakten
Proteins in der Lage sind. Bei der Verwendung der Peptide der Erfindung
Für diesen
Zweck wählt
der Anwender selbst ein Bindungsprotein mit einer helikalen Domäne aus,
die mit einem Liganden wechselwirkt, und identifiziert dann eine
Kandidatenbindungsdeterminante, die sich innerhalb einer Sequenz
aus sechs (oder mehr) zusammenhängenden
Aminosäuren
in der helikalen Bindungsdomäne
befindet. Die Kandidatenbindungsdomäne kann unter Verwendung von
Alanin-Scanning-Mutagenese identifiziert werden, um zu bestimmen,
ob die Kandidatensequenz einen oder mehrere Aminosäurereste
enthält,
die für
die Ligandenbindung entscheidend sind. Als Nächstes wird ein eingeschränktes Peptid,
das die Kandidatensequenz enthält,
entworfen, indem zwei Reste in der Kandidatensequenz (bezeichnet
als I und I+7), die durch eine dazwischenliegende Sequenz aus sechs
Aminosäuren
getrennt sind und nicht mit einem Liganden wechselwirken (wie durch
die Alanin-Scanning-Mutagenese im vorherigen Schritt bestimmt wurde)
zur Substitution mit Aminosäureresten
einer Seitenkette, die einen eine Amidbindung bildenden Substituenten
enthält,
ausgewählt
werden. Das Peptid wird synthetisiert, und die eine Amidbindung
bildenden Seitenketten-Substituenten der fremden I- und I+7-Reste werden genutzt,
um das Peptid gemäß den im
nachstehenden Abschnitt II beschriebenen Verfahren der Erfindung
in α-helikaler
Konformation festzumachen. Schließlich wird die Bindungsaktivität des arretierten Helixpeptids
mit dem Liganden getestet, beispielsweise in einem Bindungskompetitionstest
mit dem intakten Bindungsprotein, und die Ergebnisse des Tests können verwendet
werden, um zu bestimmen, ob ein peptidmimetischer oder kleinmolekularer
Antagonist unter Verwendung der Bindungsdeterminante als Strukturmodell
entwickelt werden könnte.
-
In
einer weiteren Ausführungsform
werden die arretierten Helixpeptide der Erfindung eingesetzt, um intakte
Bindungsproteine oder Proteinbindungsdomänen bei der Affinitätsreinigung
eines Liganden zu ersetzen. Beispielsweise wird Protein A im Allgemeinen
zur affinitätschromatographischen
Reinigung von IyG-Molekülen
eingesetzt. Wie im nachstehenden Beispiel 2 beschrieben wird, kann
eine arretierte Helixspezies des Peptids Phe-Asn-Met-(1)-Gln-Gln-Arg-Arg-Phe-Tyr-(2)-Ala-Leu-His
(worin die Aminosäurereste
an den Positionen (1) und (2) in der entsprechenden Z-Domänensequenz
beide durch Glutaminsäurereste
ersetzt sind), die einer Bindungsdeterminante in Helix 1 der Z-Domäne entspricht,
zur Bindung von IgG verwendet werden. Demgemäß stellt die Erfindung eingeschränkte Helixspezies
bereit, die Bindungsdeterminanten in Helix 1 der Z-Domäne in Protein
A enthalten, einschließlich Molekülen der
obigen Formel (4), worin Z Gln-Gln-Arg-Arg-Phe-Tyr ist. In einer
Ausführungsform
ist die eingeschränkte
Helixspezies ein Molekül
der Formel (4), worin Z Gln-Gln-Arg-Arg-Phe-Tyr ist, X Phe-Asn-Met
ist und Y Ala-Leu-His ist. Die IgG-Bindungsmoleküle der Erfindung werden am
besten unter Einsatz der im nachstehenden Abschnitt II beschriebenen Festphasen-Peptidsyntheseverfahren
synthetisiert, sodass die Moleküle
an Harzkügelchen
verankert sind, die für
Säulen-
oder Chargenaffinitätschromatographie
geeignet sind.
-
In
einer weiteren Ausführungsform
sind die arretierten Helixpeptide der Erfindung so aufgebaut, dass sie
Epitope in Proteinen nachahmen, und werden verwendet, um polyklonale
oder monoklonale Antikörper
gegen solche Epitope zu bilden. Da die Peptide häufig zu klein sind, um eine
Immunantwort zu erzeugen, können die
Peptide unter Verwendung eines difunktionellen oder derivatisierenden
Mittels, beispielsweise Maleinimidobenzoylsulfosuccinimidester (Konjugation über Cysteinreste),
N-Hydroxysuccinimid
(über Lysinreste), Glutaraldehyd,
Bernsteinsäureanhydrid,
SOCl2 oder R1N=C=NR,
worin R und R1 unterschiedliche Alkylgruppen
sind, an Träger
konjugiert werden, die bekanntermaßen in der zu immunisierenden
Spezies immunogen sind, beispielsweise Schlüsselloch-Napfschnecken-Hämocyanin,
Serumalbumin, Rinderthyroglabulin oder Sojabohnentrypsininhibitor.
-
Die
arretierten Helixpeptide der Erfindung sind vor allem zur Isolierung
von synthetischen Antikörperklonen
mit ausgewählter
Bindungsaktivität
aus kombinatorischen Phagendisplay-Bibliotheken nützlich.
Das arretierte Helixpeptid bringt dahingehend einen deutlichen Vorteil
gegenüber
dem intakten Protein oder der Proteindomäne mit sich, dass die Verwendung
des arretierten Helixpeptids die Isolierung von Bindungsaktivitäten für das jeweilige
Konformationsepitop von Interesse ermöglicht. Ohne die arretierten
Helixpeptide der Erfindung würde
das Konformationsepitop wahrscheinlich eine strukturelle Unterstützung von
anderen Regionen des Proteins oder der Proteindomäne erfordern,
deren Gegenwart im Liganden zur gleichzeitigen Isolierung von unerwünschten
Klonen führen
würde.
Außerdem
würde die
Synthese von arretierten Helixpeptiden, die an polymeren Harzen
verankert sind, wie in den nachste henden Abschnitten II und III
beschrieben wird, Material bereitstellen, das leicht zum Panning
von Phagendisplay-Bibliotheken in Säulen gepackt werden kann.
-
In
einem weiteren Aspekt werden die arretierten Helixpeptide der Erfindung
verwendet, um konformationsstabile Varianten von Peptiden oder Proteinen
bereitzustellen, die in nichteingeschränkter Form unter Bedingungen
von Interesse eine „wackelige" oder instabile α-helikale
Sekundärstruktur
an einer oder mehreren Stellen aufweisen. Vor allem können die
Verfahren der Erfindung Probleme einiger Antigene lösen, die
mit der instabilität
von Konformationsepitopen zusammenhängen. Manchmal ist ein Konformationsepitop
aufgrund eines oder mehrerer „wackeliger" oder instabiler α-helikaler Sekundärstrukturelemente
im Epitop nicht in der Lage, die gewünschte antigene Determinante
zu präsentieren.
Die Einschränkung
solcher „wackeliger" α-helikaler Strukturen gemäß den Verfahren
der Erfindung würde
das Konformationsepitop stabilisieren und eine Präsentation
der gewünschten
antigenen Determinante ermöglichen.
Diese Anwendung der vorliegenden Verfahren und Peptide ist beispielsweise
bei der Vakzinenentwicklung und Bildung von polyklonalen oder monoklonalen
Antikörpern
aus Wirtstieren oder Isolierung von Antikörperklonen aus Phagendisplay-Bibliotheken
besonders nützlich.
-
In
einer Ausführungsform
der Erfindung, in der die arretierten Helixpeptide der Erfindung
verwendet werden, um konformationsstabile Varianten von Peptiden
oder Proteinen bereitzustellen, die in nichteingeschränkter Form
unter Bedingungen von Interesse eine „wackelige" oder instabile α-helikale Sekundärstruktur an
einer oder mehreren Stellen aufweisen, wird eine Verbindung bereitgestellt,
die ein eingeschränktes
helikales Peptid enthält,
das als Immunogen, als Vakzine und zur Diagnose des HIV (Human Immunodeficiency
Virus) zweckdienlich ist. AIDS (Acquired Immune Deficiency Syndrome)
wird durch ein Retrovirus ausgelöst,
das als HIV (Human Immunodeficiency Virus) bezeichnet wird. Es wurden
große
Anstrengungen unternommen, eine Vakzine zu entwickeln, die eine
schützende
Immunantwort induziert, und zwar ausgehend von der Induktion von
Antikörpern
oder zellulären
Reaktionen. Neuere Bemühungen
nutzten Untereinheitenvakzinen, worin aus Sicherheitsgründen ein HIV-Protein
und kein abgeschwächtes
oder abgetötetes
Virus als Immunogen in der Vakzine eingesetzt wird.
-
Die
Human-Immunschwäche-Virus-1-(HIV-1-)
Hüllglykoproteine
gp120 und gp41 vermitteln einen viralen Tropismus für und nachfolgenden
Eintritt in Zielzellen (Freed et al., The Journal of Biological
Chemistry 270, 23883-23886 (1995)). Die Rolle von gp120 besteht
darin, Zielzellen durch Wechselwirkungen mit CD4 und einem von mehreren
Corezeptoren zu binden (D'Souza
et al., Nature Medicine 2, 1293–1300
(1996)), wonach gp41 die Fusion von Virus- und Zellmembranen fördert. Der
Mechanismus, durch den gp41 eine Membranfusion vermittelt, war vor
kurzem Gegenstand intensiver Untersuchungen. Die Ergebnisse lassen
darauf schließen,
dass der Prozess die Bildung eines Superhelix-Trimers umfassen kann,
das wahrscheinlich den Übergang
vom Ruhe- in einen fusogenen Zustand antreibt, wie am Modell von
Influenza-Hämagglutinin
gezeigt wurde (Wilson et al., Nature 289, 366–373 (1981); Carr et al., Cell
63, 823–832
(1993); Bullough et al., Nature 371, 37–43 (1994)).
-
Es
wurde herausgefunden, dass zwei von HIV-1-gp41 stammende lineare
Peptide Virusfusionen hemmen. Das erste davon, DP107, stellt eine
Abschnitt von gp41 nahe dem N-terminalen Fusionspeptid dar, und es
wurde gezeigt, dass es in Lösung
helikal ist und auf eine Weise oligomerisiert, die mit der Superhelixbildung übereinstimmt
(Gallaher et al., AIDS Res. Hum. Retraviruses 5, 431–440 (1989);
Weissenhorn et al., Nature 387, 426–430 (1997)). Ein wirksameres
Peptid, DP178, stammt aus der C-terminalen
Region der gp41-Ektodomäne
(Wild et al., PNAS 91, 9770–9774
(1994); Jiang et al., Nature 365, 113 (1993)). Obwohl von dieser Region
von gp41 vorausgesagt wurde, dass sie α-helikal sein würde (Gallaher
et al., AIDS Res. Hum. Retroviruses 5, 431–440 (1989)), fehlt DP178 selbst
in Lösung
eine erkennbare Struktur (Wild et al, PNAS 91, 9770–9774 (1994)).
Versuche, den Wirkmechanismus von DP178 zu untersuchen, wurden dadurch
erschwert, dass seine bioaktive Konformation nicht vollkommen geklärt ist.
Vor kurzem haben kristallographische Untersuchungen (Chan et al.,
Cell 89, 263–273
(1997); Weissenhorn et al., Nature 387, 426–430 (1997)) und Lösungsuntersuchungen
(Lawless et al., Biochemistry 35, 13697–13708 (1996); Lu et al., Nature
Structural Biology 2, 1075–1082
(1995); Rabenstein et al., Biochemistry 35, 13922, 13918 (1996))
gezeigt, dass losgelöste Segmente
von HIV-1-gp41,
die mit DP107 und DP178 überlappen,
sich in einem dicht gepackten helikalen Bündel verbinden. Das C-terminale
Segment, das DP178 entspricht, bildet eine verlängerte Helix, die sich antiparallel
eng an eine von einem N-terminalen (DP107) Superhelix-Trimer gebildete
Furche angelagert. Obwohl diese Daten eine mögliche Kapformation für DP178
vorschlagen, stellen sie keine schlüssigen Informationen über den
Mechanismus der Peptidhemmung während
Virusfusionsvorgängen
bereit.
-
Die
vorliegende Erfindung stellt eingeschränkte helikale Formen von DP178
und homologen Sequenzen und Varianten bereit, welche die Einschränkungen
auf dem Gebiet der Erfindung in Bezug auf DP178 überwinden und eine effizientere
Nutzung von DP178-artigen Sequenzen ermöglichen. Demgemäß wird in
einer Ausführungsform
der Erfindung ein eingeschränktes
helikales Peptid bereitgestellt, bei dem zumindest die interne Aminosäuresequenz
(und vorzugsweise auch benachbarte Aminosäuresequenzen) aus der C-terminalen
Region der HIV-1-LAI-Isolat-Transmembranprotein-gp41-Ektodomänenaminosäuren 633
bis 678 ausgewählt
sind, die mit der Sequenz überlappen,
die dem Peptid DP178 entspricht (Aminosäurereste 643 bis 673). Diese
Region ist eine 46 Aminosäuren
umfassende Sequenz (vom Amino- zum Carboxyterminus gelesen): NH2-WMEWEREIDNYTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-COOH.
-
Peptide
in einer α-helikalen
Superhelixkonformation wechselwirken miteinander auf charakteristische Weise,
die durch die Primärsequenz
der einzelnen Peptide bestimmt wird. Die Tertiärstruktur einer α-Helix sieht so
aus, dass 7 Aminosäurereste
in der Primärsequenz
etwa 2 Umkehrschleifen der α-Helix
entsprechen. Demgemäß kann eine
primäre
Aminosäuresequenz,
die eine α-helikale
Konformation ergibt, in Einheiten aus jeweils 7 Resten unterteilt
werden, die Heptaden genannt werden (und die Form abcdefg aufweisen).
Die Kernpolypeptide bestehen aus eine Reihe von hintereinander angeordneten
Heptaden. Wenn die Sequenz einer Heptade in einem bestimmen Kernpeptid
wiederholt wird, kann die Heptad als „Heptaden-Repeat" (Heptaden-Wiederholung)
oder einfach als „Repeat" bezeichnet werden.
-
Es
werden Verbindungen als Ausführungsformen
der Erfindung bereitgestellt, die ein eingeschränktes helikales Peptid enthalten,
das aus einem Peptid besteht, das eine Sequenz aus acht Aminosäureresten
umfasst, worin die Sequenz aus acht Aminosäureresten einen ersten terminalen
Rest und einen zweiten terminalen Rest aufweist, worin der erste
terminale Rest und der zweite terminale Rest eine interne Sequenz
aus sechs Aminosäureresten
flankieren, worin der erste und der zweite terminale Rest Seitenketten
aufweisen, die miteinander verbunden sind und eine arretierte Gruppierung
bilden, um das Peptid auf eine helikale Form einzuschränken. Die
interne Sequenz aus sechs Aminosäuren
weist die Form abcde, defgab oder cdefga auf und umfasst eine Sequenz
aus sechs zusammenhängenden
Aminosäuren,
die in der HIV-1LAI-Stamm-Transmembranprotein-gp41-Aminosäuresequenz
633 bis 678, in deren homologer Sequenz von einem anderen HIV-Stamm,
in einer Consensus-Sequenz
derer homologen Sequenzen aus einer beliebigen HIV-Clade oder einer
Aminosäure-Substitutionsvariante
davon gefunden wurde. Gemäß der Erfindung
ist jeder der Aminosäuren
in den oben genannten Sequenzen eine Position a, b, c, d, e, f oder
g zugeordnet. Die Zuordnungen basieren auf der Zuordnung der Aminosäure 633
der HIV-LAI-gp41-633-678-Sequenz zu Position a einer Heptaden-Grundaufstellung
abcdefg. Nachfolgende Aminosäuren
in der Sequenz sind demgemäß ihren
Positionen zugeordnet. 18 zeigt
die Heptaden-Positionszuordnung der einzelnen Aminosäuren in
der Sequenz. Die Zuordnung kann leicht auf Homologe und Consensus-Sequenzen übertragen
werden, indem ihre Aminosäuren
mit der entsprechenden Aminosäure
in der repräsentativen
HIV-LAI-Sequenz abgeglichen werden. Der Aminosäure 633 oder ihrer entsprechenden
Aminosäure
in einem Homolog oder einer Consensus-Sequenz ist die Position a
zugeordnet, mit der das Muster der zugeordneten Grundaufstellung
abcdef beginnt.
-
In
diesen in den 16–18 gezeigten
repräsentativen
Verbindungen und Sequenzen befindet sich die Arretiergruppierung
oder „Fessel" zwischen benachbarten
f-Positionen, wenn
die interne Sequenz die Form gabcde aufweist, zwischen benachbarten
c-Positionen, wenn die interne Sequenz die Form defgab aufweist,
oder zwischen benachbarten b-Positionen, wenn die interne Sequenz
die Form cdefga aufweist. In den am meisten bevorzugten Ausführungsformen
erfolgt die Arretierung zwischen benachbarten f-Positionen, in welchem
Fall die Aminosäuren
an der f-Position
durch Aminosäuren
ersetzt sind, die zur Bereitstellung einer Helix-Arretiergruppierung geeignet sind. 18 zeigt den Abgleich der abcdefg-Grundzuordnung mit
den für die
Erfindung relevanten Sequenzen. In einer bevorzugten Ausführungsform
weist die interne Sequenz aus sechs Aminosäuren die Form gabcde auf. Diese „interne
Sequenz" aus sechs
Aminosäuren
aus gp41 kann in jeglichen hierin dargelegten Verbindungen, Formeln
und Syntheseverfahren für
die Gruppierung „Z" substituiert werden.
-
Obwohl
die interne Aminosäuresequenz
vorzugsweise von einer Sequenz aus sechs zusammenhängenden
Aminosäuren
in der HIV-1LAI-Stamm-gp41-Aminosäuresequenz 633 bis 678, in
deren homologer Sequenz von einem anderen HIV-Stamm oder in einer
Consensus-Sequenz derer homologen Sequenzen aus einer beliebigen
HIV-Clade stammt, kann sie eine Aminosäure-Substitutionsvariante davon
sein. Die Sequenzen der Erfindung umfassen auch Analoge der HIV-gp41-Sequenz
633–678
und Trunkierungen, die unter anderem Peptide umfassen können, welche
die Sequenz 633–678
umfassen, die eine oder mehrere Aminosäure-Substitutionen, -Insertionen
und/oder -Deletionen enthält.
Die Analoge der Sequenz zeigen, wenn sie in eingeschränkten Peptiden
der Erfindung enthalten sind, antivirale Aktivität und können weiters auch zusätzliche vorteilhafte
Eigenschaften haben, wie z.B. erhöhte Bioverfügbarkeit und/oder Stabilität, oder
können
Antikörper
mit verbesserter HIV-Stamm-Erkennung
hervorrufen.
-
HIV-1-
und HIV-2-Hüllproteine
sind zwar strukturell unterschiedlich, es existiert jedoch eine
auffällige Aminosäurekonservierung
innerhalb der gp41-633-678 entsprechenden Regionen von HIV-1 und
HIV-2. Aminosäuresubstitutionen
können
konservativ oder nichtkonservativ sein. Konservative Aminosäuresubstitutionen bestehen
im Ersetzen einer oder mehrerer Aminosäuren der 633-678-Peptidsequenz
durch Aminosäuren ähnlicher
Ladungs-, Größen- und/oder
Hydrophobieeigenschaften, wie z.B. eine Aminosäuresubstitution von Glutaminsäure (E)
zu Asparaginsäure
(D). Nichtkonservative Substitutionen bestehen im Ersetzen einer
oder mehrerer Aminosäuren
der 633-678-Peptidsequenz durch Aminosäuren unähnlicher Ladungs-, Größen- und/oder Hydrophobieeigenschaften,
wie z.B. eine Substitution von Glutaminsäure (E) zu Valin (V).
-
Deletionen
der 633-678-Region oder deren Homologer liegen ebenfalls im Schutzumfang
der Erfindung. Derartige Deletionen bestehen in der Entfernung einer
oder mehrerer Aminosäuren,
wobei die Längenuntergrenze
der resultierenden Peptidsequenz für eine Verwendung als interne
Sequenz eines eingeschränkten
helikalen Peptids 6 Aminosäuren
beträgt.
Vorzugsweise bleiben bei der Deletion ausreichend viele Aminosäuren zurück, um zumindest
zwei Arretierungen, wie hierin beschrieben, vorsehen zu können. Beispiele
für solche
Deletionen sind das HIV35-Peptid und dessen eingeschränkte Helixverbindungen
der Erfindung, die eine Arretierung (z.B. HIV-24) bzw. zwei Arretierungen
(z.B. HIV-31) aufweisen. Insbesondere sind die Deletionen terminale
Trunkierungen, führen
jedoch in allen Fällen
zu Peptiden, die bei Einschränkung
entlang der f-b-c-Helixfläche
weiterhin von den hierin verwendeten Superhelix-Suchalgorithmen
erkannt werden, oder aber, die die a-d-Helixflächenorientierung und räumliche
Ausrichtung des Ausgangsmoleküls
beibehalten, oder aber, die antifusogene oder antivirale Aktivität aufweisen
können.
-
Am
meisten bevorzugte Verbindungen sind jene, die bei Einsatz als Immunogene
Antikörper
hervorrufen, die HIV-virale fusogene Aktivität oder Infektiosität neutralisienen.
-
Die
Peptide der Erfindung können
weiters homologe Sequenzen der Sequenz 633–678 des HIV-LAI-Stamms umfassen,
die in eingeschränkter
helikalen Form antivirale Aktivität aufweisen. Besonders bevorzugt
bilden die eingeschränkten
Peptide, wenn sie als Haptene verwendet werden, Antikörper, die
vitale Fusionsereignisse blockieren, was zu einer Hemmung der Virusinfektiosität führt. Solche
Homologe sind Peptide, deren Aminosäuresequenzen aus den Aminosäuresequenzen
der Peptidregionen anderer (d.h. keiner HIV-1LAI-) Viren bestehen,
die der gp41-Peptidregion 633–678
entsprechen. Solche Viren können
andere HIV-1-Isolate und HIV-2-Isolate umfassen, sind jedoch nicht
darauf beschränkt.
Homologe, die von der entsprechenden gp41-Peptidregion von anderen
(d.h. Nicht-HIV-1LAI-) HIV-1-Isolaten stammen, kön nen die in 16A bis 16G dargestellten
oder andere bekannte entsprechende Sequenzen umfassen. Besonders bevorzugt
sind jene, die von HIV-1SF2, HIV-1RF und HIV-1MN, GNE6, GNE8 und
vom Thai-Stamm-Isolat A244 stammen.
-
In
einer besonders bevorzugten Ausführungsform
sind die Aminosäuren
an den Positionen a und d der internen Sequenz aus sechs Aminosäuren im
helikalen Peptid nicht aminosäuresubstituiert,
sondern sind die Aminosäuren
in den bekannten Isolaten oder Consensus-Sequenzen (siehe die 16A–16G und 17).
-
Auch
sind Ausführungsformen,
bei denen die Aminosäuren
an den Positionen g und e der internen Sequenz aus sechs Aminosäuren im
helikalen Peptid nicht aminosäuresubstituiert
sind am meisten bevorzugt. Eine Aminosäure an einer der Positionen
a, d, g oder e der internen Sequenz aus sechs Aminosäuren kann
in bevorzugten Ausführungsformen
im helikalen Peptid konservativ substituiert sein. Die Positionen
a und d, und weniger direkt die Positionen g und e, sind vermutlich
jene, die auf der Fläche
der eingeschränkten
Helix liegen, die mit dem gp41-Kerntrimer wechselwirkt (siehe 19). Da vermutet wird, dass die Positionen f,
b und c nicht direkt an der Bindung beteiligt sind, sondern eher
dazu dienen, die Helixstruktur zu ermöglichen, sind bevorzugte Variationen
an den Positionen b und c der internen Sequenz aus sechs Aminosäuren im
helikalen Peptid nicht aminosäuresubstituiert,
wenn sie nicht die arretierenden (fesselnden) Reste sind. Weniger
bevorzugt sind Verbindungen, worin eine Aminosäure an einer der Positionen
b, c oder f der internen Sequenz aus sechs Aminosäuren konservativ
substituiert ist oder eine beliebige nichthelixbrechenden Aminosäure ist,
welche die Arretierungsgruppierung nicht beeinträchtigt. Bevorzugte Chimären aus
internen Sequenzen sind solche, worin eine Aminosäure an einer
der Positionen a, d, g oder e der internen Sequenz aus sechs Aminosäuren im
helikalen Peptid durch eine Aminosäure an der entsprechenden Position
eines anderen HIV-Virusstamms substituiert ist.
-
Bevorzugte
Verbindungen der Erfindung können
Sequenzen aus HIV-1-Clade-Consensus-Sequenzen umfassen:
-
Die
Aminosäuren
in diesen Sequenzen sind durch einen Einbuchstabencode dargestellt,
worin ein Kleinbuchstabe für
die angegebene Aminosäure
oder eine Substitution durch eine Aminosäure an der entsprechenden Position
in einer Sequenz innerhalb der gleichen Clade steht, und worin ein
? eine beliebige Aminosäure
an der entsprechenden Position in einer Sequenz innerhalb derselben
Clade ist. Am meisten bevorzugt sind Homologe oder Consensus-Sequenzen
aus den 16A–16G.
Die internen Sequenzen finden sich vorzugsweise in Virussequenzen
in der aus den Claden A, B, C, D, E, F, G und F/B bestehenden Gruppe
von HIV-1-Claden.
-
Verbindungen
der Erfindung können
den ersten und den zweiten terminalen Rest mit einer Seitenkette aufweisen,
die einen eine Amidbindung bildenden Substituenten enthält, die
unter Bildung einer Amidbindung miteinander verbunden sind, um ein
eingeschränktes
helikales Peptid zu bilden. Der eine Amidbindung bildende Seitenketten-Substituent
des ersten terminalen Rests und der eine Amidbindung bildende Seitenketten-Substituent
des zweiten terminalen Rests sind unabhängig voreinander aus der aus
einem Aminosubstituenten und einem Carboxysubstituenten bestehenden
Gruppe ausgewählt.
Der eine Amidbindung bildende Seitenketten-Substituent des ersten
terminalen Rests ist ein Carboxysubstituent, der eine Amidbindung
bildende Seitenketten-Substituent des zweiten terminalen Rests ist
ein Carboxysubstituent, und der difunktionelle Linker ist ein Diamin,
worin die erste und die zweite funktionelle Gruppe Aminogruppen
sind. In einer bevorzugten Form sind der erste terminate Rest und
der zweite terminale Rest unabhängig
voneinander aus der aus Asp und Glu bestehenden Gruppe ausgewählt, noch
bevorzugter sind der erste terminale Rest und der zweite terminale
Rest beide Glu. Die ersten terminalen Reste können eine D-Thio-Lysin-Seitenkette
aufweisen, und der zweite terminale Rest ein L-Thio-Lysin, die unter
Bildung einer disulfidgebundenen Arretiergruppierung miteinander
verbunden sind, um ein eingeschränktes
helikales Peptid zu bilden.
-
In
einer anderen Ausführungsform
ist das eingeschränkte
Peptid ein Hapten, das an ein Träger-Makromolekül gebunden
ist, vorzugsweise kovalent an das eingeschränkte helikale Peptid gebunden
ist, wie hierin erläutert
wird. Das Makromolekül
kann an der Arretiergruppierung oder an Aminosäuren an den Positionen f, b
oder c des eingeschränkten
helikalen Peptids an das helikale Peptid gebunden sein und kann
ein beliebiger Träger
sein, der die a-d-Fläche
des eingeschränkten
helikalen Peptids nicht stört.
Ein bevorzugter Träger
für immunogene
Zwecke ist Schlüsselloch-Napfschnecken-Hämocyanin,
oder auch andere hierin erläuterte
Träger.
-
In
anderen Ausführungsformen
enthalten die Verbindungen mehr als ein eingeschränktes helikales Peptid.
Die internen Sequenzen eines ersten und eines zweiten eingeschränkten helikalen
Peptids in diesen Ausführungsformen
sind vorzugsweise unterschiedlich. Die internen Sequenzen eines
ersten und zweiten eingeschränkten
helikalen Peptids stammen von der gleichen HIV-gp41-Sequenz oder
einer Consensus-Sequenz der gleichen HIV-Clade oder von einer Aminosäure-Substitutionsvariante
davon. Die internen Sequenzen des ersten und zweiten eingeschränkten helikalen
Peptids sind aus jenen ausgewählt,
die in der HIV-gp41-Sequenz oder einer Consensus-Sequenz der gleichen
HIV-Clade oder einer Aminosäure-Substitutionsvariante
davon durch zumindest zwei helikale Umkehrschleifen (oder sechs
Reste) getrennt sind. Die Verbindungen können weiters ein drittes eingeschränktes helikales
Peptid umfassen. Wiederum sind die internen Sequenzen des ersten,
des zweiten und des dritten eingeschränkten helikalen Peptids vorzugsweise
unterschiedlich. In einem Beispiel sind die drei Sequenzen als separate
eingeschränkte
helikale Segmente in einer Superhelix des Polypeptid-Rückgrats
einer 633-678-Sequenz vorhanden, wie sie in 18 dargestellt
ist.
-
In
anderen Ausführungsformen
enthalten die Verbindungen der Erfindung 1 bis 38, 1 bis 35 oder,
noch bevorzugter, 1 bis 19 Aminosäuren, die einen oder beide
der terminalen Reste des helikalen Peptids flankieren. Die flankierenden
Aminosäuren
sind vorzugsweise die flankierenden Aminosäuren für die interne Sequenz, wie
sie in einer Sequenz aus einer HIV-gp41-Sequenz zu finden sind.
-
In
einer weiteren Ausführungsform
umfassen die Verbindungen weiters eine Blockiergruppe, die an einen
oder an beide der terminalen Reste des helikalen Peptids gebunden
ist, um proteolytischen Abbau zu verhindern. Die Blockiergruppe
kann eine D-Aminosäure
oder eine Nichtamidbindung zwischen benachbarten flankierenden Aminosäuren enthalten.
-
Besonders
bevorzugte Verbindungen umfassen jene, in denen sich eine einfache
Arretierung innerhalb der Sequenz YTSLIHSLIEESQNQQEKNEQELLELD (Seq.-ID
Nr. 2) oder einer homologen Sequenz davon, innerhalb der Sequenz
EWDREINNYTSLIHSLIEESQNQQEKNEQE (Seq.-ID Nr. 107) oder einer homologen
Sequenz davon, innerhalb der Sequenz YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNF
(Seq.-ID Nr. 108) oder einer homologen Sequenz davon befinden, um
ein eingeschränktes
helikales Peptid zu erhalten. Mehr als eine Einschränkung, vorzugsweise
zwei, können
in diesen Sequenzen vorhanden sein, wie die in 18 gezeigten Beispiele zeigen. In 18 sind Positionen von helikalen Abschnitten der
Form gabcde dargestellt, wenn eine, zwei oder drei i-bis-i+7-Arretierungen
in einer 633-678-Sequenz oder Variante davon vorhanden sind. Die
zweifach arretierten Varianten (II), (III) und HIV31 und die einfach
arretierten Varianten HIV24, (IX) und (XI) sind bevorzugte Verbindungen,
welche die Arretierpositionen zeigen. Weniger bevorzugt sind die
dreifach arretierte Variante, die zweifach arretierten Varianten
(VI) und (VII) und die einfach arretierten Varianten VIII und XII.
Besonders bevorzugt sind die trunkierten Varianten HIV24 und HIV31
und ihre Homologe aus anderen HIV-Stämmen oder Consensus-Sequenzen
oder Substitutionsvarianten davon. Noch weniger bevorzugt sind i-bis-i+4+Arretierungen,
um ein „wackeliges" helikales Segment
einzuschränken.
-
In
einer bevorzugten Ausführungsform
gibt es zumindest zwei eingeschränkte
helikale Peptide in der Verbindung, die beispielsweise als unterschiedliche
und unabhängige
Haptene an KHL oder ein synthetisches TASP- oder Lysin-Netzwerk
gebunden sind oder als zwei oder mehr arretierte helikale Segmente
innerhalb einer längeren
Polypeptidsequenz, vorzugsweise einer, die eine Neigung zur Bildung
einer verlängerten
oder superhelikalen Struktur aufweist. Die internen Sequenzen des
ersten und des zweiten eingeschränkten
helikalen Peptids sind vorzugsweise unterschiedlich, beispielsweise
mehrere Haptene auf einem einzelnen Hapten-Träger oder zwei oder mehr arretierte
helikale Segmente innerhalb einer längeren Superhelix-Polypeptidsequenz. In
letzterem Fall stammen die internen Sequenzen des ersten und des
zweiten eingeschränkten
helikalen Peptids vorzugsweise von der gleichen IHV-gp41-Sequenz, einer Consensus-Sequenz
der gleichen HIV-Clade oder der gleichen Aminosäure-Substitutionsvariante davon.
Die beiden helikalen Peptide sind über eine trennende Aminosäuresequenz
miteinander verbunden, die 5 bis 7, 12 bis 14 oder 19 bis 21 nichthelixbrechende, natürliche oder
nichtnatürliche
Aminosäuren
umfassen können,
und worin vorzugsweise die internen Sequenzen des ersten und des
zweiten eingeschränkten
helikalen Peptids von der gleichen HIV-gp41-Sequenz, einer Consensus-Sequenz
der gleichen HIV-Clade oder der gleichen Aminosäure-Substitutionsvariante davon stammen.
Die Trennsequenz kann eine zusammenhängende Aminosäuresequenz
sein, die aus einer dazwischenliegenden Sequenz, die sich zwischen
den beiden internen Sequenzen in der gleichen HIV-gp41-Sequenz befindet,
einer Consensus-Sequenz der gleichen HIV-Clade oder der gleichen
Aminosäure-Substitutionsvariante
davon ausgewählt
ist und jene beiden Aminosäuren
ausschließt,
die den helikalen Peptid-Arretierpositionen entsprechen, die direkt
die dazwischenliegende Sequenz flankieren. Ein Beispiel ist HIV31,
worin die beiden eingeschränkten
Segmente (internen Aminosäuresequenzen)
in der Ausgangssequenz (HIV35) durch eine Sequenz aus acht Aminosäuren getrennt
sind, wobei die Aminosäuren
an benachbarten f-Positionen, die zur Arretierung verwendet werden,
nicht als Teil der dazwischenliegenden Sequenz betrachtet werden,
sodass die dazwischenliegende Sequenz eine Sequenz aus sechs Aminosäuren ist,
die als Trennsequenz aus sechs Aminosäuren in das endgültige Peptid
synthetisiert wird. Die Trennsequenz ist besonders bevorzugt 6,
13 oder 20 Aminosäuren
lang, um eine Anordnung von a-d-Flächen zwischen eingeschränkten helikalen
Peptiden aufrechtzuerhalten.
-
Die
Aminosäuren
in der Trennsequenz behalten die abcdefg-Zuordnungspositionen der
dazwischenliegenden Sequenz bei, worin vorzugsweise die Aminosäuren an
den Positionen a und d in der Trennsequenz identisch mit den ihnen
entsprechenden Aminosäuren
in der dazwischenliegenden Sequenz sind. Außerdem sind in bevorzugten
Ausführungsformen
die Aminosäuren
an den Trennsequenzpositionen g und e ebenfalls identisch mit den
ihnen entsprechenden Aminosäuren
in der dazwischenliegenden Sequenz. Weniger bevorzugt ist eine Aminosäure an einer
der Positionen a, d, g oder e in der Trennsequenz konservativ substituiert (mit
einer anderen Sequenz als in der Clade an dieser Position dargestellt).
Am meisten bevorzugt behalten die Aminosäuren in der Trennsequenz die
abcdefg-Zuordnungspositionen der dazwischenliegenden Sequenz bei,
und eine Aminosäure
an einer der Positionen a, d, g oder e ist in der Trennsequenz durch
eine entsprechende Aminosäure
aus ihrer homologen Sequenz von einem anderen HIV-Stamm, aus einer
Consensus-Sequenz ihrer homologen Sequenzen einer beliebigen anderen
HIV-Clade oder aus einer Aminosäure-Substitutionsvariante
davon substituiert. Die Aminosäuren
an den Positionen b, c oder f in der Trennsequenz können beliebige
nichthelixbrechende Aminosäuren
sein, wobei die Präferenzen
in 22 und 23A und
B angeführt
sind. Chimären
können
gebildet werden, worin eine Aminosäure an einer der Positionen
a, d, g oder e der internen Sequenz aus sechs Aminosäuren im
helikalen Peptid durch eine Aminosäure an der entsprechenden Position
eines anderen HIV-Virusstamms substituiert ist. Auf ähnliche
Weise können
Substitutionen der gleichen Art in flankierenden oder trennenden
Sequenzen vorgenommen werden. Bevorzugt sind Verbindungen, worin
die interne Aminosäuresequenz
aus einer der Peptidsequenzen aus 23A und 23B stammt. Noch bevorzugter ist die Verbindung
der Erfindung aus der aus eingeschränkten helikalen Peptiden mit
jeder möglichen
Sequenz bestehenden Gruppe ausgewählt, welche eine oder jede
beliebige Kombination von Aminosäuresubstitutionen
aufweist, die in der in den 23A und 23B dargestellten Serie der helikalen Peptide
I bis XII angeführt
sind. In anderen Ausführungsformen
ist die Verbindung aus der aus eingeschränkten helikalen Peptiden mit
jeder möglichen
Se quenz bestehenden Gruppe ausgewählt, welche eine oder jede
beliebige Kombination von Aminosäuretrunkierungen,
die in der in den 23A und 23B dargestellten
Serie der helikalen Peptide I bis XII angeführt sind, aufweist. In weiteren
Ausführungsformen
ist die Verbindung aus der aus Aminosäuresubstitutionen mit jeder
möglichen
Sequenz bestehenden Gruppe ausgewählt, welche eine oder jede
beliebige Kombination von Aminosäuresubstitutionen,
die in der in den 23A und 23B dargestellten
Serie der helikalen Peptide I bis XII angeführt sind, in Kombination mit
einer oder jeder beliebigen Kombination von Aminosäuretrunkierungen,
die in der in den 23A und 23B dargestellten
Serie der helikalen Peptide I bis XII angeführt sind, aufweist. X in diesen
Sequenzen kann jede beliebige nichthelixbrechende Aminosäure sein.
-
In
einer weiteren Ausführungsform
der Erfindung können
Peptide, welche die hierin beschriebenen Sequenzen aufweisen, mit
zusätzlichen
chemischen Gruppen synthetisiert werden, die an ihren Amino- und/oder
Carboxytermini vorhanden sind, sodass beispielsweise die Stabilität, Bioverfügbarkeit
und/oder Hemmungsaktivität
der Peptide gesteigert wird. Hydrophobe Gruppen, wie z.B. Carbobenzoxyl-,
Dansyl- oder t-Butyloxycarbonylgruppen
können
beispielsweise an den Aminotermini hinzugefügt werden. Eine Acetylgruppe
oder eine 9-Fluorenylmethoxycarbonylgruppe kann an den Aminotermini
platziert werden. Eine hydrophobe Gruppe, t-Butyloxycarbonyl- oder Amidogruppe
können
an den Carboxytermini hinzugefügt
werden. Außerdem
können
die Peptide der Erfindung so synthetisiert werden, dass ihre sterische
Konfiguration verändert wird.
Beispielsweise kann anstelle des üblichen L-Isomers das D-Isomer eines oder
mehrerer der Aminosäurereste
des Peptids verwendet werden. Die Verbindungen können zumindest eine benachbarte
Aminosäuren verbindende
Bindung enthalten, die keine Peptidbindung und vorzugsweise nichthelixbrechend
ist. Nichtpeptidbindungen zur Verwendung in flankierenden Sequenzen
umfassen eine Imino-, Ester-, Hydrazin-, Semicarbazid-, Oxim- oder
Azobindung. Außerdem
kann zumindest einer der Aminosäurereste
der Peptide der Erfindung durch einen allgemein bekannten, nicht
natürlich
vorkommenden Aminosäurerest
substituiert sein, der vorzugsweise nichthelixbrechend ist. Besonders
bevorzugt liegt die nichtnatürliche
Aminosäure
oder Nichtamidbindung, die benachbarte Aminosäuren verbindet, sofern vorhanden,
außerhalb
der internen Sequenz und ist noch bevorzugter nichthelixbrechend.
Außerdem
kann zumindest einer der Aminosäurereste
der Peptide der Erfindung durch einen allgemein bekannten, nicht
natürlich
vorkommenden Aminosäurerest
substituiert sein. Solche Veränderungen
können
zur Steigerung der Stabilität,
Bioverfügbarkeit,
Immunogenität
und/oder Hemmungsaktivität
der Peptide der Erfindung dienen.
-
Ohne
sich auf eine bestimmte Theorie festlegen zu wollen, wird davon
ausgegangen, dass die eingeschränkten
helikalen Peptide ihre Aktivität
durch eine Wechselwirkung der a-d-Fläche der Helix ableiten. Die starke
Anti-HIV-Aktivität
der Verbindungen der Erfindung rührt
von der gp41-633-678-Region her, die einer vermutlichen α-Helixregion
entspricht, die sich am C-terminalen Ende der gp41-Ektodomäne befindet
und sich mit einer distalen Stelle auf gp41 zu assoziieren schient,
deren interaktive Struktur durch das Leucin-Zipper-Motiv, eine Superhelixstruktur,
die als DP-107 bezeichnet
wird, beeinflusst wird. Die Assoziation dieser beiden Domänen kann
eine molekulare Bindung oder „molekulare
Klammer" wiederspiegeln,
die eng mit dem Fusionsprozess zusammenhängt (siehe die 18 und 19).
Die DP107-Region bildet einen Kerntrimerkomplex mit einer Furche,
welche die a-d-Fläche
der helikalen Peptide der Erfindung erkennt und bindet.
-
Wenn
sie als Peptide synthetisiert werden, sind sowohl DP-107 als auch
DP-178 starke Inhibitoren einer HIV-Infektion und fusionieren wahrscheinlich
aufgrund ihrer Fähigkeit,
Komplexe mit viralem gp41 zu bilden und dessen fusogenen Prozess
zu beeinflussen; während
eines strukturellen Übergangs
des viralen Proteins von der nativen Struktur in den fusogenen Zustand
können
das DP-107- und das DP-178-Peptid
beispielsweise Zugang zu ihren jeweiligen Bindungsstellen am viralen
gp41 gewinnen und zerstörenden
Einfluss ausüben.
-
Wenn
mehr als ein eingeschränktes
helikales Peptid vorhanden ist, als Teil einer Superhelix oder eines verlängerten
Helix-Polypeptid-Rückgrats,
dann befinden sich die Positionen a und d eines ersten eingeschränkten helikalen
Peptids folglich in der gleichen Ebene wie die Positionen a und
d des zweiten eingeschränkten
helikalen Peptids. Mit anderen Worten sind die a-d-Flächen der
beiden Helices in der gleichen Ebene angeordnet. Um diese Orientierung
zu erreichen, wenn sich die Helices in einer Polypeptid-Superhelix
befinden, sind das erste und zweite eingeschränkte helikale Peptid durch
entweder 5 bis 7, 12 bis 14 oder 19 bis 21 natürliche oder nichtnatürliche helixbildende
Aminosäuren
getrennt. Vorzugsweise sind das erste und das zweite eingeschränkte helikale
Peptid durch entweder 6, 13 oder 20 natürliche oder nichtnatürliche helixbildende
Aminosäuren
getrennt. Eine besonders bevorzugte Anordnung des ersten, zweiten
und jedes weiteren eingeschränkten
helikalen Peptids ist jene, die in DP107 zu finden ist, worin die
a-d-Flächen
in der gleichen Ebene angeordnet sind, um eine Wechselwirkung mit
der Furche im Kerntrimer zu ermöglichen.
-
Wenn
die besonders bevorzugte Fesselungschemie, wie sie hierin gelehrt
wird, eingesetzt wird, sind die Verbindungen der Erfindung aus der
aus Folgendem bestehenden Gruppe ausgewählt:
Verbindungen der
Formel (1):
worin S nicht vorhanden ist
oder ein Makromolekül
ist,
X Wasserstoff oder eine beliebige Aminosäure oder
Aminosäuresequenz
ist,
Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht
vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz
ist,
Z eine beliebige Aminosäuresequenz, bestehend aus sechs
Aminosäuren
ist, worin die interne Sequenz aus sechs Aminosäuren die Form gabcde, defgab
oder cdefga aufweist und aus einer Gruppe von Sequenzen ausgewählt ist,
die eine Sequenz aus sechs zusammenhängenden Aminosäuren in
der HIV-1LAI-Stamm-gp41-Aminosäuresequenz
633 bis 678, in deren homologer Sequenz von einem anderen HIV-Stamm,
in einer Consensus-Sequenz derer homologen Sequenzen von einer beliebigen
HIV-Clade oder einer Aminosäure-Substitutionsvariante
davon umfasst, worin der Aminosäure
633 oder ihrer entsprechenden Aminosäure in der homologen Sequenz,
Consensus-Sequenz oder Sequenzvariante die Position a einer Grundaufstellung
abcdefg zugeordnet ist;
m und p unabhängig voneinander aus den ganzen
Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt sind,
mit der Maßgabe,
dass m + p kleiner als oder gleich 6 ist, und
n eine beliebige
ganze Zahl im Bereich von (7 – (m
+ p)) bis (9 – (m
+ p)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass n größer als
1 ist;
Verbindungen der Formel (6):

worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, worin die interne Sequenz aus sechs Aminosäuren die Form gabcde, defgab
oder cdefga aufweist und aus einer Gruppe von Sequenzen ausgewählt ist,
die eine Sequenz aus sechs zusammenhängenden Aminosäuren in
der HIV-1LAI-Stamm-gp41-Aminosäuresequenz
633 bis 678, in deren homologer Sequenz von einem anderen HIV-Stamm,
in einer Consensus-Sequenz derer homologen Sequenzen von einer beliebigen
HIV-Clade oder einer Aminosäure-Substitutionsvariante
davon umfasst, worin der Aminosäure
633 oder ihrer entsprechenden Aminosäure in der homologen Sequenz,
Consensus-Sequenz oder Sequenzvariante die Position a einer Grundaufstellung
abcdefg zugeordnet ist; q aus den ganzen Zahlen von 1 bis 7, Grenzen
ein geschlossen, ausgewählt
ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze
Zahl im Bereich von (7 – (q
+ s)) bis (9 – (q
+ s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als
0 ist;
Verbindungen der Formel (11):

worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, worin die interne Sequenz aus sechs Aminosäuren die Form gabcde, defgab
oder cdefga aufweist und aus einer Gruppe von Sequenzen ausgewählt ist,
die eine Sequenz aus sechs zusammenhängenden Aminosäuren in
der HIV-1LA1-Stamm-gp41-Aminosäuresequenz
633 bis 678, in deren homologer Sequenz von einem anderen HIV-Stamm,
in einer Consensus-Sequenz derer homologen Sequenzen von einer beliebigen
HIV-Clade oder einer Aminosäure-Substitutionsvariante
davon umfasst, worin der Aminosäure
633 oder ihrer entsprechenden Aminosäure in der homologen Sequenz,
Consensus-Sequenz oder Sequenzvariante die Position a einer Grundaufstellung
abcdefg zugeordnet ist; t aus den ganzen Zahlen von 0 bis 6, Grenzen
eingeschlossen, ausgewählt
ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen,
ausgewählt
ist, mit der Maßgabe,
dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze
Zahl im Bereich von (7 – (t
+ v)) bis (9 – (t
+ v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als
0 ist; und
Verbindungen der Formel (16):

worin S nicht vorhanden ist
oder ein Makromolekül
ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz
ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden
ist, oder eine beliebige Aminosäure
oder Aminosäuresequenz
ist, Z eine beliebige Aminosäuresequenz,
bestehend aus sechs Aminosäuren
ist, worin die interne Sequenz aus sechs Aminosäuren die Form gabcde, defgab
oder cdefga aufweist und aus einer Gruppe von Sequenzen ausgewählt ist,
die eine Sequenz aus sechs zusammenhängenden Aminosäuren in
der HIV-1LAI-Stamm-gp41-Aminosäuresequenz
633 bis 678, in deren homologer Sequenz von einem anderen HIV-Stamm,
in einer Consensus-Sequenz derer homologen Sequenzen von einer beliebigen
HIV-Clade oder einer Aminosäure-Substitutionsvariante
davon umfasst, worin der Aminosäure
633 oder ihrer entsprechenden Aminosäure in der homologen Sequenz,
Consensus-Sequenz oder Sequenzvariante die Position a einer Grundaufstellung
abcdefg zugeordnet ist; w und y unabhängig voneinander aus den ganzen
Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt sind,
mit der Maßgabe,
dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze
Zahl im Bereich von (7 – (w
+ y)) bis (9 – (w
+ y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als
oder gleich 0 ist.
-
Diese
Verbindungen können
weiters S' enthalten,
wenn S nicht vorhanden ist, und X ist eine beliebige Aminosäure oder
Aminosäuresequenz,
worin S' ein an
X gebundenes Makromolekül
ist. X oder Y kann eine Blockiergruppe enthalten, die enzymatischen
Abbau verhindert. Herkömmliche
terminale Blockiergruppen, wie sie auf dem Gebiet der Erfindung
bekannt sind, sind geeignet. X oder Y kann auch eine D- Aminosäure oder eine
Nichtamidbindung zwischen benachbarten Aminosäuren enthalten, um enzymatischen
Abbau zu verhindern.
-
Die
Verbindungen können
wie hierin gelehrt mit Trägern
formuliert werden. Wenn das helikale Peptid als Hapten verwendet
werden soll, kann der Träger
ein Adjuvans sein. Typischerweise sind Zusammensetzungen der Erfindung
steril. Zusammensetzungen können
zumindest zwei Verbindungen der Erfindung, entweder frei oder kovalent
oder ionisch aneinander gebunden enthalten. Die Peptide der Erfindung,
die Virusfusionshemmwirkung aufweisen können in Kombination mit anderen
therapeutischen Wirkstoffen verwendet werden, vorzugsweise in Kombination
mit einem anderen antiviralen Mittel, um dessen antivirale Wirkung
zu steigern. Solche antiviralen Mittel umfassen, sind jedoch nicht
beschränkt
auf solche, die auf eine anderes Target-Molekül wirken, das an der Virusreplikation
beteiligt ist, beispielsweise reverse-Transkriptase-Inhibitoren, Virusprotease-Inhibitoren,
Glykosylierungsinhibitoren; solche, die auf ein anderes Target-Molekül wirken,
das an Virusübertragung
beteiligt ist; solche, die auf eine andere Stelle desselben Moleküls wirken;
und solche, die das Auftreten von Virusresistenz verhindern oder
verringern.
-
Bei
der Behandlung von Säugetieren,
einschließlich
Menschen, mit einer Virusinfektion wird eine therapeutisch wirksame
Menge der Verbindungen der Erfindung oder ein pharmazeutisch annehmbares
Derivat in einer Dosis verabreicht, die ausreicht, um die Virusreplikation
zu hemmen, entweder alleine oder in Kombination mit anderen virushemmenden
Arzneimitteln. Beispielsweise kann HIV31 oder HIV24 etwa 12 Wachen lang
in einer Menge von 0,1 mg/kg bis 1,0 mg/kg pro Tag als Infusion
verabreicht werden. Eine bevorzugte Dosis beträgt 20 mg bis 35 mg. Dosen können in
Intervallen von etwa einmal täglich
bis 4-mal täglich
und vorzugsweise etwa einmal alle zwei Tage bis einmal täglich verabreicht
werden. Eine bevorzugte Dosis wird verabreicht, um Spitzen-Plasmakonzentrationen
einer Verbindung von etwa 1 mg/ml bis 10 mg/ml zu erreichen. Dies
kann durch sterile Injektion einer etwa 2,0%igen Lösung der
verabreichten Bestandteile in gepufferter Salzlösung erreicht werden (jede
beliebige geeignete Salzlösung,
die Fachleuten auf dem Gebiet der Chemie bekannt ist, kann eingesetzt
werden). Gewünschte
Blutwerte können
durch kontinuierliche Infusi on aufrechterhalten werden, wie anhand
der durch HPLC gemessenen Plasmawerte ermittelt wird. Pharmazeutische
Zusammensetzungen, welche die Verbindungen der Erfindung enthalten,
können
einem menschlichen Patienten alleine oder in pharmazeutischen Zusammensetzungen
verabreicht werden, worin sie mit geeigneten Trägern oder einem oder mehreren
Exzipienten, wie hierin beschrieben, in Dosen zur Behandlung einer
Virusinfektion, insbesondere einer HIV-Infektion, vermischt werden.
Geeignete Verabreichungswege umfassen orale, rektale, transmukosale
oder intestinale Verabreichung; parenterale Verabreichung, einschließlich intramuskulärer, subkutaner,
intramedullärer
Injektionen, sowie intrathekalen, direkten intraventrikulären, intravenösen, intraperitonealen,
intranasalen oder intraokularen Injektionen; transdermale, topische,
vaginale und dergleichen. Dosierungsformen umfassen, sind jedoch
nicht beschränkt
auf Tabletten, Pastillen, Dispersionen, Suspensionen, Zäpfchen,
Lösungen,
Kapseln, Cremes, Pflaster, Minipumpen und dergleichen.
-
Wie
hierin erläutert
sind die Verbindungen der Erfindung vor allem als Haptene geeignet,
um einen Antikörper
hervorzurufen, der an die Verbindung bindet, wobei der Antikörper spezifisch
ein Epitop bindet, das eine Aminosäure an Position a, d, e oder
g im helikalen Peptid umfasst. Bevorzugte Antikörper sind monoklonal. Antikörper der
Erfindung erkennen nicht nur die Peptide der Erfindung, sondern
erkennen vorzugsweise auch die entsprechende Sequenz, sofern im
Virus vorhanden. Sie können
auch nichteingeschränktes
DP178 binden. Noch bevorzugter neutralisiert der Antikörper NIV-Virus-Infektiosität und/oder
neutralisiert HIV-Virus-Membranfusion. Somit können die Antikörper eine
gp41-Sequenz erkennen und binden.
-
Hierin
wird ein Verfahren zur Immunisierung eines Tiers beschrieben, welches
die Verabreichung einer immunogenen Menge einer Verbindung der Erfindung
an das Tier umfasst.
-
Außerdem wird
hierin ein Verfahren zur prophylaktischen oder therapeutischen Behandlung
eines Säugetiers
beschrieben, das HIV-gefährdet
oder -infiziert ist, umfassend das Verabreichen einer Zusammensetzung,
die eine prophylaktisch oder therapeutisch wirksame Menge einer
Verbindung der Erfindung und einen Träger umfasst.
-
Obwohl
erwartet wird, dass Antikörper
der Erfindung breite virale Aktivität aufweisen, umfasst die Zusammensetzung
vorzugsweise interne 6-Aminosäure-Sequenzen
von unterschiedlichen HIV-Stämmen
oder HIV-Claden. Die Zusammensetzungen umfassen auch Vakzinenformulierungen.
Die Formulierungen können ein
oder mehrere (mehrwertige) eingeschränkte helikale Peptide von unterschiedlichen
HIV-Stämmen
zur Verwendung als Vakzine oder Immunogen enthalten. Die Zusammensetzung
kann einem Patienten, der infektionsgefährdet ist oder eine solche
Behandlung benötigt,
prophylaktisch oder therapeutisch verabreicht werden, wobei die
Dosierungen und Verabreichungswege und -mittel eingesetzt werden,
die leicht zu bestimmen sind. Es kann jedoch auch chronische Verabreichung
bevorzugt sein, und die Dosierungen können demgemäß eingestellt werden.
-
Die
Verabreichung der Verbindungen, welche die eingeschränkten helikalen
Peptide der Erfindung enthalten, als prophylaktische Vakzine (oder
therapeutische Vakzine) kann die Verabreichung einer Konzentration
von Peptiden an einen Wirt umfassen, die wirksam ist, um eine Immunantwort
hervorzurufen, die ausreicht, um HIV zu neutralisieren, beispielsweise
durch Hemmung der Fähigkeit
von HIV, Zellen zu infizieren. Die genaue Konzentration hängt vom
spezifischen Peptid ab, das verabreicht werden soll, kann aber unter
Einsatz von Standardverfahren zum Testen der Entwicklung einer Immunantwort
bestimmt werden, die Fachleuten auf dem Gebiet der Erfindung wohlbekannt
sind. Die Peptide, die als Vakzine verwendet werden sollen, werden üblicherweise
intramuskulär
verabreicht. Die Peptide können
mit einem geeigneten Adjuvans formuliert werden, um die immunologische
Antwort zu verstärken.
Solche Adjuvantien können
Mineralgele, wie z.B. Aluminiumhydroxid; Tenside, wie z.B. Lysolecithin,
Pluronic-Polyole, Polyanione; andere Peptide; Ölemulsionen; und möglicherweise
nützliche
menschliche Adjuvantien, wie z.B. BCG und Crynebacterium parvum,
umfassen, sind jedoch nicht darauf beschränkt. Zahlreiche Verfahren sind
geeignet, um die hier beschriebenen Vakzinenformulierungen einzuführen. Diese
Verfahren umfassen, sind jedoch nicht beschränkt auf orale, intradermale,
intramuskuläre,
intraperitoneale, intravenöse,
subkutane und intranasale Wege.
-
Eine
Verbindung dieser Erfindung in einem geeigneten Träger oder
Exzipienten wird eingesetzt, um eine Vakzine herzustellen. Das Polypeptid
kann alleine verwendet werden, wird aber vorzugsweise in einer mehrwertigen
Untereinheitenvakzine verabreicht, die interne Sequenzen vom MN-Stamm
enthält.
Die Vakzine umfasst üblicherweise
eingeschränkte
Helices, die 3 bis etwa 5 unterschiedliche Stämme repräsentieren, aber 30 oder mehr
unterschiedliche, auf gp41 basierende, eingeschränkte helikale Peptide können verwendet
werden, um eine wirksamere Vakzine bereitzustellen. Von besonderem
Interesse sind gp41-Sequenzen von Breakthrough-Isolaten von HIV-Vakzinen-Versuchen.
Die Verwendung einer homologen gp41-Sequenz aus einem oder mehreren
Breakthrough-Isolaten in einer Untereinheitenvakzine, üblicherweise
zusammen mit einer Sequenz aus einem herkömmlicherweise vorhandenen Isolat
wie der MN-Sequenz, kann Schutz gegen HIV-Stämme bereitstellen, die sich
in ausreichendem Maß vom
herkömmlichen
Stamm (z.B. MN) unterscheiden, sodass die typische einzelne Untereinheitenvakzine
keinen Schutz gegen diese Stämme
verleiht.
-
Die
Herstellung von Polypeptiden zur Verwendung in einer Vakzine ist
allgemein bekannt. Die Verbindung mit dem gewünschten Reinheitsgrad und in
ausreichender Konzentration, um Antikörperbildung zu induzieren,
wird mit einem physiologisch annehmbaren Träger vermischt. Ein physiologisch
annehmbarer Träger ist
in der Dosis und Konzentration, in der er in der Vakzine eingesetzt
wird, nichttoxisch für
einen Rezipienten. Im Allgemeinen wird die Vakzine für eine Injektion,
normalerweise intramuskuläre
oder subkutane Injektion, formuliert. Geeignete Träger für die Injektion
sind steriles Wasser, bevorzugt sind aber physiologische Salzlösungen,
wie z.B. normale Salzlösung,
oder gepufferte Salzlösungen,
wie z.B. phosphatgepufferte Salzlösung oder Ringer-Laktat. Die
Vakzine enthält
im Allgemeinen ein Adjuvans. Nützliche
Adjuvantien umfassen QS21 (Quillaja saponaria, im Handel von Cambridge
Biotech, Worcester, MA, USA erhältlich),
das zytotoxische T-Zellen stimuliert, und Alaun (Aluminiumhydroxid-Adjuvans).
Formulierungen mit unterschiedlichen Adjuvantien, die zelluläre oder
lokale Immunität
erhöhen,
können
ebenfalls eingesetzt werden.
-
Weitere
Exzipienten, die in der Vakzine vorhanden sein können, umfassen niedermolekulare
Polypeptide (weniger als etwa 10 Reste), Proteine, Aminosäuren, Kohlenhydrate,
einschließlich
Glucose und Dextrane, Chelatbildner, wie z.B. EDTA, und andere Exzipienten,
die das Protein stabilisieren oder das Wachstum von Mikroorganismen
hemmen.
-
Die
Vakzine kann außerdem
noch andere HIV-Proteine enthalten. Vor allem gp120 oder der extrazelluläre Abschnitt
von gp41 oder HIV-1-Kernproteine, wie z.B. P24, P17 und P55, können in
der Vakzine vorhanden sein. Vorzugsweise stammt jegliches gp120,
das in der Vakzine vorhanden ist, von einer HIV-Isolat-Sequenz,
die in einem eingeschränkten
helikalen Peptid enthalten ist, das in der Vakzine vorhanden ist.
-
Vakzinenformulierungen
umfassen im Allgemeinen insgesamt 10 bis 5.000 μg der Verbindung, noch bevorzugter
etwa 100 bis 1000 μm,
noch bevorzugter etwa 300 bis 600 μg, am besten in etwa 1,0 ml
bis 1,5 ml des Trägers.
Die Menge der Verbindung, die ein beliebiges Isolat oder eine beliebige
Clade in der Vakzine repräsentiert,
variiert je nach Immunogenität
der Verbindung. Ein eingeschränktes
helikales Peptid mit Sequenzen von einigen HIV-Stämmen kann
beispielsweise weniger immunogen sein als jene vom MN-Stamm. Wenn
Peptide, die zwei Stämme
mit unterschiedlicher Immunogenität darstellen, in Kombination
eingesetzt werden, wird eine empirische Titration der Menge der
einzelnen Viren durchgeführt,
um den Prozentsatz des Peptids der einzelnen Stämme in der Vakzine zu bestimmen.
Für Isolate
mit ähnlicher
Immunogenität
sind etwa gleiche Peptidmengen der einzelnen Isolate in der Vakzine
vorhanden. Verfahren zur Bestimmung der relativen Menge eines immunogenen
Proteins in mehrwertigen Vakzinen sind allgemein bekannt und wurden beispielsweise
zur Bestimmung der relativen Anteile verschiedener Isolate in mehrwertigen
Poliovakzinen eingesetzt.
-
Die
Vakzinen werden, je nach Schema, im Allgemeinen im Monat 0, 1 und
6, 8 oder 12 verabreicht. Ein bevorzugtes Schema umfasst eine Verabreichung
im Monat 0, 1, 6 und 12. Nach dem Immunisierungsvorgang können jährliche
oder zweijährliche Auffrischungen
verabreicht werden, Während
des Immunisierungsvorgangs und danach können jedoch die Neutralisationsantikörperwerte
getestet und das Schema dementsprechend angepasst werden.
-
Die
Vakzine wird nichtinfizierten Individuen verabreicht. Außerdem kann
die Vakzine seropositiven Individuen verabreicht werden, um die
Immunantwort auf das Virus zu steigern.
-
Obwohl
die hierin beschriebenen Verbindungen wie oben beschrieben als Vakzine
verwendet werden können,
können
die Verbindungen auch alleine oder in Kombinationen mit demselben
Formulierungstyp als Immunogen eingesetzt werden, um Antikörper zu
induzieren, welche das/die Isolat(e) im Immunogen erkennen. Immunogene
werden auf die gleiche Weise formuliert und können dieselben Exzipienten
usw. enthalten. Antikörper,
die durch die Immunogene induziert werden, können in der Diagnostik zur
Detektion des HIV-Stamms in Patientenseren- oder Körperflüssigkeitsproben
eingesetzt werden, oder um das jeweilige gp41-Molekül oder das
Virus einer Affinitätsreinigung
zu unterziehen. Die Verbindung finden auch in diagnostischen Tests
zum Nachweis der Gegenwart von Antikörpern gegen HIV in Seren von
Individuen, die vermutlich infiziert sind, Anwendung.
-
In
einer weiteren Ausführungsform
werden die arretierten Helixpeptide der Erfindung verwendet, um kombinatorische
Bibliotheken von eingeschränkten
Peptiden herzustellen. Kombinatorische Peptidbibliotheken sind auf
einzigartige Weise dazu geeignet, eingeschränkte Peptide miteinzubeziehen.
Diese Bibliotheken werden durch ein Verfahren der „geteilten
Synthese" konstruiert,
bei dem ein fester Träger
(z.B. Kügelchen)
in gleichen Mengen aliquotiert wird und an jeden Teil separat eine
andere Aminosäure
gebunden wird. Die Teile werden gepoolt, erneut aufgeteilt, und
das Verfahren wird wiederholt. Beim „Peptid-auf-Kügelchen-Verfahren" ergibt dieses Verfahren
ein Gemisch aus Kügelchen,
von denen jedes mit einem Peptid mit einer einzigartigen Sequenz
verbunden ist. Das Kügelchengemisch
kann direkt in einer Bindungsselektion eingesetzt werden, wobei
die Bindung kolorimetrisch detektiert und positive Kügelchen
physikalisch zur Mikrosequenzierung aus dem Gemisch entfernt werden
(Clackson und Wells, Tibtech 12, 173–184 (1994)). Um eine Bibliothek
von Peptiden herzustellen, die eine Zufallssequenz aus sechs (oder
mehr) Aminosäuren
enthalten, die durch I- und I+7-Reste gemäß der Erfindung in einer helikalen
Konfurmation arretiert sind, wird das Teilungssyntheseverfahren
modifiziert, um I- und I+7-Reste
in jeder Zufalls-Aminosäuresequenz
an festgelegten Positionen zu platzieren, die durch sechs Reste
getrennt sind, und die Peptide werden zyklisiert, indem die eine
Amidbindung bildenden Seitenketten-Substituenten der I- und I+7-Reste
in den einzelnen Peptiden unter Verwendung eines der im nachstehenden
Abschnitt II beschriebenen Verfahrens verbunden werden.
-
Kombinatorische
Bibliotheken, welche die eingeschränkten Peptide der Erfindung
enthalten, sind ein besonders leistungsstarkes Werkzeug zur Identifikation
von hoch affinen Liganden beim Arzneimitteldesign. Angesichts der
Prävalenz
des α-helikalen
Motivs an aktiven Stellen von Bindungsproteinen, einschließlich DNA-Bindungsproteinen,
und der Abwesenheit von Aminosäuresequenz-Einschränkungen
im Fesselungssystem der Erfindung erhöhen die arretierten Helixpeptide
der Erfindung die Nützlichkeit
von kombinatorischen Peptidbibliotheken in Screening-Verfahren auf
spezifische Bindungsaktivitäten,
wie beispielsweise den Verfahren des US-Patents Nr. 5.306.619, das
zum Screenen auf DNA-Sequenz-spezifische Bindungsmoleküle eingesetzt
wird, deutlich.
-
II. Verfahren zur Konstruktion
von synthetischen arretierten Helixpeptiden
-
Gemäß vorliegender
Erfindung wird ein Element einer α-helikalen
Struktur aus seinem Umfeld in einem nativen Protein entfernt, indem
ein Peptid mit einer Aminosäuresequenz
konstruiert wird, das die α-helikale
Sekundärstruktur
von Interesse im nativen Protein überspannt und das kurze Peptid
in einer α-helikalen Konformation
eingeschränkt
wird, welche die α-helikale
Sekundärstruktur
von Interesse nachahmt. Die vorliegenden Erfindung ermöglicht es
dem Anwender, jedes Peptid, das sechs Aminosäuren lang ist, in einer helikalen
Struktur zu arretieren, indem eine Aminosäure mit einem eine Amidbindung
bildenden Seitenketten-Substituenten am N- Terminus des Peptids platziert wird,
und eine weitere Aminosäure
mit einem eine Amidbindung bildenden Seitenketten-Substituenten
am C-Terminus des Peptids platziert wird, und dann die eine Amidbindung
bildenden Seitenketten-Substituenten des N-terminalen und des C-terminalen
Rests verbunden werden, um eine zyklisierte Struktur zu bilden,
welche die Konformation einer α-Helix
nachahmt. Die vorliegenden Erfindung ermöglichen es dem Anwender außerdem,
jede Sequenz aus sechs Aminosäureresten
in einem größeren Peptid
in einer helikalen Konformation zu arretieren, indem zwei Reste
mit eine Amidbindung bildenden Seitenketten-Substituenten in die
N-terminale Aminosäureposition
und die C-terminale Aminosäureposition, welche
die Sequenz (bestehend aus sechs Aminosäureresten) von Interesse innerhalb
eines größeren Peptids flankieren,
importiert werden und die eine Amidbindung bildenden Seitenketten-Substituenten
des N-terminalen und C-terminalen flankierenden Rests dann verbunden
werden, um eine zyklisierte Struktur zu bilden, welche die Konformation
einer α-Helix
nachahmt.
-
Es
gibt zumindest zwei allgemeine Verfahren zur Konstruktion der eingeschränkten Helixpeptide
der Erfindung: (1) Synthese eines linearen Peptids, das ein Paar
von Resten umfasst, welche eine Aminosäuresequenz flankieren, die
sechs Reste lang ist, worin die beiden flankierenden Reste unabhängig voneinander aus
der aus Aminosäuren
mit eine Amidbindung bildenden Seitenketten-Substituenten bestehenden
Gruppe ausgewählt
sind, gefolgt von der Überbrückung der
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste mit einem difunktionellen Linker, um das Peptid zu zyklisieren;
und (2) Synthese eines linearen Peptids, das ein Paar von Resten
umfasst, welche eine Aminosäuresequenz
flankieren, die sechs Reste lang ist, worin die beiden flankierenden
Reste unabhängig
voneinander aus der aus Aminosäuren
mit eine Amidbindung bildenden Seitenketten-Substituenten bestehenden Gruppe ausgewählt ist,
und worin einer der flankierenden Reste zur Peptidkette hinzugefügt wird,
welche einen difunktionellen Linker trägt, sodass eine funktionelle
Gruppe des Linkers mit dem eine Amidbindung bildenden Seitenketten-Substituenten
des Rests verbunden wird, gefolgt vom Verbinden der freien funktionellen
Gruppe des Linkers mit dem eine Amidbindung bildenden Sei tenketten-Substituenten
am anderen flankierenden Rest, um das Peptid zu zyklisieren.
-
Jede
beliebige Aminosäure
mit einer Seitenkette, die einen Substituenten aufweist, der in
der Lage ist, eine Amidbindung zu bilden, kann hierin als flankierender
Rest eingesetzt werden. Geeignete flankierende Aminosäurereste
umfassen Aminosäuren
mit Seitenketten, die eine freie Carboxylgruppe tragen, wie z.B.
Aminoprapandisäure,
Asp, Glu, 2-Aminohexandisäure
und 2-Aminoheptandisäure,
sowie Aminasäuren
mit Seitenketten, die eine freie Aminogruppe tragen, wie z.B. 2,3-Diaminopropansäure (2,3-Diaminopropionsäure), 2,4-Diaminobutansäure (2,4-Diaminobuttersäure), 2,5-Diaminopentansäure und
Lys.
-
(1) Synthese eines linearen
Peptids ohne an einen difunktionellen Linker gebundene flankierende
Aminosäure
-
a. Peptidsynthese
-
Die
gewünschte
Peptidsequenz ist so aufgebaut, dass die Sequenz aus sechs Aminosäureresten,
aus der eine Helix gebildet werden soll, sich zwischen zwei flankierenden
Resten erstreckt, die unabhängig
voneinander aus der Aminosäureresten
mit eine Amidbindung bildenden Seitenketten-Substituenten bestehenden Gruppe
ausgewählt
ist. In einer Ausführungsform
sind die eine Amidbindung bildenden Seitenketten-Substituenten des
N-terminalen und des C-terminalen flankierenden Rests unabhängig voneinander
aus der aus einem Carboxysubstituenten und einem Aminosubstituenten
bestehenden Gruppe ausgewählt.
In einer weiteren Ausführungsform
sind die eine Amidbindung bildenden Seitenketten-Substituenten des
N-terminalen und
des C-terminalen flankierenden Rests beide Carboxysubstituenten.
In einer weiteren Ausführungsform
ist der eine Amidbindung bildende Seitenketten-Substituent eines der flankierenden
Reste ein Carboxysubstituent und der eine Amidbindung bildende Seitenketten-Substituent
des anderen flankierenden Rests ein Aminosubstituent. In einer weiteren
Ausführungsform
sind die flankierenden Reste unabhängig voneinander aus der aus
Aminopropandisäure,
Asp, Glu, 2- Aminohexandisäure, 2-Aminoheptandisäure, 2-Aminooctandisäure, 2-Aminononandisäure, 2,3-Diaminopropansäure, 2,4-Diaminobutansäure, 2,5-Diaminopentansäure, Lys,
2,7-Diaminoheptansäure,
2,8-Diaminooctansäure
und 2,9-Diaminononansäure bestehenden
Gruppe ausgewählt.
-
In
einigen Ausführungsformen
enthält
das gewünschte
Peptid eine zusätzliche
Aminosäure
oder zusätzliche
Aminosäuren,
die sich vom C-terminalen flankierenden Rest und/oder vom N-terminalen
flankierenden Rest aus erstrecken.
-
Sobald
die gewünschte
Peptidsequenz ausgewählt
ist, kann eine chemische Synthese durchgeführt werden, um das eingeschränkte helikale
Peptid der Erfindung zu konstruieren. Dies kann erreicht werden,
indem eines von verschiedenen Verfahren, die auf dem Gebiet der
Erfindung allgemein bekannt sind (siehe R.F. Kelley & M.E. Winkler,
in: Genetic Engineering Principles and Methods, J.K. Setlow, Hrsg.,
Plenum Press, N.Y., Bd. 12, S. 1–19 (1990); J.M. Steart, J.D.
Young, Solid Phase Peptide Synthesis, Pierce Chemical Co., Rockford,
IL (1984); siehe auch US-Patent Nr. 4.105.603, 3.972.859, 3.842.067;
und 3.862.925) modifiziert wird, um ein gewünschtes Peptid herzustellen.
-
Peptide
der Erfindung werden am besten unter Einsatz von Festphasen-Peptidsynthese hergestellt
(J. Merrifield, J. Am. Chem. Soc. 85, 2149 (1964); Houghten, Proc.
Natl. Acad. Sci. USA 82, 5132 (1985). Eine Festphasensynthese beginnt
am Carboxyterminus des geplanten Peptids, indem eine geschützte Aminosäure an einen
inerten festen Träger
gebunden wird. Der inerte feste Träger kann jedes beliebige Makromolekül sein, das
in der Lage ist, als Anker für
den C-Terminus der Ausgangsaminosäure zu dienen. Typischerweise
ist der makromolekulare Träger
ein vernetztes Polymerharz (z.B. ein Polyamid- oder Polystyrolharz),
wie es in den 1-1 und 1-2 auf
den Seiten 2 und 4 in Stewart und Young, s.o., gezeigt wird. In
einer Ausführungsform
wird die C-terminale Aminosäure
an ein Polystyrolharz gebunden, um einen Benzylester herzustellen.
Ein makromolekularer Träger
wird so gewählt,
dass die Peptid-Ankerbindung unter den Bedingungen stabil ist, die
zum Entschützen
der α-Aminogruppe
der blockierten Aminosäuren
während
der Peptid synthese eingesetzt werden. Wenn eine basenlabile α-Schutzgruppe
verwendet wird, ist es wünschenswert,
eine säurelabile
Verbindung zwischen dem Peptid und dem festen Träger einzusetzen. Ein säurelabiles
Etherharz ist beispielsweise wirksam für eine Synthese eines basenlabilen
Fmoc-Aminosäurepeptids,
wie dies auf Seite 16 in Stewart und Young, s.o., beschrieben wird.
Alternativ dazu können
eine Peptidankerbindung und eine α-Schutzgruppe
eingesetzt werden, die unterschiedlich labil gegenüber Azidolyse
sind. Beispielsweise funktioniert ein Aminomethylharz, wie z.B.
das Phenylacetamidomethyl- (Pam-) Harz, gut zusammen mit Boc-Aminosäurepeptidsynthese,
wie sie auf Seite 11–12
in Stewart und Young, s.o., beschrieben wird.
-
Nachdem
die Ausgangsaminosäure
an einen inerten festen Träger
gebunden wurde, wird die α-Aminoschutzgruppe
der Ausgangsaminosäure
entfernt, beispielsweise mit Trifluoressigsäure (TFA) in Methylenchlorid
und Neutralisierung in beispielsweise Triethylamin (TEA). Nach Entschützung der α-Aminogruppe
der Ausgangsaminosäure
wird die nächste α-Amino- und
Seitenketten-geschützte
Aminosäure
der Synthese hinzugefügt.
Die restlichen α-Amino-geschützten, und
falls erforderlich Seitenkettengeschützten, Aminosäuren werden
dann nacheinander in der gewünschten
Reihenfolge durch Kondensation gebunden, um eine Zwischenverbindung
zu erhalten, die an den festen Träger gebunden ist. Alternativ
dazu können
einige Aminosäuren
aneinander gebunden werden, um ein Fragment des gewünschten
Peptids zu bilden, gefolgt vom Hinzufügen des Peptidfragments zum
wachsenden Festphasen-Peptidkette.
-
Die
Kondensationsreaktion zwischen zwei Aminosäuren oder einer Aminosäure und
einem Peptid oder einem Peptid und einem Peptid kann nach herkömmlichen
Kondensationsverfahren, wie z.B. durch das Azidverfahren, gemischtes
Säureanhydridverfahren,
DCC-(N,N'-Dicyclohexylcarbodiimid-)
oder DIC-(N,N'-Diisopropylcarbodiimid-)
Verfahren, Aktivesterverfahren, p-Nitrophenylesterverfahren, BOP-(Benzotriazol-1-yloxytris[dimethylamino]phosphoniumhexafluorophosphat-)
Verfahren, N-Hydroxybernsteinsäureimidoesterverfahren
usw., sowie nach dem Woodward-Reagens-K-Verfahren
erfolgen.
-
Normalerweise
wird bei der chemischen Synthese von Peptiden jede reaktive Seitenkettengruppe
der Aminosäuren
mit geeigneten Schutzgruppen entschützt. Diese Schutzgruppen werden
schlussendlich entfernt, nachdem die gewünschte Polypeptidkette zusammengesetzt
wurde. Auch üblich
ist ein Schutz der α-Aminogruppe
an der Aminosäure
oder einem Fragment, während
das Gesamte an der Carboxygruppe reagiert, gefolgt von der selektiven
Entfernung der α-Aminoschutzgruppe,
damit an dieser Stelle eine nachfolgende Reaktion stattfinden kann.
Demgemäß wird bei
der Polypeptidsynthese häufig
eine Zwischenverbindung hergestellt, die alle Aminosäurereste
in der gewünschten
Reihenfolge in der Peptidkette enthält, wobei an einige dieser
Reste Seitenketten-Schutzgruppen gebunden sind. Diese Schutzgruppen
werden meist zum im Wesentlichen gleichen Zeitpunkt entfernt, um
das gewünschte
Produkt herzustellen, gefolgt von einer Abspaltung vom Harz. Einen Überblick über Schutzgruppen
und Verfahren zu deren Verwendung bei der Peptidsynthese findet
sich in Protective Groups in Organic Synthesis, 1. Aufl., T.W. Greene
und P.G.M. Wuts, Wiley & Sons,
New York (1991).
-
Beispiele
für geeignete
Schutzgruppen für α-Amino- und
Seitenketten-Aminogruppen sind Benzyloxycarbonyl-(abgekürzt mit
Z), Isonicotinyloxycarbonyl-(iNOC), o-Chlorbenzyloxycarbonyl-[Z(2Cl)],
p-Nitrobenzyloxycarbonyl-[Z(NO2)], p-Methoxybenzyloxycarbonyl-[Z(OMe)],
t-Butoxycarbonyl-(Boc), t-Amyloxycarbonyl-(Aoc), Isobornyloxycarbonyl-,
Adamantyloxycarbonyl-, 2-(4-Biphenyl)-2-propyloxycarbonyl-(Bpoc), 9-Fluorenylmethoxycarbonyl-(Fmoc),
Methylsulfonylethoxycarbonyl-(Msc), Trifluoracetyl-, Phthalyl-,
Formyl-, 2-Nitrophenylsulfenyl-(NPS), Diphenylthiophosphinyl-(Ppt)
und Dimethylthiophosphinyl-(Mpt) Gruppen und dergleichen.
-
Beispiele
für Schutzgruppen
für die
funktionelle Carboxygruppe sind Benzylester, (Obz), Cyclohexylester
(Chx), 4-Nitrobenzylester (Onb), t-Butylester (Obut), 4-Pyridylmethylester
(Opic) und dergleichen. Oft ist es wünschenswert, dass Aminosäuren wie
Arginin, Cystein und Serin, die eine andere funktionelle Gruppe
aufweisen als Amino- und Carboxygruppen, mit einer geeigneten Schutzgruppe
geschützt
werden. Die Guanidinogruppe von Arginin kann beispielsweise mit
Nitro, p-Toluolsulfonyl, Benzyloxycarbonyl, Adamantyloxycarbonyl,
p-Methoxybenzolsulfonyl, 4-Methoxy-2,6-dimethylbenzolsulfonyl (Nds),
1,3,5-Trimethylphenylsulfonyl (Mts) und dergleichen geschützt werden.
Die Thiolgruppe von Cystein kann mit p-Methoxybenzyl, Trityl und dergleichen
geschützt
sein.
-
In
einer Ausführungsform
werden die Peptide der Erfindung mithilfe von Blockiergruppen synthetisiert, welche
die eine Amidbindung bildenden Seitenketten-Substituenten der N-terminalen und C-terminalen
flankierenden Reste schützen.
Die Schutzgruppe oder -gruppen für
die eine Amidbindung bildenden Seitenketten-Substituenten der N-terminalen und C-terminalen
flankierenden Reste können
die gleichen wie oder andere als die Schutzgruppe oder -gruppen
sein, die zum Blockieren der funktionellen Seitenkettengruppen anderer Reste
im Peptid eingesetzt werden. In einer bevorzugten Ausführungsform
sind die Schutzgruppe oder -gruppen, die zum Blockieren der eine
Amidbindung bildenden Seitenketten-Substituenten verwendet werden,
in Bezug auf die Schutzgruppen, die für die anderen funktionellen
Seitenkettengruppe im Peptid eingesetzt werden, unterschiedlich
entfernbar, d.h. die eine Amidbindung bildenden Seitenketten-Substituenten
können
entschützt
werden, ohne dass die anderen funktionellen Seitenkettengruppen
im Peptid entschützt
werden, und zwar zusätzlich
zur unterschiedlichen Entfernbarkeit in Bezug auf die in der Peptidsynthese
eingesetzte α-Aminoschutzgruppe.
In einer anderen bevorzugten Ausführungsform werden die eine
Amidbindung bildenden Seitenketten-Substituenten der flankierenden Reste
orthogonal in Bezug aufeinander entschützt, sodass der eine Amidbindung
bildende Seitenketten-Substituent eines flankierenden Rests entschützt werden
kann, ohne dass der eine Amidbindung bildende Seitenketten-Substituent
des anderen flankierenden Rests entschützt wird.
-
Geeignete
Schutzgruppen zur Verwendung beim orthogonalen Schützen der
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste in Bezug auf andere funktionelle Gruppe und/oder in Bezug
aufeinander umfassen Paare von unterschiedlich entfernbaren Carboxyschutzgruppen,
wie beispielsweise reduktionslabilen Schutzgruppen, z.B. Allyl-
oder Benzylestern, gepaart mit einer basenlabilen Carboxyschutzgruppen,
z.B. Fluorenylmethylestern, Methyl oder anderen primä ren Alkylestern.
Fluorenylmethyl-, Methyl- oder andere primäre Alkylgruppen oder andere
basenlabile Carboxyschutzgruppen können aus ihren entsprechenden
Estern entfernt werden, um eine freie Carboxylgruppe zu erhalten
(ohne dass Allyl- oder Benzylester oder anderen reduktionslabile
Ester entschützt
werden), und zwar durch Verseifung der Ester mit einer geeigneten
Base, wie z.B. Piperidin und Natriumhydroxid, in einem geeigneten
Lösungsmittel,
wie z.B. Dimethylacetamid oder Methanol und Wasser, für einen
Zeitraum von 10 bis 120 Minuten, vorzugsweise 20 Minuten, bei 0
bis 50°C.
Der Allyl- oder Benzyl- oder andere reduktionslabile Ester kann,
falls gewünscht,
entfernt werden, und zwar durch Reduktion in Gegenwart eines geeigneten Übergangsmetallkatalysators,
wie z.B. Pd(PPh3)4,
Pd(PPh3)2Cl2, Pd(OAc)2 oder
Pd/C, mit einer Wasserstoffquelle, z.B. H2-Gas,
in einem geeigneten Lösungsmittel,
wie z.B. Dimethylacetamid, Dimethylformamid, N-Methylpyrrolidinon
oder Methanol, für einen
Zeitraum von 10 bis 500 Minuten, vorzugsweise 100 Minuten, bei 0
bis 50°C.
Der Einfachheit und Bequemlichkeit halber sind alle Carboxyschutzgruppen,
die durch Pd-katalysierte Verfahren entfernbar sind, welche zur
Reduktion des geschützten
Carboxysubstituenten führen,
in der Bezeichnung „reduktionslabile Schutzgruppen" enthalten, wie sie
hierin verwendet wird, auch wenn Pdkatalysierte Entschützungsverfahren eventuell
nicht zur Reduktion der betreffenden Schutzgruppe führen.
-
In
Ausführungsformen,
bei denen Pd-Katalyse die Bildung von Zwischenprodukten von mit
reduktionslabilen Schutzgruppen derivatisiertem Pd, wie z.B. von
Pd-Allyl-Derivaten,
umfasst, kann der Pd-Katalysator durch Umsetzung mit einem geeigneten
Nucleophil, wie z.B. Piperidin oder Tributylzinnhydrid, wiederhergestellt
werden. Wenn solche reduktionslabile Gruppen verwendet werden, um
in Kombination mit basenlabilen Schutzgruppen einen orthogonalen
Schutz bereitzustellen, wird vorzugsweise entweder (1) ein Syntheseschema
verwendet, das eine Entfernung der basenlabilen Schutzgruppen vor
der Entfernung der reduktionslabilen Schutzgruppen erfordert, oder
(2) der Pd-Katalysator mit einem Nucleophil wiederhergestellt, das
die basenlabilen Schutzgruppen nicht entschützt.
-
Alternativ
dazu können
die Carboxy-Substituenten der flankierenden Reste in Bezug auf andere
funktionelle Gruppen und/oder in Bezug aufeinander unter Einsatz
einer säurelabilen
Schutzgruppe, wie beispielsweise eines tertiären Alkylesters, z.B. t-Butylester,
in Kombination mit einer reduktionslabilen Schutzgruppe, beispielsweise
dem oben beschriebenen Allyl- oder Benzylester, orthogonal geschützt werden.
Die tertiäre
Alkyl- oder andere säurelabile
Estergruppe kann durch Azidolyse, beispielsweise mit Trifluoressigsäure in Methylenchlorid,
entfernt werden, und der Allyl- oder Benzyl- oder andere reduktionslabile
Ester kann durch Reduktion in Gegenwart eines Übergangsmetallkatalysators,
wie oben beschrieben, entfernt werden.
-
In
einer anderen Ausführungsform
können
die Carboxy-Substituenten der flankierenden Reste in Bezug auf andere
funktionelle Gruppen und/oder in Bezug aufeinander unter Einsatz
einer fluoridionenlabilen Schutzgruppe, beispielsweise 2-(Trimethylsilyl)ethyl-
und Silylester, in Kombination mit einer reduktionslabilen Schutzgruppe,
wie z.B. dem oben beschriebenen Allyl- oder Benzylester, oder in
Kombination mit einer basenlabilen Schutzgruppe, wie z.B. dem oben
beschriebenen Fluorenylmethyl-, Methyl- oder anderen primären Alkylester,
orthogonal entschützt
werden, ohne dass die reduktionslabilen oder basenlabilen Ester
entschützt werden.
Die 2-(Trimethylsilyl)ethyl-,
Silyl- oder andere fluoridlabile Estergruppe kann durch Umsetzung
mit einer geeigneten Fluoridionenquelle, wie z.B. Tetrabutylammoniumfluorid,
in Gegenwart eines geeigneten Lösungsmittels,
wie z.B. Dimethylacetamid (DMA), Dimethylformamid (DMF), Tetrahydrofuran
(THF) oder Acetonitril, entfernt werden.
-
Geeignete
Schutzgruppen zur Verwendung beim orthogonalen Schützen der
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste in Sezug auf andere funktionelle Gruppen und/oder in Bezug
aufeinander umfasst auch Paare unterschiedlich entfernbarer Aminoschutzgruppen,
wie z.B. eine Allyloxycarbonyl- oder andere reduktionslabile Aminoschutzgruppe,
die mit einer t-Butoxycarbonyl-(Boc-) oder anderen säurelabilen
Aminoschutzgruppe gepaart ist, und eine reduktionslabile Schutzgruppe, die
mit einer Fluorenylmethoxycarbonyl-(Fmoc-) oder anderen basenlabilen
Aminoschutzgruppe gepaart ist. Eine Allyloxycarbonyl-(oder mit einer
anderen reduktionslabilen Blockiergruppe) geschützte Aminogruppe kann durch
Reduktion unter Einsatz eines Übergangsmetallkatalysators
entschützt
werden, wie in den oben beschriebenen Verfahren zur Entfernung von
reduktionslabilen Carboxyschutzgruppen erläutert wird, ohne dass eine
Boc- oder Fmoc-geschützte
Aminogruppe entschützt
wird. Gleichermaßen
kann eine säurelabile Aminoschutzgruppe
und eine basenlabile Aminoschutzgruppe durch Azidolyse bzw. basische
Verseifung entfernt werden, ohne dass eine reduktionslabile Aminoschutzgruppe
entfernt wird. Der Einfachheit und Bequemlichkeit halber sind alle
Aminoschutzgruppen, die durch Pd-katalysierte Verfahren entfernbar
sind, die zur Reduktion der geschützten Aminosubstituenten führen, in
der Bezeichnung „reduktionslabile
Schutzgruppen",
wie er hierin verwendet wird, eingeschlossen, auch wenn solche Pd-katalysierte
Entschützungsverfahren
eventuell nicht zur Reduktion der betreffenden Schutzgruppe führen.
-
In
einer anderen Ausführungsform
können
die Aminosubstituenten der flankierenden Reste in Bezug auf andere
funktionelle Gruppen und/oder in Bezug aufeinander unter Einsatz
einer fluoridlabilen Schutzgruppe, wie z.B. 2-Trimethylsilylethylcarbamat
(Teoc), in Kombination mit einer reduktionslabilen Schutzgruppe,
wie z.B. Allyfoxylcarbonyl, oder in Kombination mit einer basenlabilen
Schutzgruppe, wie z.B. Fluorenylmethoxycarbonyl, wie oben beschrieben
orthogonal geschützt
werden. Die Teoc- oder
andere fluoridlabile Gruppe kann durch Umsetzung mit einer geeigneten
Fluoridionenquelle, wie z.B. Tetrabutylammoniumfluorid, wie in den oben
beschriebenen Verfahren zur Entfernung von fluoridlabilen Carboxyschutzgruppen
entfernt werden, ohne dass eine Allyloxycarboyl- oder Fluorenylmethoxycarbonyl-geschützte Aminogruppe
entschützt
wird. Gleichmaßen
kann eine reduktionslabile Aminoschutzgruppe und eine basenlabile
Aminoschutzgruppe durch Reduktion bzw. basische Verseifung entfernt
werden, ohne dass eine fluoridlabile Aminoschutzgruppe entfernt wird.
-
In
Ausführungsformen,
bei denen ein Carboxysubstituent als eine Amidbindung bildender
Seitenketten-Substituent eines flankierenden Rests eingesetzt wird
und ein Aminosubstituent als eine Amidbindung bildender Seitenketten-Substituent
des anderen flankierenden Rests eingesetzt wird, können der
Carboxysubstituent und der Aminosubstituent in Bezug aufeinander
orthogonal geschützt
werden, indem eine reduktionslabile Schutzgruppe eingesetzt wird,
um einen Substituenten zu blockieren, z.B. Allylester oder Allyloxycarbonyl, und
eine fluoridlabile, säurelabile
oder basenlabile Schutzgruppe, um einen anderen Substituenten zu
blockieren, z.B. Silylester, t-Butylester,
Fluorenylmethylester, Teoc, Boc oder Fmoc.
-
In
einer bevorzugten Ausführungsform
wird eine reduktionslabile Schutzgruppe verwendet, um den eine Amidbindung
bildenden Seitenketten-Substituenten eines flankierenden Rests zu
blockieren, und der eine Amidbindung bildende Seitenketten-Substituent des anderen
flankierenden Rests wird so gewählt,
dass er orthogonalen Schutz in Bezug auf sowohl die reduktionslabile
Schutzgruppe als auch die α-Aminoschutzgruppe, die
in der Peptidsynthese eingesetzt werden, bereitstellt. In einer
Ausführungsform
unter Anwendung von Fmoc-Chemie zur Peptidsynthese wird orthogonaler
Schutz der eine Amidbindung bildenden Seitenketten-Substituenten
beispielsweise durch eine reduktionslabile Schutzgruppe und eine
säurelabile
Schutzgruppe bereitgestellt. Gleichmaßen wird in einer Ausführungsform
unter Anwendung von Boc-Chemie zur Peptidsynthese orthogonaler Schutz
der eine Amidbindung bildenden Seitenketten-Substituenten durch
eine reduktionslabile Schutzgruppe und eine basenlabile Schutzgruppe
bereitgestellt.
-
In
einer weiteren bevorzugten Ausführungsform
werden die eine Amidbindung bildenden Seitenketten-Substituenten
der flankierenden Reste in Bezug aufeinander, in Bezug auf eine α-Aminoschutzgruppe,
die bei der Peptidsynthese eingesetzt wird, und in Bezug auf die
Schutzgruppe, die zur Blockierung anderer funktioneller Seitenkettengruppen
irr der Peptidkette orthogonal geschützt.
-
In
einer weiteren bevorzugten Ausführungsform
werden die eine Amidbindung bildenden Seitenketten-Substituenten
der flankierenden Reste in Bezug aufeinander und in Bezug auf die α-Aminoschutzgruppe, die
bei der Peptidsynthese eingesetzt wird, orthogonal geschützt, aber
nur einer der eine Amidbindung bildenden Seitenketten-Substituenten
wird in Bezug auf die Schutzgruppen, die zur Blockierung ande rer
funktioneller Seitenkettengruppen in der Peptidkette eingesetzt
werden, orthogonal geschützt.
In dieser Ausführungsform wird
der eine Amidbindung bildende Seitenketten-Substituent mit vollem
orthogonalem Schutz vorzugsweise als Target für die anfängliche Bindung des Peptids
an den difunktionellen Linker verwendet. Da der eine Amidbindung
bildende Seitenketten-Substituent mit vollem orthogonalem Schutz
entschützt
werden kann, ohne dass andere funktionelle Gruppen entschützt werden,
ist die Bindungsreaktion spezifisch für den gewünschten, eine Amidbindung bildenden
Seitenketten-Substituenten und verringert die Produktion von unerwünschten
Derivaten von Peptid/difunktionellem Linker. Obwohl eine Zyklisierung
die Entschützung
des eine Amidbindung bildenden Seitenketten-Substituenten des anderen
flankierenden Rests erfordert und eine gleichzeitige Entschützung anderer
funktioneller Seitenkettengruppen verursachen kann, ist die Wahrscheinlichkeit
geringer, dass unerwünschte
Derivate entstehen, da die Peptidketten an einem festen Träger verankert
sind und die Linker-Länge
regioselektiv eine Bindungsreaktion zwischen der ungebundenen funktionellen
Gruppe des Linkers und dem eine Amidbindung bildenden Seitenketten-Substituenten
der anderen flanierenden Rests bevorzugt. Wenn nach der Zyklisierung
weitere Peptidkettensynthese gewünscht
wird, kann jede beliebige funktionelle Seitenkettengruppe anderer
Aminosäurereste,
die durch die Zyklisierungsreaktionen entschützt worden waren, erneut geschützt werden,
bevor die Kettensynthese wiederaufgenommen wird.
-
Viele
der oben beschriebenen blockierten Aminosäuren können im Handel erworben werden,
wie beispielsweise von Novabiochem (San Diego, CA, USA), Bachem
CA (Torrence, CA, USA) oder Peninsula Labs (Belmont, CA, USA).
-
Außerdem können die
Verfahren der Erfindung auch zusammen mit Flüssigphasen-Peptidsynthese umgesetzt werden, beispielsweise
mit den in Principles of Peptide Synthesis, 2. Aufl., M. Bodansky,
Springer-Verlag (1993) oder in The Practice of Peptide Synthesis,
2. Aufl., M. Bodansky und A. Bodansky, Springer Verlag (1994) beschriebenen
Flüssigphasen-Peptidsyntheseverfahren.
Es versteht sich, dass Flüssigphasen-Peptidsyntheseverfahren
leicht so modifiziert werden können,
dass die gewünschten
flankierenden Reste, mit oder ohne orthogonal geschützte, eine
Amid bindung bildende Seitenketten-Substituenten, in die Peptidkette
von Interesse eingebaut werden, wobei ähnliche Verfahren eingesetzt
werden wie bei den hierin beschriebenen Festphasen-Peptidsyntheseverfahren.
Außerdem
versteht sich, dass alle Verweise auf Peptidsynthese hierin, sofern
nicht anders angegeben, sowohl Festphasen- als auch Flüssigphasen-(oder
Lösungsphasen-) Peptidsyntheseverfahren
umfassen.
-
b. Peptidzyklisierung
-
Nachdem
die gewünschte
Aminosäuresequenz
vervollständigt
wurde, wird das lineare Peptid zyklisiert, um das Peptid auf eine
helikale Konformation einzuschränken.
Jedes beliebige Verfahren zur Verbrückung der eine Amidbindung
bildenden Seitenketten-Substituenten der flankierenden Reste mit
einem difunktionellen Linker ist zur Herstellung der eingeschränkten helikalen
Peptide der Erfindung geeignet.
-
(i) Selektion eines difunktionellen
Linkers
-
Typischerweise
ist der zur Verwendung hierin geeignete difunktionelle Linker in
der Lage, zwei funktionelle Gruppen, die in einem Abstand von etwa
5 Å bis
etwa 30 Å,
vorzugsweise etwa 8 Å bis
etwa 14 Å,
noch bevorzugter etwa 10 Å,
voneinander getrennt sind, so zu präsentieren, dass der eine Amidbindung
bildende Seitenketten-Substituent
eines der flankierenden Reste eine Amidbindung mit einer der funktionellen
Gruppen des Linkers bilden kann und der eine Amidbindung bildende
Seitenketten-Substituent des anderen flankierenden Rests eine Amidbindung
mit der verbleibenden funktionellen Gruppe des Linkers bilden kann.
Es versteht sich, dass die Art des Molekülgerüsts, das zur Präsentation
der gewünschten
funktionellen Gruppen in der passenden räumlichen Beziehung verwendet
wird, für
die Umsetzung der Erfindung nicht entscheidend ist. Obwohl unverzweigte
und verzweigte Alkylgerüste
zur Verwendung hierin geeignet sind, ist die Erfindung nicht darauf
beschränkt.
Beispielsweise können
Alkenyl-, Alkinyl-, Cycloalkyl- oder andere aliphatische Kohlenwasserstoffspezies,
mit oder ohne Heteroatome, und Monophenyl-, Biphenyl-, Naphthyl-
und andere aromatische Kohlenwasserstoffspezies, mit oder ohne Hetero atome,
die mit den gewünschten
funktionellen Gruppen in der passenden räumlichen Beziehung substituiert
sind (z.B. para- oder meta-Substitutionen in Ringstrukturen, wie z.B.
Monophenyl, Biphenyl, Naphthyl und dergleichen), verwendet werden,
um die eine Amidbindung bildenden Seitenketten-Substituenten der
flankierenden Reste zu verbinden.
-
Die
im funktionellen Linker eingesetzten funktionellen Gruppen werden
so gewählt,
dass sie in der Lage sind, Amidbindungen mit den eine Amidbindung
bildenden Seitenketten-Substituenten der flankierenden Reste zu
bilden, die im zu zyklisierenden Peptid verwendet werden. In Ausführungsformen,
worin der eine Amidbindung bildende Seitenketten-Substituent jedes
flankierenden Rests ein Carboxysubstituent ist, wird das Peptid
am besten mit einem Diaminlinker zyklisiert. In einem Beispiel werden
die flankierenden Reste und der Diaminlinker gemäß nachstehender Tabelle 1 ausgewählt. Es
versteht sich, dass die flankierenden Reste und Linkermoleküle, die
in der nachstehenden Tabelle 1 angeführt sind, nicht nur das jeweilige
Molekül
darstellen, das dem chemischen Namen gemäß IUPAC entspricht, sondern
auch Varianten des Moleküls,
die weitere Substituenten oder modifizierte Substituenten enthalten,
welche die Funktion der Amino- und/oder Carboxygruppen im Molekül nicht
verhindern und/oder wesentlich verändern, wobei diese Funktion
zur Verwendung des Moleküls
in den Verfahren der Erfindung notwendig ist. Demgemäß versteht
sich, dass jedes nachstehend angeführte Molekül auch Molekülvarianten
umfasst, die Alkenyl-, Alkinyl- und andere ungesättigte Bindungen, Heteroatome,
Cycloalkylsubstituenten, aromatische Substituenten oder andere Substituenten
im Kohlenstoff-Rückgrat des
Moleküls
enthalten, und/oder Varianten, welche die obigen oder andere Substituenten
oder Gruppen anstelle von Wasserstoffatomen auf dem Kohlenstoff-Rückgrat des Moleküls enthalten.
-
-
-
In
Ausführungsformen,
in denen der eine Amidbindung bildende Seitenketten-Substituent jedes
flankierenden Rests ein Aminosubstituent ist, wird das Peptid am
besten mit einem Dicarbonsäurelinker
zyklisiert. In einem Beispiel werden die flankierenden Reste und
der Dicarbonsäurelinker
gemäß nachstehender
Tabelle 2 ausgewählt.
Es versteht sich, dass die flankierenden Reste und Linkermoleküle, die
in der nachstehenden Tabelle 2 angeführt sind, nicht nur das jeweilige
Molekül
darstellen, das dem chemischen Namen gemäß IUPAC entspricht, sondern
auch Varianten des Moleküls,
die weitere Substituenten oder modifizierte Substituenten enthalten,
welche die Funktion der Amino- und/oder Carboxygruppen im Molekül nicht
verhindern und/oder wesentlich verändern, wobei diese Funktion
zur Verwendung des Moleküls
in den Verfahren der Erfindung notwendig ist. Demgemäß versteht
sich, dass jedes nachstehend angeführte Molekül auch Molekülvarianten
umfasst, die Alkenyl-, Alkinyl- und andere ungesättigte Bindungen, Heteroatome,
Cycloalkylsubstituenten, aromatische Substituenten oder andere Substituenten
im Kohlenstoff-Rückgrat
des Moleküls
enthalten, und/oder Varianten, welche die obigen oder andere Substituen ten
oder Gruppen anstelle von Wasserstoffatomen auf dem Kohlenstoff-Rückgrat des
Moleküls
enthalten.
-
-
-
In
Ausführungsformen,
bei denen ein Aminosubstituent als eine Amidbindung bildender Seitenketten-Substituent
des einen fankierenden Rests und ein Carboxysubstituent als eine
Amidbindung bildender Seitenketten-Substituent des anderen flankierenden
Rests eingesetzt wird, wird das Peptid am besten mit einem aminosubstituierten
Carbonsäure-(Aminocarbonsäure-) Linker
zyklisiert. In einem Beispiel werden die flankierenden Reste und
der Aminocarbonsäure-Linker
gemäß nachstehender
Tabelle 3 ausgewählt.
Es versteht sich, dass die flankierenden Reste und Linkermoleküle, die
in der nachstehenden Tabelle 3 angeführt sind, nicht nur das jeweilige
Molekül
darstellen, das dem chemischen Namen gemäß IUPAC entspricht, sondern auch
Varianten des Moleküls,
die weitere Substituenten oder modifizierte Substituenten enthalten,
welche die Funktion der Amino- und/oder Carboxygruppen im Molekül nicht
verhindern und/oder wesentlich verändern, wobei diese Funktion
zur Verwendung des Moleküls
in den Verfahren der Erfindung notwendig ist. Demgemäß versteht
sich, dass jedes nachstehend angeführte Molekül auch Molekülvarianten
um fasst, die Alkenyl-, Alkinyl- und andere ungesättigte Bindungen, Heteroatome,
Cycloalkylsubstituenten, aromatische Substituenten oder andere Substituenten
im Kohlenstoff-Rückgrat
des Moleküls
enthalten, und/oder Varianten, welche die obigen oder andere Substituenten
oder Gruppen anstelle von Wasserstoffatomen auf dem Kohlenstoff-Rückgrat des
Moleküls
enthalten.
-
-
-
-
(ii) Zyklisierungsverfahren
-
Sobald
die flankierenden Reste und der difunktionelle Linker ausgewählt wurden
und die Peptidkette, welche die flankierenden Reste überspannt,
auf einer festen Phase synthetisiert wurde, kann der difunktionelle Linker
verwendet werden, um das festphasengebundene Peptid durch ein beliebiges
geeignetes Verfahren zu zyklisieren. Es versteht sich, dass die
Erfindung Verfahren zur Zyklisierung eines Peptids umfasst, nachdem die
endgültige
Peptidkette vollständig
synthetisiert wurde, sowie Verfahren zur Zyklisierung des Peptids
zu jedem beliebigen Zeitpunkt während
der Peptidsynthese, an dem die Peptidkette die flankierenden Reste
enthält,
die durch den difunktionellen Linker zu vernetzen sind. Verfahren
zur Zyklisierung des Peptids umfassen (1) die Entschützung der
eine Amidbindung bildenden Seitenketten- Substituenten der flankierenden Reste
und die Umsetzung des Festphasenpeptids mit dem difunktionellen
Linker, um gleichzeitig Amidbindungen zwischen den beiden funktionellen
Gruppen des Linkers und den eine Amidbindung bildenden Seitenketten-Substituenten
beider flankierender Reste zu bilden; (2) das Entschützen des
eine Amidbindung bildenden Seitenketten-Substituenten von nur einem
der flankierenden Reste (ohne dass der eine Amidbindung bildende
Seitenketten-Substituent des anderen flankierenden Rests entschützt wird),
das Umsetzen des difunktionellen Linkers mit dem Festphasenpeptid,
um eine Amidbindung zwischen einer funktionellen Gruppe auf dem
Linker und dem eine Amidbindung bildenden Seitenketten-Substituenten des
entschützten
flankierenden Rests zu bilden, und dann das intramolekulare Umsetzen
der freien funktionellen Gruppe auf dem Linker und des eine Amidbindung
bildenden Seitenketten-Substituenten des anderen flankierenden Rests,
wodurch das Peptid zyklisiert wird; und (3) das Entschützen der
eine Amidbindung bildenden Seitenketten-Substituenten beider flankierender
Reste, wodurch ein einfach geschützter
difunktionellen Linker erhalten wird, worin nur eine der beiden
eine Amidbindung bildenden funktionellen Gruppen des Linkers in
der Lage ist, mit einem entsprechenden, eine Amidbindung bildenden
Seitenketten-Substituenten in einem flankierenden Rest zu reagieren,
das Umsetzen des einfach geschützten,
difunktionellen Linkers mit dem Festphasenpeptid, um eine Amidbindung zwischen
der freien funktionellen Gruppe auf dem Linker und dem eine Amidbindung
bildenden Seitenketten-Substituenten eines der entschützten flankierenden
Rests zu bilden, das Entschützen
der blockierten funktionellen Gruppe auf dem Linker und dann das
intramolekulare Umsetzen der freien funktionellen Gruppe auf dem
Linker und des eine Amidbindung bildenden Seitenketten-Substituenten
des anderen flankierenden Rests, wodurch das Peptid zyklisiert wird.
Die orthogonalen Entschützungsreaktionen,
nichtorthogonalen Entschützungsreaktionen
und Amidbindung-Bildungs-reaktionen können wie im obigen Abschnitt
(B)(II)(1)(a) beschrieben durchgeführt werden.
-
Bei
der Umsetzung der Verfahren der Erfindung, die im Allgemeinen wie
oben als Verfahren (2) und (3) beschrieben sind, ist es wünschenswert,
Syntheseschemata einzusetzen, welche die Vorteile eines orthogonalen
Schutzes und einer orthogona len Entschützung von funktionellen Gruppen
nutzen, um die Bildung von unerwünschten
Derivaten zu vermeiden. Für
Fachleute auf dem Gebiet der Erfindung ist aus den folgenden repräsentativen
Syntheseschemata ersichtlich, dass die Schutzgruppen für die eine
Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste, das eingesetzte Peptidsyntheseverfahren und die Abfolge der
Peptidzyklisierungsreaktionen so gewählt werden können, dass
jede dieser Komponenten des Syntheseschemas die Spezifität der Reaktionen
erhöht
und die Ausbeute des gewünschten
Produkts verbessert.
-
(iii) Zyklisierung unter
Einsatz von Diaminlinkern
-
In
einem Beispiel, bei dem Carboxysubstituenten für die eine Amidbindung bildenden
Seitenketten-Substituenten beider flankierender Reste, ein Diaminlinker
zur Zyklisierung und Fmoc-Chemie zur Peptidsynthese verwendet werden,
werden die Carboxysubstituenten orthogonal in Bezug aufeinander
und in Bezug auf die Fmoc-geschützte α-Aminogruppe
des N-terminalen Rests in der Peptidkette geschützt, indem eine Allylgruppe
zum Schutz des Carboxysubstituenten des einen flankierenden Rests
und ein t-Butylester zum Schutz des Carboxysubstituenten des anderen
flankierenden Rests eingesetzt werden. In diesem Beispiel kann das
Peptid zyklisiert werden, indem (1) eine Reduktion zur Entschützung des
Allyl-geschützten
Carboxysubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der t-Butylestergeschützte
Carboxysubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder einfach geschützter
(z.B. mit Allyloxycarbonyl oder Boc einfach geschützter) Diaminlinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Aminogruppen des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) Azidolyse zur Entschützung des t-Butylester-geschützten Carboxysubstituenten
des anderen flankierenden Rests und zur Entschützung der Boc-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn ein mit Boc einfach geschützter Diaminlinker
als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyloxycarbonyl-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn ein mit Allyloxycarbonyl
einfach geschützter
Diaminlinker als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankieren den Rests mit der freien Aminogruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) Azidolyse zur
Entschützung
des t-Butyiester-geschützten
Carboxysubstituenten des einen flankierenden Rests eingesetzt wird
(ohne dass der Allyl-geschützte
Carboxysubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder einfach geschützter
(z.B. mit Allyloxycarbonyl oder Boc einfach geschützter) Diaminlinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Aminogruppen des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) eine Reduktion zur Entschützung des Allyl-geschützten Carboxysubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyloxycarbonyl-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn ein mit Allyloxycarbonyl einfach
geschützter
Diaminlinker als Linker verwendet wird; (4) Azidolyse zur Entschützung der
Boc-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn ein mit Boc einfach
geschützter
Diaminlinker als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
In
einem Beispiel, bei dem Carboxysubstituenten für die eine Amidbindung bildenden
Seitenketten-Substituenten beider flankierender Reste, ein Diaminlinker
zur Zyklisierung und Boc-Chemie für die Peptidsynthese verwendet
werden, werden die Carboxysubstituenten in Bezug aufeinander und
in Bezug auf die Boc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette orthogonal geschützt, indem eine Allylgruppe
zum Schutz des Carboxysubstituenten eines flankierenden Rests und
ein Fluorenylmethyl-(Fm-) Ester zum Schutz des Carboxysubstituenten
des anderen flankierenden Rests eingesetzt werden. In diesem Beispiel
kann das Peptid zyklisiert werden, indem (1) eine Reduktion zur
Entschützung
des Allyl-geschützten Carboxysubstituenten
eines flankierenden Rests eingesetzt wird (ohne dass der Fm-geschützte Carboxysubstituent
des anderen flankierenden Rests entschützt wird); (2) ein ungeschützter oder
einfach geschützter
(z.B. mit Allyloxycarbonyl oder Fmoc ein fach geschützter) Diaminlinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Aminogruppen des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) basische Verseifung zur Entschützung des Fm-Ester-geschützten Carboxysubstituenten des
anderen flankierenden Rests und zur Entschützung der Fmoc-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn ein mit Fmoc einfach geschützter Diaminlinker
als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyloxycarbonyl-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn ein mit Allyloxycarbonyl
einfach geschützter
Diaminlinker als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) basische Verseifung
zur Entschützung des
Fm-Ester-geschützten
Carboxysubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Allyl-geschützte
Carboxysubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter oder
einfach geschützter
(z.B. mit Allyloxycarbonyl oder Fmoc einfach geschützter) Diaminlinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Aminogruppen des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) eine Reduktion zur Entschützung des Allyl-geschützten Carboxysubstituenien
des anderen flankierenden Rests und zur Entschützung der Allyloxycarbonyl-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn ein mit Allyloxycarbonyl einfach
geschützter
Diaminlinker als Linker verwendet wird; (4) basische Verseifung
zur Entschützung
der Fmoc-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn ein mit Fmoc einfach
geschützter
Diaminlinker als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
(iv) Zyklisierung unter
Einsatz von Dicarbonsäurelinkern
-
In
einem Beispiel, bei dem Aminosubstituenten für die eine Amidbindung bildenden
Seitenketten-Substituenten beider flankierender Reste, ein Dicarbonsäurelinker
zur Zyklisierung und Fmoc-Chemie zur Peptidsynthese verwendet werden,
werden die Aminosubstituenten orthogonal in Bezug aufeinander und
in Bezug auf die Fmoc-geschützte α-Aminogruppe
des N-terminalen Rests in der Peptidkette geschützt, indem eine Allyloxycarbonylgruppe
zum Schutz des Aminosubstituenten des einen flankierenden Rests
und eine Boc-Gruppe zum Schutz des Aminosubstituenten des anderen
flankierenden Rests eingesetzt werden. In diesem Beispiel kann das
Peptid zyklisiert werden, indem (1) eine Reduktion zur Entschützung des
Allyloxycarbonylgeschützten
Aminosubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Boc-geschützte
Aminosubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder einfach geschützter
(z.B. mit Allyl- oder t-Butylester einfach geschützter) Dicarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Carboxygruppen des Linkers und dem entschützten Aminosubstituenten
zu bilden; (3) Azidolyse zur Entschützung des Boc-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der t-Butylester-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn ein mit t-Butylester einfach geschützter Dicarbonsäurelinker
als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyl-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn ein mit Allyl einfach geschützter Dicarbonsäurelinker
als Linker verwendet wird; und (5) der freie Aminosubstituent des
anderen flankierenden Rests mit der freien Carboxygruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) Azidolyse zur
Entschützung
des Boc-geschützten
Aminosubstituenten des einen flankierenden Rests eingesetzt wird
(ohne dass der Allyloxycarbonyl-geschützte Aminosubstituent des anderen
flankierenden Rests entschützt
wird); (2) ein ungeschützter
oder einfach geschützter
(z.B. mit Allyl- oder t-Butylester einfach geschützter) Dicarbonsäurelinker mit
dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Carboxygruppen des Linkers und dem entschützten Aminosubstituenten
zu bilden; (3) eine Reduktion zur Entschützung des Allyloxycarbonyl-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyl-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn ein mit Allyl einfach geschützter Dicarbonsäurelinker
als Linker verwendet wird; (4) Azidolyse zur Entschützung der
t-Butylester-geschützten
Carboxygruppe des Linkers eingesetzt wird, wenn ein mit t-Butylester
einfach geschützter
Dicarbonsäurelinker
als Linker verwendet wird; und (5) der freie Aminosubstituent des
anderen flankierenden Rests mit der freien Carboxygruppe des Linkers intramolekular
umgesetzt wird, um eine Amidbindung zu bilden, die das Peptid zyklisiert.
-
In
einem Beispiel, bei dem Aminosubstituenten für die eine Amidbindung bildenden
Seitenketten-Substituenten beider flankierender Reste, ein Dicarbonsäurelinker
zur Zyklisierung und Boc-Chemie für die Peptidsynthese verwendet
werden, werden die Aminosubstituenten in Bezug aufeinander und in
Bezug auf die Boc-geschützte α-Aminogruppe
des N-terminalen Rests in der Peptidkette orthogonal geschützt, indem
eine Allyloxycarbonylgruppe zum Schutz des Aminosubstituenten eines
flankierenden Rests und eine Fmoc-Gruppe zum Schutz des Aminosubstituenten
des anderen flankierenden Rests eingesetzt werden. In diesem Beispiel
kann das Peptid zyklisiert werden, indem (1) eine Reduktion zur
Entschützung
des Aliyloxycarbonyl-geschützten Aminosubstituenten
eines flankierenden Rests eingesetzt wird (ohne dass der Fmoc-geschützte Aminosubstituent
des anderen flankierenden Rests entschützt wird); (2) ein ungeschützter oder
einfach geschützter
(z.B. mit Allyl- oder Fm-Ester
einfach geschützter)
Dicarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Carboxygruppen des Linkers und dem entschützten Aminosubstituenten
zu bilden; (3) basische Verseifung zur Entschützung des Fmoc-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der Fm-Ester-geschützten Carboxygruppe des
Linkers eingesetzt wird, wenn ein mit Fm-Ester einfach geschützter Dicarbonsäurelinker
als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyl-geschützten
Carboxygruppe des Linkers eingesetzt wird, wenn ein mit Allyl einfach
geschützter Dicarbonsäurelinker
als Linker verwendet wird; und (5) der freie Aminosubstituent des
anderen flankierenden Rests mit der freien Carboxygruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) basische Verseifung
zur Entschützung des
Fmac-geschützten
Aminosubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Allyloxycarbonyl-geschützte
Aminosubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder einfach geschützter
(z.B. mit Allyl- oder Fm-Ester einfach geschützter) Dicarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
einer der Carboxygruppen des Linkers und dem entschützten Aminosubstituenten
zu bilden; (3) eine Reduktion zur Entschützung des Allyloxycarbonyl-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyl-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn ein mit Allyl einfach geschützter Dicarbonsäurelinker
als Linker verwendet wird; (4) basische Verseifung zur Entschützung der
Fm-Ester-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn ein mit Fmoc einfach geschützter Dicarbonsäurelinker
als Linker verwendet wird; und (5) der freie Aminosubstituent des
anderen flankierenden Rests mit der freien Carboxygruppe des Linkers
intramolekular umgesetzt wird, um eine Amidbindung zu bilden, die
das Peptid zyklisiert.
-
(v) Zyklisierung unter
Einsatz von Aminocarbonsäurelinkern
-
In
einem Beispiel, bei dem ein Aminosubstituent für den eine Amidbindung bildenden
Seitenketten-Substituenten eines flankierenden Rests, ein Garboxysubstituent
für den
eine Amidbindung bildenden Seitenketten-Substituenten des anderen
flankierenden Rests, ein Aminocarbonsäurelinker zur Zyklisierung
und Fmoc-Chemie zur Peptidsynthese verwendet werden, werden die
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste orthogonal in Bezug aufeinander und in Bezug auf die Fmoc-geschützte α-Aminogruppe
des N-terminalen Rests in der Peptidkette geschützt, indem eine Allyloxycarbonylgruppe
zum Schutz des Aminosubstituenten des einen flankierenden Rests
und ein t-Butylester zum Schutz des Carboxysubstituenten des anderen
flankierenden Rests eingesetzt werden. In diesem Beispiel kann das Peptid
zyklisiert werden, indem (1) eine Reduktion zur Entschützung des
Allyloxycarbonyl-geschützten
Aminosubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der t-Butylester-geschützte
Carboxysubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder aminoge-schützter (z.B. mit
Allyloxycarbonyl aminogeschützter
oder mit Boc aminogeschützter)
Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Carboxygruppe des Linkers und dem entschützten Aminosubstituenten zu
bilden; (3) Azidolyse zur Entschützung
des t-Butylester-geschützten
Carboxysubstituenten des anderen flankierenden Rests und zur Entschützung der
Boc-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Boc-geschützten
Aminogruppe als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyloxycarbonyl-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn eine Allyloxycarbonyl-geschützte Aminogruppe
als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) Azidolyse zur
Entschützung
des t-Butylester-geschützten
Carboxysubstituenten eines flankierenden Rests eingesetzt wird (ahne
dass der Allyloxycarbonyl-geschützte
Aminosubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder carboxygeschützter
(z.B. mit Allyl- oder t-Butylester carboxygeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Aminogruppe des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) eine Reduktion zur Entschützung des Allyloxycarbonyl-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyl-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyl-geschützten
Carboxygruppe als Linker verwendet wird; (4) Azidolyse zur Entschützung der
t-Butylester-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer t-Butylester-geschützten Carboxygruppe
als Linker verwendet wird; und (5) der freie Aminosubstituent des
anderen flankierenden Rests mit der freien Carboxygruppe des Linkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
In
einem anderen Beispiel, bei dem ein Aminosubstituent für den eine
Amidbindung bildenden Seitenketten-Substituenten eines flankierenden
Rests, ein Carboxysubstituent für
den eine Amidbindung bildenden Seitenketten-Substituenten des anderen
flankierenden Rests, ein Aminocarbonsäurelinker zur Zyklisierung und
Fmoc-Chemie für
die Peptidsynthese verwendet werden, werden die eine Amidbindung
bildenden Seitenketten-Substituenten der flankierenden Reste in
Bezug aufeinander und in Bezug auf die Fmoc-geschützte α-Aminogruppe
des N-terminalen Rests in der Peptidkette orthagonal geschützt, indem
eine Boc-Gruppe zum Schutz des Aminosubstituenten eines flankierenden
Rests und eine Allylgruppe zum Schutz des Carboxysubstituenten des
anderen flankierenden Rests eingesetzt werden. In diesem Beispiel
kann das Peptid zyklisiert werden; indem (1) Azidolyse zur Entschützung des
Boc-geschützten
Aminosubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Allyl-geschützte
Carboxysubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder aminogeschützter
(z.B. mit Allyloxycarbonyl aminogeschützter oder mit Boc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Carboxygruppe des Linkers und dem entschützten Aminosubstituenten zu
bilden; (3) eine Reduktion zur Entschützung des Allyl-geschützten Carboxysubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyloxycarbonyl-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyloxycarbonyl-geschützten
Aminogruppe als Linker verwendet wird; (4) Azidolyse zur Entschützung der
Boc-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Boc-geschützten
Aminogruppe als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) Azidolyse zur
Entschützung
des Boc-geschützten
Aminosubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Allyl-geschützte
Carboxysubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder aminogeschützter
(z.B. mit Allyloxycarbonyl aminogeschützter oder mit Boc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Carboxygruppe des Linkers und dem entschützten Aminosubstituenten zu
bilden; (3) eine Reduktion zur Entschützung des Allyl-geschützten Carboxysubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyloxycarbonyl-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyloxycarbonyl-geschützten
Aminogruppe als Linker verwendet wird; (4) Azidolyse zur Entschützung der
Boc-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Boc-geschützten
Gruppe als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
In
einem Beispiel, bei dem ein Aminosubstituent für den eine Amidbindung bildenden
Seitenketten-Substituenten eines flankierenden Rests, ein Carboxysubstituent
für den
eine Amidbindung bildenden Seitenketten-Substituenten des anderen
flankierenden Rests, ein Aminocarbonsäurelinker zur Zyklisierung
und Boc-Chemie für
die Peptidsynthese verwendet werden, werden die eine Amidbindung
bildenden Seitenketten-Substituenten der flankierenden Reste in
Bezug aufeinander und in Bezug auf die Boc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette orthogonal geschützt, indem eine Allyloxycarbonylgruppe zum
Schutz des Aminosubstituenten eines flankierenden Rests und ein
Fm-Ester zum Schutz des Carboxysubstituenten des anderen flankierenden
Rests eingesetzt werden. In diesem Beispiel kann das Peptid zyklisiert
werden, indem (1) eine Reduktion zur Entschützung des Allyloxycarbonyl-geschützten Aminosubstituenten
eines flankierenden Rests eingesetzt wird (ohne dass der Fm-Ester-geschützte Carboxysubstituent
des anderen flankierenden Rests entschützt wird); (2) ein ungeschützter oder
aminogeschützter
(z.B. mit Allyloxycarbonyl aminogeschützter oder mit Boc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Carboxygruppe des Linkers und dem entschützten Aminosubstituenten zu
bilden; (3) basische Verseifung zur Entschützung des Fm-Ester-geschützten Carboxysubstituenten
des anderen flankierenden Rests und zur Entschützung der Fmoc-geschützten Aminogruppe des
Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit einer Fmoc-geschützten Aminogruppe
als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyloxycarbonyl-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyfoxycarbonyl-geschützten Aminogruppe als
Linker verwendet wird; und (5) der freie Carboxysubstituent des
anderen flankierenden Rests mit der freien Aminogruppe des Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) basische Verseifung
zur Entschützung des
Fm-Ester-geschützten
Carboxysubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Allyloxycarbonyl-geschützte
Aminosubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder carboxygeschützter
(z.B. mit Allyl- oder Fm-Ester carboxygeschützter) Aminocarbonsäurelinker mit
dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Aminogruppe des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) eine Reduktion zur Entschützung des Allyl-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyl-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyl-geschützten Carboxygruppe als
Linker verwendet wird; (4) basische Verseifung zur Entschützung der
Fm-Ester-geschützten
Carboxygruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Fm-Ester-geschützten
Carboxygruppe als Linker verwendet wird; und (5) der freie Aminosubstituent
des anderen flankierenden Rests mit der freien Carboxygruppe des
Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
In
einem weiteren Beispiel, bei dem ein Aminosubstituent für den eine
Amidbindung bildenden Seitenketten-Substituenten eines flankierenden
Rests, ein Carboxysubstituent für
den eine Amidbindung bildenden Seitenketten-Substituenten des anderen
flankierenden Rests, ein Aminocarbonsäurelinker zur Zyklisierung und
Boc-Chemie für
die Peptidsynthese verwendet werden, werden die eine Amidbindung
bildenden Seitenketten-Substituenten der flankierenden Reste in
Bezug aufeinander und in Bezug auf die Boc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette orthogonal geschützt, indem eine Fmoc-Gruppe zum
Schutz des Aminosubstituenten eines flankierenden Rests und eine
Allylgruppe zum Schutz des Carboxysubstituenten des anderen flankierenden
Rests eingesetzt werden. In diesem Beispiel kann das Peptid zyklisiert
werden, indem (1) basische Verseifung zur Entschützung des Fmoc-geschützten Aminosubstituenten eines
flankierenden Rests eingesetzt wird (ohne dass der Allyl-geschützte Carboxysubstituent
des anderen flankierenden Rests entschützt wird); (2) ein ungeschützter oder
aminogeschützter
(z.B. mit Allyloxycarbonyl aminogeschützter oder mit Fmoc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Carboxygruppe des Linkers und dem entschützten Aminosubstituenten zu
bilden; (3) eine Reduktion zur Entschützung des Allyl-geschützten Carboxysubstituenten
des anderen flankierenden Rests und zur Entschützung der Allyloxycarbonyl-geschützten Aminogruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyloxycarbonyl-geschützten
Aminogruppe als Linker verwendet wird; (4) basische Verseifung zur
Entschützung
der Fmoc-geschützten
Aminogruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Fmoc-geschützten
Aminogruppe als Linker verwendet wird; und (5) der freie Carboxysubstituent
des anderen flankierenden Rests mit der freien Aminogruppe des Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
Alternativ
dazu kann das Peptid zyklisiert werden, indem (1) eine Reduktion
zur Entschützung
des Allyl-geschützten
Carboxysubstituenten eines flankierenden Rests eingesetzt wird (ohne
dass der Fmoc-geschützte
Aminosubstituent des anderen flankierenden Rests entschützt wird);
(2) ein ungeschützter
oder carboxygeschützter
(z.B. mit Allyl- oder Fm-Ester carboxygeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der Aminogruppe des Linkers und dem entschützten Carboxysubstituenten
zu bilden; (3) basische Verseifung zur Entschützung des Fmoc-geschützten Aminosubstituenten
des anderen flankierenden Rests und zur Entschützung der Fm-Ester-geschützten Carboxygruppe
des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Fm- Ester-geschützten Carboxygruppe
als Linker verwendet wird; (4) eine Reduktion zur Entschützung der
Allyl-geschützten
Carboxygruppe des Linkers eingesetzt wird, wenn eine Aminocarbonsäure mit
einer Allyl-geschützten
Carboxygruppe als Linker verwendet wird; und (5) der freie Aminosubstituent
des anderen flankierenden Rests mit der freien Carboxygruppe des
Aminocarbonsäurelinkers
intramolekular umgesetzt wird, um das Peptid zu zyklisieren.
-
In
einem weiteren Beispiel, bei dem ein Aminosubstituent für den eine
Amidbindung bildenden Seitenketten-Substituenten eines flankierenden
Rests, ein Carboxysubstituent für
den eine Amidbindung bildenden Seitenketten-Substituenten des anderen
flankierenden Rests, ein Aminocarbonsäurelinker zur Zyklisierung und
Fmoc-Chemie für
die Peptidsynthese verwendet werden, wird für Regioselektivität des Zyklisierungsvorgangs
gesorgt, indem die eine Amidbindung bildenden Seitenketten-Substituenten der
flankierenden Reste in Bezug auf die Fmoc-geschützte α-Aminogruppe des N-terminalen Rests in
der Peptidkette orthogonal geschützt
werden, nicht aber in Bezug aufeinander, und indem eine der funktionellen
Gruppen des Aminocarbonsäurelinkers
in Bezug auf die Fmoc-geschützte α-Aminogruppe
des N-terminalen
Rests in der Peptidkette orthogonal geschützt wird.
-
In
einem Beispiel der oben genannten Ausführungsform, bei dem ein Allyoxycarbonyl-geschützter Aminosubstituent
als eine Amidbindung bildender Seitenketten-Substituent eines flankierenden Rests,
ein Allyl-geschützter
Carboxysubstituent als eine Amidbindung bildender Seitenketten-Substituent
des anderen flankierenden Rests, ein einfach geschützter Aminocarbonsäurelinker
und Fmoc-Chemie für
die Peptidsynthese verwendet werden, kann das Peptid zyklisiert
werden, indem (1) eine Reduktion zur orthogonalen Entschützung der
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste eingesetzt wird (ohne dass die Fmoc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette entschützt
wird); (2) ein carboxygeschützter
(z.B. mit Allyl- oder t-Butylester carboxygeschützter) oder aminogeschützter (z.B.
mit Allyloxycarbonyl oder Boc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbin dung zwischen
der ungeschützten
funktionellen Gruppe des Linkers und dem entsprechenden, eine Amidbindung
bildenden Seitenketten-Substituenten auf einem der flankierenden
Reste zu bilden; (3) eine Reduktion oder Azidolyse, je nach Fall,
zur Entschützung
der geschützten
funktionellen Gruppe des Linkers eingesetzt wird; und (4) der freie,
eine Amidbindung bildende Seitenketten-Substituent des anderen flankierenden
Rests mit der freien funktionellen Gruppe des Linkers umgesetzt
wird, um das Peptid zu zyklisieren.
-
In
einem Beispiel der oben genannten Ausführungsform, bei dem ein Boc-geschützter Aminosubstituent
als eine Amidbindung bildender Seitenketten-Substituent eines flankierenden Rests,
ein t-Butylester-geschützter
Carboxysubstituent als eine Amidbindung bildender Seitenketten-Substituent
des anderen flankierenden Rests, ein einfach geschützter Aminocarbonsäurelinker
und Fmoc-Chemie für
die Peptidsynthese verwendet werden, kann das Peptid zyklisiert
werden, indem (1) Azidolyse zur orthogonalen Entschützung der eine
Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste eingesetzt wird (ohne dass die Fmoc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette entschützt
wird); (2) ein carboxygeschützter
(z.B. mit Allyl- oder t-Butylester carboxygeschützter) oder aminogeschützter (z.B.
mit Allyloxycarbonyl oder Boc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der ungeschützten
funktionellen Gruppe des Linkers und dem entsprechenden, eine Amidbindung
bildenden Seitenketten-Substituenten
auf einem der flankierenden Reste zu bilden; (3) eine Reduktion
oder Azidolyse, je nach Fall, zur Entschützung der geschützten funktionellen
Gruppe des Linkers eingesetzt wird; und (4) der freie, eine Amidbindung
bildende Seitenketten-Substituent
des anderen flankierenden Rests mit der freien funktionellen Gruppe
des Linkers umgesetzt wird, um das Peptid zu zyklisieren.
-
In
einer weiteren Ausführungsform,
bei der ein Aminosubstituent für
den eine Amidbindung bildenden Seitenketten-Substituenten eines
flankierenden Rests, ein Carboxysubstituent für den eine Amidbindung bildenden
Seitenketten-Substituenten des anderen flankierenden Rests, ein
Aminocarbonsäurelinker
zur Zyklisierung und Boc- Chemie
für die
Peptidsynthese verwendet werden, wird für Regioselektivität des Zyklisierungsvorgangs
gesorgt, indem die eine Amidbindung bildenden Seitenketten-Substituenten der
flankierenden Reste in Bezug auf die Boc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette orthogonal geschützt werden, nicht aber in Bezug
aufeinander, und indem eine der funktionellen Gruppen des Aminocarbonsäurelinkers
in Bezug auf die Boc-geschützte α-Aminogruppe
des N-terminalen
Rests in der Peptidkette orthogonal geschützt wird.
-
In
einem Beispiel für
die oben genannte Ausführungsform,
bei dem ein Allyloxycarbonyl-geschützter Aminosubstituent als
eine Amidbindung bildender Seitenketten-Substituent eines flankierenden Rests,
ein Allyl-geschützter
Carboxysubstituent als eine Amidbindung bildender Seitenketten-Substituent
des anderen flankierenden Rests, ein Aminocarbonsäurelinker
und Boc-Chemie für
die Peptidsynthese verwendet werden, kann das Peptid zyklisiert
werden, indem (1) eine Reduktion zur orthogonalen Entschützung der
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden
Reste eingesetzt wird (ohne dass die Boc-geschützte α-Aminogruppe des N-terminalen Rests in
der Peptidkette entschützt
wird); (2) ein carboxygeschützter
(z.B. mit Allyl- oder Fm-Ester carboxygeschützter) oder aminogeschützter (z.B.
mit Allyloxycarbonyl oder Fmoc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der ungeschützten
funktionellen Gruppe des Linkers und dem entsprechenden, eine Amidbindung
bildenden Seitenketten-Substituenten auf einem der flankierenden
Reste zu bilden; (3) eine Reduktion oder basische Verseifung, je
nach Fall, zur Entschützung
der geschützten
funktionellen Gruppe des Linkers eingesetzt wird; und (4) der freie,
eine Amidbindung bildende Seitenketten-Substituent des anderen flankierenden
Rests mit der freien funktionellen Gruppe des Linkers umgesetzt
wird, um das Peptid zu zyklisieren.
-
In
einem Beispiel für
die oben genannte Ausführungsform,
bei dem ein Fmoc-geschützter Aminosubstituent
als eine Amidbindung bildender Seitenketten-Substituent eines flankierenden Rests,
ein Fm-Ester-geschützter
Carboxysubstituent als eine Amidbindung bildender Seitenketten-Substituent
des anderen flankierenden Rests, ein Aminocarbonsäurelinker
und Boc-Chermie für
die Peptidsynthese verwendet werden, kann das Peptid zyklisiert
werden, indem (1) basische Verseifung zur orthogonalen Entschützung der
eine Amidbindung bildenden Seitenketten-Substituenten der flankierenden Reste
eingesetzt wird (ohne dass die Boc-geschützte α-Aminogruppe des N-terminalen
Rests in der Peptidkette entschützt
wird); (2) ein carboxygeschützter
(z.B. mit Allyl- oder Fm-Ester carboxygeschützter) oder aminogeschützter (z.B.
mit Allyloxycarbonyl oder Fmoc aminogeschützter) Aminocarbonsäurelinker
mit dem Festphasenpeptid umgesetzt wird, um eine Amidbindung zwischen
der ungeschützten
funktionellen Gruppe des Linkers und dem entsprechenden, eine Amidbindung
bildenden Seitenketten-Substituenten auf einem der flankierenden
Reste zu bilden; (3) eine Reduktion oder basische Verseifung, je
nach Fall, zur Entschützung
der geschützten
funktionellen Gruppe des Linkers eingesetzt wird; und (4) der freie,
eine Amidbindung bildende Seitenketten-Substituent des anderen flankierenden
Rests mit der freien funktionellen Gruppe des Linkers umgesetzt
wird, um das Peptid zu zyklisieren.
-
Nach
der Zyklisierung wird das eingeschränkte Helixpeptid gegebenenfalls
vom festen Träger
abgespalten, gewonnen und gereinigt. Das Peptid kann mithilfe eines
Reagens vom festen Träger
abgetrennt werden, das in der Lage ist, die Bindung zwischen Peptid
und fester Phase aufzubrechen und gegebenenfalls blockierte funktionelle
Seitenketten-Gruppen auf dem Peptid zu entschützen. In einer Ausführungsform
wird das Peptid durch Azidolyse mit flüssigem Fluorwasserstoff (Flusssäure, HF)
vorn festen Träger
abgespalten, das auch jegliche verbleibende Seitenketten-Schutzgruppen enlernt.
Um eine Alkylierung der Reste im Peptid (beispielsweise eine Alkylierung
von Methionin-, Cystein- und Tyrosinresten) zu verhindern, enthält das Azidolyse-Reaktionsgemisch
vorzugsweise Thiokresol- und Kresol als Fänger. Nach der HF-Abspaltung
wird das Harz mit Ether gewaschen, und das freie Peptid wird aus
dem Harz extrahiert, gefolgt von Waschvorgängen mit Essigsäurelösungen.
Die vereinigten Waschlösungen
werden gefriergetrocknet, und der Rückstand wird gereinigt.
-
c. Flüssigphasenzyklisierung
-
Alternativ
dazu kann das Peptid vor dem Zyklisierungsschritt vom festen Träger abgespalten
werden. In einer Ausführungsform
wird, nachdem der difunktionelle Linker an den eine Amidbindung
bildenden Seitenketten-Substituenten des ersten flankierenden Rests
im Peptid gebunden wurde, das Peptid vom festen Träger abgespalten.
Das Peptid wird gewonnen, an dem eine Amidbindung bildenden Seitenketten-Substituenten des zweiten
flankierenden Rests entblockiert (falls erforderlich) und dann in
niedrigen Konzentrationen in einem Reaktionsgemisch zyklisiert,
um intramolekulare Amidbindungsbildung zu maximieren. Typischerweise
kann unter Bedingungen, unter denen die Konzentration des Peptids
eine intramolekulare Konzentration an freien Carboxy- und Aminogruppen
bereitstellt, die höher
ist als die intermolekulare Konzentration an freien Carboxy- und
Aminogruppen im Reaktionsgemisch, der höchste Grad an intramolekularer
Amidbindungsbildung erreicht werden. In einer Ausführungsform
wird eine Peptidkonzentration von 1 nM bis 1 M, vorzugsweise 1 μM bis 1 mM,
noch bevorzugter 1 μM
bis 100 μM,
eingesetzt, um die Zyklisierung zu maximieren. Die Zyklisierung
von freiem Peptid kann mithilfe jeder beliebigen Aminosäure-Kupplungsreaktionen
durchgeführt
werden, die zur Helikalisierung eines an einen festen Träger gebundenen
Peptids wie oben beschrieben eingesetzt wird.
-
d. Syntheseschemata
-
In
einer Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (1), (1a), (1b), (1c), (1d), (1e),
(1f) und (1g) hergestellt, indem (in der Peptidsynthese, wie sie
in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die jeweilige in
der obigen Tabelle 1 angeführte
Kombination aus flankierenden Resten und Diaminlinker verwendet
wird, welche die Werte für
n, m und p bereitstellt, welche die Verbindung von Interesse charakterisieren,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iii) beschriebenen Verfahren
zyklisiert wird. Beispielsweise können alle Verbindungen der
Formeln (1), (1a), (1b), (1c), (1d), (1e), (1f) und (1g), die durch
m = 0, p = 0 und n = 7, 8 oder 9 charakterisiert sind, hergestellt
werden, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Diaminlinker verwendet werden, die in Tabelle 1 unter Nr. 1 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iii) beschriebenen Verfahren
zyklisiert wird. In einem weiteren Beispiel können alle Verbindungen der
Formeln (1), (1 a), (1b), (1c), (1d), (1e), (1f) und (1g), die durch
m = 0, p = 6 und n = 2 oder 3 charakterisiert sind, hergestellt
werden, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Diaminlinker verwendet werden, die in Tabelle 1 unter Nr. 7 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iii) beschriebenen Verfahren
zyklisiert wird. In einem weiteren Beispiel können alle Verbindungen der
Formeln (1), (1 a), (1b), (1c), (1 d), (1e), (1f) und (1g), die
durch m = 3, p = 3 und n = 2 oder 3 charakterisiert sind, hergestellt
werden, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Diaminlinker verwendet werden, die in Tabelle 1 unter Nr. 16 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iii) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (2), (2a), (2b), (2c), (2d), (2e),
(2f), (2g), (3), (3a), (3b), (3c), (3d), (3e), (3f) und (3g) hergestellt,
indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger Diaminlinker
verwendet werden, die in Tabelle 1 unter Nr. 9 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iii) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (4), (4a), (4b), (4c), (4d), (4e),
(4f) und (4g) hergestellt, indem (in der Peptidsynthese, wie sie
in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Diaminlinker verwendet werden, die in Tabelle
1 unter Nr. 13 angeführt
sind, und das resultierende Peptid gemäß den im obigen Abschnitt (B)(II)(1)(b)(ii)
oder (iii) beschriebenen Verfahren zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (5), (5a), (5b), (5c), (5d), (5e),
(5f) und (5g) hergestellt, indem (in der Peptidsynthese, wie sie
in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Diaminlinker verwendet werden, die in Tabelle
1 unter Nr. 8 angeführt
sind, und das resultierende Peptid gemäß den im obigen Abschnitt (B)(II)(1)(b)(ii)
oder (iii) beschriebenen Verfahren zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (6), (6a), (6b), (6c), (6d), (6e),
(6f), (Tg), (11), (11a), (11b), (11c), (11d), (11e), (11f) und (11g)
hergestellt, indem (in der Peptidsynthese, wie sie in Abschnitt
(B)(II)(1)(a) oben beschrieben ist) die jeweilige in der obigen
Tabelle 3 angeführte
Kombination aus flankierenden Resten und Aminocarbonsäurelinker
verwendet wird, welche die Werte für q, r und s bereitstellt,
welche die Verbindung von Interesse charakterisieren, oder die Werte
für t,
u und v, welche die Verbindung von Interesse charakterisieren, je
nach Fall, und das resultierende Peptid gemäß den im obigen Abschnitt (B)(II)(1)(b)(ii)
oder (v) beschriebenen Verfahren zyklisiert wird. Beispielsweise
können
alle Verbindungen der Formeln (6), (6a), (6b), (6c), (6d), (6e),
(6f), (6g), (11), (11a), (11b), (11c), (11d), (11e), (11f) und (11g),
die durch q = 1, s = 0 und r = 6, 7 oder 8 charakterisiert sind
oder durch t = 0, v = 1 und u = 6, 7 oder 8 charakterisiert sind,
je nach Fall, hergestellt werden, indem (in der Peptidsynthese,
wie sie in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Aminocarbonsäurelinker verwendet werden,
die in Tabelle 3 unter Nr. 1 angeführt sind, und das resultierende
Peptid gemäß den im obigen
Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren zyklisiert
wird.
-
In
einem weiteren Beispiel können
alle Verbindungen der Formeln (6), (6a), (6b), (6c), (6d), (6e),
(6f), (6g), (11), (11a), (11b), (11c), (11d), (11e), (11f) und (11g),
die durch q = 1, s = 6 und r = 1 oder 2 charakterisiert sind oder
durch t = 6, v = 1 und u = 1 oder 2 charakterisiert sind, je nach
Fall, hergestellt werden, indem (in der Peptidsynthese, wie sie
in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Aminocarbonsäurelinker verwendet werden,
die in Tabelle 3 unter Nr. 28 angeführt sind, und das resultierende
Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einem weiteren Beispiel können
alle Verbindungen der Formeln (6), (6a), (6b), (6c), (6d), (6e),
(6f), (6g), (11), (11a), (11b), (11c), (11d), (11e), (11f) und (11g),
die durch q = 7, s = 0 und r = 1 oder 2 charakterisiert sind oder
durch t = 0, v = 1 und u = 1 oder 2 charakterisiert sind, je nach
Fall, hergestellt werden, indem (in der Peptidsynthese, wie sie
in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Aminocarbonsäurelinker verwendet werden,
die in Tabelle 3 unter Nr. 7 angeführf sind, und das resultierende
Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einem weiteren Beispiel können
alle Verbindungen der Formeln (6), (6a), (6b), (6c), (6d), (6e),
(6f), (6g), (11), (11a), (11b), (11c), (11d), (11e), (11f) und (11g),
die durch q = 3, s = 4 und r = 1 oder 2 charakterisiert sind oder
durch t = 4, v = 3 und u = 1 oder 2 charakterisiert sind, je nach
Fall, hergestellt werden, indem (in der Peptidsynthese, wie sie
in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Aminocarbonsäurelinker verwendet werden,
die in Tabelle 3 unter Nr. 25 angeführt sind, und das resultierende
Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (7), (7a), (7b), (7c), (7d), (7e),
(7f), (7g), (13), (13a), (13b), (13c), (13d), (13e), (13f) und (13g)
hergestellt, indem (in der Peptidsynthese, wie sie in Abschnitt
(B)(II)(1)(a) oben beschrieben ist) die flankierenden Reste und
ein beliebiger Aminocarbonsäurelinker
verwendet werden, die in Tabelle 3 unter Nr. 14 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (8), (8a), (8b), (8c), (8d), (8e),
(8f), (8g), (12), (12a), (12b), (12c), (12d), (12e), (12f) und (12g)
hergestellt, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Aminocarbonsäurelinker
verwendet werden, die in Tabelle 3 unter Nr. 9 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (9), (9a), (9b), (9c), (9d), (9e),
(9f), (9g), (15), (15a), (15b), (15c), (15d), (15e), (15f) und (15g)
hergestellt, indem (in der Peptidsynthese, wie sie in Abschnitt
(B)(II)(1)(a) oben beschrieben ist) die flankierenden Reste und
ein beliebiger Aminocarbonsäurelinker
verwendet werden, die in Tabelle 3 unter Nr. 15 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Hefixverbindungen der Formeln (10), (10a), (10b), (10c), (10d),
(10e), (10f), (10g), (14), (14a), (14b), (14c), (14d), (14e), (14f)
und (14g) hergestellt, indem (in der Peptidsynthese, wie sie in
Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden Reste
und ein beliebiger Aminocarbonsäurelinker
verwendet werden, die in Tabelle 3 unter Nr. 8 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (v) beschriebenen Verfahren
zyklisiert wird.
-
In
einer Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (16), (16a), (16b), (16c), (16d),
(16e), (16f) und (16g) hergestellt, indem (in der Peptidsynthese,
wie sie in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die jeweilige
in der obigen Tabelle 2 angeführte
Kombination aus flankierenden Resten und Dicarbonsäurelinker
verwendet wird, welche die Werfe für w, x und y bereitstellt,
welche die Verbindung von Interesse charakterisieren, und das resultierende
Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren
zyklisiert wird. Beispielsweise können alle Verbindungen der Formeln
(16), (16a), (16b), (16c), (16d), (16e), (16f) und (16g), die durch
w = 1, y = 1 und x = 5, 6 oder 7 charakterisiert sind, hergestellt
werden, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Dicar bunsäurelinker
verwendet werden, die in Tabelle 2 unter Nr. 1 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren
zyklisiert wird. In einem weiteren Beispiel können alle Verbindungen der
Formeln (16), (16a), (16b), (16c), (16d), (16e), (16f) und (16g),
die durch w = 1, y = 7 und x = 0 oder 1charakterisiert sind oder
durch w = 7, y = 1 und x = 0 oder 1 charakterisiert sind, hergestellt
werden, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Dicarbonsäurelinker
verwendet werden, die in Tabelle 2 unter Nr. 7 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren
zyklisiert wird. In einem weiteren Beispiel können alle Verbindungen der
Formeln (16), (16a), (16b), (16c), (16d), (16e), (16f) und (16g),
die durch w = 4, y = 4 und x = 0 oder 1charakterisiert sind, hergestellt
werden, indem (in der Peptidsynthese, wie sie in Abschnitt (B)(II)(1)(a)
oben beschrieben ist) die flankierenden Reste und ein beliebiger
Dicarbonsäurelinker
verwendet werden, die in Tabelle 2 unter Nr. 16 angeführt sind,
und das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (17), (17a), (17b), (17c), (17d),
(17e), (17f), (17g), (18), (18a), (18b), (18c), (18d), (18e), (18f)
und (18g) hergestellt, indem (in der Peptidsynthese, wie sie in
Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden Reste
und ein beliebiger Dicarbonsäurelinker
verwendet werden, die in Tabelle 2 unter Nr. 2 angeführt sind, und
das resultierende Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren zyklisiert
wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (19), (19a), (19b), (19c), (19d),
(19e), (19f) und (19g) hergestellt, indem (in der Peptidsynthese,
wie sie in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Dicarbonsäurelinker verwendet werden,
die in Tabelle 2 unter Nr. 1 angeführt sind, und das resultierende
Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren
zyklisiert wird.
-
In
einer weiteren Ausführungsform
wird jede der eingeschränkten
Helixverbindungen der Formeln (20), (20a), (20b), (20c), (20d),
(20e), (20f) und (20g) hergestellt, indem (in der Peptidsynthese,
wie sie in Abschnitt (B)(II)(1)(a) oben beschrieben ist) die flankierenden
Reste und ein beliebiger Dicarbonsäurelinker verwendet werden,
die in Tabelle 2 unter Nr. 8 angeführt sind, und das resultierende
Peptid gemäß den im
obigen Abschnitt (B)(II)(1)(b)(ii) oder (iv) beschriebenen Verfahren
zyklisiert wird.
-
(2) Synthese eines linearen
Peptids mit an einen difunktionellen Linker gebundener flankierender
Aminosäure
-
Das
Peptid ist so aufgebaut, dass die Sequenz, die helikalisiert werden
soll, eine sechs Aminosäuren lange
Aminosäuresequenz
umfasst, die sich zwischen flankierenden Resten erstreckt, wie in
Abschnitt (B)(II)(1)(a) oben beschrieben. Das Peptid kann durch
Modifikation der in Abschnitt (B)(II)(1)(a) oben beschriebenen Festphasen-Syntheseverfahren
konstruiert werden, worin einer der flankierenden Reste vor der
Hinzufügung
zur Peptidkette an einen difunktionellen Linker gebunden wird. Dies
erlaubt es dem Linker, als Teil einer Standardaminosäure in das
Peptid inkorporiert zu werden.
-
Der
flankierende Rest kann mithilfe jedes beliebigen geeigneten Mittels
an den difunktionellen Linker gebunden werden. Typischerweise wird
der eine Amidbindung bildende Seitenketten-Substituent des flankierenden
Rests verwendet, um eine Amidbindung mit einer der funktionellen
Gruppen auf dem Linker zu bilden. In einer Ausführungsform, die zum Einsatz
gemeinsam mit Fmoc-Chemie bestimmt ist, wird der Linker-derivatisierte
flankierende Rest erzeugt, indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem t-Butylester-geschützten α-Carboxysubstituenten
und einem ungeschützten Seitenketten-Aminosubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Carboxygruppe des
Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der t-Butylester-geschützte α-Carboxysubstituent des derivatisierten
Aminosäureresis
wird dann durch Azidolyse entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform
zur Verwendung gemeinsam mit Fmoc-Chemie wird der Linker-derivatisierte
flankierende Rest erzeugt, indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten Seitenketten-Aminosubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Carboxygruppe des
Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer Ausführungsform,
die zum Einsatz gemeinsam mit Boc-Chemie bestimmt ist, wird der
Linker-derivatisierte flankierende Rest erzeugt, indem ein im Handel
erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Fm-Ester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Aminosubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Carboxygruppe des
Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Fm-Ester-geschützte α-Carboxysubstituent des derivatisierten
Aminosäurerests
wird dann durch basische Verseifung entfernt, um einen Einbau der
derivatisierten Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform
zur Verwendung gemeinsam mit Boc-Chemie wird der Linker-derivatisierte
flankierende Rest erzeugt, indem ein im Handel erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten Seitenketten-Aminosubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Carboxygruppe des
Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer Ausführungsform,
die zum Einsatz gemeinsam mit Fmoc-Chemie bestimmt ist, wird der
Linker-derivatisierte flankierende Rest erzeugt, indem ein im Handel
erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem t-Butylester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Carboxysubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Aminogruppe des Linkers
und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der t-Butylester-geschützte α-Carboxysubstituent des derivatisierten
Aminosäurerests wird
dann durch Azidolyse entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform
zur Verwendung gemeinsam mit Fmoc-Chemie wird der Linker-derivatisierte
flankierende Rest erzeugt, indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten Seitenketten-Carboxysubstituenten
bezogen wird und dann die α-Substituent-geschützte Amino säure mit
einem difunktionellen Linker mit einer freien Aminogruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Aminogruppe des Linkers
und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer Ausführungsform,
die zum Einsatz gemeinsam mit Boc-Chemie bestimmt ist, wird der
Linker-derivatisierte flankierende Rest erzeugt, indem ein im Handel
erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Fm-Ester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Carboxysubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Aminogruppe des Linkers
und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Fm-Ester-geschützte α-Carboxysubstituent des derivatisierten
Aminosäurerests
wird dann durch basische Verseifung entfernt, um einen Einbau der
derivatisierten Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform
zur Verwendung gemeinsam mit Boc-Chemie wird der Linker-derivatisierte
flankierende Rest erzeugt, indem ein im Handel erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten Seitenketten-Aminosubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Aminogruppe des Linkers
und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Amino säurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
Es
ist wünschenswert,
eine der funktionellen Gruppen auf dem difunktionellen Linker zu
schützen,
entweder bevor der Linker an den flankierenden Rest gebunden wird,
der dazu ausgewählt
wurde, den Linker zu tragen, oder nach der Anbindung, aber vor Hinzufügung des
linkergebundenen flankierenden Rests zur Peptidkette. Dies verbessert
die Ausbeute durch Vermeidung unerwünschter Reaktionen der freien
funktionellen Gruppe auf dem an einen flankierenden Rest gebundenen
Linker während
der Peptidsynthese. Die freie funktionelle Gruppe auf dem Linker
kann mit einer der in Abschnitt (B)(II)(1)(a) oben beschriebenen
Amino- oder Carboxy-Schutzgruppen blockiert werden. In einer Ausführungsform
sind die freie funktionelle Gruppe auf dem Linker und die α-Aminogruppen
orthogonal geschützt,
sodass die α-Aminogruppen
bei der Peptidsynthese entschützt
werden können,
ohne dass die freie funktionelle Gruppe auf dem Linker entschützt wird.
Es versteht sich, dass jedes der oben genannten Verfahren zur Bindung
von difunktionellen Linkern an flankierende Reste leicht modifiziert
werden kann, um einen bestimmten flankierenden Rest mit einem ausgewählten orthogonal einfach
geschützten
difunktionellen Linker zu derivatisieren.
-
In
einer Ausführungsform,
die zur Verwendung mit Fmoc-Chemie bestimmt ist, wird ein mit einem
orthogonal einfach geschützten
difunktionellen Linker derivatisierter flankierender Rest geschaffen,
indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem t-Butylester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Aminosubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe und
mit entweder einer Allyl-geschützten Carboxygruppe
oder einer Allyloxycarbonyl-geschützten Aminogruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Carboxygruppe des
Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensati onsverfahren
eingesetzt wird. Der t-Butylester-geschützte α-Carboxysubstituent des derivatisierten
Aminosäurerests
wird dann durch Azidolyse entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform
zur Verwendung gemeinsam mit Fmoc-Chemie wird ein mit einem orthogonal
einfach geschützten
difunktionellen Linker derivatisierter flankierender Rest geschaffen,
indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Aminosubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe und
mit entweder einer Boc-geschützten Aminogruppe
oder einer t-Butylester-geschützten
Carboxygruppe umgesetzt wird, um eine Amidbindung zwischen der freien
Carboxygruppe des Linkers und dem ungeschützten Seitenketten-Aminosubstituenten
der Aminosäure
zu bilden, wobei eines der im obigen Abschnitt (B)(II)(1)(a) beschriebenen
Kondensationsverfahren eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer Ausführungsform,
die zum Einsatz gemeinsam mit Boc-Chemie bestimmt ist, wird ein
mit einem orthogonal einfach geschützten difunktionellen Linker
derivatisierter flankierender Rest geschaffen, indem ein im Handel
erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Fm-Ester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Aminosubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe und
mit entweder einer Allyloxycarbonyl-geschützten Aminogruppe oder einer
Allyl-geschützten Carboxygruppe
umgesetzt wird, um eine Amidbindung zwischen der freien Carboxygruppe
des Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Fm-Ester-geschützte α-Carboxysubstituent des derivati sierten
Aminosäurerests
wird dann durch basische Verseifung entfernt, um einen Einbau der
derivatisierten Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform,
die zum Einsatz gemeinsam mit Boc-Chemie bestimmt ist, wird ein mit
einem orthogonal einfach geschützten
difunktionellen Linker derivatisierter flankierender Rest geschalten, indem
ein im Handel erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Aminosubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Carboxygruppe und
mit entweder einer Fmoc-geschützten
Aminogruppe oder einer Fm-Ester-geschützten Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Carboxygruppe des
Linkers und dem ungeschützten
Seitenketten-Aminosubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer Ausführungsform,
die zur Verwendung mit Fmoc-Chemie bestimmt ist, wird ein mit einem
orthogonal einfach geschützten
difunktionellen Linker derivatisierter flankierender Rest geschaffen,
indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem t-Butylester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Carboxysubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe und mit
entweder einer Allyloxycarbonyl-geschützten Aminogruppe oder einer
Allyl-geschützten Carboxygruppe
umgesetzt wird, um eine Amidbindung zwischen der freien Aminogruppe
des Linkers und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der t-Butylester-geschützte α-Carboxysubstituent des de rivatisierten
Aminosäurerests
wird dann durch Azidolyse entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform
zur Verwendung gemeinsam mit Fmoc-Chemie wird ein mit einem orthogonal
einfach geschützten
difunktionellen Linker derivatisierter flankierender Rest geschaffen,
indem ein im Handel erhältlicher
Aminosäurerest
mit einem Fmoc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Carboxysubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe und mit
entweder einer Boc-geschützten
Aminogruppe oder einer t-Butylester-geschützten Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Aminogruppe des Linkers
und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer Ausführungsform,
die zum Einsatz gemeinsam mit Boc-Chemie bestimmt ist, wird ein
mit einem orthogonal einfach geschützten difunktionellen Linker
derivatisierter flankierender Rest geschaffen, indem ein im Handel
erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Fm-Ester-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Carboxysubstituenten bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe und mit
entweder einer Allyloxycarbonylgeschützten Aminogruppe oder einer
Allyl-geschützten Carboxygruppe
umgesetzt wird, um eine Amidbindung zwischen der freien Aminogruppe
des Linkers und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Fm-Ester-geschützte α-Carboxysubstituent des derivatisierten Aminosäurerests
wird dann durch basische Verseifung entfernt, um einen Einbau der
derivatisierten Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einer weiteren Ausführungsform,
die zum Einsatz gemeinsam mit Boc-Chemie bestimmt ist, wird ein mit
einem orthogonal einfach geschützten
difunktionellen Linker derivatisierter flankierender Rest geschaffen, indem
ein im Handel erhältlicher
Aminosäurerest
mit einem Boc-geschützten α-Aminosubstituenten,
einem Allyl-geschützten α-Carboxysubstituenten
und einem ungeschützten
Seitenketten-Carboxysubstituenten
bezogen wird und dann die α-Substituent-geschützte Aminosäure mit
einem difunktionellen Linker mit einer freien Aminogruppe und mit
entweder einer Fmoc-geschützten
Aminogruppe oder einer Fm-Ester-geschützten Carboxygruppe umgesetzt
wird, um eine Amidbindung zwischen der freien Aminogruppe des Linkers
und dem ungeschützten
Seitenketten-Carboxysubstituenten der Aminosäure zu bilden, wobei eines
der im obigen Abschnitt (B)(II)(1)(a) beschriebenen Kondensationsverfahren
eingesetzt wird. Der Allyl-geschützte α-Carboxysubstituent
des derivatisierten Aminosäurerests
wird dann durch Reduktion entfernt, um einen Einbau der derivatisierten
Aminosäure
in die Peptidkette zu ermöglichen.
-
In
einem weiteren Aspekt können
die oben genannten Ausführungsformen,
bei denen ein mit einem difunktionellen Linker derivatisierter flankierender
Rest eingesetzt wird, modifiziert werden, indem der eine Amidbindung
bildende Seitenketten-Substituent
des nichtderivatisierten (nicht schon vorher an einen difunktionellen
Linker gebundenen) flankierenden Rests in Bezug auf die α-Amino-Schutzchemie,
die bei der Peptidsynthese eingesetzt wird, und in Bezug auf einen
oder alle der eine Amidbindung bildenden Substituenten, die in den
Seitenketten anderer Aminosäurereste
im Peptid vorkommen, orthogonal geschützt wird. In diesem Aspekt
kann der eine Amidbindung bildende Seitenketten-Substituent des
nichtderivatisierten flankierenden Rests selektiv entblockiert werden,
was ein Peptid ergibt, das durch eine Kondensationsreaktion zyklisiert
werden kann, die spezifisch und gezielt zwischen dem entschützten, eine
Amidbindung bildenden Seitenketten-Substituenten des nichtderivatisierten
flankierenden Rests und der freien funktionellen Gruppe des di funktionellen
Linkers erfolgt. Geeignete Verfahren für einen orthogonalen Schutz
von eine Amidbindung bildenden Seitenketten-Substituenten sind oben
in Abschnitt (B)(II)(1)(a) beschrieben.
-
Nach
vollständiger
Festphasen-Peptidsynthese kann das Peptid durch eine Kupplungsreaktion
zwischen der freien funktionellen Gruppe des difunktionellen Linkers
und dem eine Amidbindung bildenden Seitenketten-Substituenten des
nichtderivatisierten flankierenden Rests wie in Abschnitt (B)(II)(1)(b)
oben beschrieben zyklisiert werden. Jegliche Blockiergruppen, die
den eine Amidbindung bildenden Seitenketten-Substituenten des nichtderivatisierten
flankierenden Rests und/oder die freie funktionelle Gruppe des difunktionellen
Linkers schützen,
werden entfernt, und die entschützten
Gruppen werden unter Einsatz eines im obigen Abschnitt (B)(II)(1)(a)
beschriebenen Kondensationsverfahrens unter Ausbildung einer Amidbindung
verbunden. Gegebenenfalls wird das resultierende zyklisierte (eingeschränkt-helikale)
Peptid vom festen Träger
abgespalten, gewonnen und gereinigt.
-
Alternativ
dazu kann das Peptid vor dem Zyklisierungsschritt vom festen Träger abgespalten
werden. In einer Ausführungsform
wird das Peptid nach der Synthese der linearen Peptidkette vom festen
Träger
abgespalten. Das Peptid wird gewonnen, am eine Amidbindung bildenden
Seitenketten-Substituenten des nichtderivatisierten flankierenden
Rests und/oder der freien funktionellen Gruppe des difunktionellen
Linkers entblockiert und dann in niedrigen Konzentrationen in einem
Reaktionsgemisch zyklisiert, um die intramolekulare Amidbindungsbildung
zu maximieren. Typischerweise kann unter Bedingungen, unter denen
die Konzentration des Peptids eine intramolekulare Konzentration
an freien, eine Amidbindung bildenden Substituenten oder Gruppen
bereitstellt, die höher
ist als die intermolekulare Konzentration an freien, eine Amidbindung
bildenden Substituenten oder Gruppen im Reaktionsgemisch, der höchste Grad
an intramolekularer Amidbindungsbildung erreicht werden. In einer
Ausführungsform
wird eine Peptidkonzentration von 1 nM bis 1 M, vorzugsweise 1 μM bis 1 mM,
noch bevorzugter 1 μM
bis 100 μM,
eingesetzt, um die Zyklisierung zu maximieren. Die Zyklisierung
eines freien Peptids kann mithilfe jeder beliebigen Kon densationsreaktion
durchgeführt
werden, die zur Helikalisierung eines Festphasenpeptids wie oben
beschrieben eingesetzt wird.
-
III. Verfahren zur Konstruktion
von halbsynthetischen arretierten Helixproteinen
-
Außerdem werden
hierin halbsynthetische Proteine bereitgestellt, die arretierte
Helixpeptide umfassen, die an eine oder mehrere größere, rekombinant
synthetisierte Proteinmoleküle
gebunden oder darin eingebaut sind. Die halbsynthetischen arretierten
Helixpeptide der Erfindung können
durch ein beliebiges herkömmliches
Verfahren hergestellt werden, einschließlich einer Ligation der arretierten
Helixpeptide, die wie im obigen Abschnitt (B)(II) beschrieben synthetisiert
wurden, an eine oder mehrere rekombinant synthetisierte Proteinsequenzen.
Proteinligasen, wie z.B. die „Subtiligasen", können beispielsweise
zur Verknüpfung
der arretierten Helixpeptide, die wie hierin beschrieben hergestellt
wurden, zu größeren, rekombinant
synthetisierten Proteinfragmenten eingesetzt werden.
-
In
einer Ausführungsform
werden die Verfahren der Erfindung modifiziert, um ein arretiertes
Helixpeptid herzustellen, das als „erstes Ligationssubstrat" in den Subtiligase-katalysierten
Peptidligationsverfahren dient, die in der Internationalen Patentanmeldung
Nr. PCT/US91/05480 (WO 92/02615, veröffentlicht am 20. Februar 1992)
beschrieben sind, oder als „Donorester", „Donorpeptid" bzw. „Pn...P4-P3-P2-P1-glc-F-Amidester" in den Subtiligase-katalysierten
Peptidligationsverfahren, die von Abrahmsen et al., Biochem. 30,
4151–4159 (1991),
Jackson et al., Science 266, 243–247 (1994) und Chang et al.,
Proc. Natl. Acad. Sci. USA 91, 12544–12548 (1994) beschrieben werden.
Das arretierte Helixpeptid kann so synthetisiert werden, dass der C-terminale
Aminosäurerest
des zyklisierten Peptids in einer Esterbindung mit der 2-Hydroxylgruppe einer 2-Hydroxycarbonsäure, wie
z.B. Glykolsäure
oder Milchsäure,
vorliegt, um eine Abgangsgruppe zu bilden, die von der jeweiligen
Subtiligase von Interesse bevorzugt wird, d.h. sodass der 2-Hydroxycarbonsäureester,
der in 2B der WO 92/02615 als X-Rest
des „ersten
Ligationssubstrats" dargestellt
ist, dem ersten Rest ähnelt, der
sich im normalen Peptidsubstrat von Subtilisin auf der N- terminalen Seite
der hydrolysierbaren Amidbindung befindet, dargestellt als Rest
P1' des „Hydrolysesubstrats" in 2B.
-
In
einer weiteren Ausführungsform
umfasst die Abgangsgruppe einen 2-Hydroxycarbonsäure- und einen anderen Aminosäurerest,
der in 2B von WO 92/02615 als R2''-Rest des ersten
Ligationssubstrats dargestellt ist, worin die Carboxygruppe des
2-Hydroxycarbonsäurerests
in einer Amidbindung mit der α-Aminogruppe
des weiteren Aminosäurerests
vorliegt. In solchen Ausführungsformen.
kann der Aminosäurerest
in der Abgangsgruppe so gewählt
werden, dass er dem zweiten Rest ähnelt, der sich auf der N-terminalen
Seite der hydrolysierbaren Amidbindung im normalen Peptidsubstrat
von Subtilisin befindet, dargestellt als P2' des „Hydrolysesubstrats" in 2 der
WO 92/02615. in einer bevorzugten Ausführungsform ist die Abgangsgruppe eine
Glykolat-Phenylalanyl-(glc-F-) Gruppierung, wie z.B. die in Beispiel
2 der WO 920/02615 beschriebene Glykolat-Phenylalanyl-Amid-(glc-F-NN2-) Gruppierung.
-
In
einem Aspekt wird die glc-F-Abgangsgruppe an ihre richtige Position
im C-Terminus des
arretierten Helixpeptids gebracht, indem ein Boc- oder Fmoc-α-Amino-geschütztes Phenylalanin
erhalten wird, das α-Amino-geschützte Phenylalanin
mit einer α-Carboxyester-
oder Amidbindung an Festphasenharz gebunden wird, die geschützte α-Aminogruppe
entschützt
wird, ein Glykolsäurerest
in Form eines t-Butylethers hinzugefügt wird, um eine Amidbindung
zwischen der Carboxygruppe der Glykolsäure und der freien α-Aminogruppe des
Festphasenphenylalanins zu bilden, die t-Butylethergruppe mit Säure aus
dem Glykolsäurerest
entfernt wird und eine Esterbindung zwischen dem freien Hydroxyl
des Glykolsäurerests
und dem α-Carboxyl
des nächsten
Aminosäurerests
in der C-terminalen Sequenz gebildet wird, die für das arretierte Helixpeptid
gewünscht
wird. Nachfolgende Aminosäuren
können
hinzugefügt
werden, und das resultierende Peptid kann gemäß einem der oben beschriebenen
Verfahren helikalisiert werden, wobei herkömmliche Boc- oder Fmoc-Chemie
für die
Peptidsynthese eingesetzt werden. In einer Ausführungsform wird eine glc-F-NH2-Abgangsgrupe im Wesentlichen wie in Beispiel
2 der WO 92/02615 beschrieben oder wie von Jackson et al., Science
266, 243–247
(1994), beschrieben in die gewünschte
Peptidkette eingebaut.
-
In
einer weiteren Ausführungsform
umfasst das „Donorpeptid" eine flexible Linkersequenz
zwischen dem C-terminalen Rest der Sequenz des arretierten Helixpeptids
und der Abgangsgruppensequenz, wie z.B. einen Di- oder Triglycinlinker,
um die Flexibilität
und Zugänglichkeit
der Abgangsgruppe des Donorpeptids für Subtiligase zu fördern.
-
Nachdem
das Donorpeptid (mit der Helix-Arretierfesssel an seiner Position)
erhalten wurde, kann eine Subtiligase verwendet werden, um ein Peptid-
oder Proteinfragment (hergestellt durch Rekombinations- oder andere
Syntheseverfahren), das in 2C der
WO 92/02615 als „zweites
Ligationssubstrat",
in 2 auf Seite 244 von Jackson et al., Science 266,
243–247
(1994) als „Akzeptorpeptid" und im Syntheseschema
auf Seite 12545 von Chang et al., Proc. Natl. Acad. Sci. USA 91,
12544–12548
(1994) als „Nucleophil" bezeichnet wird, an
den C-Terminus des Donorpeptids zu ligieren, indem die Abgangsgruppe
gemäß einem
der oberen Subtiligasekatalysierten Peptidligationsverfahren verdrängt wird.
In Ausführungsformen,
in denen Akzeptorpeptide oder -proteine aufgrund der Proteinstruktur
höherer
Ordnung einen relativ gut zugänglichen
N-Terminus aufweisen, kann die Ligationseffizienz verbessert werden,
indem der Aufbau des Akzeptorpeptids verändert wird, um eine flexible
Linkersequenz, wie z.B. eine Di- oder Triglycinsequenz, am N-Terminus
einzubauen, um die Flexibilität
und Zugänglichkeit
des Akzeptorpeptid-N-Terminus in der Peptidligationsreaktion zu
fördern.
Alternativ dazu kann die Zugänglichkeit
des Akzeptorpeptid-N-Terminus und/oder des Donorpeptid-C-Terminus
für Subtiligase
verbessert werden, indem die Ligationsreaktion unter Denaturierungsbedingungen
durchgeführt wird,
was unerwünschte
Strukturkonformationen, die von den Peptidsubstraten eingenommen
werden könnten,
eliminiert. In solchen Ausführungsformen
wird vorzugsweise eine denaturierungsstabile Subtiligase eingesetzt,
wie z.B. die von Chang et al., s.o., beschriebene „Stabiligase" (die in 4 M Guanidin-hydrochlorid
nahezu 50 % der katalytischen Aktivität beibehalten kann).
-
Es
versteht sich, dass weitere Peptide mit einer geeigneten Abgangsgruppe
am C-Terminus synthetisiert
und anschließend
an den N-Terminus des halbsynthetischen Peptids ligiert werden können, das
die arretierte Helixgruppierung enthält, indem die obigen Verfahren
wiederholt werden, bis ein komplettes Peptid mit dem gewünschten
N-Terminus erhalten wird.
-
Für den Fall,
dass das fertige, halbsynthetische arretierte Helixprotein als Folge
der eingesetzten Syntheseverfahren in denaturierter, fehlgefalteter
oder ansonsten inaktiver Form erhalten wird, kann die inaktive Spezies
durch Renaturierungsverfahren, die auf dem Gebiet der Erfindung
wohlbekannt sind, zur nativen oder aktiven Konformation rückgefaltet
werden. Typische Renaturierungsverfahren nutzen ein Chaotrop, wie
z.B. Harnstoff bei einem hohen pH oder Guanidin-hydrochlorid, um
inaktives Material aufzufalten, gefolgt von einer Verdünnung des
Denaturierungsmittels, damit eine Rückfaltung stattfinden kann,
während
die Bildung von zufälligen
Disulfidbindungen vor der Einnahme der biologisch aktiven Konformation
durch nichtkovalente intramolekulare Wechselwirkungen verhindert
wird (siehe US-Patente Nr. 4.512.922; 4.518.256; 4.511.502; und 4.511.503).
Inverse Mizellen oder Ionenaustauschchromatograpie werden verwendet,
um die Rückfaltung
von denaturierten Proteinen zu unterstützen, indem ein einzelnes Proteinmolekül in Mizellen
eingeschlossen wird oder Proteine auf einem Harz isoliert werden
und dann das Denaturierungsmittel entfernt wird (Hagen et al., Biotechnol.
Bioeng. 35, 966–975
(1990); Creighton in Protein Structure Folding and Design, S. 249–251, D.L. Oxender,
Hrsg., Alan R. Liss, Inc., New York (1985)). Außerdem kann eine kanformationsspezifische
Rückfaltung
mit Liganden und Antikörpern
gegen die native Struktur des Proteins durchgeführt werden (Cleland und Wang
in Biotechnelogy, S. 528–555,
H.-J. Rehm und G. Reed, Hrsg., VCH, New York). Da die Wahrscheinlichkeit
höher ist,
mit dem Protein in seiner nativen Konformation wechselzuwirken,
können
diese Bindungsmoleküle
verwendet werden, um die Faltungsreaktionen in Richtung eines Proteins
in nativem Zustand zu lenken. Die oben genannten Gewinnungsverfahren
werden, mit kleineren Modifikationen, als für die Gewinnung von biologisch
aktiven rekombinanten Proteinen aus Einschlusskörpern universell einsetzbar
angesehen und sind ge nauso für
die Gewinnung von biologisch aktiven Proteinen aus den halbsynthetischen
Verfahren der Erfindung einsetzbar.
-
IV. Verfahren zur Konstruktion
von an Makromoleküle
gebundenen arretierten Helixpeptiden
-
In
einer Ausführungsform
können
die eingeschränkten
helikalen Peptide der Erfindung, die an einen makromolekularen festen
Träger
gebunden sind, erhalten werden, indem die arretierten Helixpeptide
nach den im obigen Abschnitt (II) beschriebenen Festphasensyntheseverfahren
konstruiert werden und das intakte Konjugat aus festem Träger und
Peptid gewonnen wird. Alternativ dazu kann das zyklisierte Peptid
nach der Synthese von der Festphase abgespalten und dann durch ein
auf dem Gebiet der Erfindung bekanntes, geeignetes Verfahren an
das Makromolekül
der Wahl gebunden werden. Ein häufig
zur Anbindung von Peptidliganden an Polysaccharid-Matrices, z.B.
Agarose, Dextran oder Cellulose, eingesetztes Verfahren umfasst
die Aktivierung des Trägers
mit Halogencyan und die darauf folgende Bindung der primären aliphatischen
oder aromatischen Amine des Peptids an die aktivierte Matrix. Die
Inaktivierung von Polysacchariden mit Bromcyan (CNBr) bei alkalischem
pH wurde von Axen et al., Nature 214, 1302 (1967) in die Affinitätschromatographie
eingeführt. In
einem Aspekt der Erfindung wird die Aktivierung von Polysaccharidmatrices,
insbesondere Agarosematrices, nach dem Titrationsaktivierungsverfahren
durchgeführt.
Bei diesem Verfahren werden 20 g ausgiebig gewaschener, feuchter
Agarosekuchen zu 20 ml Wasser in einem 100 ml fassenden Becherglas
zugesetzt, das mit einem Thermometer für 0–100°C, einem pH-Meter und einem
25 mm langen Magnetrührstäbchen ausgestattet
ist. Die Suspension wird langsam gerührt, die Temperatur wird durch
Zusatz von zerstoßenem
Eis auf etwa 10–15°C gesenkt,
und der pH wird durch Zusatz von 1–2 Tropfen 4 N NaOH auf 10,8 ± 0,1 eingestellt. Der
Aktivierungsvorgang wird durch Zusatz des CNBr initiiert, und der
pH der Reaktion wird durch manuelle Titration mit der 4 N NaOH auf
10,8 ± 0,1
gehalten. Das CNBr (100 g/mg Feuchtgewicht Gel) kann als kristalliner
Feststoff, zerstoßener
Feststoff, wässrige
Lösung
oder durch Zusatz einer Aliquote einer Stammlösung zu gesetzt werden. Letztere
kann hergestellt werden, indem CNBr in Acetonitril (1 g/ml) gelöst und bei –20°C in einer
dicht verschlossenen Phiole gelagert wird. Die Temperatur wird dann
auf 18–20°C ansteigen
gelassen.
-
Trotz
der relativen Einfachheit des Titrationsverfahrens ist gegebenenfalls
zu bevorzugen, das schnellere und technisch vereinfachte Verfahren
gemäß March
et al., Anal. Biochem. 60, 149 (1974) einzusetzen. Der Aktivierungsvorgang
wird in konzentriertem Carbonatpuffer durchgeführt. Die erforderliche Menge
an gewaschenem Gel wird im gleichen Volumen von 2 M NaHCO3-NaCO3-Puffer (pH
10,9) in einem Becherglas suspendiert, das mit einem Thermometer
und einem Magnetrührstäbchen ausgestattet
ist. Die Aufschlämmung wird
auf etwa 4–5°C abgekühlt, und
das aktivierte Gel wird in eine Sinternutsche gefüllt und
gewaschen.
-
Die
für die
oben beschriebenen Verfahren empfohlene CNBr-Konzentration reicht
für mäßige Peptidsubstitutionsgrade
aus. Wenn niedrigere oder höhere
Aktivierungsgrade erforderlich sind, können für die Titration 50 mg und 200–300 mg
CNBr/g Feuchtgewicht Gel zusammen mit 2 M und 8 M NaOH eingesetzt
werden.
-
Es
ist allgemein anerkannt, dass die CNBR-aktivierten intermediären funktionellen
Gruppen von Polysaccharidgelen begrenzte Stabilität aufweisen,
und deshalb sollte das Gel vorzugsweise so rasch wie möglich gewaschen
werden, bevor es auf das Kupplungsreaktionsmedium übertragen
wird. Am Ende des Aktivierungsschritts wird das Gel rasch durch
Zusatz von zerstoßenem
Eis gekühlt
und in eine große
Glassinternutsche gegossen, der vorher mit zerstoßenem Eis
gekühlt
worden war. Die Suspension wird rasch in einen Büchner-Kolben (2 Liter) filtriert,
der festes Eisen(III)sulfat enthält,
um nichtumgesetztes CNBr und Cyanide, wie z.B. harmloses Ferrocyanid,
zu entfernen. Das Gel wird anschließend durch Abnutschen mit 1
Liter eiskaltem destilliertem Wasser und 1 Liter des Puffers, der
in der Kupplungsstufe verwendet wird, typischerweise mit eiskaltem
0,1 M NaHCO3-NaCO3-Puffer
(pH 8,5–9,5),
gewaschen.
-
CNBr-aktivierte
Sepharose 4B ist im Handel von Pharmacia erhältlich, womit der gefährliche
Umgang mit CNBr umgangen wird. Das aktivierte Gel wird in Gegenwart
von Dextran und Lactose gefriergetrocknet, um die Kügelchenform
beizubehalten und in 15 g fassenden luftdichten Verpackungen bereitgestellt.
Die erforderliche Menge an gefriergetrocknetem Pulver wird in 1
mM HCl auf einem Glasfilter gequollen und mit zumindest 200 ml der
gleichen Lösung
pro Gramm Pulver gewaschen. 1 g gefriergetrocknetes Material entspricht etwa
3,5 ml an endgültigem
Gelvolumen. Die Peptidligand-Bindungskapazität des Gels wird wirksamer aufrechterhalten,
wenn mit Lösungen
mit niedrigem pH und nicht mit Lösungen
mit einem pH über
7 gewaschen wird. Das Gel ist bereit, einen Peptidliganden zu binden,
sobald der Waschvorgang abgeschlossen ist.
-
Pharmacia
vertreibt auch CNBr-aktivierte Sepharose 6 MB zur Verwendung in
der Zellbiologie und Immunologie zur Trennung von „funktionell
homogenen Zellpopulationen".
Sie wird durch Aktivierung von Sepharose-6MB-Makrokügelchen
(Durchmesser 200–300 μm) mit Bromcyan
hergestellt und auf gleiche Weise gehandhabt wie CNBr-aktivierte
Sepharose 4B.
-
Das
anzubindenden Peptid wird in einem dem Volumen des gepackten Gels
entsprechenden Volumen des kalten Puffers gelöst, zum feuchten, gewaschenen
Gel zugesetzt, und dann wird die Suspension sofort (in einer Nutsche)
mit einem Glasrührstab
gerührt.
Der gesamte Vorgang des Waschens, Zusetzens der Peptidlösung und
Vermischens dauert vorzugsweise weniger als 90 Sekunden. Die Suspension
wird aus der Nutsche in ein Becherglas übergeführt, das ein Magnetrührstäbchen enthält, und
vorsichtig bei 4°C
gerührt.
Obwohl die Reaktion im Wesentlichen nach 2 bis 3 Stunden abgeschlossen
ist, wird das Gemisch 16 bis 20 Stunden lang bei 4°C stehen
gelassen, um das vollständige
Verschwinden von reaktiven Polysaccharidgruppen sicherzustellen.
Das Gel mit angebundenen Peptiden wird dann mit großen Portionen
Wasser gewaschen, bis sichergestellt ist, dass kein Peptid mehr
entfernt wird.
-
Die
Peptidmenge, die an das Polysaccharidgel gebunden wird, kann zum
Teil- über
die zur aktivierten Matrix zugesetzte Peptidmenge gesteuert werden.
Wenn hochsubstituierte Polysaccharidgel-Derivate gewünscht werden,
sollte die zugesetzte Peptidmenge 20- bis 30-mal größer sein
als jene, die im Endprodukt gewünscht
wird. Bei herkömmlichen
Verfahren werden 100 bis 150 mg Bromcyan pro ml gepacktes Polysaccharidgel
verwendet, aber viel höhere
Ankupplungsausbeuten können
erzielt werden, wenn diese Menge auf 250 bis 300 mg erhöht wird.
Der pH, bei dem die Kupplungsreaktion durchgeführt wird, beeinflusst auch
den Ankupplungsgrad, da nur die nichtprotonierte Form der Aminogruppen
eines Peptids mit CNBr-aktivierten Polysacchariden reagiert. Vorzugsweise
wird die N-terminale α-Aminogruppe
des Peptidliganden zur Kupplung mit der aktivierten Polysaccharidmatrix
verwendet. α-Aminogruppen
binden sich bei einem pH von etwa 0,5 bis 10,0 optimal. Wenn die
Kupplung an der/den ε-Aminogruppe(n)
des ausgewählten
Peptidliganden (wie z.B. die ε-Aminogruppen
der Lysinylreste) gewünscht
wird, sollte die Kupplungsreaktion bei einem pH-Wert von etwa 10,0
durchgeführt
werden, und ein großer
Peptidüberschuss
sollte zugesetzt werden. Wenn eine Kupplung an den aromatischen
Aminogruppen in den Histidyl- oder Tryptophanylresten des ausgewählten Peptids
gewünscht
wird, kann bei pH-Werten zwischen 8 und 9 eine sehr hohe Kupplungseffizienz
erreicht werden.
-
Weitere
Details der Erfindung finden sich in den nachstehenden Beispielen,
die den Schutzumfang der Erfindung genauer definieren.
-
BEISPIELE
-
BEISPIEL 1
-
Experimenteller Teil
-
Berechnungsverfahren
-
Alle
Berechnungen wurden mit dem DISCOVER-Programm (Biosym Technologies,
San Diego, USA) unter Verwendung des AMBER-Kraftfelds aller Atome
(S.J. Weiner; P.A. Kollman; D.A. Case; U.C. Singh; C. Ghio; G. Alagona;
S. Profeta, Jr.; P. W. einer J. Am. Chem. Soc. 106, 765–784 (1984);
S.J. Weiner; P.A. Kollman; D.T. Nguyen; D.A. Case, J. Comp. Chem.
7, 230–252
(1986)) mit einer entfernungsabhängigen
Dielektrizitätskonstante
(ε = 4r).
-
Synthese
-
Materialien und Verfahren
-
Peptide
wurden unter Einsatz von herkömmlichen
Festphasen-Syntheseverfahren hergestellt (R.B. Merrifield, J. Am.
Chem. Soc., 85, 2149–2154
(1963); E. Kaiser; R.L. Colescot; C.D. Bossinger; P.I. Cook, Anal. Biochem.
34, 595–598
(1970)). Organische Chemikalien wurden von Aldrich (Milwaukee, Wis.,
USA) oder Fluka (Ronkonkoma, N.Y., USA) bezogen. Geschützte Aminosäuren wurden
bei Bachem CC (Torrance, Calif., USA) oder Peninsula Labs (Belmont,
Calif., USA) erstanden. BOP (Benzotriazol-1-yloxytris[dimethylamino]phosphoniumhexafluorophosphat)
wurde von Richelieu Biotechnologies (Montreal, Kanada) bezogen.
Lösungsmittel
stammten von Baxter (McGaw Park, Illin., USA), Baker (Phillipsburg,
N.J., USA) oder Mallinckrodt (Paris, Ky., USA). Polystyrolträger wurden
von Advanced ChemTech (Louisville, Ky., USA) erworben.
-
Mono-t-butyloxycarbonyl-(BOC-)
1,3-Propandiamin wurde wie folgt hergestellt, 2-(tert-Butoxycarbonyloxyimino)-2-phenylaceionitril
(18 g, 73 mmol) wurde über
einen Zeitraum von 10 Minuten portionsweise zu einer Lösung von
1,3-Diaminopropan (12,5 g, 184 mmol) in 100 ml Tetrahydrofuran zugesetzt,
die auf 0°C
gekühlt
war. Nach vier Stunden bei 0°C
wurde die Reaktion zwei Stunden lang auf 25°C erwärmen gelassen. Dann wurden
die Reaktion mit 150 ml Ethylacetat verdünnt und zweimal mit 100 ml
gesättigtem
wässrigem Natriumchlorid
gewaschen. Die organische Phase wurde mit dreimal 100 ml 10 % wässriger
Citronensäure
extrahiert, und die vereinigten wässrigen Phasen wurden zweimal
mit 100 ml Ethylacetat gewaschen. Die wässrige Phase wurde in einem
Eisbad gekühlt,
und der pH wurde mit 50%igem Natriumhydroxid auf etwa 13 eingestellt.
Die basische wässrige
Phase wurde dann mit dreimal 100-ml-Volumina Dichlormethan extrahiert.
Die organische Phase wurde mit Kaliumcarbonat getrocknet und filtriert.
Das Lösungsmittel
wurde durch Rotationsverdampfung entfernt, um Mono-tert-butyloxycarbonyl-1,3-diaminopropane zu
erhalten.
-
Peptide
wurden mittels RP-HPLC auf einer Vydac-C-18-säule gereinigt und mit Acetonitril/Wasser-Gradienten,
die 0,1 Vol.-% Trifluoressigsäure
(TFA) enthielten, eluiert. Dann wurden die Peptide durch Elektrospray-MS
auf einem Tripel-Quadrupol-Massenspektrometer
PE SCIEX API III+ und durch quantitative Aminosäureanalyse auf einem automatischen
Aminosäureanalysator
Beckmann 6300 charakterisiert. Organische Zwischenprodukte wurden
durch 1H- und 13C-Magnetresonanz
(NMR) auf einem Varian VXR-300S und durch hochauflösende Massenspektrometrie
(MS) auf einem Tandem-Massenspektrometer JEOL JMS-HX110HF/HX110HF
analysiert.
-
AcTNE(OFm)DLAARRE(OAllyl)QQnh-MBHA-Polystyrol
(1a):
-
Das
lineare Peptid 1 a mit der gezeigten Sequenz wurde durch herkömmliche
Kupplungszyklen unter Verwendung von drei Moläquivalenten BOC-Aminosäure, 3,3
Moläquivalenten
BOP und 3,3 Moläquivalenten N-Methylmorpholin
in Dichlormethan (CH2Cl2)
und Dimethylacetamid (DMA), falls für die Löslichkeit erforderlich, eine
Stunde lang bei Raumtemperatur auf p-MBHA-Harz (4,25 Gramm (g),
0,57 Milliäquivalente/Gramm (mÄqu./g),
2,42 Millimol (mmol)) synthetisiert. Die N-Acetyl-Kappe wurde durch
20-minütige
Behandlung mit 5 Millilitern (ml) Essigsäureanhydrid in 3%igem Triethylamin
(TEA) in CH2Cl2 bei
Raumtemperatur angebunden. Das Harz wurde getrocknet und gewogen
(4,41 g, schätzungsweise
0,22 mÄqu./g).
-
AcTNE(OFm)DLAARRE(OFm)QQnh-MBHA-Polystyrol
(1f):
-
AcAEE(OFm)AAAKFLE(OAllyl)Allyl)-AHAnh-MBHA-Polystyrol
(2a):
-
Die
angegebenen linearen Peptide 1f und 2a wurden wie oben für 1a beschrieben
synthetisiert.
-
AcTNQ(γ-NHCH3)DLAARRQ(γ-NHCH3)QQnh2 (1b):
-
Das
lineare harzgebundene Peptid 1f (0,60 g, 0,17 mmol) wurde mit 20%igem
Piperidin in DMA 20 Minuten lang doppelt entschützt. Die freien Carbonsäuren wurden
1,5 Stunden lang mit BOP (1,57 g, 3,55 mmol) und N-Methylmorpholin
(0,90 ml, 8,2 mmol) in CH2Cl2/DMA
an Methylamin (CH3NH3Cl,
0,26 g, 3,85 mmol) gebunden. Das Harz wurde gewaschen und getrocknet,
und die Peptid-Harz-Bindung wurde mit wasserfreier Fluorwasserstoffsäure (HF)
(10 ml) bei 0°C
eine Stunde lang mit Anisol (1 ml) und Ethylmethylsulfid (EtSMe)
(0,5 ml) als Fänger
gespalten. Das Harz wurde zweimal mit Ether, einmal mit Ethylacetat
und dann erneut mit Ether gewaschen. Das freie Peptid wurde dann
durch aufeinander folgende Waschvorgänge mit 10%iger Essigsäure, Eisessig,
Acetonitril, 10%iger Essigsäure
und Wasser aus dem Harz extrahiert. Die vereinigten Lösungen wurden
gefriergetrocknet, und der Rückstand
wurde gereinigt.
–CH2CH2CH2–
-
Cyclo-AcTNQ(γ-NH)DLAARRQ(γ-NH)QQnh2 (1c):
-
1. Unter Verwendung von
ungeschütztem
Propandiamin:
-
Das
lineare Peptid 1a auf dem Harz (0,51 g, 0,11 mmol) wurde am Fluorenylmethylester
mit 20%igem Piperidin in DMA 20 Minuten lang entschützt, und
das resultierende Piperidinsalz wurde durch zweimaliges Waschen
mit 1%iger TFA in CH2Cl2 neutralisiert.
Die freie Carbonsäure
wurde eine Stunde lang mit BOP (0,40 g, 0,90 mmol) und Diisopropylethylamin
(DIPEA) (0,17 ml, 0,98 mmol) in CH2Cl2 an 1,3-Propandiamin (0,12 ml, 1,44 mmol)
gebunden, gefolgt vom Zusatz von DMA und dem Fortsetzen des Kuppelns
für weitere
45 Minuten. Der Glutaminsäureallylester
wurde mit Tetrakis(triphenylphosphin)palladium(0) (Pd(PPh3)4) (0,21 g, 0,18
mmol) in 20%igem Piperidin in DMA 1,5 Stunden lang entschützt, und
das Piperidin wurde durch zweimaliges Waschen mit 1%iger TFA in
CH2Cl2 entfernt.
Die resultierende Aminosäure
wurde mit BOP (0,32 g, 0,72 mmol) und DIPEA (0,13 ml, 0,75 mmol)
in CH2Cl2 3,5 Stunden
lang zyklisiert. Ein Kaiser-Test ergab eine erkennbare purpurne
Färbung,
sodass die Zyklisierung mit BOP (0,44 g, 0,99 mmol) und DIPEA (0,19
ml, 1,09 mmol) in CH2Cl2 weitere
drei Tage lang wiederholt wurde. Das Peptid wurde vom Harz abgespalten,
wie oben beschrieben für
1b beschrieben wurde.
-
2. Unter Verwendung von
Mono-BOC-Propandiamin:
-
Das
lineare Peptid 1a auf dem Harz (0,57 g, 0,13 mmol) wurde am Fluorenylmethylester
mit 20%igem Piperidin in DMA 0,5 Stunden lang entschützt, und
das re sultierende Piperidinsalz wurde durch zweimaliges Waschen
mit 1%iger TFA in CH2Cl2 neutralisiert.
Die freie Carbonsäure
wurde eine Stunde lang mit BOP (0,52 g, 1,18 mmol) und DIPEA (0,25
ml, 1,44 mmol) in CH2Cl2/DMA
an Mono-tert-butyloxycarbonyl-1,3-propandiamin (0,23 ml, 1,32 mmol)
gebunden. Der Glutaminsäureallylester
wurde mit (Pd(PPh3)4)
(0,21 g, 0,18 mmol) in 20%igem Piperidin in DMA 1,5 Stunden lang
entschützt,
und das Piperidin wurde durch zweimaliges Waschen mit 1%iger TFA
in CH2Cl2 entfernt;
der Kaiser-Test war an diesem Punkt negativ. Die BOC-Gruppe wurde
mit TFA/CH2Cl2/Anisol/1,2-Ethandithiol
(45:45:5:5 Vol./Vol.) entfernt; das freie Amin ergab dann einen
positiven Kaiser-Test. Die resultierende Aminosäure wurde mit BOP (0,58 g,
1,31 mmol) und DIPEA (0,30 ml, 1,72 mmol) in CH2Cl2 zwei Stunden lang zyklisiert, wonach der
Kaiser-Test nur eine schwache blau-grüne Färbung ergab. Das Peptid wurde
vom Harz abgespalten, wie oben beschrieben für 1b beschrieben wurde.
-
AcAEQ(γ-NHCH3)AAAKFLQ(γ-NHCH3)AHAnh2 (2b):
-
Das
lineare Peptid 2a auf dem Harz (0,60 g, 0,19 mmol) wurde am Allyl-
und am Fluorenylmethylester mit Pd(PPh3)4 (0,1 g, 0,09 mmol) in 20%igem Piperidin
in DMA 30 Minuten lang entschützt,
und das resultierende Piperidinsalz wurde durch Waschen mit 50%iger
TFA in CH2Cl2 mit
Anisol und 1,2-Ethandithiol neutralisiert. Die freie Carbonsäure wurden
mit BOP (0,32 g, 0,72 mmol) und DIPEA (0,35 mL, 2,01 mmol) in CH2Cl2/DMA 1 Stunde
lang an Methylamin (40 % wässrig,
0,16 ml, 1,86 mmol) gebunden. Das Peptid wurde vom Harz abgespalten,
wie oben beschrieben für
1b beschrieben wurde.
-
1d, 1e, 2c, 2d, 2e:
-
1d
und 1e wurden aus 1a hergestellt, und 2c–2e wurden aus 2a hergestellt,
und zwar nach denselben Verfahren wie oben für 1c beschrieben, wobei ungeschütztes 1,3-Propandiamin,
Kupplung mit 1,4-Butandiamin für
1d und 2d und mit 1,5-Pentandiamin für 1e und 2e eingesetzt wurden.
-
AcTNk(S-Acm)DLAARRK(S-Acm)QQnh2 (3a):
-
AcAEk(S-Acm)AAAKFLK(S-Acm)AHAnh2 (4a):
-
Die
linearen Peptide 3a und 4a wurde durch herkömmliche Merrifield-Verfahren
unter Einsatz von FMOC-Chemie synthetisiert (E. Atherton; R.C. Sheppard,
J. Chem. Soc., Chem. Commum, 165–166(1985)). Fmoc-D-Thiolys(Acm)-OH
(7,k(S-Acm)) und Fmoc-L-Thiolys(Acm)-OH (10,k(S-Acm)) wurden wie
nachstehend beschrieben hergestellt.
-
AcTNk(S)DLAARRK(S)QQnh2 (3b):
-
AcAEk(S)AAAKFLK(S)AIAnh2 (4b):
-
Die
zyklischen Peptide 3b und 4b wurden durch gleichzeitige Entschützung und
Oxidation aus 3a bzw. 4a hergestellt. Etwa 16 mg Acm-geschütztes Peptid
wurden in 1,5 ml Wasser gelöst,
das 10%ige Essigsäure enthielt,
und dann mit Trifluorethanol auf 50 ml Gesamtvolumen verdünnt, um
eine Endkonzentration von etwa 200 Mikromol/Liter (μM) zu erhalten.
Insgesamt 12 ml einer 6-Millimol/Liter-(mM) Lösung von Iod (80 Milligramm
(mg), gelöst
in 3 ml Essigsäure
und mit Trifluorethanol auf 50 ml verdünnt) wurden in 1-ml-Portionen über einen
Zeitraum von 10 Stunden zugesetzt, während der Fortschritt der Reaktion
durch HPLC überwacht wurde.
Als das Ausgangsmaterial verbraucht war, wurde die Reaktion mit
Wasser verdünnt
und gefriergetrocknet, und das rohe oxidierte Material wurde gereinigt.
-
(2S,5R)-2,5-Dihydro-3,6-diethoxy-2-isopropyl-5-(4-brombutyl)pyrazin
(5):
-
n-Butyllithium
(13,3 ml einer 1,6 M Lösung
in Hexan, 21,3 mmol) wurde über
einen Zeitraum von 5 Minuten zu einer Lösung von (2S)-2,5-Dihydro-3,6-diethoxy-2-isopropylpyrazin
(Schollkopf-Reagens) (4,30 g, 20,3 mmol) in Tetrahydrofuran (THF)
zugesetzt. Die Lösung
wurde 15 Minuten lang bei –78°C gehalten,
wonach 1,4-Dibrombutan (9,75 ml, 81,2 mmol) auf einmal zugesetzt
wurde. Nach 2,5 Stunden bei –78°C wurde die
Reaktion auf Raumtemperatur erwärmen
gelassen und mit Diethylether (100 ml) verdünnt. Die organische Phase wurde
mit Wasser (100 ml) und Kochsalzlösung (100 ml) gewaschen und
dann über
Magnesiumsulfat (MgSO4) getrocknet. Nach
Filtration und Einengung wurde der Großteil des 1,4-Dibrombutans im
Hochvakuum entfernt. Das zurückbleibende Öl wurde
durch Kieselgelchromatographie (2 % Ethylacetat in Hexan) gereinigt, um
5 (3,7 g, 57 %) als farblose Flüssigkeit
bereitzustellen; [α]25 D – 1,13° (c = 4,5,
CHCl3); IR (Dünnfilm) 2957, 1689, 1456, 1364,
1304, 1230, 1144, 1038 cm–1; 1H-NMR
(300 MHz, CDCl3) δ 4,04–4,18 (m, 4H) 3,94–4,02 (m, 1H)
3,90 (t, J = 3,9, 1H) 3,39 (t, J = 6,9, 2H) 2,21–2,32 (m, 1H) 1,68–1,92 (m,
4H) 1,33–1,47
(m, 2H) 1,274 (t, J = 7,2, 3H) 1,268 (t, J = 7,2, 3H) 1,03 (d, J
= 6,9, 3H) 0,70 (d, J = 6,9, 3H); 13C-NMR
(100,6 MHz, CDCl3) δ 163,12, 163,05, 60,72, 60,48,
55,09, 33,60, 33,11, 32,66, 31,80, 23,22, 19,03, 16,6, 14,34, 14,31;
Massenspektrum (FAB+) 347,1 (MH+).
-
(2S,5R)-2,5-Dihydro-3,6-diethoxy-2-isopropyl-5-(4-(4-methoxybenzyl)thiobutyl)pyrazin
(6):
-
Kalium-tert-butoxid
(11,8 ml einer 1 M Lösung
in THF) wurde über
einen Zeitraum von fünf
Minuten bei 25°C
zu einer Lösung
von 4-Methoxy-α-toluolthiol
(1,85 ml 90%ige Lösung,
11,8 mmol) in THF (20 ml) zugesetzt, was einen schweren, weißen Niederschlag
ergab, der 25 Minuten lang gerührt
wurde. Eine Lösung
von 5 (3,7 g, 10,7 mmol) in THF (20 ml) wurde zugesetzt und das
Rühren
weitere drei Stunden fortgesetzt. Das Reaktionsgemisch wurde durch
Rotationsverdampfung eingeengt und anschließend zwischen Wasser (50 ml) und
Diethylether (100 ml) verteilt, Die organische Phase wurde mit Kochsalzlösung gewaschen,
getrocknet (MgSO4) und eingeengt. Der Rückstand
wurde durch Kieselgelchromatographie (1 %, steigend auf 2,5 % Ethylacetat
in Hexan) gereinigt, um 6 (2,90 g, 91 %) als farbloses Öl bereitzustellen;
[α]25 D – 3,77° (c = 3,5, CHCl3); IR (Dünnfilm)
2957, 1689, 1510, 1238, 1038 cm–1; 1H-NMR (300 MHz, CDCl3) δ 7,21 (d,
J = 8,7, 2H) 6,84 (d, J = 8,4, 2H) 4,02–4,20 (m, 4H) 3,93–3,99 (m,
1H) 3,88 (t, J = 3,3, 1H) 3,80 (s, 3H) 3,65 (s, 2H) 2,39 (t, J =
7,5, 2H) 2,20–2,32
(m, 1H) 1,64–1,82
(m, 2H) 1,50–1,62
(m, 2H) 1,22–1,38
(m, 2H) 1,27 (t, J = 7,2, 6H) 1,03 (d, J = 6,9, 3H) 0,70 (d, J =
6,9, 3H); 13C-NMR (100,6 MHz CDCl3) δ 163,20,
163,00, 158,50, 130,57, 129,80, 113,80, 60,67, 60,45, 60,40, 55,24,
55,20, 35,57, 33,68, 31,74, 31,14, 29,19, 23,92, 19,05, 16,60, 14,36,
14,32; Massenspektrum (FAB+) 421,2 (MH+).
-
(2R)-2-(9-Fluorenylmethoxycarbonyl)amino-6-acetamidomethylthiohexansäure (D-thio(Acm)lysin)
(7):
-
Wasser
(30 ml) und 3 N HCl (6,5 ml) wurden zu einer Lösung von 6 (2,90 g, 9,32 mmol)
in THF (50 ml) zugesetzt. Das Gemisch wurde 12 Stunden lang bei
25°C gerührt, und
dann wurde das THF durch Rotationsverdampfung entfernt. Die Lösung wurde
mit wässrigem
Kaliumcarbonat (K2CO3)
auf einen pH von 10 eingestellt und dann zweimal mit Ethylacetat
(75 ml) extrahiert. Die organischen Extrakte wurden getrocknet (MgSO4) und eingeengt, und der Rückstand
wurde durch Kieselgelchromatographie (1:1, steigend auf 2:1 Ethylacetat/Hexan
mit 0,5 % Triethylamin) gereinigt, um rohen S-Methoxybenzylthiolysinethylester
(2,65 g, 91 %) als farbloses Öl
zu erhalten, das direkt in der nächsten
Stufe eingesetzt wurde. Dieser Ester (3,02 g, 9,71 mmol) wurde bei
0°C in einem
Gemisch aus Trifluoressigsäure
(50 ml) und Anisol (1 ml) gelöst,
und Quecksilberacetat (3,09 g, 9,71 mmol) wurde zugesetzt, um eine
klare Lösung
zu erhalten. Nach 15 Minuten wurde die Trifluoressigsäure durch
Rotationsverdampfung entfernt, und der Rückstand wurde zuerst mit Wasser
(75 ml) verdünnt
und dann mit Diethylether (75 ml) gewaschen. Die wässrige Phase
wurde 30 Minuten lang mit Schwefelwasserstoff (H2S)
(der durch die Lösung
hindurchperlen gelassen wurde) behandelt, und der resultierende schwarze
Niederschlag wurde durch ein Celite-Bett abfiltriert. Das Filtrat
wurde durch Ratationsverdampfung eingeengt, erneut in Wasser (20
ml) gelöst
und durch ein 0,45-Mikrometer-(μm-)Nylonfilter
filtriert. Dann wurde die Lösung
erneut eingeengt, und der resultierende Schaum wurde über Nacht
im Hochvakuum getrocknet. Der Rückstand
wurden anschließend
in THF (20 ml) gelöst
und auf 0°C
abgekühlt.
Eine Lösung
von Lithiumhydroxid (1,22 g, 29,1 mmol) in Wasser (20 ml) wurde
in zwei Portionen in einem Abstand von 15 Minuten zugesetzt. Nach
zwei Stunden wurde die Reaktion auf Raumtemperatur erwärmen gelassen,
und der pH wurde mit 1 mol/Liter (M) wässriger Citronensäure auf
7,0 eingestellt. Das Lösungsmittel
wurde durch Rotationsverdampfung entfernt, der Rückstand wurde in Dioxan (80
ml) gelöst,
und Fmoc-H-Hydroxysuccinimid (3,3 g, 9,71 mmol) wurde zugesetzt,
gefolgt von gesättigtem
wässrigem
Natriumbicarbonat (NaHCO3) (15 ml). Nach
einer Stunde wurde das Lösungsmittel
durch Rotationsverdampfung entfernt, und der Rückstand wurde zwischen Wasser
(50 ml) und Ethylacetat (50 ml) verteilt. Die wässrige Phase wurde mit 1 M
wässriger
Citronensäure auf
einen pH von 2,5 eingestellt und dann dreimal mit Ethylacetat (jeweils
75 ml) gewaschen Die vereinigten organischen Phasen wurden getrocknet
(MgSO4) und eingeengt. Der Rückstand
wurde durch Kieselgelchromatographie (zuerst mit 2:1 Ethylacetat/Hexan
mit 0,5 % Essigsäure,
dann mit Ethylacetat mit 0,5 % Essigsäure, dann mit 5 % Methanol
in Ethylacetat mit 0,5 % Essigsäure)
gereinigt, und produkthältige
Fraktionen wurden aus Toluol (150 ml, dreimal) eingeengt, bevor
sie in Wasser mit Acetonitril gelöst und gefriergetrocknet wurden,
um 7 (3,16 g, 71 % über
vier Stufen) als weißes
Pulver bereitzustellen; [α]25 D – 1,8° (c = 2,
EtOH); IR (Dünnfilm)
2800–3400,
1709, 1536, 1260, 759, 740 cm–1; 1H-NMR
(300 MHz, DMSO-d6) δ 8,42
(t, J = 6,0, 1H) 7,88 (d, J = 7,3, 2H) 7,72 (d, J = 7,5, 2H) 7,58
(d, J = 8,1, 1H) 7,407 (t, J = 7,5, 2H) 7,32 (t, J = 7,5, 2H) 4,16–4,30 (m,
5H) 3,91 (m, 1H) 2,53 (t, J = 7,2, 2H) 1,82 (s, 3H) 1,33–1,76 (m,
6H); 13C-NMR (100,6 MHz, DMSO-d6) δ 174,00,
169,16, 156,08, 143,86, 143,80, 140,71, 127,61, 127,05, 125,28,
120,08, 65,57, 53,90, 46,68, 30,54, 30,01, 28,79, 24,91, 22,54;
hochauflösendes
Massenspektrum (FAB+) 457,1785–, Err[ppm/mmu] – 2,7/–1,2.
-
(2R,5S)-2,5-Dihydro-3,6-dimethoxy-2-isopropyl-5-(4-bromobutan)pyrazin
(8):
-
- [α]25 D + 1,73° (c = 4,4,
CNCl3); IR (Dünnfilm) 2959, 1696, 1458, 1434,
1237, 1196, 1007 cm–1; 1H-NMR
(300 MHz, CDCl3) δ 3,98–4,47 (m, 1H) 3,95 (t, J =
3,6, 1H) 3,70 (s, 3H) 3,68 (s, 3H) 3,40 (t, J = 6,9, 2H) 2,19–2,32 (m,
1H) 1,66–1,94
(m, 4H) 1,34–1,48
(m, 2H) 1,05 (d, J = 6,9, 3H) 0,69 (d, J = 6,9, 3H); 13C-NMR
(100,6 MHz, CDCl3) δ 163,62, 163,60, 60,77, 55,11,
52,31, 33,54, 33,10, 32,61, 31,73, 23,26, 19,02, 16,56; Massenspektrum
(FAB+) 319,1 (MH+).
-
(2R,5S)-2,5-Dihydro-3,6-dimethoxy-2-isopropyl-5-(4-(4-methoxybenzyl)thiobutyl)pyrazin
(9):
-
- [α]25 D + 3,96° (c = 3,63,
CHCl3); IR (Dünnfilm) 2944, 1696, 1510, 1238
cm–1; 1H-NMR (300 MHz, CDCl3) δ 7,21 (d,
J = 8,7, 2H) 6,83 (d, J = 8,4, 2H) 3,97–4,25 (m, 1H) 3,93 (t, J =
3,3, 1H) 3,79 (s, 3H) 3,68 (s, 3H) 3,67 (s, 3H) 3,65 (s, 2H) 2,39
(t, J = 7,5, 2H) 2,18–2,30
(m, 1H) 1,60–1,84
(m, 2H) 1,49–1,62
(m, 2H) 1,23–1,38
(m, 2H) 1,27 (t, J = 7,2, 6H) 1,04 (d, J = 6,9, 33H) 0,68 (d, J
= 6,9, 3H); 13C-NMR (100,6 MHz, CDCl3) δ 163,72, 163,47, 158,48,
130,55, 129,78, 113,78, 60,68, 55,22, 55,18, 52,27, 35,58, 33,63,
31,64, 3,12, 29,13, 23,88, 19,02, 16,51; Massenspektrum (FAB+) 393,2
(MH+).
-
(2S)-2-(9-Fluorenylmethoxycarbonyl)amino-6-acetamidomethylthiohexansäure: (L-Thio(Acm)lysin)
(10):
-
- [α]25 D + 1,3° (c = 2,
EtOH); IR (Dünnfilm)
2800–3400,
1709, 1536, 1260, 759, 740 cm–1; 1H-NMR
(300 MHz, DMSO-d6) 6 8,42 (t, J = 6,0, 1H) 7,88 (d, J = 7,3, 2H)
7,72 (d, J = 7,5, 2H) 7,58 (d, J = 8,1, 1H) 7,407 (t, J = 7,5, 2H)
7,32 (t, J = 7,5, 2H) 4,16–4,30
(m, 5H) 3,91 (m, 1H) 2,53 (t, J = 7,2, 2H) 1,82 (s, 3H) 1,33–1,76 (m, 6H); 13C-NMR (100,6 MHz, DMSO-d6) δ 174,00,
169,16, 156,08, 143,86, 143,80, 140,71, 127,61; 127,05, 125,28,
120,08, 65,57, 53,90, 46,68, 30,54, 30,01, 28,79, 24,91, 22,54;
hochauflösendes
Massenspektrum (FAB+) 457,1776, Err[ppm/mmu] –4,6/–2,1.
-
Diese
Materialien wurden auf die gleiche Weise wie 5, 6 und 7, ausgehend
von (2R)-2,5-Dihydro-3,6-dimethoxy-2-isopropylpyrazin
(Merck) hergestellt.
-
NMR-Spektroskopie
-
Für jedes
Peptid wurden 2–4
mg gereinigtes Material in 440 Mikroliter (μl) 25 mM d3-Natriumacetat, das
5 % Deuteriumoxid (D2O) und 0,1 Millimol/Liter
(mM) Natriumazid enthielt, zugesetzt, was eine Gesamtpeptidkonzentration
von 1–6
mM ergab; der pH wurde durch Zusatz von 1 M Natriumhydroxid (NaOH)
auf 4,5 eingestellt. Alle Spektren wurden bei 5°C oder 10°C auf einem Bruker-AMX-500-Spektrometer
aufgenommen. Zweidimensionale COSY- (W.P. Aue, E. Bartholdi und
R.R. Ernst, J. Chem. Phys. 64, 2229–2246 (1976)), ROESY- (A.A.
Bothner-By, R.L. Stephens, J.-M. Lee, C.D. Warnen und R.W. Jeanloz,
J. Am. Chem. Soc. 106, 811–813
(1984); M.J. Rance, Magn. Reson. 74, 557–564 (1987)) und TOCSY- (L.
Braunschweiler und R.R. Ernst, J. Magn. Reson. 53, 521–528 (1983);
A. Bax und D.G. Davis, J. Magn. Reson. 65, 355–360 (1985)) Spektren wurden
mit einer Phasendiskriminierung ω1 aufgenommen, die mit TPPI erreicht wurde
(D. Marion und K. Wuthrich, Biochem. Biophys. Res. Commun. 113,
967–974
(1983)). Die Gesamtaufnahmedauer betrug etwa 2, 4 und 12 Stunden
für COSY-,
TOCSY- bzw. ROESY-Spektren. Wasserunterdrückung wurde durch kohärente leistungsschwache
Bestrahlung der Wasserresonanz für
die 1,5 Sekunden Relaxationszeit erreicht. ROESY- und TOCSY-Spektren
wurden wie von M. Akke, N.J. Skelton, J. Kordel und W.J. Chazin
in Techniques in Protein Chemistry II, S. 401–408, J.J. Villafranca, Hrsg.
Academic Press, Inc.: Boca Raton, Fla., USA (1991) beschrieben erhalten;
außerdem
wurden Phasenkorrekturen erster Ordnung durch eine Aufnahme auf
sinusmodulierte Weise in ω1 vermieden. TOCSY-Vermischung wurde mit einer sauberen
DIPSI-2rc-Sequenz erreicht, die 90 Millisekunden (ms) lang angelegt
wurde (J. Cavanagh und M.J. Rance, Magn. Reson 96, 670–678 (1992)).
Die ROESY-Spektren wurden mit einem Spinlock-Impuls von 4,0 Kilohertz
(kHz) für
eine Dauer von 200 ms erhalten. Die Spektren wurde weiterbearbeitet
und mithilfe des Felix-Software-Pakets (Biosym Technologies, San
Diego, Calif., USA) analysiert. 3JHN-Hα wurden
durch Anpassen eines Antiphasenpaars von Lorentz-Linien in ω2-Abschnitte von COSY-Spektren mit hoher
digitaler Auflösung
erhalten.
-
Die
Amidprotonen-Austauschgeschwindigkeiten mit einem Lösungsmittel
wurden für
1b und 1c gemessen, indem das Peptid aus H2O
gefriergetrocknet wurde und eine Reihe von eindimensionalen (1D-) NMR-Spektren
aufgenommen wurde, gleich nachdem das Peptid in D2O
gelöst
worden war. Die Austauschgeschwindigkeitskonstanten wurden bestimmt,
indem eine Exponentialanpassung auf Basis dreier Parameter an die
Signale des zerfallenden Amids vorgenommen wurde. Schutzfaktoren
wurden als Austauschgeschwindigkeitsverhältnis im vernetzten und nichtvernetzten
Peptid berechnet.
-
Strukturberechnung
-
NOESY-
(A. Kumar, R.R. Ernst und K. Wuthrich, Biochem. Biophys. Res. Commun.
95, 1–6
(1980); G. Bodenhausen, H. Kogler und R.R. Ernst, J. Magn. Reson.
58, 370–388
(1984)) und ROESY-Daten wurden bei Mischdauern von 300 ms bzw. 200
ms aus Wasser (H2O) und D2O
unter Verwendung einer Probe von 1c (etwa 8 mM) gewonnen. Die gesamte
Aufnahmedauer betrug 24 Stunden pro Experiment. Aus diesen Daten
wurden Distanzeinschränkungen
erzeugt, indem Kreuzpeaks anhand des integrierten Peak-Volumens
als stark, schwach oder sehr schwach kategorisiert wurden und eine
obere Schwelte von 2,9, 3,5, 4,6 bzw. 5,6 Ångström (Å) zu den entsprechenden Interprotonabständen zugeordnet
wurde. Der Diederwinkel Φ war
für Reste,
für die 3JHN-Hα weniger als 6,0 Hertz
(Hz) betrug, zwischen –90° und –40° eingeschränkt. Die
Werte für 3JHN-Hα wurden anhand eines COSY-Spektrums
bestimmt, das in einer D2O-Lösung mit
einem 35°-Mischimpuls
erhalten worden war. χ1-Einschränkungen
und stereospezifische Hβ-Zuordnungen wurden auf
Basis dieser Kopplungskonstanten und der Ergebnisse der anfänglichen
Strukturberechnungen für
vier Seitenketten erhalten (N.J. Skelton, K.C. Garcia, D.V. Goeddel,
C. Quan und J.P. Burnier, Biochemistry 33, 13581–13592 (1994)).
-
Strukturen
wurden mithilfe des Programms DGII unter Einsatz der CVFF-Kraftfeldparameter
berechnet (Biosym Technologies, San Diego, Calif., USA). Eingabeeinschränkungen
bestanden aus 141-Interprotonen-Abständen, 9 Φ Diedenwinkeleinschränkungen
und 5 χ1 Diederwinkeleinschränkungen. Explizite Wasserstoffbrückenbindungen
wurden nicht inkludiert. Die Strukturen wurden mit einer Dreiecks- und Vierecksglättung vor
der perspektivischen Einbettung aller Atome erzeugt. Die eingebetteten
Strukturen wurden 10.000 Schritte lang im vierdimensionalen Raum
ausgeheilt, während
sie mit allen Atommassen auf 1000 eingestellt von 200 Kelvin (K)
ausgehend abgekühlt
wurden. Die DG-Strukturen wurden durch rMD unter Verwendung des AMBER-Kraftfelds
im DISCOVER-Programm (Biosym Technologies) verfeinert. Die Strukturen
wurden 3 Picosekunden (ps) lang bei 600 K ausgeheilt, 1,8 ps lang
auf 0 K abgekühlt
und schließlich
200 rEM-Durchgängen unterzogen.
Die Ladungen auf Glu-, Asp-, Arg-, den C-terminalen und N-terminalen
Resten wurden auf 0,2 e reduziert, und eine distanzabhängige dielektrische
pf 1/4r wurde eingesetzt. Einschränkungen wurden als Square-Well-Potentiale
mit Krafteinschränkungen
von 25 Kilokalorien/Mol/Angström2 (kcal·mol–1·Å–2)
und 100 Kilokalorien/Mol/Radiant2 (kcal·mol–1·rad–2)
für Distanzen
bzw. Diederwinkel eingesetzt. Im letzten Berechnungsdurchgang wurden
60 Strukturen in DGII eingebettet und durch rMD verfeinert.
-
Zirkulardichroismus
-
CD-Spektren
wurden auf einem Aviv-G2-Spektrometer mit einer temperaturgesteuerten
Zelle mit einer Weglänge
von 0,1 Zentimeter (cm) erhalten. Lösungen für Analysespektren wurden durch
Verdünnung
von NMR-Proben auf etwa 100 Mikromol/Liter (μM) mit zusätzlichem NMR-Puffer vorbereitet.
Punkte wurden alle 0,2 Nanometer (nm) mit einer Bandbreite von 0,2
nm und einer Mittelungszeit von 2 Sekunden genommen. Die kürzeste erreichbare
Wellenlänge
wurde durch Absorption des Acetatpuffers eingeschränkt. Die
dargestellten Kurven wurden mit Standardparametern geglättet (10-Punkt-Glättung).
-
ERGEBNISSE UND DISKUSSION
-
Überlegungen zum Aufbau
-
Ausgehend
von synthetischen und geometrischen Überlegungen wurde bestimmt,
dass die Amidchemie eingesetzt werden sollte, um die I- und I+7-Seitenketten
zu verbinden. Disulfidbindungen sind zwar synthetisch machbar, führten jedoch
eine ungewünschte
90°-Drehung
in die Bindung ein. Um die Fähigkeit
von einfachen Alkylkettenlinkern zu nutzen, eine sterische Zusammendrängung im
Bereich nahe der I+3- und I+4-Reste zu vermeiden, wurden Bindungsverfahren
zur Verbrückung
von entweder Gln oder Asn bei I und I+7 mit einer Alkandiylkette
in Betracht gezogen. Gln wurde ausgewählt, weil seine größere Länge die
Verwendung einer minimalen Fessel zur Verbindung dieser Seitenketten
erlaubt. Eine veranschaulichende Gruppe von Proteinkristallstrukturen
aus der Brookhaven-Datenbank (F.C. Bernstein; T.F. Koetzle; G.J.B.
Williams; E.F. Meyer; M.D. Brice; J.R. Rodgers; O. Kennard; T. Shimanouchi;
M. Tasumi, J. Mol. Biol. 112, 535–542 (1977)) wurde auf jegliches
Vorkommen von Glutamin in einem helikalen Kontext durchsucht (ϕ = –60° ± 30° und ψ = 45° ± 30°). Die resultierende
Datengruppe wurde verwendet, um die Seitenketten-Rotamerverteilungen von natürlich vorkommenden
helikalen Glutaminresten zu bestimmen. Im Allgemeinen weisen Aminosäurereste
in einem α-helikalen
Kontakt χ1 = –60° auf, eine
Konformation, die für
die I+7-Position eines Seitenkettenlinkers geeignet ist. Glutamin
weist eine relativ hohe Population (14,6 %) des Rotamers χ1 = 180° auf,
was eine signifikante natürlich
Konformation darstellt, welche die Seitenkette in Richtung des C-terminalen
Endes der Helix ausrichtet. Es wurden Rotamerkombinationen identifiziert,
welche die Nε2-Nε2-Distanz
zwischen der I- und I+7-Seitenkette
in einem helikalen Peptidmodell verringern. In Abhängigkeit
von den χ3-Werten
wurden Abstände
im Bereich von 5,3 Ä bis
7 Ä gefunden,
wenn das I-Glutamin χ1- und χ2-Winkel von 180° und 60° annimmt und das I+7-Glutamin χ1- und χ2-Werte von –60° bzw. 180° annimmt.
-
Der
Bau eines Modells zeigte, dass eine 4-Methylen-„Brücke" diese beiden Glutamin-Seitenketten effizient
verbinden kann, ohne dass ungünstige
Drehwechselwirkungen auftreten. Modelle von 3-, 4- und 5-Methylen-verbrückten helikalen
Peptiden wurden unter Einsatz von distanzgeometrischen Verfahren
(Quantum Chemistry Program Exchange, Program Nr. 590, mit dem Titel
DGEOM von Blaney et al.), gefolgt von Energieminimierung konstruiert.
Alle Reste mit Ausnahme der gebundenen Glutamine waren Alanin. Die
Konformationsstabilität
der helikalen Peptide wurde unter Einsatz von 1 Nanosekunde (Nucleinsäure) nichteingeschränkter molekularer
Dynamik bei 298 K nach einer anfänglichen Äquilibrierungsperiode
von 100 Picosekunden (ps) beurteilt, während derer harmonische Einschränkungen
(25 Kilokalorien/Mol/Ångström (kcal·mol–1·Å–1))
angewendet wurden, um die Helizität aufrechtzuerhalten. Als Kontrolle
wurde eine Polyalaninhelix 1 ns lang in Gegenwart von identischen
Einschränkungen
berechnet.
-
Peptide
mit einer 2-Methylenbrücke
behielten eine konstante helikale Konformation bei, weisen aber eine
deutliche „Krümmung" der Helixachse auf.
Peptide mit einer 4-Methylenbrücke
behielten die Helizität
mit geringer Verzerrung bei und wiesen mit dem Kontrollpeptid vergleichbare
Rückgrat-Diederwinkel
auf; die χ1- und χ2-Winkel der gefesselten Glutamine veränderten
sich während
der Simulation nicht. Peptide, die auf einer 5-Methylenbrücke basierten,
entkamen der helikalen Konformation vorübergehend und bildeten verschachtelte
Drehungen um den I+5-Rest. Mehrere Seitenketten-Rotamere wurden
ebenfalls im I+7-Rest gefunden. Ausgehend von diesen Beobachtungen
wurde bestimmt, dass die 4-Methylenbrücke die bevorzugte Fessellänge bereitstellen
würde.
-
Synthese und Charakterisierung
-
Aminosäuresequenzen
für Testpeptide
basierten auf der C-terminalen Helix von Apamin (E. Habermann und
K.G. Reiz, Biochem. Z 343, 192–203
(1965); G.L. Callewaert, R. Shipolini und C.A. Vernon, FEBS Lett.
1, 111–113
(1968); R. Shipolini, A.F. Bradbury, G.L. Callewaert und C.A. Vernon,
Chem. Commun. 679–680
(1967)) und auf dem S-Peptid, das vom C-Peptid von RMAse A abgeleitet
ist (J.E. Brown; W.A. Klee, Biochemistry 10, 470–476 (1971)). Die Sequenzen
dieser Peptide sind in der nachstehenden Tabelle 1 dargestellt.
-
Tabelle
1. Strukturen der Peptide 1–4
-
Die
linear geschützten
Peptide 1a, 1f und 2a wurden durch herkömmliche Merrifield-Verfahren unter Verwendung
von t-Butyloxycarbonyl-(BOC-) Chemie synthetisiert. Die Kontrollpeptide
1b und 2b wurden aus 1f und 2a durch gleichzeitige Entschützung beider
Glutamatreste nach einer Kupplung mit Methylamin hergestellt (1).
Die Synthese von 1d aus 1f durch doppelte Entschützung und Kupplung mit 1,4- Butandiamin ergab
geringe Ausbeute/Reinheit. Die eingeschränkten Peptide 1c–e und 2c–e wurden
aus 1a und 2a durch Entfernung des Fluorenylmethylesters aus Glu3,
Verbindung mit dem geeigneten Alkandiamin, Entfernung des Allylesters
aus Glu10 und Zyklisierung erhalten (1). Die
Ausbeuten wurden durch den Einsatz von einfach BOC-geschütztem Alkandiamin
im ersten Kupplungsschritt und durch den Einsatz eines Polystyrolharzes
mit 2 % Divinylbenzol-(DVB-) Vernetzer verbessert. Die vollständigen Peptide
wurden vom Harz mit Flusssäure (HF)
abgespalten und durch präparative
Hochleistungs-Flüssigchromatographie
(HPLC) gereinigt. Der Einbau der Fessel in die feste Phase erlaubte
die Vervollständigung
der Synthese mit nur einer einzigen Reinigung.
-
Die
auf Thiolysin basierenden Peptide 3a und 4a wurden unter Einsatz
herkömmlicher
Merrifield-Verfahren und FMOC-Chemie, gefolgt von der Abspaltung
vom Harz mit Trifluoressigsäure/Triethylsilan
(9:1 Vol./Vol.) und Reinigung durch präparative HPLC in der linearen,
Acetamidomethyl-geschützten
Form hergestellt. Diese wurden durch gleichzeitige Entschützung und
Oxidation mit Essigsäure
und molekularem Iod in Trifluorethanol in die Disulfidformen 3b
und 4b in Lösung übergeführt.
-
Die
Peptide 1–4
wurden durch Massenspektrometrie und durch quantitative Aminosäureanalyse
charakterisiert. Alle Peptide ergaben Ergebnisse, die mit den gewünschten
Strukturen übereinstimmten.
-
Geschütztes D-
(7) und L-Thiolysin (10) wurden wie in 2 dargestellt
hergestellt. Schöllkopf-Reagens
(U. Schöllkopf,
U. Groth, C. Deng, Angew. Chem. Int. Ed. Engl. 20, 798–799 (1981))
wurde mit n-Butyllithium gefolgt von 1,4-Dibrombutan behandelt,
um das bekannte Brombutylpyrazin 5 zu erhalten. Das Bromid wurde
mit dem Kaliumsatz von Methoxytoluolthiol ersetzt, um 6 zu erhalten.
Das Pyrazin wurde mit wässriger Salzsäure (HCl)
hydrolysiert, und das Thiol wurde mit Quecksilberacetat (Hg(OAc)2) in TFA, gefolgt von H2S entschützt. Der
rohe Thiolysinethylester wurden dann erneut mit Acetamidomethanol
in TFA geschützt.
Der Ester wurde mit Lithiumhydroxid (LiOH) hydrolysiert, und die
freie S-geschützte
Aminosäure
wurde mit Fmoc-N-Hydroxysuccinirnid in Dioxan N-geschützt, um
7 zu erhalten. Die gleichen Verfahren wurden für die Synthese von 10 eingesetzt.
-
Protonen-NMR
-
Die
Peptide 1–4
wurden durch 2D-
1H-NMR untersucht. Resonanzpositionen
wurden durch herkömmliche
Sequenzzuordnungsverfahren erhalten (K. Wüthrich, NMR of Proteins and
Nucleic Acids, Wi1ey, N.Y. (1986)) und sind in der nachstehenden
Tabelle 2 angeführt. Tabelle
2. Chemische Verschiebungen
a (Rückgrat-Kopplungskonstanten
b) der Apaminsequenzpeptide 1 und 3
- a Chemische Verschiebungen wurden bei pH
4,5 und 5°C
erhalten. Verschiebungen beziehen sich auf die interne H2O-Resonanz bei 4,96 Teilen pro Million (ppm)
und sind auf ±0,01
ppm genau.
- b 3JHN-Hα sind
in Klammern in Hertz-Einheiten (Hz) angegeben.
-
Repräsentative
TOCSY- und ROESY-Spektren eines diamideingeschränkten Peptids (1c) sind in
den 10 und 3 dargestellt.
Eine Zusammenfassung der HN-HN-
und Hα-HN-ROEs zwischen benachbarten Resten, die
für diese
Zuordnungen verwendet wurden, sind in 4 für 1b und
1c dargestellt. Der Grad der Helizität der einzelnen Peptide wurde
anhand dieser Spektren aufgrund der Gegenwart von starken sequentiellen HN-HN-ROE-Kreuzpeaks,
der Gegenwart von I-, I+3-Hα-HN-
oder -Hα-Hβ-ROE-Kreuzpeaks und einer 3JHN-Hα unter 6,0 Hz beurteilt.
Die in 4 zusammengefassten Daten zeigen,
dass das Peptid 1c zwischen den Resten Asn2 und Gln10 helikal ist.
Unter Gln10 steigt 3JHN-Hα über 6,0
Hz, aber gewisse ROEs im mittleren Bereich sind immer noch vorhanden,
was auf einen teilweisen oder vorübergehenden helikalen Charakter
hinweist. Solch ein Ausfransen an den Helixtermini tritt bei NMR-Untersuchungen von
Peptiden und Proteinen häufig
auf. Die chemischen 1H-Verschiebungen von 1c verändern sich
um weniger als 0,02 über
den Konzentrati onsbereich von 8,0–0,06 mM; dies zeigte, dass
die helikale Konformation nicht durch einen Eigenassoziationsvorgang
stabilisiert war.
-
Der
Einbau der Diamidvernetzung in die Peptide 1 und 2 verringerte den
Mittelwert von 3JHN-Hα in
der eingeschränkten
Region deutlich, steigerte die Anzahl an beobachtbaren (I,I+3)-ROES
und erhöhte
die durch Zirkulardichroismus bestimmbare prozentuelle Helizität, wie in
der nachstehenden Tabelle 3 dargestellt ist. Somit waren die Peptide
1c–1e
und 2c–2e
deutlich stärker
helikal als die Kontrollpeptide 1b und 2b. Die Ergebnisse für Peptid
4 zeigten, dass die Ausbildung der Disulfidbindung das Peptid helikal
eingeschränkte.
Ein Reihe von ROEs im mittleren Bereich konnte jedoch nicht beobachtet
werden, und die 3JHN-Hα Werfe
für die
beiden Thiolysin-Reste und für
Leu9 in 4b lagen über
6,0 Hz; dies wies auf eine Verzerrung der helikalen Struktur im Bereich
des D-Thiolysin-Rests hin, was aus einfachen Strukturüberlegungen
zu erwarten war. Die Daten in der nachstehenden Tabelle 3 zeigen,
dass der Einbau der Disulfidbindung in das Peptid 3b keinen helikalen
Charakter verlieh, was vermuten lässt, dass das Thiolysinverfahren
von der Primärsequenz
abhängt
und folglich nicht allgemein einsetzbar ist.
-
Tabelle
3. Evaluierung der Peptidhelizität
-
Werte
unter 6 für
die mittlere 3-Bindungs-NH-Na-Kopplungskonstante weisen auf Helizität hin. I-I+3-ROEs
im mittleren Bereich sind als beobachteter Anteil der Gesamtanzahl
an solchen ROEs, die möglich ist,
angegeben, wobei jedes schwache ROE halb gezählt wurde. Die durch CD bestimmte
prozentuelle Helizität
wurde wie von Lyu et al., J.C. Shermanm, A. Chen, N.R. Kallenbach,
Proc. Natl. Acad. Sci. U.S.A. 88, 5317–5320 (1991) und W.C. Johnson,
I. Tinoco Jr., J. Am. Chem. Soc., 94, 4389–4390 (1972) beschrieben erhalten.
-
Das
Peptid 1c wurde für
eine detailliertere Analyse durch NMR ausgewählt. ROESY-Spektren mit höherer Empfindlichkeit (erhöhte Gesamtaufzeichnungsdauer
und Peptidkonzentration) und NOESY-Spektren wurden erhalten und
analysiert, um Eingabeeinschränkungen
für Strukturberechnungen
bereitzustellen. Neben den oben beschriebenen ROEs wurden Hα-NN-(I,I+4)-Wechselwirkungen beobachtet, was
darauf hinweist, dass die angenommenen helikale Konformation nicht
vom 310-Typ sondern eine reguläre α-helikale
Varietät
ist (K. Wüthrich,
NMR of Proteins and Nucleic Acids., Wiley, New York, UASA (1986)).
Interprotonen-Abstandseinschränkungen
wurden aus den ROESY- und NOESY-Daten abgeleitet und als Basis für die Berechnung
einer Struktur für
1c unter Verwendung von Distanzgeometrie (DG) und eingeschränkter molekularer
Dynamik (rMD) verwendet. Nahezu die Hälfte (66) der 141 Einschränkungen
lag zwischen zwei bis vier Reste voneinander entfernten Aminosäuren in
der Primärsequenz,
was für
eine helikale Konformation zu erwarten war. Diederwinkeleinschränkungen,
die auf den erhaltenen 3JHN-Hα und 3JHN-Hβ basierten, wurden ebenfalls
in diesen Berechnungen verwendet, aber explizite Wasserstoffbrückenbindungseinschränkungen
wurden nicht eingesetzt.
-
Die
letztendendliche Zusammensetzung von 20 Strukturen ist in 5 dargestellt.
Die Strukturen stimmten gut mit den Eingabedaten überein,
ohne Verletzung der Distanzeinschränkung über 0,1 Ångström (Å), ohne Verletzung des Diederwinkels über 1,0° und mit
einem mittleren Einschränkungsverletzung-Energieterm
von 0,10 ± 0,09
Kilokalorien/Mol (kcal·mol–1).
Die verfügbaren
NMR-Daten definierten die Rückgratatome der
Reste Thr1 bis Gln10 gut (quadratisches Mittel der Abweichung von
der mittleren Struktur: 0,38 ± 0,08 Å), aber
die beiden C-terminalen Glutaminreste sind nicht gut definiert.
Die Seitenketten von Thr1, Gln3, Asp4, Leu6 und Gln10 weisen gut
definierte χ1-Werte auf, aber nur Gln1 wies einen übereinstimmenden
Wert für χ2 in allen Strukturen auf.
-
H
N(I)-O(I-4)-Wasserstoffbrückenbindungen zu den Amidprotonen
von Leu5, Ala6 und gln10 waren in mehr als 90 % der Strukturen vorhanden,
was darauf hinweist, dass diese Reste eine vorwiegend α-helikale Konformation
annahmen. Obwohl (i,i-4)-Wasserstoffbrückenbindungen
für die
Amidprotonen von Ala7, Arg8 und Arg9 in etwa 50 % der Strukturen
erkennbar waren, waren H
N(I)-O(I-3)-Wasserstoffbrückenbindungen
in 25–35
% der Strukturen vorhanden, was darauf hinweist, dass in diesem
Bereich eine leichte Verzerrung der Helix gegeben war. Die in der
nachstehenden Tabelle 4 angeführten
Daten zeigen, dass in Peptid 1c die Amidwasserstoffe von Leu5 bis
Gln10 alle vor Austausch mit einem Lösungsmittel geschützt waren,
im Vergleich zum Kontrollpeptid 1b mit Faktoren von bis zu 25. Diese
Beobachtung stimmt auch damit überein,
dass die Amidwasserstoffe dieser Reste an den Wasserstoffbrückenbindungen
beteiligt sind. Interessanterweise waren Wasserstoffbrückenbindungen
von Asp4-H
N bis Thr1-O
γ2 in
80 % der Strukturen vorhanden, was darauf hinweist, dass eine N-Kappen-Wasserstoffbrückenbindungswechselwirkung
(E.T. Harper, G.D. Rose, Biochemistry 32, 7605–7609 (1993)) sogar in diesem
kurzen Peptid vorhanden war. Das Amidproton von Asp4 war jedoch
nicht merklich vor Austausch geschützt (Tabelle 4), sodass diese
Wasserstoffbrückenbindung
von eher vorübergehendem
Charakter sein könnte. Tabelle
4. Amidwasserstoff-Austauschgeschwindigkeitskonstanten
a und
Schutzfaktoren
b für Peptid 1b und 1c
- a Die Geschwindigkeitskonstanten
sind in der Einheit Sekunden–1 (s–1)
angegeben. n.b. bedeutet, dass der Austausch schnell genug stattfand,
dass kein Peak im NMR-Spektrum
erkennbar war, das 300 Sekunden (s) nach dem Zusatz von D2O erhalten wurde. Unter der Annahme, dass
mehr als 90 % der Wasserstoffe in 300 s ausgetauscht wurden, ist
in diesen Fällen
die Berechnung einer Untergrenze von 0,013 s–1 (lokg
k = –1,89)
für die Geschwindigkeitskonstante
möglich.
- b Schutzfaktoren werden berechnet als
Geschwindigkeitskonstante für
Peptid 1c, dividiert durch jene für Peptid 1b.
-
Mit
Ausnahme der ψ-Winkel
von Ala6 und Gln10 lagen die Rückgrat-Diederwinkel
in der gesamten gefesselten Region nahe dem für eine ideale α-Helix erwarteten
(mittlerer ϕ = –63° ± 8°, mittlerer ψ = –42° ± 8°), was darauf
hinweist, dass Abweichungen vom Idealwert sehr gering waren. Der ψ für Ala6 war
15° niedriger als
für eine α-Helix erwartet und
der für
eine 310-Helix erwarteten ähnlicher;
der höhere
Wert von ψ für Gln10 spiegelte
das Ausfransen außerhalb
der gefesselten Region wider. Die leichte Verzerrung bei Ala6 könnte das Ergebnis
der kürzeren
Fessel sein, die in diesem Peptid vorhanden ist (nur drei Methylengruppen).
Obwohl die Diamidbindung durch die NMR-Daten nicht gut definiert
war, nahmen die Seitenketten von Gln3 und Gln10 Konformationen an,
die den von den oben beschriebenen Modellierungsexperimenten ähnelten
(Gln3 χ1 = –173° ± 17°; χ2 = 34° ± 47°; Gln10 χ1 = –71° ± 7°, χ2 = 174° ± 22°). Die Gesamtschlussfolgerung
war, dass 1c in einer Lösung
eine α-helikale
Struktur von Asp1 bis Gln10 mit einer N-terminalen Capping-Box und
einer sehr leichten Verzerrung in der zentralen Umkehrschleife der
Helix annahm.
-
Zirkulardichroismus (CD)
-
CD-Spektren
wurden mit wässrigen
Lösungen
von 1–4
mit zwischen 20 und 120 Mikromol/Liter (μM) bei 280 K, pH 5 erhalten.
Die Spektren der Peptide 1 und 3 (Apaminsequenz) sind in 8 dargestellt,
und jene der Peptide 2 und 4 (C-Peptidsequenz) in 8.
Die Zahlenwerte für
die prozentuelle Helizität,
die aus der molaren Elliptizität
pro Rest für
die Peptide bei 222 Nanometern (nm) berechnet wurden, sind in der
obigen Tabelle 3 angeführt.
-
Die
CD-Daten stützen
die aus den NMR-Untersuchungen abgeleiteten Schlussfolgerungen.
Beide Fesselungsverfahren steigerten die Helizität der C-Peptidsequenz deutlich
(8). Aber nur das Diamidverfahren konnte die Apaminsequenz
unter den eingesetzten Bedingungen helikal machen; das Thiolysin-eingeschränkte Peptid
3b schien nicht helikal zu sein (7). Das
CD-Spektrum von 4b wies trotz der wesentlichen negativen Elliptizität bei 222
nm mehrere Merkmale auf, die auf einen geringeren Helizitätsgrad hinweisen
als bei 2c–2c:
die niedrige minimale Wellenlänge
in 4b war von 208 nm (ein typischer Wert für eine α-Helix) zu 204 nm verschoben,
und die erkennbare Schulter des 190-nm-Maximums war viel geringer
als bei 2c–2e.
-
Hitzedenaturierungsexperimente
wurden an den Apamin-basierten Peptiden 1b–1e durchgeführt. Im ersten
Experiment wurden CD-Spektren der Peptide 1b–1e in 10-°C-Intervallen von 7°C (280 K) bis 57°C (330 K)
aufgenommen. Angesichts der Tatsache, dass 1c–1e die Helizität in diesem
Temperaturbereich gut aufrechterhielten, wurden Spektren von 1c
bis zu 97°C
(370 K) aufgenommen, wo ein gewisser Helizitätsverlust zu beobachten war
(6). Die molare Elliptizität von 1c bei 97°C und 22
nm war immer noch wesentlich stärker
negativ als die des nichthelikalen Kontrolipeptids 1b bei 7°C und 222
nm.
-
Experimente
zur Untersuchung der Auswirkungen von Erhitzen und Wiederabkühlen der
Peptide waren aufgrund mehrerer Faktoren kompliziert: Das CD-Spektrometer
zeigte bei längeren
Expreimenten eine Verschiebung der Grundlinie; die Konzentration
der Proben veränderte
sich aufgrund der Verdampfung bei höheren Betriebstemperaturen;
und es schien zu einer gewissen Schwankung des Probenverhaltens
in Abhängigkeit
von Aufheiz- und Abkühlungsgeschwindigkeit
zu kommen. Eine Gruppe von CD-Spektren von 1c wurde vor, während und
nach dem Erhitzen auf 87°C
für einen
Tag aufgenommen. Die Auswirkung der Verschiebung der Grundlinie
wurde durch eine lineare Normalisierung der Spektren basierend auf
Utas reduziert. Die Auswirkung der Konzentrationsänderung
aufgrund von Probenverdampfung wurde durch Normalisierung des Spektrums
nach dem Erhitzen auf die gleiche Amplitude wie das Spektrum vor
dem Erhitzen bei einer Wellenlänge
(λ) von
204 nm korrigiert. Diese Wellenlänge
wurde als Punkt gewählt,
an dem eine α-Helix
und ein Zufallsknäuel
gleiche Beträge
zur Elliptizität
leisten, sodass eine Interkonversion eines Peptids zwischen diesen Konformationen
das Ausmaß der
Elliptizität
nicht beeinflusst. Die resultierenden Spektren sind in 9 dargestellt.
Die starke Übereinstimmung
der Kurvenform zwischen den Spektren vor und nach dem Erhitzen weisen
darauf hin, dass die meisten der oder alle helikalen Strukturen
beim Abkühlen
nach teilweiser Denaturierung durch Erhitzen auf 87°C wiedererhalten
wurden. Der kleine Unterschied in der Gesamtamplitude könnte auf
ein geringes Maß an
bleibender Denaturierung zurückzuführen sein
oder könnte
ein Überbleibsel
des Normalisierungsvorgangs sein. Dieses Experimente zeigt, dass
die α-Helix
von 1c gegenüber
relativ rauen Bedingungen stabil war, ein Merkmal, das ihre allgemeine
Nutzbarkeit verbessert.
-
Schlussfolgerung
-
Ein
neues Verfahren zur Einschränkung
von kleinen Peptiden auf eine α-helikale
Konformation wurde entworfen. Diese I- bis I+7-basierte Fessel ist
ein erfolgreiches allgemeines Verfahren zur Induktion von α-Helizität in kleinen
Peptiden und weist mehrere wünschenswerte
Merkmale auf. Erstens erlaubt es maximal mögliche Sequenzvariabilität. Jeder
Rest, außer
den beiden Fesselungsresten selbst, kann verändert werden. Zweitens liegt
die durch dieses Verfahren induzierte Helizität in wässriger Lösung bei Raumtemperatur (RT) nahe
100 %. Der Vergleich der helikalen Peptide 1c–1e mit dem nichthelikalen
Peptid 1b zeigt, dass die Helizität durch Einführung des
Linkers erreicht wird und keine Eigenschaft der Primärsequenz
ist. Drittens werden diese gefesselten Peptide durch herkömmliche
Festphasen-(Merrifield-) Chemie synthetisiert und erfordern lediglich
kostengünstige,
im Handel erhältliche
Reagenzien. Viertens kann das Verfahren sogar für Peptide eingesetzt werden,
die nur acht Reste lang sind. Fünftens
stellt es keine chemische Anforderungen in Bezug auf die Umgebung,
und es wurde gezeigt, dass es trotz Veränderungen der Temperatur- und
Pufferbedingungen gute Helizität
induziert. Dieses Verfahren ist allgemein nützlich für Untersuchen an biologisch
aktiven helikalen Regionen von Proteinen, für die experimentelle Untersuchung
von Helixbildung, -propagation und -stabilität und für physikalische organische
Experimente bezüglich
der Wechselwirkungen von helikalen Peptiden mit ihrer Umgebung.
-
BEISPIEL 2
-
Das
zyklisierte Peptid FNM(5)QQRRFY(6)ALH (11)
wurde unter Einsatz von Fmoc-Chemie nach herkömmlichen Festphasen-Vorschriften,
worin Fmoc-Glutamin säure-δ-(5-allyloxycarbonyl-1,5-diaminopentan)
(5) (wie nachstehend beschrieben synthetisiert) und Fmoc-Glutaminsäure-δ-allylester
(6) (im Handel von Millipore erhältlich)
als Standard-Aminosäuren
in die Peptidsynthese eingebaut wurden, gefolgt von einer Zyklisierung
synthetisiert, wie in 11 dargestellt ist. Fmoc-Glutaminsäure-δ-(5-allyloxycarbonyl-1,5-diaminapentan)
(5) wurde wie im nachstehenden Schema 1 dargestellt synthetisiert.
-
-
Mono-t-butyloxycarbonyl-(BOC-)
1,5-pentandiamin wurde unter Verwendung von 1,5-Diaminopentan (12,5
g, 122 mmol) anstelle von 1,3-Diaminopropan in der Synthese von
Monoallyloxycarbonyl-1,3-diaminopropan, wie in Beispiel 1 oben beschrieben,
synthetisiert, was 10 g (49 mmol) Mono-terf-butyloxycarbonyl-1,5-diaminopentan (1)
ergab. Mono-tert-butyloxycarbonyl-1,5-diaminopentan (1) (5,8 g,
28,7 mmol) wurde in 75 ml Dichlormethan mit 7,5 ml Diisopropylethylamin
gelöst
und auf 0°C
abgekühlt.
Ein Lösung
von Allylchlorformiat (3,3 ml) in Dichlormethan (25 ml) wurde über fünf Minuten
zugesetzt. Die Reaktion wurde ein Stunde lang auf Raumtemperatur
erwärmen
gelassen, und dass wurde das Lösungsmittel
durch Rotationsverdampfung entfernt. Der Rückstand wurde in 100 ml Ethylacetat
gelöst
und mit drei 100-ml-Portionen 10%iger Citronensäure, einmal mit 100 ml gesättigtem
wässrigem
Natriumbicarbonat und einmal mit 100 ml gesättigtem wässrigem Natriumchlorid gewaschen.
Die organische Phase wurde über
Magnesiumsulfat getrocknet, und das Lösungsmittel wurde durch Rotationsverdampfung
entfernt. Das resultierende Öl
(2) wurde 30 Minuten lang mit 25 ml Trifluoressigsäure behandelt.
Die Trifluoressigsäure
wurde durch Rotationsverdampfung entfernt, und der resultierende
Rückstand
wurde zweimal in Dichlormethan gelöst und dann eingedampft, um restliches
Lösungsmittel
zu entfernen. Der Rückstand
wurde in 50 ml 3 N Salzsäure
gelöst
und mit zweimal 50 ml Dichlormethan gewaschen. Die wässrige Phase
wurde in einem Eisbad gekühlt,
und der pH wurde mit 50%igem wässrigem
Natriumhydroxid auf etwa 13 eingestellt. Die basische wässrige Phase
wurde mit dreimal 100 ml Dichlormethan extrahiert, die vereinigten
organischen Phase wurden mit 100 ml gesättigtem wässrigem Natriumchlorid gewaschen
und dann über
Kaliumcarbonat getrocknet. Das Gemisch wurde filtriert und das Lösungsmittel
zuerst durch Rotationsverdampfung und dann im Hochvakuum entfernt,
um 3,95 g Monoallyloxycarbonyl-1,5-diaminopentane(3) als farbloses Öl zu erhalten.
-
Fmoc-Glutaminsäure-α-tert-butylester,
9,0 g (21,1 mmol, Bachem Calif., USA) wurde in 100 ml Dichlormethan
gelöst.
Dicyclohexylcarbodiimid (4,4 g, 21,3 mol) und N-Hydroxybenzotriazol
(0,3 g, 2,1 mmol) wurden zu dieser Lösung zugesetzt, gefolgt vom
Monoallyloxycarbonyl-1,5-diaminopentan (3) (3,95 g, 21,2 mmol).
Die Reaktion wurde 14 Stunden lang bei 25°C gerührt, dann über eine Stunde auf 0°C abgekühlt. Unlösliches
Material wurde abfiltriert, und das Filtrat wurde durch Rotationsverdampfung
eingeengt. Der Rückstand
wurde in 150 ml Ethylacetat gelöst
und zweimal mit 100 ml 10 % wässriger
Citronensäure,
zweimal mit 100 ml gesättigtem
wässrigem
Natriumbicarbonat und einmal mit 100 ml Kochsalzlösung gewaschen.
Nachdem das Ganze über
Magnesiumsulfat getrocknet und filtriert worden war, wurde das Lösungsmittel
durch Rotationsverdampfung entfernt. Der Rückstand wurde unter Erhit zen
in etwa 75 ml Ethylacetat gelöst,
und Nexan:Ethylacetat (2:1) wurde zugesetzt, bis die Lösung trüb wurde.
Nachdem das Ganze einige Stunden lang stehen gelassen worden war,
wurde der kristalline Niederschlag abfiltriert, und die weißen Kristalle
wurden mit Hexan:Ethylacetat (2:1) gewaschen und im Vakuum getrocknet,
um 11,4 g von (4) zu erhalten (90 %).
-
Der
tert-Butylester (4) (11 g, 18,5 mmol) wurde unter Rühren in
50 ml Trifluoressigsäure
gelöst.
Nach 45 Minuten wurde die Trifluoressigsäure durch Rotationsverdampfung
entfernt; die restliche Trifuoressigsäure wurde durch dreimaliges
Eindampfen aus 50 ml Dichlormethan entfernt. Der Rückstand
wurde unter Erhitzen in 75 ml Ethylacetat gelöst, durch Celite filtriert,
und Hexan:Ethylacetat (3:1) wurden zugesetzt, bis eine Trübung auftrat.
Kristalle wurden drei Stunden lang bei 25°C wachsen gelassen, wonach das
Ganze eine Stunde lang auf 0°C
abgekühlt
wurde. Die Kristalle wurden abfiltriert, mit Hexan:Ethylacetat (3:1)
gewaschen und dann im Vakuum getrocknet, um 9,5 g (95 %) von (5)
als grauweiße
Kristalle zu erhalten.
-
Nach
der Peptidsynthese von FNM(5)QQRRFY(6)ALH wurde der N-Terminus des
Festphasenpeptids an Mono-tert-butylbernsteinsäure gebunden, die Allyl- und
Allyloxycarbonyl-Schutzgruppen wurden unter Verwendung von 500 mg
Pd(PPh3)2Cl2 in 20 ml 20%igem Piperidin in Dimethylacetamid über 1,5
Stunden bei Raumtemperatur entfernt. Das Harz wurden dann mit 20%igem
Piperidin in Dimethylacetamid, Dimethylacetamid, Dichloromethan
und schließlich
mit 0,5%iger Trifluoressigsäure
in Dichloromethan gewaschen. Das Harz wurde in Dichloromethan suspendiert,
und 1,5 Äquivalente
HATU mit 3 Äqu.
N,N-Diisopropylethylamin in 5 ml Dimethylacetamid wurden zugesetzt.
Nach zwei Stunden wurden das Harz durch den Ninhydrintest auf freie
Amine untersucht und stellte sich als negativ heraus. Das Peptid
wurde mit 95% Trifuoressigsäure,
5% Triethylsilan vom Harz abgespalten und unter Einsatz von RP-HPLC gereinigt.
-
Die
helikale Struktur des zyklisierten Peptids, die in 11 dargestellt ist, wurde durch Zirkuiardichroismus
(CD) und Magnetresonanz (NMR) bestätigt. Beide dieser Verfahren
zeigten, dass das arretierte Helixpeptid vorwiegend α-helikalen
Charakter besaß.
Durch Mikrakalorimetrie und Oberflächen-Plasmonresonanz wurde
bestimmt, dass das arretierte Helixpeptid IgG mit einer Affinität (Kd) von
etwa 100 μM
band. Bin Kontrollpeptid ohne Arretierabschnitt des Moleküls wies
keine IgG-Bindung auf, die durch Mikrokalorimetrie nachweisbar wäre.
-
BEISPIEL 3
-
Um
zu bestätigen,
dass der kovalente Arretiermechanismus voll funktionsfähig ist
und dass durch dieses Verfahren arretierte Peptide in der Lage sind,
einen Liganden mit hoher Affinität
zu binden, wurde ein 33-Aminosäure-Peptid,
das auf Helix 1 der Z-Domäne von Protein
A basierte, wie im nachstehenden Schema 2 dargestellt mit der i-
bis i+7-Bindung synthetisiert. Schema
2
worin (5) und (6) die im obigen Beispiel 2 beschriebenen
Allyloxycarbonyl- und Allylgeschützten
Aminosäuren sind.
Das Peptid wurde wie im obigen Beispiel 2 beschrieben synthetisiert
und zyklisiert. Die Helizität
des Peptids wurde durch CD und NMR verifiziert, und die durch CD überwachte
Hitzedenaturierung des Peptids zeigte, dass sich das Peptid bei
90°C nur
teilweise auffaltet, was mit der Stabilität der kovalenten Bindung übereinstimmt.
Die IgG-Bindungsaffinität
(Kd) dieses Peptids (gemessen durch Oberflächen-Plasmonresonanz) wurde
mit etwa 20 nM bestimmt.
-
BEISPIEL 4
-
Von
der Ektodomäne
des HIV-1-Hüllproteins
gp41 abgeleitete lineare Peptide hemmen bekanntermaßen virale
Fusionsvorgänge.
Die stärksten
davon (DP178) entsprechen einer proximalen Membranregion von gp41,
von der vorhergesagt wird, dass sie α-helikal ist. DP178 fehlt jedoch
in Lösung
eine erkennbare Struktur, was eine mechanistische Interpretation
seiner Aktivität äußerst schwierig
macht. Unter Anwendung der hierin gelehrten Helixarretierungschemie
wurden eingeschränkte
Versionen von DP178 hergestellt, um zu bestimmen, ob die Helizität für seine
Hemmwirkung der Infektiosität
notwendig oder ausreichend ist, und um eins wahrscheinliche Wirkungsweise
dieses Moleküls
bei einer Primärinfektion
zu definieren (gemessen durch Virusinfektiositätstests).
-
Durch
Einschränkung
von DP178-Analoga auf eine helikale Konformation haben die Erfinder
gezeigt, dass die Helizität
notwendig, aber nicht ausreichend ist, um Hemmwirkung bereitzustellen.
Die korrekte Fläche der
Helix muss auch exponiert sein. Zwei neuere Kristallstrukturen von
gp41 zeigen, dass diese Fläche
in einer Furche verborgen ist, die durch ein Superhelixtrimer gebildet
wird. Zusammen genommen zeigen diese Ergebnisse, dass DP178 die
Infektiosität
durch Blockierung dieser Furche hemmt und dass die durch Kristallographie bestimmte
Konformation von gp41 den fusogenen Zustand darstellt.
-
Eine
Reihe von Analoga von DP167, worin Segmente des Amidrückgrats
helikal eingeschränkt
waren (12), wurde vorbereitet. Da
kurze α-Helices üblicherweise
in Lösung
unstrukturiert sind (Marqusee et al., Proc. Natl. Acad. Sci. USA
86, 5286–5290
(1989)), wurde eine kovalente Vernetzung zwischen den Aminosäure-Seitenketten an den
Positionen i und i+7 der Polypeptidkette, wie hierin gelehrt (siehe
auch Phelan et al., J. Am. Chem. Soc. 119, 455–460 (1997), das durch Verweis
hierin aufgenommen ist), bereitgestellt, welche die dazwischen liegenden
Reste in einer stabilen α-helikalen
Konformation arretiert.
-
Eine
trunkierte Form von DP178, die als HIV35 (12)
bezeichnet wird, wurde als Referenz verwendet. In Ermangelung detaillierter
Informationen bezüglich
der Assoziation von DP178 mit DP107 wurde die Neigung zur Bildung
einer Superhelix (Lupas et al., Science 252, 1162–1164 (1991))
für 29
verschiedene gp160-Sequenzen berechnet, um zu bestimmen, ob die
DP178 entsprechende Region als Superhelix zählt. Die N-terminalen 27 Reste
von DP178, die für
das Referenzpeptid HIV35 ausgewählt
wurden, behielten einen hohen Gesamtpunktewert mit einer übereinstimmenden
Heptaden-Anordnung bei. Die „a–d"-Fläche, die
anhand der Punktealgorithmen vorhergesagt wurde, entsprach der Fläche, die
sich an den Trimerkern anlagerte. Diese entsprechende Region ist
in der Röntgenstruktur
vollkommen helikal und wird unter Verwendung eines mit jenen in
Superhelices verwandten 4-3-Heptaden-Repeats an den Trimerkern angelagert.
Unter Einsatz der hierin gelehrten Helixarretierungschemie und -verfahren
haben die Erfinder die Exposition dieses Repeats (Positionen „a" und „d" der Heptade) durch
Einführung
von Vernetzungen zwischen Paaren von benachbarten Resten auf der
gegenüberliegenden
Fläche
der Helix (Position „f") verstärkt. Somit
wurden Arretierungen (Fesseln) zwischen „f" und „f" gebildet, um die mögliche Helix einzuschränken. Analoga
von HIV35 (12) wurden hergestellt, die
entweder eine (HIV24) oder zwei (HIF31) Fesseln enthielten, um gesteigerte
Helizität
bereitzustellen. Ein Kontrollpeptid (HIV30) wurde hergestellt, worin
eine Fessel zwischen aufeinander folgenden „d"-Resten eingeführt wurde, um die Helizität zu stabilisieren,
während
mögliche
Bindungswechselwirkungen über
die „a–d"-Fläche blockiert
wurden.
-
Lineare
Peptide wurden gemäß herkömmliche
Festphasenverfahren unter Einsatz von Fmoc-Chemie synthetisiert
(Fields et al., Int. J. Peptide Protein Res. 35, 161–214 (1990)),
wie dies hierin gelehrt wird. Genauer gesagt wurden Helixdipoleffekte
minimiert, indem die C-Termini als Amide und die N-Termini als Succinatgruppen
blockiert wurden. Nach der Bildung der Lactam-Brücken, wie hierin gelehrt (siehe
auch Phelan et al., J. Am. Chem. Soc. 119, 455–460 (1997)), wurden die Peptide
vom Harz abgespalten und unter Einsatz von präparativer RP-HPLC mit Wasser/Acetonitril/0,1%-TFA-Gradienten
in der mobilen Phase bis zur Homogenität gereinigt. Die Identität der einzelnen
Peptide wurde durch Elektrospray- Massenspektrometrie
bestätigt: HIV-24,
berechnete Masse 3396,8, gefunden, 3396,0; HIV-30, berechnete Masse
3413,7, gefunden, 343,8; HIV-31, berechnete Masse 3520,0, gefunden,
3520,7; HIV-35, berechnete Masse 3330,8, gefunden, 3330,5.
-
Zirkulardichroismusanalyse
(13) bestätigt,
dass die Arretierungsstrategie die Nelizität der DP178-Trunkierungen deutlich
erhöht.
CD-Spektren wurden mit einem CD-Spektrometer
AVIV 62DS unter Einsatz von Küvetten
mit einer Weglänge
von 0,05 cm aufgenommen. Die Spektren wurden durch Mittelung der
Daten von drei Durchgängen
von 250 nm bis 190 nm mit Steigerungen von 1,0 nm und einer Mittelungsdauer
von 2 Sekunden bei jeder Wellenlänge
erhalten. Die Peptidkonzentrationen betrugen etwa 200 μM in einer
Lösung
von 10 mM Tris-HCl, pH 7,5 mit 6 Vol.-% Acetonitril. Zur Umrechnung
der Rohdaten in molare Elliptizitätswerte wurden genaue Konzentrationen
durch Messung von A276 und A280 (Edelhoch,
Biochemistry 6, 1948–1954
(1967)) bestimmt; diese Werfe wurden durch quantitative Aminosäureanalysen
bestätigt.
-
Das
nichteingeschränkte
Peptid HIV35 zeigte ein praktisch nichtssagendes Spektrum, ähnlich wie
das für
DP178 berichtete (Lawless et al., Biochemistry 35, 13697–13708 (1996)).
Die CD-Spektren der Peptide mit einer einzelnen Einschränkung (HIV24
und HIV30) wiesen Minima bei 209 und 222 nm auf, die charakteristisch für α-Helices
waren. Die Intensitätsverhältnisse
zwischen diesen beiden Regionen weichen vom Idealwert ab, was vermuten
lässt,
dass Regionen des Peptidrückgrats
außerhalb
des eingeschränkten
Segments fehlgeordnet sind. Im Gegensatz dazu scheint das doppelt
eingeschränkte
Analog HIV31 größtenteils
helikal zu sein, wie durch CD bestimmt wurde, der die Form und das
Intensitätsprofil
einer typischen α-Helix ergab.
-
Virusinfektiositätstest wurden
verwendet, um die helikal eingeschränkten Konstrukte zu charakterisieren.
Normale menschliche Peripherblutzellen (PBMCs) wurden 24 Stunden
lang mit Phytohämagglutinin (PGHA)
in RPMI-1640-Medium, das Interleukin 2 enthielt, stimuliert. Das
PHA-Medium wurde entfernt, und die Zellen wurden über Nacht
in RPMI 1640 mit Glutamin, 20 % hitzeaktiviertem fötalem Kälberserum
und Gentamicin gezüchtet.
Zu Beginn des Tests wurden vorgetiterte Virus-Stammlösungen eine
Stunde lang mit Peptiden äquilibriert,
bevor sie zu den PBMCs zugesetzt wurden (2,5 × 105 Zellen
pro Well). Die Zellen wurden drei Tage lang gezüchtet, zur Entfernung von extrazellulären Viren
und Peptiden gespült
und dann mit frischem Medium ergänzt
und weitere vier Tage lang gezüchtet.
Nach sieben Tagen wurden die Zellen lysiert, und p24-Antigen wurden
durch ELISA bestimmt. Die Peptide wurden in jeder Konzentration
dreimal durchlaufen gelassen. Virustiter wurden für jeden
Durchgang doppelt bestimmt. Jeder Test umfasste außerdem die
folgende Kontrolle in dreifacher Ausführung: nichtinfizierte Zellen
als negative Kontrolle, infizierte Zellen ohne Peptid als positive Kontrolle
und Virus-Inokulum ohne Zellen zum Erhalt eines p24-Grundlinienwerts.
Die Peptide wurden auf Zytotoxizität getestet, indem sie in der
höchsten
Testkonzentration (etwa 100 μM)
mit nichtinfizierten Zellen inkubiert und dann die oben beschriebenen
Zeilen gezüchtet
wurden. Nach 7 Tagen wurden die Zellzahlen mithilfe von Mikroskopie
geschätzt
und mit einer identischen Charge von Zellen verglichen, die nicht
mit dem Peptid behandelt worden war; keines der Peptide hemmte normales
Zellwachstum unter diesen Bedingungen.
-
Als
sie den Virusinfektiositätstests
unterzogen wurden wiesen die Peptide ein auffallendes Muster relativer
Wirksamkeit auf, das sich sowohl über Syncitium-induzierende (SI-)
als auch Nicht-Syncitium-induzierende (NSI-) Stämme von HIV-1 erstreckten (Zhang
et al., Nature 383, 768 (1996)), Wie in den 14A und 14B dargestellt führte eine Trunkierung des hydrophoben
C-Terminus von DP178 (HIV35) zu einem dramatischen Rückgang der
Aktivität,
die teilweise wiederhergestellt wurde, als eine einzelne Einschränkung, d.h. ein
eingeschränktes
helikales Peptid (mit teilweise α-helikalem
Charakter) eingeführt
wurde (HIV24). Der Zusatz einer zweiten Einschränkung (HIV31) verlieh starken
helikalen Charakter und steigerte die Wirksamkeit des Peptids auf
Werte, die mit DP178 vergleichbar waren. Somit startete die zusätzliche
Stabilisierung, die durch eine vorherige Anordnung von HIV31 in
einer aktiven helikalen Konformation verliehen wird, den Verlust der
Bindungsenergie durch Deletion des C-Terminus. Im Gegensatz dazu
nahm eine einzelne Einschrän kung, die
Helizität
induzierte und gleichzeitige die „a–d"-Fläche
blockierte (HIV30), die Aktivität
vollkommen weg.
-
Eine
Reihe von kürzeren
eingeschränkten
Peptiden, welche die Positionen 631–644, 643–656, 649–662, 656–669 und 663–678 von
HIV-1LAI überspannen, die zwischen benachbarten
Resten an den „f"-Positionen der Heptade
gefesselt sind, wurden vorbereitet, um zu bestimmen, ob eine Untergruppe
von HIV35 oder seine N- und C-terminalen
flankierenden Regionen ausreichten, um die Infektiosität zu blockieren.
Alle Peptide, egal ob eingeschränkt
oder nichteingeschränkt,
zeigten keine signifikante Aktivität. Die Peptide 631–644 enthalten
den hydrophoben Cluster, von dem in der Röntgenstruktur nachgewiesen
wurde, dass er sich in einen Hohlraum im Trimerkern anlagert (Chan
et al., Cell 89, 263–273
(1997)).
-
Die
relativen Aktivitäten
von HIV35, HIV24 und HIV31 zeigen eine klare Korrelation zwischen
Helizität und
Hemmwirkung auf. Die äußerst unterschiedlichen
Aktivitäten
von HIV30 und HIV24 zeigen, dass die Peptidhemmung auch die Exposition
der Fläche
der Helix erfordert, die sich, wie durch Kristallographie nachgewiesen
wurde, an den N-terminalen Trimerkern von gp41 anlagert.
-
Die
hierin präsentierten
Daten stützen
zusammen mit den vorangegangenen Modellstudien an isolierten Peptiden
und den kürzlich
veröffentlichten
Kristallstrukturen die Hypothese, dass die Peptide Virusinfektiosität durch
Bindung an die Ruheform von gp41 hemmen und die Bildung des fusogenen
Zustands verhindern. Das Peptid HIV31 ist konformationell so eingeschränkt, dass
es größtenteils
helikal ist, und die Wahrscheinlichkeit ist hoch, dass es mit einer
zugänglichen
zugehörigen
Qberfläche
in der Ruheform von gp41 wechselwirkt. Da eine Röntgenanalyse zeigt, dass die
Fläche
von HIV31, die zur Hemmung der Virusfusion erforderlich ist, in
der durch den N-terminalen Trimerkern gebildeten Furche verborgen
ist, glauben die Erfinder (ohne sich auf eine bestimmte Theorie
festzulegen), dass diese Furche die zugehörige Oberfläche für die Peptide darstellt.
-
15 ist eine schematische Darstellung eines derzeitigen
Modells zur Anordnung des fusogenen Zustands von gp41 und des Mechanismus,
durch den die eingeschränkten
Helices diesen Vorgang hemmen. Das Modell ist in keiner Weise als
Einschränkung
der Erfindung zu verstehen und soll die Erfinder auf keine bestimmte
Theorie zur Umsetzung der Erfindung einschränken. Die Ruheform von gp41
(oben links) wird vermutlich konstitutiv trimerisiert, wobei ein
Superhelixbündel
nahe dem N-terminalen
Fusionspeptid vorhanden ist (Pfeil). Die Region, die dem C-Terminus
der Ektodomänen
entspricht (dunkle Linien), ist anfangs nicht an das Trimerbündel gebunden
und weist eine unbekannte Konformation auf. Eine Konformationsverschiebung,
die aus der Bindung von gp120 an entweder CD4, einen Corezeptor
oder beides resultiert, kann dann eine Assoziation des C-terminalen
Abschnitts von gp41 mit dem N-terminalen Bündel erlauben. Die resultierende
antiparallele helikale Anordnung (oben rechts), die in den Röntgenstrukturen
erkennbar ist, ist vermutlich der fusogene Zustand von gp41. Eine
Umordnung in diesen Zustand kann blockiert werden, wenn die Trimerfurchen durch
Hemmpeptide besetzt sind (unten links). Im Falle einer Blockierung
auf diese Weise würde
eine nachfolgende Konformationsverschiebung im gp41-Cluster das
Protein aus dem Weg heraus maskieren (unten rechts).
-
Die
Peptide DP178 und HIV24 hemmen die Infektiosität von genetisch entfernten
und phänotypisch unterschiedlichen
Subtypen von HIV-1 (Gao et al., Journal of Virology 70, 1651–1667 (1996)).
Außerdem
ist die Oberfläche,
von der vorgeschlagen wird, dass sie daran binden, eine der höchstkonservierten
Regionen im HIV-1-Genom.
Die Erfinder haben DP178 gegen andere Stämme getestet und herausgefunden,
dass es ähnlich
starke Hemmwirkung gegen den im Labor angepassten Stamm MN/H9 und
die primären
Isolate 301660 und Th009 aufwiest. Der Stamm Th009 ist vom Subtyp
E und genetisch vom vorherrschenden nordamerikanischen Subtyp B
(z.B. JRCSF) (Zhang et al., Nature 383, 768 (1996)). Diese Ergebnisse
stimmen mit Beobachtungen aus anderen Laboratorien überein (Wild
et al., Prac. Natl. Acad. Sci. USA 89, 10537–10541 (1992); Wild et al.,
Proc. Natl. Acad. Sci. USA 91, 9770–9774 (1994); Jiang et al.,
Nature 365, 113 (1993)). Neben JRCSF und BZ167 haben die Erfinder
auch HIV24 gegen Th009 getestet und herausgefunden, dass es vergleichbare
Stärke
aufweist, was darauf hinweist, dass der vorgeschlage ne Membranfusionsmechanismus
auf sehr unterschiedliche Stämme
von HIV-1 zutrifft.
-
Andere
Mittel, wie beispielsweise Antikörper,
die auf diese Oberfläche
abzielen, könnten
somit viel versprechend für
die therapeutische Behandlung von AIDS sein.
-
BEISPIEL 5
-
Um
eine Vakzine herzustellen, die gegen eine HIV-Infektion wirksam
wäre, entweder
als Prophylaktikum oder als Therapeutikum nach einer Infektion (gegebenenfalls
in Kombination mit Anti-HIV-Medikamenten oder anderen Untereinheitenvakzinen),
wurden eingeschränkte α-helikale
Peptide aus der 633-678-Region von gp41 hergestellt und als Immunogene
verwendet.
-
Varianten
von HIV 24 mit der Sequenz "Gly
Gly Cys" am C-Terminus
oder "Cys Gly Gly" am N-Terminus wurden
hergestellt. Diese Peptide wurden unter Verwendung eines heterobifunktionellen
Vernetzers, wie z.B. 4-(N-Maleimidomethyl)cyclohexan-1-carbonsäure-3-sulfo-N-nydroxysuccinimidester,
der von Sigma verfügbar
ist, oder eines Äquivalents
davon (z.B. „Sulfo-MBS" von Pierce) an KLH
konjugiert. Immunisierungen wurden wie nachstehend beschrieben vorgenommen.
-
Polyklonale
Antikörper
gegen KLH-konjugierte HIV-Peptide wurden in weiblichen Meerschweinchen hergestellt
(Hartley-Stamm von Simonson Labs). Fünfzig μg Peptid in 250 μl PBS wurden
mit 250 μl
Freundschem Adjuvans (komplettes Adjuvans für die Primärinjektion und inkomplettes
Adjuvans für
alle Auffrischungen) emulgiert. Injektionen von 70–100 μg Peptid/kg
Körpergewicht
wurden über
eine Kombination aus subkutanen und intramuskulären Steilen in einem Dreiwochenzyklus
verabreicht. In der zweiten und dritten Woche wurde nach der jeweiligen
Auffrischung Blut abgenommen.
-
Die
Seren von immunisierten Tieren wurden auf eine Protein-A-Säule geladen,
um nach einer Elution gereinigtes Gesamt-Ig bereitzustellen. Für die arretierten
Helices selektive Antikörper
wurden erhalten, indem der Gesamt-Ig-Pool über eine Affinitätssäule laufen
gelassen wurden, die einen mit immobilisierten arretierten Helices
beladenen Träger
enthielt. Dieser Träger
wurde hergestellt, indem zuerst die oben beschriebenen cysteinhältigen Peptide
(HIV 2, 27, 28 und 29) mit Biotin-Maleinimid (ebenfalls von Sigma;
N-Biotinyl-N'-[6-maleinimidohexanoyl]hydrazid)
umgesetzt wurden, um Peptide zu erhalten, die an einem Terminus
biotinyliert waren. Diese Peptide wurden auf ein Harz geladen, das
mit Streptavidin (Pierce, "Ultralink
Avidin") vorbeladen worden
war, um das beschriebene Affinitätsgel
bereitzustellen.
-
Der
Gesamt-Ig-Pool aus der Protein-A-Säule wurde über die geeignete Affinitätssäule laufen
gelassen (d.h. jene mit dem passenden immobilisierten Hapten). Nichtspezifische
Antikörper
wurden im Durchfluss eluiert und als negative Kontrollen aufbewahrt.
Spezifische Antikörper
wurden aus der Protein-A-Säule
eluiert, in einen Testpuffer dialysiert und gelagert.
-
Überraschenderweise
waren die erhaltenen Antikörpertiter
für gp41-Untereinheitenpeptide
ziemlich hoch. Dies ist vor allem überraschend, weil diese Region
von gp41 (633–678)
auf dem Gebiet der Erfindung nicht dafür bekannt ist, HIV-neutralisierende
Antikörper
zu bilden.
-
Die
affinitätsgereinigten
polyklonalen Antikörper
werden dem Virusinfektiositätstest
unterzogen, der zur Beurteilung der Peptide eingesetzt wird. Die
zur Erzeugung von polyklonalen Antikörperpräparaten, welche die Infektiosität hemmen,
verwendeten Haptene sind zur Verwendung in einer Vakzine wünschenswert.
Am meisten bevorzugt sind Kandidaten, die zu breit kreuzreaktiven
Antikörpern
führen,
welche in der Lage sind, verschiedene HIV-1-Isolate in vitro zu
neutralisieren.
-
HIV-1-Vakzinen-Kandidaten
können
in verfügbaren
Tiermodellen getestet werden, beispielsweise in den von Berman et
al., J. Virol. 7, 4464–9
(1992); Haigwood et al., J. Virol. 66, 172–82 (1992) und Salmon-Ceron
et al., AIDS Res. and Human Retroviruses 11, 1479–86 (1995)
beschriebenen Schimpansen für
gp120-Untereinheitenvakzinen. Am meisten bevorzugt sind Kandidaten,
die zur breit kreuzreaktiven Anti körpern führen, welche in der Lage sind,
unterschiedliche HIV-1-Isolate in diesen Tierstudien zu neutralisieren
und so Schutz bei einer Provokation mit homologen und heterologen
Stämmen
von HIV-1 bereitstellt. Der erfolgreiche Schutz von Schimpansen
ist ermutigend und hat sich in der Geschichte als verlässlicher
Indikator für
die Wirksamkeit einer Vakzine bewährt.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-



















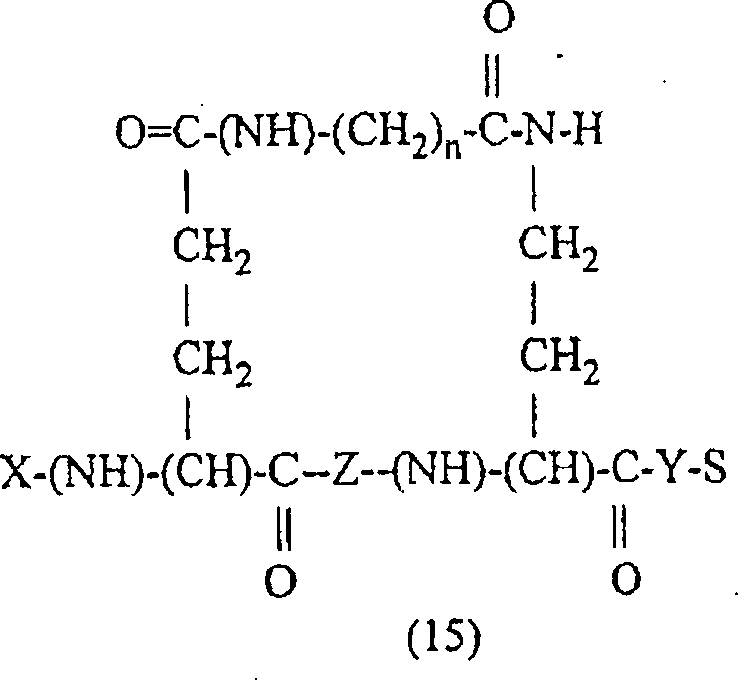































































































































































































































































 worin S nicht vorhanden ist oder ein Makromolekül ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Z eine beliebige Aminosäuresequenz bestehend aus sechs Aminosäuren ist; q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist, mit der Maßgabe, dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze Zahl im Bereich von (7-(q + s)) bis (9 – (q + s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als 0 ist; Verbindung der Formel (11):
worin S nicht vorhanden ist oder ein Makromolekül ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Z eine beliebige Aminosäuresequenz bestehend aus sechs Aminosäuren ist; q aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt ist, s aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist, mit der Maßgabe, dass q + s kleiner als oder gleich 7 ist, und r eine beliebige ganze Zahl im Bereich von (7-(q + s)) bis (9 – (q + s)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass r größer als 0 ist; Verbindung der Formel (11): worin S nicht vorhanden ist oder ein Makromolekül ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Z eine beliebige Aminosäuresequenz bestehend aus sechs Aminosäuren ist; t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt ist, mit der Maßgabe, dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze Zahl im Bereich von (7 – (t + v)) bis (9 – (t + v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als 0 ist; und Verbindung der Formel (16):
worin S nicht vorhanden ist oder ein Makromolekül ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Z eine beliebige Aminosäuresequenz bestehend aus sechs Aminosäuren ist; t aus den ganzen Zahlen von 0 bis 6, Grenzen eingeschlossen, ausgewählt ist und v aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt ist, mit der Maßgabe, dass t + v kleiner als oder gleich 7 ist, und u eine beliebige ganze Zahl im Bereich von (7 – (t + v)) bis (9 – (t + v)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass u größer als 0 ist; und Verbindung der Formel (16): worin S nicht vorhanden ist oder ein Makromolekül ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Z eine beliebige Aminosäuresequenz bestehend aus sechs Aminosäuren ist; w und y unabhängig voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt sind, mit der Maßgabe, dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze Zahl im Bereich von (7 – (w + y)) bis (9 – (w + y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als oder gleich 0 ist.
worin S nicht vorhanden ist oder ein Makromolekül ist, X Wasserstoff oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Y nicht vorhanden ist oder Hydroxyl ist, wenn S nicht vorhanden ist, oder eine beliebige Aminosäure oder Aminosäuresequenz ist, Z eine beliebige Aminosäuresequenz bestehend aus sechs Aminosäuren ist; w und y unabhängig voneinander aus den ganzen Zahlen von 1 bis 7, Grenzen eingeschlossen, ausgewählt sind, mit der Maßgabe, dass w + y kleiner als oder gleich 8 ist, und x eine beliebige ganze Zahl im Bereich von (7 – (w + y)) bis (9 – (w + y)), Grenzen eingeschlossen, ist, mit der Maßgabe, dass x größer als oder gleich 0 ist.