CN1217706A - 多孔碳材料及其制造方法 - Google Patents
多孔碳材料及其制造方法 Download PDFInfo
- Publication number
- CN1217706A CN1217706A CN97194425A CN97194425A CN1217706A CN 1217706 A CN1217706 A CN 1217706A CN 97194425 A CN97194425 A CN 97194425A CN 97194425 A CN97194425 A CN 97194425A CN 1217706 A CN1217706 A CN 1217706A
- Authority
- CN
- China
- Prior art keywords
- carbon
- compound
- treatment
- porous carbon
- producing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 title claims abstract description 111
- 229910052799 carbon Inorganic materials 0.000 title claims abstract description 104
- 239000000463 material Substances 0.000 title claims abstract description 7
- 238000000034 method Methods 0.000 title claims description 20
- 150000001722 carbon compounds Chemical class 0.000 claims abstract description 71
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 52
- 239000001257 hydrogen Substances 0.000 claims abstract description 52
- 238000001179 sorption measurement Methods 0.000 claims abstract description 43
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 38
- 239000007789 gas Substances 0.000 claims abstract description 33
- 238000010438 heat treatment Methods 0.000 claims abstract description 29
- 238000004519 manufacturing process Methods 0.000 claims abstract description 29
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 28
- 150000002367 halogens Chemical class 0.000 claims abstract description 28
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 191
- 238000011282 treatment Methods 0.000 claims description 115
- 229910052757 nitrogen Inorganic materials 0.000 claims description 91
- 239000003575 carbonaceous material Substances 0.000 claims description 58
- 238000005658 halogenation reaction Methods 0.000 claims description 55
- 230000026030 halogenation Effects 0.000 claims description 53
- 238000000197 pyrolysis Methods 0.000 claims description 45
- 239000000460 chlorine Substances 0.000 claims description 39
- 239000002994 raw material Substances 0.000 claims description 37
- 239000003990 capacitor Substances 0.000 claims description 29
- 239000005011 phenolic resin Substances 0.000 claims description 23
- 238000005695 dehalogenation reaction Methods 0.000 claims description 19
- 239000011261 inert gas Substances 0.000 claims description 16
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 12
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 12
- 229910052794 bromium Inorganic materials 0.000 claims description 12
- -1 hydrogen compound Chemical class 0.000 claims description 12
- 235000013162 Cocos nucifera Nutrition 0.000 claims description 11
- 244000060011 Cocos nucifera Species 0.000 claims description 11
- 229920005989 resin Polymers 0.000 claims description 11
- 239000011347 resin Substances 0.000 claims description 11
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 claims description 9
- 239000007849 furan resin Substances 0.000 claims description 9
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 238000010000 carbonizing Methods 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 6
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 5
- 239000000203 mixture Substances 0.000 claims description 4
- 230000002140 halogenating effect Effects 0.000 claims description 3
- 239000011257 shell material Substances 0.000 claims description 2
- 238000003763 carbonization Methods 0.000 abstract description 10
- 239000003245 coal Substances 0.000 abstract description 4
- 238000000926 separation method Methods 0.000 abstract description 4
- 238000005660 chlorination reaction Methods 0.000 description 21
- 230000000052 comparative effect Effects 0.000 description 18
- 239000008188 pellet Substances 0.000 description 18
- KXGFMDJXCMQABM-UHFFFAOYSA-N 2-methoxy-6-methylphenol Chemical compound [CH]OC1=CC=CC([CH])=C1O KXGFMDJXCMQABM-UHFFFAOYSA-N 0.000 description 16
- 229920001568 phenolic resin Polymers 0.000 description 16
- 239000011230 binding agent Substances 0.000 description 14
- 230000000694 effects Effects 0.000 description 14
- 230000031709 bromination Effects 0.000 description 13
- 238000005893 bromination reaction Methods 0.000 description 13
- 239000011148 porous material Substances 0.000 description 12
- 239000011342 resin composition Substances 0.000 description 11
- 239000010426 asphalt Substances 0.000 description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- 239000003463 adsorbent Substances 0.000 description 9
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- 229910001873 dinitrogen Inorganic materials 0.000 description 6
- 239000008187 granular material Substances 0.000 description 6
- 238000010298 pulverizing process Methods 0.000 description 6
- 239000007772 electrode material Substances 0.000 description 5
- 150000002431 hydrogen Chemical class 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 230000004584 weight gain Effects 0.000 description 5
- 235000019786 weight gain Nutrition 0.000 description 5
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 238000005469 granulation Methods 0.000 description 4
- 230000003179 granulation Effects 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 239000013081 microcrystal Substances 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 150000002240 furans Chemical class 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 239000007791 liquid phase Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000006298 dechlorination reaction Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000012983 electrochemical energy storage Methods 0.000 description 2
- 239000003077 lignite Substances 0.000 description 2
- 239000003915 liquefied petroleum gas Substances 0.000 description 2
- 229940057995 liquid paraffin Drugs 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- 229910014033 C-OH Inorganic materials 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 229910014570 C—OH Inorganic materials 0.000 description 1
- 229910014569 C—OOH Inorganic materials 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- RHZUVFJBSILHOK-UHFFFAOYSA-N anthracen-1-ylmethanolate Chemical compound C1=CC=C2C=C3C(C[O-])=CC=CC3=CC2=C1 RHZUVFJBSILHOK-UHFFFAOYSA-N 0.000 description 1
- 239000003830 anthracite Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 238000005899 aromatization reaction Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000011280 coal tar Substances 0.000 description 1
- 239000000571 coke Substances 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000010439 graphite Substances 0.000 description 1
- 229910002804 graphite Inorganic materials 0.000 description 1
- 238000005087 graphitization Methods 0.000 description 1
- 230000003450 growing effect Effects 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 239000012433 hydrogen halide Substances 0.000 description 1
- 229910000039 hydrogen halide Inorganic materials 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 238000006068 polycondensation reaction Methods 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 238000005979 thermal decomposition reaction Methods 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B38/00—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof
- C04B38/0022—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof obtained by a chemical conversion or reaction other than those relating to the setting or hardening of cement-like material or to the formation of a sol or a gel, e.g. by carbonising or pyrolysing preformed cellular materials based on polymers, organo-metallic or organo-silicon precursors
- C04B38/0025—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof obtained by a chemical conversion or reaction other than those relating to the setting or hardening of cement-like material or to the formation of a sol or a gel, e.g. by carbonising or pyrolysing preformed cellular materials based on polymers, organo-metallic or organo-silicon precursors starting from inorganic materials only, e.g. metal foam; Lanxide type products
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/02—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising inorganic material
- B01J20/20—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising inorganic material comprising free carbon; comprising carbon obtained by carbonising processes
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/515—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics
- C04B35/52—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics based on carbon, e.g. graphite
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B38/00—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B38/00—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof
- C04B38/0051—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof characterised by the pore size, pore shape or kind of porosity
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/32—Carbon-based
- H01G11/34—Carbon-based characterised by carbonisation or activation of carbon
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/13—Energy storage using capacitors
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Ceramic Engineering (AREA)
- Organic Chemistry (AREA)
- Materials Engineering (AREA)
- Structural Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Power Engineering (AREA)
- Dispersion Chemistry (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Analytical Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Carbon And Carbon Compounds (AREA)
- Electric Double-Layer Capacitors Or The Like (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
在对通过干馏碳化合物所得干馏碳实施卤化处理制造多孔碳材料的方法中,使用氢含量为0.1-5.0重量%的干馏碳,或将碳化合物在250-1200℃下干馏得到的干馏碳,或在对碳化合物实施卤化处理制造多孔碳材料的方法中,该碳化合物是原料碳化合物或/和将碳化合物在250℃以下加热的碳化合物,通过以上制造方法可以获得作为气体分离用吸附材料,具有比过去更优良的吸附性能,和电化学能量贮存性能更优良的多孔性碳材料。
Description
技术领域
本发明是关于制造具有能吸附氮和氧等小分子的微孔和/或亚微孔结构多孔碳材料的方法,可应用于工业玻璃精制或分离用吸附剂、双电荷层电容器等电极材料的碳材料。
本说明书是根据向日本专利局提出的专利申请(特愿平9-2421),该日本申请中记载的内容作为一部分内容撰写入本说明书内。
背景技术
(碳材料的原料)
作为碳材料的原料,可利用褐煤·次褐煤·无烟煤·焦炭·木炭和椰壳炭等动植物物质的碳化物,在惰性气体下加热酚醛树脂·呋喃树脂·偏氯乙烯树脂等各种树脂的生成物,等等。这些物质也应用于本发明中。本发明中把块状原料碳化合物、原料碳化合物的粉碎处理物、进一步将其成型处理物、等等碳化前的原材料,简单地统称为碳化合物。将碳化合物进行干馏得到的物质称为干馏碳。
(碳材料的用途)
由于碳材料在化学上是惰性的,虽然广泛用作吸附剂、催化剂、电极材料、机械用材料等等。但这些用途都与碳材料的结构和密集情况息息相关。
被称作多孔碳的碳,具有发达的细孔,所以也就具有不同的特殊作用。例如,利用吸附现象,进行分离和精制混合物,或用作双电荷层电容器用碳和锂二次电池用碳等贮存电化学能量的媒体。
(碳材料的构造)
碳材料的结构,根据原料或制造方法可获得各种不同的结构。
对炭等赋予活化而得到的活性炭,是由除去了微晶碳(微晶)、链状结构的碳等所形成。难以石墨化的碳、去除了微晶杂乱叠层的结构,在这些微晶的间隙中形成了由微孔到大孔的广大范围的细孔。
微晶是数层平行的碳六元环网面重叠累积成的物质,构成碳六元环的石墨碳是利用SP2混杂轨道结合而成。将碳六元环形成网面叫作基本面。
易于石墨化的碳,通过高温下加热微晶增长,最终形成石墨。
难以石墨化的碳和完全没进行石墨化的易于石墨化的碳,通常含有未组织化的碳。所谓未组织化的碳,是除只和石墨碳进行化学结合的石墨碳之外的其它碳,称作具有链状结构的碳、附着在碳六元环周边上的碳、在碳六元环的最外缘(棱柱面)上的碳、使碳六元环(微晶)彼此形成交联结构的碳。在未组织化的碳中,以C-H、C-OH、C-OOH、C=O等形式与氢原子、氧原子等相结合或形成碳的双键(-C=C-)。
将孔径为0.8nm以下的细孔称作亚微孔,将0.8~2nm范围的细孔称作微孔。这些范围内的细孔的大小具有和吸附分子和离子大小大致相同的量级,可以认为这些细孔的结构与气体分子的吸附和离子的吸藏现象和密集息息相关。
(碳吸附剂)
作为用于气体分离的碳吸附剂一般使用的活性炭,以活化法制造,作为老技术,在特开平1-242409号公报(水蒸汽活化法)、特开平5-132377号公报(二氧化碳气活化法)中有所公开。作为由空气分离氮气所使用的碳分子筛的制造方法,在特公昭52-18675号等公报中有所公开。
(双电荷层电容器)
双电荷层电容器的电极,也称作极化性电极,要求具有很大的静电容量。在双电荷层上储蓄的电容量(静电容量),大概取决于固液界面的表面积,作为电极材料,可以使用比表面积大,具有导电性的碳材料。特别是活化处理的活性炭供于实际应用。作为双电荷层电容器用的碳电极老技术,已知在特公平4-70770号公报、特公昭63-10574号公报、特开平4-175277号公报、特开平1-321620号等公报中有所公开。
在以往的多孔碳材料制造方法中,所利用的吸附剂是以氮、氧等分子直径小的气体为对象,吸附容量很不充分。用作双电荷层电容器等电池的电极材料时,电化学能量的贮存容量也不充分。
发明的公开
本发明的目的是提供一种碳孔碳材料,作为气体分离用吸附剂,具有比老产品更优良的吸附性能,而且具有优良的电化学能量贮存性能。
本发明的多孔碳材料的制造方法,其特征是对碳化合物进行干馏所得干馏碳实施卤化处理,制造多孔碳材料的方法中,该干馏碳中氢含量为0.1~5.0重量%。
或者,本发明的多孔碳材料的制造方法,其特征是对碳化合物进行干馏所得干馏碳,实施卤化处理,制造多孔碳材料的方法中,干馏碳是将碳化合物在250~1200℃下进行干燥得到的干馏碳。
或者,本发明的多孔碳材料的制造方法,其特征是对碳化合物实施卤化处理,制造多孔碳材料的方法中,该碳化合物是将原料碳化合物或/和碳化合物在250℃以下进行加热,得到的碳化合物。
本发明的多孔碳材料,其特征是由上述任何一种制造方法所制造,在25℃,1个大气压下的氮吸附量为12~20cm3/g。
或者,本发明的多孔碳材料,其特征是由上述任何一种制造方法所制造,双电荷层电容器的静电容量为30~90F/cm3。
附图的简述
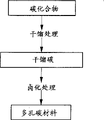
图1是本发明多孔碳材料的制造方法工序示意图。
图2是本发明多孔碳材料的另一制造方法工序示意图。
图3是制造本发明碳材料装置的简要示意图。
图4是测量静电容量的装置简要断面图。
图5是比较例和实施例的碳,对氮吸附量的比较图。
图6是比较例和实施例的碳,对静电容量的比较图。
实施本发明的最佳方案
本发明的特征是将碳化合物和对碳化合物进行干馏所得干馏碳任何一种实施卤化处理,以制造多孔碳材料。
在本发明的第1个实施方案中,其特征是如图1所示,将碳化合物进行干馏处理,对所得干馏碳实施卤化处理制造多孔碳材料,在此多孔碳材料的制造方法中,是对氢含量为0.1~5.0重量%的干馏碳实施卤化处理。
本发明的第2个实施方案中,其特征是如图1所示,对碳化合物进行干馏处理,对所得干馏碳实施卤化处理,以制造多孔碳材料,在此多孔碳材料的制造方法中,是对在250~1200℃下进行干馏的干馏碳实施卤化处理。
本发明的第3个实施方案中,其特征是如图2所示,对碳化合物实施卤化处理,在此多孔碳材料的制造方法中,该碳化合物是原料碳化合物或/和将原料碳化合物在250℃下加热的碳化合物(以下也称之为碳化合物)。
(原料碳化合物)
本发明可以使用各种形式的原料碳化合物,最好使用椰壳、酚醛树脂、呋喃树脂、偏氯乙烯树脂、沥青。当原料碳化合物的形状过大时,由于干馏或氧化难以均匀,且迅速地进行到内部,所以不理想。当原料碳化合物大小约10mm以上时,最好粗粉碎到约2~4mm后,再进行干馏处理。原料碳化合物为高分子树脂时,最好通过热交联实施固化处理后,再粉碎。原料碳化合物的粗粉碎方法,可使用早已公知的方法。当原料碳化合物的形状小于0.1mm时,在干馏或氧化处理中,由于这种原料不便于处理,所以要加入粘合剂进行造粒,例如,制成直径2mm,长4~5mm的颗粒(小圆柱状)。
(干馏处理)
干馏是从碳化合物中除去氧和氢,进行碳化的处理。作为干馏方法,最通行的方法是在氮气和氩气等惰性气体中进行加热的方法。
在本发明的第1个实施方案中,为充分呈现卤化处理的效果,干馏碳的氢含有率(干馏碳中的氢的量(g)对于干馏碳(g)的百分率)为0.1~5.0重量%,最好为0.15~3.0重量%。当不足0.1重量%时,氢和卤素的取代不充分,因此,没有充分呈现卤化处理效果。当超过5.0重量%时,卤化处理也难以获得理想效果。
在本发明的第2个实施方案中,干馏处理的温度为250~1200℃,最好300~1150℃,更好500~1100℃。当干馏温度低于250℃时,碳化不充分,不足500℃的卤化处理温度下,得不到卤化处理效果,也不能并行产生干馏效果。当在超过1200℃下进行干馏时,碳化会过度进行,残留氢的量不能保持在0.1重量%,而且,由于碳的石墨结构增加,未组织化碳减少,不能充分产生卤化反应,也就不能充分获得卤化处理效果。
干馏作业中惰性气体的供给量(空塔速度),最好为0.05~0.3L/cm2·分(L大约为大气压、室温下的气体体积。cm2为反应炉的截面积。以下相同)。
本发明的第1或第2实施方案中,供卤化处理的碳(干馏碳),依照碳化合物的碳化过程,有经由固相得到的碳,当然也有经由液相得到的碳,也包括在气相中热分解析出的碳。
将原料的碳化合物和原料碳化合物,在250℃以下的温度下进行加热的试料中,最好在500℃以上的温度下,在含卤素的惰性气体中,对其进行加热处理时,干馏和卤化同时产生,从而获得本发明的效果(本发明的第3个实施方案)。
(干馏的作用)
干馏可以以固相,也可以以液相进行。原料碳化物中所含的氢、碳或氧原子,在比较低的干馏温度下,挥发出低分子的石蜡、烯烃或芳香烃,随着温度的增高,释放出二氧化碳、一氧化碳和甲烷,或氢。在残存的固相或液相中产生碳键的分解、环化、芳香化、缩聚反应,生成石墨结构的碳。
在本发明的第1和第2实施方案中,由于卤化处理是干馏碳中未组织化碳和卤素反应,所以卤化的程度取决于未组织化碳的量。未组织化碳的量取决于干馏的程度,即,由干馏温度决定的。因此,卤化处理的效果依赖于干馏温度。本发明的第3个方案中,由于干馏和卤化同时产生,所以卤化的程度取决于卤化处理温度。
(造粒处理)
原料的碳化合物,例如,大小在0.1mm以下,为了以后容易进行处理,而加入粘合剂,最好形成颗粒。但是,0.1mm仅是示例,并不限定于此。
粘合剂可使用酚醛树脂、沥青、煤焦油等。为使造粒或分型性很好进行,也可以添加乙醇或液体石蜡。粘合剂的添加量过多时,会降低卤化处理的效果。过少时,进行造粒会降低它的物理强度。例如,在本发明的第1和第2实施方案中,对于100重量份干馏碳,或在本发明第3实施方案中,对于100重量份碳化合物,以酚醛树脂+溶剂+液体石蜡的总合计,最好添加30~60重量份。
将干馏碳或碳化合物进行造粒后,再将其干燥,干燥的目的是将颗粒加热到140~200℃,最好150~180℃,除去粘合剂中所含的低沸点挥发成分,增强颗粒的强度,以适宜后续的干馏。
将粘合剂加入到粉末干馏碳或粉末碳化合物中,进行造粒,干燥的颗粒,为使粘合剂碳化,最好接着进行再次干馏处理。再次干馏处理是在氮气等惰性气体中进行加热的处理。再次干馏处理的条件,最好是在和干馏处理原料的碳化合物相同的条件下进行。
(卤化处理)
在本发明的第1和第2实施方案中,对干馏处理碳化合物得到的干馏碳,或本发明第3实施方案中,将原料碳化合物或将原料碳化合物在250℃下加热的碳化合物实施卤化处理。
这种卤化处理包括卤化处理和脱卤处理。
卤化处理是通过将干馏碳或碳化合物在卤素或含卤素的惰性气体中进行加热,使卤素进行反应的处理。作为卤素可以使用所有的卤素,但最好使用氯或溴。
卤化处理干馏碳时的加热温度为350~1000℃,最好是450~800℃。在低于350℃下,必须延长反应时间,效率降低,超过1000℃,进行干馏,由于氢原子减少,导致卤化反应难以进行。对原料碳化合物或在250℃下对原料碳化合物进行加热的碳化合物,或者它们的混合物,进行卤化处理时,最好500~1000℃,更好600~800℃。
卤化处理时的卤素浓度,例如,氮气中的卤素气的浓度最好为1~50容量%,更好是3~30容量%,但并不仅限于此。对含卤素气体的空塔速度没有特殊限制,最好是0.05~0.3L/cm2·分。
脱卤处理,可以是(a)高温脱卤处理或(b)低温脱卤处理中任何一种,或者在(a)处理后再进行(b)处理,或者在(b)处理后进行(a)处理。
高温脱卤处理是在氮气或氩气等惰性气体中,或真空排气下,对卤化处理碳进行加热的处理。真空排气时的真空度最好是10mmHg。加热温度最好600~1300℃,650~1200℃更好。在低于600℃下,伴随着脱卤的进行,碳原子的结合或排列的反应很不充分。在超过1300℃下,热收缩的效果过大,对细孔构造的形成产生恶劣影响。虽然对惰性气体的供给量没有特殊限制,最好是0.05~0.3L/cm2·分。
低温脱卤处理是在氢化合物(含氢原子的化合物)或含氢化合物的惰性气体中,对卤化处理碳进行加热的处理。作为氢化合物,可以使用水蒸汽,或甲烷、乙烷、乙烯、丙烷、丙烯、丁烷、丁烯等低级烃类,或氢、或它们的混合气。工业上也适当地使用液化石油气(LPG)没有完全燃烧时的燃烧排气。
氢化合物的浓度,水蒸汽时,为2~10容量%,低级烃时,为5~50容量%,氢时,为10~60容量%。低温脱卤处理时气体的供给量,最好为0.05~0.3L/cm2·分。
低温脱卤处理的温度,氢化合物是水蒸汽或低级烃时,最好为600~850℃。在低于600℃下,处理时间增长,超过850℃时,水蒸汽有时产生活化,低级烃时有碳析出,不理想。氢化物为氢时,最好是600~1200℃。低于600℃时,处理时间增长,超过1200℃时,由于热收缩的作用,对细孔构造的形成产生恶劣影响。
(卤化处理的作用)
当使卤气与干馏碳或碳化合物接触时,卤气和未组织化碳进行反应。这些反应有,对碳的双键进行卤素加成反应,与未组织化碳结合的氢原子和卤原子的交换反应(产生和卤素等摩尔的卤化氢),脱氢反应等。在卤化反应和脱卤反应中产生多种形式的反应,作为代表性的反应,是以下式表示的反应,
(但是,附与C旁边的·,表示未组织化的碳)推测是由新的碳原子和碳原子的结合(碳键)形成的物质。
上述这种反应和,新形成的碳键,对碳网面或微晶的石墨结构缺陷起到修复作用,对微晶起到增长的作用,对于改变微晶的集合状态起作用,作为结果,可以推测,能很好地吸附小分子,硫酸离子等电介质离子适宜地形成双电荷层,能形成很多的微孔和/或亚微孔。
根据本发明可以获得用于氮气吸附量为12~20cm3/g的吸附剂、静电容量为30~90F/cm3的双电荷层电容器的碳材料。
实施例
以下借助实施例明确说明本发明的作用和效果,但以下实施例不过是示例而已,本发明并不仅限于以下实施例。
粉碎处理
利用日陶科学(株)制小型振动球磨机NB-O将干馏碳进行粉碎。粉碎程度可通过调节粉碎时间来进行。在粗粉碎原料碳化合物时,使用粗粉碎机粉碎到约2~5mm大小。
造粒处理
使用圆盘造粒机(不ニパゥダル(株)制PV-5型)进行造粒。以下所示实施例中,不进行特殊切断的情况下可制造出2mmΦ×5~6mm的颗粒。
干馏、卤化处理的设备
图3中简要示出了实施本发明时,干馏或卤化处理的设备。图中,标号1为加热炉、2为石英管、3为具有耐热性和通气性的搁板(碳材料的承载器)、4为碳材料、5为氮气供应管、6为卤化处理时卤素、水蒸汽的供应管、7为排气管、8为橡胶栓。加热炉具有能控制所需温度的功能。各种气体的供入压力大致为大气压。气体供应量可用浮子流量计测定。
卤化处理
在下述实施例中,没有特殊记载时,都按如下方式进行卤化处理。
进行氯处理时,在含10容量%氯气的氮气流(1L/分)下,于600℃对干馏碳试料(5~15g)进行加热1小时,使其氯化。接着,将氯化干馏碳,在氮气流(2L/分)下,于900℃,加热30分钟,进行处理,再在含25℃饱和水蒸汽的氮气流(1L/分)下,于700℃进行加热处理30分钟,脱除氯。另外,也进行了将原料碳化合物在含卤素气体的惰性气体中进行加热,实施氯化处理,然后进行脱卤素处理的实验。
溴处理时,除了在含8容量%溴气的氮气流下进行3小时溴化处理外,其它和氯处理一样。
卤化处理时的重量变化(%),以(卤化处理后的碳重量(g)÷原碳重量(g))×100表示。实际卤化处理时,伴随着碳化进行,重量同时减少,有时重量变化形成负值(以下用-号表示)。
吸附量的测定
以下实施例中氮吸附量的测定,利用容量法(机器:日本ベル(株)制ベルソ-プ28),在25℃、1个大气压下进行。测定前,将试料在100℃下进行2小时真空脱气。下述实施例的吸附量是25℃、1个大气压下的吸附气体体积。
静电容量的测定
将卤化处理的碳进行粉碎,使平均粒径约为9μm,加入30重量%的硫酸水溶液形成浆糊状,将其装入氯乙烯树脂制的型具(直径25mm、厚2mm)中,制备二个,再将它们以聚丙烯制的隔离片隔开,并相对重合,从两侧插入铂制的集电极,制作成图4所示的静电容量测定元件。图中标号11为碳电极、12为垫圈、13为集电极、14为隔离片。静电容量C(F)是在充电后,以一定电流I(A)进行放电,测定电压由V1(V)降到V2(V)的时间Δt(s),按下式求出。
C=I×Δt/(V1-V2)
在本测定中,以900mV充电24小时后,再进行恒流放电(I-4mA/cm2),使电压从V1=540mV降到V2=360mV,测定此时所用的时间,求出静电容量。静电容量是每1cm3碳的值。
氢量的分析
将约3g碳材料在氧0.1L/分和氮0.9L/分的混合气流下,进行完全燃烧,将氢变换成水蒸汽,用五氧化二磷(P2O5)吸收管吸收,由重量变化求出水蒸汽的量,计算出氢的量。氢量对碳材料的重量(氢含有率)以重量%表示。
比较例1:酚醛树脂
将酚醛树脂(群荣化学工业(株)制PGA-4560)在约160℃下固化,并粉碎,将酚醛树脂作为粘合剂加工成颗粒。将这种颗粒约15g在氮气流(1L/分)加热30分钟进行干馏,试料①在600℃、试料②在800℃、试料③在1000℃、试料④在1100℃。这些试料的氮气吸附量分别是①5.2cm3/g、②10.4cm3/g、③11.2cm3/g、④10.8cm3/g。双电荷层电容器的静电容量分别是①7.7F/cm3、②16.1F/cm3、③14.4F/cm3、④11.8F/cm3。
比较例2:酚醛树脂
将酚醛树脂固化、粉碎,作为粘合剂添加酚醛树脂,加工成颗粒。将这种颗粒约15g在氮气流下(1L/分)加热30分钟进行干馏,试料①在300℃,试料②在400℃。将这些试料分别在含10容量%氯气的氮气流(1L/分)下,加热进行氯化处理。试料①在300℃温度下处理16小时,试料②在400℃温度下处理16小时。氯化处理时重量增加,①是-0.7重量%,②是-1.8重量%。将这些试料在氮气流(2L/分)下,于900℃加热,再在含25℃饱和水蒸汽的氮气流(1L/分)下,于700℃加热20分钟,进行脱氯。这些试料的氮气吸附量,①是10.9cm3/g、②是10.9cm3/g。双电荷层电容器的静电容量,①是15.3F/cm3、②是15.2F/cm3。
比较例3:椰壳
将粗粉碎2~5mm大小的椰壳(约15g)在氮气流下(1L/分),于500℃加热20分钟进行干馏。再将该干馏碳在氮气流(1L/分)下加热30分钟次再进行干馏,试料①在600℃,试料②在700℃,试料③在800℃,试料④在1000℃、试料⑤在1100℃。
这些试料的氮气吸附量分别是①4.7cm3/g、②5.6cm3/g、③7.2cm3/g、④9.1cm3/g、⑤7.6cm3/g。双电荷层电容器的静电容量分别是①3.7F/cm3、②11.3F/cm3、③12.9F/cm3、④9.5F/cm3、⑤6.0F/cm3。
比较例4:呋喃树脂
将呋喃树脂(日立化成工业(株)制VF-302)固化、粉碎,以酚醛树脂作粘合剂加工成颗粒。将这种颗粒约15g在氮气流下(1L/分)加热30分钟进行干馏,试料①在600℃、试料②在800℃、试料③在1000℃、试料④在1100℃。这些试料的氮气吸附量分别是①4.8cm3/g、②9.9cm3/g、③10.6cm3/g、④6.8cm3/g。双电荷层电容器的静电容量分别是①5.7F/cm3、②15.3F/cm3、③11.2F/cm3、④6.3F/cm3。
比较例5:偏氯乙烯树脂
将偏氯乙烯树脂(市售的卷材)在500℃下干馏30分钟,并粉碎,以酚醛树脂作粘合剂加工成颗粒。将这种颗粒约15g在氮气流下(1L/分)加热30分钟,进行再次干馏,试料①在600℃、试料②在800℃、试料③在1000℃、试料④在1100℃。这些试料的氮气吸附量分别是①7.4cm3/g、②10.6cm3/g、③11.0cm3/g、④10.8cm3/g。双电荷层电容器的静电容量分别是①6.6F/cm3、②15.8F/cm3、③14.1F/cm3、④11.1F/cm3。
比较例6:石油沥青
将石油沥青在250℃的干燥机内保持5小时,并进行加热实施氧化处理。将这种沥青约15g在氮气流下(1L/分)加热30分钟进行干馏,试料①在600℃、试料②在800℃、试料③在1000℃、试料④在1100℃。这些试料的氮气吸附量分别是①1.9cm3/g、②7.7cm3/g、③8.9cm3/g、④7.5cm3/g。双电荷层电容器的静电容量分别是①2.1F/cm3、②12.0F/cm3、③8.2F/cm3、④5.0F/cm3。
实施例1:酚醛树脂、氯化处理
将酚醛树脂固化、粉碎、作为粘合剂添加酚醛树脂加工成颗粒。分析氢量,结果是3.00重量%。将这种试料约15g在含10容量%氯气的氮气流(1L/分)下,于500℃加热1小时进行氯化处理。氯化处理时的重量增加为-14.7重量%。将这些试料在氮气流下(2L/分),于900℃加热,再在25℃的含饱和水蒸汽的氮气流(1L/分)下,于700℃加热20分钟,进行脱氯。
这种碳材料的氮气吸附量为14.0cm3/g。双电荷层电容器的静电容量为66.4F/cm3。在500℃的氯化温度下,对原料酚醛树脂进行氯化处理时,非常有效。干馏碳的氢量为3.00重量%是有效的,能获得很大值的氮吸附量、静电容量。
实施例2:酚醛树脂、氯化处理
将酚醛树脂固化、粉碎、作为粘合剂添加酚醛树脂,加工成颗粒状(试料①)。将这种试料约15g在氮气流(1L/分)下,加热30分钟进行干馏,而试料②在300℃、试料③在600℃、试料④在800℃、试料⑤在900℃、试料⑥在1000℃,试料⑦在1100℃。分析干馏碳中所含氢量,结果是①3.00重量%、②2.95重量%、③2.35重量%、④1.00重量%、⑤0.56重量%、⑥0.48重量%、⑦0.33重量%。
将这些试料分别在含10容量%氧气的氮气流(1L/分)下,于600℃加热1小时,进行氯化处理。氯化处理时的重量增加分别是①-21.3重量%、②-15.7重量%、③20.4重量%、④22.3重量%、④18.3重量%、⑥12.4重量%、⑦9.9重量%。将这些试料在氮气流下(2L/分)于900℃加热,再在25℃的含饱和水蒸汽的气流下(1L/分)、于700℃加热20分钟进行脱氯。
这些碳材料的氮气吸附量分别是①15.5cm3/g、②15.7cm3/g、③16.2cm3/g、④14.9cm3/g、⑤14.2cm3/g、⑥13.0cm3/g、⑦12.2cm3/g。这些试料的双电荷层电容器的静电容量分别是①71.1F/cm3、②73.2F/cm3、③76.6F/cm3、④73.0F/cm3、⑤65.8F/cm3、⑥53.0F/cm3、⑦40.5F/cm3。有效的干馏温度在300~1100℃之间,对原料酚醛树脂进行氯化处理时也非常有效。干馏碳的有效含氢量在3.00~0.33重量%之间,可获得很大值的氮吸附量、和静电容量。
实施例3:椰壳、氯化处理
将椰壳破碎成2-5mm(试料①),取约15g,在氮气流(1L/分)下,试料②在600℃、试料③在800℃、试料④在900℃、试料⑤在1000℃、试料⑥在1100℃下,加热30分钟,进行再次干馏。分析干馏碳中含氢量,结果是①2.80重量%、②2.01重量%、③0.77重量%、④0.54重量%、⑤0.47重量%、⑥0.35重量%,将这些试料分别在含10容量%氯气的氮气流(1L/分)下,于600℃加热1小时,进行氯化处理。氯化处理时的重量增加分别是①-65.0重量%、②27.9重量%、③25.3重量%、④18.3重量%、⑤7.3重量%、⑥3.9重量%,将这些试料在氮气流下(2L/分)于900℃加热,再在25℃的含饱和水蒸汽的氮气流(1L/分)下,于700℃加热20分钟进行脱氯。
这些碳材料的氮气吸附量分别是①14.3cm3/g、②15.3cm3/g、③14.2cm3/g、④13.9cm3/g、⑤13.0cm3/g、⑥12.1cm3/g。这些试料的双电荷层电容器的静电容量分别是①63.0F/cm3、②64.6F/cm3、③63.9F/cm3、④54.6F/cm3、⑤42.5F/cm3、⑥30.1F/cm3。有效干馏温度在600~1100℃之间,对原料椰壳进行氯化处理时也很有效。有效的含氢量为2.80~0.35重量%之间,可获得很大值的氮气吸附量和静电容量。
实施例4:呋喃树脂、氯化处理
将呋喃树脂硬化并粉碎,添加酚醛树脂作为粘合剂加工成颗粒(试料①)。将这种试料约15g,在氮气流(1L/分)下,试料②在300℃、试料③在600℃、试料④在800℃、试料⑤在1000℃、试料⑥在1100℃,加热30分钟进行干馏。原料及干馏碳中所含氢量分别是①3.20重量%、②3.10重量%、③2.10重量%、④0.90重量%、⑤0.34重量%、⑥0.20重量%。
将这些试料分别在含10容量%氯气的氮气流(1L/分)下,于600℃加热1小时进行氯化处理。氯化处理时重量增加分别是①-45.3重量%、②-30.2重量%、③23.2重量%、④19.3重量%、⑤5.8重量%、⑥3.3重量%,将这些试料在氮气流(2L/分)下,于900℃的温度加热,再在25℃的含饱和水蒸汽的氮气流(1L/分)下,于700℃的温度加热20分钟进行脱氯。
这些碳材料的氮气吸附量分别是①14.5cm3/g、②14.8cm3/g、③15.4cm3/g、④14.1cm3/g、⑤12.9cm3/g、⑥12.4cm3/g。这些试料的双电荷层电容器的静电容量分别是①72.0F/cm3、②73.3F/cm3、③74.3F/cm3、④75.0F/cm3、⑤51.5F/cm3、⑥45.5F/cm3。有效干馏温度在300~1100℃之间,对原料呋喃树脂进行氯化处理时非常有效。有效含氢量在3.2~0.20重量%之间,可获得很大值的氮气吸附量和静电容量。
实施例5:偏氯乙烯树脂、氯化处理
将偏氯乙烯树脂在500℃干馏30分钟,并粉碎,作为粘合剂添加酚醛树脂,加工成颗粒状,将这种颗粒约15g,在氮气流(1L/分)下,试料①于600℃、试料②于800℃、试料③于1000℃、试料④于1100℃,加热30分钟,进行再次干馏。干馏碳中所含氢量分别是①2.01重量%、②0.90重量%、③0.32重量%、④0.19重量%。
将这些试料分别在含10容量%氯气的氮气流(1L/分)下,于600℃加热1小时,进行氯化处理。氯化处理时的重量增加分别是①25.2重量%、②18.5重量%、③7.1重量%、④3.8重量%。将这些试料在氮气流(2L/分)下,于900℃加热,再在25℃的含饱和水蒸汽的氮气流(1L/分)下,于700℃加热20分钟进行脱氯。
这些碳材料的氮气吸附量分别是①15.3cm3/g、②14.6cm3/g、③13.4cm3/g、④12.8cm3/g。这些试料的双电荷层电容器的静电容量分别是①72.8F/cm3、②70.4F/cm3、③51.3F/cm3、④44.9F/cm3。有效干馏温度为600~1100℃之间,有效含氢量为2.01~0.19重量%之间,可获得很大值的氮气吸附量和静电容量。
实施例6:石油沥青、氯化处理
将石油沥青在250℃的干燥机中保持5小时,进行加热实施氧化处理(试料①),将这种沥青(15g)在氮气流下(1L/分),试料②于600℃、试料③于800℃、试料④于1000℃、试料⑤于1100℃下,加热30分钟进行干馏。各试料中含氢量分别是①4.20重量%、②2.01重量%、③0.90重量%、④0.40重量%、⑤0.22重量%。接着,将这些试料分别在含10容量%氯气的氮气流下(1L/分),于600℃的温度加热1小时,进行氯化。氯化处理时重量增加分别是①-12.0重量%、②19.8重量%、③20.0重量%、④6.4重量%、⑤3.5重量%。接着,在氮气流下(2L/分),于900℃的温度加热20分钟,再在25℃的含饱和水蒸汽的氮气流下(1L/分),于700℃加热进行脱氯。
各试料的氮气吸附量分别是①14.7cm3/g、②15.5cm3/g、③14.4cm3/g、④12.7cm3/g、⑤12.0cm3/g。双电荷层电容器的静电容量分别是①67.6F/cm3、②69.5F/cm3、③67.2F/cm3、④47.4F/cm3、⑤34.5F/cm3。有效的干馏温度为600~1100℃之间,对氧化后的原料石油沥青进行氯化处理也非常有效。有效的含氢量为4.20~0.22重量%之间,可获得很大值的氮吸附量和静电容量。
实施例7:酚醛树脂,溴化处理
将酚醛树脂固化并粉碎,作为粘合剂添加酚醛树脂,加工成颗粒状。将这种颗粒约15g在氮气流下(1L/分),试料①于600℃,试料②于800℃,试料③于900℃,试料④于1000℃、试料⑤于1100℃的各温度下,进行20分钟干馏,得到干馏碳。各试料中所含氢量分别是①2.35重量%、②1.00重量%、③0.56重量%、④0.48重量%、⑤0.33重量%,将这些试料分别在含8容量%溴气的氮气流(1L/分)下,于600℃的温度加热3小时,进行溴化处理。溴化处理时的重量增加分别是①19.3重量%、②30.1重量%、③1 8.0重量%、④12.0重量%、⑤9.6重量%。接着,在氮气流下(2L/分),于900℃的温度加热20分钟,再在25℃的含饱和水蒸汽的氮气流下(1L/分),于700℃加热进行脱溴。
各试料的氮气吸附量分别是①16.2cm3/g、②14.8cm3/g、③14.0cm3/g、④13.0cm3/g、⑤12.0cm3/g。双电荷层电容器的静电容量分别是①76.6F/cm3、②72.7F/cm3、③65.0F/cm3、④52.0F/cm3、⑤40.0F/cm3。有效的干馏温度为600~1100℃之间。有效的含氢量为2.35~0.33重量%,可获得很大值的氮气吸附量和静电容量。
实施例8:椰壳、溴化处理
将粗粉碎的椰壳(约15g)在氮气流下(1L/分),试料①于600℃,试料②于800℃,试料③于900℃,试料④于1000℃、试料⑤于1100℃的各温度下,进行20分钟干馏,得到干馏碳。各试料中所含氢量分别是①2.01重量%、②0.77重量%、③0.54重量%、④0.47重量%、⑤0.35重量%。将各试料分别在含8容量%溴的氮气流下(1L/分),于600℃的温度加热3小时,进行溴化处理。溴化处理时的重量增加分别是①26.9重量%、②24.4重量%、③17.6重量%、④7.1重量%、⑤3.6重量%,接着,在氮气流下(2L/分),于900℃的温度加热20分钟,再在25℃的含饱和水蒸汽的氮气流下(1L/分),于700℃的温度加热进行脱溴。
各试料的氮气吸附量分别是①14.9cm3/g、②13.9cm3/g、③13.4cm3/g、④12.5cm3/g、⑤12.0cm3/g。双电荷层电容器的静电容量分别是①64.0F/cm3、②63.1F/cm3、③54.1F/cm3、④42.2F/cm3、⑤30.0F/cm3。有效的干馏温度为600~1100℃的广泛的范围。有效的氢含量为2.01~0.35重量%之间,可得到很大值的氮气吸附量和静电容量。
实施例9:呋喃树脂,溴化处理
将呋喃树脂固化并粉碎,作为粘合剂添加酚醛树脂,加工成颗粒状,将这种颗粒约15g在氮气流下(1L/分),试料①于600℃,试料②于800℃,试料③于1000℃,试料④于1100℃的温度下,进行20分钟干馏,得到干馏碳。各试料中所含氢量分别是①2.10重量%、②0.90重量%、③0.34重量%、④0.24重量%。将这些试料分别在含8容量%溴的氮气流下(1L/分),于600℃的温度加热3小时,进行溴化处理。溴化处理时的重量增加分别是①23.0重量%、②19.0重量%、③5.2重量%、④2.8重量%。接着,在氮气流下(2L/分),于900℃的温度加热20分钟,再在25℃的含饱和水蒸汽的氮气流下(1L/分),于700℃的温度加热进行脱溴。
各试料的氮气吸附量分别是①15.2cm3/g、②13.9cm3/g、③12.6cm3/g、④12.1cm3/g,双电荷层电容器的静电容量分别是①74.0F/cm3、②74.0F/cm3、③50.5F/cm3、④44.3F/cm3。有效的干馏温度为600~1100℃的广泛的范围。干馏碳中所含氢量为2.10~0.24重量%之间有效,可得到很大值的氮气吸附量和静电容量。
实施例10:偏氯乙烯树脂,溴化处理
将偏氯乙烯树脂在500℃下干馏30分钟,并粉碎,作为粘合剂添加酚醛树脂,加工成颗粒状,将这种颗粒约15g在氮气流下(1L/分),试料①于600℃,试料②于800℃,试料③于1000℃,试料④于1100℃的温度下,再次干馏20分钟,得到干馏碳。各试料中所含氢量分别是①2.01重量%、②0.90重量%、③0.32重量%、④0.19重量%,将各试料分别在含8容量%溴的氮气流(1L/分)下,于600℃的温度加热3小时,进行溴化处理。溴化处理时的重量增加分别是①25.0重量%、②18.0重量%、③6.6重量%、④3.2重量%。接着,在氮气流下(2L/分),于900℃的温度加热20分钟,再在25℃的含饱和水蒸汽的氮气流下(1L/分),于700℃的温度加热进行脱溴。
各试料的氮吸附量分别是①15.1cm3/g、②14.6cm3/g、③13.2cm3/g、④12.6cm3/g。双电荷层电容器的静电容量分别是①72.4F/cm3、②70.0F/cm3、③51.1F/cm3、④44.6F/cm3。有效的干馏温度为600~1100℃的广泛的范围。有效的氢含量为2.01~0.19重量%,可获得很大值的氮气吸附量和静电容量。
将比较例和实施例的结果示于表1、表2、及图5(氮吸附量)、图6(静电容量)。由这些结果可知,本发明的制造方法,可以有效获得具有高氮气吸附性能、高静电容量的多孔性碳材料。
表1
注:表中的标号如下:[1]比较例、实施例的编号,[2]原料的种类,[3]试料编号,[4]干馏温度(℃),[5]氢含量(重量%),[6]卤素气体的种类,[7]卤化处理温度(℃),[8]卤化处理时的重量增加(重量%),[9]在氮气流下的加热(高温脱卤)处理的温度(℃),[10]在水蒸汽环境下的加热(低温脱卤)处理的温度(℃),[11]氮气吸附量(cm3/g),[12]双电荷层电容器的静电容量(F/cm3),--没有进行该处理工序。
| [1] | [2] | [3] | [4]℃ | [5]重量% | [6] | [7]℃ | [8]重量% | [9]℃ | [10]℃ | [11]cm3/g | [12]F/cm3 |
| 比较例1 | 酚醛树脂 | ①②③④ | 60080010001100 | -------- | ------ | -------- | -------- | -------- | 5.210.411.210.8 | 7.716.114.411.8 | |
| 比较例2 | 酚醛树脂 | ①② | 300400 | ClCl | 300400 | -0 7-1.8 | 900900 | 700700 | 10.910.9 | 15.315.2 | |
| 比较例3 | 椰壳 | ①②③④⑤ | 60070080010001100 | ---------- | ---------- | ---------- | ---------- | ---------- | 4.75.67.29.17.6 | 3.711.312.99.56.0 | |
| 比较例4 | 呋喃树脂 | ①②③④ | 60080010001100 | -------- | -------- | -------- | -------- | -------- | 4.89.910.66.8 | 5.715.311.26.3 | |
| 比较例5 | 偏氯乙烯树脂 | ①②③④ | 60080010001100 | -------- | -------- | -------- | -------- | -------- | 7.410.611.010.8 | 6.615.814.111.1 | |
| 比较例6 | 沥青 | ①②③④ | 60080010001100 | -------- | -------- | -------- | -------- | -------- | 1.97.78.97.5 | 2.112.08.25.0 |
表2
注:表中的标号如下:[1]比较例、实施例的编号,[2]原料的种类,[3]试料编号,[4]干馏温度(℃),[5]氢含量(重量%),[6]卤素气体的种类,[7]卤化处理温度(℃),[8]卤化处理时的重量增加(重量%),[9]在氮气流下的加热(高温脱卤)处理的温度(℃),[10]在水蒸汽环境下的加热(低温脱卤)处理的温度(℃),[11]氮气吸附量(cm3/g),[12]双电荷层电容器的静电容量(F/cm3),--没有进行该处理工序。
| [1] | [2] | [3] | [4]℃ | [5]重量% | [6] | [7]℃ | [8]重量% | [9]℃ | [10]℃ | [11]cm3/g | [12]F/cm3 |
| 实施例1 | 酚醛树脂 | ① | - | 3.00 | Cl | 500 | -14.7 | 900 | 700 | 14.0 | 66.4 |
| 实施例2 | 酚醛树脂 | ①②③④⑤⑥⑦ | -30060080090010001100 | 3.002.932.351.000.560.480.33 | ClClClClClClCl | 600600600600600600600 | -21.3-15 720.422.318.312.49.9 | 900900900900900900900 | 700700700700700700700 | 15.515.716.214.914.213.012.2 | 71.173.276.673.065.853.040.5 |
| 实施例3 | 椰壳 | ①②③④⑤⑥ | -60080090010001100 | 2.802.010.770.540.470.35 | ClClClClClCl | 600600600600600600 | -65.027.925.318.37.33.9 | 900900900900900900 | 700700700700700700 | 14.315.314.213.913.012.1 | 63.064.663.954.642.530.1 |
| 实施例4 | 呋喃树脂 | ①②③④⑤⑥ | -30060080010001100 | 3.203.102.100.900.340.20 | ClClClClClCl | 600600600600600600 | -45.3-30.223.219.35.83.3 | 900900900900900900 | 700700700700700700 | 14.514.815.414.112.912.4 | 72.073.374.375.051.545.5 |
| 实施例5 | 偏氯乙烯树脂 | ①②③④ | 60080010001100 | 2.010.900.320.19 | ClClClCl | 600600600600 | 25.218.57.13.8 | 900900900900 | 700700700700 | 15.314.613.412.8 | 72.870.451.344 9 |
表3
注:表中的标号如下:[1]比较例、实施例的编号,[2]原料的种类,[3]试料编号,[4]干馏温度(℃),[5]氢含量(重量%),[6]卤素气体的种类,[7]卤化处理温度(℃),[8]卤化处理时的重量增加(重量%),[9]在氮气流下的加热(高温脱卤)处理的温度(℃),[10]在水蒸汽环境下的加热(低温脱卤)处理的温度(℃),[11]氮气吸附量(cm3/g),[12]双电荷层电容器的静电容量(F/cm3),--没有进行该处理工序。
| [1] | [2] | [3] | [4]℃ | [5]重量% | [6] | [7]℃ | [8]重量% | [9]℃ | [10]℃ | [11]cm3/g | [12]F/cm3 |
| 实施例6 | 沥青 | ①②③④⑤ | -60080010001100 | 4.202.010.900.400.22 | ClClClClCl | 600600600600600 | -12.019.820.06.43.5 | 900900900900900 | 700700700700700 | 14.715.514.412.712.0 | 67.669.567.247.434.5 |
| 实施例7 | 酚醛树脂 | ①②③④⑤ | 60080090010001100 | 2.351.000.560.480.33 | BrBrBrBrBr | 600600600600600 | 19.330.118.012.09.6 | 900900900900900 | 700700700700700 | 16.214.814.013.012.0 | 76.672.765.052.040.0 |
| 实施例8 | 椰壳 | ①②③④⑤ | 60080090010001100 | 2.010.770.540.470.35 | BrBrBrBrBr | 600600600600600 | 26.924.417.67.13.6 | 900900900900900 | 700700700700700 | 14.913.913.412.512.0 | 64.063.154.142.230.0 |
| 实施例9 | 呋喃树脂 | ①②③④ | 60080010001100 | 2.100.900.340.24 | BrBrBrBr | 600600600600 | 23.019.05.22.8 | 900900900900 | 700700700700 | 15.213.912.612.1 | 74.074.050.544.3 |
| 实施例10 | 偏氯乙烯树脂 | ①②③④ | 60080010001100 | 2.010.900.320.19 | BrBrBrBr | 600600600600 | 25.018.06.63.2 | 900900900900 | 700700700700 | 15.114.613.212.6 | 72.470.051.144.6 |
产业上的利用可能性
根据本发明,可以获得用于吸附氮气等小分子吸附量很大的吸附剂的和用于双电荷层电容器静电容量很大的多孔碳材料。
本发明获得的多孔碳材料,由于具有发达的微孔或亚微孔,所以除了上述用途外,还有多种多样的用途,例如,氩气、甲烷等小分子气体的吸附剂、锂离子二次电池等的电池的电极材料、催化剂、催化剂载体、生物化学中的用途等等。
Claims (11)
1.一种多孔碳材料的制造方法,其特征是对碳化合物、及干馏碳化合物得到的干馏碳中任何一种实施卤化处理,以制得多孔碳材料。
2.根据权利要求1记载的多孔碳材料的制造方法,其特征是在对通过干馏碳化合物所得干馏碳实施卤化处理制造多孔碳材料的方法中,上述干馏碳的氢含量为0.1~5.0重量%。
3.根据权利要求1记载的多孔碳材料的制造方法,其特征是在对通过干馏碳化合物所得干馏碳实施卤化处理制造多孔碳材料的方法中,上述干馏碳是将上述碳化合物在250~1200℃下进行干馏获得的干馏碳。
4.根据权利要求2或3记载的多孔碳材料的制造方法,其特征是上述卤化处理包括:将上述干馏碳在卤素气体和含卤素的惰性气体的任何一种气体中,进行使卤素和干馏碳反应的卤化处理,和将这种卤化处理的干馏碳,在从惰性气体中、真空排气下、氢化合物气体中、及含氢化合物的惰性气体组中,选出的1种的气氛中,进行加热脱卤处理。
5.根据权利要求1记载的多孔碳材料的制造方法,在对碳化合物实施卤化处理制造多孔碳材料的方法中,其特征是该碳化合物是选自原料碳化合物、将原料碳化合物在250℃下加热的碳化合物、及它们的混合物中的1种。
6.根据权利要求5记载的多孔碳材料的制造方法,其特征是上述卤化处理包括,将上述碳化合物在卤素气体和含卤素的惰性气体中的任一种中加热,进行使卤素和碳化合物反应的卤化处理,和将这种卤化处理的碳化合物在选自惰性气体、真空排气下、氢化合物气体及含氢化合物的惰性气体中的任一种气氛中,进行加热脱卤处理。
7.根据权利要求6中记载的多孔碳材料的制造方法,其特征是上述卤化处理是在500~1000℃的温度进行。
8.根据权利要求1、2、3、5中的任一项记载的多孔碳材料的制造方法,其特征是上述卤素是选自氯和溴中的至少一种。
9.根据权利要求1、2、3、5中任一项记载的多孔碳材料的制造方法,其特征是上述原料碳化合物是选自椰壳、酚醛树脂、呋喃树脂、偏氯乙烯树脂和沥青中的至少一种。
10.一种由权利要求1、2、3、5中任一项记载的方法制造的多孔碳材料,其特征是该材料在25℃,1个大气压下的氮吸附量为12~20cm3/g。
11.一种由权利要求1、2、3、5中任一项记载的方法制造的多孔碳材料,其特征是该材料的双电荷层电容器的静电容量为30~90F/cm3。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2421/97 | 1997-01-09 | ||
| JP242197 | 1997-01-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1217706A true CN1217706A (zh) | 1999-05-26 |
Family
ID=11528793
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN97194425A Pending CN1217706A (zh) | 1997-01-09 | 1997-11-06 | 多孔碳材料及其制造方法 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0891943A1 (zh) |
| KR (1) | KR20000064574A (zh) |
| CN (1) | CN1217706A (zh) |
| CA (1) | CA2248218A1 (zh) |
| WO (1) | WO1998030496A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101454243B (zh) * | 2006-05-31 | 2012-04-18 | 促进科学E.V.麦克斯-普朗克公司 | 多孔导电碳材料及其应用 |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8002880B2 (en) | 2002-12-10 | 2011-08-23 | Advanced Technology Materials, Inc. | Gas storage and dispensing system with monolithic carbon adsorbent |
| US6743278B1 (en) * | 2002-12-10 | 2004-06-01 | Advanced Technology Materials, Inc. | Gas storage and dispensing system with monolithic carbon adsorbent |
| KR101496934B1 (ko) | 2006-11-15 | 2015-03-03 | 유니버시티 오브 워싱톤 스루 이츠 센터 포 커머셜리제이션 | 전기 이중층 캐패시턴스 장치 |
| US8293818B2 (en) | 2009-04-08 | 2012-10-23 | Energ2 Technologies, Inc. | Manufacturing methods for the production of carbon materials |
| FI2448748T3 (fi) | 2009-07-01 | 2024-10-09 | Basf Mobile Emissions Catalysts Llc | Ultrapuhtaita synteettisiä hiilimateriaaleja |
| US8916296B2 (en) | 2010-03-12 | 2014-12-23 | Energ2 Technologies, Inc. | Mesoporous carbon materials comprising bifunctional catalysts |
| US8198210B2 (en) | 2010-05-27 | 2012-06-12 | Corning Incorporated | Halogenated activated carbon materials for high energy density ultracapacitors |
| CN103261090A (zh) | 2010-09-30 | 2013-08-21 | 艾纳G2技术公司 | 储能颗粒的增强式装填 |
| CN108538625B (zh) | 2010-12-28 | 2020-12-08 | 巴斯福股份公司 | 包含增强的电化学特性的碳材料 |
| US8679231B2 (en) | 2011-01-19 | 2014-03-25 | Advanced Technology Materials, Inc. | PVDF pyrolyzate adsorbent and gas storage and dispensing system utilizing same |
| US20120262127A1 (en) | 2011-04-15 | 2012-10-18 | Energ2 Technologies, Inc. | Flow ultracapacitor |
| CN103947017B (zh) | 2011-06-03 | 2017-11-17 | 巴斯福股份公司 | 用于混合能量存储装置中的碳‑铅共混物 |
| US8842417B2 (en) | 2011-09-23 | 2014-09-23 | Corning Incorporated | High voltage electro-chemical double layer capacitor |
| US9409777B2 (en) | 2012-02-09 | 2016-08-09 | Basf Se | Preparation of polymeric resins and carbon materials |
| KR101442198B1 (ko) * | 2012-02-27 | 2014-09-22 | 서강대학교산학협력단 | 다공성 탄소 구조체, 이의 제조 방법, 상기 다공성 탄소 구조체를 포함하는 탄소 음극 활물질, 이를 이용한 리튬 이온 배터리, 및 이의 제조 방법 |
| WO2013181295A1 (en) | 2012-05-29 | 2013-12-05 | Advanced Technology Materials, Inc. | Carbon adsorbent for hydrogen sulfide removal from gases containing same, and regeneration of adsorbent |
| CN104641499B (zh) * | 2012-08-30 | 2016-09-28 | 株式会社吴羽 | 非水电解质二次电池负极用碳质材料及其制造方法 |
| JP5681753B2 (ja) * | 2012-12-07 | 2015-03-11 | 住友ベークライト株式会社 | 負極材料、負極活物質、負極およびアルカリ金属イオン電池 |
| CN105190948B (zh) | 2013-03-14 | 2019-04-26 | 14族科技公司 | 包含锂合金化的电化学改性剂的复合碳材料 |
| US10195583B2 (en) | 2013-11-05 | 2019-02-05 | Group 14 Technologies, Inc. | Carbon-based compositions with highly efficient volumetric gas sorption |
| WO2015137980A1 (en) | 2014-03-14 | 2015-09-17 | Energ2 Technologies, Inc. | Novel methods for sol-gel polymerization in absence of solvent and creation of tunable carbon structure from same |
| WO2017031006A1 (en) | 2015-08-14 | 2017-02-23 | Energ2 Technologies, Inc. | Composites of porous nano-featured silicon materials and carbon materials |
| KR102637617B1 (ko) | 2015-08-28 | 2024-02-19 | 그룹14 테크놀로지스, 인코포레이티드 | 극도로 내구성이 우수한 리튬 인터칼레이션을 나타내는 신규 물질 및 그의 제조 방법 |
| KR101938850B1 (ko) | 2017-01-24 | 2019-01-15 | 연태헌 | 필터 재료의 제조방법 |
| CN110582823A (zh) | 2017-03-09 | 2019-12-17 | 14集团技术公司 | 含硅前体在多孔支架材料上的分解 |
| US11174167B1 (en) | 2020-08-18 | 2021-11-16 | Group14 Technologies, Inc. | Silicon carbon composites comprising ultra low Z |
| US11335903B2 (en) | 2020-08-18 | 2022-05-17 | Group14 Technologies, Inc. | Highly efficient manufacturing of silicon-carbon composites materials comprising ultra low z |
| US11639292B2 (en) | 2020-08-18 | 2023-05-02 | Group14 Technologies, Inc. | Particulate composite materials |
| KR20230082028A (ko) | 2020-09-30 | 2023-06-08 | 그룹14 테크놀로지스, 인코포레이티드 | 규소-탄소 복합재의 산소 함량 및 반응성을 제어하기 위한 부동태화의 방법 |
| CN116832774B (zh) * | 2023-07-03 | 2024-02-20 | 上海交通大学 | 一种生物质衍生炭颗粒及其制备方法和应用 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5969411A (ja) * | 1982-10-14 | 1984-04-19 | Agency Of Ind Science & Technol | 高密度炭素状物質の製造方法 |
| JP3540085B2 (ja) * | 1995-02-09 | 2004-07-07 | 呉羽化学工業株式会社 | 電池電極用炭素質材料、その製造方法、電極構造体および電池 |
| US6475461B1 (en) * | 1995-03-30 | 2002-11-05 | Nippon Sanso Corporation | Porous carbonaceous material, manufacturing method therefor and use thereof |
| JPH08268773A (ja) * | 1995-03-30 | 1996-10-15 | Nippon Sanso Kk | 多孔性炭素、その製造方法およびその用途 |
| JPH08306593A (ja) * | 1995-04-27 | 1996-11-22 | Nippon Sanso Kk | 電気二重層キャパシタ用炭素電極の製造方法及び炭素電極並びに電気二重層キャパシタ |
| US5985489A (en) * | 1995-06-20 | 1999-11-16 | Nippon Sanso Corporation | Carbon for a lithium secondary battery, lithium secondary battery, and manufacturing methods therefor |
-
1997
- 1997-11-06 EP EP97911463A patent/EP0891943A1/en not_active Ceased
- 1997-11-06 KR KR1019980707110A patent/KR20000064574A/ko not_active Application Discontinuation
- 1997-11-06 CN CN97194425A patent/CN1217706A/zh active Pending
- 1997-11-06 WO PCT/JP1997/004036 patent/WO1998030496A1/ja not_active Application Discontinuation
- 1997-11-06 CA CA002248218A patent/CA2248218A1/en not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101454243B (zh) * | 2006-05-31 | 2012-04-18 | 促进科学E.V.麦克斯-普朗克公司 | 多孔导电碳材料及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0891943A1 (en) | 1999-01-20 |
| KR20000064574A (ko) | 2000-11-06 |
| CA2248218A1 (en) | 1998-07-16 |
| EP0891943A4 (zh) | 1999-02-03 |
| WO1998030496A1 (fr) | 1998-07-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1217706A (zh) | 多孔碳材料及其制造方法 | |
| US11370665B2 (en) | Method for producing activated carbon | |
| CN100335407C (zh) | 活性炭、其制造方法及可极化电极 | |
| JP4968425B2 (ja) | 球状多孔性炭素粒子粉末及びその製造法 | |
| Maldonado-Hódar et al. | Catalytic graphitization of carbon aerogels by transition metals | |
| JP5400892B2 (ja) | 多孔質の活性炭の製造方法 | |
| KR100264400B1 (ko) | 2차전지용탄소질전극재료및그제조방법 | |
| JP5763228B2 (ja) | 高表面積炭素及びその製造方法 | |
| KR102037463B1 (ko) | 폐플라스틱과 석유계 잔사유를 활용한 고수율 활성탄 제조 방법 및 이에 의해 제조된 고효율 흡착 활성탄 | |
| KR102113719B1 (ko) | 활성탄 및 이의 제조방법 | |
| CN1265638A (zh) | 生产多孔碳制品的方法及由其生产的制品 | |
| CN1231986C (zh) | 锂二次电池的电极材料以及使用该电极材料的锂二次电池 | |
| JP2012507470A5 (zh) | ||
| KR20170030545A (ko) | 금속 공기 전지용 전극 재료 | |
| JP2006213544A (ja) | 粒状活性炭とその製造方法 | |
| US5985489A (en) | Carbon for a lithium secondary battery, lithium secondary battery, and manufacturing methods therefor | |
| JP2008050237A (ja) | 球状多孔性炭素粒子粉末及びその製造法 | |
| KR102177976B1 (ko) | 그래핀 복합체, 전극 활물질의 제조방법 및 이를 포함하는 이차전지 | |
| WO2021253010A1 (en) | Systems and methods for processing coal for use in a direct air capture system | |
| US6013208A (en) | Manufacturing method for carbon material for electrical double layer capacitor | |
| JP4615868B2 (ja) | 電気二重層キャパシタ用多孔質炭素の製造方法、該製造方法により得られた電気二重層キャパシタ用多孔質炭素、及び、該電気二重層キャパシタ用多孔質炭素を用いた電気二重層キャパシタ | |
| JP3191941B2 (ja) | 電気二重層キャパシタ用炭素材の製造方法並びに炭素電極および電気二重層キャパシタ | |
| KR102522123B1 (ko) | 메조 기공 부피와 비표면적이 큰 활성탄소의 제조방법 및 이를 통해 제조된 활성탄소 | |
| JP2828509B2 (ja) | リチウム二次電池用炭素とその製造方法並びにリチウム二次電池とその製造方法 | |
| JP2000143224A (ja) | 多孔性炭素材の製造方法及びそれにより得られた多孔性炭素材 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C01 | Deemed withdrawal of patent application (patent law 1993) | ||
| WD01 | Invention patent application deemed withdrawn after publication |