CN112469709A - 比拉斯汀的制备方法及其中间体 - Google Patents
比拉斯汀的制备方法及其中间体 Download PDFInfo
- Publication number
- CN112469709A CN112469709A CN201980049580.4A CN201980049580A CN112469709A CN 112469709 A CN112469709 A CN 112469709A CN 201980049580 A CN201980049580 A CN 201980049580A CN 112469709 A CN112469709 A CN 112469709A
- Authority
- CN
- China
- Prior art keywords
- formula
- solvate
- compound
- acid
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- ACCMWZWAEFYUGZ-UHFFFAOYSA-N bilastine Chemical compound N=1C2=CC=CC=C2N(CCOCC)C=1C(CC1)CCN1CCC1=CC=C(C(C)(C)C(O)=O)C=C1 ACCMWZWAEFYUGZ-UHFFFAOYSA-N 0.000 title claims abstract description 43
- 229960004314 bilastine Drugs 0.000 title claims abstract description 43
- 238000002360 preparation method Methods 0.000 title claims description 15
- 150000001875 compounds Chemical class 0.000 claims abstract description 133
- 239000012453 solvate Substances 0.000 claims abstract description 90
- 238000000034 method Methods 0.000 claims abstract description 53
- 230000008707 rearrangement Effects 0.000 claims abstract description 17
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 16
- 230000001590 oxidative effect Effects 0.000 claims abstract description 15
- 150000003839 salts Chemical class 0.000 claims description 68
- 238000006243 chemical reaction Methods 0.000 claims description 54
- -1 Alkyl ortho ester Chemical class 0.000 claims description 38
- 239000002253 acid Substances 0.000 claims description 25
- 239000000203 mixture Substances 0.000 claims description 22
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 21
- 229910052740 iodine Inorganic materials 0.000 claims description 21
- 229910052801 chlorine Inorganic materials 0.000 claims description 17
- 125000004185 ester group Chemical group 0.000 claims description 16
- 239000007800 oxidant agent Substances 0.000 claims description 15
- 229910052794 bromium Inorganic materials 0.000 claims description 14
- 238000005917 acylation reaction Methods 0.000 claims description 13
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 11
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 11
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 9
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 9
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 9
- 230000010933 acylation Effects 0.000 claims description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 9
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 9
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- 239000011630 iodine Substances 0.000 claims description 8
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 7
- 239000003377 acid catalyst Substances 0.000 claims description 7
- 230000003647 oxidation Effects 0.000 claims description 6
- 238000007254 oxidation reaction Methods 0.000 claims description 6
- 239000002841 Lewis acid Substances 0.000 claims description 5
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 5
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 claims description 4
- 230000003301 hydrolyzing effect Effects 0.000 claims description 4
- 150000007517 lewis acids Chemical class 0.000 claims description 4
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 claims description 4
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 claims description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims description 3
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 claims description 3
- 239000007983 Tris buffer Substances 0.000 claims description 3
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 claims description 3
- 235000015165 citric acid Nutrition 0.000 claims description 3
- 239000001630 malic acid Substances 0.000 claims description 3
- 235000011090 malic acid Nutrition 0.000 claims description 3
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 3
- 229910017604 nitric acid Inorganic materials 0.000 claims description 3
- 235000006408 oxalic acid Nutrition 0.000 claims description 3
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 claims description 2
- PEZNEXFPRSOYPL-UHFFFAOYSA-N (bis(trifluoroacetoxy)iodo)benzene Chemical compound FC(F)(F)C(=O)OI(OC(=O)C(F)(F)F)C1=CC=CC=C1 PEZNEXFPRSOYPL-UHFFFAOYSA-N 0.000 claims description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 claims description 2
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 claims description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 2
- 239000001530 fumaric acid Substances 0.000 claims description 2
- 235000011087 fumaric acid Nutrition 0.000 claims description 2
- ICIWUVCWSCSTAQ-UHFFFAOYSA-N iodic acid Chemical compound OI(=O)=O ICIWUVCWSCSTAQ-UHFFFAOYSA-N 0.000 claims description 2
- JYJVVHFRSFVEJM-UHFFFAOYSA-N iodosobenzene Chemical compound O=IC1=CC=CC=C1 JYJVVHFRSFVEJM-UHFFFAOYSA-N 0.000 claims description 2
- 239000011976 maleic acid Substances 0.000 claims description 2
- 239000000126 substance Substances 0.000 claims description 2
- 239000011975 tartaric acid Substances 0.000 claims description 2
- 235000002906 tartaric acid Nutrition 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims 1
- 230000015572 biosynthetic process Effects 0.000 abstract description 7
- 238000003786 synthesis reaction Methods 0.000 abstract description 7
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Substances COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 31
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 30
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- 239000002904 solvent Substances 0.000 description 25
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 239000003960 organic solvent Substances 0.000 description 20
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 18
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 18
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 17
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 16
- 239000000460 chlorine Substances 0.000 description 16
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 14
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- 239000002585 base Substances 0.000 description 14
- 239000000543 intermediate Substances 0.000 description 14
- HYTRYEXINDDXJK-UHFFFAOYSA-N Ethyl isopropyl ketone Chemical compound CCC(=O)C(C)C HYTRYEXINDDXJK-UHFFFAOYSA-N 0.000 description 12
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 12
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 11
- 230000007062 hydrolysis Effects 0.000 description 11
- 238000006460 hydrolysis reaction Methods 0.000 description 11
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 10
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 10
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Chemical compound [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 9
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 8
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 8
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 8
- 150000007529 inorganic bases Chemical class 0.000 description 8
- 239000003495 polar organic solvent Substances 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 229910019142 PO4 Inorganic materials 0.000 description 7
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 7
- 235000019439 ethyl acetate Nutrition 0.000 description 7
- 231100000331 toxic Toxicity 0.000 description 7
- 230000002588 toxic effect Effects 0.000 description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 125000003118 aryl group Chemical group 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 6
- 150000002825 nitriles Chemical class 0.000 description 6
- XNLICIUVMPYHGG-UHFFFAOYSA-N pentan-2-one Chemical compound CCCC(C)=O XNLICIUVMPYHGG-UHFFFAOYSA-N 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 6
- 150000003462 sulfoxides Chemical class 0.000 description 6
- 239000004215 Carbon black (E152) Substances 0.000 description 5
- 229910052783 alkali metal Inorganic materials 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- 150000002576 ketones Chemical class 0.000 description 5
- 230000035484 reaction time Effects 0.000 description 5
- ZHGNHOOVYPHPNJ-UHFFFAOYSA-N Amigdalin Chemical compound FC(F)(F)C(=O)OCC1OC(OCC2OC(OC(C#N)C3=CC=CC=C3)C(OC(=O)C(F)(F)F)C(OC(=O)C(F)(F)F)C2OC(=O)C(F)(F)F)C(OC(=O)C(F)(F)F)C(OC(=O)C(F)(F)F)C1OC(=O)C(F)(F)F ZHGNHOOVYPHPNJ-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 4
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 4
- 150000008041 alkali metal carbonates Chemical class 0.000 description 4
- 239000003849 aromatic solvent Substances 0.000 description 4
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 125000002524 organometallic group Chemical group 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- 239000007836 KH2PO4 Substances 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 229910000318 alkali metal phosphate Inorganic materials 0.000 description 3
- 229910000025 caesium bicarbonate Inorganic materials 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 3
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 3
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 3
- 229910000397 disodium phosphate Inorganic materials 0.000 description 3
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical group 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 238000009776 industrial production Methods 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 229910052808 lithium carbonate Inorganic materials 0.000 description 3
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 3
- 229910000032 lithium hydrogen carbonate Inorganic materials 0.000 description 3
- 229910052751 metal Chemical class 0.000 description 3
- 239000002184 metal Chemical class 0.000 description 3
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 239000011736 potassium bicarbonate Substances 0.000 description 3
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 3
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 3
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 3
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 3
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 3
- YBJXRWANRTYCJE-UHFFFAOYSA-N 1-(2-ethoxyethyl)-2-piperidin-4-ylbenzimidazole Chemical compound N=1C2=CC=CC=C2N(CCOCC)C=1C1CCNCC1 YBJXRWANRTYCJE-UHFFFAOYSA-N 0.000 description 2
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- HBOGHPAOOWUTLB-UHFFFAOYSA-N 2-piperidin-4-yl-1h-benzimidazole Chemical compound C1CNCCC1C1=NC2=CC=CC=C2N1 HBOGHPAOOWUTLB-UHFFFAOYSA-N 0.000 description 2
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 150000004982 aromatic amines Chemical class 0.000 description 2
- 150000001499 aryl bromides Chemical class 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 238000006880 cross-coupling reaction Methods 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000003759 ester based solvent Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N formic acid Substances OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 239000005453 ketone based solvent Substances 0.000 description 2
- 229940049920 malate Drugs 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000005580 one pot reaction Methods 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 150000002905 orthoesters Chemical class 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical group C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- MNNZINNZIQVULG-UHFFFAOYSA-N 2-chloroethylbenzene Chemical compound ClCCC1=CC=CC=C1 MNNZINNZIQVULG-UHFFFAOYSA-N 0.000 description 1
- YYEROYLAYAVZNW-UHFFFAOYSA-N 2-methyl-2-phenylpropanoic acid Chemical compound OC(=O)C(C)(C)C1=CC=CC=C1 YYEROYLAYAVZNW-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 229910015844 BCl3 Inorganic materials 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 238000005863 Friedel-Crafts acylation reaction Methods 0.000 description 1
- 229910004039 HBF4 Inorganic materials 0.000 description 1
- 229910021576 Iron(III) bromide Inorganic materials 0.000 description 1
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 101150101537 Olah gene Proteins 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 229910003910 SiCl4 Inorganic materials 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- 229910003074 TiCl4 Inorganic materials 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 208000024780 Urticaria Diseases 0.000 description 1
- BHHYHSUAOQUXJK-UHFFFAOYSA-L Zinc fluoride Inorganic materials F[Zn]F BHHYHSUAOQUXJK-UHFFFAOYSA-L 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 201000010435 allergic urticaria Diseases 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- PQLAYKMGZDUDLQ-UHFFFAOYSA-K aluminium bromide Chemical compound Br[Al](Br)Br PQLAYKMGZDUDLQ-UHFFFAOYSA-K 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical class B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- HCUYBXPSSCRKRF-UHFFFAOYSA-N diphosgene Chemical compound ClC(=O)OC(Cl)(Cl)Cl HCUYBXPSSCRKRF-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 238000006197 hydroboration reaction Methods 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 1
- 229910000359 iron(II) sulfate Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002560 ketene acetals Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZPVSMUUBFOTAV-UHFFFAOYSA-N methyl 2-[4-(2-chloroethyl)phenyl]-2-methylpropanoate Chemical compound COC(=O)C(C)(C)C1=CC=C(CCCl)C=C1 VZPVSMUUBFOTAV-UHFFFAOYSA-N 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000004344 phenylpropyl group Chemical group 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- QQKDTTWZXHEGAQ-UHFFFAOYSA-N propyl carbonochloridate Chemical compound CCCOC(Cl)=O QQKDTTWZXHEGAQ-UHFFFAOYSA-N 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- FDNAPBUWERUEDA-UHFFFAOYSA-N silicon tetrachloride Chemical compound Cl[Si](Cl)(Cl)Cl FDNAPBUWERUEDA-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 238000006561 solvent free reaction Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- XTHPWXDJESJLNJ-UHFFFAOYSA-N sulfurochloridic acid Chemical compound OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 150000003606 tin compounds Chemical class 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- FEONEKOZSGPOFN-UHFFFAOYSA-K tribromoiron Chemical compound Br[Fe](Br)Br FEONEKOZSGPOFN-UHFFFAOYSA-K 0.000 description 1
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- ZBZJXHCVGLJWFG-UHFFFAOYSA-N trichloromethyl(.) Chemical compound Cl[C](Cl)Cl ZBZJXHCVGLJWFG-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000006886 vinylation reaction Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/62—Halogen-containing esters
- C07C69/63—Halogen-containing esters of saturated acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/39—Preparation of carboxylic acid esters by oxidation of groups which are precursors for the acid moiety of the ester
- C07C67/42—Preparation of carboxylic acid esters by oxidation of groups which are precursors for the acid moiety of the ester by oxidation of secondary alcohols or ketones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/46—Friedel-Crafts reactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明涉及一种式(III)化合物的制备方法,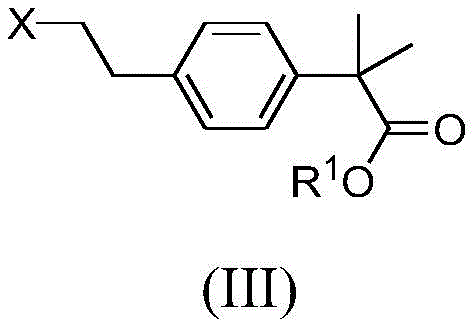 其中,X为离去基团;且R1为C1‑C6烷基;所述方法包括式(II)化合物或其溶剂化物的氧化重排,
其中,X为离去基团;且R1为C1‑C6烷基;所述方法包括式(II)化合物或其溶剂化物的氧化重排,
Description
技术领域
本发明涉及一种在合成比拉斯汀中制备关键中间体的方法,以及上述方法中的新中间体。
背景技术
比拉斯汀是第二代抗组胺药物,用于治疗过敏性鼻结膜炎和荨麻疹。

近年来,已经公开了几种使用式(Ⅲ)化合物作为关键中间体合成比拉斯汀的方法,因为它提供了一条直接合成比拉斯汀的路线,最大限度地减少了保护基团的化学反应。

例如,WO 2009/102155公开了在钯催化剂、t-Bu3P和ZnF2的存在下,通过使化合物4和甲基三甲基硅基二甲基酮缩醛(5)反应来制备关键中间体。接着,通过将关键中间体与1-(2-乙氧基乙基)-2-哌啶-4-基-1H-苯并咪唑(3a)反应并随后发生酯基水解或者与2-(4-哌啶基)-1H-苯并咪唑(7)反应并随后发生苯并咪唑的氮烷基化和酯基的水解,使其转化为比拉斯汀。然而,由于合成的关键步骤中所使用的烯酮缩醛和膦的不稳定性、可得性和成本,这种交叉偶联方法缺乏工业利益。

CN 104151160 A涉及合成关键中间体(Ⅲ)的类似化合物,但其缺少离去基团。在钯催化剂和氨基锂的存在下,通过卤代苯化合物(2)的烷基化合成上述化合物。此外,这种方法使用有机金属试剂和氨基碱,使其非常不稳定且价格昂贵。

在Synthetic Communications 2011,41(9),1394-1402中公开的比拉斯汀的制备方法,其包括钯催化的芳基溴化物(3)与乙烯基三丁基锡或乙烯基硼酸酐的乙烯基化,并随后对生成的苯乙烯(11)进行硼氢化反应。这种交替交叉偶联方法在第一步骤中使用高成本和低可得性的乙烯基合成子(并且在锡化合物中是高毒性的)。此外,反马氏羟基化步骤是使用众所周知且不受欢迎的硼烷(高毒性和易燃气体)进行的,这也破坏了其工业适用性。

CN 104326909 A和CN 102675101 A涉及一种关键中间体(Ⅲ)的合成方法,其包括在路易斯酸存在下2-甲基-2-苯基丙酸或其酯的酰化以及随后发生的氧代基团的还原。这种方法尽管在工业上适用,但在酰化和还原步骤中均使用化学计量试剂,会产生大量的残留物。

综上,根据本发明记载的现有技术公开的式(Ⅲ)化合物的制备方法,其需要使用有机金属试剂、有毒试剂、苛刻的反应条件或具有较低的生产率,因此不适合工业化生产。
因此,有必要开发一种获得式(Ⅲ)化合物的新方法,所述式(Ⅲ)化合物是合成比拉斯汀的关键中间体,该方法克服了与现有技术中的已知方法相关的全部或部分问题。
发明内容
本发明面临的问题是提供一种制备式(Ⅲ)化合物及其中间体的新方法。
与现有技术的方法相比,本发明的方法满足工业生产的需要。其可以在没有有机金属和有毒试剂的情况下,在较短的反应时间内制备式(Ⅲ)化合物,这是一种简单且低成本的方法,并且能以高产率得到目标产物。
发明人发现,通过对易得和经济的起始原料进行酰化和随后的氧化重排,可以以一种非常直接的方式获得式(Ⅲ)化合物。如实验部分所示,该方法不仅避免了使用剧毒、不稳定和/或昂贵的试剂,而且相比现有技术的方法,以更短的反应时间和更高的产率得到式(Ⅲ)化合物。所有这些特征使得本发明的方法具有高成本效益,因此非常适合工业规模生产。
通过本发明的方法获得的式(Ⅲ)化合物已经包括了随后与哌啶基化合物反应所需的离去基团,因此在制备比拉斯汀时不需要额外的反应步骤。
因此,在第一方面,本发明涉及一种式(Ⅲ)化合物

或其溶剂化物的制备方法,其中,
X为离去基团;且
R1为C1-C6烷基;
该方法包含式(II)化合物或其溶剂化物的氧化重排,

在第二方面,本发明涉及式(II)化合物

或其溶剂化物作为中间体在制备比拉斯汀中的应用,其中X为离去基团。
在第三方面,本发明涉及式(II’)化合物

或其溶剂化物,其中X选自Cl、I、OMs、OTs和OTf。
发明的详细说明
术语“烷基”是指包含1至6个(“C1-C6烷基”),优选1至3个(“C1-C3烷基”)碳原子的直链或支链烷烃衍生物,其通过单键与分子的其余部分相结合。烷基的示例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基、己基。优选为甲基。
术语“芳基”是指具有6至10个(“C6-C10芳基”),优选6个或10个碳原子,由1或2个芳香核彼此稠合而成的芳香族基团。芳基的示例包括苯基、萘基、茚基、菲基等。优选为苯基。
术语“(C6-C10)芳基(C1-C6)烷基”是指被如上定义的芳基基团取代的如上定义的烷基基团。这类基团的例子包括苄基、苯乙基、苯丙基、萘甲基等。优选为苄基。
术语“卤代烷基”是指含有1-6个(“C1-C6卤代烷基”),优选1-3个(“C1-C3卤代烷基”)碳原子的如上定义的烷基基团中至少一个氢原子被卤素取代。卤代烷基的例子包括但不限于CF3、CCl3、CHF2、CF2CF3。优选为-CF3。
术语“卤素”是指溴、氯、碘或氟。
术语“离去基团”是指在取代反应例如亲核取代反应中可被另一个官能团取代的官能团或原子。合适的离去基团在本领域是公知的。在某一具体实施方案中,所述离去基团选自卤素、C1-C6烷基磺酸盐、C1-C6卤代烷基磺酸盐和(C1-C6)烷基(C6-C10)芳基磺酸盐,例如氯、溴、碘、甲磺酸盐(OMs)、三氟甲磺酸盐(OTf)、甲苯磺酸盐(OTs)等。
本发明还涉及本说明书中所描述的化合物的“盐”。举例来说,所述盐可为酸加成盐、碱加成盐或金属盐,并且可以由含有碱性或酸性基团的母体化合物通过本领域已知的常规化学方法合成。所述盐一般是通过例如将上述化合物的游离酸或碱的形式与化学计量的合适的碱或酸在水或有机溶剂中或在两者的混合物中反应来制备。通常优选非水介质,例如醚、乙酸乙酯、乙醇、丙酮、异丙醇或乙腈。所述酸加成盐的示例包括无机酸加成盐,例如盐酸盐、氢溴酸盐、氢碘化物、硫酸盐、硝酸盐、磷酸盐等。有机酸加成盐,例如乙酸盐、马来酸盐、富马酸盐、柠檬酸盐、草酸盐、琥珀酸盐、酒石酸盐、苹果酸盐、扁桃酸盐、甲磺酸盐、对甲苯磺酸盐、三氟乙酸盐、樟脑磺酸盐等。碱加成盐的示例包括无机碱盐,例如铵盐和有机碱盐,例如乙二胺、乙醇胺、N,N-二烷基乙醇胺、三乙醇胺、谷氨酰胺、氨基酸碱性盐等。金属盐的示例包括例如钠盐、钾盐、钙盐、镁盐、铝盐和锂盐。在某一具体实施方案中,该盐是酸加成盐,例如盐酸盐、氢溴酸盐、氢碘化物、硫酸盐、硝酸盐、磷酸盐、乙酸盐、马来酸盐、富马酸盐、柠檬酸盐、草酸盐、琥珀酸盐、酒石酸盐、苹果酸盐、扁桃酸盐、甲磺酸盐、对甲苯磺酸盐、三氟乙酸盐或樟脑磺酸盐。较佳地,所述盐选自HCl、HBr、H3PO4、H2SO4、MsOH、pTsOH、TFA、柠檬酸盐和富马酸盐。
同样,本说明书中描述的化合物可以以游离化物或溶剂化物(例如水合物,醇化物等)的形式获得或使用,这两种形式均包括在本发明的范围内。溶剂化方法在现有技术中通常是已知的。溶剂化物优选为水合物。
术语“有机溶剂”包括例如环醚和无环醚(例如Et2O、iPr2O、tBu2O、MeOtBu、1,4-二氧六环,四氢呋喃,甲基四氢呋喃),烃类溶剂(例如戊烷、己烷、庚烷),卤代溶剂(例如二氯甲烷、氯仿、氯苯),芳香族溶剂(例如甲苯、二甲苯),酮类溶剂(例如丙酮、丁酮、戊酮、甲基乙基酮、乙基异丙基酮),酯类溶剂(例如EtOAc、iPrOAc),腈类溶剂(例如乙腈、苄腈),酰胺类溶剂(例如DMF、DMA、HMPA、NMP),醇类溶剂(例如甲醇、乙醇、丙醇、异丙醇、仲丁醇、叔丁醇),亚砜类溶剂(DMSO)及其混合物。
在第一方面,本发明涉及一种式(Ⅲ)化合物

或其溶剂化物的制备方法,其中,
X为离去基团;且
R1为C1-C6烷基;
该方法包括式(II)化合物或其溶剂化物的氧化重排,

在某一实施方案中,X选自Cl、Br、I、OMs、OTs和OTf。较佳地,X选自Cl、Br和I;更佳地,X选自Cl和Br;进一步更佳地X为C1。
在另一个优选实施方案中,R1为C1-C3烷基;优选为Me或Et;R1更优选为Me。
在某一具体实施方案中,本发明的方法包括:
(a)式(I)化合物或其溶剂化物的酰化,

以提供式(II)化合物或其溶剂化物,

其中X是离去基团,和
(b)式(II)化合物或其溶剂化物的氧化重排,以提供式(III)化合物或其溶剂化物,

其中R1为C1-C6烷基。
在某一实施方案中,X选自Cl、Br和I,R1为C1-C3烷基。
尽管式(II)化合物在芳环的β-位含有离去基团,但发明人发现这些化合物的氧化重排进行得非常有效(例如Olah et al.,J.Am.Chem.Soc.1982,104,5168-5172报道了这类化合物在酸性条件下的不稳定性)。因此,该方法可以以非常直接的方式制备式(III)化合物。
此外,发明人发现本发明的方法以高产率和较短的反应时间得到式(III)化合物。它不需要使用有机金属或有毒试剂,简单且成本低,并能以高产率得到目标产物。因此,该方法非常适合工业规模生产。
在某一具体实施方案中,本发明的方法进一步包括将式(III)化合物或其溶剂化物转化为比拉斯汀或其盐或其溶剂化物。
式(III)化合物可以通过现有技术中的已知方法转化成比拉斯汀(例如WO 2009/102155,CN 104326909 A,CN 102675101 A,Synthetic Communications 2011,41(9),1394-1402)。
在某一具体实施方案中,将式(III)化合物或其溶剂化物转化为比拉斯汀或其盐或其溶剂化物,包括:
(c)使式(III)化合物或其溶剂化物

和式(IV)化合物或其溶剂化物反应,


以提供式(V)化合物或其溶剂化物,

其中,X和R1的定义如前所述,R2选自H和-CH2CH2OEt;和
(d)将式(V)化合物或其盐或溶剂化物转化为比拉斯汀或其盐或其溶剂化物。
当式(IV)化合物或其盐或溶剂化物中的R2为CH2CH2OEt时,在步骤(c)之后获得R2为CH2CH2OEt的式(V)化合物。在这种情况下,步骤(d)包括将R2为CH2CH2OEt的式(V)化合物或其盐或溶剂化物中的酯基进行水解,以提供比拉斯汀,或其盐或溶剂化物。
当式(IV)化合物或其盐或溶剂化物中的R2为H时,在步骤(c)之后获得R2为H的式(V)化合物。在这种情况下,步骤(d)包括:
(d1)使R2为H的式(V)化合物,或其盐或溶剂化物和式(VI)化合物反应,

其中,Y为离去基团,
以提供R2为-CH2CH2OEt的式(V)化合物或其盐或溶剂化物;和
(d2)将R2为-CH2CH2OEt的式(V)化合物或其盐或溶剂化物中的酯基水解,以提供比拉斯汀或其盐或溶剂化物。
在某一实施方案中,Y选自Cl、Br、I、OMs、OTs和OTf。
发明人惊奇地发现,如前所述的R2为H的式(V)化合物或其盐或溶剂化物和式(VI)化合物的反应,可以在酯基水解的反应条件下进行,从而直接由R2为H的式(V)化合物或其盐或溶剂化物得到比拉斯汀或其盐或溶剂化物。
这样,当式(V)化合物或其盐或溶剂化物中的酯基水解在步骤(d1)所用的反应条件下进行时,以非常直接的方式获得比拉斯汀:从式(I)起始化合物出发,仅需4个合成步骤。
令人惊奇的是,与现有技术中的其它方法相比,所述方法以非常有效的方式进行,以高收率和纯度直接得到比拉斯汀,并且避免了额外的水解步骤。
在某一具体实施方案中,步骤(d1)和(d2)以一锅法进行。即,这两个步骤在同一反应容器或同一反应器中进行,而无需分离出R2为-CH2CH2OEt的式(V)中间体。这避免了中间体的冗长的分离过程和纯化过程。因此,所述一锅法缩短了获得比拉斯汀所需要的总反应步数,并节省了时间和资源。
在另一实施方案中,式(V)化合物或其盐或溶剂化物中的酯基水解在步骤(d1)所用的反应条件下进行。在这种情况下,比拉斯汀或其盐或溶剂化物直接从R2为H的式(V)化合物或其盐或溶剂化物获得。
因此,在本发明的另一实施方案中,步骤(d)包括使R2为H的式(V)化合物或其盐或溶剂化物和式(VI)化合物反应,

其中Y是离去基团,
并且将式(V)化合物或其盐或溶剂化物中的酯基进行水解,以提供比拉斯汀或其盐或溶剂化物。
在这种情况下,与式(VI)化合物的反应以及酯基的水解都是在单个反应步骤中进行的。
在另一实施方案中,分离步骤(d1)后获得的化合物,可在步骤(d2)之前进行纯化。
根据本发明的另一实施方案中,将式(III)化合物或其溶剂化物转化为比拉斯汀或其盐或溶剂化物,包括:
(c’)式(III)化合物或其溶剂化物的水解,其中X和R1的定义如前所述,


以提供式(III’)化合物或其盐或溶剂化物;

(d’)使式(III’)化合物或其盐或溶剂化物和式(IV)化合物或其盐或溶剂化物反应,其中R2选自H和-CH2CH2OEt;

以提供(V’)化合物或其盐或溶剂化物;和

(e’)如有需要,将式(V’)化合物或其盐或溶剂化物转化为比拉斯汀或其盐或溶剂化物。
如果式(IV)化合物中的R2为H,则需要步骤(e’)。
另外,当式(IV)化合物中的R2为-CH2CH2OEt时,在步骤(d’)后得到比拉斯汀。在这种情况下,上述将式(III)化合物转化为比拉斯汀的方法仅包括步骤(c’)和(d’)。
当式(IV)化合物或其盐或溶剂化物中的R2为H时,在步骤(d’)之后获得R2为H的式(V’)化合物。在这种情况下,步骤(e’)包括将R2为H的式(V’)化合物或其盐或溶剂化物和式(VI)化合物反应,其中Y为离去基团,

以提供比拉斯汀或其盐或溶剂化物。
在某一实施方案中,Y选自Cl、Br、I、OMs、OTs和OTf。
较佳地,根据本发明制备比拉斯汀的方法,其包括依次公开的上述步骤(步骤(a)至(d)或步骤(a)至(e′)),而没有其它的反应步骤(例如官能团的保护或去保护)。即,在某一优选实施方案中,根据本发明制备比拉斯汀的方法,其包括上述公开的反应步骤(步骤(a)至(d)或步骤(a)至(e′))。除非另有说明,该方法可以包括在部分或所有这些反应步骤之后的分离和纯化步骤。
式(II)化合物的氧化重排
在某一实施方案中,在氧化剂和三(C1-C6)烷基原酸酯或(C1-C6)烷醇或其混合物的存在下,进行式(II)化合物或其溶剂化物的氧化重排,以提供式(III)化合物或其溶剂化物。
在某一实施方案中,三(C1-C6)烷基原酸酯选自三甲基和三乙基原酸酯。
在某一具体实施方案中,三乙基-三甲基原酸酯选自三乙基-三甲基原甲酸酯。
在某一实施方案中,(C1-C6)烷醇选自MeOH和EtOH。
较佳地,以三(C1-C6)烷基原酸酯、(C1-C6)烷醇或其混合物作为反应溶剂。
合适的氧化剂包括碘氧化剂,例如I2、ICl、ICl3、HIO3、PhI(OAc)2、PhI(OCOCF3)2、PhI(OTf)2、PhI(OH)OTs、PhIO、NIS、IBX、DMP。在某一实施方案中,氧化剂选自I2、ICl、HIO3和PhI(OAc)2。
在某一具体实施方案中,相对于式(II)的化合物,氧化剂的量为1.0至10.0摩尔当量;优选1.0至5.0,更优选1.0至3.0摩尔当量。
在某一优选实施方案中,该反应是在酸的存在下进行。可用作催化剂的合适的酸包括无机酸和有机酸,例如硫酸、盐酸、氢溴酸、硝酸、磷酸、乙酸、三氟乙酸、樟脑磺酸、对甲苯磺酸、甲磺酸、马来酸、富马酸、柠檬酸、草酸、琥珀酸、酒石酸和苹果酸。在某一具体实施方案中,所述酸选自HCl、HBr、H3PO4、H2SO4、MsOH、pTsOH和TFA。所述酸优选为H2SO4或HCl。
较佳地,该酸催化剂由酸前体原位生成。因此,在某一优选实施方案中,该反应在酸前体的存在下进行。合适的酸前体是技术人员公知的,包括氯化烷基酸或烷基氯甲酸酯,例如2,2-二甲基丙酰氯、乙酰氯、丙酰氯、氯甲酸乙酯、氯甲酸甲酯、氯甲酸丙酯、氯甲酸苄酯、氯甲酸三氯甲酯。在某一实施方案中,该反应在盐酸前体如2,2-二甲基丙酰氯或氯甲酸乙酯的存在下进行。
在某一具体实施方案中,相对于式(II)化合物,该酸的量为0.01至0.9摩尔当量;优选0.05至0.6,更优选0.1至0.5摩尔当量。
在某一实施方案中,该反应是在-20℃和150℃之间的温度下进行的,优选在20℃和130℃之间的温度,优选在20℃和100℃之间的温度。
在本发明某一实施方案中,所述氧化重排在三(C1-C6)烷基原酸酯、(C1-C6)烷醇或其混合物、氧化剂和酸催化剂的存在下进行。
在某一具体实施方案中,氧化重排在三(C1-C6)烷基原酸酯、碘氧化剂和酸催化剂的存在下进行。
在某一具体实施方案中,该反应在没有溶剂(除了用于反应的试剂之外)的情况下进行。较佳地,三(C1-C6)烷基原酸酯作为反应的溶剂。
在另一实施方案中,氧化重排在有机溶剂存在下进行。在某一实施方案中,有机溶剂选自环醚和无环醚(例如Et2O、iPr2O、tBu2O、MeOtBu、1,4-二氧六环、THF、2-甲基四氢呋喃),烃类溶剂(例如戊烷、己烷、庚烷),卤代溶剂(例如二氯甲烷、氯仿、氯苯),芳香族溶剂(例如甲苯、二甲苯),醇类溶剂(例如甲醇、乙醇、丙醇、异丙醇、仲丁醇、叔丁醇)及其混合物。所述有机溶剂优选为醇类溶剂;更优选C1-6烷醇,例如甲醇、乙醇、丙醇、异丙醇、仲丁醇、叔丁醇;进一步优选为甲醇。
在某一实施方案中,所述氧化重排是在原甲酸三甲酯(TMOF)、碘氧化剂和酸催化剂存在下进行的。
在另一实施方案中,所述氧化重排是在原甲酸三甲酯(TMOF)、碘氧化剂、酸催化剂和有机溶剂的存在下进行。
在另一实施方案中,所述氧化重排是在原甲酸三甲酯(TMOF),选自I2、ICl、HIO3和PhI(OAc)2的碘氧化剂和选自HCl、HBr、H3PO4、H2SO4、MsOH、pTsOH和TFA的酸的存在下进行。
在另一实施方案中,所述氧化重排在原甲酸三甲酯(TMOF),选自I2、ICl、HIO3和PhI(OAc)2的碘氧化剂,选自HCl、HBr、H3PO4、H2SO4、MsOH、pTsOH和TFA的酸以及有机溶剂的存在下进行。
在另一实施方案中,所述氧化重排在原甲酸三甲酯(TMOF),选自I2、ICl、HIO3和PhI(OAc)2的碘氧化剂,选自H2SO4和HCl的酸的存在下进行。
在另一实施方案中,所述氧化重排在原甲酸三甲酯(TMOF),选自I2、ICl、HIO3和PhI(OAc)2的碘氧化剂,选自H2SO4和HCl的酸以及有机溶剂的存在下进行。
式(I)化合物的酰化
式(I)化合物或其溶剂化物的酰化是在合适的酰化试剂存在下进行的。在某一实施方案中,酰化试剂选自(iPrCO)2O和具有iPrCO-Z分子式的化合物,其中Z选自OH、Cl、Br和I。较佳地,酰化试剂为iPrCO-Cl。
在某一具体实施方案中,相对于式(I)化合物,所述酰化试剂的量为1.0至10.0摩尔当量;优选1.0至5.0,更优选1.0至3.0摩尔当量。
在某一实施方案中,所述反应在质子酸和/或路易斯酸的存在下进行。在现有技术中,用于傅克酰基化的合适的质子酸和路易斯酸是已知的。在某一实施方案中,所述酸选自AlCl3、AlBr3、FeCl3、FeBr3、BF3、BBr3、BCl3、ZnO、ZnCl2、ZnBr2、TiCl4、SnCl4、SiCl4、POCl3、FeSO4及其水合物或溶剂化物、HCl、H2SO4、H3PO4、HClO4、HBF4、ClSO3H、MsOH和TfOH。所述酸优选为AlCl3。
在某一具体实施方案中,相对于式(I)化合物,所述酸的量为0.1至10.0摩尔当量;优选0.1至5.0,更优选0.5至3.0摩尔当量。
在某一具体实施方案中,所述酰化反应在有机溶剂的存在下进行。合适的溶剂包括,例如醚、烃类溶剂、卤代溶剂、芳香族溶剂、酮类溶剂、酯类溶剂及其混合物。在某一实施方案中,所述溶剂为烃类溶剂,例如戊烷、己烷或庚烷。在某一实施方案中,所述反应在非极性有机溶剂的存在下进行,例如醚(例如Et2O、iPr2O、tBu2O、MeOtBu、1,4-二氧六环)、烃类溶剂(例如戊烷、己烷、庚烷)、卤代溶剂(例如二氯甲烷、氯仿)或芳香族溶剂(例如甲苯、二甲苯)。
在另一优选实施方案中,进行无溶剂反应,即所述反应在无溶剂下进行。
所述反应优选在无溶剂或在非极性有机溶剂存在下进行。
在某一实施方案中,所述反应在-78℃和100℃之间的温度下进行,优选在-20℃和60℃之间的温度,优选在0℃和40℃之间的温度。
在某一具体实施方案中,所述酰化反应在合适的酰化试剂和酸的存在下进行。
在另一实施方案中,酰化反应在酰化试剂和质子酸和/或路易斯酸的存在下进行,所述酰化试剂选自(iPrCO)2O和具有iPrCO-Z分子式的化合物,其中Z选自OH、Cl、Br和I。
在某一优选实施方案中,所述酰化反应在iPrCO-Cl和AlCl3的存在下进行。
式(III)或(III’)化合物与式(IV)化合物的反应
式(III)或(III’)化合物分别与式(IV)化合物反应以提供式(V)或(V’)化合物,其可如先前在现有技术中公开的那样进行。
在本发明的某一具体实施方案中,所述反应在碱和有机溶剂的存在下进行。
合适的碱包括无机和有机碱,例如碱金属碳酸盐或碳酸氢盐(例如Na2CO3、K2CO3、Cs2CO3、Li2CO3、NaHCO3、KHCO3、CsHCO3、LiHCO3)、碱金属磷酸盐(例如Na3PO4、K3PO4、Na2HPO4、K2HPO4、NaH2PO4、KH2PO4)、碱金属烷氧化物(例如NaOMe、KOMe、NaOEt、KOEt、NaOtBu、KOtBu)、碱金属氢氧化物(例如NaOH、KOH、LiOH、CsOH)、脂肪族或芳香族胺(例如Me2NH、Et2NH、iPr2NH、Bu2NH、Me3N、Et3N、Bu3N、iPr2EtN、N-甲基吗啉、吡啶、DMAP、苯胺、N,N-二甲基苯胺)。所述碱优选为无机碱,更优选为碱金属碳酸盐、碳酸氢盐或磷酸盐。
在某一实施方案中,所述有机溶剂为极性有机溶剂,例如THF、酮(例如丙酮、丁酮、戊酮、甲基乙基酮、乙基异丙基酮)、酯(例如EtOAc、iPrOAc)、腈(例如乙腈、苄腈)、酰胺(例如DMF、DMA、HMPA、NMP)、亚砜(DMSO)、醇(例如甲醇、乙醇、丙醇、异丙醇、仲丁醇、叔丁醇)或其混合物。在另一实施方案中,所述有机溶剂为极性非质子有机溶剂,例如THF、酮(例如丙酮、丁酮、戊酮、甲基乙基酮、乙基异丙基酮)、酯(例如EtOAc、iPrOAc)、腈(例如乙腈、苄腈)、酰胺(例如DMF、DMA、HMPA、NMP)、亚砜(DMSO)或其混合物。
在某一优选实施方案中,所述反应在无机碱和极性非质子有机溶剂存在下进行。
在某一实施方案中,所述反应在20℃和180℃之间的温度下进行,优选在20℃和160℃之间的温度,优选在50℃和150℃之间的温度。
R2为氢的式(V)或(V’)化合物与式(VI)化合物的反应
R2为氢的式(V)或(V’)化合物分别与式(VI)化合物的反应以提供R2为-CH2CH2OEt的式(V)化合物或比拉斯汀,其可如先前在现有技术中公开的那样进行。
在本发明某一具体实施方案中,所述反应在碱和有机溶剂存在下进行。
合适的碱包括无机和有机碱,例如碱金属碳酸盐或碳酸氢盐(例如Na2CO3、K2CO3、Cs2CO3、Li2CO3、NaHCO3、KHCO3、CsHCO3、LiHCO3)、碱金属磷酸盐(例如Na3PO4、K3PO4、Na2HPO4、K2HPO4、NaH2PO4、KH2PO4)、碱金属烷氧化物(例如NaOMe、KOMe、NaOEt、KOEt、NaOtBu、KOtBu)、碱金属氢氧化物(例如NaOH、KOH、LiOH、CsOH)、脂肪族或芳香族胺(例如Me2NH、Et2NH、iPr2NH、Bu2NH、Me3N、Et3N、Bu3N、iPr2EtN、N-甲基吗啉、吡啶、DMAP、苯胺、N,N-二甲基苯胺)。所述碱优选为无机碱,更优选为碱金属氢氧化物或碱金属烷氧化物;进一步优选为碱金属氢氧化物例如NaOH、KOH、LiOH或CsOH。
在某一实施方案中,所述有机溶剂为极性有机溶剂,例如THF、酮(例如丙酮、丁酮、戊酮、甲基乙基酮、乙基异丙基酮)、酯(例如EtOAc、iPrOAc)、腈(例如乙腈、苄腈)、酰胺(例如DMF、DMA、HMPA、NMP)、亚砜(DMSO)、醇(例如甲醇、乙醇、丙醇、异丙醇、仲丁醇、叔丁醇)或其混合物。在另一实施方案中,所述有机溶剂为极性非质子有机溶剂,例如THF、酮(例如丙酮、丁酮、戊酮、甲基乙基酮、乙基异丙基酮)、酯(例如EtOAc、iPrOAc)、腈(例如乙腈、苄腈)、酰胺(例如DMF、DMA、HMPA、NMP)、亚砜(DMSO)或其混合物。
在某一优选实施方案中,所述反应在无机碱和极性有机溶剂存在下进行,所述无机碱优选碱金属氢氧化物。
在某一实施方案中,所述反应在0℃和180℃之间的温度下进行,优选在20℃和160℃之间的温度,优选在30℃和100℃之间的温度。
在本发明的某一优选实施方案中,在R2为H的式(V)化合物与式(VI)化合物反应的反应条件下将酯基水解为羧酸。在这种情况下,无需额外的反应步骤直接获得比拉斯汀或其盐或溶剂化物。在某一具体实施方案中,其是在无机碱和极性有机溶剂(例如DMSO)的存在下进行,所述无机碱优选碱金属氢氧化物(例如KOH或NaOH)。
酯基的水解
R2为-CH2CH2OEt的式(V)化合物或式(III)化合物中的酯基分别水解以提供比拉斯汀或式(III’)化合物,其可如先前在现有技术中公开的那样进行。
在某一实施方案中,所述反应通过酸性或碱性水解进行。在某一具体实施方案中,通过在加热条件下用酸或碱处理来进行水解。合适的酸包括乙酸、三氟乙酸、甲磺酸、三氟甲磺酸、HCl、HBr、HF、HClO4、H2SO4、HNO3、H3PO4、甲酸、丙酸、丁酸、苹果酸、柠檬酸、苯甲酸、对甲苯磺酸、草酸和琥珀酸,优选HCl、HBr和H2SO4。合适的碱包括碱金属碳酸盐或碳酸氢盐(例如Na2CO3、K2CO3、Cs2CO3、Li2CO3、NaHCO3、KHCO3、CsHCO3、LiHCO3)、碱金属磷酸盐(例如Na3PO4、K3PO4、Na2HPO4、K2HPO4、NaH2PO4、KH2PO4)、碱金属烷氧化物(例如NaOMe、KOMe、NaOEt、KOEt、NaOtBu、KOtBu)和碱金属氢氧化物(例如NaOH、KOH、LiOH、CsOH)。所述碱优选为碱金属氢氧化物或碱金属烷氧化物;更优选为碱金属氢氧化物例如NaOH、KOH、LiOH或CsOH。
在某一实施方案中,所述反应在水、有机溶剂或其混合物存在下进行。
在某一具体实施方案中,所述反应在有机溶剂存在下进行,所述有机溶剂优选为极性有机溶剂例如THF、酮(例如丙酮、丁酮、戊酮、甲基乙基酮、乙基异丙基酮)、酯(例如EtOAc、iPrOAc)、腈(例如乙腈、苄腈)、酰胺(例如DMF、DMA、HMPA、NMP)、亚砜(DMSO)、醇(例如甲醇、乙醇、丙醇、异丙醇、仲丁醇、叔丁醇)或其混合物。
在某一优选实施方案中,所述反应在无机碱和极性有机溶剂存在下进行,所述无机碱优选为碱金属氢氧化物。
在某一实施方案中,所述反应在0℃和180℃之间的温度下进行,优选在20℃和160℃之间的温度,优选在30℃和100℃之间的温度。
在某一优选实施方案中,酯基的水解是在R2为H的式(V)化合物与式(VI)化合物反应所使用的碱性条件下进行,因此,在单个反应步骤中直接由R2为H的式(V)化合物或其盐或溶剂化物获得比拉斯汀或其盐或溶剂化物。
在另一方面,本发明涉及使用式(II)化合物或其溶剂化物作为中间体制备比拉斯汀,其中X为离去基团。

在某一实施方案中,X选自Cl、Br、I、OMs、OTs和OTf;X优选为Cl或Br,更优选为Cl。
在另一方面,本发明涉及式(II’)化合物或其溶剂化物,其中,X选自Cl、I、OMs、OTs和OTf。

在某一优选实施方案中,X为Cl。
应该理解,本文公开的范围包括本文公开的实施例的所有可能的组合。
实施方式
实施例1 1-(4-(2-氯乙基)苯基)-2-甲基丙基-1-酮的制备

在反应容器中,将iPrCOCl(8.2g)冷却至9℃,并与AlCl3(5.2g)混合。逐滴加入2-氯乙基苯(4.3g),并将混合物搅拌60分钟,在0℃下倒入HCl 1M(50mL)。加入TBME(10mL)并搅拌混合物。用NaOH 1M(25mL)洗涤有机层,分离,用无水硫酸钠干燥,然后浓缩,真空下除去溶剂。获得7.4克1-(4-(2-氯乙基)苯基)-2-甲基丙基-1-酮,产率100%,纯度90%。
实施例2 2-[4-(2-氯乙基)-苯基]-2-甲基-丙酸甲酯的制备

在装有回流冷凝器的圆底烧瓶中,向1-[4-(2-氯-乙基)-苯基]-2-甲基-丙基-1-酮(13.42g)的TMOF(55mL)的溶液中加入ICl(9.6mL)。这种混合物在放热反应中反应相当剧烈。几分钟后,当鼓泡和回流停止时,将混合液冷却,并按如下方式进行处理:用饱和碳酸氢钠溶液(120mL)淬灭,用二氯甲烷(80mL×3)萃取。用Na2S2O3溶液(10%,150mL)洗涤有机层,用无水硫酸钠干燥并浓缩。得到15.03克2-[4-(2-氯-乙基)-苯基]-2-甲基-丙酸甲酯,收率98%,纯度85%。
实施例3 2-[4-(2-氯-乙基)-苯基]-2-甲基-丙酸甲酯的制备

将I2(794mg)加入至装有1-[4-(2-氯-乙基)-苯基]-2-甲基-丙基-1-酮(200mg)和TMOF(913μL)的圆底烧瓶中。在加入硫酸(22μL)之前,将该混合物搅拌5分钟。此时,将容器置于80℃下加热。1小时后,加入碳酸氢钠溶液(5mL)进行处理,然后用二氯甲烷(5mL×3)萃取。用Na2S2O3溶液(20mL)洗涤有机层,用无水硫酸钠干燥,并在真空下浓缩。得到0.226g 2-[4-(2-氯-乙基)-苯基]-2-甲基-丙酸甲酯,收率99%,纯度86%。
实施例4 2-[4-(2-氯-乙基)-苯基]-2-甲基-丙酸甲酯的制备
将I2(290mg)溶于1.65mL的0.23M的HCl的MeOH溶液中。将其与1-[4-(2-氯-乙基)-苯基]-2-甲基-丙基-1-酮(200mg)和TMOF(400mg)混合。将该混合物在100℃下加热10分钟,冷却至室温,并倒入5mL的Na2S2O3溶液。用DCM(2×5mL)萃取混合物。用Na2S2O3溶液(5mL)洗涤有机层,用无水硫酸钠干燥,并在真空下浓缩。得到0.220克2-[4-(2-氯-乙基)-苯基]-2-甲基-丙酸甲酯,收率96%,纯度70%。
另外,[4-(2-氯-乙基)-苯基]-2-甲基-丙酸甲酯也可按实施例4的方法获得,但使用氯甲酸乙酯作为酸源,而不是HCl的MeOH溶液。
产率的比较
在下表中,将本发明的工艺的产率和工艺周期时间与现有技术中的最佳或最接近的实施例的产率和工艺周期时间进行比较。所述产率是指基于该方法中使用的原料、试剂和溶剂的总量而获得的式(III)产物的量。这是衡量一个工艺的成本效益的标准,因此对工业规模生产具有重要意义。


[a]专利CN 102675101 A不包含详细的实验过程。
[b]测量反应步骤中使用的原料、试剂和溶剂的总量。不考虑后处理过程中使用的溶剂和溶液。
[c]没有后处理,仅考虑反应时间。
[d]由于没有溶剂体积的数据,仅考虑甲苯中1M的浓度(典型的商品化描述)。
如上所示,本发明的方法可以以非常高的产率和较短的反应时间制备式(III)化合物。因此,这种方法非常节省成本并且非常适合工业化生产。此外,该方法不需要像其它现有技术方法那样使用有机金属或剧毒性试剂。最后,所得到的式(III)化合物已经包括随后与哌啶基化合物反应所需的离去基团,因此在制备比拉斯汀时不需要额外的反应步骤。
Claims (15)
1.一种式(III)化合物或其溶剂化物的制备方法,

其中,
X为离去基团;且
R1为C1-C6烷基;
所述方法包括式(II)化合物或其溶剂化物的氧化重排,

2.如权利要求1所述的制备方法,其包括:
(a)式(I)化合物或其溶剂化物的酰化,其中X是离去基团,

以提供式(II)化合物或其溶剂化物,

和
(b)式(II)化合物或其溶剂化物的氧化重排,以提供式(III)化合物或其溶剂化物,

其中R1为C1-C6烷基。
3.如权利要求1所述的制备方法,其中X选自Cl、Br、I、OMs、OTs和OTf。
4.如权利要求1-3任一项所述的制备方法,所述氧化重排在三(C1-C6)烷基原酸酯、(C1-C6)烷醇或其混合物、氧化剂和酸催化剂的存在下进行。
5.如权利要求4任一项所述的制备方法,所述氧化剂为碘氧化剂,例如I2、ICl、ICl3、HIO3、PhI(OAc)2、PhI(OCOCF3)2、PhI(OTf)2、PhI(OH)OTs、PhIO、NIS、IBX或DMP。
6.如权利要求4或5任一项所述的制备方法,所述酸催化剂选自硫酸、盐酸、氢溴酸、硝酸、磷酸、乙酸、三氟乙酸、樟脑磺酸、对甲苯磺酸、甲磺酸、马来酸、富马酸、柠檬酸、草酸、琥珀酸、酒石酸和苹果酸。
7.如权利要求2-6任一项所述的制备方法,所述酰化在酰化试剂的存在下进行,所述酰化试剂选自(iPrCO)2O和具有iPrCO-Z分子式的化合物,其中Z选自OH、Cl、Br和I。
8.如权利要求2-7任一项所述的制备方法,所述酰化在质子酸和/或路易斯酸存在下进行。
9.如权利要求1-8任一项所述的制备方法,其包括将式(III)化合物或其溶剂化物转化为比拉斯汀或其盐或溶剂化物。
10.如权利要求1-9任一项所述的制备方法,其进一步包括:
(c)使式(III)化合物或其溶剂化物

和式(IV)化合物或其溶剂化物反应,


以提供式(V)化合物或其溶剂化物,

其中,X为离去基团;且R1为C1-C6烷基;
R2选自H和-CH2CH2OEt;和
(d)将式(V)化合物或其盐或溶剂化物转化为比拉斯汀或其盐或其溶剂化物。
11.如权利要求10所述的制备方法,其中式(IV)和(V)化合物中的R2为-CH2CH2OEt,且步骤(d)包括将式(V)化合物或其盐或溶剂化物中酯基进行水解,以提供比拉斯汀,或其盐或溶剂化物。
12.如权利要求10所述的制备方法,其中式(IV)和(V)化合物中的R2为H,且步骤(d)包括:
(d1)使R2为H的式(V)化合物或其盐或溶剂化物和式(VI)化合物反应,

其中,Y为离去基团,
以提供R2为-CH2CH2OEt的式(V)化合物或其盐或溶剂化物;和
(d2)将R2为-CH2CH2OEt的式(V)化合物或其盐或溶剂化物中的酯基水解,以提供比拉斯汀或其盐或溶剂化物。
13.如权利要求10-12任一项所述的制备方法,其中步骤(d)包括在单个反应步骤中使R2为H的式(V)化合物或其盐或溶剂化物和式(VI)化合物反应,

其中Y为离去基团,
且水解式(V)化合物或其盐或溶剂化物中的酯基,以提供比拉斯汀或其盐或溶剂化物。
14.如权利要求1-9任一项所述的制备方法,其进一步包括:
(c’)水解式(III)化合物或其溶剂化物,其中X为离去基团;且R1为C1-C6烷基,

以提供式(III’)化合物或其盐或溶剂化物;

(d’)使式(III’)化合物或其盐或溶剂化物和式(IV)化合物或其盐或溶剂化物反应,其中R2选自H和-CH2CH2OEt;

以提供式(V’)化合物或其盐或溶剂化物;和

(e’)如有需要,将式(V’)化合物或其盐或溶剂化物转化为比拉斯汀或其盐或溶剂化物。
15.一种式(II’)化合物或其溶剂化物,

其中X选自Cl、I、OMs、OTs和OTf。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18382556.1 | 2018-07-24 | ||
| EP18382556.1A EP3599235A1 (en) | 2018-07-24 | 2018-07-24 | Process and intermediates for the preparation of bilastine |
| PCT/EP2019/069772 WO2020020873A1 (en) | 2018-07-24 | 2019-07-23 | Process and intermediates for the preparation of bilastine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112469709A true CN112469709A (zh) | 2021-03-09 |
Family
ID=63103892
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980049580.4A Pending CN112469709A (zh) | 2018-07-24 | 2019-07-23 | 比拉斯汀的制备方法及其中间体 |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US11370742B2 (zh) |
| EP (2) | EP3599235A1 (zh) |
| JP (1) | JP7145312B2 (zh) |
| KR (1) | KR20210038905A (zh) |
| CN (1) | CN112469709A (zh) |
| AU (1) | AU2019311251B2 (zh) |
| BR (1) | BR112021001179A2 (zh) |
| CA (1) | CA3107879A1 (zh) |
| CL (1) | CL2021000171A1 (zh) |
| CO (1) | CO2021000940A2 (zh) |
| CY (1) | CY1124909T1 (zh) |
| EA (1) | EA202190346A1 (zh) |
| EC (1) | ECSP21008461A (zh) |
| ES (1) | ES2901786T3 (zh) |
| HR (1) | HRP20211994T1 (zh) |
| HU (1) | HUE056822T2 (zh) |
| LT (1) | LT3826997T (zh) |
| MA (1) | MA53370B1 (zh) |
| MX (1) | MX2021000924A (zh) |
| PE (1) | PE20211243A1 (zh) |
| PH (1) | PH12021550144A1 (zh) |
| PL (1) | PL3826997T3 (zh) |
| PT (1) | PT3826997T (zh) |
| SG (1) | SG11202100576WA (zh) |
| SI (1) | SI3826997T1 (zh) |
| WO (1) | WO2020020873A1 (zh) |
| ZA (1) | ZA202101214B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117024313A (zh) * | 2023-08-09 | 2023-11-10 | 重庆恩联生物科技有限公司 | 一种比拉斯汀中间体2-甲基-2-{4-[2-甲基-4-磺酰氧基-乙基]苯基}丙酸甲酯的合成方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112110893A (zh) * | 2020-10-28 | 2020-12-22 | 山东齐环医药科技有限公司 | 一种比拉斯汀的制备方法 |
| WO2022214921A1 (en) * | 2021-04-10 | 2022-10-13 | Ocimum Labs Private Limited | An improved process for the preparation of intermediates used in the preparation of antihistamine agent |
| WO2023037184A2 (en) * | 2021-08-19 | 2023-03-16 | Biophore India Pharmaceuticals Pvt. Ltd | Novel process for the preparation of bilastine and intermediates thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS52108943A (en) * | 1976-03-10 | 1977-09-12 | Teikoku Hormone Mfg Co Ltd | Alkyl phenyl ketone derivative |
| CN101952273A (zh) * | 2008-02-12 | 2011-01-19 | 柳韩洋行 | 2-甲基-2′-苯基丙酸衍生物的制备方法和新的中间体化合物 |
| CN104151160A (zh) * | 2013-05-15 | 2014-11-19 | 重庆华邦制药有限公司 | 用酯类化合物合成2-甲基丙酸酯衍生物的方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009050116A2 (en) * | 2007-10-17 | 2009-04-23 | Basf Se | Adhesion promoting photoinitiators for uv cured coatings over metal surfaces |
| CN102675101B (zh) | 2012-05-16 | 2014-01-29 | 王蕾 | 2-(4-卤乙基)苯基-2-甲基丙酸酯的制备方法及合成比拉斯汀的方法 |
| CN103159674A (zh) * | 2013-04-03 | 2013-06-19 | 苏州安诺生物医药技术有限公司 | 2-苯烷酰胺类化合物及其制备方法、药物组合物和用途 |
| WO2014188453A2 (en) * | 2013-05-24 | 2014-11-27 | Msn Laboratories Private Limited | Novel process for the preparation of 2-[4-(2-{4-[1-(2-ethoxyethyl)-1h-benzimidazol-2-yl]-1-piperidinyl}ethyl) phenyl]-2-methylpropanoic acid |
| CN104326909A (zh) | 2014-09-22 | 2015-02-04 | 暨南大学 | 一种制备α,α-二甲基-4-(2-卤乙基)苯乙酸酯及合成比拉斯汀的方法 |
-
2018
- 2018-07-24 EP EP18382556.1A patent/EP3599235A1/en not_active Withdrawn
-
2019
- 2019-07-23 US US17/261,731 patent/US11370742B2/en active Active
- 2019-07-23 PE PE2021000095A patent/PE20211243A1/es unknown
- 2019-07-23 SI SI201930140T patent/SI3826997T1/sl unknown
- 2019-07-23 HR HRP20211994TT patent/HRP20211994T1/hr unknown
- 2019-07-23 CN CN201980049580.4A patent/CN112469709A/zh active Pending
- 2019-07-23 ES ES19742604T patent/ES2901786T3/es active Active
- 2019-07-23 MX MX2021000924A patent/MX2021000924A/es unknown
- 2019-07-23 CA CA3107879A patent/CA3107879A1/en not_active Abandoned
- 2019-07-23 SG SG11202100576WA patent/SG11202100576WA/en unknown
- 2019-07-23 JP JP2021503753A patent/JP7145312B2/ja active Active
- 2019-07-23 MA MA53370A patent/MA53370B1/fr unknown
- 2019-07-23 BR BR112021001179-0A patent/BR112021001179A2/pt not_active Application Discontinuation
- 2019-07-23 PL PL19742604T patent/PL3826997T3/pl unknown
- 2019-07-23 PT PT197426042T patent/PT3826997T/pt unknown
- 2019-07-23 LT LTEPPCT/EP2019/069772T patent/LT3826997T/lt unknown
- 2019-07-23 AU AU2019311251A patent/AU2019311251B2/en not_active Ceased
- 2019-07-23 EP EP19742604.2A patent/EP3826997B1/en active Active
- 2019-07-23 HU HUE19742604A patent/HUE056822T2/hu unknown
- 2019-07-23 EA EA202190346A patent/EA202190346A1/ru unknown
- 2019-07-23 KR KR1020217005292A patent/KR20210038905A/ko not_active Application Discontinuation
- 2019-07-23 WO PCT/EP2019/069772 patent/WO2020020873A1/en active Application Filing
-
2021
- 2021-01-19 PH PH12021550144A patent/PH12021550144A1/en unknown
- 2021-01-21 CL CL2021000171A patent/CL2021000171A1/es unknown
- 2021-01-28 CO CONC2021/0000940A patent/CO2021000940A2/es unknown
- 2021-02-05 EC ECSENADI20218461A patent/ECSP21008461A/es unknown
- 2021-02-23 ZA ZA2021/01214A patent/ZA202101214B/en unknown
-
2022
- 2022-01-14 CY CY20221100038T patent/CY1124909T1/el unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS52108943A (en) * | 1976-03-10 | 1977-09-12 | Teikoku Hormone Mfg Co Ltd | Alkyl phenyl ketone derivative |
| CN101952273A (zh) * | 2008-02-12 | 2011-01-19 | 柳韩洋行 | 2-甲基-2′-苯基丙酸衍生物的制备方法和新的中间体化合物 |
| CN104151160A (zh) * | 2013-05-15 | 2014-11-19 | 重庆华邦制药有限公司 | 用酯类化合物合成2-甲基丙酸酯衍生物的方法 |
Non-Patent Citations (2)
| Title |
|---|
| SHOU-RI SHENG等: "A facile conversion of aryl alkyl ketones into methyl 2-arylalkanoates using poly[4(diacetoxyiodo)styrene]", 《JOURNAL OF CHEMICAL RESEARCH》 * |
| TAMURA, YASUMITSU等: "Oxidative 1,2-aryl migration of alkyl aryl ketones by using diacetoxyphenylidine:Synthesis of arylacetate. 2-arylpropanoate and 2-arylsuccinate", 《CHEMICAL & PHARMACEUTICAL BULLETIN》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117024313A (zh) * | 2023-08-09 | 2023-11-10 | 重庆恩联生物科技有限公司 | 一种比拉斯汀中间体2-甲基-2-{4-[2-甲基-4-磺酰氧基-乙基]苯基}丙酸甲酯的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CY1124909T1 (el) | 2023-01-05 |
| US20210300856A1 (en) | 2021-09-30 |
| PH12021550144A1 (en) | 2021-09-13 |
| KR20210038905A (ko) | 2021-04-08 |
| HUE056822T2 (hu) | 2022-03-28 |
| SI3826997T1 (sl) | 2022-01-31 |
| EP3599235A1 (en) | 2020-01-29 |
| EA202190346A1 (ru) | 2021-05-20 |
| JP2021531300A (ja) | 2021-11-18 |
| AU2019311251B2 (en) | 2021-10-21 |
| CO2021000940A2 (es) | 2021-02-17 |
| CL2021000171A1 (es) | 2021-07-09 |
| MA53370A (fr) | 2021-06-02 |
| US11370742B2 (en) | 2022-06-28 |
| PT3826997T (pt) | 2022-01-21 |
| ZA202101214B (en) | 2022-09-28 |
| AU2019311251A1 (en) | 2021-03-04 |
| MA53370B1 (fr) | 2021-12-31 |
| EP3826997A1 (en) | 2021-06-02 |
| HRP20211994T1 (hr) | 2022-04-01 |
| PE20211243A1 (es) | 2021-07-13 |
| JP7145312B2 (ja) | 2022-09-30 |
| ES2901786T3 (es) | 2022-03-23 |
| ECSP21008461A (es) | 2021-03-31 |
| EP3826997B1 (en) | 2021-11-10 |
| SG11202100576WA (en) | 2021-02-25 |
| LT3826997T (lt) | 2021-12-27 |
| CA3107879A1 (en) | 2020-01-30 |
| MX2021000924A (es) | 2021-03-31 |
| WO2020020873A1 (en) | 2020-01-30 |
| PL3826997T3 (pl) | 2022-05-30 |
| BR112021001179A2 (pt) | 2021-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112469709A (zh) | 比拉斯汀的制备方法及其中间体 | |
| EP1694678A2 (en) | Process for preparing hexahydropyrimido 1,2-a¨azepine-2-carboxylates and related compounds | |
| WO2010030345A2 (en) | Processes for the preparation of aminosulfone compounds | |
| CN107922352B (zh) | 制备蛋白质脱乙酰酶抑制剂的方法 | |
| EP3412666A1 (en) | Process and intermediates for the preparation of bcl-2 inhibitors including venetoclax through reductive amination | |
| JP6174156B2 (ja) | オスペミフェンの製造方法 | |
| KR20180118054A (ko) | 의약품 합성용 중간체 화합물의 제조 방법 | |
| JP2012530075A (ja) | 二置換アミノジフルオロスルフィニウム塩、その調製プロセスおよびデオキソフッ素化試薬としての使用方法 | |
| EP3356342B1 (en) | Process and intermediates for the preparation of benzo[b]thiophene compounds | |
| AU2011319706B2 (en) | Process for the preparation of anagrelide and analogues thereof | |
| CN110035992B (zh) | 用于制备3-取代的2-乙烯基苯基磺酸酯的方法 | |
| CN113816955B (zh) | 一种ret激酶抑制剂中间体及其制备方法 | |
| HUT77742A (hu) | Eljárás (E)-2-fluor-metilén-4-(4-fluor-fenil)-butil-amin és- diformilamid-alkálifém-sói előállítására | |
| JPH0419216B2 (zh) | ||
| RU2635094C2 (ru) | Способ получения 4-(циклопропилметокси)-n-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида | |
| KR20230145461A (ko) | 리스디플람의 제조 방법 | |
| JP6459709B2 (ja) | 3,3−ジフルオロ−2−ヒドロキシプロピオン酸の実用的な製造方法 | |
| JP2022056770A (ja) | アミン誘導体若しくはその塩、並びに、その塩及び環状アミン誘導体の製造方法 | |
| JPH0717929A (ja) | N,o−ジアルキルヒドロキシルアミンの製造方法 | |
| JP2001172247A (ja) | 4−シアノ−2−フルオロベンジルクロリドの製造方法 | |
| OA16582A (en) | Process for the preparation of anagrelide and analogues thereof. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| AD01 | Patent right deemed abandoned |
Effective date of abandoning: 20240507 |
|
| AD01 | Patent right deemed abandoned |