CN1104426C - 紫杉醇和三尖杉宁碱的分离和纯化 - Google Patents
紫杉醇和三尖杉宁碱的分离和纯化 Download PDFInfo
- Publication number
- CN1104426C CN1104426C CN96193149A CN96193149A CN1104426C CN 1104426 C CN1104426 C CN 1104426C CN 96193149 A CN96193149 A CN 96193149A CN 96193149 A CN96193149 A CN 96193149A CN 1104426 C CN1104426 C CN 1104426C
- Authority
- CN
- China
- Prior art keywords
- taxol
- mixture
- cephalomannine
- residue
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images



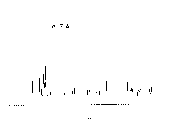

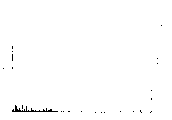


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
Abstract
本发明叙述了从天然原料如红豆杉树种的皮、针叶和树枝,组织培养物和真菌中提取和分离紫杉烷类、尤其是紫杉醇的方法,其中紫杉烷类从粗提取物中分离的过程为:在极性和非极性溶剂两相间分配,在非极性溶液中沉淀,通过卤化不饱和紫杉烷衍生物使混合物发生反应,然后层析分离并从极性和非极性溶剂混合物中结晶出紫杉烷类;更具体地说,叙述了在紫杉醇存在下卤化不饱和侧链紫杉烷衍生物、尤其是三尖杉宁碱的方法,其中溴优选加到不饱和紫杉烷的双键上,紫杉醇不发生变化,紫杉醇容易从含有较少极性的卤化紫杉烷衍生物的混合物中分离出来。
Description
发明领域
本发明是关于各种来源的紫杉醇(paclitaxel)的分离和纯化,紫杉醇的来源包括有机物质如植物原料,人工培养品和霉菌,更主要的是来源于短叶红豆杉(Taxus.brevifolia,Pacific Yew)树皮,以及欧红豆杉(T.baccata)、云南红豆杉(T.yunnanensis)和西藏红豆杉(T.Walichiana)。本发明提供的方法可分离和纯化紫杉烷(Taxane)混合物中的紫杉醇,该混合物中含有不同浓度的紫杉醇和其它各种紫杉烷,包括具有不饱和侧链与紫杉醇极其相似的紫杉烷,如三尖杉宁碱(cephlomannine)。
发明背景
紫杉醇是治疗各种转移肿瘤的有名的化疗药。它已被美国FDA(Foodand Drug Administration,食品与药品管理部门)批准用于治疗卵巢和乳腺癌,最近又正进行治疗肺癌和结肠癌的临床试验。
该化合物是天然产物,主要由短叶紫杉树皮中提取,同时在欧洲红豆杉、西藏红豆杉及云南红豆杉(T.yunnanensis)中也有发现,另外,自其它植物原料中也得到了生物量的提取物,如自T.hicksii,T.densiformis,T.gem,T.wardii,东北紫杉(T.cuspidata),T.capitata,T.brownii和T.dark green spreader它们含有紫杉烷型化合物的混合物。紫杉醇也可通过植物的细胞培养及霉菌中得到。试剂级的紫杉醇市场上可以买到,例如Aldrinch化学公司,该产品号为NO.41,701-7;Sigma化学公司,该产品号为T7002,T1912,两种编号的产品来源不一样;Fluka化学公司的产品编号为NO.86346,ICN生物医药公司的产品编号为NO.193532。
在各种原料中,紫杉醇的浓度是特别低,例如在短叶红豆杉皮中,其含量在0.0004-0.08%(重量比)之间。从含量那样低的原料中,提取并纯化供临床用的化合物,是很困难的,要推向市场,更是不现实。
目前,关于紫杉醇的提取和纯化方法,已有几个报道。Wani等,J.Am,Chem,Soc.93,9:2325-2327(1971),该文介绍,短叶红豆杉的茎皮用乙醇提取,浓缩提取液,有氯仿和水提取,紫杉醇溶于氯仿相。进一步的纯化是采用硅酸镁、交联葡聚糖和硅胶柱层析。
国立癌症研究所(NCI)的方法(1983年)是用甲醇提取短叶红豆杉的树皮,接着用二氯甲烷自提取液中萃取。蒸干二氯甲烷,加丙酮溶解,再加正己烷沉淀除杂质,溶液部分进一步用柱层析纯化。
上述两个方法产率都很低,少于0.02%。不可能得到市售数量的产品。这是因为有各种其它紫杉烷共存,例如与紫杉醇极其相似的三尖杉宁碱,它们在结构上及物理性质上都很相似,图1表明了两者的化学结构。
在Potier等(J.Nat.Prod.,47,1:131-137[1984])制订的方法里,将国立癌症研究所方法中沉淀步骤,改为溶剂对萃取,即用极性递增的各种溶剂,进行连续萃取。经氧化铝和硅胶柱层析后,紫杉醇被富集,但与三尖杉宁碱共存。最后用HPLC分离紫杉醇与三尖杉宁碱,其产率比用Wani等的方法或NCI的方法都高得多。
Potier等的方法和Wani等及NCI方法一样,存在着一个主要的障碍,就是要采用多次常规层析方法来分离具有近似分离参数的各种紫杉烷,才能得到紫杉醇纯品。为了得到足够量的达到临床要求纯度的产品,必须进行大规模的多次常规层析分离,这就要消耗大量资金,因此,大批生产实际上是不可能的。
由于紫杉醇和三尖杉宁碱在结构上和性质上的极其相似性,反复多次分离是必需的。如图1所示,两者的差别仅在于,紫杉醇侧链上的氨基是同苯甲酸进行酰化的,而三尖杉宁碱侧链上的氨基是含双键的顺芷酸酰化的。
分离紫杉醇和三尖杉宁碱的非层析方法已有报道。例如将三尖杉宁碱的侧链上双键进行化学改造。例如Kingston等(J.Nat.Prod.,55:NO.2,259-261[1992])报道,在氧化锇(OsO4)存在条件下,催化氧化三尖杉宁碱侧链双键,得到二醇,然后用层析和重结晶方法来与紫杉醇分离。这个方法的问题在于,它采用未经纯化的紫杉烷混合物,而OsO4的催化氧化是不适用于粗提物的,因为它氧化三尖杉宁碱侧链上双键的选择性太低。当然,如果这个方法能用的话,将大大降低萃取和纯化过程的价格。另外,OsO4有严重的毒性,不适用于制药工业。
在美国专利(Nos.5,334,732和5,336,648 Murray等)中,介绍了用臭氧氧化三尖杉宁碱侧链的方法。该方法用臭氧氧化粗提物,将会产生许多不希望发生的反应。臭氧分介反应是很强的、而且无选择性,它能因紫杉醇和三尖杉宁碱中的许多官能团发生不希望的氧化反应。例如同紫杉醇分子中的醛、酮、胺等反应,或者同紫杉醇及三尖杉宁碱的紫杉烷环上的双键反应。此外,臭氧发生器,设备也很昂贵。
这样,自含有复杂的紫杉烷的生物量的粗提混合物中,或者极而言之,自主要含有紫杉醇和三尖杉宁碱较纯的混合物中分离和纯化紫杉醇,当前还是局限于上述的非经济的层析技术和非选择性的氧化方法。因此,需要一个经济实用的方法。以便自紫杉醇与三尖杉宁碱混合物中以及其它非常相似的紫杉烷中分离纯化出有价值的抗癌化合物紫杉醇。
因此,本发明就是提供一个比现有方法更简单,更经济有效的方法,分离纯化重要的化疗化合物紫杉醇。
发明概要
为了达到上述目标,本发明提供一个新的、独特的方法,自含有紫杉醇的紫杉烷型复杂混合物的生物材料粗提中,分离纯化紫杉醇,尤其是自短叶红豆杉(T.brevifolia)、欧洲红豆杉(T.baccata)、云南红豆杉(T.yunnanensis)和西藏红豆杉(T.walilichiana)生树皮中,以及从植物原料如各种红豆杉的针叶和树枝中分离纯化紫杉醇。本发明方法进一步包括由紫杉的细胞培养和产生紫杉醇的霉菌中所得到的紫杉醇的进一步纯化。
一方面本发明提供了一个自有机原料中,尤其是紫杉烷混合物中分离纯化紫杉醇的方法,方法包括有机溶剂萃取,得到含有紫杉醇的组合物,接着层析分离紫杉醇和其它紫杉烷。最好再用闪式色谱(FlashChromatography),以硅胶为吸附剂的正相色谱进行分离,得到含有紫杉醇、三尖杉宁碱和其它紫杉烷的精制混合物。采用适合于三尖杉宁碱不饱和支链选择卤化的条件,将该精制混合物与卤素,最好是溴进行卤化反应,得三尖杉宁碱二卤代非对映异构体的混合物;这样,紫杉醇就很容易从混合物中分出,产率很高。
本发明的另一方面,自粗提的有机物质中分离纯化紫杉醇方法的每一步,都伴随有三尖杉宁碱的选择性卤化,不论是粗提的混合物还是主要含有紫杉醇、三尖杉宁碱和其它紫杉烷的精制混合物,用常规的层析技术提取和纯化之前或之后,都要进行三尖杉宁碱的选择性卤化。
本发明的再一个方面,是通过对三尖杉宁碱的新型的化学改造,使紫杉醇能同与之十分相似的三尖杉宁碱高效地、经济地并实际上是定量地分离出来。
本发明的紫杉醇和三尖杉宁碱的分离和纯化的方法具体如下所述:
1.一种从含有紫杉烷混合物的有机原料中分离和纯化紫杉醇的方法,所述方法包括:
(1)从上述有机原料中提取含有紫杉化合物的组合物;
(2)从所述组合物中层析分离含有紫杉醇、三尖杉宁碱和其它紫杉烷的混合物,然后
(3)选择有效条件,将混合物与卤素反应,选择性地将三尖杉宁碱转变成二卤代三尖杉宁碱的非对映异构体混合物;然后
(4)从上述混合物中,分离紫杉醇。
2.上述1的方法,其中有机原料用第一萃取溶剂萃取后,蒸发得到含紫杉醇的第一残渣;然后用第二萃取溶剂萃取第一残渣,再蒸发,得到含紫杉醇的第二残渣,接着进一步用结晶法纯化第二残渣。
3.上述2的方法,其中第一萃取溶剂是甲醇,含紫杉醇的第一残渣被分配于水相和选自下列的有机相之间:二氯甲烷、二氯乙烷和氯仿,该残渣经所述溶剂萃取后,干燥,得第二残渣,将此含紫杉醇的第二残渣溶于丙酮中,加己烷沉淀出非极性杂质,再将此丙酮-己烷溶液蒸发到原体积的1/3,得到含紫杉醇的粘稠状第三残渣。
4.上述3的方法,其中所述溶剂和水的体积比1∶1。
5.上述3的方法,其中向粘稠状第三残渣中加入10倍体积的己烷,析出浅黄色沉淀,形成含紫杉醇的固体第四残渣。
6.上述4的方法,进一步包括将第四固体残渣溶于丙酮与二氯甲烷和/或二氯乙烷的混合溶剂中,通过硅胶柱和色谱溶剂进行闪式色谱分离,将含有紫杉醇和三尖杉宁碱混合物所得的各级分合并、蒸干,得到含紫杉醇的第五残渣。
7.上述6的方法,其中所述色谱所用的溶剂是丙酮与二氯甲烷或二氯乙烷的混合物,其体积比是1∶9到3∶7。
8.上述6的方法,其中将第五残渣溶于选自下列的氯代溶剂中:CCl4、CHCl3、C2H4Cl2或CH2Cl2,并与卤素反应。
9.上述8的方法,其中所说的卤素是溴。
10.上述9的方法,其中溴溶于卤代溶剂中,其浓度为0.01M到0.1M。
11.上述10的方法,其中溴化反应在温度-20℃到20℃和黑暗条件下进行。
12.上述11的方法,其中存在于上述氯代溶剂中的三尖杉宁碱基本上完全转变成二溴代三尖杉宁碱非对映异构体混合物,然后从该反应混合物中分离紫杉醇。
13.上述12的方法,其中含紫杉醇的有机物质选自:短叶红豆杉的树皮、红豆杉各品种的植物原料,红豆杉各品种的细胞培养物以及能产生紫杉醇的霉菌。
14.上述12的方法,其中通过硅胶层析,将紫杉醇自反应混合物中分离出来,层析用的洗脱液是丙酮与二氯甲烷或二氯乙烷的混合物,其体积比是1∶9到3∶7。
15.上述12的方法,其中分离出的紫杉醇在丙酮与己烷混合物中结晶,使紫杉醇析出。
16.一种从含有紫杉醇和三尖杉宁碱混合物中分离紫杉醇的方法,所述方法包括以下步骤:a)将该混合物与卤素反应,反应温度和时间应充分保证,让三尖杉宁碱基本上完全被卤化;b)将紫杉醇与三尖杉宁碱卤代衍生物分离。
17.上述16的方法,其中所述卤素可选用氟、氯、溴或碘。
18.上述17的方法,其中所述卤素是溴。
19.上述18的方法,其中溴的浓度是0.01M到0.1M。
20.上述16的方法,其中反应a)的温度范围是-20℃至20℃。
21.上述19的方法,其中溴溶于氯代溶剂CCl4、CHCl3或CH2Cl2中。
22.上述21的方法,其中氯代溶剂是CCl4或CHCl3。
23.上述16的方法,其中反应a)在黑暗条件下进行。
24.上述16的方法,其中b)步分离是采用硅胶层析,选用适当溶剂作洗脱液。
25.上述24的方法,其中溶剂是丙酮与二氯甲烷或1,2二氯乙烷的混合物,其体积比是1∶9到3∶7。
26.上述25的方法,其中含紫杉醇的各个部分,蒸干成固体残渣。
27.上述26的方法,其中用结晶法进一步纯化紫杉醇。
28.上述27的方法,其中固体残渣被溶于丙酮中。
29.上述28的方法,其中紫杉醇从己烷内结晶析出。
30.上述25的方法,其中紫杉醇与2″,3″-二溴代三尖杉宁碱的非对映异构体混合物分离。
31.上述16的方法,其中反应a)用HPLC监测。
32.上述16的方法,其中混合物是来自含有紫杉醇的原料。
33.上述32的方法,其中含紫杉醇原料选自:短叶红豆杉树皮,红豆杉各品种的植物原料、红豆杉各品种的细胞培养物以及能产生紫杉醇的霉菌。
参考下面有关本发明的具体方案、实例和附图的详细描述,将有助于对本发明的充分理解。
附图简要说明
图1,紫杉醇和三尖杉宁碱结构的一般表达。
图2,能按本发明进行卤化的带有各种不饱和支链的几个不饱和紫杉烷和其中的官能团结构的一般表达。
图3,较好地体现本发明的一个方面,自短叶红豆杉中分离和纯化紫杉醇的工艺流程图。
图4,三尖杉宁碱选择性溴化的较好的反应路线图。
图5,用本发明方法获得的紫杉醇的紫外光谱图。
图6,用本发明方法制得的紫杉醇的红外光谱图。
图7a,用本发明方法制得的紫杉醇的质子(1H)核磁共振谱图。
图7b,用本发明方法制得的紫杉醇的碳13(13C)核磁共振谱图。
图8a和8b,用本发明方法制得的紫杉醇的电子轰击源质谱(EI-MS)图。
图9,用本发明方法制得的紫杉醇的直接化学电离源质谱(DCI-MS)图。
图10,用本发明方法制得的紫杉醇的快原子轰击源质谱(FAB-MS,正离子型)图。
图11,用本发明方法制得的紫杉醇的快原子轰击源质谱(FAB-MS,负离子型)图。
图12,用本发明方法制得的紫杉醇的高效液相色谱图(HPLC)。
图13,用本发明方法制得的紫杉醇的热重分析(TGA)图。
图14,用本发明方法制得的紫杉醇的差示扫描量热(DSC)图。
本发明优选实例的详细描述
本发明提供一个简单、经济的方法,从各种生物原料中提取和纯化紫杉醇,如从含有紫杉烷混合物的植物材料中,例如从短叶红豆杉(T.brevifolia,Pacific Yew tree)和云南红豆杉(T.yunnanensis)的树皮中,从其它品种的红豆杉针叶和树枝中,以及其它含有紫杉醇的原料,如多种红豆杉的细胞培养品及能产生紫杉醇的霉菌中来提取纯化紫杉醇。
本发明的方法包括选择性卤化,最好是溴化,未被纯化过、部分纯化过或经过纯化的混合物,混合物中含有紫杉醇、三尖杉宁碱和带有不饱和侧链的其它紫杉烷衍生物,卤化结果选择性地使某些紫杉烷的结构变换,但不破坏紫杉醇。
下面是本发明方法的一个具体实例:
含有各种紫杉烷的复杂混合物的生物材料,优选用低级醇,如甲醇、乙醇等提取,混合物最好加工成具有较大的表面体积比,以便提高紫杉醇在最初提取相中的质量转移率,例如用短叶红豆杉树皮为原料,其中含紫杉醇低于0.1%(w/w)。将树皮粉碎成细碎的混合物,加甲醇提取到认为混合物中的紫杉醇已充分被提取出来。然后将甲醇提取液浓缩(例如旋转蒸发)到原体积的二十分之一。
接着进行进一步的提取,例如将甲醇浓缩物,分配于适当的溶剂如二氯甲烷、氯仿或二氯乙烷和水两相之间,两相体积比最好是1∶1,这样水溶性成分即被萃取进水相。这些水溶性成分可能是紫杉醇的水溶性苷,和其它极性更大的化合物,这些成分留待进一步加工,制备有用的物质。在本发明的这个实施例中,将含紫杉醇的有机相蒸发成含紫杉醇的残渣,再通过沉淀去杂质进行纯化。例如,将残渣溶于丙酮中,加入等体积的己烷,则非级性杂质呈焦油状沉淀析出,并通过过滤除去。滤去沉淀后的丙酮-己烷溶液被浓缩,再加新鲜己烷,析出的沉淀在40℃下,抽高真空(1mm至2mm)进行干燥。
在此实施例中,残渣最好溶解于最小量的层析溶剂中,例如二氯甲烷或二氯乙烷中,然后通过硅胶柱,闪式层析。流动相可能是丙酮与二氯甲烷的混合物或丙酮与二氯乙烷的混合物,体积比为1∶9到3∶7。首先流出柱子的几份洗脱液,含有低级性化合物,接着的洗脱液含有不同浓度的紫杉醇和三尖杉宁碱。淋洗结束,用丙酮和甲醇洗硅胶柱子,洗脱液弃去。
合并含有紫杉醇和三尖杉宁碱的各份洗脱液,蒸发成含有紫杉醇的残渣。此残渣最好溶于氯代溶剂中,如四氯化碳、二氯甲烷、氯仿或二氯乙烷中。在此溶液中,在适合于三尖杉宁碱分子内的不饱和侧链双健(包括其它含紫杉烷的不饱和侧链)的选择性卤化条件下,进行卤化,得到一个二卤代三尖杉宁碱的非对映异构体与紫杉醇的混合物溶液,其中还含有其它侧链卤化了的紫杉烷化合物。虽然所有卤素都能适用,但最好是用溴,因为它卤化效率高,且价格低廉。最好溶液中所有的三尖杉宁碱都完全转变成三尖杉宁碱的二卤代非对映体混合物,以便紫杉醇能容易地自混合物中分离出来。
优选在接近0℃,避光条件下、并剧烈搅拌,使氯代溶剂中的紫杉烷进行溴化。
反应速率最好慢一点,以便控制氢溴酸的产生速率,减少或完全避免紫杉醇残渣的水解。通过色谱分析,例如高效液相色谱分析(HPLC),测定三尖杉宁碱反应完全或基本上反应完全后,停止加溴。主要含有紫杉醇和二溴化三尖杉宁碱异构体的溴化溶剂溶液,首先用稀亚硫酸钠溶液洗涤,接着用碳酸氢钠洗,以除去和中和剩余的溴及反应中产生的氢溴酸。
有机相再经水洗涤后,用无水硫酸钠干燥,然后蒸干。固体残渣溶于小量溶剂中,例如二氯甲烷中,通过层析法,可将含三尖杉宁碱的二溴代异构体的级分与含紫杉醇的级分分开,如层析柱为硅胶柱、洗脱液优选为丙酮-二氯乙烷(1∶9,v/v)、借助TLC(薄层层析)和HPLC(高效液相色谱)的检测,合并含有紫杉醇的各份洗脱液并蒸干。将得到的固体残渣溶于丙酮中,加己烷使紫杉醇结晶析出。滤出结晶,洗涤,干燥后得最终产物。
上述方法简便易行。紫杉醇和三尖杉宁碱的产品及副产品的分析测定,最好用TLC和HPLC,以避免产物的损失。从上面我们已经看到三尖杉宁碱的卤化,尤其是溴化,是一个新颖的、方便的方法。在将含有紫杉醇和三尖杉宁碱的混合物进行层析分离时,使用该法增加了紫杉醇分离的选择性。与过去报道的化学改造紫杉烷(taxanes)的方法不同,本发明的方法中,溴化反应能够被控制,使紫杉醇不致有明显的损失。图3是自短叶红豆杉中分离纯化紫杉醇的一个较好的流程图。图4是三尖杉宁碱选择性溴化的一个较好的反应线路图。
按照本发明,分离过程与原料中紫杉醇浓度无关、也与起始原料混合物的组成无关。因此,本发明的工艺规程也能很方便地应用于其它原料中紫杉醇的分离纯化的下游工程,例如从培养的植物细胞和产生紫杉醇的霉菌中进一步分离纯化紫杉醇。选择性卤化可以在工艺过程中任何一步应用,以利于紫杉醇的分离和纯化。
混合物中含有三尖杉宁碱和1%-99.9%的紫杉醇,卤化过程都是相似的。混合物最好溶于大量的氯代溶剂中。例如CCl4或CHCl3中,冷却到接近0℃,在搅拌情况下,加入化学计量的(是三尖杉宁碱摩尔当量数的1.2倍)溴的CCl4或CHCl3的稀溶液,直到三尖杉宁碱溴化完全。整个反应在黑暗中进行,温度不超过20℃,最好是5℃,反应进行程度通过HPLC检测控制。溴化结束后,洗涤反应混合物除去剩余的溴。
在所有情况下,溴化三尖杉宁碱和其它不饱和的紫杉烷,紫杉醇的回收率高。利用各种方法,例如层析法和结晶法分离和纯化含有紫杉醇和溴代化合物的混合物。将三尖杉宁碱转换成极性较小的二溴衍生物,使紫杉醇有可能较容易地从混合物中分离出来。
向混合物中加入溴的摩尔当量数,主要取决于三尖杉宁碱和其它不饱和化合物的浓度。一般来说,纯度较差的混合物(即相对于三尖杉宁碱量来说,不饱和紫杉烷的量较大)需要溴的摩尔数比纯度高的混合物需溴量较大,以便使不饱和紫杉烷溴化完全。图2表明了各种不饱和紫杉烷的结构。如果混合物中不饱和化合物含量较高,例如taxicin、taxicin-1、taxinin和(或)云实醇(brevifoliol,短叶紫杉醇),则要求加入溴的摩尔数较多,因为它们吸收的溴多于1摩尔当量。
卤化过程所用的溶剂,对于所用的卤素,尤其是溴一定是惰性的。按照本发明,采用的溶剂最好是氯代溶剂,如CCl4、CHCl3、CH2Cl2、C2H4Cl2,而以CCl4较好。卤化过程的温度,对溴化来说有效的是-20℃到+20℃,较好的是-5℃到+5℃。进行上述反应较好的试剂是0.01M到0.1M溴的CCl4或CHCl3溶液。这些试剂,市场上都能买到。
根据常识应当了解,利用卤素,例如用溴来溴化含有几个官能团(它们对溴或其它卤素是灵敏的)的紫杉烷化合物时,紫杉醇或其它列于表2的化合物,将会发生不希望的反应。但是实验发现,三尖杉宁碱侧链双键卤化的选择性特别高,其它含有环外双键的紫杉烷溴化也有很高的选择性。在本发明的方法里,溴化反应期间,紫杉醇既无明显的降解,也不被溴化。但反应如果曝光太长或者卤素过量太多、紫杉醇也会降解成几个未知的化合物。在卤化(溴化)期间,定期用HPLC监测,则可很容易避免紫杉醇的降解。
实例
下面实例是关于紫杉醇的纯化,是本发明工艺流程的优选实施方案。实验所用的试剂均由制造厂商或供应厂商提供。由产品或副产品得到的紫杉醇和三尖杉宁碱都用薄层色谱(采用Merck#5554,F254硅胶板)和HPLC监测。HPLC系统由Waters(沃特斯)510泵,660E系统控制器,712WISP或710E WISP自动进样器,Waters 490程序控制多波长检测器,Waters 990光敏二极管序列检测器,NEC APCIV计算机,WatersLambdaMax 481型分光光度计和数据处理单元组成。HPLC柱包括3.9mm×300mm苯基反相Waters μbondapak柱和苯基防护柱。闪式色谱所用的硅胶是购自ICM生物医学公司(Biomedicals)的32-63mm筛孔的硅胶。
应当了解,下面例子仅是为了解释本发明,而不是用来限制本发明或权利要求的范围或精神。
例1
自粗生物材料样品中纯化紫杉醇
第一步
将短叶红豆杉(Taxus brevifolia)的树皮切成2-4mm大小。其中紫杉醇含量为0.03-0.1%w/w。将45公斤树皮置于不锈钢罐内。用150升甲醇提取三次,每次提取时间为5天,令提取物频频再循环,以促进混合。采用旋转蒸发浓缩提取液到10-15升。提取温度不超过40℃。结果99%的紫杉醇能被提取到甲醇相。
第二步
由第一步得到的甲醇提取液的浓缩物,分配于两相中,例如二氯甲烷或二氯乙烷及其类似物与水两相中。将等体积的二氯甲烷和水加入到15升甲醇提取物中。混合物缓慢搅拌15分钟,静置2小时,两相分层。二氯甲烷相用于进一步分离紫杉醇。根据检测,若水相中仍含有紫杉醇,则用二氯甲烷再萃取,合并两次萃取后的二氯甲烷相。若萃取过程中发生乳化,则加0.5-1升甲醇破乳。将二氯甲烷萃取物旋转蒸发到干。固体残渣在0.9-1.1kg之间,紫杉醇的含量为1.8-2.2%w/w。在整个工艺过程中,温度不得超过40℃。
第三步
第二步的水相含有紫杉醇苷,10-脱乙酰基浆果赤霉素III(10-deacetyl baccatin III),浆果赤霉素III(baccatin III)和其它极性化合物。向35升水相中加入5升盐水溶液后,用20升乙酸乙酯萃取,分层。上层乙酸乙酯相含有紫杉醇苷,10-脱乙酰基浆果赤霉素III,浆果赤霉素III及其它极性化合物。下层水相再用乙酸乙酯萃取。合并两次的乙酸乙酯相,约30-35升,然后旋转蒸发呈粘稠状棕黑色液体,体积在2.8-3.2升之间,储存起来,可进一步从中分离出苷。
第四步
将第二步得到的二氯甲烷固体残渣,溶于2升丙酮中。在强烈搅拌下,加入等体积己烷。在此条件下,极性杂质沉淀析出。静置澄清,倾析出上清液,进一步加工。用丙酮-己烷(1∶1,v/v)洗沉淀,过滤;滤液与前面倾析出的上清液合并,旋转蒸发到原体积的1/3,得到1.5-3.0升的黄棕色的粘稠状残渣。
第5步
第4步得到的丙酮-己烷残渣滴加到10~15升己烷中,边加边强烈地搅拌。亮黄色的沉淀开始形成。经过将近8个小时,滤出沉淀在40℃下,真空(1mm到2mm)干燥,得到产物约0.5-0.6kg。
第6步
第5步得到的固体残渣,溶于0.5升丙酮-二氯甲烷(1∶9,v/v)中。利用硅胶柱,进行闪式色谱分离,上述相同溶剂作流动相。柱中装填的硅胶在3.5-4kg之间。每份收集洗脱液的体积为1升。用TLC和HPLC监测每个样品。三尖杉宁碱与紫杉醇同时洗脱下来。合并含有三尖杉宁碱和紫杉醇的级分,旋转蒸发到干。固体残渣是紫杉醇和三尖杉宁碱粗的混合物,约55克到70克,含45-55%(w/w)的紫杉醇,或36g到40g紫杉醇。
第7步
由第6步得到的紫杉醇和三尖杉宁碱混合物,经测定含28.8%(w/w)的三尖杉宁碱和5 1.2%(w/w)的紫杉醇,经化学改造后,分离紫杉醇和三尖杉宁碱。将10g粗的混合物溶于1升氯代溶剂中,如CCl4,CHCl3,CH2Cl2和二氯乙烷中。较好体现本发明的做法是,在0℃,强烈搅拌条件下,用0.01M溴的CCl4溶液与上述粗的混合物溶液,避光缓慢地进行反应。反应过程用HPLC监测。当三尖杉宁碱完全参加反应了,则终止溴化反应。用亚硫酸钠水溶液洗去痕量剩余的溴。反应期间生成的氢溴酸,用碳酸氢钠稀溶液(0.5%,w/w)洗去。最后的有机提取物经无水硫酸钠干燥后,旋转蒸发得固体残渣,重量为13.2g。
第8步
从第7步制得的溴化残渣溶于丙酮-二氯甲烷混合物(1∶9,v/v)中,通过硅胶柱进行层析分离。收集的各级分用TLC和HPLC检测。将含有紫杉醇的各部分合并,旋转蒸发到干。得到的白色固体残渣,重6.1g。
第9步
将第8步制得的残渣溶于丙酮中,加等体积的正己烷或己烷使紫杉醇结晶折出。晶体用冷丙酮-己烷(1∶1,v/v)洗涤后,在40℃真空干燥。得到的固体结晶4.84g,通过HPLC测定,其中紫杉醇含量>97%,w/w。
例2
部分纯化过的三尖杉宁碱的溴化
将0.63g含有91.5%三尖杉宁碱(0.0007摩尔)和6-7%紫杉醇的样品,溶于150ml CCl4中,加入到500ml园底三口烧瓶内,烧瓶上装有一个250ml分液漏斗。将烧瓶浸于冰盐浴内,当温度达到-5℃,将溴的(0.1221g)CCl4溶液(76.31mL,0.01M)在搅拌情况下,缓慢地加入烧瓶中,通过调整加入溴的速率,控制反应速度,使反应温度不致超过5℃。三尖杉宁碱对溴的摩尔比为1∶1.1。加溴的时间约需3小时,反应结果的溶液呈混浊的浅棕色。
利用HPLC每个小时取样分析,监测溴化反应进行的程度。根据HPLC的测定,当所有的三尖杉宁碱转变成2″,3″-二溴衍生物时,溴化就完成了,总共需要8个小时。由于溴逐渐被消耗,反应混合物呈浅黄色至无色。
接着将反应混合物转移到1升的分液漏斗中,先后用0.5%亚硫酸钠水溶液(300mL)、0.5%碳酸氢钠水溶液(300ml)洗涤,最后用去离子水(每次200ml)洗两次,使pH值达到6.5。合并水相,用CH2Cl2萃取一次。将萃取后的CH2Cl2相与前面的有机相萃取物合并,加Na2SO4干燥后,过滤,滤液蒸干,得到0.76g浅奶油色固体,根据原料计算,产率接近100%。
将得到的奶油色固体产物,在硅胶柱上(50g,ICN Silitech,32-63D,60A)进行柱层析,洗脱液为丙酮-CH2Cl2(10∶90)。收集洗脱液,每50ml一份,通过TLC(Silicagel 60 F254,Merck#5554,展开剂为丙酮-CH2Cl2(20∶80);显色剂为香草醛-硫酸的甲醇溶液)进行监测。合并仅出现一个斑点(Rf=0.64)的各部分(第26-38份),浓缩到干,得0.485g浅奶油色到白色的结晶,mp(熔点):158℃,经鉴别为2″,3″-二溴代三尖杉宁碱。以原料三尖杉宁碱为基础,产率约为70%。
例3
含有三尖杉宁碱,紫杉醇和
其它紫杉烷型化合物的粗混合物的溴化
采用与例2相似的装置,将紫杉醇粗产品2g(根据HPLC测定,含51.2%紫杉醇,28.8%三尖杉宁碱和约20%其它紫杉烷及非紫杉烷杂质)溶于150mL CCl4和150mL CH2Cl2中,溶液呈透明的浅黄色。将烧瓶浸入冰盐水中,并搅拌溶液。当温度达到-5℃,加入溴溶液(0.1332g纯溴溶于83.13mL(0.01M)CCl4中,1M三尖杉宁碱:1.2M溴),控制加溴的速度,保持反应混合物的温度低于5℃。整个加溴过程约需3小时,最后溶液呈棕黄色混浊液。加溴结束后,让反应在相同条件下,继续进行8小时,每小时用HPLC测定紫杉醇和三尖杉宁碱的含量。溶液呈浅黄色或无色后反应完成,所有的三尖杉宁碱都转变成二溴代衍生物。如果8小时后,溶液中仍有1-2%的三尖杉宁碱,则在反应起始条件下,再滴加10mL 0.01M的溴CCl4溶液,继续反应1小时,再取样用HPLC分析。
剩余在反应混合物中的溴,通过用0.5%Na2SO3水溶液(300mL),0.5%NaHCO3水溶液(200ml)和去离子水(2次,每次200mL)洗涤去除。反应混合物经无水Na2SO4干燥后,真空浓缩至干,得到2.35g浅奶油色到白色的干燥结晶残渣。将此结晶残渣,按例2条件,用硅胶柱层析纯化。被分离的混合物与硅胶的比例为1∶60,这样共需120g硅胶。硅胶柱流出的每一份用TLC检查,每第3份取样用HPLC分析。经TLC和HPLC检查,合并具有相同Rf和相同保留时间的各部分。这样得到两部分混合液。第25-39份,在TLC板上Rf=0.64的单一点的,为二溴代三尖杉宁碱。而第41-81份,Rf=0.49的单一点的为紫杉醇。
第25-39份,在约40℃下,真空浓缩到干,得到白色到浅黄色的固体(0.460g,理论产率的66.6%)。
得到的二溴代三尖杉宁碱测定结果如下:
m.p.(熔点):158-160℃(色谱纯为96.19%)
Rf=0.64(单点),在硅胶60 F254板上,
(Merck,#5554)
展开剂:丙酮-二氯甲烷(20∶80)
显色剂;香草醛-硫酸的甲醇溶液
质谱测定[FAB]+:
[M+H]+=990,992,994
[M+Na]+=1014
[M+K]+=1030
第41-81份,浓缩干燥后,得到1.16g的紫杉醇(>100%的理论产率),采用丙酮/己烷(50∶50)重结晶,过滤,用与重结晶溶剂比例相同的冷溶剂洗涤,40℃下,真空干燥24小时。得0.902g(根据原料重量,这相当于理论值的45.11%;根据原料中紫杉醇含量的HPLC测定,相当于理论值的88.1%)白色结晶。
紫杉醇的(分离和纯化)测定结果如下:
m.p.214-216℃
Rf=0.49,标准品在硅胶60 F254板上[Merck#5554]
展开剂:丙酮-CH2Cl2(20∶80)
显色剂:香草醛-硫酸的甲醇溶液紫外光谱(甲醇中): 228.4(297146.8)(λmax nm,(ε)) 206.6(26540.1)红外光谱(Kbr,cm-1):3500,1105,1070(叔和仲OH)
3430,1650,1580(-CONH-)
3070,1610,1520,780,710,
(单取代芳香环)
2950,2910,1480,1450,1370
(CH3,CH2,CH)
3020,1315,980(双键)
1730,1270(芳香酯)
1715,1240(>C=O)
1730,1180(乙酸酯)
850(环氧环)样品的紫外光谱和红外光谱谱图,均与纯的紫杉醇的谱图相符。
例4
自粗的紫杉烷(Taxanes)混合物中
分离和纯化紫杉醇,并作分析测定
将10.00g粗紫杉醇样品溶液(根据HPLC分析,此样品含28.8%三尖杉宁碱,51.2%紫杉醇及约20%的其它紫杉烷和非紫杉烷化杂质)溶于1.5升CCl4中。所得溶液置于2.0升三口烧瓶内,并将三口烧瓶浸于冰-盐浴中。三口烧瓶上装有一个500ml的分液漏斗。回流冷凝管、温度计和磁搅拌器。搅拌反应混合物,直到温度降到5℃。然后在3小时过程中滴加41.2ml的0.1M的溴(0.665g溴)的CCl4溶液。三尖杉宁碱和溴的摩尔比率为1∶1.2。反应整个过程中,温度不超过5℃。加溴结束后,继续搅拌,维持温度在-1到5℃。每小时取样,用HPLC监测反应的进行,直到所有的三尖杉宁碱都转变成二溴代衍生物(约8小时)。反应完成后溶液体积为1500-1600mL,溶液颜色为浅黄色或奶油色,这与起始的原料有关,过量的少量溴的存在亦有影响。
为除去痕量溴,先后用0.5%Na2SO3水溶液(500mL),NaHCO3水溶液(500mL)和去离子水(2×500mL)洗涤反应混合物。反应混合物经无水硫酸钠干燥后,真空浓缩到干,得到13.20g的浅奶油色到白色的结晶产物。
按照例2和3的条件,采用硅胶柱,对产物进行层析分离。玻璃柱100×5cm,将600g硅胶拌成浆状灌入玻璃柱内(比例1∶50)。洗脱液为丙酮-CH2Cl2(10∶90)。最后用1升丙酮-CH2Cl2(25∶75)淋洗。接收的每一份洗脱液用TLC检测,每第3份洗脱液用HPLC检测。第11-22份洗脱液,在TLC板上呈单个斑点,其Rf=0.64,合并后在Buchi旋转蒸发器上,40℃下,真空浓缩蒸干,得3.25g(95%)的2″,3″-二溴代三尖杉宁碱的白色到浅黄色固体。
产品分析如下:
m.p.(熔点):158-160℃。
Rf=0.64(单点),在硅胶60 F254板上[Merck#5554]
展开剂:丙酮-CH2Cl2(20;80)。
显色剂;香草醛-硫酸的甲醇溶液。
元素分析和分子量如下:(根据HR FAB+);C45H54NO14 79Br2[M+H]+:
计算值:990.191000
实测值:990.191103(Δm=0.1ppm)C45H54NO14 79Br81Br[M+H]+:
计算值:992.181000
实测值:992.189057(Δm=8.1ppm)C45H54NO14 81Br2[M+H]+:
计算值:994.175000
实测值:994.187011(Δm=12.1ppm)C45H53NO14Na79Br81Br[M+Na]+:
计算值:1014.161000
实测值:1014.172002(Δm=9.9ppm)C45H53NO14K79Br81Br[M+K]+::
计算值:1030.097000
实测值:1030.144940(Δm=46.5ppm)紫外光谱(CH3OH中):[λmax nm,(∈)]274.2(1550.8);227.1
(18610.4);221.8(18325.1)红外光谱(KBr)(cm-1): 3500,1105,1070(叔和仲OH)
3420,1670,1580(-CONH-)
3110,3060,1605,1505,770,
710(单取代芳香化合物)
3060,2960,2915,2870,1465,
1370,(-CH3,-CH2-,=CH-)
3020,1670,1310,980(双键)
1730,1270(芳香酯)
1715,1240(>C=O)
1730,1180(乙酸酯)
855(环氧环)
520(溴代化合物)质子核磁共振(CDCl3中)(300MHz): 1.94(d,3H,-COC(Br)
CH 3-5″)(ppm;仅侧链质子) 1.98(d,3H,-HC(Br)
CH 3-4″)
4.63(qt,1H,-1 CH(Br)-3″)13C核磁共振(300MHz) 170.21和170.25(C-1′)(ppm;仅侧链C) 172.26和172.32(C-1″)
72.76和72.90(C-2′)
69.71和69.88(C-2″)
54.34和54.52(C-3)
55.13和55.35(C-3″)
30.39和30.77(C-4″)
27.21和27.62(C-5″)电子轰击电离质谱:[M]+(m/z) 568,551,509,491,449,431,405,
(主要碎片) 391,386,329,326,308,278
264,245,217,200,188,159,149,
122,105,91,83,77,55,43。直接化学电离源质谱:[M+H]+(m/z) 569,552,510,492,474,450,432,
(主要碎 424,392,387,370,329,327,片)
309,279,265,264,246,218,200,
188,167,149,125,124,106,
101,100,91,83,69。FAB+-MS:(M/Z) 1030[M+K]+;1014[M+Na]+;992
[M+H]+(见元素分析);
974[M-H2O]+;932[M-AcOH]+;914
[M-AcOH-H2O]+;912[M-HBr]+;870
[M-BzOH]+;854[870-H2O-2H];
832[M-2HBr]+;705[M-243-Ac]+;
569[T]+;551[T-H2O];
509[T-AcOH]+;491[T-AcOH-H2O
]+;448[T-BzOH]+;
429;424[SH2]+;413;405[S-H2O]+;
391[S-O-H2O]+;
387[T-AcOH-BzOH]+;376;347
[S-O-CO-HCHO]+;338:
327[387-AcOH]+;315;284[327-
Ac]+;279;264[832-T]+or
[424-2HBr]+;246[264-H2O]+;231;
218[264-HCOOH]+;188;
167[S-C5H8ONBr2]+;149[167-H2O]+;
133;122[BzOH]+;113:
105[Bz]+;91[C7H7]+;83;77[C6H6]+
76;57;55;
(T=化合物中紫杉烷环;S-化合物
中酸(侧)链)HPLC:条件1: 柱CN10μ(250×4.6mm)
流动相CH3CN∶H2O(40∶60)
流速1mL/min
Waters 490紫外检测器,波长227nm
进样体积20μL
RT2″,3″-二溴代三尖杉宁碱26.06分。条件2: 柱:Curosil G6μ(250×3.2mm)
流动相CH3CN∶H2O(45∶55)
流速0.75mL/min
Waters 490检测器,波长227nm
进样体积20μL
RT2″3″-二溴代三尖杉宁碱2非对映异构
体:
RTI=23.53
RTII=24.50
热重分析(TGA):
温度(稳定性%)28.04℃
(100.0%),100.00℃(99.64%),
150.00℃(98.88%),175.00℃(95.35%),
180.00℃(86.74%),200.00℃
(60.38%),250.00℃(45.03%)。
示差扫描量热法(DSC):173.76℃,187.73℃
从第26-68份,每份经TLC检测,仅出现一个斑点(Rf=0.49,与紫杉醇标准品相符),经HPLC检测,仅出现一个峰。将这些部分合并,在40℃下,采用Buchi旋转蒸发器,真空浓缩干燥,得到6.10g白色固体。将此固体置于60mL丙酮-己烷(50∶50)结晶,过滤,用同样比例的冷溶剂洗涤,在40℃下,高真空干燥24小时,得到4.84g(92%)白色固体结晶。与紫杉醇标准品相比较,定为紫杉醇。
纯化过的紫杉醇分析如下:
m.p.(熔点):214℃-216℃
TLC
Rf=0.49(有紫杉醇标准品作对照)
硅胶60F254板(merck#5554)
展开剂:丙酮-CH2Cl2(20∶80)
显色剂:香草醛-硫酸的甲醇溶液
元素分析:C47H51O14N: %C %H %N
计算值: 66.11 6.02 1.64
实测值: 65.97 5.89 1.63图5紫外光谱(甲醇中):(λmax nm(∈)) 227.2(29824.1)图6 208.0(26256.3)红外光谱(KBr)(cm-1)
3500,1105,1070,(叔和仲.OH)
3430,1650,1580(-CONH-)
1610,1520,780,710(单取代芳环)
2950,2910,1480,1450,1370(-CH3,
-CH2-,>CH-基)
3020,1315,980(双键)
1725,1270(芳香酯)
1710,1240(>C=O)
850(环氧环)图7a质子核磁共振谱: 1.88(S,1OH,C-1);5.66(d,1H,C-2);(300MHz;CDCl3)(ppm)3.82(dd.1H,C-3);2.38(S,3H,CH3COO,C-
4);4.94(dd,1H,C-5);
1.88(ddd,1H,C-6);2.48(ddd,1H,C-6)
2.53(d,1 OH,C-7);4.38(dd,1H,C-7)
6.27(S,1H,C-10);2.23(S,3H,CH3COO,
C-10);6.20(Qt,1H,C-13)
2.27(ddd,1H,C-14);2.33(dd,1H,C-14);
1.13(S,3H,C-17);1.23(S,3H,C-18);1.78
(S,3H,C-18);1.68(S,3H,C-19);4.20(dd.
1H,C-20);4.30(S,1H,C-20);3.77(S,1H,
C-2′);4.78(ddd,1H,C-2′);5.20(ddd,1H,
C-3),7.10(d,1H,N-1);7.30÷7.53(m.10H,P-
和m-芳环上的质子A1B1,&C1);7.64(t1H,
A1-P);7.72(dd,2H,C1-O);8.11(dd,2H,A1-
O).图7b13C核磁共振谱 79.1(C-1);75.1(C-2);45.8(C-3);81.2(C-4);(300MHz,CDCl3)(ppm) 84.4(C-5);35.6(C-6);72.1(C-7);56.7(C-8);
203.6(C-9);75.6(C-10);133.3(C-11);
141.9(C-12);72.3(C-13);35.7(C-14);43.2(C-
15);21.8(C-16);26.9(C-17);14.7(C-18);
9.5(C-19);76.5(C-20);73.3(C-2′);55.1(C-3′)
20.7(
CH3COat C-10);22.6(
CH3CO,C-
4);170.3(CH3 CO,C-10);171.1(CH3 CO,C-
4);167.0(Ar
CO-A1);167.0(Ar
CO-C1);
172.7(phISCO-);129.3(aC-C1);133.8(aC-B1)
138.1(aC-C1);130.3(O-C,A1);127.0(O-C,
B1);127.0(O-C,C1);128.7(m-C,A1);图8a和8b 128.6(m-C,B1);129.0(m-C,C1);133.6(p-C,
A1);131.9(p-C,B1);128.3(p-C,C1)电子撞击电离质谱: 568[T]+;550[T-H2O]+;508[T-AcOH]+;[M]+=853(m/z,主要碎片)490[T-AcOH-H2O]+;448[T-2AcOH]+or[T-
BzOH]+
386[T-AcOH-BzOH]+;326[T-BzOH-
2AcOH]+;308[326-H2O]+;286[M-T]+或
[S]+;280;268[S-O]+;240[S-O-CO]+;210[
S-O-CO-HCOH]+;
122[BzOH]+;105[Bz]+;91[C7H7]+;77[C6H5]+;5
1;43[Ac]+。直接化 569;551;509;492;449;387;327;学电离质谱:[M+H]+ 311;287;269;240;224;222;210;=854(m/z;主要碎片) 165;149;123;105;92;71。图10快原子轰击质谱(正离子) 892[M+K]+;876[M+Na]+;854[M+H]+;569;(m/z;主要碎片) 551;523;509;495;369;327;286;240;210;
277;155;149;119;105;85;69。图11快原子轰击质谱(负离子) 852[M-H]-图12HPLC柱 苯基键合柱(μBondapak Phcnyl)流动相 CH3CN∶CH3OH∶H2O-32∶20∶48流速 1mL/min检测器 Waters 490,波长227nm进样体积 20μL图13热重分析法: 温度(稳定性):50.00℃(100.0%),205.00℃
(99.86%),215.00℃(99.10%),220.00℃
(92.19%),250.00℃(56.66%),275.00℃
(45.92%)。图14示差扫描量热法: 210.85℃水含量(%H2O): 0.90%(Karl Fischer)
Claims (33)
1.一种从含有紫杉烷混合物的有机原料中分离和纯化紫杉醇的方法,所述方法包括:
(1)从上述有机原料中提取含有紫杉化合物的组合物;
(2)从所述组合物中层析分离含有紫杉醇、三尖杉宁碱和其它紫杉烷的混合物,然后
(3)选择有效条件,将混合物与卤素反应,选择性地将三尖杉宁碱转变成二卤代三尖杉宁碱的非对映异构体混合物;然后
(4)从上述混合物中,分离紫杉醇。
2.权利要求1的方法,其中有机原料用第一萃取溶剂萃取后,蒸发得到含紫杉醇的第一残渣;然后用第二萃取溶剂萃取第一残渣,再蒸发,得到含紫杉醇的第二残渣,接着进一步用结晶法纯化第二残渣。
3.权利要求2的方法,其中第一萃取溶剂是甲醇,含紫杉醇的第一残渣被分配于水相和选自下列的有机相之间:二氯甲烷、二氯乙烷和氯仿,该残渣经所述溶剂萃取后,干燥,得第二残渣,将此含紫杉醇的第二残渣溶于丙酮中,加己烷沉淀出非极性杂质,再将此丙酮-己烷溶液蒸发到原体积的1/3,得到含紫杉醇的粘稠状第三残渣。
4.权利要求3的方法,其中所述溶剂和水的体积比1∶1。
5.权利要求3的方法,其中向粘稠状第三残渣中加入10倍体积的己烷,析出浅黄色沉淀,形成含紫杉醇的固体第四残渣。
6.权利要求4的方法,进一步包括将第四固体残渣溶于丙酮与二氯甲烷和/或二氯乙烷的混合溶剂中,通过硅胶柱和色谱溶剂进行闪式色谱分离,将含有紫杉醇和三尖杉宁碱混合物所得的各级分合并、蒸干,得到含紫杉醇的第五残渣。
7.权利要求6的方法,其中所述色谱所用的溶剂是丙酮与二氯甲烷或二氯乙烷的混合物,其体积比是1∶9到3∶7。
8.权利要求6的方法,其中将第五残渣溶于选自下列的氯代溶剂中:CCl4、CHCl3、C2H4Cl2或CH2Cl2,并与卤素反应。
9.权利要求8的方法,其中所说的卤素是溴。
10.按权利要求9的方法,其中溴溶于卤代溶剂中,其浓度为0.01M到0.1M。
11.权利要求10的方法,其中溴化反应在温度-20℃到20℃和黑暗条件下进行。
12.权利要求11的方法,其中存在于上述氯代溶剂中的三尖杉宁碱基本上完全转变成二溴代三尖杉宁碱非对映异构体混合物,然后从该反应混合物中分离紫杉醇。
13.权利要求12的方法,其中含紫杉醇的有机物质选自:短叶红豆杉的树皮、红豆杉各品种的植物原料,红豆杉各品种的细胞培养物以及能产生紫杉醇的霉菌。
14.权利要求12的方法,其中通过硅胶层析,将紫杉醇自反应混合物中分离出来,层析用的洗脱液是丙酮与二氯甲烷或二氯乙烷的混合物,其体积比是1∶9到3∶7。
15.权利要求12的方法,其中分离出的紫杉醇在丙酮与己烷混合物中结晶,使紫杉醇析出。
16.一种从含有紫杉醇和三尖杉宁碱混合物中分离紫杉醇的方法,所述方法包括以下步骤:a)将该混合物与卤素反应,反应温度和时间应充分保证,让三尖杉宁碱基本上完全被卤化;b)将紫杉醇与三尖杉宁碱卤代衍生物分离。
17.权利要求16的方法,其中所述卤素可选用氟、氯、溴或碘。
18.权利要求17的方法,其中所述卤素是溴。
19.权利要求18的方法,其中溴的浓度是0.01M到0.1M。
20.权利要求16的方法,其中反应a)的温度范围是-20℃至20℃。
21.权利要求19的方法,其中溴溶于氯代溶剂CCl4、CHCl3或CH2Cl2中。
22.权利要求21的方法,其中氯代溶剂是CCl4或CHCl3。
23.权利要求16的方法,其中反应a)在黑暗条件下进行。
24.权利要求16的方法,其中b)步分离是采用硅胶层析,选用适当溶剂作洗脱液。
25.权利要求24的方法,其中溶剂是丙酮与二氯甲烷或1,2二氯乙烷的混合物,其体积比是1∶9到3∶7。
26.权利要求25的方法,其中含紫杉醇的各个部分,蒸干成固体残渣。
27.权利要求26的方法,其中用结晶法进一步纯化紫杉醇。
28.权利要求27的方法,其中固体残渣被溶于丙酮中。
29.权利要求28的方法,其中紫杉醇从己烷内结晶析出。
30.权利要求25的方法,其中紫杉醇与2″,3″-二溴代三尖杉宁碱的非对映异构体混合物分离。
31.权利要求16的方法,其中反应a)用HPLC监测。
32.权利要求16的方法,其中混合物是来自含有紫杉醇的原料。
33.权利要求32的方法,其中含紫杉醇原料选自:短叶红豆杉树皮,红豆杉各品种的植物原料、红豆杉各品种的细胞培养物以及能产生紫杉醇的霉菌。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/572,240 US5654448A (en) | 1995-10-02 | 1995-12-13 | Isolation and purification of paclitaxel from organic matter containing paclitaxel, cephalomannine and other related taxanes |
| US08/572,240 | 1995-12-13 | ||
| ZA976834A ZA976834B (en) | 1995-10-02 | 1997-07-31 | Isolation and purification of paclitaxel from organic matter containing paclitaxel cephalomannine and other related taxanes |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1182424A CN1182424A (zh) | 1998-05-20 |
| CN1104426C true CN1104426C (zh) | 2003-04-02 |
Family
ID=27075788
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN96193149A Expired - Fee Related CN1104426C (zh) | 1995-12-13 | 1996-11-01 | 紫杉醇和三尖杉宁碱的分离和纯化 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US5654448A (zh) |
| EP (1) | EP0809639B1 (zh) |
| JP (1) | JPH11500753A (zh) |
| CN (1) | CN1104426C (zh) |
| AU (1) | AU720719B2 (zh) |
| BR (1) | BR9607137A (zh) |
| CA (1) | CA2210972C (zh) |
| NZ (1) | NZ321499A (zh) |
| WO (1) | WO1997021696A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1331859C (zh) * | 2005-01-04 | 2007-08-15 | 四川华申科技开发有限公司 | 分离紫杉醇和三尖杉磷碱的方法 |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5840748A (en) * | 1995-10-02 | 1998-11-24 | Xechem International, Inc. | Dihalocephalomannine and methods of use therefor |
| KR100266448B1 (ko) | 1997-06-26 | 2000-09-15 | 박종헌 | 식물세포 배양 중의 온도변화에 의한 택솔의 대량생산 방법 |
| AU1701699A (en) * | 1997-11-19 | 1999-06-07 | Xechem International, Inc. | Halogenated paclitaxel derivatives |
| KR100474228B1 (ko) * | 1998-10-28 | 2005-12-13 | 주식회사 보락 | 파크리탁셀의분리방법 |
| US6136989A (en) * | 1998-12-30 | 2000-10-24 | Phytogen Life Sciences, Incorporated | Method for high yield and large scale extraction of paclitaxel from paclitaxel-containing material |
| US5969165A (en) * | 1999-01-07 | 1999-10-19 | 508037 (Nb) Inc. | Isolation and purification of paclitaxel and other related taxanes by industrial preparative low pressure chromatography on a polymeric resin column |
| US6002025A (en) * | 1999-02-24 | 1999-12-14 | Bcm Developement Inc. | Method for the purification of taxanes |
| CN1111166C (zh) * | 1999-09-10 | 2003-06-11 | 云南汉德生物技术有限公司 | 水溶性三尖杉磷碱聚氨基酸酯或其盐,含它们的药物组合物及其医药用途 |
| EP1229934B1 (en) | 1999-10-01 | 2014-03-05 | Immunogen, Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| CA2395573A1 (en) * | 2000-01-13 | 2001-07-19 | Purdue Research Foundation | Production of taxol and taxanes |
| US6452024B1 (en) | 2000-02-22 | 2002-09-17 | Chaichem Pharmaceuticals International | Process for extraction and purification of paclitaxel from natural sources |
| KR100666652B1 (ko) * | 2000-02-25 | 2007-01-09 | 한화석유화학 주식회사 | 식물 원료로부터 초임계 유체를 이용하여 탁솔을 제조하는방법 |
| AU2002223319A1 (en) | 2000-11-08 | 2002-05-21 | Actipharm Inc. | Process for mass production of gmp paclitaxel and related taxanes |
| WO2002038555A1 (en) * | 2000-11-13 | 2002-05-16 | Lahu Saiji | Isolation of taxanes |
| US6759539B1 (en) | 2003-02-27 | 2004-07-06 | Chaichem Pharmaceuticals International | Process for isolation and purification of paclitaxel from natural sources |
| CN102702139B (zh) | 2006-04-03 | 2016-01-20 | 药物热化学品公司 | 热提取方法和产物 |
| US20090077871A1 (en) * | 2007-05-04 | 2009-03-26 | Alex Berg Gebert | Process for obtaining low and medium molecular weight Polyphenols and standardized solid fuel from tree wood or bark |
| CN101307040B (zh) * | 2007-05-14 | 2010-12-29 | 中国科学院大连化学物理研究所 | 一种分离制备紫杉醇的方法 |
| US7905990B2 (en) | 2007-11-20 | 2011-03-15 | Ensyn Renewables, Inc. | Rapid thermal conversion of biomass |
| US20110284359A1 (en) | 2010-05-20 | 2011-11-24 | Uop Llc | Processes for controlling afterburn in a reheater and for controlling loss of entrained solid particles in combustion product flue gas |
| US8499702B2 (en) | 2010-07-15 | 2013-08-06 | Ensyn Renewables, Inc. | Char-handling processes in a pyrolysis system |
| US9441887B2 (en) | 2011-02-22 | 2016-09-13 | Ensyn Renewables, Inc. | Heat removal and recovery in biomass pyrolysis |
| US9347005B2 (en) | 2011-09-13 | 2016-05-24 | Ensyn Renewables, Inc. | Methods and apparatuses for rapid thermal processing of carbonaceous material |
| US10400175B2 (en) | 2011-09-22 | 2019-09-03 | Ensyn Renewables, Inc. | Apparatuses and methods for controlling heat for rapid thermal processing of carbonaceous material |
| US10041667B2 (en) | 2011-09-22 | 2018-08-07 | Ensyn Renewables, Inc. | Apparatuses for controlling heat for rapid thermal processing of carbonaceous material and methods for the same |
| US9044727B2 (en) | 2011-09-22 | 2015-06-02 | Ensyn Renewables, Inc. | Apparatuses and methods for controlling heat for rapid thermal processing of carbonaceous material |
| US9109177B2 (en) | 2011-12-12 | 2015-08-18 | Ensyn Renewables, Inc. | Systems and methods for renewable fuel |
| US9670413B2 (en) | 2012-06-28 | 2017-06-06 | Ensyn Renewables, Inc. | Methods and apparatuses for thermally converting biomass |
| TWI645026B (zh) | 2013-06-26 | 2018-12-21 | 安信再生公司 | 可再生燃料之系統及方法 |
| CA2995845A1 (en) | 2015-08-21 | 2017-03-02 | Ensyn Renewables, Inc. | Liquid biomass heating system |
| MY193949A (en) | 2016-12-29 | 2022-11-02 | Ensyn Renewables Inc | Demetallization Of Liquid Biomass |
| CN113896695B (zh) * | 2021-05-26 | 2023-12-01 | 四川杉宝生物科技有限公司 | 从曼地亚红豆杉中同时提取三尖杉宁碱和紫杉醇的方法 |
| CN113512015A (zh) * | 2021-05-26 | 2021-10-19 | 上海卓鼎生物技术有限公司 | 一种工业化提纯三尖杉宁碱的方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1069965A (zh) * | 1991-04-19 | 1993-03-17 | 密西西比大学 | 分离塔三烷衍生物的方法及含其的组合物 |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2601675B1 (fr) * | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Derives du taxol, leur preparation et les compositions pharmaceutiques qui les contiennent |
| US4942184A (en) * | 1988-03-07 | 1990-07-17 | The United States Of America As Represented By The Department Of Health And Human Services | Water soluble, antineoplastic derivatives of taxol |
| US5157049A (en) * | 1988-03-07 | 1992-10-20 | The United States Of America As Represented By The Department Of Health & Human Services | Method of treating cancers sensitive to treatment with water soluble derivatives of taxol |
| FR2629818B1 (fr) * | 1988-04-06 | 1990-11-16 | Centre Nat Rech Scient | Procede de preparation du taxol |
| US4960790A (en) * | 1989-03-09 | 1990-10-02 | University Of Kansas | Derivatives of taxol, pharmaceutical compositions thereof and methods for the preparation thereof |
| US5019504A (en) * | 1989-03-23 | 1991-05-28 | The United States Of America As Represented By The Secretary Of Agriculture | Production of taxol or taxol-like compounds in cell culture |
| MY110249A (en) * | 1989-05-31 | 1998-03-31 | Univ Florida State | Method for preparation of taxol using beta lactam |
| US5175315A (en) * | 1989-05-31 | 1992-12-29 | Florida State University | Method for preparation of taxol using β-lactam |
| US5136060A (en) * | 1989-11-14 | 1992-08-04 | Florida State University | Method for preparation of taxol using an oxazinone |
| US5015744A (en) * | 1989-11-14 | 1991-05-14 | Florida State University | Method for preparation of taxol using an oxazinone |
| US5059699A (en) * | 1990-08-28 | 1991-10-22 | Virginia Tech Intellectual Properties, Inc. | Water soluble derivatives of taxol |
| US5278324A (en) * | 1990-08-28 | 1994-01-11 | Virginia Tech Intellectual Properties, Inc. | Water soluble derivatives of taxol |
| US5475120A (en) * | 1990-11-02 | 1995-12-12 | University Of Florida | Method for the isolation and purification of taxol and its natural analogues |
| US5194635A (en) * | 1991-03-18 | 1993-03-16 | Virginia Tech Intellectual Properties, Inc. | Rearranged taxol compounds and method of using in testing of in vivo activity |
| CA2071160A1 (en) * | 1991-07-31 | 1993-02-01 | Vittorio Farina | Asymmetric synthesis of taxol side chain |
| US5243045A (en) * | 1991-09-23 | 1993-09-07 | Florida State University | Certain alkoxy substituted taxanes and pharmaceutical compositions containing them |
| US5274124A (en) * | 1991-09-23 | 1993-12-28 | Florida State University | Metal alkoxides |
| US5250683A (en) * | 1991-09-23 | 1993-10-05 | Florida State University | Certain substituted taxanes and pharmaceutical compositions containing them |
| US5229526A (en) * | 1991-09-23 | 1993-07-20 | Florida State University | Metal alkoxides |
| US5283253A (en) * | 1991-09-23 | 1994-02-01 | Florida State University | Furyl or thienyl carbonyl substituted taxanes and pharmaceutical compositions containing them |
| US5227400A (en) * | 1991-09-23 | 1993-07-13 | Florida State University | Furyl and thienyl substituted taxanes and pharmaceutical compositions containing them |
| US5338872A (en) * | 1993-01-15 | 1994-08-16 | Florida State University | Process for the preparation of 10-desacetoxybaccatin III and 10-desacetoxytaxol and derivatives thereof |
| US5284864A (en) * | 1991-09-23 | 1994-02-08 | Florida State University | Butenyl substituted taxanes and pharmaceutical compositions containing them |
| US5272171A (en) * | 1992-02-13 | 1993-12-21 | Bristol-Myers Squibb Company | Phosphonooxy and carbonate derivatives of taxol |
| IT1254515B (it) * | 1992-03-06 | 1995-09-25 | Indena Spa | Tassani di interesse oncologico, loro metodo di preparazione ed uso |
| US5200534A (en) * | 1992-03-13 | 1993-04-06 | University Of Florida | Process for the preparation of taxol and 10-deacetyltaxol |
| JPH07508163A (ja) * | 1992-04-01 | 1995-09-14 | ユニオン・キャンプ・コーポレーション | イチイ属の体細胞胚形成の誘導及びそれからのタキサン環含有アルカロイドの生産 |
| US5254703A (en) * | 1992-04-06 | 1993-10-19 | Florida State University | Semi-synthesis of taxane derivatives using metal alkoxides and oxazinones |
| US5248796A (en) * | 1992-06-18 | 1993-09-28 | Bristol-Myers Squibb Company | Taxol derivatives |
| GB9213077D0 (en) * | 1992-06-19 | 1992-08-05 | Erba Carlo Spa | Polymerbound taxol derivatives |
| US5254580A (en) * | 1993-01-19 | 1993-10-19 | Bristol-Myers Squibb Company | 7,8-cyclopropataxanes |
| US5294637A (en) * | 1992-07-01 | 1994-03-15 | Bristol-Myers Squibb Company | Fluoro taxols |
| US5334732A (en) * | 1992-07-02 | 1994-08-02 | Hauser Chemical Research, Inc. | Oxidation of cephalomannine with ozone in the presence of taxol |
| US5202448A (en) * | 1992-08-14 | 1993-04-13 | Napro Biotherapeutics, Inc. | Processes of converting taxanes into baccatin III |
| US5470866A (en) * | 1992-08-18 | 1995-11-28 | Virginia Polytechnic Institute And State University | Method for the conversion of cephalomannine to taxol and for the preparation of n-acyl analogs of taxol |
| US5319112A (en) * | 1992-08-18 | 1994-06-07 | Virgnia Tech Intellectual Properties, Inc. | Method for the conversion of cephalomannine to taxol and for the preparation of N-acyl analogs of taxol |
| US5279949A (en) * | 1992-12-07 | 1994-01-18 | Board Of Trustees Operating Michigan State University | Process for the isolation and purification of taxol and taxanes from Taxus spp |
| US5475011A (en) * | 1993-03-26 | 1995-12-12 | The Research Foundation Of State University Of New York | Anti-tumor compounds, pharmaceutical compositions, methods for preparation thereof and for treatment |
| US5412092A (en) * | 1993-04-23 | 1995-05-02 | Bristol-Myers Squibb Company | N-substituted 2-azetidinones |
| US5336684A (en) * | 1993-04-26 | 1994-08-09 | Hauser Chemical Research, Inc. | Oxidation products of cephalomannine |
-
1995
- 1995-12-13 US US08/572,240 patent/US5654448A/en not_active Expired - Fee Related
-
1996
- 1996-11-01 BR BR9607137A patent/BR9607137A/pt not_active Application Discontinuation
- 1996-11-01 EP EP96937101A patent/EP0809639B1/en not_active Expired - Lifetime
- 1996-11-01 CA CA002210972A patent/CA2210972C/en not_active Expired - Fee Related
- 1996-11-01 NZ NZ321499A patent/NZ321499A/en unknown
- 1996-11-01 JP JP9522026A patent/JPH11500753A/ja not_active Ceased
- 1996-11-01 CN CN96193149A patent/CN1104426C/zh not_active Expired - Fee Related
- 1996-11-01 AU AU74848/96A patent/AU720719B2/en not_active Ceased
- 1996-11-01 WO PCT/US1996/017545 patent/WO1997021696A1/en active IP Right Grant
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1069965A (zh) * | 1991-04-19 | 1993-03-17 | 密西西比大学 | 分离塔三烷衍生物的方法及含其的组合物 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1331859C (zh) * | 2005-01-04 | 2007-08-15 | 四川华申科技开发有限公司 | 分离紫杉醇和三尖杉磷碱的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| BR9607137A (pt) | 1997-11-25 |
| CA2210972C (en) | 2003-10-21 |
| US5654448A (en) | 1997-08-05 |
| EP0809639A4 (en) | 1999-01-13 |
| AU7484896A (en) | 1997-07-03 |
| NZ321499A (en) | 1999-04-29 |
| JPH11500753A (ja) | 1999-01-19 |
| AU720719B2 (en) | 2000-06-08 |
| EP0809639A1 (en) | 1997-12-03 |
| EP0809639B1 (en) | 2003-12-10 |
| CA2210972A1 (en) | 1997-06-19 |
| WO1997021696A1 (en) | 1997-06-19 |
| CN1182424A (zh) | 1998-05-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1104426C (zh) | 紫杉醇和三尖杉宁碱的分离和纯化 | |
| CN1036342C (zh) | 9,11-脱氢甾类化合物及9-α-羟基-16,17-脱氢甾类化合物的制备方法 | |
| CN1067682C (zh) | 抗肿瘤化合物、药物组合物、其制备方法以及其用于治疗的方法 | |
| CN1046261C (zh) | 紫杉醇及其衍生物的合成 | |
| CN1080256C (zh) | 新的β-内酰胺类及制备紫杉烷的方法 | |
| CN101048394A (zh) | 太平洋紫杉醇向多烯紫杉醇的半合成转化 | |
| CN1095070A (zh) | 制备n4-酰基-5′-脱氧-5-氟胞苷衍生物的新方法 | |
| CN1942460A (zh) | 紫杉烷衍生物的一锅合成及其向太平洋紫杉醇和多烯紫杉醇的转化 | |
| CN1207292C (zh) | 制备矢车菊苷配质低聚物的合成方法 | |
| CN1300289A (zh) | 新的三尖杉烷衍生物及其制备方法 | |
| CN1016507B (zh) | 新的福斯克林衍生物的制备方法 | |
| CN1216861C (zh) | 维生素d衍生物结晶及其制备方法 | |
| CN1151740A (zh) | 7-醚-紫杉酚类似物、抗肿瘤药物用途及含有它们的药物组合物 | |
| CN1087634A (zh) | 7-异二氢氮杂茚基-喹诺酮衍生物和7-异二氢氮杂茚基-萘啶酮衍生物 | |
| CN101061133A (zh) | 制备17-羟基-6β,7β;15β,16β-双亚甲基-3-氧代-17α-孕甾-4-烯-21-羧酸γ-内酯的工业方法和用于该方法的关键中间体 | |
| CN1703409A (zh) | 新紫杉烷和用途及制备方法 | |
| CN1108657A (zh) | 雪花胺衍生物、其制备方法及其作为药物的用途 | |
| CN1886400A (zh) | 9,10-α,α-OH紫杉烷类似物及其制备方法 | |
| CN1286823C (zh) | 用于制备紫杉烷的噁唑烷中间体 | |
| CN1653081A (zh) | 甾族化合物的c-17螺甾内酯化和6,7氧化 | |
| CN1068821A (zh) | 苯并二氢吡喃衍生物 | |
| CN1192014C (zh) | Hiv蛋白酶抑制剂的制备方法 | |
| CN1896065A (zh) | 1-(3′,4′,5′-三取代苯基)-异喹啉类化合物及其制备方法和用途 | |
| CN100339373C (zh) | 紫杉醇类似物的制备和作为抗肿瘤剂的应用 | |
| CN1147491C (zh) | 新的吖啶羧酸酯化合物、其制备方法及其药物组合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20030402 Termination date: 20101101 |