CN1053901A - 制备银金属细分颗粒的方法 - Google Patents
制备银金属细分颗粒的方法 Download PDFInfo
- Publication number
- CN1053901A CN1053901A CN91100858A CN91100858A CN1053901A CN 1053901 A CN1053901 A CN 1053901A CN 91100858 A CN91100858 A CN 91100858A CN 91100858 A CN91100858 A CN 91100858A CN 1053901 A CN1053901 A CN 1053901A
- Authority
- CN
- China
- Prior art keywords
- silver
- gelatin
- acid
- group
- particle
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B11/00—Obtaining noble metals
- C22B11/04—Obtaining noble metals by wet processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F9/00—Making metallic powder or suspensions thereof
- B22F9/16—Making metallic powder or suspensions thereof using chemical processes
- B22F9/18—Making metallic powder or suspensions thereof using chemical processes with reduction of metal compounds
- B22F9/24—Making metallic powder or suspensions thereof using chemical processes with reduction of metal compounds starting from liquid metal compounds, e.g. solutions
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Mechanical Engineering (AREA)
- Metallurgy (AREA)
- Organic Chemistry (AREA)
- Manufacture Of Metal Powder And Suspensions Thereof (AREA)
- Medicinal Preparation (AREA)
- Manufacture And Refinement Of Metals (AREA)
Abstract
本发明提供了制备细分银颗粒的还原性方法,其
中银颗粒从银盐、明胶和酸式磷酸烷基酯的酸性水溶
液中沉淀析出。
Description
本发明涉及制备细分银颗粒的改进的方法。更进一步地说,本发明涉及制备具有狭窄的粒度分布的银颗粒的方法。
银粉被广泛地应用于电子工业中,用于制备导体厚膜糊。这些厚膜糊用于通过丝网印刷法形成施加于基片的导电电路图形。然后将这些电路干燥并焙烧以蒸发掉液态有机载体并使银颗粒烧结而形成导线电路图形。
印刷电路技术需要较致密的和较精确的电子电路。为了满足这些要求,导电线路的宽度已变得更窄,且线路间的距离也更小了。为了形成更紧密的堆积和更窄的线路,银粉必须尽可能地呈球形形状,并且有狭窄的粒度分布。
现有的用于制备金属粉末的很多方法可用于生产银粉。例如化学法、物理法:例如雾化或研磨、热分解,和电化学方法均可采用。
在电子应用中采用的银粉通常是采用化学沉淀方法生产的。银粉是通过化学还原制备的,其中将银的可溶性盐的水溶液与合适的还原剂在银粉可以沉淀出来的条件下进行反应。所用的最普通的银盐是硝酸银。包括肼、亚硫酸盐和甲酸盐在内的无机还原剂可用于还原硝酸银。由于聚集作用,这些方法所制得的粉末粒子很粗(大于2微米),形状无规则,并具有大的粒度分布。
有机还原剂,例如醇、糖或醛,与碱(金属)氢氧化物一起使用以创造硝酸银的还原条件,在这些条件下,还原反应非常快,且难以控制,得到的是带有残余碱离子的粉末。尽管粒度小(<1微米),但这些粉末倾向于具有不规则的形状,宽的粒度分布,不能堆积很紧。这些类型的银粉显示出烧结难以控制和在厚膜印刷导线电路中线路分辨率不够。
因而,本发明的目的是提供制备具有狭窄粒度分布的金属银的细分颗粒的还原方法。本发明特别涉及包括下面系列步骤的金属银细分颗粒的制备方法:
A.形成银盐、明胶和对应于下列结构式的酸式磷酸,烷基酯的非碱性水溶液:
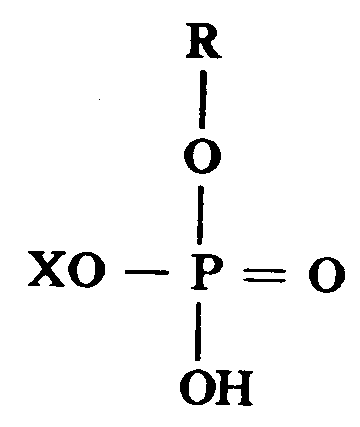
其中X独立地选自H和R基团,R是C6-20的烷基,其可选择地包括最多10个环氧乙烷(EO)部分,溶液中至少包括0.2摩尔溶解的银盐/升溶液,0.001-0.02克明胶/克金属银和0.1-0.5克酸式磷酸烷基酯/升初始溶液;
B.将化学计量超量的水溶性甲酸盐与得自步骤A的反应溶液相掺合,使得银盐完全还原,由此使离散的金属银颗粒得以沉淀并伴之形成CO2和HNO3,同时,将反应溶液保持于足以使沉淀的颗粒保持分散的速率的搅拌下,直至还原反应完成,但搅拌速率也要足够低以避免反应分散液形成泡沫;
C.从反应溶液的液体组分中分离出银颗粒;
D.用去离子水洗涤分离的银颗粒以从中除去其所吸附的物质;和
E.将经洗涤的银颗粒干燥以从中除去水。
Short的专利(U.S.2,752,237)涉及采用过量碱金属盐从含有少量残余的HNO3的AgNO3水溶液中使AgCO3沉淀而制备银的方法。然后用还原剂,例如甲醛还原Ag2CO3碱性悬浮液。
Cubra等人的专利(U.S.3,201,223)中涉及通过(1)加入碱(金属)氢氧化物使Ag2O从AgNO3溶液中沉淀,(2)用甲醛使Ag2O转化为甲酸银,和(3)加热甲酸银以离解甲酸盐残基而得到胶体保护的金属银颗粒,而制备小银颗粒的方法。
Block等人的专利(U.S.3,345,158)中揭示了通过将甲酸加入沸腾的AgNO3溶液(pH=1)中而形成银微晶的方法。
德国专利第2,219,531中揭示了通过形成银复合化合物并加入还原剂(如肼或甲酸钠),使该化合物还原而制备银粉末的方法。该过程在碱性pH下进行。
T.Kubota在Journal of Applied Physics (Japan),39(9):861-868,1970的文章“On the Control of Particle Size in Fine Silver Powders Prepared by Chemical Precipitation”中揭示了用甲醛使银颗粒从氨银溶液中沉淀而制备银颗粒的方法。加入明胶以调整银粒度。
本发明的方法是一种还原方法,其中细分的银颗粒是从银盐、明胶和酸式磷酸烷基酯的酸性水溶液中沉淀出来。该过程是按照下列酸化反应而进行的:
任何水溶性的银盐,例如Ag3PO4、Ag2SO4、硝酸银等均可用于本发明的过程中。但是,不溶性的银盐,例如AgCl则是不适宜的。银盐可以以低至0.2摩尔/升和高压正好在盐溶解度以下的浓度而被使用。为了使所制得的银颗粒不致于太小,较佳为不采用浓度低于0.2摩尔/升的银盐。业已发现,0.6摩尔/升的浓度是最佳的。
本发明的过程可以在只要使液相能维持的很宽广的温度范围内进行。因此,该过程可以在室温或更低的温度下进行。但是,反应速度比较慢并有可能反应进行不完全。因此,较佳是在一个升高的温度下(至少约为50℃)进行该反应。尽管可采用更高的温度,但并不由此而获得重大的额外的效益。因此,50-90℃温度范围是较佳的,70-80℃的温度则更佳。
因为过程的反应是以液相进行的,操作的压力不是关健变量,过程可以在大气压下最方便和经济地进行。
这里所采用的术语“明胶”是指普通的动物明胶或骨胶,它是通过在一定的压力下,用水煮沸动物组织、骨头、腱、韧带等而衍生的白蛋白。用酸抽提的明胶(A型)或用碱抽提的明胶(B型)或两者可用于本发明的过程中。食品级、技术级或U.S.P级明胶均可以采用。
明胶的主要目的是有助于控制粒度。本发明的过程中仅需非常少量的明胶,其量之小以至不会易于察觉地增加反应溶液的粘度。尤其,明胶的量应在0.001-0.02克明胶/克溶解的银离子的范围内。如果明胶用量小于0.01克,则粒度过大,且粒度分布(PSD)太宽;但如果明胶用量大于0.02克,则粒子又过小。0.005-0.018的明胶浓度是较佳的。因此,明胶浓度是过程的一个变量,它与过程的其它变量一起可以被控制,以获得所需要的粒子特性。
明胶仅是过程的一个变量,但它对于获得准确地控制由本发明的过程所制得的银粉末的粒度和PSD是重要的。本发明的实施例中所需的酸式磷酸烷基酯是与下列化学式相应的那些:

在上式中,X独立地选自H和R基团,R是C6-20烷基,它可选择地包含最多10个环氧乙烷(EO)部分。EO部分不大于4是较佳的。这些材料的很多种类都是市场上可购得的,其中R基团对H基团的比率是变化的。例如,其中R/H之比是50/50或75/25的这类化合物是可购得的。满足上述标准的所有这类材料均适合用于本发明,只要它们可以均匀地悬浮于水中。它们不是必需在反应条件下完全溶解于水中。
上述酸式磷酸烷基酯与明胶结合使用是必需的。例如,当明胶被省略,在过程中只使用酸式磷酸烷基酯时,粉末有聚集的趋势,且粒度分布太宽。此外,当既不用明胶又不用酸式磷酸烷基酯时,所得到的颗粒严重聚集,甚至呈海绵状,粒度分布相当宽,即<1微米->40微米。
为了达到生效,用于本发明的过程中的酸式磷酸烷基酯的浓度为至少0.05克/升。但是,更高的浓度也可采用,但浓度高于约10克/升不呈现任何进一步的优势。0.1-0.5克/升的浓度是较佳的。
作为本发明的过程的还原剂,任何水溶性甲酸盐,例如甲酸钠、甲酸钾或甲酸铵均可使用。所用的甲酸盐的量必须化学计量足够地能还原在反应溶液中的所有银阳离子,并且最好是摩尔过量以保证除去反应溶液中的所有银。至少0.1摩尔/摩尔的摩尔过量是较佳的,0.50则更佳。虽然过程中可采用更大过量的甲酸盐,但是它们无技术优势。为使反应溶液的泡沫形成趋势最小,以连续或间断的方式缓慢地加入甲酸盐是较佳的。通常,较慢的甲酸盐加料速率引起较大的银颗粒的形成。因此,甲酸盐的加料速率不仅应足够地慢以防止泡沫形成,而且又要足够地快以获得小尺寸的颗粒。
为了达到较低的反应速率并较好地控制反应速率,本发明的过程要在非碱性条件下进行。碱性过程对于银的沉淀作用是不利的,因为它会使所得到的银颗粒过小,且会形成作为极限溶解度的中间体的氧化银(Ag2O)。另一方面,在本发明的过程中,所有反应剂形式都是可溶的。
调节本发明的过程的pH是不必要的,因为酸式磷酸烷基酯和硝酸银的存在会使初始反应溶液呈酸性,且在过程中二氧化碳和硝酸的演变可保持反应溶液处于酸性状态下。
当过程进行时,为了提供空间均匀生长颗粒条件并由此而防止粒度分布变宽,保持沉淀出来的银颗粒分散于反应溶液中是必需的。这可通过搅拌反应溶液而达到。但是,由于表面活性的酸式磷酸烷基酯的存在而导致反应溶液发泡的趋势,保持足够低的搅拌程度以防止大量泡沫形成是必要的。
沉淀反应完成后,将颗粒从反应溶液中分离出来,洗涤以除去吸附在颗粒上的离子,然后干燥。
颗粒可以通过常用的方法,例如倾析、过滤、离心等方法,而从反应溶液中分离出来。然后用水(较佳为去离子水)洗涤所移出的带有很多水的颗粒,以除去颗粒上所吸附的离子。这可以通过在水中重复地洗涤颗粒直到洗涤溶液的电导率低于约20微西门子(一微西门子相当于一微欧姆)。在洗涤步骤之后,通过诸如烘箱干燥、冷冻干燥、真空干燥、通风干燥等技术或这些技术的组合使经洗涤的颗粒干燥。
实例
一般程序
在一个配备有挡板和船用螺旋浆型搅拌器的8升玻璃反应容器中,将磷酸酯表面活性剂分散并溶解于去离子(DI)水中。在50℃下溶解明胶。将溶液加热至80℃并将AgNO3溶解至特定的浓度。在另一个容器中,在80℃下制备特定浓度的甲酸盐溶液。在足够的搅拌速率下,以规定用于一个特定的时间周期的进料速率,开始将溶液加入反应容器,以便将固体产物均匀地悬浮于液体介质中。
加料周期结束后,在相同的搅拌速度下,将悬浮液在80℃下保持30分钟。停止加热和搅拌。过滤并用去离子(DI)水洗涤固体产物至电导率为10微欧姆。冷冻干燥。
通过前述步骤制备20批银颗粒以观察过程变量对于所沉淀的银颗粒的性能的影响。这20批的数据在下列表1中给出。
2-9栏是直接观察或计算的,栏10中表面积SA是通过BET测定得出的。栏11和12中的最小和最大粒度分布(PSD)是通过根据SEM显微照相直接测定而估算的。栏13表示冷冻干燥状态下,SEM照相中粉末是否出现块集或熔合到一起。
实例1:除另外指明外,与基本条件对照,其它情况是相似的,产品粉末是球形的,并具有在0.1-0.4微米之间的相当均匀的PSD。SEM照相中粉末未出现块集。SA是2.1米2/克。
实例2:显示了将反应剂的浓度依次从50%减少至30%,引起尺寸的很略微减小;从而,SA略微增加。可能归因于限制反应剂(AgNO3)的较少量是有效的。
实例3:显示了依次将反应剂浓度从50%增加到80%,并采用二步加料方案,其中甲酸盐在2.6X的加料速率下加入,在基本条件下历经10分钟,然后,在1.3×下保持160分钟,引起粉末中有些熔合颗粒,具有不规则的形状和较低的表面积。
实例4:显示了在70%基本速率的速率下加料达60分钟,然后再在基本速率的140%的速率下达90分钟,结果所得到的粉末本质上等同于基本条件下所得的产品。
实例5:显示了将1/10明胶浓度和1/3磷酸酯表面活性剂(TDP)浓度作为基本条件,结果得到具有较宽的PSD和只有一半SA的,即平均粒径较大的粉末。
实例6:显示了采用1/2明胶浓度和同样浓度的TDP作为基本条件,结果得到略大的PSD和略低的SA。
实例7:显示了不采用明胶,但将TDP的浓度保持相同作为基本条件,结果得到较宽得多的PSD和较低得多的SA,而SEM中粉末出现块集。
实例8:显示了不采用明胶和磷酸酯表面活性剂,结果得到高度熔合或块集的具有非常宽的PSD和非常低的SA的粉末。
实例9:显示了采用基本浓度的明胶,但不采用磷酸酯,结果得到很小的颗粒,表现出严重聚集的粉末。
实例10:显示了将明胶浓度减至一半,且仍不采用磷酸酯,结果得到具有相当宽的PSD和严重块集的粉末。
实例11:与实例5相似,除了采用1/4基本明胶浓度(与1/10不同),仍用1/3基本TDP浓度。得到具有散布于实例5和实例1之间的SA和PSD的粉末。
实例12:与实例11相似,除了明胶是在0和90分钟时,分相同两期地加入。得到较小和较均匀的(较窄的PSD)粉末。
实例13:与实例5、9和10相比较,显示了不采用磷酸酯(如实例9和10中),在1/10基本条件下的明胶浓度(如实例5中),结果得到具有非常宽的PSD(比实例5宽得多)和低的SA(从实例5的1.0变成0.4)的粉末。且粉末是相当块集的。
实例14:与实例6相似,除了用不锈钢反应容器代替玻璃的之外。得到性能与实例6中的非常相似的产品。下面的实例15-20都采用相同的不锈钢容器,因而应认为与此情况相似。
实例15:与实例14相似,除了采用具有乙氧基结构的更迭磷酸酯表面活性剂PS-121(Witco)。得到具有较宽的PSD和较低的SA(1.6米2/克,而实例14中为2.1米2/克)的粉末。
实例16:与实例14相似,除了采用与TDP(R=C13)非常相似的更迭磷酸酯PS-900(Witco)。产品粉末实际上与实例14的相同。
实例17:与实例14相似,除了采用R=C8的更迭磷酸酯PS-400(Witco)。得到较小尺寸,较高SA(2.7米2/克,而实例14中为2.1米2/克),和较宽PSD的粉末。
实例18:与实例9相似,其中采用基本浓度的明胶,不采用磷酸酯、但是,这里是将明胶溶解于甲酯盐加料液中,并与甲酸盐一起逐渐地加入反应容器中。得到非常细小,但无块集的粉末,与之相比,实例9的粉末则出现严重聚集。
实例19:与实例16相似,除了加料速率是2.2×(较短的进料时间)。粉末仅略小于实例16的(SA=2.2米2/克,而实例16中为2.1米2/克),这表明较高的进料速率仅有很小的影响。
实例20:与实例16相似,除了进料速率是实例16的1/2.25(44%),并且进料时间较长(270分钟,而实例16中为120分钟)。粉末仅比实例16的平均直径略大(SA=1.7米2/克,而实例16中为2.1米2/克)和具有略宽的PSD,这表明较低的加料速率仅有很小的影响。
Claims (9)
1、一种制备金属银细分颗粒的方法,其特征在于它包括下列一系列步骤:
A.形成银盐、明胶和对应于下列结构式的酸式磷酸烷基酯的非碱性水溶液:

其中X独立地选自H和R基团,R是C6-20的烷基,其可选择地包括最多10个环氧乙烷(EO)部分,溶液中至少包括0.2摩尔溶解的银盐/升溶液,0.001-0.02克明胶/克金属银和0.1-0.5克酸式磷酸烷基酯/升初始溶液;
B.将化学计量过量的水溶性甲酸盐与得自步骤A的反应溶液相掺合,使银盐完全还原,由此使离散的金属银颗粒得以沉淀并伴之形成CO2和HNO3,同时,将反应溶液保持于足以使沉淀的颗粒保持分散的速率的搅拌下,直至还原反应完成,但搅拌速率也要足够低以避免反应分散液形成泡沫;
C.从反应溶液的液体组分中分离出银颗粒;
D.用去离子水洗涤分离的银颗粒,从中除去其所吸附的物质,和
E.使经洗涤的银颗粒干燥以从中除去水。
2、如权利要求1所述的方法,其特征在于水溶性甲酸盐选自由Na+、K+和NH+ 4甲酸盐及其混合物所组成的基团。
3、如权利要求1所述的方法,其特征在于还原反应在60-90℃下进行。
4、如权利要求1所述的方法,其特征在于酸式磷酸烷基酯是酸式磷酸十三烷酯,其中烷基链是具有一条四个环氧乙烷基的链的乙氧基链。
5、如权利要求1所述的方法,其特征在于酸式磷酸烷基酯的X的40-80%是R,60-20%是H基。
6、如权利要求5所述的方法,其特征在于X基团的50%是R,50%是H。
7、如权利要求5所述的方法,其特征在于X基团中75%是R,25%是H。
8、如权利要求1所述的方法,其特征在于其中R是C8-15烷基。
9、如权利要求8所述的方法,其特征在于R基团是C13烷基。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US07/475,927 US4979985A (en) | 1990-02-06 | 1990-02-06 | Process for making finely divided particles of silver metal |
| US475,927 | 1990-02-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1053901A true CN1053901A (zh) | 1991-08-21 |
Family
ID=23889753
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN91100858A Pending CN1053901A (zh) | 1990-02-06 | 1991-02-06 | 制备银金属细分颗粒的方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US4979985A (zh) |
| EP (1) | EP0514473A4 (zh) |
| KR (1) | KR927003855A (zh) |
| CN (1) | CN1053901A (zh) |
| IE (1) | IE910373A1 (zh) |
| WO (1) | WO1991012347A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1072995C (zh) * | 1993-07-13 | 2001-10-17 | 纳幕尔杜邦公司 | 制备细碎密实球形银粉的方法 |
| CN101933127A (zh) * | 2008-02-01 | 2010-12-29 | 欧恩吉电子化学品有限责任公司 | 在金属表面上沉积银的方法和组合物 |
| CN101232963B (zh) * | 2005-07-25 | 2011-05-04 | 住友金属矿山株式会社 | 铜微粒分散液及其制造方法 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5188660A (en) * | 1991-10-16 | 1993-02-23 | E. I. Du Pont De Nemours And Company | Process for making finely divided particles of silver metals |
| IL106958A (en) * | 1993-09-09 | 1996-06-18 | Ultrafine Techn Ltd | Method of producing high-purity ultra-fine metal powder |
| JP3429958B2 (ja) * | 1996-08-28 | 2003-07-28 | 三井金属鉱業株式会社 | 銀コロイド液の製造方法 |
| US8425926B2 (en) † | 2003-07-16 | 2013-04-23 | Yongxing Qiu | Antimicrobial medical devices |
| JP4489389B2 (ja) * | 2003-07-29 | 2010-06-23 | 三井金属鉱業株式会社 | 微粒銀粉の製造方法 |
| JP4489388B2 (ja) * | 2003-07-29 | 2010-06-23 | 三井金属鉱業株式会社 | 微粒銀粉の製造方法 |
| EP1825940B1 (en) * | 2004-11-29 | 2012-06-13 | DIC Corporation | Method for producing surface-treated silver-containing powder |
| CN100362339C (zh) * | 2005-02-25 | 2008-01-16 | 南京师范大学 | 制备测试拉曼光谱银溶胶的方法 |
| KR101111462B1 (ko) * | 2009-09-17 | 2012-02-22 | 충남대학교산학협력단 | 암모늄 포름산을 이용한 다공성 은 분말의 제조방법 |
| KR20130035014A (ko) * | 2011-09-29 | 2013-04-08 | 삼성전기주식회사 | 금속 입자의 제조방법, 이를 이용하여 제조된 잉크 조성물 및 페이스트 조성물 |
| CN103406550B (zh) * | 2013-08-26 | 2015-07-15 | 中科铜都粉体新材料股份有限公司 | 制备压敏元件电子浆料用银微粉的方法 |
| KR102178009B1 (ko) * | 2018-11-30 | 2020-11-12 | 엘에스니꼬동제련 주식회사 | 수축률 조절이 가능한 은 분말의 제조방법 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH434588A (de) * | 1962-11-15 | 1967-04-30 | Tesla Np | Verfahren zur Herstellung von Silberpulver |
| US3345158A (en) * | 1964-08-10 | 1967-10-03 | Ibm | Electrical conductor material and method of making same |
| US3998622A (en) * | 1975-12-23 | 1976-12-21 | E. I. Du Pont De Nemours And Company | Rhodium from hydroformylation still heels |
| DE2559191C2 (de) * | 1975-12-30 | 1982-11-25 | Agfa-Gevaert Ag, 5090 Leverkusen | Verfahren zur Herstellung von Silberdispersionen für Filter- und Lichthofschutzschichten |
| US4371459A (en) * | 1981-12-17 | 1983-02-01 | E. I. Du Pont De Nemours And Company | Flexible screen-printable conductor composition |
-
1990
- 1990-02-06 US US07/475,927 patent/US4979985A/en not_active Expired - Fee Related
-
1991
- 1991-01-30 WO PCT/US1991/000472 patent/WO1991012347A1/en not_active Application Discontinuation
- 1991-01-30 EP EP19910904606 patent/EP0514473A4/en not_active Withdrawn
- 1991-01-30 KR KR1019920701867A patent/KR927003855A/ko not_active Application Discontinuation
- 1991-02-05 IE IE037391A patent/IE910373A1/en unknown
- 1991-02-06 CN CN91100858A patent/CN1053901A/zh active Pending
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1072995C (zh) * | 1993-07-13 | 2001-10-17 | 纳幕尔杜邦公司 | 制备细碎密实球形银粉的方法 |
| CN101232963B (zh) * | 2005-07-25 | 2011-05-04 | 住友金属矿山株式会社 | 铜微粒分散液及其制造方法 |
| CN101933127A (zh) * | 2008-02-01 | 2010-12-29 | 欧恩吉电子化学品有限责任公司 | 在金属表面上沉积银的方法和组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| IE910373A1 (en) | 1991-08-14 |
| US4979985A (en) | 1990-12-25 |
| EP0514473A1 (en) | 1992-11-25 |
| EP0514473A4 (en) | 1993-05-19 |
| WO1991012347A1 (en) | 1991-08-22 |
| KR927003855A (ko) | 1992-12-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1053901A (zh) | 制备银金属细分颗粒的方法 | |
| KR101193762B1 (ko) | 고분산성 구상 은 분말 입자의 제조 방법 및 그로부터 형성된 은 입자 | |
| CN1072995C (zh) | 制备细碎密实球形银粉的方法 | |
| Ducamp-Sanguesa et al. | Synthesis and characterization of fine and monodisperse silver particles of uniform shape | |
| US8084140B2 (en) | Silver platelets comprising palladium | |
| CN101065518A (zh) | 镀金液以及镀金方法 | |
| JP2013541640A (ja) | 銀粒子およびその製造方法 | |
| CN101687253A (zh) | 球形铜微粉及其制造方法 | |
| JP2012525506A (ja) | 銀粒子およびその製造方法 | |
| EP1819467A1 (en) | Method of production of high purity silver particles | |
| CN1072120A (zh) | 制造银金属细粉的方法 | |
| JP2000129318A (ja) | 銀粉およびその製造方法 | |
| US4182627A (en) | Balls containing tungsten carbide | |
| CN116689773A (zh) | 一种基于液相还原的铁尾矿制备零价铁方法 | |
| CN101380679A (zh) | 一种由前驱体配合物制备单分散超细球形镍粉的方法 | |
| RU2415707C2 (ru) | Способ приготовления платиновых катализаторов | |
| CN116441528A (zh) | 一种超细球状镍粉及其制备方法 | |
| CN112846213B (zh) | 一种低氧含量高分散纳米球形钴粉的制备方法 | |
| CN1041193C (zh) | 由元素镍制备氢氧化镍的方法 | |
| RU2738174C1 (ru) | Способ получения тонкодисперсного серебряного порошка | |
| CN1181045C (zh) | 高纯度乙二胺四乙酸四钠盐的制备方法 | |
| KR101314990B1 (ko) | 전도성 구리 분말 제조방법 | |
| CN112063017B (zh) | 一种无机助剂及其制备方法 | |
| CN1046683C (zh) | 成核生长分步进行的液相制取超细氧化锌的方法 | |
| SU1202712A1 (ru) | Способ получени порошка серебра |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C01 | Deemed withdrawal of patent application (patent law 1993) | ||
| WD01 | Invention patent application deemed withdrawn after publication |