CN102491906A - 一种2-甲基-3-三氟甲基苯胺的合成方法 - Google Patents
一种2-甲基-3-三氟甲基苯胺的合成方法 Download PDFInfo
- Publication number
- CN102491906A CN102491906A CN201110432682XA CN201110432682A CN102491906A CN 102491906 A CN102491906 A CN 102491906A CN 201110432682X A CN201110432682X A CN 201110432682XA CN 201110432682 A CN201110432682 A CN 201110432682A CN 102491906 A CN102491906 A CN 102491906A
- Authority
- CN
- China
- Prior art keywords
- nitro
- methyl
- toluene
- acid
- prepare
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
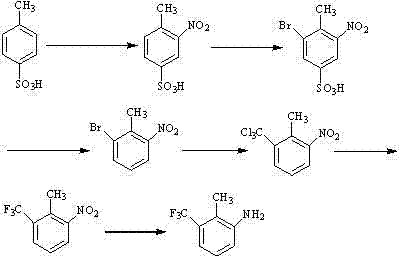
本发明公开了一种2-甲基-3-三氟甲基苯胺的合成方法,包括下述步骤:①制备3-硝基-4-甲基苯磺酸;②制备3-硝基-4-甲基-5-溴苯磺酸;③制备2-硝基-6-溴甲苯;④制备2-硝基-6-三氯甲基甲苯;⑤制备2-硝基-6-三氟甲基甲苯;⑥制备2-甲基-3-三氟甲基苯胺:在步骤⑤中加入5%钯炭及甲醇,在35℃下,氢化还原,除去催化剂,得到2-甲基-3-三氟甲基苯胺。本发明采用对甲苯磺酸为起始原料,从而解决了现有技术的不足,提高目标产物的收率。
Description
技术领域
本发明涉及禽药物,是一种动物专用药的中间体2-甲基-3-三氟甲基苯胺的合成方法。
背景技术
2-甲基-3-三氟甲基苯胺是氟尼辛葡甲胺的中间体,氟尼辛葡甲胺是目前已批准的少数动物专用非甾体类药物之一,主要通过抑制环氧化酶,减少前列腺素等炎性介质的生成而发挥解热、抗炎和镇痛作用。该药的合成路线均采用以2-甲基-3-三氟甲基苯胺为中间体与2-氯烟酸或2-氯烟酸乙酯发生吡啶环上亲核取代得到氟尼辛,然后氟尼辛与葡甲胺发生酸碱成盐反应得到氟尼辛葡甲胺。由于2-甲基-3-三氟甲基苯胺苯环上取代基相互之间位置独特,合成工艺较为复杂,生产成本较高,使2-甲基-3-三氟甲基苯胺成为制约氟尼辛葡甲胺生产成本的主要因素。而美国专利US4172095公开一种合成2-甲基-三氟甲基苯胺的方法:是以3-三氟甲基苯胺为原料,先与二甲基亚砜缩合,继而发生重排,还原得到2-甲基-3-三氟甲基苯胺,该方法的主要缺点是重排时没有位置选择性,生成两种混合物,而且量上差别不大,需要精馏提纯,而且收率特别低。美国另一项专利US4831193公开方法的缺点是:反应中用到价格较贵的三甲基溴化亚砜,而且正丁基锂用量大,收率低,成本高。另外,国外还有的方法中,没有商业化的起始原料,正丁基锂用量也大,成本高、收率低,这些方法的收率从28%-50%不等。
发明内容
本发明的目的是,提供一种2-甲基-3-三氟甲基苯胺的合成方法,它采用对甲苯磺酸为起始原料,从而解决了现有技术的不足,提高目标产物的收率。
本发明为实现上述目的,通过以下方案实现:一种2-甲基-3-三氟甲基苯胺的合成方法,包括下述步骤:
①制备3-硝基-4-甲基苯磺酸:取对甲基苯磺酸及65%硝酸在98%硫酸中反应生成3-硝基-4-甲基苯磺酸,对甲基苯磺酸、98%硫酸与65%硝酸的物料用量摩尔比为1:5.5~6.5:1.1~1.3;
②制备3-硝基-4-甲基-5-溴苯磺酸:在步骤①中的3-硝基-4-甲基苯磺酸中加入溴及冰醋酸,反应生成3-硝基-4-甲基-5-溴苯磺酸,3-硝基-4-甲基苯磺酸与溴的质量比为1:0.6~1;3-硝基-4-甲基苯磺酸与冰醋酸质量体积比为1:2~4;
③制备2-硝基-6-溴甲苯:在步骤②中的3-硝基-4-甲基-5-溴苯磺酸中加入稀硫酸、醋酸钠及水,加热至95℃-100℃,脱磺酸基得到2-硝基-6-溴甲苯,3-硝基-4-甲基-5-溴苯磺酸与醋酸钠和40%硫酸的质量比为1:0.2~0.4: 0.3~0.6;3-硝基-4-甲基-5-溴苯磺酸与水质量体积比为1:3~10;
④制备2-硝基-6-三氯甲基甲苯:在步骤③的2-硝基-6-溴甲苯中加入正丁基锂及四氯化碳反应后得到2-硝基-6-三氯甲基甲苯,2-硝基-6-溴甲苯中与正丁基锂和四氯化碳的摩尔比为1:1~1.5: 2~3;
⑤制备2-硝基-6-三氟甲基甲苯:在步骤④中的2-硝基-6-三氯甲基甲苯中加入氟化钾在N,N-二甲基甲酰胺中反应得到2-硝基-6-三氟甲基甲苯,氟化钾与2-硝基-6-三氯甲基甲苯的摩尔比为3~10:1;
⑥制备2-甲基-3-三氟甲基苯胺:在步骤⑤中加入5%钯炭及甲醇,在35℃下,氢化还原,除去催化剂,得到2-甲基-3-三氟甲基苯胺,2-硝基-6-三氟甲基甲苯和甲醇质量体积比为1:2~5;2-硝基-6-三氟甲基甲苯与5%钯炭质量比为1:0.02~0.05;合成路线为:

本发明所述步骤⑥中加入反应溶剂,所述的溶剂为甲醇、乙醇或冰醋酸的任一种,2-硝基-6-三氟甲基甲苯与溶剂的质量体积比为1:2~5。
本发明所述步骤⑥中加入催化剂,催化剂为5%钯碳、二氧化铂或亚铬酸铜的任一种,2-硝基-6-三氟甲基甲苯与催化剂的质量体积比为1:0.02~0.05。
本发明所述的一种2-甲基-3-三氟甲基苯胺的合成方法,优选的方案是:具体步骤如下:
①制备3-硝基-4-甲基苯磺酸:将100kg对甲基苯磺酸和319kg浓硫酸置入反应釜中,搅拌1.5小时,同时在搅拌中缓慢加入65%浓硝酸61.8kg,搅拌时保持温度30-40℃,加完后,保持30-40℃继续搅拌1小时,得到反应混合物;然后把反应混合物倒入100kg冰和200L水的混合液中,用二氯甲烷2×500L萃取,合并二氯甲烷层,用水2×500L洗涤,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到淡黄色油3-硝基-4-甲基苯磺酸115kg,收率92%;
②制备3-硝基-4-甲基-5-溴苯磺酸:将步骤①中的115kg 3-硝基-4-甲基苯磺酸加入到230L冰醋酸中,加热至70℃,滴加69kg液溴,保温70℃反应12小时,冷却至室温,加入600L冰水,搅拌,过滤,用冷水2×230L洗涤,干燥得到3-硝基-4-甲基-5-溴苯磺酸130kg,收率84%;
③制备2-硝基-6-溴甲苯:将步骤②中的130kg 3-硝基-4-甲基-5-溴苯磺酸,以及39kg醋酸钠,58.5kg 40%硫酸加入到390L纯化水中,90℃下反应24小时,冷却至5℃,抽滤,滤饼用冷水2×300L充分洗涤,减压干燥得淡黄色2-硝基-6-溴甲苯85.7kg,收率90%;
④制备2-硝基-6-三氯甲基甲苯:
(4.1)将步骤③的85.7kg 2-硝基-6-溴甲苯溶在900L干燥四氢呋喃溶液中,在氮气保护下冷却至0℃,然后滴加1.6M正丁基锂的己烷溶液180L,控制滴加速度,以保持内温升温不超过10℃为准,滴加完毕,在5℃下保持该温度搅拌2.5小时,得到混合液;
(4.2)在另一反应器中加入89.2kg四氯化碳和400L干燥四氢呋喃,在氮气保护下冷却至-10℃~-15℃,滴加步骤(4.1)中的混合液,控制内温不超过-5℃,缓慢升温至室温,用饱和氯化铵溶液淬灭反应,在减压下浓缩,然后用乙醚2×800L萃取,用食盐水800L洗涤乙醚层,用10kg无水硫酸镁干燥浓缩得到98.6kg2-硝基-6-三氯甲基甲苯,收率98%;
⑤制备2-硝基-6-三氟甲基甲苯:将步骤(4.2)中得到的98.6kg2-硝基-6-三氯甲基甲苯,以及67.4kg氟化钾加入到500L干燥N,N-二甲基甲酰胺中,加热至80-85℃,反应20小时,冷却至25℃;加入4000L冰水中,用二氯甲烷2×800L萃取,合并二氯甲烷层,用水2×1000L洗涤两次,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到2-硝基-6-三氟甲基甲苯 71.4kg,收率90%;
⑥制备2-甲基-3-三氟甲基苯胺:将步骤⑤中的2-硝基-6-三氟甲基甲苯 71.4kg,以及1.43kg 5%钯炭加入到142.8L甲醇中,在35℃、0.5MPa压力下氢化还原3小时,过滤除去催化剂,减压蒸出溶剂,残余物通过水蒸气蒸馏提纯,得到2-甲基-3-三氟甲基苯胺58.3kg,收率95%。
本发明提供的2-甲基-3-三氟甲基苯胺的合成方法,采用对甲苯磺酸为起始原料,该方法中的硝酸、硫酸、溴、冰醋酸等均为廉价易得物料,从而大幅降低了生产成本,本发明提供的2-甲基-3-三氟甲基苯胺的合成方法经过硝化,溴代,脱磺酸,三氯甲基化,氟化和还原得到2-甲基-3-三氟甲基苯胺,使副产物极少。并且正丁基锂的用量仅为公知技术的1/3,进一步降低了生产成本,同时通过氟化钾亲核取代氟化的方法,避免了特殊反应釜高压氟化的危险操作。本发明提供的2-甲基-3-三氟甲基苯胺的合成方法,在原料来源广泛、生产成本降低的同时,收率高达90%左右,并且不需要使用专用仪器设备,操作安全,分离方便,适于工业化生产。
具体实施方式
本发明所述的2-甲基-3-三氟甲基苯胺的合成方法的步骤如下:
①制备3-硝基-4-甲基苯磺酸:取对甲基苯磺酸及65%硝酸在98%硫酸中反应生成3-硝基-4-甲基苯磺酸,对甲基苯磺酸、98%硫酸与65%硝酸的物料用量摩尔比为1:5.5~6.5:1.1~1.3;
②制备3-硝基-4-甲基-5-溴苯磺酸:在步骤①中的3-硝基-4-甲基苯磺酸中加入溴及冰醋酸,反应生成3-硝基-4-甲基-5-溴苯磺酸,3-硝基-4-甲基苯磺酸与溴的质量比为1:0.6~1;3-硝基-4-甲基苯磺酸与冰醋酸质量体积比为1:2~4;
③制备2-硝基-6-溴甲苯:在步骤②中的3-硝基-4-甲基-5-溴苯磺酸中加入稀硫酸、醋酸钠及水,加热至95℃-100℃,脱磺酸基得到2-硝基-6-溴甲苯,3-硝基-4-甲基-5-溴苯磺酸与醋酸钠与40%硫酸的质量比为1:0.2~0.4: 0.3~0.6;3-硝基-4-甲基-5-溴苯磺酸与水质量体积比为1:3~10;
④制备2-硝基-6-三氯甲基甲苯:在步骤③的2-硝基-6-溴甲苯中加入正丁基锂及四氯化碳反应后得到2-硝基-6-三氯甲基甲苯,2-硝基-6-溴甲苯中与正丁基锂和四氯化碳的摩尔比为1:1~1.5: 2~3;
⑤制备2-硝基-6-三氟甲基甲苯:在步骤④中的2-硝基-6-三氯甲基甲苯中加入氟化钾在N,N-二甲基甲酰胺中反应得到2-硝基-6-三氟甲基甲苯,氟化钾与2-硝基-6-三氯甲基甲苯的摩尔比为3~10:1;
⑥制备2-甲基-3-三氟甲基苯胺:在步骤⑤中加入5%钯炭及甲醇,在35℃下,氢化还原,除去催化剂,得到2-甲基-3-三氟甲基苯胺,2-硝基-6-三氟甲基甲苯与甲醇质量体积比为1:2~5;2-硝基-6-三氟甲基甲苯与5%钯炭质量比为1:0.02~0.05;合成路线为:
所述步骤⑥中加入反应溶剂,所述的溶剂为甲醇、乙醇或冰醋酸的任一种,2-硝基-6-三氟甲基甲苯与溶剂的质量体积比为1:2~5。步骤⑥中加入催化剂,催化剂为5%钯碳、二氧化铂或亚铬酸铜的任一种,2-硝基-6-三氟甲基甲苯与催化剂的质量体积比为1:0.02~0.05。
本发明上述合成方法有多种具体方法:
1、一种2-甲基-3-三氟甲基苯胺的合成方法,具体步骤如下:
①制备3-硝基-4-甲基苯磺酸:将100kg对甲基苯磺酸和319kg浓硫酸置入反应釜中,搅拌1.5小时,同时在搅拌中缓慢加入65%浓硝酸61.8kg,搅拌时保持温度30-40℃,加完后,保持30~40℃继续搅拌1小时,得到反应混合物;然后把反应混合物倒入100kg冰和200L水的混合液中,用二氯甲烷2×500L萃取,合并二氯甲烷层,用水2×500L洗涤,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到淡黄色油3-硝基-4-甲基苯磺酸115kg,收率92%;
②制备3-硝基-4-甲基-5-溴苯磺酸:将步骤①中的115kg 3-硝基-4-甲基苯磺酸加入到230L冰醋酸中,加热至70℃,滴加69kg液溴,保温70℃反应12小时,冷却至室温,加入600L冰水,搅拌,过滤,用冷水2×230L洗涤,干燥得到3-硝基-4-甲基-5-溴苯磺酸130kg,收率84%;
③制备2-硝基-6-溴甲苯:将步骤②中的130kg 3-硝基-4-甲基-5-溴苯磺酸,以及39kg醋酸钠,58.5kg 40%硫酸加入到390L纯化水中,90℃下反应24小时,冷却至5℃,抽滤,滤饼用冷水2×300L充分洗涤,减压干燥得淡黄色2-硝基-6-溴甲苯85.7kg,收率90%;
④制备2-硝基-6-三氯甲基甲苯:
(4.1)将步骤③的85.7kg 2-硝基-6-溴甲苯溶在900L干燥四氢呋喃溶液中,在氮气保护下冷却至0℃,然后滴加1.6M正丁基锂的己烷溶液180L,控制滴加速度,以保持内温升温不超过10℃为准,滴加完毕,在5℃下保持该温度搅拌2.5小时,得到混合液;
(4.2)在另一反应器中加入89.2kg四氯化碳和400L干燥四氢呋喃,在氮气保护下冷却至-10℃~ -15℃,滴加步骤(4.1)中的混合液,控制内温不超过-5℃,缓慢升温至室温,用饱和氯化铵溶液淬灭反应,在减压下浓缩,然后用乙醚2×800L萃取,用食盐水800L洗涤乙醚层,用10kg无水硫酸镁干燥浓缩得到98.6kg2-硝基-6-三氯甲基甲苯,收率98%;
⑤制备2-硝基-6-三氟甲基甲苯:将步骤(4.2)中得到的98.6kg2-硝基-6-三氯甲基甲苯,以及67.4kg氟化钾加入到500L干燥N,N-二甲基甲酰胺中,加热至80-85℃,反应20小时,冷却至25℃;加入4000L冰水中,用二氯甲烷2×800L萃取,合并二氯甲烷层,用水2×1000L洗涤两次,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到2-硝基-6-三氟甲基甲苯 71.4kg,收率90%;
⑥制备2-甲基-3-三氟甲基苯胺:将步骤⑤中的2-硝基-6-三氟甲基甲苯 71.4kg,以及1.43kg 5%钯炭加入到142.8L甲醇中,在35℃、0.5MPa压力下氢化还原3小时,过滤除去催化剂,减压蒸出溶剂,残余物通过水蒸气蒸馏提纯,得到2-甲基-3-三氟甲基苯胺58.3kg,收率95%。
2、一种2-甲基-3-三氟甲基苯胺的合成方法,具体步骤如下:
①制备3-硝基-4-甲基苯磺酸:将100kg对甲基苯磺酸和348kg浓硫酸置入反应釜中,搅拌1小时,同时在搅拌中缓慢加入65%浓硝酸67.5kg,搅拌时保持温度30~40℃,加完后,保持30~40℃继续搅拌1小时,得到反应混合物;然后把反应混合物倒入100kg冰和200L水的混合液中,用二氯甲烷2×500L萃取,合并二氯甲烷层,用水2×500L洗涤,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到淡黄色油3-硝基-4-甲基苯磺酸120kg,收率96%;
②制备3-硝基-4-甲基-5-溴苯磺酸:将步骤①中的120kg 3-硝基-4-甲基苯磺酸加入到360L冰醋酸中,加热至75℃,滴加96kg液溴,保温70℃反应12小时,冷却至室温,加入600L冰水,搅拌,过滤,用冷水2×230L洗涤,干燥得到3-硝基-4-甲基-5-溴苯磺酸138kg,收率85%;
③制备2-硝基-6-溴甲苯:将步骤②中的138kg 3-硝基-4-甲基-5-溴苯磺酸,以及27.6kg醋酸钠,58.5kg 41.4%硫酸加入到897L纯化水中,100℃下反应24小时,冷却至5℃,抽滤,滤饼用冷水2×300L充分洗涤,减压干燥得淡黄色2-硝基-6-溴甲苯89.6kg,收率88.7%;
④制备2-硝基-6-三氯甲基甲苯:
(4.1)将步骤③的89.6kg 2-硝基-6-溴甲苯溶在900L干燥四氢呋喃溶液中,在氮气保护下冷却至0℃,然后滴加1.6M正丁基锂的己烷溶液236.5L,控制滴加速度,以保持内温升温不超过10℃为准,滴加完毕,在5℃下保持该温度搅拌2.5小时,得到混合液;
(4.2)在另一反应器中加入111.5kg四氯化碳和400L干燥四氢呋喃,在氮气保护下冷却至-10℃~ -15℃,滴加步骤(4.1)中的混合液,控制内温不超过-5℃,缓慢升温至室温,用饱和氯化铵溶液淬灭反应,在减压下浓缩,然后用乙醚2×800L萃取,用食盐水800L洗涤乙醚层,用10kg无水硫酸镁干燥,浓缩得到101.4kg2-硝基-6-三氯甲基甲苯,收率96%;
⑤制备2-硝基-6-三氟甲基甲苯:将步骤(4.2)中得到的101.4kg2-硝基-6-三氯甲基甲苯,以及150kg氟化钾加入到500L干燥N,N-二甲基甲酰胺中,加热至80-85℃,反应20小时,冷却至25℃;加入4000L冰水中,用二氯甲烷2×800L萃取,合并二氯甲烷层,用水2×1000L洗涤两次,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到2-硝基-6-三氟甲基甲苯 76kg,收率94.5 %;
⑥制备2-甲基-3-三氟甲基苯胺:将步骤⑤中的2-硝基-6-三氟甲基甲苯 76kg,以及1.90kg 5%钯炭加入到379.5L甲醇中,在35℃、0.5MPa压力下氢化还原3小时,过滤除去催化剂,减压蒸出溶剂,残余物通过水蒸气蒸馏提纯,得到2-甲基-3-三氟甲基苯胺60.7kg,收率93%。
3、一种2-甲基-3-三氟甲基苯胺的合成方法,具体步骤如下:
①制备3-硝基-4-甲基苯磺酸:将100kg对甲基苯磺酸和377kg浓硫酸置入反应釜中,搅拌1小时,同时在搅拌中缓慢加入65%浓硝酸77.1kg,搅拌时保持温度30~40℃,加完后,保持30~40℃继续搅拌1小时,得到反应混合物;然后把反应混合物倒入100kg冰和200L水的混合液中,用二氯甲烷2×500L萃取,合并二氯甲烷层,用水2×500L洗涤,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到淡黄色油3-硝基-4-甲基苯磺酸118kg,收率94.4%;
②制备3-硝基-4-甲基-5-溴苯磺酸:将步骤①中的118kg 3-硝基-4-甲基苯磺酸加入到472L冰醋酸中,加热至70℃,滴加118kg液溴,保温70℃反应12小时,冷却至室温,加入600L冰水,搅拌,过滤,用冷水2×230L洗涤,干燥得到3-硝基-4-甲基-5-溴苯磺酸134.5kg,收率84.5%;
③制备2-硝基-6-溴甲苯:将步骤②中的134.5kg 3-硝基-4-甲基-5-溴苯磺酸,以及53.8kg醋酸钠,80.7kg 40%硫酸加入到1345L纯化水中,100℃下反应24小时,冷却至0℃,抽滤,滤饼用冷水2×300L充分洗涤,减压干燥得淡黄色2-硝基-6-溴甲苯89.4kg,收率91%;
④制备2-硝基-6-三氯甲基甲苯:
(4.1)将步骤③的89.4kg 2-硝基-6-溴甲苯溶在894L干燥四氢呋喃溶液中,在氮气保护下冷却至0℃,然后滴加1.6M正丁基锂的己烷溶液283L,控制滴加速度,以保持内温升温不超过10℃为准,滴加完毕,在0℃下保持该温度搅拌2.5小时,得到混合液;
(4.2)在另一反应器中加入133.8kg四氯化碳和447L干燥四氢呋喃,在氮气保护下冷却至-10℃~ -15℃,滴加步骤(4.1)中的混合液,控制内温不超过-5℃,缓慢升温至室温,用饱和氯化铵溶液淬灭反应,在减压下浓缩,然后用乙醚2×800L萃取,用食盐水800L洗涤乙醚层,用10kg无水硫酸镁干燥,浓缩得到99.1kg2-硝基-6-三氯甲基甲苯,收率率94.1%;
⑤制备2-硝基-6-三氟甲基甲苯:将步骤(4.2)中得到的99.1kg2-硝基-6-三氯甲基甲苯,以及225.4kg氟化钾加入到500L干燥N,N-二甲基甲酰胺中,加热至80-85℃,反应20小时,冷却至25℃,加入4000L冰水中,用二氯甲烷2×800L萃取,合并二氯甲烷层,用水2×1000L洗涤两次,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到2-硝基-6-三氟甲基甲苯77.3kg,收率97%;
⑥制备2-甲基-3-三氟甲基苯胺:将步骤⑤中的2-硝基-6-三氟甲基甲苯77.3kg,以及2.3kg 5%钯炭加入到271L甲醇中,在35℃、0.5MPa压力下氢化还原3小时,过滤除去催化剂,减压蒸出溶剂,残余物通过水蒸气蒸馏提纯,得到2-甲基-3-三氟甲基苯胺62.4kg,收率93.8%。
Claims (4)
1. 一种2-甲基-3-三氟甲基苯胺的合成方法,其特征在于:包括下述步骤:
①制备3-硝基-4-甲基苯磺酸:取对甲基苯磺酸及65%硝酸在98%硫酸中反应生成3-硝基-4-甲基苯磺酸,对甲基苯磺酸、98%硫酸与65%硝酸的物料用量摩尔比为1:5.5~6.5:1.1~1.3;
②制备3-硝基-4-甲基-5-溴苯磺酸:在步骤①中的3-硝基-4-甲基苯磺酸中加入溴及冰醋酸,反应生成3-硝基-4-甲基-5-溴苯磺酸,3-硝基-4-甲基苯磺酸与溴的质量比为1:0.6~1; 3-硝基-4-甲基苯磺酸与冰醋酸质量体积比为1:2~4;
③制备2-硝基-6-溴甲苯:在步骤②中的3-硝基-4-甲基-5-溴苯磺酸中加入稀硫酸、醋酸钠及水,加热至95℃-100℃,脱磺酸基得到2-硝基-6-溴甲苯,3-硝基-4-甲基-5-溴苯磺酸与醋酸钠和40%硫酸的质量比为1:0.2~0.4: 0.3~0.6;3-硝基-4-甲基-5-溴苯磺酸与水质量体积比为1:3~10;
④制备2-硝基-6-三氯甲基甲苯:在步骤③的2-硝基-6-溴甲苯中加入正丁基锂及四氯化碳反应后得到2-硝基-6-三氯甲基甲苯,2-硝基-6-溴甲苯中与正丁基锂和四氯化碳的摩尔比为1:1~1.5: 2~3;
⑤制备2-硝基-6-三氟甲基甲苯:在步骤④中的2-硝基-6-三氯甲基甲苯中加入氟化钾在N,N-二甲基甲酰胺中反应得到2-硝基-6-三氟甲基甲苯,氟化钾与2-硝基-6-三氯甲基甲苯的摩尔比为3~10:1;
⑥制备2-甲基-3-三氟甲基苯胺:在步骤⑤中加入5%钯炭及甲醇,在35℃下,氢化还原,除去催化剂,得到2-甲基-3-三氟甲基苯胺,2-硝基-6-三氟甲基甲苯与甲醇质量体积比为1:2~5;2-硝基-6-三氟甲基甲苯与5%钯炭质量比为1:0.02~0.05;合成路线为:

2.根据权利要求1所述的一种2-甲基-3-三氟甲基苯胺的合成方法,其特征在于:所述步骤⑥中加入反应溶剂,所述的溶剂为甲醇、乙醇或冰醋酸的任一种,2-硝基-6-三氟甲基甲苯与溶剂的质量体积比为1:2~5。
3.根据权利要求1所述的一种2-甲基-3-三氟甲基苯胺的合成方法,其特征在于:所述步骤⑥中加入催化剂,催化剂为5%钯碳、二氧化铂或亚铬酸铜的任一种,2-硝基-6-三氟甲基甲苯与催化剂的质量体积比为1:0.02~0.05。
4.根据权利要求1所述的一种2-甲基-3-三氟甲基苯胺的合成方法,其特征在于:具体步骤如下:
①制备3-硝基-4-甲基苯磺酸:将100kg对甲基苯磺酸和319kg浓硫酸置入反应釜中,搅拌1.5小时,同时在搅拌中缓慢加入65%浓硝酸61.8kg,搅拌时保持温度30-40℃,加完后,保持30-40℃继续搅拌1小时,得到反应混合物;然后把反应混合物倒入100kg冰和200L水的混合液中,用二氯甲烷2×500L萃取,合并二氯甲烷层,用水2×500L洗涤,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到淡黄色油3-硝基-4-甲基苯磺酸115kg,收率92%;
②制备3-硝基-4-甲基-5-溴苯磺酸:将步骤①中的115kg 3-硝基-4-甲基苯磺酸加入到230L冰醋酸中,加热至70℃,滴加69kg液溴,保温70℃反应12小时,冷却至室温,加入600L冰水,搅拌,过滤,用冷水2×230L洗涤,干燥得到3-硝基-4-甲基-5-溴苯磺酸130kg,收率84%;
③制备2-硝基-6-溴甲苯:将步骤②中的130kg 3-硝基-4-甲基-5-溴苯磺酸,以及39kg醋酸钠,58.5kg 40%硫酸加入到390L纯化水中,90℃下反应24小时,冷却至5℃,抽滤,滤饼用冷水2×300L充分洗涤,减压干燥得淡黄色2-硝基-6-溴甲苯85.7kg,收率90%;
④制备2-硝基-6-三氯甲基甲苯:
(4.1)将步骤③的85.7kg 2-硝基-6-溴甲苯溶在900L干燥四氢呋喃溶液中,在氮气保护下冷却至0℃,然后滴加1.6M正丁基锂的己烷溶液180L,控制滴加速度,以保持内温升温不超过10℃为准,滴加完毕,在5℃下保持该温度搅拌2.5小时,得到混合液;
(4.2)在另一反应器中加入89.2kg四氯化碳和400L干燥四氢呋喃,在氮气保护下冷却至-10℃~ -15℃,滴加步骤(4.1)中的混合液,控制内温不超过-5℃,缓慢升温至室温,用饱和氯化铵溶液淬灭反应,在减压下浓缩,然后用乙醚2×800L萃取,用食盐水800L洗涤乙醚层,用10kg无水硫酸镁干燥浓缩得到98.6kg2-硝基-6-三氯甲基甲苯,收率98%;
⑤制备2-硝基-6-三氟甲基甲苯:将步骤(4.2)中得到的98.6kg2-硝基-6-三氯甲基甲苯,以及67.4kg氟化钾加入到500L干燥N,N-二甲基甲酰胺中,加热至80-85℃,反应20小时,冷却至25℃;加入4000L冰水中,用二氯甲烷2×800L萃取,合并二氯甲烷层,用水2×1000L洗涤两次,有机相用10kg无水硫酸镁干燥,减压蒸出溶剂得到2-硝基-6-三氟甲基甲苯 71.4kg,收率90%;
⑥制备2-甲基-3-三氟甲基苯胺:将步骤⑤中的2-硝基-6-三氟甲基甲苯 71.4kg,以及1.43kg 5%钯炭加入到142.8L甲醇中,在35℃、0.5MPa压力下氢化还原3小时,过滤除去催化剂,减压蒸出溶剂,残余物通过水蒸气蒸馏提纯,得到2-甲基-3-三氟甲基苯胺58.3kg,收率95%。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201110432682 CN102491906B (zh) | 2011-12-21 | 2011-12-21 | 一种2-甲基-3-三氟甲基苯胺的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201110432682 CN102491906B (zh) | 2011-12-21 | 2011-12-21 | 一种2-甲基-3-三氟甲基苯胺的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102491906A true CN102491906A (zh) | 2012-06-13 |
| CN102491906B CN102491906B (zh) | 2013-10-23 |
Family
ID=46183776
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 201110432682 Active CN102491906B (zh) | 2011-12-21 | 2011-12-21 | 一种2-甲基-3-三氟甲基苯胺的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102491906B (zh) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108911989A (zh) * | 2018-08-15 | 2018-11-30 | 济南悟通生物科技有限公司 | 一种2-甲基-3-三氟甲基苯胺的合成方法 |
| CN111848410A (zh) * | 2020-07-13 | 2020-10-30 | 上海万巷制药有限公司 | 一种2-硝基-苯胺的制备方法 |
| CN112159325A (zh) * | 2020-10-10 | 2021-01-01 | 浙江工业大学 | 一种合成2-氨基-3-硝基甲苯的方法 |
| CN112394130A (zh) * | 2020-12-02 | 2021-02-23 | 济南悟通生物科技有限公司 | 2-甲基-3-三氟甲基苯胺合成工艺中杂质的分析方法 |
| CN113461541A (zh) * | 2021-07-01 | 2021-10-01 | 湖北可赛化工有限公司 | 一种合成对氯邻甲苯胺的方法 |
| CN113461538A (zh) * | 2021-07-12 | 2021-10-01 | 无锡双启科技有限公司 | 一种2-氯-3-溴苯胺的制备方法 |
| CN115073298A (zh) * | 2022-05-31 | 2022-09-20 | 湖南华腾制药有限公司 | 连续流微反应器合成2-甲基-3-三氟甲基苯胺的方法 |
| CN117142987A (zh) * | 2023-08-29 | 2023-12-01 | 上海优合贝德医药科技有限公司 | 一种3-取代-4-甲基-5-硝基苯磺酸的合成方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5363324A (en) * | 1976-11-12 | 1978-06-06 | Ciba Geigy Ag | 22methyll44trifluoromethyllaniline process for preparing same and application thereof |
| US4132737A (en) * | 1978-02-27 | 1979-01-02 | Eli Lilly And Company | Trifluoromethyl substituted 1-aminoindanes |
| WO1988010247A1 (en) * | 1987-06-19 | 1988-12-29 | Schering Corporation | Novel synthesis of 3-amino-2-methylbenzotrifluoride |
-
2011
- 2011-12-21 CN CN 201110432682 patent/CN102491906B/zh active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5363324A (en) * | 1976-11-12 | 1978-06-06 | Ciba Geigy Ag | 22methyll44trifluoromethyllaniline process for preparing same and application thereof |
| US4132737A (en) * | 1978-02-27 | 1979-01-02 | Eli Lilly And Company | Trifluoromethyl substituted 1-aminoindanes |
| WO1988010247A1 (en) * | 1987-06-19 | 1988-12-29 | Schering Corporation | Novel synthesis of 3-amino-2-methylbenzotrifluoride |
Non-Patent Citations (2)
| Title |
|---|
| VINCENT J. TRAYNELIS 等: "Ylide Methylation of Aromatic Nitro Compounds", 《THE JOURANL OF ORGANIC CHEMISTRY》 * |
| 郁铭 等: "2-甲基-3-(三氟甲基)苯胺合成研究进展", 《化学文摘》 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108911989A (zh) * | 2018-08-15 | 2018-11-30 | 济南悟通生物科技有限公司 | 一种2-甲基-3-三氟甲基苯胺的合成方法 |
| CN108911989B (zh) * | 2018-08-15 | 2020-11-03 | 济南悟通生物科技有限公司 | 一种2-甲基-3-三氟甲基苯胺的合成方法 |
| CN111848410A (zh) * | 2020-07-13 | 2020-10-30 | 上海万巷制药有限公司 | 一种2-硝基-苯胺的制备方法 |
| CN112159325A (zh) * | 2020-10-10 | 2021-01-01 | 浙江工业大学 | 一种合成2-氨基-3-硝基甲苯的方法 |
| CN112394130A (zh) * | 2020-12-02 | 2021-02-23 | 济南悟通生物科技有限公司 | 2-甲基-3-三氟甲基苯胺合成工艺中杂质的分析方法 |
| CN113461541A (zh) * | 2021-07-01 | 2021-10-01 | 湖北可赛化工有限公司 | 一种合成对氯邻甲苯胺的方法 |
| CN113461541B (zh) * | 2021-07-01 | 2022-09-13 | 湖北可赛化工有限公司 | 一种合成对氯邻甲苯胺的方法 |
| CN113461538A (zh) * | 2021-07-12 | 2021-10-01 | 无锡双启科技有限公司 | 一种2-氯-3-溴苯胺的制备方法 |
| CN115073298A (zh) * | 2022-05-31 | 2022-09-20 | 湖南华腾制药有限公司 | 连续流微反应器合成2-甲基-3-三氟甲基苯胺的方法 |
| CN117142987A (zh) * | 2023-08-29 | 2023-12-01 | 上海优合贝德医药科技有限公司 | 一种3-取代-4-甲基-5-硝基苯磺酸的合成方法 |
| CN117142987B (zh) * | 2023-08-29 | 2024-09-20 | 上海优合贝德医药科技有限公司 | 一种3-取代-4-甲基-5-硝基苯磺酸的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102491906B (zh) | 2013-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102491906B (zh) | 一种2-甲基-3-三氟甲基苯胺的合成方法 | |
| CN102069014B (zh) | 一种手性α-苯乙胺锌、铜配合物的用途 | |
| CN102093194A (zh) | 3-环丙基甲氧基-4-二氟甲氧基苯甲酸的合成新方法 | |
| CN112851646B (zh) | 特戈拉赞的制备方法 | |
| CN105130926B (zh) | 一种亚甲基蓝的制备方法 | |
| CN115417797B (zh) | 一种联苯肼酯的制备方法 | |
| CN103613498A (zh) | 环丙贝特的合成方法 | |
| WO2014071545A1 (zh) | 一种2-溴-3-氟苯甲酸的制备方法 | |
| CN109896943A (zh) | 一种木豆素及其结构类似物的化学制备方法 | |
| CN102942509A (zh) | 一种酚磺乙胺的合成方法 | |
| CN107089982B (zh) | 4,5-二取代-1-氢-吡咯(2,3-f)喹啉-2,7,9-三羧酸酯化合物及应用 | |
| CN106279097A (zh) | 一种丙烯基‑1,3‑磺酸内酯的制备方法 | |
| CN106146457B (zh) | 一种5-氯-2-酰氯噻吩中间体及其制备方法 | |
| CN104326992A (zh) | 一种合成二氟甲基三唑啉酮和甲磺草胺的方法 | |
| CN102766088B (zh) | 一种合成4,4'-二溴-2,2'-联吡啶的新工艺 | |
| CN109928867A (zh) | 3,5-二羟基戊苯的合成方法 | |
| CN113387903A (zh) | 一种帕瑞昔布钠杂质的合成方法 | |
| CN102675148B (zh) | 对羟基苯乙腈的制备方法 | |
| JP6912143B2 (ja) | 4−ペンタフルオロチオフェノール類化合物と調製方法及びペンタフルオロサルファー置換ベンゾピラン化合物の調製方法 | |
| CN103819418B (zh) | 一种合成唑草酮和唑草酮中间体的方法 | |
| CN101514167B (zh) | 手性巴氯芬的制备方法 | |
| CN106748796A (zh) | 制备1,5‑二氟‑2,4‑二硝基苯的方法 | |
| CN102766053B (zh) | 3-氟-4-硝基苯酚的生产方法 | |
| CN106542958A (zh) | 一种邻碘苯胺的制备方法 | |
| CN108530297B (zh) | 2-氯-3-甲基苯甲酸及其中间体的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |