CN101962383A - 一种美罗培南的合成方法 - Google Patents
一种美罗培南的合成方法 Download PDFInfo
- Publication number
- CN101962383A CN101962383A CN 201010541665 CN201010541665A CN101962383A CN 101962383 A CN101962383 A CN 101962383A CN 201010541665 CN201010541665 CN 201010541665 CN 201010541665 A CN201010541665 A CN 201010541665A CN 101962383 A CN101962383 A CN 101962383A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- reaction
- meropenem
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D477/00—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring
- C07D477/02—Preparation
- C07D477/04—Preparation by forming the ring or condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D477/00—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring
- C07D477/02—Preparation
- C07D477/06—Preparation from compounds already containing the ring or condensed ring systems, e.g. by dehydrogenation of the ring, by introduction, elimination or modification of substituents
- C07D477/08—Modification of a carboxyl group directly attached in position 2, e.g. esterification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D477/00—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring
- C07D477/10—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 4, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2
- C07D477/12—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 4, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2 with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached in position 6
- C07D477/16—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 4, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2 with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached in position 6 with hetero atoms or carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 3
- C07D477/20—Sulfur atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
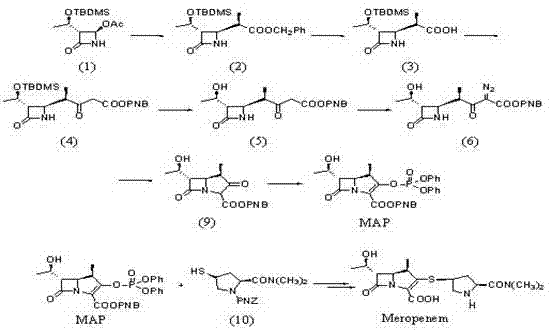
本发明涉及一种美罗培南的合成方法,具体反应路线如下:
Description
技术领域
本发明涉及一种β-甲基碳青霉烯类抗生素,尤其是涉及美罗培南的制备方法。
背景技术
碳青霉烯抗生素(Carbapenem,即培南类)是一种新型的β-内酰胺类抗生素,以抗菌谱广,抗菌作用强而著称,如美罗培南(Meropenem)、亚胺培南(Imipenem) 和比阿培南 (Biapenem) 等,在治愈重症感染方面发挥着重要的作用。
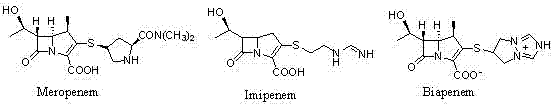
对于培南类的合成方法,以往研究较多的都是先分别合成相应的培南侧链化合物和母核MAP,两者再缩合脱除保护基得到培南产品。如美国专利USP4933333,以4-乙酰氧基氮杂环丁酮(4AA)为起始原料,经数步反应后得到母核MAP。母核再与侧链经缩合脱保护得到美罗培南。但是这种方法比较繁琐,合成步骤长、总收率低,合成化合物(9)时不可避免地要用到贵金属催化
剂铑。
中国发明专利文献CN200810142137.5曾介绍了一种合成美罗培南的方法,

这种方法路线较短,操作简便,原料简单易得,但也存在一些需要改进的地方。
发明内容
本发明在上述中国专利的基础上,进行了改进研究:以烯丙基代替对硝基苄基,具体的反应路线如下,其中涉及两个新化合物(Ⅱ)和(Ⅲ):

反应步骤如下:
1)式 (IV) 化合物和式 (V) 化合物溶于有机溶剂后在缩合剂的作用下通过缩合反应得到式(Ⅱ)化合物,反应时间为2~24小时,反应温度为0~40℃(优选室温);
2)式 (Ⅱ) 化合物和式 (Ⅵ)化合物溶于甲苯、乙酸乙酯或四氢呋喃后在碱的作用下进行反应,生成式 (Ⅲ) 化合物,反应时间为1~3小时,反应温度为-20~5℃(较佳为0℃);
3)式(Ⅲ) 化合物溶于环己烷、正庚烷、正辛烷、甲苯或二甲苯后在有机磷试剂的作用下发生Wittig关环反应得到式 (Ⅶ) 化合物,所述有机磷试剂是三苯基磷、三正丁基磷、亚磷酸三乙酯或亚磷酸三甲酯;
4)式(Ⅶ) 化合物溶于甲醇、四氢呋喃、丙酮、正戊烷、正己烷、乙醚、乙腈、二氯甲烷、氯仿或乙酸乙酯后在酸的作用下水解硅醚键,得到式 (Ⅷ) 化合物;所述酸是稀盐酸、氢氟酸、四丁基氟铵、苄基三丁基氟化铵、氟氢化铵或醋酸,酸和式(Ⅶ) 化合物的摩尔比为5~15:1;所述水解反应的温度为0~40℃,反应时间为8~24小时;
5)式(Ⅷ) 化合物溶于甲醇、乙醇、叔丁醇、异丁醇、异丙醇、四氢呋喃、二氧杂环己烷、丙酮、二氯甲烷、氯仿、乙酸乙酯以及水中的一种或几种所组成的溶剂后在钯催化剂作用下氢化脱去烯丙基得到目标产物(I),所述钯催化剂与式(Ⅷ) 化合物的摩尔比为0.0001~0.5;反应温度为0~40℃(较佳为室温),反应时间为2~24h。
步骤1)中式(IV) 化合物和式 (V) 化合物的摩尔比为1.05~1.0:1,所述缩合剂和式(IV) 化合物的摩尔比为1.50~1.05:1。
所述缩合剂是碳二亚胺类试剂或N, N’-羰基二咪唑;所述有机溶剂是丙酮、乙腈、甲苯、四氢呋喃、氯仿或二甲基甲酰胺。优选丙酮。
步骤2)中式(Ⅵ)化合物与式 (Ⅱ) 化合物的摩尔比为1.5~2.5:1(较佳为2:1),所述碱与所述式(Ⅵ)化合物的摩尔比为1.2~2:1(较佳为1.5:1)。
步骤3)中所述有机磷试剂与式 (Ⅲ) 化合物的摩尔比为2~8:1(较佳为3.5:1);所述关环反应的温度为25~100℃,反应时间为10~24小时。
所述碳二亚胺类试剂是二环己基碳二亚胺、二异丙基碳二亚胺或1-(3-二甲胺基丙基)-3-乙基碳二亚胺。
所述碱是无机碱或有机碱;当为无机碱时,是氢氧化钠、碳酸钠或碳酸氢钠;当为有机碱时是吡啶、三乙胺、二异丙基乙基胺或2, 6-二甲基吡啶。
所述钯催化剂是醋酸钯、氯化钯、硝酸钯、双三苯基膦氯化钯或四三苯基膦钯。
步骤5)中的保护基受体为吗啉、二甲基环己二酮、三丁基氢化锡、N,N-二甲基巴比士酸、2-乙基己酸或己酸。
本发明的合成方法具有如下优点:1)由于烯丙基的空间位阻比对硝基苄基小,有利于wittig环合反应的进行,提高了收率;2)反应条件温和,用四三苯基膦钯催化剂在常温常压下即可脱除烯丙基保护基;3)与传统方法相比,使用的试剂低毒、安全,环境污染少,避免了贵金属铑催化剂的使用,降低了成本;4)不使用氢气,安全环保;5)路线简短,操作简单。
具体实施方式
下面,通过以下实施例对本发明作进一步说明,它将有助于理解本发明,但并不限制本发明的内容。
实施例 1
1)(3R, 4S)-3-[(R)-1-(叔丁基二甲基硅氧基)乙基]-4-[(2’S, 4’R)- 1-(烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-2-氮杂环丁酮(Ⅱ)的合成
在500ml反应瓶中,加入22.6 g (0.075mol) (3S,4S)-3-[(R)1-(叔丁基二甲基硅氧基)乙基]-4-[(R)-1-羰乙基]-2-氮杂环丁酮(Ⅳ)、17.1 g (0.083 mol) 二环己基碳二亚胺 (DCC) 的100 ml丙酮溶液和0.76 g 4-二甲胺基吡啶 (DMAP),搅拌下滴加20.3g (0.078 mol) (2S, 4R)-2-二甲胺羰基-4-巯基-1-(烯丙氧羰基)吡咯烷 (V) 的125 ml丙酮溶液,室温下反应14小时。过滤,收集滤液,浓缩,向其中加入200 ml甲苯,用200 ml 5 %的醋酸溶液,200ml饱和碳酸氢钠溶液和150ml饱和盐水洗涤,无水硫酸镁干燥,减压蒸干得无色液体34.8g (0.064mol, 85.7%)(3R, 4S)-3-[(R)-1-(叔丁基二甲基硅氧基)乙基]-4-[(2’S, 4’R)-1-(烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-2-氮杂环丁酮(Ⅱ),不经进一步处理可直接投入下步反应。
1H-NMR (400 MHz, CDC13):
0.82(9H, s), 1.227(3H, d), 1.245(6H, m), 1.833-1.888(1H, m), 2.014(3H, s), 3.98(1H, m), 2.630-2.663(1H, m), 2.816-2.849(1H,s), 2.935-2.953(3H,m), 3.027-079(3H, d), 3.378-3.401(1H, m), 3.792-3.796 (1H, d), 3.807-3.953(1H, m), 4.042-4.160(3H, m), 4.492-4.570(2H, m), 4.670-4.739(1H,m), 5.164-5.295(1H, m), 5.807-5.921(1H, m), 6.214(1H, s)。
实施例 2
2)(3R, 4S)-3-[(R)-1-(叔丁基二甲基硅氧基)乙基]-4-[(2’S, 4’R)- 1-(烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-1-(烯丙氧草酰基)-2-氮杂环丁酮(Ⅲ)的合成
在1000ml反应瓶中,加入34.8 g(0.064mol)(3R, 4S)-3-[(R)-1-(叔丁基二甲基硅氧基)乙基]-4-[(2’S, 4’R)-1-(烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-2-氮杂环丁酮(Ⅱ)、15.0 ml三乙胺和350 ml甲苯,控制温度在-10℃以下,滴加18.9 g (0.128mol) 氯草酸对硝基苄酯 (Ⅵ),升温至0℃(-20~5℃均可)反应1~3h。然后缓慢滴入250 ml 冰水,搅拌10 min。静止分层,有机相用饱和碳酸氢钠溶液洗涤三次,每次200 ml。无水硫酸镁干燥,过滤,蒸干得白色固体40.7g (0.0622mol, 收率97.3%)(3R, 4S)-3-[(R)-1-(叔丁基二甲基硅氧基)乙基]-4-[(2’S, 4’R)-1-(烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-1-(烯丙氧草酰基)-2-氮杂环丁酮(Ⅲ) ,产物不经进一步纯化直接投入下步反应。
Mp:33~34℃
1H-NMR (300 MHz, CDC13):
0.819(9H, s), 1.167(3H, d), 1.188(4H, d), 1.693(5H, s), 1.850-1.926(1H, m), 2.631-2.700(1H, m), 2.941-2.960(3H,d), 3.029-3.080(3H,d), 3.357-3.433(1H, m), 3.506-3.545(2H, m), 3.918-3.968(1H, m), 4.054-4.123(2H, m), 4.270-4.291(1H, m), 4.391(1H,s), 4.518-4.568(2H, m), 4.588-4.779(3H, m), 5.178-5.416(3H, m), 5.861-5.982(2H,m)。
实施例 3
3)(5R,6S,8R,2’S, 4’S)-[(R)-1-(叔丁基二甲基硅氧基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸酯 (Ⅶ) 的合成
在500ml反应瓶中,加入40.7g(0.0622mol )(3R, 4S)-3-[(R)-1-(叔丁基二甲基硅氧基)乙基]-4-[(2’S, 4’R)-1-(烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-1-(烯丙氧草酰基)-2-氮杂环丁酮(Ⅲ)和150 ml 甲苯,氮气保护下加入22 ml亚磷酸三甲酯(还可再加入1 g对苯二酚),于60℃反应16小时后,减压蒸除溶剂,加入300 ml乙酸乙酯重结晶,收集固体,于40℃真空烘干得32.8g(0.0528mol, 收率85.0%)(5R,6S,8R,2’S, 4’S)-[(R)-1-(叔丁基二甲基硅氧基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸酯 (Ⅶ)。
1H-NMR (300 MHz, CDC13):
0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1H, m), 2.97-3.11(6H, m), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m)。
实施例 4
4) (5R, 6S,8R,2’S, 4’S)- [(R)-1-(羟基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸酯 (Ⅷ) 的合成
室温下,于2000ml反应瓶中,加入32.8g(0.0528mol)(5R,6S,8R,2’S, 4’S)-[(R)-1-(叔丁基二甲基硅氧基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3.2.0] -庚-2-烯-7-酮-2-羧酸酯 (Ⅶ)、27.4 ml醋酸、41.3g氟氢化胺和1000ml二氯甲烷,室温下搅拌48h。反应完毕后,冰浴下向反应液中加入500 ml 饱和碳酸氢钠水溶液搅拌10分钟,分出二氯甲烷层,加入无水硫酸镁干燥,得到黄色固体26.2g(0.0517mol,收率98.0%)(5R, 6S,8R,2’S, 4’S)- [(R)-1-(羟基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3. 2. 0]-庚-2-烯-7-酮-2-羧酸酯 (Ⅷ),产物不经进一步纯化直接投入下步反应。
1H-NMR (300 MHz, CDC13):
1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m), 4.25(2H,m), 4.47-4.87(5H, m), 5.15-5.50(4H, m), 5.94(2H, m)。
实施例 5
5) (5R,6S,8R,2’S,4’S)-3-[4-(2-二甲胺基羰基)吡咯烷基硫]-6-(1-羟基乙基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸酯 (I) 的合成
向反应瓶中,加入26.2g(0.0517mol)(5R, 6S,8R,2’S, 4’S)- [(R)-1-(羟基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3. 2. 0]-庚-2-烯-7-酮-2-羧酸酯 (Ⅷ)、21.3g(0.152mol)二甲基环己二酮和550ml乙酸乙酯,升温至30℃,再向其中滴加1.0g(0.865mmol)四三苯基膦钯的150ml二氯甲烷溶液, 氮气保护下室温反应3h完毕。向反应液中加入300ml水搅拌,分出水层,水层用乙酸乙酯洗涤,冰浴冷却下滴加500ml四氢呋喃,加入晶种搅拌,收集晶体真空干燥,得到浅黄色晶体13.4g(0.0352mol, 收率68.1%)(5R,6S,8R,2’S,4’S)-3-[4-(2-二甲胺基羰基)吡咯烷基硫]-6-(1-羟基乙基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸的三水合物 (I)—美罗培南。
IR max KBr cm-1 : 1755,1627,1393,1252,1130
NMR (D2O, 300Hz):
1.25 (3H,d),1.81-1.96 (1H,m),2.96 (3H,s),3.03 (3H,s),3.14-3.20 (3H,m),3.31-3.41 (2H,m),3.62-3.72 (1H,m),3.90-4.00 (1H,m),4.14-4.26 (2H,m),4.63 (1H,t)。
实施例 6
6) (5R,6S,8R,2’S,4’S)-3-[4-(2-二甲胺基羰基)吡咯烷基硫]-6-(1-羟基乙基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸酯 (I) 的合成
将实施例5中21.3g(0.152mol)二甲基环己二酮替换为45.1g(0.155mol)三丁基氢化锡,滴加0.125g(0.108mmol)四三苯基膦钯,其他加量和方法均相同,得到16.2g(0.0426mol, 82.5%)(5R,6S,8R,2’S,4’S)-3-[4-(2-二甲胺基羰基)吡咯烷基硫]-6-(1-羟基乙基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸的三水合物 (I)—美罗培南。
实施例 7
7) (5R,6S,8R,2’S,4’S)-3-[4-(2-二甲胺基羰基)吡咯烷基硫]-6-(1-羟基乙基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸酯 (I) 的合成
向反应瓶中,加入26.2g(0.0517mol)(5R, 6S,8R,2’S, 4’S)- [(R)-1-(羟基)乙基] -3-[4-(1-烯丙氧羰基-1-二甲胺基羰基)吡咯烷基硫]-6-(1-烯丙氧羰基乙氧基)-1-氮杂双环[3. 2. 0]-庚-2-烯-7-酮-2-羧酸酯 (Ⅷ)、6.0g(0.0387mol) N,N-二甲基巴比士酸和500ml二氯甲烷,再向其中滴加6.0g(5.2mmol)四三苯基膦钯的100ml二氯甲烷溶液, 氮气保护下室温反应5h完毕。向反应液中加入300ml水搅拌,分出水层,水层用乙酸乙酯洗涤,冰浴冷却下滴加四氢呋喃,析出晶体,收集晶体真空干燥,得到15.7g(0.0413mol, 收率80.1%)(5R,6S,8R,2’S,4’S)-3-[4-(2-二甲胺基羰基)吡咯烷基硫]-6-(1-羟基乙基)-1-氮杂双环[3.2.0]-庚-2-烯-7-酮-2-羧酸的三水合物 (I)—美罗培南。
Claims (9)
1.一种美罗培南的合成方法,其特征在于,所述合成方法的具体反应路线如下:

反应步骤如下:
1)式 (IV) 化合物和式 (V) 化合物溶于有机溶剂后在缩合剂的作用下通过缩合反应得到式(Ⅱ)化合物,反应时间为2~24小时,反应温度为0~40℃;
2)式 (Ⅱ) 化合物和式 (Ⅵ)化合物溶于甲苯、乙酸乙酯或四氢呋喃后在碱的作用下进行反应,生成式 (Ⅲ) 化合物,反应时间为1~3小时,反应温度为-20~5℃;
3)式(Ⅲ) 化合物溶于环己烷、正庚烷、正辛烷、甲苯或二甲苯后在有机磷试剂的作用下发生Wittig关环反应得到式 (Ⅶ) 化合物,所述有机磷试剂是三苯基磷、三正丁基磷、亚磷酸三乙酯或亚磷酸三甲酯;
4)式(Ⅶ) 化合物溶于甲醇、四氢呋喃、丙酮、正戊烷、正己烷、乙醚、乙腈、二氯甲烷、氯仿或乙酸乙酯后在酸的作用下水解硅醚键,得到式 (Ⅷ) 化合物;所述酸是稀盐酸、氢氟酸、四丁基氟铵、苄基三丁基氟化铵、氟氢化铵或醋酸,所述酸和式(Ⅶ) 化合物的摩尔比为5~15:1;所述水解反应的温度为0~40℃,反应时间为8~24小时;
5)式(Ⅷ) 化合物溶于甲醇、乙醇、叔丁醇、异丁醇、异丙醇、四氢呋喃、二氧杂环己烷、丙酮、二氯甲烷、氯仿以及水中的一种或几种所组成的溶剂后在钯催化剂作用下氢化脱去烯丙基得到目标产物(I),所述钯催化剂与式(Ⅷ) 化合物的摩尔比为0.0001~0.5:1;反应温度为0~40℃,反应时间为2~24h。
2.根据权利要求1所述的一种美罗培南的合成方法,其特征在于,步骤1)中式(IV) 化合物和式 (V) 化合物的摩尔比为1.05~1.0:1,所述缩合剂和式(IV) 化合物的摩尔比为1.50~1.05:1。
3.根据权利要求1或2所述的一种美罗培南的合成方法,其特征在于,所述缩合剂是碳二亚胺类试剂或N, N’-羰基二咪唑;所述有机溶剂是丙酮、乙腈、甲苯、四氢呋喃、氯仿或二甲基甲酰胺。
4.根据权利要求1所述的一种美罗培南的合成方法,其特征在于,步骤2)中式(Ⅵ)化合物与式 (Ⅱ) 化合物的摩尔比为1.5~2.5:1,所述碱与所述式(Ⅵ)化合物的摩尔比为1.2~2:1。
5.根据权利要求1所述的一种美罗培南的合成方法,其特征在于,步骤3)中
所述有机磷试剂与式 (Ⅲ) 化合物的摩尔比为2~8:1;所述关环反应的温度为25~100 ℃,反应时间为10~24小时。
6.根据权利要求3所述的一种美罗培南的合成方法,其特征在于,所述碳二亚胺类试剂是二环己基碳二亚胺、二异丙基碳二亚胺或1-(3-二甲胺基丙基)-3-乙基碳二亚胺。
7.根据权利要求1所述的一种美罗培南的合成方法,其特征在于,步骤2)中的所述碱是无机碱或有机碱;当为无机碱时,是氢氧化钠、碳酸钠或碳酸氢钠;当为有机碱时是吡啶、三乙胺、二异丙基乙基胺或2, 6-二甲基吡啶。
8.根据权利要求1所述的一种美罗培南的合成方法,其特征在于,所述钯催化剂是醋酸钯、氯化钯、硝酸钯、双三苯基膦氯化钯或四三苯基膦钯。
9.根据权利要求1所述的一种美罗培南的合成方法,其特征在于,步骤5)中的保护基受体为吗啉、二甲基环己二酮、三丁基氢化锡、N,N-二甲基巴比士酸、2-乙基己酸或己酸。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201010541665 CN101962383A (zh) | 2010-11-12 | 2010-11-12 | 一种美罗培南的合成方法 |
| PCT/CN2011/001587 WO2012062035A1 (zh) | 2010-11-12 | 2011-09-19 | 一种美罗培南的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201010541665 CN101962383A (zh) | 2010-11-12 | 2010-11-12 | 一种美罗培南的合成方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101962383A true CN101962383A (zh) | 2011-02-02 |
Family
ID=43515447
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 201010541665 Pending CN101962383A (zh) | 2010-11-12 | 2010-11-12 | 一种美罗培南的合成方法 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101962383A (zh) |
| WO (1) | WO2012062035A1 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102250096A (zh) * | 2011-09-05 | 2011-11-23 | 江西华邦药业有限公司 | 一种美罗培南的制备方法 |
| WO2012062035A1 (zh) * | 2010-11-12 | 2012-05-18 | 上海巴迪生物医药科技有限公司 | 一种美罗培南的合成方法 |
| CN104072523A (zh) * | 2014-07-14 | 2014-10-01 | 上海新亚药业有限公司 | 比阿培南的制备方法 |
| CN108191869A (zh) * | 2018-01-22 | 2018-06-22 | 重庆天地药业有限责任公司 | 美罗培南的纯化方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6475488A (en) * | 1987-09-17 | 1989-03-22 | Sumitomo Pharma | Production of beta-lactam compound |
| US4888344A (en) * | 1986-07-30 | 1989-12-19 | Sumitomo Pharmaceuticals Company, Limited | Carbapenem compound in crystalline form, and its production and use |
| CN101348486A (zh) * | 2008-08-29 | 2009-01-21 | 深圳市海滨制药有限公司 | 一种美罗培南的制备方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101962383A (zh) * | 2010-11-12 | 2011-02-02 | 上海巴迪生物医药科技有限公司 | 一种美罗培南的合成方法 |
-
2010
- 2010-11-12 CN CN 201010541665 patent/CN101962383A/zh active Pending
-
2011
- 2011-09-19 WO PCT/CN2011/001587 patent/WO2012062035A1/zh active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4888344A (en) * | 1986-07-30 | 1989-12-19 | Sumitomo Pharmaceuticals Company, Limited | Carbapenem compound in crystalline form, and its production and use |
| JPS6475488A (en) * | 1987-09-17 | 1989-03-22 | Sumitomo Pharma | Production of beta-lactam compound |
| CN101348486A (zh) * | 2008-08-29 | 2009-01-21 | 深圳市海滨制药有限公司 | 一种美罗培南的制备方法 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012062035A1 (zh) * | 2010-11-12 | 2012-05-18 | 上海巴迪生物医药科技有限公司 | 一种美罗培南的合成方法 |
| CN102250096A (zh) * | 2011-09-05 | 2011-11-23 | 江西华邦药业有限公司 | 一种美罗培南的制备方法 |
| CN102250096B (zh) * | 2011-09-05 | 2016-04-06 | 江西华邦药业有限公司 | 一种美罗培南的制备方法 |
| CN104072523A (zh) * | 2014-07-14 | 2014-10-01 | 上海新亚药业有限公司 | 比阿培南的制备方法 |
| CN104072523B (zh) * | 2014-07-14 | 2017-10-24 | 上海上药新亚药业有限公司 | 比阿培南的制备方法 |
| CN108191869A (zh) * | 2018-01-22 | 2018-06-22 | 重庆天地药业有限责任公司 | 美罗培南的纯化方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2012062035A1 (zh) | 2012-05-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102712585B (zh) | 中性内肽酶抑制剂中间体和其制备方法 | |
| CN101348486B (zh) | 一种美罗培南的制备方法 | |
| EP2388261B1 (en) | Improved process for the preparation of carbapenem using carbapenem intermediates and recovery of carbapenem | |
| CN101962383A (zh) | 一种美罗培南的合成方法 | |
| JPH07502983A (ja) | 金属アルコキシドとβ−ラクタムを使用する置換イソセリンエステルの製造 | |
| WO2012038979A2 (en) | A process for preparation of ertapenem | |
| WO2016002918A1 (ja) | インドール化合物の製造方法 | |
| JPWO2010047296A1 (ja) | 新規ピリミジン誘導体およびHMG−CoA還元酵素阻害剤中間体の製造方法 | |
| WO2015099126A1 (ja) | 5-ヒドロキシピペリジン-2-カルボン酸の製造方法 | |
| WO2007104219A1 (en) | A process for the preparation of the intermediate of β-methyl carbapenem | |
| JP2522671B2 (ja) | 結晶態のカルバペネム化合物、その製造方法およびその化合物を含有する注射用抗菌剤 | |
| WO2003042180A9 (fr) | Production d'ester d'acide oxoheptonoique optiquement actif | |
| CN101891746B (zh) | 一种美罗培南的制备方法 | |
| CN102174047A (zh) | 一种多尼培南的新型制备工艺 | |
| WO2004043961A1 (ja) | 経口投与用カルバペネム化合物の製造方法 | |
| CN111410607B (zh) | 六氢呋喃并呋喃醇衍生物的制备方法、其中间体及其制备方法 | |
| KR20160008026A (ko) | 고순도 로수바스타틴 칼슘염의 제조 방법 | |
| JP2003277390A (ja) | アゼチジノン化合物の製造方法 | |
| WO2018108130A1 (en) | Process for preparation of novel androgen receptor antagonist | |
| CN101585847A (zh) | 法罗培南钠的合成方法 | |
| EP0689543B1 (en) | Process for the preparation of condensed carbapeneme derivatives | |
| WO2017168438A1 (en) | Process for preparing pure allyl protected keto derivative | |
| CN101983187A (zh) | (s)-3-(1-氰基-1,1-二苯基甲基)吡咯烷的制备方法 | |
| CN107746396B (zh) | 一种新型化合物6,6-二甲基四氢吡喃-2-甲醇及其制备方法 | |
| ZA200300590B (en) | Process for preparing discodermolide and analogues thereof. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20110202 |