CN101870660A - 一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法 - Google Patents
一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法 Download PDFInfo
- Publication number
- CN101870660A CN101870660A CN 201010166768 CN201010166768A CN101870660A CN 101870660 A CN101870660 A CN 101870660A CN 201010166768 CN201010166768 CN 201010166768 CN 201010166768 A CN201010166768 A CN 201010166768A CN 101870660 A CN101870660 A CN 101870660A
- Authority
- CN
- China
- Prior art keywords
- reaction
- ephedrine
- hydrochloride
- propiophenone
- alpha
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

Abstract
本发明涉及一种以α-溴代苯丙酮为原料,经甲胺化、拆分、还原、酰化、酸解、水解等步骤,制备L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱。该方法具有反应条件温和、原料易得、设备投资少、三废治理简单、产率高等优点。本发明制备的盐酸麻黄碱和盐酸伪麻黄碱,属于β2肾上腺素受体激动药(β2adrenoceptor agonists),其制剂(片剂、胶囊,如:中美天津史克制药有限公司生产的新康泰克胶囊)口服后主要通过刺激交感神经末梢释放去甲肾上腺素,以间接方式起拟交感神经作用。临床上被用于感冒的辅助治疗,能减轻感冒、过敏性鼻炎、鼻炎及鼻窦炎引起的鼻粘膜充血症状。
Description
技术领域
本发明属于医药化工领域,合成麻黄素行业。
背景技术
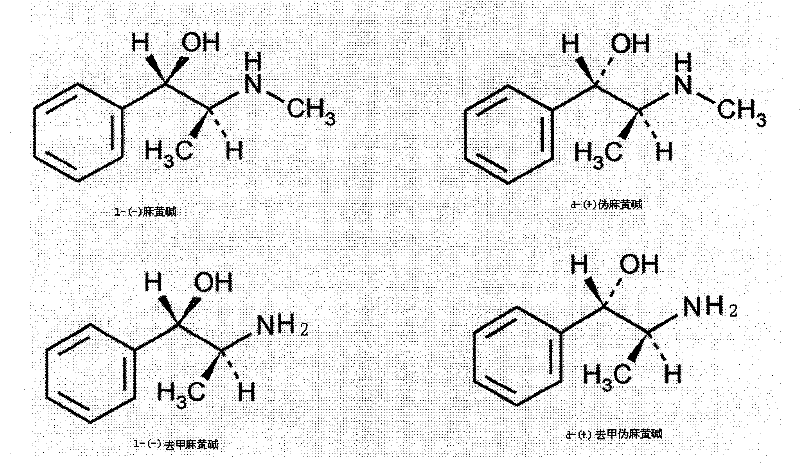
盐酸伪麻黄碱是中药麻黄的有效成分之一。麻黄是指麻黄科植物草麻黄Ephedra sinica Stapf,中麻黄Ephedra intermedia Schrenk ex C.A.Mey.或木贼麻黄Ephedra equisetina Bge.的草质茎。麻黄的主要成分为生物碱(0.8%-2%)。生物碱的80%-85%为伪麻黄碱(右旋伪麻黄碱,D-pseudo-ephedrine);其次为麻黄碱(左旋麻黄碱L-ephedrine,以及微量的L-N-甲基麻黄碱(L-N-methyl-ephedrine)、D-N-甲基伪麻黄碱(D-N-methyl-pseudo-ephedrine)、去甲基麻黄碱(L-nor-ephedrine)、去甲基伪麻黄碱(D-nor-pseudo-ephedrine)和麻黄次碱(ephedine,麻黄定)等。1887年nagai从麻黄草中分离出一种纯净的碱性物质,命名为麻黄碱(Ephedrine),1889年Merck从欧洲麻黄类植物中分离出麻黄碱,并首次获得伪麻黄碱(1),19世纪20年代,我国著名的药理学家陈克恢(2)等证明该类化合物具有拟交感活性,阐明了其潜在的药用价值。1950年,科学家通过化学方法将麻黄碱的构型与(R)-(-)-扁桃酸和L-丙氨酸的构型相关联而确定了麻黄碱和伪麻黄碱的构型。并证明以上六种麻黄生物碱中,右旋伪麻黄碱(D-pseudo-ephedrine)与麻黄碱(左旋麻黄碱L-ephedrine)互为非对映异构体。其结构式如下:
分子式:L-(-)麻黄碱C10H15ON
d-(+)伪麻黄碱C10H15ON
L-(-)去甲基麻黄碱C9H13ON
d-(+)伪麻黄碱C9H13ON
传统的麻黄素生产工艺,采用麻黄草提取法。其工艺流程要点是:麻黄草原料经精选、加水加压浸煮三次后,得到的草水提取液加碱游离至PH=11.5-12,将该游离液体输送至高位罐中,经二甲苯萃取塔逆流萃取,使游离态的麻黄碱转移至二甲苯液体中,得到二甲苯提取液,该部分二甲苯提取液经草酸水溶液反萃取后,得到含草酸麻黄总碱的水溶液。草酸麻黄总碱的水溶液真空减压浓缩,放冷、结晶、离心分离,得到固体为草酸麻黄碱粗品,液体为草酸伪麻黄碱水溶液。分别进行脱草酸根、脱铁、中和、脱色反应后,过滤、浓缩、结晶、离心分离,得到盐酸伪麻黄碱和盐酸麻黄碱半成品,再分别经过重结晶后,生产出盐酸伪麻黄碱成品。采用麻黄草提取生产盐酸伪麻黄碱,存在三个方面的缺陷:其一、野生麻黄草资源匮乏及国家关于禁采野生麻黄草的政策,使公司盐酸伪麻黄碱产量收到原料供应不足的严重制约。多年来,公司麻黄素生产线产量最高达到过5吨,仅占到设计量的10%,导致生产设施的浪费。其二、采集野生麻黄草资源破坏生态环境,据分析,以麻黄素价值与环境恢复代价相比,两者的比例高达1∶20。同时,以野生麻黄草为原料,经浸煮、二甲苯萃取工艺生产麻黄素而产生的废水,其BOD/COD指标严重超标,不能直接排放,必须经过治理达标。麻黄素废水治理问题,是国内提取麻黄素行业普遍存在的问题,目前只能采用生物降解方式才能根治,废水治理成本以每吨产品产生废水600吨计算,为12万元,使得生产企业无法承受。其三、以野生麻黄草为原料生产麻黄素,每吨产品需要消耗麻黄草200吨,吨生产成本高达人民币50万元,与市场价格倒挂。
目前,化学合成手段生产麻黄素面临的主要问题是如何低成本的得到光学纯产物;虽然不对称合成的方法已得到很大的发展,但有工业价值的方法并不多。目前文献报道的L-麻黄碱方法和伪麻黄碱生产方法主要有以下几种:
1)L-(+)-苦杏仁酸和D-(-)-苦杏仁酸为拆分剂(a),U.S.Patent:1867274;b),J.A m.Chem.Soc.,1929,51:1906.):此方法的优点是拆分效率高(75%以上);产品光学纯度高(99%以上).缺点是:L-(+)-苦杏仁酸和D-(-)苦杏仁酸不易得到,价格高,不能适应大工业化生产的要求。
2)利用D-arabonic acid为拆分剂(US Patent 34T8101)
此方法的优点是:产物的拆分率(80%以上)和光学纯度都较高,且拆分剂可以回收利用。但D-arabonic acid价格昂贵,不易得到。
本发明在优先考察合成品的安全性、有效性与已经上市产品一致的前提下,考虑到工艺简化、原辅料易得、成本低廉、具备一定的工业化生产推广价值、专利保护情况,研究出以溴代苯丙酮为起始原料合成麻黄素的方法。
发明内容
本发明的目的在于用一种易购的普通化工原料生产麻黄素,从而节约保护麻黄草资源,保护生态环境。
本发明人经实验研究发现,以α-溴代苯丙酮为起始原料,经甲胺化反应、拆分反应、还原反应、转旋反应、重结晶五步反应可以生产出符合中国药典标准的L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱。
为实现本发明的目的,本发明采用以下的技术方案:一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于包括如下步骤:以α-溴代苯丙酮或α-氯代苯丙酮为起始原料,经甲胺化反应、拆分反应、还原反应、转旋反应生产盐酸伪麻黄碱,其反应由以下步骤:
1.甲胺化反应
分子式:α-溴代苯丙酮C9H9OBr
α-甲胺基苯丙酮C10H13ON
(1)使用的原辅料按照以下重量百分比(或质量分数)配比:
以投入起始原料α-溴代苯丙酮,含量≥96%,分子量213.07g,反应过程中,α-溴代苯丙酮mol数或重量与投入其他原辅料的mol比例分别为:
α-溴代苯丙酮∶甲胺mol比=1∶1
α-溴代苯丙酮∶甲胺水溶液(含量≥30%)重量比=1∶1~1.1
α-溴代苯丙酮∶反应溶剂甲苯(含量≥96%)重量比=1∶5~5.5
α-溴代苯丙酮∶碱化游离用氢氧化钠(含量≥95%)重量比=1∶0.2~0.25
α-溴代苯丙酮∶酸化中和用盐酸(含量≥30%)重量比=1∶2.5~3
α-溴代苯丙酮∶结晶溶剂丙酮(含量≥95%)重量比=1∶2.5~3
(2)反应主要参数:
反应温度为40℃~45℃,搅拌速度为60转/分~70转/分。
2、拆分反应
(1)使用(2R,3S)-(-)二苯甲酰基酒石酸[(2R,3S)-(-).H2O]作为拆分剂,其溶解采用乙酸乙酯作为溶解试剂,其重量比为1∶0.7~0.9;
(2)拆分剂的拆分目标是上一步甲胺化反应得物α-甲胺基苯丙酮盐酸盐,α-甲胺基苯丙酮盐酸盐(含量≥98%)与拆分剂(含量≥99%)的投料mol比为1∶0.5~0.55。重量比(含结晶水的重量)为:1∶1.06~1.1;
(3)拆分反应过程中,乙酸乙酯与甲醇的比例调整至7~10∶1。
拆分反应
分子式:α-甲胺基苯丙酮C10H13ON
(2R,3S)-(-)二苯甲酰基酒石酸C18H14O8
2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-α-甲胺基苯丙酮C38H40O10N2
3.还原反应
分子式:(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-α-甲胺基苯丙酮盐C38H40O10N2
(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-麻黄碱盐C38H44O10N2

分子式:(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-麻黄碱盐C38H44O10N2
(S)-(-)-麻黄碱盐酸盐C10H15ON·HCL
(S)-(-)-麻黄碱C10H15ON
还原反应由以下步骤:
(1)使用硼氢化钾KBH4作为还原剂,使用二级反渗透纯化水作为反应溶剂;
(2)上步拆分反应得物溶解时,加入二级反渗透纯化水4~6倍水;
(3)上步拆分反应得物与还原剂硼氢化钾的mol比为1∶1.0~1.05,其重量比为1∶0.1~0.13;
(4)还原反应温度控制在25℃以内。
4.转旋反应
转旋反应由以下步骤:
(1)根据以上步骤所制得的L-(-)盐酸麻黄碱粗品,其干燥失重≤0.5%,旋光度在33~36.5时,该粗品可直接投入转旋反应。如干燥失重超过0.5%,可对粗品进行再次烘干处理,直到达标。
(2)转旋反应时,盐酸麻黄碱粗品与加入乙酸酐的的摩尔比理论量为1∶0.5~0.6,理论重量比为:1∶0.39~0.42。实际保障转旋率的投料比例为:mol比例:1∶4.6~6,重量比:1∶1.8~2.4。
(3)转旋反应时,反应温度控制在110℃~120℃,反应时间控制在4~8小时。
5.反应溶剂或助剂为:
1)试剂为:醇类试剂:甲醇、乙醇,酯类试剂:乙酸乙酯、乙酸丁酯,醚类试剂:乙醚,酸酐类试剂:乙酸酐等;
2)所用碱为:NaOH,KOH;
3)所用酸为:HCl,H2SO4;
4)脱水剂为:无水硫酸钠、无水硫酸镁;
5)所用溶剂为:甲苯、二甲苯、丙酮等有机溶剂。
转旋反应

分子式:(S)-(-)-麻黄碱盐酸盐C10H15ON·HCL
乙酰化(S)-(-)-麻黄碱C12H17O2N
盐酸伪麻黄碱C10H15ON·HCL
本发明的优点和效果是:本发明的方法具有反应条件温和、原料易得、设备投资少、三废治理简单、产率高等优点。
本发明制备的额盐酸麻黄碱和盐酸伪麻黄碱,属于β2肾上腺素受体激动药(β2 adrenoceptor agonists),其制剂(片剂、胶囊,如:中美天津史克制药有限公司生产的新康泰克胶囊)口服后主要通过刺激交感神经末梢释放去甲肾上腺素,以间接方式起拟交感神经作用。盐酸伪麻黄碱收缩血管具有一定的选择性,主要收缩上呼吸道血管,消除鼻咽部粘膜充血,是一种较好的上呼吸道粘膜的减充血剂。该产品对支气管平滑肌的扩张作用和全身血管收缩作用较盐酸麻黄碱弱,其加快心率、升高血压,中枢神经兴奋等不良反应也较轻。临床上被用于感冒的辅助治疗,能减轻感冒、过敏性鼻炎、鼻炎及鼻窦炎引起的鼻粘膜充血症状。
附图说明
以下结合附图和具体实施例对本发明作进一步详细说明。
图1是本发明的工艺流程图。
具体实施方式
以下结合反应方程式及附图1工艺流程图对本发明进行详细说明:
工序1:以α-溴代苯丙酮为起始原料进行甲胺化反应
在2000ml的旋转式玻璃蒸发反应器中,加入100-110ml甲苯溶液和15-16.5ml甲胺水溶液,水浴温度恒温在40-45℃,开动搅拌,搅拌速度控制为60-70转/分,滴加α-溴代苯丙酮21.0g,再滴加由4g氢氧化钠配置的15%的氢氧化钠水溶液,加毕,反应两小时,停止加热,冷至室温。用萃取器分离出有机相,用甲苯提取水相两次(30ml*2次),合并有机相。有机相中滴加15%盐酸水溶液100-150ml,搅拌1小时,分出水相,减压蒸发至糖浆状。加50-60ml丙酮振摇,静置一夜,滤出白色固体物,即为α-甲胺基苯丙酮的盐酸盐。
工序2:拆分反应
在500L搪玻璃反应釜中,称取加入取α-甲胺基苯丙酮盐酸盐30kg,开搅拌30转/分,加入饱和氢氧化钠60.0L调至PH=12-13,搅拌溶解。再加入乙酸乙酯15L搅拌静置分层,弃去下层碱水液,得到上层38L乙酸乙酯及α-甲胺基苯丙酮混合液体备用。另外在500L溶解罐中取拆分剂(2R,3S)-(-)二苯甲酰基酒石酸[(2R,3S)-(-)DBTA.2H2O]26kg加入乙酸乙酯25L,溶解,真空抽至200L高位计量储罐中,再缓慢加入到“乙酸乙酯及α-甲胺基苯丙酮混合液体”中,滴加过程中产生大量沉淀,滴加完毕后再加入无水甲醇4L使溶液澄清,后禁止搅拌,放置使自然结晶24小时以上。离心分离,母液待处理套用,固体用乙酸乙酯∶甲醇=10∶1溶液洗涤至产品色泽变无色或暗红色。所得固体为(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-α-甲胺基苯丙酮的盐(简称:中间体1号)。重量30.5Kg。
工序3:还原反应
取中间体30.5Kg加入4-6倍量自来水溶液,加入还原剂(加入量为中间体量的0.5mol倍量1.2KG,加入过程控制温度小于25度,缓慢加入使充分反应,量大则需要建立检测方法待进一步研究确定)加入完毕继续反应30分钟以上,后加入0.3-0.4L盐酸,调节PH=3-4,加入乙酸丁酯100L萃取一次,乙酸丁酯层留用待回收拆分剂,母液加入饱和的氢氧化钠溶液20L,调节PH=11-12.5加入甲苯100L萃取两次,母液套用于下批次生产,甲苯层中加入盐酸酸化,析出白色结晶,取下层液体及白色结晶浓缩结晶离心分离得到盐酸麻黄碱粗品,母液留用套用。
质量指标:
干燥失重:0.82%
旋光度:-32.44,-33.25
总灰分:0.03%,0.05%
熔点:219.7-221.3
性状:符合药典标准。
鉴别:符合规定
含量:98.66%
酸碱度:PH=3.10
溶液的澄清度与颜色:符合规定。
重金属:符合规定
硫酸盐:符合规定
检测结论:符合粗品质量标准。
工序4、转旋反应
将左旋麻黄碱或左旋盐酸麻黄碱20g,加入乙酸酐35mL进行乙酰化,反应温度控制在100-130℃之间回流4-6h;冷却后,加入浓盐酸16mL,回流6-8h;冷却,加入蒸馏水36mL,回流水解12小时后停止反应。浓缩除去乙酸溶液液,得到右旋麻黄碱盐酸盐的粗品。加碱游离后酸化处理精制得到D-(-)-盐酸伪麻黄碱成品。
工序5、重结晶反应
将盐酸伪麻黄碱半成品用4倍量纯水溶解,加入数量比为0.05%的针状活性炭,过滤,滤渣清洗三次至含量低于0.1%送锅炉房焚烧。滤液真空减压浓缩至波美度18时,放入冷却槽车。
实施例一
1.在2000ml的旋转式玻璃蒸发反应器中,加入1100ml甲苯溶液和160ml甲胺水溶液,水浴温度恒温在45℃,开动搅拌,搅拌速度控制为65转/分,滴加α-溴代苯丙酮210g,再滴加由50g氢氧化钠配置的15%的氢氧化钠水溶液,加毕,反应两小时,停止加热,冷至室温。用萃取器分离出有机相,用甲苯提取水相两次(300ml*2次),合并有机相。有机相中滴加15%盐酸水溶液1500ml,搅拌1小时,分出水相,减压蒸发至糖浆状。加550ml丙酮振摇,静置一夜,滤出白色固体物,即为α-甲胺基苯丙酮的盐酸盐(收率为74%)。
2、在1000ml玻璃反应器中,称取加入取α-甲胺基苯丙酮盐酸盐200g,开搅拌30转/分,加入饱和氢氧化钠400ml调至PH=12-13,搅拌溶解。再加入乙酸乙酯100L搅拌静置分层,弃去下层碱水液,得到上层260ml乙酸乙酯及α-甲胺基苯丙酮混合液体备用。另外在1000ml反应器中取拆分剂(2R,3S)-(-)二苯甲酰基酒石酸[(2R,3S)-(-)DBT A.H2O]190g加入乙酸乙酯100ml,溶解,再缓慢加入到“乙酸乙酯及α-甲胺基苯丙酮混合液体”中,滴加过程中产生大量沉淀,滴加完毕后再加入无水甲醇30ml使溶液澄清,后禁止搅拌,放置使自然结晶24小时以上。离心分离,母液待处理套用,固体用乙酸乙酯∶甲醇=7∶1溶液洗涤至产品色泽变无色或暗红色。所得固体为(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-α-甲胺基苯丙酮的盐(简称:中间体1号)。重量216g于6倍量纯水中加入0.55mol还原剂硼氢化钾,还原,后加盐酸酸化,乙酸丁酯萃取分离提取拆分剂二苯甲酰基酒石酸,酸水层加40%氢氧化钠游离甲苯萃取,萃取液加盐酸酸化结晶制备L-(-)盐酸麻黄碱一次收率为32.68%
3.将L-(-)盐酸麻黄碱100g,加入乙酸酐180mL进行乙酰化,反应温度控制在110-130℃之间回流6h;冷却后,加入浓盐酸90mL,回流8h;冷却,加入蒸馏水180mL,回流水解12小时后停止反应。浓缩除去乙酸溶液,得到右旋麻黄碱盐酸盐的粗品。加20%氢氧化钠游离后酸化处理精制得到D-(-)-盐酸伪麻黄碱成品,一次收率为75%转化率为90%以上。母液套用。
实施例二
1.在2000ml的旋转式玻璃蒸发反应器中,加入1000ml甲苯溶液和150ml甲胺水溶液,水浴温度恒温在40℃,开动搅拌,搅拌速度控制为60转/分,滴加α-溴代苯丙酮210.0g,再滴加由40g氢氧化钠配置的15%的氢氧化钠水溶液,加毕,反应两小时,停止加热,冷至室温。用萃取器分离出有机相,用甲苯提取水相两次(300ml*2次),合并有机相。有机相中滴加15%盐酸水溶液1000ml,搅拌1小时,分出水相,减压蒸发至糖浆状。加500ml丙酮振摇,静置一夜,滤出白色固体物,即为α-甲胺基苯丙酮的盐酸盐(收率为73%);
2.取α-甲胺基苯丙酮的盐酸盐100g滴加40%氢氧化钠溶液反应2小时加入乙酸乙酯溶液50ml分离上部油层备用,后在搅拌下缓慢加入二苯甲酰基酒石酸90g溶解于50ml乙酸乙酯的溶液,加甲醇10ml调节溶液至澄清,禁止搅拌,精制24小时结晶分离,母液回收套用,固体用乙酸乙酯∶甲醇=10∶1溶液洗涤至产品色泽变无色或暗红色。结晶为L(-)苯丙酮·二苯甲酰基酒石酸盐,重量212g,于4倍量纯水中加入0.5mol还原剂硼氢化钾,还原,后加盐酸酸化,乙酸丁酯萃取分离提取拆分剂二苯甲酰基酒石酸,酸水层加40%氢氧化钠游离萃取,母液回收拆分剂,萃取液加盐酸酸化结晶制备L-(-)盐酸麻黄碱一次收率为32.68%;
3.将L-(-)盐酸麻黄碱100g,加入乙酸酐200mL进行乙酰化,反应温度控制在110-130℃之间回流5h;冷却后,加入浓盐酸100mL,回流6h;冷却,加入蒸馏水180mL,回流水解8小时后停止反应。浓缩除去乙酸溶液,得到右旋麻黄碱盐酸盐的粗品。加40%氢氧化钠游离后酸化处理精制得到D-(-)-盐酸伪麻黄碱成品,一次收率为75%转化率为90%以上。母液套用。
实施例三
1.在2000ml的旋转式玻璃蒸发反应器中,加入1100ml甲苯溶液和165ml甲胺水溶液,水浴温度恒温在43℃,开动搅拌,搅拌速度控制为65转/分,滴加α-溴代苯丙酮210g,再滴加由45g氢氧化钠配置的15%的氢氧化钠水溶液,加毕,反应两小时,停止加热,冷至室温。用萃取器分离出有机相,用甲苯提取水相两次(300ml*2次),合并有机相。有机相中滴加15%盐酸水溶液1200ml,搅拌1小时,分出水相,减压蒸发至糖浆状。加600ml丙酮振摇,静置一夜,滤出白色固体物,即为α-甲胺基苯丙酮的盐酸盐(收率为76%);
2、在1000ml玻璃反应器中,称取加入取α-甲胺基苯丙酮盐酸盐100g,开搅拌30转/分,加入饱和氢氧化钠200ml调至PH=12-13,搅拌溶解。再加入乙酸乙酯50ml搅拌静置分层,弃去下层碱水液,得到上层135ml乙酸乙酯及α-甲胺基苯丙酮混合液体备用。另外在1000ml反应器中取拆分剂(2R,3S)-(-)二苯甲酰基酒石酸[(2R,3S)-(-)DBTA.H2O]95g加入乙酸乙酯100ml,溶解,再缓慢加入到“乙酸乙酯及α-甲胺基苯丙酮混合液体”中,滴加过程中产生大量沉淀,滴加完毕后再加入无水甲醇25ml使溶液澄清,后禁止搅拌,放置使自然结晶24小时以上。离心分离,母液待处理套用,固体用乙酸乙酯∶甲醇=7.5∶1溶液洗涤至产品色泽变无色或暗红色。所得固体为(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-α-甲胺基苯丙酮的盐(简称:中间体1号)。重量204g,于5倍量纯水中加入0.52mol还原剂硼氢化钾,还原,后加盐酸酸化,乙酸丁酯萃取分离提取拆分剂二苯甲酰基酒石酸,酸水层加40%氢氧化钠游离甲苯萃取,萃取液加盐酸酸化结晶制备L-(-)盐酸麻黄碱一次收率为32.05%;
3.将L-(-)盐酸麻黄碱100g,加入乙酸酐240mL进行乙酰化,反应温度控制在110-130℃之间回流4h;冷却后,加入浓盐酸90mL,回流6h;冷却,加入蒸馏水200mL,回流水解8小时后停止反应。浓缩除去乙酸溶液,得到右旋麻黄碱盐酸盐的粗品。加40%氢氧化钠游离后酸化处理精制得到D-(-)-盐酸伪麻黄碱成品,一次收率为73%转化率为90%以上。母液套用。
实施例四
1.以氯代苯丙酮为起始原料进行甲胺化反应在2000ml的旋转式玻璃蒸发反应器中,加入1000ml甲苯溶液和165ml甲胺水溶液,水浴温度恒温在43℃,开动搅拌,搅拌速度控制为65转/分,滴加α-氯代苯丙酮170g,再滴加由45g氢氧化钠配置的15%的氢氧化钠水溶液,加毕,反应两小时,停止加热,冷至室温。用萃取器分离出有机相,用甲苯提取水相两次(30ml*2次),合并有机相。有机相中滴加15%盐酸水溶液1500ml,搅拌1小时,分出水相,减压蒸发至糖浆状。加60ml丙酮振摇,静置一夜,滤出白色固体物,即为α-甲胺基苯丙酮的盐酸盐。(收率为66%);
2、在1000ml玻璃反应器中,称取加入取α-甲胺基苯丙酮盐酸盐100g,开搅拌30转/分,加入饱和氢氧化钠200ml调至PH=12-13,搅拌溶解。再加入乙酸乙酯100ml搅拌静置分层,弃去下层碱水液,得到上层180ml乙酸乙酯及α-甲胺基苯丙酮混合液体备用。另外在1000ml反应器中取拆分剂(2R,3S)-(-)二苯甲酰基酒石酸[(2R,3S)-(-)DBT A.H2O]95g加入乙酸乙酯100ml,溶解,再缓慢加入到“乙酸乙酯及α-甲胺基苯丙酮混合液体”中,滴加过程中产生大量沉淀,滴加完毕后再加入无水甲醇20ml使溶液澄清,后禁止搅拌,放置使自然结晶24小时以上。离心分离,母液待处理套用,固体用乙酸乙酯∶甲醇=10∶1溶液洗涤至产品色泽变无色或暗红色。所得固体为(2R,3S)-(-)二苯甲酰基酒石酸(S)-(-)-α-甲胺基苯丙酮的盐(简称:中间体1号)。重量196g,于5倍量纯水中加入0.55mol还原剂硼氢化钾,还原,后加盐酸酸化,乙酸丁酯萃取分离提取拆分剂二苯甲酰基酒石酸,酸水层加40%氢氧化钠游离甲苯萃取,萃取液加盐酸酸化结晶制备L-(-)盐酸麻黄碱一次收率为31.05%;
3.将L-(-)盐酸麻黄碱100g,加入乙酸酐160mL进行乙酰化,反应温度控制在110-130℃之间回流6h;冷却后,加入浓盐酸90mL,回流8h;冷却,加入蒸馏水180mL,回流水解12小时后停止反应。浓缩除去乙酸溶液,得到右旋麻黄碱盐酸盐的粗品。加40%氢氧化钠游离后酸化处理精制得到D-(-)-盐酸伪麻黄碱成品,一次收率为71%转化率为90%以上。母液套用。
Claims (8)
1.一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于包括如下步骤:以α-溴代苯丙酮或α-氯代苯丙酮为起始原料,经甲胺化反应、拆分反应、还原反应、转旋反应生产盐酸伪麻黄碱。
2.根据权利要求1所述的一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,以α-溴代苯丙酮或α-氯代苯丙酮为起始原料,其甲胺化反应由以下步骤:
(1)使用的原辅料按照以下重量百分比(或质量分数)配比:
以投入起始原料α-溴代苯丙酮,含量≥96%,分子量213.07g,反应过程中,α-溴代苯丙酮mol数或重量与投入其他原辅料的mol比例分别为:
α-溴代苯丙酮∶甲胺mol比=1∶1
α-溴代苯丙酮∶甲胺水溶液(含量≥30%)重量比=1∶1~1.1
α-溴代苯丙酮∶反应溶剂甲苯(含量≥96%)重量比=1∶5~5.5
α-溴代苯丙酮∶碱化游离用氢氧化钠(含量≥95%)重量比=1∶0.2~0.25
α-溴代苯丙酮∶酸化中和用盐酸(含量≥30%)重量比=1∶2.5~3
α-溴代苯丙酮∶结晶溶剂丙酮(含量≥95%)重量比=1∶2.5~3
(2)反应主要参数:
反应温度为40℃~45℃,搅拌速度为60转/分~70转/分。
3.根据权利要求1所述的一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,拆分反应由以下步骤:
(1)使用(2R,3S)-(-)二苯甲酰基酒石酸[(2R,3S)-(-).H2O]作为拆分剂,其溶解采用乙酸乙酯作为溶解试剂,其重量比为1∶0.7~0.9;
(2)拆分剂的拆分目标是上一步甲胺化反应得物α-甲胺基苯丙酮盐酸盐,α-甲胺基苯丙酮盐酸盐(含量≥98%)与拆分剂(含量≥99%)的投料mol比为1∶0.5~0.55,重量比(含结晶水的重量)为:1∶1.06~1.1;
(3)拆分反应过程中,乙酸乙酯与甲醇的比例调整至7~10∶1。
4.根据权利要求1所述的一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,还原反应由以下步骤:
(1)使用硼氢化钾KBH4作为还原剂,使用二级反渗透纯化水作为反应溶剂;
(2)上步拆分反应得物溶解时,加入二级反渗透纯化水4~6倍水;
(3)上步拆分反应得物与还原剂硼氢化钾的mol比为1∶1.0~1.05,其重量比为1∶0.1~0.13;
(4)还原反应温度控制在25℃以内。
5.根据权利要求1-2所述的一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,所述用甲胺化、拆分、还原、酰化,酸解、水解法制备L-麻黄碱和d一伪麻黄碱方法,其特征是步骤(1)中所说的原料是溴代苯丙酮或氯代苯丙酮。
6.根据权利要求1所述的一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,转旋反应由以下步骤:
(1)根据以上步骤所制得的L-(-)盐酸麻黄碱粗品,其干燥失重≤0.5%,旋光度在33~36.5时,该粗品可直接投入转旋反应,如干燥失重超过0.5%,可对粗品进行再次烘干处理,直到达标。
(2)转旋反应时,盐酸麻黄碱粗品与加入乙酸酐的的摩尔比理论量为1∶0.5~0.6,理论重量比为:1∶0.39~0.42,实际保障转旋率的投料比例为:mol比例:1∶4.6~6,重量比:1∶1.8~2.4;
(3)转旋反应时,反应温度控制在110℃~120℃,反应时间控制在4~8小时。
7.根据权利要求1-2所述的L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,反应溶剂或助剂为:
1)试剂为:醇类试剂,酯类试剂,醚类试剂,酸酐类试剂等;
2)所用碱为:NaOH,KOH;
3)所用酸为:HCl,H2SO4;
4)脱水剂为:无水硫酸钠、无水硫酸镁;
5)所用溶剂为:甲苯、二甲苯、丙酮等有机溶剂。
8.根据权利要求7所述的L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法,其特征在于,所述醇类试剂:甲醇、乙醇,酯类试剂:乙酸乙酯、乙酸丁酯,醚类试剂:乙醚,酸酐类试剂:乙酸酐。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201010166768 CN101870660A (zh) | 2010-05-10 | 2010-05-10 | 一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201010166768 CN101870660A (zh) | 2010-05-10 | 2010-05-10 | 一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101870660A true CN101870660A (zh) | 2010-10-27 |
Family
ID=42995763
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 201010166768 Pending CN101870660A (zh) | 2010-05-10 | 2010-05-10 | 一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101870660A (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104119240A (zh) * | 2013-04-23 | 2014-10-29 | 中国人民解放军军事医学科学院毒物药物研究所 | (S)-(-)-α-甲胺基苯丙酮的制备方法 |
| CN106008183A (zh) * | 2016-06-07 | 2016-10-12 | 浙江普洛康裕制药有限公司 | 麻黄碱或伪麻黄碱及麻黄碱或伪麻黄碱中间体的制备方法 |
| CN106957234A (zh) * | 2017-03-23 | 2017-07-18 | 陕西金冠牧业有限公司 | 一种盐酸伪麻黄碱的制备方法 |
| CN108473418A (zh) * | 2016-01-04 | 2018-08-31 | 株式会社爱茉莉太平洋 | 使用极性非质子溶剂的n-[4-(1-氨基乙基)苯基]磺酰胺衍生物的手性拆分方法 |
| CN108623485A (zh) * | 2018-06-18 | 2018-10-09 | 东莞市联洲知识产权运营管理有限公司 | 一种高纯盐酸d-伪麻黄碱微晶体的制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1583714A (zh) * | 2004-06-02 | 2005-02-23 | 上海医药工业研究院 | [(S)-(-)-α-甲胺基苯丙酮] 2·(2R,3R)-酒石酸衍生物的制备方法 |
| CN1673211A (zh) * | 2004-03-24 | 2005-09-28 | 新疆大学 | 盐酸L-麻黄碱异构化为盐酸d-伪麻黄碱的新工艺 |
| CN1706812A (zh) * | 2004-06-07 | 2005-12-14 | 上海医药工业研究院 | (1r,2s)-(-)-麻黄碱或其盐酸盐的制备方法 |
| CN101570492A (zh) * | 2009-06-08 | 2009-11-04 | 新疆大学 | 一种化学合成麻黄素的方法 |
-
2010
- 2010-05-10 CN CN 201010166768 patent/CN101870660A/zh active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1673211A (zh) * | 2004-03-24 | 2005-09-28 | 新疆大学 | 盐酸L-麻黄碱异构化为盐酸d-伪麻黄碱的新工艺 |
| CN1583714A (zh) * | 2004-06-02 | 2005-02-23 | 上海医药工业研究院 | [(S)-(-)-α-甲胺基苯丙酮] 2·(2R,3R)-酒石酸衍生物的制备方法 |
| CN1706812A (zh) * | 2004-06-07 | 2005-12-14 | 上海医药工业研究院 | (1r,2s)-(-)-麻黄碱或其盐酸盐的制备方法 |
| CN101570492A (zh) * | 2009-06-08 | 2009-11-04 | 新疆大学 | 一种化学合成麻黄素的方法 |
Non-Patent Citations (2)
| Title |
|---|
| 《JOURNAL OF PHARMACEUTICAL SCIENCES》 19810630 JOHN W. A FINDLAY STEREOSPECIFIC RADIOIMMUNOASSAYS FOR D-PSEUDOEPHEDRINE IN HUMAN PLASMA AND THEIR APPLICATION TO BIOEQUIVALENCE STUDIES 624-631 第70卷, 第6期 2 * |
| 《中国医药工业杂志》 20001231 孙同庆 dl-麻黄碱和dl-伪麻黄碱的合成 534-535 , 第12期 2 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104119240A (zh) * | 2013-04-23 | 2014-10-29 | 中国人民解放军军事医学科学院毒物药物研究所 | (S)-(-)-α-甲胺基苯丙酮的制备方法 |
| CN108473418A (zh) * | 2016-01-04 | 2018-08-31 | 株式会社爱茉莉太平洋 | 使用极性非质子溶剂的n-[4-(1-氨基乙基)苯基]磺酰胺衍生物的手性拆分方法 |
| CN106008183A (zh) * | 2016-06-07 | 2016-10-12 | 浙江普洛康裕制药有限公司 | 麻黄碱或伪麻黄碱及麻黄碱或伪麻黄碱中间体的制备方法 |
| CN106008183B (zh) * | 2016-06-07 | 2019-05-07 | 浙江普洛康裕制药有限公司 | 麻黄碱或伪麻黄碱及麻黄碱或伪麻黄碱中间体的制备方法 |
| CN106957234A (zh) * | 2017-03-23 | 2017-07-18 | 陕西金冠牧业有限公司 | 一种盐酸伪麻黄碱的制备方法 |
| CN108623485A (zh) * | 2018-06-18 | 2018-10-09 | 东莞市联洲知识产权运营管理有限公司 | 一种高纯盐酸d-伪麻黄碱微晶体的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101870660A (zh) | 一种L-(-)-盐酸麻黄碱和d-(+)-盐酸伪麻黄碱制备方法 | |
| CN100402488C (zh) | 一种达泊西汀的合成方法 | |
| CN103739504B (zh) | 一种重酒石酸间羟胺的合成方法 | |
| CN1164588C (zh) | 黄连素及其盐类的制备方法 | |
| CN102503840A (zh) | 一种地佐辛的制备方法 | |
| CN102267957B (zh) | 非布司他a晶型的制备方法 | |
| CN108059601A (zh) | 一种制备3-氨基-1-金刚烷醇的工艺 | |
| CN114573500A (zh) | 一种雷芬那辛中间体的制备方法 | |
| CN102229538B (zh) | 一种达泊西汀的合成方法 | |
| CN104478871B (zh) | 一种胆碱m受体拮抗剂阿地溴铵及其制备方法 | |
| CN101643439A (zh) | 一种乙基甲基胺甲酰氯的制备方法 | |
| CN108395437A (zh) | 氘代化合物及其医药用途 | |
| CN101570492B (zh) | 一种化学合成麻黄素的方法 | |
| CN103288630A (zh) | 一种丹参素钠的合成方法 | |
| CN110105195A (zh) | 一种从青蒿蜡油中提取二氢青蒿酸的方法 | |
| CN112830881B (zh) | 从橙皮苷废液中分离辛弗林的方法 | |
| CN102838579A (zh) | 一种制备1,3,6,7-四羟基双苯吡酮的方法 | |
| CN106632038B (zh) | 一种八氢异喹啉的拆分方法 | |
| CN101898974A (zh) | 乙二醇-双-(2-氨基乙醚)四乙酸的生产方法 | |
| CN103551191B (zh) | 一种手性两亲嵌段共聚物在手性合成中的应用 | |
| CN102079691A (zh) | 采用一种组合羟基保护基法合成反式白藜芦醇 | |
| CN114163415B (zh) | 一种盐酸度洛西汀中间体的制备方法 | |
| CN103992272B (zh) | 一种盐酸喷他佐辛酯、其制备方法及其用途 | |
| CN112110865A (zh) | 一种异烟酰胺阿西莫司共晶体ⅱ及其制备方法 | |
| CN112409195A (zh) | 一种(s)-氯胺酮盐酸盐的制备方法及其中间体、其晶型 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20101027 |