Estala 24eta “SAIS E POLIMORFOS DE UM COMPOSTO DE TETRACICLINA”
Pedido Relacionado
Este pedido reivindica o benefício de prioridade sob 35 U.S.C. 119(e) ao Pedido Provisório U.S. pendente No. 61/128.712, depositado em 23 de maio de 2008, os conteúdos dos quais estão incorporados aqui por referência.
Antecedente da Invenção
O desenvolvimento dos antibióticos de tetraciclina foi o resultado direto de uma avaliação sistemática de espécimes de solo coletadas a partir de muitas partes do mundo para evidência de microorganismos capazes de produzir composições bactericidas e/ou bacteriostáticas. O primeiro destes novos compostos foi introduzido em 1948 sob o nome clortetraciclina. Dois anos depois, oxitetraciclina tornou-se disponível. O esclarecimento da estrutura química destes compostos confirmou a similaridade e forneceu a base analítica para a produção de um terceiro membro deste grupo em 1952, tetraciclina. Uma nova família de compostos de tetraciclina, sem o grupo metila ligado a anel presente em tetraciclinas precoces, foi preparada em 1957 e tornou-se disponível publicamente em 1967; e minociclina estava em uso antes de 1972.
Recentemente, esforços de pesquisa focalizaram em desenvolver novas composições antibióticas de tetraciclina eficazes sob condições terapêuticas variadas e rotinas de administração. Novos análogos de tetraciclina foram da mesma forma investigados que pode provar ser igual a ou mais eficazes do que os compostos de tetraciclina originalmente introduzidos. Exemplos incluem Patente U.S. Nos. 2.980.584; 2.990.331; 3.062.717; 3.165.531; 3.454.697; 3.557.280; 3.674.859; 3.957.980; 4.018.889; 4.024.272; e 4.126.680. Estas patentes são representativas da faixa de tetraciclina farmaceuticamente ativa e composições de análogo de tetraciclina.
Historicamente, em seguida seu desenvolvimento inicial e introdução, as tetraciclinas foram constatadas ser altamente farmacologicamente eficaz contra rickettsiae; várias bactérias gram-positivas e gram-negativas; e os agentes responsáveis por linfogranuloma venéreo, conjuntivite de inclusão, e psitacose. Portanto, as tetraciclinas tornaram-se conhecidas como antibióticos de amplo espectro. Com o estabelecimento subsequente de sua atividade antimicrobiana in vitro, a efetividade em infecções experimentais, e propriedades farmacológicas, as tetraciclinas como uma classe rapidamente tornaram-se amplamente usadas para propósitos terapêuticos. Entretanto, este uso difundido de tetraciclinas tanto para doenças principais quanto secundárias e doenças levou diretamente ao aparecimento de resistência a estes antibióticos ainda entre as espécies bacterianas altamente suscetíveis tanto comensais quanto patogênicas (por exemplo, pneumocócicas e Salmonella). A elevação de organismos resistentes à tetraciclina resultou em um declínio geral em uso de tetraciclinas e composições de análogo de tetraciclina como o antibiótico de escolha.
Cada composto farmacêutico tem uma concentração de sangue terapêutica ideal e uma concentração letal. A biodisponibilidade do composto determina a força de dosagem na formulação de fármaco necessária para obter o nível de sangue ideal. Se o fármaco pode cristalizar-se como dois ou mais polimorfos que diferem-se na biodisponibilidade, a dose ideal dependerá do polimorfo presente na formulação. Alguns fármacos mostram uma margem estreita entre concentrações terapêuticas e letais. Cloranfenicol-3-palmitato (CAPP), por exemplo, é um antibiótico de amplo espectro conhecido por cristalizar em pelo menos três formas polimórficas e uma forma amorfa. A forma mais estável, A, é comercializada. A diferença em bioatividade entre este polimorfo e outra forma, B, é um fator de oito, desse modo criando a possibilidade de superdosagens fatais do composto se não intencionalmente administrado como a forma B devido às alterações durante o processamento e/ou armazenamento. Portanto, agências reguladoras, tal como a United State Food e Drug Administration, começaram a colocar controles rigorosos no conteúdo polimórfico do componente ativo em formas de dosagem sólidas. Em geral, para fármacos que existem em formas polimórficas, se qualquer diferente do puro, polimorfo termodinamicamente preferido será comercializado, a agência reguladora pode requerer o monitoramento batelada por batelada. Desse modo, torna-se importante igualmente por razões médicas e comerciais produzir e comercializar o fármaco puro em seu polimorfo mais termodinamicamente estável, substancialmente livre de outros polimorfos cineticamente favorecidos.
Por exemplo, formas de sal de um composto, e formas polimórficas do composto livre ou sal, são conhecidas na técnica farmacêutica para afetar, por exemplo, a solubilidade, taxa de dissolução, biodisponibilidade, estabilidade química e física, fluidez, fractabilidade, e capacidade de compressão do composto bem como a segurança e eficácia de produtos de fármaco com base no composto (veja, por exemplo, Knapman, Modem Drug Dicovery, 2000, 3(2): 53).
Desta maneira, a identificação de uma forma de sal ou base livre de um composto com propriedades físicas e químicas ideais avançará o desenvolvimento de compostos de tetraciclina como farmacêuticos. As mais úteis de tais propriedades físicas e químicas incluem: preparação fácil e reprodutível, cristalinidade, não higroscopicidade, solubilidade aquosa, estabilidade para luz visível e ultravioleta, baixa taxa de degradação sob condições de estabilidade aceleradas de temperatura e umidade, baixa taxa de isomerização entre formas isoméricas, e segurança para administração à longo prazo a humanos.
Sumário da Invenção
Em uma modalidade, a invenção pertence, pelo menos em parte, a uma forma de estado sólida estável, tal como uma forma cristalina, do composto de aminoalquil tetraciclina, Composto 1:
(4S,4AS,5AR, 12 AS)-4-7-Bis(dimetilamino)-9 {[(2,2-dimetilpropil)amino]metil}3,10,12,12A-tetraidróxi-1,11-dioxo-1,4,4A,5,5A,6,11,12A-octaidrotetraceno-2-carboxamida (9-(2,2-dimetil-propil-aminometil)-minociclina).
Em outra modalidade, a invenção pertence, pelo menos em parte, a um sal de HCI do Composto 1. Em outra modalidade, a invenção pertence, pelo menos em parte, a um sal de tosilato (p-toluenossulfonato) do Composto 1. Em outra modalidade, a invenção pertence, pelo menos em parte, para um sal de mesilato do Composto 1
Em outra modalidade, a invenção pertence, pelo menos em parte, a uma forma cristalina estável do Composto 1.
Em outra modalidade, a invenção pertence, pelo menos em parte, a uma forma cristalina estável de um sal do Composto 1. Por exemplo, a forma cristalina estável de um sal é uma forma cristalina estável de um sal de tosilato, HCI ou mesilato do Composto 1.
Em outra modalidade, a invenção pertence, pelo menos em parte, a um polimorfo do Composto 1.
Em outra modalidade, a invenção pertence, pelo menos em parte, a um polimorfo de um sal do Composto 1.
Por exemplo, a invenção refere-se a um polimorfo de um sal de tosilato do Composto 1. A invenção refere-se, em parte a um polimorfo da forma 1 do Composto 1. A invenção refere-se, em parte a uma polimorfo da forma 2 do Composto 1. A invenção refere-se, em parte a um polimorfo da forma 3 do Composto 1.
Por exemplo, um polimorfo da forma 1 de um sal de tosilato do Composto 1 tem picos de difração de pó de Raios X em aproximadamente 8,06, 13,02, e 18,83 °2Θ usando radiação de Cu Ka. Em algumas modalidades, o polimorfo da forma 1 de um sal de tosilato do Composto 1 tem picos de difração de pó de Raios X em aproximadamente 8,06, 11,41, 13,02, 18,83, 20,54, e 24,53 °20 usando radiação de Cu Ka. Em algumas modalidades, o polimorfo da forma 1 de um sal de tosilato do Composto 1 tem picos de difração de pó de Raios X em aproximadamente 5,60, 8,06, 8,57, 11,41, 13,02, 15,58, 18,83, 20,54, e 24,53 °20 usando radiação de Cu Ka.
Por exemplo, um polimorfo da forma 1 de um sal de tosilato do Composto 1 é estável a temperatura em uma faixa de aproximadamente 0°C a cerca de 70°C. Em algumas modalidades, o polimorfo da forma 1 de um sal de tosilato do Composto 1 é tabela em temperatura em uma faixa de cerca de 5°C a cerca de 50°C. Em algumas modalidades, o poli morfo da forma 1 de urn sal de tosilato do Composto 1 é tabela em temperatura em uma faixa de cerca de 20°C a cerca de 30°C.
O polimorfo da forma 1 de um sal de tosilato do Composto 1 pode ser obtido cristalizando-se o sal de tosilato do referido Composto 1 a partir de isopropanol.
Por exemplo, um polimorfo da forma 2 de um sal de tosilato do Composto 1 tem picos de padrão de difração de pó de Raios X a 7,82, 11,88, 16,12 e 21,46 °2Θ usando radiação de Cu Ka.
Por exemplo, um polimorfo da forma 3 de um sal de tosilato do Composto 1 tem picos de padrão de difração de pó de Raios X a 5,11, 8,89, 10,34, 11,76 e 15,60 °2Θ usando radiação de Cu Ka.
Em ainda outra modalidade, a invenção inclui composições farmacêuticas compreendendo uma forma cristalina do Composto 1 e um diluente farmaceuticamente aceitável, excipiente ou veículo.
Por exemplo, a composição de composto farmacêutico da invenção inclui uma composição compreendendo um polimorfo do Composto 1 e um diluente farmaceuticamente aceitável, excipiente ou veículo.
Em outra modalidade, o composto de composição farmacêutica da invenção inclui um sal do Composto 1 e um diluente farmaceuticamente aceitável, excipiente ou veículo. Por exemplo, o sal pode ser um sal de HCI, um sal de tosilato, ou um sal de mesilato.
Em uma modalidade, o composto de composição farmacêutica da invenção inclui um polimorfo de um sal do Composto 1 e um diluente farmaceuticamente aceitável, excipiente ou veículo. Por exemplo, o polimorfo pode ser um polimorfo de sal de tosilato, sal de HCI, ou sal de mesilato do Composto 1.
Em algumas modalidades, a composição farmacêutica compreende um polimorfo do Composto 1, ou sal do mesmo em uma forma pura.
Em outra modalidade, a composição farmacêutica da invenção inclui um polimorfo do sal de tosilato do Composto 1 e um diluente farmaceuticamente aceitável, excipiente ou veículo. Por exemplo, o polimorfo pode ser um polimorfo da forma 1, uma forma 2, ou uma forma 3 de sal de tosilato do Composto 1.
Em algumas modalidades, a composição farmacêutica compreende um polimorfo de sal de tosilato, sal de HCI, ou sal de mesilato do Composto 1 em uma forma pura.
Em outro aspecto da invenção, o sal do Composto 1 é mais estável do que a base livre do Composto 1.
Em outra modalidade, a invenção inclui um método para preparar uma forma cristalina estável do Composto 1.
Em outra modalidade, a invenção inclui um método para preparar uma forma cristalina estável de um sal do Composto 1. Por exemplo, a cristalina estável pode ser uma crista lina de um sal de tosilato, HCI, ou mesilato do Composto 1.
Em outra modalidade, a invenção inclui um método para preparar um polimorfo de um sal do Composto 1. Por exemplo, o polimorfo pode ser um polimorfo de um sal de tosilato, HCI, ou mesilato do Composto 1.
Em outra modalidade, a invenção inclui um método para preparar um polimorfo de um sal de tosilato do Composto 1. Por exemplo, o polimorfo pode ser um polimorfo da forma 1, um da forma 2, ou um da forma 3 de um sal de tosilato do Composto 1.
Em uma modalidade, a invenção inclui um método para preparar um polimorfo da forma 1 a partir de um sal de tosilato do Composto 1, em que o método compreende: combinar o Composto 1 com um solvente para produzir uma lama; e adicionar ácido ptoluenossulfônico. Por exemplo, o solvente pode ser um solvente alcoólico, tal como isopropanol. O ácido p-toluenossulfônico é fornecido em uma quantidade dentre 25 a 75% em peso em relação à quantidade do referido Composto 1, por exemplo, de 25 a 50% em peso, de 30 a 40% em peso, ou 33% em peso em relação à quantidade do referido Composto 1. Por exemplo, o ácido p-toluenossulfônico é fornecido em uma forma de monoidrato de ácido ptoluenossulfônico.
Por exemplo, a lama é aquecida antes da adição de ácido p-toluenossulfônico.
Por exemplo, a lama é agitada depois da adição de ácido p-toluenossulfônico. Por exemplo, a agitação é conduzida em uma temperatura em uma faixa de 20 a 25°C. Por exemplo, a agitação é conduzida durante 10 a 24 horas.
Por exemplo, a lama é secada. Por exemplo, o teor de água do sobrenadante da referida lama está em uma faixa de 0,2 a 1,0 mg/mL, ou em uma faixa de 0,4 a 0,8 mg/mL.
Em ainda outra modalidade, a invenção inclui um método para preparar um polimorfo da forma 1 a partir de um sal de tosilato do Composto 1, em que o método compreende: preparar uma solução do Composto 1 em um solvente ou uma combinação de solventes; e adicionar uma solução de ácido p-toluenossulfônico em um solvente ou uma combinação de solventes.
Por exemplo, o solvente é um solvente alcoólico, tal como metanol, etanol, ou isopropanol. Por exemplo, a combinação de solventes inclui um solvente alcoólico. Por exemplo, a combinação de solventes também compreende um segundo solvente alcoólico. Por exemplo, a combinação de solventes inclui etanol e isopropanol. Por exemplo, a combinação de solventes também inclui um antissolvente, tal como cetona, éter, e éster. Por exemplo, o éter inclui, porém não é limitado a, éter metil-t-butílico. Por exemplo, a combinação de solventes inclui um solvente alcoólico e um antissolvente. Por exemplo, a combinação de solventes inclui metanol e éter metil-t-butílico.
Por exemplo, ácido p-toluenossulfônico é fornecido em uma quantidade dentre 25 a 75% em peso, de 30 a 50% em peso, de 35 a 45% em peso, ou 40% em peso em relação à quantidade do referido Composto 1. Por exemplo, ácido p-toluenossulfônico é fornecido em uma forma de um monoidrato de ácido p-toluenossulfônico.
Por exemplo, a solução é preparada em uma temperatura em uma faixa de 0 a 60°C , em uma temperatura em uma faixa de 15 a 45°C, ou em uma temperatura em uma faixa de 20 a 25°C.
Por exemplo, a solução é aquecida depois de ser preparada. Por exemplo, a solução é mantida em uma temperatura em uma faixa de 20 a 50°C, ou a cerca de 45°C.
Por exemplo, o método também compreende adicionar um cristal de semente a partir de sal de monotosilato do Composto 1 para produzir uma lama. A lama pode ser agitada durante 10 a 24 horas ou durante cerca de 22 horas. A lama pode ser agitada em uma temperatura em uma faixa de 15 a 45°C ou a cerca de 20°C. A lama pode ser secada. Por exemplo, o teor de água da lama está em uma faixa de 1 a 10% em peso, ou em uma faixa de 2 a 6% em peso, ou cerca de 3% em peso.
Em outra modalidade, a invenção inclui um método para preparar um polimorfo da forma 1 a partir de um sal de tosilato do Composto 1, em que o método compreende: dissolver uma base livre do Composto 1 em um primeiro solvente ou combinação de solventes para formar uma primeira solução; dissolvendo ácido p-toluenossulfônico em um segundo solvente ou combinação de solventes para formar uma segunda solução; e combinar a referida primeira e segunda soluções para formar uma terceira solução.
Em uma modalidade, o primeiro e segundo solvente ou combinação de solventes podem ser os mesmos ou diferentes. Em outra modalidade, o solvente pode ser um solvente alcoólico, tal como metanol, etanol, e isopropanol. Em outra modalidade, a combinação de solventes é uma combinação de dois solventes alcoólicos, incluindo, porém não limitado a etanol e isopropanol. Em um exemplo preferido, a relação de volume para volume de etanol e isopropanol é 2 para 1. Em ainda outra modalidade, a combinação de solventes é uma combinação que inclui, porém não é limitada a, um solvente alcoólico e um antissolvente (por exemplo, uma cetona, um éter, um éster, etc.). Por exemplo, a combinação de solventes é uma combinação que inclui, porém não é limitada a, metanol e éter metil-t-butílico. Em um exemplo preferido, a relação de volume para volume de metanol e éter metil-t-butílico é 1 para 1,2.
Em outra modalidade, o método também compreende adicionar um sal de tosilato do polimorfo da forma 1 do Composto 1 à terceira solução para formar uma quarta solução. Por exemplo, a sal de tosilato do polimorfo da forma 1 é um cristal de semente. Em algumas modalidades, a quarta solução forma uma lama sob agitação. A lama pode ser lavada com um solvente ou uma combinação de solventes, que podem ser os mesmos ou diferentes do primeiro solvente ou combinação de solvente, ou o segundo solvente ou combinação de solventes. A lama pode ser secada.
Em outra modalidade, a invenção refere-se a uma composição pura compreendendo o Composto 1, onde na composição é cerca de 90-100%, preferivelmente 95-100%, mais preferivelmente 98-100% (p / p) ou 99-100% (p / p) puro; por exemplo, menos do que cerca de 10%, menos do que cerca de 5%, menos do que cerca de 2% ou menos do que cerca de 1% de impureza está presente. Tais impurezas incluem, por exemplo, produtos de degradação, produtos oxidados, epímeros, solventes, e/ou outras impurezas indesejáveis.
Em ainda outra modalidade, a invenção inclui um método para tratar um estado responsivo de tetraciclina em um indivíduo, administrando ao indivíduo uma quantidade eficaz de uma forma cristalina do Composto 1. Por exemplo, o indivíduo é um indivíduo humano.
Em contudo outra modalidade, a invenção inclui um método para tratar um estado responsivo de tetraciclina em um indivíduo, administrando ao indivíduo uma quantidade eficaz de um sal estável do Composto 1. Por exemplo, o sal estável é um sal de tosilato, um de HCI, ou um de mesilato do Composto 1.
Em ainda outra modalidade, a invenção inclui um método para tratar um estado responsivo de tetraciclina em um indivíduo, administrando ao indivíduo uma quantidade eficaz de um polimorfo do Composto 1.
Em ainda outra modalidade, a invenção inclui um método para tratar um estado responsivo de tetraciclina em um indivíduo, administrando ao indivíduo uma quantidade eficaz de um polimorfo de sal do Composto 1. Por exemplo, o polimorfo pode ser um polimorfo de um sal de tosilato, um de HCI, ou um de mesilato do Composto 1.
Em ainda outra modalidade, a invenção inclui um método para tratar um estado responsivo de tetraciclina em um indivíduo, administrando ao indivíduo uma quantidade eficaz de um polimorfo de sal de tosilato do Composto 1. Por exemplo, o polimorfo de tosilato pode ser um polimorfo da forma 1, um da forma 2, ou um da forma 3 de sal de tosilato do Composto 1.
Por exemplo, o estado responsivo de tetraciclina é uma infecção bacteriana. A infecção bacteriana pode estar associada com bactérias gram-positivas, ou bactérias gramnegativas. Em algumas modalidades, a infecção bacteriana está associada com E. coli, S. aureus, ou E. faecalis.
Em algumas modalidades, a infecção bacteriana é resistente a outros antibióticos de tetraciclina, que incluem porém não são limitados a, tetraciclina, minociclina, doxiciclina, sanciclina, clortetraciclina, demeclociclina, oxitetraciclina, celocardina, rolitetraciclina, limeciclina, metaciclina, apiciclina, clomociclina, pipaciclina, mepilciclina, megluciclina, guameciclina, penimociclina, e etamociclina.
Breve Descrição dos Desenhos
A FIG. 1 fornece um padrão de difração de pós de Raios X de uma amostra com preendendo Composto cristalino 1 a 25°C;
A FIG. 2 fornece um padrão de difração de pós de Raios X a 25°C do material de partida (E00285), forma 1 sal de tosilato, forma 2 sal de tosilato, forma 3 sal de tosilato, e a forma de sal de tosilato amorfo do Composto 1;
A FIG. 3 fornece uma comparação de padrões de difração de pós de Raios X a 25°C para Composto cristalino 1 (E00285) e uma amostra compreendendo o sal de tosilato da forma 1, obtido a partir recristalização de sal de tosilato amorfo do Composto 1 em IPA;
A FIG. 4 fornece uma comparação de padrões de difração de pós de Raios X a 25°C para amostras compreendendo sal de tosilato da forma 1, sal de tosilato da forma 2, e sal de tosilato da forma 3 que foram secados durante a noite em um vácuo;
A FIG. 5 fornece uma análise de difração de pó de Raios X de temperatura variável a partir de amostras compreendendo sal de tosilato da forma 2;
A Fig. 6 fornece uma análise de difração de pó de Raios X de temperatura variável a partir de amostras compreendendo o sal de tosilato da forma 3;
A FIG. 7 fornece uma análise de difração de pó de Raios X de temperatura variável de amostras compreendendo uma lama de uma mistura de 50:50 de sal de tosilato da forma 1 e sal de tosilato da forma 3 em IPA;
A FIG. 8 fornece um padrão de difração de pós de Raios X de alta resolução de uma amostra compreendendo sal de tosilato da forma 1 do Composto 1;
A FIG. 9 fornece um padrão de difração de pós de Raios X de alta resolução de uma amostra compreendendo sal de tosilato da forma 2 do Composto 1 (93,2% de pureza de HPLC);
A FIG. 10 fornece um padrão de difração de pós de Raios X de alta resolução de uma amostra compreendendo sal de tosilato da forma 3 do Composto 1 (96,7% de pureza de HPLC).
Descrição Detalhada da Invenção
Compostos antibióticos de tetraciclina foram por muito tempo conhecidos por ter estabilidade limitada na forma de base livre de fase sólida. Um composto análogo de tal tetraciclina não cristalina, (4S,4AS,5AR,12AS)-4-7-Bis(dimetilamino)-9-{[(2,2dimetilpropil)amino]metil}-3,10,12,12A-tetraidróxi-1,11 -dioxo-1,4,4a,5,5a,6,11,12Aoctaidrotetraceno-2-carboxamida (Composto 1; MW = 556,66, MF = C29H40N4O7), tem limitado a estabilidade na fase sólida sob exposição a ar, luz e/ou umidade.
(4S,4AS,5AR,12AS)-4-7-Bis(dimetilamino)-9{[(2,2-dimetilpropil)amino]metil}3,10,12,12A-tetraidróxi-1,11-dioxo-1,4,4A,5,5A,6,11,12A-octaidrotetraceno-2-carboxamida.
Especificamente, o Composto 1 é um sólido amorfo amarelo que é instável em temperaturas mais altas do que 0°C e quando expostas a ar. O Composto 1 deve ser armazenado em temperaturas abaixo de 0°C com exposição limitada na fase sólida a ar, luz e umidade. Fora destas condições de exposição limitadas, o Composto 1 degrada para produzir produtos de degradação incluindo produtos de degradação a ar 2, 3 e 4, bem como o 4epi-isômero 5.
Antes desta descrição, nenhuma forma cristalina estável ou sais de ácido cristalino estáveis do Composto 1 foi conhecida.
A presente invenção refere-se a Composto cristalino 1, formas de sal do Composto 1, formas polimórficas do Composto 1 ou formas polimórficas de sais do Composto 1; composições farmacêuticas compreendendo as formas cristalinas, formas de sal, formas polimórficas, ou formas polimórficas de sais do Composto 1; métodos de preparar as formas cristalinas, formas de sal, formas polimórficas, ou formas polimórficas de sais do Composto 1; e métodos de seu uso para o tratamento de estados responsivos por tetraciclina.
1. Compostos de Forma Sólida
O Composto 1 é um composto de tetraciclina. O termo “composto de tetraciclina” inclui muitos compostos com uma estrutura de anel similar a tetraciclina. Exemplos de compostos de tetraciclina incluem: tetraciclina, clortetraciclina, oxitetraciclina, demeclociclina, metaciclina, sanciclina, doxiciclina, e minociclina.
A base livre e certos sais farmaceuticamente aceitáveis do Composto 1 são descritos no pedido U.S. No. de Série 10/786.881, correspondendo a Publicação U.S. No. 2005/0026876 A1. Não há ensino ou sugestão de formas cristalinas do Composto 1, ou que quaisquer das formas de sal descritas são superiores às outras, como julgado pela lista de propriedades descritas acima.
Desse modo, a presente invenção refere-se a necessidade por compostos de tetra ciclina melhorados e a necessidade para formas de estado sólido melhoradas de compostos de tetraciclina para fabricação e biodisponibilidade.
A forma de estado sólido do composto de tetraciclina, Composto 1, pode estar em uma forma cristalina. A forma cristalina do composto pode ser uma base livre. Formas cristalinas de sais diferentes do composto de base livre podem ser formadas. Exemplos de ácidos que podem ser usados para converter a base livre em um sal incluem, porém não são limitados a, HCI, ácido p-toluenossulfônico, ácido trifluoroacético, ácido metilsulfônico, ácido benzenolsulfônico, e ácido acético.
As formas neutras dos compostos podem ser regeneradas contatando-se o sal com uma base ou ácido e isolando-se o composto origem da maneira convencional. A forma origem do composto pode diferir das várias formas de sal em certas propriedades físicas, tal como solubilidade em solventes polares.
Como descrito aqui, um processo pelo qual as formas cristalinas diferentes do Composto 1 podem ser geradas foi desenvolvido. Mais especificamente, os inventores mostraram que a forma cristalina obtida principalmente depende da natureza do solvente usado no processo. Para propósitos desta descrição o termo “forma cristalina” refere-se a uma forma polimórfica ou uma forma não amorfa, sem distinção. “Forma polimórfica” refere-se a uma estrutura organizada envolvendo apenas moléculas do soluto e tendo uma assinatura cristalina característica.
Os termos, “polimorfos” e “formas polimórficas” e termos relacionados aqui referemse a formas cristalinas da mesma molécula, e polimorfos diferentes podem ter propriedades físicas diferentes tal como, por exemplo, temperaturas de fusão, calores de fusão, solubilidades, taxa de dissolução e/ou espectros vibracionais como um resultado da disposição ou conformação das moléculas na treliça cristalina. As diferenças em propriedades físicas exibidas por polimorfos afetam parâmetros farmacêuticos tais como estabilidade de armazenamento, capacidade de compressão e densidade (importante na formulação e fabricação do produto), e taxa de dissolução (um fator importante em biodisponibilidade). Diferenças em estabilidade podem da mesma forma resultar em mudanças em reatividade química (por exemplo, oxidação diferencial, tal que uma forma de dosagem descolore mais rapidamente quando compreendida de um polimorfo do que quando compreendida de outro polimorfo) ou propriedade mecânica (por exemplo, comprimidos esfarelados em armazenamento como um polimorfo cineticamente favorecido converte o polimorfo termodinamicamente mais estável) ou ambos (por exemplo, comprimidos de um polimorfo são mais suscetíveis à decomposição em umidade alta). Como um resultado de diferenças de solubilidade/dissolução, no caso extremo, algumas transições polimórficas podem resultar na falta de potência ou, no outro extremo, toxicidade. Além disso, as propriedades físicas do cristal podem ser importantes no processamento, por exemplo, um polimorfo podería ser mais provável de formar solvatos ou poderia ser difícil de filtrar e lavar livre de impurezas (isto é, forma de partícula e distribuição de tamanho poderiam ser diferentes entre os polimorfos).
Polimorfos de uma molécula pode ser obtido por vários métodos, como conhecido na técnica. Tais métodos incluem, porém não são limitados à recristalização por fusão, resfriamento por fusão, recristalização de solvente, dessolvatação, evaporação rápida, resfriamento rápido, resfriamento lento, difusão de vapor e sublimação.
Técnicas para caracterizar polimorfos incluem, porém não são limitadas a, calorimetria de varredura diferencial (DSC), difractometria de pó de Raios X (XRPD), difractometria de Raios X de cristal único, espectroscopia vibracional, por exemplo, espectroscopia de IR e Raman, NMR de estado sólido, microscopia ótica de estágio quente, microscopia de elétron de varredura (SEM), cristalografia de elétron e análise quantitativa, análise de tamanho de partícula (PSA), análise de área de superfície, estudos de solubilidade e estudos de dissolução.
O termo, solvato, quando aqui usado, refere-se a uma forma cristalina de uma substância que contém solvente. O termo “hidrato” refere-se a um solvato em que o solvente é água.
Um solvato dessolvatado é uma forma cristalina de uma substância que pode apenas ser feita removendo-se o solvente de um solvato.
O termo, ‘forma amorfa”, quando aqui usado, refere-se a uma forma não cristalina de uma substância.
Quando aqui usado, o termo “puro” significa cerca de 90-100%, preferivelmente 95100%, mais preferivelmente 98-100% (p./p.) ou 99-100% (p./p.) de composto puro; por exemplo menos do que cerca de 10%, menos do que cerca de 5%, menos do que cerca de 2% ou menos do que cerca de 1% de impureza está presente.
Tais impurezas incluem, por exemplo, produtos de degradação, produtos oxidados, epímeros, solventes, e/ou outras impurezas indesejáveis.
Quando aqui usado, um composto é “estável” onde quantidades significantes de produtos de degradação não são observadas sob condições constantes de umidade, exposição a luz e em temperaturas mais altas do que 0°C durante um período de quatro semanas. Um composto não é considerado ser estável em uma certa condição quando impurezas de degradação aparecerem ou uma porcentagem de área de impurezas existentes começa a crescer. A quantidade de crescimento de degradação como uma função de tempo é importante determinando-se estabilidade de composto.
Todas as faixas mencionadas aqui são pretendidas abranger as metas indicadas da faixa bem como todos os valores incluídos e faixas, incluindo aqueles não especificamente mencionados.
A presente invenção é dirigida a formas cristalinas, formas de sal e polimorfos do
Composto 1; composições compreendendo as formas cristalinas, sais e polimorfos sozinhos ou em combinação com outros ingredientes ativos; métodos de preparar a cristalina, sais e polimorfos; e métodos de seu uso na modulação de estados receptivos de composto de tetraciclina. Enquanto não pretendendo ser ligado por qualquer teoria particular de operação, a estabilidade de armazenamento, capacidade de compressão, densidade ou propriedades de dissolução das formas cristalinas, sais e polimorfos são benéficos para fabricação, formulação e bio-disponibilidade do composto de tetraciclina.
Polimorfos e sais preferidos da invenção são aqueles que são caracterizados por propriedades físicas, por exemplo, estabilidade, solubilidade, higroscopicidade e taxa de dissolução, apropriadas para formas de dosagem clínicas e terapêuticas. Polimorfos preferidos da invenção são aqueles que são caracterizados por propriedades físicas, por exemplo, morfologia cristalina, capacidade de compressão e dureza, adequadas para fabricação de uma forma de dosagem sólida. Tais propriedades podem ser determinadas usando técnicas tal como difração de Raios X, microscopia, espectroscopia de IR, análise térmica e análise de higroscopicidade, como descrito aqui e conhecido na técnica.
1.1 Sais do Composto 1
Em um aspecto, a presente invenção fornece formas cristalinas de sais farmaceuticamente aceitáveis particulares do Composto 1. Este aspecto da invenção fornece formas cristalinas de HCI, mesilato e sais de tosilato do Composto 1:
(4S,4AS,5AR,12AS)-4-7-Bis(dimetilamino)-9{[(2,2-dimetilpropil)amino]metil}3,10,12,12A-tetraidróxi-1,11-dioxo-1,4,4A,5,5A,6,11,12A-octaidrotetraceno-2-carboxamida.
Cada sal da invenção pode ser feito a partir de uma preparação do Composto 1. O Composto 1 pode ser sintetizado ou obtido de acordo com qualquer método aparente para aqueles de experiência na técnica. Em modalidades preferidas, o Composto 1 é preparado de acordo com os métodos descritos em detalhes nos exemplos abaixo. Veja, por exemplo, Publicação U.S. No. 2005/0026876 A1, os teores dos quais estão aqui incorporados por referência em sua totalidade.
Alternativamente, o Composto 1 pode ser preparado isolando-se um sal particular do Composto 1 e convertendo-se um tal sal do Composto 1 à forma neutra por tratamento com uma base apropriada. Por exemplo, o Composto 1 pode ser preparado isolando-se o sal de cloridrato do Composto 1 por filtração, em seguida convertendo-se à forma neutra por tratamento com carbonato de sódio monobásico em acetato de etila, ou outra base adequa13 da.
O Composto 1 preparado por qualquer método pode estar contatado com um ácido apropriado, líquido (isto é, livre de mistura ou diluição) ou em um solvente ou solventes inertes adequados, para produzir as formas de sal da invenção. Por exemplo, o Composto 1 pode estar contatado com um ácido p-toluenossulfônico para produzir as formas de sal de tosilato da invenção.
Estudos de estabilidade foram realizados no Composto de base livre 1 e um sal de diHCI amorfo do Composto 1. Este sal foi formado dissolvendo-se o composto em solução aquosa, ajustando o pH da solução em aproximadamente 4,2, seguido por liofilização. A base livre degradada em menos do que um mês a 40°C, e aproximadamente três meses a 4°C. Em comparação, o sal de diHCI do Composto 1 foi estável durante 6 meses a 40°C, e durante dois anos em temperatura ambiente (25°C).
Como mostrado em detalhes nos exemplos abaixo, o sal de tosilato do Composto 1, e polimorfos do mesmo, exibe propriedades desejáveis.
1.2 Polimorfos do Composto 1
A presente invenção da mesma forma fornece polimorfos do Composto 1. Em certas modalidades, os polimorfos da invenção são polimorfos do sal de tosilato do Composto
1.
Cada polimorfo da invenção pode ser feito de uma preparação do Composto 1. O Composto sólido 1 pode ser dissolvido e em seguida cristalizado a partir das misturas de solvente descritas abaixo para produzir as formas polimórficas da invenção. Em modalidades particulares da invenção, um sal de tosilato do Composto 1 pode ser dissolvido e em seguida cristalizado a partir das misturas de solvente descritas abaixo para produzir certas formas polimórficas da invenção. Em algumas modalidades da invenção, a base livre do Composto 1 pode ser dissolvida e em seguida ácido é adicionado para formar um sal cristalino do Composto 1.
Em uma modalidade, a presente invenção fornece um polimorfo de um sal de tosilato do Composto 1.
Em uma outra modalidade, a invenção fornece um polimorfo da forma 1 do sai de tosilato do Composto 1, tendo um padrão de difração de pós de Raios X similar àquele da FIG. 8, as características do padrão de difração todas mostradas na Tabela 1. Por exemplo, um polimorfo da forma 1 particular da invenção tem picos de padrão de difração de pó de Raios X a 5,60, 8,06, 8,57, 11,41, 13,02, 15,58, 18,83, 20,54 e 24,53 °20 usando radiação de Cu Ka. Por exemplo, um polimorfo da forma 1 particular da invenção tem picos de padrão de difração de pó de Raios X a 8,06, 11,41, 13,02, 18,83, 20,54 e 24,53 °2Θ usando radiação de Cu Ka. Por exemplo, um polimorfo da forma 1 particular da invenção tem picos de padrão de difração de pó de Raios X at 8,06, 13,02, 18,83 e 24,53 °2Θ. Por exemplo, um polimorfo da forma 1 particular da invenção tem picos de padrão de difração de pó de Raios X principais a 8,06 e 18,83 °2Θ.
Tabela 1
|
Angulo (2-teta °) |
valor de d (Angstrom) |
Intensidade (Contagens) |
Intensidade (%) |
|
5,60 |
15,78 |
347 |
15,9 |
|
8,06 |
10,97 |
2184 |
100,0 |
|
8,57 |
10,30 |
581 |
26,6 |
|
9,80 |
9,01 |
308 |
14,1 |
|
10,89 |
8,12 |
233 |
10,7 |
|
11,41 |
7,75 |
667 |
30,5 |
|
13,02 |
6,79 |
626 |
28,7 |
|
13,78 |
6,42 |
261 |
12,0 |
|
14,92 |
5,93 |
252 |
11,5 |
|
15,58 |
5,68 |
346 |
15,8 |
|
16,10 |
5,50 |
262 |
12,0 |
|
17,07 |
5,19 |
345 |
15,8 |
|
18,83 |
4,71 |
979 |
44,8 |
|
20,54 |
4,32 |
838 |
38,4 |
|
21,83 |
4,07 |
489 |
22,4 |
|
23,00 |
3,86 |
395 |
18,1 |
|
24,53 |
3,63 |
661 |
30,3 |
|
25,10 |
3,55 |
341 |
15,6 |
|
27,82 |
3,20 |
404 |
18,5 |
|
28,48 |
3,13 |
357 |
16,3 |
|
30,26 |
2,95 |
302 |
13,8 |
|
34,82 |
2,57 |
236 |
10,8 |
|
36,19 |
2,48 |
254 |
11,6 |
|
37,54 |
2,39 |
247 |
11,3 |
|
40,49 |
2,23 |
368 |
16,8 |
Em outra modalidade, a presente invenção fornece a forma 2 do sal de tosilato do
Composto 1. Em uma modalidade, o polimorfo da forma 2 do sal de tosilato do Composto 1 tem um padrão de difração de pós de Raios X similar aquele da FIG. 9, as características do padrão de difração todas mostradas na Tabela 2. Por exemplo, um polimorfo da forma 2 particular da invenção tem picos de padrão de difração de pó de Raios X a 7,82, 11,88, 12,68, 16,12,18,63, 21,46 e 23,74 °2Θ usando radiação de Cu Ka. Por exemplo, um polimor10 fo da forma 2 particular da invenção tem picos de padrão de difração de pó de Raios X principais a 7,82, 11,88, 16,12 e 21,46 °2Θ. Por exemplo, um polimorfo da forma 2 particular da invenção tem picos de padrão de difração de pó de Raios X a 11,88 e 16,12 °20.
Tabela 2
|
Angulo (2-teta °) |
valor de d (Angstrom) |
Intensidade (Contagens) |
Intensidade (%) |
|
5,317 |
16,6224 |
135 |
10,6 |
|
6,272 |
14,09246 |
275 |
21,5 |
|
6,511 |
13,57561 |
274 |
21,5 |
|
7,108 |
12,43554 |
137 |
10,8 |
|
7,821 |
11,30413 |
827 |
64,8 |
|
9,712 |
9,10741 |
173 |
13,5 |
|
10,783 |
8,20461 |
340 |
26,6 |
|
11,875 |
7,4528 |
1258 |
98,6 |
|
12,682 |
6,97995 |
904 |
70,8 |
|
13,182 |
6,7162 |
611 |
47,9 |
|
13,985 |
6,33261 |
299 |
23,4 |
|
15,578 |
5,68838 |
512 |
40,1 |
|
16,122 |
5,49766 |
1100 |
86,2 |
|
16,635 |
5,32915 |
467 |
36,6 |
|
17,397 |
5,09763 |
697 |
54,7 |
|
18,63 |
4,76275 |
967 |
75,8 |
|
20,235 |
4,38856 |
647 |
50,7 |
|
20,666 |
4,298 |
636 |
49,8 |
|
21,456 |
4,14147 |
1276 |
100 |
|
22,51 |
3,9499 |
747 |
58,6 |
|
23,744 |
3,7473 |
1076 |
84,3 |
|
24,448 |
3,64103 |
749 |
58,7 |
|
25,651 |
3,47293 |
652 |
51,1 |
|
26,303 |
3,38824 |
638 |
50 |
|
27,225 |
3,27554 |
678 |
53,2 |
|
27,4 |
3,25505 |
747 |
58,5 |
|
27,823 |
3,20648 |
763 |
59,8 |
|
28,193 |
3,16532 |
588 |
46,1 |
|
Em ainda out |
ra modalidade, a presente invenção fornece a forma 3 do Composto 1 |
Em outra modalidade, o polimorfo da forma 3 do sal de tosilato do Composto 1 tem um pa5 drão de difração de pós de Raios X similar aquele da FIG. 10, as características do padrão de difração todas mostradas na Tabela 3. Por exemplo, um polimorfo da forma 3 particular da invenção tem picos de padrão de difração de pó de Raios X a 5,11, 8,89, 10,34, 11,76, 13,70, 14,81 e 15,60 °20 usando radiação de Cu Ka. Por exemplo, um polimorfo da forma 3 particular da invenção tem picos de padrão de difração de pó de Raios X principais a 5,11, 8,89, 10,34, 11,76 e 15,60 °20. Por exemplo, um polimorfo da forma 3 particular da invenção tem picos de padrão de difração de pó de Raios X principais a 5,11 e 15,60 °20.
Tabela 3
|
Angulo (2-Teta °) |
valor de d (Angstrom) |
Intensidade (Contgens) |
Intensidade (%) |
|
5,11 |
17,29 |
1184 |
66,4 |
|
8,89 |
9,95 |
475 |
26,6 |
|
10,34 |
8,56 |
431 |
24,2 |
|
11,76 |
7,53 |
404 |
22,7 |
|
13,70 |
6,46 |
524 |
29,4 |
|
14,81 |
5,98 |
552 |
31 |
|
15,60 |
5,68 |
17,83 |
100 |
|
17,23 |
5,15 |
661 |
37,1 |
|
17,93 |
4,95 |
1014 |
56,9 |
|
18,35 |
4,83 |
832 |
46,6 |
|
18,74 |
4,74 |
914 |
51,3 |
|
19,00 |
4,67 |
874 |
49 |
|
20,15 |
4,41 |
889 |
49,9 |
|
20,36 |
4,36 |
913 |
51,2 |
|
20,65 |
4,30 |
940 |
52,7 |
|
21,65 |
4,10 |
681 |
38,2 |
|
22,59 |
3,94 |
923 |
51,8 |
|
23,25 |
3,83 |
1206 |
67,7 |
|
23,71 |
3,75 |
872 |
48,9 |
|
24,94 |
3,57 |
718 |
40,3 |
|
25,43 |
3,50 |
551 |
30,9 |
|
26,12 |
3,41 |
745 |
41,8 |
|
26,64 |
3,35 |
709 |
39,8 |
|
27,15 |
3,28 |
689 |
38,7 |
|
27,55 |
3,24 |
754 |
42,3 |
|
0 sal de tosi |
ato do Composto 1 cris |
talizado como partículas muito pequenas, irre- |
guiares, tipicamente 5-8 microns em tamanho. A FIG. 1 descreve difração de pó de Raios X (XRPD) de um sólido cristalino do sal de tosilato do Composto 1. Este composto foi mostrado fundir a 190°C, seguido por decomposição.
Absorção de vapor gravimétrico foi realizada no Composto 1 ou seu sal de tosilato.
Foi determinado que houve 2,5 moléculas de água por molécula do Composto 1. XRPD foi realizada para comparar o material de partida (E00285) com o material desidratado. Os da17 dos não indicaram a mudança na forma.
2. Síntese do Composto 1
Diidrocloridrato de 9-(aminometil)-minociclina (200 mg, 1 eq.), DMF e trimetilacetaldeído (45 μΙ, 1 eq.) foi combinado em frascos de 40 mL e agitado. Trietilamina (150 μί, 3 eq.) foi em seguida adicionada. Depois de agitar em temperatura ambiente durante vários minutos, NaBH(OAc)3 (175 mg, 2 eq.) e lnCI3 (9 mg, 0,1 eq.) foi adicionado. Depois de uma hora, a reação foi clara e vermelha. Cromatografia líquida mostrou um único produto para a reação. A reação foi extinguida com metanol, o solvente foi removido, e o produto foi purificado usando cromatografia de coluna purificada.
O Composto 1 foi purificado por cromatografia injetando-se uma solução aquosa de pH baixo do composto em uma HPLC em um gradiente de solvente orgânico polar, e combinando-se as frações do produto, tal que o composto foi purificado. Seleção de fases móveis ácidas adequadas aumentou a estabilidade de processo e seletividade. Fases móveis de ácido orgânico e mineral foram eficazes para separar subprodutos incluindo impurezas de epímero e estritamente eluindo por produtos através de controle de pH ou escolha de ácido. Fases móveis ácidas da mesma forma protegidas contra degradação oxidativa do composto.
Por exemplo, a solução de pH baixo teve um pH de cerca de 2 - 3. Exemplos de soluções que foram usadas incluem soluções aquosas a 0,1% de ácido metano sulfônico e soluções aquosas a 0,1% de ácido trifluoroacético. Em certas modalidades, um gradiente isocrático foi usado a partir de 94% da solução aquosa e 6% de acetonitrila ou outro solvente orgânico polar para purificar o composto epimérico e intimamente eluindo por subprodutos.
As frações de produto aquosas resultantes podem ser combinadas e o pH pode ser ajustado em cerca de 4,0 - 4,5 usando uma base (por exemplo, NaOH). Impurezas hidrofóbicas e degradantes oxidativos do composto podem ser removidos lavando-se a solução aquosa com um solvente orgânico não polar (por exemplo, CH2CI2). As camadas orgânicas foram descartadas e as camadas aquosas foram combinadas e retidas.
Deveria ser notado que os solventes orgânicos, tal como cloreto de metileno, podem ser usados para seletivamente remover as impurezas hidrofóbicas de eluição tardia tal como 4-carbonila por produtos e outros degradantes oxidativos da solução aquosa ácida do composto.
O pH das camadas aquosas combinadas pode em seguida ser ajustado em pH neutro, por exemplo, cerca de 7,5 a cerca de 8,5. O pH pode ser ajustado pela adição de uma base, tal como NaOH. A solução neutra foi em seguida lavada com um solvente orgânico não polar, tal como cloreto de metileno. Deveria ser notado que o ajuste de pH seletivo em faixas de pH neutro da mesma forma permitiu o composto ser extrair no solvente orgânico enquanto mantendo o β-epímero indesejado e por produtos na fase aquosa.
Além disso, antioxidantes podem da mesma forma ser adicionados às soluções aquosas dos compostos descritos aqui. Os antioxidantes podem ser fornecidos para prevenir a degradação oxidativa dos compostos. Antioxidantes tais como sulfitos ou bissulfitos de amônio podem ser usados.
3. Métodos de Preparar Formas Polimórficas do Composto 1
A invenção da mesma forma pertence a métodos de preparar formas polimórficas do Composto cristalino 1.
Em uma modalidade, a forma 1 do sal de tosilato do Composto 1 pode ser feita por qualquer método de preparar a forma 1 aparente para aqueles de experiência na técnica com base nos ensinamentos aqui. Em certas modalidades, a forma 1 pode ser formada a partir de maturação do sal de tosilato amorfo do Composto 1 em isopropanol, acetona, acetato de etila, metil pentanona, tolueno ou solução de acetonitrila. A forma 1 pode da mesma forma ser obtida a partir de recristalização do sal de tosilato amorfo suspenso em isopropanol. A forma 1 pode da mesma forma ser obtida dissolvendo-se a base livre em um solvente apropriado ou combinação de solventes tal como dois álcoois ou um álcool e um antissolvente tal como uma cetona, éter, éster, etc. Depois da adição do ácido, o sal pode ser cristalizado lentamente na forma correta.
Sistema de solvente em que impurezas e base livre do Composto 1 são solúveis enquanto o sal cristalino estável do Composto 1 é insolúvel, por exemplo, lama cristalina pode ser formada por precipitação, pode ser selecionado.
Em outra modalidade, a forma 2 do sal de tosilato do Composto 1 pode ser feita por qualquer método de preparar a forma 2 aparente para aqueles de experiência na técnica com base nos ensinamentos aqui. Em certas modalidades, a forma 2 pode ser formada a partir de maturação do sal de tosilato amorfo do Composto 1 em diclorometano.
Em outra modalidade, a forma 3 do sal de tosilato do Composto 1 pode ser feita por qualquer método de preparar a forma 3 aparente para aqueles de experiência na técnica com base nos ensinamentos aqui. Em certas modalidades, a forma 3 pode ser formada a partir da maturação do sal de tosilato amorfo do Composto 1 em metil etil cetona, acetato de etila ou metil pentanona. A Forma 3 pode ser obtida a partir da maturação da forma 1 em metil pentanona.
Em uma outra modalidade, as formas polimórficas de sal de tosilato do Composto 1 descrito acima podem ser produzidas por métodos que incluem as etapas de combinar o Composto 1 com um solvente para produzir uma lama, e adicionar ácido p-toluenossulfônico.
Qualquer solvente adequado pode ser usado para criar a lama. Solventes que podem ser usados em modalidades incluem solventes alcoólicos, tais como isopropanol. Qualquer combinação adequada de solvente pode ser usada para criar a solução de qual o sal cristaliza. Combinações de solvente que podem ser usadas em modalidades incluem, po19 rém não são limitadas a, metanol e éter metil-t-butílico ou etanol e isopropanol.
Por exemplo, uma lama do Composto 1 em um solvente ou uma combinação de solventes pode ser produzida em uma temperatura de cerca de 0 °C a cerca de 60°C, tal como de cerca de 15°C a cerca de 45°C, ou de cerca de 20°C a cerca de 25°C. Depois que é produzida, a lama pode opcionalmente ser aquecida e/ou mantida em uma temperatura de cerca de 15°C a cerca de 60°C, tal como de cerca de 20 °C a cerca de 50°C, ou cerca de 45°C.
Uma vez que a lama é criada, o ácido p-toluenossulfônico pode ser adicionado em uma quantidade suficiente para produzir um sal de ácido p-toluenossuifônico do Composto 1. Em uma modalidade, o ácido p-toluenossulfônico é fornecido em uma quantidade de cerca de 25 a cerca de 75% em peso, de cerca de 25 a cerca de 50% em peso, de cerca de 30 a cerca de 40% em peso, ou cerca de 33% em peso em relação à quantidade do Composto 1. O ácido p-toluenossulfônico pode ser adicionado na forma de um monoidrato de ácido ptoluenossulfônico.
Formas polimórficas do sal de tosilato do Composto 1 podem ser formadas por um método de solução. Por exemplo, uma solução do Composto 1 pode ser produzida em uma temperatura de cerca de 0°C a cerca de 60°C, tal como de cerca de 15°C a cerca de 45°C, ou de cerca de 20 °C a cerca de 25 °C. Depois que é produzida, a solução pode opcionalmente ser aquecida e/ou mantida em uma temperatura de cerca de 15°C a cerca de 60 °C, tal como de cerca de 20°C a cerca de 50°C, ou cerca de 45°C.
Uma vez que a solução é criada, o ácido p-toluenossulfônico pode ser adicionado em uma quantidade suficiente para produzir um sal de ácido p-toluenossulfônico do Composto 1. Em uma modalidade, o ácido p-toluenossulfônico é fornecido em uma quantidade de cerca de 25 a cerca de 75% em peso, de cerca de 30 a cerca de 50% em peso, de cerca de 35 a cerca de 45% em peso, ou cerca de 40% em peso em relação à quantidade do Composto 1. O ácido p-toluenossulfônico pode ser adicionado na forma de um monoidrato de ácido p-toluenossulfônico.
Em uma modalidade, polimorfo da forma 1 usado para semear a solução pode ser adicionado. Qualquer solvente adequado pode ser usado para formar a solução de ácido ptoluenossulfônico. Solventes adequados incluem solventes alcoólicos, tais como isopropanol ou combinações de solvente tais como metanol e éter metil-t-butílico. Em uma modalidade preferida, a relação vol. / vol. de metanol para éter metil-t-butílico é 1:1,2. Solventes adequados incluem uma combinação de dois ou mais solventes alcoólicos, tais como uma combinação de etanol e isopropanol. Em uma modalidade preferida, a relação de vol. / vol. de etanol para isopropanol é 2:1. Em modalidades particulares, a solução de ácido ptoluenossulfônico inclui o mesmo solvente usado para criar a lama ou a solução do Composto 1.
Depois da adição de ácido p-toluenossulfônico no solvente apropriado, uma lama do polimorfo da forma 1 do sal de tosilato do Composto é formada. O teor de água do sobrenadante da lama pode ser ajustado em um nível adequado seguindo a adição do ácido ptoluenossulfônico. Tipicamente, o teor de água do sobrenadante de lama pode estar em uma faixa de cerca de 0,2 a cerca de 1,0 mg/mL, tal como de cerca de 0,4 a cerca de 0,8 mg/mL, por exemplo, cerca de 0,6 mg/mL, cerca de 0,54 mg/mL, etc.
Seguindo a adição do ácido p-toluenossulfônico, a lama ou a solução pode ser agitada para produzir uma lama cristalina. Agitação pode ser conduzida durante mais do que 48 horas. Entretanto, agitação é tipicamente conduzida durante um período dentre cerca de 5 a cerca de 36 horas, tal como de cerca de 10 a cerca de 24 horas ou cerca de 18 horas.
Agitação pode ser conduzida em qualquer temperatura adequada para produzir a lama cristalina. Por exemplo, a lama pode ser agitada em uma temperatura de cerca de 0°C a cerca de 60 °C, tal como de cerca de 15°C a cerca de 45°C ou de cerca de 20°C a cerca de 25°C.
Depois da formação de cristal, a lama cristalina pode ser filtrada para remover o sobrenadante, e os cristais podem ser lavados com qualquer solvente adequado. Em modalidades, os cristais podem ser lavados uma a quatro vezes, e o solvente pode ser qualquer solvente adequado para a preparação da lama cristalina. Em particular, o solvente usado para lavar os cristais pode ser o mesmo solvente ou solventes usados para formar a lama original ou solução, ou a solução de ácido p-toluenossulfônico.
Os cristais produzidos podem em seguida ser secados para remover solvente em excesso por qualquer método adequado. Por exemplo, secagem pode ser realizada por um ou mais métodos incluindo porém podem não limitados a temperaturas elevadas em uma faixa de cerca de 0°C a cerca de 60 °C, tal como de cerca de 15°C a cerca de 45°C; soprando nitrogênio seco sobre os cristais; e soprando nitrogênio umedecido sobre os cristais.
Estudos de maturação foram realizados em que uma amostra do sal de tosilato do Composto 1 foi suspensa em solventes diferentes, filtrados, e o sólido úmido foi analisado por XRPD. Três formas polimórficas do sal de tosilato do Composto 1 foram observadas. A FIG. 2 descreve os espectros de XRPD do material de partida (E00285), sal de tosilato da forma 1, sal de tosilato da forma 2, sal de tosilato da forma 3, e a forma amorfa do Composto 1.
A tabela 4 lista os solventes usados para experiências de manutenção. Tabela 4
|
Exemplo No. |
Solvente |
Forma |
|
1 |
água |
Forma 1 |
|
2 |
nitrometano |
Amorfa |
|
3 |
anisol |
Forma 2 |
|
4 |
2-propanol |
Forma 1 |
|
5 |
metiletil cetona |
Forma 3 |
|
6 |
acetona |
Forma 1 |
|
7 |
acetato de etila |
Forma 1 |
|
8 |
dioxano |
Amorfa |
|
9 |
acetonitrila |
Forma 1 |
|
10 |
tolueno |
Forma 1 |
|
11 |
diclorometano |
Forma 2 |
|
12 |
clorofórmio |
Amorfa |
|
13 |
TBME |
Amorfa |
|
14 |
acetato de isopropila |
Forma 2 |
|
15 |
NMP |
dissolvida |
|
16 |
4-metil-2-pentanona |
Forma 3 |
|
17 |
THF |
Goma |
|
18 |
10% de EtOAc / cicloexano |
Amorfa |
|
19 |
10% de águ/EtOH |
Amorfa |
|
20 |
10% de água/THF |
Amorfa |
|
21 |
10% de águ/ACN |
Amorfa |
|
22 |
10% de água/2-propanol |
Amorfa |
|
23 |
10% de água/acetona |
Amorfa |
|
24 |
10% de água/dioxano |
Amorfa |
Recristalização do material amorfo do Composto 1 foi realizada em vários solventes.
Apenas a recristalização em 2-propanol (álcool isopropílico, IPA) produziu o sal de tosilato da forma 1, como mostrado na Tabela 5. A FIG. 3 compara espectros de XRPD do Composto de referência 1 (E00285) e o sal de tosilato da forma recristalizada 1 de IPA.
Tabela 5
|
Experiência No. |
Solvente |
Volume |
A 50°C |
XRPD de precipitado de sólido |
|
1 |
nitrometano |
24 |
Solúvel |
|
|
2 |
anisol |
200 |
Insolúvel |
Amorfo |
|
3 |
2-propanol |
60 |
Solúvel |
Forma 1 |
|
4 |
metiletil cetona |
100 |
Solúvel |
Amorfo |
|
5 |
Acetona |
80 |
Solúvel |
Amorfo |
|
6 |
acetato de etila |
200 |
Insolúvel |
Amorfo |
|
7 |
dioxano |
120 |
Solúvel |
Amorfo |
|
8 |
acetonitrila |
80 |
Solúvel |
|
|
9 |
tolueno |
200 |
Insolúvel |
Amorfo |
|
10 |
diclorometano |
5 |
Solúvel |
Amorfo |
|
11 |
clorofórmio |
5 |
Solúvel |
Amorfo |
|
12 |
TBME |
200 |
Insolúvel |
Amorfo |
|
13 |
acetato de isopropila |
200 |
Insolúvel |
Amorfo |
|
14 |
4-metil-2-pentanona |
200 |
Insolúvel |
Amorfo |
|
15 |
THF |
100 |
Solúvel |
Amorfo |
|
16 |
10% de EtO-
Ac/cicloexano |
200 |
Insolúvel |
Amorfo |
Um sumário do polimorfismo so sal de tosilato do Composto 1 é apresentado no
Esquema 1:
Esquema
Depois da recristalização, as amostras do sal de tosilato da forma 1, forma 2, e forma 3 foram secadas durante a noite em um vácuo e analisadas por XRPD, como mostrado 5 na FIG. 4. Não houve mudança na forma depois da secagem.
XRPD de temperatura variável foi realizada no sal de tosilato da forma 2 e forma 3 do Composto 1. Veja FIG. 5 e FIG. 6, respectivamente.
As estabilidades relativas das formas polimórficas do Composto 1 foram realizadas.
Por exemplo, sal de tosilato da forma 1 foi submetido a uma experiência de maturação du10 rante 24 horas em IPA ou metil pentanona, ou semeado com sal de tosilato da forma 2 ou forma 3. Durante esta experiência, não houve mudança da forma 1 na forma 2 ou 3. Uma lama de uma mistura de 50:50 da forma 1 e forma 3 foi analisada em IPA durante 18 horas a 0°C, 25°C, 40°C, e 60°C, como mostrado na FIG. 7. Não houve mudança da forma 1 na forma 3.
Experiências de recristalização indicaram que sal de tosilato da forma 1 pode ser obtido de forma reprodutiva a partir do sal de tosilato amorfo suspendendo-se em IPA. A Forma 1 do Composto 1 pode da mesma forma ser obtido de forma reprodutiva pela adição de ácido tósico. Uma varredura de XRPD de alta resolução da forma 1 é descrita na FIG. 8 e a característica do padrão de difração é mostrada na Tabela 1.
4. Composições Farmacêuticas Compreendendo Compostos, Sais, Formas Cristalinas ou Polimorfos dos mesmos da Invenção
Em uma outra modalidade, a invenção pertence a composições farmacêuticas compreendendo um composto de tetraciclina da invenção (por exemplo, sintetizado, ou purificado pelos métodos da invenção) ou um sal farmaceuticamente aceitável, pró-fármaco ou éster do mesmo. As composições farmacêuticas podem compreender um veículo farmaceuticamente aceitável.
O termo composição quando aqui usado é pretendido abranger um produto compreendendo os ingredientes especificados (e nas quantidades especificadas, se indicado), bem como qualquer produto que resulta, diretamente ou indiretamente, de combinação dos ingredientes especificados nas quantidades especificadas. Por farmaceuticamente aceitável é pretendido que o diluente, excipiente ou veículo deve ser compatível com os outros ingredientes da formulação e não danosos ao recipiente do mesmo.
Como mencionado acima, certas modalidades dos presentes compostos podem conter um grupo funcional básico, tal como amino ou alquilamino, e são, desse modo, capazes de formar sais farmaceuticamente aceitáveis com ácidos farmaceuticamente aceitáveis. O termo sais farmaceuticamente aceitáveis são reconhecidos na técnica e iclui sais de adição de ácido inorgânico e orgânico relativamente não tóxicos de compostos da presente invenção. Estes sais podem estar preparados in situ durante o isolamento final e purificação dos compostos da invenção, ou separadamente reagindo-se um composto purificado da invenção em sua forma de base livre com um ácido orgânico ou inorgânico adequado, e isolando-se o sal desse modo formado. Sais representativos incluem o bromidrato, cloridrato, sulfato, bissulfato, fosfato, nitrato, acetato, valerato, oleato, palmitato, estearato, laurato, benzoato, lactato, fosfato, tosilato, citrato, maleato, fumarato, sucinato, tartarato, naftilato, mesilato, glicoeptonato, lactobionato, e sais de laurilsulfonato, e similares. (Veja, por exemplo, Berge e outro (1977) Pharmaceuticals Salts, J. Farm. SCI. 66:1-19).
Em outros casos, os compostos da presente invenção podem conter um ou mais grupos funcionais ácidos e, desse modo, são capazes de formar sais farmaceuticamente aceitáveis com bases farmaceuticamente aceitáveis. O termo sais farmaceuticamente aceitáveis nestes exemplos incluem sais de adição de base relativamente não tóxicos, inorgânico e orgânico de compostos da presente invenção. Estes sais podem ser igualmente preparados in situ durante o isolamento final e purificação dos compostos, ou separadamente reagindo-se o composto purificado em sua forma de ácido livre com uma base adequada, tal como o hidróxido, carbonato ou bicarbonate de um cátion de metal farmaceuticamente aceitável, com amônia, ou com uma amina primária, secundária ou terciária orgânica farmaceuticamente aceitável. Sais de álcali ou alcalinos terrosos representativos incluem os sais de lítio, sódio, potássio, cálcio, magnésio, e alumínio e similares. Aminas orgânicas representativas úteis para a formação de sais de adição de base incluem etilamina, dietilamina, etilenodiamina, etanolamina, dietanolamina, piperazina, e similares.
O termo ésteres farmaceuticamente aceitáveis se referem aos produtos de esterificados relativamente não tóxicos, dos compostos da presente invenção. Estes ésteres podem ser preparados in situ durante o isolamento final e purificação dos compostos, ou separadamente reagindo-se o composto purificado em sua forma de ácido livre ou hidroxila com um agente de esterificação adequado. Ácidos carboxílicos podem ser convertidos em ésteres por meio de tratamento com um álcool na presença de um catalisador. Hidroxilas podem ser convertidas em ésteres por meio de tratamento com um agente de esterificação tais como haletos de alcanoíla. O termo da mesma forma inclui grupos hidrocarboneto inferiores capazes de ser solvatados sob condições fisiológicas, por exemplo, ésteres alquílicos, ésteres metílicos, etílicos e propílicos. (Veja, por exemplo, Berge e outro, supra.).
A invenção da mesma forma pertence a compostos de tetraciclina, que são sintetizados e/ou purificados pelos métodos da invenção, e sais farmaceuticamente aceitáveis dos mesmos.
A frase veículo farmaceuticamente aceitável é reconhecido na técnica e inclui um material farmaceuticamente aceitável, composição ou veículo, adequado para administrar compostos da presente invenção a mamíferos. Os veículos incluem carga líquida ou sólida, diluente, excipiente, solvente ou material de encapsulação, envolvido carregando ou transportando o agente objeto de um órgão, ou porção do corpo, para outro órgão, ou porção do corpo. Cada veículo deve ser aceitável no sentido de ser compatível com os outros ingredientes da formulação e não prejudicial ao paciente. Alguns exemplos de materiais que podem servir como veículos farmaceuticamente aceitáveis incluem: açúcares, tais como lactose, glicose e sacarose; amidos, tais como amido de milho e amido de batata; celulose e seus derivados, tal como carboximetil celulose sódica, etil celulose e acetato de celulose; tragacanto pulverizado; malte; gelatina; talco; excipientes, tais como manteiga de cacau e cera de supositório; óleos, tal como óleo de amendoim, óleo de caroço de algodão, óleo de açafroa, óleo de gergelim, azeite de oliva, óleo de milho e óleo de feijão-soja; glicóis, tais como propileno glicol; polióis, tais como glicerina, sorbitol, manitol e polietileno glicol; ésteres, tais como oleato de etila e laurato de etila; ágar; agentes de tamponamento, tal como hidróxido de magnésio e hidróxido de alumínio; ácido algínico; água livre de pirogênio; solução salina isotônica; a solução de Ringer; álcool etílico; soluções de tampão de fosfato; e outras substâncias compatíveis não tóxicas empregadas em formulações farmacêuticas.
Agentes umectantes, emulsificantes e lubrificantes, tal como lauril sulfato de sódio e estearato de magnésio, bem como agentes colorantes, agentes de liberação, agentes de revestimento, agentes adoçantes, agentes flavorizantes e perfumantes, preservativos e antioxidantes podem da mesma forma estar presentes nas composições.
Exemplos de antioxidantes farmaceuticamente aceitáveis incluem: antioxidantes solúveis em água, tal como ácido ascórbico, cloridrato de cisteína, bissulfato de sódio, metabissulfito de sódio, sulfito de sódio e similares; antioxidantes solúveis em óleo, tal como palmitato de ascorbila, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), lecitina, gaiato de propila, α-tocoferol, e similares; e agentes de quelação de metal, tais como ácido cítrico, ácido tetraacético de etilenodiamina (EDTA), sorbitol, ácido tartárico, ácido fosfórico e similares.
Formulações da presente invenção incluem aquelas adequadas para administração oral, nasal, tópica, transdérmica, bucal, sublingual, retal, vaginal e/ou parenteral. As formulações podem convenientemente ser apresentadas em forma de dosagem unitária e podem ser preparadas por quaisquer métodos bem conhecidos na técnica de farmácia. A quantidade de ingrediente ativo que pode ser combinada com um material de veículo para produzir uma forma de dosagem simples geralmente será aquela quantidade do composto que produz um efeito terapêutico. Geralmente fora de cem por cento, esta quantidade variará de cerca de 1 por cento a cerca de noventa e nove por cento de ingrediente ativo, preferivelmente de cerca de 5 por cento a cerca de 70 por cento, mais preferivelmente de cerca de 10 por cento a cerca de 30 por cento.
Métodos de preparar estas formulações ou composições incluem a etapa de trazer em associação um composto da presente invenção com o veículo e, opcionalmente , um ou mais ingredientes adicionais. Em geral, as formulações são preparadas uniformemente e intimamente trazendo-se em associação um composto da presente invenção com veículos líquidos, ou veículos sólidos finamente divididos, ou ambos, e em seguida, se necessário, moldar o produto.
Formulações da invenção adequadas para administração oral podem estar na forma de cápsulas, sinetes, pílulas, comprimidos, pastilhas (usando uma base flavorizada, normalmente sacarose e acácia ou tragacanto), pós, grânulos, ou como uma solução ou uma suspensão em um líquido aquoso ou não aquoso, ou como uma emulsão líquida de óleo-em-água ou água-em-óleo, ou como um elixir ou xarope, ou como pastilhas (usando uma base inerte, tal como gelatina e glicerina, ou sacarose e acácia) e/ou como antissépticos bucais e similares, cada qual contendo uma quantidade pré-determinada de um composto da presente invenção como um ingrediente ativo. Um composto da presente invenção pode da mesma forma ser administrado como um bolo, eletuário ou pasta.
Em formas de dosagem sólidas da invenção para administração oral (cápsulas, comprimidos, pílulas, drágeas, pós, grânulos e similares), o ingrediente ativo é misturado com um ou mais veículos farmaceuticamente aceitáveis, tais como citrato de sódio ou fosfato de dicálcio e/ou qualquer do seguinte: cargas ou extensores, tais como amidos, lactose, sacarose, glicose, manitol e/ou ácido silícico; aglutinantes, tais como carboximetilcelulose, alginatos, gelatina, polivinil pirrolidona, sacarose e/ou acácia; umectantes, tais como glicerol; agentes desintegrantes, tais como ágar-ágar, carbonato de cálcio, batata ou amido de tapioca, ácido algínico, certos silicatos, e carbonato de sódio; agentes retardantes de solução, tal como parafina; aceleradores de absorção, tal como compostos de amônio quaternários; agentes umectantes, tais como álcool cetílico e monoestearato de glicerol; absorventes, tal como caulim e argila de bentonita; lubrificantes, tais como talco, estearato de cálcio, estearato de magnésio, polietileno glicóis sólidos, lauril sulfato de sódio, e misturas dos mesmos; e agentes colorantes. No caso de cápsulas, comprimidos e pílulas, as composições farmacêuticas podem da mesma forma compreender agentes de tamponamento. Composições sólidas de um tipo similar podem da mesma forma ser empregadas como cargas em cápsulas de gelatina carregadas macias e duras usando tais excipientes como lactose ou açúcares de leite, bem como polietileno glicóis de peso molecular alto e similares.
Um comprimido pode ser feito por compressão ou moldando, opcionalmente com um ou ingredientes mais adicionais. Comprimidos comprimidos podem ser preparados usando aglutinante (por exemplo, gelatina ou hidroxipropilmetil celulose), lubrificante, diluente inerte, preservativo, desintegrante (por exemplo, glicolato de amido de sódio ou carboximetil celulose sódica reticulada), surfactante ou agente de dispersão. Comprimidos moldados podem ser feitos moldando-se em uma máquina adequada uma mistura do composto pulverizado umedecido com um diluente líquido inerte.
Os comprimidos, e outras formas de dosagem sólida das composições farmacêuticas da presente invenção, tais como drágeas, cápsulas, pílulas e grânulos, podem opcionalmente ser marcados ou preparados com revestimentos e cascas, tal como revestimentos entéricos e outros revestimentos bem conhecidas na técnica de formulação farmacêutica. Eles podem da mesma forma ser formulados para fornecer liberação lenta ou controlada do ingrediente ativo aqui usando, por exemplo, hidroxipropilmetil celulose em proporções variadas para fornecer o perfil de liberação desejado, outras matrizes de polímero, lipossomas e/ou microsferas. Eles podem ser esterilizados por, por exemplo, filtração através de um filtro de retenção de bactéria, ou incorporando agentes de esterilização na forma de composições sólidas estéreis, que podem ser dissolvidas em água estéril, ou algum outro meio injetável estéril imediatamente antes do uso. Estas composições podem da mesma forma opcionalmente conter agentes de opacificação e podem ser de uma composição que eles liberam o(s) ingrediente(s) ativo(s) apenas, ou preferencialmente, em uma certa porção do trato gastrointestinal, opcionalmente, de uma maneira atrasada. Exemplos de composições de inclusão que podem ser usadas incluem substâncias poliméricas e ceras. O ingrediente ativo pode da mesma forma estar na forma microencapsulada, se apropriado, com um ou mais dos excipientes descritos acima.
Formas de dosagem líquida para administração oral dos compostos da invenção incluem emulsões farmaceuticamente aceitáveis, microemulsões, soluções, suspensões, xaropes e elixires. Além do ingrediente ativo, as formas de dosagem líquidas podem conter diluente inerte geralmente usado na técnica, tal como água ou outros solventes, agentes de solubilização e emulsificantes, tal como álcool etílico, álcool isopropílico, carbonato de etila, acetato de etila, álcool benzílico, benzoato de benzila, propileno glicol, 1,3-butileno glicol, óleos (em particular, óleos de caroço de algodão, amendoim, milho, germe, azeitona, rícino e gergelim), glicerol, álcool tetraidrofurílico, polietileno glicóis e ésteres de ácido graxo de sorbitano, e misturas dos mesmos.
Além de diluentes inertes, as composições orais podem da mesma forma incluir adjuvantes tais como agentes umectantes, agentes emulsificante e de suspensão, agentes adoçantes, agentes flavorizantes, agentes colorantes, agentes preservativos e perfumantes.
Suspensões, além dos compostos ativos, podem conter agentes de suspensão como, por exemplo, álcoois isostearílico etoxilados, polioxietileno sorbitol e ésteres de sorbitano, celulose microcristalina, metaidróxido de alumínio, bentonita, ágar-ágar e tragacanto, e misturas dos mesmos.
Formulações das composições farmacêuticas da invenção para administração retal ou vaginal podem ser apresentadas como um supositório que pode ser preparado misturando-se um ou mais compostos da invenção com um ou mais excipientes não irritantes adequados ou veículos compreendendo, por exemplo, manteiga de cacau, polietileno glicol, uma cera de supositório ou um salicilato, e que é sólido em temperatura ambiente, porém líquido em temperatura corporal e, portanto, derreterá no reto ou cavidade vaginal e liberará o composto ativo.
Formulações da presente invenção que são adequadas para administração vaginal da mesma forma incluem pessários, tampões, cremes, géis, pastas, espumas ou formulações de spray contendo tais veículos como são conhecidos na técnica será apropriado.
Formas de dosagem para a administração tópica ou transdérmica de um composto desta invenção inclui pós, sprays, unguentos, pastas, cremes, loções, géis, soluções, emplastros e inalantes. O composto ativo pode ser misturado sob condições estéreis com um veículo farmaceuticamente aceitável, e com quaisquer preservativos, tampões ou propelentes que podem ser requeridos.
Os unguentos, pastas, cremes e géis podem conter, além de um composto ativo desta invenção, excipientes, tais como gorduras animais e vegetais, óleos, ceras, parafinas, amido, tragacanto, derivados de celulose, polietileno glicóis, silicones, bentonitas, ácido silí28 cico, talco e òxido de zinco, ou misturas dos mesmos.
Pós e sprays podem conter, além de um composto desta invenção, excipientes tais como lactose, talco, ácido silícico, hidróxido de alumínio, silicatos de cálcio e pó de poliamida, ou misturas destas substâncias. Sprays podem adicionalmente conter propelentes habituais, tais como clorofluoroidrocarbonetos e hidrocarbonetos não substituídos voláteis, tais como butano e propano.
Emplastros transdérmicos têm a vantagem adicionada de fornecer liberação controlada de um composto da presente invenção ao corpo. Tais formas de dosagem podem ser feitas dissolvendo-se ou dispersando-se o composto no próprio meio. Realçadores de absorção podem da mesma forma ser usados para aumentar o fluxo do composto através da pele. A taxa de tal fluxo pode ser controlada fornecendo uma membrana de controle de taxa ou dispersando o composto ativo em uma matriz de polímero ou gel.
Formulações oftálmicas, unguentos de olho, pós, soluções e similares, são da mesma forma contempladas como estando dentro do escopo desta invenção.
Composições farmacêuticas desta invenção adequadas para administração parenteral compreendem um ou mais compostos da invenção em combinação com uma ou mais soluções não aquosas ou aquosas isotônicas estéreis farmaceuticamente aceitáveis, dispersões, suspensões ou emulsões, ou pós estéreis que podem ser reconstituídos em soluções injetáveis estéreis ou dispersões justamente antes do uso, que pode conter antioxidantes, tampões, bacteriostatos, solutos que torna a formulação isotônica com o sangue do recipiente pretendido ou agentes de suspensão ou espessantes.
Exemplos de veículos aquosos e não aquosos adequados que podem ser empregados nas composições farmacêuticas da invenção incluem água, etanol, polióis (tais como glicerol, propileno glicol, polietileno glicol, e similares), e misturas adequadas dos mesmos, óleos vegetais, tais como azeite de oliva, e ésteres orgânicos injetáveis, tais como oleato de etila. A própria fluidez pode ser mantida, por exemplo, pelo uso de materiais de revestimento, tais como lecitina, pela manutenção do tamanho de partícula requerido no caso de dispersões, e pelo uso de surfactantes.
Estas composições podem da mesma forma conter adjuvantes tais como preservativos, agentes umectantes, agentes emulsificantes e agentes de dispersão. Prevenção da ação de microorganismos pode ser garantida pela inclusão de vários agentes antibacterianos e antifúngicos, por exemplo, parabeno, clorobutanol, ácido sórbico de fenol, e similares. Pode da mesma forma ser desejável incluir agentes isotônicos, tais como açúcares, cloreto de sódio, e similares nas composições. Além disso, absorção prolongada da forma farmacêutica injetável pode ser provocada pela inclusão de agentes que atrasam absorção tal como monoestearato de alumínio e gelatina.
Em alguns casos, para prolongar o efeito de um fármaco, é desejável reduzir a ab sorção do fármaco de injeção subcutânea ou intramuscular. Isto pode ser realizado pelo uso de uma suspensão líquida de material cristalino ou amorfo tendo solubilidade de água pobre. A taxa de absorção do fármaco em seguida depende de sua taxa de dissolução que, sucessivamente, pode depender do tamanho de cristal e forma cristalina. Alternativamente, absorção atrasada de uma forma de fármaco parenteralmente administrada pode ser realizada dissolvendo-se ou suspendendo-se o fármaco em um veículo de óleo.
Formas de depósito injetáveis são feitas formando-se matrizes de microencápsula dos compostos objeto em polímeros biodegradáveis tais como polilactídeo-poliglicolídeo. Dependendo da relação de fármaco para polímero, e a natureza do polímero particular empregado, a taxa de liberação de fármaco pode ser controlada. Exemplos de outros polímeros biodegradáveis incluem poli(ortoésteres) e poli(anidridos). Formulações injetáveis de depósito são da mesma forma preparadas atraindo-se o fármaco em lipossomas ou microemulsões que são compatíveis com o tecido corporal.
As preparações da presente invenção podem ser determinadas oralmente, parenteralmente, topicamente ou retalmente. Elas são determinadas por formas adequadas para cada rotina de administração. Por exemplo, elas são administradas em comprimidos ou forma de cápsula. Por exemplo, elas são administradas por injeção, infusão, inalação, loção, unguento, supositório, etc. Administração oral é preferida.
As frases “administração parenteral” e “parenteralmente administrado” como usado aqui significa modos de administração diferentes de enteral e administração tópica, normalmente por injeção, e inclui, sem limitação, injeção intravenosa, intramuscular, intraarterial, intratecal, intracapsular, intraorbital, intracardíaca, intradérmica, intraperitoneal, transtraqueal, subcutânea, subcuticular, intraarticular, subcapsular, subaraquinoide, intraspinal e intrasternal e infusão.
As frases administração sistêmica, administrada sistemicamente, administração periférica e perifericamente administrada quando usadas aqui significam a administração de um composto, fármaco ou outro material diferente de diretamente no sistema nervoso central, tal que entra no sistema do paciente e, desse modo, é submetido a metabolismo e outros processos semelhantes, por exemplo, administração subcutânea.
Estes compostos podem ser administrados aos humanos e outros animais para terapia por qualquer rotina adequada de administração, incluindo oralmente, nasalmente (como por, por exemplo, um sprafl, retalmente, intravaginalmente, parenteralmente, intracisternalmente e topicamente (como por pós, unguentos ou gotas, incluindo bucalmente e sublingualmente).
Independente da rotina de administração selecionada, os compostos da presente invenção que podem ser usados em uma forma hidratada adequada e/ou as composições farmacêuticas da presente invenção, são formulados em formas de dosagem farmacêutica mente aceitáveis por métodos convencionais conhecidos a aqueles de experiência na técnica.
O termo quantidade terapeuticamente eficaz refere-se à quantidade do sal objeto ou polimorfo que extrairá a resposta biológica ou médica de um tecido, sistema, animal ou humano que está sendo buscado pelo pesquisador, veterinário, doutor médico ou outro clínico ou isso é suficiente para prevenir desenvolvimento de ou aliviar até certo ponto um ou mais dos sintomas da doença sendo tratada.
Níveis de dosagem atuais dos ingredientes ativos nas composições farmacêuticas desta invenção podem ser variados para obter uma quantidade do ingrediente ativo que é eficaz para obter a resposta terapêutica desejada para um paciente particular, composição, e modo de administração, sem ser tóxico ao paciente.
O nível de dosagem selecionado dependerá de uma variedade de fatores incluindo a atividade do composto particular da presente invenção empregada, ou o éster, sal ou amida do mesmo, a rotina de administração, o tempo de administração, a taxa de excreção do composto particular sendo empregado, a duração do tratamento, outros fármacos, compostos e/ou materiais usados em combinação com o composto particular empregado, a idade, sexo, peso, condição, saúde geral e história médica anterior do paciente a ser tratado, e fatores semelhantes bem conhecidos nas técnicas médicas.
Um médico ou veterinário tendo experiência ordinária na técnica pode facilmente determinar e prescrever a quantidade eficaz da composição farmacêutica requerida. Por exemplo, o médico ou veterinário podería começar doses dos compostos da invenção empregadas na composição farmacêutica em níveis mais baixos do que aqueles requeridos para obter o efeito terapêutico desejado e gradualmente aumentar a dosagem até que o efeito desejado seja obtido.
Em geral, uma dose diária adequada de um composto da invenção será aquela quantidade do composto que é a dose eficaz mais baixa para produzir um efeito terapêutico. Uma tal dose eficaz geralmente dependerá dos fatores descritos acima. Geralmente, doses intravenosas e subcutâneas dos compostos desta invenção para um paciente, quando usado para os efeitos analgésicos indicados, variará de cerca de 0,0001 a cerca de 100 mg por quilograma de peso corporal por dia, mais preferivelmente de cerca de 0,01 a cerca de 50 mg por kg por dia, e ainda mais preferivelmente de cerca de 0,1 a cerca de 10 mg por kg por dia. Por exemplo, em algumas modalidades as doses estão entre 0,5 e 4,0 mg por kg por dia. Se desejado, a dose diária eficaz do composto ativo pode ser administrada como um, dois, três, quatro, cinco, seis ou mais subdoses administradas separadamente em intervalos apropriados ao longo do dia ou semana ou outro período de tempo adequado, opcionalmente, em formas de dosagem unitárias.
Enquanto for possível para um composto da presente invenção ser administrado sozinho, é preferível administrar o composto como uma composição farmacêutica.
5. Métodos de Usar os Compostos de Tetraciclina da Invenção
A invenção da mesma forma pertence a um método para tratar um estado responsive de tetraciclina em um indivíduo, administrando ao indivíduo uma quantidade eficaz de uma composição compreendendo um Composto 1 de acordo com a invenção ou um sal farmaceuticamente aceitável do mesmo, tal que o estado é tratado.
Os termos tratar, tratando ou tratamento, quando aqui usado, referem-se a um método de aliviar ou ab-rogar uma doença ou distúrbio (por exemplo, o estado sensível ao composto de tetraciclina) e/ou seus sintomas auxiliares. Os termos prevenir, prevenindo ou prevenção, quando aqui usados, referem-se a um método de impedir um indivíduo de adquirir uma doença ou distúrbio. O indivíduo, quando aqui usado, inclui um mamífero. O mamífero pode ser por exemplo, qualquer mamífero, por exemplo, um humano, primata, camundongo, rato, cachorro, gato, vaca, cavalo, cabra, camelo, ovelha ou um porco. Preferivelmente, o mamífero é um humano.
A linguagem estado sensível ao composto de tetraciclina ou estado responsivo de tetraciclina inclui estados que podem ser tratados, prevenidos, ou de outra maneira melhorados pela administração de um composto de tetraciclina da invenção. Estados sensíveis ao composto de tetraciclina incluem infecções bacterianas, virais, e fúngicas (incluindo aquelas que são resistentes a outros compostos de tetraciclina), cânceres (por exemplo, câncer de próstata, câncer de mama, câncer de cólon, câncer do pulmão, melanoma, linfo cânceres e outros distúrbios caracterizados por proliferação celular indesejada, enquanto incluindo, porém não limitado a, aqueles descritos em U.S. 6.100.248), artrite, osteoporose, diabete, e outros estados para os quais compostos de tetraciclina foram constatados ser ativos (veja, por exemplo, Patente U.S. Nos. 5.789.395; 5.834.450; 6.277.061 e 5.532.227 cada das quais está expressamente incorporada aqui por referência). Compostos da invenção podem ser usados para prevenir ou controlar doenças mamíferas e veterinárias importantes tais como diarréia, infecções do trato urinário, infecções de pele e estrutura de pele, orelha, nariz e infecções de garganta, infecção de ferida, mastite e similares. Além disso, métodos para tratar neoplasmas usando compostos de tetraciclina da invenção são da mesma forma incluídos (van der Bozert e outro, Cancer Res., 1998, 48:6686-6690). Em uma modalidade, o estado responsivo de tetraciclina não é uma infecção bacteriana, em outra modalidade, os compostos de tetraciclina da invenção são essencialmente não antibacterianos. Por exemplo, compostos de tetraciclina não antibacterianos da invenção podem ter valores de MIC maiores do que cerca de 4 pg/ml quando medidos por ensaios conhecidos na técnica.
Estados sensíveis ao composto de tetraciclina da mesma forma incluem estados associados a processo inflamatório (IPAS). O termo estado associado a processo inflamatório inclui estados em que a inflamação ou fatores inflamatórios (por exemplo, metalopro teinases matriz (MMPs), óxido nítrico (NO), TNF, interleucinas, proteínas plasma, sistemas de defesa celulares, citocinas, metabólitos de lipídio, protease, radicais tóxicos, moléculas de adesão, etc.) estão envolvidos ou presentes em uma área em quantidades aberrantes. O processo inflamatório é a resposta de tecido vivo ao dano. A causa de inflamação pode ser devido a dano físico, substâncias químicas, micro-organismos, necrose de tecido, câncer ou outros agentes. Inflamação aguda é duração curta durante apenas alguns dias. Se for duração mais longa, entretanto, em seguida pode ser referido como inflamação crônica.
IPAS’s incluem distúrbios inflamatórios. Distúrbios inflamatórios são geralmente caracterizados por calor, vermelhidão, inchaço, dor ou perda de função. Exemplos de causas de distúrbios inflamatórios incluem, porém não são limitados à infecções microbianas (por exemplo, infecções bacterianas e fúngicas), agentes físicos (por exemplo, queimaduras, radiação e trauma), agentes químicos (por exemplo, toxinas e substâncias cáusticas), necrose de tecido e vários tipos de reações imunológicas. Em uma outra modalidade, os IPAS incluem distúrbios descritos na Patente U.S. Nos. 5.929.055 e 5.532.227, incorporadas aqui por referência em sua totalidade.
Exemplos de distúrbios inflamatórios incluem, porém não são limitados a, osteoartrite, artrite reumatoide, infecções agudas e crônicas (bacterianas e fúngicas, incluindo difteria e coqueluche), bronquites agudas e crônicas, sinusite, infecções respiratórias superiores (resfriado comum, etc.), gastroenterite aguda e crônica e colite, cistite aguda e crônica e uretrite, dermatite aguda e crônica, conjuntivite aguda e crônica, serosite aguda e crônica (pericardite, peritonite, sinovite, pleurite e tendinite), pericardite urêmica, coleciste aguda e crônica, vaginite aguda e crônica, uveíte aguda e crônica, reações de fármaco, mordida de inseto, queimas (térmicas, químicas e elétricas) e queimadura de sol.
O termo estado associado a processo inflamatório inclui, em uma modalidade, estados associados a NO. O termo estados associados a NO inclui estados que envolvem ou estão associados com óxido nítrico (NO) ou óxido nítrico sintase induzível (iNOS). Estados associado a NO inclui estados que são caracterizados por quantidades aberrantes de NO e/ou iNOS. Preferivelmente, o estado associado a NO pode ser tratado administrando-se compostos de tetraciclina da invenção. Os distúrbios, doenças e estados descritos em Patentes U.S. Nos. 6.231.894; 6.015.804; 5.919.774; e 5,789,395 estão da mesma forma incluídos como estados associados a NO. Os teores totais de cada uma destas patentes estão aqui incorporadas por referência.
Outros exemplos de estados associados a NO incluem, porém não são limitados a, malária, senescência, diabetes, acidente vascular, distúrbios neurodegenerativos (doença de Alzheimer, doença de Huntington e doença de Parkinson), doença cardíaca (lesão associada a reperfusão após infarto), diabetes juvenil, distúrbios inflamatórios, osteoartrite, artrite reumatoide, infecções agudas, recorrentes e crônicas (bacterianas, viras e fúngicas), bron quite aguda e crônica, sinusite, e infecções respiratórias (resfriado comum, etc.), gastroenterite aguda e crônica e colite, cistite aguda e crônica e uretrite, dermatite aguda e crônica, conjuntivite aguda e crônica, serosite aguda e crônica (pericardite, peritonite, sinovite, pleurite e tendinite), pericardite urêmica, coleciste aguda e crônica, fibrose cística, vaginite aguda e crônica, uveíte aguda e crônica, reações de fármaco, mordida de inseto, queimaduras (térmica, química e elétrica), e queimadura de sol.
O termo estado associado a processo inflamatório da mesma forma inclui, em uma modalidade, estados associados à metaloproteinase matriz (MMPAS). MMPAS incluem estados caracterizados por quantidades aberrantes de atividade de MMPs ou MMP. Estes estão da mesma forma incluídos como estados sensíveis ao composto de tetraciclina que podem ser tratados usando compostos da invenção.
Exemplos de estados associados de metaloproteinase matriz (MMPAS’s) incluem, porém não são limitados à arteriosclerose, ulceração córnea, enfisema, osteoartrite, esclerose múltipla (Liedtke e outro, Ann. Neurol. 1998, 44:35-46; Chandler e outro, J., NeuroimmunolASQl, 72:155-71), osteossarcoma, osteomielite, bronquiectasia, doença obstrutiva pulmonar crônica, doenças de pele e olho, periodontite, osteoporose, artrite reumatoide, colites ulcerativas, distúrbios inflamatórios, crescimento de tumor e invasão (Stetler - Stevenson e outro, Annu. Rev. Cell Biol. 1993, 9:541-73; Tryggvason e outro, Biochim. Biophys. Acta 1987, 907:191-217; Li e outro, Mol. Carcinog. 1998, 22:84-89), metástase, lesão pulmonar aguda, acidente vascular cerebral, isquemia, diabetes, aneurismas aórticos ou vasculares, feridas de tecido de pele, olho seco, osso e degradação de cartilagem (Greenwald e outro, Bone 1998, 22:33-38; Ryan e outro, Curr. Op. Rheumatol 1996, 8:238-247). Outros MMPAS incluem aqueles descritos na Patente U.S. Nos. 5.459.135; 5.321.017; 5.308.839; 5.258.371; 4.935.412; 4.704.383; 4.666.897 e RE 34.656, incorporado aqui por referência em sua totalidade.
Em outra modalidade, o estado sensível a composto de tetraciclina é câncer. Exemplos de cânceres que os compostos de tetraciclina da invenção podem ser úteis para tratar incluem todos os tumores sólidos, isto é, carcinomas por exemplo, adenocarcinomas e sarcomas. Adenocarcinomas são carcinomas derivados de tecido glandular ou em que as células de tumor formam estruturas glandulares reconhecíveis. Sarcomas amplamente incluem tumores cujas células são incrustadas em uma substância fibrilar ou homogênea semelhante a tecido conjuntivo embrionário. Exemplos de carcinomas que podem ser tratadas usando os compostos da invenção incluem, porém não são limitados a, carcinomas da próstata, mama, ovário, testículo, pulmão, cólon e mama. Os métodos da invenção não são limitados ao tratamento destes tipos de tumor, porém estende-se em qualquer tumor sólido derivado de qualquer sistema de órgão. Exemplos de cânceres tratáveis incluem, porém não são limitados a, câncer de cólon, câncer de bexiga, câncer de mama, melanoma, carcinoma ovariano, carcinoma prostático, câncer de pulmão, e uma variedade de outros cânceres igualmente. Os métodos da invenção da mesma forma causam a inibição de crescimento de câncer em adenocarcinomas, tal como, por exemplo, aqueles da próstata, mama, rim, ovário, testículos e cólon.
Em uma modalidade, a invenção pertence a um método para tratar um indivíduo sofrendo ou em risco de sofrer de câncer, administrando-se uma quantidade eficaz de um composto de tetraciclina substituído tal que inibição de crescimento de célula de câncer ocorre, isto é, proliferação celular, capacidade de invasão, metástase, ou incidência de tumor é diminuída, é diminuída, ou parada. A inibição pode resultar em inibição de um processo inflamatório, sub-regulação de um processo inflamatório, algum outro mecanismo ou uma combinação de mecanismos. Altemativamente, os compostos de tetraciclina podem ser úteis para prevenir retomo de câncer, por exemplo, para tratar câncer residual seguindo reseção cirúrgica ou radioterapia. Os compostos de tetraciclina úteis de acordo com a invenção são especialmente vantajosos quando eles são substancialmente não tóxicos comparados a outros tratamentos de câncer. Em uma outra modalidade, os compostos da invenção são administrados em combinação com terapia de câncer padrão, tal como, porém não limitado a, quimioterapia.
Exemplos de estados sensíveis à tetraciclina da mesma forma incluem distúrbios neurológicos que incluem igualmente distúrbios neuropsiquiátricos e neurodegenerativos, porém não são limitados a, a doença de Alzheimer, demências relacionadas à doença de Alzheimer tal como a doença de Pick’s, doenças de Parkinson e outras doenças corporais difusas de Lewy, demência senil, doença de Huntington, síndrome de Gilles de la Tourette, esclerose múltipla, esclerose lateral amiotrófica (ALS), paralisia supranuclear progressiva, epilepsia, e doença de Creutzfeldt-Jakob, distúrbios de função autônoma tal como hipertensão e transtorno do sono, distúrbios neuropsiquiátricos (depressão, esquizofrenia, transtorno esquizoafetivo, psicose de Korsakoff’s, mania, transtorno de ansiedade, transtornos fóbicos, etc.), distúrbios de aprendizado e memória (amnésia ou perda de memória relacionada a idade, transtorno de déficit de atenção, etc.), transtorno distímico, transtorno depressivo principal, transtorno obsessivo compulsivo, distúrbios de uso de substância psicoativas, ansiedade, fobias, transtorno do pânico, bem como transtorno afetivo bipolar (transtorno bipolar severo (humor) (BP-1), transtornos neurológicos afetivos bipolares, por exemplo, enxaqueca e obesidade, etc.). Outros distúrbios neurológicos incluem aqueles listados no American Psychiatric Association’s Diagnostic and Statistical manual of Mental Disorders (DSM), a versão mais atual da qual está incorporada aqui por referência em sua totalidade.
Em outra modalidade, o estado sensível ao composto de tetraciclina é diabetes, por exemplo, diabete juvenil, diabetes melito, diabetes tipo I, ou diabetes tipo II. Em uma outra modalidade, glicosilação de proteína não é afetada pela administração dos compostos de tetraciclina da invenção. Em outra modalidade, o composto de tetraciclina da invenção é administrado em combinação com terapias diabéticas padrões, tal como, porém não limitadas à terapia de insulina.
Em outra modalidade, o estado sensível ao composto de tetraciclina é um distúrbio de massa óssea. Distúrbios de massa óssea incluem distúrbios onde ossos de indivíduos são distúrbios e/ou estados onde a formação, reparo ou remodelagem óssea é vantajoso. Por exemplo, distúrbios de massa óssea incluem osteoporose (por exemplo, uma diminuição na densidade e resistência óssea), fraturas ósseas, formação óssea associada com procedimentos cirúrgicos (por exemplo, reconstrução facial), osteogênese imperfeita (doença de osso frágil), hipofosfatasia, doença de Paget, displasia fibrosa, osteoporose, doença de osso de mieloma, e a depleção de cálcio em osso, tal como aquela que está relacionada ao hiperparatireoidismo primário. Distúrbios de massa óssea incluem todos os estados em que a formação, reparo ou remodelagem óssea é vantajoso ao indivíduo bem como todos os outros distúrbios associados com os ossos ou sistema de esqueleto de um indivíduo que pode ser tratado com os compostos de tetraciclina da invenção, Em uma outra modalidade, os distúrbios de massa óssea incluem aqueles descritos na Patente U.S. Nos. 5.459.135; 5.231.017; 5.998.390; 5.770.588; RE 34.656; 5.308.839; 4.925.833; 3.304.227 e 4.666.897 cada das quais está incorporada aqui por referência em sua totalidade.
Em outra modalidade, o estado sensível ao composto de tetraciclina é lesão pulmonar aguda. Lesões pulmonares agudas incluem síndrome da angústia respiratória de adulto (ARDS), síndrome de pós-bomba (PPS) e trauma. Trauma inclui qualquer lesão a tecido vivo causado por um agente extrínseco ou evento. Exemplos de trauma incluem, porém não são limitados a, lesões por esmagamento, contato com uma superfície dura, ou corte ou outra lesão aos pulmões.
A invenção da mesma forma pertence a um método para tratar lesão pulmonar aguda administrando-se um composto de tetraciclina substituído da invenção.
Os estados sensíveis à tetraciclina da invenção da mesma forma incluem distúrbios pulmonares crônicas. A invenção pertence a métodos para tratar distúrbios pulmonares crônicos administrando um composto de tetraciclina, tal como aqueles descritos aqui. O método inclui administrar a um indivíduo uma quantidade eficaz de um composto de tetraciclina substituído tal que o distúrbio pulmonar crônico é tratado. Exemplos de distúrbios pulmonares crônicos incluem, porém não são limitados, a asma, fibrose cística e enfisema. Em uma outra modalidade, os compostos de tetraciclina da invenção usados para tratar distúrbios pulmonares crônicos e/ou agudos tal como aqueles descritos na Patente U.S. Nos. 5.977.091; 6.043.231; 5.523.297; e 5.773.430 cada das quais está incorporada aqui por referência em sua totalidade.
Em ainda outra modalidade, o estado sensível ao composto de tetraciclina é isque mia, acidente vascular cerebral ou acidente vascular cerebral isquêmico. A invenção da mesma forma pertence a um método para tratar isquemia, acidente vascular cerebral ou acidente vascular cerebral isquêmico administrando-se uma quantidade eficaz de um composto de tetraciclina substituído da invenção. Em uma outra modalidade, os compostos de tetraciclina da invenção são usados para tratar tais distúrbios como descrito na Patente U.S. Nos. 6.231.894; 5.773.430; 5.919.775 e 5.789.395, incorporada aqui por referência.
Em outra modalidade, o estado sensível ao composto de tetraciclina é uma ferida de pele. A invenção da mesma forma pertence, pelo menos em parte, a um método para melhorar a resposta de cura do tecido epitelializado (por exemplo, pele, mucosa, etc.) para lesão traumática aguda (por exemplo, corte, queimadura, arranhão, etc.). O método pode incluir usar um composto de tetraciclina da invenção (que pode ou não pode ter atividade antibacteriana) para melhorar a capacidade do tecido epitelializado para curar ferimentos agudos. O método pode aumentar a taxa de acúmulo de colágeno do tecido curativo. O método pode da mesma forma diminuir a atividade proteolítica no tecido epitelializado diminuindo-se a atividade colagenolítica e/ou gelatinolítica de MMPs. Em uma outra modalidade, o composto de tetraciclina da invenção é administrado à superfície da pele (por exemplo, topicamente). Em uma outra modalidade, o composto de tetraciclina da invenção usado para tratar uma ferida de pele, e outro tal distúrbio como descrito em, por exemplo, Patente U.S. Nos. 5.827.840; 4.704.383; 4.935.412; 5.258.371; 5.308.8391 5.459.135; 5.532.227 e 6.015.804 cada das quais está incorporada aqui por referência em sua totalidade.
Em ainda outra modalidade, o estado sensível ao composto de tetraciclina é um aneurisma aórtico ou vascular em tecido vascular de um indivíduo (por exemplo, um indivíduo tendo ou em risco de ter um aneurisma aórtico ou vascular, etc.). O composto de tetraciclina pode ser eficaz para reduzir o tamanho do aneurisma vascular ou pode ser administrado ao indivíduo antes do começo do aneurisma vascular tal que o aneurisma é prevenido. Em uma modalidade, o tecido vascular é uma artéria, por exemplo, a aorta, por exemplo, a aorta abdominal. Em uma outra modalidade, os compostos de tetraciclina da invenção são usados para tratar distúrbios descritos na Patente U.S. Nos. 6.043.225 e 5.834.449, incorporada aqui por referência em sua totalidade.
Infecções bacterianas podem ser causadas por uma ampla variedade de bactérias gram-positivas e gram-negativas. Os compostos da invenção são úteis como antibióticos contra organismos que são resistentes a outros compostos de tetraciclina. A atividade antibiótica dos compostos de tetraciclina da invenção pode ser determinada usando o método de diluição de caldo padrão in vitro descrito em Waltz, J.A., National Commission for Clinical Laboratory Standards, Document M7-A2, vol. 10, no. 8, pp. 13-20, 2a edição, Villanova, PA (1990).
Os compostos de tetraciclina podem da mesma forma ser usados para tratar infec ções tradicionalmente tratadas com compostos de tetraciclina tais como, por exemplo, infecção por rickettsiae, várias infecções bacterianas gram-positivas e gram-negativas, linfogranuloma venéreo, conjuntivite de inclusão e psitacose. Os compostos de tetraciclina podem ser usados para tratar infecções, por exemplo, por K. pneumoniae, Salmonella, E., hirae, A., baumanii, B., catarrhalis, H., influenzae, P. aeruginosa, E. faecium, E., coll, S. aureus ou E. faecalis. Em uma modalidade, o composto de tetraciclina é usado para tratar uma infecção bacteriana que é resistente a outros compostos antibióticos de tetraciclina. O composto de tetraciclina da invenção pode ser administrado com um veículo farmaceuticamente aceitável
A linguagem em combinação com outro agente terapêutico ou tratamento inclui co-administração do composto de tetraciclina, (por exemplo, inibidor) e com o outro agente terapêutico ou tratamento, administração do composto de tetraciclina primeiro, seguido pelo outro agente terapêutico ou tratamento e administração do outro agente terapêutico ou tratamento primeiro, seguido pelo composto de tetraciclina. O outro agente terapêutico pode ser qualquer agente que é conhecido na técnica para tratar, prevenir, ou reduzir os sintomas de uma doença ou distúrbio, tal como IPAS. Além disso, o outro agente terapêutico pode ser qualquer agente de benefício ao paciente quando administrado em combinação com a administração de um composto de tetraciclina. Em uma modalidade, as doenças, tais como câncer, tratados pelos métodos da invenção incluem aqueles descritos na Patente U.S. Nos. 6.100.248; 5.843.925; 5.837.696 e 5.668.122, incorporada aqui por referência em sua totalidade.
A linguagem quantidade eficaz do composto é aquela quantidade necessária ou suficiente para tratar ou prevenir um estado sensível ao composto de tetraciclina. A quantidade eficaz pode variar, dependendo de tais fatores como o tamanho e peso do indivíduo, o tipo de doença, ou o composto de tetraciclina particular. Por exemplo, a escolha do composto de tetraciclina pode afetar o que constitui uma quantidade eficaz. Alguém de experiência ordinária na técnica seria capaz de estudar os fatores anteriormente mencionados e fazer a determinação em relação à quantidade eficaz do composto de tetraciclina sem experiência indevida.
Nos métodos terapêuticos da invenção, um ou mais compostos de tetraciclina da invenção pode ser administrado sozinho a um indivíduo, ou mais tipicamente um composto da invenção será administrado como parte de uma composição farmacêutica em mistura com excipiente convencional, isto é, substâncias de veículo orgânicas ou inorgânicas farmaceuticamente aceitáveis adequadas para parenteral, oral ou outra administração desejada e que não reagem danosamente com os compostos ativos e não são danosas ao recipiente do mesmo.
A invenção é também ilustrada pelos seguintes exemplos, que não deveríam ser interpretados como também limitando.
6. Exemplificacão da Invenção
Exemplo 1: Síntese de 9-Alguil Aminometil Minociclina
Bis-alquilado R,=R,= CHjPht, R2=H
Tris-alquilado R,=R3=R2=CH2Pht
CHjNHj
THF/EtOH
R & R’ =H ou grupo alquila
OH O OH O O base livre
R & R’ =H ou grupo alquila
Esquema 2
Cloridrato de minociclina (composto 2) foi dissolvido em ácido metilsulfônico ou ácido fluorídrico com anidrido metilsulfônico ou ácido recuperador de água similar tal como áci5 do tríflico. N-hidroximetil ftalimida foi adicionado à mistura reacional. A mistura foi agitada a 20-35°C até que a reação foi concluída. A solução de ácido foi adicionada a uma mistura de gelo/água, e sal tríflico pode ser facilmente precipitado, filtrado e coletado. O sólido foi redissolvido em acetona e trazido em um pH neutro com base. O produto foi precipitado pela adição de água. Se o ácido tríflico estiver presente como recuperador, o produto pode ser 10 precipitado sem neutralização. O produto foi isolado como uma mistura do produto bis e tris alquilado. O material isolado desta reação foi enriquecido na relação de bis desejada (90%).
O sólido foi suspenso no EtOH ou MeOH. Aminólise foi realizada usando metilamina. Um subproduto de ftalamida precipitada como a reação progrediu e foi removido por 15 filtração. O produto de sólido amarelo claro foi precipitado pela adição de cerca de 1,5 volume de t-butilmetiléter para a mistura reacional, e coletado através de uma filtração simples que deixou muitas impurezas pequenas e reagente de metilamina na solução. Outra purifi cação do composto foi realizada por re-suspensão com um álcool alifático inferior tal como metanol.
O composto 4 como base livre foi transferido em um vaso de hidrogenação que foi carregado com metanol e aldeído. Um catalisador de Pd/C inativado foi carregado e o vaso foi pressurizado com gás hidrogênio. A mistura reacional foi hidrogenada sob pressão de hidrogênio em torno de 30 Psi durante cerca de 24 horas. Quando a conversão do composto 4 a 1 foi completo, a solução foi filtrada e lavada por uma almofada de Celite. Neste momento a mistura reacional conteve epímero β C-4 muito baixo, em torno de 3-7%.
O produto (1) foi trabalhado como segue para isolar o produto seletivamente de suas impurezas. O pH da solução foi ajustado em cerca de 4,5 com HCI concentrado e a solução foi extraída com diclorometano. A camada aquosa foi extraída com diclorometano para seletivamente recuperar o produto de epímero preferido (por exemplo, a). As camadas de diclorometano foram combinadas e concentradas, e 2 L de n-heptano foram adicionados para precipitar o produto. Outra purificação foi obtida repetindo-se o procedimento de preparação com ou sem f-butilmetiléter para dissolver o produto cru.
Exemplo 2: Purificação do Composto 1
Base livre de 9-(2',2,-dimetilpropil aminometil) minociclina crua (40 g) foi dissolvida em 150 mL de tampão A (solução aquosa a 0,1% de ácido metanossulfônico - MSA) e o pH foi ajustado em 2-3 com MSA.
A solução foi filtrada e injetada em uma HPLC e o produto foi eluído com um gradiente isocrático de 94% de tampão A e 6% de acetonitrila. A coleção de fração de produto foi iniciada quando o pico de produto foi detectado. Cada fração foi analisada e um critério de aceitação maior do que 80% de AUG do pico principal foi usado para as frações de produto precoces. Ao combinar frações, o nível de impurezas e concentração relativa das frações agrupadas foi fatorado nos critérios de seleção que encontram as especificações de produto final. Às frações de produto foi adicionada uma solução aquosa a 10% de sulfito de sódio igual a 10% do volume original das frações coletadas.
O seguinte exemplo representa a produção de uma única injeção. A produção de injeções múltiplas pode da mesma forma ser combinada e preparada. Um volume de fração de produto de 3,5 L (incluindo sulfito de sódio) foi coletado e o pH foi ajustado em 4,0 - 4,5 usando uma solução de hidróxido de sódio. A solução aquosa foi lavada com 2 L de diclorometano e a camada orgânica foi separada e descartada.
O pH da camada aquosa é ajustado em 7,5 - 8,5 usando hidróxido de sódio e o produto foi extraído quatro vezes com 2,4 L de diclorometano. O pH foi reajustado em 7,5 8,5 com hidróxido de sódio ou MSA, antes de cada extração.
As quatro camadas de diclorometano foram combinadas e concentradas em cerca de 200 mL, que foram em seguida adicionadas lentamente (durante um período de cerca de minutos) em um n-heptano vigorosamente agitado (2,5 L). A suspensão foi agitada durante cerca de 10 minutos em temperatura ambiente e diluída lentamente (durante um período de 5 minutos) com n-heptano 1,5 L. A lama é resfriada a 0-5°C e agitada durante 1-2 horas. O sólido suspenso foi filtrado e lavado com porções de 3 x 150 mL de n-heptano. O produto foi secado sob vácuo a 40°C durante pelo menos 24 horas até que um peso constante foi obtido e os níveis de todos os solventes residuais estavam dentro da especificação. Aproximadamente 13,6 g de base livre de 9-(2',2,-dimetilpropil aminometil) minociclina foi isolado como um sólido amarelo.
Exemplo 3: Preparação de sal de HCI Cristalino do Composto 1
O Composto 1 (13 g) foi dissolvido em acetona (300 mL), filtrado e o filtro foi adicionalmente lavado com acetona. O filtrado combinado e as lavagens foram resfriados a 5°C. Ao filtrado combinado e às lavagens, uma solução de HCI concentrada (3,9 mL) em acetona (79 mL) foi lentamente adicionada com agitação vigorosa. A lama resultante foi agitada em um banho de gelo durante 15 minutos e filtrada.
A primeira colheita de sólido foi lavada com acetona fria e pentano e secada em vácuo durante 48 horas, rendendo 13,7 g de sólido amorfo amarelo. O frasco com filtrado saturado foi revestido com chapa de alumínio e deixado durante 2 semanas durante as quais o crescimento de únicos cristais foi observado. Cristais foram coletados por fiitração e lavados com hexano.
Exemplo 4: Preparação de Sal de Ácido Metanossulfõnico Cristalino do Composto 1
Em um frasco de 25 - mL 3 - gargalos sob uma atmosfera de nitrogênio inerte, 3 mL de isopropanol (IPA) foi carregado. Uma lama foi preparada adicionando-se 225 mg de base livre amorfa do Composto 1 ao frasco. A lama foi aquecida em uma temperatura de 45°C. Hidrato de ácido metanossulfõnico (98,0 mg) foi em seguida acrescentado à lama. A lama foi agitada a 45 °C durante uma hora, em seguida resfriada a 22 °C para produzir uma lama cristalina grossa. A lama foi filtrada e lavada com IPA (2x1 mL). Excesso de IPA foi removido da massa cristalina secando-se a 55°C durante mais do que duas horas para obter um peso constante. Sal de mesilato cristalino (ácido metanossulfõnico) do Composto 1 (180 mg) foi isolado. Foi determinado que o sal de mesilato cristalino foi instável a 5°C.
Exemplo 5: Preparação de Sal de Tosilato Cristalino do Composto 1 (usando um método de lama)
Em um frasco de de 3 gargalos de 5-L sob uma atmosfera de nitrogênio inerte, 2,0 L de isopropanol (IPA) foi carregado. Uma lama foi preparada adicionando-se 289 g da base livre amorfa do Composto 1 ao frasco. Uma solução de hidrato de ácido p-toluenossulfônico (97,0 g) em EPA (400 mL) foi em seguida adicionada à lama. O teor de água do sobrenadante de lama foi ajustado a 0,6 g/L com a adição de água (9 mL), e a lama foi agitada a 2025°C durante 18 horas para produzir uma lama cristalina grossa. A lama foi filtrada e lavada com I PA (2 x 500 mL). Excesso de I PA foi removido da massa cristalina soprando-se nitrogênio seco através da massa durante 24 horas. Com os sólidos contendo 3 por cento em peso (% em peso) de IPA, a massa foi também secada soprando-se nitrogênio umedecido através da massa em uma umidade relativa de 70-75% por 24 horas. A massa reteve 0,9% em peso de IPA que não foi também reduzido por este método. Água em excesso foi em seguida removida da massa soprando-se nitrogênio seco através da massa durante 24 horas. O sal de tosilato do Composto 1 foi isolado como um pó laranja (337 g). O sal de tosilato isolado do Composto 1 foi cristalino com apenas uma forma observada: um hemiidrato não esteiquiométrico, como determinado por difração de pó de Raios X (XPRD) e análise termogravimétrica (TG).
Exemplo 6: Preparação de Sal de Tosilato Cristalino do Composto 1 (usando um método de solução)
Em um frasco de 3 gargalos de 5-L sob uma atmosfera de nitrogênio inerte, 1,7 L de metanol e 1,7 L de éter metil-t-butílico foram carregados. Monoidrato de ácido P-tolueno sulfônico (209 g) e a base livre amorfa do Composto 1 (556g) foram adicionados com agitação ao frasco para obter uma solução clara. Uma quantidade de semente (3 g) do sal de monotosilato do composto 1 foi adicionada para iniciar a cristalização e uma outra quantidade de metanol (0,1 L) e éter metil-t-butílico (0,5 L) foi adicionada. A lama resultante foi agitada a cerca de 20°C durante 22 horas para produzir uma lama cristalina grossa. A lama foi filtrada e lavada com uma mistura de 1,1 L de metanol e 1,3 L de éter de metil-t-butílico seguido por éter metil-t-butílico (2 x 2,4 L). O sal de tosilato do Composto 1 foi isolado como um pó laranja. Solvente em excesso foi removido da massa cristalina soprando-se nitrogênio seco através da massa durante 24 horas. A massa foi em seguida secada sob vácuo a cerca de 30°C até que os sólidos contivessem cerca de 6 por cento em peso (% em peso) de solvente. A massa foi também secada sob vácuo a cerca de 45°C até que os sólidos contivessem menos do que 3 por cento em peso (% em peso) de solvente. O sal de tosilato isolado do Composto 1 foi cristalino com a forma 1 observada como determinado por difração de pó de Raios X (XPRD).
Exemplo 7: Caracterização de Sal de Tosilato do Composto 1
O padrão de XRPD do sal de tosilato isolado do Composto 1 compreendeu valores 2Θ em graus de 5,6, 8,0, 8,6,11,4, 13,0, 15,5,18,8, 20,4 e 24,5.
O sal de tosilato cristalino foi submetido à análise termogravimétrica (TG) sob fluxo de nitrogênio em uma taxa de aquecimento de 10 °C/minuto. Uma perda de peso de 3,9% foi observada até 81,3°C, que foi devido à perda de água.
Ao secar, teor de água no sal de tosilato cristalino do Composto 1 foi calculado ser 0,5%. Depois de repousar em temperatura ambiente durante 24 horas, este valor aumentou para 5%. Em comparação com o Composto 1 amorfo, o sal de tosilato cristalino do Compos to 1 foi estável durante semanas e meses em temperatura ambiente, mantendo um teor de água de cerca de 5%.
A higroscopicidade do sal de tosilato cristalino objeto foi determinada usando um analisador de absorção de vapor simétrico, e relatado como % em peso ganho como uma função de umidade relativa percentual (% de RH) de intervalos de 5% a 95% a 1%, a 5% a 25,0°C. Um ganho de peso máximo de 12% em peso em 95% de RH, com histerese leve em desabsorção, foi observado, e uma perda de 2% em peso devido a IPA foi observada.
O sal de tosilato cristalino foi determinado ter as solubilidades resumidas na Tabela 6 abaixo. As solubilidades foram determinadas misturando-se sólidos em excesso com solvente, em temperatura ambiente durante duas horas, seguido por filtração do sobrenadante. A concentração de sobrenadante foi determinada por cromatografia líquida de alto desempenho (HPLC). Equilíbrio de 56,8 mg do sal de tosilato cristalino objeto em 0,5 mL de água e 122,1 mg do sal de tosilato cristalino objeto em 1,0 mL de água resultou em soluções claras. O pH de 122,1 mg do sal de tosilato cristalino objeto em 1,0 mL de água foi determinado ser 5,70.
Tabela 6
|
Solvente |
Solubilidade (mg/mL) |
|
Água |
>100 |
|
Acetonitrila |
5,9 |
|
Metanol |
86,1 |
|
IPA |
5,8 |
Exemplo 8: XRPD do Composto 1
Padrões de Difração de Pó de Raios X foram coletados em um difractômetro Siemens D5000 usando radiação de Cu Ka (40kV, 40mA), Θ-Θ goniômetro, divergência de V20 e fendas receptoras, uma monocromador secundário de grafite e uma contadora de cintilação. O instrumento é conferido por desempenho usando um padrão de Corundum certificado (NIST 1976). O software usado para coleta de dados foi Diffrac Plus XRD Commander v2.3.1 e os dados foram analisados e apresentados usando Diffrac Plus EVA v 11,0.0.2 ou v 13.0.0.2.
Amostras de pó foram preparadas como uso de espécimes de placa lisa. Aproximadamente 35 mg da amostra foram suavemente empacotadas em um corte de cavidade em pastilha de silício de base zero, polida (510). A amostra foi girada em seu próprio plano durante a análise. Os detalhes da coleta de dados são:
Faixa angular: 2 a 42 °20
Tamanho da Etapa: 0,05 °2Θ
Tempo de coleção: 4 s/etapa.
Padrões de Difração de Pó de Raios X de Alta resolução foram coletados em um di fractômetro Bruker AXS C2 GADDS usando radiação de Cu Ka (40 kV, 40 MA), estágio XYZ automático, microscópio de video a laser para posicionamento de auto-amostra e urn detector de área HiStar 2-dimensional. Óticas de Raios X consiste em um único espelho de multicamadas de Gõbel acoplado com um colimador de furo de alfinete de 0,3 mm.
A divergência de feixe, isto é o tamanho eficaz do feixe de Raios X na amostra, foi de aproximadamente 4 mm. Um modo de varredura contínuo Θ-Θ foi empregado com uma distância de amostra - detector de 20 cm que produz uma faixa 20 eficaz de 3,2 ° - 29,7 °. Tipicamente, a amostra seria exposta à faixa de Raios X durante 120 segundos. O software usado para coleta de dados foi GADDS para WNT 4.1.16 e os dados foram analisados e apresentados usando Diffrac Plus EVA v 9.0.0.2 ou v 13.0.0.2.
Exemplo 9: Estudo de Estabilidade de Temperatura e Umidade
Um estudo de estabilidade foi conduzido em sal de tosilato e sal de mesilato do Composto 1, monitorando-se as mudanças nos perfis de impureza de cromatografia líquida de alto desempenho (HPLC) para cada salgado quando exposto a várias condições de temperaturas e/ou umidade. Cada amostra foi colocada em um recipiente fechado e exposta a três ambientes controlados: refrigerada (5°C), 20°C e 60% de umidade relativa, e 40°C e 75% de umidade relativa. Depois de duas semanas, o sal de tosilato cristalino foi determinado ser a forma cristalina mais estável do Composto 1. O estudo de estabilidade do sal de tosilato cristalino foi continuado.
As amostras foram analisadas pelo seguinte método de perfil de impureza de HPLC fase reversa em pontos de tempo 0, 1, 2, 4 semanas (Tabela 7) e 3 meses (Tabela 8) para o sal de tosilato; e 0, 1 e 2 semanas para o sal de mesilato (Tabela 9).
Análise de HPLC de fase reversa foi conduzida usando uma coluna de SYMMETRY SHIELD RP18 (4,6 x 250 mm, 5 pm de tamanho de partícula), embora análise usando uma coluna similar ou equivalente seria esperada render resultados similares. Os componentes de fase móvel foram 0,01 M de acetato de amônio em água em pH 3,3 (A) e acetonitrila (B). A composição de fase móvel foi um gradiente que aumentou de 6% para 15% B em cinco minutos, sustentada a 15% B durante 15 minutos, aumentou de 15% a 60% de B em dez minutos, em seguida de 60% a 90% de B em dois minutos, e em seguida o sistema foi reequilibrado a 6% de B durante quatro minutos. A taxa de fluxo foi 1,0 mUminuto, e o volume de injeção foi 10,0 pL. A temperatura de coluna foi mantida a 30°C. A detecção foi por luz ultravioleta (UV) a 280 nm. O tempo de retenção para o Composto 1 foi de aproximadamente 15,7 minutos. A solução de amostra foi preparada na fase móvel A em uma concentração final de 2,0 mg/mL.
Tabela 7
|
Ponto de Tempo |
Impurezas totais** |
RRT
0,52 |
RRT
0,77 |
RRT
0,93 |
RRT
1,19 |
RRT***
1,26 |
RRT
1,77 |
Composto 1
(%) |
| |
|
(%) |
(%) |
(%) |
(%) |
(%) |
(%) |
|
|
Inicial |
1,60 |
0,06 |
ND |
ND |
0,24 |
1,2 |
0,10 |
98,4 |
|
1 semana
5°C |
1,51 |
0,09 |
ND |
ND |
0,21 |
1,1 |
0,11 |
98,5 |
|
20°C & 60% RH* |
1,63 |
0,09 |
ND |
ND |
0,21 |
1,2 |
0,12 |
98,3 |
|
40°C % 75% RH |
1,80 |
0,09 |
0,05 |
ND |
0,21 |
1,3 |
0,14 |
98,2 |
|
2 semanas
5°C |
1,62 |
0,08 |
ND |
0,08 |
0,24 |
1,1 |
0,12 |
98,4 |
|
20°C & 60% RH |
1,66 |
0,08 |
ND |
0,08 |
0,25 |
1,1 |
0,15 |
98,4 |
|
40°C & 75 RH |
1,68 |
0,08 |
ND |
0,09 |
0,25 |
1,1 |
0,16 |
98,3 |
|
4 semanas
5°C |
1,61 |
0,09 |
0,08 |
0,07 |
0,23 |
1,0 |
0,14 |
98,3 |
|
20°C & 60% RH |
1,84 |
0,10 |
0,10 |
0,12 |
0,24 |
1,1 |
0,18 |
98,2 |
|
40°C & 75% RH |
1,96 |
0,10 |
0,17 |
0,13 |
0,26 |
1,1 |
0,20 |
98,1 |
*RH denota umidade relativa ** Impurezas totais incluíram todas as impurezas em uma batelada específica *** RRT 1,26 denota beta-epímero.
Tabela 8
|
Ponto de tempo |
20°C & 60% RH* |
40°C & 75% RH |
|
Betaepimero
RRT-
1,26 |
Composto
4-Ceto
RRT-1,57 |
Impureza de M-2 RRT=0,84 |
Impureza Total** |
Betaepimero RRT=
1,26 |
Composto 4-Ceto RRT-1,57 |
Impureza de M-
2
RRT=
0,84 |
Impureza Total |
|
0 mês |
1,76% |
0,05% |
- |
2,12% |
1,76% |
0,05% |
- |
2,12% |
|
3 meses |
1,74% |
0,10% |
0,18% |
2,86% |
1,66% |
0,14% |
0,31% |
3,12% |
*RH denota umidade relativa ** Impurezas totais incluíram todas as impurezas em uma batelada específica Tabela 9
|
Ponto de tempo |
Impurezas Totais
(%) |
RRT
0,52 (%) |
RRT
0,77 (%) |
RRT
0,93 (%) |
RRT
1,19
(%) |
RRT
1,26
(%) |
RRT
1,77 (%) |
Composto 1 (%) |
|
Inicial |
1,70 |
ND |
ND |
ND |
0,20 |
1,5 |
ND |
98,3 |
|
1 semana
5°C |
1,85 |
ND |
ND |
ND |
0,17 |
1,6 |
0,08 |
98,2 |
|
20°C & |
2,06 |
ND |
ND |
ND |
0,17 |
1,8 |
0,09 |
98,0 |
|
60% RH* |
2,97 |
0,07 |
0,05 |
ND |
0,16 |
2,6 |
0,09 |
97,1 |
|
40°C
75% RH |
& |
|
2 semanas |
|
|
|
|
|
|
|
|
|
5°C |
|
1,65 |
ND |
ND |
0,06 |
0,20 |
1,3 |
0,09 |
98,3 |
|
20°C |
& |
1,87 |
ND |
ND |
0,06 |
0,20 |
0,11 |
0,11 |
98,1 |
|
60% RH
40°C
75% RH |
& |
3,38 |
0,05 |
0,07 |
0,06 |
0,19 |
0,11 |
0,11 |
96,6 |
| |
Os dados acima demonstram que o sa |
de tosila |
to do Composto 1 é estável quando |
armazenados refrigerados e/ou 20°C & 60% de condições de RH durante pelo menos quatro semanas. O sal de tosilato do Composto 1 é estável em temperatura em uma faixa de 0°C 70°C, ou em uma faixa de 5°C - 50°C, ou em uma faixa de 20 °C - 30°C.
Exemplo 10: Estudo de Fotoestabilidade
Um estudo de fotoestabilidade foi conduzido no sal de tosilato do Composto 1. Duas amostras foram preparadas em placas de Petri de vidro claras. Uma amostra foi embrulhada em chapa de alumínio para servir como uma amostra de controle. Ambas as amostras foram colocadas na Câmara ES 2000 Environmental Light 10 e expostas a 12 kilolux de luz fluorescente branca resfriada durante um total de 47 horas. Amostras foram em seguida analisadas por método de perfil de impureza de HPLC, e os resultados são resumidos na Tabela 10.
Tabela 10
|
Amostra |
RRT
0,52 (%) |
RRT
0,0,64
(%) |
RRT
0,77 (%) |
RRT
0,83 (%) |
RRT
0,93 (%) |
RRT
1,19 (%) |
RRT
1,26
(%) |
RRT
1,77
(%) |
Composto 1 (%) |
|
Controle |
0,06 |
ND |
0,08 |
ND |
0,12 |
0,26 |
1,2 |
0,16 |
98,2 |
|
Exposta |
0,06 |
ND |
0,09 |
ND |
0,13 |
0,26 |
1,1 |
0,16 |
98,2 |
Equivalentes
Aqueles versados na técnica reconhecerão, ou serão capazes de verificar usando não mais do que experimentação rotineira, muitos equivalentes para as modalidades específicas e métodos descritos aqui. Tais equivalentes são pretendidos ser abrangidos pelo escopo das seguintes reivindicações.
Todas as patentes, pedidos de patente, e referências de literatura citadas aqui estão por meio destes incorporados expressamente por referência.










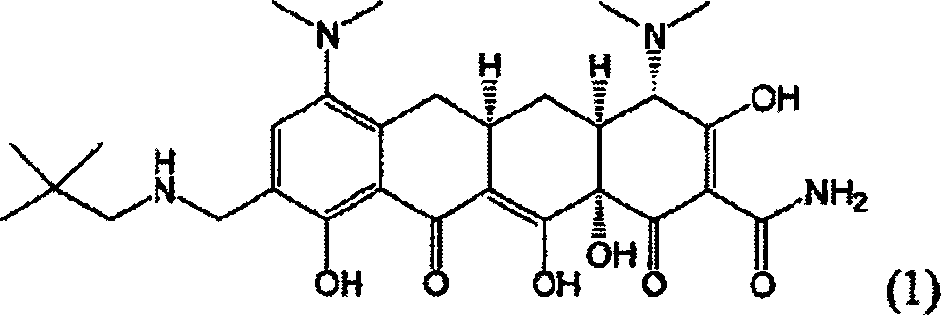
 a partir de isopropanol.
a partir de isopropanol. o método sendo CARACTERIZADO pelo fato de compreender:dissolver uma base livre do Composto 1 em um primeiro solvente ou combinação de solventes para formar uma primeira solução;dissolver ácido p-toluenossulfônico em um segundo solvente ou combinação de solventes para formar uma segunda solução; e combinar as referidas primeira e segunda solução para formar uma terceira solução.
o método sendo CARACTERIZADO pelo fato de compreender:dissolver uma base livre do Composto 1 em um primeiro solvente ou combinação de solventes para formar uma primeira solução;dissolver ácido p-toluenossulfônico em um segundo solvente ou combinação de solventes para formar uma segunda solução; e combinar as referidas primeira e segunda solução para formar uma terceira solução. CARACTERIZADO pelo fato de ser obtido de acordo com o método de acordo com a reivindicação 9.
CARACTERIZADO pelo fato de ser obtido de acordo com o método de acordo com a reivindicação 9.