REFERÊNCIA CRUZADA A PEDIDOS DE PATENTE RELACIONADOS
[001] Este pedido de patente reivindica o benefício de prioridade deste ao Pedido de Patente U.S. No de Série 61/919 347, depositado em 20 de dezembro de 2013, cujo conteúdo é aqui incorporado, em sua totalidade, por referência neste pedido de patente.
CAMPO DA INVENÇÃO
[002] A presente invenção provê, entre outros, métodos, kits e composições farmacêuticas para tratar ou melhorar os efeitos de um câncer em um indivíduo, utilizando (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1, como o dabrafenib ou um sal farmaceuticamente aceitável deste, para tratar ou melhorar os efeitos do câncer.
INCORPORAÇÃO POR REFERÊNCIA DA LISTAGEM DE SEQUÊNCIA
[003] Este pedido de patente contém referências a sequências de aminoácidos e/ou de ácidos nucleicos as quais foram depositadas simultaneamente ao mesmo como o arquivo em texto da listagem de sequências "0375604.txt", com tamanho de 255 KB, criado em 19 de dezembro de 2014. A listagem de sequências citada acima é aqui incorporada, em sua totalidade, por referência neste pedido de patente, segundo 37 C.F.R. § 1.52(e)(5).
ANTECEDENTES DA INVENÇÃO
[004] Fármacos inibidores direcionados contra componentes da via de sinalização das proteínas quinases ativadas por mitógenos (MAPK) mostram eficácia em uma variedade de cânceres, especialmente aqueles portadores de mutações na proteína quinase BRAF. Há inibidores aprovados de RAF e de MEK quinase para uso como agente isolado no melanoma metastático avançado com mutação no gene BRAF, e a agência Food and Drug Administration (FDA) dos Estados Unidos está atualmente submetendo a combinação de dabrafenib com trametinib à revisão para essa indicação. Seja isoladamente ou combinados, inibidores de BRAF e de MEK mostram atividade variável em outros cânceres, com eficácia promissora no câncer da tireoide e do pulmão com mutação em BRAF, bem como atividade marginal no câncer colorretal com mutação em BRAF.
[005] Padrões variáveis de eficácia clínica são vistos com os inibidores de BRAF e MEK. A extensão e a penetrância da regressão tumoral inicial, bem como a duração de resposta antes da regressão da doença variam de modo único de acordo com a classe de cada fármaco isoladamente, ou quando administrados em estratégias de combinações sequenciais ou simultâneas. Até o momento, a terapia combinada de dabrafenib e trametinib parece ser a intervenção preferida para o melanoma com mutação em BRAF.
[006] Tal como com outras terapias direcionadas, os padrões de resposta da doença aos inibidores de RAF e MEK parecem ser influenciados pela heterogeneidade genética intrínseca presente nos cânceres em que os fármacos são utilizados. Por exemplo, demonstrou-se que certas alterações genéticas, incluindo alterações no gene PTEN e outras que ativam a sinalização de PI3K para crescimento celular, podem predizer resposta insatisfatória inicial e/ou progressão relativamente no melanoma com mutação em BRAF tratado com o inibidor de RAF, vemurafenib. Do mesmo modo, mutações diretas em loci do gene MEK parecem emergir em tumores que progrediram após o tratamento com BRAF, MEK ou de fármacos combinados. Diversos exemplos adicionais, resultantes de mutações por amplificações e splicing dos genes RAS e RAF, sugerem que a resistência adquirida a fármacos é produzida quando a pleiotropia oncogênica encontra a pressão seletiva de tratamentos medicamentosos direcionados.
[007] Por conseguinte, novos agentes direcionados inibiriam idealmente diversos nós de vias oncogênicos, e também seriam eficazes em combinações ao induzirem uma carga de pressão seletiva que excedesse a capacidade adaptativa de diversos genomas de câncer. O presente pedido de patente visa atender, entre outros, à necessidade de novos agentes direcionados.
SUMÁRIO DA INVENÇÃO
[008] Uma modalidade da presente invenção é um método para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse método compreende administrar ao indivíduo uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD- 523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste, para tratar ou melhorar os efeitos do câncer.
[009] Outra modalidade da presente invenção é um método para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse método compreende administrar ao indivíduo uma quantidade eficaz de (i) BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é o dabrafenib ou um sal farmaceuticamente aceitável deste, para tratar ou melhorar os efeitos do câncer.
[0010] Uma modalidade adicional da presente invenção é um método para causar a morte de células cancerosas. Esse método compreende contatar a célula cancerosa com uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste.
[0011] Mais uma modalidade da presente invenção é um kit para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse kit compreende uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste, embalados junto com instruções para o seu uso.
[0012] Uma modalidade adicional da presente invenção é uma composição farmacêutica para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Essa composição farmacêutica compreende um diluente ou veículo farmaceuticamente aceitável e uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste, em que a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
[0013] Outra modalidade da presente invenção é um método para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse método compreende administrar ao indivíduo uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD- 523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ-736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF (Liu et al., 2007), CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF-365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes, para tratar ou melhorar os efeitos do câncer.
[0014] Uma modalidade adicional da presente invenção é um método para causar a morte de células cancerosas. Esse método compreende contatar a célula cancerosa com uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ- 736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF- 365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes.
[0015] Mais uma modalidade da presente invenção é um kit para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse kit compreende uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ- 736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF- 365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes, embalados junto com instruções para o seu uso.
[0016] Outra modalidade da presente invenção é uma composição farmacêutica para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Essa composição farmacêutica compreende um diluente ou veículo farmaceuticamente aceitável e uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ- 736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF- 365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes, em que a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
DESCRIÇÃO RESUMIDA DOS DESENHOS
[0017] O arquivo da patente ou pedido de patente contém pelo menos um desenho executado em cores. Cópias da publicação desta patente ou pedido de patente com desenho(s) em cores serão fornecidas pelo Escritório mediante solicitação e pagamento da taxa necessária.
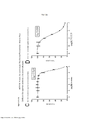
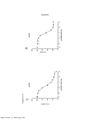
[0018] As Figuras 1A-C mostram o progresso de um estudo de aumento da dose em uma linhagem de células de melanoma maligno humano (células A375) durante o Mês 1. Os vários tratamentos (trametinib (um inibidor de MEK tipo 2), dabrafenib (um inibidor de BRAF) e BVD-523 (um inibidor de ERK1/2)) são como indicados.
[0019] As Figuras 2A-H mostram os resultados de um ensaio de proliferação que rastreia as mudanças na sensibilidade ao(s) agente(s) aumentado(s) gradualmente no Mês 1. Os vários tratamentos (trametinib, dabrafenib, BVD-523 e paclitaxel) são como indicados na parte superior do gráfico. A legenda à direita do gráfico mostra os vários tipos de células geradas a partir do estudo de aumento da dose. Por exemplo, "dabrafenib" refere-se às células que foram tratadas com a dose mais alta de dabrafenib a partir do Mês 1 do estudo de aumento da dose. Parental refere-se às células controle que não foram tratadas com fármacos. As Figuras 2A-2C e 2G são normalizadas para o controle, enquanto que as Figuras 2D-2F e 2H mostram os dados não processados.
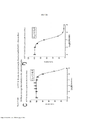
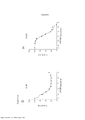
[0020] As Figuras 3A-3D mostram o progresso de um estudo de aumento da dose em células A375 durante o Mês 2. OS vários tratamentos (trametinib, dabrafenib e BVD-523) são como indicados.
[0021] As Figuras 4A-H mostram os resultados de um ensaio de proliferação que rastreia as mudanças na sensibilidade ao(s) agente(s) aumentado(s) gradualmente no Mês 2. Os vários tratamentos (trametinib, dabrafenib, BVD-523 e paclitaxel) são como indicados na parte superior do gráfico. A legenda à direita do gráfico mostra os vários tipos de células gerados pelo estudo de aumento da dose. Por exemplo, "dabrafenib" refere-se às células que foram tratadas com a dose mais alta de dabrafenib a partir do Mês 2 do estudo de aumento da dose. Parental refere-se às células controle que não foram tratadas com fármacos. As Figuras 4A-4C e 4G são normalizadas para o controle, enquanto que as Figuras 4D-4F e 4H mostram os dados não processados.
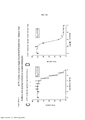
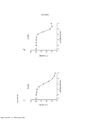
[0022] As Figuras 5A-H mostram somente os dados da linhagem de células parentais e de BVD-523 da Figura 4. Os vários tratamentos (trametinib, dabrafenib, BVD-523 e paclitaxel) são como indicados. As Figuras 5A-5C e 5G são normalizadas para o controle, enquanto que as Figuras 5D-5F e 5H mostram os dados não processados.
[0023] As Figuras 6A-D mostram o progresso do estudo de aumento da dose em uma linhagem de células malignas humanas (células A375) durante o Mês 3. Os vários tratamentos (trametinib, dabrafenib e BVD-523) são como indicados.
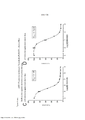
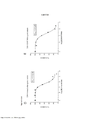
[0024] A Figura 7 é um histograma mostrando os resultados de um ensaio de proliferação conforme aplicado a células cultivadas nas cavidades controle com DMSO do ensaio de aumento da dose.
[0025] As Figuras 8A-D constituem um conjunto de gráficos de linhas mostrando ensaios de proliferação durante o Mês 3 do estudo. Os vários tratamentos (trametinib, dabrafenib, BVD-523 e paclitaxel) são como indicados na parte superior do gráfico. A legenda à direita do gráfico mostra os vários tipos de células gerados pelo estudo de aumento da dose. Por exemplo, "dabrafenib" refere-se às células que foram tratadas com a dose mais alta de dabrafenib a partir do Mês 3 do estudo de aumento da dose. Parental refere-se às células controle que não foram tratadas com fármacos.
[0026] As Figuras 9A-D mostram somente os dados da linhagem de células parentais, dabrafenib e BVD-523 da Figura 8.
[0027] A Figura 10A é uma matriz de doses mostrando a % de inibição da combinação trametinib/dabrafenib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 10B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação trametinib/dabrafenib. As Figuras 10C e 10D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e trametinib como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 10E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib com trametinib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0028] A Figura 11A é uma matriz de doses mostrando a % de inibição da combinação trametinib/dabrafenib em células A375 utilizando o ensaio de viabilidade celular com CellTiter-Glo. A Figura 11B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação trametinib/dabrafenib. As Figuras 11C e 11D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e trametinib como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com CellTiter- Glo. A Figura 11E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib com trametinib em células A375 utilizando o ensaio de viabilidade celular com CellTiter-Glo.
[0029] A Figura 12A é uma matriz de doses mostrando a % de inibição da combinação BVD-523/dabrafenib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 12B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação BVD-523/dabrafenib. As Figuras 12C e 12D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e BVD-523 como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 12E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib com BVD-523 em células A375 utilizando o ensaio de viabilidade com Alamar Blue.
[0030] A Figura 13A é uma matriz de doses mostrando a % de inibição da combinação BVD-523/dabrafenib em células A375 utilizando o ensaio de viabilidade celular com CellTiter-Glo. A Figura 13B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação BVD-523/dabrafenib. As Figuras 13C e 13D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e BVD-523 como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com CellTiter- Glo. A Figura 13E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib com BVD-523 em células A375 utilizando o ensaio de viabilidade celular com CellTiter-Glo.
[0031] A Figura 14A é uma matriz de doses mostrando a % de inibição da combinação trametinib/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 14B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação trametinib/BVD-523. As Figuras 14C e 14D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com BVD-523 e trametinib como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 14E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de BVD-523 com trametinib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0032] A Figura 15A é uma matriz de doses mostrando a % de inibição da combinação trametinib/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com CellTiter-Glo. A Figura 15B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação trametinib/BVD-523. As Figuras 15C e 15D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com BVD-523 e trametinib como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com CellTiter- Glo. A Figura 15E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de BVD-523 com trametinib em células A375 utilizando o ensaio de viabilidade celular com CellTiter-Glo.
[0033] As Figuras 16A-D constituem um conjunto de imagens mostrando a análise por Western Blotting da sinalização MAPK em células A375 após um tratamento de 4 horas com várias concentrações (em nM) de BVD-523, dabrafenib (Dab) e trametinib (Tram). 40 μg de proteína total foram carregados em cada raia exceto quando indicado de outra forma. Nesse experimento, coletaram-se amostras em duplicata. As Figuras 16A e 16B mostram resultados de amostras em duplicata. Do mesmo modo, as Figuras 16C e 16D também mostram resultados de amostras em duplicata. Nas Figuras 16A e 16B, um sinal relativamente fraco de PRSK1 foi detectado em células A375 quando comparado a outros marcadores. Um anticorpo diferente pRSK1-S380, da Cell Signaling (cat. no 11989) foi testado, mas não forneceu um sinal detectável (dados não mostrados). Nas Figuras. 16C e 16D, pCRAF-338 forneceu um sinal mínimo.
[0034] As Figuras 17A-D constituem um conjunto de imagens mostrando a análise por Western Blotting da sinalização MAPK em uma linhagem de células de carcinoma colorretal humano (células HCT116) após um tratamento de 4 horas com várias concentrações (em nM) de BVD-523, dabrafenib (Dab) e Trametinib (Tram). 40 μg de proteína total foram carregados em cada raia exceto quando indicado de outra forma. Nesse experimento, coletaram-se amostras em duplicata. As Figuras 17A e 17B mostram resultados de amostras em duplicata. Do mesmo modo, as Figuras 17C e 17D também mostram resultados de amostras em duplicata. Nas Figuras 17A-17B, os níveis de pRSK1 parecem ser muito baixos em células HCT116 e, nas Figuras 17C e 17D, o sinal de pCRAF-338 foi também muito fraco.
[0035] As Figuras 18A-D constituem um conjunto de imagens mostrando a análise por Western Blotting de proteínas envolvidas no ciclo celular e apoptose em células A375 de melanoma após um tratamento de 24 horas com várias concentrações (em nM) de BVD- 523 ("BVD523"), trametinib ("tram") e/ou dabrafenib ("Dab"), conforme indicado. 50 μg de proteína total foram carregados em cada raia exceto quando indicado de outra forma. Nesse experimento, coletaram-se amostras em duplicata. As Figuras 18A e 18B mostram resultados de amostras em duplicata. Do mesmo modo, as Figuras 18C e 18D também mostram resultados de amostras em duplicata. Nas Figuras 18A e 18B, não se evidenciou banda de tamanho correspondente ao de PARP clivada (89 kDa).
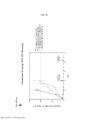
[0036] A Figura 19 é um histograma mostrando a viabilidade de células A375 após 96 horas de incubação com várias quantidades de BVD-523 ou BVD-523 combinado com AZ628 30 nM (um inibidor de RAF) ou dabrafenib 3 nM. Os Escores Bliss são mostrados nas caixas amarelas.
[0037] A Figura 20 é um histograma mostrando a atividade de caspase em células A375 após 24 horas de incubação com várias quantidades de BVD-523 ou BVD-523 combinado com AZ628 30 nM ou dabrafenib 3 nM.
[0038] A Figura 21 é um histograma mostrando a atividade de caspase em células A375 após 48 horas de incubação com várias quantidades de BVD-523 ou BVD-523 combinado com AZ628 30 nM ou dabrafenib 3 nM.
[0039] A Figura 22 é um histograma mostrando a viabilidade de células HCT116 após 96 horas de incubação com várias quantidades de BVD-523 ou BVD-523 combinado com ABT-263 3 μM. Os Escores Bliss são mostrados nas caixas amarelas.
[0040] A Figura 23 é um histograma mostrando a atividade de caspase em células HCT116 após 24 horas de incubação com várias quantidades de BVD-523 ou BVD-523 combinado com ABT-263 3 μM.
[0041] A Figura 24 é um histograma mostrando a atividade de caspase em células HCT116 após 48 horas de incubação com várias quantidades de BVD-523 ou BVD-523 combinado com ABT-263 3 μM.
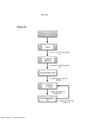
[0042] A Figura 25 é um fluxograma mostrando o protocolo de aumento da dose utilizado no presente.
[0043] A Figura 26 mostra os tempos individuais dos camundongos até o desfecho no estudo.
[0044] A Figura 27 mostra o crescimento tumoral médio (Figura 27A) e o gráfico de Kaplan-Meier (Figura 27B) do estudo.
[0045] As Figuras 28A-28D mostram o crescimento tumoral médio para vários grupos de camundongos que receberam combinações dabrafenib/BVD-523 em comparação às monoterapias.
[0046] A Figura 29 mostra a alteração percentual no peso corporal em relação ao Dia 1 no estudo in vivo.
[0047] A Figura 30 mostra um desenho esquemático da via de proteínas quinases ativadas por mitógenos (MAPK).
[0048] A Figura 31A é uma matriz de doses mostrando a % de inibição da combinação AZ628/trametinib em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 31B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação AZ628/trametinib. As Figuras 31C e 31D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com AZ628 e trametinib como agentes isolados em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 31E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de AZ628/trametinib em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0049] A Figura 32A é uma matriz de doses mostrando a % de inibição da combinação AZ628/BVD-523 em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 32B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação AZ628/BVD-523. As Figuras 32C e 32D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com AZ628 e BVD-523 como agentes isolados em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 32E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de AZ628/BVD-523 em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0050] A Figura 33A é uma matriz de doses mostrando a % de inibição da combinação sorafenib/trametinib em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 33B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação sorafenib/trametinib. As Figuras 33C e 33D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com sorafenib e trametinib como agentes isolados em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 33E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de sorafenib/trametinib em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0051] A Figura 34A é uma matriz de doses mostrando a % de inibição da combinação sorafenib/BVD-523 em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 34B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação sorafenib/BVD-523. As Figuras 34C e 34D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com sorafenib e BVD-523 como agentes isolados em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 34E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de sorafenib/BVD-523 em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0052] A Figura 35A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/trametinib em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 35B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação dabrafenib/trametinib. As Figuras 35C e 35D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e trametinib como agentes isolados em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 35E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib/trametinib em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0053] A Figura 36A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/BVD-523 em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 36B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação dabrafenib/BVD-523. As Figuras 36C e 36D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e BVD-523 como agentes isolados em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 36E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib/BVD-523 em células HCT116 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0054] A Figura 37A é uma matriz de doses mostrando a % de inibição da combinação AZ628/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 37B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação AZ628/BVD-523. As Figuras 37C e 37D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com AZ628 e BVD-523 como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 37E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de AZ628/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0055] A Figura 38A é uma matriz de doses mostrando a % de inibição da combinação sorafenib/trametinib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 38B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação sorafenib/trametinib. As Figuras 38C e 38D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com sorafenib e trametinib como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 38E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de sorafenib/trametinib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0056] A Figura 39A é uma matriz de doses mostrando a % de inibição da combinação sorafenib/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 39B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação sorafenib/BVD-523. As Figuras 39C e 39D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com sorafenib e BVD-523 como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 39E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de sorafenib/BVD- 523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0057] A Figura 40A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/trametinib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 40B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação dabrafenib/trametinib. As Figuras 40C e 40D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e trametinib como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 40E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib/trametinib em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0058] A Figura 41A é uma matriz de doses mostrando a % de inibição da combinação de dabrafenib/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 41B é uma matriz de doses mostrando o excesso sobre Bliss para a combinação dabrafenib/BVD-523. As Figuras 41C e 41D mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e BVD-523 como agentes isolados em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue. A Figura 41E mostra a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos combinados de dabrafenib/BVD-523 em células A375 utilizando o ensaio de viabilidade celular com Alamar Blue.
[0059] A Figura 42 mostra os resultados de ensaios de proliferação com agentes isolados em células A375 (Figura 42A - Figura 42F) e G-361 (Figura 42G - Figura 42L). Os resultados de proliferação são mostrados para o tratamento com dabrafenib (Figura 42A e Figura 42G), vemurafenib (Figura 42B e Figura 42H), TAK-632 (Figura 42C e Figura 42I), BVD-523 (Figura 42D e Figura 42J), SCH772984 (Figura 42E e Figura 42K) e Paclitaxel (Figura 42F e Figura 42L).
[0060] A Figura 43A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/BVD-523 em células A375. A Figura 43B é uma matriz de doses mostrando o excesso de Loewe para a combinação dabrafenib/BVD-523. A Figura 43C é uma matriz de doses mostrando o excesso de Bliss para a combinação de dabrafenib/BVD-523. As Figuras 43D e 43E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e BVD-523 como agentes isolados em células A375.
[0061] A Figura 44A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/SCH772984 em células A375. A Figura 43B é uma matriz de doses mostrando o excesso de Loewe para a combinação dabrafenib/SCH772984. A Figura 43C é uma matriz de doses mostrando o excesso de Bliss para a combinação dabrafenib/SCH772984. As Figuras 43D e 43E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e SCH772984 como agentes isolados em células A375.
[0062] A Figura 45A é uma matriz de doses mostrando a % de inibição da combinação vemurafenib/BVD-523 em células A375. A Figura 45B é uma matriz de doses mostrando o excesso de Loewe para a combinação vemurafenib/BVD-523. A Figura 45C é uma matriz de doses mostrando o excesso de Bliss para a combinação vemurafenib/BVD-523. As Figuras 45D e 45E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com vemurafenib e BVD-523 como agentes isolados em células A375.
[0063] A Figura 46A é uma matriz de doses mostrando a % de inibição da combinação vemurafenib/SCH772984 em células A375. A Figura 46B é uma matriz de doses mostrando o excesso de Loewe para a combinação vemurafenib/SCH772984. A Figura 46C é uma matriz de doses mostrando o excesso de Bliss para a combinação vemurafenib/SCH772984. As Figuras 46D e 46E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com vemurafenib e SCH772984 como agentes isolados em células A375.
[0064] A Figura 47A é uma matriz de doses mostrando a % de inibição da combinação TAK-632/BVD-523 em células A375. A Figura 47B é uma matriz de doses mostrando o excesso de Loewe para a combinação TAK-632/BVD-523. A Figura 47C é uma matriz de doses mostrando o excesso de Bliss para a combinação TAK-632/BVD-523. As Figuras 47D e 47E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com TAK-632 e BVD-523 como agentes isolados em células A375.
[0065] A Figura 48A é uma matriz de doses mostrando a % de inibição da combinação TAK-632/SCH772984 em células A375. A Figura 48B é uma matriz de doses mostrando o excesso de Loewe para a combinação TAK-632/SCH772984. A Figura 48C é uma matriz de doses mostrando o excesso de Bliss para a combinação TAK- 632/SCH772984. As Figuras 48D e 48E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com TAK-632 e SCH772984 como agentes isolados em células A375.
[0066] A Figura 49A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/BVD-523 em células G-361. A Figura 49B é uma matriz de doses mostrando o excesso de Loewe para a combinação dabrafenib/BVD-523. A Figura 49C é uma matriz de doses mostrando o excesso de Bliss para a combinação dabrafenib/BVD-523. As Figuras 49D e 49E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e BVD-523 como agentes isolados em células G-361.
[0067] A Figura 50A é uma matriz de doses mostrando a % de inibição da combinação dabrafenib/SCH772984 em células G-361. A Figura 50B é uma matriz de doses mostrando o excesso de Loewe para a combinação dabrafenib/SCH772984. A Figura 50C é uma matriz de doses mostrando o excesso de Bliss para a combinação dabrafenib/SCH772984. As Figuras 50D e 50E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com dabrafenib e SCH772984 como agentes isolados em células G-361.
[0068] A Figura 51A é uma matriz de doses mostrando a % de inibição da combinação vemurafenib/BVD-523 em células G-361. A Figura 51B é uma matriz de doses mostrando o excesso de Loewe para a combinação vemurafenib/BVD-523. A Figura 51C é uma matriz de doses mostrando o excesso de Bliss para a combinação vemurafenib/BVD-523. As Figuras 51D e 51E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com vemurafenib e BVD-523 como agentes isolados em células G-361.
[0069] A Figura 52A é uma matriz de doses mostrando a % de inibição da combinação vemurafenib/SCH772984 em células G-361. A Figura 52B é uma matriz de doses mostrando o excesso de Loewe para a combinação vemurafenib/SCH772984. A Figura 52C é uma matriz de doses mostrando o excesso de Bliss para a combinação vemurafenib/SCH772984. As Figuras 52D e 52E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com vemurafenib e SCH772984 como agentes isolados em células G-361.
[0070] A Figura 53A é uma matriz de doses mostrando a % de inibição da combinação TAK-632/BVD-523 em células G-361. A Figura 53B é uma matriz de doses mostrando o excesso de Loewe para a combinação TAK-632/BVD-523. A Figura 53C é uma matriz de doses mostrando o excesso de Bliss para a combinação TAK-632/BVD-523. As Figuras 53D e 53E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com TAK-632 e BVD-523 como agentes isolados em células G-361.
[0071] A Figura 54A é uma matriz de doses mostrando a % de inibição da combinação TAK-632/SCH772984 em células G-361. A Figura 54B é uma matriz de doses mostrando o excesso de Loewe para a combinação TAK-632/SCH772984. A Figura 54C é uma matriz de doses mostrando o excesso de Bliss para a combinação TAK- 632/SCH772984. As Figuras 54D and 54E, respectivamente, mostram a % de viabilidade em relação a controles tratados somente com DMSO de tratamentos com TAK-632 e SCH772984 como agentes isolados em células G-361.
[0072] A Figura 55A mostra escores de sinergia para as combinações testadas em células A375 e células G-361. A Figura 55B mostra um gráfico dos valores apresentados na Figura 55A.
[0073] A Figura 56A mostra volumes de Loewe para as combinações testadas em células A375 e células G-361. A Figura 56B mostra um gráfico dos valores apresentados na Figura 56A.
[0074] A Figura 57A mostra volumes de Bliss para as combinações testadas em células A375 e células G-361. A Figura 57B mostra um gráfico dos valores apresentados Figura 57A.
[0075] A Figura 58 mostra os resultados da combinação de BVD- 523 e SCH772984. A Figura 58A apresenta uma matriz de doses mostrando a inibição para a combinação em células A375. As Figuras 58B - 58C mostram os resultados de ensaios de proliferação com os agentes isolados para a combinação na Figura 58A. A Figura 58D mostra o excesso de Loewe para a combinação na Figura 58A e a Figura 58E mostra o excesso de Bliss para a combinação na Figura 58A.
DESCRIÇÃO DETALHADA DA INVENÇÃO
[0076] Uma modalidade da presente invenção é um método para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse método compreende administrar ao indivíduo uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD- 523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste, para tratar ou melhorar os efeitos do câncer.
[0077] Neste relatório descritivo, os termos "tratar", "trata", "tratamento" e variações gramaticais destes significam submeter um indivíduo a um protocolo, regime, processo ou remédio, em que se deseja obter uma resposta fisiológica ou resultado naquele indivíduo, por exemplo, um paciente. Especificamente, os métodos e as composições da presente invenção podem ser utilizados para tornar mais lento o desenvolvimento de sintomas da doença ou retardar o aparecimento da doença ou condição, ou deter a progressão de desenvolvimento da doença. No entanto, pelo fato de todo indivíduo tratado poder não responder a um determinado protocolo de tratamento, regime, processo ou remédio, tratar não requer que a resposta fisiológica ou resultado desejado seja alcançado por cada e todo indivíduo ou toda população de indivíduos, por exemplo, população de pacientes. Desse modo, um dado indivíduo ou população de indivíduos, por exemplo, população de pacientes pode deixar de responder ou responder inadequadamente ao tratamento.
[0078] Neste relatório descritivo, os termos "melhorar", "melhora" e variações gramaticais destes significam reduzir a gravidade dos sintomas de uma doença em um indivíduo.
[0079] Neste relatório descritivo, um "indivíduo" é um mamífero, de preferência, um ser humano. Além de humanos, as categorias de mamíferos no âmbito da presente invenção incluem, por exemplo, animais agrícolas, animais domésticos, animais de laboratório, etc. Alguns exemplos de animais agrícolas incluem vacas, suínos, cavalos, cabras, etc. Alguns exemplos de animais domésticos incluem cães, gatos, etc. Alguns exemplos de animais de laboratório incluem primatas, ratos, camundongos, coelhos, cobaias, etc.
[0080] Na presente invenção, BVD-523 é um composto de acordo com a Fórmula (I):
[0081] e seus sais farmaceuticamente aceitáveis. BVD-523 pode ser sintetizado de acordo com os métodos descritos em, por exemplo, a Patente U.S. No 7 354 939. Os enantiômeros e as misturas racêmicas de ambos os enantiômeros de BVD-523 são também contempladas pelo âmbito da presente invenção. BVD-523 é um inibidor de ERK1/2 com um mecanismo de ação, o qual se acredita ser, por exemplo, único e distinto de certos outros inibidores de ERK1/2, como SCH772984. Por exemplo, outros inibidores de ERK1/2, como SCH772984, inibm a autofosforilação de ERK (Morris et al., 2013), enquanto que BVD-523 permite a autofosforilação de ERK, embora sem deixar de inibir ERK (Figura 18).
[0082] Neste relatório descritivo, "inibidor de RAF" significa aquelas substâncias que (i) interagem diretamente com RAF, por exemplo, ao se ligarem a RAF e (ii) reduzem a expressão ou a atividade de RAF. Os inibidores de RAF podem ser classificados em dois tipos de acordo com seus respectivos modos de ligação. Neste relatório descritivo, inibidores de RAF "Tipo 1" são aqueles inibidores direcionados aos sítios de ligação de ATP da quinase em sua conformação ativa. Os inibidores de RAF "Tipo 2" RAF são aqueles inibidores que se ligam a uma conformação inativa da quinase. Exemplos não limitantes de inibidores de RAF do tipo 1 incluem:
(GlaxoSmithKline), GDC-0879 (Genentech), L-779450 B-Raf (Merck), PLX3202 (Plexxikon), PLX4720 (Plexxikon), SB-590885 (GlaxoSmithKline), SB-699393 (GlaxoSmithKline), vemurafenib (Plexxikon), seus sais farmaceuticamente aceitáveis e combinações destes. De preferência, o inibidor de RAF do tipo 1, é o dabrafenib ou um sal farmaceuticamente aceitável deste.
[0083] Em um aspecto desta modalidade, o indivíduo com cancer possui uma mutação somática em BRAF ou é refratário ao tratamento com inibidores da via MAPK. De preferência, o indivíduo é refratário ao tratamento com inibidores da via MAPK não de ERK.
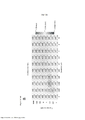
[0084] Neste relatório descritivo, "mutação somática" significa uma alteração ocorrida em qualquer célula não destinada a se tornar uma célula germinativa. A mutação pode ser, por exemplo, uma substituição, deleção, inserção ou uma fusão. A Tabela 1 abaixo mostra uma visão geral da distribuição de mutações no gene BRAF, como apresentadas no banco de dados de Sanger. TABELA 1 - Visão geral da distribuição de mutações no gene BRAF
[0085] Mutações no gene BRAF são encontradas em aproximadamente 66% dos melanomas (Davies et al., 2002; Brose et al., 2002; Hocket et al., 2007), e em uma porcentagem relativamente menor em outros cânceres, 36% dos tumores da tireoide e 10% dos cânceres do cólon (Xu et al., 2003; Fransen et al., 2004). A mutação de BRAF mais prevalente ocorre no aminoácido 600 da proteína quinase do tipo selvagem (SEQ ID NO:2), que leva à substituição de valina por ácido glutâmico resultando na B-RafV600E mutante, que representa cerca de 80% das mutações BRAF (Davies et al., 2002; Hocker et al., 2007). A atividade quinase do domínio quinase de B- RafV600E é 500 vezes mais alta em comparação à atividade basal de B-Raf do tipo selvagem (Wan et al., 2004). Entre as outras mutações em BRAF identificadas no melanoma, V600K e V600D/R são também comuns e representam 16% e 3% de todas as mutações no gene BRAF, respectivamente (Long et al., 2011). Além do melanoma, as mutações em BRAF são também comuns em muitos outros tipos de câncer, incluindo carcinoma papilar da tireoide, carcinoma de ovário e carcinoma colorretal (Wellbrock et al., 2004). Em um estudo, encontraram-se variantes de splicing do gene BRAF (eliminação dos exons 14 e 15) em 5/24 (21%) das linhagens celulares de câncer colorretal (Seth et al., 2009).
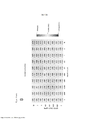
[0086] A Tabela 2 abaixo, com base no banco de dados de Sanger, mostra a distribuição e a frequência de mutações no gene BRAF em tumores humanos. TABELA 2
[0087] A Tabela 3 abaixo mostra sequências selecionadas de ácidos nucleicos e aminoácidos de BRAF. Essas sequências podem ser utilizadas em métodos para identificar indivíduos com genótipo de BRAF mutante (tais como nos métodos apresentados abaixo). TABELA 3
[0088] Métodos para identificar mutações em ácidos nucleicos, como os genes BRAF identificados acima, são conhecidos na técnica. Os ácidos nucleicos podem ser obtidos de amostras biológicas. Na presente invenção, amostras biológicas incluem, entre outros, de sangue, plasma, urina, pele, saliva e de biópsias. As amostras biológicas são obtidas de um indivíduo por procedimentos e métodos rotineiros que são conhecidos na técnica.
[0089] Exemplos não limitantes de métodos para identificar mutações incluem PCR, sequenciamento, captura de híbridos, captura em solução, sondas moleculares de inversão, ensaios de hibridização fluorescente in situ (FISH) e combinações destes.
[0090] Vários métodos de sequenciamento são conhecidos na técnica. Esses incluem, entre outros, sequenciamento de Sanger (também referido como sequenciamento didesoxi) e vários métodos de sequenciamento por síntese (SBS) como os descritos em, por exemplo, Metzker 2005, sequenciamento por hibridização, por ligação (por exemplo, WO 2005021786), por degradação (por exemplo, Patente U.S. Nos 5 622 824 e 6 140 053) e sequenciamento em nanoporos (os quais são disponibilizados comercialmente pela Oxford Nanopore Technologies, Reino Unido). Em técnicas de sequenciamento profundo, um determinado nucleotídeo na sequência é lido mais de uma vez durante o processo de sequenciamento. Técnicas do sequenciamento profundo são reveladas em, por exemplo, Publicação do Pedido de Patente U.S. No 20120264632 e Publicação da Patente Internacional No WO2012125848.
[0091] Os métodos à base de PCR para detectar mutações são conhecidos na técnica e empregam amplificação por PCR, em que cada sequência alvo na amostra possui um par correspondente de primers únicos, específicos para a sequência. Por exemplo, o método de polimorfismos no comprimento de fragmentos de restrição-reação em cadeia da polimerase (PCR-RFLP) permite a detecção rápida de mutações depois que sequências genômicas são amplificadas por PCR. A mutação é discriminada por digestão com endonucleases de restrição específicas e é identificada por eletroforese. Vide, por exemplo, Ota et al., 2007. As mutações podem também ser detectadas utilizando PCR em tempo real. Vide, por exemplo, a publicação do Pedido de Patente Internacional No WO2012046981.
[0092] Métodos de captura de híbridos são conhecidos na técnica e são revelados em, por exemplo, a Publicação do Pedido de Patente U.S. No 20130203632 e nas Patentes U.S. Nos 8 389 219 e 8 288 520. Esses métodos baseiam-se na hibridização seletiva de regiões genômicas alvo com os oligonucleotídeos criados pelo usuário. A hibridização pode ser com oligonucleotídeos imobilizados em microarranjos de alta ou baixa densidade (captura em arranjo) ou pode ser hibridização em fase solúvel com oligonucleotídeos modificados com um ligante (por exemplo, biotina) os quais podem ser imobilizados subsequentemente em uma superfície sólida, como uma microesfera (captura em solução).
[0093] Os métodos de sondas moleculares de inversão (MIP) são conhecidos na técnica e são descritos em, por exemplo, Absalan et al., 2008. Tais métodos utilizam moléculas MIP, que são sondas especiais "padlock" (cadeado) (Nilsson et al., 1994) para genotipagem. Uma molécula MIP é um oligonucleotídeo linear que contém regiões específicas, sequências universais, sítios de restrição e uma sequência Tag (indexação) (16-22 bp). Em tais métodos, uma sonda MIP hibridiza-se diretamente em volta do marcador genético/SNP de interesse. O método MIP pode também utilizar diversos conjuntos de sondas "padlock" que se hibridizam com o DNA genômico em paralelo (Hardenbol et al., 2003). Em caso de pareamento perfeito, regiões com homologia genômica são ligadas, passando por uma inversão na configuração (conforme sugerido pelo nome da técnica) e criando uma molécula circular. Depois da primeira restrição, todas as moléculas são amplificadas com primers universais. Os amplicons sofrem nova restrição para assegurar a criação de fragmentos curtos para hibridização em um microarranjo. Os fragmentos curtos gerados são marcados e, por meio de uma sequência Tag, hibridizados com um cTag (fita complementar para indexação) em um arranjo. Após a formação de um duplex Tag-cTag, detecta-se um sinal.
[0094] Neste relatório descritivo, ser "refratário" ao tratamento com inibidores da via MAPK significa que um ou mais inibidores da via MAPK têm eficácia reduzida no tratamento de câncer.
[0095] Neste relatório descritivo, um "inibidor da via de proteínas quinases ativadas por mitógenos (MAPK)" é qualquer substância que reduz a atividade, a expressão ou a fosforilação de proteínas na via MAPK que resultam em redução do crescimento celular ou aumento na morte celular.
[0096] Uma visão geral das cascatas das MAPK de mamíferos é mostrada na Figura 30. Os detalhes das vias MAPK são revisados em, por exemplo, Akinleye et al., 2013. Resumidamente, com respeito ao módulo de ERK1/2 na Figura 30 (caixa lilás), a cascata de sinalização de MAPK 1/2 é ativada pela ligação do ligante aos receptores tirosina quinase (RTK). Os receptores ativados recrutam e fosforilam as proteínas adaptadoras Grb2 e SOS, as quais então interagem com a Ras GTPase ligada à membrana e provoca a sua ativação. Em sua forma ativada GTP ligada, a Ras recruta e ativa Raf quinases (A-Raf, B-Raf e C-Raf/RaF-1). As Raf quinases ativadas ativam MAPK 1/2 (MKK1/2), a qual, por sua vez, catalisa a fosforilação de resíduos de treonina e tirosina na sequência de ativação Thr-Glu-Tyr de ERK1/2. Com respeito ao módulo de JNK/p38 (caixa amarela na Figura 30), as quinases a montante, MAP3Ks, como MEKK1/4, ASK1/2 e MLK1/2/3, ativam MAP2K3/6 (MKK3/6), MAP2K4 (MKK4) e MAP2K7 (MKK7). Essas MAP2Ks ativam a seguir as proteínas quinases JNK, incluindo JNK1, JNK2 e JNK3, bem como p38 α/β/Y/δ. Para executarem suas funções, as JNKs ativam diversos fatores de transcrição, incluindo c- Jun, ATF-2, NF-ATc1, HSF-1 e STAT3. Com respeito ao módulo de ERK5 (caixa azul na Figura 30), as quinases acima de MAP2K5 (MKK5) são MEKK2 e MEKK3. O alvo mais bem caracterizado abaixo da MEK5 é a ERK5, conhecida também como grande MAP quinase 1 (BMK1) por seu tamanho ser o dobro daquele de outras MAPKs.
[0097] Exemplos não limitantes de inibidores da via MAPK incluem inibidores de RAS, inibidores de RAF, inibidores de MEK, inibidores de ERK1/2, seus sais farmaceuticamente aceitáveis e combinações destes.
[0098] Neste relatório descritivo, um "inibidor de RAS" significa aquelas substâncias que (i) interagem diretamente com RAS, por exemplo, ligando-se à proteína RAS e (ii) diminuem a expressão ou a atividade de RAS. Exemplos não limitantes de inibidores de RAS incluem, entre outros, inibidores da farnesil transferase (como, por exemplo, tipifarnib e lonafarnib), pequenas moléculas contendo grupo farnesila (como, por exemplo, salirasib e TLN-4601), DCAI, como revelado por Maurer (Maurer et al., 2012), Kobe0065 e Kobe2602, como revelados por Shima (Shima et al., 2013), HBS 3 (Patgiri et al., 2011) e AIK-4 (Allinky).
[0099] Neste relatório descritivo, um "inibidor de RAF" significa aquelas substâncias que (i) interagem diretamente com RAF, por exemplo, ligando-se à proteína RAS e (ii) diminuem a expressão ou a atividade de RAS. Exemplos não limitantes de inibidores de RAF, como, por exemplo, A-RAF, B-RAF e C-RAF (Raf-1). Exemplos não limitantes de inibidores de RAF incluem:
[00100] AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ-736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene- 283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 523 (cctatcgttagagtcttcctg) de BRAF (Liu et al., 2007), CTT239065 (Institute of Cancer Research), dabrafenib (GSK2118436), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GDC-0879 (Genentech), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), L779450 (Merck), LBT613 (Novartis), LErafAON (NeoPharm, Inc.), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX3202 (Plexxikon), PLX4720 (Plexxikon), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF-365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), SB-590885 (GlaxoSmithKline), SB699393 (GlaxoSmithKline), sorafenib (Onyx Pharmaceuticals), TAK 632 (Takeda), TL-241 (Teligene), vemurafenib (RG7204 ou PLX4032) (Daiichi Sankyo), XL-281 (Exelixis), ZM-336372 (AstraZeneca), seus sais farmaceuticamente aceitáveis e combinações destes.
[00101] Neste relatório descritivo, um "inibidor de MEK" significa aquelas substâncias que (i) interagem diretamente com MEK, por exemplo, ligando-se à proteína MEK e (ii) diminuem a expressão ou a atividade de MEK. Dessa forma, os inibidores que atuam acima de MEK, como os inibidores de RAS e os inibidores de RAF, não são inibidores de MEF de acordo com a presente invenção. Exemplos não limitantes de inibidores de MEK incluem a toxina do antraz, antroquinonol (Golden Biotechnology), ARRY-142886 ((2-hidróxi- etoxi)-amida do ácido 6-(4-bromo-2-cloro-fenilamino)-7-fluoro-3-metil- 3H-benzoimidazol-5-carboxílico) (Array BioPharma), ARRY-438162 (Array BioPharma), AS-1940477 (Astellas), AS-703988 (Merck KGaA), bentamapimode (Merck KGaA), BI-847325 (Boehringer Ingelheim), E- 6201 (Eisai), GDC-0623 (Hoffmann-La Roche), GDC-0973 (cobimetinib) (Hoffmann-La Roche), L783277 (Merck), fator letal, parte da toxina do antraz, MEK162 (Array BioPharma), PD 098059 (2-(2'- amino-3'-metoxifenil)-oxanaftalen-4-ona) (Pfizer), PD 184352 (CI-1040) (Pfizer), PD-0325901 (Pfizer), pimasertib (Santhera Pharmaceuticals), RDEA119 (Ardea Biosciences/Bayer), refametinib (AstraZeneca), RG422 (Chugai Pharmaceutical Co.), RO092210 (Roche), RO4987655 (Hoffmann-La Roche), RO5126766 (Hoffmann-La Roche), selumetinib (AZD6244) (AstraZeneca), SL327 (Sigma), TAK-733 (Takeda), trametinib (Japan Tobacco), U0126 (1,4-diamino-2,3-diciano-1,4-bis(2- aminofeniltio)butadieno) (Sigma), WX-554 (Wilex), polipeptídeo YopJ (Mittal et al., 2010), seus sais farmaceuticamente aceitáveis e combinações destes.
[00102] Neste relatório descritivo, um "inibidor de ERK1/2" significa aquelas substâncias que (i) interagem diretamente com ERK1 e/ou ERK2, por exemplo, ligando-se à proteína ERK1/2 e (ii) diminuem a expressão ou a atividade das proteínas quinases ERK1 e/ou ERK2. Portanto, os inibidores que atuam a montante de ERK1/2, como os inibidores de MEK e os inibidores de RAF, não são inibidores de ERK1/2 de acordo com a presente invenção. Exemplos não limitantes de um inibidor de ERK1/2 incluem AEZS-131 (Aeterna Zentaris), AEZS-136 (Aeterna Zentaris), BVD-523, SCH-722984 (Merck & Co.), SCH-772984 (Merck & Co.), SCH-900353 (MK-8353) (Merck & Co.), seus sais farmaceuticamente aceitáveis e combinações destes.
[00103] Em outro aspecto desta modalidade, o método compreende ainda administrar ao indivíduo pelo menos um agente terapêutico adicional eficaz para tratar ou melhorar os efeitos do câncer. O agente terapêutico adicional pode ser selecionado a partir do grupo constituído por um anticorpo ou seu fragmento, um agente citotóxico, uma toxina, um radionuclídeo, um imunomodulador, um agente terapêutico fotoativo, um agente radiossensibilizante, um hormônio, um agente antiangiogênico e combinações destes.
[00104] Neste relatório descritivo, "anticorpo" abrange imunoglobulinas de ocorrência natural bem como imunoglobulinas de ocorrência não natural, incluindo, por exemplo, anticorpos de cadeia única, anticorpos quiméricos (por exemplo, anticorpos murinos humanizados) e anticorpos heteroconjugados (por exemplo, anticorpos biespecíficos). Os fragmentos de anticorpos incluem aqueles que se ligam ao antígeno (por exemplo, Fab', F(ab')2, Fab, Fv e rIgG). Vide também, por exemplo, Pierce Catalog and Handbook, 1994-1995 (Pierce Chemical Co., Rockford, Ill.); Kuby, J., Immunology, 3rd Ed., W.H. Freeman & Co., Nova York (1998). O termo anticorpo também inclui moléculas bivalentes ou biespecíficas, diabodies, triabodies e tetrabodies. O termo "anticorpo" inclui ainda anticorpos policlonais e monoclonais.
[00105] Exemplos de anticorpos terapêuticos que podem ser utilizados na presente invenção incluem rituximabe (Rituxan), cetuximabe (Erbitux), bevacizumabe (Avastin) e ibritumomabe (Zevalin).
[00106] Os agentes citotóxicos de acordo com a presente invenção incluem agentes que danificam o DNA, antimetabólitos, agentes antimicrotúbulos, agentes antibióticos, etc. Os agentes que danificam o DNA incluem agentes alquilantes, agentes à base de platina, agentes intercalantes e inhibidores da replicação de DNA. Exemplos não limitantes de agentes alquilantes de DNA incluem ciclofosfamida, mecloretamina, uramustina, melfalano, clorambucil, ifosfamida, carmustina, lomustina, estreptozocina, bussulfano, temozolomida, seus sais farmaceuticamente aceitáveis, pró-fármacos e combinações destes. Exemplos não limitantes de agentes à base de platina incluem cisplatina, carboplatina, oxaliplatina, nedaplatina, satraplatina, tetranitrato de triplatina, seus sais farmaceuticamente aceitáveis, pró-fármacos e combinações destes. Exemplos não limitantes de agentes intercalantes incluem doxorrubicina, daunorrubicina, idarrubicina, mitoxantrona, seus sais farmaceuticamente aceitáveis, pró-fármacos e combinações destes. Exemplos não limitantes de inibidores da replicação de DNA incluem irinotecano, topotecano, ansacrina, etoposídeo, fosfato de etoposídeo, teniposídeo, seus sais farmaceuticamente aceitáveis, pró-fármacos e combinações destes. Os antimetabólitos incluem antagonista do folato, como metotrexato e pemetrexede, antagonistas da purina, como 6- mercaptopurina, dacarbazina e fludarabina, e antagonistas da pirimidina como 5-fluorouracila, arabinosilcitosina, capecitabina, gencitabina, decitabina, seus sais farmaceuticamente aceitáveis, pró- fármacos e combinações destes. Os agentes antimicrotúbulos incluem, entre outros, alcaloides da vinca, paclitaxel (Taxol®), docetaxel (Taxotere®) e ixabepilona (Ixempra®). Os agentes antibióticos incluem, entre outros, actinomicina, antraciclinas, valrubicina, epirrubicina, bleomicina, plicamicina, mitomicina, seus sais farmaceuticamente aceitáveis, pró-fármacos e combinações destes.
[00107] Os agentes citotóxicos de acordo com a presente invenção também incluem um inibidor da via PI3K/Akt. Exemplos não limitantes de um inibidor da via PI3K/Akt incluem A-674563 (CAS no 552325-73-2), AGL 2263, AMG-319 (Amgen, Thousand Oaks, CA), AS-041164 (5-benzo[1,3]dioxol-5-ilmetileno-tiazolidina-2,4-diona), AS- 604850 (5-(2,2-diflúor-benzo[1,3]dioxol-5-ilmetileno)-tiazolidina-2,4- diona), AS-605240 (5-quinoxilin-6-metileno-1,3-tiazolidina-2,4-diona), AT7867 (CAS no 857531-00-1), série de benzimidazóis, Genentech (Roche Holdings Inc., South San Francisco, CA), BML-257 (CAS no 32387-96-5), CAL-120 (Gilead Sciences, Foster City, CA), CAL-129 (Gilead Sciences), CAL-130 (Gilead Sciences), CAL-253 (Gilead Sciences), CAL-263 (Gilead Sciences), CAS no 612847-09-3, CAS no 681281-88-9, CAS no 75747-14-7, CAS no 925681-41-0, CAS no 98510-80-6, CCT128930 (CAS no 885499-61-6), CH5132799 (CAS no 1007207-67-1), CHR-4432 (Chroma Therapeutics, Ltd., Abingdon, Reino Unido), FPA 124 (CAS no 902779-59-3), GS-1101 (CAL-101) (Gilead Sciences), GSK 690693 (CAS no 937174-76-0), H-89 (CAS no 127243-85-0), Honokiol, IC87114 (Gilead Science), IPI-145 (Intellikine Inc.), KAR-4139 (Karus Therapeutics, Chilworth, UK), KAR-4141 (Karus Therapeutics), KIN-1 (Karus Therapeutics), KT 5720 (CAS no 108068-98-0), Miltefosina, MK-2206 dicloridrato (CAS no 1032350-132), ML-9 (CAS no 105637-50-1), cloridrato de naltrindol, OXY-111A (NormOxys Inc., Brighton, MA), perifosina, PHT-427 (CAS no 119195157-1), inibidor da isoformas delta de PI3 quinase, Merck KGaA (Merck & Co., Whitehouse Station, NJ), inibidores de PI3 quinase delta, Genentech (Roche Holdings Inc.), inibidores de PI3 quinase delta, Incozen (Incozen Therapeutics, Pvt. Ltd., Hydrabad, Índia), inibidores-2 de PI3 quinase delta, Incozen (Incozen Therapeutics), inibidor de PI3 quinase, Roche-4 (Roche Holdings Inc.), inibidores de PI3 quinase, Roche (Roche Holdings Inc.), inibidores de PI3 quinase, Roche-5 (Roche Holdings Inc.), inibidores de PI3-alfa/delta, Pathway Therapeutics (Pathway Therapeutics Ltd., South San Francisco, CA), inibidores de PI3-delta, Cellzome (Cellzome AG, Heidelberg, Alemanha), inibidores de PI3-delta, Intellikine (Intellikine Inc., La Jolla, CA), inibidores de PI3-delta, Pathway Therapeutics-1 (Pathway Therapeutics Ltd.), inibidores de PI3-delta, Pathway Therapeutics-2 (Pathway Therapeutics Ltd.), inibidores de PI3-delta/gama, Cellzome (Cellzome AG), inibidores de PI3-delta/gama, Cellzome (Cellzome AG), inibidores de PI3-delta/gama, Intellikine (Intellikine Inc.), inibidores de PI3-delta/gama, Intellikine (Intellikine Inc.), inibidores de PI3-delta/gama, Pathway Therapeutics (Pathway Therapeutics Ltd.), inibidores de PI3-delta/gama, Pathway Therapeutics (Pathway Therapeutics Ltd.), inibidor de PI3-gama, Evotec (Evotec), inibidor de PI3-gama, Cellzome (Cellzome AG), inibidores de PI3-gama, Pathway Therapeutics (Pathway Therapeutics Ltd.), inibidores de PI3K delta/gama, Intellikine-1 (Intellikine Inc.), inibidores de PI3K delta/gama, Intellikine-1 (Intellikine Inc.), pictilisib (Roche Holdings Inc.), PIK-90 (CAS no 677338-12-4), SC-103980 (Pfizer, New York, NY), SF-1126 (Semafore Pharmaceuticals, Indianapolis, IN), SH-5, SH-6, tetra-hidro curcumina, TG100-115 (Targegen Inc., San Diego, CA), triciribina, X-339 (Xcovery, West Palm Beach, FL), XL-499 (Evotech, Hamburgo, Alemanha), seus sais farmaceuticamente aceitáveis e combinações destes.
[00108] Na presente invenção, o termo "toxina" significa um veneno antigênico ou veneno de origem vegetal ou animal. Um exemplo constitui a toxina diftérica ou partes desta.
[00109] Na presente invenção, o termo "radionuclídeo" significa uma substância radioativa administrada ao paciente, por exemplo, por via intravenosa ou oral, após o qual, penetra mediante o metabolismo normal do paciente no órgão ou tecido alvo, onde libra radiação local por um curto período de tempo. Os exemplos de radionuclídeos incluem, entre outros, I-125, At-211, Lu-177, Cu-67, I-131, Sm-153, Re-186, P-32, Re-188, In-114m e Y-90.
[00110] Na presente invenção, o termo "imunomodulador" significa uma substância que altera a resposta imune ao aumentar ou reduzir a capacidade do sistema imune para produzir anticorpos ou células sensibilizadas que reconhecem e reagem com o antígeno que iniciou a produção deles. Os imunomoduladores podem ser preparados recombinantes, sintéticos ou naturais e incluem citocinas, corticosteroides, agentes citotóxicos, timosina e imunoglobulinas. Alguns imunomoduladores estão presentes naturalmente no corpo, e certos destes estão disponíveis em preparados farmacológicos. Os exemplos de imunomoduladores incluem, entre outros, fator estimulador de colônias de granulócitos (G-CSF), interferons, imiquimode e frações da membrana celular de bactérias, IL-2, IL-7, IL- 12, CCL3, CCL26, CXCL7 e citosina-fosfato-guanosina sintética (CpG).
[00111] Na presente invenção, o termo "agente terapêutico fotoativo" significa aqueles compostos e composições que ficam ativos quando da exposição à luz. Certos exemplos de agentes terapêuticos fotoativos são revelados, por exemplo, no Pedido de Patente U.S. No de Série 2011/0152230 A1, "Photoactive Metal Nitrosyls For Blood Pressure Regulation And Cancer Therapy".
[00112] Na presente invenção, o termo "agente radiossensibilizante" significa um composto que faz com que as células tumorais se tornem mais sensíveis à radioterapia. Os exemplos de agentes radiossensibilizantes incluem misonidazol, metronidazol, tirapazamina e trans crocetinato de sódio.
[00113] Na presente invenção, o termo "hormônio" significa uma substância librada por células em uma parte do corpo que afeta a células em outra parte do corpo. Os exemplos de hormônios incluem, entre outros, prostaglandinas, leucotrienos, prostaciclina, tromboxano, amilina, hormônio antimülleriano, adiponectina, hormônio adrenocorticotrófico, angiotensinogênio, angiotensina, vasopressina, atriopeptina, peptídeo natriurético cerebral, calcitonina, colecistoquinina, hormônio librador de corticotrofina, encefalina, endotelina, eritropoietina, hormônio folículo-estimulante, galanina, gastrina, grelina, glucagon, hormônio librador de gonadotrofina, hormônio librador do hormônio do crescimento, gonadotrofina coriônica humana, lactogênio placentário humano, hormônio do crescimento, inibina, insulina, somatomedina, leptina, liptropina, hormônio luteinizante, hormônio estimulador de melanócitos, motilina, orexina, oxitocina, polipeptídeo pancreático, hormônio da paratireoide, prolactina, hormônio librador de prolactina, relaxina, renina, secretina, somatostaina, trombopoietina, hormônio tireoestimulante, testosterona, desidroepiandrosterona, androstenediona, dihidrotestosterona, aldosterona, estradiol, estrona, estriol, cortisol, progesterona, calcitriol e calcidiol.
[00114] Alguns compostos interferem com a atividade de certos hormônios ou interrompem a produção de certos hormônios. Esses compostos interferentes hormonais incluem, entre outros, tamoxifeno (Nolvadex®), anastrozol (Arimidex®), letrozol (Femara®) e fulvestrant (Faslodex®). Tais compostos estão também dentro do significado de hormônio na presente invenção.
[00115] Neste relatório descritivo, um agente "antiangiogênico" significa uma substância que reduz ou inib o crescimento de novos vasos sanguíneos, como, por exemplo, um inibidor do fator de crescimento endotelial vascular (VEGF) e um inibidor da migração de células endoteliais. Os agentes antiangiogênicos incluem, entre outros, 2-metoxiestradiol, angiostatina, bevacizumabe, fator inibidor de angiogênese derivado de cartilagem, endostatina, IFN-α, IL-12, itraconazol, linomida, fator plaquetário 4, prolactina, SU5416, suramina, tasquinimode, tecogalano, tetratiomolibdato, talidomida, trombospondina, TNP-470, ziv-aflibrcept, seus sais farmaceuticamente aceitáveis, pró-fármacos e combinações destes.
[00116] Em um aspecto adicional desta modalidade, a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente. Neste relatório descritivo, "sinérgico" significa mais do que aditivo. Os efeitos sinérgicos podem ser medidos por vários ensaios conhecidos na técnica, incluindo, entre outros, aqueles aqui descritos, tais como o ensaio do excesso sobre Bliss.
[00117] Outra modalidade da presente invenção é um método para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse método compreende administrar ao indivíduo uma quantidade eficaz de (i) BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é o dabrafenib ou um sal farmaceuticamente aceitável deste, para tratar ou melhorar os efeitos do câncer.
[00118] Indivíduos adequados e preferidos são aqueles aqui descritos. Nesta modalidade, os métodos podem ser utilizados para tratar os cânceres descritos acima, incluindo aqueles cânceres com os perfis mutacionais identificados acima. Os métodos para identificar tais mutações são também apresentados acima.
[00119] Em um aspecto desta modalidade, o BVD-523 ou um sal farmaceuticamente aceitável deste é administrado na forma de uma composição farmacêutica compreendendo ainda um veículo ou diluente farmaceuticamente aceitável.
[00120] Em mais um aspecto desta modalidade, p dabrafenib ou um sal farmaceuticamente aceitável deste é administrado na forma de uma composição farmacêutica compreendendo ainda um veículo ou diluente farmaceuticamente aceitável.
[00121] Em mais um aspecto desta modalidade, o método compreende ainda administrar ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme descritos no presente.
[00122] Em um aspecto adicional desta modalidade, a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
[00123] Outra modalidade da presente invenção é um método para causar a morte de células cancerosas. Esse método compreende contatar a célula cancerosa com uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste.
[00124] Inibidores de RAF tipo 1 adequados e preferidos são aqueles aqui descritos. Nesta modalidade, causar a morte de células cancerosas pode ser realizado em células cancerosas com vários perfis mutacionais e/ou que são caracterizadas como aquelas descritas acima. Os métodos para identificar tais mutações são também apresentados acima.
[00125] Em um aspecto desta modalidade, os métodos podem ser realizados in vitro ou in vivo, e podem ser utilizados para causar a morte de células cancerosas, por exemplo, eliminando as células cancerosas, em células dos tipos de câncer aqui descritos.
[00126] Em outro aspecto desta modalidade, a célula cancerosa é uma célula cancerosa de mamíferos. De preferência, a célula cancerosa de mamíferos é obtida de um mamífero selecionado a partir do grupo constituído por seres humanos, primatas, animais agrícolas e animais domésticos. Mais preferivelmente, a célula cancerosa de mamíferos é uma célula cancerosa humana.
[00127] Em mais um aspecto desta modalidade, o contato da célula cancerosa com o primeiro e o segundo agente anticâncer proporciona um efeito sinérgico quando comparado ao contato da célula cancerosa com qualquer um dos agentes anticâncer isoladamente.
[00128] Em outro aspecto desta modalidade, o método compreende ainda o contato da célula cancerosa com ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descrito.
[00129] Em mais um aspecto desta modalidade, o contato da célula cancerosa com o primeiro e o segundo agente anticâncer proporciona um efeito sinérgico quando comparado ao contato da célula cancerosa com qualquer um dos agentes anticâncer isoladamente. Nesta modalidade, "contatar" significa levar o BVD-523 e os inibidores de RAF tipo 1 e, opcionalmente, um ou mais agentes terapêuticos adicionais a uma grande proximidade com as células cancerosas. Isso pode ser realizado utilizando técnicas convencionais de libração de fármacos a mamíferos ou, na situação in vitro, por exemplo, colocando BVD-523 e os inibidores de RAF tipo 1 e, opcionalmente outros agentes terapêuticos em um meio de cultura no qual as células cancerosas se encontram.
[00130] Mais uma modalidade da presente invenção constitui um kit para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse kit compreende uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste, embalados junto com instruções para o seu uso.
[00131] Esses kits podem também incluir recipientes adequados para o armazenamento, por exemplo, ampolas, frascos-ampolas, tubos, etc., para cada agente anticâncer da presente invenção (os quais, podem estar na forma de composições farmacêuticas) e outros reagentes, por exemplo, tampões, soluções salinas balanceadas, etc. para uso na administração dos agentes anticâncer aos indivíduos. Os agentes anticâncer da invenção e outros reagentes podem estar presentes nos kits em qualquer forma conveniente, tais como, por exemplo, em forma de solução ou de pó. Os kits podem incluir ainda um recipiente de embalagem, opcionalmente dispondo de um ou mais compartimentos para abrigar a composição farmacêutica e os outros reagentes opcionais.
[00132] Os indivíduos e inibidores de RAF tipo 1 adequados e preferidos são como aqui descritos. Nesta modalidade, o kit pode ser utilizado para tratar os cânceres descritos acima, incluindo aqueles cânceres com os perfis mutacionais identificados neste relatório descritivo. Os métodos para identificar tais mutações são apresentados acima.
[00133] Em mais um aspecto desta modalidade, o kit compreende ainda ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descritos.
[00134] Em um aspecto adicional desta modalidade, a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
[00135] Outra modalidade da presente invenção é uma composição farmacêutica para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Essa composição farmacêutica compreende um diluente ou veículo farmaceuticamente aceitável e uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um tipo de inibidor de RAF do tipo 1 ou um sal farmaceuticamente aceitável deste, em que a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente. Essa composição farmacêutica pode compreender ainda um diluente ou veículo farmaceuticamente aceitável.
[00136] Os indivíduos e inibidores de RAF tipo 1 adequados e preferidos são como aqui descritos. As composições farmacêuticas da invenção podem ser utilizadas para tratar os cânceres descritos acima, incluindo aqueles cânceres com os perfis mutacionais identificados neste relatório descritivo. Os métodos para identificar tais mutações são apresentados acima.
[00137] Em mais um aspecto desta modalidade, a composição farmacêutica compreende ainda ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descritos.
[00138] Outra modalidade da presente invenção é um método para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse método compreende administrar ao indivíduo uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD- 523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ-736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX- 818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF-365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes, para tratar ou melhorar os efeitos do câncer. De preferência, o segundo agente anticâncer é o regorafenib ou um sal farmaceuticamente aceitável deste.
[00139] Nesta modalidade, os indivíduos adequados e preferidos são como aqui descritos. Nesta modalidade, os métodos podem ser utilizados para tratar os cânceres descritos acima, incluindo aqueles cânceres com perfis mutacionais identificados acima. Os métodos para identificar tais mutações são também como apresentados acima.
[00140] Em mais um aspecto desta modalidade, o método compreende ainda administrar ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descritos.
[00141] Em outro aspecto desta modalidade, a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
[00142] Uma modalidade adicional da presente invenção é um método para causar a morte de células cancerosas. Esse método compreende contatar a célula cancerosa com uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ- 736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF- 365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes. De preferência, o segundo agente anticâncer é o regorafenib ou um sal farmaceuticamente aceitável deste.
[00143] As células cancerosas adequadas e preferidas são como aqui descritas. Nesta modalidade, causar a morte da célula cancerosa pode ser realizado em células cancerosas com vários perfis mutacionais e/ou que são caracterizadas conforme descrito acima. Métodos para identificar tais mutações são também como apresentados acima.
[00144] Os métodos desta modalidade, os quais podem ser realizados in vitro ou in vivo, podem ser empregados para causar a morte de células cancerosas, por exemplo, eliminando as células cancerosas, em células dos tipos de câncer aqui descritos.
[00145] Em um aspecto desta modalidade, a célula cancerosa é uma célula cancerosa de mamíferos. De preferência, a célula cancerosa de mamíferos é obtida de um mamífero selecionado a partir do grupo constituído por seres humanos, primatas, animais agrícolas e animais domésticos. Mais preferivelmente, a célula cancerosa de mamíferos é uma célula cancerosa humana.
[00146] Em outro aspecto desta modalidade, o método compreende ainda administrar ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descritos.
[00147] Em mais um aspecto desta modalidade, o contato da célula cancerosa com o primeiro e o segundo agente anticâncer proporciona um efeito sinérgico quando comparado ao contato da célula cancerosa com qualquer um dos agentes anticâncer isoladamente.
[00148] Nesta modalidade, "contatar" significar levar o BVD-523 e inibidores de RAF e, opcionalmente, um ou mais agentes terapêuticos adicionais a uma grande proximidade com as células cancerosas. Isso pode ser realizado utilizando técnicas convencionais de libração de fármacos a mamíferos ou, na situação in vitro, por exemplo, colocando BVD-523 e inibidores de RAF e, opcionalmente, outros agentes terapêuticos em um meio de cultura no qual se encontram as células cancerosas.
[00149] Mais uma modalidade da presente invenção é um kit para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Esse kit compreende uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ- 736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX-818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF- 365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes, embalados junto com instruções para o seu uso. De preferência, o segundo agente anticâncer é o regorafenib ou um sal farmaceuticamente aceitável deste.
[00150] Nesta modalidade, os indivíduos adequados e preferidos são como aqui descritos. Nesta modalidade, o kit pode ser utilizado para tratar os cânceres descritos acima, incluindo aqueles cânceres com os perfis mutacionais definidos neste relatório descritivo. Métodos para identificar tais mutações são como apresentados acima.
[00151] Em mais um aspecto desta modalidade, o kit compreende ainda ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descritos.
[00152] Em outro aspecto desta modalidade, a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
[00153] Outra modalidade da presente invenção é uma composição farmacêutica para tratar ou melhorar os efeitos de um câncer em um indivíduo que o necessita. Essa composição farmacêutica compreende um diluente ou veículo farmaceuticamente aceitável e uma quantidade eficaz de (i) um primeiro agente anticâncer, que é o BVD-523 ou um sal farmaceuticamente aceitável deste, e de (ii) um segundo agente anticâncer, que é um inibidor de RAF selecionado a partir do grupo constituído por AAL881 (Novartis); AB-024 (Ambit Biosciences), ARQ-736 (ArQule), ARQ-761 (ArQule), AZ628 (Axon Medchem BV), BeiGene-283 (BeiGene), BIIB-024 (MLN 2480) (Sunesis & Takeda), inibidor de b-raf (Sareum), inibidor de BRAF quinase (Selexagen Therapeutics), siRNA 313 (tacaccagcaagctagatgca) e 253 (cctatcgttagagtcttcctg) de BRAF, CTT239065 (Institute of Cancer Research), DP-4978 (Deciphera Pharmaceuticals), HM-95573 (Hanmi), GW-5074 (Sigma Aldrich), ISIS 5132 (Novartis), LErafAON (NeoPharm, Inc.), LBT613 (Novartis), LGX- 818 (Novartis), pazopanib (GlaxoSmithKline), PLX5568 (Plexxikon), RAF-265 (Novartis), RAF-365 (Novartis), regorafenib (Bayer Healthcare Pharmaceuticals, Inc.), RO 5126766 (Hoffmann-La Roche), TAK 632 (Takeda), TL-241 (Teligene), XL-281 (Exelixis), seus sais farmaceuticamente aceitáveis e combinações destes, em que a administração do primeiro e do segundo agente anticâncer proporciona um efeito sinérgico quando comparada à administração de qualquer um desses agentes anticâncer isoladamente.
[00154] Nesta modalidade, os indivíduos adequados e preferidos são como aqui descritos. As composições farmacêuticas da invenção podem ser utilizadas para tratar os cânceres descritos acima, incluindo aqueles cânceres com os perfis mutacionais identificados neste relatório descritos. Métodos para identificar tais mutações são também como apresentados acima.
[00155] Em mais um aspecto desta modalidade, a composição farmacêutica compreende ainda ao menos um agente terapêutico adicional, de preferência um inibidor da via PI3K/Akt, conforme aqui descritos.
[00156] As composições farmacêuticas de acordo com a presente invenção podem estar em uma forma de dose unitária compreendendo ambos os agentes anticâncer. Em outro aspecto desta modalidade, o primeiro agente anticâncer esta em uma primeira forma de dose unitária e o segundo agente anticâncer está em uma segunda forma de dose unitária, separada da primeira.
[00157] O primeiro e o segundo agente anticâncer podem ser coadministrados ao indivíduo, seja simultaneamente ou em momentos diferentes, conforme considerado adequado por um médico. Se o primeiro e o segundo agente anticâncer forem administrados em momentos diferentes, por exemplo, uma administração em série, o primeiro agente anticâncer pode ser administrado ao indivíduo antes do segundo agente anticâncer. Alternativamente, o segundo agente anticâncer pode ser administrado ao indivíduo antes do primeiro agente anticâncer.
[00158] Na presente invenção, uma "quantidade eficaz" ou uma "quantidade terapeuticamente eficaz" de um agente anticâncer da invenção, incluindo composições farmacêuticas o contendo que são descritas no presente, é uma quantidade de tal agente ou composição que é suficiente para serem obtidos resultados benéficos ou desejados, como os aqui descritos, quando administrada a um indivíduo. Formas farmacêuticas, modos de administração e as doses eficazes podem ser determinados empiricamente, e fazer tais determinações está ao alcance da competência no assunto. Os técnicos no assunto entendem que a dose variará com a via de administração, a taxa de excreção, a duração do tratamento, a identidade de quaisquer outros fármacos administrados, a idade, o tamanho e a espécie de mamífero, por exemplo, paciente humano, e fatores semelhantes bem conhecidos nas áreas da Medicina e Medicina Veterinária. Em geral, uma dose adequada de um agente ou composição de acordo com a invenção será aquela quantidade do agente ou composição, que é a dose mais baixa eficaz para produzir o efeito desejado. A dose eficaz de um agente ou composição da presente invenção pode ser administrada em duas, três, quatro, cinco ou mais subdoses, administradas separadamente em intervalos apropriados ao longo do dia.
[00159] Um exemplo adequado, não limitante de uma dose de BVD-523, um inibidor de RAF ou de outro agente anticâncer aqui descrito é entre aproximadamente 1 mg/kg e 2400 mg/kg ao dia, como entre aproximadamente 1 mg/kg e 1200 mg/kg ao dia, 75 mg/kg ao dia e 300 mg/kg ao dia, incluindo entre aproximadamente 1 mg/kg e 100 mg/kg ao dia. Outras doses representativas de tais agentes incluem aproximadamente 1 mg/kg, 5 mg/kg, 10 mg/kg, 15 mg/kg, 20 mg/kg, 25 mg/kg, 30 mg/kg, 35 mg/kg, 40 mg/kg, 45 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 75 mg/kg, 80 mg/kg, 90 mg/kg, 100 mg/kg, 125 mg/kg, 150 mg/kg, 175 mg/kg, 200 mg/kg, 250 mg/kg, 300 mg/kg, 400 mg/kg, 500 mg/kg, 600 mg/kg, 700 mg/kg, 800 mg/kg, 900 mg/kg, 1000 mg/kg, 1100 mg/kg, 1200 mg/kg, 1300 mg/kg, 1400 mg/kg, 1500 mg/kg, 1600 mg/kg, 1700 mg/kg, 1800 mg/kg, 1900 mg/kg, 2000 mg/kg, 2100 mg/kg, 2200 mg/kg e 2300 mg/kg ao dia. A dose eficaz de BVD-523, de inibidores de RAF ou de outros agentes anticâncer aqui descritos pode ser administrada em duas, três, quatro, cinco ou mais subdoses, administradas separadamente em intervalos apropriados ao longo do dia.
[00160] O BVD-523, os inibidores de RAF ou outros agentes anticâncer ou composições farmacêuticas os contendo da presente invenção podem ser administrado de qualquer maneira desejada e eficaz; por ingestão oral ou como pomada ou gotas para administração local aos olhos, ou por administração parenteral ou outra de qualquer maneira adequada como intraperitoneal, subcutânea, tópica, intradérmica, por inalação, intrapulmonar, retal, vaginal, sublingual, intramuscular, intravenosa, intra-arterial, intratecal ou intralinfática. Além disso, o BVD-523, os inibidores de RAF ou outros agentes anticâncer ou composições farmacêuticas os contendo da presente invenção podem ser administrados em conjunto com outros tratamentos. O BVD-523, os inibidores de RAF ou outros agentes anticâncer ou composições farmacêuticas os contendo podem ser encapsulados ou de outra forma protegidos contra secreção gástrica ou outras secreções, se desejado.
[00161] As composições farmacêuticas da invenção compreendem um ou mais ingredientes ativos, por exemplo, agentes anticâncer, em mistura com um ou mais diluentes ou veículos farmaceuticamente aceitáveis e, opcionalmente, um ou mais outros compostos, fármacos, ingredientes e/ou materiais. Independentemente da via de administração selecionada, os agentes/compostos da presente invenção são formulados em formas de apresentação farmaceuticamente aceitáveis por métodos convencionais conhecidos pelos técnicos no assunto. Vide, por exemplo, Remington, The Science and Practice of Pharmacy (21a edição, Lippincott Williams e Wilkins, Filadélfia, PA.).
[00162] Diluentes ou veículos farmaceuticamente aceitáveis são bem conhecidos na técnica (vide exemplo, Remington, The Science and Practice of Pharmacy (21a edição, Lippincott Williams e Wilkins, Filadélfia, PA.) e o National Formulary (American Pharmaceutical Association, Washington, D.C.)) e incluem açúcares (por exemplo, lactose, sacarose, manitol e sorbitol), amidos, preparados celulósicos, fosfatos de cálcio (por exemplo, fosfato dicálcico, fosfato tricálcico e hidrogenofosfato de cálcio), citrato de sódio, água, soluções aquosas (por exemplo, solução salina, injetável de cloreto de sódio, injetável de Ringer, injetável de dextrose, injetável de dextrose e cloreto de sódio, injetável de Ringer com lactato), alcoóis (por exemplo, álcool etílico, álcool propílico e álcool benzílico), polióis (por exemplo, glicerol, propilenoglicol e polietilenoglicol), ésteres orgânicos (por exemplo, oleato de etila e triglicerídeos), polímeros biodegradáveis (por exemplo, polilactídeo-poliglicolídeo, poli(ortoésteres) e poli(anidridos)), matrizes elastoméricas, lipossomos, microesferas, óleos (por exemplo, de milho, gérmen, oliva, rícino, gergelim, semente de algodão e amendoim), manteiga de cacau, ceras (por exemplo, ceras para supositórios), parafinas, silicones, talco, silicilato, etc. Cada diluente ou veículo farmaceuticamente aceitável utilizado em uma composição farmacêutica da invenção precisa ser "aceitável" no sentido de ser compatível com os outros ingredientes da formulação e não ser nocivo para o indivíduo. Os diluentes ou veículos adequados para uma forma farmacêutica selecionada e uma via de administração pretendida são bem conhecidos na técnica, e diluentes ou veículos aceitáveis para uma forma farmacêutica e um método de administração podem ser determinados por qualquer técnico no assunto.
[00163] As composições farmacêuticas da invenção podem, opcionalmente, conter ingredientes e/ou materiais adicionais, comumente utilizados em composições farmacêuticas. Esses ingredientes e materiais são bem conhecidos na técnica e incluem (1) materiais de preenchimento ou extensores, como amidos, lactose, sacarose, glicose, manitol e ácido silícico; (2) aglutinantes, como carboximetilcelulose, alginatos, gelatina, polivinilpirrolidona, hidroxipropilmetilcelulose, sacarose e acácia; (3) umectantes, como glicerol; (4) agentes desintegrantes, como ágar-ágar, carbonato de cálcio, amido de batata ou de tapioca, ácido algínico, certos silicatos, amido glicolato de sódio, carboximetilcelulose sódica reticulada e carbonato de sódio; (5) agentes retardadores de solução, como parafina; (6) acelerados da absorção, como compostos de amônio quaternário; (7) agentes hidratantes, como álcool cetílico e monoestearato de glicerol; (8) absorventes, tais como caulim e argila bentonita; (9) lubrificantes, como talco, estearato de cálcio, estearato de magnésio, polietilenoglicóis sólidos e lauril sulfato de sódio; (10) agentes de suspensão, como alcoóis isoestearílicos etoxilados, polioxietileno sorbitol e ésteres de sorbitana, celulose microcristalina, metahidróxido de alumínio, bentonita, ágar-ágar e tragacanta; (11) agentes de tamponamento; (12) excipientes, como lactose, açúcares do leite, polietilenoglicóis, gorduras animais e vegetais, óleos, ceras, parafinas, manteiga de cacau, amidos, tragacanta, derivados celulósicos, polietilenoglicol, silicones, bentonitas, ácido silícico, talco, salicilato, óxido de zinco, hidróxido de alumínio, silicatos de cálcio e pó de poliamida; (13) diluentes inertes, como água ou outros solventes; (14) conservantes; (15) agentes de superfície ativa; (16) agentes dispersantes; (17) agentes controladores da libração ou retardadores da absorção, como hidroxipropilmetilcelulose, outras matrizes poliméricas, polímeros biodegradáveis, lipossomos, microesferas, monoestearato de alumínio, gelatina e ceras; (18) agentes opacificantes; (19) adjuvantes; (20) agentes hidratantes; (21) agentes emulsificantes e de suspensão; (22), agentes solubilizantes e emulsificantes, como álcool etílico, álcool isopropílico, carbonato de etila e acetato de etila, álcool benzílico, benzoato de benzila, propilenoglicol, 1,3-butilenoglicol, óleos (especialmente, óleo de semente de algodão, amendoim, milho, gérmen, oliva, rícino e gergelim), glicerol, álcool tetra-hidrofurílico, polietilenoglicóis e ésteres de ácidos graxos de sorbitana; (23) propelentes, como clorofluorohidrocarbonetos e hidrocarbonetos não substituídos voláteis, como butano e propano; (24) antioxidantes; (25) agentes que tornam a formulação isotônica com o sangue do receptor pretendido, como açúcares e cloreto de sódio; (26) agentes espessantes; (27) materiais de revestimento, como lecitina; e (28) agentes adoçantes, aromatizantes, corantes, perfumantes e conservantes. Cada um de tais ingredientes ou materiais precisa ser "aceitável" no sentido de ser compatível com os outros ingredientes da formulação e não ser nocivo ao indivíduo. Os ingredientes e materiais adequados para uma forma farmacêutica selecionada e uma via de administração pretendida são bem conhecidos na técnica, e ingredientes e materiais aceitáveis para uma forma farmacêutica escolhida e um método de administração podem ser determinados por qualquer técnico no assunto.
[00164] As composições farmacêuticas da presente invenção adequadas para administração oral podem estar na forma de cápsulas, sachês, pílulas, comprimidos, pós, grânulos ou de solução ou suspensão em líquido aquoso ou não aquoso, de emulsão líquida óleo em água ou água em óleo, de elixir ou xarope, pastilha, bolus, electuário ou de pasta. Essas formulações podem ser preparadas por métodos conhecidos na técnica, por exemplo, por meio de processos convencionais de revestimento em panela, mistura, granulação ou liofilização.
[00165] Formas farmacêuticas sólidas para administração oral (cápsulas, comprimidos, pílulas, pastilhas, pós, grânulos e os semelhantes) podem ser preparadas, por exemplo, misturando o(s) ingrediente(s) ativo(s) com um ou mais diluentes ou veículos farmaceuticamente aceitáveis e, opcionalmente, um ou mais materiais de preenchimento, extensores, aglutinantes, umectantes, agentes desintegrantes, agentes retardadores de solução, aceleradores da absorção, agentes hidratantes, absorventes, lubrificantes e/ou agentes corantes. Composições sólidas de tipo semelhante podem ser empregadas como materiais de preenchimento em cápsulas gelatinosas moles e duras utilizando um excipiente adequado. Um comprimido pode ser produzido por compressão ou moldagem, opcionalmente com um ou mais ingredientes acessórios. Comprimidos prensados podem ser preparados utilizando um aglutinante adequado, lubrificante, diluente inerte, conservante, agente desintegrantes, tensoativo ou dispersante. Comprimidos moldados podem ser produzidos por moldagem em uma máquina adequada. Os comprimidos e outras formas farmacêuticas sólidas, como drágeas, cápsulas, pílulas e grânulos, podem ser opcionalmente classificados ou preparados com revestimentos e invólucros, tais como revestimentos entéricos e outros revestimentos bem conhecidos na técnica da formulação farmacêutica. Além disso, podem também ser formulados de modo a proporcionar libração controlada ou lenta do ingrediente ativo contido no interior. Podem ser esterilizados, por exemplo, por filtração através de um filtro retentor de bactérias. Essas composições podem conter opcionalmente agentes opacificantes e podem ter tal composição que librem o ingrediente ativo somente ou, de preferência, em certa parte do trato gastrointestinal, opcionalmente, de maneira retardada. O ingrediente ativo pode também estar em forma microencapsulada.
[00166] As formas farmacêuticas líquidas para administração oral incluem emulsões, microemulsões, soluções, suspensões, xaropes e elixires farmaceuticamente aceitáveis. As formas farmacêuticas líquidas podem conter diluentes inertes adequados comumente utilizados na técnica. Além de diluentes inertes, as composições orais podem também incluir adjuvantes, como agentes hidratantes, agentes emulsificantes e de suspensão, agentes adoçantes, aromatizantes, corantes, perfumantes e conservantes. As suspensões podem conter agentes de suspensão.
[00167] As composições farmacêuticas da presente invenção para administração retal ou vaginal podem ser apresentadas como supositório, o qual pode ser preparado misturando um ou mais ingredientes ativos com um ou mais diluentes ou veículos não irritantes adequados que são sólidos à temperatura ambiente, mas líquidos à temperatura corporal e que, portanto, se derreterão no reto ou na cavidade vaginal e librarão o composto ativo. As composições farmacêuticas da presente invenção que são adequadas para administração vaginal também incluem pessários, tampões, cremes, géis, pastas, espumas ou formulações em spray contendo tais diluentes ou veículos farmaceuticamente aceitáveis que sejam conhecidos na técnica como apropriados.
[00168] As formas farmacêuticas para administração tópica ou transdérmica incluem pós, sprays, pomadas, pastas, cremes, loções, géis, soluções, adesivos, gotas e soluções para inalação. O(s) agente(s)/composto(s) ativo(s) pode(m) ser misturado(s) em condições estéreis com um diluente ou veículo farmaceuticamente aceitável adequado. As pomadas, pastas, cremes e géis podem conter excipientes. Os pós e sprays podem conter excipientes e propelentes.
[00169] As composições farmacêuticas da presente invenção adequadas para administração parenteral podem compreender um ou mais agentes/compostos em combinação com uma ou mais soluções aquosas ou não aquosas isotônicas, dispersões, suspensões ou emulsões estéreis ou com pós estéreis que podem ser reconstituídos em soluções injetáveis estéreis ou dispersões imediatamente antes do uso, os quais podem conter antioxidantes adequados, tampões, soluções que tornam a formulação isotônica com o sangue do receptor pretendido, ou agentes de suspensão ou espessantes. A fluidez adequada pode ser mantida, por exemplo, pelo uso de materiais de revestimento, pela manutenção do tamanho de partículas necessário no caso de dispersões e pelo uso de surfactantes. Essas composições farmacêuticas podem também conter adjuvantes adequados, como agentes hidratantes, agentes emulsificantes e agentes dispersantes. Pode também ser conveniente incluir agentes isotônicos. Além disso, a absorção prolongada da forma farmacêutica injetável pode ser ocasionada pela inclusão de agentes que retardam a absorção.
[00170] Em alguns casos, a fim de prolongar o efeito de um fármaco (por exemplo, formulação farmacêutica), é desejável desacelerar sua absorção da injeção subcutânea ou intramuscular. Isso pode ser realizado pelo uso de uma suspensão líquida de um material cristalino ou amorfo dispondo de baixa solubilidade em água.
[00171] A taxa de absorção do agente ativo/fármaco depende então de sua taxa de dissolução que, por sua vez, pode depender do tamanho de cristal e da forma cristalina. Alternativamente, a absorção retardada de um agente/fármaco administrado por via parenteral pode ser realizada dissolvendo ou suspendendo o agente ativo/fármaco em um veículo oleoso. Formas injetáveis de depósito podem ser produzidas formando matrizes microencapsuladas do ingrediente ativo em polímeros biodegradáveis. Dependendo da proporção do ingrediente ativo para polímero e da natureza do polímero específico empregado, a taxa de libração do ingrediente ativo pode ser controlada. Formulações injetáveis de depósito são também preparadas aprisionando o fármaco em lipossomos ou microemulsões que são compatíveis com tecidos corporais. Os materiais injetáveis podem ser esterilizados, por exemplo, por filtração através de um filtro retentor de bactérias.
[00172] As formulações podem ser apresentadas em dose unitária ou em recipientes vedados de doses múltiplas, por exemplo, ampolas e frascos-ampolas, e podem ser armazenadas em condição liofilizada requerendo somente a adição do diluente ou veículo líquido estéril, por exemplo, água para injetáveis, imediatamente antes do uso. Soluções e suspensões injetáveis extemporâneas podem ser preparadas a partir de pós, grânulos ou comprimidos estéreis do tipo descrito acima.
[00173] A presente invenção provê as combinações mostradas para reforçar os efeitos de inibidores de ERK. Os requerentes também demonstraram, neste pedido de patente, que a combinação de diferentes inibidores de ERK é do mesmo modo sinérgica. Por conseguinte, contempla-se que os efeitos das combinações aqui descritas possam ser melhorados ainda mais pelo uso de um ou mais inibidores de ERK adicionais. Consequentemente, algumas modalidades da presente invenção incluem um ou mais inibidores de ERK adicionais.
[00174] Os exemplos a seguir são fornecidos para ilustrar mais detalhadamente os métodos da presente invenção. Esses exemplos são ilustrativos apenas e não se destinam a servir de limitação ao âmbito da invenção de maneira alguma.
EXEMPLOS
EXEMPLO 1
MATERIAIS E MÉTODOS
[00175] Linhagens de células cancerosas foram mantidas em cultura celular sob condições padrão de meio e soro. Para estudos de aumento da dose, células A375 foram divididas, cultivadas até cerca de 40-60% de confluência e, a seguir, tratadas com a dose inicial do fármaco especificado. A Tabela 4 mostra um resumo dos tratamentos com fármaco em escalada. TABELA 4 - Resumo de tratamentos em escalada
[00176] Os aumentos da dose de agentes únicos foram realizados com base em Little et al., 2011 e está descrito na Figura 25. As células foram então deixadas crescer até 70-90% de confluência e foram divididas. As taxas de divisão foram mantidas tão "normais" quanto possível e razoavelmente constantes entre os tratamentos (por exemplo, no mínimo 50% da taxa de divisão normal das parentais). O meio era renovado a cada 3-4 dias. Quando as células atingiam novamente cerca de 40-60% de confluência, a dose era aumentada. No caso de perda da janela de 40-60%, as células eram divididas de novo e recebiam a dose assim que tivessem atingido 40-60% de confluência. Mais uma vez, o meio era renovado a cada 3-4 dias. O processo foi repetido conforme necessário (Figura 25).
[00177] Para tratamentos com agentes únicos, as concentrações iniciais e os aumentos da dose foram conduzidos a partir da IC50 aproximada, aumentando em pequenos incrementos ou suavemente, para as 4-5 doses iniciais, dobrando a dose, aumentando no mesmo incremento, para as 4 doses seguintes, depois mudando para aumentos de 1,5 vezes na concentração das doses subsequentes.
[00178] Para os tratamentos combinados, as concentrações iniciais e os aumentos da dose foram conduzidos a partir da metade da IC50 aproximada de cada composto (o ensaio da combinação sugere que esse valor resultará em cerca de 40-70% da faixa de inibição), aumentando conforme efetuado para os agentes únicos (ou seja, dobrando a dose inicial e depois aumentando no mesmo incremento para as 4 doses seguintes, a seguir, mudando para aumentos de 1,5 vezes na concentração). A Tabela 5 mostra os aumentos projetados para a dose utilizando esses esquemas. TABELA 5 - Aumentos projetados para a dose - Mês 1
[00179] Populações celulares de clones resistente foram derivadas a partir de pools de células resistentes limitando a diluição.
[00180] Utilizaram-se ensaios de proliferação para rastrear as alterações na sensibilidade ao(s) agente(s) aumentado(s) gradualmente em intervalos de tempo apropriados (por exemplo, cada mês, embora o momento seja dependente de números adequados das células então disponíveis). Para os ensaios de proliferação, as células foram semeadas em placas de 96 cavidades a 3000 células por cavidade em meio DMEM sem fármaco, contendo FBS 10%, e deixadas que se aderissem durante a noite antes da adição do composto ou veículo controle. Os compostos foram preparados a partir de estoques de DMSO para fornecer uma faixa de concentração final como mostrada nas Figuras 2A-H. A concentração final de DMSO ficou constante em 0,1%. Os compostos em teste foram incubados com as células por 96 horas a 37 °C e CO2 5% em atmosfera umidificada. Alamar Blue 10% (v/v) foi então adicionado e incubado por 4 horas, e o produto fluorescente foi detectado utilizando um leitor de placas BMG FLUOstar. O valor médio de fundo com meio apenas foi subtraído e os dados analisados utilizando uma equação logística de 4 parâmetros no GraphPad Prism. Paclitaxel foi utilizado como controle positivo.
[00181] Os ensaios de proliferação de 1 mês foram iniciados no Dia 28 utilizando células cultivadas nas concentrações de cada agente indicada na Tabela 6. TABELA 6 - Concentrações iniciais dos fármacos utilizados nos ensaios de proliferação - Mês 1
[00182] Os ensaios de proliferação de 2 meses foram iniciados no Dia 56 utilizando células cultivadas nas concentrações de cada agente indicada na Tabela 7. TABELA 7 - Concentrações iniciais dos fármacos utilizados nos ensaios de proliferação - Mês 2
[00183] No final do período de aumento da dose de 3 meses, as culturas foram mantidas na concentração mais alta por 2 semanas antes da rodada final de ensaios de proliferação e clonagem potencial de células isoladas. Como os ensaios de proliferação/clonagem de células isoladas requeriam células que se proliferassem ativamente, para os tratamentos nos quais as células estavam se proliferando muito lentamente na concentração mais alta ou com a concentração aumentada apenas recentemente, uma cultura de apoio (backup) foi também mantida a uma concentração mais baixa (Tabela 8). Para o tratamento com BVD-523, no qual as células aparentavam ter parado de crescer quase que completamente e pareciam especialmente frágeis na concentração mais alta (1,8 μM), foram mantidas culturas a uma concentração mais baixa por um período de duas semanas. TABELA 8 - Detalhes de tratamentos em cultura a uma concentração fixa por 2 semanas
[00184] Os ensaios de proliferação de 3 meses utilizaram células cultivadas nas concentrações de cada agente indicada na Tabela 9. TABELA 9 - Concentrações iniciais dos fármacos utilizados nos ensaios de proliferação - Mês 3
[00185] Para os estudos de combinações, células A375 (ATCC) foram semeadas em placas de 96 cavidades, em triplicata, a uma densidade celular de 3000 células/cavidade em DMEM mais FBS 10% e deixadas que se aderissem durante a noite antes da adição do composto teste ou veículo controle. As combinações foram testadas utilizando uma matriz de dose 10x8 com concentração final de DMSO de 0,2%. Seguiu-se um período de incubação de 96 horas no ensaio, com adição subsequente de Alamar Blue 10% (v/v) e 4 horas de incubação antes da leitura em um leitor de placas fluorescentes. Depois da leitura de Alamar Blue, o meio/mistura de Alamar Blue foi removido, 100 μL de CellTiter-Glo/PBS (1:1) foram adicionados e as placas processadas segundo as instruções do fabricante (Promega). Os valores de fundo do meio apenas foram subtraídos antes que os dados fossem analisados. O modelo de aditividade de Bliss foi então aplicado.
[00186] Resumidamente, valores preditos de inibições fracionadas para a inibição combinação foram calculados utilizando a equação: Cbiiss = A + B - (A x B), onde A e B são as inibições fracionadas obtidas para o fármaco A isoladamente ou o fármaco B isoladamente a concentrações específicas. Cbliss é a inibição fracionada que seria esperada se a combinação dos dois fármacos fosse exatamente aditiva. Os valores de Cbliss são subtraídos dos valores de inibição fracionada observados experimentalmente para fornecer um valor de "excesso sobre Bliss". Valores de excesso sobre Bliss superiores a 0 indicam sinergia, enquanto que valores inferiores a 0 indicam antagonismo. Os valores de excesso sobre Bliss são traçados em mapas de calor ± desvio padrão (SD).
[00187] Os dados individuais e combinados são também apresentados como curvas de dose-resposta geradas no GraphPad Prism (traçadas utilizando % de viabilidade em relação a controles tratados apenas com DMSO).
[00188] Para estudos de combinações focalizadas, foram realizados ensaios de viabilidade com Alamar Blue como descritos acima para os estudos de combinações. Adicionalmente, realizaram- se ensaios com Caspase-Glo 3/7. Resumidamente, células HCT116 foram semeadas em triplicata em placas brancas de 96 cavidades a uma densidade celular de 5000 células/cavidade em meio de McCoy 5A mais FBS 10%. Células A375 foram semeadas a uma densidade celular de 5000 células/cavidade em DMEM mais FBS 10%. Deixou-se que as células se aderissem durante a noite antes da adição da quantidade indicada do composto teste ou veículo controle. A concentração final de DMSO foi 0,2%, e estauporina 800 nM foi incluída como controle positivo. Períodos de incubação de 24 e 48 horas foram utilizados no ensaio. Em seguida, Caspase-Glo® 3/7 50% (v/v) foi adicionado, as placas foram misturadas por 5 minutos em um agitador orbital e incubadas por 1 hora à temperatura ambiente antes da leitura em um leitor de placa luminescente. Os valores de fundo apenas com meio foram subtraídos antes que os dados fossem analisados.
EXEMPLO 2
AUMENTO DA DOSE E ENSAIOS DE PROLIFERAÇÃO - MÊS 1 PROGRESSO DO AUMENTO DA DOSE - MÊS 1
[00189] Células A375 sofreram aumento da dose de BVD-523, dabrafenib e trametinib, seja como agentes isolados ou combinados. As doses foram aumentadas em incrementos pequenos durante o primeiro mês. A não ser por uma redução acentuada na taxa de crescimento, as células em geral toleraram bem os aumentos, e planejou-se que as doses seriam aumentadas mais agressivamente no Mês 2. As Figuras 1A-C mostram o progresso em 1 mês para os estudos de aumento da dose.
RESULTADOS DO ENSAIO DE PROLIFERAÇÃO - MÊS 1
[00190] Realizaram-se ensaios de proliferação para avaliar a resposta das linhagens celulares que sofreram aumento da dose, em comparação à linhagem celular, a tratamentos com BVD-523, dabrafenib e trametinib.
[00191] As Figuras 2A-H mostram resultados normalizados e não processados do ensaio de proliferação obtidos no Mês 1 dos estudos. Notar que as diferenças em sinais máximos em controles com DMSO entre os diferentes tratamentos (Figuras 2D-F, 2H) sugerem taxas diferenciais de crescimento entre os tratamentos. Essas diferenças podem influenciar as respostas das linhagens a inibidores nos ensaios de proliferação.
[00192] A Tabela 10 mostra dados de IC50 para o Mês 1 dos estudos. TABELA 10 - Dados de IC50 - Mês 1
*Par = Linhagem celular parental Composto Linhagem cellular, IC50 relativa (nM) Par* / Tram./ Dab / BVD-523 / Dab/Tram / Dab/523 / Tram/523 Dabrafenib 6 29 about 161 8 58 68 11 Trametinib 0.5 2.2 2.5 0.7 3.9 3.1 2.5 BVD-523 189 335 350 268 300 412 263 Paclitaxel 2.2 3.0 3.3 3.4 3.5 3.4 3.4 *Par = Linhagem celular parental
[00193] Houve indícios precoces de que as células cultivadas na presença de doses crescentes de dabrafenib ou trametinib, seja como agentes isolados ou combinados, estavam exibindo respostas diminuídas a esses dois agentes nos ensaios de proliferação.
[00194] Nos estágios iniciais do Mês 2, a taxa de crescimento de células no tratamento somente com dabrafenib aumentou consideravelmente em relação aos estágios iniciais do Mês 1. Isso possibilitou aumentar a taxa de progressão e sugeriu que a resistência estava se tornando evidente.
EXEMPLO 3
AUMENTO DA DOSE E ENSAIOS DE PROLIFERAÇÃO - MÊS 2 PROGRESSO DO AUMENTO DA DOSE - MÊS 2
[00195] O segundo mês de estudos viu a maioria dos tratamentos mudarem para uma fase na qual as doses eram aumentadas em incrementos maiores (1,5 vezes) quando comparada à fase inicial de aumento suave. O aumento de dabrafenib e trametinib, como agente isolado, foi mais rápido, com as células crescendo em concentrações equivalentes a 100x a IC50 da célula parental (Figuras 3A, B). O aumento de BVD-523, como agente isolado, progrediu mais lentamente quando comparado aos de dabrafenib e trametinib (Figura 3C). Vide a Figura 3D para uma comparação dos aumentos dos agentes isolados. As células que sofreram aumento de BVD-523 tinham uma aparência mais "frágil" e houve um número maior de células flutuantes em comparação às populações com aumento de dabrafenib e de trametinib.
[00196] O aumento de agentes combinados progrediu mais lentamente do que os tratamentos com os agentes isolados. A combinação BVD-523/trametinib foi especialmente eficaz em impedir que as células progredissem.
RESULTADOS DO ENSAIO DE PROLIFERAÇÃO - MÊS 2
[00197] Os ensaios de proliferação em populações celulares com dose aumentada de dabrafenide e trametinib como agentes isolados revelaram desvios modestos nas curvas de resposta à dose, sugerindo que um período adicional de aumento gradual seria benéfico para enriquecimento de células resistentes. Curiosamente, nos ensaios de proliferação, houve evidência para sugerir que células expostas ao BVD-523 crescem menos quando da retirada do inibidor, talvez indicando um nível de dependência.
[00198] As Figuras 4A-H mostram resultados normalizados e não processados do ensaio de proliferação, obtidos no Mês 2 dos estudos. Observar que as diferenças nos sinais máximos em controles por DMSO entre os diferentes tratamentos (Figuras 4D-F, 4H) sugerem taxas diferenciais de crescimento entre os tratamentos. Essas diferenças podem influenciar as respostas das linhagens aos inibidores nos ensaios de proliferação.
[00199] As Figuras 5A-H mostram resultados normalizados e não processados do ensaio de proliferação, obtidos no Mês 2 dos estudos com ênfase nos dados somente da linhagem parental e a do BVD-523.
[00200] A Tabela 11 mostra dados de IC50 para o Mês 2 dos estudos. As IC50s relativas foram determinadas a partir de ajustes na curva de 4 parâmetros no Prism. TABELA 11 - Dados de IC50 - Mês 2
*Par = Linhagem celular parental
[00201] As Figuras 6A-C mostram o aumento de agentes isolados e combinados para o Mês 3 dos estudos. A Figura 6D mostra uma comparação de aumentos de agentes isolados.
RESULTADOS DO ENSAIO DE PROLIFERAÇÃO - MÊS 3
[00202] A Figura 7 mostra uma avaliação do crescimento durante os ensaios de proliferação em cavidades controle com DMSO. As Figuras 8A-D mostram resultados do Mês 3 dos estudos. As Figuras 9A-D mostram resultados do Mês 3 dos estudos com ênfase em linhagens celulares tratadas pelos agentes isolados.
[00203] A Tabela 12 mostra dados de IC50 para o Mês 3 dos estudos. As IC50s relativas foram determinadas a partir de ajustes na curva de 4 parâmetros no Prism. Os valores de IC50 não foram determinados para a linhagem celular com aumento da dose de trametinib pela ausência de crescimento durante o ensaio (ND: não realizado). TABELA 12 - Dados de IC50 - Mês 3
*Par = Linhagem celular parental
[00204] Como previsto, as células A375, portadoras de uma mutação em BRAF (V600E), foram sensíveis ao dabrafenib. Os valores da IC50 dos agentes isolados, calculados utilizando Alamar Blue (Figuras 10, 12, 14), foram em geral ligeiramente menores para Dabrafenib e BVD-523 quando comparados àqueles derivados utilizando CellTiter-Glo (Figuras 11, 13, 15). Os valores de IC50 publicados para Dabrafenib e Trametinib em um ensaio CellTiter-Glo de 72 horas eram 28 ± 16 nM e 5 ± 3 nM respectivamente (Greger et al., 2012; King et al., 2013) - os resultados para os agentes isolados aqui relatados são compatíveis com esses valores. Houve alguma evidência para uma janela de sinergia em todos os tratamentos. A variação entre três replicas foi baixa, contudo, houve alguma evidência de efeitos de borda que provavelmente explica o crescimento aparente reforçado observado em alguns tratamentos em comparação ao controle sem fármaco (por exemplo, especialmente aparente na combinação Trametinib/BVD-523). Isso torna a interpretação da análise de Bliss mais desafiadora, pois, em alguns tratamentos, pode ter resultado o reforço no nível de sinergia este pode ter resultado de artefato.
[00205] Os ensaios de combinações foram repetidos para células A375. Adicionalmente, células HCT116 foram utilizadas em um ensaio de acompanhamento das combinações. Os resultados desses experimentos são mostrados nas Figuras 31-41. As potências dos agentes únicos, BVD-523, Trametinib e Dabrafenib, foram compatíveis com aquelas relatadas nos estudos anteriores.
[00206] Células HCT116 são células de câncer colorretal humano com mutações em KRAS. Dabrafenib e Trametinib foram antagonistas em concentrações relevantes no alvo. Por outro lado, Trametinib exibiu sinergia com AZ628 em uma ampla faixa de combinações, e com concentrações mais altas de Sorafenib. BVD-523 exibiu janelas de sinergia com AZ628 e com Sorafenib.
[00207] Em células A375, trametinib exibiu áreas de sinergia em concentrações mais baixas de Dabrafenib e AZ628. BVD-523 exibiu uma janela de sinergia com as concentrações mais baixas de Sorafenib.
EXEMPLO 6
MARCADORES ALTERADOS POR BVD-523 DA ATIVIDADE DE MAPK QUINASE E DA FUNÇÃO EFETORA
[00208] Para os estudos por Western Blotting, células HCT116 (5 x 106) foram semeadas em placas de 10 cm em meio McCoy 5A mais FBS 10%. Células A375 (2,5 x 106) foram semeadas em placas de 10 cm em meio DMEM mais FBS 10%. Deixou-se que as células se aderissem durante a noite antes da adição da quantidade indicada do composto teste (BVD-523) ou veículo controle. As células foram tratadas por 4 ou 24 horas antes do isolamento de lisados proteicos de células inteiras, conforme especificado abaixo. As células foram colhidas por tripsinização, centrifugadas para obtenção do pellet e congeladas imediatamente. Os lisados foram preparados com tampão RIPA (Ensaio de radioimunoprecipitação), clarificados por centrifugação e quantificados com ensaio do ácido bicinconínico (BCA). 20-50 μg de proteína foram resolvidos por eletroforese com SDS-PAGE, transferidos para uma membrana de PVDF e sondados utilizando os anticorpos detalhados na Tabela 13 (para o tratamento de 4 horas) e na Tabela 14 (para o tratamento de 24 horas) abaixo. TABELA 13 -Detalhes de anticorpos


Tabela 14 – Detalhes de anticorpos
[00209] As Figuras 16-18 mostram análises por Western Blotting de células tratadas com BVD-523 em várias concentrações para o seguinte: 1) componentes da sinalização de MAPK em células A375 após 4 horas; 2) sinalização do ciclo celular e apoptose em A375 tratadas por 24 horas com várias quantidades de BVD-523; e 3) sinalização de MAPK em células HCT-116 tratadas por 4 horas. Os resultados mostram que o tratamento agudo e prolongado com BVD- 523 em células cancerosas com mutações em RAF e RAS in vitro afeta tanto a fosforilação de substratos como alvos efetores de ERK quinases. As concentrações de BVD-523 necessárias para induzir essas alterações estão tipicamente na faixa de baixo micromolar.
[00210] As alterações em diversos marcadores específicos da atividade são notáveis. Inicialmente, a abundância de isoformas de ERK quinase que migram lentamente aumenta após o tratamento com BVD-523; alterações modestas podem ser observadas agudamente, e aumentam após o tratamento prolongado. Embora isso pudesse indicar um aumento em formas fosforilados enzimaticamente ativas de ERK, ainda assim é notável que múltiplas proteínas submetidas à regulação direta e indireta por ERK permaneçam "desligadas" após o tratamento com BVD-523. Em primeiro lugar, as proteínas RSK1/2 exibm fosforilação reduzida em resíduos que são rigorosamente dependentes de ERK para modificação proteica (T359/S363). Em segundo lugar, o tratamento com BVD-523 induz alterações complexas na fosfatase reguladora de MAPK por feedback, DUSP6: isoformas da proteína de migração lenta são reduzidas após o tratamento agudo, enquanto os níveis de proteínas totais são reduzidos em grande medida após o tratamento prolongado com BVD- 523. Esses dois achados são compatíveis com atividade reduzida de ERK quinases, as quais controlam a função de DUSP6 através de mecanismos pós-traducionais e transcricionais. No global, apesar de aumentos em formas celulares de ERK que são consideradas tipicamente ativas, parece provável que a atividade celular enzimática de ERK é totalmente inibida após o tratamento agudo ou o prolongado com BVD-523.
[00211] De modo condizente com essas observações, genes efetores que requerem a sinalização de MAPK são alterados após o tratamento com BVD-523. O mecanismo do ciclo celular G1/S é regulado em nível pós-traducional e transcricional pela sinalização de MAPK, e os níveis proteicos de ciclina-D1 são reduzidos em grande medida após o tratamento prolongado com BVD-523. Do mesmo modo, a expressão gênica e a abundância de proteínas efetoras de apoptose requerem sinalização intacta de MAPK, sendo que os níveis totais do Bim-EL aumentam após o tratamento prolongado com BVD- 523. Como observado acima, contudo, a clivagem proteolítica de PARP e maior apoptose não foram percebidas no fundo de células A375; sugerindo que fatores adicionais podem influenciar se alterações na sinalização de efetoras dependentes de BVD-523/ERK serão traduzidas em eventos definitivos como morte celular e parada do ciclo celular
[00212] Coerentemente com a atividade celular de BVD-523, a análise de marcadores sugere que a inibição de ERK altera uma variedade de eventos de sinalização molecular em células cancerosas, tornando-as suscetíveis à redução da proliferação e sobrevida celular.
[00213] Em suma, as Figuras 16-18 mostram que BVD-523 inib a via de sinalização de MAPK e que pode ser mais favorável quando comparado à inibição de RAF ou MEK nesse cenário.
[00214] Finalmente, as propriedades de BVD-523 podem torná-lo um agente preferido para uso como inibidor de ERK, quando comparado a outros agentes com atividade similar. Sabe-se que fármacos inibidores de quinases exibm interações únicas e específicas com seus alvos enzimáticos, e que a eficácia do fármaco é fortemente influenciada pelo modo de inibição direta, bem como pela suscetibilidade a mudanças adaptativas que ocorrem após o tratamento. Por exemplo, inibidores de ABL, KIT, EGFR e ALK quinases são eficazes somente quando seu alvo cognato é encontrado em configurações ativas ou inativas. Do mesmo modo, certos desses inibidores são sensíveis de modo único à mutação genética secundária, ou a mudanças adaptativas pós-traducionais, do alvo proteico. Finalmente, inibidores de RAF mostram potência diferenciada perante RAF quinases presentes em certos complexos proteicos e/ou localizações subcelulares. Em suma, considerando que as ERK quinases são conhecidas por igualmente existirem em estados bioquímicos diversos, variáveis e complexos, parece provável que BVD-523 possa interagir com esses alvos e inibi-los de maneira distinta e altamente preferível à de outros agentes.
EXEMPLO 7
ENSAIO IN VIVO
CAMUNDONGOS
[00215] Camundongos nu atímicos do sexo feminino (Crl:NU(Ncr)- Foxn/nu, Charles River) tinham nove semanas de idade com uma faixa de peso corporal (PC) de 17,5 a 26,2 gramas no Dia 1 do estudo. Os animais foram alimentados e consumiram ad libitum água (osmose reversa, Cl 1 ppm) e a dieta NIH 31 Modified and Irradiated Lab Diet®, que consistia em proteína bruta 18,0%, gordura bruta 5,0% e fibra bruta 5,0%. Os camundongos foram mantidos em Enrich-o'cobsTM Laboratory Animal Bedding irradiados em microisoladores estáticos em um ciclo de luz de 12 horas a 20-22 °C (68-72 °F) e 40-60% de umidade. As recomendações do Guide for Care and Use of Laboratory Animals, referentes a restrições, sistemas de criação, procedimentos cirúrgicos, regulamentos para alimentos e líquidos e cuidados veterinários, foram cumpridas.
IMPLANTAÇÃO IN VIVO E CRESCIMENTO TUMORAL
[00216] Os xenoenxertos dos tumores foram iniciados com melanomas humanos da linhagem A375 por transplante subcutâneo seriado em camundongos nu atímicos. No dia de implante do tumor, cada camundongo em teste recebeu um fragmento de 1 mm3 com células A375 implantado por via subcutânea no flanco direito, e o crescimento tumoral foi monitorado como o tamanho médio que se aproximava da faixa alvo de 80 a 120 mm3. Os tumores eram medidos em duas dimensões utilizando paquímetros, e o volume foi calculado utilizando a fórmula:
[00217] onde w = largura e l = comprimento, em mm, do tumor. O peso do tumor pode ser estimado com base no pressuposto de que 1 mg é equivalente a 1 mm3 de volume tumoral.
[00218] Dez dias depois da implantação do tumor, designado como Dia 1 do estudo, os animais foram separados em nove grupos (Grupos 1-9), cada um sendo constituído por quinze camundongos, e um grupo (Grupo 10) formado por dez camundongos. Os volumes tumorais individuais variaram de 75 a 144 mm3 e os volumes tumorais médios dos grupos eram 110 ou 111 mm3.
AGENTES TERAPÊUTICOS
[00219] BVD-523 e dabrafenib foram fornecidos em forma de pó seco e armazenados à temperatura ambiente protegidos da luz.
[00220] As doses de BVD-523 foram preparadas suspendendo a quantidade necessária do pó de BVD-523 em água deionizada com carboximetilcelulose 1% ("CMC 1%"). Preparou-se um estoque de BVD-523 10 mg/mL que foi utilizado para a dose do grupo de BVD-523 100 mg/kg. Alíquotas do estoque foram diluídas com o veículo até uma concentração de 5,0 mg/mL para fornecer a dose de BVD-523 50 mg/kg em um volume de administração de 10 mL/kg. As doses de BVD-523 foram armazenadas a 4 °C e protegidas da luz por até uma semana.
[00221] O pó seco de dabrafenib consistia em 84,5% do composto ativo, o que foi levado em conta ao se preparar as doses. Dabrafenib foi formulado em CMC 1% a concentrações de 11,834 e 5,917 mg/mL para produzir as doses de composto ativo de 100 e 50 mg/kg, respectivamente, em um volume de administração de 10 mL/kg. As doses de dabrafenib foram armazenadas protegidas da luz a 4 °C por até uma semana.
[00222] O veículo CMC 1% ("Veículo") foi utilizado na dose para o grupo controle.
[00223] As dose de temozolomida (Temodar®, Schering Corporation, Lote no 2RSA013) foram preparadas suspendendo o conteúdo do número necessário de cápsulas de 100 mg de Temodar® em água deionizada a uma concentração de 15 mg/mL, o que forneceu uma dose de 150 mg/kg em um volume de administração de 10 mL/kg. A temozolomida foi armazenada protegida da luz a 4 °C durante o período de administração de 5 dias.
TRATAMENTO
[00224] No Dia 1 do estudo, os camundongos foram separados em nove grupos (Grupos 1-9), cada um sendo constituído por quinze camundongos, e um grupo (Grupo 10) formado por dez camundongos, e a administração foi iniciada de acordo com o plano de tratamento resumido na Tabela 15 abaixo. As doses foram fornecidas por gavagem oral (v.o.) em um volume de administração de 10 mL/kg (0,2 mL por 20 gramas de peso corporal), aumentadas até o peso corporal de cada animal individual. As doses de veículo e dabrafenib deveriam ser fornecidas uma vez diariamente até o final do estudo (1x/dia até o final), enquanto que as doses de BVD-523 deveriam ser fornecidas duas vezes diariamente até o final do estudo (2x/dia até o final). Para as doses duas vezes ao dia, a administração foi iniciada à tarde no Dia 1, de modo que uma dose foi fornecida no primeiro dia ("dose 1 do primeiro dia"). TABELA 15 - Desenho do protocolo para o estudo A375 in vivo

Veículo = carboximetilcelulose 1% (CMC) em água DI. Para doses duas vezes ao dia, uma dose foi fornecida à tarde no primeiro dia e uma dose na manhã do dia último dia.
[00225] A administração nos grupos de combinações foi modificada durante o estudo conforme descrito abaixo.
CONTROLES
[00226] O Grupo 1 recebeu o veículo CMC 1% CMC e serviu como o grupo controle para o cálculo de % TGD. O Grupo 10 recebeu temozolomida a 150 mg/kg uma vez ao dia por cinco dias (1x/dia x 5) e serviu como grupo de referência.
TRATAMENTOS EM MONOTERAPIA
[00227] Os Grupos 6 e 7 receberam dabrafenib 50 e 100 mg/kg, respectivamente. Os Grupos 8 e 9 receberam BVD-523 50 e 100 mg/kg, respectivamente.
TRATAMENTOS COMBINADOS
[00228] Os Grupos 2 e 3 receberam as combinações de dabrafenib 50 mg/kg com BVD-523 50 ou 100 mg/kg, respectivamente. Os Grupos 4 e 5 receberam as combinações de dabrafenib 100 mg/kg dabrafenib com BVD-523 50 ou 100 mg/kg, respectivamente. Em decorrência da resposta surpreendente ao tratamento combinado, a administração nos Grupos 2-5 foi interrompida no Dia 20 a fim de monitorar novo crescimento do tumor. A administração deveria ser reiniciada em um grupo quando a carga tumoral média atingisse 1000 mm3. No Dia 42, a carga tumoral média de 1000 mm3 não havia sido atingida em qualquer um dos grupos de combinações. A administração foi reiniciada a fim de permitir a amostragem de soro e do tumor após a dose final para análises farmacocinéticas. Com início no Dia 42, os Grupos 2-5 receberam dabrafenib, fornecido uma vez ao dia por quatro dias, e BVD-523, fornecido duas vezes ao dia por três dias, seguido por uma dose de BVD-523 na manhã do Dia 45. Os esquemas da administração final são mostrados abaixo na Tabela 16. TABELA 16 - Resumo das respostas no Estudo A375 in vivo


n = número de animais em um grupo cuja morte não foi por causas acidentais ou desconhecidas; Veículo = carboximetilcelulose 1% (CMC) em água DI. Para doses 2x/dia x 19 e 2x/dia até o final, uma dose foi fornecida à tarde no primeiro dia e uma dose na manhã do último dia. O T-C máximo nesse estudo é 35,8 dias (389%), em comparação ao Grupo 1. Significância estatística (Teste logrank): ne = não avaliado, ns = não significante, * = P < 0,05, ** = P < 0,01, *** = P < 0,001, em comparação ao grupo indicado. MTV (n) = mediana do volume tumoral (imd) para o número de animais no dia da análise de TGD (exclui animais atingindo o desfecho para volume tumoral). PR = regressões parciais; CR = número total de regressões completas; TFS = sobreviventes livres de tumor, ou seja, CRs no final do estudo; Nadir do PC médio = peso corporal mais baixo médio do grupo, como % de mudança em relação ao Dia 1; --indica que não se observou diminuição no peso corporal médio; TR = morte relacionada ao tratamento; NTR = morte não relacionada ao tratamento.
[00229] Os tumores eram medidos utilizando paquímetros duas vezes por semana, e cada animal era sacrificado quando seu tumor atingia o desfecho predeterminado para volume tumoral de 2000 mm3 ou no dia final, o que acontecesse antes. Animais que saíram do estudo, para o desfecho de volume tumoral, foram documentados como sacrificados por progressão tumoral (TP), com a data do sacrifício. O tempo até o desfecho (TTE) para análise foi calculado para cada camundongo pela seguinte equação:
[00230] onde TTE é expresso em dias, volume de desfecho é expresso em mm3, b é o intercepto e m é a inclinação da linha obtida por regressão linear de um conjunto de dados de crescimento tumoral transformados em logaritmo. O conjunto de dados consiste na primeira observação acima do volume de desfecho utilizado na análise e as três observações consecutivas imediatamente precedentes ao atingimento desse volume de desfecho. O TTE calculado é geralmente inferior à data de TP, o dia no qual o animal foi sacrificado pelo tamanho do tumor. Para animais com tumores que não atingiram o volume de desfecho, atribuiu-se um valor de TTE igual ao último dia do estudo. Qualquer animal classificado como morto por causas NTR (não relacionadas ao tratamento) em decorrência de acidente (NTRa) ou por etiologia desconhecida (NTRu) foi excluído dos cálculos de TTE (e de todas as análises posteriores). Animais classificados como mortes TR (relacionadas ao tratamento) ou NTRm (morte não relacionada ao tratamento em decorrência de metástase) receberam um valor de TTE igual ao dia da morte.
[00231] O resultado do tratamento foi avaliado pelo retardo no crescimento tumoral (TGD), definido como o aumento na mediana de tempo até o desfecho (TTE) em um grupo de tratamento em comparação ao grupo controle: TGD = T - C,
[00232] expresso em dias ou como uma porcentagem da mediana de TTE do grupo controle:
[00233] onde:
[00234] T = mediana de TTE para um grupo de tratamento e
[00235] C = mediana de TTE para o grupo controle designado.
CRITÉRIOS PARA RESPOSTAS DE REGRESSÃO
[00236] A eficácia do tratamento pode ser determinada a partir da incidência e magnitude das respostas de regressão observadas durante o estudo. O tratamento pode provocar regressão parcial (PR) ou regressão completa (CR) do tumor em um animal. Em uma resposta PR, o volume tumoral tinha 50% ou menos de seu volume do Dia 1 em três medições consecutivas no transcorrer do estudo, e igual ou maior do que 135 mm3 em uma ou mais dessas três medições. Em uma resposta CR, o volume tumoral era inferior a 135 mm3 em três medições consecutivas no transcorrer do estudo. Um animal com resposta CR no encerramento de um estudo era classificado adicionalmente como sobrevivente livre de tumor (TFS). Os animais foram monitorados para respostas de regressão.
TOXICIDADE
[00237] Os animais foram pesados diariamente nos Dias 1-5, depois duas vezes por semana até a conclusão do estudo. Os camundongos foram observados frequentemente para detectar sinais evidentes de quaisquer efeitos colaterais adversos relacionados ao tratamento (TR), e os sinais clínicos eram registrados quando observados. A perda de peso corporal individual foi monitorada segundo o protocolo, e qualquer animal que excedesse os limites para perda aceitável do peso corporal era sacrificado. A perda de peso corporal média do grupo foi também monitorada de acordo com o protocolo. A administração deveria ser suspensa em qualquer grupo que excedesse os limites para perda aceitável do peso corporal médio. Caso o peso corporal médio se recuperasse, então a administração deveria ser reiniciada naquele grupo, mas em um esquema de administração de dose mais baixa ou menos frequente.
[00238] Toxicidade aceitável para a dose máxima tolerada (MTD) foi definida como perda do peso corporal médio do grupo inferior a 20% durante o estudo e no máximo 10% das mortes relacionadas ao tratamento (TR). Uma morte era classificada como TR se atribuível a efeitos colaterais do tratamento, evidenciados por sinais clínicos e/ou na necropsia, ou era também classificada como TR se decorrente de causas desconhecidas durante o período de administração ou dentro de 14 dias da última dose. Uma morte era classificada como não relacionada ao tratamento (NTR) se não houvesse evidência de que a morte estava relacionada a efeitos colaterais do tratamento. As mortes NTR foram caracterizadas ainda com base na causa de morte. Uma morte era classificada como NTRa se resultasse de um acidente ou erro humano. Uma morte era classificada como NTRm se a necropsia indicasse que esta pudesse ter resultado da disseminação do tumor por invasão e/ou metástase. Uma morte era classificada como NTRu se a causa de morte fosse desconhecida e não houvesse evidência disponível de morte relacionada a efeitos colaterais do tratamento, metástases, acidente ou erro humano, embora a morte em decorrência de efeitos colaterais do tratamento não possa ser excluída.
AMOSTRAGEM
[00239] Quando disponível, sacrificou-se cinco camundongos por grupo por punção cardíaca terminal sob a anestesia com dióxido de carbono em 3, 6 e 12 horas após a dose final, e o volume total de sangue de cada animal foi coletado. O soro foi separado e armazenado congelado a -80 °C até a remessa. Além disso, os tumores desses camundongos foram colhidos e divididos em duas partes. Uma parte foi imediatamente congelada e armazenada a -80 °C. A outra parte foi fixada por 16-24 horas em formol neutro tamponado 10% e depois transferida para etanol 70%. Para grupos com camundongos sem tumor detectável, o sítio do implante, incluindo a espessura toda de pele e músculo, foi coletado de três camundongos por grupo.
ANÁLISES ESTATÍSTICAS E GRÁFICAS
[00240] O programa Prism (GraphPad) para Windows 3.03 foi utilizado nas representações gráficas e análises estatísticas.
[00241] O teste logrank, que avalia a experiência de sobrevida global, foi utilizado para analisar a significância das diferenças entre os valores de TTE de dois grupos. A análise por logrank inclui os dados de todos os animais em um grupo, exceto aqueles avaliados como mortes NTR. Análises estatísticas bicaudais foram conduzidas no nível de significância P = 0,05. Os testes estatísticos não foram ajustados para comparações múltiplas. O programa Prism resume os resultados dos testes como não significante (ns) em P > 0,05, significante (simbolizado por "*") em 0,01 < P < 0,05, como muito significante ("**") em 0,001 < P <0,01 e extremamente significante ("***") em P < 0,001. Como os testes de significância estatística não fornecerem uma estimativa da magnitude da diferença entre grupos, todos os níveis de significância foram descritos como significante ou não significante no texto deste relatório.
[00242] Construiu-se um gráfico de dispersão para mostrar os valores de TTE de cada camundongo, por grupo. Os volumes tumorais médios dos grupos foram traçados em função do tempo. Quando um animal saía do estudo pelo tamanho do tumor, o volume tumoral final registrado para o animal foi incluído com os dados utilizados para calcular o volume médio em momentos subsequentes. As barras de erro (quando presentes) indicam um erro padrão da média (SEM). Gráficos de Kaplan-Meier mostram a porcentagem de animais em cada grupo que ainda permaneciam no estudo versus o tempo. O gráfico de Kaplan-Meier e o teste logrank compartilham os mesmos conjuntos de dados de TTE. Alterações percentuais no peso corporal médio em relação ao Dia 1 foram calculadas de cada grupo para cada dia de medição do peso corporal, e foram representadas no gráfico em função do tempo. Os gráficos de crescimento tumoral e peso corporal excluíram os dados para mortes NTR, e foram truncados depois que 50% dos animais avaliáveis em um grupo haviam saído do estudo.
RESULTADOS
[00243] Os grupos no estudo A375 in vivo foram tratados de acordo com o protocolo modificado descrito na Tabela 15. O experimento foi encerrado no Dia 45. A Tabela 16 apresenta um resumo das respostas ao tratamento de cada grupo. A Figura 26 é um gráfico de dispersão mostrando os TTEs individuais de cada grupo. A Figura 27 apresenta gráficos de crescimento tumoral médio (Figura 27A) e de sobrevida de Kaplan-Meier (Figura 27B) para cada grupo no estudo. As Figuras 28A-D apresentam gráficos de crescimento tumoral médio para as quatro combinações, quando comparadas às suas respectivas monoterapias. A Figura 29 apresenta gráficos de alterações percentuais no peso corporal médio em relação ao Dia 1 para cada grupo.
EFICÁCIA - CRESCIMENTO DE MELANOMAS HUMANOS DA LINHAGEM A375 EM CAMUNDONGOS CONTROLE (GRUPO 1)
[00244] No Grupo 1, um camundongo controle foi encontrado morto para além da necropsia no Dia 4, e a morte foi avaliada como NTRu. Os outros catorze tumores em controles progrediram rapidamente e de modo uniforme até o desfecho de 2000 mm3 com mediana de TTE de 9,2 dias, estabelecendo um TGD máximo possível de 35,8 dias (389%) para o estudo de 45 dias (Tabela 15). O gráfico de dispersão mostra um agrupamento de TTEs de controles (Figura 26). O gráfico de crescimento tumoral médio do Grupo 1 ilustrou o crescimento tumoral rápido de controle (Figura 27A e Figuras 28A-D).
EFICÁCIA - RESPOSTA AO DABRAFENIB EM MONOTERAPIA (GRUPOS 6 E 7)
[00245] Os Grupos 6 e 7 receberam dabrafenib em monoterapia a 50 e 100 mg/kg, respectivamente, via oral, 1x/dia até o final. As medianas de TTE para os Grupos 6 e 7 foram 16,1 e 28,5 dias, respectivamente, correspondendo a TGDs relacionados à dose de 6,9 dias (75%) e 19,3 dias (210%), com uma diferença significativa na sobrevida para cada um, quando comparados aos controles (Grupo 1 versus 6 ou 7, P < 0,001). Uma PR foi registrada no grupo dabrafenib 100 mg/kg (Tabela 16). Todos os tumores do Grupo 6 atingiram o volume tumoral do desfecho de 2000 mm3, enquanto que 13/15 tumores do Grupo 7 atingiram o desfecho e dois permaneceram no Dia com um MTV de 282 mm3 (Tabela 16). Os gráficos de crescimento tumoral médio para os Grupos 6 e 7 ilustraram os retardos relacionados à dose, embora tumores em ambos os grupos tenham progredido durante o tratamento (Figura 27A).
EFICÁCIA - RESPOSTA AO BVD-523 EM MONOTERAPIA (GRUPOS 8 E 9)
[00246] Os Grupos 8 e 9 receberam BVD-523 em monoterapia a 50 e 100 mg/kg, respectivamente, via oral, 2x/dia até o final. As medianas de TTE para os Grupos 8 e 9 foram 8,6 e 18,5 dias, respectivamente, os quais corresponderam a nenhum TGD para o grupo BVD-523 50 mg/kg e TGD de 9,3 dias (101%) para o grupo BVD-523 100 mg/kg (Tabela 16). As análises por logrank detectaram uma diferença significativa na sobrevida somente para BVD-523 100 mg/kg quando comparado a controles (Grupo 1 versus 8, P > 0,05; Grupo 1 versus 9, P < 0,001). O Grupo 8 teve uma CR que permanecia um TFS no Dia 45, enquanto no Grupo 9 houve dois CRs/TFSs, e todos os outros tumores nesses dois grupos atingiram o volume tumoral do desfecho de 2000 mm3 (Tabela 16). O gráfico de crescimento tumoral médio para o grupo BVD-523 50 mg/kg foi comparável àquele de controles, enquanto que o grupo BVD-523 100 mg/kg mostrou retardo marginal com tumores que progrediram durante o tratamento (Figura 27A).
EFICÁCIA - RESPOSTA AO TRATAMENTO COM COMBINAÇÕES DE DABRAFENIB E BVD-523 (GRUPOS 2-5)
[00247] Os Grupos 2 e 3 receberam dabrafenib 50 mg/kg com BVD-523 50 ou 100 mg/kg, respectivamente, enquanto que os Grupos 4 e 5 receberam dabrafenib 100 mg/kg com BVD-523 50 ou 100 mg/kg, respectivamente. Como indicado na Tabela 16, os regimes combinados foram modificados de modo que a administração foi finalizada depois do Dia 20 e depois reiniciada no Dia 42 (Tabela 16).
[00248] As medianas de TTE para os Grupos 2-5 foram cada 45,0 dias, correspondendo ao TGD máximo possível para o estudo (35,8 dias, 389%) e um benefício significativo à sobrevida global em comparação a controles (Grupo 1 versus 2-5, P < 0,001).
[00249] Cinco tumores no Grupo 2 atingiram o volume do desfecho de 2000 mm3, enquanto que, nos Grupos 3-5, nenhum tumor cresceu até o volume do desfecho. No Grupo 2, houve três PRs e oito CRs, com sete camundongos que permaneciam TFSs no Dia 45 (Tabela 16). No Grupo 3, houve uma morte NTRu no Dia 31, e os outros catorze camundongos obtiveram CRs e permaneciam TFSs no final do estudo. No Grupo 4, houve uma PR e catorze CRs que permaneciam TFSs, enquanto que, no Grupo 5, 100% foram TFSs.
[00250] As cargas tumorais médias não eram detectáveis nos Grupos 2-5 no Dia 20 quando a administração foi interrompida (Figura 27A). O crescimento tumoral médio reiniciou somente no grupo da combinação na dose mais baixa (Grupo 2), tendo permanecido não detectável até o final do estudo nos outros três grupos de combinações (Figura 27Al). O gráfico de crescimento tumoral de cada grupo de combinação mostrou atividade notável quando comparado às suas respectivas monoterapias (Figuras 28A-D).
EFICÁCIA - RESPOSTA AO TRATAMENTO COM TEMOZOLOMIDA (GRUPO 10)
[00251] O tratamento de referência com temozolomida resultou em uma mediana de TTE de 10,5 dias, que correspondeu a TGD descartável (1,3 dias, 14%), sem regressões (Tabela 16). As análises por logrank não detectaram diferença significativa na sobrevida para o grupo temozolomida quando comparado a controles (Grupo 1 versus 10, P = 0,052). O gráfico de crescimento tumoral médio para esse grupo mostrou retardo desprezível quando comparado ao gráfico para controles do Grupo 1 (Figura 27A).
EFEITOS COLATERAIS
[00252] A Tabela 16 fornece um resumo de perdas máximas de PC médio e de mortes TR e NTR. A Figura 29 apresenta gráficos de alterações percentuais de PC médio em relação ao Dia 1 para cada grupo.
[00253] Não foram registradas mortes TR no estudo, mas duas mortes NTRu foram avaliadas (Tabela 16). Uma morte NTRu foi registrada no Grupo 1 no Dia 4, e uma segunda morte NTRu foi registrada no Grupo 3 no Dia 31. O animal do Grupo 1 foi encontrado morto para além da necropsia sem observações clínicas prévias, enquanto que o camundongo do Grupo 3 estava magro, encurvado e letárgico um pouco antes da morte, e a necropsia revelou uma massa de nódulos brancos no fígado sugerindo que doença metastática era uma possível causa de morte. Houve perdas desprezíveis ou nenhuma no PC médio entre os grupos no estudo (Tabela 16 e Figura 29), e nenhum sinal notável de efeitos colaterais relacionados ao tratamento entre os grupos de BVD-523 e dabrafenib em monoterapia e em terapia combinada.
SUMÁRIO
[00254] O estudo in vivo avaliou combinações de BVD-523 com dabrafenib quanto à eficácia no modelo de xenoenxerto de melanoma humano da linhagem A375 em camundongos nu. BVD-523 foi administrado por via oral a 50 ou 100 mg/kg em um esquema de duas vezes ao dia, e dabrafenib foi fornecido por via oral a 50 ou 100 mg/kg em um esquema diário, isoladamente e combinados. Devido à resposta surpreendente ao tratamento combinado, a administração nos grupos de combinações foi interrompida no Dia 20 para monitorar novo crescimento do tumor, e foi reiniciada no Dia 42 para a coleta de amostras no final do estudo no Dia 45.
[00255] Os tumores da linhagem A375 de controles progrediram rapidamente e de modo uniforme até o desfecho do volume tumoral. A mediana de TTE para controles foi 9,2 dias, estabelecendo um TGD máximo possível de 35,8 dias (389%) para o estudo de 45 dias. Uma faixa estreita de TTEs controles, que refletiu o crescimento tumoral controle uniforme, permitiu que o teste logrank detectasse pequenas diferenças entre camundongos controle e tratados. O tratamento de referência com temozolomida resultou em TGD desprezível (1,3 dias, 14%) e sem regressões, compatíveis com resultados anteriores para temozolomida nesse modelo de tumor.
[00256] Monoterapias de dabrafenib a 50 e 100 mg/kg produziram eficácia relacionada à dose, com TGDs de 6,9 dias (75%) e 19,3 dias (210%), respectivamente, e uma PR no grupo dabrafenib 100 mg/kg. A monoterapia de BVD-523 50 mg/kg foi inativa, não produzindo TGD nem diferença significativa na sobrevida dos controles (P > 0,05). O único TFS nesse grupo poderia ter sido em decorrência do tratamento ou uma regressão espontânea. A monoterapia de BVD-523 100 mg/kg marginalmente ativa, resultando em TGD de 9,3 dias (101%), uma diferença significativa na sobrevida em comparação a controles (P < 0,001), e dois TFSs que poderiam ter sido em decorrência do tratamento ou uma regressão espontânea.
[00257] Cada uma das quatro combinações de dabrafenib com BVD-523 testadas nesse estudo foi altamente ativa, produzindo o TGD máximo possível, respostas de regressão notáveis e sobrevida global estatisticamente superior quando comparadas às suas monoterapias correspondentes (P < 0,001). O grupo da combinação com a dose mais baixa (Grupo 2) produziu um número notável de 7/15 TFSs. As três combinações com doses mais altas (Grupos 3-5) alcançaram 43/44 sobreviventes livres de tumor no final do estudo, incluindo 15/15 TFSs no grupo da combinação com a dose mais alta (Grupo 5). É ainda importante enfatizar que, dado um tempo médio de duplicação inferior a 3 dias para tumores controle, não ocorreu novo crescimento tumoral em 43/44 camundongos entre os Grupos 3-5 durante o descanso da administração dos Dias 21 a 42, cuja duração de tempo corresponde a aproximadamente dobrar 7 vezes o tumor. Esses resultados foram compatíveis com atividade curativa ou próxima à curativa.
[00258] Em suma, dabrafenib e BVD-523 produziram individualmente uma eficácia marginal relacionada à dose em monoterapias, mas atividade acentuada quando combinados. As combinações de dabrafenib com BVD-523 testadas nesse estudo produziram sobrevida livre de tumor notável e eficácia superior em relação a qualquer um dos agentes fornecido isoladamente.
[00259] Demonstra-se que a inibição de ERK quinase, que é exemplificada utilizando BVD-523, é eficaz em combinação com o inibidor de RAF dabrafenib em um modelo de melanoma com mutação em BRAF. Em células, o tratamento combinado de BVD-523 e dabrafenib induz janelas de inibição sinérgica da proliferação celular. Quando administrados junto em um modelo de xenoenxerto, o tratamento combinado provoca regressão proeminente e durável do tumor quando combinado à terapia com agentes isolados.
[00260] Adicionalmente, quando células A375 são induzidas para exibir resistência adquirida a fármacos após a exposição prolongada aos inibidores da cascata de MAPK, a inibição de ERK utilizando BVD- 523 revela propriedades atraentes. Dentro de semanas após o tratamento com dabrafenib ou trametinib, é possível isolar células A375 que crescem rapidamente em concentrações 10 vezes maiores do que a concentração inibitória IC50 de crescimento do respectivo composto. Depois de 2 meses, células expostas ao BVD-523 isolado cresceram pouco, e somente suportaram o tratamento quando a exposição ao fármaco representava aumentos inferiores a 10 vezes acima a IC50. As células tratadas com a combinação de BVD-523 e dabrafenib exibiram igualmente baixo crescimento, e somente puderam ser cultivados em níveis discretamente aumentados do dabrafenib quando em combinação.
[00261] Finalmente, BVD-523 foi testado em um modelo de melanoma derivado de biópsias obtidas de um paciente que exibia progressão da doença após resposta inicial ao vemurafenib. Curiosamente, esse modelo in vivo revelou resistência cruzada adquirida, parecendo insensível ao dabrafenib e ao trametinib. BVD- 523 aparentemente é eficaz no modelo, contudo, e induziu uma resposta antitumoral potente, quer isoladamente ou em combinação com dabrafenib.
[00262] Em conjunto, esses resultados sugerem que o tratamento combinado de inibidores de ERK e de RAF é eficaz no contexto de melanoma com mutação no gene BRAF. BVD-523 dispõe de um novo modo de ação farmacológica e possivelmente exib duração prolongada em modelos que mostram sensibilidade intrínseca ou resistência adquirida aos inibidores de BRAF e de MEK. A combinação de inibidores de RAF e de ERK para cânceres com mutação no gene BRAF inib uma via oncogênica em dois pontos de controle, o que, por sua vez, parece criar uma barreira difícil contra subversão e resistência adquirida a fármacos.
[00263] Esses achados indicam que a terapia com a combinação de inibidores de ERK e de RAF pode ser eficaz em uma variedade de cânceres, especialmente aqueles que abrigam alterações oncogênicas no gene BRAF, incluindo melanoma, cânceres da tireoide, do pulmão e do cólon.
EXEMPLO 8
ESTUDOS DE COMBINAÇÕES ADICIONAIS
ENSAIOS DE PROLIFERAÇÃO COM AGENTE ISOLADO
[00264] As células foram semeadas em placas de 96 cavidades nas densidades e nas condições de meio indicadas na Tabela 17 e deixadas que se aderissem durante a noite antes da adição do composto ou veículo controle. Os compostos foram preparados com estoques de DMSO para fornecer as concentrações finais desejadas. A concentração final de DMSO foi mantida constante em 0,1%. Os compostos em teste foram incubados com as células por 72 horas a 37 °C, CO2 5% em atmosfera umidificada. O reagente CellTiter-Glo® (Promega, Madison, WI) foi adicionado de acordo com as instruções do fabricante e a luminescência detectada utilizando o leitor de placas BMG FLUOstar (BMG Labtech, Ortenberg, Alemanha). O valor médio de fundo com meio apenas foi subtraído e os dados analisados utilizando uma equação logística com 4 parâmetros no programa GraphPad Prism (GraphPad Software, La Jolla, CA).
ENSAIOS DE PROLIFERAÇÃO DE COMBINAÇÕES
[00265] As células foram semeadas, em triplicata, em placas de 96 cavidades nas densidades e nas condições de meio indicadas na Tabela 17 e deixadas que se aderissem durante a noite antes da adição do composto ou veículo controle. Os compostos foram preparados com estoques de DMSO para fornecer as concentrações finais desejadas. A concentração final de DMSO foi mantida constante em 0,2%. As combinações foram testadas utilizando uma matriz de dose 10 x 8. Os compostos em teste foram incubados com as por 72 horas a 37 °C, CO2 5% em atmosfera umidificada. O reagente CellTiter-Glo® (Promega, Madison, WI) foi adicionado de acordo com as instruções do fabricante e a luminescência detectada utilizando o leitor de placas BMG FLUOstar (BMG Labtech, Ortenberg, Alemanha). O valor médio de fundo com meio apenas foi subtraído e os dados analisados.
[00266] Para os ensaios 10 x 8 de combinações, as interações das combinações na matriz de dose foram determinadas pelos modelos de Aditividade de Loewe e independência de Bliss utilizando o software para análise de combinações Chalice™ (Horizon Discovery Group, Cambridge, MA), conforme delineado no manual do usuário (disponível em chalice.horizondiscovery.com/chalice- portal/documentation/analyzer/home.jsp). A sinergia é determinada comparando o nível de inibição observado experimentalmente em cada ponto da combinação com o valor previsto para aditividade, o qual é derivado a partir das respostas dos agentes isolados ao longo das bordas da matriz. Interações sinérgicas potenciais foram identificadas exibindo o excesso de inibição calculado sobre aquele previsto como aditivo na matriz de dose em um mapa de calor, e relatando um "Escore de Sinergia" quantitativo com base no modelo de Loewe. Os dados de agentes isolados derivados das placas de ensaio das combinações foram apresentados como curvas de dose-resposta geradas no GraphPad Prism (GraphPad Software, La Jolla, CA) (traçadas utilizando a porcentagem de viabilidade em relação a controles tratados somente com DMSO). TABELA 17 - Densidade de semeadura da linhagem celular e meios de crescimento

[00267] O objetivo desse estudo era avaliar os efeitos de se combinar inibidores de ERK com inibidores de RAF Tipo I. Um novo inibidor de ERK, BVD-523, com dois inibidores de RAF Tipo I, Dabrafenib (GSK2118436) e Vemurafenib (PLX4032), e um inibidor Tipo II, TAK-632, em duas linhagens celulares de melanoma com mutação no gene BRAF, V600E, as células A375 e G-361. Um segundo inibidor de ERK, com mecanismo distinto (SCH772984), foi também testado em combinação com Dabrafenib (GSK2118436) e Vemurafenib (PLX4032).
[00268] Primeiramente, foram realizados ensaios de proliferação com os agentes isolados para selecionar uma faixa adequada de concentrações para os estudos das combinações. Embora as duas linhagens celulares tivessem um nível de sensibilidade frente ao paclitaxel semelhante, as células G-361 aparentavam ser 4 a 6 vezes menos sensíveis à inibição de ERK e de RAF, quando comparadas às células A375 (Figura 42). Os resultados de IC50 estão resumidos na Tabela 18. TABELA 18 -IC50 como agentes isolados dos compostos testados
*Os valores para o dabrafenib devem ser considerados como aproximações, pois o topo das curvas não ficou bem definido pela faixa de doses testadas.
[00269] As interações de combinações entre dois compostos foram avaliadas em uma matriz 8x10 de concentrações utilizando os Modelos de Aditividade de Loewe e Independência de Bliss com o software de bioinformática Chalice™ (Horizon Discovery Group, Cambridge, MA). O Chalice™ permite identificar interações sinérgicas potenciais exibindo o excesso de inibição calculado sobre aquele previsto como aditivo na matriz de dose em um mapa de calor, e relatando um "Escore de Sinergia" quantitativo com base no modelo de Loewe.
[00270] Em células A375 (Figura 43 - Figura 48), a análise utilizando o modelo de Loewe indicou que as combinações com BVD- 523 pareceram principalmente aditivas. Os resultados utilizando o método de Bliss foram semelhantes, embora esse método sugerisse a presença de uma região de antagonismo leve em concentrações mais elevadas de cada combinação. Por outro lado, em células G-361 (Figura 49 - Figura 54), embora a maior parte das interações na matriz de dose tivesse sido também aditiva, os dois modelos de análise também revelaram pequenos bolsões de sinergia modesta nas concentrações médias. Resultados semelhantes foram obtidos com um segundo inibidor de ERK com mecanismo distinto (SCH772984). Isso respalda a noção de que provavelmente as sinergias observadas em G-361 estejam relacionadas especificamente à inibição de ERK e não se devam a efeitos fora do alvo.
[00271] Em suma, esses resultados sugerem que as interações entre BVD-523 e inibidores de RAF tipo I e tipo II RAF são ao menos aditivas e, em alguns casos, sinérgicas, em linhagens celulares de melanoma portadoras de uma mutação V600E no gene BRAF.
[00272] As interações sinérgicas foram pontuadas de duas maneiras (Figura 55 - Figura 57). A atividade em excesso sobre aquela prevista se uma combinação fosse aditiva pode ser calculada utilizando uma pontuação simples de volume, que calcula o volume entre a superfície de resposta medida e a prevista. Essa pontuação de volume mostra se a resposta global a uma combinação é sinérgica (valores positivos), antagonista (valores negativos) ou aditiva (valores ~ 0). Adicionalmente, um "Escore de Sinergia" é um volume ponderado pela inibição dependente de valores positivos acima da aditividade de Loewe. Esse escore propicia uma priorização adicional em favor de combinações cuja sinergia ocorre em níveis elevados de efeito, ignorando partes antagonistas da superfície de resposta.
EXEMPLO 9
INTERAÇÕES DE COMBINAÇÕES ENTRE INIBIDORES DE ERK
[00273] Células A375 da linhagem celular de melanoma com mutação em RAF foram cultivadas em DMEM com FBS 10% e semeadas, em triplicata, em placas de 96 cavidades a uma densidade inicial de 2000 células por cavidade. As interações de combinações entre inibidores de ERK e BVD-523 e SCH772984 foram analisadas depois de 72 horas como descrito acima no Exemplo 8. A viabilidade foi determinada utilizando o reagente CellTiter-Glo® (Promega, Madison, WI) de acordo com as instruções do fabricante e a luminescência detectada utilizando o leitor de placas BMG FLUOstar (BMG Labtech, Ortenberg, Alemanha).
[00274] A visualização dos mapas de calor de "excesso de inibição" de Loewe e Bliss sugeriu que a combinação de BVD-523 com SCH772984 foi principalmente aditiva com janelas de sinergia potencial em doses na faixa intermediária (Figura 58).
[00275] Em suma, esses resultados sugerem que interações entre BVD-523 e SCH772984 são ao menos aditivas e, em alguns casos, sinérgicas. DOCUMENTOS AVRUCH, J.; et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res., 2001, 127-155. BROSE, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res., 2002, 62, 6997-7000. DAVIES et al., Mutations of the BRAF gene in human cancer. Nature, 2002, 417, 949-954. FRANSEN et al., Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis, 2004, 25, 527-533. GARNETT, M.J.; et al. Wildtype and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell, 2005, 20, 963-969. GREGER, James G., et al. "Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations." Molecular cancer therapeutics 11.4 (2012): 909-920. HOCKER et al., Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants. Hum. Mutat., 2007, 28, 578-588. KING, Alastair J., et al. "Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions." PloS one 8.7 (2013): e67583. LI et al., Recent advances in the research and development of B-Raf Inhibitors. Current Medicinal Chemistry, 2010, 17:1618-1634. LIU, Dingxie, et al. "BRAF V600E maintains proliferation, transformation, and tumorigenicity of BRAF-mutant papillary thyroid cancer cells." Journal of Clinical Endocrinology & Metabolism 92.6 (2007): 2264-2271. LITTLE, A.S., et al., Amplification of the Driving Oncogene, KRAS or BRAF, Underpins Acquired Resistance to MEK1/2 Inhibitors in Colorectal Cancer Cells. Sci. Signal. 4, ra17 (2011). LONG GV, Menzies AM, Nagrial AM, et al. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J Clin Oncol. 2011. MANANDHAR SP, Hildebrandt ER, Schmidt WK. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screen. 2007;12(7):983-993. MAURER, T, Garrenton, LS, Oh, A, Pitts, K, Anderson, DJ, Skelton, NJ, Fauber, BP, Pan, B, Malek, S, Stokoe, D, Ludlam, MJC, Bowman, KK, Wu, J, Giannetti, AM, Starovasnik, MA, Mellman, I, Jackson, PK, Rudolph, J, Wang, W, Fang, G. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. PNAS. 2012;109(14):5299-304. MITTAL, Rohit et al. "The acetyltransferase activity of the bacterial toxin YopJ of Yersinia is activated by eukaryotic host cell inositol hexakisphosphate." Journal of Biological Chemistry 285.26 (2010): 19927-19934. PATGIRI, A, Yadav, KK, Arora, PS, Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7:585-587. PORTER SB, Hildebrandt ER, Breevoort SR, Mokry DZ, Dore TM, Schmidt WK. Inhibition of the CaaX proteases Rce1p and Ste24p by peptidyl (acyloxy)methyl ketones. Biochim Biophys Acta.2007;1773(6):853-862. RUSHWORTH, L.K.; et al. Regulation and role of Raf-1/B- Raf heterodimerization. Mol. Cell Biol., 2006, 26, 2262-2272. SETH et al., Concomitant mutations and splice variants in KRAS and BRAF demonstrate complex perturbation of the Ras/Raf signalling pathway in advanced colorectal cancer, Gut 2009;58:1234- 1241 SHIMA, F, Yoshikawa, Y, Ye, M, Araki, M, Matsumoto, S, Liao, J, Hu, L, Sugimoto, T, Ijiri, Y, Takeda, A, Nishiyama, Y, Sato, C, Muraoka, S, Tamura, A, Osoda, T, Tsuda, K-I, Miyakawa, T, Fukunishi, H, Shimada, J, Kumasaka, Yamamoto, M, Kataoka, T. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. PNAS. 2013;110(20):8182-7. WAN, et al., Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell, 2004, 116, 855867. WEBER, C.K.; et al. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res., 2001, 61, 3595-3598. Wellbrock et al. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol., 2004, 5, 875-885. XU et al., High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res., 2003, 63, 4561-4567.
[00276] Todos os documentos citados neste pedido de patente são aqui incorporados por referência conforme seria caso recitados integralmente no presente documento.
[00277] Embora modalidades ilustrativas da presente invenção tenham sido descritas neste pedido de patente, deve-se entender que a invenção não se limita àquelas descritas, e que várias outras alterações ou modificações podem ser efetuadas pelo técnico no assunto sem que, contudo, se desviem do âmbito ou espírito da invenção.