BR112014000443B1 - Métodos para determinação do prognóstico de um indivíduo com câncer - Google Patents
Métodos para determinação do prognóstico de um indivíduo com câncer Download PDFInfo
- Publication number
- BR112014000443B1 BR112014000443B1 BR112014000443-9A BR112014000443A BR112014000443B1 BR 112014000443 B1 BR112014000443 B1 BR 112014000443B1 BR 112014000443 A BR112014000443 A BR 112014000443A BR 112014000443 B1 BR112014000443 B1 BR 112014000443B1
- Authority
- BR
- Brazil
- Prior art keywords
- cancer
- treatment
- methylated
- individual
- methylation
- Prior art date
Links
Images














Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/118—Prognosis of disease development
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
Abstract
métodos e ácidos nucléicos para determinação do prognóstico de um indivíduo com câncer. a invenção fornece métodos, ácidos nucléicos e kits para determinação do prognóstico de um indivíduo que tem câncer. a invenção revela sequências genômicas cujos padrões de metilação possuem utilidade para a detecção aprimorada do referido distúrbio permitindo, dessa forma, o diagnóstico e tratamento aprimorados de pacientes.
Description
[001] A presente invenção está relacionada aos marcadores de DNA genômico úteis na determinação do prognóstico de um indivíduo com câncer, determinação do tratamento médico para um indivíduo com câncer, determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo, detecção de uma forma agressiva de câncer em um indivíduo, seleção de um indivíduo com câncer para tratamento de câncer ou determinação da carga tumoral ou carga de câncer em um indivíduo. Modalidades particulares fornecem métodos, ácidos nucléicos, arranjos de ácido nucléico e kits úteis para determinação do prognóstico de um indivíduo que tem câncer.
[002] Métodos para determinação do prognóstico e, dessa forma, métodos e agentes para determinação de tratamento, de um paciente com câncer incluem a determinação do estadiamento do tumor com base em vários critérios. Frequentemente essa determinação inclui procedimentos invasivos para observar alterações histológicas na morfologia tecidual e no nível de invasão do tumor no tecido vizinho e metástase.
[003] Em particular, o câncer cólon-retal é o segundo câncer mais frequente na Europa e nos Estados Unidos (412.900 e 150.000 indivíduos em 2006, respectivamente). Em 75% dos casos a doença é removida por cirurgia. No entanto, há recorrência em 30-40% dos cânceres cólon-retais em estágio II-III, a maioria dentro de 3-5 anos do diagnóstico inicial. Além disso, somente 16-66% dos pacientes são sintomáticos no diagnóstico de recorrência e desses tumores apenas 1,7-7% são ressecáveis. Dessa forma, 93-98,3% de casos recorrentes são identificados muito além do momento em que a ressecção é suficiente para remover todo o tumor ou células tumorais. Veja Fakih, M.G. MD, CEA “Monitoring in Colorectal Cancer, What You Should Know”, Volume 20: Número 6: 2006.
[004] As diretrizes práticas atuais para vigilância pós-ressecção para tumores em Estágio II e maior incluem o monitoramento de CEA (antígeno carcinoembrionário) a cada 3-6 meses por 2 anos e depois a cada 6 meses por um total de 5 anos, e/ou colonoscopia após 1 ano, opcionalmente repetida a cada segundo ano. Para pacientes com câncer cólon-retal em Estágio I e II que são positivos por CEA antes da cirurgia, apenas 3% a 32% dos pacientes podem ser monitorados por monitoramento baseado em CEA, deixando 68- 97% de pacientes em Estágio I e II que não podem ser monitorados de forma alguma com CEA. Além disso, a sensibilidade do CEA depende do local de recorrência, de tal forma que apenas uma porção dos 3-32% de pacientes que podem ser monitorados podem se beneficiar.
[005] Atualmente, o único marcador prognóstico válido na previsão do resultado final da pacientes com câncer cólon-retal (CRC) é o sistema de estadiamento TumorLinfonodo-Metástase (TNM) (Tumor-Node-Metastasis). Os parâmetros desse sistema são geralmente qualitativos e não são informativos para diferenciação adicional do risco em pacientes de risco padrão, que constituem a maioria dos cânceres de cólon em estágio II. Aproximadamente 30% de pacientes com câncer de cólon possuem uma doença em estágio II. As diretrizes atuais do “National Comprehensive Cancer Network” (NCCN) não recomendam o uso de rotina de quimioterapia adjuvante para todos os pacientes com câncer de cólon em estágio II, mas sim consideram o tratamento adjuvante no quadro de risco elevado de recorrência. A taxa de sobrevida em cinco anos para a população global de pacientes em estágio II foi estimada como sendo de 75-80%. Apesar dessas taxas de cura relativamente elevadas somente com cirurgia, em uma proporção significante de pacientes de câncer em estágio II haverá recidiva. A identificação de marcadores que distinguem aqueles pacientes em risco baixo daqueles em risco elevado de recorrência da doença será útil para identificar aqueles pacientes que seriam candidatos para quimioterapia adjuvante. Os biomarcadores no câncer de cólon em estágio II até hoje estavam limitados ao diagnóstico clínico, mas não eram usados no prognóstico ou resultado clínico final.
[006] Foram descritas várias proteínas e marcadores genéticos em uma tentativa de aprimorar a informação prognóstica e prever o benefício do tratamento sistêmico. Diferentemente de outros tipos de câncer, com a exceção da mutação de KRAS, nenhum dos marcadores estudados entrou na conduta clínica do câncer cólon-retal até agora.
[007] Metilação de ilha CpG: foi demonstrado que a metilação aberrante de ilhas CpG leva ao silenciamento transcricional de certos genes que foram ligados previamente à patogênese de vários distúrbios proliferativos celulares, incluindo câncer. Ilhas CpG são sequências que são ricas em dinucleotídeos CpG e podem normalmente ser encontradas na região 5’ de aproximadamente 50% de todos os genes humanos. A metilação das citosinas nessas ilhas leva à perda de expressão gênica e foi relatada na inativação do cromossomo X e imprinting genômico.
[008] Metilação de DNA e prognóstico de doença: foi demonstrado que a metilação de DNA está associada ao prognóstico do paciente em diversas publicações como, por exemplo, EP 1692316 e WO 2007/085497.
[009] Há necessidade de um meio melhor para determinar o prognóstico, resultado clínico final, carga tumoral, carga de câncer e/ou inclusão de um paciente em um grupo de tratamento, em qualquer ponto, começando no diagnóstico inicial e continuando durante o período de tratamento, incluindo a habilidade para determinar o estado de recidiva, remissão ou recorrência, usando técnicas de testagem minimamente invasivas.
[010] A invenção fornece um método para determinação do prognóstico de um indivíduo com câncer, que compreende as etapas de: medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra prétratamento indica tratamento de câncer adicional para o indivíduo. Em uma modalidade, uma quantidade aumentada do DNA genômico metilado ou fragmento na amostra póstratamento comparada com a amostra pré-tratamento indica que o câncer é agressivo ou possui potencial metastático ou tempo de sobrevida reduzido para o indivíduo. Em uma modalidade preferida, o método da invenção fornece um método para determinação do prognóstico de um indivíduo com câncer, que compreende as etapas de: a) medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento quando comparada com a amostra pré-tratamento indica um prognóstico ruim e, dessa forma, uma necessidade para tratamento de câncer adicional para o indivíduo. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível prétratamento medido de DNA metilado e d) determinação do prognóstico de um indivíduo com câncer com base no resultado da comparação da etapa c), de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento quando comparada com a amostra pré-tratamento indica um prognóstico ruim e, dessa forma, uma necessidade para tratamento de câncer adicional para o indivíduo.
[011] Níveis estáveis ou até mesmo aumentados do DNA genômico metilado, preferivelmente, indicam que o tratamento escolhido falhou em remover as células de câncer descarregando o fragmento de DNA metilado ou que seu número aumentou apesar do tratamento, ou seja, o câncer cresceu ainda mais. Em contraste com isso, níveis diminuídos do DNA genômico metilado, preferivelmente, indicam que o número de células de câncer diminuiu, ou seja, que o tratamento foi bem sucedido na redução da carga tumoral do paciente. Em particular, uma diminuição a níveis que estão abaixo do nível de detecção, indica que todas as células de câncer podem ter sido erradicadas do paciente, ou seja, uma cura do câncer. Tipicamente, um câncer que responde de forma pobre ao tratamento é considerado agressivo.
[012] Se o tratamento de câncer aplicado é tratamento localizado, uma diminuição do nível do DNA genômico metilado até um nível abaixo do limite de detecção, preferivelmente indica uma cura do câncer. Será subentendido por aqueles habilitados na técnica que - dependendo do resultado final de estudos clínicos – outros níveis limiares para definição de uma “cura” de um paciente podem ser definidos. O estabelecimento desses níveis limiares pode ser obtido por métodos estatísticos convencionais no campo de estatística (médica).
[013] No entanto, se o nível do DNA genômico metilado medido após tratamento localizado está acima do nível de detecção, isso, preferivelmente, indica que o tratamento localizado foi insuficiente para obter uma cura completa. Este é tipicamente o caso se o câncer já se espalhou além da área afetada pelo tratamento localizado. Portanto, até mesmo no caso de uma diminuição do nível do DNA genômico metilado, a presença continuada de níveis detectáveis do DNA genômico metilado indica um prognóstico ruim, porque um câncer que se espalha além de seu local de origem é, tipicamente, bem mais difícil de tratar.
[014] A seleção de tratamento adicional de um paciente com câncer depende de seu prognóstico. Se o prognóstico é bom, o tratamento subsequente não precisa ser tão agressivo como em casos com um prognóstico ruim. Na medida em que o prognóstico do paciente é um parâmetro importante para a seleção de tratamento adicional de um paciente com câncer, a invenção fornece um método para determinação do tratamento médico para um indivíduo com câncer, que compreende as etapas de: medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra prétratamento indica tratamento de câncer adicional para o indivíduo. Em uma modalidade preferida, a invenção também fornece um método para determinação de qual tipo de tratamento médico é adequado para um indivíduo com câncer, que compreende as etapas de: a) medição do nível prétratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica tratamento de câncer adicional para o indivíduo. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível prétratamento de DNA metilado medido e d) determinação, com base no resultado da comparação da etapa c), de qual tipo de tratamento médico é adequado para um indivíduo com câncer, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra prétratamento indica tratamento de câncer adicional para o indivíduo.
[015] De preferência, um nível pós-tratamento do DNA genômico metilado que diminuiu abaixo do nível de detecção indica que não é necessário nenhum tratamento médico adicional. Nesses casos, um monitoramento do paciente quanto a recidivas pode ser suficiente. No entanto, se o nível pós-tratamento do DNA genômico metilado não diminui ou até mesmo aumenta, pode ser necessário tratamento médico adicional. Na medida em que um nível crescente do DNA genômico metilado indica um insucesso do tratamento, essa situação, preferivelmente, indica a necessidade de mudar para um tipo diferente de tratamento.
[016] Será subentendido por aqueles habilitados na técnica que a escolha de um tratamento adequado de um paciente com câncer não pode ser baseado exclusivamente no resultado de um único teste laboratorial. Essa decisão se baseia, preferivelmente, na avaliação médica da condição do paciente. A referida avaliação preferivelmente inclui resultados de métodos diagnósticos convencionais como, por exemplo, métodos de imagem, bem como o estado de saúde geral do paciente particular em adição aos resultados obtidos por aplicação do método da presente invenção.
[017] A invenção fornece um método para determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo que compreende: medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra prétratamento indica que o câncer é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo. Em uma modalidade preferida, a invenção fornece um método para determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo que compreende: a) medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível pós- tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado e d) determinação, com base no resultado da comparação da etapa c), se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo.
[018] Além disso, a invenção fornece um método para determinação se um tumor de um indivíduo com câncer é agressivo e/ou possui potencial metastático, que compreende as etapas de: a) medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o tumor é agressivo e/ou possui potencial metastático. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado e d) determinação, com base no resultado da comparação da etapa c), se um tumor de um indivíduo com câncer é agressivo e/ou possui potencial metastático, de forma que uma quantidade aumentada ou equivalente do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o tumor é agressivo e/ou possui potencial metastático.
[019] A invenção fornece um método para detecção de uma forma agressiva de câncer em um indivíduo, que compreende: a) medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível póstratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo, de forma que uma quantidade aumentada do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o câncer é uma forma agressiva. Em uma modalidade preferida, a invenção fornece um método para detecção de uma forma agressiva de câncer em um indivíduo, que compreende: a) medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que uma quantidade aumentada do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o câncer é uma forma agressiva. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado e d) detecção, com base no resultado da comparação da etapa c), de uma forma agressiva de câncer em um indivíduo, de forma que uma quantidade aumentada do DNA genômico metilado ou fragmento na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o câncer é uma forma agressiva.
[020] A invenção fornece um método para seleção de um indivíduo com câncer para tratamento de câncer que compreende: medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo, de forma que um aumento na quantidade de DNA genômico metilado do gene na amostra pós-tratamento comparada com a amostra pré-tratamento indica tratamento de câncer adicional. Em uma modalidade preferida, a invenção fornece um método para seleção de um indivíduo com câncer para tratamento de câncer adicional que compreende: medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que um aumento na quantidade de DNA genômico metilado do gene, ou do fragmento deste, ou uma quantidade equivalente do referido DNA, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra pré-tratamento indica a necessidade de tratamento de câncer adicional. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado e d) seleção, com base no resultado da comparação da etapa c), de um indivíduo com câncer para tratamento de câncer adicional, de forma que um aumento na quantidade de DNA genômico metilado do gene, ou do fragmento deste, ou uma quantidade equivalente do referido DNA, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra pré-tratamento, indica a necessidade de tratamento de câncer adicional.
[021] Consequentemente, a presente invenção fornece um método para determinação do sucesso de um tratamento contra câncer em um indivíduo, que compreende as etapas de: a) medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e b) medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado, de forma que (i) uma diminuição na quantidade de DNA genômico metilado do gene, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o tratamento foi bem sucedido e (ii) um aumento na quantidade de DNA genômico metilado do gene, ou do fragmento deste, ou uma quantidade equivalente do referido DNA, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra prétratamento indica que o tratamento não foi bem sucedido. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado e d) determinação, com base no resultado da comparação da etapa c), do sucesso de um tratamento contra câncer em um indivíduo, de forma que (i) uma diminuição na quantidade de DNA genômico metilado do gene, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o tratamento foi bem sucedido e (ii) um aumento na quantidade de DNA genômico metilado do gene, ou do fragmento deste, ou uma quantidade equivalente do referido DNA, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra prétratamento indica que o tratamento não foi bem sucedido.
[022] De preferência, um tratamento que foi “bem sucedido” obteve pelo menos um dos seguintes efeitos: remissão do câncer, aumento do tempo até a recorrência do câncer, aumento do tempo até a progressão do tumor, alívio dos sintomas do câncer, redução da massa tumoral e diminuição do número de tumores. Mais preferivelmente, um “tratamento bem sucedido”, é caracterizado por uma cura do câncer, ou seja, a erradicação completa de células tumorais detectáveis e não detectáveis. Um indicador preferido da cura do câncer é a sobrevida livre de recorrência do paciente por pelo menos 5 anos ou, mais preferivelmente, pelo menos 10 anos.
[023] Um tratamento que “não foi bem sucedido”, preferivelmente, falhou em obter qualquer um dos objetivos descritos acima.
[024] A invenção fornece um método para determinação da carga tumoral ou carga de câncer em um indivíduo que compreende: medição do nível pré-tratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; de forma que um aumento na quantidade de DNA genômico metilado do gene na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o indivíduo possui carga tumoral ou carga de câncer aumentada ou equivalente ou que a carga tumoral ou carga de câncer não foi diminuída pelo tratamento. Em uma modalidade preferida, a invenção fornece um método para determinação do desenvolvimento de carga tumoral ou carga de câncer em um indivíduo que compreende: a) medição do nível prétratamento de DNA genômico metilado de um gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; b) medição do nível pós-tratamento de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica obtida do indivíduo; e c) comparação do nível pós-tratamento medido com o nível pré-tratamento medido de DNA metilado, de forma que um aumento na quantidade de DNA genômico metilado do gene, ou do fragmento deste, ou uma quantidade equivalente do referido DNA, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o indivíduo possui carga tumoral ou carga de câncer aumentada ou equivalente ou que a carga tumoral ou carga de câncer não foi diminuída pelo tratamento. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível pré-tratamento medido de DNA metilado e d) determinação, com base no resultado da comparação da etapa c), do desenvolvimento de carga tumoral ou carga de câncer em um indivíduo, de forma que um aumento na quantidade de DNA genômico metilado do gene, ou do fragmento deste, ou uma quantidade equivalente do referido DNA, ou do fragmento deste, na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o indivíduo possui carga tumoral ou carga de câncer aumentada ou equivalente ou que a carga tumoral ou carga de câncer não foi diminuída pelo tratamento.
[025] A invenção fornece um método para determinação da carga tumoral ou carga de câncer em um indivíduo que compreende a comparação do nível pós-tratamento de DNA genômico metilado de um gene, ou um fragmento deste, em uma amostra biológica obtida do indivíduo com o nível prétratamento de DNA genômico metilado do gene ou fragmento, de forma que um aumento na quantidade de DNA genômico metilado do gene na amostra pós-tratamento comparada com a amostra pré-tratamento indica que o indivíduo possui carga tumoral ou carga de câncer aumentada ou equivalente ou que a carga tumoral ou carga de câncer não foi diminuída pelo tratamento. Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação do nível pós-tratamento medido e do nível prétratamento medido de DNA metilado e d) determinação, com base no resultado da comparação da etapa c), da carga tumoral ou carga de câncer em um indivíduo, de forma que um aumento na quantidade de DNA genômico metilado do gene na amostra pós-tratamento comparada com a amostra prétratamento indica que o indivíduo possui carga tumoral ou carga de câncer aumentada ou equivalente ou que a carga tumoral ou carga de câncer não foi diminuída pelo tratamento.
[026] O nível de DNA metilado dos genes da presente invenção é geralmente útil como um marcador para propriedades de um câncer como, por exemplo, agressividade ou carga tumoral. Uma comparação dos níveis de DNA metilado obtidos em pontos no tempo diferentes, portanto, indica, independentemente do tratamento em andamento, como as propriedades do câncer se desenvolvem ao longo do tempo.
[027] Por essa razão, a presente invenção fornece um método para o monitoramento de uma propriedade de um câncer selecionada do grupo que consiste em carga tumoral, carga de câncer, agressividade de um câncer e o prognóstico de um indivíduo com câncer, que compreende as etapas de: a) medição do nível de DNA genômico metilado de um gene, ou de um fragmento deste, em uma primeira amostra biológica obtida de um indivíduo que sofre de câncer; b) medição do nível de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica adicional obtida do indivíduo; e c) comparação dos níveis medidos de DNA metilado na amostra adicional e na primeira amostra, em que um nível aumentado de DNA metilado do gene, ou do fragmento deste, na amostra adicional indica que a carga tumoral, massa tumoral ou a agressividade do câncer aumentou ou que o prognóstico do paciente piorou e (ii) um nível diminuído de DNA metilado do gene, ou do fragmento deste, na amostra adicional indica que a carga tumoral, massa tumoral ou a agressividade do câncer diminuiu ou que o prognóstico do paciente melhorou.
[028] Em uma modalidade preferida do método, o método compreende as etapas c) e d) da seguinte forma: c) comparação dos níveis medidos de DNA metilado na amostra adicional e na primeira amostra e d) determinação, com base no resultado da comparação da etapa c), se a carga tumoral, massa tumoral ou a agressividade do câncer aumentou ou diminuiu ou que o prognóstico do paciente piorou ou melhorou.
[029] A primeira e a segunda amostras podem ser retiradas em qualquer tempo, desde que a segunda amostra seja retirada após a primeira amostra. De preferência, a segunda amostra é retirada pelo menos 1 mês, pelo menos 2 meses, pelo menos 3 meses, pelo menos 6 meses, pelo menos 9 meses ou pelo menos 12 meses após a primeira amostra.
[030] O método descrito acima para o monitoramento de uma propriedade do câncer é especialmente adequado para o monitoramento de um paciente cujo câncer já foi tratado antes para recorrência e/ou progressão do câncer. Dessa forma, em uma modalidade particularmente preferida da presente invenção, o paciente é um paciente com câncer cujo tratamento aparentemente curou o câncer. A determinação da metilação dos genes da presente invenção em pelo menos 2 amostras retiradas em pontos no tempo diferentes após tratamento pode ser usada para detectar uma recorrência do câncer. É um problema geral no campo de terapia do câncer que um tratamento pode ser aparentemente eficaz, ou seja, ele diminui a massa tumoral do paciente abaixo do nível que é detectável com os métodos diagnósticos disponíveis, em particular métodos de imagem. No entanto, umas poucas células de câncer podem permanecer, apesar o tratamento aparentemente bem sucedido. Essas células podem proliferar e causar uma recidiva do câncer até mesmo anos após um tratamento aparentemente bem sucedido. Portanto, um acompanhamento de pacientes tratados por algum período de tempo após tratamento é boa prática médica a fim de detectar uma recidiva o mais cedo possível. Como o método da presente invenção é tanto sensível (Septina 9, em particular, pode ser usada para detectar estágios iniciais de carcinoma do cólon), de fácil realização e não invasivo, ele é particularmente adequado para monitorar pacientes tratados com câncer durante acompanhamento.
[031] Em um aspecto dos métodos da invenção, o gene é SEPTIN9 (ID. DE SEQ. Nº: 1) ou RASSF2a (ID. DE SEQ. Nº: 16).
[032] Em outro aspecto dos métodos da invenção, o gene é SEPTIN9 (ID. DE SEQ. Nº: 1).
[033] Em outro aspecto dos métodos da invenção, o gene é RASSF2A (ID. DE SEQ. Nº: 16).
[034] Em uma modalidade preferida adicional da invenção, os métodos descritos acima se baseiam na medição do nível de DNA metilado tanto de SEPTIN9 quanto de RASSF2A.
[035] Em outro aspecto do método da invenção, o câncer é selecionado do grupo que consiste em: câncer do cólon; e câncer cólon-retal. Em uma modalidade, o estágio do câncer é Estágio I câncer cólon-retal. Em outra modalidade, o estágio do câncer é câncer cólon-retal em Estágio II. Em outra modalidade, o câncer é câncer cólon-retal em Estágio III. Em outra modalidade, o câncer é câncer cólon-retal em Estágio IV.
[036] Em outro aspecto dos métodos da invenção, o tratamento é selecionado do grupo que consiste em: cirurgia ou ressecção; imunoterapia; radiação; quimioterapia; terapia dirigida a tumores sólidos; terapia dirigida a tumores de tecido mole; e terapia dirigida a células sanguíneas.
[037] Em outro aspecto dos métodos da invenção, o tratamento é localizado na região do câncer/tumor no indivíduo. Em outro aspecto dos métodos da invenção, o tratamento não está localizado na região do câncer/tumor no indivíduo.
[038] O termo “tratamento localizado” preferivelmente se refere à ressecção cirúrgica do tumor e/ou radioterapia. O termo “tratamento não localizado” é equivalente a tratamento sistêmico e, preferivelmente, se refere à quimioterapia e/ou imunoterapia.
[039] Em outro aspecto dos métodos da invenção, a amostra biológica é selecionada do grupo que consiste em: tecido, sangue, fezes, urina e fluido de lavagem pulmonar, mama, próstata, cólon, reto, ou uma combinação desses tecidos. Em uma modalidade, a amostra é soro ou plasma. O uso de soro ou plasma é preferido.
[040] Em outro aspecto dos métodos da invenção, DNA genômico metilado ou fragmento deste é medido quantitativamente ou medido quantitativamente em parte. Em outro aspecto dos métodos da invenção, DNA genômico metilado ou fragmento é medido qualitativamente ou medido qualitativamente em parte. Em outro aspecto dos métodos da invenção, DNA genômico metilado ou fragmento é medido quantitativamente em parte e qualitativamente em parte ou semiquantitativamente.
[041] Em outro aspecto dos métodos da invenção, a medição do DNA genômico metilado ou fragmento compreende o contato do DNA genômico da amostra biológica com pelo menos um reagente, ou série de reagentes, que distingue entre dinucleotídeos CpG metilados e não metilados dentro de pelo menos uma região-alvo do DNA genômico, em que a região-alvo compreende, ou hibridiza sob condições rigorosas para, uma sequência de pelo menos 9, pelo menos 16 ou pelo menos 25 nucleotídeos contíguos de IDS. DE SEQ. Nos: 1, 2, 3 ou 16 em que os referidos nucleotídeos contíguos compreendem pelo menos uma sequência de dinucleotídeo CpG. Em uma modalidade, o contato do DNA genômico, ou do fragmento deste, em b), compreende o uso de um reagente selecionado do grupo que compreende bissulfeto, sulfeto de hidrogênio, dissulfeto, e combinações destes.
[042] Em outro aspecto, os métodos da invenção compreendem: a) extração ou algum outro modo de isolamento do DNA genômico ou fragmento deste das amostras biológicas; b) tratamento do DNA genômico extraído ou isolado ou de um fragmento deste com um ou mais reagentes para converter bases de citosina que são não metiladas na posição 5 destas em uracil ou em outra base que é diferente de forma detectável à citosina em termos de propriedades de hibridização; c) contato do DNA genômico tratado, ou do fragmento tratado, com uma enzima de amplificação e pelo menos um iniciador que compreende uma sequência contígua de pelo menos 9, pelo menos 10, pelo menos 11, pelo menos 12, pelo menos 13, pelo menos 14, pelo menos 15, pelo menos 16, pelo menos 17, pelo menos 19, pelo menos 20, pelo menos 25 ou pelo menos 50 nucleotídeos que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para a sequência tratada ou para um complemento desta, em que o DNA genômico tratado, ou o fragmento deste, é amplificado para produzir pelo menos um produto amplificado, ou não é amplificado; e d) determinação, com base na presença, ausência ou quantidade, ou em uma propriedade do referido produto amplificado, do estado ou nível de metilação de pelo menos um dinucleotídeo CpG do gene, ou uma média, ou um valor que reflete um estado ou nível médio de metilação de diversos dinucleotídeos CpG do gene. O DNA genômico tratado citado acima é preferivelmente selecionado do grupo que consiste nos IDS. DE SEQ. Nos: 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19 e 20.
[043] Em outro aspecto, os métodos da invenção compreendem: a) extração ou algum outro modo de isolamento do DNA genômico ou fragmento deste das amostras biológicas; b) digestão do DNA genômico extraído ou isolado, ou de um fragmento deste, com uma ou mais enzimas de restrição sensíveis à metilação; c) o contato do digesto da enzima de restrição de DNA de b), com uma enzima de amplificação e pelo menos dois iniciadores adequados para a amplificação de uma sequência que compreende pelo menos um dinucleotídeo CpG do gene; e d) determinação, com base na presença, ausência ou classe de um produto amplificado, do estado ou nível de metilação de pelo menos um dinucleotídeo CpG do gene.
[044] Informações adicionais sobre métodos preferidos para medição do nível de um DNA genômico metilado podem ainda ser encontradas abaixo no relatório descritivo. E uma modalidade especialmente preferida da presente invenção, o método para medição de níveis de metilação de DNA genômico é MethyLightTM, HeavyMethlTM ou PCR metilação-específica.
[045] Em outro aspecto, a invenção fornece um ácido nucléico genômico metilado de SEPTIN9 ou um fragmento que compreende pelo menos 9, pelo menos 10, pelo menos 11, pelo menos 12, pelo menos 13, pelo menos 14, pelo menos 15, pelo menos 16, pelo menos 17, pelo menos 19, pelo menos 20, pelo menos 25 ou pelo menos 50 nucleotídeos contíguos do ácido nucléico e sequências complementares a este para uso na determinação do prognóstico de um indivíduo com câncer. Outra modalidade da presente invenção fornece um ácido nucléico genômico metilado de RASSF2A ou um fragmento que compreende pelo menos 9, pelo menos 16, pelo menos 25 ou pelo menos 50 nucleotídeos contíguos do ácido nucléico e sequências complementares a este para uso na determinação do prognóstico de um indivíduo com câncer. Em uma modalidade, o indivíduo possui câncer cólon-retal.
[046] Em outro aspecto, a invenção fornece o uso de ácido nucléico genômico metilado de SEPTIN9 ou um fragmento que compreende pelo menos 9, pelo menos 16, pelo menos 25 ou pelo menos 50 nucleotídeos contíguos do ácido nucléico e sequências complementares a este para determinação do prognóstico de um indivíduo com câncer. Em uma modalidade, o indivíduo possui câncer cólon-retal.
[047] Em outro aspecto, a invenção fornece um ácido nucléico de DNA genômico de SEPTIN9 ou RASSF2A tratado com bissulfeto que compreende pelo menos 9, pelo menos 10, pelo menos 1 1, pelo menos 12, pelo menos 13, pelo menos 14, pelo menos 15, pelo menos 16, pelo menos 17, pelo menos 19, pelo menos 20, pelo menos 25 ou pelo menos 50 nucleotídeos contíguos, ou um complemento para este, para uso na determinação do prognóstico de um indivíduo com câncer. De preferência, a sequência do DNA de SEPTIN9 ou RASSF2A tratado com bissulfeto é definido pelo ID. DE SEQ. Nº: 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19 ou 20. Em uma modalidade, a sequência de base contígua compreende pelo menos uma sequência de dinucleotídeo CpG, TpG ou CpA.
[048] Em outro aspecto, a invenção fornece um kit para determinação do prognóstico de um indivíduo com câncer, determinação do tratamento médico para um indivíduo com câncer, para determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo, para detecção de uma forma agressiva de câncer em um indivíduo, para seleção de um indivíduo com câncer para tratamento de câncer, ou para determinação da carga tumoral ou carga de câncer em um indivíduo que compreende: a) diversos oligonucleotídeos ou polinucleotídeos capazes de hibridizar sob condições rigorosas ou moderadamente rigorosas para os produtos de transcrição do gene ou DNA genômico metilado; e b) meios para detectar a hibridização. Em uma modalidade, o gene ou DNA genômico metilado é SEPTIN9. Em uma modalidade, o gene ou DNA genômico metilado é RASSF2A.
[049] Em outro aspecto, a invenção fornece um kit para determinação do prognóstico de um indivíduo com câncer, determinação do tratamento médico para um indivíduo com câncer, para determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo, para detecção de uma forma agressiva de câncer em um indivíduo, para seleção de um indivíduo com câncer para tratamento de câncer, ou para determinação da carga tumoral ou carga de câncer em um indivíduo, que compreende: (a) um reagente de bissulfeto; (b) pelo menos um conjunto de oligonucleotídeos contendo dois oligonucleotídeos cujas sequências em cada caso são idênticas, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com comprimento de pelo menos 9, pelo menos 10, pelo menos 11, pelo menos 12, pelo menos 13, pelo menos 14, pelo menos 15, pelo menos 16, pelo menos 17, pelo menos 19, pelo menos 20, pelo menos 25 ou pelo menos 50 nucleotídeos de uma sequência de SEPTIN9 ou de um gene de RASSF2A.
[050] Em outros aspectos, a invenção fornece o uso dos métodos aqui descritos, dos ácidos nucléicos como aqui descritos e/ou um kit como aqui descritos para determinação do prognóstico de um indivíduo com câncer.
[051] A presente invenção fornece um método para determinação do prognóstico de um indivíduo que tem câncer em um indivíduo que compreende a determinação dos níveis de expressão de pelo menos um gene ou sequência genômica em que a sequência genômica está metilada em cânceres e não metilada um tecidos não cancerosos. A metilação do DNA genômico que codifica um gene ou, em particular, a metilação de sua região promotora diminui a expressão do referido gene. Consequentemente, a metilação do gene em questão gera uma informação diagnóstica similar à sua subexpressão. Dessa forma, o nível ou quantidade de metilação/ou expressão do gene em uma amostra biológica isolada do referido indivíduo é indicativo do prognóstico do referido indivíduo. Vários aspectos da presente invenção fornecem marcadores genéticos, de forma que análise da expressão do referido marcador permite a determinação do prognóstico de um indivíduo que tem câncer. Em uma modalidade, o referido nível de expressão é determinado por detecção da presença, ausência ou nível de mRNA transcrito pelo referido gene. Em uma modalidade adicional, o referido nível de expressão é determinado por detecção da presença, ausência ou nível de um polipeptídeo codificado pelo referido gene ou sequência deste.
[052] A presente invenção fornece um método para determinação do prognóstico de um indivíduo que tem câncer cólon-retal (CRC) ou câncer de cólon em um indivíduo que compreende a determinação dos níveis de metilação de DNA de Septina 9 (Septina 9) ou de RASSF2A em plasma isolado do referido indivíduo em que, após ressecção do tumor primário, o estado de metilação é indicativo do prognóstico do referido indivíduo. Em uma modalidade, a ressecção é curativa.
[053] Os exemplos aqui descritos mostraram que o biomarcador de Septina 9 diminui em aproximadamente 73% do CRC Estágio II investigado e somente em 20 % dos pacientes em Estágio III após ressecção do tumor primário. A presença de Septina 9 em pacientes com CRC após tratamento com intenção curativa pode ser usada como um indicador prognóstico precoce de recorrência da doença. O fato de que Septina 9 ainda é detectável após ressecção do tumor primário, indica um risco elevado da presença de células tumorais (por exemplo, micrometástase) que ainda estão no corpo do paciente e que podem ser detectadas de forma sensível por Septina 9.
[054] Em modalidades adicionais, a referida expressão é determinada por detecção da presença, ausência ou quantidade de metilação de CpG dentro do referido gene, e a partir daí dedução do prognóstico do referido indivíduo que possui câncer. O referido método compreende as seguintes etapas: i) o contato do DNA genômico isolado de uma amostra biológica obtida do indivíduo com pelo menos um reagente, ou série de reagentes, que distingue entre dinucleotídeos CpG metilados e não metilados dentro de pelo menos uma região-alvo do DNA genômico, em que a sequência de nucleotídeos da referida região-alvo compreende pelo menos uma sequência de dinucleotídeo CpG de pelo menos um gene ou sequência genômica desse grupo de genes e ii) determinação do prognóstico de um indivíduo que tem câncer. De preferência, a região-alvo compreende, ou hibridiza sob condições rigorosas para, uma sequência de pelo menos 16, pelo menos 25 ou pelo menos 50 nucleotídeos contíguos.
[055] O referido uso do gene pode ser habilitado por meio de qualquer análise da expressão do gene, por meio de análise da expressão de mRNA ou análise da expressão de proteína. Em uma modalidade, a determinação do prognóstico de um indivíduo que tem câncer é habilitada por meio de análise do estado de metilação de pelo menos um gene ou sequência genômica que está metilada em tecido canceroso, mas não metilada em tecido não canceroso, incluindo isoformas, fragmentos, elementos promotores ou reguladores, e versões antissenso destes.
[056] A invenção fornece um método para a análise de amostras biológicas quanto às características associadas à progressão de câncer, o método caracterizado pelo fato de que o ácido nucléico, ou um fragmento deste, é colocado em contato com um reagente ou série de reagentes capaz de distinguir entre dinucleotídeos CpG metilados e não metilados dentro da sequência genômica. Em uma modalidade, o gene é SEPTIN9 ou RASSF2A.
[057] De preferência, a sequência de SEPTIN9 é definida pelo ID. DE SEQ. Nº: 1, 2 ou 3. Mais preferivelmente, a sequência de SEPTIN9 é definida pelo ID. DE SEQ. Nº: 2 ou 3. A sequência de RASSF2A é, preferivelmente, definida pelo ID. DE SEQ. Nº: 16.
[058] Em uma modalidade preferida da presente invenção, o estado de metilação da região promotora de SEPTIN9 e/ou RASSF2A é determinado. Em uma modalidade mais preferida, o estado de metilação de pelo menos uma citosina composta pela sequência genômica definida pelo ID. DE SEQ. Nº: 32 e/ou 34 é determinado. Em uma modalidade ainda mais preferida da invenção, o estado de metilação de pelo menos uma citosina selecionada do grupo que consiste nas citosinas nas posições 21, 28, 30, 37 e 39 do ID. DE SEQ. Nº: 32 e posições 25, 29, 46, 52, 58, 70, 74, 79 e 89 do ID. DE SEQ. Nº: 34 é determinado. Na modalidade mais preferida, o estado de metilação de todas as posições de citosina mencionadas anteriormente no ID. DE SEQ. Nº: 32 e/ou 34 é determinado.
[059] A presente invenção fornece um método para verificação de parâmetros epigenéticos de DNA genômico associados ao desenvolvimento de câncer.
[060] A fonte da amostra de teste é um tecido ou fluido corporal como, por exemplo, tecidos e fluidos corporais selecionados do grupo que consiste em tecido, sangue, plasma, soro, urina, fluido de lavagem pulmonar, fezes, pulmão, mama, cólon, reto, intestino e combinações destes.
[061] Especificamente, a presente invenção fornece um método para determinação do prognóstico de um indivíduo que tem câncer adequado para uso em uma ferramenta prognóstica, que compreende: obtenção de uma amostra biológica que compreende ácido(s) nucléico(s) genômico(s); contato do(s) ácido(s) nucléico(s), ou um fragmento deste(s), com um reagente ou diversos reagentes suficientes para distinção entre sequências de dinucleotídeo CpG metiladas e não metiladas dentro de uma sequência-alvo do ácido nucléico em questão, em que a sequência-alvo compreende, ou hibridiza sob condições rigorosas para, uma sequência que compreende pelo menos 16, pelo menos 25 ou pelo menos 50 nucleotídeos contíguos do gene, os referidos nucleotídeos contíguos compreendendo pelo menos uma sequência de dinucleotídeo CpG; e determinação, com base pelo menos em parte na referida distinção, do estado de metilação de pelo menos uma sequência de dinucleotídeo CpG-alvo, ou uma média, ou um valor que reflete um estado de metilação médio de diversas sequências de dinucleotídeo CpG-alvo.
[062] A distinção entre sequências de dinucleotídeo CpG metiladas e não metiladas dentro da sequência-alvo compreende a conversão ou não conversão dependente do estado de metilação de pelo menos uma sequência de dinucleotídeo CpG desse tipo na sequência de dinucleotídeos convertida ou não convertida correspondente dentro de uma sequência selecionada do grupo que consiste em fitas senso e antissenso convertidas por bissulfeto dos genes e regiões contíguas destes que correspondem à sequência-alvo.
[063] Modalidades adicionais fornecem um método para a determinação do prognóstico de um indivíduo que tem câncer que compreende: obtenção de uma amostra biológica que possui DNA genômico do indivíduo; extração do DNA genômico; tratamento do DNA genômico, ou um de fragmento deste, com um ou mais reagentes para converter bases de citosina não metiladas da posição 5 em uracil ou em outra base que é diferente de forma detectável da citosina em termos de propriedades de hibridização; contato do DNA genômico tratado, ou do fragmento tratado deste, com uma enzima de amplificação e pelo menos dois iniciadores que compreendem, em cada caso, uma sequência contígua com pelo menos 9 nucleotídeos de comprimento que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para uma sequência selecionada do grupo que consiste em fitas senso e antissenso convertidas por bissulfeto, e complementos destas, em que o DNA tratado, ou o fragmento deste, é amplificado para produzir um produto amplificado, ou não é amplificado; e determinação, com base na presença, ausência ou classe, ou em uma propriedade do referido produto amplificado, do estado de metilação ou uma média, ou um valor que reflete uma média do nível de metilação de pelo menos um, mas, mais preferivelmente, diversos dinucleotídeos CpG das sequências genômicas.
[064] Os métodos aqui descritos compreendem o uso de pelo menos um método selecionado do grupo que consiste em: i) hibridização de pelo menos uma molécula de ácido nucléico que compreende uma sequência contígua com pelo menos 9, pelo menos 25 ou pelo menos 50 nucleotídeos de comprimento que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para uma sequência selecionada do grupo que consiste em fitas senso e antissenso convertidas por bissulfeto, e complementos destas; ii) hibridização de pelo menos uma molécula de ácido nucléico, ligada a uma fase sólida, que compreende uma sequência contígua com pelo menos 9 nucleotídeos pelo menos 25 ou pelo menos 50 de comprimento que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para uma sequência selecionada do grupo que consiste em fitas senso e antissenso convertidas por bissulfeto, e complementos destas; iii) hibridização de pelo menos uma molécula de ácido nucléico que compreende uma sequência contígua com pelo menos 9, pelo menos 25 ou pelo menos 50 nucleotídeos de comprimento que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para uma sequência selecionada do grupo que consiste em fitas senso e antissenso convertidas por bissulfeto, e complementos destas, e extensão de pelo menos uma dessas moléculas de ácido nucléico hibridizadas por pelo menos uma base de nucleotídeo; e iv) sequenciamento do produto amplificado.
[065] Modalidades adicionais fornecem um método para a análise (ou seja, determinação da progressão da doença e/ou prognóstico do paciente) de um câncer, que compreende: obtenção de uma amostra biológica que possui DNA genômico do indivíduo; extração do DNA genômico; contato do DNA genômico, ou de um fragmento deste, que compreende uma ou mais sequências selecionadas do grupo que consiste nas sequências genômicas ou uma sequência que hibridiza sob condições rigorosas para elas, com uma ou mais enzimas de restrição sensíveis à metilação, em que o DNA genômico é digerido produzindo, dessa forma, fragmentos de digestão, ou não é digerido; e determinação, com base na presença, ausência ou classe de, ou na propriedade de pelo menos um desses fragmentos, do estado de metilação de pelo menos uma sequência de dinucleotídeo CpG das sequências genômicas ou uma média, ou um valor que reflete um estado de metilação médio de diversas sequências de dinucleotídeo CpG destas. O DNA genômico digerido ou não digerido pode ser amplificado antes da referida determinação. Modalidades adicionais fornecem novas sequências de ácidos nucléicos genômicas e quimicamente modificadas, além de oligômeros de oligonucleotídeos e/ou PNA para análise de padrões de metilação de citosina dentro das sequências genômicas.
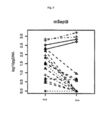
[066] As Figuras 1-4 mostram Boxplots de proporções de DNA de Septina 9 pós-/pré-cirurgia (pg de DNA de Sept9 metilado pós-cirurgia dividido por pg de DNA de Sept9 metilado pré-cirurgia) em pacientes com câncer cólon-retal separados por estágio do câncer (eixo x). As proporções só foram tabuladas para pacientes que exibem níveis de DNA de Septina 9 ˃ 0 pré-cirurgia. Os quatro números no topo da tabela são valores p de testes t unicaudados usando níveis pós- versus pré-cirurgia pareados por pacientes. Nas Figs. 1 e 3: Estágio I: linha pontilhada e círculos, Estágio II: linha tracejada e triângulo, Estágio III: linha tracejada/pontilhada e cruzes e Estágio IV: linha fechada e losangos.
[067] As Figuras 5 e 6 mostram níveis de DNA de Septina 9 (eixo y: log10 de pg de DNA de Sept9 metilado) em pacientes com câncer cólon-retal pré- e pós-cirurgia (eixo x). Os diferentes estágios do câncer foram visualizados da seguinte forma: Figura 5: Estágio I (4 pacientes): linha pontilhada e círculos; estágio II (9 pacientes): linha tracejada e triângulo; Estágio III (4 pacientes): linha tracejada/pontilhada e cruzes; Estágio IV (2 pacientes): linha fechada e losango.
[068] As Figuras 7 e 8 mostram Boxplots de proporções de DNA de RASSF2A pós-/pré-cirurgia (pg de DNA de Sept9 metilado pós-cirurgia dividido por pg de DNA de RASSF2A metilado pré-cirurgia) em pacientes com câncer cólon-retal separados por estágio do câncer (eixo x). As proporções só foram tabuladas para pacientes que exibem níveis de DNA de RASSF2A ˃ 0 pré-cirurgia. Os quatro números no topo da tabela são valores p de testes t pareados unicaudados usando níveis pós- versus pré-cirurgia pareados por pacientes. Figura 5: Estágio I (4 pacientes): linha pontilhada e círculos; estágio II (9 pacientes): linha tracejada e triângulo; Estágio III (4 pacientes): linha tracejada/pontilhada e cruzes; Estágio IV (2 pacientes): linha fechada e losango.
[069] As Figuras 9-12 mostram níveis de DNA de RASSF2A metilado (eixo y: log10 de pg DNA de RASSF2A metilado) em pacientes com câncer cólon-retal pré- e pós-cirurgia (como apresentado no eixo x). Os diferentes estágios do câncer foram visualizados da seguinte forma: Estágio I (4 pacientes): linha pontilhada e círculos; estágio II (9 pacientes): linha tracejada e triângulo; Estágio III (4 pacientes): linha tracejada/pontilhada e cruzes; Estágio IV (2 pacientes): linha fechada e losango.
[070] A Figura 13 mostra a localização do gene de SEPT9 dentro do genoma humano no cromossomo 17q25 (elencado em julho de 2005). Setas indicam a localização dos IDS. DE SEQ. Nos: 2 e 3.
[071] A Figura 14 mostra a análise quantitativa de metilação de Septina 9 em plasma pré- e pós-cirurgia de pacientes com CRC.
[072] A Figura 15 mostra a análise quantitativa de metilação de RASSF2A em plasma pré- e pós-cirurgia de pacientes com CRC.
[073] O termo “proporção observada/esperada” (“proporção O/E”) se refere à frequência de dinucleotídeos CpG dentro de uma sequência de DNA particular, e corresponde ao [número de sítios de CpG / (número de C bases x número de bases G)] /comprimento de banda para cada fragmento.
[074] O termo “ilha CpG” se refere a uma região contígua de DNA genômico que satisfaz os critérios de (1) que possui uma frequência de dinucleotídeos CpG que corresponde a uma “proporção observada/esperada” ˃0,6, e (2) que possui um “teor de GC” ˃0,5. Ilhas CpG têm tipicamente, mas nem sempre, entre cerca de 0,2 até cerca de 1 KB, ou até cerca de 2 kb de comprimento.
[075] O termo “estado de metilação” se refere à presença, ausência ou classe de 5-metilcitosina (“5-mCyt”) em um ou diversos dinucleotídeos CpG dentro de uma sequência de DNA. Estados de metilação em um ou mais sítios de metilação de CpG particulares (cada um tendo duas sequências de dinucleotídeo CpG) dentro de uma sequência de DNA incluem “não metilado”, “totalmente metilado” e “hemimetilado”.
[076] O termo “hemimetilação” se refere ao estado de metilação de um DNA de fita dupla em que apenas uma fita deste é metilada.
[077] O termo “AUC”, como aqui usado, é uma abreviação para a área sob uma curva. Em particular, ele se refere à área sob uma curva de Característica de Operação do Receptor (ROC). A curva ROC é um gráfico da taxa de positivo verdadeiro contra a taxa de falso-positivo para os diferentes pontos de corte possíveis de um teste diagnóstico. Ela mostra o trade-off entre sensibilidade e especificidade dependendo do ponto de corte selecionado (qualquer aumento na sensibilidade será acompanhado por uma diminuição na especificidade). A área sob uma curva ROC (AUC) é uma medida para a precisão de um teste (quanto maior a área melhor, o ótimo é 1, um teste aleatório teria uma curva ROC situada na diagonal com uma área de 0,5; para referência: J.P. Egan. “Signal Detection Theory and ROC Analysis”, Academic Press, Nova York, 1975).
[078] O termo “microarranjo” se refere de forma ampla tanto aos “microarranjos de DNA” quanto aos “chip(s) de DNA”, como reconhecidos na técnica, e engloba todos os suportes sólidos reconhecidos pela técnica, e engloba todos métodos para afixação de moléculas de ácido nucléico a eles ou a síntese de ácidos nucléicos sobre eles.
[079] “Parâmetros genéticos” são mutações e polimorfismos de genes e sequências ainda necessárias para sua regulação. São designadas como mutações, em particular, inserções, deleções, mutações pontuais, inversões e polimorfismos e, particularmente preferidos, SNPs (polimorfismos de nucleotídeo único).
[080] “Parâmetros epigenéticos” são, em particular, metilação de citosina. Parâmetros epigenéticos adicionais incluem, por exemplo, a acetilação de histonas que, no entanto, não podem ser diretamente analisados usando o método descrito mas que, por sua vez, se correlacionam com a metilação de DNA.
[081] O termo “reagente de bissulfeto” se refere a um reagente que compreende bissulfeto, dissulfeto, sulfeto de hidrogênio ou combinações destes, úteis como aqui revelado para distinguir entre sequências de dinucleotídeo CpG metiladas e não metiladas.
[082] O termo “ensaio de metilação” se refere a qualquer ensaio para determinação do estado de metilação de uma ou mais sequências de dinucleotídeo CpG dentro de uma sequência de DNA.
[083] O termo “MS.AP-PCR” (Reação em Cadeia de Polimerase Inicializada Arbitrariamente Sensível à Metilação) se refere à tecnologia reconhecida pela técnica que permite uma varredura global do genoma usando iniciadores ricos em CG para se concentrar nas regiões com maior probabilidade de conter dinucleotídeos CpG, e descrita por Gonzalgo e cols., Cancer Research 57: 594-599, 1997.
[084] O termo “MethyLightTM” se refere à metodologia de PCR em tempo real baseada em fluorescência reconhecida pela técnica descrita por Eads e cols., Cancer Res. 59: 2.302- 2.306, 1999.
[085] O termo ensaio “HeavyMethylTM”, na sua modalidade aqui implementada, se refere a um ensaio, em que sondas de bloqueio metilação-específicas (também aqui denominadas bloqueadores) que cobrem posições de CpG entre, ou cobertas pelos iniciadores de amplificação, permitem a amplificação seletiva metilação-específica de uma amostra de ácido nucléico.
[086] O termo ensaio “HeavyMethylTM MethyLightTM”, na sua modalidade aqui implementada, se refere a um ensaio HeavyMethylTM MethyLightTM, que é uma variação do ensaio MethyLightTM, em que o ensaio MethyLightTM é combinado com sondas de bloqueio metilação-específicas que cobrem posições de CpG entre os iniciadores de amplificação.
[087] O termo “Ms-SNuPE” (Extensão de Iniciador de Nucleotídeo Único sensível à metilação) se refere ao ensaio reconhecido pela técnica descrito por Gonzalgo e Jones, Nucleic Acids Res. 25: 2.529-2.531, 1997.
[088] O termo “MSP” (PCR metilação-específica) se refere ao ensaio de metilação reconhecido pela técnica descrito por Herman e cols. Proc. Natl. Acad. Sci. USA 93: 9.821-9.826, 1996, e pela Patente U.S. Nº 5.786.146.
[089] O termo “COBRA” (Análise Combinada de Restrição de Bissulfeto) se refere ao ensaio de metilação reconhecido pela técnica descrito por Xiong e Laird, Nucleic Acids Res. 25: 2.532-2.534, 1997.
[090] O termo “MCA” (Amplificação de Ilha CpG Metilada) se refere ao ensaio de metilação descrito por Toyota e cols., Cancer Res. 59: 2.307-12, 1999, e em WO 00/26401 A1.
[091] O termo “hibridização” deve ser subentendido como uma ligação de um oligonucleotídeo a uma sequência complementar ao longo das linhas dos pareamentos de bases de Watson-Crick no DNA da amostra, formando a duplex estrutura.
[092] “Condições rigorosas de hibridização”, como aqui definidas, envolvem a hibridização a 68°C em 5x SSC/5x solução de Denhardt/SDS 1,0% e lavagem em 0,2x SSC/SDS 0,1% em temperatura ambiente, ou envolvem o seu equivalente reconhecido pela técnica (por exemplo, condições nas quais uma hibridização é realizada a 60°C em 2,5 x tampão SSC, seguido por várias etapas de lavagem a 37°C em uma concentração baixa de tampão, e permanece estável). Condições moderadamente rigorosas, como aqui definidas, envolvem a inclusão de lavagem em 3x SSC a 42°C, ou o seu equivalente reconhecido pela técnica. Os parâmetros de concentração de sal e temperatura podem ser variados para obter o nível ótimo de identidade entre a sonda e o ácido nucléico-alvo. Diretrizes em relação a essas condições estão disponíveis na técnica, por exemplo, por Sambrook e cols., 1989, “Molecular Cloning, A Laboratory Manual”, Cold Spring Harbor Press, N.Y.; e Ausubel e cols. (eds.), 1995, “Current Protocols in Molecular Biology”, (John Wiley & Sons, N.Y.) na Unidade 2.10.
[093] Os termos “enzimas de restrição metilaçãoespecíficas” ou “enzimas de restrição sensíveis à metilação” devem ser considerados como significando uma enzima que digere seletivamente um ácido nucléico dependendo do estado de metilação de seu sítio de reconhecimento. No caso de enzimas de restrição que cortam especificamente se o sítio de reconhecimento não está metilado ou hemimetilado, o corte não ocorrerá, ou ocorrerá com uma eficiência significantemente reduzida, se o sítio de reconhecimento estiver metilado. No caso de enzimas de restrição que cortam especificamente se o sítio de reconhecimento está metilado, o corte não ocorrerá, ou ocorrerá com uma eficiência significantemente reduzida, se o sítio de reconhecimento não está metilado. São preferidas enzimas de restrição metilação-específicas, cuja sequência de reconhecimento de contém um dinucleotídeo CG (por exemplo, cgcg ou cccggg). Ainda mais preferidas para algumas modalidades são enzimas de restrição que não cortam se a citosina nesse dinucleotídeo está metilada no átomo de carbono C5.
[094] “Enzimas de restrição não-metilação-específicas” ou “enzimas de restrição não sensíveis à metilação” são enzimas de restrição que cortam uma sequência de ácidos nucléicos independentemente do estado de metilação com eficiência quase idêntica. Elas também são denominadas “enzimas de restrição metilação-inespecíficas”.
[095] Em referência às sequências de arranjo composto, a frase “nucleotídeos contíguos” se refere à uma região contígua de sequência de qualquer sequência contígua individual do arranjo composto, mas não incluem uma região da sequência de arranjo composto que inclui um “nodo”, como aqui definido acima.
[096] A descrição de um biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, como um indicador prognóstico de câncer deve ser considerado como incluindo todas as variantes de transcrito deste e todos os elementos promotores e reguladores deste. Além disso, como são conhecidos diversos SNPs dentro do biomarcador ou gene, o termo deve ser considerado como incluindo todas as variantes de sequência deste.
[097] A presente invenção fornece um método para determinação do prognóstico de um indivíduo que tem câncer, que compreende a determinação dos níveis de metilação e/ou de expressão de pelo menos um biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, em uma amostra biológica isolada do referido indivíduo em que metilação e/ou o estado de expressão é indicativo do prognóstico do referido indivíduo que possui câncer.
[098] Métodos para determinação do prognóstico e, dessa forma, os métodos e agentes para tratamento de um paciente com câncer, incluem a determinação do estadiamento do tumor com base em vários critérios. Frequentemente essa determinação inclui procedimentos invasivos para observar alterações histológicas na morfologia tecidual e nível de invasão do tumor no tecido vizinho e metástase. Vários métodos de estadiamento ou classificação de câncer são usados para avaliar a progressão ou o estado do câncer usando critérios de classificação padronizados.
[099] No câncer cólon-retal, dois desses métodos de estadiamento são o estadiamento Tumor-Linfonodo-Metástase (TNM) (Estágios I-IV) como desenvolvido pelo “American Joint Committee on Cancer” (“AJCC Cancer Staging Manual”, 6ª Edição, Springer-Verlag, Nova York, 2002), aqui incorporado por referência, e o sistema de estadiamento de Duke ou Astler-Coller modificado (Estágios A-D) (Astler V.B., Coller F.A., Ann. Surg. 1954; 139: 846-52). Ambos os métodos estão relacionados a medições da disseminação do tumor primário através de camadas da parede do cólon ou retal aos órgãos adjacentes, linfonodos e locais distantes para avaliar a progressão do tumor. Estimativas do risco de recorrência e decisões de tratamento no câncer de cólon se baseiam atualmente primariamente no estadiamento do tumor.
[100] A invenção fornece métodos e kits para determinação do prognóstico de um indivíduo com câncer, determinação do tratamento médico para um indivíduo com câncer, determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo, detecção de uma forma agressiva de câncer em um indivíduo, seleção de um indivíduo com câncer para tratamento de câncer, ou determinação da carga tumoral ou carga de câncer em um indivíduo que compreende a determinação dos níveis de metilação e/ou de expressão de pelo menos um biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, em uma amostra biológica isolada do referido indivíduo, em que o estado de metilação e/ou de expressão é indicativo do prognóstico do referido indivíduo que possui câncer. Os métodos compreendem a extração ou algum outro modo de isolamento do DNA genômico ou fragmento deste das amostras biológicas; tratamento do DNA genômico extraído ou isolado ou de um fragmento deste com um ou mais reagentes para converter bases de citosina que são não metiladas na posição 5 destas em uracil ou em outra base que é diferente de forma detectável à citosina em termos de propriedades de hibridização; contato do DNA genômico tratado, ou do fragmento tratado, com uma enzima de amplificação e pelo menos um iniciador que compreende uma sequência contígua de pelo menos 9, pelo menos 18, pelo menos 25 ou pelo menos 50 nucleotídeos que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para a sequência tratada ou para um complemento desta, em que o DNA genômico tratado, ou o fragmento deste, é amplificado para produzir pelo menos um produto amplificado, ou não é amplificado; e determinação, com base na presença, ausência ou quantidade, ou em uma propriedade do referido produto amplificado, do estado ou nível de metilação de pelo menos um dinucleotídeo CpG do gene, ou uma média, ou um valor que reflete um estado ou nível médio de metilação de diversos dinucleotídeos CpG do gene.
[101] Métodos de tratamento do DNA extraído, amplificação do DNA e detecção do DNA, e análise do DNA, são adicionalmente aqui descritos.
[102] A invenção fornece a detecção em uma amostra biológica isolada de um indivíduo com câncer de um biomarcador ou gene que está metilado no câncer, mas não metilado em tecido não canceroso, e ainda o prognóstico, determinação do resultado clínico final ou determinação de tratamento médico para o indivíduo com câncer.
[103] De preferência, o método da invenção compreende as etapas de: a) medição do nível de DNA genômico metilado de um gene, ou de um fragmento deste, em uma primeira amostra biológica obtida de um indivíduo que sofre de câncer; b) medição do nível de DNA genômico metilado do gene, ou de um fragmento deste, em uma amostra biológica adicional obtida do indivíduo; e c) comparação dos níveis medidos de DNA metilado na amostra adicional e na primeira amostra.
[104] Em uma modalidade, a detecção e análise são realizadas em uma amostra pré-tratamento e novamente em uma amostra pós-tratamento, em que o tratamento é qualquer tratamento do paciente (ou tecido do paciente) com um procedimento ou administração que diminuísse, removesse, encolhesse, minimizasse ou retirasse o tumor. Esses métodos incluem, sem limitação, ressecção cirúrgica, imunoterapia, radioterapia, quimioterapia, terapias dirigidas de tumor sólido, terapia com laser, terapias dirigidas a tecidos moles e tratamentos sanguíneos de câncer. Nessa modalidade, a “amostra pré-tratamento” corresponde à “primeira amostra” e a “amostra pós-tratamento” corresponde à “amostra posterior”.
[105] A amostra pré-tratamento pode ser retirada em qualquer momento antes que o tratamento se inicie. No entanto, ela é retirada preferivelmente no máximo 1 semana, no máximo 2 semanas, no máximo 4 semanas ou no máximo 8 semanas antes que o tratamento se inicie. A amostra póstratamento é retirada preferivelmente em qualquer momento após o início do tratamento. Caso o tratamento seja quimioterapia, é explicitamente previsto que a amostra póstratamento seja retirada antes que o período de tratamento do paciente seja completado, desde que o paciente tenha recebido pelo menos uma dosagem de pelo menos um composto farmacêutico usado para quimioterapia.
[106] O tratamento recomendado do câncer de cólon depende do estadiamento do tumor. Os Estágios I, II e III são caracterizados pela ausência de metástases distantes. Portanto, a ressecção cirúrgica do tumor é o tratamento de escolha. Para os Estágios II B, II C, III e II A de alto risco, quimioterapia adjuvante pode ser recomendada. Para tumores em estágio IV, a ressecção cirúrgica é o único tratamento recomendado se o número e localização de metástases distantes indicam a chance de uma cura completa por remoção de todos os tumores. Na doença em estágio IV, a ressecção cirúrgica é acompanhada por quimioterapia adjuvante e/ou neoadjuvante.
[107] Em uma modalidade preferida da presente invenção, o tumor é carcinoma do cólon em estágio I, II ou III. Nesse caso, um nível do DNA genômico metilado na amostra póstratamento que indica a remoção completa do tumor, preferivelmente um nível abaixo do limite de detecção, indica que o tratamento do câncer por ressecção cirúrgica foi bem sucedido. Isso é equivalente a um bom prognóstico do paciente e quimioterapia adjuvante como tratamento adicional não é, preferivelmente, recomendada.
[108] Em outra modalidade preferida da presente invenção, o tratamento é quimioterapia adjuvante ou neoadjuvante de um tumor em estágio I, II ou III ou um tumor em estágio IV operável ou quimioterapia sem cirurgia adicional como tratamento sistêmico de um tumor em estágio IV inoperável. Nesse caso, um nível diminuído do DNA genômico metilado na amostra pós-tratamento quando comparada com a amostra pré-tratamento, preferivelmente, indica que o regime de tratamento quimioterápico selecionado foi bem sucedido na redução da massa tumoral do paciente. Isso é equivalente à indicação de que o regime de tratamento quimioterápico não precisa ser adaptado. No entanto, se o nível da sequência genômica metilada na amostra pós-tratamento permanece constante ou até mesmo aumenta, isso indica, preferivelmente, que o tratamento atual não é bem sucedido e que o regime de tratamento precisa ser adaptado. Nessa modalidade, a retirada de mais de uma amostra pós-tratamento em pontos de tempo diferentes durante quimioterapia é preferida a fim de determinar constantemente se o regime de tratamento quimioterápico ainda é ou não bem sucedido.
[109] A invenção fornece um método de prognóstico de um indivíduo com câncer que engloba a detecção das células tumorais quando o tumor primário foi removido, por exemplo, pelos métodos descritos acima. Dessa forma, a invenção permite determinar se os procedimentos empregados para remover o tumor primário foram bem sucedidos e completos. Além disso, a invenção fornece métodos para determinar se o tumor se disseminou. Historicamente, métodos para determinar se o tumor se disseminou se baseiam em métodos patológicos e histológicos de determinação do envolvimento de linfonodos e metástase, por exemplo, os métodos de estadiamento de câncer descritos acima. Com a presente invenção, parâmetros como, por exemplo, carga tumoral, carga de câncer, disseminação e/ou metástase do tumor, podem ser determinados retirando-se uma primeira amostra, da qual o DNA genômico é avaliado quanto à presença de um gene ou biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, e retirando-se uma segunda amostra, da qual o DNA genômico é avaliado quanto à presença de um gene ou biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, e determinando-se se o tumor ou as células de câncer permanecem no indivíduo e, dessa forma, indicam ainda uma necessidade para tratamento clínico.
[110] Em uma modalidade preferida, a presença de níveis detectáveis do biomarcador após remoção do tumor primário indica que o tumor não foi removido completamente. Mais preferivelmente, essa situação indica que o tumor já se disseminou localmente no tecido circundante ou linfonodos ou sistemicamente em órgãos além do cólon, reto ou apêndice.
[111] Em certas modalidades, os métodos de detecção são realizados quantitativamente, quantitativamente em parte, qualitativamente, qualitativamente em parte, ou quantitativamente em parte e qualitativamente em parte.
[112] Em certas modalidades, o gene ou biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso é Septina 9. Em certas modalidades, o gene ou biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso é RASSF2A.
[113] O gene de Septina 9 humana (também conhecida como proteína de fusão MLL septina-like, proteína de fusão MLL septina-like MSF-A, Slpa, Eseptin, Msf, proteína septinalike septina ovariana/de mama (septina Ov/Br) e Septina D1) está localizado no cromossomo 17q25 dentro do contig AC068594.15.1.168501 e é um membro da família do gene de Septina. A Figura 13 fornece a anotação Ensembl do gene de Septina 9, e mostra 4 variantes de transcrito, as variantes de Septina 9 e as variantes Q9HC74 (que são versões truncadas dos transcritos de Septina 9). O ID. DE SEQ. Nº: 1 fornece a sequência do referido gene, que compreende regiões de ambos os transcritos de Septina 9 e Q9HC74 e regiões promotoras. O ID. DE SEQ. Nº: 2 e o ID. DE SEQ. Nº: 3 são subregiões destes que fornecem a sequência de regiões promotoras ricas em CpG de transcritos de Septina 9 e Q9HC74, respectivamente. Os IDS. DE SEQ. Nos: 4 e 5 são sequências para as fita senso e fita antissenso do DNA de Septina 9 tratadas quimicamente (bissulfeto), respectivamente, que correspondem à sequência do ID. DE SEQ. Nº: 1 (ou seja, em que dinucleotídeos CpG estão metilados), como mostrado na Tabela 1. Os IDS. DE SEQ. Nos: 10 e 11 são sequências para a fita senso e fita antissenso do DNA de Septina 9 tratadas quimicamente (bissulfeto), respectivamente, que correspondem à sequência do ID. DE SEQ. Nº: 1 (ou seja, em que dinucleotídeos CpG estão não metilados), como mostrado na Tabela 1. Os IDS. DE SEQ. Nos: 6 e 7 são sequências para a fita senso e fita antissenso do DNA de Septina 9 tratadas quimicamente (bissulfeto), respectivamente, que correspondem ao ID. DE SEQ. Nº: 2 (ou seja, em que dinucleotídeos CpG estão metilados), como mostrado na Tabela 1. Os IDS. DE SEQ. Nos: 12 e 13 são sequências para a fita senso e fita antissenso do DNA de Septina 9 tratadas quimicamente (bissulfeto), respectivamente, que correspondem ao ID. DE SEQ. Nº: 2 (ou seja, em que dinucleotídeos CpG estão não metilados), como mostrado na Tabela 1. Os IDS. DE SEQ. Nos: 8 e 9 são sequências para a fita senso e a fita antissenso do DNA de Q9HC74 tratadas quimicamente (bissulfeto), respectivamente, que correspondem à sequência do ID. DE SEQ. Nº: 3 (ou seja, em que dinucleotídeos CpG estão metilados), como mostrado na Tabela 1. Os IDS. DE SEQ. Nos: 14 e 15 são sequências para a fita senso e fita antissenso do DNA de Septina 9 tratadas quimicamente (bissulfeto), respectivamente, que correspondem à sequência do ID. DE SEQ. Nº: 3 (ou seja, em que dinucleotídeos CpG estão não metilados), como mostrado na Tabela 1. Septina 9 e essas variantes também foram descritas no Pedido de Patente U.S. publicado Nº US-2009- 0075260, emitido como Patente U.S. Nº 7.951.563; Pedido de Patente U.S. publicado Nº 2006-0286576, emitido como Patente U.S. No: 7.749.702; e no Pedido de Patente U.S. publicado Nº US-2011-0039719, todos aqui incorporados por referência para a descrição e informação de sequência gene SEPTIN9. Sequências adicionais relacionadas ao gene de Septina 9 são descritas nos exemplos e descrição aqui apresentados.
[114] Em certas modalidades, o gene ou biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, é RASSF2A (ID. DE SEQ. Nº: 16). O gene RASSF2 está localizado na localização cromossômica 20p13, e codifica múltiplas isoformas do transcrito de mRNA. Membros da família da proteína Ras estão associados com câncer, RASSF2 se liga à K-Ras, e a expressão de RASSF2 está associada com crescimento celular controlado. A perda da expressão resulta em proliferação celular não inibida e, consequentemente, RASSF2 é um gene supressor de tumor (Vos e cols. J. Biol. Chem., Vol. 278, Número 30, 28.045-28.051, 25 de julho de 2003). O gene RASSF2 compreende uma região densa em CpG no promotor do gene, que transpõe os primeiros 2 éxons não codificadores. Essa região foi caracterizada como sendo co-metilada e, além disso, sua metilação foi associada ao desenvolvimento de carcinomas gástricos e do cólon. Hesson e cols. (Oncogene, Junho de 2005, 2; 24(24): 3.987-94) caracterizaram a ilha CpG como sendo co-metilada, por meio de análise COBRA e sequenciamento de bissulfeto de linhagens de células de câncer de cólon. Além disso, eles confirmaram por análise MSP que 21/30 (70%) de linhagens de células de câncer de cólon analisadas estavam metiladas dentro da região promotora de RASSF2A. Pesquisas adicionais indicaram que a metilação de RASSF2 pode estar associada ao câncer gástrico (Endoh e cols. Br. J. Cancer. 12 de dezembro de 2005; 93 (12): 1.395-9) e câncer nasofaríngeo (Zhang e cols. Int. J. Cancer. 1º de janeiro de 2007; 120 (1): 32-8). ID. DE SEQ. Nº: 16 fornece a sequência de RASSF2A. IDS. DE SEQ. Nos: 17 e 18 são sequências para a fita senso e a fita antissenso de DNA de RASSF2A tratadas quimicamente (bissulfeto), respectivamente, que correspondem ao ID. DE SEQ. Nº: 16 (ou seja, em que dinucleotídeos CpG estão metilados), como mostrado na Tabela 1. Os IDS. DE SEQ. Nos: 19 e 20 são sequências para a fita senso e a fita antissenso do DNA de RASSF2A tratadas quimicamente (bissulfeto), respectivamente, que correspondem à sequência do ID. DE SEQ. Nº: 16 (ou seja, em que dinucleotídeos CpG estão não metilados), como mostrado na Tabela 1. A sequência genômica do gene inteiro é mostrada no ID. DE SEQ. Nº: 16, que foi descrita no Pedido de Patente U.S. publicado Nº US-2010-0092953, que é aqui incorporado por referência quanto à descrição e informação de sequência do gene de RASSF2A.
[115] Usando os métodos da invenção, a presença, determinada quantitativamente, quantitativamente em parte, qualitativamente, qualitativamente em parte ou quantitativamente em parte e qualitativamente em parte, do gene ou biomarcador na amostra pós-tratamento em um indivíduo em Estágio I ou II indica um prognóstico ruim ou a necessidade de um tratamento de câncer mais agressivo.
[116] Usando os métodos da invenção, um nível equivalente ou maior do gene ou biomarcador na amostra póstratamento em um indivíduo em Estágio III indica a necessidade de continuar o monitoramento da utilização dos métodos aqui descritos para observar se há uma tendência do nível do gene ou biomarcador para aumentar.
[117] Os métodos da invenção permitem não somente a detecção de uma alteração no nível do gene ou biomarcador na amostra pós-tratamento, mas também continuação do monitoramento de ou vigilância da resposta ao tratamento ou eficácia de não tratamento do indivíduo e podem ser usados para determinar se o câncer ou tumor está em remissão ou recorrência.
[118] A modificação com bissulfeto de DNA é uma ferramenta reconhecida pela técnica usada para avaliar o estado de metilação de CpG. O método usado mais frequentemente para análise de DNA quanto à presença de 5- metilcitosina se baseia na reação de bissulfeto com citosina de forma que, após hidrólise alcalina subsequente, a citosina é convertida em uracil que corresponde à timina em seu comportamento de pareamento de bases. Significantemente, no entanto, 5-metilcitosina permanece não modificada sob essas condições. Consequentemente, o DNA original é convertido de tal forma que metilcitosina, que originalmente não poderia ser distinguida de citosina por seu comportamento de hibridização, pode agora ser detectada como a única citosina restante usando metodologias padronizadas de biologia molecular reconhecidas pela técnica, por exemplo, por amplificação e hibridização, ou por sequenciamento. Todas essas técnicas se baseiam nas propriedades diferenciais de pareamento de bases, que podem agora ser totalmente exploradas.
[119] Uma visão geral de métodos reconhecidos pela técnica para detecção de 5-metilcitosina é fornecida por Rein, T., e cols., Nucleic Acids Res., 26: 2.255, 1998.
[120] A técnica de bissulfeto, com poucas exceções (por exemplo, Zeschnigk M., e cols., Eur. J. Hum. Genet. 5: 94- 98, 1997), só é usada atualmente em pesquisa. Em geral, fragmentos curtos específicos de um gene conhecido são amplificados subsequente a um tratamento com bissulfeto, e completamente sequenciados (Olek e Walter, Nat. Genet. 1997 17: 275-6, 1997), submetidos a uma ou mais reações de extensão de iniciador (Gonzalgo e Jones, Nucleic Acids Res., 25: 2.529-31, 1997; WO 95/00669; Patente U.S. Nº 6.251.594) para analisar posições individuais de citosina, ou tratados por digestão enzimática (Xiong e Laird, Nucleic Acids Res., 25: 2.532-4, 1997). A detecção por hibridização também foi descrita na técnica (Olek e cols., WO 99/28498). Adicionalmente, o uso da técnica de bissulfeto para detecção de metilação com relação a genes individuais foi descrito (Grigg e Clark, Bioessays, 16: 431-6, 1994; Zeschnigk M., e cols., Hum. Mol. Genet., 6: 387-95, 1997; Feil R., e cols., Nucleic Acids Res., 22: 695, 1994; Martin V., e cols., Gene, 157: 261-4, 1995; WO 9746705 e WO 9515373).
[121] A presente invenção fornece o uso da técnica de bissulfeto, em combinação com um ou mais ensaios de metilação, para determinação do estado de metilação de sequências de dinucleotídeo CpG dentro das sequências genômicas. Dinucleotídeos CpG genômicos podem ser metilados ou não metilados (alternativamente conhecidos como supra- e inframetilados, respectivamente). No entanto, os métodos da presente invenção são adequados para a análise de amostras biológicas de uma natureza heterogênea, por exemplo, uma concentração baixa de células tumorais dentro de um fundo de sangue ou ejaculado. Consequentemente, quando se analisa o estado de metilação de uma posição de CpG dentro de uma amostra desse tipo, aqueles habilitados na técnica podem usar um ensaio quantitativo para determinação do nível (por exemplo, percentual, fração, proporção, proporção ou grau) de metilação em uma posição de CpG particular, oposto a um estado de metilação. Consequentemente, o termo “estado de metilação” deve ser considerado como significando um valor que reflete o grau de metilação em uma posição de CpG. A menos que especificamente estabelecido, os termos “hipermetilado” ou “suprametilado” devem ser considerados como significando um nível de metilação acima daquele de um ponto de corte especificado, em que o referido valor de corte pode ser um valor que representa o nível médio ou mediano de metilação para certa população, ou é preferivelmente um nível de corte otimizado. O “valor de corte” também é aqui denominado um “limiar”. No contexto da presente invenção os termos “metilados”, “hipermetilados” ou “suprametilados” devem ser considerados como incluindo um nível de metilação acima do valor de corte de metilação zero (0)% (ou equivalentes deste) para todas as posições de CpG dentro e associadas com (por exemplo, em regiões promotoras ou reguladoras) pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso.
[122] De acordo com a presente invenção, a determinação do estado de metilação de sequências de dinucleotídeo CpG dentro das sequências genômicas possui utilidade na determinação do prognóstico de um indivíduo que tem câncer.
[123] Procedimentos do ensaio de metilação. Vários procedimentos de ensaio de metilação são conhecidos na técnica, e podem ser usados em conjunto com a presente invenção. Esses ensaios permitem a determinação do estado de metilação de um ou diversos dinucleotídeos CpG (por exemplo, ilhas CpG) dentro de uma sequência de DNA. Esses ensaios envolvem, entre outras técnicas, sequenciamento de DNA de DNA tratado com bissulfeto, PCR (para amplificação sequência-específica), análise Southern blot e uso de enzimas de restrição sensíveis à metilação.
[124] Por exemplo, o sequenciamento genômico foi simplificado para análise de padrões de metilação de DNA e distribuição de 5-metilcitosina por utilização de tratamento com bissulfeto (Frommer e cols., Proc. Natl. Acad. Sci. USA 89: 1.827-1.831, 1992). Adicionalmente, é usada a digestão com enzima de restrição de produtos de PCR amplificados a partir de DNA convertido por bissulfeto, por exemplo, o método descrito por Sadri e Hornsby (Nucl. Acids Res. 24: 5.058-5.059, 1996) ou COBRA (Análise Combinada de Restrição de Bissulfeto) (Xiong e Laird, Nucleic Acids Res. 25: 2.532-2.534, 1997).
[125] COBRA. A análise COBRATM é um ensaio quantitativo de metilação útil para determinação dos níveis de metilação de DNA em loci gênicos específicos em pequenas quantidades de DNA genômico (Xiong e Laird, Nucleic Acids Res. 25: 2.532-2.534, 1997). Resumidamente, a digestão com enzima de restrição é usada para revelar diferenças de sequência metilação-dependentes em produtos de PCR de DNA tratado com bissulfeto de sódio. Diferenças de sequência metilaçãodependentes são primeiro introduzidas no DNA genômico por tratamento com bissulfeto padronizado de acordo com o procedimento descrito por Frommer e cols. (Proc. Natl. Acad. Sci. USA 89: 1.827-1.831, 1992). A amplificação por PCR do DNA convertido por bissulfeto é então realizada usando iniciadores específicos para as ilhas CpG de interesse, seguida por digestão com endonuclease de restrição, eletroforese em gel, e detecção usando sondas de hibridização marcadas específicas. Os níveis de metilação na amostra de DNA original são representados pelas quantidades relativas de produto de PCR digerido e não digerido de modo linearmente quantitativo através de um amplo espectro de níveis de metilação de DNA. Além disso, essa técnica pode ser aplicada de forma confiável ao DNA obtido de amostras de tecido microdissecadas embebidas em parafina.
[126] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em COBRATM típico) para análise COBRATM podem incluir, sem limitação: iniciadores de PCR para gene específico (ou sequência de DNA ou ilha CpG tratada com bissulfeto); enzima de restrição e tampão apropriados; oligonucleotídeo de hibridização de gene; oligonucleotídeo de hibridização de controle; kit de marcação de quinase para sonda de oligonucleotídeo; e nucleotídeos marcados. Adicionalmente, reagentes de conversão de bissulfeto podem incluir: tampão de desnaturação de DNA; tampão de sulfonação; reagentes ou kits de recuperação de DNA (por exemplo, precipitação, ultrafiltração, coluna de afinidade); tampão de dessulfonação; e componentes de recuperação de DNA.
[127] De preferência, ensaios como, por exemplo, “MethyLightTM” (uma técnica de PCR em tempo real baseada em fluorescência) (Eads e cols., Cancer Res. 59: 2.302-2.306, 1999), reações Ms-SNuPETM (Extensão de Iniciador de Nucleotídeo Único sensível à metilação) (Gonzalgo e Jones, Nucleic Acids Res. 25: 2.529-2.531, 1997), PCR metilaçãoespecífica (“MSP”; Herman e cols., Proc. Natl. Acad. Sci. USA 93: 9.821-9.826, 1996; Patente U.S. Nº 5.786.146) e amplificação de ilha CpG metilada (“MCA”; Toyota e cols., Cancer Res. 59: 2.307-12, 1999) são usados isoladamente ou em combinação com outros desses métodos.
[128] A técnica do ensaio “HeavyMethylTM” é um método quantitativo para avaliação de diferenças de metilação baseado na amplificação metilação-específica de DNA tratado com bissulfeto. Sondas de bloqueio metilação-específicas (também aqui denominadas bloqueadores) que cobrem posições de CpG entre, ou cobertas pelos iniciadores de amplificação permitem a amplificação seletiva metilação-específica de uma amostra de ácido nucléico.
[129] O termo ensaio “HeavyMethylTM MethyLightTM”, na sua modalidade aqui implementada, se refere a um ensaio HeavyMethylTM MethyLightTM, que é uma variação do ensaio MethyLightTM, em que o ensaio MethyLightTM é combinado com sondas de bloqueio metilação-específicas que cobrem posições de CpG entre os iniciadores de amplificação. O ensaio HeavyMethylTM também pode ser usado em combinação com iniciadores de amplificação metilação-específicos.
[130] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em MethyLightTM típico) para análise HeavyMethylTM podem incluir, sem limitação: iniciadores de PCR para genes específicos (ou sequência de DNA ou ilha CpG tratada com bissulfeto); oligonucleotídeos de bloqueio; tampões de PCR otimizados e desoxinucleotídeos; e Taq polimerase.
[131] MSP. MSP (PCR metilação-específica) permite a avaliação do estado de metilação de praticamente qualquer grupo de sítios de CpG dentro de uma ilha CpG, independente do uso de enzimas de restrição sensíveis à metilação (Herman e cols. Proc. Natl. Acad. Sci. USA 93: 9.821-9.826, 1996; Patente U.S. Nº 5,786,146). Resumidamente, DNA é modificado por conversão com bissulfeto de sódio de todas as citosinas não metiladas em uracil, e subsequentemente amplificação com iniciadores específicos para DNA metilado versus não metilado. MSP necessita somente de pequenas quantidades de DNA, é sensível a alelos 0,1% metilados de certo lócus de ilha CpG, e pode ser realizada em DNA extraído de amostras embebidas em parafina. Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em MSP típico) para análise MSP podem incluir, sem limitação: iniciadores de PCR metilados e não metilados para um gene específico (ou sequência de DNA ou ilha CpG tratada com bissulfeto), tampões de PCR otimizados e desoxinucleotídeos, e sondas específicas.
[132] MethyLightTM. O ensaio MethyLightTM é um ensaio quantitativo de metilação de alto rendimento que utiliza tecnologia de PCR em tempo real baseada em fluorescência (TaqManTM) que não necessita de manipulações adicionais após a etapa de PCR (Eads e cols., Cancer Res. 59: 2.302- 2.306, 1999). Resumidamente, o processo de MethyLightTM começa com uma amostra mista de DNA genômico que é convertida, em uma reação de bissulfeto de sódio, em um pool misto de diferenças de sequência metilação-dependentes de acordo com procedimentos padronizados (o processo de bissulfeto converte resíduos de citosina não metilados em uracil). PCR baseada em fluorescência é então realizada em uma reação “distorcida” (com iniciadores de PCR que se sobrepõem a dinucleotídeos CpG conhecidos). A discriminação de sequência pode ocorre tanto no nível do processo de amplificação quanto no nível do processo de detecção de fluorescência.
[133] O ensaio MethyLightTM pode ser usado como um teste quantitativo para padrões de metilação na amostra de DNA genômico, em que a discriminação de sequência ocorre no nível de sonda hibridização. Nessa versão quantitativa, a reação de PCR permite a amplificação metilação-específica na presença de uma sonda fluorescente que se sobrepõe a um suposto sítio de metilação particular. Um controle não distorcido para a quantidade de DNA inserido é fornecido por uma reação na qual nem os iniciadores, nem a sonda se sobrepõe a quaisquer dinucleotídeos CpG. Alternativamente, um teste qualitativo para metilação genômica é obtido por sondagem do pool de PCR distorcido com oligonucleotídeos de controle que não “cobrem” sítios de metilação conhecidos (uma versão baseada em fluorescência das técnicas HeavyMethylTM e MSP), ou com oligonucleotídeos que cobrem sítios de metilação potenciais.
[134] O processo de MethyLightTM pode ser usado com quaisquer sondas adequadas, por exemplo, “TaqMan®”, Lightcycler® etc. Por exemplo, DNA genômico de fita dupla é tratado com bissulfeto de sódio e submetido a um de dois conjuntos de reações de PCR usando sondas TaqMan®;, por exemplo, com oligonucleotídeos iniciadores de MSP e/ou bloqueadores de HeavyMethyl e sonda TaqMan®. A sonda TaqMan® é marcada duplamente com moléculas fluorescentes “repórteres” e “extintoras”, e é projetada para ser específica para uma região com teor relativamente elevado de GC, de forma que ela derrete em uma temperatura em torno de 10°C maior no ciclo de PCR do que os iniciadores diretos ou reversos. Isso permite que a sonda TaqMan® permaneça totalmente hibridizada durante a etapa de anelamento/extensão de PCR. À medida que a Taq polimerase sintetiza enzimaticamente uma nova fita durante PCR, ela irá eventualmente alcançar a sonda TaqMan® anelada. A atividade de endonuclease 5’ a 3’ de Taq polimerase irá então deslocar a sonda TaqMan® por sua digestão para liberar a molécula repórter fluorescente para detecção quantitativa de seu sinal agora não extinto usando um sistema de detecção fluorescente em tempo real.
[135] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em MethyLightTM típico) para análise MethyLightTM podem incluir, sem limitação: iniciadores de PCR para um gene específico (ou sequência de DNA ou ilha CpG tratada com bissulfeto); sondas TaqMan® ou Lightcycler®; tampões de PCR otimizados e desoxinucleotídeos; e Taq polimerase.
[136] O ensaio QMTM (metilação quantitativa) é um teste quantitativo alternativo para padrões de metilação em amostras de DNA genômico, em que a discriminação de sequência ocorre no nível da sonda hibridização. Nessa versão quantitativa, a reação de PCR permite a amplificação não distorcida na presença de uma sonda fluorescente que se sobrepõe a um suposto sítio de metilação particular. Um controle não distorcido para a quantidade de DNA inserido é fornecido por uma reação na qual nem os iniciadores, nem a sonda se sobrepõe a quaisquer dinucleotídeos CpG. Alternativamente, um teste qualitativo para metilação genômica é obtido por sondagem do pool de PCR distorcido com oligonucleotídeos de controle que não “cobrem” sítios de metilação conhecidos (uma versão baseada em fluorescência das técnicas HeavyMethylTM e MSP), ou com oligonucleotídeos que cobrem sítios de metilação potenciais.
[137] O processo de QMTM pode ser usado com quaisquer sondas adequadas, por exemplo, “TaqMan®”, Lightcycler® etc. no processo de amplificação. Por exemplo, DNA genômico de fita dupla é tratado com bissulfeto de sódio e submetido aos iniciadores não distorcidos e à sonda TaqMan®. A sonda TaqMan® é marcada duplamente com moléculas fluorescentes “repórteres” e “extintoras”, e é projetada para ser específica para uma região com teor relativamente elevado de GC, de forma que ela derrete em uma temperatura em torno de 10°C maior no ciclo de PCR do que os iniciadores diretos ou reversos. Isso permite que a sonda TaqMan® permaneça totalmente hibridizada durante a etapa de anelamento/extensão de PCR. À medida que a Taq polimerase sintetiza enzimaticamente uma nova fita durante PCR, ela irá eventualmente alcançar a sonda TaqMan® anelada. A atividade de endonuclease 5’ a 3’ de Taq polimerase irá então deslocar a sonda TaqMan® por sua digestão para liberar a molécula repórter fluorescente para detecção quantitativa de seu sinal agora não extinto usando um sistema de detecção fluorescente em tempo real.
[138] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em QMTM típico) para análise QMTM podem incluir, sem limitação: iniciadores de PCR para um gene específico (ou sequência de DNA ou ilha CpG tratada com bissulfeto); sondas TaqMan® ou Lightcycler®; tampões de PCR otimizados e desoxinucleotídeos; e Taq polimerase.
[139] Ms-SNuPE. A técnica Ms-SNuPETM é um método quantitativo para avaliação de diferenças de metilação em sítios de CpG específicos com base no tratamento de DNA com bissulfeto, seguido por extensão de iniciador de nucleotídeo único (Gonzalgo e Jones, Nucleic Acids Res. 25: 2.529-2.531, 1997). Resumidamente, DNA genômico é reagido com bissulfeto de sódio para converter citosina não metilada em uracil deixando, ao mesmo tempo, 5- metilcitosina inalterada. A amplificação da sequência-alvo desejada é então realizada usando iniciadores de PCR específicos para o DNA convertido por bissulfeto, e o produto resultante é isolado e usado como um modelo para análise de metilação no sítio de CpG(s) de interesse. Pequenas quantidades de DNA podem ser analisadas (por exemplo, cortes de patologia microdissecados), e evita utilização de enzimas de restrição para determinação do estado de metilação em sítios de CpG.
[140] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em Ms-SNuPETM típico) para análise Ms-SNuPETM podem incluir, sem limitação: iniciadores de PCR para um gene específico (ou sequência de DNA ou ilha CpG tratada com bissulfeto); tampões de PCR otimizados e desoxinucleotídeos; kit de extração de gel; iniciadores de controle positivo; iniciadores de Ms-SNuPETM para um gene específico; tampão de reação (para a reação de Ms-SNuPE); e nucleotídeos marcados. Adicionalmente, reagentes de conversão de bissulfeto podem incluir: tampão de desnaturação de DNA; tampão de sulfonação; reagentes ou kit de recuperação de DNA (por exemplo, precipitação, ultrafiltração, coluna de afinidade); tampão de dessulfonação; e componentes de recuperação de DNA.
[141] Nova utilidade para a detecção de um biomarcador que está metilado no câncer, mas não metilado em tecido não canceroso, como um indicador prognóstico de câncer/tumor no sangue.
[142] Em um aspecto, o método da invenção compreende as seguintes etapas: i) determinação da metilação e/ou expressão de pelo menos um gene ou sequência genômica que está metilada em tecido canceroso, mas não metilada em tecido não canceroso; e ii) determinação do prognóstico de um indivíduo que tem câncer. Em uma modalidade, as etapas são realizadas em tecido corpóreo ou sangue. Em uma modalidade, o gene é SEPTIN9 (IDS. DE SEQ. Nos: 1-15, e outras sequências aqui descritas), cuja sequência genômica está não metilada em tecido não canceroso e metilada em tecido canceroso. Em outra modalidade, o gene é RASSF2A (IDS. DE SEQ. Nos: 16-20, e outras sequências aqui descritas), cuja sequência genômica está não metilada em tecido não canceroso e metilada em tecido canceroso.
[143] O método da invenção pode ser habilitado por meio de qualquer análise da expressão de um RNA transcrito a partir daí ou polipeptídeo ou proteína traduzida a partir do referido RNA, preferivelmente por meio de análise da expressão de mRNA ou análise da expressão de polipeptídeo. No entanto, na modalidade mais preferida da invenção, a determinação do prognóstico de um indivíduo que tem câncer é habilitada por meio de análise do estado de metilação de pelo menos um gene ou sequência genômica, e/ou elementos promotores ou reguladores da sequência genômica que estão não metilados em tecido não canceroso, e metilados em tecido canceroso. Em outras modalidades, a presente invenção também fornece ensaios e métodos prognósticos, tanto quantitativos quanto qualitativos, para detecção da expressão de um ou mais dos genes em um indivíduo e determinação, a partir daí, do prognóstico de um indivíduo que tem câncer no referido indivíduo. Em outras modalidades, a hipermetilação e/ou subexpressão de um ou mais dos genes está associada à progressão e agressividade do câncer.
[144] Em uma modalidade preferida, a presença de DNA de Septina 9 metilado em uma amostra antes da cirurgia acima de 3 pg/ml é indicativa da presença de câncer. De preferência, após cirurgia, um sinal negativo de metilação de Septina 9 é indicativo de bom prognóstico (0 pg/ml de Septina 9 metilado). De preferência, após cirurgia, a presença de Septina 9 metilado acima de 0 a 3 pg/ml de amostra indica um risco baixo para a recorrência de câncer. De preferência, após cirurgia, um nível de Septina 9 metilado de 3 a 30 pg/ml de plasma é indicativo de um risco médio para a recorrência de câncer. De preferência, após cirurgia, a presença de Septina 9 metilado acima de 30 pg/ml de amostra indica um risco elevado ou recorrência. Em uma modalidade preferida, a presença de DNA de RASSF2A metilado em uma amostra antes da cirurgia acima de 3 pg/ml é indicativa da presença de câncer. De preferência, após cirurgia, um sinal negativo de metilação de RASSF2A é indicativo de bom prognóstico (0 pg/ml de RASSF2A metilado). De preferência, após cirurgia, a presença de RASSF2A metilado acima de 0 a 3 pg/ml de amostra indica um risco baixo para a recorrência de câncer. De preferência, após cirurgia, um nível de RASSF2A metilado de 3 a 30 pg/ml de plasma é indicativo de um risco médio para a recorrência de câncer. De preferência, após cirurgia, a presença de RASSF2A metilado acima de 30 pg/ml de amostra indica um risco elevado ou recorrência. Amostras preferidas são sangue, tecido tumoral e plasma. De preferência, o câncer é câncer cólon-retal.
[145] Para detectar a presença de mRNA que codifica um gene ou sequência genômica, uma amostra é obtida do indivíduo. A amostra pode ser qualquer amostra adequada que compreenda matéria celular do tumor. Tipos de amostra adequados incluem tecido, sangue, plasma ou soro e todas as combinações possíveis destes. Prefere-se que os referidos tipos de amostra sejam sangue. A amostra pode ser tratada para extrair o RNA nela contido. O ácido nucléico resultante da amostra é então analisado. Muitas metodologias são conhecidas na técnica para determinação de níveis absolutos e relativos de expressão gênica, técnicas comumente usadas adequadas para uso na presente invenção incluem hibridização in situ (por exemplo, FISH), análise Northern, ensaios de proteção de RNase (RPA), microarranjos e técnicas à base de PCR, por exemplo, PCR quantitativa e PCR de exibição diferencial ou qualquer outro método de detecção de ácido nucléico. A técnica de transcrição reversa/de reação de cadeia de polimerização (RT-PCR) pode ser usada. O método de RT-PCR é bem conhecido na técnica (por exemplo, veja Watson e Fleming, supra).
[146] O método de RT-PCR pode ser realizado da seguinte forma. RNA celular total é isolado, por exemplo, pelo método-padrão de isotiocianato de guanidínio e o RNA total é transcrito de forma reversa. O método de transcrição reversa envolve síntese de DNA em um modelo de RNA usando uma enzima transcriptase reversa e um iniciador de oligonucleotídeo dT da extremidade 3’ e/ou iniciadores de hexâmero aleatórios. O cDNA assim produzido é então amplificado por meio de PCR (Belyavsky e cols., Nucl. Acid Res. 17: 2.919-2.932, 1989; Krug e Berger, “Methods in Enzymology”, Academic Press, N.Y., Vol. 152, páginas 316- 325, 1987, que são incorporados por referência). É ainda preferida a variante “em tempo real” de RT-PCR, em que o produto de PCR é detectado por meio de sondas de hibridização (por exemplo, TaqMan, LightCycler, Molecular Beacons & Scorpion) ou verde SYBR. O sinal detectado das sondas ou verde SYBR é então quantificado por referência a uma curva-padrão ou por comparação dos valores de Ct com aquele de um padrão de calibração. A análise de genes de manutenção é frequentemente usada para normalizar os resultados.
[147] Na análise Northern blot, mRNA total ou poli(A)+ é processado em um gel de agarose desnaturante e detectado por hibridização para uma sonda marcada no próprio gel seco ou em uma membrana. O sinal resultante é proporcional à quantidade de RNA-alvo na população de RNA.
[148] A comparação dos sinais de duas ou mais populações de células ou tecidos revela diferenças relativas nos níveis de expressão gênica. A quantificação absoluta pode ser realizada por comparação do sinal com uma curva-padrão gerada usando quantidades conhecidas de um transcrito in vitro que corresponde ao RNA-alvo. A análise de genes de manutenção, genes cujos níveis de expressão supostamente permanecem relativamente constantes independentemente das condições, é frequentemente usada para normalizar os resultados, eliminando quaisquer diferenças aparentes causadas por transferência desigual de RNA para a membrana ou carregamento desigual de RNA no gel.
[149] A primeira etapa na análise Northern é o isolamento de RNA intacto, puro, das células ou tecido de interesse. Como Northern blots distinguem RNAs por tamanho, a integridade da amostra influencia o grau até o qual um sinal é localizado em uma banda única. Amostras de RNA parcialmente degradado farão com que o sinal seja borrado ou distribuído sobre várias bandas com uma perda global na sensibilidade e possivelmente uma interpretação errônea dos dados. Na análise Northern blot, sondas de DNA, RNA e oligonucleotídeos podem ser usadas, e essas sondas são preferivelmente marcadas (por exemplo, marcadores radioativos, marcadores de massa ou marcadores fluorescentes). O tamanho do RNA-alvo, não da sonda, determinará o tamanho da banda detectada e, portanto, métodos como, por exemplo, marcação random-primed, que gera sondas de comprimentos variáveis, são adequados para síntese da sonda. A atividade específica da sonda determinará o nível de sensibilidade e, portanto, preferese que sondas com atividades específicas elevadas sejam usadas.
[150] Em um ensaio de proteção de RNase, o RNA-alvo e uma sonda de RNA de um comprimento definido são hibridizados em solução. Após hibridização, o RNA é digerido com RNases específicas para ácidos nucléicos de fita simples para remover qualquer RNA-alvo de fita simples não hibridizado e a sonda. As RNases são inativadas e o RNA é separado, por exemplo, por eletroforese em gel de poliacrilamida desnaturante. A quantidade de sonda de RNA intacta é proporcional à quantidade de RNA-alvo na população de RNA. RPA pode ser usada para quantificação relativa e absoluta da expressão gênica e também para mapeamento da estrutura do RNA, por exemplo, fronteiras íntron/éxon e sítio de início de transcrição. O ensaio de proteção de RNase é preferível à análise Northern blot, na medida em que ele geralmente possui um limite de detecção mais baixo.
[151] As sondas de RNA antissenso usadas em RPA são geradas por transcrição in vitro de um modelo de DNA com um ponto final definido, e estão tipicamente na faixa de 50- 600 nucleotídeos. O uso de sondas de RNA que incluem sequências adicionais não homólogas ao RNA-alvo permite que o fragmento protegido seja distinguido da sonda de comprimento total. Sondas de RNA são tipicamente usadas ao invés de sondas de DNA em função da facilidade de geração de sondas de RNA de fita simples e da reprodutibilidade e confiabilidade da digestão do duplexo RNA:RNA com RNases (Ausubel e cols. 2003), são particularmente preferidas sondas com atividades específicas elevadas.
[152] É particularmente preferido o uso de microarranjos. O processo de análise de microarranjo pode ser dividido em duas partes principais. A primeira é a imobilização de sequências gênicas conhecidas sobre lâminas de vidro ou outro suporte sólido, seguida por hibridização do cDNA marcado de forma fluorescente (que compreende as sequências a serem interrogadas) para os genes conhecidos imobilizados na lâmina de vidro (ou outra fase sólida). Após hibridização, os arranjos são escaneados usando um scanner de microarranjo fluorescente. A análise da intensidade fluorescente relativa de genes diferentes fornece uma medição das diferenças na expressão gênica.
[153] Os arranjos de DNA podem ser gerados por imobilização de oligonucleotídeos pré-sintetizados sobre lâminas de vidro preparadas ou outras superfícies sólidas. Nesse caso, sequências gênicas representativas são fabricadas e preparadas usando síntese de oligonucleotídeos padronizados e métodos de purificação. Essas sequências gênicas sintetizadas são complementares ao(s) transcrito(s) de RNA de pelo menos um gene que está metilado no câncer, mas não metilado em tecido não canceroso, e tendem a ser sequências mais curtas na faixa de 25-70 nucleotídeos. Alternativamente, óligos imobilizados podem ser quimicamente sintetizados in situ na superfície da lâmina. A síntese de oligonucleotídeos in situ envolve a adição consecutiva dos nucleotídeos apropriados aos spots no microarranjo; spots que não recebem um nucleotídeo são protegidos durante cada estágio do processo usando máscaras físicas ou virtuais. De preferência, os referidos ácidos nucléicos sintetizados são ácidos nucléicos travados.
[154] Em experimentos de microarranjo de definição de perfil de expressão, os modelos de RNA usados são representativos do perfil de transcrição das células ou tecidos sob estudo. Primeiro o RNA é isolado das populações de células ou tecidos a serem comparados. Cada amostra de RNA é então usada como um modelo para gerar cDNA marcado de forma fluorescente por meio de uma reação de transcrição reversa. A marcação fluorescente do cDNA pode ser obtida por métodos de marcação direta ou de marcação indireta. Durante a marcação direta, os nucleotídeos modificados de forma fluorescente (por exemplo, Cy®3- ou Cy®5-dCTP) são incorporados diretamente no cDNA durante a transcrição reversa. Alternativamente, a marcação indireta pode ser obtida por incorporação de nucleotídeos modificados com aminoalil durante a síntese de cDNA, e depois conjugando-se um corante de N-hidroxisuccinimida (NHS)-éster ao cDNA modificado com aminoalil após o término da reação de transcrição reversa. Alternativamente, a sonda pode ser não marcada, mas pode ser detectável por ligação específica com um ligante que é marcado, direta ou indiretamente. Marcadores e métodos adequados para marcação de ligantes (e sondas) são conhecidos na técnica, e incluem, por exemplo, marcadores radioativos que podem ser incorporados por métodos conhecidos (por exemplo, tradução nick ou kinasing). Outros marcadores adequados incluem, sem limitação, biotina, grupos fluorescentes, grupos quimioluminescentes (por exemplo, dioxetanos, particularmente dioxetanos desencadeados), enzimas, anticorpos, e semelhantes.
[155] Para realizar análise diferencial da expressão gênica, cDNA gerado por amostras de RNA diferentes é marcado com Cy®3. O cDNA marcado resultante é purificado para remover nucleotídeos não incorporados, corante livre e RNA residual. Após purificação, as amostras de cDNA marcado são hibridizadas para o microarranjo. O rigor de hibridização é determinado por diversos fatores durante hibridização e durante o procedimento de lavagem, incluindo temperatura, força iônica, duração de tempo e concentração de formamida. Esses fatores são descritos em, por exemplo, Sambrook e cols. (“Molecular Cloning: A Laboratory Manual”, 2ª Edição, 1989). O microarranjo é escaneado póshibridização usando um scanner de microarranjo fluorescente. A intensidade fluorescente de cada spot indica o nível de expressão do gene analisado; spots brilhantes correspondem aos genes fortemente expressos, enquanto spots turvos indicam expressão fraca.
[156] Após obtenção das imagens, os dados brutos devem ser analisados. Primeiro, a fluorescência de fundo deve ser subtraída da fluorescência de cada spot. Os dados são então normalizados para uma sequência de controle, por exemplo, ácidos nucléicos adicionados de forma exógena (preferivelmente RNA ou DNA), ou um painel de genes de manutenção para considerar qualquer hibridização não específica, imperfeições do arranjo ou variabilidade na montagem do arranjo, marcação de cDNA, hibridização ou lavagem. A normalização dos dados permite que sejam comparados os resultados de múltiplos arranjos.
[157] Outro aspecto da invenção está relacionado a um kit para uso na determinação do prognóstico de um indivíduo que tem câncer de acordo com os métodos da presente invenção, o referido kit compreendendo: um meio para medição do nível de transcrição de pelo menos um gene ou um gene da sequência genômica que está metilado no câncer, mas não metilado em tecido não canceroso. Em uma modalidade preferida, os meios para medição do nível de transcrição compreendem oligonucleotídeos ou polinucleotídeos capazes de hibridizar sob condições rigorosas ou moderadamente rigorosas para os produtos de transcrição de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso. Na modalidade mais preferida, o nível de transcrição é determinado por técnicas selecionadas do grupo de análise Northern Blot, PCR com transcriptase reversa, PCR em tempo real, proteção de RNAse e microarranjo. Em outra modalidade da invenção, o kit ainda compreende meios para obtenção e/ou armazenamento de uma amostra biológica do indivíduo. É preferido um kit que ainda compreenda um recipiente que é mais preferivelmente adequado para contenção dos meios para medição do nível de transcrição e da amostra biológica do indivíduo, e, mais preferivelmente ainda, ainda compreende instruções para uso e interpretação dos resultados do kit.
[158] Em uma modalidade preferida, o kit compreende: (a) diversos oligonucleotídeos ou polinucleotídeos capazes de hibridizar sob condições rigorosas ou moderadamente rigorosas para os produtos de transcrição de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso; (b) um recipiente, preferivelmente adequado para contenção dos oligonucleotídeos ou polinucleotídeos e de uma amostra biológica do indivíduo que compreende os produtos de transcrição, em que os oligonucleotídeos ou polinucleotídeos podem hibridizar sob condições rigorosas ou moderadamente rigorosas para os produtos de transcrição, (c) meios para detectar a hibridização de (b); e, opcionalmente, (d) instruções para uso e interpretação dos resultados do kit.
[159] O kit também pode conter outros componentes como, por exemplo, tampão de hibridização (em que os oligonucleotídeos devem ser usados como uma sonda) embalado em um recipiente separado. Alternativamente, quando os oligonucleotídeos devem ser usados para amplificar uma região-alvo, o kit pode conter, embalados em recipientes separados, uma polimerase e um tampão de reação otimizados para extensão de iniciador mediada pela polimerase, por exemplo, PCR. De preferência, a referida polimerase é uma transcriptase reversa. É ainda preferido que o referido kit ainda contenha um reagente de RNAse.
[160] A presente invenção ainda fornece métodos para a detecção da presença do polipeptídeo codificado pelas referidas sequências gênicas em uma amostra obtida do referido indivíduo.
[161] Níveis aberrantes de expressão de polipeptídeo dos polipeptídeos codificados por pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, estão associados com o prognóstico de um indivíduo que tem câncer.
[162] De acordo com a presente invenção, a subexpressão dos referidos polipeptídeos está associada com um prognóstico negativo de um indivíduo que tem câncer.
[163] Qualquer método conhecido na técnica para detecção de polipeptídeos pode ser usado. Esses métodos incluem, sem limitação, espectrometria de massa, imunodifusão, imunoeletroforese, métodos imunoquímicos, ensaios de aglutinante-ligante, técnicas imunoistoquímicas, ensaios de aglutinação e complemento (por exemplo, veja “Basic and Clinical Immunology”, Sites e Terr, eds., Appleton & Lange, Norwalk, Conn, páginas 217-262, 1991 que é incorporado por referência). São preferidos métodos de imunoensaio de aglutinante-ligante, incluindo a reação de anticorpos com um epitopo ou epitopos e o deslocamento competitivo de um polipeptídeo marcado ou derivado deste.
[164] Certas modalidades da presente invenção compreendem o uso de anticorpos específicos para o(s) polipeptídeo(s) codificado(s) por pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso.
[165] Esses anticorpos são úteis para determinação do prognóstico de um indivíduo que tem câncer. Em certas modalidades, a produção de anticorpos monoclonais ou policlonais pode ser induzida pelo uso de um epitopo codificado por um polipeptídeo de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, como um antigene. Esses anticorpos podem, por sua vez, ser usados para detectar polipeptídeos expressos. Os níveis desses polipeptídeos presentes podem ser quantificados por métodos convencionais. A ligação anticorpo-polipeptídeo pode ser detectada e quantificada por diversos meios conhecidos na técnica, por exemplo, marcação com ligantes fluorescentes ou radioativos. A invenção ainda compreende kits para realização dos procedimentos mencionados acima, em que esses kits contêm anticorpos específicos para os polipeptídeos investigados.
[166] Diversos imunoensaios de ligação competitiva e não competitiva de polipeptídeo são bem conhecidos na técnica. Os anticorpos empregados nesses ensaios podem ser não marcados, por exemplo, como usado em testes de aglutinação, ou marcados para uso em uma ampla variedade de métodos de ensaio. Marcadores que podem ser usados incluem radionuclídeos, enzimas, substâncias fluorescentes, substâncias quimioluminescentes, substratos ou co- fatores de enzima, inibidores de enzima, partículas, corantes e semelhantes. Ensaios preferidos incluem sem limitação, radioimunoensaio (RIA), imunoensaios de enzimas, por exemplo, ensaio imunoabsorvente ligado à enzima (ELISA), imunoensaios fluorescentes e semelhantes. Anticorpos policlonais ou monoclonais ou seus epitopos podem ser feitos para uso em imunoensaios por qualquer um de diversos métodos conhecidos na técnica.
[167] Em uma modalidade alternativa do método, as proteínas podem ser detectadas por meio de análise Western blot. A referida análise é padrão na técnica; resumidamente, proteínas são separadas por meio de eletroforese, por exemplo, SDS-PAGE. As proteínas separadas são então transferidas para uma membrana adequada (ou papel), por exemplo, nitrocelulose, retendo a separação espacial obtida por eletroforese. A membrana é então incubada com um agente de bloqueio para se ligar aos lugares pegajosos restantes na membrana, agentes comumente usados incluem proteínas genéricas (por exemplo, proteína do leite). Um anticorpo específico para a proteína de interesse é então adicionado, o referido anticorpo sendo marcado de forma detectável, por exemplo, por corantes ou meios enzimáticos (por exemplo, fosfatase alcalina ou peroxidase de raiz-forte). A localização do anticorpo na membrana é então detectada.
[168] Em uma modalidade alternativa do método, as proteínas podem ser detectadas por meio de imunoistoquímica (o uso de anticorpos para antígenos sonda-específicos em uma amostra). A referida análise é padrão na técnica, em que a detecção de antígenos em tecidos é conhecida como imunoistoquímica, enquanto a detecção em células cultivadas é geralmente denominada imunocitoquímica. Resumidamente, o anticorpo primário deve ser detectado por ligação ao seu antígeno específico. O complexo antígeno-anticorpo é então ligado por um anticorpo secundário ligado à enzima. Na presença do substrato e cromógeno necessários, a enzima ligada é detectada de acordo com depósitos coloridos nos sítios de ligação de anticorpo-antígeno. Há uma ampla gama de tipos de amostra, afinidade antígeno-anticorpo, tipos de anticorpo, e métodos de aumento da detecção adequados. Dessa forma, as condições ótimas para detecção imunoistoquímica ou imunocitoquímica devem ser determinadas por aqueles habilitados na técnica para cada caso individual.
[169] Uma abordagem para a preparação de anticorpos para um polipeptídeo é a seleção e preparação de uma sequência de aminoácidos de todo ou parte do polipeptídeo, síntese química da sequência de aminoácidos e sua injeção em um animal apropriado, normalmente um coelho ou um camundongo (Milstein e Kohler, Nature 256: 495-497, 1975; Gulfre e Milstein, “Methods in Enzymology: Immunochemical Techniques” 73: 1-46, Langone e Banatis eds., Academic Press, 1981, que são incorporados por referência em sua totalidade). Métodos para preparação do polipeptídeos ou seus epitopos incluem, sem limitação, síntese química, técnicas de DNA recombinante ou isolamento de amostras biológicas.
[170] Na etapa final do método, o prognóstico do indivíduo é determinado, de forma que a subexpressão (de mRNA ou polipeptídeos) é indicativa do prognóstico de um indivíduo que tem câncer. O termo “subexpressão” deve ser considerado como significando a expressão em um nível detectado menor do que um valor de corte predeterminado que pode ser selecionado do grupo que consiste no valor médio, mediano ou um valor limiar otimizado. O termo “superexpressão” deve ser considerado como significando expressão em um nível detectado maior do que um valor de corte predeterminado que pode ser selecionado do grupo que consiste no valor médio, mediano ou um valor limiar otimizado.
[171] Outro aspecto da invenção fornece um kit para uso na determinação do prognóstico de um indivíduo que tem câncer de acordo com os métodos da presente invenção, que compreende: um meio para detecção de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em polipeptídeos de tecido não canceroso. Os meios para detecção dos polipeptídeos compreendem preferivelmente anticorpos, derivados de anticorpo ou fragmentos de anticorpo. Os polipeptídeos são detectados mais preferivelmente por meio de Western Blotting utilizando um anticorpo marcado. Em outra modalidade da invenção, o kit ainda compreende meios para obtenção de uma amostra biológica do indivíduo. É preferido um kit que ainda compreenda um recipiente adequado à contenção dos meios para detecção dos polipeptídeos na amostra biológica do indivíduo e, mais preferivelmente ainda, ainda compreenda instruções para uso e interpretação dos resultados do kit. Em uma modalidade preferida o kit compreende: (a) um meio para detecção de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em polipeptídeos de tecido não canceroso; (b) um recipiente adequado à contenção dos referidos meios e da amostra biológica do indivíduo que compreende os polipeptídeos, em que os meios podem formar complexos com os polipeptídeos; (c) um meio para detectar os complexos de (b); e, opcionalmente, (d) instruções para uso e interpretação dos resultados do kit.
[172] O kit também pode conter outros componentes como, por exemplo, tampões ou soluções adequadas ao bloqueio, lavagem ou revestimento, embalados em um recipiente separado.
[173] Modalidades particulares da presente invenção fornecem uma nova aplicação da análise de níveis e/ou padrões de metilação dentro de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, que permite a determinação do prognóstico de um indivíduo que tem câncer.
[174] Em uma modalidade do método, o prognóstico de um indivíduo que tem câncer é determinado por análise do estado de metilação de um ou mais dinucleotídeos CpG de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso.
[175] Em uma modalidade da invenção, o referido método compreende as seguintes etapas: i) o contato do DNA genômico (preferivelmente isolado de tecido, sangue, plasma ou soro) obtido do indivíduo com pelo menos um reagente, ou série de reagentes, que distingue entre dinucleotídeos CpG metilados e não metilados dentro de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso (incluindo regiões promotoras e reguladoras desta) e ii) determinação do prognóstico do referido indivíduo que possui câncer.
[176] Prefere-se que os referidos (um ou mais) dinucleotídeos CpG de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, estejam compreendidos dentro de uma respectiva sequência genômica-alvo deste, como fornecido nas sequências genômicas e complementos destas. A presente invenção ainda fornece um método para verificação de parâmetros genéticos e/ou epigenéticos de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, e/ou da sequência genômica de acordo com as sequências genômicas dentro de um indivíduo por análise da metilação de citosina. O referido método compreendendo o contato de um ácido nucléico que compreende as sequências genômicas em uma amostra biológica obtida do referido indivíduo com pelo menos um reagente ou uma série de reagentes, em que o referido reagente, ou série de reagentes, distingue entre dinucleotídeos CpG metilados e não metilados dentro do ácido nucléico-alvo.
[177] Em uma modalidade preferida, o referido método compreende as seguintes etapas: na primeira etapa, uma amostra do tecido a ser analisada é obtida. A fonte pode ser qualquer fonte adequada, por exemplo, tecido, sangue, plasma ou soro, e todas as combinações possíveis destes. Prefere-se que as referidas fontes de DNA sejam tecido, sangue, plasma ou soro.
[178] O DNA genômico é então isolado da amostra. O DNA genômico pode ser isolado por qualquer meio padronizado na técnica, incluindo o uso de kits comercialmente disponíveis. Resumidamente, quando o DNA de interesse está encapsulado por uma membrana celular, a amostra biológica deve ser rompida e lisada por meios enzimáticos, químicos ou mecânicos. A solução de DNA pode ser então depurada de proteínas e outros contaminantes, por exemplo, por digestão com proteinase K. O DNA genômico é então recuperado da solução. Isso pode ser realizado por meio de diversos métodos, incluindo remoção de sal, extração orgânica ou ligação do DNA a um suporte de fase sólida. A escolha do método será afetada por vários fatores, incluindo o tempo, custos e a quantidade de DNA necessária.
[179] Quando o DNA da amostra não está contido em uma membrana (por exemplo, DNA circulante de uma amostra de sangue), podem ser usados métodos-padrão na técnica para o isolamento e/ou purificação de DNA. Esses métodos incluem o uso de um reagente de degeneração de proteína, por exemplo, sal caotrópico, por exemplo, cloridrato de guanidina ou uréia; ou um detergente, por exemplo, dodecil sulfato de sódio (SDS), brometo de cianogênio. Métodos alternativos incluem, sem limitação, precipitação com etanol ou precipitação com propanol, concentração a vácuo, entre outros, por meio de uma centrífuga. Aqueles habilitados na técnica também podem utilizar dispositivos como, por exemplo, dispositivos de filtro, por exemplo, ultrafiltração, superfícies ou membranas de sílica, partículas magnéticas, partículas de poliestirol, superfícies de poliestirol, superfícies carregadas positivamente e membranas carregadas positivamente, membranas carregadas, superfícies carregadas, membranas com mudança de carga, superfícies com mudança de carga.
[180] Após os ácidos nucléicos terem sido extraídos, o DNA genômico de fita dupla é usado na análise, análise de metilação pode ser realizada por qualquer meio conhecido na técnica incluindo, sem limitação, análise de enzima de restrição sensível à metilação e análise de reagente químico.
[181] Na segunda etapa do método, a amostra de DNA genômico é tratada de tal forma que bases de citosina que não estão metiladas na posição 5’ são convertidas em uracil, timina, ou outra base que seja dissimilar à citosina em termos de comportamento de hibridização. Isso será aqui subentendido como “pré-tratamento” ou “tratamento”.
[182] Isso é preferivelmente obtido por meio de tratamento com um reagente de bissulfeto. O termo “reagente de bissulfeto” se refere a um reagente que compreende bissulfeto, dissulfeto, sulfeto de hidrogênio ou combinações destes, útil como aqui revelado para distinguir entre sequências de dinucleotídeo CpG metiladas e não metiladas. Métodos do referido tratamento são conhecidos na técnica (por exemplo, PCT/EP2004/011715, que é incorporada por referência em sua totalidade). Prefere-se que o tratamento com bissulfeto seja realizado na presença de solventes desnaturantes como, por exemplo, sem limitação, n-alquilenoglicol, particularmente dietileno glicol dimetil éter (DME), ou na presença de dioxano ou derivados de dioxano. Em uma modalidade preferida, os solventes desnaturantes são usados em concentrações entre 1% e 35% (v/v). Também prefere-se que a reação de bissulfeto seja realizada na presença de depuradores como, por exemplo, sem limitação, derivados de cromano, por exemplo, ácido 6- hidróxi-2,5,7,8-tetrametilcromano-2-carboxílico ou ácido trihidroxibenzoato e derivados destes, por exemplo, ácido gálico (veja: PCT/EP2004/011715 que é incorporada por referência em sua totalidade). A conversão de bissulfeto é realizada preferivelmente em uma temperatura de reação entre 30°C e 70°C, de forma que a temperatura seja aumentada até acima de 85°C por períodos de tempos curtos durante a reação (veja: PCT/EP2004/011715 que é incorporada por referência em sua totalidade). O DNA tratado com bissulfeto é preferivelmente purificado antes da quantificação. Isso pode ser realizado por qualquer meio conhecido na técnica como, por exemplo, sem limitação, ultrafiltração, preferivelmente realizada por meio de colunas Microcon (TM) (fabricadas por Millipore (TM)). A purificação é realizada de acordo com um protocolo do fabricante modificado (veja: PCT/EP2004/011715 que é incorporada por referência em sua totalidade).
[183] Na terceira etapa do método, fragmentos do DNA tratado são amplificados, usando conjuntos de oligonucleotídeos iniciadores de acordo com a presente invenção, e uma enzima de amplificação. A amplificação de vários segmentos de DNA pode ser realizada simultaneamente em um e no mesmo vaso de reação. Tipicamente, a amplificação é realizada usando uma reação em cadeia de polimerase (PCR). De preferência, os referidos produtos amplificados possuem 100 a 2.000 pares de bases de comprimento. O conjunto de oligonucleotídeos iniciadores inclui pelo menos dois oligonucleotídeos cujas sequências são, cada uma, complementares reversas, idênticas ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 16 pares de bases de comprimento das sequências de base de uma das sequências de bissulfeto e sequências a elas complementares.
[184] Em uma modalidade alternativa do método, o estado de metilação de posições de CpG pré-selecionadas dentro de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, e preferivelmente dentro das sequências de ácidos nucléicos de acordo com as sequências genômicas, pode ser detectado por uso de oligonucleotídeos iniciadores metilaçãoespecíficos. Essa técnica (MSP) foi descrita na Patente U.S. Nº 6.265.171 para Herman. O uso de iniciadores estado de metilação-específicos para a amplificação de DNA tratado com bissulfeto permite a diferenciação entre ácidos nucléicos metilados e não metilados. Pares de iniciadores de MSP contêm pelo menos um iniciador que hibridiza para um dinucleotídeo CpG tratado com bissulfeto. Portanto, a sequência dos referidos iniciadores compreende pelo menos um dinucleotídeo CpG. Iniciadores de MSP específicos para DNA não metilado contêm uma “T” na posição da posição de C no CpG. De preferência, portanto, é necessário que a sequência de base dos referidos iniciadores compreenda uma sequência que possui um comprimento de pelo menos 9 nucleotídeos que hibridiza para uma sequência de ácidos nucléicos tratada de acordo com uma das sequências de bissulfeto e sequências a elas complementares, em que a sequência de base dos referidos oligômeros compreende pelo menos um dinucleotídeo CpG. Uma modalidade preferida adicional do método compreende o uso de oligonucleotídeos bloqueadores (o ensaio HeavyMethylTM). O uso desses oligonucleotídeos bloqueadores foi descrito por Yu e cols., BioTechniques 23: 714-720, 1997. Oligonucleotídeos de bloqueio da sonda são hibridizados para o ácido nucléico tratado com bissulfeto concomitantemente com os iniciadores de PCR. A amplificação por PCR do ácido nucléico é terminada na posição 5’ da sonda de bloqueio, de tal forma que a amplificação de um ácido nucléico seja suprimida onde a sequência complementar à sonda de bloqueio está presente. As sondas podem ser projetadas para hibridizar para o ácido nucléico tratado com bissulfeto de uma forma estado de metilação-específica. Por exemplo, para detecção de ácidos nucléicos metilados dentro de uma população de ácidos nucléicos não metilados, a supressão da amplificação de ácidos nucléicos que não estão metilado na posição em questão seria efetuada pelo uso de sondas de bloqueio que compreendem um “CpA” ou “TpA” na posição em questão, em oposição a um “CpG”, se a supressão de amplificação de ácidos nucléicos metilados é desejada.
[185] Para métodos de PCR que utilizam oligonucleotídeos bloqueadores, a ruptura eficiente da amplificação mediada por polimerase exige que os oligonucleotídeos bloqueadores não sejam alongados pela polimerase. De preferência, isso é obtido por meio do uso de bloqueadores que são 3’-desoxioligonucleotídeos, ou oligonucleotídeos derivatizados na posição 3’ com um grupo diferente de um grupo hidroxil “livre”. Por exemplo, 3’-Oacetil oligonucleotídeos são representativos de uma classe preferida de molécula bloqueadora.
[186] Adicionalmente, a decomposição mediada por polimerase dos oligonucleotídeos bloqueadores deve ser excluída. De preferência, essa exclusão compreende o uso de uma polimerase desprovida de atividade de 5’-3’ exonuclease, ou o uso de oligonucleotídeos bloqueadores modificados que possuem, por exemplo, pontes de tioato nos seus terminais 5’ que tornam a molécula bloqueadora nuclease-resistente. Aplicações particulares podem não necessitar dessas modificações de 5’ do bloqueador. Por exemplo, se os sítios de ligação de bloqueador e de iniciador se superpõem excluindo, dessa forma, a ligação do iniciador (por exemplo, com bloqueador em excesso), a degradação do oligonucleotídeo bloqueador será substancialmente excluída. Isso ocorre porque a polimerase não estenderá o iniciador em direção e através do bloqueador (na direção 5’-3’) – um processo que normalmente resulta em degradação do oligonucleotídeo bloqueador hibridizado.
[187] Uma modalidade de bloqueador/PCR particularmente preferida, para os objetivos da presente invenção e como aqui implementada, compreende o uso de oligômeros de ácido nucléico peptídico (PNA) como oligonucleotídeos de bloqueio. Esses oligômeros bloqueadores de PNA são idealmente adequados, pois não são decompostos nem estendidos pela polimerase.
[188] De preferência, portanto, é necessário que a sequência de base dos referidos oligonucleotídeos de bloqueio compreenda uma sequência que possui um comprimento de pelo menos 9 nucleotídeos que hibridiza para uma sequência de ácidos nucléicos tratada de acordo com uma das sequências de bissulfeto e sequências a elas complementares, em que a sequência de base dos referidos oligonucleotídeos compreende pelo menos um dinucleotídeo CpG, TpG ou CpA. É particularmente preferido que a sequência de base dos referidos oligonucleotídeos de bloqueio compreenda uma sequência que possui um comprimento de pelo menos 9 nucleotídeos que hibridiza para uma sequência de ácidos nucléicos tratada de acordo com um dos IDS. DE SEQ. Nos: 5, 6, 9 ou 10 e sequências a eles complementares, em que a sequência de base dos referidos oligonucleotídeos compreende pelo menos um dinucleotídeo TpG ou CpA.
[189] Os fragmentos obtidos por meio da amplificação podem carregar um marcador detectável direta ou indiretamente. São preferidos marcadores na forma de marcadores de fluorescência, radionuclídeos, ou fragmentos destacáveis da molécula que possuem uma massa típica que pode ser detectada em um espectrômetro de massa. Quando os referidos marcadores são marcadores de massa, prefere-se que os produtos amplificados marcados tenham uma única carga líquida positiva ou negativa, permitindo uma melhor capacidade de detecção no espectrômetro de massa. A detecção pode ser realizada e visualizada por meio de, por exemplo, espectrometria de massa por dessorção/ionização a laser com auxílio de matriz (MALDI) ou usando espectrometria de massa de eletrospray (ESI).
[190] A espectrometria de massa por dessorção/ionização a laser com auxílio de matriz (MALDI-TOF) é um desenvolvimento muito eficiente para a análise de biomoléculas (Karas e Hillenkamp, Anal. Chem., 60: 2.299- 301, 1988). Um analito é embebido em uma matriz de absorção luminosa. A matriz é evaporada por um pulso de laser curto transportando, assim, a molécula do analito para dentro da fase de vapor de forma não fragmentada. O analito é ionizado por colisões com moléculas de matriz. A voltagem aplicada acelera os íons em um tubo de vôo de campo livre. Em função de suas massas diferentes, os íons são acelerados em taxas diferentes. Íons menores alcançam o detector mais cedo do que aqueles maiores. A espectrometria MALDI-TOF é bem adequada à análise de peptídeos e proteínas. A análise de ácidos nucléicos é um pouco mais difícil (Gut e Beck, Current Innovations and Future Trends, 1: 147-57, 1995). A sensibilidade com relação à análise de ácido nucléico é aproximadamente 100 vezes menor do que para peptídeos, e diminui desproporcionalmente com o aumento do tamanho do fragmento. Além disso, para ácidos nucléicos que possuem múltiplos arcabouços negativamente carregados, o processo de ionização por meio da matriz é consideravelmente menos eficiente. Na espectrometria MALDI-TOF, a seleção da matriz desempenha um papel eminentemente importante. Para dessorção de peptídeos, foram encontradas várias matrizes muito eficientes que produzem uma cristalização muito fina. Há agora várias matrizes responsivas para DNA; no entanto, a diferença na sensibilidade entre peptídeos e ácidos nucléicos não foi reduzida. Essa diferença na sensibilidade pode ser reduzida, no entanto, modificando-se quimicamente o DNA de forma a torná-lo mais similar a um peptídeo. Por exemplo, ácidos nucléicos de fosforotioato, nos quais os fosfatos comuns do arcabouço são substituídos com tiofosfatos, podem ser convertidos em um DNA de carga neutra usando química de alquilação simples (Gut e Beck, Nucleic Acids Res. 23: 1.367-73, 1995). O acoplamento de um tag de carga a esse DNA modificado resulta em um aumento na sensibilidade de MALDI-TOF até o mesmo nível encontrado para peptídeos. Uma vantagem adicional do tagging de carga é a estabilidade aumentada da análise contra impurezas, o que torna a detecção de substratos não modificados consideravelmente mais difícil.
[191] Na quarta etapa do método, os produtos amplificados obtidos durante a terceira etapa do método são analisados a fim de verificar o estado de metilação dos dinucleotídeos CpG antes do tratamento.
[192] Em modalidades nas quais os produtos amplificados foram obtidos por meio de amplificação MSP, a presença, ausência ou classe de um produto amplificado é, por ela própria, indicativa do estado de metilação das posições de CpG cobertas pelo iniciador, de acordo com a sequências de base do referido iniciador.
[193] Os produtos amplificados obtidos tanto por meio de PCR-padrão quanto por meio de PCR metilação-específica podem ser ainda analisados por meio de métodos based-based como, por exemplo, sem limitação, tecnologia de arranjo e tecnologias baseadas em sondas, bem como por meio de técnicas como, por exemplo, sequenciamento e extensão dirigida por modelo.
[194] Em uma modalidade do método, os produtos amplificados sintetizados na etapa três são subsequentemente hibridizados para um arranjo ou um conjunto de sondas de oligonucleotídeos e/ou PNA. Nesse contexto, a hibridização ocorre da seguinte forma: o conjunto de sondas usado durante a hibridização é preferivelmente composto por pelo menos 2 oligômeros de oligonucleotídeos ou PNA; no processo, os produtos amplificados servem como sondas que hibridizam para oligonucleotídeos previamente ligados a uma fase sólida; os fragmentos não hibridizados são subsequentemente removidos; os referidos oligonucleotídeos contêm pelo menos uma sequência de base que possui um comprimento de pelo menos 9 nucleotídeos que é complementar ou idêntica reversa a um segmento das sequências de base especificadas na presente Listagem de Sequências; e o segmento compreende pelo menos um dinucleotídeo CpG, TpG ou CpA. A porção hibridizante dos ácidos nucléicos hibridizantes possui tipicamente pelo menos 9, 15, 20, 25, 30 ou 35 nucleotídeos de comprimento. No entanto, moléculas mais longas possuem utilidade na invenção e estão, dessa forma, dentro do escopo da presente invenção.
[195] Em uma modalidade preferida, o referido dinucleotídeo está presente no terço central do oligômero. Por exemplo, em que o oligômero compreende um dinucleotídeo CpG, o referido dinucleotídeo é preferivelmente o quinto a nono nucleotídeo da extremidade 5’ de um 13-mer. Existe um oligonucleotídeo para a análise de cada dinucleotídeo CpG dentro de uma sequência selecionada do grupo que consiste nas sequências genômicas, e nas posições equivalentes dentro das sequências de bissulfeto. Os referidos oligonucleotídeos também podem estar presentes na forma de ácidos nucléicos peptídicos. Os produtos amplificados não hibridizados são então removidos. Os produtos amplificados hibridizados são então detectados. Nesse contexto, preferese que marcadores anexados aos produtos amplificados sejam identificáveis em cada posição da fase sólida na qual uma sequência de oligonucleotídeos está localizada.
[196] Ainda em outra modalidade adicional do método, o estado de metilação genômica das posições de CpG pode ser verificado por meio de sondas de oligonucleotídeo (como detalhado acima) que são hibridizadas para o DNA tratado com bissulfeto concomitantemente com os iniciadores de PCR de amplificação (em que os referidos iniciadores podem ser metilação-específicos ou padronizados).
[197] Uma modalidade particularmente preferida desse método é o uso de PCR quantitativa em tempo real baseada em fluorescência (Heid e cols., Genome Res. 6: 986-994, 1996; veja também a Patente U.S. Nº 6.331.393) que emprega uma sonda de oligonucleotídeo fluorescente com marcação dupla (PCR TaqManTM, usando um Sistema de Detecção de Sequência ABI Prism 7700, Perkin Elmer Applied Biosystems, Foster City, Califórnia). A reação de PCR TaqManTM emprega o uso de um oligonucleotídeo de interrogação não extensível, denominado uma sonda TaqManTM, que, em modalidades preferidas, é projetada para hibridizar para uma sequência rica em CpG localizada entre os iniciadores de amplificação diretos e reversos. A sonda TaqManTM ainda compreende uma “porção repórter” e uma “porção extintora” fluorescentes ligadas covalentemente a porções vinculadoras (por exemplo, fosforamiditas) anexadas aos nucleotídeos do oligonucleotídeo TaqManTM. Para análise de metilação dentro de ácidos nucléicos subsequente ao tratamento com bissulfeto, é necessário que a sonda seja metilaçãoespecífica, como descrito na Patente U.S. Nº 6.331.393, (aqui incorporada por referência em sua totalidade), também conhecido como o ensaio MethyLightTM. Variações sobre a metodologia de detecção TaqManTM que também são adequadas para uso com a invenção descrita incluem o uso de tecnologia de sonda dupla (LightCyclerTM) ou iniciadores de amplificação fluorescentes (tecnologia SunriseTM). Ambas essas técnicas podem ser adaptadas de uma forma adequada para uso com DNA tratado com bissulfeto e, além disso, para análise de metilação dentro de dinucleotídeos CpG.
[198] Em uma modalidade preferida adicional do método, a quarta etapa do método compreende o uso de extensão de oligonucleotídeo dirigida por modelo, por exemplo, MSSNuPE, como descrito por Gonzalgo e Jones, Nucleic Acids Res. 25: 2.529-2.531, 1997.
[199] Ainda em outra modalidade adicional do método, a quarta etapa do método compreende sequenciamento e subsequente análise de sequência do produto amplificado gerado na terceira etapa do método (Sanger F., e cols., Proc. Natl. Acad. Sci. USA 74: 5.463-5.467, 1977).
[200] Em uma modalidade do método, os ácidos nucléicos genômicos são isolados e tratados de acordo com as três primeiras etapas do método descrito acima, especificamente:
- a) obtenção, de um indivíduo, de uma amostra biológica que possui DNA genômico do indivíduo;
- b) extração ou algum outro modo de isolamento do DNA genômico;
- c) tratamento do DNA genômico de b), ou um fragmento deste, com um ou mais reagentes para converter bases de citosina que são não metiladas na posição 5 destas em uracil ou em outra base que é diferente de forma detectável à citosina em termos de propriedades de hibridização; e em que:
- d) a amplificação subsequente ao tratamento em c) é realizada de forma metilação-específica, especificamente por uso de iniciadores ou oligonucleotídeos de bloqueio metilação-específicos, e ainda em que:
- e) a detecção dos produtos amplificados é realizada por meio de uma sonda de detecção em tempo real, como descrito acima, de
- f) determinação de um prognóstico para o indivíduo.
- g) De preferência, quando a amplificação subsequente de d) é realizada por meio de iniciadores metilaçãoespecíficos, como descrito acima, os referidos iniciadores metilação-específicos compreendem uma sequência que possui um comprimento de pelo menos 9, pelo menos 6, pelo menos 25 ou pelo menos 50 nucleotídeos que hibridiza para uma sequência de ácidos nucléicos tratada de acordo com uma das sequências de bissulfeto e sequências a elas complementares, em que a sequência de base dos referidos oligômeros compreende pelo menos um dinucleotídeo CpG.
[201] A etapa e) do método, especificamente a detecção dos produtos amplificados específicos indicativa do estado de metilação de uma ou mais posições de CpG de acordo com as sequências genômicas é realizada por meio de métodos de detecção em tempo real, como descrito acima.
[202] Em uma modalidade alternativa da invenção, a segunda etapa descrita acima pode ser realizada por meio de análise de enzima de restrição sensível à metilação ou metilação-específica. São conhecidos métodos na técnica em que um reagente de enzima de restrição sensível à metilação, ou uma série de reagentes de enzima de restrição que compreendem reagentes de enzima de restrição sensível à metilação que distingue entre dinucleotídeos CpG metilados e não metilados dentro de uma região-alvo são utilizados na determinação da metilação, por exemplo, sem limitação, DMH.
[203] Em uma modalidade preferida, o DNA pode ser clivado antes do tratamento com enzimas de restrição sensíveis à metilação. Tais métodos são conhecidos na técnica e podem incluir meios tanto físicos quanto enzimáticos. É particularmente preferido o uso de uma ou diversas enzimas de restrição que não são sensíveis à metilação, e cujos sítios de reconhecimento são ricos em AT e não compreendem dinucleotídeos CG. O uso dessas enzimas permite a conservação de regiões ricas em ilhas CpG e em CpG no DNA fragmentado. As enzimas de restrição não metilação-específicas são preferivelmente selecionadas do grupo que consiste em MseI, BfaI, Csp6I, Tru1I, Tvu1I, Tru9I, Tvu9I, MaeI e XspI. É particularmente preferido o uso de duas ou três dessas enzimas. É particularmente preferido o uso de uma combinação de MseI, BfaI e Csp6I.
[204] O DNA fragmentado pode então ser ligado aos oligonucleotídeos adaptadores a fim de facilitar a amplificação enzimática subsequente. A ligação de oligonucleotídeos a fragmentos de DNA de extremidade romba e pegajosa é conhecida na técnica, e é realizada por meio de desfosforilação das extremidades (por exemplo, usando fosfatase alcalina de bezerro ou camarão) e ligação subsequente usando enzimas ligase (por exemplo, T4 DNA ligase) na presença de dATPs. Os oligonucleotídeos adaptadores possuem tipicamente pelo menos 18 pares de bases de comprimento.
[205] Na terceira etapa, o DNA (ou fragmentos deste) é então digerido com uma ou mais enzimas de restrição sensíveis à metilação. A digestão é realizada de tal forma que a hidrólise do DNA no sítio de restrição seja informativa do estado de metilação de um dinucleotídeo CpG específico de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso.
[206] De preferência, a enzima de restrição metilaçãoespecífica é selecionada do grupo que consiste em Bsi E1, Hga I HinPL, Hpy99I, Ava I, Bee AI, Bsa HI, BisI, BstUI, BshI236I, AccII, BstFNI, McrBC, GlaI, MvnI, HpaII (HapII), HhaI, AciI, SmaI, HinP1I, HpyCH4IV, EagI e misturas de duas ou mais das enzimas acima. É preferida uma mistura que contém as enzimas de restrição BstUI, HpaII, HpyCH4IV e HinP1I.
[207] Na quarta etapa, que é opcional, mas uma modalidade preferida, os fragmentos de restrição são amplificados. Isso é realizado preferivelmente usando uma reação em cadeia de polimerase, e os referidos produtos amplificados podem carregar marcadores detectáveis adequados, como discutido acima, especificamente marcadores fluoróforos, radionuclídeos e marcadores de massa. É particularmente preferida a amplificação por meio de uma enzima de amplificação e pelo menos dois iniciadores que compreendem, em cada caso, uma sequência contígua com pelo menos 16 nucleotídeos de comprimento que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para uma sequência selecionada do grupo que consiste nas sequências genômicas, e complementos destas. De preferência, a referida sequência contígua possui pelo menos 16, 20 ou 25 nucleotídeos de comprimento. Em uma modalidade alternativa, os referidos iniciadores podem ser complementares para qualquer um dos adaptadores ligados aos fragmentos.
[208] Na quinta etapa, os produtos amplificados são detectados. A detecção pode ser por qualquer meio padronizado na técnica, por exemplo, sem limitação, análise por eletroforese em gel, análise de hibridização, incorporação de tags detectáveis dentro dos produtos de PCR, análise de arranjo de DNA, análise MALDI ou ESI. De preferência, a referida detecção é realizada por hibridização para pelo menos um ácido nucléico ou ácido nucléico peptídico que compreende, em cada caso, uma sequência contígua com pelo menos 16 nucleotídeos de comprimento que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para uma sequência selecionada do grupo que consiste nas sequências genômicas, e complementos destas. De preferência, a referida sequência contígua possui pelo menos 16, 20 ou 25 nucleotídeos de comprimento.
[209] Subsequente à determinação do estado ou nível de metilação dos ácidos nucléicos genômicos, o prognóstico de um indivíduo que tem câncer é deduzido com base no estado ou nível de metilação de pelo menos uma sequência de dinucleotídeo CpG que está metilada no câncer, mas não metilada em tecido não canceroso, ou uma média, ou um valor que reflete um estado de metilação médio de diversas sequências de dinucleotídeo CpG das sequências genômicas, em que a metilação está associada ao prognóstico de um indivíduo que tem câncer. Em que a referida metilação é determinada por meios quantitativos e o ponto de corte para determinação da referida presença de metilação é preferivelmente zero (ou seja, em que uma amostra que exibe qualquer grau de metilação é determinada como tendo um estado metilado na posição de CpG analisada). Todavia, é previsto que aqueles habilitados na técnica possam desejar ajustar o referido valor de corte a fim de fornecer um ensaio de uma sensibilidade ou especificidade particularmente preferida. Consequentemente, o referido valor de corte pode ser aumentado (aumentando, dessa forma, a especificidade), o referido valor de corte pode estar dentro de uma faixa selecionada do grupo que consiste em 0%-5%, 5%-10%, 10%-15%, 15%-20%, 20%-30% e 30%-50%. São particularmente preferidos valores de corte que são de pelo menos 0,1%, 1%, 10%, 15%, 25% e 30%.
[210] Como aqui usado, o termo “prognóstico” deve ser considerado como significando um indicador da progressão prevista da doença (incluindo, sem limitação, agressividade e potencial metastático) e/ou tempo de sobrevida previsto do paciente.
[211] No contexto da presente invenção, o termo “agressividade” significa um ou mais de probabilidade elevada de recidiva pós-cirurgia; sobrevida abaixo da média ou abaixo da mediana do paciente; sobrevida livre de doença abaixo da média ou abaixo da mediana; sobrevida livre de recidiva abaixo da média ou abaixo da mediana; complicações relacionadas ao tumor acima da média; progressão rápida do tumor ou metástases.
[212] A menos que estabelecido de forma diferente, como aqui usado, o termo “sobrevida” deve ser considerado como incluindo todos os seguintes: sobrevida até mortalidade, também conhecida como sobrevida global (em que a referida mortalidade pode ser independentemente de causa ou relacionada ao tumor); “sobrevida livre de recorrência” (em que o termo “recorrência” deve incluir recorrência tanto localizada quanto distante); sobrevida livre de metástase; sobrevida livre de doença (em que o termo “doença” deve incluir câncer e doenças a ele associadas). A duração da referida sobrevida pode ser calculada por referência a um ponto de definido partida (por exemplo, momento do diagnóstico ou início de tratamento) e ponto final (por exemplo, morte, recorrência ou metástase).
[213] A invenção revelada fornece ácidos nucléicos tratados, derivados das sequências genômicas, em que o tratamento é adequado para converter pelo menos uma base de citosina não metilada da sequência do DNA genômico em uracil ou em outra base que é diferente de forma detectável à citosina em termos de hibridização, para uso na determinação de prognóstico de um indivíduo que tem câncer ou um tumor. As sequências genômicas em questão podem compreender uma ou mais posições consecutivas de CpG metilado. O referido tratamento dos ácidos nucleicos preferivelmente compreende o uso de um reagente selecionado do grupo que consiste em bissulfeto, sulfeto de hidrogênio, dissulfeto, e combinações destes. Em uma modalidade preferida da invenção, a invenção fornece um ácido nucléico modificado de ocorrência não natural que compreende uma sequência com pelo menos 16 bases de nucleotídeos contíguas de comprimento de uma sequência selecionada do grupo que consiste nas sequências de bissulfeto, em particular das sequências como definidas pelos IDS. DE SEQ. Nos: 5, 7, 10 a 13 e 18 a 20. Em modalidades preferidas adicionais da invenção, o referido ácido nucléico possui pelo menos 50, 100, 150, 200, 250 ou 500 pares de bases de comprimento de um segmento da sequência de ácidos nucléicos revelada nas sequências de bissulfeto. É particularmente preferida uma molécula de ácido nucléico que é idêntica ou complementar a todas ou a uma porção das sequências das sequências de bissulfeto, mas não às sequências genômicas ou a outro DNA de ocorrência natural.
[214] Prefere-se que a referida sequência compreenda pelo menos um dinucleotídeo CpG, TpA ou CpA e sequências complementares a ele. As sequências das sequências de bissulfeto fornecem versões modificadas de ocorrência não natural do ácido nucléico de acordo com as sequências genômicas, em que a modificação de cada sequência genômica resulta na síntese de um ácido nucléico que possui uma sequência que é única e distinta da referida sequência genômica da seguinte forma: para cada DNA genômico de fita senso, quatro versões convertidas são reveladas. Uma primeira versão em que “C” é convertido em “T”, mas “CpG” permanece “CpG” (ou seja, corresponde ao caso quando, para a sequência genômica, todos os resíduos “C” de sequências de dinucleotídeo CpG estão metilados e, dessa forma, não são convertidos); uma segunda versão revela o complemento da sequência do DNA genômico revelada (ou seja, fita antissenso), em que “C” é convertido em “T”, mas “CpG” permanece “CpG” (ou seja, corresponde ao caso quando, para todos os resíduos “C” de sequências de dinucleotídeo CpG estão metilados e, dessa forma, não são convertidos). As sequências convertidas “suprametiladas” das sequências genômicas correspondem aos IDS. DE SEQ. Nos: 4-9 para SEPTIN9 e aos IDS. DE SEQ. Nos: 17 e 18 para RASSF2A. Uma terceira versão quimicamente convertida de cada sequências genômicas é fornecida, em que “C” é convertido em “T” para todos os resíduos “C”, incluindo aqueles de sequências de dinucleotídeos “CpG” (ou seja, corresponde ao caso quando, para as sequências genômicas, todos os resíduos “C” de sequências de dinucleotídeo CpG são não metilados); uma versão quimicamente convertida final de cada sequência revela o complemento da sequência do DNA genômico revelada (ou seja, fita antissenso), em que “C” é convertido em “T” para todos os resíduos “C”, incluindo aqueles de sequências de dinucleotídeos “CpG” (ou seja, corresponde ao caso quando, para o complemento (fita antissenso) de cada sequência genômica, todos os resíduos “C” de sequências de dinucleotídeo CpG são não metilados). As sequências convertidas “inframetiladas” das sequências genômicas correspondem aos IDS. DE SEQ. Nos: 10-15 para SEPTIN9 e aos IDS. DE SEQ. Nos: 19 e 20 para RASSF2A.
[215] Significantemente, até agora, as sequências de ácidos nucléicos e moléculas de acordo com as sequências de bissulfeto nas foram implicadas ou conectas ao prognóstico de um indivíduo que tem câncer.
[216] Em uma modalidade alternativa, a invenção ainda fornece oligonucleotídeos ou oligômeros adequados para uso nos métodos da invenção para detecção do estado de metilação de citosina dentro do DNA genômico ou tratado (modificado quimicamente). O referido oligonucleotídeo ou ácidos nucléicos do oligômero fornecem novos meios prognósticos. O referido oligonucleotídeo ou oligômero que compreende uma sequência de ácidos nucléicos que possui um comprimento de pelo menos nove (9) nucleotídeos que é idêntica, hibridiza, sob condições moderadamente rigorosas ou rigorosas (como aqui definidas acima) para uma sequência de ácidos nucléicos tratada de acordo com as sequências de bissulfeto e/ou sequências complementares a elas, ou para uma sequência genômica de acordo com as sequências genômicas e/ou sequências complementares a elas.
[217] Dessa forma, a presente invenção inclui moléculas de ácido nucléico (por exemplo, oligonucleotídeos e moléculas de ácido nucléico peptídico (PNA) (oligômeros de PNA)) que hibridizam sob condições de hibridização moderadamente rigorosas e/ou rigorosas para todas ou para uma porção das sequências ou para os complementos destas. É particularmente preferida uma molécula de ácido nucléico que hibridiza sob condições de hibridização moderadamente rigorosas e/ou rigorosas para todas ou para uma porção das sequências das sequências de bissulfeto, mas não para as sequências genômicas ou outro DNA genômico humano.
[218] A porção idêntica ou hibridizante dos ácidos nucléicos hibridizantes possui tipicamente pelo menos 9, 16, 20, 25, 30 ou 35 nucleotídeos de comprimento. No entanto, moléculas mais longas possuem utilidade na invenção e estão, portanto, dentro do escopo da presente invenção.
[219] De preferência, a porção hibridizante dos ácidos nucléicos hibridizantes da invenção é pelo menos 95% ou pelo menos 98% ou 100% idêntica à sequência, ou a uma porção desta, ou aos complementos desta.
[220] Ácidos nucléicos hibridizantes do tipo aqui descrito podem ser usados, por exemplo, como um iniciador (por exemplo, um iniciador de PCR), ou uma sonda ou iniciador prognóstico. De preferência, a hibridização da sonda de oligonucleotídeo para uma amostra de ácido nucléico é realizada sob condições rigorosas, e a sonda é 100% idêntica à sequência-alvo. A estabilidade do duplexo ou híbrido de ácido nucléico é expressa como a temperatura de fusão ou Tm, que é a temperatura na qual uma sonda se dissocia de um DNA-alvo. Essa temperatura de fusão é usada para definir as condições de rigor necessárias.
[221] Para sequências-alvo que estão relacionadas e são substancialmente idênticas à sequência correspondente das sequências genômicas (por exemplo, variantes alélicas e SNPs), e não idênticas, é útil primeiro estabelecer a menor temperatura na qual só ocorre hibridização homóloga com uma concentração de sal particular (por exemplo, SSC ou SSPE). A seguir, presumindo-se que 1% de não combinação resulta em uma diminuição de 1°C na Tm, a temperatura da lavagem final na reação de hibridização é reduzida consequentemente (por exemplo, caso se busque sequências que possuem ˃ 95% de identidade com a sonda, a temperatura da lavagem final é diminuída em 5°C). Na prática, a alteração na Tm pode ser entre 0,5°C e 1,5°C por 1% de não combinação.
[222] Exemplos de oligonucleotídeos da invenção de comprimento X (em nucleotídeos), como indicado por posições de polinucleotídeos com referência às sequências genômicas e convertidas aqui descritas, incluem aqueles que correspondem a conjuntos (conjuntos senso e antissenso) de oligonucleotídeos consecutivamente superpostos de comprimento X, em que os oligonucleotídeos dentro de cada conjunto consecutivamente superposto (que corresponde a certo valor X) são definidos como o conjunto finito de oligonucleotídeos Z de posições de nucleotídeos:
[223] n a (n + (X-1));
[224] em que n = 1, 2, 3, ...(Y-(X-1));
[225] em que Y é igual ao comprimento (nucleotídeos ou pares de bases) dos IDS. DE SEQ. Nos: 1-20;
[226] em que X é igual ao comprimento comum (em nucleotídeos) de cada oligonucleotídeo no conjunto (por exemplo, X = 20 para um conjunto de 20-mers consecutivamente superpostos); e
[227] em que o número (Z) de consecutivamente oligômeros superpostos de comprimento X para certo ID. DE SEQ. Nº: de comprimento Y é igual a Y - (X-1).
[228] De preferência, o conjunto é limitado àqueles oligômeros que compreendem pelo menos um dinucleotídeo CpG, TpG ou CpA.
[229] Exemplos de oligonucleotídeos 20-mer da invenção incluem os oligômeros aqui descritos (e o conjunto antissenso complementara a eles), indicados por posições de polinucleotídeos com referência aos IDS. DE SEQ. Nos: 1 a 20:
[230] 1-20, 2-21, 3-22, 4-23, 5-24, etc.
[231] De preferência, o conjunto é limitado àqueles oligômeros que compreendem pelo menos um dinucleotídeo CpG, TpG ou CpA.
[232] Da mesma forma, exemplos de oligonucleotídeos 25- mer da invenção incluem o seguinte conjunto de oligômeros xxx (e o conjunto antissenso complementar a eles), indicados por posições de polinucleotídeos com referência aos IDS. DE SEQ. Nos: 1 a 20:
[233] 1-25, 2-26, 3-27, 4-28, 5-29, etc.
[234] De preferência, o conjunto é limitado àqueles oligômeros que compreendem pelo menos um dinucleotídeo CpG, TpG ou CpA.
[235] A presente invenção engloba, para cada uma das sequências que está metilada no câncer, mas não metilada em tecido não canceroso (senso e antissenso), múltiplos conjuntos de oligonucleotídeos consecutivamente superpostos ou oligonucleotídeos modificados de comprimento X, em que, por exemplo, X = 9, 10, 17, 20, 22, 23, 25, 27, 30 ou 35 nucleotídeos.
[236] Os oligonucleotídeos ou oligômeros de acordo com a presente invenção constituem ferramentas eficazes úteis para verificar parâmetros genéticos e epigenéticos da sequência genômica que corresponde às sequências genômicas. Conjuntos desses oligonucleotídeos ou oligonucleotídeos modificados são aqueles conjuntos de oligômeros consecutivamente superpostos que correspondem a uma sequência que está metilada no câncer, mas não metilada em tecido não canceroso (e aos complementos desta). De preferência, os referidos oligômeros compreendem pelo menos um dinucleotídeo CpG, TpG ou CpA.
[237] Oligonucleotídeos ou oligômeros particularmente preferidos de acordo com a presente invenção são aqueles nos quais a citosina das sequências de dinucleotídeo CpG (ou do dinucleotídeo TpG ou CpA convertido correspondente) está dentro do terço médio do oligonucleotídeo; ou seja, quando o oligonucleotídeo possui, por exemplo, 13 bases de comprimento, o dinucleotídeo CpG, TpG ou CpA está posicionado dentro do quinto ao nono nucleotídeo a partir da extremidade 5’.
[238] Os oligonucleotídeos da invenção também podem ser modificados ligando-se quimicamente o oligonucleotídeo a uma ou mais porções ou conjugados para aumentar a atividade, estabilidade ou detecção do oligonucleotídeo. Essas porções ou conjugados incluem porções de cromóforos, fluoróforos, lipídeos como, por exemplo, colesterol, ácido cólico, tioéter, cadeias alifáticas, fosfolipídeos, poliaminas, polietileno glicol (PEG), palmitil e outras, como revelado, por exemplo, nas Patentes U.S. Nos 5.514.758, 5.565.552, 5.567.810, 5.574.142, 5.585.481, 5.587.371, 5.597.696 e 5.958.773. As sondas também podem existir na forma de um PNA (ácido nucléico peptídico) que possui propriedades de pareamento particularmente preferidas. Dessa forma, o oligonucleotídeo pode incluir outros grupos anexados como, por exemplo, peptídeos, e pode incluir agentes de clivagem desencadeada por hibridização (Krol e cols., BioTechniques 6: 958-976, 1988) ou agentes intercalantes (Zon, Pharm. Res. 5: 539-549, 1988). Para essa finalidade, o oligonucleotídeo pode ser conjugado a outra molécula, por exemplo, um cromóforo, fluoróforo, peptídeo, um agente reticulação desencadeada por hibridização, agente de transporte, agente de clivagem desencadeada por hibridização, etc.
[239] O oligonucleotídeo também pode compreender pelo menos uma porção de açúcar e/ou base modificada reconhecida pela técnica, ou pode compreender um arcabouço modificado ou ligação internucleosídeo não natural.
[240] Os oligonucleotídeos ou oligômeros de acordo com modalidades particulares da presente invenção são tipicamente usados em “conjuntos”, que contêm pelo menos um oligômero para análise de cada um dos dinucleotídeos CpG de uma sequência genômica selecionada do grupo que consiste nas sequências genômicas e sequências complementares a elas, ou ao dinucleotídeo CpG, TpG ou CpA correspondente dentro de uma sequência dos ácidos nucléicos tratados de acordo com as sequências de bissulfeto e sequências a elas complementares. No entanto, prevê-se que, por fatores econômicos ou por outros fatores, pode ser preferível analisar uma seleção limitada dos dinucleotídeos CpG dentro das referidas sequências, e o teor do conjunto de oligonucleotídeos é alterado de acordo.
[241] Portanto, em modalidades particulares, a presente invenção fornece um conjunto de pelo menos dois (2) (oligômeros de oligonucleotídeos e/ou PNA) úteis para detecção do estado de metilação de citosina em DNA genômico tratado (as sequências de bissulfeto), ou em DNA genômico (as sequências genômicas e sequências complementares a elas). Essas sondas permitem a determinação do prognóstico de um indivíduo que tem câncer. O conjunto de oligômeros também pode ser usado para detecção de polimorfismos de nucleotídeo único (SNPs) em DNA genômico tratado (as sequências de bissulfeto), ou em DNA genômico (as sequências genômicas e sequências complementares a elas).
[242] Em modalidades preferidas, pelo menos um e, mais preferivelmente, todos os membros de um conjunto de oligonucleotídeos estão ligados a uma fase sólida.
[243] Em modalidades adicionais, a presente invenção fornece um conjunto de pelo menos dois (2) oligonucleotídeos que são usados como oligonucleotídeos “iniciadores” para amplificação de sequências de DNA de uma sequência que está metilada no câncer, mas não metilada em tecido não canceroso, e sequências complementares a ela, ou segmentos desta.
[244] Prevê-se que os oligonucleotídeos possam constituir todo ou parte de um “arranjo” ou “chip de DNA” (ou seja, um arranjo de diferentes oligômeros de oligonucleotídeos e/ou PNA ligado a uma fase sólida). Um arranjo desse tipo de diferentes sequências de oligômeros de oligonucleotídeos e/ou PNA pode ser caracterizado, por exemplo, pelo fato de que ele está arranjado na fase sólida na forma de uma treliça retangular ou hexagonal. A superfície de fase sólida pode ser composta por silício, vidro, poliestireno, alumínio, aço, ferro, cobre, níquel, prata ou ouro. Nitrocelulose, bem como plásticos como, por exemplo, náilon, que podem existir na forma de péletes ou também como matrizes de resina, também podem ser usados. Uma visão geral da técnica estabelecida sobre a fabricação de arranjo de oligômeros pode ser reunida a partir de uma edição especial de “Nature Genetics” (Nature Genetics Supplement, Volume 21, Janeiro de 1999, e a partir da literatura nele citada). Sondas marcadas de forma fluorescente são frequentemente usadas para a varredura de arranjos de DNA imobilizados. A simples adesão de corantes Cy3 e Cy5 ao 5’-OH da sonda específica é particularmente adequada para marcadores de fluorescência. A detecção da fluorescência das sondas hibridizadas pode ser realizada, por exemplo, por meio de um microscópio confocal. Corantes Cy3 e Cy5, além de muitos outros, são disponíveis comercialmente.
[245] Prevê-se também que os oligonucleotídeos, ou sequências particulares destes, possam constituir todo ou parte de um “arranjo virtual”, em que os oligonucleotídeos, ou sequências particulares destes, são usados, por exemplo, como “especificadores” como parte de, ou em combinação com, uma população diversa de sondas marcadas únicas para analisar uma mistura complexa de analitos. Tal método, por exemplo, é descrito em US 2003/0013091 (Patente U.S., Número de Série 09/898.743, publicada em 16 de janeiro de 2003). Em tais métodos, são gerados marcadores suficientes, de tal modo que cada ácido nucléico na mistura complexa (ou seja, cada analito) possa ser ligado unicamente a um marcador único e, dessa forma, detectado (cada marcador é contado diretamente, resultando em uma leitura digital de espécie cada na mistura).
[246] É particularmente preferido que os oligômeros de acordo com a invenção sejam utilizados para determinação do prognóstico de um indivíduo que tem câncer.
[247] Além disso, um aspecto adicional da presente invenção é um kit que compreende: um meio para determinação da metilação de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso. Os meios para determinação da metilação de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, compreendem preferivelmente um reagente que contém bissulfeto; um ou diversos oligonucleotídeos cujas sequências em cada caso são idênticas, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 9, pelo menos 18, pelo menos 25 ou pelo menos 50 bases de comprimento de uma sequência selecionada das sequências de bissulfeto; e, opcionalmente, instruções para realização e avaliação do método descrito de análise de metilação. Em uma modalidade, a sequência de base dos referidos oligonucleotídeos compreende pelo menos um dinucleotídeo CpG, CpA ou TpG.
[248] Em uma modalidade adicional, o referido kit pode ainda compreender reagentes-padrão para a realização de uma análise de metilação posição de CpG-específica, em que a referida análise compreende uma ou mais das seguintes técnicas: MS-SNuPE, MSP, MethyLightTM, HeavyMethyl, COBRA e sequenciamento de ácido nucléico. No entanto, um kit de acordo com a presente invenção também pode conter somente parte dos componentes mencionados anteriormente.
[249] Em uma modalidade preferida, o kit pode compreender reagentes de conversão de bissulfeto adicionais selecionados do grupo que consiste em: tampão de desnaturação de DNA; tampão de sulfonação; reagentes ou kits de recuperação de DNA (por exemplo, precipitação, ultrafiltração, coluna de afinidade); tampão de dessulfonação; e componentes de recuperação de DNA.
[250] Em uma modalidade alternativa adicional, o kit pode conter, embalados em recipientes separados, uma polimerase e um tampão de reação otimizados para extensão de iniciador mediada pela polimerase, por exemplo, PCR. Em outra modalidade da invenção o kit ainda compreende meios para obtenção e/ou armazenamento de uma amostra biológica do indivíduo. É preferido um kit que ainda compreende um recipiente adequado para contenção dos meios para determinação da metilação de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, na amostra biológica do indivíduo e, mais preferivelmente ainda, ainda compreende instruções para uso e interpretação dos resultados do kit. Em uma modalidade preferida, o kit compreende: (a) um reagente de bissulfeto; (b) um recipiente adequado à contenção do referido reagente de bissulfeto e da amostra biológica do indivíduo; (c) pelo menos um conjunto de oligonucleotídeos iniciadores contendo dois oligonucleotídeos cujas sequências em cada caso são idênticas, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 9 ou mais preferivelmente 18 bases de comprimento de uma sequência selecionada das sequências de bissulfeto; e, opcionalmente, (d) instruções para uso e interpretação dos resultados do kit. Em uma modalidade alternativa preferida o kit compreende: (a) um reagente de bissulfeto; (b) um recipiente adequado à contenção do referido reagente de bissulfeto e da amostra biológica do indivíduo; (c) pelo menos um oligômero de oligonucleotídeo e/ou PNA que possui um comprimento de pelo menos 9 ou 16 nucleotídeos que é idêntico ou hibridiza para uma sequência de ácidos nucléicos pré-tratados de acordo com uma das sequências de bissulfeto e sequências a elas complementares; e, opcionalmente, (d) instruções para uso e interpretação dos resultados do kit.
[251] Em uma modalidade alternativa, o kit compreende: (a) um reagente de bissulfeto; (b) um recipiente adequado à contenção do referido reagente de bissulfeto e da amostra biológica do indivíduo; (c) pelo menos um conjunto de oligonucleotídeos iniciadores contendo dois oligonucleotídeos cujas sequências em cada caso são idênticas, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 9 ou mais preferivelmente 18 bases de comprimento de uma sequência selecionada das sequências de bissulfeto; (d) pelo menos um oligômero de oligonucleotídeo e/ou PNA que possui um comprimento de pelo menos 9 ou 16 nucleotídeos que é idêntico ou hibridiza para uma sequência de ácidos nucléicos pré-tratados de acordo com uma das sequências de bissulfeto e sequências a elas complementares; e, opcionalmente, (e) instruções para uso e interpretação dos resultados do kit.
[252] O kit também pode conter outros componentes como, por exemplo, tampões ou soluções adequadas ao bloqueio, lavagem ou revestimento, embalados em um recipiente separado.
[253] Outro aspecto da invenção está relacionada a um kit para uso na determinação do prognóstico de um indivíduo que tem câncer, o referido kit compreendendo: um meio para medição do nível de transcrição de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, e um meio para determinação da metilação de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso.
[254] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em COBRATM típico) para análise COBRATM podem incluir, sem limitação: iniciadores de PCR para pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, enzima de restrição e tampão apropriados; óligo de hibridização de gene; óligo de hibridização de controle; kit de marcação de quinase para sonda de óligo; e nucleotídeos marcados. Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em MethyLightTM típico) para análise MethyLightTM podem incluir, sem limitação: iniciadores de PCR para a sequência convertida por bissulfeto de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, sondas específicas de bissulfeto (por exemplo, TaqManTM ou LightCyclerTM); tampões de PCR otimizados e desoxinucleotídeos; e Taq polimerase.
[255] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em Ms-SNuPETM típico) para análise Ms-SNuPETM podem incluir, sem limitação: iniciadores de PCR para um gene específico (ou sequência de DNA ou ilha CpG tratada com bissulfeto); tampões de PCR otimizados e desoxinucleotídeos; kit de extração de gel; iniciadores de controle positivo; iniciadores de Ms-SNuPETM para a sequência convertida por bissulfeto de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso tampão de reação (para a reação de Ms-SNuPE); e nucleotídeos marcados.
[256] Reagentes típicos (por exemplo, como podem ser encontrados em um kit baseado em MSP típico) para análise MSP podem incluir, sem limitação: iniciadores de PCR metilados e não metilados para a sequência convertida por bissulfeto de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, tampões de PCR otimizados e desoxinucleotídeos, e sondas específicas.
[257] Além disso, um aspecto adicional da presente invenção é um kit alternativo que compreende um meio para determinação da metilação de pelo menos um gene ou sequência genômica que está metilada no câncer, mas não metilada em tecido não canceroso, em que os referidos meios compreendem preferivelmente pelo menos uma enzima de restrição metilação-específica; um ou diversos oligonucleotídeos iniciadores (preferivelmente um ou diversos pares de iniciadores) adequados à amplificação de uma sequência que compreende pelo menos um dinucleotídeo CpG de uma sequência selecionada das sequências genômicas; e, opcionalmente, instruções para realização e avaliação do método descrito de análise de metilação. Em uma modalidade, as sequências de base dos referidos oligonucleotídeos são idênticas, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 18 bases de comprimento de uma sequência selecionada das sequências genômicas.
[258] Em uma modalidade adicional, o referido kit pode compreender uma ou diversas sondas de oligonucleotídeo para a análise dos fragmentos do digesto, preferivelmente os referidos oligonucleotídeos são idênticos, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento de pelo menos 16 bases de comprimento de uma sequência selecionada das sequências genômicas.
[259] Em uma modalidade preferida o kit pode compreender reagentes adicionais selecionados do grupo que consiste em: tampão (por exemplo, enzima de restrição, PCR, tampões de armazenamento ou lavagem); reagentes ou kits de recuperação de DNA (por exemplo, precipitação, ultrafiltração, coluna de afinidade) e componentes de recuperação de DNA.
[260] Em uma modalidade alternativa adicional, o kit pode conter, embalados em recipientes separados, uma polimerase e um tampão de reação otimizados para extensão de iniciador mediada pela polimerase, por exemplo, PCR. Em outra modalidade da invenção o kit ainda compreende meios para obtenção e/ou armazenamento de uma amostra biológica do indivíduo. Em uma modalidade preferida, o kit compreende: (a) um reagente de enzima de restrição sensível à metilação; (b) um recipiente adequado à contenção do referido reagente e da amostra biológica do indivíduo; (c) pelo menos um conjunto de oligonucleotídeos, um ou diversos ácidos nucléicos ou ácidos nucléicos peptídicos que são idênticos, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 9 bases de comprimento de uma sequência selecionada das sequências genômicas; e, opcionalmente, (d) instruções para uso e interpretação dos resultados do kit. Em uma modalidade alternativa preferida, o kit compreende: (a) um reagente de enzima de restrição sensível à metilação; (b) um recipiente adequado à contenção do referido reagente e da amostra biológica do indivíduo; (c) pelo menos um conjunto de oligonucleotídeos iniciadores adequado à amplificação de uma sequência que compreende pelo menos um dinucleotídeo CpG de uma sequência selecionada das sequências genômicas; e, opcionalmente, (d) instruções para uso e interpretação dos resultados do kit.
[261] Em uma modalidade alternativa, o kit compreende: (a) um reagente de enzima de restrição sensível à metilação; (b) um recipiente adequado à contenção do referido reagente e da amostra biológica do indivíduo; (c) pelo menos um conjunto de oligonucleotídeos iniciadores adequado à amplificação de uma sequência que compreende pelo menos um dinucleotídeo CpG de uma sequência selecionada das sequências genômicas ; (d) pelo menos um conjunto de oligonucleotídeos um ou diversos ácidos nucléicos ou ácidos nucléicos peptídicos que são idênticos, são complementares ou hibridizam sob condições rigorosas ou altamente rigorosas para um segmento com pelo menos 9 bases de comprimento de uma sequência selecionada das sequências genômicas e opcionalmente (e) instruções para uso e interpretação dos resultados do kit.
[262] O kit também pode conter outros componentes como, por exemplo, tampões ou soluções adequadas ao bloqueio, lavagem ou revestimento, embalados em um recipiente separado.
[263] A invenção ainda está relacionada a um kit para uso na determinação em um indivíduo do prognóstico de um indivíduo que tem câncer por meio de análise de enzima de restrição sensível à metilação. O referido kit compreende um recipiente e um componente de microarranjo de DNA. O referido componente de microarranjo de DNA sendo uma superfície sobre a qual diversos oligonucleotídeos são imobilizados em posições designadas e em que o oligonucleotídeo compreende pelo menos um sítio de metilação de CpG. Pelo menos um dos referidos oligonucleotídeos é específico para pelo menos um gene ou sequência genômica selecionada do grupo que consiste em dos genes e compreende uma sequência de pelo menos 15 pares de bases de comprimento, mas no máximo 200 bp, de uma sequência de acordo com uma das sequências genômicas. De preferência, a referida sequência possui pelo menos 15 pares de bases de comprimento, mas no máximo 80 bp, de uma sequência de acordo com uma das sequências genômicas. É ainda preferido que a referida sequência tenha pelo menos 20 pares de bases de comprimento, mas no máximo 30 bp, de uma sequência de acordo com uma das sequências genômicas. O referido kit de teste preferivelmente ainda compreende um componente de enzima de restrição que compreende uma ou diversas enzimas de restrição sensíveis à metilação.
[264] Em uma modalidade adicional, o referido kit de teste é ainda caracterizado por compreender pelo menos uma enzima de restrição metilação-específica, e em que os oligonucleotídeos compreendem um local de restrição de pelo menos uma das referidas enzimas de restrição metilaçãoespecíficas.
[265] O kit pode ainda compreender um ou vários dos seguintes componentes que são conhecidos na técnica para enriquecimento de DNA: um componente de proteína, as referida proteína se ligando seletivamente ao DNA metilado; um componente de ácido nucléico formador de tríplex, um ou diversos vinculadores, opcionalmente em uma solução adequada; substâncias ou soluções para a realização de uma ligação, por exemplo, ligases, tampões; substâncias ou soluções para a realização de uma cromatografia em coluna; substâncias ou soluções para a realização de um enriquecimento baseado em imunologia (por exemplo, imunoprecipitação); substâncias ou soluções para a realização de uma amplificação de ácido nucléico, por exemplo, PCR; um corante ou vários corantes, se aplicável com um reagente de acoplamento, se aplicável em uma solução; substâncias ou soluções para a realização de uma hibridização; e/ou substâncias ou soluções para a realização de uma etapa de lavagem.
[266] A invenção descrita ainda fornece uma composição de matéria útil para determinação do prognóstico de um indivíduo que tem câncer. A referida composição compreendendo pelo menos um ácido nucléico com 18 pares de bases de comprimento de um segmento da sequência de ácidos nucléicos revelada nas sequências de bissulfeto, e uma ou mais substâncias escolhidas do grupo que compreende: 1-5 mM de cloreto de magnésio, 100-500 μΜ de dNTP, 0,5-5 unidades de Taq polimerase, albumina sérica bovina, um oligômero, em particular um oligômero de oligonucleotídeos ou ácidos nucléicos peptídicos (PNA), o referido oligômero compreendendo, em cada caso, pelo menos uma sequência de base que possui um comprimento de pelo menos 9 nucleotídeos que é complementar ou hibridiza sob condições moderadamente rigorosas ou rigorosas para um DNA genômico pré-tratado de acordo com uma das sequências de bissulfeto e sequências a elas complementares. Prefere-se que a referida composição de matéria compreenda uma solução-tampão adequada à estabilização do referido ácido nucléico em uma solução aquosa e permita reações baseadas em polimerase dentro da referida solução. Tampões adequados são conhecidos na técnica e comercialmente disponíveis.
[267] A presente invenção também está relacionada ao uso de um kit ou um oligonucleotídeo, como definido acima, para determinação do prognóstico de um indivíduo com câncer, determinação do tratamento médico para um indivíduo com câncer, determinação se um tumor de um indivíduo com câncer indica que o tumor é agressivo ou possui potencial metastático ou indica um tempo de sobrevida reduzido para o indivíduo, detecção de uma forma agressiva de câncer em um indivíduo, seleção de um indivíduo com câncer para tratamento de câncer, ou determinação da carga tumoral ou carga de câncer em um indivíduo, compreendendo um indivíduo com câncer.
[268] Em modalidades preferidas adicionais da invenção, o referido pelo menos um ácido nucléico possui pelo menos 50, 100, 150, 200, 250 ou 500 pares de bases de comprimento de um segmento da sequência de ácidos nucléicos revelada nas sequências de bissulfeto.

[269] Os níveis de Septina 9 e RASSF2A metilado como exemplos de genes ou sequências genômicas que estão metiladas em tecido canceroso, mas não metiladas em tecido não canceroso, foram investigados em amostras de plasma combinadas de pacientes com CRC pré- e pós-ressecção cirúrgica.
[270] Os níveis de Septina 9 e RASSF2A foram determinados pelo método de ensaio de tríplex que mede genes de Septina 9, RASSF2A metilado e HB14. Ensaios similares podem ser executados em formato singleplex, duplex, tríplex, quadriplex ou multiplex.
[271] Métodos para medição da metilação ou do estado de metilação de genes e sequências genômicas são conhecidos na técnica. Veja, por exemplo, Patente U.S. Nº 7.229.759, ou Patente Européia Nº EP 1370691, ambas aqui incorporadas por referência a esse ensaio de metilação e métodos de detecção. A metilação ou o estado de metilação de genes e sequências genômicas foi aqui medido. O DNA do plasma foi convertido com bissulfeto e o nível de DNA metilado em posições nas sequências genômicas foi detectado em um ensaio tríplex.
[272] Prateleiras magnéticas Invitrogen (DynaMag-15 e DynaMag-2) foram usadas. A Lavagem A foi preparada por adição de 45 ml de etanol (Merck, A000920; 99,8%) do Concentrado de Lavagem A Epi proColonUS. A Lavagem B foi preparada por adição de 28 ml de etanol (Merck, A000920; 99,8%) ao Concentrado de Lavagem B Epi proColonUS. A um tubo de Falcon de 15 ml rotulado, 3,5 ml de plasma sanguíneo foram combinados com 3,5 ml de tampão de liseligação, misturados por turbilhonamento, e incubados em temperatura ambiente por 10 min. A essas reação de lise, 90 μl de glóbulos magnéticos (Dynabeads MyOne SILANE, recém suspensos por turbilhonamento por 30 segundos) e 2,5 ml de etanol foram adicionados até que o volume total fosse de aproximadamente 10 ml. O tubo foi misturado por sua inversão manual 5-6 vezes e incubado com uma agitadora rotatória (Rotator) por 45 min em temperatura ambiente. A primeira lavagem foi realizada por colocação de tubos de 15 ml em uma prateleira magnética por pelo menos 5 min, e depois o tampão foi descartado e os tubos foram transferidos em uma prateleira não magnética. Mil e quinhentos μl de Lavagem A (como descrito acima, tampão de lise-ligação + etanol f.d. Molekularbiologie) foram adicionados e os glóbulos foram ressuspensos por turbilhonamento por 10 segundos. A suspensão de glóbulos foi transferida em um tubo rotulado de 2 ml com uma pipeta de transferência. Tubos SafeLock de 2 ml apenas para assegurar fechamento seguro durante incubação a 80°C foram usados para centrifugação. A pipeta de transferência foi colocada de volta no tubo de 15 ml por pelo menos 2 min para coletar os glóbulos restantes e eles foram colocados no tubo de 2 ml com a mesma pipeta de transferência. Os tubos de 2 ml foram colocados na prateleira magnética por 2 min, e depois o máximo possível de tampão de lavagem foi retirado por pipeta tomando-se cuidado para não remover os glóbulos. Os tubos foram colocados em uma centrífuga e centrifugados por 10 segundos a 1.000 rcf para coletar glóbulos no fundo, colocados na prateleira magnética por 2 min e remoção de tampão residual.
[273] O DNA do plasma foi eluído por adição de 100 μl de tampão de eluição (10 mM de Tris pH 8,0) a cada tubo e os glóbulos foram ressuspensos por turbilhonamento por 10 segundos, assegurando-se que o pélete foi ressuspenso completamente, e depois incubado a 80°C por 15 min em uma agitadora térmica a 1.000 rpm. Os tubos foram submetidos à centrifugação pulsada para remoção de gotas da tampa, e colocados na prateleira magnética por 2 min. O eluato completo (aproximadamente 100 μl) foi transferido em tubos pré-rotulados de 2,0 ml.
[274] O DNA do plasma foi convertido com bissulfeto por adição dos seguintes reagentes ao eluato: 150 μl de solução de bissulfeto (ABS, solução de bissulfeto de amônio, usar somente tubos não abertos, descartar tubos usados) e 25 μl de tampão de proteção (contém THFA) (5g de Trolox + 40 ml de THFA). Os tubos foram fechados e turbilhonados por 10 segundos para misturar cuidadosamente. Os tubos foram submetidos à centrifugação pulsada para evitar líquido na tampa, e depois colocados em um bloco ou agitadora térmica e incubados por 45 min a 80°C sem agitação. Os tubos foram submetidos à centrifugação pulsada para remover gotas da tampa, e depois os glóbulos foram ressuspensos por turbilhonamento por 10 segundos, assegurando-se que todos os glóbulos estão cuidadosamente suspensos. Os componentes seguintes foram adicionados em cada reação de bissulfeto na ordem: 1.000 μl de Lavagem A e 20 μl de glóbulos magnéticos (Dynabeads MyOne SILANE) até um volume total de 300 µl e misturados cuidadosamente por turbilhonamento, e depois foram incubados na agitadora térmica e agitados a 1.000 rpm por 45 min em temperatura ambiente. Os tubos foram submetidos à centrifugação pulsada para remover gotas da tampa e colocados na prateleira magnética por 2 min para capturar as partículas. A seguir, uma pipeta fresca foi usada para remover o máximo de líquido possível sem tocar nas partículas capturadas. Os tubos foram removidos da prateleira magnética para o procedimento de lavagem. Oitocentos μl de Lavagem A foram adicionados, e os glóbulos foram enxaguados da parede, e depois ressuspensos por turbilhonamento, o tubo submetido à centrifugação pulsada para a remoção de gotas da tampa, e colocado na prateleira magnética por 2 min. Usando uma pipeta fresca, o máximo de líquido possível foi removido, sem tocar as partículas capturadas. Os tubos foram retirados da prateleira magnética para o procedimento de lavagem. Oitocentos μl de Lavagem B foram adicionados, e os glóbulos foram enxaguados da parede, ressuspensos por turbilhonamento e o tubo submetido à centrifugação pulsada para a remoção de gotas da tampa, e depois foram colocados na prateleira magnética por 2 min. Usando uma pipeta fresca, o máximo de líquido possível foi removido, sem tocar as partículas capturadas. Os tubos foram retirados da prateleira magnética para o procedimento de lavagem. Quatrocentos μl de Lavagem B foram adicionados, e os glóbulos foram enxaguados da parede, ressuspensos por turbilhonamento, submetidos à centrifugação pulsada para a remoção de gotas da tampa e colocados na prateleira magnética por 2 min. Usando uma pipeta fresca, o máximo de líquido possível foi removido, sem tocar as partículas capturadas. Os tubos foram brevemente centrifugados para coletar as gotas restantes até o fundo, colocados na prateleira magnética por 2 min, e depois líquido residual foi removido com uma pipeta. Foi permitido que o pélete secasse por 10 min em temperatura ambiente com tubos abertos no magneto.
[275] Os tubos foram transferidos em uma prateleira não magnética e 55 μl de tampão de eluição (10 mM de Tris pH 8,0) foram adicionados a cada tubo. Os glóbulos foram então ressuspensos por turbilhonamento 20 segundos/min, e depois os tubos foram incubados a 80°C por 5 min em uma agitadora térmica a 1.000 rpm, e depois turbilhonados novamente por 10 segundos, brevemente centrifugados para coletar todo o líquido até o fundo. Os tubos foram colocados na prateleira magnética por 2 min, e o eluato completo foi transferido para uma placa de PCR de 96 poços (ou tubos pré-rotulados de 0,5 ml).
[276] As sequências de sondas e iniciadores para o ensaio tríplex realizado são mostradas na Tabela 2.
[277] Ctgcccaccagccatcatgtcggaccccgcggtcaacgcgcagctggat gggatcattt (ID. DE SEQ. Nº: 32)
[278] Ttgtttattagttattatgtcggatttcgcggttaacgcgtagttggat gggattattt (ID. DE SEQ. Nº: 33)
[279] Acttagagctgaatgcaaagtaagcgctcgaaatgcagaagtagccggg gccgcccacggcacctgcctcgctcggggcgagagaagacgccaggctgaggtcccag (ID. DE SEQ. Nº: 34)
[280] Atttagagttgaatgtaaagtaagcgttcgaaatgtagaagtagtcggg gtcgtttacggtatttgtttcgttcggggcgagagaagacgttaggttgaggttttag (ID. DE SEQ. Nº: 35)
[281] Os resultados mostram que pacientes com cânceres em estágio I e II tendem a perder o sinal de Septina 9 e RASF2A após a cirurgia, pacientes com estágio III tendem a reter o sinal de Septina 9 e RASF2A. Os 2 pacientes com CRC em estágio IV, que já possuem doença metastática, “retêm” o sinal de Septina 9 e RASF2A após cirurgia. Isso indica que as metástases podem ser detectadas por Septina 9 e RASF2A até mesmo se o tumor primário foi ressecado. Os resultados são revelados nas Figuras 1-12. As Figuras 14 e 15 exibem os resultados discutidos de Septina 9 e RASSF2A de forma quantitativa. Os valores são indicados em pg de DNA de Septina 9/RASSF2A metilado por ml de plasma.
[282] A detecção de Septina 9 pode ser feita por várias tecnologias da técnica estabelecida que podem detectar a metilação de DNA no sangue/plasma. Uma análise qualitativa, semiquantitativa e/ou quantitativa de mSeptina 9 é possível e está altamente conectada ao uso desejado e à população de pacientes/tumores de interesse.
[283] Determinação da amostra de análise
[284] Para análise qualitativa:
[285] Paciente com tumor CRC em Estágio I.
[286] Sinal de Septina 9 positivo antes da cirurgia
[287] Sinal de Septina 9 negativo após cirurgia = bom prognóstico
[288] Sinal de Septina 9 positivo após cirurgia = prognóstico ruim (risco de metástase)
[289] Bom prognóstico significa, em modalidades preferidas da invenção, que o indivíduo que foi submetido à cirurgia é monitorado, ou seja, um ou mais testes para a recorrência de câncer são repetidos em intervalos de tempo. Em uma modalidade preferida particular, um teste desse tipo é a detecção de DNA de Septina 9 metilado como aqui revelado, e em US 2006-0286576 ou WO 2006/113466.
Sinal de RASSF2A positivo antes de cirurgia
Sinal de RASSF2A negativo após cirurgia = bom prognóstico
Sinal de RASSF2A positivo após cirurgia = prognóstico ruim (risco de metástase)
Bom prognóstico significa, em modalidades preferidas da invenção, que o indivíduo que foi submetido à cirurgia é monitorado, ou seja, um ou mais testes para a recorrência de câncer são repetidos em intervalos de tempo.
Para análise semiquantitativa:
Pacientes com tumor CRC em Estágios I, II e III.
Sinal de Septina 9 positivo antes de cirurgia (1 de 3 medições replicadas) = presença de tumor
Sinal de Septina 9 negativo após cirurgia = bom prognóstico (3 de 3 medições replicadas)
1 de 3 sinal de Septina positivo 9 após cirurgia = risco baixo
2 de 3 sinal de Septina 9 positivo após cirurgia = risco médio
3 de 3 sinal de Septina 9 positivo após cirurgia = alto risco
Pacientes com tumor CRC em Estágios I, II e III.
Sinal de RASSF2A positivo antes de cirurgia (1 de 3 medições replicadas) = presença de tumor
Sinal de RASSF2A negativo após cirurgia = bom prognóstico (3 de 3 medições replicadas)
1 de 3 sinal de RASSF2A positivo após cirurgia = risco baixo
2 de 3 sinal de RASSF2A positivo após cirurgia = risco médio
3 de 3 sinal de RASSF2A positivo após cirurgia = alto risco
Para análise quantitativa:
Pacientes com tumor CRC em Estágios II e III e (IV).
Detecção da magnitude de Septina 9 antes e depois de cirurgia, por exemplo, por utilização de um padrão interno. Pacientes com tumor CRC em Estágios I, II e III.
Septina 9 antes da cirurgia acima de 3 pg/ml de plasma = presença de tumor
Sinal de Septina 9 negativo após cirurgia = bom prognóstico (0 pg/ml Septina 9)
Septina 9 ˃ 0 a 3 pg/ml de plasma após cirurgia = risco baixo
Septina 9 de 3 a 30 pg/ml de plasma após cirurgia = risco médio
Septina 9 acima de 30 pg/ml de plasma após cirurgia = alto risco
Detecção da magnitude de RASSF2A antes e depois de cirurgia, por exemplo, por utilização de um padrão interno. Pacientes com tumor CRC em Estágios I, II e III.
RASSF2A antes da cirurgia acima de 3 pg/ml de plasma = presença de tumor
Sinal de RASSF2A negativo após cirurgia = bom prognóstico (0 pg/ml de RASSF2A no plasma)
RASSF2A após cirurgia -˃ 0 a 3 pg/ml de plasma = risco baixo
RASSF2A de 3 a 30 pg/ml de plasma após cirurgia = risco médio
RASSF2A acima de 30 pg/ml de plasma de RASSF2A após cirurgia = alto risco.
Sinal de RASSF2A positivo antes de cirurgia
Sinal de RASSF2A negativo após cirurgia = bom prognóstico
Sinal de RASSF2A positivo após cirurgia = prognóstico ruim (risco de metástase)
Bom prognóstico significa, em modalidades preferidas da invenção, que o indivíduo que foi submetido à cirurgia é monitorado, ou seja, um ou mais testes para a recorrência de câncer são repetidos em intervalos de tempo.
Para análise semiquantitativa:
Pacientes com tumor CRC em Estágios I, II e III.
Sinal de Septina 9 positivo antes de cirurgia (1 de 3 medições replicadas) = presença de tumor
Sinal de Septina 9 negativo após cirurgia = bom prognóstico (3 de 3 medições replicadas)
1 de 3 sinal de Septina positivo 9 após cirurgia = risco baixo
2 de 3 sinal de Septina 9 positivo após cirurgia = risco médio
3 de 3 sinal de Septina 9 positivo após cirurgia = alto risco
Pacientes com tumor CRC em Estágios I, II e III.
Sinal de RASSF2A positivo antes de cirurgia (1 de 3 medições replicadas) = presença de tumor
Sinal de RASSF2A negativo após cirurgia = bom prognóstico (3 de 3 medições replicadas)
1 de 3 sinal de RASSF2A positivo após cirurgia = risco baixo
2 de 3 sinal de RASSF2A positivo após cirurgia = risco médio
3 de 3 sinal de RASSF2A positivo após cirurgia = alto risco
Para análise quantitativa:
Pacientes com tumor CRC em Estágios II e III e (IV).
Detecção da magnitude de Septina 9 antes e depois de cirurgia, por exemplo, por utilização de um padrão interno. Pacientes com tumor CRC em Estágios I, II e III.
Septina 9 antes da cirurgia acima de 3 pg/ml de plasma = presença de tumor
Sinal de Septina 9 negativo após cirurgia = bom prognóstico (0 pg/ml Septina 9)
Septina 9 ˃ 0 a 3 pg/ml de plasma após cirurgia = risco baixo
Septina 9 de 3 a 30 pg/ml de plasma após cirurgia = risco médio
Septina 9 acima de 30 pg/ml de plasma após cirurgia = alto risco
Detecção da magnitude de RASSF2A antes e depois de cirurgia, por exemplo, por utilização de um padrão interno. Pacientes com tumor CRC em Estágios I, II e III.
RASSF2A antes da cirurgia acima de 3 pg/ml de plasma = presença de tumor
Sinal de RASSF2A negativo após cirurgia = bom prognóstico (0 pg/ml de RASSF2A no plasma)
RASSF2A após cirurgia -˃ 0 a 3 pg/ml de plasma = risco baixo
RASSF2A de 3 a 30 pg/ml de plasma após cirurgia = risco médio
RASSF2A acima de 30 pg/ml de plasma de RASSF2A após cirurgia = alto risco.
Claims (9)
- Método caracterizado pelo fato de ser para:
(i) determinação do prognóstico de um indivíduo com câncer de cólon ou cólon-retal após cirurgia ou remoção por ressecção do tumor primário e antes de um tratamento seguinte a cirurgia ou ressecção do tumor primário,
(ii) determinar a necessidade de tratamento médico adicional para um indivíduo com câncer de cólon ou cólonretal após o tratamento de remoção do tumor primário e antes de um tratamento seguinte a cirurgia ou ressecção do tumor e antes de um tratamento seguinte a cirurgia ou ressecção,
(iii) selecionar um indivíduo com câncer de cólon ou cólon-retal para tratamento adicional de câncer após tratamento de remoção do tumor primário e antes de um tratamento seguinte a cirurgia ou ressecção, ou
(iv) determinar, após o tratamento de remoção do tumor primário e antes de um tratamento seguinte a cirurgia ou ressecção, se o câncer de cólon ou cólon-retal de um paciente com câncer é metastático, compreendendo as etapas de:- a. medição do nível de DNA genômico metilado de SEPTIN9 (ID. DE SEQ. Nº 1) que é metilado no câncer de cólon ou cólon-retal, mas não metilado no tecido não canceroso, ou de um fragmento deste, em uma amostra sanguínea obtida do indivíduo antes da remoção do tumor primário por cirurgia ou ressecção (amostra de prétratamento);
- b. medição do nível de DNA genômico metilado de SEPTIN9 (ID. DE SEQ. Nº 1) que é metilado no câncer de cólon ou cólon-retal, mas não metilado no tecido não canceroso, ou de um fragmento deste, em uma amostra sanguínea obtida do indivíduo após o tumor primário ter sido removido por cirurgia ou ressecção e antes de um tratamento seguinte a cirurgia ou ressecção do tumor primário (amostra pós-tratamento),
(i) um mau prognóstico,
(ii) a necessidade de tratamento adicional contra o câncer para o indivíduo,
(iii) selecionar o indivíduo com câncer para tratamento adicional, ou
(iv) que o câncer é metastático. - Método, de acordo com a reivindicação 1, caracterizado pelo fato de que adicionalmente o nível do DNA genômico metilado de RASSF2A (ID. DE SEQ. Nº 16), é medido, preferencialmente, a metilação do DNA genômico é medida por pelo menos a uma citosina selecionada do grupo que consiste em posições 21, 28, 30, 37 e 39 do ID. DE SEQ. Nº: 32 para SEPTI9 (ID. DE SEQ. Nº 1) e posições 25, 29, 46, 52, 58, 70, 74, 79 e 89 do ID. DE SEQ. Nº: 34 para RASSF2A (ID. DE SEQ. Nº 16).
- Método, de acordo com a reivindicação 1, caracterizado pelo fato de que o estágio do câncer é câncer cólon-retal em Estágio I, Estágio II ou Estágio III.
- Método, de acordo com qualquer uma das reivindicações 1 a 3, caracterizado pelo fato de que a amostra sanguínea é soro ou plasma.
- Método, de acordo com qualquer uma das reivindicações 1 a 4, caracterizado pelo fato de que o DNA genômico metilado ou um fragmento é medido quantitativamente, ou quantitativamente em parte, quantitativamente em parte e qualitativamente em parte ou semiquantitativamente.
- Método, de acordo com qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que o DNA genômico metilado ou um fragmento é medido por um ensaio no qual sondas de bloqueio metilação-específicas que cobrem as posições CpG entre ou cobertas pelos iniciadores de amplificação, permitem a amplificação seletiva metilaçãoespecífica, ou por PCR metilação-específica.
- Método, de acordo com qualquer uma das reivindicações 1 a 6, caracterizado pelo fato de que a medição do DNA genômico metilado ou um fragmento compreende o contato do DNA genômico da amostra sanguínea com pelo menos um reagente, ou série de reagentes, que distingue entre dinucleotídeos CpG metilados e não metilados dentro de pelo menos uma região-alvo do DNA genômico, em que a região-alvo compreende, ou hibridiza sob condições rigorosas para, uma sequência de pelo menos 9, pelo menos 16 ou pelo menos 25 nucleotídeos contíguos do ID. DE SEQ. Nº: 1, em que os referidos nucleotídeos contíguos compreendem pelo menos uma sequência de dinucleotídeo CpG, em que o referido reagente é preferencialmente selecionado do grupo que compreende bissulfeto, sulfeto de hidrogênio, dissulfeto e combinações dos mesmos.
- Método, de acordo com qualquer uma das reivindicações 1 a 7, caracterizado pelo fato de que o nível de DNA genômico metilado é medido para SEPTIN9 (ID. DE SEQ. Nº 1) e opcionalmente para RASSF2a (ID. DE SEQ. Nº 16), e em que o DNA genômico metilado ou fragmento é medido por um ensaio no qual sondas de bloqueio metilaçãoespecíficas que cobrem as posições CpG entre ou cobertas pelos iniciadores de amplificação, permitem a amplificação seletiva metilação-específica, ou por PCR metilaçãoespecífica.
- Método caracterizado pelo fato de ser para:
(i) determinação do prognóstico de um indivíduo com câncer de cólon ou cólon-retal após cirurgia ou remoção por ressecção do tumor primário e antes do tratamento seguinte a remoção do tumor primário,
(ii) determinar a necessidade de tratamento médico adicional para um indivíduo com câncer após remoção do cólon ou cólon-retal do tumor primário e antes de um tratamento seguinte a cirurgia ou ressecção,
(iii) selecionar um indivíduo com câncer de cólon ou cólon-retal para tratamento adicional de câncer após tratamento de remoção do tumor primário e antes de um tratamento seguinte a remoção do tumor primário, ou
(iv) determinar, após cirurgia e ressecção de remoção do tumor primário se o cólon ou cólon-retal de um paciente com câncer é metastático e antes do tratamento seguinte a cirurgia ou ressecção, compreendendo as etapas de:- a. medição do nível da expressão gênica de SEPTIN9 e/ou de seus elementos promotores ou reguladores ou um fragmento destes e opcionalmente do gene de RASSF2A e/ou de seus elementos promotores ou reguladores ou um fragmento destes, em uma amostra sanguínea obtida do indivíduo; antes da remoção do tumor primário por cirugia ou ressecção (amostra pré-tratamento);
- b. medição do nível da expressão gênica de SEPTIN9 e/ou seus elementos promotores ou reguladores ou um fragmento destes e opcionalmente ou do gene de RASSF2A e/ou de seus elementos promotores ou reguladores ou um fragmento destes, em uma amostra sanguínea obtida do indivíduo, após o tumor primário ter sido removido por cirurgia ou ressecção e antes de um tratamento seguinte a cirurgia ou ressecção (amostra pós-tratamento),
(i) um mau prognóstico,
(ii) a necessidade de tratamento adicional contra o câncer para o indivíduo,
(iii) selecionar o indivíduo com câncer para tratamento adicional, ou
(iv) que o câncer é metastático.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161505919P | 2011-07-08 | 2011-07-08 | |
| US61/505,919 | 2011-07-08 | ||
| PCT/EP2012/063436 WO2013007702A1 (en) | 2011-07-08 | 2012-07-09 | Methods and nucleic acids for determining the prognosis of a cancer subject |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BR112014000443A2 BR112014000443A2 (pt) | 2017-02-14 |
| BR112014000443B1 true BR112014000443B1 (pt) | 2021-03-23 |
Family
ID=46458553
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR112014000443-9A BR112014000443B1 (pt) | 2011-07-08 | 2012-07-09 | Métodos para determinação do prognóstico de um indivíduo com câncer |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US10626462B2 (pt) |
| EP (1) | EP2729579B1 (pt) |
| JP (1) | JP6397762B2 (pt) |
| KR (1) | KR102046668B1 (pt) |
| CN (2) | CN108048573A (pt) |
| AU (1) | AU2012282528B2 (pt) |
| BR (1) | BR112014000443B1 (pt) |
| CA (1) | CA2840149C (pt) |
| DK (1) | DK2729579T3 (pt) |
| EA (1) | EA034232B1 (pt) |
| ES (1) | ES2654561T3 (pt) |
| HR (1) | HRP20180001T1 (pt) |
| HU (1) | HUE036036T2 (pt) |
| IL (1) | IL230303B (pt) |
| LT (1) | LT2729579T (pt) |
| MX (1) | MX358117B (pt) |
| NO (1) | NO2729579T3 (pt) |
| SI (1) | SI2729579T1 (pt) |
| WO (1) | WO2013007702A1 (pt) |
| ZA (1) | ZA201400089B (pt) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9732390B2 (en) | 2012-09-20 | 2017-08-15 | The Chinese University Of Hong Kong | Non-invasive determination of methylome of fetus or tumor from plasma |
| US10706957B2 (en) | 2012-09-20 | 2020-07-07 | The Chinese University Of Hong Kong | Non-invasive determination of methylome of tumor from plasma |
| CA2951141C (en) * | 2014-06-04 | 2021-11-30 | Quest Diagnostics Investments Incorporated | Methylated markers for colorectal cancer |
| CN104745575B (zh) | 2014-08-08 | 2019-03-12 | 博诚研究中心 | 用于检测细胞增殖性异常或疾病程度分级的基因组合物及其用途 |
| CN115181802A (zh) * | 2015-04-23 | 2022-10-14 | 博尔诚(北京)科技有限公司 | 一种检测Septin9基因甲基化的试剂盒及其应用 |
| WO2017009322A1 (en) * | 2015-07-15 | 2017-01-19 | Universiteit Gent | Probes and a methylation in situ hybridization assay |
| JP6897970B2 (ja) * | 2015-09-08 | 2021-07-07 | 国立大学法人山口大学 | 大腸腫瘍の有無を検査する方法 |
| CN109804086B (zh) | 2016-08-10 | 2023-06-13 | 格里尔公司 | 制备双标签dna库用于亚硫酸盐转化定序的方法 |
| US11965157B2 (en) | 2017-04-19 | 2024-04-23 | Singlera Genomics, Inc. | Compositions and methods for library construction and sequence analysis |
| CN107385056A (zh) * | 2017-08-10 | 2017-11-24 | 上海时代基因医学科技有限公司 | 一种检测SEPT9基因不同Cpg岛甲基化的试剂盒及应用 |
| WO2019076949A1 (en) * | 2017-10-18 | 2019-04-25 | Deutsches Krebsforschungszentrum | MEANS AND METHODS FOR CLASSIFYING COLORECTAL CANCERS |
| US20200370126A1 (en) * | 2017-11-29 | 2020-11-26 | The Regents Of The University Of Michigan | Compositions and methods for characterizing cancer |
| CN108796080B (zh) * | 2018-06-06 | 2022-06-07 | 苏州唯善生物科技有限公司 | 用于结直肠癌诊断、检测或筛查的引物和探针组 |
| CN109055552A (zh) * | 2018-08-23 | 2018-12-21 | 北京迈基诺基因科技股份有限公司 | 检测Septin9基因启动子是否甲基化的方法及其专用成套试剂 |
| CN112195243A (zh) * | 2020-09-22 | 2021-01-08 | 北京华大吉比爱生物技术有限公司 | 一种检测多基因甲基化的试剂盒及其应用 |
| KR102404750B1 (ko) * | 2021-10-14 | 2022-06-07 | 주식회사 엔도믹스 | 세포 유리 dna를 이용한 대장암 진단을 위한 메틸화 마커 유전자 및 이의 용도 |
| CN114561452A (zh) * | 2022-03-10 | 2022-05-31 | 天津市人民医院 | mSEPT9作为预测结直肠癌患者术后复发风险标志物及用于评价化疗方案有效性的应用 |
| KR102559496B1 (ko) * | 2022-04-26 | 2023-07-25 | 연세대학교 산학협력단 | 핵산의 메틸화 여부 확인용 조성물 및 핵산의 메틸화 여부를 확인하는 방법 |
| CN115029462B (zh) * | 2022-08-10 | 2022-11-08 | 中国农业科学院农业质量标准与检测技术研究所 | 一种动植物疫病的核酸标准物质、形貌模拟方法及其应用 |
| WO2024064369A1 (en) * | 2022-09-23 | 2024-03-28 | Flagship Pioneering Innovations Vi, Llc | Methods for amplifying nucleic acids |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US150000A (en) | 1874-04-21 | Improvement in cigar mouth - pieces | ||
| US412900A (en) | 1889-10-15 | roaers | ||
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US5457183A (en) | 1989-03-06 | 1995-10-10 | Board Of Regents, The University Of Texas System | Hydroxylated texaphyrins |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| SE501439C2 (sv) | 1993-06-22 | 1995-02-13 | Pharmacia Lkb Biotech | Sätt och anordning för analys av polynukleotidsekvenser |
| US6184211B1 (en) | 1993-11-30 | 2001-02-06 | Methylgene Inc. | Inhibition of DNA methyltransferase |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5514758A (en) | 1994-09-30 | 1996-05-07 | The Goodyear Tire & Rubber Company | Process for making latex for high performance masking tape |
| US5786146A (en) | 1996-06-03 | 1998-07-28 | The Johns Hopkins University School Of Medicine | Method of detection of methylated nucleic acid using agents which modify unmethylated cytosine and distinguishing modified methylated and non-methylated nucleic acids |
| US6017704A (en) | 1996-06-03 | 2000-01-25 | The Johns Hopkins University School Of Medicine | Method of detection of methylated nucleic acid using agents which modify unmethylated cytosine and distinguishing modified methylated and non-methylated nucleic acids |
| US6251594B1 (en) | 1997-06-09 | 2001-06-26 | Usc/Norris Comprehensive Cancer Ctr. | Cancer diagnostic method based upon DNA methylation differences |
| DE19754482A1 (de) | 1997-11-27 | 1999-07-01 | Epigenomics Gmbh | Verfahren zur Herstellung komplexer DNA-Methylierungs-Fingerabdrücke |
| US7700324B1 (en) | 1998-11-03 | 2010-04-20 | The Johns Hopkins University School Of Medicine | Methylated CpG island amplification (MCA) |
| US5958773A (en) | 1998-12-17 | 1999-09-28 | Isis Pharmaceuticals Inc. | Antisense modulation of AKT-1 expression |
| US6331393B1 (en) | 1999-05-14 | 2001-12-18 | University Of Southern California | Process for high-throughput DNA methylation analysis |
| CN1150317C (zh) * | 1999-08-10 | 2004-05-19 | 中国医学科学院肿瘤医院肿瘤研究所 | 一种食管癌相关新基因 |
| AU2002224328A1 (en) * | 2000-09-20 | 2002-04-02 | Case Western Reserve University | Methods and compositions for detecting cancers associated with methylation of hmlh1promoter dna |
| DE10112515B4 (de) | 2001-03-09 | 2004-02-12 | Epigenomics Ag | Verfahren zum Nachweis von Cytosin-Methylierungsmustern mit hoher Sensitivität |
| US7473767B2 (en) | 2001-07-03 | 2009-01-06 | The Institute For Systems Biology | Methods for detection and quantification of analytes in complex mixtures |
| CN1187456C (zh) * | 2001-11-27 | 2005-02-02 | 暨南大学 | CpG岛甲基化检测试剂盒及其应用 |
| WO2005059172A2 (en) | 2003-12-11 | 2005-06-30 | Epigenomics Ag | Method and nucleic acids for the improved treatment of breast cell proliferative disorders |
| DE10348407A1 (de) * | 2003-10-17 | 2005-05-19 | Widschwendter, Martin, Prof. | Prognostische und diagnostische Marker für Zell-proliferative Erkrankungen von Brustgeweben |
| JP4765058B2 (ja) | 2004-12-13 | 2011-09-07 | 国立大学法人 岡山大学 | 核酸修飾前の検体の前処理方法 |
| SG183708A1 (en) * | 2005-04-15 | 2012-09-27 | Epigenomics Ag | Methods and nucleic acids for analyses of cellular proliferative disorders |
| WO2007085497A2 (en) | 2006-01-30 | 2007-08-02 | Epigenomics Ag | Markers for the prediction of outcome of anthracycline treatment |
| WO2007118704A2 (en) * | 2006-04-17 | 2007-10-25 | Epigenomics Ag | Methods and nucleic acids for the detection of colorectal cell proliferative disorders |
| US9939441B2 (en) * | 2006-07-21 | 2018-04-10 | Epigenomics Ag | Methods and nucleic acids for analyses for cellular proliferative disorders |
| WO2008100913A2 (en) * | 2007-02-12 | 2008-08-21 | The Johns Hopkins University | Early detection and prognosis of colon cancers |
| JP5378687B2 (ja) | 2007-03-02 | 2013-12-25 | エフ.ホフマン−ラ ロシュ アーゲー | Basp1遺伝子および/またはsrd5a2遺伝子中のメチル化シトシンを利用する、肝臓癌、肝臓癌発症リスク、肝臓癌再発リスク、肝臓癌悪性度および肝臓癌の経時的進展の検出方法 |
| US20110177511A1 (en) | 2008-09-19 | 2011-07-21 | Noriaki Yamamoto | Method for determining breast cancer metastasis and method for evaluating serum |
| WO2010065916A1 (en) | 2008-12-04 | 2010-06-10 | Rush University Medical Center | Dna methylation based test for monitoring efficacy of treatment |
| EP2309005B1 (en) | 2009-08-03 | 2015-03-04 | Epigenomics AG | Methods for preservation of genomic DNA sequence complexity |
-
2012
- 2012-07-09 MX MX2014000116A patent/MX358117B/es active IP Right Grant
- 2012-07-09 CN CN201810020458.1A patent/CN108048573A/zh active Pending
- 2012-07-09 NO NO12732686A patent/NO2729579T3/no unknown
- 2012-07-09 US US14/131,445 patent/US10626462B2/en active Active
- 2012-07-09 ES ES12732686.6T patent/ES2654561T3/es active Active
- 2012-07-09 BR BR112014000443-9A patent/BR112014000443B1/pt active IP Right Grant
- 2012-07-09 KR KR1020147003119A patent/KR102046668B1/ko active IP Right Grant
- 2012-07-09 EP EP12732686.6A patent/EP2729579B1/en active Active
- 2012-07-09 CN CN201280033813.XA patent/CN103732759A/zh active Pending
- 2012-07-09 HU HUE12732686A patent/HUE036036T2/hu unknown
- 2012-07-09 CA CA2840149A patent/CA2840149C/en active Active
- 2012-07-09 SI SI201231155T patent/SI2729579T1/en unknown
- 2012-07-09 AU AU2012282528A patent/AU2012282528B2/en active Active
- 2012-07-09 JP JP2014517844A patent/JP6397762B2/ja active Active
- 2012-07-09 EA EA201400117A patent/EA034232B1/ru not_active IP Right Cessation
- 2012-07-09 WO PCT/EP2012/063436 patent/WO2013007702A1/en active Application Filing
- 2012-07-09 LT LTEP12732686.6T patent/LT2729579T/lt unknown
- 2012-07-09 DK DK12732686.6T patent/DK2729579T3/en active
-
2014
- 2014-01-02 IL IL230303A patent/IL230303B/en active IP Right Grant
- 2014-01-06 ZA ZA2014/00089A patent/ZA201400089B/en unknown
-
2018
- 2018-01-02 HR HRP20180001TT patent/HRP20180001T1/hr unknown
-
2020
- 2020-03-10 US US16/814,321 patent/US11261499B2/en active Active
-
2022
- 2022-01-18 US US17/577,836 patent/US20220243277A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US20220243277A1 (en) | 2022-08-04 |
| CN103732759A (zh) | 2014-04-16 |
| LT2729579T (lt) | 2018-02-26 |
| HUE036036T2 (hu) | 2018-06-28 |
| MX2014000116A (es) | 2014-03-27 |
| BR112014000443A2 (pt) | 2017-02-14 |
| MX358117B (es) | 2018-08-06 |
| IL230303B (en) | 2018-05-31 |
| ES2654561T3 (es) | 2018-02-14 |
| CA2840149C (en) | 2021-10-26 |
| US20150031021A1 (en) | 2015-01-29 |
| HRP20180001T1 (hr) | 2018-03-09 |
| EA034232B1 (ru) | 2020-01-20 |
| SI2729579T1 (en) | 2018-07-31 |
| EA201400117A1 (ru) | 2014-06-30 |
| JP2014520520A (ja) | 2014-08-25 |
| EP2729579A1 (en) | 2014-05-14 |
| KR102046668B1 (ko) | 2019-11-19 |
| NO2729579T3 (pt) | 2018-03-03 |
| WO2013007702A1 (en) | 2013-01-17 |
| EP2729579B1 (en) | 2017-10-04 |
| JP6397762B2 (ja) | 2018-09-26 |
| US11261499B2 (en) | 2022-03-01 |
| US20200308656A1 (en) | 2020-10-01 |
| DK2729579T3 (en) | 2018-01-08 |
| KR20140044385A (ko) | 2014-04-14 |
| CA2840149A1 (en) | 2013-01-17 |
| US10626462B2 (en) | 2020-04-21 |
| AU2012282528A1 (en) | 2014-01-23 |
| CN108048573A (zh) | 2018-05-18 |
| AU2012282528B2 (en) | 2017-04-13 |
| ZA201400089B (en) | 2022-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11261499B2 (en) | Methods and nucleic acids for determining the prognosis of a cancer subject | |
| JP5721950B2 (ja) | 選択された遺伝子のエピジェネティックな変化及び癌 | |
| ES2486190T3 (es) | Métodos y ácidos nucleicos para análisis de trastornos proliferativos celulares | |
| BRPI0607508B1 (pt) | método para detecção e/ou classificação de distúrbios proliferativos celulares colorretais ou hepatocelulares, e uso do mesmo | |
| ES2587068T3 (es) | Métodos y ácidos nucleicos para la detección de desórdenes celulares proliferativos colorrectales | |
| JP2008522590A (ja) | 前立腺細胞増殖障害の予後に関連する遺伝子発現分析のための方法および核酸 | |
| US9939441B2 (en) | Methods and nucleic acids for analyses for cellular proliferative disorders | |
| US9850532B2 (en) | Methods and nucleic acids for the analysis of gene expression associated with the development of prostate cell proliferative disorders | |
| EP2634264B1 (en) | Methods and nucleic acids related to the gene GLI3 for analyses of cellular proliferative disorders | |
| US20170067119A1 (en) | Methods and nucleic acids for the analysis of gene expression associated with the development of prostate cell proliferative disorders | |
| WO2009092597A2 (en) | Methods and nucleic acids for analyses of prostate cancer | |
| US20110104695A1 (en) | Methods of predicting therapeutic efficacy of cancer therapy | |
| US20060024684A1 (en) | Prognostic markers for prediction of treatment response and/or survival of breast cell proliferative disorder patients | |
| EP1561821A2 (en) | Prognostic markers for prediction of treatment response and/or survival of breast cell proliferative disorder patients | |
| WO2007047699A1 (en) | Method and nucleic acids for the improved treatment of breast cancers | |
| WO2007137873A1 (en) | Method and nucleic acids for the improved treatment of breast cancers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07G | Grant request does not fulfill article 229-c lpi (prior consent of anvisa) [chapter 7.7 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 09/07/2012, OBSERVADAS AS CONDICOES LEGAIS. |