WO2023140165A1 - 多価アルコール類の製造方法 - Google Patents
多価アルコール類の製造方法 Download PDFInfo
- Publication number
- WO2023140165A1 WO2023140165A1 PCT/JP2023/000562 JP2023000562W WO2023140165A1 WO 2023140165 A1 WO2023140165 A1 WO 2023140165A1 JP 2023000562 W JP2023000562 W JP 2023000562W WO 2023140165 A1 WO2023140165 A1 WO 2023140165A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- producing
- reaction
- crude reaction
- mass
- polyhydric alcohols
- Prior art date
Links
- 150000005846 sugar alcohols Polymers 0.000 title claims abstract description 65
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 46
- 239000003054 catalyst Substances 0.000 claims abstract description 53
- 239000012295 chemical reaction liquid Substances 0.000 claims abstract description 52
- 238000004821 distillation Methods 0.000 claims abstract description 50
- 229910052751 metal Inorganic materials 0.000 claims abstract description 47
- 239000002184 metal Substances 0.000 claims abstract description 38
- 238000006722 reduction reaction Methods 0.000 claims abstract description 29
- 239000001257 hydrogen Substances 0.000 claims abstract description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 18
- 125000002723 alicyclic group Chemical group 0.000 claims abstract description 12
- 239000007858 starting material Substances 0.000 claims abstract description 11
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 94
- 238000005984 hydrogenation reaction Methods 0.000 claims description 35
- 238000000746 purification Methods 0.000 claims description 24
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical compound C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 claims description 21
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 19
- 238000009835 boiling Methods 0.000 claims description 14
- 239000010948 rhodium Substances 0.000 claims description 14
- 229910052703 rhodium Inorganic materials 0.000 claims description 13
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 claims description 13
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 11
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 10
- 150000001298 alcohols Chemical class 0.000 claims description 10
- FGUUNXDRMVKHCF-UHFFFAOYSA-N bis(hydroxymethyl)tricyclo[5.2.1.0(2,6)]decane Chemical compound C12CCCC2(CO)C2(CO)CC1CC2 FGUUNXDRMVKHCF-UHFFFAOYSA-N 0.000 claims description 10
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 claims description 8
- 229910052707 ruthenium Inorganic materials 0.000 claims description 7
- 239000003463 adsorbent Substances 0.000 claims description 5
- 229910052759 nickel Inorganic materials 0.000 claims description 5
- 229910052763 palladium Inorganic materials 0.000 claims description 5
- 230000000737 periodic effect Effects 0.000 claims description 3
- 229910052723 transition metal Inorganic materials 0.000 claims description 3
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 claims 2
- 238000007670 refining Methods 0.000 abstract description 5
- 125000004435 hydrogen atom Chemical class [H]* 0.000 abstract description 2
- 229920001744 Polyaldehyde Polymers 0.000 abstract 1
- 239000007788 liquid Substances 0.000 description 45
- 238000007037 hydroformylation reaction Methods 0.000 description 44
- 238000006243 chemical reaction Methods 0.000 description 40
- 238000000605 extraction Methods 0.000 description 38
- 239000002904 solvent Substances 0.000 description 33
- 150000001299 aldehydes Chemical class 0.000 description 27
- 239000007795 chemical reaction product Substances 0.000 description 27
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 238000000034 method Methods 0.000 description 24
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 24
- 239000000243 solution Substances 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 239000007810 chemical reaction solvent Substances 0.000 description 14
- 239000000047 product Substances 0.000 description 13
- 239000007789 gas Substances 0.000 description 12
- 238000010438 heat treatment Methods 0.000 description 12
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- -1 alicyclic diols Chemical class 0.000 description 10
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 9
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 9
- 229910002091 carbon monoxide Inorganic materials 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- 150000002903 organophosphorus compounds Chemical class 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- OTLDLKLSNZMTTA-UHFFFAOYSA-N octahydro-1h-4,7-methanoindene-1,5-diyldimethanol Chemical compound C1C2C3C(CO)CCC3C1C(CO)C2 OTLDLKLSNZMTTA-UHFFFAOYSA-N 0.000 description 8
- 238000005979 thermal decomposition reaction Methods 0.000 description 8
- 238000004817 gas chromatography Methods 0.000 description 7
- 239000002994 raw material Substances 0.000 description 7
- 239000002253 acid Substances 0.000 description 6
- 239000000654 additive Substances 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 5
- 230000000052 comparative effect Effects 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 125000000217 alkyl group Chemical group 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- GGRQQHADVSXBQN-FGSKAQBVSA-N carbon monoxide;(z)-4-hydroxypent-3-en-2-one;rhodium Chemical compound [Rh].[O+]#[C-].[O+]#[C-].C\C(O)=C\C(C)=O GGRQQHADVSXBQN-FGSKAQBVSA-N 0.000 description 4
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- 150000003003 phosphines Chemical class 0.000 description 4
- AQSJGOWTSHOLKH-UHFFFAOYSA-N phosphite(3-) Chemical class [O-]P([O-])[O-] AQSJGOWTSHOLKH-UHFFFAOYSA-N 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 230000002194 synthesizing effect Effects 0.000 description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- JKIJEFPNVSHHEI-UHFFFAOYSA-N Phenol, 2,4-bis(1,1-dimethylethyl)-, phosphite (3:1) Chemical compound CC(C)(C)C1=CC(C(C)(C)C)=CC=C1OP(OC=1C(=CC(=CC=1)C(C)(C)C)C(C)(C)C)OC1=CC=C(C(C)(C)C)C=C1C(C)(C)C JKIJEFPNVSHHEI-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 239000010941 cobalt Substances 0.000 description 3
- 229910017052 cobalt Inorganic materials 0.000 description 3
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 150000003284 rhodium compounds Chemical class 0.000 description 3
- 238000001179 sorption measurement Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 150000003464 sulfur compounds Chemical class 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- GWHJZXXIDMPWGX-UHFFFAOYSA-N 1,2,4-trimethylbenzene Chemical compound CC1=CC=C(C)C(C)=C1 GWHJZXXIDMPWGX-UHFFFAOYSA-N 0.000 description 2
- KVNYFPKFSJIPBJ-UHFFFAOYSA-N 1,2-diethylbenzene Chemical compound CCC1=CC=CC=C1CC KVNYFPKFSJIPBJ-UHFFFAOYSA-N 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical class OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 2
- QAFOOMOQTYFCFJ-UHFFFAOYSA-N [P]C=O Chemical compound [P]C=O QAFOOMOQTYFCFJ-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000001342 alkaline earth metals Chemical class 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 238000010923 batch production Methods 0.000 description 2
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 2
- 238000010924 continuous production Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 238000005260 corrosion Methods 0.000 description 2
- 230000007797 corrosion Effects 0.000 description 2
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical compound CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- MRELNEQAGSRDBK-UHFFFAOYSA-N lanthanum(3+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[La+3].[La+3] MRELNEQAGSRDBK-UHFFFAOYSA-N 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 150000003138 primary alcohols Chemical class 0.000 description 2
- 238000004904 shortening Methods 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- VIDOPANCAUPXNH-UHFFFAOYSA-N 1,2,3-triethylbenzene Chemical compound CCC1=CC=CC(CC)=C1CC VIDOPANCAUPXNH-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- UNEATYXSUBPPKP-UHFFFAOYSA-N 1,3-Diisopropylbenzene Chemical compound CC(C)C1=CC=CC(C(C)C)=C1 UNEATYXSUBPPKP-UHFFFAOYSA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- SPPWGCYEYAMHDT-UHFFFAOYSA-N 1,4-di(propan-2-yl)benzene Chemical compound CC(C)C1=CC=C(C(C)C)C=C1 SPPWGCYEYAMHDT-UHFFFAOYSA-N 0.000 description 1
- RJWLXGOSIRVRAR-UHFFFAOYSA-N 2,4-dimethylbenzene-1,3-diol Chemical compound CC1=CC=C(O)C(C)=C1O RJWLXGOSIRVRAR-UHFFFAOYSA-N 0.000 description 1
- RGUZWBOJHNWZOK-UHFFFAOYSA-N 3,6-dimethylbenzene-1,2-diol Chemical compound CC1=CC=C(C)C(O)=C1O RGUZWBOJHNWZOK-UHFFFAOYSA-N 0.000 description 1
- VVMPMRQKEVGPQB-UHFFFAOYSA-N CC(C)(C)C1=C(C(C(C)(C)P(O)(O)O)C2=C(C(C)(C)C)C=CC=C2)C=CC=C1 Chemical compound CC(C)(C)C1=C(C(C(C)(C)P(O)(O)O)C2=C(C(C)(C)C)C=CC=C2)C=CC=C1 VVMPMRQKEVGPQB-UHFFFAOYSA-N 0.000 description 1
- ODINCKMPIJJUCX-UHFFFAOYSA-N Calcium oxide Chemical compound [Ca]=O ODINCKMPIJJUCX-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 229910019603 Rh2O3 Inorganic materials 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical class CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- LUSFFPXRDZKBMF-UHFFFAOYSA-N [3-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1CCCC(CO)C1 LUSFFPXRDZKBMF-UHFFFAOYSA-N 0.000 description 1
- YIMQCDZDWXUDCA-UHFFFAOYSA-N [4-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1CCC(CO)CC1 YIMQCDZDWXUDCA-UHFFFAOYSA-N 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001341 alkaline earth metal compounds Chemical class 0.000 description 1
- 150000004996 alkyl benzenes Chemical class 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- IZALUMVGBVKPJD-UHFFFAOYSA-N benzene-1,3-dicarbaldehyde Chemical compound O=CC1=CC=CC(C=O)=C1 IZALUMVGBVKPJD-UHFFFAOYSA-N 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- GPMUMMNTAZMBEC-UHFFFAOYSA-N bis(oxomethylidene)rhodium Chemical compound [Rh].[O+]#[C-].[O+]#[C-] GPMUMMNTAZMBEC-UHFFFAOYSA-N 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- BMRWNKZVCUKKSR-UHFFFAOYSA-N butane-1,2-diol Chemical compound CCC(O)CO BMRWNKZVCUKKSR-UHFFFAOYSA-N 0.000 description 1
- OWBTYPJTUOEWEK-UHFFFAOYSA-N butane-2,3-diol Chemical compound CC(O)C(C)O OWBTYPJTUOEWEK-UHFFFAOYSA-N 0.000 description 1
- 239000000292 calcium oxide Substances 0.000 description 1
- 235000012255 calcium oxide Nutrition 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012018 catalyst precursor Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 239000007809 chemical reaction catalyst Substances 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229910052570 clay Inorganic materials 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- DDTBPAQBQHZRDW-UHFFFAOYSA-N cyclododecane Chemical compound C1CCCCCCCCCCC1 DDTBPAQBQHZRDW-UHFFFAOYSA-N 0.000 description 1
- WHKHKMGAZGBKCK-UHFFFAOYSA-N cyclohexane-1,3-dicarbaldehyde Chemical compound O=CC1CCCC(C=O)C1 WHKHKMGAZGBKCK-UHFFFAOYSA-N 0.000 description 1
- QWKLKVRIQGSSKF-UHFFFAOYSA-N cyclohexane-1,4-dicarbaldehyde Chemical compound O=CC1CCC(C=O)CC1 QWKLKVRIQGSSKF-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- WJTCGQSWYFHTAC-UHFFFAOYSA-N cyclooctane Chemical compound C1CCCCCCC1 WJTCGQSWYFHTAC-UHFFFAOYSA-N 0.000 description 1
- 239000004914 cyclooctane Substances 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- HXEQSCUBDIKNLN-UHFFFAOYSA-N ditert-butyl ethanediperoxoate Chemical compound CC(C)(C)OOC(=O)C(=O)OOC(C)(C)C HXEQSCUBDIKNLN-UHFFFAOYSA-N 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000006266 etherification reaction Methods 0.000 description 1
- 150000005194 ethylbenzenes Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- ACCCMOQWYVYDOT-UHFFFAOYSA-N hexane-1,1-diol Chemical class CCCCCC(O)O ACCCMOQWYVYDOT-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 150000005172 methylbenzenes Chemical class 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical class OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 1
- 229910000484 niobium oxide Inorganic materials 0.000 description 1
- URLJKFSTXLNXLG-UHFFFAOYSA-N niobium(5+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Nb+5].[Nb+5] URLJKFSTXLNXLG-UHFFFAOYSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical class OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 1
- UWJJYHHHVWZFEP-UHFFFAOYSA-N pentane-1,1-diol Chemical class CCCCC(O)O UWJJYHHHVWZFEP-UHFFFAOYSA-N 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000011027 product recovery Methods 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- ULWHHBHJGPPBCO-UHFFFAOYSA-N propane-1,1-diol Chemical compound CCC(O)O ULWHHBHJGPPBCO-UHFFFAOYSA-N 0.000 description 1
- ODLMAHJVESYWTB-UHFFFAOYSA-N propylbenzene Chemical class CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- KUCOHFSKRZZVRO-UHFFFAOYSA-N terephthalaldehyde Chemical compound O=CC1=CC=C(C=O)C=C1 KUCOHFSKRZZVRO-UHFFFAOYSA-N 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- NBHHVFNVXFCMSA-UHFFFAOYSA-N tri(cycloheptyl)phosphane Chemical compound C1CCCCCC1P(C1CCCCCC1)C1CCCCCC1 NBHHVFNVXFCMSA-UHFFFAOYSA-N 0.000 description 1
- MPULMSAVZAPUQG-UHFFFAOYSA-N tri(cyclooctyl)phosphane Chemical compound C1CCCCCCC1P(C1CCCCCCC1)C1CCCCCCC1 MPULMSAVZAPUQG-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- DHWBYAACHDUFAT-UHFFFAOYSA-N tricyclopentylphosphane Chemical compound C1CCCC1P(C1CCCC1)C1CCCC1 DHWBYAACHDUFAT-UHFFFAOYSA-N 0.000 description 1
- UPWXXWXICHRHEN-UHFFFAOYSA-N tricyclopropylphosphane Chemical compound C1CC1P(C1CC1)C1CC1 UPWXXWXICHRHEN-UHFFFAOYSA-N 0.000 description 1
- QJPBQMYVNLSGJS-UHFFFAOYSA-N tris(2-tert-butyl-5-methoxyphenyl) phosphite Chemical compound COC1=CC=C(C(C)(C)C)C(OP(OC=2C(=CC=C(OC)C=2)C(C)(C)C)OC=2C(=CC=C(OC)C=2)C(C)(C)C)=C1 QJPBQMYVNLSGJS-UHFFFAOYSA-N 0.000 description 1
- BMHHAKYFQUVKJF-UHFFFAOYSA-N tris(2-tert-butyl-5-methylphenyl) phosphite Chemical compound CC1=CC=C(C(C)(C)C)C(OP(OC=2C(=CC=C(C)C=2)C(C)(C)C)OC=2C(=CC=C(C)C=2)C(C)(C)C)=C1 BMHHAKYFQUVKJF-UHFFFAOYSA-N 0.000 description 1
- SZPHBONKPMLMCA-UHFFFAOYSA-N tris(2-tert-butylphenyl) phosphite Chemical compound CC(C)(C)C1=CC=CC=C1OP(OC=1C(=CC=CC=1)C(C)(C)C)OC1=CC=CC=C1C(C)(C)C SZPHBONKPMLMCA-UHFFFAOYSA-N 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/49—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reaction with carbon monoxide
- C07C45/50—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reaction with carbon monoxide by oxo-reactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/132—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group
- C07C29/136—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH
- C07C29/14—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of a —CHO group
- C07C29/141—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of a —CHO group with hydrogen or hydrogen-containing gases
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/74—Separation; Purification; Use of additives, e.g. for stabilisation
- C07C29/76—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/74—Separation; Purification; Use of additives, e.g. for stabilisation
- C07C29/76—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment
- C07C29/80—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment by distillation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C35/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C35/22—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system
- C07C35/37—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system with a hydroxy group on a condensed system having three rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/56—Ring systems containing bridged rings
- C07C2603/58—Ring systems containing bridged rings containing three rings
- C07C2603/60—Ring systems containing bridged rings containing three rings containing at least one ring with less than six members
- C07C2603/66—Ring systems containing bridged rings containing three rings containing at least one ring with less than six members containing five-membered rings
Definitions
- the present invention relates to a method for producing polyhydric alcohols, which comprises distilling and purifying a crude reaction liquid obtained by subjecting a polyhydric aldehyde having an alicyclic structure as a starting material to a reduction reaction.
- a method of producing polyhydric alcohols by conducting a reduction reaction in the presence of a hydrogenation catalyst and hydrogen using a polyhydric aldehyde as a starting material is known.
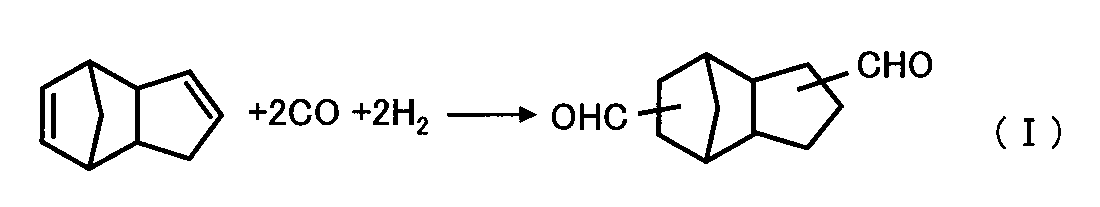
- a method of synthesizing tricyclodecanedimethanol as a polyhydric alcohol by hydrogenating (hydrogenating) dicyclopentadiene as a polyhydric alcohol after hydroformylating it to obtain tricyclodecane dicarbaldehyde as a polyhydric aldehyde Patent Document 1.
- Distillation is used industrially as a method for purifying the target high-boiling-point alcohol from crude products containing high-boiling-point alcohols as polyhydric alcohols.
- the number of stages in the distillation column is increased in order to separate and recover high-purity polyhydric alcohols from the crude product, and the bottom temperature of the distillation column must be increased.
- the polyhydric alcohol is thermally decomposed, resulting in a decrease in the product recovery rate or contamination of the product with thermally decomposed products.
- Patent Document 1 discloses a technique of adding a sulfur compound to the crude product to suppress the thermal decomposition of the high boiling point alcohol.
- Patent Document 2 discloses a technique of adding an alkaline earth metal compound to the crude product to make the aldehyde condensate heavy and suppressing distillation in order to separate the aldehyde condensate and the high boiling point alcohol, which have similar boiling points, thereby shortening the heating time and suppressing the thermal decomposition of the high boiling point alcohol.
- Patent Document 3 discloses a technique in which, in order to separate aldehyde condensates and high boiling point alcohols having similar boiling points, an acid is added to the crude product to make the aldehyde condensates heavy to suppress distillation, thereby shortening the heating time and suppressing the thermal decomposition of the high boiling point alcohols.
- Patent Documents 4 and 5 disclose techniques for suppressing the decomposition of high-boiling-point alcohols by filtering the hydrogenation catalyst in the crude reaction liquid using a filter.
- Patent Document 3 With the technology disclosed in Patent Document 3, there is a risk that the added acid will corrode the reactor and process piping.
- additives such as sulfur compounds, alkaline earth metals, acids, etc. may be mixed into high-boiling-point alcohol products after distillation to contaminate the products. Furthermore, the use of the additive requires equipment for supplying the additive and, in some cases, equipment for removing the additive, which poses problems of complicating the manufacturing process and increasing the manufacturing cost.
- the purpose of the present invention is to solve these problems.
- An object of the present invention is to provide a method for producing polyhydric alcohols that can separate and recover high-purity polyhydric alcohols at a high yield by suppressing thermal decomposition of the polyhydric alcohols when distilling and refining the crude reaction liquid obtained by performing a reduction reaction using polyhydric aldehydes having an alicyclic structure as starting materials.
- the present inventor found that the above problem can be solved by setting the content of the metal element contained in the crude reaction liquid before distillation to a predetermined value or less.
- the gist of the present invention is as follows.
- a method for producing a polyhydric alcohol comprising distilling and purifying a crude reaction liquid obtained by subjecting a polyhydric aldehyde having an alicyclic structure as a starting material to a reduction reaction, A method for producing polyhydric alcohols, wherein the distillation purification is performed after the content of the metal element in the crude reaction liquid is adjusted to 30 ppm by mass or less.
- the present invention it is possible to suppress thermal decomposition of polyhydric alcohols and to separate and recover high-purity polyhydric alcohols at a high yield when distilling and refining a crude reaction liquid obtained by performing a reduction reaction using polyhydric aldehydes having an alicyclic structure as a starting material. Furthermore, according to the present invention, it is possible to perform distillation purification without using additives such as sulfur compounds, alkaline earth metals, acids, etc. disclosed in Patent Documents 1 to 3, so that the product polyhydric alcohols can be prevented from being mixed with additives, corrosion of manufacturing equipment, and an increase in manufacturing costs.
- additives such as sulfur compounds, alkaline earth metals, acids, etc.
- the method for producing polyhydric alcohols of the present invention is a method for producing polyhydric alcohols, which comprises distilling and refining a crude reaction liquid obtained by performing a reduction reaction using polyhydric aldehydes having an alicyclic structure as a starting material.
- polyhydric alcohols refer to alcohols having two or more hydroxyl groups in the molecule.
- Polyhydric aldehydes having an alicyclic structure which are starting materials for synthesizing polyhydric alcohols in the present invention, are polyhydric aldehydes having a structure having one or more cyclic hydrocarbon groups.
- polyhydric aldehydes refer to aldehydes having two or more aldehyde groups in the molecule.
- polyhydric aldehydes include polyhydric aldehydes having 6 to 20 carbon atoms, more preferably 8 to 12 carbon atoms, optionally having substituents or heteroatoms, and having an alicyclic or aromatic skeleton. More specifically, alicyclic dialdehydes such as 1,3-cyclohexanedicarbaldehyde or 1,4-cyclohexanedicarbaldehyde, 3(4),8(9)-tricyclo[5.2.1.0]decanedicarbaldehyde, 2(3),5(6)-bicyclo[2.2.1]heptanedicarbaldehyde; or aromatic dialdehydes such as terephthalaldehyde and isophthalaldehyde. Polyhydric aldehydes are not limited to these.
- corresponding alicyclic diols such as 1,3-cyclohexanedimethanol or 1,4-cyclohexanedimethanol, 3(4),8(9)-tricyclo[5.2.1.0]decanedimethanol, 2(3),5(6)-bicyclo[2.2.1]heptanedimethanol; or aromatic diols such as p-xylenediol and m-xylenediol are produced.
- Aromatic diols to be produced are not limited to these.
- polyhydric alcohols having a boiling point of 300°C or higher under normal pressure are effective as the above polyhydric alcohols having an alicyclic structure.
- the reduction reaction can be carried out in the presence of a hydrogenation catalyst and hydrogen.
- transition metal catalysts belonging to the 4th and/or 5th periods of the long period periodic table are preferred.
- Raney catalysts such as Raney nickel, Raney cobalt, and Raney copper
- supported catalysts in which hydrogenation-active metals such as nickel, cobalt, platinum, palladium, rhodium, ruthenium, and copper are supported on carriers such as diatomaceous earth, silica, alumina, silica-alumina, clay, titania, zirconia, magnesia, calcia, lanthanum oxide, niobium oxide, and carbon
- metal complex catalysts composed of metals such as nickel, cobalt, platinum, palladium, rhodium, ruthenium, and copper, and organic or inorganic ligands.
- hydrogenation catalysts containing ruthenium, rhodium, palladium, and nickel are particularly
- the hydrogenation catalyst contained in the reaction product liquid containing polyhydric alcohols obtained by such a hydrogenation-reduction reaction is removed by general methods such as filtration, adsorption, and extraction.
- the liquid after removal of the hydrogenation catalyst is used as a crude reaction liquid for distillation and purification.
- metal elements Prior to this distillation purification, metal elements are removed so that the content of metal elements in the crude reaction liquid is 30 mass ppm or less, preferably 10 mass ppm or less.
- the crude reaction liquid obtained by the hydrogenation reduction reaction according to the present invention contains metal elements derived from the hydrogenation catalyst.
- the content of the metal element in the crude reaction liquid varies depending on the amount of the hydrogenation catalyst used in the hydrogenation reduction reaction, but is usually about 10 to 100 mass ppm.
- the present inventors have found that the metal element derived from the hydrogenation catalyst decomposes the polyhydric alcohol by heating in the distillation purification step, causing a decrease in the yield and purity of the polyhydric alcohol.
- thermal decomposition of polyhydric alcohols caused by metal elements in the distillation and purification step is suppressed by removing the metal elements from the crude reaction liquid prior to distillation and purification.
- the method for removing metal elements from the crude reaction solution to reduce its content there are no particular restrictions on the method for removing metal elements from the crude reaction solution to reduce its content.
- a method of treating using a known adsorbent can be mentioned.
- Specific examples of the treatment with an adsorbent include activated carbon treatment, cation exchange resin, silica gel adsorption, and the like, but the activated carbon treatment is preferred because of its removal efficiency and ability to reuse the adsorbent.
- the method of activated carbon treatment may be a batch process in which activated carbon is added to the crude reaction liquid and stirred, and then solid-liquid separation is performed by filtration or the like, or a continuous process in which the crude reaction liquid is passed through an activated carbon packed tower.
- the amount of activated carbon to be added to the crude reaction liquid is appropriately determined according to the metal element adsorption capacity of the activated carbon, the metal element content in the crude reaction liquid, and so on.
- the processing flow rate is not particularly limited, but it may be processed at a space velocity (LHSV) of 1 to 10.
- LHSV space velocity
- Such activated carbon treatment may be performed multiple times. That is, the activated carbon-treated solution obtained by subjecting the crude reaction solution to activated carbon treatment may be subjected to activated carbon treatment again. In this case, the type and amount of activated carbon used, treatment conditions, etc. may be changed between the first activated carbon treatment and the second activated carbon treatment.
- the crude reaction liquid is subjected to activated carbon treatment or the like so that the metal element content in the crude reaction liquid is 30 mass ppm or less, preferably 10 mass ppm or less, and then purified by distillation. From the viewpoint of suppressing the thermal decomposition of polyhydric alcohols, it is preferable that the metal element content of the crude reaction liquid subjected to this distillation purification is as low as possible.
- the metal element content of the crude reaction liquid to be subjected to distillation purification is preferably 5 ppm by mass or less, more preferably 1 mass ppm or less.
- the pH of the crude reaction liquid to be subjected to distillation purification is preferably within the range of 6-8. If the lower limit of the pH is 6 or more, the by-production of low-boiling-point compounds that are likely to be caused by dehydration of the product alcohol and the by-production of high-boiling-point compounds that are likely to be caused by dimerization such as etherification are suppressed. On the other hand, if the upper limit of the pH is 8 or less, the distillation purification equipment is less susceptible to alkali corrosion, which is preferable.
- the pH of the reaction product obtained by the hydrogenation-reduction reaction of polyhydric aldehydes is 6 to 8, and even if this is subjected to hydrogenation catalyst removal treatment and metal element removal treatment, the pH hardly changes.
- the pH may deviate from the range of 6 to 8 due to acid and alkaline components eluted from the hydrogenation catalyst.
- the conditions for this distillation purification are not particularly limited, but usually a distillation column having a theoretical plate number of 1 to 30, a bottom temperature of the distillation column of 150 to 250°C, a pressure of 0.1 to 100 kPa, and a reflux ratio of 1 to 30 can be used by a person skilled in the art to appropriately optimize the distillation purification conditions according to the purpose.
- Such a method for producing polyhydric alcohols of the present invention is a high-boiling compound and requires high-temperature conditions during distillation, is not solid at room temperature, and is difficult to perform another purification such as crystallization. Therefore, it is particularly effective for producing tricyclo[5.2.1.0(2,6)]decanedimethanol by hydrogenation reduction of tricyclodecanedicarbaldehyde.
- the method for producing tricyclo[5.2.1.0(2,6)]decane dimethanol comprises a step of hydroformylating dicyclopentadiene as a polyhydric aldehyde as a starting material to obtain tricyclodecanedicarbaldehyde, and a step of reducing the tricyclodecanedicarbaldehyde with a hydrogenation catalyst in the presence of hydrogen to obtain a crude reaction liquid containing tricyclo[5.2.1.0(2,6)]decanedimethanol.
- the content of metal elements in the crude reaction solution is adjusted to 30 mass ppm or less, preferably 10 mass ppm or less, and then purified by distillation as described above.
- the hydroformylation method of dicyclopentadiene is not particularly limited and can be carried out according to a conventional method.
- tricyclodecane dicarbaldehyde can be produced by hydroformylating dicyclopentadiene using hydrogen and carbon monoxide in the presence of a catalyst comprising a rhodium compound and an organophosphorus compound in a hydroformylation reaction solvent comprising a hydrocarbon compound, as shown in the following reaction formula (I).
- the rhodium compound used in this hydroformylation step does not depend on the form of its precursor as long as it forms a complex with the organophosphorus compound and exhibits hydroformylation activity in the presence of hydrogen and carbon monoxide.
- a catalyst precursor material such as Rh(acac)(CO) 2 , Rh2O3, Rh4 (CO) 12 , Rh6 (CO) 16 , Rh( NO3 ) 3 may be introduced into the reaction mixture together with the organophosphorus compound to form a rhodium metal hydridocarbonyl phosphorus complex having catalytic activity in the reaction vessel, or a rhodium metal hydridocarbonyl phosphorus complex catalyst may be prepared in advance and introduced into the reaction vessel. Good.
- Rh(acac)(CO) 2 is used as a rhodium precursor material and is reacted with an organophosphorus compound in the presence of a solvent and then introduced into a reactor along with excess free organophosphorus compound to provide a catalytically active rhodium-organophosphorus complex catalyst.
- Organophosphorus compounds that form catalysts for hydroformylation reactions with rhodium compounds include phosphites and phosphines.
- the phosphite is preferably a compound represented by the general formula P(--OR 1 ) (--OR 2 ) (--OR 3 ) (wherein R 1 , R 2 and R 3 each represent an optionally substituted aryl group or alkyl group) because it is effective for the hydroformylation reaction of dicyclopentadiene.
- R 1 , R 2 and R 3 include aryl groups such as phenyl and naphthyl groups optionally substituted with methyl, ethyl, isopropyl, n-butyl, t-butyl, methoxy and the like; aliphatic alkyl groups such as methyl, ethyl, isopropyl, n-butyl and t-butyl; cyclopentyl groups optionally substituted with lower alkyl such as methyl, ethyl, isopropyl, n-butyl and t-butyl; An alicyclic alkyl group such as a cyclohexyl group and the like are included.
- aryl groups such as phenyl and naphthyl groups optionally substituted with methyl, ethyl, isopropyl, n-butyl, t-butyl, methoxy and the like
- aliphatic alkyl groups such as methyl
- Suitable phosphites include tris(2-t-butylphenyl)phosphite, tris(3-methyl-6-t-butylphenyl)phosphite, tris(3-methoxy-6-t-butylphenyl)phosphite, tris(2,4-di-t-butylphenyl)phosphite, di(2-t-butylphenyl)(t-butyl)phosphite and the like.
- Phosphites are not limited to these. These phosphites may be used alone or in combination of two or more.
- phosphines sterically hindered alkylphosphines are particularly effective for the hydroformylation reaction of dicyclopentadiene.
- Typical examples include tricyclopropylphosphine, tricyclobutylphosphine, tricyclopentylphosphine, tricyclohexylphosphine, tricycloheptylphosphine, tricyclooctylphosphine and the like.
- Phosphines are not limited to these. These phosphines may be used alone or in combination of two or more.
- tricyclodecane dicarbaldehyde can be obtained at a sufficient hydroformylation reaction rate as long as the organic phosphorus compound is present in the hydroformylation reaction solution in a range of 1 to 400 times by moles, preferably 3 to 200 times by moles, the rhodium metal.
- hydroformylation reaction of dicyclopentadiene can be carried out without using a solvent, it can be carried out more preferably by using an organic solvent that is inert to the reaction.
- the hydroformylation reaction solvent is preferably one that separates into layers with the alcohol.
- solvents include aromatic hydrocarbon compounds, aliphatic hydrocarbon compounds, and alicyclic hydrocarbon compounds.
- aromatic hydrocarbon compound benzene, methylbenzenes such as toluene, xylene, mesitylene and pseudocumene, ethylbenzenes such as ethylbenzene, diethylbenzene and triethylbenzene, propylbenzenes such as isopropylbenzene, 1,3-diisopropylbenzene and 1,4-diisopropylbenzene, and various other alkylbenzenes can be suitably used.
- methylbenzenes such as toluene, xylene, mesitylene and pseudocumene
- ethylbenzenes such as ethylbenzene, diethylbenzene and triethylbenzene
- propylbenzenes such as isopropylbenzene, 1,3-diisopropylbenzene and 1,4-diisopropylbenzene, and
- aliphatic hydrocarbon compounds examples include pentane, hexane, heptane, octane, isooctane, dodecane, and decane. Aliphatic hydrocarbon compounds are not limited as long as they are liquid at standard temperature and pressure. As the alicyclic hydrocarbon compound, cyclohexane, cyclooctane, cyclododecane, decalin, methylcyclohexane and the like are preferably used.
- the amount of the rhodium catalyst used is usually 10 to 5000 ppm by mass, preferably 50 to 2000 ppm by mass as rhodium metal, relative to the raw material dicyclopentadiene. When rhodium is used above 50 ppm, recovery of the catalyst is required.
- the temperature and pressure of the hydroformylation reaction of dicyclopentadiene are generally 40 to 160°C, preferably 80 to 140°C, and generally 1 to 15 MPa.
- the hydroformylation reaction is slow, and when it is higher than 160°C, side reactions from dicyclopentadiene and hydroformylation reaction products in the reaction solution proceed, resulting in a decrease in aldehyde yield.
- the pressure is lower than 1 MPa, the hydroformylation reaction is slow, and when the pressure is higher than 15 MPa, a high-pressure reactor is used, resulting in high equipment cost.
- the molar ratio of hydrogen and carbon monoxide in the hydrogen/carbon monoxide mixed gas used for the reaction can be selected from the range of 0.2 to 5.0 for the introduced gas composition (hydrogen/carbon monoxide). If the composition of the hydrogen/carbon monoxide mixed gas is out of this range, the hydroformylation reaction activity or aldehyde selectivity will decrease.
- hydroformylation reaction method a continuous feed method is adopted in which dicyclopentadiene as a starting material alone or as a mixed solution of dicyclopentadiene and a solvent is supplied to a reactor in which a rhodium-organophosphorus complex catalyst, a solvent, and a mixed gas of hydrogen and carbon monoxide exist.
- a reactor in which a rhodium-organophosphorus complex catalyst, a solvent, and a mixed gas of hydrogen and carbon monoxide exist.
- the dicyclopentadiene In order to maintain the fluidity of the dicyclopentadiene, it is preferable to dilute it with the above-mentioned solvent and supply it to the reactor at a temperature at which the dicyclopentadiene is not depolymerized to produce cyclopentadiene.
- the reaction product solution is directly or after being diluted with the hydrocarbon compound or other hydrocarbon compound used in the reaction as a hydroformylation reaction solvent, brought into contact with alcohol to extract the product tricyclodecane dicarbaldehyde while leaving the catalyst component in the hydroformylation reaction solvent layer, and separate the layers.
- Examples of alcohols include primary alcohols having 1 to 3 carbon atoms and polyhydric alcohols having 2 to 6 carbon atoms.
- Examples of primary alcohols having 1 to 3 carbon atoms include methanol, ethanol and propanol.
- Examples of polyhydric alcohols having 2 to 6 carbon atoms include ethylene glycol, 1,3-propanediol, 1,2-propanediol, 1,4-butanediol, 1,2-butanediol, 1,3-butanediol, 2,3-butanediol, isomers of pentanediol, neopentyl glycol, hexanediol, glycerin, pentaerythritol, and trimethylolpropane.
- methanol, ethylene glycol, propanediol, and butanediol are preferably used because they have relatively low boiling points, are inexpensive, and are easy to handle as liquids.
- extraction solvents may be used alone or in combination of two or more. Alternatively, the extraction may be carried out in the presence of water in the alcohol. Addition of water facilitates the distribution of aldehydes and catalyst components to each layer.
- reaction solvent and extraction solvent used in the hydroformylation reaction have different densities in order to achieve effective layer separation.
- One preferred combination of hydroformylation reaction solvent and extraction solvent containing tricyclodecane dicarbaldehyde is a combination of methylcyclohexane and ethylene glycol, or methylcyclohexane and methanol, and water.
- the volume ratio between the extraction solvent and the reaction product solution to be used is determined by the solubility of tricyclodecane dicarbaldehyde in the extraction solvent and the amount of tricyclodecane dicarbaldehyde to be extracted.
- tricyclodecane dicarbaldehyde to be separated exhibits high solubility in the extraction solvent and is present at a low concentration in the reaction solution
- tricyclodecane dicarbaldehyde can be practically extracted by using an extraction solvent with a low volume ratio (extraction solvent/reaction solution).
- the volume ratio can vary from 10:1 to 1:10.
- a hydroformylation reaction solvent such as methylcyclohexane may be added in an amount of about 5 to 20% by mass relative to the reaction product solution. Addition of a hydroformylation reaction solvent can improve the removal rate of the catalyst.
- the temperature at which the extraction operation is performed is not particularly limited, but it is practical to carry it out below the hydroformylation reaction temperature.
- an extraction solvent may be added to carry out an extraction operation.
- the hydroformylation reaction product liquid may be withdrawn from the hydroformylation reactor and an extraction operation may be performed in an extraction tank. It is also possible to add the extraction solvent directly to the hydroformylation reactor to carry out the extraction operation and to carry out the subsequent hydroformylation reaction while retaining the catalyst components in the hydroformylation reactor.
- the hydroformylation reaction product liquid is withdrawn and the operation is carried out in an extraction tank, the hydrocarbon solvent layer containing the catalyst is returned to the hydroformylation reactor and used again for the reaction.
- the process can be performed as a batch process or as a continuous process.
- a tricyclodecane dicarbaldehyde-containing solution containing 10 to 90% by mass of tricyclodecane dicarbaldehyde and 10 to 90% by mass of the extraction solvent can be obtained. Further, when a reaction solvent is added, a tricyclodecane dicarbaldehyde-containing solution containing 5 to 90% by mass of tricyclodecane dicarbaldehyde, 5 to 90% by mass of extraction solvent, and 5 to 90% by mass of reaction solvent can be obtained.
- the alcohol of the extraction solvent reacts with a portion of the hydroformylation product tricyclodecane dicarbaldehyde to form an acetal compound in which tricyclodecane dicarbaldehyde is acetalized.
- the content of the acetal compound in tricyclodecane dicarbaldehyde is usually about 0.1 to 50% by mass, more preferably about 1 to 25% by mass.
- Tricyclodecanedimethanol can be produced in high yield by hydrogenating tricyclodecane dicarbaldehyde converted from the acetal compound, which is preferable.
- the amount of water present in the hydrogenation-reduction reaction is preferably equal to or greater than the amount of the acetal compound in the hydrogenation-reduction reaction liquid, and is an amount that does not cause phase separation of the reaction liquid.
- the water content in the hydrogenation reduction reaction is 2% by mass or more, preferably 2 to 30% by mass, more preferably 5 to 25% by mass, particularly preferably 10 to 20% by mass, based on the total reaction solution. When the water content is within the above range, it is possible to effectively obtain the above effects due to the presence of water in the hydrogenation reaction system without layer separation between water and the reaction solvent. This water may be added in the extraction step of separating the catalyst components and polyhydric aldehydes from the hydroformylation reaction product liquid, or may be added to the reaction system immediately before the hydrogenation reduction reaction.
- the reaction mode a method in which the catalyst is charged as a slurry in a stirring reactor, the reaction is carried out in batch mode, and after the reaction the catalyst is sedimented and filtered to separate it from the product liquid; a perfusion-type reaction in which the shaped catalyst is charged in a tubular reactor and the product liquid and hydrogen gas are flowed over the catalyst;
- the amount of catalyst used is not particularly limited as long as tricyclodecanedimethanol can be produced with industrially advantageous productivity.
- the reaction temperature and pressure of the hydrogenation reduction reaction are usually 40-200°C, preferably 70-150°C, and usually 15 MPa or less.
- the temperature is lower than 40°C, the hydrogenation-reduction reaction is slow, and when it is higher than 200°C, a side reaction from the target tricyclodecanedimethanol proceeds and the yield of tricyclodecanedimethanol decreases.
- the pressure is higher than 15 MPa, a high-pressure reactor is used, resulting in high equipment cost.
- the crude reaction solution containing tricyclodecanedimethanol obtained in this way is subjected to a metal element content reduction treatment such as activated carbon treatment after removing the hydrogenation catalyst as described above, and then subjected to distillation purification.
- a metal element content reduction treatment such as activated carbon treatment
- the reaction liquid in the reactor was cooled to room temperature, and the residual gas in the reactor was released to obtain 12.5 kg of hydroformylation reaction product liquid.
- the amount of dicyclopentadiene as a raw material compound contained in the reaction solution before the reaction and the amount of tricyclodecane dicarbaldehyde produced as a product in the reaction product solution after the reaction were analyzed by gas chromatography, and the yield of tricyclodecane dicarbaldehyde was determined to be 99%.
- ⁇ Extraction operation> 3.77 kg of methanol and 2 kg of water were added to 12.5 kg of the obtained hydroformylation reaction product liquid, and the mixture was stirred for 30 minutes under a nitrogen atmosphere. After that, the mixture was allowed to stand for 30 minutes to separate into two phases for extraction. 0.4 kg of methylcyclohexane was added to the obtained lower phase (a1) and stirred for 30 minutes. After that, it was allowed to stand for 30 minutes, separated into two phases, and an extraction operation was performed to obtain a lower phase (a2) of 13.8 kg.
- the composition of the obtained lower phase (a2) was analyzed by gas chromatography and found to be 47% by mass of tricyclodecane dicarbaldehyde, 27% by mass of methanol, 14% by mass of water, 7% by mass of methylcyclohexane, and 5% by mass of other components.
- reaction liquid in the reactor was cooled to room temperature, the residual gas in the reactor was released, and the ruthenium-carrying carbon was separated by filtration using a 5 ⁇ m filter to obtain 14.5 kg of reaction product liquid.
- the amount of tricyclodecanedicarbardehyd which is a raw material compound contained in the reaction solution before the reaction
- the amount of tricyclo[5.2.1.0(2,6)]decanedimethanol (hereinafter referred to as "TCDDM”), which is a product in the reaction product solution after the reaction, were analyzed by gas chromatography.
- TCDDM tricyclo[5.2.1.0(2,6)]decanedimethanol
- the Ru concentration of the reaction product liquid analyzed by fluorescent X-ray analysis was 36 ppm by mass, and the pH was 7 as measured with pH test paper. This reaction product liquid is called "crude reaction product liquid".
- Example 1 Powdered activated carbon A was added to 100 g of the crude reaction product liquid obtained in Reference Example 1 so that the content concentration was 1% by mass, and the mixture was stirred at room temperature for 3 hours. Next, the reaction product liquid after stirring was filtered to remove the powdered activated carbon A, and 95 g of an activated carbon-treated liquid was obtained. As a result of analysis by gas chromatography, the composition of this activated carbon-treated liquid was 52% by mass of TCDDM, 25% by mass of methanol, 14% by mass of water, 4% by mass of methylcyclohexane, and 5% by mass of other components. The Ru concentration of the activated carbon-treated liquid analyzed by fluorescent X-ray analysis was 3.5 ppm by mass, and the pH measured by pH test paper was 7.
- the solvent of the obtained activated carbon-treated liquid was distilled off under reduced pressure, heated at 230°C, which is the distillation temperature for distilling TCDDM, for 5 hours, and then cooled to room temperature.
- the residual rate of TCDDM was 94% by mass.
- Example 2 Powdered activated carbon B was added to 100 g of the crude reaction product liquid obtained in Reference Example 1 so that the content concentration was 1% by mass, and the mixture was stirred at room temperature for 3 hours. Next, the reaction product liquid after stirring was filtered to remove the powdered activated carbon B, and 95 g of an activated carbon-treated liquid was obtained.
- the composition of this activated carbon-treated liquid analyzed in the same manner as in Example 1 was 52% by weight of TCDDM, 25% by weight of methanol, 14% by weight of water, 4% by weight of methylcyclohexane, and 5% by weight of other components.
- the Ru concentration of the activated carbon treatment liquid was 0.7 ppm by mass, and the pH was 7.
- Example 1 the content of the metal element (Ru) contained in the activated carbon treatment liquid before the solvent was distilled off under reduced pressure was 30 ppm by mass or less, so the TCDDM residual rate after heating for 5 hours at 230°C, which is the distillation temperature for distilling TCDDM, was high.
- Comparative Example 1 since the content of the metal element (Ru) contained in the crude reaction product liquid exceeded 30 mass ppm, the TCDDM residual rate after heating for 5 hours at 230°C, which is the distillation temperature for distilling TCDDM, was low. Moreover, from the results of Examples 1 and 2 and Comparative Example 1, it was confirmed that the lower the content of the metal element (Ru), the higher the TCDDM residual ratio.
- Example 3 0.15 kg of powdered activated carbon B was added to 14.5 kg of the crude reaction product liquid obtained in Reference Example 1, and after stirring for 3.5 hours, the stirring was stopped and filtered to obtain 13 kg of the first activated carbon-treated liquid. 0.13 kg of powdered activated carbon B was added again to this first activated carbon-treated liquid, and after stirring for 3 hours, the mixture was filtered to obtain 12 kg of a second activated carbon-treated liquid.
- the Ru concentration of the second activated carbon treatment liquid analyzed in the same manner as in Example 1 was 0.4 ppm by mass, and the pH was 7.
- composition of the second activated carbon-treated liquid was 45% by mass of tricyclodecanedimethanol, 29% by mass of methanol, 14% by mass of water, 7% by mass of methylcyclohexane, and 5% by mass of other components.
- the content of the metal element (Ru) in the crude reaction liquid to be distilled is 30 ppm by mass or less, preferably 10 ppm by mass or less, thereby reducing loss due to decomposition of TCDDM and producing TCDDM with high yield and high purity.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
具体的には、アルケン類としてジシクロペンタジエンを用いて、これをヒドロホルミル化することにより、多価アルデヒド類としてトリシクロデカンジカルバルデヒドを得た後、これを水素化(水添)することによって、多価アルコール類としてトリシクロデカンジメタノールを合成する方法が挙げられる(特許文献1)。
さらに、前記添加物を使用するには、該添加剤の供給設備や、場合によっては添加剤の除去設備等が必要となり、製造工程の複雑化や製造コストの増加が課題となる。
前記粗反応液中の金属元素の含有量を30質量ppm以下とした後に前記蒸留精製を行う、多価アルコール類の製造方法。
該トリシクロデカンジカルバルデヒドを、水素化触媒と水素の存在下に還元反応して、トリシクロ[5.2.1.0(2,6)]デカンジメタノールを含む粗反応液を得る工程を含む、[1]~[12]のいずれかに記載の多価アルコール類の製造方法。
さらに、本発明によれば、特許文献1~3に開示された、硫黄化合物、アルカリ土類金属、酸等の添加剤を用いることなく蒸留精製することが可能であるため、製品多価アルコール類への前記添加剤の混入、製造設備の腐食、及び製造コストの増加を抑制できる。
本発明において多価アルデヒド類とは、分子中に2以上のアルデヒド基を有するアルデヒド類のことをいう。
これらのうち、特に、ルテニウム、ロジウム、パラジウム、ニッケルを含む水素化触媒が、水素化触媒活性、触媒コスト及び触媒分離性の観点から好ましく、とりわけルテニウム触媒が好ましい。
本発明者は、この水素化触媒に由来する金属元素が、蒸留精製工程における加熱で多価アルコール類を分解し、多価アルコール類の収率及び純度を低下させる原因となることを知見した。
本発明では、蒸留精製に先立ち、粗反応液中の金属元素を除去することで、蒸留精製工程における金属元素に起因する多価アルコール類の熱分解を抑制する。
この蒸留精製に供する粗反応液の金属元素含有量は、低いほど、多価アルコール類の熱分解抑制の観点から好ましい。蒸留精製に供する粗反応液の金属元素含有量は、特に5質量ppm以下、とりわけ1質量ppm以下とすることが好ましい。
本発明によるトリシクロ[5.2.1.0(2,6)]デカンジメタノールの製造方法は、出発原料としての多価アルデヒド類として、ジシクロペンタジエンをヒドロホルミル化してトリシクロデカンジカルバルデヒドを得る工程、及び、該トリシクロデカンジカルバルデヒドを水素化触媒と水素の存在下に還元反応して、トリシクロ[5.2.1.0(2,6)]デカンジメタノールを含む粗反応液を得る工程を有する。前述の通り、この粗反応液中の金属元素の含有量を30質量ppm以下、好ましくは10質量ppm以下とした後、前述の通り蒸留精製する。
ジシクロペンタジエンのヒドロホルミル化の方法には特に制限はなく、常法に従って行うことができる。
Rh(acac)(CO)2,Rh2O3,Rh4(CO)12,Rh6(CO)16,Rh(NO3)3などの触媒前駆体物質を有機リン化合物と共に反応混合物中に導入し反応容器内で触媒活性を持つロジウム金属ヒドリドカルボニルリン錯体を形成させてもよいし、あらかじめロジウム金属ヒドリドカルボニルリン錯体触媒を調製してそれを反応容器内に導入してもよい。
このうち、ホスファイトとしては、ジシクロペンタジエンのヒドロホルミル化反応に有効であることから、一般式P(-OR1)(-OR2)(-OR3)(式中、R1,R2およびR3はそれぞれ置換されていてもよいアリール基又はアルキル基を表す。)で示される化合物が好ましい。R1,R2およびR3の具体例としては、メチル基、エチル基、イソプロピル基、n-ブチル基、t-ブチル基、メトキシ基などで置換されていてもよいフェニル基およびナフチル基などのアリール基;メチル基、エチル基、イソプロピル基、n-ブチル基、t-ブチル基などの脂肪族アルキル基;メチル基、エチル基、イソプロピル基、n-ブチル基、t-ブチル基などの低級アルキル基で置換されていてもよいシクロペンチル基、シクロヘキシル基などの脂環式アルキル基等が挙げられる。
脂環式炭化水素化合物としては、シクロヘキサン、シクロオクタン、シクロドデカン、デカリン、メチルシクロヘキサンなどが好適に使用される。
ヒドロホルミル化反応終了後、反応生成液をそのまま、又は、ヒドロホルミル化反応溶媒として反応で使用した炭化水素化合物もしくは他の炭化水素化合物で希釈した後、アルコールと接触させて、触媒成分をヒドロホルミル化反応溶媒層に残したまま、生成物であるトリシクロデカンジカルバルデヒドをアルコールに抽出し、層分離を行う。
炭素数1~3の第1級アルコールとしてはメタノール、エタノール、プロパノールが挙げられる。
炭素数2~6の多価アルコールとしてはエチレングリコール、1,3-プロパンジオール、1,2-プロパンジオール、1,4-ブタンジオール、1,2-ブタンジオール、1,3-ブタンジオール、2,3-ブタンジオール、ペンタンジオールの各異性体、ネオペンチルグリコール、ヘキサンジオール、グリセリン、ペンタエリスリトール、トリメチロールプロパンなどが使用される。
この中で、メタノールやエチレングリコール、プロパンジオール、ブタンジオールが比較的沸点が低く、価格も安く、液体として取扱もしやすいので好適に使用される。
これらの抽出溶媒は単独で使用してもよいし、2種以上を組合わせて使用しても良い。
また、アルコールに水を共存させて抽出を行ってもよい。水の添加によりアルデヒドや触媒成分が各層へ分配し易くなる。
生成物の濃度が高いほど、反応生成液からトリシクロデカンジカルバルデヒドを抽出するための体積比率(抽出溶媒/反応生成液)は高くなる。トリシクロデカンジカルバルデヒドが抽出溶液に比較的低い溶解度を示す場合は、体積比率は10:1~1:10の範囲で変動し得る。
少ない抽出溶媒使用量でトリシクロデカンジカルバルデヒドの抽出量を多くするため、抽出溶媒を分け、数回の抽出操作を行うことが有効である。
この場合、最終段階の抽出操作では、メチルシクロヘキサン等のヒドロホルミル化反応溶媒を反応生成液に対して5~20質量%程度添加してもよい。ヒドロホルミル化反応溶媒の添加で触媒の除去率を向上させることができる。
抽出溶媒のアルコールは、ヒドロホルミル化生成物であるトリシクロデカンジカルバルデヒドの一部と反応してトリシクロデカンジカルバルデヒドをアセタール化したアセタール化合物を生成する。
トリシクロデカンジカルバルデヒド中のアセタール化合物の含有率は、通常0.1~50質量%程度であり、更に1~25質量%程度である。
上記の抽出操作により得られたトリシクロデカンジカルバルデヒドを含む抽出液(トリシクロデカンジカルバルデヒド含有溶液)は、次いで、前述の水素化触媒、好ましくは、ルテニウム(Ru)触媒の存在下に水素化還元を行い、下記反応式(II)の通り、トリシクロデカンジメタノールを製造する。
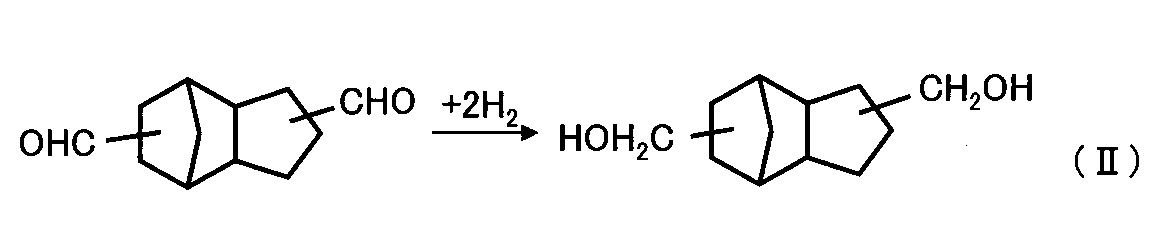
・アセチルアセトナートジカルボニルロジウム(商品名:Rh(acac)(CO)2
、エヌ・イー ケムキャット株式会社製)
・ルテニウム担持炭素(ドライベースRu含有率5%、含水率56%)(商品名:Ru/C、エヌ・イーケムキャット株式会社製)
・トリス(2,4-ジ-tert-ブチルフェニル)ホスファイト(商品名:DBPO、東京化成工業株式会社製)
・メチルシクロヘキサン(商品名:メチルシクロヘキサン、富士フィルム和光純薬株式会社製)
・ジシクロペンタジエン(富士フィルム和光純薬株式会社製)
・粉末活性炭A(商品名:白鷺ANO-2、大阪ガスケミカル株式会社製)
・粉末活性炭B(商品名:特性白鷺、大阪ガスケミカル株式会社製)
<ヒドロホルミル化反応>
内容量20Lのオートクレーブ反応器に、窒素雰囲気下で、ヒドロホルミル化反応触媒の原料化合物として、Rh(acac)(CO)2 2.51g(9.73mmol)、トリス(2,4-ジ-t-ブチルフェニル)ホスファイト188.6g(0.291mol)を量り取り、有機溶媒としてメチルシクロヘキサン3.9kgを仕込んだ。その後、120rpmで攪拌しつつ、反応器内の反応液の温度を70℃まで昇温した。次いで、ガス導入バルブより速やかに水素と一酸化炭素の混合ガス(水素:一酸化炭素=1:1(モル比))を反応器内の圧力が5MPaGとなるように圧入し、この圧力を維持したまま、反応液の温度を100℃まで昇温した。その後、さらに原料化合物としてジシクロペンタジエン4.9kgを5時間かけてフィードし、さらに3時間反応させた。反応中は反応で消費された量の混合ガスを、反応器内の圧力を5MPaGに維持しながら、反応器内に導入し続けた。
得られたヒドロホルミル化反応生成液12.5kgに対し、メタノール3.77kg、水2kgを加え、窒素雰囲気下で30分間攪拌した。その後、30分間静置し2相に分離させ抽出操作を行った。得られた下相(a1)にメチルシクロヘキサンを0.4kg加え30分間攪拌した。その後、30分間静置し2相に分離させ抽出操作を行い、13.8kgの下相(a2)を得た。
得られた下相(a2)の組成をガスクロマトグラフィーにより分析したところ、トリシクロデカンジカルバルデヒド47質量%、メタノール27質量%、水14質量%、メチルシクロヘキサン7質量%、その他成分5質量%であった。
内容量20Lのオートクレーブ反応器に、上述した抽出操作により得られた下相(a2)13.8kg、ルテニウム担持炭素0.14kgを仕込んだ後、120rpmで攪拌しつつ、反応器内の反応液の温度を160℃まで昇温した。次いで、ガス導入バルブより水素ガスを反応器内の圧力が5MPaGとなるとなるように圧入し、この圧力と反応液の温度を維持したまま、8時間反応させた。反応中は反応で消費された量の水素ガスを、反応器内の圧力を5MPaGに維持しながら、反応器内に導入し続けた。
反応前の反応液に含まれる原料化合物のトリシクロデカンジカルバルデヒドの量と、反応後の反応生成液中の生成物であるトリシクロ[5.2.1.0(2,6)]デカンジメタノール(以下、「TCDDM」という。)の生成量をガスクロマトグラフィーにて分析した結果、TCDDMの収率は94%であった。
蛍光X線分析により分析した反応生成液のRu濃度は36質量ppmであり、pH試験紙により測定したpHは7であった。この反応生成液を「粗反応生成液」という。
参考例1で得られた粗反応生成液100gに、粉末活性炭Aを含有濃度が1質量%となるように添加し、室温で3時間攪拌した。次いで、攪拌後の反応生成液をろ過して、前記粉末活性炭Aを除去し、活性炭処理液95gを得た。
ガスクロマトグラフィーにて分析した結果、この活性炭処理液の組成はTCDDM52質量%、メタノール25質量%、水14質量%、メチルシクロヘキサン4質量%、その他成分5質量%であった。
蛍光X線分析により分析した活性炭処理液のRu濃度は3.5質量ppmであり、pH試験紙により測定したpHは7であった。
参考例1で得られた粗反応生成液100gに、粉末活性炭Bを含有濃度が1質量%となるように添加し、室温で3時間攪拌した。次いで、攪拌後の反応生成液をろ過して、前記粉末活性炭Bを除去し、活性炭処理液95gを得た。
実施例1におけると同様に分析したこの活性炭処理液の組成はTCDDM52質量%、メタノール25質量%、水14質量%、メチルシクロヘキサン4質量%、その他成分5質量%であった。活性炭処理液のRu濃度は0.7質量ppmであり、pHは7であった。
参考例1で得られたRu濃度36ppmの粗反応生成液100gを活性炭処理することなしに、実施例1と同様に、蒸留のモデル実験として溶媒を減圧留去した後加熱して加熱後のTCDDMの残存率を分析したところ、TCDDMの残存率は56質量%であった。

一方、比較例1では、粗反応生成液に含まれる金属元素(Ru)の含有量が30質量ppmを超えるため、TCDDMを蒸留する際の蒸留温度である230℃で5時間加熱した後のTCDDM残存率は低位となった。
また、実施例1及び実施例2並びに比較例1の結果から、金属元素(Ru)の含有量が低いほど、TCDDM残存率は高くなる傾向が確認された。
参考例1で得られた粗反応生成液14.5kgに対し、粉末活性炭Bを0.15kg添加し3.5時間攪拌した後、攪拌を停止し、ろ過して第1の活性炭処理液13kgを得た。この第1の活性炭処理液に再度0.13kgの粉末活性炭Bを添加し3時間攪拌した後、ろ過して第2の活性炭処理液12kgを得た。実施例1におけると同様に分析した第2の活性炭処理液のRu濃度は0.4質量ppm、pHは7であった。
ガスクロマトグラフィーにて分析したところ、第2の活性炭処理液の組成はトリシクロデカンジメタノール45質量%、メタノール29質量%、水14質量%、メチルシクロヘキサン7質量%、その他成分5質量%であった。
一連の蒸留において、加熱前の蒸留原料液(第2の活性炭処理液)および加熱後の留出液と塔底液に含まれるTCDDMの量をガスクロマトグラフィーにて分析した結果、TCDDMの残存率は99質量%であった。
本出願は、2022年1月20日付で出願された日本特許出願2022-007310に基づいており、その全体が引用により援用される。
Claims (13)
- 脂環式構造を有する多価アルデヒド類を出発原料として、還元反応を行うことにより得られた粗反応液を蒸留精製することを含む多価アルコール類の製造方法であって、
前記粗反応液中の金属元素の含有量を30質量ppm以下とした後に前記蒸留精製を行う、多価アルコール類の製造方法。 - 前記粗反応液中の金属元素の含有量を10質量ppm以下とした後に前記蒸留精製を行う、請求項1に記載の多価アルコール類の製造方法。
- 前記還元反応を、水素化触媒と水素の存在下で行う、請求項1又は2に記載の多価アルコール類の製造方法。
- 前記多価アルコール類が、常圧下における沸点が300℃以上である、請求項1~3のいずれか一項に記載の多価アルコール類の製造方法。
- 前記金属元素が、前記水素化触媒に由来する金属である、請求項3又は4に記載の多価アルコール類の製造方法。
- 前記金属元素が、長周期型周期表第4及び/又は第5周期に属する遷移金属元素である、請求項1~5のいずれか一項に記載の多価アルコール類の製造方法。
- 前記金属元素が、ルテニウム、ロジウム、パラジウム及びニッケルからなる群より選択される少なくとも1種である、請求項6に記載の多価アルコール類の製造方法。
- 前記粗反応液のpHを6~8の範囲内とした後に、前記蒸留精製を行う、請求項1~7のいずれか一項に記載の多価アルコール類の製造方法。
- 前記粗反応液を、吸着剤を用いて処理することで、該粗反応液中の金属元素の含有量を10質量ppm以下とする、請求項1~8のいずれか一項に記載のアルコール類の製造方法。
- 前記粗反応液を活性炭処理することで、該粗反応液中の金属元素の含有量を10質量ppm以下とする、請求項9に記載の多価アルコール類の製造方法。
- 前記粗反応液中の金属元素の含有量を1質量ppm以下とした後、前記蒸留精製を行う、請求項1~10のいずれか一項に記載の多価アルコール類の製造方法。
- 前記多価アルコール類がトリシクロ[5.2.1.0(2,6)]デカンジメタノールである、請求項1~11のいずれか一項に記載の多価アルコール類の製造方法。
- ジシクロペンタジエンをヒドロホルミル化して、前記多価アルデヒド類としてのトリシクロデカンジカルバルデヒドを得る工程、及び
該トリシクロデカンジカルバルデヒドを、水素化触媒と水素の存在下に還元反応して、トリシクロ[5.2.1.0(2,6)]デカンジメタノールを含む粗反応液を得る工程を含む、請求項1~12のいずれか一項に記載の多価アルコール類の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020247024557A KR20240125650A (ko) | 2022-01-20 | 2023-01-12 | 다가 알코올류의 제조 방법 |
| CN202380017896.1A CN118574806A (zh) | 2022-01-20 | 2023-01-12 | 多元醇类的制造方法 |
| JP2023504436A JP7501778B2 (ja) | 2022-01-20 | 2023-01-12 | 多価アルコール類の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022007310 | 2022-01-20 | ||
| JP2022-007310 | 2022-04-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023140165A1 true WO2023140165A1 (ja) | 2023-07-27 |
Family
ID=87348785
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/000562 WO2023140165A1 (ja) | 2022-01-20 | 2023-01-12 | 多価アルコール類の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP7501778B2 (ja) |
| KR (1) | KR20240125650A (ja) |
| CN (1) | CN118574806A (ja) |
| TW (1) | TW202337877A (ja) |
| WO (1) | WO2023140165A1 (ja) |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02188540A (ja) * | 1989-01-13 | 1990-07-24 | Sumitomo Chem Co Ltd | メタノールの精製法 |
| JPH1112209A (ja) * | 1997-04-30 | 1999-01-19 | Asahi Chem Ind Co Ltd | 環状アルコールの分離取得方法 |
| JPH1180056A (ja) * | 1997-07-18 | 1999-03-23 | Asahi Chem Ind Co Ltd | 環状アルコールの分離取得方法 |
| JP2000007595A (ja) * | 1998-06-26 | 2000-01-11 | New Japan Chem Co Ltd | シクロヘキサンジメタノールの製造方法 |
| JP2001010999A (ja) | 1999-07-02 | 2001-01-16 | Mitsubishi Gas Chem Co Inc | トリシクロデカンジメタノール及び/又はペンタシクロペンタデカンジメタノールの製造法 |
| JP2001172214A (ja) * | 1999-12-16 | 2001-06-26 | Mitsubishi Gas Chem Co Inc | ジオールの製造方法 |
| JP2002047225A (ja) | 2000-07-27 | 2002-02-12 | Mitsubishi Gas Chem Co Inc | 多環式ジオール類の蒸留方法 |
| JP2002284771A (ja) * | 2001-03-28 | 2002-10-03 | Nof Corp | グリシドールの精製方法 |
| JP2003192621A (ja) | 2001-12-14 | 2003-07-09 | Celanese Chemicals Europe Gmbh | 脂環式アルコールの精製方法 |
| JP2004196778A (ja) | 2002-12-04 | 2004-07-15 | Mitsubishi Chemicals Corp | アルコールの製造方法 |
| JP2006514087A (ja) * | 2003-02-24 | 2006-04-27 | シエル・インターナシヨナル・リサーチ・マートスハツペイ・ベー・ヴエー | 蒸留による1,3−プロパンジオールの精製 |
| JP2010235516A (ja) * | 2009-03-31 | 2010-10-21 | Kuraray Co Ltd | 精製ジオールの製造方法 |
| WO2011064184A1 (de) | 2009-11-26 | 2011-06-03 | Basf Se | Verfahren zur herstellung von kunststoffen mit 1,6-hexandiol mit einem aldehydanteil von kleiner 500ppm |
| JP2013501035A (ja) * | 2009-08-07 | 2013-01-10 | ランクセス・ドイチュランド・ゲーエムベーハー | トリメチロールプロパンの色数を改善する方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1160525A (ja) | 1997-08-21 | 1999-03-02 | Kuraray Co Ltd | ジオール類の製造方法 |
-
2023
- 2023-01-12 JP JP2023504436A patent/JP7501778B2/ja active Active
- 2023-01-12 WO PCT/JP2023/000562 patent/WO2023140165A1/ja active Application Filing
- 2023-01-12 CN CN202380017896.1A patent/CN118574806A/zh active Pending
- 2023-01-12 KR KR1020247024557A patent/KR20240125650A/ko unknown
- 2023-01-17 TW TW112101968A patent/TW202337877A/zh unknown
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02188540A (ja) * | 1989-01-13 | 1990-07-24 | Sumitomo Chem Co Ltd | メタノールの精製法 |
| JPH1112209A (ja) * | 1997-04-30 | 1999-01-19 | Asahi Chem Ind Co Ltd | 環状アルコールの分離取得方法 |
| JPH1180056A (ja) * | 1997-07-18 | 1999-03-23 | Asahi Chem Ind Co Ltd | 環状アルコールの分離取得方法 |
| JP2000007595A (ja) * | 1998-06-26 | 2000-01-11 | New Japan Chem Co Ltd | シクロヘキサンジメタノールの製造方法 |
| JP2001010999A (ja) | 1999-07-02 | 2001-01-16 | Mitsubishi Gas Chem Co Inc | トリシクロデカンジメタノール及び/又はペンタシクロペンタデカンジメタノールの製造法 |
| JP2001172214A (ja) * | 1999-12-16 | 2001-06-26 | Mitsubishi Gas Chem Co Inc | ジオールの製造方法 |
| JP2002047225A (ja) | 2000-07-27 | 2002-02-12 | Mitsubishi Gas Chem Co Inc | 多環式ジオール類の蒸留方法 |
| JP2002284771A (ja) * | 2001-03-28 | 2002-10-03 | Nof Corp | グリシドールの精製方法 |
| JP2003192621A (ja) | 2001-12-14 | 2003-07-09 | Celanese Chemicals Europe Gmbh | 脂環式アルコールの精製方法 |
| JP2004196778A (ja) | 2002-12-04 | 2004-07-15 | Mitsubishi Chemicals Corp | アルコールの製造方法 |
| JP2006514087A (ja) * | 2003-02-24 | 2006-04-27 | シエル・インターナシヨナル・リサーチ・マートスハツペイ・ベー・ヴエー | 蒸留による1,3−プロパンジオールの精製 |
| JP2010235516A (ja) * | 2009-03-31 | 2010-10-21 | Kuraray Co Ltd | 精製ジオールの製造方法 |
| JP2013501035A (ja) * | 2009-08-07 | 2013-01-10 | ランクセス・ドイチュランド・ゲーエムベーハー | トリメチロールプロパンの色数を改善する方法 |
| WO2011064184A1 (de) | 2009-11-26 | 2011-06-03 | Basf Se | Verfahren zur herstellung von kunststoffen mit 1,6-hexandiol mit einem aldehydanteil von kleiner 500ppm |
| JP2013512293A (ja) * | 2009-11-26 | 2013-04-11 | ビーエーエスエフ ソシエタス・ヨーロピア | アルデヒド含分500ppm未満を有する1,6−ヘキサンジオールを用いたプラスチックの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN118574806A (zh) | 2024-08-30 |
| TW202337877A (zh) | 2023-10-01 |
| JPWO2023140165A1 (ja) | 2023-07-27 |
| JP7501778B2 (ja) | 2024-06-18 |
| KR20240125650A (ko) | 2024-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1065194B2 (en) | Production of tricyclodecane dicarbaldehyde, pentacyclopentadecane dicarbaldehyde and corresponding dimethanols | |
| EP3696160B1 (en) | Extraction process for high-boiling aldehyde product separation and catalyst recovery | |
| CN100400490C (zh) | 制备三环癸烷二醛的方法 | |
| WO2004050591A1 (ja) | アルコールの製造方法 | |
| CN103687833B (zh) | 制备1,4-环己烷二甲醇的方法 | |
| JP7250039B2 (ja) | ノルマルブタノール、イソ-ブタノール、及び2-アルキルアルカノールを供給する方法 | |
| KR101200288B1 (ko) | Tcd-알코올 dm의 제조방법 | |
| KR20150031277A (ko) | 2-에틸헥산올로부터 출발하는, 구조적으로 분지형인 c9 모노카복실산들의 혼합물의 카복실산 에스테르들의 제조방법, 상기 혼합물의 트리에틸렌 글리콜, 네오펜틸 글리콜 및 1,3-부탄디올의 카복실산 에스테르들 및 이들의 용도 | |
| KR20190072568A (ko) | 2-알킬알칸올의 제조 방법 | |
| JP4573003B2 (ja) | トリシクロデカンジメタノール及び/又はペンタシクロペンタデカンジメタノールの製造法 | |
| JP7501778B2 (ja) | 多価アルコール類の製造方法 | |
| JP2022147136A (ja) | トリシクロデカンジメタノールの製造方法 | |
| JP2672473B2 (ja) | α−位においてアルキル残基により置換されたアルデヒドの製法 | |
| JP2009506105A (ja) | 界面活性剤用アルコールの製造方法 | |
| JP4754058B2 (ja) | イソプロピルアルコールの製造方法 | |
| US20100069678A1 (en) | Hydroformylation process | |
| JP2013523799A (ja) | 液相中での不斉第二級tert−ブチルアミンの製造方法 | |
| JP4573002B2 (ja) | トリシクロデカンジカルバルデヒド及び/又はペンタシクロペンタデカンジカルバルデヒドの製造法 | |
| JP4431844B2 (ja) | ジオールの製造方法 | |
| JP2010235516A (ja) | 精製ジオールの製造方法 | |
| JP2005281255A (ja) | 精製アルコールの製造方法 | |
| JP3864617B2 (ja) | アルコールの製造方法 | |
| CN116947611A (zh) | 一种氢甲酰化反应的方法和装置 | |
| JP4779294B2 (ja) | アルコールの製造方法 | |
| KR20040065236A (ko) | 알데히드의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023504436 Country of ref document: JP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23743160 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20247024557 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023743160 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023743160 Country of ref document: EP Effective date: 20240820 |