WO2022270611A1 - 抗ctla-4抗体 - Google Patents
抗ctla-4抗体 Download PDFInfo
- Publication number
- WO2022270611A1 WO2022270611A1 PCT/JP2022/025220 JP2022025220W WO2022270611A1 WO 2022270611 A1 WO2022270611 A1 WO 2022270611A1 JP 2022025220 W JP2022025220 W JP 2022025220W WO 2022270611 A1 WO2022270611 A1 WO 2022270611A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- sequence
- antibody
- amino acid
- ctla
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 438
- 102000004196 processed proteins & peptides Human genes 0.000 claims abstract description 428
- 229920001184 polypeptide Polymers 0.000 claims abstract description 427
- 229940045513 CTLA4 antagonist Drugs 0.000 claims abstract description 201
- 150000001413 amino acids Chemical class 0.000 claims abstract description 173
- 238000000034 method Methods 0.000 claims abstract description 154
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 57
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 56
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 56
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 361
- 230000027455 binding Effects 0.000 claims description 246
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 117
- 102000008203 CTLA-4 Antigen Human genes 0.000 claims description 112
- 108010021064 CTLA-4 Antigen Proteins 0.000 claims description 112
- 230000004075 alteration Effects 0.000 claims description 90
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 57
- 229960005305 adenosine Drugs 0.000 claims description 57
- 150000001875 compounds Chemical class 0.000 claims description 55
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 claims description 33
- 239000008194 pharmaceutical composition Substances 0.000 claims description 26
- 230000001472 cytotoxic effect Effects 0.000 claims description 20
- 241000282412 Homo Species 0.000 claims description 16
- 102000043321 human CTLA4 Human genes 0.000 claims description 16
- 241000699670 Mus sp. Species 0.000 claims description 10
- 239000003937 drug carrier Substances 0.000 claims description 10
- 230000001419 dependent effect Effects 0.000 claims description 9
- 238000012258 culturing Methods 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 229910052739 hydrogen Inorganic materials 0.000 claims description 2
- 229910052721 tungsten Inorganic materials 0.000 claims description 2
- 229910052727 yttrium Inorganic materials 0.000 claims description 2
- 230000004048 modification Effects 0.000 abstract description 63
- 238000012986 modification Methods 0.000 abstract description 63
- 238000004519 manufacturing process Methods 0.000 abstract description 31
- 210000004027 cell Anatomy 0.000 description 208
- 235000001014 amino acid Nutrition 0.000 description 188
- 229940024606 amino acid Drugs 0.000 description 173
- 241000282414 Homo sapiens Species 0.000 description 143
- 230000000694 effects Effects 0.000 description 80
- 206010028980 Neoplasm Diseases 0.000 description 79
- 239000000427 antigen Substances 0.000 description 71
- 108091007433 antigens Proteins 0.000 description 66
- 102000036639 antigens Human genes 0.000 description 66
- 108010073807 IgG Receptors Proteins 0.000 description 60
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 58
- 239000012636 effector Substances 0.000 description 56
- 102000009490 IgG Receptors Human genes 0.000 description 52
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 description 51
- 108010021468 Fc gamma receptor IIA Proteins 0.000 description 50
- 238000006467 substitution reaction Methods 0.000 description 50
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 48
- 230000006870 function Effects 0.000 description 47
- 101710099301 Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 44
- 102100029193 Low affinity immunoglobulin gamma Fc region receptor III-A Human genes 0.000 description 43
- 108090000623 proteins and genes Proteins 0.000 description 43
- 210000001519 tissue Anatomy 0.000 description 42
- 230000004481 post-translational protein modification Effects 0.000 description 41
- 238000003556 assay Methods 0.000 description 38
- 235000018102 proteins Nutrition 0.000 description 36
- 102000004169 proteins and genes Human genes 0.000 description 36
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 34
- 108010021472 Fc gamma receptor IIB Proteins 0.000 description 32
- 102100029205 Low affinity immunoglobulin gamma Fc region receptor II-b Human genes 0.000 description 32
- 210000003289 regulatory T cell Anatomy 0.000 description 32
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 31
- 239000013598 vector Substances 0.000 description 31
- 210000001744 T-lymphocyte Anatomy 0.000 description 30
- 125000000539 amino acid group Chemical group 0.000 description 30
- 230000002401 inhibitory effect Effects 0.000 description 30
- 239000000203 mixture Substances 0.000 description 27
- 230000005888 antibody-dependent cellular phagocytosis Effects 0.000 description 26
- 230000003213 activating effect Effects 0.000 description 23
- 230000014509 gene expression Effects 0.000 description 22
- 108060003951 Immunoglobulin Proteins 0.000 description 21
- 241001465754 Metazoa Species 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 21
- 239000003814 drug Substances 0.000 description 21
- 102000018358 immunoglobulin Human genes 0.000 description 21
- -1 nobuenbiquin Chemical compound 0.000 description 21
- 102000009109 Fc receptors Human genes 0.000 description 20
- 108010087819 Fc receptors Proteins 0.000 description 20
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 20
- 201000011510 cancer Diseases 0.000 description 20
- 238000000338 in vitro Methods 0.000 description 20
- 238000001727 in vivo Methods 0.000 description 20
- 238000005259 measurement Methods 0.000 description 19
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 19
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 18
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 18
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 18
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 18
- 239000012634 fragment Substances 0.000 description 18
- 230000001976 improved effect Effects 0.000 description 18
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 17
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 17
- LNQVTSROQXJCDD-UHFFFAOYSA-N adenosine monophosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(CO)C(OP(O)(O)=O)C1O LNQVTSROQXJCDD-UHFFFAOYSA-N 0.000 description 17
- 210000004408 hybridoma Anatomy 0.000 description 17
- 102000005962 receptors Human genes 0.000 description 17
- 108020003175 receptors Proteins 0.000 description 17
- 230000004071 biological effect Effects 0.000 description 16
- 229940127089 cytotoxic agent Drugs 0.000 description 16
- 238000005516 engineering process Methods 0.000 description 16
- 210000003462 vein Anatomy 0.000 description 16
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 15
- 201000010099 disease Diseases 0.000 description 15
- 230000001965 increasing effect Effects 0.000 description 14
- 238000003780 insertion Methods 0.000 description 14
- 230000037431 insertion Effects 0.000 description 14
- 239000003446 ligand Substances 0.000 description 14
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 13
- 230000004913 activation Effects 0.000 description 13
- 230000000259 anti-tumor effect Effects 0.000 description 13
- 238000010586 diagram Methods 0.000 description 13
- 238000010494 dissociation reaction Methods 0.000 description 13
- 230000005593 dissociations Effects 0.000 description 13
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 13
- 102100026120 IgG receptor FcRn large subunit p51 Human genes 0.000 description 12
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 12
- 241000699666 Mus <mouse, genus> Species 0.000 description 12
- 238000012217 deletion Methods 0.000 description 12
- 230000037430 deletion Effects 0.000 description 12
- 229940127121 immunoconjugate Drugs 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- 101710177940 IgG receptor FcRn large subunit p51 Proteins 0.000 description 11
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 11
- 230000001404 mediated effect Effects 0.000 description 11
- 230000003472 neutralizing effect Effects 0.000 description 11
- 239000003795 chemical substances by application Substances 0.000 description 10
- 239000000562 conjugate Substances 0.000 description 10
- 231100000599 cytotoxic agent Toxicity 0.000 description 10
- 210000002443 helper t lymphocyte Anatomy 0.000 description 10
- 238000010172 mouse model Methods 0.000 description 10
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 9
- 208000023275 Autoimmune disease Diseases 0.000 description 9
- 241000282693 Cercopithecidae Species 0.000 description 9
- 206010035226 Plasma cell myeloma Diseases 0.000 description 9
- 230000000295 complement effect Effects 0.000 description 9
- 239000002254 cytotoxic agent Substances 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 235000013922 glutamic acid Nutrition 0.000 description 9
- 239000004220 glutamic acid Chemical group 0.000 description 9
- 201000000050 myeloid neoplasm Diseases 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 238000002560 therapeutic procedure Methods 0.000 description 9
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 8
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical group OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- 238000004113 cell culture Methods 0.000 description 8
- 230000005779 cell damage Effects 0.000 description 8
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 8
- 230000002285 radioactive effect Effects 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 210000004881 tumor cell Anatomy 0.000 description 8
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 7
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 7
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 7
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 7
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 7
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 7
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 7
- 239000004472 Lysine Substances 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 7
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 208000037887 cell injury Diseases 0.000 description 7
- 230000006378 damage Effects 0.000 description 7
- 230000028993 immune response Effects 0.000 description 7
- 229960005386 ipilimumab Drugs 0.000 description 7
- 230000000670 limiting effect Effects 0.000 description 7
- 230000035772 mutation Effects 0.000 description 7
- 210000000822 natural killer cell Anatomy 0.000 description 7
- 150000002482 oligosaccharides Chemical class 0.000 description 7
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 7
- 238000003752 polymerase chain reaction Methods 0.000 description 7
- 230000002829 reductive effect Effects 0.000 description 7
- 238000012552 review Methods 0.000 description 7
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 7
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 6
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 6
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 6
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 6
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 6
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical group OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 6
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 6
- 108010029485 Protein Isoforms Proteins 0.000 description 6
- 102000001708 Protein Isoforms Human genes 0.000 description 6
- 108091008874 T cell receptors Proteins 0.000 description 6
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 230000004663 cell proliferation Effects 0.000 description 6
- 235000018417 cysteine Nutrition 0.000 description 6
- 230000003013 cytotoxicity Effects 0.000 description 6
- 231100000135 cytotoxicity Toxicity 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 6
- 230000013595 glycosylation Effects 0.000 description 6
- 238000006206 glycosylation reaction Methods 0.000 description 6
- 239000001963 growth medium Substances 0.000 description 6
- 230000016784 immunoglobulin production Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 230000003993 interaction Effects 0.000 description 6
- 108010068617 neonatal Fc receptor Proteins 0.000 description 6
- 229920001542 oligosaccharide Polymers 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 210000000952 spleen Anatomy 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 235000000346 sugar Nutrition 0.000 description 6
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 6
- 229960005486 vaccine Drugs 0.000 description 6
- 102000006306 Antigen Receptors Human genes 0.000 description 5
- 108010083359 Antigen Receptors Proteins 0.000 description 5
- 239000004475 Arginine Substances 0.000 description 5
- 241000894006 Bacteria Species 0.000 description 5
- 238000002965 ELISA Methods 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- 101001014668 Homo sapiens Glypican-3 Proteins 0.000 description 5
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 5
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 5
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 5
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 5
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 5
- 241000283973 Oryctolagus cuniculus Species 0.000 description 5
- 206010057249 Phagocytosis Diseases 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 239000012491 analyte Substances 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 5
- 210000004899 c-terminal region Anatomy 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 5
- 231100000433 cytotoxic Toxicity 0.000 description 5
- 230000003247 decreasing effect Effects 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 229960004679 doxorubicin Drugs 0.000 description 5
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 5
- 239000000710 homodimer Substances 0.000 description 5
- 102000048373 human GPC3 Human genes 0.000 description 5
- 210000002865 immune cell Anatomy 0.000 description 5
- 230000001900 immune effect Effects 0.000 description 5
- 210000000987 immune system Anatomy 0.000 description 5
- 210000004698 lymphocyte Anatomy 0.000 description 5
- 210000002540 macrophage Anatomy 0.000 description 5
- 229960000485 methotrexate Drugs 0.000 description 5
- 238000002823 phage display Methods 0.000 description 5
- 230000008782 phagocytosis Effects 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 238000000159 protein binding assay Methods 0.000 description 5
- 238000003127 radioimmunoassay Methods 0.000 description 5
- 230000004044 response Effects 0.000 description 5
- 229960004355 vindesine Drugs 0.000 description 5
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 5
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 4
- APLNAFMUEHKRLM-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)N=CN2 APLNAFMUEHKRLM-UHFFFAOYSA-N 0.000 description 4
- XTWYTFMLZFPYCI-KQYNXXCUSA-N 5'-adenylphosphoric acid Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XTWYTFMLZFPYCI-KQYNXXCUSA-N 0.000 description 4
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 4
- XTWYTFMLZFPYCI-UHFFFAOYSA-N Adenosine diphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(O)=O)C(O)C1O XTWYTFMLZFPYCI-UHFFFAOYSA-N 0.000 description 4
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 description 4
- 241000699800 Cricetinae Species 0.000 description 4
- 201000004624 Dermatitis Diseases 0.000 description 4
- 241000196324 Embryophyta Species 0.000 description 4
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 4
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 4
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 4
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 4
- 101100215928 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) ALY1 gene Proteins 0.000 description 4
- 230000006044 T cell activation Effects 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- 239000012472 biological sample Substances 0.000 description 4
- 150000001720 carbohydrates Chemical class 0.000 description 4
- 230000010261 cell growth Effects 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 230000024203 complement activation Effects 0.000 description 4
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 4
- 230000001086 cytosolic effect Effects 0.000 description 4
- 238000002784 cytotoxicity assay Methods 0.000 description 4
- 231100000263 cytotoxicity test Toxicity 0.000 description 4
- 239000013604 expression vector Substances 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 4
- 230000036039 immunity Effects 0.000 description 4
- 230000002163 immunogen Effects 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- 108091008042 inhibitory receptors Proteins 0.000 description 4
- 238000001155 isoelectric focusing Methods 0.000 description 4
- 210000003292 kidney cell Anatomy 0.000 description 4
- 210000000265 leukocyte Anatomy 0.000 description 4
- 210000004962 mammalian cell Anatomy 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000003094 microcapsule Substances 0.000 description 4
- 210000001616 monocyte Anatomy 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 241000894007 species Species 0.000 description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 4
- 108700012359 toxins Proteins 0.000 description 4
- 230000009261 transgenic effect Effects 0.000 description 4
- BKKWZCSSYWYNDS-JEDNCBNOSA-N 2-aminoacetic acid;(2s)-2,6-diaminohexanoic acid Chemical compound NCC(O)=O.NCCCC[C@H](N)C(O)=O BKKWZCSSYWYNDS-JEDNCBNOSA-N 0.000 description 3
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 3
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 3
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 3
- 206010003445 Ascites Diseases 0.000 description 3
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- 241000699802 Cricetulus griseus Species 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- 206010071602 Genetic polymorphism Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102000003886 Glycoproteins Human genes 0.000 description 3
- 108090000288 Glycoproteins Proteins 0.000 description 3
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 3
- 101000971533 Homo sapiens Killer cell lectin-like receptor subfamily G member 1 Proteins 0.000 description 3
- 101000917826 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-a Proteins 0.000 description 3
- 101001056707 Homo sapiens Proepiregulin Proteins 0.000 description 3
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 3
- 102100021457 Killer cell lectin-like receptor subfamily G member 1 Human genes 0.000 description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical group OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 3
- OVRNDRQMDRJTHS-RTRLPJTCSA-N N-acetyl-D-glucosamine Chemical compound CC(=O)N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-RTRLPJTCSA-N 0.000 description 3
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 3
- 102000004316 Oxidoreductases Human genes 0.000 description 3
- 108090000854 Oxidoreductases Proteins 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- 102000035195 Peptidases Human genes 0.000 description 3
- 229920001213 Polysorbate 20 Polymers 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 3
- 241000283984 Rodentia Species 0.000 description 3
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 3
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 3
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 230000009824 affinity maturation Effects 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 229940049595 antibody-drug conjugate Drugs 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 235000014633 carbohydrates Nutrition 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 229960004630 chlorambucil Drugs 0.000 description 3
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 230000021615 conjugation Effects 0.000 description 3
- 238000010276 construction Methods 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- 238000006911 enzymatic reaction Methods 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 229930013356 epothilone Natural products 0.000 description 3
- 229960002949 fluorouracil Drugs 0.000 description 3
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 3
- 235000008191 folinic acid Nutrition 0.000 description 3
- 239000011672 folinic acid Substances 0.000 description 3
- 230000033581 fucosylation Effects 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 239000000833 heterodimer Substances 0.000 description 3
- 102000043415 human EREG Human genes 0.000 description 3
- 230000008105 immune reaction Effects 0.000 description 3
- 230000003053 immunization Effects 0.000 description 3
- 230000005847 immunogenicity Effects 0.000 description 3
- 229940072221 immunoglobulins Drugs 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 230000008595 infiltration Effects 0.000 description 3
- 238000001764 infiltration Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 238000004255 ion exchange chromatography Methods 0.000 description 3
- 229960001691 leucovorin Drugs 0.000 description 3
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 230000003211 malignant effect Effects 0.000 description 3
- 238000013507 mapping Methods 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 210000000440 neutrophil Anatomy 0.000 description 3
- 210000001672 ovary Anatomy 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 3
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 230000001737 promoting effect Effects 0.000 description 3
- 238000010188 recombinant method Methods 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 238000010254 subcutaneous injection Methods 0.000 description 3
- 239000007929 subcutaneous injection Substances 0.000 description 3
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 3
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 231100000765 toxin Toxicity 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 238000011830 transgenic mouse model Methods 0.000 description 3
- 229930013292 trichothecene Natural products 0.000 description 3
- 150000003327 trichothecene derivatives Chemical class 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 2
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 2
- KHWCHTKSEGGWEX-RRKCRQDMSA-N 2'-deoxyadenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(O)=O)O1 KHWCHTKSEGGWEX-RRKCRQDMSA-N 0.000 description 2
- YJLUBHOZZTYQIP-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)NN=N2 YJLUBHOZZTYQIP-UHFFFAOYSA-N 0.000 description 2
- VPFUWHKTPYPNGT-UHFFFAOYSA-N 3-(3,4-dihydroxyphenyl)-1-(5-hydroxy-2,2-dimethylchromen-6-yl)propan-1-one Chemical compound OC1=C2C=CC(C)(C)OC2=CC=C1C(=O)CCC1=CC=C(O)C(O)=C1 VPFUWHKTPYPNGT-UHFFFAOYSA-N 0.000 description 2
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- 108091035707 Consensus sequence Proteins 0.000 description 2
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 101710140859 E3 ubiquitin ligase TRAF3IP2 Proteins 0.000 description 2
- 102100026620 E3 ubiquitin ligase TRAF3IP2 Human genes 0.000 description 2
- 102000001301 EGF receptor Human genes 0.000 description 2
- 108060006698 EGF receptor Proteins 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 108091006020 Fc-tagged proteins Proteins 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 2
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 2
- 101000917824 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-b Proteins 0.000 description 2
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 2
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 2
- 208000028622 Immune thrombocytopenia Diseases 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 2
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 108060001084 Luciferase Proteins 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 101000859077 Mus musculus Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 102000057297 Pepsin A Human genes 0.000 description 2
- 108090000284 Pepsin A Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Natural products OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 2
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 2
- 241000219061 Rheum Species 0.000 description 2
- 108010039491 Ricin Proteins 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- CYQFCXCEBYINGO-UHFFFAOYSA-N THC Natural products C1=C(C)CCC2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3C21 CYQFCXCEBYINGO-UHFFFAOYSA-N 0.000 description 2
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- DKJJVAGXPKPDRL-UHFFFAOYSA-N Tiludronic acid Chemical compound OP(O)(=O)C(P(O)(O)=O)SC1=CC=C(Cl)C=C1 DKJJVAGXPKPDRL-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- IBXPAFBDJCXCDW-MHFPCNPESA-A [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@H]2C[C@H](O)[C@@H](COP([S-])(=O)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3CO)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@H]2C[C@H](O)[C@@H](COP([S-])(=O)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3CO)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O IBXPAFBDJCXCDW-MHFPCNPESA-A 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 238000007792 addition Methods 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 238000012867 alanine scanning Methods 0.000 description 2
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 2
- 229960003896 aminopterin Drugs 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000005809 anti-tumor immunity Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940125644 antibody drug Drugs 0.000 description 2
- 239000000611 antibody drug conjugate Substances 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- 102000015736 beta 2-Microglobulin Human genes 0.000 description 2
- 108010081355 beta 2-Microglobulin Proteins 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- QZPQTZZNNJUOLS-UHFFFAOYSA-N beta-lapachone Chemical compound C12=CC=CC=C2C(=O)C(=O)C2=C1OC(C)(C)CC2 QZPQTZZNNJUOLS-UHFFFAOYSA-N 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 238000005251 capillar electrophoresis Methods 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 230000022534 cell killing Effects 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 238000002983 circular dichroism Methods 0.000 description 2
- HJKBJIYDJLVSAO-UHFFFAOYSA-L clodronic acid disodium salt Chemical compound [Na+].[Na+].OP([O-])(=O)C(Cl)(Cl)P(O)([O-])=O HJKBJIYDJLVSAO-UHFFFAOYSA-L 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000004590 computer program Methods 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- COFJBSXICYYSKG-OAUVCNBTSA-N cph2u7dndy Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 COFJBSXICYYSKG-OAUVCNBTSA-N 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- DAEAPNUQQAICNR-RRKCRQDMSA-K dADP(3-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP([O-])(=O)OP([O-])([O-])=O)O1 DAEAPNUQQAICNR-RRKCRQDMSA-K 0.000 description 2
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 230000006806 disease prevention Effects 0.000 description 2
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 2
- 229930188854 dolastatin Natural products 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 210000003754 fetus Anatomy 0.000 description 2
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- 101150034785 gamma gene Proteins 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 238000012239 gene modification Methods 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 2
- 238000005734 heterodimerization reaction Methods 0.000 description 2
- 238000013537 high throughput screening Methods 0.000 description 2
- 230000013632 homeostatic process Effects 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 229960001101 ifosfamide Drugs 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 230000005965 immune activity Effects 0.000 description 2
- 230000037451 immune surveillance Effects 0.000 description 2
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000000099 in vitro assay Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 238000002595 magnetic resonance imaging Methods 0.000 description 2
- 230000008774 maternal effect Effects 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 210000004379 membrane Anatomy 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 2
- 239000002105 nanoparticle Substances 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 229940111202 pepsin Drugs 0.000 description 2
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229940068977 polysorbate 20 Drugs 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 235000019419 proteases Nutrition 0.000 description 2
- 230000005180 public health Effects 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 2
- 238000003571 reporter gene assay Methods 0.000 description 2
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 229960003787 sorafenib Drugs 0.000 description 2
- PVYJZLYGTZKPJE-UHFFFAOYSA-N streptonigrin Chemical compound C=1C=C2C(=O)C(OC)=C(N)C(=O)C2=NC=1C(C=1N)=NC(C(O)=O)=C(C)C=1C1=CC=C(OC)C(OC)=C1O PVYJZLYGTZKPJE-UHFFFAOYSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 229920003169 water-soluble polymer Polymers 0.000 description 2
- 229960004276 zoledronic acid Drugs 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- JKHVDAUOODACDU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(2,5-dioxopyrrol-1-yl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCN1C(=O)C=CC1=O JKHVDAUOODACDU-UHFFFAOYSA-N 0.000 description 1
- PVGATNRYUYNBHO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(2,5-dioxopyrrol-1-yl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O PVGATNRYUYNBHO-UHFFFAOYSA-N 0.000 description 1
- BQWBEDSJTMWJAE-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[(2-iodoacetyl)amino]benzoate Chemical compound C1=CC(NC(=O)CI)=CC=C1C(=O)ON1C(=O)CCC1=O BQWBEDSJTMWJAE-UHFFFAOYSA-N 0.000 description 1
- PMJWDPGOWBRILU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCC(C=C1)=CC=C1N1C(=O)C=CC1=O PMJWDPGOWBRILU-UHFFFAOYSA-N 0.000 description 1
- VLARLSIGSPVYHX-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-(2,5-dioxopyrrol-1-yl)hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCN1C(=O)C=CC1=O VLARLSIGSPVYHX-UHFFFAOYSA-N 0.000 description 1
- WCMOHMXWOOBVMZ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[3-(2,5-dioxopyrrol-1-yl)propanoylamino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)CCN1C(=O)C=CC1=O WCMOHMXWOOBVMZ-UHFFFAOYSA-N 0.000 description 1
- IHVODYOQUSEYJJ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]amino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)C(CC1)CCC1CN1C(=O)C=CC1=O IHVODYOQUSEYJJ-UHFFFAOYSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- RIWLPSIAFBLILR-WVNGMBSFSA-N (2s)-1-[(2s)-2-[[(2s,3s)-2-[[(2s)-2-[[(2s,3r)-2-[[(2r,3s)-2-[[(2s)-2-[[2-[[2-[acetyl(methyl)amino]acetyl]amino]acetyl]amino]-3-methylbutanoyl]amino]-3-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]pentanoyl]amino]-3-methylpentanoyl]amino]-5-(diaminomethy Chemical compound CC(=O)N(C)CC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1CCC[C@H]1C(=O)NCC RIWLPSIAFBLILR-WVNGMBSFSA-N 0.000 description 1
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 1
- YXTKHLHCVFUPPT-YYFJYKOTSA-N (2s)-2-[[4-[(2-amino-5-formyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid;(1r,2r)-1,2-dimethanidylcyclohexane;5-fluoro-1h-pyrimidine-2,4-dione;oxalic acid;platinum(2+) Chemical compound [Pt+2].OC(=O)C(O)=O.[CH2-][C@@H]1CCCC[C@H]1[CH2-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 YXTKHLHCVFUPPT-YYFJYKOTSA-N 0.000 description 1
- FLWWDYNPWOSLEO-HQVZTVAUSA-N (2s)-2-[[4-[1-(2-amino-4-oxo-1h-pteridin-6-yl)ethyl-methylamino]benzoyl]amino]pentanedioic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1C(C)N(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FLWWDYNPWOSLEO-HQVZTVAUSA-N 0.000 description 1
- TVIRNGFXQVMMGB-OFWIHYRESA-N (3s,6r,10r,13e,16s)-16-[(2r,3r,4s)-4-chloro-3-hydroxy-4-phenylbutan-2-yl]-10-[(3-chloro-4-methoxyphenyl)methyl]-6-methyl-3-(2-methylpropyl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetrone Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H](O)[C@@H](Cl)C=2C=CC=CC=2)C/C=C/C(=O)N1 TVIRNGFXQVMMGB-OFWIHYRESA-N 0.000 description 1
- XRBSKUSTLXISAB-XVVDYKMHSA-N (5r,6r,7r,8r)-8-hydroxy-7-(hydroxymethyl)-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrobenzo[f][1,3]benzodioxole-6-carboxylic acid Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H](CO)[C@@H]2C(O)=O)=C1 XRBSKUSTLXISAB-XVVDYKMHSA-N 0.000 description 1
- IEUUDEWWMRQUDS-UHFFFAOYSA-N (6-azaniumylidene-1,6-dimethoxyhexylidene)azanium;dichloride Chemical compound Cl.Cl.COC(=N)CCCCC(=N)OC IEUUDEWWMRQUDS-UHFFFAOYSA-N 0.000 description 1
- AESVUZLWRXEGEX-DKCAWCKPSA-N (7S,9R)-7-[(2S,4R,5R,6R)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione iron(3+) Chemical compound [Fe+3].COc1cccc2C(=O)c3c(O)c4C[C@@](O)(C[C@H](O[C@@H]5C[C@@H](N)[C@@H](O)[C@@H](C)O5)c4c(O)c3C(=O)c12)C(=O)CO AESVUZLWRXEGEX-DKCAWCKPSA-N 0.000 description 1
- INAUWOVKEZHHDM-PEDBPRJASA-N (7s,9s)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-7-[(2r,4s,5s,6s)-5-hydroxy-6-methyl-4-morpholin-4-yloxan-2-yl]oxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1 INAUWOVKEZHHDM-PEDBPRJASA-N 0.000 description 1
- RCFNNLSZHVHCEK-IMHLAKCZSA-N (7s,9s)-7-(4-amino-6-methyloxan-2-yl)oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound [Cl-].O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)C1CC([NH3+])CC(C)O1 RCFNNLSZHVHCEK-IMHLAKCZSA-N 0.000 description 1
- NOPNWHSMQOXAEI-PUCKCBAPSA-N (7s,9s)-7-[(2r,4s,5s,6s)-4-(2,3-dihydropyrrol-1-yl)-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCC=C1 NOPNWHSMQOXAEI-PUCKCBAPSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- AGNGYMCLFWQVGX-AGFFZDDWSA-N (e)-1-[(2s)-2-amino-2-carboxyethoxy]-2-diazonioethenolate Chemical compound OC(=O)[C@@H](N)CO\C([O-])=C\[N+]#N AGNGYMCLFWQVGX-AGFFZDDWSA-N 0.000 description 1
- FQERLIOIVXPZKH-UHFFFAOYSA-N 1,2,4-trioxane Chemical compound C1COOCO1 FQERLIOIVXPZKH-UHFFFAOYSA-N 0.000 description 1
- VILFTWLXLYIEMV-UHFFFAOYSA-N 1,5-difluoro-2,4-dinitrobenzene Chemical compound [O-][N+](=O)C1=CC([N+]([O-])=O)=C(F)C=C1F VILFTWLXLYIEMV-UHFFFAOYSA-N 0.000 description 1
- KZEVSDGEBAJOTK-UHFFFAOYSA-N 1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-2-[5-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]ethanone Chemical compound N1N=NC=2CN(CCC=21)C(CC=1OC(=NN=1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)=O KZEVSDGEBAJOTK-UHFFFAOYSA-N 0.000 description 1
- DIYPCWKHSODVAP-UHFFFAOYSA-N 1-[3-(2,5-dioxopyrrol-1-yl)benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=CC(N2C(C=CC2=O)=O)=C1 DIYPCWKHSODVAP-UHFFFAOYSA-N 0.000 description 1
- CULQNACJHGHAER-UHFFFAOYSA-N 1-[4-[(2-iodoacetyl)amino]benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=C(NC(=O)CI)C=C1 CULQNACJHGHAER-UHFFFAOYSA-N 0.000 description 1
- MQLACMBJVPINKE-UHFFFAOYSA-N 10-[(3-hydroxy-4-methoxyphenyl)methylidene]anthracen-9-one Chemical compound C1=C(O)C(OC)=CC=C1C=C1C2=CC=CC=C2C(=O)C2=CC=CC=C21 MQLACMBJVPINKE-UHFFFAOYSA-N 0.000 description 1
- YBBNVCVOACOHIG-UHFFFAOYSA-N 2,2-diamino-1,4-bis(4-azidophenyl)-3-butylbutane-1,4-dione Chemical compound C=1C=C(N=[N+]=[N-])C=CC=1C(=O)C(N)(N)C(CCCC)C(=O)C1=CC=C(N=[N+]=[N-])C=C1 YBBNVCVOACOHIG-UHFFFAOYSA-N 0.000 description 1
- BTOTXLJHDSNXMW-POYBYMJQSA-N 2,3-dideoxyuridine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(=O)NC(=O)C=C1 BTOTXLJHDSNXMW-POYBYMJQSA-N 0.000 description 1
- KGLPWQKSKUVKMJ-UHFFFAOYSA-N 2,3-dihydrophthalazine-1,4-dione Chemical compound C1=CC=C2C(=O)NNC(=O)C2=C1 KGLPWQKSKUVKMJ-UHFFFAOYSA-N 0.000 description 1
- BOMZMNZEXMAQQW-UHFFFAOYSA-N 2,5,11-trimethyl-6h-pyrido[4,3-b]carbazol-2-ium-9-ol;acetate Chemical compound CC([O-])=O.C[N+]1=CC=C2C(C)=C(NC=3C4=CC(O)=CC=3)C4=C(C)C2=C1 BOMZMNZEXMAQQW-UHFFFAOYSA-N 0.000 description 1
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- FZDFGHZZPBUTGP-UHFFFAOYSA-N 2-[[2-[bis(carboxymethyl)amino]-3-(4-isothiocyanatophenyl)propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)C(C)CN(CC(O)=O)CC(N(CC(O)=O)CC(O)=O)CC1=CC=C(N=C=S)C=C1 FZDFGHZZPBUTGP-UHFFFAOYSA-N 0.000 description 1
- FBUTXZSKZCQABC-UHFFFAOYSA-N 2-amino-1-methyl-7h-purine-6-thione Chemical compound S=C1N(C)C(N)=NC2=C1NC=N2 FBUTXZSKZCQABC-UHFFFAOYSA-N 0.000 description 1
- QCXJFISCRQIYID-IAEPZHFASA-N 2-amino-1-n-[(3s,6s,7r,10s,16s)-3-[(2s)-butan-2-yl]-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-10-propan-2-yl-8-oxa-1,4,11,14-tetrazabicyclo[14.3.0]nonadecan-6-yl]-4,6-dimethyl-3-oxo-9-n-[(3s,6s,7r,10s,16s)-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-3,10-di(propa Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N=C2C(C(=O)N[C@@H]3C(=O)N[C@H](C(N4CCC[C@H]4C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]3C)=O)[C@@H](C)CC)=C(N)C(=O)C(C)=C2O2)C2=C(C)C=C1 QCXJFISCRQIYID-IAEPZHFASA-N 0.000 description 1
- YIMDLWDNDGKDTJ-QLKYHASDSA-N 3'-deamino-3'-(3-cyanomorpholin-4-yl)doxorubicin Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1C#N YIMDLWDNDGKDTJ-QLKYHASDSA-N 0.000 description 1
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 1
- AZKSAVLVSZKNRD-UHFFFAOYSA-M 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Chemical compound [Br-].S1C(C)=C(C)N=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 AZKSAVLVSZKNRD-UHFFFAOYSA-M 0.000 description 1
- PWMYMKOUNYTVQN-UHFFFAOYSA-N 3-(8,8-diethyl-2-aza-8-germaspiro[4.5]decan-2-yl)-n,n-dimethylpropan-1-amine Chemical compound C1C[Ge](CC)(CC)CCC11CN(CCCN(C)C)CC1 PWMYMKOUNYTVQN-UHFFFAOYSA-N 0.000 description 1
- QGJZLNKBHJESQX-UHFFFAOYSA-N 3-Epi-Betulin-Saeure Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C(=C)C)C5C4CCC3C21C QGJZLNKBHJESQX-UHFFFAOYSA-N 0.000 description 1
- CLOUCVRNYSHRCF-UHFFFAOYSA-N 3beta-Hydroxy-20(29)-Lupen-3,27-oic acid Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C(O)=O)CCC5(C)CCC(C(=C)C)C5C4CCC3C21C CLOUCVRNYSHRCF-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- ZMRMMAOBSFSXLN-UHFFFAOYSA-N 4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanehydrazide Chemical compound C1=CC(CCCC(=O)NN)=CC=C1N1C(=O)C=CC1=O ZMRMMAOBSFSXLN-UHFFFAOYSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- WYXSYVWAUAUWLD-SHUUEZRQSA-N 6-azauridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 WYXSYVWAUAUWLD-SHUUEZRQSA-N 0.000 description 1
- 229960005538 6-diazo-5-oxo-L-norleucine Drugs 0.000 description 1
- YCWQAMGASJSUIP-YFKPBYRVSA-N 6-diazo-5-oxo-L-norleucine Chemical compound OC(=O)[C@@H](N)CCC(=O)C=[N+]=[N-] YCWQAMGASJSUIP-YFKPBYRVSA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- CJIJXIFQYOPWTF-UHFFFAOYSA-N 7-hydroxycoumarin Natural products O1C(=O)C=CC2=CC(O)=CC=C21 CJIJXIFQYOPWTF-UHFFFAOYSA-N 0.000 description 1
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 description 1
- FUXVKZWTXQUGMW-FQEVSTJZSA-N 9-Aminocamptothecin Chemical compound C1=CC(N)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 FUXVKZWTXQUGMW-FQEVSTJZSA-N 0.000 description 1
- HDZZVAMISRMYHH-UHFFFAOYSA-N 9beta-Ribofuranosyl-7-deazaadenin Natural products C1=CC=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O HDZZVAMISRMYHH-UHFFFAOYSA-N 0.000 description 1
- 108010066676 Abrin Proteins 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000026872 Addison Disease Diseases 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-N Adenosine triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-N 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 206010050245 Autoimmune thrombocytopenia Diseases 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 108091008875 B cell receptors Proteins 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 230000003844 B-cell-activation Effects 0.000 description 1
- 239000012664 BCL-2-inhibitor Substances 0.000 description 1
- 108090000363 Bacterial Luciferases Proteins 0.000 description 1
- 229940123711 Bcl2 inhibitor Drugs 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- DIZWSDNSTNAYHK-XGWVBXMLSA-N Betulinic acid Natural products CC(=C)[C@@H]1C[C@H]([C@H]2CC[C@]3(C)[C@H](CC[C@@H]4[C@@]5(C)CC[C@H](O)C(C)(C)[C@@H]5CC[C@@]34C)[C@@H]12)C(=O)O DIZWSDNSTNAYHK-XGWVBXMLSA-N 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 210000001239 CD8-positive, alpha-beta cytotoxic T lymphocyte Anatomy 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 101710158575 Cap-specific mRNA (nucleoside-2'-O-)-methyltransferase Proteins 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 231100000023 Cell-mediated cytotoxicity Toxicity 0.000 description 1
- 206010057250 Cell-mediated cytotoxicity Diseases 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- 108010023736 Chondroitinases and Chondroitin Lyases Proteins 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 108700032819 Croton tiglium crotin II Proteins 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- 229920000832 Cutin Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- 206010011878 Deafness Diseases 0.000 description 1
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 1
- XXGMIHXASFDFSM-UHFFFAOYSA-N Delta9-tetrahydrocannabinol Natural products CCCCCc1cc2OC(C)(C)C3CCC(=CC3c2c(O)c1O)C XXGMIHXASFDFSM-UHFFFAOYSA-N 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- AUGQEEXBDZWUJY-ZLJUKNTDSA-N Diacetoxyscirpenol Chemical compound C([C@]12[C@]3(C)[C@H](OC(C)=O)[C@@H](O)[C@H]1O[C@@H]1C=C(C)CC[C@@]13COC(=O)C)O2 AUGQEEXBDZWUJY-ZLJUKNTDSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- CYQFCXCEBYINGO-DLBZAZTESA-N Dronabinol Natural products C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@H]21 CYQFCXCEBYINGO-DLBZAZTESA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- OBMLHUPNRURLOK-XGRAFVIBSA-N Epitiostanol Chemical compound C1[C@@H]2S[C@@H]2C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 OBMLHUPNRURLOK-XGRAFVIBSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 1
- 101710082714 Exotoxin A Proteins 0.000 description 1
- 206010073306 Exposure to radiation Diseases 0.000 description 1
- 206010015866 Extravasation Diseases 0.000 description 1
- 108010021470 Fc gamma receptor IIC Proteins 0.000 description 1
- 108090000331 Firefly luciferases Proteins 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- MPJKWIXIYCLVCU-UHFFFAOYSA-N Folinic acid Natural products NC1=NC2=C(N(C=O)C(CNc3ccc(cc3)C(=O)NC(CCC(=O)O)CC(=O)O)CN2)C(=O)N1 MPJKWIXIYCLVCU-UHFFFAOYSA-N 0.000 description 1
- 108010015133 Galactose oxidase Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 108010073178 Glucan 1,4-alpha-Glucosidase Proteins 0.000 description 1
- 102100022624 Glucoamylase Human genes 0.000 description 1
- 239000004366 Glucose oxidase Substances 0.000 description 1
- 108010015776 Glucose oxidase Proteins 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 208000003807 Graves Disease Diseases 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 101000819490 Homo sapiens Alpha-(1,6)-fucosyltransferase Proteins 0.000 description 1
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 1
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 1
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 1
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 1
- 108010003272 Hyaluronate lyase Proteins 0.000 description 1
- 102000001974 Hyaluronidases Human genes 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical class C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 1
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 108010041012 Integrin alpha4 Proteins 0.000 description 1
- 238000012695 Interfacial polymerization Methods 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 102000004889 Interleukin-6 Human genes 0.000 description 1
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 201000010743 Lambert-Eaton myasthenic syndrome Diseases 0.000 description 1
- 229920001491 Lentinan Polymers 0.000 description 1
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 1
- MEPSBMMZQBMKHM-UHFFFAOYSA-N Lomatiol Natural products CC(=C/CC1=C(O)C(=O)c2ccccc2C1=O)CO MEPSBMMZQBMKHM-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 1
- 102100029206 Low affinity immunoglobulin gamma Fc region receptor II-c Human genes 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 208000005777 Lupus Nephritis Diseases 0.000 description 1
- 229910052765 Lutetium Inorganic materials 0.000 description 1
- 239000004907 Macro-emulsion Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- VJRAUFKOOPNFIQ-UHFFFAOYSA-N Marcellomycin Natural products C12=C(O)C=3C(=O)C4=C(O)C=CC(O)=C4C(=O)C=3C=C2C(C(=O)OC)C(CC)(O)CC1OC(OC1C)CC(N(C)C)C1OC(OC1C)CC(O)C1OC1CC(O)C(O)C(C)O1 VJRAUFKOOPNFIQ-UHFFFAOYSA-N 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 244000302512 Momordica charantia Species 0.000 description 1
- 235000009811 Momordica charantia Nutrition 0.000 description 1
- 102000016943 Muramidase Human genes 0.000 description 1
- 108010014251 Muramidase Proteins 0.000 description 1
- 206010028424 Myasthenic syndrome Diseases 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 1
- 102100031688 N-acetylgalactosamine-6-sulfatase Human genes 0.000 description 1
- 108010072915 NAc-Sar-Gly-Val-(d-allo-Ile)-Thr-Nva-Ile-Arg-ProNEt Proteins 0.000 description 1
- 206010029240 Neuritis Diseases 0.000 description 1
- SYNHCENRCUAUNM-UHFFFAOYSA-N Nitrogen mustard N-oxide hydrochloride Chemical compound Cl.ClCC[N+]([O-])(C)CCCl SYNHCENRCUAUNM-UHFFFAOYSA-N 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 241000238413 Octopus Species 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241000609499 Palicourea Species 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 206010034277 Pemphigoid Diseases 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 208000031845 Pernicious anaemia Diseases 0.000 description 1
- 240000007643 Phytolacca americana Species 0.000 description 1
- 235000009074 Phytolacca americana Nutrition 0.000 description 1
- 101100413173 Phytolacca americana PAP2 gene Proteins 0.000 description 1
- 206010036105 Polyneuropathy Diseases 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Porfiromycine Chemical compound O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 1
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 1
- 102100024924 Protein kinase C alpha type Human genes 0.000 description 1
- 101710109947 Protein kinase C alpha type Proteins 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 102220612914 Putative uncharacterized protein PIK3CD-AS1_Y12W_mutation Human genes 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 208000003782 Raynaud disease Diseases 0.000 description 1
- 208000012322 Raynaud phenomenon Diseases 0.000 description 1
- 101710138747 Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- NSFWWJIQIKBZMJ-YKNYLIOZSA-N Roridin A Chemical compound C([C@]12[C@]3(C)[C@H]4C[C@H]1O[C@@H]1C=C(C)CC[C@@]13COC(=O)[C@@H](O)[C@H](C)CCO[C@H](\C=C\C=C/C(=O)O4)[C@H](O)C)O2 NSFWWJIQIKBZMJ-YKNYLIOZSA-N 0.000 description 1
- CIEYTVIYYGTCCI-UHFFFAOYSA-N SJ000286565 Natural products C1=CC=C2C(=O)C(CC=C(C)C)=C(O)C(=O)C2=C1 CIEYTVIYYGTCCI-UHFFFAOYSA-N 0.000 description 1
- 240000003946 Saponaria officinalis Species 0.000 description 1
- 229920002305 Schizophyllan Polymers 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- 241000191940 Staphylococcus Species 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- BXFOFFBJRFZBQZ-QYWOHJEZSA-N T-2 toxin Chemical compound C([C@@]12[C@]3(C)[C@H](OC(C)=O)[C@@H](O)[C@H]1O[C@H]1[C@]3(COC(C)=O)C[C@@H](C(=C1)C)OC(=O)CC(C)C)O2 BXFOFFBJRFZBQZ-QYWOHJEZSA-N 0.000 description 1
- WFWLQNSHRPWKFK-UHFFFAOYSA-N Tegafur Chemical compound O=C1NC(=O)C(F)=CN1C1OCCC1 WFWLQNSHRPWKFK-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 241000473945 Theria <moth genus> Species 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- FYAMXEPQQLNQDM-UHFFFAOYSA-N Tris(1-aziridinyl)phosphine oxide Chemical compound C1CN1P(N1CC1)(=O)N1CC1 FYAMXEPQQLNQDM-UHFFFAOYSA-N 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 101710162629 Trypsin inhibitor Proteins 0.000 description 1
- 229940122618 Trypsin inhibitor Drugs 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 1
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 description 1
- VGQOVCHZGQWAOI-UHFFFAOYSA-N UNPD55612 Natural products N1C(O)C2CC(C=CC(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-UHFFFAOYSA-N 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 108010092464 Urate Oxidase Proteins 0.000 description 1
- 101150117115 V gene Proteins 0.000 description 1
- 244000000188 Vaccinium ovalifolium Species 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 240000001866 Vernicia fordii Species 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 241000863480 Vinca Species 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 108010093894 Xanthine oxidase Proteins 0.000 description 1
- 102100033220 Xanthine oxidase Human genes 0.000 description 1
- SPJCRMJCFSJKDE-ZWBUGVOYSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] 2-[4-[bis(2-chloroethyl)amino]phenyl]acetate Chemical compound O([C@@H]1CC2=CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)C(=O)CC1=CC=C(N(CCCl)CCCl)C=C1 SPJCRMJCFSJKDE-ZWBUGVOYSA-N 0.000 description 1
- MBHRHUJRKGNOKX-UHFFFAOYSA-N [(4,6-diamino-1,3,5-triazin-2-yl)amino]methanol Chemical class NC1=NC(N)=NC(NCO)=N1 MBHRHUJRKGNOKX-UHFFFAOYSA-N 0.000 description 1
- XZSRRNFBEIOBDA-CFNBKWCHSA-N [2-[(2s,4s)-4-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]-2-oxoethyl] 2,2-diethoxyacetate Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)C(OCC)OCC)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 XZSRRNFBEIOBDA-CFNBKWCHSA-N 0.000 description 1
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 1
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 1
- 108010023617 abarelix Proteins 0.000 description 1
- 229960002184 abarelix Drugs 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 229940028652 abraxane Drugs 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 229940037127 actonel Drugs 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- NLTUCYMLOPLUHL-KQYNXXCUSA-N adenosine 5'-[gamma-thio]triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=S)[C@@H](O)[C@H]1O NLTUCYMLOPLUHL-KQYNXXCUSA-N 0.000 description 1
- 229960001456 adenosine triphosphate Drugs 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229940062527 alendronate Drugs 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 108010001818 alpha-sarcin Proteins 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229940037003 alum Drugs 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960002749 aminolevulinic acid Drugs 0.000 description 1
- 210000004381 amniotic fluid Anatomy 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- BBDAGFIXKZCXAH-CCXZUQQUSA-N ancitabine Chemical compound N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 BBDAGFIXKZCXAH-CCXZUQQUSA-N 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- VGQOVCHZGQWAOI-HYUHUPJXSA-N anthramycin Chemical compound N1[C@@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-HYUHUPJXSA-N 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000009830 antibody antigen interaction Effects 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 238000010913 antigen-directed enzyme pro-drug therapy Methods 0.000 description 1
- 210000000612 antigen-presenting cell Anatomy 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045686 antimetabolites antineoplastic purine analogs Drugs 0.000 description 1
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 description 1
- 229940045713 antineoplastic alkylating drug ethylene imines Drugs 0.000 description 1
- 229940045688 antineoplastic antimetabolites pyrimidine analogues Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000008209 arabinosides Chemical class 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 229950011321 azaserine Drugs 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 1
- QGJZLNKBHJESQX-FZFNOLFKSA-N betulinic acid Chemical compound C1C[C@H](O)C(C)(C)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CC[C@@H](C(=C)C)[C@@H]5[C@H]4CC[C@@H]3[C@]21C QGJZLNKBHJESQX-FZFNOLFKSA-N 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 238000013357 binding ELISA Methods 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- HUTDDBSSHVOYJR-UHFFFAOYSA-H bis[(2-oxo-1,3,2$l^{5},4$l^{2}-dioxaphosphaplumbetan-2-yl)oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O HUTDDBSSHVOYJR-UHFFFAOYSA-H 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- 229960005520 bryostatin Drugs 0.000 description 1
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 description 1
- MUIWQCKLQMOUAT-AKUNNTHJSA-N bryostatin 20 Natural products COC(=O)C=C1C[C@@]2(C)C[C@]3(O)O[C@](C)(C[C@@H](O)CC(=O)O[C@](C)(C[C@@]4(C)O[C@](O)(CC5=CC(=O)O[C@]45C)C(C)(C)C=C[C@@](C)(C1)O2)[C@@H](C)O)C[C@H](OC(=O)C(C)(C)C)C3(C)C MUIWQCKLQMOUAT-AKUNNTHJSA-N 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N butyl alcohol Substances CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 1
- 108700002839 cactinomycin Proteins 0.000 description 1
- 229950009908 cactinomycin Drugs 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- XREUEWVEMYWFFA-CSKJXFQVSA-N carminomycin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XREUEWVEMYWFFA-CSKJXFQVSA-N 0.000 description 1
- 229930188550 carminomycin Natural products 0.000 description 1
- XREUEWVEMYWFFA-UHFFFAOYSA-N carminomycin I Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XREUEWVEMYWFFA-UHFFFAOYSA-N 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 229950001725 carubicin Drugs 0.000 description 1
- BBZDXMBRAFTCAA-AREMUKBSSA-N carzelesin Chemical compound C1=2NC=C(C)C=2C([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)C3=CC4=CC=C(C=C4O3)N(CC)CC)=C2C=C1OC(=O)NC1=CC=CC=C1 BBZDXMBRAFTCAA-AREMUKBSSA-N 0.000 description 1
- 229950007509 carzelesin Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 230000005890 cell-mediated cytotoxicity Effects 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 230000012085 chronic inflammatory response Effects 0.000 description 1
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 238000005354 coacervation Methods 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 238000007398 colorimetric assay Methods 0.000 description 1
- 210000003022 colostrum Anatomy 0.000 description 1
- 235000021277 colostrum Nutrition 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 108010047295 complement receptors Proteins 0.000 description 1
- 102000006834 complement receptors Human genes 0.000 description 1
- 230000004154 complement system Effects 0.000 description 1
- 230000009827 complement-dependent cellular cytotoxicity Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000011970 concomitant therapy Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 201000003278 cryoglobulinemia Diseases 0.000 description 1
- 108010089438 cryptophycin 1 Proteins 0.000 description 1
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 1
- 108010090203 cryptophycin 8 Proteins 0.000 description 1
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 239000002619 cytotoxin Substances 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 125000001295 dansyl group Chemical group [H]C1=C([H])C(N(C([H])([H])[H])C([H])([H])[H])=C2C([H])=C([H])C([H])=C(C2=C1[H])S(*)(=O)=O 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 229950003913 detorubicin Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- PZXJOHSZQAEJFE-UHFFFAOYSA-N dihydrobetulinic acid Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C(C)C)C5C4CCC3C21C PZXJOHSZQAEJFE-UHFFFAOYSA-N 0.000 description 1
- 125000005442 diisocyanate group Chemical group 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 206010013023 diphtheria Diseases 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000009300 dissolved air flotation Methods 0.000 description 1
- ZWIBGKZDAWNIFC-UHFFFAOYSA-N disuccinimidyl suberate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCCC(=O)ON1C(=O)CCC1=O ZWIBGKZDAWNIFC-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229940115080 doxil Drugs 0.000 description 1
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 1
- 229960004242 dronabinol Drugs 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- FSIRXIHZBIXHKT-MHTVFEQDSA-N edatrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CC(CC)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FSIRXIHZBIXHKT-MHTVFEQDSA-N 0.000 description 1
- 229950006700 edatrexate Drugs 0.000 description 1
- 210000003162 effector t lymphocyte Anatomy 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 229950000549 elliptinium acetate Drugs 0.000 description 1
- 229940120655 eloxatin Drugs 0.000 description 1
- JOZGNYDSEBIJDH-UHFFFAOYSA-N eniluracil Chemical compound O=C1NC=C(C#C)C(=O)N1 JOZGNYDSEBIJDH-UHFFFAOYSA-N 0.000 description 1
- 229950010213 eniluracil Drugs 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229950002973 epitiostanol Drugs 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- QCYAXXZCQKMTMO-QFIPXVFZSA-N ethyl (2s)-2-[(2-bromo-3-oxospiro[3.5]non-1-en-1-yl)amino]-3-[4-(2,7-naphthyridin-1-ylamino)phenyl]propanoate Chemical compound N([C@@H](CC=1C=CC(NC=2C3=CN=CC=C3C=CN=2)=CC=1)C(=O)OCC)C1=C(Br)C(=O)C11CCCCC1 QCYAXXZCQKMTMO-QFIPXVFZSA-N 0.000 description 1
- 229940009626 etidronate Drugs 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 229960004945 etoricoxib Drugs 0.000 description 1
- MNJVRJDLRVPLFE-UHFFFAOYSA-N etoricoxib Chemical compound C1=NC(C)=CC=C1C1=NC=C(Cl)C=C1C1=CC=C(S(C)(=O)=O)C=C1 MNJVRJDLRVPLFE-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000036251 extravasation Effects 0.000 description 1
- 230000008713 feedback mechanism Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 238000005558 fluorometry Methods 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 150000002224 folic acids Chemical class 0.000 description 1
- 229940001490 fosamax Drugs 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229940020967 gemzar Drugs 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000005017 genetic modification Effects 0.000 description 1
- 235000013617 genetically modified food Nutrition 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940116332 glucose oxidase Drugs 0.000 description 1
- 235000019420 glucose oxidase Nutrition 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 239000003966 growth inhibitor Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 102000053350 human FCGR3B Human genes 0.000 description 1
- 229960002773 hyaluronidase Drugs 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- KNOSIOWNDGUGFJ-UHFFFAOYSA-N hydroxysesamone Natural products C1=CC(O)=C2C(=O)C(CC=C(C)C)=C(O)C(=O)C2=C1O KNOSIOWNDGUGFJ-UHFFFAOYSA-N 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 150000002463 imidates Chemical class 0.000 description 1
- 230000005934 immune activation Effects 0.000 description 1
- 229940126546 immune checkpoint molecule Drugs 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 229940124452 immunizing agent Drugs 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940125798 integrin inhibitor Drugs 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 230000009878 intermolecular interaction Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000002563 ionic surfactant Substances 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- SIUGQQMOYSVTAT-UHFFFAOYSA-N lapachol Natural products CC(=CCC1C(O)C(=O)c2ccccc2C1=O)C SIUGQQMOYSVTAT-UHFFFAOYSA-N 0.000 description 1
- CWPGNVFCJOPXFB-UHFFFAOYSA-N lapachol Chemical compound C1=CC=C2C(=O)C(=O)C(CC=C(C)C)=C(O)C2=C1 CWPGNVFCJOPXFB-UHFFFAOYSA-N 0.000 description 1
- 229910052745 lead Inorganic materials 0.000 description 1
- 229940115286 lentinan Drugs 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 description 1
- 229950001750 lonafarnib Drugs 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- RVFGKBWWUQOIOU-NDEPHWFRSA-N lurtotecan Chemical compound O=C([C@]1(O)CC)OCC(C(N2CC3=4)=O)=C1C=C2C3=NC1=CC=2OCCOC=2C=C1C=4CN1CCN(C)CC1 RVFGKBWWUQOIOU-NDEPHWFRSA-N 0.000 description 1
- 229950002654 lurtotecan Drugs 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- 239000004325 lysozyme Substances 0.000 description 1
- 229960000274 lysozyme Drugs 0.000 description 1
- 235000010335 lysozyme Nutrition 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229940099262 marinol Drugs 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- VJRAUFKOOPNFIQ-TVEKBUMESA-N methyl (1r,2r,4s)-4-[(2r,4s,5s,6s)-5-[(2s,4s,5s,6s)-5-[(2s,4s,5s,6s)-4,5-dihydroxy-6-methyloxan-2-yl]oxy-4-hydroxy-6-methyloxan-2-yl]oxy-4-(dimethylamino)-6-methyloxan-2-yl]oxy-2-ethyl-2,5,7,10-tetrahydroxy-6,11-dioxo-3,4-dihydro-1h-tetracene-1-carboxylat Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=C(O)C=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1C[C@H](O)[C@H](O)[C@H](C)O1 VJRAUFKOOPNFIQ-TVEKBUMESA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 108010010621 modeccin Proteins 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 1
- 229950006780 n-acetylglucosamine Drugs 0.000 description 1
- 239000002088 nanocapsule Substances 0.000 description 1
- 210000000581 natural killer T-cell Anatomy 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 230000017095 negative regulation of cell growth Effects 0.000 description 1
- 230000035407 negative regulation of cell proliferation Effects 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- MQYXUWHLBZFQQO-UHFFFAOYSA-N nepehinol Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C)CCC(C(=C)C)C5C4CCC3C21C MQYXUWHLBZFQQO-UHFFFAOYSA-N 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 229960000435 oblimersen Drugs 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 230000003571 opsonizing effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 229940046231 pamidronate Drugs 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229940046159 pegylated liposomal doxorubicin Drugs 0.000 description 1
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 229950010632 perifosine Drugs 0.000 description 1
- SZFPYBIJACMNJV-UHFFFAOYSA-N perifosine Chemical compound CCCCCCCCCCCCCCCCCCOP([O-])(=O)OC1CC[N+](C)(C)CC1 SZFPYBIJACMNJV-UHFFFAOYSA-N 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- NUKCGLDCWQXYOQ-UHFFFAOYSA-N piposulfan Chemical compound CS(=O)(=O)OCCC(=O)N1CCN(C(=O)CCOS(C)(=O)=O)CC1 NUKCGLDCWQXYOQ-UHFFFAOYSA-N 0.000 description 1
- 229950001100 piposulfan Drugs 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229960001237 podophyllotoxin Drugs 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 description 1
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920001583 poly(oxyethylated polyols) Polymers 0.000 description 1
- 108010054442 polyalanine Proteins 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 208000005987 polymyositis Diseases 0.000 description 1
- 230000007824 polyneuropathy Effects 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001902 propagating effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- 231100000654 protein toxin Toxicity 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- WOLQREOUPKZMEX-UHFFFAOYSA-N pteroyltriglutamic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(=O)NC(CCC(=O)NC(CCC(O)=O)C(O)=O)C(O)=O)C(O)=O)C=C1 WOLQREOUPKZMEX-UHFFFAOYSA-N 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000007420 radioactive assay Methods 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 229940099538 rapamune Drugs 0.000 description 1
- 229910052761 rare earth metal Inorganic materials 0.000 description 1
- 150000002910 rare earth metals Chemical class 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 150000004508 retinoic acid derivatives Chemical class 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 229940089617 risedronate Drugs 0.000 description 1
- IMUQLZLGWJSVMV-UOBFQKKOSA-N roridin A Natural products CC(O)C1OCCC(C)C(O)C(=O)OCC2CC(=CC3OC4CC(OC(=O)C=C/C=C/1)C(C)(C23)C45CO5)C IMUQLZLGWJSVMV-UOBFQKKOSA-N 0.000 description 1
- 102220068344 rs199989979 Human genes 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 238000013391 scatchard analysis Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 229940112726 skelid Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- MKNJJMHQBYVHRS-UHFFFAOYSA-M sodium;1-[11-(2,5-dioxopyrrol-1-yl)undecanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCCCCCCCCN1C(=O)C=CC1=O MKNJJMHQBYVHRS-UHFFFAOYSA-M 0.000 description 1
- ULARYIUTHAWJMU-UHFFFAOYSA-M sodium;1-[4-(2,5-dioxopyrrol-1-yl)butanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O ULARYIUTHAWJMU-UHFFFAOYSA-M 0.000 description 1
- VUFNRPJNRFOTGK-UHFFFAOYSA-M sodium;1-[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]oxy-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)C1CCC(CN2C(C=CC2=O)=O)CC1 VUFNRPJNRFOTGK-UHFFFAOYSA-M 0.000 description 1
- MIDXXTLMKGZDPV-UHFFFAOYSA-M sodium;1-[6-(2,5-dioxopyrrol-1-yl)hexanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCCCN1C(=O)C=CC1=O MIDXXTLMKGZDPV-UHFFFAOYSA-M 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 229950006315 spirogermanium Drugs 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- ICXJVZHDZFXYQC-UHFFFAOYSA-N spongistatin 1 Natural products OC1C(O2)(O)CC(O)C(C)C2CCCC=CC(O2)CC(O)CC2(O2)CC(OC)CC2CC(=O)C(C)C(OC(C)=O)C(C)C(=C)CC(O2)CC(C)(O)CC2(O2)CC(OC(C)=O)CC2CC(=O)OC2C(O)C(CC(=C)CC(O)C=CC(Cl)=C)OC1C2C ICXJVZHDZFXYQC-UHFFFAOYSA-N 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 239000013595 supernatant sample Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- 210000004243 sweat Anatomy 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 210000001179 synovial fluid Anatomy 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229940099419 targretin Drugs 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical compound O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 210000001138 tear Anatomy 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 229940019375 tiludronate Drugs 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- RUELTTOHQODFPA-UHFFFAOYSA-N toluene 2,6-diisocyanate Chemical compound CC1=C(N=C=O)C=CC=C1N=C=O RUELTTOHQODFPA-UHFFFAOYSA-N 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- 229950010147 troxacitabine Drugs 0.000 description 1
- RXRGZNYSEHTMHC-BQBZGAKWSA-N troxacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)OC1 RXRGZNYSEHTMHC-BQBZGAKWSA-N 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- HDZZVAMISRMYHH-LITAXDCLSA-N tubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@@H]1O[C@@H](CO)[C@H](O)[C@H]1O HDZZVAMISRMYHH-LITAXDCLSA-N 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 230000005951 type IV hypersensitivity Effects 0.000 description 1
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- HFTAFOQKODTIJY-UHFFFAOYSA-N umbelliferone Natural products Cc1cc2C=CC(=O)Oc2cc1OCC=CC(C)(C)O HFTAFOQKODTIJY-UHFFFAOYSA-N 0.000 description 1
- ORHBXUUXSCNDEV-UHFFFAOYSA-N umbelliferone Chemical compound C1=CC(=O)OC2=CC(O)=CC=C21 ORHBXUUXSCNDEV-UHFFFAOYSA-N 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 229940002005 zometa Drugs 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
Images





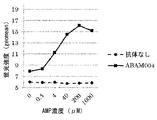


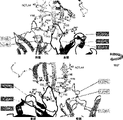















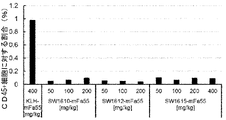

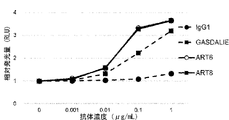



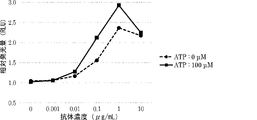
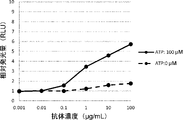


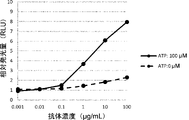
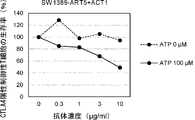
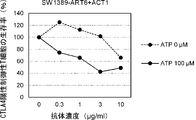







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/74—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/80—Vectors or expression systems specially adapted for eukaryotic hosts for fungi
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/80—Vectors or expression systems specially adapted for eukaryotic hosts for fungi
- C12N15/81—Vectors or expression systems specially adapted for eukaryotic hosts for fungi for yeasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/02—Libraries contained in or displayed by microorganisms, e.g. bacteria or animal cells; Libraries contained in or displayed by vectors, e.g. plasmids; Libraries containing only microorganisms or vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/524—CH2 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/526—CH3 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/72—Increased effector function due to an Fc-modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
Definitions
- the present invention relates to anti-CTLA-4 antibodies and methods of using them.
- the present invention also relates to polypeptides comprising variant Fc regions containing amino acid alterations in the parental Fc region, and methods for producing said polypeptides.
- Immune checkpoints are thought to play an important role in maintaining homeostasis in the immune system. On the other hand, it has become clear that some tumors use immune checkpoints to evade the immune system.
- CTLA-4 cytotoxic T-lymphocyte-associated antigen 4
- PD-1 programmed cell death 1
- PD-L1 programmed cell death ligand 1
- regulatory T cells which is thought to play an important role in the immunosuppressive function of regulatory T cells (e.g., non-regulatory T cells).
- regulatory T cells e.g., non-regulatory T cells.
- Patent Document 5 The infiltration of regulatory T cells into tumor tissue is thought to result in the attenuation or inhibition of immune surveillance against tumors. In fact, it has been revealed that regulatory T cells are increased in many human carcinomas (see, for example, Non-Patent Document 6), and the local infiltration of regulatory T cells into tumors in cancer patients It has been reported that it can be a poor prognostic factor. Conversely, removal or reduction of regulatory T cells from tumor tissue is expected to lead to enhancement of anti-tumor immunity.
- ipilimumab an anti-CTLA-4 antibody
- ipilimumab an anti-CTLA-4 antibody
- autoimmune diseases develop due to systemic enhancement of immune activity.
- 60% of patients treated with ipilimumab had adverse events, most of which were skin or gastrointestinal autoimmune diseases.
- Other clinical trials have also reported that about half of patients treated with ipilimumab developed similar autoimmune diseases. To reduce these side effects, some patients treated with ipilimumab are given immunosuppressants.
- Non-Patent Document 12 Antibodies are attracting attention as pharmaceuticals because they are highly stable in the blood and have few side effects.
- Most antibody drugs currently on the market are antibodies of the human IgG1 subclass.
- Numerous studies have been conducted on antibody-dependent cytotoxicity (hereafter referred to as ADCC) and complement-dependent cytotoxicity (hereafter referred to as CDC), which are the effector functions of IgG class antibodies. It has been reported that, among human IgG classes, antibodies of the IgG1 subclass have the highest ADCC activity and CDC activity (Non-Patent Document 14).
- ADCP Antibody-dependent cell-mediated phagocytosis
- Fc ⁇ R activated macrophages
- isoforms of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIb, Fc ⁇ RIIIa, and Fc ⁇ RIIIb have been reported in the Fc ⁇ R protein family, and their allotypes have also been reported (Non-Patent Document 17).
- Non-Patent Documents 7 and 8 The importance of Fc ⁇ R-mediated effector functions for the antitumor effects of antibodies has been reported using mouse models (Non-Patent Documents 7 and 8). In addition, a correlation was observed between clinical efficacy in humans and the high-affinity polymorphic allotype (V158) and low-affinity polymorphic allotype (F158) of Fc ⁇ RIIIa (Non-Patent Document 18). Similarly, it has been shown that Fc ⁇ RIIa allotypes (H131 and R131) have different clinical effects (Non-Patent Document 19). These reports indicate that antibodies with Fc regions optimized for binding to specific Fc ⁇ Rs mediate stronger effector functions and thereby exert effective anti-tumor effects.
- the balance of binding activity of antibodies to activating receptors composed of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIIa, and Fc ⁇ RIIIb and inhibitory receptors composed of Fc ⁇ RIIb is an important factor in optimizing antibody effector functions.
- an Fc region with enhanced binding activity to activating receptors and reduced binding activity to inhibitory receptors it may be possible to impart optimal effector functions to antibodies (Non-Patent Document 20).
- Non-Patent Document 20 Regarding the binding of the Fc region and Fc ⁇ R, several amino acid residues in the hinge region and CH2 domain of the antibody and the sugar chain added to Asn at EU numbering 297 bound to the CH2 domain are important.
- Non-Patent Document 14 Non-Patent Document 21, Non-Patent Document 22. Focusing on this binding site, Fc region mutants with various Fc ⁇ R-binding properties have been studied so far, and Fc region mutants with higher activating Fc ⁇ R-binding activity have been obtained (Patent Document 5, Patent Document 6, Non-Patent Document 9, Non-Patent Document 10). For example, Lazar et al., by replacing the 239th Ser, 330th Ala, and 332nd Ile of human IgG1 with Asp, Leu, and Glu, respectively, increased the binding to human Fc ⁇ RIIIa (V158) by about 370 times. (Non-Patent Document 9, Patent Document 6). Shinkawa et al.
- Non-Patent Document 23 proposes the same modifications to the Fc regions of both antibody H chains, or introduce the same sugar chain modifications.
- the Fc of the antibody is a homodimer, it has been reported that it binds to Fc ⁇ R at 1:1 and recognizes Fc ⁇ R asymmetrically in the lower hinge and CH2 regions (non Patent document 11). Considering that the Fc region interacts asymmetrically with Fc ⁇ R, introduction of different modifications to each H chain may enable more precise optimization of the interaction between IgG and Fc ⁇ R.
- Patent Document 7 Patent Document 8
- Patent Document 9 Patent Document 10
- ADCP activity is also an important effector function of antibodies, and has been reported to contribute to antitumor effects (Non-Patent Document 24).
- ADCP activity can be enhanced by inhibiting the "Don't eat me" signal typified by CD47 (Non-Patent Document 24), and can also be enhanced by enhancing the binding ability to Fc ⁇ RIIa (Non-Patent Document 25).
- Fc ⁇ RIIa which is an activating Fc ⁇ R
- inhibitory Fc ⁇ RIIb have a very high homology in the amino acid sequence of the extracellular region, and it is difficult to selectively enhance the binding ability to Fc ⁇ RIIa (Non-Patent Document 26 ).
- the binding ability to Fc ⁇ RIIa is enhanced, the binding ability to Fc ⁇ RIIb, which is an inhibitory receptor, is also enhanced, possibly resulting in weakening of effector function.
- variants with significantly improved Fc ⁇ RIIa-binding ability also have enhanced Fc ⁇ RIIb-binding ability as compared to native IgG1 (Patent Documents 9 and 10). Therefore, in order to exhibit high ADCC / ADCP activity, it is preferable that the binding ability to Fc ⁇ RIIb is not enhanced, and the binding to Fc ⁇ RIIIa and Fc ⁇ RIIa can be enhanced as much as possible, but such variants have not been reported. .
- the present invention provides anti-CTLA-4 antibodies and methods of use thereof.
- the invention also provides polypeptides comprising variant Fc regions and methods for their production.
- the present invention provides the following [1] to [26].
- [1] (A) variable regions with CTLA-4 binding activity dependent on the concentration of adenosine-containing compounds, and (B) An anti-CTLA-4 antibody comprising a mutated Fc region containing multiple amino acid alterations in the parent Fc region, wherein the parent Fc region is composed of two polypeptide chains, and the mutated Fc region comprises: An anti-CTLA-4 antibody comprising amino acid alterations in: (i) positions 234, 235, 236, 239, 268, 270, 298, and 330, represented by EU numbering, in the first polypeptide of the parent Fc region, and (ii) positions 270, 298, 326, 330 and 334, represented by EU numbering, in the second polypeptide of the parent Fc region; [2] The anti-CTLA-4 antibody of [1], wherein the variable region has at least one characteristic selected from (a) to (i) below: (a) the binding activity in the presence of 100 ⁇ M a
- [5] (a) HVR-H1 (SEQ ID NO: 223) comprising the amino acid sequence SX1TMN , wherein X1 is H, A , R, or K, ( b) the amino acid sequence SISX1X2SX3YIYYAX4 HVR - H2 ( sequence _ number: 224), and (c) HVR-H3 (SEQ ID NO: 225) comprising the amino acid sequence YGX1REDMLWVFDY , wherein X1 is K or A, according to any one of [1] to [4] Anti-CTLA-4 antibody as described.
- [ 6 ] ( a) comprising the amino acid sequence X1GX2STX3VGDYX4X5VX6 , where X1 is T, D, Q, or E, X2 is T or P , X3 is D or G, and X HVR-L1 (SEQ ID NO: 226), wherein 4 is N or T, X 5 is Y or W, and X 6 is S or H, (b) comprising the amino acid sequence X 1 TX 2 X 3 KPX 4 , and X 1 is HVR-L2 (SEQ ID NO: 227), wherein E, F, or Y, X2 is S or I , X3 is K or S, X4 is S, E, or K, and (c) the amino acid sequence X1
- a heavy chain variable domain FR1 comprising any one of the amino acid sequences of SEQ ID NO: 229 to 232, FR2 comprising the amino acid sequence of SEQ ID NO: 233, FR3 comprising the amino acid sequence of SEQ ID NO: 234, and SEQ ID NO:
- the anti-CTLA-4 antibody of [6] further comprising FR3 comprising one amino acid sequence and FR4 comprising any one amino acid sequence of SEQ ID NOs: 245-246.
- the variant Fc region further comprises amino acid alterations at positions 250 and 307, represented by EU numbering, in the second polypeptide of the parent Fc region; of anti-CTLA-4 antibodies.
- Trp at position 366 represented by EU numbering, in the first polypeptide of the parental Fc region
- Ser at position 366 represented by EU numbering, position 368, in the second polypeptide of the parental Fc region.
- Ala at and Val at position 407 (d) Ser at position 366, Ala at position 368, and Val at position 407 represented by EU numbering in the first polypeptide of the parent Fc region and EU numbering in the second polypeptide of the parent Fc region.
- Trp at position 366 represented by (e) Cys at position 349, represented by EU numbering, and Trp at position 366, in the first polypeptide of the parent Fc region, and position 356, represented by EU numbering, in the second polypeptide of the parent Fc region. Cys at position 366, Ser at position 366, Ala at position 368, and Val at position 407, (f) Cys at position 356, Ser at position 366, Ala at position 368, and Val at position 407, represented by EU numbering, in the first polypeptide of the parent Fc region, and the second polypeptide of the parent Fc region; Cys at position 349 and Trp at position 366 in the peptide, represented by EU numbering.
- the variant Fc region further comprises any of the following amino acid alterations (a) to (d) in the first polypeptide and/or the second polypeptide of the parent Fc region:
- the anti-CTLA-4 antibody of [20] comprising [22] (1) the first H chain polypeptide of SEQ ID NO:335, the second H chain polypeptide of SEQ ID NO:336, and the L chain polypeptide of SEQ ID NO:161, or (2) the first H chain polypeptide of SEQ ID NO: 337, the second H chain polypeptide of SEQ ID NO: 338, and the L chain polypeptide of SEQ ID NO: 161;
- An anti-CTLA-4 antibody comprising: [23] an isolated nucleic acid encoding the anti-CTLA-4 antibody of any
- a method for producing an anti-CTLA-4 antibody comprising culturing the host cell of [24] so that the anti-CTLA-4 antibody is produced.
- a pharmaceutical preparation comprising the anti-CTLA-4 antibody of any one of [1] to [22] and a pharmaceutically acceptable carrier.
- the disclosure provides the following.
- a polypeptide comprising a mutant Fc region containing amino acid alterations in the parent Fc region, wherein the parent Fc region is composed of two polypeptide chains, and the mutant Fc region contains amino acid alterations at the following positions: , a polypeptide: (i) positions 234, 235, 236, 239, 268, 270 and 298, represented by EU numbering, in the first polypeptide of the parent Fc region, and (ii) positions 270, 298, 326 and 334, represented by EU numbering, in the second polypeptide of the parent Fc region; [102] the polypeptide of [101], wherein the variant Fc region further comprises an amino acid alteration at position 326, represented by EU numbering, in the first polypeptide of the parent Fc region; [103] the polypeptide of [101] or [102], wherein the variant Fc region further comprises an amino acid alteration at position 236 (EU numbering) in the second polypeptide of the parent F
- variant Fc region further comprises amino acid alterations at positions 250 and 307, represented by EU numbering, in the second polypeptide of the parent Fc region; peptide.
- mutant Fc region further comprises any of the following amino acid alterations (a) to (f): (a) Lys at position 356, represented by EU numbering, in the first polypeptide of the parental Fc region, and Glu at position 439, represented by EU numbering, in the second polypeptide of the parental Fc region; (b) Glu at position 439, represented by EU numbering, in the first polypeptide of the parental Fc region, and Lys at position 356, represented by EU numbering, in the second polypeptide of the parental Fc region.
- Trp at position 366 represented by EU numbering, in the first polypeptide of the parental Fc region
- Ser at position 366 represented by EU numbering, position 368, in the second polypeptide of the parental Fc region.
- Ala at and Val at position 407 (d) Ser at position 366, Ala at position 368, and Val at position 407 represented by EU numbering in the first polypeptide of the parent Fc region and EU numbering in the second polypeptide of the parent Fc region.
- Trp at position 366 represented by (e) Cys at position 349, represented by EU numbering, and Trp at position 366, in the first polypeptide of the parent Fc region, and position 356, represented by EU numbering, in the second polypeptide of the parent Fc region. Cys at position 366, Ser at position 366, Ala at position 368, and Val at position 407, (f) Cys at position 356, Ser at position 366, Ala at position 368, and Val at position 407, represented by EU numbering, in the first polypeptide of the parent Fc region, and the second polypeptide of the parent Fc region; Cys at position 349 and Trp at position 366 in the peptide, represented by EU numbering.
- variant Fc region further comprises any of the following amino acid alterations (a) to (d) in the first polypeptide and/or the second polypeptide of the parent Fc region:
- the mutant Fc region has enhanced binding activity to at least one Fc ⁇ receptor selected from the group consisting of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIb, and Fc ⁇ RIIIa compared to the parent Fc region, [101] to [117] ]
- the polypeptide of [118] wherein the mutant Fc region has enhanced binding activity to Fc ⁇ RIIa and Fc ⁇ RIIIa compared to the parent Fc region; [120] any one of [101] to [119], wherein the mutant Fc region has improved selectivity between activating Fc ⁇ receptors and inhibitory Fc ⁇ receptors compared to the parent Fc region of polypeptides.
- the binding activity to activating Fc ⁇ receptors is selectively enhanced as compared to the binding activity to inhibitory Fc ⁇ receptors, compared to the parental Fc region, [101] to [119].
- the ratio of activating Fc ⁇ receptor-binding activity to inhibitory Fc ⁇ receptor-binding activity is greater in the mutant Fc region than in the parent Fc region, [101] to [119].
- the ratio (A/I ratio) in the polypeptide containing the mutant Fc region is 1.1-fold or more, 1.2-fold or more, 1.3-fold or more, 1.4-fold or more compared to the polypeptide containing the parent Fc region, 1.5 times or more, 1.6 times or more, 1.7 times or more, 1.8 times or more, 1.9 times or more, 2 times or more, 3 times or more, 4 times or more, 5 times or more, 6 times or more, 7 times or more, 8 times or more, 9 times 10 times or more, 20 times or more, 30 times or more, 40 times or more, 50 times or more, 60 times or more, 70 times or more, 80 times or more, 90 times or more, 100 times or more, 200 times or more, 300 times or more, 400x or more, 500x or more, 600x or more, 700x or more, 800x or more, 900x or more, 1000x or more, 2000x or more, 3000x or more, 4000x or more, 5000x or more, 6000x
- the ratio (A/I ratio) in the polypeptide comprising a mutant Fc region is 10 or more, 20 or more, 30 or more, 40 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, 600 or more, 700 or more, 800 or more, 900 or more, 1000 or more, 2000 or more, 3000 or more, 4000 or more, 5000 or more, 6000 or more, 7000 or more,
- the polypeptide of [120-3] which is 8000 or more, 9000 or more, 10000 or more, 11000 or more, 12000 or more, 13000 or more, 14000 or more, or 15000 or more.
- the activating Fc ⁇ receptor is at least one Fc ⁇ receptor selected from the group consisting of Fc ⁇ RIa, Fc ⁇ RIIa, and Fc ⁇ RIIIa
- the inhibitory Fc ⁇ receptor is Fc ⁇ RIIb A polypeptide according to any of the preceding claims.
- a method for producing a polypeptide comprising a mutant Fc region comprising the step of introducing amino acid alterations into the parent Fc region, wherein the parent Fc region is composed of two polypeptide chains, and A method wherein an amino acid modification is introduced: (i) positions 234, 235, 236, 239, 268, 270 and 298, represented by EU numbering, in the first polypeptide of the parent Fc region, and (ii) positions 270, 298, 326 and 334, represented by EU numbering, in the second polypeptide of the parent Fc region; [124] A method for producing a polypeptide comprising a mutant Fc region, comprising the step of introducing amino acid alterations into the parent Fc region, wherein the parent Fc region is composed of two polypeptide chains, and A method wherein an amino acid modification is introduced: (i) positions 234, 235, 23
- the ratio (A/I ratio) is 1.1-fold or more, 1.2-fold or more, 1.3-fold or more, 1.4-fold or more, 1.5-fold or more, 1.6-fold or more, or 1.7-fold compared to the polypeptide comprising the parent Fc region 1.8 times or more, 1.9 times or more, 2 times or more, 3 times or more, 4 times or more, 5 times or more, 6 times or more, 7 times or more, 8 times or more, 9 times or more, 10 times or more, 20 times or more, 30 times or more, 40 times or more, 50 times or more, 60 times or more, 70 times or more, 80 times or more, 90 times or more, 100 times or more, 200 times or more, 300 times or more, 400 times or more, 500 times or more, 600
- the alteration of function is enhancement of ADCC activity, CDC activity, or ADCP activity;
- FIG. 1 shows the in vitro antibody-dependent cellular cytotoxicity (ADCC) activity of the anti-CTLA4 switch antibody SW1610-ART5+ACT1 against CD4-positive T cells in which CTLA4 expression was induced, as described in Example 2-1.
- FIG. 4 shows the results of measurements in the presence and absence of ATP.
- Figure 2 shows the in vitro antibody-dependent cellular cytotoxicity (ADCC) activity of anti-CTLA4 switch antibody SW1610-ART12 against CD4-positive T cells in which CTLA4 expression was induced, as described in Example 2-1.
- FIG. 4 shows the results of measurements in the presence and absence of each.
- FIG. 3 shows the in vitro antibody-dependent cellular cytotoxicity (ADCC) activity of the anti-CTLA4 switch antibody SW1610-ART4 against CD4-positive T cells in which CTLA4 expression was induced, as described in Example 2-1.
- FIG. 4 shows the results of measurements in the presence and absence of each.
- Figure 4 shows CD4 positivity in human peripheral blood mononuclear cells (PBMC) when anti-CTLA4 switch antibody SW1610-ART12 was added in the presence and absence of ATP, as described in Example 2-2.
- FIG. 4 is a diagram showing the abundance ratio of regulatory T (Treg) cells.
- FIG. 5 shows the CTLA-4 binding activity of the anti-CTLA-4 antibody ABAM004 depending on the ATP, ADP or AMP concentrations, as described in Reference Examples 1-9.
- FIG. 6 shows the AMP concentration-dependent binding activity of ABAM004, an anti-CTLA-4 antibody, to CTLA-4-expressing cells, as described in Reference Examples 1-10.
- FIG. 7 shows the ADCC activity of the anti-CTLA-4 antibody ABAM004 against CTLA-4-expressing cells in the presence and absence of AMP, as described in Reference Example 1-11.
- FIG. 8 shows the mode of binding of ABAM004 Fab fragment and AMP, as described in Reference Examples 2-13.
- FIG. 9 shows the mode of binding of the ABAM004 Fab fragment, AMP, and human CTLA4 (hCTLA4), as described in Reference Example 2-14.
- the heavy chain of the antibody is shown in black, the light chain in gray, hCTLA4 in white, and AMP in a ball-and-stick model.
- FIG. 10 is a diagram mapping the epitope of the ABAM004 Fab fragment into the amino acid sequence of hCTLA4, as described in Reference Examples 2-14.
- amino acid residues shown in black indicate amino acid residues of hCTLA4 containing one or more non-hydrogen atoms located within 4.2 ⁇ of either portion of ABAM004 or AMP in the crystal structure.
- Amino acid residues shown in gray indicate residues for which no models were constructed because they were disordered in the crystal structure.
- FIG. 10 is a diagram mapping the epitope of the ABAM004 Fab fragment into the amino acid sequence of hCTLA4, as described in Reference Examples 2-14.
- amino acid residues shown in black indicate amino acid residues of hCTLA4 containing one or more non-hydrogen atoms located within 4.2 ⁇ of either portion of ABAM004 or AMP in the crystal structure.
- Amino acid residues shown in gray indicate residues for which no models were constructed because they were disordered in the crystal
- FIG. 11 shows the structures of the antibody and AMP extracted from the crystal structures of the complex of the ABAM004 Fab fragment alone and AMP, and the ternary complex of AMP and CTLA4, as described in Reference Example 2-15. is a superimposed diagram.
- the heavy chain of the antibody is shown in black, the light chain in gray, and AMP in a ball-and-stick model.
- the thin line indicates the structure of the ABAM004 Fab fragment alone
- the medium thick line indicates the structure of the 2-part complex with AMP
- the thick line indicates the structure of the 3-part complex.
- FIG. 12 shows the CTLA-4 binding activity of the anti-CTLA-4 antibody ABAM004 and its variant 04H0150/04L0072 depending on the ATP, ADP or AMP concentrations, as described in Reference Example 3-2. It is a diagram. As notations in the figure, WT indicates ABAM004, and H150L072 indicates 04H0150/04L0072.
- FIG. 13 shows the ATP concentration-dependent neutralizing activity of anti-CTLA-4 antibody SW1077 against CTLA-4, as described in Reference Example 3-6.
- FIG. 14 is a diagram showing the antitumor effect of the anti-CTLA-4 antibody mNS-mFa55 (control antibody) in a mouse model implanted with FM3A cell line, as described in Reference Example 3-7-4. .
- Figure 15 shows the anti-tumor effect of the anti-CTLA-4 antibody SW1208-mFa55 (switch antibody) in a mouse model implanted with FM3A cell lines, as described in Reference Example 3-7-4. It is a diagram.
- FIG. 16 shows anti-CTLA-4 antibody mNS-mFa55 (control antibody) and SW1208-mFa55 (switch antibody) administration in a mouse model transplanted with FM3A cell line, as described in Reference Example 3-7-7.
- FIG. 3 shows changes in the ratio of effector Treg cells in tumors over time.
- mNS-mFa55 was administered via the tail vein at 0.1 mg/kg, 1 mg/kg, 10 mg/kg and 100 mg/kg
- SW1208-mFa55 was administered at 0.1 mg/kg, 1 mg/kg and 10 mg/kg. 100 mg/kg and 500 mg/kg were administered through the tail vein.
- Six days after administration tumors were sampled and the increase or decrease in effector Tregs was evaluated by FACS analysis.
- Figure 17 shows anti-CTLA-4 antibody mNS-mFa55 (control antibody) and SW1208-mFa55 (switch antibody) administration in a mouse model transplanted with FM3A cell line, as described in Reference Example 3-7-8.
- FIG. 10 is a diagram showing changes in the ratio of activated helper T cells in the spleen over time.
- mNS-mFa55 was administered at 0.1 mg/kg, 1 mg/kg, 10 mg/kg, and 100 mg/kg via the tail vein, and SW1208-mFa55 was administered at 0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg and 500 mg/kg were administered through the tail vein.
- Six days after the administration spleens were collected, and the increase or decrease in activated helper T cells was evaluated by FACS analysis.
- FIG. 18 shows the anti-tumor effect of the anti-CTLA-4 antibody SW1389-mFa55 (switch antibody) in a mouse model transplanted with Hepa1-6/hGPC3 cell lines, as described in Reference Example 4-3-5.
- Figure 19 shows the antitumor effect of the anti-CTLA-4 antibody hNS-mFa55 (control antibody) in a mouse model transplanted with Hepa1-6/hGPC3 cell lines, as described in Reference Example 4-3-5.
- Figure 19 shows the antitumor effect of the anti
- Figure 20 shows anti-CTLA-4 antibody hNS-mFa55 (control antibody) and SW1389-mFa55 ( FIG. 10 shows changes in the ratio of effector Treg cells in tumors upon administration of switch antibody).
- SW1389-mFa55 at 0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, 500 mg /kg via the tail vein.
- FIG. 10 shows changes in the percentage of activated helper T cells in the spleen upon administration of switch antibody).
- hNS-mFa55 at 0.1 mg/kg, 1 mg/kg, 10 mg/kg, 30 mg/kg, SW1389-mFa55 at 0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, 500 mg /kg via the tail vein.
- spleens were collected, and the increase or decrease in activated helper T cells was evaluated by FACS analysis.
- FIG. 22 shows the antitumor effect of the anti-CTLA-4 antibody SW1610-mFa55 (switch antibody) in a mouse model transplanted with Hepa1-6/hGPC3 cell lines, as described in Reference Example 5-4-5.
- Figure 23 shows the anti-tumor effect of the anti-CTLA-4 antibody SW1612-mFa55 (switch antibody) in a mouse model transplanted with Hepa1-6/hGPC3 cell lines, as described in Reference Example 5-4-5.
- Figure 23 shows the anti-tumor effect of the anti-CTLA-4 antibody SW1612-
- Figure 24 shows the antitumor effect of the anti-CTLA-4 antibody SW1615-mFa55 (switch antibody) in a mouse model transplanted with Hepa1-6/hGPC3 cell lines, as described in Reference Example 5-4-5.
- FIG. 25 shows anti-CTLA-4 antibodies SW1610-mFa55, SW1612-mFa55 and SW1615-mFa55 ( All of these figures show changes in the proportion of effector Treg cells in tumors upon administration of a switch antibody.
- negative control antibody KLH-mFa55 at 400 mg/kg were administered through the tail vein.
- Six days after administration tumors were sampled and the increase or decrease in effector Tregs was evaluated by FACS analysis.
- Figure 26 shows anti-CTLA-4 antibodies SW1610-mFa55, SW1612-mFa55 and SW1615-mFa55 ( All of these figures show changes in the ratio of activated helper T cells in the spleen upon administration of switch antibody.
- SW1610-mFa55 at 50 mg/kg, 100 mg/kg and 200 mg/kg, SW1612-mFa55 at 50 mg/kg, 100 mg/kg and 200 mg/kg, SW1615-mFa55 at 50 mg/kg and 100 mg/kg kg, 200 mg/kg, 400 mg/kg, and negative control antibody KLH-mFa55 at 400 mg/kg were administered through the tail vein.
- Six days after the administration spleens were collected, and the increase or decrease in activated helper T cells was evaluated by FACS analysis.
- IgG1 is MDX10D1H-G1m/MDX10D1L-k0MT
- GASDALIE is MDX10D1H-GASDALIE/MDX10D1L-k0MT
- ART6 is MDX10D1H-Kn462/MDX10D1H-Hl445/MDX10D1L-k0MT
- ART8 is MDX10D1H-KnDX10D461/MDX1HDX10D461 -Hl443/MDX10D1L-k0MT respectively.
- IgG1 is an antibody having a control constant region
- GASDALIE is an antibody having a constant region described in the prior art
- ART6 and ART8 are antibodies having a modified constant region produced in Reference Example 6-1. is.
- FIG. 28 shows a comparison of in vitro ADCP activities of antibodies having various modified constant regions with enhanced binding to Fc ⁇ R, as described in Reference Example 6-3.
- IgG1 is MDX10D1H-G1m/MDX10D1L-k0MT
- GASDIE is MDX10D1H-GASDIE/MDX10D1L-k0MT
- ART6 is MDX10D1H-Kn462/MDX10D1H-Hl445/MDX10D1L-k0MT
- ART8 is MDX10D1H-Kn461/MDX10MT. -Hl443/MDX10D1L-k0MT respectively.
- IgG1 is an antibody having a control constant region
- GASDIE is an antibody having a constant region described in a prior document
- ART6 and ART8 are antibodies having a modified constant region produced in Reference Example 6-1. is.
- FIG. 29 shows in vitro ADCC activity of the anti-CTLA4 switch antibody SW1389-ART6, which has a modified constant region with enhanced binding to Fc ⁇ R, as described in Reference Example 6-4.
- FIG. 30 shows in vitro ADCC activity of anti-CTLA4 switch antibody SW1610-ART6 having a modified constant region with enhanced binding to Fc ⁇ R, as described in Reference Example 6-4.
- FIG. 31 shows in vitro ADCC activity of anti-CTLA4 switch antibody SW1612-ART6 having a modified constant region with enhanced binding to Fc ⁇ R, as described in Reference Example 6-4.
- FIG. 32 shows the neutralizing activity of the anti-CTLA4 switch antibody SW1389 against CTLA4 (the activity of canceling the signal of CTLA4 that suppresses the activation of effector cells), as described in Reference Example 6-5. It is a figure which shows.
- FIG. 33 shows the neutralizing activity of the anti-CTLA4 switch antibody SW1610 against CTLA4 (the activity of canceling the signal of CTLA4 that suppresses the activation of effector cells), as described in Reference Example 6-5. It is a figure which shows.
- FIG. 34 shows the neutralizing activity of the anti-CTLA4 switch antibody SW1612 against CTLA4 (the activity of canceling the signal of CTLA4 that suppresses the activation of effector cells), as described in Reference Example 6-5. It is a figure which shows.
- FIG. 33 shows the neutralizing activity of the anti-CTLA4 switch antibody SW1610 against CTLA4 (the activity of canceling the signal of CTLA4 that suppresses the activation of effector cells), as described in Reference Example 6-5.
- FIG. 35 shows the neutralizing activity of the anti-CTLA4 switch antibody SW1615 against CTLA4 (activity to release CTLA4 signaling that suppresses activation of effector cells), as described in Reference Example 6-5. It is a figure which shows.
- Figure 36 shows the in vitro cytotoxic activity of the anti-CTLA4 switch antibody SW1389-ART5+ACT1 against CTLA4-positive regulatory T cells, as described in Reference Example 6-6.
- Figure 37 shows the in vitro cytotoxic activity of the anti-CTLA4 switch antibody SW1389-ART6+ACT1 against CTLA4-positive regulatory T cells, as described in Reference Example 6-6.
- Figure 38 shows the in vitro cytotoxic activity of the anti-CTLA4 switch antibody SW1610-ART5+ACT1 against CTLA4-positive regulatory T cells, as described in Reference Example 6-6.
- Figure 39 shows the in vitro cytotoxic activity of the anti-CTLA4 switch antibody SW1610-ART6+ACT1 against CTLA4-positive regulatory T cells, as described in Reference Example 6-6.
- FIG. 43 is a diagram showing the hC1q-binding activity of each antibody having a modified Fc, as described in Reference Example 12.
- 44 is a continuation showing the binding activity to hC1q of each antibody with modified Fc, as described in Reference Example 12.
- an "acceptor human framework” for the purposes of this specification is a light chain variable domain (VL) framework or heavy chain variable domain (VH ) a framework containing the amino acid sequence of the framework.
- Acceptor human frameworks "derived from” human immunoglobulin frameworks or human consensus frameworks may contain those same amino acid sequences or may contain alterations in the amino acid sequences. In some embodiments, the number of amino acid changes is 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less.
- the VL acceptor human framework is identical in sequence to a VL human immunoglobulin framework sequence or a human consensus framework sequence.
- Antibody-dependent cell-mediated cytotoxicity refers to the activity of secreted immunoglobulins on specific cytotoxic cells (e.g., NK cells, neutrophils and macrophages). binds to Fc receptors (FcR) present in the cytotoxic effector cells, thereby enabling these cytotoxic effector cells to specifically bind to antigen-bearing target cells and subsequently kill the target cells with cytotoxins
- FcR Fc receptors
- FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol 9: 457-92 (1991).
- an in vitro ADCC assay such as those described in US Pat. Nos. 5,500,362 or 5,821,337 or US Pat. No. 6,737,056 (Presta) can be performed.
- Useful effector cells for such assays include PBMC and NK cells.
- ADCC activity of a molecule of interest may be assessed in vivo in an animal model, such as that disclosed in Clynes et al. PNAS (USA) 95: 652-656 (1998). .
- cytotoxic activity examples include, for example, the antibody-dependent cell-mediated cytotoxicity (ADCC) activity described above, the complement-dependent cytotoxicity (CDC) activity described below, and cytotoxic activity by T cells.
- CDC activity means cytotoxic activity by the complement system.
- ADCC activity means an activity in which an antibody binds to an antigen present on the cell surface of a target cell, and the effector cell binds to the antibody, causing the effector cell to damage the target cell.
- ADCC activity means an activity in which an antibody binds to an antigen present on the cell surface of a target cell, and the effector cell binds to the antibody, causing the effector cell to damage the target cell.
- ADCC activity means an activity in which an antibody binds to an antigen present on the cell surface of a target cell, and the effector cell binds to the antibody, causing the effector cell to damage the target cell.
- Whether the antibody of interest has ADCC activity or whether it has CDC activity can be measured by a known
- Neutralizing activity refers to the activity of inhibiting a biological activity by binding an antibody to a molecule involved in that biological activity.
- the biological activity is provided by binding of the ligand and receptor.
- binding of the antibody to the ligand or receptor inhibits binding of the ligand to the receptor.
- Antibodies with such neutralizing activity are called neutralizing antibodies.
- the neutralizing activity of a test substance can be measured by comparing the biological activity in the presence of ligand between conditions in the presence or absence of the test substance.
- ADCP antibody-dependent cellular phagocytosis
- phagocytic immune cells e.g., macrophages, neutrophils, and dendritic cells.
- binding activity and “binding capacity” are used interchangeably herein and refer to one or more binding sites (e.g. , variable region or Fc region) and the binding partner of a molecule (eg, antigen or Fc ⁇ receptor).
- binding activity is not strictly limited to 1:1 interactions between members of a binding pair (eg, antibody and antigen, or Fc region and Fc ⁇ receptor).
- avidity refers to the intrinsic binding affinity (“affinity”) when the members of a binding pair reflect a 1:1 interaction with monovalence. When members of a binding pair are capable of both monovalent and multivalent binding, avidity is the sum of their avidity.
- binding activity of a molecule X for its partner Y can generally be expressed by the dissociation constant (KD) or "amount of analyte bound per unit amount of ligand". Binding activity can be measured by routine methods known in the art, including those described herein. Specific illustrative and exemplary embodiments for measuring binding activity are described below.
- An “affinity matured” antibody has one or more modifications in one or more hypervariable regions (HVRs) that confer improved affinity for the antibody's antigen as compared to a parent antibody that does not possess the modifications.
- HVRs hypervariable regions
- anti-CTLA-4 antibody or "antibody that binds CTLA-4" is an antibody capable of binding CTLA-4 with sufficient affinity such that the antibody targets CTLA-4.
- the extent of binding of the anti-CTLA-4 antibody to an unrelated non-CTLA-4 protein is determined (e.g., by radioimmunoassay (RIA)) when the antibody's binding to CTLA-4 less than about 10% of
- the antibody that binds CTLA-4 is ⁇ 1 ⁇ M, ⁇ 100 nM, ⁇ 10 nM, ⁇ 1 nM, ⁇ 0.1 nM, ⁇ 0.01 nM, or ⁇ 0.001 nM (e.g., 10 ⁇ 8 M or less , eg 10 ⁇ 8 M to 10 ⁇ 13 M, eg 10 ⁇ 9 M to 10 ⁇ 13 M).
- the anti-CTLA-4 antibody binds to an epitope of CT
- antibody is used in the broadest sense, and as long as it exhibits the desired antigen-binding activity, it is not limited to, but is not limited to, monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., It encompasses a variety of antibody structures, including bispecific antibodies) and antibody fragments.
- antibody fragment refers to a molecule other than an intact antibody that contains a portion of the intact antibody that binds to the antigen to which the intact antibody binds.
- antibody fragments include, but are not limited to, Fv, Fab, Fab', Fab'-SH, F(ab') 2 ; diabodies; linear antibodies; single chain antibody molecules (e.g., scFv ); and multispecific antibodies formed from antibody fragments.
- an "antibody that binds to the same epitope" as a reference antibody refers to an antibody that blocks the binding of the reference antibody to its own antigen in a competition assay, e.g. block the binding of the aforementioned antibody to its own antigen in, for example, 50% or more.
- a competition assay e.g. block the binding of the aforementioned antibody to its own antigen in, for example, 50% or more.
- autoimmune disease refers to a non-malignant disease or disorder arising from and directed against the individual's own tissues.
- autoimmune disease specifically excludes malignant or cancerous diseases or conditions, in particular B-cell lymphoma, acute lymphoblastic leukemia (ALL), chronic lymphocytic Exclude chronic lymphocytic leukemia (CLL), hairy cell leukemia, and chronic myeloblastic leukemia.
- ALL acute lymphoblastic leukemia
- CLL chronic lymphocytic leukemia
- hairy cell leukemia and chronic myeloblastic leukemia.
- autoimmune diseases or disorders include, but are not limited to: inflammatory reactions such as inflammatory skin diseases including psoriasis and dermatitis (e.g., atopic dermatitis); scleroderma and sclerosis; inflammatory bowel disease-related reactions (e.g., Crohn's disease and ulcerative colitis); respiratory distress syndrome (including adult respiratory distress syndrome: ARDS); dermatitis; colitis; glomerulonephritis; allergic conditions such as eczema and asthma and other conditions with T-cell infiltration and chronic inflammatory responses; atherosclerosis; rheumatoid arthritis; systemic lupus erythematosus (SLE) (including but not limited to lupus nephritis, cutaneous lupus); diabetes (e.g., type I diabetes or insulin-dependent diabetes); multiple sclerosis; Raynaud's syndrome; allergic encephalomyelitis; Sjogren's syndrome; juvenile-onset diabetes; and by inflammatory
- cancer and “cancerous” refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth/proliferation. Examples of cancer include breast cancer and liver cancer.
- complement dependent cytotoxicity refers to the Fc effector domain of a target-bound antibody that activates a series of enzymatic reactions that result in the formation of holes in the target cell's membrane. means a mechanism for inducing Typically, antigen-antibody complexes formed on target cells bind and activate complement component C1q, which in turn activates the complement cascade leading to target cell death. bring. Complement activation may also lead to the deposition of complement components on the surface of target cells, which promote ADCC by binding complement receptors (e.g. CR3) on leukocytes. .
- complement receptors e.g. CR3
- “Chemotherapeutic agent” refers to a chemical compound useful in the treatment of cancer.
- chemotherapeutic agents include: alkylating agents such as thiotepa and cyclophosphamide (CYTOXAN®); alkyl sulfonates such as busulfan, improsulfan, and piposulfan; benzodopa, carbocones aziridines such as , meturedopa, and uredopa; ethyleneimines and methylolmelamines, including altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide, and trimethylomelamine; delta-9-tetrahydrocannabinol (dronabinol, MARINOL®); beta-lapachone; lapachol; colchicine; betulinic acid; (irinotecan, CAMPTOSAR®), acetylcamptothecin, scopolectin, and 9-aminocamptothecin
- Nitrogen mustards such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, and ranimustine; enediyne antibiotics ⁇ e.g. et al., Angew. Chem Intl. Ed.
- purine analogues pyrimidine analogues, such as ancitabine, azacytidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens, such as carsterone, dromostanolone propionate, epithiostanol, mepithiostane, testolactone anti-adrenal agents such as aminoglutethimide, mitotane, trilostane; enals); folic acid supplements, such as folinic acid; acegratone; aldophosphamide glycosides; aminolevulinic acid; eniluracil; amsacrine; elliptinium acetate; epothilone; etogluside; gallium nitrate; hydrocyurea; lentinan; 2-Ethylhydrazide; Procarbazine; PSK
- celecoxib or etoricoxib proteosome inhibitors
- proteosome inhibitors e.g. PS341); VELCADE®); CCI-779; tipifarnib (R11577); sorafenib, ABT510; Bcl-2 inhibitors such as oblimersen sodium (GENASENSE®); tyrosine kinase inhibitors (see definition below); serine-threonine kinase inhibitors, such as rapamycin (sirolimus, RAPAMUNE®); lonafarnib (SCH 6636, Farnesyltransferase inhibitors, such as SARASARTM); and pharmaceutically acceptable salts, acids, or derivatives of any of the above; and combinations of two or more of the above, such as cyclophosphamide, doxorubicin.
- proteosome inhibitors e.g. PS341
- VELCADE® CCI-779
- tipifarnib R11577
- CHOP an abbreviation for combination therapy of vincristine and prednisolone
- FOLFOX an abbreviation for treatment regimens with oxaliplatin (ELOXANTINTM) in combination with 5-FU and leucovorin.
- chimeric antibody means that a portion of the heavy and/or light chains are derived from a particular source or species, while the remainder of the heavy and/or light chains are derived from a different source or species. Refers to antibodies.
- the "class" of an antibody refers to the type of constant domain or constant region found in the heavy chain of an antibody.
- the heavy-chain constant domains that correspond to the different classes of immunoglobulins are called ⁇ , ⁇ , ⁇ , ⁇ , and ⁇ , respectively.
- cytotoxic agent refers to a substance that inhibits or prevents the function of cells and/or causes the death or destruction of cells.
- Cytotoxic agents include, but are not limited to, radioactive isotopes such as 211 At, 131 I, 125 I, 90 Y, 186 Re, 188 Re, 153 Sm, 212 Bi, 32 P, 212 Pb and radioisotopes of Lu); chemotherapeutic agents or agents (e.g., methotrexate, adriamycin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin, or other intercalating growth inhibitors; enzymes such as nucleases and fragments thereof; antibiotics; and the various chemotherapeutic agents disclosed above.
- radioactive isotopes such as 211 At, 131 I, 125 I, 90 Y, 186 Re, 188 Re
- Effective cells refer to leukocytes that express one or more FcRs and exert effector functions. In certain embodiments, the cell expresses at least Fc ⁇ RIII and exerts ADCC effector function. Examples of leukocytes that mediate ADCC include peripheral blood mononuclear cells (PBMCs), natural killer (NK) cells, monocytes, cytotoxic T cells and neutrophils. Effector cells can be isolated from natural sources, such as blood. In certain embodiments, the effector cells can be human effector cells.
- “Effector function” refers to the biological activity that is attributed to the Fc region of an antibody and differs depending on the antibody isotype.
- Examples of antibody effector functions include: C1q binding and complement-dependent cytotoxicity (CDC); Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (CDC); cell-mediated cytotoxicity (ADCC); antibody-dependent cell-mediated phagocytosis (ADCP); downregulation of cell surface receptors (e.g., B cell receptors); and B cell activation .
- epitope includes any determinant capable of being bound by an antibody.
- An epitope is the region of an antigen that is bound by an antibody that targets it, and includes specific amino acids that directly contact the antibody.
- Epitopic determinants can include chemically active surface groupings of molecules such as amino acids, sugar side chains, phosphoryl or sulfonyl groups, and possess specific three dimensional structural characteristics, and/or specific charge characteristics. can be done.
- antibodies specific for a particular target antigen preferentially recognize epitopes on that target antigen in a complex mixture of proteins and/or macromolecules.
- Fc receptor refers to a receptor that binds to the Fc region of an antibody.
- the FcR is a native human FcR.
- the FcR is one that binds IgG antibodies (gamma receptors), and receptors of the Fc ⁇ RI, Fc ⁇ RII, and Fc ⁇ RIII subclasses are allelic variants and alternatively spliced forms of these receptors. including, including.
- Fc ⁇ RII receptors include Fc ⁇ RIIA (an “activating receptor”) and Fc ⁇ RIIB (an “inhibiting receptor”), which have similar amino acid sequences that differ primarily in their cytoplasmic domains.
- Activating receptor Fc ⁇ RIIA contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic domain.
- the inhibitory receptor Fc ⁇ RIIB contains an immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic domain (see, e.g., Daeron, Annu. Rev. Immunol. 15: 203-234 (1997)). matter).
- FcR for example, Ravetch and Kinet, Annu. Rev. Immunol 9: 457-492 (1991); Capel et al., Immunomethods 4: 25-34 (1994); and de Haas et al., J. Lab. Clin Reviewed in Med 126: 330-341 (1995).
- Other FcRs including those identified in the future, are also encompassed by the term "FcR" herein.
- Fc receptor or “FcR” also refers to the transfer of maternal IgG to the fetus (Guyer et al., J. Immunol. 117: 587 (1976) and Kim et al., J. Immunol. 24: 249 (1994)) as well as the neonatal receptor FcRn, which is responsible for regulating immunoglobulin homeostasis. Methods for measuring binding to FcRn are known (e.g., Ghetie and Ward., Immunol. Today 18(12): 592-598 (1997); Ghetie et al., Nature Biotechnology, 15(7): 637- 640 (1997); Hinton et al., J. Biol. Chem. 279(8): 6213-6216 (2004); see WO2004/92219 (Hinton et al.)).
- Binding to human FcRn in vivo and serum half-life of human FcRn high-affinity binding polypeptides can be determined, for example, in transgenic mice or transfected human cell lines expressing human FcRn, or in which polypeptides with mutated Fc regions are It can be measured in administered primates.
- WO2000/42072 (Presta) describes antibody variants with improved or decreased binding to FcRs. See, for example, Shields et al. J. Biol. Chem. 9(2): 6591-6604 (2001).
- Fc region is used herein to define the C-terminal region of an immunoglobulin heavy chain that includes at least a portion of the constant region.
- the term includes native sequence Fc regions and variant Fc regions.
- the human IgG heavy chain Fc region extends from Cys226 or from Pro230 to the carboxyl terminus of the heavy chain.
- lysine (Lys447) or glycine-lysine (Gly446-Lys447) at the C-terminus of the Fc region may or may not be present.
- the numbering of amino acid residues in the Fc region or constant region is according to Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, According to the EU numbering system (also known as the EU index), as described in MD 1991.
- Fc region-containing antibody refers to an antibody containing an Fc region.
- the C-terminal lysine of the Fc region (residue 447 according to the EU numbering system) or the C-terminal glycine-lysine of the Fc region (residues 446-447), for example during purification of the antibody or nucleic acids encoding the antibody can be removed by recombinant manipulation of
- a composition comprising an antibody having an Fc region according to the present invention may be an antibody with G446-K447, an antibody with G446 without K447, an antibody with G446-K447 completely removed, or an antibody of the above three types may contain mixtures of
- variable region refers to the domains of the heavy or light chains of an antibody that are involved in binding the antibody to the antigen.
- the heavy and light chain variable domains (VH and VL, respectively) of native antibodies are usually similar, with each domain containing four conserved framework regions (FR) and three hypervariable regions (HVR). structure (see, for example, Kindt et al. Kuby Immunology, 6th ed., W.H. Freeman and Co., page 91 (2007)).
- a single VH or VL domain may be sufficient to confer antigen binding specificity.
- antibodies that bind a particular antigen may be isolated using the VH or VL domains from the antibody that binds the antigen to screen a complementary library of VL or VH domains, respectively. See, e.g., Portolano et al., J. Immunol. 150: 880-887 (1993); Clarkson et al., Nature 352: 624-628 (1991).
- FR Framework or "FR” refers to variable domain residues other than hypervariable region (HVR) residues.
- the FRs of variable domains usually consist of four FR domains: FR1, FR2, FR3 and FR4. Accordingly, HVR and FR sequences usually appear in VH (or VL) in the following order: FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4.
- full length antibody “whole antibody” and “whole antibody” are used interchangeably herein and have a structure substantially similar to that of a naturally occurring antibody or as defined herein. It refers to an antibody having a heavy chain containing an Fc region that
- a “functional Fc region” has an "effector function” of a native-sequence Fc region.
- effector functions include C1q binding; CDC; Fc receptor binding; ADCC; phagocytosis; Such effector functions generally require the Fc region to be combined with a binding domain (e.g., an antibody variable domain) and are assessed using, for example, various assays disclosed within the definitions herein. can be
- human antibody is an antibody with an amino acid sequence that corresponds to that of an antibody produced by a human or human cells or derived from a human antibody repertoire or other non-human source using human antibody coding sequences. This definition of human antibody specifically excludes humanized antibodies that contain non-human antigen-binding residues.
- a "human consensus framework” is a framework that represents the most commonly occurring amino acid residues in a selected group of human immunoglobulin VL or VH framework sequences.
- the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences.
- the subgroup of sequences is the subgroup in Kabat et al., Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3.
- the subgroup is subgroup ⁇ I according to Kabat et al., supra.
- the subgroup is subgroup III according to Kabat et al., supra.
- a “humanized” antibody refers to a chimeric antibody that contains amino acid residues from non-human HVRs and amino acid residues from human FRs.
- a humanized antibody comprises substantially all of at least one, typically two, variable domains in which all or substantially all HVRs (e.g., CDRs) are non- Corresponds to that of a human antibody, and all or substantially all FRs correspond to those of a human antibody.
- a humanized antibody optionally may comprise at least a portion of an antibody constant region derived from a human antibody.
- a "humanized form" of an antibody (eg, a non-human antibody) refers to an antibody that has undergone humanization.
- hypervariable region is hypervariable in sequence (“complementarity determining region” or “CDR”) and/or is structurally defined. Refers to the regions of the variable domain of an antibody that form loops (“hypervariable loops”) and/or contain antigen-contacting residues (“antigen-contacting”). Antibodies typically contain six HVRs: three in VH (H1, H2, H3) and three in VL (L1, L2, L3).
- Exemplary HVRs herein include: (a) at amino acid residues 26-32 (L1), 50-52 (L2), 91-96 (L3), 26-32 (H1), 53-55 (H2), and 96-101 (H3); resulting hypervariable loops (Chothia and Lesk, J. Mol. Biol. 196: 901-917 (1987)); (b) at amino acid residues 24-34 (L1), 50-56 (L2), 89-97 (L3), 31-35b (H1), 50-65 (H2), and 95-102 (H3); the resulting CDRs (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed.
- HVR residues and other residues in the variable domain are numbered herein according to Kabat et al., supra.
- immunoconjugate is an antibody conjugated to one or more heterologous molecules (heterologous molecules include, but are not limited to, cytotoxic agents).
- an “isolated” antibody is one that has been separated from a component of its original environment.
- antibodies are analyzed electrophoretically (e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis) or chromatographically (e.g., ion-exchange or reverse-phase HPLC). Purified to greater than 95% or 99% purity as measured.
- electrophoretically e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis
- chromatographically e.g., ion-exchange or reverse-phase HPLC
- isolated nucleic acid refers to a nucleic acid molecule that has been separated from components of its original environment.
- An isolated nucleic acid includes a nucleic acid molecule contained within cells that normally contain the nucleic acid molecule, but the nucleic acid molecule is present extrachromosomally or on a chromosome other than its native chromosomal location. exists in position.
- isolated nucleic acid encoding an antibody refers to one or more nucleic acid molecules encoding the heavy and light chains (or fragments thereof) of an antibody, whether on a single vector or on separate vectors. and nucleic acid molecules present in one or more locations in a host cell.
- first polypeptide and second polypeptide refer to polypeptides that constitute the Fc region of an antibody.
- First polypeptide and second polypeptide mean that they differ in sequence from each other, preferably at least in the CH2 region. Furthermore, the CH3 regions may have different sequences.
- the polypeptide may be, for example, a polypeptide that constitutes the Fc region of native IgG, or a polypeptide obtained by modifying the polypeptide that constitutes the Fc region of native IgG. .
- Native IgG means a polypeptide that includes the same amino acid sequence as IgG found in nature and belongs to the class of antibodies that are substantially encoded by the immunoglobulin gamma gene.
- native human IgG means native human IgG1, native human IgG2, native human IgG3, native human IgG4, and the like. Natural-type IgG includes naturally occurring mutants and the like.
- Polypeptide in the present invention usually refers to peptides and proteins having a length of about 10 amino acids or more. Moreover, although it is usually a polypeptide of biological origin, it is not particularly limited, and for example, it may be a polypeptide consisting of an artificially designed sequence. Also, it may be a natural polypeptide, a synthetic polypeptide, a recombinant polypeptide, or the like. In addition, the protein molecule in the present invention refers to a molecule containing the polypeptide.
- a preferred example of the polypeptide of the present invention is an antibody. More preferred examples include natural IgG and antibodies obtained by modifying natural IgG.
- native IgG include, in particular, native human IgG.
- Native IgG refers to a polypeptide that contains the same amino acid sequence as IgG found in nature and belongs to the class of antibodies substantially encoded by the immunoglobulin gamma gene.
- native human IgG means native human IgG1, native human IgG2, native human IgG3, native human IgG4, and the like.
- Natural-type IgG includes naturally occurring mutants and the like.
- polypeptide comprising an Fc region is not particularly limited as long as it is a polypeptide comprising an Fc region, but for example, an antibody comprising an Fc region.
- composition comprising a polypeptide having an Fc region is a polypeptide comprising an Fc region with G446-K447, a polypeptide comprising an Fc region with G446 and without K447, a polypeptide comprising an Fc region with G446 and without K447, or a mixture of the above three types of polypeptides.
- an "isolated" polypeptide is one that has been separated from a component of its original environment.
- the polypeptide is electrophoretically (e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis) or chromatographically (e.g., ion exchange or reverse phase HPLC). Purified to greater than 95% or 99% purity as measured by For a review of methods for assessment of polypeptide purity, see, eg, Flatman et al., J. Chromatogr. B 848: 79-87 (2007).
- isolated nucleic acid encoding a polypeptide refers to one or more nucleic acid molecules that encode the polypeptide (e.g., the Fc region of an antibody, or antibody heavy and light chains or fragments thereof). It includes nucleic acid molecules on one vector or on separate vectors and nucleic acid molecules present in one or more locations in a host cell.
- vector refers to a nucleic acid molecule capable of propagating another nucleic acid to which it has been linked.
- the term includes vectors as self-replicating nucleic acid structures and vectors that integrate into the genome of a host cell into which they are introduced.
- Certain vectors are capable of directing the expression of nucleic acids to which they are operably linked.
- Such vectors are also referred to herein as "expression vectors".
- a vector can be introduced into a host cell by a method using a virus, an electroporation method, or the like, but the introduction of the vector is not limited to ex vivo introduction, and the vector can also be directly introduced into the living body. be.
- host cell refers to a cell (including progeny of such a cell) into which exogenous nucleic acid has been introduced.
- a host cell includes “transformants” and “transformed cells,” including the primary transformed cell and progeny derived therefrom at any passage number. Progeny may not be completely identical in nucleic acid content to the parent cell, but may contain mutations. Mutant progeny that have the same function or biological activity as with which the originally transformed cell was screened or selected are also included herein.
- the term "monoclonal antibody” as used herein refers to an antibody obtained from a substantially homogeneous population of antibodies. That is, the individual antibodies that make up the population are mutated antibodies that may arise (e.g., mutated antibodies that contain naturally occurring mutations or that arise during the manufacture of monoclonal antibody preparations. are identical and/or bind to the same epitope, except that they are present in greater amounts). In contrast to polyclonal antibody preparations, which typically include different antibodies directed against different determinants (epitopes), each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on the antigen.
- monoclonal indicates the character of the antibody as being obtained from a population of substantially homogeneous antibodies and should not be construed as requiring production of the antibody by any particular method.
- monoclonal antibodies used in accordance with the present invention include, but are not limited to, hybridoma technology, recombinant DNA technology, phage display technology, transgenic animals containing all or part of the human immunoglobulin locus. and such methods and other exemplary methods for making monoclonal antibodies are described herein.
- naked antibody refers to an antibody that is not conjugated to a heterologous moiety (eg, a cytotoxic moiety) or a radiolabel.
- a naked antibody may be present in a pharmaceutical formulation.
- Native antibodies refer to immunoglobulin molecules with various structures occurring in nature.
- native IgG antibodies are heterotetrameric glycoproteins of approximately 150,000 daltons composed of two identical light chains and two identical heavy chains that are disulfide-bonded. From N-terminus to C-terminus, each heavy chain has a variable region (VH), also called a variable heavy domain or heavy chain variable domain, followed by three constant domains (CH1, CH2 and CH3). Similarly, from N-terminus to C-terminus, each light chain has a variable region (VL), also called a variable light domain or light chain variable domain, followed by a constant light (CL) domain.
- VH variable region
- VL variable region
- CL constant light domain
- the light chains of antibodies can be assigned to one of two types, called kappa ( ⁇ ) and lambda ( ⁇ ), based on the amino acid sequences of their constant domains.
- a “native sequence Fc region” contains an amino acid sequence identical to the amino acid sequence of an Fc region found in nature.
- Native sequence human Fc regions include native sequence human IgG1 Fc regions (non-A and A allotypes); native sequence human IgG2 Fc regions; native sequence human IgG3 Fc regions; including those variants present in
- a “variant Fc region” comprises an amino acid sequence that differs from that of a native sequence Fc region by at least one amino acid modification (alteration), preferably one or more amino acid substitutions.
- the variant Fc region has at least one amino acid substitution, such as about 1 to about 30 amino acid substitutions, in the native sequence Fc region or parental Fc region compared to the native sequence Fc region or parental Fc region. , preferably from about 1 to about 20 amino acid substitutions, more preferably from about 1 to about 10 amino acid substitutions, and most preferably from about 1 to about 5 amino acid substitutions.
- Variant Fc regions herein are preferably at least about 80% homologous to native sequence Fc regions or parental Fc regions, preferably at least about 85% homologous thereto, more preferably at least about 90% homologous to them. % homology, most preferably at least about 95% homology therewith.
- a "percent (%) amino acid sequence identity" to a reference polypeptide sequence is obtained after aligning the sequences to obtain the maximum percent sequence identity and introducing gaps if necessary, and without any conservative substitutions. Defined as the percentage of amino acid residues in a candidate sequence that are identical to amino acid residues in the reference polypeptide sequence, not considered part of the identity. Alignments for purposes of determining percent amino acid sequence identity may be performed by a variety of methods within the skill in the art, such as BLAST, BLAST-2, ALIGN, Megalign (DNASTAR) software, or GENETYX® (stock This can be accomplished by using publicly available computer software, such as the company Genetics. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the entire length of the sequences being compared.
- the ALIGN-2 sequence comparison computer program is the work of Genentech, Inc. and its source code has been filed with the US Copyright Office, Washington DC, 20559 with user documentation, under US Copyright Registration No. TXU510087. Registered.
- the ALIGN-2 program is publicly available from Genentech, Inc., South San Francisco, Calif., or may be compiled from source code.
- ALIGN-2 programs are compiled for use on UNIX operating systems, including Digital UNIX V4.0D. All sequence comparison parameters were set by the ALIGN-2 program and do not vary.
- the % amino acid sequence identity of a given amino acid sequence A to, with, or to a given amino acid sequence B is calculated as follows: Hundredfold. where X is the number of amino acid residues scored as identical matches in the alignment of A and B by the sequence alignment program ALIGN-2, and Y is the total number of amino acid residues in B. . It is understood that the % amino acid sequence identity of A to B does not equal the % amino acid sequence identity of B to A if the length of amino acid sequence A does not equal the length of amino acid sequence B. deaf. Unless otherwise specified, all % amino acid sequence identity values used herein are obtained using the ALIGN-2 computer program as described in the immediately preceding paragraph.
- pharmaceutical formulation and “pharmaceutical composition” are used interchangeably and refer to a preparation in a form such that the biological activity of the active ingredients contained therein can be exerted. and that does not contain additional elements that are unacceptably toxic to the subjects to whom it is administered.
- “Individuals” or “subjects” are mammals. Mammals include, but are not limited to, domestic animals (eg, cows, sheep, cats, dogs, horses), primates (eg, humans and non-human primates such as monkeys), rabbits, and , including rodents (eg, mice and rats). In certain aspects, the individual or subject is human.
- “Pharmaceutically acceptable carrier” refers to an ingredient other than the active ingredient in pharmaceutical formulations and pharmaceutical compositions that is non-toxic to a subject.
- Pharmaceutically acceptable carriers include, but are not limited to, buffers, excipients, stabilizers, or preservatives.
- an “effective amount” of an agent refers to that amount, at dosages necessary and for periods of time necessary, effective to achieve the desired therapeutic or prophylactic result. .
- package insert is commonly included in commercial packaging of therapeutic products and includes information about indications, usage, dosage, method of administration, concomitant therapies, contraindications, and/or warnings regarding the use of such therapeutic products. Used to refer to instructions for use.
- CTLA-4" may be from any vertebrate source, including mammals such as primates (e.g., humans) and rodents (e.g., mice and rats). Any native CTLA-4 of The term encompasses "full length" unprocessed CTLA-4 as well as any form of CTLA-4 that results from processing in the cell. The term also includes naturally occurring variants of CTLA-4, such as splice variants and allelic variants.
- Exemplary human CTLA-4 amino acid sequence is SEQ ID NO: 214
- mouse CTLA-4 amino acid sequence is SEQ ID NO: 247
- monkey CTLA-4 amino acid sequence is SEQ ID NO: 248,
- human CTLA-4 amino acid sequence is SEQ ID NO: 248.
- the amino acid sequence of the extracellular domain is shown in SEQ ID NO:28.
- CTLA-4 is sometimes referred to as CTLA4.
- Treg cells are a subpopulation of T cells that regulate the immune system, maintain tolerance to self-antigens, and prevent autoimmune diseases. These cells generally suppress or downregulate the induction and proliferation of effector T cells.
- the best understood Treg cells are those that express CD4, CD25 and Foxp3 (CD4 + CD25 + Treg cells). These Tregs are different from helper T cells.
- Several different methods are used to identify and monitor Treg cells. As defined by CD4 and CD25 expression (CD4 + CD25 + cells), Treg cells constitute approximately 5-10% of the mature CD4 + T cell subpopulation in mice and humans, whereas approximately 1-2% of Tregs It can be measured in whole blood.
- Foxp3 expression may additionally be measured (CD4 + CD25 + Foxp3 + cells). Absence or low level expression of CD127 may also be used in combination with the presence of CD4 and CD25 as another marker.
- Treg cells also express high levels of CTLA-4 and GITR. Tregs can also be identified by the methods described in the Examples below.
- the terms “substantially similar,” “substantially equal,” or “substantially the same” refer to between two numbers (e.g., with respect to the antibodies of the invention, see/compare The similarity between those two values (for example, the KD value) is defined by those skilled in the art as having little or no biological and/or no similarity in terms of biological characteristics measured by the value (e.g., KD value). or high enough to be considered statistically insignificant.
- treatment refers to clinical treatments intended to alter the natural course of the individual being treated.
- intervention which can be performed both for prophylaxis and during the course of a clinical condition.
- Desirable effects of treatment include, but are not limited to, prevention of disease onset or recurrence, relief of symptoms, attenuation of any direct or indirect pathological effects of disease, prevention of metastasis, prevention of disease Includes reduced rate of progression, amelioration or amelioration of disease state, and remission or improved prognosis.
- the antibodies of the invention are used to delay the onset of disease or slow the progression of disease.
- polypeptides comprising variant Fc regions of the invention are used to delay disease onset or slow disease progression.
- tumor refers to all neoplastic cell growth and proliferation and all precancerous and cancerous cells and tissues, whether malignant or benign.
- cancer refers to all neoplastic cell growth and proliferation and all precancerous and cancerous cells and tissues, whether malignant or benign.
- cancer refers to all neoplastic cell growth and proliferation and all precancerous and cancerous cells and tissues, whether malignant or benign.
- cancer refers to all neoplastic cell growth and proliferation and all precancerous and cancerous cells and tissues, whether malignant or benign.
- tumor tissue means tissue containing at least one tumor cell.
- Tumor tissue is usually composed of a mass of tumor cells (parenchyma) that constitutes the main body of the tumor, and connective tissue and blood vessels (stroma) that exist between them and support the tumor. In some cases, the distinction between the two is clear, and in others, the two are mixed. Immune cells and the like may infiltrate into the tumor tissue.
- non-tumor tissue means tissue other than tumor tissue in vivo. Healthy/normal tissue without disease is a representative example of non-tumor tissue.
- the invention is based in part on anti-CTLA-4 antibodies and their uses.
- antibodies are provided that bind to CTLA-4.
- Antibodies of the invention are useful, for example, for the diagnosis or treatment of cancer.
- the invention provides isolated antibodies that bind to CTLA-4.
- the anti-CTLA-4 antibodies of the invention have CTLA-4 binding activity that is dependent on the concentration of the adenosine-containing compound.
- the binding activity to CTLA-4 is higher in the presence of the adenosine-containing compound than in the absence of the adenosine-containing compound.
- the binding activity to CTLA-4 is higher in the presence of high concentrations of adenosine-containing compounds than in the presence of low concentrations of adenosine-containing compounds.
- the difference in binding activity to CTLA-4 is, for example, 2-fold or more, 3-fold or more, 5-fold or more, 10-fold or more, 20-fold or more, 30-fold or more, 50-fold or more, 100-fold or more, 200-fold. 300 times or more, 500 times or more, 1 x 10 3 times or more, 2 x 10 3 times or more, 3 x 10 3 times or more, 5 x 10 3 times or more, 1 x 10 4 times or more, 2 x 10 4 times 3 ⁇ 10 4 times or more, 5 ⁇ 10 4 times or more, or 1 ⁇ 10 5 times or more.
- the binding activity of an anti-CTLA-4 antibody can be expressed as a KD (Dissociation constant) value.
- the anti-CTLA-4 antibody has a lower KD value in the presence of the adenosine-containing compound than in the absence of the adenosine-containing compound.
- the anti-CTLA-4 antibody has a lower KD value in the presence of high concentrations of adenosine-containing compound than in the presence of low concentrations of adenosine-containing compound.
- the difference in KD values of anti-CTLA-4 antibodies is, for example, 2-fold or more, 3-fold or more, 5-fold or more, 10-fold or more, 20-fold or more, 30-fold or more, 50-fold or more, 100-fold or more, 200-fold or more.
- the KD value of the anti-CTLA-4 antibody in the presence of an adenosine-containing compound or in the presence of a high concentration of an adenosine-containing compound is, for example, 9 ⁇ 10 ⁇ 7 M or less, 8 ⁇ 10 ⁇ 7 M or less, 7 ⁇ 10 ⁇ 7 M or less, 6 ⁇ 10 -7 M or less, 5 ⁇ 10 -7 M or less, 4 ⁇ 10 -7 M or less, 3 ⁇ 10 -7 M or less, 2 ⁇ 10 -7 M or less, 1 ⁇ 10 -7 M or less , 9 ⁇ 10 -8 M or less, 8 ⁇ 10 -8 M or less, 7 ⁇ 10 -8 M or less, 6 ⁇ 10 -8 M or less, 5 ⁇ 10 -8 M or less, 4 ⁇ 10 -8 M or less, 3 ⁇ 10 -8 M or less, 2 ⁇ 10 -8 M or less, 1 ⁇ 10 -8 M or less, 9 ⁇ 10 -9 M or less, 8 ⁇ 10 -9 M or less, 7 ⁇ 10 -9 M or less, 6 ⁇ 10
- the KD value of the anti-CTLA-4 antibody in the absence of an adenosine-containing compound or in the presence of a low concentration of an adenosine-containing compound is, for example, 1 ⁇ 10 ⁇ 8 M or more, 2 ⁇ 10 ⁇ 8 M or more, 3 ⁇ 10 ⁇ 8 M or higher, 4 ⁇ 10 -8 M or higher, 5 ⁇ 10 -8 M or higher, 6 ⁇ 10 -8 M or higher, 7 ⁇ 10 -8 M or higher, 8 ⁇ 10 -8 M or higher, 9 ⁇ 10 -8 M or higher 1 ⁇ 10 -7 M or more, 2 ⁇ 10 -7 M or more, 3 ⁇ 10 -7 M or more, 4 ⁇ 10 -7 M or more, 5 ⁇ 10 -7 M or more, 6 ⁇ 10 -7 M or more, 7 ⁇ 10 -7 M or more, 8 ⁇ 10 -7 M or more, 9 ⁇ 10 -7 M or more, 1 ⁇ 10 -6 M or more, 2 ⁇ 10 -6 M or more, 3 ⁇ 10 -6 M or more, 4 ⁇ 10 ⁇ 6 M or more, 5 ⁇
- the binding activity of an anti-CTLA-4 antibody may be expressed as a kd (dissociation rate constant) value instead of a KD value.
- the binding activity of an anti-CTLA-4 antibody may be expressed as the amount of CTLA-4 bound per unit amount of antibody.
- the binding amount of an antibody immobilized on a sensor chip and the binding amount of an antigen further bound thereon are each measured as a response unit (RU).
- a value obtained by dividing the antigen binding amount by the antibody binding amount can be defined as the antigen binding amount per unit amount of antibody.
- CTLA-4 binds more in the presence of the adenosine-containing compound than in the absence of the adenosine-containing compound.
- CTLA-4 binds more in the presence of high concentrations of adenosine-containing compounds than in the presence of low concentrations of adenosine-containing compounds.
- the difference in the amount of CTLA-4 binding is, for example, 2-fold or more, 3-fold or more, 5-fold or more, 10-fold or more, 20-fold or more, 30-fold or more, 50-fold or more, 100-fold or more, 200-fold or more.
- the value of the CTLA-4 binding amount in the presence of an adenosine-containing compound or in the presence of a high concentration of an adenosine-containing compound is, for example, 0.01 or more, 0.02 or more, 0.03 or more, 0.04 or more, 0.05 or more, 0.06 or more, 0.07 or more, It can be 0.08 or greater, 0.09 or greater, 0.1 or greater, 0.2 or greater, 0.3 or greater, 0.4 or greater, 0.5 or greater, 0.6 or greater, 0.7 or greater, 0.8 or greater, 0.9 or greater, or 1 or greater.
- the value of CTLA-4 binding in the absence of an adenosine-containing compound or in the presence of a low concentration of an adenosine-containing compound is, for example, 0.5 or less, 0.4 or less, 0.3 or less, 0.2 or less, 0.1 or less, 0.09 or less, or 0.08 or less.
- ⁇ 0.07, ⁇ 0.06, ⁇ 0.05, ⁇ 0.04, ⁇ 0.03, ⁇ 0.02, ⁇ 0.01, ⁇ 0.009, ⁇ 0.008, ⁇ 0.007, ⁇ 0.006, ⁇ 0.005, ⁇ 0.004, ⁇ 0.003, ⁇ 0.002, or ⁇ 0.001 can be.
- the KD value, kd value, binding amount value, etc. referred to herein are measured or calculated by performing a surface plasmon resonance assay at 25°C or 37°C (e.g., See Reference Example 3 herein).
- concentrations include, for example, 1 nM or higher, 3 nM or higher, 10 nM or higher, 30 nM or higher, 100 nM or higher , 300 nM or higher, 1 ⁇ M or higher, 3 ⁇ M or higher, 10 ⁇ M or higher, 30 ⁇ M or higher, 100 ⁇ M or higher, 300 ⁇ M or higher, 1 mM or higher, 3 mM or higher, 10 mM or higher, 30 mM or higher, 100 mM or higher, 300 mM or Higher, can be mentioned.
- the high concentration here can be sufficient for each anti-CTLA-4 antibody to exhibit maximal binding activity.
- 1 ⁇ M, 10 ⁇ M, 100 ⁇ M, 1 mM, or an amount sufficient for each anti-CTLA-4 antibody to exhibit maximal binding activity can be selected as the high concentration herein.
- low concentrations include, for example, 1 mM or lower concentrations, 300 ⁇ M or lower concentrations, 100 ⁇ M or lower concentrations, 30 ⁇ M or lower concentrations, 10 ⁇ M or lower concentrations , 3 ⁇ M or less, 1 ⁇ M or less, 300 nM or less, 100 nM or less, 30 nM or less, 10 nM or less, 3 nM or lower concentrations, 1 nM or lower concentrations, 300 pM or lower concentrations, 100 pM or lower concentrations, 30 pM or lower concentrations, 10 pM or lower concentrations, 3 pM or lower Lower Concentrs, such as 1 pM or lower, can be mentioned.
- the low concentration can be the concentration at which each anti-CTLA-4 antibody exhibits minimal binding activity.
- a substantially zero concentration (absence of an adenosine-containing compound) can also be selected as one aspect of low concentration. In one embodiment, 1 mM, 100 ⁇ M, 10 ⁇ M, 1 ⁇ M, the concentration at which the respective anti-CTLA-4 antibody exhibits minimal binding activity, or the absence of an adenosine compound is selected as the low concentration herein. be able to.
- the ratio of high concentration to low concentration is, for example, 3-fold or more, 10-fold or more, 30-fold or more, 100-fold or more, 300-fold or more, 1 ⁇ 10 3 times or more, 3 ⁇ 10 3 times or more, 1 ⁇ 10 4 times or more, 3 ⁇ 10 4 times or more, 1 ⁇ 10 5 times or more, 3 ⁇ 10 5 times or more, 1 ⁇ 10 6 times or more, 3 ⁇ 10 6 times or more, 1 ⁇ 10 7 times or more, 3 ⁇ 10 7 times or more, 1 ⁇ 10 8 times or more, 3 ⁇ 10 8 times or more, 1 x 10 9 times or more, 3 x 10 9 times or more, 1 x 10 10 times or more, 3 x 10 10 times or more, 1 x 10 11 times or more, 3 Values of 1 ⁇ 10 11 times or more, 1 ⁇ 10 12 times or more can be selected.
- the anti-CTLA-4 antibody of the present invention also has binding activity for adenosine-containing compounds.
- the binding amount of the adenosine-containing compound per unit amount of antibody can be calculated and used as the binding activity of the anti-CTLA-4 antibody to the adenosine-containing compound.
- a specific method for measuring and calculating such a binding amount is described in Examples below.
- the binding amount of the adenosine-containing compound per unit amount of the anti-CTLA-4 antibody of the present invention is, for example, 0.0001 or more, 0.0002 or more, 0.0003 or more, 0.0004 or more, 0.0005 or more, 0.0006 or more, 0.0007 or more, 0.0008 or more, 0.0009. 0.001 or more, 0.002 or more, 0.003 or more, 0.004 or more, 0.005 or more, 0.006 or more, 0.007 or more, 0.008 or more, 0.009 or more, or 0.01 or more.
- an anti-CTLA-4 antibody of the invention forms a ternary complex with an adenosine-containing compound and CTLA-4.
- the anti-CTLA-4 antibody binds the adenosine-containing compound via heavy chain CDR1, CDR2, CDR3.
- the anti-CTLA-4 antibody has a binding motif for an adenosine-containing compound. Binding motifs for adenosine-containing compounds are present, for example, at positions 33, 52, 52a, 53, 56, 58, 95, 96, 100a, 100b, 100c as represented by Kabat numbering. It can consist of at least one amino acid.
- the anti-CTLA-4 antibody is at positions 33, 52, 52a, 53, 56, 58, 95, 96, 100a, 100b, 100c, e.g., Kabat numbering. binds to the adenosine-containing compound through at least one amino acid selected from the group consisting of
- the anti-CTLA-4 antibody comprises Thr at position 33, Ser at position 52, Ser at position 52a, Arg at position 53, Tyr at position 56, Tyr at position 58, Tyr at position 95, as represented by Kabat numbering.
- Tyr at position 96 Gly at position 100a, Met at position 100a, Leu at position 100b, and Trp at position 100c.
- CTLA-4 may further bind to a complex formed by binding an anti-CTLA-4 antibody and an adenosine-containing compound.
- the adenosine-containing compound may also be present at the interface where the anti-CTLA-4 antibody and CTLA-4 interact and bind to both of them.
- the fact that the anti-CTLA-4 antibody forms a ternary complex with an adenosine-containing compound and CTLA-4 can be confirmed, for example, by techniques such as crystal structure analysis described below (see Examples).
- the anti-CTLA-4 antibody of the invention is human CTLA-4 (extracellular domain, SEQ ID NO: 28) at amino acids 3 (Met), 33 (Glu), 35 (Arg), 53rd amino acid (Thr), 97th amino acid (Glu), 99th amino acid (Met), 100th amino acid (Tyr), 101st amino acid (Pro), 102nd amino acid (Pro ), the 103rd amino acid (Pro), the 104th amino acid (Tyr), the 105th amino acid (Tyr), and the 106th amino acid (Leu). These amino acids may constitute the epitope of the anti-CTLA-4 antibodies of the invention.
- the anti-CTLA-4 antibodies of the invention bind to the region from amino acid 97 (Glu) to amino acid 106 (Leu) of human CTLA-4 (extracellular domain, SEQ ID NO:28). . In another embodiment, the anti-CTLA-4 antibodies of the invention bind to the region from amino acid 99 (Met) to amino acid 106 (Leu) of human CTLA-4 (extracellular domain, SEQ ID NO:28). .
- the anti-CTLA-4 antibodies of the invention are ABAM004 (VH, SEQ ID NO: 10; VL, SEQ ID NO: 11; HVR-H1, SEQ ID NO: 100; HVR- H2, SEQ ID NO: 101; HVR-H3, SEQ ID NO: 102; HVR-Ll, SEQ ID NO: 113; HVR-L2, SEQ ID NO: 114; HVR-L3, SEQ ID NO: 115).
- an anti-CTLA-4 antibody of the invention binds to the same epitope as ABAM004. When anti-CTLA-4 antibodies are present in excess, binding of ABAM004 to CTLA-4 is reduced, e.g.
- the anti-CTLA-4 antibody of the present invention exhibits cytotoxic activity against CTLA-4-expressing cells.
- CTLA-4 When CTLA-4 is expressed on the surface of a target cell and an anti-CTLA-4 antibody binds to it, the cell can be damaged.
- Cell damage is caused by antibody-bound effector cells, such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). It can also be caused by antibody-bound complement, such as complement-dependent cytotoxicity (CDC).
- ADCC antibody-dependent cellular cytotoxicity
- ADCP antibody-dependent cellular phagocytosis
- CDC complement-dependent cytotoxicity
- it may be induced by a cytotoxic agent (eg, radioisotope, chemotherapeutic agent, etc.) attached to an antibody, such as an immunoconjugate.
- a cytotoxic agent eg, radioisotope, chemotherapeutic agent, etc.
- cytotoxicity may include an action of inducing cell death, an action of suppressing cell proliferation, an action of impairing cell function, and the like. If anti-CTLA-4 antibodies are present in sufficient amounts, e.g. , ⁇ 55%, ⁇ 60%, ⁇ 65%, ⁇ 70%, ⁇ 75%, ⁇ 80%, ⁇ 85%, ⁇ 90%, or ⁇ 95% of CTLA-4 expressing cells can be done. Measurement of such cytotoxic activity can be performed in comparison with measurement in the absence of antibody or in the presence of a negative control antibody. An exemplary cytotoxicity assay is provided herein.
- the anti-CTLA-4 antibody of the present invention exhibits neutralizing activity against CTLA-4.
- CTLA-4 is known to function by interacting with its ligand, CD80 (B7-1) or CD86 (B7-2).
- anti-CTLA-4 antibodies inhibit the interaction of CTLA-4 with CD80 (B7-1) or CD86 (B7-2).
- anti-CTLA-4 antibodies are present in sufficient amounts, the interaction of CTLA-4 with CD80 (B7-1) or CD86 (B7-2) is reduced e.g.
- Measurement of such inhibitory activity can be performed in comparison with measurement in the absence of antibody or in the presence of a negative control antibody.
- a specific method of measuring neutralizing activity is provided herein.
- the anti-CTLA-4 antibodies of the present invention bind CTLA-4 from multiple animal species.
- animal species include mammals such as humans, monkeys, mice, rats, hamsters, guinea pigs, rabbits, pigs, cows, goats, horses, sheep, camels, dogs, cats, and the like.
- anti-CTLA-4 antibodies bind CTLA-4 from humans and non-humans (eg, monkeys, mice, rats, etc.).
- the amino acid sequence of human CTLA-4 is shown in SEQ ID NO:214, that of monkey CTLA-4 is shown in SEQ ID NO:247, and that of mouse CTLA-4 is shown in SEQ ID NO:248.
- Amino acid sequences of CTLA-4 derived from other animal species can also be appropriately determined by methods known to those skilled in the art.
- adenosine-containing compounds in the present invention include, for example, adenosine (ADO), adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), cyclic adenosine monophosphate (cAMP), deoxyadenosine (dADO), deoxyadenosine triphosphate (dATP), deoxyadenosine diphosphate (dADP), deoxyadenosine monophosphate (dAMP), adenosine gamma thiotriphosphate (ATP ⁇ S), etc.
- ADO adenosine
- ATP adenosine triphosphate
- ADP adenosine diphosphate
- AMP adenosine monophosphate
- cAMP cyclic adenosine monophosphate
- deoxyadenosine triphosphate dATP
- deoxyadenosine diphosphate d
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:223; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:224; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:223; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:224; and (c) the amino acid sequence of SEQ ID NO:225. and HVR-H3 containing sequences.
- the present invention provides (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:226; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:227; Antibodies are provided comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:226; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:227; and (c) the amino acid sequence of SEQ ID NO:228. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO:223, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO:224 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 225; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO:226, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO:227, and (iii) the amino acid sequence of SEQ ID NO:228 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:223; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:224; and (c) SEQ ID NO:225. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO:226; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:227; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 228 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:100; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:101; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:100; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:101; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:113; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:114; Antibodies are provided comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:113; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:114; and (c) the amino acid sequence of SEQ ID NO:115. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 100, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 101 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 113, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 114, and (iii) the amino acid sequence of SEQ ID NO: 115 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:100; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:101; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 113; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 114; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 115 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 100; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 104; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:100; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:104; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:116; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; Antibodies are provided comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:116; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:115. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO:100, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO:104 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 116, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 115 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:100; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:104; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO:116; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 115 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:105; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:106; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:105; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:106; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides a Antibodies comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:122; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 105, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 106 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 122, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:105; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:106; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 122; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 108; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:108; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:121; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:123; Antibodies are provided comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:121; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:123; and (c) the amino acid sequence of SEQ ID NO:153. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 108 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 121, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 123, and (iii) the amino acid sequence of SEQ ID NO: 153 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:108; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 121; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 123; (f) SEQ ID NO: and HVR-L3 comprising a 153 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:110; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:110; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides a Antibodies comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:122; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 110 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 122, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:110; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 122; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:112; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:112; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides a Antibodies comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:128; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 112 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 128, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:112; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 128; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 111; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:111; and (c) the amino acid sequence of SEQ ID NO:152. and HVR-H3 containing sequences.
- the present invention provides a Antibodies comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:128; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 111 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 152; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 128, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:111; and (c) SEQ ID NO:152. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 128; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:112; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:112; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the present invention provides a Antibodies comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:129; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 112 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 129, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:112; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 129; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 111; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:111; and (c) the amino acid sequence of SEQ ID NO:152. and HVR-H3 containing sequences.
- the present invention provides a Antibodies comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:129; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 111 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 152; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 129, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:111; and (c) SEQ ID NO:152. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 129; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:109; Antibodies are provided comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising sequences.
- the antibody comprises (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:109; and (c) the amino acid sequence of SEQ ID NO:102. and HVR-H3 containing sequences.
- the invention provides (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:130; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; Antibodies are provided comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3 comprising amino acid sequences.
- the antibody comprises (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO:130; (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO:117; and (c) the amino acid sequence of SEQ ID NO:133. and HVR-L3 containing sequences.
- the antibody of the invention comprises (a) a VH domain, (i) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 107, (ii) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 109 H2, and (iii) a VH domain comprising at least one, at least two, or all three VH HVR sequences selected from HVR-H3 comprising the amino acid sequence of SEQ ID NO: 102; and (b) VL a domain comprising (i) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 130, (ii) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (iii) the amino acid sequence of SEQ ID NO: 133 and a VL domain comprising at least one, at least two, or all three VL HVR sequences selected from HVR-L3.
- the present invention provides (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO:107; (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO:109; and (c) SEQ ID NO:102. (d) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 130; (e) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117; (f) SEQ ID NO: Antibodies are provided, including HVR-L3, which comprises a 133 amino acid sequence.
- any one or more amino acids of the anti-CTLA-4 antibody described above are substituted at the following HVR positions: - in HVR-H1 (SEQ ID NO: 223): position 2 - in HVR-H2 (SEQ ID NO: 224): positions 4, 5, 7, 13, and 16 - in HVR-H3 (SEQ ID NO: 225): position 3 - in HVR-L1 (SEQ ID NO: 226): positions 1, 3, 6, 11, 12, and 14 - in HVR-L2 (SEQ ID NO: 227): positions 1, 3, 4, and 7 - in HVR-L3 (SEQ ID NO: 228): positions 1 and 10
- substitutions provided herein are conservative substitutions.
- any one or more of the following substitutions may be made in any combination: -In HVR-H1 (SEQ ID NO: 100): H2A, R or K -In HVR-H2 (SEQ ID NO: 101): S4T; R5Q; G7H; D13E or R;
- the anti-CTLA-4 antibody is humanized.
- the anti-CTLA-4 antibody comprises the HVR of any of the above embodiments and further comprises an acceptor human framework (eg, human immunoglobulin framework or human consensus framework).
- the anti-CTLA-4 antibody comprises the HVR of any of the above embodiments and further comprises a VH or VL comprising FR sequences.
- the anti-CTLA-4 antibody comprises the following heavy and/or light chain variable domain FR sequences: For the heavy chain variable domain, FR1 has the amino acid sequence of any one of SEQ ID NOs: 229-232.
- FR2 comprises the amino acid sequence of SEQ ID NO:233
- FR3 comprises the amino acid sequence of SEQ ID NO:23
- FR4 comprises the amino acid sequence of SEQ ID NO:235.
- FR1 comprises the amino acid sequence of any one of SEQ ID NOs: 236-238
- FR2 comprises the amino acid sequence of any one of SEQ ID NOs: 240-241
- FR3 comprises SEQ ID NOs: 242-241.
- FR4 comprises the amino acid sequence of any one of SEQ ID NOS:245-246.
- the anti-CTLA-4 antibody is at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, Contains heavy chain variable domain (VH) sequences with 99% or 100% sequence identity.
- VH heavy chain variable domain
- a total of 1 to 10, 11, 12, 13, 14, or 15 amino acids are substituted, inserted, and/or deleted in SEQ ID NO:10.
- the anti-CTLA-4 antibody comprises the VH sequence in SEQ ID NO: 10, including post-translational modifications of that sequence.
- the VH is (a) HVR-H1 comprising the amino acid sequence of SEQ ID NO: 100, (b) HVR-H2 comprising the amino acid sequence of SEQ ID NO: 101, and (c) SEQ ID NO: 102. 1, 2, or 3 HVRs selected from HVR-H3 containing amino acid sequences.
- Post-translational modifications include, but are not limited to, modification of heavy or light chain N-terminal glutamine or glutamic acid to pyroglutamic acid by pyroglutamylation.
- an anti-CTLA-4 antibody comprises a light chain variable domain (VL) with sequence identity.
- VL light chain variable domain
- a VL sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to a reference sequence is , substitutions (eg, conservative substitutions), insertions, or deletions, but anti-CTLA-4 antibodies containing such sequences retain the ability to bind CTLA-4.
- the anti-CTLA-4 antibody comprises the VL sequence in SEQ ID NO: 11, including post-translational modifications of that sequence.
- the VL is (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 113, (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 114, and (c) SEQ ID NO: 115. 1, 2, or 3 HVRs selected from HVR-L3 containing amino acid sequences.
- Post-translational modifications include, but are not limited to, modification of heavy or light chain N-terminal glutamine or glutamic acid to pyroglutamic acid by pyroglutamylation.
- an anti-CTLA-4 antibody comprises a light chain variable domain (VL) with sequence identity.
- VL light chain variable domain
- a VL sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to a reference sequence is , substitutions (eg, conservative substitutions), insertions, or deletions, but anti-CTLA-4 antibodies containing such sequences retain the ability to bind CTLA-4.
- the anti-CTLA-4 antibody comprises the VL sequence in SEQ ID NO: 149, including post-translational modifications of that sequence.
- the VL is (a) HVR-L1 comprising the amino acid sequence of SEQ ID NO: 130, (b) HVR-L2 comprising the amino acid sequence of SEQ ID NO: 117, and (c) SEQ ID NO: 133.
- HVRs selected from HVR-L3 containing amino acid sequences.
- Post-translational modifications include, but are not limited to, modification of heavy or light chain N-terminal glutamine or glutamic acid to pyroglutamic acid by pyroglutamylation.
- an anti-CTLA-4 antibody comprising a VH in any of the above embodiments and a VL in any of the above embodiments.
- the antibody comprises the VH and VL sequences in SEQ ID NO:10 and SEQ ID NO:11, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO:98 and SEQ ID NO:99, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:88, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:89, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:90, respectively, including post-translational modifications of the sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:91, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:92, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:93, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:94, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:83 and SEQ ID NO:95, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO:84 and SEQ ID NO:97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:85 and SEQ ID NO:97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:86 and SEQ ID NO:97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:86 and SEQ ID NO:134, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO: 136 and SEQ ID NO: 97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO: 135 and SEQ ID NO: 97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO: 136 and SEQ ID NO: 95, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO: 137 and SEQ ID NO: 97, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO: 138 and SEQ ID NO: 97, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:138 and SEQ ID NO:144, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:138 and SEQ ID NO:145, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:138 and SEQ ID NO:146, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO: 139 and SEQ ID NO: 146, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO: 140 and SEQ ID NO: 146, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO: 141 and SEQ ID NO: 146, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:140 and SEQ ID NO:147, respectively, including post-translational modifications of said sequences.
- the antibody comprises the VH and VL sequences in SEQ ID NO:141 and SEQ ID NO:147, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:140 and SEQ ID NO:148, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO: 141 and SEQ ID NO: 148, respectively, including post-translational modifications of said sequences. In one embodiment, the antibody comprises the VH and VL sequences in SEQ ID NO:136 and SEQ ID NO:149, respectively, including post-translational modifications of said sequences.
- heterologous anti-CTLA-4 antibodies comprise at least two different variable regions selected from among the variable regions comprising the VH and VL sequences described above.
- the antibody comprised the VH and VL sequences in SEQ ID NO: 140 and SEQ ID NO: 146, respectively, and the VH and VL sequences in SEQ ID NO: 141 and SEQ ID NO: 146, and post-translational modifications of the sequences. Including things, including things.
- the antibody comprised the VH and VL sequences in SEQ ID NO: 140 and SEQ ID NO: 147, respectively, and the VH and VL sequences in SEQ ID NO: 141 and SEQ ID NO: 147, and post-translational modifications of the sequences.
- Post-translational modifications include, but are not limited to, modification of heavy or light chain N-terminal glutamine or glutamic acid to pyroglutamic acid by pyroglutamylation.
- the amino acid may be substituted with glutamic acid.
- the heavy or light chain N-terminal amino acid of the anti-CTLA-4 antibody provided herein is glutamic acid, the amino acid may be substituted with glutamine.
- the present invention provides antibodies that bind to the same epitope as the anti-CTLA-4 antibodies provided herein.
- antibodies are provided that bind to the same epitope as the antibodies listed in Tables 7, 12, 17, and 22.
- an antibody that binds to an epitope in a fragment of CTLA-4 comprising at least one amino acid selected from the group consisting of the amino acid at position (Tyr) and the amino acid at position 106 (Leu).
- antibodies are provided that bind to an epitope in a fragment of CTLA-4 consisting of amino acids 97 (Glu) through 106 (Leu) of SEQ ID NO:28. In a specific embodiment, antibodies are provided that bind to an epitope in a fragment of CTLA-4 consisting of amino acids 99 (Met) through 106 (Leu) of SEQ ID NO:28.
- the anti-CTLA-4 antibody is a monoclonal antibody, including chimeric, humanized or human antibodies.
- the anti-CTLA-4 antibody is an antibody fragment, eg, an Fv, Fab, Fab', scFv, diabody, or F(ab') 2 fragment.
- the antibody is a full-length antibody, eg, a complete IgG1 antibody, a complete IgG4 antibody, or other antibody classes or isotypes as defined herein.
- the anti-CTLA-4 antibody of the present invention comprises an Fc region.
- the anti-CTLA-4 antibodies of the invention comprise a constant region.
- the constant regions may be heavy chain constant regions (including Fc regions), light chain constant regions, or both.
- the Fc region is a native sequence Fc region.
- Exemplary heavy chain constant regions derived from native antibodies include human IgG1 (SEQ ID NO: 249), human IgG2 (SEQ ID NO: 250), human IgG3 (SEQ ID NO: 251), human IgG4 (SEQ ID NO: 252 ) and other heavy chain constant regions.
- heavy chain constant regions include heavy chain constant regions such as SEQ ID NO:82, SEQ ID NO:158, SEQ ID NO:334.
- exemplary light chain constant regions derived from native antibodies include human ⁇ chain (SEQ ID NO: 33, SEQ ID NO: 63, SEQ ID NO: 159), human ⁇ chain (SEQ ID NO: 53, SEQ ID NO: 87). and light chain constant regions such as
- the Fc region is a mutant Fc region produced by amino acid alterations to the native-sequence Fc region.
- the variant Fc region has enhanced binding activity to at least one Fc ⁇ receptor selected from the group consisting of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIb, Fc ⁇ RIIIa, as compared to a native sequence Fc region.
- the variant Fc region has enhanced binding activity to Fc ⁇ RIIa and Fc ⁇ RIIIa compared to a native sequence Fc region.
- heavy chain constant regions comprising such mutated Fc regions include heavy chain constant regions listed in Tables 29 to 33, SEQ ID NOs: 31, 32, 41-46, 65, 66, 81, 207, 239. , 253-271, 276, 277, 278, 308, 309, 311-333, 358-367, and the like.
- the Fc region of the native sequence is usually constructed as a homodimer consisting of two identical polypeptide chains.
- the mutant Fc regions may be homodimers composed of polypeptide chains with the same sequence, or heterodimers composed of polypeptide chains with different sequences. good.
- the heavy chain constant region containing the Fc region may be a homodimer composed of polypeptide chains with the same sequence, or a heterodimer composed of polypeptide chains with different sequences. There may be.
- heterologous heavy chain constant regions include, for example, a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:31 and the polypeptide chain of SEQ ID NO:32, the polypeptide chain of SEQ ID NO:43 and the polypeptide chain of SEQ ID NO:44.
- a heavy chain constant region comprising a peptide chain, the polypeptide chain of SEQ ID NO:45 and a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:46, the polypeptide chain of SEQ ID NO:254 and the polypeptide chain of SEQ ID NO:256 a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:257 and a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:258, the polypeptide chain of SEQ ID NO:259 and the polypeptide chain of SEQ ID NO:260
- a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:261 and the polypeptide chain of SEQ ID NO:263, the polypeptide chain of SEQ ID NO:262 and the polypeptide chain of SEQ ID NO:264 a constant region, a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:265 and the polypeptide chain of SEQ ID NO:267, a heavy chain constant region comprising
- an anti-CTLA-4 antibody may incorporate any of the features listed in items 1-7 below.
- the binding activity of the antibodies provided herein is ⁇ 10 ⁇ M, ⁇ 1 ⁇ M, ⁇ 100 nM, ⁇ 10 nM, ⁇ 1 nM, ⁇ 0.1 nM, A dissociation constant (KD) of ⁇ 0.01 nM, or ⁇ 0.001 nM (eg, 10 ⁇ 8 M or less, such as 10 ⁇ 8 M to 10 ⁇ 13 M, such as 10 ⁇ 9 M to 10 ⁇ 13 M).
- KD dissociation constant
- antibody binding activity is measured by a radiolabeled antigen binding assay (RIA).
- RIA radiolabeled antigen binding assay
- the RIA is performed using the Fab version of the antibody of interest and its antigen.
- the in-solution binding affinity of a Fab for an antigen can be determined by equilibrating the Fab with a minimal concentration of ( 125 I)-labeled antigen in the presence of an increasing dose series of unlabeled antigen and then coating the bound antigen with an anti-Fab antibody. 293:865-881 (1999)).
- the Fab of interest is then incubated overnight, although this incubation can be continued for a longer time (eg, about 65 hours) to ensure equilibrium is reached.
- the mixture is then transferred to a capture plate for incubation (eg, 1 hour) at room temperature.
- the solution is then removed and the plate washed 8 times with 0.1% polysorbate 20 (TWEEN-20®) in PBS. Once the plates are dry, 150 ⁇ l/well scintillant (MICROSCINT-20TM, Packard) is added and the plates are counted for 10 minutes in a TOPCOUNTTM gamma counter (Packard). Concentrations of each Fab that give 20% or less of maximal binding are selected for use in competitive binding assays.
- the binding activity of the antibody is measured by surface plasmon resonance spectroscopy, for example, using BIACORE (trademark registered) T200 or BIACORE (trademark registered) 4000 (GE Healthcare, Uppsala, Sweden).
- a ligand capture method is used.
- BIACORE® Control Software is used for instrument operation.
- ligand-capturing molecules e.g. Tag antibodies, anti-IgG antibodies, protein A, etc. are immobilized.
- Ligand capture molecules are diluted with a 10 mM sodium acetate solution of appropriate pH and injected at appropriate flow rates and injection times.
- a buffer solution containing 0.05% polysorbate 20 (other names are Tween (registered trademark)-20) is used as the measurement buffer, the flow rate is 10-30 ⁇ L/min, and the measurement temperature is preferably 25°C. Measured at 37°C.
- the buffer solution for measurement was used to prepare Serial dilutions of antibody (analyte) are injected.
- the measurement results are analyzed using BIACORE® Evaluation Software. Calculation of the kinetics parameters was performed by fitting the binding and dissociation sensorgrams simultaneously using a model of 1:1 Binding, and the binding rate (kon or ka), dissociation rate (koff or kd), the equilibrium dissociation constant (KD) can be calculated. If the binding activity is weak, especially if the dissociation is rapid and calculation of kinetic parameters is difficult, the steady state model may be used to calculate the equilibrium dissociation constant (KD). As another parameter of binding activity, the amount of analyte bound (RU) at a specific concentration is divided by the amount of ligand captured (RU) to calculate the amount of analyte bound per unit amount of ligand. good.
- the antibodies provided herein are antibody fragments.
- Antibody fragments include, but are not limited to, Fab, Fab', Fab'-SH, F(ab') 2 , Fv, and scFv fragments, as well as other fragments described below.
- Fab fragment antigen
- Fab' fragment antigen-specific antibody fragment
- Fab'-SH fragment antigen-specific antibody fragment
- Fv fragment antigen-specific antibody fragment
- scFv fragments see, for example, Pluckthun, in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., (Springer-Verlag, New York), pp. 269-315 (1994); 16185; and U.S. Pat. Nos.
- a diabody is an antibody fragment with two antigen-binding sites that may be bivalent or bispecific.
- WO1993/01161 Hudson et al., Nat. Med. 9: 129-134 (2003); Hollinger et al., Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993 ) reference.
- Triabodies and tetrabodies are also described in Hudson et al., Nat. Med. 9: 129-134 (2003).
- a single domain antibody is an antibody fragment that contains all or part of the heavy chain variable domain or all or part of the light chain variable domain of an antibody.
- the single domain antibody is a human single domain antibody (Domantis, Inc., Waltham, MA; see, eg, US Pat. No. 6,248,516 B1).
- Antibody fragments are produced in a variety of ways, including, but not limited to, proteolytic digestion of intact antibodies, production by recombinant host cells (e.g., E. coli or phage), as described herein. can be made by the method of
- the antibodies provided herein are chimeric antibodies.
- Certain chimeric antibodies are described, for example, in US Pat. No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA, 81: 6851-6855 (1984).
- a chimeric antibody comprises a non-human variable region (eg, a variable region derived from a non-human primate such as mouse, rat, hamster, rabbit, or monkey) and a human constant region.
- a chimeric antibody is a "class-switched" antibody that has an altered class or subclass from that of the parent antibody. Chimeric antibodies also include antigen-binding fragments thereof.
- the chimeric antibody is a humanized antibody.
- non-human antibodies are humanized to reduce immunogenicity in humans while retaining the specificity and affinity of the parent non-human antibody.
- a humanized antibody comprises one or more variable domains in which HVRs (e.g. CDRs (or portions thereof)) are derived from non-human antibodies and FRs (or portions thereof) are derived from human antibody sequences.
- HVRs e.g. CDRs (or portions thereof)
- FRs or portions thereof
- a humanized antibody optionally will also comprise at least a portion of a human constant region.
- a non-human antibody e.g., the antibody from which the HVR residues were derived
- Human framework regions that can be used for humanization were selected using, but not limited to: the "best fit” method (see Sims et al. J. Immunol. 151: 2296 (1993)) Framework regions; framework regions derived from the consensus sequences of human antibodies of a particular subgroup of light or heavy chain variable regions (Carter et al. Proc. Natl. Acad. Sci. USA, 89: 4285 (1992) and Presta et al. J. Immunol., 151: 2623 (1993)); human mature (somatic mutation) framework regions or human germline framework regions (e.g. Almagro and Fransson, Front. Biosci.
- the antibodies provided herein are human antibodies.
- Human antibodies can be produced by various techniques known in the art. Human antibodies are reviewed in van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-374 (2001) and Lonberg, Curr. Opin. Immunol. 20: 450-459 (2008).
- Human antibodies may be prepared by administering an immunogen to transgenic animals that have been engineered to produce fully human antibodies or fully antibodies with human variable regions in response to an antigenic challenge.
- Such animals typically contain all or part of a human immunoglobulin locus, wherein all or part of the human immunoglobulin locus replaces the endogenous immunoglobulin locus, or is extrachromosomally or It exists in a state of being randomly incorporated into the chromosome of the animal.
- the endogenous immunoglobulin loci are usually inactivated.
- Human antibodies can also be made by hybridoma-based methods. Human myeloma and mouse-human heteromyeloma cell lines for the production of human monoclonal antibodies have been previously described (e.g. Kozbor J. Immunol., 133: 3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications, pp.51-63 (Marcel Dekker, Inc., New York, 1987); and Boerner et al., J. Immunol., 147: 86 (1991)). Human antibodies generated via human B-cell hybridoma technology are also described in Li et al., Proc. Natl. Acad. Sci. USA, 103: 3557-3562 (2006).
- Additional methods include, for example, US Pat. No. 7,189,826 (describing production of monoclonal human IgM antibodies from hybridoma cell lines) and Ni, Xiandai Mianyixue, 26(4): 265-268 (2006) (human - describing human hybridomas).
- Human hybridoma technology (trioma technology) is also described in Vollmers and Brandlein, Histology and Histopathology, 20(3): 927-937 (2005) and Vollmers and Brandlein, Methods and Findings in Experimental and Clinical Pharmacology, 27(3): 185-191 (2005) .
- Human antibodies can also be generated by isolating selected Fv clone variable domain sequences from human-derived phage display libraries. Such variable domain sequences can then be combined with the desired human constant domain. Techniques for selecting human antibodies from antibody libraries are described below.
- Antibodies of the invention may be isolated by screening combinatorial libraries for antibodies with the desired activity or activities. For example, various methods are known in the art for generating phage display libraries and screening such libraries for antibodies with desired binding characteristics. Such methods are reviewed in Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, 2001) and see, for example, McCafferty et al., Nature 348: 552-554; Clackson et al., Nature 352: 624-628 (1991); Marks et al., J. Mol. Biol.
- repertoires of VH and VL genes are separately cloned by the polymerase chain reaction (PCR) and randomly recombined into a phage library, which is derived from Winter et al. ., Ann. Rev. Immunol., 12: 433-455 (1994).
- Phage typically display antibody fragments, either as single-chain Fv (scFv) fragments or as Fab fragments.
- Libraries from immunized sources provide high affinity antibodies to the immunogen without the need to construct hybridomas.
- a naive repertoire can be cloned (e.g., from humans) to provide a wide range of non-self and It can also provide a single source of antibodies to self antigens.
- naive library was cloned from stem cells with pre-rearranged V-gene segments and hypervariable CDR3 as described in Hoogenboom and Winter, J. Mol. Biol., 227: 381-388 (1992). It can also be made synthetically by using PCR primers that encode the region and contain randomized sequences to accomplish the rearrangement in vitro.
- Patent literature describing human antibody phage libraries includes, for example: U.S. Patent No. 5,750,373, and U.S. Patent Application Publication Nos. 2005/0079574, 2005/0119455, 2005/0266000, 2007/0117126, 2007 /0160598, 2007/0237764, 2007/0292936, and 2009/0002360.
- Antibodies or antibody fragments isolated from human antibody libraries are considered human antibodies or human antibody fragments herein.
- the antibodies provided herein are multispecific antibodies (eg, bispecific antibodies).
- Multispecific antibodies are monoclonal antibodies that have binding specificities for at least two different sites.
- one of the binding specificities is for CTLA-4 and the other is for any other antigen.
- bispecific antibodies may bind to two different epitopes of CTLA-4.
- Bispecific antibodies may be used to localize cytotoxic agents to cells that express CTLA-4.
- Bispecific antibodies can be prepared as full length antibodies or as antibody fragments.
- Multispecific antibodies include, but are not limited to, recombinant co-expression of two immunoglobulin heavy-light chain pairs with different specificities (Milstein and Cuello, Nature 305: 537 (1983), WO93/08829, and Traunecker et al., EMBO J. 10:3655 (1991)), and knob-in-hole technology (see, e.g., U.S. Patent No. 5,731,168).
- Multispecific antibodies are engineered by electrostatic steering effects to create Fc heterodimeric molecules (WO2009/089004A1); bridging two or more antibodies or fragments (U.S.
- Antibodies or fragments herein also include “dual-acting Fabs” or “DAFs”, which contain one antigen-binding site that binds CTLA-4 and another, different antigen (e.g., US Patent Application Publication No. 2008 /0069820).
- amino acid sequence variants of the antibodies provided herein are also contemplated. For example, it may be desirable to improve the binding affinity and/or other biological properties of an antibody.
- Amino acid sequence variants of the antibody may be prepared by introducing appropriate modifications into the nucleotide sequence encoding the antibody, or by peptide synthesis. Such modifications include, for example, deletions from and/or insertions into and/or substitutions of residues in the antibody amino acid sequence. Any combination of deletion, insertion, and substitution may be made to arrive at the final construct, provided that the final construct possesses the desired characteristics (eg, antigen binding).
- antibody variants with one or more amino acid substitutions are provided.
- Target sites for substitutional mutagenesis include HVR and FR.
- Conservative substitutions are shown in Table 1 under the heading of "preferred substitutions”. More substantial changes are provided in Table 1 under the heading "Exemplary Substitutions" and are detailed below with reference to classes of amino acid side chains.
- Amino acid substitutions may be introduced into the antibody of interest and the product screened for the desired activity, e.g., retained/improved antigen binding, decreased immunogenicity, or improved ADCC or CDC. good.
- Amino acids can be divided into groups according to common side chain properties: (1) Hydrophobic: Norleucine, Methionine (Met), Alanine (Ala), Valine (Val), Leucine (Leu), Isoleucine (Ile); (2) Neutral hydrophilicity: Cysteine (Cys), Serine (Ser), Threonine (Thr), Asparagine (Asn), Glutamine (Gln); (3) acidic: aspartic acid (Asp), glutamic acid (Glu); (4) basic: histidine (His), lysine (Lys), arginine (Arg); (5) residues affecting chain orientation: glycine (Gly), proline (Pro); (6) Aromaticity: Tryptophan (Trp), Tyrosine (Tyr), Phenylalanine (Phe).
- Non-conservative substitutions refer to exchanging a member of one of these classes for another class.
- substitutional variant involves substituting one or more hypervariable region residues of a parent antibody (eg, a humanized or human antibody).
- a parent antibody eg, a humanized or human antibody
- the resulting variants selected for further study exhibit modifications (e.g., improvements) in certain biological properties (e.g., increased affinity, decreased immunogenicity) compared to the parent antibody. ) and/or substantially retain certain biological properties of the parent antibody.
- exemplary substitutional variants are affinity matured antibodies, which may suitably be generated using, for example, phage display-based affinity maturation techniques such as those described herein. Briefly, one or more HVR residues are mutated and the mutated antibodies are displayed on phage and screened for a particular biological activity (eg, binding affinity).
- HVRs Modifications (eg, substitutions) can be made in HVRs, for example, to improve antibody affinity. Such alterations may occur at HVR "hotspots", i.e., residues encoded by codons that are frequently mutated during the somatic maturation process (e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)) and/or at antigen-contacting residues, and the resulting mutated VH or VL can be tested for binding affinity.
- HVR "hotspots” i.e., residues encoded by codons that are frequently mutated during the somatic maturation process (e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)) and/or at antigen-contacting residues, and the resulting mutated VH or VL can be tested for binding affinity.
- Affinity maturation by construction and reselection from secondary libraries has been described, for example, by Hoogenboom
- affinity maturation diversity is introduced into the variable gene selected for maturation by any of a variety of methods (eg, error-prone PCR, chain shuffling or oligonucleotide-directed mutagenesis).
- a secondary library is then created. This library is then screened to identify any antibody variants with the desired affinity.
- Another method of introducing diversity involves an HVR-directed approach that randomizes several HVR residues (eg, 4-6 residues at a time).
- HVR residues involved in antigen binding can be specifically identified using, for example, alanine scanning mutagenesis or modeling. In particular, CDR-H3 and CDR-L3 are often targeted.
- substitutions, insertions, or deletions may be made within one or more HVRs, so long as such modifications do not substantially reduce the ability of the antibody to bind antigen.
- conservative modifications eg, conservative substitutions as provided herein
- modifications can be, for example, outside the antigen-contacting residues of the HVR.
- each HVR is unmodified or contains no more than 1, 2, or 3 amino acid substitutions.
- a useful method for identifying residues or regions of an antibody that can be targeted for mutagenesis is "alanine scanning mutagenesis," as described by Cunningham and Wells (1989) Science, 244: 1081-1085. is called.
- a residue or group of target residues e.g., charged residues such as arginine, aspartate, histidine, lysine, and glutamic acid
- neutral or negatively charged amino acids e.g., alanine or polyalanine
- Further substitutions may be introduced at amino acid positions that have shown functional sensitivity to this initial substitution.
- a crystal structure of the antigen-antibody complex can be analyzed to identify contact points between the antibody and antigen. Such contact residues and neighboring residues may be targeted as replacement candidates or may be excluded from replacement candidates. Mutants can be screened to determine whether they contain the desired property.
- Amino acid sequence insertions range in length from one residue to more than 100 residues at the amino and/or carboxyl terminus of the polypeptide, as well as insertions of single or multiple amino acid residues within the sequence. Including fusion.
- Examples of terminal insertions include antibodies with a methionyl residue at the N-terminus.
- Other insertional variants of antibody molecules include fusions to the N- or C-terminus of the antibody with enzymes (eg, for ADEPT) or polypeptides that increase the plasma half-life of the antibody.
- the antibodies provided herein have been modified to increase or decrease the degree to which the antibody is glycosylated.
- the addition or deletion of glycosylation sites to the antibody can be conveniently accomplished by altering the amino acid sequence to create or remove one or more glycosylation sites.
- the carbohydrate added to it may be modified.
- Native antibodies produced by mammalian cells typically contain branched, biantennary oligosaccharides, which are usually attached by N-linkage to Asn297 of the CH2 domain of the Fc region. See, for example, Wright et al. TIBTECH 15: 26-32 (1997).
- Oligosaccharides include various carbohydrates such as mannose, N-acetylglucosamine (GlcNAc), galactose, and sialic acid, as well as fucose attached to the GlcNAc in the "stem" of the biantennary oligosaccharide structure.
- modifications of oligosaccharides in antibodies of the invention may be made to generate antibody variants with certain improved properties.
- antibody variants are provided that have carbohydrate structures that lack fucose attached (directly or indirectly) to the Fc region.
- the amount of fucose in such antibodies can be 1%-80%, 1%-65%, 5%-65% or 20%-40%.
- the amount of fucose is the sum of all sugar structures (e.g. complex, hybrid and high mannose structures) attached to Asn297 measured by MALDI-TOF mass spectrometry as described in e.g. WO2008/077546. is determined by calculating the average amount of fucose within the glycan at Asn297 relative to Asn297 represents the asparagine residue located around position 297 of the Fc region (EU numbering of Fc region residues).
- Asn297 could be located ⁇ 3 amino acids upstream or downstream of position 297, ie between positions 294-300.
- Such fucosylation variants may have improved ADCC function. See, for example, U.S. Patent Application Publication Nos. 2003/0157108 (Presta, L.); 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd).
- a cell line capable of producing defucosylated antibodies is Lec13 CHO cells, which lack protein fucosylation (Ripka et al. Arch. Biochem. Biophys. 249: 533-545 (1986); US Patent Application Publication No. US2003/0157108 A1, Presta, L; and WO2004/056312 A1, Adams et al., especially Example 11) and knockout cell lines such as alpha-1,6-fucosyltransferase gene FUT8 knockout CHO cells (e.g. Yamane-Ohnuki). 87: 614 (2004); Kanda, Y. et al., Biotechnol. Bioeng., 94(4): 680-688 (2006); and WO2003/085107). .
- Antibody variants with bisected oligosaccharides are further provided, for example, where a biantennary oligosaccharide attached to the Fc region of the antibody is bisected by GlcNAc. Such antibody variants may have reduced fucosylation and/or improved ADCC function. Examples of such antibody variants are described, for example, in WO2003/011878 (Jean-Mairet et al.); US Pat. No. 6,602,684 (Umana et al.); and US2005/0123546 (Umana et al.). there is Antibody variants with at least one galactose residue in the oligosaccharide attached to the Fc region are also provided. Such antibody variants may have improved CDC function. Such antibody variants are described, for example, in WO1997/30087 (Patel et al.); WO1998/58964 (Raju, S.); and WO1999/22764 (Raju, S.).
- Fc Region Variants may be introduced into the Fc region of the antibodies provided herein, thereby generating Fc region variants.
- An Fc region variant may comprise a human Fc region sequence (eg, a human IgG1, IgG2, IgG3, or IgG4 Fc region) containing amino acid modifications (eg, substitutions) at one or more amino acid positions.
- antibody variants that possess some, but not all, effector functions are also within the contemplation of the present invention, which effector functions are associated with the antibody, although its in vivo half-life is important, Certain effector functions (such as complement and ADCC) make them desirable candidates for applications where they are unnecessary or deleterious.
- In vitro and/or in vivo cytotoxicity assays can be performed to confirm reduction/deficiency of CDC and/or ADCC activity.
- Fc receptor (FcR) binding assays can be performed to confirm that the antibody lacks Fc ⁇ R binding (and thus likely lacks ADCC activity) while maintaining FcRn binding capacity.
- NK cells the primary cells that mediate ADCC, express only Fc ⁇ RIII, whereas monocytes express Fc ⁇ RI, Fc ⁇ RII, and Fc ⁇ RIII.
- FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol. 9: 457-492 (1991).
- in vitro assays for assessing ADCC activity of a molecule of interest are described in US Pat. No. 5,500,362 (e.g., Hellstrom, I. et al. Proc. 83: 7059-7063 (1986)) and Hellstrom, I et al., Proc. Nat'l Acad. Sci.
- non-radioactive assays may be used (e.g., ACT1TM non-radioactive cytotoxicity assay for flow cytometry (CellTechnology, Inc. Mountain View, Calif.); and CytoTox 96TM non-radioactive cytotoxicity assays (Promega, Madison, WI)).
- Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and natural killer (NK) cells.
- PBMC peripheral blood mononuclear cells
- NK natural killer cells.
- the ADCC activity of the molecule of interest is assessed in vivo, e.g., in an animal model as described in Clynes et al. Proc.
- Nat'l Acad. Sci. may be A C1q binding assay may also be performed to confirm that the antibody is incapable of binding C1q and thus lacks CDC activity. See, for example, C1q and C3c binding ELISAs in WO2006/029879 and WO2005/100402. CDC measurements may also be performed to assess complement activation (e.g., Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996); Cragg, M.S. et al., Blood 101 : 1045-1052 (2003); and Cragg, M.S. and M.J. Glennie, Blood 103: 2738-2743 (2004)).
- complement activation e.g., Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996); Cragg, M.S. et al., Blood 101 : 1045-1052 (2003); and Cragg, M.S. and M
- FcRn binding and in vivo clearance/half-life can also be performed using methods known in the art (e.g. Petkova, S.B. et al., Int'l. Immunol. 18(12): 1759-1769 (2006)).
- Antibodies with reduced effector function include those with one or more substitutions of Fc region residues 238, 265, 269, 270, 297, 327, and 329 (US Pat. No. 6,737,056).
- Fc variants include amino acid positions 265, 269, 270, 297, and 327, including the so-called "DANA" Fc variant with substitutions of residues 265 and 297 to alanine (U.S. Pat. No. 7,332,581).
- the antibody variant has one or more amino acid substitutions that improve ADCC (e.g., substitutions at positions 298, 333, and/or 334 (residues in EU numbering) of the Fc region). Including the associated Fc region.
- modified i.e., increased or Modifications are made in the Fc region that result in C1q binding (either decreased) and/or complement dependent cytotoxicity (CDC).
- FcRn Increased half-life and neonatal Fc receptors (FcRn: responsible for the transfer of maternal IgGs to the fetus (Guyer et al., J. Immunol. 117: 587 (1976); Kim et al., J. Immunol. 24: 249 (1994)))
- FcRn responsible for the transfer of maternal IgGs to the fetus
- These antibodies comprise an Fc region with one or more substitutions therein that increase the binding of the Fc region to FcRn.
- Such Fc variants have the following Fc region residues: , 413, 424, or 434 (eg, substitution of Fc region residue 434 (US Pat. No. 7,371,826)).
- cysteine engineered antibodies eg, "thioMAbs”
- residues undergoing substitution occur at accessible sites of the antibody.
- reactive thiol groups are placed at accessible sites of the antibody, which reactive thiol groups connect the antibody to other moieties (drug moieties or linker-drug moieties). etc.) to create immunoconjugates as further detailed herein.
- any one or more of the following residues may be replaced with cysteine: V205 of the light chain (Kabat numbering); A118 of the heavy chain (EU numbering); and S400 of the heavy chain Fc region. (EU numbering).
- Cysteine engineered antibodies may be generated, for example, as described in US Pat. No. 7,521,541.
- the antibodies provided herein may be further modified to contain additional non-protein moieties that are known in the art and readily available. Suitable moieties for antibody derivatization include, but are not limited to, water-soluble polymers.
- Non-limiting examples of water-soluble polymers include, but are not limited to, polyethylene glycol (PEG), copolymers of ethylene glycol/propylene glycol, carboxymethylcellulose, dextran, polyvinyl alcohol, polyvinylpyrrolidone, poly 1,3 Dioxolane, poly 1,3,6 trioxane, ethylene/maleic anhydride copolymer, polyamino acid (either homopolymer or random copolymer), and dextran or poly(n-vinylpyrrolidone) polyethylene glycol, polypropylene glycol homopolymer, polypropylene Including oxide/ethylene oxide copolymers, polyoxyethylated polyols (eg, glycerol), polyvinyl alcohol, and mixtures thereof.
- PEG polyethylene glycol
- copolymers of ethylene glycol/propylene glycol carboxymethylcellulose
- dextran polyvinyl alcohol
- polyvinylpyrrolidone poly 1,3 Diox
- Polyethylene glycol propionaldehyde may have manufacturing advantages due to its stability in water.
- the polymer can be of any molecular weight and can be branched or unbranched.
- the number of polymers attached to the antibody can vary, and if more than one polymer is attached they can be the same or different molecules. Generally, the number and/or types of polymers used for derivatization include, but are not limited to, the specific property or function of the antibody to be improved, the The decision can be made based on considerations such as whether to be used for therapy.
- conjugates of antibodies and non-protein moieties that can be selectively heated by exposure to radiation are provided.
- the non-protein portion is a carbon nanotube (Kam et al., Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)).
- the radiation can be of any wavelength, including but not limited to, such that it heats the non-protein moiety to a temperature that kills cells in close proximity to the antibody-non-protein moiety while not harming normal cells. Including wavelength.
- the anti-CTLA-4 antibody of this specification can be combined with various existing technologies.
- One non-limiting embodiment of such a combination of techniques is the generation of chimeric antigen receptor (CAR)-expressing cells using an anti-CTLA-4 antibody.
- CAR chimeric antigen receptor
- Examples of cells here include T cells, ⁇ T cells, NK cells, NKT cells, cytokine-induced killer (CIK) cells, and macrophages (Int J Mol Sci. (2019) 20(11), 2839, Nat Rev Drug Discov. (2020) 19(5), 308).
- CAR-T One of the non-limiting methods for preparing T cells expressing the CAR (CAR-T) is the antigen-binding domain of an anti-CTLA-4 antibody (e.g., scFv), the transmembrane domain of TCR, and T cells.
- an anti-CTLA-4 antibody e.g., scFv
- TCR TCR
- a method of introducing a CAR containing a signal domain of a co-stimulatory molecule such as CD28 for enhancing activation into an effector cell such as a T cell by genetic modification technology can be mentioned.
- Non-limiting examples of techniques that can be combined with the anti-CTLA-4 antibody of the present specification include production of a T cell-redirecting antibody using an anti-CTLA-4 antibody (Nature (1985) 314 (6012) , 628-31, Int J Cancer (1988) 41 (4), 609-15, Proc Natl Acad Sci USA (1986) 83 (5), 1453-7).
- a non-limiting embodiment of the T cell redirecting antibody includes a binding domain for any of the constituent subunits of the T cell receptor (TCR) complex on T cells, particularly a binding domain for the CD3 epsilon chain among CD3, Bispecific antibodies comprising the antigen binding domain of an anti-CTLA-4 antibody.
- Antibodies can be produced using recombinant methods and constructions, for example, as described in US Pat. No. 4,816,567.
- an isolated nucleic acid encoding an anti-CTLA-4 antibody described herein is provided.
- Such nucleic acids may encode VL-comprising amino acid sequences and/or VH-comprising amino acid sequences of an antibody (eg, antibody light and/or heavy chains).
- one or more vectors eg, expression vectors
- host cells containing such nucleic acids are provided.
- the host cell comprises (1) a vector comprising a nucleic acid encoding an amino acid sequence comprising the VL of the antibody and an amino acid sequence comprising the VH of the antibody; or (2) an amino acid sequence comprising the VL of the antibody.
- a first vector containing nucleic acid encoding the sequence and a second vector containing nucleic acid encoding the amino acid sequence comprising the VH of the antibody are included (eg, transformed).
- the host cells are eukaryotic (eg, Chinese Hamster Ovary (CHO) cells) or lymphoid cells (eg, Y0, NS0, Sp2/0 cells).
- Methods of making anti-CTLA-4 antibodies comprising recovering from culture medium).
- the antibody-encoding nucleic acid (e.g., as described above) is isolated and subjected to one or more methods for further cloning and/or expression in a host cell. Insert into a vector.
- nucleic acids may be readily isolated and sequenced using conventional procedures (e.g., oligonucleotide probes capable of specifically binding to the genes encoding the heavy and light chains of the antibody). by using).
- Suitable host cells for cloning or expression of antibody-encoding vectors include prokaryotic or eukaryotic cells described herein.
- antibodies may be produced in bacteria, particularly if glycosylation and Fc effector functions are not required.
- For expression of antibody fragments and polypeptides in bacteria see, e.g., U.S. Pat. Biology, Vol. 248 (see also B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp.245-254). After expression, the antibody may be isolated from the bacterial cell paste in the soluble fraction and may be further purified.
- filamentous fungi or yeast including strains of fungi and yeast in which the glycosylation pathway has been "humanized", resulting in the production of antibodies with a partial or complete human glycosylation pattern.
- Nuclear microbes are preferred cloning or expression hosts for antibody-encoding vectors. See Gerngross, Nat. Biotech. 22: 1409-1414 (2004) and Li et al., Nat. Biotech. 24: 210-215 (2006).
- invertebrates and vertebrates are also suitable host cells for the expression of glycosylated antibodies.
- invertebrate cells include plant and insect cells.
- a number of baculovirus strains have been identified for use in conjugation with insect cells, particularly for transformation of Spodoptera frugiperda cells.
- Plant cell cultures can also be used as hosts. See, for example, U.S. Patent Nos. 5,959,177, 6,040,498, 6,420,548, 7,125,978, and 6,417,429 (describing PLANTIBODIESTM technology for producing antibodies in transgenic plants).
- Vertebrate cells can also be used as hosts.
- mammalian cell lines adapted to grow in suspension would be useful.
- Other examples of useful mammalian host cell lines are the SV40-transformed monkey kidney CV1 strain (COS-7); the human embryonic kidney strain (Graham et al., J. Gen Virol. 36: 59 (1977 ) 293 or 293 cells described in Etc.); baby hamster kidney cells (BHK); mouse Sertoli cells (TM4 cells described in Mather, Biol. Reprod.
- monkey kidney cells (CV1 ); African green monkey kidney cells (VERO-76); human cervical carcinoma cells (HELA); canine kidney cells (MDCK); Buffalo rat hepatocytes (BRL 3A); Hep G2); mouse mammary carcinoma (MMT 060562); TRI cells (described, for example, in Mather et al., Annals N.Y. Acad. Sci. 383: 44-68 (1982)); MRC5 cells;
- Other useful mammalian host cell lines are Chinese Hamster Ovary (CHO) cells, including DHFR-CHO cells (Urlaub et al., Proc. Natl. Acad. Sci.
- Polyclonal antibodies are preferably raised in animals by multiple subcutaneous (sc) or intraperitoneal (ip) injections of the relevant antigen and an adjuvant.
- the animal (usually a non-human mammal) is injected intradermally at multiple sites with, for example, 100 ⁇ g or 5 ⁇ g of protein or conjugate (for rabbits or mice, respectively) in combination with 3 volumes of Freund's complete adjuvant. to immunize against the antigen, immunogenic conjugate, or derivative.
- the animals are boosted with 1/5 to 1/10 the original amount of peptide or conjugate in Freund's complete adjuvant by subcutaneous injection at multiple sites.
- the animals are bled and the serum is assayed for antibody titer. Animals are boosted until titers plateau.
- the animal is boosted with a conjugate of the same antigen but conjugated to another protein and/or via another cross-linking reagent.
- Conjugates also can be made in recombinant cell culture as protein fusions. Aggregating agents such as alum are also preferably used to enhance the immune response.
- Monoclonal antibodies are obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies that make up the population may be present in small amounts with potential naturally occurring mutations and/or post-translational modifications (e.g., isomerization, amidation).
- the modifier "monoclonal” indicates the character of the antibody as not being a mixture of separate antibodies.
- monoclonal antibodies can be made using the hybridoma method first described in Kohler et al., Nature 256(5517): 495-497 (1975).
- a mouse or other suitable host animal such as a hamster, is immunized as described herein above to produce or have the ability to produce antibodies that specifically bind to the protein used for immunization.
- induce certain lymphocytes induce certain lymphocytes.
- lymphocytes can be immunized in vitro.
- Immunizing agents typically include antigen proteins or fusion variants thereof.
- PBL peripheral blood lymphocytes
- spleen cells or lymph node cells are used when non-human mammalian sources are desired. Lymphocytes are then fused with an immortalized cell line using a suitable fusing agent such as polyethylene glycol to form hybridoma cells (Goding, Monoclonal Antibodies: Principles and Practice, Academic Press (1986), pp. 59- 103).
- Immortalized cell lines are usually transformed mammalian cells, especially myeloma cells of rodent, bovine and human origin. Usually, rat or mouse myeloma cell lines are utilized.
- the hybridoma cells thus produced are seeded and grown in a suitable culture medium that preferably contains one or more substances that inhibit the growth or survival of the unfused, parental myeloma cells.
- the culture medium for the hybridomas typically contains hypoxanthine, aminopterin, hypoxanthine, aminopterin, a substance that prevents the growth of HGPRT-deficient cells. and thymidine (HAT medium).
- hypoxanthine guanine phosphoribosyltransferase HGPRT or HPRT
- the culture medium for the hybridomas typically contains hypoxanthine, aminopterin, hypoxanthine, aminopterin, a substance that prevents the growth of HGPRT-deficient cells. and thymidine (HAT medium).
- Preferred immortalized myeloma cells are cells that fuse efficiently, support stable high-level production of antibody by selected antibody-producing cells, and are sensitive to a medium such as HAT medium.
- mouse myeloma strains such as those derived from MOPC-21 and MPC-11 mouse tumors available from the Salk Institute Cell Distribution Center, San Diego, CA, USA, and from the American Type Culture Collection, Manassas, VA, USA. SP-2 cells (and derivatives thereof such as X63-Ag8-653) are preferred.
- Human myeloma cell lines and mouse-human heteromyeloma cell lines have also been described for the production of human monoclonal antibodies (Kozbor et al., J Immunol. 133(6): 3001-3005 (1984); Brodeur et al. ., Monoclonal Antibody Production Techniques and Applications, Marcel Dekker, Inc., New York, pp. 51-63 (1987)).
- the culture medium in which the hybridoma cells are growing is assayed for the production of monoclonal antibodies against the antigen.
- the binding specificity of monoclonal antibodies produced by hybridoma cells is determined by immunoprecipitation or by an in vitro binding assay, such as a radioimmunoassay (RIA) or enzyme-linked immunosorbent assay (ELISA).
- RIA radioimmunoassay
- ELISA enzyme-linked immunosorbent assay
- the clones can be subcloned by limiting dilution and grown by standard methods (Goding, supra). .
- Suitable culture media for this purpose include, for example, D-MEM or RPMI-1640 medium.
- Hybridoma cells can also be grown in vivo as tumors in mammals.
- Monoclonal antibodies secreted by the subclones are purified from culture medium, ascites fluid, or serum by conventional immunoglobulin purification methods such as protein A-sepharose, hydroxyapatite chromatography, gel electrophoresis, dialysis, or affinity chromatography. , are properly separated.
- Antibodies may be produced by immunizing a suitable host animal against the antigen.
- the antigen is a polypeptide comprising full-length CTLA-4.
- the antigen is a polypeptide comprising soluble CTLA-4.
- the antigen is a polypeptide comprising a region corresponding to amino acids 97 (Glu) to 106 (Leu) of human CTLA-4 (extracellular domain, SEQ ID NO:28).
- the antigen is a polypeptide comprising a region corresponding to amino acids 99 (Met) to 106 (Leu) of human CTLA-4 (extracellular domain, SEQ ID NO:28).
- the invention also includes antibodies produced by immunizing an animal against an antigen. Antibodies may incorporate any of the features described above for exemplary anti-CTLA-4 antibodies, alone or in combination.
- Anti-CTLA-4 antibodies provided herein can be identified, screened, or demonstrated for physical/chemical properties and/or biological activity by various assays known in the art. may be made
- the antibodies of the invention are tested for their antigen-binding activity by known methods such as, for example, ELISA, Western blot, surface plasmon resonance assays, and the like.
- the antibody competes for binding to CTLA-4 with an anti-CTLA-4 antibody described herein (e.g., an anti-CTLA-4 antibody described in Tables 7, 12, 17, and 22) Competition assays can be used to identify antibodies that do.
- competition assays can be used to identify antibodies that do.
- when such competing antibodies are present in excess at least 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60% , 65%, 70%, 75% or more block (eg, reduce) binding of the reference antibody to CTLA-4.
- binding is inhibited by at least 80%, 85%, 90%, 95%, or more.
- such competing antibodies are bound by an anti-CTLA-4 antibody described herein (e.g., an anti-CTLA-4 antibody described in Tables 7, 12, 17, and 22). binds to the same epitope (eg, linear or conformational epitope).
- an anti-CTLA-4 antibody described herein e.g., an anti-CTLA-4 antibody described in Tables 7, 12, 17, and 22.
- binds to the same epitope eg, linear or conformational epitope.
- mapping epitopes bound by antibodies are provided in Morris (1996) "Epitope Mapping Protocols," in Methods in Molecular Biology vol. 66 (Humana Press, Totowa, NJ).
- immobilized CTLA-4 is tested for its ability to compete with a first labeled antibody for binding to CTLA-4 and the first antibody for binding to CTLA-4. Incubate in a solution containing a second unlabeled antibody. A second antibody may be present in the hybridoma supernatant. As a control, immobilized CTLA-4 is incubated in a solution containing the first labeled antibody but no second unlabeled antibody. After incubation under conditions permissive for binding of the first antibody to CTLA-4, excess unbound antibody is removed and the amount of label bound to immobilized CTLA-4 is measured.
- assays are provided for identifying those anti-CTLA-4 antibodies that have biological activity.
- Biological activity may include, for example, cell growth inhibitory activity, cytotoxic activity (eg, ADCC/CDC activity, ADCP activity), immunostimulatory activity, CTLA-4 inhibitory activity.
- cytotoxic activity eg, ADCC/CDC activity, ADCP activity
- CTLA-4 inhibitory activity e.g., ADCC/CDC activity, ADCP activity
- antibodies that possess such biological activity in vivo and/or in vitro are also provided.
- antibodies of the invention are tested for such biological activity.
- the antibodies of the invention are tested for their ability to inhibit cell growth or proliferation in vitro.
- Methods for measuring inhibition of cell growth or proliferation are well known in the art.
- Certain cell proliferation assays exemplified by the "cell killing” assays described herein, measure cell viability.
- One such assay is the CellTiter-GloTM Luminescent Cell Viability Assay, commercially available from Promega (Madison, Wis.). This assay determines the number of viable cells in culture based on the abundance of ATP, an indicator of metabolically active cells. See Crouch et al (1993) J. Immunol. Meth. 160: 81-88, U.S. Patent No. 6,602,677.
- Assays may be performed in 96- or 384-well format making them amenable to automated high-throughput screening (HTS). See Cree et al (1995) AntiCancer Drugs 6: 398-404.
- the assay procedure involves adding a single reagent (CellTiter-Glo® Reagent) directly to cultured cells. This lyses the cells and the luciferase reaction produces a luminescent signal.
- the luminescence signal is proportional to the abundance of ATP which is directly proportional to the number of viable cells present in the culture. Data can be recorded by luminometer or CCD camera imaging device. Luminescence values are represented by relative light units (RLU).
- Another cell proliferation assay is a colorimetric assay that measures the oxidation of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to formazan by mitochondrial reductase, "MTT ” assay. Similar to the CellTiter-GloTM Assay, this assay indicates the number of metabolically active cells present in the cell culture. See, for example, Mosmann (1983) J. Immunol. Meth. 65: 55-63 and Zhang et al. (2005) Cancer Res. 65: 3877-3882.
- Cells used in any of the above in vitro assays include cells or cell lines that naturally express CTLA-4 or that have been engineered to express CTLA-4. Such cells also include cell lines that express CTLA-4 and cell lines that do not normally express CTLA-4 but have been transfected with a nucleic acid encoding CTLA-4.
- anti-CTLA-4 antibodies are tested for their ability to inhibit cell growth or proliferation in vivo.
- anti-CTLA-4 antibodies are tested for their ability to inhibit tumor growth in vivo.
- In vivo model systems such as xenograft models, can be used for such testing.
- human tumor cells are introduced into appropriately immunocompromised non-human animals, such as athymic "nude" mice.
- An antibody of the invention is administered to this animal.
- the ability of antibodies to inhibit or reduce tumor growth is measured.
- the human tumor cells are tumor cells derived from a human patient.
- human tumor cells are introduced into a suitably immunocompromised non-human animal by subcutaneous injection or by implantation into a suitable site, such as the mammary fat pad.
- a representative assay for measuring ADCC activity of therapeutic antibodies is based on the 51 Cr release assay and includes the steps of: labeling target cells with [ 51 Cr]Na 2 CrO 4 ; Opsonizing target cells that express antigen thereon with an antibody; combining opsonized radiolabeled target cells and effector cells in a suitable ratio in a microtiter plate in the presence or absence of a test antibody. incubating the mixture of cells, preferably for 16-18 hours, preferably at 37° C.; collecting the supernatant; and analyzing the radioactivity in the supernatant sample.
- Graphs can be generated by varying the target cell: effector cell ratio or antibody concentration.
- a complement dependent cytotoxicity (CDC) assay is performed, eg, as described in Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996). can do. Briefly, various concentrations of the polypeptide variant and human complement are diluted in buffer. Cells expressing the antigen to which the polypeptide variant binds are diluted to a density of ⁇ 1 ⁇ 10 6 cells/ml. A mixture of polypeptide variants, diluted human complement and antigen-expressing cells is added to flat-bottom tissue culture 96-well plates and allowed to incubate at 37° C. and 5% CO 2 for 2 hours to promote complement-mediated cell lysis.
- CDC complement dependent cytotoxicity
- Exemplary assays for ADCP activity can include: coating target bioparticles, such as E. coli-labeled FITC (Molecular Probes) or Staphylococcus aureus-FITC, with a test antibody; the opsonized particles described above to THP-1 effector cells (monocytic cell line, available from ATCC) at 1:1, 10:1, 30:1, 60:1, 75:1, or 100:1 to allow Fc ⁇ R-mediated phagocytosis to occur; preferably incubating the cells with E. coli-FITC/antibody at 37° C.
- target bioparticles such as E. coli-labeled FITC (Molecular Probes) or Staphylococcus aureus-FITC
- THP-1 effector cells monocytic cell line, available from ATCC
- the ADCP assay comprises E. coli-FITC (control), E. coli-FITC and THP-1 cells (used as Fc ⁇ R-independent ADCP activity), E. coli-FITC, THP-1 cells and test It is performed using an antibody (used as Fc ⁇ R-dependent ADCP activity).
- the cytotoxic activity of antibodies is usually accompanied by the binding of antibodies to the cell surface. Whether or not the antigen is expressed on the surface of the target cell can be appropriately confirmed by a technique known to those skilled in the art such as FACS.
- Activation of immunity can be detected using cell-mediated immune reaction or humoral immune reaction as an index.
- cytokines eg, IL-6, G-CSF, IL-12, TNF ⁇ and IFN ⁇
- immune cells eg, B cells, T cells, NK cells
- macrophages e.g., monocytes, etc.
- enhancement of activation state e.g, enhancement of function, enhancement of cytotoxic activity, and the like.
- T cell activation can be detected by measuring upregulation of activation markers such as CD25, CD69 and ICOS.
- activation markers such as CD25, CD69 and ICOS.
- patients treated with the anti-CTLA-4 antibody ipilimumab are known to have increased ICOS + CD4 + T cells in the peripheral blood after administration. It is thought to be an effect of activation of immune status (Cancer Immunol. Res. (2013) 1(4): 229-234).
- T cells Activation of T cells requires not only antigen receptor (TCR)-mediated stimulation but also CD28-mediated co-stimulation.
- TCR antigen receptor
- CD28 on the surface of T cells binds to B7-1 (CD80) or B7-2 (CD86) present on the surface of antigen-presenting cells, an accessory signal is transmitted within the T cell and the T cell is activated.
- CTLA-4 is expressed on the surface of activated T cells. Since CTLA-4 binds to CD80 and CD86 with stronger affinity than CD28, it preferentially interacts with CD80 and CD86 over CD28, resulting in suppression of T cell activation.
- an assay for measuring inhibitory activity against CTLA-4 comprises the following steps: binding purified CTLA-4 protein to a support such as a microtiter plate or magnetic beads; adding CD80 or CD86, washing away unbound components, and quantifying the bound labeled CD80 or CD86. Whether a subject antibody cross-reacts with CD28 can be determined by performing a similar assay, substituting CD28 for CTLA-4.
- inhibitory activity against CTLA-4 can also be measured using functional assays that detect T cell activation as described above. For example, in a system in which a T cell population is stimulated with cells expressing CD80 or CD86 and T cell activation is measured, when a test antibody having CTLA-4 inhibitory activity is added thereto, T cell A further enhancement of activation is provided.
- the present invention also includes one or more cytotoxic agents (e.g. chemotherapeutic agents or chemotherapeutic agents, growth inhibitory agents, toxins (e.g. protein toxins of bacterial, fungal, plant or animal origin, enzymatically active Immunoconjugates are provided comprising an anti-CTLA-4 antibody herein conjugated to a toxin (or fragment thereof) or radioisotope).
- cytotoxic agents e.g. chemotherapeutic agents or chemotherapeutic agents, growth inhibitory agents, toxins (e.g. protein toxins of bacterial, fungal, plant or animal origin, enzymatically active Immunoconjugates are provided comprising an anti-CTLA-4 antibody herein conjugated to a toxin (or fragment thereof) or radioisotope).
- the immunoconjugate is an antibody-drug conjugate (ADC), wherein the antibody is conjugated to one or more drugs, including but not limited to There are: maytansinoids (see U.S. Pat. Nos. 5,208,020, 5,416,064, and EP 0,425,235 B1); e.g. monomethylauristatin drug moieties DE and DF (MMAE and MMAF) (U.S. Pat. Nos. 5,635,483 and 5,780,588) dolastatin; calicheamicin or derivatives thereof (U.S. Pat. Nos.
- ADC antibody-drug conjugate
- the immunoconjugate comprises an antibody described herein conjugated to an enzymatically active toxin or fragment thereof, including but not limited to: Diphtheria A chain , non-binding active fragment of diphtheria toxin, exotoxin A chain (from Pseudomonas aeruginosa), ricin A chain, abrin A chain, modeccin A chain, alpha-sarcin, Aleurites fordii protein, diantin Proteins, Phytolacca americana protein (PAPI, PAPII and PAP-S), momordica charantia inhibitor, curcin, crotin, saponaria officinalis inhibitor, gelonin, mitogellin, restrictocins, phenomycins, enomycins, and trichothecenes.
- Diphtheria A chain non-binding active fragment of diphtheria toxin
- exotoxin A chain from Pseudomonas aeruginosa
- an immunoconjugate comprises an antibody described herein conjugated to a radioactive atom to form a radioconjugate.
- radioisotopes are available for the production of radioconjugates. Examples include radioactive isotopes of 211 At, 131 I, 125 I, 90 Y, 186 Re, 188 Re, 153 Sm, 212 Bi, 32 P, 212 Pb and Lu.
- the radioactive conjugate can be used for scintigraphic examination (e.g. Tc-99m or 123 I) or for nuclear magnetic resonance (NMR) imaging (magnetic resonance imaging, as MRI). (also known as .
- Conjugates of antibodies and cytotoxic agents can be made using a variety of bifunctional protein linking agents. e.g. N-succinimidyl-3-(2-pyridyldithio)propionate (SPDP), succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC), iminothiolane (IT), bifunctional derivatives of imidoesters (e.g. dimethyl adipimidate HCl), active esters (e.g. disuccinimidyl suberate), aldehydes (e.g. glutaraldehyde), bis-azido compounds (e.g.
- SPDP N-succinimidyl-3-(2-pyridyldithio)propionate
- SMCC succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate
- IT iminothiolane
- bis(p-azidobenzoyl)hexanediamine), bis- diazonium derivatives (e.g. bis-(p-diazoniumbenzoyl)-ethylenediamine), diisocyanates (e.g. toluene 2,6-diisocyanate), and bis-active fluorine compounds (e.g. 1,5-difluoro-2,4-dinitrobenzene) is.
- a ricin immunotoxin can be prepared as described in Vitetta et al., Science 238: 1098 (1987).
- Carbon-14 labeled 1-isothiocyanatobenzyl-3-methyldiethylenetriaminepentaacetic acid is an exemplary chelating agent for conjugation of radionuclides to antibodies. See WO94/11026.
- the linker can be a "cleavable linker" that facilitates intracellular release of the cytotoxic agent.
- acid-labile linkers, peptidase-sensitive linkers, photolabile linkers, dimethyl linkers, or disulfide-containing linkers (Chari et al., Cancer Res. 52: 127-131 (1992); U.S. Patent No. 5,208,020) have been used. obtain.
- Immunoconjugates or ADCs herein are commercially available (e.g. from Pierce Biotechnology, Inc., Rockford, IL., U.S.A.) BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, MPBH, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, sulfo-EMCS, sulfo-GMBS, sulfo-KMUS, sulfo-MBS, sulfo-SIAB, sulfo-SMCC, and sulfo-SMPB, and SVSB (succinimidyl-(4-vinylsulfone) Conjugates prepared using cross-linking reagents including, but not limited to, benzoate) are expressly contemplated.
- cross-linking reagents including, but not limited to, benzoate
- any of the anti-CTLA-4 antibodies provided herein are useful for detecting the presence of CTLA-4 in a biological sample.
- the term "detection" as used herein includes quantitative or qualitative detection.
- the biological sample is a cell or tissue, e.g., serum, whole blood, plasma, biopsy sample, tissue sample, cell suspension, saliva, sputum, oral fluid, cerebrospinal fluid, amniotic fluid, Contains ascites, milk, colostrum, mammary gland secretions, lymph, urine, sweat, tears, gastric fluid, synovial fluid, ascites, eye fluid, or mucus.
- anti-CTLA-4 antibodies are provided for use in diagnostic or detection methods.
- methods are provided for detecting the presence of CTLA-4 in a biological sample.
- the method comprises contacting the biological sample with an anti-CTLA-4 antibody described herein under conditions permissive for binding of the anti-CTLA-4 antibody to CTLA-4; and Including detecting whether a complex is formed between an anti-CTLA-4 antibody and CTLA-4.
- anti-CTLA-4 antibodies are used to select subjects amenable to treatment with anti-CTLA-4 antibodies, eg, where CTLA-4 is a biomarker for selecting patients.
- the antibody of the present invention can be used, for example, to check the state of immune response and diagnose dysfunction of the immune system.
- Labels include labels or moieties that are detected directly (e.g., fluorescent labels, chromogenic labels, electron-dense labels, chemiluminescent labels, and radioactive labels) and indirectly detected through, e.g., enzymatic reactions or intermolecular interactions. including, but not limited to, moieties such as enzymes or ligands.
- Exemplary labels include, but are not limited to: the radioisotopes 32 P, 14 C, 125 I, 3 H and 131 I, fluorophores such as rare earth chelates or fluorescein and its derivatives.
- luciferases such as dansyl, umbelliferone, firefly luciferase and bacterial luciferase (U.S. Pat. No. 4,737,456), luciferin, 2,3-dihydrophthalazinedione, horseradish peroxidase (HRP), alkaline Phosphatases, ⁇ -galactosidase, glucoamylase, lysozyme, monosaccharide oxidases (e.g.
- glucose oxidase galactose oxidase and glucose-6-phosphate dehydrogenase
- heterocyclic oxidases such as uricase and xanthine oxidase
- dye precursors using hydrogen peroxide biotin/avidin spin labels, bacteriophage labels, stable free radicals, and the like.
- compositions of the anti-CTLA-4 antibodies described herein comprise antibodies of desired purity in one or more of any pharmaceutically acceptable carriers (Remington's Pharmaceutical Sciences 16th edition, Osol , A. Ed. (1980)) in the form of lyophilized formulations or aqueous solutions.
- Pharmaceutically acceptable carriers are generally nontoxic to recipients at the dosages and concentrations employed and include, but are not limited to: phosphates, citrates, buffers such as acid salts and other organic acids; antioxidants, including ascorbic acid and methionine; preservatives (octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride; benzethonium chloride; phenol, butyl, or benzyl alcohol; alkylparabens such as methyl or propylparaben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); proteins such as immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates,
- Exemplary pharmaceutically acceptable carriers herein further include interstitial drug dispersing agents such as soluble neutrally active hyaluronidase glycoprotein (sHASEGP).
- interstitial drug dispersing agents such as soluble neutrally active hyaluronidase glycoprotein (sHASEGP).
- sHASEGP soluble neutrally active hyaluronidase glycoprotein
- Certain exemplary sHASEGPs and methods of their use are described in US Patent Application Publication Nos. 2005/0260186 and 2006/0104968.
- sHASEGP is combined with one or more additional glycosaminoglycanase, such as chondroitinase.
- Aqueous antibody formulations include those described in US Pat. No. 6,171,586 and WO2006/044908, the latter formulation containing a histidine-acetate buffer.
- compositions herein may contain more than one active ingredient if required for the particular indication being treated. Those with complementary activities that do not adversely affect each other are preferred. Such active ingredients are present in any suitable combination in amounts that are effective for the purpose intended.
- the active ingredient is incorporated into microcapsules prepared, for example, by droplet formation (coacervation) techniques or by interfacial polymerization (for example, hydroxymethylcellulose or gelatin microcapsules, and poly(methyl methacrylate) microcapsules, respectively).
- microcapsules prepared, for example, by droplet formation (coacervation) techniques or by interfacial polymerization (for example, hydroxymethylcellulose or gelatin microcapsules, and poly(methyl methacrylate) microcapsules, respectively).
- colloidal drug delivery systems eg, liposomes, albumin microspheres, microemulsions, nanoparticles, and nanocapsules
- macroemulsions eg, incorporated into macroemulsions.
- sustained release formulation may be prepared.
- sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing the antibody, which matrices are in the form of shaped articles, eg films or microcapsules.
- the formulations used for in vivo administration are usually sterile. Sterility is readily accomplished, for example, by filtration through sterile filtration membranes.
- anti-CTLA-4 antibodies may be used in therapeutic methods.
- an anti-CTLA-4 antibody is provided for use as a pharmaceutical.
- anti-CTLA-4 antibodies are provided for use in treating tumors.
- anti-CTLA-4 antibodies are provided for use in therapeutic methods.
- the invention provides an anti-CTLA-4 antibody for use in a method of treating an individual with a tumor comprising administering to the individual an effective amount of an anti-CTLA-4 antibody. I will provide a.
- the invention provides anti-CTLA-4 antibodies for use in cell killing.
- the present invention provides for use in a method of injuring cells in an individual comprising administering to said individual an effective amount of an anti-CTLA-4 antibody to injure the cells.
- Anti-CTLA-4 antibodies are provided.
- An "individual” according to any of the above aspects is preferably a human.
- immune cells such as lymphocytes are infiltrated into tumor tissue in vivo, and they also constitute part of the tumor tissue.
- the tumor tissue is infiltrated with immune cells, particularly regulatory T (Treg) cells.
- damage to cells is caused by ADCC activity, CDC activity, or ADCP activity.
- cells that express CTLA-4 on their cell surface are injured.
- the injured cells are Treg cells.
- Treg cells infiltrating within the tumor tissue are injured.
- the degree of efficacy of the anti-CTLA-4 antibody of the present invention as a drug varies depending on the tissue within the individual. In certain embodiments, the degree varies depending on the concentration of the adenosine-containing compound in the tissue. In further embodiments, the effect is increased in tissues with high concentrations of adenosine-containing compounds relative to tissues with low concentrations of adenosine-containing compounds. Tissues with high concentrations of adenosine-containing compounds include, for example, tumor tissue. Tissues with low concentrations of adenosine-containing compounds include, for example, non-tumor tissues such as normal tissues. In some embodiments, immune activation is stronger in tumor tissue as compared to non-tumor tissue.
- immunity is activated at lower doses in tumor tissue compared to non-tumor tissue.
- therapeutic effects are observed at doses lower than those at which side effects are observed.
- the therapeutic effect is the expression of an anti-tumor effect (e.g., tumor regression, induction of cell death or growth inhibition of tumor cells, etc.), and the side effect is an autoimmune disease (normal tissue induced by excessive immune response). (including injury to the body).
- the tumor is selected from the group consisting of breast cancer and liver cancer.
- the present invention provides use of anti-CTLA-4 antibodies in the manufacture or preparation of medicaments.
- the medicament is for treatment of a tumor.
- the medicament is for use in a method of treating a tumor comprising administering to an individual having a tumor an effective amount of the medicament.
- the medicament is for cell injury.
- the medicament is for use in a method of injuring cells in an individual comprising administering to said individual an effective amount of the medicament to injure the cells.
- An "individual" according to any of the above aspects may be a human.
- the invention provides methods of treating tumors.
- the method comprises administering to an individual with such a tumor an effective amount of an anti-CTLA-4 antibody.
- An "individual” according to any of the above aspects may be a human.
- the invention provides methods for damaging cells in an individual.
- the method comprises administering to the individual an effective amount of an anti-CTLA-4 antibody to kill the cell.
- the "individual" is a human.
- the invention provides pharmaceutical formulations (pharmaceutical compositions) comprising any of the anti-CTLA-4 antibodies provided herein (e.g., in any of the therapeutic methods described above). for use).
- a pharmaceutical formulation comprises any of the anti-CTLA-4 antibodies provided herein and a pharmaceutically acceptable carrier.
- the present invention provides pharmaceutical formulations (pharmaceutical compositions) for use in treating tumors.
- the present invention provides pharmaceutical formulations (pharmaceutical compositions) for use in cell injury.
- the invention provides a method for preparing a medicament or pharmaceutical formulation comprising mixing any of the anti-CTLA-4 antibodies provided herein with a pharmaceutically acceptable carrier. Methods are provided (eg, for use in any of the therapeutic methods described above).
- the antibodies of the present invention may be administered by any suitable means, including parenteral, intrapulmonary, and nasal administration, and intralesional administration if desired for local treatment.
- Parenteral injection includes intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration.
- Dosing may be by any suitable route, eg, by injection, such as intravenous or subcutaneous injection, depending in part on whether the administration is short or long term.
- Various dosing schedules are contemplated herein, including, but not limited to, single doses or repeated doses over various time points, bolus doses, and pulse infusions.
- the antibodies of the invention are formulated, dosed, and administered in a manner consistent with good medical practice.
- Factors to be considered in this regard are the particular disorder being treated, the particular mammal being treated, the clinical presentation of the individual patient, the cause of the disorder, the site of delivery of the agent, the method of administration, the schedule, and other factors known to health care practitioners.
- an appropriate dose of an antibody of the invention will depend on the type of disease being treated, the type of antibody, the severity and course of the disease, whether the antibody is administered for prophylactic or therapeutic purposes. It will depend on the medication history, the patient's clinical history and response to antibodies, and the discretion of the attending physician.
- the antibody is suitably administered to the patient at one time or over a series of treatments. Depending on the type and severity of the disease, for example, about 1 ⁇ g/kg to 15 mg/kg (e.g., 0.1 mg/kg to 10 mg/kg) of antibody may be the first candidate dose for administration to a patient.
- One typical daily dosage might range from about 1 ⁇ g/kg to 100 mg/kg or more, depending on the factors mentioned above.
- One exemplary dose of antibody is within the range of about 0.05 mg/kg to about 10 mg/kg.
- one or more doses (or any combination thereof) of about 0.5 mg/kg, 2.0 mg/kg, 4.0 mg/kg, or 10 mg/kg may be administered to the patient.
- Such doses may be administered intermittently, eg, every week or every three weeks (eg, such that the patient receives from about 2 to about 20, or eg, about 6 doses of the antibody).
- a high loading dose may be followed by one or more lower doses. The progress of this therapy is easily monitored by conventional techniques and measurements.
- the anti-CTLA-4 antibody of the present specification can be administered by administering or incorporating a nucleic acid encoding the anti-CTLA-4 antibody into the body using a vector or the like, and directly expressing the anti-CTLA-4 antibody in the body. , may be administered without a vector.
- vectors include virus vectors, plasmid vectors, and adenovirus vectors.
- a nucleic acid encoding an anti-CTLA-4 antibody may be directly administered to a living body, or cells transfected with a nucleic acid encoding an anti-CTLA-4 antibody may be administered to a living body.
- a method in which mRNA encoding an anti-CTLA-4 antibody is chemically modified to increase the stability of the mRNA in vivo, and the mRNA is directly administered to humans to express an anti-CTLA-4 antibody in vivo see EP2101823B, WO2013/120629.
- B cells into which a nucleic acid encoding an anti-CTLA-4 antibody has been introduced may be administered (Sci Immunol. (2019) 4(35), eaax0644).
- Bacteria into which a nucleic acid encoding an anti-CTLA-4 antibody has been introduced may also be administered (Nature Reviews Cancer (2016) 18, 727-743).
- Non-limiting examples of techniques that can be combined with the anti-CTLA-4 antibody of the present specification include generation of T cells that secrete T cell-redirecting antibodies using anti-CTLA-4 antibodies (Trends Immunol. (2019) 40(3) 243-257).
- One of the non-limiting preparation methods is a binding domain for any of the constituent subunits of the T cell receptor (TCR) complex on T cells, particularly a binding domain for the CD3 epsilon chain among CD3, and anti-CTLA-4.
- TCR T cell receptor
- Examples include a method of introducing a nucleic acid encoding a bispecific antibody containing an antigen-binding domain of an antibody into effector cells such as T cells by gene modification technology.
- an article of manufacture contains a device useful for the treatment, prevention, and/or diagnosis of the disorders described above.
- the product includes the container and any label on or accompanying the container.
- Preferred containers include, for example, bottles, vials, syringes, IV (intravenous) solution bags, and the like.
- Containers may be formed from a variety of materials, such as glass or plastic.
- the container may hold the composition alone or in combination with another composition effective for treating, preventing, and/or diagnosing a condition, and may have a sterile access port.
- the container may be an intravenous solution bag or vial having a stopper pierceable by a hypodermic injection needle).
- At least one active ingredient in the composition is an antibody of the invention.
- the label or package insert indicates that the composition is used for treating the condition of choice.
- Articles of manufacture in this aspect of the invention may further include a package insert indicating that the composition may be used to treat a particular condition.
- the product further comprises a second container comprising a pharmaceutically acceptable buffer, such as Bacterial Bacterial Water for Injection (BWFI), Phosphate Buffered Saline, Ringer's Solution, and Dextrose Solution. may contain. It may further include other commercially desirable or user-desirable equipment such as other buffers, diluents, filters, needles, and syringes.
- BWFI Bacterial Bacterial Water for Injection
- Phosphate Buffered Saline Phosphate Buffered Saline
- Ringer's Solution Ringer's Solution
- Dextrose Solution Dextrose Solution
- any of the products described above may comprise an immunoconjugate of the invention instead of or in addition to an anti-CTLA-4 antibody.
- the invention provides an isolated polypeptide comprising a variant Fc region.
- the polypeptide is an antibody.
- the polypeptide is an Fc fusion protein.
- a variant Fc region has at least one Includes amino acid residue alterations (eg, substitutions).
- the native sequence Fc region is typically organized as a homodimer composed of two identical polypeptide chains. Amino acid alterations in the variant Fc regions of the present invention may be introduced into either one of the two polypeptide chains of the parent Fc region, or into both of the two polypeptide chains.
- the invention provides variant Fc regions with altered function compared to the parental Fc region.
- the variant Fc regions of the invention have enhanced Fc ⁇ receptor binding activity compared to the parental Fc region.
- the variant Fc regions of the present invention have enhanced binding activity to at least one Fc ⁇ receptor selected from the group consisting of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIb, Fc ⁇ RIIIa, as compared to the parental Fc region.
- the variant Fc regions of the invention have enhanced binding activity to Fc ⁇ RIIa.
- the variant Fc regions of the invention have enhanced binding activity to Fc ⁇ RIIIa.
- variant Fc regions of the invention have enhanced binding activity to Fc ⁇ RIIa and Fc ⁇ RIIIa.
- the variant Fc regions of the invention have enhanced ADCC, CDC, or ADCP activity compared to the parental Fc region.
- the variant Fc regions of the invention have at least one position selected from the group consisting of positions 234, 235, 236, 298, 330, 332, and 334, represented by EU numbering. Contains amino acid modifications. Alternatively, amino acid alterations and the like described in International Publications WO2013/002362 and WO2014/104165 can be similarly used in the present invention.
- the binding activity of parental Fc regions and variant Fc regions can be expressed as KD (Dissociation constant) values.
- the ratio of [KD value of parental Fc region for Fc ⁇ RIIa]/[KD value of mutant Fc region for Fc ⁇ RIIa] is, for example, 1.5 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 15 or more, 20 or more, 25 or more, 30 or more, 40 or more, or 50 or more.
- the Fc ⁇ RIIa can be Fc ⁇ RIIa R or Fc ⁇ RIIa H. Alternatively, it may be both.
- the Fc region KD value for Fc ⁇ RIIa may be the Fc region KD value for Fc ⁇ RIIa R or the Fc region KD value for Fc ⁇ RIIa H. Alternatively, it may be the sum or average value of both.
- the value of the ratio [binding activity of parental Fc region to Fc ⁇ RIIIa]/[binding activity of mutant Fc region to Fc ⁇ RIIIa] is, for example, 2 or more, 3 or more, 5 or more, 10 or more, 20 or more, 30 or more , 50 or more, 100 or more, 200 or more, 300 or more, 500 or more, 1 ⁇ 10 3 or more, 2 ⁇ 10 3 or more, 3 ⁇ 10 3 or more, or 5 ⁇ 10 3 or more.
- the Fc ⁇ RIIIa can be Fc ⁇ RIIIa F or Fc ⁇ RIIIa V. Alternatively, it may be both. Therefore, the Fc region KD value for Fc ⁇ RIIIa may be the Fc region KD value for Fc ⁇ RIIIa F or the Fc region KD value for Fc ⁇ RIIIa V. Alternatively, it may be the sum or average value of both.
- the KD value of the mutant Fc region for Fc ⁇ RIIa is, for example, 1.0 ⁇ 10 ⁇ 6 M or less, 5.0 ⁇ 10 ⁇ 7 M or less, 3.0 ⁇ 10 ⁇ 7 M or less, 2.0 ⁇ 10 ⁇ 7 M or less, 1.0 ⁇ 10 -7 M or less, 5.0 ⁇ 10 -8 M or less, 3.0 ⁇ 10 -8 M or less, 2.0 ⁇ 10 -8 M or less, 1.0 ⁇ 10 -8 M or less, 5.0 ⁇ 10 -9 M or less, 3.0 ⁇ 10 - 9 M or less, 2.0 ⁇ 10 ⁇ 9 M or less, or 1.0 ⁇ 10 ⁇ 9 M or less.
- the Fc ⁇ RIIa can be Fc ⁇ RIIa R or Fc ⁇ RIIa H.
- the KD value of the mutant Fc region for Fc ⁇ RIIIa is, for example, 1.0 ⁇ 10 ⁇ 6 M or less, 5.0 ⁇ 10 ⁇ 7 M or less, 3.0 ⁇ 10 ⁇ 7 M or less, 2.0 ⁇ 10 ⁇ 7 M or less, 1.0 ⁇ 10 -7 M or less, 5.0 ⁇ 10 -8 M or less, 3.0 ⁇ 10 -8 M or less, 2.0 ⁇ 10 -8 M or less, 1.0 ⁇ 10 -8 M or less, 5.0 ⁇ 10 -9 M or less, 3.0 ⁇ 10 - 9 M or less, 2.0 ⁇ 10 -9 M, 1.0 ⁇ 10 -9 M or less, 5.0 ⁇ 10 -10 M or less, 3.0 ⁇ 10 -10 M or less, 2.0 ⁇ 10 -10 M or 1.0 ⁇ 10 -10 M or less is.
- the Fc ⁇ RIIIa can be Fc ⁇ RIIIa F or Fc ⁇ RIIIa V. Alternatively, it may be both.
- the binding activity of the parental Fc region and the mutant Fc region may be represented by kd (Dissociation rate constant) values instead of KD values.
- the binding activity of the parental Fc region and the variant Fc region may be expressed as the amount of Fc ⁇ receptor binding of the Fc region per unit amount.
- the amount of binding of the Fc region immobilized on the sensor chip and the amount of binding of the Fc ⁇ receptor further bound thereto are each measured as a response unit (RU).
- the value obtained by dividing the binding amount of the Fc ⁇ receptor by the binding amount of the Fc region can be defined as the binding amount of the Fc region to the Fc ⁇ receptor per unit amount.
- a specific method for measuring and calculating such a binding amount is described in Examples below.
- the ratio [Fc ⁇ RIIa-binding amount of the mutant Fc region]/[Fc ⁇ RIIa-binding amount of the parental Fc region] is, for example, 1.5 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 7 or more, 8 or more, 9 or more, 10 or more, 15 or more, 20 or more, 25 or more, 30 or more, 40 or more, or 50 or more.
- the ratio of [Fc ⁇ RIIIa binding amount of mutant Fc region]/[Fc ⁇ RIIIa binding amount of parental Fc region] is, for example, 2 or more, 3 or more, 5 or more, 10 or more, 20 or more, 30 50 or more, 100 or more, 200 or more, 300 or more, 500 or more, 1 ⁇ 10 3 or more, 2 ⁇ 10 3 or more, 3 ⁇ 10 3 or more, or 5 ⁇ 10 3 or more.
- the KD value, kd value, binding amount value, and the like represented herein are measured or calculated by performing a surface plasmon resonance assay at 25°C or 37°C (e.g., this See Reference Example 8 of the specification).
- the mutant Fc regions of the present invention have improved selectivity between activating and inhibitory Fc ⁇ receptors compared to the parental Fc region. In other words, the mutant Fc regions of the present invention have significantly enhanced activating Fc ⁇ receptor-binding activity over inhibitory Fc ⁇ receptor-binding activity compared to the parent Fc region.
- the activating Fc ⁇ receptor is at least one Fc ⁇ receptor selected from the group consisting of Fc ⁇ RIa, Fc ⁇ RIIa R, Fc ⁇ RIIa H, Fc ⁇ RIIIa F, Fc ⁇ RIIIa V, and the inhibitory Fc ⁇ receptor is Fc ⁇ RIIb. .
- the variant Fc regions of the invention have improved selectivity between Fc ⁇ RIIa and Fc ⁇ RIIb. In some embodiments, the variant Fc regions of the invention have improved selectivity between Fc ⁇ RIIIa and Fc ⁇ RIIb. In further embodiments, the variant Fc regions of the invention have improved selectivity between Fc ⁇ RIIa and Fc ⁇ RIIb and selectivity between Fc ⁇ RIIIa and Fc ⁇ RIIb.
- the variant Fc regions of the invention comprise at least one amino acid modification at at least one position selected from the group consisting of positions 236, 239, 268, 270, and 326 as represented by EU numbering. .
- amino acid alterations described in International Publication WO2013/002362 and WO2014/104165 can also be used in the present invention.
- the binding activity of parental Fc regions and mutant Fc regions can be expressed as KD (Dissociation constant) values. Aspects of binding activity to Fc ⁇ RIIa and Fc ⁇ RIIIa are as described above.
- the value of the ratio [KD value of the parental Fc region for Fc ⁇ RIIb]/[KD value of the mutant Fc region for Fc ⁇ RIIb] is, for example, 10 or less, 5 or less, 3 or less, 2 or less, 1 or less, 0.5 or less. , 0.3 or less, 0.2 or less, or 0.1 or less.
- the binding activity of the parental Fc region and the mutant Fc region may be expressed by kd (Dissociation rate constant) values instead of KD values.
- the binding activity of the parental Fc region and the mutant Fc region may be expressed as the amount of Fc ⁇ receptor binding of the Fc region per unit amount described above.
- the ratio of [Fc ⁇ RIIb binding amount of mutant Fc region]/[Fc ⁇ RIIb binding amount of parental Fc region] is, for example, 10 or less, 5 or less, 3 or less, 2 or less, 1 or less, 0.5 less than or equal to 0.3, less than or equal to 0.2, or less than or equal to 0.1.
- the binding amount of the mutant Fc region to Fc ⁇ RIIb is, for example, 0.5 or less, 0.3 or less, 0.2 or less, 0.1 or less, 0.05 or less, 0.03 or less, 0.02 or less, 0.01 or less, 0.005 or less, 0.003 or less, 0.002 or less, or less than 0.001.
- the improved selectivity between activating Fc ⁇ receptors and inhibitory Fc ⁇ receptors is selective for binding activity to activating Fc ⁇ receptors compared to binding activity to inhibitory Fc ⁇ receptors. In other words, it is an increase in the ratio of binding activity to activating Fc ⁇ receptors to binding activity to inhibitory Fc ⁇ receptors (A/I ratio).
- A/I ratio is an index for exhibiting excellent effector functions, and polypeptides with a high A/I ratio can be evaluated as having excellent effector functions.
- the Fc ⁇ receptor-binding activity of the parental Fc region and the mutant Fc region can be expressed by the KD value, the kd value, or the Fc ⁇ receptor-binding amount of the Fc region per unit amount.
- the A / I ratio can be expressed as follows using the KD value, kd value, or binding amount, respectively: [KD value for inhibitory Fc ⁇ receptor] / [KD value for activating Fc ⁇ receptor] , [kd value for inhibitory Fc ⁇ receptor]/[kd value for activating Fc ⁇ receptor], or [binding amount for activating Fc ⁇ receptor]/[binding amount for inhibitory Fc ⁇ receptor].
- the A/I ratio of the mutated Fc region of the present invention is 1.1-fold or more, 1.2-fold or more, 1.3-fold or more, 1.4-fold or more, 1.5-fold or more, 1.6-fold or more, 1.7-fold compared to the parental Fc region.
- the A/I ratio value of the mutant Fc region of the present invention is 300 or more, 400 or more, 500 or more, 600 or more, 700 or more, 800 or more, 900 or more, 1000 or more, 2000 or more, 3000 or more, 4000 or more, 5000 or more, 6000 or more, 7000 or more, 8000 or more, 9000 or more, 10000 or more, 11000 or more, 12000 or more, 13000 or more, 14000 or more, or 15000 or more.
- the A/I ratio is the ratio of Fc ⁇ RIa-binding activity to Fc ⁇ RIIb-binding activity, the ratio of Fc ⁇ RIIa-binding activity to Fc ⁇ RIIb-binding activity, and the ratio of Fc ⁇ RIIIa-binding activity to Fc ⁇ RIIb-binding activity. or the ratio of [the sum or average value of two or three of Fc ⁇ RIa-binding activity, Fc ⁇ RIIa-binding activity, and Fc ⁇ RIIIa-binding activity] to Fc ⁇ RIIb-binding activity.
- the Fc ⁇ RIIa is Fc ⁇ RIIa R, Fc ⁇ RIIa H, or both, thus the avidity to Fc ⁇ RIIa is the sum or average of avidity to Fc ⁇ RIIa R, avidity to Fc ⁇ RIIa H, or both .
- Fc ⁇ RIIIa is Fc ⁇ RIIIa F, Fc ⁇ RIIIa V, or both, and thus binding activity to Fc ⁇ RIIIa is the sum or average of binding activity to Fc ⁇ RIIIa F, binding activity to Fc ⁇ RIIIa V, or both .
- variant Fc regions of the invention comprise amino acid alterations at the following positions: (i) positions 234, 235, 236, 239, 268, 270 and 298, represented by EU numbering, in the first polypeptide of the parent Fc region, and (ii) positions 270, 298, 326 and 334, represented by EU numbering, in the second polypeptide of the parent Fc region;
- the variant Fc region of the invention further comprises an amino acid modification at position 326, represented by EU numbering, in the first polypeptide of the parent Fc region.
- the variant Fc region of the invention further comprises an amino acid modification at position 236, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the invention further comprises an amino acid alteration at position 332, represented by EU numbering, in the first polypeptide of the parent Fc region. In a particular embodiment, the variant Fc region of the invention further comprises an amino acid alteration at position 330, represented by EU numbering, in the first polypeptide of the parent Fc region. In a particular embodiment, the variant Fc region of the invention further comprises an amino acid modification at position 332, represented by EU numbering, in the second polypeptide of the parental Fc region. In certain embodiments, the variant Fc region of the invention further comprises an amino acid modification at position 330, represented by EU numbering, in the second polypeptide of the parental Fc region. Alternatively, amino acid alterations described in International Publication WO2013/002362 and WO2014/104165 can also be used in the present invention.
- variant Fc regions of the invention comprise amino acid alterations at the following positions: (i) positions 234, 235, 236, 239, 268, 270, 298, and 330, represented by EU numbering, in the first polypeptide of the parent Fc region, and (ii) positions 270, 298, 326, 330 and 334, represented by EU numbering, in the second polypeptide of the parent Fc region;
- the variant Fc region of the invention further comprises an amino acid modification at position 326, represented by EU numbering, in the first polypeptide of the parent Fc region.
- the variant Fc region of the invention further comprises an amino acid modification at position 236, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the invention further comprises an amino acid alteration at position 332, represented by EU numbering, in the first polypeptide of the parent Fc region.
- the variant Fc region of the invention further comprises an amino acid modification at position 332, represented by EU numbering, in the second polypeptide of the parental Fc region.
- amino acid alterations described in International Publication WO2013/002362 and WO2014/104165 can also be used in the present invention.
- the variant Fc regions of the present invention have improved stability compared to the parental Fc region.
- the stability is thermodynamic stability.
- Thermodynamic stability of a polypeptide can be determined using, for example, the Tm value as an indicator.
- the Tm value can be measured using methods known to those skilled in the art, such as CD (circular dichroism), DSC (differential scanning calorimeter), and DSF (differential scanning fluorometry).
- the mutant Fc region of the present invention has a CH2 region Tm value of 0.1 degrees or more, 0.2 degrees or more, 0.3 degrees or more, 0.4 degrees or more, 0.5 degrees or more, 1 degree or more, 2 degrees or more, compared to the parent Fc region. 3 degrees or more, 4 degrees or more, 5 degrees or more, 10 degrees or more.
- variant Fc regions of the invention are selected from the group consisting of positions 250 and 307, represented by EU numbering, in the first and/or second polypeptide of the parent Fc region It contains at least one amino acid modification at at least one position.
- amino acid modifications described in International Publication WO2013/118858 can also be used in the present invention.
- the mutant Fc regions of the present invention are composed of two polypeptide chains that differ in sequence from each other.
- the variant Fc regions of the invention have enhanced heterodimerization between a first polypeptide and a second polypeptide.
- different peptide chains preferentially associate to form heterodimers rather than identical polypeptide chains to associate to form homodimers. is preferably formed.
- the heterodimerization of the mutated Fc region is promoted, for example, homodimers and heterodimers are separated from the produced mutated Fc region by a technique such as chromatography, and the component ratio of each can be determined by asking for
- the variant Fc region of the invention comprises positions 349, 356, 366, 368, 407, represented by EU numbering, in the first polypeptide and/or the second polypeptide of the parent Fc region. and at least one amino acid modification at at least one position selected from the group consisting of 439.
- amino acid alterations described in International Publication WO2006/106905 and WO1996/027011 can also be used in the present invention.
- the mutant Fc regions of the present invention have enhanced FcRn-binding activity under acidic pH.
- acidic pH means pH 4.0-6.5.
- the acidic pH is pH 4.0, 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0 , 6.1, 6.2, 6.3, 6.4, and 6.5.
- the acidic pH is pH 5.8.
- variant Fc regions of the invention are located at positions 428, 434, 436, 438, and 440, represented by EU numbering, in the first and/or second polypeptide of the parental Fc region.
- the variant Fc regions of the invention are at positions 234, 235, 236, 239, 250, 268, 270, 298, 307, 326, 330, 332, 334, 349, 356, 366 as represented by EU numbering. , 368, 407, 428, 434, 436, 438, 439, and 440.
- the variant Fc region of the present invention comprises amino acid alterations at positions 234, 235, 236, 239, 268, 270, 298, 326, and 334, represented by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 268, 270, and 298, represented by EU numbering, in the first polypeptide of the parental Fc region, and (ii) containing amino acid alterations at positions 270, 298, 326, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region;
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 268, 270, 298, represented by EU numbering, in the first polypeptide of the parental Fc region; and 326, and (ii) amino acid alterations at positions 236, 270, 298, 326, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the present invention comprises amino acid alterations at positions 234, 235, 236, 239, 268, 270, 298, 326, 330, and 334 represented by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 268, 270, 298, and 330, represented by EU numbering, in the first polypeptide of the parental Fc region. and (ii) amino acid alterations at positions 270, 298, 326, 330, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the present invention comprises amino acid alterations at positions 234, 235, 236, 239, 250, 268, 270, 298, 307, 326, and 334 represented by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, 298, represented by EU numbering, in the first polypeptide of the parental Fc region; and 307, and (ii) amino acid alterations at positions 250, 270, 298, 307, 326, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, represented by EU numbering, in the first polypeptide of the parental Fc region; 298, 307, and 326, and (ii) amino acid alterations at positions 236, 250, 270, 298, 307, 326, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- a variant Fc region of the invention comprises amino acid alterations at positions 234, 235, 236, 239, 268, 270, 298, 326, 330, 332, and 334 as indicated by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 268, 270, 298, 330, represented by EU numbering, in the first polypeptide of the parent Fc region; and 332, and (ii) amino acid alterations at positions 236, 270, 298, 326, 330, 332, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- a variant Fc region of the invention comprises amino acid alterations at positions 234, 235, 236, 239, 250, 268, 270, 298, 307, 326, 330, 332, and 334 as indicated by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, 298, represented by EU numbering, in the first polypeptide of the parent Fc region; Amino acid alterations at positions 236, 250, 270, 298, 307, 326, 330, 332, and 334, represented by EU numbering, in 307, 330, and 332 and (ii) the second polypeptide of the parental Fc region including.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, represented by EU numbering, in the first polypeptide of the parent Fc region; Amino acid alterations at positions 298, 307 and 326 and (ii) the second polypeptide of the parental Fc region at positions 236, 250, 270, 298, 307, 326, 330, 332 and 334 represented by EU numbering including.
- the variant Fc region of the present invention comprises amino acid alterations at positions 234, 235, 236, 239, 250, 268, 270, 298, 307, 326, 332, and 334, represented by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, 298, represented by EU numbering, in the first polypeptide of the parental Fc region; 307, 326, and 332 and (ii) amino acid alterations at positions 236, 250, 270, 298, 307, 326, 332, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the present invention comprises amino acid alterations at positions 234, 235, 236, 239, 250, 268, 270, 298, 307, 326, 332, and 334, represented by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, 298, represented by EU numbering, in the first polypeptide of the parental Fc region; 307, and 332 and (ii) amino acid alterations at positions 250, 270, 298, 307, 326, 332, and 334, represented by EU numbering, in the second polypeptide of the parental Fc region.
- the variant Fc region of the present invention comprises amino acid alterations at positions 234, 235, 236, 239, 250, 268, 270, 298, 307, 326, 330, and 334 represented by EU numbering.
- the variant Fc region of the invention comprises (i) positions 234, 235, 236, 239, 250, 268, 270, 298, represented by EU numbering, in the first polypeptide of the parental Fc region; 307, and 330, and (ii) amino acid alterations at positions 250, 270, 298, 307, 326, 330, and 334, represented by EU numbering, in the second polypeptide of the parent Fc region.
- the variant Fc region of the invention comprises (i) Tyr or Phe at position 234, Gln or Tyr at position 235, position 236, as indicated by EU numbering, in the first polypeptide of the parent Fc region; Met at position 239, Val at position 250, Asp at position 268, Glu at position 270, Ala at position 298, Pro at position 307, Asp at position 326, Met at position 330, Glu at position 332, and ( ii) Ala at position 236, Val at position 250, Glu at position 270, Ala at position 298, Pro at position 307, Asp at position 326, represented by EU numbering, in the second polypeptide of the parent Fc region It contains at least one amino acid modification selected from the group consisting of Met or Lys at position 330, Asp or Glu at position 332, Glu at position 334.
- the variant Fc regions of the invention further comprise any of the following amino acid alterations (a)-(f): (a) Lys at position 356, represented by EU numbering, in the first polypeptide of the parental Fc region, and Glu at position 439, represented by EU numbering, in the second polypeptide of the parental Fc region; (b) Glu at position 439, represented by EU numbering, in the first polypeptide of the parental Fc region, and Lys at position 356, represented by EU numbering, in the second polypeptide of the parental Fc region.
- Trp at position 366 represented by EU numbering, in the first polypeptide of the parental Fc region
- Ser at position 366 represented by EU numbering, position 368, in the second polypeptide of the parental Fc region.
- Ala at and Val at position 407 (d) Ser at position 366, Ala at position 368, and Val at position 407 represented by EU numbering in the first polypeptide of the parent Fc region and EU numbering in the second polypeptide of the parent Fc region.
- Trp at position 366 represented by (e) Cys at position 349, represented by EU numbering, and Trp at position 366, in the first polypeptide of the parent Fc region, and position 356, represented by EU numbering, in the second polypeptide of the parent Fc region. Cys at position 366, Ser at position 366, Ala at position 368, and Val at position 407, (f) Cys at position 356, Ser at position 366, Ala at position 368, and Val at position 407, represented by EU numbering, in the first polypeptide of the parent Fc region, and the second polypeptide of the parent Fc region; Cys at position 349 and Trp at position 366 in the peptide, represented by EU numbering.
- the variant Fc region of the present invention further comprises any of the following amino acid alterations (a) to (d) in the first polypeptide and/or the second polypeptide of the parent Fc region: (a) Ala at position 434 represented by EU numbering, (b) Ala at position 434, Thr at position 436, Arg at position 438, Glu at position 440, represented by EU numbering; (c) Leu at position 428, Ala at position 434, Thr at position 436, Arg at position 438, Glu at position 440, represented by EU numbering; (d) Leu at position 428, Ala at position 434, Arg at position 438, Glu at position 440, represented by EU numbering.
- a polypeptide comprising a variant Fc region of the present invention is an antibody heavy chain constant region.
- the present invention provides a polypeptide comprising the amino acid sequence of any one of SEQ ID NOs: 280, 281, 283-305, 308, 309, 311-333.
- the invention provides a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:308 and the polypeptide chain of SEQ ID NO:309, the polypeptide chain of SEQ ID NO:311 and the polypeptide chain of SEQ ID NO:312.
- a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:313 and a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:314, the polypeptide chain of SEQ ID NO:315 and a heavy chain comprising the polypeptide chain of SEQ ID NO:316 a heavy chain constant region comprising a chain constant region, the polypeptide chain of SEQ ID NO:317 and a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:318, the polypeptide chain of SEQ ID NO:319 and the polypeptide chain of SEQ ID NO:320 a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:321 and the polypeptide chain of SEQ ID NO:322, a heavy chain constant region comprising the polypeptide chain of SEQ ID NO:323 and the polypeptide chain of SEQ ID NO:324, a heavy chain constant region comprising the polypeptide chain of SEQ ID NO: 325 and the polypeptide chain of SEQ ID NO: 3
- Fc ⁇ receptor refers to a receptor that can bind to the Fc region of IgG1, IgG2, IgG3, and IgG4 monoclonal antibodies.
- Fc ⁇ receptor refers to a receptor that can bind to the Fc region of IgG1, IgG2, IgG3, and IgG4 monoclonal antibodies.
- this family includes Fc ⁇ RI (CD64), which includes isoforms Fc ⁇ RIa, Fc ⁇ RIb, and Fc ⁇ RIc; -2), and Fc ⁇ RII (CD32), including Fc ⁇ RIIc; and Fc ⁇ RIII (CD16), including isoforms Fc ⁇ RIIIa (including allotypes V158 and F158) and Fc ⁇ RIIIb (including allotypes Fc ⁇ RIIIb-NA1 and Fc ⁇ RIIIb-NA2), and , any undiscovered human Fc ⁇ R, Fc ⁇ R isoform or allotype, including but not limited to.
- Fc ⁇ RIIb1 and Fc ⁇ RIIb2 have been reported as splice variants of human Fc ⁇ RIIb.
- human Fc ⁇ RIIb3 includes all splice variants registered with NCBI, NP_001002273.1, NP_001002274.1, NP_001002275.1, NP_001177757.1, and NP_003992.3.
- human Fc ⁇ RIIb in addition to Fc ⁇ RIIb, has all genetic polymorphisms reported in the past (Arthritis Rheum. 48: 3242-3252 (2003); Kono et al., Hum. Mol. Genet. 14: 2881- 2892 (2005); and Kyogoju et al., Arthritis Rheum. 46: 1242-1254 (2002)) and all genetic polymorphisms that may be reported in the future.
- Fc ⁇ RIIa There are two allotypes of Fc ⁇ RIIa, one in which the amino acid at position 131 of Fc ⁇ RIIa is histidine (H form) and the other in which the amino acid at position 131 is substituted with arginine (R form) (Warrmerdam, J. .Exp. Med. 172: 19-25 (1990)).
- Fc ⁇ Rs include, but are not limited to, human, mouse, rat, rabbit, and monkey-derived Fc ⁇ Rs, and may be derived from any organism.
- Mouse Fc ⁇ Rs include, but are not limited to, Fc ⁇ RI (CD64), Fc ⁇ RII (CD32), Fc ⁇ RIII (CD16), and Fc ⁇ RIII-2 (CD16-2), and any mouse Fc ⁇ R or Fc ⁇ R isoform No.
- the amino acid sequence of human Fc ⁇ RI is set forth in SEQ ID NO: 131 (NP_000557.1); the amino acid sequence of human Fc ⁇ RIIa is SEQ ID NO: 132 (AAH20823.1), SEQ ID NO: 142, SEQ ID NO: 143, or SEQ ID NO: 150 the amino acid sequence of human Fc ⁇ RIIb is set forth in SEQ ID NO: 151 (AAI46679.1), SEQ ID NO: 169, or SEQ ID NO: 172; the amino acid sequence of human Fc ⁇ RIIIa is set forth in SEQ ID NO: 174 (AAH33678.1), set forth in SEQ ID NO:175, SEQ ID NO:176, or SEQ ID NO:177; and the amino acid sequence of human Fc ⁇ RIIIb is set forth in SEQ ID NO:178 (AAI28563.1).
- human FcRn is structurally similar to major histocompatibility complex (MHC) class I polypeptides, sharing 22-29% sequence identity with class I MHC molecules.
- MHC major histocompatibility complex
- FcRn is expressed as a heterodimer consisting of a transmembrane ⁇ or heavy chain complexed with a soluble ⁇ or light chain ( ⁇ 2 microglobulin).
- ⁇ -chain of FcRn consists of three extracellular domains ( ⁇ 1, ⁇ 2, ⁇ 3) and a short cytoplasmic domain anchors the protein to the cell surface.
- the ⁇ 1 and ⁇ 2 domains interact with the FcRn binding domain in the Fc region of antibodies (Raghavan et al. (Immunity (1994) 1, 303-315).
- the amino acid sequence of human FcRn is SEQ ID NO: 179 (NP_004098.1),
- the amino acid sequence of ⁇ 2 microglobulin is set forth in SEQ ID NO: 180 (NP_004039.1).
- the parental Fc region refers to the Fc region prior to the introduction of the amino acid alterations described herein.
- the parental Fc region is a native sequence Fc region (or a native antibody Fc region).
- Antibodies include, for example, IgA (IgA1, IgA2), IgD, IgE, IgG (IgG1, IgG2, IgG3, IgG4), IgM, and the like.
- Antibodies can be human or simian (eg, cynomolgus, rhesus, marmoset, chimpanzee, or baboon).
- a native antibody may contain naturally occurring mutations.
- sequences of proteins of immunological interest are described in "Sequences of proteins of immunological interest," NIH Publication No. 91-3242, any of which can be used in the present invention.
- the amino acid sequence at positions 356-358 can be either DEL or EEM.
- the amino acid at position 214 can be either K or R.
- the parent Fc region is a heavy chain constant of human IgG1 (SEQ ID NO:249), human IgG2 (SEQ ID NO:250), human IgG3 (SEQ ID NO:251), or human IgG4 (SEQ ID NO:252).
- the parental Fc region is an Fc region derived from the heavy chain constant region of SEQ ID NO:82, SEQ ID NO:158, or SEQ ID NO:334.
- the parental Fc region is an Fc region (Fc region of the reference variant sequence) generated by making amino acid modifications other than those described herein to the Fc region of the native sequence (Fc region of the reference variant sequence) good too.
- the native sequence Fc region is typically organized as a homodimer composed of two identical polypeptide chains.
- amino acid modifications performed for other purposes can be combined in the variant Fc regions described herein.
- amino acid substitutions that enhance FcRn binding activity Hinton et al., J. Immunol. 176(1): 346-356 (2006); Dall'Acqua et al., J. Biol. Chem. 281(33): 23514-23524 (2006); Petkova et al., Intl. Immunol. 18(12): 1759-1769 (2006); Zalevsky et al., Nat. Biotechnol. /019447; WO2006/053301; and WO2009/086320) and amino acid substitutions to improve antibody heterogeneity or stability (WO2009/041613) may be added.
- Polypeptides having the property of repeatedly binding to multiple antigen molecules as described in WO2009/125825, WO2012/073992, or WO2013/047752 can be combined with the variant Fc regions described herein.
- the amino acid modifications disclosed in EP1752471 and EP1772465 may be combined in CH3 of the variant Fc regions described herein for the purpose of conferring the ability to bind to other antigens.
- amino acid modifications that lower the pI of the constant region may be combined in the variant Fc regions described herein for the purpose of increasing plasma retention.
- amino acid modifications that increase the pI of the constant region may be combined in the variant Fc regions described herein for the purpose of facilitating cellular uptake.
- amino acid alterations that increase the pI of the constant region may be combined in the variant Fc regions described herein for the purpose of promoting plasma clearance of the target molecule.
- such modifications may include substitutions at at least one position selected from the group consisting of positions 311, 343, 384, 399, 400, and 413, e.g., represented by EU numbering. .
- substitutions may be replacement of the amino acid at each position with Lys or Arg.
- Techniques for making heterodimeric antibodies include, but are not limited to, the knob-in-hole technique (e.g., Nat. Biotechnol., (16); 677-681 (1998) and US Patent No. 5,731,168) and by manipulating electrostatic steering effects (WO2006/106905, WO2009/089004A1, J. Biol. Chem., (285), 19637-19646 (2010), etc.) good too.
- the knob-in-hole technique e.g., Nat. Biotechnol., (16); 677-681 (1998) and US Patent No. 5,731,168
- electrostatic steering effects WO2006/106905, WO2009/089004A1, J. Biol. Chem., (285), 19637-19646 (2010), etc.
- homologous polypeptides comprising variant Fc regions For binding heterologous polypeptides comprising variant Fc regions, homologous polypeptides comprising variant Fc regions by introducing electrostatic repulsion at the interface of the CH2 or CH3 domains of the Fc region, as described in WO2006/106905. It is possible to apply a technique for suppressing unintended binding of
- amino acid residues contacting the CH2 or CH3 domain interface of the Fc region include residues at position 356 (EU numbering), residues at position 439 (EU numbering), residues at position 357 (EU numbering), residue at position 370 (EU numbering), residue at position 399 (EU numbering), and residue at position 409 (EU numbering).
- an Fc region can be generated in which 1 to 3 pairs of amino acid residues selected from (1) to (3) shown below have the same charge: (1) in the CH3 domain; (2) amino acid residues at positions 357 and 370 (EU numbering) in the CH3 domain; and (3) positions 399 and 409 (EU numbering) in the CH3 domain. amino acid residue.
- 1 to 3 pairs of amino acid residues selected from (1) to (3) above have the same charge in the CH3 domain of the first Fc region, and are selected in the first Fc region generating a heterologous polypeptide comprising a variant Fc region, wherein said pair of amino acid residues have the same charge in the CH3 domain of the second Fc region, but the charges of the first and second Fc regions are diametrically opposite can do.
- the negatively charged amino acid residues are preferably selected from glutamic acid (E) and aspartic acid (D), and the positively charged amino acid residues are lysine (K), arginine (R ), and histidine (H).
- polypeptides comprising homologously variant Fc regions are usually also present as impurities. , is produced.
- polypeptides comprising heterologous variant Fc regions can be efficiently obtained by separation and purification from polypeptides comprising homologously variant Fc regions using known techniques. Homodimerization using ion exchange chromatography by introducing amino acid modifications that cause a difference in the isoelectric point between the homodimeric antibody and the heterodimeric antibody into the variable regions of the two antibody heavy chains A method for efficiently separating and purifying heterodimeric antibodies from antibodies has been reported (WO2007/114325).
- heterodimeric antibodies using protein A chromatography by constructing heterodimeric antibodies comprising two heavy chains derived from mouse IgG2a that binds to protein A and rat IgG2b that does not bind to protein A. Another method has been reported (WO1998/050431 and WO1995/033844).
- protein A binding can be achieved by replacing amino acid residues at positions 435 and 436 (EU numbering) located in the protein A binding site of the antibody heavy chain with amino acids such as Tyr or His to confer different protein A binding affinities. Chromatography can be used to efficiently purify heterodimeric antibodies.
- amino acid alteration means any substitution, deletion, addition, insertion and modification, or a combination thereof.
- amino acid modification can be rephrased as amino acid mutation.
- the number of amino acid modifications introduced into the Fc region is not limited. In certain embodiments, the It can be less than or equal to 28, or less than or equal to 30.
- the present invention provides methods for producing polypeptides comprising mutant Fc regions.
- the invention provides a method of producing a polypeptide comprising a variant Fc region with altered function.
- the invention provides methods of modifying the function of a polypeptide comprising an Fc region.
- the polypeptide is an antibody.
- the polypeptide is an Fc fusion protein.
- those methods comprise introducing at least one amino acid modification into the parental Fc region.
- those methods comprise (i) providing a polypeptide comprising a parental Fc region, and (ii) introducing at least one amino acid modification into the parental Fc region.
- those methods may further comprise (iii) measuring the function of the polypeptide comprising the mutated Fc region.
- a native Fc region is usually composed of two identical polypeptide chains. Amino acid alterations to the parental Fc region may be introduced into either one of the two polypeptide chains of the parental Fc region, or into both of the two polypeptide chains.
- a method for producing a polypeptide comprising a variant Fc region comprises the steps of: (i) providing one or more nucleic acids encoding a polypeptide comprising a parent Fc region; introducing at least one mutation in the coding region, (iii) introducing the nucleic acid produced in (ii) into a host cell, and (iv) expressing a polypeptide comprising the mutated Fc region (iii ) and culturing the cells described in ).
- the method may further comprise recovering the polypeptide comprising the mutated Fc region from the host cell culture of (v)(iv).
- the nucleic acid produced in (ii) may be contained in one or more vectors (eg, expression vectors, etc.).
- the amino acid modification used in the present production method is any single modification selected from among the amino acid modifications that can be contained in the mutant Fc region described above, a combination of single modifications, or Table 29 selected from the combination modifications listed in Table 34.
- the Fc region may be obtained by partially digesting IgG1, IgG2, IgG3, IgG4 monoclonal antibodies, etc. with a protease such as pepsin, and then re-eluting the fraction adsorbed to the protein A column.
- the protease is not particularly limited as long as it can digest the full-length antibody and thereby produce Fab and F(ab')2 in a restrictive manner by appropriately setting the enzymatic reaction conditions such as pH. , examples include pepsin and papain.
- a polypeptide comprising a mutant Fc region of the present invention may be produced by other methods known in the art, in addition to the production method described above. Polypeptides comprising variant Fc regions produced by the production methods described herein are also included in the present invention.
- an isolated nucleic acid that encodes a polypeptide comprising a variant Fc region of the invention.
- Such nucleic acids may encode an amino acid sequence comprising the first polypeptide and/or an amino acid sequence comprising the second polypeptide of the variant Fc region.
- one or more vectors eg, expression vectors
- host cells containing such nucleic acids are provided.
- the host cell comprises (1) a vector comprising a nucleic acid encoding an amino acid sequence comprising a first polypeptide of a variant Fc region and an amino acid sequence comprising a second polypeptide of a variant Fc region; or (2) a first vector comprising a nucleic acid encoding an amino acid sequence comprising a first polypeptide of a variant Fc region and a second vector comprising a nucleic acid encoding an amino acid sequence comprising a second polypeptide of a variant Fc region It contains (eg has been transformed with) two vectors.
- host cells are eukaryotic (eg, Chinese Hamster Ovary (CHO) cells) or lymphoid (eg, Y0, NS0, Sp2/0 cells).
- culturing a host cell containing a nucleic acid encoding a polypeptide comprising a variant Fc region of the invention under conditions suitable for expression of the polypeptide, and, optionally, expressing the polypeptide in the host cell or A method is provided for producing a polypeptide comprising a variant Fc region of the invention, comprising recovering from the host cell culture medium).
- nucleic acid encoding the polypeptide is isolated and placed into one or more vectors for further cloning and/or expression in a host cell. insert.
- nucleic acids may be readily isolated and sequenced using conventional procedures (e.g., using oligonucleotide probes capable of specifically binding to the gene encoding the Fc region of the antibody). ).
- Suitable host cells for cloning or expressing vectors encoding polypeptides comprising a mutated Fc region of the present invention include prokaryotic or eukaryotic cells.
- filamentous fungi or yeast including strains of fungi and yeast in which the glycosylation pathway has been "humanized", resulting in the production of antibody Fc regions with a partially or fully human glycosylation pattern.
- Eukaryotic microbes such as are suitable cloning or expression hosts for vectors encoding polypeptides comprising variant Fc regions of the invention. See Gerngross, Nat. Biotech. 22: 1409-1414 (2004) and Li et al., Nat. Biotech. 24: 210-215 (2006).
- invertebrates and vertebrates are also suitable host cells for the expression of glycosylated antibody Fc regions.
- invertebrate cells include plant and insect cells.
- a number of baculovirus strains have been identified for use in conjugation with insect cells, particularly for transformation of Spodoptera frugiperda cells.
- Plant cell cultures can also be used as hosts. See, for example, U.S. Patent Nos. 5,959,177, 6,040,498, 6,420,548, 7,125,978, and 6,417,429.
- Vertebrate cells can also be used as hosts.
- mammalian cell lines adapted to grow in suspension would be useful.
- Other examples of useful mammalian host cell lines are the SV40-transformed monkey kidney CV1 strain (COS-7); the human embryonic kidney strain (Graham et al., J. Gen Virol. 36: 59 (1977 ) 293 or 293 cells described in Etc.); baby hamster kidney cells (BHK); mouse Sertoli cells (TM4 cells described in Mather, Biol. Reprod.
- monkey kidney cells (CV1 ); African green monkey kidney cells (VERO-76); human cervical carcinoma cells (HELA); canine kidney cells (MDCK); Buffalo rat hepatocytes (BRL 3A); Hep G2); mouse mammary carcinoma (MMT 060562); TRI cells (described, for example, in Mather et al., Annals N.Y. Acad. Sci. 383: 44-68 (1982)); MRC5 cells;
- Other useful mammalian host cell lines are Chinese Hamster Ovary (CHO) cells, including DHFR-CHO cells (Urlaub et al., Proc. Natl. Acad. Sci. USA 77: 4216 (1980)); Includes myeloma cell lines such as NS0, and Sp2/0.
- the assays or techniques described herein For the purpose of identifying or screening the mutant Fc regions provided herein, or for the purpose of demonstrating their physical or chemical properties, or biological activity, the assays or techniques described herein Various measurement methods known in the art may be used.
- binding assays for determining the binding activity of a polypeptide comprising a variant Fc region to one or more FcR family members are described herein or known in the art.
- binding assays include, but are not limited to, surface plasmon resonance assays, amplified luminescence proximity homogeneous assay (ALPHA) screening, ELISA, and fluorescence-activated cell sorting (FACS) (Lazar et al., Proc. Natl. Acad. Sci. USA (2006) 103(11): 4005-4010).
- the binding activity of a polypeptide comprising a mutated Fc region to FcR family members can be measured using a surface plasmon resonance assay.
- immobilization or Various FcRs are allowed to interact as analytes with the captured mutated Fc region-containing polypeptide.
- FcRs may be immobilized or captured on a sensor chip and polypeptides containing variant Fc regions used as analytes. Such interactions result in binding sensorgrams that can be analyzed to calculate the dissociation constant (KD) value for the binding.
- KD dissociation constant
- the difference in the resonance unit (RU) value (that is, the amount of FcR binding) in the sensorgram before and after the interaction with the FcR was evaluated as the binding activity of the polypeptide containing the mutant Fc region to the FcR. It can also be used as an index.
- the difference in the RU value in the sensorgram (that is, the binding amount of the polypeptide containing the mutant Fc region)
- a correction value obtained by dividing the FcR binding amount that is, the FcR binding amount per unit amount of the polypeptide containing the mutant Fc region) may be used as an indicator of the binding activity.
- polypeptides comprising variant Fc regions may be used in therapeutic methods.
- polypeptides comprising variant Fc regions are provided for use as pharmaceuticals.
- polypeptides comprising mutated Fc regions are provided for use in treating tumors.
- polypeptides comprising variant Fc regions are provided for use in therapeutic methods.
- the invention provides a mutant Fc region for use in a method of treating an individual with a tumor comprising administering to the individual an effective amount of a polypeptide comprising the mutant Fc region.
- a polypeptide is provided comprising:
- the invention provides polypeptides comprising variant Fc regions for use in cell killing.
- the present invention is for use in a method of killing cells in an individual comprising administering to said individual an effective amount of a polypeptide comprising a mutated Fc region to injure the cells.
- a polypeptide comprising a mutated Fc region of An "individual" according to any of the above aspects is preferably a human.
- injury to cells is induced by ADCC activity, CDC activity, or ADCP activity.
- the present invention provides use of a polypeptide comprising a mutated Fc region in the manufacture or preparation of a medicament.
- the medicament is for treatment of a tumor.
- the medicament is for use in a method of treating a tumor comprising administering to an individual having a tumor an effective amount of the medicament.
- the medicament is for cell injury.
- the medicament is for use in a method of injuring cells in an individual comprising administering to said individual an effective amount of the medicament to injure the cells.
- An "individual" according to any of the above aspects may be a human.
- the invention provides methods of treating tumors.
- the method comprises administering to an individual with such a tumor an effective amount of a polypeptide comprising a mutated Fc region.
- An "individual” according to any of the above aspects may be a human.
- the invention provides methods for damaging cells in an individual.
- the method comprises administering to the individual an effective amount of a polypeptide comprising a mutated Fc region to kill the cell.
- the "individual" is a human.
- the invention provides pharmaceutical formulations (pharmaceutical compositions) comprising a polypeptide comprising a variant Fc region provided herein.
- the pharmaceutical formulation (pharmaceutical composition) further comprises a pharmaceutically acceptable carrier.
- the present invention provides pharmaceutical formulations (pharmaceutical compositions) for use in treating tumors.
- the present invention provides pharmaceutical formulations (pharmaceutical compositions) for use in cell injury. ⁇ Anti-CTLA-4 antibody containing a mutated Fc region>
- the invention provides an isolated antibody that includes a variant Fc region and that binds CTLA-4.
- the anti-CTLA-4 antibodies of the invention comprise variable regions that have CTLA-4 binding activity that is dependent on the concentration of the adenosine-containing compound.
- the anti-CTLA-4 antibodies of the invention are variant Fc regions comprising multiple amino acid alterations in the parental Fc region, at positions 234, 235, 236, 239, 268, as represented by EU numbering. Includes variant Fc regions containing amino acid alterations at 270, 298, 326, 330, and 334.
- the invention provides: (A) variable regions with CTLA-4 binding activity dependent on the concentration of adenosine-containing compounds, and (B) An anti-CTLA-4 antibody comprising a mutated Fc region containing multiple amino acid alterations in the parent Fc region, wherein the parent Fc region is composed of two polypeptide chains, and the mutated Fc region comprises: An anti-CTLA-4 antibody comprising amino acid alterations in: (i) positions 234, 235, 236, 239, 268, 270, 298, and 330, represented by EU numbering, in the first polypeptide of the parent Fc region, and (ii) positions 270, 298, 326, 330 and 334 represented by EU numbering in the second polypeptide of the parent Fc region I will provide a.
- the present invention provides an anti-CTLA-1 comprising a first H chain polypeptide of SEQ ID NO:335, a second H chain polypeptide of SEQ ID NO:336, and a L chain polypeptide of SEQ ID NO:161.
- the present invention provides an anti-CTLA-1 comprising a first H chain polypeptide of SEQ ID NO:337, a second H chain polypeptide of SEQ ID NO:338, and a L chain polypeptide of SEQ ID NO:161.
- Example 0 Concept of switch antibody that exerts antibody-dependent cytotoxic activity against cell surface markers of regulatory T cells only in cancer microenvironment Ipilimumab activates effector T cells by CTLA4 expressed on the surface of effector T cells It was thought that inhibition of CTLA4-expressing T cells exerted its antitumor effect, but recently, it has been reported that antibody-dependent cytotoxicity (ADCC activity) against CTLA4-expressing T cells is also important. Depletion of regulatory T cells and ADCC activity have been found to be key mechanisms of antitumor efficacy of anti-CTLA4 antibodies.
- ADCC activity antibody-dependent cytotoxicity
- the ADCC activity by IgG1 antibody induces cytotoxic activity by binding the constant region of the antibody to Fc ⁇ R of NK cells and macrophages, and antibodies with constant regions modified to enhance the binding are more It is also known to induce strong cytotoxic activity and exhibit antitumor effects.
- Example 1 Production of anti-CTLA4 switch antibody with modified Fc region (1-1) Expression and Purification of Anti-CTLA4 Switch Antibody Having Modified Fc Region
- Anti-CTLA4 switch antibody with modified Fc region (04H1654-KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1 abbreviation: SW1610-ART5+ACT1, 04H1654-KT456/ 04L1610-lam1//04H1656-HT446/04L1610-lam1 (abbreviation: SW1610-ART4) and 04H1654-KT498/04L1610-lam1//04H1656-HT518/04L1610-lam1 (abbreviation: SW1610-ART12) were prepared.
- the SW1610-ART5+ACT1 antibody was produced by combining the heavy chain and light chain genes produced in Reference Example 6. Specifically, 04H1654-KT473 (SEQ ID NO: 184) as one heavy chain, 04H1656-HT451 (SEQ ID NO: 272) as the other heavy chain, and 04L1610-lam1 (SEQ ID NO: 161) as the light chain. and expressed and purified by methods known to those skilled in the art.
- the SW1610-ART4 antibody has the C-terminal Gly and Lys of human IgG1 (IGHG1*03) removed as one heavy chain, and the CH2 region has the same modification as Kn456 described in Reference Example 7, and the CH3 region has An antibody heavy chain 04H1654-KT456 (SEQ ID NO: 335) gene was constructed having the modification E356K that promotes heterodimerization described in WO2006/106905 and 04H1654 (SEQ ID NO: 140) as the heavy chain variable region. .
- the CH2 region has the same modification as Hl446 described in Reference Example 7, and the CH3 region has the heterodivision described in WO2006/106905.
- the antibody heavy chain 04H1656-HT446 (SEQ ID NO:336) gene was generated with the modification K439E to facilitate quantification and with 04H1656 (SEQ ID NO:141) as the heavy chain variable region.
- 04L1610-lam1 (SEQ ID NO: 161) was used as a light chain, expressed and purified by methods known to those skilled in the art.
- the SW1610-ART12 antibody similarly removes the C-terminal Gly and Lys of human IgG1 (IGHG1*03) as one heavy chain, and has the same modification as Kn498 described in Reference Example 7 in the CH2 region, and CH3 A gene of antibody heavy chain 04H1654-KT498 (SEQ ID NO: 337) having a modified E356K that promotes heterodimerization described in WO2006/106905 in the region and 04H1654 (SEQ ID NO: 140) as the heavy chain variable region was prepared. was done.
- the CH2 region has the same modification as Hl518 described in Reference Example 7, and the CH3 region has the heterodivision described in WO2006/106905.
- the antibody heavy chain 04H1656-HT518 (SEQ ID NO:338) gene was generated with the modification K439E to facilitate quantification and with 04H1656 (SEQ ID NO:141) as the heavy chain variable region.
- 04L1610-lam1 (SEQ ID NO: 161) was used as a light chain, expressed and purified by methods known to those skilled in the art.
- heterodimeric antibodies antibodies having two different heavy chain polypeptides and/or two different light chain polypeptides
- heavy chain variable region - (first heavy chain constant region) / (first light chain variable region) - (first light chain constant region) / / (second heavy chain variable region) - (second two heavy chain constant regions)/(second light chain variable region)-(second light chain constant region).
- the first heavy chain variable region of this antibody is 04H1654, the first heavy chain constant region is KT456, the first the light chain variable region is 04L1610, the first light chain constant region is lam1, the second heavy chain variable region is 04H1656, the second heavy chain constant region is HT446, the second light chain variable region is 04L1610, the second light chain constant region is lam1.
- Binding activity was evaluated at 25°C using 50 mM phosphate buffer, 150 mM NaCl, 0.05 w/v%-P20, pH 7.4 as a running buffer.
- CaptureSelect (trademark) Human Fab-lambda Kinetics Biotin Conjugate (ThermoFisher scientific) was immobilized on a Series S Sensor chip SA sensor chip (Cytiva) as a ligand-capturing molecule. About 250 RU, 500 RU or about 1000 RU of the antibody was captured by allowing the SA sensor chip to interact with an antibody solution prepared with a running buffer. hFc ⁇ Rs and cyFc ⁇ Rs diluted in running buffer were then allowed to bind to the captured antibodies.
- hFc ⁇ Rs and cyFc ⁇ Rs were diluted to a concentration of 5 points or more at a common ratio of 2, with the concentration shown in Table 2 as the maximum concentration, and used for measurement.
- the chip was regenerated with 10 mM Glycine-HCl (pH 1.5) and repeatedly captured antibody and measured.
- the binding activity of each antibody to each Fc ⁇ R was evaluated by calculating the KD value with a Steady state model or 1:1 binding model using Biacore Insight Evaluation Software.
- hFc ⁇ Rs and cyFc ⁇ Rs used as analytes were prepared as described below.
- the gene sequences of the extracellular domain of hFc ⁇ R are hFc ⁇ RIa (NCBI reference sequence: NM_000566.3), hFc ⁇ RIIa (NCBI reference sequence: NM_001136219.1), hFc ⁇ RIIb (NCBI reference sequence: NM_004001.3), hFc ⁇ RIIIa (NCBI reference sequence: NM_001127593 .1), and the polymorphic site was designed with reference to the following documents (for hFc ⁇ RIIaR: Warmerdam, PAM et al., 1990, J. Exp. Med.
- hFc ⁇ RIa SEQ ID NO: 341
- hFc ⁇ RIIa(H) SEQ ID NO: 342
- hFc ⁇ RIIa(R) SEQ ID NO: 343
- hFc ⁇ RIIb SEQ ID NO: 344
- hFc ⁇ RIIIa(F) SEQ ID NO: 345
- hFc ⁇ RIIIa(V) SEQ ID NO: 346
- a His tag was added to the C-terminus, and each resulting gene was inserted into an expression vector designed for mammalian cell expression by methods known to those skilled in the art.
- the expression vector was introduced into human embryonic kidney cell-derived FreeStyle293 cells (Invitrogen) to express the target protein.
- the resulting culture supernatant was filtered and purified in principle through the following four steps, or three steps excluding the first anion exchange chromatography step.
- Anion exchange chromatography was performed using Q Sepharose Fast Flow (GE Healthcare) as the first step.
- affinity chromatography against His-tag was performed using HisTrap HP (GE Healthcare).
- gel filtration column chromatography was performed using HiLoad 26/600 Superdex 200 pg (GE Healthcare). Sterile filtration was performed as a fourth step.
- the absorbance of the purified protein at 280 nm was measured using a spectrophotometer and the concentration of the purified protein was determined using the extinction coefficient calculated by the method of PACE (Protein Science 4:2411-2423 (1995)). was done.
- the cyFc ⁇ R extracellular domain gene was constructed by cloning the cyFc ⁇ R cDNA using methods known to those skilled in the art.
- the amino acid sequences of the extracellular domains of cyFc ⁇ R are shown in the sequence listing as follows: cyFc ⁇ RIa (SEQ ID NO:347), cyFc ⁇ RIIa1 (SEQ ID NO:348), cyFc ⁇ RIIa2 (SEQ ID NO:349), cyFc ⁇ RIIa3 (SEQ ID NO:350).
- cyFc ⁇ RIIb SEQ ID NO: 351
- cyFc ⁇ RIIIa(R) SEQ ID NO: 352
- cyFc ⁇ RIIIa(S) SEQ ID NO: 353
- a gene sequence encoding a His tag was then added to the 3' end of each gene.
- Each gene obtained was inserted into an expression vector designed for mammalian cell expression by a method known to those skilled in the art.
- the expression vector was introduced into human embryonic kidney cell-derived FreeStyle293 cells (Invitrogen) to express the target protein. After culturing, the culture supernatant obtained was, in principle, filtered and purified through the following four steps.
- Cation exchange chromatography was performed using SP Sepharose FF as a first step.
- affinity chromatography against His-tag HisTrapHP
- gel filtration column chromatography Superdex200
- Sterile filtration was performed as a fourth step.
- the absorbance of the purified protein at 280 nm was measured using a spectrophotometer and the concentration of the purified protein was determined using the extinction coefficient calculated by the method of PACE (Protein Science 4:2411-2423 (1995)). was done.
- Table 3 shows the KD values of SW1610-ART12 and Tremfya for each hFc ⁇ R
- Table 4 shows the KD values of SW1610-ART12 and Tremfya for cyFc ⁇ R.
- the KD values for hFc ⁇ RIIa(H), hFc ⁇ RIIa(R), hFc ⁇ RIIIa(V), and hFc ⁇ RIIIa(F) in SW1610-ART12 were smaller than those in Tremfya.
- Example 2 In vitro pharmacological activity evaluation of anti-CTLA4 switch antibody with modified Fc region (2-1) ADCC activity of anti-CTLA4 switch antibody with modified Fc region using human peripheral blood mononuclear cells as effector cells -KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1 (SW1610-ART5+ACT1), 04H1654-KT456/04L1610-lam1//04H1656-HT446/04L1610-lam1 (SW1610-ART4), 04H1654-KT410/04H1654-KT410 Antibody concentration-dependent ADCC activity of -lam1//04H1656-HT518/04L1610-lam1 (SW1610-ART12) was measured according to the following method. At this time, human peripheral blood mononuclear cells (hereinafter referred to as human PBMC) were used as effector cells, and the ADCC activity of the test antibody was measured as follows.
- a human PBMC solution was prepared. Using a syringe in which 200 ⁇ L of 1000 units/mL heparin solution (Novo Heparin Injection 5,000 Units, Novo Nordisk) was injected in advance, 50 mL of peripheral blood was collected from a healthy human volunteer (adult male). The peripheral blood diluted 2-fold with PBS (-) was divided into 4 equal parts and centrifuged with 15 ml of Ficoll-Paque PLUS in Leucosep lymphocyte separation tubes (Greiner bio-one ) was added. The separation tube into which the peripheral blood was dispensed was centrifuged at room temperature for 10 minutes at a speed of 1000 rpm, and then the mononuclear cell fraction layer was collected.
- heparin solution Novo Heparin Injection 5,000 Units, Novo Nordisk
- the cells contained in the fraction layer were washed once with RPMI-1640 (Sigma) containing 10% FBS (hereinafter referred to as 10% FBS/RPMI), the cells were washed in 10% FBS/RPMI. Suspended to a cell density of 1 ⁇ 10 7 cells/mL. The cell suspension was used as a human PBMC solution for subsequent experiments.
- RPMI-1640 Sigma
- 10% FBS/RPMI 10% FBS/RPMI
- CD4+ cells were then isolated from frozen PBMCs to use anti-CD3/28 antibody-coated beads-stimulated CD4-positive T cells (CD4+ cells) as target cells.
- CD4+ cells were isolated by negative selection using a CD4 T cell isolation kit (Milteny biotec). After that, 10% FBS/RPMI was used to adjust the concentration to 4 ⁇ 10 5 cells/mL. Incubated for 4 days. After 4 days of stimulation, the cells were harvested and used for experiments.
- ADCC activity was assessed by Cr51 (chromium-51) (J-RAM) release.
- Target cells were pre-labeled by adding 90 ⁇ L of 10% FBS/RPMI and 10 ⁇ L of Cr51 solution to 2 ⁇ 10 6 cells and culturing in a 5% carbon dioxide incubator at 37° C. for 2 hours. . Labeled target cells were then washed with 10% FBS/RPMI and adjusted to 2 ⁇ 10 5 cells/mL with 10% FBS/RPMI.
- A represents the average count rate in wells to which each test antibody was added.
- B represents the average count rate in wells to which 50 ⁇ L of 4% NP-40 solution was added to wells to which only target cells were added.
- C represents the mean count rate in wells containing only 10% FBS/RPMI.
- Tests were conducted using PBMC from 3 donors for SW1610-ART5+ACT1, and PBMC from 2 donors for SW1610-ART4 and SW1610-ART12, respectively, as effector cells. (%) was calculated. The results are shown in Figures 1, 2 and 3. This result confirmed the ATP-dependent ADCC activity of the anti-CTLA4 switch antibody with altered Fc.
- human PBMCs (Cryopreserved Human PBMCs, ASTARTE) were freeze-thawed and added to CD3/CD28 Ab Dynabeads (1:1)/OpTmizer/L-Gln/Penicillin-Streptomycin to a cell density of 4 ⁇ 10 5 cells/mL. and cultured at 37° C. for 7 days in a 5% CO 2 incubator.
- PBMCs were collected, washed twice with auto MACS Rinsing Solution (Milteny), reacted with the following antibodies, and the fraction of immune cells present was analyzed by FACS analysis.
- Viability determination reagent Biolegend, Zombie Aqua
- anti-CD3 antibody (BD, clone: UCHT1)
- anti-CD4 antibody (BD, clone: RPA-T4)
- anti-CD8 antibody (BD, clone: SK1)
- anti-CD45RA antibody Biolegend, clone: 5Hg
- anti-CD25 antibody BD, clone: 2A3
- anti-CTLA4 antibody Biolegend, clone: BNI3
- the CH2 region has the same modification as Hl076 described in Reference Example 7, and the modification Y349C that promotes heterodimerization in the CH3 region
- a gene for antibody heavy chain 04H1389-Hp076 (SEQ ID NO: 340) was generated having /T366S/L368A/Y407V and 04H1389 (SEQ ID NO: 29) as the heavy chain variable region.
- 04L1305-k0MT (SEQ ID NO: 275) was used as a light chain, expressed and purified by methods known to those skilled in the art.
- SW1389-ART5 + ACT1 antibody, human IgG1 (IGHG1 * 03) C-terminal Gly and Lys are removed as one heavy chain, the CH2 region has the same modification as Kn462 described in Reference Example 7, CH3 A gene for antibody heavy chain 04H1389-Kp462 (SEQ ID NO: 354) having a modified S354C/T366W that promotes heterodimerization in the region and 04H1389 (SEQ ID NO: 29) as a heavy chain variable region is prepared, and A combination of modifications was introduced that improved the blood kinetics of the antibody as described in Mabs, 2017, 9, 844-853.
- N434A / Y436T / Q438R / S440E which is a combination of modifications that enhance binding to human FcRn under acidic conditions and modifications that reduce binding to Rheumatoid factor, was introduced into the CH3 region of 04H1389-Kp462.
- a gene of 04H1389-Kp473 (SEQ ID NO: 355) was constructed.
- the CH2 region has the same modification as Hl441 described in Reference Example 7, and the modification Y349C that promotes heterodimerization in the CH3 region /T366S/L368A/Y407V, 04H1389-Hp451 ( SEQ ID NO:357) was constructed. Furthermore, 04L1305-k0MT (SEQ ID NO: 275) was used as a light chain, expressed and purified by methods known to those skilled in the art.
- the SW1610-ART4 antibody and SW1610-ART12 antibody used were the antibodies produced in Example 1-1.
- SW1610-ART12 antibody which are 1 and 10 times the estimated therapeutic dose, respectively, were similarly administered to cynomolgus monkeys (3 males and 3 females) intravenously once a week for a total of 5 doses.
- general condition observation, body weight measurement, blood/blood chemical test, urinalysis, blood autoantibody measurement, immunophenotyping, blood cytokine measurement, bone marrow examination, pathological examination, and plasma drug concentration measurement were performed.
- the development of anti-drug antibodies and accompanying decrease in exposure were observed in many cases during the administration period.
- Antigen proteins were expressed and purified using the following method. Human embryonic kidney cell-derived FreeStyle 293-F strain (Invitrogen) suspended in FreeStyle 293 Expression Medium (Invitrogen) at a cell density of 1.33 ⁇ 10 6 cells/mL and seeded in flasks. Plasmids were introduced by the lipofection method. Biotin was added to a final concentration of 100 ⁇ M 3 hours after the introduction of the plasmid, and cultured in a CO 2 incubator (37°C, 8% CO 2 , 125 rpm) for 4 days. The antigen was purified in Absorbance at 280 nm of the purified antigen solution was measured using a spectrophotometer.
- the concentration of the purified antigen was calculated from the measured values obtained using the extinction coefficient calculated by the PACE method (Protein Science (1995) 4, 2411-2423).
- mCTLA4-His (Sino Biologicals Inc. 50503-M08H, Accession No. NP_033973.2), in which a His tag is fused to the extracellular region of mCTLA4, and Abatacept (Alfresa Corporation), in which a human IgG1 constant region is fused to hCTLA4, Biotinylated by amine coupling method (PIERCE Cat.No.21329).
- Antibodies whose binding activity to mouse CTLA4 extracellular region (mCTLA4) changes in the presence and absence of small molecules were screened from the constructed naive human antibody phage display library. That is, phages displaying antibodies exhibiting binding activity to mCTLA4 captured on beads in the presence of small molecules were collected. Phages were recovered from the phage eluate eluted from the beads in the absence of small molecules. In this acquisition method, biotin-labeled mCTLA4 (mCTLA4-His-Biotin) was used as an antigen.
- the phage produced from E. coli retaining the constructed phage display phagemid was purified by a general method. After that, a phage library solution dialyzed against TBS was obtained. Panning with antigen immobilized on magnetic beads was performed. NeutrAvidin coated beads (Sera-Mag SpeedBeads NeutrAvidin-coated) or Streptavidin coated beads (Dynabeads M-280 Streptavidin) were used as magnetic beads.
- Purified phage with TBS added was subjected to ELISA according to the following procedure. StreptaWell 96 microtiter plates (Roche) were coated overnight with 100 ⁇ L TBS containing mCTLA4-His-Biotin. After removing mCTLA4-His-Biotin not bound to the plate by washing each well of the plate with TBST, the wells were blocked with 250 ⁇ L of 2% skimmed milk-TBS for 1 hour or longer. 2% skimmed milk-TBS was removed, and the plate, to which the prepared purified phage was added to each well, was then allowed to stand at room temperature for 1 hour, so that mCTLA4-His- Biotin was allowed to bind in the absence or presence of ATP.
- clones with an absorbance higher than 0.2 in the presence of ATP are determined to be positive, and clones with an absorbance ratio higher than 2 in the presence/absence of ATP have ATP-dependent antigen-binding ability (switch clones).
- switch clones a small molecule such as ATP.
- Phages were produced by a general method from E. coli harboring the constructed phage display phagemid.
- a phage library solution was obtained by diluting the phage population precipitated by adding 2.5 M NaCl/10% PEG to the E. coli culture medium in which the phage production was performed with TBS.
- BSA was added to the phage library solution to a final concentration of 4%.
- Panning was performed with antigen immobilized on magnetic beads.
- NeutrAvidin coated beads Sera-Mag SpeedBeads NeutrAvidin-coated
- Streptavidin coated beads Dynabeads M-280 Streptavidin
- As antigen biotinylated Abatacept-Biotin was used.
- the NucleoFast 96 with 100 ⁇ L of H 2 O added to each well was washed again by centrifugation (4,500 g, 30 minutes). Finally, 100 ⁇ L of TBS was added, and the phage liquid contained in the supernatant of each well of the NucleoFast 96 was collected at room temperature for 5 minutes.
- Purified phages added with TBS or TBS containing ATP or its metabolites were subjected to ELISA by the following procedure.
- a StreptaWell 96 microtiter plate (Roche) was coated overnight with 100 ⁇ L of TBS containing the biotin-labeled antigen (Abatacept-Biotin) prepared in Reference Example 1-1. After removing free Abatacept-Biotin by washing each well of the plate with TBST, the wells were blocked with 250 ⁇ L of 2% SkimMilk-TBS for 1 hour or more.
- clones with an absorbance S/N ratio higher than 2 in the presence of ATP or its metabolites are determined positive, and those with an absorbance ratio higher than 2 in the presence/absence of ATP or its metabolites was determined to be a clone (switch clone) having an antigen-binding ability dependent on ATP or its metabolites.
- Antibodies were purified from the culture supernatant after 4 days of culture in a CO 2 incubator (37°C, 8% CO 2 , 90 rpm) using rProtein A SepharoseTM Fast Flow (Amersham Biosciences) by a method known to those skilled in the art. Absorbance at 280 nm of the purified antibody solution was measured using a spectrophotometer. The concentration of the purified antibody was calculated using the extinction coefficient calculated by the PACE method from the measured values obtained (Protein Science (1995) 4, 2411-2423).
- a StreptaWell 96 microtiter plate (Roche) was coated with 100 ⁇ L of TBS containing hCTLA4-His-Biotin for over 1 hour at room temperature. After removing hCTLA4-His-Biotin not bound to the plate by washing each well of the plate with Wash buffer, the wells were blocked with 250 ⁇ L of Blocking Buffer for 1 hour or more. 100 ⁇ L each of purified IgG adjusted to 2.5 ⁇ g/mL in Sample Buffer containing AMP at a final concentration of 1 mM was added to each well from which Blocking Buffer was removed, and the plate was allowed to stand at room temperature for 1 hour.
- Each IgG was allowed to bind to hCTLA4-His-Biotin present in each well. After washing with Wash Buffer containing AMP at a final concentration of 1 mM, HRP-conjugated anti-human IgG antibody (BIOSOURCE) diluted with Sample Buffer was added to each well and the plate was incubated for 1 hour. After washing with Wash Buffer containing each small molecule, the color development reaction of the solution in each well to which TMB single solution (ZYMED) was added was stopped by adding sulfuric acid, and the color development was measured by absorbance at 450 nm. was done. A buffer containing the composition shown in Table 8 was used as the buffer.
- BIOSOURCE HRP-conjugated anti-human IgG antibody
- Table 9 shows the measured results. For the wells where the value overflowed, it was tentatively set to 5.00. All clones ABAM001, ABAM002, ABAM003, ABAM004, ABAM005 and ABAM006 showed significantly lower absorbance in the absence of AMP than in the presence of AMP. From these results, it was confirmed that all clones ABAM001, ABAM002, ABAM003, ABAM004, ABAM005, and ABAM006 have the property that antigen binding changes depending on the presence or absence of small molecules.
- ABAM004 suspended in TBS 500 nM hCTLA4-His-BAP and 10 concentrations of ATP, ADP or AMP diluted by a factor of 4 from 4000 ⁇ M and 2 mM MgCl2 were added. containing solution was injected into each flow cell at a flow rate of 10 ⁇ L/min for 3 minutes. This 3-minute period was defined as the binding phase of hCTLA4-His-BAP, and after the completion of the binding phase, the 2-minute period during which the buffer was switched to the running buffer was defined as the dissociation phase of hCTLA4-His-BAP.
- the regeneration solution was injected for 30 seconds at a flow rate of 30 ⁇ l/min.
- the above was defined as the ABAM004 binding activity measurement cycle.
- the amount of hCTLA-4-His-BAP that interacted with ABAM004 in the binding phase was corrected for the amount of antibody captured.
- Biacore T200 Evaluation Software Version: 2.0 and Microsoft Excel 2013 (Microsoft) were used for data analysis and plotting.
- Figure 5 shows the binding amounts of ABAM004 and hCTLA4-His-BAP in the presence of ATP and its metabolites obtained in this measurement.
- ABAM004 has the property of binding to hCTLA4 using not only ATP but also ATP metabolites as switches. In addition, it was shown to be an antibody with the strongest binding activity especially in the presence of AMP.
- AMP was added to the FACS buffer at final concentrations of 0, 0.4, 4, 40, 200, and 1000 ⁇ M, and this was used as the wash buffer.
- An antibody (Goat F(ab'2) Anti-Human IgG Mouse ads-FITC, Beckman 732598) was added, and allowed to stand again at 4°C for 30 minutes in the dark. After another washing operation, measurement and analysis were performed using a flow cytometer (FACS CyAn TM ADP). The results are shown in FIG.
- a human PBMC solution was prepared. Using a syringe in which 200 ⁇ L of 1000 units/mL heparin solution (Novo Heparin Injection 5,000 Units, Novo Nordisk) was previously injected, 50 mL of peripheral blood was collected from a healthy volunteer (adult male). The peripheral blood diluted 2-fold with PBS (-) was divided into 4 equal parts, and 15 ml of Ficoll-Paque PLUS was pre-filled and centrifuged in Leucosep lymphocyte separation tubes (Greiner bio-one ). The separation tube into which the peripheral blood was dispensed was centrifuged at room temperature for 10 minutes at a speed of 2150 rpm, and then the mononuclear cell fraction layer was collected.
- heparin solution Novo Heparin Injection 5,000 Units, Novo Nordisk
- the cells After washing the cells contained in the fraction layer once with RPMI-1640 (Nacalai Tesque) containing 10% FBS (hereinafter referred to as 10% FBS/RPMI), the cells were diluted with 10% FBS/RPMI. Suspended to a cell density of 1 ⁇ 10 7 cells/mL. The cell suspension was used as a human PBMC solution for subsequent experiments.
- RPMI-1640 Nacalai Tesque
- 10% FBS/RPMI 10% FBS/RPMI
- hCTLA4-CHO cells in which the human CTLA4 extracellular domain was forcedly expressed in CHO cells as target cells were suspended in 10% FBS/RPMI at a concentration of 2 ⁇ 10 5 cells/mL. Furthermore, AMP (sigma) diluted to 4 mM using RPMI was used as an AMP solution for subsequent tests.
- ADCC activity was assessed by LDH (lactate dehydrogenase) release.
- 50 ⁇ L of an antibody solution prepared at each concentration (0, 0.04, 0.4, 4, 40 ⁇ g/mL) was added to each well of a 96-well U-bottom plate, and 50 ⁇ L of target cells were seeded ( 1 ⁇ 10 4 cells/well).
- 50 ⁇ l of AMP solution was added to each plate and allowed to stand at room temperature for 15 minutes.
- A represents the average value of LDH activity (OD492nm) in wells to which each test antibody was added.
- B represents the average value of LDH activity (OD492 nm) in wells to which 10 ⁇ L of 20% Triton-X aqueous solution was added to wells after reaction.
- C represents the average value of LDH activity (OD492nm) in wells in which target cells were added with 150 ⁇ L of 10% FBS/RPMI or 100 ⁇ L of 10% FBS/RPMI and 50 ⁇ L of AMP solution.
- D represents the mean value of LDH activity (OD492nm) in wells containing only 10% FBS/RPMI. The test was performed in duplicate, and the average ADCC activity (%) in the test reflecting the ADCC activity of the test antibody was calculated. The results are shown in FIG.
- the antibody ABAM004 has antigen-binding activity in the presence of AMP and has the ability to kill target cells by exhibiting ADCC activity.
- the structure was determined by molecular replacement using the program Phaser (J. Appl. Cryst. (2007) 40, 658-674).
- the Fab fragment search model was derived from the published crystal structure of Fab (PDB code: 4NKI).
- the model was built with the Coot program (Acta Cryst. D66: 486-501 (2010)) and refined with the program Refmac5 (Acta Cryst. D67: 355-467 (2011)).
- the crystallographic reliability factor (R) of the diffraction intensity data from 52.92-1.70 ⁇ was 16.92% and the Free R value was 21.22%. Structure refinement statistics are shown in Table 10.
- the structure was determined by molecular replacement using the program Phaser (J. Appl. Cryst. (2007) 40, 658-674).
- the Fab fragment search model was derived from the published crystal structure of Fab (PDB code: 4NKI).
- the model was built with the Coot program (Acta Cryst. D66: 486-501 (2010)) and refined with the program Refmac5 (Acta Cryst. D67: 355-367 (2011)).
- the crystallographic reliability factor (R) of the diffraction intensity data from 47.33-2.89 ⁇ was 19.97% and the Free R value was 25.62%. Structure refinement statistics are shown in Table 10.
- hCTLA4 Extracellular Domain The extracellular domain of hCTLA4 was obtained by restriction digestion of abatacept with Endoproteinase Lys-C (Roche, Catalog No. 11047825001) followed by protein A column (MabSelect) to remove the Fc fragment. SuRe, GE Healthcare) and loading on a gel filtration column (Superdex200 10/300, GE Healthcare). Fractions containing the extracellular domain of hCTLA4 were pooled and stored at -80°C.
- the structure was determined by molecular replacement using the program Phaser (J. Appl. Cryst. (2007) 40, 658-674).
- the Fab fragment search model was derived from the published Fab crystal structure (PDB code: 4NKI) and the hCTLA4 extracellular domain search model was derived from the published human CTLA4 crystal structure (PDB code: 3OSK, J. Biol. Chem. 286: 6685-6696 (2011)).
- the model was built with the Coot program (Acta Cryst. D66: 486-501 (2010)) and refined with the program Refmac5 (Acta Cryst. D67: 355-367 (2011)).
- the crystallographic reliability factor (R) of the diffraction intensity data from 52.81-3.09 ⁇ was 23.49% and the Free R value was 31.02%. Structure refinement statistics are shown in Table 10.
- the adenine ring portion of AMP includes T33 belonging to the heavy chain CDR1 of the antibody, Y95 belonging to CDR3, L100B and W100C side chains, and G96 and M100A main chains.
- the main chain carbonyl oxygens of G96 and M100A form a hydrogen bond with the N at the 6-position of AMP
- the main chain amide NH group of W100C forms a hydrogen bond with the N at the 1-position
- Y95, L100B, and W100C It was found that the antibody strongly recognizes the adenine ring portion by forming an interaction using the pi electrons of the adenine ring portion in the side chain.
- the ribose moiety is recognized by the side chains of T33 belonging to heavy chain CDR1, Y56 belonging to CDR2, and Y58 through van der Waals interactions and interactions with pi electrons possessed by Y56.
- the phosphate group portion is recognized by the side chains of T33 belonging to heavy chain CDR1, S52, S52A and R53 belonging to CDR2, and the main chain of S52A.
- the hydrogen bond formed by the side chain of T33 and the backbone amide NH group of S52A with the phosphate moiety and the van der Waals interaction between S52 and R53 play an important role in the recognition of the phosphate moiety. It is thought that there are Note that the amino acid residue numbering of Fabs is based on the Kabat numbering scheme.
- the epitopes of the ABAM004 Fab contact region are mapped in the hCTLA4 crystal structure and amino acid sequence, respectively.
- Epitopes include those amino acid residues of hCTLA4 that contain one or more non-hydrogen atoms located within 4.2 ⁇ of either portion of the ABAM004 Fab or AMP in the crystal structure.
- At least antigens M3, E33, R35, T53, E97, M99, Y100, P101, P102, P103, Y104, Y105, L106 are recognized by heavy chain CDR2, CDR3, light chain CDR1, CDR3 and AMP of the antibody. It was revealed from the crystal structure that In particular, the loop consisting of M99 to Y104 of the antigen is strongly recognized by the antibody as if buried in the CDR loop of the antibody, and is thought to play a major role in antigen recognition by the antibody.
- AMP-dependent antigen binding mechanism Fig. 11 shows the crystal structure of ABAM004 Fab alone, the crystal structure of the complex of ABAM004 Fab and AMP, and the crystal of the tertiary complex composed of ABAM004 Fab, AMP and CTLA4. It is a drawing in which the variable region of the antibody is extracted from the structure and superimposed around the heavy chain.
- AMP-dependent antigen binding not only the direct interaction between AMP and CTLA4 shown in Reference Example 2-14 but also the conformational change of the antibody associated with AMP binding is considered to be important.
- the point mutants were measured for human CTLA4 (Abatacept and hCTLA4-His-BAP) binding activity in the absence of ATP and in the presence of ATP, ADP, or AMP using Biacore T200 or Biacore 4000 (GE Healthcare), and the binding activity was improved.
- were screened for mutations that cause Mutants were prepared by combining mutations with improved binding activity, and KD values were calculated using Biacore.
- H32A, S52aT for the heavy chain of ABAM004 and T24D, T26P, E50F for the light chain (the numbers are Kabat numbers) improved the binding properties of ABAM004.
- This variant is called 04H0150/04L0072 (VH SEQ ID NO: 47, VL SEQ ID NO: 48).
- Protein A/G (Pierce) suspended in 10 mM sodium acetate pH 4.0 was added at 10 ⁇ L/min for 30 minutes to bind. After that, excess active groups on the flow cells were blocked by adding 1 M ethanolamine-HCl at 10 ⁇ L/min for 10 minutes.
- the set temperature was 25°C and TBS was used as the running buffer. 10 mM Glycine-HCl (pH 1.5) was used as the regeneration solution. After the antibody suspended in TBS was captured, a TBS solution containing 500 nM hCTLA4-His-BAP, 10 concentrations of ATP, ADP or AMP diluted from 4000 ⁇ M with a common ratio of 4, and 2 mM MgCl was added to each It was injected into the flow cell at a flow rate of 10 ⁇ L/min for 3 minutes. This 3 minute period was defined as the hCTLA4-His-BAP binding phase.
- the dissociation phase was set for 2 minutes after switching to the running buffer.
- the regeneration solution was injected at a flow rate of 30 ⁇ L/min for 30 seconds, and the above was defined as the binding activity measurement cycle.
- the amount of hCTLA4-His-BAP that interacted with ABAM004 and 04H0150/04L0072 in the binding phase, corrected for the amount of antibody captured, is shown in FIG.
- the small molecule concentration in the binding phase was kept at 62.5 ⁇ M or 1 mM, and 8 concentrations of hCTLA4-His-BAP diluted by a factor of 2 from 2000 nM were used in the binding phase.
- Table 11 shows the KD values obtained from the hCTLA4-His-BAP binding amount analysis.
- Biacore T200 Evaluation Software Version: 2.0 and Microsoft Excel 2013 (Microsoft) were used for data analysis and plotting.
- a steady state affinity model was used to calculate the KD value.
- SM represents the small molecule (ATP, ADP or AMP) used in each assay.
- a human CTLA4 solution with or without a small molecule (ATP, ADP, or AMP) was allowed to interact with the antibody in the presence and absence of the small molecule.
- the ability to bind to human CTLA4 was evaluated. Measurement was performed at 25°C using Tris buffered saline, 0.02% PS20 as a running buffer.
- a combination of a modification that enhances binding to human CTLA4 in the presence of a small molecule and a modification that reduces binding to human CTLA4 in the absence of a small molecule, found using the above method, yields superior A profiled anti-human CTLA4 antibody was generated.
- Gene of antibody heavy chain 04H0150-G1m (SEQ ID NO: 209) having 04H0150 as the heavy chain variable region and G1m (SEQ ID NO: 82) from which Gly and Lys at the C-terminal of human IgG1 are removed as the heavy chain constant region
- Antibody heavy chain genes were constructed by combining modifications found by comprehensive modification introduction and modifications to Framework.
- the gene of antibody heavy chain MDX10D1H-G1m (SEQ ID NO: 210) having the heavy chain variable region MDX10D1H (SEQ ID NO: 154) of the existing anti-human CTLA4 antibody described in WO0114424 and the light chain variable region MDX10D1L (SEQ ID NO: 210)
- the gene for the antibody light chain MDX10D1L-k0MT (SEQ ID NO:211) with SEQ ID NO:155) was generated.
- antibodies were expressed and purified by methods known to those skilled in the art to produce the desired anti-CTLA4 antibodies.
- Table 12 is a list of sequence numbers of heavy chain variable regions, light chain variable regions, heavy chain constant regions, light chain constant regions, and hyper variable regions of antibodies produced.
- the antibodies herein are named according to the following convention: (heavy chain variable region)-(heavy chain constant region)/(light chain variable region)-(light chain constant region). For example, if the antibody name is 04H0150-G1m/04L0072-lam1, the heavy chain variable region of this antibody is 04H0150, the heavy chain constant region is G1m, the light chain variable region is 04L0072, and the light chain constant region is lam1. means.
- Binding assays of the produced antibodies to human CTLA4 were performed using a Biacore T200. 20 mM ACES (pH 7.4), 150 mM NaCl, 2 mM MgCl 2 , 0.05% Tween20 with ATP added to the desired concentration was used as the running buffer, and the measurement was performed at 37°C. . First, antibodies were captured by allowing an antibody solution prepared in a running buffer containing no ATP to interact with Series S Sensor Chip CM3 (GE Healthcare) on which Protein G (CALBIOCHEM) was immobilized.
- Series S Sensor Chip CM3 GE Healthcare
- human CTLA4 solution prepared in running buffer to which ATP was added to achieve the desired concentration, or human CTLA4 solution prepared in running buffer without ATP was allowed to interact with the presence of ATP and The ability of the antibody to bind human CTLA4 in the absence of ATP was evaluated.
- the chip was regenerated with 25 mM NaOH and 10 mM Glycine-HCl (pH 1.5), and repeated antibody captures and measurements were performed.
- the dissociation constant for CTLA4 of each antibody was calculated using Biacore T200 Evaluation Software 2.0. Specifically, the binding rate constant ka (L/mol/s) and the dissociation rate constant kd (1/s) were calculated by global fitting the sensorgram obtained by the measurement with a 1:1 Langmuir binding model.
- the dissociation constant KD (mol/L) was calculated from the value.
- the dissociation constant KD (mol/L) was calculated using a steady state model.
- the amount of CTLA4 bound per unit amount of antibody was calculated by correcting the amount of CTLA4 binding determined from the sensorgram obtained by measurement with the amount of antibody captured on the chip surface. Table 13 shows the results of these measurements.
- the value of "binding to human CTLA4" in the table indicates the binding amount of human CTLA4 per unit amount of antibody when interacting with human CTLA4 at 1000 nM under each ATP concentration condition described.
- KD (M) for ” indicates the dissociation constant to human CTLA4 under each ATP concentration condition.
- the KD values marked with * in the table were calculated using the steady state model. All variants prepared using 04H0150-G1m/04L0072-lam1 as a parent antibody showed enhanced binding in the presence of ATP compared to 04H0150-G1m/04L0072-lam1.
- 04H0150-G1m / 04L0072-lam1 and their variants had a higher binding amount in the presence of 10 ⁇ M ATP than in the presence of 1 ⁇ M, and a higher binding amount in the presence of 100 ⁇ M. It was shown to bind to human CTLA4 in an ATP concentration-dependent manner. On the other hand, MDX10D1H-G1m/MDX10D1L-k0MT for comparison did not exhibit such ATP concentration-dependent binding to human CTLA4.
- 04H1077-G1m/04L1305-k0MT in which the light chain framework and constant region of 04H1077-G1m/04L1086-lam1 are replaced with human ⁇ chain, binds to human CTLA4 in the absence of ATP compared to 04H1077-G1m/04L1086-lam1 was enhanced, but ATP concentration-dependent binding was also enhanced. These results indicated that the ATP-dependent binding to human CTLA4 was maintained even after substitution with the human ⁇ chain sequence.
- 04H1077-G1m/04L1066-lam1, 04H1077-G1m/04L1305-k0MT, and 04H1207-G1m/04L1086-lam1 are existing anti-human CTLA4 antibodies in the presence of 100 ⁇ M ATP. It showed almost the same binding activity as MDX10D1H-G1m/MDX10D1L-k0MT, and 04H1208-G1m/04L1407-k0MT showed stronger binding activity than MDX10D1H-G1m/MDX10D1L-k0MT in the presence of 10 ⁇ M or more of ATP.
- Mouse CTLA4 was prepared as follows. A mouse CTLA4 extracellular region ligated with a His tag (mCTLA4-His) (SEQ ID NO: 49) was gene-synthesized and inserted into an animal expression plasmid. Human embryonic kidney cell-derived FreeStyle 293-F strain (Invitrogen) suspended in FreeStyle 293 Expression Medium (Invitrogen) at a cell density of 1.33 ⁇ 10 6 cells/mL and seeded in flasks. Plasmids were introduced by the lipofection method.
- mCTLA4-His His tag
- binding to mouse CTLA4 in the table indicates the binding amount of mouse CTLA4 per unit amount of antibody when interacting with mouse CTLA4 at 1000 nM under each ATP concentration condition described.
- KD (M) for CTLA4" indicates the dissociation constant for mouse CTLA4 under each ATP concentration condition.
- hUH02-G1d/hUL01-k0 binds mouse CTLA4 to the same extent regardless of ATP concentration, whereas both 04H1077-G1m/04L1086-lam1 and 04H1208-G1m/04L1407-k0MT are ATP concentration dependent. was shown to bind to mouse CTLA4.
- Anti-mCTLA4 control antibody (hUH02-mFa55/hUL01-mk1 abbreviation: mNS-mFa55) and anti-CTLA4 switch (04H1077-mFa55/04L1086-ml0r abbreviation: SW1077-mFa55, 04H1208-mFa55/04L1407s-mk1 (abbreviation: SW1208-mFa55) were produced.
- the mNS-mFa55 antibody uses the heavy chain variable region hUH02 (SEQ ID NO: 16) and the light chain variable region hUL01 (SEQ ID NO: 17), and the constant region is the mouse heavy chain constant region mFa55 (SEQ ID NO: 18) and the wild-type Mouse light chain constant region mk1 (SEQ ID NO: 19) was used.
- a mouse heavy chain constant region modified to enhance Fc ⁇ receptor binding was used and expressed and purified by a method known to those skilled in the art.
- the SW1077-mFa55 antibody uses heavy chain variable region 04H1077 (SEQ ID NO: 20) and light chain variable region 04L1086 (SEQ ID NO: 21), and constant regions are mouse heavy chain constant region mFa55 (SEQ ID NO: 18) and wild type Mouse light chain constant region ml0r (SEQ ID NO: 22) was used.
- a mouse heavy chain constant region modified to enhance Fc ⁇ receptor binding was used and expressed and purified by a method known to those skilled in the art.
- the SW1208-mFa55 antibody uses heavy chain variable region 04H1208 (SEQ ID NO: 23) and light chain variable region 04L1407s (SEQ ID NO: 24), and constant regions are mouse heavy chain constant region mFa55 (SEQ ID NO: 18) and wild-type Mouse light chain constant region mk1 (SEQ ID NO: 19) was used.
- a mouse heavy chain constant region modified to enhance Fc ⁇ receptor binding was used and expressed and purified by a method known to those skilled in the art.
- mCD86-Fc-His (Sino Biologics Inc. 50068-M03H, Accession No. NP_062261.3), mouse CD86 fused with human constant region and His tag diluted in TBS to a final concentration of 55 nM, final concentration SW1077-mFa55 antibody solutions diluted to 6.25, 1.56, 0.390, 0.0977, 0.0061, and 0 ⁇ g/mL, and ATP solutions diluted to final concentrations of 0, 1, 10, and 100 ⁇ M, respectively.
- Tumor volume major axis x minor axis x minor axis/2
- the drugs to be administered to the FM3A cell transplantation model are anti-mouse CTLA4 control antibody (mNS-mFa55) and anti-CTLA4 switch antibody (SW1208-mFa55) prepared in Reference Example 3-4. ).
- SW1208-mFa55 was 0.005 mg/mL , 0.05 mg/mL, 0.5 mg/mL, 5 mg/mL, and 25 mg/mL, respectively, using His-buffer (20 mM His-HCl, 150 mM NaCl, pH 6.0).
- mNS-mFa55 was 0.01 mg/kg, 0.1 mg/kg, 0.25 mg/kg, 1 mg/kg, 10 mg/kg , 30 mg/kg, and 100 mg/kg, and SW1208-mFa55 was administered to mice at doses of 0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, and 500 mg/kg, respectively. did. Administration was performed via the tail vein at a dose of 20 mL/kg of the prepared administration solution. Table 15 shows the details of the drug treatments used to measure antitumor efficacy.
- TGI Tumor Growth Inhibition
- Anti-mouse CTLA4 control antibody (mNS-mFa55) at 0.1 mg/kg and 1 mg on day 7 after transplantation for evaluation of Treg cells in tumor and verification of systemic effect in spleen /kg, 10 mg/kg, 100 mg/kg were administered via the tail vein, and anti-CTLA4 switch antibody (SW1208-mFa55) was administered at 0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, It was administered through the tail vein at 500 mg/kg.
- Table 16 provides details regarding drug treatment during evaluation of Treg cells within the tumor and validation of systemic effects in the spleen.
- Anti-CD45 antibody (BD, clone: 30-F11), anti-CD3 antibody (BD, clone: 145-2C11), anti-CD4 antibody (BD, clone: RM4-5), anti-FoxP3 antibody (eBioscience, clone: FJK-16s ), anti-ICOS antibody (eBioscience, clone: 7E17G9), anti-KLRG1 antibody (Biolegend, clone: 2F1/KLRG1). FACS analysis was performed on a BD LSRFortessa X-20 (BD).
- 04H1389-G1m/04L1086- in which R53Q and G55H modifications were introduced into the heavy chain variable regions of 04H1207-G1m/04L1086-lam1 and 04H1208-G1m/04L1086-lam1 prepared in Reference Example 3 lam1 and 04H1382-G1m/04L1086-lam1 were generated.
- 04H1389-G1m/04L1305-k0MT was prepared by replacing the light chain of 04H1389-G1m/04L1086-lam1 with the human ⁇ chain sequence.
- Table 17 lists the SEQ ID NOs of the heavy chain variable regions, light chain variable regions, heavy chain constant regions, light chain constant regions, and Hyper Variable Regions of these antibodies.
- Binding measurements for ATP were performed at 37° C. using 20 mM ACES (pH 7.4), 150 mM NaCl, 2 mM MgCl 2 , 0.05% Tween20 as running buffer.
- Second, the ability to bind to the antibody was evaluated by allowing an ATP solution prepared with a running buffer to interact. The chip was regenerated with 25 mM NaOH and 10 mM Glycine-HCl (pH 1.5) and repeated antibody captures and measurements were performed.
- the binding amount of each antibody to ATP was calculated by correcting the amount of binding when ATP was injected at a concentration of 100 nM with the amount of antibody captured on the chip surface, thereby calculating the amount of ATP binding per unit amount of antibody. rice field. Binding to human CTLA4 was measured using Biacore T200 by the method described in Reference Example 3-3. Table 18 shows the results of these measurements.
- 04H1389-G1m/04L1086-lam1 and 04H1382-G1m/04L1086-lam1 have enhanced ATP-binding ability compared to parent antibodies 04H1207-G1m/04L1086-lam1 and 04H1208-G1m/04L1086-lam1 before introduction of R53Q/G55H It had been.
- the binding abilities of 04H1389-G1m/04L1086-lam1 and 04H1382-G1m/04L1086-lam1 to human CTLA4 were compared with the parental antibodies 04H1207-G1m/04L1086-lam1 and 04H1208-G1m/04L1086-lam1 before introduction of R53Q/G55H.
- 04H1389-G1m/04L1305-k0MT in which the light chain of 04H1389-G1m/04L1086-lam1 is replaced with the human ⁇ chain sequence, also has the same ATP-binding ability as 04H1389-G1m/04L1086-lam1, and ATP-dependent human CTLA4 binding. was shown to have a binding capacity of
- the hNS-mFa55 antibody uses the heavy chain variable region MDX10D1H (SEQ ID NO: 26) and the light chain variable region MDX10D1L (SEQ ID NO: 27), and the constant region is the mouse heavy chain constant region mFa55 (SEQ ID NO: 18) and the wild-type Mouse light chain constant region mk1 (SEQ ID NO: 19) is used.
- a mouse heavy chain constant region modified to enhance Fc ⁇ receptor binding was used and expressed and purified by a method known to those skilled in the art.
- the SW1389-mFa55 antibody uses heavy chain variable region 04H1389 (SEQ ID NO: 29) and light chain variable region 04L1305 (SEQ ID NO: 30), and constant regions are mouse heavy chain constant region mFa55 (SEQ ID NO: 18) and wild type Mouse light chain constant region mk1 (SEQ ID NO: 19) is used.
- a mouse heavy chain constant region modified to enhance Fc ⁇ receptor binding was used and expressed and purified by a method known to those skilled in the art.
- the binding of the produced hNS-mFa55 and SW1389-mFa55 to human CTLA4 was evaluated.
- 20 mM ACES (pH 7.4), 150 mM NaCl, 2 mM MgCl 2 , 0.05% Tween20 with ATP added to the desired concentration was used as the running buffer, and the measurement was performed at 37°C. .
- SW1389-mFa55 was also shown to bind human CTLA4 in an ATP-dependent manner comparable to 04H1389-G1m/04L1305-k0MT made with the same variable regions and human constant regions listed in Table 18.
- Tumor volume major axis x minor axis x minor axis/2
- TGI tumor growth inhibition
- both hNS-mFa55 and SW1389-mFa55 showed efficacy with a TGI of 60 or higher 18 days after administration at a dose of 1 mg/kg or higher ( Figures 18 and 19).
- anti-CD45 antibody (BD, clone: 30-F11), anti-CD3 antibody (BD, clone: UCHT1), anti-CD4 antibody (BD, clone: RM4-5), anti-FoxP3 antibody (eBioscience, clone: FJK-16s), Anti-ICOS antibody (eBioscience, clone: 7E17G9), anti-CCR7 antibody (Biolegend, clone: 4B12), anti-KLRG1 antibody (Biolegend, clone: 2F1/KLRG1). FACS analysis was performed on a BD LSRFortessa X-20 (BD).
- Light chains 04L1594-lam1, 04L1581-lam1, 04L1610-lam1, 04L1612-lam1, and 04L1610-lam1 in Table 22 have been introduced with CDR and framework modifications to the light chain 04L1086-lam1 of the parent antibody. It has a ⁇ chain germline sequence framework and a constant region.
- 04L1615-k0MT, 04L1616-k0MT, and 04L1617-k0MT have CDR modifications introduced to 04L1086-lam1 and have the framework and constant region of the germline sequence of human ⁇ chain.
- Heavy chain variable region 04H1389v373 is a parent antibody heavy chain variable region 04H1389 CDR modified, 04H1637, 04H1643, 04H1654, 04H1656, 04H1642, 04H1735 are modified CDRs of 04H1389, Framework A heavy chain variable region in which the sequence is replaced with a different germline.
- the KD values marked with * in the table were calculated using a steady state model. All variants prepared using 04H1389-G1m/04L1086-lam1 as a parent antibody bind to human CTLA4 in an ATP-dependent manner and exhibit a KD of 3.7 ⁇ 10 ⁇ 8 M in the presence of 10 ⁇ M ATP. It was shown to have stronger binding ability than antibody. In addition, all of these antibodies were shown to have a stronger binding ability than the existing anti-human CTLA4 antibody MDX10D1H-G1m/MDX10D1L-k0MT in the presence of 10 ⁇ M ATP.
- the binding ability of the prepared variants to human CTLA4 in the presence of ADP and AMP was evaluated using the Biacore T200 and compared with the binding ability in the presence of ATP.
- the binding ability to human CTLA4 in the presence of ADP and AMP was performed using the method described in Reference Example 3-3, as in the evaluation of the binding ability in the presence of ATP, and ADP was used instead of ATP. , or with AMP (Table 24).
- ATP-dependent anti-CTLA4 antibodies also binds to human CTLA4 not only in the presence of ATP, but also in the presence of ADP and AMP. it was high. Therefore, these antibodies were shown to bind to CTLA4 in an ATP-, ADP-, and AMP-dependent manner. These antibodies had the highest binding capacity in the presence of ATP, followed by the highest binding capacity in the presence of ADP, and the lowest binding capacity in the presence of AMP.
- the binding ability in the presence of ADP was about 3 times stronger than the binding ability in the presence of AMP, and the binding ability in the presence of ATP was about 5 times stronger than the binding ability in the presence of AMP.
- the ka values were similar in the presence of all small molecules, but the kd values differed. dissociation was rapid, indicating that the difference in KD values for different small molecules was due to the difference in dissociation rates.
- Cynomolgus CTLA4 was prepared as follows. CyCTLA4-His-BAP (SEQ ID NO: 50), in which a His tag and a BAP tag are fused to the C-terminus of the cynomolgus CTLA4 extracellular region, was gene-synthesized and inserted into an animal expression plasmid.
- the SW1615-mFa55 antibody uses heavy chain variable region 04H1389 (SEQ ID NO: 29) and light chain variable region 04L1615 (SEQ ID NO: 34), and the constant regions are mouse heavy chain constant region mFa55 (SEQ ID NO: 18) and wild-type Mouse light chain constant region mk1 (SEQ ID NO: 19) is used.
- a mouse heavy chain constant region modified to enhance Fc ⁇ receptor binding was used and expressed and purified by a method known to those skilled in the art.
- one heavy chain variable region 04H1654 (SEQ ID NO: 35) has a mouse heavy chain constant region mFa55m2P1 (SEQ ID NO: 36) as a constant region, and the other heavy chain variable region 04H1656 (SEQ ID NO: 37 ) linked the mouse heavy chain constant region mFa55m2N1 (SEQ ID NO: 38), and the light chain variable region 04L1610 (SEQ ID NO: 39) used the wild-type mouse light chain constant region ml0r (SEQ ID NO: 22). It was expressed and purified by a method known to the trader.
- one heavy chain variable region 04H1654 (SEQ ID NO: 35) has a mouse heavy chain constant region mFa55m2P1 (SEQ ID NO: 36) as a constant region, and the other heavy chain variable region 04H1656 (SEQ ID NO: 37 ) linked the mouse heavy chain constant region mFa55m2N1 (SEQ ID NO: 38), and the light chain variable region 04L1612 (SEQ ID NO: 40) used the wild-type mouse light chain constant region ml0r (SEQ ID NO: 22). It was expressed and purified by a method known to the trader.
- the heavy chain variable region IC17Hdk (SEQ ID NO: 51) was linked to the mouse heavy chain constant region mFa55 (SEQ ID NO: 18) as a constant region, and the light chain variable region IC17L (SEQ ID NO: 52) was wild-type.
- type mouse light chain constant region mk1 (SEQ ID NO: 19) was used and expressed and purified by methods known to those skilled in the art.
- Tumor volume major axis x minor axis x minor axis/2
- the drug to be administered to the Hepa1-6/hGPC3 cell transplantation model is the anti-CTLA4 switch antibody prepared in Reference Example 5-2 (SW1610-mFa55, SW1612-mFa55, SW1615- mFa55).
- the administered drug was prepared using His-buffer (20 mM His-HCl, 150 mM NaCl, pH 6.0) so as to be 0.03 mg/mL, 0.1 mg/mL and 0.3 mg/mL.
- TGI Tumor Growth Inhibition
- anti-CD45 antibody (BD, clone: 30-F11), anti-CD3 antibody (BD, clone: UCHT1), anti-CD4 antibody (BD, clone: RM4-5), anti-FoxP3 antibody (eBioscience, clone: FJK-16s), Anti-ICOS antibody (eBioscience, clone: 7E17G9), anti-CCR7 antibody (Biolegend, clone: 4B12), anti-KLRG1 antibody (Biolegend, clone: 2F1/KLRG1). FACS analysis was performed on a BD LSRFortessa X-20 (BD).
- G1d (SEQ ID NO: 158) having 04H1637 (SEQ ID NO: 138) as one heavy chain variable region and having C-terminal Gly and Lys removed from the human IgG1 heavy chain constant region
- L234Y / L235Q / G236W / S239M / H268D / D270E / S298A are introduced, and the antibody heavy chain 04H1637-Kn125 (SEQ ID NO: 162) gene having a modification Y349C / T366W that promotes heterodimerization in the CH3 region is created. was done.
- 04L1610-lam1 (SEQ ID NO: 161) as the antibody light chain
- a heterodimer 04H1637-Kn125/04L1610-lam1//04H1637-Hl076/04L1610-lam1 was prepared by a method known to those skilled in the art.
- L235Q, G236W, S239M, H268D, D270E, S298A, K326D, in addition to K334E, L234F reported as a modification that changes the binding to Fc ⁇ R in WO2013 / 002362 , A330K, Mol.
- the C-terminal Gly and Lys of human IgG1 are removed, the CH2 region has the same modification as Kn462, and the CH3 region has the modification E356K that promotes heterodimerization described in WO2006/106905.
- a gene for antibody heavy chain 04H1654-KT462 SEQ ID NO: 182 having 04H1654 (SEQ ID NO: 140) as the heavy chain variable region was constructed.
- the C-terminal Gly and Lys of human IgG1 (IGHG1 * 03) are removed, the CH2 region has the same modification as Hl441, and the CH3 region has the modification K439E that promotes heterodimerization described in WO2006/106905.
- a gene for antibody heavy chain 04H1656-HT441 (SEQ ID NO: 170) was constructed having 04H1656 (SEQ ID NO: 141) as the heavy chain variable region.
- 04H1656-HT445 (SEQ ID NO: 171)
- 04H1654-KT461 (SEQ ID NO: 183)
- 04H1656-HT443 (SEQ ID NO: 173) genes were constructed.
- combinations of modifications that improve blood kinetics of antibodies described in Mabs, 2017, 9, 844-853 were investigated.
- N434A / Y436T / a combination of modifications that enhance binding to human FcRn under acidic conditions and modifications that reduce binding to Rheumatoid factor
- a gene of 04H1654-KT473 (SEQ ID NO: 184) into which Q438R/S440E was introduced was constructed.
- a gene of 04H1656-HT482 (SEQ ID NO: 185) was prepared by introducing N434A/Y436T/Q438R/S440E into 04H1656-HT445 (SEQ ID NO: 171).
- antibody heavy chain 04H1654-KT481 (SEQ ID NO: 186) and antibody heavy chain 04H1656-HT498 (SEQ ID NO: 187) were prepared by introducing the same modification into 04H1654-KT461 and 04H1656-HT443. These heavy chains were combined and 04L1610-lam1 or 04L1612-lam1 (SEQ ID NO: 188) was used as the light chain to prepare the desired heterodimeric antibody.
- the extracellular domain of Fc ⁇ R was prepared by the following method. First, the Fc ⁇ R extracellular domain gene was synthesized by a method known to those skilled in the art. At that time, the sequence of each Fc ⁇ R was produced based on the information registered with NCBI. Specifically, the sequence of NCBI accession # NM_000566.3 for Fc ⁇ RI, the sequence of NCBI accession # NM_001136219.1 for Fc ⁇ RIIa, the sequence of NCBI accession # NM_004001.3 for Fc ⁇ RIIb, and the NCBI accession for Fc ⁇ RIIIa # Built based on the sequence of NM_001127593.1 with a C-terminal His tag added.
- 1st step is cation exchange column chromatography (SP Sepharose FF)
- 2nd step is affinity column chromatography against His tag (HisTrap HP)
- 3rd step is gel filtration column chromatography (Superdex200)
- 4th step is sterile Filtration was performed.
- anion exchange column chromatography using Q sepharose FF was performed in the first step.
- the concentration of the purified protein was calculated by measuring the absorbance at 280 nm using a spectrophotometer and using the extinction coefficient calculated from the obtained value by a method such as PACE (Protein Science, 1995 , 4, 2411-2423).
- Human FcRn was prepared by the method described in WO2010/107110.
- the chip was regenerated with 10 mM Glycine-HCl (pH 1.5) and 25 mM NaOH, and repeated antibody captures and measurements were performed.
- the dissociation constant KD (mol/L) for Fc ⁇ R of each antibody was calculated using Biacore T200 Evaluation Software 2.0, the dissociation constant for Fc ⁇ RIa and Fc ⁇ RIIIa was calculated using the 1:1 Langmuir binding model, and the dissociation constant for Fc ⁇ RIIa was calculated using the Steady state affinity model. was done.
- Fc ⁇ RIIb the amount of Fc ⁇ RIIb bound per unit amount of antibody was calculated by correcting the amount of binding of Fc ⁇ RIIb determined from the sensorgram obtained by measurement with the amount of antibody captured on the chip surface.
- KD (M) for hFcRn and “KD (M) for hFc ⁇ Rs” in the table respectively indicate the dissociation constants for hFcRn and each Fc ⁇ R, and the "binding amount” is when Fc ⁇ RIIb is allowed to interact at 1000 nM shows the binding amount of Fc ⁇ RIIb per unit amount of antibody.
- G1m and hFcRn relative to KD and "G1m and hFc ⁇ Rs relative to KD” are the values obtained by dividing the KD value of 04H1637-G1m/04L1610-lam1 for hFcRn and each Fc ⁇ R by the KD value of each variant
- “relative binding amount” indicates the value obtained by dividing the binding amount of each variant to Fc ⁇ RIIb by the binding amount of 04H1637-G1m/04L1610-lam1.
- the amino acid sequences of antibody heavy chain 04H1637-G1m and antibody light chain 04L1610-lam1 are shown in SEQ ID NOs: 160 and 161, respectively.
- 04H1656-HT441 (sequence No.: 272) gene was constructed.
- the amino acid sequence of antibody heavy chain HT451 is shown in SEQ ID NO:276.
- a heterodimeric antibody was produced by combining 04H1654-KT473 and 04H1656-HT451 and using 04L1610-lam1 as the antibody light chain.
- Table 30 shows the results of interaction analysis between the prepared antibodies and human FcRn and human Fc ⁇ R.
- the generated heterodimeric antibodies 04H1654-KT462/04L1610-lam1//04H1656-HT441/04L1610-lam1 and 04H1654-KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1 are both It was shown that binding to activating Fc ⁇ Rs Fc ⁇ RIIa and Fc ⁇ RIIIa was enhanced compared to 04H1656-G1m/04L1610-lam1 having a constant region. In addition, all of these antibodies maintained binding to the inhibitory Fc ⁇ R Fc ⁇ RIIb to the same extent as 04H1656-G1m/04L1610-lam1.
- 04H1654-KT462/04L1610-lam1//04H1656-HT441/04L1610-lam1 introduced N434A/Y436T/Q438R/S440E for 04H1654-KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1 before and It was shown that the binding ability to human FcRn was enhanced in comparison.
- Antibody heavy chain 04H1389-Ks462 (SEQ ID NO: 191) in which T366W was introduced as a heterodimerization modification in the CH3 region, and 04H1389-Km462 (SEQ ID NO: 199) in which Y349C/T366W was used as a heterodimerization modification gene was created.
- Antibody heavy chain 04H1389-Ks473 (SEQ ID NO: 195), 04H1389-Hs482 (SEQ ID NO: 196), 04H1389-Ks481 (SEQ ID NO: 197), 04H1389-Hs498 (SEQ ID NO: 198), 04H1389-Km473 (SEQ ID NO: 203), 04H1389-Hm482 (SEQ ID NO: 204), 04H1389-Km481 (SEQ ID NO: 205), and 04H1389-Hm498 (SEQ ID NO: 206) were constructed. 04L1615-k0MT (SEQ ID NO: 190) was used as the light chain to generate the desired heterodimer.
- a homodimer 04H1389-G1m/04L16150k0MT having 04H1389-G1m was also prepared for comparison.
- Interaction analysis between the produced antibody and human Fc ⁇ R was performed using Biacore T200. 50 mM Na-Phosphate, 150 mM NaCl, 0.05% Tween20 (pH 7.4) was used as running buffer, and measurement was performed at 25°C.
- the sensor chip used was Series S SA (GE Healthcare) with CaptureSelect Human Fab-kappa Kinetics Biotin Conjugate (Thermo Fisher Scientific) immobilized thereon. Antibodies of interest were captured on this chip and allowed to interact with each Fc ⁇ R diluted in running buffer.
- the chip was regenerated with 10 mM Glycine-HCl (pH 1.5) and 25 mM NaOH, and repeated antibody captures and measurements were performed.
- the dissociation constant KD (mol/L) for Fc ⁇ R of each antibody was calculated using Biacore T200 Evaluation Software 2.0, the dissociation constant for Fc ⁇ RIa and Fc ⁇ RIIIa was calculated using the 1:1 Langmuir binding model, and the dissociation constant for Fc ⁇ RIIa was calculated using the Steady state affinity model. was done.
- Fc ⁇ RIIb the amount of Fc ⁇ RIIb bound per unit amount of antibody was calculated by correcting the amount of binding of Fc ⁇ RIIb determined from the sensorgram obtained by measurement with the amount of antibody captured on the chip surface.
- FcRn binding was measured using 50 mM Na-Phosphate, 150 mM NaCl, 0.05% Tween20 (pH 6.0) as a running buffer, and the dissociation constant KD (mol/L) was calculated using a steady state model (Table 31 ).
- the values indicated with "*" are values calculated by the Steady state affinity model.
- All of the heterodimeric antibodies produced here had enhanced binding activity to Fc ⁇ RIIa and Fc ⁇ RIIIa compared to 04H1389-G1m/04L1615-k0MT.
- 04H1389-KS473/04L1615-K0MT // 04H1389-HS482/04L1615-K0MT
- 04H1389-KS481/04L1615-K0MT // K0MT // 04H1389-HS4H1389-K04L1615-K0MT
- Km473/04L1615-k0MT//04H1389-Hm482/04L1615-k0MT and 04H1389-Km481/04L1615-k0MT//04H1389-Hm498/04L1615-k0MT bind to human FcRn compared to the parental antibody before modification It had enhanced activity and a binding profile similar to that of the parent antibody in terms of Fc ⁇ R binding activity.
- 04H1389 (SEQ ID NO: 136) as a heavy chain variable region, the same modification as HT441 was introduced into the heavy chain CH2 region, and Y349C / T366W was used as a heterodimerization modification in the CH3 region 04H1389-Hm441 ( SEQ ID NO: 273) was generated.
- N434A / Y436T / Q438R / S440E which is a combination of modifications that enhance binding to human FcRn under acidic conditions and modifications that reduce binding to Rheumatoid factor, was introduced into 04H1389-Hm451 (sequence Number: 274).
- the amino acid sequences of antibody heavy chains Hm441 and Hm451 are shown in SEQ ID NOs: 277 and 278, respectively.
- a heterodimeric antibody was prepared using 04H1389-Km473, 04H1389-Hm451 or 04H1389-Hm482 as the antibody heavy chain and 04L1305-k0MT as the antibody light chain.
- Table 32 shows the results of interaction analysis between the prepared antibodies and human FcRn and human Fc ⁇ R.
- the generated heterodimeric antibodies 04H1389-Km473/04L1305-k0MT//04H1389-Hm451/04L1305-k0MT and 04H1389-Km473/04L1305-k0MT//04H1389-Hm482/04L1305-k0MT are both It was shown that binding to activating Fc ⁇ Rs Fc ⁇ RIIa and Fc ⁇ RIIIa was enhanced compared to 04H1389-G1m/04L1305-k0MT having a constant region. All of these antibodies were also shown to have enhanced human FcRn-binding ability compared to 04H1389-G1m/04L1305-k0MT.
- Antibody heavy chain MDX10D1H-Kn125 (SEQ ID NO: 217), MDX10D1H-Hl076 (SEQ ID NO: 218) having MDX10D1H (SEQ ID NO: 154) as a heavy chain variable region and heavy chain constant regions listed in Table 29 , MDX10D1H-Kn462 (SEQ ID NO: 219), MDX10D1H-Hl445 (SEQ ID NO: 220), MDX10D1H-Kn461 (SEQ ID NO: 221), MDX10D1H-Hl443 (SEQ ID NO: 222), and the CH2 region of native human IgG1 A gene for MDX10D1H-G1m (SEQ ID NO: 210) was generated.
- Antibody heavy chain MDX10D1H-GASDIE (SEQ ID NO:: 215) genes were generated. Also, as a variant with enhanced binding to Fc ⁇ RIIIa, an antibody heavy chain MDX10D1H-GASDALIE ( SEQ ID NO: 216) was constructed. MDX10D1L-k0MT (SEQ ID NO: 211) was used as the antibody light chain to produce the antibody of interest. These human Fc ⁇ R-binding activities were measured by the method using the aforementioned CaptureSelect Human Fab-kappa Kinetics Biotin Conjugate (Table 33).
- KD (M) for hFc ⁇ R indicates the dissociation constant for each Fc ⁇ R listed, and the "binding amount” is Fc ⁇ RIIb per unit amount of antibody when interacting with Fc ⁇ RIIb at 1000 nM. Indicates the amount of binding.
- Relative value for KD of G1m and hFc ⁇ Rs shows the value obtained by dividing the KD value of MDX10D1H-G1m / MDX10D1L-k0MT for each Fc ⁇ R by the KD value of each variant
- “relative binding amount” is each variant for Fc ⁇ RIIb The value obtained by dividing the binding amount of MDX10D1H-G1m/MDX10D1L-k0MT by the binding amount is shown.
- the generated heterodimers MDX10D1H-Kn125/MDX10D1H-Hl076/MDX10D1L-k0MT, MDX10D1H-Kn462/MDX10D1H-Hl445/MDX10D1L-k0MT, MDX10D1H-Kn461/MDX10D1H-Hl443/MDX10D1L-k0MT, none of the existing Fc ⁇ R binding
- the binding to Fc ⁇ RIIIa was enhanced compared to the enhanced antibodies MDX10D1H-GASDIE/MDX10D1L-k0MT and MDX10D1H-GASDALIE/MDX10D1L-k0MT.
- MDX10D1H-Kn462 / MDX10D1H-Hl445 / MDX10D1L-k0MT showed that the binding to Fc ⁇ RIIaH was enhanced about twice as compared to MDX10D1H-GASDIE / MDX10D1L-k0MT, which is an existing Fc ⁇ RIIa-enhancing antibody.
- one heavy chain variable region 04H1654 (SEQ ID NO: 35) has a human heavy chain constant region Kn462 (SEQ ID NO: 43) as a constant region, and the other heavy chain variable region 04H1656 (SEQ ID NO: 37 ) ligates the human heavy chain constant region Hl445 (SEQ ID NO: 44), and the light chain variable region 04L1610 (SEQ ID NO: 39) uses the wild-type human light chain constant region lam1 (SEQ ID NO: 53). It was expressed and purified by a method known to the trader.
- one heavy chain variable region 04H1654 (SEQ ID NO: 35) has a human heavy chain constant region Kn462 (SEQ ID NO: 43) as a constant region, and the other heavy chain variable region 04H1656 (SEQ ID NO: 37 ) ligates the human heavy chain constant region Hl445 (SEQ ID NO: 44), and the light chain variable region 04L1612 (SEQ ID NO: 40) uses the wild-type human light chain constant region lam1 (SEQ ID NO: 53). It was expressed and purified by a method known to the trader.
- the SW1389-ART6 antibody has two heavy chain variable regions 04H1389 (SEQ ID NO: 29) with human heavy chain constant region Kn462 (SEQ ID NO: 43) and human heavy chain constant region Hl445 (SEQ ID NO: 44) as constant regions, respectively.
- the light chain variable region 04L1305 (SEQ ID NO: 30) was expressed and purified using the wild-type human light chain constant region k0MT (SEQ ID NO: 33) by methods known to those skilled in the art.
- hFc ⁇ RIIIaV ADCC Reporter Bioassay, Core Kit (Promega) was used for in vitro ADCC activity measurement. 12.5 ⁇ L of hCTLA4-CHO cells adjusted to 2 ⁇ 10 6 /mL were added to each well of a 96-well plate as target cells, and assay buffer (4% Low IgG Serum in RPMI1640) was added to the medium. Used.
- ATP solution diluted with assay buffer to final concentrations of 0, 100 ⁇ M, SW1389-ART6 diluted with assay buffer to final concentrations of 0, 0.001, 0.01, 0.1, 1, 10 ⁇ g/mL , SW1610-ART6 and SW1612-ART6 antibody solutions were sequentially added, and finally 25 ⁇ L of hFc ⁇ RIIIaV-expressing Jurkat cells (supplied with the kit) adjusted to 3 ⁇ 10 6 /mL in medium were added as an effector cell solution. After being mixed to 75 ⁇ L, it was allowed to stand at 37° C. for 6 hours in a 5% CO 2 incubator.
- SW1389 antibody uses 04H1389 (SEQ ID NO: 29) as the heavy chain variable region and 04L1305 (SEQ ID NO: 30) as the light chain variable region, is ligated to a human constant region, and then expressed by a method known to those skilled in the art. refined.
- the SW1610 antibody uses 04H1654 (SEQ ID NO: 35) and 04H1656 (SEQ ID NO: 37) as the heavy chain variable region and 04L1610 (SEQ ID NO: 39) as the light chain variable region, and is linked to the human constant region.
- the SW1612 antibody uses 04H1654 (SEQ ID NO: 35) and 04H1656 (SEQ ID NO: 37) as the heavy chain variable region and 04L1612 (SEQ ID NO: 40) as the light chain variable region, and is linked to the human constant region. It was expressed and purified by methods known to those skilled in the art.
- the SW1615 antibody uses 04H1389 (SEQ ID NO: 29) as the heavy chain variable region and 04L1615 (SEQ ID NO: 34) as the light chain variable region, is ligated to a human constant region, and then expressed by a method known to those skilled in the art. refined.
- CTLA-4 Blockade Bioassay (Promega) was used for in vitro neutralizing activity measurement.
- 25 ⁇ L of aAPC-Raji cells supplied with the kit adjusted to a concentration of 1 ⁇ 10 6 /mL with medium, were added as target cells, and assay buffer (10% FBS in RPMI1640) was added to the medium. Used.
- ATP solution diluted with assay buffer to final concentrations of 0, 100 ⁇ M, SW1389 and SW1610 diluted with assay buffer to final concentrations of 0, 0.001, 0.01, 0.1, 1, 10 ⁇ g/mL , SW1612 and SW1615 variable region-containing antibody solutions were sequentially added, and finally 25 ⁇ L of IL2-luc2-CTLA4-Jurkat cells (supplied with the kit) adjusted to 2 ⁇ 10 6 /mL with medium were added as effector cell solutions. After being added and mixed to a total volume of 75 ⁇ L, it was allowed to stand at 37° C. for 6 hours in a 5% CO 2 incubator.
- the neutralizing activity of the anti-CTLA4 switch antibody against hCTLA4-expressing cells differed in the presence and absence of ATP, confirming the existence of ATP-dependent neutralizing activity.
- the SW1610-ART5+ACT1 antibody has 04H1654-KT473 (SEQ ID NO: 184) as one heavy chain, 04H1656-HT451 (SEQ ID NO: 272) as the other heavy chain, and 04L1610-lam1 (SEQ ID NO: : 161), expressed and purified by methods known to those skilled in the art.
- the SW1610-ART6+ACT1 antibody has 04H1654-KT473 (SEQ ID NO: 184) as one heavy chain, 04H1656-HT482 (SEQ ID NO: 185) as the other heavy chain, and 04L1610-lam1 (SEQ ID NO: 185) as the light chain. 161), expressed and purified by methods known to those skilled in the art.
- the SW1389-ART5+ACT1 antibody has 04H1389-Km473 (SEQ ID NO: 203) as one heavy chain, 04H1389-Hm451 (SEQ ID NO: 274) as the other heavy chain, and 04L1305-k0MT (SEQ ID NO: : 275), expressed and purified by methods known to those skilled in the art.
- the SW1389-ART6+ACT1 antibody has 04H1389-Km473 (SEQ ID NO: 203) as one heavy chain, 04H1389-Hm482 (SEQ ID NO: 204) as the other heavy chain, and 04L1305-k0MT (SEQ ID NO: 204) as the light chain. 275), expressed and purified by methods known to those skilled in the art.
- ATP solution adjusted to 0 or 400 ⁇ M with RPMI/10% FBS was added, well suspended, and then allowed to stand at 37°C for 6 hours in a CO 2 incubator (every 2 hours 5 ⁇ L of ATP solution adjusted to 0 or 4000 ⁇ M was added in total twice).
- PBMCs were collected, washed twice with auto MACS Rinsing Solution (Milteny), reacted with the following antibodies, and the fraction of immune cells present was analyzed by FACS analysis.
- Viability determination reagent Biolegend, Zombie Aqua
- anti-CD3 antibody BD, clone: UCHT1
- anti-CD4 antibody BD, clone: RPA-T4
- anti-CD8 antibody BD, clone: SK1
- anti-CD45RA antibody Biolegend, clone: HI100
- anti-CD25 antibody BD, clone: 2A3
- anti-CD16 antibody Biolegend, clone: 3G8
- anti-CD56 antibody Biolegend, clone: HCD56
- anti-CTLA4 antibody Biolegend, clone: BNI3
- the Fc region variant Kn125/Hl076 (abbreviation in this specification: ART1) described in WO2013002362 and WO2014104165 has a strongly enhanced binding ability to Fc ⁇ RIIIa, but the binding to Fc ⁇ RIIa As compared with IgG1, the activity was enhanced only several times, and it was considered that further enhancement was necessary in order to exhibit strong ADCP activity.
- Kn120/Hl068 (abbreviation in this specification: ART2) has enhanced binding to both Fc ⁇ RIIa and Fc ⁇ RIIIa, and strong activity is expected for both ADCC and ADCP, but it also has the ability to bind to inhibitory Fc ⁇ RIIb.
- ART1 which is an Fc region variant with enhanced binding to Fc ⁇ RIIIa described in WO2013002362 and WO2014104165, was constructed as follows.
- Binding enhancement modifications to Fc ⁇ R for the CH2 region of H240-G1d by introducing L234Y / L235Q / G236W / S239M / H268D / D270E / S298A, by introducing Y349C / T366W for the CH3 region H240- Kn125 (SEQ ID NO:280) was generated.
- H240-Hl076 (SEQ ID NO: 281) was prepared by introducing D270E/K326D/A330M/K334E into the CH2 region of H240-G1d and introducing D356C/T366S/L368A/Y407V into the CH3 region. rice field.
- H240-Kn125, H240-Hl076, and a plasmid having the gene of L73-k0 (SEQ ID NO: 282), which is the light chain of anti-human Epiregulin antibody, were mixed, and human embryonic kidney cell-derived Expi 293 strain (Invitrogen) was subjected to lipofection.
- human embryonic kidney cell-derived Expi 293 strain (Invitrogen) was subjected to lipofection.
- an Fc modified antibody against human Epiregulin H240-Kn125/L73-k0//H240 -Hl076/L73-k0: Antibody abbreviation EGL-ART1
- Absorbance at 280 nm of the purified antibody solution was measured using a spectrophotometer.
- the concentration of the purified antibody was calculated using the extinction coefficient calculated by the PACE method from the measured values obtained (Protein Science (1995) 4, 2411-2423).
- EGL-ART2 H240-Kn120/L73-k0//H240-Hl068/L73-k0
- EGL-ART2 H240-Kn120/L73-k0//H240-Hl068/L73-k0
- an Fc region variant with enhanced binding to both Fc ⁇ RIIa and Fc ⁇ RIIIa described in WO2013002362 and WO2014104165 made.
- Antibodies lacking fucose can also be obtained by producing antibodies in cells in which beta 1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidae II are forcibly expressed (Biotechnol, Bioeng. (2006) 93 (5), 851-861).
- EGL-afucosyl H240-G1d/L73-k_glycomab was prepared by these methods known to those skilled in the art.
- new Fc region variants ART3, ART4, ART5, ART6, ART8, ART10, ART11, ART12 listed in Table 34 were generated. rice field. These variants commonly introduce L234F, L235Q, G236W, S239M, H268D, D270E, and S298A in one heavy chain and D270E, S298A, K326D, and K334E in the other heavy chain. It was prepared by introducing a combination of modifications that change the binding to Fc ⁇ R to the asymmetric modification group.
- Antibodies of interest were captured on this chip and allowed to interact with each Fc ⁇ R diluted in running buffer.
- the chip was regenerated with 10 mM Glycine-HCl (pH 1.5) and repeated antibody captures and measurements were performed.
- the dissociation constant KD (mol/L) of each antibody for Fc ⁇ R was calculated using Biacore Insight Evaluation Software, the dissociation constant for Fc ⁇ RIIb was calculated using the Steady state affinity model, and the dissociation constant for other Fc ⁇ Rs was calculated using the 1:1 Langmuir binding model. (Table 35).
- the "relative value for KD of G1d and hFc ⁇ Rs" in the table is the value obtained by dividing the KD value for each Fc ⁇ R of G1d by the KD value for each Fc ⁇ R of each antibody, and how much each antibody is enhanced against G1d It is a value that indicates whether
- the "A / I ratio” is the value obtained by dividing the KD of each antibody for Fc ⁇ RIIb by the KD for each Fc ⁇ R, and how selectively binding to activating Fc ⁇ R is enhanced relative to binding to inhibitory Fc ⁇ R A value that indicates whether the
- ART3, ART4, ART5, ART6, ART8, ART10, ART11, and ART12 produced in the present invention were all enhanced against Fc ⁇ RIIIaF and Fc ⁇ RIIIaV than G1d.
- These variants are symmetrically modified existing Fc ⁇ R-enhanced antibodies GASDALIE, SDALIE, GASDIE and Afucosyl antibodies compared to both Fc ⁇ RIIIaF and Fc ⁇ RIIIaV Fc ⁇ R It was enhanced more.
- ART4 2519.9-fold
- ART6 986.7-fold
- ART8 1966.7-fold
- ART12 577.5-fold
- ART4, ART8, and ART10 had enhanced binding to Fc ⁇ RIIIaF even when compared with ART1 (1170.2-fold), which had stronger binding to Fc ⁇ RIIIaF than ART2.
- ART4 462.2-fold
- ART6 321.9-fold
- ART8 694.9-fold
- ART10 565.5-fold
- the binding of ART8 and ART10 was further enhanced compared to ART1 (322.8-fold).
- ART6 enhanced Fc ⁇ RIIIaV to the same extent as ART1.
- ART3 (32.1 times), ART4 (6.3 times), ART5 (33.9 times), ART6 (118.0 times), ART8 (15.0 times), ART10 (2.7 times), ART11 (4.9 times), ART12 (3.1 times)
- Fc ⁇ RIIaH especially ART3, ART5, ART6, and ART8 compared to ART2 (14.7-fold)
- ART3, ART5, and ART6 are more strongly enhanced than GASDIE (16.4-fold), which is an Fc ⁇ RIIa-enhancing antibody that has been introduced with existing symmetrical modifications, and exhibits stronger ADCP activity than any existing variant. expected to show.
- GASDIE GAA
- ART3 (13.7-fold), ART4 (3.2-fold), ART5 (10.4-fold), ART6 (24.1-fold), ART8 (13.2-fold), ART10 (1.5-fold), and ART11 (1.3-fold) was also enhanced compared to G1d, but the existing variants GASDIE (24.7-fold) and ART2 (49.0-fold) were more enhanced than these variants.
- Fc ⁇ RIIb which is an inhibitory receptor, induces intracellular signals that suppress immune responses in contrast to activating Fc ⁇ Rs, and thus is expected to inhibit signals from activating Fc ⁇ Rs. It has actually been reported that the antitumor effect of the antibody is enhanced in Fc ⁇ RIIb knockout mice (Nature Medicine 2000, 6, 443-436). A correlation has also been observed between the difference in antitumor effects of mouse IgG subclasses and the ratio of binding to activating Fc ⁇ R and inhibitory Fc ⁇ R (A/I ratio) (Science 2005, 310, 1510-1512).
- Fc ⁇ RIIaR has high sequence homology with Fc ⁇ RIIb, it is difficult to confer selectivity, and it is difficult to say that the variants reported so far actually have excellent selectivity.
- ART10 (A / I ratio 6.6), ART11 (A / I ratio 11.5), ART12 (A / I ratio 12.8), ART4 (A / I ratio 22.5), ART8 (A / I ratio 28.1), ART6 (A/I ratio 42.4), ART5 (A/I ratio 49.9), ART3 (A/I ratio 52.4) all had G1d (A/I ratio 6.1) Among them, ART4, ART8, ART6, ART5, and ART3 were shown to be superior to ART2 (A/I ratio 13.0) and GASDIE (A/I ratio 18.6).
- ART10 A/I ratio 17.3
- ART8 A/I ratio 47.2
- ART12 A/I ratio 60.3
- ART11 A/I ratio 63.3
- ART4 A/I I ratio 65.2
- ART3 A/I ratio 180.7
- ART5 A/I ratio 240.1
- ART6 A/I ratio 307.0
- ART4, ART8, ART3, ART5, and ART6 are antibodies with a better A/I ratio than existing enhancing antibodies against Fc ⁇ RIIaR.
- ART3, ART5, and ART6 can be said to be antibodies with binding ability and A/I ratio superior to existing enhancing antibodies against Fc ⁇ RIIaH.
- the A/I ratio of Fc ⁇ RIIIaF was ART3 (A/I ratio 396.1), ART5 (A/I ratio 398.8), ART11 (A/I ratio 694.1), ART6 (A/I ratio 975.5), ART8 (A/I ratio 2350.3), ART10 (A/I ratio 3159.9), ART12 (A/I ratio 4309.9), and ART4 (A/I ratio 9943.2), all of which were superior to G1d (A/I ratio 3.4).
- ART4 showed a better A/I ratio than ART1 (A/I ratio 4947.2), which is an Fc ⁇ RIIIa-specific enhanced variant described in WO2014104165.
- the A/I ratio of Fc ⁇ RIIIaV was ART3 (A/I ratio 2003.7), ART5 (A/I ratio 2064.8), ART6 (A/I ratio 2625.7), ART11 (A/I ratio 3974.6), ART8 (A/I ratio ratio 6852.0), ART12 (A/I ratio 9721.0), ART10 (A/I ratio 11436.1), and ART4 (A/I ratio 15047.3), all of which were G1d (A/I ratio 28.1) and the existing enhanced variant Afucosyl ( A/I ratio of 298.1).
- ART4 and ART10 showed a better A/I ratio than ART1 (A/I ratio 11261.3), which is an Fc ⁇ RIIIa-specific enhanced variant described in WO2014104165. From the above results, it can be said that ART4 is an antibody that has even better binding ability and A/I ratio than the existing enhancing antibody ART1 for both Fc ⁇ RIIIaF and Fc ⁇ RIIIaV.
- ADCC Reporter Bioassay hFc ⁇ RIIIaV ADCC Reporter Bioassay, Effector cells, Propagation Model (Promega) was used for in vitro ADCC activity measurement. The concentration was adjusted to 5 ⁇ 10 5 /mL in each well of a 384-well plate with medium. 10 ⁇ L of Hepa1-6/hEREG cells were added as target cells, and Assay Buffer (96% RPMI, 4% FBS) was used as the medium.
- the antibody prepared in Reference Example 7 and EGL-G4d (heavy chain SEQ ID NO: 306, light chain SEQ ID NO: 282) having a human IgG4 sequence as a negative control are each at a final concentration of 1 ⁇ g / mL 10 ⁇ L was added after being diluted with assay buffer so that there were 11 points at a common ratio of 10, and finally 10 ⁇ L of hFc ⁇ RIIIaV-expressing Jurkat cells adjusted to 3 ⁇ 10 6 /mL with medium was added as an effector cell solution, After being mixed to a total volume of 30 ⁇ L, it was allowed to stand overnight at 37° C. in a 5% CO 2 incubator.
- Bio-Glo reagent used Bio-glo Luciferase Assay System (Buffer and Substrate). The luminescence of each well was then measured with a plate reader.
- the fold induction was obtained by dividing the luminescence value of each well by the luminescence value of the antibody-free well, and was used as an index for evaluating the ADCC of each antibody. The results obtained are shown in FIG.
- the EC50 value of each sample was calculated by JMP 11.2.1 (SAS Institute Inc.) and shown in Table 36.
- hFc ⁇ RIIaH-expressing Jurkat cells As a cell solution, 10 ⁇ L of hFc ⁇ RIIaH-expressing Jurkat cells attached to the kit were added, mixed to make a total of 30 ⁇ L, and then allowed to stand at 37° C. for 6 hours in a 5% CO 2 incubator.
- the hFc ⁇ RIIaH-expressing Jurkat cells had a cell sap density of 9.68 ⁇ 10 5 /mL. After that, the plate was allowed to stand at room temperature for 15 minutes, and 30 ⁇ L of Bio-Glo reagent was added to each well. Bio-Glo reagent used Bio-glo Luciferase Assay System (Buffer and Substrate). The luminescence of each well was then measured with a plate reader.
- a value obtained by dividing the luminescence value of each well by the luminescence value of the antibody-free well was defined as fold induction, and was used as an index for evaluating the ADCP of each antibody.
- the results obtained are shown in FIG.
- the EC50 value of each sample was calculated using JMP 11.2.1 (SAS Institute Inc.) and shown in Table 37.
- the reporter gene induction activity for Hepa1-6/hEREG cells of the antibody with the modified Fc produced this time has a stronger activity than that of the wild-type human IgG1 constant region.
- the results in Table 37 showed that the antibody exhibited activity at a lower concentration than the variant with symmetrical engineering of the CH2 region and the low-fucose antibody produced by sugar chain modification.
- ART2, ART3, ART5, ART6, and ART8 were shown to have activity at lower concentrations than ART1. Among them, ART3, ART6, and ART8 showed activity at lower concentrations than ART2, which had enhanced binding to hFc ⁇ RIIaH.
- EGL-ART6 Preparation of drug to be administered EGL-ART6 produced in the present invention was the strongest antibacterial agent from the results of A/I ratio in Reference Example 8 and the intensity of reporter gene induction activity in Reference Examples 9 and 10. Tumor activity was expected. Therefore, as drugs to be administered to the Hepa1-6/hEREG cell transplantation model, an anti-hEREG control antibody (EGL-G1d) prepared in the same manner as in Reference Example 7 and an anti-hEREG antibody having Fc with enhanced Fc ⁇ R binding (EGL-afucosyl, EGL-ART6) were each prepared using His buffer (150 mM NaCl, 20 mM His-HCl buffer pH 6.0) so as to be 1 mg/mL.
- His buffer 150 mM NaCl, 20 mM His-HCl buffer pH 6.0
- TGI tumor growth inhibition
- the anti-human Epiregulin antibodies produced in Reference Examples 7 and 9 were subjected to ELISA. Buffers shown in Table 39 were prepared as appropriate. Human C1q protein (hC1q) was used as antigen.
- composition of buffer used for human C1q ELISA Composition of buffer used for human C1q ELISA
- 96-well maxisorp plates (Thermo fisher) were coated overnight at 4°C with 50 ⁇ L of each antibody prepared in PBS at 30, 10, 3, 1, 0.3, 0.1, 0.03 ⁇ g/mL. .
- the well was blocked with 200 ⁇ L of Blocking/dilution Buffer at room temperature for 2 hours or more.
- 50 ⁇ L of hC1q (Calbiochem) adjusted to a final concentration of 3 ⁇ g/mL in Blocking/dilution Buffer was added.
- the plate was allowed to stand at room temperature for 1 hour to allow hC1q to bind to each antibody present in each well. After washing with Wash Buffer, 50 ⁇ L of HRP-conjugated anti-hC1q antibody (AbDSerotec) diluted with Blocking/dilution Buffer was added to each well, and the plate was incubated for 1 hour. After washing with Wash Buffer, TMB single solution (Invitrogen) was added. After the color development reaction of the solution in each well was stopped by adding Stop Buffer, the color development was measured by absorbance at 450 nm and 690 nm. As the buffer, a buffer containing the composition shown in Table 39 was used. The measured results are shown in FIGS. 43 and 44. FIG.
- ART3, ART5, and ART11 had the same binding ability as G1d.
- Afucosyl and ART8 had the same binding ability as G1d.
- ART1, ART2, ART4, ART6, ART10, ART12, GASDALIE, SDALIE, and GASDIE had lower binding to C1q than G1d.
- ART1, ART2, ART4, ART6, ART12, GASDALIE, SDALIE, and GASDIE had their C1q-binding ability attenuated to the same level as that of G4d having a human IgG4 sequence.
- ART3, ART5, and ART11 which had enhanced binding to C1q compared to G1d, did not introduce modifications at positions 330 and 332, and the S298A modification that is said to improve binding to C1q. It is thought that the binding was enhanced by the effect of modification to and position 326 (Science, 2018, 359, 794-797).
- the anti-CTLA-4 antibodies of the present disclosure have immune cell activating action, cytotoxic activity, and/or antitumor activity, but have low action on non-tumor tissues such as normal tissues. , development, manufacture, provision, use, etc. of drugs with few side effects.
- the polypeptide comprising the mutant Fc region of the present disclosure and the method for producing and using the same strongly binds to activating Fc ⁇ RIIa and Fc ⁇ RIIIa, and suppresses binding to inhibitory Fc ⁇ RIIb, resulting in high ADCC/ADCP activity. and/or development, manufacture, provision, use, etc. of a medicament having anti-tumor activity.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- General Engineering & Computer Science (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Plant Pathology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmacology & Pharmacy (AREA)
- Physics & Mathematics (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Mycology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cell Biology (AREA)
- Epidemiology (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description
CTLA-4は1987年にマウスに由来するキラーT細胞クローンのcDNAライブラリから遺伝子がクローニングされた、免疫グロブリンスーパーファミリーに属する糖タンパク質である(例えば非特許文献2を参照)。CTLA-4を介してT細胞の免疫応答が抑制されることが知られている。CTLA-4の機能を抑制してT細胞の活性化を促進させることが癌の退縮につながるとの考えから、1996年には担癌マウスへの抗CTLA-4抗体の投与により腫瘍の退縮効果が観察されたことが報告されている(例えば非特許文献3を参照)。2000年から、ヒトにおける抗CTLA-4抗体の有効性の評価が進められ、2011年には抗ヒトCTLA-4モノクローナル抗体(イピリムマブ)が米国Food and Drug Administration(FDA,食品医薬品局)から世界初の免疫活性化抗体医薬として承認を受けた。イピリムマブ以外にも多数の抗CTLA-4モノクローナル抗体が作製され(例えば特許文献1、特許文献2、特許文献3、特許文献4を参照)、それらの医薬品としての開発が試みられている。免疫チェックポイントを阻害することでその免疫抑制機構を解除し、結果的に免疫活性を高めるこうした薬剤は、免疫チェックポイント阻害剤と呼ばれている。
一方で、T細胞の中には免疫抑制機能を有する細胞が一部存在することが以前より知られていたが、それが1995年にCD25陽性CD4陽性のT細胞として同定され、制御性T細胞と名付けられた(例えば非特許文献4を参照)。2003年には、制御性T細胞に特異的に発現して、その発生および機能を制御するマスター遺伝子であるFoxp3遺伝子が同定された。Foxp3は転写因子として様々な免疫応答関連遺伝子の発現を制御している。Foxp3は、なかでも、制御性T細胞におけるCTLA-4の恒常的な発現に関与しており、それが制御性T細胞による免疫抑制機能に重要な役割を果たしていると考えられている(例えば非特許文献5を参照)。
腫瘍組織に制御性T細胞が浸潤することで、それが腫瘍に対する免疫監視機構を減弱あるいは阻害する結果につながっていると考えられている。実際に、ヒトの多くの癌腫において制御性T細胞が増加していることが明らかにされており(例えば非特許文献6を参照)、制御性T細胞の腫瘍の局所への浸潤が癌患者の予後不良因子となり得ることが報告されている。逆に、腫瘍組織から制御性T細胞を除去あるいは減少させることができれば、抗腫瘍免疫の増強につながると期待される。現在、制御性T細胞を標的とした癌免疫療法の開発が精力的に進められつつある。
抗CTLA-4抗体であるイピリムマブの投与により抗腫瘍免疫は増強されるが、一方で、免疫活性を全身的に増強するために自己免疫疾患を発症することが報告されている。ある臨床試験においてはイピリムマブを投与した患者の60%に有害事象が見られ、その多くが皮膚あるいは消化管に関する自己免疫疾患であった。他の臨床試験においてもイピリムマブを投与した患者のうち約半数が同様の自己免疫疾患を発症したと報告されている。このような副作用を抑えるため、イピリムマブを投与した患者に免疫抑制剤が投与されるケースもある。こうした免疫チェックポイント阻害剤の副作用を抑えつつ抗腫瘍免疫応答を維持することが可能な新たな薬剤の開発が望まれている。
治療用抗体を生体内に投与した場合、その標的となる抗原が病変部位にのみ特異的に発現していることが望ましいが、多くの場合、非病変部位である正常組織にも同じ抗原が発現しており、それが治療の観点からは望ましくない副作用の原因となり得る。例えば、腫瘍抗原に対する抗体は、ADCC等によって腫瘍細胞に対する傷害活性を発揮し得る一方で、正常組織にも同じ抗原が発現していた場合、正常細胞をも傷害してしまう可能性がある。上記のような問題を解決するために、標的となる組織(例えば腫瘍組織)に特定の化合物が多量に存在する現象に着目し、そうした化合物の濃度に応じて抗原に対する結合活性が変化する抗原結合分子を創作する技術が開発された(例えば特許文献11を参照)。
〔1〕
(A) アデノシン含有化合物の濃度に依存したCTLA-4結合活性を有する可変領域、および
(B) 親Fc領域において複数のアミノ酸改変を含む変異Fc領域
を含む抗CTLA-4抗体であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、抗CTLA-4抗体:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および330、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、330、および334。
〔2〕 可変領域が、以下の(a)から(i)より選択される少なくとも一つの特徴を有する、〔1〕に記載の抗CTLA-4抗体:
(a) 100 μMのアデノシン含有化合物の存在下での結合活性が、アデノシン含有化合物の非存在下での結合活性と比べて、2倍以上高い、
(b) 100 μMのアデノシン含有化合物の存在下でのKD値が5×10-7 M以下である、
(c) アデノシン含有化合物の非存在下でのKD値が1×10-6 M以上である、
(d) アデノシン含有化合物およびCTLA-4とともに三者複合体を形成する、
(e) ヒトCTLA-4(細胞外ドメイン、配列番号:28)の97番目のアミノ酸から106番目のアミノ酸の領域に結合する、
(f) CTLA-4への結合に関して、ABAM004(VH、配列番号:10;およびVL、配列番号:11)と競合する、
(g) ABAM004(VH、配列番号:10;およびVL、配列番号:11)によって結合されるのと同じエピトープに結合する、
(h) CTLA-4発現細胞に対して細胞傷害活性を示す、および
(i) ヒトおよびマウス由来のCTLA-4に結合する。
〔3〕 モノクローナル抗体である、〔1〕または〔2〕に記載の抗CTLA-4抗体。
〔4〕 ヒト抗体、ヒト化抗体、またはキメラ抗体である、〔1〕から〔3〕のいずれか一項に記載の抗CTLA-4抗体。
〔5〕 (a) アミノ酸配列SX1TMNを含み、X1はH、A、R、またはKであるHVR-H1(配列番号:223)、(b) アミノ酸配列SISX1X2SX3YIYYAX4SVX5Gを含み、X1はSまたはT、X2はRまたはQ、X3はGまたはH、X4はD、E、またはR、X5はKまたはRであるHVR-H2(配列番号:224)、および (c) アミノ酸配列YGX1REDMLWVFDYを含み、X1はKまたはAであるHVR-H3(配列番号:225)を含む、〔1〕から〔4〕のいずれか一項に記載の抗CTLA-4抗体。
〔6〕 (a) アミノ酸配列X1GX2STX3VGDYX4X5VX6を含み、X1はT、D、Q、またはE、X2はTまたはP、X3はDまたはG、X4はNまたはT、X5はYまたはW、X6はSまたはHであるHVR-L1(配列番号:226)、(b) アミノ酸配列X1TX2X3KPX4を含み、X1はE、F、またはY、X2はSまたはI、X3はKまたはS、X4はS、E、またはKであるHVR-L2(配列番号:227)、および (c) アミノ酸配列X1TYAAPLGPX2を含み、X1はSまたはQ、X2はMまたはTであるHVR-L3(配列番号:228)をさらに含む、〔5〕に記載の抗CTLA-4抗体。
〔7〕 配列番号:229~232のいずれか1つのアミノ酸配列を含む重鎖可変ドメインFR1、配列番号:233のアミノ酸配列を含むFR2、配列番号:234のアミノ酸配列を含むFR3、および配列番号:235のアミノ酸配列を含むFR4をさらに含む、〔5〕に記載の抗CTLA-4抗体。
〔8〕 配列番号:236~238のいずれか1つのアミノ酸配列を含む軽鎖可変ドメインFR1、配列番号:240~241のいずれか1つのアミノ酸配列を含むFR2、配列番号:242~244のいずれか1つのアミノ酸配列を含むFR3、および配列番号:245~246のいずれか1つのアミノ酸配列を含むFR4をさらに含む、〔6〕に記載の抗CTLA-4抗体。
〔9〕 (a) 配列番号:83~86、98、135~141のいずれか1つのアミノ酸配列と少なくとも95%の配列同一性を有するVH配列;(b) 配列番号:88~95、97、99、134、144~149のいずれか1つのアミノ酸配列と少なくとも95%の配列同一性を有するVL配列;または (c) 配列番号:83~86、98、135~141のいずれか1つのアミノ酸配列を有するVH配列、および配列番号:88~95、97、99、134、144~149のいずれか1つのアミノ酸配列を有するVL配列を含む、〔1〕から〔4〕のいずれか一項に記載の抗CTLA-4抗体。
〔10〕
(1) 配列番号:98のVH配列および配列番号:99のVL配列、
(2) 配列番号:83のVH配列および配列番号:88のVL配列、
(3) 配列番号:83のVH配列および配列番号:89のVL配列、
(4) 配列番号:83のVH配列および配列番号:90のVL配列、
(5) 配列番号:83のVH配列および配列番号:91のVL配列、
(6) 配列番号:83のVH配列および配列番号:92のVL配列、
(7) 配列番号:83のVH配列および配列番号:93のVL配列、
(8) 配列番号:83のVH配列および配列番号:94のVL配列、
(9) 配列番号:83のVH配列および配列番号:97のVL配列、
(10) 配列番号:83のVH配列および配列番号:95のVL配列、
(11) 配列番号:84のVH配列および配列番号:97のVL配列、
(12) 配列番号:85のVH配列および配列番号:97のVL配列、
(13) 配列番号:86のVH配列および配列番号:97のVL配列、
(14) 配列番号:86のVH配列および配列番号:134のVL配列、
(15) 配列番号:136のVH配列および配列番号:97のVL配列、
(16) 配列番号:135のVH配列および配列番号:97のVL配列、
(17) 配列番号:136のVH配列および配列番号:95のVL配列、
(18) 配列番号:137のVH配列および配列番号:97のVL配列、
(19) 配列番号:138のVH配列および配列番号:97のVL配列、
(20) 配列番号:138のVH配列および配列番号:144のVL配列、
(21) 配列番号:138のVH配列および配列番号:145のVL配列、
(22) 配列番号:138のVH配列および配列番号:146のVL配列、
(23) 配列番号:139のVH配列および配列番号:146のVL配列、
(24) 配列番号:140のVH配列および配列番号:146のVL配列、
(25) 配列番号:141のVH配列および配列番号:146のVL配列、
(26) 配列番号:140のVH配列および配列番号:147のVL配列、
(27) 配列番号:141のVH配列および配列番号:147のVL配列、
(28) 配列番号:140のVH配列および配列番号:148のVL配列、
(29) 配列番号:141のVH配列および配列番号:148のVL配列、
(30) 配列番号:136のVH配列および配列番号:149のVL配列、
(31) 配列番号:140のVH配列および配列番号:146のVL配列を含む第一の可変領域、ならびに配列番号:141のVH配列および配列番号:146のVL配列を含む第二の可変領域、または
(32) 配列番号:140のVH配列および配列番号:147のVL配列を含む第一の可変領域、ならびに配列番号:141のVH配列および配列番号:147のVL配列を含む第二の可変領域、
を含む、〔9〕に記載の抗CTLA-4抗体。
〔11〕 全長IgG1抗体である、〔1〕から〔10〕のいずれか一項に記載の抗CTLA-4抗体。
〔12〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む、〔1〕から〔11〕のいずれか一項に記載の抗CTLA-4抗体。
〔13〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む、〔1〕から〔12〕のいずれか一項に記載の抗CTLA-4抗体。
〔14〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236におけるアミノ酸改変を含む、〔1〕から〔13〕のいずれか一項に記載の抗CTLA-4抗体。
〔15〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置250および307におけるアミノ酸改変を含む、〔1〕から〔14〕のいずれか一項に記載の抗CTLA-4抗体。
〔16〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置250および307におけるアミノ酸改変を含む、〔1〕から〔15〕のいずれか一項に記載の抗CTLA-4抗体。
〔17〕 変異Fc領域が、以下に記載のアミノ酸改変の中から選択される少なくとも1つのアミノ酸改変を含む、〔1〕から〔16〕のいずれか一項に記載の抗CTLA-4抗体:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234におけるPhe、位置235におけるGln、位置236におけるTrp、位置239におけるMet、位置250におけるVal、位置268におけるAsp、位置270におけるGlu、位置298におけるAla、位置307におけるPro、位置330におけるMet、位置332におけるGlu、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236におけるAla、位置250におけるVal、位置270におけるGlu、位置298におけるAla、位置307におけるPro、位置326におけるAsp、位置330におけるMet、位置332におけるGlu、位置334におけるGlu。
〔18〕 変異Fc領域が、さらに以下の(a)~(f)のいずれかのアミノ酸改変を含む、〔1〕から〔17〕のいずれか一項に記載の抗CTLA-4抗体:
(a) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置356におけるLys、および親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置439におけるGlu、
(b) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置439におけるGlu、および親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置356におけるLys。
(c) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置366におけるTrp、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置366におけるSer、位置368におけるAla、および位置407におけるVal、
(d) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置366におけるSer、位置368におけるAla、および位置407におけるVal、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置366におけるTrp、
(e) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置349におけるCysおよび位置366におけるTrp、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置356におけるCys、位置366におけるSer、位置368におけるAla、および位置407におけるVal、
(f) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置356におけるCys、位置366におけるSer、位置368におけるAla、および位置407におけるVal、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置349におけるCysおよび位置366におけるTrp。
〔19〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドおよび/または第二のポリペプチドにおける、以下の(a)~(d)のいずれかのアミノ酸改変を含む、〔1〕から〔18〕のいずれか一項に記載の抗CTLA-4抗体:
(a) EUナンバリングで表される位置434におけるAla、
(b) EUナンバリングで表される位置434におけるAla、位置436におけるThr、位置438におけるArg、および位置440におけるGlu、
(c) EUナンバリングで表される位置428におけるLeu、位置434におけるAla、位置436におけるThr、位置438におけるArg、および位置440におけるGlu、
(d) EUナンバリングで表される位置428におけるLeu、位置434におけるAla、位置438におけるArg、および位置440におけるGlu。
〔20〕 変異Fc領域を含む重鎖定常領域を含む、〔1〕から〔19〕のいずれか一項に記載の抗CTLA-4抗体。
〔21〕 重鎖定常領域が、
(1) 配列番号:358の第一のポリペプチド、および配列番号:359の第二のポリペプチド、または
(2) 配列番号:360の第一のポリペプチド、および配列番号:361の第二のポリペプチド、
を含む、〔20〕に記載の抗CTLA-4抗体。
〔22〕
(1) 配列番号:335の第一のH鎖ポリペプチド、配列番号:336の第二のH鎖ポリペプチド、および配列番号:161のL鎖ポリペプチド、または
(2) 配列番号:337の第一のH鎖ポリペプチド、配列番号:338の第二のH鎖ポリペプチド、および配列番号:161のL鎖ポリペプチド、
を含む、抗CTLA-4抗体。
〔23〕 〔1〕から〔22〕のいずれか一項に記載の抗CTLA-4抗体をコードする、単離された核酸。
〔24〕 〔23〕に記載の核酸を含む、宿主細胞。
〔25〕 抗CTLA-4抗体を製造する方法であって、抗CTLA-4抗体が製造されるように〔24〕に記載の宿主細胞を培養することを含む、方法。
〔26〕 〔1〕から〔22〕のいずれか一項に記載の抗CTLA-4抗体および薬学的に許容される担体を含む、薬学的製剤。
〔101〕 親Fc領域においてアミノ酸改変を含む変異Fc領域を含むポリペプチドであって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、ポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、および298、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、および334。
〔102〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置326におけるアミノ酸改変を含む、〔101〕に記載のポリペプチド。
〔103〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236におけるアミノ酸改変を含む、〔101〕または〔102〕に記載のポリペプチド。
〔104〕 親Fc領域においてアミノ酸改変を含む変異Fc領域を含むポリペプチドであって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、ポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および326、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、270、298、326、および334。
〔105〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む、〔101〕から〔104〕のいずれかに記載のポリペプチド。
〔106〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置330におけるアミノ酸改変を含む、〔101〕から〔105〕のいずれかに記載のポリペプチド。
〔107〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む、〔101〕から〔106〕のいずれかに記載のポリペプチド。
〔108〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置330におけるアミノ酸改変を含む、〔101〕から〔107〕のいずれかに記載のポリペプチド。
〔109〕 親Fc領域においてアミノ酸改変を含む変異Fc領域を含むポリペプチドであって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、ポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、330、および332、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、270、298、326、330、332、および334。
〔110〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置250および307におけるアミノ酸改変を含む、〔101〕から〔109〕のいずれかに記載のポリペプチド。
〔111〕 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置250および307におけるアミノ酸改変を含む、〔101〕から〔110〕のいずれかに記載のポリペプチド。
〔112〕 親Fc領域においてアミノ酸改変を含む変異Fc領域を含むポリペプチドであって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、ポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、250、268、270、298、および307、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置250、270、298、307、326、および334。
〔113〕 親Fc領域においてアミノ酸改変を含む変異Fc領域を含むポリペプチドであって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、ポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、250、268、270、298、307、および326、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、250、270、298、307、326、および334。
〔114〕 親Fc領域においてアミノ酸改変を含む変異Fc領域を含むポリペプチドであって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、ポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、250、268、270、298、307、330、および332、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、250、270、298、307、326、330、332、および334。
〔115〕 以下に記載のアミノ酸改変の中から選択される少なくとも1つのアミノ酸改変を含む、〔101〕から〔114〕のいずれかに記載のポリペプチド:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234におけるTyr、もしくはPhe、位置235におけるGln、もしくはTyr、位置236におけるTrp、位置239におけるMet、位置250におけるVal、位置268におけるAsp、位置270におけるGlu、位置298におけるAla、位置307におけるPro、位置326におけるAsp、位置330におけるMet、位置332におけるGlu、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236におけるAla、位置250におけるVal、位置270におけるGlu、位置298におけるAla、位置307におけるPro、位置326におけるAsp、位置330におけるMet、もしくはLys、位置332におけるAsp、もしくはGlu、位置334におけるGlu。
〔116〕 変異Fc領域が、さらに以下の(a)~(f)のいずれかのアミノ酸改変を含む、〔101〕から〔115〕のいずれかに記載のポリペプチド:
(a) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置356におけるLys、および親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置439におけるGlu、
(b) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置439におけるGlu、および親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置356におけるLys。
(c) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置366におけるTrp、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置366におけるSer、位置368におけるAla、および位置407におけるVal、
(d) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置366におけるSer、位置368におけるAla、および位置407におけるVal、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置366におけるTrp、
(e) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置349におけるCysおよび位置366におけるTrp、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置356におけるCys、位置366におけるSer、位置368におけるAla、および位置407におけるVal、
(f) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置356におけるCys、位置366におけるSer、位置368におけるAla、および位置407におけるVal、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置349におけるCysおよび位置366におけるTrp。
〔117〕 変異Fc領域が、さらに親Fc領域の第一のポリペプチドおよび/または第二のポリペプチドにおける、以下の(a)~(d)のいずれかのアミノ酸改変を含む、〔101〕から〔116〕のいずれかに記載のポリペプチド:
(a) EUナンバリングで表される位置434におけるAla、
(b) EUナンバリングで表される位置434におけるAla、位置436におけるThr、位置438におけるArg、および位置440におけるGlu、
(c) EUナンバリングで表される位置428におけるLeu、位置434におけるAla、位置436におけるThr、位置438におけるArg、および位置440におけるGlu、
(d) EUナンバリングで表される位置428におけるLeu、位置434におけるAla、位置438におけるArg、および位置440におけるGlu。
〔118〕 親Fc領域に比べて、変異Fc領域において、FcγRIa、FcγRIIa、FcγRIIb、FcγRIIIaからなる群より選択される少なくとも一つのFcγ受容体に対する結合活性が増強している、〔101〕から〔117〕のいずれかに記載のポリペプチド。
〔119〕 親Fc領域に比べて、変異Fc領域において、FcγRIIaおよびFcγRIIIaに対する結合活性が増強している、〔118〕に記載のポリペプチド。
〔120〕 親Fc領域に比べて、変異Fc領域において、活性型Fcγ受容体と抑制型Fcγ受容体との間の選択性が向上している、〔101〕から〔119〕のいずれかに記載のポリペプチド。
〔120-2〕 親Fc領域に比べて、変異Fc領域において、活性型Fcγ受容体に対する結合活性が、抑制型Fcγ受容体に対する結合活性に比べて、選択的に増強されている、〔101〕から〔119〕のいずれかに記載のポリペプチド。
〔120-3〕 親Fc領域に比べて、変異Fc領域において、抑制型Fcγ受容体への結合活性に対する活性型Fcγ受容体への結合活性の比率(A/I比)が大きい、〔101〕から〔119〕のいずれかに記載のポリペプチド。
〔120-4〕 変異Fc領域を含むポリペプチドにおける前記比率(A/I比)が、親Fc領域を含むポリペプチドと比べて、1.1倍以上、1.2倍以上、1.3倍以上、1.4倍以上、1.5倍以上、1.6倍以上、1.7倍以上、1.8倍以上、1.9倍以上、2倍以上、3倍以上、4倍以上、5倍以上、6倍以上、7倍以上、8倍以上、9倍以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍以上、60倍以上、70倍以上、80倍以上、90倍以上、100倍以上、200倍以上、300倍以上、400倍以上、500倍以上、600倍以上、700倍以上、800倍以上、900倍以上、1000倍以上、2000倍以上、3000倍以上、4000倍以上、5000倍以上、6000倍以上、7000倍以上、8000倍以上、9000倍以上、又は10000倍以上、大きい、〔120-3〕に記載のポリペプチド。
〔120-5〕 変異Fc領域を含むポリペプチドにおける前記比率(A/I比)の値が、10以上、20以上、30以上、40以上、50以上、60以上、70以上、80以上、90以上、100以上、200以上、300以上、400以上、500以上、600以上、700以上、800以上、900以上、1000以上、2000以上、3000以上、4000以上、5000以上、6000以上、7000以上、8000以上、9000以上、10000以上、11000以上、12000以上、13000以上、14000以上、又は15000以上である、〔120-3〕に記載のポリペプチド。
〔121〕 活性型Fcγ受容体がFcγRIa、FcγRIIa、FcγRIIIaからなる群より選択される少なくとも1つのFcγ受容体であり、抑制型Fcγ受容体がFcγRIIbである、〔120〕~〔120-5〕のいずれかに記載のポリペプチド。
〔122〕 変異Fc領域を含むポリペプチドが抗体である、〔101〕から〔121〕のいずれかに記載のポリペプチド。
〔123〕 親Fc領域にアミノ酸改変を導入する工程を含む、変異Fc領域を含むポリペプチドを製造する方法であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ以下の位置にアミノ酸改変が導入される、方法:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、および298、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、および334。
〔124〕 親Fc領域にアミノ酸改変を導入する工程を含む、変異Fc領域を含むポリペプチドを製造する方法であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ以下の位置にアミノ酸改変が導入される、方法:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および326、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、270、298、326、および334。
〔125〕 親Fc領域にアミノ酸改変を導入する工程を含む、変異Fc領域を含むポリペプチドを製造する方法であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ以下の位置にアミノ酸改変が導入される、方法:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、330、および332、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、270、298、326、330、332、および334。
〔127〕 〔126〕に記載の核酸を含む、宿主細胞。
〔128〕 ポリペプチドを製造する方法であって、ポリペプチドが製造されるように〔127〕に記載の宿主細胞を培養することを含む、方法。
〔129〕 腫瘍の治療における使用のための、〔101〕から〔122〕のいずれかに記載のポリペプチド。
〔130〕 細胞の傷害における使用のための、〔101〕から〔122〕のいずれかに記載のポリペプチド。
〔131〕 細胞の傷害がADCC活性、CDC活性、またはADCP活性による、〔130〕に記載のポリペプチド。
〔132〕 〔101〕から〔122〕のいずれかに記載のポリペプチドおよび薬学的に許容される担体を含む、医薬組成物。
〔133〕 腫瘍治療用の医薬組成物である、〔132〕に記載の医薬組成物。
〔134〕 細胞傷害用の医薬組成物である、〔132〕に記載の医薬組成物。
〔135〕 細胞の傷害がADCC活性、CDC活性、またはADCP活性による、〔134〕に記載の医薬組成物。
〔136〕 〔101〕から〔122〕のいずれかに記載のポリペプチドまたは〔132〕に記載の医薬組成物を投与することを含む、腫瘍を治療する方法。
〔137〕 〔101〕から〔122〕のいずれかに記載のポリペプチドまたは〔132〕に記載の医薬組成物を投与することを含む、細胞を傷害する方法。
〔138〕 細胞の傷害がADCC活性、CDC活性、またはADCP活性による、〔137〕に記載の方法。
〔139〕 腫瘍治療剤の製造における、〔101〕から〔122〕のいずれかに記載のポリペプチドの使用。
〔140〕 細胞傷害剤の製造における、〔101〕から〔122〕のいずれかに記載のポリペプチドの使用。
〔141〕 細胞の傷害がADCC活性、CDC活性、またはADCP活性による、〔140〕に記載の使用。
〔142〕 親Fc領域にアミノ酸改変を導入する工程を含む、Fc領域を含むポリペプチドの機能を改変する方法であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ以下の位置にアミノ酸改変が導入される、方法:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、および298、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、および334。
〔143〕 親Fc領域にアミノ酸改変を導入する工程を含む、Fc領域を含むポリペプチドの機能を改変する方法であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ以下の位置にアミノ酸改変が導入される、方法:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および326、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、270、298、326、および334。
〔144〕 親Fc領域にアミノ酸改変を導入する工程を含む、Fc領域を含むポリペプチドの機能を改変する方法であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ以下の位置にアミノ酸改変が導入される、方法:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、330、および332、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、270、298、326、330、332、および334。
〔145〕 前記機能の改変が、FcγRIIaおよびFcγRIIIaに対する結合活性の増強である、〔142〕から〔144〕のいずれかに記載の方法。
〔146〕 前記機能の改変が、活性型Fcγ受容体と抑制型Fcγ受容体との間の選択性の向上である、〔142〕から〔144〕のいずれかに記載の方法。
〔147〕 前記機能の改変が、抑制型Fcγ受容体に対する結合活性に比べた場合の活性型Fcγ受容体に対する結合活性の選択的な増強である、〔142〕から〔144〕のいずれかに記載の方法。
〔148〕 前記機能の改変が、抑制型Fcγ受容体への結合活性に対する活性型Fcγ受容体への結合活性の比率(A/I比)の増大である、〔142〕から〔144〕のいずれかに記載の方法。
〔149〕 前記比率(A/I比)が、親Fc領域を含むポリペプチドと比べて、1.1倍以上、1.2倍以上、1.3倍以上、1.4倍以上、1.5倍以上、1.6倍以上、1.7倍以上、1.8倍以上、1.9倍以上、2倍以上、3倍以上、4倍以上、5倍以上、6倍以上、7倍以上、8倍以上、9倍以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍以上、60倍以上、70倍以上、80倍以上、90倍以上、100倍以上、200倍以上、300倍以上、400倍以上、500倍以上、600倍以上、700倍以上、800倍以上、900倍以上、1000倍以上、2000倍以上、3000倍以上、4000倍以上、5000倍以上、6000倍以上、7000倍以上、8000倍以上、9000倍以上、又は10000倍以上、増大する、〔148〕に記載の方法。
〔150〕 活性型Fcγ受容体がFcγRIa、FcγRIIa、FcγRIIIaからなる群より選択される少なくとも1つのFcγ受容体であり、抑制型Fcγ受容体がFcγRIIbである、〔142〕から〔149〕のいずれかに記載の方法。
〔151〕 前記機能の改変が、ADCC活性、CDC活性、またはADCP活性の増強である、〔142〕から〔144〕のいずれかに記載の方法。
別途定義しない限り、本明細書で使用される技術用語および科学用語は、本発明が属する技術分野の当業者によって一般的に理解されるのと同じ意味を有する。Singleton et al., Dictionary of Microbiology and Molecular Biology 2nd ed., J. Wiley & Sons (New York, N.Y. 1994)、およびMarch, Advanced Organic Chemistry Reactions, Mechanisms and Structure 4th ed., John Wiley & Sons (New York, N.Y. 1992)は、本出願において使用される用語の多くに対する一般的指針を当業者に提供する。特許出願および刊行物を含む、本明細書に引用される全ての参考文献は、その全体が参照により本明細書に組み入れられる。
(a) アミノ酸残基26-32 (L1)、50-52 (L2)、91-96 (L3)、26-32 (H1)、53-55 (H2)、および96-101 (H3)のところで生じる超可変ループ (Chothia and Lesk, J. Mol. Biol. 196: 901-917 (1987));
(b) アミノ酸残基24-34 (L1)、50-56 (L2)、89-97 (L3)、31-35b (H1)、50-65 (H2)、および95-102 (H3)のところで生じるCDR (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991));
(c) アミノ酸残基27c-36 (L1)、46-55 (L2)、89-96 (L3)、30-35b (H1)、47-58 (H2)、および93-101 (H3) のところで生じる抗原接触 (MacCallum et al. J. Mol. Biol. 262: 732-745 (1996));ならびに、
(d) HVRアミノ酸残基46-56 (L2)、47-56 (L2)、48-56 (L2)、49-56 (L2)、26-35 (H1)、26-35b (H1)、49-65 (H2)、93-102 (H3)、および94-102 (H3)を含む、(a)、(b)、および/または(c)の組合せ。
別段示さない限り、HVR残基および可変ドメイン中の他の残基(例えば、FR残基)は、本明細書では上記のKabatらにしたがって番号付けされる。
アミノ酸配列比較にALIGN-2が用いられる状況では、所与のアミノ酸配列Aの、所与のアミノ酸配列Bへの、またはそれとの、またはそれに対する%アミノ酸配列同一性(あるいは、所与のアミノ酸配列Bへの、またはそれとの、またはそれに対する、ある%アミノ酸配列同一性を有するまたは含む所与のアミノ酸配列A、ということもできる)は、次のように計算される:分率X/Yの100倍。ここで、Xは配列アラインメントプログラムALIGN-2によって、当該プログラムのAおよびBのアラインメントにおいて同一である一致としてスコアされたアミノ酸残基の数であり、YはB中のアミノ酸残基の全数である。アミノ酸配列Aの長さがアミノ酸配列Bの長さと等しくない場合、AのBへの%アミノ酸配列同一性は、BのAへの%アミノ酸配列同一性と等しくないことが、理解されるであろう。別段特に明示しない限り、本明細書で用いられるすべての%アミノ酸配列同一性値は、直前の段落で述べたとおりALIGN-2コンピュータプログラムを用いて得られるものである。
一局面において、本発明は、抗CTLA-4抗体およびそれらの使用に一部基づくものである。特定の態様において、CTLA-4に結合する抗体が提供される。本発明の抗体は、例えば、癌の診断または治療のために、有用である。
一局面において、本発明はCTLA-4に結合する、単離された抗体を提供する。特定の態様において、本発明の抗CTLA-4抗体は、アデノシン含有化合物の濃度に依存したCTLA-4結合活性を有する。いくつかの態様において、アデノシン含有化合物の非存在下に比べて、アデノシン含有化合物の存在下におけるCTLA-4への結合活性がより高い。あるいは別の態様において、低濃度のアデノシン含有化合物の存在下に比べて、高濃度のアデノシン含有化合物の存在下におけるCTLA-4への結合活性がより高い。さらなる態様において、CTLA-4への結合活性の差は、例えば2倍以上、3倍以上、5倍以上、10倍以上、20倍以上、30倍以上、50倍以上、100倍以上、200倍以上、300倍以上、500倍以上、1×103倍以上、2×103倍以上、3×103倍以上、5×103倍以上、1×104倍以上、2×104倍以上、3×104倍以上、5×104倍以上、または1×105倍以上である。
-HVR-H1(配列番号:223)における:ポジション2
-HVR-H2(配列番号:224)における:ポジション4、5、7、13、および16
-HVR-H3(配列番号:225)における:ポジション3
-HVR-L1(配列番号:226)における:ポジション1、3、6、11、12、および14
-HVR-L2(配列番号:227)における:ポジション1、3、4、および7
-HVR-L3(配列番号:228)における:ポジション1、および10
-HVR-H1(配列番号:100)において: H2A、RまたはK
-HVR-H2(配列番号:101)において: S4T;R5Q;G7H;D13EまたはR;K16R
-HVR-H3(配列番号:102)において: K3A
-HVR-L1(配列番号:113)において: T1D、QまたはE;T3P;D6G;N11T;Y12W;S14H
-HVR-L2(配列番号:114)において: E1FまたはY;S3I;K4S;S7EまたはK
-HVR-L3(配列番号:115)において: S1Q;M10T
特定の態様において、本明細書で提供される抗体の結合活性(binding activity)は、≦10 μM、≦1 μM、≦100 nM、≦10 nM、≦1 nM、≦0.1 nM、≦0.01 nM、または≦0.001 nM(例えば、10-8 M以下、例えば10-8 M~10-13 M、例えば10-9 M~10-13 M)の解離定数 (KD) である。
特定の態様において、本明細書で提供される抗体は、抗体断片である。抗体断片は、これらに限定されるものではないが、Fab、Fab'、Fab'-SH、F(ab')2、Fv、および scFv断片、ならびに、後述する他の断片を含む。特定の抗体断片についての総説として、Hudson et al. Nat. Med. 9: 129-134 (2003) を参照のこと。scFv断片の総説として、例えば、Pluckthun, in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., (Springer-Verlag, New York), pp. 269-315 (1994);加えて、WO93/16185;ならびに米国特許第5,571,894号および第5,587,458号を参照のこと。サルベージ受容体結合エピトープ残基を含みインビボ (in vivo) における半減期の長くなったFabおよびF(ab')2断片についての論説として、米国特許第5,869,046号を参照のこと。
特定の態様において、本明細書で提供される抗体は、キメラ抗体である。特定のキメラ抗体が、例えば、米国特許第4,816,567号;および、Morrison et al., Proc. Natl. Acad. Sci. USA, 81: 6851-6855 (1984) に記載されている。一例では、キメラ抗体は、非ヒト可変領域(例えば、マウス、ラット、ハムスター、ウサギ、またはサルなどの非ヒト霊長類に由来する可変領域)およびヒト定常領域を含む。さらなる例において、キメラ抗体は、親抗体のものからクラスまたはサブクラスが変更された「クラススイッチ」抗体である。キメラ抗体は、その抗原結合断片も含む。
特定の態様において、本明細書で提供される抗体は、ヒト抗体である。ヒト抗体は、当該技術分野において知られる種々の手法によって製造され得る。ヒト抗体は、van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-374 (2001) および Lonberg, Curr. Opin. Immunol. 20: 450-459 (2008) に、概説されている。
本発明の抗体は、所望の1つまたは複数の活性を伴う抗体についてコンビナトリアルライブラリをスクリーニングすることによって単離してもよい。例えば、ファージディスプレイライブラリの生成や、所望の結合特性を備える抗体についてそのようなライブラリをスクリーニングするための、様々な方法が当該技術分野において知られている。そのような方法は、Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, 2001) において総説されており、さらに例えば、McCafferty et al., Nature 348: 552-554;Clackson et al., Nature 352: 624-628 (1991);Marks et al., J. Mol. Biol. 222: 581-597 (1992);Marks and Bradbury, in Methods in Molecular Biology 248: 161-175 (Lo, ed., Human Press, Totowa, NJ, 2003);Sidhu et al., J. Mol. Biol. 338(2): 299-310 (2004);Lee et al., J. Mol. Biol. 340(5): 1073-1093 (2004);Fellouse, Proc. Natl. Acad. Sci. USA 101(34): 12467-12472 (2004);およびLee et al., J. Immunol. Methods 284(1-2): 119-132 (2004) に記載されている。
特定の態様において、本明細書で提供される抗体は、多重特異性抗体(例えば、二重特異性抗体)である。多重特異性抗体は、少なくとも2つの異なる部位に結合特異性を有する、モノクローナル抗体である。特定の態様において、結合特異性の1つは、CTLA-4に対するものであり、もう1つは他の任意の抗原へのものである。特定の態様において、二重特異性抗体は、CTLA-4の異なった2つのエピトープに結合してもよい。二重特異性抗体は、CTLA-4を発現する細胞に細胞傷害剤を局在化するために使用されてもよい。二重特異性抗体は、全長抗体としてまたは抗体断片として調製され得る。
特定の態様において、本明細書で提供される抗体のアミノ酸配列変異体も、考慮の内である。例えば、抗体の結合アフィニティおよび/または他の生物学的特性を改善することが、望ましいこともある。抗体のアミノ酸配列変異体は、抗体をコードするヌクレオチド配列に適切な修飾を導入すること、または、ペプチド合成によって、調製されてもよい。そのような修飾は、例えば、抗体のアミノ酸配列からの欠失、および/または抗体のアミノ酸配列中への挿入、および/または抗体のアミノ酸配列中の残基の置換を含む。最終構築物が所望の特徴(例えば、抗原結合性)を備えることを前提に、欠失、挿入、および置換の任意の組合せが、最終構築物に至るために行われ得る。
特定の態様において、1つまたは複数のアミノ酸置換を有する抗体変異体が提供される。置換的変異導入の目的部位は、HVRおよびFRを含む。保存的置換を、表1の「好ましい置換」の見出しの下に示す。より実質的な変更を、表1の「例示的な置換」の見出しの下に提供するとともに、アミノ酸側鎖のクラスに言及しつつ下で詳述する。アミノ酸置換は目的の抗体に導入されてもよく、産物は、例えば、保持/改善された抗原結合性、減少した免疫原性、または改善したADCCまたはCDCなどの、所望の活性についてスクリーニングされてもよい。

(1) 疎水性:ノルロイシン、メチオニン (Met)、アラニン (Ala)、バリン (Val)、ロイシン (Leu)、イソロイシン (Ile);
(2) 中性の親水性:システイン (Cys)、セリン (Ser)、トレオニン (Thr)、アスパラギン (Asn)、グルタミン (Gln);
(3) 酸性:アスパラギン酸 (Asp)、グルタミン酸 (Glu);
(4) 塩基性:ヒスチジン (His)、リジン (Lys)、アルギニン (Arg);
(5) 鎖配向に影響する残基:グリシン (Gly)、プロリン (Pro);
(6) 芳香族性:トリプトファン (Trp)、チロシン (Tyr)、フェニルアラニン (Phe)。
非保存的置換は、これらのクラスの1つのメンバーを、別のクラスのものに交換することをいう。
特定の態様において、本明細書で提供される抗体は、抗体がグリコシル化される程度を増加させるまたは減少させるように改変されている。抗体へのグリコシル化部位の追加または削除は、1つまたは複数のグリコシル化部位を作り出すまたは取り除くようにアミノ酸配列を改変することにより、簡便に達成可能である。
特定の態様において、本明細書で提供される抗体のFc領域に1つまたは複数のアミノ酸修飾を導入して、それによりFc領域変異体を生成してもよい。Fc領域変異体は、1つまたは複数のアミノ酸ポジションのところでアミノ酸修飾(例えば、置換)を含む、ヒトFc領域配列(例えば、ヒトIgG1、IgG2、IgG3、またはIgG4のFc領域)を含んでもよい。
特定の態様において、抗体の1つまたは複数の残基がシステイン残基で置換された、システイン改変抗体(例えば、「thioMAbs」)を作り出すことが望ましいだろう。特定の態様において、置換を受ける残基は、抗体の、アクセス可能な部位に生じる。それらの残基をシステインで置換することによって、反応性のチオール基が抗体のアクセス可能な部位に配置され、当該反応性のチオール基は、当該抗体を他の部分(薬剤部分またはリンカー‐薬剤部分など)にコンジュゲートして本明細書でさらに詳述するようにイムノコンジュゲートを作り出すのに使用されてもよい。特定の態様において、以下の残基の任意の1つまたは複数が、システインに置換されてよい:軽鎖のV205(Kabatナンバリング);重鎖のA118(EUナンバリング);および重鎖Fc領域のS400(EUナンバリング)。システイン改変抗体は、例えば、米国特許第7,521,541号に記載されるようにして生成されてもよい。
特定の態様において、本明細書で提供される抗体は、当該技術分野において知られておりかつ容易に入手可能な追加の非タンパク質部分を含むように、さらに修飾されてもよい。抗体の誘導体化に好適な部分は、これに限定されるものではないが、水溶性ポリマーを含む。水溶性ポリマーの非限定的な例は、これらに限定されるものではないが、ポリエチレングリコール (PEG)、エチレングリコール/プロピレングリコールのコポリマー、カルボキシメチルセルロース、デキストラン、ポリビニルアルコール、ポリビニルピロリドン、ポリ1, 3ジオキソラン、ポリ1, 3, 6トリオキサン、エチレン/無水マレイン酸コポリマー、ポリアミノ酸(ホモポリマーまたはランダムコポリマーのいずれでも)、および、デキストランまたはポリ(n-ビニルピロリドン)ポリエチレングリコール、ポリプロピレングリコールホモポリマー、ポリプロピレンオキシド/エチレンオキシドコポリマー、ポリオキシエチル化ポリオール類(例えばグリセロール)、ポリビニルアルコール、および、これらの混合物を含む。ポリエチレングリコールプロピオンアルデヒドは、その水に対する安定性のために、製造において有利であるだろう。ポリマーは、いかなる分子量でもよく、枝分かれしていてもしていなくてもよい。抗体に付加されるポリマーの数には幅があってよく、1つ以上のポリマーが付加されるならそれらは同じ分子であってもよいし、異なる分子であってもよい。一般的に、誘導体化に使用されるポリマーの数および/またはタイプは、これらに限定されるものではないが、改善されるべき抗体の特定の特性または機能、抗体誘導体が規定の条件下での療法に使用されるか否か、などへの考慮に基づいて、決定することができる。
例えば、米国特許第4,816,567号に記載されるとおり、抗体は組み換えの方法や構成を用いて製造することができる。一態様において、本明細書に記載の抗CTLA-4抗体をコードする、単離された核酸が提供される。そのような核酸は、抗体のVLを含むアミノ酸配列および/またはVHを含むアミノ酸配列(例えば、抗体の軽鎖および/または重鎖)をコードしてもよい。さらなる態様において、このような核酸を含む1つまたは複数のベクター(例えば、発現ベクター)が提供される。さらなる態様において、このような核酸を含む宿主細胞が提供される。このような態様の1つでは、宿主細胞は、(1) 抗体のVLを含むアミノ酸配列および抗体のVHを含むアミノ酸配列をコードする核酸を含むベクター、または、(2) 抗体のVLを含むアミノ酸配列をコードする核酸を含む第一のベクターと抗体のVHを含むアミノ酸配列をコードする核酸を含む第二のベクターを含む(例えば、形質転換されている)。一態様において、宿主細胞は、真核性である(例えば、チャイニーズハムスター卵巣 (CHO) 細胞)またはリンパ系の細胞(例えば、Y0、NS0、Sp2/0細胞))。一態様において、抗CTLA-4抗体の発現に好適な条件下で、上述のとおり当該抗体をコードする核酸を含む宿主細胞を培養すること、および任意で、当該抗体を宿主細胞(または宿主細胞培養培地)から回収することを含む、抗CTLA-4抗体を作製する方法が提供される。
本明細書で提供される抗CTLA-4抗体は、当該技術分野において知られている種々の測定法によって、同定され、スクリーニングされ、または物理的/化学的特性および/または生物学的活性について明らかにされてもよい。
一局面において、本発明の抗体は、例えばELISA、ウエスタンブロット、表面プラズモン共鳴アッセイ等の公知の方法によって、その抗原結合活性に関して試験される。
一局面において、生物学的活性を有する抗CTLA-4抗体のそれを同定するための測定法が提供される。生物学的活性は、例えば、細胞増殖阻害活性、細胞傷害活性(例えば、ADCC/CDC活性、ADCP活性)、免疫賦活化活性、CTLA-4阻害活性を含んでよい。また、このような生物学的活性をインビボおよび/またはインビトロで有する抗体が、提供される。
本発明はまた、1つまたは複数の細胞傷害剤(例えば化学療法剤または化学療法薬、増殖阻害剤、毒素(例えば細菌、真菌、植物もしくは動物起源のタンパク質毒素、酵素的に活性な毒素、もしくはそれらの断片)または放射性同位体)にコンジュゲートされた本明細書の抗CTLA-4抗体を含むイムノコンジュゲートを提供する。
特定の態様において、本明細書で提供される抗CTLA-4抗体のいずれも、生物学的サンプルにおけるCTLA-4の存在を検出するのに有用である。本明細書で用いられる用語「検出」は、定量的または定性的な検出を包含する。特定の態様において、生物学的サンプルは、細胞または組織、例えば、血清、全血、血奬、生検試料、組織試料、細胞懸濁液、唾液、痰、口腔液、脳脊髄液、羊水、腹水、乳汁、初乳、乳腺分泌物、リンパ液、尿、汗、涙液、胃液、関節液、腹水、眼液、または粘液を含む。
本明細書に記載の抗CTLA-4抗体の薬学的製剤は、所望の純度を有する抗体を、1つまたは複数の任意の薬学的に許容される担体 (Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)) と混合することによって、凍結乾燥製剤または水溶液の形態で、調製される。薬学的に許容される担体は、概して、用いられる際の用量および濃度ではレシピエントに対して非毒性であり、これらに限定されるものではないが、以下のものを含む:リン酸塩、クエン酸塩、および他の有機酸などの緩衝液;アスコルビン酸およびメチオニンを含む、抗酸化剤;保存料(オクタデシルジメチルベンジル塩化アンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム;塩化ベンゼトニウム;フェノール、ブチル、またはベンジルアルコール;メチルまたはプロピルパラベンなどのアルキルパラベン;カテコール;レソルシノール;シクロヘキサノール;3-ペンタノール;およびm-クレゾールなど);低分子(約10残基未満)ポリペプチド;血清アルブミン、ゼラチン、または免疫グロブリンなどのタンパク質;ポリビニルピロリドンなどの親水性ポリマー;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン、またはリジンなどのアミノ酸;グルコース、マンノース、またはデキストリンを含む、単糖、二糖、および他の炭水化物;EDTAなどのキレート剤;スクロース、マンニトール、トレハロース、ソルビトールなどの、砂糖類;ナトリウムなどの塩形成対イオン類;金属錯体(例えば、Zn-タンパク質錯体);および/またはポリエチレングリコール (PEG) などの非イオン系表面活性剤。本明細書の例示的な薬学的に許容される担体は、さらに、可溶性中性活性型ヒアルロニダーゼ糖タンパク質 (sHASEGP)などの間質性薬剤分散剤を含む。特定の例示的sHASEGPおよびその使用方法は、米国特許出願公開第2005/0260186号および第2006/0104968号に記載されている。一局面において、sHASEGPは、コンドロイチナーゼなどの1つまたは複数の追加的なグリコサミノグリカナーゼと組み合わせられる。
本明細書で提供される抗CTLA-4抗体のいずれも、治療的な方法において使用されてよい。
一局面において、医薬品としての使用のための、抗CTLA-4抗体が提供される。さらなる局面において、腫瘍の治療における使用のための、抗CTLA-4抗体が提供される。特定の態様において、治療方法における使用のための、抗CTLA-4抗体が提供される。特定の態様において、本発明は、腫瘍を有する個体を治療する方法であって、当該個体に抗CTLA-4抗体の有効量を投与する工程を含む方法における使用のための、抗CTLA-4抗体を提供する。さらなる態様において、本発明は、細胞の傷害における使用のための抗CTLA-4抗体を提供する。特定の態様において、本発明は、個体において細胞を傷害する方法であって、細胞を傷害するために当該個体に抗CTLA-4抗体の有効量を投与する工程を含む方法における使用のための、抗CTLA-4抗体を提供する。上記態様の任意のものによる「個体」は、好適にはヒトである。
本発明の別の局面において、上述の障害の治療、予防、および/または診断に有用な器材を含んだ製品が、提供される。製品は、容器、および当該容器上のラベルまたは当該容器に付属する添付文書を含む。好ましい容器としては、例えば、ボトル、バイアル、シリンジ、IV(intravenous)溶液バッグなどが含まれる。容器類は、ガラスやプラスチックなどの、様々な材料から形成されていてよい。容器は組成物を単体で保持してもよいし、症状の治療、予防、および/または診断のために有効な別の組成物と組み合わせて保持してもよく、また、無菌的なアクセスポートを有していてもよい(例えば、容器は、皮下注射針によって突き通すことのできるストッパーを有する静脈内投与用溶液バッグまたはバイアルであってよい)。組成物中の少なくとも1つの有効成分は、本発明の抗体である。ラベルまたは添付文書は、組成物が選ばれた症状を治療するために使用されるものであることを示す。本発明のこの態様における製品は、さらに、組成物が特定の症状を治療するために使用され得ることを示す、添付文書を含んでもよい。あるいはまたは加えて、製品はさらに、注射用制菌水 (BWFI)、リン酸緩衝生理食塩水、リンガー溶液、およびデキストロース溶液などの、薬学的に許容される緩衝液を含む、第二の容器を含んでもよい。他の緩衝液、希釈剤、フィルター、針、およびシリンジなどの、他の商業的観点またはユーザの立場から望ましい器材をさらに含んでもよい。
<変異Fc領域を含むポリペプチド>
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、および298、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、および334。
特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置326におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置330におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置330におけるアミノ酸改変を含む。あるいは、国際公開WO2013/002362やWO2014/104165に記載のアミノ酸改変も本発明において同様に用いられ得る。
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および330、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、330、および334。
特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置326におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置332におけるアミノ酸改変を含む。特定の態様において、あるいは、国際公開WO2013/002362やWO2014/104165に記載のアミノ酸改変も本発明において同様に用いられ得る。
一態様において、本発明の変異Fc領域は、EUナンバリングで表される位置234、235、236、239、250、268、270、298、307、326、330、332、および334におけるアミノ酸改変を含む。特定の態様において、本発明の変異Fc領域は、(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、250、268、270、298、307、330、および332、ならびに(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、250、270、298、307、326、330、332、および334におけるアミノ酸改変を含む。別の特定の態様において、本発明の変異Fc領域は、(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、250、268、270、298、307、および326、ならびに(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置236、250、270、298、307、326、330、332、および334におけるアミノ酸改変を含む。
(a) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置356におけるLys、および親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置439におけるGlu、
(b) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置439におけるGlu、および親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置356におけるLys。
(c) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置366におけるTrp、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置366におけるSer、位置368におけるAla、および位置407におけるVal、
(d) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置366におけるSer、位置368におけるAla、および位置407におけるVal、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置366におけるTrp、
(e) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置349におけるCysおよび位置366におけるTrp、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置356におけるCys、位置366におけるSer、位置368におけるAla、および位置407におけるVal、
(f) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置356におけるCys、位置366におけるSer、位置368におけるAla、および位置407におけるVal、ならびに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置349におけるCysおよび位置366におけるTrp。
(a) EUナンバリングで表される位置434におけるAla、
(b) EUナンバリングで表される位置434におけるAla、位置436におけるThr、位置438におけるArg、位置440におけるGlu、
(c) EUナンバリングで表される位置428におけるLeu、位置434におけるAla、位置436におけるThr、位置438におけるArg、位置440におけるGlu、
(d) EUナンバリングで表される位置428におけるLeu、位置434におけるAla、位置438におけるArg、位置440におけるGlu。
一局面において、医薬品としての使用のための、変異Fc領域を含むポリペプチドが提供される。さらなる局面において、腫瘍の治療における使用のための、変異Fc領域を含むポリペプチドが提供される。特定の態様において、治療方法における使用のための、変異Fc領域を含むポリペプチドが提供される。特定の態様において、本発明は、腫瘍を有する個体を治療する方法であって、当該個体に変異Fc領域を含むポリペプチドの有効量を投与する工程を含む方法における使用のための、変異Fc領域を含むポリペプチドを提供する。さらなる態様において、本発明は、細胞の傷害における使用のための、変異Fc領域を含むポリペプチドを提供する。特定の態様において、本発明は、個体において細胞を傷害する方法であって、細胞を傷害するために当該個体に変異Fc領域を含むポリペプチドの有効量を投与する工程を含む方法における使用のための、変異Fc領域を含むポリペプチドを提供する。上記態様の任意のものによる「個体」は、好適にはヒトである。
<変異Fc領域を含む抗CTLA-4抗体>
(A) アデノシン含有化合物の濃度に依存したCTLA-4結合活性を有する可変領域、および
(B) 親Fc領域において複数のアミノ酸改変を含む変異Fc領域
を含む抗CTLA-4抗体であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、抗CTLA-4抗体:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および330、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、330、および334
を提供する。
イピリムマブはエフェクターT細胞表面に発現するCTLA4によるエフェクターT細胞の活性化抑制を阻害することで抗腫瘍効果が発揮されていると考えられていたが、最近、CTLA4発現T細胞に対する抗体依存的細胞傷害活性(ADCC活性)も重要であることが報告され、腫瘍中の制御性T細胞の除去とADCC活性が抗CTLA4抗体の抗腫瘍効果の重要な作用機序であることが見出されている。
(1-1)改変されたFc領域を有する抗CTLA4スイッチ抗体の発現と精製
参考実施例6および7で見出された改変を導入したFc領域と、参考実施例4で作製された抗CTLA4スイッチ抗体の可変領域を組み合わせることで、改変されたFc領域を有する抗CTLA4スイッチ抗体(04H1654-KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1 略名:SW1610-ART5+ACT1、04H1654-KT456/04L1610-lam1//04H1656-HT446/04L1610-lam1 略名:SW1610-ART4、および04H1654-KT498/04L1610-lam1//04H1656-HT518/04L1610-lam1 略名:SW1610-ART12)が各種作製された。
SW1610-ART4抗体は、一方の重鎖としてヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のKn456と同じ改変を有し、CH3領域にWO2006/106905に記載のヘテロ二量化を促進する改変E356Kを有し、重鎖可変領域として04H1654(配列番号:140)をもつ抗体重鎖04H1654-KT456(配列番号:335)の遺伝子が作製された。同様に、ヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のHl446と同じ改変を有し、CH3領域にWO2006/106905に記載のヘテロ二量化を促進する改変K439Eを有し、重鎖可変領域として04H1656(配列番号:141)をもつ抗体重鎖04H1656-HT446(配列番号:336)の遺伝子が作製された。さらに軽鎖として04L1610-lam1(配列番号:161)を使用し、当業者公知の方法で発現、精製された。
SW1610-ART12抗体は、同様に一方の重鎖としてヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のKn498と同じ改変を有し、CH3領域にWO2006/106905に記載のヘテロ二量化を促進する改変E356Kを有し、重鎖可変領域として04H1654(配列番号:140)をもつ抗体重鎖04H1654-KT498(配列番号:337)の遺伝子が作製された。同様に、ヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のHl518と同じ改変を有し、CH3領域にWO2006/106905に記載のヘテロ二量化を促進する改変K439Eを有し、重鎖可変領域として04H1656(配列番号:141)をもつ抗体重鎖04H1656-HT518(配列番号:338)の遺伝子が作製された。さらに軽鎖として04L1610-lam1(配列番号:161)を使用し、当業者公知の方法で発現、精製された。
実施例1-1で作製された抗体04H1654-KT498/04L1610-lam1//04H1656-HT518/04L1610-lam1(SW1610-ART12)の各ヒトFcγR(以下hFcγRと示す)および各カニクイザルFcγR (以下cyFcγRと示す)に対する結合活性が Biacore 8k+(Cytiva)を用いて評価された。この時、L鎖がλ鎖となっているIgG1抗体であるTremfya(Janssen Pharmaceutical K.K)もコントロールとして評価された。結合活性にはランニングバッファーとして50 mMリン酸緩衝液、150 mM NaCl、0.05 w/v%-P20、pH7.4が用いられ、25℃で評価が実施された。リガンド捕捉用分子としてCaptureSelect(商標)Human Fab-lambda Kinetics Biotin Conjugate(ThermoFisher scientific)をSeries S Sensor chip SA センサーチップ(Cytiva)に固相化した。ランニングバッファーで調製した抗体溶液をこのSAセンサーチップに相互作用させることで、抗体を約250RUもしくは500RUもしくは約1000 RU捕捉させた。その後、ランニングバッファーで希釈したhFcγRおよびcyFcγRを、捕捉させた抗体に結合させた。この時、hFcγRsおよびcyFcγRsは、表2に記載の濃度を最大濃度として、公比2にて5点以上の濃度に希釈され、測定に用いられた。チップは10 mM Glycine-HCl (pH 1.5) を用いて再生され、繰り返し抗体を捕捉して測定が行われた。各抗体の各FcγRに対する結合活性は、Biacore Insight Evaluation Softwareを用いてSteady stateモデルもしくは1:1 bindingモデルにてKD値を算出することにより評価された。

hFcγRの細胞外ドメインの遺伝子配列は、hFcγRIa(NCBIリファレンス配列:NM_000566.3)、hFcγRIIa(NCBIリファレンス配列:NM_001136219.1)、hFcγRIIb(NCBIリファレンス配列:NM_004001.3)、hFcγRIIIa(NCBIリファレンス配列:NM_001127593.1)から取得されて、多型部位に関しては次の文献を参考にして設計された(hFcγRIIaRについては Warmerdam, P. A. M. et al., 1990, J. Exp. Med. 172:19-25、hFcγRIIIaRについては Wu, J. et al., 1997, J. Clin. Invest. 100(5):1059-1070)。発現および精製に使用したhFcγRの細胞外ドメインのアミノ酸配列は配列表に次のように示された:hFcγRIa(配列番号:341)、hFcγRIIa(H)(配列番号:342)、hFcγRIIa(R)(配列番号:343)、hFcγRIIb(配列番号:344)、hFcγRIIIa(F)(配列番号:345)、hFcγRIIIa(V)(配列番号:346)。次に、HisタグをC末端に付加し、得られた各遺伝子は哺乳動物細胞発現用に設計された発現ベクターに当業者公知の方法によって挿入された。発現ベクターはヒト胎児腎臓細胞由来のFreeStyle293細胞(Invitrogen)に導入されて、標的タンパク質は発現させられた。培養後、得られた培養上清は、原則として以下の4工程、あるいは最初の陰イオン交換クロマトグラフィーの工程を除いた3工程でろ過・精製された。最初のステップとしてQ Sepharose Fast Flow(GE Healthcare)を使用した陰イオン交換クロマトグラフィーが実行された。2番目のステップとして、HisTrap HP(GE Healthcare)を使用して Hisタグに対するアフィニティークロマトグラフィーが実行された。3番目のステップとして、HiLoad 26/600 Superdex 200 pg(GE Healthcare)を用いてゲルろ過カラムクロマトグラフィーが実行された。4番目のステップとして滅菌ろ過が実行された。精製タンパク質の280nmでの吸光度は分光光度計を使用して測定されて、精製タンパク質の濃度はPACEの方法(Protein Science 4:2411-2423(1995))によって計算された吸光係数を使用して決定された。


(2-1)ヒト末梢血単核球をエフェクター細胞として用いた、改変された Fc領域を有する抗CTLA4スイッチ抗体のADCC活性
実施例1で作製された、ATP依存的に抗原に結合する抗体04H1654-KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1(SW1610-ART5+ACT1)、04H1654-KT456/04L1610-lam1//04H1656-HT446/04L1610-lam1(SW1610-ART4)、04H1654-KT498/04L1610-lam1//04H1656-HT518/04L1610-lam1(SW1610-ART12)は以下の方法に従って抗体濃度依存的なADCC活性が測定された。この時、ヒト末梢血単核球(以下、ヒトPBMCと指称する。)をエフェクター細胞として用いて被験抗体のADCC活性が以下のように測定された。
その後ラベル化標的細胞は10 % FBS/ RPMIで洗浄された後、2×105細胞/mLになるように10 % FBS/ RPMIを用いて調製された。
ADCC活性(%)={(A-C)-(B-C)}×100
実施例1-1で作製された04H1654-KT498/04L1610-lam1//04H1656-HT518/04L1610-lam1(SW1610-ART12)のCTLA4陽性制御性T細胞(CD3+ CD4+ CD25+ CD45RA- CTLA4+)に対するIn vitro細胞傷害活性が評価された。まずヒトPBMC(Cryopreserved Human PBMC, ASTARTE)が凍結融解され、CD3/CD28 Ab Dynabeads (1:1)/ OpTmizer/L-Gln/Penicillin-Streptomycin中に細胞密度が4×105細胞/ mLとなるように懸濁され、5 % CO2インキュベーター内にて7日間37 ℃で培養された。7日後、細胞を回収しRPMI/ 10 % FBSで2回洗浄された後に、96ウェルV底プレートの各ウェル中に50 μLずつ播種され(1×106 細胞/ウェル)、続いて各濃度(0.0003、0.003、0.03、0.3、3、30 μg/mL)にRPMI/ 10 % FBSで調製した抗体溶液が96ウェルV底プレートの各ウェル中に50 μLずつ添加された。さらに、0あるいは300 μMに10 % FBS/ RPMIで調製されたATP溶液が50 μLずつ添加され、よく懸濁された後にCO2インキュベーター内にて6時間37 ℃で静置された。6時間後にPBMCを回収しauto MACS Rinsing Solution(Milteny)で2回洗浄した後に、以下の抗体と反応させ、FACS解析によって存在する免疫細胞の画分が解析された。生死判定用試薬(Biolegend、Zombie Aqua)、抗CD3抗体(BD、クローン: UCHT1)、抗CD4抗体(BD、クローン: RPA-T4)、抗CD8抗体(BD、クローン:SK1)、抗CD45RA抗体(Biolegend、クローン:5Hg)、抗CD25抗体(BD、クローン: 2A3)、抗CTLA4抗体(Biolegend、クローン:BNI3)。FACS解析はBD LSR Fortessa X-20(BD)にて行われた。得られた結果を図4に示す。
(3-1)カニクイザル毒性試験に供する、抗CTLA4コントロール抗体および抗CTLA4スイッチ抗体の作製
SW1389-ART1抗体は、一方の重鎖としてヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のKn125と同じ改変を有し、CH3領域にヘテロ二量化を促進する改変S354C/T366Wを有し、重鎖可変領域として04H1389(配列番号:29)をもつ抗体重鎖04H1389-Kp125(配列番号:339)の遺伝子が作製された。同様に、ヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のHl076と同じ改変を有し、CH3領域にヘテロ二量化を促進する改変Y349C/T366S/L368A/Y407Vを有し、重鎖可変領域として04H1389(配列番号:29)をもつ抗体重鎖04H1389-Hp076(配列番号:340)の遺伝子が作製された。さらに軽鎖として04L1305-k0MT(配列番号:275)を使用し、当業者公知の方法で発現、精製された。
SW1389-ART5+ACT1抗体は、一方の重鎖としてヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のKn462と同じ改変を有し、CH3領域にヘテロ二量化を促進する改変S354C/T366Wを有し、重鎖可変領域として04H1389(配列番号:29)をもつ抗体重鎖04H1389-Kp462(配列番号:354)の遺伝子が作製され、さらに、Mabs, 2017, 9, 844-853に記載されている抗体の血中動態を改善する改変の組み合わせが導入された。具体的には04H1389-Kp462のCH3領域に対して、酸性条件下においてヒトFcRnに対する結合を増強する改変およびRheumatoid factorに対する結合を低減する改変の組み合わせであるN434A/Y436T/Q438R/S440Eが導入された04H1389-Kp473(配列番号:355)の遺伝子が作製された。同様に、ヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域に参考実施例7に記載のHl441と同じ改変を有し、CH3領域にヘテロ二量化を促進する改変Y349C/T366S/L368A/Y407Vを有し、重鎖可変領域として04H1389(配列番号:29)をもつ抗体重鎖04H1389-Hp441(配列番号:356)にも同様の改変が導入された、04H1389-Hp451(配列番号:357)の遺伝子が作製された。さらに軽鎖として04L1305-k0MT(配列番号:275)を使用し、当業者公知の方法で発現、精製された。
SW1610-ART4抗体およびSW1610-ART12抗体は、実施例1-1で作製された抗体が使用された。
全身反応をはじめとする毒性を評価・比較する目的で,実施例3-1で調製されたSW1389-ART1、SW1389-ART5+ACT1、SW1610-ART4、およびSW1610-ART12抗体をカニクイザル(雌雄各3例)に毎週1回、計5回投与した。投与用量は1回目投与7日後の血漿中濃度が各抗体で一定になる用量として、それぞれ60 mg/kg、20 mg/kg、60 mg/kg、および30 mg/kgを設定した。投与はシリンジポンプを用いて低速静脈内投与し、一般状態観察、体重測定、血液・血液化学的検査、血中自己抗体測定、イムノフェノタイピング、血中サイトカイン測定、骨髄検査、病理学的検査、および血漿中薬物濃度測定を実施した。
0.3 mg/kgおよび3 mg/kgのSW1610-ART12抗体投与群においても、多くの例で投与期間中から抗薬物抗体の発現およびそれに伴う曝露低下が認められた。本試験においても一般状態や体重等に重篤な所見はみられなかったが、0.3 mg/kgからは自己抗体の発現、血中の炎症パラメータの増加、腎臓における軽微~軽度の病理組織学的変化が認められ、これらの毒性変化の発現率・重症度には用量依存性があった。以上より、SW1610-ART12抗体を用いたカニクイザルの毒性試験において、推定薬効用量の100倍にあたる30 mg/kgまで各種毒性変化の発現率・重症度の用量依存性、及び忍容性が認められた。
(1-1)低分子存在下において抗原に結合する抗体の取得のための抗原調製
抗原として、ビオチン化されたマウスCTLA4細胞外領域(mCTLA4)、ヒトCTLA4細胞外領域(hCTLA4)、アバタセプトが調製された。具体的にはhCTLA4細胞外領域はC末端にHisタグおよびBAPタグが融合したものhCTLA4-His-BAP(配列番号:1)が遺伝子合成され、動物発現用プラスミドへ挿入された。抗原タンパク質は以下の方法を用いて発現、精製された。FreeStyle 293 Expression Medium培地(Invitrogen)に1.33×106細胞/mLの細胞密度で懸濁されて、フラスコに播種されたヒト胎児腎細胞由来FreeStyle 293-F株(Invitrogen)に対して、調製されたプラスミドがリポフェクション法により導入された。プラスミド導入後3時間後にビオチンが終濃度100 μMとなるように添加され、CO2インキュベーター(37℃、8% CO2、125 rpm)で4日間培養された培養上清から、当業者公知の方法で抗原が精製された。分光光度計を用いて、精製された抗原溶液の280 nmでの吸光度が測定された。得られた測定値からPACE法により算出された吸光係数を用いて精製された抗原の濃度が算出された(Protein Science (1995) 4, 2411-2423)。一方mCTLA4の細胞外領域にHisタグが融合されたmCTLA4-His(Sino Biologics Inc. 50503-M08H, Accession No. NP_033973.2)およびhCTLA4にヒトIgG1定常領域が融合したアバタセプト(アルフレッサ株式会社)は、アミンカップリング法によりビオチン化された(PIERCE Cat.No.21329)。
ヒトPBMCから作成したポリA RNAや、市販されているヒトポリA RNAなどを鋳型として当業者に公知な方法に従い、互いに異なるヒト抗体配列のFabドメインを提示する複数のファージからなるヒト抗体ファージディスプレイライブラリが構築された。
(1-2)で得られた大腸菌のシングルコロニーから、常法(Methods Mol. Biol. (2002) 178, 133-145)に習い、ファージ含有培養上清が回収された。NucleoFast 96 (MACHERY-NAGEL)を用いて回収された培養上清が限外濾過された。培養上清各100 μLがNucleoFast 96の各ウェルにアプライされ、4,500 g、45分間遠心分離を行いフロースルーが除去された。H2O 100 μLを加え、再度4,500 g、30分間遠心分離による洗浄が行われた。その後、TBS 100 μLを加え、室温で5分間静置した後、上清に含まれるファージ液が回収された。

先行特許WO2015/083764において構築されたラショナルデザイン抗体ファージディスプレイライブラリから、ATPまたはATP代謝物(たとえばADP、AMP、アデノシン(ADO)など)存在条件下で抗原に対する結合活性を示す抗体が取得された。取得のために、ATPまたはATP代謝物存在下でビーズにキャプチャーされた抗原に対して結合能を示す抗体を提示しているファージが回収され、その後ATPまたはATP代謝物の非存在条件下でビーズから溶出された溶出液からファージが回収された。
上記の方法によって得られた大腸菌のシングルコロニーから、常法(Methods Mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。NucleoFast 96(MACHEREY-NAGEL)を用いて、回収された培養上清が限外ろ過された。培養上清各100 μLが各ウェルにアプライされたNucleoFast 96を遠心分離(4,500 g、45分間)することによってフロースルーが除去された。100 μLのH2Oが各ウェルに加えられた当該NucleoFast 96が、再度遠心分離(4,500 g、30分間)によって洗浄された。最後にTBS 100 μLが加えられ、室温で5分間静置された当該NucleoFast 96の各ウェルの上清に含まれるファージ液が回収された。

ファージELISAの結果、ATPおよびその代謝物が存在する条件下で抗原に対する結合活性があると判断されたクローンから特異的なプライマーlacPF(配列番号:2), G1seqR(配列番号:3)を用いて増幅された遺伝子の塩基配列が解析された。解析の結果、ATPおよびその代謝物存在下でビオチン標識アバタセプトに対する結合活性を有すると判断されたクローンABADh11-4_020、ABADh11-4_086、ABADh12-4_014、ABADh12-5_001、ABADh12-5_046、ABADh5-5_041が取得され、それぞれABAM001、ABAM002、ABAM003、ABAM004、ABAM005、ABAM006とクローン名が再付与された(表7)。

ヒトラショナルデザインファージライブラリから取得されたABAM001、ABAM002、ABAM003、ABAM004、ABAM005、ABAM006の可変領域をコードする遺伝子はヒトIgG1/Lambdaの動物発現用プラスミドへ挿入された。以下の方法を用いて抗体が発現された。FreeStyle 293 Expression Medium培地(Invitrogen)に1.33×106細胞/mLの細胞密度で懸濁されて、6well plateの各ウェルへ3 mLずつ播種されたヒト胎児腎細胞由来FreeStyle 293-F株(Invitrogen)に対して、調製されたプラスミドがリポフェクション法により導入された。CO2インキュベーター(37℃、8 % CO2、90 rpm)で4日間培養された培養上清から、rProtein A SepharoseTM Fast Flow(Amersham Biosciences)を用いて当業者公知の方法で抗体が精製された。分光光度計を用いて、精製された抗体溶液の280 nmでの吸光度が測定された。得られた測定値からPACE法により算出された吸光係数を用いて精製された抗体の濃度が算出された(Protein Science (1995) 4, 2411-2423)。
取得されたABAM001、ABAM002、ABAM003、ABAM004、ABAM005、ABAM006の6抗体がIgG ELISAに供された。また表8に示すBufferが適宜調製された。抗原としてビオチン標識されたヒトCTLA4(hCTLA4-His-Biotin)が用いられた。


CTLA4スイッチ抗体としてABAM004の更なる評価が行われた。
Biacore T200(GE Healthcare)を用いて、ABAM004とhCTLA4-His-BAPとの抗原抗体反応の相互作用が解析された。アミンカップリング法でprotein A/G(Pierce)が適当量固定化されたSensor chip CM5(GE Healthcare)にABAM004をキャプチャーさせ、抗原である参考実施例1-1で調製されたhCTLA4-His-BAPを相互作用させた。ランニングバッファーにはTBSが用いられ、再生溶液としては10 mM Glycine-HCl (pH 1.5)が用いられた。
Flow cytometerを用いて、ABAM004とヒトCTLA4との抗原抗体相互作用がAMP存在下、非存在下でどのように変化するのか評価された。ヒトCTLA4を安定発現するCHO細胞(hCTLA4-CHO細胞)が適切な濃度で準備された。この時懸濁には0.1 % BSA入りPBS(FACS Buffer)が用いられた。当該細胞溶液100 μLに対し、終濃度10 mg/mLとなるように抗体が添加されたのち、AMPが終濃度0, 0.4, 4, 40, 200, 1000 μMになるように添加され、4℃で30分間静置された。その後FACS BufferにAMPが終濃度0, 0.4, 4, 40, 200, 1000 μMになるように添加されたものをWash Bufferとして使用し、当該細胞株が洗浄されたのち、FITC標識された二次抗体(Goat F(ab'2) Anti-Human IgG Mouse ads-FITC、Beckman 732598)が添加され、再び4℃で30分間遮光下に静置された。再度洗浄操作が行われた後、Flow cytometer (FACS CyAnTM ADP)で測定、解析された。結果を図6に示す。
ATP依存的に抗原に結合する抗体は以下の方法に従って抗体濃度依存的なADCC活性が測定された。この時、ヒト末梢血単核球(以下、ヒトPBMCと指称する。)をエフェクター細胞として用いて被験抗体のADCC活性が以下のように測定された。
ADCC活性(%)={(A-D)-(C-D)}×100/{(B-D)-(C-D)}
(2-1)AMP をスイッチとする抗CTLA4結合抗体ABAM004のX線結晶構造解析
参考実施例1においてライブラリより取得されたAMPをスイッチとするhCTLA4結合抗体ABAM004のFab断片単独、ABAM004のFab断片とAMPの複合体、並びにABAM004のFab断片とAMP、hCTLA4の細胞外ドメインの複合体の結晶構造が解析された。
結晶化用ABAM004全長抗体の調製及び精製は当業者公知の方法により行われた。
ABAM004のFab断片を、rLys-C(Promega、カタログ番号V1671)による制限消化、続いて、Fc断片を除去するためのプロテインAカラム(MabSelect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex200 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
2-3の方法で精製した結晶化用ABAM004のFab断片を約13 mg/mLに濃縮し、シッティングドロップ蒸気拡散法により20℃で結晶化を行った。リザーバー溶液は、0.1 M MES pH 6.5、25 %w/v ポリエチレングリコール4000から成るものであった。これにより得られた結晶を0.08 M MES pH 6.5、20% w/v ポリエチレングリコール4000、20% エチレングリコールの溶液に浸漬した。
X線回折データを、高エネルギー加速器研究機構の放射光施設フォトンファクトリーのBL-17Aで測定した。測定中、結晶を常に-178℃の窒素流下に置いて凍結状態を維持し、ビームラインに接続されたQuantum 270 CCD検出器(ADSC)を用いて、結晶を1回に0.5°回転させながら、合計360枚のX線回折画像を収集した。セルパラメータの決定、回折スポットのインデックス付け、および回折像から得られた回折データの処理を、Xia2プログラム(J. Appl. Cryst. 43: 186-190 (2010))、XDSパッケージ(Acta. Cryst. D66: 125-132 (2010))、およびScala(Acta. Cryst. D62: 72-82 (2006))を用いて行い、最終的に分解能1.70Åまでの回折強度データを取得した。結晶学的データ統計値が表10に示される。

ABAM004のFab断片を、パパイン(Roche Diagnostics、カタログ番号1047825)による制限消化、続いて、Fc断片を除去するためのプロテインAカラム(MabSelect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex200 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製した。Fab断片を含む画分をプールして、-80℃で保存した。
2-6の方法で精製した結晶化用ABAM004のFab断片を約13 mg/mLに濃縮したものに対して、終濃度が2 mMになるようにAMPを添加し、シッティングドロップ蒸気拡散法により20℃で結晶化を行った。リザーバー溶液は、0.1 M Morpheus buffer 2 pH 7.5、37.5% w/v MPD_P1K_P3350、10% Morpheus Carboxylic acids(Morpheus、Molecular Dimensions)から成るものであった。
X線回折データを、高エネルギー加速器研究機構の放射光施設フォトンファクトリーのBL-1Aで測定した。測定中、結晶を常に-178℃の窒素流下に置いて凍結状態を維持し、ビームラインに接続されたPilatus 2M 検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計720枚のX線回折画像を収集した。セルパラメータの決定、回折スポットのインデックス付け、および回折像から得られた回折データの処理を、Xia2プログラム(J. Appl. Cryst. 43: 186-190 (2010))、XDSパッケージ(Acta. Cryst. D66: 125-132 (2010))、およびScala(Acta. Cryst. D62: 72-82 (2006))を用いて行い、最終的に分解能1.70Åまでの回折強度データを取得した。結晶学的データ統計値が表10に示される。
hCTLA4の細胞外ドメインは、アバタセプトにEndoproteinase Lys-C(Roche、カタログ番号11047825001)による制限消化、続いて、Fc断片を除去するためのプロテインAカラム(MabSelect SuRe、GE Healthcare)、およびゲル濾過カラム(Superdex200 10/300、GE Healthcare)へのローディングを用いるという方法により調製した。hCTLA4の細胞外ドメインを含む画分をプールして、-80℃で保存した。
2-9の方法で精製したhCTLA4細胞外ドメインを、2-6の方法で精製したABAM004のFab断片と1.5:1のモル比で混合し、終濃度が2 mMになるようにAMPを添加した。複合体を、25 mM HEPES pH7.5、100 mM NaCl、2 mM AMPで平衡化したカラムを用いたゲル濾過クロマトグラフィー(Superdex200 10/300、GE Healthcare)により精製した。
精製した複合体を約8 mg/mLに濃縮し、シーディング法と組み合わせたシッティングドロップ蒸気拡散法により20℃で結晶化を行った。リザーバー溶液は、0.1 M Morpheus buffer 1 pH 6.5、37.5% w/v M1K3350、10% halogens(Morpheus、Molecular Dimensions)から成るものであった。
X線回折データを、SPring-8のBL32XUで測定した。測定中、結晶を常に-178℃の窒素流下に置いて凍結状態を維持し、ビームラインに接続されたMX-225HS CCD検出器(RAYONIX)を用いて、結晶を1回に1.0°回転させながら、合計180枚のX線回折画像を収集した。セルパラメータの決定、回折スポットのインデックス付け、および回折像から得られた回折データの処理を、Xia2プログラム(J. Appl. Cryst. 43: 186-190 (2010))、XDSパッケージ(Acta. Cryst. D66: 125-132 (2010))、およびScala(Acta. Cryst. D62: 72-82 (2006))を用いて行い、最終的に分解能1.70Åまでの回折強度データを取得した。結晶学的データ統計値が表10に示される。
本結晶構造より、AMPは、主に当該抗体の重鎖によって認識されることがわかった。
AMPのアデニン環部分は重鎖CDR1、3によって、リボース部分並びにリン酸基部分は、CDR1、2によって認識される。
図9および、図10では、ABAM004 Fab接触領域のエピトープが、それぞれ、hCTLA4結晶構造中およびアミノ酸配列中にマッピングされている。エピトープは、結晶構造においてABAM004 FabまたはAMPのいずれかの部分から4.2Å以内の距離に位置する非水素原子を1個以上含むhCTLA4のアミノ酸残基を含んでいる。
少なくとも、抗原のM3、E33、R35、T53、E97、M99、Y100、P101、P102、P103、Y104、Y105、L106は、当該抗体の重鎖CDR2、CDR3、軽鎖CDR1、CDR3ならびに、AMPによって認識されることが結晶構造から明らかになった。特に、抗原のM99からY104から成るループが、当該抗体のCDRループの中に埋もれるように当該抗体に強く認識されており、抗体による抗原認識に大きな役割を果たしていると考えられる。
図11は、ABAM004 Fab単独の結晶構造、ABAM004 FabとAMPの複合体の結晶構造、並びにABAM004 FabとAMP及びCTLA4から成る3者複合体の結晶構造から、当該抗体の可変領域を抽出し、重鎖を中心にして重ね合わせた図である。AMP依存的な抗原結合は、参考実施例2-14で示したAMPとCTLA4の間の直接的な相互作用だけではなく、AMPの結合に伴う当該抗体の構造変化が重要であると考えられる。
(3-1)ABAM004抗体のCTLA4結合活性増強改変体の作製
参考実施例1に記載のヒトラショナルデザインファージライブラリから取得されたABAM004(VH配列番号:10、VL配列番号:11)はアミノ酸配列が改変され、当該配列とATP類似体非存在下のCTLA4結合活性が低下され、ATP類似体存在下でのヒトCTLA4結合活性が向上され、ATPおよびATP類似体への結合が向上された。このために参考実施例2に記載の方法で取得されたABAM004とAMPの共結晶構造及びABAM004とAMP、ヒトCTLA4の共結晶構造を基に当該結合に関与することが予想される残基の点変異体が作製された。さらにCDRに含まれる全アミノ酸をAlaあるいはProで置換した改変体も作製された。当該点変異体はBiacore T200あるいはBiacore 4000(GE Healthcare)によりATP非存在下およびATP、ADPあるいはAMP存在下のヒトCTLA4(AbataceptとhCTLA4-His-BAP)結合活性が測定され、当該結合活性を向上させる変異がスクリーニングされた。結合活性を向上させた変異を組み合わせた変異体が作製され、BiacoreによりKD値算出が行われた。その結果、ABAM004の重鎖をH32A、S52aT、軽鎖をT24D、T26P、E50F(番号はKabat番号)に置換するとABAM004の当該結合特性を向上させることが明らかになった。当該変異体を04H0150/04L0072(VH配列番号:47、VL配列番号:48)と呼称する。
最初にBiacore T200測定用にチップが作成された。Biacore T200の設定温度は25℃、流速は10 μL/minとされた。ランニングバッファーにはHBS-EP+が用いられた。Sensor chip CM5(GE Healthcare)に対し、NHS(N-hydroxysuccinimide)とEDC(N-ethyl-N'-(dimethylaminopropyl) carbodiimide)の等量混合液が流速10 μL/minで10分間添加されflow cellが活性化された。次に10 mM sodium acetate pH 4.0に懸濁した25 μg/mLのProtein A/G(Pierce)が10 μL/minで30分間添加され結合された。その後、flow cell上の過剰の活性基は1 M ethanolamine-HClが10 μL/minで10分間添加されブロックされた。

より優れた抗CTLA4抗体を作製するため、参考実施例3-1において作製された抗ヒトCTLA4抗体の可変領域である04H0150/04L0072に対してアミノ酸改変が網羅的に導入された。PCR等当業者公知の方法により04H0150および04L0072の全てのCDRに対してシステインを除く全18アミノ酸への置換体が作製された。作製された約1200個の改変体のヒトCTLA4に対する結合測定がBiacore 4000を用いて実施された。Series S Sensor Chip CM5(GE Healthcare)にProtein A/G(Thermo Fisher Scientific)を固定化したチップに対して、抗体改変体の培養上清を相互作用させて抗体がキャプチャーされた。次に小分子(ATP、ADP、あるいはAMP)を添加したヒトCTLA4溶液、あるいは小分子を添加していないヒトCTLA4溶液を相互作用させ、小分子存在下、および小分子非存在下での抗体とヒトCTLA4との結合能が評価された。ランニングバッファーとしてTris buffered saline, 0.02% PS20を用い、25℃で測定が行われた。
なお、本明細書における抗体は、以下の規則に従い命名されている;(重鎖可変領域)-(重鎖定常領域)/(軽鎖可変領域)-(軽鎖定常領域)。例えば、04H0150-G1m/04L0072-lam1という抗体名であれば、本抗体の重鎖可変領域は04H0150、重鎖定常領域はG1m、軽鎖可変領域は04L0072、軽鎖定常領域はlam1であることを意味する。


マウスCTLA4細胞外領域にHisタグが連結されたもの(mCTLA4-His)(配列番号:49)が遺伝子合成され、動物発現用プラスミドへ挿入された。FreeStyle 293 Expression Medium培地(Invitrogen)に1.33×106細胞/mLの細胞密度で懸濁されて、フラスコに播種されたヒト胎児腎細胞由来FreeStyle 293-F株(Invitrogen)に対して、調製されたプラスミドがリポフェクション法により導入された。分光光度計を用いて、精製された抗原溶液の280 nmでの吸光度が測定された。得られた測定値からPACE法により算出された吸光係数を用いて精製された抗原の濃度が算出された(Protein Science (1995) 4, 2411-2423)。

抗mCTLA4コントロール抗体(hUH02-mFa55/hUL01-mk1 略名:mNS-mFa55)および抗CTLA4スイッチ(04H1077-mFa55/04L1086-ml0r 略名:SW1077-mFa55、04H1208-mFa55/04L1407s-mk1 略名:SW1208-mFa55)が作製された。mNS-mFa55抗体は、重鎖可変領域hUH02(配列番号:16)および軽鎖可変領域hUL01(配列番号:17)を用い、定常領域はマウス重鎖定常領域mFa55(配列番号:18)および野生型マウス軽鎖定常領域mk1(配列番号:19)を使用した。この時Fcγ受容体への結合を増強するよう改変を加えたマウス重鎖定常領域を使用し、当業者公知の方法で発現、精製された。
SW1077-mFa55抗体は、重鎖可変領域04H1077(配列番号:20)および軽鎖可変領域04L1086(配列番号:21)を用い、定常領域はマウス重鎖定常領域mFa55(配列番号:18)および野生型マウス軽鎖定常領域ml0r(配列番号:22)を使用した。この時Fcγ受容体への結合を増強するよう改変を加えたマウス重鎖定常領域を使用し、当業者公知の方法で発現、精製された。
SW1208-mFa55抗体は、重鎖可変領域04H1208(配列番号:23)および軽鎖可変領域04L1407s(配列番号:24)を用い、定常領域はマウス重鎖定常領域mFa55(配列番号:18)および野生型マウス軽鎖定常領域mk1(配列番号:19)を使用した。この時Fcγ受容体への結合を増強するよう改変を加えたマウス重鎖定常領域を使用し、当業者公知の方法で発現、精製された。
参考実施例3-4で調製された抗CTLA4スイッチ抗体(SW1077-mFa55)の中和活性は競合ELISA法により評価された。96ウェルのプレートに、mCTLA4にヒト定常領域を連結したmCTLA4-Fc(配列番号:25)を0.1 M NaHCO3, 0.05% NaN3で5 μg/mL (55 nM) に希釈し、作製されたmCTLA4-Fc溶液を100 μLずつ加え、4℃で一晩静置し、プレート表面に固定化した。TBS, 0.1% Tween20で3回washしたのちに、TBSで2%に希釈したBSA溶液を各ウェルに250 μL加え、プレート表面をブロッキングした。その後、3回washした。最終濃度が55 nMになるようにTBSで希釈したマウスCD86にヒト定常領域およびHisタグが融合されたmCD86-Fc-His(Sino Biologics Inc. 50068-M03H, Accession No. NP_062261.3)、最終濃度が6.25, 1.56, 0.390, 0.0977, 0.0061, 0 μg/mLとなるように希釈したSW1077-mFa55抗体溶液、および最終濃度が0, 1, 10, 100 μMとなるように希釈したATP溶液とをそれぞれ合計100 μLとなるように混合し、各ウェルに加えて、1時間37℃で静置した。その後、各ウェルに加えられた溶液のATP濃度と同じATP濃度を含むように調製されたTBS, 0.1% Tween20で各ウェルを3回washした。各ウェルに加えられた溶液のATP濃度と同じATP濃度を含むようにブロッキングバッファーで10000倍に希釈したanti-His-tag mAb-HRP-Direct(MBLライフサイエンス)を各ウェルに100 μL加え1時間37℃で静置した。その後、各ウェルに加えられた溶液のATP濃度と同じATP濃度を含むように調製されたTBS, 0.1% Tween20で各ウェルを3回washした。そこへTMB溶液を各ウェルに100 μL加え1時間37℃で静置し、1M H2SO4を各ウェルに50 μL加え反応を停止し、吸光マイクロプレートリーダー(Wako Sunrise)で450 nmの吸光度を検出した。
同ATP濃度条件下での、抗体未添加ウェルの吸光度の値をmCTLA4-mCD86結合率100%とし、抗体添加でその結合率がどの程度低下するかを評価した。その結果を図13に示す。
(3-7-1)細胞株および同系腫瘍株移植マウスモデルの作製
細胞は理化学研究所より購入したマウス乳癌株FM3A細胞を用いた。FM3A細胞は10% Bovine serum(Thermo Fisher Scientific)を含むRPMI1640培地(SIGMA)にて維持継代した。マウスは日本チャールス・リバー社から購入したC3H/HeNマウス(7週齢、♀)を用いた。マウスの腹部皮下にFM3A細胞を移植し、移植腫瘍の体積がおよそ150 mm3から300 mm3になった時点でモデル成立とした。
腫瘍体積=長径×短径×短径/2
FM3A細胞移植モデルへの投与薬剤は、参考実施例3-4で調製された抗マウスCTLA4コントロール抗体(mNS-mFa55)および抗CTLA4スイッチ抗体(SW1208-mFa55)とした。mNS-mFa55は0.0005 mg/mL, 0.005 mg/mL, 0.0125 mg/mL, 0.05 mg/mL, 0.5 mg/mL, 1.5 mg/mL, 5 mg/mLになるよう、SW1208-mFa55は0.005 mg/mL, 0.05 mg/mL, 0.5 mg/mL, 5 mg/mL, 25 mg/mLになるよう、それぞれHis-buffer(20 mM His-HCl, 150 mM NaCl, pH 6.0)を用いて調製した。
移植後9日目に、mNS-mFa55は0.01 mg/kg, 0.1 mg/kg, 0.25 mg/kg, 1 mg/kg, 10 mg/kg, 30 mg/kg, 100 mg/kgの用量で、SW1208-mFa55は0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, 500 mg/kgの用量で、それぞれマウスに投与した。投与は、調製した投与液を20 mL/kgの用量で、尾静脈より行った。
抗腫瘍効果測定に際しての薬剤処置に関する詳細を表15に示した。

抗腫瘍効果については、(3-7-1)に記した計算式にて算出した腫瘍体積で評価した。
TGI(%)=(1-(測定時における着目する群の腫瘍体積の平均値-初回投与時における着目する群の腫瘍体積の平均値)÷(測定時におけるcontrol群の腫瘍体積の平均値-初回投与時のcontrol群の腫瘍体積の平均値))×100
移植後7日目に抗マウスCTLA4コントロール抗体(mNS-mFa55)を0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kgにて尾静脈より投与し、抗CTLA4スイッチ抗体(SW1208-mFa55)を0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, 500 mg/kgにて尾静脈より投与した。腫瘍内のTreg細胞の評価及び脾臓での全身性作用検証に際しての薬剤処置に関する詳細を表16に示した。

抗体投与後6日目に、麻酔下でマウスを安楽死処置し、腫瘍と脾臓を摘出した。摘出した脾臓より、10% FBS(SIGMA)を含むRPMI-1640培地(SIGMA)を用いて細胞懸濁液を調製した後、Mouse Erythrocyte Lysing kit(R&D)を用いて溶血処理し、脾臓細胞を調製した。摘出した腫瘍は、Tumor dissociation kit, mouse(miltenyi)を用いて破砕した。脾臓細胞、腫瘍を破砕したものをともに以下の抗体と反応させ、FACS解析によって存在する免疫細胞の画分を解析した。抗CD45抗体(BD、クローン: 30-F11)、抗CD3抗体(BD、クローン: 145-2C11)、抗CD4抗体(BD、クローン: RM4-5)、抗FoxP3抗体(eBioscience、クローン: FJK-16s)、抗ICOS抗体(eBioscience、クローン: 7E17G9)、抗KLRG1抗体(Biolegend、クローン: 2F1/KLRG1)。FACS解析はBD LSRFortessa X-20(BD)にて行った。
抗マウスCTLA4コントロール抗体(mNS-mFa55)及び抗CTLA4スイッチ抗体(SW1208-mFa55)投与時の腫瘍内のeffector Treg細胞(CD4+ FoxP3+ KLRG1+)の変化を評価した。結果、mNS-mFa55もSW1208-mFa55も、1 mg/kg以上のdoseにおいて、effector Tregの割合がCD45陽性細胞の0.2%未満まで減少した(図16)。
mNS-mFa55及びSW1208-mFa55投与時の脾臓内の活性化helper T 細胞(CD4+ Foxp3- ICOS+)の変化を、FACS解析にて評価した。結果、mNS-mFa55投与時には脾臓での活性化helper T 細胞のCD45陽性細胞に対する割合が顕著に増加したが、SW1208-mFa55は投与doseを上げていっても、脾臓での活性化helper T 細胞のCD45陽性細胞に対する割合を顕著に増加させることは無かった(図17)。当該スイッチ抗体はコントロール抗体と同等の薬効を示す一方で、腫瘍以外の組織では反応を起こさないことが確認され、腫瘍局所でのみ活性を示すというコンセプトがマウス生体内で証明された。
参考実施例3において作製された抗CTLA4スイッチ抗体の更なる改変と評価が実施された。
参考実施例2-1において実施された構造解析の結果より、抗体重鎖のCDR2がAMPのリン酸基と相互作用していることが示された。小分子がATPの場合には、γリン酸基が重鎖CDR2と立体障害を起こす可能性が考えられたため、この領域のアミノ酸を置換し、ATPとの結合能の増強が検討された。具体的には、参考実施例3において作製された04H1207-G1m/04L1086-lam1および04H1208-G1m/04L1086-lam1の重鎖可変領域に対してR53QとG55Hの改変を導入した04H1389-G1m/04L1086-lam1と、04H1382-G1m/04L1086-lam1が作製された。また04H1389-G1m/04L1086-lam1の軽鎖をヒトκ鎖の配列に置換した04H1389-G1m/04L1305-k0MTが作製された。表17はこれらの抗体の重鎖可変領域、軽鎖可変領域、重鎖定常領域、軽鎖定常領域、および超可変領域(Hyper Variable Region)の配列番号の一覧を示す。


抗ヒトCTLA4コントロール抗体(MDX10D1H-mFa55/MDX10D1L-mk1 略名:hNS-mFa55)および抗CTLA4スイッチ抗体(04H1389-mFa55/04L1305-mk1 略名:SW1389-mFa55)が作製された。hNS-mFa55抗体は、重鎖可変領域MDX10D1H(配列番号:26)および軽鎖可変領域MDX10D1L(配列番号:27)を用い、定常領域はマウス重鎖定常領域mFa55(配列番号:18)および野生型マウス軽鎖定常領域mk1(配列番号:19)を使用している。この時Fcγ受容体への結合を増強するよう改変を加えたマウス重鎖定常領域を使用し、当業者公知の方法で発現、精製された。
SW1389-mFa55抗体は、重鎖可変領域04H1389(配列番号:29)および軽鎖可変領域04L1305(配列番号:30)を用い、定常領域はマウス重鎖定常領域mFa55(配列番号:18)および野生型マウス軽鎖定常領域mk1(配列番号:19)を使用している。この時Fcγ受容体への結合を増強するよう改変を加えたマウス重鎖定常領域を使用し、当業者公知の方法で発現、精製された。

(4-3-1)細胞株
Hepa1-6/hGPC3細胞を用いた。この細胞株は、マウス肝癌株Hepa1-6細胞をATCCより購入し、human Glypican 3(hGPC3、配列番号:181)遺伝子をtransfectionによって恒常発現させ、クローン化したものである。Hepa1-6/hGPC3細胞を10% FBS(SIGMA)および600 μg/mL GENETICIN(gibco)を含むD-MEM(high glucose)培地(SIGMA)にて維持継代した。
Human CTLA4 knock-in マウス(Blood (2005) 106(9): 3127-3133)と社内で作製したhuman CD3 EDG replaced mouse(Sci Rep (2017) 7: 45839)の交雑系統である、human CTLA4 KI, human CD3 EDG replacedマウス(hCTLA4 KI hCD3 EDG replacedマウス)を用いた。hCTLA4 KI hCD3 EDG replacedマウスの皮下にHepa1-6/hGPC3細胞を移植し、移植腫瘍の体積の平均がおよそ200 mm3から400 mm3になった時点でモデル成立とした。
腫瘍体積=長径×短径×短径/2
Hepa1-6/hGPC3細胞移植モデルへの投与薬剤としては、参考実施例4-2で調製された抗CTLA4スイッチ抗体(SW1389-mFa55)または、抗ヒトCTLA4コントロール抗体(hNS-mFa55)をそれぞれ 0.01, 0.1, 1, 5, 10, 20 mg/mL, 0.01, 0.1, 1, 3 mg/mLになるようにHis buffer(150 mM NaCl/ 20 mM His-HCl buffer pH 6.0)を用いて調製した。
移植後7日目にSW1389-mFa55を0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg、hNS-mFa55を0.1 mg/kg, 1 mg/kg, 10 mg/kg, 30 mg/kgにてマウスに尾静脈より投与した。抗腫瘍効果測定に際しての薬剤処置に関する詳細を表20に示した。

抗腫瘍効果については、(4-3-2)に記した計算式にて算出した腫瘍体積で評価した。統計解析にはJMP 11.2.1(SAS Institute Inc.)を用いた。
TGI=(1-(測定時における着目する群の腫瘍体積の平均値-抗体投与前の時点での腫瘍体積の平均値)÷(測定時におけるcontrol群腫瘍体積の平均値-抗体投与前の時点での腫瘍体積の平均値))×100
移植後7日目にSW1389-mFa55を0.1 mg/kg, 1 mg/kg, 10 mg/kg, 100 mg/kg, 500 mg/kgにてマウスに尾静脈より投与し、hNS-mFa55を0.1 mg/kg, 1 mg/kg, 10 mg/kg, 30 mg/kgにて尾静脈より投与した。腫瘍内のTreg細胞の評価及び脾臓での全身性作用検証に際しての薬剤処置に関する詳細を表21に示した。

抗体投与後6日目に、麻酔下でマウスを安楽死処置し、腫瘍と脾臓を摘出した。摘出した脾臓より、10% FBS(SIGMA)を含むRPMI-1640培地(SIGMA)を用いて細胞懸濁液を調製した後、Mouse Erythrocyte Lysing kit(R&D)を用いて溶血処理し、脾臓細胞を調製した。摘出した腫瘍は、Tumor dissociation kit, mouse(miltenyi)を用いて破砕した。脾臓細胞、腫瘍を破砕したものをともに以下の抗体と反応させ、FACS解析によって存在する免疫細胞の画分を解析した。抗CD45抗体(BD、クローン: 30-F11)、抗CD3抗体(BD、クローン: UCHT1)、抗CD4抗体(BD、クローン: RM4-5)、抗FoxP3抗体(eBioscience、クローン: FJK-16s)、抗ICOS抗体(eBioscience、クローン: 7E17G9)、抗CCR7抗体(Biolegend、クローン: 4B12)、抗KLRG1抗体(Biolegend、クローン: 2F1/KLRG1)。FACS解析はBD LSRFortessa X-20(BD)にて行った。
SW1389-mFa55投与時および、hNS-mFa55投与時の腫瘍内のeffector Treg細胞(CD4+ FoxP3+ CCR7lowKLRG1+)の変化を評価した。結果、hNS-mFa55 もSW1389-mFa55も、1 mg/kg以上のdoseにおいて、effector Tregの割合がCD45陽性細胞の0.2%未満まで減少した(図20)。
hNS-mFa55またはSW1389-mFa55投与時の、脾臓内の活性化helper T細胞(CD4+ Foxp3- ICOS+)の変化を、FACS解析にて評価した。結果、hNS-mFa55投与時には脾臓での活性化helper T細胞のCD45陽性細胞に対する割合が顕著に増加したが、SW1389-mFa55投与時には投与doseを上げていっても脾臓における活性化helper T細胞の顕著な増加は見られなかった(図21)。当該スイッチ抗体はコントロール抗体と同等の薬効を示す一方で、腫瘍以外の組織では反応を起こさないことが確認され、腫瘍局所でのみ活性を示すというコンセプトがヒトCTLA4 KIマウス生体内で証明された。
参考実施例4において作製された抗CTLA4スイッチ抗体の更なる改変と評価が実施された。
04H1389-G1m/04L1086-lam1のCDRに対して、アミノ酸改変が網羅的に導入され、より優れたプロファイルを有する改変が探索された。アミノ酸改変の網羅的導入および評価は参考実施例3-3に記載の方法を用いて行われた。ここで見出された改変の組み合わせおよびFrameworkを置換した改変体が作製された。表22はこれらの抗体の重鎖可変領域、軽鎖可変領域、重鎖定常領域、軽鎖定常領域、および超可変領域(Hyper Variable Region)の配列番号の一覧を示す。表22中の軽鎖04L1594-lam1、04L1581-lam1、04L1610-lam1、04L1612-lam1、04L1610-lam1は親抗体の軽鎖04L1086-lam1に対してCDRおよびframeworkへの改変が導入されており、ヒトλ鎖のgermline配列のframeworkと定常領域を有する。また04L1615-k0MT、 04L1616-k0MT、 04L1617-k0MTは、04L1086-lam1に対してCDRへの改変が導入され、かつヒトκ鎖のgermline配列のframeworkと定常領域を有する。重鎖可変領域04H1389v373は親抗体の重鎖可変領域04H1389のCDRへの改変が導入されたもの、04H1637、04H1643、04H1654、04H1656、04H1642、04H1735は04H1389のCDRに改変が導入されるとともに、Frameworkの配列を異なるgermlineに置換した重鎖可変領域である。



カニクイザルCTLA4細胞外領域C末端にHisタグおよびBAPタグが融合したものcyCTLA4-His-BAP(配列番号:50)が遺伝子合成され、動物発現用プラスミドへ挿入された。FreeStyle 293 Expression Medium培地(Invitrogen)に1.33×106細胞/mLの細胞密度で懸濁されて、フラスコに播種されたヒト胎児腎細胞由来FreeStyle 293-F株(Invitrogen)に対して、調製されたプラスミドがリポフェクション法により導入され、プラスミド導入後3時間後にビオチンが終濃度100 μMとなるように添加され、CO2インキュベーター(37℃、8% CO2, 125 rpm)で4日間培養された。各検体の培養上清から、当業者公知の方法で抗原が精製された。分光光度計を用いて、精製された抗原溶液の280 nmでの吸光度が測定された。得られた測定値からPACE法により算出された吸光係数を用いて精製された抗原の濃度が算出された(Protein Science (1995) 4, 2411-2423)。

改変された抗CTLA4スイッチ抗体(04H1654-mFa55m2P1/04L1610-ml0r//04H1656-mFa55m2N1/04L1610-ml0r 略名:SW1610-mFa55、04H1654-mFa55m2P1/04L1612-ml0r//04H1656-mFa55m2N1/04L1612-ml0r 略名:SW1612-mFa55、および04H1389-mFa55/04L1615-mk1 略名:SW1615-mFa55)、および陰性コントロール抗体(IC17Hdk-mFa55/IC17L-mk1 略名:KLH-mFa55)が作製された。
SW1615-mFa55抗体は、重鎖可変領域04H1389(配列番号:29)および軽鎖可変領域04L1615(配列番号:34)を用い、定常領域はマウス重鎖定常領域mFa55(配列番号:18)および野生型マウス軽鎖定常領域mk1(配列番号:19)を使用している。この時Fcγ受容体への結合を増強するよう改変を加えたマウス重鎖定常領域を使用し、当業者公知の方法で発現、精製された。
SW1610-mFa55抗体は、一方の重鎖可変領域04H1654(配列番号:35)は定常領域としてマウス重鎖定常領域mFa55m2P1(配列番号:36)を、もう一方の重鎖可変領域04H1656(配列番号:37)はマウス重鎖定常領域mFa55m2N1(配列番号:38)を連結し、さらに軽鎖可変領域04L1610(配列番号:39)は野生型マウス軽鎖定常領域ml0r(配列番号:22)を使用し、当業者公知の方法で発現、精製された。
SW1612-mFa55抗体は、一方の重鎖可変領域04H1654(配列番号:35)は定常領域としてマウス重鎖定常領域mFa55m2P1(配列番号:36)を、もう一方の重鎖可変領域04H1656(配列番号:37)はマウス重鎖定常領域mFa55m2N1(配列番号:38)を連結し、さらに軽鎖可変領域04L1612(配列番号:40)は野生型マウス軽鎖定常領域ml0r(配列番号:22)を使用し、当業者公知の方法で発現、精製された。
陰性コントロール抗体は、重鎖可変領域IC17Hdk(配列番号:51)は定常領域としてマウス重鎖定常領域mFa55(配列番号:18)を連結し、さらに軽鎖可変領域IC17L(配列番号:52)は野生型マウス軽鎖定常領域mk1(配列番号:19)を使用し、当業者公知の方法で発現、精製された。
マウス定常領域を有する抗CTLA4抗体の抗原への結合能が参考実施例4-2に記載の方法で評価された(表26)。マウス定常領域を有するこれらの抗体はいずれも、同じ可変領域でかつヒト定常領域を有する表25に記載の抗体と同等のATP依存的なヒトCTLA4への結合能を有することが示された。

(5-4-1)細胞株
Hepa1-6/hGPC3細胞を用いた。この細胞株は、マウス肝癌株Hepa1-6細胞をATCCより購入し、human Glypican 3(hGPC3)遺伝子をtransfectionによって恒常発現させ、クローン化したものである。Hepa1-6/hGPC3細胞は10% FBS(SIGMA)および0.6 mg/mL G418(ナカライテスク)を含むD-MEM(high glucose)培地(SIGMA)にて維持継代した。
Human CTLA4 knock-in マウス(Blood (2005) 106(9): 3127-3133)と社内で作製したhuman CD3 EDG replaced mouse(Sci Rep (2017) 7: 45839)の交雑系統である、human CTLA4 KI, human CD3 EDG replacedマウス(hCTLA4 KI hCD3 EDG replacedマウス)を用いた。hCTLA4 KI hCD3 EDG replacedマウスの皮下にHepa1-6/hGPC3細胞を移植し、移植腫瘍の体積の平均がおよそ200 mm3から400 mm3になった時点でモデル成立とした。
腫瘍体積=長径×短径×短径/2
Hepa1-6/hGPC3細胞移植モデルへの投与薬剤は、参考実施例5-2で調製された抗CTLA4スイッチ抗体(SW1610-mFa55、SW1612-mFa55、SW1615-mFa55)とした。投与薬剤は0.03 mg/mL, 0.1 mg/mL, 0.3 mg/mLになるようにHis-buffer(20 mM His-HCl, 150 mM NaCl, pH 6.0)を用いて調製した。
移植後8日目に抗CTLA4スイッチ抗体3検体をそれぞれ0.3 mg/kg, 1 mg/kg, 3 mg/kgにてマウスに尾静脈より投与した。抗腫瘍効果測定に際しての薬剤処置に関する詳細を表27に示した。

抗腫瘍効果については、(5-4-2)に記した計算式にて算出した腫瘍体積で評価した。
TGI(%)=(1-(測定時における着目する群の腫瘍体積の平均値-初回投与時における着目する群の腫瘍体積の平均値)÷(測定時におけるcontrol群の腫瘍体積の平均値-初回投与時のcontrol群の腫瘍体積の平均値))×100
移植後10日目にSW1610-mFa55を50 mg/kg, 100 mg/kg, 200 mg/kg、SW1612-mFa55を50 mg/kg, 100 mg/kg, 200 mg/kg、SW1615-mFa55を50 mg/kg, 100 mg/kg, 200 mg/kg, 400mg/kgにてマウスに尾静脈より投与した。また、コントロール群には、陰性コントロール抗体であるIC17Hdk-mFa55/IC17L-mk1(略名:KLH-mFa55)を400 mg/kgにて尾静脈より投与した。腫瘍内のTreg細胞の評価及び脾臓での全身性作用検証に際しての薬剤処置に関する詳細を表28に示した。

抗体投与後6日目に、麻酔下でマウスを安楽死処置し、腫瘍と脾臓を摘出した。摘出した脾臓より、10% FBS(SIGMA)を含むRPMI-1640培地(SIGMA)を用いて細胞懸濁液を調製した後、Mouse Erythrocyte Lysing kit(R&D)を用いて溶血処理し、脾臓細胞を調製した。摘出した腫瘍は、Tumor dissociation kit, mouse(miltenyi)を用いて破砕した。脾臓細胞、腫瘍を破砕したものをともに以下の抗体と反応させ、FACS解析によって存在する免疫細胞の画分を解析した。抗CD45抗体(BD、クローン: 30-F11)、抗CD3抗体(BD、クローン: UCHT1)、抗CD4抗体(BD、クローン: RM4-5)、抗FoxP3抗体(eBioscience、クローン: FJK-16s)、抗ICOS抗体(eBioscience、クローン: 7E17G9)、抗CCR7抗体(Biolegend、クローン: 4B12)、抗KLRG1抗体(Biolegend、クローン: 2F1/KLRG1)。FACS解析はBD LSRFortessa X-20(BD)にて行った。
抗CTLA4スイッチ抗体投与時の腫瘍内のeffector Treg細胞(CD4+ FoxP3+ CCR7lowKLRG1+)の変化を評価した。結果、SW1610-mFa55、SW1612-mFa55およびSW1615-mFa55は投与した全てのdoseにおいて、effector Tregの割合がCD45陽性細胞の0.2%未満まで減少した(図25)。
抗CTLA4スイッチ抗体投与時の脾臓内の活性化helper T細胞(CD4+ Foxp3- ICOS+)の変化を、FACS解析にて評価した。結果、SW1610-mFa55およびSW1612-mFa55は評価したdoseのうち、50 mg/kgのdoseにおいて、SW1615-mFa55は評価したdoseのうち、200 mg/kg以下のdoseにおいて脾臓での活性化helper T細胞のCD45陽性細胞に対する割合を、有意に増加させることは無かった。有意差検定はJMP 11.2.1(SAS Institute Inc.)を用い、KLH-mFa55投与群に対してDunnettの検定を行った(図26)。当該スイッチ抗体はいずれも薬効を示す一方で、腫瘍以外の組織では反応を起こさない事が確認され、腫瘍局所でのみ活性を示すという性質を有することが確認された。
細胞傷害性エフェクター機能であるADCCおよびADCPを増強した抗体を作製するため、活性型FcγRであるFcγRIIIaおよびFcγRIIaに対する結合能を増強したFc領域改変体の作製が検討された。
WO2013/002362に記載されているFcγRへの結合能を増強した重鎖定常領域Kn125およびHl076を有し、重鎖可変領域として04H1637を、軽鎖として04L1610-lam1を有するヘテロ二量化抗体、04H1637-Kn125/04L1610-lam1//04H1637-Hl076/04L1610-lam1が作製された。具体的には、一方の重鎖可変領域として04H1637(配列番号:138)を有し、ヒトIgG1重鎖定常領域のC末端のGlyおよびLysが除去されたG1d(配列番号:158)に対して、L234Y/L235Q/G236W/S239M/H268D/D270E/S298Aが導入され、さらにCH3領域にヘテロ二量化を促進する改変Y349C/T366Wを有する抗体重鎖04H1637-Kn125(配列番号:162)の遺伝子が作製された。同様に、もう一方の重鎖可変領域として04H1637(配列番号:138)を有し、ヒトIgG1重鎖定常領域G1d(配列番号:158)に対して、D270E/K326D/A330M/K334Eが導入され、さらにCH3領域にヘテロ二量化を促進する改変D356C/T366S/L368A/Y407Vを有する抗体重鎖04H1637-Hl076(配列番号:163)の遺伝子が作製された。抗体軽鎖として04L1610-lam1(配列番号:161)を用い、当業者公知の方法によりヘテロ二量体04H1637-Kn125/04L1610-lam1//04H1637-Hl076/04L1610-lam1が作製された。ここでCH2領域に導入されている改変のうち、L235Q、G236W、S239M、H268D、D270E、S298A、K326D、K334Eに加えて、WO2013/002362においてFcγRへの結合を変化させる改変として報告されているL234F、A330K、Mol. Cancer Ther., 2008, 7, 2517-2527およびWO2004/029207に報告されているG236A、I332E、I332D、WO2013/118858において安定性を向上させる改変として報告されているT250V、T307Pを組み合わせた抗体重鎖04H1637-Kn462(配列番号:164)、04H1637-Hl441(配列番号:165)、04H1637-Hl445(配列番号:166)、04H1637-Kn461(配列番号:167)、04H1637-Hl443(配列番号:168)の遺伝子が作製された。また、ヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域にKn462と同じ改変を有し、CH3領域にWO2006/106905に記載のヘテロ二量化を促進する改変E356Kを有し、重鎖可変領域として04H1654(配列番号:140)をもつ抗体重鎖04H1654-KT462(配列番号:182)の遺伝子が作製された。同様に、ヒトIgG1(IGHG1*03)のC末端のGlyおよびLysが除去され、CH2領域にHl441と同じ改変を有し、CH3領域にWO2006/106905に記載のヘテロ二量化を促進する改変K439Eを有し、重鎖可変領域として04H1656(配列番号:141)をもつ抗体重鎖04H1656-HT441(配列番号:170)の遺伝子が作製された。以下同様に、04H1656-HT445(配列番号:171)、04H1654-KT461(配列番号:183)、04H1656-HT443(配列番号:173)の遺伝子が作製された。さらに、Mabs, 2017, 9, 844-853に記載されている抗体の血中動態を改善する改変の組み合わせが検討された。具体的には、04H1654-KT462(配列番号:182)のCH3領域に対して、酸性条件下においてヒトFcRnに対する結合を増強する改変およびRheumatoid factorに対する結合を低減する改変の組み合わせであるN434A/Y436T/Q438R/S440Eが導入された04H1654-KT473(配列番号:184)の遺伝子が作製された。同様に04H1656-HT445(配列番号:171)に対して、N434A/Y436T/Q438R/S440Eが導入された04H1656-HT482(配列番号:185)の遺伝子が作製された。同様に、04H1654-KT461, 04H1656-HT443に対して同じ改変を導入した抗体重鎖04H1654-KT481(配列番号:186)、抗体重鎖04H1656-HT498(配列番号:187)がそれぞれ作製された。これらの重鎖を組み合わせ、04L1610-lam1あるいは04L1612-lam1(配列番号:188)が軽鎖として用いられ、目的とするヘテロ二量化抗体が作製された。
表29はこれらの測定結果を示す。





作製されたヘテロ二量体、MDX10D1H-Kn125/MDX10D1H-Hl076/MDX10D1L-k0MT、MDX10D1H-Kn462/MDX10D1H-Hl445/MDX10D1L-k0MT、MDX10D1H-Kn461/MDX10D1H-Hl443/MDX10D1L-k0MTはいずれも既存のFcγR結合増強抗体であるMDX10D1H-GASDIE/MDX10D1L-k0MT、MDX10D1H-GASDALIE/MDX10D1L-k0MTと比較してFcγRIIIaへの結合が増強されていた。またMDX10D1H-Kn462/MDX10D1H-Hl445/MDX10D1L-k0MTは既存のFcγRIIa増強抗体であるMDX10D1H-GASDIE/MDX10D1L-k0MTと比較して、FcγRIIaHへの結合が約2倍増強されていることが示された。
In vitro ADCC活性測定にはhFcγRIIIaV ADCC Reporter Bioassay, Core Kit(Promega)が用いられた。96ウェルプレートの各ウェルに、培地により2×106/mLに濃度を調製した hCTLA4-CHO細胞をターゲット細胞として25 μLずつ加えられ、培地にはAssay Buffer(90% RPMI1640, 10% FBS)が使用された。次に最終濃度が0, 0.001, 0.01, 0.1, 1 μg/mLとなるようにassay bufferで希釈した各抗体溶液がそれぞれ25 μL加えられ、最後にエフェクター細胞溶液として培地により6×106/mLに調製されたhFcγRIIIaV 発現Jurkat細胞(キットに付属)が25 μL加えられ、合計75 μLとなるように混合された後、5% CO2インキュベーター内にて37℃で一晩静置された。その後プレートは室温で15分静置され、各ウェルにBio-Glo reagentが75 μLずつ加えられた。Bio-Glo reagentにはBio-glo Luciferase Assay System (Buffer and Substrate)が使用された。その後各ウェルの発光はプレートリーダーで測定された。各ウェルの発光の値を抗体未添加ウェルの発光の値で割った値をFold inductionとし、各抗体のADCCを評価する指標とした。得られた結果を図27に示す。図中では、Fold inductionを相対発光量(RLU)と表記する。
In vitro ADCP活性測定にはhFcγRIIaH ADCP Reporter Bioassay, Core Kit(Promega)を用いた。96ウェルプレートの各ウェルに、培地により1×106/mLに濃度を調製した hCTLA4-CHO細胞をターゲット細胞として25 μLずつ加えられ、培地にはAssay Buffer(4% Low IgG serum in RPMI1640)が使用された。次に最終濃度が0, 0.001, 0.01, 0.1, 1 μg/mLとなるようにassay bufferで希釈した各抗体溶液がそれぞれ25 μL加えられ、最後にエフェクター細胞溶液としてキットに付属のhFcγRIIaH 発現Jurkat細胞が25 μL加えられ、合計75 μLとなるように混合された後、5% CO2インキュベーター内にて37℃で一晩静置された。hFcγRIIaH 発現Jurkat細胞はその細胞液密度が8.25×105/mLであった。その後プレートは室温で15分静置され、各ウェルにBio-Glo reagentが75 μlずつ加えられた。Bio-Glo reagentにはBio-glo Luciferase Assay System (Buffer and Substrate)が使用された。その後各ウェルの発光はプレートリーダーで測定された。各ウェルの発光の値を抗体未添加ウェルの発光の値で割った値をFold inductionとし、各抗体のADCPを評価する指標とした。得られた結果を図28に示す。図中では、Fold inductionを相対発光量(RLU)と表記する。
改変されたFcを有する抗CTLA4スイッチ抗体(04H1654-Kn462/04L1610-lam1//04H1656-Hl445/04L1610-lam1 略名:SW1610-ART6、04H1654-Kn462/04L1612-lam1//04H1656-Hl445/04L1612-lam1 略名:SW1612-ART6、および04H1389-Kn462/04L1305-k0MT//04H1389-Hl445/04L1305-k0MT 略名:SW1389-ART6)が作製された。
SW1610-ART6抗体は、一方の重鎖可変領域04H1654(配列番号:35)は定常領域としてヒト重鎖定常領域Kn462(配列番号:43)を、もう一方の重鎖可変領域04H1656(配列番号:37)はヒト重鎖定常領域Hl445(配列番号:44)を連結し、さらに軽鎖可変領域04L1610(配列番号:39)は野生型ヒト軽鎖定常領域lam1(配列番号:53)を使用し、当業者公知の方法で発現、精製された。
SW1612-ART6抗体は、一方の重鎖可変領域04H1654(配列番号:35)は定常領域としてヒト重鎖定常領域Kn462(配列番号:43)を、もう一方の重鎖可変領域04H1656(配列番号:37)はヒト重鎖定常領域Hl445(配列番号:44)を連結し、さらに軽鎖可変領域04L1612(配列番号:40)は野生型ヒト軽鎖定常領域lam1(配列番号:53)を使用し、当業者公知の方法で発現、精製された。
SW1389-ART6抗体は、二つの重鎖可変領域04H1389(配列番号:29)は定常領域としてヒト重鎖定常領域Kn462(配列番号:43)およびヒト重鎖定常領域Hl445(配列番号:44)をそれぞれ連結し、さらに軽鎖可変領域04L1305(配列番号:30)は野生型ヒト軽鎖定常領域k0MT(配列番号:33)を使用し、当業者公知の方法で発現、精製された。
改変された抗CTLA4スイッチ抗体として、SW1389, SW1610, SW1612, SW1615の可変領域を持つヒト抗体が作成された。
SW1389抗体は、重鎖可変領域として04H1389(配列番号:29)、軽鎖可変領域として04L1305(配列番号:30)が使用され、ヒト定常領域に連結されたのち、当業者公知の方法で発現、精製された。
SW1610抗体は、重鎖可変領域として04H1654(配列番号:35)および04H1656(配列番号:37)、軽鎖可変領域として04L1610(配列番号:39)が使用され、ヒト定常領域に連結されたのち、当業者公知の方法で発現、精製された。
SW1612抗体は、重鎖可変領域として04H1654(配列番号:35)および04H1656(配列番号:37)、軽鎖可変領域として04L1612(配列番号:40)が使用され、ヒト定常領域に連結されたのち、当業者公知の方法で発現、精製された。
SW1615抗体は、重鎖可変領域として04H1389(配列番号:29)、軽鎖可変領域として04L1615(配列番号:34)が使用され、ヒト定常領域に連結されたのち、当業者公知の方法で発現、精製された。
In vitro 中和活性測定にはCTLA-4 Blockade Bioassay(Promega)が用いられた。96ウェルプレートの各ウェルに、培地により1×106/mLに濃度を調製した Kit付属のaAPC-Raji細胞がターゲット細胞として25μLずつ加えられ、培地にはAssay Buffer(10% FBS in RPMI1640)が使用された。次に最終濃度が0, 100 μMとなるようにassay bufferで希釈したATP溶液、最終濃度が0, 0.001, 0.01, 0.1, 1 ,10 μg/mLとなるようにassay bufferで希釈したSW1389、SW1610, SW1612およびSW1615の可変領域を持つ抗体溶液が順次加えられ、最後にエフェクター細胞溶液として培地により2×106/mLに調製されたIL2-luc2-CTLA4-Jurkat細胞(キットに付属)が25 μL加えられ、合計75 μLとなるように混合された後、5% CO2インキュベーター内にて6時間37℃で静置された。その後プレートは室温で15分静置され、各ウェルにBio-Glo reagentが75 μLずつ加えられた。Bio-Glo reagentにはBio-glo Luciferase Assay System (Buffer and Substrate)が使用された。その後各ウェルの発光はプレートリーダーで測定された。各ウェルの発光の値を抗体未添加ウェルの発光の値で割った値をFold inductionとし、各抗体の中和活性を評価する指標とした。得られた結果を図32(SW1389)、図33(SW1610)、図34(SW1612)および図35(SW1615)に示す。図中では、Fold inductionを相対発光量(RLU)と表記する。
改変されたFcを有する抗CTLA4スイッチ抗体(04H1654-KT473/04L1610-lam1//04H1656-HT451/04L1610-lam1 略名:SW1610-ART5+ACT1、04H1654-KT473/04L1610-lam1//04H1656-HT482/04L1610-lam1 略名:SW1610-ART6+ACT1、04H1389-Km473/04L1305-k0MT//04H1389-Hm451/04L1305-k0MT 略名:SW1389-ART5+ACT1)、04H1389-Km473/04L1305-k0MT//04H1389-Hm482/04L1305-k0MT 略名:SW1389-ART6+ACT1)が作製された。
SW1610-ART5+ACT1抗体は、一方の重鎖として04H1654-KT473(配列番号:184)、もう一方の重鎖として04H1656-HT451(配列番号:272)、さらに軽鎖として04L1610-lam1(配列番号:161)を使用し、当業者公知の方法で発現、精製された。
SW1610-ART6+ACT1抗体は、一方の重鎖として04H1654-KT473(配列番号:184)、もう一方の重鎖として04H1656-HT482(配列番号:185)、さらに軽鎖として04L1610-lam1(配列番号:161)を使用し、当業者公知の方法で発現、精製された。
SW1389-ART5+ACT1抗体は、一方の重鎖として04H1389-Km473(配列番号:203)、もう一方の重鎖として04H1389-Hm451(配列番号:274)、さらに軽鎖として04L1305-k0MT(配列番号:275)を使用し、当業者公知の方法で発現、精製された。
SW1389-ART6+ACT1抗体は、一方の重鎖として04H1389-Km473(配列番号:203)、もう一方の重鎖として04H1389-Hm482(配列番号:204)、さらに軽鎖として04L1305-k0MT(配列番号:275)を使用し、当業者公知の方法で発現、精製された。
この結果から改変されたFcを有する抗CTLA4スイッチ抗体のCTLA4陽性制御性T細胞に対する細胞傷害活性はATP存在下・非存在下で異なり、ATP依存的なCTLA4陽性制御性T細胞に対する細胞傷害活性が存在することが確認された。
これまでに細胞傷害性エフェクター機能であるADCCおよびADCPを増強するFc改変体は報告されているが、CH2領域を対称にエンジニアリングした改変体や、糖鎖改変によって作られた低フコース抗体はいずれもFcγRへの結合をさらに増強する余地が残されていた。またWO2013002362、WO2014104165に記載されている、CH2領域が非対称にエンジニアリングされた改変体は、対称にエンジニアリングされたFc領域改変体と比較して大きくFcγRへの結合能が増強されていたが、更なる改善の余地があった。具体的には、WO2013002362、WO2014104165に記載されているFc領域改変体Kn125/Hl076(本明細書中での略称:ART1)は、FcγRIIIaへの結合能が強く増強されているが、FcγRIIaへの結合能がIgG1と比較して数倍しか増強されておらず、強いADCP活性を示すためにはさらなる増強が必要であると考えられた。またKn120/Hl068(本明細書中での略称:ART2)はFcγRIIa、FcγRIIIa両方への結合が増強されており、ADCC、ADCP共に強い活性が期待されるが、抑制型のFcγRIIbへの結合能も増強されており、優れたエフェクター機能を発揮するための指標であるA/I比が小さいという課題があった。つまり既存の方法では、最も理想的なプロファイルである、「より強く活性型FcγRIIaおよびFcγRIIIaに結合し、かつ抑制型FcγRIIbへの結合が抑えられた抗体改変技術」は達成されていない。そこで本発明ではさらなる組み合わせ改変を検討し、これらの課題を克服する優れたプロファイルのFc領域改変体の作製が検討された。


FcγRの細胞外ドメインはWO2014104165に記載の方法で調製された。作製された抗体とヒトFcγRとの相互作用解析はBiacore 8K+を用いて以下の方法で実施された。ランニングバッファーには50 mM Na-Phosphate、150 mM NaCl、0.05% Tween20 (pH 7.4)が用いられ、25℃で測定が実施された。センサーチップには、Series S SA(GE Healthcare)に対しCaptureSelect Human Fab-kappa Kinetics Biotin Conjugate(Thermo Fisher Scientific)が固定化されたチップが用いられた。このチップに対して目的の抗体がキャプチャーされ、ランニングバッファーで希釈した各FcγRが相互作用させられた。チップは10 mM Glycine-HCl (pH 1.5)を用いて再生され、繰り返し抗体をキャプチャーして測定が行われた。各抗体のFcγRに対する解離定数KD (mol/L)は、Biacore Insight Evaluation Softwareを用い、FcγRIIbに対する解離定数はSteady state affinity model、その他のFcγRに対する解離定数は1:1 Langmuir binding modelを用いて算出された(表35)。


(9-1)ヒトEpiregulin発現細胞(Hepa1-6/hEREG)の作製
マウス肝癌株Hepa1-6細胞はATCC社より購入され、ヒトEREG(hEREG)遺伝子をトランスフェクションによって導入し、恒常発現するクローンが選抜された。hEREG遺伝子はZeocinにて選抜される。Hepa1-6 /hEREG細胞は10%FBS(SIGMA社製)、400 μg/mL Zeocinを含むD-MEM(high glucose)培地(SIGMA社製)にて維持継代された。
In vitro ADCC活性測定にはhFcγRIIIaV ADCC Reporter Bioassay, Effector cells, Propagation Model(Promega)が用いられた。384ウェルプレートの各ウェルに、培地により5×105/mLに濃度を調製した Hepa1-6/hEREG細胞をターゲット細胞として10 μLずつ加えられ、培地にはAssay Buffer (96% RPMI, 4% FBS)が使用された。次に参考実施例7で作製された抗体および陰性対照としてヒトIgG4の配列を有するEGL-G4d(重鎖配列番号:306、軽鎖配列番号:282)がそれぞれ,最終濃度が1 μg/mLから公比10で11点となるようにassay bufferで希釈された後10 μL加えられ、最後にエフェクター細胞溶液として培地により3×106/mLに調製されたhFcγRIIIaV 発現Jurkat細胞が10 μL加えられ、合計30 μLとなるように混合された後、5% CO2インキュベーター内にて37℃で一晩静置された。その後プレートは室温で15分静置され、各ウェルにBio-Glo reagentが30 μLずつ加えられた。Bio-Glo reagentにはBio-glo Luciferase Assay System(Buffer and Substrate)が使用された。その後各ウェルの発光はプレートリーダーで測定された。

In vitro ADCP活性測定にはhFcγRIIaH ADCP Reporter Bioassay, Core Kit(Promega)を用いた。384ウェルプレートの各ウェルに、培地により1×106/mLに濃度を調製した Hepa1-6/hEREG細胞をターゲット細胞として10 μLずつ加えられ、培地にはAssay Buffer (96% RPMI, 4% FBS)が使用された。次に参考実施例7で作製された抗体がそれぞれ,最終濃度が0、0.001、0.01、0.1、1、10 μg/mLとなるようにassay bufferで希釈された後10 μL加えられ、最後にエフェクター細胞溶液としてキットに付属のhFcγRIIaH 発現Jurkat細胞が10 μL加えられ、合計30 μLとなるように混合された後、5% CO2インキュベーター内にて37℃で6時間静置された。hFcγRIIaH 発現Jurkat細胞はその細胞液密度が9.68×105/mLであった。その後プレートは室温で15分静置され、各ウェルにBio-Glo reagentが30 μLずつ加えられた。Bio-Glo reagentにはBio-glo Luciferase Assay System(Buffer and Substrate)が使用された。その後各ウェルの発光はプレートリーダーで測定された。各ウェルの発光の値を抗体未添加ウェルの発光の値で割った値をFold inductionとし、各抗体のADCPを評価する指標とされた。得られた結果は図41に示された。また各検体のEC 50値はJMP 11.2.1(SAS Institute Inc.)にて算出され、表37に示された。

(11-1)細胞株
参考実施例9-1で作製されたHepa1-6/hEREG細胞を10%FBS(SIGMA)、400 μg/mL Zeocinを含むD-MEM(high glucose)培地(SIGMA)にて維持継代された。
薬効試験にはヒトFcγRトランスジェニックマウス(Proc Natl Acad Sci U S A. 2012 Apr 17; 109(16): 6181-6186.)が用いられた。16週令の雄マウスに細胞の生着率を向上させる目的で抗アシアロGM1抗体(aGM1, WAKO)が100 μL/headで腹腔内投与された。aGM1投与翌日、皮下にHepa1-6/hEREG細胞とmatrigel(CORNING)が1:1で混合された細胞溶液が、細胞数として1×107 cells移植され、移植腫瘍の体積の平均がおよそ300 mm3から500 mm3になった時点でモデル成立とした。
移植腫瘍の体積は以下の式にて算出した。
腫瘍体積=長径×短径×短径/2
本発明で作製されたEGL-ART6は参考実施例8のA/I比の結果および参考実施例9、10のレポーター遺伝子誘導活性の強さから、最も強い抗腫瘍活性が期待された。そこで、Hepa1-6/hEREG細胞移植モデルへの投与薬剤としては、参考実施例7と同一の方法で調製された抗hEREGコントロール抗体(EGL-G1d)およびFcγR結合を増強したFcを有する抗hEREG抗体(EGL-afucosyl、EGL-ART6)がそれぞれ1 mg/mLになるようにHis buffer (150 mM NaCl、20 mM His-HCl buffer pH6.0)を用いて調製された。
移植後7日目にEGL-G1d、EGL-afucosyl、EGL-ART6が10 mg/kgになるように尾静脈より投与された。
抗腫瘍効果測定に際しての薬剤処置に関する詳細を表38に示した。

抗腫瘍効果については、(11-2)に記した計算式にて算出した腫瘍体積で評価した。
TGI(tumor growth inhibition)値については、以下の計算式より算出した。
TGI=(1-(測定時における着目する群の腫瘍体積の平均値-抗体投与前の時点での腫瘍体積の平均値)÷(測定時におけるcontrol群腫瘍体積の平均値-抗体投与前の時点での腫瘍体積の平均値))×100
抗体のCDC活性は抗腫瘍効果に寄与するという報告がある一方で(Nat. Immunol., 2017, 18, 889)、infusion-related reactionに代表されるCDC活性由来の副作用を引き起こすことが知られている(J. Immunol. 2008, 180, 2294-2298およびBr. J. Haematol. 2001, 115, 807-811)。従ってADCC活性やADCP活性を増強した抗体医薬品を開発する場合であっても、標的疾患によってCDC活性の程度を選択できることが好ましい。補体とFcとの相互作用はC1qによって仲介される。C1qとFcの相互作用を解析した報告によると(Science, 2018, 359, 794-797およびMolecular Immunology 2012, 51, 66-72)、Fc領域上のFcγRとC1qの相互作用部位は一部重複していると考えられる。これらの文献において、Fc上のポジションのうち、EUナンバリング329番目、330番目、331番目の残基がC1qとの相互作用に重要であり、その他にもEUナンバリング268番目や270番目、298番目の残基もC1qとの結合に寄与していると報告されている。これらのポジションおよびその周辺残基は、本発明でFcγRへの結合を増強するために改変されている部位であることから、作製されたFc領域改変体はC1qへの結合も増強あるいは減弱されている可能性が高く、抗体医薬品の開発においてCDC活性を制御できる可能性がある。そこで作製されたFc領域改変体のC1qへの結合評価が実施された。

Claims (15)
- (A) アデノシン含有化合物の濃度に依存したCTLA-4結合活性を有する可変領域、および
(B) 親Fc領域において複数のアミノ酸改変を含む変異Fc領域
を含む抗CTLA-4抗体であって、親Fc領域が二本のポリペプチド鎖によって構成され、かつ変異Fc領域が、以下の位置におけるアミノ酸改変を含む、抗CTLA-4抗体:
(i) 親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置234、235、236、239、268、270、298、および330、ならびに
(ii) 親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置270、298、326、330、および334。 - 可変領域が、以下の(a)から(i)より選択される少なくとも一つの特徴を有する、請求項1に記載の抗CTLA-4抗体:
(a) 100 μMのアデノシン含有化合物の存在下での結合活性が、アデノシン含有化合物の非存在下での結合活性と比べて、2倍以上高い、
(b) 100 μMのアデノシン含有化合物の存在下でのKD値が5×10-7 M以下である、
(c) アデノシン含有化合物の非存在下でのKD値が1×10-6 M以上である、
(d) アデノシン含有化合物およびCTLA-4とともに三者複合体を形成する、
(e) ヒトCTLA-4(細胞外ドメイン、配列番号:28)の97番目のアミノ酸から106番目のアミノ酸の領域に結合する、
(f) CTLA-4への結合に関して、ABAM004(VH、配列番号:10;およびVL、配列番号:11)と競合する、
(g) ABAM004(VH、配列番号:10;およびVL、配列番号:11)によって結合されるのと同じエピトープに結合する、
(h) CTLA-4発現細胞に対して細胞傷害活性を示す、および
(i) ヒトおよびマウス由来のCTLA-4に結合する。 - (a) アミノ酸配列SX1TMNを含み、X1はH、A、R、またはKであるHVR-H1(配列番号:223)、(b) アミノ酸配列SISX1X2SX3YIYYAX4SVX5Gを含み、X1はSまたはT、X2はRまたはQ、X3はGまたはH、X4はD、E、またはR、X5はKまたはRであるHVR-H2(配列番号:224)、および (c) アミノ酸配列YGX1REDMLWVFDYを含み、X1はKまたはAであるHVR-H3(配列番号:225)を含む、請求項1または2に記載の抗CTLA-4抗体。
- (a) アミノ酸配列X1GX2STX3VGDYX4X5VX6を含み、X1はT、D、Q、またはE、X2はTまたはP、X3はDまたはG、X4はNまたはT、X5はYまたはW、X6はSまたはHであるHVR-L1(配列番号:226)、(b) アミノ酸配列X1TX2X3KPX4を含み、X1はE、F、またはY、X2はSまたはI、X3はKまたはS、X4はS、E、またはKであるHVR-L2(配列番号:227)、および (c) アミノ酸配列X1TYAAPLGPX2を含み、X1はSまたはQ、X2はMまたはTであるHVR-L3(配列番号:228)をさらに含む、請求項3に記載の抗CTLA-4抗体。
- (a) 配列番号:83~86、98、135~141のいずれか1つのアミノ酸配列と少なくとも95%の配列同一性を有するVH配列;(b) 配列番号:88~95、97、99、134、144~149のいずれか1つのアミノ酸配列と少なくとも95%の配列同一性を有するVL配列;または (c) 配列番号:83~86、98、135~141のいずれか1つのアミノ酸配列を有するVH配列、および配列番号:88~95、97、99、134、144~149のいずれか1つのアミノ酸配列を有するVL配列を含む、請求項1または2に記載の抗CTLA-4抗体。
- (1) 配列番号:98のVH配列および配列番号:99のVL配列、
(2) 配列番号:83のVH配列および配列番号:88のVL配列、
(3) 配列番号:83のVH配列および配列番号:89のVL配列、
(4) 配列番号:83のVH配列および配列番号:90のVL配列、
(5) 配列番号:83のVH配列および配列番号:91のVL配列、
(6) 配列番号:83のVH配列および配列番号:92のVL配列、
(7) 配列番号:83のVH配列および配列番号:93のVL配列、
(8) 配列番号:83のVH配列および配列番号:94のVL配列、
(9) 配列番号:83のVH配列および配列番号:97のVL配列、
(10) 配列番号:83のVH配列および配列番号:95のVL配列、
(11) 配列番号:84のVH配列および配列番号:97のVL配列、
(12) 配列番号:85のVH配列および配列番号:97のVL配列、
(13) 配列番号:86のVH配列および配列番号:97のVL配列、
(14) 配列番号:86のVH配列および配列番号:134のVL配列、
(15) 配列番号:136のVH配列および配列番号:97のVL配列、
(16) 配列番号:135のVH配列および配列番号:97のVL配列、
(17) 配列番号:136のVH配列および配列番号:95のVL配列、
(18) 配列番号:137のVH配列および配列番号:97のVL配列、
(19) 配列番号:138のVH配列および配列番号:97のVL配列、
(20) 配列番号:138のVH配列および配列番号:144のVL配列、
(21) 配列番号:138のVH配列および配列番号:145のVL配列、
(22) 配列番号:138のVH配列および配列番号:146のVL配列、
(23) 配列番号:139のVH配列および配列番号:146のVL配列、
(24) 配列番号:140のVH配列および配列番号:146のVL配列、
(25) 配列番号:141のVH配列および配列番号:146のVL配列、
(26) 配列番号:140のVH配列および配列番号:147のVL配列、
(27) 配列番号:141のVH配列および配列番号:147のVL配列、
(28) 配列番号:140のVH配列および配列番号:148のVL配列、
(29) 配列番号:141のVH配列および配列番号:148のVL配列、
(30) 配列番号:136のVH配列および配列番号:149のVL配列、
(31) 配列番号:140のVH配列および配列番号:146のVL配列を含む第一の可変領域、ならびに配列番号:141のVH配列および配列番号:146のVL配列を含む第二の可変領域、または
(32) 配列番号:140のVH配列および配列番号:147のVL配列を含む第一の可変領域、ならびに配列番号:141のVH配列および配列番号:147のVL配列を含む第二の可変領域、
を含む、請求項5に記載の抗CTLA-4抗体。 - 変異Fc領域が、さらに親Fc領域の第一のポリペプチドにおける、EUナンバリングで表される位置250および307におけるアミノ酸改変を含む、請求項1から6のいずれか一項に記載の抗CTLA-4抗体。
- 変異Fc領域が、さらに親Fc領域の第二のポリペプチドにおける、EUナンバリングで表される位置250および307におけるアミノ酸改変を含む、請求項1から7のいずれか一項に記載の抗CTLA-4抗体。
- 変異Fc領域を含む重鎖定常領域を含む、請求項1から8のいずれか一項に記載の抗CTLA-4抗体。
- 重鎖定常領域が、
(1) 配列番号:358の第一のポリペプチド、および配列番号:359の第二のポリペプチド、または
(2) 配列番号:360の第一のポリペプチド、および配列番号:361の第二のポリペプチド、
を含む、請求項9に記載の抗CTLA-4抗体。 - (1) 配列番号:335の第一のH鎖ポリペプチド、配列番号:336の第二のH鎖ポリペプチド、および配列番号:161のL鎖ポリペプチド、または
(2) 配列番号:337の第一のH鎖ポリペプチド、配列番号:338の第二のH鎖ポリペプチド、および配列番号:161のL鎖ポリペプチド、
を含む、抗CTLA-4抗体。 - 請求項1から11のいずれか一項に記載の抗CTLA-4抗体をコードする、単離された核酸。
- 請求項12に記載の核酸を含む、宿主細胞。
- 抗CTLA-4抗体を製造する方法であって、抗CTLA-4抗体が製造されるように請求項13に記載の宿主細胞を培養することを含む、方法。
- 請求項1から11のいずれか一項に記載の抗CTLA-4抗体および薬学的に許容される担体を含む、薬学的製剤。
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3221833A CA3221833A1 (en) | 2021-06-25 | 2022-06-24 | Anti-ctla-4 antibody |
| BR112023022992A BR112023022992A2 (pt) | 2021-06-25 | 2022-06-24 | Anticorpo anti-ctla-4 |
| CN202280044970.4A CN117616123A (zh) | 2021-06-25 | 2022-06-24 | 抗ctla-4抗体 |
| IL309115A IL309115A (en) | 2021-06-25 | 2022-06-24 | Anti-CTLA-4 antibodies |
| MX2023013897A MX2023013897A (es) | 2021-06-25 | 2022-06-24 | Anticuerpo anti-antigeno 4 del linfocito t citotoxico (anti-ctla-4). |
| AU2022299846A AU2022299846B2 (en) | 2021-06-25 | 2022-06-24 | Anti–ctla-4 antibody |
| EP22828527.6A EP4361273A1 (en) | 2021-06-25 | 2022-06-24 | Anti-ctla-4 antibody |
| CR20240026A CR20240026A (es) | 2021-06-25 | 2022-06-24 | Anticuerpo anti-ctla-4 |
| KR1020247002157A KR102690141B1 (ko) | 2021-06-25 | 2022-06-24 | 항ctla-4 항체 |
| JP2023530135A JP7472405B2 (ja) | 2021-06-25 | 2022-06-24 | 抗ctla-4抗体 |
| CONC2024/0000518A CO2024000518A2 (es) | 2021-06-25 | 2024-01-22 | Anticuerpo anti-ctla-4 |
| JP2024063159A JP2024095774A (ja) | 2021-06-25 | 2024-04-10 | 抗ctla-4抗体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-105804 | 2021-06-25 | ||
| JP2021105804 | 2021-06-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022270611A1 true WO2022270611A1 (ja) | 2022-12-29 |
Family
ID=84544417
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/025220 WO2022270611A1 (ja) | 2021-06-25 | 2022-06-24 | 抗ctla-4抗体 |
Country Status (15)
| Country | Link |
|---|---|
| EP (1) | EP4361273A1 (ja) |
| JP (2) | JP7472405B2 (ja) |
| KR (1) | KR102690141B1 (ja) |
| CN (1) | CN117616123A (ja) |
| AR (1) | AR126220A1 (ja) |
| AU (1) | AU2022299846B2 (ja) |
| BR (1) | BR112023022992A2 (ja) |
| CA (1) | CA3221833A1 (ja) |
| CL (1) | CL2023003836A1 (ja) |
| CO (1) | CO2024000518A2 (ja) |
| CR (1) | CR20240026A (ja) |
| IL (1) | IL309115A (ja) |
| MX (1) | MX2023013897A (ja) |
| TW (1) | TW202317627A (ja) |
| WO (1) | WO2022270611A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11912989B2 (en) | 2013-12-04 | 2024-02-27 | Chugai Seiyaku Kabushiki Kaisha | Antigen-binding molecules, the antigen-binding activity of which varies according to the concentration of compounds, and libraries of said molecules |
Citations (131)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| EP0404097A2 (de) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispezifische und oligospezifische, mono- und oligovalente Rezeptoren, ihre Herstellung und Verwendung |
| EP0425235A2 (en) | 1989-10-25 | 1991-05-02 | Immunogen Inc | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| WO1994029351A2 (en) | 1993-06-16 | 1994-12-22 | Celltech Limited | Antibodies |
| WO1995033844A1 (de) | 1994-06-03 | 1995-12-14 | GSF - Forschungszentrum für Umwelt und Gesundheit GmbH | Verfahren zur herstellung von heterologen bispezifischen antikörpern |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| WO1996027011A1 (en) | 1995-03-01 | 1996-09-06 | Genentech, Inc. | A method for making heteromultimeric polypeptides |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5739116A (en) | 1994-06-03 | 1998-04-14 | American Cyanamid Company | Enediyne derivatives useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US5750373A (en) | 1990-12-03 | 1998-05-12 | Genentech, Inc. | Enrichment method for variant proteins having altered binding properties, M13 phagemids, and growth hormone variants |
| US5770710A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Antitumor and antibacterial substituted disulfide derivatives prepared from compounds possessing a methlytrithio group |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5770701A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Process for preparing targeted forms of methyltrithio antitumor agents |
| US5780588A (en) | 1993-01-26 | 1998-07-14 | Arizona Board Of Regents | Elucidation and synthesis of selected pentapeptides |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| WO1998050431A2 (en) | 1997-05-02 | 1998-11-12 | Genentech, Inc. | A method for making multispecific antibodies having heteromultimeric and common components |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO2000042072A3 (en) | 1999-01-15 | 2000-11-30 | Genentech Inc | Polypeptide variants with altered effector function |
| US6171586B1 (en) | 1997-06-13 | 2001-01-09 | Genentech, Inc. | Antibody formulation |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO2001014424A2 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| WO2001029246A1 (fr) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Procede de production d'un polypeptide |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| US6267958B1 (en) | 1995-07-27 | 2001-07-31 | Genentech, Inc. | Protein formulation |
| WO2002031140A1 (fr) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cellules produisant des compositions d'anticorps |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6602677B1 (en) | 1997-09-19 | 2003-08-05 | Promega Corporation | Thermostable luciferases and methods of production |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| US6630579B2 (en) | 1999-12-29 | 2003-10-07 | Immunogen Inc. | Cytotoxic agents comprising modified doxorubicins and daunorubicins and their therapeutic use |
| WO2003084570A1 (fr) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicament contenant une composition d'anticorps appropriee au patient souffrant de polymorphisme fc$g(g)riiia |
| WO2003085107A1 (fr) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cellules à génome modifié |
| WO2003085119A1 (fr) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Procede d'amelioration de l'activite d'une composition d'anticorps de liaison avec le recepteur fc$g(g) iiia |
| WO2004029207A2 (en) | 2002-09-27 | 2004-04-08 | Xencor Inc. | Optimized fc variants and methods for their generation |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| WO2004092219A2 (en) | 2003-04-10 | 2004-10-28 | Protein Design Labs, Inc | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050079574A1 (en) | 2003-01-16 | 2005-04-14 | Genentech, Inc. | Synthetic antibody phage libraries |
| WO2005035778A1 (ja) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | α1,6-フコシルトランスフェラーゼの機能を抑制するRNAを用いた抗体組成物の製造法 |
| WO2005035586A1 (ja) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | 融合蛋白質組成物 |
| US20050119455A1 (en) | 2002-06-03 | 2005-06-02 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (ja) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | 抗体組成物を含有する医薬 |
| WO2005100402A1 (en) | 2004-04-13 | 2005-10-27 | F.Hoffmann-La Roche Ag | Anti-p-selectin antibodies |
| US20050260186A1 (en) | 2003-03-05 | 2005-11-24 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20050266000A1 (en) | 2004-04-09 | 2005-12-01 | Genentech, Inc. | Variable domain library and uses |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US20060025576A1 (en) | 2000-04-11 | 2006-02-02 | Genentech, Inc. | Multivalent antibodies and uses therefor |
| WO2006019447A1 (en) | 2004-07-15 | 2006-02-23 | Xencor, Inc. | Optimized fc variants |
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| WO2006044908A2 (en) | 2004-10-20 | 2006-04-27 | Genentech, Inc. | Antibody formulation in histidine-acetate buffer |
| US7041870B2 (en) | 2000-11-30 | 2006-05-09 | Medarex, Inc. | Transgenic transchromosomal rodents for making human antibodies |
| WO2006053301A2 (en) | 2004-11-12 | 2006-05-18 | Xencor, Inc. | Fc variants with altered binding to fcrn |
| US20060104968A1 (en) | 2003-03-05 | 2006-05-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminogly ycanases |
| WO2006067913A1 (ja) | 2004-12-22 | 2006-06-29 | Chugai Seiyaku Kabushiki Kaisha | フコーストランスポーターの機能が阻害された細胞を用いた抗体の作製方法 |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| WO2006106905A1 (ja) | 2005-03-31 | 2006-10-12 | Chugai Seiyaku Kabushiki Kaisha | 会合制御によるポリペプチド製造方法 |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| EP1752471A1 (en) | 2005-01-05 | 2007-02-14 | f-star Biotechnologische Forschungs- und Entwicklungsges.m.b.H. | Synthetic immunoglobulin domains with binding properties engineered in regions of the molecule different from the complementarity determining regions |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US20070061900A1 (en) | 2000-10-31 | 2007-03-15 | Murphy Andrew J | Methods of modifying eukaryotic cells |
| US20070117126A1 (en) | 1999-12-15 | 2007-05-24 | Genentech, Inc. | Shotgun scanning |
| US20070160598A1 (en) | 2005-11-07 | 2007-07-12 | Dennis Mark S | Binding polypeptides with diversified and consensus vh/vl hypervariable sequences |
| US20070237764A1 (en) | 2005-12-02 | 2007-10-11 | Genentech, Inc. | Binding polypeptides with restricted diversity sequences |
| WO2007114325A1 (ja) | 2006-03-31 | 2007-10-11 | Chugai Seiyaku Kabushiki Kaisha | 二重特異性抗体を精製するための抗体改変方法 |
| US20070292936A1 (en) | 2006-05-09 | 2007-12-20 | Genentech, Inc. | Binding polypeptides with optimized scaffolds |
| US20080069820A1 (en) | 2006-08-30 | 2008-03-20 | Genentech, Inc. | Multispecific antibodies |
| WO2008077546A1 (en) | 2006-12-22 | 2008-07-03 | F. Hoffmann-La Roche Ag | Antibodies against insulin-like growth factor i receptor and uses thereof |
| WO2008119353A1 (en) | 2007-03-29 | 2008-10-09 | Genmab A/S | Bispecific antibodies and methods for production thereof |
| US20090002360A1 (en) | 2007-05-25 | 2009-01-01 | Innolux Display Corp. | Liquid crystal display device and method for driving same |
| US7498298B2 (en) | 2003-11-06 | 2009-03-03 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| WO2009041613A1 (ja) | 2007-09-26 | 2009-04-02 | Chugai Seiyaku Kabushiki Kaisha | 抗体定常領域改変体 |
| US7521541B2 (en) | 2004-09-23 | 2009-04-21 | Genetech Inc. | Cysteine engineered antibodies and conjugates |
| US7527791B2 (en) | 2004-03-31 | 2009-05-05 | Genentech, Inc. | Humanized anti-TGF-beta antibodies |
| WO2009086320A1 (en) | 2007-12-26 | 2009-07-09 | Xencor, Inc | Fc variants with altered binding to fcrn |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2009125825A1 (ja) | 2008-04-11 | 2009-10-15 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2010107110A1 (ja) | 2009-03-19 | 2010-09-23 | 中外製薬株式会社 | 抗体定常領域改変体 |
| WO2011028952A1 (en) | 2009-09-02 | 2011-03-10 | Xencor, Inc. | Compositions and methods for simultaneous bivalent and monovalent co-engagement of antigens |
| WO2011122011A2 (en) | 2010-03-30 | 2011-10-06 | Chugai Seiyaku Kabushiki Kaisha | Antibodies with modified affinity to fcrn that promote antigen clearance |
| WO2011131746A2 (en) | 2010-04-20 | 2011-10-27 | Genmab A/S | Heterodimeric antibody fc-containing proteins and methods for production thereof |
| WO2012016227A2 (en) | 2010-07-29 | 2012-02-02 | Xencor, Inc. | Antibodies with modified isoelectric points |
| WO2012058768A1 (en) | 2010-11-05 | 2012-05-10 | Zymeworks Inc. | Stable heterodimeric antibody design with mutations in the fc domain |
| WO2012073992A1 (ja) | 2010-11-30 | 2012-06-07 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2012132067A1 (ja) | 2011-03-30 | 2012-10-04 | 中外製薬株式会社 | 抗原結合分子の血漿中滞留性と免疫原性を改変する方法 |
| WO2013002362A1 (ja) | 2011-06-30 | 2013-01-03 | 中外製薬株式会社 | ヘテロ二量化ポリペプチド |
| WO2013046704A2 (en) | 2011-09-30 | 2013-04-04 | Chugai Seiyaku Kabushiki Kaisha | THERAPEUTIC ANTIGEN-BINDING MOLECULE WITH A FcRn-BINDING DOMAIN THAT PROMOTES ANTIGEN CLEARANCE |
| WO2013047752A1 (ja) | 2011-09-30 | 2013-04-04 | 中外製薬株式会社 | 抗原の消失を促進する抗原結合分子 |
| WO2013118858A1 (ja) | 2012-02-09 | 2013-08-15 | 中外製薬株式会社 | 抗体のFc領域改変体 |
| WO2013120629A1 (en) | 2012-02-15 | 2013-08-22 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded therapeutic protein |
| WO2013180201A1 (ja) | 2012-05-30 | 2013-12-05 | 中外製薬株式会社 | 会合化した抗原を消失させる抗原結合分子 |
| WO2013180200A1 (ja) | 2012-05-30 | 2013-12-05 | 中外製薬株式会社 | 標的組織特異的抗原結合分子 |
| WO2014104165A1 (ja) | 2012-12-27 | 2014-07-03 | 中外製薬株式会社 | ヘテロ二量化ポリペプチド |
| WO2014145159A2 (en) | 2013-03-15 | 2014-09-18 | Permeon Biologics, Inc. | Charge-engineered antibodies or compositions of penetration-enhanced targeting proteins and methods of use |
| WO2015083764A1 (ja) | 2013-12-04 | 2015-06-11 | 中外製薬株式会社 | 化合物の濃度に応じて抗原結合能の変化する抗原結合分子及びそのライブラリ |
| WO2015083674A1 (ja) | 2013-12-06 | 2015-06-11 | 株式会社ニコン | 撮像素子および撮像装置 |
| WO2016125495A1 (en) | 2015-02-05 | 2016-08-11 | Chugai Seiyaku Kabushiki Kaisha | Antibodies comprising an ion concentration dependent antigen-binding domain, fc region variants, il-8-binding antibodies, and uses therof |
| EP2101823B1 (en) | 2007-01-09 | 2016-11-23 | CureVac AG | Rna-coded antibody |
| JP2020073557A (ja) * | 2012-02-24 | 2020-05-14 | 中外製薬株式会社 | FcγRIIBを介して抗原の消失を促進する抗原結合分子 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CZ303703B6 (cs) | 1998-12-23 | 2013-03-20 | Pfizer Inc. | Monoklonální protilátka nebo její antigen-vázající fragment, farmaceutická kompozice obsahující tuto protilátku nebo fragment, bunecná linie produkující tuto protilátku nebo fragment, zpusob prípravy této protilátky, izolovaná nukleová kyselina kóduj |
| KR100704984B1 (ko) * | 2005-09-27 | 2007-04-09 | 삼성전기주식회사 | 이미지 신뢰성 확보를 위한 데이터 처리 시스템 |
| GB0624500D0 (en) * | 2006-12-07 | 2007-01-17 | Istituto Superiore Di Sanito | A novel passive vaccine for candida infections |
| GB201103955D0 (en) | 2011-03-09 | 2011-04-20 | Antitope Ltd | Antibodies |
| JP5972915B2 (ja) | 2011-03-16 | 2016-08-17 | アムジエン・インコーポレーテツド | Fc変異体 |
| CA2874083C (en) * | 2014-12-05 | 2024-01-02 | Universite Laval | Tdp-43-binding polypeptides useful for the treatment of neurodegenerative diseases |
| MY183415A (en) * | 2014-12-19 | 2021-02-18 | Chugai Pharmaceutical Co Ltd | Anti-c5 antibodies and methods of use |
| MA53355A (fr) | 2015-05-29 | 2022-03-16 | Agenus Inc | Anticorps anti-ctla-4 et leurs procédés d'utilisation |
| JP7501991B2 (ja) * | 2016-03-15 | 2024-06-18 | キャンサー・リサーチ・テクノロジー・リミテッド | 抗体及び関連分子並びにその使用 |
| BR112021002037A2 (pt) * | 2018-08-10 | 2021-05-04 | Chugai Seiyaku Kabushiki Kaisha | molécula de ligação de antígeno anti-cd137 e uso da mesma |
-
2022
- 2022-06-24 CA CA3221833A patent/CA3221833A1/en active Pending
- 2022-06-24 IL IL309115A patent/IL309115A/en unknown
- 2022-06-24 CN CN202280044970.4A patent/CN117616123A/zh active Pending
- 2022-06-24 CR CR20240026A patent/CR20240026A/es unknown
- 2022-06-24 KR KR1020247002157A patent/KR102690141B1/ko active IP Right Grant
- 2022-06-24 TW TW111123617A patent/TW202317627A/zh unknown
- 2022-06-24 WO PCT/JP2022/025220 patent/WO2022270611A1/ja active Application Filing
- 2022-06-24 EP EP22828527.6A patent/EP4361273A1/en active Pending
- 2022-06-24 AU AU2022299846A patent/AU2022299846B2/en active Active
- 2022-06-24 MX MX2023013897A patent/MX2023013897A/es unknown
- 2022-06-24 AR ARP220101659A patent/AR126220A1/es unknown
- 2022-06-24 JP JP2023530135A patent/JP7472405B2/ja active Active
- 2022-06-24 BR BR112023022992A patent/BR112023022992A2/pt unknown
-
2023
- 2023-12-21 CL CL2023003836A patent/CL2023003836A1/es unknown
-
2024
- 2024-01-22 CO CONC2024/0000518A patent/CO2024000518A2/es unknown
- 2024-04-10 JP JP2024063159A patent/JP2024095774A/ja active Pending
Patent Citations (143)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5648260A (en) | 1987-03-18 | 1997-07-15 | Scotgen Biopharmaceuticals Incorporated | DNA encoding antibodies with altered effector functions |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5770710A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Antitumor and antibacterial substituted disulfide derivatives prepared from compounds possessing a methlytrithio group |
| US5770701A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Process for preparing targeted forms of methyltrithio antitumor agents |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| EP0404097A2 (de) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispezifische und oligospezifische, mono- und oligovalente Rezeptoren, ihre Herstellung und Verwendung |
| US5416064A (en) | 1989-10-25 | 1995-05-16 | Immunogen, Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| EP0425235A2 (en) | 1989-10-25 | 1991-05-02 | Immunogen Inc | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US6417429B1 (en) | 1989-10-27 | 2002-07-09 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5750373A (en) | 1990-12-03 | 1998-05-12 | Genentech, Inc. | Enrichment method for variant proteins having altered binding properties, M13 phagemids, and growth hormone variants |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5780588A (en) | 1993-01-26 | 1998-07-14 | Arizona Board Of Regents | Elucidation and synthesis of selected pentapeptides |
| WO1994029351A2 (en) | 1993-06-16 | 1994-12-22 | Celltech Limited | Antibodies |
| WO1995033844A1 (de) | 1994-06-03 | 1995-12-14 | GSF - Forschungszentrum für Umwelt und Gesundheit GmbH | Verfahren zur herstellung von heterologen bispezifischen antikörpern |
| US5773001A (en) | 1994-06-03 | 1998-06-30 | American Cyanamid Company | Conjugates of methyltrithio antitumor agents and intermediates for their synthesis |
| US5767285A (en) | 1994-06-03 | 1998-06-16 | American Cyanamid Company | Linkers useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US5739116A (en) | 1994-06-03 | 1998-04-14 | American Cyanamid Company | Enediyne derivatives useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US5877296A (en) | 1994-06-03 | 1999-03-02 | American Cyanamid Company | Process for preparing conjugates of methyltrithio antitumor agents |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| WO1996027011A1 (en) | 1995-03-01 | 1996-09-06 | Genentech, Inc. | A method for making heteromultimeric polypeptides |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US6267958B1 (en) | 1995-07-27 | 2001-07-31 | Genentech, Inc. | Protein formulation |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| WO1998050431A2 (en) | 1997-05-02 | 1998-11-12 | Genentech, Inc. | A method for making multispecific antibodies having heteromultimeric and common components |
| US6171586B1 (en) | 1997-06-13 | 2001-01-09 | Genentech, Inc. | Antibody formulation |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| US6602677B1 (en) | 1997-09-19 | 2003-08-05 | Promega Corporation | Thermostable luciferases and methods of production |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US7332581B2 (en) | 1999-01-15 | 2008-02-19 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US7371826B2 (en) | 1999-01-15 | 2008-05-13 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000042072A3 (en) | 1999-01-15 | 2000-11-30 | Genentech Inc | Polypeptide variants with altered effector function |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| WO2001014424A2 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| WO2001029246A1 (fr) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Procede de production d'un polypeptide |
| US20070117126A1 (en) | 1999-12-15 | 2007-05-24 | Genentech, Inc. | Shotgun scanning |
| US6630579B2 (en) | 1999-12-29 | 2003-10-07 | Immunogen Inc. | Cytotoxic agents comprising modified doxorubicins and daunorubicins and their therapeutic use |
| US20060025576A1 (en) | 2000-04-11 | 2006-02-02 | Genentech, Inc. | Multivalent antibodies and uses therefor |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| WO2002031140A1 (fr) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cellules produisant des compositions d'anticorps |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| US20070061900A1 (en) | 2000-10-31 | 2007-03-15 | Murphy Andrew J | Methods of modifying eukaryotic cells |
| US7041870B2 (en) | 2000-11-30 | 2006-05-09 | Medarex, Inc. | Transgenic transchromosomal rodents for making human antibodies |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| WO2003085119A1 (fr) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Procede d'amelioration de l'activite d'une composition d'anticorps de liaison avec le recepteur fc$g(g) iiia |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2003085107A1 (fr) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cellules à génome modifié |
| WO2003084570A1 (fr) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicament contenant une composition d'anticorps appropriee au patient souffrant de polymorphisme fc$g(g)riiia |
| US20040110704A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells of which genome is modified |
| US20050119455A1 (en) | 2002-06-03 | 2005-06-02 | Genentech, Inc. | Synthetic antibody phage libraries |
| WO2004029207A2 (en) | 2002-09-27 | 2004-04-08 | Xencor Inc. | Optimized fc variants and methods for their generation |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20050079574A1 (en) | 2003-01-16 | 2005-04-14 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20050260186A1 (en) | 2003-03-05 | 2005-11-24 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20060104968A1 (en) | 2003-03-05 | 2006-05-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminogly ycanases |
| WO2004092219A2 (en) | 2003-04-10 | 2004-10-28 | Protein Design Labs, Inc | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| WO2005035586A1 (ja) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | 融合蛋白質組成物 |
| WO2005035778A1 (ja) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | α1,6-フコシルトランスフェラーゼの機能を抑制するRNAを用いた抗体組成物の製造法 |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| US7498298B2 (en) | 2003-11-06 | 2009-03-03 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| WO2005053742A1 (ja) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | 抗体組成物を含有する医薬 |
| US7527791B2 (en) | 2004-03-31 | 2009-05-05 | Genentech, Inc. | Humanized anti-TGF-beta antibodies |
| US20050266000A1 (en) | 2004-04-09 | 2005-12-01 | Genentech, Inc. | Variable domain library and uses |
| WO2005100402A1 (en) | 2004-04-13 | 2005-10-27 | F.Hoffmann-La Roche Ag | Anti-p-selectin antibodies |
| WO2006019447A1 (en) | 2004-07-15 | 2006-02-23 | Xencor, Inc. | Optimized fc variants |
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| US7521541B2 (en) | 2004-09-23 | 2009-04-21 | Genetech Inc. | Cysteine engineered antibodies and conjugates |
| WO2006044908A2 (en) | 2004-10-20 | 2006-04-27 | Genentech, Inc. | Antibody formulation in histidine-acetate buffer |
| WO2006053301A2 (en) | 2004-11-12 | 2006-05-18 | Xencor, Inc. | Fc variants with altered binding to fcrn |
| WO2006067913A1 (ja) | 2004-12-22 | 2006-06-29 | Chugai Seiyaku Kabushiki Kaisha | フコーストランスポーターの機能が阻害された細胞を用いた抗体の作製方法 |
| EP1772465A1 (en) | 2005-01-05 | 2007-04-11 | f-star Biotechnologische Forschungs- und Entwicklungsges.m.b.H. | Synthetic immunoglobulin domains with binding properties engineered in regions of the molecule different from the complementarity determining regions |
| EP1752471A1 (en) | 2005-01-05 | 2007-02-14 | f-star Biotechnologische Forschungs- und Entwicklungsges.m.b.H. | Synthetic immunoglobulin domains with binding properties engineered in regions of the molecule different from the complementarity determining regions |
| WO2006106905A1 (ja) | 2005-03-31 | 2006-10-12 | Chugai Seiyaku Kabushiki Kaisha | 会合制御によるポリペプチド製造方法 |
| US20070160598A1 (en) | 2005-11-07 | 2007-07-12 | Dennis Mark S | Binding polypeptides with diversified and consensus vh/vl hypervariable sequences |
| US20070237764A1 (en) | 2005-12-02 | 2007-10-11 | Genentech, Inc. | Binding polypeptides with restricted diversity sequences |
| WO2007114325A1 (ja) | 2006-03-31 | 2007-10-11 | Chugai Seiyaku Kabushiki Kaisha | 二重特異性抗体を精製するための抗体改変方法 |
| US20070292936A1 (en) | 2006-05-09 | 2007-12-20 | Genentech, Inc. | Binding polypeptides with optimized scaffolds |
| US20080069820A1 (en) | 2006-08-30 | 2008-03-20 | Genentech, Inc. | Multispecific antibodies |
| WO2008077546A1 (en) | 2006-12-22 | 2008-07-03 | F. Hoffmann-La Roche Ag | Antibodies against insulin-like growth factor i receptor and uses thereof |
| EP2101823B1 (en) | 2007-01-09 | 2016-11-23 | CureVac AG | Rna-coded antibody |
| WO2008119353A1 (en) | 2007-03-29 | 2008-10-09 | Genmab A/S | Bispecific antibodies and methods for production thereof |
| US20090002360A1 (en) | 2007-05-25 | 2009-01-01 | Innolux Display Corp. | Liquid crystal display device and method for driving same |
| WO2009041613A1 (ja) | 2007-09-26 | 2009-04-02 | Chugai Seiyaku Kabushiki Kaisha | 抗体定常領域改変体 |
| WO2009086320A1 (en) | 2007-12-26 | 2009-07-09 | Xencor, Inc | Fc variants with altered binding to fcrn |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2009125825A1 (ja) | 2008-04-11 | 2009-10-15 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2010107110A1 (ja) | 2009-03-19 | 2010-09-23 | 中外製薬株式会社 | 抗体定常領域改変体 |
| WO2011028952A1 (en) | 2009-09-02 | 2011-03-10 | Xencor, Inc. | Compositions and methods for simultaneous bivalent and monovalent co-engagement of antigens |
| WO2011122011A2 (en) | 2010-03-30 | 2011-10-06 | Chugai Seiyaku Kabushiki Kaisha | Antibodies with modified affinity to fcrn that promote antigen clearance |
| WO2011131746A2 (en) | 2010-04-20 | 2011-10-27 | Genmab A/S | Heterodimeric antibody fc-containing proteins and methods for production thereof |
| WO2012016227A2 (en) | 2010-07-29 | 2012-02-02 | Xencor, Inc. | Antibodies with modified isoelectric points |
| WO2012058768A1 (en) | 2010-11-05 | 2012-05-10 | Zymeworks Inc. | Stable heterodimeric antibody design with mutations in the fc domain |
| WO2012073992A1 (ja) | 2010-11-30 | 2012-06-07 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2012132067A1 (ja) | 2011-03-30 | 2012-10-04 | 中外製薬株式会社 | 抗原結合分子の血漿中滞留性と免疫原性を改変する方法 |
| WO2013002362A1 (ja) | 2011-06-30 | 2013-01-03 | 中外製薬株式会社 | ヘテロ二量化ポリペプチド |
| WO2013047752A1 (ja) | 2011-09-30 | 2013-04-04 | 中外製薬株式会社 | 抗原の消失を促進する抗原結合分子 |
| WO2013046704A2 (en) | 2011-09-30 | 2013-04-04 | Chugai Seiyaku Kabushiki Kaisha | THERAPEUTIC ANTIGEN-BINDING MOLECULE WITH A FcRn-BINDING DOMAIN THAT PROMOTES ANTIGEN CLEARANCE |
| WO2013118858A1 (ja) | 2012-02-09 | 2013-08-15 | 中外製薬株式会社 | 抗体のFc領域改変体 |
| WO2013120629A1 (en) | 2012-02-15 | 2013-08-22 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded therapeutic protein |
| JP2020073557A (ja) * | 2012-02-24 | 2020-05-14 | 中外製薬株式会社 | FcγRIIBを介して抗原の消失を促進する抗原結合分子 |
| WO2013180201A1 (ja) | 2012-05-30 | 2013-12-05 | 中外製薬株式会社 | 会合化した抗原を消失させる抗原結合分子 |
| WO2013180200A1 (ja) | 2012-05-30 | 2013-12-05 | 中外製薬株式会社 | 標的組織特異的抗原結合分子 |
| JP2020040975A (ja) * | 2012-05-30 | 2020-03-19 | 中外製薬株式会社 | 標的組織特異的抗原結合分子 |
| WO2014104165A1 (ja) | 2012-12-27 | 2014-07-03 | 中外製薬株式会社 | ヘテロ二量化ポリペプチド |
| WO2014145159A2 (en) | 2013-03-15 | 2014-09-18 | Permeon Biologics, Inc. | Charge-engineered antibodies or compositions of penetration-enhanced targeting proteins and methods of use |
| WO2015083764A1 (ja) | 2013-12-04 | 2015-06-11 | 中外製薬株式会社 | 化合物の濃度に応じて抗原結合能の変化する抗原結合分子及びそのライブラリ |
| WO2015083674A1 (ja) | 2013-12-06 | 2015-06-11 | 株式会社ニコン | 撮像素子および撮像装置 |
| WO2016125495A1 (en) | 2015-02-05 | 2016-08-11 | Chugai Seiyaku Kabushiki Kaisha | Antibodies comprising an ion concentration dependent antigen-binding domain, fc region variants, il-8-binding antibodies, and uses therof |
Non-Patent Citations (169)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1980 |
| ACTA CRYST, vol. D66, 2010, pages 486 - 501 |
| ACTA CRYST, vol. D67, 2011, pages 355 - 367 |
| ACTA. CRYST., vol. D62, 2006, pages 72 - 82 |
| ACTA. CRYST., vol. D66, 2010, pages 125 - 132 |
| ALMAGROFRANSSON, FRONT. BIOSCI., vol. 13, 2008, pages 1619 - 1633 |
| ARTHRITIS RHEUM., vol. 48, 2003, pages 3242 - 3252 |
| BACA ET AL., J. BIOL. CHEM., vol. 272, 1997, pages 10678 - 10684 |
| BIOTECHNOL, BIOENG., vol. 93, no. 5, 2006, pages 851 - 861 |
| BLOOD |
| BOERNER ET AL., J. IMMUNOL., vol. 147, 1991, pages 3001 |
| BR. J. HAEMATOL., vol. 115, 2001, pages 807 - 811 |
| BRENNAN ET AL., SCIENCE, vol. 229, 1985, pages 81 |
| BRUGGEMANN, M. ET AL., J. EXP. MED., vol. 172, 1990, pages 1351 - 1361 |
| BRUNET ET AL., NATURE, vol. 328, 1987, pages 267 - 270 |
| CANCER IMMUNOL. RES. |
| CAPEL ET AL., IMMUNOMETHODS, vol. 4, 1994, pages 25 - 34 |
| CARTER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 4285 |
| CARTON ET AL., BLOOD, vol. 99, 2002, pages 754 - 758 |
| CHARI ET AL., CANCER RES., vol. 52, 1992, pages 127 - 131 |
| CHEN ET AL., J. MOL. BIOL., vol. 293, 1999, pages 865 - 881 |
| CHOTHIALESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CHOWDHURY, METHODS MOL. BIOL., vol. 207, 2008, pages 179 - 196 |
| CHU ET AL., MOL IMMUNOL, vol. 45, 2008, pages 3926 - 3933 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLARK, CHEM IMMUNOL, vol. 65, 1997, pages 88 - 110 |
| CLYNES ET AL., NAT MED, vol. 6, 2000, pages 443 - 446 |
| CLYNES ET AL., PNAS (USA, vol. 95, pages 652 - 656 |
| CLYNES ET AL., PROC NATL ACAD SCI U S A, vol. 95, 1998, pages 652 - 656 |
| CLYNES ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 95, 1998, pages 652 - 656 |
| CRAGG, M.S. ET AL., BLOOD, vol. 101, 2003, pages 1045 - 1052 |
| CRAGG, M.S.M.J. GLENNIE, BLOOD, vol. 103, 2004, pages 2738 - 2743 |
| CREE ET AL., ANTICANCER DRUGS, vol. 6, 1995, pages 398 - 404 |
| CROUCH ET AL., J. IMMUNOL. METH., vol. 160, 1993, pages 81 - 88 |
| CUNNINGHAMWELLS, SCIENCE, vol. 244, 1989, pages 1081 - 1085 |
| DAERON, ANNU. REV. IMMUNOL., vol. 15, 1997, pages 203 - 234 |
| DALL'ACQUA ET AL., J. BIOL. CHEM., vol. 281, no. 33, 2006, pages 23514 - 23524 |
| DALL'ACQUA ET AL., METHODS, vol. 36, 2005, pages 61 - 68 |
| DAVIS ET AL., PROT. ENG. DES. & SEL., vol. 23, 2010, pages 195 - 202 |
| DE HAAS ET AL., J. LAB. CLIN. MED., vol. 126, pages 330 - 341 |
| DUBOWCHIK ET AL., BIOORG. & MED. CHEM. LETTERS, vol. 12, 2002, pages 1529 - 1532 |
| FELLOUSE, PROC. NATL. ACAD. SCI. USA, vol. 101, no. 34, 2004, pages 12467 - 12472 |
| FLATMAN ET AL., J. CHROMATOGR. B, vol. 848, 2007, pages 79 - 87 |
| GAZZANO-SANTORO ET AL., J. IMMUNOL. METHODS, vol. 202, 1996, pages 163 |
| GERNGROSS, NAT. BIOTECH., vol. 22, 2004, pages 1409 - 1414 |
| GHETIE ET AL., IMMUNOL. TODAY, vol. 18, no. 12, 1997, pages 592 - 598 |
| GHETIE ET AL., NATURE BIOTECHNOLOGY, vol. 15, no. 7, 1997, pages 637 - 640 |
| GLYCOBIOL, vol. 17, no. 1, 2006, pages 104 - 118 |
| GODING: "Monoclonal Antibodies: Principles and Practice", 1986, ACADEMIC PRESS, pages: 59 - 103 |
| GRAHAM ET AL., J. GEN VIROL., vol. 36, 1977, pages 59 |
| GREENWOOD ET AL., EUR J IMMUNOL, vol. 23, 1993, pages 1098 - 1104 |
| GRIFFITHS ET AL., EMBO J, vol. 12, 1993, pages 725 - 734 |
| GRUBER ET AL., J. IMMUNOL., vol. 152, 1994, pages 5368 |
| GUYER ET AL., J. IMMUNOL., vol. 117, 1976, pages 587 |
| HARLOWLANE: "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY |
| HELLSTROM, I ET AL., PROC. NAT 7ACAD. SCI. USA, vol. 82, 1985, pages 1499 - 1502 |
| HELLSTROM, I. ET AL., PROC. NAT 7ACAD. SCI. USA, vol. 83, 1986, pages 7059 - 7063 |
| HINMAN ET AL., CANCER RES., vol. 53, 1993, pages 3336 - 3342 |
| HINTON ET AL., J. BIOL. CHEM., vol. 279, no. 8, 2004, pages 6213 - 6216 |
| HINTON ET AL., J. IMMUNOL., vol. 176, no. 1, 2006, pages 346 - 356 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 102, 2005, pages 11600 - 11605 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HOOGENBOOMWINTER, J. MOL. BIOL., vol. 227, 1992, pages 381 - 388 |
| HORTON ET AL., CANCER RES, vol. 68, 2008, pages 8049 - 8057 |
| HUDSON ET AL., NAT. MED., vol. 9, 2003, pages 129 - 134 |
| IDUSOGIE ET AL., J. IMMUNOL., vol. 164, 2000, pages 4178 - 4184 |
| IMMUNOL. METHODS, vol. 202, 1996, pages 163 |
| INT J CANCER, vol. 41, 1988 |
| INT J MOL SCI., vol. 20, no. 11, 2019, pages 2839 |
| J EXP MED, vol. 170, 1989, pages 1369 - 1385 |
| J. APPL. CRYST., vol. 40, 2007, pages 658 - 674 |
| J. APPL. CRYST., vol. 43, 2010, pages 186 - 190 |
| J. BIOL. CHEM., vol. 286, 2011, pages 6685 - 6696 |
| J. IMMUNOL. METHODS, vol. 306, 2005, pages 151 - 160 |
| J. IMMUNOL., vol. 180, 2008, pages 2294 - 2298 |
| J. STRUCT. BIOL., vol. 194, 2016, pages 78 - 89 |
| JEFFERISLUND, IMMUNOL LETT, vol. 82, 2002, pages 57 - 65 |
| JEFFREY ET AL., BIOORGANIC & MED. CHEM. LETTERS, vol. 16, 2006, pages 358 - 362 |
| KANDA, Y. ET AL., BIOTECHNOL. BIOENG., vol. 94, no. 4, 2006, pages 680 - 688 |
| KING ET AL., J. MED. CHEM., vol. 45, 2002, pages 4336 - 4343 |
| KLIMKA ET AL., BR. J. CANCER, vol. 83, 2000, pages 252 - 260 |
| KOHLER ET AL., NATURE, vol. 256, no. 5517, 1975, pages 495 - 497 |
| KONO ET AL., HUM. MOL. GENET., vol. 14, 2005, pages 2881 - 2892 |
| KOSTELNY ET AL., J. IMMUNOL., vol. 148, no. 5, 1992, pages 1547 - 1553 |
| KOZBOR ET AL., J. IMMUNOL., vol. 133, no. 6, 1984, pages 3001 - 3005 |
| KRATZ ET AL., CURRENT MED. CHEM., vol. 13, 2006, pages 477 - 523 |
| KYOGOJU ET AL., ARTHRITIS RHEUM, vol. 46, 2002, pages 1242 - 1254 |
| LAZAR ET AL., PROC NATL ACAD SCI U S A, vol. 103, 2006, pages 4005 - 4010 |
| LAZAR ET AL., PROC. NATL. ACAD. SCI. USA, vol. 103, no. 11, 2006, pages 4005 - 4010 |
| LEACH ET AL., SCIENCE, vol. 271, 1996, pages 1734 - 1736 |
| LEE ET AL., J. IMMUNOL. METHODS, vol. 284, no. 1-2, 2004, pages 119 - 132 |
| LI ET AL., NAT. BIOTECH., vol. 24, 2006, pages 210 - 215 |
| LODE ET AL., CANCER RES., vol. 58, 1998, pages 2925 - 2928 |
| LONBERG, CURR. OPIN. IMMUNOL., vol. 20, 2008, pages 450 - 459 |
| LONBERG, NAT. BIOTECH., vol. 23, 2005, pages 1117 - 1125 |
| MABS, vol. 9, 2017, pages 844 - 853 |
| MATHER ET AL., ANNALS N.Y. ACAD. SCI., vol. 383, 1982, pages 44 - 68 |
| MATHER, BIOL. REPROD., vol. 23, 1980, pages 243 - 251 |
| MCCAFFERTY ET AL., NATURE, vol. 305, 1983, pages 537 - 554 |
| MERCHANT ET AL., NAT.BIOTECH., vol. 16, 1998, pages 677 - 681 |
| METHODS MOL. BIOL., vol. 178, 2002, pages 133 - 145 |
| MIMOTO ET AL., PROTEIN ENG DES SEL, vol. 26, 2013, pages 589 - 598 |
| MOL. CANCER THER., vol. 7, 2008, pages 2517 - 2527 |
| MOLECULAR IMMUNOLOGY, vol. 51, 2012, pages 66 - 72 |
| MORGAN ET AL., IMMUNOLOGY, vol. 86, 1995, pages 319 - 324 |
| MORRISON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 - 6855 |
| MOSMANN, J. IMMUNOL. METH., vol. 65, 1983, pages 55 - 63 |
| MUNSON, ANAL. BIOCHEM., vol. 107, no. 1, 1980, pages 220 - 239 |
| NAGY ET AL., PROC. NATL. ACAD. SCI. USA, vol. 97, 2000, pages 829 - 834 |
| NAT REV DRUG DISCOV, vol. 19, no. 5, 2020, pages 308 |
| NAT. BIOTECHNOL., vol. 16, 1998, pages 677 - 681 |
| NAT. IMMUNOL., vol. 18, 2017, pages 889 |
| NATURE MEDICINE, vol. 6, 2000, pages 443 - 436 |
| NATURE REVIEWS CANCER, vol. 18, 2018, pages 727 - 743 |
| NATURE, vol. 314, no. 6012, 1985, pages 628 - 31 |
| NI, XIANDAI MIANYIXUE, vol. 26, no. 4, 2006, pages 265 - 268 |
| NICOLAOU ET AL., ANGEW. CHEM, vol. 33, 1994, pages 183 - 186 |
| NIMMERJAHNRAVETCH, SCIENCE, vol. 310, 2005, pages 1510 - 1512 |
| NISHIKAWASAKAGUCHI, INT J CANCER, vol. 127, 2010, pages 759 - 767 |
| OKAZAKI ET AL., J. MOL. BIOL., vol. 336, no. 5, pages 1239 - 1249 |
| PADLAN, MOL. IMMUNOL., vol. 28, 1991, pages 489 - 498 |
| PARDOLL, NAT REV CANCER, vol. 12, 2012, pages 252 - 264 |
| PAVLOUBELSEY, EUR J PHARM BIOPHARM, vol. 59, 2005, pages 389 - 396 |
| PETKOVA ET AL., INTL. IMMUNOL., vol. 18, no. 12, 2006, pages 1759 - 1769 |
| PETKOVA, S.B. ET AL., INT'L. IMMUNOL., vol. 18, no. 12, 2006, pages 1759 - 1769 |
| PORTOLANO ET AL., J. IMMUNOL., vol. 151, 1993, pages 2623 - 887 |
| PRESTA ET AL., CANCER RES., vol. 57, 1997, pages 4593 - 4599 |
| PROC NATL ACAD SCI USA, vol. 83, no. 5, 1986, pages 1453 - 7 |
| PROC NATL ACAD SCI USA., vol. 109, no. 16, 17 April 2012 (2012-04-17), pages 6181 - 6186 |
| PROTEIN SCIENCE |
| PROTEIN SCIENCE, vol. 4, 1995, pages 2411 - 2423 |
| QUEEN ET AL., PROC. NATL ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| RADAEV ET AL., J BIOL CHEM, vol. 276, 2001, pages 16469 - 16477 |
| RAGHAVAN ET AL., IMMUNITY, vol. 1, 1994, pages 303 - 315 |
| RAVETCHKINET, ANNU. REV. IMMUNOL, vol. 9, 1991, pages 457 - 492 |
| RAVETCHKINET, ANNU. REV. IMMUNOL., vol. 9, 1991, pages 457 - 492 |
| REICHERT ET AL., NAT BIOTECHNOL, vol. 23, 2005, pages 1073 - 1078 |
| RICHARDS ET AL., MOL CANCER THER, vol. 7, 2008, pages 2517 - 2527 |
| RIDGWAY ET AL., PROT. ENG., vol. 9, 1996, pages 617 - 621 |
| RIECHMANN ET AL., NATURE, vol. 322, 1988, pages 738 - 740 |
| RIPKA ET AL., ARCH. BIOCHEM. BIOPHYS., vol. 249, 1986, pages 533 - 545 |
| ROSOK ET AL., J. BIOL. CHEM., vol. 271, 1996, pages 22611 - 22618 |
| SAKAGUCHI ET AL., J IMMUNOL, vol. 155, 1995, pages 1151 - 1164 |
| SCI IMMUNOL, vol. 4, no. 35, 2019, pages eaax0644 |
| SCI REP, vol. 7, 2017, pages 45839 |
| SCIENCE, vol. 359, 2018, pages 794 - 797 |
| SHIELDS ET AL., J. BIOL. CHEM., vol. 9, no. 2, 2001, pages 6591 - 6604 |
| SHINKAWA ET AL., J BIOL CHEM, vol. 278, 2003, pages 3466 - 3473 |
| SIDHU ET AL., J. MOL. BIOL., vol. 338, no. 2, 2004, pages 299 - 310 |
| TAKAHASHI ET AL., J EXP MED, vol. 192, 2000, pages 303 - 310 |
| TAMURA ET AL., ANN ONCOL, vol. 22, 2011, pages 1302 - 1307 |
| TORGOV ET AL., BIOCONJ. CHEM., vol. 16, 2005, pages 717 - 721 |
| TRAUNECKER ET AL., EMBO J., vol. 10, 1991, pages 3655 |
| TRENDS IMMUNOL., vol. 40, no. 3, 2019, pages 243 - 257 |
| TSAO ET AL., JCI INSIGHT, vol. 4, 2019, pages e131882 |
| URLAUB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4216 |
| VAN DIJKVAN DE WINKEL, CURR. OPIN. PHARMACOL., vol. 5, 2001, pages 368 - 374 |
| VITETTA ET AL., SCIENCE, vol. 238, 1987, pages 1098 - 63 |
| VOLLMERSBRANDLEIN, HISTOLOGY AND HISTOPATHOLOGY, vol. 20, no. 3, 2005, pages 927 - 937 |
| VOLLMERSBRANDLEIN, METHODS AND FINDINGS IN EXPERIMENTAL AND CLINICAL PHARMACOLOGY, vol. 27, no. 3, 2005, pages 185 - 191 |
| WARMERDAM, P. A. M. ET AL., J. EXP. MED., vol. 172, pages 19 - 25 |
| WINTER ET AL., ANN. REV. IMMUNOL., vol. 113, 1994, pages 433 - 455 |
| WRIGHT ET AL., TIBTECH, vol. 15, 1997, pages 26 - 32 |
| WU, J. ET AL., J. CLIN. INVEST., vol. 100, no. 5, 1997, pages 1059 - 1070 |
| YAMANE-OHNUKI ET AL., BIOTECH. BIOENG., vol. 87, 2004, pages 614 |
| YAZAKIWU: "Methods in Molecular Biology", vol. 248, 1996, HUMANA PRESS, article "Epitope Mapping Protocols", pages: 255 - 268 |
| ZALEVSKY ET AL., BLOOD, vol. 113, 2009, pages 3735 - 3743 |
| ZALEVSKY ET AL., NAT. BIOTECHNOL., vol. 28, no. 2, 2010, pages 157 - 159 |
| ZHANG ET AL., CANCER RES., vol. 65, 2005, pages 3877 - 3882 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11912989B2 (en) | 2013-12-04 | 2024-02-27 | Chugai Seiyaku Kabushiki Kaisha | Antigen-binding molecules, the antigen-binding activity of which varies according to the concentration of compounds, and libraries of said molecules |
Also Published As
| Publication number | Publication date |
|---|---|
| IL309115A (en) | 2024-02-01 |
| CO2024000518A2 (es) | 2024-04-18 |
| JP7472405B2 (ja) | 2024-04-22 |
| EP4361273A1 (en) | 2024-05-01 |
| TW202317627A (zh) | 2023-05-01 |
| JPWO2022270611A1 (ja) | 2022-12-29 |
| AU2022299846A1 (en) | 2023-12-07 |
| KR20240024213A (ko) | 2024-02-23 |
| JP2024095774A (ja) | 2024-07-10 |
| AU2022299846A9 (en) | 2023-12-14 |
| CA3221833A1 (en) | 2022-12-29 |
| CL2023003836A1 (es) | 2024-05-24 |
| CN117616123A (zh) | 2024-02-27 |
| AU2022299846B2 (en) | 2024-08-15 |
| CR20240026A (es) | 2024-03-14 |
| BR112023022992A2 (pt) | 2024-01-23 |
| MX2023013897A (es) | 2023-12-11 |
| KR102690141B1 (ko) | 2024-07-30 |
| AR126220A1 (es) | 2023-09-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10323094B2 (en) | Humanized and affinity matured antibodies to FcRH5 and methods of use | |
| US20240059774A1 (en) | Anti-ctla-4 antibody and use thereof | |
| KR102645629B1 (ko) | 항ctla-4 항체 및 그의 사용 | |
| TWI820484B (zh) | 抗hla-dq2.5抗體及其用於治療乳糜瀉的用途 | |
| JP2024095774A (ja) | 抗ctla-4抗体 | |
| JP2024099585A (ja) | 抗ctla-4抗体の使用 | |
| TW202216764A (zh) | 抗ctla-4抗體及其之用途 | |
| EA045931B1 (ru) | Анти-ctla-4 антитело и его применение |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22828527 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023530135 Country of ref document: JP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112023022992 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2301007553 Country of ref document: TH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022299846 Country of ref document: AU Ref document number: AU2022299846 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2023/013897 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 309115 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2022299846 Country of ref document: AU Date of ref document: 20220624 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3221833 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 003419-2023 Country of ref document: PE Ref document number: P6003307/2023 Country of ref document: AE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280044970.4 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 140250140003006663 Country of ref document: IR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202393536 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202447003016 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20247002157 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020247002157 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 112023022992 Country of ref document: BR Kind code of ref document: A2 Effective date: 20231103 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022828527 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022828527 Country of ref document: EP Effective date: 20240125 |