WO2022234855A1 - 中枢毒性が低減したオリゴヌクレオチドの設計方法 - Google Patents
中枢毒性が低減したオリゴヌクレオチドの設計方法 Download PDFInfo
- Publication number
- WO2022234855A1 WO2022234855A1 PCT/JP2022/019561 JP2022019561W WO2022234855A1 WO 2022234855 A1 WO2022234855 A1 WO 2022234855A1 JP 2022019561 W JP2022019561 W JP 2022019561W WO 2022234855 A1 WO2022234855 A1 WO 2022234855A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oligonucleotide
- present
- formula
- toxicity
- group
- Prior art date
Links
- 108091034117 Oligonucleotide Proteins 0.000 title claims abstract description 185
- 238000000034 method Methods 0.000 title claims abstract description 92
- 230000001988 toxicity Effects 0.000 title claims abstract description 87
- 231100000419 toxicity Toxicity 0.000 title claims abstract description 87
- 230000002829 reductive effect Effects 0.000 title claims abstract description 29
- 239000002777 nucleoside Substances 0.000 claims abstract description 61
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 43
- 150000003833 nucleoside derivatives Chemical class 0.000 claims abstract description 42
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 39
- 238000012986 modification Methods 0.000 claims abstract description 30
- 230000004048 modification Effects 0.000 claims abstract description 30
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims abstract description 27
- 235000000346 sugar Nutrition 0.000 claims abstract description 22
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 18
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 9
- 125000005843 halogen group Chemical group 0.000 claims abstract description 9
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims description 42
- 239000003153 chemical reaction reagent Substances 0.000 claims description 23
- 125000003835 nucleoside group Chemical group 0.000 claims description 20
- 230000014509 gene expression Effects 0.000 claims description 18
- 201000010099 disease Diseases 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 14
- 238000004519 manufacturing process Methods 0.000 claims description 12
- 229910052740 iodine Inorganic materials 0.000 claims description 5
- 230000002194 synthesizing effect Effects 0.000 claims description 3
- 229940126585 therapeutic drug Drugs 0.000 claims 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 76
- 239000000074 antisense oligonucleotide Substances 0.000 description 66
- 108020004999 messenger RNA Proteins 0.000 description 36
- 125000003729 nucleotide group Chemical group 0.000 description 27
- 230000000692 anti-sense effect Effects 0.000 description 22
- 239000003814 drug Substances 0.000 description 21
- 230000000694 effects Effects 0.000 description 20
- 210000004027 cell Anatomy 0.000 description 19
- 150000007523 nucleic acids Chemical group 0.000 description 19
- 108020004707 nucleic acids Proteins 0.000 description 18
- 102000039446 nucleic acids Human genes 0.000 description 18
- 238000013461 design Methods 0.000 description 16
- 239000002773 nucleotide Substances 0.000 description 16
- 108090000623 proteins and genes Proteins 0.000 description 16
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 13
- 229940124597 therapeutic agent Drugs 0.000 description 13
- 238000011156 evaluation Methods 0.000 description 12
- 238000001668 nucleic acid synthesis Methods 0.000 description 12
- 102000004169 proteins and genes Human genes 0.000 description 11
- 241000699670 Mus sp. Species 0.000 description 10
- 125000003277 amino group Chemical group 0.000 description 10
- 125000003545 alkoxy group Chemical group 0.000 description 8
- 125000000217 alkyl group Chemical group 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 8
- 125000005842 heteroatom Chemical group 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 6
- 230000003542 behavioural effect Effects 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000002502 liposome Substances 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 230000001629 suppression Effects 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 5
- 206010019851 Hepatotoxicity Diseases 0.000 description 5
- 210000003169 central nervous system Anatomy 0.000 description 5
- 230000002708 enhancing effect Effects 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 230000007686 hepatotoxicity Effects 0.000 description 5
- 231100000304 hepatotoxicity Toxicity 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 4
- 125000004414 alkyl thio group Chemical group 0.000 description 4
- 125000003710 aryl alkyl group Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 230000001276 controlling effect Effects 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 210000003757 neuroblast Anatomy 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 150000004713 phosphodiesters Chemical class 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 description 4
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- -1 2-oxo-1,2-dihydropyrimidin-1-yl group Chemical group 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101000891579 Homo sapiens Microtubule-associated protein tau Proteins 0.000 description 3
- 102100034343 Integrase Human genes 0.000 description 3
- 101710203526 Integrase Proteins 0.000 description 3
- 239000000232 Lipid Bilayer Substances 0.000 description 3
- 102100040243 Microtubule-associated protein tau Human genes 0.000 description 3
- 206010029350 Neurotoxicity Diseases 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 102000006602 glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 3
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 2
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 2
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 208000004130 Blepharoptosis Diseases 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 206010010904 Convulsion Diseases 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 206010015995 Eyelid ptosis Diseases 0.000 description 2
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 102000006382 Ribonucleases Human genes 0.000 description 2
- 108010083644 Ribonucleases Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 206010044221 Toxic encephalopathy Diseases 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- 230000007059 acute toxicity Effects 0.000 description 2
- 231100000403 acute toxicity Toxicity 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 125000001369 canonical nucleoside group Chemical group 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 230000036461 convulsion Effects 0.000 description 2
- 239000013256 coordination polymer Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 238000003633 gene expression assay Methods 0.000 description 2
- 238000009396 hybridization Methods 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 238000000185 intracerebroventricular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 231100000228 neurotoxicity Toxicity 0.000 description 2
- 230000007135 neurotoxicity Effects 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 150000008300 phosphoramidites Chemical class 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 201000003004 ptosis Diseases 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- YKBGVTZYEHREMT-KVQBGUIXSA-N 2'-deoxyguanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 YKBGVTZYEHREMT-KVQBGUIXSA-N 0.000 description 1
- MXHRCPNRJAMMIM-SHYZEUOFSA-N 2'-deoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-SHYZEUOFSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- GVZJRBAUSGYWJI-UHFFFAOYSA-N 2,5-bis(3-dodecylthiophen-2-yl)thiophene Chemical compound C1=CSC(C=2SC(=CC=2)C2=C(C=CS2)CCCCCCCCCCCC)=C1CCCCCCCCCCCC GVZJRBAUSGYWJI-UHFFFAOYSA-N 0.000 description 1
- CKTSBUTUHBMZGZ-ULQXZJNLSA-N 4-amino-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-tritiopyrimidin-2-one Chemical compound O=C1N=C(N)C([3H])=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-ULQXZJNLSA-N 0.000 description 1
- LUCHPKXVUGJYGU-XLPZGREQSA-N 5-methyl-2'-deoxycytidine Chemical compound O=C1N=C(N)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 LUCHPKXVUGJYGU-XLPZGREQSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 229910002012 Aerosil® Inorganic materials 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006102 Bradypnoea Diseases 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 1
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 1
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 1
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 108050001049 Extracellular proteins Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 101001066129 Homo sapiens Glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- VQAYFKKCNSOZKM-IOSLPCCCSA-N N(6)-methyladenosine Chemical compound C1=NC=2C(NC)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O VQAYFKKCNSOZKM-IOSLPCCCSA-N 0.000 description 1
- DYSDOYRQWBDGQQ-XLPZGREQSA-N N6-Methyl-2'-deoxyadenosine Chemical compound C1=NC=2C(NC)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 DYSDOYRQWBDGQQ-XLPZGREQSA-N 0.000 description 1
- DYSDOYRQWBDGQQ-UHFFFAOYSA-N N6-Methyldeoxyadenosine Natural products C1=NC=2C(NC)=NC=NC=2N1C1CC(O)C(CO)O1 DYSDOYRQWBDGQQ-UHFFFAOYSA-N 0.000 description 1
- VQAYFKKCNSOZKM-UHFFFAOYSA-N NSC 29409 Natural products C1=NC=2C(NC)=NC=NC=2N1C1OC(CO)C(O)C1O VQAYFKKCNSOZKM-UHFFFAOYSA-N 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- 238000002123 RNA extraction Methods 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 208000034799 Tauopathies Diseases 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000000848 adenin-9-yl group Chemical group [H]N([H])C1=C2N=C([H])N(*)C2=NC([H])=N1 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000003444 anaesthetic effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 108010045569 atelocollagen Proteins 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 208000024336 bradypnea Diseases 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- GMTYREVWZXJPLF-AFHUBHILSA-N butorphanol D-tartrate Chemical compound OC(=O)[C@@H](O)[C@H](O)C(O)=O.N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 GMTYREVWZXJPLF-AFHUBHILSA-N 0.000 description 1
- 229960001590 butorphanol tartrate Drugs 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 231100000153 central nervous system (CNS) toxicity Toxicity 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000005159 cyanoalkoxy group Chemical group 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- MXHRCPNRJAMMIM-UHFFFAOYSA-N desoxyuridine Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 230000003370 grooming effect Effects 0.000 description 1
- 125000003738 guanin-9-yl group Chemical group O=C1N([H])C(N([H])[H])=NC2=C1N=C([H])N2[*] 0.000 description 1
- 229940029575 guanosine Drugs 0.000 description 1
- 102000047486 human GAPDH Human genes 0.000 description 1
- 208000013403 hyperactivity Diseases 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000006742 locomotor activity Effects 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 229960004882 medetomidine hydrochloride Drugs 0.000 description 1
- VPNGEIHDPSLNMU-UHFFFAOYSA-N medetomidine hydrochloride Chemical compound Cl.C=1C=CC(C)=C(C)C=1C(C)C1=CNC=N1 VPNGEIHDPSLNMU-UHFFFAOYSA-N 0.000 description 1
- 229940041616 menthol Drugs 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 1
- 229960003793 midazolam Drugs 0.000 description 1
- 239000007758 minimum essential medium Substances 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000010355 oscillation Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000004545 purin-9-yl group Chemical group N1=CN=C2N(C=NC2=C1)* 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 230000014493 regulation of gene expression Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- DWRXFEITVBNRMK-JXOAFFINSA-N ribothymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 DWRXFEITVBNRMK-JXOAFFINSA-N 0.000 description 1
- 238000009781 safety test method Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 229940079827 sodium hydrogen sulfite Drugs 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 238000005987 sulfurization reaction Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 125000003294 thymin-1-yl group Chemical group [H]N1C(=O)N(*)C([H])=C(C1=O)C([H])([H])[H] 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000041 toxicology testing Toxicity 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7125—Nucleic acids or oligonucleotides having modified internucleoside linkage, i.e. other than 3'-5' phosphodiesters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/11—Antisense
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/315—Phosphorothioates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/323—Chemical structure of the sugar modified ring structure
- C12N2310/3231—Chemical structure of the sugar modified ring structure having an additional ring, e.g. LNA, ENA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/33—Chemical structure of the base
- C12N2310/334—Modified C
- C12N2310/3341—5-Methylcytosine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/50—Methods for regulating/modulating their activity
- C12N2320/53—Methods for regulating/modulating their activity reducing unwanted side-effects
Definitions
- the present invention relates to a method for designing oligonucleotides with reduced central toxicity, oligonucleotides synthesized based on the design results, therapeutic agents containing the oligonucleotides, and the like.
- Antisense oligonucleotides exert their effects by hybridizing to target nucleic acid sequences and suppressing gene expression itself.
- Various artificial nucleic acids have been developed and introduced for the purpose of improving the nuclease resistance of antisense oligonucleotides and improving the binding affinity and specificity for target nucleic acids, and various formulations have been marketed.
- a new problem that has recently emerged is how to avoid the potential toxicity of artificial nucleic acids.
- Toxicity of antisense oligonucleotides is classified into so-called “toxicity caused by hybridization with RNA (off-target toxicity)" and "toxicity caused by binding to intracellular and extracellular proteins and metal ions, etc., independent of hybridization with RNA. can be categorized as toxic (non-off-target toxicity). And various approaches have been taken to avoid these toxicities.
- Patent Document 1 discloses that non-off-target toxicity can be avoided by appropriately chemically modifying the base and sugar moieties of nucleic acids.
- Patent Document 2 non-Watson-Crick It is disclosed that the formation of type base pairs can be suppressed and avoided.
- phosphorothioate modification between nucleosides is the cause of hepatotoxicity (for example, Non-Patent Document 1). It is disclosed that hepatotoxicity can be reduced while maintaining the activity by substituting an artificial nucleic acid having a structure.
- toxicity evaluation related to antisense oligonucleotide toxicity reduction techniques so far has mainly been performed using hepatotoxicity as the main indicator, and no deep knowledge has been obtained about toxicity evaluation in other tissues.
- central toxicity is an important endpoint in drug safety testing, but little is known about central toxicity of antisense oligonucleotides.
- Non-Patent Document 2 The relationship between antisense oligonucleotides and central toxicity, especially the reduction of central toxicity, is found in Patent Document 4. Correlation between antisense oligonucleotide sequence and central toxicity (oscillation of intracellular free calcium concentration in nerve cells) is disclosed. In addition, as far as the present inventors know, there is only one document that evaluates central toxicity due to phosphorothioate modification in antisense oligonucleotides (Non-Patent Document 2).
- Non-Patent Document 2 discloses that central toxicity can be reduced by replacing the phosphorothioate bond of the wing portion with a phosphodiester bond in a gapmer-type oligonucleotide having a phosphorothioate modification.
- Non-Patent Document 2 the phosphorothioate modification is removed in an antisense oligonucleotide (ASO) in which a part of the phosphorothioate modification is removed and the 2'-position of the sugar moiety is modified to contribute to stability.
- ASO antisense oligonucleotide
- antisense activity can be reduced compared to ASOs without the antisense activity. The reduction in antisense activity is believed to be due to the reduction in ASO stability resulting from the removal of the phosphorothioate modification.
- an object of the present invention is to modify a site other than the 2'-position of the sugar moiety of ASO, preferably by further removing a part of the phosphorothioate modification of ASO, thereby reducing the central
- An object of the present invention is to provide a method of designing ASOs capable of reducing toxicity, preferably further maintaining or enhancing antisense activity, and ASOs synthesized based on the design results.
- Non-Patent Document 2 the modification at the 5'-position of the sugar moiety, not the modification at the 2'-position or the 4'-position of the sugar moiety, which is widely used in nucleic acid modification (for example, Non-Patent Document 2).
- Patent Document 3 by introducing a specific structure such as a cyclopropane structure at the 5'-position of the sugar moiety of a phosphorothioate-modified nucleoside, non-specific We got the idea that it might be possible to reduce central toxicity through suppression of binding to proteins.
- Patent Document 3 discloses that hepatotoxicity is reduced in ASOs in which a modification is introduced at the 5′ position of the sugar moiety of a nucleoside and a phosphorothioate bond near the modification is replaced with a phosphodiester bond.
- ASOs in which a modification is introduced at the 5′ position of the sugar moiety of a nucleoside and a phosphorothioate bond near the modification is replaced with a phosphodiester bond.
- the mechanism of occurrence of hepatotoxicity by ASO and central toxicity by ASO is considered to be largely different. Based on this idea, the present inventors have made further intensive studies and completed the present invention.
- a method of designing oligonucleotides with reduced central toxicity comprising: (1) The carbon atom at the 5′-position of the sugar portion of at least one nucleoside constituting an oligonucleotide having a phosphorothioate modification is represented by the following formula I
- R6 and R7 are each independently a hydrogen atom, a halogen atom, or a methyl group) or the following formula I'
- R 4 and R 5 are each independently a hydrogen atom, a methyl group, or an ethyl group (except when both R 4 and R 5 are hydrogen atoms)) and optionally (2) replacing at least one phosphorothioate bond of said oligonucleotide with a phosphodiester bond.
- the oligonucleotide is a gapmer-type oligonucleotide.
- at least one of the nucleosides substituted in step (1) is located in the gap region.
- a method for evaluating the degree of central toxicity reduction of oligonucleotides having phosphorothioate modifications comprising the following steps (1)-(3): (1) providing an oligonucleotide having a phosphorothioate modification; (2) a step of preparing an oligonucleotide designed by the method according to any one of [1] to [5]; and (3) central toxicity of the oligonucleotide prepared in step (2); ) to compare the central toxicity of the oligonucleotides prepared in ).
- An oligonucleotide having a phosphorothioate linkage with reduced central toxicity, wherein the 5′-position carbon atom of the sugar moiety of at least one nucleoside constituting the oligonucleotide is represented by the following formula I
- R6 and R7 are each independently a hydrogen atom, a halogen atom, or a methyl group) or the following formula I'
- R 4 and R 5 are each independently a hydrogen atom, a methyl group, or an ethyl group (except when both R 4 and R 5 are hydrogen atoms)) optionally at least one internucleoside bond is a phosphodiester bond.
- the oligonucleotide of [8] wherein at least one of the nucleosides having the structure represented by formula I or I' above is located in the gap region.
- a reagent for controlling gene expression comprising the oligonucleotide of any one of [7] to [11].
- a therapeutic agent for diseases comprising the oligonucleotide according to any one of [7] to [11].
- a method for controlling gene expression comprising administering the oligonucleotide of any one of [7] to [11].
- a method for treating a disease in a mammal which comprises administering to the mammal an effective amount of the oligonucleotide according to any one of [7] to [11].
- FIG. 1 shows the measurement results of behavioral scores when antisense oligonucleotide (LX-A4285) was intracerebroventricularly administered to mice.
- Figure 2 shows the evaluation results of the antisense effect when antisense oligonucleotides (LX-A0070, LX-A4285, LX-A5106, LX-A5108 and LX-A5113) were administered to mice to human neuroblasts.
- FIG. 3 shows the measurement results of behavioral scores when antisense oligonucleotides (LX-A4285, LX-A5108 and LX-A5113) were intracerebroventricularly administered to mice.
- oligonucleotides with reduced central toxicity can be designed by using a nucleoside having a 5'-cyclopropane structure as a modified nucleotide instead of a phosphorothioate-modified nucleotide. It is an invention completed based on
- a method for designing oligonucleotides with reduced central toxicity comprising: (1) The 5′-position carbon atom of the sugar portion of at least one nucleoside constituting an oligonucleotide having a phosphorothioate modification (hereinafter sometimes referred to as “PS modified nucleotide”) is represented by the following formula I
- R6 and R7 are each independently a hydrogen atom, a halogen atom, or a methyl group) or the following formula I'
- oligonucleotides shall also include pharmacologically acceptable salts of oligonucleotides.
- the design method of the present invention may include (2) a step of substituting at least one phosphorothioate bond of the oligonucleotide with a phosphodiester bond.
- a method for reducing central toxicity of oligonucleotides comprising: (1′) The 5′-position carbon atom of the sugar portion of at least one nucleoside constituting the PS-modified nucleotide is represented by the following formula I
- R6 and R7 are each independently a hydrogen atom, a halogen atom, or a methyl group) or the following formula I'
- R 4 and R 5 are each independently a hydrogen atom, a methyl group, or an ethyl group (except when both R 4 and R 5 are hydrogen atoms)
- a method (hereinafter sometimes referred to as "the toxicity reduction method of the present invention") is provided, comprising the step of substituting a structure represented by: In one aspect, both R6 and R7 are hydrogen atoms.
- the toxicity reduction method of the present invention includes: (2') A step of replacing at least one phosphorothioate bond of the oligonucleotide with a phosphodiester bond may be included.
- the "oligonucleotide having a phosphorothioate modification” means that the bond between at least one nucleoside of the oligonucleotide is a phosphorothioate bond (i.e., the oxygen atom of the phosphate group between the nucleosides is substituted with a sulfur atom. ) means an oligonucleotide.
- Phosphorothioate modification is generally used to enhance the in vivo stability of natural oligonucleotides such as DNA and RNA, especially enzyme resistance.
- these phosphorothioate-modified oligonucleotides have a common problem of toxicity. The method of the present invention was completed as a result of research aimed at reducing central toxicity among the toxicity of phosphorothioate-modified oligonucleotides.
- Central toxicity in the present invention is also referred to as "central nervous system toxicity", and toxicity findings derived from the central nervous system identified by general condition changes and brain histopathological changes in rodents, non-human primates and humans point to For example, in rodents, general condition changes including reversible minor symptoms of irritability, hypolocomotion, bradypnea and blepharoptosis to severe symptoms such as convulsions and death.
- central toxicity in the present invention can be evaluated by behavioral scores obtained by a modified method of Irwin (Irwin S., Psychopharmacologia. 1968; 13(3): 222-257).
- reduced central toxicity refers to the central toxicity of the PS-modified nucleotide (hereinafter sometimes referred to as "target oligonucleotide”) that is the target (design source) of the method of the present invention.
- target oligonucleotide the oligonucleotide after performing step (1) or (1′) of the method of the present invention (i.e., the designed oligonucleotide or the oligonucleotide with reduced toxicity)
- the present invention hereinafter, “after performing the present invention (sometimes referred to as "oligonucleotide”
- oligonucleotide has low central toxicity, or is expected to have low central toxicity.
- the median behavioral score in the group administered with the subject oligonucleotide and the oligonucleotide after administration of the present invention were calculated using the modified Irwin method. If the difference from the median behavior score in the group tested is 1 or more (e.g., 1, 5, 10, 20 or more), it can be evaluated that central toxicity has been reduced, and such a value indicates central The degree of toxicity can also be assessed.
- step (1) of the design method of the present invention it is not necessary to actually synthesize oligonucleotides, and it is sufficient to imagine them in your head.
- the imaged oligonucleotides are embodied in a program that operates on a computer (eg, oligonucleotide design software, graphic design tools, office software, etc.) or on paper. Therefore, as described in Example 1 below, for example, the act of designing oligonucleotides that are expected to reduce central toxicity and compiling the oligonucleotides in a tabular format is also a practice of the designing method of the present invention. correspond to
- the target oligonucleotide contains a sequence complementary to the sequence of the oligonucleotide (in this specification, mRNA shall also include pre-mRNA.) function (post-transcriptional modification, translation, etc.) (hereinafter sometimes referred to as “inhibitory ASO”), or enhance the function (hereinafter sometimes referred to as "enhancing ASO”) good.
- Inhibitory ASOs typically form a double-stranded region with a target mRNA, and the double-stranded region is cleaved by ribonuclease H (RNase H), thereby suppressing the function of the target mRNA. .
- enhanced ASO typically forms a double-stranded region with a pre-mRNA splicing-promoting sequence, masks the sequence, and skips splicing, thereby enhancing (recovering) the function of mRNA. (which results in increased cellular abundance of functional proteins).
- the function of the mRNA can be enhanced by forming a double-stranded region with the binding sequence of the enzyme that degrades the mRNA contained in the target mRNA, thereby masking the sequence and suppressing the degradation of the mRNA. can.
- the inhibitory ASO is preferably a gapmer-type oligonucleotide.
- the term "gapmer-type oligonucleotide” means a plurality of (eg, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 or more) nucleotides recognized by RNase H.
- ap region is at least one modified to confer resistance to ribonucleases (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) nucleotides (herein, the 3′ external region is referred to as the “3′ wing region” and the 5′ external region as the “5′ (sometimes referred to as "wing regions”).
- ribonucleases e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more
- the 3′ external region is referred to as the “3′ wing region” and the 5′ external region as the “5′ (sometimes referred to as "wing regions”).
- At least one nucleoside constituting each of the 3' wing region and the 5' wing region is preferably a bridging nucleoside.
- the enhanced ASO preferably has at least one nucleoside modified to impart resistance to RNase.
- Such ASOs may be those in which all nucleoside residues are modified, or those in which some nucleoside residues are modified (ie, Mixmer-type oligonucleotides).
- a bridged nucleoside is preferred as such a modified nucleoside.
- nucleosides that make up the target oligonucleotides include nucleosides represented by the following formula II.
- Base represents a purin-9-yl group or a 2-oxo-1,2-dihydropyrimidin-1-yl group optionally having one or more substituents selected from the ⁇ group, wherein
- the ⁇ group is a hydroxyl group, a hydroxyl group protected by a protecting group for nucleic acid synthesis, a linear alkyl group having 1 to 6 carbon atoms, a linear alkoxy group having 1 to 6 carbon atoms, a mercapto group, or a protecting group for nucleic acid synthesis.
- a mercapto group a linear alkylthio group having 1 to 6 carbon atoms, an amino group, a linear alkylamino group having 1 to 6 carbon atoms, an amino group protected by a protecting group for nucleic acid synthesis, and a halogen atom
- R 8 is a hydrogen atom
- R 9 is a hydrogen atom, a halogen atom, or a linear alkoxy group having 1 to 6 carbon atoms which may be substituted with a linear alkoxy group having 1 to 6 carbon atoms.
- R 8 and R 9 together have the following formula:
- R 21 is selected from a hydrogen atom, an optionally branched or ring-forming alkyl group having 1 to 6 carbon atoms, an optionally branched or ring-forming alkenyl group having 2 to 6 carbon atoms, and the above ⁇ group an aryl group having 3 to 10 carbon atoms which may have one or more optional substituents and may contain a heteroatom, and one or more optional substituents selected from the ⁇ group an aralkyl group having an aryl moiety of 3 to 12 carbon atoms which may optionally contain a heteroatom, or an amino group-protecting group for nucleic acid synthesis;
- R 22 and R 23 are each independently a hydrogen atom; optionally substituted with an aryl group having 3 to 12 carbon atoms which may contain a heteroatom, and optionally branched or forming a ring; an alkyl group having 1 to 6 carbon atoms; or an aralkyl group having an aryl moiety having 3 to 12 carbon atoms
- examples of the crosslinked nucleoside include nucleosides having the following structural formula.
- R is a hydrogen atom, an optionally branched or ring-forming alkyl group having 1 to 7 carbon atoms, an optionally branched or ring-forming alkenyl group having 2 to 7 carbon atoms, a hetero atom an aryl group having 3 to 12 carbon atoms which may contain , an aralkyl group having an aryl moiety having 3 to 12 carbon atoms which may contain a hetero atom, or an amino group-protecting group for nucleic acid synthesis.
- R is a hydrogen atom, a methyl group, an ethyl group, an n-propyl group, an isopropyl group, a phenyl group, or a benzyl group, more preferably R is a hydrogen atom or a methyl group.
- the definition of Base is: Same definition as in Formula II above.)
- the nucleosides constituting the subject oligonucleotides may be natural nucleosides, and examples of such natural nucleosides include adenosine, N6-methyladenosine, guanosine, uridine, 5-methyluridine, cytidine, deoxyadenosine, N6- methyl-2'-deoxyadenosine, deoxyguanosine, thymidine, deoxyuridine, deoxycytidine, 5-methyl-2'-deoxycytidine and the like.
- the length of the target oligonucleotide is not particularly limited as long as it has antisense activity, but is typically 10 to 50 nucleotides long, preferably 10 to 30 nucleotides long, more preferably 13 to 30 nucleotides long. Yes, more preferably 15-20 nucleotides in length.
- At least one nucleoside constituting an oligonucleotide after implementing the present invention can be represented by the following formula III or III'.
- R 6 , R 7 , R 8 , R 9 and Base are the same as in Formula I and Formula II above.
- both R 6 and R 7 are hydrogen atoms.
- the nucleoside having the structure represented by formula I or formula I' may be present at any position of the target PS-modified nucleotide. It is preferred, however, that at least one (eg 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) nucleosides are located in the gap region.
- the phosphorothioate linkage to be replaced as a result of step (2) or (2') of the method of the invention may be present between any of the nucleosides of the PS modified nucleotide of interest, but at least one (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) internucleoside phosphorothioate linkages are preferably located in the gap region.
- the phosphorothioate linkage to be substituted as a result of step (2) or (2′) of the method of the invention is 5′ of the nucleoside substituted in step (1) or (1′) of the method of the invention. It may be a laterally or 3′ adjacent bond, or it may be a non-adjacent bond. That is, the target nucleotide residues in step (1) or (1') and step (2) or (2') of the present invention can be arbitrarily selected.
- modifications may be made in addition to substitution of the 5'-position carbon atom of the sugar portion of the nucleoside and substitution of the phosphorothioate bond.
- modifications include, for example, 2′-O-methoxyethyl modification of the sugar moiety, 2′-O-methyl modification of the sugar moiety, 2′-fluoro modification of the sugar moiety, and modification of the 2′-position and 4′-position of the sugar moiety.
- examples include, but are not limited to, bridging (substitution to a bridging nucleoside), substitution of a bridging nucleoside to another bridging nucleotide, methylation or acetylation of Base (base portion), substitution of Base, and the like.
- the base pair may be not only a Watson-Crick base pair but also a Hoogsteen base pair or a Wobble base pair.
- Examples of the crosslinked nucleosides include nucleosides having the following structural formulas.
- R is a hydrogen atom, an optionally branched or ring-forming alkyl group having 1 to 7 carbon atoms, an optionally branched or ring-forming alkenyl group having 2 to 7 carbon atoms, a hetero atom an aryl group having 3 to 12 carbon atoms which may contain , an aralkyl group having an aryl moiety having 3 to 12 carbon atoms which may contain a hetero atom, or an amino group-protecting group for nucleic acid synthesis.
- R is a hydrogen atom, a methyl group, an ethyl group, an n-propyl group, an isopropyl group, a phenyl group, or a benzyl group, more preferably R is a hydrogen atom or a methyl group.
- the definition of Base is: Same definition as in Formula II above.)
- oligonucleotides with reduced central toxicity compared to PS-modified nucleotides are designed, or The central toxicity of PS modified nucleotides is reduced.
- the reduction in central neurotoxicity of oligonucleotides following the practice of the present invention may actually be confirmed, and the degree of reduction in central neurotoxicity of oligonucleotides following the practice of the present invention may be assessed.
- a method of assessing the degree of central toxicity reduction of a PS-modified nucleoside comprising the following steps (I)-(III): (I) preparing a PS-modified nucleotide; (II) a step of preparing an oligonucleotide designed by the design method of the present invention, and (III) comparing the central toxicity of the oligonucleotide prepared in step (II) and the central toxicity of the oligonucleotide prepared in step (I) A method (hereinafter sometimes referred to as the "evaluation method of the present invention") is provided.
- the PS-modified nucleotide prepared in step (I) of the evaluation method of the present invention can be obtained by purchasing a commercially available product, producing it by a known synthesis method, or having it produced by a third party outsourced synthesis. can.
- the oligonucleotides prepared in step (II) of the evaluation method of the present invention are prepared by a known synthesis method described below based on the structure of the oligonucleotide designed by the design method of the present invention, or the synthesis is commissioned. It can be obtained by having someone else produce it.
- step (II) of the evaluation method of the present invention can be performed using the method of calculating the score by the modified Irwin method described above or other known methods.
- Step (III) of the evaluation method of the present invention can be carried out by comparing the scores and the like.
- Oligonucleotides designed by the method of the present invention not only have reduced central toxicity, but can also maintain antisense activity.
- "maintenance of antisense activity” means that the antisense activity of the oligonucleotide after carrying out the present invention is maintained or increased compared to the antisense activity of the target oligonucleotide.
- SH-SY5Y cells are contacted with the same amount of each antisense oligonucleotide, and after 24 hours or more, It can be performed by recovering mRNA and comparing the amount of said mRNA.
- the oligonucleotide after carrying out the present invention if there is no statistically significant difference in the amount of target mRNA compared to the group contacted with the target oligonucleotide, or if it is reduced, anti- It can be evaluated that the sense activity is maintained.
- enhanced ASO the same amount of each antisense oligonucleotide is brought into contact with SH-SY5Y cells, and after 24 hours or more, proteins encoded by target mRNAs are collected from the cells, and functional proteins among the proteins are collected. can be done by comparing the amounts of
- the method of the present invention may further comprise a step of evaluating the antisense activity of the oligonucleotide after carrying out the present invention.
- a step can be performed using the method for measuring the amount of the target mRNA or the protein encoded by the mRNA, or other known methods.
- a step of designing an oligonucleotide by the design method of the present invention and (2) a method for producing a low central toxicity oligonucleotide (hereinafter referred to as "the present invention may be referred to as the "method of manufacturing”.) is provided.
- low toxicity oligonucleotide means an oligonucleotide that has low central toxicity or is expected to have low central toxicity compared to the target oligonucleotide.
- the median behavioral score in the group administered with the subject oligonucleotide and the oligonucleotide after administration of the present invention were calculated using the modified Irwin method.
- a low central toxicity oligonucleotide can be evaluated if the difference from the median behavior score in the group tested is 1 or more (e.g., 1, 5, 10, 20 or more), and such a value can also be used to assess the degree of central toxicity.
- the method for synthesizing oligonucleotides in step (2) of the production method of the present invention is not particularly limited, and known methods can be adopted. Examples of such known methods include chemical synthesis methods such as the phosphoramidite method and the H-phosphonate method. In the chemical synthesis method, a commercially available automatic nucleic acid synthesizer can be used. In addition, the chemical synthesis method generally uses amidite.
- the amidite is not particularly limited, and may be produced by a known method (eg, the method described in Patent Document 3 and International Publication No. 2020/166551), or a commercially available amidite may be used.
- the central toxicity may be actually evaluated by performing the same step as step (III) of the evaluation method of the present invention for the oligonucleotide produced by the production method of the present invention.
- an oligonucleotide having phosphorothioate linkages with reduced central toxicity comprising: The carbon atom at the 5′-position of the sugar portion of at least one nucleoside constituting the oligonucleotide has the following formula I
- an oligonucleotide having a phosphorothioate modification (hereinafter sometimes referred to as "the ASO of the present invention") is provided, which is characterized by forming a structure represented by:
- both R6 and R7 are hydrogen atoms.
- at least one internucleoside bond is preferably a phosphodiester bond from the viewpoint of further reducing central toxicity.
- At least one nucleoside that constitutes the ASO of the present invention can be represented by the following formula III or III'.
- R 6 , R 7 , R 8 , R 9 and Base are the same as the definitions in Formula I and Formula II of 1.
- both R 6 and R 7 are hydrogen atoms; be.
- the ASO of the present invention may be an inhibitory ASO or an enhancing ASO.
- an inhibitory ASO it is preferably a gapmer oligonucleotide, but is not limited to this.
- an enhanced ASO it is preferably a mixmer-type oligonucleotide, but is not limited to this.
- a nucleoside having a structure represented by Formula I or Formula I′ above may be present at any position in the ASO of the present invention, but at least one (eg, 1, 2, 3, 4, 5, 6 , 7, 8, 9, 10 or more) are preferably located in the gap region.
- the phosphodiester linkages may be present between any of the nucleosides of the ASOs of the invention, but at least one (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) internucleoside phosphodiester linkages are preferably located in the gap region.
- the phosphodiester bond is adjacent to the 5' side or 3' side of the nucleoside having the structure represented by Formula I or Formula I' above. They may be in the same position or may not be adjacent to each other.
- At least one nucleoside constituting each of the 3' wing region and the 5' wing region is preferably a bridging nucleoside.
- Examples of such crosslinked nucleosides include the above 1. The same as the bridged nucleoside described in .
- the ASO of the present invention can suppress or enhance the function of target mRNA, thereby controlling gene expression. Therefore, the ASO of the present invention can be used as a reagent for regulating gene expression (hereinafter sometimes referred to as "reagent of the present invention").
- reagent of the present invention includes both the terms “reagent for suppressing gene expression” and “reagent for enhancing gene expression”.
- the reagent of the present invention contains two or more ASOs or other reagents
- the reagent can be provided as a reagent kit containing each ASO and reagents in separate reagents.
- the term "gene expression” includes at least "production of a functional protein encoded by target mRNA", but also includes “production of target mRNA". used in the sense Therefore, the suppression of gene expression means not only the reduction in the amount of functional protein encoded by the gene in cells, but also the reduction of mRNA transcribed from the gene by administration of the ASO of the present invention. It may also include decreasing its abundance in a cell.
- the enhancement of gene expression means not only that the administration of the ASO of the present invention increases the abundance in cells of the functional protein encoded by the gene, but also that the mRNA transcribed from the gene may also include increasing the abundance of in cells.
- the ASO can be administered to a subject alone or together with a pharmacologically acceptable carrier.
- the introduction target include cells, tissues, organs, etc. of mammals including humans. Accordingly, a method for regulating gene expression in a subject, which comprises administering the ASO of the present invention to the subject, is also provided.
- the reagent of the present invention may further contain a reagent for nucleic acid introduction.
- the reagent for nucleic acid introduction includes calcium chloride, calcium enrichment reagent, atelocollagen; liposome; nanoparticle; Cationic lipids such as ethyleneimine (PEI) and the like can be used.
- the ASO of the present invention is particularly suitable for use as a pharmaceutical because it has reduced central toxicity. Therefore, therapeutic agents for diseases (hereinafter sometimes referred to as “therapeutic agents of the present invention") containing the ASO of the present invention are provided.
- treatment includes alleviation or improvement of symptoms, prevention, delay or cessation of progression of disease or symptoms, or manifestation of symptoms.
- diseases include diseases caused by overexpression or overaccumulation of the protein encoded by the ASO target mRNA of the present invention, or by deletion or reduction of the protein encoded by the ASO target mRNA of the present invention. mentioned.
- diseases include neurological diseases (eg, neurodegenerative diseases such as tauopathy).
- an effective amount of the ASO of the present invention may be used alone, or may be formulated as a pharmaceutical composition together with any carrier, for example, a pharmaceutically acceptable carrier (formulated are also referred to as therapeutic agents).
- a pharmaceutically acceptable carrier formulated are also referred to as therapeutic agents.
- Pharmaceutically acceptable carriers include, for example, excipients such as sucrose and starch, binders such as cellulose and methylcellulose, disintegrants such as starch and carboxymethylcellulose, lubricants such as magnesium stearate and aerosil, citric acid, Fragrance agents such as menthol, preservatives such as sodium benzoate and sodium hydrogen sulfite, stabilizers such as citric acid and sodium citrate, suspending agents such as methyl cellulose and polyvinyl pyrrolid, dispersing agents such as surfactants, water, Examples include, but are not limited to, diluents such as physiological saline, base waxes, and the like.
- the therapeutic agent of the present invention may further contain a reagent for nucleic acid introduction.
- a reagent for nucleic acid introduction the same reagents as described above can be used.
- the therapeutic agent of the present invention may also be a pharmaceutical composition in which the ASO of the present invention is encapsulated in liposomes.
- Liposomes are closed microscopic vesicles having an internal phase surrounded by one or more lipid bilayers, and can typically hold water-soluble substances in the internal phase and fat-soluble substances within the lipid bilayers.
- the ASOs of the present invention may be retained within the liposome internal phase or within the lipid bilayer.
- the liposomes used in the present invention may be monolayered or multilayered, and the particle size can be appropriately selected, for example, in the range of 10 to 1000 nm, preferably 50 to 300 nm. Considering deliverability to the target tissue, the particle size is, for example, 200 nm or less, preferably 100 nm or less.
- Methods for encapsulating water-soluble compounds such as oligonucleotides in liposomes include lipid film method (vortex method), reverse phase evaporation method, surfactant removal method, freeze-thaw method, remote loading method and the like. is not limited to, and any known method can be selected as appropriate.
- the medicament of the present invention can be administered orally or parenterally to mammals (e.g. humans, rats, mice, guinea pigs, rabbits, sheep, horses, pigs, cows, dogs, cats, monkeys). is possible, but parenteral administration is preferred. Also provided, therefore, is a method of treating a disease in a mammal comprising administering to the mammal an effective amount of an ASO of the present invention.
- mammals e.g. humans, rats, mice, guinea pigs, rabbits, sheep, horses, pigs, cows, dogs, cats, monkeys.
- Formulations suitable for parenteral administration include aqueous and non-aqueous isotonic sterile injectable solutions, which may contain antioxidants, buffers, bacteriostatic agents, tonicity agents, and the like. Also included are aqueous and non-aqueous sterile suspensions, which may contain suspending agents, solubilizers, thickeners, stabilizers, preservatives, and the like.
- the formulation can be enclosed in a container such as an ampoule or a vial in units of unit doses or multiple doses.
- the active ingredient and a pharmaceutically acceptable carrier can be lyophilized and stored in such a manner that they can be dissolved or suspended in an appropriate sterile vehicle just prior to use.
- Other formulations suitable for parenteral administration can include sprays and the like.
- the content of the ASO of the present invention in the pharmaceutical composition is, for example, about 0.1 to 100% by weight of the entire pharmaceutical composition.
- the dosage of the therapeutic agent of the present invention varies depending on the purpose of administration, administration method, type of target disease, severity, and conditions of administration target (sex, age, body weight, etc.).
- a single dose of the ASO of the present invention is generally 0.01 mg/kg or more and 1000 mg/kg or less. It is desirable to administer such doses 1 to 10 times, more preferably 5 to 10 times.
- the therapeutic agent of the present invention can also be used, for example, in combination with therapeutic agents for diseases already on the market.
- These concomitant drugs can be formulated together with the medicament of the present invention and administered as a single formulation, or can be formulated separately from the medicament of the present invention and administered via the same or different route as the medicament of the present invention. , can be administered simultaneously or staggered.
- the dose of these concomitant drugs may be the amount usually used when the drug is administered alone, or the dose can be reduced from the amount usually used.
- LX-A4285 has a phosphorothioate bond as an internucleoside bond and is a sequence known to exhibit central toxicity according to WO 2016/127000 A1.
- 5 is 5-methylcytosine
- Ln is LNA
- ⁇ is a phosphorothioate bond
- Cp is DNA introduced with a cyclopropane structure at the 5' position of the sugar moiety (herein referred to as "5'-cyclopropane DNA ) are shown respectively.
- Oligonucleotides related to the present invention were synthesized by the method described in Tetrahedron Letters 22, 1859-1862 (1981), International Publication No. 2011/052436, etc.
- LNA Locked Nucleic Acid
- 5'-cyclopropane DNA 5'-CP-DNA
- Base is 5-methylcytosin-1-yl group, thymin-1-yl group, adenin-9-yl group or guanin-9-yl group.
- Oligonucleotides containing 5'-cyclopropane DNA were synthesized with reference to the method described in WO2020/158910.
- Oligonucleotides containing Locked Nucleic Acid (LNA) or 5'-cyclopropane DNA (5'-CP-DNA) were synthesized using an automatic nucleic acid synthesizer (nS-8 type, manufactured by Gene Design, Inc.). Chain length extension was performed using standard phosphoramidite protocols (solid support: CPG, iodine for oxidation to form phosphorodiester (PO) backbone, DDTT for sulfurization to form phosphorothioate (PS) backbone (( (dimethylamino-methylidene)amino)-3H-1,2,4-dithiazaoline-3-thione), etc.).
- LNA Locked Nucleic Acid
- 5'-CP-DNA 5'-cyclopropane DNA
- LNA Locked Nucleic Acid
- 5'-CP 5'-cyclopropane DNA
- Example 2 Construction of Method for Evaluating Toxicity of Antisense Nucleotide in Mouse Central Nervous System BALB/cCrSlc (male, 7 weeks old, Japan SLC Co., Ltd.) was used to evaluate central toxicity of antisense oligonucleotides in mice.
- the animals were kept in open racks under an environment of temperature: 18-28°C, humidity: 30-80%, lighting time: 12 hours/day, and all animals were given feed and water ad libitum.
- Antisense oligonucleotides were administered to mice intracerebroventricularly to evaluate central toxicity.
- Mice were placed in a stereotaxic apparatus under deep anesthesia by intraperitoneal administration of a triple anesthetic (butorphanol tartrate 5 mg/kg, midazolam 4 mg/kg, medetomidine hydrochloride 0.3 mg/kg) at a volume of 10 mL/kg. Fixed. The skull was exposed, and a hole was drilled at the administration site (brain coordinates: -0.2 mm caudally from bregma, -1.0 mm to the right, and -2.5 mm deep) using a dental drill. A single dose was administered intracerebroventricularly using a syringe and polyethylene tube. LX-A4285 shown in Table 1 was used as the antisense oligonucleotide.
- mice 1 to 24 hours after a single intracerebroventricular administration of LX-A4285, behavioral analysis of mice was performed by Irwin's modified method.
- evaluation items items such as locomotor activity, standing behavior, hyperactivity, walking, grooming, blepharoptosis, vocalization, tremor, and convulsion were scored.
- Example 3 In Vitro Suppression of mRNA Expression by Antisense Nucleotides in Human Neuroblasts Using the antisense oligonucleotides used in Example 1 above, MAPT mRNA expression suppression effects in human neuroblasts were evaluated. A control without addition of oligonucleotide was used. Also, for comparison, a negative control oligonucleotide (also referred to as Negative Control; NC): 5(Y) ⁇ A(Y) ⁇ T(Y) ⁇ t ⁇ t ⁇ a ⁇ g ⁇ a ⁇ a ⁇ g ⁇ t ⁇ c ⁇ 5(Y) ⁇ T(Y) ⁇ c (LX-A0070; SEQ ID NO: 6) was used. In the above sequences, 5 represents 5-methylcytosine and Y represents AmNA.
- NC Negative Control
- SH-SY5Y cells (ECACC, EC94030304-F0) were used as human neuroblasts.
- Each antisense oligonucleotide (LX-A0070, LX-A4285, LX-A5106, LX-A5108 and LX-A5113) was transfected into SH-SY5Y cells using a commercially available transfection reagent (ThermoFisher Scientific, Lipofectamine 3000). After incorporation, the mRNA expression level was measured by qRT-PCR to examine knockdown activity (mRNA expression suppression). The procedure is shown below.
- RNA extraction reagent (MACHEREY-NAGEL, Nucleo ZOL).
- a reverse transcription reaction and a PCR amplification reaction were performed using a reagent for nucleic acid amplification reaction (QIAGEN, QuantiFast Probe RT-PCR kit).
- the nucleic acid amplification reaction was carried out by temperature cycling of 50°C 10 minutes ⁇ 95°C 5 minutes ⁇ [(95°C 10 seconds ⁇ 60°C 30 seconds) x 40 cycles].
- the amount of mRNA by each antisense oligonucleotide is shown as a relative value when the amount of mRNA in the oligonucleotide-free cells is set to 1.
- the primer sets used are as follows: (Primer set for MAPT detection) TaqMan Gene Expression Assay Hs00902194_m1_4331182 (ThermoFisher Scientific) (Primer set for GAPDH detection) TaqMan Gene Expression Assay Hs02786624_g1_4331182 (ThermoFisher Scientific)
- Example 4 Acute Toxicity of Antisense Nucleotides in Mouse Central Nervous System From among the antisense nucleotides listed in Tables 1 and 2, representative ones with low cytotoxicity while maintaining knockdown activity (data not shown) were selected. Two (LX-A5108 and LX-A5113) were selected and evaluated for acute toxicity in the central nervous system of mice. The evaluation method followed the method described in Example 2. The results are shown in FIG. Figure 3 confirms that sequences with a 5'-CP and an increased proportion of phosphodiester linkages have reduced central toxicity compared to antisense nucleotides with only phosphorothioate linkages.
- antisense nucleotides having 5'-modified nucleosides retain mRNA expression-suppressing activity while reducing central toxicity.
- oligonucleotides with reduced central toxicity can be designed. Oligonucleotides synthesized based on the results of the design can be used as therapeutic agents for diseases, because administration to cells or individuals enables regulation of gene expression while suppressing side effects.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biotechnology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Epidemiology (AREA)
- Plant Pathology (AREA)
- Microbiology (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Saccharide Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
しかしながら、これまでのアンチセンスオリゴヌクレオチドの毒性低減技術に係る毒性評価は肝毒性を主な指標になされ、それ以外の組織における毒性評価について深い知見は得られていない。例えば、中枢毒性は医薬品の安全性試験において重要な評価項目であるが、アンチセンスオリゴヌクレオチドの中枢毒性に関してはほとんど知見がない。
[1]
中枢毒性が低減したオリゴヌクレオチドを設計する方法であって、
(1)ホスホロチオエート修飾を有するオリゴヌクレオチドを構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子を、下記式I

R6およびR7は、それぞれ独立して、水素原子、ハロゲン原子、またはメチル基である)
または下記式I’

R4およびR5は、それぞれ独立して、水素原子、メチル基またはエチル基である(ただし、R4およびR5が共に水素原子である場合を除く))
で表される構造に置換する工程、および任意により
(2)該オリゴヌクレオチドが有する少なくとも一つのホスホロチオエート結合をホスホジエステル結合に置換する工程
を含むことを特徴とする、方法。
[2]
オリゴヌクレオチドがギャップマー型オリゴヌクレオチドである、[1]に記載の方法。
[3]
上記工程(1)で置換されるヌクレオシドの少なくとも一つがギャップ領域に位置する、[2]に記載の方法。
[4]
上記工程(2)で置換されるホスホロチオエート結合の少なくとも一つがギャップ領域に位置する、[2]または[3]に記載の方法。
[5]
3’ウイング領域および5’ウイング領域を構成する少なくとも一つのヌクレオシドが架橋型ヌクレオシドである、[2]~[4]のいずれか一つに記載の方法。
[6]
ホスホロチオエート修飾を有するオリゴヌクレオチドの中枢毒性の低減の程度を評価する方法であって、以下の工程(1)~(3):
(1)ホスホロチオエート修飾を有するオリゴヌクレオチドを準備する工程、
(2)[1]~[5]のいずれか一つに記載の方法により設計したオリゴヌクレオチドを準備する工程、および
(3)工程(2)で準備したオリゴヌクレオチドの中枢毒性と、工程(1)で準備したオリゴヌクレオチドの中枢毒性とを比較する工程
を含む、方法。
[7]
中枢毒性が低減したホスホロチオエート結合を有するオリゴヌクレオチドであって、オリゴヌクレオチドを構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子が、下記式I
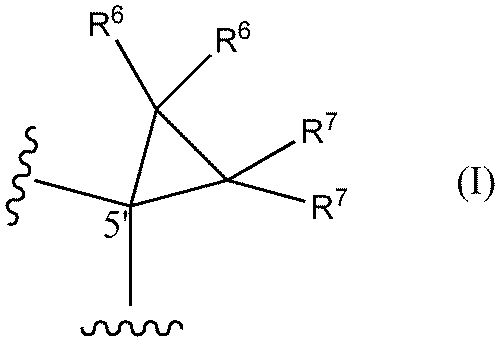
R6およびR7は、それぞれ独立して、水素原子、ハロゲン原子、またはメチル基である)
または下記式I’

R4およびR5は、それぞれ独立して、水素原子、メチル基またはエチル基である(ただし、R4およびR5が共に水素原子である場合を除く))
で表される構造を形成しており、任意により少なくとも一つのヌクレオシド間結合がホスホジエステル結合である、オリゴヌクレオチド。
[8]
ギャップマー型オリゴヌクレオチドである、[7]に記載のオリゴヌクレオチド。
[9]
上記式IまたはI’で表される構造を有するヌクレオシドの少なくとも一つがギャップ領域に位置する、[8]に記載のオリゴヌクレオチド。
[10]
前記ホスホジエステル結合の少なくとも一つがギャップ領域に位置する、[8]または[9]に記載のオリゴヌクレオチド。
[11]
3’ウイング領域および5’ウイング領域を構成する少なくとも一つのヌクレオシドが架橋型ヌクレオシドである、[8]~[10]のいずれか一つに記載のオリゴヌクレオチド。
[12]
[7]~[11]のいずれか一つに記載のオリゴヌクレオチドを含む、遺伝子の発現制御用試薬。
[13]
[7]~[11]のいずれか一つに記載のオリゴヌクレオチドを含む、疾患の治療薬。
[14]
(1)[1]~[5]のいずれか一つに記載の方法によりオリゴヌクレオチドを設計する工程、および
(2)工程(1)で設計したオリゴヌクレオチドを合成する工程
を含む、低中枢毒性オリゴヌクレオチドの製造方法。
[15]
[7]~[11]のいずれか一つに記載のオリゴヌクレオチドを投与することを含む、遺伝子の発現制御方法。
[16]
哺乳動物に対し、[7]~[11]のいずれか一つに記載のオリゴヌクレオチドの有効量を投与することを特徴とする、該哺乳動物における疾患の治療方法。
[17]
疾患の治療における使用のための[7]~[11]のいずれか一つに記載のオリゴヌクレオチド。
[18]
疾患の治療薬を製造するための[7]~[11]のいずれか一つに記載のオリゴヌクレオチドの使用。
本発明は、ホスホロチオエート修飾ヌクレオチドに代わる修飾ヌクレオチドとして5’-シクロプロパン構造を有するヌクレオシドを用いることで、中枢毒性が低減したオリゴヌクレオチドを設計できることを発見したことに基づいて完成した発明である。
(1)ホスホロチオエート修飾を有するオリゴヌクレオチド(以下、「PS修飾ヌクレオチド」と称することがある。)を構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子を、下記式I
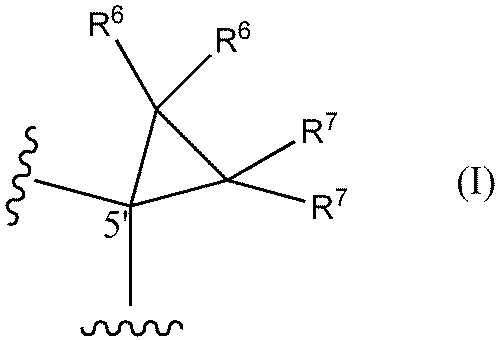
R6およびR7は、それぞれ独立して、水素原子、ハロゲン原子、またはメチル基である)
または下記式I’
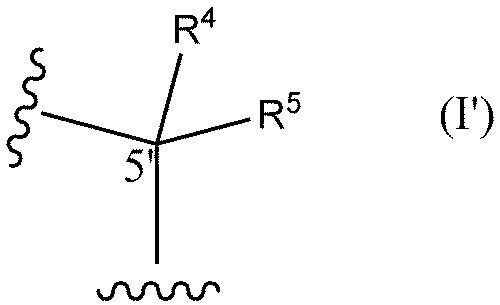
R4およびR5は、それぞれ独立して、水素原子、メチル基またはエチル基である(ただし、R4およびR5が共に水素原子である場合を除く))
で表される構造に置換する工程を含むことを特徴とする、方法(以下、「本発明の設計方法」と称することがある。)が提供される。一態様において、R6およびR7は共に水素原子である。本明細書において、オリゴヌクレオチドには、薬理学上許容されるオリゴヌクレオチドの塩も含まれるものとする。
(1’)PS修飾ヌクレオチドを構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子を、下記式I
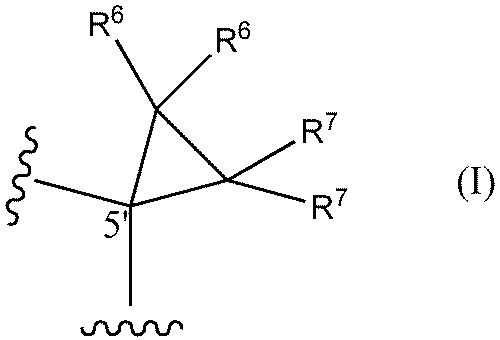
R6およびR7は、それぞれ独立して、水素原子、ハロゲン原子、またはメチル基である)
または下記式I’

R4およびR5は、それぞれ独立して、水素原子、メチル基またはエチル基である(ただし、R4およびR5が共に水素原子である場合を除く))
で表される構造に置換する工程を含むことを特徴とする、方法(以下、「本発明の毒性低減方法」と称することがある。)が提供される。一態様において、R6およびR7は共に水素原子である。
(2’)上記オリゴヌクレオチドが有する少なくとも一つのホスホロチオエート結合をホスホジエステル結合に置換する工程、が含まれていてもよい。
以下では、本発明の設計方法と本発明の毒性低減方法の両方に適用する事項については、これらの方法をまとめて単に「本発明の方法」と称することがある。
本発明の方法は、ホスホロチオエート修飾オリゴヌクレオチドが有する毒性のうち、中枢毒性に注目しそれを低減する目的で研究を進めた結果完成したものである。
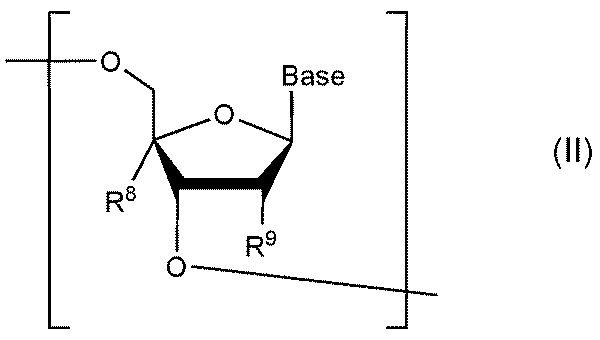
Baseは、α群から選択される任意の置換基を1以上有していてもよいプリン-9-イル基または2-オキソ-1,2-ジヒドロピリミジン-1-イル基を表し、ここで、該α群は、水酸基、核酸合成の保護基で保護された水酸基、炭素数1から6の直鎖アルキル基、炭素数1から6の直鎖アルコキシ基、メルカプト基、核酸合成の保護基で保護されたメルカプト基、炭素数1から6の直鎖アルキルチオ基、アミノ基、炭素数1から6の直鎖アルキルアミノ基、核酸合成の保護基で保護されたアミノ基、およびハロゲン原子からなり、そして
R8は水素原子でありかつR9は水素原子、ハロゲン原子、または炭素数1から6の直鎖アルコキシ基で置換されていてもよい炭素数1から6の直鎖アルコキシ基である。)
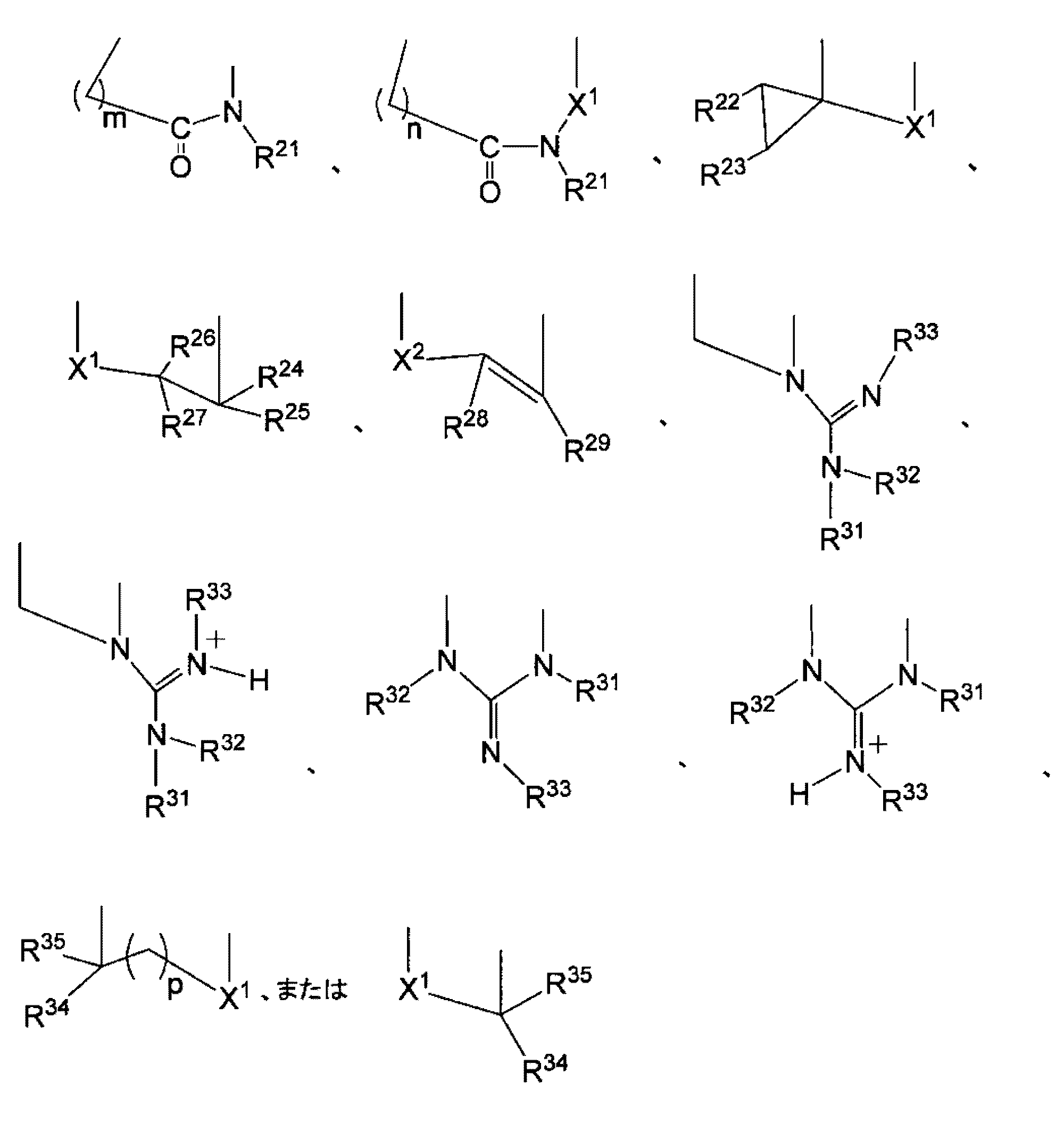
R21は、水素原子、分岐または環を形成していてもよい炭素数1から6のアルキル基、分岐または環を形成していてもよい炭素数2から6のアルケニル基、前記α群から選択される任意の置換基を1以上有していてもよくそしてヘテロ原子を含んでいてもよい炭素数3から10のアリール基、該α群から選択される任意の置換基を1以上有していてもよくそしてヘテロ原子を含んでいてもよい炭素数3から12のアリール部分を有するアラルキル基、または核酸合成のアミノ基の保護基であり;
R22およびR23は、それぞれ独立して、水素原子;ヘテロ原子を含んでいてもよい炭素数3から12のアリール基で置換されていてもよく、かつ分岐または環を形成していてもよい炭素数1から6のアルキル基;またはヘテロ原子を含んでいてもよい炭素数3から12のアリール部分を有するアラルキル基;であるか、あるいは
R22およびR23は一緒になって、-(CH2)q-[式中、qは2から5の整数である]を表し;
R24およびR25は、それぞれ独立して、水素原子;水酸基;分岐または環を形成していてもよい炭素数1から6のアルキル基;分岐または環を形成していてもよい炭素数1から6のアルコキシ基;アミノ基;および核酸合成の保護基で保護されたアミノ基;からなる群から選択される基であるか、あるいは、
R24およびR25は一緒になって、=C(R36)R37[式中、R36およびR37は、それぞれ独立して、水素原子、水酸基、核酸合成の保護基で保護された水酸基、メルカプト基、核酸合成の保護基で保護されたメルカプト基、アミノ基、炭素数1から6の直鎖または分岐鎖アルコキシ基、炭素数1から6の直鎖または分岐鎖アルキルチオ基、炭素数1から6のシアノアルコキシ基、または炭素数1から6の直鎖または分岐鎖アルキルアミノ基を表す]を表し;
R26およびR27は、それぞれ独立して、水素原子、分岐または環を形成していてもよい炭素数1から6のアルキル基、分岐または環を形成していてもよい炭素数1から6のアルコキシ基、または炭素数1から6の直鎖または分岐鎖アルキルチオ基であり;
R28は、水素原子、分岐または環を形成していてもよい炭素数1から6のアルキル基、分岐または環を形成していてもよい炭素数1から6のアルコキシ基、または炭素数1から6の直鎖または分岐鎖アルキルチオ基であり;
R29は、水素原子、水酸基、分岐または環を形成していてもよい炭素数1から6のアルキル基、分岐または環を形成していてもよい炭素数1から6のアルコキシ基、アミノ基、あるいは核酸合成の保護基で保護されたアミノ基であり;
R31、R32、およびR33は、それぞれ独立して、水素原子、分岐または環を形成していてもよい炭素数1から6のアルキル基、あるいは核酸合成のアミノ基の保護基であり;
R34およびR35は、それぞれ独立して、水素原子、水酸基、分岐または環を形成していてもよい炭素数1から6のアルキル基、分岐または環を形成していてもよい炭素数1から6のアルコキシ基、アミノ基、または核酸合成の保護基で保護されたアミノ基であり;
mは0から2の整数であり;
nは0から1の整数であり;
pは0から1の整数であり;
X1は、酸素原子、硫黄原子、またはアミノ基であり;そして
X2は酸素原子または硫黄原子である]
で表される二価の基を表す。)を構成してもよく、かかる構成を有するヌクレオシドを架橋型ヌクレオシドと称することがある。


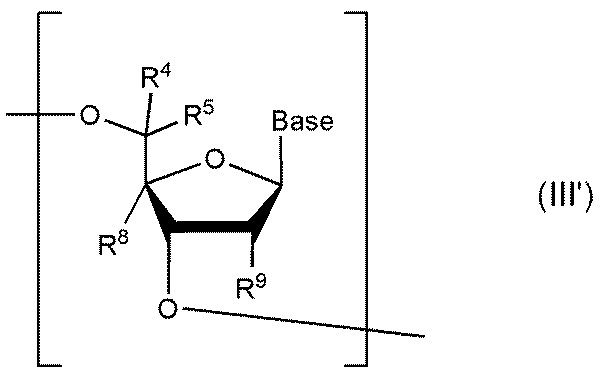

(I)PS修飾ヌクレオチドを準備する工程、
(II)本発明の設計方法により設計したオリゴヌクレオチドを準備する工程、および
(III)工程(II)で準備したオリゴヌクレオチドの中枢毒性と、工程(I)で準備したオリゴヌクレオチドの中枢毒性を比較する工程
を含む、方法(以下、「本発明の評価方法」と称する場合がある。)が提供される。
別の態様において、本発明により、
(1)本発明の設計方法によりオリゴヌクレオチドを設計する工程、および
(2)工程(1)で設計したオリゴヌクレオチドを合成する工程
を含む、低中枢毒性オリゴヌクレオチドの製造方法(以下、「本発明の製法」と称することがある。)が提供される。
前述のとおり、本発明により、中枢毒性が低減したオリゴヌクレオチドを設計または製造することができる。したがって、別の態様において、中枢毒性が低減したホスホロチオエート結合を有するオリゴヌクレオチドであって、
オリゴヌクレオチドを構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子が、下記式I

または下記式I’
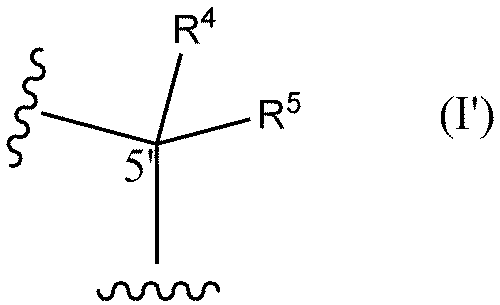
で表される構造を形成していることを特徴とする、ホスホロチオエート修飾を有するオリゴヌクレオチド(以下、「本発明のASO」と称することがある)が提供される。一態様において、R6およびR7は共に水素原子である。本発明のASOは、中枢毒性をさらに低減させる観点からは、少なくとも一つのヌクレオシド間結合がホスホジエステル結合であることが好ましい。


アンチセンスオリゴヌクレオチドによる中枢毒性を評価するために、以下のオリゴヌクレオチドを設計および合成した。LX-A4285はヌクレオシド間結合がホスホロチオエート結合であり、WO 2016/127000 A1により中枢毒性を示すことが知られている配列である。表中、5は5-メチルシトシンを、LnはLNAを、^はホスホロチオエート結合を、Cpは糖部5’位にシクロプロパン構造を導入したDNA(本明細書中では「5’-シクロプロパンDNA」と表記する)をそれぞれ示す。



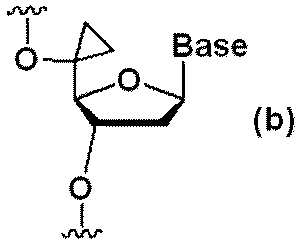
アンチセンスオリゴヌクレオチドによるマウス中枢毒性の評価には、BALB/cCrSlc(オス、7週齢、日本エスエルシー株式会社)を用いた。動物は温度:18~28℃、湿度:30~80%、照明時間:12時間/日の環境下のオープンラックにて飼育し、全ての動物は飼料および水分を自由摂取させた。
上記実施例1において使用したアンチセンスオリゴヌクレオチドを用いて、ヒト神経芽細胞におけるMAPT mRNA発現抑制効果を評価した。
オリゴヌクレオチドを添加しないものをコントロールとした。また、比較のために、ネガティブコントロールのオリゴヌクレオチド(Negative Control; NCとも称する):5(Y)^A(Y)^T(Y)^t^t^a^g^a^a^g^t^c^5(Y)^T(Y)^c(LX-A0070;配列番号6)を用いた。前記配列中、5は5-メチルシトシンを、YはAmNAをそれぞれ示す。
対数増殖期の SH-SY5Y 細胞を3.0×104個/ウェルにて、24ウェルプレートのウェル(10%ウシ胎児血清(FBS)を含むハムF12(Ham's F12)およびイーグル最小必須培地(E-MEM)の等比混合培地を含む)中に播いた。24時間後、各アンチセンスオリゴヌクレオチドをウェル内に添加し(培地中でのアンチセンスオリゴヌクレオチドの濃度:1、3、10、30または100nM)、24時間以上インキュベートした。
(MAPT検出用プライマーセット)
TaqMan Gene Expression Assay Hs00902194_m1_4331182(ThermoFisher Scientific社)
(GAPDH検出用プライマーセット)
TaqMan Gene Expression Assay Hs02786624_g1_4331182(ThermoFisher Scientific社)
表1及び表2に記載のアンチセンスヌクレオチドの中から、代表としてノックダウン活性を維持しつつ細胞毒性が低いもの(データは示さず)を2つ(LX-A5108及びLX-A5113)選択し、該ヌクレオチドについて、マウス中枢神経における急性毒性を評価した。評価方法は実施例2に記載の方法に従った。結果を図3に示す。図3から、ホスホロチオエート結合のみを有するアンチセンスヌクレオチドと比較して、5’-CPを有しホスホジエステル結合の割合が増加した配列では中枢毒性が低減することを確認した。
Claims (14)
- 中枢毒性が低減したオリゴヌクレオチドを設計する方法であって、
(1)ホスホロチオエート修飾を有するオリゴヌクレオチドを構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子を、下記式I
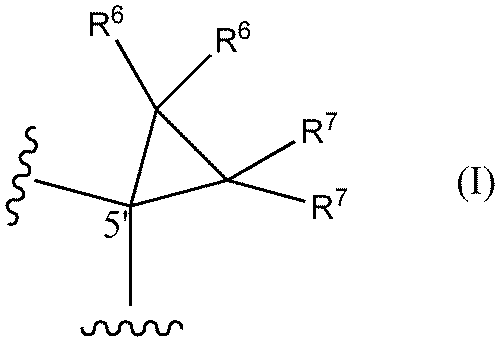
R6およびR7は、それぞれ独立して、水素原子、ハロゲン原子、またはメチル基である)
または下記式I’
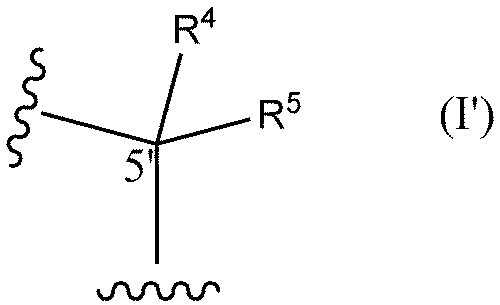
R4およびR5は、それぞれ独立して、水素原子、メチル基またはエチル基である(ただし、R4およびR5が共に水素原子である場合を除く))
で表される構造に置換する工程、および任意により
(2)該オリゴヌクレオチドが有する少なくとも一つのホスホロチオエート結合をホスホジエステル結合に置換する工程
を含むことを特徴とする、方法。 - オリゴヌクレオチドがギャップマー型オリゴヌクレオチドである、請求項1に記載の方法。
- 上記工程(1)で置換されるヌクレオシドの少なくとも一つがギャップ領域に位置する、請求項2に記載の方法。
- 上記工程(2)で置換されるホスホロチオエート結合の少なくとも一つがギャップ領域に位置する、請求項2または3に記載の方法。
- 3’ウイング領域および5’ウイング領域を構成する少なくとも一つのヌクレオシドが架橋型ヌクレオシドである、請求項2~4のいずれか1項に記載の方法。
- ホスホロチオエート修飾を有するオリゴヌクレオチドの中枢毒性の低減の程度を評価する方法であって、以下の工程(1)~(3):
(1)ホスホロチオエート修飾を有するオリゴヌクレオチドを準備する工程、
(2)請求項1~5のいずれか1項に記載の方法により設計したオリゴヌクレオチドを準備する工程、および
(3)工程(2)で準備したオリゴヌクレオチドの中枢毒性と、工程(1)で準備したオリゴヌクレオチドの中枢毒性とを比較する工程
を含む、方法。 - 中枢毒性が低減したホスホロチオエート結合を有するオリゴヌクレオチドであって、オリゴヌクレオチドを構成する少なくとも一つのヌクレオシドの糖部5’位炭素原子が、下記式I
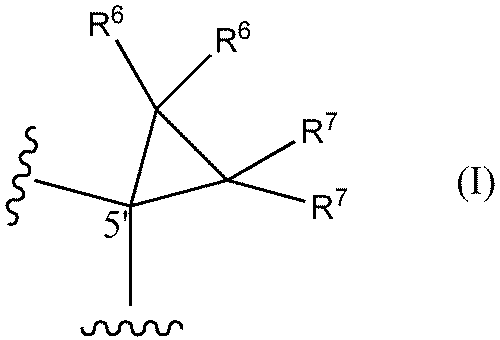
R6およびR7は、それぞれ独立して、水素原子、ハロゲン原子、またはメチル基である)
または下記式I’
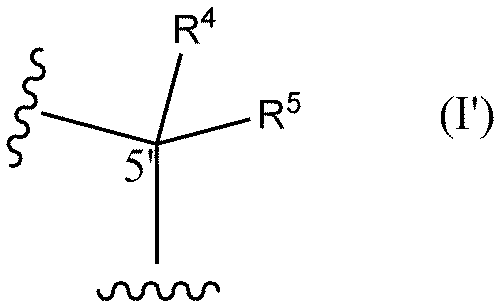
R4およびR5は、それぞれ独立して、水素原子、メチル基またはエチル基である(ただし、R4およびR5が共に水素原子である場合を除く))
で表される構造を形成しており、任意により少なくとも一つのヌクレオシド間結合がホスホジエステル結合である、オリゴヌクレオチド。 - ギャップマー型オリゴヌクレオチドである、請求項7に記載のオリゴヌクレオチド。
- 上記式IまたはI’で表される構造を有するヌクレオシドの少なくとも一つがギャップ領域に位置する、請求項8に記載のオリゴヌクレオチド。
- 前記ホスホジエステル結合の少なくとも一つがギャップ領域に位置する、請求項8または9に記載のオリゴヌクレオチド。
- 3’ウイング領域および5’ウイング領域を構成する少なくとも一つのヌクレオシドが架橋型ヌクレオシドである、請求項8~10のいずれか1項に記載のオリゴヌクレオチド。
- 請求項7~11のいずれか1項に記載のオリゴヌクレオチドを含む、遺伝子の発現制御用試薬。
- 請求項7~11のいずれか1項に記載のオリゴヌクレオチドを含む、疾患の治療薬。
- (1)請求項1~5のいずれか1項に記載の方法によりオリゴヌクレオチドを設計する工程、および
(2)工程(1)で設計したオリゴヌクレオチドを合成する工程
を含む、低中枢毒性オリゴヌクレオチドの製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22798959.7A EP4335859A1 (en) | 2021-05-06 | 2022-05-06 | Method for designing oligonucleotide having reduced central toxicity |
| JP2023518702A JPWO2022234855A1 (ja) | 2021-05-06 | 2022-05-06 | |
| CN202280034660.4A CN117529488A (zh) | 2021-05-06 | 2022-05-06 | 中枢毒性降低的寡核苷酸的设计方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021078507 | 2021-05-06 | ||
| JP2021-078507 | 2021-05-06 | ||
| JP2021151543 | 2021-09-16 | ||
| JP2021-151543 | 2021-09-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022234855A1 true WO2022234855A1 (ja) | 2022-11-10 |
Family
ID=83932781
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/019561 WO2022234855A1 (ja) | 2021-05-06 | 2022-05-06 | 中枢毒性が低減したオリゴヌクレオチドの設計方法 |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP4335859A1 (ja) |
| JP (1) | JPWO2022234855A1 (ja) |
| WO (1) | WO2022234855A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024071099A1 (ja) * | 2022-09-29 | 2024-04-04 | 国立大学法人東京医科歯科大学 | 5'-シクロプロピレン修飾を含む核酸分子 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011052436A1 (ja) | 2009-10-29 | 2011-05-05 | 国立大学法人大阪大学 | 架橋型人工ヌクレオシドおよびヌクレオチド |
| WO2016127000A1 (en) | 2015-02-04 | 2016-08-11 | Bristol-Myers Squibb Company | Methods of selecting therapeutic molecules |
| WO2018155451A1 (ja) | 2017-02-21 | 2018-08-30 | 国立大学法人大阪大学 | 核酸化合物およびオリゴヌクレオチド |
| WO2018155450A1 (ja) | 2017-02-21 | 2018-08-30 | 国立大学法人大阪大学 | アンチセンスオリゴ核酸 |
| WO2020158910A1 (ja) | 2019-02-01 | 2020-08-06 | 国立大学法人大阪大学 | 5'位修飾ヌクレオシドおよびそれを用いたヌクレオチド |
| WO2020166551A1 (ja) | 2019-02-13 | 2020-08-20 | 国立大学法人大阪大学 | 5'位修飾ヌクレオシドおよびそれを用いたヌクレオチド |
| JP2021078507A (ja) | 2015-09-29 | 2021-05-27 | ジョンソン・アンド・ジョンソン・コンシューマー・インコーポレイテッドJohnson & Johnson Consumer Inc. | シマレンガタケを培養するための方法 |
| JP2021151543A (ja) | 2017-07-03 | 2021-09-30 | 株式会社藤商事 | 遊技機 |
-
2022
- 2022-05-06 EP EP22798959.7A patent/EP4335859A1/en active Pending
- 2022-05-06 JP JP2023518702A patent/JPWO2022234855A1/ja active Pending
- 2022-05-06 WO PCT/JP2022/019561 patent/WO2022234855A1/ja active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011052436A1 (ja) | 2009-10-29 | 2011-05-05 | 国立大学法人大阪大学 | 架橋型人工ヌクレオシドおよびヌクレオチド |
| WO2016127000A1 (en) | 2015-02-04 | 2016-08-11 | Bristol-Myers Squibb Company | Methods of selecting therapeutic molecules |
| JP2021078507A (ja) | 2015-09-29 | 2021-05-27 | ジョンソン・アンド・ジョンソン・コンシューマー・インコーポレイテッドJohnson & Johnson Consumer Inc. | シマレンガタケを培養するための方法 |
| WO2018155451A1 (ja) | 2017-02-21 | 2018-08-30 | 国立大学法人大阪大学 | 核酸化合物およびオリゴヌクレオチド |
| WO2018155450A1 (ja) | 2017-02-21 | 2018-08-30 | 国立大学法人大阪大学 | アンチセンスオリゴ核酸 |
| JP2021151543A (ja) | 2017-07-03 | 2021-09-30 | 株式会社藤商事 | 遊技機 |
| WO2020158910A1 (ja) | 2019-02-01 | 2020-08-06 | 国立大学法人大阪大学 | 5'位修飾ヌクレオシドおよびそれを用いたヌクレオチド |
| WO2020166551A1 (ja) | 2019-02-13 | 2020-08-20 | 国立大学法人大阪大学 | 5'位修飾ヌクレオシドおよびそれを用いたヌクレオチド |
Non-Patent Citations (4)
| Title |
|---|
| IRWIN S., PSYCHOPHARMACOLOGIA, vol. 13, no. 3, 1968, pages 222 - 257 |
| MOAZAMI MICHAEL P., REMBETSY-BROWN JULIA M., WANG FENG, KRISHNAMURTHY PRANATHI MEDA, WEISS ALEXANDRA, MAROSFOI MIKLOS, KING ROBERT: "Quantifying and Mitigating Motor Phenotypes Induced by Antisense Oligonucleotides in the Central Nervous System", BIORXIV, 15 February 2021 (2021-02-15), XP055983072, Retrieved from the Internet <URL:https://www.biorxiv.org/content/10.1101/2021.02.14.431096v1.full.pdf> [retrieved on 20221118], DOI: 10.1101/2021.02.14.431096 * |
| TETRAHEDRON LETTERS, vol. 22, 1981, pages 1859 - 1862 |
| VASQUEZ GUILLERMO, FREESTONE GRAEME C, WAN W BRAD, LOW AUDREY, DE HOYOS CHERYL LI, YU JINGHUA, PRAKASH THAZHA P, ǾSTERGAARD MICHAE: "Site-specific incorporation of 5′-methyl DNA enhances the therapeutic profile of gapmer ASOs", NUCLEIC ACIDS RESEARCH, OXFORD UNIVERSITY PRESS, GB, vol. 49, no. 4, 26 February 2021 (2021-02-26), GB , pages 1828 - 1839, XP055983071, ISSN: 0305-1048, DOI: 10.1093/nar/gkab047 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024071099A1 (ja) * | 2022-09-29 | 2024-04-04 | 国立大学法人東京医科歯科大学 | 5'-シクロプロピレン修飾を含む核酸分子 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2022234855A1 (ja) | 2022-11-10 |
| EP4335859A1 (en) | 2024-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4977035B2 (ja) | 腫瘍の治療における、TGF−β、VEGF、インターロイキン−10、C−JUN、C−FOSまたはプロスタグランジンE2遺伝子にアンチセンスなオリゴヌクレオチドの低適用量の使用 | |
| US20150232846A1 (en) | Pseudocircularization oligonucleotides for modulating rna | |
| WO2021178237A2 (en) | Oligonucleotide compositions and methods thereof | |
| JP2022526419A (ja) | 中枢神経系における遺伝子発現を阻害するための組成物及び方法 | |
| JP2015518712A (ja) | Mecp2発現を調節するための組成物及び方法 | |
| JP2015523854A (ja) | Smn遺伝子ファミリー発現を調節するための組成物及び方法 | |
| JP2015523853A (ja) | Atp2a2発現を調節するための組成物及び方法 | |
| JP2015518713A (ja) | Utrn発現を調節するための組成物及び方法 | |
| KR20240036132A (ko) | 대상에게서 smn2 스플라이싱을 조정하기 위한 조성물 및 방법 | |
| JP2015523855A (ja) | Apoa1及びabca1発現を調節するための組成物及び方法 | |
| JP2016521556A (ja) | Foxp3発現を調節するための組成物及び方法 | |
| JP2015518711A (ja) | Bdnf発現を調節するための組成物及び方法 | |
| JP4787409B2 (ja) | 治療用ホスホジエステラーゼ阻害剤 | |
| BR122020018622A2 (pt) | Molécula de ácido nucleico para redução de papd5 e papd7 de mrna para o tratamento de infecção hepatite b | |
| WO2022234855A1 (ja) | 中枢毒性が低減したオリゴヌクレオチドの設計方法 | |
| WO2021157730A1 (ja) | 核酸医薬とその使用 | |
| WO2021187540A1 (ja) | Scn1a遺伝子の発現及び/又は機能調節剤 | |
| WO2021039879A1 (ja) | 胃癌分子標的核酸医薬 | |
| WO2021187392A1 (ja) | モルホリノ核酸を含むヘテロ核酸 | |
| US20230041016A1 (en) | Anti-slc6a1 oligonucleotides and related methods | |
| JP6934695B2 (ja) | 核酸医薬とその使用 | |
| CN117529488A (zh) | 中枢毒性降低的寡核苷酸的设计方法 | |
| JP2023528435A (ja) | 遺伝子転写物のモジュレーターを使用する神経学的疾患の処置 | |
| TW202130809A (zh) | 用於抑制β-ENaC表現之RNAi藥劑、其組合物及使用方法 | |
| JP2021517909A (ja) | 胆管減少症関連状態を治療するための方法及び組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22798959 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023518702 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18558056 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280034660.4 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022798959 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022798959 Country of ref document: EP Effective date: 20231206 |