WO2019208571A1 - アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 - Google Patents
アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 Download PDFInfo
- Publication number
- WO2019208571A1 WO2019208571A1 PCT/JP2019/017249 JP2019017249W WO2019208571A1 WO 2019208571 A1 WO2019208571 A1 WO 2019208571A1 JP 2019017249 W JP2019017249 W JP 2019017249W WO 2019208571 A1 WO2019208571 A1 WO 2019208571A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- group
- compound
- hydrogen atom
- amidite
- Prior art date
Links
- 0 CC(C)OCC(C)(C)OCC(C)(C)O*(*)(*)CC#N Chemical compound CC(C)OCC(C)(C)OCC(C)(C)O*(*)(*)CC#N 0.000 description 13
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon, or a metal, e.g. chelates, vitamin B12
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/16—Preparation of carboxylic acid nitriles by reaction of cyanides with lactones or compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/11—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound oxygen atoms bound to the same saturated acyclic carbon skeleton
- C07C255/13—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound oxygen atoms bound to the same saturated acyclic carbon skeleton containing cyano groups and etherified hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/02—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols
- C07C319/12—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols by reactions not involving the formation of mercapto groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/10—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C323/11—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/12—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/1892—Preparation; Treatments not provided for in C07F7/20 by reactions not provided for in C07F7/1876 - C07F7/1888
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/067—Pyrimidine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/167—Purine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to an amidite compound and a method for producing a polynucleotide using the compound. Furthermore, this invention relates to the intermediate compound of the said amidite compound, and the manufacturing method of this intermediate compound.
- RNA is a useful material that can be used as an RNA probe, antisense RNA, ribozyme, siRNA, aptamer, and the like.
- RNA can be synthesized by a solid phase synthesis method or the like.
- a nucleoside phosphoramidite hereinafter referred to as “amidite”
- examples of such a protecting group for the hydroxyl group at the 2 ′ position of amidite include TBDMS (t-butyldimethylsilyl), TOM (triisopropylsilyloxymethyl), ACE (bis (2-acetoxyethoxy) methyl) and the like. It has been.
- the protecting groups disclosed in Patent Documents 1 and 2 have been reported as protecting groups for the hydroxyl group at the 2 ′ position of the amidite. A method for synthesizing RNA using these amidites having these protecting groups is disclosed in It is not always satisfactory in terms of yield and purity.
- An object of the present invention is to provide an amidite compound capable of synthesizing RNA with high purity and a method for producing a polynucleotide using the compound. Furthermore, this invention aims at providing the intermediate compound of the said amidite compound, and the manufacturing method of this intermediate compound.
- RNA with high purity by using the following groups as the protecting group for the hydroxyl group at the 2 ′ position of the amidite. I got the knowledge.
- R a and R b are the same or different and represent a methyl group, an ethyl group or a hydrogen atom. However, R a and R b do not represent a hydrogen atom at the same time.
- n represents an integer of 1 to 5.
- the present invention has been completed based on these findings, and has been completed and provides the following amidite compound, a method for producing a polynucleotide using the compound, an ether compound, and a method for producing the ether compound. Is.
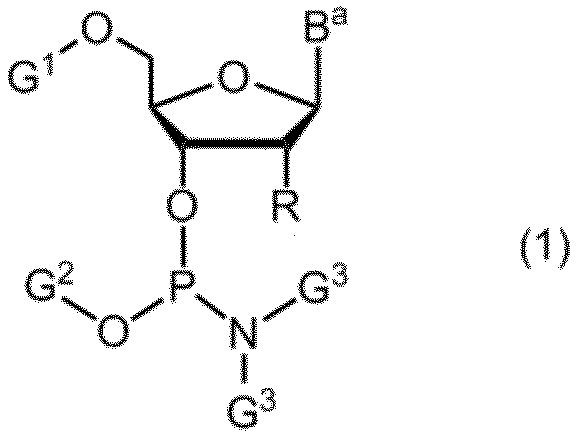
- Item 1 An amidite compound represented by formula (1).
- R is the formula: (In the formula, R a and R b are the same or different and represent a methyl group, an ethyl group or a hydrogen atom. However, R a and R b do not represent a hydrogen atom at the same time.
- n represents an integer of 1 to 5.
- B a represents a group having the nucleic acid base skeleton may be protected
- G 1 and G 2 are the same or different and represent a hydroxyl-protecting group
- G 3 is the same or different and represents an alkyl group.
- Item 2. Item 2.
- the amidite compound according to Item 1 wherein R a is a methyl group or an ethyl group, and R b is a hydrogen atom.
- Item 3. Item 3. The amidite compound according to Item 1 or 2, wherein G 1 is the following group. (Wherein R 1 , R 2 and R 3 are the same or different and represent hydrogen or an alkoxy group.)
- Item 4. Item 4. The amidite compound according to any one of Items 1 to 3, wherein G 2 is the following group.
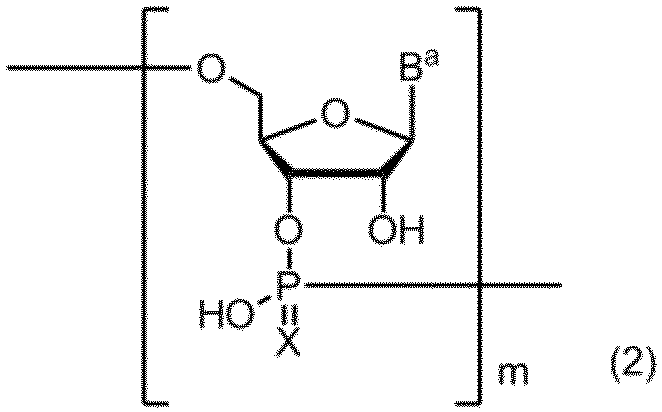
- Item (2) comprising the step of subjecting the amidite compound according to any one of Items 1 to 5 to a solid phase synthesis reaction: (Wherein, B a represents a group having the nucleic acid base skeleton may be protected by the same or different, X represents an oxygen atom or a sulfur atom, m represents a positive integer. ) The manufacturing method of the compound containing the polynucleotide skeleton shown by these. Item 7.
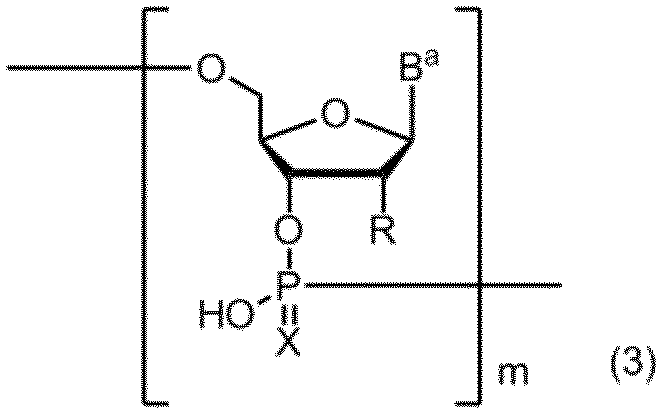
- a compound containing a polynucleotide skeleton of formula (2) is produced by a solid phase synthesis reaction using the amidite compound (3):
- B a represents a group having the nucleic acid base skeleton may be protected by the same or different
- X represents an oxygen atom or a sulfur atom
- R is the same or different and has the formula:
- R a and R b are the same or different and represent a methyl group, an ethyl group or a hydrogen atom. However, R a and R b do not represent a hydrogen atom at the same time.
- n represents an integer of 1 to 5.
- m represents a positive integer.
- R a , R b and n are as defined in item 1, and R c represents a C1-C6 alkyl group or a phenyl group.
- Item 11. The ether compound according to Item 10, wherein R a is a methyl group or an ethyl group, R b is a hydrogen atom, and R c is methyl.
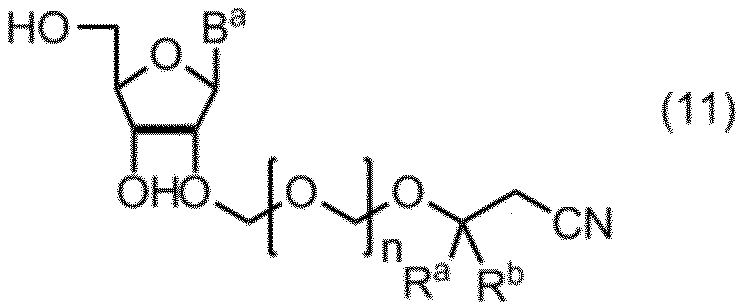
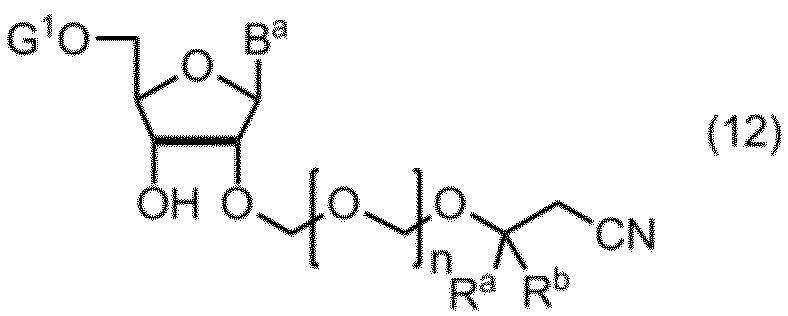
- the compound of formula (10) is further deprotected to give formula (11): (In the formula, B a , R a , R b and n are as defined above.) Obtaining a compound represented by: By selectively protecting the 5 ′ hydroxyl group of the compound of formula (11), the formula (12): (In the formula, B a , R a , R b and n are as defined above, and G 1 represents a hydroxyl-protecting group.) Obtaining a compound represented by: A compound of formula (12) is represented by formula (13): (In the formula, G 2 represents a hydroxyl-protecting group, and G 3 is the same or different and represents an alkyl group.) A step of reacting with a phosphorodiamidite represented by: The manufacturing method of the compound of Formula (1) of claim
- G 4 are process according to claim 19 having a G4-1 or G4-2 structure.
- n represents an integer of 1 to 5, B a represents a group having the nucleic acid base skeleton may be protected, G 4 represents a protecting group. )
- Formula (12) (In the formula, B a , R a , R b and n are as defined in item 21, and G 1 represents a hydroxyl-protecting group.)
- a compound represented by Item 24. Use of the amidite compound of formula (1) in the production of RNA.
- RNA can be easily produced with high purity in a solid phase synthesis method.
- the amidite compound of the present invention is represented by the formula (1).
- R is (In the formula, R a and R b are the same or different and represent a methyl group, an ethyl group or a hydrogen atom. However, R a and R b do not represent a hydrogen atom at the same time.
- n represents an integer of 1 to 5.
- B a represents a group having the nucleic acid base skeleton may be protected
- G 1 and G 2 are the same or different and represent a hydroxyl-protecting group
- G 3 is the same or different and represents an alkyl group.
- Nucleobases in B a is not particularly limited.
- the nucleobase include adenine, cytosine, guanine, uracil, thymine, 5-methylcytosine, pseudouracil, 1-methyl pseudouracil and the like.
- the nucleobase may be substituted with a substituent.
- substituents include halogen atoms, acyl groups, alkyl groups, arylalkyl groups, alkoxy groups, alkoxyalkyl groups, cyanoalkyl groups, hydroxy groups, hydroxymethyl groups, acyloxymethyl groups, amino groups, monoalkyls. Examples thereof include an amino group, a dialkylamino group, a carboxy group, a cyano group, a nitro group, and the like, and combinations of two or more kinds of these substituents.
- the protecting group for the amino group is not particularly limited, and a protecting group used in known nucleic acid chemistry can be used.
- a protecting group For example, methyl group, benzoyl group, 4-methoxybenzoyl group, acetyl group, propionyl group, butyryl group, isobutyryl group, phenylacetyl group, phenoxyacetyl group, 4-tert-butylphenoxyacetyl group, 4-isopropylphenoxyacetyl group, And (dimethylamino) methylene groups, and combinations of two or more of these protecting groups.
- R 4 represents a hydrogen atom, a methyl group, a phenoxyacetyl group, a 4-tert-butylphenoxyacetyl group, a 4-isopropylphenoxyacetyl group, a phenylacetyl group, an acetyl group or a benzoyl group
- R 5 represents a hydrogen atom, an acetyl group, an isobutyryl group or a benzoyl group
- R 6 represents a hydrogen atom, a phenoxyacetyl group, a 4-tert-butylphenoxyacetyl group, a 4-isopropylphenoxyacetyl group, a phenylacetyl group, an acetyl group, or an isobutyryl group
- R 7 represents a 2-cyanoethyl group
- R 8 represents a hydrogen atom, a methyl group, a benzoyl group, a 4-me
- G 1 can be used without particular limitation as long as it can function as a protecting group, and known protecting groups used in amidite compounds can be widely used.
- G 1 is preferably the following group. (Wherein R 1 , R 2 and R 3 are the same or different and represent hydrogen or an alkoxy group.)
- R 1 , R 2 and R 3 is preferably hydrogen, and the remaining two are preferably alkoxy groups, and the alkoxy group is particularly preferably a methoxy group.
- G 2 can be used without particular limitation as long as it can function as a protecting group, and known protecting groups used in amidite compounds can be widely used.
- Examples of G 2 include a hydrogen atom, an alkyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, a haloalkyl group, an aryl group, a heteroaryl group, an arylalkyl group, a cycloalkenyl group, a cycloalkylalkyl group, and a cyclylalkyl group.
- Hydroxyalkyl group aminoalkyl group, alkoxyalkyl group, heterocyclylalkenyl group, heterocyclylalkyl group, heteroarylalkyl group, silyl group, silyloxyalkyl group, mono, di or trialkylsilyl group, mono, di or trialkylsilyl And oxyalkyl groups, which may be substituted with one or more electron withdrawing groups.
- G 2 is preferably an alkyl group substituted with an electron withdrawing group.
- the electron withdrawing group include a cyano group, a nitro group, an alkylsulfonyl group, a halogen, an arylsulfonyl group, a trihalomethyl group, and a trialkylamino group, and a cyano group is preferable.
- G 2 is particularly preferably the following group.
- G 3 two G 3 may be bonded to each other to form a cyclic structure.
- the alkyl group may be linear or branched, and is preferably an alkyl group having 1 to 12 carbon atoms, more preferably an alkyl group having 1 to 6 carbon atoms.
- Examples of the alkyl group include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, and hexyl.
- the alkyl group here also includes an alkyl moiety such as an alkoxy group.
- R a is preferably methyl.
- n is preferably an integer of 1 to 4, more preferably an integer of 1 to 3, still more preferably 1 or 2, and particularly preferably 1.
- the amidite compound of the present invention can be used in a free state or a salt state.
- the salt of the amidite compound of the present invention is not particularly limited, and examples thereof include salts with inorganic bases such as sodium salts, magnesium salts, potassium salts, calcium salts, and aluminum salts; organic bases such as methylamine, ethylamine, and ethanolamine. And salts with basic amino acids such as lysine, ornithine and arginine, and ammonium salts.
- the salt may be an acid addition salt.
- the salt include hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid and other mineral acids; formic acid, acetic acid, Organic acids such as propionic acid, oxalic acid, malonic acid, malic acid, tartaric acid, fumaric acid, succinic acid, lactic acid, maleic acid, citric acid, methanesulfonic acid, trifluoromethanesulfonic acid, ethanesulfonic acid; and aspartic acid, Examples include acid addition salts with acidic amino acids such as glutamic acid.
- the amidite compound of the present invention includes salts, hydrates, solvates, crystal polymorphs and the like.
- the amidite compound of the present invention can be appropriately modified in accordance with known methods described in Japanese Patent No. 5157168, Japanese Patent No. 5554811, etc., or the methods described in Examples described later, or if necessary. It can be manufactured by the added method.
- the present invention also includes an intermediate compound for producing the amidite compound represented by the formula (1).
- an intermediate compound include an ether compound represented by the formula (4).
- R a and R b are the same or different and represent a methyl group, an ethyl group or a hydrogen atom. However, R a and R b do not represent a hydrogen atom at the same time.
- n represents an integer of 1 to 5.
- R c is a C1-C6 alkyl group or a phenyl group.
- the ether compound represented by the formula (4) is obtained by reacting, for example, bis (alkylthiomethyl) ether or bis (phenylthiomethyl) ether with 3-hydroxy-3-alkylpropanenitrile in a solvent in the presence of an oxidizing agent and an acid.
- Bis (alkylthiomethyl) ether or bis (phenylthiomethyl) ether is obtained, for example, by reacting bischloromethylether or bis (aryloxymethyl) ether with the corresponding alkylmercaptan or phenylmercaptan as shown in the following formula be able to.
- Examples of bis (aryloxymethyl) ether include bis (2,4,6-trichlorophenyloxymethyl) ether.
- it can manufacture also by the method which added the change suitably to these methods according to the method as described in the Example mentioned later or if needed.
- Step a will be described below.
- a cyanide ion for example, a cyanide ion derived from sodium cyanide, potassium cyanide, copper cyanide, trimethylsilyl cyanide and the like can be used.
- Examples of the base in this reaction include alkali metal hydroxide, alkaline earth metal hydroxide, ammonium hydroxide, and combinations of two or more thereof.
- lithium hydroxide and lithium hydroxide monohydrate are preferably used.
- the amount of the base is usually 0.01 to 1 equivalent, preferably 0.1 to 0.3 equivalent, relative to the epoxy compound represented by the formula (5).
- the amount of cyanide ions is usually 0.3 to 2 equivalents, preferably 0.6 to 0.8 equivalents, relative to the compound represented by the formula (5).
- the reaction temperature of this reaction is usually ⁇ 20 to 40 ° C., preferably 0 to 35 ° C.
- the reaction time of this reaction is usually 0.5 to 24 hours, preferably 1 to 5 hours.
- Step b will be described below.
- the oxidizing agent examples include N-halogenated succinimides such as N-chlorosuccinimide, N-bromosuccinimide and N-iodosuccinimide, N-halogenated hydantoins such as 1,3-diiodo-5,5-dimethylhydantoin, and Examples include halogens such as chlorine, bromine and iodine, and combinations of two or more thereof.
- N-halogenated succinimide is preferably used, and N-iodosuccinimide is more preferably used.
- the acid is not particularly limited, and examples thereof include perfluoroalkyl carboxylic acids and salts thereof, perfluoroalkyl sulfonic acids and salts thereof, alkyl sulfonic acids and salts thereof, and combinations of two or more thereof.
- the salt include a copper salt and a silver salt.
- Specific examples of the acid include methanesulfonic acid, p-toluenesulfonic acid, camphorsulfonic acid, trifluoromethanesulfonic acid, and silver trifluoromethanesulfonate, and combinations of two or more thereof. In the present invention, trifluoromethanesulfonic acid is preferably used.
- solvent examples include tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentylmethyl ether, dioxane, dichloromethane, toluene, and the like, and combinations of two or more thereof.
- tetrahydrofuran is preferably used.
- the amount of 3-hydroxyalkyl nitrile is usually 0.5 to 2.0 equivalents, preferably 0.8 to 1.5 equivalents relative to bis (alkylthiomethyl) ether or bis (phenylthiomethyl) ether. is there.
- the amount of the oxidizing agent is usually 0.5 to 2 equivalents, preferably 0.7 to 1.2 equivalents relative to bis (alkylthiomethyl) ether or bis (phenylthiomethyl) ether.
- the amount of the acid is usually 0.001 to 2.0 equivalents, preferably 0.01 to 0.1 equivalents, relative to bis (alkylthiomethyl) ether or bis (phenylthiomethyl) ether.
- the reaction temperature of this reaction is usually ⁇ 80 ° C. to 0 ° C., preferably ⁇ 50 ° C. to ⁇ 30 ° C.
- the reaction time of this reaction is usually 1 to 24 hours, preferably 2 to 6 hours.
- the completion of the reaction can be confirmed by, for example, sampling a part of the reaction mass and analyzing it by GC, TLC, LC or the like. After completion of the reaction, the reaction may be stopped by adding a base such as triethylamine to the reaction mass.
- the residue containing the ether compound represented by the formula (7) can be obtained by pouring the reaction mass into water and subjecting it to usual post-treatment operations such as organic solvent extraction, washing and concentration. By subjecting the residue to a purification operation such as distillation or column chromatography, a high purity ether compound represented by the formula (7) can be obtained.
- R a is a methyl group or an ethyl group
- R b is a hydrogen atom
- R c is a methyl group. More preferably, R a is a methyl group, R b is a hydrogen atom, and R c is a methyl group.
- the compound of the formula (8) can be produced by a production method including the following steps A to C.
- Step A Step of hydrolyzing 3-aminocrotononitrile
- Step B Step of reducing cyanoacetone obtained in Step A
- Step C 3-hydroxybutanenitrile and bis (methylthiomethyl) ether obtained in Step B in a solvent in the presence of an oxidizing agent and an acid. The step of reacting.
- cyanoacetone can be obtained by hydrolyzing 3-aminocrotononitrile.
- Hydrolysis can be performed, for example, by mixing 3-aminocrotononitrile and an acid in the presence of water.
- the acid may be a hydrous acid or an acid anhydride, and examples thereof include hydrochloric acid, sulfuric acid, methanesulfonic acid and the like.
- hydrochloric acid is preferably used. This reaction is carried out in the presence of water by adding water when an acid anhydride is used as the acid, while water may or may not be added when using a hydrous acid as the acid. Also good.
- the amount of acid is usually 1 to 10 equivalents, preferably 1 to 1.5 equivalents, relative to 3-aminocrotononitrile.
- the reaction temperature of this reaction is usually ⁇ 20 to 100 ° C., preferably 0 to 85 ° C.
- the reaction time of this reaction is usually 1 to 24 hours, preferably 1 to 4 hours.
- Examples of the reducing agent used in the reduction step of Step B include metal reducing agents such as sodium borohydride.
- the amount of the reducing agent is usually 0.25 to 2 equivalents, preferably 0.5 to 1 equivalent, relative to cyanoacetone.
- This reaction can be carried out in a solvent.
- the solvent include ether solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, 4-methyltetrahydropyran, cyclopentylmethyl ether, dioxane, and methanol, ethanol and the like.
- ether solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, 4-methyltetrahydropyran, cyclopentylmethyl ether, dioxane, and methanol, ethanol and the like.
- alcohol solvents and combinations of two or more of these solvents are examples of these solvents.
- tetrahydrofuran is preferably used.
- this reduction step can also be performed by a biological method such as using yeast.
- the reaction temperature of this reaction is usually ⁇ 20 to 60 ° C., preferably 0 to 35 ° C.
- the reaction time of this reaction is usually 0.5 to 24 hours, preferably 1 to 4 hours.
- the reaction in Step C is performed by reacting bis (methylthiomethyl) ether with alcohol in the presence of an oxidizing agent (for example, a halogenating agent).
- an oxidizing agent for example, a halogenating agent
- the halogenating agent include N-halogenated succinimides such as N-chlorosuccinimide, N-bromosuccinimide and N-iodosuccinimide, and N-halogenated hydantoins such as 1,3-diiodo-5,5-dimethylhydantoin.
- Halogens such as chlorine, bromine and iodine.
- N-halogenated succinimide is preferably used, and N-iodosuccinimide is more preferably used.
- the type of acid used in this reaction is not particularly limited, and examples thereof include perfluoroalkylcarboxylic acid and its salt, perfluoroalkylsulfonic acid and its salt, and alkylsulfonic acid and its salt.
- the salt include a copper salt and a silver salt.
- Specific examples of the acid include methanesulfonic acid, paratoluenesulfonic acid, trifluoromethanesulfonic acid, and silver trifluoromethanesulfonate. In the present invention, trifluoromethanesulfonic acid is preferably used.
- solvent examples include tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, 4-methyltetrahydropyran, cyclopentylmethyl ether, dioxane, dichloromethane, toluene and the like.
- tetrahydrofuran is preferably used.
- the amount of 3-hydroxybutanenitrile is usually 0.5 to 2.0 equivalents, preferably 0.8 to 1.5 equivalents, relative to bis (methylthiomethyl) ether.
- the amount of the oxidizing agent is usually 0.5 to 2 equivalents, preferably 0.7 to 1.2 equivalents, relative to bis (methylthiomethyl) ether.
- the amount of the acid is usually 0.001 to 2.0 equivalents, preferably 0.01 to 0.1 equivalents, relative to bis (methylthiomethyl) ether.
- the reaction temperature of this reaction is usually ⁇ 80 ° C. to 0 ° C., preferably ⁇ 50 ° C. to ⁇ 30 ° C.
- the reaction time of this reaction is usually 1 to 24 hours, preferably 2 to 6 hours.
- the completion of the reaction can be confirmed by, for example, sampling a part of the reaction mass and analyzing it by GC, TLC, LC or the like. After completion of the reaction, the reaction may be stopped by adding a base such as triethylamine to the reaction mass.
- the residue containing the ether compound represented by formula (8) can be obtained by pouring the reaction mass into water and subjecting it to usual post-treatment operations such as organic solvent extraction, washing and concentration. By subjecting the residue to a purification operation such as distillation or column chromatography, a high purity ether compound represented by the formula (8) can be obtained.
- the amidite compound of the present invention can be used as a material for producing RNA in a solid phase synthesis method.
- RNA can be produced with high purity.
- the method for producing a compound containing a polynucleotide skeleton represented by the following formula (2) of the present invention includes a step of performing a solid-phase synthesis reaction using the amidite compound described above.
- B a represents a group having the nucleic acid base skeleton may be protected by the same or different
- X represents an oxygen atom or a sulfur atom
- m represents a positive integer.
- the production method of the present invention includes a step of obtaining a compound having an oligonucleotide skeleton represented by formula (2) by treating a compound having an oligonucleotide skeleton represented by formula (3) with tetraalkylammonium fluoride.
- B a represents a group having the nucleic acid base skeleton may be protected by the same or different
- X represents an oxygen atom or a sulfur atom
- R is the same or different and has the formula:
- R a and R b are the same or different and represent a methyl group, an ethyl group or a hydrogen atom. However, R a and R b do not represent a hydrogen atom at the same time.
- n represents an integer of 1 to 5.
- Represents m represents a positive integer.
- M is not particularly limited, and is preferably an integer of 2 to 300.
- the “compound containing a polynucleotide skeleton” means a compound containing at least one RNA, and preferably a compound consisting only of RNA.
- the solid phase synthesis reaction can be performed according to a known method such as a phosphoramidite method (for example, the methods described in Japanese Patent Nos. 5157168 and 5554881). Moreover, it can implement using the commercially available automatic synthesizer of a nucleic acid etc.
- the method for producing a compound containing a polynucleotide skeleton represented by the formula (2) includes (A) a hydroxyl group at the 5 ′ position of the first amidite compound supported on a solid phase carrier (for example, the formula (1) ) G 1 ) deprotection, (B) a step of condensing the deprotected amidite compound produced in step (A) with a second amidite compound, (C) an unreacted compound in step (B) An optional step of capping the hydroxyl group at the 5 ′ position, a step of converting the phosphite group of the condensate produced in (D), (B) or (C) into a phosphoric acid group or a thiophosphoric acid group, (E) step ( The step obtained by cutting out the compound obtained in D) from the solid phase carrier and deprotecting the 2′-position and the hydroxyl group of the nucleobase, (F) deprotecting the 5′-position hydroxyl group, and
- the compound having the oligonucleotide skeleton represented by the formula (3) is preferably treated with tetraalkylammonium fluoride to remove the protecting group at the 2′-position, whereby the oligonucleotide skeleton represented by the formula (2) is removed.
- Conditions according to a known method can be adopted as the reaction conditions (reaction temperature, reaction time, amount of reagent, etc.) of the reaction.
- RNA can be isolated and purified as necessary.
- HPLC high performance liquid chromatography
- the production method of the present invention makes it possible to produce RNA with higher purity than before.
- the reaction conditions for producing the amidite compound of the present invention and the ether compound represented by formula (4) are not particularly limited.
- the ether compound represented by the formula (4) can also be synthesized using a flow reactor.
- a reduction step with hydrogen or a reduction step with magnesium or the like can be added in the presence of a transition metal catalyst such as palladium.
- a compound of formula (1) can be prepared from a compound of formula (9) by steps 1, 2, 3 and 4 of Scheme 1 below.
- B a represents the same meaning as defined above
- G 4 typically has a G 4 -1 or G 4 -2 structure below.
- Process 1 (etherification process)
- the etherification step is performed by reacting the compound of formula (9) with the compound of formula (4). This reaction is usually carried out by adding an oxidizing agent.
- the oxidizing agent used in this step is not particularly limited, but is a group consisting of N-chlorosuccinimide, N-bromosuccinimide, N-iodosuccinimide, iodine, 1,3-diiodo-5,5′-dimethylhydantoin, bromine and chlorine. It is preferable that it is at least one selected from.
- an acid can be added, and the acid to be used is not particularly limited, but at least one selected from the group consisting of perfluoroalkylcarboxylic acids, perfluoroalkylsulfonic acids, alkylsulfonic acids and salts thereof. It is preferable that
- the reaction solvent used in this step is not particularly limited.
- ethers such as diethyl ether, THF (tetrahydrofuran), 2-methyltetrahydrofuran, tetrahydropyran, 4-methyltetrahydropyran, dimethoxyethane, diglyme, cyclopentylmethyl ether, dioxane and the like.
- nitriles such as acetonitrile, aromatic hydrocarbons such as toluene, chlorobenzene and dichlorobenzene, dichloromethane and the like, and combinations of two or more of these solvents.
- Preferred solvents include ethers such as diethyl ether, THF (tetrahydrofuran), 2-methyltetrahydrofuran, tetrahydropyran, 4-methyltetrahydropyran, dimethoxyethane, diglyme, cyclopentylmethyl ether, dioxane and the like. More preferred solvents include tetrahydropyran and 4-methyltetrahydropyran.
- the reaction time in this step is not particularly limited, but is, for example, 10 minutes to 12 hours, preferably 10 minutes to 6 hours.
- the reaction temperature is not particularly limited, but is, for example, ⁇ 80 to 30 ° C., preferably ⁇ 60 to 10 ° C.
- the concentration of the ether compound represented by the formula (4) is not particularly limited, and can be set as appropriate.
- the number of moles of the ether compound represented by the formula (4) is, for example, 0.5 to 2 times, preferably 0.8 to 1.times the mole number of the compound represented by the formula (9). 5 times.
- the number of moles of the oxidizing agent is, for example, 0.5 to 10 times, preferably 0.8 to 6 times the number of moles of the compound represented by the formula (9).
- Process 2 (deprotection process)
- the compound of the formula (10) obtained in the step 1 is subjected to a deprotection reaction to be converted into a compound of the formula (11).
- the deprotection step can be carried out by a known method.
- the deprotection can be performed by reacting hydrogen fluoride / triethylamine or hydrogen fluoride / pyridine in a solvent.
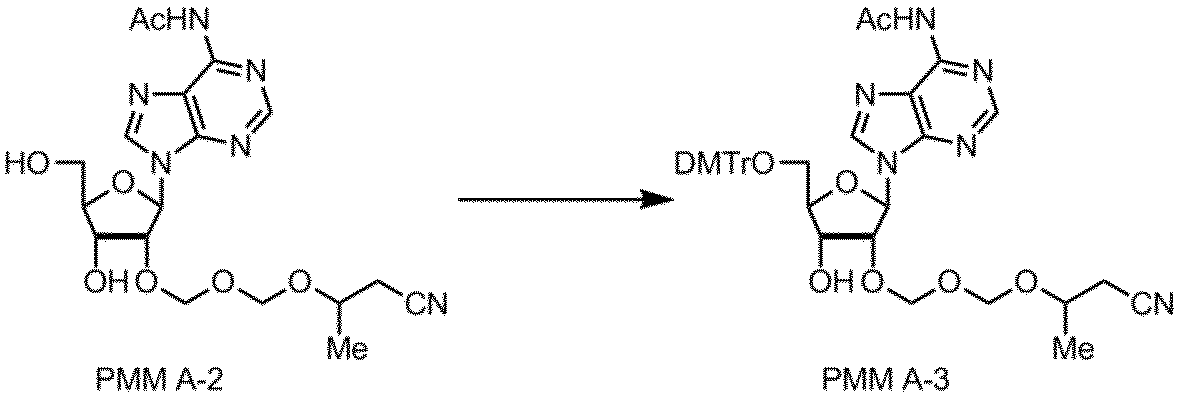
- Step 3 The compound of the formula (11) obtained in the above step is subjected to a protection step, and the introduction of the protecting group can be carried out by a known method. Typically, the compound (11) is added to the compound (11) in 4,4. A protecting group is introduced by reacting '-dimethoxytrityl chloride to produce compound (12).
- PMM (((1-cyanopropan-2-yl) oxy) methoxy) methyl group
- BMM (((1-cyanobutan-2-yl) oxy) methoxy) methyl group
- TBM ((((1-cyano- 2-methylpropan-2-yl) oxy) methoxy) methyl group
- PMM2 ((2-cyanopropoxy) methoxy) methyl group
- CPM ((1-cyanopropan-2-yl) oxy) methyl group
- A Adenine
- G guanine
- C cytosine
- U uracil.
- Production Example 2 1) Production of cyanoacetone 3-Aminocrotononitrile (50 g, 0.609 mol) and 6N hydrochloric acid (143 mL) were mixed, stirred at a bath temperature of 60 ° C. for 1 hour, allowed to cool to room temperature, and the precipitate was removed by filtration. The reaction solution was extracted with dichloromethane (250 mL), and the organic layer was dried over magnesium sulfate. After distilling off the solvent with a rotary evaporator, the precipitate was removed by filtration and dried with a vacuum pump to obtain cyanoacetone (41.4 g, yield 81%, GC purity 99.4%).
- the solvent was distilled off with a rotary evaporator and a vacuum pump to obtain a yellow liquid (38 g).
- the resulting liquid was purified by distillation under reduced pressure (70 to 80 Pa, bath temperature 70 ° C.) to obtain 3-hydroxybutanenitrile (29.5 g, yield 69.5%, GC purity 99.1%).
- a 10 ° C. aqueous solution prepared in advance (21.8 g of sodium thiosulfate pentahydrate, 7.7 g of sodium bicarbonate, and 165 mL of water)
- the reaction solution was poured into the solution.
- Ethyl acetate (50 mL) was added, and the mixture was stirred at 10-15 ° C. for 30 minutes, filtered through celite (7.5 g), and separated.
- Production Example 8 Production of CPM agent A 2000 ml eggplant flask was charged with 51.0 g (600 mmol) of 3-hydroxybutanenitrile (3-HBN) and 640 ml (9 mol) of dimethyl sulfoxide, 350 ml (6.1 mol) of acetic acid, and 600 ml of acetic anhydride (6. 3 mol) was added. After stirring in nitrogen gas at room temperature for 15 minutes, the mixture was heated and stirred in an oil bath (50 ° C.) for 12 hours. After confirming disappearance of the raw material 3-HBN by GC, it was allowed to cool to room temperature.
- 3-HBN 3-hydroxybutanenitrile
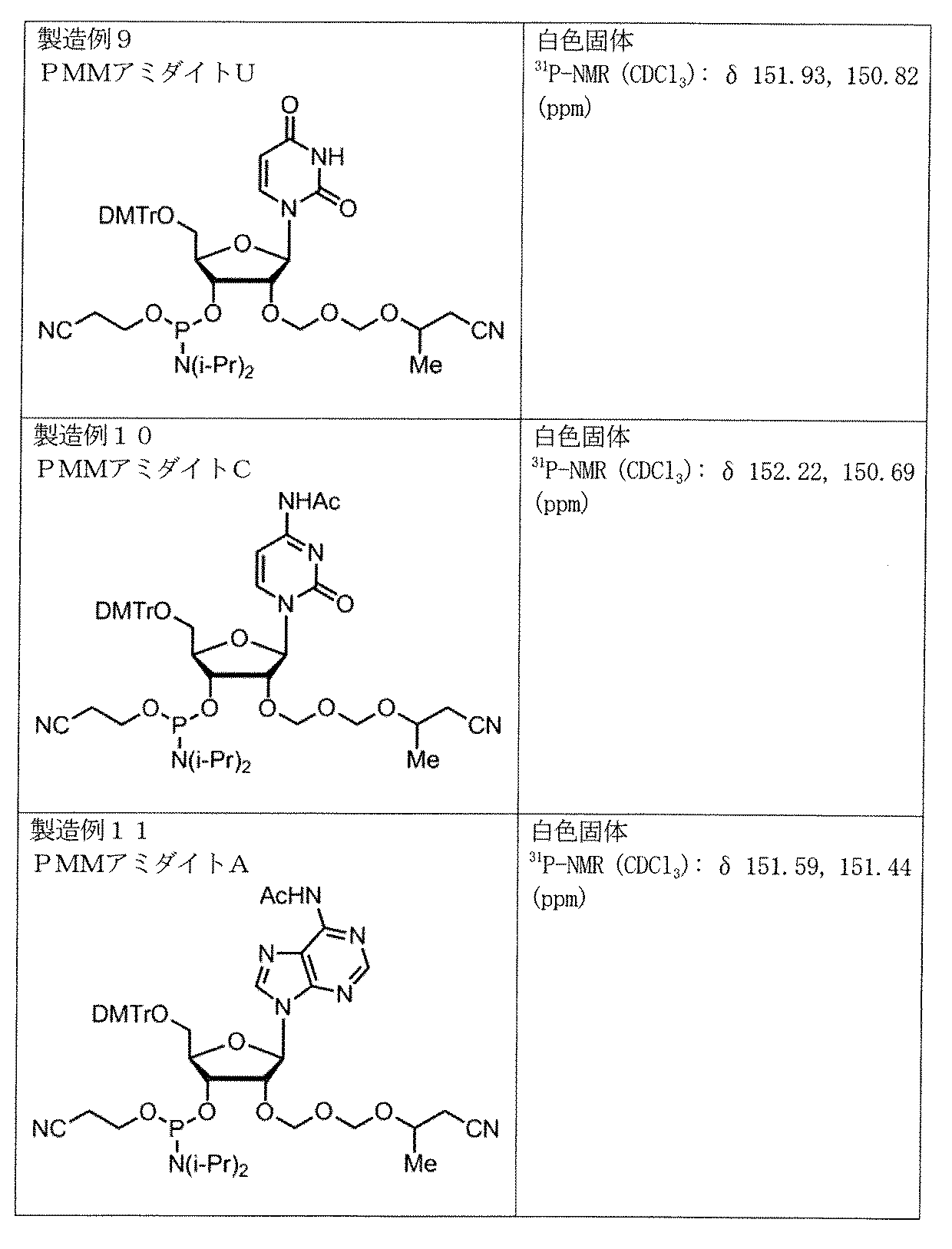
- Production and production example 9 of amidite A production example of PMM amidite U whose nucleobase moiety is uracil is shown below.
- TIPS-U (20.0 g), THF (40 ml) and toluene (60 ml) were charged into a container and concentrated to 74 ml for dehydration.
- the reaction solution was cooled to ⁇ 50 ° C., and a PMM agent (10.80 g), TfOH (12.48 g), and a NIS (12.4 g) / THF (26 ml) solution were added dropwise. After stirring at ⁇ 50 ° C. for 2 hours, the reaction solution was poured into an ice-cooled sodium bicarbonate (7.0 g) / sodium thiosulfate (20.0 g) / water (130 ml) solution, and the solution was separated at room temperature.
- the crude PMM-U-1 was dissolved in acetone (40 mL), hydrogen trifluoride / triethylamine (7.28 g) was added, and the mixture was stirred at 20 ° C. for 18 hours.
- the reaction mixture was poured into tert-butyl methyl ether (200 ml) and stirred for 30 minutes.
- the reaction mixture was filtered, and the solid remaining on the filter paper was washed with tert-butyl methyl ether (40 ml).
- the resulting precipitate was collected by filtration and dried under reduced pressure to give a crude product (14.56 g) containing the target compound.
- Production Example 10 A production example of PMM amidite C whose nucleobase moiety is cytosine is shown below.
- TIPS-C 50.0 g
- toluene 250 ml
- the solution was concentrated to 150 ml.
- THF 110 ml
- the reaction solution was cooled to ⁇ 50 ° C.
- a PMM agent (24.9 g)
- N-iodosuccinimide 28.8 g
- THF 65 ml
- trifluoromethanesulfonic acid 21 .3 g
- Acetonitrile (264 ml) was added to the PMM C-3 (44.0 g), and the mixture was concentrated to 132 ml.
- Diisopropylamine tetrazolide (12.1 g) and 2-cyanoethyl N, N, N ′, N′-tetraisopropyl phosphorodiamidite (22.3 g) were added at 25 ° C., and the mixture was stirred at 35 ° C. for 2 hours.
- the reaction solution was poured into a solution consisting of toluene (440 ml), water (220 ml) and sodium hydrogen carbonate (22 g), and separated at room temperature.
- Production Example 11 A production example of PMM amidite A in which the nucleobase moiety is adenine is shown below.
- TIPS-A (60.0 g) and toluene (350 ml) were added to the flask, and the solution was concentrated to 180 ml. After adding THF (120 ml), the reaction solution was cooled to ⁇ 10 ° C., and iodine (165.54 g) and a PMM agent (28.6 g) were added. After stirring at 0 ° C.
- the organic layer was washed 4 times with a 50% aqueous DMF solution (400 ml), twice with water (220 ml) and once with an aqueous sodium chloride solution (200 ml).
- Sodium sulfate (20 g) was added to the organic layer, filtered, and concentrated to 120 ml to obtain a crude product containing the target compound. Purification by silica gel column chromatography was performed to obtain the target compound PMM A amidite (46.35 g).
- Nucleic acid production example 1 Using the PMM amidite U, BMM amidite U, and TBM amidite U prepared in the above production example, a uridine 50-mer represented by the sequence of SEQ ID NO: 1 was synthesized. 5'-UUUUUUUUUUU UUUUUUUU UUUUUUUU UUUUUUUU UUUUUUUUU UUUUUUUUUU UUUUUUUUUU UUUUUUUUUUUU-3 '(sequence number 1) (In the formula, U means sodium uridine monophosphate)
- Synthesis was performed from the 3 'side to the 5' side using NTS M-4MX-E (manufactured by Nippon Techno Service Co., Ltd.) as a nucleic acid synthesizer.
- NTS M-4MX-E manufactured by Nippon Techno Service Co., Ltd.
- porous glass is used as the solid support
- high-purity trichloroacetic acid toluene solution is used as the deblocking solution
- 5-benzylmercapto-1H-tetrazole is used as the condensing agent
- iodine solution is used as the oxidizing agent
- a phenoxyacetic acid solution and an N-methylimidazole solution were used as a capping solution.
- the purity of the crude oligonucleotide product after solid phase synthesis was measured by HPLC.
- the crude product was separated into each component by HPLC (wavelength 260 nm, column ACQUITY UPLC Oigonucleotide BHE C18, 2.1 mm ⁇ 100 mm), and the purity of the oligonucleotide from the area value of the main product in the total area value of the obtained chromatogram was calculated.
- Example 1 As a result of synthesizing uridine 50-mer (molecular weight 15246.53) using PMM amidite U, OD 260 per 0.406 ⁇ mol was 97.48 OD, and the purity was 84.3%. From the value of OD 260 , the yield per ⁇ mol was calculated to be 9604 ⁇ g / ⁇ mol. The results are shown in Table 4.
- Example 2 Similarly, as a result of synthesizing uridine 50-mer (molecular weight 15246.53) using BMM amidite U, OD 260 per 0.200 ⁇ mol was 48.51 OD, and the purity was 72.5%. From the value of OD 260 , the yield per 1 mol was calculated to be 9702 ⁇ g / ⁇ mol. The results are shown in Table 4.
- Example 3 Similarly, uridine 50-mer (molecular weight 15246.53) was synthesized using TBM amidite U. As a result, OD 260 per 1.074 ⁇ mol was 249.74 OD, and the purity was 66.2%. From the value of OD 260 , the yield per 1 mol was calculated to be 9301 ⁇ g / ⁇ mol. The results are shown in Table 4.
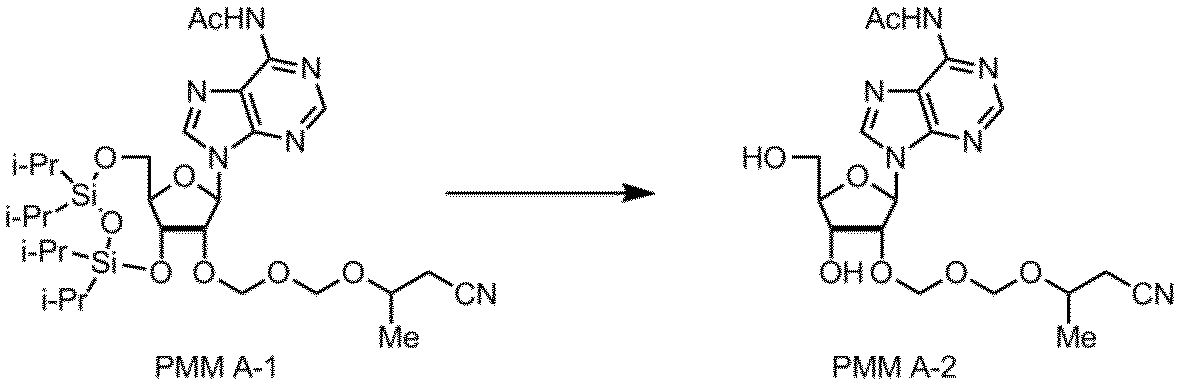
- Example 4 A method for producing a synthetic intermediate PMM A-1 for PMM amidite A in which the nucleobase moiety is adenine is shown below.
- TIPS-A 5.00 g
- toluene 25 ml
- Toluene (10 ml) was added to the flask and the solution was concentrated to 15 ml.
- 4-Methyltetrahydropyran (10 ml) was added as a solvent, and then the reaction solution was cooled to ⁇ 10 ° C., and iodine (13.8 g) and a PMM agent (2.38 g) were added. After stirring at 0 ° C.
- Example 5 PMM A-1 was obtained in the same manner as in Example 4 except that the same amount of tetrahydrofuran (THF) was used instead of 4-methyltetrahydropyran as the solvent. The results are shown in Table 5.
- THF tetrahydrofuran
- Example 6 In the method of Example 4, the same amount of dioxane was used as the solvent in place of 4-methyltetrahydropyran, and the reaction was performed in the same manner except that the stirring time was 3.5 hours to obtain PMM A-1. It was. The results are shown in Table 5. As shown in Table 5 above, good results in the production yield of the synthetic intermediate can be obtained.
- Nucleic acid production example 2 Sequence (A): 5′-AGCAGAGUACACACAGCAUAUACC-P-GGUAUAUGCUGUGUGUACUCUGCUUC-PG-3 ′ (SEQ ID NO: 2, 3) 53mer
- P is represented by a partial structure separated by a wavy line in the following chemical formula.
- AGCAGAGUAC ACACAGCAUA UACC SEQ ID NO: 2) GGUAUAUGCU GUGUGUACUC UGCUUC (SEQ ID NO: 3)
- AKTA Oligopilot 100plus manufactured by GE Healthcare
- the oligonucleotide consisting of the sequence (A) was synthesized from the 3 'side toward the 5' side by the phosphoramidite solid phase synthesis method.
- porous glass is used as a solid support
- a high-purity trichloroacetic acid toluene solution is used as a deblocking solution
- 5-benzylmercapto-1H-tetrazole is used as a condensate
- boron is used as an oxidizing agent.
- the solution was used, and phenoxyacetic acid solution and N-methylimidazole solution were used as capping solution.
- Example 7 and Comparative Example 3 the relative yield per 1 mol relative to the yield of Example 7 was determined, and the results are shown in Table 6.
- Example 7 Using the above PMM amidite U, PMM amidite C, PMM amidite A, PMM amidite G and compound (3) described in WO2017 / 188042, an oligonucleotide comprising the above sequence (A) was synthesized. As a result, the purity was 60.8. %Met. The results of purity and relative yield per ⁇ mol are shown in Table 6.
- Sequence number 1 of a sequence table shows the base sequence of uridine 50-mer.
- SEQ ID Nos. 2 and 3 in the sequence listing represent the nucleotide sequences of the oligonucleotides constituting the sequence produced in Nucleic Acid Production Example 2.
Abstract
高純度でのRNAの合成が可能となる式(1)で示されるアミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法を提供する。(式中、Rは(2)(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。nは1~5の整数を表す。)を表し、Baは保護されていてもよい核酸塩基骨格を有する基を表し、G1及びG2は同一又は相異なって水酸基の保護基を表し、G3は同一又は相異なってアルキル基を表す。)
Description
本特許出願は、日本国特許出願2018-083148号(2018年4月24日出願)および日本国特許出願2018-141559号(2018年7月27日出願)に基づくパリ条約上の優先権および利益を主張するものであり、ここに引用することによって、上記出願に記載された内容の全体が本明細書中に組み込まれるものとする。
本発明は、アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法に関する。さらに、本発明は、上記アミダイト化合物の中間体化合物及び該中間体化合物の製造方法に関する。
RNAは、RNAプローブ、アンチセンスRNA、リボザイム、siRNA、アプタマーなどとして利用可能であり、有用な素材である。
RNAは固相合成法などにより合成可能であり、固相合成法ではヌクレオシドのホスホロアミダイト(以下、「アミダイト」と称する)が原料として用いられる。このようなアミダイトの2’位の水酸基の保護基としては、例えば、TBDMS(t-ブチルジメチルシリル)、TOM(トリイソプロピルシリルオキシメチル)、ACE(ビス(2-アセトキシエトキシ)メチル)等が知られている。さらにアミダイトの2’位の水酸基の保護基として、特許文献1及び2が開示する保護基が報告されているが、これらの保護基を有するアミダイトを使用するRNAの合成方法は、得られるRNAの収率や純度の点で必ずしも満足のいくものではない。
本発明は、高純度でのRNAの合成が可能となるアミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法を提供することを目的とする。さらに、本発明は、上記アミダイト化合物の中間体化合物及び該中間体化合物の製造方法を提供することを目的とする。
本発明者らは、上記目的を達成すべく鋭意研究を重ねた結果、アミダイトの2’位の水酸基の保護基として、以下の基を使用することで高純度でのRNAの合成が可能となるという知見を得た。
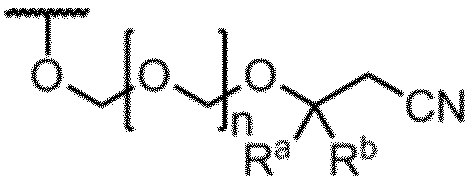
(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
nは1~5の整数を表す。)
nは1~5の整数を表す。)
本発明は、これら知見に基づき、更に検討を重ねて完成されたものであり、以下のアミダイト化合物、該化合物を用いたポリヌクレオチドの製造方法、エーテル化合物、及び該エーテル化合物の製造方法を提供するものである。
本発明は以下の項に記載する実施態様を含むが、これらに限定されるものではない。
項1. 式(1)で示されるアミダイト化合物。

(式中、Rは、式:

(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
nは1~5の整数を表す。)を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G1及びG2は同一又は相異なって水酸基の保護基を表し、
G3は同一又は相異なってアルキル基を表す。)
項2. Raがメチル基またはエチル基であり、Rbが水素原子である、項1に記載のアミダイト化合物。
項3. G1が以下の基である、項1又は2に記載のアミダイト化合物。

(式中、R1、R2及びR3は同一又は相異なって水素又はアルコキシ基を表す。)
項4. G2が以下の基である、項1~3のいずれか一項に記載のアミダイト化合物。

項5. G3がイソプロピル基である、項1~4のいずれか一項に記載のアミダイト化合物。
項6. 項1~5のいずれか一項に記載のアミダイト化合物を固相合成反応に供する工程を含む、式(2):
(式中、Baは同一又は相異なって保護されていてもよい核酸塩基骨格を有する基を表し、
Xは酸素原子又は硫黄原子を表し、
mは正の整数を表す。)
で示されるポリヌクレオチド骨格を含有する化合物の製造方法。
項7. 式(2)のポリヌクレオチド骨格を含有する化合物が、前記アミダイト化合物を用いる固相合成反応で生成する式(3):
(式中、Baは同一又は相異なって保護されていてもよい核酸塩基骨格を有する基を表し、
Xは酸素原子又は硫黄原子を表し、
Rは同一又は相異なって、式:

(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
nは1~5の整数を表す。)を表し、
mは正の整数を表す。)
で示されるオリゴヌクレオチド骨格を有する化合物にテトラアルキルアンモニウムフルオライドを反応させる工程で生成した化合物である、項6に記載の製造方法。
項8. Raがメチル基またはエチル基であり、Rbが水素原子である、項7に記載の製造方法。
項9. n=1である、項6~8のいずれか一項に記載の製造方法。
項10. 式(4)で示されるエーテル化合物。

(式中、Ra、Rbおよびnは、項1に定義のとおりであり、RcはC1~C6アルキル基、又はフェニル基を表す。)
項11. Raがメチル基またはエチル基であり、Rbが水素原子であり、Rcがメチルである、項10に記載のエーテル化合物。
項12. n=1である、項10または11に記載の化合物。
項13. 工程a:式(5):

(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。)
で示される化合物とシアン化物イオンとを反応させる工程、及び
工程b:酸化剤及び酸存在下、溶媒中で、工程aで得られた式(6):
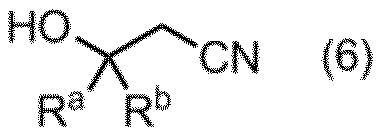
(式中、RaおよびRbは前記定義のとおりである。)
で示される3-ヒドロキシアルキルニトリルとビス(メチルチオメチル)エーテルとを反応させる工程、
を含む、式(7):

(式中、RaおよびRbは前記定義のとおりである。)
で示されるエーテル化合物の製造方法。
項14. Raがメチル基またはエチル基であり、Rbが水素原子である、項13に記載の製造方法。
項15. Raがメチル基であり、Rbが水素原子である、項13に記載の製造方法。
項16. 工程A:3-アミノクロトノニトリルを加水分解する工程、
工程B:工程Aで得られたシアノアセトンを還元して、3-ヒドロキシブタンニトリルを得る工程、および
工程C:酸化剤及び酸存在下、溶媒中で、工程Bで得られた3-ヒドロキシブタンニトリルとビス(メチルチオメチル)エーテルとを反応させる工程、
を含む、式(8):
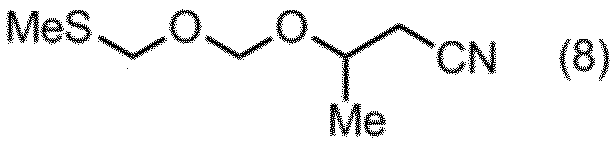
で示される項15に記載の製造方法。
項17. 式(9):
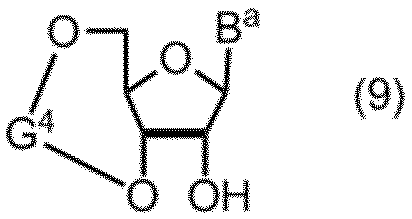
(式中、Baは、保護されていてもよい核酸塩基骨格を有する化合物を表し、G4は、水酸基の保護基を表す。)
で示される化合物を、酸化剤の存在下、式(4):

(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
RcはC1~C6アルキル基、又はフェニル基である。
nは、1~4の整数を表す。)
で示される化合物と、反応させることを特徴とする、式(10):

(式中、Ba、Ra、Rb、nおよびG4は、前記の定義のとおりである。)
で示される化合物の製造方法。
項18. 反応溶媒として、テトラヒドロピランまたは4-メチルテトラヒドロピランを用いる、項17に記載の製造方法。
項19. 式(10)の化合物をさらに脱保護して、式(11):

(式中、Ba、Ra、Rbおよびnは、前記の定義のとおりである。)
で示される化合物を得る工程、
式(11)の化合物の5’の水酸基を選択的に保護して、式(12):
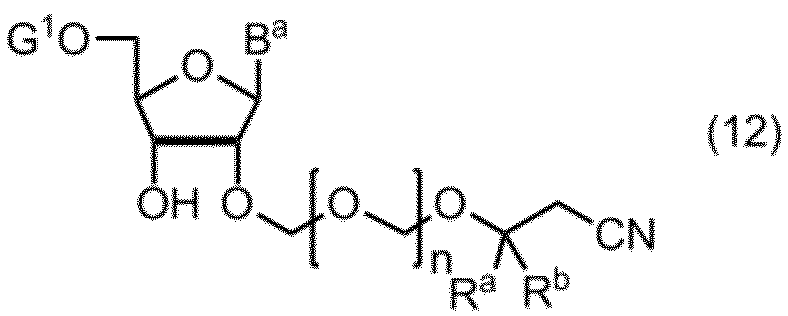
(式中、Ba、Ra、Rbおよびnは、前記の定義のとおりであり、G1は、水酸基の保護基を表す。)
で示される化合物を得る工程、
式(12)の化合物を式(13):

(式中、G2は水酸基の保護基を表し、G3は同一又は相異なってアルキル基を表す。)
で表されるホスホロジアミダイトと反応させる工程、
を含む、項1に記載の式(1)の化合物の製造方法。
項20. G4は、G4-1またはG4-2構造を有する項19に記載の製造方法。
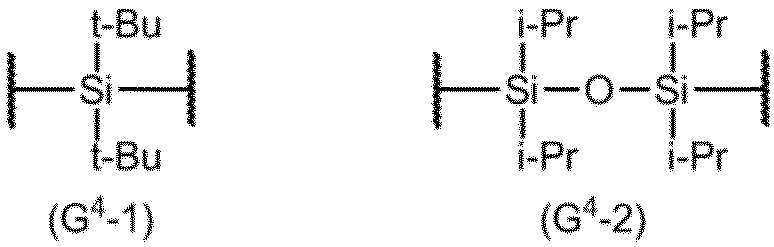
項21. 式(10):

(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
nは1~5の整数を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G4は、保護基を表す。)
で示される化合物。
項22. 式(11):
(式中、Ba、Ra、Rbおよびnは、項21に定義のとおりである。)
で示される化合物。
項23. 式(12):
(式中、Ba、Ra、Rbおよびnは、項21に定義のとおりであり、G1は、水酸基の保護基を表す。)
で示される化合物。
項24.前記式(1)のアミダイト化合物のRNAの製造における使用。
項1. 式(1)で示されるアミダイト化合物。
nは1~5の整数を表す。)を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G1及びG2は同一又は相異なって水酸基の保護基を表し、
G3は同一又は相異なってアルキル基を表す。)
項2. Raがメチル基またはエチル基であり、Rbが水素原子である、項1に記載のアミダイト化合物。
項3. G1が以下の基である、項1又は2に記載のアミダイト化合物。
項4. G2が以下の基である、項1~3のいずれか一項に記載のアミダイト化合物。
項6. 項1~5のいずれか一項に記載のアミダイト化合物を固相合成反応に供する工程を含む、式(2):
Xは酸素原子又は硫黄原子を表し、
mは正の整数を表す。)
で示されるポリヌクレオチド骨格を含有する化合物の製造方法。
項7. 式(2)のポリヌクレオチド骨格を含有する化合物が、前記アミダイト化合物を用いる固相合成反応で生成する式(3):
Xは酸素原子又は硫黄原子を表し、
Rは同一又は相異なって、式:
nは1~5の整数を表す。)を表し、
mは正の整数を表す。)
で示されるオリゴヌクレオチド骨格を有する化合物にテトラアルキルアンモニウムフルオライドを反応させる工程で生成した化合物である、項6に記載の製造方法。
項8. Raがメチル基またはエチル基であり、Rbが水素原子である、項7に記載の製造方法。
項9. n=1である、項6~8のいずれか一項に記載の製造方法。
項10. 式(4)で示されるエーテル化合物。
項11. Raがメチル基またはエチル基であり、Rbが水素原子であり、Rcがメチルである、項10に記載のエーテル化合物。
項12. n=1である、項10または11に記載の化合物。
項13. 工程a:式(5):
で示される化合物とシアン化物イオンとを反応させる工程、及び
工程b:酸化剤及び酸存在下、溶媒中で、工程aで得られた式(6):
で示される3-ヒドロキシアルキルニトリルとビス(メチルチオメチル)エーテルとを反応させる工程、
を含む、式(7):
で示されるエーテル化合物の製造方法。
項14. Raがメチル基またはエチル基であり、Rbが水素原子である、項13に記載の製造方法。
項15. Raがメチル基であり、Rbが水素原子である、項13に記載の製造方法。
項16. 工程A:3-アミノクロトノニトリルを加水分解する工程、
工程B:工程Aで得られたシアノアセトンを還元して、3-ヒドロキシブタンニトリルを得る工程、および
工程C:酸化剤及び酸存在下、溶媒中で、工程Bで得られた3-ヒドロキシブタンニトリルとビス(メチルチオメチル)エーテルとを反応させる工程、
を含む、式(8):
項17. 式(9):
で示される化合物を、酸化剤の存在下、式(4):
RcはC1~C6アルキル基、又はフェニル基である。
nは、1~4の整数を表す。)
で示される化合物と、反応させることを特徴とする、式(10):
で示される化合物の製造方法。
項18. 反応溶媒として、テトラヒドロピランまたは4-メチルテトラヒドロピランを用いる、項17に記載の製造方法。
項19. 式(10)の化合物をさらに脱保護して、式(11):
で示される化合物を得る工程、
式(11)の化合物の5’の水酸基を選択的に保護して、式(12):
で示される化合物を得る工程、
式(12)の化合物を式(13):
で表されるホスホロジアミダイトと反応させる工程、
を含む、項1に記載の式(1)の化合物の製造方法。
項20. G4は、G4-1またはG4-2構造を有する項19に記載の製造方法。
nは1~5の整数を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G4は、保護基を表す。)
で示される化合物。
項22. 式(11):
で示される化合物。
項23. 式(12):
で示される化合物。
項24.前記式(1)のアミダイト化合物のRNAの製造における使用。
本発明のアミダイト化合物によれば、固相合成法においてRNAを高い純度で簡便に製造することが可能となる。
以下、本発明について詳細に説明する。
なお、本明細書において「含む(comprise)」とは、「本質的にからなる(essentially consist of)」という意味と、「のみからなる(consist of)」という意味をも包含する。
本発明のアミダイト化合物は、式(1)で表されることを特徴とする。
(式中、Rは、
(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
nは1~5の整数を表す。)を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G1及びG2は同一又は相異なって水酸基の保護基を表し、
G3は同一又は相異なってアルキル基を表す。)
nは1~5の整数を表す。)を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G1及びG2は同一又は相異なって水酸基の保護基を表し、
G3は同一又は相異なってアルキル基を表す。)
Baにおける核酸塩基は、特に限定されない。当該核酸塩基としては、アデニン、シトシン、グアニン、ウラシル、チミン、5-メチルシトシン、シュードウラシル、1-メチルシュードウラシルなどが挙げられる。また、核酸塩基は、置換基により置換されていてもよい。そのような置換基としては、例えば、ハロゲン原子、アシル基、アルキル基、アリールアルキル基、アルコキシ基、アルコキシアルキル基、シアノアルキル基、ヒドロキシ基、ヒドロキシメチル基、アシルオキシメチル基、アミノ基、モノアルキルアミノ基、ジアルキルアミノ基、カルボキシ基、シアノ基、およびニトロ基など、並びにそれらの2種類以上の置換基の組み合わせが挙げられる。
核酸塩基が環外にアミノ基を有する場合、当該アミノ基の保護基としては、特に限定されず、公知の核酸化学で用いられる保護基を使用することができ、そのような保護基としては、例えば、メチル基、ベンゾイル基、4-メトキシベンゾイル基、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、フェニルアセチル基、フェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、4-イソプロピルフェノキシアセチル基、および(ジメチルアミノ)メチレン基など、並びにそれらの2種類以上の保護基の組み合わせが挙げられる。
Baは、より具体的には下記式
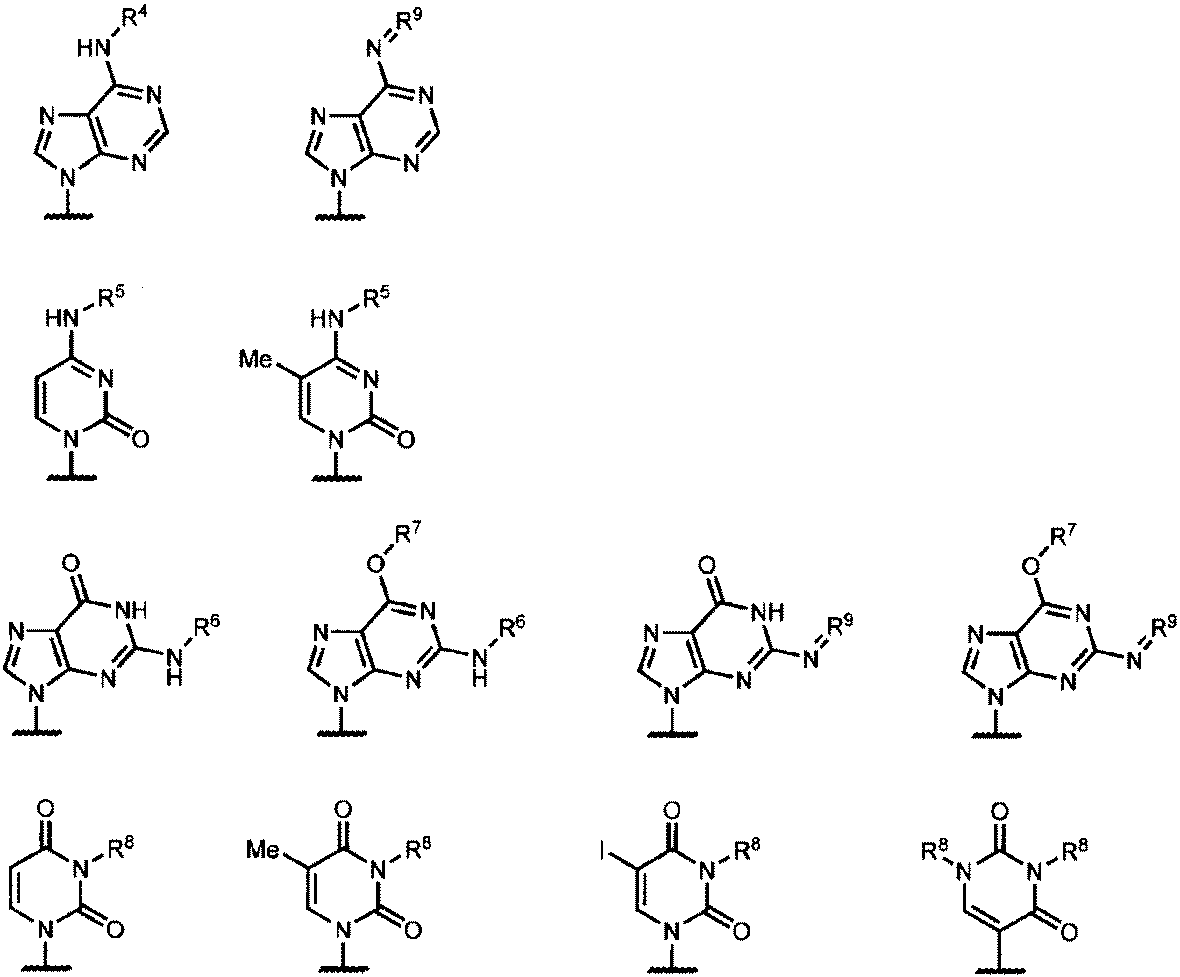
(式中、R4は水素原子、メチル基、フェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、4-イソプロピルフェノキシアセチル基、フェニルアセチル基、アセチル基又はベンゾイル基を表し、
R5は水素原子、アセチル基、イソブチリル基又はベンゾイル基を表し、
R6は水素原子、フェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、4-イソプロピルフェノキシアセチル基、フェニルアセチル基、アセチル基又はイソブチリル基を表し、
R7は2-シアノエチル基を表し、
R8は水素原子、メチル基、ベンゾイル基、4-メトキシベンゾイル基又は4-メチルベンゾイル基を表し、
R9はジメチルアミノメチレン基を表す。)
のいずれかで表される基を表す。
R5は水素原子、アセチル基、イソブチリル基又はベンゾイル基を表し、
R6は水素原子、フェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、4-イソプロピルフェノキシアセチル基、フェニルアセチル基、アセチル基又はイソブチリル基を表し、
R7は2-シアノエチル基を表し、
R8は水素原子、メチル基、ベンゾイル基、4-メトキシベンゾイル基又は4-メチルベンゾイル基を表し、
R9はジメチルアミノメチレン基を表す。)
のいずれかで表される基を表す。
G1としては、保護基として機能し得るものであれば特に制限なく使用することができ、アミダイト化合物で使用される公知の保護基を広く使用することができる。
G1としては、好ましくは、以下の基である。
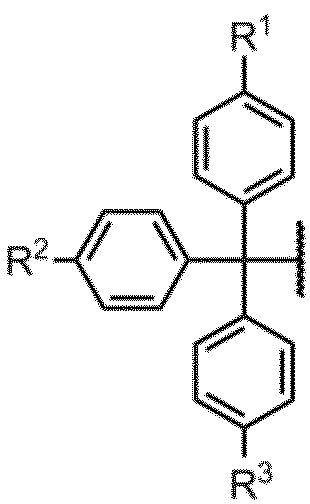
(式中、R1、R2及びR3は同一又は相異なって水素又はアルコキシ基を表す。)
R1、R2及びR3は、1つが水素であり、残りの2つがアルコキシ基であることが好ましく、アルコキシ基としてはメトキシ基が特に好ましい。
G2としては、保護基として機能し得るものであれば特に制限なく使用することができ、アミダイト化合物で使用される公知の保護基を広く使用することができる。G2としては、例えば、水素原子、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、ハロアルキル基、アリール基、ヘテロアリール基、アリールアルキル基、シクロアルケニル基、シクロアルキルアルキル基、シクリルアルキル基、ヒドロキシアルキル基、アミノアルキル基、アルコキシアルキル基、ヘテロシクリルアルケニル基、ヘテロシクリルアルキル基、ヘテロアリールアルキル基、シリル基、シリルオキシアルキル基、モノ、ジ又はトリアルキルシリル基、モノ、ジ又はトリアルキルシリルオキシアルキル基などが挙げられ、これらは1つ以上の電子求引基で置換されていてもよい。
G2としては、好ましくは、電子求引基で置換されたアルキル基である。当該電子求引基としては、例えば、シアノ基、ニトロ基、アルキルスルホニル基、ハロゲン、アリールスルホニル基、トリハロメチル基、トリアルキルアミノ基などが挙げられ、好ましくはシアノ基である。
G2としては、特に好ましくは、以下の基である。

G3は、2つのG3が互いに結合して環状構造を形成していてもよい。G3としては、両方がイソプロピル基であることが好ましい。
アルキル基は、直鎖状又は分岐鎖状のいずれでもよく、好ましくは炭素数1~12のアルキル基、より好ましくは炭素数1~6のアルキル基である。アルキル基としては、例えば、メチル、エチル、n-プロビル、イソプロピル、n-ブチル、イソブチル、tert-ブチル、n-ペンチル、イソペンチル、及びヘキシルが挙げられる。ここでのアルキル基には、アルコキシ基などのアルキル部分も含まれる。
Raは、好ましくはメチルである。nは、好ましくは1~4の整数、より好ましくは1~3の整数、更に好ましくは1又は2、特に好ましくは1である。
また、本発明のアミダイト化合物は、フリーの状態又は塩の状態で使用することができる。本発明のアミダイト化合物の塩としては、特に制限されず、例えば、ナトリウム塩、マグネシウム塩、カリウム塩、カルシウム塩、アルミニウム塩等の無機塩基との塩;メチルアミン、エチルアミン、エタノールアミン等の有機塩基との塩;リジン、オルニチン、アルギニン等の塩基性アミノ酸との塩及びアンモニウム塩が挙げられる。当該塩は、酸付加塩であってもよく、かかる塩としては、具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等の鉱酸;ギ酸、酢酸、プロピオン酸、シュウ酸、マロン酸、リンゴ酸、酒石酸、フマル酸、コハク酸、乳酸、マレイン酸、クエン酸、メタンスルホン酸、トリフルオロメタンスルホン酸、エタンスルホン酸等の有機酸;および、アスパラギン酸、グルタミン酸等の酸性アミノ酸との酸付加塩が挙げられる。本発明のアミダイト化合物には、塩、水和物、溶媒和物、結晶多形なども含まれる。
本発明のアミダイト化合物は、特許第5157168号公報、特許第5554881号公報などに記載された公知の方法や、後述する実施例に記載の方法に則して又は必要によりこれらの方法に適宜変更を加えた方法により製造することができる。
また、本発明のアミダイト化合物の具体例としては、実施例の表1に示す化合物が挙げられる。
本発明には、式(1)で示されるアミダイト化合物の製造中間体化合物も包含される。そのような中間体化合物としては、式(4)で示されるエーテル化合物が挙げられる。
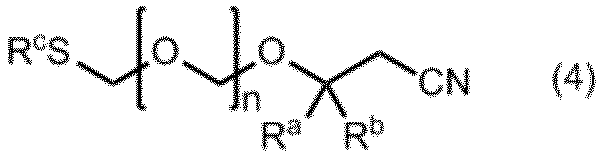
(式中、RaおよびRbは同一又は相異なってメチル基、エチル基又は水素原子を表す。ただしRaおよびRbが同時に水素原子を表すことはない。
nは1~5の整数を表す。
RcはC1~C6アルキル基、又はフェニル基である。)
nは1~5の整数を表す。
RcはC1~C6アルキル基、又はフェニル基である。)
式(4)で示されるエーテル化合物は、例えばビス(アルキルチオメチル)エーテルまたはビス(フェニルチオメチル)エーテルと3-ヒドロキシ-3-アルキルプロパンニトリルとを、酸化剤及び酸存在下、溶媒中で反応させることにより製造することができる。
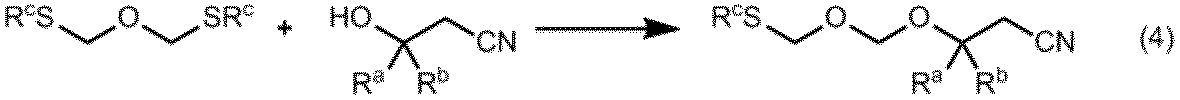
ビス(アルキルチオメチル)エーテルまたはビス(フェニルチオメチル)エーテルは、例えば下式に示す通り、ビスクロロメチルエーテルまたはビス(アリールオキシメチル)エーテルと対応するアルキルメルカプタンまたはフェニルメルカプタンとを反応させることによって得ることができる。ビス(アリールオキシメチル)エーテルとしては例えば、ビス(2,4,6-トリクロロフェニルオキシメチル)エーテルが挙げられる。
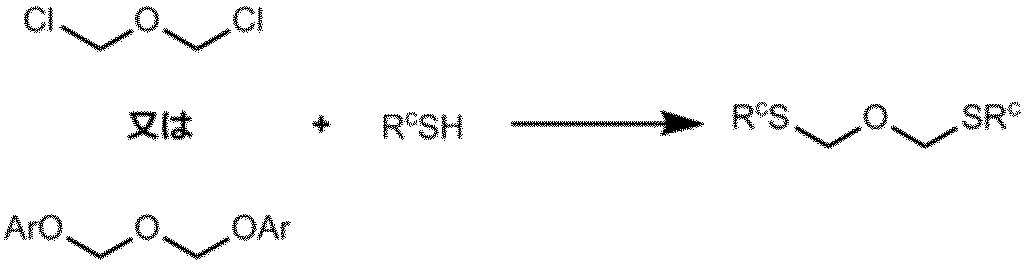
また後述する実施例に記載の方法に則して又は必要によりこれらの方法に適宜変更を加えた方法によっても製造することができる。
式(4)のエーテル化合物の一例である式(7)の化合物を製造する工程aおよび工程bについて、説明する。
工程aについて以下説明する。
シアン化物イオンとしては、例えば、シアン化ナトリウム、シアン化カリウム、シアン化銅、トリメチルシリルシアニドなどに由来するシアン化物イオンを使用することができる。
シアン化物イオンとしては、例えば、シアン化ナトリウム、シアン化カリウム、シアン化銅、トリメチルシリルシアニドなどに由来するシアン化物イオンを使用することができる。
トリメチルシリルシアニドを用いる場合、塩基を添加することが好ましい。
本反応における塩基としては、例えば、アルカリ金属水酸化物、アルカリ土類金属水酸化物、および水酸化アンモニウムなど、並びにこれらの2種類以上の組み合わせが挙げられる。本発明においては、水酸化リチウム及び水酸化リチウム一水和物が好ましく用いられる。塩基の量は、式(5)で示されるエポキシ化合物に対して、通常0.01~1当量であり、好ましくは0.1~0.3当量である。
シアン化物イオンの量は、式(5)で示される化合物に対して、通常0.3~2当量であり、好ましくは0.6~0.8当量である。
本反応の反応温度は、通常-20~40℃であり、好ましくは0~35℃である。本反応の反応時間は、通常0.5~24時間であり、好ましくは通常1~5時間である。
工程bについて、以下説明する。
酸化剤としては、例えば、N-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミド等のN-ハロゲン化スクシンイミド、1,3-ジヨード-5,5-ジメチルヒダントイン等のN-ハロゲン化ヒダントイン、および塩素、臭素、ヨウ素等のハロゲン等、並びにこれらの2種類以上の組み合わせが挙げられる。本発明においては、N-ハロゲン化スクシンイミドが好ましく用いられ、N-ヨードスクシンイミドが更に好ましく用いられる。
酸化剤としては、例えば、N-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミド等のN-ハロゲン化スクシンイミド、1,3-ジヨード-5,5-ジメチルヒダントイン等のN-ハロゲン化ヒダントイン、および塩素、臭素、ヨウ素等のハロゲン等、並びにこれらの2種類以上の組み合わせが挙げられる。本発明においては、N-ハロゲン化スクシンイミドが好ましく用いられ、N-ヨードスクシンイミドが更に好ましく用いられる。
酸は、特に限定されないが、例えば、パーフルオロアルキルカルボン酸及びその塩、パーフルオロアルキルスルホン酸及びその塩、並びにアルキルスルホン酸及びその塩、並びにこれらの2種類以上の組み合わせが挙げられる。塩としては、例えば、銅塩及び銀塩が挙げられる。酸としては、具体的には、メタンスルホン酸、パラトルエンスルホン酸、カンファースルホン酸、トリフルオロメタンスルホン酸、およびトリフルオロメタンスルホン酸銀など、並びにこれらの2種類以上の組み合わせが挙げられる。本発明においては、トリフルオロメタンスルホン酸が好ましく用いられる。
溶媒としては、例えば、テトラヒドロフラン、2-メチルテトラヒドロフラン、シクロペンチルメチルエーテル、ジオキサン、ジクロロメタン、およびトルエンなど、並びにこれらの2種類以上の組み合わせが挙げられる。本発明においては、テトラヒドロフランが好ましく用いられる。
3-ヒドロキシアルキルニトリルの量は、ビス(アルキルチオメチル)エーテルもしくはビス(フェニルチオメチル)エーテルに対して、通常0.5~2.0当量であり、好ましくは0.8~1.5当量である。酸化剤の量は、ビス(アルキルチオメチル)エーテルもしくはビス(フェニルチオメチル)エーテルに対して、通常0.5~2当量であり、好ましくは0.7~1.2当量である。酸の量は、ビス(アルキルチオメチル)エーテルもしくはビス(フェニルチオメチル)エーテルに対して、通常0.001~2.0当量であり、好ましくは0.01~0.1当量である。
本反応の反応温度は、通常-80℃~0℃であり、好ましくは-50℃~-30℃である。本反応の反応時間は、通常1~24時間であり、好ましくは2~6時間である。
反応の終了は例えば、反応マスの一部をサンプリングし、GC、TLC、LC等の分析法により確認することができる。反応終了後は、反応マスにトリエチルアミン等の塩基を加えて反応を停止させてもよい。反応マスを水に注加し、有機溶媒抽出、洗浄、濃縮等の通常の後処理操作に付すことにより、式(7)で示されるエーテル化合物を含む残渣を得ることができる。当該残渣を、蒸留やカラムクロマトグラフィー等の精製操作に付すことにより、高純度の式(7)で示されるエーテル化合物を得ることができる。
前記態様において、好ましくは、Raは、メチル基またはエチル基であり、Rbは、水素原子であり、Rcはメチル基である。より好ましくは、Raは、メチル基であり、Rbは、水素原子であり、Rcはメチル基である。
以下のような方法で製造することもできる。
これらの化合物のうち、式(8)の化合物は、以下の工程A~Cを含む製造方法により製造することができる。

工程A:3-アミノクロトノニトリルを加水分解する工程、
工程B:工程Aで得られたシアノアセトンを還元する工程、及び
工程C:酸化剤及び酸存在下、溶媒中で、工程Bで得られた3-ヒドロキシブタンニトリルとビス(メチルチオメチル)エーテルとを反応させる工程。
工程B:工程Aで得られたシアノアセトンを還元する工程、及び
工程C:酸化剤及び酸存在下、溶媒中で、工程Bで得られた3-ヒドロキシブタンニトリルとビス(メチルチオメチル)エーテルとを反応させる工程。
工程A:

本反応では、3-アミノクロトノニトリルを加水分解することでシアノアセトンを得ることができる。
加水分解は、例えば、3-アミノクロトノニトリルと酸を水の存在下、混合することにより行うことができる。酸としては、含水酸であっても、または無水酸であってもよく、例えば、塩酸、硫酸、メタンスルホン酸などが挙げられる。本発明においては、塩酸が好ましく用いられる。本反応は、酸として無水酸を使用する場合には、水を添加することによる水の存在下で行い、一方で、酸として含水酸を使用する場合には、水を添加してもしなくてもよい。
酸の量は、3-アミノクロトノニトリルに対して、通常1~10当量であり、好ましくは1~1.5当量である。
本反応の反応温度は、通常-20~100℃であり、好ましくは0~85℃である。本反応の反応時間は、通常1~24時間であり、好ましくは1~4時間である。
工程B:

工程Bの還元工程で用いる還元剤としては、例えば、水素化ホウ素ナトリウムなどの金属還元剤が挙げられる。
還元剤の量は、シアノアセトンに対して、通常0.25~2当量であり、好ましくは0.5~1当量である。
本反応は溶媒中で行うことができ、溶媒としては、例えば、テトラヒドロフラン、2-メチルテトラヒドロフラン、テトラヒドロピラン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、ジオキサンなどのエーテル系溶媒、及びメタノール、エタノールなどのアルコール系溶媒、並びにこれら溶媒の2種類以上の組み合わせが挙げられる。本発明においては、テトラヒドロフランが好ましく用いられる。
また、本還元工程は、酵母を用いるなどの生物学的な方法によっても行うことができる。
本反応の反応温度は、通常-20~60℃であり、好ましくは0~35℃である。本反応の反応時間は、通常0.5~24時間であり、好ましくは通常1~4時間である。
工程C:
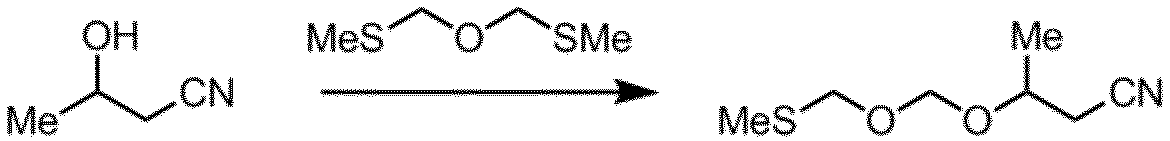
工程Cの反応は、酸化剤(例えば、ハロゲン化剤)存在下にアルコールにビス(メチルチオメチル)エーテルを反応させて実施される。かかるハロゲン化剤としては、例えば、N-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミド等のN-ハロゲン化スクシンイミド、1,3-ジヨード-5,5-ジメチルヒダントイン等のN-ハロゲン化ヒダントイン、塩素、臭素、ヨウ素等のハロゲン等が挙げられる。本発明においては、N-ハロゲン化スクシンイミドが好ましく用いられ、N-ヨードスクシンイミドが更に好ましく用いられる。
この反応において用いる酸の種類は、特に限定されないが、例えば、パーフルオロアルキルカルボン酸及びその塩、パーフルオロアルキルスルホン酸及びその塩、並びにアルキルスルホン酸及びその塩が挙げられる。塩としては、例えば、銅塩及び銀塩が挙げられる。酸としては、具体的には、メタンスルホン酸、パラトルエンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロメタンスルホン酸銀などが挙げられる。本発明においては、トリフルオロメタンスルホン酸が好ましく用いられる。
溶媒としては、例えば、テトラヒドロフラン、2-メチルテトラヒドロフラン、テトラヒドロピラン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、ジオキサン、ジクロロメタン、トルエンなどが挙げられる。本発明においては、テトラヒドロフランが好ましく用いられる。
3-ヒドロキシブタンニトリルの量は、ビス(メチルチオメチル)エーテルに対して、通常0.5~2.0当量であり、好ましくは0.8~1.5当量である。酸化剤の量は、ビス(メチルチオメチル)エーテルに対して、通常0.5~2当量であり、好ましくは0.7~1.2当量である。酸の量は、ビス(メチルチオメチル)エーテルに対して、通常0.001~2.0当量であり、好ましくは0.01~0.1当量である。
本反応の反応温度は、通常-80℃~0℃であり、好ましくは-50℃~-30℃である。本反応の反応時間は、通常1~24時間であり、好ましくは2~6時間である。
反応の終了は例えば、反応マスの一部をサンプリングし、GC、TLC、LC等の分析法により確認することができる。反応終了後は、反応マスにトリエチルアミン等の塩基を加えて反応を停止させてもよい。反応マスを水に注加し、有機溶媒抽出、洗浄、濃縮等の通常の後処理操作に付すことにより、式(8)で示されるエーテル化合物を含む残渣を得ることができる。当該残渣を、蒸留やカラムクロマトグラフィー等の精製操作に付すことにより、高純度の式(8)で示されるエーテル化合物を得ることができる。
本発明のアミダイト化合物は、固相合成法においてRNAを製造するための材料として使用することができる。本発明のアミダイト化合物を固相合成法において使用することで、高い純度でRNAを製造することが可能となる。
本発明の下記式(2)で示されるポリヌクレオチド骨格を含有する化合物の製造方法は、上記のアミダイト化合物を用いて固相合成反応を行う工程を含むことを特徴とする。
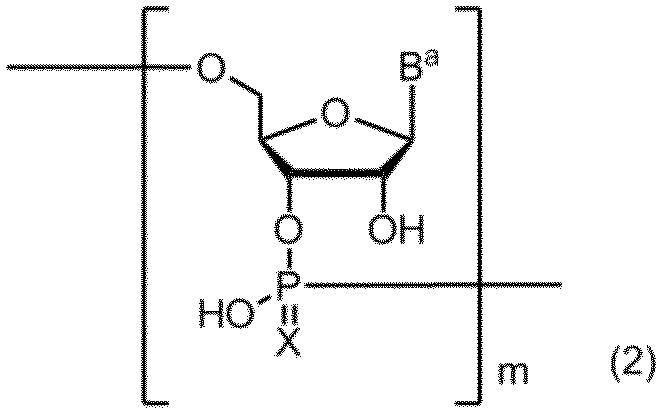
(式中、Baは同一又は相異なって保護されていてもよい核酸塩基骨格を有する基を表し、
Xは酸素原子又は硫黄原子を表し、
mは正の整数を表す。)
Xは酸素原子又は硫黄原子を表し、
mは正の整数を表す。)
また、本発明の製造方法は、式(3)で示されるオリゴヌクレオチド骨格を有する化合物をテトラアルキルアンモニウムフルオライドにより処理して式(2)で示されるオリゴヌクレオチド骨格を有する化合物を得る工程を含むこともできる。
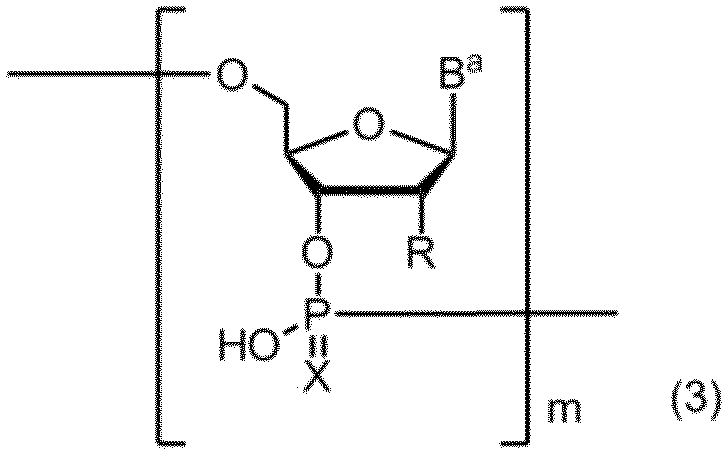
(式中、Baは同一又は相異なって保護されていてもよい核酸塩基骨格を有する基を表し、
Xは酸素原子又は硫黄原子を表し、
Rは同一又は相異なって、式:
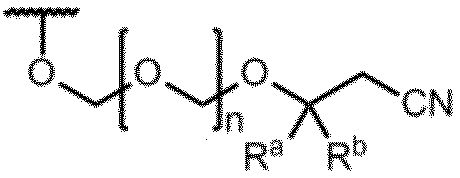
(式中、RaおよびRbは、同一又は相異なって、メチル基、エチル基または水素原子を表す。ただし、RaおよびRbが、同時に水素原子を表すことはない。
nは1~5の整数を表す。)
を表し、
mは正の整数を表す。)
Xは酸素原子又は硫黄原子を表し、
Rは同一又は相異なって、式:
nは1~5の整数を表す。)
を表し、
mは正の整数を表す。)
式(2)及び(3)のBa及びmは、式(1)のものと同様である。
mは、特に制限されず、好ましくは2~300の整数である。
本発明において「ポリヌクレオチド骨格を含有する化合物」とは、少なくとも1つのRNAを含む化合物であって、好ましくはRNAのみからなる化合物を意味する。
固相合成反応は、ホスホロアミダイト法などの公知の方法(例えば、特許第5157168号公報及び特許第5554881号公報に記載された方法)に従い実施することができる。また、市販されている核酸の自動合成装置等を用いて実施することができる。
式(2)で示されるポリヌクレオチド骨格を含有する化合物の製造方法は、具体的には、(A)固相担体に担持した第1のアミダイト化合物の5’位の水酸基(例えば、式(1)のG1)を脱保護する工程、(B)工程(A)で生成した脱保護したアミダイト化合物を第2のアミダイト化合物と縮合させる工程、(C)工程(B)における未反応の化合物の5’位の水酸基をキャッピングする任意の工程、(D)(B)あるいは(C)で生成した縮合物の亜リン酸基をリン酸基又はチオリン酸基に変換する工程、(E)工程(D)で得られた化合物を固相担体から切り出し、2’位及び核酸塩基の水酸基を脱保護する工程、(F)5’位の水酸基を脱保護する工程などの工程を含む。(A)~(D)の工程を繰り返すことにより、所望の鎖長のポリヌクレオチド骨格を含有する化合物(例えば、式(3)の化合物)を製造することができる。
式(3)で示されるオリゴヌクレオチド骨格を有する化合物を、好ましくはテトラアルキルアンモニウムフルオライドにより処理することにより、2’位の保護基が脱離され、式(2)で示されるオリゴヌクレオチド骨格を有する化合物を製造することができる。当該反応の反応条件(反応温度、反応時間、試薬の量など)は公知の方法に従った条件を採用することができる。
本発明の製造方法で得られた式(2)で示されるオリゴヌクレオチド骨格を有する化合物は、必要により単離及び精製を行い得る。通常、RNAを沈殿、抽出及び精製する方法を用いることで、単離することができる。具体的には、反応後の溶液にエタノール、イソプロピルアルコールなどのRNAに対して溶解性の低い溶媒を加えることでRNAを沈殿させる方法や、フェノール/クロロホルム/イソアミルアルコール(例えば、フェノール/クロロホルム/イソアミルアルコール=25/24/1)の溶液を反応溶液に加え、RNAを水層に抽出させる方法が採用される。その後、逆相カラムクロマトグラフィー、陰イオン交換カラムクロマトグラフィー、アフィニティカラムクロマトグラフィー等の公知の高速液体クロマトグラフィー(HPLC)の手法などにより単離、精製することができる。
本発明の製造方法により、従来より高純度でRNAを製造することが可能となる。
本発明アミダイト化合物および式(4)で表されるエーテル化合物の製造における反応条件は特に限定されない。式(4)で表されるエーテル化合物はフローリアクターを用いて合成することもできる。
本発明アミダイト化合物に含まれる不純物を削減する目的で、パラジウムなどの遷移金属触媒存在下、水素による還元工程あるいはマグネシウム等による還元工程を追加することもできる。
本発明アミダイト化合物および式(4)で表されるエーテル化合物の製造における反応条件は特に限定されない。式(4)で表されるエーテル化合物はフローリアクターを用いて合成することもできる。
本発明アミダイト化合物に含まれる不純物を削減する目的で、パラジウムなどの遷移金属触媒存在下、水素による還元工程あるいはマグネシウム等による還元工程を追加することもできる。
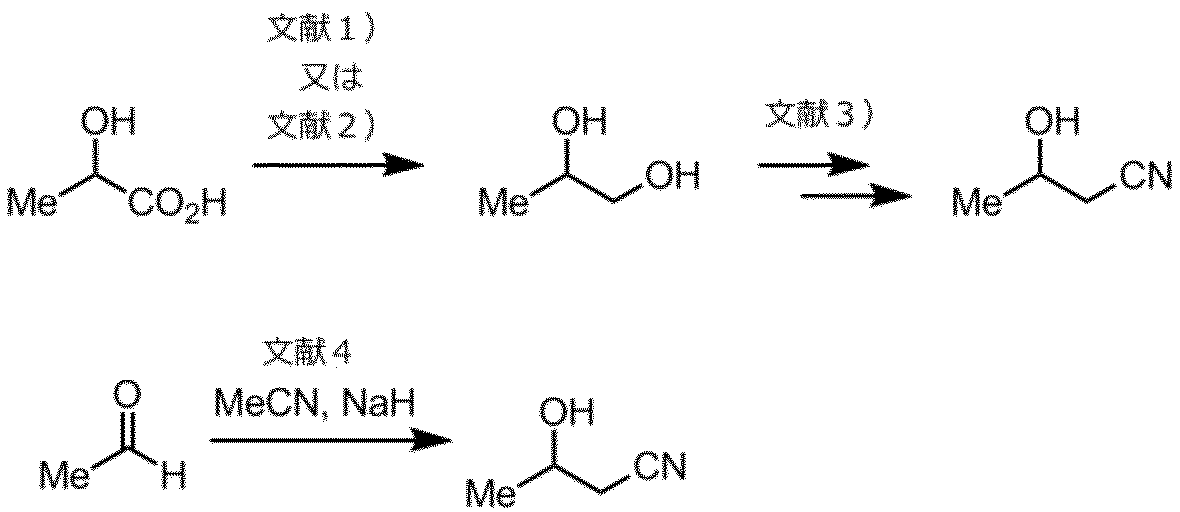
3-ヒドロキシブタンニトリルは、文献1)Chemical Communications, 2011, 47,(12), 3613、文献2)Applied Microbiology and Biotechnology, 2016, 100, (3), 1241、文献3)Journal of Organic Chemistry, 2010, 75(21), 7092、および文献4)中国特許 CN 107602497を参考に、乳酸あるいはアセトアルデヒドを原料とする下記の製造ルートによっても合成することができる。
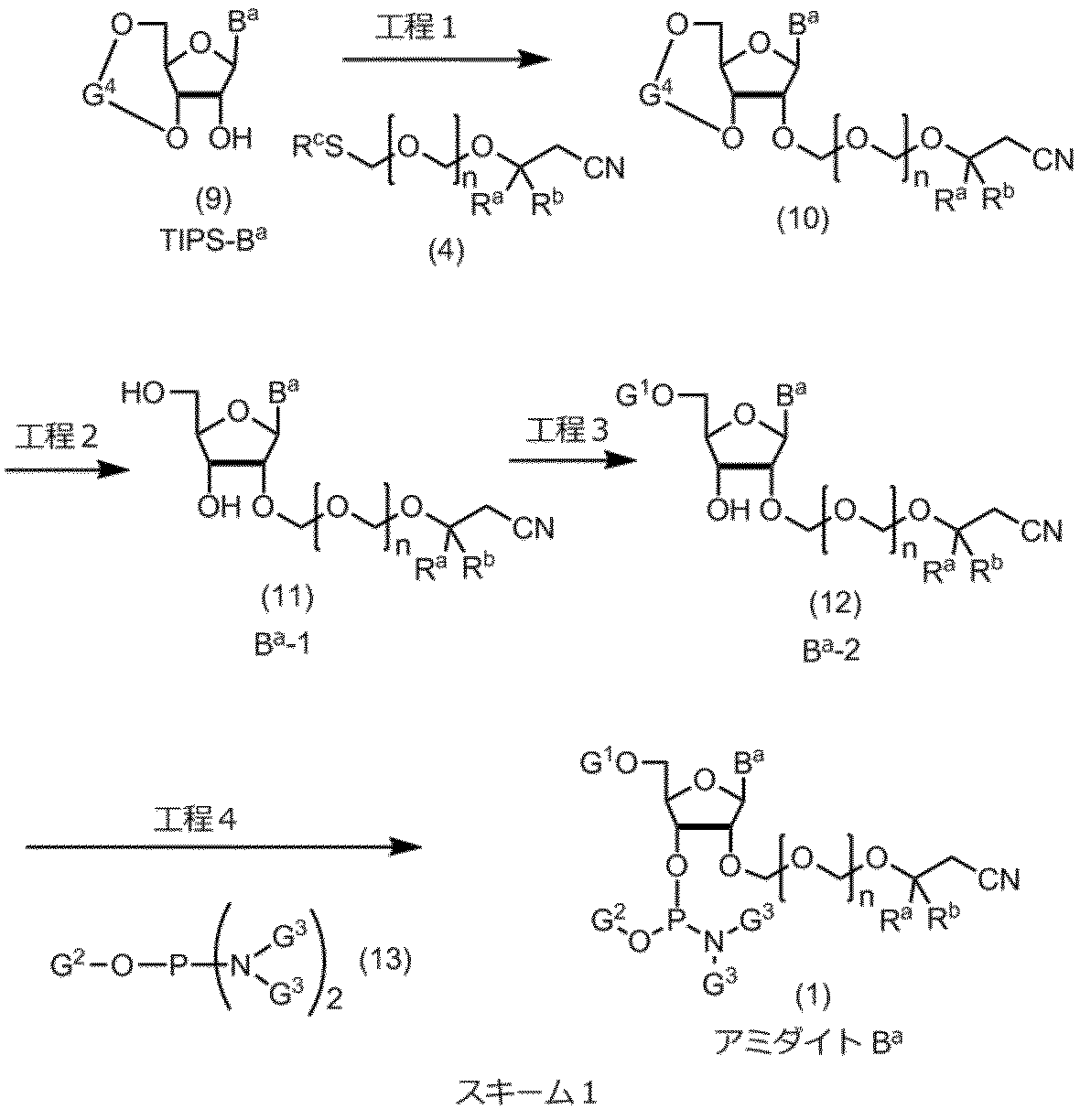
式(1)の化合物は、下記スキーム1の工程1、2、3および4により、式(9)の化合物から製造することができる。
式(9)の化合物において、Baは、前記と同じ意味を表し、G4は、典型的には、下記のG4-1またはG4-2構造を有する。

これらの化合物は、市販品を購入することもできるし、例えば、Tetrahedron Letters, 2005, 46, 2961に記載の方法により製造することもできる。
式(9)の化合物において、Baは、前記と同じ意味を表し、G4は、典型的には、下記のG4-1またはG4-2構造を有する。
工程1(エーテル化工程)
エーテル化工程は、式(9)の化合物を式(4)の化合物と反応させて実施される。この反応は、通常、酸化剤を添加して実施される。この工程において用いる酸化剤は、特に限定されないが、N-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミド、ヨウ素、1,3-ジヨード-5、5‘-ジメチルヒダントイン、臭素および塩素からなる群から選択される少なくとも一つであることが好ましい。
エーテル化工程は、式(9)の化合物を式(4)の化合物と反応させて実施される。この反応は、通常、酸化剤を添加して実施される。この工程において用いる酸化剤は、特に限定されないが、N-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミド、ヨウ素、1,3-ジヨード-5、5‘-ジメチルヒダントイン、臭素および塩素からなる群から選択される少なくとも一つであることが好ましい。
この工程においては、酸を添加することも可能であり、用いる酸は特に限定されないが、ペルフルオロアルキルカルボン酸、ペルフルオロアルキルスルホン酸、アルキルスルホン酸およびそれらの塩からなる群から選択される少なくとも一つであることが好ましい。
この工程において用いる反応溶媒は、特に限定されないが、例えば、ジエチルエーテル、THF(テトラヒドロフラン)、2-メチルテトラヒドロフラン、テトラヒドロピラン、4-メチルテトラヒドロピラン、ジメトキシエタン、ジグリム、シクロペンチルメチルエーテル、ジオキサン等のエーテル、またはアセ卜ニトリル等のニトリル、トルエン、クロロベンゼン、ジクロロベンゼン等の芳香族炭化水素、ジクロロメタン等、並びにこれら溶媒の2種類以上の組み合わせが挙げられる。好ましい溶媒としては、ジエチルエーテル、THF(テトラヒドロフラン)、2-メチルテトラヒドロフラン、テトラヒドロピラン、4-メチルテトラヒドロピラン、ジメトキシエタン、ジグリム、シクロペンチルメチルエーテル、ジオキサン等のエーテルが挙げられる。より好ましい溶媒は、テトラヒドロピラン、4-メチルテトラヒドロピランが挙げられる。
この工程において反応時聞は、特に限定されないが、例えば、10分~12時間、好ましくは10分~6時間である。
この工程において反応温度は、特に限定されないが、例えば-80~30℃、好ましくは、-60~10℃である。
この工程において前記式(4)で表されるエーテル化合物の濃度も、特に限定されず、適宜設定可能である。
この工程において前記式(4)で表されるエーテル化合物のモル数は、式(9)で表される化合物のモル数に対し、例えば0.5~2倍、好ましくは0.8~1.5倍である。
この工程において前記酸化剤のモル数は、式(9)で表される化合物のモル数に対し、例えば0.5~10倍、好ましくは0.8~6倍である。
工程2(脱保護工程)
前記工程1で得られた式(10)の化合物は、脱保護反応に供して式(11)の化合物に変換される。脱保護工程は、公知の方法で実施できるが、典型的には、溶媒中、フッ化水素/トリエチルアミンあるいはフッ化水素/ピリジンを作用させ、脱保護することができる。
前記工程1で得られた式(10)の化合物は、脱保護反応に供して式(11)の化合物に変換される。脱保護工程は、公知の方法で実施できるが、典型的には、溶媒中、フッ化水素/トリエチルアミンあるいはフッ化水素/ピリジンを作用させ、脱保護することができる。
工程3(5’水酸基の保護工程)
前記工程で得られた式(11)の化合物は、保護工程に供され、保護基の導入は、公知の方法で実施できるが、典型的には、ピリジン中、化合物(11)に4,4’-ジメトキシトリチルクロリドを反応させて保護基が導入され、化合物(12)が製造される。
前記工程で得られた式(11)の化合物は、保護工程に供され、保護基の導入は、公知の方法で実施できるが、典型的には、ピリジン中、化合物(11)に4,4’-ジメトキシトリチルクロリドを反応させて保護基が導入され、化合物(12)が製造される。
工程4(アミダイト化工程)
この工程は前記工程で得られた式(12)の化合物に、式(13)の化合物を反応させることによって実施される。典型的には、ジイソプロピルアンモニウムテトラゾリドの存在下、式(13)の化合物として2-シアノエチルーN,N,N’,N’-テトライソプロピルホスホロジアミダイトを反応させて行われる。アミダイト化は、特許第5554881号公報の実施例2~5に記載された方法に準じて行うことができる。
以上説明のとおり、式(9)、(10)、(11)および(12)の化合物は、式(1)のアミダイト化合物の製造に使用することができる。これらの化合物の好ましい態様においては、n=1である。
この工程は前記工程で得られた式(12)の化合物に、式(13)の化合物を反応させることによって実施される。典型的には、ジイソプロピルアンモニウムテトラゾリドの存在下、式(13)の化合物として2-シアノエチルーN,N,N’,N’-テトライソプロピルホスホロジアミダイトを反応させて行われる。アミダイト化は、特許第5554881号公報の実施例2~5に記載された方法に準じて行うことができる。
以下、本発明を更に詳しく説明するため実施例を挙げる。しかし、本発明はこれら実施例等になんら限定されるものではない。
本明細書中、以下の略号を使用する。
PMM=(((1-シアノプロパン-2-イル)オキシ)メトキシ)メチル基、BMM=(((1-シアノブタン-2-イル)オキシ)メトキシ)メチル基、TBM=(((1-シアノ-2-メチルプロパン-2-イル)オキシ)メトキシ)メチル基、PMM2=((2-シアノプロポキシ)メトキシ)メチル基、CPM=((1-シアノプロパン-2-イル)オキシ)メチル基;A=アデニン、G=グアニン、C=シトシン、U=ウラシル。
本明細書中、以下の略号を使用する。
PMM=(((1-シアノプロパン-2-イル)オキシ)メトキシ)メチル基、BMM=(((1-シアノブタン-2-イル)オキシ)メトキシ)メチル基、TBM=(((1-シアノ-2-メチルプロパン-2-イル)オキシ)メトキシ)メチル基、PMM2=((2-シアノプロポキシ)メトキシ)メチル基、CPM=((1-シアノプロパン-2-イル)オキシ)メチル基;A=アデニン、G=グアニン、C=シトシン、U=ウラシル。
PMMアミダイトの製造
製造例1
1)3-ヒドロキシブタンニトリルの製造
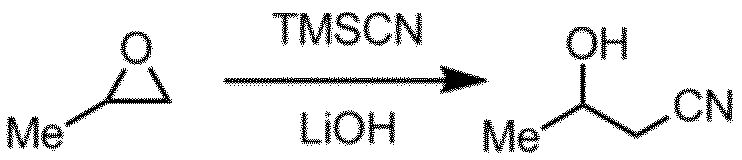
酸化プロピレン(12.4g、0.21mol)に水酸化リチウム一水和物(1.8g、42.8mmol)を加え、4℃に冷却し、トリメチルシリルシアニド(15.5g、0.15mol)をゆっくり滴下した。滴下完了し数十分経過後、内温が35℃まで上昇した。そのまま氷浴(内温5℃)で30分間、10~15℃にて1時間、更に室温(25℃)にて30分間攪拌した。反応液に水(15mL)を加え室温にて30分間攪拌した。その後、ジエチルエーテル(50mL×3回)抽出、飽和食塩水洗浄、無水硫酸ナトリウム乾燥を行い、溶媒を留去したところ、無色透明液状の粗3-ヒドロキシブタンニトリル10.6g(収率84%)を得た。
製造例1
1)3-ヒドロキシブタンニトリルの製造
2)PMM化剤の製造

ビス(メチルチオメチル)エーテル(32.41g、0.234mol、2.0eq.)をDry THF(300mL)に溶解、モレキュラーシーブス4A(32g)を加え10分間攪拌した。混合物を-50℃まで冷却し、トリフルオロメタンスルホン酸(TfOH)(0.88mL、5.85mmol、0.05eq.)とN-ヨードスクシンイミド(NIS)(31.5g、0.140mol、1.2eq.)とを添加、そこに粗3-ヒドロキシブタンニトリル(10g、0.117mol)を滴下し、-50~-45℃付近にて2時間攪拌した。反応液に飽和亜硫酸ナトリウム水溶液(150mL)、飽和炭酸水素ナトリウム(150mL)及び酢酸エチル(300mL)を加え、0~10℃で10分間攪拌した。分液後、有機層を飽和食塩水(150mL)にて洗浄し、無水硫酸マグネシウムにて乾燥後、減圧下溶媒を留去した。残渣をシリカゲルクロマトグラフィー精製(Hexane/AcOEt=10/1~5/1、シリカ800mL)し、黄色液状物のPMM化剤を5.9g(収率28%)得た。GC/FIDにて純度分析した結果、純度97.2%であった。
PMM化剤
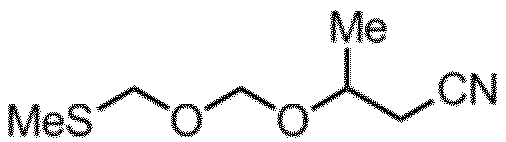
淡黄色透明液体
1H-NMR (CDCl3): δ 4.93-4.84 (m, 2H) 4.75(s, 2H) 4.06-4.00 (m, 1H) 2.57 (t, 2H), 2.17(s, 3H) 1.35 (d, 3H)
1H-NMR (CDCl3): δ 4.93-4.84 (m, 2H) 4.75(s, 2H) 4.06-4.00 (m, 1H) 2.57 (t, 2H), 2.17(s, 3H) 1.35 (d, 3H)
なお、ビス(メチルチオメチル)エーテルの製造方法は、特開2016-50203号公報を参照。
製造例2
1)シアノアセトンの製造
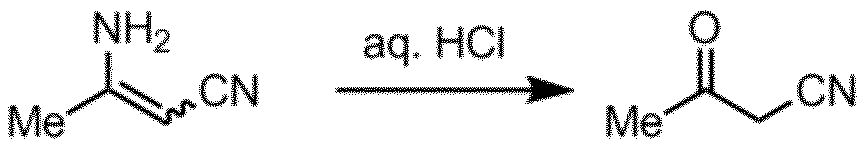
3-アミノクロトノニトリル(50g、0.609mol)と6N塩酸(143mL)を混合し、バス温度60℃にて1時間撹拌後、室温まで放冷、析出物をろ過により除去した。反応溶液をジクロロメタン(250mL)で抽出後、有機層を硫酸マグネシウムによって乾燥した。ロータリーエバポレーターで溶媒を留去後、析出物をろ過により除き、真空ポンプで乾燥してシアノアセトン(41.4g、収率81%、GC純度99.4%)を得た。
1)シアノアセトンの製造
2)3-ヒドロキシブタンニトリル
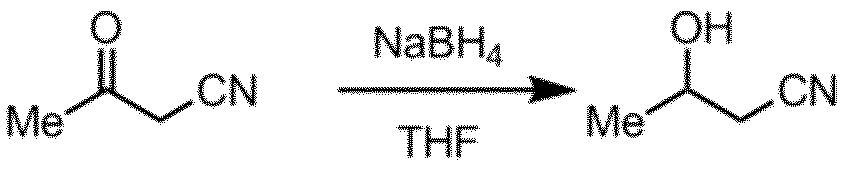
THF(207mL)に水素化ホウ素ナトリウム(11.35g、0.30mol)を添加し、氷浴で冷却後、シアノアセトン(41.4g、0.50mol)のTHF(41mL)溶液を滴下した(内温0~8℃、滴下時間35分)。氷浴下で更に30分間撹拌後、水(100mL)を添加し、酢酸エチル(200mL)で抽出した。有機層を飽和食塩水処理及び硫酸マグネシウムで乾燥後、析出した固体をろ過により除いた。ロータリーエバポレーター及び真空ポンプにより溶媒を留去し黄色液体(38g)を得た。得られた液体を減圧蒸留により精製し(70~80Pa、バス温度70℃)、3-ヒドロキシブタンニトリル(29.5g、収率69.5%、GC純度99.1%)を得た。
3)PMM化剤の製造

ビスメチルチオメチルエーテル(7.45g、53.9mmol)、モレキュラーシーブス4A(7.5g)及びTHF(111mL)を混合し、冷却後(-60~-55℃)、N-ヨードスクシンイミド(NIS)(14.4g、1.19equiv.)、トリフルオロメタンスルホン酸(TfOH)(143μL、0.030equiv.)及び3-ヒドロキシブタンニトリル(5.0g、1.09equiv.)を添加した。-50~-45℃にて4時間撹拌後、トリエチルアミン(5.1mL)を滴下した。反応容器を水浴に漬けて反応液を10℃付近まで昇温した後、事前に調整しておいた10℃の水溶液(チオ硫酸ナトリウム5水和物21.8gと重曹7.7gと水165mL)に反応液を注いだ。酢酸エチル(50mL)を加えて、10~15℃にて30分間撹拌し、セライト(7.5g)でろ過後分液し、有機層を飽和食塩水(30mL)にて洗浄、硫酸マグネシウム(3.8g)で乾燥、ロータリーエバポレーターで濃縮し、11.5gの粗生成物(11.5g,GC純度49.8%)を得た。シリカゲルカラム(シリカゲル300mL、ヘキサン/酢酸エチル=10/1)を用いて精製し、3-((メチルチオメトキシ)メトキシ)ブタンニトリル(淡黄色透明液体、5.32g、収率56%、純度99.5%)を得た。
製造例3~8(2‘位保護化剤の製造)
上記の実施例と同様にして(R)-PMM化剤、(S)-PMM化剤、BMM化剤、TBM化剤、PMM2化剤を製造した。また下記に記載の方法によってCPM化剤を製造した。
上記の実施例と同様にして(R)-PMM化剤、(S)-PMM化剤、BMM化剤、TBM化剤、PMM2化剤を製造した。また下記に記載の方法によってCPM化剤を製造した。
製造例3
(R)-PMM化剤(シグマアルドリッチ社(R)-(+)-プロピレンオキシドより合成)
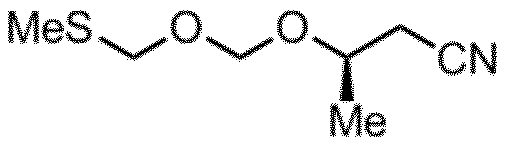
淡黄色透明液体
1H-NMR (CDCl3): δ4.92 (d,1H) 4.85(d,1H) 4.75 (s,2H) 4.03 (m,1H) 2.57 (m,2H) 2.17 (s,3H) 1.35 (d,3H)
(R)-PMM化剤(シグマアルドリッチ社(R)-(+)-プロピレンオキシドより合成)
1H-NMR (CDCl3): δ4.92 (d,1H) 4.85(d,1H) 4.75 (s,2H) 4.03 (m,1H) 2.57 (m,2H) 2.17 (s,3H) 1.35 (d,3H)
製造例4
(S)-PMM化剤(シグマアルドリッチ社(S)-(-)-プロピレンオキシドより合成)
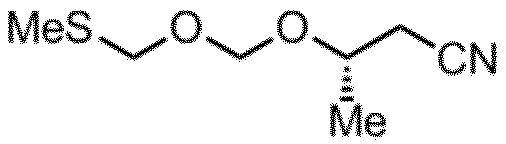
淡黄色透明液体
1H-NMR (CDCl3): δ4.92 (d,1H) 4.85 (d,1H) 4.75 (s,2H) 4.03 (m,1H) 2.57 (m,2H) 2.17 (s,3H) 1.34 (d,3H)
(S)-PMM化剤(シグマアルドリッチ社(S)-(-)-プロピレンオキシドより合成)
1H-NMR (CDCl3): δ4.92 (d,1H) 4.85 (d,1H) 4.75 (s,2H) 4.03 (m,1H) 2.57 (m,2H) 2.17 (s,3H) 1.34 (d,3H)
製造例5
BMM化剤

淡黄色透明液体
1H-NMR (CDCl3): δ4.95 (d,1H) 4.84 (d,1H) 4.76 (s,2H) 3.79 (m,1H) 2.60 (m,2H) 2.17 (s,3H) 1.70 (m,2H) 0.97 (t,3H)
BMM化剤
1H-NMR (CDCl3): δ4.95 (d,1H) 4.84 (d,1H) 4.76 (s,2H) 3.79 (m,1H) 2.60 (m,2H) 2.17 (s,3H) 1.70 (m,2H) 0.97 (t,3H)
製造例6
TBM化剤
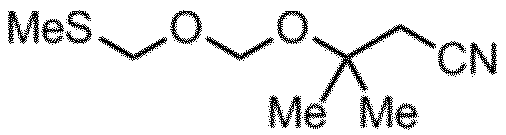
淡黄色透明液体
1H-NMR (CDCl3): δ 4.94 (s,2H) 4.77 (s,2H) 2.58 (s,2H) 2.16 (s,3H) 1.42 (s,3H)
TBM化剤
1H-NMR (CDCl3): δ 4.94 (s,2H) 4.77 (s,2H) 2.58 (s,2H) 2.16 (s,3H) 1.42 (s,3H)
製造例7
PMM2化剤

淡黄色透明液体
1H-NMR (CDCl3): δ4.86 (s, 2H) 4.73 (s, 2H) 3.71-3.61 (m, 2H) 2.94-2.87 (m, 1H) 2.18 (s, 3H) 1.35 (d, 3H)
PMM2化剤
1H-NMR (CDCl3): δ4.86 (s, 2H) 4.73 (s, 2H) 3.71-3.61 (m, 2H) 2.94-2.87 (m, 1H) 2.18 (s, 3H) 1.35 (d, 3H)
製造例8
CPM化剤の製造
2000mlのナスフラスコに3-ヒドロキシブタンニトリル(3-HBN)51.0g(600mmol)、ジメチルスルホキシド640ml(9mol)を入れ、酢酸 350ml(6.1mol)、無水酢酸 600ml(6.3mol)を加えた。窒素ガス中室温で15分間撹拌した後、油浴(50℃)で12時間加熱撹拌した。GCで原料3-HBNの消失を確認して、室温まで放冷した。重曹 1600g(19.5mol)をイオン交換水8Lに懸濁して、上記の反応液を徐々に添加した。発泡が治まってから、分液ロートに移して酢酸エチル 500mlで抽出した。有機層をイオン交換水 1Lで洗浄し、硫酸マグネシウムで乾燥、減圧濃縮して91.0gの淡黄褐色油状物を得た。
当該油状物をシリカゲルカラムで精製した(SiO2 1000ml、n-Hexane:AcOEt=20:1でスラリーを作り、充填した。)。サンプルをカラムにチャージして、n-Hexane:AcOEt=20:1で溶出した。1000ml溶出後、200ml分取(15本)した。n-Hexane:AcOEt=3:1で溶出後、200ml分取(15本)した。目的物のフラクションを合わせて減圧濃縮し、真空ラインで減圧乾燥した。53.28gの無色油状物を得た。
CPM化剤の製造
2000mlのナスフラスコに3-ヒドロキシブタンニトリル(3-HBN)51.0g(600mmol)、ジメチルスルホキシド640ml(9mol)を入れ、酢酸 350ml(6.1mol)、無水酢酸 600ml(6.3mol)を加えた。窒素ガス中室温で15分間撹拌した後、油浴(50℃)で12時間加熱撹拌した。GCで原料3-HBNの消失を確認して、室温まで放冷した。重曹 1600g(19.5mol)をイオン交換水8Lに懸濁して、上記の反応液を徐々に添加した。発泡が治まってから、分液ロートに移して酢酸エチル 500mlで抽出した。有機層をイオン交換水 1Lで洗浄し、硫酸マグネシウムで乾燥、減圧濃縮して91.0gの淡黄褐色油状物を得た。
当該油状物をシリカゲルカラムで精製した(SiO2 1000ml、n-Hexane:AcOEt=20:1でスラリーを作り、充填した。)。サンプルをカラムにチャージして、n-Hexane:AcOEt=20:1で溶出した。1000ml溶出後、200ml分取(15本)した。n-Hexane:AcOEt=3:1で溶出後、200ml分取(15本)した。目的物のフラクションを合わせて減圧濃縮し、真空ラインで減圧乾燥した。53.28gの無色油状物を得た。
CPM化剤
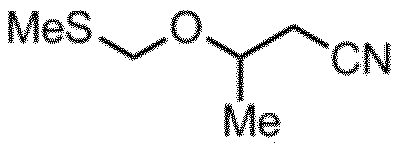
淡黄色透明液体
1H-NMR (CDCl3): δ4.72 (d,1H) 4.64 (d,1H) 4.15 (m,1H) 2.56 (m,2H) 2.20 (s,3H) 1.31 (d,3H)
1H-NMR (CDCl3): δ4.72 (d,1H) 4.64 (d,1H) 4.15 (m,1H) 2.56 (m,2H) 2.20 (s,3H) 1.31 (d,3H)
アミダイトの製造
製造例9
核酸塩基部分がウラシルであるPMMアミダイトUの製造例を以下に示す。
製造例9
核酸塩基部分がウラシルであるPMMアミダイトUの製造例を以下に示す。
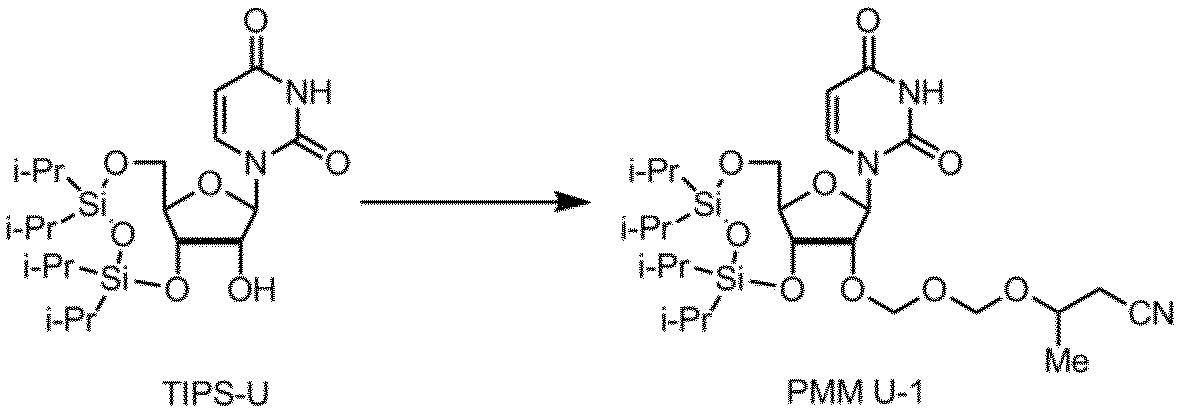
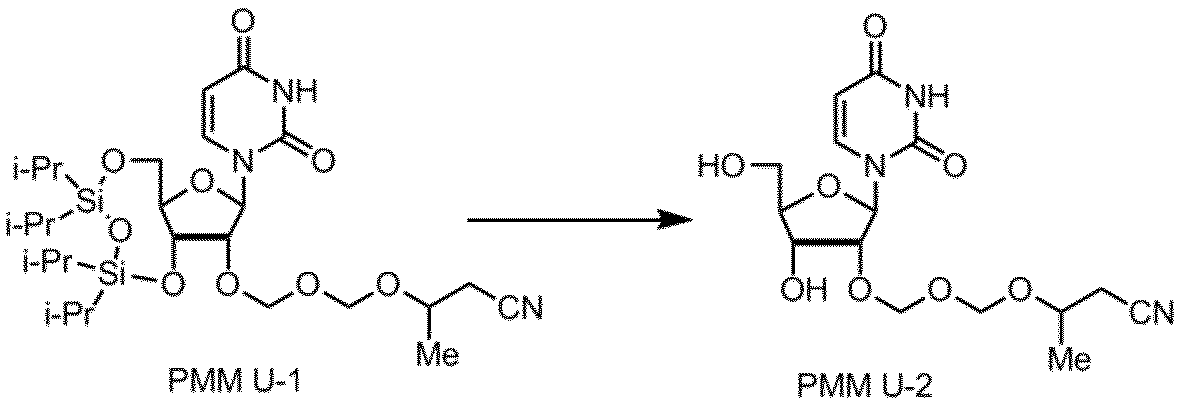
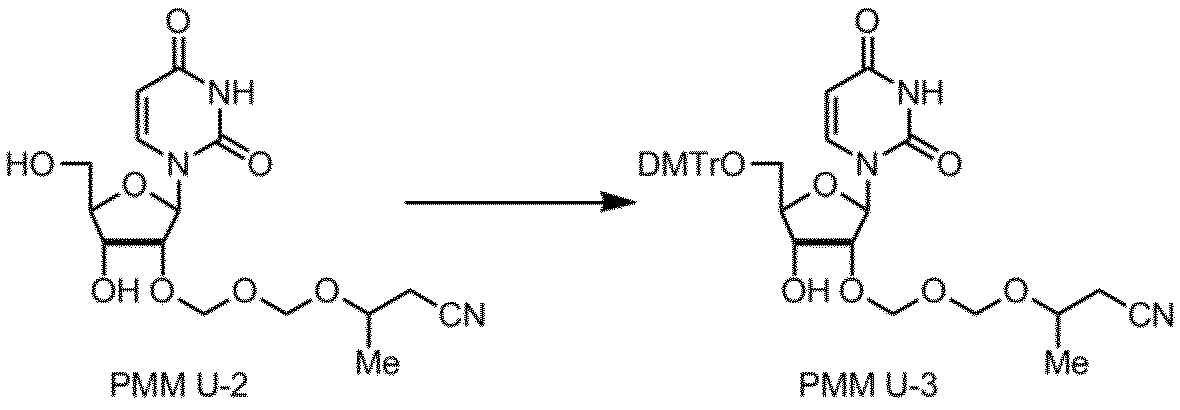

製造例10
核酸塩基部分がシトシンであるPMMアミダイトCの製造例を以下に示す。

TIPS-C(50.0g)、トルエン(250ml)をフラスコに加え、溶液を150mlまで濃縮した。THF(110ml)を加えた後、反応液を-50℃まで冷却し、PMM化剤(24.9g)、N-ヨードスクシンイミド(28.8g)のTHF(65ml)溶液、トリフルオロメタンスルホン酸(21.3g)の順に滴下した。-50℃で30分間攪拌後、反応液を炭酸水素ナトリウム(15.0g)、チオ硫酸ナトリウム(42.5g)、水(275ml)、トルエン(170ml)からなる氷冷した溶液へ注加し、室温にて分液した。次に有機層を炭酸水素ナトリウム(9.0g)、チオ硫酸ナトリウム(25.0g)および水(165ml)溶液で洗浄した。さらに有機層を塩化ナトリウム(25.0g)および水(250ml)溶液で洗浄し、有機層を150mlまで濃縮した。THF(200ml)を加えて150mlまで濃縮する操作を2回行い、目的化合物PMM C-1を含む粗生成物を得た。Exact Mass:654.3、Actual Mass:653.4(ESI(-))
核酸塩基部分がシトシンであるPMMアミダイトCの製造例を以下に示す。
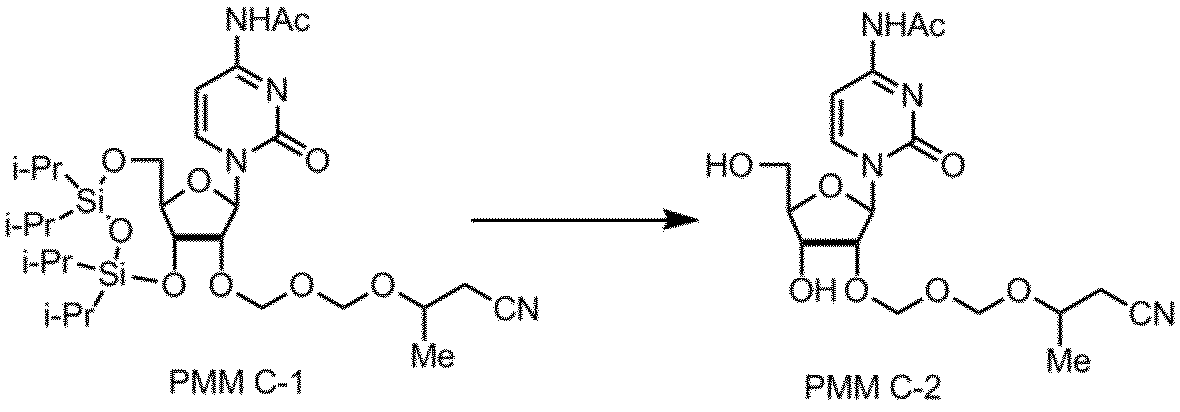

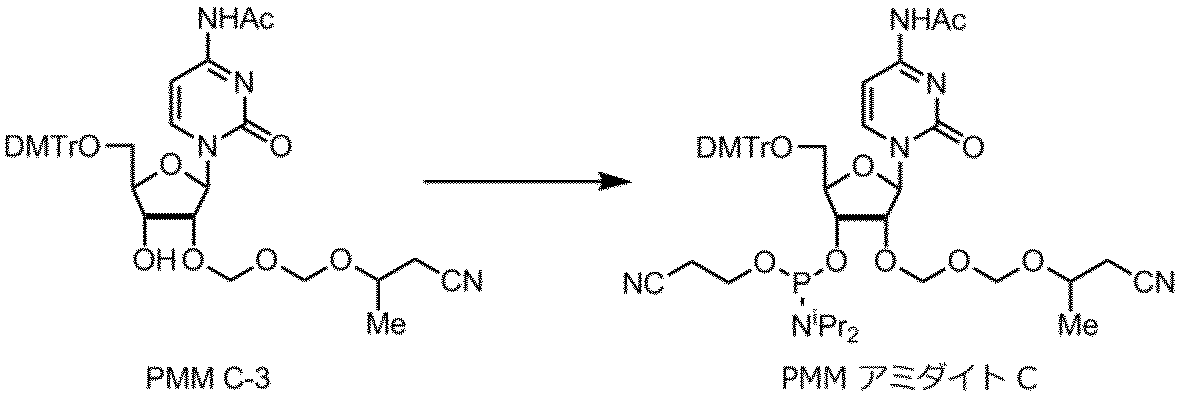
製造例11
核酸塩基部分がアデニンであるPMMアミダイトAの製造例を以下に示す。

TIPS-A(60.0g)、トルエン(350ml)をフラスコに加え、溶液を180mlまで濃縮した。THF(120ml)を加えた後、反応液を-10℃まで冷却し、ヨウ素(165.54g)、PMM化剤(28.6g)を添加した。0℃で2時間攪拌後、反応液を炭酸水素ナトリウム(33.6g)、チオ硫酸ナトリウム(336g)、水(480ml)、トルエン(180ml)からなる氷冷した溶液へ注加し、室温にて分液した。次に有機層を炭酸水素ナトリウム(16.8g)、チオ硫酸ナトリウム(168g)および水(240ml)溶液で洗浄した。さらに有機層を塩化ナトリウム(30.0g)および水(300ml)溶液で洗浄し、有機層を180mlまで濃縮し、目的化合物を含む粗生成物を得た。シリカゲルカラムクロマトグラフィーで精製して、目的物PMM A-1(51.62g)を得た。Exact Mass:678.3、Actual Mass:677.4(ESI(-))
核酸塩基部分がアデニンであるPMMアミダイトAの製造例を以下に示す。

製造例
製造例9~11のアミダイトのスペクトルデータを以下に示す。
製造例9~11のアミダイトのスペクトルデータを以下に示す。
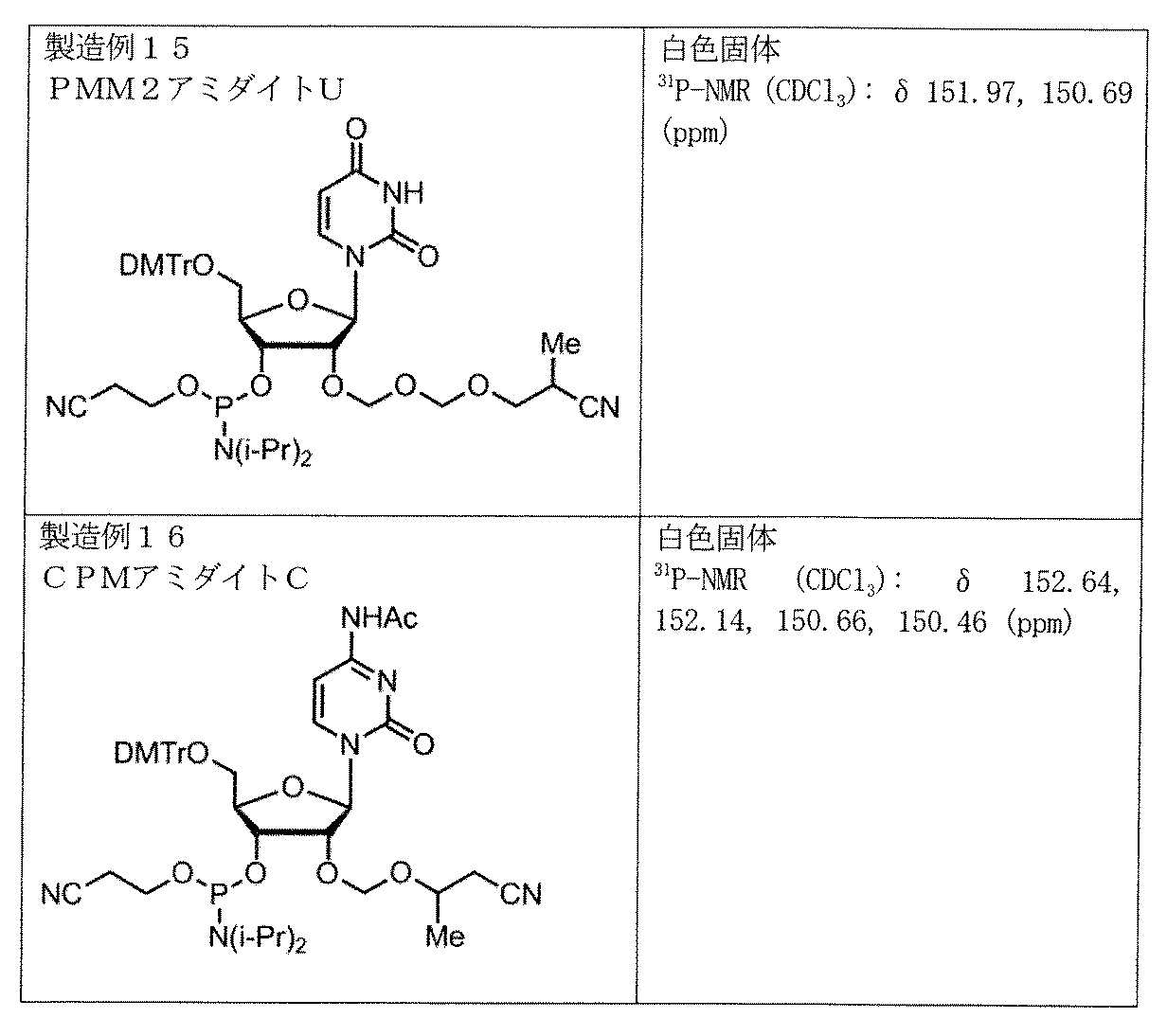
製造例12~16
上記の実施例と同様にしてPMMアミダイトG、BMMアミダイトU、TBMアミダイトU、及び参考化合物であるPMM2アミダイトU、CPMアミダイトCを製造した。
製造したアミダイトのスペクトルデータを以下に示す。

上記の実施例と同様にしてPMMアミダイトG、BMMアミダイトU、TBMアミダイトU、及び参考化合物であるPMM2アミダイトU、CPMアミダイトCを製造した。
製造したアミダイトのスペクトルデータを以下に示す。
核酸の製造例1
上記製造例で作製したPMMアミダイトU、BMMアミダイトUおよびTBMアミダイトUを用いて、下記配列番号1の配列で示されるウリジン50量体を合成した。
5’-UUUUUUUUUU UUUUUUUUUU UUUUUUUUUU UUUUUUUUUU UUUUUUUUUU-3’(配列番号1)
(式中、Uはウリジンモノリン酸ナトリウム塩を意味する)
上記製造例で作製したPMMアミダイトU、BMMアミダイトUおよびTBMアミダイトUを用いて、下記配列番号1の配列で示されるウリジン50量体を合成した。
5’-UUUUUUUUUU UUUUUUUUUU UUUUUUUUUU UUUUUUUUUU UUUUUUUUUU-3’(配列番号1)
(式中、Uはウリジンモノリン酸ナトリウム塩を意味する)
核酸合成機としてNTS M-4MX-E(日本テクノサービス株式会社製)を用いて3’側から5’側に向かって合成した。合成には固相担体として多孔質ガラスを使用し、デブロッキング溶液として高純度トリクロロ酢酸トルエン溶液を使用し、縮合剤として5-ベンジルメルカプト-1H-テトラゾールを使用し、酸化剤としてヨウ素溶液を使用し、キャッピング溶液としてフェノキシ酢酸溶液とN-メチルイミダゾール溶液とを使用して行った。
固相合成後のオリゴヌクレオチド粗生成物の純度の測定は、HPLCにより行った。粗生成物をHPLC(波長260nm、カラムACQUITY UPLC Oigonucleotide BHE C18,2.1mm×100mm)によって各成分に分離し、得られたクロマトグラムの総面積値における主生成物の面積値からオリゴヌクレオチドの純度を算出した。
実施例1
PMMアミダイトUを用いてウリジン50量体(分子量15246.53)を合成した結果、0.406μmolあたりのOD260は97.48ODであり、純度は84.3%であった。OD260の値から、1μmolあたりの収量は9604μg/μmolと算出された。結果を表4に示す。
PMMアミダイトUを用いてウリジン50量体(分子量15246.53)を合成した結果、0.406μmolあたりのOD260は97.48ODであり、純度は84.3%であった。OD260の値から、1μmolあたりの収量は9604μg/μmolと算出された。結果を表4に示す。
(OD260とは1mL溶液(pH=7.5)における10mm光路長あたりのUV260nmの吸光度である。一般的にRNAでは1OD=40μgであることが知られているから、吸光度よりRNAの生成量が算出できる。)
実施例2
同様にBMMアミダイトUを用いてウリジン50量体(分子量15246.53)を合成した結果、0.200μmolあたりのOD260は48.51ODであり、純度は72.5%であった。OD260の値から、1μmolあたりの収量は9702μg/μmolと算出された。結果を表4に示す。
同様にBMMアミダイトUを用いてウリジン50量体(分子量15246.53)を合成した結果、0.200μmolあたりのOD260は48.51ODであり、純度は72.5%であった。OD260の値から、1μmolあたりの収量は9702μg/μmolと算出された。結果を表4に示す。
実施例3
同様にTBMアミダイトUを用いてウリジン50量体(分子量15246.53)を合成した結果、1.074μmolあたりのOD260は249.74ODであり、純度は66.2%であった。OD260の値から、1μmolあたりの収量は9301μg/μmolと算出された。結果を表4に示す。
同様にTBMアミダイトUを用いてウリジン50量体(分子量15246.53)を合成した結果、1.074μmolあたりのOD260は249.74ODであり、純度は66.2%であった。OD260の値から、1μmolあたりの収量は9301μg/μmolと算出された。結果を表4に示す。
比較例1
一方、参考製造例で得られたPMM2アミダイトUを使用して、同様に固相合成を行い、ウリジン50量体を製造した結果、0.406μmolあたりのOD260は100.81ODであり、純度は50.4%であった。OD260の値から、1μmolあたりの収量は9932μg/μmolと算出された。結果を表4に示す。
一方、参考製造例で得られたPMM2アミダイトUを使用して、同様に固相合成を行い、ウリジン50量体を製造した結果、0.406μmolあたりのOD260は100.81ODであり、純度は50.4%であった。OD260の値から、1μmolあたりの収量は9932μg/μmolと算出された。結果を表4に示す。
比較例2
特許第5554881号公報実施例2に記載されているウリジンEMMアミダイトを用いてウリジン50量体(分子量15246.53)を合成した結果、1.028μmolあたりのOD260は245.91ODであり、純度は55.8%であった。OD260の値から、1μmolあたりの収量は9568μg/μmolと算出された。結果を表4に示す。
特許第5554881号公報実施例2に記載されているウリジンEMMアミダイトを用いてウリジン50量体(分子量15246.53)を合成した結果、1.028μmolあたりのOD260は245.91ODであり、純度は55.8%であった。OD260の値から、1μmolあたりの収量は9568μg/μmolと算出された。結果を表4に示す。
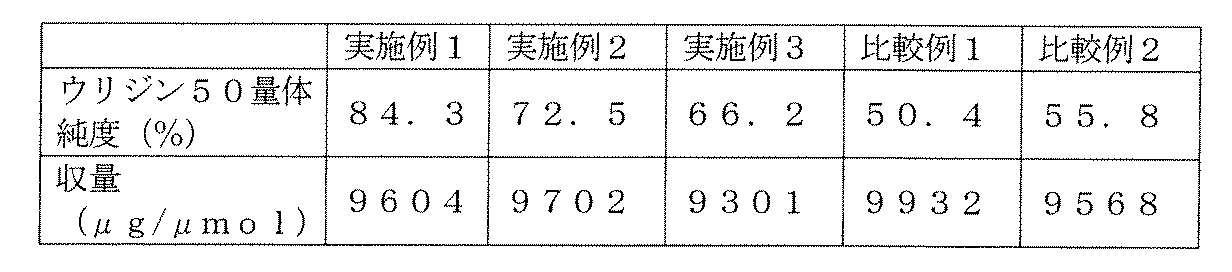
以上の表4に示すように、ウリジン50量体の純度の良好な結果を得ることができる。
実施例4
核酸塩基部分がアデニンであるPMMアミダイトAの合成中間体PMM A-1の製造方法を以下に示す。
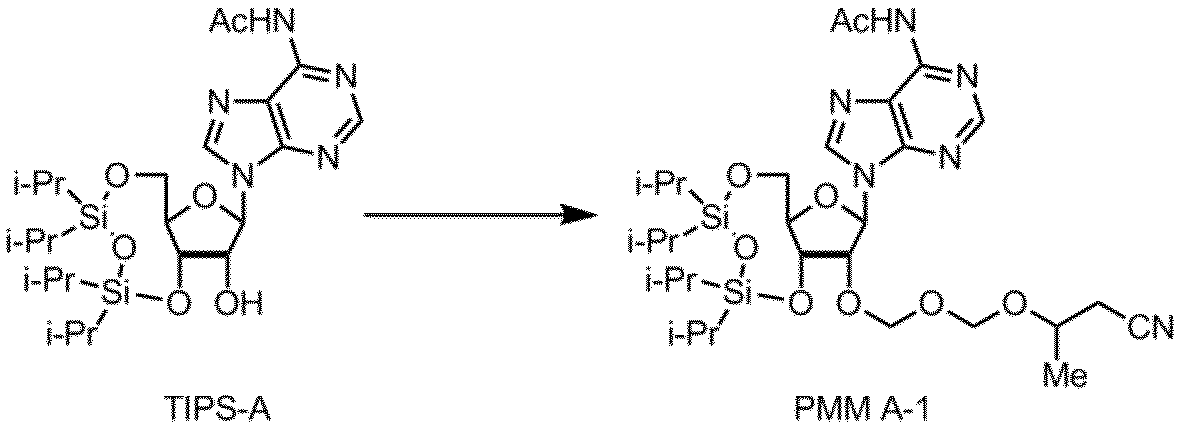
TIPS-A(5.00g)、トルエン(25ml)をフラスコに加え、溶液を15mlまで濃縮した。トルエン(10ml)をフラスコに加え、溶液を15mlまで濃縮した。4-メチルテトラヒドロピラン(10ml)を溶媒として加えた後、反応液を-10℃まで冷却し、ヨウ素(13.8g)、PMM化剤(2.38g)を添加した。0℃で1時間攪拌後、反応液を、炭酸水素ナトリウム(2.8g)、チオ硫酸ナトリウム(28g)、水(40ml)、トルエン(25ml)からなる氷冷した溶液へ注加し、室温にて分液した。有機層をLCで分析し、目的物であるPMM A-1の面積百分率を求めた。Exact Mass:678.3、Actual Mass:677.4(ESI(-))結果を表5に示す。
核酸塩基部分がアデニンであるPMMアミダイトAの合成中間体PMM A-1の製造方法を以下に示す。
実施例5
実施例4の方法において、溶媒として、4-メチルテトラヒドロピランの代わりに同量のテトラヒドロフラン(THF)を用いる以外は、同様の方法で反応させ、PMM A-1を得た。結果を表5に示す。
実施例4の方法において、溶媒として、4-メチルテトラヒドロピランの代わりに同量のテトラヒドロフラン(THF)を用いる以外は、同様の方法で反応させ、PMM A-1を得た。結果を表5に示す。
実施例6
実施例4の方法において、溶媒として、4-メチルテトラヒドロピランの代わりに同量のジオキサンを用い、攪拌時間を3.5時間とする以外は、同様の方法で反応させ、PMM A-1を得た。結果を表5に示す。

以上の表5に示すように、合成中間体の製造収量の良好な結果を得ることができる。
実施例4の方法において、溶媒として、4-メチルテトラヒドロピランの代わりに同量のジオキサンを用い、攪拌時間を3.5時間とする以外は、同様の方法で反応させ、PMM A-1を得た。結果を表5に示す。
以上の表5に示すように、合成中間体の製造収量の良好な結果を得ることができる。
核酸の製造例2
配列(A):5’-AGCAGAGUACACACAGCAUAUACC-P-GGUAUAUGCUGUGUGUACUCUGCUUC-P-G-3’(配列番号2,3) 53mer
前記配列(A)において、Pは、以下の化学式において波線で区切られる部分構造で示される。
AGCAGAGUAC ACACAGCAUA UACC(配列番号2)
GGUAUAUGCU GUGUGUACUC UGCUUC(配列番号3)
配列(A):5’-AGCAGAGUACACACAGCAUAUACC-P-GGUAUAUGCUGUGUGUACUCUGCUUC-P-G-3’(配列番号2,3) 53mer
前記配列(A)において、Pは、以下の化学式において波線で区切られる部分構造で示される。
AGCAGAGUAC ACACAGCAUA UACC(配列番号2)
GGUAUAUGCU GUGUGUACUC UGCUUC(配列番号3)
核酸合成機としてAKTA Oligopilot100plus(GEヘルスケア社製)を用いて、ホスホロアミダイト固相合成法により、上記配列(A)からなるオリゴヌクレオチドを3’側から5’側に向かって合成した。合成には固相担体として多孔質ガラスを使用し、デブロッキング溶液として高純度トリクロロ酢酸トルエン溶液を使用し、縮ヨ合剤として5-ベンジルメルカプト-1H-テトラゾールを使用し、酸化剤としてウ素溶液を使用し、キャッピング溶液としてフェノキシ酢酸溶液とN-メチルイミダゾール溶液を使用した。
[オリゴヌクレオチド収量の測定]
前記粗生成物のOD260を測定した。OD260とは1mL溶液(pH=7.5)における10mm光路長あたりのUV260nmの吸光度を表す。一般的にRNAでは1OD=40μgであることが知られていることから、前記OD260の測定値に基づき、収量を算出した。実施例7及び比較例3については実施例7の収量に対する1μmolあたりの相対収量を求め、その結果を表6に示した。
前記粗生成物のOD260を測定した。OD260とは1mL溶液(pH=7.5)における10mm光路長あたりのUV260nmの吸光度を表す。一般的にRNAでは1OD=40μgであることが知られていることから、前記OD260の測定値に基づき、収量を算出した。実施例7及び比較例3については実施例7の収量に対する1μmolあたりの相対収量を求め、その結果を表6に示した。
実施例7
上記PMMアミダイトU、PMMアミダイトC、PMMアミダイトA、PMMアミダイトGおよびWO2017/188042に記載の化合物(3)を使用し、上記配列(A)からなるオリゴヌクレオチドを合成した結果、純度は60.8%であった。純度と1μmolあたりの相対収量の結果を表6に示す。
上記PMMアミダイトU、PMMアミダイトC、PMMアミダイトA、PMMアミダイトGおよびWO2017/188042に記載の化合物(3)を使用し、上記配列(A)からなるオリゴヌクレオチドを合成した結果、純度は60.8%であった。純度と1μmolあたりの相対収量の結果を表6に示す。
比較例3
US2012/0035246の実施例2に記載のEMMアミダイトU、実施例3に記載のEMMアミダイトC、実施例4に記載のEMMアミダイトA、実施例5に記載のEMMアミダイトGおよびWO2017/188042に記載の化合物(3)を使用し、上記配列(A)からなるオリゴヌクレオチドを合成した結果、純度は53.0%であった。純度と1μmolあたりの相対収量の結果を表6に示す。
US2012/0035246の実施例2に記載のEMMアミダイトU、実施例3に記載のEMMアミダイトC、実施例4に記載のEMMアミダイトA、実施例5に記載のEMMアミダイトGおよびWO2017/188042に記載の化合物(3)を使用し、上記配列(A)からなるオリゴヌクレオチドを合成した結果、純度は53.0%であった。純度と1μmolあたりの相対収量の結果を表6に示す。
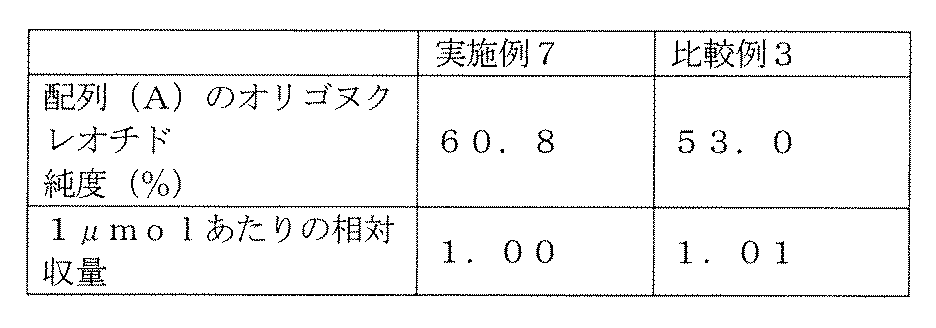
以上の表6に示すように、配列(A)のオリゴヌクレオチドの純度の良好な結果を得ることができる。
配列表の配列番号1は、ウリジン50量体の塩基配列を示す。
配列表の配列番号2,3は、核酸の製造例2で製造した配列を構成するオリゴヌクレオチドの塩基配列を表す。
配列表の配列番号2,3は、核酸の製造例2で製造した配列を構成するオリゴヌクレオチドの塩基配列を表す。
Claims (24)
- 式(1)で示されるアミダイト化合物。

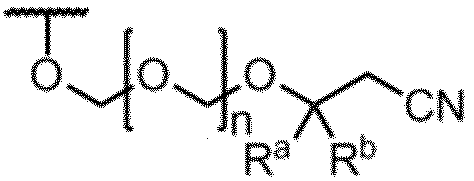
nは1~5の整数を表す。)を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G1及びG2は同一又は相異なって水酸基の保護基を表し、
G3は同一又は相異なってアルキル基を表す。) - Raがメチル基またはエチル基であり、Rbが水素原子である、請求項1に記載のアミダイト化合物。
- G1が以下の基である、請求項1又は2に記載のアミダイト化合物。

- G2が以下の基である、請求項1~3のいずれか一項に記載のアミダイト化合物。

- G3がイソプロピル基である、請求項1~4のいずれか一項に記載のアミダイト化合物。
- 請求項1~5のいずれか一項に記載のアミダイト化合物を固相合成反応に供する工程を含む、式(2):

Xは酸素原子又は硫黄原子を表し、
mは正の整数を表す。)
で示されるポリヌクレオチド骨格を含有する化合物の製造方法。 - 式(2)のポリヌクレオチド骨格を含有する化合物が、前記アミダイト化合物を用いる固相合成反応で生成する式(3):
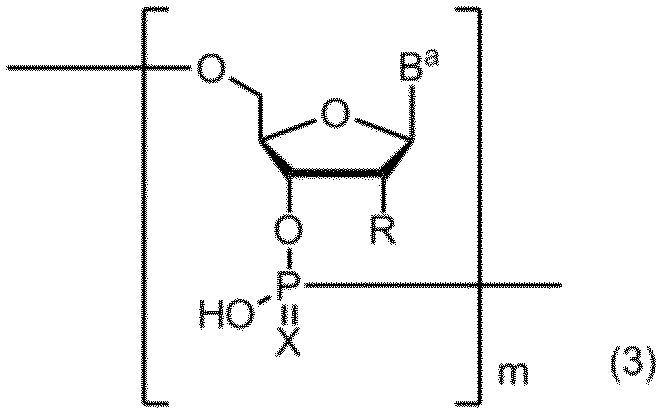
Xは酸素原子又は硫黄原子を表し、
Rは同一又は相異なって、式:
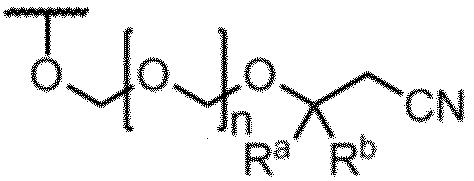
nは1~5の整数を表す。)を表し、
mは正の整数を表す。)
で示されるオリゴヌクレオチド骨格を有する化合物にテトラアルキルアンモニウムフルオライドを反応させる工程で生成した化合物である、請求項6に記載の製造方法。 - Raがメチル基またはエチル基であり、Rbが水素原子である、請求項7に記載の製造方法。
- n=1である、請求項6~8のいずれか一項に記載の製造方法。
- 式(4)で示されるエーテル化合物。

- Raがメチル基またはエチル基であり、Rbが水素原子であり、Rcがメチルである、項10に記載のエーテル化合物。
- n=1である、請求項10または11に記載の化合物。
- 工程a:式(5):
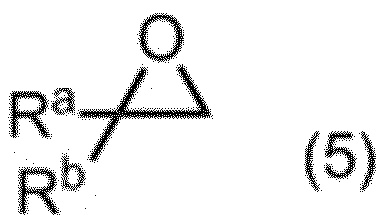
で示される化合物とシアン化物イオンとを反応させる工程、及び
工程b:酸化剤及び酸存在下、溶媒中で、工程aで得られた式(6):

で示される3-ヒドロキシアルキルニトリルとビス(メチルチオメチル)エーテルとを反応させる工程、
を含む、式(7):

で示されるエーテル化合物の製造方法。 - Raがメチル基またはエチル基であり、Rbが水素原子である、請求項13に記載の製造方法。
- Raがメチル基であり、Rbが水素原子である、請求項13に記載の製造方法。
- 工程A:3-アミノクロトノニトリルを加水分解する工程、
工程B:工程Aで得られたシアノアセトンを還元して、3-ヒドロキシブタンニトリルを得る工程、および
工程C:酸化剤及び酸存在下、溶媒中で、工程Bで得られた3-ヒドロキシブタンニトリルとビス(メチルチオメチル)エーテルとを反応させる工程、
を含む、式(8):
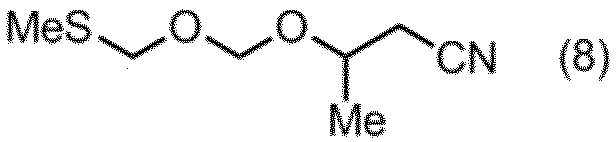
- 式(9):
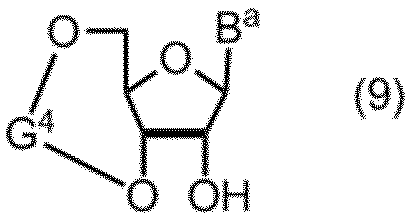
で示される化合物を、酸化剤の存在下、式(4):
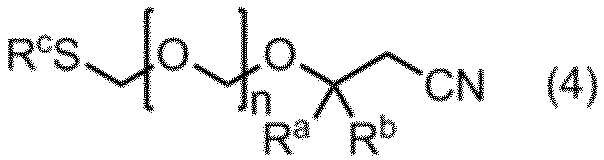
RcはC1~C6アルキル基、又はフェニル基である。
nは、1~4の整数を表す。)
で示される化合物と、反応させることを特徴とする、式(10):

で示される化合物の製造方法。 - 反応溶媒として、テトラヒドロピランまたは4-メチルテトラヒドロピランを用いる、請求項17に記載の製造方法。
- 式(10)の化合物をさらに脱保護して、式(11):
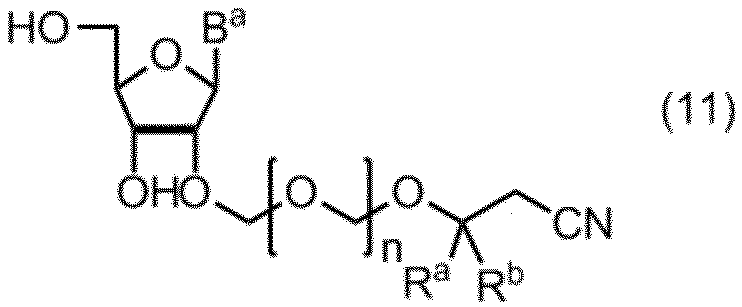
で示される化合物を得る工程、
式(11)の化合物の5’の水酸基を選択的に保護して、式(12):

で示される化合物を得る工程、
式(12)の化合物を式(13):
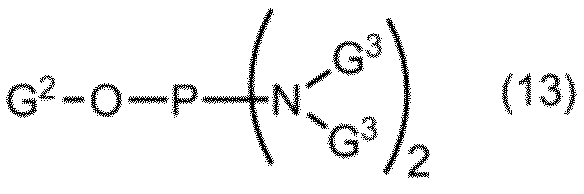
で表されるホスホロジアミダイトと反応させる工程、
を含む、請求項1に記載の式(1)の化合物の製造方法。 - G4は、G4-1またはG4-2構造を有する請求項19に記載の製造方法。
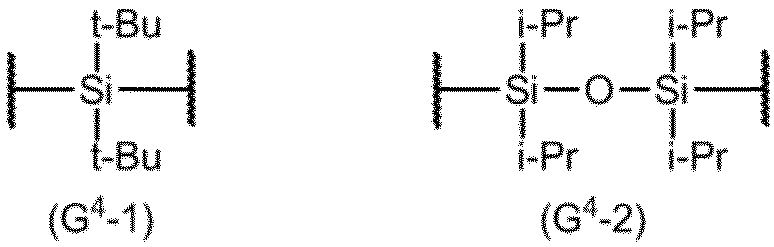
- 式(10):

nは1~5の整数を表し、
Baは保護されていてもよい核酸塩基骨格を有する基を表し、
G4は、保護基を表す。)
で示される化合物。 - 式(11):
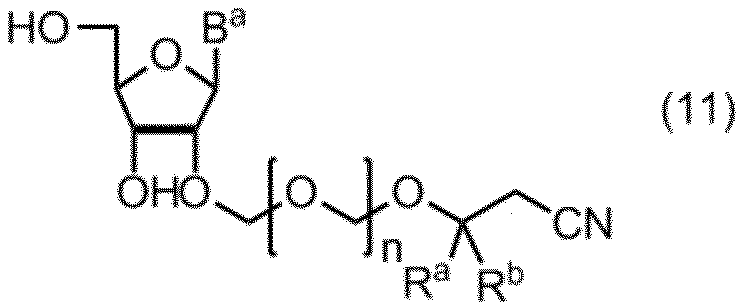
で示される化合物。 - 式(12):
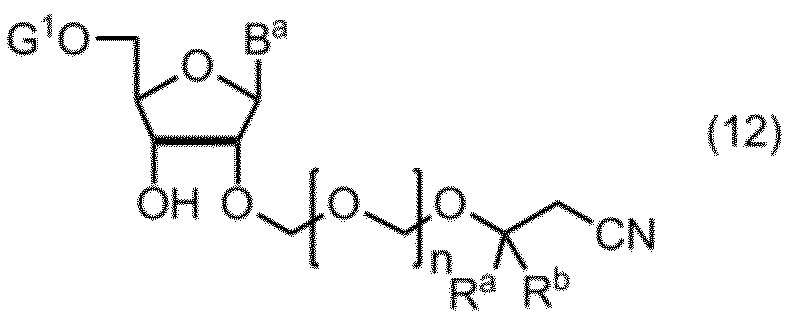
で示される化合物。 - 前記式(1)のアミダイト化合物のRNAの製造における使用。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020207029839A KR20210004986A (ko) | 2018-04-24 | 2019-04-23 | 아미다이트 화합물 및 그 화합물을 사용한 폴리뉴클레오티드의 제조 방법 |
| US17/049,905 US11401296B2 (en) | 2018-04-24 | 2019-04-23 | Amidite compound and method for producing polynucleotide using said compound |
| EP19792159.6A EP3786173A4 (en) | 2019-04-23 | Amidite compound and method for producing polynucleotide using said compound | |
| CN201980027335.3A CN112020507B (zh) | 2018-04-24 | 2019-04-23 | 酰胺化合物及使用该化合物的多核苷酸的制造方法 |
| JP2020515487A JP7280248B2 (ja) | 2018-04-24 | 2019-04-23 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-083148 | 2018-04-24 | ||
| JP2018083148 | 2018-04-24 | ||
| JP2018-141559 | 2018-07-27 | ||
| JP2018141559 | 2018-07-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019208571A1 true WO2019208571A1 (ja) | 2019-10-31 |
Family
ID=68295061
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/017249 WO2019208571A1 (ja) | 2018-04-24 | 2019-04-23 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11401296B2 (ja) |
| JP (1) | JP7280248B2 (ja) |
| KR (1) | KR20210004986A (ja) |
| CN (1) | CN112020507B (ja) |
| WO (1) | WO2019208571A1 (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021070507A1 (ja) * | 2019-10-08 | 2021-04-15 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021079617A1 (ja) * | 2019-10-23 | 2021-04-29 | 住友化学株式会社 | 配糖体化合物、アミダイト化合物、およびこれら化合物を用いたポリヌクレオチドの製造方法 |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021193954A1 (ja) | 2020-03-27 | 2021-09-30 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021210409A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
| WO2021210408A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022009959A1 (ja) | 2020-07-09 | 2022-01-13 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022147223A3 (en) * | 2020-12-31 | 2022-09-09 | Alnylam Pharmaceuticals, Inc. | 2'-modified nucleoside based oligonucleotide prodrugs |
| CN115551805A (zh) * | 2020-05-13 | 2022-12-30 | 住友化学株式会社 | 无机多孔质基材、无机多孔质担载体、及核酸的制造方法 |
| WO2023282120A1 (ja) | 2021-07-06 | 2023-01-12 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
| WO2024024766A1 (ja) * | 2022-07-25 | 2024-02-01 | 東レ株式会社 | アルキルハライド、それを用いたアルキル化剤及びヌクレオシド誘導体の製造方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020050411A1 (ja) * | 2018-09-07 | 2020-03-12 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| JP7469360B2 (ja) | 2022-04-06 | 2024-04-16 | 大阪新薬株式会社 | 3,5-ジ-ターシャリーブチルサリチル酸の製造方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011034072A1 (ja) * | 2009-09-16 | 2011-03-24 | 株式会社キラルジェン | Rna及びその誘導体合成のための新規保護基 |
| US20120035246A1 (en) | 2010-08-03 | 2012-02-09 | Bonac Corporation | Single-stranded nucleic acid molecule having nitrogen-containing alicyclic skeleton |
| WO2012073857A1 (ja) * | 2010-11-30 | 2012-06-07 | 株式会社キラルジェン | 2'-o-修飾rna |
| WO2013027843A1 (ja) * | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| JP5157168B2 (ja) | 2004-08-26 | 2013-03-06 | 日本新薬株式会社 | ホスホロアミダイト化合物及びオリゴrnaの製法 |
| JP2016050203A (ja) | 2014-08-29 | 2016-04-11 | 住友化学株式会社 | エーテル化合物の製造方法 |
| WO2017188042A1 (ja) | 2016-04-26 | 2017-11-02 | 住友化学株式会社 | 一本鎖核酸分子用モノマーの製造方法 |
| CN107602497A (zh) | 2017-10-10 | 2018-01-19 | 浦拉司科技(上海)有限责任公司 | 一种3‑氨基‑5‑烷基异噁唑的制备方法 |
| JP2018083148A (ja) | 2016-11-22 | 2018-05-31 | 旭サナック株式会社 | 静電塗装装置 |
| JP2018141559A (ja) | 2012-06-29 | 2018-09-13 | サン−ゴバン パフォーマンス プラスティックス レンコール リミティド | 構成要素係合構造を有するトレランスリング |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE373464A (ja) * | 1929-09-18 | |||
| JPS5554881Y2 (ja) | 1977-02-03 | 1980-12-19 | ||
| JP2008174524A (ja) | 2007-01-22 | 2008-07-31 | Nippon Shinyaku Co Ltd | リボ核酸化合物の製造方法 |
-
2019
- 2019-04-23 JP JP2020515487A patent/JP7280248B2/ja active Active
- 2019-04-23 US US17/049,905 patent/US11401296B2/en active Active
- 2019-04-23 KR KR1020207029839A patent/KR20210004986A/ko not_active Application Discontinuation
- 2019-04-23 WO PCT/JP2019/017249 patent/WO2019208571A1/ja unknown
- 2019-04-23 CN CN201980027335.3A patent/CN112020507B/zh active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5157168B2 (ja) | 2004-08-26 | 2013-03-06 | 日本新薬株式会社 | ホスホロアミダイト化合物及びオリゴrnaの製法 |
| WO2011034072A1 (ja) * | 2009-09-16 | 2011-03-24 | 株式会社キラルジェン | Rna及びその誘導体合成のための新規保護基 |
| US20120035246A1 (en) | 2010-08-03 | 2012-02-09 | Bonac Corporation | Single-stranded nucleic acid molecule having nitrogen-containing alicyclic skeleton |
| WO2012073857A1 (ja) * | 2010-11-30 | 2012-06-07 | 株式会社キラルジェン | 2'-o-修飾rna |
| WO2013027843A1 (ja) * | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| JP5554881B2 (ja) | 2011-08-25 | 2014-07-23 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| JP2018141559A (ja) | 2012-06-29 | 2018-09-13 | サン−ゴバン パフォーマンス プラスティックス レンコール リミティド | 構成要素係合構造を有するトレランスリング |
| JP2016050203A (ja) | 2014-08-29 | 2016-04-11 | 住友化学株式会社 | エーテル化合物の製造方法 |
| WO2017188042A1 (ja) | 2016-04-26 | 2017-11-02 | 住友化学株式会社 | 一本鎖核酸分子用モノマーの製造方法 |
| JP2018083148A (ja) | 2016-11-22 | 2018-05-31 | 旭サナック株式会社 | 静電塗装装置 |
| CN107602497A (zh) | 2017-10-10 | 2018-01-19 | 浦拉司科技(上海)有限责任公司 | 一种3‑氨基‑5‑烷基异噁唑的制备方法 |
Non-Patent Citations (4)
| Title |
|---|
| APPLIED MICROBIOLOGY AND BIOTECHNOLOGY, vol. 100, no. 3, 2016, pages 1241 |
| CHEMICAL COMMUNICATIONS, vol. 47, no. 12, 2011, pages 3613 |
| JOURNAL OF ORGANIC CHEMISTRY, vol. 75, no. 21, 2010, pages 7092 |
| TETRAHEDRON LETTERS, vol. 46, 2005, pages 2961 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021070507A1 (ja) * | 2019-10-08 | 2021-04-15 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021079617A1 (ja) * | 2019-10-23 | 2021-04-29 | 住友化学株式会社 | 配糖体化合物、アミダイト化合物、およびこれら化合物を用いたポリヌクレオチドの製造方法 |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021193954A1 (ja) | 2020-03-27 | 2021-09-30 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021210409A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
| WO2021210408A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| CN115551805A (zh) * | 2020-05-13 | 2022-12-30 | 住友化学株式会社 | 无机多孔质基材、无机多孔质担载体、及核酸的制造方法 |
| CN115551805B (zh) * | 2020-05-13 | 2023-10-20 | 住友化学株式会社 | 无机多孔质基材、无机多孔质担载体、及核酸的制造方法 |
| WO2022009959A1 (ja) | 2020-07-09 | 2022-01-13 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022147223A3 (en) * | 2020-12-31 | 2022-09-09 | Alnylam Pharmaceuticals, Inc. | 2'-modified nucleoside based oligonucleotide prodrugs |
| WO2023282120A1 (ja) | 2021-07-06 | 2023-01-12 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
| WO2024024766A1 (ja) * | 2022-07-25 | 2024-02-01 | 東レ株式会社 | アルキルハライド、それを用いたアルキル化剤及びヌクレオシド誘導体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20210238217A1 (en) | 2021-08-05 |
| EP3786173A1 (en) | 2021-03-03 |
| JPWO2019208571A1 (ja) | 2021-04-30 |
| KR20210004986A (ko) | 2021-01-13 |
| CN112020507B (zh) | 2023-10-10 |
| US11401296B2 (en) | 2022-08-02 |
| CN112020507A (zh) | 2020-12-01 |
| JP7280248B2 (ja) | 2023-05-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7280248B2 (ja) | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 | |
| KR101281836B1 (ko) | 포스포라미다이트 화합물 및 올리고 rna의 제조 방법 | |
| JP7045669B2 (ja) | リボ核酸h-ホスホネートモノマーの合成方法および本モノマーを用いたオリゴヌクレオチド合成 | |
| WO2007099896A1 (ja) | 核酸保護基の脱離方法 | |
| WO2021079617A1 (ja) | 配糖体化合物、アミダイト化合物、およびこれら化合物を用いたポリヌクレオチドの製造方法 | |
| WO2020032152A1 (ja) | 4'-置換ヌクレオシド誘導体の立体選択的合成法 | |
| JP4691101B2 (ja) | 1−α−ハロ−2,2−ジフルオロ−2−デオキシ−D−リボフラノース誘導体及びその製造方法 | |
| WO2007097447A1 (ja) | 核酸保護基の脱離方法 | |
| JP2021534174A (ja) | Sglt阻害剤の合成に有用な中間体の製造 | |
| US8158774B2 (en) | Method for introducing a nucleic-acid protecting group | |
| KR101259648B1 (ko) | 2′,2′-디플루오로뉴클레오시드 및 중간체의 새로운 제조방법 | |
| JP7423533B2 (ja) | 配糖体化合物の製造方法 | |
| JP6429264B2 (ja) | ボラノホスフェート化合物、及び核酸オリゴマー | |
| WO2022255469A1 (ja) | ホスホロチオエート及びボラノホスフェートを含むキメラ型核酸オリゴマー、及びその製造方法 | |
| JP2009256335A (ja) | 2’位にアルキル型保護基を有するリボ核酸の製造法 | |
| JPWO2010079813A1 (ja) | イノシン誘導体の製造方法 | |
| KR20220079832A (ko) | 핵산 올리고머의 제조 방법 | |
| JP2007077047A (ja) | 2−フルオロ−6−o−置換ケトライド誘導体の製造方法およびその中間体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19792159 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020515487 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019792159 Country of ref document: EP Effective date: 20201124 |