WO2018174250A1 - プリプレグおよび繊維強化複合材料 - Google Patents
プリプレグおよび繊維強化複合材料 Download PDFInfo
- Publication number
- WO2018174250A1 WO2018174250A1 PCT/JP2018/011715 JP2018011715W WO2018174250A1 WO 2018174250 A1 WO2018174250 A1 WO 2018174250A1 JP 2018011715 W JP2018011715 W JP 2018011715W WO 2018174250 A1 WO2018174250 A1 WO 2018174250A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- component
- particles
- prepreg
- mass
- epoxy resin
- Prior art date
Links
Images



Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/38—Layered products comprising a layer of synthetic resin comprising epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/0405—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres
- C08J5/042—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres with carbon fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/20—Layered products comprising a layer of synthetic resin characterised by the use of special additives using fillers, pigments, thixotroping agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/10—Reinforcing macromolecular compounds with loose or coherent fibrous material characterised by the additives used in the polymer mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/249—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs characterised by the additives used in the prepolymer mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
- C08J2363/02—Polyglycidyl ethers of bis-phenols
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2377/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2379/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen, or carbon only, not provided for in groups C08J2361/00 - C08J2377/00
- C08J2379/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08J2379/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2477/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2477/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2477/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2477/06—Polyamides derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2479/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen, or carbon only, not provided for in groups C08J2461/00 - C08J2477/00
- C08J2479/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08J2479/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
Definitions
- the present invention relates to prepregs and fiber reinforced composite materials.
- This application is filed in Japanese Patent Application No. 2017-058507 filed in Japan on March 24, 2017, Japanese Patent Application No. 2017-058508 filed in Japan on March 24, 2017, and February 23, 2018.
- Japanese Patent Application No. 2018-030289 filed in Japan the contents of which are incorporated herein.
- Fiber-reinforced composite materials are widely used in the sports / leisure field, the automobile field, the aircraft field, and other general industrial fields because of their light weight, high strength, and high rigidity. Recently, fiber reinforced composite materials having lighter weight, higher strength, and higher rigidity are often used in the automotive field, the aircraft field, and the like.
- the fiber reinforced composite material is a material having reinforced fibers and a matrix resin as essential components.
- the fiber reinforced composite material is an anisotropic material in which the strength and elastic modulus in the fiber axis direction of the reinforcing fiber are extremely high, but the strength and elastic modulus in the direction perpendicular to the fiber axis direction are low.
- the fiber-reinforced composite material is manufactured, for example, by laminating a prepreg impregnated with an uncured thermosetting resin composition on a reinforcing fiber base, thermoforming the resin, and curing the thermosetting resin composition.
- a fiber reinforced composite material by using a prepreg using a woven fabric of reinforcing fibers or by laminating the fiber axis directions of prepregs using reinforcing fibers arranged in one direction in different directions.
- the physical properties in each direction of the fiber-reinforced composite material that is an anisotropic material are controlled.
- the cured product of the thermosetting resin composition when a cured product of a thermosetting resin composition is used as a matrix resin, the cured product of the thermosetting resin composition has various disadvantages such as cost, productivity, and heat resistance, but has a disadvantage of poor toughness. Therefore, the toughness of the interlayer region of the fiber reinforced composite material becomes insufficient.
- a fiber-reinforced composite material in which fine particles such as polyamide having high toughness are arranged in an interlayer region (Patent Documents 1 and 2).
- a specific fiber-reinforced composite material in which fine particles having a specific particle size distribution index, sphericity and glass transition temperature are arranged in an interlayer region, and an elastomer component is included in a matrix resin (Patent Document 3).
- the fiber reinforced composite material of Patent Document 1 since the interlaminar fracture toughness when progressing by the mode II interlaminar fracture toughness is high, the compressive strength after impact is high, and damage due to falling weight impact on the member surface is suppressed.
- the fiber reinforced composite material of Patent Document 1 does not sufficiently consider the morphology of the microparticles after molding and the interfacial properties between the microparticles and the matrix resin composition.
- the mode I interlaminar fracture toughness and the mode II interlaminar fracture toughness necessary for such complication are insufficient.
- the morphology related to the fine particles in the fiber reinforced composite material is controlled by the melting point and molding temperature of the fine particles arranged in the interlayer region.
- the fiber reinforced composite material (2) has a high mode II interlaminar fracture toughness that is effective for improving the compressive strength after impact and a high mode I interlaminar fracture toughness that is necessary for complications such as an increase in size and a three-dimensional curved shape.
- the morphology of the fine particles after molding and the interfacial properties between the fine particles and the matrix resin composition are not sufficiently considered, and depending on the manufacturing conditions of the fiber reinforced composite material, the interlayer region of the fiber reinforced composite material is similar to Patent Document 2.
- the cracks caused by the peeling stress in the out-of-plane direction cannot remain stably in the interlayer region, the effect of the fine particle arrangement cannot be sufficiently obtained.
- a fiber-reinforced composite material excellent in mode I interlayer fracture toughness and mode II interlayer fracture toughness is produced by arranging fine tough polyamide particles in the interlayer region to form a unique morphology.
- a possible method is disclosed. However, if the manufacturing conditions such as the curing temperature and the heating rate are restricted and the unique morphology cannot be formed, sufficient mode I interlayer fracture toughness and mode II interlayer fracture toughness are not exhibited.
- An object of the present invention is to provide a prepreg and a fiber-reinforced composite material that are stable and excellent in mode I interlayer fracture toughness and mode II interlayer fracture toughness regardless of manufacturing conditions such as curing temperature and heating rate.
- a prepreg comprising a component (A), a component (B) and a component (C),
- (A) is a reinforcing fiber substrate
- (B) is an epoxy resin composition
- the component (C) is the component (c1) or the component (c2)
- the component (c1) includes polyamide particles and thermosetting polyimide particles
- the component (C) is the component (c1), The prepreg according to [1] or [2], wherein the mass ratio represented by [polyamide particles]: [thermosetting polyimide particles] is 60:40 to 95: 5.
- the component (C) is the component (c1), The prepreg according to any one of [1] to [3], wherein the polyamide particles in the component (c1) have a melting point of 140 ° C to 175 ° C.
- the component (C) is the component (c1), The prepreg according to any one of [1] to [4], wherein the polyamide particles in (c1) are crystalline copolymer nylon particles.
- the component (C) is the component (c1), The prepreg according to any one of [1] to [4], wherein the polyamide particles in (c1) are true spherical particles made of a copolymer of nylon 12 and nylon 6.
- the prepreg according to [7], wherein the true spherical particles of polyamide having a melting point of 140 to 175 ° C. are crystalline copolymer nylon particles.
- the prepreg according to [7] wherein the true spherical particles of polyamide having a melting point of 140 to 175 ° C.
- thermosetting polyimide particles are true spherical particles made of a copolymer of nylon 12 and nylon 6.
- the component (C) is the component (c2), The prepreg according to any one of [1] and [7] to [9], wherein (c2) further includes thermosetting polyimide particles.
- the component (C) is the component (c2), (C2) further contains thermosetting polyimide particles, The prepreg according to any one of [1] and [7] to [10], wherein the thermosetting polyimide particles include a chemical structure represented by the following general formula (1) or the following general formula (2).
- R represents a divalent linking group.
- the component (B) contains an epoxy resin and an aromatic polyamine,
- the epoxy resin contains an epoxy resin having a naphthalene skeleton
- a fiber-reinforced composite material obtained by laminating two or more prepregs according to any one of [1] to [15] and heating at a temperature not lower than the curing temperature of the component (B).
- a fiber-reinforced composite material comprising a component (A), a component (B ′) and a component (C),
- the component (A) is a reinforcing fiber substrate
- the component (B ′) is a cured product of the epoxy resin composition
- the component (C) is the component (c1) or the component (c2)
- the component (c1) includes polyamide particles and thermosetting polyimide particles
- the component (c2) includes polyamide spherical particles having a melting point of 140 to 175 ° C.
- the component (B) contains an epoxy resin, The prepreg according to any one of [1] to [15], wherein the epoxy resin contains an epoxy resin having an oxazolidone ring skeleton. [19] The prepreg according to [18], wherein a ratio of the epoxy resin having an oxazolidone ring skeleton is 10 to 40% by mass with respect to 100% by mass of the epoxy resin in the component (B). [20] The component (B) contains an aromatic polyamine, The prepreg according to any one of [1] to [15], [18], and [19], wherein the aromatic polyamine is 4,4′-diaminodiphenylsulfone.
- the component (B) contains an epoxy resin, [1] to [15], wherein the epoxy resin includes at least one epoxy resin selected from the group consisting of a bisphenol A type epoxy resin, a bisphenol F type epoxy resin, and a trifunctional or higher functional epoxy resin; 18] to [20].
- the epoxy resin includes at least one epoxy resin selected from the group consisting of a bisphenol A type epoxy resin, a bisphenol F type epoxy resin, and a trifunctional or higher functional epoxy resin; 18] to [20].
- the present invention it is possible to provide a prepreg and a fiber-reinforced composite material that are stably excellent in mode I interlayer fracture toughness and mode II interlayer fracture toughness regardless of manufacturing conditions such as a curing temperature and a heating rate.
- the prepreg of the present invention is a prepreg comprising the following component (A), component (B) and component (C).
- the component (c2) includes polyamide spherical particles having a melting point of 140 to 175 ° C.
- the fiber-reinforced composite material of the present invention is a fiber-reinforced composite material containing the cured product of the above component (A), the component (B ′) and the component (C), and a plurality of the components (A) are laminated. And a fiber reinforced composite material in which the component (C) is present on the surface layer of the component (A). Furthermore, the fiber reinforced composite material of the present invention is a cured product of a laminate in which two or more of the above prepregs are laminated.
- Crystalline polyamide resin means a resin that exhibits a melting point in differential scanning calorimetry (hereinafter referred to as DSC).
- Amorphous polyamide resin means a resin having no melting point in DSC.
- Interlayer means a region in the fiber-reinforced composite material produced by laminating prepregs, where the fraction of reinforcing fibers in the vicinity of the boundary between the prepregs to be laminated is small.
- “Spherical particles” means that spherical particles have a short diameter and a long diameter of 10 randomly selected particles using a scanning electron microscope (manufactured by JEOL Ltd., JSM-6390). Measured and refers to particles having an average value of the ratio of the minor axis to the major axis (minor axis / major axis) of the 10 particles of 0.95 or more.
- “Melting point” is the melting peak temperature of the DSC curve of the crystalline resin obtained as follows. The crystalline resin was heated from room temperature to about 30 ° C. above the estimated melting point at 10 ° C./min and held at a temperature about 30 ° C. above the estimated melting point for 10 minutes.
- Glass transition temperature is a midpoint glass transition temperature determined from DSC measurement of an amorphous resin as follows. The amorphous resin is heated from room temperature to about 30 ° C. higher than the estimated glass transition temperature at 10 ° C./min, and kept at a temperature about 30 ° C. higher than the estimated glass transition temperature for 10 minutes. It was. The amorphous resin, which was rapidly cooled to a temperature about 50 ° C.
- the “average particle size” means a particle size (D50) corresponding to a cumulative frequency of 50% in a volume-based cumulative distribution obtained by particle size distribution measurement.
- Epoxy resin means a compound having two or more epoxy groups in the molecule.
- the “interlaminar fracture toughness” means a limit value of energy necessary for generating an interlaminar delamination crack per unit area.
- “GIC” means the mode I interlaminar fracture toughness value at the beginning of crack growth.
- GIIC means the mode II interlaminar fracture toughness value at the beginning of crack growth.
- “Mode I” means a deformation mode in which the direction of crack opening displacement is perpendicular to the crack surface (opening shape).
- “Mode II” means a deformation mode in which the direction of the crack opening displacement is parallel to the crack surface and perpendicular to the crack leading edge (longitudinal shear type).
- “Crack opening displacement” refers to the relative displacement of the upper and lower surfaces of the crack.
- the fiber-reinforced composite material according to an embodiment of the present invention is a fiber-reinforced composite material obtained by laminating and curing two or more prepregs that satisfy specific conditions.
- the prepreg includes a component (A), a component (B), and a component (C).
- a component is a reinforced fiber base material and it is preferable that it is a sheet form.
- the reinforcing fiber substrate may be one in which reinforcing fibers are arranged in a single direction, or may be arranged in a random direction.
- Examples of the form of component (A) include woven fabrics and nonwoven fabrics of reinforcing fibers, and sheets in which long fibers of reinforcing fibers are aligned in one direction.
- the component (A) is a sheet composed of a bundle of reinforcing fibers in which long fibers are aligned in a single direction from the viewpoint that a fiber-reinforced composite material having a high specific strength and high specific modulus can be formed. From the viewpoint of easy handling, a woven fabric of reinforcing fibers is preferable.
- the reinforcing fiber may be a long fiber, and the long fiber may be in the form of a strand.
- the reinforcing fibers may be pulverized (milled), long fibers, or those obtained by cutting strands thereof (chopped).
- Examples of the material of the reinforcing fiber include glass fiber, carbon fiber (including graphite fiber), aramid fiber, and boron fiber.
- the reinforced fiber base material is preferably a carbon fiber base material from the viewpoint of mechanical properties and weight reduction of the fiber reinforced composite material.
- the tensile strength of carbon fiber based on ASTM D4018 is preferably 3500 MPa or more, more preferably 5000 MPa or more, and further preferably 6000 MPa or more.
- the tensile elastic modulus is preferably 150 GPa or more, more preferably 200 GPa or more, and further preferably 250 GPa or more.
- the carbon fiber used in the fiber reinforced composite material preferably has a high strand strength, and conforms to ASTM D4018 of carbon fiber.
- the compliant strand strength is preferably 6000 MPa or more.
- the fiber diameter of the carbon fiber is preferably 3 ⁇ m or more, and preferably 12 ⁇ m or less. If the fiber diameter of the carbon fiber is 3 ⁇ m or more, for example, in a process such as comb or roll for processing the carbon fiber, the carbon fiber moves laterally and the carbon fibers rub against each other, the carbon fiber and the roll surface, etc. When carbon fiber rubs, the carbon fibers are less likely to break or fluff. For this reason, the fiber reinforced composite material of the stable intensity
- a component is an epoxy resin composition.
- the component (B) preferably contains an epoxy resin and an epoxy resin curing agent.
- a component may contain other components other than an epoxy resin and a hardening
- Epoxy resin As the epoxy resin, usually a bifunctional or higher functional epoxy resin having two or more epoxy groups in the molecule is used. As the epoxy resin, an epoxy resin having an oxazolidone ring skeleton is preferable from the viewpoint of improving toughness while maintaining the heat resistance and rigidity of the cured product of component (B).
- the epoxy resin has a comparatively low viscosity and does not adversely affect the properties such as heat resistance and toughness of the cured product of the component (B).
- a bisphenol A type epoxy resin that is liquid at 25 ° C. Either or both of liquid bisphenol F type epoxy resins are preferred.
- epoxy resin either or both of a bisphenol A type epoxy resin solid at 25 ° C. and a bisphenol F type epoxy resin solid at 25 ° C. are preferable from the viewpoint of imparting toughness to the cured product of component (B). .
- the epoxy resin is preferably a trifunctional or higher functional epoxy resin having three or more epoxy groups in the molecule from the viewpoint of improving the heat resistance of the cured product (B).
- Epoxy resin having oxazolidone ring skeleton is also called a urethane-modified epoxy resin or an isocyanate-modified epoxy resin.
- Commercially available epoxy resins having an oxazolidone ring skeleton include EPICLON (registered trademark) TSR-400 manufactured by DIC, Epototo (registered trademark) YD-952 manufactured by Nippon Steel & Sumikin Chemical Co., Ltd., and D.C. E. R. (Registered Trademark) 858, LSA3301 manufactured by Asahi Kasei E-Materials, and the like.
- the proportion of the epoxy resin having an oxazolidone ring skeleton is preferably 5 to 70% by mass and more preferably 10 to 60% by mass with respect to 100% by mass of the total epoxy resin in the component (B). If the ratio of the epoxy resin having an oxazolidone ring skeleton is not less than the lower limit of the above range, the toughness can be sufficiently improved while sufficiently maintaining the heat resistance and rigidity of the cured product of component (B). If the ratio of the epoxy resin having an oxazolidone ring skeleton is less than or equal to the upper limit of the above range, the viscosity of the component (B) does not become too high, the handleability of the component (B) is good, and the preparation of the prepreg is easy. , The tackiness and draping properties of the prepreg are improved.
- Bisphenol A type epoxy resin and bisphenol F type epoxy resin When the epoxy resin having a solid oxazolidone ring skeleton is used, since the viscosity of the component (B) increases, either a bisphenol A type epoxy resin that is liquid at 25 ° C. or a bisphenol F type epoxy resin that is liquid at 25 ° C. It is preferable to use with one or both.
- the bisphenol F type epoxy resin that is liquid at 25 ° C. is slightly inferior in heat resistance to the liquid bisphenol A type epoxy resin that is liquid at 25 ° C., but has a lower viscosity than the liquid bisphenol A type epoxy resin, and component (B) It is preferable from the viewpoint that a relatively high elastic modulus can be imparted to the cured product.
- liquid bisphenol A type epoxy resin at 25 ° C. examples include jER (registered trademark) 828 manufactured by Mitsubishi Chemical Co., Ltd. and D. E. R. (Registered trademark) 331, Nippon Steel & Sumikin Chemical Co., Ltd. Epototo (registered trademark) YD-128, DIC Corporation EPICLON (registered trademark) 850, and the like.
- liquid bisphenol F type epoxy resin at 25 ° C. examples include jER (registered trademark) 807 manufactured by Mitsubishi Chemical Co., Ltd. and D. E. R. (Registered trademark) 354, Epototo (registered trademark) YD-170 manufactured by Nippon Steel & Sumikin Chemical Co., Ltd., EPICLON (registered trademark) 830 manufactured by DIC, and the like.
- the total ratio of the bisphenol A type epoxy resin that is liquid at 25 ° C. and the bisphenol F type epoxy resin that is liquid at 25 ° C. is preferably 10 to 80% by mass with respect to 100% by mass of the total epoxy resin in component (B). More preferably, it is 20 to 60% by mass. If the total ratio of the bisphenol A type epoxy resin that is liquid at 25 ° C. and the bisphenol F type epoxy resin that is liquid at 25 ° C. is not less than the lower limit of the above range, the component (B) can have an appropriate viscosity, The handleability of the component (B) can be improved, and the impregnation of the component (A) can be facilitated. If the total ratio of the bisphenol A type epoxy resin that is liquid at 25 ° C.
- the viscosity of the component (B) is suppressed from becoming excessively low. It is possible to suppress a large amount of the component (B) from flowing out of the system when the prepreg produced by impregnating the component (B) into the component (A) is heated and cured, and a fiber-reinforced composite material It is possible to prevent the possibility of adversely affecting the shape and mechanical properties.
- the bisphenol A type epoxy resin solid at 25 ° C. and the bisphenol F type epoxy resin solid at 25 ° C. are more heat resistant than the bisphenol A type epoxy resin that is liquid at 25 ° C. and the bisphenol F type epoxy resin that is liquid at 25 ° C. Although slightly reduced, it is possible to impart toughness to the viscosity adjustment of the component (B) and the cured product of the component (B).
- a bisphenol F-type epoxy resin solid at 25 ° C. is slightly inferior in heat resistance to a solid bisphenol A-type epoxy resin at 25 ° C., but is preferable because it can impart a relatively high elastic modulus to the cured product of component (B). .
- the total proportion of the bisphenol A type epoxy resin solid at 25 ° C. and the bisphenol F type epoxy resin solid at 25 ° C. is preferably 1 to 60% by mass with respect to 100% by mass of the total epoxy resin in the component (B). 5 to 40% by mass is more preferable. If the total ratio of the bisphenol A type epoxy resin solid at 25 ° C. and the bisphenol F type epoxy resin solid at 25 ° C. is not less than the lower limit of the above range, sufficient toughness can be imparted to the cured product of component (B). . If the total ratio of the bisphenol A type epoxy resin solid at 25 ° C. and the bisphenol F type epoxy resin solid at 25 ° C.
- the viscosity of the component (B) is suppressed from becoming excessively high. It is possible to suppress deterioration of handleability of the component (B) and difficulty in impregnation of the component (A).
- Trifunctional or higher epoxy resin examples include triazine skeleton-containing epoxy resin, aminophenol type epoxy resin, aminocresol type epoxy resin, and the like.
- Examples of the tetrafunctional or higher functional epoxy resin include diaminodiphenylmethane type epoxy resin, cresol novolac type epoxy resin, phenol novolac type epoxy resin, aromatic glycidylamine type epoxy resin and the like.
- the proportion of the trifunctional or higher functional epoxy resin is preferably less than 50% by mass and more preferably 5 to 40% by mass with respect to 100% by mass of the total epoxy resin in the component (B).
- the ratio of the trifunctional or higher functional epoxy resin is within the above range, the reactivity of the component (B) can be adjusted to an appropriate range, and the heat resistance of the cured matrix resin assembly can be improved.
- the reactivity of the component (B) is in an appropriate range, the adhesion of the interface between the component (C) described later, particularly the component (C) and the component (B) is prevented from becoming too weak, and the component (C) is blended. The effect of imparting excellent interlaminar fracture toughness to the fiber-reinforced composite material can be sufficiently exhibited.
- the ratio of the trifunctional or higher functional epoxy resin is within the above range, the crosslinking density of the cured product of the component (B) can be suppressed from being excessively increased, and the toughness of the cured product of the component (B) is significantly reduced. This can also be suppressed.
- trifunctional epoxy resins include jER (registered trademark) 630 manufactured by Mitsubishi Chemical Corporation, Araldite (registered trademark) MY0500, MY0510, MY0600, MY0610 manufactured by HUNTSMAN, and TEPIC (registered trademark) manufactured by Nissan Chemical Co., Ltd. G, -S, -SP, -VL and the like.
- jER registered trademark
- jER registered trademark
- jER manufactured by Mitsubishi Chemical Co.
- MY0500, MY0510, MY0600, MY0610 manufactured by HUNTSMAN and jER manufactured by Mitsubishi Chemical Co. which is a diaminodiphenylmethane type epoxy resin.
- (Registered trademark) 604, Arundite (registered trademark) MY720 manufactured by HUNTSMAN, and YH434L manufactured by Nippon Steel & Sumikin Chemical Co., Ltd. can be blended to increase the heat resistance and elasticity of the cured matrix resin without greatly increasing the viscosity of the matrix resin composition.
- a diaminodiphenylmethane type epoxy resin is more preferable because it does not excessively increase the water absorption of the cured matrix resin.
- Component (B) is, if necessary, other epoxy resins such as bisphenol S type, naphthalene type, dicyclopentadiene type, resorcin type, hydroquinone type, bisphenoxyethanol fluorene type, bisphenol fluorene type, biscresol fluorene type epoxy resin May be included.
- Curing agent Any curing agent may be used as long as it can cure the epoxy resin.
- the curing agent examples include amines, acid anhydrides (carboxylic acid anhydrides), phenols (novolak resins, etc.), mercaptans, Lewis acid amine complexes, onium salts, imidazoles, and the like.
- the epoxy resin curing agent is preferably an aromatic polyamine and more preferably diaminodiphenyl sulfone from the viewpoint of excellent heat resistance and toughness of the cured product of the component (B).
- the amount of curing agent added varies depending on the type of curing agent.
- the addition amount of diaminodiphenyl sulfone is preferably such that the number of active hydrogen equivalents of diaminodiphenyl sulfone is 0.9 to 1.5 times the equivalent of epoxy group of the epoxy resin. The amount of 1.1 to 1.3 times is more preferable.
- the amount of diaminodiphenylsulfone added is within the above range, the heat resistance and toughness of the cured product of component (B) are further improved.
- 3,3′-diaminodiphenylsulfone can give a cured product with higher elasticity compared to 4,4′-diaminodiphenylsulfone, but the heat resistance of the cured product is inferior, and the following ( In some cases, adhesion at the interface between the component (C), particularly the component (C) and the component (B) is weakened, and the interlaminar fracture toughness of the fiber-reinforced composite material is lowered.
- 4,4′-diaminodiphenyl sulfone is inferior in elasticity of the resulting cured product as compared with 3,3′-diaminodiphenyl sulfone, but is excellent in heat resistance of the cured product, and component (C) described below, particularly In some cases, the adhesion at the interface between the component (C) and the component (B) is difficult to weaken, and the interlaminar fracture toughness of the fiber-reinforced composite material can be expressed more highly.
- 3,3′-diaminodiphenylsulfone is more preferable for applications where the compression properties of fiber reinforced composite materials are required, and 4,4′-diamino is used for applications where the heat resistance and interlaminar fracture toughness of fiber reinforced composite materials are required.
- Diphenyl sulfone is more preferred.
- 3,3'-diaminodiphenylsulfone and 4,4'-diaminodiphenylsulfone can be used together.
- Additives include thermoplastic elastomers, elastomer fine particles (excluding component (C)), core-shell type elastomer fine particles, block copolymers composed of acrylic resins, compounds having one epoxy group in the molecule, dilution Agents, inorganic particles (such as silica), carbonaceous components (such as carbon nanotubes), flame retardants (such as phosphorus compounds), and defoaming agents.
- Examples of commercially available core-shell type elastomer fine particles include METABRENE (registered trademark) manufactured by Mitsubishi Rayon Co., Ltd., Staphyloid manufactured by Aika Kogyo Co., Ltd. and Paraloid (registered trademark) manufactured by Dow Chemical.
- the core-shell type elastomer fine particles may be dispersed in the epoxy resin in advance.
- Examples of commercially available core-shell type elastomer fine particle-dispersed epoxy resins include Kane Ace (registered trademark) manufactured by Kaneka Corporation and Acryset (registered trademark) BP series manufactured by Nippon Shokubai Co., Ltd.
- the core-shell type elastomer fine particle-dispersed epoxy resin is preferably used because it not only facilitates the preparation of the component (B) but also makes it possible to improve the dispersion state of the core-shell type elastomer fine particles in the component (B).
- Nanostrength (registered trademark) series manufactured by Arkema for example, Nanostrength (registered trademark) M52N, Nanostrength (registered trademark) M22N.
- the component (B) an epoxy resin composition containing an epoxy resin, 60 to 100% by mass of the epoxy resin having a naphthalene skeleton with respect to 100% by mass of the epoxy resin, and further containing an aromatic polyamine as a curing agent is also included. Can be mentioned.
- This (B) component may contain other components other than an epoxy resin and a hardening
- the component (B) is an epoxy resin containing an epoxy resin, 60 to 100% by mass of the epoxy resin having a naphthalene skeleton with respect to 100% by mass of the epoxy resin, and further containing an aromatic polyamine as a curing agent. The case will be described.
- Epoxy resin having naphthalene skeleton The epoxy resin is usually a bifunctional or higher functional epoxy resin having two or more epoxy groups in the molecule, and the epoxy resin having a naphthalene skeleton has a naphthalene ring in the molecule and two or more epoxy resins. It is a bifunctional or higher functional epoxy resin having a group. Compared with a normal bisphenol A type epoxy resin or a bisphenol F type epoxy resin, an epoxy resin having a naphthalene skeleton can provide good heat resistance to a cured product because of its rigid skeleton.
- the bifunctional epoxy resin having a naphthalene skeleton is a bifunctional epoxy resin having a naphthalene ring in the molecule and two epoxy groups.
- the bifunctional epoxy resin having a naphthalene skeleton can impart good heat resistance to the cured matrix resin because of its rigid skeleton.
- the toughness of the matrix resin after curing is reduced because the crosslink density of the cured product is not increased. It can prevent that the adhesiveness of an interface falls and it cannot fully obtain the effect of the above-mentioned fine particle arrangement.
- the bifunctional epoxy resin having a naphthalene skeleton also prevents an increase in the water absorption of the matrix resin after curing. be able to.
- Examples of commercially available bifunctional epoxy resins having a naphthalene skeleton include EPICLON (registered trademark) HP-4032D and HP-4032SS manufactured by DIC.
- the ratio of the bifunctional epoxy resin having a naphthalene skeleton is preferably 50 to 100% by mass, more preferably 55 to 90% by mass, and more preferably 60 to 85% by mass with respect to 100% by mass of the total epoxy resin in the component (B). Is even more preferable.
- the ratio of the bifunctional epoxy resin having a naphthalene skeleton is not less than the lower limit of the above range, the toughness of the cured product (B), the adhesion at the interface between the reinforcing fiber and the matrix resin, and the water absorption amount are maintained.
- the heat resistance of the cured product can be sufficiently improved.
- the bifunctional epoxy resin having a naphthalene skeleton is usually in a liquid state, by using a thermoplastic resin soluble in an epoxy resin such as a solid epoxy resin or polyethersulfone together with the component (B) The handleability is improved, making the prepreg easy to manufacture, and improving the tackiness and drapeability of the prepreg.
- Tri- to tetra-functional epoxy resin having a naphthalene skeleton is a tri- to tetra-functional epoxy resin having a naphthalene skeleton in the molecule and having 3 to 4 epoxy groups.
- the tri- to tetrafunctional epoxy resin having a naphthalene skeleton can give good heat resistance to the cured matrix resin because of its rigid skeleton, and it can also be cured by increasing the crosslink density of the cured product. Good heat resistance can be imparted to the later matrix resin.
- tri- to tetra-functional epoxy resins having a naphthalene skeleton include EPICLON (registered trademark) HP-4700, HP-4710, HP-4770 manufactured by DIC Corporation, NC-7000L manufactured by Nippon Kayaku Co., Ltd. NC-7300L etc. are mentioned.
- the proportion of the tri- to tetrafunctional epoxy resin having a naphthalene skeleton is preferably 5 to 50% by mass, more preferably 10 to 40% by mass, and more preferably 15 to 35% with respect to 100% by mass of the total epoxy resin in the component (B). Mass% is even more preferable.
- the heat resistance of the cured product of the component (B) can be sufficiently improved.
- the toughness of the cured product of the component (B), the adhesion of the interface between the reinforcing fiber and the matrix resin, the water absorption can be sufficiently maintained, and the viscosity of the component (B) does not become too high. Therefore, the handleability of the component (B) is good, the preparation of the prepreg is easy, and the tackiness and draping properties of the prepreg are improved.
- the component (B) is optionally an epoxy resin having an oxazolidone ring skeleton, a bisphenol A type epoxy resin, a bisphenol F type epoxy resin, a bisphenol S type, a triazine skeleton-containing epoxy resin.
- Aminophenol type epoxy resin aminocresol type epoxy resin, cresol novolac type epoxy resin, phenol novolac type epoxy resin, aromatic glycidylamine type epoxy resin, dicyclopentadiene type, resorcin type, hydroquinone type, bisphenoxyethanol fluorene type, bisphenol Other epoxy resins such as fluorene type and biscresol fluorene type epoxy resins may be included.
- the ratio of the epoxy resin having an oxazolidone ring skeleton is preferably 10 to 40% by mass, more preferably 10 to 35% by mass, and further preferably 15 to 30% by mass with respect to 100% by mass of the total epoxy resin in the component (B). preferable. If the ratio of the epoxy resin having an oxazolidone ring skeleton is equal to or greater than the lower limit of the above range, the toughness and the interface between the reinforcing fiber and the matrix resin can be maintained while sufficiently maintaining the heat resistance and rigidity of the cured product (B). Adhesion can be improved.
- the ratio of the epoxy resin having an oxazolidone ring skeleton is less than or equal to the upper limit of the above range, the viscosity of the component (B) does not become too high, the handleability of the component (B) is good, and the preparation of the prepreg is easy. , The tackiness and draping properties of the prepreg are improved.
- component (B) bisphenol A type epoxy resin that is liquid at 25 ° C. and bisphenol F type that is liquid at 25 ° C.
- the epoxy resin can be effectively used for adjusting the viscosity of the component (B).
- Commercially available products of liquid bisphenol A type epoxy resin at 25 ° C. include jER (registered trademark) 828 manufactured by Mitsubishi Chemical Co., Ltd. and D. E. R. (Registered trademark) 331, Nippon Steel & Sumikin Chemical Co., Ltd.
- Commercially available products of liquid bisphenol F type epoxy resin at 25 ° C. include jER (registered trademark) 807 manufactured by Mitsubishi Chemical Corporation, and D.C. E. R. (Registered trademark) 354, Epototo (registered trademark) YD-170 manufactured by Nippon Steel & Sumikin Chemical Co., Ltd., EPICLON (registered trademark) 830 manufactured by DIC, and the like.
- bisphenol A type epoxy resin solid at 25 ° C. Commercially available products of bisphenol A type epoxy resin solid at 25 ° C. include jER (registered trademark) 1001, jER (registered trademark) 1002, jER (registered trademark) 1003, jER (registered trademark) 1004, manufactured by Mitsubishi Chemical Corporation, Nippon Steel Corporation. Examples include Epototo (registered trademark) YD-903 manufactured by Sumikin Chemical Co., Ltd., EPICLON (registered trademark) 1050, EPICLON (registered trademark) 2050, EPICLON (registered trademark) 3050, EPICLON (registered trademark) 4050 manufactured by DIC.
- bisphenol F type epoxy resin solid at 25 ° C. examples include jER (registered trademark) 4004P, jER (registered trademark) 4005P, jER (registered trademark) 4007P, jER (registered trademark) 4010P manufactured by Mitsubishi Chemical Corporation, Nippon Steel Corporation Examples include Epototo (registered trademark) YD-2001 and Epototo (registered trademark) YD-2004 manufactured by Sumikin Chemical.
- triazine skeleton-containing epoxy resin aminophenol type epoxy resin, aminocresol type epoxy resin, cresol novolac type epoxy resin, phenol novolac type epoxy resin, aromatic
- a polyfunctional epoxy resin such as a glycidylamine type epoxy resin may be included.
- the component (B) preferably contains an aromatic polyamine as a curing agent.
- an aromatic polyamine as a curing agent
- examples of the aromatic polyamine include diaminodiphenylmethane, diaminodiphenylsulfone, metaxylenediamine, metaphenylenediamine, and derivatives and isomers of these alkyl-substituted products as the curing agent.
- diaminodiphenylsulfone is preferred from the viewpoint that good heat resistance and toughness can be imparted by the cured product of component (B).
- the addition amount of the curing agent varies depending on the type of the curing agent.
- the addition amount of diaminodiphenyl sulfone is preferably such that the number of active hydrogen equivalents of diaminodiphenyl sulfone is 0.9 to 1.5 times the equivalent of epoxy group of the epoxy resin.
- the amount of 1.05 to 1.4 times is more preferable, and the amount of 1.1 to 1.3 times is even more preferable.
- the amount of diaminodiphenylsulfone added is within the above range, the heat resistance and toughness of the cured product of component (B) are further improved.
- 3,3′-diaminodiphenylsulfone can give a cured product with higher elasticity compared to 4,4′-diaminodiphenylsulfone, but the heat resistance of the cured product is inferior, and the following ( In some cases, adhesion at the interface between the component (C), particularly the component (C) and the component (B) is weakened, and the interlaminar fracture toughness of the fiber-reinforced composite material is lowered.
- 4,4′-diaminodiphenyl sulfone is inferior in elasticity of the resulting cured product as compared with 3,3′-diaminodiphenyl sulfone, but is excellent in heat resistance of the cured product, and component (C) described below, particularly In some cases, the adhesion at the interface between the component (C) and the component (B) is difficult to weaken, and the interlaminar fracture toughness of the fiber-reinforced composite material can be expressed more highly.
- 3,3′-diaminodiphenylsulfone is more preferable for applications where the compression properties of fiber reinforced composite materials are required, and 4,4′-diamino is used for applications where the heat resistance and interlaminar fracture toughness of fiber reinforced composite materials are required.
- Diphenyl sulfone is more preferred.
- 3,3′-diaminodiphenylsulfone and 4,4′-diaminodiphenylsulfone can be used together. Examples of commercially available 3,3′-diaminodiphenylsulfone include 3,3′-DAS from Mitsui Chemicals Fine Co., Ltd. S is mentioned.
- Additives include block copolymers composed of thermoplastic resins soluble in epoxy resins such as polyethersulfone, elastomer fine particles (excluding the component (C)), core-shell type elastomer fine particles, acrylic resins, etc. Examples thereof include compounds having one epoxy group, diluents, inorganic particles (such as silica), carbonaceous components (such as carbon nanotubes), flame retardants (such as phosphorus compounds), and defoaming agents.
- the block copolymer comprised from a core-shell type elastomer fine particle, an acrylic resin, etc.
- Examples of commercially available core-shell type elastomer fine particles include Metablene (registered trademark) manufactured by Mitsubishi Rayon Co., Ltd., Staphyloid manufactured by Aika Industries, Ltd., Paraloid (registered trademark) manufactured by Dow Chemical Company, and the like.
- the core-shell type elastomer fine particles may be dispersed in advance in the epoxy resin.
- Examples of commercially available core-shell type elastomer fine particle-dispersed epoxy resins include Kane Ace (registered trademark) manufactured by Kaneka Corporation and Acryset (registered trademark) BP series manufactured by Nippon Shokubai Co., Ltd.
- the core-shell type elastomer fine particle-dispersed epoxy resin is preferably used because it not only facilitates the preparation of the component (B) but also makes it possible to improve the dispersion state of the core-shell type elastomer fine particles in the component (B).
- Commercially available block copolymers composed of acrylic resins and the like include Nanostrength (registered trademark) series manufactured by Arkema, for example, Nanostrength (registered trademark) M52N, Nanostrength (registered trademark) M22N.
- the component (B) can be prepared by various known methods.
- (B) As a preparation method of a component the method of heating and kneading each component with a planetary mixer or a kneader is mentioned, for example.
- the particulate curing agent When a particulate curing agent such as diaminodiphenylsulfone is used as the curing agent, the particulate curing agent may aggregate and become poorly dispersed, so the particulate curing agent is pre-kneaded with a liquid epoxy resin. It is preferable to form a master batch. For preliminary kneading, it is preferable to use a kneading apparatus such as a three-roll mill or a ball mill. By making the particulate curing agent into a master batch in advance, uneven physical properties and poor curing in the cured product of component (B) due to poor dispersion, and poor impregnation of component (B) into component (A) can be suppressed.
- a kneading apparatus such as a three-roll mill or a ball mill.
- the component (C) is the component (c1) or the component (c2), the component (c1) includes polyamide particles and thermosetting polyimide particles, and the component (c2) has a spherical shape with a melting point of 140 to 175 ° C. Contains polyamide particles.
- the (c1) component polyamide particles impart excellent interlaminar fracture toughness to the fiber-reinforced composite material.
- the polyamide resin constituting the component (c1) polyamide particles is not particularly limited as long as it has an amide bond in the repeating structure.
- the polyamide resin may be polyamide resin particles made of one kind of polyamide resin or polyamide resin particles made of two or more kinds of polyamide resins. In the case of polyamide resin particles composed of two or more kinds of polyamide resins, each polyamide resin may be present uniformly in the particles or may be non-uniformly present as in a layer structure.
- the polyamide resin can be obtained by methods such as ring-opening polymerization of lactams, polycondensation of diamine and dicarboxylic acid, polycondensation of aminocarboxylic acid, and the like.
- Specific examples of the polyamide resin include nylon 6, nylon 46, nylon 66, nylon 11, nylon 12, nylon 610, nylon 612, nylon 6T, nylon 6I, nylon 9T, nylon M5T, and Daicel Evonik's trogamide. (Registered trademark) T5000, trogamide (registered trademark) CX7323, and polyamide resins containing an aromatic ring and an alicyclic ring are exemplified.
- both a crystalline polyamide resin and an amorphous polyamide resin can be preferably used, and the crystalline polyamide resin and the amorphous polyamide resin may be used alone or in combination.
- the polyamide particles (c1) crystalline copolymer nylon particles are preferable, and spherical particles made of a copolymer of nylon 12 and nylon 6 are more preferable.
- Examples of commercially available polyamide resins include VESTOSINT series (VESTOSINT (registered trademark) 2158, VESTOSINT (registered trademark) 2159, etc.) manufactured by Daicel-Evonik, Inc.
- the melting point of the polyamide particles is preferably 140 ° C. to 175 ° C., more preferably 155 to 170 ° C.
- the interface between the matrix resin and the polyamide particles can be sufficiently strengthened in the process of curing the prepreg, and a fiber-reinforced composite material having high interlaminar fracture toughness can be obtained. .
- the average particle diameter of the polyamide particles as component (c1) is preferably 2 to 50 ⁇ m, and more preferably 5 to 35 ⁇ m.
- the average particle diameter of the polyamide particles is not less than the lower limit of the range, the polyamide particles are less likely to enter the component (A) when producing a prepreg. Therefore, it is easy to obtain a prepreg that satisfies the condition that 70% by mass or more of all the components (C) described below are present in the surface layer of the component (A), and as a result, the fiber reinforced composite material can be provided with further superior interlaminar fracture toughness.
- the component (C) present on the surface layer can be determined with the uneven distribution rate described later.
- the increase in the viscosity at the time of mixing (B) component and (C) component can be suppressed.
- the average particle diameter of the polyamide particles is less than or equal to the upper limit of the above range, the component (C) in the fiber-reinforced composite material can be prevented from harming the straightness of the reinforcing fiber of the component (A).
- the deterioration of the mechanical properties of the fiber reinforced composite material can be suppressed in the interlayer region due to the peeling stress in the out-of-plane direction.
- clogging may occur in equipment such as roll coaters and die coaters. Is suppressed.
- thermosetting polyimide particles of component (c1)) (C1)
- the polyimide resin which comprises the thermosetting polyimide particle of a component is a high molecular compound which has an imide bond in a repeating structure.
- thermosetting polyimide resins have a relatively small molecular weight of the main chain and have reactive end groups. Since it has a reactive end group, the interfacial property between the thermosetting polyimide particles and the matrix resin can be strengthened in the fiber reinforced composite material, and the effect of the thermosetting polyimide particles described later can be fully expressed. be able to.
- thermosetting polyimide resin with relatively high molecular weight and almost no reactive end groups cannot strengthen the interfacial properties with the matrix resin, and it is relatively tougher than thermosetting polyimide.
- the effect of the thermosetting polyimide particles described below cannot be sufficiently exhibited.
- thermosetting polyimide resin that can be preferably used as the constituent resin of the thermosetting polyimide particles is a thermosetting polyimide resin having a chemical structure of the general formula (1), and more specifically, benzophenone tetracarboxylic dianhydride ( Thermosetting polyimides prepared from BTDA), 4,4′-methylenedianiline (MDA), and 2,4-toluenediamine (TDA) and having a non-phthalimide carbon content containing 90 to 92 percent aromatic carbon Resin.
- thermosetting polyimide resin particles include P84 (registered trademark) Polyimide manufactured by HP Polymer.
- Examples of the divalent linking group include —Ph— and —Ph—CH 2 —Ph—.
- -Ph- represents a phenylene group.
- thermosetting polyimide resin having a chemical structure of the general formula (2), more specifically, a thermosetting polyimide resin prepared from pyromellitic dianhydride and 4,4′-diaminodiphenyl ether (c1).
- the thermosetting polyimide particles of the component can be preferably used. Examples of the commercially available thermosetting polyimide resin particles include polyimide powder PIP series, PIP-3, and PIP-25 manufactured by Sanso Micron Corporation.
- thermoplastic polyimide resin (Matrimid (registered trademark) 5218 manufactured by Huntsman Co., Ltd.) is a commercially available thermoplastic polyimide resin that cannot be preferably used as the polyimide resin constituting the thermosetting polyimide particles of the component (c1).
- thermosetting polyimide particles of component (c1) The average particle size of the thermosetting polyimide particles (c1) is preferably 2 to 50 ⁇ m, more preferably 5 to 35 ⁇ m.
- the (C) component is less likely to enter the (A) component when producing the prepreg. Therefore, it is easy to obtain a prepreg that satisfies the condition that 70% by mass or more of all the above-described (C) components exist in the surface layer of the (A) component, and as a result, the fiber reinforced composite material can be provided with further superior interlaminar fracture toughness.
- the increase in the viscosity at the time of mixing (B) component and (C) component can be suppressed. Further, it is possible to prevent further aggregation and poor dispersion. If the average particle diameter of the thermosetting polyimide particles is less than or equal to the upper limit of the above range, the (C) component in the fiber reinforced composite material can be prevented from harming the straightness of the reinforced fiber of the (A) component. The deterioration of the mechanical properties of the reinforced composite material can be suppressed, and the crack generated in the interlayer region of the fiber reinforced composite material due to the peeling stress in the out-of-plane direction can be retained in the interlayer region.
- the average particle size of the thermosetting polyimide particles of component (c1) is preferably in the range of 0.5 to 10 times the average particle size of the polyamide particles of component (c1).
- the average particle size of the thermosetting polyimide particles of the component (c1) is larger than 0.5 times the average particle size of the polyamide particles of the component (c1), the heat of the polyamide particles (component c1) and the heat of the component (c1) described later. The combined effect of the curable polyimide particles can be sufficiently exhibited.
- the thickness of the interlayer region is more uniform, that is, the reinforcing fibers of the component (A) Since it can suppress damage to straightness, it can suppress degradation of the mechanical properties of the fiber reinforced composite material, and can keep the crack generated in the interlayer region of the fiber reinforced composite material due to the peeling stress in the out-of-plane direction in the interlayer region I can do it.
- thermosetting polyimide resins have relatively poor toughness, and when used alone, excellent interlaminar fracture toughness cannot be imparted to fiber-reinforced composite materials.
- the crack generated in the interlayer region of the fiber reinforced composite material due to the peeling stress in the out-of-plane direction has a property of selectively progressing at a portion having poor toughness, for example, an interface between the matrix resin and the reinforced fiber bundle.
- the cracks generated in the interlayer region gradually move to the interface between the matrix resin with less toughness and the reinforcing fiber bundle as it progresses, and finally In particular, it has completely transferred from the interlayer region to the reinforcing fiber layer, and it has not been possible to stably provide excellent interlayer fracture toughness.
- the cracks generated in the interlayer region stably progress in the interlayer region (c1) component polyamide particles and (c1) component
- the cracks generated in the interlayer region can be stably advanced and the interlayer region can be further advanced, and further, the interlaminar fracture toughness can be imparted by the addition of the polyamide particles of the component (c1). It can be stably obtained regardless of the morphology of the interlayer region of the composite material and the interface property between the polyamide particles of the component (c1) and the matrix resin composition.
- compositions of (c1) component polyamide particles and (c1) component thermosetting polyimide particles The total content of the polyamide particles of the component (c1) and the thermosetting polyimide particles of the component (c1) is 5 to 25 parts by mass, more preferably 10 to 25 parts by mass with respect to 100 parts by mass of the component (B). 12 to 25 parts by mass is more preferable, and 15 to 20 parts by mass is even more preferable.
- the prepreg contains (B) component, (c1) component polyamide particles, and (c1) component thermosetting polyimide particles. It can suppress that (B) component which occupies in resin becomes too low.
- the deterioration of the mechanical properties of the fiber reinforced composite material due to the shortage of the component (B) can be prevented, the viscosity of the matrix resin composition is suppressed from becoming too high, and the component (A) The matrix resin composition can be sufficiently impregnated.
- thermosetting polyimide particles in component (c1) The proportion of the thermosetting polyimide particles in the component (c1) is preferably 10 to 40% by mass. If the ratio of the thermosetting polyimide particles is equal to or higher than the lower limit of the above range, the effect of the thermosetting polyimide particles can be sufficiently obtained, and the interlaminar fracture toughness excellent in the fiber-reinforced composite material is stabilized for the above-mentioned reason. Can be granted. If the ratio of the thermosetting polyimide particles is not more than the upper limit of the above range, it is possible to suppress the adverse effect of the thermosetting polyimide particles having poor toughness on the interlaminar fracture toughness of the fiber reinforced composite material.
- the more preferable ratio of the thermosetting polyimide particles greatly depends on the combination of components. For example, when the interface between the component (A) and the component (B) is weaker, cracks generated in the interlayer region more easily progress to the interface between the component (A) and the component (B).
- the ratio of the thermosetting polyimide particles is preferably 20 to 40% by mass with respect to the total mass of the component (c1).
- the proportion of the thermosetting polyimide particles is preferably less than 10 to 25% by mass with respect to the total mass of the component (c1).
- the ratio of the curable polyimide particles is preferably 20 to 40% by mass with respect to the total mass of the component (c1).
- the proportion of the thermosetting polyimide particles is preferably less than 10 to 25% by mass with respect to the total mass of the component (c1).
- the ratio of thermosetting polyimide particles in component (c1) is (c1) It is preferable to further increase to 25 to 40% by mass with respect to the total mass of the components.
- the ratio of the thermosetting polyimide particles in the component (c1) is (c1) It is preferably further reduced to 10 to 20% by mass based on the total mass of the components.
- the weak interface between the component (A) and the component (B) means that the strength of the 90 ° bending test of the fiber reinforced composite material in one direction (the reinforcing fibers are all oriented in the same direction) in accordance with ASTM D790. Is less than 125 MPa.
- the strong interface between the component (A) and the component (B) means that the strength of the 90 ° bending test of the fiber reinforced composite material in one direction (the reinforcing fibers are all oriented in the same direction) in accordance with ASTM D790 is 125 MPa. That's it.
- the weak interface between the component (B) and the component (C) means that the fracture surface of the DCB (Double Cantilever Beam) test of the fiber reinforced composite material implemented in accordance with ASTM D5528 is a scanning electron microscope (SEM). ), And the interface of the component (C) 30 is exposed as shown in FIG.
- the strong interface between the component (B) and the component (C) means that the fracture surface of the DCB (Double Cantilever Beam) test of the fiber reinforced composite material carried out in accordance with ASTM D5528 is a scanning electron microscope (SEM). ), And the interface of the component (C) is not exposed as shown in FIG.
- the polyamide particles are polyamide resins having a particle shape and can impart excellent interlaminar fracture toughness to the fiber-reinforced composite material.
- a polyamide resin is a compound having an amide bond in a repeating structure.
- the polyamide resin may be a polyamide resin made of one kind of polyamide resin or a polyamide resin made of two or more kinds of polyamide resins. In the case of polyamide resin particles composed of two or more kinds of polyamide resins, each polyamide resin may be present uniformly in the particles or may be non-uniformly present as in a layer structure.
- the polyamide resin can be obtained by methods such as ring-opening polymerization of lactams, polycondensation of diamine and dicarboxylic acid, polycondensation of aminocarboxylic acid, and the like.
- Specific examples of the polyamide resin include nylon 6, nylon 46, nylon 66, nylon 11, nylon 12, nylon 610, nylon 612, nylon 6T, nylon 6I, nylon 9T, nylon M5T, and Daicel Evonik's trogamide. (Registered trademark) T5000, trogamide (registered trademark) CX7323, and polyamide resins containing an aromatic ring and an alicyclic ring are exemplified.
- Examples of commercially available polyamide resins include VESTOSINT series (VESTOSINT (registered trademark) 2158, VESTOSINT (registered trademark) 2159, etc.) manufactured by Daicel-Evonik, Inc. And TOROGAMID (registered trademark) CX7233 manufactured by Daicel-Evonik, TOROGAMID (registered trademark) T5000, and the like.
- the polyamide resin of the component (c2) is preferably a crystalline polyamide resin.
- the crystalline polyamide resin include nylon 6, nylon 46, nylon 66, nylon 11, nylon 12, nylon 610, nylon 612, nylon 6T, nylon 6I, nylon 9T, and nylon M5T. Since most of the melting point does not fall within the range of 140 to 175 ° C. due to its high crystallinity, the polyamide resin of component (C) is preferably a copolymer that can have a lower melting point due to crystallinity inhibition.
- crystalline copolymer nylon particles are preferable, and spherical particles made of a copolymer of nylon 12 and nylon 6 are more preferable.
- the copolymer of nylon 12 and nylon 6 mainly composed of nylon 12 has high toughness and low moisture absorption, and its melting point is lower than that of nylon 12, so that it can be used as a fiber-reinforced composite material in a wider range of molding conditions. It is preferable because toughness can be suitably imparted.
- the molar ratio of nylon 12 to nylon 6 in the copolymer of nylon 12 and nylon 6 is preferably 9.8: 0.2 to 6.5: 3.5 for nylon 12: nylon 6, and 9.5 : 0.5 to 7: 3 is more preferable.
- the shape of a true spherical polyamide particle having a melting point of 140 to 175 ° C. is a true sphere.
- the true spherical particles are obtained by measuring the short diameter and the long diameter of 10 randomly selected particles using a scanning electron microscope (manufactured by JEOL Ltd., JSM-6390). This refers to particles having an average value of the ratio of minor axis to major axis (minor axis / major axis) of 0.9 or more.
- the average particle size of the true spherical polyamide particles having a melting point of 140 to 175 ° C. is preferably 2 to 50 ⁇ m, more preferably 5 to 35 ⁇ m, even more preferably 6 to 25 ⁇ m.
- the component (C) is less likely to enter the component (A) when the prepreg is produced.
- the fiber reinforced composite material can be provided with further superior interlaminar fracture toughness. Moreover, the increase in the viscosity at the time of mixing (B) component and (C) component can be suppressed.
- the average particle diameter of the true spherical polyamide particles is not more than the upper limit of the above range, in the fiber-reinforced composite material, it is possible to suppress the (C) component from impairing the straightness of the reinforcing fiber of the (A) component, The deterioration of the mechanical properties of the fiber reinforced composite material can be suppressed, and the crack generated in the interlayer region of the fiber reinforced composite material due to the peeling stress in the out-of-plane direction can be retained in the interlayer region. Also, in the production of prepreg, when coating the mixture of component (B) and component (C) on the surface of the release paper with a uniform thickness, clogging may occur in equipment such as roll coaters and die coaters. Is suppressed. In addition, it is preferable that (c2) of this invention contains a thermosetting polyimide particle further.
- thermosetting polyimide particles that can be included in the component (c2) are thermosetting polyimide particles that can be used together as the component (C) together with true-spherical polyamide particles having a melting point of 140 to 175 ° C. as the component (c2). Means.
- the thermosetting polyimide particles that can be included in the component (c2) are thermosetting polyimide particles.
- a polyimide resin is a polymer compound having an imide bond in a repeating structure. Among polyimide resins, thermosetting polyimide resins have a relatively small molecular weight of the main chain and have reactive end groups.
- thermosetting polyimide particles Since it has a reactive end group, the interfacial property between the thermosetting polyimide particles and the matrix resin can be strengthened in the fiber-reinforced composite material, and the thermosetting polyimide can be included in the component (c2) described later.
- the effect of the particles can be sufficiently expressed.
- Thermoplastic polyimide resin with relatively high molecular weight and almost no reactive end groups cannot strengthen the interfacial properties with the matrix resin, and it is relatively tougher than thermosetting polyimide.
- the effect of thermosetting polyimide particles that can be included in the component (c2) described later cannot be sufficiently exhibited.
- thermosetting polyimide resin that can be preferably used as the thermosetting polyimide particles that can be included in the component (c2) is a thermosetting polyimide resin having a chemical structure of the general formula (1), and more specifically, benzophenone tetra Non-phthalimide carbon content prepared from carboxylic dianhydride (BTDA), 4,4'-methylenedianiline (MDA), and 2,4-toluenediamine (TDA) and containing 90-92 percent aromatic carbon Is a thermosetting polyimide resin.
- BTDA carboxylic dianhydride
- MDA 4,4'-methylenedianiline
- TDA 2,4-toluenediamine
- thermosetting polyimide resin examples include P84 (registered trademark) Polyimide manufactured by HP Polymer.
- Examples of the divalent linking group include —Ph— and —Ph—CH 2 —Ph—.
- -Ph- represents a phenylene group.
- thermosetting polyimide resin having a chemical structure of the general formula (2), more specifically, a thermosetting polyimide resin prepared from pyromellitic dianhydride and 4,4′-diaminodiphenyl ether (c2 ) Can be preferably used as a component.
- thermosetting polyimide resin examples include polyimide powder PIP series manufactured by Sanso Micron Corporation, such as PIP-3 and PIP-25.
- thermoplastic polyimide resin that cannot be preferably used as the thermosetting polyimide particles that can be included in the component (c2)
- Matrimid (registered trademark) 5218 manufactured by Huntsman.
- the average particle size of the thermosetting polyimide particles that can be included in the component (c2) is preferably 2 to 50 ⁇ m, and more preferably 5 to 35 ⁇ m.
- the thermosetting polyimide particles are less likely to enter the component (A) when a prepreg is produced. Therefore, it is easy to obtain a prepreg that satisfies the condition that 70% by mass or more of all the above-described (C) components exist in the surface layer of the (A) component, and as a result, the fiber reinforced composite material can be provided with further superior interlaminar fracture toughness.
- the increase in the viscosity at the time of mixing (B) component and the said thermosetting polyimide particle can be suppressed. Further, it is possible to prevent further aggregation and poor dispersion.
- thermosetting polyimide particles can be prevented from harming the straightness of the reinforcing fibers of the component (A) in the fiber reinforced composite material.
- clogging is caused by equipment such as a roll coater and a die coater. It is suppressed from waking up.
- the average particle diameter of the thermosetting polyimide particles is preferably in the range of 0.5 to 10 times the average particle diameter of true spherical polyamide particles having a melting point of 140 to 175 ° C.
- the average particle size of the thermosetting polyimide particles is larger than 0.5 times the average particle size of the true spherical polyamide particles having a melting point of 140 to 175 ° C., the true spherical polyamide particles having a melting point of 140 to 175 ° C., which will be described later, The combined effect of the thermosetting polyimide particles can be sufficiently exhibited.
- the thickness of the interlayer region is more uniform, that is, the reinforcing fiber of component (A) Can prevent the deterioration of the mechanical properties of the fiber reinforced composite material, and can prevent cracks generated in the interlayer region of the fiber reinforced composite material due to the peeling stress in the out-of-plane direction. It can be fastened.
- thermosetting polyimide resins have relatively poor toughness, and when used alone, excellent interlaminar fracture toughness cannot be imparted to fiber-reinforced composite materials.
- the crack generated in the interlayer region of the fiber reinforced composite material due to the peeling stress in the out-of-plane direction has a property of selectively progressing at a portion having poor toughness, for example, an interface between the matrix resin and the reinforced fiber bundle.
- the cracks generated in the interlayer region gradually move to the interface between the matrix resin with less toughness and the reinforcing fiber bundle as it progresses, and finally In particular, it has completely transferred from the interlayer region to the reinforcing fiber layer, and it has not been possible to stably provide excellent interlayer fracture toughness.
- thermosetting polyimide particles having poor toughness are arranged in the interlayer region of the fiber reinforced composite material, cracks generated in the interlayer region proceed stably in the interlayer region.
- composition of (c2) component spherical particle having a melting point of 140 to 175 ° C. The content of the true spherical polyamide particles having a melting point of 140 to 175 ° C. is 5 to 25 parts by mass, more preferably 10 to 25 parts by mass, and more preferably 12 to 25 parts by mass with respect to 100 parts by mass of the component (B). Preferably, 15 to 20 parts by mass is even more preferable. If the content of the component (C) is not less than the lower limit of the above range, the amount of the true spherical polyamide particles unevenly distributed in the interlayer region increases, and the interlaminar fracture toughness that is excellent in fiber-reinforced composite materials for the reasons described above. Can be stably provided.
- the component (B) in the matrix resin composed of the component (B) and the component (C) included in the prepreg is too low. Can be suppressed. That is, the deterioration of the mechanical properties of the fiber reinforced composite material due to the shortage of the component (B) can be prevented, the viscosity of the matrix resin composition is suppressed from becoming too high, and the component (A) The matrix resin composition can be sufficiently impregnated.
- composition of (c2) component of spherical particles having a melting point of 140 to 175 ° C. and thermosetting polyimide particles When the component (c2) further contains thermosetting polyimide particles, the total content of the sphere-shaped polyamide particles having a melting point of 140 to 175 ° C. and the thermosetting polyimide particles is based on 100 parts by mass of the component (B). 5 to 25 parts by mass, further preferably 10 to 25 parts by mass, more preferably 12 to 25 parts by mass, and still more preferably 15 to 20 parts by mass. If the total content of true spherical polyamide particles having a melting point of 140 to 175 ° C.
- thermosetting polyimide particles is not less than the lower limit of the above range, a true spherical polyamide having a melting point of 140 to 175 ° C. unevenly distributed in the interlayer region The amount of particles and thermosetting polyimide particles increases, and the interlaminar fracture toughness can be stably imparted to the fiber-reinforced composite material for the reasons described above. If the content of true-spherical polyamide particles having a melting point of 140 to 175 ° C.
- thermosetting polyimide particles in the content of component (c2), true spherical polyamide particles having a melting point of 140 to 175 ° C., and thermosetting polyimide particles The ratio of the thermosetting polyimide particles in the mixture of the true spherical polyamide particles having a melting point of 140 to 175 ° C. and the thermosetting polyimide particles is such that the true spherical polyamide particles and the thermosetting polyimide particles having a melting point of 140 to 175 ° C.
- the total content of is preferably 5 to 40% by mass.
- the ratio of the thermosetting polyimide particles is equal to or higher than the lower limit of the range, the effect of the thermosetting polyimide particles can be sufficiently obtained, and the interlaminar fracture toughness excellent in the fiber-reinforced composite material for the above-described reason. Can be applied stably. If the ratio of the said thermosetting polyimide particle is below the upper limit of the said range, it can suppress that the bad influence which the said thermosetting polyimide particle with poor toughness has on the interlaminar fracture toughness of a fiber reinforced composite material becomes large.
- the more preferable ratio of the thermosetting polyimide particles largely depends on the combination of the constituent elements. For example, when the interface between the component (A) and the component (B) is weaker, cracks generated in the interlayer region are more likely to proceed to the interface between the component (A) and the component (B). The ratio is preferably 20 to 40% by mass. On the other hand, when the interface between the component (A) and the component (B) is stronger, the cracks generated in the interlayer region are less likely to proceed to the interface between the component (A) and the component (B). The ratio is preferably 5 to 25% by mass. In addition, when the interface between the (B) component and the true spherical polyamide particles having a melting point of 140 to 175 ° C.
- thermosetting polyimide particles is preferably increased to 20 to 40% by mass.
- the ratio of the thermosetting polyimide particles is preferably 5 to 25% by mass.
- the ratio of the thermosetting polyimide particles is 25. It is preferable to further increase it to ⁇ 40% by mass.
- the proportion is preferably further reduced to 5 to 20% by mass.
- the weak interface between the component (A) and the component (B) means that the strength of the 90 ° bending test of the fiber-reinforced composite material in one direction (the reinforcing fibers are all oriented in the same direction) in accordance with ASTM D790. Is less than 125 MPa.
- the interface between the (B) component and the true spherical polyamide particles having a melting point of 140 to 175 ° C. and the interface between the thermosetting polyimide particles that can be included in the component (B) and the component (c2) are weak according to ASTM D5528.
- the fracture surface of the DCB (Double Cantilever Beam) test of the fiber-reinforced composite material was observed using a scanning electron microscope (SEM), and a true spherical polyamide having a melting point of 140 to 175 ° C. In this state, the particle interface and the thermosetting polyimide particle interface are exposed.
- SEM scanning electron microscope
- the fiber basis weight of the prepreg (content of reinforcing fiber per 1 m 2 : FAW) may be appropriately set according to the use of the prepreg, and is usually 50 to 300 g / m 2 .
- the resin content in the prepreg (the total ratio of the component (B) and the component (C)) is preferably 25 to 50% by mass and more preferably 30 to 40% by mass with respect to the total mass of the prepreg. If the resin content rate in a prepreg is more than the lower limit of the said range, it can suppress that the tack of a prepreg becomes low too much and can make it a tack suitable for handling. Furthermore, the mechanical characteristic fall of the fiber reinforced composite material resulting from (B) component shortage can also be prevented.
- the tack of the prepreg can be suppressed from becoming too high, and a tack suitable for handling can be obtained. Furthermore, the mechanical characteristic fall of the fiber reinforced composite material accompanying the improvement of Vf (volume ratio containing the reinforced fiber in the fiber reinforced composite material) resulting from the component (B) excess can also be prevented.
- the thickness of the prepreg may be appropriately set according to the use of the prepreg, and is usually 0.05 to 0.3 mm.
- the thickness of the prepreg can be measured with a thickness gauge.
- the prepreg in the present invention can be produced by the method disclosed in Patent Document 2, its application, and the like.
- the method ( ⁇ ), the method ( ⁇ ), A method ( ⁇ ) and a method ( ⁇ ) are preferred, and the (C) component can be unevenly distributed in the interlayer region more uniformly, and many (C) in the production process
- the method ( ⁇ ) or the method ( ⁇ ) is more preferable from the viewpoint that the components can be prevented from splashing and deteriorating the production environment.
- the resin film (F1) can be produced by coating the component (B) on the surface of a release paper or the like. Examples of the method for impregnating the component (B) with the component (A) include known methods such as a method of heating and pressing with a heated press roll.
- Resin film (F1) and base prepreg (P1) can be produced in the same manner as in method ( ⁇ ).
- the resin film (F2) can be produced by coating the component (B) on the surface of a release paper or the like and spraying the component (C) on the surface of the component (B).
- Examples of a method of bonding the resin film (F2) to the base prepreg (P1) include known methods such as a method of heating and pressurizing with a heating press roll. If the temperature is too high, most of the component (B) contained in the resin film (F2) is impregnated in the component (A) of the base prepreg (P1), and the tackiness of the prepreg is almost lost, and the fiber-reinforced composite material Problems may arise when manufacturing If the pressure is too high, most of the component (C) contained in the resin film (F2) enters the component (A) of the base prepreg (P1), and the straightness of the reinforcing fiber may be impaired. ) Component (C) component is almost lost on the surface of the component.
- the component (B) contained in the base prepreg (P1) and the component (B) contained in the resin film (F2) may have the same resin composition or different resin compositions. Due to the nature of the method ( ⁇ ) in which the resin film (F2) is further bonded to the base prepreg (P1), the content of the component (B) in the base prepreg (P1) should be lower than that in the method ( ⁇ ). preferable.
- a base prepreg (P1) is produced by laminating a resin film (F1) composed of the component (B) on one or both surfaces of the component (A) and impregnating the component (B) with the component (A).
- the resin film (F3) containing the component (B) and the component (C) is bonded to one side or both sides of the base prepreg (P1).
- the base prepreg (P1) can be produced in the same manner as in the method ( ⁇ ).
- Resin film (F3) can be produced by applying a mixture of component (B) and component (C) to the surface of a release paper or the like.
- Examples of a method of bonding the resin film (F3) to the base prepreg (P1) include known methods such as a method of heating and pressurizing with a hot press roll. If the temperature is too high, most of the component (B) contained in the resin film (F3) is impregnated in the component (A) of the base prepreg (P1), and the tackiness of the prepreg is almost lost, and the fiber-reinforced composite material Problems may arise when manufacturing If the pressure is too high, most of the component (C) contained in the resin film (F3) enters the component (A) of the base prepreg (P1), and the straightness of the reinforcing fiber may be impaired. ) Component (C) component is almost lost on the surface of the component.
- the component (B) contained in the base prepreg (P1) and the component (B) contained in the resin film (F3) may have the same resin composition or different resin compositions.
- the content of the component (B) in the base prepreg (P1) should be lower than that in the method ( ⁇ ). preferable.
- the resin film (F3) can be produced in the same manner as in the method ( ⁇ ).
- the (C) component is filtered on the (A) component, and the (C) component is unevenly distributed near the surface of the prepreg. Since the component (C) is filtered on the component (A), the average particle size of the component (C) is preferably larger than that of other methods.
- the average particle diameter of the (c1) component polyamide particles and (c2) the true spherical particles of polyamide having a melting point of 140 to 175 ° C. is preferably 5 to 50 ⁇ m, more preferably 10 to 50 ⁇ m, and even more preferably 15 to 50 ⁇ m.
- the average particle diameter of the thermosetting polyimide particles (c1) and the thermosetting polyimide particles that can be contained in the component (c2) is preferably 15 to 50 ⁇ m, more preferably 20 to 50 ⁇ m, and further preferably 25 to 50 ⁇ m. preferable.
- the fiber-reinforced composite material according to the first aspect of the present invention is obtained by laminating two or more prepregs and heating at or above the curing temperature of the component (B) to cure the component (B).
- the temperature at which the laminated prepreg is heat-molded may be any temperature as long as the component (B) can be appropriately cured, but the time required for curing the component (B) of the equipment used for the thermoforming To 170-190 ° C. If the temperature at the time of heat molding is 170 ° C. or higher, the component (B) is sufficiently cured, and a cured product having higher heat resistance can be obtained. If the temperature at the time of heat forming is 190 ° C. or less, it is possible to use cheaper equipment and auxiliary materials used for heat forming.
- the interface between the component (B) and the component (C) is further strengthened, and cracks generated in the interlayer region of the cured product are more difficult to progress to the interface between the component (A) and the component (B).
- the component is crystalline, it is preferably cured at a temperature equal to or higher than the melting point of the component (C), and when the component (C) is amorphous, it is cured at a temperature equal to or higher than the glass transition temperature of the component (C).
- the heat molding time may be any time that can sufficiently cure the component (B) and is suitable for the heat molding method described later.
- the heat molding time is preferably 1 to 4 hours. If the heat molding time is 1 hour or longer, the curing of the component (B) is sufficient. If the thermoforming exceeds 4 hours, the production cost becomes higher.
- thermoforming method examples include known methods such as an autoclave molding method, an oven molding method, and a press molding method.
- an autoclave molding method is preferable because a fiber-reinforced composite material having more excellent mechanical properties can be obtained.
- the fiber-reinforced composite material according to the second aspect of the present invention includes a component (A), a component (B ′) and a component (C), the component (A) is a reinforcing fiber base, and the component (B ′) It is a cured product of an epoxy resin composition, the component (C) is the component (c1) or the component (c2), the component (c1) includes polyamide particles and thermosetting polyimide particles, and the component (c2) has a melting point. It contains true spherical particles of polyamide at 140-175 ° C.
- a plurality of (A) components are laminated, and the (C) component exists between the layers of the (A) component.
- the component (A) and the component (C) include the same components as the component (A) and the component (C) described in the description of the prepreg.
- the component (B ′) include those obtained by curing the component (B) described in the description of the prepreg.
- the component (B) is preferably heated to 170 to 190 ° C. The heating time is preferably 1 to 4 hours.
- the average particle diameter of the resin particles was determined as follows. The resin particles were subjected to particle size distribution measurement using a laser scattering particle size measuring machine (manufactured by JGC Corporation, MODEL: 7340 Microtrac FRA) to obtain a cumulative distribution. The particle diameter (D50) at which the cumulative frequency on a volume basis in the cumulative distribution was 50% was defined as the average particle diameter.
- the laminated prepreg was heated from room temperature to 180 ° C. at a heating rate of 2 ° C./min or 0.5 ° C./min using an autoclave and held for 2 hours.
- the laminated prepreg was held in the autoclave at a temperature lowering speed of 3 ° C./min until it became 50 ° C. or lower.
- the evaluation molded plate 1 was taken out from the autoclave.
- the pressure in the autoclave was 0.6 MPa from the start of heating to the removal. (Uniformity ratio of heat-deformed resin particles)
- a 20 mm square test piece was cut out from the evaluation molded plate.
- the cross section of the test piece was polished using a polishing machine (REFINE-POLISHER APM-122, manufactured by Refine Tech Co., Ltd.). Using a digital microscope (manufactured by KEYENCE, VHX-5000), a photograph in which the cross section of the test piece was magnified 500 times was obtained. From the photograph, the heat-deformable resin particles in the interlayer region between the reinforcing fiber substrates and the heat-deformable resin particles in the reinforcing fiber substrates are cut out, the mass of the cut-out photograph is measured, and unevenly distributed from the following formula (3) The rate was calculated.
- a polishing machine REFINE-POLISHER APM-122, manufactured by Refine Tech Co., Ltd.
- Uneven distribution rate mass of thermally deformable resin particles present in the interlayer region / (mass of thermally deformable resin particles present in the interlayer region + mass of thermally deformable resin particles in the reinforcing fiber substrate) ⁇ 100 Formula ( 3)
- GIC Measurement of GIC
- GIIC Measurement of GIIC
- ⁇ Raw material> ((A) component) (Reinforced fiber bundle) MR70: Carbon fiber bundle (manufactured by Mitsubishi Chemical Corporation, PYROFIL (registered trademark) MR70 12P, strand strength: 7000 MPa, carbon fiber diameter: 5 ⁇ m, number of carbon fibers: 12000).
- jER807 Bisphenol F type liquid epoxy resin (manufactured by Mitsubishi Chemical Corporation, jER (registered trademark) 807).
- jER604 diaminodiphenylmethane type semi-solid epoxy resin (manufactured by Mitsubishi Chemical Corporation, jER (registered trademark) 604).
- VESTOSINT 2158 Polyamide 12 particles (manufactured by Daicel-Evonik, VESTOSINT (registered trademark) 2158 natural, melting point: 177 ° C., average particle size: 21 ⁇ m).
- p84 Thermosetting polyimide particles (manufactured by HP Polymer, P84 (registered trademark) Polyimide, average particle size: 17.7 ⁇ m).
- PIP-3 thermosetting polyimide particles (manufactured by Sanso Micron, polyimide powder PIP-3, average particle size: 2.8 ⁇ m).
- PIP-25 Thermosetting polyimide particles (manufactured by Sanso Micron, polyimide powder PIP-25, average particle size: 12.9 ⁇ m). (Resin particles not included in component (c1))
- P84NT1 Polyimide particles (manufactured by Daicel-Evonik, polyimide P84 (registered trademark) NT1, average particle diameter: 8.9 ⁇ m).
- P84NT2 polyimide particles (manufactured by Daicel-Evonik, polyimide P84 (registered trademark) NT2, average particle diameter: 15.9 ⁇ m)
- Example 1> (Preparation of component (B)) To the planetary mixer, 35 parts by mass of TSR-400, 49 parts by mass of jER807, 16 parts by mass of jER604, and 2.8 parts by mass of E2020P were added. The jacket temperature of the planetary mixer was set to 140 to 160 ° C., and mixing was performed until the raw materials became uniform. It stood to cool until the temperature of the content became 60 degrees C or less, and 40.6 mass parts of Seikacure S was added to the planetary mixer. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain the component (B).
- the mixture (BC) was applied to the surface of the release paper with a uniform thickness to produce a resin film (F3).
- the resin film (F3) is bonded to both sides of the component (A) in which a plurality of MR70s are aligned to form a sheet, and the component (A) is impregnated into the component (A) using a fusing press.
- the component (C) was filtered on the component (A), and the component (C) was unevenly distributed near the surface of the prepreg to obtain a prepreg.
- Table 1 shows the composition and the production method of the prepreg. (Manufacture and evaluation of fiber reinforced composite materials) In accordance with the method described above, a molded plate for evaluation was produced. Evaluation was performed on the evaluation molded plate. The results are shown in Table 1.
- Example 2 (Preparation of component (B)) In the same manner as in Example 1, component (B) was obtained. (Preparation of mixture (BC)) 18.6 parts by mass of VESTINTINT 2158 and 9.3 parts by mass of p84 are added to 143.4 parts by mass of component (B) in the planetary mixer, and the total content of VESTOSINT 2158 and p84 is added to 100 parts by mass of component (B). The proportion of p84 in the total content of 19.5 parts by mass and VESTOSINT 2158 and p84 was 33.3% by mass. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC).
- Example 3 (Preparation of component (B)) In the same manner as in Example 1, component (B) was obtained. (Preparation of mixture (BC)) VESTOSINT 2158 (14.3 parts by mass) and p84 (4.2 parts by mass) are added to 143.4 parts by mass of component (B) in the planetary mixer, and the total content of VESTOSINT 2158 and p84 with respect to 100 parts by mass of component (B). The proportion of p84 in the total content of 12.9 parts by mass and VESTOSINT 2158 and p84 was 22.7% by mass. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC).
- Example 4 (Preparation of component (B)) In the same manner as in Example 1, component (B) was obtained. (Preparation of mixture (BC)) 14.5 parts by weight of VESTINTINT 2158 and 2.0 parts by weight of p84 are added to 143.4 parts by weight of the component (B) in the planetary mixer, and the total content of VESTINTINT 2158 and p84 with respect to 100 parts by weight of the component (B). The proportion of p84 in the total content of 12.9 parts by mass and VESTOSINT 2158 and p84 was 10.8% by mass. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC).
- Example 5 (Preparation of component (B)) In the same manner as in Example 1, component (B) was obtained. (Preparation of mixture (BC)) 12.3 parts by mass of VESTOSINT 2158 and 6.2 parts by mass of PIP-3 are added to 143.4 parts by mass of the component (B) in the planetary mixer, and the total of VESTOSINT 2158 and PIP-3 with respect to 100 parts by mass of the component (B) The content of PIP-3 in the total content of VESTOSINT 2158 and PIP-3 was 33.5% by mass. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC).
- Example 6 (Preparation of component (B)) In the same manner as in Example 1, component (B) was obtained. (Preparation of mixture (BC)) 12.3 parts by mass of VESTOSINT 2158 and 6.2 parts by mass of PIP-25 are added to 143.4 parts by mass of the component (B) in the planetary mixer, and the total of VESTOSINT 2158 and PIP-25 with respect to 100 parts by mass of the component (B) The content of PIP-25 in the total content of VESTOSINT 2158 and PIP-25 was 33.5% by mass. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC).
- the fiber reinforced composite materials of Examples 1 to 6 exhibit a GIIc of 1.0 kJ / m 2 or more under any molding conditions of 2.0 ° C./min and 0.5 ° C./min, and have mode II interlaminar fracture toughness. It was excellent.
- the fiber reinforced composite materials of Comparative Examples 1 to 3 that do not contain thermosetting polyimide particles exhibit a GIIc of less than 1.0 kJ / m 2 under molding conditions of 0.5 ° C./min, and have a Mode II interlaminar fracture toughness. inferior.
- Example 4 containing only thermosetting polyimide particles, the values were as low as less than 1.0 kJ / m 2 under any molding conditions of 2.0 ° C./min and 0.5 ° C./min.
- Example 2 which contained more (C) components showed higher GIIc.
- the total content of the component (C) with respect to 100 parts by mass of the component (B) is more preferable in Example 2.
- Example 1 is compared with Examples 3 and 4
- GIIc under the molding conditions of 0.5 ° C./min is higher in the order of Example 4, Example 3, and Example 1.
- the ratio of the thermosetting polyimide particles occupying the component (C) ((c1) polyamide particles and thermosetting polyimide particles) is higher than that in Example 4 than in Example 3.
- Example 1 is preferable to Example 3.
- the fiber-reinforced composite material obtained by the method for producing a fiber-reinforced composite material of the present invention is to provide a fiber-reinforced composite material that is stably excellent in mode I interlayer fracture toughness and mode II interlayer fracture toughness regardless of the production conditions. It is useful for aircraft and sports / leisure applications, automobile applications, other general industrial applications (tension materials), and the like.
- ⁇ Raw material> ((A) component) (Reinforced fiber bundle) MR70: Carbon fiber bundle (manufactured by Mitsubishi Chemical Corporation, PYROFIL (registered trademark) MR70 12P, strand strength: 7000 MPa, carbon fiber diameter: 5 ⁇ m, number of carbon fibers: 12000).
- TSR-400 An epoxy resin having an oxazolidone ring skeleton (manufactured by DIC, EPICLON TSR-400).
- jER807 Bisphenol F type liquid epoxy resin (manufactured by Mitsubishi Chemical Corporation, jER (registered trademark) 807).
- jER604 diaminodiphenylmethane type semi-solid epoxy resin (manufactured by Mitsubishi Chemical Corporation, jER (registered trademark) 604).
- ((C) component) Component (c2) Polyamide Particles MW-330: Spherical Copolymerized Polyamide Particles (manufactured by Sumika Environmental Science Co., Ltd., MW-330, melting point: 166 ° C., average particle size: 8 ⁇ m, average value of short diameter / long diameter: 0 .96).
- Thermosetting polyimide particles that can be included in the component p84 Thermosetting polyimide particles (manufactured by HP Polymer, P84 (registered trademark) Polyimide, average particle size: 18 ⁇ m).
- VESTOSINT 2158 Non-spherical polyamide 12 particles (manufactured by Daicel-Evonik Co., Ltd., VESTOSINT (registered trademark) 2158 natural, melting point: 177 ° C., average particle diameter: 21 ⁇ m, average value of short diameter / long diameter: 0.65).
- Ny12 particle A True spherical polyamide 12 particle (melting point: 177 ° C., average particle diameter: 21 ⁇ m, average value of short diameter / long diameter: 0.94).
- TR55 True spherical amorphous polyamide particles (Milamide (registered trademark) TR55, manufactured by M Chemie, melting point: 160 ° C., average particle diameter: 10 ⁇ m, average value of short diameter / long diameter: 0.91).
- Example 7 (Preparation of component (B)) To the planetary mixer, 35 parts by mass of TSR-400, 49 parts by mass of jER807, 16 parts by mass of jER604, and 2.8 parts by mass of E2020P were added. The jacket temperature of the planetary mixer was set to 140 to 160 ° C., and mixing was performed until the raw materials became uniform. It stood to cool until the temperature of the content became 60 degrees C or less, and 40.6 mass parts of Seikacure S was added to the planetary mixer. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain the component (B).
- a prepreg was produced by the method ( ⁇ ).
- the component (B) and the mixture (BC) were respectively applied to the surface of the release paper with a uniform thickness to produce resin films (F1) and (F3).
- a resin film (F1) is laminated on both sides of the component (A), which is formed by aligning a plurality of MR70 sheets, and a base prepreg is impregnated with the component (B) using a fusing press.
- (P1) was produced, and a resin film (F3) was bonded to both sides of the base prepreg (P1) to obtain a prepreg.
- Content of (C) component with respect to 100 mass parts of (B) component of the obtained prepreg will be 13.0 mass parts.
- Table 4 shows the composition and the production method of the prepreg.
- Example 8 (Preparation of component (B)) In the same manner as in Example 1, component (B) was obtained.
- a prepreg was produced by the method ( ⁇ ).
- the mixture (BC ′) was applied to the surface of the release paper with a uniform thickness to produce a resin film (F3).
- the resin film (F3) is laminated on both sides of the component (A) in which a plurality of MR70s are aligned to form a sheet, and the component (B) is added to the component (A) using a fusing press.
- VESTOSINT 2158 was filtered on the component (A), and VESTOSINT 2158 was unevenly distributed in the vicinity of the surface of the prepreg to obtain a prepreg.
- the total content of VESTOSINT 2158 with respect to 100 parts by mass of component (B) of the obtained prepreg is 13.0 parts by mass.
- Table 4 shows the composition and the production method of the prepreg.
- a prepreg was produced by the method ( ⁇ ). Using a hot melt coater, the mixture (BC ′) was applied to the surface of the release paper with a uniform thickness to produce a resin film (F3).
- the resin film (F3) is bonded to both sides of the component (A) in which a plurality of MR70s are aligned to form a sheet, and the component (A) is impregnated into the component (A) using a fusing press.
- VESTOSINT 2158 and p84 were filtered on the component (A), and VESTOSINT 2158 and p84 were unevenly distributed near the surface of the prepreg to obtain a prepreg.
- the total content of VESTOSINT 2158 and p84 with respect to 100 parts by mass of component (B) of the obtained prepreg is 12.9 parts by mass.
- Table 4 shows the composition and the production method of the prepreg.
- a prepreg was produced by the method ( ⁇ ). Using a hot melt coater, the mixture (BC ′) was applied to the surface of the release paper with a uniform thickness to produce a resin film (F3).
- the resin film (F3) is bonded to both sides of the component (A) in which a plurality of MR70s are aligned to form a sheet, and the component (A) is impregnated into the component (A) using a fusing press.
- Ny12 particles A are filtered on the component (A) and Ny12 particles 12 particles A were unevenly distributed to obtain a prepreg.
- Component (B) of the obtained prepreg 1 The content of Ny12 particles A with respect to 00 parts by mass is 12.9 parts by mass.
- Table 4 shows the composition and the production method of the prepreg.
- a prepreg was produced by the method ( ⁇ ). Using a hot melt coater, the mixture (BC ′) was applied to the surface of the release paper with a uniform thickness to produce a resin film (F3).
- the resin film (F3) is bonded to both sides of the component (A) in which a plurality of MR70s are aligned and formed into a sheet, and the component (A) is impregnated into the component (A) using a fusing press, TR55 was filtered on the component (A), and TR55 was unevenly distributed near the surface of the prepreg to obtain a prepreg.
- grains A with respect to 100 mass parts of (B) component of the obtained prepreg will be 13.0 mass parts.
- Table 4 shows the composition and the production method of the prepreg.
- the fiber-reinforced composite materials of Examples 7 and 8 exhibited a GIc of 0.7 kJ / m 2 or more under both molding conditions of 2.0 ° C./min and 0.5 ° C./min, and 1.5 kJ / m. GIIc of 2 or more was exhibited, and the interlaminar fracture toughness was stable and excellent.
- the fiber reinforced composite materials of Comparative Examples 5 to 8 containing no component (C) exhibited GIc and GIIc which were greatly inferior under molding conditions of 0.5 ° C./min, and had unstable interlaminar fracture toughness.
- thermosetting polyimide particles that can be further included in component (c2) (component (C) together with true spherical polyamide particles having a melting point of 140 to 175 ° C. of component (c2))
- component (c2) component (C) together with true spherical polyamide particles having a melting point of 140 to 175 ° C. of component (c2)
- the difference between GIc and GIIc was smaller than that due to molding conditions, and more stable interlaminar fracture toughness was exhibited.
- the glass transition temperature was measured according to ASTM D4065 using ARES-RDA (manufactured by TA Instruments).
- ASTM D4065 the temperature of the intersection of the tangent in the glass state and the tangent at the inflection point in the transition state is defined as the glass transition temperature.
- the width perpendicular to the fiber axis direction of the reinforcing fiber was 12.7 mm.
- the heating rate was 5 ° C./min and the frequency was 1 Hz.
- the moisture absorption process was performed by immersing the test specimen for a measurement for 2 weeks in 71 degreeC warm water before a measurement.
- ⁇ Raw material> ((A) component) (Reinforced fiber bundle) MR70: Carbon fiber bundle (manufactured by Mitsubishi Chemical Corporation, PYROFIL (registered trademark) MR70 12P, strand strength: 7000 MPa, carbon fiber diameter: 5 ⁇ m, number of carbon fibers: 12000).
- HP-4032SS Bifunctional naphthalene type epoxy resin (manufactured by DIC, EPICLON (registered trademark) HP-4032SS)
- HP-4700 Tetrafunctional naphthalene type epoxy resin (manufactured by DIC, EPICLON (registered trademark) HP-4700)
- jER807 Bisphenol F type liquid epoxy resin (manufactured by Mitsubishi Chemical Corporation, jER (registered trademark) 807)
- jER604 diaminodiphenylmethane type semi-solid epoxy resin (manufactured by Mitsubishi Chemical Corporation, jER (registered trademark) 604)
- Curing agent Seikacure S: 4,4'-diaminodiphenylsulfone (Wakayama Seika Kogyo Co., Ltd., Seikacure S)
- E2020P Polyethersulfone (manufactured by BASF Japan, ULTRASON (registered trademark) E2020 P MICRO) (
- Example 9> (Preparation of component (B)) To a planetary mixer, 35 parts by mass of TSR-400, 65 parts by mass of HP-4032SS, and 2.8 parts by mass of E2020P were added. The jacket temperature of the planetary mixer was set to 140 to 160 ° C., and mixing was performed until the raw materials became uniform. The contents were allowed to cool to 60 ° C. or less, and 43 parts by mass of Seikacure S was added to the planetary mixer. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain the component (B).
- a resin film (F1) is laminated on both sides of the component (A), which is formed by aligning a plurality of MR70 sheets, and a base prepreg is impregnated with the component (B) using a fusing press.
- (P1) was produced, and a resin film (F3) was bonded to both sides of the base prepreg (P1) to obtain a prepreg.
- the sum total of content of (C) component with respect to 100 mass parts of (B) component of the obtained prepreg will be 13.0 mass parts.
- Table 5 shows the composition and the production method of the prepreg. (Manufacture and evaluation of fiber reinforced composite materials) In accordance with the method described above, a molded plate for evaluation was produced. Evaluation was performed on the evaluation molded plate. The results are shown in Table 5.
- Example 10 (Preparation of component (B)) In the same manner as in Example 9, component (B) was obtained. (Preparation of mixture (BC ′)) 38.6 parts by mass of MW-330 and 4.7 parts by mass of p84 were added to 143.4 parts by mass of the component (B) in the planetary mixer. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC ′). (Preparation of prepreg) Using a hot melt coater, the component (B) and the mixture (BC ′) were respectively applied to the surface of the release paper with a uniform thickness to prepare resin films (F1) and (F3).
- a resin film (F1) is laminated on both sides of the component (A), which is formed by aligning a plurality of MR70 sheets, and a base prepreg is impregnated with the component (B) using a fusing press.
- (P1) was produced, and a resin film (F3) was bonded to both sides of the base prepreg (P1) to obtain a prepreg.
- the total content of the component (c2) component with respect to 100 parts by mass of the component (B) of the obtained prepreg is 13.0 parts by mass.
- Table 5 shows the composition and the production method of the prepreg. (Manufacture and evaluation of fiber reinforced composite materials) In the same manner as in Example 9, a molded plate for evaluation was produced and evaluated. The results are shown in Table 5.
- Example 11> 35 parts by mass of HP-4700, 65 parts by mass of HP-4032SS, and 2.8 parts by mass of E2020P were added to the planetary mixer.
- the jacket temperature of the planetary mixer was set to 140 to 160 ° C., and mixing was performed until the raw materials became uniform. The contents were allowed to cool to 60 ° C. or less, and 51.5 parts by mass of Seika Cure S was added to the planetary mixer.
- the jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain the component (B).
- Preparation of mixture (BC) 45.9 parts by mass of MW-330 was added to 154.3 parts by mass of the component (B) in the planetary mixer.
- Example 12 (Preparation of component (B)) In the same manner as in Example 11, component (B) was obtained. (Preparation of mixture (BC ′)) 35.4 parts by mass of MW-330 and 10.5 parts by mass of p84 were added to 154.3 parts by mass of the component (B) in the planetary mixer. The jacket temperature was set to 55 to 70 ° C., and the mixture was mixed until the raw materials became uniform to obtain a mixture (BC ′). (Preparation of prepreg) In the same manner as in Example 10, a prepreg was produced by the method ( ⁇ ). The total content of the component (c2) with respect to 100 parts by mass of the component (B) of the obtained prepreg is 13.0 parts by mass. Table 5 shows the composition and the production method of the prepreg. (Manufacture and evaluation of fiber reinforced composite materials) In the same manner as in Example 9, a molded plate for evaluation was produced and evaluated. The results are shown in Table 5.
- the present invention it is possible to provide a prepreg and a fiber-reinforced composite material that are stably excellent in mode I interlayer fracture toughness and mode II interlayer fracture toughness regardless of manufacturing conditions such as a curing temperature and a heating rate.
Abstract
Description
本願は、2017年3月24日に、日本に出願された特願2017-058507号、2017年3月24日に、日本に出願された特願2017-058508号、及び2018年2月23日に、日本に出願された特願2018-030289号に基づき優先権を主張し、その内容をここに援用する。
(1)層間領域に高靱性なポリアミド等の微粒子を配置した繊維強化複合材料(特許文献1、2)。
(2)層間領域に特定の粒子径分布指数、真球度およびガラス転移温度を有する微粒子を配置し、マトリックス樹脂にエラストマー成分を含ませた特定の繊維強化複合材料(特許文献3)。
(3)層間領域に高靱性なポリアミドの微粒子を配置し特異なモルホロジーを形成させた繊維強化複合材料、および製造方法。(特許文献4、5)。
近年、航空機の構造材、風車のブレード等の大型かつ3次元的な曲面形状を有する部材への繊維強化複合材料の適応が進められている。大型部材や3次元的な曲面形状を有する部材に引張や圧縮の応力が負荷された場合、繊維強化複合材料の層間領域に面外方向への引き剥がし応力が発生する。よって、繊維強化複合材料の層間領域において生じた亀裂の進行を抑制するモードI層間破壊靱性およびモードII層間破壊靱性が重要な特性となっている。
(2)の繊維強化複合材料は、衝撃後圧縮強度の向上に有効なモードII層間破壊靱性および大型化や3次元的な曲面形状のような複雑化に必要なモードI層間破壊靱性が高い。しかし、成型後の微粒子のモルホロジーや微粒子とマトリックス樹脂組成物との界面性状に関して十分に考慮されておらず、繊維強化複合材料の製造条件によっては特許文献2と同様に繊維強化複合材料の層間領域に面外方向への引き剥がし応力により生じた亀裂が層間領域に安定して留まることが出来ないため、微粒子配置の効果が十分に得ることが出来ない。
(3)の特許文献には、層間領域に高靱性なポリアミドの微粒子を配置し特異なモルホロジーを形成させることで、モードI層間破壊靱性およびモードII層間破壊靱性に優れた繊維強化複合材料を製造できる方法が開示されている。しかし、硬化温度や昇温速度等の製造条件に制約がかかり、前記特異なモルホロジーを形成させることができなければ、十分なモードI層間破壊靱性およびモードII層間破壊靱性は発現しない。
[1] (A)成分、(B)成分および(C)成分を含むプリプレグであって、
前記(A)が強化繊維基材であり、
前記(B)がエポキシ樹脂組成物であり、
前記(C)成分が(c1)成分または(c2)成分であり、
前記(c1)成分がポリアミド粒子と熱硬化性ポリイミド粒子とを含み、
前記(c2)成分が融点140~175℃のポリアミドの真球形状の粒子を含む、プリプレグ。
[2] 前記(C)成分が前記(c1)成分である、[1]に記載のプリプレグ。
[3] 前記(C)成分が前記(c1)成分であり、
[ポリアミド粒子]:[熱硬化性ポリイミド粒子]で表される質量比が60:40~95:5である、[1]または[2]に記載のプリプレグ。
[4] 前記(C)成分が前記(c1)成分であり、
前記(c1)成分中のポリアミド粒子の融点が140℃~175℃である、[1]から[3]のいずれか一項に記載のプリプレグ。
[5] 前記(C)成分が前記(c1)成分であり、
前記(c1)中のポリアミド粒子が結晶性共重合ナイロン粒子である、[1]から[4]のいずれか一項に記載のプリプレグ。
[6] 前記(C)成分が前記(c1)成分であり、
前記(c1)中のポリアミド粒子が、ナイロン12とナイロン6との共重合体からなる真球形状の粒子である、[1]から[4]のいずれか一項に記載のプリプレグ。
[7] 前記(C)成分が前記(c2)成分である、[1]に記載のプリプレグ。
[8] 前記融点140~175℃のポリアミドの真球形状の粒子が、結晶性共重合ナイロン粒子である、[7]に記載のプリプレグ。
[9] 前記融点140~175℃のポリアミドの真球形状の粒子が、ナイロン12とナイロン6との共重合体からなる真球形状の粒子である、[7]に記載のプリプレグ。
[10] 前記(C)成分が前記(c2)成分であり、
前記(c2)が、更に熱硬化性ポリイミド粒子を含む、[1]、および[7]から[9]のいずれかに記載のプリプレグ。
[11]
前記(C)成分が前記(c2)成分であり、
前記(c2)が、更に熱硬化性ポリイミド粒子を含み、
前記熱硬化性ポリイミド粒子が、下記一般式(1)または下記一般式(2)の化学構造を含む、[1]、および[7]~[10]のいずれか一項に記載のプリプレグ。


[13] 前記(A)成分が強化繊維を含み、前記強化繊維が炭素繊維である、[1]から[12]のいずれか一項に記載のプリプレグ。
[14] 前記(B)成分が、エポキシ樹脂、及び芳香族ポリアミンを含有し、
前記エポキシ樹脂が、ナフタレン骨格を有するエポキシ樹脂を含有し、
前記ナフタレン骨格を有するエポキシ樹脂の含有量が、前記エポキシ樹脂の総質量に対し、60~100質量%である、[1]から[13]のいずれか一項に記載のプリプレグ。
[15] 前記(C)成分の含有量が、前記(B)成分100質量部に対し5~25質量部である、[1]~[14]のいずれか一項に記載のプリプレグ。
[16] [1]から[15]のいずれか一項に記載のプリプレグを2枚以上積層し、前記(B)成分の硬化温度以上で加熱してなる繊維強化複合材料。
[17] (A)成分、(B’)成分および(C)成分を含む繊維強化複合材料であって、
前記(A)成分が強化繊維基材であり、
前記(B’)成分がエポキシ樹脂組成物の硬化物であり、
前記(C)成分が(c1)成分又は(c2)成分であり、
前記(c1)成分がポリアミド粒子と熱硬化性ポリイミド粒子とを含み、
前記(c2)成分が融点140~175℃のポリアミドの真球形状の粒子を含み、
前記(A)成分が複数枚積層されており、かつ前記(C)成分が前記(A)成分の層間に存在する、繊維強化複合材料。
前記エポキシ樹脂が、オキサゾリドン環骨格を有するエポキシ樹脂を含有する、[1]から[15]のいずれか一項に記載のプリプレグ。
[19] 前記オキサゾリドン環骨格を有するエポキシ樹脂の割合は、前記(B)成分中の前記エポキシ樹脂100質量%に対し、10~40質量%である、[18]に記載のプリプレグ。
[20] 前記(B)成分が、芳香族ポリアミンを含有し、
前記芳香族ポリアミンが4,4’-ジアミノジフェニルスルホンである、[1]から[15]、[18]、及び[19]のいずれか一項に記載のプリプレグ。
[21] 前記(B)成分が、エポキシ樹脂を含有し、
前記エポキシ樹脂が、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、及び3官能以上のエポキシ樹脂からなる群から選択される少なくとも1種のエポキシ樹脂を含む、[1]から[15]、および[18]から[20]に記載のプリプレグ。
本発明のプリプレグは、下記(A)成分、(B)成分および(C)成分を含むプリプレグである。
(A)成分:強化繊維基材
(B)成分:エポキシ樹脂組成物
(C)成分:(c1)成分又は(c2)成分であり、前記(c1)成分がポリアミド粒子と熱硬化性ポリイミド粒子を含み、前記(c2)成分が融点140~175℃のポリアミドの真球形状の粒子を含む。
また、本発明の繊維強化複合材料は、上記(A)成分、(B’)成分の硬化物および(C)成分を含む繊維強化複合材料であって、(A)成分が複数枚積層されており、かつ(C)成分が(A)成分の表層に存在する、繊維強化複合材料である。
更に本発明の繊維強化複合材料は、上記のプリプレグの2枚以上が積層された積層体の硬化物である。
本発明および本発明に関する説明において使用する用語は、以下の通りである。
「結晶性ポリアミド樹脂」とは、示差走査熱量測定(以下、DSCと記載する。)において融点が現れる樹脂のことを意味する。
「非晶性ポリアミド樹脂」とは、DSCにおいて融点が現れない樹脂のことを意味する。
・「層間」とはプリプレグを積層して製造された繊維強化複合材料において、積層されるプリプレグとプリプレグとの境界近傍の強化繊維の分率が小さくなっている領域を意味する。
「真球形状の粒子」とは、真球状の粒子とは、走査型電子顕微鏡(日本電子社製、JSM-6390)を用い、無作為に選ばれた10個の粒子について短径および長径を測定し、当該10個の粒子の長径に対する短径の比(短径/長径)の平均値が0.95以上である粒子のことを指す。
「融点」は、以下のように得られた結晶性樹脂のDSC曲線の融解ピーク温度である。結晶性樹脂を、室温から、推測される融点よりも約30℃高い温度まで、10℃/分で加熱し、推測される融点よりも約30℃高い温度にて10分間保った。次に結晶性樹脂を、推測される融点よりも約50℃低い温度まで10℃/分で冷却した。そして結晶性樹脂を、推測される融点よりも約30℃高い温度まで10℃/分で加熱した。
「ガラス転移温度」は、以下の通り非結晶性樹脂のDSC測定から求めた中間点ガラス転移温度である。非晶性樹脂を、室温から、推測されるガラス転移温度よりも約30℃高い温度まで、10℃/分で加熱し、推測されるガラス転移温度よりも約30℃高い温度にて10分間保った。非晶性樹脂を、推測されるガラス転移温度よりも約50℃低い温度まで急冷した非晶性樹脂を、推測されるガラス転移温度よりも約30℃高い温度まで20℃/分で加熱した。得られたDSC曲線のガラス転移温度に伴うベースラインの転移箇所において、低温側のベースラインを延長した直線と高温側のベースラインを延長した直線とから縦軸方向に等距離にある直線とベースラインの転移部分の曲線が交わる点をガラス転移温度とした。
「平均粒子径」は、粒子径分布測定によって得られた体積基準による累積分布において累積頻度50%にあたる粒子径(D50)のことを意味する。
「エポキシ樹脂」とは、分子内に2つ以上のエポキシ基を有する化合物のことを意味する。
「層間破壊靱性」とは、単位面積辺りの層間剥離亀裂を生じる際に必要なエネルギーの限界値のことを意味する。
「GIC」とは、亀裂進展初期のモードI層間破壊靱性値のことを意味する。
「GIIC」とは、亀裂進展初期のモードII層間破壊靱性値のことを意味する。
「モードI」とは、亀裂開口変位の方向が各々亀裂面に対して垂直な(開口形)変形モードのことを意味する。
「モードII」とは、亀裂開口変位の方向が亀裂面に平行で、亀裂前縁に垂直な(縦せん断形)変形モードのことを意味する。
「亀裂開口変位」とは、亀裂上下面の相対的変位をいう。
本発明の一実施形態に係る繊維強化複合材料は、特定の条件を満たす2つ以上のプリプレグを積層して硬化させてなる繊維強化複合材料である。
プリプレグは、(A)成分、(B)成分、および(C)成分を含む。
(A)成分は、強化繊維基材であり、シート状であることが好ましい。強化繊維基材は、強化繊維が単一方向に配列したものであってもよく、ランダム方向に配列したものであってもよい。
(A)成分の形態としては強化繊維の織物、および不織布、並びに強化繊維の長繊維が一方向に引き揃えられたシート等が挙げられる。
(A)成分は、比強度や比弾性率が高い繊維強化複合材料を成形することができるという観点からは、長繊維が単一方向に引き揃えられた強化繊維の束からなるシートであることが好ましく、取り扱いが容易であるという観点からは、強化繊維の織物であることが好ましい。
強化繊維の材質としては、ガラス繊維、炭素繊維(黒鉛繊維を包含する。)、アラミド繊維、およびボロン繊維等が挙げられる。強化繊維基材は、繊維強化複合材料の機械的物性および軽量化の観点から、炭素繊維基材が好ましい。
炭素繊維束における炭素繊維の本数は、1,000~70,000本が好ましい。
(B)成分は、エポキシ樹脂組成物である。
(B)成分は、エポキシ樹脂およびエポキシ樹脂の硬化剤を含むことが好ましい。(B)成分は、必要に応じてエポキシ樹脂および硬化剤以外の他の成分を含んでもよい。
エポキシ樹脂としては、通常、分子内に2つ以上のエポキシ基を有する2官能以上のエポキシ樹脂が用いられる。エポキシ樹脂としては、(B)成分の硬化物の耐熱性、剛性を維持したまま、靱性を向上できる点からは、オキサゾリドン環骨格を有するエポキシ樹脂が好ましい。
オキサゾリドン環骨格を有するエポキシ樹脂は、ウレタン変性エポキシ樹脂またはイソシアネート変性エポキシ樹脂とも呼ばれる。オキサゾリドン環骨格を有するエポキシ樹脂の市販品としては、DIC社製のEPICLON(登録商標)TSR-400、新日鉄住金化学社製のエポトート(登録商標)YD-952、DOW社製のD.E.R.(登録商標)858、旭化成イーマテリアルズ社製のLSA3301等が挙げられる。
固形であるオキサゾリドン環骨格を有するエポキシ樹脂を用いた場合、(B)成分の粘度が高くなるため、25℃で液状のビスフェノールA型エポキシ樹脂および25℃で液状のビスフェノールF型エポキシ樹脂のいずれか一方または両方と併用することが好ましい。
ら好ましい。
3官能のエポキシ樹脂としては、トリアジン骨格含有エポキシ樹脂、アミノフェノール型エポキシ樹脂、アミノクレゾール型エポキシ樹脂等が挙げられる。
中でもアミノフェノール型エポキシ樹脂である三菱ケミカル社製のjER(登録商標)630、HUNTSMAN製のAraldite(登録商標)MY0500、MY0510、MY0600、MY0610やジアミノジフェニルメタン型エポキシ樹脂である三菱化学社製のjER(登録商標)604、HUNTSMAN製のAraldite(登録商標)MY720、新日鐵住金化学製のYH434Lは、配合することでマトリックス樹脂組成物の粘度を大きく上げることなく、硬化したマトリックス樹脂の耐熱性や弾性率を上げることができるため好ましく、さらにジアミノジフェニルメタン型エポキシ樹脂は上記の点に加え、硬化したマトリックス樹脂の吸水率を過度に上げることが無いためより好ましい。
(B)成分は、必要に応じて、ビスフェノールS型、ナフタレン型、ジシクロペンタジエン型、レゾルシン型、ヒドロキノン型、ビスフェノキシエタノールフルオレン型、ビスフェノールフルオレン型、ビスクレゾールフルオレン型エポキシ樹脂等の他のエポキシ樹脂を含んでいてもよい。
硬化剤は、エポキシ樹脂を硬化させ得るものであればよい。
(B)成分に含まれ得る他の成分としては、公知の各種添加剤が挙げられる。
(B)成分としては、エポキシ樹脂を含み、前記エポキシ樹脂100質量%に対し、60~100質量%がナフタレン骨格を有するエポキシ樹脂であり、さらに硬化剤として芳香族ポリアミンを含むエポキシ樹脂組成物も挙げられる。この(B)成分は、必要に応じてエポキシ樹脂および硬化剤以外の他の成分を含んでもよい。以下、(B)成分が、エポキシ樹脂を含み、エポキシ樹脂100質量%に対し60~100質量%がナフタレン骨格を有するエポキシ樹脂であり、さらに硬化剤として芳香族ポリアミンを含むエポキシ樹脂組成物である場合について説明する。
エポキシ樹脂とは、通常、分子内に2つ以上のエポキシ基を有する2官能以上のエポキシ樹脂であり、ナフタレン骨格を有するエポキシ樹脂とは分子内にナフタレン環を有し、かつ2つ以上のエポキシ基を有する2官能以上のエポキシ樹脂である。通常のビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂と比較してナフタレン骨格を有するエポキシ樹脂はその剛直な骨格のため、硬化物に良好な耐熱性を付与することができる。
ナフタレン骨格を有する2官能のエポキシ樹脂とは、分子内にナフタレン環を有し、かつ2つのエポキシ基を有する2官能のエポキシ樹脂である。ナフタレン骨格を有する2官能のエポキシ樹脂は前述のとおり、その剛直な骨格のため硬化後のマトリックス樹脂に良好な耐熱性を付与することができる。通常、硬化物の耐熱性付与に使用される多官能エポキシ樹脂と異なり、硬化物の架橋密度を上げないため、硬化後のマトリックス樹脂の靭性が低下してしまったり、強化繊維とマトリックス樹脂との界面の密着性が低下してしまったりして、上述の微粒子配置の効果が十分に得ることが出来ないことを防ぐことができる。またさらに多量の多官能エポキシを使用すると硬化後のマトリックス樹脂の吸水量が多くなる傾向があるが、ナフタレン骨格を有する2官能のエポキシ樹脂は硬化後のマトリックス樹脂の吸水量を多くすることも防ぐことができる。ナフタレン骨格を有する2官能のエポキシ樹脂の市販品としては、DIC社製のEPICLON(登録商標)HP-4032D、HP-4032SS等が挙げられる。
ナフタレン骨格を有する2官能のエポキシ樹脂の割合は、(B)成分中の全エポキシ樹脂100質量%に対し、50~100質量%が好ましく、55~90質量%がより好ましく、60~85質量%がより一層好ましい。ナフタレン骨格を有する2官能のエポキシ樹脂の割合が前記範囲の下限値以上であれば、(B)成分の硬化物の靱性、強化繊維とマトリックス樹脂との界面の密着性、吸水量を維持したまま、硬化物の耐熱性を十分に向上できる。またナフタレン骨格を有する2官能のエポキシ樹脂は通常、液状であるため固形のエポキシ樹脂やポリエーテルスルホンのようなエポキシ樹脂に可溶な熱可塑性樹脂を一緒に使用することで、(B)成分の取扱性がよくなり、プリプレグの作製が容易になったり、プリプレグのタック性およびドレープ性がよくなったりする。
ナフタレン骨格を有する3~4官能のエポキシ樹脂とは、分子内にナフタレン骨格を有し、かつ3~4個のエポキシ基を有する3~4官能のエポキシ樹脂である。ナフタレン骨格を有する3~4官能のエポキシ樹脂は前述のとおり、その剛直な骨格のため硬化後のマトリックス樹脂に良好な耐熱性を付与することができ、さらに硬化物の架橋密度を上げることでも硬化後のマトリックス樹脂に良好な耐熱性を付与することができる。ナフタレン骨格を有する3~4官能のエポキシ樹脂の市販品としては、DIC株式会社製のEPICLON(登録商標)HP-4700、HP-4710、HP-4770、日本化薬株式会社製のNC-7000L、NC-7300L等が挙げられる。
ナフタレン骨格を有する3~4官能のエポキシ樹脂の割合は、(B)成分中の全エポキシ樹脂100質量%に対し、5~50質量%が好ましく、10~40質量%がより好ましく、15~35質量%がより一層好ましい。ナフタレン骨格を有する3~4官能のエポキシ樹脂の割合が前記範囲の下限値以上であれば、(B)成分の硬化物の耐熱性を十分に向上できる。一方で上限値以下であれば(B)成分の硬化物の靱性、強化繊維とマトリックス樹脂との界面の密着性、吸水量を十分に維持でき、さらに(B)成分の粘度が高くなりすぎないため、(B)成分の取扱性がよく、プリプレグの作製が容易であり、プリプレグのタック性およびドレープ性がよくなる。
(B)成分は、ナフタレン骨格を有するエポキシ樹脂に加えて、必要に応じて、オキサゾリドン環骨格を有するエポキシ樹脂、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールS型、トリアジン骨格含有エポキシ樹脂、アミノフェノール型エポキシ樹脂、アミノクレゾール型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、芳香族グリシジルアミン型エポキシ樹脂、ジシクロペンタジエン型、レゾルシン型、ヒドロキノン型、ビスフェノキシエタノールフルオレン型、ビスフェノールフルオレン型、ビスクレゾールフルオレン型エポキシ樹脂等の他のエポキシ樹脂を含んでいてもよい。
25℃で固形のビスフェノールA型エポキシ樹脂および25℃で固形のビスフェノールF型エポキシ樹脂は、25℃で液状のビスフェノールA型エポキシ樹脂および25℃で液状のビスフェノールF型エポキシ樹脂に比べ、耐熱性がやや低下するものの、(B)成分の粘度調整や(B)成分の硬化物に靱性を付与することができる。
25℃で固形のビスフェノールF型エポキシ樹脂は、25℃で固形のビスフェノールA型エポキシ樹脂に比べやや耐熱性が劣るものの、(B)成分の硬化物に比較的高い弾性率を付与できる点から好ましい。25℃で固形のビスフェノールF型エポキシ樹脂の市販品としては、三菱ケミカル社製のjER(登録商標)4004P、jER(登録商標)4005P、jER(登録商標)4007P、jER(登録商標)4010P、新日鉄住金化学社製のエポトート(登録商標)YD-2001、エポトート(登録商標)YD-2004等が挙げられる。
より一層、(B)成分の硬化物の耐熱性を向上させるためにトリアジン骨格含有エポキシ樹脂、アミノフェノール型エポキシ樹脂、アミノクレゾール型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、芳香族グリシジルアミン型エポキシ樹脂等の多官能エポキシ樹脂を含んでもよい。
(B)成分は、硬化剤として芳香族ポリアミンを含むことが好ましい。芳香族ポリアミンを硬化剤として使用することで良好な耐熱性の硬化物を得ることができる。
芳香族ポリアミンとしては、硬化剤としては、ジアミノジフェニルメタン、ジアミノジフェニルスルホン、メタキシレンジアミン、メタフェニレンジアミンおよびこれらのアルキル置換体等の誘導体や異性体などがある。中でも(B)成分の硬化物により良好な耐熱性および靭性を付与できる点からジアミノジフェニルスルホンが好ましい。
硬化剤の添加量は、硬化剤の種類によって異なる。硬化剤がジアミノジフェニルスルホンである場合、ジアミノジフェニルスルホンの添加量は、エポキシ樹脂のエポキシ基1当量に対してジアミノジフェニルスルホンの活性水素当量数が0.9~1.5倍となる量が好ましく、1.05~1.4倍となる量がより好ましく1.1~1.3倍となる量がより一層好ましい。ジアミノジフェニルスルホンの添加量が前記範囲内であれば、(B)成分の硬化物の耐熱性および靱性がさらに優れる。
3,3’-ジアミノジフェニルスルホンは、4,4’-ジアミノジフェニルスルホンと比較して、より高弾性の硬化物を得ることができるが、硬化物の耐熱性の面は劣ったり、後述の(C)成分、特に(C)成分と(B)成分との界面の接着が弱くなり、繊維強化複合材料の層間破壊靱性が低下したりする場合もある。4,4’-ジアミノジフェニルスルホンは、3,3’-ジアミノジフェニルスルホンと比較して、得られる硬化物の弾性が劣るが、硬化物の耐熱性に優れ、かつ後述の(C)成分、特に(C)成分と(B)成分との界面の接着が弱くなりにくく、繊維強化複合材料の層間破壊靱性をより高く発現できる場合がある。よって、繊維強化複合材料の圧縮特性が求められる用途においては3,3’-ジアミノジフェニルスルホンがより好ましく、繊維強化複合材料の耐熱性や層間破壊靱性が求められる用途においては4,4’-ジアミノジフェニルスルホンがより好ましい。また用途によっては、3,3’-ジアミノジフェニルスルホンと4,4’-ジアミノジフェニルスルホンとを共に使用することも可能である。3,3’-ジアミノジフェニルスルホンの市販品としては三井化学ファイン株式会社の3,3’-DASが挙げられ、4,4’-ジアミノジフェニルスルホンの市販品としては和歌山精化工業株式会社のセイカキュアSが挙げられる。
(B)成分に含まれ得る他の成分としては、公知の各種添加剤が挙げられる。
添加剤としては、ポリエーテルスルホン等のエポキシ樹脂に可溶な熱可塑性樹脂、エラストマー微粒子((C)成分を除く。)、コアシェル型エラストマー微粒子、アクリル樹脂等から構成されるブロック共重合体、分子中に1つのエポキシ基を有する化合物、希釈剤、無機粒子(シリカ等)、炭素質成分(カーボンナノチューブ等)、難燃剤(リン化合物等)、脱泡剤等が挙げられる。添加剤としては、(B)成分の硬化物の耐熱性を低下させることなく靱性を向上させる点から、コアシェル型エラストマー微粒子やアクリル樹脂等から構成されるブロック共重合体が好ましい。
コアシェル型エラストマー微粒子の市販品としては、三菱レイヨン社製のメタブレン(登録商標)、アイカ工業社製のスタフィロイド、ダウケミカル社製のパラロイド(登録商標)等が挙げられる。
コアシェル型エラストマー微粒子は、エポキシ樹脂にあらかじめ分散されていてもよい。コアシェル型エラストマー微粒子分散エポキシ樹脂の市販品としては、カネカ社製のカネエース(登録商標)、日本触媒社製のアクリセット(登録商標)BPシリーズ等が挙げられる。コアシェル型エラストマー微粒子分散エポキシ樹脂は、(B)成分の調製を容易にするだけでなく、(B)成分中のコアシェル型エラストマー微粒子の分散状態を良好にすることができる点から、好ましく用いられる。
アクリル樹脂等から構成されるブロック共重合体の市販品としては、Arkema社製のNanostrength(登録商標)シリーズ、例えばNanostrength(登録商標)M52N、Nanostrength(登録商標)M22Nが挙げられる。
(B)成分は、様々な公知の方法で調製できる。(B)成分の調製方法としては、例えば、各成分をプラネタリーミキサーやニーダーにて加熱、混練する方法が挙げられる。
(C)成分は、(c1)成分又は(c2)成分であり、(c1)成分が、ポリアミド粒子と熱硬化性ポリイミド粒子を含み、(c2)成分が融点140~175℃の真球形状のポリアミド粒子を含む。
((c1)成分のポリアミド粒子)
(c1)成分のポリアミド粒子は、繊維強化複合材料により優れた層間破壊靱性を付与するものである。(c1)成分のポリアミド粒子を構成するポリアミド樹脂は、繰り返し構造中にアミド結合を有していれば、特に限定されない。前記ポリアミド樹脂は1種のポリアミド樹脂からなるポリアミド樹脂粒子でも、2種以上のポリアミド樹脂からなるポリアミド樹脂粒子でもよい。2種以上のポリアミド樹脂からなるポリアミド樹脂粒子である場合は、各ポリアミド樹脂が粒子中で均一に存在していても、層構造のように不均一に存在していてもよい。ポリアミド樹脂は例えばラクタム類の開環重合、ジアミンとジカルボン酸の重縮合、アミノカルボン酸の重縮合等の方法により得ることができる。前記ポリアミド樹脂の具体的な例としてはナイロン6、ナイロン46、ナイロン66、ナイロン11、ナイロン12、ナイロン610、ナイロン612、ナイロン6T、ナイロン6I、ナイロン9T、ナイロンM5T、またダイセル・エボニック社のトロガミド(登録商標)T5000、トロガミド(登録商標)CX7323のように芳香族環や脂環を含むポリアミド樹脂等が挙げられる。
また結晶性ポリアミド樹脂、非晶性ポリアミド樹脂、いずれも好ましく使用することができ、結晶性ポリアミド樹脂、非晶性ポリアミド樹脂をいずれか単独で使用しても、混合して使用してもよい。
(c1)のポリアミド粒子としては、結晶性共重合ナイロン粒子が好ましく、ナイロン12とナイロン6との共重合体からなる真球形状の粒子がより好ましい。
ポリアミド樹脂の市販品としては、ダイセル・エボニック社製のVESTOSINTシリーズ(VESTOSINT(登録商標)2158、VESTOSINT(登録商標)2159等)、エムスケミー社製のグリルアミド(登録商標)TR90NZ、グリルアミド(登録商標)TR55、ダイセル・エボニック社製のTOROGAMID(登録商標)CX7323、TOROGAMID(登録商標)T5000等が挙げられる。
ポリアミド粒子の融点は、140℃~175℃が好ましく、155~170℃がより好ましい。
ポリアミド粒子の融点が上記範囲内であると、プリプレグを硬化する過程でマトリックス樹脂とポリアミド粒子との界面を十分に強くすることができ、高い層間破壊靭性を持つ繊維強化複合材料とすることができる。
(c1)成分のポリアミド粒子の平均粒子径は、2~50μmが好ましく、5~35μmがより好ましい。前記ポリアミド粒子の平均粒子径が前記範囲の下限値以上であれば、プリプレグを製造する際に、前記ポリアミド粒子が(A)成分に入り込みにくくなる。そのため、後述の全ての(C)成分の70質量%以上が(A)成分の表層に存在する条件を満たすプリプレグを得やすく、結果として繊維強化複合材料にさらに優れた層間破壊靱性を付与できる。なお表層に存在する(C)成分は後述の偏在化率を持って判断できる。また、(B)成分と(C)成分とを混合した際の粘度の増加を抑制できる。前記ポリアミド粒子の平均粒子径が前記範囲の上限値以下であれば、繊維強化複合材料において(C)成分が(A)成分の強化繊維の真直性を害することを抑制できるため、繊維強化複合材料の機械特性の低下を抑えることができたり、面外方向への引き剥がし応力により繊維強化複合材料の層間領域生じた亀裂を層間領域に留めることができたりする。また、プリプレグの製造において、(B)成分と(C)成分との混合物を離型紙の表面に均一な厚さで塗工する際に、ロールコーター、ダイコーター等の設備で目詰まりを起こすことが抑えられる。
(c1)成分の熱硬化性ポリイミド粒子を構成するポリイミド樹脂は、繰り返し構造中にイミド結合を有す高分子化合物である。ポリイミド樹脂の中で、主鎖の分子量が比較的小さく、かつ反応性末端基を有すものが熱硬化性ポリイミド樹脂である。反応性末端基を有すため、繊維強化複合材料中で熱硬化性ポリイミド粒子とマトリックス樹脂との界面性状を強固にすることができ、後述の前記熱硬化性ポリイミド粒子の効果を十分に発現させることができる。分子量が比較的大きく、かつほぼ反応性末端基を有さない熱可塑性ポリイミド樹脂はマトリックス樹脂との界面性状を強固にすることができず、さらに熱硬化性ポリイミドと比較的して靭性に優れるため、後述の熱硬化性ポリイミド粒子の効果を十分に発現させることができない。
熱硬化性ポリイミド粒子の構成樹脂として好ましく使用できる熱硬化性ポリイミド樹脂は、一般式(1)の化学構造を含む熱硬化性ポリイミド樹脂であり、さらに具体的にはベンゾフェノンテトラカルボン酸二無水物(BTDA)、4,4’-メチレンジアニリン(MDA)、および2,4-トルエンジアミン(TDA)から調製され、90~92パーセントの芳香族炭素を含む非フタルイミド炭素含有量を有する熱硬化性ポリイミド樹脂である。市販の前記熱硬化性ポリイミド樹脂の粒子としては、HP Polymer社製のP84(登録商標)Polyimideがある。


(c1)成分の熱硬化性ポリイミド粒子の平均粒子径は、2~50μmが好ましく、5~35μmがより好ましい。前記熱硬化性ポリイミド粒子の平均粒子径が前記範囲の下限値以上であれば、プリプレグを製造する際に、(C)成分が(A)成分に入り込みにくくなる。そのため、前述の全ての(C)成分の70質量%以上が(A)成分の表層に存在する条件を満たすプリプレグを得やすく、結果として繊維強化複合材料にさらに優れた層間破壊靱性を付与できる。また、(B)成分と(C)成分とを混合した際の粘度の増加を抑制できる。そして、さらに凝集して分散不良となることを防ぐこともできる。
前記熱硬化性ポリイミド粒子の平均粒子径が前記範囲の上限値以下であれば、繊維強化複合材料において(C)成分が(A)成分の強化繊維の真直性を害することを抑制できるため、繊維強化複合材料の機械特性の低下を抑えることができたり、面外方向への引き剥がし応力により繊維強化複合材料の層間領域生じた亀裂を層間領域に留めることができたりする。また、プリプレグの製造において、(B)成分と(C)成分との混合物を離型紙の表面に均一な厚さで塗工する際に、ロールコーター、ダイコーター等の設備で目詰まりを起こすことが抑えられる。
(c1)成分の熱硬化性ポリイミド粒子の平均粒子径は、(c1)成分のポリアミド粒子の平均粒子径の0.5~10倍の範囲であることが好ましい。(c1)成分の熱硬化性ポリイミド粒子の平均粒子径が(c1)成分のポリアミド粒子の平均粒子径の0.5倍より大きくすると後述の(c1)成分のポリアミド粒子と(c1成分)の熱硬化性ポリイミド粒子の併用の効果が十分に発現することができる。また(c1)成分の熱硬化性ポリイミド粒子の平均粒子径が(c1)成分のポリアミド粒子の平均粒子径の10倍以下にすると層間領域の厚みがより均一、すなわち(A)成分の強化繊維の真直性を害することを抑制できるため、繊維強化複合材料の機械特性の低下を抑えることができたり、面外方向への引き剥がし応力により繊維強化複合材料の層間領域生じた亀裂を層間領域に留めることができたりする。
一般的に熱硬化性ポリイミド樹脂は比較的靭性に乏しく、単独の使用では繊維強化複合材料により優れた層間破壊靱性を付与することはできない。一方で面外方向への引き剥がし応力により繊維強化複合材料の層間領域に生じた亀裂はより靭性に乏しい部分、例えばマトリックス樹脂と強化繊維束の界面を選択的に進行する性質がある。そのため、従来の高靭性のマトリックス樹脂や微粒子を層間領域に配置した場合、前記層間領域に生じた亀裂は進行に伴い徐々により靭性の乏しいマトリックス樹脂と強化繊維束の界面へ転移してゆき、最終的には層間領域から強化繊維層へ完全に転移してしまっており、安定して優れた層間破壊靱性を付与することはできなかった。しかし靭性に乏しい熱硬化性ポリイミド粒子を繊維強化複合材料の層間領域に配置した場合、前記層間領域に生じた亀裂は安定して層間領域を進行する(c1)成分のポリアミド粒子と(c1)成分の熱硬化性ポリイミド粒子を併用することで、層間領域に生じた亀裂を安定して層間領域を進行させ、さらに(c1)成分のポリアミド粒子添加による層間破壊靱性付与することができるため、繊維強化複合材料の層間領域のモルホロジーや(c1)成分のポリアミド粒子とマトリックス樹脂組成物との界面性状に関わらず安定して得ることができる。
(c1)成分のポリアミド粒子と(c1)成分の熱硬化性ポリイミド粒子の含有量の合計は(B)成分100質量部に対し5~25質量部であり、さらに10~25質量部が好ましく、12~25質量部がより好ましく、15~20質量部がより一層好ましい。(c1)成分のポリアミド粒子と(c1)成分の熱硬化性ポリイミド粒子の含有量の合計が前記範囲の下限値以上であれば、層間領域に偏在する(c1)成分のポリアミド粒子と(c1)成分の熱硬化性ポリイミド粒子の量が多くなり、上述の理由から繊維強化複合材料に優れた層間破壊靱性を安定して付与できる。(c1)成分のポリアミド粒子の含有量が前記範囲の上限値以下であれば、プリプレグが含む(B)成分、(c1)成分のポリアミド粒子および(c1)成分の熱硬化性ポリイミド粒子からなるマトリックス樹脂中に占める(B)成分が低くなりすぎることを抑制することができる。すなわち(B)成分不足に起因する繊維強化複合材料の機械特性低下を防ぐことができたり、マトリックス樹脂組成物の粘度が高くなりすぎることを抑制し、プリプレグを作製する際に(A)成分にマトリックス樹脂組成物を十分に含浸させることができたりする。
(c1)成分中の熱硬化性ポリイミド粒子の割合は、10~40質量%が好ましい。熱硬化性ポリイミド粒子の割合が前記範囲の下限値以上であれば、熱硬化性ポリイミド粒子の効果を十分に得ることができ、上述の理由から繊維強化複合材料に優れた層間破壊靱性を安定して付与できる。熱硬化性ポリイミド粒子の割合が前記範囲の上限値以下であれば、靭性の乏しい熱硬化性ポリイミド粒子が繊維強化複合材料の層間破壊靱性に与える悪影響が大きくなることを抑えることが出来る。
(A)成分と(B)成分の界面が強いとは、ASTM D790に準拠して実施の一方向(強化繊維が全て同じ方向に配向する)繊維強化複合材料の90°曲げ試験の強度が125MPa以上であることをいう。
(B)成分と(C)成分の界面が弱いとは、ASTM D5528に準拠して実施の繊維強化複合材料のDCB(Double Cantilever Beam)試験の破面を走査型電子顕微鏡(SEM:Scanning Electron Microscope)を使用して観察して、図1に示すように(C)成分30の界面が露出している状態である。
(B)成分と(C)成分の界面が強いとは、ASTM D5528に準拠して実施の繊維強化複合材料のDCB(Double Cantilever Beam)試験の破面を走査型電子顕微鏡(SEM:Scanning Electron Microscope)を使用して観察して、図2に示すように(C)成分の界面が露出していない状態である。
ポリアミド粒子とは粒子形状のポリアミド樹脂のことであり、繊維強化複合材料により優れた層間破壊靱性を付与できる。ポリアミド樹脂とは、繰り返し構造中にアミド結合を有している化合物である。前記ポリアミド樹脂は1種のポリアミド樹脂からなるポリアミド樹脂でも、2種以上のポリアミド樹脂からなるポリアミド樹脂でもよい。2種以上のポリアミド樹脂からなるポリアミド樹脂粒子である場合は、各ポリアミド樹脂が粒子中で均一に存在していても、層構造のように不均一に存在していてもよい。ポリアミド樹脂は例えばラクタム類の開環重合、ジアミンとジカルボン酸の重縮合、アミノカルボン酸の重縮合等の方法により得ることができる。前記ポリアミド樹脂の具体的な例としてはナイロン6、ナイロン46、ナイロン66、ナイロン11、ナイロン12、ナイロン610、ナイロン612、ナイロン6T、ナイロン6I、ナイロン9T、ナイロンM5T、またダイセル・エボニック社のトロガミド(登録商標)T5000、トロガミド(登録商標)CX7323のように芳香族環や脂環を含むポリアミド樹脂等が挙げられる。 ポリアミド樹脂の市販品としては、ダイセル・エボニック社製のVESTOSINTシリーズ(VESTOSINT(登録商標)2158、VESTOSINT(登録商標)2159等)、エムスケミー社製のグリルアミド(登録商標)TR90NZ、グリルアミド(登録商標)TR55、ダイセル・エボニック社製のTOROGAMID(登録商標)CX7323、TOROGAMID(登録商標)T5000等が挙げられる。
融点140~175℃の真球形状のポリアミド粒子の形状は真球状である。具体的に真球状の粒子とは、走査型電子顕微鏡(日本電子社製、JSM-6390)を用い、無作為に選ばれた10個の粒子について短径および長径を測定し、前記10個の粒子の長径に対する短径の比(短径/長径)の平均値が0.9以上である粒子のことを指す。
融点140~175℃の真球形状のポリアミド粒子の平均粒子径は、2~50μmが好ましく、5~35μmがより好ましく、6~25μmがより一層好ましい。前記真球形状のポリアミド粒子の平均粒子径が前記範囲の下限値以上であれば、プリプレグを製造する際に、(C)成分が(A)成分に入り込みにくくなる。そのため、後述の全ての(C)成分の70質量%以上が(A)成分の表層に存在する条件を満たすプリプレグを得やすく、結果として繊維強化複合材料にさらに優れた層間破壊靱性を付与できる。また、(B)成分と(C)成分とを混合した際の粘度の増加を抑制できる。前記真球形状のポリアミド粒子の平均粒子径が前記範囲の上限値以下であれば、繊維強化複合材料において(C)成分が(A)成分の強化繊維の真直性を害することを抑制できるため、繊維強化複合材料の機械特性の低下を抑えることができたり、面外方向への引き剥がし応力により繊維強化複合材料の層間領域生じた亀裂を層間領域に留めることができたりする。また、プリプレグの製造において、(B)成分と(C)成分との混合物を離型紙の表面に均一な厚さで塗工する際に、ロールコーター、ダイコーター等の設備で目詰まりを起こすことが抑えられる。
なお、本発明の(c2)は、更に熱硬化性ポリイミド粒子を含むことが好ましい。
(c2)成分に含めることができる熱硬化性ポリイミド粒子とは、(c2)成分の融点140~175℃の真球形状のポリアミド粒子とともに(C)成分として併用することができる熱硬化性ポリイミド粒子を意味する。(c2)成分に含めることができる熱硬化性ポリイミド粒子は、熱硬化性ポリイミド粒子である。ポリイミド樹脂とは繰り返し構造中にイミド結合を有す高分子化合物である。ポリイミド樹脂の中で、主鎖の分子量が比較的小さく、かつ反応性末端基を有すものが熱硬化性ポリイミド樹脂である。反応性末端基を有すため、繊維強化複合材料中で熱硬化性ポリイミド粒子とマトリックス樹脂との界面性状を強固にすることができ、後述の(c2)成分に含めることができる熱硬化性ポリイミド粒子の効果を十分に発現させることができる。分子量が比較的大きく、かつほぼ反応性末端基を有さない熱可塑性ポリイミド樹脂はマトリックス樹脂との界面性状を強固にすることができず、さらに熱硬化性ポリイミドと比較的して靭性に優れるため、後述の(c2)成分に含めることができる熱硬化性ポリイミド粒子の効果を十分に発現させることができない。


(c2)成分に含めることができる熱硬化性ポリイミド粒子の平均粒子径は、2~50μmが好ましく、5~35μmがより好ましい。前記熱硬化性ポリイミド粒子の平均粒子径が前記範囲の下限値以上であれば、プリプレグを製造する際に、前記熱硬化性ポリイミド粒子が(A)成分に入り込みにくくなる。そのため、前述の全ての(C)成分の70質量%以上が(A)成分の表層に存在する条件を満たすプリプレグを得やすく、結果として繊維強化複合材料にさらに優れた層間破壊靱性を付与できる。また、(B)成分と前記熱硬化性ポリイミド粒子とを混合した際の粘度の増加を抑制できる。そして、さらに凝集して分散不良となることを防ぐこともできる。
一般的に熱硬化性ポリイミド樹脂は比較的靭性に乏しく、単独の使用では繊維強化複合材料により優れた層間破壊靱性を付与することはできない。一方で面外方向への引き剥がし応力により繊維強化複合材料の層間領域に生じた亀裂はより靭性に乏しい部分、例えばマトリックス樹脂と強化繊維束の界面を選択的に進行する性質がある。そのため、従来の高靭性のマトリックス樹脂や微粒子を層間領域に配置した場合、前記層間領域に生じた亀裂は進行に伴い徐々により靭性の乏しいマトリックス樹脂と強化繊維束の界面へ転移してゆき、最終的には層間領域から強化繊維層へ完全に転移してしまっており、安定して優れた層間破壊靱性を付与することはできなかった。しかし靭性に乏しい熱硬化性ポリイミド粒子を繊維強化複合材料の層間領域に配置した場合、前記層間領域に生じた亀裂は安定して層間領域を進行する。融点140~175℃の真球形状のポリアミド粒子と熱硬化性ポリイミド粒子を併用することで、層間領域に生じた亀裂を安定して層間領域を進行させ、さらに融点140~175℃の真球形状のポリアミド粒子添加による層間破壊靱性付与することができるため、繊維強化複合材料の層間領域のモルホロジーや融点140~175℃の真球形状のポリアミド粒子とマトリックス樹脂組成物との界面性状に関わらず安定して得ることができる。
融点140~175℃の真球形状のポリアミド粒子の含有量は、(B)成分100質量部に対し5~25質量部であり、さらに10~25質量部が好ましく、12~25質量部がより好ましく、15~20質量部がより一層好ましい。(C)成分の含有量が前記範囲の下限値以上であれば、層間領域に偏在する前記真球形状のポリアミド粒子の量が多くなり、上述の理由から繊維強化複合材料に優れた層間破壊靱性を安定して付与できる。前記真球形状のポリアミド粒子の含有量が前記範囲の上限値以下であれば、プリプレグが含む(B)成分と(C)成分からなるマトリックス樹脂中に占める(B)成分が低くなりすぎることを抑制することができる。すなわち(B)成分不足に起因する繊維強化複合材料の機械特性低下を防ぐことができたり、マトリックス樹脂組成物の粘度が高くなりすぎることを抑制し、プリプレグを作製する際に(A)成分にマトリックス樹脂組成物を十分に含浸させることができたりする。
(c2)成分が熱硬化性ポリイミド粒子を更に含有する場合、融点140~175℃の真球形状のポリアミド粒子と熱硬化性ポリイミド粒子との含有量の合計は(B)成分100質量部に対し5~25質量部であり、さらに10~25質量部が好ましく、12~25質量部がより好ましく、15~20質量部がより一層好ましい。融点140~175℃の真球形状のポリアミド粒子と熱硬化性ポリイミド粒子の含有量の合計が前記範囲の下限値以上であれば、層間領域に偏在する融点140~175℃の真球形状のポリアミド粒子と熱硬化性ポリイミド粒子の量が多くなり、上述の理由から繊維強化複合材料に優れた層間破壊靱性を安定して付与できる。融点140~175℃の真球形状のポリアミド粒子の含有量が前記範囲の上限値以下であれば、プリプレグが含む(B)成分、融点140~175℃の真球形状のポリアミド粒子および熱硬化性ポリイミド粒子からなるマトリックス樹脂中に占める(B)成分が低くなりすぎることを抑制することができる。すなわち(B)成分不足に起因する繊維強化複合材料の機械特性低下を防ぐことができたり、マトリックス樹脂組成物の粘度が高くなりすぎることを抑制し、プリプレグを作製する際に(A)成分にマトリックス樹脂組成物を十分に含浸させることができたりする。
融点140~175℃の真球形状のポリアミド粒子と熱硬化性ポリイミド粒子の混合物に占める熱硬化性ポリイミド粒子の割合は、融点140~175℃の真球形状のポリアミド粒子と熱硬化性ポリイミド粒子との含有量の合計に対し、5~40質量%が好ましい。前記熱硬化性ポリイミド粒子の割合が前記範囲の下限値以上であれば、前記熱硬化性ポリイミド粒子の効果を十分に得ることができ、上述の理由から繊維強化複合材料に優れた層間破壊靱性を安定して付与できる。前記熱硬化性ポリイミド粒子の割合が前記範囲の上限値以下であれば、靭性の乏しい前記熱硬化性ポリイミド粒子が繊維強化複合材料の層間破壊靱性に与える悪影響が大きくなることを抑えることが出来る。
プリプレグの繊維目付(1m2あたりの強化繊維の含有量:FAW)は、プリプレグの用途に応じて適宜設定すればよく、通常、50~300g/m2である。
プリプレグにおける樹脂含有率((B)成分および(C)成分の合計の割合)は、プリプレグの総質量に対し、25~50質量%が好ましく、30~40質量%がより好ましい。プリプレグにおける樹脂含有率が前記範囲の下限値以上であれば、プリプレグのタックが低くなりすぎることを抑制し、取扱に適したタックとすることができる。さらに(B)成分不足に起因する繊維強化複合材料の機械特性低下を防ぐこともできる。プリプレグにおける樹脂含有率が前記範囲の上限値以下であれば、プリプレグのタックが高くなりすぎることを抑制し、取扱に適したタックとすることができる。さらに(B)成分過剰に起因するVf(繊維強化複合材料中の強化繊維含有の体積率)向上に伴う繊維強化複合材料の機械特性低下を防ぐこともできる。
プリプレグの厚さは、プリプレグの用途に応じて適宜設定すればよく、通常、0.05~0.3mmである。
なお、プリプレグの厚さはシックネスゲージで測定できる。
本発明におけるプリプレグは、特許文献2に開示された方法、その応用等によって製造できる。
ら、方法(α)、方法(β)、方法(γ)および方法(δ)からなる群から選ばれる1つの方法が好ましく、より均一に層間領域に(C)成分を偏在させることができる点、および製造過程で多くの(C)成分が舞い散り製造環境を悪化させることを防ぐことができる点から、方法(γ)または方法(δ)がより好ましい。
方法(α)は、(B)成分からなる樹脂フィルム(F1)を(A)成分の片面または両面に貼り合わせ、(B)成分を(A)成分に含浸させてベースプリプレグ(P1)を作製し、ベースプリプレグ(P1)の片面または両面に(C)成分を散布する方法である。
樹脂フィルム(F1)は、(B)成分を離型紙等の表面に塗工することによって作製できる。(B)成分を(A)成分に含浸させる方法としては、加熱プレスロールで加熱、加圧する方法等の公知の方法が挙げられる。
方法(β)は、(B)成分からなる樹脂フィルム(F1)を(A)成分の片面または両面に貼り合わせ、(B)成分を(A)成分に含浸させてベースプリプレグ(P1)を作製し、(B)成分の表面に(C)成分が散布された樹脂フィルム(F2)を、ベースプリプレグ(P1)の片面または両面に貼り合わせる方法である。
ベースプリプレグ(P1)にさらに樹脂フィルム(F2)を貼り合わせる方法(β)の性質上、ベースプリプレグ(P1)における(B)成分の含有率は、方法(α)に比べ低くしておくことが好ましい。
方法(γ)は、(B)成分からなる樹脂フィルム(F1)を(A)成分の片面または両面に貼り合わせ、(B)成分を(A)成分に含浸させてベースプリプレグ(P1)を作製し、(B)成分、(C)成分を含む樹脂フィルム(F3)を、ベースプリプレグ(P1)の片面、または両面に貼り合わせる方法である。
ベースプリプレグ(P1)は、方法(α)と同様にして作製できる。
方法(δ)は、(B)成分および(C)成分を含む樹脂フィルム(F3)、もしくは(B)成分、(C)成分を含む樹脂フィルム(F3)を、(A)成分の片面または両面に貼り合わせ、(B)成分を(A)成分に含浸させる方法である。
樹脂フィルム(F3)は、方法(γ)と同様にして作製できる。
本発明の第一の態様における繊維強化複合材料は、プリプレグを2つ以上積層し、(B)成分の硬化温度以上で加熱して(B)成分を硬化することによって得られる。
積層したプリプレグを加熱成形する際の温度は、(B)成分を適切に硬化可能な温度であればいずれの温度でもよいが、加熱成形に用いる設備の(B)成分の硬化に要する時間の点から170~190℃が好ましい。加熱成形する際の温度が170℃以上であれば、(B)成分が十分に硬化し、より高い耐熱性を有する硬化物を得ることができる。加熱成形する際の温度が190℃以下であれば、加熱成形に用いる設備や副資材としてより安価なものを用いることができる。
本発明の第二の態様における繊維強化複合材料では、(A)成分は複数枚積層されており、かつ(C)成分が(A)成分の層間に存在している。
(A)成分、(C)成分としては、プリプレグの説明において述べた(A)成分、及び(C)成分と同様のものが挙げられる。
(B’)成分としては、プリプレグの説明において述べた(B)成分を硬化させたものが挙げられる。
(B)成分を硬化させる方法としては、(B)成分を170~190℃に加熱することが好ましい。加熱時間は1~4時間が好ましい。
(平均粒子径)
樹脂粒子の平均粒子径は、以下のようにして求めた。
樹脂粒子について、レーザー散乱式粒度測定機(日揮装社製、MODEL:7340 マイクロトラックFRA)を用いて粒子径分布測定を行い、累積分布を得た。累積分布にける体積基準での累積頻度が50%となる粒子径(D50)を平均粒子径とした。
図3に示す通り、プリプレグ2を、強化繊維の繊維軸方向が揃うように20枚積層した。図3において、矢印は強化繊維の繊維軸方向を表している。10枚目のプリプレグ2と11枚目のプリプレグ2との間に、強化繊維の繊維軸方向に対して長手方向が直角になるように、厚さ50μmの長尺のフッ素樹脂フィルム3を、フッ素樹脂フィルム3の幅70mm程度がプリプレグ2と重複するように挟んだ。積層されたプリプレグに隙間のないよう真空バッグを被せた。積層されたプリプレグを、オートクレーブを用いて2℃/分、または0.5℃/分の昇温速度で室温から180℃まで加熱し、2時間保持した。積層されたプリプレグを、3℃/minの降温スピードで50℃以下となるまでオートクレーブ内に保持した。オートクレーブ内から評価用成形板1を取り出した。加熱開始から取り出しまでの間、オートクレーブ内の圧力は0.6MPaとした。
(熱変形樹脂粒子の偏在化率)
評価用成形板から20mm角の試験片を切り出した。研磨機(リファインテック社製、REFINE-POLISHER APM-122)を用いて、試験片の断面を研磨した。デジタルマイクロスコープ(KEYENCE社製、VHX-5000)を用いて試験片の断面を500倍に拡大した写真を得た。写真から、強化繊維基材間の層間領域にある熱変形樹脂粒子と、強化繊維基材内にある熱変形樹脂粒子とを切り抜き、切り抜いた写真の質量を測定し下記式(3)から偏在化率を算出した。
偏在化率=層間領域に存在する熱変形樹脂粒子の質量/(層間領域に存在する熱変形樹脂粒子の質量+強化繊維基材内にある熱変形樹脂粒子の質量)×100 ・・・式(3)
評価用成形板について、インストロン万能試験機(インストロン社製)を用い、ASTM D5528に準拠してGICを測定した。
評価用成形板について、インストロン万能試験機(インストロン社製)を用い、ASTM D7905に準拠してGIICを測定した。
((A)成分)
(強化繊維束)
MR70:炭素繊維束(三菱ケミカル社製、PYROFIL(登録商標)MR70 12P、ストランド強度:7000MPa、炭素繊維の繊維径:5μm、炭素繊維の本数:12000本)。
((B)成分)
(エポキシ樹脂)
TSR-400:オキサゾリドン環骨格を有するエポキシ樹脂(DIC社製、EPICLON TSR-400)。
jER807:ビスフェノールF型液状エポキシ樹脂(三菱ケミカル社製、jER(登録商標)807)。
jER604:ジアミノジフェニルメタン型半固形エポキシ樹脂(三菱ケミカル社製、jER(登録商標)604)。
(硬化剤)
セイカキュアーS:4,4’-ジアミノジフェニルスルホン(和歌山精化工業社製、セイカキュアーS)。
(任意成分)
E2020P:ポリエーテルスルホン(BASFジャパン社製、ULTRASON(登録商標)E2020 P SR MICRO)。
((C)成分)
((c1)成分のポリアミド粒子)
VESTOSINT2158:ポリアミド12粒子(ダイセル・エボニック社製、VESTOSINT(登録商標)2158 natural、融点:177℃、平均粒子径:21μm)。
((c1)成分の熱硬化性ポリイミド粒子)
p84:熱硬化性ポリイミド粒子(HP Polymer社製、P84(登録商標)Polyimide、平均粒子径:17.7μm)。
PIP-3:熱硬化性ポリイミド粒子(山曹ミクロン社製、ポリイミドパウダーPIP-3、平均粒子径:2.8μm)。
PIP-25:熱硬化性ポリイミド粒子(山曹ミクロン社製、ポリイミドパウダーPIP-25、平均粒子径:12.9μm)。
((c1)成分に含まれない樹脂粒子)
P84NT1:ポリイミド粒子(ダイセル・エボニック社製、ポリイミドP84(登録商標)NT1、平均粒子径:8.9μm)。
P84NT2:ポリイミド粒子(ダイセル・エボニック社製、ポリイミドP84(登録商標)NT2、平均粒子径:15.9μm)
((B)成分の調製)
プラネタリミキサーに、TSR-400の35質量部、jER807の49質量部、jER604の16質量部、E2020Pの2.8部質量を加えた。プラネタリミキサーのジャケット温度を140~160℃に設定し、原料が均一となるまで混合した。内容物の温度が60℃以下となるまで放冷し、プラネタリミキサーにセイカキュアーSの40.6質量部を加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を12.3質量部、およびp84を6.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とp84の合計の含有量を12.9質量部、そしてVESTOSINT2158とp84の合計の含有量に占めるp84の割合を33.5質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
方法(δ)によってプリプレグを作製した。
ホットメルトコーターを用いて、混合物(BC)を離型紙の表面に均一な厚さで塗工して樹脂フィルム(F3)を作製した。
樹脂フィルム(F3)を、MR70の複数本を引き揃えてシート状にした(A)成分の両面に貼り合わせ、ヒュージングプレス機を用いて (B)成分を構成要素(A)に含浸させ、(C)成分は(A)成分上で濾し、プリプレグの表面近傍に(C)成分を偏在化させ、プリプレグを得た。プリプレグの組成、作製方法を表1に示す。
(繊維強化複合材料の製造、および評価)
上述した方法にしたがい、評価用成形板を作製した。評価用成形板について評価を行った。結果を表1に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を18.6質量部、およびp84を9.3質量部加え、(B)成分の100質量部に対するVESTOSINT2158とp84の合計の含有量を19.5質量部、そしてVESTOSINT2158とp84の合計の含有量に占めるp84の割合を33.3質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表1に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を14.3質量部、およびp84を4.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とp84の合計の含有量を12.9質量部、そしてVESTOSINT2158とp84の合計の含有量に占めるp84の割合を22.7質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表1に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を16.5質量部、およびp84を2.0質量部加え、(B)成分の100質量部に対するVESTOSINT2158とp84の合計の含有量を12.9質量部、そしてVESTOSINT2158とp84の合計の含有量に占めるp84の割合を10.8質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混
合物(BC)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表1に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を12.3質量部、およびPIP-3を6.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とPIP-3の合計の含有量を12.9質量部、そしてVESTOSINT2158とPIP-3の合計の含有量に占めるPIP-3の割合を33.5質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表1に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を12.3質量部、およびPIP-25を6.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とPIP-25の合計の含有量を12.9質量部、そしてVESTOSINT2158とPIP-25の合計の含有量に占めるPIP-25の割合を33.5質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表1に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC’)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT215
8を18.6質量部加え、(B)成分の100質量部に対するVESTOSINT2158の含有量を13.0質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表2に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC’)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を12.3質量部、およびP84NT1を6.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とP84NT1の合計の含有量を12.9質量部、そしてVESTOSINT2158とP84NT1の合計の含有量に占めるP84NT1の割合を33.5質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表2に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC’)の調製)
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を12.3質量部、およびP84NT2を6.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とP84NT2の合計の含有量を12.9質量部、そしてVESTOSINT2158とP84NT1の合計の含有量に占めるP84NT2の割合を33.5質量%とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表2に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
(混合物(BC’)の調製)
プラネタリミキサー中の(B)成分143.4質量部にP84を18.6質量部加え、(B)成分の100質量部に対するP84の合計の含有量を13.0質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
(プリプレグの作製)
実施例1と同様に、方法(δ)によってプリプレグを作製した。
(繊維強化複合材料の製造、および評価)
実施例1と同様に、評価用成形板を作製、および評価を行った。結果を表2に示す。

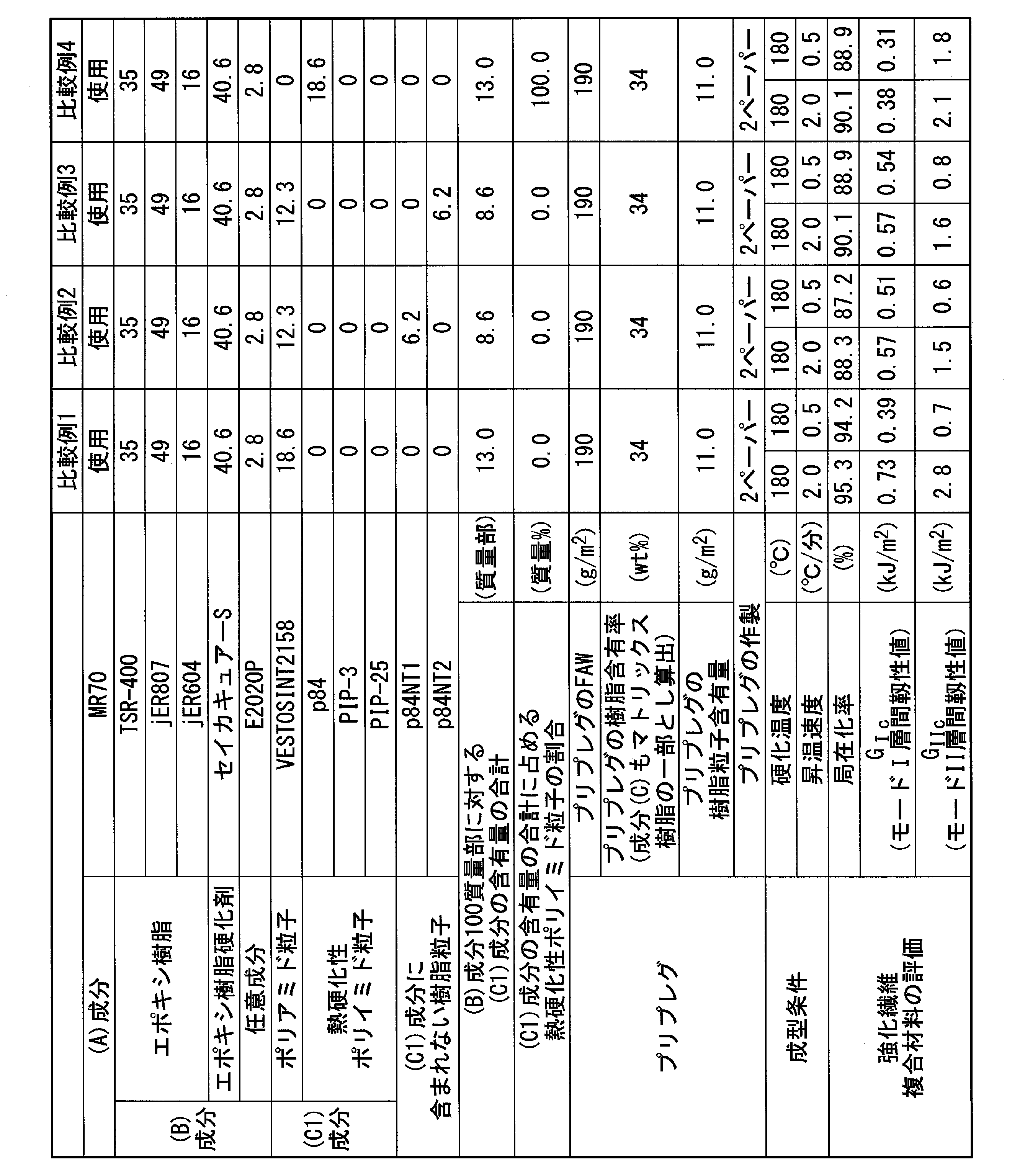

実施例1と実施例2とを比較すると、より多くの(C)成分を含む、実施例2の方がより高いGIIcを示した。優れたモードII層間破壊靱性の発現の観点において、(B)成分100質量部に対する(C)成分の含有量の合計は、実施例2の方がより好ましい。
また実施例1と実施例3、4とを比較すると、0.5℃/分の成型条件のGIIcは、実施例4、実施例3、実施例1の順で高くなっており、これらの実施例の(A)成分~(C)の組み合わせにおいては、(C)成分((c1)ポリアミド粒子と熱硬化性ポリイミド粒子)占める熱硬化性ポリイミド粒子の割合は、実施例4よりも実施例3が好ましく、実施例3よりも実施例1が好ましい。
本発明の繊維強化複合材料の製造方法によって得られた繊維強化複合材料は、製造条件に関わらず安定してモードI層間破壊靱性およびモードII層間破壊靱性に優れた繊維強化複合材料を提供するため、航空機用途をはじめスポーツ・レジャー用途、自動車用途、他の一般産業用途(緊張材)等として有用である。
((A)成分)
(強化繊維束)
MR70:炭素繊維束(三菱ケミカル社製、PYROFIL(登録商標)MR70 12P、ストランド強度:7000MPa、炭素繊維の繊維径:5μm、炭素繊維の本数:12000本)。
(エポキシ樹脂)
TSR-400:オキサゾリドン環骨格を有するエポキシ樹脂(DIC社製、EPICLON TSR-400)。
jER807:ビスフェノールF型液状エポキシ樹脂(三菱ケミカル社製、jER(登録商標)807)。
(硬化剤)
セイカキュアーS:4,4’-ジアミノジフェニルスルホン(和歌山精化工業社製、セイカキュアーS)。
(任意成分)
E2020P:ポリエーテルスルホン(BASFジャパン社製、ULTRASON(登録商標)E2020 P SR MICRO)。
(c2)成分のポリアミド粒子
MW-330:真球状共重合ポリアミド粒子(住化エンバイロメンタルサイエンス社製、MW-330、融点:166℃、平均粒子径:8μm、短径/長径の平均値:0.96)。
p84:熱硬化性ポリイミド粒子(HP Polymer社製、P84(登録商標)Polyimide、平均粒子径:18μm)。
VESTOSINT2158:非真球状ポリアミド12粒子(ダイセル・エボニック社製、VESTOSINT(登録商標)2158 natural、融点:177℃、平均粒子径:21μm、短径/長径の平均値:0.65)。
Ny12粒子A:真球状ポリアミド12粒子(融点:177℃、平均粒子径:21μm、短径/長径の平均値:0.94)。
TR55:真球状非晶性ポリアミド粒子(エムスケミー社製グリルアミド(登録商標)TR55、融点:160℃、平均粒子径:10μm、短径/長径の平均値:0.91)。
特開平10-316750記載の真球状ポリアミドの製造方法とおり、真球状のポリアミド12粒子を作製した。
((B)成分の調製)
プラネタリミキサーに、TSR-400の35質量部、jER807の49質量部、jER604の16質量部、E2020Pの2.8部質量を加えた。プラネタリミキサーのジャケット温度を140~160℃に設定し、原料が均一となるまで混合した。内容物の温度が60℃以下となるまで放冷し、プラネタリミキサーにセイカキュアーSの40.6質量部を加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にMW-330を37.2質量部加え、(B)成分の100質量部に対するMW-330の含有量を25.9質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
方法(γ)によってプリプレグを作製した。
樹脂フィルム(F1)を、MR70の複数本を引き揃えてシート状にした(A)成分の両面に貼り合わせ、ヒュージングプレス機を用いて (B)成分を(A)成分に含浸させベースプリプレグ(P1)を作製し、さらにベースプリプレグ(P1)の両面に樹脂フィルム(F3)を張り合わせることで、プリプレグを得た。得られたプリプレグの(B)成分 100質量部に対する(C)成分の含有量は13.0質量部となる。プリプレグの組成、作製方法を表4に示す。
上述した方法にしたがい、評価用成形板を作製した。評価用成形板について評価を行った。結果を表4に示す。
((B)成分の調製)
実施例1と同様にして(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にMW-330を33.0質量部、およびp84を4.0質量部加え、(B)成分の100質量部に対するMW-330およびp84の合計の含有量を12.9質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
実施例7と同様に、方法(γ)によってプリプレグを作製した。得られたプリプレグの(B)成分 100質量部に対する(C)成分の含有量は12.9質量部となる。プリプレグの組成、作製方法を表4に示す。
実施例7と同様に、評価用成形板を作製、および評価を行った。結果を表4に示す。
((B)成分の調製)
実施例7と同様にして(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を18.6質量部加え、(B)成分の100質量部に対するVESTOSINT2158の含有量を13.0質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
方法(δ)によってプリプレグを作製した。
樹脂フィルム(F3)を、MR70の複数本を引き揃えてシート状にした(A)成分の両面に貼り合わせ、ヒュージングプレス機を用いて (B)成分を構成要素(A
)に含浸させ、VESTOSINT2158は(A)成分上で濾し、プリプレグの表面近傍にVESTOSINT2158を偏在化させ、プリプレグを得た。得られたプリプ
レグの(B)成分 100質量部に対するVESTOSINT2158の含有量の合
計は13.0質量部となる。プリプレグの組成、作製方法を表4に示す。
実施例7と同様に、評価用成形板を作製、および評価を行った。結果を表4に示す。
((B)成分の調製)
実施例7と同様にして(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にVESTOSINT2158を12.3質量部、およびp84を6.2質量部加え、(B)成分の100質量部に対するVESTOSINT2158とp84の合計の含有量を12.9質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
方法(δ)によってプリプレグを作製した。
ホットメルトコーターを用いて、混合物(BC’)を離型紙の表面に均一な厚さで塗工して樹脂フィルム(F3)を作製した。
樹脂フィルム(F3)を、MR70の複数本を引き揃えてシート状にした(A)成分の両面に貼り合わせ、ヒュージングプレス機を用いて (B)成分を構成要素(A)に含浸させ、VESTOSINT2158およびp84は(A)成分上で濾し、プリプレグの表面近傍にVESTOSINT2158およびp84を偏在化させ、プリプレグを得た。得られたプリプレグの(B)成分 100質量部に対す
るVESTOSINT2158とp84の含有量の合計は12.9質量部となる。プリプレグの組成、作製方法を表4に示す。
実施例7と同様に、評価用成形板を作製、および評価を行った。結果を表4に示す。
((B)成分の調製)
実施例7と同様にして(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にNy12粒子Aを18.6質量部加え、(B)成分の100質量部に対するNy12粒子Aの含有量を13.0質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
方法(δ)によってプリプレグを作製した。
ホットメルトコーターを用いて、混合物(BC’)を離型紙の表面に均一な厚さで塗工して樹脂フィルム(F3)を作製した。
12粒子Aを偏在化させ、プリプレグを得た。得られたプリプレグの(B)成分 1
00質量部に対するNy12粒子Aの含有量は12.9質量部となる。プリプレグの組成、作製方法を表4に示す。
実施例7と同様に、評価用成形板を作製、および評価を行った。結果を表4に示す。
((B)成分の調製)
実施例7と同様にして(B)成分を得た。
プラネタリミキサー中の(B)成分143.4質量部にTR55を18.6質量部加え、(B)成分の100質量部に対するTR55粒子Aの含有量を13.0質量部とした。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
方法(δ)によってプリプレグを作製した。
ホットメルトコーターを用いて、混合物(BC’)を離型紙の表面に均一な厚さで塗工して樹脂フィルム(F3)を作製した。
実施例7と同様に、評価用成形板を作製、および評価を行った。結果を表4に示す。

(繊維強化複合材料からなるガラス転移温度評価用成形板の作製)
プリプレグを、強化繊維の繊維軸方向が揃うように12枚積層した。積層されたプリプレグに隙間のないよう真空バッグを被せた。積層されたプリプレグを、オートクレーブを用いて2℃/分の昇温速度で室温から180℃まで加熱し、2時間保持した。積層されたプリプレグを、3℃/minの降温スピードで50℃以下となるまでオートクレーブ内に保持した。オートクレーブ内から評価用成形板を取り出した。加熱開始から取り出しまでの間、オートクレーブ内の圧力は0.6MPaとした。
ガラス転移温度評価用成形板について、ARES-RDA(TAインスツルメント社製)を用い、ASTM D4065に準拠してガラス転移温度を測定した。貯蔵弾性率G’曲線における、ガラス状態での接線と転移状態の変曲点での接線との交点の温度をガラス転移温度とした、測定用試験体のサイズは強化繊維の繊維軸方向の長さが55mm、強化繊維の繊維軸方向と垂直となる幅が12.7mmとした。昇温速度は5℃/分、周波数は1Hzとした。また測定用試験体を測定前に71℃の温水に2週間浸漬させることで吸湿処理を行った。
((A)成分)
(強化繊維束)
MR70:炭素繊維束(三菱ケミカル社製、PYROFIL(登録商標)MR70 12P、ストランド強度:7000MPa、炭素繊維の繊維径:5μm、炭素繊維の本数:12000本)。
((B)成分)
(エポキシ樹脂)
TSR-400:オキサゾリドン環骨格を有するエポキシ樹脂(DIC社製、EPICLON(登録商標)TSR-400)。
HP-4032SS:2官能ナフタレン型エポキシ樹脂(DIC社製、EPICLON(登録商標)HP-4032SS)
HP-4700:4官能ナフタレン型エポキシ樹脂(DIC社製、EPICLON(登録商標)HP-4700)
jER807:ビスフェノールF型液状エポキシ樹脂(三菱ケミカル社製、jER(登録商標)807)
jER604:ジアミノジフェニルメタン型半固形エポキシ樹脂(三菱ケミカル社製、jER(登録商標)604)
(硬化剤)
セイカキュアーS:4,4’-ジアミノジフェニルスルホン(和歌山精化工業社製、セイカキュアーS)
(任意成分)
E2020P:ポリエーテルスルホン(BASFジャパン社製、ULTRASON(登録商標)E2020 P SR MICRO)
((C)成分)
(c2)成分のポリアミド粒子
MW-330:真球状共重合ポリアミド粒子(住化エンバイロメンタルサイエンス社製、MW-330、融点:166℃、平均粒子径:8μm、短径/長径の平均値:0.96)
(c2)成分に含めることができる熱硬化性ポリイミド粒子
p84:熱硬化性ポリイミド粒子(HP Polymer社製、P84(登録商標)Polyimide、平均粒子径:18μm)
((B)成分の調製)
プラネタリミキサーに、TSR-400の35質量部、HP-4032SSの65質量部、E2020Pの2.8部質量を加えた。プラネタリミキサーのジャケット温度を140~160℃に設定し、原料が均一となるまで混合した。内容物の温度が60℃以下となるまで放冷し、プラネタリミキサーにセイカキュアーSの43質量部を加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分145.8質量部にMW-330を43.3質量部加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
方法(γ)によってプリプレグを作製した。
ホットメルトコーターを用いて、(B)成分および混合物(BC)をそれぞれ離型紙の表面に均一な厚さで塗工して樹脂フィルム(F1)および(F3)を作製した。
樹脂フィルム(F1)を、MR70の複数本を引き揃えてシート状にした(A)成分の両面に貼り合わせ、ヒュージングプレス機を用いて (B)成分を(A)成分に含浸させベースプリプレグ(P1)を作製し、さらにベースプリプレグ(P1)の両面に樹脂フィルム(F3)を張り合わせることで、プリプレグを得た。得られたプリプレグの(B)成分100質量部に対する(C)成分の含有量の合計は13.0質量部となる。プリプレグの組成、作製方法を表5に示す。
(繊維強化複合材料の製造、および評価)
上述した方法にしたがい、評価用成形板を作製した。評価用成形板について評価を行った。結果を表5に示す。
((B)成分の調製)
実施例9と同様にして(B)成分を得た。
(混合物(BC’)の調製)
プラネタリミキサー中の(B)成分143.4質量部にMW-330を38.6質量部、およびp84を4.7質量部加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
(プリプレグの作製)
ホットメルトコーターを用いて、(B)成分および混合物(BC’)をそれぞれ離型紙の表面に均一な厚さで塗工して樹脂フィルム(F1)および(F3)を作製した。
樹脂フィルム(F1)を、MR70の複数本を引き揃えてシート状にした(A)成分の両面に貼り合わせ、ヒュージングプレス機を用いて (B)成分を(A)成分に含浸させベースプリプレグ(P1)を作製し、さらにベースプリプレグ(P1)の両面に樹脂フィルム(F3)を張り合わせることで、プリプレグを得た。得られたプリプレグの(B)成分100質量部に対する構成要素(c2)成分の含有量の合計は13.0質量部となる。プリプレグの組成、作製方法を表5に示す。
(繊維強化複合材料の製造、および評価)
実施例9と同様に、評価用成形板を作製、および評価を行った。結果を表5に示す。
((B)成分の調製)
プラネタリミキサーに、HP-4700の35質量部、HP-4032SSの65質量部、E2020Pの2.8部質量を加えた。プラネタリミキサーのジャケット温度を140~160℃に設定し、原料が均一となるまで混合した。内容物の温度が60℃以下となるまで放冷し、プラネタリミキサーにセイカキュアーSの51.5質量部を加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、(B)成分を得た。
(混合物(BC)の調製)
プラネタリミキサー中の(B)成分154.3質量部にMW-330を45.9質量部加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC)を得た。
(プリプレグの作製)
方法(γ)によってプリプレグを作製した。
実施例9と同様に、方法(γ)によってプリプレグを作製した。得られたプリプレグの(B)成分 100質量部に対する(C)成分の含有量の合計は13.0質量部となる。プリプレグの組成、作製方法を表5に示す。
(繊維強化複合材料の製造、および評価)
上述した方法にしたがい、評価用成形板を作製した。評価用成形板について評価を行った。結果を表5に示す。
((B)成分の調製)
実施例11と同様にして(B)成分を得た。
(混合物(BC’)の調製)
プラネタリミキサー中の(B)成分154.3質量部にMW-330を35.4質量部、およびp84を10.5質量部加えた。ジャケット温度を55~70℃に設定し、原料が均一となるまで混合し、混合物(BC’)を得た。
(プリプレグの作製)
実施例10と同様に、方法(γ)によってプリプレグを作製した。得られたプリプレグの(B)成分100質量部に対する構成要素(c2)めるの含有量の合計は13.0質量部となる。プリプレグの組成、作製方法を表5に示す。
(繊維強化複合材料の製造、および評価)
実施例9と同様に、評価用成形板を作製、および評価を行った。結果を表5に示す。

2 プリプレグ
3 フッ素樹脂フィルム
20 (B)成分
30 (C)成分
Claims (17)
- (A)成分、(B)成分および(C)成分を含むプリプレグであって、
前記(A)が強化繊維基材であり、
前記(B)がエポキシ樹脂組成物であり、
前記(C)成分が(c1)成分または(c2)成分であり、
前記(c1)成分がポリアミド粒子と熱硬化性ポリイミド粒子とを含み、
前記(c2)成分が融点140~175℃のポリアミドの真球形状の粒子を含む、プリプレグ。 - 前記(C)成分が前記(c1)成分である、請求項1に記載のプリプレグ。
- 前記(C)成分が前記(c1)成分であり、
[ポリアミド粒子]:[熱硬化性ポリイミド粒子]で表される質量比が60:40~95:5である、請求項1または2に記載のプリプレグ。 - 前記(C)成分が前記(c1)成分であり、
前記(c1)成分中のポリアミド粒子の融点が140℃~175℃である、請求項1から3のいずれか一項に記載のプリプレグ。 - 前記(C)成分が前記(c1)成分であり、
前記(c1)中のポリアミド粒子が結晶性共重合ナイロン粒子である、請求項1から4のいずれか一項に記載のプリプレグ。 - 前記(C)成分が前記(c1)成分であり、
前記(c1)中のポリアミド粒子が、ナイロン12とナイロン6との共重合体からなる真球形状の粒子である、請求項1から4のいずれか一項に記載のプリプレグ。 - 前記(C)成分が前記(c2)成分である、請求項1に記載のプリプレグ。
- 前記融点140~175℃のポリアミドの真球形状の粒子が、結晶性共重合ナイロン粒子である、請求項7に記載のプリプレグ。
- 前記融点140~175℃のポリアミドの真球形状の粒子が、ナイロン12とナイロン6との共重合体からなる真球形状の粒子である、請求項7に記載のプリプレグ。
- 前記(C)成分が前記(c2)成分であり、
前記(c2)が、更に熱硬化性ポリイミド粒子を含む、請求項1、および7から9のいずれかに記載のプリプレグ。 - 前記(C)成分が前記(c2)成分であり、
前記(c2)が、更に熱硬化性ポリイミド粒子を含み、
前記熱硬化性ポリイミド粒子が、下記一般式(1)または下記一般式(2)の化学構造を含む、請求項1、7~10のいずれか一項に記載のプリプレグ。


- 前記(C)成分の70質量%以上が前記(A)成分の表層に存在する、請求項1から11のいずれか一項に記載のプリプレグ。
- 前記(A)成分が強化繊維を含み、前記強化繊維が炭素繊維である、請求項1から12のいずれか一項に記載のプリプレグ。
- 前記(B)成分が、エポキシ樹脂、及び芳香族ポリアミンを含有し、
前記エポキシ樹脂が、ナフタレン骨格を有するエポキシ樹脂を含有し、
前記ナフタレン骨格を有するエポキシ樹脂の含有量が、前記エポキシ樹脂の総質量に対し、60~100質量%である、請求項1から13のいずれか一項に記載のプリプレグ。 - 前記(C)成分の含有量が、前記(B)成分100質量部に対し5~25質量部である、請求項1~14のいずれか一項に記載のプリプレグ。
- 請求項1から15のいずれか一項に記載のプリプレグを2枚以上積層し、前記(B)成分の硬化温度以上で加熱してなる繊維強化複合材料。
- (A)成分、(B’)成分および(C)成分を含む繊維強化複合材料であって、
前記(A)成分が強化繊維基材であり、
前記(B’)成分がエポキシ樹脂組成物の硬化物であり、
前記(C)成分が(c1)成分又は(c2)成分であり、
前記(c1)成分がポリアミド粒子と熱硬化性ポリイミド粒子とを含み、
前記(c2)成分が融点140~175℃のポリアミドの真球形状の粒子を含み、
前記(A)成分が複数枚積層されており、かつ前記(C)成分が前記(A)成分の層間に存在する、繊維強化複合材料。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18771916.6A EP3604406A4 (en) | 2017-03-24 | 2018-03-23 | PREMIX AND FIBER REINFORCED COMPOSITE MATERIAL |
| CA3056671A CA3056671C (en) | 2017-03-24 | 2018-03-23 | Prepreg and fiber-reinforced composite material |
| JP2018518751A JP6673474B2 (ja) | 2017-03-24 | 2018-03-23 | プリプレグおよび繊維強化複合材料 |
| US16/572,164 US10920031B2 (en) | 2017-03-24 | 2019-09-16 | Prepreg and fiber-reinforced composite material |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017-058507 | 2017-03-24 | ||
| JP2017058507 | 2017-03-24 | ||
| JP2017-058508 | 2017-03-24 | ||
| JP2017058508 | 2017-03-24 | ||
| JP2018030289 | 2018-02-23 | ||
| JP2018-030289 | 2018-02-23 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/572,164 Continuation-In-Part US10920031B2 (en) | 2017-03-24 | 2019-09-16 | Prepreg and fiber-reinforced composite material |
| US16/572,164 Continuation US10920031B2 (en) | 2017-03-24 | 2019-09-16 | Prepreg and fiber-reinforced composite material |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018174250A1 true WO2018174250A1 (ja) | 2018-09-27 |
Family
ID=63585481
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/011715 WO2018174250A1 (ja) | 2017-03-24 | 2018-03-23 | プリプレグおよび繊維強化複合材料 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10920031B2 (ja) |
| EP (1) | EP3604406A4 (ja) |
| JP (1) | JP6673474B2 (ja) |
| CA (1) | CA3056671C (ja) |
| WO (1) | WO2018174250A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2019208040A1 (ja) * | 2018-04-23 | 2021-05-27 | 三菱ケミカル株式会社 | 炭素繊維強化複合材料用エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料 |
| WO2021132091A1 (ja) | 2019-12-23 | 2021-07-01 | 東レ株式会社 | 熱硬化性樹脂組成物、熱硬化性樹脂硬化物、プリプレグ及び繊維強化複合材料 |
| WO2022039104A1 (ja) * | 2020-08-20 | 2022-02-24 | 日鉄ケミカル&マテリアル株式会社 | エポキシ樹脂組成物及び硬化物 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117069987B (zh) * | 2023-10-17 | 2023-12-29 | 天津爱思达航天科技股份有限公司 | 一种碳纤维预浸料及其制备方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63162732A (ja) | 1986-12-25 | 1988-07-06 | Toray Ind Inc | プリプレグ |
| JPH10316750A (ja) | 1997-03-18 | 1998-12-02 | Ube Ind Ltd | 球状ポリアミドおよびその製法 |
| JPH11323087A (ja) * | 1989-01-04 | 1999-11-26 | Cytec Technol Corp | 強化熱硬化性構造材料 |
| JP2009286895A (ja) | 2008-05-29 | 2009-12-10 | Mitsubishi Rayon Co Ltd | プリプレグおよび繊維強化複合材料の成形方法 |
| WO2012102201A1 (ja) | 2011-01-28 | 2012-08-02 | 東レ株式会社 | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| WO2015033998A1 (ja) * | 2013-09-06 | 2015-03-12 | ダイセル・エボニック株式会社 | 繊維強化樹脂およびその製造方法並びに成形品 |
| JP2015098536A (ja) * | 2013-11-19 | 2015-05-28 | Jx日鉱日石エネルギー株式会社 | 繊維強化複合材料の製造方法、プリプレグ、粒子含有樹脂組成物及び繊維強化複合材料 |
| JP2015098533A (ja) * | 2013-11-19 | 2015-05-28 | Jx日鉱日石エネルギー株式会社 | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
| JP2015098535A (ja) * | 2013-11-19 | 2015-05-28 | Jx日鉱日石エネルギー株式会社 | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
| JP2016199682A (ja) | 2015-04-10 | 2016-12-01 | 東邦テナックス株式会社 | 繊維強化複合材料 |
| JP2017058508A (ja) | 2015-09-16 | 2017-03-23 | 富士ゼロックス株式会社 | 現像装置、画像形成ユニット、画像形成装置 |
| JP2017058507A (ja) | 2015-09-16 | 2017-03-23 | 日本電信電話株式会社 | 音声認識装置、音声認識方法、プログラム |
| JP2017206615A (ja) | 2016-05-18 | 2017-11-24 | 三菱ケミカル株式会社 | 繊維強化複合材料の製造方法 |
| JP2018030289A (ja) | 2016-08-24 | 2018-03-01 | 三菱鉛筆株式会社 | 摩擦具及び摩擦具セット |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0274899B1 (en) | 1986-12-25 | 1994-02-09 | Toray Industries, Inc. | Highly tough composite materials |
| GB0619401D0 (en) * | 2006-10-02 | 2006-11-08 | Hexcel Composites Ltd | Composite materials with improved performance |
| GB2460050A (en) * | 2008-05-14 | 2009-11-18 | Hexcel Composites Ltd | Epoxy composite |
| US9187636B2 (en) * | 2012-08-26 | 2015-11-17 | Hexcel Corporation | Composite material with polyamide particle mixtures |
| ES2905454T3 (es) * | 2014-09-22 | 2022-04-08 | Cytec Ind Inc | Materiales compuestos con alta conductividad eléctrica en la dirección Z |
| US10208176B2 (en) * | 2016-06-22 | 2019-02-19 | Hexcel Corporation | Composite material with thermoplastic toughened novolac-based epoxy resin matrix |
| WO2018066600A1 (ja) * | 2016-10-04 | 2018-04-12 | 三菱ケミカル株式会社 | プリプレグ、プリプレグ積層体、および繊維強化複合材料 |
-
2018
- 2018-03-23 EP EP18771916.6A patent/EP3604406A4/en active Pending
- 2018-03-23 WO PCT/JP2018/011715 patent/WO2018174250A1/ja active Application Filing
- 2018-03-23 JP JP2018518751A patent/JP6673474B2/ja active Active
- 2018-03-23 CA CA3056671A patent/CA3056671C/en active Active
-
2019
- 2019-09-16 US US16/572,164 patent/US10920031B2/en active Active
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63162732A (ja) | 1986-12-25 | 1988-07-06 | Toray Ind Inc | プリプレグ |
| JPH11323087A (ja) * | 1989-01-04 | 1999-11-26 | Cytec Technol Corp | 強化熱硬化性構造材料 |
| JPH10316750A (ja) | 1997-03-18 | 1998-12-02 | Ube Ind Ltd | 球状ポリアミドおよびその製法 |
| JP2009286895A (ja) | 2008-05-29 | 2009-12-10 | Mitsubishi Rayon Co Ltd | プリプレグおよび繊維強化複合材料の成形方法 |
| WO2012102201A1 (ja) | 2011-01-28 | 2012-08-02 | 東レ株式会社 | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| WO2015033998A1 (ja) * | 2013-09-06 | 2015-03-12 | ダイセル・エボニック株式会社 | 繊維強化樹脂およびその製造方法並びに成形品 |
| JP2015098536A (ja) * | 2013-11-19 | 2015-05-28 | Jx日鉱日石エネルギー株式会社 | 繊維強化複合材料の製造方法、プリプレグ、粒子含有樹脂組成物及び繊維強化複合材料 |
| JP2015098533A (ja) * | 2013-11-19 | 2015-05-28 | Jx日鉱日石エネルギー株式会社 | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
| JP2015098535A (ja) * | 2013-11-19 | 2015-05-28 | Jx日鉱日石エネルギー株式会社 | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
| JP2016199682A (ja) | 2015-04-10 | 2016-12-01 | 東邦テナックス株式会社 | 繊維強化複合材料 |
| JP2017058508A (ja) | 2015-09-16 | 2017-03-23 | 富士ゼロックス株式会社 | 現像装置、画像形成ユニット、画像形成装置 |
| JP2017058507A (ja) | 2015-09-16 | 2017-03-23 | 日本電信電話株式会社 | 音声認識装置、音声認識方法、プログラム |
| JP2017206615A (ja) | 2016-05-18 | 2017-11-24 | 三菱ケミカル株式会社 | 繊維強化複合材料の製造方法 |
| JP2018030289A (ja) | 2016-08-24 | 2018-03-01 | 三菱鉛筆株式会社 | 摩擦具及び摩擦具セット |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3604406A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2019208040A1 (ja) * | 2018-04-23 | 2021-05-27 | 三菱ケミカル株式会社 | 炭素繊維強化複合材料用エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料 |
| JP7167980B2 (ja) | 2018-04-23 | 2022-11-09 | 三菱ケミカル株式会社 | 炭素繊維強化複合材料用エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料 |
| US11745485B2 (en) | 2018-04-23 | 2023-09-05 | Mitsubishi Chemical Corporation | Epoxy resin composition for carbon-fiber-reinforced composite materials, prepreg, and carbon-fiber-reinforced composite material |
| WO2021132091A1 (ja) | 2019-12-23 | 2021-07-01 | 東レ株式会社 | 熱硬化性樹脂組成物、熱硬化性樹脂硬化物、プリプレグ及び繊維強化複合材料 |
| WO2022039104A1 (ja) * | 2020-08-20 | 2022-02-24 | 日鉄ケミカル&マテリアル株式会社 | エポキシ樹脂組成物及び硬化物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3056671C (en) | 2021-06-15 |
| US10920031B2 (en) | 2021-02-16 |
| EP3604406A4 (en) | 2020-03-18 |
| EP3604406A1 (en) | 2020-02-05 |
| US20200010634A1 (en) | 2020-01-09 |
| CA3056671A1 (en) | 2018-09-27 |
| JP6673474B2 (ja) | 2020-03-25 |
| JPWO2018174250A1 (ja) | 2019-03-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11292883B2 (en) | Prepreg, prepreg laminate, and fiber-reinforced composite material | |
| JP6673474B2 (ja) | プリプレグおよび繊維強化複合材料 | |
| KR102081662B1 (ko) | 에폭시 수지 조성물, 프리프레그 및 탄소 섬유 강화 복합 재료 | |
| CA2682549C (en) | Composite material with blend of thermoplastic particles | |
| RU2605424C2 (ru) | Композиция на основе эпоксидных смол и пленка, препрег и армированный волокнами пластик, полученные с использованием такой композиции | |
| JP5532549B2 (ja) | プリプレグおよび繊維強化複合材料の成形方法 | |
| EP2134764A1 (en) | Composite material with blend of thermoplastic particles | |
| TWI735810B (zh) | 樹脂組成物之製造方法 | |
| JP6970755B2 (ja) | プリプレグ及びその製造方法、並びに繊維強化複合材料の製造方法 | |
| JP2019038939A (ja) | プリプレグおよび繊維強化複合材料 | |
| JP2010095557A (ja) | プリプレグおよび繊維強化複合材料 | |
| JP6907468B2 (ja) | 繊維強化複合材料の製造方法 | |
| TWI815628B (zh) | 碳纖維束、預浸體、纖維強化複合材料 | |
| WO2019208040A1 (ja) | 炭素繊維強化複合材料用エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料 | |
| JP6555006B2 (ja) | エポキシ樹脂組成物、樹脂硬化物、プリプレグおよび繊維強化複合材料 | |
| JP2008050587A (ja) | プリプレグおよび複合材料 | |
| JP2019156981A (ja) | プリプレグ、繊維強化複合材料、及びそれらの製造方法 | |
| WO2021117461A1 (ja) | プリプレグ、積層体および一体化成形品 | |
| US20150174872A1 (en) | Toughened composite materials and methods of manufacturing thereof | |
| JP2020176249A (ja) | エポキシ樹脂組成物、プリプレグ、プリプレグの製造方法、及び繊維強化複合材料の製造方法 | |
| JP2011057907A (ja) | 熱硬化性樹脂組成物とそれを用いたプリプレグ | |
| JPH04292635A (ja) | プリプレグ | |
| JP2023067347A (ja) | 積層体 | |
| WO2024005111A1 (ja) | 強化繊維ステッチ基材、プリフォーム材、及び繊維強化複合材料、並びにこれらの製造方法 | |
| JP2023048264A (ja) | 積層体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018518751 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18771916 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3056671 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018771916 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2018771916 Country of ref document: EP Effective date: 20191024 |