WO2017010389A1 - ポリアミド樹脂繊維、ポリアミド樹脂繊維の製造方法、ポリアミド樹脂組成物、織物および編物 - Google Patents
ポリアミド樹脂繊維、ポリアミド樹脂繊維の製造方法、ポリアミド樹脂組成物、織物および編物 Download PDFInfo
- Publication number
- WO2017010389A1 WO2017010389A1 PCT/JP2016/070080 JP2016070080W WO2017010389A1 WO 2017010389 A1 WO2017010389 A1 WO 2017010389A1 JP 2016070080 W JP2016070080 W JP 2016070080W WO 2017010389 A1 WO2017010389 A1 WO 2017010389A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyamide resin
- resin fiber
- derived
- fiber
- dicarboxylic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/06—Polyamides derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/13—Phenols; Phenolates
- C08K5/134—Phenols containing ester groups
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/78—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from copolycondensation products
- D01F6/80—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from copolycondensation products from copolyamides
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/88—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from mixtures of polycondensation products as major constituent with other polymers or low-molecular-weight compounds
- D01F6/90—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from mixtures of polycondensation products as major constituent with other polymers or low-molecular-weight compounds of polyamides
-
- D—TEXTILES; PAPER
- D03—WEAVING
- D03D—WOVEN FABRICS; METHODS OF WEAVING; LOOMS
- D03D15/00—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used
- D03D15/20—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used characterised by the material of the fibres or filaments constituting the yarns or threads
- D03D15/283—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used characterised by the material of the fibres or filaments constituting the yarns or threads synthetic polymer-based, e.g. polyamide or polyester fibres
-
- D—TEXTILES; PAPER
- D03—WEAVING
- D03D—WOVEN FABRICS; METHODS OF WEAVING; LOOMS
- D03D15/00—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used
- D03D15/40—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used characterised by the structure of the yarns or threads
- D03D15/41—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used characterised by the structure of the yarns or threads with specific twist
-
- D—TEXTILES; PAPER
- D04—BRAIDING; LACE-MAKING; KNITTING; TRIMMINGS; NON-WOVEN FABRICS
- D04B—KNITTING
- D04B1/00—Weft knitting processes for the production of fabrics or articles not dependent on the use of particular machines; Fabrics or articles defined by such processes
- D04B1/14—Other fabrics or articles characterised primarily by the use of particular thread materials
- D04B1/16—Other fabrics or articles characterised primarily by the use of particular thread materials synthetic threads
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/14—Polymer mixtures characterised by other features containing polymeric additives characterised by shape
- C08L2205/16—Fibres; Fibrils
Definitions
- the present invention relates to a polyamide resin fiber, a method for producing a polyamide resin fiber, a polyamide resin composition, a woven fabric and a knitted fabric.
- Polyamide resin is a resin excellent in heat resistance and mechanical strength, and has various applications.
- Patent Document 1 (A) 5 to 90 parts by weight of a polyphenylene ether-based resin and (B) 95 to 10 parts by weight of a polyamide resin are combined with 100 parts by weight of (C) a compatibilizing agent 0.01
- a thermoplastic resin composition comprising: 30 parts by weight, (D) 0.1 to 100 parts by weight of a plasticizer that imparts flexibility to polyamide, and (E) 0 to 100 parts by weight of a rubber-like substance. It is disclosed. Further, Patent Document 1 describes that such a thermoplastic resin composition has flexibility and high tensile strength at the same time. Patent Document 1 describes that such a thermoplastic resin composition is used for pipes and tubes.
- Patent Document 2 describes a hollow structure comprising a layer made of a resin composition comprising one or more semi-aromatic polyamides and one or more functional polyolefins. ing. Such hollow structures are described to exhibit a balance of properties regarding flexibility, permeability barrier to refrigerant fluid, and retention of properties upon heating and exposure to refrigerant fluid.
- Patent Document 3 (A) 2 to 60 parts by weight of a compound represented by the general formula (1) with respect to 100 parts by weight of a polyamide resin selected from polyamide 11, polyamide 12 or a mixture thereof, (In the formula, m is an integer of 7 or more and 10 or less.) (B) A polyamide resin composition comprising 0.05 to 5 parts by weight of a monohydric alcohol having 16 to 24 carbon atoms having a branched chain is disclosed. Examples of applications of such polyamide resin compositions include industrial robots, construction machines, fuel hoses for automobiles, hydraulic and pneumatic tubes, and injection molded parts for automobile interiors.
- the polyamide resin is processed into various shapes. Furthermore, in recent years, polyamide resin fibers in which a polyamide resin or a polyamide resin composition is made into a fiber form are also used. Among such polyamide resin fibers, xylylenediamine-based polyamide resin fibers are useful because of their low water absorption and excellent chemical resistance. However, since the polyamide resin fibers are fibers, high durability is particularly required as compared with polyamide resin molded products having other shapes.
- the object of the present invention is to provide a polyamide resin fiber having excellent durability. Moreover, it aims at providing the manufacturing method of a polyamide resin fiber, a polyamide resin composition, a textile fabric, and knitting.
- the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid, 50 mol% or more of the structural unit derived from diamine is derived from xylylenediamine, and 50 mol% or more of the structural unit derived from dicarboxylic acid is an ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms.
- Polyamide resin fiber derived from General formula (1) R 1 is an alkyl group having 1 to 10 carbon atoms, R 2 is an alkyl group having 2 to 12 carbon atoms, and n is an integer of 1 to 3.
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- ⁇ 3> The polyamide when the polyamide resin fiber is fixed and the ceramic plate derived from aluminum oxide is brought into contact with the polyamide resin fiber while rotating a ceramic plate derived from aluminum oxide at a friction plate rotation speed of 120 rpm so that an average load of 150 g is applied.
- ⁇ 6> The polyamide resin fiber according to any one of ⁇ 1> to ⁇ 4>, wherein 50 mol% or more of the structural units derived from the dicarboxylic acid are derived from adipic acid.
- ⁇ 7> The polyamide resin fiber according to any one of ⁇ 1> to ⁇ 6>, wherein a phosphorus atom concentration in the polyamide resin fiber is 0.1 to 10 ppm.
- ⁇ 8> The polyamide resin fiber according to any one of ⁇ 1> to ⁇ 7>, wherein the tensile strength measured according to JIS L 1013 is 2.0 cN / dtex or more.
- ⁇ 9> The polyamide resin fiber according to any one of ⁇ 1> to ⁇ 8>, wherein the polyamide resin fiber is stretched.
- the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid.
- 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine
- 50 mol% or more of the structural unit derived from the dicarboxylic acid is an ⁇ , ⁇ -linear aliphatic having 4 to 20 carbon atoms.
- a process for producing a polyamide resin fiber comprising drawing a fiber comprising a polyamide resin composition derived from dicarboxylic acid;
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- ⁇ 12> The method for producing a polyamide resin fiber according to ⁇ 10> or ⁇ 11>, wherein the overall draw ratio is 4.1 times or more.
- ⁇ 13> The method for producing a polyamide resin fiber according to any one of ⁇ 10> to ⁇ 12>, wherein the fiber before stretching has a fineness of 100 to 5000 dtex.
- ⁇ 14> 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin, and the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid. 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine, and 50 mol% or more of the structural unit derived from the dicarboxylic acid is an ⁇ , ⁇ -linear aliphatic having 4 to 20 carbon atoms.
- a polyamide resin composition derived from a dicarboxylic acid General formula (1)
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- ⁇ 17> A woven fabric using the fiber according to any one of ⁇ 1> to ⁇ 9>.
- ⁇ 18> A knitted fabric using the fiber according to any one of ⁇ 1> to ⁇ 9>.
- a polyamide resin fiber excellent in durability a method for producing a polyamide resin fiber, a polyamide resin composition, a woven fabric and a knitted fabric.
- the polyamide resin fiber of the present invention contains 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin, and the polyamide resin is derived from a diamine-derived structural unit and a dicarboxylic acid.
- 50 mol% or more of the structural unit derived from diamine is derived from xylylenediamine
- 50 mol% or more of the structural unit derived from dicarboxylic acid is ⁇ , ⁇ having 4 to 20 carbon atoms. -Derived from linear aliphatic dicarboxylic acids.
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- Patent Document 1 The blending of a plasticizer such as a compound represented by the general formula (1) with a polyamide resin is described in Patent Document 1, Patent Document 2, and Patent Document 3, for example.
- a plasticizer such as a compound represented by the general formula (1)
- Patent Document 2 Patent Document 3
- it was composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid, and 50 mol% or more of the structural unit derived from the diamine was derived from xylylenediamine, and derived from the dicarboxylic acid.
- a polyamide resin in which 50 mol% or more of the structural unit is derived from an ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms (hereinafter sometimes referred to as “XD polyamide resin”) has a general formula ( By blending the compound represented by 1) at a predetermined ratio, it was possible to achieve remarkably excellent durability. That is, when a plasticizer is added, the crystallization temperature of the polyamide resin at the time of temperature rise generally tends to decrease. When the crystallization temperature is lowered, crystallization is likely to proceed, and stretching is usually difficult.
- the compound represented by the general formula (1) was blended with the XD-based polyamide resin, the crystallization temperature at the time of the temperature rise decreased.
- the polyamide resin fiber of the present invention contains a compound represented by the general formula (1), which is a kind of plasticizer, it has a high draw ratio and a synergistic effect of plasticizing effect by the plasticizer. A polyamide resin fiber excellent in abrasion is obtained. Based on the above, the present invention has succeeded in providing a polyamide resin fiber capable of achieving durability. Details of the present invention will be described below.
- the polyamide resin fiber of the present invention comprises a polyamide resin composition containing 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the XD polyamide resin. More preferably, it is formed by stretching a fiber made of the polyamide resin composition.
- the details of the polyamide resin composition of the present invention will be described.
- the XD-based polyamide resin used as an essential component in the present invention is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid, and 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine, More than 50 mol% of the structural unit derived from the acid is derived from the ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms.
- the XD-based polyamide resin is preferably derived from at least one mol of xylylenediamine, more preferably at least 70 mol%, more preferably at least 80 mol%, more preferably at least 90 mol% of the structural unit derived from diamine.
- the diamine component that is the raw material of the XD-based polyamide resin contains 70% by mole or more of metaxylylenediamine, more preferably 80% by mole or more, and still more preferably 90% by mole or more.
- the metaxylylenediamine in the diamine component is 70 mol% or more
- the polyamide resin obtained therefrom can exhibit excellent gas barrier properties.
- the xylylenediamine is preferably composed of 30 to 100 mol% metaxylylenediamine and 0 to 70 mol% paraxylylenediamine, and preferably 70 to 100 mol% metaxylylenediamine and 0. More preferably, it consists of ⁇ 30 mol% paraxylylenediamine.
- diamines other than xylylenediamine that can be used as the raw material diamine component of the XD polyamide resin include tetramethylenediamine, pentamethylenediamine, 2-methylpentanediamine, hexamethylenediamine, heptamethylenediamine, octamethylenediamine, and nonamethylene.
- Aliphatic diamines such as diamine, decamethylenediamine, dodecamethylenediamine, 2,2,4-trimethyl-hexamethylenediamine, 2,4,4-trimethylhexamethylenediamine, 1,3-bis (aminomethyl) cyclohexane, 1 , 4-bis (aminomethyl) cyclohexane, 1,3-diaminocyclohexane, 1,4-diaminocyclohexane, bis (4-aminocyclohexyl) methane, 2,2-bis (4-aminocyclohexyl) propyl Aliphatic diamines such as bread, bis (aminomethyl) decalin, bis (aminomethyl) tricyclodecane, diamines having aromatic rings such as bis (4-aminophenyl) ether, paraphenylenediamine, bis (aminomethyl) naphthalene Etc., and one kind or a mixture of two or more kinds can be used.
- a diamine other than xylylenediamine is used as the diamine component, it is used in an amount of 30 mol% or less, preferably 1 to 25 mol%, particularly preferably 5 to 20 mol% of the structural unit derived from the diamine.
- the ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms that is preferably used as the raw material dicarboxylic acid component of the XD-based polyamide resin includes ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 6 to 16 carbon atoms. ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 6 to 10 carbon atoms is more preferable.
- ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms examples include succinic acid, glutaric acid, pimelic acid, suberic acid, azelaic acid, adipic acid, sebacic acid, undecanedioic acid, and dodecanedioic acid.
- Aliphatic dicarboxylic acids can be exemplified, and one or more can be mixed and used, but among these, the melting point of the polyamide resin is in an appropriate range for molding, so at least one of adipic acid and sebacic acid One is preferred, and adipic acid is particularly preferred.
- dicarboxylic acid component other than the ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms examples include phthalic acid compounds such as isophthalic acid, terephthalic acid and orthophthalic acid, 1,2-naphthalenedicarboxylic acid, 3-naphthalenedicarboxylic acid, 1,4-naphthalenedicarboxylic acid, 1,5-naphthalenedicarboxylic acid, 1,6-naphthalenedicarboxylic acid, 1,7-naphthalenedicarboxylic acid, 1,8-naphthalenedicarboxylic acid, 2,3- Examples thereof include naphthalenedicarboxylic acids such as isomers such as naphthalenedicarboxylic acid, 2,6-naphthalenedicarboxylic acid, and 2,7-naphthalenedicarboxylic acid, and one kind or a mixture of two or more kinds can be used.
- phthalic acid compounds such as isophthalic acid,
- terephthalic acid or isophthalic acid may be used from the viewpoint of molding processability and barrier properties. preferable.
- the proportion of terephthalic acid and isophthalic acid is preferably 30 mol% or less, more preferably 1 to 30 mol%, particularly preferably 5 to 20 mol% of the structural unit derived from dicarboxylic acid.
- a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid means that the amide bond constituting the XD-based polyamide resin is formed by a bond between the dicarboxylic acid and the diamine.
- XD type polyamide resin contains other site
- a repeating unit having an amide bond that is not derived from a bond between a dicarboxylic acid and a diamine, a trace amount of impurities, and the like are included.
- the XD polyamide resin is a component constituting the polyamide resin in addition to the diamine component and the dicarboxylic acid component, and lactams such as ⁇ -caprolactam and laurolactam, aminocapron as long as the effects of the present invention are not impaired.
- Aliphatic aminocarboxylic acids such as acids and aminoundecanoic acids can also be used as copolymerization components.
- 90% by weight or more of the XD polyamide resin is preferably a structural unit derived from diamine or a structural unit derived from dicarboxylic acid.
- the XD polyamide resin used in the present invention preferably has a phosphorus atom concentration of 0.1 to 10 ppm, more preferably 1 to 8 ppm. By setting it as such a range, the improvement of continuous productivity by prevention of yellowing of a fiber and suppression of clogging of a polymer filter is compatible, and the effect of the present invention is more effectively exhibited.
- the polyamide resin fibers used in the present invention preferably have a phosphorus atom concentration in a predetermined range, but such phosphorus atoms are usually derived from a polyamide resin.
- the XD polyamide resin used in the present invention preferably has a number average molecular weight (Mn) of 6,000 to 30,000, more preferably 8,000 to 28,000, still more preferably 9,000 to 26,000. When it is in such a range, the moldability becomes better.
- the XD polyamide resin used in the present invention preferably has a molecular weight distribution (weight average molecular weight / number average molecular weight (Mw / Mn)) of 1.8 to 3.1.
- the molecular weight distribution is more preferably 1.9 to 3.0, still more preferably 2.0 to 2.9.
- the molecular weight distribution of the XD polyamide resin can be adjusted, for example, by appropriately selecting the polymerization reaction conditions such as the type and amount of the initiator and catalyst used during the polymerization, and the reaction temperature, pressure, and time. It can also be adjusted by mixing a plurality of types of XD polyamide resins having different average molecular weights obtained under different polymerization conditions or by separately precipitating the polymerized XD polyamide resins.
- the molecular weight distribution can be obtained by GPC measurement. Specifically, using “HLC-8320GPC” manufactured by Tosoh Corporation as an apparatus and two “TSK gel Super HM-H” manufactured by Tosoh Corporation as a column, eluent Measured under conditions of hexafluoroisopropanol (HFIP) having a sodium trifluoroacetate concentration of 10 mmol / l, a resin concentration of 0.02% by weight, a column temperature of 40 ° C., a flow rate of 0.3 ml / min, and a refractive index detector (RI). It can obtain
- a calibration curve is prepared by dissolving 6 levels of PMMA in HFIP.
- the XD polyamide resin has a terminal amino group concentration ([NH 2 ]) of preferably less than 100 ⁇ equivalent / g, more preferably 5 to 75 ⁇ equivalent / g, and still more preferably 10 to 60 ⁇ equivalent / g.
- the carboxyl group concentration ([COOH]) is preferably less than 150 ⁇ eq / g, more preferably 10 to 120 ⁇ eq / g, and still more preferably 10 to 100 ⁇ eq / g.
- the viscosity is easily stabilized when the XD-based polyamide resin is processed into a film or fiber, and the reactivity with a carbodiimide compound described later is good. Tend to be.
- the ratio of the terminal amino group concentration to the terminal carboxyl group concentration is preferably 0.7 or less, more preferably 0.6 or less, particularly preferably 0. .5 or less. When this ratio is larger than 0.7, it may be difficult to control the molecular weight when polymerizing the XD polyamide resin.
- the terminal amino group concentration can be measured by dissolving 0.5 g of XD polyamide resin in 30 ml of a phenol / methanol (4: 1) mixed solution with stirring at 20-30 ° C. and titrating with 0.01 N hydrochloric acid. .
- the terminal carboxyl group concentration was determined by dissolving 0.1 g of XD polyamide resin in 30 ml of benzyl alcohol at 190 ° C., cooling in a 40 ° C. water bath, and titrating 0.01 N (normative) KOH solution. Can be measured.
- the description of paragraphs 0052 to 0053 of JP-A-2014-173196 can be referred to for the production method of the XD polyamide resin, and the contents thereof are incorporated in the present specification.
- the melting point of the XD polyamide resin is preferably 150 to 350 ° C., more preferably 180 to 300 ° C., and still more preferably 180 to 250 ° C. Further, the glass transition point of the XD polyamide resin is preferably 50 to 100 ° C., more preferably 55 to 100 ° C., and particularly preferably 60 to 100 ° C. Within this range, the heat resistance tends to be good.
- the melting point in the present invention is the temperature at the peak top of the endothermic peak at the time of temperature rise observed by the DSC (Differential Scanning Calorimetry) method. Specifically, it is measured by the method described in the examples described later. Value. When the equipment used in the examples is not available due to reasons such as out of print, it can be measured using other equipment having equivalent performance. Hereinafter, the same applies to other measurement methods.
- the glass transition point refers to a glass transition point that is measured by once heating and melting a sample to eliminate the influence of the thermal history on crystallinity and then raising the temperature again.
- DSC-60 manufactured by Shimadzu Corporation
- the sample amount is about 1 mg
- nitrogen is flowed at 30 ml / min as the atmospheric gas
- the heating rate is 10 ° C./min.
- the polyamide resin heated and melted from room temperature to a temperature higher than the expected melting point under dry conditions is rapidly cooled with dry ice, and the temperature is raised again to a temperature higher than the melting point at a rate of 10 ° C./min to obtain the glass transition point.
- Can do
- the lower limit of the crystallization temperature at the time of temperature rise of the XD polyamide resin is preferably 50 ° C. or higher, more preferably 80 ° C. or higher, further preferably 100 ° C. or higher, particularly preferably 120 ° C. or higher, and 140 ° C. or higher. Even more preferred.
- the upper limit of the crystallization temperature of the XD polyamide resin at the time of temperature rise is preferably 180 ° C. or less, more preferably 170 ° C. or less, further preferably 162 ° C. or less, particularly preferably 155 ° C. or less, and preferably 148 ° C. or less. Even more preferred.
- the XD-based polyamide resin used in the present invention is a compound in which the crystallization temperature at the time of temperature rise when the compound represented by 5% by weight of the general formula (1) is blended is represented by the general formula (1) Is preferably lower than the crystallization temperature at the time of temperature rise when not blended, the difference is more preferably 3 ° C. or more, further preferably 5 ° C. or more, and preferably 10 ° C. or more. Particularly preferred.
- the upper limit of the difference in crystallization temperature at the time of temperature rise is not particularly defined, but can be, for example, 40 ° C. or lower, further 35 ° C. or lower, and particularly 30 ° C. or lower. it can.
- the measuring method of the crystallization temperature at the time of temperature rising in this invention follows the method as described in the Example mentioned later.
- the proportion of the XD polyamide resin in the polyamide resin composition of the present invention is 50% by weight or more, preferably 60% by weight or more, more preferably 70% by weight or more, and 80% by weight or more. You can also.
- the polyamide resin composition of the present invention may contain a polyamide resin other than the XD polyamide resin.
- examples of such other polyamide resins include polyamide 4, polyamide 6, polyamide 11, polyamide 12, polyamide 46, polyamide 66, polyamide 610, polyamide 612, polyhexamethylene terephthalamide (polyamide 6T), polyhexamethylene isophthalate.
- Examples include ramid (polyamide 6I), polyamide 66 / 6T, polyamide 9T, polyamide 9MT, polyamide 6I / 6T, and the like.
- the content of the other polyamide resin in the polyamide resin composition of the present invention is preferably 1 to 50 parts by weight, more preferably 5 to 40 parts by weight, based on 100 parts by weight of the XD polyamide resin.
- the polyamide resin composition of this invention contains the compound represented by General formula (1).
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- the OH group part improves the familiarity with the XD polyamide resin.
- the OH group moiety may be substituted at any of the ortho, para and meta positions, but the para position is more preferred.
- the group represented by — (CH 2 ) n — is presumed to play the role of a linking group that connects the hydroxyphenyl ester group and —CR 1 R 2. . n is preferably 1 or 2, and more preferably 1.
- R 1 is preferably an alkyl group having 1 to 9 carbon atoms, more preferably an alkyl group having 2 to 9 carbon atoms, and 2 to 8 carbon atoms.
- the alkyl group is more preferably an alkyl group having 3 to 7 carbon atoms, particularly preferably an alkyl group having 4 to 6 carbon atoms.
- the alkyl group as R 1 is preferably a linear or branched alkyl group, and more preferably a linear alkyl group.
- R 2 is preferably an alkyl group having 2 to 10 carbon atoms, more preferably an alkyl group having 3 to 9 carbon atoms, and 5 to 9 carbon atoms. Are more preferable, and an alkyl group having 6 to 8 carbon atoms is particularly preferable.
- the alkyl group as R 2 is preferably a linear or branched alkyl group, and more preferably a linear alkyl group.
- the number of carbon atoms constituting R 2 is preferably 2 or more, and preferably 2 to 4 larger than the number of carbon atoms constituting R 1. More preferred. By adopting such a configuration, the effect of the present invention is more effectively exhibited.
- the compound represented by the general formula (1) contains 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin in the polyamide resin composition.
- the lower limit of the compound represented by the general formula (1) is 0.5 parts by weight or more, preferably 0.6 parts by weight or more, more preferably 0.8 parts by weight or more, and 1.0 part by weight or more. Is more preferable, and 2.0 parts by weight or more is particularly preferable.
- the upper limit is 15 parts by weight or less, preferably 10 parts by weight or less, more preferably 8 parts by weight or less, and further preferably 7 parts by weight or less. Only 1 type may be sufficient as the compound represented by General formula (1), and 2 or more types may be sufficient as it.
- the total amount is preferably within the above range.
- the compound represented by the general formula (1) is blended in an amount of preferably 8 to 15 parts by weight, more preferably 9 to 13 parts by weight, based on 100 parts by weight of the polyamide resin, the melt viscosity can be lowered.
- the polyamide resin composition containing the compound represented by the general formula (1) at the above ratio has a melt viscosity of 500 Pa ⁇ s at 260 ° C. and an apparent shear rate of 122 (S ⁇ 1 ). It can be:
- the lower limit value of the melt viscosity in this case is not particularly defined, but can be set to, for example, 400 Pa ⁇ s or more.
- the temperature at which the weight is increased at a rate of 10 ° C./min and the weight is reduced by 3% by weight with respect to the weight at 150 ° C. of the polyamide resin composition containing the compound represented by the general formula (1) at the above ratio. Can be high. In particular, these effects are remarkable as compared with the case where a plasticizer other than the compound represented by the general formula (1) is blended. Only 1 type may be sufficient as the compound represented by General formula (1), and 2 or more types may be sufficient as it. When 2 or more types are included, the total amount is preferably within the above range.
- the polyamide resin composition of the present invention may contain one or more plasticizers other than the compound represented by the general formula (1).
- plasticizers include the plasticizers described in paragraph 0039 of JP-A No. 7-11131, the contents of which are incorporated herein.
- the polyamide resin composition of the present invention preferably has a configuration that does not substantially contain a plasticizer other than the compound represented by the general formula (1). “Substantially free” means that, for example, in the polyamide resin composition of the present invention, the content of the other plasticizer is 0.1% by weight or less of the weight of the compound represented by the general formula (1). Say.
- the polyamide resin composition of the present invention may contain a resin component other than the polyamide resin.
- resin components other than polyamide resins include polyolefin resins such as polyethylene and polypropylene, polyester resins such as polyethylene terephthalate and polybutylene terephthalate, polycarbonate resins, polyoxymethylene resins, polyether ketones, polyether sulfones, and thermoplastic polyethers.
- a thermoplastic resin such as imide is exemplified.
- the polyamide resin composition of the present invention can be configured to contain substantially no thermoplastic resin other than the polyamide resin. “Substantially not contained” means that, for example, in the polyamide resin composition of the present invention, the content of the thermoplastic resin other than the polyamide resin is 5% by weight or less of the weight of the polyamide resin.
- the polyamide resin composition of the present invention includes an antioxidant, a stabilizer such as a heat stabilizer, a hydrolysis resistance improver, a weather resistance stabilizer, and a matting agent as long as the objects and effects of the present invention are not impaired.
- Additives such as ultraviolet absorbers, nucleating agents, plasticizers, dispersants, flame retardants, antistatic agents, anti-coloring agents, anti-gelling agents, coloring agents, release agents, and the like can be added. Details of these can be referred to the description of paragraphs 0130 to 0155 of Japanese Patent No. 4894982, the contents of which are incorporated herein.
- the polyamide resin composition is an XD polyamide resin and the compound represented by the general formula (1), and preferably accounts for 80% by weight or more, more preferably 90% by weight or more.
- the polyamide resin composition of this invention may contain fillers, such as carbon fiber, it is preferable not to contain substantially. “Substantially free” means, for example, that the blending amount of the filler is 3% by weight or less of the polyamide resin composition of the present invention.
- the polyamide resin composition of the present invention is preferably used for the production of polyamide resin fibers, and more preferably for the production of stretched polyamide resin fibers. Moreover, you may use for another use.
- the melting point of the polyamide resin composition of the present invention is preferably 150 to 350 ° C., more preferably 180 to 300 ° C., and further preferably 180 to 250 ° C.
- the lower limit of the crystallization temperature at the time of heating of the polyamide resin composition of the present invention is preferably 50 ° C. or higher, more preferably 80 ° C. or higher, further preferably 100 ° C. or higher, particularly preferably 120 ° C. or higher, 140 It can also be set to ° C or lower.
- the upper limit of the crystallization temperature at the time of temperature rising of the polyamide resin composition of the present invention is preferably 180 ° C. or less, more preferably 170 ° C. or less, further preferably 162 ° C.
- the polyamide resin composition used in the present invention is a compound in which the crystallization temperature at the time of temperature rise when the compound represented by 5% by weight of the general formula (1) is blended is represented by the general formula (1) Is preferably lower than the crystallization temperature at the time of temperature rise when not blended, the difference is more preferably 3 ° C. or more, further preferably 5 ° C. or more, and preferably 10 ° C. or more. Particularly preferred.
- the upper limit of the difference in crystallization temperature at the time of temperature rise is not particularly defined, but can be, for example, 40 ° C. or lower, further 35 ° C. or lower, and particularly 30 ° C. or lower. it can.
- the polyamide resin fiber of the present invention fixes the polyamide resin fiber to the polyamide resin fiber while rotating the ceramic plate derived from aluminum oxide at a friction plate rotation speed of 120 rpm so that an average load of 150 g is applied.
- a polyamide resin fiber having a rotation speed (hereinafter sometimes referred to as “abrasion-resistant rotation speed”) of 100 or more until the polyamide resin fiber is cut when brought into contact with each other can be obtained.
- Such high wear resistance is achieved by blending the XD polyamide resin with the compound represented by the general formula (1) and stretching.
- the rotational speed of the abrasion resistance can be measured by passing the fiber to be measured through each yarn guide, fixing both ends of the fiber with a terminal and a clip carrying an arbitrary load, holding a recording pen over the fiber, and turning on the switch to turn the friction disc. Measured as the number of revolutions until the fiber breaks, using a device that is loaded on a friction piece, which is a ceramic plate derived from 11 differently shaped aluminum oxides attached to the friction disc .
- FIG. 1 on page 564 of a conjugation force testing machine (textile engineering, Vol. 26, No. 7 1973) manufactured by Hiruta Riken Co., LTD. Examples of the described yarn-bonding force tester).
- the wear-resistant rotational speed in the present invention is a value measured by the method described in the examples. Since the polyamide resin fiber of the present invention can be drawn at a high draw ratio and contains a compound (plasticizer) represented by the general formula (1), high wear resistance can be achieved.
- the wear-resistant rotational speed in the present invention is preferably 100 or more.
- the upper limit of the wear-resistant rotational speed is not particularly defined and is preferably higher. For example, when a polyamide resin in which 50 mol% or more of the structural unit derived from dicarboxylic acid is derived from adipic acid is used, even if it is 250 or less, it is practically used. , Excellent level. Moreover, when using the polyamide resin from which 50 mol% or more of structural units derived from dicarboxylic acid derive from sebacic acid, even if it is 1500 or less, it is a practically excellent level.
- the polyamide resin fiber of the present invention can have a tensile strength measured according to JIS L 1013 of 2.0 cN / dtex or more, and further 2.2 cN / dtex or more.
- the upper limit of the tensile strength is not particularly defined and is preferably higher.
- a polyamide resin in which 50 mol% or more of the structural units derived from dicarboxylic acid are derived from adipic acid is used, 4.0 cN / dtex. Even below, it is a practically excellent level.
- the polyamide resin from which 50 mol% or more of structural units derived from dicarboxylic acid derive from sebacic acid even if it is 8.0 cN / dtex or less, it is a practically excellent level.
- the phosphorus atom concentration of the polyamide resin fiber of the present invention is preferably 0.1 to 10 ppm, and more preferably 1 to 8 ppm. By setting it as such a range, the prevention of yellowing of a fiber and the improvement of continuous productivity are compatible, and the effect of this invention is exhibited more effectively.
- the phosphorus component contained in the polyamide resin fiber includes a phosphorus component derived from a polyamide resin, a phosphorus component derived from various additives (for example, a flame retardant), and the like.
- the length (weight average fiber length) of the polyamide resin fiber of the present invention is not particularly defined, but is usually 2 cm or more, preferably in the range of 0.1 m to 20,000 m, more preferably 1 m to It is 10,000 m, more preferably 100 m to 7,000 m.
- the lower limit of the fineness of the polyamide resin fiber of the present invention is preferably 50 dtex or more, more preferably 60 dtex or more, further preferably 70 dtex or more, particularly preferably 80 dtex or more, and 110 dtex. The above is more preferable.
- the upper limit value is preferably 10,000 dtex or less, more preferably 1000 dtex or less, further preferably 800 dtex or less, and particularly preferably 600 dtex or less.
- Such a polyamide resin fiber may be a monofilament, but is usually a multifilament composed of two or more monofilaments.
- the number of fibers is preferably 10 to 1000 f, more preferably 10 to 500 f, and further preferably 20 to 100 f.
- 60 dtex to 120 dtex is preferable for apparel use.
- 100 to 1500 dtex is preferable for use for fishing nets, and 1000 to 3000 dtex is preferable for use for tire cords.
- the cross section of the polyamide resin fiber of the present invention is usually circular.
- the term “circular” as used herein includes not only a circular in the mathematical sense, but also includes those that are generally recognized as circular in the technical field of the present invention.
- the cross section of the polyamide resin fiber in the present invention may be a shape other than a circle, and may be a flat shape such as an ellipse or an oval, for example.
- polyamide resin fiber in the present invention is a multifilament. It goes without saying that the present invention is not limited to these forms.
- a step of forming monofilament polyamide resin fibers into a bundle may be included in the course of the production steps of the polyamide resin fibers of the present invention.
- polyamide resin fibers are bundled with at least one of a sizing agent and a surface treatment agent (sometimes referred to as oil agent, sizing agent, etc.). It is done.
- the sizing agent and the surface treatment agent are not particularly limited as long as they have a function of converging polyamide resin fibers, but are not limited to oil agents such as mineral oil and animal / vegetable oil, and nonionic surfactants. And surfactants such as anionic surfactants and amphoteric surfactants. More specifically, ester compounds, alkylene glycol compounds, polyolefin compounds, phenyl ether compounds, polyether compounds, silicone compounds, polyethylene glycol compounds, amide compounds, sulfonate compounds, phosphate compounds, A carboxylate compound and a combination of two or more of these are preferred.
- the total amount of the sizing agent and the surface treating agent for the polyamide resin fiber is preferably 0.1 to 5.0% by weight, more preferably 0.5 to 2.0% by weight, based on the polyamide resin fiber.
- the treatment method of the polyamide resin fiber with the sizing agent and / or the surface treatment agent is not particularly defined as long as the intended purpose can be achieved.
- a sizing agent and / or a surface treatment agent dissolved in a solution is added to a polyamide resin fiber, and the sizing agent and / or the surface treatment agent is attached to the surface of the polyamide resin fiber.
- the sizing agent and / or the surface treatment agent can be air blown to the surface of the polyamide resin fiber.
- the second embodiment of the multifilament in the present invention is exemplified by a method of twisting into a bundle.
- twisting only the polyamide resin fibers may be twisted, or other resin fibers or fibers other than resin fibers may be twisted together.
- fibers other than resin fibers include carbon fibers.
- the twist is preferably applied after stretching, and further after stretching and heat setting.
- means for treating with a sizing agent, a surface treatment agent, or the like may be used in combination.
- the method of twisting is not particularly defined, and a known method can be adopted.
- the number of twists can be appropriately determined according to the number of fibers of the polyamide resin fiber, the fineness, and the like. For example, it can be 1 to 200 times / m (fiber length), and more preferably 1 to 100 times / m. More preferably, it can be 1 to 70 times / m, and particularly 1 to 50 times / m.
- the third embodiment of the multifilament in the present invention is exemplified by a core-sheath structure.
- the core-sheath structure refers to, for example, a form consisting of two or more regions, such as a pencil core and a sheath.
- a monofilament or multifilament polyamide resin fiber is used as a core, and the periphery thereof is formed. It is preferable to provide a layer structure that serves as a sheath. By setting it as such a structure, the characteristic of a polyamide resin fiber can be changed and it becomes possible to use it for many kinds of uses.
- the core and sheath portions do not have to be completely distinguished as regions.
- the core portion when the core portion is a multifilament composed of a plurality of polyamide resin fibers, a part of the material constituting the sheath may penetrate between the polyamide resin fibers constituting the multifilament. Further, the core portion may be made of only polyamide resin fibers or may contain other fibers. Further, the core portion may be the multifilament of the first embodiment or the second embodiment.
- the sheath portion can be appropriately determined according to the application. For example, when an adhesive is used for the sheath portion, it preferably serves as an adhesive when a nonwoven fabric or the like is produced using such multifilaments.
- an adhesive used for the sheath portion for example, a resin composition having a melting point lower than that of the core polyamide resin fiber, for example, a polyamide resin (MXD6) for the core and a polypropylene resin (PP) for the sheath, the sheath is heated by heating.
- a resin composition having a melting point lower than that of the core polyamide resin fiber for example, a polyamide resin (MXD6) for the core and a polypropylene resin (PP) for the sheath
- MXD6 polyamide resin
- PP polypropylene resin
- the method for producing a polyamide resin fiber of the present invention comprises 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin, It is composed of a structural unit derived from diamine and a structural unit derived from dicarboxylic acid, 50 mol% or more of the structural unit derived from diamine is derived from xylylenediamine, and 50 mol% or more of the structural unit derived from dicarboxylic acid is carbon.
- the method comprises drawing a fiber comprising a polyamide resin composition derived from an ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 atoms.
- the polyamide resin fiber excellent in durability is obtained. Moreover, since the manufacturing method of the polyamide resin fiber of this invention is excellent in durability, even if it produces continuously, it is hard to cut
- the said polyamide resin (XD type polyamide resin) and the compound represented by General formula (1) are synonymous with the above-mentioned, and a preferable range is also the same.
- the polyamide resin fiber is usually produced according to a melt spinning method.
- a step of melt spinning a polyamide resin composition a step of passing a fiber comprising a polyamide resin composition after melt spinning through a cooling zone, and drawing a fiber comprising a polyamide resin composition
- the method including the process to do is mentioned.
- the process which makes the fiber which consists of the polyamide resin composition which carried out the melt spinning pass the hot zone may be included before the process which passes a cooling zone.
- a step of applying a sizing agent and / or a surface treating agent to the fibers of the polyamide resin composition before the step of drawing the fibers of the polyamide resin composition after the step of passing through the cooling zone. You may go out.
- a step of heat-setting a fiber made of the polyamide resin composition may be included.
- the process which twists the fiber which consists of a polyamide resin composition may be included.
- a polyamide resin composition containing an XD-based polyamide resin, a compound represented by the general formula (1), other additives, and the like is melted and discharged from a die (1 in FIG. 1).
- the temperature at which the nozzle 1 is discharged is defined as the spinning temperature.
- the number of holes in the base may be one, or two or more.
- a base having 10 to 100 holes is exemplified.
- the lower limit of the temperature (spinning temperature) of the polyamide resin composition at the time of discharge is preferably a temperature equal to or higher than the melting point of the XD-based polyamide resin, more preferably the melting point of the XD-based polyamide resin + 5 ° C or higher. More preferable is a melting point of + 10 ° C. or higher.
- the upper limit of the temperature of the polyamide resin composition at the time of discharge is not particularly defined, it can be, for example, the melting point of the XD polyamide resin + 60 ° C. or lower.
- the temperature of the XD polyamide resin at the time of discharge may be set based on the melting point of the XD polyamide resin having the lowest melting point.
- the temperature may be set based on the lowest melting point.
- the fiber composed of the spun polyamide resin composition usually passes through a hot zone and a cooling zone. When the molten polyamide resin composition passes through the die, the polyamide resin composition is oriented.
- the hot zone plays a role of relaxing the orientation of the polyamide resin composition, and is usually a region having a length of about several tens of centimeters.
- the cooling zone plays a role of cooling the fiber made of the polyamide resin composition to such an extent that it can be stretched.
- cold air is blown to the polyamide resin composition in a region having a length of about several meters.
- the cooling air in the cooling zone is preferably in the range of 10 to 100 ° C. and 10 to 50 m / min.
- the temperature of the fiber that has passed through the cooling zone is preferably about room temperature (eg, 10 to 40 ° C.).
- Fibers made from the spun polyamide resin composition can be bundled.
- the first and second embodiments of the multifilament described above are preferable.
- a bundling agent and / or a surface treatment agent may be added to form a bundle.
- the sizing agent is immersed in the solution 2 containing the sizing agent and / or the surface treatment agent, but other means may be used.
- the polyamide resin fiber to be produced may be a monofilament, and in this case, such a step is unnecessary.
- the fiber made of the melt-spun polyamide resin composition is preferably stretched. Stretching may be performed by stretching a fiber made of a melt-spun polyamide resin composition as it is, or may be stretched in a bundled state, but is usually stretched in a bundled state.
- the stretching may be one-stage stretching or two-stage or more stretching, preferably two-stage stretching, preferably 2 to 4 stage stretching, and more preferably 2 or 3 stage stretching.
- stretching in two steps or more it is preferable to make the draw ratio of extending
- the stretching ratio of the first stretching is preferably 1.03 or more, more preferably 1.03 to 3.0, and even more preferably 1.03 to 2.0, although it depends on the application.
- the draw ratio of the second drawing is preferably 1.5 to 10.0 times, more preferably 2.0 to 6.0 times, and even more preferably 2.5 to 5.0 times.
- This embodiment is particularly preferable when a polyamide resin in which 50 mol% or more of the structural units derived from dicarboxylic acid are derived from adipic acid is used.
- the stretching ratio of stretching in the second and subsequent stages can be sequentially reduced as compared with stretching in the first stage.
- the draw ratio of the first drawing is preferably 1.5 to 10.0 times, more preferably 2.0 to 6.0 times, and even more preferably 2.5 to 5.0 times.
- the stretching ratio of the second stretching is preferably 1.03 or more, more preferably 1.03 to 3.0, and even more preferably 1.03 to 2.0, although it depends on the application.
- This embodiment is particularly preferable when a polyamide resin in which 50 mol% or more of the structural units derived from dicarboxylic acid are derived from sebacic acid is used.
- a slight difference in peripheral speed occurs between the rolls, and some stretching (for example, a stretch of less than 1.06, May stretch less than 1.03 times).
- the stretching is preferably performed by using two rolls (stretching rolls) having different peripheral speeds of the rolls. The ratio of the peripheral speeds of the two rolls is the draw ratio.
- the draw ratio between the two rolls is expressed as the peripheral speed of the roll through which the fiber passes / the peripheral speed of the roll through which the fiber passes first.
- the film is slightly stretched (stretched) between the roll PR and the roll GR1, the first stretching is performed between the roll GR1 and the roll GR2, and the second stretching is performed between the roll GR2 and the roll GR3.
- the roll GR1 has a higher peripheral speed of the roll than the roll PR
- the roll GR2 has a higher peripheral speed of the roll than the roll GR1
- the roll GR3 has a higher peripheral speed of the roll than the roll GR2.
- the lower limit of the total draw ratio of the fibers made of the polyamide resin composition is preferably 1.5 times or more, more preferably 4.1 times or more, and 4.2 times or more. It is particularly preferred that it is 4.5 times or more, more preferably 5.0 times or more.
- the upper limit value is more preferably 15 times or less, further preferably 10 times or less, particularly preferably 8 times or less, and further preferably 5.5 times or less.
- the total draw ratio refers to the product of the draw ratios. When stretching using a roll, the total stretching ratio can be calculated from the following formula.
- Total draw ratio (times) roll speed A / roll speed B The roll speed A is the speed of the roll through which the fiber finally passes among the drawn rolls, and the roll speed B is the speed of the roll through which the fiber passes first among the drawn rolls.
- heating may be performed, and heating is preferable. Heating is preferably performed by adjusting the surface temperature of the roll.
- the surface temperature of a roll it can set suitably according to the kind of XD type polyamide resin, a desired draw ratio, etc.
- the surface temperature of the roll GR1 is the melting point of the XD-based polyamide resin ⁇ (180 to 130 ° C.)
- the surface temperature of the roll GR2 is the surface temperature of the roll GR1 + (5 to 25 ° C.)
- the surface temperature of the roll GR3 can be set to the surface temperature of the roll GR2 + (26 to 100) ° C.
- the plasticizer may volatilize and adhere to the roll during manufacture of the polyamide resin fiber, making it difficult to stretch.
- a compound represented by the general formula (1) is used, There is no such problem.
- the fineness of the fiber before drawing is preferably 50 to 10,000 dtex, more preferably 100 to 8000 dtex, further preferably 100 to 5000 dtex, and 200 to 4000 dtex. Is more preferable, and 300 to 3000 dtex is even more preferable.
- the number of fibers is preferably 10 to 1000 f, more preferably 10 to 500 f, and further preferably 20 to 100 f.
- relaxation may be performed after stretching.
- the relaxation can be performed, for example, between the roll GR3 and the roll GR4 and between the roll GR4 and the roll WD in FIG. That is, it is preferable to relax between two rolls (relaxation rolls) having different peripheral speeds.
- the relaxation rate can be calculated from the following formula when relaxing using a roll, for example, and is preferably 1 to 10%, more preferably 2 to 5%.
- Relaxation rate (%) ⁇ 1 ⁇ (roll speed C / roll speed D) ⁇ ⁇ 100
- Roll speed C refers to the roll speed of the roll through which the fibers pass last among the relaxing rolls
- roll speed D refers to the roll speed of the roll through which the fibers first pass among the relaxing rolls.
- the relaxation roll through which the fiber first passes is generally a drawing roll through which the fiber passes last. That is, one roll may serve as both a relaxation roll and a stretching roll.
- the surface temperature of the roll during relaxation for example, in the example shown in the figure, the surface temperature of the roll GR4 can be set to the surface temperature of the roll GR3 ⁇ (20 to 80) ° C.
- the relaxed polyamide resin fiber is usually wound and stored on a roll or the like. Moreover, the obtained polyamide resin fiber can be cut and used in various applications.
- the final draw ratio after relaxation of the polyamide resin fiber of the present invention is preferably 4.0 times or more, more preferably 4.2 times or more, and even 5.0 or more. Good. Moreover, although the upper limit of the final draw ratio is not particularly defined, it can be, for example, 6.0 times or less.
- the final draw ratio means the draw ratio of the finally obtained polyamide resin fiber based on the total draw ratio and the relaxation rate compared with before the draw, and is a value calculated from the following formula.
- Final draw ratio (total draw ratio) ⁇ ⁇ (100 ⁇ relaxation rate) / 100 ⁇
- polyamide resin fiber of the present invention may be used as a fiber as it is, or may be processed into a molding material such as a mixed yarn or a braided string. Further, it is also preferably used as a molding material such as woven fabric, knitted fabric, and non-woven fabric. These may use only polyamide resin fibers as fiber components, but it is also preferable to use them in combination with other resin fibers or reinforcing fibers. In particular, examples of reinforcing fibers include carbon fibers and glass fibers.
- the polyamide resin fiber and molding material of the present invention are used for transport parts such as automobiles, general machine parts, precision machine parts, electronic / electric equipment parts, OA equipment parts, building materials / residential equipment parts, medical devices, leisure sports goods (for example, , Fishing line), play equipment, medical products, food packaging films, clothing and other daily necessities, defense and aerospace products.
- parts such as automobiles, general machine parts, precision machine parts, electronic / electric equipment parts, OA equipment parts, building materials / residential equipment parts, medical devices, leisure sports goods (for example, , Fishing line), play equipment, medical products, food packaging films, clothing and other daily necessities, defense and aerospace products.
- the obtained pellets were charged into a tumbler (rotary vacuum tank) having a heating medium heating mantle, and heated at 200 ° C. for 1 hour in a reduced pressure state (0.5 to 10 Torr), thereby solidifying the obtained pellets.
- Phase polymerization was performed to obtain a polyamide resin (MXD6, melting point: 237 ° C., relative viscosity: 2.65, moisture content: 0.05%).
- the contents were taken out in a strand shape and pelletized with a pelletizer to obtain 15 kg pellets.
- the obtained pellets were placed in a tumbler (rotary vacuum tank) having a heating medium heating mantle, and heated at 195 ° C. for 1 hour in a reduced pressure state (0.5 to 10 Torr). Phase polymerization was performed to obtain a polyamide resin (MP10, melting point: 213 ° C., relative viscosity: 2.60, moisture content: 0.03%).
- HD-PB Hexyldecyl p-hydroxybenzoate, manufactured by Kao Corporation
- EH-PB ethyl hexyl p-hydroxybenzoate, obtained from Tokyo Chemical Industry Co., Ltd.
- EH-OB ethyl hexyl o-hydroxybenzoate, obtained from Tokyo Chemical Industry Co., Ltd.
- BBSA N-butylbenzenesulfonamide, Daihachi Chemical Industry Co., Ltd. Company made, BM-4
- Example 1 Provide method of polyamide resin fiber> A polyamide resin composition in which the plasticizer shown in Table 1 was blended in the proportion (unit: parts by weight) shown in Table 1 with respect to 100 parts by weight of the polyamide resin shown in Table 1 was melt-spun according to FIG. Specifically, a polyamide resin composition obtained by melting and kneading each component was prepared by using a spinning temperature of 260 ° C., a discharge rate of 24 g / min, a die width of 0.7 mm, and a die having 48 nozzle holes (the total number of fibers during spinning is Spinning was performed under the conditions of 48f).
- Fibers made of a polyamide resin composition that has been spun are allowed to pass through a hot zone and a cooling zone, and then fibers made of a polyamide resin composition that has become approximately room temperature (hereinafter sometimes referred to as “fibers before stretching”) are bundled. It was immersed in an agent (Takemoto Yushi Co., Ltd., Delion PP-807) (2 in FIG. 1) to form a bundle. Thereafter, each roll was passed at the roll speed and roll surface temperature shown in Table 1. Here, the roll PR is not heated.
- Table 1 the stretch ratio between rolls PR and GR1 is a stretch
- the stretch between GR1 and GR2 is a first stretch
- the stretch between GR2 and GR3 is a second stretch.
- the overall draw ratio is the product of these three draw ratios. Thereafter, relaxation was performed between the rolls GR3 and GR4 and between the rolls GR4 and WD to obtain polyamide resin fibers. The obtained polyamide resin fiber was wound up on a roll. Various characteristics of the obtained polyamide resin fiber were evaluated. The results are shown in Table 1 below.
- ⁇ Difference in crystallization temperature at the time of temperature rise of the fiber before drawing ( ⁇ temperature rise crystallization temperature)> The difference of the crystallization temperature at the time of temperature rising of the polyamide resin fiber of each Example and a comparative example and the crystallization temperature at the time of temperature rising of the polyamide resin fiber in the comparative example 1 were computed.
- ICP inductively coupled plasma
- the fineness (positive fineness) of the polyamide resin fiber was measured in accordance with JIS L 1013: 2010.
- ⁇ Abrasion resistance test> A ceramic plate derived from aluminum oxide was manufactured by friction plate rotation speed (Hiruta Riken Co., LTD.), And a conjugation force tester (textile engineering, Vol. 26, No. 7 1973) so that an average load of 150 g was applied. Using a yarn binding force tester described in FIG. 1 on page 564 of the yearly issue), while rotating at 120 rpm, the number of rotations until the polyamide resin fibers were cut was measured.
- Example 6 A polyamide resin composition in which the plasticizer shown in Table 1 was blended in the proportion (unit: parts by weight) shown in Table 1 with respect to 100 parts by weight of the polyamide resin shown in Table 1 was melt-spun according to FIG. Specifically, a polyamide resin composition obtained by melt-kneading each component is a spin temperature of 260 ° C., a discharge amount of 5 g / min, a base width of 0.7 mm, a base having 34 holes in the base (the total number of fibers during spinning is Spinning was performed under the conditions of 34f).
- Fibers made of a polyamide resin composition that has been spun are allowed to pass through a hot zone and a cooling zone, and then fibers made of a polyamide resin composition that has become approximately room temperature (hereinafter sometimes referred to as “fibers before stretching”) are bundled. It was immersed in an agent (Takemoto Yushi Co., Ltd., Delion PP-807) (2 in FIG. 1) to form a bundle. Thereafter, each roll was passed at the roll speed and roll surface temperature shown in Table 1. Here, the roll PR is not heated.
- Table 1 the stretch ratio between rolls PR and GR1 is a stretch
- the stretch between GR1 and GR2 is a first stretch
- the stretch between GR2 and GR3 is a second stretch.
- the overall draw ratio is the product of these three draw ratios. Thereafter, relaxation was performed between the rolls GR3 and GR4 and between the rolls GR4 and WD to obtain polyamide resin fibers. The obtained polyamide resin fiber was wound up on a roll. The obtained polyamide resin fiber was evaluated for various characteristics in the same manner as in Example 1. The results are shown in Table 1 below.
- Example 7 A polyamide resin composition in which the plasticizer shown in Table 1 was blended in the proportion (unit: parts by weight) shown in Table 1 with respect to 100 parts by weight of the polyamide resin shown in Table 1 was melt-spun according to FIG. Specifically, a polyamide resin composition obtained by melt-kneading each component is a spin temperature of 260 ° C., a discharge amount of 36 g / min, a base width of 0.7 mm, a base having 72 holes in the base (the total number of fibers during spinning is 72f).
- Fibers made of a polyamide resin composition that has been spun are allowed to pass through a hot zone and a cooling zone, and then fibers made of a polyamide resin composition that has become approximately room temperature (hereinafter sometimes referred to as “fibers before stretching”) are bundled. It was immersed in an agent (Takemoto Yushi Co., Ltd., Delion PP-807) (2 in FIG. 1) to form a bundle. Thereafter, each roll was passed at the roll speed and roll surface temperature shown in Table 1. Here, the roll PR is not heated.
- Table 1 the stretch ratio between rolls PR and GR1 is a stretch
- the stretch between GR1 and GR2 is a first stretch
- the stretch between GR2 and GR3 is a second stretch.
- the overall draw ratio is the product of these three draw ratios. Thereafter, relaxation was performed between the rolls GR3 and GR4 and between the rolls GR4 and WD to obtain polyamide resin fibers. The obtained polyamide resin fiber was wound up on a roll. The obtained polyamide resin fiber was evaluated for various characteristics in the same manner as in Example 1. The results are shown in Table 1 below.
- Example 1 ⁇ Examples 2 to 5, 8 and Comparative Examples 1 to 5>
- Example 1 As shown in Table 1, raw materials and stretching conditions were changed, and the others were performed in the same manner. The results are shown in Table 1 below.
- Comparative Example 2 the fiber made of the polyamide resin composition was cut during stretching. Moreover, in the comparative example 3, the fiber which consists of a polyamide resin composition stuck to the roll at the time of extending
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Textile Engineering (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polyamides (AREA)
- Artificial Filaments (AREA)
- Knitting Of Fabric (AREA)
- Woven Fabrics (AREA)
Abstract
Description
例えば、特許文献1には、(A)ポリフェニレンエーテル系樹脂 5~90重量部、および(B)ポリアミド樹脂 95~10重量部の合計100重量部に対し、(C)相容化剤 0.01~30重量部、(D)ポリアミドに柔軟性を与える可塑剤0.1重量部~100重量部、(E)ゴム様物質0~100重量部からなることを特徴とする熱可塑性樹脂組成物が開示されている。さらに、特許文献1には、このような熱可塑性樹脂組成物が柔軟性と高い引っ張り強度を同時に有することが記載されている。また、特許文献1には、このような熱可塑性樹脂組成物が、パイプやチューブに用いられることが記載されている。
一方、特許文献2には、1種またはそれ以上の半芳香族ポリアミドおよび1種またはそれ以上の官能性ポリオレフィンを含んでなる樹脂組成物でできた層を含んでなる、中空構造体が記載されている。このような中空構造体は、可撓性、冷媒流体に対する透過バリア、ならびに加熱および冷媒流体暴露時の特性の保持率に関する特性のバランスを示すことが記載されている。
さらに、特許文献3には、ポリアミド11、ポリアミド12またはこれらの混合物から選ばれるポリアミド樹脂 100重量部に対して、(A)一般式(1)で表される化合物2~60重量部と、
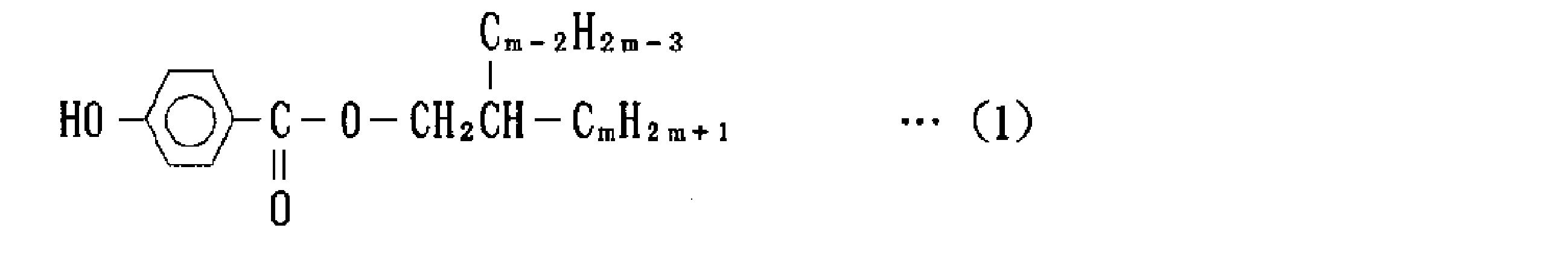
(B)分岐鎖を有する炭素数16~24の1価のアルコール0.05~5重量部を配合してなるポリアミド樹脂組成物が開示されている。また、このようなポリアミド樹脂組成物の用途として、産業用ロボット、建設機械、自動車等の燃料ホース、油圧、空圧チューブ、また自動車内装用の射出成形部品が例示されている。
本発明は、かかる課題を解決することを目的としたものであって、耐久性に優れたポリアミド樹脂繊維を提供することを目的とする。また、ポリアミド樹脂繊維の製造方法、ポリアミド樹脂組成物、織物および編み物を提供することを目的とする。
<1>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂繊維;
一般式(1)
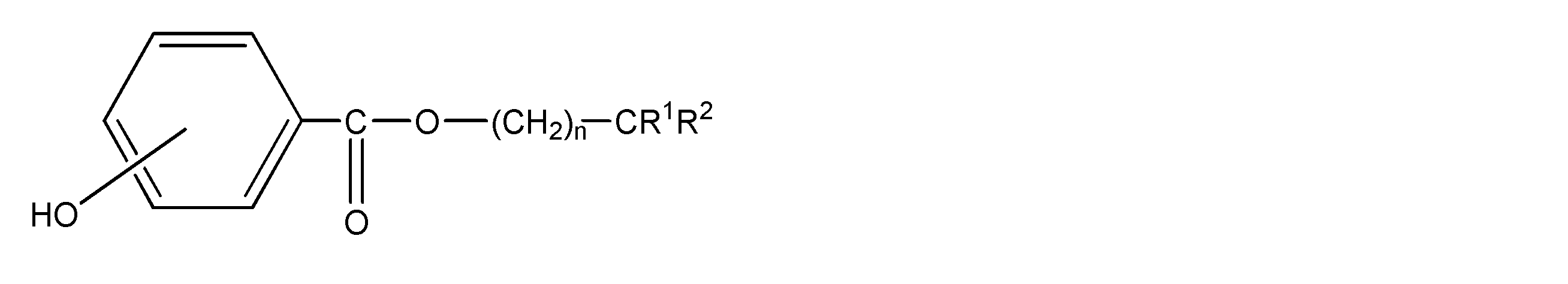
<2>前記ポリアミド樹脂繊維の繊度が、50~800dtexである、<1>に記載のポリアミド樹脂繊維。
<3>前記ポリアミド樹脂繊維を固定し、平均150gの荷重がかかるように、酸化アルミニウム由来のセラミック板を摩擦板回転速度120rpmで回転させながら、前記ポリアミド樹脂繊維に接触させたときの、前記ポリアミド樹脂繊維が切れるまでの回転数が100以上である、<2>に記載のポリアミド樹脂繊維。
<4>前記キシリレンジアミンが、30~100モル%のメタキシリレンジアミンと0~70モル%のパラキシリレンジアミンからなる、<1>~<3>のいずれかに記載のポリアミド樹脂繊維。
<5>前記ジカルボン酸由来の構成単位の50モル%以上が、セバシン酸およびアジピン酸の少なくとも1種に由来する、<1>~<4>のいずれかに記載のポリアミド樹脂繊維。
<6>前記ジカルボン酸由来の構成単位の50モル%以上が、アジピン酸に由来する、<1>~<4>のいずれかに記載のポリアミド樹脂繊維。
<7>前記ポリアミド樹脂繊維中のリン原子濃度が0.1~10ppmである、<1>~<6>のいずれかに記載のポリアミド樹脂繊維。
<8>JIS L 1013に従って測定した引張強さが、2.0cN/dtex以上である、<1>~<7>のいずれかに記載のポリアミド樹脂繊維。
<9>前記ポリアミド樹脂繊維が延伸されている、<1>~<8>のいずれかに記載のポリアミド樹脂繊維。
<10>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂組成物からなる繊維を、延伸することを含む、ポリアミド樹脂繊維の製造方法;
一般式(1)
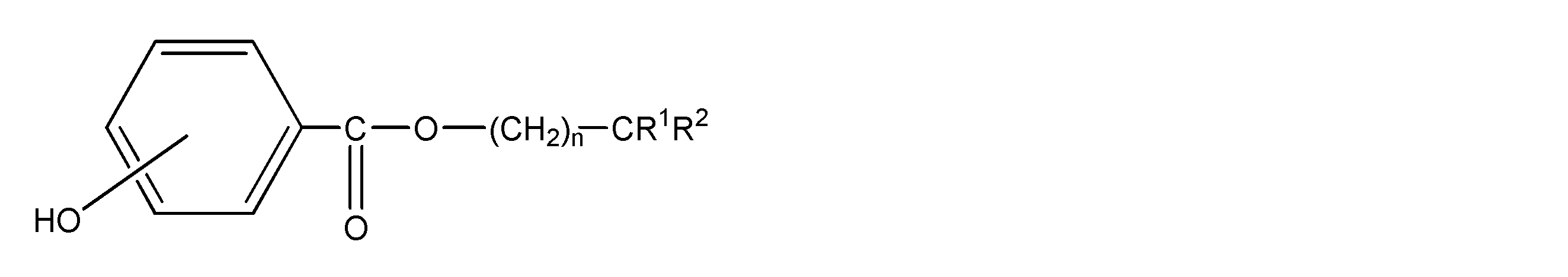
<11>前記延伸を2段階以上で行う、<10>に記載のポリアミド樹脂繊維の製造方法。
<12>総合延伸倍率が、4.1倍以上である、<10>または<11>に記載のポリアミド樹脂繊維の製造方法。
<13>前記延伸前の繊維の繊度が、100~5000dtexである、<10>~<12>のいずれかに記載のポリアミド樹脂繊維の製造方法。
<14>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、ポリアミド樹脂組成物;
一般式(1)

<15>前記ポリアミド樹脂組成物中のリン原子濃度が0.1~10ppmである、<14>に記載のポリアミド樹脂組成物。
<16>延伸されたポリアミド樹脂繊維を製造するために用いる、<14>または<15>に記載のポリアミド樹脂組成物。
<17><1>~<9>のいずれかに記載の繊維を用いた織物。
<18><1>~<9>のいずれかに記載の繊維を用いた編み物。
一般式(1)

このような構成とすることにより、耐久性に優れたポリアミド樹脂繊維が得られる。
すなわち、可塑剤を添加すると、ポリアミド樹脂の昇温時の結晶化温度は、通常、低下する傾向にある。結晶化温度が低下すると結晶化が進行しやすくなり、通常は、延伸が困難になる。ここで、XD系ポリアミド樹脂に、一般式(1)で表される化合物を配合した場合も、昇温時の結晶化温度が低下した。しかしながら、昇温時の結晶化温度が低下したにもかかわらず、XD系ポリアミド樹脂と一般式(1)で表される化合物の組み合わせからなるポリアミド樹脂組成物からなる繊維は、容易に延伸が可能であった。さらには、ポリアミド樹脂組成物からなる繊維の場合、一般式(1)で表される化合物を添加しない場合に比べても、延伸倍率を高めることが可能であることを見出した。さらに、本発明のポリアミド樹脂繊維は、高い延伸倍率で延伸した結果、優れた機械的強度(引張強さや切断時の強さ)を達成している。特に後述するように、他の可塑剤を配合しても起きない現象である点で、驚くべきものである。さらに、本発明のポリアミド樹脂繊維は、可塑剤の1種である、一般式(1)で表される化合物を含んでいるため、高延伸倍率と可塑剤による可塑化効果の相乗効果により、耐摩耗性に優れたポリアミド樹脂繊維が得られる。以上に基づき、本発明では、耐久性を達成することが可能なポリアミド樹脂繊維の提供に成功したものである。
以下、本発明の詳細について説明する。
本発明で必須成分として用いるXD系ポリアミド樹脂は、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する。
より具体的には、キシリレンジアミンが、30~100モル%のメタキシリレンジアミンと0~70モル%のパラキシリレンジアミンからなることが好ましく、70~100モル%のメタキシリレンジアミンと0~30モル%のパラキシリレンジアミンからなることがさらに好ましい。
ジアミン成分として、キシリレンジアミン以外のジアミンを用いる場合は、ジアミン由来の構成単位の30モル%以下であり、好ましくは1~25モル%、特に好ましくは5~20モル%の割合で用いる。
尚、後述するとおり、本発明で用いるポリアミド樹脂繊維は、リン原子濃度が所定の範囲であることが好ましいが、かかるリン原子は、通常、ポリアミド樹脂に由来する。
数平均分子量(Mn)=2,000,000/([COOH]+[NH2])
XD系ポリアミド樹脂の分子量分布は、例えば、重合時に使用する開始剤や触媒の種類、量および反応温度、圧力、時間等の重合反応条件などを適宜選択することにより調整できる。また、異なる重合条件によって得られた平均分子量の異なる複数種のXD系ポリアミド樹脂を混合したり、重合後のXD系ポリアミド樹脂を分別沈殿させることにより調整することもできる。
XD系ポリアミド樹脂の製造方法は、特開2014-173196号公報の段落0052~0053の記載を参酌でき、これらの内容は本明細書に組み込まれる。
また、XD系ポリアミド樹脂のガラス転移点は、50~100℃が好ましく、55~100℃がより好ましく、特に好ましくは60~100℃である。この範囲であると、耐熱性が良好となる傾向にある。
ガラス転移点とは、試料を一度加熱溶融させ熱履歴による結晶性への影響をなくした後、再度昇温して測定されるガラス転移点をいう。測定には、例えば、島津製作所社(SHIMADZU CORPORATION)製「DSC-60」を用い、試料量は約1mgとし、雰囲気ガスとしては窒素を30ml/分で流し、昇温速度は10℃/分の条件で室温から予想される融点以上の温度まで加熱し溶融させたポリアミド樹脂を、ドライアイスで急冷し、10℃/分の速度で融点以上の温度まで再度昇温し、ガラス転移点を求めることができる。
さらに、本発明で用いるXD系ポリアミド樹脂は、5重量%の一般式(1)で表される化合物を配合したときの昇温時の結晶化温度が、一般式(1)で表される化合物を配合しない場合の昇温時の結晶化温度よりも低いことが好ましく、その差が、3℃以上であることがより好ましく、5℃以上であることがさらに好ましく、10℃以上であることが特に好ましい。昇温時の結晶化温度の差の上限値については、特に定めるものでは無いが、例えば、40℃以下とでき、さらには35℃以下とすることもでき、特には30℃以下とすることもできる。
本発明における昇温時の結晶化温度の測定方法は、後述する実施例に記載の方法に従う。
本発明のポリアミド樹脂組成物は、上記XD系ポリアミド樹脂以外のポリアミド樹脂を含んでいても良い。このような他のポリアミド樹脂としては、ポリアミド4、ポリアミド6、ポリアミド11、ポリアミド12、ポリアミド46、ポリアミド66、ポリアミド610、ポリアミド612、ポリヘキサメチレンテレフタラミド(ポリアミド6T)、ポリヘキサメチレンイソフタラミド(ポリアミド6I)、ポリアミド66/6T、ポリアミド9T、ポリアミド9MT、ポリアミド6I/6T等が挙げられる。この中でも、他のポリアミド樹脂を含む場合、ポリアミド6およびポリアミド66の少なくとも1種が好ましい。
本発明のポリアミド樹脂組成物における、他のポリアミド樹脂の含有量は、配合する場合、XD系ポリアミド樹脂100重量部に対し、1~50重量部が好ましく、5~40重量部がより好ましい。
本発明のポリアミド樹脂組成物は、一般式(1)で表される化合物を含む。

一般式(1)で表される化合物において、-(CH2)n-で表される基は、上記ヒドロキシフェニルエステル基と、-CR1R2をつなぐ連結基の役割を果たすと推定される。nは1または2が好ましく、1がさらに好ましい。
一般式(1)で表される化合物において、R1は、炭素数1~9のアルキル基であることが好ましく、炭素数2~9のアルキル基であることがより好ましく、炭素数2~8のアルキル基であることがさらに好ましく、炭素数3~7のアルキル基であることが特に好ましく、炭素数4~6のアルキル基であることが一層好ましい。R1としてのアルキル基は、直鎖または分岐アルキル基が好ましく、直鎖アルキル基がより好ましい。
一般式(1)で表される化合物において、R2は、炭素数2~10のアルキル基であることが好ましく、炭素数3~9のアルキル基であることがより好ましく、炭素数5~9のアルキル基であることがさらに好ましく、炭素数6~8のアルキル基であることが特に好ましい。R2としてのアルキル基は、直鎖または分岐アルキル基が好ましく、直鎖アルキル基がより好ましい。
また、本発明では、一般式(1)で表される化合物において、R2を構成する炭素数が、R1を構成する炭素数よりも、2以上大きいことが好ましく、2~4大きいことがより好ましい。このような構成とすることにより、本発明の効果がより効果的に発揮される。

一般式(1)で表される化合物は、1種のみであってもよいし、2種以上であってもよい。2種以上含む場合、合計量が上記範囲であることが好ましい。
また、一般式(1)で表される化合物を、ポリアミド樹脂100重量部に対し、好ましくは8~15重量部、より好ましくは9~13重量部配合すると、溶融粘度を低くできる。より具体的には、上記割合で、一般式(1)で表される化合物を含むポリアミド樹脂組成物の、260℃、見かけのせん断速度122(S-1)のときの溶融粘度を500Pa・s以下にすることができる。この場合の溶融粘度の下限値は特に定めるものではないが、例えば、400Pa・s以上とすることができる。さらに、上記割合で、一般式(1)で表される化合物を含むポリアミド樹脂組成物の150℃における重量に対し、10℃/分の速度で昇温して3重量%重量が減少する温度を高くすることができる。特にこれらの効果は、可塑剤として、一般式(1)で表される化合物以外を配合した場合に比べて顕著である。
一般式(1)で表される化合物は、1種のみであってもよいし、2種以上であってもよい。2種以上含む場合、合計量が上記範囲であることが好ましい。
本発明のポリアミド樹脂組成物は、ポリアミド樹脂以外の他の樹脂成分を含んでいても良い。ポリアミド樹脂以外の他の樹脂成分としては、ポリエチレン、ポリプロピレン等のポリオレフィン樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート等のポリエステル樹脂、ポリカーボネート樹脂、ポリオキシメチレン樹脂、ポリエーテルケトン、ポリエーテルスルフォン、熱可塑性ポリエーテルイミド等の熱可塑性樹脂が例示される。
また、本発明のポリアミド樹脂組成物では、ポリアミド樹脂以外の熱可塑性樹脂を実質的に含まない構成とすることができる。実質的に含まないとは、例えば、本発明のポリアミド樹脂組成物において、ポリアミド樹脂以外の熱可塑性樹脂の含有量が、ポリアミド樹脂の重量の5重量%以下であることをいう。
さらに、本発明の目的・効果を損なわない範囲で、本発明のポリアミド樹脂組成物には、酸化防止剤、熱安定剤等の安定剤、耐加水分解性改良剤、耐候安定剤、艶消剤、紫外線吸収剤、核剤、可塑剤、分散剤、難燃剤、帯電防止剤、着色防止剤、ゲル化防止剤、着色剤、離型剤等の添加剤等を加えることができる。これらの詳細は、特許第4894982号公報の段落0130~0155の記載を参酌でき、これらの内容は本明細書に組み込まれる。
しかしながら、ポリアミド樹脂組成物は、XD系ポリアミド樹脂と一般式(1)で表される化合物で、全体の80重量%以上を占めることが好ましく、90重量%以上を占めることがより好ましい。
また、本発明のポリアミド樹脂組成物は、炭素繊維等の充填剤を含んでいてもよいが、実質的に含まないことが好ましい。実質的に含まないとは、例えば、充填剤の配合量が、本発明のポリアミド樹脂組成物の3重量%以下であることをいう。
本発明のポリアミド樹脂組成物は、上述のとおり、好ましくはポリアミド樹脂繊維の製造に用いられ、さらに好ましくは延伸されたポリアミド樹脂繊維の製造に用いられる。また、他の用途に用いても良い。
また、本発明のポリアミド樹脂組成物の昇温時の結晶化温度の下限値は、50℃以上が好ましく、80℃以上がより好ましく、100℃以上がさらに好ましく、120℃以上が特に好ましく、140℃以とすることもできる。また、本発明のポリアミド樹脂組成物の昇温時の結晶化温度の上限値は、180℃以下が好ましく、170℃以下がより好ましく、162℃以下がさらに好ましく、155℃以下が特に好ましく、148℃以下がより一層好ましい。
さらに、本発明で用いるポリアミド樹脂組成物は、5重量%の一般式(1)で表される化合物を配合したときの昇温時の結晶化温度が、一般式(1)で表される化合物を配合しない場合の昇温時の結晶化温度よりも低いことが好ましく、その差が、3℃以上であることがより好ましく、5℃以上であることがさらに好ましく、10℃以上であることが特に好ましい。昇温時の結晶化温度の差の上限値については、特に定めるものでは無いが、例えば、40℃以下とでき、さらには35℃以下とすることもでき、特には30℃以下とすることもできる。
本発明のポリアミド樹脂繊維は、ポリアミド樹脂繊維を固定し、平均150gの荷重がかかるように、酸化アルミニウム由来のセラミック板を摩擦板回転速度120rpmで回転させながら、ポリアミド樹脂繊維に接触させたときの、ポリアミド樹脂繊維が切れるまでの回転数(以下、「耐摩耗回転数」ということがある)が100以上であるポリアミド樹脂繊維とすることができる。このような高い耐摩耗性は、XD系ポリアミド樹脂に、一般式(1)で表される化合物を配合し、かつ、延伸することによって達成される。
耐摩耗回転数は、例えば、測定される繊維を各ヤーンガイドに通し、繊維両端をターミナルと任意の荷重を乗せるクリップで固定し、繊維に記録ペンをはさみ、スイッチを入れると摩擦盤が回転し、繊維が、摩擦盤に取り付けられている11種類の形状の違う酸化アルミニウム由来のセラミック板である摩擦片の上で、負荷がかけられる装置を用い、繊維が切れるまでの回転数として測定される。このような試験を行う試験機としては、蛭田理研(Hiruta Riken Co., LTD.)製、抱合力試験機(繊維工学、Vol.26,No.7 1973年発行の564頁の第1図に記載の糸抱合力試験機)が例示される。本発明における耐摩耗回転数は、より詳細には、実施例に記載の方法で測定された値とする。
本発明のポリアミド樹脂繊維は、高い延伸倍率で延伸することができ、かつ、一般式(1)で表される化合物(可塑剤)を配合しているため、高い耐摩耗性を達成できる。本発明における耐摩耗回転数は、100以上であることが好ましい。耐摩耗回転数の上限は特に定めるものでは無く、高い方がよいが、例えば、ジカルボン酸由来の構成単位の50モル%以上がアジピン酸に由来するポリアミド樹脂を用いる場合、250以下でも、実用上、優れたレベルである。また、ジカルボン酸由来の構成単位の50モル%以上がセバシン酸に由来するポリアミド樹脂を用いる場合、1500以下でも、実用上、優れたレベルである。
特に、衣料用用途としては、60dtex~120dtexが好ましく、工業材料用途としては、例えば、漁網用用途としては100~1500dtexが好ましく、タイヤコード用用途としては1000~3000dtexが好ましい。
本発明における、マルチフィラメントの第一の実施形態としては、集束剤および表面処理剤(油剤、サイズ剤等と呼ばれることもある)の少なくとも1種で、ポリアミド樹脂繊維を束状にすることが挙げられる。この場合の集束剤および表面処理剤としては、ポリアミド樹脂繊維を収束する機能を有するものであれば、その種類は特に定めるものではないが、鉱油および動・植物油などの油剤、非イオン界面活性剤、アニオン界面活性剤および両性界面活性剤などの界面活性剤を例示できる。より具体的には、エステル系化合物、アルキレングリコール系化合物、ポリオレフィン系化合物、フェニルエーテル系化合物、ポリエーテル系化合物、シリコーン系化合物、ポリエチレングリコール系化合物、アミド系化合物、スルホネート系化合物、ホスフェート系化合物、カルボキシレート系化合物およびこれらを2種以上組み合わせたものが好ましい。
ポリアミド樹脂繊維の集束剤および表面処理剤の量は、合計で、ポリアミド樹脂繊維の0.1~5.0重量%が好ましく、0.5~2.0重量%がより好ましい。
ポリアミド樹脂繊維の集束剤および/または表面処理剤による処理方法は、所期の目的を達成できる限り特に定めるものではない。例えば、ポリアミド樹脂繊維に、集束剤および/または表面処理剤を溶液に溶解させたものを付加し、ポリアミド樹脂繊維の表面に集束剤および/または表面処理剤を付着させることが挙げられる。あるいは集束剤および/または表面処理剤をポリアミド樹脂繊維の表面に対してエアブローすることによってもできる。
また、撚りをかけると共に、上記マルチフィラメントの第一の実施形態で述べたように、集束剤や表面処理剤等で処理する手段を併用してもよい。
撚りのかけ方は、特に定めるものではなく、公知の方法を採用できる。撚りの回数としては、ポリアミド樹脂繊維の繊維数、繊度等に応じて適宜定めることができるが、例えば1~200回/m(繊維長)とすることができ、さらには1~100回/mとすることができ、よりさらには1~70回/mとすることができ、特には1~50回/mとすることができる。
尚、芯と鞘の部分は、完全に領域として区別されている必要はない。例えば、芯の部分が、複数のポリアミド樹脂繊維からなるマルチフィラメントの場合、鞘を構成する材料の一部が、マルチフィラメントを構成するポリアミド樹脂繊維間に浸透する場合もあろう。
また、芯の部分は、ポリアミド樹脂繊維のみからなっていてもよいし、他の繊維等を含んでいても良い。また、芯の部分は、上記第一の実施形態または第二の実施形態のマルチフィラメントであってもよい。
一方、鞘の部分については、用途に応じて適宜定めることができる。例えば、鞘の部分に接着剤を用いると、かかるマルチフィラメントを用いて不織布等を製造する際に、接着剤の役割を果たし好ましい。鞘の部分に用いる接着剤として、例えば、芯のポリアミド樹脂繊維よりも低融点の樹脂組成物、例えば、芯にポリアミド樹脂(MXD6)を、鞘にポリプロピレン樹脂(PP)を用いると、加熱によって鞘が接着剤の役割を果たし好ましい。
本発明のポリアミド樹脂繊維の製造方法は、ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂組成物からなる繊維を、延伸することを含むことを特徴とする。このような構成とすることにより、耐久性に優れたポリアミド樹脂繊維が得られる。また、本発明のポリアミド樹脂繊維の製造方法は、耐久性に優れることから、連続生産しても繊維が切れにくく、連続生産(特に、100m以上の連続生産)に優れた製造方法となる。ここで、上記ポリアミド樹脂(XD系ポリアミド樹脂)および一般式(1)で表される化合物は、上述と同義であり、好ましい範囲も同様である。
ポリアミド樹脂繊維は、通常、溶融紡糸法に従って、製造される。ポリアミド樹脂繊維の製造方法の一例としては、ポリアミド樹脂組成物を溶融紡糸する工程、溶融紡糸後のポリアミド樹脂組成物からなる繊維を、冷却ゾーンを通過させる工程、ポリアミド樹脂組成物からなる繊維を延伸する工程を含む方法が挙げられる。
また、溶融紡糸したポリアミド樹脂組成物からなる繊維を、冷却ゾーンを通過させる工程前に、ホットゾーンを通過させる工程を含んでいてもよい。さらに、前記冷却ゾーンを通過する工程の後、ポリアミド樹脂組成物からなる繊維を延伸する工程の前に、ポリアミド樹脂組成物からなる繊維に、集束剤および/または表面処理剤を付与する工程を含んでいてもよい。加えて、延伸工程の後、ポリアミド樹脂組成物からなる繊維を熱固定する工程を含んでいてもよい。さらに、熱固定した後に、ポリアミド樹脂組成物からなる繊維に撚りをかける工程を含んでいてもよい。
以下、図1に従って、本発明のポリアミド樹脂繊維の製造方法について具体的に説明するが、本発明の製造方法はこれに限定されるものではない。
この時の口金の孔数は、1つであっても良いし、2つ以上であっても良い。例えば、マルチフィラメントを製造する場合は、その繊維数に応じた孔の数を設けることが好ましい。一例として、10~100個の孔を有する口金が例示される。吐出時のポリアミド樹脂組成物の温度(紡糸温度)の下限値は、XD系ポリアミド樹脂の融点以上の温度であることが好ましく、XD系ポリアミド樹脂の融点+5℃以上がより好ましく、XD系ポリアミド樹脂の融点+10℃以上がさらに好ましい。吐出時のポリアミド樹脂組成物の温度の上限値は、特に定めるものでは無いが、例えば、XD系ポリアミド樹脂の融点+60℃以下とすることができる。尚、XD系ポリアミド樹脂を2種以上含む場合、最も融点の低いXD系ポリアミド樹脂の融点を基準に、吐出時のXD系ポリアミド樹脂の温度を設定するとよい。また、XD系ポリアミド樹脂が融点を2つ以上有する場合についても、最も低い融点を基準に温度を設定すると良い。
紡糸されたポリアミド樹脂組成物からなる繊維は、通常、ホットゾーンおよび冷却ゾーンを通過する。溶融したポリアミド樹脂組成物が口金を通過するときに、ポリアミド樹脂組成物が配向する。ホットゾーンは、ポリアミド樹脂組成物の配向を緩和する役割を果たし、通常、数十センチメートル程度の長さの領域である。冷却ゾーンは、延伸できる程度にポリアミド樹脂組成物からなる繊維を冷却する役割を果たし、通常、数メートル程度の長さの領域で、ポリアミド樹脂組成物に冷風を吹き付ける。冷却ゾーンの冷風は10~100℃、10~50m/分の範囲が好ましい。冷却ゾーンを通過した繊維の温度は、室温程度(例えば、10~40℃)であることが好ましい。
2段階以上で延伸する場合、1段階目の延伸よりも、2段階目以降の延伸の延伸倍率を順次高くすることが好ましい。第1の延伸の延伸倍率としては、用途等にもよるが、1.03倍以上が好ましく、1.03~3.0倍がより好ましく、1.03~2.0倍がさらに好ましい。第2の延伸の延伸倍率は、1.5~10.0倍が好ましく、2.0~6.0倍がより好ましく、2.5~5.0倍がさらに好ましい。本実施形態は、ジカルボン酸由来の構成単位の50モル%以上が、アジピン酸に由来するポリアミド樹脂を用いる場合に特に好ましい。
他の実施形態として、2段階以上で延伸する場合、1段階目の延伸よりも、2段階目以降の延伸の延伸倍率を順次低くすることもできる。この場合、第1の延伸の延伸倍率は、1.5~10.0倍が好ましく、2.0~6.0倍がより好ましく、2.5~5.0倍がさらに好ましい。第2の延伸の延伸倍率としては、用途等にもよるが、1.03倍以上が好ましく、1.03~3.0倍がより好ましく、1.03~2.0倍がさらに好ましい。本実施形態は、ジカルボン酸由来の構成単位の50モル%以上が、セバシン酸に由来するポリアミド樹脂を用いる場合に特に好ましい。
また、ロールを用いて製造する場合、例えば、図1のPRロールとGR1の間でも、ロールの間にわずかな周速速度差が生じ、多少の延伸(例えば、1.06未満のストレッチ、さらには1.03倍未満のストレッチ)がされることがあるであろう。
延伸は、ロールの周速速度の異なる2つのロール(延伸ロール)を用いて延伸することが好ましい。2つのロールの周速速度の比が延伸倍率になる。すなわち、2つのロール間の延伸倍率は、繊維が後に通過するロールの周速速度/繊維が先に通過するロールの周速速度として表される。図1では、紡糸後、ロールPRとロールGR1の間で微延伸(ストレッチ)し、ロールGR1とロールGR2の間で第1の延伸を行い、ロールGR2とロールGR3の間で第2の延伸を行っている。すなわち、ロールGR1は、ロールPRよりもロールの周速速度が速く、ロールGR2は、ロールGR1よりもロールの周速速度が速く、ロールGR3は、ロールGR2よりも、ロールの周速速度が速い。
本発明では、ポリアミド樹脂組成物からなる繊維の総合延伸倍率は、下限値は、1.5倍以上であることが好ましく、4.1倍以上であることがさらに好ましく、4.2倍以上であることが特に好ましく、4.5倍以上が一層好ましく、5.0倍以上であってもよい。上限値は、15倍以下であることがより好ましく、10倍以下であることがさらに好ましく、8倍以下であることが特に好ましく、5.5倍以下が一層好ましい。ここで、総合延伸倍率とは各延伸倍率の積をいう。
ロールを用いて、延伸する場合、総合延伸倍率は下記式より算出できる。
総合延伸倍率(倍)= ロール速度A/ロール速度B
ロール速度Aは、延伸ロールのうち、繊維が最後に通過するロールの速度であり、ロール速度Bは、延伸ロールのうち、繊維が最初に通過するロールの速度をいう。
可塑剤の種類によっては、ポリアミド樹脂繊維の製造時に、可塑剤が揮発して、ロールに付着してしまい、延伸しにくくなる場合があるが、一般式(1)で表される化合物を用いると、このような問題もない。
緩和率は、例えば、ロールを用いて、緩和する場合、下記式より算出でき、1~10%が好ましく、2~5%がさらに好ましい。
緩和率(%)= {1-(ロール速度C/ロール速度D)}×100
ロール速度Cは、緩和ロールのうち、繊維が最後に通過するロールのロール速度をいい、ロール速度Dは、緩和ロールのうち、繊維が最初に通過するロールのロール速度をいう。尚、繊維が最初に通過する緩和ロールは、一般的には、繊維が最後に通過する延伸ロールとなる。すなわち、1つのロールが、緩和ロールと延伸ロールを兼ねる場合もある。
緩和時のロールの表面温度は、例えば、図の例では、ロールGR4の表面温度を、ロールGR3の表面温度-(20~80)℃とすることができる。
本発明のポリアミド樹脂繊維の緩和等を行った後の最終延伸倍率は、4.0倍以上であることが好ましく、4.2倍以上であることがより好ましく、5.0以上であってもよい。また、最終延伸倍率の上限値は特に定めるものではないが、例えば、6.0倍以下とすることができる。
最終延伸倍率とは、総合延伸倍率と緩和率に基づき、最終的に得られるポリアミド樹脂繊維の、延伸前と比較した延伸倍率を意味し、下記式から算出される値である。
最終延伸倍率=(総合延伸倍率)x{(100-緩和率)/100}
本発明のポリアミド樹脂繊維は、繊維としてそのまま用いてもよいが、混繊糸、組紐等の成形材料に加工してもよい。また、織物、編み物、不織布等の成形材料としても好ましく用いられる。
これらは、繊維成分として、ポリアミド樹脂繊維のみを用いても良いが、他の樹脂繊維や強化繊維などと組み合わせて用いることも好ましい。特に、強化繊維としては、炭素繊維、ガラス繊維等が例示される。
本発明のポリアミド樹脂繊維および成形材料は、自動車等輸送機部品、一般機械部品、精密機械部品、電子・電気機器部品、OA機器部品、建材・住設関連部品、医療装置、レジャースポーツ用品(例えば、釣り糸)、遊戯具、医療品、食品包装用フィルム、衣類等の日用品、防衛および航空宇宙製品等に広く用いられる。
<<ポリアミド樹脂 MXD6の合成>>
アジピン酸8.9kgに次亜リン酸ナトリウム一水和物0.3gおよび酢酸ナトリウム0.1gを加え、反応缶内で0.1MPaAにおいて170℃にて加熱し溶融した後、内容物を撹拌しながら、メタキシリレンジアミン8.3kgを2時間かけて徐々に滴下し、温度を250℃まで上昇させた。温度上昇後、1時間かけて圧力を0.08MPaAまで緩やかに低下させ、0.5時間保持した。反応終了後、内容物をストランド状に取り出し、ペレタイザーにてペレット化して、15kgのペレットを得た。得られたペレットを熱媒加熱の外套を有するタンブラー(回転式の真空槽)に仕込み、減圧状態(0.5~10Torr)において200℃で1時間加熱を続けることで、得られたペレットの固相重合を行い、ポリアミド樹脂(MXD6、融点:237℃、相対粘度:2.65、水分率:0.05%)を得た。
セバシン酸10.1kgに次亜リン酸ナトリウム一水和物0.3gおよび酢酸ナトリウム0.1gを加え、反応缶内で0.1MPaAにおいて170℃にて加熱し溶融した後、内容物を撹拌しながら、メタキシリレンジアミンとパラキシリレンジアミンの混合ジアミン(メタキシリレンジアミン/パラキシリレンジアミン=70/30(重量%))6.7kgを2時間かけて徐々に滴下し、温度を250℃まで上昇させた。温度上昇後、1時間かけて圧力を0.08MPaAまで緩やかに低下させ、0.5時間保持した。反応終了後、内容物をストランド状に取り出し、ペレタイザーにてペレット化して、15kgのペレットを得た。得られたペレットを熱媒加熱の外套を有するタンブラー(回転式の真空槽)に仕込み、減圧状態(0.5~10Torr)において195℃で1時間加熱を続けることで、得られたペレットの固相重合を行い、ポリアミド樹脂(MP10、融点:213℃、相対粘度:2.60、水分率:0.03%)を得た。
HD-PB:p-ヒドロキシ安息香酸ヘキシルデシル、花王株式会社製、エキセパール HD-PB
EH-PB:p-ヒドロキシ安息香酸エチルヘキシル、東京化成工業株式会社より入手
EH-OB:o-ヒドロキシ安息香酸エチルヘキシル、東京化成工業株式会社より入手
BBSA:N-ブチルベンゼンスルホンアミド、大八化学工業株式会社製、BM-4
<ポリアミド樹脂繊維の製造方法>
表1に示すポリアミド樹脂100重量部に対し、表1に示す可塑剤を、表1に示す割合(単位:重量部)で配合したポリアミド樹脂組成物を、図1に従って溶融紡糸した。具体的には、各成分を溶融混練したポリアミド樹脂組成物を、紡糸温度260℃、吐出量24g/分、口金幅0.7mm、口金の孔数48個の口金(紡糸時の合計繊維数は48f)の条件で紡糸した。紡糸したポリアミド樹脂組成物からなる繊維をホットゾーンおよび冷却ゾーンを通過させた後、概ね室温となったポリアミド樹脂組成物からなる繊維(以下、「延伸前繊維」と呼ぶことがある)を、集束剤(竹本油脂株式会社製、デリオン PP-807)(図1の2)に浸漬させて、束状にした。その後、表1に示すロール速度およびロールの表面温度で、各ロールを通過させた。ここで、ロールPRは非加熱である。表1において、ロールPRとGR1の間の延伸倍率がストレッチであり、GR1とGR2の間の延伸が第1の延伸であり、GR2とGR3の間の延伸が第2の延伸である。総合延伸倍率はこれらの3つの延伸倍率の積である。その後、ロールGR3~GR4間、およびロールGR4~WD間に緩和を行い、ポリアミド樹脂繊維を得た。得られたポリアミド樹脂繊維は、ロールに巻き取った。
得られたポリアミド樹脂繊維について、各種特性の評価を行った。結果を下記表1に示す。
島津製作所(SHIMADZU CORPORATION)製、DSC-60を用い、試料量は1mgとし、雰囲気ガスとしては窒素を30ml/分で流し、昇温速度は10℃/分の条件で室温(25℃)から予想される融点以上の温度まで加熱した際に観測される発熱ピークのピークトップの温度をポリアミド樹脂繊維の昇温時の結晶化温度とした。
各実施例および比較例のポリアミド樹脂繊維の昇温時の結晶化温度と、比較例1におけるポリアミド樹脂繊維の昇温時の結晶化温度の差を算出した。
島津製作所(SHIMADZU CORPORATION)製、DSC-60を用い、試料量は1mgとし、雰囲気ガスとしては窒素を30ml/分で流し、昇温速度は10℃/分の条件で室温(25℃)から予想される融点以上の温度まで加熱し溶融させ次いで、溶融したポリアミド樹脂繊維を、ドライアイスで急冷し、10℃/分の速度で融点以上の温度まで再度昇温した際に観測される吸熱ピークのピークトップの温度をポリアミド樹脂繊維の融点とした。
ポリアミド樹脂繊維0.5gを秤量し、濃硫酸を20ml加え、ヒーター上で湿式分解した。冷却後、過酸化水素5mlを加え、ヒーター上で加熱し、全量が2~3mlになるまで濃縮した。再び冷却し、純水で500mlとした。Thermo Jarrell Ash社製 IRIS/IPを用いて、高周波誘導結合プラズマ(ICP)発光分析により、波長213.618nmにて定量した。
JIS L 1013:2010の規定に従い、ポリアミド樹脂繊維の繊度(正量繊度)を測定した。
JIS L 1013に従って、ポリアミド樹脂繊維を23℃、50%RHの環境で調湿した後、チャック間距離20cm、引張速度20cm/分の条件で測定し、ポリアミド樹脂繊維が切断した時の荷重を切断時の強さとして測定した。また、引張強さは、JIS L 1013に従い、切断時の強さをポリアミド樹脂繊維の繊度(正量繊度)で除して算出した。
平均150gの荷重がかかるように、酸化アルミニウム由来のセラミック板を摩擦板回転速度(蛭田理研(Hiruta Riken Co., LTD.)製、抱合力試験機(繊維工学、Vol.26,No.7 1973年発行の564頁の第1図に記載の糸抱合力試験機)を用い、120rpmで回転させながら、ポリアミド樹脂繊維に接触させたときの、ポリアミド樹脂繊維が切れるまでの回転数を測定した。
表1に示すポリアミド樹脂100重量部に対し、表1に示す可塑剤を、表1に示す割合(単位:重量部)で配合したポリアミド樹脂組成物を、図1に従って溶融紡糸した。具体的には、各成分を溶融混練したポリアミド樹脂組成物を、紡糸温度260℃、吐出量5g/分、口金幅0.7mm、口金の孔数34個の口金(紡糸時の合計繊維数は34f)の条件で紡糸した。紡糸したポリアミド樹脂組成物からなる繊維をホットゾーンおよび冷却ゾーンを通過させた後、概ね室温となったポリアミド樹脂組成物からなる繊維(以下、「延伸前繊維」と呼ぶことがある)を、集束剤(竹本油脂株式会社製、デリオン PP-807)(図1の2)に浸漬させて、束状にした。その後、表1に示すロール速度およびロールの表面温度で、各ロールを通過させた。ここで、ロールPRは非加熱である。表1において、ロールPRとGR1の間の延伸倍率がストレッチであり、GR1とGR2の間の延伸が第1の延伸であり、GR2とGR3の間の延伸が第2の延伸である。総合延伸倍率はこれらの3つの延伸倍率の積である。その後、ロールGR3~GR4間、およびロールGR4~WD間に緩和を行い、ポリアミド樹脂繊維を得た。得られたポリアミド樹脂繊維は、ロールに巻き取った。
得られたポリアミド樹脂繊維について、実施例1と同様に、各種特性の評価を行った。結果を下記表1に示す。
表1に示すポリアミド樹脂100重量部に対し、表1に示す可塑剤を、表1に示す割合(単位:重量部)で配合したポリアミド樹脂組成物を、図1に従って溶融紡糸した。具体的には、各成分を溶融混練したポリアミド樹脂組成物を、紡糸温度260℃、吐出量36g/分、口金幅0.7mm、口金の孔数72個の口金(紡糸時の合計繊維数は72f)の条件で紡糸した。紡糸したポリアミド樹脂組成物からなる繊維をホットゾーンおよび冷却ゾーンを通過させた後、概ね室温となったポリアミド樹脂組成物からなる繊維(以下、「延伸前繊維」と呼ぶことがある)を、集束剤(竹本油脂株式会社製、デリオン PP-807)(図1の2)に浸漬させて、束状にした。その後、表1に示すロール速度およびロールの表面温度で、各ロールを通過させた。ここで、ロールPRは非加熱である。表1において、ロールPRとGR1の間の延伸倍率がストレッチであり、GR1とGR2の間の延伸が第1の延伸であり、GR2とGR3の間の延伸が第2の延伸である。総合延伸倍率はこれらの3つの延伸倍率の積である。その後、ロールGR3~GR4間、およびロールGR4~WD間に緩和を行い、ポリアミド樹脂繊維を得た。得られたポリアミド樹脂繊維は、ロールに巻き取った。
得られたポリアミド樹脂繊維について、実施例1と同様に、各種特性の評価を行った。結果を下記表1に示す。
実施例1において、表1に示す通り、原料や延伸条件を変更し、他は同様に行った。
結果を下記表1に示す。

上記ポリアミド樹脂繊維を経糸および緯糸に用い、目付が300g/m2となるように平織で調整した。実施例の織物は耐久性に優れていることを確認した。
<編物の製造>
上記ポリアミド樹脂繊維を用い、目付が300g/m2となるようにメリヤス編みで調整した。実施例の編物は耐久性に優れていることを確認した。
2 集束剤
11 ホットゾーン
12 冷却ゾーン
Claims (18)
- ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂繊維;
一般式(1)

- 前記ポリアミド樹脂繊維の繊度が、50~800dtexである、請求項1に記載のポリアミド樹脂繊維。
- 前記ポリアミド樹脂繊維を固定し、平均150gの荷重がかかるように、酸化アルミニウム由来のセラミック板を摩擦板回転速度120rpmで回転させながら、前記ポリアミド樹脂繊維に接触させたときの、該ポリアミド樹脂繊維が切れるまでの回転数が100以上である、請求項2に記載のポリアミド樹脂繊維。
- 前記キシリレンジアミンが、30~100モル%のメタキシリレンジアミンと0~70モル%のパラキシリレンジアミンからなる、請求項1~3のいずれか1項に記載のポリアミド樹脂繊維。
- 前記ジカルボン酸由来の構成単位の50モル%以上が、セバシン酸およびアジピン酸の少なくとも1種に由来する、請求項1~4のいずれか1項に記載のポリアミド樹脂繊維。
- 前記ジカルボン酸由来の構成単位の50モル%以上が、アジピン酸に由来する、請求項1~4のいずれか1項に記載のポリアミド樹脂繊維。
- 前記ポリアミド樹脂繊維中のリン原子濃度が0.1~10ppmである、請求項1~6のいずれか1項に記載のポリアミド樹脂繊維。
- JIS L 1013に従って測定した引張強さが、2.0cN/dtex以上である、請求項1~7のいずれか1項に記載のポリアミド樹脂繊維。
- 前記ポリアミド樹脂繊維が延伸されている、請求項1~8のいずれか1項に記載のポリアミド樹脂繊維。
- ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂組成物からなる繊維を、延伸することを含む、ポリアミド樹脂繊維の製造方法;
一般式(1)

- 前記延伸を2段階以上で行う、請求項10に記載のポリアミド樹脂繊維の製造方法。
- 総合延伸倍率が、4.1倍以上である、請求項10または11に記載のポリアミド樹脂繊維の製造方法。
- 前記延伸前の繊維の繊度が、100~5000dtexである、請求項10~12のいずれか1項に記載のポリアミド樹脂繊維の製造方法。
- ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、ポリアミド樹脂組成物;
一般式(1)

- 前記ポリアミド樹脂組成物中のリン原子濃度が0.1~10ppmである、請求項14に記載のポリアミド樹脂組成物。
- 延伸されたポリアミド樹脂繊維を製造するために用いる、請求項14または15に記載のポリアミド樹脂組成物。
- 請求項1~9のいずれか1項に記載の繊維を用いた織物。
- 請求項1~9のいずれか1項に記載の繊維を用いた編み物。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016569866A JP6164379B2 (ja) | 2015-07-16 | 2016-07-07 | ポリアミド樹脂繊維、ポリアミド樹脂繊維の製造方法、ポリアミド樹脂組成物、織物および編物 |
| US15/737,085 US20180171142A1 (en) | 2015-07-16 | 2016-07-07 | Polyamide resin fiber, method for manufacturing polyamide resin fiber, polyamide resin composition, woven fabric, and knitted fabric |
| EP16824369.9A EP3323914B1 (en) | 2015-07-16 | 2016-07-07 | Polyamide resin fiber, production method for polyamide resin fiber, polyamide resin composition, woven fabric, and knitted fabric |
| RU2017144019A RU2017144019A (ru) | 2015-07-16 | 2016-07-07 | Полиамидное полимерное волокно, способ изготовления полиамидного полимерного волокна, полиамидная полимерная композиция, тканое полотно и трикотажное полотно |
| CA2978918A CA2978918C (en) | 2015-07-16 | 2016-07-07 | Polyamide resin fiber, method for manufacturing polyamide resin fiber, polyamide resin composition, woven fabric, and knitted fabric |
| CN201680034825.2A CN107735514B (zh) | 2015-07-16 | 2016-07-07 | 聚酰胺树脂纤维、聚酰胺树脂纤维的制造方法、聚酰胺树脂组合物、织物和编织物 |
| KR1020177024764A KR101872002B1 (ko) | 2015-07-16 | 2016-07-07 | 폴리아미드 수지 섬유, 폴리아미드 수지 섬유의 제조방법, 폴리아미드 수지 조성물, 직물 및 편물 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015141871 | 2015-07-16 | ||
| JP2015-141871 | 2015-07-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017010389A1 true WO2017010389A1 (ja) | 2017-01-19 |
Family
ID=57757317
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/070080 Ceased WO2017010389A1 (ja) | 2015-07-16 | 2016-07-07 | ポリアミド樹脂繊維、ポリアミド樹脂繊維の製造方法、ポリアミド樹脂組成物、織物および編物 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20180171142A1 (ja) |
| EP (1) | EP3323914B1 (ja) |
| JP (1) | JP6164379B2 (ja) |
| KR (1) | KR101872002B1 (ja) |
| CN (1) | CN107735514B (ja) |
| CA (1) | CA2978918C (ja) |
| RU (1) | RU2017144019A (ja) |
| TW (1) | TWI604013B (ja) |
| WO (1) | WO2017010389A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107201030A (zh) * | 2017-06-07 | 2017-09-26 | 巢湖市鼎盛渔具有限公司 | 一种易着色渔网线的制备方法 |
| WO2018092623A1 (ja) * | 2016-11-18 | 2018-05-24 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| JP2018087319A (ja) * | 2016-11-18 | 2018-06-07 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| JP2020075383A (ja) * | 2018-11-06 | 2020-05-21 | 三菱瓦斯化学株式会社 | 繊維強化樹脂材料の製造方法 |
| JPWO2020250564A1 (ja) * | 2019-06-11 | 2020-12-17 | ||
| WO2022074966A1 (ja) | 2020-10-08 | 2022-04-14 | 三菱瓦斯化学株式会社 | フィラメント、構造体、樹脂組成物、および、フィラメントの製造方法 |
| JPWO2022074965A1 (ja) * | 2020-10-08 | 2022-04-14 | ||
| JP2022082179A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形体、および、ポリアミド樹脂の廃棄方法 |
| JP2022082178A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
| JP2022082177A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
| JP2022082180A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 生分解性樹脂組成物、生分解性成形体、ポリアミド樹脂の廃棄方法、および、組成物 |
| WO2023032408A1 (ja) | 2021-09-03 | 2023-03-09 | 三菱瓦斯化学株式会社 | 樹脂組成物および成形品 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019226445A1 (en) * | 2018-05-23 | 2019-11-28 | Corning Incorporated | Vacuum slow cooling device for optical fiber draw |
| CN111118659B (zh) * | 2019-12-31 | 2022-06-24 | 神马实业股份有限公司 | 一种染色聚酰胺色丝的制备方法及其制备的染色聚酰胺色丝 |
| CN116253015B (zh) * | 2022-11-30 | 2026-04-10 | 榆林天盛缘玻璃纤维科技有限公司 | 玻璃纤维多段运输分段配重装置及控制方法 |
| CN118880535B (zh) * | 2024-09-23 | 2025-01-21 | 比音勒芬服饰股份有限公司 | 改性锦纶纤维增强的结构响应型针织面料的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01185362A (ja) * | 1988-01-18 | 1989-07-24 | Kao Corp | ポリアミド樹脂組成物 |
| JP2000192331A (ja) * | 1998-12-25 | 2000-07-11 | Mitsubishi Engineering Plastics Corp | ポリアミド・モノフィラメント |
| JP5694610B2 (ja) * | 2012-07-05 | 2015-04-01 | 帝人株式会社 | 成形用材料、その成形体、および該成形体の製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5415694B2 (ja) * | 1971-12-03 | 1979-06-16 | ||
| JPS62283151A (ja) * | 1986-05-31 | 1987-12-09 | Daiseru Hiyurusu Kk | ポリアミド樹脂成形品 |
| US20150045532A1 (en) * | 2012-01-12 | 2015-02-12 | Mitsubishi Gas Chemical Company, Inc. | Polyether polyamide elastomer |
| WO2014027649A1 (ja) * | 2012-08-14 | 2014-02-20 | 三菱瓦斯化学株式会社 | ポリエーテルポリアミド樹脂組成物 |
| JP5884676B2 (ja) | 2012-08-14 | 2016-03-15 | 三菱瓦斯化学株式会社 | ポリエーテルポリアミド繊維 |
| KR20150042194A (ko) * | 2012-08-14 | 2015-04-20 | 미쯔비시 가스 케미칼 컴파니, 인코포레이티드 | 폴리에테르폴리아미드 섬유 |
| RU2648086C2 (ru) * | 2012-09-25 | 2018-03-22 | Мицубиси Гэс Кемикал Компани, Инк. | Армированный волокном полиамидный полимерный материал |
| JP6376122B2 (ja) * | 2013-03-01 | 2018-08-22 | 三菱瓦斯化学株式会社 | 複合繊維、織物、編み物および複合材料 |
-
2016
- 2016-07-07 KR KR1020177024764A patent/KR101872002B1/ko active Active
- 2016-07-07 CA CA2978918A patent/CA2978918C/en active Active
- 2016-07-07 JP JP2016569866A patent/JP6164379B2/ja active Active
- 2016-07-07 RU RU2017144019A patent/RU2017144019A/ru not_active Application Discontinuation
- 2016-07-07 CN CN201680034825.2A patent/CN107735514B/zh active Active
- 2016-07-07 WO PCT/JP2016/070080 patent/WO2017010389A1/ja not_active Ceased
- 2016-07-07 US US15/737,085 patent/US20180171142A1/en not_active Abandoned
- 2016-07-07 EP EP16824369.9A patent/EP3323914B1/en active Active
- 2016-07-15 TW TW105122320A patent/TWI604013B/zh active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01185362A (ja) * | 1988-01-18 | 1989-07-24 | Kao Corp | ポリアミド樹脂組成物 |
| JP2000192331A (ja) * | 1998-12-25 | 2000-07-11 | Mitsubishi Engineering Plastics Corp | ポリアミド・モノフィラメント |
| JP5694610B2 (ja) * | 2012-07-05 | 2015-04-01 | 帝人株式会社 | 成形用材料、その成形体、および該成形体の製造方法 |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11034836B2 (en) | 2016-11-18 | 2021-06-15 | Mitsubishi Gas Chemical Company, Inc. | Resin composition, molded article and method for manufacturing molded article |
| WO2018092623A1 (ja) * | 2016-11-18 | 2018-05-24 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| JP2018087319A (ja) * | 2016-11-18 | 2018-06-07 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| CN107201030B (zh) * | 2017-06-07 | 2019-10-25 | 巢湖市鼎盛渔具有限公司 | 一种易着色渔网线的制备方法 |
| CN107201030A (zh) * | 2017-06-07 | 2017-09-26 | 巢湖市鼎盛渔具有限公司 | 一种易着色渔网线的制备方法 |
| JP2020075383A (ja) * | 2018-11-06 | 2020-05-21 | 三菱瓦斯化学株式会社 | 繊維強化樹脂材料の製造方法 |
| JP7138015B2 (ja) | 2018-11-06 | 2022-09-15 | 三菱瓦斯化学株式会社 | 繊維強化樹脂材料の製造方法 |
| JPWO2020250564A1 (ja) * | 2019-06-11 | 2020-12-17 | ||
| JP7593313B2 (ja) | 2019-06-11 | 2024-12-03 | 三菱瓦斯化学株式会社 | フィラメントおよび釣り糸 |
| WO2020250564A1 (ja) | 2019-06-11 | 2020-12-17 | 三菱瓦斯化学株式会社 | フィラメントおよび釣り糸 |
| WO2022074966A1 (ja) | 2020-10-08 | 2022-04-14 | 三菱瓦斯化学株式会社 | フィラメント、構造体、樹脂組成物、および、フィラメントの製造方法 |
| JPWO2022074965A1 (ja) * | 2020-10-08 | 2022-04-14 | ||
| WO2022074965A1 (ja) | 2020-10-08 | 2022-04-14 | 三菱瓦斯化学株式会社 | フィラメント、材料および材料の製造方法 |
| JP7775832B2 (ja) | 2020-10-08 | 2025-11-26 | 三菱瓦斯化学株式会社 | フィラメント、材料および材料の製造方法 |
| JP2022082180A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 生分解性樹脂組成物、生分解性成形体、ポリアミド樹脂の廃棄方法、および、組成物 |
| JP2022082177A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
| JP7585735B2 (ja) | 2020-11-20 | 2024-11-19 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形体、および、ポリアミド樹脂の廃棄方法 |
| JP7585736B2 (ja) | 2020-11-20 | 2024-11-19 | 三菱瓦斯化学株式会社 | 生分解性樹脂組成物、生分解性成形体、ポリアミド樹脂の廃棄方法、および、組成物 |
| JP2022082178A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
| JP7613062B2 (ja) | 2020-11-20 | 2025-01-15 | 三菱瓦斯化学株式会社 | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
| JP7615632B2 (ja) | 2020-11-20 | 2025-01-17 | 三菱瓦斯化学株式会社 | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
| JP2022082179A (ja) * | 2020-11-20 | 2022-06-01 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形体、および、ポリアミド樹脂の廃棄方法 |
| WO2023032408A1 (ja) | 2021-09-03 | 2023-03-09 | 三菱瓦斯化学株式会社 | 樹脂組成物および成形品 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107735514B (zh) | 2020-03-10 |
| CN107735514A (zh) | 2018-02-23 |
| CA2978918C (en) | 2018-01-09 |
| TWI604013B (zh) | 2017-11-01 |
| RU2017144019A3 (ja) | 2019-08-16 |
| US20180171142A1 (en) | 2018-06-21 |
| KR101872002B1 (ko) | 2018-06-27 |
| KR20170108159A (ko) | 2017-09-26 |
| JPWO2017010389A1 (ja) | 2017-07-13 |
| EP3323914A4 (en) | 2019-04-03 |
| JP6164379B2 (ja) | 2017-07-19 |
| EP3323914A1 (en) | 2018-05-23 |
| RU2017144019A (ru) | 2019-08-16 |
| TW201708391A (zh) | 2017-03-01 |
| CA2978918A1 (en) | 2017-01-09 |
| EP3323914B1 (en) | 2020-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6164379B2 (ja) | ポリアミド樹脂繊維、ポリアミド樹脂繊維の製造方法、ポリアミド樹脂組成物、織物および編物 | |
| RU2654418C2 (ru) | Композитные волокна, тканые полотна, трикотажные полотна и композитные материалы | |
| TWI681865B (zh) | 複合材料、複合材料之製造方法及成形品之製造方法 | |
| CA2904496C (en) | Commingled yarn, method for manufacturing the commingled yarn, and, woven fabric | |
| JP2014173196A (ja) | 混繊糸、織物および編み物、複合材料、並びに、複合材料の製造方法 | |
| KR102109905B1 (ko) | 직물 및 이를 성형하여 이루어지는 성형품 | |
| WO2016039242A1 (ja) | 混繊糸の製造方法、混繊糸、巻取体、および、織物 | |
| KR102385582B1 (ko) | 복합재료, 복합재료의 제조방법 및 성형품의 제조방법 | |
| CN113906172B (zh) | 长丝及钓鱼线 | |
| JP7757973B2 (ja) | フィラメント、構造体、樹脂組成物、および、フィラメントの製造方法 | |
| JP7615632B2 (ja) | 繊維、成形体、繊維の廃棄方法および成形体の廃棄方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016569866 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16824369 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20177024764 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2978918 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15737085 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017144019 Country of ref document: RU Ref document number: 2016824369 Country of ref document: EP |