WO2016159374A1 - 配糖体化合物の製造方法 - Google Patents
配糖体化合物の製造方法 Download PDFInfo
- Publication number
- WO2016159374A1 WO2016159374A1 PCT/JP2016/060971 JP2016060971W WO2016159374A1 WO 2016159374 A1 WO2016159374 A1 WO 2016159374A1 JP 2016060971 W JP2016060971 W JP 2016060971W WO 2016159374 A1 WO2016159374 A1 WO 2016159374A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound represented
- general formula
- formula
- glycoside compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)(CO*(C1C(CO)(C2)[C@@]1N=O)C2S)OCC(C)(*)OC* Chemical compound CC(*)(CO*(C1C(CO)(C2)[C@@]1N=O)C2S)OCC(C)(*)OC* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/02—Phosphorylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/067—Pyrimidine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/167—Purine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon or a metal, e.g. chelates or vitamin B12
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/34—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from hydroxy compounds or their metallic derivatives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
Definitions
- the present invention relates to a method for producing a glycoside compound.
- a phosphoramidite method or the like As a method for producing (synthesizing) a nucleic acid such as DNA or RNA, for example, a phosphoramidite method or the like is used.
- a nucleoside phosphoramidite (hereinafter sometimes simply referred to as “phosphoramidite”) is used.
- the protecting group at the 2′-position of the phosphoramidite include a TBDMS (tertiarybutyldimethylsilyl) group, a TOM (triisopropylsilyloxymethyl) group, an ACE (bis (2-acetoxyethoxy) methyl) group, and the like. A number of protecting groups have been used.
- TBDMS amidite may not have a very high nucleic acid yield and purity when a nucleic acid is synthesized by a coupling (condensation) reaction. Then, development of the protecting group which can be manufactured at low cost and can provide the phosphoramidite which can manufacture a nucleic acid with a high yield and high purity is tried (patent documents 1 and 2).
- An object of the present invention is to provide a method for producing a phosphoramidite suitable for nucleic acid production (synthesis) more efficiently and with high purity.
- the present inventors have changed the selection of the reagent for introducing the protecting group, the order of addition thereof, and the reaction conditions, and also a method for preparing the starting material (amidite reagent). (E.g., improving the purity of the amidite reagent by examining the distillation method (selecting additives)) and finding that phosphoramidites can be produced more efficiently and with higher purity than conventional methods.
- the present invention has been completed.
- R and R ′ are the same or different and are an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cyclo An alkenylalkyl group or an alkoxyalkyl group)
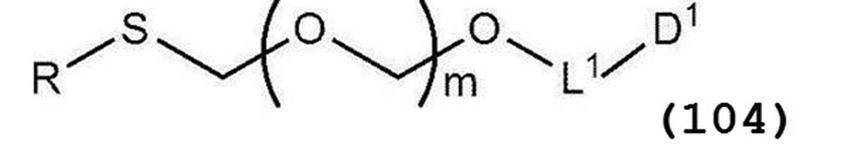
- a glycoside compound represented by general formula (104) and a thioether compound represented by general formula (104) are subjected to a coupling reaction to form general formula (Ib):
- R 1 represents a hydroxyl-protecting group
- R 2a and R 2b are the same or different and represent a hydrogen atom or a substituent
- R 2c is substituted with a hydrogen atom, an electron-withdrawing group or an electron-withdrawing group.
- R 2a and R 2b together with the nitrogen atom to which they are attached may form a ring; the definitions of the other symbols are as described above
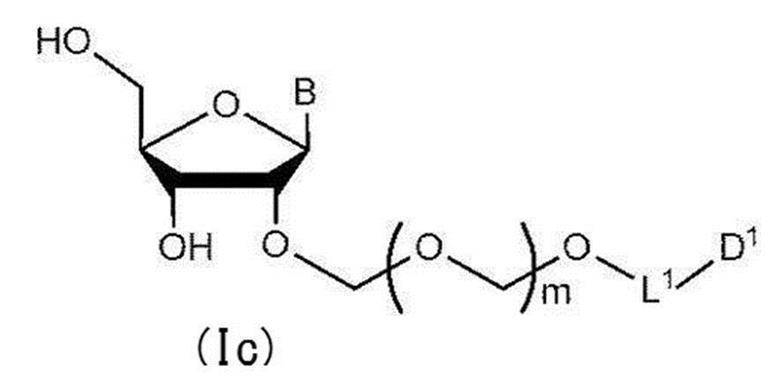
- the compound represented by general formula (Ib) is deprotected to give general formula (Ic):
- R and R ′ are the same or different and are an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cyclo An alkenylalkyl group or an alkoxyalkyl group)
- a glycoside compound represented by general formula (104) and a thioether compound represented by general formula (104) are subjected to a coupling reaction to form general formula (Ib):
- R and R ′ are the same or different and are an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cyclo An alkenylalkyl group or an alkoxyalkyl group)
- a glycoside compound represented by general formula (104) and a thioether compound represented by general formula (104) are subjected to a coupling reaction to form general formula (Ib):
- R and R ′ are the same or different and are an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cyclo An alkenylalkyl group or an alkoxyalkyl group)
- a glycoside compound represented by general formula (104) and a thioether compound represented by general formula (104) are subjected to a coupling reaction to form general formula (Ib):
- R and R ′ are the same or different and are an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cyclo An alkenylalkyl group or an alkoxyalkyl group)
- a glycoside compound represented by general formula (104) and a thioether compound represented by general formula (104) are subjected to a coupling reaction to form general formula (Ib):
- R 1 represents a hydroxyl-protecting group; the definitions of the other symbols are as described above.
- R 1 represents a protecting group for a hydroxyl group
- R 2a and R 2b are the same or different and each is a hydrogen atom or a substituent
- R 2c is substituted with hydrogen atom, an electron withdrawing group or an electron withdrawing group
- R 2a and R 2b together with the nitrogen atom to which they are attached may form a ring; the definitions of the other symbols are as described above
- the manufacturing method of the glycoside compound represented by general formula (I) including the process (process 4) of obtaining the glycoside compound represented by these.
- [9] The method according to any one of [1] to [8] above, wherein the additive in the distillation in Step 0 is 4,4′-bismaleimide diphenylmethane.
- n is 1.
- step 1 Any one of [1] to [14] above, wherein in step 1, a halogenating agent is added after the addition of the desiccant, the Lewis acid and the thioether compound represented by the general formula (104). The method described in 1. [17] The method according to [15] above, wherein in step 1, a Lewis acid is added after the addition of the halogenating agent. [18] The method according to [16] above, wherein in step 1, the thioether compound represented by formula (104) is added after the addition of the Lewis acid. [19] The method according to any one of [1] to [18] above, wherein the halogenating agent in step 1 is an iodinating agent. [20] The method described in [19] above, wherein the iodinating agent is 1,3-diiodo-5,5-dimethylhydantoin or iodine.
- phosphoramidite can be produced more efficiently and with high purity.
- the “atomic group having a nucleobase skeleton” means a functional group having a nucleobase skeleton in all or part of its structure.
- the “nucleobase skeleton” may be a natural nucleobase skeleton or an artificial nucleobase skeleton, but is preferably a natural nucleobase skeleton.
- the natural nucleobase is adenine, cytosine, guanine, uracil, thymine, or other nitrogen-containing aromatic ring (eg, 5-alkylpyrimidine, 5-halogenopyrimidine, deazapurine, deazapyrimidine, azapurine, azapyrimidine) It is more preferable.
- Halogen includes fluorine atom, chlorine atom, bromine atom and iodine atom.
- alkyl group means a linear or branched alkyl group having 1 to 30, preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 4 carbon atoms. , Ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl , Nonadecyl, icosyl and the like.
- Preferable examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl and the like.
- alkenyl group means a straight or branched alkenyl group having 2 to 30, preferably 2 to 12, and more preferably 2 to 8 carbon atoms. In the alkyl group, one or more double bonds are formed. And the like. Specific examples include vinyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 1,3-butadienyl, 3-methyl-2-butenyl and the like.
- Alkynyl group means a linear or branched alkynyl group having 2 to 30, preferably 2 to 12, and more preferably 2 to 8 carbon atoms, and has one or more triple bonds in the alkyl group. And the like. Specific examples include ethynyl, propynyl, propargyl, butynyl, pentynyl, hexynyl and the like. The alkynyl group may further have one or more double bonds.
- Alkoxy group means a linear or branched alkoxy group having 1 to 30, preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 4 carbon atoms.
- Examples include oxy, n-hexyloxy, 2-hexyloxy and the like.
- Aryl group means an aryl group having 6 to 24 carbon atoms, preferably 6 to 10 carbon atoms, and is a monocyclic aromatic hydrocarbon group such as phenyl, 1-naphthyl, 2-naphthyl, 1-anthryl, 2- And polycyclic aromatic hydrocarbon groups such as anthryl, 9-anthryl, 1-phenanthryl, 2-phenanthryl, 3-phenanthryl, 4-phenanthryl, and 9-phenanthryl.
- “Aralkyl group” means an aralkyl group having 7 to 30 carbon atoms, preferably 7 to 11 carbon atoms, and specific examples thereof include benzyl, 2-phenethyl, naphthalenylmethyl, and the like.
- Cycloalkyl group means a cycloalkyl group having 3 to 24 carbon atoms, preferably 3 to 15 carbon atoms. Specifically, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, bridged ring And a hydrocarbon group such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and a bridged cyclic hydrocarbon group.
- bridged cyclic hydrocarbon group examples include bicyclo [2.1.0] pentyl, bicyclo [2.2.1] heptyl, bicyclo [2.2.2] octyl, bicyclo [3.2.1]. Examples include octyl, tricyclo [2.2.1.0] heptyl, bicyclo [3.3.1] nonane, 1-adamantyl, 2-adamantyl and the like. Examples of the “spiro hydrocarbon group” include spiro [3.4] octyl.
- Cycloalkenyl group means a cycloalkenyl group having 3 to 24 carbon atoms, preferably 3 to 7 carbon atoms, which contains at least one, preferably 1 or 2 double bonds, specifically cyclopropenyl. , Cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl and the like.
- the cycloalkenyl group also includes a bridged cyclic hydrocarbon group and a spiro hydrocarbon group having an unsaturated bond in the ring.
- Examples of the “bridged cyclic hydrocarbon group having an unsaturated bond in the ring” include bicyclo [2.2.2] octenyl, bicyclo [3.2.1] octenyl, and tricyclo [2.2.1.0]. And heptenyl.
- Examples of the “spiro hydrocarbon group having an unsaturated bond in the ring” include spiro [3.4] octenyl.
- cycloalkylalkyl group means an alkyl group (described above) substituted with the cycloalkyl group, preferably a cycloalkylalkyl group having 4 to 30 carbon atoms, more preferably 4 to 11 carbon atoms. Specific examples include cyclopropylmethyl, 2-cyclobutylethyl, cyclopentylmethyl, 3-cyclopentylpropyl, cyclohexylmethyl, 2-cyclohexylethyl, cycloheptylmethyl and the like.
- Alkoxyalkyl group means an alkyl group substituted with the alkoxy group (described above), preferably a linear or branched alkoxyalkyl group having 2 to 30 carbon atoms, more preferably 2 to 12 carbon atoms. . Specific examples include methoxymethyl, methoxyethyl, ethoxymethyl, ethoxyethyl and t-butoxymethyl.
- alkylene group means a linear or branched alkylene group having 1 to 30, preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 4 carbon atoms. , Ethylene, propylene, and the like.
- heteroaryl group includes, for example, a monocyclic aromatic heterocyclic group and a condensed aromatic heterocyclic group.
- heteroaryl include furyl (eg, 2-furyl, 3-furyl), thienyl (eg, 2-thienyl, 3-thienyl), pyrrolyl (eg, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), Imidazolyl (eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl), pyrazolyl (eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl), triazolyl (eg, 1,2,4-triazol-1-yl, 1,2,4-triazol-3-yl, 1,2,4-triazol-4-yl), tetrazolyl (eg 1-tetrazolyl, 2-tetrazolyl, 5-tetrazolyl), oxazolyl (eg 2-oxazo
- Electrode-withdrawing group refers to a group that attracts electrons more easily than a hydrogen atom, and specifically includes cyano, nitro, alkylsulfonyl (eg, methylsulfonyl, ethylsulfonyl), halogen ( Fluorine atom, chlorine atom, bromine atom or iodine atom), arylsulfonyl (eg, phenylsulfonyl, naphthylsulfonyl), trihalomethyl (eg, trichloromethyl, trifluoromethyl) and the like.
- alkylsulfonyl eg, methylsulfonyl, ethylsulfonyl
- halogen Fluorine atom, chlorine atom, bromine atom or iodine atom
- arylsulfonyl eg, phenylsulfonyl, naphthylsul
- hydroxyl-protecting group means a general hydroxyl-protecting group known to those skilled in the art, which is introduced to prevent the reaction of a hydroxyl group.
- acyl protecting groups such as acetyl and benzoyl
- alkyl protecting groups such as trityl, 4-methoxytrityl, 4,4′-dimethoxytrityl and benzyl
- silyl protecting groups such as tert-butyldimethylsilyl and tert-butyldiphenylsilyl.
- Halogen fluorine atom, chlorine atom, bromine atom or iodine atom
- (2) an alkyl group (described above); (3) an alkenyl group (described above); (4) an alkynyl group (described above); (5) Haloalkyl groups (eg, chloromethyl, fluoromethyl, dichloromethyl, difluoromethyl, dichlorofluoromethyl, trifluoromethyl, pentafluoroethyl, etc.); (6) aryl group (described above); (7) heteroaryl group (described above); (8) an aralkyl group (described above); (9) a cycloalkyl group (described above); (10) a cycloalkenyl group (described above); (11) a cycloalkylalkyl group (described above); (12) cycloalkenylalkyl groups (eg, cyclopentenylethyl, cycl
- the present invention provides a method for producing a glycoside compound represented by the general formula (I) (hereinafter also referred to as glycoside compound (I)) or a salt thereof (hereinafter also referred to as the production method of the present invention). .
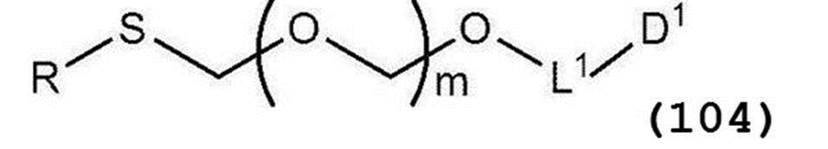
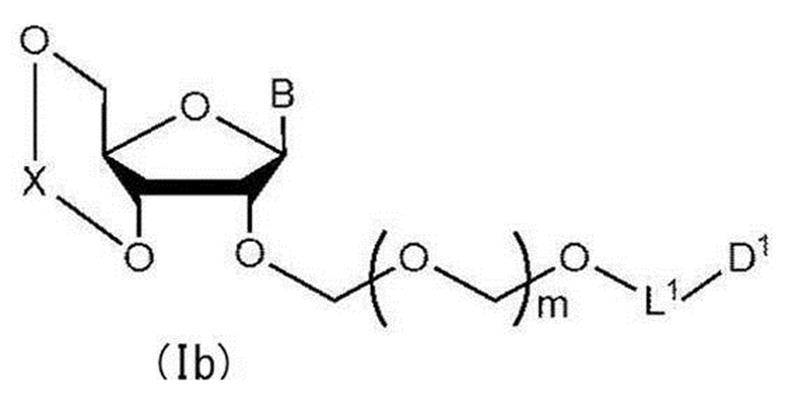
- B represents an atomic group having a nucleobase skeleton; m is a positive integer; L 1 represents an alkylene group; D 1 represents an electron-withdrawing group; R 1 represents a hydroxyl-protecting group.
- R 2a and R 2b are the same or different and each represents a hydrogen atom or a substituent; R 2c represents a hydrogen atom, an electron withdrawing group or a substituent optionally substituted with an electron withdrawing group; 2a and R 2b together with the nitrogen atom to which they are attached may form a ring]
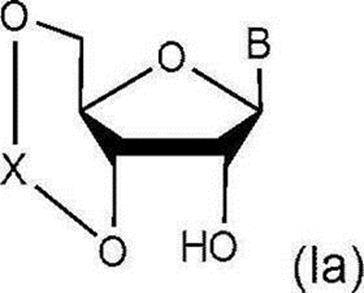
- the production method of the present invention includes the following steps (step 1). [Step 1] In the presence of a halogenating agent, a drying agent and a Lewis acid, the general formula (Ia):
- glycoside compound (Ia) A glycoside compound represented by the formula (hereinafter also referred to as glycoside compound (Ia)) and general formula (104):
- R is an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cycloalkenylalkyl group or alkoxyalkyl group.
- L 1 represents an alkylene group;
- D 1 represents an electron-attracting group)
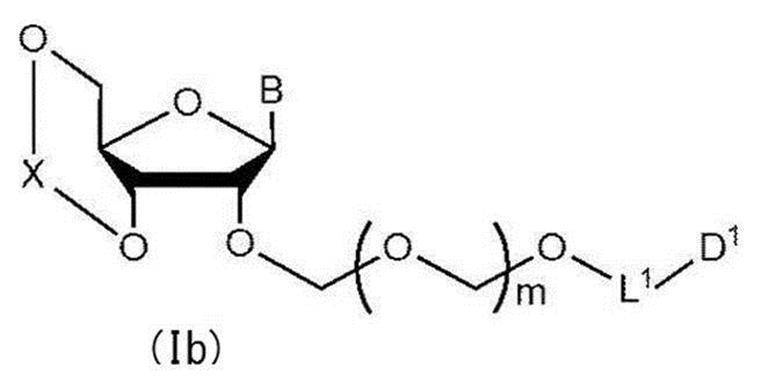
- glycoside compound (Ib) A glycoside compound represented by the formula (hereinafter also referred to as glycoside compound (Ib)). Therefore, the present invention also provides a method for producing glycoside compound (Ib) by Step 1.
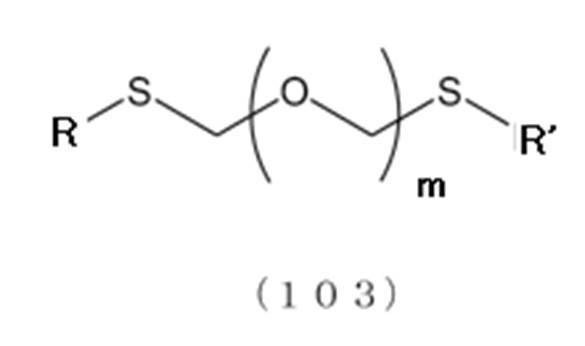
- the thioether compound (104) is prepared by the following step (step 0).
- step 0 [Step 0] Formula (103):
- R and R ′ are the same or different and are an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cyclo An alkenylalkyl group or an alkoxyalkyl group)
- a thioether compound hereinafter also referred to as a thioether compound (103)
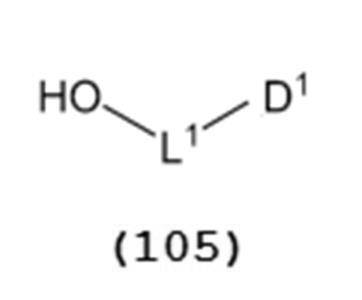
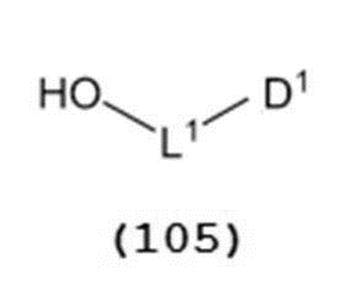
- a general formula (105) a general formula (105):
- the present invention also provides a method for producing the thioether compound (104) by Step 0 and a method for producing the glycoside compound (Ib) by Step 0 and Step 1.
- B is 9-position nitrogen for adenine, 1-position nitrogen for cytosine, 9-position nitrogen for guanine, 1-position nitrogen for uracil, or 1-position for thymine. More preferably, it is bonded to the D-ribose skeleton in the chemical formula (I) with nitrogen. Moreover, the nucleobase in B may be substituted with an arbitrary substituent or may not be substituted.
- substituents examples include a halogen, an acyl group (synonymous with an acyl group as a protecting group for the following amino group), an alkyl group, an aralkyl group, an alkoxy group, an alkoxyalkyl group, a hydroxy group, an amino group, and a monoalkylamino group. (Eg, methylamino, ethylamino, etc.), dialkylamino group (eg, dimethylamino, diethylamino, etc.), carboxy group, cyano group, nitro group and the like.
- substituents may be zero, one, or plural (for example, two to three), and in the case of plural, one kind or plural kinds may be used.
- B may or may not have a protecting group.
- the amino group when the nucleobase in B has an amino group (amino substituent) outside the ring, the amino group may be protected by a protecting group.
- the amino-protecting group is not particularly limited, and may be, for example, the same protecting group used in known nucleic acid chemistry.
- Examples of the amino-protecting group include an acyl group.
- Examples of the acyl group include benzoyl group, 4-methoxybenzoyl group, acetyl group, propionyl group, butyryl group, isobutyryl group, phenylacetyl group, phenoxyacetyl group, 4-tert-butylphenoxyacetyl group, 4-isopropylphenoxy group.
- acetyl group etc. are mentioned.
- (dimethylamino) methylene group and the like can be mentioned.
- B is preferably adenine, cytosine, guanine, uracil and thymine.
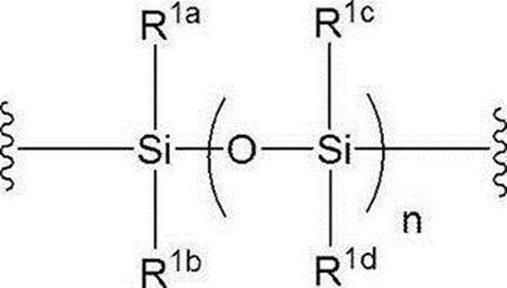
- N represents 0 or 1, preferably n is 1.
- R 1a to R 1d are the same or different and each represents a hydrogen atom, an alkyl group or an alkoxy group, preferably all alkyl groups, more preferably an alkyl group having 1 to 6 carbon atoms, particularly preferably an isopropyl group. is there.
- M is a positive integer, preferably an integer of 1 to 10, more preferably 1 or 2, and particularly preferably 1.
- R and R ′ are the same or different and each represents an alkyl group, alkenyl group, alkynyl group, aryl group, aralkyl group, cycloalkyl group, cycloalkenyl group, cycloalkylalkyl group, cycloalkenylalkyl group or alkoxyalkyl group, preferably An alkyl group, more preferably an alkyl group having 1 to 6 carbon atoms, and particularly preferably any methyl group.
- L 1 is an alkylene group, preferably an alkylene group having 1 to 6 carbon atoms, more preferably an ethylene group.
- D 1 is an electron withdrawing group, preferably cyano, nitro, alkylsulfonyl, halogen, arylsulfonyl, trihalomethyl, etc., more preferably a cyano group.
- R 2a and R 2b are the same or different and each represents a hydrogen atom or a substituent, preferably the same and a substituent, more preferably an alkyl group (preferably a linear or branched alkyl group having 1 to 4 carbon atoms, more Preferably, it is an isopropyl group.
- R 2a and R 2b may form a ring together with the nitrogen atom to which they are bonded, but the ring is a nitrogen-containing heterocyclic ring, and a nitrogen atom other than the nitrogen atom, an oxygen atom or a sulfur atom It may or may not have, and may or may not have a substituent.
- R 2a and R 2b are each preferably a linear or branched alkyl group having 1 to 4 carbon atoms, and more preferably an isopropyl group.
- R 2c represents a hydrogen atom, an electron withdrawing group or a substituent that may be substituted with an electron withdrawing group, preferably an electron withdrawing group or a substituent that may be substituted with an electron withdrawing group, More preferred is a substituent which may be substituted with an electron withdrawing group.
- R 2c is an alkyl group (preferably a linear or branched alkyl group having 1 to 4 carbon atoms, more preferably an ethyl group) which may be substituted with an electron withdrawing group (preferably a cyano group). And particularly preferred is a cyanoethyl group.
- the halogenating agent used in Step 0 is not particularly limited, but chloro such as chlorine, hydrochloric acid, hydrogen chloride, thionyl chloride, sulfuryl chloride, mesyl chloride, phosphorus oxychloride, phosphorus trichloride, phosphorus pentachloride, N-chlorosuccinimide and the like.
- Brominating agents such as N-bromosuccinimide, N-bromophthalimide, 1,3-dibromohydantoin, 5,5-dimethyl-1,3-dibromohydantoin; iodine, sodium iodide, potassium iodide, N- And an iodinating agent such as iodo-succinimide and 1,3-diiodo-5,5-dimethylhydantoin. Preferred is an iodinating agent, and more preferred is N-iodo-succinimide.
- the desiccant used in Step 0 those usually used in the art can be used without particular limitation, but molecular sieve is preferable.
- Lewis acid used in Step 1 those usually used in the field can be used without particular limitation, and can be appropriately selected depending on the raw materials and reagents used, but preferably a perfluoroalkylcarboxylic acid.
- perfluoroalkylsulfonic acid eg, trifluoromethanesulfonic acid
- alkylsulfonic acid eg, methanesulfonic acid, ethanesulfonic acid
- at least one selected from the group thereof is there.
- methanesulfonic acid More preferred is methanesulfonic acid, trifluoromethanesulfonic acid or a salt thereof (preferably a silver salt), and particularly preferred is methanesulfonic acid or a silver salt of trifluoromethanesulfonic acid.
- the thioether compound (103) used in Step 0 is commercially available, or can be synthesized with reference to the method described in WO2013 / 027843.
- the alcohol compound (105) used in Step 0 is commercially available, or can be synthesized by a method known per se.
- the conditions for the coupling reaction between the thioether compound (103) and the alcohol compound (105) are not particularly limited.
- the reaction solvent for the coupling reaction is not particularly limited, and examples thereof include ketones such as acetone, methyl ethyl ketone, and acetophenone; ethers such as diethyl ether, THF (tetrahydrofuran), and dioxane; and nitriles such as acetonitrile.
- the reaction time of the coupling reaction is not particularly limited, but is, for example, 1 to 12 hours, preferably 1 to 8 hours, more preferably 1 to 4 hours.
- the reaction temperature for the coupling reaction is not particularly limited, but preferably optimal conditions are set as appropriate depending on the raw materials and reagents used.
- the concentrations of the thioether compound (103) and the alcohol compound (105) are also not particularly limited and can be set as appropriate.
- the substance amount ratio between the thioether compound (103) and the alcohol compound (105) is not particularly limited, and may be, for example, a stoichiometric ratio or any other ratio.
- the amount of other reactants used is not particularly limited.
- the number of moles of the alcohol compound (105) is, for example, 0.2 to 3 times, preferably 0.2 to 1.5 times, more preferably 0.5 to 1 times the number of moles of the thioether compound (103). is there.
- the number of moles of the halogenating agent is, for example, 0.5 to 3 times, preferably 0.5 to 1.5 times, and more preferably 0.8 times the number of moles of the thioether compound (103). 5 to 1 times.
- the number of moles of the Lewis acid is, for example, 1 to 3 times, preferably 1 to 2 times, and more preferably 1 to 1.5 times the number of moles of the thioether compound (103).
- the number of moles of the Lewis acid is, for example, 0.005 to 0.5 times, preferably 0.01 to 0.1 times, more than the number of moles of the thioether compound (103). Preferably it is 0.025 to 0.035 times.
- the amount of the desiccant (preferably molecular sieve) used is not particularly limited, but it is preferably used in excess with respect to each of the reactants.
- the reaction conditions for the coupling reaction may be set as appropriate with reference to conditions such as coupling reactions between known thioether compounds and alcohol compounds, or may be set as appropriate with reference to the conditions of Examples described later. Also good.
- step 0 distillation is performed after the coupling reaction. The distillation can be carried out by a method known per se, but since a high-purity thioether compound (104) is obtained, at least one selected from a sulfur-containing antioxidant and a maleimide group-containing compound as an additive. It is preferable to carry out repeatedly under reduced pressure in the presence of.
- the sulfur-containing antioxidant is a compound having a sulfur atom in the molecule having an antioxidant action, and examples thereof include phenothiazine.
- the maleimide group-containing compound is a compound having a maleimide group in the molecule, and examples thereof include maleimide, 4,4′-bismaleimide diphenylmethane, and bis (3-ethyl-5-methyl-4-maleimidophenyl) methane.
- 4,4′-bismaleimide diphenylmethane is preferably used as an additive in distillation.
- the amount of the additive used is not particularly limited, but it is used in a weight of 0.01 to 0.5 times, preferably 0.05 to 0.4 times, more preferably 0.1 to 0.3 times that of the reactant. . Specifically, it can be carried out with reference to the conditions of Examples described later.
- the distillation can increase the thioether compound (104) to a purity of 80% or more, preferably 90%, more preferably 95% or more, and even more preferably 98% or more.
- the halogenating agent used in Step 1 is not particularly limited, but chloro such as chlorine, hydrochloric acid, hydrogen chloride, thionyl chloride, sulfuryl chloride, mesyl chloride, phosphorus oxychloride, phosphorus trichloride, phosphorus pentachloride, N-chlorosuccinimide and the like.
- Brominating agents such as N-bromosuccinimide, N-bromophthalimide, 1,3-dibromohydantoin, 5,5-dimethyl-1,3-dibromohydantoin; iodine, sodium iodide, potassium iodide, N- And an iodinating agent such as iodo-succinimide and 1,3-diiodo-5,5-dimethylhydantoin.
- it is an iodinating agent, and more preferably 1,3-diiodo-5,5-dimethylhydantoin or iodine.
- the iodinating agent when the nucleobase in the atomic group having the nucleobase skeleton of B is cytosine, uracil and guanine, the iodinating agent is 1,3-diiodo-5,5-dimethylhydantoin. is there. In another preferred embodiment of the present invention, when the nucleobase in the atomic group having the nucleobase skeleton of B is adenine, the iodinating agent is iodine.
- two or more halogenating agents may be used.
- an iodinating agent eg, 1,3-diiodo-5,5-dimethylhydantoin
- iodine may or may not be used in combination.
- the desiccant used in step 1 those usually used in the art can be used without particular limitation, but molecular sieve is preferable.
- Lewis acid used in Step 1 those usually used in the art can be used without particular limitation, and can be appropriately selected depending on the raw materials and reagents used, but preferably a perfluoroalkylcarboxylic acid.
- perfluoroalkylsulfonic acid eg, trifluoromethanesulfonic acid
- alkylsulfonic acid eg, methanesulfonic acid, ethanesulfonic acid
- at least one selected from the group thereof is there.
- methanesulfonic acid trifluoromethanesulfonic acid or a salt thereof (preferably a silver salt), and particularly preferred is methanesulfonic acid or trifluoromethanesulfonic acid.
- the Lewis acid when the nucleobase in the atomic group having the nucleobase skeleton of B is cytosine, uracil and guanine, the Lewis acid is trifluoromethanesulfonic acid.
- the Lewis acid is methanesulfonic acid.
- the glycoside compound (Ia) used in Step 1 is commercially available or can be synthesized by a method known per se.
- the conditions for the coupling reaction between glycoside compound (Ia) and thioether compound (104) are not particularly limited.
- the reaction solvent for the coupling reaction is not particularly limited, and examples thereof include ketones such as acetone, methyl ethyl ketone, and acetophenone; ethers such as diethyl ether, THF (tetrahydrofuran), and dioxane; and nitriles such as acetonitrile.
- the reaction time of the coupling reaction is not particularly limited, and is, for example, 15 minutes to 6 hours, preferably 15 minutes to 2 hours, more preferably 30 minutes to 1 hour.
- the reaction temperature (flask temperature) of the coupling reaction is not particularly limited, but preferably optimal conditions are appropriately set depending on the raw materials, reagents, etc. used. For example, it is ⁇ 90 to 0 ° C., preferably ⁇ 70 to ⁇ 20 ° C., more preferably ⁇ 60 to ⁇ 30 ° C.
- concentrations of glycoside compound (Ia) and thioether compound (104) are not particularly limited and can be set as appropriate.
- the substance amount ratio between the glycoside compound (Ia) and the thioether compound (104) is not particularly limited, and may be, for example, a stoichiometric ratio or any other ratio.
- the amount of other reactants used is not particularly limited.
- the number of moles of the thioether compound (104) is, for example, 1 to 5 times, preferably 1 to 3 times, and 1 to 2 times the number of moles of the glycoside compound (Ia).
- the number of moles of the halogenating agent preferably iodinating agent
- the halogenating agent is 1,3-diiodo-5,5-dimethylhydantoin, it is, for example, 1 to 5 times, 1 to 3 times, preferably 1 to 2 times the number of moles of glycoside compound (Ia). Times, more preferably 1 to 1.5 times.
- the halogenating agent is iodine
- it is, for example, 1 to 10 times, preferably 2 to 10 times, more preferably 4 to 8 times, more preferably 5 to 6 times the number of moles of glycoside compound (Ia).
- the number of moles of the Lewis acid is, for example, 1 to 3 times, preferably 1 to 2 times, more preferably 1 to 1.5 times the number of moles of the glycoside compound (Ia).
- the reaction conditions for the coupling reaction may be appropriately set with reference to, for example, conditions such as amidite synthesis of known glycoside compounds, or may be appropriately set with reference to the conditions of Examples described later. good.
- step 1 the order of adding the halogenating agent, Lewis acid, thioether compound (104) and desiccant is not particularly limited.
- the thioether compound (104) is added last. preferable. That is, it is preferable to add the halogenating agent to the reaction system in the presence of the desiccant, and then add the Lewis acid, and then add the thioether compound (104).
- the halogenating agent is preferably added last. That is, it is preferable to add a Lewis acid to the reaction system in the presence of a desiccant, and then add a thioether compound (104) followed by a halogenating agent.
- the addition of the thioether compound (104), the halogenating agent, and the Lewis acid to the reaction system can be carried out at an arbitrary interval as long as the order of addition is followed, and is appropriately set depending on the amount charged.
- the production method of the present invention preferably further includes the following step (step 2).
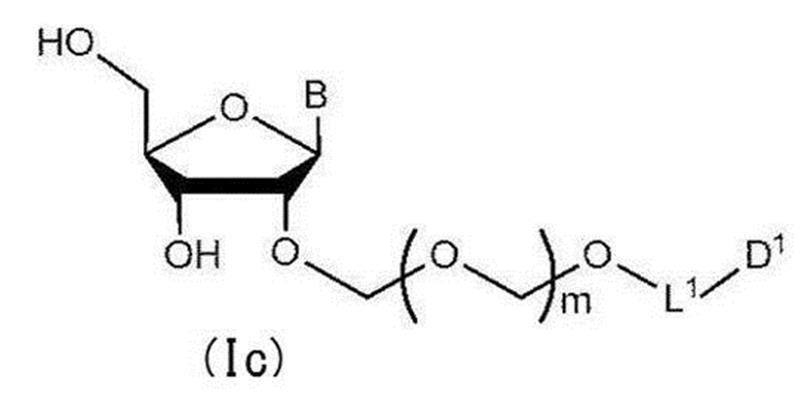
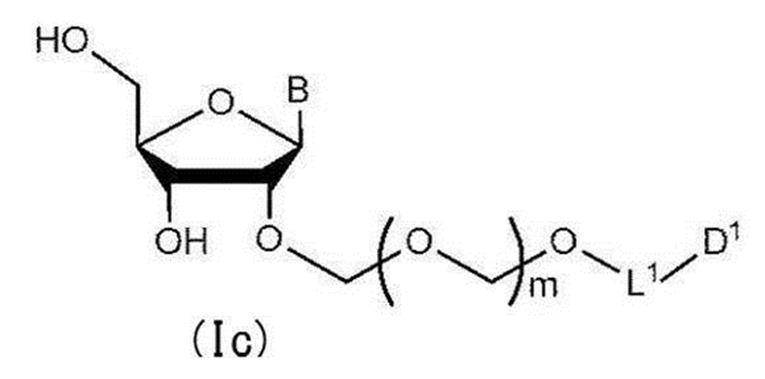
- step 2 The glycoside compound (Ib) is deprotected to give the general formula (Ic):
- glycoside compound (Ic) A glycoside compound represented by the formula (hereinafter also referred to as glycoside compound (Ic)). Therefore, the present invention also provides a method for producing glycoside compound (Ic) by the above step 2 (more specifically, step 0, step 1 and subsequent step 2).
- the deprotection reaction conditions are not particularly limited, and for example, a known deprotecting agent can be used.
- the deprotecting agent is not particularly limited, and examples thereof include hydrogen fluoride pyridine, triethylamine hydrogen trifluoride, ammonium fluoride, hydrofluoric acid, and tetrabutylammonium fluoride.
- the reaction solvent for the deprotection reaction is not particularly limited, and examples thereof include ketones such as acetone; ethers such as diethyl ether and THF (tetrahydrofuran); alcohols such as methanol and ethanol; and nitriles such as acetonitrile.
- the reaction time of the deprotection reaction is not particularly limited, but is, for example, 30 minutes to 24 hours, preferably 2 to 12 hours, more preferably 2 to 4 hours.
- the reaction temperature for the deprotection reaction is not particularly limited, but is, for example, 0 to 100 ° C., preferably 20 to 60 ° C., more preferably 20 to 50 ° C.
- the concentrations of the glycoside compound (Ib) and the deprotecting agent are not particularly limited and can be set as appropriate.
- the substance amount ratio between the glycoside compound (Ib) and the deprotecting agent is not particularly limited, and may be, for example, a stoichiometric ratio or any other ratio.
- the amount of other reactants used is not particularly limited.
- the number of moles of the deprotecting agent is, for example, 0.1 to 20 times, preferably 0.2 to 10 times, more preferably 1 to 5 times the number of moles of the glycoside compound (Ib).
- the reaction conditions for the deprotection reaction may be appropriately set with reference to conditions such as a similar deprotection reaction in known glycosides, or may be set as appropriate with reference to the conditions of Examples described later. Also good.
- the production method of the present invention preferably further includes the following step (Step 3).
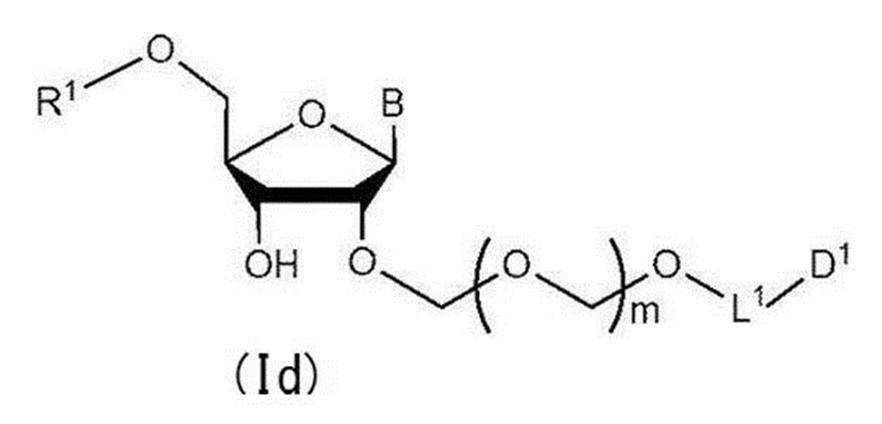
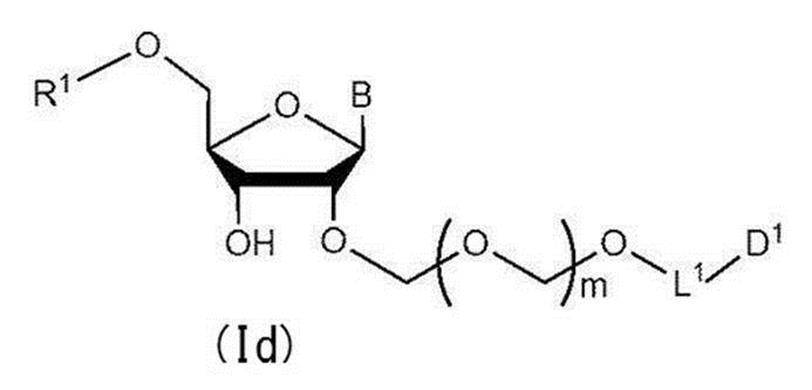
- Step 3 A hydroxyl-protecting group is introduced into the glycoside compound (Ic) to give a general formula (Id):
- R 1 represents a hydroxyl-protecting group, and the definitions of other symbols are as described in Step 1
- a glycoside compound represented by the formula hereinafter also referred to as glycoside compound (Id)
- the present invention also provides a method for producing a glycoside compound (Id) by the above step 3 (more specifically, the above step 0, step 1, step 2 and subsequent step 3).
- the protecting group for the hydroxyl group is not particularly limited, but is preferably an alkyl protecting group such as a methyl group, a tert-butyl group, a benzyl group, a trityl group; a tetrahydropyranyl group, a methoxymethyl group.
- Ether protecting groups such as: carbonate protecting groups such as methyl carbonate group and ethyl carbonate group; silicon protecting groups such as trimethylsilyl group, tert-butyldimethylsilyl group, tert-butyldiphenylsilyl group, etc., preferably 4,4′-dimethoxytrityl (DMTr).
- the reaction conditions for the protecting group introduction step are not particularly limited, and may be appropriately set with reference to, for example, similar reactions in known glycoside compounds.
- the protecting group introducing agent may be appropriately selected according to R 1 .
- the protecting group is 4,4′-dimethoxytrityl
- 4,4′-dimethoxytrityl chloride can be used as the protecting group introducing agent.
- the reaction solvent is not particularly limited, and examples thereof include polar solvents such as pyridine; nitriles such as acetonitrile; ethers such as tetrahydrofuran.
- the reaction time is not particularly limited, but is, for example, 30 minutes to 24 hours, preferably 2 to 12 hours, and more preferably 2 to 4 hours.
- the reaction temperature is not particularly limited and is, for example, 0 to 100 ° C., preferably 10 to 60 ° C., more preferably 20 to 30 ° C.
- the concentrations of the glycoside compound (Ic) and the protecting group introducing agent to be used are not particularly limited and can be appropriately set.
- the substance amount ratio between the glycoside compound (Ic) and the protecting group introducing agent is not particularly limited, and may be, for example, a stoichiometric ratio or any other ratio.
- the amount of other reactants used is not particularly limited.
- the number of moles of the protecting group introducing agent is, for example, 1 to 100 times, preferably 1 to 20 times, more preferably 1 to 5 times the number of moles of the glycoside compound (Ic).
- the reaction conditions for the introduction reaction of the protecting group R 1 may be appropriately set with reference to, for example, similar reaction conditions in known glycosides, or may be appropriately set with reference to the conditions of the examples described later. May be.
- the manufacturing method of the present invention further includes the following step (step 4).
- step 4 A step of obtaining a glycoside compound (I) by phosphorylating the glycoside compound (Id). Therefore, the present invention also provides a method for producing glycoside compound (I) by the above step 4 (more specifically, the above step 0, step 1, step 2, step 3 and subsequent step 4).
- the phosphorylation of the glycoside compound (Id) in Step 4 is performed by reacting with the following compound.
- R 2a and R 2b are the same or different and each represents a hydrogen atom or a substituent;
- R 2c represents a hydrogen atom, an electron withdrawing group or a substituent which may be substituted with an electron withdrawing group;
- R 2a and R 2b may form a ring together with the nitrogen atom to which they are bonded)
- Two R 2a may be the same or different, but are preferably the same.
- Two R 2b may be the same or different, but are preferably the same.
- R 2a and R 2b may be the same or different, but are preferably the same, more preferably a substituent, and particularly preferably an alkyl group, particularly an isopropyl group.
- R 2c is preferably an electron withdrawing group or a substituent which may be substituted with an electron withdrawing group, more preferably a substituent which may be substituted with an electron withdrawing group, particularly preferably an electron withdrawing group.
- cyano is preferable.
- R 2c is preferably a cyanoethyl group.
- a glycoside compound, a thioether or the like compound used in the present invention or provided by the present invention may be enantiomer, tautomer or steric compound.
- isomers such as isomers (eg, geometric isomers, conformational isomers, and optical isomers) exist, any isomers are included in the compound of the present invention.
- the chemical formula representing the glycoside compound of the present invention is drawn so that the sugar skeleton of the glycoside is D-ribose, but it may be an enantiomer thereof, that is, L-ribose.
- the salt of the compound of the present invention may be an acid addition salt or a base addition salt.
- the acid forming the acid addition salt may be an inorganic acid or an organic acid
- the base forming the base addition salt may be an inorganic base or an organic base.
- the inorganic acid is not particularly limited.
- the organic acid is not particularly limited, and examples thereof include p-toluenesulfonic acid, methanesulfonic acid, oxalic acid, p-bromobenzenesulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, and acetic acid.
- the inorganic base is not particularly limited, and examples thereof include ammonium hydroxide, alkali metal hydroxides, alkaline earth metal hydroxides, carbonates and hydrogen carbonates, and more specifically, for example, Examples thereof include sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, calcium hydroxide and calcium carbonate.
- the organic base is not particularly limited, and examples thereof include ethanolamine, triethylamine, and tris (hydroxymethyl) aminomethane. The method for producing these salts is not particularly limited.
- EMM cyanoethoxymethoxymethyl DIH: 1,3-diiodo-5,5-dimethylhydantoin
- TfOH trifluoromethanesulfonic acid
- MS4A molecular sieves 4A
- THF TEA ⁇ 3HF: triethylamine trihydrofluoride
- DMTr 4,4'-dimethoxytrityl iPr: isopropyl
- NIS N-iodosuccinimide
- AgOTf silver trifluoromethanesulfonate
- EtOH ethanol
- EMM reagent (1004) Synthesis of EMM reagent (1004) According to the following scheme, an EMM reagent (1004) was synthesized. “EMM” is an abbreviation for “cyanoethoxymethoxymethyl” (the same applies hereinafter).
- the reaction solution was cooled to 0 ° C., and the resulting precipitate was filtered. After vacuum drying, the target compound (1c) (15 g, purity 97.7%, two-stage yield 66%) was obtained as a white precipitate.
- the instrumental analysis value of the compound (1c) is shown below.
- Uridine EMM amidite (I-2) was synthesized according to the following scheme.
- diisopropylammonium tetrazolide (205 mg, 1.2 mmol) and acetonitrile (5 mL) were added, and 2-cyanoethyl-N, N, N ′, N′-tetraisopropyl phosphorodiamidite (362 mg, 1.2 mL) was added. mmol) was added and stirred at 40 ° C. for 2 hours. After completion of the reaction, the solvent was distilled off under reduced pressure, dichloromethane was added, and each was washed once with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution.
- N 2 -phenoxyacetyl-3 ′, 5′-O- (tetraisopropyldisiloxane-1,3-diyl) guanosine (3a) (1.0 g, 1.5 mmol) was dissolved in tetrahydrofuran, and toluene was added to add 2 It was azeotroped once and azeotroped once with tetrahydrofuran and vacuum-dried.
- the reaction mixture was immersed in an ice bath at 0 ° C., water (4 mL) was added and stirred for 10 minutes, isopropyl ether (30 mL) was added and stirred for 30 minutes.
- the white precipitate was filtered under reduced pressure and vacuum dried to obtain the target compound (3c) (0.63 g, purity 89.76%).
- the instrumental analysis value of the compound (3c) is shown below.
- N 2 -phenoxyacetyl-5′-O- (4,4′-dimethoxytrityl) -2′-O- (2-cyanoethoxymethoxymethyl) guanosine (3d) N 2 -phenoxyacetyl-2′-O- (2-cyanoethoxymethoxymethyl) guanosine (3c) (630 mg, 1.2 mmol) was azeotroped with pyridine and dried in vacuo. It was dissolved in pyridine (6.0 mL), 4,4′-dimethoxytrityl chloride (600 mg, 1.8 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 2 hours.
- diisopropylammonium tetrazolide 180 mg, 1.1 mmol
- acetonitrile 3 mL
- 2-cyanoethyl-N, N, N ′, N′-tetra Isopropyl phosphorodiamidite 0.61 mL, 1.9 mmol
- the solvent was distilled off under reduced pressure, ethyl acetate was added, and each was washed once with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution.
- Example 4 Synthesis of adenosine EMM amidite (I-4) Adenosine EMM amidite (I-4) was synthesized according to the following scheme.
- N 6 -acetyl-5′-O- (4,4′-dimethoxytrityl) -2′-O- (2-cyanoethoxymethoxymethyl) adenosine (4d) N 6 -acetyl-2′-O- (2-cyanoethoxymethoxymethyl) adenosine (4c) was azeotroped with pyridine and dried in vacuo. It was dissolved in pyridine (10 mL), 4,4′-dimethoxytrityl chloride (1.2 g, 3.5 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 3 hours.
- diisopropylammonium tetrazolide (170 mg, 1.0 mmol) and acetonitrile (5.5 mL) were added, and 2-cyanoethyl-N, N, N ′, N′-tetraisopropylphosphorodiamidite (0.32 mL, 1.0 mL) was added. mmol) was added and stirred at 40 ° C. for 3 hours. After completion of the reaction, the solvent was distilled off under reduced pressure, ethyl acetate was added, and each was washed once with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution.
- phosphoramidite as a raw material for nucleic acid synthesis can be produced efficiently and with high purity. Accordingly, it becomes possible to produce nucleic acids with high purity and high yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Saccharide Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
しかし、TOMアミダイト、ACEアミダイト等の従来のホスホロアミダイトは、その製造コストが高いため、医薬品等の合成原料としては用いにくい。また、TBDMSアミダイトは、カップリング(縮合)反応により核酸を合成する際の、核酸の収率および純度があまり高くない場合がある。
そこで、低コストに製造できて、かつ、核酸を高収率および高純度で製造可能なホスホロアミダイトを提供可能な、保護基の開発が試みられている(特許文献1及び2)。
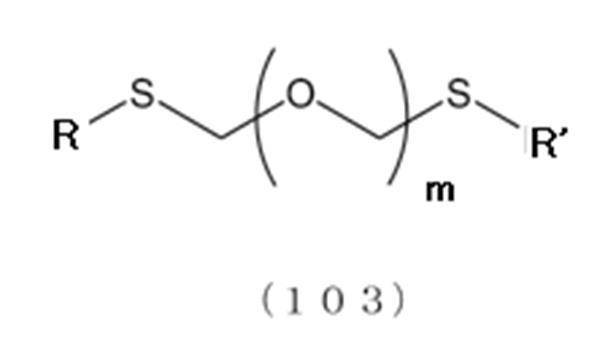
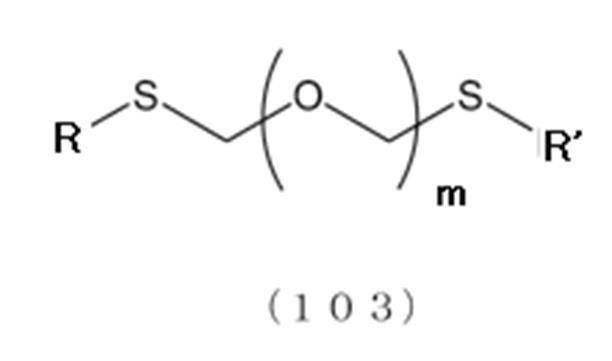
[1]一般式(103):

で表されるチオエーテル化合物と、一般式(105):
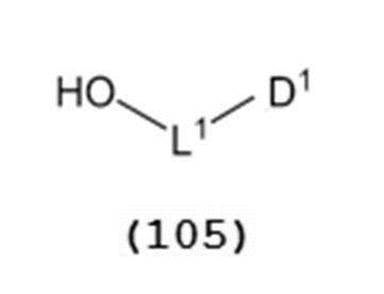
で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):
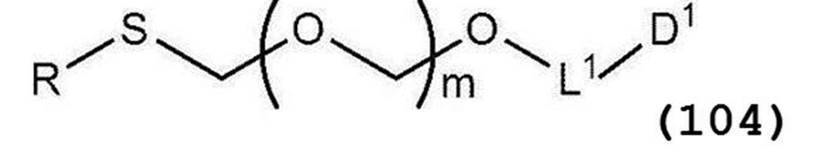
で表されるチオエーテル化合物を得る工程(工程0);および
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

Xは下記式:

で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

で表される配糖体化合物を得る工程(工程1)を含む、一般式(I):

[2]さらに一般式(Ib)で表される化合物を脱保護して一般式(Ic):

で表される配糖体化合物を得る工程(工程2)を含む、上記[1]記載の製造方法。
[3]さらに一般式(Ic)で表される配糖体化合物に水酸基の保護基を導入して一般式(Id):

で表される配糖体化合物を得る工程(工程3)を含む、上記[2]記載の製造方法。
[4]さらに一般式(Id)で表される配糖体化合物をリン酸化して一般式(I)で表される配糖体化合物を得る工程(工程4)を含む、上記[3]記載の製造方法。
[5]一般式(103):

で表されるチオエーテル化合物と、一般式(105):

で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):

で表されるチオエーテル化合物を得る工程(工程0);および
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

Xは下記式:

で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

で表される配糖体化合物を得る工程(工程1)を含む、一般式(Ib)で表される配糖体化合物の製造方法。
[6]一般式(103):
で表されるチオエーテル化合物と、一般式(105):

で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):
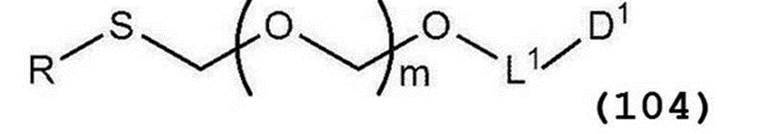
で表されるチオエーテル化合物を得る工程(工程0);
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

Xは下記式:

で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

で表される配糖体化合物を得る工程(工程1);および
一般式(Ib)で表される配糖体化合物を脱保護して一般式(Ic):

で表される配糖体化合物を得る工程(工程2)を含む、一般式(Ic)で表される配糖体化合物の製造方法。
[7]一般式(103):

で表されるチオエーテル化合物と、一般式(105):

で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):
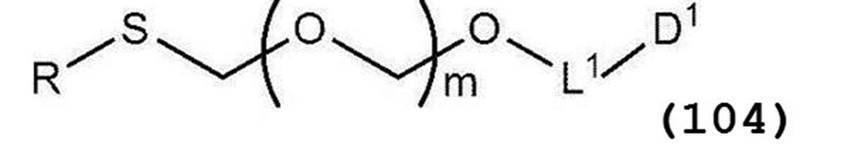
で表されるチオエーテル化合物を得る工程(工程0);
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):
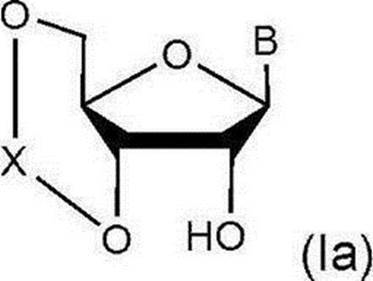
Xは下記式:
で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

で表される配糖体化合物を得る工程(工程1);
一般式(Ib)で表される配糖体化合物を脱保護して一般式(Ic):

で表される配糖体化合物を得る工程(工程2);および
一般式(Ic)で表される配糖体化合物に水酸基の保護基を導入して一般式(Id):

で表される配糖体化合物を得る工程(工程3)を含む、一般式(Id)で表される配糖体化合物の製造方法。
[8]一般式(103):
で表されるチオエーテル化合物と、一般式(105):

で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):
で表されるチオエーテル化合物を得る工程(工程0);
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

Xは下記式:

で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

で表される配糖体化合物を得る工程(工程1);
一般式(Ib)で表される配糖体化合物を脱保護して一般式(Ic):

で表される配糖体化合物を得る工程(工程2);
一般式(Ic)で表される配糖体化合物に水酸基の保護基を導入して一般式(Id):

で表される配糖体化合物を得る工程(工程3);および
一般式(Id)で表される配糖体化合物をリン酸化して一般式(I):

[9]工程0の蒸留における該添加剤が、4,4’-ビスマレイミドジフェニルメタンである、上記[1]~[8]のいずれかに記載の方法。
[10]nが1である、上記[1]~[9]のいずれかに記載の方法。
[12]mが1である、上記[1]~[11]のいずれかに記載の方法。
[13]L1がエチレン基である、上記[1]~[12]のいずれかに記載の方法。
[14]D1がシアノ基である、上記[1]~[13]のいずれかに記載の方法。
[15]工程1において、ハロゲン化剤、乾燥剤およびルイス酸の添加後に一般式(104)で表されるチオエーテル化合物を添加することを特徴とする、上記[1]~[14]のいずれかに記載の方法。
[16]工程1において、乾燥剤、ルイス酸および一般式(104)で表されるチオエーテル化合物の添加後にハロゲン化剤を添加することを特徴とする、上記[1]~[14]のいずれかに記載の方法。
[17]工程1において、ハロゲン化剤の添加後にルイス酸を添加することを特徴とする、上記[15]に記載の方法。
[18]工程1において、ルイス酸の添加後に一般式(104)で表されるチオエーテル化合物を添加することを特徴とする、上記[16]に記載の方法。
[19]工程1におけるハロゲン化剤がヨウ素化剤である、上記[1]~[18]のいずれかに記載の方法。
[20]ヨウ素化剤が1,3-ジヨード-5,5-ジメチルヒダントイン又はヨウ素である、上記[19]に記載の方法。
[22]工程1におけるルイス酸がトリフルオロメタンスルホン酸である、上記[1]~[21]のいずれかに記載の方法。
[23]工程1におけるルイス酸がメタンスルホン酸である、上記[1]~[21]のいずれかに記載の方法。
「核酸塩基骨格を有する原子団」とは、その構造の全てあるいは一部に核酸塩基骨格を有する官能基を意味する。ここで「核酸塩基骨格」とは天然核酸塩基骨格でも人工核酸塩基骨格でもよいが、好ましくは天然核酸塩基骨格である。
該天然核酸塩基としては、アデニン、シトシン、グアニン、ウラシル、チミン、またはその他の含窒素芳香環(例、5-アルキルピリミジン、5-ハロゲノピリミジン、デアザプリン、デアザピリミジン、アザプリン、アザピリミジン)であることがより好ましい。
(1)ハロゲン(フッ素原子、塩素原子、臭素原子またはヨウ素原子);
(2)アルキル基(上述);
(3)アルケニル基(上述);
(4)アルキニル基(上述);
(5)ハロアルキル基(例、クロロメチル、フルオロメチル、ジクロロメチル、ジフルオロメチル、ジクロロフルオロメチル、トリフルオロメチル、ペンタフルオロエチル等);
(6)アリール基(上述);
(7)ヘテロアリール基(上述);
(8)アラルキル基(上述);
(9)シクロアルキル基(上述);
(10)シクロアルケニル基(上述);
(11)シクロアルキルアルキル基(上述);
(12)シクロアルケニルアルキル基(例、シクロペンテニルエチル、シクロヘキセニルエチル、シクロヘキセニルブチル等);
(13)ヒドロキシアルキル基(例、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、ヒドロキシブチル等);
(14)アルコキシアルキル基(上述);
(15)アミノアルキル基(例、アミノメチル、アミノエチル、アミノプロピル等);
(16)ヘテロシクリル基(例、1-ピロリニル、2-ピロリニル、3-ピロリニル、1-ピロリジニル、2-ピロリジニル、3-ピロリジニル、ピロリジノン、1-イミダゾリニル、2-イミダゾリニル、4-イミダゾリニル、1-イミダゾリジニル、2-イミダゾリジニル、4-イミダゾリジニル、イミダゾリジノン、1-ピラゾリニル、3-ピラゾリニル、4-ピラゾリニル、1-ピラゾリジニル、3-ピラゾリジニル、4-ピラゾリジニル、ピペリジノン、ピペリジノ、2-ピペリジニル、3-ピペリジニル、4-ピペリジニル、1-ピペラジニル、2-ピペラジニル、ピペラジノン、2-モルホリニル、3-モルホリニル、モルホリノ、テトラヒドロピラニル、テトラヒドロフラニル等);
(17)ヘテロシクリルアルケニル基(例、2-ピペリジニルエテニル等);
(18)ヘテロシクリルアルキル基(例、ピペリジニルメチル、ピペラジニルメチル等);
(19)ヘテロアリールアルキル基(例、ピリジルメチル、キノリン-3-イルメチル等);
(20)シリル基;
(21)シリルオキシアルキル基(例、シリルオキシメチル、シリルオキシエチル等);
(22)モノ・ジもしくはトリアルキルシリル基(例、メチルシリル、エチルシリル等);及び
(23)モノ・ジもしくはトリアルキルシリルオキシアルキル基(例、トリメチルシリルオキシメチル等)
等が挙げられる。これらの各置換基は、さらに電子求引基(上述)で置換されていてもされていなくても良い。

本発明の製造方法は、下記工程(工程1)を含む。
[工程1]
ハロゲン化剤、乾燥剤及びルイス酸の存在下、一般式(Ia):

Xは下記式:

で表される配糖体化合物(以下、配糖体化合物(Ia)とも称する)と一般式(104):

で表されるチオエーテル化合物(以下、チオエーテル化合物(104)とも称する)をカップリング反応させて一般式(Ib):

で表される配糖体化合物(以下、配糖体化合物(Ib)とも称する)を得る工程。
従って、本発明は工程1により、配糖体化合物(Ib)を製造する方法をも提供する。
[工程0]
一般式(103):
で表されるチオエーテル化合物(以下、チオエーテル化合物(103)とも称する)と、一般式(105):

で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留してチオエーテル化合物(104)を得る工程。
従って、本発明は工程0により、チオエーテル化合物(104)を製造する方法、工程0及び工程1により配糖体化合物(Ib)を製造する方法をも提供する。
Bは、好ましくは、アデニン、シトシン、グアニン、ウラシル及びチミンである。
工程0においては、前記カップリング反応後に蒸留を行うことを特徴とする。前記蒸留は、自体公知の方法により実施することができるが、高純度のチオエーテル化合物(104)が得られることから、添加剤として硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の存在下、減圧下で繰り返し実施することが好ましい。硫黄含有抗酸化剤は、抗酸化作用を有する分子内に硫黄原子を有する化合物であって、フェノチアジン等が挙げられる。マレイミド基含有化合物は、分子内にマレイミド基を有する化合物であって、マレイミド、4,4’-ビスマレイミドジフェニルメタン、ビス(3-エチル-5-メチル-4-マレイミドフェニル)メタン等が挙げられる。好ましくは4,4’-ビスマレイミドジフェニルメタンである。本発明では4,4’-ビスマレイミドジフェニルメタンが蒸留の際の添加剤として好ましく用いられる。添加剤の使用量は特に限定されないが、反応物質に対し0.01~0.5倍、好ましくは0.05~0.4倍、さらに好ましくは0.1~0.3倍の重量で用いる。具体的には後述の実施例の条件を参考にして実施することができる。当該蒸留により、チオエーテル化合物(104)を、純度80%以上、好ましくは90%、より好ましくは95%以上、さらに好ましくは98%以上にまで高めることができる。
工程1において、ハロゲン化剤、ルイス酸、チオエーテル化合物(104)及び乾燥剤の添加順序は特に限定されない。本発明の好ましい一実施態様として、Bの核酸塩基骨格を有する原子団における核酸塩基がシトシン、ウラシル及びグアニンの場合には、工程1においては、チオエーテル化合物(104)の添加は最後に行うことが好ましい。即ち、乾燥剤存在下の反応系にハロゲン化剤を添加し、続いてルイス酸を添加した後にチオエーテル化合物(104)を添加することが好ましい。本発明の別の好ましい一実施態様として、Bの核酸塩基骨格を有する原子団における核酸塩基がアデニンの場合には、工程1においては、ハロゲン化剤の添加は最後に行うことが好ましい。即ち、乾燥剤存在下の反応系にルイス酸を添加し、続いてチオエーテル化合物(104)を添加した後にハロゲン化剤を添加することが好ましい。チオエーテル化合物(104)、ハロゲン化剤、及びルイス酸の反応系への添加は、上記添加順序に従う限り任意の間隔で行なうことができ、それらの仕込み量等によって適宜設定される。
[工程2]
配糖体化合物(Ib)を脱保護して一般式(Ic):
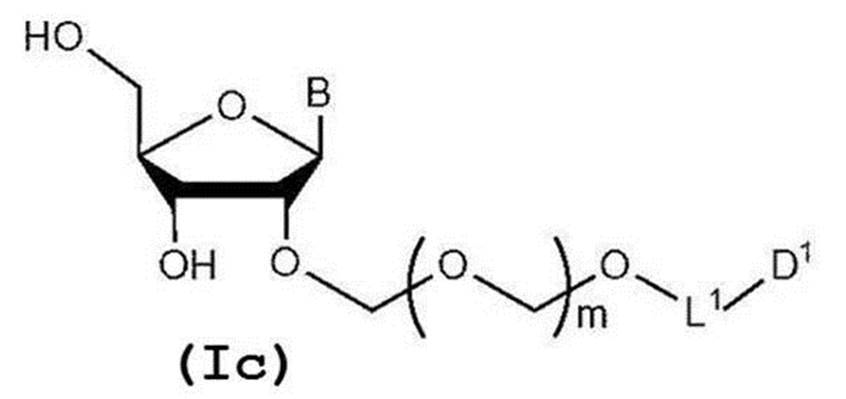
で表される配糖体化合物(以下、配糖体化合物(Ic)とも称する)を得る工程。
従って、本発明は上記工程2(より詳細には上記工程0、工程1及びそれに続く工程2)により、配糖体化合物(Ic)を製造する方法をも提供する。
工程2において、脱保護反応の条件は特に限定されないが、例えば、公知の脱保護剤を用いることができる。前記脱保護剤は、特に限定されないが、例えば、フッ化水素ピリジン、トリエチルアミン三フッ化水素、フッ化アンモニウム、フッ化水素酸、テトラブチルアンモニウムフロリド等が挙げられる。脱保護反応の反応溶媒は、特に限定されないが、例えば、アセトン等のケトン;ジエチルエーテル、THF(テトラヒドロフラン)等のエーテル;メタノール、エタノール等のアルコール;またはアセトニトリル等のニトリル等が挙げられる。脱保護反応の反応時間は、特に限定されないが、例えば、30分~24時間、好ましくは2~12時間、より好ましくは2~4時間である。脱保護反応の反応温度は、特に限定されないが、例えば、0~100℃、好ましくは、20~60℃、より好ましくは20~50℃である。配糖体化合物(Ib)および脱保護剤の濃度も、特に限定されず、適宜設定可能である。配糖体化合物(Ib)と脱保護剤との物質量比も特に限定されず、例えば、化学量論比でも良いし、他の任意の比でも良い。その他の反応物質の使用量も、特に限定されない。脱保護剤のモル数は、配糖体化合物(Ib)のモル数に対し、例えば0.1~20倍、好ましくは0.2~10倍、より好ましくは1~5倍である。脱保護反応の反応条件は、例えば、公知の配糖体における類似の脱保護反応等の条件を参考にして適宜設定しても良いし、後述の実施例の条件を参考にして適宜設定しても良い。
[工程3]
配糖体化合物(Ic)に水酸基の保護基を導入して一般式(Id):

で表される配糖体化合物(以下、配糖体化合物(Id)とも称する)を得る工程を含む。
従って、本発明は上記工程3(より詳細には上記工程0、工程1、工程2及びそれらに続く工程3)により、配糖体化合物(Id)を製造する方法をも提供する。
保護基R1の導入反応において、保護基導入剤は、R1に応じて適宜選択すればよい。例えば保護基が4,4’-ジメトキシトリチルである場合、保護基導入剤は4,4’-ジメトキシトリチルクロライドを用いることができる。
反応溶媒は、特に限定されないが、例えば、ピリジン等の極性溶媒;またはアセトニトリル等のニトリル;テトラヒドロフラン等のエーテル等が挙げられる。反応時間は、特に限定されないが、例えば、30分~24時間、好ましくは2~12時間、より好ましくは2~4時間である。反応温度は、特に限定されないが、例えば、0~100℃、好ましくは、10~60℃、より好ましくは20~30℃である。用いる配糖体化合物(Ic)および保護基導入剤の濃度も、特に限定されず、適宜設定可能である。配糖体化合物(Ic)と保護基導入剤との物質量比も特に限定されず、例えば、化学量論比でも良いし、他の任意の比でも良い。その他の反応物質の使用量も、特に限定されない。保護基導入剤のモル数は、配糖体化合物(Ic)のモル数に対し、例えば1~100倍、好ましくは1~20倍、より好ましくは1~5倍である。保護基R1の導入反応の反応条件は、例えば、公知の配糖体における類似の反応の条件を参考にして適宜設定しても良いし、後述の実施例の条件を参考にして適宜設定しても良い。
[工程4]
配糖体化合物(Id)をリン酸化して配糖体化合物(I)を得る工程。
従って、本発明は上記工程4(より詳細には上記工程0、工程1、工程2、工程3及びそれに続く工程4)により、配糖体化合物(I)を製造する方法をも提供する。

2つのR2aは同一でも異なっていてもよいが好ましくは同一である。2つのR2bは同一でも異なっていてもよいが好ましくは同一である。R2a及びR2bは同一でも異なっていてもよいが、好ましくは同一であり、より好ましくは置換基であり、特に好ましくはアルキル基、特にイソプロピル基であることが好ましい。
R2cは、好ましくは電子求引基又は電子求引基で置換されていてもよい置換基であり、より好ましくは電子求引基で置換されていてもよい置換基、特に好ましくは電子求引基で置換されていてもよい炭素数1~4の直鎖又は分岐鎖のアルキル基(例、エチル)である。電子求引基としてはシアノが好ましい。
R2cは、好ましくはシアノエチル基である。
略語一覧:
EMM: cyanoethoxymethoxymethyl
DIH: 1,3-diiodo-5,5-dimethylhydantoin
TfOH: trifluoromethanesulfonic acid
MS4A: molecular sieves 4A
THF: tetrahydrofuran
TEA・3HF: triethylamine trihydrofluoride
DMTr: 4,4'-dimethoxytrityl
iPr: isopropyl
NIS: N-iodosuccinimide
AgOTf: silver trifluoromethanesulfonate
EtOH: ethanol
下記スキームに従い、EMM化試薬(1004)を合成した。なお、「EMM」は、「シアノエトキシメトキシメチル(cyanoethoxymethoxymethyl)」の略である(以下において同じ)。

市販のビス(メチルチオメチル)エーテル(1003)(30g,217mmol)を、アルゴン雰囲気下、テトラヒドロフラン(300mL)に溶解させた。その溶液に、シアノエタノール(15g,217mmol、減圧蒸留により精製)およびモレキュラーシーブス4A(30g)を加え、10分間撹拌した。この混合物に、さらにN-ヨードスクシンイミド(49g,217mmol)を加えて溶解させ、直ぐに-45℃に冷却した。冷却後、さらにトリフルオロメタンスルホン酸銀(1.7g, 6.5mmol)を加え3時間撹拌した。撹拌後、氷冷した10%チオ硫酸ナトリウム水溶液を加え、-45℃で激しく撹拌した。10分後にクーリングバスから取り出し、酢酸エチルを加え30分間激しく撹拌した。反応溶液をセライトろ過した。ろ液を10%チオ硫酸ナトリウム水溶液、飽和炭酸水素ナトリウム水溶液、および飽和塩化ナトリウム水溶液を前記順序で用いて洗浄した。その後、有機層を分取し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。上記の反応を5回行い、粗生成物(150g)を得た。この粗生成物に4,4’-ビスマレイミドジフェニルメタン(33g)を加え0.50mmHgで減圧蒸留し、主留として粗精製物(71g、沸点93~96℃)を得た。得られた粗精製物(71g)に4,4’-ビスマレイミドジフェニルメタン(15g)を加え0.25mmHgで減圧蒸留し、主留として目的化合物(50g、沸点83~86℃、純度98.46%)を得た。
1H-NMR(400MHz, CDCl3) δ: 4.86(2H, s), 4.73(2H, s), 3.80(2H, t, J=6.3Hz), 2.64(2H, t, J=6.3Hz), 2.18(3H, s).
下記スキームに従い、シチジンEMMアミダイト(I-1)を合成した。

市販のN4-アセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)シチジン(1a)(30 g, 57 mmol)をトルエンにて2回、テトラヒドロフランにて1回共沸し、真空乾燥させた。アルゴン雰囲気下、テトラヒドロフラン(180 mL)に溶解させ、モレキュラーシーブス4A(30 g)、1,3-ジヨード-5,5-ジメチルヒダントイン(33 g, 85 mmol)を加えて撹拌し、その混合液を-60℃に冷却した。-60℃でトリフルオロメタンスルホン酸(7.5 mL, 85 mmol)を滴下した。5分間撹拌した後、フラスコ内温度-60℃にて2-シアノエトキシメチルメチルチオメチルエーテル(1004)(14 g, 85 mmol)を加え-60℃にて30分間撹拌した。反応終了後、冷却した飽和チオ硫酸ナトリウム水溶液と飽和炭酸水素ナトリウム水溶液の混合溶液を反応溶液に加え、茶褐色が消失するまで激しく撹拌した。酢酸エチルにて抽出し、有機層を飽和チオ硫酸ナトリウム水溶液で2回、飽和炭酸水素ナトリウム水溶液で1回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、目的化合物(1b)の粗生成物(45g,純度93.8%)を得た。以下に、化合物(1b)の機器分析値を示す。
化合物(1b):
1H-NMR(400 MHz, CDCl3) δ: 9.17(1H, s), 8.30(1H, d, J=7.2Hz), 7.41(1H, d, J= 7.8Hz), 5.79(1H, s), 5.18(1H, d, J= 6.8 Hz), 5.03(d, 1H, J= 7.4 Hz), 4.29(1H, d, J=13.7 Hz), 4.23-4.10(5H, m), 4.03-3.96(2H, m), 3.87-3.75(1H, m), 2.76-2.65(2H, m), 2.24(3H, s), 1.11-0.89(28H, m).
N4-アセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)-2’-O-(2-シアノエトキシメトキシメチル)シチジン(1b)(45 g)を、アルゴン雰囲気下、テトラヒドロフラン(240 mL)に溶解させた。その溶液に、トリエチルアミン三フッ化水素(13 mL, 81 mmol)を加えて、室温で2時間撹拌した。反応溶液を0℃に冷却して、生じた沈殿をろ過した。真空乾燥後、白色沈殿の目的化合物(1c)(15g,純度97.7%,2段階収率66%)を得た。以下に、化合物(1c)の機器分析値を示す。
化合物(1c):
1H-NMR(400 MHz, D2O) δ: 8.24(1H, d, J= 7.3 Hz), 7.24(1H, d, J= 7.8 Hz), 5.92(1H, d, J= 2.4 Hz), 5.02(1H, d, J= 6.8 Hz), 4.89(1H, d, J= 6.8 Hz), 4.79-4.74(2H, m), 4.29(1H, dd, J= 4.9, 2.9 Hz), 4.17(1H, t, J= 6.3 Hz), 4.09-4.05(1H, m), 3.90-3.85(1H, m), 3.77-3.70(3H, m), 2.67(2H, t, J= 6.1 Hz), 2.12(3H, s).
融点:171-172℃
N4-アセチル-2’-O-(2-シアノエトキシメトキシメチル)シチジン(1c)(15 g, 38 mmol)をピリジンで共沸し、真空乾燥させた。ピリジン(200 mL)に溶解させ、0℃で4,4’-ジメトキシトリチルクロライド(19 g, 57 mmol)を加えて室温にて4時間撹拌した。反応終了後、メタノールを加えて30分間撹拌し、減圧下溶媒留去した。残渣にジクロロメタンを加えて、飽和炭酸水素ナトリウム水溶液で2回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル、0.05%ピリジン含有→酢酸エチル:アセトン=1:1、0.05%ピリジン含有)にて精製し、目的化合物(1d)(23g,純度99.2%,収率86%)を得た。以下に、化合物(1d)の機器分析値を示す。
化合物(1d):
1H-NMR (400 MHz, CDCl3) δ: 8.61(1H, br.s), 8.49(1H, d, J= 7.8 Hz), 7.42-7.26(9H, m), 7.09(1H, d, J= 7.3 Hz), 6.88-6.86(4H, m), 5.94(1H, s), 5.35(1H, d, J= 6.8 Hz), 5.11(1H, d, J= 6.8 Hz), 4.92(1H, d, J= 7.3 Hz), 4.87(1H, d, J= 7.3 Hz), 4.49-4.40(1H, m), 4.29(1H, d, J= 4.9 Hz), 4.15-4.08(1H, m), 3.86(t, 2H, J= 6.2 Hz), 3.82(s, 6H), 3.63(dd, 1H, J= 10.6, 2.6 Hz), 3.55(dd, 1H, J= 10.6, 2.6 Hz), 2.64(2H, t, J= 6.3 Hz), 2.56(d, 1H, J= 8.8 Hz), 2.21(3H, s).
N4-アセチル-5’-O-(4,4’-ジメトキシトリチル)-2’-O-(2-シアノエトキシメトキシメチル)シチジン(1d)(23 g, 31 mmol)をアセトニトリルで共沸し、真空乾燥させた。この操作を2回行った。アルゴン雰囲気下、ジイソプロピルアンモニウムテトラゾリド(6.5g, 38 mmol)とアセトニトリル(80 mL)を加え、さらに2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(12 mL, 38 mmol)を加え、40℃で3時間撹拌した。反応終了後、減圧下溶媒留去し、酢酸エチルを加えて飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液でそれぞれ1回ずつ洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:アセトン=2:1、0.1%トリエチルアミン含有)で精製し、目的化合物(I-1)(25g,純度98.8%,収率88%)を得た。以下に、化合物(I-1)の機器分析値を示す。
化合物(I-1):
31P-NMR(162MHz, CDCl3) δ: 151.8, 150.3. MS(ESI+): m/z 923[M+Na]+
下記スキームに従い、ウリジンEMMアミダイト(I-2)を合成した。
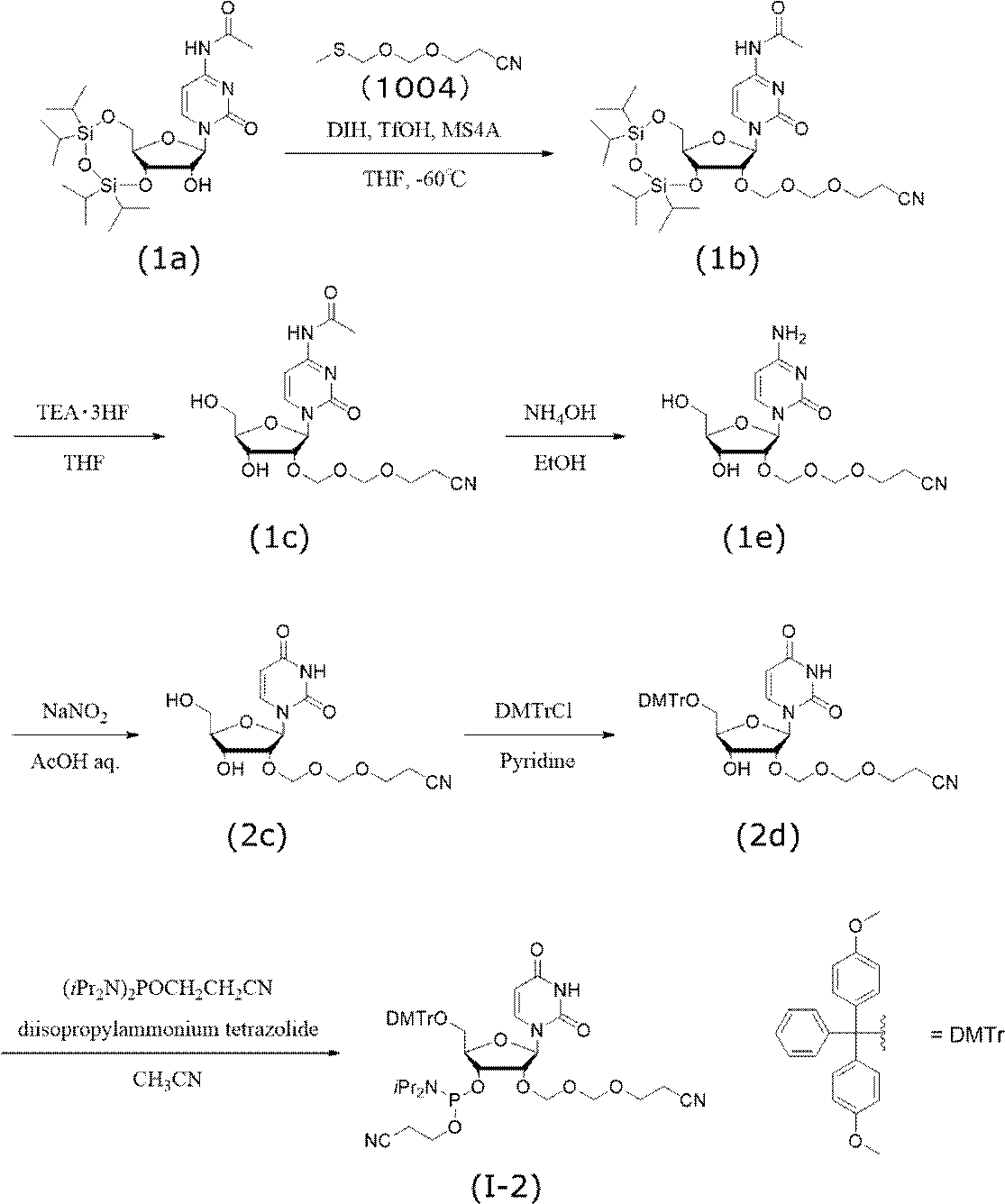
市販のN4-アセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)シチジン(1a)(1.0 g, 1.9 mmol)をトルエンにて2回、テトラヒドロフランにて1回共沸し、真空乾燥させた。アルゴン雰囲気下、テトラヒドロフラン(6.0 mL)に溶解させ、モレキュラーシーブス4A(1.0 g)、1,3-ジヨード-5,5-ジメチルヒダントイン(1.1 g, 2.9 mmol)を加えて撹拌し、その混合液を-60℃に冷却した。-60℃でトリフルオロメタンスルホン酸(0.25 mL, 2.9 mmol)を滴下した。5分間撹拌した後、フラスコ内温度-60℃にて2-シアノエトキシメチルメチルチオメチルエーテル(1004)(0.46 g, 2.9 mmol)を加え-60℃にて30分間撹拌した。反応終了後、冷却した飽和チオ硫酸ナトリウム水溶液と飽和炭酸水素ナトリウム水溶液の混合溶液を反応溶液に加え、茶褐色が消失するまで激しく撹拌した。酢酸エチルにて抽出し、有機層を飽和チオ硫酸ナトリウム水溶液で2回、飽和炭酸水素ナトリウム水溶液で1回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、目的化合物(1b)の粗生成物(1.2g,純度95.9%)を得た。
N4-アセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)-2’-O-(2-シアノエトキシメトキシメチル)シチジン(1b)(1.2 g)を、アルゴン雰囲気下、テトラヒドロフラン(8.0 mL)に溶解させた。その溶液に、トリエチルアミン三フッ化水素(0.40 mL, 2.5 mmol)を加えて、室温で2時間撹拌した。反応溶液を0℃に冷却して、生じた沈殿をろ過した。真空乾燥後、白色沈殿の目的化合物(1c)(0.46g、純度98.7%,2段階収率61%)を得た。
N4-アセチル-2’-O-(2-シアノエトキシメトキシメチル)シチジン(1c)(0.46 g, 1.2 mmol)を、エタノール(8.4 mL)に溶解させた。その溶液に、水(6.3 mL)および濃アンモニア水(2.1 mL)を加えて4℃で一晩静置した。反応終了後、減圧下溶媒留去し、目的化合物(1e)の粗生成物0.53gを得た。
2’-O-(2-シアノエトキシメトキシメチル)シチジン(1e)(0.53 g, 1.5 mmol)を、水(10 mL)に溶解させた。その溶液に、亜硝酸ナトリウム(1.6 g, 22.5 mmol)および酢酸(2.5 mL)を加えて40℃で4時間撹拌し、室温に戻して一晩撹拌した。反応終了後、減圧下溶媒留去し、残渣にピリジンを加えて減圧ろ過した。母液を減圧留去し、目的化合物(2c)の粗生成物0.8gを得た。以下に、化合物(2c)の機器分析値を示す。
化合物(2c):
1H-NMR (400 MHz, CDCl3) d: 10.23 (1H, br.s), 7.90 (1H, d, J = 7.8 Hz), 5.84 (1H, d, J = 2.9 Hz), 5.59 (1H, d, J = 8.3 Hz), 5.09 (1H, d, J = 7.0 Hz), 4.98 (1H, d, J = 6.7 Hz), 4.87 (2H, s), 4.25-4.22 (3H, m), 3.99 (1H, s), 3.83-3.69 (5H, m), 2.70-2.61 (2H, m).
融点:162-163℃
2’-O-(2-シアノエトキシメトキシメチル)ウリジン(2c)(0.8g, 2.2 mmol)をピリジンで共沸し、真空乾燥させた。ピリジン(13 mL)に溶解させ、4,4’-ジメトキシトリチルクロライド(895 mg, 2.6 mmol)を加えて室温にて2時間撹拌した。反応終了後、溶媒を半分だけ減圧下溶媒留去した。残渣にジクロロメタンを加えて、飽和炭酸水素ナトリウム水溶液で2回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=2:1、0.05%ピリジン含有)で精製し、目的物690mg(純度99.89%,3段階収率90%)を得た。以下に、化合物(2d)の機器分析値を示す。
化合物(2d):
1H-NMR(400 MHz,CDCl3) δ: 8.62(1H, br.s), 7.99(1H, d, J= 7.8 Hz), 7.40-7.25(9H, m), 6.90-6.84(4H, m), 5.96(1H, d, J= 2.0 Hz), 5.28(1H, d, J= 8.3 Hz), 5.18(1H, d, J= 6.8 Hz), 5.03(1H, d, J= 7.3 Hz), 4.87(2H, d, J= 7.3 Hz), 4.48(1H, q, J= 5.4 Hz), 4.29(1H, dd, J= 5.1, 2.2 Hz), 4.11-4.07(1H, m), 3.87(2H, t, J= 6.0 Hz), 3.84(6H, s), 3.55(2H, dd, J= 9.0, 2.2 Hz), 2.76(1H, d, J= 7.8 Hz), 2.65(2H, t, J= 6.6 Hz).
5’-O-(4,4’-ジメトキシトリチル)-2’-O-(2-シアノエトキシメトキシメチル)ウリジン(2d)(690 mg, 1.0 mmol)をアセトニトリルで共沸し、真空乾燥させた。この操作を2回行った。アルゴン雰囲気下、ジイソプロピルアンモニウムテトラゾリド(205 mg, 1.2 mmol)とアセトニトリル(5 mL)を加え、さらに2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(362 mg, 1.2 mmol)を加え、40℃で2時間撹拌した。反応終了後、減圧下溶媒留去し、ジクロロメタンを加えて飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液でそれぞれ1回ずつ洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:アセトン=1:1、0.1%トリエチルアミン含有)で精製し、目的化合物(I-2)(804mg,純度98.4%,収率89%)を得た。以下に、化合物(I-2)の機器分析値を示す。
化合物(I-2):
31P-NMR(162 MHz, CDCl3) δ: 153.5, 151.9.
下記スキームに従い、グアノシンEMMアミダイト(I-3)を合成した。
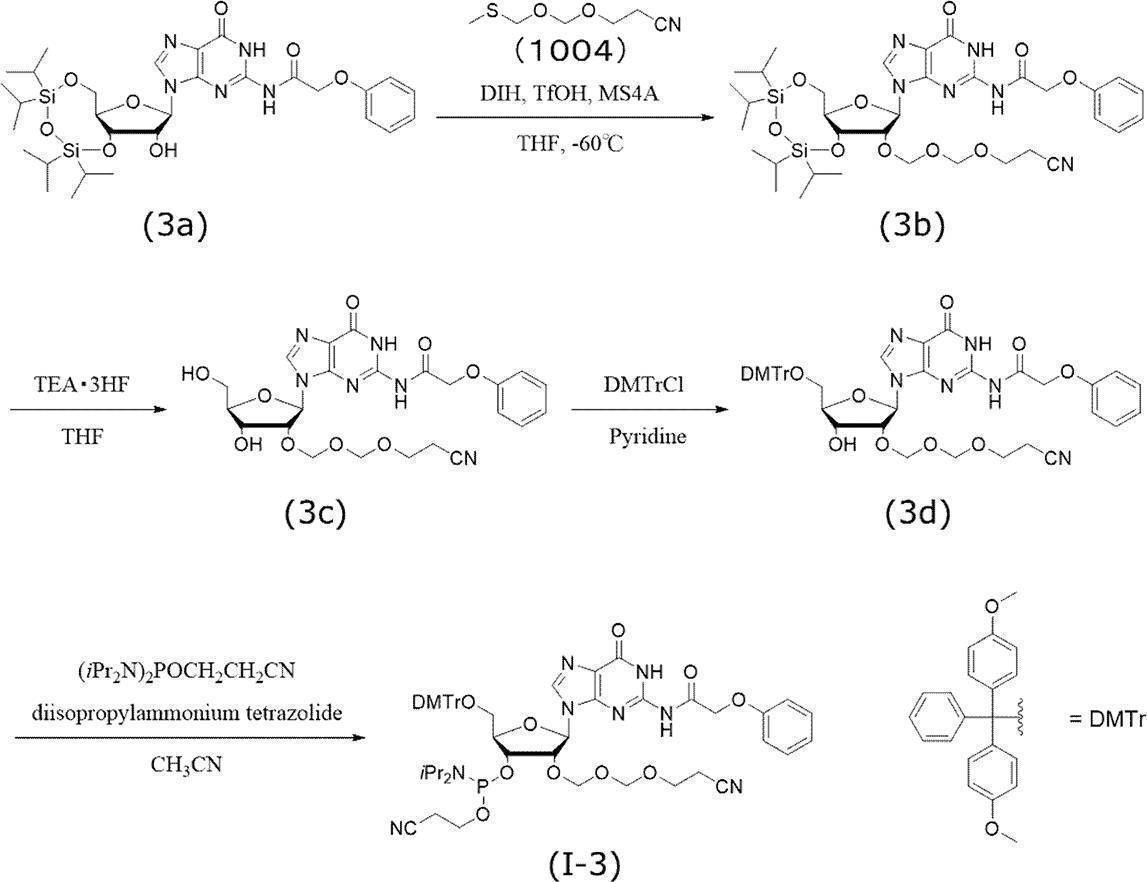
市販のN2-フェノキシアセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)グアノシン(3a)(1.0 g, 1.5 mmol)をテトラヒドロフランに溶解させ、トルエンを加えて2回共沸、テトラヒドロフランにて1回共沸し、真空乾燥させた。アルゴン雰囲気下、テトラヒドロフラン(6 mL)に溶解させ、モレキュラーシーブス4A(1.0 g)を加え、室温にて1,3-ジヨード-5,5-ジメチルヒダントイン(0.87 g, 2.3 mmol)を加えて撹拌した。その混合液を-60℃に冷却し、5分間撹拌した後、トリフルオロメタンスルホン酸(0.2 mL, 2.3 mmol)を滴下した。5分間撹拌した後、フラスコ内温度-60℃にて2-シアノエトキシメチルメチルチオメチルエーテル(1004)(0.37 g, 2.3 mmol)を加えて-60℃で30分間撹拌した。反応終了後、冷却した飽和チオ硫酸ナトリウム水溶液と飽和炭酸水素ナトリウム水溶液の混合溶液を反応溶液に加え、茶褐色が消失するまで激しく撹拌した。酢酸エチルにて抽出し、有機層を飽和チオ硫酸ナトリウム水溶液で2回、飽和炭酸水素ナトリウム水溶液で1回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、目的化合物(3b)の粗生成物(1.3g,純度88.9%)を得た。以下に、化合物(3b)の機器分析値を示す。
化合物(3b):
1H-NMR (400 MHz, CDCl3) δ: 11.79(1H, s), 9.11(1H, s), 8.04(1H, s), 7.41-7.34(2H, m), 7.13-6.97(3H, m), 5.94(1H, s), 5.08, 4.97(2H, 2d, J= 7.2 Hz), 4.87-4.67(2H, m), 4.51-4.46(1H, dd, J= 9.3, 4.9 Hz), 4.33-4.24(2H, m), 4.15(1H, d, J= 9.3 Hz), 4.02(1H, dd, J= 13.2, 2.4 Hz), 3.77-3.71(2H, m), 2.76-2.53(2H, m), 1.11-0.94(28H, m).
N2-フェノキシアセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)-2’-O-(2-シアノエトキシメトキシメチル)グアノシン(3b)(1.3 g, 1.7 mmol)をアルゴン雰囲気下テトラヒドロフラン(5 mL)に溶解させた。その溶液に、トリエチルアミン三フッ化水素(0.33 mL, 2.0 mmol)を加えて35℃にて2時間撹拌した。反応混合物を0℃の氷浴に浸し、水(4 mL)を加え10分間撹拌した後、イソプロピルエーテル(30 mL)を加え30分間撹拌した。白色沈澱を減圧ろ過し、真空乾燥させることで、目的化合物(3c)(0.63g,純度89.76%)を得た。以下に、化合物(3c)の機器分析値を示す。
化合物(3c):
1H-NMR (400 MHz, DMSO-d6) δ: 11.78(2H, br.s), 8.32(1H, s), 7.41-7.31(2H, m), 7.07-6.98(3H, m), 6.00(1H, d, J= 5.8 Hz), 5.37(1H, s), 5.18(1H, s), 4.88(2H, s), 4.85-4.78(2H, m), 4.72-4.59(3H, m), 4.34(1H, m), 4.00(1H, m), 3.75-3.56(3H, m), 2.79-2.69(2H, m).
N2-フェノキシアセチル-2’-O-(2-シアノエトキシメトキシメチル)グアノシン(3c)(630 mg, 1.2 mmol)をピリジンで共沸し、真空乾燥させた。ピリジン(6.0 mL)に溶解させ、0℃で4,4’-ジメトキシトリチルクロライド(600 mg, 1.8 mmol)を加えて室温にて2時間撹拌した。反応終了後、メタノールを加えて10分間撹拌し、減圧下溶媒留去した。残渣にジクロロメタンを加えて、飽和炭酸水素ナトリウム水溶液で2回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ジクロロメタン:酢酸エチル=1:1、0.05%ピリジン含有)で精製し、目的化合物(3d)(800mg,純度99.0%,収率80%)を得た。以下に、化合物(3d)の機器分析値を示す。
化合物(3d):
1H-NMR (400 MHz, CDCl3) δ: 11.82(1H, s), 8.63(1H, s), 7.84(1H, s), 7.43-7.21(9H, m), 6.86-6.82(4H, m), 6.06(1H, d, J= 5.9 Hz), 4.95(1H, t, J=5.7Hz), 4.78(2H, m), 4.67-4.63(2H, m), 4.50-4.45(1H, m), 4.30-4.26(1H, m), 3.81(6H, s), 3.79-3.67(2H, m), 3.44(2H, dd, J= 10.6, 3.7 Hz), 2.91(1H, s), 2.64-2.56(2H, m), 1.66(3H, s).
N2-フェノキシアセチル-5’-O-(4,4’-ジメトキシトリチル)-2’-O-(2-シアノエトキシメトキシメチル)グアノシン(3d)(800 mg, 0.96 mmol)をアセトニトリルで共沸し、真空乾燥させた。この操作を2回行った。そうして得られた混合物に、アルゴン雰囲気下、ジイソプロピルアンモニウムテトラゾリド(180 mg, 1.1 mmol)とアセトニトリル(3 mL)を加え、さらに2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.61 mL, 1.9 mmol)を加え、室温にて18時間撹拌した。反応終了後、減圧下溶媒留去し、酢酸エチルを加えて飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液でそれぞれ1回ずつ洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:10、0.1%トリエチルアミン含有)で精製し、目的化合物(I-3)(803mg,純度98.3%,収率81%)を得た。以下に、化合物(I-3)の機器分析値を示す。
化合物(I-3):
31P-NMR (162 MHz, CDCl3) δ: 152.7, 152.6.
下記スキームに従い、アデノシンEMMアミダイト(I-4)を合成した。
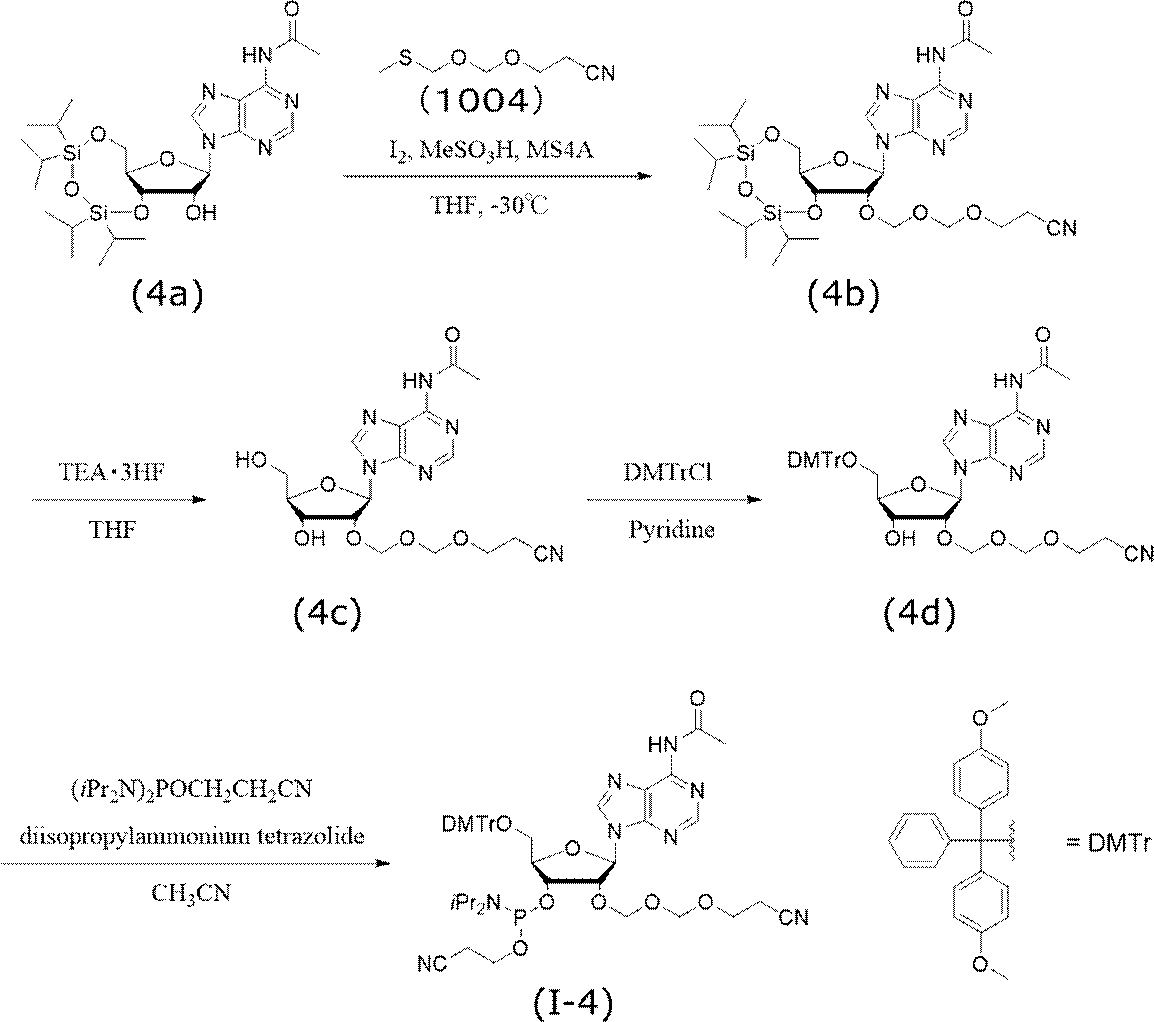
市販のN6-アセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)アデノシン(4a)(1.0 g, 1.8 mmol)をトルエンにて2回、テトラヒドロフランにて1回共沸し、真空乾燥させた。アルゴン雰囲気下、テトラヒドロフラン(1.8 mL)に溶解させ、モレキュラーシーブス4A(1.0 g)を加えて、-30℃で10分間撹拌した。-30℃にてメタンスルホン酸(0.26 g, 2.7 mmol)を加えて10分間撹拌した後、2-シアノエトキシメチルメチルチオメチルエーテル(1004)(580 mg, 3.6 mmol)を加えて10分間撹拌したのち、ヨウ素(2.76 g, 10.9 mmol)を加えて、-30℃にて30分間撹拌した。反応終了後、冷却した飽和チオ硫酸ナトリウム水溶液と飽和炭酸水素ナトリウム水溶液の混合溶液を反応溶液に加え、茶褐色が消失するまで激しく撹拌した。酢酸エチルにて抽出し、有機層を飽和チオ硫酸ナトリウム水溶液で2回、飽和炭酸水素ナトリウム水溶液で1回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、目的化合物(4b)の粗生成物(1.32g,純度83.8%)を得た。以下に、化合物(4b)の機器分析値を示す。
化合物(4b):
1H-NMR (400 MHz, CDCl3) δ: 8.68(1H, s), 8.66(1H, s), 8.33(1H, s), 6.12(1H, s), 5.08(1H, d, J= 7.0 Hz), 4.91-4.80(3H, m), 4.67(1H, d, J= 7.8 Hz), 4.52(1H, d, J= 4.3 Hz), 4.25(1H, d, J= 13.0 Hz), 4.17(1H, d, J= 9.4 Hz), 4.09-4.02(2H, m), 3.89-3.80(1H, m), 2.67(2H, m), 2.63(3H, s), 1.11-0.98(28H, m).
N6-アセチル-3’,5’-O-(テトライソプロピルジシロキサン-1,3-ジイル)-2’-O-(2-シアノエトキシメトキシメチル)アデノシン(4b)(1.32 g, 2.0 mmol)をアルゴン雰囲気下テトラヒドロフラン(6.0 mL)に溶解させた。その溶液に、トリエチルアミン三フッ化水素(0.39 mL, 2.4 mmol)を加えて室温にて2時間撹拌した。ジイソプロピルエーテルを加えトリチュレーションし、デカンテーションにより溶媒を除去した。この操作を3回行った。得られた黄色固体~シロップ状物質を真空乾燥し、目的化合物(4c)(1.07 g)を得た。以下に、化合物(4c)の機器分析値を示す。
化合物(4c):
1H-NMR (400 MHz, DMSO-d6) δ: 10.71(1H, s), 8.71(1H, s), 8.66(1H, s), 6.17(1H, d, J= 5.8 Hz), 5.41(1H, d, J= 5.4 Hz), 5.20(2H, m), 4.80-4.73(3H, m), 4.65-4.60(2H, m), 4.37-4.33(1H, m), 4.00-4.01(1H, m), 3.73-3.64(1H, m), 3.61-3.51(2H, m), 2.79-2.64(2H, m), 2.22(3H, s).
N6-アセチル-2’-O-(2-シアノエトキシメトキシメチル)アデノシン(4c)(1.07 g, 2.5 mmol)をピリジンで共沸し、真空乾燥させた。ピリジン(10 mL)に溶解させ、0℃で4,4’-ジメトキシトリチルクロライド(1.2 g, 3.5 mmol)を加え、室温にて3時間撹拌した。反応終了後、メタノールを加えて10分間撹拌し、減圧下溶媒留去した。残渣に酢酸エチルを加えて、飽和炭酸水素ナトリウム水溶液で2回、飽和塩化ナトリウム水溶液で1回洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル、0.05%ピリジン含有)で精製し、目的化合物(4d)(670mg,純度99.6%)を得た。以下に、化合物(4d)の機器分析値を示す。
化合物(4d):
1H-NMR (400 MHz, CDCl3) δ: 8.62-8.58(2H, m), 8.17(1H, s), 7.46-7.39(2H, m), 7.37-7.20(7H, m), 6.87-6.79(4H, m), 6.20(1H, d, J= 4.9 Hz), 5.03-4.75(3H, m), 4.52(1H, s), 4.30-4.23(1H, m), 4.12(2H, d, J= 7.3 Hz), 3.79(6H, s), 3.79-3.69(2H, m), 3.52-3.44(2H, m), 2.61(3H, s), 2.58(1H, d, J= 5.5 Hz), 2.51(2H, t, J= 5.9 Hz).
N6-アセチル-5’-O-(4,4’-ジメトキシトリチル)-2’-O-(2-シアノエトキシメトキシメチル)アデノシン(4d)(670 mg, 0.9 mmol)をアセトニトリルで共沸し、真空乾燥させた。この操作を2回行った。アルゴン雰囲気下、ジイソプロピルアンモニウムテトラゾリド(170 mg, 1.0 mmol)とアセトニトリル(5.5 mL)を加え、さらに2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト(0.32mL, 1.0 mmol)を加え、40℃で3時間撹拌した。反応終了後、減圧下溶媒留去し、酢酸エチルを加えて飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液でそれぞれ1回ずつ洗浄した。洗浄した有機層を無水硫酸ナトリウムにて乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル:アセトン=10:1、0.1%トリエチルアミン含有)で精製し、目的化合物(I-4)(700mg,純度98.3%,収率82%)を得た。以下に、化合物(I-4)の機器分析値を示す。
化合物(I-4):
31P-NMR (162 MHz, CDCl3) δ: 152.7, 152.6.
Claims (23)
- 一般式(103):

(式中、mは正の整数であり;R及びR’は同一又は異なってアルキル基、アルケニル基、アルキニル基、アリール基、アラルキル基、シクロアルキル基、シクロアルケニル基、シクロアルキルアルキル基、シクロアルケニルアルキル基またはアルコキシアルキル基を示す)
で表されるチオエーテル化合物と、一般式(105):
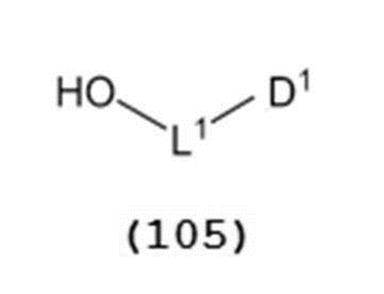
(式中、L1はアルキレン基を示し;D1は電子求引基を示す)
で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):
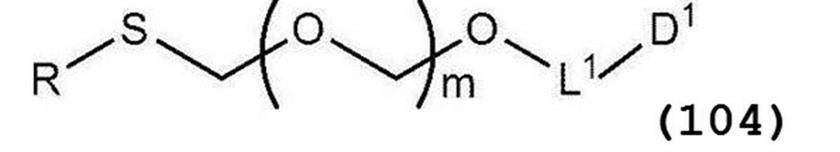
(式中、各記号の定義は上述の通りである)
で表されるチオエーテル化合物を得る工程(工程0);および
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):
[式中、Bは核酸塩基骨格を有する原子団を示し;
Xは下記式:
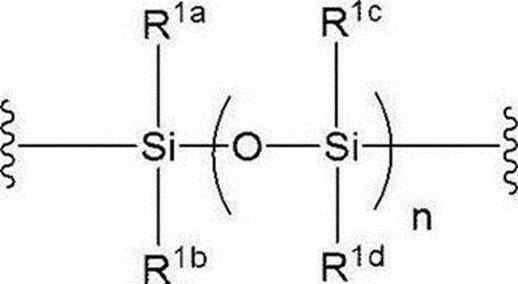
(式中、nは0又は1を示し;R1a~R1dは同一又は異なって水素原子、アルキル基又はアルコキシ基を示す)で表される基を示す]
で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):
(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程1)を含む、一般式(I):

(式中、R1は水酸基の保護基を示し;R2aおよびR2bは同一又は異なって水素原子若しくは置換基を示し;R2cは水素原子、電子求引基又は電子求引基で置換されていてもよい置換基を示し;ここでR2aおよびR2bはそれらが結合する窒素原子と一緒になって環を形成してもよく;それ以外の各記号の定義は上述の通りである)で表される配糖体化合物又はその塩の製造方法。 - さらに一般式(Ib)で表される化合物を脱保護して一般式(Ic):
(式中、各記号の定義は請求項1に記載の通りである)
で表される配糖体化合物を得る工程(工程2)を含む、請求項1記載の製造方法。 - さらに一般式(Ic)で表される配糖体化合物に水酸基の保護基を導入して一般式(Id):

(式中、各記号の定義は請求項1に記載の通りである)
で表される配糖体化合物を得る工程(工程3)を含む、請求項2記載の製造方法。 - さらに一般式(Id)で表される配糖体化合物をリン酸化して一般式(I)で表される配糖体化合物を得る工程(工程4)を含む、請求項3記載の製造方法。
- 一般式(103):

(式中、mは正の整数であり;R及びR’は同一又は異なってアルキル基、アルケニル基、アルキニル基、アリール基、アラルキル基、シクロアルキル基、シクロアルケニル基、シクロアルキルアルキル基、シクロアルケニルアルキル基またはアルコキシアルキル基を示す)
で表されるチオエーテル化合物と、一般式(105):
(式中、L1はアルキレン基を示し;D1は電子求引基を示す)
で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):

(式中、各記号の定義は上述の通りである)
で表されるチオエーテル化合物を得る工程(工程0);および
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

[式中、Bは核酸塩基骨格を有する原子団を示し;
Xは下記式:

(式中、nは0又は1を示し;R1a~R1dは同一又は異なって水素原子、アルキル基又はアルコキシ基を示す)で表される基を示す]
で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程1)を含む、一般式(Ib)で表される配糖体化合物の製造方法。 - 一般式(103):

(式中、mは正の整数であり;R及びR’は同一又は異なってアルキル基、アルケニル基、アルキニル基、アリール基、アラルキル基、シクロアルキル基、シクロアルケニル基、シクロアルキルアルキル基、シクロアルケニルアルキル基またはアルコキシアルキル基を示す)
で表されるチオエーテル化合物と、一般式(105):

(式中、L1はアルキレン基を示し;D1は電子求引基を示す)
で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):

(式中、各記号の定義は上述の通りである)
で表されるチオエーテル化合物を得る工程(工程0);
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

[式中、Bは核酸塩基骨格を有する原子団を示し;
Xは下記式:
(式中、nは0又は1を示し;R1a~R1dは同一又は異なって水素原子、アルキル基又はアルコキシ基を示す)で表される基を示す]
で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程1);および
一般式(Ib)で表される配糖体化合物を脱保護して一般式(Ic):

(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程2)を含む、一般式(Ic)で表される配糖体化合物の製造方法。 - 一般式(103):

(式中、mは正の整数であり;R及びR’は同一又は異なってアルキル基、アルケニル基、アルキニル基、アリール基、アラルキル基、シクロアルキル基、シクロアルケニル基、シクロアルキルアルキル基、シクロアルケニルアルキル基またはアルコキシアルキル基を示す)
で表されるチオエーテル化合物と、一般式(105):

(式中、L1はアルキレン基を示し;D1は電子求引基を示す)
で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):
(式中、各記号の定義は上述の通りである)
で表されるチオエーテル化合物を得る工程(工程0);
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

[式中、Bは核酸塩基骨格を有する原子団を示し;
Xは下記式:

(式中、nは0又は1を示し;R1a~R1dは同一又は異なって水素原子、アルキル基又はアルコキシ基を示す)で表される基を示す]
で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):
(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程1);
一般式(Ib)で表される配糖体化合物を脱保護して一般式(Ic):
(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程2);および
一般式(Ic)で表される配糖体化合物に水酸基の保護基を導入して一般式(Id):
(式中、R1は水酸基の保護基を示し;それ以外の各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程3)を含む、一般式(Id)で表される配糖体化合物の製造方法。 - 一般式(103):

(式中、mは正の整数であり;R及びR’は同一又は異なってアルキル基、アルケニル基、アルキニル基、アリール基、アラルキル基、シクロアルキル基、シクロアルケニル基、シクロアルキルアルキル基、シクロアルケニルアルキル基またはアルコキシアルキル基を示す)
で表されるチオエーテル化合物と、一般式(105):
(式中、L1はアルキレン基を示し;D1は電子求引基を示す)
で表されるアルコール化合物とを、ハロゲン化剤、乾燥剤及びルイス酸の存在下でカップリング反応させ、次いで硫黄含有抗酸化剤及びマレイミド基含有化合物から選択される少なくとも1種の添加剤の存在下に蒸留して一般式(104):

(式中、各記号の定義は上述の通りである)
で表されるチオエーテル化合物を得る工程(工程0);
ハロゲン化剤、乾燥剤及びルイス酸の存在下、
一般式(Ia):

[式中、Bは核酸塩基骨格を有する原子団を示し;
Xは下記式:

(式中、nは0又は1を示し;R1a~R1dは同一又は異なって水素原子、アルキル基又はアルコキシ基を示す)で表される基を示す]
で表される配糖体化合物と一般式(104)で表されるチオエーテル化合物をカップリング反応させて一般式(Ib):

(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程1);
一般式(Ib)で表される配糖体化合物を脱保護して一般式(Ic):
(式中、各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程2);
一般式(Ic)で表される配糖体化合物に水酸基の保護基を導入して一般式(Id):
(式中、R1は水酸基の保護基を示し;それ以外の各記号の定義は上述の通りである)
で表される配糖体化合物を得る工程(工程3);および
一般式(Id)で表される配糖体化合物をリン酸化して一般式(I):

(式中、R1は水酸基の保護基を示し;R2aおよびR2bは同一又は異なって水素原子若しくは置換基を示し;R2cは水素原子、電子求引基又は電子求引基で置換されていてもよい置換基を示し;ここでR2aおよびR2bはそれらが結合する窒素原子と一緒になって環を形成してもよく;それ以外の各記号の定義は上述の通りである)で表される配糖体化合物を得る工程(工程4)を含む、一般式(I)で表される配糖体化合物の製造方法。 - 工程0の蒸留における該添加剤が、4,4’-ビスマレイミドジフェニルメタンである、請求項1~8のいずれか1項に記載の方法。
- nが1である、請求項1~9のいずれか1項に記載の方法。
- R1a~R1dがイソプロピル基である、請求項1~10のいずれか1項に記載の方法。
- mが1である、請求項1~11のいずれか1項に記載の方法。
- L1がエチレン基である、請求項1~12のいずれか1項に記載の方法。
- D1がシアノ基である、請求項1~13のいずれか1項に記載の方法。
- 工程1において、ハロゲン化剤、乾燥剤およびルイス酸の添加後に一般式(104)で表されるチオエーテル化合物を添加することを特徴とする、請求項1~14のいずれか1項に記載の方法。
- 工程1において、乾燥剤、ルイス酸および一般式(104)で表されるチオエーテル化合物の添加後にハロゲン化剤を添加することを特徴とする、請求項1~14のいずれか1項に記載の方法。
- 工程1において、ハロゲン化剤の添加後にルイス酸を添加することを特徴とする、請求項15に記載の方法。
- 工程1において、ルイス酸の添加後に一般式(104)で表されるチオエーテル化合物を添加することを特徴とする、請求項16に記載の方法。
- 工程1におけるハロゲン化剤がヨウ素化剤である、請求項1~18のいずれか1項に記載の方法。
- ヨウ素化剤が1,3-ジヨード-5,5-ジメチルヒダントイン又はヨウ素である、請求項19に記載の方法。
- 乾燥剤がモレキュラーシーブである、請求項1~20のいずれか1項に記載の方法。
- 工程1におけるルイス酸がトリフルオロメタンスルホン酸である、請求項1~21のいずれか1項に記載の方法。
- 工程1におけるルイス酸がメタンスルホン酸である、請求項1~21のいずれか1項に記載の方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017510266A JP6481022B2 (ja) | 2015-04-02 | 2016-04-01 | 配糖体化合物の製造方法 |
| US15/563,841 US10377788B2 (en) | 2015-04-02 | 2016-04-01 | Method for producing glycoside compounds |
| EP16773254.4A EP3301103A4 (en) | 2015-04-02 | 2016-04-01 | METHOD FOR PRODUCING GLYCOSIDE COMPOUNDS |
| CN201680020364.3A CN107428793A (zh) | 2015-04-02 | 2016-04-01 | 配糖体化合物的制造方法 |
| KR1020177031029A KR20170129264A (ko) | 2015-04-02 | 2016-04-01 | 글리코시드 화합물의 제조 방법 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015076170 | 2015-04-02 | ||
| JP2015-076170 | 2015-04-02 | ||
| JP2015-122009 | 2015-06-17 | ||
| JP2015122009 | 2015-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016159374A1 true WO2016159374A1 (ja) | 2016-10-06 |
Family
ID=57004788
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/060971 Ceased WO2016159374A1 (ja) | 2015-04-02 | 2016-04-01 | 配糖体化合物の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10377788B2 (ja) |
| EP (1) | EP3301103A4 (ja) |
| JP (1) | JP6481022B2 (ja) |
| KR (1) | KR20170129264A (ja) |
| CN (1) | CN107428793A (ja) |
| WO (1) | WO2016159374A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019022257A1 (ja) | 2017-07-28 | 2019-01-31 | 杏林製薬株式会社 | 線維症治療剤 |
| WO2019189722A1 (ja) | 2018-03-30 | 2019-10-03 | 東レ株式会社 | ヘアピン型一本鎖rna分子の製造方法 |
| WO2020050411A1 (ja) * | 2018-09-07 | 2020-03-12 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| CN110947338A (zh) * | 2019-12-09 | 2020-04-03 | 南京科技职业学院 | 一种烷基糖苷磺基甜菜碱型两性表面活性剂及其制备方法 |
| WO2020152869A1 (ja) | 2019-01-25 | 2020-07-30 | 杏林製薬株式会社 | 線維症治療剤 |
| WO2021070507A1 (ja) | 2019-10-08 | 2021-04-15 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| WO2021079617A1 (ja) * | 2019-10-23 | 2021-04-29 | 住友化学株式会社 | 配糖体化合物、アミダイト化合物、およびこれら化合物を用いたポリヌクレオチドの製造方法 |
| WO2023097308A1 (en) | 2021-11-29 | 2023-06-01 | Hongene Biotech Corporation | Synthesis of 2' acetyl-ester protected nucleosides |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102899410B1 (ko) * | 2019-06-19 | 2025-12-12 | 야마사 쇼유 가부시키가이샤 | 가교형 뉴클레오시드 중간체의 결정 및 그의 제조 방법, 그리고 가교형 뉴클레오시드 아미다이트의 제조 방법 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4880508A (ja) * | 1972-01-14 | 1973-10-29 | ||
| JPH0680662A (ja) * | 1992-09-04 | 1994-03-22 | Sumitomo Chem Co Ltd | アルコキシメチルチオメチルシランとそれを用いるチオラン化合物の製造方法 |
| JPH07149779A (ja) * | 1993-08-17 | 1995-06-13 | Bristol Myers Squibb Co | タキサンのホスホノオキシメチルエーテル誘導体 |
| JP2001199936A (ja) * | 2000-01-19 | 2001-07-24 | Mitsubishi Chemicals Corp | ビス(メタ)アクリレート化合物の製造方法 |
| WO2013027843A1 (ja) * | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2930815A (en) * | 1959-01-27 | 1960-03-29 | Rohm & Haas | Preparation of thioethers |
| US5294637A (en) | 1992-07-01 | 1994-03-15 | Bristol-Myers Squibb Company | Fluoro taxols |
| US5478854A (en) | 1992-10-01 | 1995-12-26 | Bristol-Myers Squibb Company | Deoxy taxols |
| US5646176A (en) | 1992-12-24 | 1997-07-08 | Bristol-Myers Squibb Company | Phosphonooxymethyl ethers of taxane derivatives |
| JPWO2007097447A1 (ja) | 2006-02-27 | 2009-07-16 | 日本新薬株式会社 | 核酸保護基の脱離方法 |
| JP2008174524A (ja) | 2007-01-22 | 2008-07-31 | Nippon Shinyaku Co Ltd | リボ核酸化合物の製造方法 |
| WO2010079813A1 (ja) | 2009-01-07 | 2010-07-15 | 日本新薬株式会社 | イノシン誘導体の製造方法 |
-
2016
- 2016-04-01 US US15/563,841 patent/US10377788B2/en active Active
- 2016-04-01 EP EP16773254.4A patent/EP3301103A4/en not_active Withdrawn
- 2016-04-01 KR KR1020177031029A patent/KR20170129264A/ko not_active Withdrawn
- 2016-04-01 WO PCT/JP2016/060971 patent/WO2016159374A1/ja not_active Ceased
- 2016-04-01 JP JP2017510266A patent/JP6481022B2/ja not_active Expired - Fee Related
- 2016-04-01 CN CN201680020364.3A patent/CN107428793A/zh active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4880508A (ja) * | 1972-01-14 | 1973-10-29 | ||
| JPH0680662A (ja) * | 1992-09-04 | 1994-03-22 | Sumitomo Chem Co Ltd | アルコキシメチルチオメチルシランとそれを用いるチオラン化合物の製造方法 |
| JPH07149779A (ja) * | 1993-08-17 | 1995-06-13 | Bristol Myers Squibb Co | タキサンのホスホノオキシメチルエーテル誘導体 |
| JP2001199936A (ja) * | 2000-01-19 | 2001-07-24 | Mitsubishi Chemicals Corp | ビス(メタ)アクリレート化合物の製造方法 |
| WO2013027843A1 (ja) * | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP3301103A4 * |
| SERGEEV, V. A. ET AL.: "Reaction of dithiophenols with dicarboxylic acid bisimides, Vysokomolekulyarnye Soedineniya, Seriya B: Kratkie Soobshcheniya", CHEM. ABSTR., vol. 21, no. 7, 1979, pages 499 - 502, XP009510037 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11326163B2 (en) | 2017-07-28 | 2022-05-10 | Kyorin Pharmaceutical Co., Ltd. | Therapeutic agent for fibrosis |
| WO2019022257A1 (ja) | 2017-07-28 | 2019-01-31 | 杏林製薬株式会社 | 線維症治療剤 |
| KR20200136363A (ko) | 2018-03-30 | 2020-12-07 | 도레이 카부시키가이샤 | 헤어핀형 1개쇄 rna 분자의 제조 방법 |
| WO2019189722A1 (ja) | 2018-03-30 | 2019-10-03 | 東レ株式会社 | ヘアピン型一本鎖rna分子の製造方法 |
| US11920131B2 (en) | 2018-03-30 | 2024-03-05 | Toray Industries, Inc. | Method of producing hairpin single-stranded RNA molecule |
| JPWO2020050411A1 (ja) * | 2018-09-07 | 2021-08-30 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| US12173028B2 (en) | 2018-09-07 | 2024-12-24 | Sumitomo Chemical Company, Limited | Method for producing glycoside compound |
| WO2020050411A1 (ja) * | 2018-09-07 | 2020-03-12 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| JP7423533B2 (ja) | 2018-09-07 | 2024-01-29 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| WO2020152869A1 (ja) | 2019-01-25 | 2020-07-30 | 杏林製薬株式会社 | 線維症治療剤 |
| WO2021070507A1 (ja) | 2019-10-08 | 2021-04-15 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| KR20220079540A (ko) | 2019-10-08 | 2022-06-13 | 스미또모 가가꾸 가부시끼가이샤 | 배당체 화합물의 제조 방법 |
| JPWO2021070507A1 (ja) * | 2019-10-08 | 2021-04-15 | ||
| JP7522753B2 (ja) | 2019-10-08 | 2024-07-25 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| US12559512B2 (en) | 2019-10-08 | 2026-02-24 | Sumitomo Chemical Company, Limited | Method for producing glycoside compound |
| WO2021079617A1 (ja) * | 2019-10-23 | 2021-04-29 | 住友化学株式会社 | 配糖体化合物、アミダイト化合物、およびこれら化合物を用いたポリヌクレオチドの製造方法 |
| JPWO2021079617A1 (ja) * | 2019-10-23 | 2021-04-29 | ||
| JP7606972B2 (ja) | 2019-10-23 | 2024-12-26 | 住友化学株式会社 | 配糖体化合物、アミダイト化合物、およびこれら化合物を用いたポリヌクレオチドの製造方法 |
| US12509481B2 (en) | 2019-10-23 | 2025-12-30 | Sumitomo Chemical Company, Limited | Glycoside compound, amidite compound, and production method for polynucleotide using said compounds |
| CN110947338A (zh) * | 2019-12-09 | 2020-04-03 | 南京科技职业学院 | 一种烷基糖苷磺基甜菜碱型两性表面活性剂及其制备方法 |
| WO2023097308A1 (en) | 2021-11-29 | 2023-06-01 | Hongene Biotech Corporation | Synthesis of 2' acetyl-ester protected nucleosides |
| US11897914B2 (en) | 2021-11-29 | 2024-02-13 | Hongene Biotech Corporation | Synthesis of 2′ protected nucleosides |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2016159374A1 (ja) | 2017-11-16 |
| EP3301103A4 (en) | 2019-01-23 |
| JP6481022B2 (ja) | 2019-03-13 |
| CN107428793A (zh) | 2017-12-01 |
| US10377788B2 (en) | 2019-08-13 |
| KR20170129264A (ko) | 2017-11-24 |
| US20180079768A1 (en) | 2018-03-22 |
| EP3301103A1 (en) | 2018-04-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6481022B2 (ja) | 配糖体化合物の製造方法 | |
| JP5554881B2 (ja) | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 | |
| JPWO2017086397A1 (ja) | オリゴヌクレオチドの製造方法 | |
| WO2018070543A1 (ja) | 新規な配糖体化合物及びその製造方法 | |
| HK1229337B (en) | Thioether compound for the protection of the 2'-hydroxy group in nucleosides to be used in oligonucleotide synthesis | |
| HK1229337A (en) | Thioether compound for the protection of the 2'-hydroxy group in nucleosides to be used in oligonucleotide synthesis | |
| HK1229337A1 (en) | Thioether compound for the protection of the 2'-hydroxy group in nucleosides to be used in oligonucleotide synthesis | |
| HK1199456B (en) | Nucleoside phosphoramidates for producing nucleic acids | |
| BR112014004313B1 (pt) | Composto glicosídeo, e, métodos de produção de um composto glicosídeo e de um ácido nucleico | |
| HK40013293A (en) | Novel glycoside compound and production method therefor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: IDP00201607922 Country of ref document: ID |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16773254 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017510266 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15563841 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20177031029 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016773254 Country of ref document: EP |