WO2016010002A1 - タンパク質のエピトープを同定するための方法 - Google Patents
タンパク質のエピトープを同定するための方法 Download PDFInfo
- Publication number
- WO2016010002A1 WO2016010002A1 PCT/JP2015/070072 JP2015070072W WO2016010002A1 WO 2016010002 A1 WO2016010002 A1 WO 2016010002A1 JP 2015070072 W JP2015070072 W JP 2015070072W WO 2016010002 A1 WO2016010002 A1 WO 2016010002A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- protein
- peptide
- dendritic
- Prior art date
Links
- 108090000623 proteins and genes Proteins 0.000 title claims abstract description 188
- 102000004169 proteins and genes Human genes 0.000 title claims abstract description 167
- 238000000034 method Methods 0.000 title claims abstract description 159
- 210000004027 cell Anatomy 0.000 claims abstract description 387
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 187
- 210000000130 stem cell Anatomy 0.000 claims abstract description 115
- 108700018351 Major Histocompatibility Complex Proteins 0.000 claims abstract description 6
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 claims abstract description 6
- 210000004443 dendritic cell Anatomy 0.000 claims description 141
- 230000005847 immunogenicity Effects 0.000 claims description 48
- 210000001616 monocyte Anatomy 0.000 claims description 47
- 239000000203 mixture Substances 0.000 claims description 29
- 108010058597 HLA-DR Antigens Proteins 0.000 claims description 27
- 102000006354 HLA-DR Antigens Human genes 0.000 claims description 27
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 claims description 27
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 claims description 27
- -1 CD86 Proteins 0.000 claims description 26
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 claims description 25
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 claims description 24
- 230000002163 immunogen Effects 0.000 claims description 24
- 239000012679 serum free medium Substances 0.000 claims description 22
- 238000004519 manufacturing process Methods 0.000 claims description 21
- 210000002966 serum Anatomy 0.000 claims description 21
- 102000004127 Cytokines Human genes 0.000 claims description 20
- 108090000695 Cytokines Proteins 0.000 claims description 20
- 102000004388 Interleukin-4 Human genes 0.000 claims description 20
- 108090000978 Interleukin-4 Proteins 0.000 claims description 20
- 229940028885 interleukin-4 Drugs 0.000 claims description 20
- 108010062347 HLA-DQ Antigens Proteins 0.000 claims description 18
- 230000008569 process Effects 0.000 claims description 15
- 108010010378 HLA-DP Antigens Proteins 0.000 claims description 14
- 102000015789 HLA-DP Antigens Human genes 0.000 claims description 14
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 claims description 13
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 claims description 13
- 210000003716 mesoderm Anatomy 0.000 claims description 13
- 201000010099 disease Diseases 0.000 claims description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 12
- 210000003714 granulocyte Anatomy 0.000 claims description 12
- 230000002829 reductive effect Effects 0.000 claims description 12
- 239000003102 growth factor Substances 0.000 claims description 10
- 230000002757 inflammatory effect Effects 0.000 claims description 9
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims description 7
- 230000001939 inductive effect Effects 0.000 claims description 7
- 210000004504 adult stem cell Anatomy 0.000 claims description 6
- 230000004936 stimulating effect Effects 0.000 claims description 6
- 239000004480 active ingredient Substances 0.000 claims description 5
- 210000001671 embryonic stem cell Anatomy 0.000 claims description 5
- 229940088597 hormone Drugs 0.000 claims description 5
- 239000005556 hormone Substances 0.000 claims description 5
- 238000012546 transfer Methods 0.000 claims description 5
- 102000004190 Enzymes Human genes 0.000 claims description 4
- 108090000790 Enzymes Proteins 0.000 claims description 4
- 102000019034 Chemokines Human genes 0.000 claims description 3
- 108010012236 Chemokines Proteins 0.000 claims description 3
- 101710172711 Structural protein Proteins 0.000 claims description 3
- 239000012634 fragment Substances 0.000 claims description 3
- 102000004196 processed proteins & peptides Human genes 0.000 abstract description 54
- 239000002243 precursor Substances 0.000 abstract 1
- 235000018102 proteins Nutrition 0.000 description 115
- 150000001413 amino acids Chemical group 0.000 description 64
- 238000004458 analytical method Methods 0.000 description 53
- 210000000612 antigen-presenting cell Anatomy 0.000 description 46
- 239000002609 medium Substances 0.000 description 37
- 230000014509 gene expression Effects 0.000 description 33
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 32
- 239000000427 antigen Substances 0.000 description 24
- 210000002443 helper t lymphocyte Anatomy 0.000 description 24
- 108091007433 antigens Proteins 0.000 description 22
- 102000036639 antigens Human genes 0.000 description 22
- 230000004069 differentiation Effects 0.000 description 19
- 229960000598 infliximab Drugs 0.000 description 18
- 239000011324 bead Substances 0.000 description 16
- 230000003394 haemopoietic effect Effects 0.000 description 14
- 210000002540 macrophage Anatomy 0.000 description 14
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 13
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 13
- 102100022338 Integrin alpha-M Human genes 0.000 description 13
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 13
- 210000001082 somatic cell Anatomy 0.000 description 13
- 108010010803 Gelatin Proteins 0.000 description 12
- 102100022297 Integrin alpha-X Human genes 0.000 description 12
- 229920000159 gelatin Polymers 0.000 description 12
- 239000008273 gelatin Substances 0.000 description 12
- 235000019322 gelatine Nutrition 0.000 description 12
- 235000011852 gelatine desserts Nutrition 0.000 description 12
- 230000006698 induction Effects 0.000 description 12
- 239000006228 supernatant Substances 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 11
- 241000282412 Homo Species 0.000 description 11
- 206010028980 Neoplasm Diseases 0.000 description 11
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 11
- 102100039037 Vascular endothelial growth factor A Human genes 0.000 description 11
- 201000011510 cancer Diseases 0.000 description 11
- 239000012091 fetal bovine serum Substances 0.000 description 11
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 10
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 10
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 10
- 229960000074 biopharmaceutical Drugs 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 201000006417 multiple sclerosis Diseases 0.000 description 10
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 9
- 238000012258 culturing Methods 0.000 description 9
- 230000008672 reprogramming Effects 0.000 description 9
- 241000124008 Mammalia Species 0.000 description 8
- 238000012300 Sequence Analysis Methods 0.000 description 8
- 210000001744 T-lymphocyte Anatomy 0.000 description 8
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 8
- 230000030741 antigen processing and presentation Effects 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 239000003550 marker Substances 0.000 description 8
- 229940024606 amino acid Drugs 0.000 description 7
- 235000001014 amino acid Nutrition 0.000 description 7
- 230000002265 prevention Effects 0.000 description 7
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 6
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 6
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 6
- 108010033276 Peptide Fragments Proteins 0.000 description 6
- 102000007079 Peptide Fragments Human genes 0.000 description 6
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 6
- 230000001464 adherent effect Effects 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- 210000002257 embryonic structure Anatomy 0.000 description 6
- 239000002158 endotoxin Substances 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 229920006008 lipopolysaccharide Polymers 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 239000012498 ultrapure water Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 5
- 208000023275 Autoimmune disease Diseases 0.000 description 5
- 102100024505 Bone morphogenetic protein 4 Human genes 0.000 description 5
- 108010074328 Interferon-gamma Proteins 0.000 description 5
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 5
- 229930182555 Penicillin Natural products 0.000 description 5
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 210000003719 b-lymphocyte Anatomy 0.000 description 5
- 230000000875 corresponding effect Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 235000013601 eggs Nutrition 0.000 description 5
- 238000010828 elution Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 229940049954 penicillin Drugs 0.000 description 5
- 210000001778 pluripotent stem cell Anatomy 0.000 description 5
- 229960005322 streptomycin Drugs 0.000 description 5
- 229910021642 ultra pure water Inorganic materials 0.000 description 5
- 238000000108 ultra-filtration Methods 0.000 description 5
- 239000013598 vector Substances 0.000 description 5
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 4
- 108020004414 DNA Proteins 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 208000009292 Hemophilia A Diseases 0.000 description 4
- 101000762379 Homo sapiens Bone morphogenetic protein 4 Proteins 0.000 description 4
- 102100037850 Interferon gamma Human genes 0.000 description 4
- 108010002386 Interleukin-3 Proteins 0.000 description 4
- 102000000646 Interleukin-3 Human genes 0.000 description 4
- 229930182816 L-glutamine Natural products 0.000 description 4
- 102000004058 Leukemia inhibitory factor Human genes 0.000 description 4
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 4
- 101150042431 SOX gene Proteins 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- 230000006044 T cell activation Effects 0.000 description 4
- 102000036693 Thrombopoietin Human genes 0.000 description 4
- 108010041111 Thrombopoietin Proteins 0.000 description 4
- 125000000539 amino acid group Chemical group 0.000 description 4
- 210000004102 animal cell Anatomy 0.000 description 4
- 210000002459 blastocyst Anatomy 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000006166 lysate Substances 0.000 description 4
- 210000001161 mammalian embryo Anatomy 0.000 description 4
- 238000004949 mass spectrometry Methods 0.000 description 4
- 206010028417 myasthenia gravis Diseases 0.000 description 4
- 210000004940 nucleus Anatomy 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 4
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 3
- 108090000145 Bacillolysin Proteins 0.000 description 3
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 3
- 235000009109 Betula pendula Nutrition 0.000 description 3
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 3
- 101150013553 CD40 gene Proteins 0.000 description 3
- 102100035793 CD83 antigen Human genes 0.000 description 3
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 3
- 102100026735 Coagulation factor VIII Human genes 0.000 description 3
- 206010009944 Colon cancer Diseases 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 101000968532 Dictyocaulus viviparus DVA-1 polyprotein Proteins 0.000 description 3
- 201000003542 Factor VIII deficiency Diseases 0.000 description 3
- 102100020715 Fms-related tyrosine kinase 3 ligand protein Human genes 0.000 description 3
- 101710162577 Fms-related tyrosine kinase 3 ligand protein Proteins 0.000 description 3
- 102100028976 HLA class I histocompatibility antigen, B alpha chain Human genes 0.000 description 3
- 108010058607 HLA-B Antigens Proteins 0.000 description 3
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 3
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 3
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 3
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 description 3
- 102000015696 Interleukins Human genes 0.000 description 3
- 108010063738 Interleukins Proteins 0.000 description 3
- 210000004322 M2 macrophage Anatomy 0.000 description 3
- 102000035092 Neutral proteases Human genes 0.000 description 3
- 108091005507 Neutral proteases Proteins 0.000 description 3
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 3
- 102100035194 Placenta growth factor Human genes 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229920002684 Sepharose Polymers 0.000 description 3
- 108091008874 T cell receptors Proteins 0.000 description 3
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 3
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 3
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 3
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 3
- 229940125644 antibody drug Drugs 0.000 description 3
- 230000000890 antigenic effect Effects 0.000 description 3
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 3
- 239000013575 birch pollen allergen Substances 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000013592 cell lysate Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 210000003690 classically activated macrophage Anatomy 0.000 description 3
- 238000005345 coagulation Methods 0.000 description 3
- 230000015271 coagulation Effects 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 210000002950 fibroblast Anatomy 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 210000004602 germ cell Anatomy 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 102000057593 human F8 Human genes 0.000 description 3
- 238000001114 immunoprecipitation Methods 0.000 description 3
- 238000000126 in silico method Methods 0.000 description 3
- 229940047122 interleukins Drugs 0.000 description 3
- 238000000534 ion trap mass spectrometry Methods 0.000 description 3
- 239000012139 lysis buffer Substances 0.000 description 3
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 3
- 229960004857 mitomycin Drugs 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 210000004681 ovum Anatomy 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 229940104230 thymidine Drugs 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 2
- 102100035248 Alpha-(1,3)-fucosyltransferase 4 Human genes 0.000 description 2
- 208000028185 Angioedema Diseases 0.000 description 2
- 101710145634 Antigen 1 Proteins 0.000 description 2
- 241000219430 Betula pendula Species 0.000 description 2
- 108010049955 Bone Morphogenetic Protein 4 Proteins 0.000 description 2
- 210000001239 CD8-positive, alpha-beta cytotoxic T lymphocyte Anatomy 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 102000003951 Erythropoietin Human genes 0.000 description 2
- 108090000394 Erythropoietin Proteins 0.000 description 2
- 108010011459 Exenatide Proteins 0.000 description 2
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 2
- 102100038000 F-box only protein 15 Human genes 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical group NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 102100028972 HLA class I histocompatibility antigen, A alpha chain Human genes 0.000 description 2
- 102100028971 HLA class I histocompatibility antigen, C alpha chain Human genes 0.000 description 2
- 108010075704 HLA-A Antigens Proteins 0.000 description 2
- 108010052199 HLA-C Antigens Proteins 0.000 description 2
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 2
- 102000003964 Histone deacetylase Human genes 0.000 description 2
- 108090000353 Histone deacetylase Proteins 0.000 description 2
- 101001022185 Homo sapiens Alpha-(1,3)-fucosyltransferase 4 Proteins 0.000 description 2
- 101000878635 Homo sapiens F-box only protein 15 Proteins 0.000 description 2
- 101000746373 Homo sapiens Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 2
- 101001002709 Homo sapiens Interleukin-4 Proteins 0.000 description 2
- 101001094700 Homo sapiens POU domain, class 5, transcription factor 1 Proteins 0.000 description 2
- 101000713275 Homo sapiens Solute carrier family 22 member 3 Proteins 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 102000013462 Interleukin-12 Human genes 0.000 description 2
- 108010065805 Interleukin-12 Proteins 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- 102000004889 Interleukin-6 Human genes 0.000 description 2
- 101150039798 MYC gene Proteins 0.000 description 2
- 101150076359 Mhc gene Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 241000746983 Phleum pratense Species 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 238000010847 SEQUEST Methods 0.000 description 2
- 241000235070 Saccharomyces Species 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 206010043276 Teratoma Diseases 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 206010052779 Transplant rejections Diseases 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- 208000006682 alpha 1-Antitrypsin Deficiency Diseases 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 2
- 238000010835 comparative analysis Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 238000012136 culture method Methods 0.000 description 2
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical group BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 229940105423 erythropoietin Drugs 0.000 description 2
- 239000003797 essential amino acid Substances 0.000 description 2
- 235000020776 essential amino acid Nutrition 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 230000004720 fertilization Effects 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 229960002743 glutamine Drugs 0.000 description 2
- 102000055229 human IL4 Human genes 0.000 description 2
- 229960000900 human factor viii Drugs 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 230000016784 immunoglobulin production Effects 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- IZWSFJTYBVKZNK-UHFFFAOYSA-N lauryl sulfobetaine Chemical compound CCCCCCCCCCCC[N+](C)(C)CCCS([O-])(=O)=O IZWSFJTYBVKZNK-UHFFFAOYSA-N 0.000 description 2
- 238000001638 lipofection Methods 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 108010082117 matrigel Proteins 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 2
- 238000003752 polymerase chain reaction Methods 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 210000001236 prokaryotic cell Anatomy 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 229950010131 puromycin Drugs 0.000 description 2
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 238000004885 tandem mass spectrometry Methods 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- 241000712461 unidentified influenza virus Species 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- MRXDGVXSWIXTQL-HYHFHBMOSA-N (2s)-2-[[(1s)-1-(2-amino-1,4,5,6-tetrahydropyrimidin-6-yl)-2-[[(2s)-4-methyl-1-oxo-1-[[(2s)-1-oxo-3-phenylpropan-2-yl]amino]pentan-2-yl]amino]-2-oxoethyl]carbamoylamino]-3-phenylpropanoic acid Chemical compound C([C@H](NC(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C=O)C1NC(N)=NCC1)C(O)=O)C1=CC=CC=C1 MRXDGVXSWIXTQL-HYHFHBMOSA-N 0.000 description 1
- LOGFVTREOLYCPF-KXNHARMFSA-N (2s,3r)-2-[[(2r)-1-[(2s)-2,6-diaminohexanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxybutanoic acid Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)[C@H]1CCCN1C(=O)[C@@H](N)CCCCN LOGFVTREOLYCPF-KXNHARMFSA-N 0.000 description 1
- HNSDLXPSAYFUHK-UHFFFAOYSA-N 1,4-bis(2-ethylhexyl) sulfosuccinate Chemical group CCCCC(CC)COC(=O)CC(S(O)(=O)=O)C(=O)OCC(CC)CCCC HNSDLXPSAYFUHK-UHFFFAOYSA-N 0.000 description 1
- KSXTUUUQYQYKCR-LQDDAWAPSA-M 2,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC KSXTUUUQYQYKCR-LQDDAWAPSA-M 0.000 description 1
- FADQCEBBTITJBI-UHFFFAOYSA-N 2-[(2-methoxyphenyl)methoxymethyl]oxirane Chemical compound COC1=CC=CC=C1COCC1OC1 FADQCEBBTITJBI-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- MJZJYWCQPMNPRM-UHFFFAOYSA-N 6,6-dimethyl-1-[3-(2,4,5-trichlorophenoxy)propoxy]-1,6-dihydro-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCCCOC1=CC(Cl)=C(Cl)C=C1Cl MJZJYWCQPMNPRM-UHFFFAOYSA-N 0.000 description 1
- KKJUPNGICOCCDW-UHFFFAOYSA-N 7-N,N-Dimethylamino-1,2,3,4,5-pentathiocyclooctane Chemical compound CN(C)C1CSSSSSC1 KKJUPNGICOCCDW-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 241000228245 Aspergillus niger Species 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108091008875 B cell receptors Proteins 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- 108060000903 Beta-catenin Proteins 0.000 description 1
- 102000015735 Beta-catenin Human genes 0.000 description 1
- 244000274847 Betula papyrifera Species 0.000 description 1
- 235000009113 Betula papyrifera Nutrition 0.000 description 1
- 235000010928 Betula populifolia Nutrition 0.000 description 1
- 235000002992 Betula pubescens Nutrition 0.000 description 1
- 102100024506 Bone morphogenetic protein 2 Human genes 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- OLVPQBGMUGIKIW-UHFFFAOYSA-N Chymostatin Natural products C=1C=CC=CC=1CC(C=O)NC(=O)C(C(C)CC)NC(=O)C(C1NC(N)=NCC1)NC(=O)NC(C(O)=O)CC1=CC=CC=C1 OLVPQBGMUGIKIW-UHFFFAOYSA-N 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 206010013883 Dwarfism Diseases 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 102100028072 Fibroblast growth factor 4 Human genes 0.000 description 1
- 108010029961 Filgrastim Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 102100022871 GTPase ERas Human genes 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Chemical group 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 102100028970 HLA class I histocompatibility antigen, alpha chain E Human genes 0.000 description 1
- 102100028966 HLA class I histocompatibility antigen, alpha chain F Human genes 0.000 description 1
- 102100028967 HLA class I histocompatibility antigen, alpha chain G Human genes 0.000 description 1
- 108010037799 HLA-DQ1 antigen Proteins 0.000 description 1
- 108010024164 HLA-G Antigens Proteins 0.000 description 1
- 102100021888 Helix-loop-helix protein 1 Human genes 0.000 description 1
- 208000031220 Hemophilia Diseases 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 108010027412 Histocompatibility Antigens Class II Proteins 0.000 description 1
- 102000011787 Histone Methyltransferases Human genes 0.000 description 1
- 108010036115 Histone Methyltransferases Proteins 0.000 description 1
- 101000762366 Homo sapiens Bone morphogenetic protein 2 Proteins 0.000 description 1
- 101001060274 Homo sapiens Fibroblast growth factor 4 Proteins 0.000 description 1
- 101000620820 Homo sapiens GTPase ERas Proteins 0.000 description 1
- 101000986085 Homo sapiens HLA class I histocompatibility antigen, alpha chain E Proteins 0.000 description 1
- 101000986080 Homo sapiens HLA class I histocompatibility antigen, alpha chain F Proteins 0.000 description 1
- 101000897691 Homo sapiens Helix-loop-helix protein 1 Proteins 0.000 description 1
- 101000716729 Homo sapiens Kit ligand Proteins 0.000 description 1
- 101001139146 Homo sapiens Krueppel-like factor 2 Proteins 0.000 description 1
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 1
- 101001109685 Homo sapiens Nuclear receptor subfamily 5 group A member 2 Proteins 0.000 description 1
- 101001081555 Homo sapiens Plasma protease C1 inhibitor Proteins 0.000 description 1
- 101000652321 Homo sapiens Protein SOX-15 Proteins 0.000 description 1
- 101000984042 Homo sapiens Protein lin-28 homolog A Proteins 0.000 description 1
- 101000984033 Homo sapiens Protein lin-28 homolog B Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 101000740205 Homo sapiens Sal-like protein 1 Proteins 0.000 description 1
- 101000740178 Homo sapiens Sal-like protein 4 Proteins 0.000 description 1
- 101000851696 Homo sapiens Steroid hormone receptor ERR2 Proteins 0.000 description 1
- 101000666775 Homo sapiens T-box transcription factor TBX3 Proteins 0.000 description 1
- 101000652332 Homo sapiens Transcription factor SOX-1 Proteins 0.000 description 1
- 101000652324 Homo sapiens Transcription factor SOX-17 Proteins 0.000 description 1
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 1
- 101000687911 Homo sapiens Transcription factor SOX-3 Proteins 0.000 description 1
- 101000808011 Homo sapiens Vascular endothelial growth factor A Proteins 0.000 description 1
- 101000976622 Homo sapiens Zinc finger protein 42 homolog Proteins 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 108090000193 Interleukin-1 beta Proteins 0.000 description 1
- 102000003777 Interleukin-1 beta Human genes 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- 108010002616 Interleukin-5 Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 102100020675 Krueppel-like factor 2 Human genes 0.000 description 1
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical group OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 244000207740 Lemna minor Species 0.000 description 1
- 235000006439 Lemna minor Nutrition 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 108090000542 Lymphotoxin-alpha Proteins 0.000 description 1
- 102000004083 Lymphotoxin-alpha Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- 101000804949 Mus musculus Developmental pluripotency-associated protein 2 Proteins 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 101150012532 NANOG gene Proteins 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 102100022669 Nuclear receptor subfamily 5 group A member 2 Human genes 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 102100040681 Platelet-derived growth factor C Human genes 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- HCBIBCJNVBAKAB-UHFFFAOYSA-N Procaine hydrochloride Chemical compound Cl.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 HCBIBCJNVBAKAB-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Chemical group OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 1
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 1
- 102100030244 Protein SOX-15 Human genes 0.000 description 1
- 102100025460 Protein lin-28 homolog A Human genes 0.000 description 1
- 102100025459 Protein lin-28 homolog B Human genes 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 102100037204 Sal-like protein 1 Human genes 0.000 description 1
- 102100037192 Sal-like protein 4 Human genes 0.000 description 1
- 241001138501 Salmonella enterica Species 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 241000580858 Simian-Human immunodeficiency virus Species 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 102100036831 Steroid hormone receptor ERR2 Human genes 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102100038409 T-box transcription factor TBX3 Human genes 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GYDJEQRTZSCIOI-UHFFFAOYSA-N Tranexamic acid Chemical compound NCC1CCC(C(O)=O)CC1 GYDJEQRTZSCIOI-UHFFFAOYSA-N 0.000 description 1
- 102100030248 Transcription factor SOX-1 Human genes 0.000 description 1
- 102100030243 Transcription factor SOX-17 Human genes 0.000 description 1
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 1
- 102100024276 Transcription factor SOX-3 Human genes 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 108010073925 Vascular Endothelial Growth Factor B Proteins 0.000 description 1
- 108010073923 Vascular Endothelial Growth Factor C Proteins 0.000 description 1
- 108010073919 Vascular Endothelial Growth Factor D Proteins 0.000 description 1
- 102100038217 Vascular endothelial growth factor B Human genes 0.000 description 1
- 102100038232 Vascular endothelial growth factor C Human genes 0.000 description 1
- 102100038234 Vascular endothelial growth factor D Human genes 0.000 description 1
- 241000269370 Xenopus <genus> Species 0.000 description 1
- 102100023550 Zinc finger protein 42 homolog Human genes 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 229940060205 adagen Drugs 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229940031675 advate Drugs 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000013566 allergen Substances 0.000 description 1
- 229960004784 allergens Drugs 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 1
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 238000003277 amino acid sequence analysis Methods 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 238000005571 anion exchange chromatography Methods 0.000 description 1
- 230000014102 antigen processing and presentation of exogenous peptide antigen via MHC class I Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229940038528 aralast Drugs 0.000 description 1
- 229950005725 arcitumomab Drugs 0.000 description 1
- 210000004507 artificial chromosome Anatomy 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 229960004669 basiliximab Drugs 0.000 description 1
- 229940031422 benefix Drugs 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 210000001109 blastomere Anatomy 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- 229940084891 byetta Drugs 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229960001838 canakinumab Drugs 0.000 description 1
- 238000005277 cation exchange chromatography Methods 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 210000003855 cell nucleus Anatomy 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 230000007969 cellular immunity Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940049197 cerezyme Drugs 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 108010086192 chymostatin Proteins 0.000 description 1
- 229940088949 cinryze Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000001332 colony forming effect Effects 0.000 description 1
- 230000002281 colonystimulating effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 238000010219 correlation analysis Methods 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000003968 dna methyltransferase inhibitor Substances 0.000 description 1
- 108010067396 dornase alfa Proteins 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229960000284 efalizumab Drugs 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 239000012645 endogenous antigen Substances 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 229940058249 evithrom Drugs 0.000 description 1
- 229960001519 exenatide Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 108091006047 fluorescent proteins Proteins 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 239000005350 fused silica glass Substances 0.000 description 1
- 229960000578 gemtuzumab Drugs 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 238000012252 genetic analysis Methods 0.000 description 1
- 102000054766 genetic haplotypes Human genes 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229960001743 golimumab Drugs 0.000 description 1
- 229940046528 grass pollen Drugs 0.000 description 1
- 239000013574 grass pollen allergen Substances 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 102000046148 human BMP4 Human genes 0.000 description 1
- 102000054189 human CD80 Human genes 0.000 description 1
- 102000055151 human KITLG Human genes 0.000 description 1
- 102000044507 human SERPING1 Human genes 0.000 description 1
- 102000058223 human VEGFA Human genes 0.000 description 1
- 229940065770 humatrope Drugs 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 108010039650 imiglucerase Proteins 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 230000002998 immunogenetic effect Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229920000831 ionic polymer Polymers 0.000 description 1
- 230000005865 ionizing radiation Effects 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 101150072261 large T gene Proteins 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 229940087875 leukine Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 1
- 108010052968 leupeptin Proteins 0.000 description 1
- 101150111214 lin-28 gene Proteins 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 239000003697 methyltransferase inhibitor Substances 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 210000003003 monocyte-macrophage precursor cell Anatomy 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 210000000472 morula Anatomy 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 229940029345 neupogen Drugs 0.000 description 1
- 229940112216 novoseven Drugs 0.000 description 1
- HEGSGKPQLMEBJL-RKQHYHRCSA-N octyl beta-D-glucopyranoside Chemical compound CCCCCCCCO[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HEGSGKPQLMEBJL-RKQHYHRCSA-N 0.000 description 1
- 229960002450 ofatumumab Drugs 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 229960000402 palivizumab Drugs 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 230000008186 parthenogenesis Effects 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 108010091212 pepstatin Proteins 0.000 description 1
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical compound OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 108010017992 platelet-derived growth factor C Proteins 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 210000004986 primary T-cell Anatomy 0.000 description 1
- 229960001309 procaine hydrochloride Drugs 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 229940107568 pulmozyme Drugs 0.000 description 1
- 238000004451 qualitative analysis Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 239000000941 radioactive substance Substances 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 238000004725 rapid separation liquid chromatography Methods 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108010013773 recombinant FVIIa Proteins 0.000 description 1
- 108010025139 recombinant factor VIII SQ Proteins 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 229940013175 recothrom Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 229940116176 remicade Drugs 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000002629 repopulating effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 229940116243 retavase Drugs 0.000 description 1
- 108010051412 reteplase Proteins 0.000 description 1
- 229940053182 rhophylac Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 108010038379 sargramostim Proteins 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 229940082569 selenite Drugs 0.000 description 1
- MCAHWIHFGHIESP-UHFFFAOYSA-L selenite(2-) Chemical compound [O-][Se]([O-])=O MCAHWIHFGHIESP-UHFFFAOYSA-L 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 239000004017 serum-free culture medium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000007974 sodium acetate buffer Substances 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229960003824 ustekinumab Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 229940018782 wilate Drugs 0.000 description 1
- 229940032528 zemaira Drugs 0.000 description 1
Images


































Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6872—Intracellular protein regulatory factors and their receptors, e.g. including ion channels
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/15—Cells of the myeloid line, e.g. granulocytes, basophils, eosinophils, neutrophils, leucocytes, monocytes, macrophages or mast cells; Myeloid precursor cells; Antigen-presenting cells, e.g. dendritic cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/415—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from plants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/745—Blood coagulation or fibrinolysis factors
- C07K14/755—Factors VIII, e.g. factor VIII C (AHF), factor VIII Ag (VWF)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/241—Tumor Necrosis Factors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0068—General culture methods using substrates
- C12N5/0075—General culture methods using substrates using microcarriers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0639—Dendritic cells, e.g. Langherhans cells in the epidermis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0645—Macrophages, e.g. Kuepfer cells in the liver; Monocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5047—Cells of the immune system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/22—Colony stimulating factors (G-CSF, GM-CSF)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2304—Interleukin-4 (IL-4)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
Definitions
- the present invention relates to a method for identifying a protein having immunogenicity and the like, for example, a method for identifying an epitope that may play a causative role in induction of immunogenicity. Also related.
- biopharmaceuticals antibody drugs, biologics, hormones, proteins, etc.
- the immunogenicity of these biopharmaceuticals is a problem.
- a biopharmaceutical can act as an antigen and induce the production of antibodies in the patient's body.
- neutralizing antibodies may be produced against the biopharmaceutical and treatment efficiency may be reduced.
- allergic reactions, leaching reactions, infusion reactions and the like can be caused.
- antibodies that produce autoimmune diseases and the like by neutralizing endogenous self-proteins corresponding to biopharmaceuticals can be produced.
- an antigen is presented on a major histocompatibility complex (also referred to as MHC molecule) present on the cell surface of an antigen-presenting cell (APC) (this is referred to as “antigen presentation”).
- MHC molecules involved in antigen presentation MHCI molecules (class I) and MHC II molecules (class II) are known.
- MHCI molecules act on killer T cells (CD8 positive T cells), and MHCII molecules act on helper T cells (CD4 positive T cells).
- MHCI molecules act on endogenous antigens in their own cells, while MHCII molecules act on foreign antigens.
- an antigen-antibody reaction or the like can be caused by an antigen presentation via an MHCI molecule for a cancer antigen produced in a cancer cell.
- antigen-antibody reaction and the like can be caused by antigen presentation via MHCII molecules.
- the endogenous protein in its own cell is degraded into small peptides by the proteasome.
- the peptide then binds to MHCI molecules synthesized in the endoplasmic reticulum to form a complex. Thereafter, the complex is transported to the cell surface, so that the peptide is presented as an epitope on the MHCI molecule.
- the MHCII molecule when used, first, the foreign protein is taken up into the antigen-presenting cell by endocytosis. The incorporated protein is then broken down into small peptides by lysosomes and then combined with MHCII molecules to form a complex. Thereafter, the complex is transported to the cell surface, so that the peptide is presented as an epitope on the MHCII molecule. The T cell receptor of helper T cells can then bind to the antigen presenting cell via the peptide.
- peptide sequences presented on MHC molecules In order to avoid the immunogenicity of antibody drugs and the like, studies have been conducted to identify peptide sequences presented on MHC molecules. This makes it possible to predict the immunogenicity of a protein or peptide intended to be administered to a living body. Further, for example, based on the information of the epitope sequence, the epitope can be modified by site-directed mutagenesis for the purpose of producing a non-immunogenic protein.
- Known methods for identifying peptide sequences include methods using in silico prediction algorithms and T cell proliferation assays (for example, measuring the proliferation ability of helper T cells by incorporating tritium-labeled thymidine). .
- the protein is brought into contact with an antigen-presenting cell such as a dendritic cell (DC) to induce antigen presentation, and a peptide derived from the protein is presented on the MHC molecule on the cell.
- an antigen-presenting cell such as a dendritic cell (DC)
- DC dendritic cell
- a peptide derived from the protein is presented on the MHC molecule on the cell.
- PBMC peripheral blood mononuclear cells
- monocytes monocytes
- peptide sequences derived from proteins in serum may also be detected.
- the amount of PBMC that can be obtained is limited, so often PBMCs from multiple donors are often pooled and used in bulk. It was not easy to determine if it was involved in induction of patient immunogenicity.
- antigen-presenting cells specifically, cells expressing major histocompatibility complex (MHC molecule)
- MHC molecule major histocompatibility complex
- a method for identifying an epitope of a protein comprising: The following steps: (A) A step of bringing a target protein into contact with a cell expressing a major histocompatibility complex (MHC molecule) differentiated from a stem cell or a progenitor cell derived therefrom; (B) isolating the complex of the peptide contained in the target protein and the MHC molecule from the cell expressing the MHC molecule; and (C) eluting the peptide from the complex and identifying it. Including the method.
- MHC molecule major histocompatibility complex
- the method according to [1] comprising the step of verifying whether the identified peptide is an epitope that induces immunogenicity.
- the stem cells are selected from the group consisting of induced pluripotent stem cells (iPS cells), embryonic stem cells (ES cells), nuclear transfer ES cells (ntES cells), embryonic germ stem cells (EG cells) and adult stem cells The method according to [1] or [2].
- iPS cells induced pluripotent stem cells
- ES cells embryonic stem cells
- ntES cells nuclear transfer ES cells
- EG cells embryonic germ stem cells
- adult stem cells The method according to [1] or [2].
- [4] The method according to any one of [1] to [3], wherein the MHC molecule is an MHCII molecule.
- the MHCII molecule is HLA-DR, HLA-DQ, or HLA-DP.
- the dendritic cell is The following steps: (A) a step of differentiating a stem cell or a progenitor cell derived therefrom to obtain a mesoderm progenitor cell; (B) differentiating the mesodermal progenitor cells to obtain monocytic cells; and (c) differentiating the monocyte cells to obtain immature dendritic cells, and optionally, immature dendritic cells Further produced by a method comprising the step of stimulating to obtain mature dendritic cells, Furthermore, the method according to any one of [1] to [10], wherein a serum-free medium is used in at least the step (c) among the steps (a) to (c).
- the mesoderm progenitor cells are differentiated in a serum-free medium containing granulocyte / macrophage colony stimulating factor (GM-CSF) and macrophage colony stimulating factor (M-CSF).
- GM-CSF granulocyte / macrophage colony stimulating factor
- M-CSF macrophage colony stimulating factor
- the dendritic cell is an immature dendritic cell, and the immature dendritic cell is induced into the mature dendritic cell by contacting with a target protein having immunogenicity.
- the target protein is selected from one or more members selected from the group consisting of cytokines, chemokines, growth factors, antibodies, enzymes, structural proteins, hormones, and fragments thereof.
- [1] The method according to any one of [14] to [14].
- [16] A method for producing a protein with reduced or eliminated immunogenicity, The following steps: (1) identifying a protein epitope according to the method of any one of [1] to [15]; (2) modifying the epitope such that binding to MHC molecules is reduced or eliminated; and (3) producing a protein having the modified epitope.
- [17] A protein obtainable according to the method of [16].
- a method for predicting whether a protein is immunogenic in a subject (I) providing a cell that expresses one or more allotypes of an MHC molecule of interest intended to be administered a target protein, wherein the cell is differentiated from a stem cell or a progenitor cell derived therefrom A process characterized by: (II) a step of bringing a target protein into contact with the “cell expressing one or more allotypes of MHC molecules”; (III) isolating a complex of a peptide contained in the target protein and an MHC molecule from the “cell expressing one or more allotypes of the MHC molecule”; (IV) eluting and identifying the peptide from the complex; and (V) optionally verifying whether the identified peptide is an epitope that induces immunogenicity, A method that indicates that the target protein is immunogenic in the subject when the identified peptide is an epitope that induces immunogenicity.
- a method for producing dendritic cells from stem cells or progenitor cells derived therefrom The following steps: (A ′) a step of differentiating a stem cell or a progenitor cell derived therefrom to obtain a mesoderm progenitor cell; (B ′) a step of obtaining monocytic cells by differentiating the mesoderm progenitor cells under a serum-free medium containing granulocyte / macrophage colony stimulating factor (GM-CSF) and macrophage colony stimulating factor (M-CSF); And (c ′) a step of differentiating the monocyte cells under serum-free medium to obtain immature dendritic cells, and optionally further stimulating the immature dendritic cells to obtain mature dendritic cells.
- GM-CSF granulocyte / macrophage colony stimulating factor
- M-CSF macrophage colony stimulating factor
- the step (c ′) (C1 ′) a step of obtaining immature dendritic cells by differentiating the monocyte cells in a serum-free medium containing granulocyte / macrophage colony stimulating factor (GM-CSF) and interleukin 4 (IL-4). Including, and in some cases, (C2 ′) comprising inducing the immature dendritic cells into mature dendritic cells by contacting the immunogen and optionally inflammatory cytokines, The method according to [23]. [25] A dendritic cell obtainable by the method according to [23] or [24].
- GM-CSF granulocyte / macrophage colony stimulating factor
- IL-4 interleukin 4
- the stem cells that express different MHC molecule allotypes
- the present invention uses stem cells or progenitor cells derived therefrom as starting material as a starting material for antigen-presenting cells for MAPPs, as compared to a system using PBMC as the starting material. It is suggested.
- stem cells are not limited in the number of cell divisions, and methods for proliferation and maintenance have been established, so that antigen-presenting cells that express allotypes of necessary MHC molecules can be produced and supplied in a stable manner in large quantities. It is also possible from the viewpoint of manufacturing cost and simplicity.
- DC refers to dendritic cells.
- An example of a scheme for differentiating human iPS cells to obtain dendritic cell-like cells is shown.
- numerator expressed on the cell surface of the monocyte-like cell produced from Ticline obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the monocyte-like cell produced from Ticline obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the monocyte-like cell produced from 201B7 line obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the monocyte-like cell produced from 201B7 line obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the dendritic cell-like cell produced from Ticline obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the dendritic cell-like cell produced from Ticline obtained by the flow cytometer analysis is shown.
- the amino acid sequence of Bet v1a is also shown.
- peptides were broadly detected at the positions corresponding to 4 positions.
- Analysis results of amino acid sequences of peptides detected when exposed to Bet v1a in MAPPs using dendritic cell-like cells derived from human iPS cells (201B7 line) (FIG. 8 (a)), and Bet v1a
- the analysis result (FIG. 8 (b)) of the amino acid sequence of the peptide detected even when exposed to Bet v1a detected under no treatment conditions is shown.
- the amino acid sequence of Bet v1a is also shown. In the amino acid sequence of Bet v1a, peptides were detected at roughly three locations.
- the analysis result (a) of the amino acid sequence of the peptide detected when exposed to Infliximab in MAPPs using dendritic cell-like cells derived from human iPS cells is shown.
- the amino acid sequences of H chain and L chain of Infliximab are also shown.
- Analysis result of amino acid sequence of peptide detected even when exposed to Infliximab, detected under Infliximab untreated conditions in MAPPs using dendritic cell-like cells derived from human iPS cells (Tic line) (b) Indicates.
- the amino acid sequences of H chain and L chain of Infliximab are also shown.
- FIG. 10B is a continuation of FIG. 10A.
- 10B is a continuation of FIG. 10B.
- 10C is a continuation of FIG. 10C.
- 10D is a continuation of FIG. 10D.
- FIG. 10E shows a continuation of FIG. 10E.
- FIG. 10F shows a continuation of FIG. 10F.
- FIG. 10G shows a continuation of FIG. 10G.
- the analysis result of the amino acid sequence of the peptide detected when exposed to Phl p1 in MAPPs using dendritic cell-like cells derived from human iPS cells is shown.
- numerator expressed on the cell surface of the monocyte cell obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the dendritic cell obtained by the flow cytometer analysis is shown.
- numerator expressed on the cell surface of the dendritic cell obtained by the flow cytometer analysis is shown.
- the analysis result of the amino acid sequence of the peptide detected in the MAPPs using PBMC derived dendritic cells of human donor under the Bet v1a addition condition and non-addition condition (control) is shown.
- the analysis result of the amino acid sequence of the peptide detected in the MAPPs using PBMC derived dendritic cells of human donor under the Bet v1a addition condition and non-addition condition (control) is shown.
- the analysis result of the amino acid sequence of the peptide detected in the MAPPs using PBMC derived dendritic cells of human donor under the Bet v1a addition condition and non-addition condition (control) is shown.
- the analysis result of the amino acid sequence of the peptide detected in the MAPPs using PBMC derived dendritic cells of human donor under the Bet v1a addition condition and non-addition condition (control) is shown.
- the analysis result of the amino acid sequence of the peptide detected in the MAPPs using PBMC derived dendritic cells of human donor under the Bet v1a addition condition and non-addition condition (control) is shown.
- the analysis result of the amino acid sequence of the peptide detected in the MAPPs using PBMC derived dendritic cells of human donor under the Bet v1a addition condition and non-addition condition (control) is shown.
- FIG. 21B is a continuation of FIG. 21A.
- the protein may be, for example, a natural protein, a recombinant protein, or a synthetic peptide prepared by artificially combining amino acids. It is understood that a protein may be a single protein or a mixture of different proteins. The protein may include unnatural amino acids. For example, it may be glycosylated when produced in vivo.
- the protein is preferably a protein (eg, antibody, hormone, etc.) associated with the treatment or prevention of an animal (preferably human).
- the protein may be selected from one or more, preferably from the group consisting of cytokines, chemokines, growth factors, antibodies, enzymes, structural proteins, hormones, and fragments of any of these.
- Proteins are presented as antigens in complex with MHC molecules after being taken up by cells and broken down, or after being taken up by cells but without being broken down or after being broken down in cells.
- the length of the amino acid sequence of the protein is not particularly problematic.
- the protein may be a peptide itself that forms a complex with an MHC molecule and presents an antigen.
- an epitope refers to a specific structural unit of an antigen that is recognized and bound by an antibody.
- An epitope is the smallest unit for antigenicity and is also called an antigenic determinant.
- differentiation refers to a state or aspect in which individual cells or cell populations that were originally single or identical are complicated or heterogeneous due to structural and / or functional changes. You may point to.
- differentiation may be used interchangeably with differentiation induction, and includes a state where differentiation induction is started, a state where differentiation induction is continued, a state where differentiation induction is completed, and the like. It is understood that a state in which a cell or a cell population that has completed is proliferating is naturally included.
- the induction may mean an action that promotes differentiation of a certain cell or cell population into another cell or cell population structurally and / or functionally, and is not particularly limited as long as differentiation can be achieved.
- the stem cell means a pluripotent stem cell and is not particularly limited as long as it has differentiation pluripotency and self-replication ability.
- stem cells include artificial pluripotent stem cells (iPS cells), embryonic stem cells (ES cells), nuclear transplant ES cells (ntES cells), embryonic germ stem cells (EG cells), and adult stem cells (WO2012 / 115276). These stem cells are preferably derived from mammals, more preferably from humans.
- ES cells are embryonic stem cells derived from the 8-cell stage of a fertilized egg, the inner cell mass of a blastocyst, which is an embryo after the morula.
- ES cells can be established by taking an inner cell mass from a blastocyst of a fertilized egg of a target animal and culturing the inner cell mass on a fibroblast feeder, and its establishment and maintenance method is known. (For example, US Patent No. 5,843,780 etc.).
- the selection of ES cells may be performed by Real-Time PCR using, for example, the expression of gene markers such as alkaline phosphatase, OCT-3 / 4, NANOG as an index.
- the expression of gene markers such as OCT-3 / 4, NANOG, FBX15, FGF4, REX1, ECAD may be used as an index (E. Kroon et al. (2008), Nat. Biotechnol., 26: 443-452).
- embryos used in producing human ES cells are, for example, returned to the mother in infertility treatment by in vitro fertilization.
- Unfertilized eggs may be used that have no intrinsic ability to grow into humans and whose cell division and growth is based on parthenogenesis.
- ES cells may be produced using only a single blastomere of the cleavage stage before the blastocyst stage without destroying the embryo's developmental potential and without destroying the fertilized egg (Chung Y , Klimanskaya I, Becker S, Marh J, Lu SJ, Johnson J, Meisner L, Lanza R. (2006).
- ES cells may be generated from human embryos that have stopped developing (Zhang X, Stojkovic P, Przyborski S, Cooke M, Armstrong L, Lako M, Stojkovic M. (2006). Stem Cells 24: 2669- 2676.).
- iPS cells can be produced by introducing specific reprogramming factors into somatic cells in the form of DNA or protein, and have almost the same properties as ES cells, such as differentiation pluripotency and self-renewal ability.
- Artificial stem cells derived from somatic cells K. Takahashi and S. Yamanaka (2006) Cell, 126: 663-676; K. Takahashi et al. (2007), Cell, 131: 861-872; J. Yu et. al. (2007), Science, 318: 1917-1920; Nakagawa, M. et al., Nat. Biotechnol. 26: 101-106 (2008); WO2007 / 069666).
- the somatic cell may refer to any animal cell (preferably, a mammalian cell including a human) excluding germ line cells and pluripotent stem cells.)
- the reprogramming factor is a gene specifically expressed in ES cells, its gene product or non-cording RNA, or a gene that plays an important role in maintaining undifferentiation of ES cells, its gene product or non-cording RNA Alternatively, it may be a low molecular compound.
- OCT3 / 4 SOX2, SOX1, SOX3, SOX15, SOX17, KLF4, KLF2, c-MYC, N-MYC, L-MYC, NANOG, LIN28, FBX15, ERAS, ECAT15-2, TCLL , Beta-catenin, LIN28B, SALL1, SALL4, ESRRB, NR5A2, TBX3.
- initialization factors may be used alone or in combination. Examples of combinations of initialization factors include the following combinations.
- OCT gene, KLF gene, SOX gene or combinations of reprogramming factors include, for example, WO2007 / 069666, WO2008 / 118820, WO2009 / 007852, WO2009 / 032194, WO2009 / 058413, WO2009 / 057831, WO2009 / 075119 , WO2009 / 079007, WO2009 / 091659, WO2009 / 101084, WO2009 / 101407, WO2009 / 102983, WO2009 / 114949, WO2009 / 117439, WO2009 / 126250, WO2009 / 126251, WO2009
- the reprogramming factor or the factor that promotes reprogramming examples include MEK inhibitors, DNA methyltransferase inhibitors, histone deacetylase (HDAC) inhibitors, and histone methyltransferase inhibitors that are known to those skilled in the art. , P53 inhibitors may be mentioned.
- the reprogramming factor is used in a somatic cell according to a method known to those skilled in the art such as a calcium phosphate method, a lipofection method, a microinjection method, or the like using a vector (for example, a viral vector, a plasmid vector, an artificial chromosome vector) or the like. May be introduced.
- DMEM DMEM / F12 or DME medium containing 10-15% FBS (these are leukemia inhibitory factor (LIF), penicillin / streptomycin, puromycin, L-glutamine, non-essential) Amino acids, ⁇ -mercaptoethanol and the like may further be included as appropriate.
- LIF leukemia inhibitory factor
- penicillin / streptomycin penicillin / streptomycin
- puromycin puromycin
- L-glutamine non-essential Amino acids
- ⁇ -mercaptoethanol a commercially available medium known to those skilled in the art may be used as appropriate.
- iPS cell culture may be appropriately set according to the composition of the medium. For example, in the presence of 5% CO 2 at 37 ° C., using 10% FBS-containing DMEM or DMEM / F12 medium, somatic cells are brought into contact with reprogramming factors and cultured for about 4 to 7 days.
- Prime ES cell culture containing basic fibroblast growth factor (bFGF) about 10 days after contact with somatic cells and reprogramming factors after repopulating on cells (eg, mitomycin C-treated STO cells, SNL cells)
- the iPS-like colonies may be generated from about 30 to about 45 days after the contact and cultured in a working medium.
- 10% FBS-containing DMEM medium (these are LIF, penicillin / streptomycin, puromycin, L-glutamine, on a feeder cell (for example, mitomycin C-treated STO cell, SNL cell) in the presence of 5% CO 2 at 37 ° C. It may further contain non-essential amino acids, ⁇ -mercaptoethanol, etc.), and may give rise to iPS-like colonies from about 25 to about 30 days or later.
- feeder cells reprogrammed somatic cells themselves may be used, or an extracellular matrix or Matrigel (BD) may be used.
- cultivate using a serum-free medium (Sun N, et al. (2009), Proc Natl Acad Sci USA 106.15720-15725).
- IPS cells may be selected according to the shape of the formed colonies (for example, can a cell cluster showing a shape close to a sphere be obtained).
- a drug resistance gene that is expressed in conjunction with a gene that is expressed when somatic cells are initialized for example, alkaline phosphatase, OCT3 / 4, NANOG
- a medium containing the corresponding drug for example, alkaline phosphatase, OCT3 / 4, NANOG
- the established iPS cells can be selected by culturing in.
- the marker gene is a fluorescent protein gene
- iPS cells can also be selected by observing with a fluorescence microscope.
- the cells may be cultured in vitro by a known differentiation method, and determined as iPS cells using the ability to differentiate into desired cells as an index.
- the cells are transplanted subcutaneously in immunodeficient mice, and the tumor tissue formed after a lapse of a predetermined period is analyzed to confirm that teratomas (teratomas) that contain various tissues are formed. It may be determined that Alternatively, it may be determined to be an iPS cell by confirming that a marker gene specifically expressed in the ES cell is expressed. Alternatively, a gene-wide gene expression pattern may be detected by a microarray or the like, and a cell highly correlated with an ES cell expression pattern may be determined as an iPS cell.
- the established iPS cells may be sold and used.
- ntES cell is an ES cell derived from a cloned embryo produced by nuclear transfer technology, and has almost the same characteristics as an ES cell derived from a fertilized egg (T. Wakayama et al. (2001), Science, 292: 740-743; S. Wakayama et al. (2005), Biol. Reprod., 72: 932-936; J. Byrne et al. (2007), Nature, 450: 497-502). That is, an ES cell established from an inner cell mass of a blastocyst derived from a cloned embryo obtained by replacing the nucleus of an unfertilized egg with the nucleus of a somatic cell is an ntES cell.
- ntES cells For the production of ntES cells, a known nuclear transfer technology (for example, JB Cibelli et al. (1998), Nature Biotechnol., 16: 642-646) may be combined with a known ES cell production technology ( Wakayama Kiyoka et al. (2008), Experimental Medicine, 26, 5 (extra number), 47-52).
- somatic cell nuclei may be injected into an enucleated unfertilized egg of a mammal and initialized by culturing for several hours.
- EG cells are cells that are established from embryonic primordial germ cells and have the same pluripotency as ES cells (Y. Matsui et al. (1992), Cell, 70: 841-847). It may be established by culturing primordial germ cells in the presence of LIF, bFGF, Stem Cell Factor (STF), etc. (Y. Matsui et al. (1992), Cell, 70: 841-847).
- Adult stem cells are cells that are not terminally differentiated and are found in vivo, and exist as a source of progenitor cells to terminally differentiated cells.
- Adult stem cells exist in each tissue in the living body, and the types of cells that can be differentiated are usually limited.
- hematopoietic stem cells that can be differentiated into monocytes, macrophages, dendritic cells and the like are particularly preferable as examples of adult stem cells.
- a hematopoietic progenitor cell refers to a cell differentiated from a hematopoietic stem cell.
- a stem cell-derived progenitor cell refers to any cell (for example, mesoderm progenitor) observed in the process of obtaining an antigen-presenting cell (specifically, a cell that expresses an MHC molecule) by differentiating the stem cell.
- Cell hematopoietic progenitor cell, granulocyte / macrophage colony forming cell, lymphoblast, monoblast, pre-monocyte or monocyte).
- MHCI molecules Peter Parham (2007), Essential Immunology; The Human Protein Atlas, http://www.proteinatlas.org/
- Proteins can be presented to killer T cells via MHCI molecules.
- specific cells have MHCII molecules in addition to MHCI molecules, and foreign antigens can be presented to helper T cells via MHCII molecules (also called professional antigen-presenting cells).
- antigen presenting cells may include both of these cell types.
- the antigen-presenting cell is the latter type, for example, dendritic cells, macrophages, monocytes, and B cells are preferable.
- cytokines such as interferon and MHCII molecules are induced, thyroid follicular cells, fibroblasts, vascular endothelial cells, etc. also function as antigen-presenting cells, so these cells are also listed as the latter type. May be.
- the antigen-presenting cell specifically, a cell that expresses an MHC molecule, such as a dendritic cell, a macrophage, a monocyte, or a B cell
- MHCI molecule and / Or cells expressing MHCII molecules may be used as an indicator, and at least one of CD11a, CD11b, CD11c, CD14, CD15, CD40, CD80, CD83, CD86, CD123, CD205, CD206, CD209, CCR7 It is more preferable to use as an index a cell expressing.
- dendritic cells are advantageous as antigen-presenting cells because they have strong antigen-presenting ability and helper T-cell activation ability.
- dendritic cells are cells having cell processes and exhibiting a dendritic or dendritic morphology.
- at least one of CD11b, CD11c, CD40, CD80, CD83, CD86, CD123, CD205, CD206, CD209, and CCR7 can be used as a criterion for determining whether or not the dendritic cell has characteristics. May be used as an index, and it is more preferable to use as an index whether all of the MHCII molecules, CD80, CD86, CD206, and CD209 are expressed.
- dendritic cells that express all of the MHCII molecules, CD80, CD86, CD206, and CD209 and are negative for CD14.
- a criterion for determining whether or not it has characteristics as a macrophage cell for example, it may be used as an index whether CD11b is further expressed in addition to the MHCII molecule.
- CD80 and CD86 are known to transmit signals to helper T cells to activate the cells.
- the dendritic cell-like cells obtained in this example have similar properties to monocyte-derived dendritic cells in terms of cell shape, cell surface molecule expression, and helper T cell stimulation ability. It may be included in the dendritic cells in the specification. Similarly, the monocyte-like cells obtained in this example may be included in the monocyte cells herein.
- the antigen-presenting cell is preferably derived from a mammal, more preferably from a human.
- the MHC molecule may be either an MHCI molecule or an MHCII molecule, but an MHCII molecule is more preferable.
- Human MHC is called human leukocyte antigen (HLA).
- MHCI molecules are further divided into classical class I molecules (class Ia) and nonclassical class I molecules (class Ib).
- Classical class I molecules include HLA-A, HLA-B, and HLA-C in humans, and nonclassical class I molecules include HLA-E, HLA-F, and HLA-G in humans.
- examples of MHCII molecules include HLA-DR, HLA-DQ, and HLA-DP in humans.
- MHC molecules are slightly different in individual amino acid sequences even among homologous animals, and can be divided into several types called allotypes. For example, in the case of HLA-DR, many allotypes such as DR1, DR2, DR3, DR4... Are known. Since allotypes are linked to each other on the MHC gene, they are inherited from a parent to a child as a pair unless genetic recombination occurs in this region. This unit is called the halotype. In patient-derived stem cells (for example, iPS cells), even if they are subcultured and differentiated, the patient's MHC gene sequence is basically retained as it is. Thus, antigen-presenting cells obtained by differentiating the stem cells The allotype of the MHC molecule possessed by is maintained.
- allotypes for example, in the case of HLA-DR, many allotypes such as DR1, DR2, DR3, DR4... are known. Since allotypes are linked to each other on the MHC gene, they are
- Each allotype can form a complex with a different antigenic peptide fragment (epitope) and present the epitope on the cell surface, so for each allotype set, in other words, for each patient with that allotype set.
- epitope an antigenic peptide fragment
- the presence or absence of immunogenicity and side effects on the target protein varies. Since allotypes and haplotypes have patterns characteristic of race and ethnicity, they can be used for analysis of the presence or absence of side effects and other immunogenicity to target proteins for each race and ethnicity.
- allotypes possessed by individuals can be determined by genetic analysis (for example, DNA amplified by polymerase chain reaction (PCR) is hybridized with beads to which probes are immobilized, the fluorescence intensity is digitized, and data analysis is performed. Therefore, it is possible to determine whether the target protein has immunogenicity, side effects, or the like based on the specified allotype information.
- genetic analysis for example, DNA amplified by polymerase chain reaction (PCR) is hybridized with beads to which probes are immobilized, the fluorescence intensity is digitized, and data analysis is performed. Therefore, it is possible to determine whether the target protein has immunogenicity, side effects, or the like based on the specified allotype information.
- Other specific examples of the genetic diagnosis include the method described in International Journal of Immunogenetics, 2011; 38: 6, pp.463-473.
- the antigen-presenting cell may express one or more allotypes of the MHC molecule of a subject (eg, a mammal, preferably a human) intended to be administered the target protein.
- a subject eg, a mammal, preferably a human
- the stem cell or progenitor cell derived therefrom in the present invention is a cell that expresses one or more allotypes of MHC molecules possessed by a subject (eg, a human patient or a healthy human subject) intended for analysis.
- a subject eg, a human patient or a healthy human subject
- one or more cells expressing one or more allotypes of the MHC molecule of the subject may be used so that all sets of allotypes of the MHC molecule possessed by the subject are included.
- cells that express one or more allotypes of MHC molecules that are highly expressed in the race or ethnic group that is intended for analysis may be prepared.
- the race, Immunogenicity may be analyzed by covering a certain percentage of the population (for example, 30% to 80% or more). As appropriate, for example, it is advantageous to perform comparative analysis of human patients, human patients and healthy human subjects, and healthy human subjects.
- the method for differentiating stem cells or progenitor cells derived therefrom into antigen-presenting cells is not particularly limited as long as it is a method known to those skilled in the art.
- methods for differentiating stem cells such as ES cells and iPS cells into monocytes, macrophages, B cells, or dendritic cells include WO2009 / 120891; WO2009 / 074341; Regen. Med.
- BMP-4 Bone Morphogenetic Protein-4
- GM-CSF Granulocyte Macrophage-Colony Stimulating Factor
- SCF Stem Cell Factor
- VEGF Vascular Endothelial Growth Factor
- monocytes Differentiating into monocytes; then the monocytes are further differentiated into immature dendritic cells using GM-CSF and Interleukin-4 (IL-4); Is differentiated into mature dendritic cells using a maturation cocktail consisting of GM-MSF, TNF- ⁇ , Interleukin-1 ⁇ (IL-1 ⁇ ), Interferon- ⁇ (IFN- ⁇ ), and PGE2. Is disclosed. Moreover, PLoS One, April 2013, Vol. 8, Issue 4, e59243 discloses that functional macrophages and dendritic cells were obtained based on monocytes differentiated from ES cells and iPS cells. Yes.
- NATURE IMMUNOLOGY Vol.5, No.4, 2004, pp.410-417 describes the method of producing T cells from ES cells, but B cells can also be produced during the production process.
- B cells can also be produced during the production process.
- WO2012 / 115276 may preferably be referred to as a specific method for differentiating stem cells or progenitor cells derived therefrom into antigen-presenting cells.
- the antigen-presenting cell is a dendritic cell
- following the step of providing a stem cell or a progenitor cell derived therefrom the following step: (A) a step of differentiating a stem cell or a progenitor cell derived therefrom to obtain a mesoderm progenitor cell; (B) differentiating the mesodermal progenitor cells to obtain monocytic cells; and (c) differentiating the monocyte cells to obtain immature dendritic cells, and optionally, immature dendritic cells
- a step of further stimulating to obtain mature dendritic cells may be included.
- a serum-free medium may be used at least in the step (c), and a serum-free medium may be used in both the steps (b) and (c).
- a serum-free medium is used in all of the steps (a) to (c).
- the serum may refer to mammal-derived serum such as human serum, monkey serum, fetal bovine serum, sheep serum, rabbit serum, rat serum, guinea pig serum, mouse serum.
- the serum-free medium refers to a medium to which no serum is added and no commercially available serum substitute such as B-27 is added, preferably albumin or albumin substitute, transferrin or transferrin substitute , Insulin or an insulin substitute, and a medium containing at least one of selenite. More preferably, it may be a medium containing Insulin-Transferrin-Selenium-X Supplement (ITS).
- ITS Insulin-Transferrin-Selenium-X Supplement
- Preferred serum-free media include, for example, minimal essential medium (MEM), Dulbecco's modified Eagle medium (DMEM), Iskov's modified Dulbecco medium (IMDM), StemPro-34 medium (Life Tech), Stemline II (SIGMA), or Primate ES cell Examples include a medium in which ITS is added to medium (ReproCELL) or the like.
- MEM minimal essential medium
- DMEM Dulbecco's modified Eagle medium
- IMDM Iskov's modified Dulbecco medium
- SIGMA Stemline II
- Primate ES cell Examples include a medium in which ITS is added to medium (ReproCELL) or the like.
- stem cells or progenitor cells derived therefrom are cultured in a medium containing a BMP family protein and then cultured in a medium containing growth factors and hematopoietic factors, or in a medium containing VEGF.
- a step of obtaining mesoderm progenitor cells by culturing in a medium containing a hematopoietic factor after culturing may be included.
- the step (b) may include a step of culturing in a medium containing a hematopoietic factor to obtain monocytic cells by differentiating the mesodermal progenitor cells.
- the step (a) and the step (b) can be performed continuously.
- the BMP family protein may refer to a cytokine having about 20 subtypes belonging to the TGF- ⁇ superfamily.
- a preferred BMP family protein in the present invention is BMP2 and / or BMP4, more preferably BMP4.
- the growth factor may be preferably VEGF, specifically, VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, PlGF (placental growth factor) -1, PlGF-2 Or these alternative splicing variants (for example, variants of 121, 165, 189 or 206 amino acids are known for VEGF-A).
- a preferred VEGF in the present invention is VEGF-A.
- the growth factor may contain bFGF in addition to VEGF.
- the hematopoietic factor is a factor that promotes the differentiation and proliferation of blood cells.
- SCF Stem Cell Factor
- G-CSF granulocyte colony stimulating factor
- GM-CSF granulocyte / macrophage colony stimulating factor
- M-CSF macrophage colony stimulating factor
- EPO erythropoietin
- TPO thrombopoietin
- IL interleukins
- the interleukins may be IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, or IL-9.
- the preferred hematopoietic factor in the step (b) may be selected from the group consisting of SCF, TPO, IL-3, Flt3-ligand, GM-CSF and M-CSF. Hematopoietic factors may be used alone or in combination.
- the step (a) using a combination of VEGF as a growth factor and SCF as a hematopoietic factor may be performed.
- the step (b) it is preferable to culture by changing the medium every several days (for example, every 3 to 4 days).
- non-adherent cells (monocyte cells (like cells)) are obtained by the step (b), they may be used as monocytes in the step (c).
- the non-adherent cells have the characteristics of monocytes, for example, using flow cytometry, etc., in addition to the expression of MHCII molecules, CD14, CD45 hi , CD11a, CD11b, which are markers of monocytes, The expression of CD15 etc. can be determined as an index.
- monocytes used for induction into dendritic cells by separating only CD14 positive cells from non-adherent cells using the magnetic bead method or the like. Can increase the percentage of cells.
- the step (c) further includes: (Ci) culturing the monocytes (floating) in a medium containing a hematopoietic factor and obtaining immature dendritic cells (like cells) by differentiation, and optionally, (Cii) a step of inducing the resulting immature dendritic cell (like cell) into an mature dendritic cell (like cell) by further contacting with an immunogen and optionally an inflammatory cytokine. It's okay.
- the immature dendritic cell-like cell or the mature dendritic cell-like cell has the characteristics of a dendritic cell, for example, using flow cytometry or the like, in addition to the expression of MHCII molecule, Whether or not at least one of a certain CD11b, CD11c, CD40, CD80, CD83, CD86, CD123, CD205, CD206, CD209, and CCR7 is further expressed may be used as an index. Furthermore, whether dendritic cells have the characteristics of immature dendritic cells or mature dendritic cells is verified using, for example, the expression variation of MHCII molecules (HLA-DR, etc.) as an index. can do.
- the hematopoietic factor may be the aforementioned factor.
- a combination of GM-CSF, IL-3 and IL-4, or a combination of GM-CSF and IL-4 may be used.
- the immunogen and the inflammatory cytokine When the immunogen and the inflammatory cytokine are brought into contact with immature dendritic cells, they can be induced into mature dendritic cells by pulsing the cells. While immature dendritic cells have high antigen phagocytic ability but low antigen presenting ability, they mature into mature dendritic cells due to invasion of antigen into the living body, and proteins such as MHCII molecules required for antigen presentation Expression can be enhanced to improve antigen presenting ability.
- the immunogen may be any substance that causes an immune response when introduced into a living body, and includes, for example, lipopolysaccharide (LPS, present in pathogens).
- LPS lipopolysaccharide
- the protein to be evaluated has immunogenicity, it can be understood by those skilled in the art that the protein can act as an immunogen.
- the immature dendritic cells are induced into mature dendritic cells by contacting the immunogenic target protein.
- the inflammatory cytokine may be, for example, Tumor Necrosis Factor- ⁇ (TNF- ⁇ ), TNF- ⁇ , IL-12, or IFN- ⁇ .
- TNF- ⁇ Tumor Necrosis Factor- ⁇
- TNF- ⁇ Tumor Necrosis Factor- ⁇
- IL-12 IL-12
- IFN- ⁇ IFN- ⁇ .
- the immunogen and the inflammatory cytokine may be used alone or in combination as appropriate.
- step (D) You may perform the process of differentiating the said monocyte cell and obtaining a macrophage.
- the monocyte cells can be differentiated into macrophages, preferably using GM-CSF or M-CSF as a hematopoietic factor.
- M1 macrophages by adding, for example, IFN- ⁇ or LPS
- M2 macrophages by adding, for example, IL-4 or IL-13
- Macrophages are It is known to be activated by receiving cytokines produced by helper T cells, and classical activation (M1 macrophage) and selective activation (M2 macrophage) are known.
- the concentrations of growth factors, hematopoietic factors, cytokines, and the like used in the above-described steps may be any concentrations that can obtain the target antigen-presenting cells, and can be appropriately determined by those skilled in the art.
- the concentration of BMP4 may be, for example, 5 to 150 ng / ml, more preferably 10 to 100 ng / ml, and further preferably 20 to 80 ng / ml.
- the concentration of VEGF may be, for example, 20 to 100 ng / ml, more preferably 30 to 70 ng / ml, and further preferably 40 to 50 ng / ml.
- the concentration of bFGF may be, for example, 10 to 100 ng / ml, and more preferably 20 to 50 ng / ml.
- the concentration of SCF may be, for example, 20 to 100 ng / ml, more preferably 30 to 70 ng / ml, and further preferably 40 to 50 ng / ml.
- the concentration of IL-3 may be, for example, 5 to 100 ng / ml, and more preferably 30 to 70 ng / ml.
- the concentration of TPO may be, for example, 1 to 25 ng / ml, and more preferably 1 to 10 ng / ml.
- the concentration of Flt3-ligand may be, for example, 10 to 100 ng / ml, and more preferably 30 to 70 ng / ml.
- the concentration of GM-CSF may be, for example, 5 to 250 ng / ml, and more preferably 50 to 200 ng / ml.
- the concentration of M-CSF may be, for example, 5 to 100 ng / ml, and more preferably 30 to 70 ng / ml.
- the concentration of IL-4 may be, for example, 3 to 100 ng / ml, and more preferably 10 to 70 ng / ml.
- the concentration of TNF- ⁇ may be, for example, 0.05 to 50 ng / ml, and more preferably 0.1 to 20 ng / ml. In the case of LPS, for example, 0.01 to 100 ⁇ g / ml may be used, and 0.1 to 10 ⁇ g / ml is more preferable.
- growth factors, hematopoietic factors, cytokines and the like may be used in appropriate combinations according to the purpose, and those skilled in the art can appropriately determine the optimum concentration.
- the concentration of the (target) protein to be evaluated may be a concentration that can identify the epitope of the protein, for example, depending on the purpose, or the protein is a target (for example, a mammal, preferably a human). Any concentration that can evaluate whether or not it has immunogenicity may be used, or any concentration that can induce immature dendritic cells to be induced into mature dendritic cells. Those skilled in the art appropriately determine the concentration. it can. Such a concentration may be, for example, 0.01 to 1000 ⁇ g / ml, and more preferably 0.1 to 100 ⁇ g / ml.
- the period of the step (a) may be, for example, 2 days or more, preferably 2 to 10 days, and more preferably 5 to 8 days.
- the period of the step (b) may be, for example, 1 day or longer, preferably 20 to 200 days, more preferably 50 to 150 days.
- the period of the step (ci) may be, for example, 1 day or more, preferably 1 to 10 days, and more preferably 4 to 6 days.
- the period of the step (cii) may be, for example, 12 hours or more, preferably 12 to 36 hours, and more preferably 24 hours (1 day).
- the period of the step (d) may be, for example, 1 day or more, preferably 1 to 20 days.
- the macrophages may be further differentiated into M1 macrophages or M2 macrophages.
- those skilled in the art can appropriately determine the optimum culture period in consideration of each culture condition.
- the present invention also includes a method for producing a dendritic cell (in vitro) from a stem cell or a progenitor cell derived therefrom, including the steps (a) to (c), and whether or not the method has been obtained or obtained.
- Good for dendritic cells that can be made. That is, dendritic cells produced by this method express not only MHCII molecules, but also CD80 and CD86, which are costimulatory molecules for helper T cells, and CD206 and CD209 that are sugar chain receptors. It was suggested that it has the ability to activate helper T cells and resistance to viruses and the like. Analysis of protein epitopes using dendritic cells obtained by this method contributes to the development of proteins with low immunogenicity, and is an excellent material for research on antigen-presenting cells against autoimmune diseases and viruses. Is expected to be.
- a method for producing a dendritic cell (in vitro) from a stem cell or a progenitor cell derived therefrom comprising the following steps: (A ′) a step of differentiating a stem cell or a progenitor cell derived therefrom to obtain a mesoderm progenitor cell; (B ′) a step of obtaining monocytic cells by differentiating the mesoderm progenitor cells under a serum-free medium containing granulocyte / macrophage colony stimulating factor (GM-CSF) and macrophage colony stimulating factor (M-CSF); And (c ′) a step of differentiating the monocyte cells under serum-free medium to obtain immature dendritic cells, and optionally further stimulating the immature dendritic cells to obtain mature dendritic cells.
- a ′ a step of differentiating a stem cell or a progenitor cell derived therefrom to obtain a mesoderm progenitor cell
- the step (c ′) (C1 ′) a step of obtaining immature dendritic cells by differentiating the monocyte cells in a serum-free medium containing granulocyte / macrophage colony stimulating factor (GM-CSF) and interleukin 4 (IL-4). Including, and in some cases, (C2 ′) comprising inducing the immature dendritic cells into mature dendritic cells by contacting the immunogen and optionally inflammatory cytokines, The method according to [23]. [25] A dendritic cell obtainable by the method according to [23] or [24].
- GM-CSF granulocyte / macrophage colony stimulating factor
- IL-4 interleukin 4
- a cell composition comprising the dendritic cell according to any one of [25] to [27].
- the dendritic cell or the cell composition is used for controlling immune response for the purpose of performing immune cell therapy against infectious diseases or malignant tumors, or treating rejection associated with autoimmune disease or organ transplantation. May be used as a cell medicine.
- the cell medicine may be used in an appropriate combination of auxiliaries such as a medium for the purpose of stably holding dendritic cells.
- this invention is the method for identifying the epitope of protein in one aspect
- mode Comprising: The following processes: (A) A step of bringing a target protein into contact with a cell expressing a major histocompatibility complex (MHC molecule) differentiated from a stem cell or a progenitor cell derived therefrom; (B) isolating the complex of the peptide contained in the target protein and the MHC molecule from the cell expressing the MHC molecule; and (C) eluting the peptide from the complex and identifying it.
- Including a method the method comprises the following steps: (D) A step of verifying whether the identified peptide is an epitope that induces immunogenicity may be included. The method can perform the entire process in vitro.
- the step (A) is preferably performed in the absence of serum.
- the amount of cells that express MHC molecules necessary to obtain 100 ng MHC molecules can depend on the number of cells, the expression intensity of MHC molecules, and the degree of expression.
- the amount of cells can be determined.
- Each allotype of the MHCII molecule (eg, HLA-DQ1) can hold about 500 to 1000 different peptide fragments (Chicz R M et al., J Exp. Med. 1993, 178, 27-47; Chicz R M & Urban R G, Immunol. Today, 1993, 15, 155-160).
- the majority of these different peptides only reach very low copy numbers and are therefore not very likely to play a physiological role in vivo.
- peptide fragments that are involved in immunogenicity for example, activate helper T cells, reach moderate to high copy numbers (Latek R & Unanue E R, Immunol. Rev. 1999, 172: 209-228) .
- These medium to high copy number peptides account for about 40-50% of the total amount of peptides eluted from MHCII molecules and may correspond to about 10-200 individual peptides.
- the peptide is a peptide that is derived from the target protein (its amino acid sequence) and can form a complex with an MHC molecule on the surface of an antigen-presenting cell (specifically, a cell that expresses an MHC molecule).
- the peptide may be bound to intracellular or extracellular MHC molecules.
- Each allotype of the MHCII molecule can form a complex with various peptides, and the amount of peptide required for sequencing each eluted peptide may be, for example, only a femtomole amount.
- an approximately femtomolar amount of a peptide fragment bound to the molecule can be isolated, and the sequence of the peptide can be identified.
- the cell membrane of the cell may be solubilized.
- the lysis may be performed by methods known to those skilled in the art, such as freeze-thawing, use of a surfactant, or a combination thereof.
- a surfactant for example, Triton X-100 (TX100), Nonidet P-40 (NP-40), Tween 20, Tween 80, n-octyl glucoside, ZWITTERGENT, Lubrol, or CHAPS may be used.
- TX100 Triton X-100
- NP-40 Nonidet P-40
- Tween 20 Tween 80
- n-octyl glucoside ZWITTERGENT
- Lubrol Lubrol
- CHAPS CHAPS
- the cell lysate containing the solubilized MHC molecule-peptide complex may be subjected to immunoprecipitation or immunoaffinity chromatography to purify the MHC molecule-peptide complex.
- immunoprecipitation or immunoaffinity chromatography antibodies specific for MHC molecules and suitable for these methods (anti-MHCI molecular antibodies such as anti-HLA-A antibody, anti-HLA-B antibody, anti-HLA-C Antibody or anti-HLA-ABC antibody; or anti-MHCII molecule antibody, preferably anti-HLA-DR antibody, anti-HLA-DQ antibody or anti-HLA-DP antibody) may be used.
- the specific antibody is preferably a monoclonal antibody and may be covalently or non-covalently bound to beads (eg, Sepharose beads or agarose beads) via, for example, Protein A.
- beads eg, Sepharose beads or agarose beads
- the amino group of an antibody may be covalently bonded to CNBr-activated sepharose to form a solid layer.
- the monoclonal antibody may be purchased commercially or may be purified from the corresponding supernatant of each hybridoma cell using protein A- or protein G-affinity chromatography.
- immunoisolation of MHC molecules may be performed by incubating antibody-beads with cell lysate while rotating for several hours.
- the washing of the antibody-bead bound with the MHC molecule-peptide complex may be performed in an Eppendorf tube.
- the result of immunoprecipitation may be analyzed by SDS-PAGE and Western blotting using an antibody that recognizes denatured MHC molecules.
- the peptide can be obtained by methods known to those skilled in the art, for example, diluted acid, such as diluted acetonitrile (Jardetzky T S et al., Nature 1991 353, 326-329), diluted acetic acid and heated (Rudensky A Y et al. ., Nature 1991, 353, 622-626; Chicz® M et al, Nature 1992, 358, 764-768, or diluted trifluoroacetic acid (Kropshofer H et al., J Exp Med 1992, 175, 1799- 1803) may be used for elution.
- Peptides are preferably eluted with diluted trifluoroacetic acid, for example at 37 ° C.
- the MHC molecule-peptide complex Before the peptide is eluted from the MHC molecule-peptide complex, it may be washed with water or a low salt buffer to remove the remaining surfactant.
- a low salt buffer a Tris buffer solution, a phosphate buffer solution, or an acetate buffer solution having a concentration of 0.5 to 10 mM may be used.
- the MHC molecule-peptide complex may be washed with ultra high purity water for HPLC.
- the washing may be performed by ultrafiltration. Ultrafiltration may be performed, for example, in an ultrafiltration tube having a cutoff value of 30 kD, 20 kD, 10 kD, or 5 kD and a tube volume of 0.5 to 1.0 ml. Washing in the ultrafiltration tube may be performed 4 to 12 times, for example, in a volume several tens of times the volume of the beads holding the MHC molecule-peptide complex.
- the peptide may be eluted from the MHC molecule-peptide complex using this ultrafiltration tube.
- the eluted peptide may then be lyophilized or dried by a centrifugal evaporator.
- Each peptide (its amino acid sequence) may be identified by fractionating and analyzing the sequence of the eluted peptide mixture using liquid chromatography mass spectrometry (LC / MS).
- LC / MS liquid chromatography mass spectrometry
- amino acid sequence of each peptide in the peptide mixture can be revealed by known methods sufficient to sequence femtomolar amounts of the peptide.
- Identification reveals the protein from which the peptide is derived and to which sequence in the protein the peptide is derived.
- the eluted peptide mixture is preferably fractionated by, for example, a combination of reverse phase chromatography and anion exchange chromatography or cation exchange chromatography, or by reverse phase chromatography alone. Fractionation uses a fused-silica micro-capillary column connected to either the nanoflow electrospray source of the mass spectrometer or to a micro fractionator that spots the fraction on a MALDI analysis plate It may be performed in the HPLC mode.
- ESI-MS electrospray ionization tandem mass spectrometry
- PSD MALDI-post source decay
- amino acid sequence analysis of each peptide may be determined using various means known to those skilled in the art. Sequence analysis may be performed by computer analysis of peptide fragment spectra, for example using the MASCOT algorithm or SEQUEST algorithm. These algorithms preferably use protein and nucleotide sequence databases to perform cross-correlation analysis of experimentally and theoretically generated tandem mass spectra. This allows for automated high-throughput sequence analysis.
- MALDI-TOF matrix-assisted laser desorption / ionization time-of-flight mass spectrometry
- the passage through the microcapillary column may be analyzed using a UV detector at a detection wavelength of 214 nm.
- the amount of peptide may be estimated by comparing the peak area of the peptide to be analyzed to the peak area of a graded amount of standard peptide (control).
- Elution of peptides from MHC molecules provides a set of peptides derived from the target protein and naturally fragmented in the antigen-presenting cells.
- To identify and eliminate false positive peptides in addition to a set of antigen-presenting cells exposed to the target protein, as a negative control, prepare a set of antigen-presenting cells that are not exposed to the target protein for comparative analysis. Is preferred. Peptides that can only be detected in antigen-presenting cells exposed to the target protein compared to antigen-presenting cells that are not exposed to the target protein may function as epitopes of the protein from which the peptide is derived and may be identified as antigenic .
- MHC binding motif means a structural feature common to peptides that bind to a specific MHC molecule (allelic polymorphism), which is necessary to form a stable complex with the MHC molecule.
- the peptide length varies from 12 to 18 amino acids, and even longer peptides can be bound because both ends of the peptide binding groove are open.
- Many MHCII molecules contain up to four residues (called “anchor residues”) related to binding in the relative positions contained in the 9-mer nonameric core region: P1, P4 , P6, and P9. However, this core region can vary in distance from the N-terminus of the peptide. Often 2 to 4 N-terminal residues precede the core region.
- the P1 anchor residue is located at position 3, 4, or 5 of many peptides capable of forming a complex with the MHCII molecule.
- peptides eluted from HLA-DR molecules may share a hydrophobic P1 anchor, such as tyrosine, phenylalanine, tryptophan, methionine, leucine, isoleucine, or valine.
- the position and type of anchor residues can be deduced from the peptide binding motifs of frequent MHC molecules.
- a computer algorithm capable of verifying a peptide sequence motif can be obtained from, for example, “Tepitope” (www.vaccinome.com, J. Hammer, Nutley, USA).
- the MHC binding ability may be verified by a method known to those skilled in the art using the detected peptide itself (for example, a synthetic peptide may be used) and a desired MHC molecule (Kropshofer H et al., J Exp. Med. 1992, 175, 1799-1803; Vogt A B et al., J. Immunol. 1994, 153, 1665-1673; Sloan V S et al., Nature 1995, 375, 802-806).
- the MHC binding ability may be verified using a cell binding assay that utilizes a cell line expressing MHC molecules and a biotinylated peptide (Arndt S O et al, EMBO J., 2000, 19 , 1241-1251).
- the relative binding capacity of the peptide to MHC may be determined by measuring the concentration (IC50) required to reduce the binding of the labeled reporter peptide by 50%.
- IC50 concentration required to reduce the binding of the labeled reporter peptide by 50%.
- each identified peptide may be used, or a peptide having a sequence (core sequence) common to the identified peptides may be used.
- the peptide to be detected is considered to depend on the type of allotype of the MHC molecule, the strength of binding affinity for the MHC molecule, and the like.
- helper T cells The ability to stimulate helper T cells is particularly important in verifying whether the identified peptide functions as an epitope.
- the identified peptide When the identified peptide stimulates helper T cells, it may be one of the indicators for determining that the peptide has immunogenicity. As the determination method, it may be tested whether the peptide identified by the method of the present invention is capable of activating helper T cells.
- each identified peptide may be used, or a peptide having a sequence (core sequence) common to the identified peptides may be used.
- helper T cells The cellular response of helper T cells may be measured by various in vitro methods known to those skilled in the art. For example, in the presence of the peptide to be evaluated, cells that express MHC molecules (for example, monocytes, macrophages, dendritic cells, etc.) are cultured together with helper T cells, and DNA proliferation is used as an indicator of cell proliferation of helper T cells. Whether thymidine (T) labeled with a radioactive substance is incorporated during replication may be measured. Alternatively, 5-bromo-2'-deoxyuridine (BrdU) may be used instead of thymidine.
- MHC molecules for example, monocytes, macrophages, dendritic cells, etc.
- the amount of BrdU incorporation may be measured using an enzyme or fluorescently labeled secondary antibody (for example, 5-Bromo-2'-deoxyuridine Labeling & Detection Kit III, Roch-Biochem, Cat No. 1 444 611). Alternatively, it is measured by Naive Primary T cell Assay (Proimmune) using a flow cytometry method with the indicator that the fluorescent dye label 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) is diluted by helper T cell proliferation. Also good.
- an enzyme or fluorescently labeled secondary antibody for example, 5-Bromo-2'-deoxyuridine Labeling & Detection Kit III, Roch-Biochem, Cat No. 1 444 611).
- it is measured by Naive Primary T cell Assay (Proimmune) using a flow cytometry method with the indicator that the fluorescent dye label 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) is diluted by helper T
- helper T cells may be evaluated by measuring various cytokines produced from helper T cells.
- cytokines include IL-2, IL-4, IL-6, IL-10, IL-12, IFN- ⁇ , or transforming growth factor- ⁇ (TGF- ⁇ ).
- Cytokine measurement methods include various methods known to those skilled in the art, such as ELISA or ELISPOT.
- dendritic cells produced by a method of producing dendritic cells from stem cells or progenitor cells derived therefrom may be used.
- Cells that express MHC molecules may be rendered non-proliferative by, for example, treatment with ionizing radiation or mitomycin C prior to assay.
- the present invention provides a method for producing a protein with reduced or eliminated immunogenicity, comprising the following steps: (1) identifying a protein epitope according to the method described above; (2) a method comprising modifying the epitope such that binding to MHC molecules is reduced or eliminated; and (3) producing a protein having the modified epitope.
- the present invention also relates to a protein obtained or obtainable in accordance with the above production method in still another embodiment.
- the “obtainable protein” may mean a protein that can be obtained by using the production method.
- the peptide in which the epitope has been identified is modified so that the binding to the MHC molecule is reduced or eliminated, or the immunogenicity can be reduced or eliminated. Or it can be modified.
- the reduction or disappearance of immunogenicity is not particularly limited as long as it is a method known to those skilled in the art.
- the above-described MHC binding motif, MHC binding ability, or recognition by helper T cells is determined as an index. Also good.
- in silico epitope prediction algorithms may be combined as appropriate.
- alteration or modification may be performed according to a method known to those skilled in the art.
- a DNA nucleotide sequence encoding the amino acid sequence of a protein containing an epitope one or more desired nucleotides of the DNA sequence can be inserted, substituted, for example, using site-directed mutagenesis or homologous recombination, or It may be deleted.
- one or more anchor residues important for binding to the MHC molecule can be changed to other amino acid residues, thereby reducing or eliminating immunogenicity.
- an amino acid residue important for epitope recognition by a T cell receptor of a helper T cell or a B cell receptor of a B cell may be changed to another amino acid residue.
- the P1 anchor of the HLA-DR1-restricted T cell epitope may be substituted with alanine, proline, glycine or a charged amino acid residue (Kropshofer et al., EMBO J. 15, 1996, 6144-6154).
- the protein having a modified epitope may be chemically synthesized, or genetically or biologically synthesized.
- host cells or animals that temporarily or permanently retain the gene of a protein having a modified epitope may be used.
- Host cells and animals can be used, for example, as production systems for protein production and expression.
- a host cell a eukaryotic cell or a prokaryotic cell may be used.
- Examples of eukaryotic cells that can be used as host cells include animal cells, plant cells, and fungal cells.
- Animal cells include mammalian cells such as CHO (Puck et al., (1958) J. Exp. Med. 108 (6): 945-956), COS, HEK293, 3T3, myeloma, BHK (baby hamster kidney) And amphibian cells such as Xenopus oocytes (Valle et al., Nature (1981) 291: 338-340) and insect cells such as Sf9, Sf21, and Tn5.
- CHO cells are preferred for mass expression purposes.
- a vector having a gene of a protein having a modified epitope for a host cell for example, calcium phosphate method, DEAE dextran method, method using cationic ribosome DOTAP (Boehringer Mannheim), electroporation method, lipofection, etc. It may be introduced by this method.
- plant cells for example, cells derived from Nicotiana tabacum and Lemna minor are known as protein production systems, and proteins may be produced by a method of culturing these cells.
- fungal cells examples include yeast, for example, cells of the genus Saccharomyces (Saccharomyces cerevisiae, Saccharomyces pombe, etc.), or filamentous fungi, for example, cells of the genus Aspergillus. -A protein expression system using niger (Aspergillus niger, etc.) may be used.
- prokaryotic cells a production system using bacterial cells may be used.
- bacterial cells for example, E. coli and Bacillus subtilis may be used.
- animals include genetically modified animals and transgenic animals.
- the type of animal is not limited, and for example, cows, sheep, mice, and the like may be used. In such a case, for example, a protein secreted in a body fluid such as milk may be collected.
- a protein having a modified epitope may be mass-produced continuously or commercially.
- the produced protein or a composition (for example, pharmaceutical composition) containing the protein is also included in the present invention.
- the present invention in another aspect, is a method for predicting whether a protein is immunogenic in a subject, (I) providing a cell that expresses one or more allotypes of an MHC molecule of interest intended to be administered a target protein, wherein the cell is differentiated from a stem cell or a progenitor cell derived therefrom
- a process characterized by: (II) a step of bringing a target protein into contact with the “cell expressing one or more allotypes of MHC molecules”; (III) isolating a complex of a peptide contained in the target protein and an MHC molecule from the “cell expressing one or more allotypes of the MHC molecule”; (IV) eluting and identifying the peptide from the complex; and (V) optionally verifying whether the identified peptide is an epitope that induces immunogenicity, When the identified peptide is an epitope that induces immunogenicity, it relates to a method that indicates that the target protein is immunogenic
- the prediction method also makes it possible to compare the presence or absence or degree of immunogenicity based on each allotype or allotype set of MHC molecules for a given protein.
- the prediction method further provides one or a plurality of cells expressing one or more allotypes of the target MHC molecule so that all sets of allotypes of the MHC molecule possessed by the target are included.
- the stem cells are still more preferably stem cells (eg, iPS cells or ES cells) derived from the subject (eg, mammals, preferably humans). This is because antigen-presenting cells obtained by differentiating stem cells maintain the allotype of the MHC molecule.
- stem cells derived from the subject antigen presentation with all the set of allotypes of the MHC molecule that the subject has Based on the ability to produce cells.
- the prediction method also relates to a method for selecting a subject (for example, a patient) having immunogenicity or a method for selecting a subject (for example, a patient) having no immunogenicity.
- the prediction method also relates to a method that indicates that one or more specific allotypes of the MHC molecule are involved in immunogenicity in relation to the protein intended for administration.
- stem cells eg, iPS cells or ES cells
- stem cells derived from the subject (eg, mammals, preferably humans) may be prepared from any somatic cell of the subject as long as the MHC molecule allotype is maintained.
- iPS cells can also be produced from PBMC isolate
- Such reports include, for example, Soares FA, Pedersen RA, Vallier L., Generation of Human Induced Pluripotent Stem Cells from Peripheral Blood Mononuclear Cells, Using Sendai Virus, Methods QuartiB: 2015 ., Generation of Patient-Specific induced Pluripotent Stem Cell from Peripheral Blood Mononuclear Cells by Sendai Reprogramming Vectors, Methods Mol Biol. 2014 Dec 19; Su RJ, Neisses A, ZhangXB. Episomal Vectors, Methods Mol Biol.
- the cell can be used in the prediction method.
- the cell derived from the subject used for the production of stem cells such as iPS cells is a cell that can identify an MHC molecule allotype (HLA genotype in humans) or an MHC molecule allotype. Cells (or predicted). Alternatively, iPS cells in which the allotype of the MHC molecule is already known (or predicted) may be used.
- the present invention provides a composition for treating and / or preventing a disease associated with the protein in a subject, comprising the protein as an active ingredient, wherein the subject is It relates to a composition, characterized in that it is selected from (only) subjects that are predicted to be non-immunogenic to the protein.
- the “therapeutic and / or prophylactic composition” preferably contains a therapeutically and / or prophylactically effective amount of the protein, and those skilled in the art can appropriately determine the effective amount of the protein.
- the “treatment and / or prevention composition” may include one or more other agents.
- protein was predicted to be non-immunogenic means that the target protein does not elicit immunogenicity in the subject or is immunized only to an extent that is acceptable from the standpoint of efficacy and safety. It may mean not to cause primality.
- the composition is preferably a pharmaceutical composition.
- the protein when used in a (pharmaceutical) composition, it can be formulated by a known pharmaceutical manufacturing method.
- the protein is contained orally as a tablet, capsule, elixir, or microcapsule with sugar coating as necessary, or in water or other pharmaceutically acceptable liquid It can be used parenterally (eg, transdermally, intranasally, transbronchially, intramuscularly, or intravenously) in the form of a solution or suspension.
- the (pharmaceutical) composition may be produced by appropriately including a pharmaceutically acceptable carrier, flavoring agent, excipient, vehicle, preservative, stabilizer, or binder.
- a liquid carrier such as fats and oils can be further contained.
- Sterile solutions for injection can be formulated according to methods well known to those skilled in the art using a vehicle such as distilled water for injection.
- Aqueous solutions for injection include, for example, isotonic solutions containing physiological saline, glucose and other adjuvants such as D-sorbitol, D-mannose and D-mannitol.
- a suitable solubilizing agent such as an alcohol such as ethanol, a polyalcohol such as propylene glycol or polyethylene glycol, a polyionic salt such as polysorbate 80 (TM) or HCO-50 may be used in combination.
- the oily liquid include sesame oil and soybean oil.
- a solubilizing agent for example, benzyl benzoate or benzyl alcohol may be used in combination.
- the prepared injection solution may be used by filling a suitable ampoule.
- the dosage, administration method, administration interval, etc. of the protein or the therapeutic and / or prophylactic composition containing the protein as an active ingredient vary depending on the body weight, age, symptoms, etc. of the patient. It can be selected and determined as appropriate.
- the protein and the disease related to the protein are not particularly limited. It is still preferred if the protein is a protein that can cause problems with immunogenicity, efficacy or safety when administered in vivo.
- Diseases are not limited, but include, for example, autoimmune diseases (for example, Rheumatoid arthritis, type I diabetes, multiple sclerosis (MS), coeliac disease, myasthenia gravis (MG) or systemic lupus erythematosus (SLE)), cancer (eg Examples include melanoma, breast cancer, B cell lymphomas, prostate cancer, renal cancer) or infectious diseases (eg diseases diseased by HIV, hepatitis C virus, measles virus, mycobacteria).
- autoimmune diseases for example, Rheumatoid arthritis, type I diabetes, multiple sclerosis (MS), coeliac disease, myasthenia gravis (MG) or systemic lupus erythematosus (SLE)
- cancer eg Examples include melanom
- a specific combination of a protein and a disease related to the protein is not limited, but Muromanab and Allograft rejection; Abciximab and PTCA adjunct; Rituximab and Non-Hodgkin lymphoma; Daclizumab and Transplant rejection; Trastuzumab and Breast, respectively.
- the present invention provides: [30] A method for the treatment and / or prevention of a disease associated with a protein, the method comprising the step of administering the protein to a subject in need of the treatment and / or prevention, and the subject comprising the aforementioned prediction method
- the protein is selected from (only) a subject that is predicted not to be immunogenic.
- the present invention provides: [31] Use of the protein for the manufacture of a medicament for treatment and / or prevention of a disease associated with the protein, and the subject of the treatment and / or prevention is the protein according to the prediction method described above For use, characterized in that it is selected from (only) subjects that are predicted to be non-immunogenic.
- the present invention relates to the use of stem cells, progenitor cells derived therefrom, or cells that express differentiated MHC molecules in the various methods of the present invention described above.
- Method-Cells used- Human iPS cells Tic (JCRB1331), introduced from JCRB cell bank; 201B7, introduced from iPS Academia Japan.
- Feeder cells EmbryoMax Primary Mouse Embryonic Fibroblasts (MEF), Hygro resistant, C57BL / 6 (purchased from Nihon Millipore, Cat.:PMEF-HL); SNL 76/7 feeder cells (SNL) (purchased from Cell Biolabs, Inc, Cat .: CBA-316).
- Feeder cells MEF
- FBS Embryonic Stem Cell Fetal Bovine Serum
- 10% L-glutamine
- Penicillin / Suspend it in DMEM Gibco, Cat.:10569-010
- Streptomycin Invitrogen, Cat.:15140-122
- dilute to 1–2 ⁇ 10 5 cells / mL and add to gelatin-coated dishes. 4 mL each was seeded and cultured at 37 ° C under 5% CO 2 for 1 day. 3.
- iPSellon cardiac, Cat.:007101
- bFGF basic fibroblast growth factor
- Feeder cells were adhered to the bottom of the dish and separated from human iPS cell colonies. 4. Collect all non-adherent colonies of human iPS cells from gelatin-coated dishes and dilute insulin-transferrin-selenium-X 100X (ITS) (Life Tech, Cat .: 51500-056) to 1/100 times. Suspended in Primate ES cell medium added as above, seeded 3 mL each in MG dishes from which the supernatant was removed, and cultured for 1 day at 37 ° C and 5% CO 2 (see the upper left figure in Fig. 2) . 5.
- ITS insulin-transferrin-selenium-X 100X
- ITS was reduced to 1/100 times, recombinant human Vascular Endothelial Growth Factor 165 (rhVEGF165) (R & D Systems, Cat.:293-VE) was 40 ng / mL, recombinant human Stem Cell Factor (rhSCF) (R & D Systems, Cat.:255-SC) was added at a rate of 4 mL of Prime ES cell medium supplemented at 50 ng / mL, and the cells were cultured for 2 days at 37 ° C and 5% CO 2 . (Refer to the photo on the right in Fig. 2). 7.
- rhVEGF165 Vascular Endothelial Growth Factor 165
- rhSCF recombinant human Stem Cell Factor
- ITS is reduced to 1/100 times, recombinant human Granulocyte Macrophage colony-stimulating Factor (rhGM-CSF) (Humanzyme, Cat.:HZ-1082) is 100 ng / mL, recombinant human Macrophage colony-stimulating StemPro-34 medium (Life Tech., Cat .: 10640) with factor (rhM-CSF) (Humanzyme, Cat .: HZ-1039) added to 50 ng / mL was added in 5 mL portions, at 37 ° C, 5 ° C. The cells were cultured under% CO 2 conditions, and the culture solution was changed every 3 to 4 days (see the middle photo in FIG. 2). 8.
- rhGM-CSF Granulocyte Macrophage colony-stimulating Factor
- Non-adherent cells appeared around 50 days in culture, and the non-adherent cells in the dish were collected once every 7 to 14 days to form monocyte-like cells. 9. Collect some monocyte-like cells, anti-HLA-DR antibody (BD biosciences, Cat.:347364), anti-human HLA-DQ antibody (BD biosciences, Cat.:555563), anti-human HLA- DP antibody (Santa Cruz Biotechnology, Cat.:sc-53308), anti-human HLA-ABC antibody (BD biosciences, Cat.:555552), anti-human CD14 antibody (BD biosciences, Cat.:558121), anti-human CD80 antibody ( BD biosciences, Cat.:561134), anti-human CD86 antibody (BD biosciences, Cat.:561128), anti-human CD206 antibody (BD biosciences, Cat.:551135), anti-human CD209 antibody (BD biosciences, Cat.:551545) , Anti-human CD
- Betula verrucosa birch pollen allergen 1, Isoform a (Bet v1a) (#Bet v 1.0101; Biomay) (amino acid sequence: SEQ ID NO: 1)
- Infliximab (trade name: REMICADE (registered trademark) (Mitsubishi Tanabe Pharma Corporation) (amino acid sequence: Heavy chain variable region: SEQ ID NO: 2; Heavy chain constant region: SEQ ID NO: 3; Light chain variable region: SEQ ID NO: 4; Light chain constant region: SEQ ID NO: 5)
- Infliximab has been confirmed to be an anti-drug antibody (ADA) in clinical practice and has an epitope sequence (Self / Nonself 2010; 1 (4) pp.314-322; Current Rheumatology Report 2005; 7: 3-9; Current Opinion in Monoclonal Thrapeutics 2003; 5 (2): 172-179).
- RhFVIII Recombinant Human Factor VIII (trade name: ADVATE (registered trademark) (Baxter) (amino acid sequence: SEQ ID NO: 112)
- rhFVIII has been confirmed to be an anti-drug antibody (ADA) in clinical practice and has an epitope sequence (Simon D. Van Haren et al, Mol Cell Proteomics 2011: 10: M110 .002246).
- RhFVIII has a molecular weight about twice that of a normal IgG antibody.
- Phl p1 Phleum pretense, timothy grass pollen allergen 1 (Phl p1) (trade name: Phl p 1.0102 (Biomay) (amino acid sequence: SEQ ID NO: 113) Phl p1 is a grass pollen antigen, and an epitope sequence has been reported (Carla Oseroff et al, J of immunol 2010: 185 (2): 943-955).
- rhGM-CSF 200 ng / mL
- rhIL-4 Human Interleukin-4 (Humanzyme, Cat .: HZ-1075) Suspended in StemPro-34 medium with 10 ng / mL at a cell concentration of 1 ⁇ 10 5 cells / mL, seeded in 3 mL each in a 6-well plate, and cultured at 37 ° C under 5% CO 2 for 5 days. Culture was performed. 2.
- rhFVIII and Phl p1 remove 2 mL of culture supernatant from each well, then add rhFVIII 30 ⁇ g / mL or Phl p1 10 ⁇ g / mL, followed by rhTNF- ⁇ 10 ng / mL, 37 ° C., 5
- the cells were cultured for 1 day under% CO2 conditions to obtain dendritic cell-like cells. 3. All the dendritic cell-like cells were collected from the 6-well plate, spun down at 1200 rpm for 5 minutes at 4 ° C, and all the supernatant was removed and suspended in 1 mL of DPBS at 4 ° C.
- Anti-HLA-DR antibody G46-6 (BD Biosciences, Cat .: 555809) was solidified on CNBr-activated Sepharose beads (GE Healthcare, Cat .: 17-0430-01) at a final concentration of 1 mg / mL, Anti-HLA-DR antibody immobilized beads were used. 2. Anti-HLA-DR antibody solidified beads were stored in PBS (Wako, Cat .: 041-20211) containing 0.02% sodium azide (Wako, Cat .: 190-14901).
- Lysis Buffer was added to a frozen pellet of dendritic cell-like cells in a 10-fold amount under ice-cooling conditions, and shaken at 1100 rpm for 1 hour at 4 ° C in Thermomixer Confort (Eppendorf) to obtain a lysate. 3. Spin down at 14000 rpm for 10 minutes at 4 ° C to separate the lysate from cell debris and cell nuclei. 4. Add 5-10 ⁇ L of anti-HLA-DR antibody-immobilized beads to 100 ⁇ L of lysate, shake at 1100 rpm, 4 ° C. overnight with a horizontal shaker, and HLA-DR-peptide in lysate The complex was bound to anti-HLA-DR antibody immobilized beads. 5.
- HLA-DR-peptide complex bound to HLA-DR antibody-immobilized beads is suspended in 400 ⁇ L of ultrapure water, transferred to Ultrafree-MC filter (Durapore PVDF, 0.22 um) (Millipore), 14000 rpm, 10 Spin down at 4 ° C for 2 seconds. 2. The ultrapure water dropped on the bottom of the tube was removed, and 400 ⁇ L of ultrapure water was added onto the filter, and the washing operation was repeated 10 times at 14000 rpm for 10-30 seconds at 4 ° C. 3.
- LC analysis conditions are as described in EP1715343A1, or similar conditions known to those skilled in the art, using a combination of reversed-phase material and ion-exchange material, or a column of reversed-phase material alone and appropriate buffering. It can carry out using a liquid.
- the HPLC column was connected to an Orbitrap Elite (Thermo) equipped with a nano-LC electrospray ionization source, and full scan precision mass spectrometry and mass spectrometry by MS-MS were performed according to the manufacturer's protocol. 2.
- Peptide sequence analysis was performed using the SEQUEST algorithm.
- FIG. 3A and FIG. 3B show the results of examining molecules expressed on the cell surface of monocyte-like cells prepared using Tic obtained by flow cytometer analysis.
- expression of CD14 which is a specific marker of monocytes, was observed, as well as expression of CD80, CD86, which are T cell activation molecules, and CD11b, CD11c, which are adhesion molecules.
- CD14 which is a specific marker of monocytes
- CD80, CD86 which are T cell activation molecules
- CD11b, CD11c which are adhesion molecules.
- FIG. 4A and FIG. 4B show the results of examining molecules expressed on the cell surface of monocyte-like cells prepared using 201B7, obtained by flow cytometer analysis.
- the monocyte-like cells obtained by this example were not only the monocyte-like cells produced using Tic, but also the expression of CD14, which is a specific monocyte marker, and T cell activation molecules. Expression of certain CD80, CD86, and adhesion molecules CD11b, CD11c was observed.
- FIG. 5A and FIG. 5B show the results of examining molecules expressed on the cell surface of dendritic cell-like cells prepared using Tic obtained by flow cytometer analysis.
- expression of antigen-presenting molecules HLA-DR, HLA-DQ, HLA-DP and HLA-ABC, and dendritic cell specific markers CD206 and CD209 were observed.
- expression of T80 activation molecules CD80 and CD86 and adhesion molecules CD11b and CD11c was observed.
- Increased expression of CD11c was observed compared to monocyte-like cells.
- the expression of CD14 a specific monocyte marker, decreased. Since each marker had a single peak, it was considered that cells having the characteristics of dendritic cells were uniformly produced.
- FIG. 6A and FIG. 6B show the results of examining the molecules expressed on the cell surface of dendritic cell-like cells prepared using 201B7, obtained by flow cytometer analysis.
- the antigen-presenting molecules HLA-DR, HLA-DQ, HLA-DP and HLA-ABC, as well as dendritic cell specificities similar to the dendritic cell-like cells prepared from Tic
- CD206 and CD209 which are genetic markers, was observed.
- CD80 and CD86 which are T cell activation molecules
- CD11b and CD11c which are adhesion molecules
- FIG. 7 shows the analysis result of the amino acid sequence of the peptide detected when dendritic cell-like cells prepared from Tic were exposed to Bet v1a (a).
- a part of the amino acid sequence of Bet v1a was detected from the peptide separated from the HLA-DR molecule extracted from the dendritic cell-like cells exposed to Bet v1a in this example.
- FIG. 7 shows the analysis result of the amino acid sequence of the peptide detected when exposed to Bet v1a, which was detected under the condition (control) when Bet v1a was not treated (b).
- epitope number indicates a group of detected peptides that are seen in order from the N-terminus in the amino acid sequence of Bet v1a. For example, 18 types of peptides were detected in epitope number 1.
- FIG. 8 shows the analysis result of the amino acid sequence of the peptide detected when dendritic cell-like cells prepared from 201B7 were exposed to Bet v1a (a).
- a part of the amino acid sequence of Bet v1a was detected from the peptide separated from the HLA-DR molecule extracted from the dendritic cell-like cells exposed to Bet v1a in this example.
- FIG. 8 shows the analysis result of the amino acid sequence of the peptide detected when exposed to Bet v1a, which was detected under the condition (control) when Bet v1a was not treated (b).
- FIG. 9A shows the analysis result of the amino acid sequence of the peptide detected when dendritic cell-like cells prepared from Tic were exposed to Infliximab (a).
- a part of the amino acid sequence found in the H chain and L chain of Infliximab was detected from the peptide separated from the HLA-DR molecule extracted from the dendritic cell-like cells exposed to Infliximab in this Example.
- FIG. 9B shows the analysis result of the amino acid sequence of the peptide detected when exposed to Infliximab, which was detected under the condition (control) when Infliximab was not treated (b). Only part of the amino acid sequence found in the H chain of Infliximab was detected.
- FIGS. 10A to 10H show the analysis results of the amino acid sequences of peptides detected when dendritic cell-like cells prepared from Tic were exposed to rhFVIII.
- a part of the amino acid sequence of rhFVIII was detected from the peptide separated from the HLA-DR molecule extracted from the dendritic cell-like cells exposed to rhFVIII in this Example.
- FIG. 11 shows the analysis result of the amino acid sequence of the peptide detected when dendritic cell-like cells prepared from Tic were exposed to Phl p1. In this example, a part of the amino acid sequence of Phl p1 was detected.
- the peptide presented on the HLA-DR molecule of the dendritic cell-like cell exposed to the antigen is separated as an HLA-DR-peptide complex using anti-HLA-DR beads.
- the sequence of each presented peptide was identified by ion trap MS / MS mass spectrometry.
- HLA-DQ molecules, HLA-DP molecules, HLA-A molecules, HLA are present in antigen-presenting cells such as monocyte-like cells and dendritic cell-like cells.
- -B molecule and HLA-C molecule were confirmed to be expressed.
- MHCII molecules for example, HLA-DQ molecule or HLA-DP molecule
- MHCI molecules instead of HLA-DR molecules, as well as peptides that are presented as antigens. Can be detected and identified.
- MAPPs are also differentiated from stem cells in the present invention or progenitor cells derived therefrom in cells expressing other MHCII molecules such as HLA-DQ molecules and HLA-DP molecules.
- MHCII molecules such as HLA-DQ molecules and HLA-DP molecules.
- MAPPs using MHCI molecules are also disclosed in, for example, Wahl A, Schafer F, Bardet W, Buchli R, Air GM, Hildebrand WH., HLA class I molecules consistently present internal influenza epitopes.Proc Natl Acad Sci US A. 2009 Jan 13; 106 (2): 540-5.
- a cell line expressing a specific allotype of HLA-B molecule is sensitized to influenza virus, and detection of the influenza virus-derived peptide sequence presented on the HLA-B molecule by MAPPs is performed.
- MHCI molecules are known to be expressed in many cell types. From the viewpoint of easy detection of MHCI molecule-peptide complexes, cells in which MHCI molecules are highly expressed It is desirable to use
- the MAPPs using MHCI molecules may use dendritic cells differentiated from the stem cells in the present invention or progenitor cells derived therefrom.
- PBMC peripheral blood mononuclear cells
- human peripheral blood mononuclear cells After 15 minutes of standing, human peripheral blood mononuclear cells add 20mL of DPBS containing 0.5% Human Serum Alubmin low IgG and 2mM EDTA 0.5M stock solution pH8.0, and spin at 1200rpm for 5 minutes at 4 °C After down, all the supernatant was removed. This operation was performed twice. 3. To human peripheral blood mononuclear cells from which the supernatant has been removed, add DPBS containing 0.5% Human Serum Alubmin low IgG and 2 mM EDTA 0.5M stock solution pH 8.0 to 1.2 ⁇ 10 8 cells / mL. The collected human peripheral blood mononuclear cells were passed through a separation magnet LS Column (Miltenyi, Cat .: 130-042-401) and used as monocyte cells.
- white birch pollen allergens Betula verrucosa, birch pollen allergen 1, Isoform a (Bet v1a) (#Bet v 1.0101; Biomay) (amino acid sequence: SEQ ID NO: 1) were used.
- Mononuclear cells from which the supernatant was removed were treated with 10% Fetal Bovine Serum (FBS) (Gibco, Cat .: 10270, 26140), 1% Non Essential amino acids (Gibco, Cat .: 11140-035), Na-Pyruvate (Gibco, Cat .: 11360-039) 1%, Kanamycine (Gibco, Cat .: 15160-047) 1%, recombinant human Granulocyte Macrophage colony-stimulating Factor (rhGM-CSF) (R & Dsystems, Cat.
- FBS Fetal Bovine Serum
- rhGM-CSF Granulocyte Macrophage colony-stimulating Factor
- RPMI 1640 Life Tech, Cat .: 11875
- 50 ng / mL and recombinant human Interleukin-4 (rhIL-4) R & Dsystems, Cat.:204-IL
- rhIL-4 R & Dsystems, Cat.:204-IL
- FIG. 12 shows the results of examining molecules expressed on the cell surface of monocytes obtained by flow cytometer analysis.
- the monocyte cells obtained by the comparative example showed expression of CD14, which is a specific marker for monocytes, and expression of HLA-DR, which is an antigen-presenting molecule, as well as expression of CD86, which is an activation molecule for T cells. Admitted.
- 13A and 13B show the results of examining molecules expressed on the cell surface of dendritic cells obtained by flow cytometer analysis.
- the expression of antigen-presenting molecules HLA-DR, HLA-DQ and HLA-ABC, specific markers of dendritic cells CD206 and CD209, and T cell Expression of CD80 and CD86 as activation molecules and CD11b and CD11c as adhesion molecules was observed.
- CD14 expression was not observed.
- FIG. 14 shows analysis results of amino acid sequences of peptides detected in (control) (FIG. 14, FIG. 15, FIG. 16, FIG. 17, FIG. 18, FIG. 19, FIG. 20 (b)). These detected specific amino acid sequences are also shown in Tables 2 to 8 (corresponding to FIGS. 14 to 20, respectively). In the table, the epitope number indicates a group of detected peptides that are seen in order from the N-terminus in the amino acid sequence of Bet v1a.
- FIG. 21A and FIG. 21B show the case of using two types of human iPS cell-derived dendritic cell-like cells and the case of using PBMC-derived dendritic cells for the amino acid sequences of peptides detected under the Bet v1a addition conditions. A comparison of is shown. In the analysis using dendritic cells derived from PBMC, it was considered that there was a difference in the amino acid sequence of the detected peptide between donors because it had different MHCII molecules between donors.
- the detected sequences differ between the lines because the donor had different types of MHCII molecules. It is thought that occurred. Nevertheless, most of the common sequences in peptides detected under Bet v1a addition conditions using human iPS cell-derived dendritic cell-like cells were detected when using PBMC-derived dendritic cells. Was consistent with the sequence.
- the detected peptide sequence 140-155 coincided with the sequence reported as the epitope sequence part in S. Mutschlener et al., Journal of Allergy and Clinical Immunology Vol. 125 (3), 2010.
- MAPPs using stem cells or progenitor cells derived therefrom were suggested to be more effective than MAPPs using PBMC, and were considered to be useful means for reducing protein immunogenicity.
- the present invention can be used in various biopharmaceutical diagnosis and medical fields by applying, for example, protein epitope sequence analysis.
- Bet v1a and Phl p1 which are foreign proteins derived from pollen
- Infliximab which is an antibody drug
- rhFVIII which is a drug that has the same amino acid sequence as the endogenous protein
- SEQ ID NO: 133 to 150 Partial peptide of H chain of Infliximab
- SEQ ID NO: 151 Partial peptide of L chain of Infliximab
- SEQ ID NO: 152 to SEQ ID NO: 153 Partial peptide of H chain of Infliximab
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Zoology (AREA)
- Immunology (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Hematology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Gastroenterology & Hepatology (AREA)
- Urology & Nephrology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Pathology (AREA)
- Food Science & Technology (AREA)
- Toxicology (AREA)
- General Physics & Mathematics (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Developmental Biology & Embryology (AREA)
- Botany (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Tropical Medicine & Parasitology (AREA)
Abstract
Description
[1]タンパク質のエピトープを同定するための方法であって、
以下の工程:
(A)幹細胞又はこれに由来する前駆細胞から分化された、主要組織適合遺伝子複合体(MHC分子)を発現する細胞に、標的タンパク質を接触させる工程;
(B)前記MHC分子を発現する細胞から、前記標的タンパク質に含まれるペプチドとMHC分子との複合体を単離する工程;及び
(C)前記複合体から、前記ペプチドを溶出し、同定する工程
を含む、方法。
[2]さらに、以下の工程:
(D)同定したペプチドが免疫原性を誘導するエピトープであるか否かを検証する工程
を含む、[1]に記載の方法。
[3]前記幹細胞は、人工多能性幹細胞(iPS細胞)、胚性幹細胞(ES細胞)、核移植ES細胞(ntES細胞)、胚性生殖幹細胞(EG細胞)及び成体幹細胞からなる群から選択される、[1]又は[2]に記載の方法。
[4]前記MHC分子は、MHCII分子である、[1]~[3]のいずれか一項に記載の方法。
[5]前記MHCII分子はHLA-DR、HLA-DQ又はHLA-DPである、[4]に記載の方法。
[6]前記MHC分子を発現する細胞は、さらに、CD80、CD86、CD206及びCD209の少なくとも1つを発現している、[1]~[5]のいずれか一項に記載の方法。
[7]前記MHC分子を発現する細胞は、CD80、CD86、CD206及びCD209の全てを発現している、[6]に記載の方法。
[8]前記MHC分子を発現する細胞は、樹状細胞である、[1]~[7]のいずれか一項に記載の方法。
[9]前記MHC分子を発現する細胞は、前記標的タンパク質を投与することが意図される対象のMHC分子の1又は複数のアロタイプを発現する、[1]~[8]のいずれか一項に記載の方法。
[10]前記工程(A)を無血清下で行う、[1]~[9]のいずれか一項に記載の方法。
[11]前記樹状細胞は、
以下の工程:
(a)幹細胞又はこれに由来する前駆細胞を分化させて中胚葉前駆細胞を得る工程;
(b)前記中胚葉前駆細胞を分化させて単球細胞を得る工程;及び
(c)前記単球細胞を分化させて未成熟樹状細胞を得、及び場合により、前記未成熟樹状細胞をさらに刺激して成熟樹状細胞を得る工程
を含む方法によって製造され、
さらに、前記工程(a)~(c)のうち、少なくとも前記工程(c)において無血清培地が用いられる、[1]~[10]のいずれか一項に記載の方法。
[12]前記工程(b)が、顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びマクロファージコロニー刺激因子(M-CSF)を含む無血清培地下で、前記中胚葉前駆細胞を分化させて単球細胞を得る工程を含む、[11]に記載の方法。
[13]前記工程(c)が、
(c1)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びインターロイキン4(IL-4)を含む無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得る工程を含み、及び場合により、
(c2)前記未成熟樹状細胞を免疫原、及び、場合により炎症性サイトカインに接触させることで、成熟樹状細胞へと誘導する工程を含む、[11]又は[12]に記載の方法。
[14]前記樹状細胞が未成熟樹状細胞であり、前記未成熟樹状細胞は、免疫原性を有する標的タンパク質に接触することで、成熟樹状細胞に誘導される、[8]~[13]のいずれか一項に記載の方法。
[15]前記標的タンパク質が、サイトカイン、ケモカイン、成長因子、抗体、酵素、構造タンパク質、ホルモン、及び、これらのいずれかの断片からなる群の1種又は1種以上から選択される、[1]~[14]のいずれか一項に記載の方法。
[16]免疫原性が減少又は消失したタンパク質の製造方法であって、
以下の工程:
(1)[1]~[15]のいずれか一項に記載の方法に従って、タンパク質のエピトープを同定する工程;
(2)MHC分子への結合が減少又は消失するように、前記エピトープを修飾する工程;及び
(3)修飾されたエピトープを有するタンパク質を製造する工程
を含む、方法。
[17][16]に記載の方法に従って得られうる、タンパク質。
[18]タンパク質が対象において免疫原性を有するか否かを予測する方法であって、
(I)標的タンパク質を投与することが意図される対象のMHC分子の1又は複数のアロタイプを発現する細胞を提供する工程であって、前記細胞は幹細胞又はこれに由来する前駆細胞から分化されることを特徴とする、工程;
(II)前記「MHC分子の1又は複数のアロタイプを発現する細胞」に、標的タンパク質を接触させる工程;
(III)前記「MHC分子の1又は複数のアロタイプを発現する細胞」から、前記標的タンパク質に含まれるペプチドとMHC分子との複合体を単離する工程;
(IV)前記複合体から、前記ペプチドを溶出し、同定する工程;及び
(V)場合により、同定したペプチドが免疫原性を誘導するエピトープであるか否かを検証する工程
を含み、
前記同定したペプチドが免疫原性を誘導するエピトープである場合に、前記標的タンパク質が前記対象において免疫原性を有することを指し示す、方法。
[19]前記対象が有するMHC分子のアロタイプの全てのセットが含まれるように、前記対象のMHC分子の1又は複数のアロタイプを発現する細胞を1又は複数提供する、[18]に記載の方法。
[20]前記幹細胞が、前記対象に由来する人工多能性幹細胞(iPS細胞)である、[18]又は[19]に記載の方法。
[21]タンパク質を有効成分として含む、対象における、前記タンパク質に関連した疾患の治療及び/又は予防用組成物であって、
前記対象は、[18]~[20]のいずれか一項に記載の方法に従って、前記タンパク質に免疫原性を有さないことが予測された対象から選択されることを特徴とする、組成物。
[22]幹細胞もしくはこれに由来する前駆細胞、又は、これから分化されたMHC分子を発現する細胞の、[1]~[16]及び[18]~[20]のいずれか一項に記載の方法における使用。
[23]幹細胞又はこれに由来する前駆細胞から樹状細胞を製造する方法であって、
以下の工程:
(a’)幹細胞又はこれに由来する前駆細胞を分化させて中胚葉前駆細胞を得る工程;
(b’)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びマクロファージコロニー刺激因子(M-CSF)を含む無血清培地下で、前記中胚葉前駆細胞を分化させて単球細胞を得る工程;及び
(c’)無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得、及び場合により、前記未成熟樹状細胞をさらに刺激して成熟樹状細胞を得る工程
を含む、方法。
[24]前記工程(c’)が、
(c1’)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びインターロイキン4(IL-4)を含む無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得る工程を含み、及び場合により、
(c2’)前記未成熟樹状細胞を、免疫原、及び、場合により炎症性サイトカインに接触させることで、成熟樹状細胞へと誘導する工程を含む、
[23]に記載の方法。
[25][23]又は[24]に記載の方法によって得られうる、樹状細胞。
[26]MHCII分子に加えて、さらに、CD80、CD86、CD206及びCD209の少なくとも1つを発現している、[25]に記載の樹状細胞。
[27]CD80、CD86、CD206及びCD209の全てを発現している、[26]に記載の樹状細胞。
[28][25]~[27]のいずれかに記載の樹状細胞を含む、細胞組成物。
[29]また、上記に記載の1又は複数の態様を任意に組み合わせたものも、当業者の技術常識に基づいて技術的に矛盾しない限り、本発明に含まれることが当業者には当然に理解される。
一態様において、ヒトになり得る受精卵や胚を破壊する事に対して生じ得る倫理的問題から、ヒトES細胞を作製する際に用いる胚としては、例えば、体外受精による不妊治療において母体に戻されなかった凍結保存されている胚のうち、破棄されることが決定した余剰胚を利用したり、体外受精プロセスで得られる発生過程が停止してしまった胚を利用したり、あるいは、それ自体でヒトへと成長する内在的能力を備えない、細胞分裂と成長が単為生殖に基づく未授精卵を利用してもよい。あるいは、胚盤胞期以前の卵割期の胚の単一割球のみを用いて、胚の発生能を損なうことなく、受精卵を破壊せずにES細胞を作製してもよい(Chung Y, Klimanskaya I, Becker S, Marh J, Lu SJ, Johnson J, Meisner L, Lanza R. (2006). Nature 439: 216-219.;Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. (2006). Nature 444: 481-485.;Chung Y, Klimanskaya I, Becker S, Li T, Maserati M, Lu SJ, Zdravkovic T, Ilic D, Genbacev O, Fisher S, Krtolica A, Lanza R. (2008). Cell Stem Cell 2: 113-117.)。あるいは、発生が停止したヒトの胚からES細胞を作製してもよい(Zhang X, Stojkovic P, Przyborski S, Cooke M, Armstrong L, Lako M, Stojkovic M. (2006). Stem Cells 24: 2669-2676.)。
初期化因子は、ES細胞に特異的に発現している遺伝子、その遺伝子産物もしくはnon-cording RNA、又は、ES細胞の未分化維持に重要な役割を果たす遺伝子、その遺伝子産物もしくはnon-cording RNA、あるいは低分子化合物であってもよい。初期化因子として、例えば、OCT3/4、SOX2、SOX1、SOX3、SOX15、SOX17、KLF4、KLF2、c-MYC、N-MYC、L-MYC、NANOG、LIN28、FBX15、ERAS、ECAT15-2、TCLL、beta-catenin、LIN28B、SALL1、SALL4、ESRRB、NR5A2、TBX3を挙げてもよい。これらの初期化因子は、単独又は組み合わせて用いてもよい。初期化因子の組み合わせとしては、例えば以下の組み合わせを挙げることができる。
(i)OCT遺伝子、KLF遺伝子、SOX遺伝子、MYC遺伝子;
(ii)OCT遺伝子、SOX遺伝子、NANOG遺伝子、LIN28遺伝子;
(iii)OCT遺伝子、KLF遺伝子、SOX遺伝子、MYC遺伝子、hTERT遺伝子、SV40 large T遺伝子;
(iv)OCT遺伝子、KLF遺伝子、SOX遺伝子
あるいは、初期化因子の組み合わせとしては、例えば、WO2007/069666、WO2008/118820、WO2009/007852、WO2009/032194、WO2009/058413、WO2009/057831、WO2009/075119、WO2009/079007、WO2009/091659、WO2009/101084、WO2009/101407、WO2009/102983、WO2009/114949、WO2009/117439、WO2009/126250、WO2009/126251、WO2009/126655、WO2009/157593、WO2010/009015、WO2010/033906、WO2010/033920、WO2010/042800、WO2010/050626、WO2010/056831、WO2010/068955、WO2010/098419、WO2010/102267、WO2010/111409、WO2010/111422、WO2010/115050、WO2010/124290、WO2010/147395、WO2010/147612、WO2012/115276に記載の組み合わせを用いてもよい。初期化因子又は初期化を促進する因子としては、例えば、当業者に公知の阻害剤である、MEK阻害剤、DNAメチルトランスフェラーゼ阻害剤、ヒストンデアセチラーゼ(HDAC)阻害剤、ヒストンメチルトランスフェラーゼ阻害剤、p53阻害剤を挙げてもよい。初期化因子は、場合によりベクター(例えば、ウイルスベクター、プラスミドベクター、人工染色体ベクター)等を用いて、例えば、リン酸カルシウム法、リポフェクション法、マイクロインジェクション法等の当業者に公知の方法に従って体細胞内に導入してよい。iPS細胞誘導のための培地としては、例えば、10~15%FBSを含有するDMEM、DMEM/F12又はDME培地(これらは白血病抑制因子(LIF)、penicillin/streptomycin、puromycin、L-glutamine、非必須アミノ酸類、β-メルカプトエタノール等をさらに適宜含んでよい。)、又は、当業者に公知の市販の培地を適宜用いてもよい。
また、特に、樹状細胞は抗原提示能が強く、ヘルパーT細胞の活性化能が強いことから、抗原提示細胞として有利である。また、抗原提示細胞としては、未成熟樹状細胞が最も好ましい。樹状細胞は、細胞突起を有し、樹状又は樹枝状の形態を呈する細胞である。樹状細胞としての特性を有しているかの判定基準としては、例えば、MHCII分子に加えて、CD11b、CD11c、CD40、CD80、CD83、CD86、CD123、CD205、CD206、CD209、CCR7の少なくとも1つをさらに発現しているかを指標としてもよく、MHCII分子、CD80、CD86、CD206、CD209の全てを発現しているかを指標とするのがより好ましい。MHCII分子、CD80、CD86、CD206、CD209の全てを発現していて、かつ、CD14陰性の樹状細胞がさらに好ましい。マクロファージ細胞としての特性を有しているかの判定基準としては、例えば、MHCII分子に加えて、CD11bをさらに発現しているかを指標としてもよい。なお、CD80及びCD86は、ヘルパーT細胞にシグナルを伝達して当該細胞を活性化させることで知られる。本実施例で得られた樹状細胞様細胞は、細胞の形状、細胞表面分子の発現、ヘルパーT細胞刺激能力という点で単球に由来する樹状細胞と類似した性質を有することから、本明細書における樹状細胞に含まれてよい。同様に、本実施例で得られた単球様細胞は、本明細書における単球細胞に含まれてよい。
(a)幹細胞又はこれに由来する前駆細胞を分化させて中胚葉前駆細胞を得る工程;
(b)前記中胚葉前駆細胞を分化させて単球細胞を得る工程;及び
(c)前記単球細胞を分化させて未成熟樹状細胞を得、及び場合により、前記未成熟樹状細胞をさらに刺激して成熟樹状細胞を得る工程
を含んでよい。また、前記工程(a)及び前記工程(b)は連続して行うことができる。さらに、前記工程(a)~(c)のうち、少なくとも前記工程(c)において無血清培地が用いられてよく、前記工程(b)と(c)の両方で無血清培地が用いられることが好ましく、前記工程(a)~(c)の全てで無血清培地が用いられることがより好ましい。
(ci)造血因子を含む培地で前記単球を(浮遊)培養し、分化させることで未成熟樹状細胞(様細胞)を得る工程を含み、及び場合により、
(cii)得られた未成熟樹状細胞(様細胞)を、さらに、免疫原、及び、場合により炎症性サイトカインに接触させることで、成熟樹状細胞(様細胞)へと誘導する工程
を含んでよい。
(d)前記単球細胞を分化させてマクロファージを得る工程
を行ってもよい。かかる場合、造血因子として好ましくはGM-CSF又はM-CSFを用いて、前記単球細胞をマクロファージへと分化させることができる。さらに、例えばIFN-γ又はLPSを添加することによりM1マクロファージへと分化させることができ、あるいは、例えばIL-4又はIL-13を添加することによりM2マクロファージへと分化させることができる(マクロファージはヘルパーT細胞の産生するサイトカインを受け取ることにより活性化することが知られており、古典的活性化(M1マクロファージ)と選択的活性化(M2マクロファージ)が知られている。)。
[23]幹細胞又はこれに由来する前駆細胞から(in vitroで)樹状細胞を製造する方法であって、以下の工程:
(a’)幹細胞又はこれに由来する前駆細胞を分化させて中胚葉前駆細胞を得る工程;
(b’)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びマクロファージコロニー刺激因子(M-CSF)を含む無血清培地下で、前記中胚葉前駆細胞を分化させて単球細胞を得る工程;及び
(c’)無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得、及び場合により、前記未成熟樹状細胞をさらに刺激して成熟樹状細胞を得る工程
を含む、方法。
[24]前記工程(c’)が、
(c1’)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びインターロイキン4(IL-4)を含む無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得る工程を含み、及び場合により、
(c2’)前記未成熟樹状細胞を、免疫原、及び、場合により炎症性サイトカインに接触させることで、成熟樹状細胞へと誘導する工程を含む、
[23]に記載の方法。
[25][23]又は[24]に記載の方法によって得られうる、樹状細胞。
[26]MHCII分子に加えて、さらに、CD80、CD86、CD206及びCD209の少なくとも1つを発現している、[25]に記載の樹状細胞。
[27]CD80、CD86、CD206及びCD209の全てを発現している、[26]に記載の樹状細胞。
[28][25]~[27]のいずれかに記載の樹状細胞を含む、細胞組成物。
(A)幹細胞又はこれに由来する前駆細胞から分化された、主要組織適合遺伝子複合体(MHC分子)を発現する細胞に、標的タンパク質を接触させる工程;
(B)前記MHC分子を発現する細胞から、前記標的タンパク質に含まれるペプチドとMHC分子との複合体を単離する工程;及び
(C)前記複合体から、前記ペプチドを溶出し、同定する工程
を含む、方法に関する。さらに、当該方法は、以下の工程:
(D)同定したペプチドが免疫原性を誘導するエピトープであるか否かを検証する工程
を含んでよい。当該方法は、全行程をin vitroで実施できる。
(1)上述の方法に従って、タンパク質のエピトープを同定する工程;
(2)MHC分子への結合が減少又は消失するように、前記エピトープを修飾する工程;及び
(3)修飾されたエピトープを有するタンパク質を製造する工程
を含む、方法に関する。また、本発明は、さらなる別の態様において、前記製造方法に従って得られた(obtained)タンパク質、又は得られ得る(obtainable)タンパク質にも関する。「得られ得る(obtainable)タンパク質」とは、前記製造方法を用いれば得ることが可能なタンパク質を意味してよい。
(I)標的タンパク質を投与することが意図される対象のMHC分子の1又は複数のアロタイプを発現する細胞を提供する工程であって、前記細胞は幹細胞又はこれに由来する前駆細胞から分化されることを特徴とする、工程;
(II)前記「MHC分子の1又は複数のアロタイプを発現する細胞」に、標的タンパク質を接触させる工程;
(III)前記「MHC分子の1又は複数のアロタイプを発現する細胞」から、前記標的タンパク質に含まれるペプチドとMHC分子との複合体を単離する工程;
(IV)前記複合体から、前記ペプチドを溶出し、同定する工程;及び
(V)場合により、同定したペプチドが免疫原性を誘導するエピトープであるか否かを検証する工程
を含み、
前記同定したペプチドが免疫原性を誘導するエピトープである場合に、前記標的タンパク質が前記対象において免疫原性を有することを指し示す、方法に関する。
[30]タンパク質に関連した疾患の治療及び/又は予防方法であって、前記治療及び/又は予防を必要とする対象に前記タンパク質を投与する工程を含み、かつ、前記対象は、前述の予測方法に従って、前記タンパク質に免疫原性を有さないことが予測された対象(のみ)から選択されることを特徴とする、治療及び/又は予防方法に関する。
[31]タンパク質に関連した疾患の治療及び/又は予防用医薬の製造のための、前記タンパク質の使用であって、かつ、前記治療及び/又は予防の対象は、前述の予測方法に従って、前記タンパク質に免疫原性を有さないことが予測された対象(のみ)から選択されることを特徴とする、使用に関する。
-使用細胞-
ヒトiPS細胞:Tic(JCRB1331), JCRB細胞バンクより導入;201B7,iPSアカデミアジャパン株式会社より導入。
フィーダー細胞:EmbryoMax Primary Mouse Embryonic Fibroblasts(MEF), Hygro resistant, C57BL/6(日本ミリポアより購入,Cat.:PMEF-HL);SNL 76/7 feeder cells (SNL)(Cell Biolabs, Incより購入,Cat.:CBA-316)。
1. 蒸留水により0.1%に希釈されたgelatin from porcine skin(SIGMA,Cat.:G1890)を温めゾル状にし,60mmディッシュに2mLずつ添加し37℃,5%CO2条件下に30~180分置き,ゼラチンコートディッシュとした。
2. フィーダー細胞(MEF)を,Embryonic Stem Cell Fetal Bovine Serum(FBS)(Gibco,Cat.:10439-024)を10%,L-glutamine(Invitrogen,Cat.:25030-081)を2mM,Penicillin/Streptomycin(Invitrogen,Cat.:15140-122)を0.5%加えたDMEM(Gibco,Cat.:10569-010)に懸濁し,1~2×105cells/mLとなるよう希釈し,ゼラチンコートディッシュに4mLずつ播種し,37℃,5%CO2条件下で1日培養した。
3. basic fibroblast growth factor(bFGF)(WAKO,Cat.:064-04541)を10ng/mLとなるように加えたiPSellon(cardio,Cat.:007101)を用いて,フィーダー細胞を播種した60mmディッシュにて,37℃,5%CO2条件下で,ヒトiPS細胞の未分化を維持した培養を実施した。
4. 細胞の増殖に応じて,分化が生じたコロニーをスクレーパーにより除去し,Neutral protease, grade I(Roche Applied Science,Cat.:04 942 086 001)2U/mLと反応させ,先に剥がれるフィーダー細胞をディッシュより除去した後,ヒトiPS細胞のコロニーをスクレーパーを用いてディッシュより回収し,bFGFを10ng/mLとなるように加えたiPSellonに懸濁し,新たにフィーダー細胞を播種した60mmディッシュに播種し,37℃,5%CO2条件下で培養を継続した。
1. 蒸留水により0.1%に希釈されたgelatin from porcine skinを温めゾル状にし,60mmディッシュに2mLずつ添加し37℃,5%CO2条件下に30~180分置き,ゼラチンコートディッシュとした。
2. フィーダー細胞(SNL)を,FBSを7%,L-glutamineを2mM,Penicillin/Streptomycinを0.5%加えたDMEM(Gibco,Cat.:10569-010)に懸濁し,1~2×105cells/mLとなるよう希釈し,ゼラチンコートディッシュに4mLずつ播種し,37℃,5%CO2条件下で1日培養した。
3. bFGFを4ng/mLとなるように加えたPrimate ES cell medium(ReproCELL,Cat.:RCHEMD001)を用いて,フィーダー細胞を播種した60mmディッシュにて,37℃,5%CO2条件下で,ヒトiPS細胞の未分化を維持した培養を実施した。
4. 細胞の増殖に応じて,分化が生じたコロニーをスクレーパーにより除去し,Neutral protease, grade I 2U/mLと反応させ,先に剥がれるフィーダー細胞をディッシュより除去した後,ヒトiPS細胞のコロニーをスクレーパーを用いてディッシュより回収し,bFGFを4ng/mLとなるように加えたPrimate ES cell mediumに懸濁し,新たにフィーダー細胞を播種した60mmディッシュに播種し,37℃,5%CO2条件下で培養を継続した。
1. DMEM(Gibco,Cat.:10569-010)により40倍に希釈したMatrigel, growth-factor reduced(BD biosciences,Cat.:356230)を60mmディッシュに2mLずつ添加し,37℃,5%CO2条件下に12~72時間置き,MGディッシュとした。
2. 蒸留水により0.1%に希釈されたgelatin from porcine skin(SIGMA,Cat.:G1890)を温めゾル状にし,60mmディッシュに2mLずつ添加し37℃,5%CO2条件下に30~180分置き,ゼラチンコートディッシュとした。
3. 未分化を維持したまま培養されているヒトiPS細胞のコロニーについて,Neutral protease, grade I(Roche Applied Science,Cat.:04 942 086 001)2U/mLをディッシュに加え,先に剥がれるフィーダー細胞をディッシュより除去した後,ヒトiPS細胞の未分化コロニーをスクレーパーを用いてディッシュより回収し,Fetal Bovine Serum, embryonic stem cell-qualified (FBS)(Life Tech,Cat.:16141)を20%, L-glutamine(Invitrogen,Cat.:25030-081)を1%,Penicillin/Streptomycin(Invitrogen,Cat.:15140-122)を0.5%,2-mercaptoethanol(Invitrogen,Cat.:21985-023)を55μM加えたMEM Alpha 1x + GlutamaxI(Life Tech,Cat.:32561-037)に懸濁し,上清を除去したゼラチンコートディッシュに4mLずつ播き,1時間,37℃,5%CO2条件下で培養を行い,フィーダー細胞をディッシュ底面に接着させヒトiPS細胞のコロニーと分離した。
4. ヒトiPS細胞の未接着コロニーをゼラチンコートディッシュより全量回収し,Insulin-Transferrin-Selenium-X 100X(ITS)(Life Tech,Cat.:51500-056)を1/100倍の希釈倍率となるように加えたPrimate ES cell mediumに懸濁し,上清を除去したMGディッシュに3mLずつ播種し,37℃,5%CO2条件下で1日培養を実施した(図2上段左の写真参照)。
5. ディッシュより培地を全量除去した後,ITSを1/100倍,recombinant human bone morphogenetic protein 4(rhBMP4)(Humanzyme,Cat.:314-BP)を50ng/mLとなるように加えたPrimate ES cell mediumを7mLずつ添加し,37℃,5%CO2条件下で4日間の培養を実施した(図2上段中央の写真参照)。
6. ディッシュより培地を全量除去した後,ITSを1/100倍,recombinant human Vascular Endothelial Growth Factor 165(rhVEGF165)(R&D Systems,Cat.:293-VE)を40ng/mL,recombinant human Stem Cell Factor(rhSCF)(R&D Systems,Cat.:255-SC)を50ng/mLとなるように加えたPrimate ES cell mediumを4mLずつ添加し,37℃,5%CO2条件下で2日間の培養を実施した(図2上段右の写真参照)。
7. 培地を全量除去した後,ITSを1/100倍,recombinant human Granulocyte Macrophage colony-stimulating Factor(rhGM-CSF)(Humanzyme,Cat.:HZ-1082)を100ng/mL,recombinant human Macrophage colony-stimulating factor(rhM-CSF)(Humanzyme,Cat.:HZ-1039)を50ng/mLとなるように加えたStemPro-34 medium(Life Tech.,Cat.:10640)を5mLずつ添加し,37℃,5%CO2条件下で培養し3~4日間置きに培養液を交換した(図2下段中央の写真参照)。
8. 7.項の操作を120日間継続した。培養50日頃より非接着性細胞が出現し,7日~14日に1度の頻度でディッシュ中の非接着細胞を回収し,単球様細胞とした。
9. 作製した単球様細胞の一部を回収し,抗HLA-DR抗体(BD biosciences,Cat.:347364),抗ヒトHLA-DQ抗体(BD biosciences,Cat.:555563),抗ヒトHLA-DP抗体(Santa Cruz Biotechnology, Cat.:sc-53308),抗ヒトHLA-ABC抗体(BD biosciences,Cat.:555552),抗ヒトCD14抗体(BD biosciences,Cat.:558121),抗ヒトCD80抗体(BD biosciences,Cat.:561134),抗ヒトCD86抗体(BD biosciences,Cat.:561128),抗ヒトCD206抗体(BD biosciences,Cat.:551135),抗ヒトCD209抗体(BD biosciences,Cat.:551545),抗ヒトCD11b抗体(BD biosciences,Cat.:555388),及び抗ヒトCD11c抗体(BD biosciences,Cat.:340544)を用いて染色し,フローサイトメーター解析装置BD FACSCanto(商標)II(BD Bioscience)を用いて分析した。
免疫原性を有するタンパク質として,陽性対照として以下のタンパク質を使用した。
(1)白樺花粉アレルゲンであるBetula verrucosa, birch pollen allergen 1, Isoform a(Bet v1a)(#Bet v 1.0101; Biomay)(アミノ酸配列:配列番号1)
(2)Infliximab(商品名:REMICADE(登録商標)(田辺三菱製薬)(アミノ酸配列:Heavy chain variable region:配列番号2;Heavy chain constant region:配列番号3;Light chain variable region:配列番号4;Light chain constant region:配列番号5)
なお,Infliximabは,臨床において抗薬物抗体(Anti-drug antibody; ADA)が確認されており,エピトープ配列が存在すると考えられている(Self/Nonself 2010;1(4) pp.314-322;Current Rheumatology Report 2005;7:3-9;Current Opinion in Monoclonal Thrapeutics 2003;5(2):172-179)。
(3)Recombinant Human Factor VIII(rhFVIII)(商品名:ADVATE(登録商標)(Baxter)(アミノ酸配列:配列番号112)
なお,rhFVIIIは,臨床において抗薬物抗体(Anti-drug antibody; ADA)が確認されており,エピトープ配列が存在すると考えられている(Simon D.Van Haren et al, Mol Cell Proteomics 2011:10:M110.002246)。また,rhFVIIIは通常のIgG抗体の2倍程度の分子量を有する。
(4)Phleum pretense, timothy grass pollen allergen 1 (Phl p1)(商品名:Phl p 1.0102 (Biomay)(アミノ酸配列:配列番号113)
なお,Phl p1はイネ科花粉抗原であり,エピトープ配列が報告されている(Carla Oseroff et al, J of immunol 2010:185(2):943-955)。
1. 回収した単球様細胞について,培地を除去し,ITSを1/100倍,rhGM-CSFを200ng/mL,recombinant human Interleukin-4(rhIL-4)(Humanzyme,Cat.:HZ-1075)を10ng/mLとなるように加えたStemPro-34 mediumに細胞濃度1×105cells/mLで懸濁し,6ウェルプレートに3mLずつ播種し,37℃,5%CO2条件下で5日間の培養を実施した。
2. 各ウェルにBet v1a 3.3μg/mL又はInfiximab 10μg/mLを加え,続いてrecombinant human Tumor Necrosis Factor-α(rhTNF-α)(Humanzyme,Cat.:HZ-1014)10ng/mLを加え,37℃,5%CO2条件下で1日培養し,樹状細胞様細胞とした。rhFVIII,Phl p1の添加においては,各ウェルより培養上清を2mL除去した後,rhFVIII 30μg/mL又は,Phl p1 10μg/mLを加え,続いてrhTNF-α 10ng/mLを加え,37℃,5%CO2条件下で1日培養し,樹状細胞様細胞とした。
3. 樹状細胞様細胞を6ウェルプレートより全量回収し,1200rpm,5分,4℃でスピンダウンした後,上清を全て除去し,4℃のDPBS 1mLに懸濁した。次いで,全量をエッペンドルフチューブに移し,2500rpm,5分,4℃でスピンダウンし,上清を全て除去し,細胞のペレットを作製し,-80℃で保管した。
4. 作製した樹状細胞様細胞の一部を回収し,抗ヒトHLA-DR抗体,抗ヒトHLA-DQ抗体,抗ヒトHLA-DP抗体,抗ヒトHLA-ABC抗体,抗ヒトCD14抗体,抗ヒトCD80抗体,抗ヒトCD86抗体,抗ヒトCD206抗体,抗ヒトCD209抗体,抗ヒトCD11b抗体,及び抗ヒトCD11c抗体を用いて染色し,フローサイトメーター解析装置BD FACSCanto(商標)IIを用いて分析した。
1. 抗HLA-DR抗体G46-6(BD Biosciences,Cat.:555809)を,CNBr-activated Sepharose beads(GE Healthcare,Cat.:17-0430-01)に終濃度1mg/mLで固層化し,抗HLA-DR抗体固層化ビーズとした。
2. 抗HLA-DR抗体固層化ビーズについては0.02%アジ化ナトリウム(Wako,Cat.:190-14901)を含むPBS(Wako,Cat.:041-20211)中で保管した。
1. Tris(SIGMA,Cat.:T1503-1KG)20mM, MgCl2(MERCK,Cat.:1.05833.0250)5mMを加え,HCl(MERCK,Cat.:1.00316.1000)を用いてpH7.8に調製した超純水(Wako,Cat.:210-01303)溶液に,10% TritonX-100(Roche Diag,Cat.:11332481001)を1/10倍, protease inhibitor mix(11.6mg/mL PMSF(nakalai,Cat.:27327-94), 1.7mg/mL pepstatin A(SIGMA,Cat.:P4265-25MG), 1.7mg/mL chymostatin(Roche Diag,Cat.:11004638001), 0.8mg/mL leupeptin(SIGMA,Cat.:L9783-25MG),及び133mg/mLアジ化ナトリウム(Wako,Cat.:190-14901)のmixture)を17/5000倍となるよう加え,Lysis Bufferを調製した。
2. 樹状細胞様細胞の凍結ペレットにLysis Bufferを氷冷条件下で10倍量加え,Thermomixer Confort(Eppendorf)にて1100rpm,1時間,4℃で振とうし,ライセートを取得した。
3. 14000rpm,10分,4℃でスピンダウンし,ライセートを細胞片や細胞核と分離した。
4. 抗HLA-DR抗体固層化ビーズをライセート100μLに対して5-10μL加え,水平振とう機(horizontal shaker)で1100rpm,4℃,一晩振とうし,ライセート中のHLA-DR-ペプチド複合体を抗HLA-DR抗体固層化ビーズに結合させた。
5. 抗HLA-DR抗体固層化ビーズに結合したHLA-DR-ペプチド複合体を3000rpm,1分,4℃でスピンダウンした後,Lysis buffer 500μLで1回,さらに0.1% Zwittergent 3-12(Calbiochem,Cat.:693015)を含むPBS 500μLで2回洗浄した。
1. HLA-DR抗体固層化ビーズに結合したHLA-DR-ペプチド複合体を400μLの超純水に懸濁し,Ultrafree-MC filter(Durapore PVDF, 0.22um)(Millipore)に移し,14000rpm,10秒,4℃でスピンダウンした。
2. チューブ底に落とした超純水を除去し,400μLの超純水をフィルタ上に加え14000rpm,10-30秒,4℃でスピンダウンする洗浄操作を10回繰り返し実施した。
3. 0.1% trifluoracetic acid(Thermo Fisher Scientific,Cat.:28904)を含む超純水60uLを加え,37℃,30分インキュベートし,HLA-DR-ペプチド複合体よりペプチド混合物を溶出させた後,14000rpm,3分,18℃でスピンダウンし,vacuum centrifuge 5305C(Eppendorf)により溶出したペプチド混合物を乾燥した。
1. 乾燥したペプチド混合物を2% acetonitrile(Wako,Cat.:018-19853),0.5% acetic acid(MERCK,Cat.:1.00066.0250),1% formic acid(MERCK,Cat.:1.11670.1000)を含む超純水15μLに再溶解し,そのうち5μLをMSに接続したnano-LC Ultimate 3000 RSLCnano system(Dionex)に注入した。LC分析条件としては,EP1715343A1に記載の条件,又はこれに類似する当業者に公知の条件で,逆相材料及びイオン交換材料を組み合わせたカラム,あるいは逆相材料単独のカラムを用い,適切な緩衝液を用いて行うことができる。HPLCカラムをナノ-LC エレクトロスプレーイオン化源を設置したOrbitrap Elite(Thermo)に連結し,製造元のプロトコルに従い,full scan精密質量分析及びMS-MSによる質量分析を実施した。
2. ペプチドの配列解析をSEQUESTアルゴリズムにより実施した。
-分化した細胞の特性-
図3A、図3Bに、フローサイトメーター解析により得られた、Ticを用いて作成した単球様細胞の細胞表面に発現する分子を調べた結果を示す。本実施例により得られた単球様細胞は単球の特異的マーカーであるCD14の発現が認められた他、T細胞の活性化分子であるCD80,CD86,接着分子であるCD11b,CD11cの発現が認められた。
図7に、Ticより作製した樹状細胞様細胞をBet v1aに暴露した場合に検出されたペプチドのアミノ酸配列の解析結果を示す(a)。本実施例でBet v1aに暴露させた樹状細胞様細胞より抽出したHLA-DR分子より分離されるペプチドからは、Bet v1aのアミノ酸配列の一部が検出された。


2回の測定(duplicate)でほぼ同様の結果が得られ、再現性が得られた(2回目のデータ示さず)。また、異なったタイミング(非接着性細胞を回収してGM-CSF及びIL-4を添加するタイミングを変えた場合)で樹状細胞様細胞への分化を行い、HLA-DR等の発現の上昇を促した場合でも、同様のペプチド配列が検出された(データ示さず)。


2回の測定(duplicate)でほぼ同様の結果が得られ、再現性が得られた(2回目のデータ示さず)。
図9Aに、Ticより作製した樹状細胞様細胞をInfliximabに暴露した場合に検出されたペプチドのアミノ酸配列の解析結果を示す(a)。本実施例でInfliximabに暴露させた樹状細胞様細胞より抽出したHLA-DR分子より分離されるペプチドからは、InfliximabのH鎖及びL鎖に認められるアミノ酸配列の一部が検出された。
図10A~図10Hに、Ticより作製した樹状細胞様細胞をrhFVIIIに暴露した場合に検出されたペプチドのアミノ酸配列の解析結果を示す。本実施例でrhFVIIIに暴露させた樹状細胞様細胞より抽出したHLA-DR分子より分離されるペプチドからはrhFVIIIのアミノ酸配列の一部が検出された。
図11に、Ticより作製した樹状細胞様細胞をPhl p1に暴露した場合に検出されたペプチドのアミノ酸配列の解析結果を示す。本実施例でPhl p1のアミノ酸配列の一部が検出された。
例えば、Karbach J, Pauligk C, Bender A, Gnjatic S, Franzmann K, Wahle C, Jager D, Knuth A, Old LJ, Jager E., Identification of new NY-ESO-1 epitopes recognized by CD4+ T cells and presented by HLA-DQ B1 03011, Int J Cancer. 2006 Feb 1;118(3):668-74.では、癌抗原の一つであるNY-ESO-1を提示させた樹状細胞を用いて、抗原特異的T細胞を作製し、同様の抗原を提示させたHLA-DQ分子発現EBV-B細胞株との再刺激により、HLA-DQ分子に提示されるNY-ESO-1中のペプチド配列を検出できたことを示しており、抗原提示されたT細胞エピトープをHLA-DQ分子を用いて同定できることが示されている。また、例えば、Duquesnoy RJ, Marrari M, Tambur AR, Mulder A, Sousa LC, da Silva AS, do Monte SJ, First report on the antibody verification of HLA-DR, HLA-DQ and HLA-DP epitopes recorded in the HLA Epitope Registry, Hum Immunol. 2014 Nov;75(11):1097-103.では、データベースからHLA-DR分子、HLA-DQ分子、HLA-DP分子に提示されやすい配列が予測されており、HLA-DQ分子やHLA-DP分子が、HLA-DR分子と同様に、特定の配列を抗原提示することが示されている。これらの技術常識に基づいて、MAPPsを、本発明における幹細胞又はこれに由来する前駆細胞から分化された、HLA-DQ分子やHLA-DP分子などの他のMHCII分子を発現する細胞にも同様に適用できることが、当業者には理解されよう。
また、MHCI分子を用いたMAPPsも、例えば、Wahl A, Schafer F, Bardet W, Buchli R, Air GM, Hildebrand WH., HLA class I molecules consistently present internal influenza epitopes. Proc Natl Acad Sci U S A. 2009 Jan 13;106(2):540-5.において既に報告されている。当該文献では、特定のアロタイプのHLA-B分子を発現させた細胞株をインフルエンザウイルスに感作させ、MAPPsによりHLA-B分子上に提示されるインフルエンザウイルス由来ペプチド配列の検出を実施している。なお、上述の通り、MHCI分子は多くの細胞種で発現していることが知られているところ、MHCI分子-ペプチド複合体の検出の容易さの観点から、MHCI分子が高発現している細胞を利用することが望ましい。一実施態様において、MHCI分子を用いたMAPPsには、本発明における幹細胞又はこれに由来する前駆細胞から分化された、樹状細胞を用いてよい。
A.方法
-使用細胞-
ヒト末梢血単核球細胞(PBMC):(Lonza co.,ltd.より購入)。
1. ヒト末梢血単核球細胞に対し,Human Serum Alubmin low IgG(SIGMA,Cat.:A3782)を0.5%、EDTA 0.5M stock solution pH8.0(Invitrogen,Cat.:15575)を2mMとなるよう加えたDPBS(Invitrogen,Cat:14190)を80μL/107cells, CD14 micro beads(Miltenyi,Cat.:130-050-201)を20μL/107cellsとなるよう加え,ボルテックスミキサーにより懸濁し,4℃,遮光下の条件で15分静置した。
2. 15分静置後のヒト末梢血単核球細胞にHuman Serum Alubmin low IgGを0.5%、EDTA 0.5M stock solution pH8.0を2mM含むDPBSを20mL加え,1200rpm,5分,4℃でスピンダウンした後,上清を全て除去した。本操作は2回行った。
3. 上清を除去したヒト末梢血単核球細胞にHuman Serum Alubmin low IgGを0.5%、EDTA 0.5M stock solution pH8.0を2mM含むDPBSを1.2×108cells/mLとなるよう加え,細胞分離用磁石LS Column(Miltenyi,Cat.:130-042-401)に通し,回収したヒト末梢血単核球細胞を単球細胞とした。
本発明の実施例と同様に,白樺花粉アレルゲンであるBetula verrucosa, birch pollen allergen 1, Isoform a(Bet v1a)(#Bet v 1.0101; Biomay)(アミノ酸配列:配列番号1)を用いた。
1. 回収した単球細胞にHuman Serum Alubmin low IgGを0.5%、EDTA 0.5M stock solution pH8.0を2mM含むDPBSを20mL加え,1200rpm,5分,4℃でスピンダウンした後,上清を全て除去した。
2. 上清を除去した単球細胞を,Fetal Bovine Serum (FBS) (Gibco,Cat.:10270,26140)を10%、Non Essential amino acids(Gibco,Cat.:11140-035)を1%,Na-Pyruvate(Gibco,Cat.:11360-039)を1%,Kanamycine(Gibco,Cat.:15160-047)を1%,recombinant human Granulocyte Macrophage colony-stimulating Factor(rhGM-CSF)(R&Dsystems,Cat.:215-GM)を50ng/mL,recombinant human Interleukin-4(rhIL-4)(R&Dsystems,Cat.:204-IL)を3ng/mLとなるよう加えたRPMI 1640(Life Tech,Cat.:11875)を用いて,3×105cells/mLの細胞密度に懸濁し,6ウェルプレートに3mLずつ播種し,37℃,5%CO2条件下で5日間の培養を実施し,培養5日後の単球細胞を樹状細胞とした。
3. 培養5日後の各ウェルより上清を2mL/ウェル除去し,Bet v1a 3.3μg/mLを加え,続いてLipopolysaccharides from Salmonella enterica serotype abortusequi(LPS)(SIGMA,Cat.:L5886)を1μg/mL加え,37℃,5%CO2条件下で1日培養した。
4. 1日培養後の樹状細胞を6ウェルプレートより全量回収し,1200rpm,5分,4℃でスピンダウンした後,上清を全て除去し,4℃のDPBS 1mLに懸濁した。次いで,全量をエッペンドルフチューブに移し,2500rpm,5分,4℃でスピンダウンし,上清を全て除去し,細胞のペレットを作製し,-80℃で保管した。
5. 樹状細胞の一部を回収し,抗ヒトHLA-DR抗体,抗ヒトHLA-DQ抗体,抗ヒトHLA-DP抗体,抗ヒトHLA-ABC抗体,抗ヒトCD14抗体,抗ヒトCD80抗体,抗ヒトCD86抗体,抗ヒトCD206抗体,抗ヒトCD209抗体,抗ヒトCD11b抗体,及び抗ヒトCD11c抗体を用いて染色し,フローサイトメーター解析装置BD FACSCantoTMIIを用いて分析した。
本発明の実施例と同様に調整した。
本発明の実施例と同様に調整した。
本発明の実施例と同様に調整した。
本発明の実施例と同様に調整した。
-分化した細胞の特性-
図12に、フローサイトメーター解析により得られた単球細胞の細胞表面に発現する分子を調べた結果を示す。比較例により得られた単球細胞は単球の特異的マーカーであるCD14の発現及び抗原提示分子であるHLA-DRの発現が認められた他、T細胞の活性化分子であるCD86の発現が認められた。
図14~図20に,評価に供した各ヒトドナー毎のBet v1a添加条件(図14,図15,図16,図17,図18,図19,図20の(a))及び非添加条件(対照)(図14,図15,図16,図17,図18,図19,図20の(b))で検出されたペプチドのアミノ酸配列の解析結果を示す。また、これらの検出された具体的なアミノ酸配列を、それぞれ、表2~表8にも示す(図14~図20にそれぞれ対応する。)。表中、エピトープ番号は、Bet v1aのアミノ酸配列において、N末端から順に見られた、検出されたペプチドの群を示す。




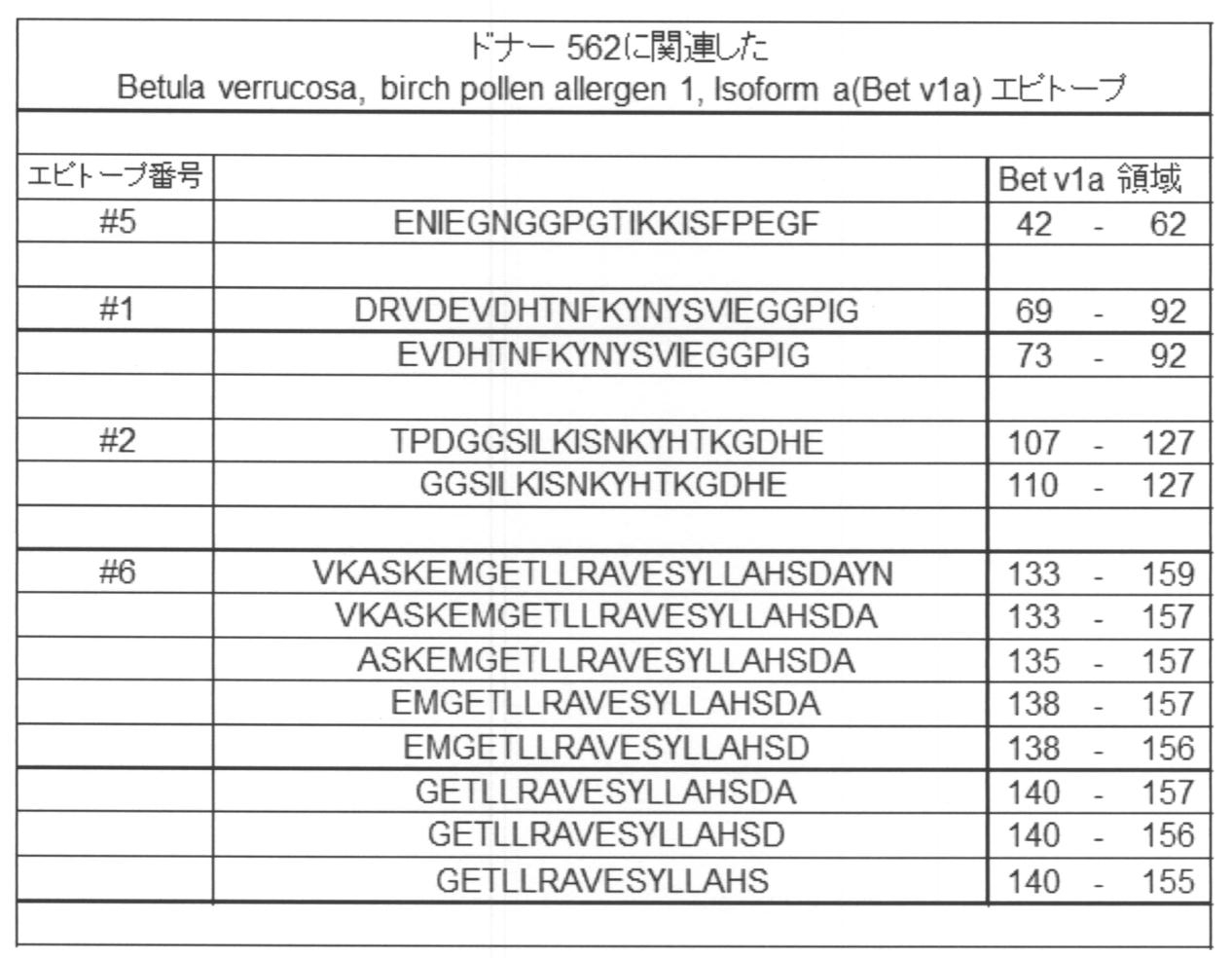


Bet v1a非添加条件で検出されたペプチド(対照)からは、Bet v1a添加条件で検出されたペプチドの一部が検出された。しかしながら,Bet v1a添加条件では,Bet v1a非添加条件よりも多くのペプチドが検出された。
図21A、図21Bに、Bet v1a添加条件において検出されたペプチドのアミノ酸配列について、2種類のヒトiPS細胞由来の樹状細胞様細胞を用いた場合とPBMC由来の樹状細胞を用いた場合との比較を示す。PBMC由来の樹状細胞を用いた解析では、ドナー間で異なるMHCII分子を有していたために、検出されるペプチドのアミノ酸配列にドナー間で違いが見られたと考えられる。また、2種類のiPS細胞由来の樹状細胞様細胞間で検出されたアミノ酸配列の違いについても、元となったドナー間で異なるタイプのMHCII分子を有していたためLine間で検出配列に違いが生じたと考えられる。それにも関わらず、ヒトiPS細胞由来の樹状細胞様細胞を用いてBet v1a添加条件において検出されたペプチドで共通した配列の多くは、PBMC由来の樹状細胞を用いた場合に検出されたペプチドの配列と一致していた。検出されたペプチド配列140-155に関しては、S. Mutschlener et al., Journal of Allergy and Clinical Immunology Vol.125 (3), 2010でエピトープ配列部分として報告された配列と一致した。
配列番号151:InfliximabのL鎖の部分ペプチド
配列番号152~配列番号153: InfliximabのH鎖の部分ペプチド
Claims (28)
- タンパク質のエピトープを同定するための方法であって、
以下の工程:
(A)幹細胞又はこれに由来する前駆細胞から分化された、主要組織適合遺伝子複合体(MHC分子)を発現する細胞に、標的タンパク質を接触させる工程;
(B)前記MHC分子を発現する細胞から、前記標的タンパク質に含まれるペプチドとMHC分子との複合体を単離する工程;及び
(C)前記複合体から、前記ペプチドを溶出し、同定する工程
を含む、方法。 - さらに、以下の工程:
(D)同定したペプチドが免疫原性を誘導するエピトープであるか否かを検証する工程
を含む、請求項1に記載の方法。 - 前記幹細胞は、人工多能性幹細胞(iPS細胞)、胚性幹細胞(ES細胞)、核移植ES細胞(ntES細胞)、胚性生殖幹細胞(EG細胞)及び成体幹細胞からなる群から選択される、請求項1又は2に記載の方法。
- 前記MHC分子は、MHCII分子である、請求項1~3のいずれか一項に記載の方法。
- 前記MHCII分子はHLA-DR、HLA-DQ又はHLA-DPである、請求項4に記載の方法。
- 前記MHC分子を発現する細胞は、さらに、CD80、CD86、CD206及びCD209の少なくとも1つを発現している、請求項1~5のいずれか一項に記載の方法。
- 前記MHC分子を発現する細胞は、CD80、CD86、CD206及びCD209の全てを発現している、請求項6に記載の方法。
- 前記MHC分子を発現する細胞は、樹状細胞である、請求項1~7のいずれか一項に記載の方法。
- 前記MHC分子を発現する細胞は、前記標的タンパク質を投与することが意図される対象のMHC分子の1又は複数のアロタイプを発現する、請求項1~8のいずれか一項に記載の方法。
- 前記工程(A)を無血清下で行う、請求項1~9のいずれか一項に記載の方法。
- 前記樹状細胞は、
以下の工程:
(a)幹細胞又はこれに由来する前駆細胞を分化させて中胚葉前駆細胞を得る工程;
(b)前記中胚葉前駆細胞を分化させて単球細胞を得る工程;及び
(c)前記単球細胞を分化させて未成熟樹状細胞を得、及び場合により、前記未成熟樹状細胞をさらに刺激して成熟樹状細胞を得る工程
を含む方法によって製造され、
さらに、前記工程(a)~(c)のうち、少なくとも前記工程(c)において無血清培地が用いられる、
請求項1~10のいずれか一項に記載の方法。 - 前記工程(b)が、顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びマクロファージコロニー刺激因子(M-CSF)を含む無血清培地下で、前記中胚葉前駆細胞を分化させて単球細胞を得る工程を含む、請求項11に記載の方法。
- 前記工程(c)が、
(c1)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びインターロイキン4(IL-4)を含む無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得る工程を含み、及び場合により、
(c2)前記未成熟樹状細胞を免疫原、及び、場合により炎症性サイトカインに接触させることで、成熟樹状細胞へと誘導する工程を含む、
請求項11又は12に記載の方法。 - 前記樹状細胞が未成熟樹状細胞であり、前記未成熟樹状細胞は、免疫原性を有する標的タンパク質に接触することで、成熟樹状細胞に誘導される、請求項8~13のいずれか一項に記載の方法。
- 前記標的タンパク質が、サイトカイン、ケモカイン、成長因子、抗体、酵素、構造タンパク質、ホルモン、及び、これらのいずれかの断片からなる群の1種又は1種以上から選択される、請求項1~14のいずれか一項に記載の方法。
- 免疫原性が減少又は消失したタンパク質の製造方法であって、
以下の工程:
(1)請求項1~15のいずれか一項に記載の方法に従って、タンパク質のエピトープを同定する工程;
(2)MHC分子への結合が減少又は消失するように、前記エピトープを修飾する工程;及び
(3)修飾されたエピトープを有するタンパク質を製造する工程
を含む、方法。 - 請求項16に記載の方法に従って得られうる、タンパク質。
- タンパク質が対象において免疫原性を有するか否かを予測する方法であって、
(I)標的タンパク質を投与することが意図される対象のMHC分子の1又は複数のアロタイプを発現する細胞を提供する工程であって、前記細胞は幹細胞又はこれに由来する前駆細胞から分化されることを特徴とする、工程;
(II)前記「MHC分子の1又は複数のアロタイプを発現する細胞」に、標的タンパク質を接触させる工程;
(III)前記「MHC分子の1又は複数のアロタイプを発現する細胞」から、前記標的タンパク質に含まれるペプチドとMHC分子との複合体を単離する工程;
(IV)前記複合体から、前記ペプチドを溶出し、同定する工程;及び
(V)場合により、同定したペプチドが免疫原性を誘導するエピトープであるか否かを検証する工程
を含み、
前記同定したペプチドが免疫原性を誘導するエピトープである場合に、前記標的タンパク質が前記対象において免疫原性を有することを指し示す、方法。 - 前記対象が有するMHC分子のアロタイプの全てのセットが含まれるように、前記対象のMHC分子の1又は複数のアロタイプを発現する細胞を1又は複数提供する、請求項18に記載の方法。
- 前記幹細胞が、前記対象に由来する人工多能性幹細胞(iPS細胞)である、請求項18又は19に記載の方法。
- タンパク質を有効成分として含む、対象における、前記タンパク質に関連した疾患の治療及び/又は予防用組成物であって、
前記対象は、請求項18~20のいずれか一項に記載の方法に従って、前記タンパク質に免疫原性を有さないことが予測された対象から選択されることを特徴とする、組成物。 - 幹細胞もしくはこれに由来する前駆細胞、又は、これから分化されたMHC分子を発現する細胞の、請求項1~16及び18~20のいずれか一項に記載の方法における使用。
- 幹細胞又はこれに由来する前駆細胞から樹状細胞を製造する方法であって、
以下の工程:
(a’)幹細胞又はこれに由来する前駆細胞を分化させて中胚葉前駆細胞を得る工程;
(b’)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びマクロファージコロニー刺激因子(M-CSF)を含む無血清培地下で、前記中胚葉前駆細胞を分化させて単球細胞を得る工程;及び
(c’)無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得、及び場合により、前記未成熟樹状細胞をさらに刺激して成熟樹状細胞を得る工程
を含む、方法。 - 前記工程(c’)が、
(c1’)顆粒球・マクロファージコロニー刺激因子(GM-CSF)及びインターロイキン4(IL-4)を含む無血清培地下で、前記単球細胞を分化させて未成熟樹状細胞を得る工程を含み、及び場合により、
(c2’)前記未成熟樹状細胞を、免疫原、及び、場合により炎症性サイトカインに接触させることで、成熟樹状細胞へと誘導する工程を含む、
請求項23に記載の方法。 - 請求項23又は24に記載の方法によって得られうる、樹状細胞。
- MHCII分子に加えて、さらに、CD80、CD86、CD206及びCD209の少なくとも1つを発現している、請求項25に記載の樹状細胞。
- CD80、CD86、CD206及びCD209の全てを発現している、請求項26に記載の樹状細胞。
- 請求項25~27のいずれか一項に記載の樹状細胞を含む、細胞組成物。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016534422A JP7012432B2 (ja) | 2014-07-14 | 2015-07-13 | タンパク質のエピトープを同定するための方法 |
| US15/326,295 US10718781B2 (en) | 2014-07-14 | 2015-07-13 | Method for identifying epitope on protein |
| EP15822419.6A EP3170901B1 (en) | 2014-07-14 | 2015-07-13 | Method for producing dendritic cells from stem cells |
| EP21171749.1A EP3929302A1 (en) | 2014-07-14 | 2015-07-13 | Method for identifying epitope on protein |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014144217 | 2014-07-14 | ||
| JP2014-144217 | 2014-07-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016010002A1 true WO2016010002A1 (ja) | 2016-01-21 |
Family
ID=55078494
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/070072 WO2016010002A1 (ja) | 2014-07-14 | 2015-07-13 | タンパク質のエピトープを同定するための方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10718781B2 (ja) |
| EP (2) | EP3170901B1 (ja) |
| JP (1) | JP7012432B2 (ja) |
| TW (1) | TWI719939B (ja) |
| WO (1) | WO2016010002A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20220389113A1 (en) * | 2019-11-06 | 2022-12-08 | Commonwealth Scientific And Industrial Research Organisation | Binding proteins to cub domain-containing protein (cdcp1) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006300945A (ja) * | 2005-04-20 | 2006-11-02 | F Hoffmann La Roche Ag | 生物薬剤の免疫原性に関するエピトープの同定のための方法 |
| JP2011515110A (ja) * | 2008-03-27 | 2011-05-19 | ジェロン・コーポレーション | 霊長類多能性幹細胞の造血系統細胞への分化 |
| JP2012530496A (ja) * | 2009-06-17 | 2012-12-06 | アボット バイオセラピューティクス コーポレイション | 抗vegf抗体とその使用 |
| JP2014506447A (ja) * | 2011-02-23 | 2014-03-17 | 国立大学法人京都大学 | 多能性幹細胞から樹状細胞を製造する方法 |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| AU2002950778A0 (en) | 2002-08-15 | 2002-09-12 | The Corporation Of The Trustees Of The Order Of The Sisters Of Mercy In Queensland | A method of characterizing dendritic cells |
| ATE474851T1 (de) | 2002-10-02 | 2010-08-15 | Hoffmann La Roche | Verfahren zur identifizierung antigener peptide |
| US9453219B2 (en) | 2003-05-15 | 2016-09-27 | Mello Biotech Taiwan Co., Ltd. | Cosmetic designs and products using intronic RNA |
| EP1715343B1 (en) | 2005-04-20 | 2009-12-09 | F. Hoffmann-La Roche Ag | Method for the identification of epitopes related to immunogenicity in biopharmaceuticals |
| US20090253203A1 (en) | 2005-08-01 | 2009-10-08 | Nupotential, Inc. | Reprogramming a Cell by Inducing a Pluripotent Gene Through Use of a Small Molecule Modulator |
| EP4223769A3 (en) | 2005-12-13 | 2023-11-01 | Kyoto University | Nuclear reprogramming factor |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| JP2008307007A (ja) | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | 出生後のヒト組織由来未分化幹細胞から誘導したヒト多能性幹細胞 |
| EP3078738B1 (en) | 2007-08-31 | 2020-05-20 | Whitehead Institute for Biomedical Research | Wnt pathway stimulation in reprogramming somatic cells |
| KR101564044B1 (ko) | 2007-10-31 | 2015-10-28 | 고쿠리츠 다이가쿠 호진 교토 다이가쿠 | 핵초기화 방법 |
| JP5558097B2 (ja) | 2007-12-10 | 2014-07-23 | 国立大学法人京都大学 | 効率的な核初期化方法 |
| EP2072617A1 (en) | 2007-12-12 | 2009-06-24 | Trimed Biotech GmbH | Method for producing dendritic cells |
| CA2709566A1 (en) | 2007-12-17 | 2009-06-25 | Gliamed, Inc. | Stem-like cells and method for reprogramming adult mammalian somatic cells |
| JP5945385B2 (ja) | 2008-01-16 | 2016-07-05 | リン、シー−ランLIN,Shi−Lung | 誘導性組換えrna因子を用いた腫瘍のない多能性胚性幹様細胞の生成 |
| US20100330677A1 (en) | 2008-02-11 | 2010-12-30 | Cambridge Enterprise Limited | Improved Reprogramming of Mammalian Cells, and Cells Obtained |
| EP2090649A1 (en) | 2008-02-13 | 2009-08-19 | Fondazione Telethon | Method for reprogramming differentiated cells |
| WO2009102983A2 (en) | 2008-02-15 | 2009-08-20 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| EP2279243B1 (en) | 2008-03-17 | 2016-01-06 | The Scripps Research Institute | Combined chemical and genetic approaches for generation of induced pluripotent stem cells |
| WO2009114949A1 (en) | 2008-03-20 | 2009-09-24 | UNIVERSITé LAVAL | Methods for deprogramming somatic cells and uses thereof |
| ES2755736T3 (es) | 2008-03-26 | 2020-04-23 | Biocon Ltd | Proceso de fermentación mejorado para un coeficiente de rendimiento más alto de inhibidor de lipasa respecto al ácido graso consumido |
| CA2695590C (en) | 2008-06-27 | 2018-01-09 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| EP2313494A2 (en) | 2008-07-14 | 2011-04-27 | Oklahoma Medical Research Foundation | Production of pluripotent cells through inhibition of bright/arid3a function |
| WO2010147612A1 (en) | 2009-06-18 | 2010-12-23 | Lixte Biotechnology, Inc. | Methods of modulating cell regulation by inhibiting p53 |
| WO2010033906A2 (en) | 2008-09-19 | 2010-03-25 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| US20120034192A1 (en) | 2008-09-19 | 2012-02-09 | Young Richard A | Compositions and methods for enhancing cell reprogramming |
| WO2010042800A1 (en) | 2008-10-10 | 2010-04-15 | Nevada Cancer Institute | Methods of reprogramming somatic cells and methods of use for such cells |
| EP2342333A4 (en) | 2008-10-30 | 2013-05-08 | Univ Kyoto | METHOD FOR THE PRODUCTION OF INDUCED PLURIPOTENTAL STEM CELLS |
| US20110059526A1 (en) | 2008-11-12 | 2011-03-10 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| EP2376626A4 (en) | 2008-12-13 | 2012-10-17 | Dna Microarray | MICRO-ENVIRONMENTAL NICHE ASSAY FOR SCREENING OF INDUCED PLURIPOTENT STEM CELLS (CIPS) |
| KR101764100B1 (ko) | 2009-02-27 | 2017-08-02 | 고쿠리츠 다이가쿠 호진 교토 다이가쿠 | 신규한 핵 재프로그래밍 물질 |
| WO2010102267A2 (en) | 2009-03-06 | 2010-09-10 | Ipierian, Inc. | Tgf-beta pathway inhibitors for enhancement of cellular reprogramming of human cells |
| WO2010111409A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Pluripotent stem cells |
| US20120076762A1 (en) | 2009-03-25 | 2012-03-29 | The Salk Institute For Biological Studies | Induced pluripotent stem cell generation using two factors and p53 inactivation |
| US8852940B2 (en) | 2009-04-01 | 2014-10-07 | The Regents Of The University Of California | Embryonic stem cell specific microRNAs promote induced pluripotency |
| US20110088107A1 (en) | 2009-04-24 | 2011-04-14 | Yaqub Hanna | Compositions and methods for deriving or culturing pluripotent cells |
| WO2010147395A2 (en) | 2009-06-16 | 2010-12-23 | Korea Research Institute Of Bioscience And Biotechnology | Medium composition comprising neuropeptide y for the generation, maintenance, prologned undifferentiated growth of pluripotent stem cells and method of culturing pluripotent stem cell using the same |
| JP5861191B2 (ja) | 2010-09-30 | 2016-02-16 | 国立大学法人 熊本大学 | ミエロイド系血液細胞の製造方法 |
-
2015
- 2015-07-13 TW TW104122486A patent/TWI719939B/zh active
- 2015-07-13 JP JP2016534422A patent/JP7012432B2/ja active Active
- 2015-07-13 WO PCT/JP2015/070072 patent/WO2016010002A1/ja active Application Filing
- 2015-07-13 EP EP15822419.6A patent/EP3170901B1/en active Active
- 2015-07-13 EP EP21171749.1A patent/EP3929302A1/en not_active Withdrawn
- 2015-07-13 US US15/326,295 patent/US10718781B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006300945A (ja) * | 2005-04-20 | 2006-11-02 | F Hoffmann La Roche Ag | 生物薬剤の免疫原性に関するエピトープの同定のための方法 |
| JP2011515110A (ja) * | 2008-03-27 | 2011-05-19 | ジェロン・コーポレーション | 霊長類多能性幹細胞の造血系統細胞への分化 |
| JP2012530496A (ja) * | 2009-06-17 | 2012-12-06 | アボット バイオセラピューティクス コーポレイション | 抗vegf抗体とその使用 |
| JP2014506447A (ja) * | 2011-02-23 | 2014-03-17 | 国立大学法人京都大学 | 多能性幹細胞から樹状細胞を製造する方法 |
Non-Patent Citations (4)
| Title |
|---|
| SATORU SENJU: "Anti-Cancer Therapy with Pluripotent Stem Cell -Derived Dendritic Cells", BIOTHERAPY, vol. 24, no. 2, 2010, pages 87 - 94, XP008185206 * |
| SATORU SENJU: "Establishment of pluripotent stem cell -derived dendritic cells for clinical application", THE JAPANESE JOURNAL OF CLINICAL HEMATOLOGY, vol. 51, no. 11, 2010, pages 1668 - 1673, XP008185205 * |
| SATORU SENJU: "Saisei Iryo no Genjo to Shinpo - ES Saibo, iPS Saibo to Taisei Kansaibo no Rinsho eno Oyo- 6. Tanosei Saibo Yurai no Jujo Saibo o Mochiita Men'eki Ryoho", HEMATOLOGY FRONTIER, vol. 19, no. 11, 2009, pages 1685 - 1692, XP008185220 * |
| See also references of EP3170901A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2016010002A1 (ja) | 2017-04-27 |
| TWI719939B (zh) | 2021-03-01 |
| TW201608240A (zh) | 2016-03-01 |
| EP3929302A1 (en) | 2021-12-29 |
| JP7012432B2 (ja) | 2022-01-28 |
| EP3170901A1 (en) | 2017-05-24 |
| US10718781B2 (en) | 2020-07-21 |
| EP3170901B1 (en) | 2021-06-02 |
| US20170219607A1 (en) | 2017-08-03 |
| EP3170901A4 (en) | 2018-01-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Tan et al. | Amnion epithelial cell-derived exosomes restrict lung injury and enhance endogenous lung repair | |
| Ungerer et al. | Galectin-9 is a suppressor of T and B cells and predicts the immune modulatory potential of mesenchymal stromal cell preparations | |
| CN107849536B (zh) | 用于得到调节性t细胞的方法及其用途 | |
| AU767756C (en) | Activation of regulatory T cells by alpha-melanocyte stimulating hormone | |
| JP5861191B2 (ja) | ミエロイド系血液細胞の製造方法 | |
| JP6876746B2 (ja) | 免疫療法のための医薬の組合わせ | |
| Kool et al. | Facilitated antigen uptake and timed exposure to TLR ligands dictate the antigen-presenting potential of plasmacytoid DCs | |
| JP2023058687A (ja) | ガンマデルタt細胞の増幅、組成物およびその使用 | |
| KR20100014871A (ko) | 조절 t 세포 에피토프, 그의 조성물 및 용도 | |
| WO2018135646A1 (ja) | CD8α+β+細胞傷害性T細胞の製造方法 | |
| US8323965B2 (en) | Identification of antigenic peptides from multiple myeloma cells | |
| Buerger et al. | The ubiquitin-like modifier FAT10 is selectively expressed in medullary thymic epithelial cells and modifies T cell selection | |
| WO2016090035A2 (en) | Modulators of activin and methods for modulating immune responses and t follicular helper cells | |
| JP2017516752A (ja) | 単離されたドナーmhc由来ペプチド及びその使用 | |
| Filipović et al. | Inhibition of Notch signaling stimulates osteoclastogenesis from the common trilineage progenitor under inflammatory conditions | |
| JP7012432B2 (ja) | タンパク質のエピトープを同定するための方法 | |
| WO2017034459A1 (en) | New use | |
| US20230218678A1 (en) | Cord blood plasma-derived exosome or mimetic thereof and pharmaceutical use thereof | |
| JP2008509878A (ja) | Bob−1特異的t細胞及び使用法 | |
| Meloni et al. | Dendritic cells loaded with apoptotic oligodendrocytes as a source of myelin T-cell epitopes in multiple sclerosis | |
| Sarraf et al. | Wnt5A supports antigen cross-presentation and CD8 T cell activation | |
| Hooper | PGE2 and IL-27: Novel Proinflammatory Mechanisms Involving Dendritic Cells and Type 1 Regulatory T Cells | |
| Facci | The immunomodulation of porcine immune cells by innate and synthetic host defense peptides | |
| Taner | Dendritic cells, rapamycin and transplant tolerance |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15822419 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016534422 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15326295 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015822419 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015822419 Country of ref document: EP |