WO2015115548A1 - トリフルオロエチレンの製造方法 - Google Patents
トリフルオロエチレンの製造方法 Download PDFInfo
- Publication number
- WO2015115548A1 WO2015115548A1 PCT/JP2015/052527 JP2015052527W WO2015115548A1 WO 2015115548 A1 WO2015115548 A1 WO 2015115548A1 JP 2015052527 W JP2015052527 W JP 2015052527W WO 2015115548 A1 WO2015115548 A1 WO 2015115548A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- solid reactant
- hfc
- differential pressure
- fluidized bed
- flow rate
- Prior art date
Links
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/25—Preparation of halogenated hydrocarbons by splitting-off hydrogen halides from halogenated hydrocarbons
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/20—Carbon compounds
- B01J27/232—Carbonates
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J8/00—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes
- B01J8/18—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes with fluidised particles
- B01J8/1818—Feeding of the fluidising gas
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J8/00—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes
- B01J8/18—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes with fluidised particles
- B01J8/1818—Feeding of the fluidising gas
- B01J8/1827—Feeding of the fluidising gas the fluidising gas being a reactant
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K5/00—Heat-transfer, heat-exchange or heat-storage materials, e.g. refrigerants; Materials for the production of heat or cold by chemical reactions other than by combustion
- C09K5/02—Materials undergoing a change of physical state when used
- C09K5/04—Materials undergoing a change of physical state when used the change of state being from liquid to vapour or vice versa
- C09K5/041—Materials undergoing a change of physical state when used the change of state being from liquid to vapour or vice versa for compression-type refrigeration systems
- C09K5/044—Materials undergoing a change of physical state when used the change of state being from liquid to vapour or vice versa for compression-type refrigeration systems comprising halogenated compounds
- C09K5/045—Materials undergoing a change of physical state when used the change of state being from liquid to vapour or vice versa for compression-type refrigeration systems comprising halogenated compounds containing only fluorine as halogen
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00017—Controlling the temperature
- B01J2208/00026—Controlling or regulating the heat exchange system
- B01J2208/00035—Controlling or regulating the heat exchange system involving measured parameters
- B01J2208/0007—Pressure measurement
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00017—Controlling the temperature
- B01J2208/00389—Controlling the temperature using electric heating or cooling elements
- B01J2208/00407—Controlling the temperature using electric heating or cooling elements outside the reactor bed
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00017—Controlling the temperature
- B01J2208/00389—Controlling the temperature using electric heating or cooling elements
- B01J2208/00415—Controlling the temperature using electric heating or cooling elements electric resistance heaters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2205/00—Aspects relating to compounds used in compression type refrigeration systems
- C09K2205/10—Components
- C09K2205/12—Hydrocarbons
- C09K2205/126—Unsaturated fluorinated hydrocarbons
Definitions
- the present invention relates to a method for producing trifluoroethylene, and more particularly to a method for efficiently producing trifluoroethylene from 1,1,1,2-tetrafluoroethane.
- trifluoroethylene Since trifluoroethylene (HFO-1123) has a low global warming potential (GWP), it is a greenhouse gas such as difluoromethane (HFC-32) and 1,1,1,2,2-pentafluoroethane (HFC-). In recent years, great expectations have been given as a new refrigerant replacing 125). In addition, in this specification, about halogenated hydrocarbon, the abbreviation (refrigerant number etc.) of the compound is described in the parenthesis after the compound name. Moreover, the abbreviation is used instead of the compound name as necessary.
- Patent Document 1 describes a method of dehydrofluorinating HFC-134a in a gas phase using a metal fluoride as a catalyst.
- Patent Document 2 describes a method of reacting HFC-134a with a metal hydroxide such as calcium hydroxide in a gas phase.
- both of the methods described in Patent Document 1 and Patent Document 2 are those in which a gas phase HFC-134a is brought into contact with a solid reactant having a fixed bed to cause a reaction. there were. That is, (1) Since the solid reactant particles and HFC-134a are difficult to uniformly mix and contact, the conversion rate of the solid reactant is low. In addition, since the reactivity of the reaction for producing HFO-1123 from HFC-134a is low, it is necessary to contact HFC-134a with the solid reactant for a long time. (2) Since the heat removal efficiency is poor on a fixed bed, hot spots are likely to occur.
- JP 2010-533151 A International Publication No. 2011/157907
- HFC-134a which is an inexpensive raw material, is used as a solid reactant and suppresses the formation of by-products such as low-molecular hydrocarbons and high-molecular carbons. It is an object of the present invention to provide a method for producing HFO-1123 stably with high selectivity.
- a raw material gas containing HFC-134a is circulated in a layer made of particulate solid reactant having an average particle diameter of 1 ⁇ m to 5000 ⁇ m, and the layer made of solid reactant flows.
- the solid reactant is brought into contact with the HFC-134a in a converted state.
- a layer made of a solid reactant (hereinafter also referred to as a solid reactant layer) is in a “fluidized state” when a fluid such as a raw material gas is ejected upward (opposite to the direction of gravity). It is a state made by circulating, and refers to a state in which solid reactant particles are suspended and suspended in a fluid.
- the upward drag due to the fluid flow acting on the solid particles balances with gravity and buoyancy, and the entire solid reactant layer behaves like a uniform fluid.
- the pressure loss when the fluid passes through the solid reactant layer is equal to the difference between gravity and buoyancy, and the solid reactant layer can be changed even if the fluid flow rate is changed as long as the fluidized state is maintained.
- the pressure loss is always constant due to the difference between gravity and buoyancy.
- the fluidized solid reactant layer is referred to as a fluidized bed or a fluidized bed.
- the particles of the solid reactant constituting the layer float and flow in the fluid. Is used for both the solid reactant layer and the solid reactant particles.
- the conversion rate of 134a and the selectivity of HFO-1123 can be sufficiently increased, and the generation of hot spots in the reaction field can be prevented.
- the by-product of low-molecular hydrocarbons and polymer carbons can be suppressed, and HFO-1123 can be obtained efficiently and stably.
- the production method of the present invention has the following advantages as compared with the method in which HFC-134a is reacted with the solid reactant in the fixed bed in the gas phase. That is, in the fluidized bed reaction, since the heat removal efficiency of reaction heat is high and hot spots are not easily generated, the progress of the side reaction (carbon-carbon bond breaking) of HFC-134a can be suppressed. Therefore, low-molecular hydrocarbons and high-molecular carbon (graphite) are hardly produced as by-products, and the selectivity for the formation reaction of R-1123 is improved.
- HFO-1123 obtained by the production method of the present invention is used as a refrigerant to replace greenhouse gases HFC-32 and HFC-125, and as a raw material monomer for synthesis of functional materials such as piezoelectric elements and films, and for synthesis. Useful as an intermediate.
- HFC-134a conversion rate and HFO -1123 has a high selectivity and can be manufactured by an efficient method with little loss due to impurity generation.
- side reactions such as carbon-carbon bond cleavage of HFC-134a can be suppressed, and polymer carbons are hardly generated as a by-product, so that a decrease in HFC-134a conversion rate with time is prevented, and HFO-1123 is maintained for a long time. Can be manufactured stably over a long period of time.
- fluidization example 1 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 2 it is the graph which plotted the differential pressure with respect to the linear velocity of the mixed gas of HFC-134a and nitrogen.
- fluidization example 3 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 4 it is the graph which plotted the differential pressure with respect to the linear velocity of the mixed gas of HFC-134a and nitrogen.
- fluidization example 5 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 6 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 7 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- the fluidization comparative example 1 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- the fluidization comparative example 2 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization comparative example 3 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization comparative example 4 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization comparative example 5 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 8 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 9 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 10 it is the graph which plotted the differential pressure with respect to the linear velocity of HFC-134a.
- fluidization example 11 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- fluidization example 12 it is the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas.
- a raw material gas containing HFC-134a is circulated in a layer made of particulate solid reactant having an average particle diameter of 1 ⁇ m to 5000 ⁇ m, and the layer made of solid reactant is fluidized.
- the HFC-134a is brought into contact with the fluidized solid reactant particles.
- This is a method for producing HFO-1123 by causing the dehydrofluorination reaction of HFC-134a to proceed by this contact.
- the reaction by the contact between the fluidized solid reactant and HFC-134a is performed using a fluidized bed reactor that forms a fluidized bed (fluidized bed) made of the solid reactant in the reactor.
- reaction formula (1) represents the reaction when the solid reactant acts as a catalyst (Cat.)
- reaction formula (2) represents the solid reactant as the basic reactant (MOH: M represents a metal).
- Represents the reaction when acting as CF 3 —CH 2 F + solid reactant (Cat.) ⁇ CF 2 CHF + HF ......... « (1) CF 3 —CH 2 F + solid reactant (MOH) ⁇ CF 2 ⁇ CHF + MF + H 2 O (2)
- HFC-134a When HFC-134a is brought into contact with the solid reactant, of the two carbon atoms of HFC-134a, one of the fluorine atoms bonded to the carbon atom bonded with three fluorine atoms and the other carbon atom A dehydrofluorination reaction in which one of the bonded hydrogen atoms is eliminated at the same time occurs. Then, HFO-1123 is generated by such a dehydrofluorination reaction of HFC-134a. At this time, the detached fluorine atom and hydrogen atom generate hydrogen fluoride when the solid reactant acts as a catalyst. On the other hand, when the solid reactant acts as a basic reactant, metal fluoride (MF) and water are generated simultaneously.
- MF metal fluoride
- a fluidized bed is formed by the particulate solid reactant having an average particle diameter of 1 ⁇ m to 5000 ⁇ m, and the solid reactant layer is fluidized.
- a fluidized bed reactor is used as the reactor in the embodiment of the present invention.
- a fluid bed reactor or a riser reactor can be used. From the viewpoint of efficiently and stably producing HFO-1123, it is preferable to use a fluid bed reactor.
- a fluid bed type reactor heats a cooling coil for heat removal or the inside of a reactor (hereinafter also referred to as a fluidized bed reactor) forming a fluidized bed (fluidized bed) as necessary.
- a fluidized bed reactor heats a cooling coil for heat removal or the inside of a reactor (hereinafter also referred to as a fluidized bed reactor) forming a fluidized bed (fluidized bed) as necessary.
- a fluidized bed reactor heats a cooling coil for heat removal or the inside of a reactor (hereinafter also referred to as a fluidized bed reactor) forming a fluidized bed (fluidized bed) as necessary.
- a reactor hereinafter also referred to as a fluidized bed reactor
- separating source gas or reaction gas, and a solid reactant is provided in the upper part in a reactor.
- the cyclone can also be installed outside the reactor.
- a gas dispersion device for supplying the raw material gas is provided at
- the supply of the source gas containing HFC-134a and the solid reactant to the fluidized bed reactor may both be performed continuously, or only the supply of the source gas containing HFC-134a. May be carried out continuously and the solid reactant may be fed batchwise.
- the method of the present invention is applied to an embodiment in which only the raw material gas containing HFC-134a is continuously supplied and the solid reactant is supplied to the fluidized bed reactor in a batch manner will be described. It is not limited.
- HFC-134a may be HFC-134a having a purity of 100% (mol%), or it may contain 1,1,2,2-tetrafluoroethane (HFC-134), which is an impurity derived from the production method. May be.
- HFC-134 the purity of HFC-134a is preferably 50 mol% or more. That is, the source gas may contain HFC-134a with a purity of 100% (mol%), or contain HFC-134a with a purity of 50 mol% or more containing impurities such as HFC-134. Also good.
- the raw material gas should contain inert gas such as nitrogen, argon, helium in addition to HFC-134a having a purity of 50 mol% or more. Is preferred.
- inert gas such as nitrogen, argon, helium
- the reaction component HFC-134a can be diluted.
- these gases are referred to as dilution gases.
- the inclusion of such a dilution gas is also preferable from the viewpoint of easy supply of HFC-134a to the reactor and adjustment of the flow rate.
- the content ratio of the diluent gas is preferably 95 mol% or less with respect to the whole raw material gas containing HFC-134a from the viewpoint of reaction efficiency, suppression of side reactions, etc. 50 mol% or less is particularly preferable.
- the content ratio of HFC-134a with respect to the entire raw material gas is preferably 5 mol% or more and less than 100 mol%, particularly preferably 50 mol% or more and less than 100 mol%.
- the flow rate of each component (HFC-134a and dilution gas) constituting the raw material gas is controlled.
- the content molar ratio of each component in source gas can be controlled.
- the solid reactant used in the present invention is in the form of particles having an average particle size of 1 ⁇ m to 5000 ⁇ m.
- the average particle diameter is a value measured by a laser diffraction / scattering particle size analyzer.
- the heat removal efficiency in the solid reactant layer which is a reaction field, is poor, and side reactions such as carbonization are likely to occur due to the occurrence of hot spots, so that the conversion rate of HFC-134a increases over time due to the influence of carbon compound adhesion. descend.
- the average particle diameter of the solid reactant exceeds 5000 ⁇ m, the flow rate of the raw material gas necessary for fluidizing the particles of the solid reactant becomes too large. Therefore, in order to ensure a sufficient contact time for the reaction with HFC-134a, a large reactor is required and the production efficiency is poor.
- the average particle diameter of the solid reactant is out of the range of 1 ⁇ m to 5000 ⁇ m, even if HFC-134a is circulated through the solid reactant layer, the solid reaction is sufficiently performed to ensure uniform contact with HFC-134a. It is difficult to fluidize the agent layer. Therefore, it is difficult to make the conversion rate of HFC-134a sufficiently high and to stably produce HFO-1123 with high selectivity.
- the average particle size of the solid reactant is preferably in the range of 40 ⁇ m to 5000 ⁇ m, more preferably in the range of 40 ⁇ m to 500 ⁇ m.
- the fluidization state of the solid reactant layer can be examined by, for example, (a) a method of visually observing or (b) a method of measuring a differential pressure.
- A Method of visually observing The upper and lower mixing of the solid reactant layer is visually observed.
- the criteria for determining the fluidization state are as follows. Fully fluidized state ... Upper and lower mixing is observed in the entire solid reactant layer Partially fluidized state ... Upper and lower mixing is observed in a part of the solid reactant layer Non-fluidized state ... No mixing of the upper and lower solid reactant layers
- (b ) Method of measuring differential pressure Measure the difference in gas pressure between the inlet side and outlet side of the reactor (hereinafter referred to as differential pressure).
- the solid reactant When the solid reactant is brought into contact with the source gas containing HFC-134a, the solid reactant may be a solid phase or may be dispersed in a liquid phase medium.
- the solvent in which the solid reactant is dispersed include water, alcohol solvents such as methanol and ethanol, and chlorine solvents such as carbon tetrachloride.
- the reaction system pressure becomes too high and the reaction at a high temperature is difficult. It is preferable to contact with source gas.
- the specific surface area of the solid reactant is preferably 1 ⁇ 400m 2 / g, 1 ⁇ 200m 2 / g is more preferable.
- the specific surface area is a value measured by the BET method (BET specific surface area).
- BET specific surface area When the specific surface area of the solid reactant is less than 1 m 2 / g, the reaction rate decreases and the reaction efficiency is poor. On the other hand, if the specific surface area exceeds 400 m 2 / g, the density of the solid reactant particles becomes too small, and the particles are likely to be scattered, resulting in poor handling.
- the bulk density of the solid reactant is preferably 0.2 ⁇ 3.0g / cm 3, more preferably 0.5 ⁇ 2.9g / cm 3, particularly preferably 0.7 ⁇ 2.5g / cm 3.
- the bulk density of the solid reactant is less than 0.2 g / cm 3 , the volume at the same mass is increased, and the reactor is not only enlarged, but also the solid reactant particles are easily scattered and the handling property is deteriorated. Therefore, manufacturing efficiency is bad.
- the bulk density of the solid reactant is larger than 3.0 g / cm 3 , the speed of the raw material gas required for fluidizing the particles of the solid reactant becomes too high. Therefore, a large reactor is required to ensure a sufficient contact time for the reaction with HFC-134a, and the production efficiency is poor.
- the solid reactant used in the present invention contains a compound involved in the reaction mechanism shown by the above reaction formula (1) or (2) as a representative example.
- Examples of the compound that can participate in the reaction mechanism represented by the reaction formula (1) or (2) are at least selected from metal oxides, metal hydroxides, metal carbonates, metal sulfates, and metal halides.
- One compound is mentioned.
- Metal oxides and metal carbonates are preferred because HFC-134a can be efficiently converted to HFO-1123.
- One solid reactant may be used alone, or two or more solid reactants may be used in combination.
- examples of the metal species contained in the metal compound include alkali metals, alkaline earth metals, transition metals, Group 12 metals, Group 13 metals, and Group 14 metals.
- alkali metals, alkaline earth metals, Group 13 metals, and Group 14 metals are preferable, and sodium, potassium, calcium, magnesium, aluminum, and silicon are particularly preferable.
- the metal oxide may be one kind of the above-mentioned metal oxide or a complex oxide of two or more kinds of metals.
- the metal hydroxide may be one kind of the above-mentioned metal hydroxide or a composite hydroxide of two or more kinds of metals.
- the metal carbonate may be a single carbonate of the above-described metal or a composite carbonate of two or more metals.
- the metal sulfate may be a single sulfate of the above-described metal or a composite sulfate of two or more metals.
- the metal halide may be a single halide of the above-described metal or a composite halide of two or more metals.
- solid reactant examples include potassium carbonate, calcium hydroxide, calcium oxide, magnesium oxide, aluminum fluoride, and aluminum oxide (alumina).
- potassium carbonate and calcium oxide are particularly preferable because HFC-134a can be efficiently converted to HFO-1123.
- the solid reactant in the present invention may be composed of only the compound that can participate in the reaction mechanism represented by the reaction formula (1) or (2), or may contain other components. Good.
- Other components that can be contained in the solid reactant include, for example, a carrier for supporting the compound that can participate in the reaction mechanism represented by the reaction formula (1) or (2).
- the carrier include an alumina carrier, a zirconia carrier, a silica carrier, a silica alumina carrier, a carbon carrier represented by activated carbon, a barium sulfate carrier, and a calcium carbonate carrier.
- the activated carbon include activated carbon prepared from raw materials such as wood, charcoal, fruit glass, coconut shell, peat, lignite and coal.
- the source gas containing HFC-134a may be introduced into the reactor (for example, a fluidized bed reactor) at room temperature, but is heated (preheated) before being introduced into the reactor in order to increase the reactivity. It is preferable to supply from.
- the raw material gas is preferably heated to a temperature of 80 to 450 ° C. and then supplied to the reactor.
- Each component (HFC-134a and dilution gas) in the raw material gas containing HFC-134a is preheated to the above temperature and then mixed, and the mixed raw material gas at the above temperature is supplied to the reactor.
- the components may be mixed first and then heated to the above temperature and supplied to the reactor.
- each component in the raw material gas may be preheated to the above temperature and then supplied separately to the reactor.
- the flow rate of HFC-134a and the dilution gas per unit time (hereinafter simply referred to as flow rate) so that the linear velocity of the raw material gas in the reactor falls within a predetermined range. ) Is preferably set.
- the linear velocity of the source gas is preferably 1 cm / s to 1000 cm / s, and more preferably 1 cm / s to 20 cm / s.
- the linear velocity means the superficial velocity
- the reactor in which the raw material gas flows is an empty column in which no packing is contained, and the flow rate at the temperature and pressure in the reactor (volumetric flow rate). Is divided by the cross-sectional area of the empty reactor.
- Linear velocity (superficial velocity) (cm / s) flow rate (cm 3 / s) / cross-sectional area (cm 2 )
- the raw material gas containing HFC-134a introduced into the reactor comes into contact with the fluidized solid reactant forming a fluidized bed (fluidized bed) in the reactor for a predetermined time.
- the temperature during the contact is preferably 50 to 500 ° C., more preferably 100 to 500 ° C., and particularly preferably 350 to 500 ° C. as the temperature in the reactor from the viewpoint of improving the reactivity.
- the pressure in the reactor is preferably 0 to 5 MPa as gauge pressure, and more preferably 0 to 1 MPa.
- the contact time between HFC-134a and the solid reactant in the reactor is preferably 0.1 to 500 seconds, more preferably 0.1 to 100 seconds, and further preferably 0.1 to 20 seconds.
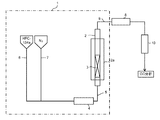
- a fluidized bed reactor 1 shown in FIG. 1 includes a vertical fluidized bed reactor 2 in an electric furnace or an electric heater 2a.
- installation of heating means such as an electric furnace or an electric heater 2a is not essential.
- a solid reactant layer 3 such as potassium carbonate is accommodated so as to form a vertical fluidized bed.
- a preheating mixer 4 equipped with heating means such as an electric heater is connected to the lower part of the fluidized bed reactor 2 through a raw material gas supply line 5.
- the source gas supply line 5 is also preferably provided with heating means such as an electric heater.
- the preheating mixer 4 is connected to an HFC-134a supply line 6 for supplying gaseous HFC-134a at normal temperature and a dilution gas supply line 7 for supplying dilution gas.
- the HFC-134a and the dilution gas are supplied to the preheating mixer 4 through the HFC-134a supply line 6 and the dilution gas supply line 7, respectively, mixed in the preheating mixer 4 and heated to a predetermined temperature, and then the raw material gas It is supplied to the fluidized bed reactor 2 by a supply line 5.
- the HFC-134a supply line 6 and the dilution gas supply line 7 are connected in front of the preheating mixer 4, and after mixing the HFC-134a and the dilution gas, the mixed gas supply line
- the HFC-134a supply line 6 and the dilution gas supply line 7 may be connected to the preheating mixer 4, respectively, so that the HFC-134a and the dilution gas are separately supplied.
- the preheating mixer 4 may be supplied.
- a preheater (preheater) (not shown) equipped with an electric heater or the like is installed in at least one of the HFC-134a supply line 6 and the dilution gas supply line 7, and the HFC-134a and dilution gas supplied through the line are installed. Alternatively, at least one of these may be preheated and then introduced into the preheating mixer 4.
- the outlet line 9 provided with heating means 8 such as an electric heater is connected to the outlet of the upper part of the fluidized bed reactor 2, and the hydrogen fluoride capturing pipe 10 is installed in the outlet line 9. Then, after the hydrogen fluoride is removed by the hydrogen fluoride capturing pipe 10, the gas led out from the outlet of the fluidized bed reactor 2 (hereinafter referred to as outlet gas) is collected in a sampling bag, and gas chromatograph The content component is analyzed and quantified by an analyzer such as (GC).
- GC analyzer
- HFO-1123 can be obtained as a component of the outlet gas.
- compounds other than HFO-1123 and unreacted raw material component (HFC-134a) contained in the outlet gas hydrogen fluoride, E / Z-1,2-difluoroethylene (E / Z-HFO-1132), 1,1-difluoroethylene (VdF), 1,1,2-trifluoroethane (HFC-143), methane, ethane, ethylene, propane, propylene, normal butane, isobutane, 1-normal butene, 2-normal butene, Isobutene, fluoroethylene (HFO-1141), 3,3-difluoropropene (HFO-1252zf), 3,3,3-trifluoropropene (HFO-1243zf), 2,3,3,3-tetrafluoropropene (HFO) -1234yf), E / Z-1,3,3,3-tetrafluoropropene (HFO) -1234yf), E
- the compound obtained as the outlet gas component can be used for various purposes as it is, but it is preferable to increase the purity of the target component HFO-1123 by purification.
- the purification method include distillation, adsorption, washing with an acidic aqueous solution, a basic aqueous solution, or a neutral aqueous solution.
- the above components other than HFO-1123 contained in the outlet gas can be separated by the above-mentioned means and removed to the desired extent.
- a method of distillation under normal pressure, increased pressure, or reduced pressure is preferable.
- High-purity HFO-1123 can be obtained by distillation under these pressures.
- the HFC-134a separated from the outlet gas can be recycled as a part of the raw material gas.
- the preheating temperature of HFC-134a, the temperature and pressure in the reactor are set values.
- reaction apparatus 1 As the reaction apparatus 1, a fluidized bed reaction apparatus 11 shown in FIG. 2 was used.
- the fluidized bed reactor 11 shown in FIG. 2 has a structure in which the fluidized bed reactor 1 shown in FIG. 1 is provided with a differential pressure measuring unit for measuring the differential pressure between the inlet side and the outlet side of the fluidized bed reactor 2.
- a vertical fluidized bed reactor made of stainless steel (SUS316) and having an inner diameter of 21.4 mm ⁇ height of 600 mm is used.
- a 1 mm SUS316 insertion tube was introduced, a K-type thermocouple was inserted therein, and the temperature in the reactor was measured.
- an eye plate and glass wool were installed at a height of 100 mm from the lower part of the fluidized bed reactor 2, and a solid reactant was filled thereon to form a solid reactant layer 3.
- the inside of the fluidized bed reactor 2 was heated by an electric furnace 2a.
- a preheating mixer 4 was connected to the lower part of the fluidized bed reactor 2 through a raw material gas supply line 5.
- the source gas supply line 5 and the preheating mixer 4 were each heated to 100 ° C. by a ribbon heater.
- the HFC-134a and the dilution gas nitrogen are mixed with mass flow controllers 6a and 7a installed in the HFC-134a supply line 6 and the dilution gas supply line 7, respectively, and then mixed by preheating with the mixed gas supply line 12. It was comprised so that the container 4 might be supplied.
- the outlet gas containing the reaction product was continuously taken out from the upper part of the fluidized bed reactor 2, passed through a hydrogen fluoride capturing tube 10 filled with 28 g of 1/16 inch sodium fluoride pellets, and then polyvinylidene fluoride (The sample was collected in a PVdF) sampling bag (hereinafter referred to as a PVdF bag), and composition analysis was performed using gas chromatography (GC).
- a PVdF polyvinylidene fluoride
- the differential pressure measuring unit was configured as follows, that is, a vertical height of 600 mm between the inlet side pipe connected to the lower part of the fluidized bed reactor 2 and the outlet side pipe connected to the upper part.
- a translucent PFA tube 13a with an inner diameter of 4.35 mm processed into a U-shape is inserted, and fluorine oil (density 1.85 g / mL (25 ° C.)) is introduced into the tube at a height of 300 mm.
- the differential pressure gauge 13 was used.
- the fluidized bed reactor 1 in FIG. 1 has the same inner diameter and height as the fluidized bed reactor 2 (inner diameter 21.4 mm ⁇ height 600 mm), and is made of a transparent acrylic resin so that the inner fluid state can be seen.
- a visualization tester 14 was also provided, and a flow visualization test device 15 was obtained.
- an eye plate and glass wool are installed at a height of 100 mm from the lower part, and a solid reactant is filled thereon, Reactant layer 3 was formed.
- a mixed gas supply line 12 of HFC-134a and dilution gas was connected to the lower part of the visualization tester 14.
- the HFC-134a and nitrogen are mixed with mass flow controllers 6a and 7a installed in the HFC-134a supply line 6 and the dilution gas supply line 7, respectively, and then mixed by the mixed gas supply line 12. Configured to supply.
- differential pressure measuring unit in order to measure the differential pressure between the inlet side and the outlet side of the visualization tester 14, a differential pressure measuring unit was provided. That is, the differential pressure gauge 13 was provided between the inlet side pipe connected to the lower part of the visualization tester 14 and the outlet side pipe connected to the upper part, similarly to the fluidized bed reactor 11 shown in FIG.
- the linear velocity of nitrogen gas or a mixed gas of nitrogen and HFC-134a is determined by determining the flow rate (volume flow rate) per unit time at the reaction temperature and reaction pressure of each gas, and the cross-sectional area of the fluidized bed reactor 2 or the visualization tester 14. It was calculated by dividing by.
- Reagent filling example 1 In the above-described visualization tester of the flow visualization test apparatus, particulate potassium carbonate (manufactured by Asahi Glass Co., Ltd., trade name: potassium carbonate FG, average particle size: 300 ⁇ m, bulk density: 0.9 g / cm 3 ) as a solid reactant. 55 g of a specific surface area: 1.2 m 2 / g (hereinafter referred to as potassium carbonate FG) was filled to a height of 150 mm.
- Reagent filling example 2 The fluidized bed reactor of the fluidized bed reactor 11 described above was charged with 55 g of particulate potassium carbonate FG to a height of 150 mm.
- particulate potassium carbonate manufactured by Asahi Glass Co., Ltd., trade name: potassium carbonate FG R-10, average particle size: 10 ⁇ m, bulk density: 0.3 g / cm 3 , ratio Surface area: 24 g of 1.4 m 2 / g (hereinafter referred to as potassium carbonate FG R-10) was filled to a height of 150 mm.
- Reagent filling example 4 The fluidized bed reactor of the fluidized bed reactor 11 was charged with 24 g of particulate potassium carbonate FG R-10 to a height of 150 mm.
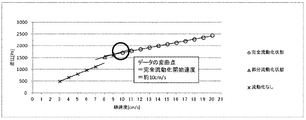
- Fluidization example 1 As shown in Reactant Packing Example 1, nitrogen gas was flowed at a flow rate of 151 mmol / min (line) at room temperature (25 ° C.) and normal pressure in a flow visualization test apparatus packed with a solid reactant (potassium carbonate FG having an average particle size of 300 ⁇ m). The flow rate was 17 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the visualization tester measured with a differential pressure gauge was 1960 Pa. Moreover, in the visualization tester, upper and lower mixing was observed in the entire layer of the solid reactant. That is, the complete fluidization state was confirmed visually.
- the visual fluidization state of the solid reactant layer in the visualization tester was determined according to the following criteria. ⁇ ... Upper and lower mixing is observed in the whole layer of the filled solid reactant (completely fluidized state) ⁇ : Upper and lower mixing is observed only in a part of the packed solid reactant layer (partially fluidized state) X: No mixing of upper and lower layers of the packed solid reactant (not fluidized)
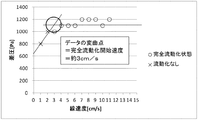
- the inflection point where the slope of the graph changes is the starting point of fluidization of the solid reactant layer, and the linear velocity at this point may be the fluidization starting velocity. it can.
- the linear velocity at the inflection point on the low linear velocity side is the partial fluidization start velocity
- the linear velocity at the inflection point on the high linear velocity side is Let it be the complete fluidization start speed. From Table 1 and FIG. 4, in fluidization example 1, it can be determined that the partial fluidization start speed of the solid reactant layer is 3 to 6 cm / s, and the complete fluidization start speed is 13 cm / s. .
- Fluidization example 2 In the flow visualization test device filled with the solid reactant (potassium carbonate FG) as shown in Reactant Packing Example 1, nitrogen gas was flowed at a flow rate of 121 mmol / min and HFC-134a was flowed at normal pressure at room temperature (25 ° C.). It was mixed and flowed at 30 mmol / min. That is, nitrogen gas 80 mol% and HFC-134a 20 mol% were mixed and flowed (mixed gas linear velocity 17 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the visualization tester measured with a differential pressure gauge was 2395 Pa. Moreover, in the visualization tester, upper and lower mixing was observed in the entire layer of the solid reactant, and the complete fluidization state was visually confirmed.
- the nitrogen gas flow rate and the HFC-134a flow rate are gradually decreased.
- the differential pressure between the inlet side and the outlet side of the visualization tester was measured with a differential pressure gauge, and the fluidization state of the solid reactant in the visibility tester was visually examined.
- Table 2 shows a summary of the flow rate of nitrogen gas, the flow rate of HFC-134a, the linear velocity of the mixed gas, the measured value of the differential pressure, and the fluidization state of the solid reactant visually.
- the graph which plotted the differential pressure with respect to the linear velocity of mixed gas is shown in FIG. From Table 2 and FIG. 5, in fluidization example 2, it can be determined that the partial fluidization start speed of the solid reactant layer is 4 to 8 cm / s and the complete fluidization start speed is 15 cm / s. .
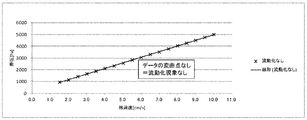
- Fluidization example 3 In the fluidized bed reactor filled with the solid reactant (potassium carbonate FG) as shown in Reactant Packing Example 2, nitrogen gas was flowed at a flow rate of 152 mmol / min (linear velocity 17 cm / s) at room temperature (25 ° C.) and normal pressure. ). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 2631 Pa.
- Table 3 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. From Table 3 and FIG. 6, in fluidization example 3, it can be determined that the partial fluidization start speed of the solid reactant layer is 6 cm / s and the complete fluidization start speed is 15 cm / s.
- Fluidization example 4 As shown in Reactant Packing Example 2, in a fluidized bed reactor filled with solid reactant, nitrogen gas was mixed at a flow rate of 143 mmol / min and HFC-134a at a flow rate of 36 mmol / min at room temperature (25 ° C.) and normal pressure. And washed away. That is, 80 mol% nitrogen gas and 20 mol% HFC-134a were mixed and flowed (mixed gas linear velocity 20 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 3248 Pa.
- Fluidization example 5 As shown in Reactant Packing Example 2, the inside of the fluidized bed reactor of the fluidized bed reactor filled with the solid reactant was heated to 310 ° C. in an electric furnace. Then, nitrogen gas was allowed to flow into the apparatus at a normal pressure and a flow rate of 91 mmol / min (linear velocity: 20 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 2558 Pa.
- Table 5 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. From Table 5 and FIG. 8, in fluidization example 5, it can be determined that the partial fluidization start speed of the solid reactant layer is 6 cm / s and the complete fluidization start speed is 10 cm / s.
- Fluidization example 6 As shown in Reactant Packing Example 2, the inside of the fluidized bed reactor of the fluidized bed reactor filled with the solid reactant was heated to 360 ° C. in an electric furnace. Then, nitrogen gas was allowed to flow into the apparatus at a normal pressure and a flow rate of 84 mmol / min (linear velocity: 20 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 2431 Pa.
- Table 6 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. From Table 6 and FIG. 9, in fluidization example 6, it can be determined that the partial fluidization start speed of the solid reactant layer is 7 cm / s and the complete fluidization start speed is 10 cm / s.
- Fluidization example 7 As shown in Reactant Packing Example 2, the inside of the fluidized bed reactor of the fluidized bed reactor filled with the solid reactant was heated to 410 ° C. in an electric furnace. Then, nitrogen gas was allowed to flow at a flow rate of 78 mmol / min (linear velocity: 20 cm / s) in the apparatus at normal pressure. At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 2431 Pa.
- Table 7 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. From Table 7 and FIG. 10, in fluidization example 7, it can be determined that the partial fluidization start speed of the solid reactant layer is 8 cm / s and the complete fluidization start speed is 10 cm / s.
- Fluidization Comparative Example 1 In the flow visualization test apparatus filled with the solid reactant (potassium carbonate FG R-10 having an average particle size of 10 ⁇ m) as shown in Reactant Packing Example 3, nitrogen gas was supplied at a flow rate of 90 mmol / hour at room temperature (25 ° C.) and normal pressure. It flowed at min (linear velocity 10 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the visualization tester measured with a differential pressure gauge was 4990 Pa. Further, in the visualization tester, the upper and lower layers of the solid reactant layer were not observed, and the flow channel was formed and was in a single flow state. That is, it was not fluidized.
- the solid reactant potassium carbonate FG R-10 having an average particle size of 10 ⁇ m
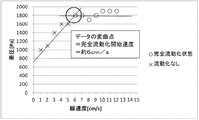
- Fluidization Comparative Example 2 In the fluidized bed reactor filled with the solid reactant (potassium carbonate FG R-10) as shown in Reactant Packing Example 4, nitrogen gas was flowed at 89 mmol / min (linear velocity) at room temperature (25 ° C.) and normal pressure. 10 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 1942 Pa.
- Table 9 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. In the graph of FIG. 12, the inflection point of the differential pressure indicating the start of fluidization of the solid reactant layer was not found. From Table 9 and FIG. 12, in fluidization comparative example 2, it can be judged that the fluidization phenomenon including partial fluidization does not occur in the linear velocity range of 10 cm / s or less.
- Fluidization Comparative Example 3 As shown in Reactant Packing Example 4, the inside of the fluidized bed reactor of the fluidized bed reactor packed with the solid reactant was heated to 310 ° C. in an electric furnace. Then, nitrogen gas was flowed into the apparatus at normal pressure and at a flow rate of 89 mmol / min (linear velocity: 8.5 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 1688 Pa.
- Table 10 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. In the graph of FIG. 13, the inflection point of the differential pressure indicating the start of fluidization of the solid reactant layer was not found. From Table 10 and FIG. 13, in fluidization comparative example 3, it can be judged that the fluidization phenomenon including partial fluidization does not occur in the linear velocity range of 8.5 cm / s or less.
- Fluidization Comparative Example 4 As shown in Reactant Packing Example 4, the inside of the fluidized bed reactor of the fluidized bed reactor filled with the solid reactant was heated to 360 ° C. in an electric furnace. Then, nitrogen gas was allowed to flow at a normal flow rate in the apparatus at a flow rate of 36 mmol / min (linear velocity: 8.5 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 2087 Pa.
- Table 11 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. In the graph of FIG. 14, the inflection point of the differential pressure indicating the start of fluidization of the solid reactant layer was not seen. From Table 11 and FIG. 14, in fluidization comparative example 4, it can be judged that the fluidization phenomenon including partial fluidization does not occur in the linear velocity range of 8.5 cm / s or less.
- Fluidization Comparative Example 5 As shown in Reactant Packing Example 4, the inside of the fluidized bed reactor of the fluidized bed reactor filled with the solid reactant was heated to 410 ° C. in an electric furnace. Then, nitrogen gas was flowed into the apparatus at a normal pressure and a flow rate of 39 mmol / min (linear velocity: 10 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 1579 Pa.
- Table 12 shows a summary of the measured values of nitrogen gas flow rate, linear velocity, and differential pressure. Moreover, the graph which plotted the differential pressure with respect to the linear velocity of nitrogen gas is shown in FIG. In the graph of FIG. 15, the inflection point of the differential pressure indicating the start of fluidization of the solid reactant layer was not seen. From Table 12 and FIG. 15, in fluidization comparative example 5, it can be judged that the fluidization phenomenon including partial fluidization does not occur in the linear velocity range of 10 cm / s or less.
- Table 13 shows the results of the fluidization tests obtained in the above fluidization examples 1 to 7 and fluidization comparative examples 1 to 5. From Table 13, potassium carbonate having an average particle diameter of 300 ⁇ m has good fluidity, and a fluidized state can be obtained by circulating gas at a predetermined linear velocity. However, potassium carbonate having an average particle diameter of 10 ⁇ m has fluidity. Unfortunately, it does not fluidize at a linear velocity of several cm / s to several tens cm / s.
- Example 1 the inside of a fluidized bed reactor of a fluidized bed reactor filled with a solid reactant (potassium carbonate FG) as shown in Reactant Filling Example 2 was heated to 360 ° C. in an electric furnace. Then, nitrogen gas was flowed into the fluidized bed reactor at normal pressure at a flow rate of 50.3 mmol / min (linear velocity 12 cm / s). From the results of the above fluidization test (fluidization example 6), it is considered that the layer of potassium carbonate FG is in a completely fluidized state at this linear velocity.
- a solid reactant potassium carbonate FG
- HFC-134a was started to flow at a flow rate of 2.5 mmol / min. After reacting by flowing HFC-134a for 10 minutes from the start of distribution of HFC-134a, the supply of HFC-134a was stopped without changing the flow rate of nitrogen gas, and the reaction in Example 1 was completed. The outlet gas from 5 minutes after the start of distribution of HFC-134a until the end of the reaction was continuously collected in a PVdF bag.
- Example 2 was carried out as it was without replacing potassium carbonate in the fluidized bed reactor.
- HFC-134a was allowed to react with the solid reactant in the same manner as in Example 1 except that the reaction conditions were set as shown in Table 14.
- the composition of the outlet gas collected in the PVdF bag was analyzed by gas chromatography (GC).
- GC gas chromatography
- the analysis results are shown as reaction conditions (nitrogen flow rate before reaction, reaction temperature, HFC-134a flow rate during reaction, nitrogen flow rate during reaction, composition during reaction (HFC-134a: nitrogen (molar ratio)), linear velocity during reaction, contact during reaction.
- Table 14 shows the time, the presence or absence of a fluidized state during the reaction, and the time during which HFC-134a was flowed (hereinafter referred to as the reaction time).
- Comparative Examples 1 and 2 First, in Comparative Example 1, the inside of a fluidized bed reactor of a fluidized bed reactor filled with a solid reactant (potassium carbonate FG R-10) as shown in Reactant Filling Example 4 was heated to 360 ° C. in an electric furnace. However, nitrogen gas was passed through the fluidized bed reactor at normal pressure at a flow rate of 6.24 mmol / min (linear velocity: 1.5 cm / s). From the results of the fluidization test (fluidization comparative example 4), it is considered that potassium carbonate FG R-10 is not fluidized at this linear velocity.
- a solid reactant potassium carbonate FG R-10
- HFC-134a was started to flow at a flow rate of 0.31 mmol / min. After reacting by flowing HFC-134a for 15 minutes after starting the flow of HFC-134a, only the supply of HFC-134a was stopped without changing the flow rate of nitrogen gas, and the reaction in Comparative Example 1 was completed. The outlet gas from 5 minutes after the start of distribution of HFC-134a until the end of the reaction was continuously collected in a PVdF bag.
- Comparative Example 2 HFC-134a was contacted with the solid reactant and reacted in the same manner as in Comparative Example 1 except that the reaction conditions were set as shown in Table 15. Then, the composition of the outlet gas collected in the PVdF bag was analyzed by gas chromatography (GC). The analysis results are shown as reaction conditions (nitrogen flow rate before reaction, reaction temperature, HFC-134a flow rate during reaction, nitrogen flow rate during reaction, composition during reaction (HFC-134a: nitrogen (molar ratio)), linear velocity during reaction, contact during reaction. The results are shown in Table 15 together with the time, the presence or absence of a fluidized state during reaction, and the reaction time.
- GC gas chromatography
- the fluidized bed reactor 16 shown in FIG. 16 has a structure in which the fluidized bed reactor 1 shown in FIG. 1 is provided with a differential pressure measuring unit for measuring the differential pressure between the inlet side and the outlet side of the fluidized bed reactor 2.
- a vertical fluidized bed reactor made of stainless steel (SUS316) having an inner diameter of 106.3 mm and a height of 550 mm was used, and the diameter of the reactor was 6 mm in the vertical direction.
- a SUS316 insertion tube was introduced, a K-type thermocouple was inserted therein, and the temperature in the reactor was measured.
- an eye plate was installed at the bottom of the fluidized bed reactor 2, and a solid reactant was filled thereon to form a solid reactant layer 3.
- the fluidized bed reactor 2 was heated by an electric heater 2a.
- a preheating mixer 4 was connected to the lower part of the fluidized bed reactor 2 through a raw material gas supply line 5.
- the source gas supply line 5 and the preheating mixer 4 were each heated to 200 to 450 ° C. by a ribbon heater.
- the HFC-134a and the dilution gas nitrogen are mixed with mass flow controllers 6a and 7a installed in the HFC-134a supply line 6 and the dilution gas supply line 7, respectively, and then mixed by preheating with the mixed gas supply line 12. It was comprised so that the container 4 might be supplied.
- the outlet gas containing the reaction product is continuously taken out from the upper part of the fluidized bed reactor 2, collected in a sampling bag made of polyvinylidene fluoride (PVdF) (hereinafter referred to as PVdF back), and gas chromatography (GC).
- PVdF polyvinylidene fluoride
- GC gas chromatography
- the differential pressure measuring unit was configured as follows, that is, a digital differential pressure gauge 17 was installed between the inlet side pipe connected to the lower part of the fluidized bed reactor 2 and the outlet side pipe connected to the upper part. .
- the linear velocity of nitrogen gas, HFC-134a, or a mixed gas of nitrogen and HFC-134a is determined by calculating the flow rate (volume flow rate) per unit time at the reaction temperature and reaction pressure of each gas by the cross-sectional area of the fluidized bed reactor 2. It was determined by dividing.
- Blank differential pressure measurement example 1 In the fluidized bed reactor 16 described above, nitrogen gas was allowed to flow at a flow rate of 3.92 mol / min (linear velocity: 18 cm / s) at room temperature (25 ° C.) and normal pressure into the empty fluidized bed reactor 2 before filling the reactants. The differential pressure was measured. At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 10900 Pa. Thereafter, the nitrogen gas flow rate was gradually decreased, and at each flow rate, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor was measured with a differential pressure gauge.
- Blank differential pressure measurement example 2 With respect to the fluidized bed reactor 16 described above, HFC-134a was allowed to flow at a flow rate of 2.61 mol / min (linear velocity: 12 cm / s) at room temperature (25 ° C.) and normal pressure into the empty fluidized bed reactor 2 before filling the reactants. The differential pressure was measured. At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 11500 Pa. Thereafter, the HFC-134a flow rate was gradually decreased, and the differential pressure between the inlet side and the outlet side of the fluidized bed reactor was measured with a differential pressure gauge at each flow rate.
- Blank differential pressure measurement example 3 Regarding the fluidized bed reactor 16 described above, the difference when flowing nitrogen gas at a flow rate of 2.47 mol / min (linear velocity: 18 cm / s) at 200 ° C. and normal pressure into the empty fluidized bed reactor 2 before filling the reactants. The pressure was measured. At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 11700 Pa. Thereafter, the nitrogen gas flow rate was gradually decreased, and at each flow rate, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor was measured with a differential pressure gauge.
- Blank differential pressure measurement example 4 Regarding the fluidized bed reactor 16 described above, the difference when flowing nitrogen gas at a flow rate of 1.25 mol / min (linear velocity: 11 cm / s) at an ordinary pressure of 300 ° C. into the empty fluidized bed reactor 2 before filling the reactants. The pressure was measured. At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 6500 Pa. Thereafter, the nitrogen gas flow rate was gradually decreased, and at each flow rate, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor was measured with a differential pressure gauge.
- Reagent filling example 5 In the fluidized bed reactor 2 of the fluidized bed reactor 16 described above, particulate calcium oxide (average particle size: 100 ⁇ m, bulk density: 1.2 g / cm 3 , specific surface area: 2.9 m 2 / g as a solid reactant). 2099 g (37.42 mol) (hereinafter referred to as calcium oxide) was filled to a height of 200 mm.
- particulate calcium oxide (average particle size: 100 ⁇ m, bulk density: 1.2 g / cm 3 , specific surface area: 2.9 m 2 / g as a solid reactant). (Hereinafter referred to as calcium oxide) 3143 g (56.05 mol) was filled to a height of 300 mm.
- the blank differential pressure before filling the reactants is subtracted from the differential pressure after filling the reactants (hereinafter referred to as differential pressure after filling) under the same conditions (temperature, pressure, gas type, flow rate).
- the fluidization start speed was determined based on the converted differential pressure.
- the inflection point where the slope of the graph changes is the starting point of fluidization of the solid reactant layer, and the linear velocity at this point is the fluidization starting velocity. be able to.
- the linear velocity at the inflection point on the low linear velocity side is the partial fluidization start velocity
- the linear velocity at the inflection point on the high linear velocity side is Let it be the complete fluidization start speed
- Fluidization example 8 In a fluidized bed reactor packed with 200 mm of solid reactant (calcium oxide having an average particle size of 100 ⁇ m) as shown in Reactant Packing Example 5, nitrogen gas was supplied at a flow rate of 3.05 mol / min at room temperature (25 ° C.) and normal pressure. (Linear speed 14 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 10900 Pa.
- solid reactant calcium oxide having an average particle size of 100 ⁇ m
- Table 16 shows the results of summarizing the converted differential pressure obtained by calculating the difference from the nitrogen gas flow rate, the linear velocity, the measured value of the differential pressure after filling, and the blank differential pressure measurement example 1. Moreover, the graph which plotted the conversion differential pressure
- Fluidization example 9 In a fluidized bed reactor packed with 300 mm of a solid reactant (calcium oxide having an average particle size of 100 ⁇ m) as shown in Reactant Packing Example 6, nitrogen gas was supplied at a flow rate of 2.83 mol / min at room temperature (25 ° C.) and normal pressure. (Linear speed 13 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 10200 Pa.
- a solid reactant calcium oxide having an average particle size of 100 ⁇ m
- Table 17 shows the results of collecting the converted differential pressure obtained by calculating the difference between the flow rate of the nitrogen gas, the linear velocity, the measured value of the differential pressure after filling, and the blank differential pressure measurement example 1. Moreover, the graph which plotted the conversion differential pressure
- Fluidization example 10 In a fluidized bed reactor packed with 200 mm of solid reactant (calcium oxide having an average particle size of 100 ⁇ m) as shown in Reactant Packing Example 5, HFC-134a was supplied at a flow rate of 2.61 mol / hour at room temperature (25 ° C.) and normal pressure. It flowed at min (linear velocity 12 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 13400 Pa.
- solid reactant calcium oxide having an average particle size of 100 ⁇ m
- Table 18 shows the results of summarizing HFC-134a flow rate, linear velocity, measured value of post-filling differential pressure, and converted differential pressure calculated from the difference from the blank differential pressure measurement example 2.
- FIG. 19 shows a graph in which the converted differential pressure is plotted against the linear velocity of HFC-134a. From Table 18 and FIG. 19, in fluidization example 10, it can be determined that the solid reactant layer complete fluidization start speed is 6 cm / s.
- Fluidization example 11 In a fluidized bed reactor filled with 200 mm of a solid reactant (calcium oxide having an average particle diameter of 100 ⁇ m) as shown in Reactant Packing Example 5, nitrogen gas was flowed at a normal pressure of 200 ° C. at a flow rate of 2.19 mol / min (linear velocity). 16 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 11200 Pa.
- a solid reactant calcium oxide having an average particle diameter of 100 ⁇ m
- Table 19 shows the results of summarizing the converted differential pressure obtained by calculating the difference from the nitrogen gas flow rate, the linear velocity, the measured value of the differential pressure after filling, and the blank differential pressure measurement example 3. Moreover, the graph which plotted the conversion differential pressure
- Fluidization example 12 In a fluidized bed reactor filled with 200 mm of a solid reactant (calcium oxide having an average particle diameter of 100 ⁇ m) as shown in Reactant Packing Example 5, nitrogen gas was supplied at a flow rate of 1.25 mol / min (linear velocity) at 300 ° C. and normal pressure. 11 cm / s). At this time, the differential pressure between the inlet side and the outlet side of the fluidized bed reactor measured with a differential pressure gauge was 7700 Pa.
- a solid reactant calcium oxide having an average particle diameter of 100 ⁇ m
- Table 20 shows the results of summarizing the converted differential pressure obtained by calculating the difference from the nitrogen gas flow rate, the linear velocity, the measured value of the differential pressure after filling, and the blank differential pressure measurement example 4. Moreover, the graph which plotted the conversion differential pressure
- Table 21 shows the results of fluidization tests obtained in the above fluidization examples 8-12. From Table 21, calcium oxide having an average particle size of 100 ⁇ m has good fluidity, and a fluidized state can be obtained by flowing gas at a linear velocity of 7 cm / s or more regardless of the gas type and filling height. Recognize. It can also be seen that the fluidity increases as the temperature rises.
- Example 3 the inside of a fluidized bed reactor of a fluidized bed reactor packed with 300 mm of a solid reactant (calcium oxide having an average particle size of 100 ⁇ m) as shown in Reactant Packing Example 6 was heated at 300 ° C. in an electric furnace. Heated to. Then, nitrogen gas was flowed into the fluidized bed reactor at normal pressure at a flow rate of 0.79 mol / min (linear velocity: 7 cm / s). From the results of fluidization examples 8 to 12, the calcium oxide layer is considered to be in a completely fluidized state at this linear velocity.
- a solid reactant calcium oxide having an average particle size of 100 ⁇ m
- the flow rate of nitrogen gas was reduced to 0.71 mol / min, and at the same time, HFC-134a started to flow at a flow rate of 0.08 mmol / min.
- the supply of HFC-134a was stopped, and at the same time, the flow rate of nitrogen gas was changed to 0.79 mol / min, and the reaction in Example 3 was conducted.
- the outlet gas from 2 minutes after the start of circulation of HFC-134a to the end of the reaction for about 10 seconds was continuously collected in a PVdF bag.
- Examples 4 to 10 were performed as they were without replacing the calcium oxide in the fluidized bed reactor.
- HFC-134a was reacted with a solid reactant in the same manner as in Example 3 except that the reaction conditions were the same as those shown in Table 22.
- the composition of the outlet gas collected in the PVdF bag was analyzed by gas chromatography (GC). The analysis results are shown as reaction conditions (nitrogen flow rate before reaction, reaction temperature, HFC-134a flow rate during reaction, nitrogen flow rate during reaction, composition during reaction (HFC-134a: nitrogen (molar ratio)), linear velocity during reaction, contact during reaction.
- Table 22 shows the time, the presence or absence of a fluidized state during the reaction, and the reaction time.
- Example 11-15 First, in Example 11, the inside of a fluidized bed reactor of a fluidized bed reactor filled with 300 mm of a solid reactant (calcium oxide having an average particle diameter of 100 ⁇ m) as shown in Example 6 of filling of the reactants is 350 ° C. in an electric furnace. Heated to. Then, nitrogen gas was flowed into the fluidized bed reactor at a normal pressure and a flow rate of 0.73 mol / min (linear velocity: 7 cm / s). From the results of fluidization examples 8 to 12, the calcium oxide layer is considered to be in a completely fluidized state at this linear velocity.
- a solid reactant calcium oxide having an average particle diameter of 100 ⁇ m
- HFC-134a started to flow at a flow rate of 0.73 mol / min.
- the supply of HFC-134a was stopped, and at the same time, the flow rate of nitrogen gas was changed to 0.73 mol / min, and the reaction in Example 11 was performed.
- the exit gas from 3 minutes after the start of circulation of HFC-134a to the end of the reaction for about 10 seconds was continuously collected in a PVdF bag.
- Examples 12 to 15 were carried out as they were without replacing the calcium oxide in the fluidized bed reactor.
- HFC-134a was reacted with a solid reactant in the same manner as in Example 11 except that the reaction conditions were set as shown in Table 23.
- the composition of the outlet gas collected in the PVdF bag was analyzed by gas chromatography (GC). The analysis results are shown as reaction conditions (nitrogen flow rate before reaction, reaction temperature, HFC-134a flow rate during reaction, nitrogen flow rate during reaction, composition during reaction (HFC-134a: nitrogen (molar ratio)), linear velocity during reaction, contact during reaction.
- Table 23 shows the time, presence or absence of a fluidized state during reaction, and reaction time.
- HFC-134a As can be seen from Tables 14 and 15, according to Example 1 and Example 2 in which HFC-134a was reacted with potassium carbonate in a fluidized state, HFC-134a was reacted with potassium carbonate not in a fluidized state. Compared with Comparative Example 1 and Comparative Example 2, the selectivity of HFO-1123 can be increased. Further, as can be seen from Tables 21 and 22, it is possible to obtain HFO-1123 with a high reaction rate and a sufficiently high selectivity by reacting HFC-134a with calcium oxide in a fluidized state.
- HFO-1123 can be efficiently and stably produced from HFC-134a. Moreover, it is useful as an industrial production method from the point of using HFC-134a which is an inexpensive raw material.
- HFC-134a which is an inexpensive raw material.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Combustion & Propulsion (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
なお、本明細書において、ハロゲン化炭化水素については、化合物名の後の括弧内にその化合物の略称(冷媒番号等)を記す。また、必要に応じて化合物名に代えて、その略称を用いる。
(1)固体反応剤の粒子とHFC-134aとを均一に混合して接触させることが難しいため、固体反応剤の転化率が低い。また、HFC-134aからHFO-1123を生成する反応の反応性が低いため、HFC-134aを長い時間固体反応剤と接触させる必要がある。
(2)固定床では除熱効率が悪いため、ホットスポットが発生しやすい。そのため、HFC-134aの炭素―炭素結合の切断等の副反応が生起しやすく、メタン、エチレン、プロピレン等の低分子炭化水素類や高分子カーボン(黒鉛)類のような副生物が生成しやすい。
(3)高分子カーボン類の副生が多くなるため、固体反応剤表面への高分子カーボン類の付着により、HFC-134aの転化率が経時的に著しく低下する。そのため、HFO-1123を安定して製造することが困難である
などの問題があった。
なお、固体反応剤層が「流動化した状態(流動化状態ともいう。)」にある場合、層を構成する固体反応剤の粒子は流体中を浮遊し流動するので、流動化状態なる語については、固体反応剤層と固体反応剤の粒子の両方について使用する。
すなわち、流動床の反応では、反応熱の除熱効率が高くホットスポットが発生しにくいため、HFC-134aの副反応(炭素―炭素結合切断)の進行を抑制できる。そのため、低分子炭化水素類や高分子カーボン(黒鉛)類が副生しにくく、R-1123の生成反応の選択率が向上する。また、高分子カーボン類が副生しにくいので、高分子カーボン類の固体反応剤表面への付着によるHFC-134a転化率の経時的な低下が防止され、R-1123を安定的に得ることができる。
CF3-CH2F + 固体反応剤(Cat.) → CF2=CHF + HF
……………(1)
CF3-CH2F + 固体反応剤(MOH) → CF2=CHF + MF + H2O ……………(2)
本発明の実施形態における反応装置として、流動床反応装置が用いられる。流動床反応装置としては、フルードベッド型反応装置またはライザー型反応装置を用いることができる。HFO-1123を効率よく安定的に製造する観点から、フルードベッド型反応装置の使用が好ましい。
本発明に使用されるHFC-134aを含む原料ガスは、後述する反応条件において常に気相で存在する。HFC-134aは、純度100%(モル%)のHFC-134aであってもよいし、製法由来の不純物である1,1,2,2-テトラフルオロエタン(HFC-134)を含むものであってもよい。HFC-134を含む場合、HFC-134aの純度は50モル%以上であることが好ましい。すなわち、原料ガスは、純度100%(モル%)のHFC-134aを含むものであってもよいし、HFC-134等の不純物を含む純度50モル%以上のHFC-134aを含むものであってもよい。
本発明に使用される固体反応剤は、平均粒子径が1μm~5000μmの粒子状のものである。なお、本明細書において、平均粒子径は、レーザー回折・散乱式粒度分析計により測定した値である。
固体反応剤の平均粒子径が1μm未満の場合には、粒子同士の付着性が高くなり、HFC-134aを含む原料ガスを固体反応剤層に流通させて接触させる際に、固体反応剤層が流動化しにくくなるため、固体反応剤の粒子とHFC-134aとの均一な混合および接触が困難となり、HFC-134aの転化率が低くなる。また、反応場である固体反応剤層における除熱効率が悪く、ホットスポットの発生によりカーボン化等の副反応が起きやすいため、カーボン化合物の付着の影響により、HFC-134aの転化率が経時的に低下する。一方、固体反応剤の平均粒子径が5000μmを超えると、固体反応剤の粒子の流動化のために必要な原料ガスの流通速度が大きくなり過ぎる。そのため、HFC-134aとの反応に十分な接触時間を確保するためには、大型の反応器を必要とし、製造効率が悪い。
(a)目視で観察する方法
固体反応剤層の上下混合を目視で観察する。流動化状態の判定基準は、下記の通りである。
完全流動化状態…固体反応剤層全体に上下混合が見られる
部分流動化状態…固体反応剤層の一部に上下混合が見られる
流動化していない状態…固体反応剤層の上下混合なし
(b)差圧を測定する方法
反応器の入口側と出口側のガス圧の差(以下、差圧という。)を測定する。そして、ガスの流通速度(例えば、後述する線速度)に対して差圧をプロットしたグラフを作成し、変曲点の存在により流動化の開始を判定する。
なお、このような固体反応剤層の流動化状態の判定方法については、さらに実施例で具体的に説明する。
金属水酸化物は、上記した金属の1種の水酸化物であってもよく、2種以上の金属の複合水酸化物であってもよい。
金属炭酸塩は、上記した金属の1種の炭酸塩であってもよく、2種以上の金属の複合炭酸塩であってもよい。
金属硫酸塩は、上記した金属の1種の硫酸塩であってもよく、2種以上の金属の複合硫酸塩であってもよい。
金属ハロゲン化物は、上記した金属の1種のハロゲン化物であってもよく、2種以上の金属の複合ハロゲン化物であってもよい。
HFC-134aを含む原料ガスは、常温のまま反応器(例えば、流動床反応器)に導入してもよいが、反応性を高めるために、反応器に導入する前に加熱(予熱)してから供給することが好ましい。予熱を行う場合、原料ガスは80~450℃の温度に加熱してから反応器に供給することが好ましい。そして、HFC-134aを含む原料ガス中の各成分(HFC-134aおよび希釈ガス)は、それぞれ上記温度に予熱してから混合し、混合された上記温度の原料ガスを反応器に供給してもよいし、先に各成分を混合してから上記温度に加熱して反応器に供給してもよい。さらに、原料ガス中の各成分をそれぞれ上記温度に予熱してから、別々に反応器に供給してもよい。
線速度(空塔速度)(cm/s)=流量(cm3/s)/断面積(cm2)
本発明において、HFO-1123の製造に使用される反応装置の一例を、図1に示す。図1に示す流動床反応装置1は、電気炉または電気ヒーター2a内に垂直型流動床反応器2を備える。なお、電気炉または電気ヒーター2aなどの加熱手段の設置は必須ではない。
本発明の製造方法においては、HFO-1123を上記出口ガスの成分として得ることができる。出口ガスに含有されるHFO-1123と未反応の原料成分(HFC-134a)以外の化合物としては、フッ化水素、E/Z-1,2-ジフルオロエチレン(E/Z-HFO-1132)、1,1-ジフルオロエチレン(VdF)、1,1,2-トリフルオロエタン(HFC-143)、メタン、エタン、エチレン、プロパン、プロピレン、ノルマルブタン、イソブタン、1-ノルマルブテン、2-ノルマルブテン、イソブテン、フルオロエチレン(HFO-1141)、3,3-ジフルオロプロペン(HFO-1252zf)、3,3,3-トリフルオロプロペン(HFO-1243zf)、2,3,3,3-テトラフルオロプロペン(HFO-1234yf)、E/Z-1,3,3,3-テトラフルオロプロペン(E/Z-HFO-1234ze)、ヘキサフルオロプロピレン(HFP)、HFC-125、HFC-134、1,1,1-トリフルオロエタン(HFC-143a)、1,1,1,2,2,3,3-ヘプタフルオロプロパン(HFC-227ca)、1,1,1,2,3,3,3-ヘプタフルオロプロパン(HFC-227ea)、1,1,1,3,3,3-ヘキサフルオロプロパン(HFC-236fa)、1,1,1,2,3,3-ヘキサフルオロプロパン(HFC-236ea)、HFC-32、トリフルオロメタン(HFC-23)およびフルオロメタン(HFC-41)、一酸化炭素、二酸化炭素、水等が挙げられる。なお、上記において、E/ZはE体とZ体の混合物を意味する。
(分析条件)
出口ガスの組成分析には、ガスクロマトグラフィー(GC)を用いた。カラムは、DB-1(アジレント・テクノロジー株式会社製、長さ60m×内径250μm×厚さ1μm)を用いた。
反応装置1としては、図2に示す流動床反応装置11を用いた。図2に示す流動床反応装置11は、図1に示す流動床反応装置1に、流動床反応器2の入口側と出口側の差圧を測定するための差圧測定部を設けた構造を有する。
この流動床反応装置11において、流動床反応器2としては、ステンレス(SUS316)製で内径21.4mm×高さ600mmの垂直型流動床用の反応器を用い、反応器の中心に直径3.1mmのSUS316製差込管を導入し、その中にK型熱電対を挿入して、反応器内の温度を測定した。また、流動床反応器2の下部から100mmの高さに目皿とグラスウールを設置し、その上に固体反応剤を充填し、固体反応剤層3を形成した。流動床反応器2内は、電気炉2aにより加熱した。
図1の流動床反応装置1に、その流動床反応器2と内径および高さが同一(内径21.4mm×高さ600mm)で、透明アクリル樹脂製で内部の流動状態が見えるように構成された可視化試験器14を併設し、流動可視化試験装置15とした。可視化試験器14内には、流動床反応装置1の流動床反応器2内と同様に、下部から100mmの高さに目皿とグラスウールを設置し、その上に固体反応剤を充填し、固体反応剤層3を形成した。また、この可視化試験器14の下部にHFC-134aと希釈ガスとの混合ガス供給ライン12を接続した。そして、HFC-134aおよび窒素は、それぞれHFC-134a供給ライン6および希釈ガス供給ライン7に設置されたマスフローコントローラー6a、7aで流量を調整し混合した後、混合ガス供給ライン12により可視化試験器14に供給するように構成した。
窒素ガス、または窒素とHFC-134aの混合ガスの線速度は、各ガスの反応温度、反応圧力における単位時間当たりの流量(体積流量)を、流動床反応器2または可視化試験器14の断面積で割って求めた。
前記した流動可視化試験装置の可視化試験器に、固体反応剤として粒子状の炭酸カリウム(旭硝子株式会社製、商品名:炭酸カリウムFG、平均粒子径:300μm、嵩密度:0.9g/cm3、比表面積:1.2m2/g(以下、炭酸カリウムFGという))の55gを150mmの高さに充填した。
前記した流動床反応装置11の流動床反応器に、粒子状の炭酸カリウムFGの55gを150mmの高さに充填した。
前記した流動可視化試験装置の可視化試験器に、粒子状の炭酸カリウム(旭硝子株式会社製、商品名:炭酸カリウムFG R-10、平均粒子径:10μm、嵩密度:0.3g/cm3、比表面積:1.4m2/g(以下、炭酸カリウムFG R-10という))の24gを150mmの高さに充填した。
前記した流動床反応装置11の流動床反応器に、粒子状の炭酸カリウムFG R-10の24gを150mmの高さに充填した。
反応剤充填例1で示すように固体反応剤(平均粒子径300μmの炭酸カリウムFG)が充填された流動可視化試験装置内に、室温(25℃)常圧で窒素ガスを流量151mmol/min(線速度17cm/s)で流した。このとき、差圧計で測定された可視化試験器の入口側と出口側との差圧は、1960Paであった。また、可視化試験器内では、固体反応剤の層全体に上下混合が見られた。すなわち、完全流動化状態が目視で確認された。
○…充填された固体反応剤の層全体に上下混合が見られる(完全流動化状態)
△…充填された固体反応剤の層の一部にのみ上下混合が見られる(部分流動化状態)
×…充填された固体反応剤の層の上下混合なし(流動化していない状態)

反応剤充填例1で示すように固体反応剤(炭酸カリウムFG)が充填された流動可視化試験装置内に、室温(25℃)常圧で窒素ガスを流量121mmol/minで、HFC-134aを流量30mmol/minで混合して流した。すなわち、窒素ガス80mol%とHFC-134a20mol%を混合して流した(混合ガスの線速度17cm/s)。このとき、差圧計で測定された可視化試験器の入口側と出口側との差圧は、2395Paであった。また、可視化試験器内では固体反応剤の層全体に上下混合が見られ、完全流動化状態が目視で確認された。

反応剤充填例2で示すように固体反応剤(炭酸カリウムFG)が充填された流動床反応装置内に、室温(25℃)常圧で窒素ガスを152mmol/minの流量(線速度17cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、2631Paであった。

反応剤充填例2で示すように固体反応剤が充填された流動床反応装置内に、室温(25℃)常圧で窒素ガスを流量143mmol/minで、HFC-134aを流量36mmol/minで混合して流した。すなわち、窒素ガス80mol%とHFC-134a20mol%を混合して流した(混合ガスの線速度20cm/s)。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、3248Paであった。

反応剤充填例2で示すように固体反応剤が充填された流動床反応装置の流動床反応器内を、電気炉で310℃に加熱した。そして、この装置内に常圧で窒素ガスを91mmol/minの流量(線速度20cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、2558Paであった。

反応剤充填例2で示すように固体反応剤が充填された流動床反応装置の流動床反応器内を、電気炉で360℃に加熱した。そして、この装置内に常圧で窒素ガスを84mmol/minの流量(線速度20cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、2431Paであった。

反応剤充填例2で示すように固体反応剤が充填された流動床反応装置の流動床反応器内を、電気炉で410℃に加熱した。そして、この装置内に常圧で窒素ガスを78mmol/minの流量(線速度20cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、2431Paであった。

反応剤充填例3で示すように固体反応剤(平均粒子径10μmの炭酸カリウムFG R-10)が充填された流動可視化試験装置内に、室温(25℃)常圧で窒素ガスを流量90mmol/min(線速度10cm/s)で流した。このとき、差圧計で測定された可視化試験器の入口側と出口側との差圧は、4990Paであった。また、可視化試験器内では、固体反応剤の層の上下混合は見られず、流路を形成し片流れしている状態であった。すなわち流動化していなかった。

反応剤充填例4で示すように固体反応剤(炭酸カリウムFG R-10)が充填された流動床反応装置内に、室温(25℃)常圧で窒素ガスを89mmol/minの流量(線速度10cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、1942Paであった。

反応剤充填例4で示すように固体反応剤が充填された流動床反応装置の流動床反応器内を、電気炉で310℃に加熱した。そして、この装置内に常圧で窒素ガスを89mmol/minの流量(線速度8.5cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、1688Paであった。

反応剤充填例4で示すように固体反応剤が充填された流動床反応装置の流動床反応器内を、電気炉で360℃に加熱した。そして、この装置内に常圧で窒素ガスを36mmol/minの流量(線速度8.5cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、2087Paであった。

反応剤充填例4で示すように固体反応剤が充填された流動床反応装置の流動床反応器内を、電気炉で410℃に加熱した。そして、この装置内に常圧で窒素ガスを39mmol/minの流量(線速度10cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、1579Paであった。


まず、実施例1において、反応剤充填例2で示すように固体反応剤(炭酸カリウムFG)が充填された流動床反応装置の流動床反応器内を、電気炉で360℃に加熱した。そして、流動床反応装置内に、常圧で窒素ガスを50.3mmol/minの流量(線速度12cm/s)で流した。なお、前記した流動化試験(流動化例6)の結果から、この線速度において、炭酸カリウムFGの層は完全流動化状態であると考えられる。
まず、比較例1において、反応剤充填例4で示すように固体反応剤(炭酸カリウムFG R-10)が充填された流動床反応装置の流動床反応器内を、電気炉で360℃に加熱しながら、流動床反応装置内に常圧で窒素ガスを6.24mmol/minの流量(線速度1.5cm/s)で流した。なお、前記した流動化試験(流動化比較例4)の結果から、この線速度で炭酸カリウムFG R-10は流動化していないと考えられる。
これらの結果を、実施例1,2については表14の下欄に、比較例1,2については表15の下欄にそれぞれ示す。
出口ガス中のHFC-134a由来成分のうちで、HFC-134a以外の成分の割合をいう。出口ガス中の{100-(HFC-134a)}/100×100(%)で計算される
反応したHFC-134aのうちで、HFO-1123に転化したのは何%かをいう。出口ガス中の(HFO-1123)/{100-(HFC-134a)}×100(%)で計算される。
反応したHFC-134aのうちで、HFO-1123以外の化合物に転化したのは何%かをいう。出口ガス中の{100-(HFC-134a)-(HFO-1123)}/{100-(HFC-134a)}×100(%)で計算される。


(分析条件)
出口ガスの組成分析は、実施例1と同様の条件で行った。
反応装置2としては、図16に示す流動床反応装置16を用いた。図16に示す流動床反応装置16は、図1に示す流動床反応装置1に、流動床反応器2の入口側と出口側の差圧を測定するための差圧測定部を設けた構造を有する。
この流動床反応装置16において、流動床反応器2としては、ステンレス(SUS316)製で内径106.3mm×高さ550mmの垂直型流動床用の反応器を用い、反応器の垂直方向に直径6mmのSUS316製差込管を導入し、その中にK型熱電対を挿入して、反応器内の温度を測定した。また、流動床反応器2の最下部に目皿を設置し、その上に固体反応剤を充填し、固体反応剤層3を形成した。流動床反応器2内は、電気ヒーター2aにより加熱した。
窒素ガス、HFC-134a、または窒素とHFC-134aの混合ガスの線速度は、各ガスの反応温度、反応圧力における単位時間当たりの流量(体積流量)を、流動床反応器2の断面積で割って求めた。
前記した流動床反応装置16について、反応剤充填前の空の流動床反応器2に、室温(25℃)常圧で窒素ガスを流量3.92mol/min(線速度18cm/s)で流した際の差圧を測定した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、10900Paであった。その後、窒素ガス流量を徐々に減少させていき、各流量で、流動床反応器の入口側と出口側との差圧を差圧計により測定した。
前記した流動床反応装置16について、反応剤充填前の空の流動床反応器2に、室温(25℃)常圧でHFC-134aを流量2.61mol/min(線速度12cm/s)で流した際の差圧を測定した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、11500Paであった。その後、HFC-134a流量を徐々に減少させていき、各流量で、流動床反応器の入口側と出口側との差圧を差圧計により測定した。
前記した流動床反応装置16について、反応剤充填前の空の流動床反応器2に、200℃常圧で窒素ガスを流量2.47mol/min(線速度18cm/s)で流した際の差圧を測定した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、11700Paであった。その後、窒素ガス流量を徐々に減少させていき、各流量で、流動床反応器の入口側と出口側との差圧を差圧計により測定した。
前記した流動床反応装置16について、反応剤充填前の空の流動床反応器2に、300℃常圧で窒素ガスを流量1.25mol/min(線速度11cm/s)で流した際の差圧を測定した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、6500Paであった。その後、窒素ガス流量を徐々に減少させていき、各流量で、流動床反応器の入口側と出口側との差圧を差圧計により測定した。
前記した流動床反応装置16の流動床反応器2に、固体反応剤として粒子状の酸化カルシウム(平均粒子径:100μm、嵩密度:1.2g/cm3、比表面積:2.9m2/g(以下、酸化カルシウムという))の2099g(37.42mol)を200mmの高さに充填した。
前記した流動床反応装置16の流動床反応器2に、固体反応剤として粒子状の酸化カルシウム(平均粒子径:100μm、嵩密度:1.2g/cm3、比表面積:2.9m2/g(以下、酸化カルシウムという))の3143g(56.05mol)を300mmの高さに充填した。
反応剤充填例5で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が200mm充填された流動床反応装置内に、室温(25℃)常圧で窒素ガスを流量3.05mol/min(線速度14cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、10900Paであった。

反応剤充填例6で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が300mm充填された流動床反応装置内に、室温(25℃)常圧で窒素ガスを流量2.83mol/min(線速度13cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、10200Paであった。

反応剤充填例5で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が200mm充填された流動床反応装置内に、室温(25℃)常圧でHFC-134aを流量2.61mol/min(線速度12cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、13400Paであった。

反応剤充填例5で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が200mm充填された流動床反応装置内に、200℃常圧で窒素ガスを流量2.19mol/min(線速度16cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、11200Paであった。

反応剤充填例5で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が200mm充填された流動床反応装置内に、300℃常圧で窒素ガスを流量1.25mol/min(線速度11cm/s)で流した。このとき、差圧計で測定された流動床反応器の入口側と出口側との差圧は、7700Paであった。


まず、実施例3において、反応剤充填例6で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が300mm充填された流動床反応装置の流動床反応器内を、電気炉で300℃に加熱した。そして、流動床反応装置内に、常圧で窒素ガスを0.79mol/minの流量(線速度7cm/s)で流した。なお、前記した流動化例8から12の結果から、この線速度において、酸化カルシウムの層は完全流動化状態であると考えられる。
まず、実施例11において、反応剤充填例6で示すように固体反応剤(平均粒子径100μmの酸化カルシウム)が300mm充填された流動床反応装置の流動床反応器内を、電気炉で350℃に加熱した。そして、流動床反応装置内に、常圧で窒素ガスを0.73mol/minの流量(線速度7cm/s)で流した。なお、前記した流動化例8から12の結果から、この線速度において、酸化カルシウムの層は完全流動化状態であると考えられる。
これらの結果を、実施例3~10については表22の下欄に、実施例11~15については表23の下欄にそれぞれ示す。
出口ガス中のHFC-134a由来成分のうちで、HFC-134a以外の成分の割合をいう。出口ガス中の{100-(HFC-134a)}/100×100(%)で計算される
反応したHFC-134aのうちで、HFO-1123に転化したのは何%かをいう。出口ガス中の(HFO-1123)/{100-(HFC-134a)}×100(%)で計算される。
反応したHFC-134aのうちで、HFO-1123以外の化合物に転化したのは何%かをいう。出口ガス中の{100-(HFC-134a)-(HFO-1123)}/{100-(HFC-134a)}×100(%)で計算される。


なお、2014年1月30日に出願された日本特許出願2014-15962号の明細書、特許請求の範囲、要約書および図面の全内容をここに引用し、本発明の明細書の開示として、取り入れるものである。
Claims (15)
- 平均粒子径が1μm~5000μmの粒子状の固体反応剤からなる層中に1,1,1,2-テトラフルオロエタンを含む原料ガスを流通させ、前記固体反応剤からなる層が流動化した状態で、前記固体反応剤と前記1,1,1,2-テトラフルオロエタンを接触させることを特徴とするトリフルオロエチレンの製造方法。
- 前記固体反応剤の平均粒子径が40μm~500μmである、請求項1に記載のトリフルオロエチレンの製造方法。
- 前記1,1,1,2-テトラフルオロエタンを含む原料ガスの流通速度が、線速度で1cm/s~1000cm/sである、請求項1または2に記載のトリフルオロエチレンの製造方法。
- 前記1,1,1,2-テトラフルオロエタンを含む原料ガスの流通速度が、線速度で1cm/s~20cm/sである、請求項3に記載のトリフルオロエチレンの製造方法。
- 前記固体反応剤が、金属酸化物、金属水酸化物、金属炭酸塩、金属硫酸塩および金属ハロゲン化物からなる群から選ばれる少なくとも1種の金属化合物を含む、請求項1~4のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記金属化合物に含まれる金属種が、アルカリ金属、アルカリ土類金属、13族金属および14族金属からなる群から選ばれる少なくとも1種である、請求項5に記載のトリフルオロエチレンの製造方法。
- 前記固体反応剤が、炭酸カリウムおよび/または酸化カルシウムである、請求項1~6のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記1,1,1,2-テトラフルオロエタンと前記固体反応剤とを接触させる温度が、100℃~500℃である、請求項1~7のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記1,1,1,2-テトラフルオロエタンを前記固体反応剤と接触させる温度が、350℃~500℃である、請求項8に記載のトリフルオロエチレンの製造方法。
- 前記固体反応剤と接触させる前記1,1,1,2-テトラフルオロエタンの圧力が、ゲージ圧で0~5MPaである、請求項1~9のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記1,1,1,2-テトラフルオロエタンと前記固体反応剤の接触時間が、0.1秒~100秒間である、請求項1~10のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記1,1,1,2-テトラフルオロエタンと前記固体反応剤の接触時間は、0.1秒~20秒間である、請求項11に記載のトリフルオロエチレンの製造方法。
- 前記原料ガス中の前記1,1,1,2-テトラフルオロエタンの含有割合が、1モル%~100モル%である、請求項1~12のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記原料ガスがさらに希釈ガスを含み、その含有量が前記原料ガスの全体に対して95モル%以下である、請求項1~13のいずれか1項に記載のトリフルオロエチレンの製造方法。
- 前記原料ガスがさらに1,1,2,2-テトラフルオロエタンを含み、その含有量が、1,1,1,2-テトラフルオロエタンと1,1,2,2-テトラフルオロエタンの合計量に対して50モル%未満である、請求項1~14のいずれか1項に記載のトリフルオロエチレンの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201580006537.1A CN105939984B (zh) | 2014-01-30 | 2015-01-29 | 三氟乙烯的制造方法 |
| EP15743584.3A EP3100998B1 (en) | 2014-01-30 | 2015-01-29 | Method for producing trifluoroethylene |
| JP2015560012A JP6432526B2 (ja) | 2014-01-30 | 2015-01-29 | トリフルオロエチレンの製造方法 |
| US15/218,120 US9802878B2 (en) | 2014-01-30 | 2016-07-25 | Method for producing trifluoroethylene |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014015962 | 2014-01-30 | ||
| JP2014-015962 | 2014-01-30 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/218,120 Continuation US9802878B2 (en) | 2014-01-30 | 2016-07-25 | Method for producing trifluoroethylene |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015115548A1 true WO2015115548A1 (ja) | 2015-08-06 |
Family
ID=53757118
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/052527 WO2015115548A1 (ja) | 2014-01-30 | 2015-01-29 | トリフルオロエチレンの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9802878B2 (ja) |
| EP (1) | EP3100998B1 (ja) |
| JP (1) | JP6432526B2 (ja) |
| CN (2) | CN105939984B (ja) |
| WO (1) | WO2015115548A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019194214A1 (ja) * | 2018-04-03 | 2019-10-10 | ダイキン工業株式会社 | フルオロオレフィンの製造方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6379391B2 (ja) * | 2013-04-30 | 2018-08-29 | Agc株式会社 | トリフルオロエチレンを含む組成物 |
| CN107243352B (zh) * | 2017-07-11 | 2019-12-20 | 上海三爱富新材料科技有限公司 | 用于合成三氟乙烯的催化剂及其制备方法 |
| CN111479910A (zh) * | 2017-12-18 | 2020-07-31 | 大金工业株式会社 | 制冷剂用或制冷剂组合物用的制冷机油、制冷机油的使用方法、以及作为制冷机油的用途 |
| JP6673413B2 (ja) * | 2018-05-08 | 2020-03-25 | ダイキン工業株式会社 | フルオロオレフィンの製造方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0517425A (ja) * | 1991-07-08 | 1993-01-26 | Mitsui Toatsu Chem Inc | アクリロニトリルの製造法 |
| JPH0656705A (ja) * | 1992-06-18 | 1994-03-01 | Exxon Res & Eng Co | 炭素質触媒を用いる炭化水素の改善された脱水素化方法 |
| JPH10505337A (ja) * | 1994-08-08 | 1998-05-26 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | 含弗素オレフィン類の製造方法 |
| JP2010070489A (ja) * | 2008-09-18 | 2010-04-02 | Unimatec Co Ltd | フルオロオレフィンアイオダイド混合物およびその製造法 |
| JP2010533151A (ja) | 2007-07-13 | 2010-10-21 | ゾルファイ フルーオル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 金属フッ化物触媒上でのハロゲンおよび水素を有するアルケンの製造 |
| WO2011157907A1 (fr) | 2010-06-15 | 2011-12-22 | Arkema France | Procédé de préparation de trifluoroéthylène |
| JP2014015962A (ja) | 2012-07-06 | 2014-01-30 | Aisin Aw Co Ltd | 車両用駆動装置 |
| WO2014178353A1 (ja) * | 2013-04-30 | 2014-11-06 | 旭硝子株式会社 | 熱サイクル用作動媒体 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7592494B2 (en) * | 2003-07-25 | 2009-09-22 | Honeywell International Inc. | Process for the manufacture of 1,3,3,3-tetrafluoropropene |
| US8373010B2 (en) * | 2010-09-03 | 2013-02-12 | Honeywell International Inc. | Methods to produce 3,3,3-trifluoropropene |
| CN103044190B (zh) * | 2012-12-21 | 2014-12-24 | 巨化集团技术中心 | 一种三氟乙烯的制备方法 |
| CN103288589A (zh) * | 2013-06-04 | 2013-09-11 | 同济大学 | 一种生产三氟乙烯联产氟化氢的方法 |
| CN105939985B (zh) * | 2014-01-30 | 2019-07-05 | Agc株式会社 | 三氟乙烯的制造方法 |
-
2015
- 2015-01-29 CN CN201580006537.1A patent/CN105939984B/zh active Active
- 2015-01-29 WO PCT/JP2015/052527 patent/WO2015115548A1/ja active Application Filing
- 2015-01-29 JP JP2015560012A patent/JP6432526B2/ja active Active
- 2015-01-29 EP EP15743584.3A patent/EP3100998B1/en active Active
- 2015-01-29 CN CN201811581676.9A patent/CN109665936A/zh active Pending
-
2016
- 2016-07-25 US US15/218,120 patent/US9802878B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0517425A (ja) * | 1991-07-08 | 1993-01-26 | Mitsui Toatsu Chem Inc | アクリロニトリルの製造法 |
| JPH0656705A (ja) * | 1992-06-18 | 1994-03-01 | Exxon Res & Eng Co | 炭素質触媒を用いる炭化水素の改善された脱水素化方法 |
| JPH10505337A (ja) * | 1994-08-08 | 1998-05-26 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | 含弗素オレフィン類の製造方法 |
| JP2010533151A (ja) | 2007-07-13 | 2010-10-21 | ゾルファイ フルーオル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 金属フッ化物触媒上でのハロゲンおよび水素を有するアルケンの製造 |
| JP2010070489A (ja) * | 2008-09-18 | 2010-04-02 | Unimatec Co Ltd | フルオロオレフィンアイオダイド混合物およびその製造法 |
| WO2011157907A1 (fr) | 2010-06-15 | 2011-12-22 | Arkema France | Procédé de préparation de trifluoroéthylène |
| JP2014015962A (ja) | 2012-07-06 | 2014-01-30 | Aisin Aw Co Ltd | 車両用駆動装置 |
| WO2014178353A1 (ja) * | 2013-04-30 | 2014-11-06 | 旭硝子株式会社 | 熱サイクル用作動媒体 |
Non-Patent Citations (1)
| Title |
|---|
| TAKASHI SHIRAI: "Ryudo Shokubai, Ryudoso no Shikenho", CHEMICAL ENGINEERING OF JAPAN, vol. 27, no. 5, 1963, pages 378 - 381 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019194214A1 (ja) * | 2018-04-03 | 2019-10-10 | ダイキン工業株式会社 | フルオロオレフィンの製造方法 |
| JP2019182755A (ja) * | 2018-04-03 | 2019-10-24 | ダイキン工業株式会社 | フルオロオレフィンの製造方法 |
| US11198662B2 (en) | 2018-04-03 | 2021-12-14 | Daikin Industries, Ltd. | Fluoroolefin production method |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105939984A (zh) | 2016-09-14 |
| US9802878B2 (en) | 2017-10-31 |
| JP6432526B2 (ja) | 2018-12-05 |
| CN109665936A (zh) | 2019-04-23 |
| CN105939984B (zh) | 2020-08-14 |
| US20160332938A1 (en) | 2016-11-17 |
| EP3100998A4 (en) | 2017-08-23 |
| JPWO2015115548A1 (ja) | 2017-03-23 |
| EP3100998B1 (en) | 2020-02-26 |
| EP3100998A1 (en) | 2016-12-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9802878B2 (en) | Method for producing trifluoroethylene | |
| JP6885447B2 (ja) | トリフルオロエチレンを含む流体の精製方法、およびトリフルオロエチレンの製造方法 | |
| US10093600B2 (en) | Method for producing trifluoroethylene | |
| JP7304681B2 (ja) | ハイドロフルオロオレフィンの製造方法 | |
| ES2616232T3 (es) | Fluoración catalítica en fase gaseosa de 1233XF hasta 1234YF | |
| JP5947337B2 (ja) | 2,2,3,3−テトラフルオロ−1−プロペンの製造方法 | |
| JP6358324B2 (ja) | トリフルオロエチレンの製造方法 | |
| US20120172637A1 (en) | CATALYTIC GAS PHASE FLUORINATION OF 1230xa to 1234yf | |
| WO2011162341A1 (ja) | 2,3,3,3-テトラフルオロプロペンの製造方法 | |
| US10399916B2 (en) | Method of producing hydrofluoroolefin | |
| WO2016163522A1 (ja) | ハイドロフルオロオレフィンの製造方法 | |
| WO2024180856A1 (ja) | 組成物、システム、組成物入り容器、及び組成物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15743584 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015560012 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015743584 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015743584 Country of ref document: EP |