WO2014188924A1 - 金属酸化物多孔質粒子、その製造方法、及びその用途 - Google Patents
金属酸化物多孔質粒子、その製造方法、及びその用途 Download PDFInfo
- Publication number
- WO2014188924A1 WO2014188924A1 PCT/JP2014/062812 JP2014062812W WO2014188924A1 WO 2014188924 A1 WO2014188924 A1 WO 2014188924A1 JP 2014062812 W JP2014062812 W JP 2014062812W WO 2014188924 A1 WO2014188924 A1 WO 2014188924A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- metal oxide
- group
- particles
- water
- hydrogen atom
- Prior art date
Links
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
- C08K7/24—Expanded, porous or hollow particles inorganic
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B13/00—Oxygen; Ozone; Oxides or hydroxides in general
- C01B13/14—Methods for preparing oxides or hydroxides in general
- C01B13/32—Methods for preparing oxides or hydroxides in general by oxidation or hydrolysis of elements or compounds in the liquid or solid state or in non-aqueous solution, e.g. sol-gel process
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
- C01B33/113—Silicon oxides; Hydrates thereof
- C01B33/12—Silica; Hydrates thereof, e.g. lepidoic silicic acid
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B37/00—Compounds having molecular sieve properties but not having base-exchange properties
- C01B37/02—Crystalline silica-polymorphs, e.g. silicalites dealuminated aluminosilicate zeolites
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
- C08K7/24—Expanded, porous or hollow particles inorganic
- C08K7/26—Silicon- containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D11/00—Inks
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D11/00—Inks
- C09D11/02—Printing inks
- C09D11/03—Printing inks characterised by features other than the chemical nature of the binder
- C09D11/037—Printing inks characterised by features other than the chemical nature of the binder characterised by the pigment
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D201/00—Coating compositions based on unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F16—ENGINEERING ELEMENTS AND UNITS; GENERAL MEASURES FOR PRODUCING AND MAINTAINING EFFECTIVE FUNCTIONING OF MACHINES OR INSTALLATIONS; THERMAL INSULATION IN GENERAL
- F16L—PIPES; JOINTS OR FITTINGS FOR PIPES; SUPPORTS FOR PIPES, CABLES OR PROTECTIVE TUBING; MEANS FOR THERMAL INSULATION IN GENERAL
- F16L59/00—Thermal insulation in general
- F16L59/02—Shape or form of insulating materials, with or without coverings integral with the insulating materials
- F16L59/028—Composition or method of fixing a thermally insulating material
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/01—Particle morphology depicted by an image
- C01P2004/04—Particle morphology depicted by an image obtained by TEM, STEM, STM or AFM
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/30—Particle morphology extending in three dimensions
- C01P2004/32—Spheres
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/30—Particle morphology extending in three dimensions
- C01P2004/45—Aggregated particles or particles with an intergrown morphology
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/51—Particles with a specific particle size distribution
- C01P2004/52—Particles with a specific particle size distribution highly monodisperse size distribution
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/62—Submicrometer sized, i.e. from 0.1-1 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/64—Nanometer sized, i.e. from 1-100 nanometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/16—Pore diameter
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/16—Pore diameter
- C01P2006/17—Pore diameter distribution
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/60—Additives non-macromolecular
- C09D7/61—Additives non-macromolecular inorganic
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/66—Additives characterised by particle size
Definitions
- the present invention relates to metal oxide porous particles, a method for producing metal oxide porous particles, and uses thereof.
- porous metal oxides have been newly applied to optical materials, low dielectric constant materials, heat insulating materials, pharmaceuticals (DDS: drug delivery systems), molecular probes, catalysts, adsorbing materials, sensors, paints, inks, etc.
- DDS drug delivery systems
- a porous material having an air layer inside for the purpose of improving heat insulation performance in particular, a porous material having pores smaller than the mean free path value of 68 nm of air at atmospheric pressure is added as a filler in a resin film or paint. In this case, it is considered that a good heat insulating effect can be obtained.
- Patent Document 1 describes a method for producing metal oxide porous particles using a surfactant micelle as a template, and has a mesopore with a pore size of 1 to 10 nm and a metal oxide with a particle size of 20 to 200 nm. It is described that porous particles can be obtained.
- Patent Document 2 describes a method for synthesizing a mesoporous metal oxide having a three-dimensional cubic phase structure using a surfactant micelle as a template, and that a mesoporous metal oxide having an average pore diameter of 5 nm can be obtained. (Example 2, FIG. 6).
- Patent Document 3 describes a method for producing a metal oxide porous body using water-insoluble polymer particles, in which mesopores form a cubic phase and the pore diameter is substantially uniform within a range of 5 to 30 nm. It is described that a certain metal oxide porous body is obtained.
- An example of producing porous particles having a particle diameter of 1 to 10 ⁇ m by spray drying is described (Example a12 and the like).
- Image display surfaces in image display devices such as liquid crystal displays, CRT displays, projection displays, plasma displays, electroluminescent displays, and reflective screens are required to be provided with scratch resistance so that they are not damaged during handling.
- the hard coat layer is formed using an active energy such as ultraviolet rays or electron beams, or a hard coat agent that is cured by heat.
- the hard coat agent comprises a binder component and high-hardness inorganic fine particles (for example, Patent Document 4). ).
- the refractive index of the hard coat layer portion is made lower than that of the display surface of the image display device. Or the method of making the refractive index of the hard coat layer surface part lower than a hard coat layer is mentioned.
- a method for lowering the refractive index of the hard coat layer a method using a fluorine-based coating agent having a low refractive index can be considered, but it cannot be said that the curability is sufficient.
- a method of adding inorganic particles having a low refractive index to the binder component can be considered.
- inorganic particles having a low refractive index include silica particles, fluoride particles such as magnesium fluoride, lithium fluoride, aluminum fluoride, fluorinated lucium, sodium fluoride, and the like.
- the refractive index of silica is 1.44. The effect of adding is small because it is not so low.
- Patent Document 5 describes a method using hollow silica particles as inorganic particles having a low refractive index. By providing an air layer having a low refractive index inside, the refractive index of the particles becomes low.
- Patent Document 6 describes a method for producing hollow silica particles using calcium carbonate as a template.
- Patent Documents 1-3 have problems in the following points.
- the mesopore diameter of the obtained metal oxide (silica) porous particles is at most 10 nm, it is unsuitable for the adsorption and encapsulation of functional molecules having a relatively large molecular size.
- the proportion of mesopores present in one particle cannot be freely changed, and it is difficult to change various characteristics such as optical characteristics, dielectric constant characteristics, and heat insulation characteristics.
- the metal oxide porous particles are cylindrical (hexagonal) structure mesopores, the wall thickness between the mesopores is generally thinner than the three-dimensional cubic phase structure, and the mechanical strength is insufficient. It becomes.
- the particle diameter of the metal oxide porous particles is large and the particle diameters are not uniform, for example, when the particles having an average particle diameter of 100 nm (0.1 ⁇ m) or more are mixed with a resin, they are likely to become opaque, It is not preferable for optical applications.
- Patent Document 2 does not describe a method for producing particles made of a metal oxide porous body.
- Patent Document 3 describes metal oxide porous particles in which mesopores form a cubic phase and the pore diameter is substantially uniform in the range of 5 to 30 nm, but the average particle diameter is small and the particle diameter is small. The aligned particles are not described.
- the hollow particle obtained by the method of patent document 6 uses calcium carbonate as a casting_mold
- Development of porous oxide particles and establishment of a production method thereof are desired. However, no satisfactory production method has been developed yet.
- the present invention has been made in view of the problems as described above, and has an average particle diameter in the range of 50 nm to 300 nm, a uniform particle diameter, and a mesopore having a pore diameter of 5 nm to 30 nm.
- a metal oxide porous particle having a three-dimensional cubic phase structure having a regular pore structure and a method for producing the same is provided.
- the 50% volume average particle diameter is 50 nm or more and 300 nm or less
- the ratio of the volume 90% average particle diameter to the volume 50% average particle diameter (D90 / D50) is 2.0 or less
- the pore diameter by the BJH method is Metal oxide porous particles having mesopores of 5 nm to 30 nm and having a three-dimensional cubic phase structure.
- the 50% volume average particle diameter is 50 nm or more and 100 nm or less, the ratio of the volume 90% average particle diameter to the volume 50% average particle diameter (D90 / D50) is 1.5 or less, and the pore diameter by the BJH method is The metal oxide porous particle according to [1], having mesopores of 5 nm or more and 30 nm or less and having a three-dimensional cubic phase structure.
- [3] A step of obtaining a mixture comprising water and / or an organic solvent that dissolves part or all of water, water-insoluble polymer particles having a volume 50% average particle diameter of 5 nm to 30 nm, and a base catalyst.
- a sol-gel reaction of the metal oxide precursor to obtain organic-inorganic composite particles; Removing the water-insoluble polymer particles from the organic-inorganic composite particles; The manufacturing method of the metal oxide porous particle as described in [1] or [2] containing this.
- the water-insoluble polymer particles are particles composed of a terminal branched polyolefin copolymer represented by the following general formula (1) and having a number average molecular weight of 2.5 ⁇ 10 4 or less.
- A represents a polyolefin chain.
- R 1 and R 2 represent a hydrogen atom or an alkyl group having 1 to 18 carbon atoms, and at least one of them is a hydrogen atom.
- X 1 and X 2 are the same. Or, differently, it represents a group containing a linear or branched polyalkylene glycol group.
- X 1 and X 2 of the terminally branched polyolefin copolymer represented by the general formula (1) are the same or different, and the general formula (2) (In the formula, E represents an oxygen atom or a sulfur atom, and X 3 represents a polyalkylene glycol group or a general formula (3) (Wherein R 3 represents an m + 1 valent hydrocarbon group; G is the same or different and is represented by —OX 4 , —NX 5 X 6 (X 4 to X 6 represent a polyalkylene glycol group); M represents the number of bonds between R 3 and G, and represents an integer of 1 to 10.) )
- general formula (4) wherein X 7 and X 8 are the same or different and each represents a polyalkylene glycol group or a group represented by the above general formula (3)).
- R 6 and R 7 represent a hydrogen atom or a methyl group, At least one of them is a hydrogen atom, R 8 and R 9 represent a hydrogen atom or a methyl group, and at least one of them is a hydrogen atom, l + m represents an integer of 2 or more and 450 or less, n is 20 Represents an integer of 300 or more.)
- R 4 and R 5 represent a hydrogen atom or an alkyl group having 1 to 18 carbon atoms, at least one of which is a hydrogen atom.
- R 6 and R 7 represent a hydrogen atom or a methyl group, At least one of them is a hydrogen atom, R 8 and R 9 represent a hydrogen atom or a methyl group, at least one of them is a hydrogen atom, R 10 and R 11 represent a hydrogen atom or a methyl group, (At least one of them is a hydrogen atom.
- L + m + o represents an integer of 3 to 450, and n represents an integer of 20 to 300.
- the step of obtaining a mixture includes [3] to [6] comprising a step of mixing the water and / or the organic solvent that dissolves a part or all of the water, the water dispersion of the water-insoluble polymer particles, and the base catalyst.
- the manufacturing method of the metal oxide porous particle as described in any one.
- the metal oxide precursor is mixed in a state diluted in advance with an organic solvent that dissolves part or all of water, [3] to [7] The manufacturing method of the metal oxide porous particle as described in any one of these.
- a resin composition containing the metal oxide porous particles according to [1] or [2] and a binder resin [9] A resin composition containing the metal oxide porous particles according to [1] or [2] and a binder resin.
- a drug delivery system (DDS) drug comprising the metal oxide porous particles according to [1] or [2], wherein the drug is encapsulated in mesopores.
- a lens having the coating film according to [19] on a surface portion [21] A lens having the coating film according to [19] on a surface portion.
- the metal oxide porous particles of the present invention have a small average particle size and a uniform particle size, they can be used for various applications and can exhibit desired characteristics effectively.
- the metal oxide porous particles of the present invention can be easily dispersed uniformly in a binder resin and can provide a highly transparent resin composition.
- the metal oxide porous particles of the present invention have larger mesopore diameters than conventional ones, and have large pores inside the particles. Therefore, it has excellent adsorptivity and can contain a desired substance.
- a large air layer is provided inside the particles, it can contribute to characteristics such as weight reduction, heat insulation, low refractive index, and low dielectric constant. Since the metal oxide porous particles of the present invention have such characteristics, they can be used for various applications.
- the metal oxide porous particle production method of the present invention has a high degree of freedom in particle design, and the ratio of mesopores (porosity) present in one particle can be freely changed. The metal oxide porous particles can be obtained efficiently.
- metal oxide porous particles of the present invention the production method thereof, and the use thereof will be described based on the first embodiment and the second embodiment.
- the metal oxide porous particles of the present embodiment have a volume 50% average particle diameter of 50 nm to 300 nm and a ratio of the volume 90% average particle diameter to the volume 50% average particle diameter (D90 / D50) of 2.0 or less. It has mesopores having a pore diameter of 5 nm or more and 30 nm or less by the Barrett-Joyner-Halenda method (BJH method), and the pore structure is a three-dimensional cubic phase structure.
- BJH method Barrett-Joyner-Halenda method
- the metal oxide porous particles of the present embodiment are so-called monodisperse particles having a substantially uniform size and high dispersibility. As shown in FIG. 1, the metal oxide porous particles have substantially uniform mesopores inside, and the mesopores constitute a mutually connected three-dimensional cubic phase structure.
- the 50% volume average particle size of the metal oxide porous particles of the present embodiment has an outer diameter of 50 nm to 300 nm, preferably 50 nm to 100 nm, and more preferably 60 nm to 90 nm. Within this range, particles can be easily produced, and can be used for various applications, and desired characteristics can be effectively expressed. For example, when used as a resin composition to be described later, a resin composition that can be uniformly dispersed in the binder resin and has high transparency can be obtained.
- the ratio of the volume 90% average particle diameter to the volume 50% average particle diameter (D90 / D50) of the metal oxide porous particles of the present embodiment is 2.0 or less, preferably 1.5 or less. More preferably, it is 1.0 or more and 1.4 or less. Within this range, the particle size distribution is narrow and the number of coarse particles is small, so that the handleability is excellent and desired characteristics can be effectively expressed. For example, when used as a resin composition described later, it is preferable in terms of uniform dispersibility in the binder resin and transparency of the resulting resin.
- the metal oxide porous particle of this embodiment has mesopores, and the BJH pore diameter is 5 nm or more and 30 nm or less, preferably 10 nm or more and 25 nm or less.
- the pore structure is a regular three-dimensional cubic phase structure. Since the metal oxide porous particles of the present embodiment are monodisperse particles, they can be easily dispersed uniformly in a film or paint, and the transparency is increased because the outer diameter is small. Since the BJH has a mesopore having a pore diameter of 5 nm or more and 30 nm or less and the pore structure is a regular three-dimensional cubic phase structure, the pores are provided with large pores. Therefore, high adsorptivity can be expected.
- the specific surface area of the metal oxide porous particles of the present embodiment is preferably at 80 m 2 / g or more, more preferably 100 m 2 / g or more, particularly preferably 150 meters 2 / g or more. Further, from the viewpoint that the thermal conductivity is preferably free from anisotropy due to the shape, it is preferable to have a spherical shape and uniform mesopores.
- the outer diameter of the metal oxide porous particles can be confirmed with a particle size distribution meter (DLS) by dynamic light scattering of a sample dispersed in water.
- the diameter of the mesopores can be observed from a TEM image photograph, and can be calculated by the BJH method from the isotherm on the adsorption side of nitrogen gas adsorption. Further, the diameter of the hole connecting the mesopores can be calculated by the BJH method from the isotherm on the desorption side of the nitrogen gas adsorption method.
- a three-dimensional cubic phase structure is taken, and when the peaks are at substantially the same position, it can be judged that a two-dimensional cylinder structure is taken.
- the metal means not only a typical metal but also a semimetal such as Si.
- the metal oxide of this embodiment silicon (Si), aluminum (Al), zinc (Zn), zirconium (Zr), indium (In), tin (Sn), titanium (Ti), lead (Pb), A metal oxide selected from hafnium (Hf), cobalt (Co), lithium (Li), barium (Ba), iron (Fe), and manganese (Mn) is preferable, and the refractive index and thermal conductivity of the substance itself are metals. From the viewpoint of being relatively low among oxides, silicon oxide (silica) is particularly preferable.
- the metal oxide may be a complex oxide containing a plurality of metals.
- the organic-inorganic composite particles obtained in the step (1) include water-insoluble polymer particles in particles made of a metal oxide body, and the water-insoluble polymer particles that are templates in the step (2) are removed.
- the metal oxide porous particles of the present embodiment are produced.
- the step (1) further includes a step (1-1) and a step (1-2).
- Step (1-1) A step of adding a water-insoluble polymer particle having a volume 50% average particle diameter of 5 nm to 30 nm and a base catalyst to water and / or an organic solvent in which a part or all of water is dissolved to obtain a mixture.
- Step (1-2) A step of mixing the mixture obtained in the step (1-1) and a metal oxide precursor, and performing a sol-gel reaction of the metal oxide precursor to obtain organic-inorganic composite particles.
- the organic / inorganic composite particles are suitable for the base catalyst because the sol-gel reaction proceeds rapidly and the metal oxide precursor forms a three-dimensional dense gel so as to enclose the water-insoluble polymer particles. Obtainable.
- each process is demonstrated in order.
- Step (1-1) In the step (1-1), specifically, the water-insoluble polymer particles (X) (hereinafter appropriately referred to as “component (X)”), water and / or an organic solvent that dissolves part or all of water (Y) (hereinafter appropriately referred to as “component (Y)”) and a base catalyst (Z) (hereinafter appropriately referred to as “component (Z)”) are mixed to prepare a mixture.
- component (X) water-insoluble polymer particles
- Y water and / or an organic solvent that dissolves part or all of water
- Z base catalyst
- the water-insoluble polymer particles (X) will be described in detail.
- the water-insoluble polymer particles preferably have a 50% volume average particle diameter of 5 nm to 30 nm.
- the metal oxide porous particles obtained from such water-insoluble polymer particles highly transparent and transparent films and coating films can be realized.
- a method for measuring the outer diameter for example, a method of measuring using a particle size distribution meter (DLS), a method of directly observing using a transmission electron microscope (TEM), a scanning electron microscope (SEM), etc. can be applied. .
- DLS particle size distribution meter
- TEM transmission electron microscope
- SEM scanning electron microscope
- water-insoluble polymer particles (X) used in the present embodiment include polyolefin-based, poly (meth) acrylic acid ester-based, polystyrene-based, polyurethane-based, polyacrylonitrile-based, polyvinyl chloride-based, polydispersible in an aqueous medium.
- Polymeric particles based on vinylidene chloride, polyvinyl acetate, or polybutadiene are preferred.
- polyolefin-based water-insoluble polymer particles are preferably used because they easily form water-insoluble polymer particles having an outer diameter of 30 nm or less. More preferable are terminal branched polyolefin copolymer particles represented by the following general formula (1).
- A represents a polyolefin chain.
- R 1 and R 2 are a hydrogen atom or an alkyl group having 1 to 18 carbon atoms and at least one of them is a hydrogen atom, and X 1 and X 2 are the same or different and are linear or branched polyalkylene Represents a group containing a glycol group.
- the number average molecular weight of the terminal branched polyolefin copolymer particles represented by the general formula (1) is 2.5 ⁇ 10 4 or less, preferably 1.5 ⁇ 10 4 or less, more preferably 4.0 ⁇ 10 3. It is as follows. Moreover, Preferably it is 5.5 * 10 ⁇ 2 > or more, More preferably, it is 8 * 10 ⁇ 2 > or more.
- the number average molecular weight is the number average molecular weight of the polyolefin chain represented by A, the number average molecular weight of the group containing a polyalkylene glycol group represented by X 1 and X 2 , and R 1 , R 2 and C 2 H min. It is expressed as the sum of molecular weights.
- the stability of the particles in the dispersion when the terminal branched polyolefin copolymer particles are used as a dispersoid, water and / or water The dispersibility in an organic solvent that dissolves a part or all of the above tends to be good, and the dispersion is easy to prepare, which is preferable.
- the polyolefin chain represented by A in the general formula (1) is obtained by polymerizing an olefin having 2 to 20 carbon atoms.
- the olefin having 2 to 20 carbon atoms include ⁇ -olefins such as ethylene, propylene, 1-butene and 1-hexene.
- the homopolymer or copolymer of these olefins may be sufficient, and the thing copolymerized with other polymerizable unsaturated compounds in the range which does not impair a characteristic may be sufficient.
- ethylene, propylene, and 1-butene are particularly preferable.
- the number average molecular weight of the polyolefin chain represented by A measured by GPC is 400 to 8000, preferably 500 to 4000, more preferably 500 to 2000. It is.
- the number average molecular weight is a value in terms of polystyrene.
- the polyolefin portion has high crystallinity, the dispersion tends to be stable, and the melt viscosity is low and the preparation of the dispersion is easy. This is preferable.
- the ratio of the weight average molecular weight (Mw) and the number average molecular weight (Mn) measured by GPC of the polyolefin chain represented by A in the general formula (1), that is, the molecular weight distribution (Mw / Mn) is not particularly limited. However, it is usually 1.0 to several tens, more preferably 4.0 or less, and still more preferably 3.0 or less.
- the weight average molecular weight (Mw), number average molecular weight (Mn) and molecular weight distribution (Mw / Mn) of the polyolefin chain represented by A by GPC are, for example, under the following conditions using GPC-150 manufactured by Millipore It can be measured.
- the molecular weight of the polyolefin chain represented by A can be measured by measuring the molecular weight of a polyolefin having an unsaturated group at one end, which will be described later, and subtracting the molecular weight corresponding to the end.
- R 1 and R 2 are a hydrogen atom or a hydrocarbon group having 1 to 18 carbon atoms, which is a substituent bonded to a double bond of the polyolefin constituting A, and preferably a hydrogen atom or a carbon group having 1 to 18 carbon atoms. It is an alkyl group. As the alkyl group, a methyl group, an ethyl group, and a propyl group are preferable.
- X 1 and X 2 are the same or different and each represents a linear or branched group containing a polyalkylene glycol group having a number average molecular weight of 50 to 10,000.
- the branching mode of the branched polyalkylene glycol group includes branching via a polyvalent carbon-valent hydrogen group or a nitrogen atom. For example, branching by a hydrocarbon group bonded to two or more nitrogen atoms, oxygen atoms or sulfur atoms in addition to the main skeleton, branching by a nitrogen atom bonded to two alkylene groups in addition to the main skeleton, and the like can be given.
- the number average molecular weight of the group containing a polyalkylene glycol group is in the above range since the dispersibility of the dispersion tends to be good and the melt viscosity is low and the preparation of the dispersion is easy.
- E represents an oxygen atom or a sulfur atom
- X 3 represents a polyalkylene glycol group, or a general formula (3)
- R 3 represents an m + 1 valent hydrocarbon group
- G is the same or different, and is represented by —OX 4 , —NX 5 X 6 (X 4 to X 6 represent a polyalkylene glycol group).
- M represents the number of bonds between R 3 and G, and represents an integer of 1 to 10.
- X 7 and X 8 are the same or different and represent a polyalkylene glycol group or a group represented by the above general formula (3)).
- the group represented by R 3 is an m + 1 valent hydrocarbon group having 1 to 20 carbon atoms.
- m is an integer of 1 to 10, preferably an integer of 1 to 6, and particularly preferably an integer of 1 to 2.
- a terminal branched polyolefin copolymer in which one of X 1 and X 2 is a group represented by the general formula (4) in the general formula (1) can be mentioned. More preferable examples include terminal branched polyolefin copolymers in which one of X 1 and X 2 is represented by the general formula (4) and the other is a group represented by the general formula (2). It is done.
- one of X 1 and X 2 is a group represented by the general formula (2), and more preferably X 1 and X 2. And a terminal branched polyolefin copolymer in which both are groups represented by the general formula (2).
- the divalent hydrocarbon group represented by Q 1 and Q 2 is preferably a divalent alkylene group, and more preferably an alkylene group having 2 to 20 carbon atoms.
- the alkylene group having 2 to 20 carbon atoms may or may not have a substituent.
- a preferable alkylene group is a hydrocarbon-based alkylene group, particularly preferably an ethylene group or a methylethylene group, and still more preferably an ethylene group.
- Q 1 and Q 2 may be one kind of alkylene group, or two or more kinds of alkylene groups may be mixed.
- the polyalkylene glycol group represented by X 3 to X 11 is a group obtained by addition polymerization of alkylene oxide.
- alkylene oxide constituting the polyalkylene glycol group represented by X 3 to X 11 include ethylene oxide, propylene oxide, butylene oxide, styrene oxide, cyclohexene oxide, epichlorohydrin, epibromohydrin, methyl glycidyl ether, allyl A glycidyl ether etc. are mentioned. Of these, propylene oxide, ethylene oxide, butylene oxide, and styrene oxide are preferable. More preferred are propylene oxide and ethylene oxide, and particularly preferred is ethylene oxide.
- the polyalkylene glycol group represented by X 3 to X 11 may be a group obtained by homopolymerization of these alkylene oxides or a group obtained by copolymerization of two or more kinds.
- Examples of preferred polyalkylene glycol groups are polyethylene glycol groups, polypropylene glycol groups, or groups obtained by copolymerization of polyethylene oxide and polypropylene oxide, and particularly preferred groups are polyethylene glycol groups.
- X 1 and X 2 in the general formula (1) have the above structure, water and / or a part or all of the water when the terminal branched polyolefin copolymer particles of the present embodiment are used as a dispersoid are dissolved. This is preferable because the dispersibility in an organic solvent is improved.
- terminal branched polyolefin copolymer particles that can be used in the present embodiment, it is preferable to use a polymer represented by the following general formula (1a) or (1b).
- R 4 and R 5 represent a hydrogen atom or an alkyl group having 1 to 18 carbon atoms, and at least one of them is a hydrogen atom.
- the alkyl group is preferably an alkyl group having 1 to 9 carbon atoms.
- R 6 and R 7 each represents a hydrogen atom or a methyl group, and at least one of them is a hydrogen atom, and R 8 and R 9 represent a hydrogen atom or a methyl group. And at least one of them is a hydrogen atom.
- l + m represents an integer of 2 to 450, preferably 5 to 200.
- n represents an integer of 20 to 300, preferably 25 to 200.
- R 4 and R 5 represents a hydrogen atom or an alkyl group having 1 to 18 carbon atoms, and at least one of a hydrogen atom.
- the alkyl group is preferably an alkyl group having 1-9 carbon atoms Further, an alkyl group having 1 to 3 carbon atoms is more preferable.
- R 6 and R 7 represent a hydrogen atom or a methyl group, and at least one of them is a hydrogen atom.
- R 8 and R 9 represent a hydrogen atom or a methyl group, and at least one of them is a hydrogen atom.
- R 10 and R 11 represent a hydrogen atom or a methyl group, and at least one of them is a hydrogen atom.
- l + m + o represents an integer of 3 to 450, preferably 5 to 200.
- n represents an integer of 20 to 300, preferably 25 to 200.
- polymer represented by the general formula (1b) it is more preferable to use a polymer represented by the following general formula (1c).
- the number of ethylene units (n) in the polyethylene chain was calculated by dividing the number average molecular weight (Mn) of the polyolefin chain represented by A in the general formula (1) by the molecular weight of the ethylene unit.
- the total number of ethylene glycol units (l + m or l + m + o) of the polyethylene glycol chain is such that the weight ratio of the polymer raw material to the ethylene oxide used during the polyethylene glycol group addition reaction is the number average molecular weight of the polymer raw material and the polyethylene glycol group (Mn ) And the ratio can be calculated.
- N, l + m or l + m + o can also be measured by 1 H-NMR.
- the terminal methyl group of the polyolefin chain represented by A in the general formula (1) shift value: 0) .88 ppm
- shift value: 1.06-1.50 ppm the integral value of the methylene group
- PEG polyethylene glycol
- the ethylene glycol of the PEG chain The total number of units (l + m or l + m + o) can be calculated.
- the polyolefin chain represented by A is composed of an ethylene-propylene copolymer
- the content of propylene that can be measured by IR, 13 C-NMR, and the integral value in 1 H-NMR n and l + m or l + m + o can be calculated.
- 1 H-NMR a method using an internal standard is also effective.
- the terminally branched polyolefin copolymer particles can be produced, for example, by the method described in WO2010 / 103856.
- the polymer particles of this embodiment comprising such a terminally branched polyolefin copolymer have a structure in which the polyolefin chain portion represented by A in the general formula (1) is oriented in the inward direction. It is a rigid particle with chain part having crystallinity.
- the terminal branched polyolefin copolymer particles of this embodiment can be dispersed again in a liquid such as a solvent after the removal of the particles by drying the dispersion because the polyolefin chain portion has crystallinity.
- the terminal branched polyolefin copolymer particles of the present embodiment are rigid particles in which the melting point of the polyolefin chain portion contained in the particles is preferably 80 ° C. or higher, more preferably 90 ° C. or higher.
- the melting point of the polyolefin chain portion When the melting point of the polyolefin chain portion is at or above the above temperature, it becomes a rigid particle having good crystallinity, and even when heated at a higher temperature, the particle collapse is suppressed. For this reason, in the manufacturing process and use scenes in various applications described later, since the collapse of the particles is suppressed, the characteristics of the terminal branched polyolefin copolymer particles of the present embodiment are not lost, and the yield of the product can be reduced. Product quality is more stable.
- the particle diameter of the terminal branched polyolefin copolymer particles of the present embodiment is constant regardless of the dilution concentration even when dispersed in a solvent or the like. That is, since it has redispersibility and a uniform dispersed particle size, it is different from micelle particles dispersed in a liquid.
- the dispersion of the present embodiment contains the water-insoluble polymer particles, preferably the terminally branched polyolefin copolymer particles, in the dispersoid, and the dispersoid dissolves water and / or a part or all of the water. Dispersed as particles in an organic solvent.
- the dispersion liquid in which the water-insoluble polymer particles are dispersed is, for example, (1) A dispersion containing the polymer particles, obtained when producing water-insoluble polymer particles, (2) A dispersion obtained by dispersing or dissolving other dispersoids and additives in the dispersion containing the polymer particles obtained when producing the water-insoluble polymer particles, (3) A dispersion obtained by dispersing water-insoluble polymer particles in water or an organic solvent having an affinity for water, and dispersing or dissolving other dispersoids or additives, Any of these are included.
- the content ratio of the water-insoluble polymer particles in the dispersion of the present embodiment is preferably 0.1 to 50% by mass, more preferably 1 to 40% by mass, and more preferably 1 to 40% by mass when the total dispersion is 100% by mass.
- the content is preferably 1 to 20% by mass.
- the content ratio of the water-insoluble polymer particles is in the above range because the practicality of the dispersion liquid is good, the viscosity can be properly maintained, and handling becomes easy.
- the 50% volume average particle diameter of the particles in the dispersion liquid of this embodiment is preferably 5 nm or more and 30 nm or less.
- the 50% volume average particle diameter of the particles can be controlled by changing the molecular weight of the water-insoluble polymer, the ratio of hydrophilic groups to lipophilic groups, the degree of branching, and the like. For example, it can be adjusted by changing the structure of the polyolefin portion and the structure of the terminal branch portion of the terminal branched polyolefin copolymer.
- the 50% volume average particle diameter in the present embodiment refers to the diameter of a particle when the total volume is 100% when the total volume is 100%, and is a dynamic light scattering particle size distribution measuring device or a microtrack. It can be measured using a particle size distribution measuring device. The shape can be observed with a transmission electron microscope (TEM) after negative staining with, for example, phosphotungstic acid.
- TEM transmission electron microscope
- the dispersion in this embodiment is obtained by dispersing water-insoluble polymer particles in water and / or an organic solvent that dissolves part or all of water.
- Dispersion in the present embodiment can be performed by a method in which water-insoluble polymer particles are physically dispersed in water and / or an organic solvent that dissolves part or all of water by mechanical shearing force.
- the dispersion method is not particularly limited, but various dispersion methods can be used. Specifically, after mixing water-insoluble polymer particles with water and / or an organic solvent that dissolves part or all of water, the mixture is melted to obtain a high-pressure homogenizer, high-pressure homomixer, extrusion kneader, autoclave, etc. And a method of spraying and pulverizing at a high pressure. The water-insoluble polymer particles are dissolved in a solvent other than water in advance, and then mixed with water and / or an organic solvent that dissolves part or all of the water, and dispersed by a high-pressure homogenizer, a high-pressure homomixer, or the like. A method is also possible.
- the solvent used for dissolving the water-insoluble polymer particles is not particularly limited as long as the water-insoluble polymer particles are dissolved, and examples thereof include toluene, cyclohexane and an organic solvent having an affinity for water.
- an organic solvent other than water it can be removed by an operation such as distillation.
- the water-insoluble polymer particles are in a molten state and are not easily deteriorated by heating.
- a dispersion can be obtained by heating and stirring at a temperature of 100 ° C. or higher, preferably 120 to 200 ° C. while applying a shearing force.
- the time required for dispersion varies depending on the dispersion temperature and other dispersion conditions, but is about 1 to 300 minutes.
- the above stirring time is preferable because the dispersion can be sufficiently performed and the water-insoluble polymer particles are hardly deteriorated.
- After the reaction it is preferable to maintain a state in which shearing force is applied until the temperature in the dispersion becomes 100 ° C. or lower, preferably 60 ° C. or lower.
- a surfactant is not indispensable, but for example, an anionic surfactant, a cationic surfactant, an amphoteric surfactant, a nonionic surfactant, etc. may coexist. .
- Anionic surfactants include, for example, carboxylates, simple alkyl sulfonates, modified alkyl sulfonates, alkyl allyl sulfonates, alkyl sulfate esters, sulfated oils, sulfate esters, sulfated fatty acid monoglycerides, sulfated alkanol amides. Sulphated ethers, alkyl phosphate esters, alkyl benzene phosphonates, naphthalene sulfonic acid / formalin condensates.
- Examples of cationic surfactants include simple amine salts, modified amine salts, tetraalkyl quaternary ammonium salts, modified trialkyl quaternary ammonium salts, trialkyl benzyl quaternary ammonium salts, and modified trialkyl benzyl quaternary salts.
- Examples include quaternary ammonium salts, alkyl pyridinium salts, modified alkyl pyridinium salts, alkyl quinolinium salts, alkyl phosphonium salts, and alkyl sulfonium salts.
- amphoteric surfactants examples include betaine, sulfobetaine, and sulfate betaine.
- Nonionic surfactants include, for example, fatty acid monoglycerin ester, fatty acid polyglycol ester, fatty acid sorbitan ester, fatty acid sucrose ester, fatty acid alkanol amide, fatty acid polyethylene glycol glycol condensate, fatty acid amide polyethylene glycol condensate, Fatty acid alcohol / polyethylene / glycol condensate, fatty acid amine / polyethylene / glycol condensate, fatty acid mercaptan / polyethylene / glycol condensate, alkyl / phenol / polyethylene / glycol condensate, polypropylene / glycol / polyethylene / glycol condensate . These surfactants can be used alone or in combination of two or more.
- a filtration step may be provided in the process for the purpose of removing foreign substances and the like.
- a stainless steel filter wire diameter 0.035 mm, plain weave
- pressure filtration air pressure 0.2 MPa
- the dispersion obtained by the above method has a pH of 1 by adding various acids and bases, for example, acids such as hydrochloric acid, sulfuric acid and phosphoric acid, and bases such as potassium hydroxide, sodium hydroxide and calcium hydroxide. No change or aggregation occurs from 13 to 13. In addition, the dispersion does not aggregate or precipitate even in a wide temperature range in which heating and refluxing or freezing and thawing are repeated under normal pressure.
- acids and bases for example, acids such as hydrochloric acid, sulfuric acid and phosphoric acid, and bases such as potassium hydroxide, sodium hydroxide and calcium hydroxide.
- the water in the above method is not particularly limited, and distilled water, ion exchange water, city water, industrial water, and the like can be used, but it is preferable to use distilled water or ion exchange water.
- the organic solvent having an affinity for water in the above method is not particularly limited as long as the dispersoid such as water-insoluble polymer particles and surfactant can be dispersed.
- the dispersoid such as water-insoluble polymer particles and surfactant can be dispersed.
- the dispersoid such as water-insoluble polymer particles and surfactant can be dispersed.
- the organic solvent can be removed by distillation or the like after preparing a dispersion containing the dispersoid.
- the dispersion in the present embodiment is preferably 0.001 to 20 parts by mass of dispersoids other than the terminal branched polyolefin copolymer particles when the water-insoluble polymer particles are 100 parts by mass.
- the content is preferably 0.01 to 10 parts by mass, more preferably 0.1 to 5 parts by mass.
- Component (Y) includes a solvent used when obtaining an aqueous dispersion using a water-insoluble polymer, the aqueous dispersion, a metal alkoxide and / or a partial hydrolysis condensate thereof, and a sol-gel reaction described later.
- the solvent used when mixing the component (Z) of the catalyst for use and the solvent used when mixing the metal oxide precursor (W) described later are included.
- Water is not particularly limited, and distilled water, ion exchange water, city water, industrial water, and the like can be used, but it is preferable to use distilled water or ion exchange water.
- the organic solvent that dissolves part or all of water is not particularly limited as long as it is an organic solvent having an affinity for water and can disperse the water-insoluble polymer.
- methanol, ethanol, propyl alcohol, isopropyl alcohol, acetonitrile, dimethyl sulfoxide, dimethylformamide, acetone, tetrahydrofuran, and dioxane are preferable because of their high affinity with water.
- the particle diameter and shape can be controlled when the metal oxide precursor is condensed, and it can be approximated to spherical fine particles of uniform size.
- component (W) described later tetraethoxysilane (TEOS) and tetramethoxysilane (TMOS) are preferable, and therefore alcohols such as ethanol and methanol are particularly preferable.
- a base catalyst is preferably used in that the condensation rate of the metal oxide precursor is controlled to form a spherical metal oxide porous body.
- quaternary compounds such as alkali metal hydroxides such as ammonia, ammonium hydroxide (ammonia water), potassium hydroxide and sodium hydroxide, tetramethylammonium hydroxide, tetraethylammonium hydroxide and tetrabutylammonium hydroxide.
- Ammonium hydroxide triethylamine, tributylamine, morpholine, pyridine, piperidine, ethylenediamine, diethylenetriamine, amines such as ethanolamine, diethanolamine, triethanolamine, 3-aminopropyltriethoxysilane, N- (2-aminoethyl)- Examples include aminosilanes such as 3-aminopropyltrimethoxysilane.
- ammonia and ammonium hydroxide ammonia water
- ammonia water ammonia water
- ammonium hydroxide ammonia water
- step (1-2) the metal oxide precursor (W) is mixed in the mixture obtained in step (1-1) and a sol-gel reaction is performed to obtain organic-inorganic composite particles.
- Metal oxide precursor (W) examples include a metal alkoxide and / or a partial hydrolysis condensate thereof, a metal halide, a metal acetate, a metal nitrate, and a metal sulfate.
- the metal alkoxide in this embodiment points to what is represented by following formula (12). (R 12 ) x 1 M (OR 13 ) y 1 (12)
- R 12 represents a hydrogen atom, an alkyl group (methyl group, ethyl group, propyl group, etc.), an aryl group (phenyl group, tolyl group, etc.), a carbon-carbon double bond-containing organic group (acryloyl group, methacryloyl group). And vinyl groups), halogen-containing groups (halogenated alkyl groups such as chloropropyl group and fluoromethyl group), and the like.
- R 13 represents a lower alkyl group having 1 to 6 carbon atoms, preferably 1 to 4 carbon atoms.
- M includes Li, Na, Mg, Al, Si, K, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Rb, Sr, Y, Nb, Zr, Mo, Ag, Cd, In, Sn, Sb, Cs, Ba, La, Ta, Hf, W, Ir, Tl, Pb, Bi, rare earth metals, etc. are mentioned.
- Si, Al A metal (alkoxide) that becomes a colorless metal oxide by a sol-gel reaction such as Zn, Zr, In, Sn, Ti, Pb, and Hf is preferable.
- silicon (Si), aluminum (Al), zirconium (Zr), titanium (Ti) and the like are preferably used, and these may be used in combination.
- silicon compounds are relatively inexpensive and easy to obtain, and the reaction proceeds slowly, so that the industrial utility value is high.
- the metal alkoxide and / or the hydrolysis condensate thereof may be a compound that becomes a metal oxide described later by performing a sol-gel reaction by adding water and a catalyst.
- TMOS tetramethoxysilane
- TEOS tetraethoxysilane
- tetrapropoxysilane tetraisopropoxysilane, methyltrimethoxysilane, methyltriethoxysilane, methyltripropoxysilane, methyltributoxysilane, ethyl Trimethoxysilane, ethyltriethoxysilane, n-propyltrimethoxysilane, n-propyltriethoxysilane, isopropyltrimethoxysilane, isopropyltriethoxysilane, dimethyldimethoxysilane, dimethyldiethoxysilane, diphenyldimethoxysilane, diphenyldiethoxysilane , Vinyltrimethoxysilane, vinyltriethoxysilane, phenyltrimethoxysilane, phenyltriethoxysilane
- metal alkoxides having various functional groups at R 12 as shown in the following 1) to 4) can also be used.
- M is alkoxysilane in which silicon (Si) is M, alkoxyzirconium in which M is zirconium (Zr), alkoxyaluminum in which M is aluminum (Al), and Alkoxy titanium where M is titanium (Ti) is preferred.
- a partially hydrolyzed condensate of metal alkoxide is a compound obtained by polycondensation of one or more metal alkoxides partially hydrolyzed using a base catalyst (Z). It is a partially hydrolyzed polycondensation compound of alkoxide.
- the partial hydrolysis condensate of metal alkoxide is preferably an alkoxysilane condensate, an alkoxyzirconium condensate, an alkoxyaluminum condensate, or an alkoxytitanium condensate.
- R 14 represents a hydrogen atom, an alkyl group (such as a methyl group, an ethyl group, or a propyl group), an alkoxy group (such as a methoxy group, an ethoxy group, a propoxy group, or a butoxy group), an aryl group (such as a phenyl group or a tolyl group). ), A carbon-carbon double bond-containing organic group (acryloyl group, methacryloyl group, vinyl group and the like), a halogen-containing group (halogenated alkyl group such as chloropropyl group and fluoromethyl group) and the like.
- Z represents F, Cl, Br, or I.
- M includes Li, Na, Mg, Al, Si, K, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Rb, Sr, Y, Nb, Zr, Mo, Ag, Cd, In, Sn, Sb, Cs, Ba, La, Ta, Hf, W, Ir, Tl, Pb, Bi, rare earth metals, etc. are mentioned.
- Si, Al, A metal (alkoxide) that becomes a colorless metal oxide by a sol-gel reaction such as Zn, Zr, In, Sn, Ti, Pb, and Hf is preferable.
- silicon (Si), aluminum (Al), zirconium (Zr), titanium (Ti) and the like are preferably used, and these may be used in combination.
- Specific examples include tetrachloro-dimethyldisilane, chloropropyldichloromethylsilane, chloromethyl (dichloro) methylsilane, di-tert-butyldichlorosilane, dibutyldichlorosilane, dichloro (methyl) -n-octylsilane, dichloro (methyl).
- Phenylsilane dichlorocyclohexylmethylsilane, dichlorodiethylsilane, dichlorodihexylsilane, dichlorodiisopropylsilane, dichlorodimethylsilane, dichlorodiphenylsilane, dichloroethylsilane, dichlorohexylmethylsilane, dichloromethylsilane, dichloromethylvinylsilane, tetrachlorosilane, 1 , 2-bis (trichlorosilyl) ethane, 3-chloropropyltrichlorosilane, allyltrichlorosilane, butyltrichlorosilane , Cyclohexyltrichlorosilane, ethyltrichlorosilane, hexachlorodisilane, hexachlorodisilane, phenyltrichlorosilane,
- Fluorosilanes, bromosilanes, iodosilanes, and the corresponding aluminum halides, zirconium halides, titanium halides, cobalt halides, lithium halides, barium halides, iron halides, manganese halides and their Hydrates are mentioned.
- examples of the metal acetate include cobalt acetate, cobalt acetoacetate, lithium acetate, lithium acetoacetate, iron acetate, iron acetoacetate, manganese acetate, manganese acetoacetate, and hydrates thereof.
- examples of the metal nitrate include cobalt nitrate, lithium nitrate, iron nitrate, manganese nitrate, and hydrates thereof.
- metal sulfate examples include titanium sulfate, zirconium sulfate, indium sulfate, zinc sulfate, selenium sulfate, antimony sulfate, tin sulfate, yttrium sulfate, and hydrates thereof.
- a metal alkoxide and / or a partially hydrolyzed condensate thereof is preferable in the application of the present embodiment.
- the metal alkoxide an alkoxysilane is more preferable, and tetraethoxysilane (TEOS) is particularly easy to handle. ), Tetramethoxysilane (TMOS) is particularly preferred.
- the mixing ratio of the water-insoluble polymer particles (X) and the metal oxide precursor (W) is not particularly limited, but is preferably 1:10 to 10: 1.
- the amount of the component (X) is large, the abundance ratio of the metal oxide is lowered, so that it is difficult to control the porous structure, and the wall connecting the pores becomes thin, so that the mechanical strength is lowered.
- the component (W) is large, the proportion of pores is low, and the surface area and porosity are small, so that the performance as a porous material cannot be expected.
- the organic solvent (Y) that dissolves water and / or a part or all of the water is preferably 30 parts by weight or more and 100000 parts by weight or less, more preferably 50 parts by weight with respect to 100 parts by weight of the metal oxide precursor (W). It is 50000 parts by weight or less. If the proportion of the component (Y) is low, the particles tend to aggregate, and if it is large, it is not preferable from the viewpoint of production efficiency.
- the ratio of water and solvent in component (Y) is not particularly limited, but is preferably 0.1: 100 to 1:50. When the amount of water is small, the sol-gel reaction rate of the metal oxide precursor condensate is remarkably reduced. When the amount is large, the reaction rate is high and it is difficult to control the particle size and shape.
- the sol-gel reaction catalyst (Z) is preferably 10 parts by weight or more and 1000 parts by weight or less with respect to 100 parts by weight of the metal oxide precursor (W). If the ratio of the component (Z) is low, the sol-gel reaction rate of the metal oxide precursor condensate is remarkably reduced.
- an organic solvent in which the component (W) dissolves part or all of water in advance is used. It is preferable to mix in a state diluted with a solvent. By diluting with an organic solvent, local reactions in the liquid containing the sol-gel reaction catalyst (Z) can be suppressed, so the particle size and shape of the metal oxide porous body can be controlled. It becomes easy to do.
- the ratio of dilution with the organic solvent is, for example, from 10 parts by weight to 10,000 parts by weight, and more preferably from 100 parts by weight to 1000 parts by weight with respect to 100 parts by weight of the component (W).
- TEOS tetraethoxysilane
- TMOS tetramethoxysilane
- the preferable reaction temperature for the sol-gel reaction of the component (W) is 1 ° C. or higher and 200 ° C. or lower, more preferably 10 ° C. or higher and 100 ° C. or lower, and 20 ° C. or higher and 50 ° C. or lower.
- the reaction time is 10 minutes or more and 72 hours or less, more preferably 1 hour or more and 24 hours or less from the viewpoint of yield and production efficiency.
- the sol-gel reaction of component (W) proceeds under atmospheric pressure, but may be performed under high pressure using an autoclave or the like.
- FIG. 2 is a schematic cross-sectional view showing the organic-inorganic composite particles according to the present embodiment.
- the obtained organic-inorganic composite particles are taken out from the reaction solution by a method such as centrifugation.
- the extracted organic-inorganic composite particles are washed and removed with a solvent for the sol-gel reaction and water with an organic solvent, and then sufficiently dried.
- the state in which the sol-gel reaction is completed is ideally a state in which all of them form MOM bonds, but some alkoxyl groups (M-OR 2 ) and M-OH groups are partially formed. Although it remains, it includes a state in which it has shifted to a solid (gel) state.
- Step (2) In the step (2), the water-insoluble polymer particles are removed from the organic-inorganic composite particles to prepare metal oxide porous particles.
- Methods for removing water-insoluble polymer particles include decomposition and removal by firing, VUV light (vacuum ultraviolet light), far-infrared rays, microwaves, irradiation and removal by plasma, extraction using solvents and water.
- VUV light vacuum ultraviolet light
- far-infrared rays far-infrared rays
- microwaves irradiation and removal by plasma
- extraction using solvents and water The method of removing is mentioned.
- a preferable temperature is 200 ° C. to 1000 ° C., more preferably 300 ° C. to 700 ° C. If the calcination temperature is too low, the water-insoluble polymer particles are not removed, while if it is too high, the pores may collapse due to being close to the melting point of the metal oxide. Firing may be performed at a constant temperature, or may be gradually raised from room temperature.
- the firing time can be changed according to the temperature, but it is preferably performed in the range of 1 hour to 24 hours. Firing may be performed in air or in an inert gas such as nitrogen or argon. Moreover, you may carry out under reduced pressure or in a vacuum.
- a VUV lamp, an excimer laser, or an excimer lamp can be used. May be used in combination with oxidation action of ozone (O 3) generated at the time of irradiating the VUV light in air.
- O 3 oxidation action of ozone
- any frequency of 2.45 GHz or 28 GHz may be used.
- the output of the microwave is not particularly limited, and a condition for removing the water-insoluble polymer particles is selected.
- a solvent or water for example, ethylene glycol, tetraethylene glycol, isopropyl alcohol, acetone, acetonitrile, methanol, ethanol, cyclohexane, dimethyl sulfoxide, dimethylformamide, dimethylimidazolidinone, xylene, toluene , Chloroform, dichloromethane and the like can be used.
- the extraction operation may be performed under heating. Further, ultrasonic (US) treatment may be used in combination.
- US ultrasonic
- the metal oxide porous particles are anionic surfactants, cationic surfactants, and amphoteric interfaces described in the method for dispersing terminal branched polyolefin copolymer particles.
- An activator, a nonionic surfactant, and the like may coexist.
- Metal oxide porous particles are typified by silane coupling agents in order to improve dispersion stability in solvents and water, or to improve compatibility with binder resins and improve mechanical strength and water resistance. You may surface-treat with an organosilicon compound (surface treatment agent).
- the surface treatment may be carried out by a known method.
- the silane coupling agent include methyltrimethoxysilane, methyltriethoxysilane, methyltrichlorosilane, dimethyldichlorosilane, trimethylchlorosilane, vinyltrimethoxysilane, vinyltriethoxysilane.
- the polymerizable monomer is a cationic polysynthetic monomer
- 3-glycidyloxypropyltrimethoxysilane, 3-glycidyloxypropylmethyldiethoxysilane which is a silane coupling agent having a cationic polymerizable functional group, 2- (3,4-epoxycyclohexyl) ethyltrimethoxysilane, 5,6-epoxyhexyltriethoxysilane, 3-ethyl-3- [3- (triethoxysilyl) propoxymethyl] oxetane and the like are desirable.
- the silane coupling agent can be used alone or in combination of two or more.
- the confirmation of the structure and the pore diameter of the metal oxide porous particles obtained in this embodiment can be performed with a transmission electron microscope (TEM / transmission electron microscope JEM-2010F manufactured by JEOL Ltd.) at 200 kV. .
- the average particle size and particle distribution can be measured by a dynamic light scattering method (particle size distribution meter / Nanotrack WAVE) for particles dispersed in water.
- the BET specific surface area of the particles can be calculated by the nitrogen adsorption method, and the pore diameter and the diameter of the hole connecting the mesopores can also be calculated by the BJH method (BELSORP-max surface area measuring device BELSORP-max).
- the metal oxide porous particles of the present embodiment can be used as they are for various applications described below, and can also be used as a resin composition containing metal oxide porous particles and a binder resin.
- the resin composition will be described.
- the binder resin refers to a resin that can bind between metal oxide porous particles or can be a medium for uniformly dispersing metal oxide porous particles.
- binder resin which can be used by this embodiment. Examples thereof include a thermosetting resin that is cured by heating, a photocurable resin that is cured by irradiation with light such as ultraviolet rays, a thermoplastic resin, and a water-soluble resin.
- polyolefin-based, poly (meth) acrylic acid ester-based, polystyrene-based, polyurethane-based, polyvinyl alcohol-based, and polyvinyl acetal-based resins having film-forming properties are preferable.
- thermosetting resin and the photocurable resin examples include an epoxy resin, an unsaturated polyester resin, a phenol resin, a urea / melamine resin, a polyurethane resin, a silicone resin, a diallyl phthalate resin, and a thermosetting polyimide resin.
- Examples of the epoxy resin include various epoxy resins such as glycidyl ether type, glycidyl ester type, glycidyl amine type, cycloaliphatic type, novolak type, naphthalene type, dicyclopentadiene type such as bisphenol A type epoxy resin.
- Unsaturated polyester resins include orthophthalic acid, isophthalic acid, terephthalic acid, alicyclic unsaturated acid, fatty saturated acid, bisphenol, halogenated acid, and halogenated bisphenol.
- a polyester resin is mentioned.
- Examples of the phenolic resin include phenolic resins such as a resol type and a novolac type.
- Thermoplastic resins include polyolefin resin, polyvinyl chloride resin, vinylidene chloride resin, polystyrene resin, acrylonitrile / butadiene / styrene copolymer resin, acrylonitrile / styrene copolymer resin, styrene block copolymer resin, methacrylic resin, polyvinyl Alcohol resin (PVA), polyvinyl acetal resin (PVB), polyacetal resin, polyamide resin, polycarbonate resin, modified polyphenylene ether resin, thermoplastic polyester resin, fluororesin, polyphenylene sulfide resin, polysulfone resin, amorphous arylate resin, polyetherimide Resin, polyethersulfone resin, polyetherketone resin, liquid crystal polymer resin, polyamideimide resin, thermoplastic polyimide resin, syndiopoly Styrene resins.
- Polyolefin resins include polyethylene resin, polypropylene resin, ⁇ -olefin copolymer resin, polybutene-1 resin, polymethylpentene resin, cyclic olefin polymer resin, ethylene / vinyl acetate copolymer resin, ethylene / methacrylic acid copolymer. Examples thereof include resins and ionomers.
- polyamide resin examples include nylon 6, nylon 66, nylon 11, and nylon 12.
- thermoplastic polyester resin examples include polyethylene terephthalate resin, polybutylene terephthalate resin, polybutylene succinate resin, and polylactic acid resin.
- Fluororesin includes polytetrafluoroethylene resin, perfluoroalkoxyalkane resin, perfluoroethylene propene copolymer resin, ethylene / tetrafluoroethylene copolymer resin, polyvinylidene fluoride resin, polychlorotrifluoroethylene resin, ethylene / chlorotrifluoroethylene Examples thereof include a copolymer resin, a tetrafluoroethylene / perfluorodioxole copolymer resin, and a polyvinyl fluoride resin.
- water-soluble resin examples include polyvinyl alcohol (PVA), polyvinyl pyrrolidone (PVP), polyethylene glycol (PEG), and derivatives thereof.
- PVA polyvinyl alcohol
- PVP polyvinyl pyrrolidone
- PEG polyethylene glycol
- Polyolefin, poly (meth) acrylic acid ester, polystyrene, and polyurethane resins having film-forming properties are polymer particles having a particle diameter of 10 to 300 ⁇ m, and are transparent after heating to room temperature or 100 ° C. or less. What forms a thick coating film is preferable.
- Binder resin from the viewpoint of dispersibility and versatility of the metal oxide porous particles, epoxy resins, phenol resins, polyimide resins, polyolefin resins, water-soluble resins, and resins having the above-mentioned film-forming properties are included. preferable. Binder resin can be used individually by 1 type or in mixture of 2 or more types.
- the weight average molecular weight of the binder resin is preferably 200 to 100,000, more preferably 500 to 80,000.
- the content of the binder resin is preferably 30 to 95% by mass, more preferably 40 to 90% by mass, and the metal oxide porous particles of the present invention
- the content of is preferably 70 to 5% by mass, more preferably 60 to 10% by mass.
- the dispersion method of the metal oxide porous particles in the binder resin is not particularly limited, and a known method can be applied.
- the following dispersion methods can be used.
- a binder resin and a dispersion medium such as an organic solvent and water may be mixed to form an emulsion.
- a binder resin (or emulsion thereof) and metal oxide porous particles are melt-kneaded by a kneader in the presence of a solvent and / or a dispersant as required, and the metal oxide porous particles ( A method for obtaining a master batch in which a lighter filler) is dispersed.
- a bead mill mixer, a three-roll mill mixer, a homogenizer mixer, a lab plast mill mixer, or the like can be used.
- the solvent-substituted metal oxide porous particle organosol is added to the binder resin (or its emulsion).
- the treatment agent include organic silicon compounds (surface treatment agents) typified by the above-mentioned silane coupling agents, anionic surfactants, cationic surfactants, amphoteric surfactants, and nonionic surfactants.
- a film or a coating film can be obtained from the resin composition of the present embodiment.
- the thermal conductivity of this film or coating film is preferably 0.1 W / mK or less, more preferably 0.05 W / mK or less. Thereby, the heat insulation efficiency can be improved.
- the HAZE value when the thickness of the film or coating film when dried is 10 ⁇ m is preferably 10% or less, more preferably 5% or less. Thereby, a highly transparent film or coating film is obtained.
- the method for creating the film or coating film is not particularly limited, and a known method can be used.
- the film or coating film is formed as follows. A coating containing metal oxide porous particles is coated on a glass substrate by adjusting the thickness using a bar coater. After drying in an oven at a temperature of 50 to 100 ° C. for 1 to 24 hours, the formed film is peeled off from the glass substrate to obtain a metal oxide porous particle-containing film or coating film.
- the thermal conductivity of the film or coating film of this embodiment can be measured by a laser flash method. Further, the HAZE value of the film or coating film of the present embodiment can be measured with a NDH4000 manufactured by Nippon Denshoku Industries Co., Ltd., with the thickness when the film or coating film is dried being 10 ⁇ m.
- the refractive index of the film can be measured by an Abbe refractometer, and the refractive index of a thin coating film can be measured by an ellipsometer.
- the metal oxide porous body of the present embodiment can be used for medicines (DDS: drug delivery system), molecular probes, catalysts, adsorbing materials, sensors, paints, inks, and the like.
- the resin composition containing the metal oxide porous body of this embodiment can be used for a low dielectric constant material such as a printed circuit board, a special paint or ink containing functional molecules, and the like.
- the film or coating film obtained from the resin composition of the present embodiment can be used for heat insulating materials such as window glass heat insulating films or heat insulating paints for automobiles, houses, buildings, etc., antireflection films for displays, touch panels and the like.
- the coating agent containing the following component (A) and component (B) is provided.
- This embodiment provides a coating agent that contains the metal oxide porous particles described in the first embodiment and exhibits sufficient scratch resistance, and uses thereof.
- the coating agent of the present embodiment can be adjusted to have a low refractive index and can impart hard coat properties depending on the properties of the binder of the component (B), so that it can be used for various purposes and effectively achieves desired properties. Can be expressed. For example, by providing a coating film obtained from the coating agent of the present embodiment on the surface of the image display device, good visibility and scratch resistance can be imparted.
- the metal oxide porous particle manufacturing method used in the present invention has a large degree of freedom in particle design and can freely change the proportion of mesopores (porosity) in one particle. The rate can be adjusted.
- the coating agent of the present embodiment is composed of the metal oxide porous particles described in the first embodiment, a compound that cures by active energy rays such as ultraviolet rays or heat, and has a low refractive index and a transparent coating layer. Can be formed.
- active energy rays such as ultraviolet rays or heat
- Metal oxide porous particles As the component (A) in the present embodiment, the metal oxide porous particles described in the first embodiment are used. Moreover, what is necessary is just to employ
- the metal oxide porous particles of the component (A) of this embodiment are mixed with the binder of the component (B) made of a curable functional group-containing compound and used as a coating agent.
- the component (B) composed of the curable functional group-containing compound an active energy ray-curable functional group-containing compound or a thermosetting functional group-containing silicon compound is particularly preferably used.
- the binder component will be described in detail.
- active energy ray-curable functional group-containing compound examples include (meth) acrylate compounds and poly (methyl) glycidyl ether compounds.
- the (meth) acrylate compound will be described.
- a preferred (meth) acrylate compound is a (meth) acrylic oligomer / monomer having two or more (meth) acryloyloxy groups in one molecule. By having two or more (meth) acryloyloxy groups in one molecule, it is cured by active energy rays such as ultraviolet rays and electron beams to form a coat layer having good scratch resistance.
- triethylene glycol di (meth) acrylate triethylene glycol di (meth) acrylate, polyethylene glycol di (meth) acrylate, neopentyl glycol di (meth) acrylate, 1,6-hexanediol di (meth) acrylate, 1,9-nonanediol di (Meth) acrylate, dimethylol-tricyclodecane di (meth) acrylate, trimethylolpropane tri (meth) acrylate, ethylene oxide modified trimethylolpropane tri (meth) acrylate, pentaerythritol tri (meth) acrylate, pentaerythritol tetra (Meth) acrylate, dipentaerythritol hexa (meth) acrylate, bisphenol A diglycidyl ether (meth) acrylic acid adduct, 1,1,3,3,5,5-hexa ((meth) acrylic) Carboxymethyl
- a (meth) acrylate compound having a urethane bond is preferably added.
- it is obtained by the reaction of diisocyanate and hydroxy (meth) acrylate.
- diisocyanate propane diisocyanate, hexamethylene diisocyanate, isophorone diisocyanate, methylene bis (cyclohexyl isocyanate), trimethylhexamethylene diisocyanate, tolylene diisocyanate 4,4-diphenylmethane diisocyanate, xylene diisocyanate, norbornene diisocyanate, methylnorbornene diisocyanate, hydroxy (meth) acrylate, 2-hydroxyethyl (meth) acrylate, 2-hydroxypropyl acrylate, glycidol methacrylate, pentaerythritol tri (meth) acrylate , Pentaerythritol t
- the number of functional groups is preferably 2 or more, more preferably 3 or more, and those using pentaerythritol tri (meth) acrylate for hydroxy (meth) acrylate. Particularly preferred.
- a (meth) acrylic monomer having one (meth) acryloyl group per molecule may be blended for the purpose of adjusting the viscosity.
- reactive monomers having several vinyl groups and thiol groups may be added for the purpose of viscosity adjustment and curability control.
- N-vinylpyrrolidone N-vinylcarbazole, vinyl acetate, trimethylolpropane bis (2-mercaptoacetate), trimethylolpropane bis (3-mercaptopropionate), pentaerythritol tetrakis (2-mercaptoacetate) ), Pentaerythritol (3-mercaptopropionate) and the like.
- a light or thermal polymerization initiator may be blended in order to promote curing by ultraviolet rays or heat.
- the photopolymerization initiator may be a commercially available one, but specific examples include benzophenone, 2,2-dimethoxy-1,2-diphenylethane-1-one (BASF Corporation product Irgacure 651). 1-hydroxy-cyclohexyl-phenyl-ketone (BASF product Irgacure 184), 2-hydroxy-2-methyl-1-phenyl-propan-1-one (BASF product Darocur 1173, Lamberti product Esacure KL200), oligo ( 2-Hydroxy-2-methyl-1-phenyl-1-propan-1-one) (Lamberti product Esacure KIP150), (2-hydroxyethyl) -phenyl) -2-hydroxy-2-methyl-1-propane-1 -On) (BASF Products Irgacure 959), 2-methyl-1 (4- (methylthio) phenyl) -2-morpholinopropan-1-one (BASF product Irgacure 907), 2-benzyl-2-di
- tertiary amines such as triethanolamine, ethyl-4-dimethylaminobenzoate, isopentylmethylaminobenzoate, etc. may be added for the purpose of photosensitization.
- BPO benzoyl peroxide
- AIBN azobisisobutylnitrile
- the poly (methyl) glycidyl ether compound will be described.
- a preferred poly (methyl) glycidyl ether compound is an oligomer / monomer having two or more (methyl) glycidyl ether groups in one molecule.
- active energy rays such as ultraviolet rays and electron beams to form a coat layer having good scratch resistance.
- Specific examples include the following compounds.
- Examples of the compound having two (methyl) glycidyl ether groups include ethylene glycol (methyl) diglycidyl ether, triethylene glycol di (methyl) glycidyl ether, tetraethylene glycol di (methyl) glycidyl ether, polyethylene glycol di (methyl) glycidyl.
- Examples include ether, glycerin di (methyl) glycidyl ether, 1,4-butanediol di (methyl) glycidyl ether, 1,6-hexanediol di (methyl) glycidyl ether, neopentyl glycol (methyl) glycidyl ether, and the like.
- compounds having 3 or more glycidyloxy groups include glycerin tri (methyl) glycidyl ether, trimethylolpropane tri (methyl) glycidyl ether, pentaerythritol tri (methyl) glycidyl ether, pentaerythritol tetra (methyl) glycidyl ether, dipenta Examples include erythritol hexa (methyl) glycidyl ether, dipentaerythritol penta (methyl) glycidyl ether, dipentaerythritol tetra (methyl) glycidyl ether, carbitol polyglycidyl ether, and the like.
- a light or thermal polymerization initiator may be blended. Specifically, there is no particular limitation as long as it is a compound that initiates cationic polymerization by light or heat, and any of them can be used.
- the cationic photopolymerization initiator may be a commercially available one, but specific examples include Uvacure 1590 and 1591 (both trade names manufactured by Daicel UCB), Adeka Optomer-SP-100, SP-170, SP- 172, SP-150 and SP-152 (both trade names manufactured by Asahi Denka Co., Ltd.) and Rhodosil-2074 (trade names manufactured by Rhodia Co., Ltd.) can be preferably used. These photocationic polymerization initiators can be used singly or in combination of two or more.
- the addition amount is desirably in the range of 0.01 to 10.0 parts by weight with respect to 100 parts by weight of the poly (methyl) glycidyl ether compound.
- a photocationic polymerization accelerator can be used in combination.
- Specific examples include 9,10-dimethoxy-2-ethyl-anthracene, 9,10-diethoxyanthracene, and 2,4-diethylthioxanthone.
- a compound that generates a cationic species or a Lewis acid by heat for example, a thermal latent cationic polymerization initiator can be used in combination.
- a thermal latent cationic polymerization initiator can be used in combination.
- thermal latent cationic polymerization initiators examples include Sun-Aid SI-60L, Sun-Aid SI-80L, Sun-Aid SI-100L, Sun-Aid SI-80, Sun-Aid SI-100, Sun-Aid SI-145, Sun-Aid SI-150, and Sun-Aid SI. -160 (trade name, manufactured by Sanshin Chemical Industry Co., Ltd.).
- the above initiators may be used alone or in combination of two or more. Further, the curing can be further promoted by using heat after the light irradiation.
- thermosetting group-containing organosilicon compound examples include 3-glycidoxypropyltrimethoxysilane, 3-glycidoxypropylmethyldimethoxysilane, 3-glycidoxypropyldimethylmethoxysilane, and 3-glycidoxypropyl.
- These compounds can form a three-dimensional network structure by heat, and can improve hard coat properties.
- These silane compounds may be used as they are, but in order to further increase the reactivity, the alkoxysilyl group is previously hydrolyzed using an acidic catalyst such as hydrochloric acid water or a basic catalyst such as aqueous ammonia. It is more preferable to use it in a silanol (Si—OH) state or in a state where it is partially condensed to form a siloxane bond (Si—O—Si).
- preferred metal chelate compounds are Cu (II), Zn (II), Co (II), Ni (II), Be (II), Ce (III), Ta (III), Ti (III), Mn (III), La (III), Cr (III), V (III), Co (III), Fe (III), Al (III), Ce (IV), Zr (IV),
- Examples include acetylacetonate having V (IV) as a central metal atom, amino acids such as amine and glycine, Lewis acid, organic acid metal salts and the like. Of these, acetylacetonate of Al (III) and Fe (III) is more preferable in terms of curing conditions, pot life of the coating liquid, and the like.
- the addition amount is desirably in the range of 0.01 to 10.0 parts by weight with respect to 100 parts by weight of the thermosetting functional group-containing organosilicon compound.
- perchloric acids can be used in combination. Preferred perchloric acids include perchloric acid, ammonium perchlorate, magnesium perchlorate and the like.
- various additives such as ultraviolet absorbers, antioxidants, silicone surfactants, and silicone oils can be blended in the coating composition according to the purpose.
- the ratio of the metal oxide porous particles (component (A)) to the curable functional group-containing compound (component (B)) depends on the type of each component, but preferably the component (A) is (A). 1 and 60 parts by weight, more preferably 5 parts by weight and 50 parts by weight, and still more preferably 10 parts by weight and 40 parts by weight with respect to a total of 100 parts by weight of (B). Within this range, it is easy to obtain a coating film having a low refractive index and good scratch resistance.
- the coating agent may be prepared by a known method, and is not particularly limited.
- the coating agent is as follows.
- a desired amount of components are mixed in a light-shielding brown glass container or a plastic container, and if necessary, the hard coat agent composition is warmed (approximately 50 ° C. or lower) and mixed thoroughly. If necessary, add other ingredients and mix well. Furthermore, it left still sufficient deaeration and it was set as the hard-coat agent composition.
- a magnetic stirrer or a stirrer was used, but a mixer, a shaker, etc. may be selected according to the amount and viscosity.
- DMF dimethylformamide
- N N'-dimethylacetamide
- N-methyl-2-pyrrolidone 2-methoxyethanol
- 2-ethoxyethanol 2-ethoxyethanol
- 2-butoxyethanol butyl cellosolve
- PEGME polyethylene glycol methyl ether
- PEGMEA polyethylene glycol methyl ether acetate
- DAA diacetone
- the viscosity of the coating solution is adjusted by the method of coating on the substrate, but is preferably 0.1 cp to 10000 cp, more preferably 0.5 cp to 500 cp, and still more preferably 1 cp to 100 cp.
- ⁇ Formation of coat film> As a method for coating the substrate, methods such as dip, spin coating, and spraying can be employed.
- the time for performing photopolymerization is preferably 1 second to 10 minutes. When the time is shorter than 1 second, the photocuring is not sufficiently performed, and when the time is longer than 10 minutes, the coat film and the base material may be deteriorated to cause coloration or cracking.
- the solvent is dried if necessary. The drying temperature and time are determined by the boiling point of the solvent used.
- the temperature conditions necessary for the thermal polymerization are generally 50 ° C. or higher, preferably 80 ° C. or higher, more preferably 100 ° C. or higher. It is determined.
- the characteristics of the coated film after curing are not particularly limited, but the refractive index is preferably 1.45 or less, more preferably 1.40 or less, in the Na D line (589.6 nm).
- the low refractive index coating material of this embodiment is a liquid crystal display, a CRT display, a projection display, a plasma display, an electroluminescence display, an image display device such as a reflective screen, a coating agent for an antireflection film such as a touch panel, and a reflective material such as a spectacle lens. It can be used for prevention coatings and the like.
- Example A (Synthesis example of terminal branched polyolefin copolymer) Number average molecular weight (Mn), weight average molecular weight (Mw), and molecular weight distribution (Mw / Mn) were measured by GPC using the method described in the text. The melting point (Tm) was measured by using DSC (differential scanning calorimetry), and the peak top temperature obtained by measurement was adopted. In addition, although melting
- the shape of the particles in the dispersion was observed by diluting the sample 200 to 500 times, negatively staining with phosphotungstic acid, and then using a transmission electron microscope (TEM / H-7650 manufactured by Hitachi, Ltd.) at 100 kV. I did it.
- a terminal epoxy group-containing ethylene polymer (E) was synthesized according to the following procedure (for example, see Synthesis Example 2 of JP-A-2006-131870).
- a stainless steel autoclave with an internal volume of 2000 ml sufficiently purged with nitrogen was charged with 1000 ml of heptane at room temperature, and the temperature was raised to 150 ° C. Subsequently, 30 kg / cm 2 G was pressurized with ethylene in the autoclave to maintain the temperature.
- a hexane solution (1.00 mmol / ml in terms of aluminum atom) of MMAO (manufactured by Tosoh Finechem) was injected with 0.5 ml (0.5 mmol), and then a toluene solution (0.0002 mmol) of a compound represented by the following general formula (14) / Ml) 0.5 ml (0.0001 mmol) was injected to initiate the polymerization.
- the polymerization was stopped by press-fitting a small amount of methanol.
- the obtained polymer solution was added to 3 liters of methanol containing a small amount of hydrochloric acid to precipitate a polymer. After washing with methanol, it was dried under reduced pressure at 80 ° C. for 10 hours to obtain a single-end double bond-containing ethylene polymer (P).
- the solid was stirred in 400 g of methanol, collected by filtration and washed with methanol.
- 96.3 g of a white solid of the terminal epoxy group-containing ethylene polymer (E) was obtained (yield 99%, polyolefin conversion rate 100%).
- a 1000 mL flask was charged with 84 parts by weight of a terminal epoxy group-containing ethylene polymer (E), 39.4 parts by weight of diethanolamine, and 150 parts by weight of toluene, and stirred at 150 ° C. for 4 hours. Thereafter, acetone was added while cooling to precipitate the reaction product, and the solid was collected by filtration. The obtained solid was stirred and washed once with an aqueous acetone solution and further three times with acetone, and then the solid was collected by filtration.
- E terminal epoxy group-containing ethylene polymer
- diethanolamine diethanolamine
- a 500 mL flask equipped with a nitrogen introduction tube, a thermometer, a cooling tube, and a stirrer is charged with 20.0 parts by weight of polymer (I) and 100 parts by weight of toluene and heated in an oil bath at 125 ° C. while stirring to obtain a solid. Was completely dissolved.
- After cooling to 90 ° C. 0.323 parts by weight of 85% KOH previously dissolved in 5.0 parts by weight of water was added to the flask and mixed for 2 hours under reflux conditions. Thereafter, water and toluene were distilled off while gradually raising the temperature in the flask to 120 ° C.
- the pressure in the flask was reduced, and the internal temperature was raised to 150 ° C. and maintained for 4 hours to further distill off water and toluene in the flask. After cooling to room temperature, the solidified solid in the flask was crushed and taken out.
- T terminal branched polyolefin copolymer
- the obtained dispersion had a volume 50% average particle size of 0.018 ⁇ m (volume 10% average particle size 0.014 ⁇ m, volume 90% average particle size 0.022 ⁇ m).
- the particle diameter measured from the transmission electron microscope observation result of the obtained dispersion was 0.015-0.030 ⁇ m.
- Example a1 (Synthesis of silica porous particles 1) Add 1 mL of a branched branched polyolefin copolymer (T) aqueous dispersion diluted to 1 wt% and 0.4 mL of 28% aqueous ammonia solution to an ethanol / water (10 mL / 2.5 mL) mixture until uniform. Stir. TEOS 20 ⁇ L was added using a micropipette. Then, it stirred at room temperature for 6 hours. The obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol. The obtained powder was dried using a vacuum dryer.
- T branched branched polyolefin copolymer
- the temperature was raised from room temperature to 550 ° C. at 3.5 ° C./min, and further calcined at 550 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles to obtain porous silica particles.
- Measurement by DLS revealed that 50% volume average particle diameter was 90 nm, D90 / D50 was 1.38, and TEM observation showed that silica porous particles having 10-20 nm pores inside were obtained. .
- the BET specific surface area was 108 m 2 / g
- the value (BJH pore diameter) determined by the BJH method from the adsorption side isotherm was 13 nm, from the desorption side isotherm. Since the value (connection part) calculated
- DLS is measured by dispersing in water using a particle size distribution meter / Nanotrack WAVE. Further, since silica is used as the porous particles and water is used as the dispersion solvent, the refractive index of silica is 1.44 and the refractive index of water is 1.0.
- Example a2 (Synthesis of porous silica particles 2) Silica porous particles were obtained by the same method as in Synthesis 1 except that TEOS was changed to 12.5 ⁇ L and the stirring time was changed to 4 hours. Measurement by DLS revealed that 50% volume average particle diameter was 70 nm, D90 / D50 was 1.32, and TEM observation showed that silica porous particles having pores of 10-30 nm inside were obtained. .
- the BET specific surface area of 105 m 2 / g, the value obtained by the BJH method from the adsorption side isotherm (BJH pore diameter) is 14 nm, and the value obtained by the BJH method from the desorption side isotherm (connection part) Since it is 4 nm or less, it was found that the structure was a three-dimensional cubic phase structure in which pores were connected to each other.
- Example a3 (Synthesis of porous silica particles 3) To 400 mL of ethanol, 20 mL of a 15 wt% terminally branched polyolefin copolymer (T) aqueous dispersion and 5 mL of 28% aqueous ammonia solution were added and stirred until uniform. 3 mL of TEOS was added using a micropipette. Then, it stirred at room temperature for 48 hours. The obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol. The obtained powder was dried using a vacuum dryer. The temperature was raised from room temperature to 550 ° C.
- Example a4 (Synthesis of porous silica particles 4) To 150 mL of ethanol, 40 mL of a 15 wt% terminal branched polyolefin copolymer (T) aqueous dispersion and 3 mL of 28% aqueous ammonia solution were added and stirred until uniform. TEOS / ethanol (8.7 mL / 35 mL) was added in one portion. Then, it stirred at room temperature for 24 hours. The obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol. The obtained powder was dried using a vacuum dryer. The temperature was raised from room temperature to 550 ° C.
- silica porous particles having a volume 50% average particle diameter of 80 nm, D90 / D50 of 1.30, and pores of 10-30 nm inside were obtained from TEM observation. It was.
- the BET specific surface area of 105 m 2 / g, the value obtained by the BJH method from the adsorption side isotherm (BJH pore diameter) is 14 nm, and the value obtained by the BJH method from the desorption side isotherm (connection part) Since it is 4 nm or less, it was found that the structure was a three-dimensional cubic phase structure in which pores were connected to each other.
- Example a5 Synthesis of porous silica particles 5
- T terminally branched polyolefin copolymer
- T terminally branched polyolefin copolymer
- TEOS / ethanol 36 mL / 144 mL
- ethyltriethoxysilane Triethoxy (ethyl) silane
- ethanol 3.6 mL / 14.4 mL
- the obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol.
- the obtained powder was dried using a vacuum dryer.
- the temperature was raised from room temperature to 550 ° C. at 3.5 ° C./min, and further calcined at 550 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles to obtain porous silica particles. From the measurement by DLS, it was found that silica porous particles having a volume 50% average particle diameter of 80 nm and D90 / D50 of 1.32 and having pores of 10-30 nm inside were obtained from TEM observation. (FIG. 3).
- the BET specific surface area was 194 m 2 / g
- the value obtained by the BJH method from the adsorption side isotherm (BJH pore diameter) was 11 nm
- the value obtained by the BJH method from the desorption side isotherm (connection part) was 4 nm or less, and it was found that the structure was a three-dimensional cubic phase structure in which pores were connected to each other.
- Example a6 Synthesis of silica porous particles 6
- ethanol 1.8 mL of deionized water
- 0.4 mL of 28% aqueous ammonia solution was added to a 50 mL flask
- a solution obtained by diluting 1.1 mL of TEOS with 4.4 mL of ethanol was added, and the mixture was stirred at room temperature for 24 hours.
- silica / terminally branched olefin copolymer composite particles were collected by centrifugation (11000 rpm, 15 minutes), washed three times with ethanol, and dried overnight at 80 ° C. The temperature was raised from room temperature to 600 ° C. in 2 hours, and further calcined at 600 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles, thereby obtaining porous silica particles. Measurement by DLS showed that 50% volume average particle diameter was 123 nm, D90 / D50 was 1.35, and TEM observation showed that silica porous particles having 10-20 nm pores inside were obtained. .
- the BET specific surface area was 183 m 2 / g, and the value obtained by the BJH method from the adsorption side isotherm (BJH pore diameter) was 15 nm, from the desorption side isotherm. Since the value (connection part) calculated
- Example a7 Synthesis of porous silica particles 7
- ethanol 9.72 mL
- deionized water 1.8 mL
- terminal branched polyolefin copolymer T
- ammonia aqueous solution 0.4 mL
- a solution obtained by diluting 1.32 mL of TEOS with 5.28 mL of ethanol was added, and the mixture was stirred at room temperature for 24 hours.
- silica / terminally branched olefin copolymer composite particles were collected by centrifugation (11000 rpm, 15 minutes), washed three times with ethanol, and dried overnight at 80 ° C. The temperature was raised from room temperature to 600 ° C. in 2 hours, and further calcined at 600 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles, thereby obtaining porous silica particles.
- Measurement by DLS revealed that 50% volume average particle diameter was 142 nm, D90 / D50 was 1.41, and TEM observation showed that silica porous particles having 10-20 nm pores inside were obtained. .
- the BET specific surface area was 153 m 2 / g
- the value (BJH pore diameter) obtained from the adsorption side isotherm by the BJH method was 12 nm, from the desorption side isotherm. Since the value (connection part) calculated
- Example a8 (Synthesis of porous silica particles 8) Silica porous particles were prepared in the same manner as in Example a6 except that in Example a6, 0.6 mL of a terminally branched polyolefin copolymer (T) aqueous dispersion in which deionized water was adjusted to 2 mL and 15% by weight was used. Got. Measurement by DLS revealed that 50% volume average particle diameter was 242 nm, D90 / D50 was 1.74, and TEM observation revealed that silica porous particles having 10-20 nm pores inside were obtained. .
- T terminally branched polyolefin copolymer
- the BET specific surface area was 102 m 2 / g, and the value obtained by the BJH method from the adsorption side isotherm (BJH pore diameter) was 14 nm, from the desorption side isotherm. Since the value (connection part) calculated
- Example a9 Synthesis of porous silica particles 9
- Silica porous particles were prepared in the same manner as in Example a7, except that in Example a7, 0.6 mL of an end-branched polyolefin copolymer (T) aqueous dispersion in which deionized water was adjusted to 2 mL and 15% by weight was used.
- T end-branched polyolefin copolymer
- the BET specific surface area was 88 m 2 / g
- the value (BJH pore diameter) obtained from the adsorption side isotherm by the BJH method was 13 nm, from the desorption side isotherm. Since the value (connection part) calculated
- Reference example a1 Synthesis of silica particles 0.1 mL of 28% aqueous ammonia was added to 5 mL of ethanol and stirred, and TEOS / ethanol (0.1 mL / 0.4 mL) was added and stirred for 4 hours. This mixture was dried to obtain silica particles. From the measurement by DLS, it was found that silica particles having a 50% volume average particle diameter of 150 nm and a D90 / D50 of 1.2 were obtained. In TEM observation, pores could not be confirmed inside the silica particles (FIG. 4). The BET specific surface area was 20 m 2 / g.
- Comparative Example a1 Synthesis of silica porous particles 10
- a cationic surfactant CTAB cetyltrimethylammonium bromide
- CTAB cetyltrimethylammonium bromide
- TEOS 0.1mL was added and it stirred for 4 hours.
- the obtained silica / CTAB composite particles were collected by centrifugation and further washed with ethanol.
- the obtained powder was dried using a vacuum dryer. The temperature was raised from room temperature to 550 ° C. at 3.5 ° C./min, and further calcined at 550 ° C. for 4 hours to remove CTAB to obtain porous silica particles.
- silica porous particles having a volume 50% average particle diameter of 300 nm and D90 / D50 of 1.3 were obtained. Further, in this comparative example a1, it was difficult to determine the pore diameter inside the particle by TEM observation.
- the BET specific surface area was 32 m 2 / g, and the pore diameter obtained by the BJH method from the adsorption side and the desorption side was 2 nm. The pores were assumed to have a two-dimensional cylinder structure.
- Comparative Example a2 Synthesis of porous silica particles 11
- Silica porous particles were obtained by the same method as in Synthesis 10 except that the amount of CTAB was changed to 10.2 mg. From the measurement by DLS, it was found that porous silica particles having a volume 50% average particle diameter of 410 nm and D90 / D50 of 1.3 were obtained. Moreover, the observation result of the particle
- Comparative Example a3 Synthesis of porous silica particles 12
- Silica porous particles were obtained by the same method as in Synthesis 10 except that the amount of CTAB was changed to 20.5 mg. In TEM observation, only irregular particles that were not spherical were obtained (FIG. 6).
- Example a10 Preparation of silica porous particle aqueous dispersion
- T terminally branched polyolefin copolymer
- 14 mL 28% aqueous ammonia solution
- TEOS / ethanol 36 mL / 150 mL
- ethyltriethoxysilane Triethoxy (ethyl) silane
- ethanol 3.6 mL / 14.4 mL
- the obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol.
- the obtained powder was dried using a vacuum dryer. The temperature was raised from room temperature to 450 ° C. at 3.5 ° C./min, and further calcined at 550 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles to obtain porous silica particles.
- 10 g of silica porous particle powder was added to 500 ml of water, and dispersion treatment was performed using a bead mill. After the dispersion treatment, a uniform dispersion without precipitation was obtained.
- Example a11 Preparation of silica porous particle ethanol dispersion
- T terminally branched polyolefin copolymer
- T terminally branched polyolefin copolymer
- TEOS / ethanol 36 mL / 150 mL
- ethyltriethoxysilane Triethoxy (ethyl) silane
- ethanol 3.6 mL / 14.4 mL
- the obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol.
- the obtained powder was dried using a vacuum dryer. The temperature was raised from room temperature to 450 ° C. at 3.5 ° C./min, and further calcined at 550 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles to obtain porous silica particles.
- 10 g of silica porous particle powder was added to 437 ml of ethanol and subjected to ultrasonic treatment (US) for 30 minutes for dispersion treatment. After the dispersion treatment, a uniform dispersion without precipitation was obtained.
- US ultrasonic treatment
- Comparative Example a4 15 parts by weight of methanol as a solvent was added to 10 parts by weight of tetramethoxysilane (TMOS) and stirred at room temperature. Further, 1 part by weight of a 1N hydrochloric acid aqueous solution of the catalyst was added dropwise, followed by stirring at 50 ° C. for 1 hour to obtain a dehydrated condensate of TMOS. To the obtained dehydrated condensate of TMOS, 3.4 g of 1N-hydrochloric acid aqueous solution was further dropped (to make the pH after addition of the terminal branched polyolefin copolymer 3), followed by stirring at room temperature and further terminal branching.
- TMOS tetramethoxysilane
- silica porous particle powder was added to 500 ml of water, and dispersion treatment was performed using a bead mill. After the dispersion treatment, a uniform dispersion without precipitation was obtained. Part of the dispersion was dried, and TEM observation of the obtained particles confirmed that the porous structure was retained. From the measurement by DLS, silica porous particles having a volume 50% average particle diameter of 3.8 ⁇ m, D90 / D50 of 5.2, and pores of 10-30 nm inside are obtained from TEM observation. all right.
- the BET specific surface area was 680 m 2 / g
- the value obtained by the BJH method from the adsorption side isotherm (BJH pore diameter) was 11 nm
- Example a12 The aqueous dispersion obtained in Example a10 was concentrated to 2.9% by weight by ultrafiltration. 8 g of acrylic resin water emulsion Almatex A9083 (solid content concentration: 50% by weight manufactured by Mitsui Chemicals, Inc.) was mixed with 35 g of this dispersion, poured into a petri dish and dried in an oven at 70 ° C., and a uniform and transparent film having a thickness of 50 ⁇ m. Got. The HAZE value was 1.7, the refractive index of the film at the D line (589 nm) was 1.38, and the thermal conductivity was 0.04 W / mK.
- Example a13 35 g of the ethanol dispersion (2.9% by weight) obtained in Example a11 and PVB resin ethanol solution (polyvinyl butyral, weight average molecular weight: 50,000 to 80,000) previously adjusted to 10% were mixed, Then, it was dried in an oven at 70 ° C. to obtain a uniform and transparent film having a thickness of 70 ⁇ m.
- the HAZE value was 0.4
- the refractive index at the D line (589 nm) of the film was 1.33
- the thermal conductivity 0.03 W / mK.
- Comparative Example a5 A membrane was prepared in the same manner as in Example a12 except that the dispersion was not used, and a membrane of only the acrylic resin water emulsion Almatex A9083 was obtained.
- the film had a HAZE value of 0.3, a refractive index of the film at the D-line (589 nm) of 1.47, and a thermal conductivity of 0.58 W / mK.
- Comparative Example a6 The silica porous particles obtained in Comparative Example a1 were dispersed by a bead mill in the same manner as in Example a10, and a membrane was prepared in the same manner as in Example a12 using the obtained dispersion.
- the HAZE value was 12, and the refractive index was not measurable.
- the thermal conductivity was 0.88 W / mK.
- Comparative Example a7 The silica porous particles obtained in Comparative Example a2 were dispersed by a bead mill in the same manner as in Example a10, and a membrane was prepared in the same manner as in Example a12 using the obtained dispersion.
- the HAZE value was 20, and the refractive index was not measurable.
- the thermal conductivity was 0.92 W / mK.
- Comparative Example a8 Using the silica porous particle dispersion obtained in Comparative Example a4, a membrane was prepared in the same manner as in Example a12. The HAZE value was 36, and the refractive index was not measurable. The thermal conductivity was 0.07 W / mK.
- Comparative Example a9 A membrane was prepared in the same manner as in Example a13 except that the dispersion was not used, and a PVB-only membrane was obtained.
- the film had a HAZE value of 0.1, a refractive index of the film at the D-line (589 nm) of 1.49, and a thermal conductivity of 0.22 W / mK.
- Example B (Synthesis example of terminal branched polyolefin copolymer) Number average molecular weight (Mn), weight average molecular weight (Mw), and molecular weight distribution (Mw / Mn) were measured by GPC using the method described in the text. The melting point (Tm) was measured by using DSC (differential scanning calorimetry), and the peak top temperature obtained by measurement was adopted. In addition, although melting
- the shape of the particles in the dispersion was observed by diluting the sample 200 to 500 times, negatively staining with phosphotungstic acid, and then using a transmission electron microscope (TEM / H-7650 manufactured by Hitachi, Ltd.) at 100 kV. I did it.
- a terminal epoxy group-containing ethylene polymer (E) was synthesized according to the following procedure (for example, see Synthesis Example 2 of JP-A-2006-131870).
- a stainless steel autoclave with an internal volume of 2000 ml sufficiently purged with nitrogen was charged with 1000 ml of heptane at room temperature, and the temperature was raised to 150 ° C. Subsequently, 30 kg / cm 2 G was pressurized with ethylene in the autoclave to maintain the temperature.
- the obtained polymer solution was added to 3 liters of methanol containing a small amount of hydrochloric acid to precipitate a polymer. After washing with methanol, it was dried under reduced pressure at 80 ° C. for 10 hours to obtain a single-end double bond-containing ethylene polymer (P).
- the solid was stirred in 400 g of methanol, collected by filtration and washed with methanol.
- 96.3 g of a white solid of the terminal epoxy group-containing ethylene polymer (E) was obtained (yield 99%, polyolefin conversion rate 100%).
- a 1000 mL flask was charged with 84 parts by weight of a terminal epoxy group-containing ethylene polymer (E), 39.4 parts by weight of diethanolamine, and 150 parts by weight of toluene, and stirred at 150 ° C. for 4 hours. Thereafter, acetone was added while cooling to precipitate the reaction product, and the solid was collected by filtration. The obtained solid was stirred and washed once with an aqueous acetone solution and further three times with acetone, and then the solid was collected by filtration.
- E terminal epoxy group-containing ethylene polymer
- diethanolamine diethanolamine
- a 500 mL flask equipped with a nitrogen introduction tube, a thermometer, a cooling tube, and a stirrer is charged with 20.0 parts by weight of polymer (I) and 100 parts by weight of toluene and heated in an oil bath at 125 ° C. while stirring to obtain a solid. Was completely dissolved.
- After cooling to 90 ° C. 0.323 parts by weight of 85% KOH previously dissolved in 5.0 parts by weight of water was added to the flask and mixed for 2 hours under reflux conditions. Thereafter, water and toluene were distilled off while gradually raising the temperature in the flask to 120 ° C.
- the pressure in the flask was reduced, and the internal temperature was raised to 150 ° C. and maintained for 4 hours to further distill off water and toluene in the flask. After cooling to room temperature, the solidified solid in the flask was crushed and taken out.
- T terminal branched polyolefin copolymer
- the obtained dispersion had a volume 50% average particle size of 0.018 ⁇ m (volume 10% average particle size 0.014 ⁇ m, volume 90% average particle size 0.022 ⁇ m).
- the particle diameter measured from the transmission electron microscope observation result of the obtained dispersion was 0.015-0.030 ⁇ m.
- the obtained silica / terminally branched olefin copolymer composite particles were collected by centrifugation and further washed with ethanol.
- the obtained powder was dried using a vacuum dryer. The temperature was raised from room temperature to 450 ° C. at 3.5 ° C./min, and further calcined at 550 ° C. for 4 hours to remove the terminal branched olefin copolymer composite particles to obtain porous silica particles.
- 10 g of silica porous particle powder was added to 437 ml of diacetone alcohol and subjected to ultrasonic treatment (US) for 30 minutes for dispersion treatment. After the dispersion treatment, a uniform dispersion without precipitation was obtained.
- US ultrasonic treatment
- a part of the dispersion was dried, and TEM observation of the obtained particles confirmed that the porous structure was maintained.
- Measurement by DLS shows that 50% volume average particle diameter is 80 nm, D90 / D50 is 1.3, and TEM observation shows that silica porous particles with 10-30 nm pores inside are obtained. It was. Further, the BET specific surface area was 235 m 2 / g, the value (BJH pore diameter) determined from the adsorption side isotherm by the BJH method was 11 nm, and the value determined from the desorption side isotherm by the BJH method (connection part). Since it was 4 nm or less, it turned out that it is a three-dimensional cubic phase structure which the pore connected mutually.
- the obtained silica porous particles are dispersed in water, and DLS measurement is performed using a particle size distribution meter / Nanotrack WAVE. Further, since silica is used as the porous particles and water is used as the dispersion solvent, the refractive index of silica is 1.44 and the refractive index of water is 1.0.
- silica porous particle powder 10 g was added to 437 ml of diacetone alcohol and subjected to ultrasonic treatment (US) for 30 minutes for dispersion treatment. After the dispersion treatment, a uniform dispersion without precipitation was obtained. From the measurement by DLS, it was found that silica porous particles having a volume 50% average particle diameter of 300 nm and D90 / D50 of 1.3 were obtained. Also, it was difficult to determine the internal pore diameter from TEM observation. The BET specific surface area was 32 m 2 / g, and the pore diameter obtained by the BJH method from the adsorption side and the desorption side was 2 nm. The pores were assumed to have a two-dimensional cylinder structure.
- Example b1 20 g of silica porous particle diacetone alcohol dispersion of Preparation Example b1 concentrated to 10% by weight, 3.0 g of trimethylolpropane triacrylate, pentaerythritol triacrylate hexamethylene diisocyanate urethane prepolymer (trade name UA manufactured by Kyoeisha Chemical Co., Ltd.) After mixing 1.0 g of -306H), 3 g of polyethylene glycol methyl ether was added.
- the refractive index of the coating material of this embodiment was measured with an Abbe refractometer manufactured by Atago Co., Ltd. for a thin film formed on a Si-wafer.
- the transparency of the present embodiment was determined by measuring the transmittance between 400 nm and 600 nm using a membrane transmittance meter (UV2200, manufactured by Shimadzu Corporation).
- the scratch resistance of the present embodiment is determined by applying a load of 1000 g and 500 g with # 0000 steel wool (manufactured by Nippon Steel Wool Co., Ltd.) on a sample formed on quartz glass, and rubbing the surface for 10 reciprocations. The degree of the mark was visually judged at the next stage.
- B 1 to 9 scratches are in the range rubbed with a load of 500 g.
- C 10 to 30 scratches are in the range rubbed with a load of 500 g.
- D Innumerable (more than 30) scratches are within the range rubbed with a load of 500 g.
- Example b2 After mixing 20 g of the silica porous particle diacetone alcohol dispersion of Preparation Example b1 concentrated to 10% by weight, 1.0 g of 1,6-hexanediol diglycidyl ether and 3.0 g of pentaerythritol triglycidyl ether, polyethylene glycol 3 g of methyl ether was added. Further, 0.15 g of Adekaoptoma-SP-150 and 0.01 g of Si-based surfactant (trade name FZ-2110 manufactured by Nippon Unicar Co., Ltd.) were added as photoinitiators, and the mixture was sufficiently stirred to prepare a coating composition. . The prepared coating composition was coated on a Si wafer and quartz glass, and irradiated with a high pressure mercury lamp (intensity 100 W / cm) for 60 seconds to form a coating film. The evaluation results are listed in Table 1.
- Example b3 20 g of the silica porous particle diacetone alcohol dispersion of Preparation Example b1 concentrated to 10% by weight was added to 4.2 g of 3-glycidoxypropyltrimethoxysilane as a silane compound and / or a partially condensed compound thereof. A 0.1N aqueous hydrochloric acid solution (1.3 g) was added with stirring, and the mixture was further stirred for 2 hours.
- Comparative Example b1 Instead of the silica porous particle diacetone alcohol dispersion of Preparation Example b1, a coating film was prepared in the same manner as in Example b3 except that 20 g of the silica porous particle diacetone alcohol dispersion of Preparation Example b2 concentrated to 10% by weight was added. Formed. The evaluation results are listed in Table 1. In addition, since the coating film obtained by Comparative Example b1 had low film transparency, the refractive index could not be measured accurately.
- Comparative Example b2 A coated film was formed in the same manner as in Example b1, except that non-porous silica particles (organosilica sol 70-100 nm ZL type, manufactured by Nissan Chemical Industries, Ltd.) were used. The evaluation results are listed in Table 1.
- Comparative Example b3 A coat film was formed in the same manner as in Example b2, except that non-porous silica particles (organosilica sol 70-100 nm ZL type, manufactured by Nissan Chemical Industries, Ltd.) were used. The evaluation results are listed in Table 1.
- Comparative Example b4 A coating film was formed in the same manner as in Example b3 except that non-porous silica particles (organosilica sol 70-100 nm ZL type, manufactured by Nissan Chemical Industries, Ltd.) were used. The evaluation results are listed in Table 1.
- Comparative Example b5 A coat film was formed in the same manner as in Example b1 except that silica porous particles were not added. The evaluation results are listed in Table 1.
- Comparative Example b6 A coat film was formed in the same manner as in Example b2 except that silica porous particles were not added. The evaluation results are listed in Table 1.
- Comparative Example b7 A coat film was formed in the same manner as in Example b3 except that silica porous particles were not added. The evaluation results are listed in Table 1.
- the coating films obtained in Examples b1-b3 exhibited high performance in all evaluation items of refractive index, transparency, and scratch resistance.
- the metal oxide porous body obtained in the present invention has a mesopore having a small particle diameter and a pore diameter of 5 nm or more, and has a regular three-dimensional cubic phase structure. Excellent transparency even when mixed in, it can be applied to optical materials, low dielectric constant materials, and heat insulation materials.
- the particles themselves are drugs (DDS: drug delivery system), molecular probes, catalysts, adsorbing materials, sensors, It is expected as a material that can be newly applied to paints and inks.
- the coating agent obtained in the present invention can adjust the refractive index of the resulting coating film to be low and can impart hard coat properties due to the properties of the binder, it can be used for liquid crystal displays, CRT displays, projection displays, plasma displays, It can be used for image display devices such as luminescence displays and reflection screens, coating agents for antireflection films such as touch panels, and antireflection coatings such as eyeglass lenses.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Polymers & Plastics (AREA)
- General Engineering & Computer Science (AREA)
- Geology (AREA)
- General Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Mechanical Engineering (AREA)
- Pigments, Carbon Blacks, Or Wood Stains (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Paints Or Removers (AREA)
- Silicon Compounds (AREA)
- Silicates, Zeolites, And Molecular Sieves (AREA)
Abstract
Description
特許文献2には、界面活性剤のミセルをテンプレートとした、3次元キュービック相構造のメソポーラス金属酸化物の合成法が記載されており、平均細孔径が5nmであるメソポーラス金属酸化物が得られる旨記載されている(実施例2、図6)。
特許文献3には、非水溶性ポリマー粒子を用いた金属酸化物多孔質体の製造方法が記載されており、メソ孔がキュービック相を形成し、かつ孔径が5~30nmの範囲で略均一である金属酸化物多孔質体が得られる旨記載されている。そして、スプレードライにより粒子径1~10μmの多孔質粒子を製造した例が、記載されている(実施例a12等)。
特許文献1の方法では、得られる金属酸化物(シリカ)多孔質粒子のメソ孔径が最大でも10nmであるため、比較的分子サイズの大きな機能性分子の吸着や内包には不向きであった。また、一粒子中に存在するメソ孔の割合を自由に変えることができず、光学特性、誘電率特性、断熱特性などの各特性を変化させることが難しい。さらに、金属酸化物多孔質粒子は、シリンダー状(ヘキサゴナル状)構造のメソ孔であるため、3次元キュービック相構造に比べ、メソ孔間の壁厚みが全体的に薄くなり、機械強度が不十分となる。その結果、塗料等に用いる場合、メソ孔構造が崩壊する可能性があった。さらに、金属酸化物多孔質粒子の粒子径が大きく、かつ粒子径が揃っていない場合、例えば、平均粒子径100nm(0.1μm)以上の当該粒子を樹脂と混合した場合、不透明になりやすく、光学用途としては好ましくない。
特許文献3には、メソ孔がキュービック相を形成し、かつ孔径が5~30nmの範囲で略均一である金属酸化物多孔質粒子が記載されているものの、平均粒子径が小さくかつ粒子径の揃った粒子は記載されていない。
また、特許文献6の方法で得られる中空粒子は炭酸カルシウムを鋳型としているので、粒径、粒子内部の中空構造のコントロールが困難と考えられる。
[1] 体積50%平均粒子径が50nm以上300nm以下、体積90%平均粒子径と体積50%平均粒子径との比(D90/D50)が2.0以下であり、BJH法による細孔径が5nm以上30nm以下のメソ孔を有し、その細孔構造が3次元キュービック相構造であることを特徴とする、金属酸化物多孔質粒子。
前記混合物に、金属酸化物前駆体を混合し、該金属酸化物前駆体のゾル-ゲル反応を行って有機無機複合体粒子を得る工程と、
前記有機無機複合体粒子から前記非水溶性ポリマー粒子を除去する工程と、
を含む、[1]又は[2]に記載の金属酸化物多孔質粒子の製造方法。

または、一般式(4)



前記水および/または水の一部または全部を溶解する前記有機溶媒と、前記非水溶性ポリマー粒子の水分散体と、前記塩基触媒とを混合する工程を含む、[3]ないし[6]のいずれか1つに記載の金属酸化物多孔質粒子の製造方法。
(A)[1]又は[2]に記載の金属酸化物多孔質粒子。
(B)硬化性官能基含有化合物。
さらに、本発明の金属酸化物多孔質粒子は、従来のものと比較してメソ孔の孔径が大きく、粒子内部に大きな空孔を備える。そのため、吸着性に優れ、所望の物質を内包することができる。また、粒子内部に大きな空気層を備えるため、軽量化、断熱性、低屈折率、低誘電率などの特性に寄与することもできる。本発明の金属酸化物多孔質粒子は、このような特性を備えるため、様々な用途に用いることができる。
また、本発明の金属酸化物多孔質粒子の製造方法は、粒子設計の自由度が大きく、一粒子中に存在するメソ孔の割合(空孔率)を自由に変えることができるため、本発明の金属酸化物多孔質粒子を効率良く得ることができる。
本実施形態の金属酸化物多孔質粒子は、体積50%平均粒子径が50nm以上300nm以下、体積90%平均粒子径と体積50%平均粒子径との比(D90/D50)が2.0以下であり、Barrett-Joyner-Halenda法(BJH法)による細孔径が5nm以上30nm以下のメソ孔を有し、その細孔構造が3次元キュービック相構造である。
以下、本実施形態について、適宜図面を用いて説明する。尚、すべての図面において、同様な構成要素には同様の符号を付し、適宜説明を省略する。
本実施形態の金属酸化物多孔質粒子は、大きさが略均一で、かつ分散性が高い、いわゆる単分散粒子である。
図1に示すように、金属酸化物多孔質粒子は内部に略均一なメソ孔を有しており、メソ孔は相互に連結した3次元キュービック相構造を構成する。本実施形態の金属酸化物多孔質粒子の体積50%平均粒子径は外径が50nm以上300nm以下であり、好ましくは50nm以上100nm以下であり、さらに好ましくは、60nm以上90nm以下である。この範囲であると粒子製造が容易であり、かつ、様々な用途に用いることができ、所望の特性を効果的に発現することができる。例えば、後述する樹脂組成物として用いる場合、バインダー樹脂中に均一に分散させることができ、透明性が高い樹脂組成物を得ることができる。
本実施形態の金属酸化物多孔質粒子は、単分散粒子であるためフィルムや塗料に均一に分散させることが容易であり、さらに外径が小さいため透明性が高くなる。BJH細孔径が5nm以上30nm以下のメソ孔を有し、その細孔構造が定形な3次元キュービック相構造となるため、粒子内部に大きな空孔を備える。そのため、高吸着性が期待できる。また、粒子内部に大きな空気層を備えるため、軽量、断熱性、低屈折率、低誘電率などの特性が期待できる。
本実施形態の金属酸化物多孔質粒子の比表面積は、80m2/g以上であることが好ましく、100m2/g以上であることがより好ましく、150m2/g以上であることが特に好ましい。
また、熱伝導性は形状に起因した異方性が無い方が好ましいという観点から、球形の形状を有し、均一なメソ孔を有することが好ましい。
本実施形態の金属酸化物としては、ケイ素(Si)、アルミニウム(Al)、亜鉛(Zn)、ジルコニウム(Zr)、インジウム(In)、スズ(Sn)、チタン(Ti)、鉛(Pb)、ハフニウム(Hf)、コバルト(Co)、リチウム(Li)、バリウム(Ba)、鉄(Fe)、マンガン(Mn)から選ばれる金属の酸化物が好ましく、物質自身の屈折率、熱伝導率が金属酸化物の中で比較的低いという観点から、ケイ素酸化物(シリカ)が特に好ましい。
また金属酸化物は複数の金属を含む複合酸化物であってもよい。
本実施形態の金属酸化物多孔質粒子の製造方法は、
水系媒体に分散可能な非水溶性ポリマー粒子の存在下に、金属酸化物前駆体のゾル-ゲル反応を行い、有機無機複合体粒子を得る工程(1)と、
有機無機複合体粒子から前記非水溶性ポリマー粒子を除去して、金属酸化物多孔質粒子を得る工程(2)と、を含む。
工程(1-1)
水および/または水の一部または全部を溶解する有機溶媒に、体積50%平均粒子径が5nm以上30nm以下である非水溶性ポリマー粒子と塩基触媒とを添加し、混合物を得る工程。
工程(1-2)
前記工程(1-1)で得られた混合物と金属酸化物前駆体を混合し、該金属酸化物前駆体のゾル-ゲル反応を行って有機無機複合体粒子を得る工程。
以下、各工程を順に説明する。
工程(1-1)においては、具体的に、非水溶性ポリマー粒子(X)(以下、適宜「成分(X)」とする)、水および/または水の一部または全部を溶解する有機溶媒(Y)(以下、適宜「成分(Y)」とする)、塩基触媒(Z)(以下、適宜「成分(Z)」とする)を混合して混合物を調製する。
工程(1-1)において、後述の非水溶性ポリマー粒子の水分散体、水および/または水の一部または全部を溶解する有機溶媒、塩基触媒を混合することにより混合物を得るのが好ましい。
非水溶性ポリマー粒子は、体積50%平均粒子径が5nm以上30nm以下であることが好ましい。このような非水溶性ポリマー粒子から得られた金属酸化物多孔質粒子を用いることにより、高断熱で透明なフィルム、塗膜が実現できる。外径の測定方法は、例えば、粒度分布計(DLS)を用いて測定する方法、透過型電子顕微鏡(TEM)、走査型電子顕微鏡(SEM)を用いて直接観察する方法等が適用可能である。
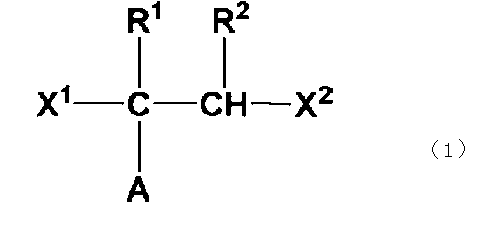
分離カラム:TSK GNH HT(カラムサイズ:直径7.5mm,長さ:300mm)
カラム温度:140℃
移動相:オルトジクロルベンゼン(和光純薬社製)
酸化防止剤:ブチルヒドロキシトルエン(武田薬品工業社製)0.025質量%
移動速度:1.0ml/分
試料濃度:0.1質量%
試料注入量:500マイクロリットル
検出器:示差屈折計
または、一般式(4)

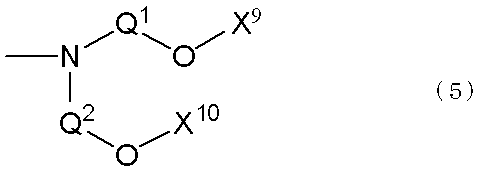
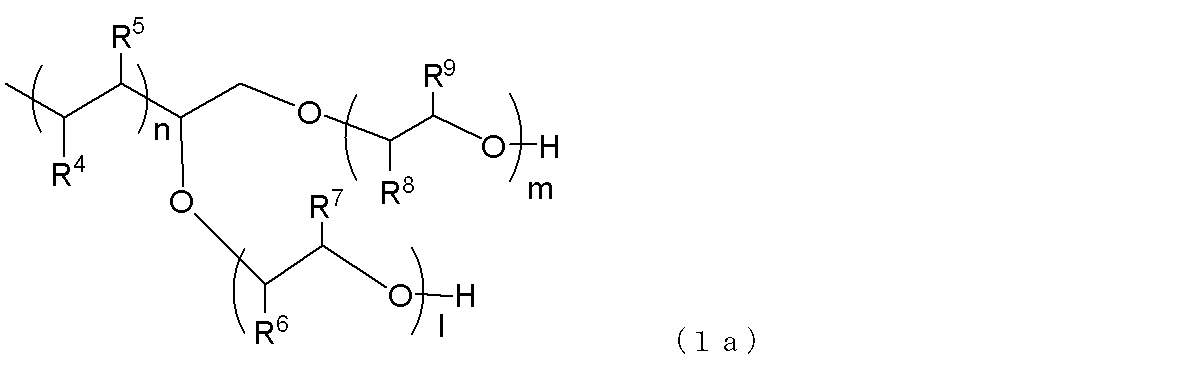
l+mは2以上450以下、好ましくは5以上200以下の整数を表す。
nは、20以上300以下、好ましくは25以上200以下の整数を表す。)

R6およびR7は、水素原子あるいはメチル基を表し、少なくともどちらか一方は水素原子である。R8およびR9は、水素原子あるいはメチル基を表し、少なくともどちらか一方は水素原子である。R10およびR11は、水素原子あるいはメチル基を表し、少なくともどちらか一方は水素原子である。
l+m+oは3以上450以下、好ましくは5以上200以下の整数を表す。
nは、20以上300以下、好ましくは25以上200以下の整数を表す。)

ポリエチレン鎖のエチレンユニット数(n)は、一般式(1)においてAで表されるポリオレフィン鎖の数平均分子量(Mn)をエチレンユニットの分子量で割ることにより算出した。また、ポリエチレングリコール鎖のエチレングリコールユニット総数(l+mもしくはl+m+o)は、ポリエチレングリコール基付加反応時の重合体原料と使用したエチレンオキシドとの重量比が、重合体原料とポリエチレングリコール基の数平均分子量(Mn)との比に同じであると仮定して算出できる。
このため、後述する各種用途における製造工程や使用場面において、粒子の崩壊が抑制されるので、本実施形態の末端分岐型ポリオレフィン系共重合体粒子が有する特性を失うことがなく、製品の歩留まりや製品の品質がより安定する。
本実施形態の分散液は前記の非水溶性ポリマー粒子、好ましくは前記末端分岐型ポリオレフィン系共重合体粒子を分散質に含み、該分散質が水および/または水の一部または全部を溶解する有機溶媒に粒子として分散している。
(1)非水溶性ポリマー粒子を製造する際に得られた、該重合体粒子を含む分散液、
(2)非水溶性ポリマー粒子を製造する際に得られた該重合体粒子を含む分散液に、さらに他の分散質や添加剤等を分散または溶解してなる分散液、
(3)非水溶性ポリマー粒子を水や水と親和性を有する有機溶媒に分散させるとともに、他の分散質や添加剤等を分散または溶解してなる分散液、
の何れをも含む。
例えば前記末端分岐型ポリオレフィン系共重合体のポリオレフィン部分の構造および末端分岐部分の構造を変えることにより調節可能である。
なお、本実施形態における体積50%平均粒子径とは、全体積を100%としたときの累積体積が50%時の粒子の直径を指し、動的光散乱式粒子径分布測定装置やマイクロトラック粒度分布測定装置を使用して測定することができる。
また、その形状は、例えばリンタングステン酸によりネガティブ染色を施した後、透過型電子顕微鏡(TEM)により観察することができる。
上記の撹拌時間では分散化を十分に行うことができ、かつ非水溶性ポリマー粒子が劣化しにくいため好ましい。反応後は、分散液中の温度が100℃以下になるまで、好ましくは60℃以下になるまでせん断力をかけた状態を保つことが好ましい。
これら界面活性剤は、単独または2種以上を併用することができる。
該分散質の含有量が上記範囲にあると、分散液の物性が実用面で良好であり、且つ分散液が凝集、沈殿を生じにくいため好ましい。
本実施形態における成分(Y)は、金属酸化物前駆体(W)(以下、適宜「成分(W)」とする)を、さらに加水分解させる目的で添加される。
本実施形態で用いる混合組成物において、金属酸化物前駆体の縮合速度を制御し、球状体の金属酸化物多孔質体を形成させる点において塩基触媒が好適に使用される。具体的には、アンモニア、水酸化アンモニウム(アンモニア水)、水酸化カリウム、水酸化ナトリウムなどのアルカリ金属水酸化物、テトラメチルアンモニウムヒドロキシド、テトラエチルアンモニウムヒドロキシド、テトラブチルアンモニウムヒドロキシドなどの4級アンモニウム水酸化物、トリエチルアミン、トリブチルアミン、モルホリン、ピリジン、ピペリジン、エチレンジアミン、ジエチレントリアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミンなどのアミン類、3-アミノプロピルトリエトキシシラン、N-(2-アミノエチル)-3-アミノプロピルトリメトキシシランなどのアミノシラン類などが挙げられる。最も好適には金属酸化物多孔質体の粒子径や形状を制御し易い点から、アンモニア、水酸化アンモニウム(アンモニア水)が用いられ、安全性の点から水酸化アンモニウム(アンモニア水)がより好ましい。
工程(1-2)においては前記工程(1-1)において得られた混合物中に、金属酸化物前駆体(W)を混合しゾル-ゲル反応を行い、有機無機複合体粒子を得る。
金属酸化物前駆体としては、金属アルコキシドおよび/またはその部分加水分解縮合物、金属ハロゲン化物、金属アセテート、金属硝酸塩、金属硫酸塩等が挙げられる。
(R12)x1M(OR13)y1 (12)
1)3-アミノプロピルトリメトキシシラン、3-アミノプロピルトリエトキシシラン、3-アミノプロピルメチルジメトキシシラン、3-アミノプロピルメチルジエトキシシラン、N-2-(アミノエチル)-3-アミノプロピルメチルジメトキシシラン、N-2-(アミノエチル)-3-アミノプロピルトリメトキシシラン、2-アミノエチルアミノメチルトリメトキシシラン、3-アミノプロピルジメチルエトキシシラン、2-(2-アミノエチルチオエチル)トリエトキシシラン、p-アミノフェニルトリメトキシシラン、N-フェニル-3-アミノプロピルメチルジメトキシシラン、N-フェニル-3-アミノプロピルメチルジエトキシシラン、N-フェニル-3-アミノプロピルトリメトキシシラン、N-フェニル-3-アミノプロピルトリエトキシシラン等のアミノ基とアルコキシシリル基とを有する化合物
2)3-グリシドキシプロピルプロピルトリメトキシシラン、3-グリシドキシプロピルプロピルトリエトキシシラン、3-グリシドキシプロピルメチルジエトキシシラン等のグリシジル基とアルコキシシリル基とを有する化合物
3)3-メルカプトプロピルメチルジメトキシシラン、3-メルカプトプロピルトリメトキシシラン等のチオール基とアルコキシシリル基とを有する化合物
4)3-ウレイドプロピルトリメトキシシラン等のウレイド基とアルコキシシリル基とを有する化合物
本実施形態において、金属アルコキシドの部分加水分解縮合物としては、アルコキシシランの縮合物、アルコキシジルコニウムの縮合物、アルコキシアルミニウムの縮合物、およびアルコキシチタンの縮合物が好ましい。
(R14)x2MZy2 (13)
非水溶性ポリマー粒子(X)と金属酸化物前駆体(W)の混合比率は特に制限されるものでは無いが、1:10~10:1が好ましい。成分(X)が多い場合、金属酸化物の存在比が低下するため多孔質構造の制御が難しくなり、細孔間をつなぐ壁が薄くなるため機械的強度が低下する。成分(W)が多い場合は孔の存在する割合が低くなるため、表面積、空孔率が小さくなり多孔質としての性能が期待できない。水および/または水の一部または全部を溶解する有機溶媒(Y)は金属酸化物前駆体(W)100重量部に対して30重量部以上100000重量部以下が好ましく、より好ましくは50重量部以上50000重量部以下である。成分(Y)の割合が低いと粒子が凝集しやすくなり、多い場合は生産効率の点から好ましくない。なお、成分(Y)中の水と溶媒の割合は特に制限されるものでは無いが0.1:100~1:50が好ましい。水が少ないと金属酸化物前駆体縮合物のゾル-ゲル反応速度が著しく低下し、多いと反応速度が速くなり粒子径や形状の制御が難しくなる。ゾル-ゲル反応用触媒(Z)は金属酸化物前駆体(W)100重量部に対して10重量部以上1000重量部以下が好ましい。成分(Z)の割合が低いと金属酸化物前駆体縮合物のゾル-ゲル反応速度が著しく低下し、割合が高いと粒子径が大きくなり300nm以下の粒子を得るのが難しくなる。
水の一部または全部を溶解する有機溶媒としてはメタノール、エタノール、プロピルアルコール、イソプロピルアルコールが好ましい。金属アルコキシドとしてテトラエトキシシラン(TEOS)、テトラメトキシシラン(TMOS)を用いる場合、エタノール、メタノールが特に好ましい。
得られた有機無機複合体粒子は遠心分離などの方法により反応液中から取り出される。取り出された有機無機複合体粒子はゾル-ゲル反応を完結させるため、ゾル-ゲル反応用触媒、水分を有機溶媒により洗浄除去し、その後充分に乾燥する。なお、ゾル-ゲル反応が完結した状態とは、理想的には全てがM-O-Mの結合を形成した状態であるが、一部アルコキシル基(M-OR2)、M-OH基を残すものの、固体(ゲル)の状態に移行した状態を含むものである。
工程(2)では有機無機複合体粒子から非水溶性ポリマー粒子を除去し、金属酸化物多孔質粒子を調製する。
本実施形態で得られた金属酸化物多孔質粒子の構造確認および細孔径の確認は透過電子顕微鏡(TEM/日本電子社製透過型電子顕微鏡JEM-2010F)で200kVの条件にて行うことができる。平均粒子径および粒子分布は水中に分散させた粒子を動的光散乱法(粒度分布計/ナノトラックWAVE)にて測定することができる。粒子のBET比表面積は窒素吸着法で、細孔径およびメソ孔を連結する孔の径はBJH法にて計算することもできる(日本ベル社製表面積測定装置BELSORP-max)。
本実施形態の金属酸化物多孔質粒子は、後述する様々な用途にそのまま用いることができ、さらに金属酸化物多孔質粒子とバインダー樹脂とを含む樹脂組成物として用いることもできる。以下、樹脂組成物について説明する。
本実施形態においてバインダー樹脂は、金属酸化物多孔質粒子間を結合しうる、あるいは金属酸化物多孔質粒子を均一に分散させる媒体となりうるものをいう。
本実施形態で使用しうるバインダー樹脂に特に制限はない。例えば、加熱により硬化する熱硬化性樹脂、紫外線等の光の照射により硬化する光硬化性樹脂、熱可塑性樹脂、水溶性樹脂が挙げられる。なかでも造膜性を有するポリオレフィン系、ポリ(メタ)アクリル酸エステル系、ポリスチレン系、ポリウレタン系、ポリビニルアルコール系、ポリビニルアセタール系の樹脂が好ましい。
光学特性、誘電率特性、断熱性等の性能発現の観点から、バインダー樹脂の含有量は、30~95質量%が好ましく、40~90質量%がより好ましく、本発明の金属酸化物多孔質粒子の含有量は70~5質量%が好ましく、60~10質量%がより好ましい。
なおバインダー樹脂と有機溶媒、水等の分散媒を混合してエマルジョンにして用いてもよい。
混練機としては、ビーズミル混合機、3本ロールミル混合機、ホモジナイザー混合機、ラボプラストミル混合機などが使用できる。
(2)水中に分散している金属酸化物多孔質粒子を、処理剤を添加して湿式処理を行なった後、溶剤置換した金属酸化物多孔質粒子オルガノゾルをバインダー樹脂(またはそのエマルジョン)に添加・混合する方法。
処理剤としては、前記のシランカップリング剤に代表される有機珪素化合物(表面処理剤)あるいはアニオン界面活性剤、カチオン界面活性剤、両性界面活性剤、ノニオン界面活性剤などが挙げられる。
本実施形態の樹脂組成物からフィルムまたは塗膜を得ることができる。このフィルムまたは塗膜の熱伝導率は、0.1W/mK以下であることが好ましく、より好ましくは0.05W/mK以下である。これにより、断熱効率が向上できる。また、このフィルムまたは塗膜の乾燥時厚みが10μmの時のHAZE値は、10%以下が好ましく、より好ましくは5%以下である。これにより、透明性の高いフィルム、または塗膜が得られる。
金属酸化物多孔質粒子を含む塗料を、ガラス基盤上にバーコーターを用い厚みを調節しコートする。オーブンにより50℃~100℃の温度で1時間~24時間乾燥させた後、形成されたフィルムをガラス基盤から剥がし取り、金属酸化物多孔質粒子含有フィルムまたは塗膜を得る。
本実施形態の金属酸化物多孔質体は、医薬(DDS:ドラッグデリバリーシステム)、分子プローブ、触媒、吸着材料、センサー、塗料、インク等に用いることができる。
本実施形態の金属酸化物多孔質体を含む樹脂組成物は、プリント基板等の低誘電率材料、機能性分子を内包した特殊塗料またはインク等に用いることができる。
本実施形態の樹脂組成物から得られるフィルムまたは塗膜は、自動車、住宅、ビル等の窓ガラス断熱フィルムまたは断熱塗料等の断熱材料、ディスプレイ、タッチパネル等の反射防止フィルム等に用いることができる。
本実施形態によれば、下記成分(A)及び成分(B)を含有するコート剤が提供される。
(A)第1実施形態に記載の金属酸化物多孔質粒子。
(B)硬化性官能基含有化合物。
以下、本実施形態について、説明する。
本実施形態における成分(A)としては、第1実施形態に記載の金属酸化物多孔質粒子が用いられる。また、当該粒子の製造方法についても第1実施形態に記載のものを採用すればよい。
本実施形態の成分(A)の金属酸化物多孔質粒子は、硬化性官能基含有化合物からなる成分(B)のバインダーと混合しコート剤として用いられる。硬化性官能基含有化合物からなる成分(B)としては、特に好ましくは活性エネルギー線硬化性官能基含有化合物または熱硬化性官能基含有珪素化合物が用いられる。以下、バインダー成分について詳細に説明する。
活性エネルギー線硬化性官能基含有化合物としては、具体的には(メタ)アクリレート化合物及びポリ(メチル)グリシジルエーテル化合物が挙げられる。
(メタ)アクリレート化合物について説明する。好ましい(メタ)アクリレート化合物は1分子中に2個以上(メタ)アクリロイルオキシ基を有する(メタ)アクリル系オリゴマー/モノマーである。1分子中に2個以上(メタ)アクリロイルオキシ基を有することにより、紫外線、電子線等の活性エネルギー線により硬化し、耐擦傷性が良好なコート層を形成する。
また、耐擦傷性を向上させる目的で、ウレタン結合を有する(メタ)アクリレート化合物が好適に添加される。一般的には、ジイソシアネートとヒドロキシ(メタ)アクリレートの反応により得られ、具体的には、ジイソシアネートとして、プロパンジイソシアネート、ヘキサメチレンジイソシアネート、イソホロンジイソシアネート、メチレンビス(シクロヘキシルイソシアネート)、トリメチルヘキサメチレンジイソシアネート、トリレンジイソシアネート、4,4-ジフェニルメタンジイソシアネート、キシレンジイソシアネート、ノルボルネンジイソシアネート、メチルノルボルネンジイソシアネート、ヒドロキシ(メタ)アクリレートとして、2-ヒドロキシエチル(メタ)アクリレート、2-ヒドロキシプロピルアクリレート、グリシドールメタクリレート、ペンタエリスリトールトリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、エチレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコールなどをそれぞれ組合せた反応により得られるウレタン(メタ)アクリレートオリゴマーなどが挙げられる。
これらのうち、硬化後の硬度を上げるためには、官能基は2つ以上が好ましく、さらに好ましくは3つ以上であり、ヒドロキシ(メタ)アクリレートにペンタエリスリトールトリ(メタ)アクリレートを用いたものが特に好ましい。
好ましいポリ(メチル)グリシジルエーテル化合物は、1分子中に2個以上(メチル)グリシジルエーテル基を有するオリゴマー/モノマーである。1分子中に2個以上の(メチル)グリシジルエーテル基を有することにより、紫外線、電子線等の活性エネルギー線により硬化し、耐擦傷性が良好なコート層を形成する。具体的には、例えば以下の化合物が挙げられる。
(メチル)グリシジルエーテル基を2個有する化合物としては、エチレングリコール(メチル)ジグリシジルエーテル、トリエチレングリコールジ(メチル)グリシジルエーテル、テトラエチレングリコールジ(メチル)グリシジルエーテル、ポリエチレングリコールジ(メチル)グリシジルエーテル、グリセリンジ(メチル)グリシジルエーテル、1,4-ブタンジオールジ(メチル)グリシジルエーテル、1,6-ヘキサンジオールジ(メチル)グリシジルエーテル、ネオペンチルグリコール(メチル)グリシジルエーテル等が挙げられる。さらにグリシジルオキシ基を3個以上有する化合物としてはグリセリントリ(メチル)グリシジルエーテル、トリメチロールプロパントリ(メチル)グリシジルエーテル、ペンタエリスリトールトリ(メチル)グリシジルエーテル、ペンタエリスリトールテトラ(メチル)グリシジルエーテル、ジペンタエリスリトールヘキサ(メチル)グリシジルエーテル、ジペンタエリスリトールペンタ(メチル)グリシジルエーテル、ジペンタエリスリトールテトラ(メチル)グリシジルエーテル、カルビトールポリグリシジルエーテル等が挙げられる。
紫外線や熱による硬化を促進させるため、光または熱重合開始剤を配合してもよい。
具体的には光または熱によりカチオン重合を開始する化合物であれば特に限定はなく、いずれでも使用することができる。
これらの光カチオン重合開始剤は、1種単独であるいは2種以上を組み合わせて使用することができる。添加量はポリ(メチル)グリシジルエーテル化合物100重量部に対して0.01~10.0重量部の範囲内が望ましい。
さらに、必要に応じて、光カチオン重合促進剤を併用することができる。具体的には、9,10-ジメトキシ-2-エチル-アントラセン、9,10-ジエトキシアントラセン、2,4-ジエチルチオキサントン等が挙げられる。
以上の開始剤は、単独で使用しても2種類以上組み合わせて使用しても構わない。また、光照射後に熱を併用しさらに硬化を進めることもできる。
熱硬化性基含有有機珪素化合物として具体的には、3-グリシドキシプロピルトリメトキシシラン、3-グリシドキシプロピルメチルジメトキシシラン、3-グリシドキシプロピルジメチルメトキシシラン、3-グリシドキシプロピルトリエトキシシラン、3-グリシドキシプロピルエチルジエトキシシラン、3-グリシドキシプロピルジエチルエトキシシラン、3-エチル-3-{[3-(トリメトキシシリル)プロポキシ]メチル}オキセタン、3-エチル-3-{[3-(トリエトキシシリル)プロポキシ]メチル}オキセタンなどが挙げられる。
熱による硬化性を促進させるため、好ましい金属キレート化合物は、Cu(II),Zn(II),Co(II),Ni(II),Be(II),Ce(III),Ta(III),Ti(III),Mn(III),La(III),Cr(III),V(III),Co(III),Fe(III),Al(III),Ce(IV),Zr(IV),V(IV)等を中心金属原子とするアセチルアセトネート、アミン、グリシン等のアミノ酸、ルイス酸、有機酸金属塩等が挙げられる。この中でも、硬化条件、塗液のポットライフなどにおいて、Al(III),Fe(III)のアセチルアセトネートがより好ましい。添加量は、熱硬化性官能基含有有機珪素化合物100重量部に対して0.01~10.0重量部の範囲内が望ましい。さらに過塩素酸類を併用して使用することも可能である。好ましい過塩素酸類としては、過塩素酸、過 塩素酸アンモニウム、過塩素酸マグネシウム等が挙げられる。
コート剤組成物には目的に応じて、これらの他に、紫外線吸収剤、酸化防止剤、シリコ-ン系界面活性剤、シリコーンオイルなどの各種添加剤を配合することができる。
コート剤の調製は、公知の方法で行えばよく、特に限定されるものではないが、例えば以下の通りである。まず所望量の成分を遮光性の褐色ガラス容器あるいはポリ容器中で混合、必要に応じてハードコート剤組成物を温め(概ね50℃以下)、完全に混合させる。さらに必要に応じその他成分を添加し充分に混合する。さらに充分静置脱気してハードコート剤組成物とした。混合はマグネチックスターラーや攪拌器を用いたが、量や粘度に応じて、ミキサー、シェーカーなどを選択すればよい。
溶剤を加える場合は、メチルアルコール、エチルアルコール、イソプロピルアルコール、ジメチルホルムアミド(DMF)、N,N′-ジメチルアセトアミド、N-メチル-2-ピロリドン、2-メトキシエタノール(メチルセロソルブ)、2-エトキシエタノール(エチルセロソルブ)、2-ブトキシエタノール(ブチルセロソルブ)、ポリエチレングリコールメチルエーテル(PEGME)、ポリエチレングリコールメチルエーテルアセテート(PEGMEA)、ダイアセトンアルコール(DAA)、エチレングリコール、テトラヒドロフラン、ジオキサン、トルエンなどが挙げられる。コーティング液の粘度は基材へのコーティングの方法により調整されるが、好ましくは、0.1cp~10000cpでありより好ましくは、0.5cp~500cpであり、さらに好ましくは1cp~100cpである。
基材へのコート方法としては、ディップ、スピンコート、スプレーなどの方法をとることができる。
硬化後のコート膜の特性は特に限定されるものではないが、屈折率がNaのD線(589.6nm)において1.45以下であることが好ましく、より好ましくは1.40以下である。
本実施形態の低屈折率コート材は、液晶ディスプレイ、CRTディスプレイ、プロジェクションディスプレイ、プラズマディスプレイ、エレクトロルミネッセンスディスプレイ、反射スクリーン等の画像表示装置、タッチパネル等の反射防止フィルム用コート剤、眼鏡レンズ等の反射防止コート等に用いることができる。
(末端分岐型ポリオレフィン系共重合体の合成例)
数平均分子量(Mn)、重量平均分子量(Mw)および分子量分布(Mw/Mn)はGPCを用い、本文中に記載した方法で測定した。また、融点(Tm)はDSC(示差走査熱量測定)を用い、測定して得られたピークトップ温度を採用した。なお、測定条件によりポリアルキレングリコール部分の融点も確認されるが、ここでは特に断りのない場合ポリオレフィン部分の融点のことを指す。1H-NMRについては、測定サンプル管中で重合体を、ロック溶媒と溶媒を兼ねた重水素化-1,1,2,2-テトラクロロエタンに完全に溶解させた後、120℃において測定した。ケミカルシフトは、重水素化-1,1,2,2-テトラクロロエタンのピークを5.92ppmとして、他のピークのケミカルシフト値を決定した。分散液中の粒子の粒子径はマイクロトラックUPA(HONEYWELL社製)にて、体積50%平均粒子径を測定した。分散液中の粒子の形状観察は、試料を200倍から500倍に希釈し、リンタングステン酸によりネガティブ染色した後、透過型電子顕微鏡(TEM/日立製作所製H-7650)で100kVの条件にて行なった。
以下の手順(例えば、特開2006-131870号公報の合成例2参照)に従って、末端エポキシ基含有エチレン重合体(E)を合成した。
充分に窒素置換した内容積2000mlのステンレス製オートクレーブに、室温でヘプタン1000mlを装入し、150℃に昇温した。続いてオートクレーブ内をエチレンで30kg/cm2G加圧し、温度を維持した。MMAO(東ソーファインケム社製)のヘキサン溶液(アルミニウム原子換算1.00mmol/ml)0.5ml(0.5mmol)を圧入し、次いで下記一般式(14)で示される化合物のトルエン溶液(0.0002mmol/ml)0.5ml(0.0001mmol)を圧入し、重合を開始した。エチレンガス雰囲気下、150℃で30分間重合を行った後、少量のメタノールを圧入することにより重合を停止した。得られたポリマー溶液を、少量の塩酸を含む3リットルのメタノール中に加えてポリマーを析出させた。メタノールで洗浄後、80℃にて10時間減圧乾燥し、片末端二重結合含有エチレン系重合体(P)を得た。
得られた末端エポキシ基含有エチレン重合体(E)は、Mw=2058、Mn=1118、Mw/Mn=1.84(GPC)であった(末端エポキシ基含有率:90mol%)。
1H-NMR:δ(C2D2Cl4)0.88(t,3H,J=6.92Hz),1.18-1.66(m),2.38(dd,1H,J=2.64,5.28Hz),2.66(dd,1H,J=4.29,5.28Hz),2.80-2.87(m,1H)
融点(Tm)121℃
1H-NMR:δ(C2D2Cl4)0.88(t,3H,J=6.6Hz),0.95-1.92(m),2.38-2.85(m,6H),3.54-3.71(m,5H)
融点(Tm)121℃
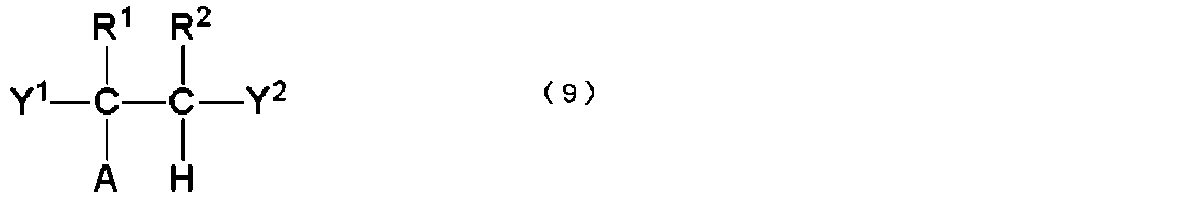
1H-NMR:δ(C2D2Cl4)0.88(3H,t,J=6.8Hz),1.06-1.50(m),2.80-3.20(m),3.33-3.72(m)
融点(Tm)-16℃(ポリエチレングリコール)、116℃
(20重量%末端分岐型ポリオレフィン系共重合体(T)水分散液の調製)
(A)前記合成例で得られた末端分岐型ポリオレフィン系共重合体(T)10重量部と溶媒(C)の蒸留水40重量部を100mlのオートクレーブに装入し、140℃、800rpmの速度で30分間加熱撹拌の後、撹拌を保ったまま室温まで冷却した。得られた分散系の体積50%平均粒子径は0.018μmであった(体積10%平均粒子径0.014μm、体積90%平均粒子径0.022μm)。得られた分散系の透過型電子顕微鏡観察結果から測定した粒子径は0.015-0.030μmであった。
(シリカ多孔質粒子の合成1)
エタノール/水(10mL/2.5mL)混合液に、1重量%に希釈した末端分岐型ポリオレフィン系共重合体(T)水分散液1mLおよび28%アンモニア水溶液0.4mLを加えて均一になるまで攪拌した。TEOS 20μLをマイクロピペットを用いて添加した。その後、室温で6時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が90nm、D90/D50が1.38であり、TEM観察から内部に10-20nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。窒素吸着法にて細孔構造を調べたところ、BET比表面積108m2/g、また、吸着側の等温線からBJH法にて求めた値(BJH細孔径)が13nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
なお、本実施例A項において、DLSの測定は粒度分布計/ナノトラックWAVEを用い水に分散させて行っている。また、多孔質粒子としてシリカを用い、分散溶媒として水を用いているので、シリカの屈折率を1.44、水の屈折率を1.0として測定している。
(シリカ多孔質粒子の合成2)
TEOSを12.5μLに変更し、攪拌時間を4時間にする以外は合成1と同様の方法によりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が70nm、D90/D50が1.32であり、TEM観察から内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。またBET比表面積105m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が14nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成3)
エタノール400mLに、15重量%末端分岐型ポリオレフィン系共重合体(T)水分散液20mLおよび28%アンモニア水溶液5mLを加えて均一になるまで攪拌した。TEOS3mLをマイクロピペットを用いて添加した。その後、室温で48時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を、減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が75nm、D90/D50が1.32であり、TEM観察から内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。またBET比表面積102m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が14nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成4)
エタノール150mLに、15重量%末端分岐型ポリオレフィン系共重合体(T)水分散液40mLおよび28%アンモニア水溶液3mLを加えて均一になるまで攪拌した。TEOS/エタノール(8.7mL/35mL)を一度に添加した。その後、室温で24時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が80nmで、D90/D50が1.30であり、TEM観察から内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。またBET比表面積105m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が14nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成5)
エタノール500mLに、15重量%末端分岐型ポリオレフィン系共重合体(T)水分散液72mLおよび28%アンモニア水溶液14.4mLを加えて均一になるまで攪拌した。TEOS/エタノール(36mL/144mL)、エチルトリエトキシシラン(Triethoxy(ethyl)silane)/エタノール(3.6mL/14.4mL)を一度に添加した。その後、室温で4時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が80nmで、D90/D50が1.32であり、TEM観察から内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることが分かった(図3)。またBET比表面積194m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が11nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成6)
50mLのフラスコに、エタノール10.6mL、脱イオン水1.8mL、15重量%に調整した末端分岐型ポリオレフィン系共重合体(T)水分散液0.8mLおよび28%アンモニア水溶液0.4mLを加えて15分間攪拌した。その後TEOS 1.1mLをエタノール4.4mLで希釈した溶液を加え、室温で24時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離(11000rpm、15分)にて分取し、エタノールを用いて3回洗浄した後、80℃で一晩乾燥させた。室温から600℃まで2時間で昇温し、さらに600℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が123nm、D90/D50が1.35であり、TEM観察から内部に10-20nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。窒素吸着法にて細孔構造を調べたところ、BET比表面積183m2/g、また、吸着側の等温線からBJH法にて求めた値(BJH細孔径)が15nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成7)
50mLのフラスコに、エタノール9.72mL、脱イオン水1.8mL、15重量%に調整した末端分岐型ポリオレフィン系共重合体(T)水分散液0.8mLおよび28%アンモニア水溶液0.4mLを加えて15分間攪拌した。その後TEOS 1.32mLをエタノール5.28mLで希釈した溶液を加え、室温で24時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離(11000rpm、15分)にて分取し、エタノールを用いて3回洗浄した後、80℃で一晩乾燥させた。室温から600℃まで2時間で昇温し、さらに600℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が142nm、D90/D50が1.41であり、TEM観察から内部に10-20nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。窒素吸着法にて細孔構造を調べたところ、BET比表面積153m2/g、また、吸着側の等温線からBJH法にて求めた値(BJH細孔径)が12nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成8)
実施例a6において、脱イオン水を2mL、15重量%に調整した末端分岐型ポリオレフィン系共重合体(T)水分散液0.6mLとした以外は実施例a6と同様の方法によりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が242nm、D90/D50が1.74であり、TEM観察から内部に10-20nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。窒素吸着法にて細孔構造を調べたところ、BET比表面積102m2/g、また、吸着側の等温線からBJH法にて求めた値(BJH細孔径)が14nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ多孔質粒子の合成9)
実施例a7において、脱イオン水を2mL、15重量%に調整した末端分岐型ポリオレフィン系共重合体(T)水分散液0.6mLとした以外は実施例a7と同様の方法によりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が251nm、D90/D50が1.36であり、TEM観察から内部に10-20nmの細孔を有するシリカ多孔質粒子が得られていることが分かった。窒素吸着法にて細孔構造を調べたところ、BET比表面積88m2/g、また、吸着側の等温線からBJH法にて求めた値(BJH細孔径)が13nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
(シリカ粒子の合成)
エタノール5mLに28%アンモニア水を0.1mLを加えて攪拌し、TEOS/エタノール(0.1mL/0.4mL)を加え4時間攪拌した。この混合物を乾燥させることにより、シリカ粒子を得た。
DLSによる測定から、得られた粒子の体積50%平均粒子径が150nmであり、D90/D50が1.2であるシリカ粒子が得られていることが分かった。なお、TEM観察において、このシリカ粒子の内部に細孔は確認できなかった(図4)。BET比表面積は20m2/gであった。
(シリカ多孔質粒子の合成10)
陽イオン界面活性剤CTAB(臭化セチルトリメチルアンモニウム)8.2mgをエタノール/水(10mL/2mL)に溶解し、28%アンモニウム水を0.2mL加え攪拌した。TEOS0.1mLを加え4時間攪拌した。得られたシリカ/CTAB複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、CTABを除去することによりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が300nm、D90/D50が1.3であるシリカ多孔質粒子が得られていることが分かった。また、本比較例a1においてはTEM観察で、この粒子内部の細孔径を判別することは難しかった。またBET比表面積32m2/g、吸着側、脱着側からBJH法で得られる細孔径は共に2nmであった。細孔は2次元シリンダー構造を有しているものと推定された。
(シリカ多孔質粒子の合成11)
CTABの量を10.2mgに変更する以外合成10と同様の方法によりシリカ多孔質粒子を得た。
DLSによる測定から、体積50%平均粒子径が410nm、D90/D50が1.3であるシリカ多孔質粒子が得られていることがわかった。また、TEMによる粒子の観察結果を図5に示す。BET比表面積64m2/g、吸着側、脱着側からBJH法で得られる細孔径は共に2nmであった。細孔は2次元シリンダー構造を有しているものと推定された。
(シリカ多孔質粒子の合成12)
CTABの量を20.5mgに変更する以外、合成10と同様の方法によりシリカ多孔質粒子を得た。TEM観察ではいずれも球状ではない不定形の粒子しか得られなかった(図6)。
(シリカ多孔質粒子水分散液の調製)
エタノール500mLに、15重量%末端分岐型ポリオレフィン系共重合体(T)水分散液72mLおよび28%アンモニア水溶液14.4mLを加えて均一になるまで攪拌した。TEOS/エタノール(36mL/150mL)、エチルトリエトキシシラン(Triethoxy(ethyl)silane)/エタノール(3.6mL/14.4mL)を一度に添加した。その後、室温で4時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を、減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて450℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。シリカ多孔質粒子の粉体10gを500mlの水に加え、ビーズミルを用いて分散処理を行った。分散処理後、沈殿のない均一な分散液が得られた。分散液の一部を乾燥させ、得られた粒子のTEM観察より、多孔質構造が保持されていることを確かめられた(図7、図8)。
DLSによる測定から、体積50%平均粒子径が80nmで、D90/D50が1.3であり、TEM観察により内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることがわかった。またBET比表面積235m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が11nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった(図9(a)(b))。
(シリカ多孔質粒子エタノール分散液の調製)
エタノール500mLに、15重量%末端分岐型ポリオレフィン系共重合体(T)水分散液72mLおよび28%アンモニア水溶液14.4mLを加えて均一になるまで攪拌した。TEOS/エタノール(36mL/150mL)、エチルトリエトキシシラン(Triethoxy(ethyl)silane)/エタノール(3.6mL/14.4mL)を一度に添加した。その後、室温で4時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を、減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて450℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。シリカ多孔質粒子の粉体10gを437mlのエタノールに加え、超音波(US)処理を30分行い、分散処理を行った。分散処理後、沈殿のない均一な分散液が得られた。分散液の一部を乾燥させ、得られた粒子のTEM観察より、多孔質構造が保持されていることを確かめられた。
DLSによる測定から、体積50%平均粒子径が80nmで、D90/D50が1.3であり、TEM観察により内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることがわかった。またBET比表面積235m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が11nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
テトラメトキシシラン(TMOS)10重量部に溶媒のメタノール15重量部を添加し、室温で攪拌した。さらに触媒の1N―塩酸水溶液1重量部を滴下した後、50℃で1時間攪拌し、TMOSの脱水縮合物を得た。
得られたTMOSの脱水縮合物に、1N―塩酸水溶液をさらに3.4g滴下した後(末端分岐型ポリオレフィン系共重合体添加後のpHを3とするため)、室温で攪拌し、さらに末端分岐型ポリオレフィン共重合体(T)の水性分散体(固形分10重量%)を72.4重量部滴下し、室温で攪拌し、末端分岐型ポリオレフィン共重合体/TMOS脱水縮合物溶液を調製した。この組成物をスプレードライヤー装置に流量6cc/minで流し込み、ノズル出口温度120℃で加圧(2.6kg/cm2)し、噴霧することで末端分岐型ポリオレフィン系共重合体/シリカの複合微粒子を得た。得られた粉体を減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。
シリカ多孔質粒子の粉体10gを500mlの水に加え、ビーズミルを用いて分散処理を行った。分散処理後、沈殿のない均一な分散液が得られた。分散液の一部を乾燥させ、得られた粒子のTEM観察より、多孔質構造が保持されていることを確かめられた。
DLSによる測定から、体積50%平均粒子径が3.8μmで、D90/D50が5.2であり、TEM観察から内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることわかった。またBET比表面積680m2/g、また、吸着側の等温線からBJH法にて得求めた値(BJH細孔径)が11nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
実施例a10で得られた水分散液を限外ろ過により2.9重量%まで濃縮した。この分散液35gにアクリル系樹脂水エマルションAlmatexA9083(三井化学社製 固形分濃度:50重量%)8gを混合し、シャーレに流し入れ70℃のオーブン中で乾燥し、厚さ50μmの均一かつ透明な膜を得た。HAZE値1.7、膜のD線(589nm)における屈折率は1.38であり、熱伝導率は0.04W/mKであった。
実施例a11で得られたエタノール分散液(2.9重量%)35gと予め10%に調整したPVB樹脂エタノール溶液(ポリビニルブチラール、重量平均分子量:50,000~80,000)を混合し、シャーレに流し入れ70℃のオーブン中で乾燥し、厚さ70μmの均一かつ透明な膜を得た。HAZE値0.4、膜のD線(589nm)における屈折率は1.33であり、熱伝導率は0.03W/mKであった。
分散液を用いなかった以外は、実施例a12と同様に膜を調製し、アクリル系樹脂水エマルションAlmatexA9083のみの膜を得た。この膜のHAZE値は0.3、膜のD線(589nm)における屈折率は1.47、熱伝導率は0.58W/mKであった。
比較例a1で得られたシリカ多孔質粒子を実施例a10と同様にビーズミルにより分散化処理し、得られた分散液を用いて実施例a12と同様の方法で膜を作成した。HAZE値は12であり、屈折率は測定不能であった。熱伝導率は0.88W/mKであった。
比較例a2で得られたシリカ多孔質粒子を実施例a10と同様にビーズミルにより分散化処理し、得られた分散液を用いて実施例a12と同様の方法で膜を作成した。HAZE値は20であり、屈折率は測定不能であった。熱伝導率は0.92W/mKであった。
比較例a4で得られたシリカ多孔質粒子分散液を用い、実施例a12と同様の方法で膜を作成した。HAZE値は36であり、屈折率は測定不能であった。熱伝導率は0.07W/mKであった。
分散液を用いなかった以外は、実施例a13と同様に膜を調製し、PVBのみの膜を得た。この膜のHAZE値は0.1、膜のD線(589nm)における屈折率は1.49、熱伝導率は0.22W/mKであった。
(末端分岐型ポリオレフィン系共重合体の合成例)
数平均分子量(Mn)、重量平均分子量(Mw)および分子量分布(Mw/Mn)はGPCを用い、本文中に記載した方法で測定した。また、融点(Tm)はDSC(示差走査熱量測定)を用い、測定して得られたピークトップ温度を採用した。なお、測定条件によりポリアルキレングリコール部分の融点も確認されるが、ここでは特に断りのない場合ポリオレフィン部分の融点のことを指す。1H-NMRについては、測定サンプル管中で重合体を、ロック溶媒と溶媒を兼ねた重水素化-1,1,2,2-テトラクロロエタンに完全に溶解させた後、120℃において測定した。ケミカルシフトは、重水素化-1,1,2,2-テトラクロロエタンのピークを5.92ppmとして、他のピークのケミカルシフト値を決定した。分散液中の粒子の粒子径はマイクロトラックUPA(HONEYWELL社製)にて、体積50%平均粒子径を測定した。分散液中の粒子の形状観察は、試料を200倍から500倍に希釈し、リンタングステン酸によりネガティブ染色した後、透過型電子顕微鏡(TEM/日立製作所製H-7650)で100kVの条件にて行なった。
以下の手順(例えば、特開2006-131870号公報の合成例2参照)に従って、末端エポキシ基含有エチレン重合体(E)を合成した。
充分に窒素置換した内容積2000mlのステンレス製オートクレーブに、室温でヘプタン1000mlを装入し、150℃に昇温した。続いてオートクレーブ内をエチレンで30kg/cm2G加圧し、温度を維持した。MMAO(東ソーファインケム社製)のヘキサン溶液(アルミニウム原子換算 1.00mmol/ml)0.5ml(0.5mmol)を圧入し、次いで下記一般式(14)で示される化合物のトルエン溶液(0.0002mmol/ml)0.5ml(0.0001mmol)を圧入し、重合を開始した。エチレンガス雰囲気下、150℃で30分間重合を行った後、少量のメタノールを圧入することにより重合を停止した。得られたポリマー溶液を、少量の塩酸を含む3リットルのメタノール中に加えてポリマーを析出させた。メタノールで洗浄後、80℃にて10時間減圧乾燥し、片末端二重結合含有エチレン系重合体(P)を得た。
得られた末端エポキシ基含有エチレン重合体(E)は、Mw=2058、Mn=1118、Mw/Mn=1.84(GPC)であった(末端エポキシ基含有率:90mol%)。
1H-NMR:δ(C2D2Cl4)0.88(t,3H,J=6.92Hz),1.18-1.66(m),2.38(dd,1H,J=2.64,5.28Hz),2.66(dd,1H,J=4.29,5.28Hz),2.80-2.87(m,1H)
融点(Tm)121℃
1H-NMR:δ(C2D2Cl4)0.88(t,3H,J=6.6Hz),0.95-1.92(m),2.38-2.85(m,6H),3.54-3.71(m,5H)
融点(Tm)121℃
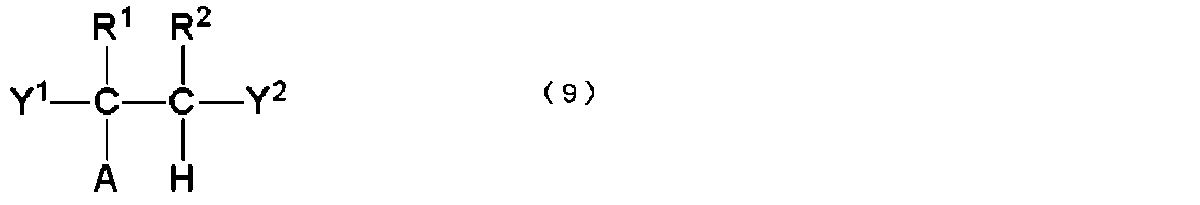
1H-NMR:δ(C2D2Cl4)0.88(3H,t,J=6.8Hz),1.06-1.50(m),2.80-3.20(m),3.33-3.72(m)
融点(Tm)-16℃(ポリエチレングリコール)、116℃
(20重量%末端分岐型ポリオレフィン系共重合体(T)水分散液の調製)
(A)前記合成例で得られた末端分岐型ポリオレフィン系共重合体(T)10重量部と溶媒(C)の蒸留水40重量部を100mlのオートクレーブに装入し、140℃、800rpmの速度で30分間加熱撹拌の後、撹拌を保ったまま室温まで冷却した。得られた分散系の体積50%平均粒子径は0.018μmであった(体積10%平均粒子径0.014μm、体積90%平均粒子径0.022μm)。得られた分散系の透過型電子顕微鏡観察結果から測定した粒子径は0.015-0.030μmであった。
(シリカ多孔質粒子ダイアセトンアルコール分散液の調製例b1)
エタノール500mLに、15重量%末端分岐型ポリオレフィン系共重合体(T)水分散液72mLおよび28%アンモニア水溶液14.4mLを加えて均一になるまで攪拌した。TEOS/エタノール(36mL/150mL)、エチルトリエトキシシラン(Triethoxy(ethyl)silane)/エタノール(3.6mL/14.4mL)を一度に添加した。その後、室温で4時間攪拌した。得られたシリカ/末端分岐型オレフィン共重合体複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を、減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて450℃まで昇温し、さらに550℃で4時間焼成し、末端分岐型オレフィン共重合体複合粒子を除去することによりシリカ多孔質粒子を得た。シリカ多孔質粒子の粉体10gを437mlのダイアセトンアルコールに加え、超音波(US)処理を30分行い、分散処理を行った。分散処理後、沈殿のない均一な分散液が得られた。分散液の一部を乾燥させ、得られた粒子のTEM観察より、多孔質構造が保持されていることを確かめた。
DLSによる測定から、体積50%平均粒子径が80nmで、D90/D50が1.3であり、TEM観察から内部に10-30nmの細孔を有するシリカ多孔質粒子が得られていることがわかった。またBET比表面積235m2/g、また、吸着側の等温線からBJH法にて求めた値(BJH細孔径)が11nm、脱着側の等温線からBJH法にて求めた値(連結部)が4nm以下であることから細孔が相互に連結した3次元キュービック相構造であることがわかった。
なお、本実施例B項においては、得られたシリカ多孔質粒子を水に分散させ、粒度分布計/ナノトラックWAVEを用いてDLS測定を行っている。また、多孔質粒子としてシリカを用い、分散溶媒として水を用いているので、シリカの屈折率を1.44、水の屈折率を1.0として測定している。
陽イオン界面活性剤CTAB(臭化セチルトリメチルアンモニウム)4.1gをエタノール/水(5L/1L)に溶解し、28%アンモニウム水を100mL加え攪拌した。TEOS 50mLを加え4時間攪拌した。得られたシリカ/CTAB複合粒子を遠心分離にて分取し、さらにエタノールを用いて洗浄処理した。得られた粉体を減圧乾燥機を用いて乾燥処理した。室温から3.5℃/minにて550℃まで昇温し、さらに550℃で4時間焼成し、CTABを除去することによりシリカ多孔質粒子を得た。シリカ多孔質粒子の粉体10gを437mlのダイアセトンアルコールに加え、超音波(US)処理を30分行い、分散処理を行った。分散処理後、沈殿のない均一な分散液が得られた。
DLSによる測定から、体積50%平均粒子径が300nm、D90/D50が1.3であるシリカ多孔質粒子が得られていることがわかった。また、TEM観察から内部の細孔径を判別することは難しかった。またBET比表面積32m2/g、吸着側、脱着側からBJH法で得られる細孔径は共に2nmであった。細孔は2次元シリンダー構造を有しているものと推定された。
10重量%まで濃縮した調製例b1のシリカ多孔質粒子ダイアセトンアルコール分散液を20g、トリメチロールプロパントリアクリレートを3.0g、ペンタエリスリトールトリアクリレートヘキサメチレンジイソシアネートウレタンプレポリマー(共栄社化学社製 商品名UA―306H)を1.0g混合後、ポリエチレングリコールメチルエーテル3gを加えた。さらに光開始剤として2,4,6-トリメチルベンゾイルージフェニルーフォスフィンオキサイドを0.15g、Si系界面活性剤(日本ユニカー(株)製 商品名FZ-2110)を0.01g添加し、充分攪拌しコーティング用組成物を調製した。調製したコーティング用組成物を用い、Si-ウェハー上、石英ガラス上にコートし、高圧水銀ランプ(強度100W/cm)を60秒間照射し、コート膜を形成した。評価結果を表1に載せた。
本実施形態のコート材の屈折率はSi-ウェハー上に形成した薄膜についてアタゴ社製アッベ屈折計により測定した。
(2)透明性
本実施形態の透明性は、石英ガラス上に形成した透過率を膜透過率計((株)島津製作所製 UV2200)により400nm~600nm間の透過率を測定した。
A:400nm~600nm間で透過率が90%以上
B:400nm~600nm間での透過率80%以上~90%未満
C:400nm~600nm間での透過率80%未満
(3)耐擦傷性試験
本実施形態の耐擦傷性は、石英ガラス上に形成したサンプルについて、♯0000のスチールウール(日本スチールウール(株)製)で1000g及び500gの荷重をかけ、10往復、表面を摩擦し、傷のついた程度を目視で次の段階で判断した。
A:500g荷重で摩擦した範囲に全く傷がつかない。
B:500g荷重で摩擦した範囲内に1~9本の傷がつく。
C:500g荷重で摩擦した範囲内に10~30本の傷がつく。
D:500g荷重で摩擦した範囲内に無数の(30本を超える)傷がつく。
10重量%まで濃縮した調製例b1のシリカ多孔質粒子ダイアセトンアルコール分散液を20g、1,6-ヘキサンジオールジグリシジルエーテルを1.0g、ペンタエリスリトールトリグリシジルエーテルを3.0g混合後、ポリエチレングリコールメチルエーテル3gを加えた。さらに光開始剤としてアデカオプトマ-SP-150を0.15g、Si系界面活性剤(日本ユニカー(株)製 商品名FZ-2110)を0.01g添加し、充分攪拌しコーティング用組成物を調製した。調製したコーティング用組成物を用い、Si-ウェハー上、石英ガラス上にコートし、高圧水銀ランプ(強度100W/cm)を60秒間照射し、コート膜を形成した。評価結果を表1に載せた。
10重量%まで濃縮した調製例b1のシリカ多孔質粒子ダイアセトンアルコール分散液を20g、シラン化合物および/またはそれが部分的に縮合した化合物として、3-グリシドキシプロピルトリメトキシシラン4.2gに0.1N塩酸水1.3gを攪拌しながら添加し、さらに2時間攪拌した。ポリエチレングリコールメチルエーテル7gを加え充分攪拌した後、金属キレート化合物として、Al(III)アセチルアセトネート0.2g、ジメチルシロキサン骨格を有する化合物として、ポリエーテル変性ポリジメチルシロキサンBYK333(BYKケミー製、製品名)0.01gを添加し4時間攪拌後、一昼夜熟成させてハードコート剤組成物を調製した。調製したコーティング用組成物を用い。Si-ウェハー上、石英ガラス上にコートし、120℃で120分間焼成を行い、コート膜を形成した。評価結果を表1に載せた。
調製例b1のシリカ多孔質粒子ダイアセトンアルコール分散液の代わりに、10重量%まで濃縮した調製例b2のシリカ多孔質粒子ダイアセトンアルコール分散液を20g加える以外実施例b3と同様の方法でコート膜を形成した。評価結果を表1に載せた。
なお、比較例b1によって得られたコート膜は、膜の透明性が低いため、正確に屈折率を測定することができなかった。
多孔質ではないシリカ粒子(日産化学社製 オルガノシリカゾル 70-100nm ZLタイプ)を用いる以外実施例b1と同様の方法でコート膜を形成した。評価結果を表1に載せた。
多孔質ではないシリカ粒子(日産化学社製 オルガノシリカゾル 70-100nm ZLタイプ)を用いる以外実施例b2と同様の方法でコート膜を形成した。評価結果を表1に載せた。
多孔質ではないシリカ粒子(日産化学社製 オルガノシリカゾル 70-100nm ZLタイプ)を用いる以外実施例b3と同様の方法でコート膜を形成した。評価結果を表1に載せた。
シリカ多孔質粒子を加えない以外実施例b1と同様の方法でコート膜を形成した。評価結果を表1に載せた。
シリカ多孔質粒子を加えない以外実施例b2と同様の方法でコート膜を形成した。評価結果を表1に載せた。
シリカ多孔質粒子を加えない以外実施例b3と同様の方法でコート膜を形成した。評価結果を表1に載せた。

Claims (22)
- 体積50%平均粒子径が50nm以上300nm以下、体積90%平均粒子径と体積50%平均粒子径との比(D90/D50)が2.0以下であり、BJH法による細孔径が5nm以上30nm以下のメソ孔を有し、その細孔構造が3次元キュービック相構造であることを特徴とする、金属酸化物多孔質粒子。
- 体積50%平均粒子径が50nm以上100nm以下、体積90%平均粒子径と体積50%平均粒子径との比(D90/D50)が1.5以下であり、BJH法による細孔径が5nm以上30nm以下のメソ孔を有し、その細孔構造が3次元キュービック相構造であることを特徴とする、請求項1に記載の金属酸化物多孔質粒子。
- 水および/または水の一部または全部を溶解する有機溶媒と、体積50%平均粒子径が5nm以上30nm以下である非水溶性ポリマー粒子と、塩基触媒とを含む、混合物を得る工程と、
前記混合物に、金属酸化物前駆体を混合し、該金属酸化物前駆体のゾル-ゲル反応を行って有機無機複合体粒子を得る工程と、
前記有機無機複合体粒子から前記非水溶性ポリマー粒子を除去する工程と、
を含む、請求項1又は2に記載の金属酸化物多孔質粒子の製造方法。 - 前記非水溶性ポリマー粒子が、下記一般式(1)で表され、数平均分子量が2.5×104以下である末端分岐型ポリオレフィン系共重合体からなる粒子である、請求項3に記載の金属酸化物多孔質粒子の製造方法。

- 前記一般式(1)で表される末端分岐型ポリオレフィン系共重合体のX1およびX2が、同一または相異なり、一般式(2)


または、一般式(4)

- 前記末端分岐型ポリオレフィン系共重合体が下記一般式(1a)または一般式(1b)で表されることを特徴とする請求項4又は5に金属酸化物多孔質粒子の製造方法。

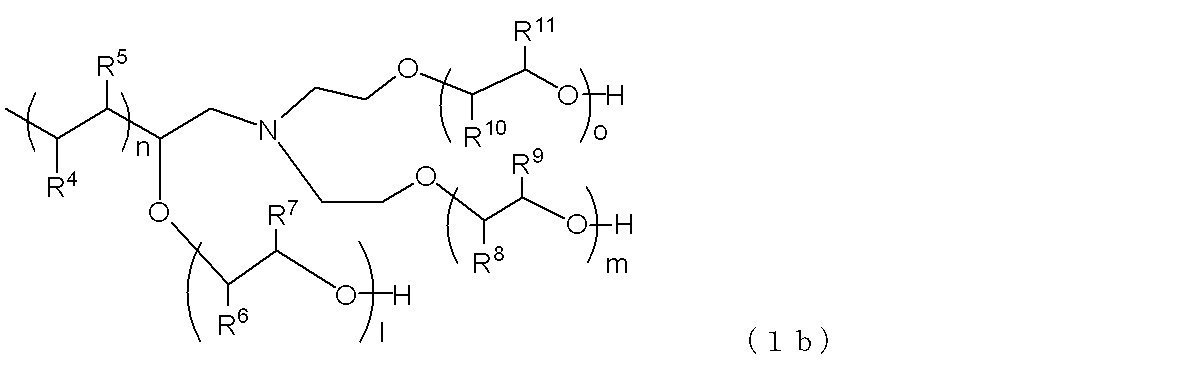
- 混合物を得る前記工程は、
前記水および/または水の一部または全部を溶解する前記有機溶媒と、前記非水溶性ポリマー粒子の水分散体と、前記塩基触媒とを混合する工程を含む、請求項3ないし6のいずれか1項に記載の金属酸化物多孔質粒子の製造方法。 - 有機無機複合体粒子を得る前記工程において、前記金属酸化物前駆体は予め水の一部または全部を溶解する有機溶媒で希釈された状態で混合される、請求項3ないし7のいずれか1項に記載の金属酸化物多孔質粒子の製造方法。
- 請求項1又は2に記載の金属酸化物多孔質粒子とバインダー樹脂とを含有する樹脂組成物。
- 請求項1又は2に記載の金属酸化物多孔質粒子を含有するフィルム。
- 請求項1又は2に記載の金属酸化物多孔質粒子を含む塗料。
- 請求項1又は2に記載の金属酸化物多孔質粒子を含む断熱材料。
- 請求項1又は2に記載の金属酸化物多孔質粒子を含む低誘電率材料。
- 請求項1又は2に記載の金属酸化物多孔質粒子を含むインク。
- メソ孔内に薬剤が内包された請求項1又は2に記載の金属酸化物多孔質粒子を含むドラッグデリバリーシステム(DDS)薬剤。
- 下記成分(A)及び成分(B)を含有するコート剤。
(A)請求項1又は2に記載の金属酸化物多孔質粒子。
(B)硬化性官能基含有化合物。 - 成分(B)が活性エネルギー線硬化性官能基含有化合物または熱硬化性官能基含有珪素化合物である請求項16に記載のコート剤。
- 成分(A)と(B)の合計100重量部に対する成分(A)割合が1重量部以上60重量部以下である請求項16または17に記載のコート剤。
- 請求項16ないし18のいずれか1項に記載のコート剤を硬化して得られるコート膜。
- 請求項19に記載のコート膜を表面部に有するフィルム。
- 請求項19に記載のコート膜を表面部に有するレンズ。
- 請求項19に記載のコート膜を表面部に有する画像表示装置。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14801195.0A EP3000785B1 (en) | 2013-05-22 | 2014-05-14 | Porous metal oxide particles, production method thereof and application thereof |
| JP2015518201A JP5964507B2 (ja) | 2013-05-22 | 2014-05-14 | 金属酸化物多孔質粒子、その製造方法、及びその用途 |
| SG11201509534VA SG11201509534VA (en) | 2013-05-22 | 2014-05-14 | Porous metal oxide particles, production method thereof andapplication thereof |
| US14/892,385 US10045942B2 (en) | 2013-05-22 | 2014-05-14 | Porous metal oxide particles, production method thereof and application thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-107963 | 2013-05-22 | ||
| JP2013107963 | 2013-05-22 | ||
| JP2013213548 | 2013-10-11 | ||
| JP2013-213548 | 2013-10-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014188924A1 true WO2014188924A1 (ja) | 2014-11-27 |
Family
ID=51933485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/062812 WO2014188924A1 (ja) | 2013-05-22 | 2014-05-14 | 金属酸化物多孔質粒子、その製造方法、及びその用途 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10045942B2 (ja) |
| EP (1) | EP3000785B1 (ja) |
| JP (1) | JP5964507B2 (ja) |
| SG (1) | SG11201509534VA (ja) |
| TW (1) | TWI633060B (ja) |
| WO (1) | WO2014188924A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017081789A (ja) * | 2015-10-29 | 2017-05-18 | 三井化学株式会社 | 球状メソポーラス金属酸化物粒子の製造方法 |
| JPWO2017026425A1 (ja) * | 2015-08-10 | 2018-07-12 | 国立大学法人京都大学 | シリカゲル製多孔性粒子およびその製造方法 |
| JP2020525375A (ja) * | 2017-06-02 | 2020-08-27 | アモーレパシフィック コーポレーションAmorepacific Corporation | 多孔性無機粒子の製造方法 |
| US10858491B2 (en) | 2015-08-10 | 2020-12-08 | Kyoto University | Porous particle made of organic polymer, method for producing porous particle made of organic polymer, and block copolymer |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101790553B1 (ko) * | 2014-05-30 | 2017-10-26 | (주)석경에이티 | 중공실리카 입자의 제조방법, 중공실리카 입자 및 그를 포함하는 조성물 및 단열 시트 |
| KR101757770B1 (ko) * | 2015-07-02 | 2017-07-13 | (주)석경에이티 | 기능성 원단 및 그 제조방법 |
| US10954393B2 (en) * | 2016-05-17 | 2021-03-23 | University Of South Carolina | Tunable nanomaterials by templating from kinetically trapped polymer micelles |
| KR101902618B1 (ko) * | 2016-05-31 | 2018-09-28 | 연세대학교 산학협력단 | 다공성 구조체 및 그 제조 방법 |
| US10428226B2 (en) | 2016-07-20 | 2019-10-01 | The Boeing Company | Sol-gel coating compositions and related processes |
| US10246594B2 (en) | 2016-07-20 | 2019-04-02 | The Boeing Company | Corrosion inhibitor-incorporated layered double hydroxide and sol-gel coating compositions and related processes |
| US10246593B2 (en) | 2016-07-20 | 2019-04-02 | The Boeing Company | Sol-gel coating compositions including corrosion inhibitor-encapsulated layered double hydroxide and related processes |
| US10421869B2 (en) | 2017-01-09 | 2019-09-24 | The Boeing Company | Sol-gel coating compositions including corrosion inhibitor-encapsulated layered metal phosphates and related processes |
| AU2018329173B2 (en) | 2017-09-11 | 2023-10-05 | Basf Se | Porous metal oxide microspheres |
| EP3681685A4 (en) | 2017-09-11 | 2021-06-16 | President And Fellows Of Harvard College | MICROBALLS WITH POLYDISPERSIC POLYMER NANOCBALLS AND POROUS METAL OXIDE MICROBALLS |
| TWI654269B (zh) | 2017-12-19 | 2019-03-21 | 財團法人工業技術研究院 | 黏著組合物 |
| MX2021000897A (es) * | 2018-07-26 | 2021-03-31 | Sumitomo Chemical Co | Composicion de resina. |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003335506A (ja) | 2002-05-16 | 2003-11-25 | Japan Science & Technology Corp | 3次元の高規則性を有するメソポーラス酸化物の合成法 |
| JP2004525846A (ja) * | 2000-11-14 | 2004-08-26 | セカ ソシエテ アノニム | メソ細孔無機固体と、その製造方法と、その触媒および吸着剤としての使用 |
| JP2005263550A (ja) | 2004-03-18 | 2005-09-29 | Nagoya Kogyo Univ | 高分散シリカナノ中空粒子及びそれを製造する方法 |
| JP2006131870A (ja) | 2004-01-30 | 2006-05-25 | Mitsui Chemicals Inc | ビシナル置換型官能基含有重合体及びその用途 |
| WO2007060884A1 (ja) | 2005-11-25 | 2007-05-31 | Catalysts & Chemicals Industries Co., Ltd. | 中空シリカ微粒子、それを含む透明被膜形成用組成物、および透明被膜付基材 |
| JP2008165040A (ja) | 2006-12-28 | 2008-07-17 | Dainippon Printing Co Ltd | ハードコート層用硬化性樹脂組成物、及びハードコートフィルム |
| JP2008280193A (ja) | 2007-05-08 | 2008-11-20 | Matsushita Electric Works Ltd | メソポーラスシリカ微粒子の製造方法、シリカ系被膜形成用塗布液、シリカ系被膜 |
| JP2009091211A (ja) * | 2007-10-10 | 2009-04-30 | Toyota Central R&D Labs Inc | 球状シリカ系多孔体及びその製造方法、並びに球状カーボン系多孔体 |
| WO2010103856A1 (ja) | 2009-03-12 | 2010-09-16 | 三井化学株式会社 | 新規な金属酸化物多孔質体、その製造方法および用途 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1479651B2 (en) | 2003-05-21 | 2017-07-19 | Toyota Jidosha Kabushiki Kaisha | Method for production of porous composite oxide |
| WO2012092361A2 (en) * | 2010-12-28 | 2012-07-05 | Saint-Gobain Ceramics & Plastics, Inc. | Polishing slurry including zirconia particles and a method of using the polishing slurry |
| JP5897303B2 (ja) | 2011-04-20 | 2016-03-30 | 三井化学株式会社 | 水性インク用顔料 |
-
2014
- 2014-05-14 WO PCT/JP2014/062812 patent/WO2014188924A1/ja active Application Filing
- 2014-05-14 JP JP2015518201A patent/JP5964507B2/ja active Active
- 2014-05-14 SG SG11201509534VA patent/SG11201509534VA/en unknown
- 2014-05-14 US US14/892,385 patent/US10045942B2/en active Active
- 2014-05-14 EP EP14801195.0A patent/EP3000785B1/en active Active
- 2014-05-19 TW TW103117513A patent/TWI633060B/zh active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004525846A (ja) * | 2000-11-14 | 2004-08-26 | セカ ソシエテ アノニム | メソ細孔無機固体と、その製造方法と、その触媒および吸着剤としての使用 |
| JP2003335506A (ja) | 2002-05-16 | 2003-11-25 | Japan Science & Technology Corp | 3次元の高規則性を有するメソポーラス酸化物の合成法 |
| JP2006131870A (ja) | 2004-01-30 | 2006-05-25 | Mitsui Chemicals Inc | ビシナル置換型官能基含有重合体及びその用途 |
| JP2005263550A (ja) | 2004-03-18 | 2005-09-29 | Nagoya Kogyo Univ | 高分散シリカナノ中空粒子及びそれを製造する方法 |
| WO2007060884A1 (ja) | 2005-11-25 | 2007-05-31 | Catalysts & Chemicals Industries Co., Ltd. | 中空シリカ微粒子、それを含む透明被膜形成用組成物、および透明被膜付基材 |
| JP2008165040A (ja) | 2006-12-28 | 2008-07-17 | Dainippon Printing Co Ltd | ハードコート層用硬化性樹脂組成物、及びハードコートフィルム |
| JP2008280193A (ja) | 2007-05-08 | 2008-11-20 | Matsushita Electric Works Ltd | メソポーラスシリカ微粒子の製造方法、シリカ系被膜形成用塗布液、シリカ系被膜 |
| JP2009091211A (ja) * | 2007-10-10 | 2009-04-30 | Toyota Central R&D Labs Inc | 球状シリカ系多孔体及びその製造方法、並びに球状カーボン系多孔体 |
| WO2010103856A1 (ja) | 2009-03-12 | 2010-09-16 | 三井化学株式会社 | 新規な金属酸化物多孔質体、その製造方法および用途 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3000785A4 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2017026425A1 (ja) * | 2015-08-10 | 2018-07-12 | 国立大学法人京都大学 | シリカゲル製多孔性粒子およびその製造方法 |
| US10858491B2 (en) | 2015-08-10 | 2020-12-08 | Kyoto University | Porous particle made of organic polymer, method for producing porous particle made of organic polymer, and block copolymer |
| JP2017081789A (ja) * | 2015-10-29 | 2017-05-18 | 三井化学株式会社 | 球状メソポーラス金属酸化物粒子の製造方法 |
| JP2020525375A (ja) * | 2017-06-02 | 2020-08-27 | アモーレパシフィック コーポレーションAmorepacific Corporation | 多孔性無機粒子の製造方法 |
| US11390530B2 (en) | 2017-06-02 | 2022-07-19 | Amorepacific Cornoration | Method for preparing porous inorganic particles |
| JP7110242B2 (ja) | 2017-06-02 | 2022-08-01 | アモーレパシフィック コーポレーション | 多孔性無機粒子の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3000785A4 (en) | 2017-01-18 |
| TW201512095A (zh) | 2015-04-01 |
| EP3000785B1 (en) | 2019-03-06 |
| SG11201509534VA (en) | 2015-12-30 |
| US20160089334A1 (en) | 2016-03-31 |
| US10045942B2 (en) | 2018-08-14 |
| EP3000785A1 (en) | 2016-03-30 |
| JP5964507B2 (ja) | 2016-08-03 |
| JPWO2014188924A1 (ja) | 2017-02-23 |
| TWI633060B (zh) | 2018-08-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5964507B2 (ja) | 金属酸化物多孔質粒子、その製造方法、及びその用途 | |
| JP5859390B2 (ja) | 断熱組成物の製造方法 | |
| WO2012032868A1 (ja) | 表面修飾チタニア粒子の製造方法、チタニア粒子分散液およびチタニア粒子分散樹脂 | |
| WO2019163918A1 (ja) | 高耐久防曇塗膜およびコーティング組成物 | |
| EP2752392A1 (en) | Inorganic oxide transparent dispersion and resin composition for forming transparent composite, and transparent composite and optical member | |
| CN107406690B (zh) | 硬涂层形成用树脂组合物及其固化物 | |
| KR20140048082A (ko) | 중합가능한 조성물, 이에 의해 수득된 경화물, 및 그 물질의 용도 | |
| JPWO2017159564A1 (ja) | 高耐久防曇塗膜およびコーティング組成物 | |
| JP6339889B2 (ja) | 金属酸化物中空粒子の製造方法 | |
| JP6637534B2 (ja) | 水系組成物、水系塗料、塗膜、及び塗装製品 | |
| JP2001281475A (ja) | 光導波路用有機・無機複合材料及びそれを用いた光導波路の製造方法 | |
| JP5514463B2 (ja) | 耐熱用コーティング組成物 | |
| JP7415230B2 (ja) | 反応性シリコーン組成物およびその硬化物 | |
| JP2021091859A (ja) | 親水性ポリマーを含む防曇剤 | |
| JP5305707B2 (ja) | 樹脂組成物及び光学部材 | |
| JP6509757B2 (ja) | 水系組成物、水系塗料、塗膜、及び塗装製品 | |
| EP2774942A1 (en) | Bivalent-trivalent metal organic-inorganic layered composite, and manufacturing method for same | |
| JP7328829B2 (ja) | コーティング組成物及び防曇塗膜 | |
| TWI824122B (zh) | 混合組成物 | |
| JP6929701B2 (ja) | 水性組成物、水性塗料、塗膜、及び塗装製品 | |
| JP5804788B2 (ja) | 多層反射防止膜の製造方法 | |
| TW202302653A (zh) | 中空粒子及其用途 | |
| JP6239763B2 (ja) | 新規な末端分岐型ポリオレフィン系重合体およびその用途 | |
| JP2020097698A (ja) | 水系組成物、水系塗料、塗膜、複合塗膜、及び塗装製品 | |
| JP2010235683A (ja) | アンチグレアコーティング組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14801195 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14892385 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2015518201 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014801195 Country of ref document: EP |