WO2014178270A1 - キシリレンジアミン組成物及びポリアミド樹脂の製造方法 - Google Patents
キシリレンジアミン組成物及びポリアミド樹脂の製造方法 Download PDFInfo
- Publication number
- WO2014178270A1 WO2014178270A1 PCT/JP2014/060346 JP2014060346W WO2014178270A1 WO 2014178270 A1 WO2014178270 A1 WO 2014178270A1 JP 2014060346 W JP2014060346 W JP 2014060346W WO 2014178270 A1 WO2014178270 A1 WO 2014178270A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- xylylenediamine
- polyamide resin
- bis
- methylbenzyl
- amine
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/17—Amines; Quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/5033—Amines aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
- C08G69/265—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids from at least two different diamines or at least two different dicarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
- C08G69/28—Preparatory processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
- C08G69/28—Preparatory processes
- C08G69/30—Solid state polycondensation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/06—Polyamides derived from polyamines and polycarboxylic acids
Definitions
- the present invention relates to a method for producing a xylylenediamine composition and a polyamide resin.
- Xylylenediamine is a compound useful as a raw material for polyamide resin raw materials, epoxy resin curing agents, and isocyanate compounds. However, it is known that xylylenediamine is altered by light, heat, oxygen, and the like and easily colored.
- Polyamide resins using xylylenediamine as the main diamine component are useful as molding materials containing glass fibers and inorganic fillers because they are excellent in mechanical properties such as strength and elastic modulus. In addition, it is also useful as a packaging material because it has excellent barrier properties against gases such as oxygen and carbon dioxide.
- the polyamide resin has a problem that it tends to be colored yellow when it is exposed to a high temperature atmosphere in a molten state or a solid state, and its use may be restricted depending on applications.
- Patent Document 1 discloses that the purity of xylylenediamine obtained by polycondensation of a diamine containing xylylenediamine and a specific aliphatic dicarboxylic acid is 99.99. There is disclosed a polyamide resin that is 9% by weight or more and has a difference in yellowness (YI value) before and after heating within 5 when heated under predetermined conditions.
- Patent Document 2 In order to improve the workability of the polyamide resin, it is known to add a crystal nucleating agent to the polyamide resin to improve the crystallization speed (Patent Document 2). However, depending on the type and amount of the crystal nucleating agent used, the mechanical properties of the polyamide resin may be reduced.
- the present invention can be used as a raw material for a polyamide resin, an epoxy resin curing agent, an isocyanate compound, and the like.
- a raw material for a polyamide resin it can obtain a polyamide resin with little coloring and quick crystallization.
- An object is to provide a xylylenediamine composition that can be produced.
- Another object of the present invention is to provide a method for producing a polyamide resin that uses xylylenediamine as a diamine component and has little coloring and quick crystallization.
- the present inventors can obtain a polyamide resin with less coloring when used as a raw material for a polyamide resin by containing a specific amount of a specific compound in xylylenediamine. It has been found that the crystallization rate of can be improved. The present inventors have also found that a polyamide resin with little coloring and quick crystallization can be produced by reacting a xylylenediamine-containing diamine with a dicarboxylic acid under specific conditions.
- the present invention is a xylylenediamine composition containing [1] xylylenediamine and bis (methylbenzyl) amine, wherein the content of bis (methylbenzyl) amine is 0 with respect to 100 parts by mass of xylylenediamine.
- a method for producing a polyamide resin which comprises a step of introducing a mass part of bis (methylbenzyl) amine into a reaction system and performing a polycondensation reaction.
- the xylylenediamine composition of the present invention is used as a raw material for a polyamide resin, it is possible to obtain a polyamide resin with little coloring and quick crystallization.
- the xylylenediamine composition of the present invention is also suitable for various uses such as an epoxy resin curing agent, or as a raw material for isocyanate compounds.
- the polyamide resin obtained by the production method of the present invention is less colored, it is suitably used as a material for packaging films, hollow containers, various molding materials, fibers and the like.
- the polyamide resin obtained by the production method of the present invention is rapidly crystallized and excellent in transparency and moldability.
- the xylylenediamine composition of the present invention (hereinafter also simply referred to as “the composition of the present invention” or “composition”) contains xylylenediamine and bis (methylbenzyl) amine, and bis (methylbenzyl).
- the amine content is 0.0005 to 0.1 parts by mass with respect to 100 parts by mass of xylylenediamine.
- the xylylenediamine used in the present invention is preferably metaxylylenediamine, paraxylylenediamine or a mixture thereof, and more preferably metaxylylenediamine from the viewpoint of gas barrier properties of the resulting polyamide resin.
- a mixture of metaxylylenediamine and paraxylylenediamine is more preferable from the viewpoint that when the material for injection molding is used, the molding cycle is fast and the strength and appearance of the molded product are improved.
- the composition of the present invention is mainly composed of xylylenediamine, and the content of xylylenediamine in the composition is preferably 99.5% by mass or more, and 99.9% by mass or more. More preferably.
- the xylylenediamine content in the composition can be measured, for example, by gas chromatography (GC) analysis.
- xylylenediamine used in the present invention
- industrially available xylylenediamine can be preferably used.
- Such xylylenediamine may contain a trace amount of impurities, but there is no particular problem in the present invention.
- Industrially available xylylenediamine can be produced using known methods. For example, when xylylenediamine is metaxylylenediamine, the production method thereof includes reacting metaxylene, ammonia and an oxygen-containing gas in a continuous reaction or batch reaction in the presence of a catalyst, and producing the produced isophthalonitrile. Examples include a hydrogenation method.
- the composition of the present invention further contains bis (methylbenzyl) amine.
- the content of bis (methylbenzyl) amine is 0.0005 to 0.1 parts by mass, preferably 0.0005 to 0.08 parts by mass, more preferably 0.0005 to 0.1 parts by mass with respect to 100 parts by mass of xylylenediamine. 0.04 parts by mass, more preferably 0.0005 to 0.02 parts by mass, still more preferably 0.001 to 0.015 parts by mass, still more preferably 0.001 to 0.01 parts by mass, and still more preferably. Is 0.001 to 0.005 parts by mass.
- the resulting polyamide resin is less colored and has a fast crystallization.
- the molding processability is improved as the crystallization speed of the polyamide resin is improved, the crystallization process time at the time of molding can be shortened, and the productivity of the molded product can be improved.
- problems such as deterioration in mechanical properties and transparency caused by adding a crystal nucleating agent to improve the molding processability of polyamide resin and the like can be avoided. Can do. Further, a polyamide resin capable of easily producing a molded product with little coloring is obtained.
- bis (methylbenzyl) amine is a compound of diamine and dicarboxylic acid. It has an effect of scavenging radicals generated from the polyamide resin obtained by the condensation reaction, and is considered to be due to suppressing degradation of the polyamide resin by the radicals.
- bis (methylbenzyl) amine promotes crystal nucleation in polyamide resin, or bis (methylbenzyl) amine itself is the starting point for crystal nucleation. It is thought that.
- xylylenediamine may produce a small amount of ammonia due to degradation of the amino group in the molecule during storage, but it has also been found that the amount of ammonia generated decreases due to the presence of bis (methylbenzyl) amine. It was done. From this, the effect that the storage stability of xylylenediamine is improved can also be expected when the xylylenediamine composition contains bis (methylbenzyl) amine. The reason for the decrease in the amount of ammonia generated is not clear, but it is presumed that some interaction between xylylenediamine and bis (methylbenzyl) amine has the effect of preventing the generation of radicals and their chaining.
- the xylylenediamine composition of the present invention can be obtained using a commercially available xylylenediamine and bis (methylbenzyl) amine and adjusting the amount of bis (methylbenzyl) amine with respect to the xylylenediamine within a predetermined range. . Also, in the production of xylylenediamine, the catalyst to be used and the production conditions are specified, and if it is possible to carry out a reaction that produces a predetermined amount of bis (methylbenzyl) amine in parallel, it is utilized. And the like. In this case, the content of bis (methylbenzyl) amine in the composition can be determined by gas chromatography analysis or the like.
- GC measurement of a xylylenediamine composition containing bis (methylbenzyl) amine is performed, and the ratio of the peak value derived from xylylenediamine to the peak value derived from bis (methylbenzyl) amine is determined to be bis ( And a method for determining the amount of methylbenzyl) amine.
- the xylylenediamine composition of the present invention can be suitably used as a raw material for polyamide resins, an epoxy resin curing agent, a raw material for isocyanate compounds, and the like.
- a polyamide resin with little coloring and quick crystallization can be produced.
- a diamine component containing the xylylenediamine composition of the present invention and a dicarboxylic acid component are introduced into a reaction system, and a known method is used.
- a polyamide resin can be produced by performing a polycondensation reaction.
- the xylylenediamine composition of the present invention When the xylylenediamine composition of the present invention is used as an epoxy resin curing agent, the xylylenediamine composition of the present invention may be used as a curing agent as it is, and the xylylenediamine composition of the present invention and a carboxylic acid or A reaction product obtained by reacting a carbonyl group-containing compound such as a derivative thereof with a known method may be used as an epoxy resin curing agent.
- the carboxylic acid derivatives include carboxylic acid anhydrides and acid chlorides.
- the xylylenediamine composition of the present invention is also suitable as a raw material for isocyanate compounds.
- the isocyanate compound is used as a raw material for urethane resins and urea resins.
- the method for producing a polyamide resin of the present invention comprises a diamine containing xylylenediamine, a dicarboxylic acid, and 0.0005 to 0.1 parts by mass of bis (methylbenzyl) amine with respect to 100 parts by mass of the xylylenediamine. It has the process of introduce
- the diamine used in the present invention is a diamine containing xylylenediamine (hereinafter also simply referred to as “diamine”).
- the xylylenediamine is preferably metaxylylenediamine, paraxylylenediamine or a mixture thereof, and more preferably metaxylylenediamine from the viewpoint of gas barrier properties of the resulting polyamide resin.
- the resulting polyamide resin has excellent melt moldability, mechanical properties, and gas barrier properties.
- the content of xylylenediamine in the diamine is preferably 70 mol% or more, more preferably 80 to 100 mol%, still more preferably 90 to 100 mol%.
- the obtained polyamide resin has excellent melt moldability, mechanical properties, and gas barrier properties.
- diamine compounds other than xylylenediamine contained in the diamine include tetramethylenediamine, pentamethylenediamine, 2-methylpentanediamine, hexamethylenediamine, heptamethylenediamine, octamethylenediamine, nonamethylenediamine, decamethylenediamine, dodecadiamine.
- Aliphatic diamines such as methylenediamine, 2,2,4-trimethyl-hexamethylenediamine, 2,4,4-trimethylhexamethylenediamine; 1,3-bis (aminomethyl) cyclohexane, 1,4-bis (aminomethyl) ) Cyclohexane, 1,3-diaminocyclohexane, 1,4-diaminocyclohexane, bis (4-aminocyclohexyl) methane, 2,2-bis (4-aminocyclohexyl) propane, bis (aminomethyl) decal
- alicyclic diamines such as bis (aminomethyl) tricyclodecane; diamines having an aromatic ring such as bis (4-aminophenyl) ether, paraphenylenediamine, and bis (aminomethyl) naphthalene. However, it is not limited to these. These diamines can be used alone or in combination of two or more.
- the dicarboxylic acid used in the present invention is not particularly limited, but at least one selected from aliphatic dicarboxylic acids having 4 to 20 carbon atoms, terephthalic acid and isophthalic acid from the viewpoints of moldability, gas barrier properties, and mechanical properties. It is preferably a seed, more preferably an aliphatic dicarboxylic acid having 4 to 20 carbon atoms, and still more preferably an aliphatic dicarboxylic acid having 4 to 12 carbon atoms.
- Examples of the aliphatic dicarboxylic acid having 4 to 20 carbon atoms include succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, 1,10-decanedicarboxylic acid, 1,11-undecanedicarboxylic acid, Examples thereof include 1,12-dodecanedicarboxylic acid, 1,14-tetradecanedicarboxylic acid, 1,16-hexadecanedicarboxylic acid, 1,18-octadecanedicarboxylic acid, and among these, adipic acid is preferred from the viewpoint of crystallinity and high elasticity. And at least one selected from sebacic acid is preferably used. These dicarboxylic acids may be used alone or in combination of two or more.
- the content of the aliphatic dicarboxylic acid having 4 to 20 carbon atoms in the dicarboxylic acid is preferably 50 mol% or more, more preferably 70 to 100 mol%, still more preferably 85 to 100 mol%.
- the obtained polyamide resin is excellent in molding processability, gas barrier property, and mechanical properties.
- the method for producing a polyamide resin of the present invention includes a step of introducing a diamine containing xylylenediamine, a dicarboxylic acid, and a predetermined amount of bis (methylbenzyl) amine into a reaction system to perform a polycondensation reaction.
- a diamine containing xylylenediamine, a dicarboxylic acid, and a predetermined amount of bis (methylbenzyl) amine into a reaction system to perform a polycondensation reaction.
- the amount of bis (methylbenzyl) amine introduced into the reaction system is 0.0005 to 0.1 parts by mass, preferably 0.0005 with respect to 100 parts by mass of xylylenediamine in the diamine.
- the amount of bis (methylbenzyl) amine introduced into the reaction system is less than 0.0005 parts by mass or more than 0.1 parts by mass with respect to 100 parts by mass of xylylenediamine in the diamine, the YI of the polyamide resin The value increases. In addition, the crystallization rate decreases, and as a result, the processability of the polyamide resin becomes low.
- the polycondensation reaction of diamine and dicarboxylic acid is not particularly limited, and any method such as a pressurization method or an atmospheric pressure dropping method can be used.
- a method of performing melt polycondensation Specifically, a method comprising heating a salt comprising a diamine and a dicarboxylic acid in the presence of water at normal pressure or under pressure, and polycondensing in a molten state while removing added water and water produced by polycondensation. Is mentioned.
- the method of adding diamine directly to molten dicarboxylic acid and performing polycondensation under a normal pressure or pressurization is also mentioned.
- diamine is continuously added to the dicarboxylic acid, while the reaction system is heated up so that the reaction temperature does not fall below the melting point of the generated oligoamide and polyamide.
- the polycondensation proceeds.
- the method for introducing bis (methylbenzyl) amine into the reaction system includes a method of directly introducing bis (methylbenzyl) amine into the polycondensation reaction system and a method of introducing a mixture of raw material diamine or dicarboxylic acid and bis (methylbenzyl) amine into the reaction system.
- a method of directly introducing bis (methylbenzyl) amine into the polycondensation reaction system and a method of introducing a mixture of raw material diamine or dicarboxylic acid and bis (methylbenzyl) amine into the reaction system.
- xylylenediamine used in the present invention if the catalyst to be used and the production conditions have a specific configuration and a reaction that produces a predetermined amount of bis (methylbenzyl) amine can be performed in parallel, The method of using it etc. is mentioned.
- the content of bis (methylbenzyl) amine in xylylenediamine can be determined by gas chromatography (GC) analysis or the like.
- the molar ratio of diamine to dicarboxylic acid is preferably in the range of 0.9 to 1.1, more preferably in the range of 0.93 to 1.07, and still more preferably in the range of 0.95 to 1. It is in the range of 05, more preferably in the range of 0.97 to 1.02. If the molar ratio is within the above range, high molecular weight tends to proceed.
- a phosphorus atom-containing compound may be added to the polycondensation reaction system.
- the phosphorus atom-containing compound include phosphinic acid compounds such as dimethylphosphinic acid and phenylmethylphosphinic acid; hypophosphorous acid, sodium hypophosphite, potassium hypophosphite, lithium hypophosphite, magnesium hypophosphite, Diphosphite compounds such as calcium hypophosphite and ethyl hypophosphite; phosphonic acid, sodium phosphonate, potassium phosphonate, lithium phosphonate, potassium phosphonate, magnesium phosphonate, calcium phosphonate, phenylphosphonic acid, ethylphosphone Phosphonic acid compounds such as acid, sodium phenylphosphonate, potassium phenylphosphonate, lithium phenylphosphonate, diethyl phenylphosphonate, sodium ethylphosphonate, potassium
- Phosphonic acid compounds Phosphonic acid compounds; phosphorous acid, sodium hydrogen phosphite, sodium phosphite, lithium phosphite, potassium phosphite, magnesium phosphite, calcium phosphite, triethyl phosphite, triphenyl phosphite, pyro-subite
- phosphorous acid compounds such as phosphoric acid.
- metal hypophosphites such as sodium hypophosphite, potassium hypophosphite and lithium hypophosphite are particularly preferably used for promoting the amidation reaction, and sodium hypophosphite is particularly preferred. preferable.
- the phosphorus atom containing compound which can be used by this invention is not limited to these compounds.
- the addition amount of the phosphorus atom-containing compound added to the polycondensation reaction system is preferably 0.1 to 1,000 ppm, more preferably 1 to 600 ppm, still more preferably in terms of the phosphorus atom concentration in the polyamide resin. 5 to 400 ppm.
- an alkali metal compound may be allowed to coexist in the polycondensation reaction system.
- alkali metal compound alkali metal hydroxides and alkali metal acetates are usually used.
- the above phosphorus atom-containing compound containing an alkali metal is excluded. Examples include lithium hydroxide, sodium hydroxide, potassium hydroxide, rubidium hydroxide, cesium hydroxide, lithium acetate, sodium acetate, potassium acetate, rubidium acetate, cesium acetate, etc., selected from sodium hydroxide and sodium acetate At least one is preferred. These can be used alone or in combination of two or more.
- the said alkali metal compound may be added in a polycondensation reaction system, and may originate from the dicarboxylic acid which is a raw material of a polyamide resin.
- the amount of the alkali metal compound used is preferably 1 to 500 ppm, more preferably 5 to 300 ppm, still more preferably 6 to 250 ppm, and still more preferably 10 to 200 ppm in terms of alkali metal atom concentration in the polyamide resin.
- the amount used is the total amount of the alkali metal compound added in the polycondensation system and the alkali metal compound derived from dicarboxylic acid which is a raw material of the polyamide resin.
- the amount of alkali metal compound used is such that the value obtained by dividing the number of moles of the alkali metal compound by the number of moles of the aforementioned phosphorus atom-containing compound is usually in the range of 0.5 to 1.0, preferably The amount is in the range of 0.55 to 0.95, more preferably 0.6 to 0.9. Within the above range, the amidation reaction proceeds at an appropriate rate.
- the phosphorus atom concentration and sodium atom concentration in the polyamide resin can be measured by known methods such as ICP emission spectroscopic analysis, ICP mass spectrometry, and X-ray photoelectron spectroscopic analysis.
- the temperature of the polycondensation reaction is preferably 150 to 300 ° C, more preferably 160 to 280 ° C, and still more preferably 170 to 270 ° C.
- the polymerization temperature is within the above range, the polymerization reaction proceeds rapidly.
- the thermal decomposition of monomers, oligomers in the middle of polymerization, polymers and the like hardly occurs, the properties of the resulting polyamide resin are good.
- the time for the polycondensation reaction is usually 1 to 5 hours after the start of dropwise addition of the diamine.
- the polyamide resin obtained as described above is taken out from the polymerization tank, pelletized, and then dried and crystallized as necessary.
- the production method of the present invention may further include a step of performing solid phase polymerization.
- Solid phase polymerization can be carried out by a known method, for example, a method of heating at a temperature of 100 ° C. or higher and lower than the melting point of polyamide for 1 to 24 hours in a nitrogen atmosphere.
- a heating device used in drying or solid phase polymerization a continuous heating drying device, a tumble dryer, a conical dryer, a rotary drum type heating device called a rotary dryer, etc., and a rotary blade inside a nauta mixer
- a continuous heating drying device a tumble dryer, a conical dryer, a rotary drum type heating device called a rotary dryer, etc.
- a rotary blade inside a nauta mixer a heating device used in drying or solid phase polymerization
- the present invention is not limited thereto, and a known device can be used.
- the relative viscosity of the polyamide resin produced as described above is preferably in the range of 1.0 to 5.0, more preferably in the range of 1.5 to 4.0, from the viewpoint of moldability and mechanical properties. is there. Specifically, the relative viscosity of the polyamide resin can be measured by the method described in Examples.
- the number average molecular weight (Mn) of the polyamide resin obtained by the production method of the present invention is preferably 10,000 to 50,000, more preferably 12,000 to 40,000 from the viewpoint of melt moldability and mechanical properties. Range.
- the number average molecular weight of a polyamide resin can be specifically measured by the method as described in an Example.
- the polyamide resin obtained by the production method of the present invention is less colored compared to the case where the amount of bis (methylbenzyl) amine introduced into the reaction system is outside the range specified in the present application.
- the YI value of the polyamide resin measured according to JIS K7373 is preferably in the range of ⁇ 20 to 5, more preferably ⁇ 20 to 2.
- the YI value of the polyamide resin can be specifically measured by the method described in the examples.
- the polyamide resin obtained by the production method of the present invention has a faster crystallization compared to the case where the amount of bis (methylbenzyl) amine introduced into the reaction system is outside the range specified in the present application. For this reason, the molding processability of the polyamide resin is improved, so that the crystallization process time at the time of molding can be shortened, that is, the molding cycle becomes faster and the productivity of the molded product can be improved. Moreover, problems such as a decrease in transparency and a decrease in mechanical properties due to the addition of a crystal nucleating agent can be avoided. Accordingly, the haze value when the polyamide resin is a film having a thickness of 100 ⁇ m is preferably 10% or less, more preferably 5% or less. The haze value can be measured using, for example, a haze value measuring apparatus (manufactured by Nippon Denshoku Industries Co., Ltd., model: COH-300A), and specifically can be measured by the method described in the examples.
- a haze value measuring apparatus
- the crystallization speed of the polyamide resin can be evaluated by measuring the half crystallization time.
- the half crystallization time represents the time required for crystallization to progress by half when a certain crystalline material shifts from the molten state to the crystallized state. Can be said to have a high crystallization rate.
- the semi-crystallization time of the obtained polyamide resin can be preferably 100 seconds or less, more preferably 90 seconds or less, and still more preferably 85 seconds or less.
- the half crystallization time can be measured by the method described in Examples.
- the polyamide resin has a matting agent, a heat stabilizer, a weathering agent, an ultraviolet absorber, a plasticizer, a flame retardant, an antistatic agent, an anti-coloring agent, an anti-gelling agent, etc., as long as the properties are not impaired.
- An additive can be mix
- the polyamide resin obtained by the production method of the present invention can be molded into various forms by a conventionally known molding method.
- the molding method include injection molding, blow molding, extrusion molding, compression molding, vacuum molding, press molding, direct blow molding, rotational molding, sandwich molding, and two-color molding. Since the polyamide resin obtained by the production method of the present invention has a high crystallization speed, the crystallization process time at the time of molding can be shortened, that is, the molding cycle becomes faster and the productivity can be improved.
- Molded products containing the polyamide resin are suitable as packaging films, hollow containers, various molding materials, fibers and the like. Further, since the molded product is less colored and the transparency is not impaired, it is suitable for a packaging film, a hollow container or the like that requires particularly high transparency.
- ⁇ Relative viscosity> 0.2 g of the polyamide resin obtained in Examples and Comparative Examples was precisely weighed and dissolved in 20 ml of 96% sulfuric acid at 20-30 ° C. with stirring. After complete dissolution, 5 ml of the solution was quickly taken into a Cannon Fenceke viscometer, and left for 10 minutes in a thermostatic bath at 25 ° C., and then the drop time (t) was measured. Further, the dropping time (t 0 ) of 96% sulfuric acid itself was measured in the same manner. The relative viscosity was calculated from t and t 0 according to the following formula. Relative viscosity t / t 0
- ⁇ Number average molecular weight (Mn)> The number average molecular weights of the polyamide resins obtained in Examples and Comparative Examples are as follows. First, the sample was dissolved in a phenol / ethanol mixed solvent and a benzyl alcohol solvent, and the carboxyl end group concentration and amino end group concentration were adjusted to hydrochloric acid and sodium hydroxide. It was determined by neutralization titration of the aqueous solution. The number average molecular weight was determined by the following formula from the quantitative values of the amino end group concentration and the carboxyl end group concentration.
- the YI value was measured according to JIS K7373. Using the polyamide resin pellets obtained in the examples and comparative examples, the YI value was measured using a color difference measuring device (manufactured by Nippon Denshoku Industries Co., Ltd., model: Z- ⁇ 80 Color Measuring System).
- ⁇ Haze value> The polyamide resin pellets obtained in the examples and comparative examples were dried, and the dried pellets were extruded using a single screw extruder under the condition of melting point + 20 ° C. to produce a film having a thickness of 100 ⁇ m.
- the haze value was measured by a transmission method using a haze value measuring apparatus (manufactured by Nippon Denshoku Industries Co., Ltd., model: COH-300A).
- ⁇ Semi-crystallization time> Using the polyamide resin pellets obtained in the examples and comparative examples, a film having a thickness of 100 ⁇ m was prepared in the same manner as described above, immediately after being held for 3 minutes at the melting point of the polyamide resin + 30 ° C. after being sandwiched between cover glasses. And cooled in an oil bath at 160 ° C. Using a crystallization rate measuring apparatus (manufactured by Kotaki Seisakusho, model: MK701), the half crystallization time was measured by the depolarization intensity method.
- metaxylylenediamine (MXDA) and paraxylylenediamine (PXDA) manufactured by Tokyo Chemical Industry Co., Ltd. were used.
- As bis (3-methylbenzyl) amine bis (3-methylbenzyl) amine produced in Production Example 1 below was used.
- Production Example 1 (Production of bis (3-methylbenzyl) amine) A 1 L autoclave was charged with 511.5 g of 3-methylbenzonitrile and 75 g of 0.5% Pd / Al 2 O 3 catalyst (manufactured by Clariant Catalyst Co., Ltd.), the inside of the reactor was replaced with hydrogen, and the pressure was increased to 0.6 MPa. . The temperature of the reaction solution was increased while stirring under a hydrogen flow, and the reaction was started when the temperature reached 100 ° C. and reacted for 110 hours.
- Pd / Al 2 O 3 catalyst manufactured by Clariant Catalyst Co., Ltd.
- the catalyst was filtered through a membrane filter (0.2 ⁇ m: Merck Millipore), and the filtrate was purified by distillation to obtain a distillate at a pressure of 0.77 kPa and a temperature of 174 to 176 ° C.
- the obtained distillate was confirmed to be bis (3-methylbenzyl) amine by GC-MS and 1 H-NMR (purity 97%).
- Example 1 (Preparation of xylylenediamine composition) A xylylenediamine composition was prepared so that the content of bis (3-methylbenzyl) amine was 0.001 part by mass with respect to 100 parts by mass of metaxylylenediamine.
- the internal temperature was raised, and when the temperature reached 250 ° C., the pressure in the reaction vessel was reduced, and the internal temperature was further raised to continue the melt polycondensation reaction at 255 ° C. for 20 minutes. . Thereafter, the inside of the system was pressurized with nitrogen, and the obtained polymer was taken out of the strand die and pelletized to obtain a polyamide resin. Said evaluation was performed about the obtained polyamide resin. The results are shown in Table 1.
- Examples 2-3 and Comparative Examples 1-2 A xylylenediamine composition was prepared in the same manner as in Example 1 except that the content of bis (3-methylbenzyl) amine in the xylylenediamine composition was changed as shown in Table 1. Was prepared. Further, a polyamide resin was produced using this xylylenediamine composition, and the evaluation was performed. The results are shown in Table 1.
- Example 10 Xylylenediamine so that bis (3-methylbenzyl) amine is 0.01 parts by mass with respect to 100 parts by mass of mixed xylylenediamine having a mass ratio of metaxylylenediamine and paraxylylenediamine of 70/30. A composition was prepared.
- Comparative Example 4 A polyamide resin was produced in the same manner as in Example 10 except that bis (3-methylbenzyl) amine was not used, and the evaluation was performed. The results are shown in Table 3.
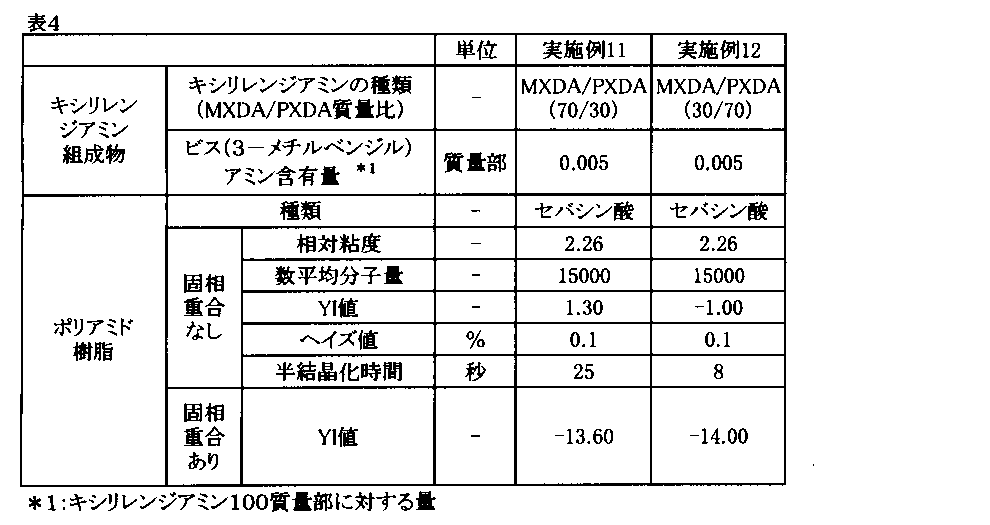
- Example 11 The content of bis (3-methylbenzyl) amine is 0.005 parts by mass with respect to 100 parts by mass of mixed xylylenediamine having a mass ratio of metaxylylenediamine and paraxylylenediamine of 70/30. A xylylenediamine composition was prepared.
- Example 12 The content of bis (3-methylbenzyl) amine is 0.005 parts by mass with respect to 100 parts by mass of the mixed xylylenediamine having a mass ratio of metaxylylenediamine to paraxylylenediamine of 30/70. A xylylenediamine composition was prepared.
- the polyamide resin obtained using the xylylenediamine composition of the present invention and obtained by the production method of the present invention is less colored and faster in crystallization than the polyamide resin of the comparative example. I understand that.
- the xylylenediamine composition of the present invention is used as a raw material for a polyamide resin, it is possible to obtain a polyamide resin with little coloring and quick crystallization.
- the xylylenediamine composition of the present invention is also suitable for various uses such as an epoxy resin curing agent, or as a raw material for isocyanate compounds.
- the polyamide resin obtained by the production method of the present invention is less colored, it is suitably used as a material for packaging films, hollow containers, various molding materials, fibers and the like.
- the polyamide resin obtained by the production method of the present invention is rapidly crystallized and excellent in transparency and moldability.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyamides (AREA)
- Epoxy Resins (AREA)
Abstract
Description
上記問題を解決する方法として、例えば特許文献1には、キシリレンジアミンを含むジアミンと特定の脂肪族ジカルボン酸とを重縮合して得られ、原料ジアミンとして使用するキシリレンジアミンの純度が99.9重量%以上であり、所定の条件下で加熱を行った際の加熱前後の黄色度(YI値)の差が5以内であるポリアミド樹脂が開示されている。
また本発明は、ジアミン成分としてキシリレンジアミンを用いた、着色が少なく、かつ結晶化が速いポリアミド樹脂の製造方法を提供することを課題とする。
また本発明者らは、特定の条件下でキシリレンジアミン含有ジアミンとジカルボン酸とを反応させることにより、着色が少なく、かつ結晶化が速いポリアミド樹脂を製造できることを見出した。
すなわち本発明は、[1]キシリレンジアミンとビス(メチルベンジル)アミンとを含有するキシリレンジアミン組成物であって、ビス(メチルベンジル)アミンの含有量がキシリレンジアミン100質量部に対し0.0005~0.1質量部であるキシリレンジアミン組成物、及び[2]キシリレンジアミンを含有するジアミン、ジカルボン酸、及び、該キシリレンジアミン100質量部に対して0.0005~0.1質量部のビス(メチルベンジル)アミンを反応系に導入し、重縮合反応を行う工程を有する、ポリアミド樹脂の製造方法を提供する。
さらに本発明の製造方法により得られたポリアミド樹脂は、着色が少ないため、包装用フィルム、中空容器、各種成形材料、繊維等の材料として好適に用いられる。本発明の製造方法により得られたポリアミド樹脂は結晶化が速く、透明性及び成形加工性にも優れる。
本発明のキシリレンジアミン組成物(以下、単に「本発明の組成物」又は「組成物」ともいう。)は、キシリレンジアミンとビス(メチルベンジル)アミンとを含有し、ビス(メチルベンジル)アミンの含有量がキシリレンジアミン100質量部に対し0.0005~0.1質量部であることを特徴とする。
本発明の組成物はキシリレンジアミンを主成分とするものであり、該組成物中のキシリレンジアミンの含有量は、99.5質量%以上であることが好ましく、99.9質量%以上であることがより好ましい。
なお、組成物中のキシリレンジアミンの含有量は、例えばガスクロマトグラフィー(GC)分析等により測定することができる。
工業的に入手可能なキシリレンジアミンは、公知の方法を用いて製造することができる。例えば、キシリレンジアミンがメタキシリレンジアミンである場合、その製造方法としては、触媒存在下、連続反応あるいはバッチ反応にて、メタキシレン、アンモニア及び酸素含有ガスを反応させ、生成したイソフタロニトリルを水素化する方法等が挙げられる。
組成物中のビス(メチルベンジル)アミンの含有量が上記範囲内であれば、該組成物をポリアミド樹脂の原料として用いると、得られるポリアミド樹脂は着色が少なく、かつ、結晶化が速いものとなる。ポリアミド樹脂の結晶化速度向上に伴い成形加工性が向上するので、成形時の結晶化工程時間を短縮することができ、成形品の生産性を向上させることができる。
本発明によれば、上記効果が得られることから、ポリアミド樹脂の成形加工性等を向上させるために結晶核剤を添加することによる、機械的物性や透明性の低下等の問題を回避することができる。また着色が少ない成形品を容易に生産しうるポリアミド樹脂が得られる。
また、キシリレンジアミンは、保存時に分子中のアミノ基が劣化してアンモニアが微量発生することがあるが、ビス(メチルベンジル)アミンが存在することによってアンモニアの発生量が減少することも見出された。このことから、キシリレンジアミン組成物がビス(メチルベンジル)アミンを含有することで、キシリレンジアミンの保存安定性が向上するという効果も期待できる。
アンモニアの発生量が減少する理由は定かではないが、キシリレンジアミンとビス(メチルベンジル)アミンとの何らかの相互作用により、ラジカルの発生やその連鎖を防ぐ効果があるものと推定される。
特に、本発明のキシリレンジアミン組成物をポリアミド樹脂の原料に用いた場合には、着色が少なく、かつ結晶化が速いポリアミド樹脂を製造することができる点で好ましい。
なお、上記エポキシ樹脂硬化剤の製造には、必要に応じその他の成分を併用してもよい。
本発明のポリアミド樹脂の製造方法は、キシリレンジアミンを含有するジアミン、ジカルボン酸、及び、該キシリレンジアミン100質量部に対して0.0005~0.1質量部のビス(メチルベンジル)アミンを反応系に導入し、重縮合反応を行う工程を有することを特徴とする。
なお、本発明のポリアミド樹脂の製造方法には上述した本発明のキシリレンジアミン組成物を用いることが好ましいが、これに限られない。
本発明に用いられるジアミンは、キシリレンジアミンを含有するジアミン(以下、単に「ジアミン」ともいう。)である。キシリレンジアミンとしては、メタキシリレンジアミン、パラキシリレンジアミン又はこれらの混合物であることが好ましく、得られるポリアミド樹脂のガスバリア性の観点からは、メタキシリレンジアミンであることがより好ましい。キシリレンジアミン含有ジアミンを用いることにより、得られるポリアミド樹脂は溶融成形性、機械的特性、及びガスバリア性に優れたものとなる。
本発明に用いられるジカルボン酸は、特に制限されないが、成形加工性、ガスバリア性、及び機械的特性の観点から、炭素数4~20の脂肪族ジカルボン酸、テレフタル酸及びイソフタル酸から選ばれる少なくとも1種であることが好ましく、炭素数4~20の脂肪族ジカルボン酸であることがより好ましく、炭素数4~12の脂肪族ジカルボン酸であることが更に好ましい。
炭素数4~20の脂肪族ジカルボン酸としては、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、1,10-デカンジカルボン酸、1,11-ウンデカンジカルボン酸、1,12-ドデカンジカルボン酸、1,14-テトラデカンジカルボン酸、1,16-ヘキサデカンジカルボン酸、1,18-オクタデカンジカルボン酸等を例示できるが、これらの中でも結晶性、高弾性の観点からアジピン酸及びセバシン酸から選ばれる少なくとも1種が好ましく使用される。これらのジカルボン酸は、1種又は2種以上を組み合わせて用いてもよい。
所定量のビス(メチルベンジル)アミンを反応系に導入して重縮合反応を行うことで上記効果が得られる理由については定かではないが、ポリアミド樹脂の着色の低減に関しては、ビス(メチルベンジル)アミンが、前記ジアミンとジカルボン酸との重縮合反応により得られるポリアミド樹脂から発生するラジカルを捕捉する効果を有するため、該ラジカルによりポリアミド樹脂が劣化するのを抑制することによると考えられる。また、ポリアミド樹脂の結晶化速度向上効果については、ビス(メチルベンジル)アミンがポリアミド樹脂中で結晶核生成を促進しているか、又はビス(メチルベンジル)アミンそのものが結晶核生成の起点になることによると考えられる。
具体的には、ジアミンとジカルボン酸とからなる塩を、水の存在下、常圧又は加圧状態で加熱し、添加した水及び重縮合により生成する水を除きながら溶融状態で重縮合させる方法が挙げられる。また、ジアミンを溶融状態のジカルボン酸に直接加えて、常圧又は加圧下で重縮合する方法も挙げられる。この場合、反応系を均一な液状態で保つために、ジアミンをジカルボン酸に連続的に加え、その間、反応温度が生成するオリゴアミド及びポリアミドの融点よりも下回らないように反応系を昇温しつつ、重縮合が進められる。
上記のうち、常圧又は加圧下で溶融させたジカルボン酸中にジアミンを滴下し、縮合水を除きながら溶融状態で重合させる溶融重合法を用いることが、ポリアミド樹脂の分子量分布を小さくできることから好ましい。
また、本発明で用いるキシリレンジアミンの製造において、使用する触媒や製造条件を特定の構成とし、ビス(メチルベンジル)アミンを所定量生成するような反応を並行して行うことが可能であればそれを利用する等の方法が挙げられる。この場合には、キシリレンジアミン中のビス(メチルベンジル)アミンの含有量は、ガスクロマトグラフィー(GC)分析等により求めることができる。例えば、ビス(メチルベンジル)アミンを含有するキシリレンジアミンのGC測定を行い、キシリレンジアミン由来のピーク値とビス(メチルベンジル)アミン由来のピーク値の比から、ビス(メチルベンジル)アミンの含有量を求める方法等が挙げられる。
これらの中でも、特に次亜リン酸ナトリウム、次亜リン酸カリウム、次亜リン酸リチウム等の次亜リン酸金属塩が、アミド化反応を促進するため好ましく用いられ、特に次亜リン酸ナトリウムが好ましい。なお、本発明で使用できるリン原子含有化合物はこれらの化合物に限定されない。
アルカリ金属化合物としては、アルカリ金属水酸化物やアルカリ金属酢酸塩が通常使用される。但し、アルカリ金属を含む上記リン原子含有化合物は除く。例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化ルビジウム、水酸化セシウム、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸ルビジウム、酢酸セシウム等が挙げられ、水酸化ナトリウム及び酢酸ナトリウムから選ばれる少なくとも1種が好ましい。これらは1種又は2種以上を組み合わせて用いることができる。
なお、上記アルカリ金属化合物は、重縮合反応系内に添加してもよく、ポリアミド樹脂の原料であるジカルボン酸由来であってもよい。
また、アルカリ金属化合物の使用量は、アルカリ金属化合物のモル数を前述のリン原子含有化合物のモル数で除した値が、通常0.5~1.0の範囲となる量であり、好ましくは0.55~0.95、より好ましくは0.6~0.9の範囲となる量である。上記範囲内であると、アミド化反応が適度な速度で進行する。
また、ポリアミド樹脂の重合度を高めるために、本発明の製造方法は、更に固相重合を行う工程を有していてもよい。固相重合は公知の方法により行うことができ、例えば、窒素雰囲気下、100℃以上でかつポリアミドの融点を下回る温度で1~24時間加熱する方法が挙げられる。
乾燥ないし固相重合で用いられる加熱装置としては、連続式の加熱乾燥装置やタンブルドライヤー、コニカルドライヤー、ロータリードライヤー等と称される回転ドラム式の加熱装置及びナウタミキサーと称される内部に回転翼を備えた円錐型の加熱装置が好適に使用できるが、これらに限定されることなく公知の装置を使用することができる。
JIS K7373に準拠して測定したポリアミド樹脂のYI値は、好ましくは-20~5、より好ましくは-20~2の範囲とすることができる。なお、ポリアミド樹脂のYI値は、具体的には実施例に記載の方法により測定できる。
本発明の製造方法では、得られるポリアミド樹脂の半結晶化時間を、好ましくは100秒以下、より好ましくは90秒以下、更に好ましくは85秒以下とすることができる。半結晶化時間は、具体的には実施例に記載の方法により測定できる。
本発明の製造方法により得られたポリアミド樹脂は、結晶化速度が速くなることから、成形時の結晶化工程時間を短縮することができ、すなわち成形サイクルが速くなり生産性を向上させることができる。上記ポリアミド樹脂を含む成形品は、包装用フィルム、中空容器、各種成形材料、繊維等として好適である。また成形品の着色が少なく、透明性も損なうことがないため、特に高い透明性が要求される包装用フィルム、中空容器等に好適である。
実施例及び比較例で得られたポリアミド樹脂0.2gを精秤し、96%硫酸20mlに20~30℃で攪拌溶解した。完全に溶解した後、速やかにキャノンフェンスケ型粘度計に溶液5mlを取り、25℃の恒温槽中で10分間放置後、落下時間(t)を測定した。また、96%硫酸そのものの落下時間(t0)も同様に測定した。t及びt0から次式により相対粘度を算出した。
相対粘度=t/t0
実施例及び比較例で得られたポリアミド樹脂の数平均分子量は、まず試料をフェノール/エタノール混合溶媒、及びベンジルアルコール溶媒にそれぞれ溶解させ、カルボキシル末端基濃度とアミノ末端基濃度を塩酸及び水酸化ナトリウム水溶液の中和滴定により求めた。数平均分子量は、アミノ末端基濃度及びカルボキシル末端基濃度の定量値から次式により求めた。
数平均分子量=2×1,000,000/([NH2]+[COOH])
[NH2]:アミノ末端基濃度(μeq/g)
[COOH]:カルボキシル末端基濃度(μeq/g)
YI値の測定はJIS K7373に準じて行った。実施例及び比較例で得られたポリアミド樹脂ペレットを用い、色差測定装置(日本電色工業(株)製、型式:Z-Σ80 Color Measuring System)を使用してYI値を測定した。
実施例及び比較例で得られたポリアミド樹脂ペレットを乾燥させ、乾燥したペレットを単軸押出機にて融点+20℃の条件で押出し、厚さ100μmのフィルムを作製した。曇価測定装置(日本電色工業(株)製、型式:COH-300A)を使用して透過法によりヘイズ値を測定した。
実施例及び比較例で得られたポリアミド樹脂ペレットを用いて、前記と同様に厚さ100μmのフィルムを作製し、カバーガラスに挟んだ後、ポリアミド樹脂の融点+30℃で3分間溶融保持した直後に、160℃のオイルバスで冷却した。結晶化速度測定装置((株)コタキ製作所製、型式:MK701)を用いて、脱偏光強度法により半結晶化時間を測定した。
1Lオートクレーブに、3-メチルベンゾニトリル511.5gと0.5%Pd/Al2O3触媒(クラリアント触媒(株)製)75gを仕込み、反応器内を水素で置換し0.6MPaまで昇圧した。水素流通下で反応液を攪拌しながら昇温し、100℃に達したところで反応を開始し、110時間反応させた。反応終了後、触媒をメンブレンフィルター(0.2μm:メルクミリポア)でろ過し、そのろ液を蒸留精製し、圧力0.77kPa、温度174~176℃における留出物を得た。得られた留出物はGC-MS及び1H-NMRによりビス(3-メチルベンジル)アミンであることを確認した(純度97%)。
(キシリレンジアミン組成物の調製)
メタキシリレンジアミン100質量部に対して、ビス(3-メチルベンジル)アミンの含有量が0.001質量部となるようにキシリレンジアミン組成物を調製した。
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた反応容器に、アジピン酸(ローディア社製)10kg(68.43mol)を仕込み、十分に窒素置換した後、更に少量の窒素気流下で系内を撹搾しながら170℃まで加熱溶融した。上記のようにして得られたキシリレンジアミン組成物9.273kg(メタキシリレンジアミン68.08mol含有)を溶融したアジピン酸に攪拌下で滴下し、生成する縮合水を系外に排出しながら、内温を連続的に2.5時間かけて240℃まで昇温した。
キシリレンジアミン組成物の滴下終了後、内温を上昇させ、250℃に達した時点で反応容器内を減圧にし、更に内温を上昇させて255℃で20分間、溶融重縮合反応を継続した。その後、系内を窒素で加圧し、得られた重合物をストランドダイから取り出して、これをペレット化することにより、ポリアミド樹脂を得た。得られたポリアミド樹脂について、前記評価を行った。結果を表1に示す。
また、上記ポリアミド樹脂500gを2Lのナス型フラスコに入れ、十分に窒素置換した後、減圧しながら、オイルバス中において190℃で4時間加熱し、固相重合を行った。固相重合後のポリアミド樹脂について、前記と同様にYI値の測定を行った。結果を表1に示す。
実施例1において、キシリレンジアミン組成物中のビス(3-メチルベンジル)アミンの含有量を各々表1に記載のとおりに変更したこと以外は、実施例1と同様にしてキシリレンジアミン組成物を調製した。また、このキシリレンジアミン組成物を用いてポリアミド樹脂を製造し、前記評価を行った。結果を表1に示す。

実施例1において、キシリレンジアミン組成物中のビス(3-メチルベンジル)アミンの含有量を各々表2に記載のとおりに変更したこと以外は、実施例1と同様にしてキシリレンジアミン組成物を調製した。また、ポリアミド樹脂の製造において、アジピン酸の仕込みと同時に次亜リン酸ナトリウム・一水和物/酢酸ナトリウム(モル比=1.5/1)を0.438g添加して溶融重縮合反応を行ったこと以外は、実施例1と同様にしてポリアミド樹脂を製造し、前記評価を行った。結果を表2に示す。

メタキシリレンジアミンとパラキシリレンジアミンの質量比が70/30である混合キシリレンジアミン100質量部に対して、ビス(3-メチルベンジル)アミンが0.01質量部となるようにキシリレンジアミン組成物を調製した。
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた反応容器に、アジピン酸(ローディア社製)10kg(68.43mol)及び次亜リン酸ナトリウム・一水和物/酢酸ナトリウム(モル比=1.5/1)13.14gを仕込み、十分に窒素置換した後、更に少量の窒素気流下で系内を撹搾しながら170℃まで加熱溶融した。上記のようにして得られたキシリレンジアミン組成物9.274kg(メタキシリレンジアミン47.66mol、パラキシリレンジアミン20.42mol)を溶融したアジピン酸に攪拌下で滴下し、生成する縮合水を系外に排出しながら、内温を連続的に2.5時間かけて260℃まで昇温した。
前記キシリレンジアミン組成物の滴下終了後、内温を上昇させ、270℃に達した時点で反応容器内を減圧にし、更に内温を上昇させて275℃で20分間、溶融重縮合反応を継続した。その後、系内を窒素で加圧し、得られた重合物をストランドダイから取り出して、これをペレット化することにより、ポリアミド樹脂を得た。得られたポリアミド樹脂について、前記評価を行った。結果を表3に示す。
また、実施例1と同様にして固相重合を行い、固相重合後のポリアミド樹脂について、前記と同様にYI値の測定を行った。結果を表3に示す。
ビス(3-メチルベンジル)アミンを用いなかったこと以外は、実施例10と同様にしてポリアミド樹脂を製造し、前記評価を行った。結果を表3に示す。

メタキシリレンジアミンとパラキシリレンジアミンの質量比が70/30である混合キシリレンジアミン100質量部に対して、ビス(3-メチルベンジル)アミンの含有量が0.005質量部となるようにキシリレンジアミン組成物を調製した。
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた反応容器に、セバシン酸(伊藤製油(株)製TAグレード)10kg(49.4mol)及び次亜リン酸ナトリウム・一水和物/酢酸ナトリウム(モル比=1.5/1)11.66gを仕込み、十分に窒素置換した後、更に少量の窒素気流下で系内を撹搾しながら170℃まで加熱溶融した。上記のようにして得られたキシリレンジアミン組成物6.680kg(メタキシリレンジアミン34.33mol、パラキシリレンジアミン14.71mol)を溶融したセバシン酸に攪拌下で滴下し、生成する縮合水を系外に排出しながら、内温を連続的に2.5時間かけて240℃まで昇温した。
キシリレンジアミン組成物の滴下終了後、内温を上昇させ、250℃に達した時点で反応容器内を減圧にし、更に内温を上昇させて255℃で20分間、溶融重縮合反応を継続した。その後、系内を窒素で加圧し、得られた重合物をストランドダイから取り出して、これをペレット化することにより、ポリアミド樹脂を得た。得られたポリアミド樹脂について、前記評価を行った。結果を表4に示す。
また、実施例1と同様にして固相重合を行い、固相重合後のポリアミド樹脂について、前記と同様にYI値の測定を行った。結果を表4に示す。
メタキシリレンジアミンとパラキシリレンジアミンの質量比が30/70である混合キシリレンジアミン100質量部に対して、ビス(3-メチルベンジル)アミンの含有量が0.005質量部となるようにキシリレンジアミン組成物を調製した。
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた反応容器に、セバシン酸(伊藤製油(株)製TAグレード)10kg(49.4mol)及び次亜リン酸ナトリウム・一水和物/酢酸ナトリウム(モル比=1.5/1)11.66gを仕込み、十分に窒素置換した後、更に少量の窒素気流下で系内を撹搾しながら170℃まで加熱溶融した。上記のようにして得られたキシリレンジアミン組成物6.680kg(メタキシリレンジアミン14.71mol、パラキシリレンジアミン34.33mol)を溶融したセバシン酸に攪拌下で滴下し、生成する縮合水を系外に排出しながら、内温を連続的に2.5時間かけて262℃まで昇温した。
キシリレンジアミン組成物の滴下終了後、内温を上昇させ、265℃に達した時点で反応容器内を減圧にし、更に内温を上昇させて275℃で20分間、溶融重縮合反応を継続した。その後、系内を窒素で加圧し、得られた重合物をストランドダイから取り出して、これをペレット化することにより、ポリアミド樹脂を得た。得られたポリアミド樹脂について、前記評価を行った。結果を表4に示す。
また、実施例1と同様にして固相重合を行い、固相重合後のポリアミド樹脂について、前記と同様にYI値の測定を行った。結果を表4に示す。
また本発明の製造方法で得られたポリアミド樹脂は、着色が少ないため、包装用フィルム、中空容器、各種成形材料、繊維等の材料として好適に用いられる。本発明の製造方法で得られたポリアミド樹脂は結晶化が速く、透明性、成形加工性にも優れる。
Claims (13)
- キシリレンジアミンとビス(メチルベンジル)アミンとを含有するキシリレンジアミン組成物であって、ビス(メチルベンジル)アミンの含有量がキシリレンジアミン100質量部に対し0.0005~0.1質量部である、キシリレンジアミン組成物。
- キシリレンジアミンがメタキシリレンジアミン、パラキシリレンジアミン又はこれらの混合物である、請求項1に記載のキシリレンジアミン組成物。
- キシリレンジアミンがメタキシリレンジアミンである、請求項1に記載のキシリレンジアミン組成物。
- キシリレンジアミンの含有量が99.5質量%以上である、請求項1~3のいずれかに記載のキシリレンジアミン組成物。
- ポリアミド樹脂の原料に用いられる、請求項1~4のいずれかに記載のキシリレンジアミン組成物。
- エポキシ樹脂硬化剤に用いられる、請求項1~4のいずれかに記載のキシリレンジアミン組成物。
- キシリレンジアミンを含有するジアミン、ジカルボン酸、及び、該キシリレンジアミン100質量部に対して0.0005~0.1質量部のビス(メチルベンジル)アミンを反応系に導入し、重縮合反応を行う工程を有する、ポリアミド樹脂の製造方法。
- ジカルボン酸が炭素数4~20の脂肪族ジカルボン酸、テレフタル酸及びイソフタル酸から選ばれる少なくとも1種である、請求項7に記載のポリアミド樹脂の製造方法。
- 前記ジアミン中のキシリレンジアミンの含有量が70モル%以上であり、ジカルボン酸中の炭素数4~20の脂肪族ジカルボン酸の含有量が50モル%以上である、請求項7又は8に記載のポリアミド樹脂の製造方法。
- 炭素数4~20の脂肪族ジカルボン酸がアジピン酸及びセバシン酸から選ばれる少なくとも1種である、請求項8又は9に記載のポリアミド樹脂の製造方法。
- キシリレンジアミンがメタキシリレンジアミン、パラキシリレンジアミン又はこれらの混合物である、請求項7~10のいずれかに記載のポリアミド樹脂の製造方法。
- キシリレンジアミンがメタキシリレンジアミンである、請求項7~11のいずれかに記載のポリアミド樹脂の製造方法。
- 更に固相重合を行う工程を有する、請求項7~12のいずれかに記載のポリアミド樹脂の製造方法。
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES14791010.3T ES2648104T3 (es) | 2013-04-30 | 2014-04-09 | Composición de xililendiamina y método de producción de resina de poliamida |
| CN201480003141.7A CN104812803B (zh) | 2013-04-30 | 2014-04-09 | 苯二甲胺组合物及聚酰胺树脂的制造方法 |
| MX2015014887A MX2015014887A (es) | 2013-04-30 | 2014-04-09 | Composicion de xililendiamina y metodo para producir una resina de poliamida. |
| US14/648,006 US9562144B2 (en) | 2013-04-30 | 2014-04-09 | Xylylenediamine composition, and method for producing polyamide resin |
| JP2014536055A JP5700177B1 (ja) | 2013-04-30 | 2014-04-09 | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 |
| RU2015146550A RU2650891C2 (ru) | 2013-04-30 | 2014-04-09 | Композиция ксилилендиамина и способ получения полиамидной смолы |
| AU2014260891A AU2014260891A1 (en) | 2013-04-30 | 2014-04-09 | Xylylenediamine composition, and method for producing polyamide resin |
| EP14791010.3A EP2993198B1 (en) | 2013-04-30 | 2014-04-09 | Xylylenediamine composition, and method for producing polyamide resin |
| BR112015025512A BR112015025512A2 (pt) | 2013-04-30 | 2014-04-09 | composição de xililenodiamina e método para a produção de resina de poliamida |
| KR1020157011932A KR101563250B1 (ko) | 2013-04-30 | 2014-04-09 | 자일릴렌디아민 조성물 및 폴리아미드수지의 제조방법 |
| SG11201508864YA SG11201508864YA (en) | 2013-04-30 | 2014-04-09 | Xylylenediamine composition, and method for producing polyamide resin |
| PH12015502352A PH12015502352A1 (en) | 2013-04-30 | 2015-10-09 | Xylylenediamine composition, and method for producing polyamide resin |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013095708 | 2013-04-30 | ||
| JP2013095705 | 2013-04-30 | ||
| JP2013-095708 | 2013-04-30 | ||
| JP2013-095705 | 2013-04-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014178270A1 true WO2014178270A1 (ja) | 2014-11-06 |
Family
ID=51843405
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/060346 WO2014178270A1 (ja) | 2013-04-30 | 2014-04-09 | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US9562144B2 (ja) |
| EP (1) | EP2993198B1 (ja) |
| JP (1) | JP5700177B1 (ja) |
| KR (1) | KR101563250B1 (ja) |
| CN (1) | CN104812803B (ja) |
| AU (1) | AU2014260891A1 (ja) |
| BR (1) | BR112015025512A2 (ja) |
| ES (1) | ES2648104T3 (ja) |
| MX (1) | MX2015014887A (ja) |
| PH (1) | PH12015502352A1 (ja) |
| RU (1) | RU2650891C2 (ja) |
| SG (1) | SG11201508864YA (ja) |
| TW (1) | TWI502026B (ja) |
| WO (1) | WO2014178270A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022080048A1 (ja) * | 2020-10-15 | 2022-04-21 | 三菱瓦斯化学株式会社 | エポキシ樹脂硬化剤、エポキシ樹脂組成物、塗料及び接着剤 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6098391B2 (ja) * | 2013-06-21 | 2017-03-22 | 三菱瓦斯化学株式会社 | ポリアミド樹脂の製造方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11158370A (ja) | 1997-11-25 | 1999-06-15 | Yasuhara Chemical Co Ltd | ポリアミド用結晶核剤およびポリアミド組成物 |
| JP2003026797A (ja) | 2001-07-19 | 2003-01-29 | Mitsubishi Gas Chem Co Inc | キシリレン基含有ポリアミド樹脂 |
| JP2008050403A (ja) * | 2006-08-22 | 2008-03-06 | Mitsubishi Gas Chem Co Inc | 酸素吸収性樹脂 |
| WO2011030911A1 (ja) * | 2009-09-14 | 2011-03-17 | 三菱瓦斯化学株式会社 | 難燃性ポリアミド樹脂組成物 |
| JP2011236285A (ja) * | 2010-05-07 | 2011-11-24 | Mitsubishi Gas Chemical Co Inc | 酸素吸収樹脂組成物の製造方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5017756B2 (ja) * | 2001-07-16 | 2012-09-05 | 三菱瓦斯化学株式会社 | 高純度メタキシリレンジアミンの製造方法 |
| JP2005132736A (ja) * | 2003-10-28 | 2005-05-26 | Mitsubishi Gas Chem Co Inc | 高品位キシリレンジアミンの製造方法 |
| CN101379021B (zh) * | 2006-02-01 | 2011-12-28 | 巴斯夫欧洲公司 | 纯苯二甲胺(xda)的制备方法 |
| DE602007003135D1 (de) * | 2006-05-18 | 2009-12-24 | Mitsubishi Gas Chemical Co | Verfahren zur Herstellung von Xylylenediamin |
| KR101700987B1 (ko) * | 2009-06-09 | 2017-02-13 | 미츠비시 가스 가가쿠 가부시키가이샤 | 폴리아미드 수지 조성물 및 성형품 |
| KR101535321B1 (ko) | 2009-06-15 | 2015-07-08 | 미츠비시 가스 가가쿠 가부시키가이샤 | 산소 흡수 수지 조성물 |
-
2014
- 2014-04-09 KR KR1020157011932A patent/KR101563250B1/ko active IP Right Grant
- 2014-04-09 ES ES14791010.3T patent/ES2648104T3/es active Active
- 2014-04-09 WO PCT/JP2014/060346 patent/WO2014178270A1/ja active Application Filing
- 2014-04-09 AU AU2014260891A patent/AU2014260891A1/en not_active Abandoned
- 2014-04-09 EP EP14791010.3A patent/EP2993198B1/en active Active
- 2014-04-09 MX MX2015014887A patent/MX2015014887A/es active IP Right Grant
- 2014-04-09 CN CN201480003141.7A patent/CN104812803B/zh active Active
- 2014-04-09 RU RU2015146550A patent/RU2650891C2/ru active
- 2014-04-09 SG SG11201508864YA patent/SG11201508864YA/en unknown
- 2014-04-09 US US14/648,006 patent/US9562144B2/en active Active
- 2014-04-09 JP JP2014536055A patent/JP5700177B1/ja active Active
- 2014-04-09 BR BR112015025512A patent/BR112015025512A2/pt not_active IP Right Cessation
- 2014-04-29 TW TW103115390A patent/TWI502026B/zh active
-
2015
- 2015-10-09 PH PH12015502352A patent/PH12015502352A1/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11158370A (ja) | 1997-11-25 | 1999-06-15 | Yasuhara Chemical Co Ltd | ポリアミド用結晶核剤およびポリアミド組成物 |
| JP2003026797A (ja) | 2001-07-19 | 2003-01-29 | Mitsubishi Gas Chem Co Inc | キシリレン基含有ポリアミド樹脂 |
| JP2008050403A (ja) * | 2006-08-22 | 2008-03-06 | Mitsubishi Gas Chem Co Inc | 酸素吸収性樹脂 |
| WO2011030911A1 (ja) * | 2009-09-14 | 2011-03-17 | 三菱瓦斯化学株式会社 | 難燃性ポリアミド樹脂組成物 |
| JP2011236285A (ja) * | 2010-05-07 | 2011-11-24 | Mitsubishi Gas Chemical Co Inc | 酸素吸収樹脂組成物の製造方法 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022080048A1 (ja) * | 2020-10-15 | 2022-04-21 | 三菱瓦斯化学株式会社 | エポキシ樹脂硬化剤、エポキシ樹脂組成物、塗料及び接着剤 |
| KR20230087471A (ko) | 2020-10-15 | 2023-06-16 | 미쯔비시 가스 케미칼 컴파니, 인코포레이티드 | 에폭시 수지 경화제, 에폭시 수지 조성물, 도료 및 접착제 |
Also Published As
| Publication number | Publication date |
|---|---|
| PH12015502352A1 (en) | 2016-02-22 |
| KR101563250B1 (ko) | 2015-10-26 |
| RU2650891C2 (ru) | 2018-04-18 |
| EP2993198B1 (en) | 2017-09-27 |
| RU2015146550A (ru) | 2017-06-01 |
| US9562144B2 (en) | 2017-02-07 |
| TWI502026B (zh) | 2015-10-01 |
| SG11201508864YA (en) | 2015-11-27 |
| EP2993198A4 (en) | 2016-12-14 |
| CN104812803A (zh) | 2015-07-29 |
| ES2648104T3 (es) | 2017-12-28 |
| JP5700177B1 (ja) | 2015-04-15 |
| KR20150063564A (ko) | 2015-06-09 |
| CN104812803B (zh) | 2016-08-17 |
| MX2015014887A (es) | 2016-03-07 |
| US20160039993A1 (en) | 2016-02-11 |
| JPWO2014178270A1 (ja) | 2017-02-23 |
| TW201500460A (en) | 2015-01-01 |
| AU2014260891A1 (en) | 2015-11-05 |
| EP2993198A1 (en) | 2016-03-09 |
| BR112015025512A2 (pt) | 2017-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5700177B1 (ja) | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 | |
| JP5776865B1 (ja) | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 | |
| JP5804225B1 (ja) | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 | |
| JP5790902B1 (ja) | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 | |
| JP5804226B1 (ja) | キシリレンジアミン組成物及びポリアミド樹脂の製造方法 | |
| JP6098391B2 (ja) | ポリアミド樹脂の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014536055 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14791010 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20157011932 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14648006 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12015502352 Country of ref document: PH |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014791010 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014791010 Country of ref document: EP Ref document number: MX/A/2015/014887 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P1462/2015 Country of ref document: AE Ref document number: IDP00201506989 Country of ref document: ID |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15257779 Country of ref document: CO |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2014260891 Country of ref document: AU Date of ref document: 20140409 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2015146550 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015025512 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112015025512 Country of ref document: BR Kind code of ref document: A2 Effective date: 20151006 |