WO2014157140A1 - グリコリドの製造方法 - Google Patents
グリコリドの製造方法 Download PDFInfo
- Publication number
- WO2014157140A1 WO2014157140A1 PCT/JP2014/058187 JP2014058187W WO2014157140A1 WO 2014157140 A1 WO2014157140 A1 WO 2014157140A1 JP 2014058187 W JP2014058187 W JP 2014058187W WO 2014157140 A1 WO2014157140 A1 WO 2014157140A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- glycolide
- gao
- glycolic acid
- organic solvent
- polar organic
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/12—1,4-Dioxanes; Hydrogenated 1,4-dioxanes not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
Definitions
- the present invention relates to a method for efficiently and economically producing high-purity glycolide for a long period of time by depolymerization of glycolic acid oligomers.
- Polyglycolic acid is a resin material with excellent biodegradability, hydrolyzability, gas barrier properties, strength, etc., and medical polymer materials such as sutures and artificial skin; packaging materials such as bottles and films; injection molded products It is used in a wide range of technical fields, such as resin materials for various industrial products such as fibers, vapor-deposited films, and fishing lines; resin materials for well drilling.
- Polyglycolic acid is a polymer having a repeating unit having a structure formed by dehydration polycondensation of glycolic acid.
- polyglycolic acid with a low degree of polymerization has insufficient strength, melt processability, gas barrier properties, etc., and its degradation rate in natural environments and in vivo is too fast, so it is durable when applied in many applications. Can not satisfy the demands of.
- glycolide which is a cyclic dimer of glycolic acid as a starting material
- Glycolide is a cyclic ester compound having a cyclic dimer structure in which two molecules of water are eliminated from two molecules of glycolic acid.
- glycolide cannot be synthesized even if glycolic acid is subjected to a dehydration reaction, and only a glycolic acid oligomer having a low polymerization degree is obtained.
- glycolic acid is polycondensed to synthesize a glycolic acid oligomer having a low polymerization degree. Then, the following reaction formula 2
- glycolide is synthesized by depolymerizing the glycolic acid oligomer.
- reaction formula 3 When ring-opening polymerization of glycolide, the following reaction formula 3
- polyglycolic acid can be produced.
- a solution phase depolymerization method has been proposed as a method suitable for mass production of glycolide.
- the solution phase depolymerization method is a method in which a mixture containing a glycolic acid oligomer and a polar organic solvent is heated to form a solution phase of the glycolic acid oligomer, and heating is continued in that state to perform depolymerization. .
- a solubilizer is contained in the mixture.
- Patent Document 1 an ⁇ -hydroxycarboxylic acid oligomer such as glycolic acid oligomer is dissolved by heating in a high-boiling polar organic solvent, and heating is continued in this state for depolymerization to produce a cyclic dimer.
- a method for producing a cyclic dimer ester in which an ester is distilled together with a polar organic solvent and a cyclic dimer ester such as glycolide is recovered from the distillate is disclosed.
- Patent Document 2 a mixture containing an aliphatic polyester such as low molecular weight polyglycolic acid and a specific polyalkylene glycol ether is heated to a temperature at which the depolymerization of the aliphatic polyester occurs to form a uniform solution phase.
- a method for producing a cyclic ester in which the aliphatic polyester is depolymerized in the state, the cyclic ester produced by the depolymerization is distilled together with the polyalkylene glycol ether, and cyclic esters such as glycolide are recovered from the distillate. Has been.
- Patent Document 3 a depolymerization reaction system containing a glycolic acid oligomer and a polar organic solvent is charged continuously or intermittently with a glycolic acid oligomer or a mixture of a glycolic acid oligomer and a polar organic solvent, and an alcoholic hydroxyl group.
- a process for producing glycolide in which a depolymerization reaction is continuously carried out by controlling the amount of a compound having a diol is disclosed.
- the depolymerization reaction in addition to enabling mass production of glycolide, the depolymerization reaction can be carried out stably.
- the production rate of glycolide is reduced due to impurities accumulated in the depolymerization reaction system. And the formation of heavy materials can be suppressed.
- the mixture containing the glycolic acid oligomer and the polar organic solvent in the reaction tank is heated for depolymerization, and the produced glycolide is co-distilled with the polar organic solvent.
- the distillate is led out of the depolymerization reaction system through lines such as piping and heat exchangers.
- the depolymerization reaction is usually carried out under reduced pressure.
- the distillate is cooled by a heat exchanger and liquefied.
- Glycolide is recovered from the liquid co-distillate.
- the polar organic solvent contained in the co-distillate is refluxed into the depolymerization reaction system.
- a new glycolic acid oligomer is added to the depolymerization reaction system.
- glycolide obtained by depolymerization of glycolic acid oligomers is not sufficiently high in purity and is called crude glycolide.
- Glycolide used as a monomer for ring-opening polymerization is required to have a high purity of 99.9% or more. Therefore, the crude glycolide obtained by depolymerization is highly purified by a purification treatment such as recrystallization or washing. If the purity of the crude glycolide is low, the purification cost cannot be reduced, and in addition, the purification process may cause blockage of the line.
- the main cause of line blockage in the depolymerization reaction is that impurities contained in the fraction distilled from the depolymerization reaction system act as a polymerization initiator, and glycolide produced and distilled by depolymerization is an oligomer in the middle of the line. It is estimated that this oligomer is attached to the surface of each part of the apparatus. In fact, the crude glycolide obtained by depolymerization contains various impurities.
- Patent Document 4 discloses that a high purity glycolide is obtained by adopting a method in which a glycolic acid oligomer and a high-boiling polar organic solvent-containing mixture are totally refluxed and then depolymerized. ing. That is, a mixture containing a glycolic acid oligomer and a high-boiling polar organic solvent having a boiling point in the range of 230 to 450 ° C.
- a process for producing glycolide comprising a step of carrying out a total reflux treatment for a reflux time within a range of 0.1 to 20 hours under the condition that substantially the entire amount of the distillate distilled from is refluxed into the reflux system
- the amount of impurities in the distillate distilled from the depolymerization reaction system is reduced, line blockage due to oligomerization of glycolide due to impurities is suppressed, and high purity glycolide is obtained by depolymerization. Is.
- the total reflux treatment means that all fractions distilled during the reflux treatment are cooled, and substantially all of the distillate is returned to the reflux system composed of the original mixture. Therefore, distillates such as polar organic solvents are not discharged out of the reflux system during the total reflux treatment.
- the mixture containing the glycolic acid oligomer and the polar organic solvent is heated to distill, then the entire distillate is cooled, so that substantially the entire amount of the distillate is reduced to the original mixture.
- a new thermal history may be added to the glycolic acid oligomer and the polar organic solvent, and there is a problem that the consumption of thermal energy increases due to distillation. there were.
- poly ( ⁇ -oxyacid) having a reduced viscosity of 0.1 dl / g or more and a carboxyl group concentration of 200 equivalents / 10 6 g or less (same as “200 eq / t or less”).
- a method for producing an aliphatic polyester is disclosed in which a cyclic dimer (lactide) obtained by producing a precursor polymer is heated and depolymerized and then subjected to ring-opening polymerization.
- a depolymerization method for obtaining lactide or glycolide as a cyclic dimer from a precursor polymer of (oxyacid) the temperature is 210 ° C. or 230 ° C., and the pressure is 0.05 mmHg (corresponding to 0.0067 kPa). It required high vacuum conditions and was not practical.
- JP-A-9-328481 (corresponding to US Pat. No. 5,830,991) International Publication No. 2002/014303 (corresponding to US Patent Application Publication No. 2003/0191326) JP 2004-523596 (corresponding to US Patent Application Publication No. 2004/0122240) International Publication No. 2010/073512 (corresponding to US Patent Application Publication No. 2011/0263875) Japanese Patent Laid-Open No. 5-287056
- the problem of the present invention is that a new heat history as in the total reflux treatment is not required, and while contributing to energy saving, the amount of impurities in the distillate distilled from the depolymerization reaction system is reduced, resulting from impurities.
- An object of the present invention is to provide a method for producing glycolide in which blockage of lines due to oligomerization of glycolide is suppressed and high-purity glycolide is obtained by depolymerization.
- glycols obtained when producing glycolic acid oligomers as raw materials for depolymerization. It has been found that high purity glycolide can be obtained continuously by reducing the terminal carboxyl group concentration of the acid oligomer, and the present invention has been completed.
- the glycolic acid oligomer is heated and depolymerized.
- a process for producing glycolide is provided, comprising the steps of:
- the following methods (1) to (8) for producing glycolide are provided as embodiments.
- (2) A glycolic acid oligomer having a terminal carboxyl group concentration of 400 eq / t or less is heated under normal pressure or reduced pressure together with a condensation step in which glycolic acid is heated under normal pressure or reduced pressure to perform a condensation reaction, and a polar organic solvent.
- the method for producing glycolide as described above which is prepared by a method for producing a glycolic acid oligomer, comprising a dehydration step in which the condensation reaction of glycolic acid is continued.
- a condensation step in which a glycolic acid oligomer having a terminal carboxyl group concentration of 400 eq / t or less is heated under normal pressure or reduced pressure to cause a condensation reaction, and a depolymerization reaction solution obtained from a depolymerization reaction system
- the glycolide prepared by the method for producing a glycolic acid oligomer further comprising a dehydration step of continuing the condensation reaction of glycolic acid by continuing heating under normal pressure or reduced pressure together with a polar organic solvent as desired. Production method.
- a glycolic acid oligomer in which a glycolic acid oligomer is heated and depolymerized, I. Heating the mixture containing a glycolic acid oligomer having a terminal carboxyl group concentration of 400 eq / t or less and a polar organic solvent to a temperature at which the glycolic acid oligomer is depolymerized under normal pressure or reduced pressure; II. Further heating is continued at the above temperature to depolymerize the glycolic acid oligomer, and glycolide produced by depolymerization from the depolymerization reaction system containing the mixture is co-distilled with the polar organic solvent outside the depolymerization reaction system. Step 2; and III.
- the glycolide production method of the present invention is a glycolide production method in which a glycolic acid oligomer is heated and depolymerized.
- the glycolic acid oligomer has a terminal carboxyl group concentration of 400 eq. It is characterized in that it is a glycolic acid oligomer of / t or less.
- the glycolic acid oligomer (hereinafter sometimes referred to as “GAO”) as a starting material in the glycolide production method of the present invention has a weight average molecular weight of 3000 or more, preferably 5000 or more, more preferably 7000 or more. (Co) polymer.
- the upper limit of the weight average molecular weight of GAO is usually about 20000, and in many cases about 15000.
- the weight average molecular weight is a value measured using gel permeation chromatography (GPC).
- GAO can be obtained by subjecting glycolic acid (which may be an ester or salt thereof) to a condensation reaction.
- the produced GAO can be used as it is as a raw material for producing glycolide, or it can be washed with an insoluble solvent such as benzene or toluene to give an unreacted product, a low polymer or a catalyst. It can also be used after removing.
- GAO desirably has a melting point (Tm) of usually 140 ° C. or higher, preferably 160 ° C. or higher, more preferably 180 ° C. or higher from the viewpoint of the yield of glycolide in the depolymerization reaction.
- the melting point is a temperature detected using a differential scanning calorimeter (DSC).
- the upper limit of the melting point of GAO is about 220 ° C.
- GAO used in the present invention is GAO having a terminal carboxyl group concentration of 400 eq / t or less.
- the terminal carboxyl group concentration of GAO is measured by the following method. That is, 0.1 g of GAO is completely dissolved in 10 ml of special grade dimethyl sulfoxide in an oil bath at a temperature of 150 ° C. over about 3 minutes. After adding 2 drops of an indicator (0.1% by mass of bromothymol blue / alcohol solution) to the solution, add 0.05 normal diazabicycloundecene / dimethylsulfoxide solution. The point where the color changes from yellow to blue is the end point.
- the terminal carboxyl group concentration of GAO is calculated as the equivalent (eq) per GAOt (ton).
- concentration of GAO contained in the mixture containing GAO and a polar organic solvent is measured with the following method. That is, 0.1 g of the mixture is completely dissolved in 10 ml of special grade dimethyl sulfoxide in an oil bath at a temperature of 150 ° C. for about 3 minutes. After adding 2 drops of the indicator to the solution, a 0.009N diazabicycloundecene / dimethyl sulfoxide solution is added, and the point at which the color of the solution visually changes from yellow to blue is the end point.
- the equivalent (eq) per 1 t (ton) of the mixture is first calculated, and then divided by the mass ratio of GAO in the mixture, thereby obtaining the equivalent (eq) per 1 t (ton) of GAO as GAO.
- the terminal carboxyl group concentration is calculated.
- the terminal carboxyl group concentration of GAO is preferably 360 eq / t or less, more preferably 250 eq / t or less, and still more preferably 245 eq / t or less.
- the terminal carboxyl group concentration of GAO can be 160 eq / t or less, further 150 eq / t or less, and if desired 140 eq / t or less.
- the lower limit of the terminal carboxyl group concentration of GAO is not particularly limited, but is usually about 50 eq / t, and in many cases about 70 eq / t. If the terminal carboxyl group concentration of GAO is too high, high-purity glycolide cannot be obtained in the glycolide production method in which GAO is heated and depolymerized, and the long-term continuous operation is caused by blockage of the glycolide production line. May be difficult.
- GAO having a terminal carboxyl group concentration of 400 eq / t or less is a glycol step in which glycolic acid is heated under normal pressure or reduced pressure to undergo a condensation reaction, and with a polar organic solvent, heating is continued under normal pressure or reduced pressure. It can be prepared by a method for producing a glycolic acid oligomer including a dehydration step in which the acid condensation reaction is continued.
- glycolic acid which may be an ester or a salt thereof
- an aqueous solution a commercially available 70% aqueous solution is available
- the condensation step is carried out by heating at a temperature of 140 to 230 ° C. for 30 minutes to 15 hours, preferably 1 to 10 hours to conduct a condensation reaction while distilling water or the like.
- a condensation catalyst or a transesterification catalyst may not be used, but a condensation reaction may be performed in the presence of a catalyst such as a tin compound or an antimony compound.
- the condensation step can be carried out under normal pressure or reduced pressure, specifically 0.1 to 90 kPa, preferably 1 to 60 kPa, but preferably under normal pressure.
- a GAO having a terminal carboxyl group concentration of 400 eq / t or less is prepared by performing a dehydration step in which the condensation reaction of glycolic acid is continued with a polar organic solvent under normal pressure or reduced pressure. can do.
- polar organic solvent As the polar organic solvent used in the dehydration step, those used as a solvent for the depolymerization reaction of GAO can be used. Specifically, a polar organic solvent having a boiling point under normal pressure of 230 to 450 ° C., preferably 255 to 430 ° C., more preferably 280 to 420 ° C. can be used. The molecular weight of the polar organic solvent is preferably in the range of 150 to 450, more preferably 180 to 420, and still more preferably 200 to 400. The amount of the polar organic solvent used in the dehydration step is usually 10 to 100 parts by mass, preferably 15 to 80 parts by mass, and more preferably 18 to 60 parts by mass with respect to 100 parts by mass of glycolic acid. By performing the dehydration step in the presence of a polar organic solvent, the condensation reaction or dehydration reaction of glycolic acid proceeds efficiently.
- polar organic solvents include aromatic dicarboxylic acid diesters, aromatic carboxylic acid esters, aliphatic dicarboxylic acid diesters, polyalkylene glycol diethers, aromatic dicarboxylic acid dialkoxyalkyl esters, aliphatic dicarboxylic acid dialkoxyalkyl esters, Examples include polyalkylene glycol diesters and aromatic phosphate esters.
- aromatic dicarboxylic acid diesters aromatic carboxylic acid esters, aliphatic dicarboxylic acid diesters, and polyalkylene glycol diethers are preferred, and polyalkylene glycol diethers are more preferred because they are less susceptible to thermal degradation.
- Polyalkylene glycol diether having a molecular weight of 150 to 450 is preferred.
- aromatic dicarboxylic acid diester examples include phthalic acid esters such as dibutyl phthalate, dioctyl phthalate, dibenzyl phthalate, and benzyl butyl phthalate.
- aromatic carboxylic acid ester examples include benzoic acid esters such as benzyl benzoate.
- aliphatic dicarboxylic acid diester examples include adipic acid esters such as dioctyl adipate and sebacic acid esters such as dibutyl sebacate.
- Particularly preferably used polyalkylene glycol diethers having a molecular weight of 150 to 450 include diethylene glycol dibutyl ether, diethylene glycol dihexyl ether, diethylene glycol dioctyl ether, diethylene glycol butyl 2-chlorophenyl ether, triethylene glycol diethyl ether, triethylene glycol dipropyl ether, Triethylene glycol dibutyl ether, triethylene glycol dihexyl ether, triethylene glycol dioctyl ether, triethylene glycol butyl octyl ether, triethylene glycol butyl decyl ether, tetraethylene glycol diethyl ether, tetraethylene glycol dipropyl ether, tetraethylene glycol Dibutyl ether, tetraethylene glycol dihexyl ether, tetraethylene glycol dioctyl ether, diethylene glycol butyl hexyl
- a polyalkylene glycol alkyl aryl ether such as a polypropylene glycol alkyl aryl ether or a polybutylene glycol alkyl aryl ether containing a propyleneoxy group or a butyleneoxy group; diethylene glycol diphenyl ether; Polyethylene glycol diaryl ethers such as triethylene glycol diphenyl ether, tetraethylene glycol diphenyl ether or compounds in which the phenyl group of these compounds is substituted with alkyl, alkoxy, halogen, etc .; in the polyethylene glycol diaryl ether, propylene instead of ethyleneoxy group Oxy group or butyleneoxy group Polyalkylene glycol diaryl ethers such as free polypropylene glycol diaryl ethers or polybutylene glycol diaryl ethers; and the like.
- solubilizer In the dehydration step, a polar organic solvent can be used alone, but since the purity of glycolide obtained by depolymerization is further improved, the dehydration step is preferably carried out in the presence of a solubilizing agent.
- the solubilizer is preferably a compound that satisfies any one or more of the following requirements.
- the compound is compatible or soluble in a polar organic solvent such as polyalkylene glycol diether. Any compound that is compatible or soluble in a polar organic solvent may be liquid or solid at room temperature.
- a compound having a functional group such as OH group, COOH group, and CONH group.
- Affinity with glycolic acid oligomer is higher than that of polar organic solvent.
- monohydric alcohol and polyhydric alcohol are particularly effective.
- a monohydric or polyhydric alcohol having a boiling point of 180 ° C. or higher, preferably 200 ° C. or higher, more preferably 230 ° C. or higher, particularly preferably 250 ° C. or higher can be used.
- monohydric or polyhydric alcohols examples include aliphatic alcohols such as decanol, tridecanol, decanediol, ethylene glycol, propylene glycol, and glycerin; aromatic alcohols such as cresol, chlorophenol, and naphthyl alcohol; polyethylene glycol, polypropylene glycol, and polybutylene.
- polyalkylene glycol such as glycol; polyalkylene glycol monoether and the like. Polyalkylene glycol monoether having a boiling point of 180 ° C. or higher is particularly preferable.
- polyalkylene glycol monoether examples include polyethylene glycol monomethyl ether, polyethylene glycol monoethyl ether, polyethylene glycol monopropyl ether, polyethylene glycol monobutyl ether, polyethylene glycol monohexyl ether, polyethylene glycol monooctyl ether, polyethylene glycol mono Polyethylene glycol monoethers such as decyl ether and polyethylene glycol monolauryl ether; in the polyethylene glycol monoether, polyalkylenes such as polypropylene glycol monoether and polybutylene glycol monoether in which ethyleneoxy group is replaced with propyleneoxy group or butyleneoxy group Glycol monoe Ether; and the like.
- the polyalkylene glycol monoether preferably has an alkyl group having 1 to 18 carbon atoms as its ether group, and more preferably has an alkyl group having 6 to 18 carbon atoms. These can be used alone or in combination of two or more.
- polyalkylene glycol monoethers polyethylene glycol monoalkyl ethers such as triethylene glycol monooctyl ether (octyltriethylene glycol) are preferable.
- the amount of solubilizing agent used is usually 10 to 100 parts by mass, preferably 15 to 80 parts by mass, more preferably 18 to 100 parts by mass of glycolic acid. ⁇ 60 parts by mass.
- a condensation catalyst or a transesterification catalyst may not be used, but may be performed in the presence of a catalyst such as a tin compound such as tin chloride or an antimony compound.
- the amount of the catalyst used is usually 10 to 100 mg, preferably 15 to 80 mg, more preferably 18 to 60 mg with respect to 100 parts by mass of glycolic acid.
- a mixture containing glycolic acid and a polar organic solvent and, if necessary, a solubilizer is added under normal pressure or reduced pressure, preferably 0.1 to 90 kPa, more preferably 1 to 60 kPa, still more preferably 1
- the heating is continued under a reduced pressure of 5 to 40 kPa, particularly preferably 2 to 30 kPa, to continue the condensation reaction of glycolic acid.
- the temperature at which the dehydration step is performed is a temperature at which the condensation reaction continues, and is usually a temperature of 100 to 250 ° C., preferably 120 to 230 ° C., more preferably 130 to 224 ° C., and still more preferably 140 to 221 ° C.
- the temperature is lower than the boiling point of the polar organic solvent, and low boiling point components such as water and unreacted raw material are boiled and discharged.
- the time for performing the dehydration step may be continued until the terminal carboxyl group concentration of the obtained GAO is 400 eq / t or less, preferably 250 eq / t or less, usually 30 minutes to 12 hours, preferably 1 to 10 hours, More preferably, it is 2 to 9 hours.
- the dehydration step can be carried out using a depolymerization reaction solution described in detail later in addition to the polar organic solvent or in place of the polar organic solvent. That is, GAO having a terminal carboxyl group concentration of 400 eq / t or less is desired together with a condensation step in which glycolic acid is heated under normal pressure or reduced pressure to undergo a condensation reaction, and a depolymerization reaction solution obtained from the depolymerization reaction system. Further, it can be prepared by a GAO production method including a dehydration step in which the condensation reaction of glycolic acid is continued by continuing heating under normal pressure or reduced pressure together with a polar organic solvent.
- a reaction solution and a depolymerization reaction solution after the condensation step containing glycolic acid, and a mixture containing a polar organic solvent and / or a solubilizing agent as necessary are subjected to normal pressure or reduced pressure. Then, heating is continued and the condensation reaction of glycolic acid is continued.
- the amount of depolymerization reaction solution used, the type and amount of polar organic solvent and / or solubilizing agent used are the same as in the case of using a polar organic solvent.
- the polar organic solvent used in the dehydration step becomes unnecessary or the amount used is reduced. This is economically advantageous.
- the depolymerization reaction solution used in the dehydration step usually contains GAO, and the depolymerization reaction solution is in a high-temperature state usually exceeding 200 ° C., the terminal carboxyl group concentration is 400 eq / It is possible to economically prepare GAO below t, and it is also efficient in terms of energy consumption.
- the present invention relates to a method for producing glycolide for depolymerization by heating GAO, I.
- II. The process of further heating at the temperature to depolymerize GAO and co-distilling glycolide produced by depolymerization from the depolymerization reaction system containing the mixture together with a polar organic solvent to the outside of the depolymerization reaction system 2; and III.
- a process for producing glycolide comprising the steps of:
- Step 1 The method for producing glycolide in which GAO of the present invention is depolymerized by heating is prepared by dissolving GAO having a terminal carboxyl group concentration of 400 eq / t or less and a polar organic solvent under normal pressure or reduced pressure. It includes step 1 of heating to the temperature for polymerization.
- the GAO having a terminal carboxyl group concentration of 400 eq / t or less is as described above, and as the polar organic solvent, the solvents mentioned above for the dehydration step can be used.
- the polar organic solvent is usually used in a proportion of 30 to 5000 parts by mass, preferably 50 to 2000 parts by mass with respect to 100 parts by mass of GAO.
- the ratio of the polar organic solvent is too small, the ratio of the GAO solution phase in the mixture containing GAO and the polar organic solvent decreases under the GAO depolymerization temperature condition (the ratio of the GAO melt phase).
- the depolymerization reactivity of GAO decreases.
- the proportion of the polar organic solvent becomes too large, the thermal efficiency in the depolymerization reaction is lowered, and the productivity of glycolide by the depolymerization reaction is lowered.
- the usage-amount of a polar organic solvent means the total amount with the remaining polar organic solvent, when the polar organic solvent used at the above-mentioned dehydration process remains in GAO to depolymerize.
- the mixture in step 1 may further contain a solubilizer.
- a solubilizer the solubilizers mentioned above for the dehydration step can be used.
- the solubilizer is usually used at a ratio of 0.1 to 500 parts by mass, preferably 1 to 300 parts by mass with respect to 100 parts by mass of GAO.
- GAO molar ratio of GAO to solubilizer
- the mixture in step 1 may further contain a catalyst.
- a tin compound such as tin chloride, an antimony compound, or the like can be used.
- step 1 the mixture containing GAO and a polar organic solvent and, if necessary, a solubilizer and / or a catalyst is heated to a temperature at which the GAO is depolymerized under normal pressure or reduced pressure.
- the temperature at which GAO is depolymerized varies depending on the degree of pressure reduction and the type of polar organic solvent, but is generally a temperature of 200 ° C. or higher. Therefore, the heating temperature is usually 200 to 350 ° C., preferably 210 to 310 ° C. Preferably it is in the range of 222 to 300 ° C, particularly preferably 226 to 290 ° C.
- step 1 of heating to a temperature at which GAO is depolymerized it is preferable to form a solution phase of GAO, and the residual ratio of the melt phase of GAO is 0.5 or less, more preferably 0.3 or less, particularly preferably zero. It is desirable to carry out the depolymerization reaction under the following conditions. That is, the depolymerization reaction is carried out in a substantially homogeneous solution phase state in which the residual ratio of the melt phase of GAO is substantially zero, particularly in order to efficiently obtain high purity glycolide. preferable.
- Step 2 The method for producing glycolide in which GAO of the present invention is depolymerized by heating is further heated at the heating temperature in step 1 to depolymerize GAO and depolymerize from the depolymerization reaction system containing the mixture.
- the heating in step 2 is performed under normal pressure or reduced pressure, but it is preferable to perform under reduced pressure of 0.1 to 90 kPa (0.75 to 675 mmHg) because the depolymerization reaction can be performed at a low temperature.
- the pressure is preferably 1 to 60 kPa (7.5 to 450 mmHg), more preferably 1.5 to 40 kPa (11.3 to 300 mmHg), and particularly preferably 2 to 30 kPa (15 to 225 mmHg).
- the amount of glycolide co-distilled out of the depolymerization reaction system together with the polar organic solvent can be adjusted by adjusting the amount of heat input.
- the process for producing glycolide by heating and depolymerizing GAO according to the present invention is the process 3 for obtaining glycolide from the co-distillate following the process 2 for co-distilling glycolide with the polar organic solvent outside the depolymerization reaction system.
- the distillate is cooled by a heat exchanger (cooler) to be liquefied, and glycolide and the polar organic solvent are liquid-phase separated.
- a glycolide phase (glycolide layer) is formed in the lower layer, and the upper layer becomes a polar organic solvent phase (a layer containing a polar organic solvent).
- Glycolide can be obtained by separating and recovering the glycolide in the lower layer in a liquid state. As described above, the amount of glycolide obtained can be adjusted by adjusting the amount of heat input.
- the cooling temperature is usually controlled within the range of 70 to 180 ° C., preferably 75 to 150 ° C., more preferably 80 to 120 ° C. If the cooling temperature is too high, side reactions such as ring-opening reactions tend to occur in the glycolide phase during the separation and recovery operation. If the cooling temperature is too low, it is difficult to separate the phases in a liquid state.
- glycolide co-distilled with the polar organic solvent passes through the solvent phase of the upper layer of the co-distillate as droplets, and the lower layer. Condensed to the glycolide phase.
- the remaining polar organic solvent phase from which glycolide has been removed from the distillate can be discharged out of the depolymerization reaction system and reused.
- the polar organic solvent may be purified by adsorption on activated carbon, or may be reused after purification by distillation.
- step 2 the glycolide produced by depolymerization is co-distilled with the polar organic solvent outside the depolymerization reaction system, and the depolymerization reaction solution remaining in the depolymerization reaction system contains a polar organic solvent and, if necessary, In addition to solubilizing agents, GAO and various by-products (impurities) are contained. As described above, GAO having a terminal carboxyl group concentration of 400 eq / t or less can be prepared by using the depolymerization reaction solution obtained from this depolymerization reaction system in the dehydration step.
- a depolymerization reaction solution obtained from a depolymerization reaction system after performing the glycolide production method including step 2 once or a plurality of times can be used.
- a depolymerization reaction solution obtained from another depolymerization reaction system can also be used.
- high-purity glycolide can be obtained without performing a total reflux treatment that adds a new heat history.
- the obtained glycolide may be purified by further recrystallization or washing, if desired, but since a high-purity glycolide is obtained, a large amount of solvent is used for purification. There is no need, and the separation operation of the solvent and glycolide is simplified.
- the mother liquor excluding glycolide (the fraction containing a polar organic solvent) can be reused almost without passing through purification or the like.
- a fraction containing a polar organic solvent may be purified by adsorbing it on activated carbon, or may be reused after being purified by distillation.
- the glycolide obtained by the glycolide production method of the present invention (hereinafter sometimes referred to as “crude glycolide”) has a purity of 85% or more from the initial stage of the depolymerization reaction.
- the terminal carboxyl group concentration is 400 eq /
- it is preferably 90% or more, more preferably 91% or more of high purity glycolide.
- the production rate of glycolide does not decrease and the distillation line is not blocked, continuous operation for the production of glycolide for a long time becomes possible.
- the measuring method of the physical property of GAO or glycolide is as follows.
- Terminal carboxyl group concentration of glycolic acid oligomer The terminal carboxyl group concentration of GAO was measured and calculated by the following method. Specifically, 0.1 g of a GAO sample was completely dissolved in 10 ml of special grade dimethyl sulfoxide in an oil bath at 150 ° C. for about 3 minutes to obtain a solution. After adding 2 drops of an indicator (0.1% by mass of bromothymol blue / alcohol solution) to the solution, add 0.05 normal diazabicycloundecene / dimethylsulfoxide solution. The point at which the color changed from yellow to blue was taken as the end point.
- the terminal carboxyl group concentration was calculated as an equivalent (eq) per GAOt (ton).
- concentration of GAO contained in the mixture containing GAO and a polar organic solvent was measured with the following method. That is, 0.1 g of the mixture was completely dissolved in 10 ml of special grade dimethyl sulfoxide in an oil bath at a temperature of 150 ° C. for about 3 minutes to obtain a solution. After adding 2 drops of the indicator to the solution, a 0.009N diazabicycloundecene / dimethylsulfoxide solution was added, and the point at which the color of the solution changed from yellow to blue was determined as the end point.
- the equivalent (eq) per 1 t (ton) of the mixture is first calculated, and then divided by the mass ratio of GAO in the mixture, thereby obtaining the equivalent (eq) per 1 t (ton) of GAO as GAO.
- the terminal carboxyl group concentration of was calculated.
- GAO production method 400 g of a 70% aqueous solution of glycolic acid (manufactured by DuPont) is poured into a 500 ml flask, heated at room temperature to 210 ° C. with stirring at normal pressure over 4 hours, and condensed while distilling water. To produce GAO (condensation step). Next, after adding 60 g of tetraethylene glycol dibutyl ether as a polar organic solvent and 60 g of octyltriethylene glycol as a solubilizer, the pressure is applied from normal pressure to 3 kPa (22.5 mmHg) for 1 hour while maintaining the temperature at 210 ° C. And slowly depressurized.
- the condensation reaction was further continued for 8 hours, and low boiling components such as water and unreacted raw materials were distilled off to prepare 312 g of a mixture containing 192 g of GAO and a polar organic solvent (dehydration step).
- the GAO contained in the obtained mixture had a terminal carboxyl group concentration of 240 eq / t and a melting point (measured with respect to the mixture. The same applies hereinafter) was 206 ° C.
- GAO production method 400 g of a 70% glycolic acid aqueous solution (manufactured by DuPont) is poured into a 500 ml flask, heated at room temperature to 215 ° C. with stirring at normal pressure over 4 hours, and condensed while distilling water. To produce GAO (condensation step). Subsequently, after adding 60 g of tetraethylene glycol dibutyl ether and 60 g of octyltriethylene glycol, the pressure was slowly reduced from normal pressure to 3 kPa over 1 hour while maintaining the temperature at 215 ° C.
- condensation reaction was continued for 6 hours to distill off low-boiling components such as water and unreacted raw materials, thereby preparing 312 g of a mixture containing 192 g of GAO and a polar organic solvent (dehydration step).
- GAO contained in the obtained mixture had a terminal carboxyl group concentration of 213 eq / t and a melting point of 212 ° C.
- GAO production method 400 g of a 70% aqueous solution of glycolic acid (manufactured by DuPont) is poured into a 500 ml volumetric flask, heated at room temperature to a temperature of 220 ° C. with stirring at normal pressure for 4 hours, and condensed while distilling water. To produce GAO (condensation step). Subsequently, after adding 60 g of tetraethylene glycol dibutyl ether and 60 g of octyltriethylene glycol, the pressure was slowly reduced from normal pressure to 3 kPa over 1 hour while maintaining the temperature at 220 ° C.
- condensation reaction was continued for 6 hours to distill off low-boiling components such as water and unreacted raw materials, thereby preparing 312 g of a mixture containing 192 g of GAO and a polar organic solvent (dehydration step).
- GAO contained in the obtained mixture had a terminal carboxyl group concentration of 187 eq / t and a melting point of 210 ° C.
- GAO production method 400 g of a 70% aqueous solution of glycolic acid (manufactured by DuPont) is poured into a 500 ml volumetric flask, heated at room temperature to a temperature of 220 ° C. with stirring at normal pressure for 4 hours, and condensed while distilling water. To produce GAO (condensation step). Next, after adding 60 g of tetraethylene glycol dibutyl ether, 60 g of octyltriethylene glycol and 64.5 mg of tin chloride as a catalyst, the pressure was slowly reduced from normal pressure to 3 kPa over 1 hour while maintaining the temperature at 220 ° C. .
- condensation reaction was continued for 6 hours to distill off low-boiling components such as water and unreacted raw materials, thereby preparing 312 g of a mixture containing 192 g of GAO and a polar organic solvent (dehydration step).
- GAO contained in the obtained mixture had a terminal carboxyl group concentration of 114 eq / t and a melting point of 213 ° C.
- GAO production method 400 g of a 70% aqueous solution of glycolic acid (manufactured by DuPont) is poured into a 500 ml volumetric flask, heated at room temperature to a temperature of 220 ° C. with stirring at normal pressure for 4 hours, and condensed while distilling water. To generate GAO. Next, while maintaining the temperature at 220 ° C., the pressure was slowly reduced from normal pressure to 3 kPa over 1 hour. Further, the condensation reaction was continued for 3 hours, and low-boiling components such as water and unreacted raw materials were distilled off to prepare 192 g of GAO. The terminal carboxyl group concentration of the obtained GAO was 527 eq / t, and the melting point was 217 ° C.
- Example 1 150 g of the mixture containing GAO and the polar organic solvent prepared in Reference Example 1, 150 g of tetraethylene glycol dibutyl ether as the polar organic solvent, and 40 g of octyltriethylene glycol as the solubilizer were injected into a 500 ml flask under normal pressure. To a temperature of 235 ° C. to make the reaction system a uniform solution (step 1). This solution was depressurized to a pressure of 4.5 kPa (33.8 mmHg) while maintaining the temperature at 235 ° C., and tetraethylene glycol dibutyl ether and the produced glycolide were co-distilled.
- Example 2 Instead of GAO prepared in Reference Example 1, tetraethylene glycol dibutyl ether and 150 g of a mixture containing GAO prepared in Reference Example 2 and a polar organic solvent were used. The produced glycolide was co-distilled, and 12.8 g of glycolide was obtained from the co-distillate. There was no blockage of the line and the purity of glycolide was 92.4%.
- Example 3 Instead of GAO prepared in Reference Example 1, tetraethylene glycol dibutyl ether and 150 g of a mixture containing GAO prepared in Reference Example 3 and a polar organic solvent were used. The produced glycolide was co-distilled to obtain 13.1 g of glycolide from the co-distillate. There was no blockage of the line and the purity of glycolide was 93.3%.
- Example 4 In place of GAO prepared in Reference Example 1, tetraethylene glycol dibutyl ether and 150 g of the mixture containing GAO prepared in Reference Example 4 and a polar organic solvent were used, except that The produced glycolide was co-distilled to obtain 17.4 g of glycolide from the co-distillate. There was no blockage of the line and the glycolide purity was 96.2%.
- terminal carboxyl group concentration of GAO used for depolymerization hereinafter sometimes referred to as “terminal COOH”
- melting point the terminal carboxyl group concentration of GAO used for depolymerization
- GAO production method the total reflux treatment Table 1 shows the presence or absence, the depolymerization conditions, and the yield and purity of the resulting glycolide (hereinafter sometimes referred to as “GL”).
- the distillate distilled from the depolymerization reaction system does not require a new heat history due to the total reflux treatment and contributes to energy saving. It was found that the amount of impurities in the product was reduced, the blockage of the line due to oligomerization of glycolide due to the impurities was suppressed, and high purity glycolide having a purity of 85% or more, preferably 90% or more could be obtained by depolymerization.
- a condensation step in which glycolic acid is heated under normal pressure or under reduced pressure for a condensation reaction
- a dehydration step in which heating is continued under normal pressure or under reduced pressure with a polar organic solvent to continue the condensation reaction of glycolic acid.
- GAO having a terminal carboxyl group concentration of 400 eq / t or less, preferably 250 eq / t or less can be efficiently prepared by the GAO production methods of Reference Examples 1 to 4.
- the GAO prepared by the GAO manufacturing method of Reference Examples 1 to 4 does not have to be subjected to the total reflux process without performing the total reflux process for applying a thermal history to the GAO once prepared. It was confirmed that high-purity glycolide having the same or higher purity as glycolide obtained by heating and depolymerization can be obtained in high yield.
- Example 4 Furthermore, from Example 4, it was found that by preparing a GAO by carrying out a dehydration step in the presence of a catalyst and depolymerizing it, an extremely high purity glycolide can be obtained in a higher yield.
- the resulting glycolide has a low purity, so that the purity of glycolide is increased. It was found that there was a possibility that the depolymerization of the blocked GAO could not be continued for a long period of time because the amount of impurities other than glycolide was large and the line was blocked due to oligomerization of glycolide.
- GAO production method 300 g of a 70% aqueous solution of glycolic acid (manufactured by DuPont) is poured into a 500 ml volumetric flask, heated at room temperature to a temperature of 220 ° C. over 4 hours with stirring at normal pressure, and condensed while distilling water. To produce GAO (condensation step). Next, while maintaining the temperature at 220 ° C., the pressure was reduced from normal pressure to 10 kPa, and then 165 g of the depolymerization reaction solution obtained from the depolymerization reaction system after repeating step 2 in the glycolide production method of Example 1. Then, the pressure was slowly reduced to 3 kPa over 1 hour.
- the condensation reaction was further continued for 3 hours to distill off low-boiling components such as water and unreacted raw materials, thereby preparing 309 g of a mixture containing GAO containing 210 g of GAO and a polar organic solvent (dehydration step).
- GAO contained in the obtained mixture had a terminal carboxyl group concentration of 390 eq / t.
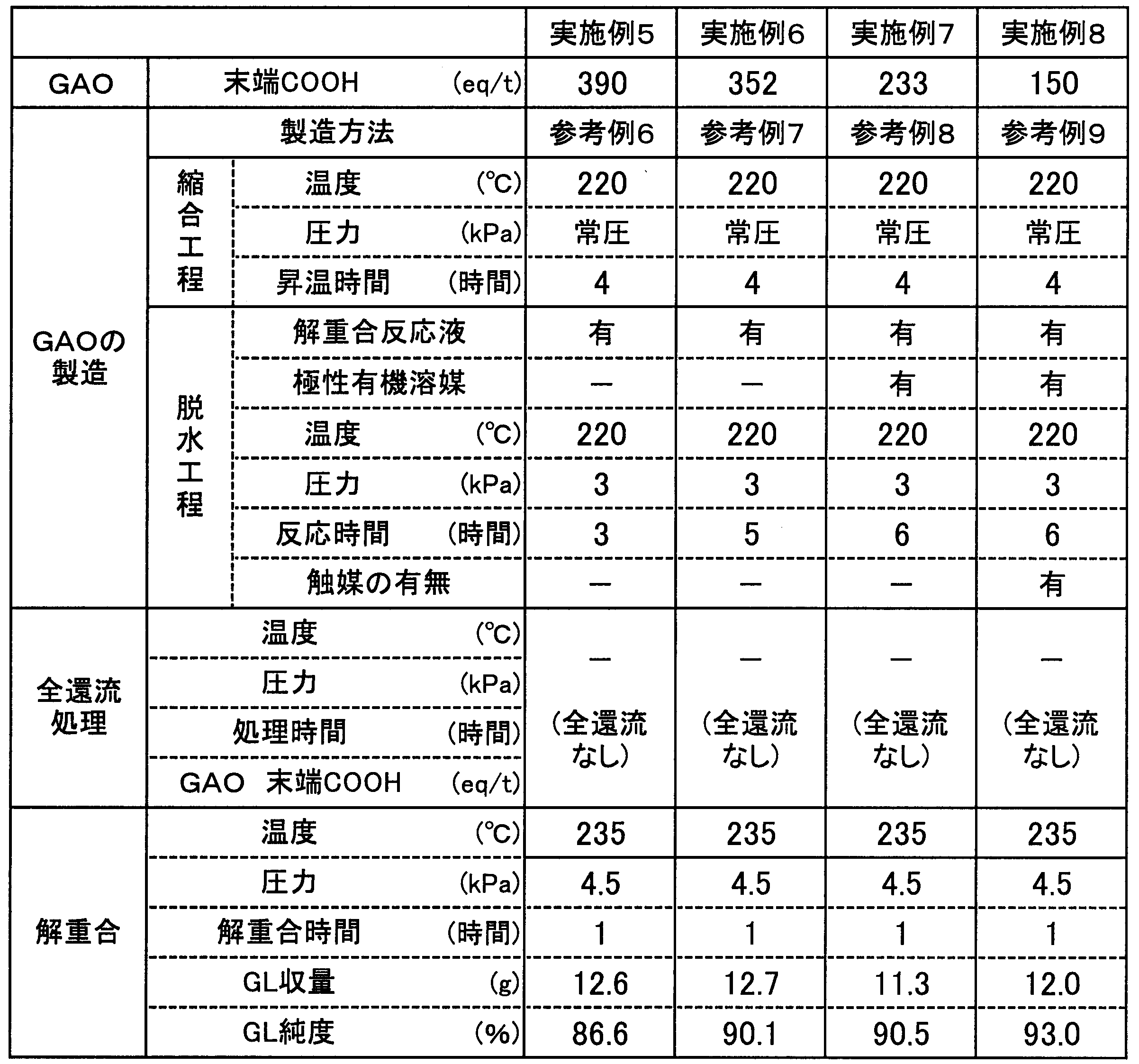
- Example 5 150 g of the mixture containing GAO prepared in Reference Example 6 and a polar organic solvent, 60 g of tetraethylene glycol dibutyl ether as a polar organic solvent, and 60 g of octyltriethylene glycol as a solubilizer were injected into a 500 ml flask under normal pressure. To a temperature of 235 ° C. to make the reaction system a uniform solution (step 1). This solution was depressurized to a pressure of 4.5 kPa (33.8 mmHg) while maintaining the temperature at 235 ° C., and tetraethylene glycol dibutyl ether and the produced glycolide were co-distilled.
- Example 6 In place of GAO prepared in Reference Example 6, tetraethylene glycol dibutyl ether and 150 g of a mixture containing GAO prepared in Reference Example 7 and a polar organic solvent were used. The produced glycolide was co-distilled to obtain 12.7 g of glycolide from the co-distillate. There was no blockage of the line and the purity of glycolide was 90.1%.
- Example 7 Instead of GAO prepared in Reference Example 6, 150 g of a mixture containing GAO prepared in Reference Example 8 and a polar organic solvent was used, and 55 g of tetraethylene glycol dibutyl ether as a polar organic solvent and solubilizer Tetraethylene glycol dibutyl ether and the produced glycolide were co-distilled in the same manner as in Example 5 except that 55 g of octyl triethylene glycol was injected, and 11.3 g of glycolide was obtained from the co-distillate. There was no blockage of the line and the purity of glycolide was 90.5%.
- Example 8 In place of GAO prepared in Reference Example 8 and in the same manner as in Example 7 except that 150 g of a mixture containing GAO prepared in Reference Example 9 and a polar organic solvent was used, tetraethylene glycol dibutyl ether and The produced glycolide was co-distilled to obtain 12.0 g of glycolide from the co-distillate. There was no blockage of the line, and the glycolide purity was 93.0%.
- Table 2 shows the terminal COOH of GAO used for depolymerization, the method for producing GAO, the presence or absence of total reflux treatment, the depolymerization conditions, and the yield and purity of the resulting glycolide (GL). Show.
- glycolide from the distillate 3 According to the method for producing glycolide of Examples 5 to 8 including the steps described above, impurities in the distillate distilled from the depolymerization reaction system while contributing to energy saving without requiring a heat history by total reflux treatment It was found that the amount was reduced, line blockage due to oligomerization of glycolide due to impurities was suppressed, and high purity glycolide having a purity of 85% or more could be obtained by depolymerization.
- a condensation step in which glycolic acid is heated under normal pressure or under reduced pressure for a condensation reaction, and a depolymerization reaction solution obtained from the depolymerization reaction system, and optionally with a polar organic solvent, under normal pressure or under reduced pressure.
- GAO having a terminal carboxyl group concentration of 400 eq / t or less can be efficiently prepared by the method for producing GAO of Reference Examples 6 to 9 including a dehydration step in which the condensation reaction of glycolic acid is continued by continuing heating.
- the GAO prepared by the GAO production method of Reference Examples 6 to 9 can produce high-purity glycolide in a high yield without performing a total reflux treatment in which once prepared GAO is subjected to a thermal history again. It was confirmed that it can be obtained.
- the present invention relates to a method for producing glycolide in which a glycolic acid oligomer is heated and depolymerized.
- I Heating the mixture containing a glycolic acid oligomer having a terminal carboxyl group concentration of 400 eq / t or less and a polar organic solvent to a temperature at which the glycolic acid oligomer is depolymerized under normal pressure or reduced pressure;
- II. Further heating is continued at the above temperature to depolymerize the glycolic acid oligomer, and glycolide produced by depolymerization from the depolymerization reaction system containing the mixture is co-distilled with the polar organic solvent outside the depolymerization reaction system.
- III III.
- glycolide from the distillate 3 In the distillate distilled from the depolymerization reaction system while contributing to energy saving without requiring a new heat history due to the total reflux treatment by being a glycolide production method characterized by comprising each step of The production of glycolide can be provided by reducing the amount of impurities, suppressing the blockage of the line due to oligomerization of glycolide due to impurities, and obtaining high-purity glycolide by depolymerization. Is expensive.
- the present invention includes a condensation step in which a glycolic acid oligomer having a terminal carboxyl group concentration of 400 eq / t or less is heated under normal pressure or reduced pressure to perform a condensation reaction, and a polar organic solvent or a depolymerization reaction solution, Glycolic acid oligomer production method that includes a dehydration step in which heating is continued under normal pressure or reduced pressure to continue the condensation reaction of glycolic acid, and can be efficiently prepared. Even if it does not perform a reflux process, since the manufacturing method of glycolide which can obtain a high purity glycolide with a high yield can be provided, industrial applicability is high.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Polyesters Or Polycarbonates (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description


I.末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、グリコール酸オリゴマーが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、グリコール酸オリゴマーの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含むことを特徴とするグリコリドの製造方法が提供される。
(1)グリコール酸オリゴマーは、末端カルボキシル基濃度が250eq/t以下のグリコール酸オリゴマーである前記のグリコリドの製造方法。
(2)末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーが、グリコール酸を常圧下または減圧下で加熱して縮合反応させる縮合工程、及び、極性有機溶媒とともに、常圧下または減圧下で加熱を継続してグリコール酸の縮合反応を継続させる脱水工程を含むグリコール酸オリゴマーの製造方法により調製されたものである前記のグリコリドの製造方法。
(3)末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーが、グリコール酸を常圧下または減圧下で加熱して縮合反応させる縮合工程、及び、解重合反応系から取得される解重合反応液とともに、所望により更に極性有機溶媒とともに、常圧下または減圧下で加熱を継続してグリコール酸の縮合反応を継続させる脱水工程を含むグリコール酸オリゴマーの製造方法により調製されたものである前記のグリコリドの製造方法。
(4)脱水工程が、可溶化剤の存在下で実施される前記のグリコリドの製造方法。
(5)工程1における混合物が、可溶化剤を含有する前記のグリコリドの製造方法。
(6)脱水工程が、触媒の存在下で実施される前記のグリコリドの製造方法。
(7)極性有機溶媒が、分子量150~450のポリアルキレングリコールジエーテルである前記のグリコリドの製造方法。
(8)可溶化剤が、沸点が180℃以上のポリアルキレングリコールモノエーテルである前記のグリコリドの製造方法。
I.末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、グリコール酸オリゴマーが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、グリコール酸オリゴマーの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含むことを特徴とするグリコリドの製造方法であることによって、全還流処理による新たな熱履歴を要せず、省エネルギーに寄与しながら、解重合反応系から留出する留出物中の不純物量が低減し、不純物に起因するグリコリドのオリゴマー化によるラインの閉塞が抑制され、解重合によって高純度グリコリドが得られるグリコリドの製造方法を提供できるという効果が奏される。
本発明のグリコリドの製造方法は、グリコール酸オリゴマーを加熱して解重合させるグリコリドの製造方法であり、グリコール酸オリゴマーが、末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーである点に特徴を有する。
本発明で使用するGAOは、末端カルボキシル基濃度が400eq/t以下のGAOである。GAOの末端カルボキシル基濃度は、以下の方法により測定する。すなわち、GAO 0.1gを、特級ジメチルスルホキシド10mlに温度150℃のオイルバス中で約3分間かけて完全に溶解させる。その溶液に指示薬(0.1質量%のブロモチモールブルー/アルコール溶液)を2滴加えた後、0.05規定のジアザビシクロウンデセン/ジメチルスルホキシド溶液を加えていき、目視で溶液の色が黄色から青色に変わった点を終点とする。その時の指示薬の滴下量から、GAO1t(トン)あたりの当量(eq)として、GAOの末端カルボキシル基濃度を算出する。また、GAOと極性有機溶媒を含有する混合物に含有されるGAOの末端カルボキシル基濃度は、以下の方法により測定する。すなわち、該混合物 0.1gを、特級ジメチルスルホキシド10mlに温度150℃のオイルバス中で約3分間かけて完全に溶解させる。その溶液に前記指示薬を2滴加えた後、0.009規定のジアザビシクロウンデセン/ジメチルスルホキシド溶液を加えていき、目視で溶液の色が黄色から青色に変わった点を終点とする。その時の指示薬の滴下量から、混合物1t(トン)あたりの当量(eq)をまず算出し、次いで混合物におけるGAOの質量比で割ることで、GAO 1t(トン)あたりの当量(eq)として、GAOの末端カルボキシル基濃度を算出する。GAOの末端カルボキシル基濃度は、好ましくは360eq/t以下、より好ましくは250eq/t以下、更に好ましくは245eq/t以下である。後に説明するように、脱水工程を、更に触媒の存在下で実施すると、GAOの末端カルボキシル基濃度を160eq/t以下、更には150eq/t以下、所望により140eq/t以下とすることができる。GAOの末端カルボキシル基濃度の下限は、特にないが、通常50eq/t程度であり、多くの場合70eq/t程度である。GAOの末端カルボキシル基濃度が大きすぎると、GAOを加熱して解重合させるグリコリドの製造方法において、高純度のグリコリドを得ることができず、また、グリコリドの製造ラインの閉塞により長期間の連続運転が困難となることがある。
末端カルボキシル基濃度が400eq/t以下のGAOは、グリコール酸を常圧下または減圧下で加熱して縮合反応させる縮合工程、及び、極性有機溶媒とともに、常圧下または減圧下で加熱を継続してグリコール酸の縮合反応を継続させる脱水工程を含むグリコール酸オリゴマーの製造方法により調製することができる。
脱水工程において使用する極性有機溶媒としては、GAOの解重合反応の溶媒として使用されているものを使用することができる。具体的には、常圧下の沸点が230~450℃、好ましくは255~430℃、より好ましくは280~420℃の範囲内である極性有機溶媒を用いることができる。また、極性有機溶媒の分子量は、好ましくは150~450、より好ましくは180~420、更に好ましくは200~400の範囲内である。脱水工程における極性有機溶媒の使用量は、グリコール酸100質量部に対して、通常10~100質量部、好ましくは15~80質量部、より好ましくは18~60質量部である。脱水工程を極性有機溶媒の存在下で行うことにより、グリコール酸の縮合反応や脱水反応が効率的に進行する。
脱水工程においては、極性有機溶媒を単独で使用することができるが、解重合によって得られるグリコリドの純度が更に向上することから、脱水工程が、可溶化剤の存在下で実施することが好ましい。可溶化剤は、次の要件のいずれか1つ以上を満たす化合物であることが好ましい。
(2)ポリアルキレングリコールジエーテルなどの極性有機溶媒に相溶性または可溶性の化合物であること。極性有機溶媒に相溶性または可溶性の化合物であれば、常温で液体でも固体でもよい。
(3)沸点が180℃以上、好ましくは200℃以上、より好ましくは230℃以上、特に好ましくは250℃以上の化合物であること。
(4)例えば、OH基、COOH基、CONH基などの官能基を有する化合物であること。
(5)極性有機溶媒よりもグリコール酸オリゴマーとの親和性が高いこと。
本発明は、GAOを加熱して解重合させるグリコリドの製造方法において、
I.末端カルボキシル基濃度が400eq/t以下のGAOと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、GAOが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、GAOの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含むことを特徴とするグリコリドの製造方法である。
本発明のGAOを加熱して解重合させるグリコリドの製造方法は、末端カルボキシル基濃度が400eq/t以下のGAOと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、該GAOが解重合する温度に加熱する工程1を含む。末端カルボキシル基濃度が400eq/t以下のGAOは先に説明したとおりのものであり、また、極性有機溶媒としては、先に脱水工程について挙げた溶媒を使用することができる。極性有機溶媒は、GAO100質量部に対して、通常30~5000質量部、好ましくは50~2000質量部の割合で使用する。極性有機溶媒の割合が小さすぎると、GAOの解重合温度条件下で、GAOと極性有機溶媒とを含有する混合物中での該GAOの溶液相の比率が低下し(GAOの融液相の比率が増大し)、GAOの解重合反応性が低下する。極性有機溶媒の割合が大きくなりすぎると、解重合反応における熱効率が低下し、解重合反応によるグリコリドの生産性が低下する。なお、極性有機溶媒の使用量は、解重合させるGAOに前記した脱水工程で使用した極性有機溶媒が残存している場合、残存している極性有機溶媒との合計量を意味する。
本発明のGAOを加熱して解重合させるグリコリドの製造方法は、工程1の加熱温度で更に加熱を継続して、GAOの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2を含む。すなわち、工程2においては、生成したグリコリド(大気圧下での沸点=240~241℃)が極性有機溶媒と共に留出することにより、留出ラインの内壁にグリコリドが析出して付着することによりラインが閉塞することがなくなり、解重合反応の長時間の継続が可能となる。GAOの解重合反応は可逆反応であるため、グリコリドを解重合反応系から留出させて、解重合反応系外に排出させると、GAOの解重合反応が効率的に進行する。
本発明のGAOを加熱して解重合させるグリコリドの製造方法は、グリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2に続いて、共留出物からグリコリドを取得する工程3を含む。具体的には、共留出物を熱交換器(冷却器)により冷却して液状化し、グリコリドと極性有機溶媒を液状で相分離させる。共留出物を相分離させると、下層にグリコリド相(グリコリド層)が形成され、上層は、極性有機溶媒相(極性有機溶媒を含む層)となる。下層のグリコリドを液状のままで分離回収して、グリコリドを取得することができる。なお、先に説明したように、入熱量を調整することによって、取得されるグリコリド量を調整することができる。液状でグリコリドと極性有機溶媒を相分離させるには、冷却温度を、通常70~180℃、好ましくは75~150℃、より好ましくは80~120℃の範囲内に制御する。冷却温度が高すぎると、分離回収操作の間にグリコリド相において開環反応などの副反応が起こりやすくなる。冷却温度が低すぎると、液状で相分離させることが困難になる。
工程2において、解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させた後に、解重合反応系に残る解重合反応液には、極性有機溶媒や必要に応じて含有する可溶化剤のほかに、GAOや、種々の副生成物(不純物)などが含有されている。先に説明したように、この解重合反応系から取得される解重合反応液を、脱水工程において使用することによって、末端カルボキシル基濃度が400eq/t以下のGAOを調製することができる。脱水工程において使用する解重合反応液としては、工程2を含むグリコリドの製造方法を1回または複数回実施した後の解重合反応系から取得される解重合反応液を使用することができるが、別の解重合反応系から取得される解重合反応液を使用することもできる。
GAOの融点は、示差走査熱量計(DSC)を用いて、不活性ガス雰囲気下、10℃/分の速さで昇温加熱して測定した。GAOを含有する混合物についても同様である。
GAOの末端カルボキシル基濃度は、以下の方法によって測定、算出した。すなわち、GAO試料0.1gを、特級ジメチルスルホキシド10mlに150℃のオイルバス中で約3分間かけて完全に溶解させ溶液とした。その溶液に指示薬(0.1質量%のブロモチモールブルー/アルコール溶液)を2滴加えた後、0.05規定のジアザビシクロウンデセン/ジメチルスルホキシド溶液を加えていき、目視で溶液の色が黄色から青色に変わった点を終点とした。その時の指示薬の滴下量から、GAO1t(トン)あたりの当量(eq)として末端カルボキシル基濃度を算出した。また、GAOと極性有機溶媒を含有する混合物に含有されるGAOの末端カルボキシル基濃度は、以下の方法により測定した。すなわち、該混合物 0.1gを、特級ジメチルスルホキシド10mlに温度150℃のオイルバス中で約3分間かけて完全に溶解させ溶液とした。その溶液に前記指示薬を2滴加えた後、0.009規定のジアザビシクロウンデセン/ジメチルスルホキシド溶液を加えていき、目視で溶液の色が黄色から青色に変わった点を終点とした。その時の指示薬の滴下量から、混合物1t(トン)あたりの当量(eq)をまず算出し、次いで混合物におけるGAOの質量比で割ることで、GAO 1t(トン)あたりの当量(eq)として、GAOの末端カルボキシル基濃度を算出した。
解重合反応により生成したグリコリドの純度は、ガスクロマトグラフィー(GC)により測定した。具体的には、グリコリドの試料200mg及び内部標準物質のp-クロロベンゾフェノン(東京化成工業株式会社製)40mgを、アセトン10mlに溶解させ、その2μlを採取し、ガスクロマトグラフィー装置に注入して、以下の条件でグリコリド量の測定を行い、あらかじめ、グリコリドと標準物質のp-クロロベンゾフェノンとを用いて作成した検量線を用いて、グリコリドの純度を求めた。
<GC条件>
測定装置: 株式会社島津製作所製「GC-2010」
カラム: キャピラリーカラムTC-17、0.25mmφ×30mm
カラム温度: 280℃
インジェクション温度: 150℃
グリコール酸70%水溶液(デュポン社製)400gを、容積500mlのフラスコに注入し、常圧で撹拌しながら室温から温度210℃まで4時間かけて昇温加熱し、水を留出させながら縮合反応を行ってGAOを生成させた(縮合工程)。次いで、極性有機溶媒としてテトラエチレングリコールジブチルエーテル60g及び可溶化剤としてオクチルトリエチレングリコール60gを加えた後、温度210℃を維持したまま、圧力を常圧から3kPa(22.5mmHg)まで1時間かけてゆっくり減圧した。更に8時間縮合反応を続けて、水及び未反応原料等の低沸分を留去させて、GAO192gを含むGAOと極性有機溶媒を含有する混合物312gを調製した(脱水工程)。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が240eq/tであり、融点(混合物について測定した。以下同様である。)は206℃であった。
グリコール酸70%水溶液(デュポン社製)400gを、容積500mlのフラスコに注入し、常圧で撹拌しながら室温から温度215℃まで4時間かけて昇温加熱し、水を留出させながら縮合反応を行ってGAOを生成させた(縮合工程)。次いで、テトラエチレングリコールジブチルエーテル60g及びオクチルトリエチレングリコール60gを加えた後、温度215℃を維持したまま、圧力を常圧から3kPaまで1時間かけてゆっくり減圧した。更に6時間縮合反応を続けて、水及び未反応原料等の低沸分を留去させて、GAO192gを含むGAOと極性有機溶媒を含有する混合物312gを調製した(脱水工程)。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が213eq/tであり、融点は212℃であった。
グリコール酸70%水溶液(デュポン社製)400gを、容積500mlのフラスコに注入し、常圧で撹拌しながら室温から温度220℃まで4時間かけて昇温加熱し、水を留出させながら縮合反応を行ってGAOを生成させた(縮合工程)。次いで、テトラエチレングリコールジブチルエーテル60g及びオクチルトリエチレングリコール60gを加えた後、温度220℃を維持したまま、圧力を常圧から3kPaまで1時間かけてゆっくり減圧した。更に6時間縮合反応を続けて、水及び未反応原料等の低沸分を留去させて、GAO192gを含むGAOと極性有機溶媒を含有する混合物312gを調製した(脱水工程)。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が187eq/tであり、融点は210℃であった。
グリコール酸70%水溶液(デュポン社製)400gを、容積500mlのフラスコに注入し、常圧で撹拌しながら室温から温度220℃まで4時間かけて昇温加熱し、水を留出させながら縮合反応を行ってGAOを生成させた(縮合工程)。次いで、テトラエチレングリコールジブチルエーテル60g、オクチルトリエチレングリコール60g、及び触媒として塩化錫64.5mgを加えた後、温度220℃を維持したまま、圧力を常圧から3kPaまで1時間かけてゆっくり減圧した。更に6時間縮合反応を続けて、水及び未反応原料等の低沸分を留去させて、GAO192gを含むGAOと極性有機溶媒を含有する混合物312gを調製した(脱水工程)。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が114eq/tであり、融点は213℃であった。
グリコール酸70%水溶液(デュポン社製)400gを、容積500mlのフラスコに注入し、常圧で撹拌しながら室温から温度220℃まで4時間かけて昇温加熱し、水を留出させながら縮合反応を行ってGAOを生成させた。次いで、温度220℃を維持したまま、圧力を常圧から3kPaまで1時間かけてゆっくり減圧した。更に3時間縮合反応を続けて、水及び未反応原料等の低沸分を留去させて、GAO192gを調製した。得られたGAOの末端カルボキシル基濃度は、527eq/tであり、融点は217℃であった。
容積500mlのフラスコに、参考例1で調製したGAOと極性有機溶媒を含有する混合物の150g、極性有機溶媒としてテトラエチレングリコールジブチルエーテル40g及び可溶化剤としてオクチルトリエチレングリコール40gを注入し、常圧下で温度235℃まで加熱して、反応系を均一な溶液にした(工程1)。この溶液を、温度235℃を維持したまま圧力4.5kPa(33.8mmHg)に減圧し、テトラエチレングリコールジブチルエーテルと生成したグリコリドとを共留出させた。解重合反応を入熱量を調整しながら1時間継続(工程2)したところ、ラインの閉塞はみられなかった。共留出物からグリコリド12.8gを取得した(工程3)。グリコリドの純度は91.5%であった。
参考例1で調製したGAOに代えて、参考例2で調製したGAOと極性有機溶媒を含有する混合物の150gを使用したことを除いて、実施例1と同様にして、テトラエチレングリコールジブチルエーテルと生成したグリコリドを共留出させ、共留出物からグリコリド12.8gを取得した。ラインの閉塞はみられず、グリコリドの純度は92.4%であった。
参考例1で調製したGAOに代えて、参考例3で調製したGAOと極性有機溶媒を含有する混合物の150gを使用したことを除いて、実施例1と同様にして、テトラエチレングリコールジブチルエーテルと生成したグリコリドを共留出させ、共留出物からグリコリド13.1gを取得した。ラインの閉塞はみられず、グリコリドの純度は93.3%であった。
参考例1で調製したGAOに代えて、参考例4で調製したGAOと極性有機溶媒を含有する混合物の150gを使用したことを除いて、実施例1と同様にして、テトラエチレングリコールジブチルエーテルと生成したグリコリドを共留出させ、共留出物からグリコリド17.4gを取得した。ラインの閉塞はみられず、グリコリドの純度は96.2%であった。
容積500mlのフラスコに、参考例5で調製したGAOの160g、テトラエチレングリコールジブチルエーテル100g、及びオクチルトリエチレングリコール89gを注入し、常圧下で温度230℃まで加熱して、反応系を均一な溶液にした(工程1)。この溶液を、温度230℃を維持したまま圧力4.5kPa(33.8mmHg)に減圧し、テトラエチレングリコールジブチルエーテルと生成したグリコリドを共留出させた。反応を1時間継続(工程2)したところ、ラインの閉塞がやや見受けられた。共留出物からグリコリド12.5gを取得した(工程3)。グリコリドの純度は81.2%であった。
容積500mlのフラスコに、参考例5で調製したGAOの160g、テトラエチレングリコールジブチルエーテル100g、及びオクチルトリエチレングリコール89gを注入し、常圧下で温度225℃まで加熱して、反応系を均一な溶液にした。この溶液を、温度225℃を維持したまま圧力3kPaまで1時間かけてゆっくり減圧し、5時間全還流処理を行って、GAO158gを含むGAOと極性有機溶媒を含有する混合物347gを調製した。この混合物に含有されるGAOは、末端カルボキシル基濃度が170eq/tであり、融点は211℃であった。

I.末端カルボキシル基濃度が400eq/t以下のGAOと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、GAOが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、GAOの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含む実施例1~4のグリコリドの製造方法によれば、全還流処理による新たな熱履歴を要せず、省エネルギーに寄与しながら、解重合反応系から留出する留出物中の不純物量が低減し、不純物に起因するグリコリドのオリゴマー化によるラインの閉塞が抑制され、解重合によって純度85%以上、好ましくは90%以上の高純度グリコリドを得ることができることが分かった。
グリコール酸70%水溶液(デュポン社製)300gを、容積500mlのフラスコに注入し、常圧で撹拌しながら室温から温度220℃まで4時間かけて昇温加熱し、水を留出させながら縮合反応を行ってGAOを生成させた(縮合工程)。次いで、温度220℃を維持したまま、圧力を常圧から10kPaまで減圧させた後、実施例1のグリコリドの製造方法における工程2を繰り返した後の解重合反応系から取得した解重合反応液165gを加え、その後更に3kPaまで1時間かけてゆっくり減圧した。更に3時間縮合反応を続けて、水及び未反応原料等の低沸分を留去させて、GAO210gを含むGAOと極性有機溶媒を含有する混合物309gを調製した(脱水工程)。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が390eq/tであった。
脱水工程における縮合反応を5時間続けたことを除いて、参考例6と同様にして、GAO210gを含むGAOと極性有機溶媒を含有する混合物309gを調製した。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が352eq/tであった。
脱水工程において、前記の解重合反応液165gと、テトラエチレングリコールジブチルエーテル24g及びオクチルトリエチレングリコール24gとを加えたこと、並びに、縮合反応を6時間続けたことを除いて、参考例6と同様にして、GAO210gを含むGAOと極性有機溶媒を含有する混合物357gを調製した。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が233eq/tであった。
脱水工程において、前記の解重合反応液165gと、テトラエチレングリコールジブチルエーテル24g及びオクチルトリエチレングリコール24gとに加えて、触媒として塩化錫59.4mgとを加えたこと、並びに、縮合反応を6時間続けたことを除いて、参考例6と同様にして、GAO210gを含むGAOと極性有機溶媒を含有する混合物357gを調製した。得られた混合物に含有されるGAOは、末端カルボキシル基濃度が150eq/tであった。
容積500mlのフラスコに、参考例6で調製したGAOと極性有機溶媒を含有する混合物の150g、極性有機溶媒としてテトラエチレングリコールジブチルエーテル60g及び可溶化剤としてオクチルトリエチレングリコール60gを注入し、常圧下で温度235℃まで加熱して、反応系を均一な溶液にした(工程1)。この溶液を、温度235℃を維持したまま圧力4.5kPa(33.8mmHg)に減圧し、テトラエチレングリコールジブチルエーテルと生成したグリコリドとを共留出させた。解重合反応を入熱量を調整しながら1時間継続(工程2)したところ、ラインの閉塞はみられなかった。共留出物からグリコリド12.6gを取得した(工程3)。グリコリドの純度は86.6%であった。
参考例6で調製したGAOに代えて、参考例7で調製したGAOと極性有機溶媒を含有する混合物の150gを使用したことを除いて、実施例5と同様にして、テトラエチレングリコールジブチルエーテルと生成したグリコリドとを共留出させ、共留出物からグリコリド12.7gを取得した。ラインの閉塞はみられず、グリコリドの純度は90.1%であった。
参考例6で調製したGAOに代えて、参考例8で調製したGAOと極性有機溶媒を含有する混合物の150gを使用したこと、並びに、極性有機溶媒としてテトラエチレングリコールジブチルエーテル55g及び可溶化剤としてオクチルトリエチレングリコール55gを注入したことを除いて、実施例5と同様にして、テトラエチレングリコールジブチルエーテルと生成したグリコリドを共留出させ、共留出物からグリコリド11.3gを取得した。ラインの閉塞はみられず、グリコリドの純度は90.5%であった。
参考例8で調製したGAOに代えて、参考例9で調製したGAOと極性有機溶媒を含有する混合物の150gを使用したことを除いて、実施例7と同様にして、テトラエチレングリコールジブチルエーテルと生成したグリコリドを共留出させ、共留出物からグリコリド12.0gを取得した。ラインの閉塞はみられず、グリコリドの純度は93.0%であった。
I.末端カルボキシル基濃度が400eq/t以下のGAOと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、GAOが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、GAOの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含む実施例5~8のグリコリドの製造方法によれば、全還流処理による熱履歴を要せず、省エネルギーに寄与しながら、解重合反応系から留出する留出物中の不純物量が低減し、不純物に起因するグリコリドのオリゴマー化によるラインの閉塞が抑制され、解重合によって純度85%以上の高純度グリコリドを得ることができることが分かった。
I.末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、グリコール酸オリゴマーが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、グリコール酸オリゴマーの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含むことを特徴とするグリコリドの製造方法であることによって、全還流処理による新たな熱履歴を要せず、省エネルギーに寄与しながら、解重合反応系から留出する留出物中の不純物量が低減し、不純物に起因するグリコリドのオリゴマー化によるラインの閉塞が抑制され、解重合によって高純度グリコリドが得られるグリコリドの製造方法を提供することができるので、産業上の利用可能性が高い。
Claims (9)
- グリコール酸オリゴマーを加熱して解重合させるグリコリドの製造方法において、
I.末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーと極性有機溶媒とを含有する混合物を、常圧下または減圧下に、グリコール酸オリゴマーが解重合する温度に加熱する工程1;
II.前記温度で更に加熱を継続して、グリコール酸オリゴマーの解重合を行うとともに、該混合物を含有する解重合反応系から解重合により生成したグリコリドを極性有機溶媒と共に解重合反応系外に共留出させる工程2;並びに
III.共留出物からグリコリドを取得する工程3;
の各工程を含むことを特徴とするグリコリドの製造方法。 - グリコール酸オリゴマーは、末端カルボキシル基濃度が250eq/t以下のグリコール酸オリゴマーである請求項1記載のグリコリドの製造方法。
- 末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーが、グリコール酸を常圧下または減圧下で加熱して縮合反応させる縮合工程、及び、極性有機溶媒とともに、常圧下または減圧下で加熱を継続してグリコール酸の縮合反応を継続させる脱水工程を含むグリコール酸オリゴマーの製造方法により調製されたものである請求項1または2記載のグリコリドの製造方法。
- 末端カルボキシル基濃度が400eq/t以下のグリコール酸オリゴマーが、グリコール酸を常圧下または減圧下で加熱して縮合反応させる縮合工程、及び、解重合反応系から取得される解重合反応液とともに、常圧下または減圧下で加熱を継続してグリコール酸の縮合反応を継続させる脱水工程を含むグリコール酸オリゴマーの製造方法により調製されたものである請求項1乃至3のいずれか1項に記載のグリコリドの製造方法。
- 脱水工程が、可溶化剤の存在下で実施される請求項3または4記載のグリコリドの製造方法。
- 工程1における混合物が、可溶化剤を含有する請求項1乃至5のいずれか1項に記載のグリコリドの製造方法。
- 脱水工程が、触媒の存在下で実施される請求項3乃至6のいずれか1項に記載のグリコリドの製造方法。
- 極性有機溶媒が、分子量150~450のポリアルキレングリコールジエーテルである請求項1乃至7のいずれか1項に記載のグリコリドの製造方法。
- 可溶化剤が、沸点が180℃以上のポリアルキレングリコールモノエーテルである請求項5乃至8のいずれか1項に記載のグリコリドの製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14775676.1A EP2980084B1 (en) | 2013-03-26 | 2014-03-25 | Method for producing glycolide |
| CN201480004299.6A CN104903306B (zh) | 2013-03-26 | 2014-03-25 | 乙交酯的制备方法 |
| US14/766,829 US9512100B2 (en) | 2013-03-26 | 2014-03-25 | Method for producing glycolide |
| KR1020157018421A KR101729877B1 (ko) | 2013-03-26 | 2014-03-25 | 글리콜리드의 제조방법 |
| JP2015508513A JP6230597B2 (ja) | 2013-03-26 | 2014-03-25 | グリコリドの製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013063841 | 2013-03-26 | ||
| JP2013-063841 | 2013-03-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014157140A1 true WO2014157140A1 (ja) | 2014-10-02 |
Family
ID=51624111
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/058187 WO2014157140A1 (ja) | 2013-03-26 | 2014-03-25 | グリコリドの製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9512100B2 (ja) |
| EP (1) | EP2980084B1 (ja) |
| JP (1) | JP6230597B2 (ja) |
| KR (1) | KR101729877B1 (ja) |
| CN (1) | CN104903306B (ja) |
| WO (1) | WO2014157140A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019172361A1 (ja) | 2018-03-07 | 2019-09-12 | 株式会社クレハ | 環状エステルの製造方法 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10266512B2 (en) | 2017-08-10 | 2019-04-23 | Novus International, Inc. | Processes for preparing heteroatom containing cyclic dimers |
| JP7039345B2 (ja) | 2018-03-20 | 2022-03-22 | 株式会社クレハ | グリコリドの製造方法 |
| JP7064913B2 (ja) * | 2018-03-20 | 2022-05-11 | 株式会社クレハ | グリコリドの製造方法 |
| CN108707134A (zh) * | 2018-07-06 | 2018-10-26 | 天津市艾普伦生物科技有限公司 | 乙交酯的合成方法 |
| CN114478469B (zh) * | 2020-10-26 | 2023-08-04 | 中国石油化工股份有限公司 | 一种低含水量粗乙交酯的制备方法及其所得乙交酯 |
| CN112480063A (zh) * | 2020-11-28 | 2021-03-12 | 万华化学(四川)有限公司 | 一种制备低酸分丙交酯的反应工艺 |
| CN113603671A (zh) * | 2021-08-13 | 2021-11-05 | 山东谷雨春生物科技有限公司 | 一种提高交酯收率的方法 |
| CN114437020B (zh) * | 2022-02-23 | 2023-03-24 | 中国科学院长春应用化学研究所 | 一种乙交酯的制备方法 |
| CN114805283A (zh) * | 2022-04-29 | 2022-07-29 | 内蒙古久泰新材料有限公司 | 一种连续稳定制备高品质乙交酯的方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05287056A (ja) | 1992-04-07 | 1993-11-02 | Toyobo Co Ltd | 脂肪族ポリエステルの製造方法 |
| JPH06287278A (ja) * | 1993-04-07 | 1994-10-11 | Toyobo Co Ltd | 脂肪族ポリエステルの製造方法 |
| JPH07309862A (ja) * | 1994-05-17 | 1995-11-28 | Toyobo Co Ltd | ラクチドの製造方法 |
| JPH09328481A (ja) | 1996-02-09 | 1997-12-22 | Kureha Chem Ind Co Ltd | α−ヒドロキシカルボン酸2量体環状エステルの製造方法及び精製方法 |
| US5830991A (en) | 1996-02-09 | 1998-11-03 | Kureha Kagaku Kagyo Kk | Preparation process and purification process of dimeric cyclic ester of hydroxycarboxylic acid |
| WO2002014303A1 (fr) | 2000-08-11 | 2002-02-21 | Kureha Kagaku Kogyo K.K. | Procede de preparation d'esters cycliques et procede de purification desdits esters |
| US20040122240A1 (en) | 2001-04-12 | 2004-06-24 | Kazuyuki Yamane | Glycolide production process, and glycolic acid oligomer for glycolide production |
| WO2010073512A1 (ja) | 2008-12-26 | 2010-07-01 | 株式会社クレハ | グリコリドの製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5618911A (en) | 1993-08-19 | 1997-04-08 | Toyo Boseki Kabushiki Kaisha | Polymer containing lactic acid as its constituting unit and method for producing the same |
| CN100412190C (zh) | 2001-03-15 | 2008-08-20 | 复旦大学 | 乙肝病毒全基因组多聚酶嵌合体的组建方法和应用 |
| WO2011132537A1 (ja) * | 2010-04-20 | 2011-10-27 | 株式会社クレハ | 水中防汚材、溶融成形物及び塗料 |
| JP5438615B2 (ja) * | 2010-07-20 | 2014-03-12 | 株式会社クレハ | ポリグリコール酸を含有する油捕集材料 |
| JP1446209S (ja) | 2011-10-12 | 2015-07-06 |
-
2014
- 2014-03-25 US US14/766,829 patent/US9512100B2/en active Active
- 2014-03-25 JP JP2015508513A patent/JP6230597B2/ja active Active
- 2014-03-25 CN CN201480004299.6A patent/CN104903306B/zh active Active
- 2014-03-25 KR KR1020157018421A patent/KR101729877B1/ko active IP Right Grant
- 2014-03-25 WO PCT/JP2014/058187 patent/WO2014157140A1/ja active Application Filing
- 2014-03-25 EP EP14775676.1A patent/EP2980084B1/en active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05287056A (ja) | 1992-04-07 | 1993-11-02 | Toyobo Co Ltd | 脂肪族ポリエステルの製造方法 |
| JPH06287278A (ja) * | 1993-04-07 | 1994-10-11 | Toyobo Co Ltd | 脂肪族ポリエステルの製造方法 |
| JPH07309862A (ja) * | 1994-05-17 | 1995-11-28 | Toyobo Co Ltd | ラクチドの製造方法 |
| JPH09328481A (ja) | 1996-02-09 | 1997-12-22 | Kureha Chem Ind Co Ltd | α−ヒドロキシカルボン酸2量体環状エステルの製造方法及び精製方法 |
| US5830991A (en) | 1996-02-09 | 1998-11-03 | Kureha Kagaku Kagyo Kk | Preparation process and purification process of dimeric cyclic ester of hydroxycarboxylic acid |
| WO2002014303A1 (fr) | 2000-08-11 | 2002-02-21 | Kureha Kagaku Kogyo K.K. | Procede de preparation d'esters cycliques et procede de purification desdits esters |
| US20030191326A1 (en) | 2000-08-11 | 2003-10-09 | Kazuyuki Yamane | Process for the preparation of cyclic esters and method for purification of the same |
| US20040122240A1 (en) | 2001-04-12 | 2004-06-24 | Kazuyuki Yamane | Glycolide production process, and glycolic acid oligomer for glycolide production |
| JP2004523596A (ja) | 2001-04-12 | 2004-08-05 | 呉羽化学工業株式会社 | グリコリドの製造方法及びグリコリド製造用グリコール酸オリゴマー |
| WO2010073512A1 (ja) | 2008-12-26 | 2010-07-01 | 株式会社クレハ | グリコリドの製造方法 |
| US20110263875A1 (en) | 2008-12-26 | 2011-10-27 | Kureha Corporation | Production Process of Glycolide |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2980084A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019172361A1 (ja) | 2018-03-07 | 2019-09-12 | 株式会社クレハ | 環状エステルの製造方法 |
| JPWO2019172361A1 (ja) * | 2018-03-07 | 2020-07-30 | 株式会社クレハ | 環状エステルの製造方法 |
| US11078179B2 (en) | 2018-03-07 | 2021-08-03 | Kureha Corporation | Method for producing cyclic ester |
Also Published As
| Publication number | Publication date |
|---|---|
| US20160002196A1 (en) | 2016-01-07 |
| JPWO2014157140A1 (ja) | 2017-02-16 |
| EP2980084A1 (en) | 2016-02-03 |
| US9512100B2 (en) | 2016-12-06 |
| EP2980084A4 (en) | 2016-08-17 |
| JP6230597B2 (ja) | 2017-11-15 |
| EP2980084B1 (en) | 2018-12-05 |
| CN104903306B (zh) | 2017-12-22 |
| KR101729877B1 (ko) | 2017-04-24 |
| KR20150107736A (ko) | 2015-09-23 |
| CN104903306A (zh) | 2015-09-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6230597B2 (ja) | グリコリドの製造方法 | |
| JP5584628B2 (ja) | グリコリドの製造方法 | |
| JP5813516B2 (ja) | グリコリドの製造方法 | |
| JP4394352B2 (ja) | グリコリドの製造方法 | |
| JP5235311B2 (ja) | 環状エステルの精製方法 | |
| CN101495440B (zh) | 羟基羧酸的纯化方法、环状酯的制造方法和聚羟基羧酸的制造方法 | |
| JP4954616B2 (ja) | グリコリドの製造方法およびグリコリド製造用グリコール酸オリゴマー | |
| JP6912655B2 (ja) | グリコリドの製造方法 | |
| JP6326373B2 (ja) | 気液の向流接触による精留工程を備えるグリコリドの製造方法、及び、粗グリコリドの精製方法 | |
| JP4750256B2 (ja) | グリコリドの精製方法 | |
| JP7064913B2 (ja) | グリコリドの製造方法 | |
| JP2006342344A (ja) | ポリエーテルポリオールの製造方法 | |
| JP2014185116A (ja) | グリコリドの製造方法 | |
| WO2006121111A1 (ja) | ポリエーテルポリオールの製造方法 | |
| WO2013039038A1 (ja) | グリコリドの製造方法 | |
| JP2014185117A (ja) | α−ヒドロキシカルボン酸オリゴマーの解重合反応を行った後の反応液からの環状エステル及び溶媒の回収方法、並びに、環状エステルの製造方法 | |
| JP7039345B2 (ja) | グリコリドの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14775676 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015508513 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20157018421 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14766829 Country of ref document: US Ref document number: 2014775676 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |